User login
Illuminating the Role of Visible Light in Dermatology
Illuminating the Role of Visible Light in Dermatology
Visible light is part of the electromagnetic spectrum and is confined to a range of 400 to 700 nm. Visible light phototherapy can be delivered across various wavelengths within this spectrum, with most research focusing on blue light (BL)(400-500 nm) and red light (RL)(600-700 nm). Blue light commonly is used to treat acne as well as actinic keratosis and other inflammatory disorders,1,2 while RL largely targets signs of skin aging and fibrosis.2,3 Because of its shorter wavelength, the clinically meaningful skin penetration of BL reaches up to1 mm and is confined to the epidermis; in contrast, RL can access the dermal adnexa due to its penetration depth of more than 2 mm.4 Therapeutically, visible light can be utilized alone (eg, photobiomodulation [PBM]) or in combination with a photosensitizing agent (eg, photodynamic therapy [PDT]).5,6
Our laboratory’s prior research has contributed to a greater understanding of the safety profile of visible light at various wavelengths.1,3 Specifically, our work has shown that BL (417 nm [range, 412-422 nm]) and RL (633 nm [range, 627-639 nm]) demonstrated no evidence of DNA damage—via no formation of cyclobutane pyrimidine dimers and/or 6-4 photoproducts, the hallmark photolesions caused by UV exposure—in human dermal fibroblasts following visible light exposure at all fluences tested.1,3 This evidence reinforces the safety of visible light at clinically relevant wavelengths, supporting its integration into dermatologic practice. In this editorial, we highlight the key clinical applications of PBM and PDT and outline safety considerations for visible light-based therapies in dermatologic practice.
Photobiomodulation
Photobiomodulation is a noninvasive treatment in which low-level lasers or light-emitting diodes deliver photons from a nonionizing light source to endogenous photoreceptors, primarily cytochrome C oxidase.7-9 On the visible light spectrum, PBM primarily encompasses RL.7-9 Photoactivation leads to production of reactive oxygen species as well as mitochondrial alterations, with resulting modulation of cellular activity.7-9 Upregulation of cellular activity generally occurs at lower fluences (ie, energy delivered per unit area) of light, whereas higher fluences cause downregulation of cellular activity.5
Recent consensus guidelines, established with expert colleagues, define additional key parameters that are crucial to optimizing PBM treatment, including distance from the light source, area of the light beam, wavelength, length of treatment time, and number of treatments.5 Understanding the effects of different parameter combinations is essential for clinicians to select the best treatment regimen for each patient. Our laboratory has conducted National Institutes of Health–funded phase 1 and phase 2 clinical trials to determine the safety and efficacy of red-light PBM.10-13 Additionally, we completed several pilot phase 2 clinical studies with commercially available light-emitting diode face masks using PBM technology, which demonstrated a favorable safety profile and high patient satisfaction across multiple self-reported measures.14,15 These findings highlight PBM as a reliable and well-tolerated therapeutic approach that can be administered in clinical settings or by patients at home.
Adverse effects of PBM therapy generally are mild and transient, most commonly manifesting as slight irritation and erythema.5 Overall, PBM is widely regarded as safe with a favorable and nontoxic profile across treatment settings. Growing evidence supports the role of PBM in managing wound healing, acne, alopecia, and skin aging, among other dermatologic concerns.8
Photodynamic Therapy
Photodynamic therapy is a noninvasive procedure during which a photosensitizer—typically 5-aminolevulinic acid (5-ALA) or a derivative, methyl aminolevulinate—reacts with a light source and oxygen, resulting in reactive oxygen species.6,16 This reaction ultimately triggers targeted cellular destruction of the intended lesional skin but with negligible effects on adjacent nonlesional tissue.6 The efficacy of PDT is determined by several parameters, including composition and concentration of the photosensitizer, photosensitizer incubation temperature, and incubation time with the photosensitizer. Methyl aminolevulinate is a lipophilic molecule and may promote greater skin penetration and cellular uptake than 5-ALA, which is a hydrophilic molecule.6
Our research further demonstrated that apoptosis increases in a dose- and temperature-dependent manner following 5-ALA exposure, both in cutaneous and mucosal squamous cell carcinoma cells and in human dermal fibroblasts.17,18 Our mechanistic insights have clinical relevance, as evidenced by an independent pilot study demonstrating that temperature-modulated PDT significantly improved actinic keratosis lesion clearance rates (P<.0001).19 Additionally, we determined that even short periods of incubation with 5-ALA (ie, 15-30 minutes) result in statistically significant increases in apoptosis (P<.05).20 Thus, these findings highlight that the choice of photosensitizing agent and the administration parameters are critical in determining PDT efficacy as well as the need to optimize clinical protocols.
Photodynamic therapy also has demonstrated general clinical and genotoxic safety, with the most common potential adverse events limited to temporary inflammation, erythema, and discomfort.21 A study in murine skin and human keratinocytes revealed that 5-ALA PDT had a photoprotective effect against previous irradiation with UVB (a known inducer of DNA damage) via removal of cyclobutane pyrimidine dimers.22 Thus, PDT has been recognized as a safe and effective therapeutic modality with broad applications in dermatology, including treatment of actinic keratosis and nonmelanoma skin cancers.16
Clinical Safety, Photoprotection, and Precautions
While visible light has shown substantial therapeutic potential in dermatology, there are several safety measures and precautions to be aware of. Visible light constitutes approximately 44% of the solar output; therefore, precautions against both UV and visible light are recommended for the general population.23 Cumulative exposure to visible light has been shown to trigger melanogenesis, resulting in persistent erythema, hyperpigmentation, and uneven skin tones across all Fitzpatrick skin types.24 Individuals with skin of color are more photosensitive to visible light due to increased baseline melanin levels.24 Similarly, patients with pigmentary conditions such as melasma and postinflammatory hyperpigmentation may experience worsening of their dermatologic symptoms due to underlying visible light photosensitivity.25
Patients undergoing PBM or PDT could benefit from visible light protection. The primary form of photoprotection against visible light is tinted sunscreen, which contains iron oxides and titanium dioxide.26 Iron (III) oxide is capable of blocking nearly all visible light damage.26 Use of physical barriers such as wavelength-specific sunglasses and wide-brimmed hats also is important for preventing photodamage from visible light.26
Final Thoughts
Visible light has a role in the treatment of a variety of skin conditions, including actinic keratosis, nonmelanoma skin cancers, acne, wound healing, skin fibrosis, and photodamage. Photobiomodulation and PDT represent 2 noninvasive phototherapeutic options that utilize visible light to enact cellular changes necessary to improve skin health. Integrating visible light phototherapy into standard clinical practice is important for enhancing patient outcomes. Clinicians should remain mindful of the rare pigmentary risks associated with visible light therapy devices. Future research should prioritize optimization of standardized protocols and expansion of clinical indications for visible light phototherapy.
- Kabakova M, Wang J, Stolyar J, et al. Visible blue light does not induce DNA damage in human dermal fibroblasts. J Biophotonics. 2025;18:E202400510. doi:10.1002/jbio.202400510
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163. doi:10.2147/CCID.S35334
- Wang JY, Austin E, Jagdeo J. Visible red light does not induce DNA damage in human dermal fibroblasts. J Biophotonics. 2022;15:E202200023. doi:10.1002/jbio.202200023
- Opel DR, Hagstrom E, Pace AK, et al. Light-emitting diodes: a brief review and clinical experience. J Clin Aesthet Dermatol. 2015;8:36-44.
- Maghfour J, Mineroff J, Ozog DM, et al. Evidence-based consensus on the clinical application of photobiomodulation. J Am Acad Dermatol. 2025;93:429-443. doi:10.1016/j.jaad.2025.04.031
- Ozog DM, Rkein AM, Fabi SG, et al. Photodynamic therapy: a clinical consensus guide. Dermatol Surg. 2016;42:804-827. doi:10.1097/DSS.0000000000000800
- Maghfour J, Ozog DM, Mineroff J, et al. Photobiomodulation CME part I: overview and mechanism of action. J Am Acad Dermatol. 2024;91:793-802. doi:10.1016/j.jaad.2023.10.073
- Mineroff J, Maghfour J, Ozog DM, et al. Photobiomodulation CME part II: clinical applications in dermatology. J Am Acad Dermatol. 2024;91:805-815. doi:10.1016/j.jaad.2023.10.074
- Mamalis A, Siegel D, Jagdeo J. Visible red light emitting diode photobiomodulation for skin fibrosis: key molecular pathways. Curr Dermatol Rep. 2016;5:121-128. doi:10.1007/s13671-016-0141-x
- Kurtti A, Nguyen JK, Weedon J, et al. Light emitting diode-red light for reduction of post-surgical scarring: results from a dose-ranging, split-face, randomized controlled trial. J Biophotonics. 2021;14:E202100073. doi:10.1002/jbio.202100073
- Nguyen JK, Weedon J, Jakus J, et al. A dose-ranging, parallel group, split-face, single-blind phase II study of light emitting diode-red light (LED-RL) for skin scarring prevention: study protocol for a randomized controlled trial. Trials. 2019;20:432. doi:10.1186/s13063-019-3546-6
- Ho D, Kraeva E, Wun T, et al. A single-blind, dose escalation, phase I study of high-fluence light-emitting diode-red light (LED-RL) on human skin: study protocol for a randomized controlled trial. Trials. 2016;17:385. doi:10.1186/s13063-016-1518-7
- Wang EB, Kaur R, Nguyen J, et al. A single-blind, dose-escalation, phase I study of high-fluence light-emitting diode-red light on Caucasian non-Hispanic skin: study protocol for a randomized controlled trial. Trials. 2019;20:177. doi:10.1186/s13063-019-3278-7
- Wang JY, Kabakova M, Patel P, et al. Outstanding user reported satisfaction for light emitting diodes under-eye rejuvenation. Arch Dermatol Res. 2024;316:511. doi:10.1007/s00403-024-03254-z
- Mineroff J, Austin E, Feit E, et al. Male facial rejuvenation using a combination 633, 830, and 1072 nm LED face mask. Arch Dermatol Res. 2023;315:2605-2611. doi:10.1007/s00403-023-02663-w
- Wang JY, Zeitouni N, Austin E, et al. Photodynamic therapy: clinical applications in dermatology. J Am Acad Dermatol. Published online February 20, 2025. doi:10.1016/j.jaad.2024.12.050
- Austin E, Koo E, Jagdeo J. Thermal photodynamic therapy increases apoptosis and reactive oxygen species generation in cutaneous and mucosal squamous cell carcinoma cells. Sci Rep. 2018;8:12599. doi:10.1038/s41598-018-30908-6
- Mamalis A, Koo E, Sckisel GD, et al. Temperature-dependent impact of thermal aminolaevulinic acid photodynamic therapy on apoptosis and reactive oxygen species generation in human dermal fibroblasts. Br J Dermatol. 2016;175:512-519. doi:10.1111/bjd.14509
- Willey A, Anderson RR, Sakamoto FH. Temperature-modulated photodynamic therapy for the treatment of actinic keratosis on the extremities: a pilot study. Dermatol Surg. 2014;40:1094-1102. doi:10.1097/01.DSS.0000452662.69539.57
- Koo E, Austin E, Mamalis A, et al. Efficacy of ultra short sub-30 minute incubation of 5-aminolevulinic acid photodynamic therapy in vitro. Lasers Surg Med. 2017;49:592-598. doi:10.1002/lsm.22648
- Austin E, Wang JY, Ozog DM, et al. Photodynamic therapy: overview and mechanism of action. J Am Acad Dermatol. Published online February 20, 2025. doi:10.1016/j.jaad.2025.02.037
- Hua H, Cheng JW, Bu WB, et al. 5-aminolaevulinic acid-based photodynamic therapy inhibits ultraviolet B-induced skin photodamage. Int J Biol Sci. 2019;15:2100-2109. doi:10.7150/ijbs.31583
- Liebel F, Kaur S, Ruvolo E, et al. Irradiation of skin with visible light induces reactive oxygen species and matrix-degrading enzymes. J Invest Dermatol. 2012;132:1901-1907. doi:10.1038/jid.2011.476
- Austin E, Geisler AN, Nguyen J, et al. Visible light. part I: properties and cutaneous effects of visible light. J Am Acad Dermatol. 2021;84:1219-1231. doi:10.1016/j.jaad.2021.02.048
- Fatima S, Braunberger T, Mohammad TF, et al. The role of sunscreen in melasma and postinflammatory hyperpigmentation. Indian J Dermatol. 2020;65:5-10. doi:10.4103/ijd.IJD_295_18
- Geisler AN, Austin E, Nguyen J, et al. Visible light. part II: photoprotection against visible and ultraviolet light. J Am Acad Dermatol. 2021;84:1233-1244. doi:10.1016/j.jaad.2020.11.074
Visible light is part of the electromagnetic spectrum and is confined to a range of 400 to 700 nm. Visible light phototherapy can be delivered across various wavelengths within this spectrum, with most research focusing on blue light (BL)(400-500 nm) and red light (RL)(600-700 nm). Blue light commonly is used to treat acne as well as actinic keratosis and other inflammatory disorders,1,2 while RL largely targets signs of skin aging and fibrosis.2,3 Because of its shorter wavelength, the clinically meaningful skin penetration of BL reaches up to1 mm and is confined to the epidermis; in contrast, RL can access the dermal adnexa due to its penetration depth of more than 2 mm.4 Therapeutically, visible light can be utilized alone (eg, photobiomodulation [PBM]) or in combination with a photosensitizing agent (eg, photodynamic therapy [PDT]).5,6
Our laboratory’s prior research has contributed to a greater understanding of the safety profile of visible light at various wavelengths.1,3 Specifically, our work has shown that BL (417 nm [range, 412-422 nm]) and RL (633 nm [range, 627-639 nm]) demonstrated no evidence of DNA damage—via no formation of cyclobutane pyrimidine dimers and/or 6-4 photoproducts, the hallmark photolesions caused by UV exposure—in human dermal fibroblasts following visible light exposure at all fluences tested.1,3 This evidence reinforces the safety of visible light at clinically relevant wavelengths, supporting its integration into dermatologic practice. In this editorial, we highlight the key clinical applications of PBM and PDT and outline safety considerations for visible light-based therapies in dermatologic practice.
Photobiomodulation
Photobiomodulation is a noninvasive treatment in which low-level lasers or light-emitting diodes deliver photons from a nonionizing light source to endogenous photoreceptors, primarily cytochrome C oxidase.7-9 On the visible light spectrum, PBM primarily encompasses RL.7-9 Photoactivation leads to production of reactive oxygen species as well as mitochondrial alterations, with resulting modulation of cellular activity.7-9 Upregulation of cellular activity generally occurs at lower fluences (ie, energy delivered per unit area) of light, whereas higher fluences cause downregulation of cellular activity.5
Recent consensus guidelines, established with expert colleagues, define additional key parameters that are crucial to optimizing PBM treatment, including distance from the light source, area of the light beam, wavelength, length of treatment time, and number of treatments.5 Understanding the effects of different parameter combinations is essential for clinicians to select the best treatment regimen for each patient. Our laboratory has conducted National Institutes of Health–funded phase 1 and phase 2 clinical trials to determine the safety and efficacy of red-light PBM.10-13 Additionally, we completed several pilot phase 2 clinical studies with commercially available light-emitting diode face masks using PBM technology, which demonstrated a favorable safety profile and high patient satisfaction across multiple self-reported measures.14,15 These findings highlight PBM as a reliable and well-tolerated therapeutic approach that can be administered in clinical settings or by patients at home.
Adverse effects of PBM therapy generally are mild and transient, most commonly manifesting as slight irritation and erythema.5 Overall, PBM is widely regarded as safe with a favorable and nontoxic profile across treatment settings. Growing evidence supports the role of PBM in managing wound healing, acne, alopecia, and skin aging, among other dermatologic concerns.8
Photodynamic Therapy
Photodynamic therapy is a noninvasive procedure during which a photosensitizer—typically 5-aminolevulinic acid (5-ALA) or a derivative, methyl aminolevulinate—reacts with a light source and oxygen, resulting in reactive oxygen species.6,16 This reaction ultimately triggers targeted cellular destruction of the intended lesional skin but with negligible effects on adjacent nonlesional tissue.6 The efficacy of PDT is determined by several parameters, including composition and concentration of the photosensitizer, photosensitizer incubation temperature, and incubation time with the photosensitizer. Methyl aminolevulinate is a lipophilic molecule and may promote greater skin penetration and cellular uptake than 5-ALA, which is a hydrophilic molecule.6
Our research further demonstrated that apoptosis increases in a dose- and temperature-dependent manner following 5-ALA exposure, both in cutaneous and mucosal squamous cell carcinoma cells and in human dermal fibroblasts.17,18 Our mechanistic insights have clinical relevance, as evidenced by an independent pilot study demonstrating that temperature-modulated PDT significantly improved actinic keratosis lesion clearance rates (P<.0001).19 Additionally, we determined that even short periods of incubation with 5-ALA (ie, 15-30 minutes) result in statistically significant increases in apoptosis (P<.05).20 Thus, these findings highlight that the choice of photosensitizing agent and the administration parameters are critical in determining PDT efficacy as well as the need to optimize clinical protocols.
Photodynamic therapy also has demonstrated general clinical and genotoxic safety, with the most common potential adverse events limited to temporary inflammation, erythema, and discomfort.21 A study in murine skin and human keratinocytes revealed that 5-ALA PDT had a photoprotective effect against previous irradiation with UVB (a known inducer of DNA damage) via removal of cyclobutane pyrimidine dimers.22 Thus, PDT has been recognized as a safe and effective therapeutic modality with broad applications in dermatology, including treatment of actinic keratosis and nonmelanoma skin cancers.16
Clinical Safety, Photoprotection, and Precautions
While visible light has shown substantial therapeutic potential in dermatology, there are several safety measures and precautions to be aware of. Visible light constitutes approximately 44% of the solar output; therefore, precautions against both UV and visible light are recommended for the general population.23 Cumulative exposure to visible light has been shown to trigger melanogenesis, resulting in persistent erythema, hyperpigmentation, and uneven skin tones across all Fitzpatrick skin types.24 Individuals with skin of color are more photosensitive to visible light due to increased baseline melanin levels.24 Similarly, patients with pigmentary conditions such as melasma and postinflammatory hyperpigmentation may experience worsening of their dermatologic symptoms due to underlying visible light photosensitivity.25
Patients undergoing PBM or PDT could benefit from visible light protection. The primary form of photoprotection against visible light is tinted sunscreen, which contains iron oxides and titanium dioxide.26 Iron (III) oxide is capable of blocking nearly all visible light damage.26 Use of physical barriers such as wavelength-specific sunglasses and wide-brimmed hats also is important for preventing photodamage from visible light.26
Final Thoughts
Visible light has a role in the treatment of a variety of skin conditions, including actinic keratosis, nonmelanoma skin cancers, acne, wound healing, skin fibrosis, and photodamage. Photobiomodulation and PDT represent 2 noninvasive phototherapeutic options that utilize visible light to enact cellular changes necessary to improve skin health. Integrating visible light phototherapy into standard clinical practice is important for enhancing patient outcomes. Clinicians should remain mindful of the rare pigmentary risks associated with visible light therapy devices. Future research should prioritize optimization of standardized protocols and expansion of clinical indications for visible light phototherapy.
Visible light is part of the electromagnetic spectrum and is confined to a range of 400 to 700 nm. Visible light phototherapy can be delivered across various wavelengths within this spectrum, with most research focusing on blue light (BL)(400-500 nm) and red light (RL)(600-700 nm). Blue light commonly is used to treat acne as well as actinic keratosis and other inflammatory disorders,1,2 while RL largely targets signs of skin aging and fibrosis.2,3 Because of its shorter wavelength, the clinically meaningful skin penetration of BL reaches up to1 mm and is confined to the epidermis; in contrast, RL can access the dermal adnexa due to its penetration depth of more than 2 mm.4 Therapeutically, visible light can be utilized alone (eg, photobiomodulation [PBM]) or in combination with a photosensitizing agent (eg, photodynamic therapy [PDT]).5,6
Our laboratory’s prior research has contributed to a greater understanding of the safety profile of visible light at various wavelengths.1,3 Specifically, our work has shown that BL (417 nm [range, 412-422 nm]) and RL (633 nm [range, 627-639 nm]) demonstrated no evidence of DNA damage—via no formation of cyclobutane pyrimidine dimers and/or 6-4 photoproducts, the hallmark photolesions caused by UV exposure—in human dermal fibroblasts following visible light exposure at all fluences tested.1,3 This evidence reinforces the safety of visible light at clinically relevant wavelengths, supporting its integration into dermatologic practice. In this editorial, we highlight the key clinical applications of PBM and PDT and outline safety considerations for visible light-based therapies in dermatologic practice.
Photobiomodulation
Photobiomodulation is a noninvasive treatment in which low-level lasers or light-emitting diodes deliver photons from a nonionizing light source to endogenous photoreceptors, primarily cytochrome C oxidase.7-9 On the visible light spectrum, PBM primarily encompasses RL.7-9 Photoactivation leads to production of reactive oxygen species as well as mitochondrial alterations, with resulting modulation of cellular activity.7-9 Upregulation of cellular activity generally occurs at lower fluences (ie, energy delivered per unit area) of light, whereas higher fluences cause downregulation of cellular activity.5
Recent consensus guidelines, established with expert colleagues, define additional key parameters that are crucial to optimizing PBM treatment, including distance from the light source, area of the light beam, wavelength, length of treatment time, and number of treatments.5 Understanding the effects of different parameter combinations is essential for clinicians to select the best treatment regimen for each patient. Our laboratory has conducted National Institutes of Health–funded phase 1 and phase 2 clinical trials to determine the safety and efficacy of red-light PBM.10-13 Additionally, we completed several pilot phase 2 clinical studies with commercially available light-emitting diode face masks using PBM technology, which demonstrated a favorable safety profile and high patient satisfaction across multiple self-reported measures.14,15 These findings highlight PBM as a reliable and well-tolerated therapeutic approach that can be administered in clinical settings or by patients at home.
Adverse effects of PBM therapy generally are mild and transient, most commonly manifesting as slight irritation and erythema.5 Overall, PBM is widely regarded as safe with a favorable and nontoxic profile across treatment settings. Growing evidence supports the role of PBM in managing wound healing, acne, alopecia, and skin aging, among other dermatologic concerns.8
Photodynamic Therapy
Photodynamic therapy is a noninvasive procedure during which a photosensitizer—typically 5-aminolevulinic acid (5-ALA) or a derivative, methyl aminolevulinate—reacts with a light source and oxygen, resulting in reactive oxygen species.6,16 This reaction ultimately triggers targeted cellular destruction of the intended lesional skin but with negligible effects on adjacent nonlesional tissue.6 The efficacy of PDT is determined by several parameters, including composition and concentration of the photosensitizer, photosensitizer incubation temperature, and incubation time with the photosensitizer. Methyl aminolevulinate is a lipophilic molecule and may promote greater skin penetration and cellular uptake than 5-ALA, which is a hydrophilic molecule.6
Our research further demonstrated that apoptosis increases in a dose- and temperature-dependent manner following 5-ALA exposure, both in cutaneous and mucosal squamous cell carcinoma cells and in human dermal fibroblasts.17,18 Our mechanistic insights have clinical relevance, as evidenced by an independent pilot study demonstrating that temperature-modulated PDT significantly improved actinic keratosis lesion clearance rates (P<.0001).19 Additionally, we determined that even short periods of incubation with 5-ALA (ie, 15-30 minutes) result in statistically significant increases in apoptosis (P<.05).20 Thus, these findings highlight that the choice of photosensitizing agent and the administration parameters are critical in determining PDT efficacy as well as the need to optimize clinical protocols.
Photodynamic therapy also has demonstrated general clinical and genotoxic safety, with the most common potential adverse events limited to temporary inflammation, erythema, and discomfort.21 A study in murine skin and human keratinocytes revealed that 5-ALA PDT had a photoprotective effect against previous irradiation with UVB (a known inducer of DNA damage) via removal of cyclobutane pyrimidine dimers.22 Thus, PDT has been recognized as a safe and effective therapeutic modality with broad applications in dermatology, including treatment of actinic keratosis and nonmelanoma skin cancers.16
Clinical Safety, Photoprotection, and Precautions
While visible light has shown substantial therapeutic potential in dermatology, there are several safety measures and precautions to be aware of. Visible light constitutes approximately 44% of the solar output; therefore, precautions against both UV and visible light are recommended for the general population.23 Cumulative exposure to visible light has been shown to trigger melanogenesis, resulting in persistent erythema, hyperpigmentation, and uneven skin tones across all Fitzpatrick skin types.24 Individuals with skin of color are more photosensitive to visible light due to increased baseline melanin levels.24 Similarly, patients with pigmentary conditions such as melasma and postinflammatory hyperpigmentation may experience worsening of their dermatologic symptoms due to underlying visible light photosensitivity.25
Patients undergoing PBM or PDT could benefit from visible light protection. The primary form of photoprotection against visible light is tinted sunscreen, which contains iron oxides and titanium dioxide.26 Iron (III) oxide is capable of blocking nearly all visible light damage.26 Use of physical barriers such as wavelength-specific sunglasses and wide-brimmed hats also is important for preventing photodamage from visible light.26
Final Thoughts
Visible light has a role in the treatment of a variety of skin conditions, including actinic keratosis, nonmelanoma skin cancers, acne, wound healing, skin fibrosis, and photodamage. Photobiomodulation and PDT represent 2 noninvasive phototherapeutic options that utilize visible light to enact cellular changes necessary to improve skin health. Integrating visible light phototherapy into standard clinical practice is important for enhancing patient outcomes. Clinicians should remain mindful of the rare pigmentary risks associated with visible light therapy devices. Future research should prioritize optimization of standardized protocols and expansion of clinical indications for visible light phototherapy.
- Kabakova M, Wang J, Stolyar J, et al. Visible blue light does not induce DNA damage in human dermal fibroblasts. J Biophotonics. 2025;18:E202400510. doi:10.1002/jbio.202400510
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163. doi:10.2147/CCID.S35334
- Wang JY, Austin E, Jagdeo J. Visible red light does not induce DNA damage in human dermal fibroblasts. J Biophotonics. 2022;15:E202200023. doi:10.1002/jbio.202200023
- Opel DR, Hagstrom E, Pace AK, et al. Light-emitting diodes: a brief review and clinical experience. J Clin Aesthet Dermatol. 2015;8:36-44.
- Maghfour J, Mineroff J, Ozog DM, et al. Evidence-based consensus on the clinical application of photobiomodulation. J Am Acad Dermatol. 2025;93:429-443. doi:10.1016/j.jaad.2025.04.031
- Ozog DM, Rkein AM, Fabi SG, et al. Photodynamic therapy: a clinical consensus guide. Dermatol Surg. 2016;42:804-827. doi:10.1097/DSS.0000000000000800
- Maghfour J, Ozog DM, Mineroff J, et al. Photobiomodulation CME part I: overview and mechanism of action. J Am Acad Dermatol. 2024;91:793-802. doi:10.1016/j.jaad.2023.10.073
- Mineroff J, Maghfour J, Ozog DM, et al. Photobiomodulation CME part II: clinical applications in dermatology. J Am Acad Dermatol. 2024;91:805-815. doi:10.1016/j.jaad.2023.10.074
- Mamalis A, Siegel D, Jagdeo J. Visible red light emitting diode photobiomodulation for skin fibrosis: key molecular pathways. Curr Dermatol Rep. 2016;5:121-128. doi:10.1007/s13671-016-0141-x
- Kurtti A, Nguyen JK, Weedon J, et al. Light emitting diode-red light for reduction of post-surgical scarring: results from a dose-ranging, split-face, randomized controlled trial. J Biophotonics. 2021;14:E202100073. doi:10.1002/jbio.202100073
- Nguyen JK, Weedon J, Jakus J, et al. A dose-ranging, parallel group, split-face, single-blind phase II study of light emitting diode-red light (LED-RL) for skin scarring prevention: study protocol for a randomized controlled trial. Trials. 2019;20:432. doi:10.1186/s13063-019-3546-6
- Ho D, Kraeva E, Wun T, et al. A single-blind, dose escalation, phase I study of high-fluence light-emitting diode-red light (LED-RL) on human skin: study protocol for a randomized controlled trial. Trials. 2016;17:385. doi:10.1186/s13063-016-1518-7
- Wang EB, Kaur R, Nguyen J, et al. A single-blind, dose-escalation, phase I study of high-fluence light-emitting diode-red light on Caucasian non-Hispanic skin: study protocol for a randomized controlled trial. Trials. 2019;20:177. doi:10.1186/s13063-019-3278-7
- Wang JY, Kabakova M, Patel P, et al. Outstanding user reported satisfaction for light emitting diodes under-eye rejuvenation. Arch Dermatol Res. 2024;316:511. doi:10.1007/s00403-024-03254-z
- Mineroff J, Austin E, Feit E, et al. Male facial rejuvenation using a combination 633, 830, and 1072 nm LED face mask. Arch Dermatol Res. 2023;315:2605-2611. doi:10.1007/s00403-023-02663-w
- Wang JY, Zeitouni N, Austin E, et al. Photodynamic therapy: clinical applications in dermatology. J Am Acad Dermatol. Published online February 20, 2025. doi:10.1016/j.jaad.2024.12.050
- Austin E, Koo E, Jagdeo J. Thermal photodynamic therapy increases apoptosis and reactive oxygen species generation in cutaneous and mucosal squamous cell carcinoma cells. Sci Rep. 2018;8:12599. doi:10.1038/s41598-018-30908-6
- Mamalis A, Koo E, Sckisel GD, et al. Temperature-dependent impact of thermal aminolaevulinic acid photodynamic therapy on apoptosis and reactive oxygen species generation in human dermal fibroblasts. Br J Dermatol. 2016;175:512-519. doi:10.1111/bjd.14509
- Willey A, Anderson RR, Sakamoto FH. Temperature-modulated photodynamic therapy for the treatment of actinic keratosis on the extremities: a pilot study. Dermatol Surg. 2014;40:1094-1102. doi:10.1097/01.DSS.0000452662.69539.57
- Koo E, Austin E, Mamalis A, et al. Efficacy of ultra short sub-30 minute incubation of 5-aminolevulinic acid photodynamic therapy in vitro. Lasers Surg Med. 2017;49:592-598. doi:10.1002/lsm.22648
- Austin E, Wang JY, Ozog DM, et al. Photodynamic therapy: overview and mechanism of action. J Am Acad Dermatol. Published online February 20, 2025. doi:10.1016/j.jaad.2025.02.037
- Hua H, Cheng JW, Bu WB, et al. 5-aminolaevulinic acid-based photodynamic therapy inhibits ultraviolet B-induced skin photodamage. Int J Biol Sci. 2019;15:2100-2109. doi:10.7150/ijbs.31583
- Liebel F, Kaur S, Ruvolo E, et al. Irradiation of skin with visible light induces reactive oxygen species and matrix-degrading enzymes. J Invest Dermatol. 2012;132:1901-1907. doi:10.1038/jid.2011.476
- Austin E, Geisler AN, Nguyen J, et al. Visible light. part I: properties and cutaneous effects of visible light. J Am Acad Dermatol. 2021;84:1219-1231. doi:10.1016/j.jaad.2021.02.048
- Fatima S, Braunberger T, Mohammad TF, et al. The role of sunscreen in melasma and postinflammatory hyperpigmentation. Indian J Dermatol. 2020;65:5-10. doi:10.4103/ijd.IJD_295_18
- Geisler AN, Austin E, Nguyen J, et al. Visible light. part II: photoprotection against visible and ultraviolet light. J Am Acad Dermatol. 2021;84:1233-1244. doi:10.1016/j.jaad.2020.11.074
- Kabakova M, Wang J, Stolyar J, et al. Visible blue light does not induce DNA damage in human dermal fibroblasts. J Biophotonics. 2025;18:E202400510. doi:10.1002/jbio.202400510
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163. doi:10.2147/CCID.S35334
- Wang JY, Austin E, Jagdeo J. Visible red light does not induce DNA damage in human dermal fibroblasts. J Biophotonics. 2022;15:E202200023. doi:10.1002/jbio.202200023
- Opel DR, Hagstrom E, Pace AK, et al. Light-emitting diodes: a brief review and clinical experience. J Clin Aesthet Dermatol. 2015;8:36-44.
- Maghfour J, Mineroff J, Ozog DM, et al. Evidence-based consensus on the clinical application of photobiomodulation. J Am Acad Dermatol. 2025;93:429-443. doi:10.1016/j.jaad.2025.04.031
- Ozog DM, Rkein AM, Fabi SG, et al. Photodynamic therapy: a clinical consensus guide. Dermatol Surg. 2016;42:804-827. doi:10.1097/DSS.0000000000000800
- Maghfour J, Ozog DM, Mineroff J, et al. Photobiomodulation CME part I: overview and mechanism of action. J Am Acad Dermatol. 2024;91:793-802. doi:10.1016/j.jaad.2023.10.073
- Mineroff J, Maghfour J, Ozog DM, et al. Photobiomodulation CME part II: clinical applications in dermatology. J Am Acad Dermatol. 2024;91:805-815. doi:10.1016/j.jaad.2023.10.074
- Mamalis A, Siegel D, Jagdeo J. Visible red light emitting diode photobiomodulation for skin fibrosis: key molecular pathways. Curr Dermatol Rep. 2016;5:121-128. doi:10.1007/s13671-016-0141-x
- Kurtti A, Nguyen JK, Weedon J, et al. Light emitting diode-red light for reduction of post-surgical scarring: results from a dose-ranging, split-face, randomized controlled trial. J Biophotonics. 2021;14:E202100073. doi:10.1002/jbio.202100073
- Nguyen JK, Weedon J, Jakus J, et al. A dose-ranging, parallel group, split-face, single-blind phase II study of light emitting diode-red light (LED-RL) for skin scarring prevention: study protocol for a randomized controlled trial. Trials. 2019;20:432. doi:10.1186/s13063-019-3546-6
- Ho D, Kraeva E, Wun T, et al. A single-blind, dose escalation, phase I study of high-fluence light-emitting diode-red light (LED-RL) on human skin: study protocol for a randomized controlled trial. Trials. 2016;17:385. doi:10.1186/s13063-016-1518-7
- Wang EB, Kaur R, Nguyen J, et al. A single-blind, dose-escalation, phase I study of high-fluence light-emitting diode-red light on Caucasian non-Hispanic skin: study protocol for a randomized controlled trial. Trials. 2019;20:177. doi:10.1186/s13063-019-3278-7
- Wang JY, Kabakova M, Patel P, et al. Outstanding user reported satisfaction for light emitting diodes under-eye rejuvenation. Arch Dermatol Res. 2024;316:511. doi:10.1007/s00403-024-03254-z
- Mineroff J, Austin E, Feit E, et al. Male facial rejuvenation using a combination 633, 830, and 1072 nm LED face mask. Arch Dermatol Res. 2023;315:2605-2611. doi:10.1007/s00403-023-02663-w
- Wang JY, Zeitouni N, Austin E, et al. Photodynamic therapy: clinical applications in dermatology. J Am Acad Dermatol. Published online February 20, 2025. doi:10.1016/j.jaad.2024.12.050
- Austin E, Koo E, Jagdeo J. Thermal photodynamic therapy increases apoptosis and reactive oxygen species generation in cutaneous and mucosal squamous cell carcinoma cells. Sci Rep. 2018;8:12599. doi:10.1038/s41598-018-30908-6
- Mamalis A, Koo E, Sckisel GD, et al. Temperature-dependent impact of thermal aminolaevulinic acid photodynamic therapy on apoptosis and reactive oxygen species generation in human dermal fibroblasts. Br J Dermatol. 2016;175:512-519. doi:10.1111/bjd.14509
- Willey A, Anderson RR, Sakamoto FH. Temperature-modulated photodynamic therapy for the treatment of actinic keratosis on the extremities: a pilot study. Dermatol Surg. 2014;40:1094-1102. doi:10.1097/01.DSS.0000452662.69539.57
- Koo E, Austin E, Mamalis A, et al. Efficacy of ultra short sub-30 minute incubation of 5-aminolevulinic acid photodynamic therapy in vitro. Lasers Surg Med. 2017;49:592-598. doi:10.1002/lsm.22648
- Austin E, Wang JY, Ozog DM, et al. Photodynamic therapy: overview and mechanism of action. J Am Acad Dermatol. Published online February 20, 2025. doi:10.1016/j.jaad.2025.02.037
- Hua H, Cheng JW, Bu WB, et al. 5-aminolaevulinic acid-based photodynamic therapy inhibits ultraviolet B-induced skin photodamage. Int J Biol Sci. 2019;15:2100-2109. doi:10.7150/ijbs.31583
- Liebel F, Kaur S, Ruvolo E, et al. Irradiation of skin with visible light induces reactive oxygen species and matrix-degrading enzymes. J Invest Dermatol. 2012;132:1901-1907. doi:10.1038/jid.2011.476
- Austin E, Geisler AN, Nguyen J, et al. Visible light. part I: properties and cutaneous effects of visible light. J Am Acad Dermatol. 2021;84:1219-1231. doi:10.1016/j.jaad.2021.02.048
- Fatima S, Braunberger T, Mohammad TF, et al. The role of sunscreen in melasma and postinflammatory hyperpigmentation. Indian J Dermatol. 2020;65:5-10. doi:10.4103/ijd.IJD_295_18
- Geisler AN, Austin E, Nguyen J, et al. Visible light. part II: photoprotection against visible and ultraviolet light. J Am Acad Dermatol. 2021;84:1233-1244. doi:10.1016/j.jaad.2020.11.074
Illuminating the Role of Visible Light in Dermatology
Illuminating the Role of Visible Light in Dermatology
Dermatology on Duty: Pathways to a Career in Military Medicine
Dermatology on Duty: Pathways to a Career in Military Medicine
Serving those who serve has been one of the most meaningful parts of my career. A career in military medicine offers dermatologists not only a chance to practice within a unique and diverse patient population but also an opportunity to contribute to something larger than themselves. Whether working with active-duty service members and their families within the Military Health System (MHS) or caring for veterans through the Department of Veterans Affairs (VA), the experience can be both enriching and rewarding. This article will explore the various pathways available to dermatologists to serve military communities, whether they are at the start of their careers or are looking for a change of pace within their established practice.
Care Pathways for Military and Veterans
To care for uniformed service members, their families, and retired personnel, dermatologists typically serve within the MHS—a global, integrated network of military hospitals and clinics dedicated to delivering health care to this population.1 TRICARE is the health insurance program that covers those eligible for care within the system, including active-duty and retired service members.2 In this context, it is important to clarify what the term retired actually means, as it differs from the term veteran when it comes to accessing health care options, and these terms frequently are conflated. A retired service member is an individual who completed at least 20 years of active-duty service or who has been medically retired because of a condition or injury incurred while on active duty.3 In contrast, a veteran may not have completed 20 years of service but has separated honorably after serving at least 24 continuous months.4 Veterans typically receive care through the VA system.5
Serving on Active Duty
In general, there are 2 main pathways to serve as a dermatologist within the MHS. The first is to commission in the military and serve on active duty. Most often, this pathway begins with a premedical student applying to medical school. Those considering military service typically explore scholarship programs such as the Health Professions Scholarship Program (HPSP)(https://www.medicineandthemilitary.com/applying-and-what-to-expect/medical-school-programs/hpsp) or the Health Services Collegiate Program (HSCP), or they apply to the Uniformed Services University of the Health Sciences (USU)(https://www.usuhs.edu/about). The HPSP and HSCP programs financially support medical students training at civilian medical schools, though in different ways—the HPSP covers tuition and fees, while the HSCP provides a salary during training but does not cover tuition.6 In contrast, students of USU attend the nation’s only military medical school, serving in uniform for 4 years while earning the pay and benefits of a junior officer in their respective service branch. Any premedical student considering the HPSP, HSCP or USU routes for service must meet the commissioning standards of their chosen branch—Army, Navy, or Air Force—and enter service as an officer before beginning medical school.
While direct commission prior to medical school is the most common route to active-duty service, board-certified dermatologists also can join a military branch later through what is called Direct Accession or Direct Commission; for example, the Navy offers a Residency to Direct Accession program, which commissions residents in their final year of training to join the Navy upon graduation. In some cases, commissioning at this stage includes a bonus of up to $600,000 in exchange for a 4-year active-duty commitment.7 The Army and Air Force offer similar direct commission programs, though specific incentives vary.8 Interested residents or practitioners can contact a local recruiting office within their branch of interest to learn more. Direct accession is open at many points in a dermatologist’s career—after residency, after fellowship, or even as an established civilian practitioner—and the initial commissioning rank and bonus generally reflect one’s level of experience.
Serving as a Civilian
Outside of uniformed service, dermatologists can find opportunities to provide care for active-duty service members, veterans, and military families through employment as General Schedule (GS) employees. The GS is a role classification and pay system that covers most federal employees in professional, administrative, and technical positions (eg, physicians). The GS system classifies most of these employees based on the complexity, responsibility, and qualifications required for their role.9 Such positions often are at the highest level of the GS pay scale, reflecting the expertise and years of education required to become a dermatologist, though pay varies by location and experience. In contrast, physicians employed through the VA system are classified as Title 38 federal employees, governed by a different pay structure and regulatory framework under the US Code of Federal Regulations.10 These regulations govern the hiring, retention, and firing guidelines for VA physicians, which differ from those of GS physicians. A full explanation is outside of the scope of this article, however.
Final Thoughts
In summary, uniformed or federal service as a dermatologist offers a meaningful and impactful way to give back to those who have served our country. Opportunities exist throughout the United States for dermatologists interested in serving within the MHS or VA. The most transparent and up-to-date resource for identifying open positions in both large metropolitan areas and smaller communities is USAJOBS.gov. While financial compensation may not always match that of private practice, the intangible benefits are considerable—stable employment, comprehensive benefits, malpractice coverage, and secure retirement, among others. There is something deeply fulfilling about using one’s medical skills in service of a larger mission. The relationships built with service members, the sense of shared purpose, and the opportunity to contribute to the readiness and well-being of those who serve all make this career path profoundly rewarding. For dermatologists seeking a practice that combines professional growth with purpose and patriotism, military medicine offers a truly special calling.
- Military Health System. Elements of the military health system. Accessed October 11, 2025. https://www.health.mil/About-MHS/MHS-Elements
- TRICARE. Plans and eligibility. Accessed October 11, 2025. https://tricare.mil/Plans/Eligibility
- Military Benefit. TRICARE for retirees. Accessed October 11, 2025. https://www.militarybenefit.org/get-educated/tricareforretirees/
- US Department of Veterans Affairs. Eligibility for VA health care. Accessed October 11, 2025. https://www.va.gov/health-care/eligibility/
- US Department of Veterans Affairs. VA priority groups. Accessed October 11, 2025. https://www.va.gov/health-care/eligibility/priority-groups/
- Navy Medicine. Health Professions Scholarship Program (HPSP) and Financial Assistance Program (FAP). Accessed October 12, 2025. https://www.med.navy.mil/Accessions/Health-Professions-Scholarship-Program-HPSP-and-Financial-Assistance-Program-FAP/
- US Navy. Navy Medicine R2DA program. Accessed October 12, 2025. https://www.navy.com/navy-medicine
- US Army Medical Department. Student programs. Accessed October 12, 2025. https://goamedd.com/student-programs
- US Office of Personnel Management. General Schedule. Accessed October 12, 2025. https://www.opm.gov/policy-data-oversight/pay-leave/pay-systems/general-schedule/
- Pines Federal Employment Attorneys. Title 38 employees: medical professionals. Accessed October 12, 2025. https://www.pinesfederal.com/va-federal-employees/title-38-employees-medical-professionals/
Serving those who serve has been one of the most meaningful parts of my career. A career in military medicine offers dermatologists not only a chance to practice within a unique and diverse patient population but also an opportunity to contribute to something larger than themselves. Whether working with active-duty service members and their families within the Military Health System (MHS) or caring for veterans through the Department of Veterans Affairs (VA), the experience can be both enriching and rewarding. This article will explore the various pathways available to dermatologists to serve military communities, whether they are at the start of their careers or are looking for a change of pace within their established practice.
Care Pathways for Military and Veterans
To care for uniformed service members, their families, and retired personnel, dermatologists typically serve within the MHS—a global, integrated network of military hospitals and clinics dedicated to delivering health care to this population.1 TRICARE is the health insurance program that covers those eligible for care within the system, including active-duty and retired service members.2 In this context, it is important to clarify what the term retired actually means, as it differs from the term veteran when it comes to accessing health care options, and these terms frequently are conflated. A retired service member is an individual who completed at least 20 years of active-duty service or who has been medically retired because of a condition or injury incurred while on active duty.3 In contrast, a veteran may not have completed 20 years of service but has separated honorably after serving at least 24 continuous months.4 Veterans typically receive care through the VA system.5
Serving on Active Duty
In general, there are 2 main pathways to serve as a dermatologist within the MHS. The first is to commission in the military and serve on active duty. Most often, this pathway begins with a premedical student applying to medical school. Those considering military service typically explore scholarship programs such as the Health Professions Scholarship Program (HPSP)(https://www.medicineandthemilitary.com/applying-and-what-to-expect/medical-school-programs/hpsp) or the Health Services Collegiate Program (HSCP), or they apply to the Uniformed Services University of the Health Sciences (USU)(https://www.usuhs.edu/about). The HPSP and HSCP programs financially support medical students training at civilian medical schools, though in different ways—the HPSP covers tuition and fees, while the HSCP provides a salary during training but does not cover tuition.6 In contrast, students of USU attend the nation’s only military medical school, serving in uniform for 4 years while earning the pay and benefits of a junior officer in their respective service branch. Any premedical student considering the HPSP, HSCP or USU routes for service must meet the commissioning standards of their chosen branch—Army, Navy, or Air Force—and enter service as an officer before beginning medical school.
While direct commission prior to medical school is the most common route to active-duty service, board-certified dermatologists also can join a military branch later through what is called Direct Accession or Direct Commission; for example, the Navy offers a Residency to Direct Accession program, which commissions residents in their final year of training to join the Navy upon graduation. In some cases, commissioning at this stage includes a bonus of up to $600,000 in exchange for a 4-year active-duty commitment.7 The Army and Air Force offer similar direct commission programs, though specific incentives vary.8 Interested residents or practitioners can contact a local recruiting office within their branch of interest to learn more. Direct accession is open at many points in a dermatologist’s career—after residency, after fellowship, or even as an established civilian practitioner—and the initial commissioning rank and bonus generally reflect one’s level of experience.
Serving as a Civilian
Outside of uniformed service, dermatologists can find opportunities to provide care for active-duty service members, veterans, and military families through employment as General Schedule (GS) employees. The GS is a role classification and pay system that covers most federal employees in professional, administrative, and technical positions (eg, physicians). The GS system classifies most of these employees based on the complexity, responsibility, and qualifications required for their role.9 Such positions often are at the highest level of the GS pay scale, reflecting the expertise and years of education required to become a dermatologist, though pay varies by location and experience. In contrast, physicians employed through the VA system are classified as Title 38 federal employees, governed by a different pay structure and regulatory framework under the US Code of Federal Regulations.10 These regulations govern the hiring, retention, and firing guidelines for VA physicians, which differ from those of GS physicians. A full explanation is outside of the scope of this article, however.
Final Thoughts
In summary, uniformed or federal service as a dermatologist offers a meaningful and impactful way to give back to those who have served our country. Opportunities exist throughout the United States for dermatologists interested in serving within the MHS or VA. The most transparent and up-to-date resource for identifying open positions in both large metropolitan areas and smaller communities is USAJOBS.gov. While financial compensation may not always match that of private practice, the intangible benefits are considerable—stable employment, comprehensive benefits, malpractice coverage, and secure retirement, among others. There is something deeply fulfilling about using one’s medical skills in service of a larger mission. The relationships built with service members, the sense of shared purpose, and the opportunity to contribute to the readiness and well-being of those who serve all make this career path profoundly rewarding. For dermatologists seeking a practice that combines professional growth with purpose and patriotism, military medicine offers a truly special calling.
Serving those who serve has been one of the most meaningful parts of my career. A career in military medicine offers dermatologists not only a chance to practice within a unique and diverse patient population but also an opportunity to contribute to something larger than themselves. Whether working with active-duty service members and their families within the Military Health System (MHS) or caring for veterans through the Department of Veterans Affairs (VA), the experience can be both enriching and rewarding. This article will explore the various pathways available to dermatologists to serve military communities, whether they are at the start of their careers or are looking for a change of pace within their established practice.
Care Pathways for Military and Veterans
To care for uniformed service members, their families, and retired personnel, dermatologists typically serve within the MHS—a global, integrated network of military hospitals and clinics dedicated to delivering health care to this population.1 TRICARE is the health insurance program that covers those eligible for care within the system, including active-duty and retired service members.2 In this context, it is important to clarify what the term retired actually means, as it differs from the term veteran when it comes to accessing health care options, and these terms frequently are conflated. A retired service member is an individual who completed at least 20 years of active-duty service or who has been medically retired because of a condition or injury incurred while on active duty.3 In contrast, a veteran may not have completed 20 years of service but has separated honorably after serving at least 24 continuous months.4 Veterans typically receive care through the VA system.5
Serving on Active Duty
In general, there are 2 main pathways to serve as a dermatologist within the MHS. The first is to commission in the military and serve on active duty. Most often, this pathway begins with a premedical student applying to medical school. Those considering military service typically explore scholarship programs such as the Health Professions Scholarship Program (HPSP)(https://www.medicineandthemilitary.com/applying-and-what-to-expect/medical-school-programs/hpsp) or the Health Services Collegiate Program (HSCP), or they apply to the Uniformed Services University of the Health Sciences (USU)(https://www.usuhs.edu/about). The HPSP and HSCP programs financially support medical students training at civilian medical schools, though in different ways—the HPSP covers tuition and fees, while the HSCP provides a salary during training but does not cover tuition.6 In contrast, students of USU attend the nation’s only military medical school, serving in uniform for 4 years while earning the pay and benefits of a junior officer in their respective service branch. Any premedical student considering the HPSP, HSCP or USU routes for service must meet the commissioning standards of their chosen branch—Army, Navy, or Air Force—and enter service as an officer before beginning medical school.
While direct commission prior to medical school is the most common route to active-duty service, board-certified dermatologists also can join a military branch later through what is called Direct Accession or Direct Commission; for example, the Navy offers a Residency to Direct Accession program, which commissions residents in their final year of training to join the Navy upon graduation. In some cases, commissioning at this stage includes a bonus of up to $600,000 in exchange for a 4-year active-duty commitment.7 The Army and Air Force offer similar direct commission programs, though specific incentives vary.8 Interested residents or practitioners can contact a local recruiting office within their branch of interest to learn more. Direct accession is open at many points in a dermatologist’s career—after residency, after fellowship, or even as an established civilian practitioner—and the initial commissioning rank and bonus generally reflect one’s level of experience.
Serving as a Civilian
Outside of uniformed service, dermatologists can find opportunities to provide care for active-duty service members, veterans, and military families through employment as General Schedule (GS) employees. The GS is a role classification and pay system that covers most federal employees in professional, administrative, and technical positions (eg, physicians). The GS system classifies most of these employees based on the complexity, responsibility, and qualifications required for their role.9 Such positions often are at the highest level of the GS pay scale, reflecting the expertise and years of education required to become a dermatologist, though pay varies by location and experience. In contrast, physicians employed through the VA system are classified as Title 38 federal employees, governed by a different pay structure and regulatory framework under the US Code of Federal Regulations.10 These regulations govern the hiring, retention, and firing guidelines for VA physicians, which differ from those of GS physicians. A full explanation is outside of the scope of this article, however.
Final Thoughts
In summary, uniformed or federal service as a dermatologist offers a meaningful and impactful way to give back to those who have served our country. Opportunities exist throughout the United States for dermatologists interested in serving within the MHS or VA. The most transparent and up-to-date resource for identifying open positions in both large metropolitan areas and smaller communities is USAJOBS.gov. While financial compensation may not always match that of private practice, the intangible benefits are considerable—stable employment, comprehensive benefits, malpractice coverage, and secure retirement, among others. There is something deeply fulfilling about using one’s medical skills in service of a larger mission. The relationships built with service members, the sense of shared purpose, and the opportunity to contribute to the readiness and well-being of those who serve all make this career path profoundly rewarding. For dermatologists seeking a practice that combines professional growth with purpose and patriotism, military medicine offers a truly special calling.
- Military Health System. Elements of the military health system. Accessed October 11, 2025. https://www.health.mil/About-MHS/MHS-Elements
- TRICARE. Plans and eligibility. Accessed October 11, 2025. https://tricare.mil/Plans/Eligibility
- Military Benefit. TRICARE for retirees. Accessed October 11, 2025. https://www.militarybenefit.org/get-educated/tricareforretirees/
- US Department of Veterans Affairs. Eligibility for VA health care. Accessed October 11, 2025. https://www.va.gov/health-care/eligibility/
- US Department of Veterans Affairs. VA priority groups. Accessed October 11, 2025. https://www.va.gov/health-care/eligibility/priority-groups/
- Navy Medicine. Health Professions Scholarship Program (HPSP) and Financial Assistance Program (FAP). Accessed October 12, 2025. https://www.med.navy.mil/Accessions/Health-Professions-Scholarship-Program-HPSP-and-Financial-Assistance-Program-FAP/
- US Navy. Navy Medicine R2DA program. Accessed October 12, 2025. https://www.navy.com/navy-medicine
- US Army Medical Department. Student programs. Accessed October 12, 2025. https://goamedd.com/student-programs
- US Office of Personnel Management. General Schedule. Accessed October 12, 2025. https://www.opm.gov/policy-data-oversight/pay-leave/pay-systems/general-schedule/
- Pines Federal Employment Attorneys. Title 38 employees: medical professionals. Accessed October 12, 2025. https://www.pinesfederal.com/va-federal-employees/title-38-employees-medical-professionals/
- Military Health System. Elements of the military health system. Accessed October 11, 2025. https://www.health.mil/About-MHS/MHS-Elements
- TRICARE. Plans and eligibility. Accessed October 11, 2025. https://tricare.mil/Plans/Eligibility
- Military Benefit. TRICARE for retirees. Accessed October 11, 2025. https://www.militarybenefit.org/get-educated/tricareforretirees/
- US Department of Veterans Affairs. Eligibility for VA health care. Accessed October 11, 2025. https://www.va.gov/health-care/eligibility/
- US Department of Veterans Affairs. VA priority groups. Accessed October 11, 2025. https://www.va.gov/health-care/eligibility/priority-groups/
- Navy Medicine. Health Professions Scholarship Program (HPSP) and Financial Assistance Program (FAP). Accessed October 12, 2025. https://www.med.navy.mil/Accessions/Health-Professions-Scholarship-Program-HPSP-and-Financial-Assistance-Program-FAP/
- US Navy. Navy Medicine R2DA program. Accessed October 12, 2025. https://www.navy.com/navy-medicine
- US Army Medical Department. Student programs. Accessed October 12, 2025. https://goamedd.com/student-programs
- US Office of Personnel Management. General Schedule. Accessed October 12, 2025. https://www.opm.gov/policy-data-oversight/pay-leave/pay-systems/general-schedule/
- Pines Federal Employment Attorneys. Title 38 employees: medical professionals. Accessed October 12, 2025. https://www.pinesfederal.com/va-federal-employees/title-38-employees-medical-professionals/
Dermatology on Duty: Pathways to a Career in Military Medicine
Dermatology on Duty: Pathways to a Career in Military Medicine
PRACTICE POINTS
- Dermatologists have diverse pathways to serve the military and veteran communities, either in uniform or as civilians.
- For those considering a military career, options include medical school scholarships or direct commission after residency.
- Those who prefer to remain civilians can find employment opportunities with the Military Heath System or the Department of Veterans Affairs that provide a way to care for this population without a service commitment.
The Habit of Curiosity: How Writing Shapes Clinical Thinking in Medical Training
The Habit of Curiosity: How Writing Shapes Clinical Thinking in Medical Training
I was accepted into my fellowship almost 1 year ago: major milestones on my curriculum vitae are now met, fellowship application materials are complete, and the stress of the match is long gone. At the start of my fellowship, I had 2 priorities: (1) to learn as much as I could about dermatologic surgery and (2) to be the best dad possible to my newborn son, Jay. However, most nights I still find myself up late editing a manuscript draft or chasing down references, long after the “need” to publish has passed. Recently, my wife asked me why—what’s left to prove?
I’ll be the first to admit it: early on, publishing felt almost purely transactional. Each project was little more than a line on an application or a way to stand out or meet a new mentor. I have reflected before on how easily that mindset can slip into a kind of research arms race, in which productivity overshadows purpose.1 This time, I wanted to explore the other side of that equation: the “why” behind it all.
I have learned that writing forces me to slow down and actually think about what I am seeing every day. It turns routine work into something I must understand well enough to explain. Even a small write-up can make me notice details I would otherwise skim past in clinic or surgery. These days, most of my projects start small: a case that taught me something, an observation that made me pause and think. Those seemingly small questions are what eventually grow into bigger ones. The clinical trial I am designing now did not begin as a grand plan—it started because I could not stop thinking about how we manage pain and analgesia after Mohs surgery. That curiosity, shaped by the experience of writing those earlier “smaller” papers, evolved into a study that might actually help improve patient care one day. Still, most of what I write will not revolutionize the field. It is not cutting-edge science or paradigm-shifting data; it is mostly modest analyses with a few interesting conclusions or surgical pearls that might cut down on a patient’s procedural time or save a dermatologist somewhere a few sutures. But it still feels worth doing.
While rotating with Dr. Anna Bar at Oregon Health & Science University, Portland, I noticed a poster hanging on the wall titled, “Top 10 Reasons Why Our Faculty Are Dedicated to Academics and Teaching,” based on the wisdom of Dr. Jane M. Grant-Kels.2 My favorite line on the poster reads, “Residents make us better by asking questions.” I think this philosophy is the main reason why I still write. Even though I am not a resident anymore, I am still asking questions. But if I had to sum up my “why” into a neat list, here is what it might look like:
Because asking questions keeps your brain wired for curiosity. Even small projects train us to remain curious, and this curiosity can mean the difference between just doing your job and continuing to evolve within it. As Dr. Rodolfo Neirotti reminds us, “Questions are useful tools—they open communication, improve understanding, and drive scientific research. In medicine, doing things without knowing why is risky.”3
Because the small stuff builds the culture. Dermatology is a small world. Even short case series, pearls, or “how we do it” pieces can shape how we practice. They may not change paradigms, but they can refine them. Over time, those small practical contributions become part of the field’s collective muscle memory.
Because it preserves perspective. Residency, fellowship, and early practice can blur together. A tiny project can become a timestamp of what you were learning or caring about at that specific moment. Years later, you may remember the case through the paper.
Because the act of writing is the point. Writing forces clarity. You cannot hide behind saying, “That’s just how I do things,” when you have to explain it to others. The discipline of organizing your thoughts sharpens your clinical reasoning and keeps you honest about what you actually know.
Because sometimes it is simply about participating. Publishing, even small pieces, is a way of staying in touch with your field. It says, “I’m still here. I’m still paying attention.”
I think about how Dr. Frederic Mohs developed the technique that now bears his name while he was still a medical student.4 He could have said, “I already made it into medical school. That’s enough.” But he did not. I guess my point is not that we are all on the verge of inventing something revolutionary; it is that innovation happens only when curiosity keeps moving us forward. So no, I do not write to check boxes anymore. I write because it keeps me curious, and I have realized that curiosity is a habit I never want to outgrow.
Or maybe it’s because Jay keeps me up at night, and I have nothing better to do.
- Jeha GM. A roadmap to research opportunities for dermatology residents. Cutis. 2024;114:E53-E56.
- Grant-Kels J. The gift that keeps on giving. UConn Health Dermatology. Accessed November 24, 2025. https://health.uconn.edu/dermatology/education/
- Neirotti RA. The importance of asking questions and doing things for a reason. Braz J Cardiovasc Surg. 2021;36:I-II.
- Trost LB, Bailin PL. History of Mohs surgery. Dermatol Clin. 2011;29:135-139, vii.
I was accepted into my fellowship almost 1 year ago: major milestones on my curriculum vitae are now met, fellowship application materials are complete, and the stress of the match is long gone. At the start of my fellowship, I had 2 priorities: (1) to learn as much as I could about dermatologic surgery and (2) to be the best dad possible to my newborn son, Jay. However, most nights I still find myself up late editing a manuscript draft or chasing down references, long after the “need” to publish has passed. Recently, my wife asked me why—what’s left to prove?
I’ll be the first to admit it: early on, publishing felt almost purely transactional. Each project was little more than a line on an application or a way to stand out or meet a new mentor. I have reflected before on how easily that mindset can slip into a kind of research arms race, in which productivity overshadows purpose.1 This time, I wanted to explore the other side of that equation: the “why” behind it all.
I have learned that writing forces me to slow down and actually think about what I am seeing every day. It turns routine work into something I must understand well enough to explain. Even a small write-up can make me notice details I would otherwise skim past in clinic or surgery. These days, most of my projects start small: a case that taught me something, an observation that made me pause and think. Those seemingly small questions are what eventually grow into bigger ones. The clinical trial I am designing now did not begin as a grand plan—it started because I could not stop thinking about how we manage pain and analgesia after Mohs surgery. That curiosity, shaped by the experience of writing those earlier “smaller” papers, evolved into a study that might actually help improve patient care one day. Still, most of what I write will not revolutionize the field. It is not cutting-edge science or paradigm-shifting data; it is mostly modest analyses with a few interesting conclusions or surgical pearls that might cut down on a patient’s procedural time or save a dermatologist somewhere a few sutures. But it still feels worth doing.
While rotating with Dr. Anna Bar at Oregon Health & Science University, Portland, I noticed a poster hanging on the wall titled, “Top 10 Reasons Why Our Faculty Are Dedicated to Academics and Teaching,” based on the wisdom of Dr. Jane M. Grant-Kels.2 My favorite line on the poster reads, “Residents make us better by asking questions.” I think this philosophy is the main reason why I still write. Even though I am not a resident anymore, I am still asking questions. But if I had to sum up my “why” into a neat list, here is what it might look like:
Because asking questions keeps your brain wired for curiosity. Even small projects train us to remain curious, and this curiosity can mean the difference between just doing your job and continuing to evolve within it. As Dr. Rodolfo Neirotti reminds us, “Questions are useful tools—they open communication, improve understanding, and drive scientific research. In medicine, doing things without knowing why is risky.”3
Because the small stuff builds the culture. Dermatology is a small world. Even short case series, pearls, or “how we do it” pieces can shape how we practice. They may not change paradigms, but they can refine them. Over time, those small practical contributions become part of the field’s collective muscle memory.
Because it preserves perspective. Residency, fellowship, and early practice can blur together. A tiny project can become a timestamp of what you were learning or caring about at that specific moment. Years later, you may remember the case through the paper.
Because the act of writing is the point. Writing forces clarity. You cannot hide behind saying, “That’s just how I do things,” when you have to explain it to others. The discipline of organizing your thoughts sharpens your clinical reasoning and keeps you honest about what you actually know.
Because sometimes it is simply about participating. Publishing, even small pieces, is a way of staying in touch with your field. It says, “I’m still here. I’m still paying attention.”
I think about how Dr. Frederic Mohs developed the technique that now bears his name while he was still a medical student.4 He could have said, “I already made it into medical school. That’s enough.” But he did not. I guess my point is not that we are all on the verge of inventing something revolutionary; it is that innovation happens only when curiosity keeps moving us forward. So no, I do not write to check boxes anymore. I write because it keeps me curious, and I have realized that curiosity is a habit I never want to outgrow.
Or maybe it’s because Jay keeps me up at night, and I have nothing better to do.
I was accepted into my fellowship almost 1 year ago: major milestones on my curriculum vitae are now met, fellowship application materials are complete, and the stress of the match is long gone. At the start of my fellowship, I had 2 priorities: (1) to learn as much as I could about dermatologic surgery and (2) to be the best dad possible to my newborn son, Jay. However, most nights I still find myself up late editing a manuscript draft or chasing down references, long after the “need” to publish has passed. Recently, my wife asked me why—what’s left to prove?
I’ll be the first to admit it: early on, publishing felt almost purely transactional. Each project was little more than a line on an application or a way to stand out or meet a new mentor. I have reflected before on how easily that mindset can slip into a kind of research arms race, in which productivity overshadows purpose.1 This time, I wanted to explore the other side of that equation: the “why” behind it all.
I have learned that writing forces me to slow down and actually think about what I am seeing every day. It turns routine work into something I must understand well enough to explain. Even a small write-up can make me notice details I would otherwise skim past in clinic or surgery. These days, most of my projects start small: a case that taught me something, an observation that made me pause and think. Those seemingly small questions are what eventually grow into bigger ones. The clinical trial I am designing now did not begin as a grand plan—it started because I could not stop thinking about how we manage pain and analgesia after Mohs surgery. That curiosity, shaped by the experience of writing those earlier “smaller” papers, evolved into a study that might actually help improve patient care one day. Still, most of what I write will not revolutionize the field. It is not cutting-edge science or paradigm-shifting data; it is mostly modest analyses with a few interesting conclusions or surgical pearls that might cut down on a patient’s procedural time or save a dermatologist somewhere a few sutures. But it still feels worth doing.
While rotating with Dr. Anna Bar at Oregon Health & Science University, Portland, I noticed a poster hanging on the wall titled, “Top 10 Reasons Why Our Faculty Are Dedicated to Academics and Teaching,” based on the wisdom of Dr. Jane M. Grant-Kels.2 My favorite line on the poster reads, “Residents make us better by asking questions.” I think this philosophy is the main reason why I still write. Even though I am not a resident anymore, I am still asking questions. But if I had to sum up my “why” into a neat list, here is what it might look like:
Because asking questions keeps your brain wired for curiosity. Even small projects train us to remain curious, and this curiosity can mean the difference between just doing your job and continuing to evolve within it. As Dr. Rodolfo Neirotti reminds us, “Questions are useful tools—they open communication, improve understanding, and drive scientific research. In medicine, doing things without knowing why is risky.”3
Because the small stuff builds the culture. Dermatology is a small world. Even short case series, pearls, or “how we do it” pieces can shape how we practice. They may not change paradigms, but they can refine them. Over time, those small practical contributions become part of the field’s collective muscle memory.
Because it preserves perspective. Residency, fellowship, and early practice can blur together. A tiny project can become a timestamp of what you were learning or caring about at that specific moment. Years later, you may remember the case through the paper.
Because the act of writing is the point. Writing forces clarity. You cannot hide behind saying, “That’s just how I do things,” when you have to explain it to others. The discipline of organizing your thoughts sharpens your clinical reasoning and keeps you honest about what you actually know.
Because sometimes it is simply about participating. Publishing, even small pieces, is a way of staying in touch with your field. It says, “I’m still here. I’m still paying attention.”
I think about how Dr. Frederic Mohs developed the technique that now bears his name while he was still a medical student.4 He could have said, “I already made it into medical school. That’s enough.” But he did not. I guess my point is not that we are all on the verge of inventing something revolutionary; it is that innovation happens only when curiosity keeps moving us forward. So no, I do not write to check boxes anymore. I write because it keeps me curious, and I have realized that curiosity is a habit I never want to outgrow.
Or maybe it’s because Jay keeps me up at night, and I have nothing better to do.
- Jeha GM. A roadmap to research opportunities for dermatology residents. Cutis. 2024;114:E53-E56.
- Grant-Kels J. The gift that keeps on giving. UConn Health Dermatology. Accessed November 24, 2025. https://health.uconn.edu/dermatology/education/
- Neirotti RA. The importance of asking questions and doing things for a reason. Braz J Cardiovasc Surg. 2021;36:I-II.
- Trost LB, Bailin PL. History of Mohs surgery. Dermatol Clin. 2011;29:135-139, vii.
- Jeha GM. A roadmap to research opportunities for dermatology residents. Cutis. 2024;114:E53-E56.
- Grant-Kels J. The gift that keeps on giving. UConn Health Dermatology. Accessed November 24, 2025. https://health.uconn.edu/dermatology/education/
- Neirotti RA. The importance of asking questions and doing things for a reason. Braz J Cardiovasc Surg. 2021;36:I-II.
- Trost LB, Bailin PL. History of Mohs surgery. Dermatol Clin. 2011;29:135-139, vii.
The Habit of Curiosity: How Writing Shapes Clinical Thinking in Medical Training
The Habit of Curiosity: How Writing Shapes Clinical Thinking in Medical Training
Practice Points
- Writing about everyday clinical experiences forces trainees to slow down, think more carefully, and better understand why they do what they do. Being able to write clearly about a clinical scenario reflects true understanding.
- The act of writing sharpens clinical judgment by requiring clarity, honesty, and reflection rather than relying on habit or routine.
- Writing fosters habits of curiosity that support continued professional growth and ongoing engagement with one’s field beyond formal training milestones.
Primary Care Clinician and Patient Knowledge, Interest, and Use of Integrative Treatment Options for Chronic Low Back Pain Management
Primary Care Clinician and Patient Knowledge, Interest, and Use of Integrative Treatment Options for Chronic Low Back Pain Management
More than 50 million US adults report experiencing chronic pain, with nearly 7% experiencing high-impact chronic pain.1-3 Chronic pain negatively affects daily function, results in lost productivity, is a leading cause of disability, and is more prevalent among veterans compared with the general population.1,2,4-6 Estimates from 2021 suggest the prevalence of chronic pain among veterans exceeds 30%; > 11% experienced high-impact chronic pain.1
Primary care practitioners (PCPs) have a prominent role in chronic pain management. Pharmacologic options for treating pain, once a mainstay of therapy, present several challenges for patients and PCPs, including drug-drug interactions and adverse effects.7 The US opioid epidemic and shift to a biopsychosocial model of chronic pain care have increased emphasis on nonpharmacologic treatment options.8,9 These include integrative modalities, which incorporate conventional approaches with an array of complementary health approaches.10-12
Integrative therapy is a prominent feature in whole person care, which may be best exemplified by the US Department of Veterans Affairs (VA) Whole Health System of care.13-14 Whole health empowers an individual to take charge of their health and well-being so they can “live their life to the fullest.”14 As implemented in the Veterans Health Administration (VHA), whole health includes the use of evidence-based
METHODS
Using a cross-sectional survey design, PCPs and patients with chronic back pain affiliated with the VA Ann Arbor Healthcare System were invited to participate in separate but similar surveys to assess knowledge, interest, and use of nonpharmacologic integrative modalities for the treatment of chronic pain. In May, June, and July 2023, 78 PCPs received 3 email
Both survey instruments are available upon request, were developed by the study team, and included a mix of yes/no questions, “select all that apply” items, Likert scale response items, and open-ended questions. For one question about which modalities they would like available, the respondent was instructed to select up to 5 modalities. The instruments were extensively pretested by members of the study team, which included 2 PCPs and a nonveteran with chronic back pain.
The list of integrative modalities included in the survey was derived from the tier 1 and tier 2 complementary and integrative health modalities identified in a VHA Directive on complementary and integrative health.15,16 Tier 1 approaches are considered to have sufficient evidence and must be made available to veterans either within a VA medical facility or in the community. Tier 2 approaches are generally considered safe and may be made available but do not have sufficient evidence to mandate their provision. For participant ease, the integrative modalities were divided into 5 subgroups: manual therapies, energy/biofield therapies, mental health therapies, nutrition counseling, and movement therapies. The clinician survey assessed clinicians’ training and interest, clinical and personal use, and perceived barriers to providing integrative modalities for chronic pain. Professional and personal demographic data were also collected. Similarly, the patient survey assessed use of integrative therapies, perceptions of and interest in integrative modalities, and potential barriers to use. Demographic and health-related information was also collected.
Data analysis included descriptive statistics (eg, frequency counts, means, medians) and visual graphic displays. Separate analyses were conducted for clinicians and patients in addition to a comparative analysis of the use and potential interest in integrative modalities. Analysis were conducted using R software. This study was deemed nonresearch quality improvement by the VA Ann Arbor Healthcare System facility research oversight board and institutional review board approval was not solicited.
RESULTS
Twenty-eight clinicians completed the survey, yielding a participation rate of 36%. Participating clinicians had a median (IQR) age of 48 years (9.5), 15 self-identified as White (54%), 8 as Asian (29%), 15 as female (54%), 26 as non-Hispanic (93%), and 25 were medical doctors or doctors of osteopathy (89%). Nineteen (68%) worked at the main hospital outpatient clinic, and 9 practiced at community-based outpatient clinics (CBOCs). Thirteen respondents (46%) reported having no formal education or training in integrative approaches. Among those with prior training, 8 clinicians had nutrition counseling (29%) and 7 had psychologic therapy training (25%). Thirteen respondents (46%) also reported using integrative modalities for personal health needs: 8 used psychological therapies, 8 used movement therapies, 10 used integrative modalities for stress management or relaxation, and 8 used them for physical symptoms (Table 1).
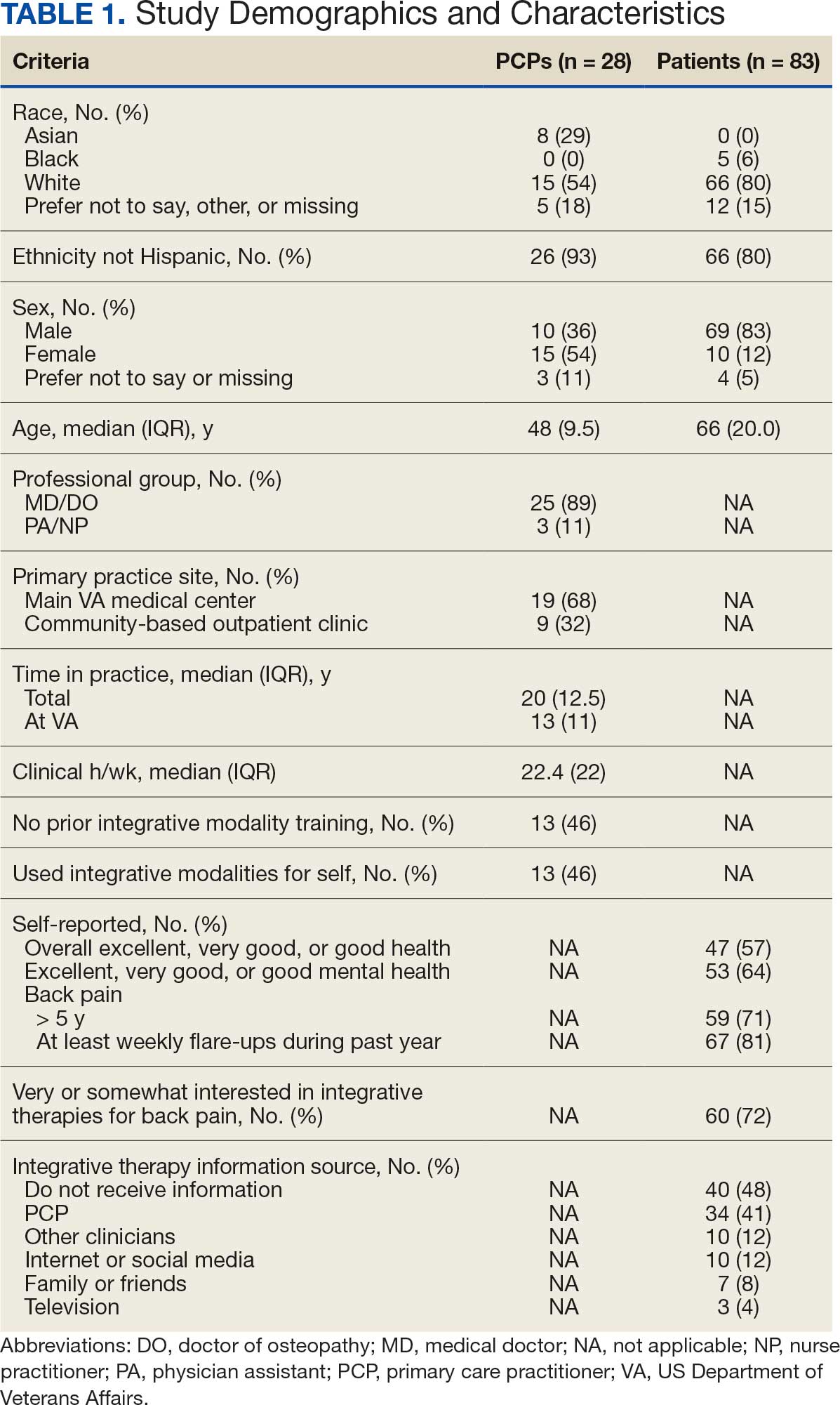
Overall, 85 of 200 patients (43%) responded to the study survey. Two patients indicated they did not have chronic back pain and were excluded. Patients had a median (IQR) age of 66 (20) years, with 66 self-identifying as White (80%), 69 as male (83%), and 66 as non-Hispanic (80%). Forty-four patients (53%) received care at CBOCs. Forty-seven patients reported excellent, very good, or good overall health (57%), while 53 reported excellent, very good, or good mental health (64%). Fifty-nine patients reported back pain duration > 5 years (71%), and 67 (81%) indicated experiencing back pain flare-ups at least once per week over the previous 12 months. Sixty patients (72%) indicated they were somewhat or very interested in using integrative therapies as a back pain treatment; however, 40 patients (48%) indicated they had not received information about these therapies. Among those who indicated they had received information, the most frequently reported source was their PCP (41%). Most patients (72%) also reported feeling somewhat to very comfortable discussing integrative medicine therapies with their PCP.
Integrative Therapy Recommendations and Use
PCPs reported recommending multiple integrative modalities: 23 (82%) recommended cognitive-behavioral therapy, 22 (79%) recommended acupuncture, 21 (75%) recommended chiropractic, 19 (68%) recommended battlefield acupuncture, recommended massage 18 (64%), 17 (61%) recommended meditation or mindfulness, and 15 (54%) recommended movement therapies such as yoga or tai chi/qigong (Figure 1). The only therapies used by at least half of the patients were chiropractic used by 59 patients (71%) and acupuncture by 42 patients (51%). Thirty-eight patients (46%) reported massage use and 21 patients (25%) used cognitive-behavioral therapy (Table 2).
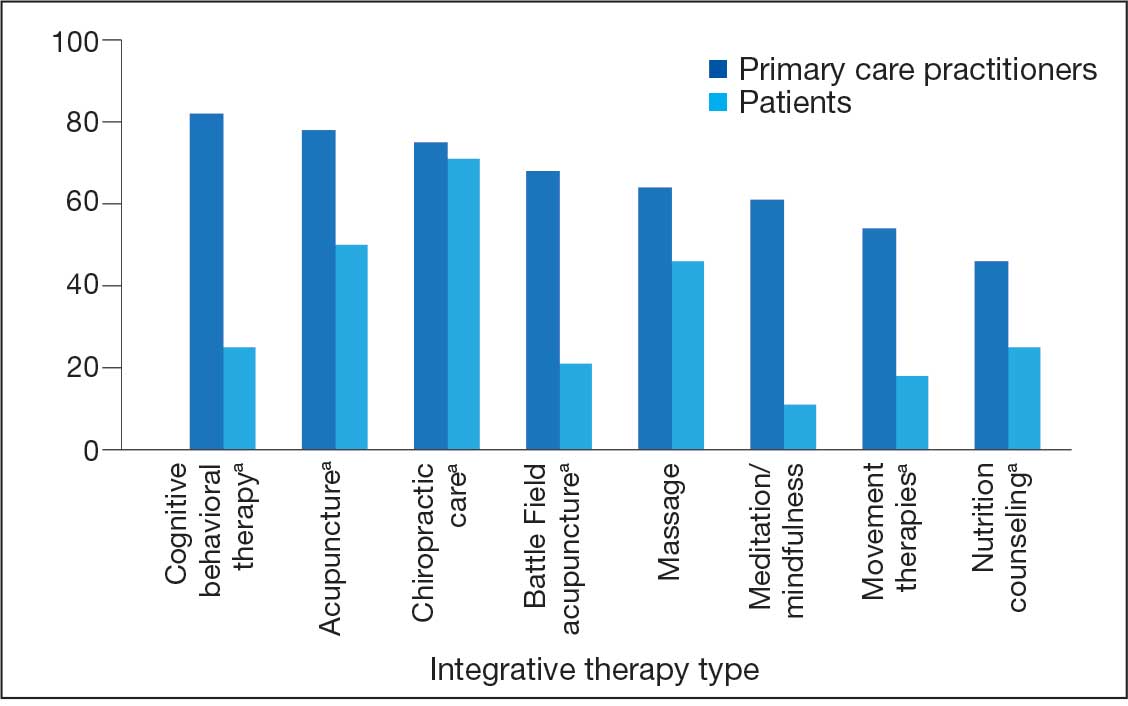
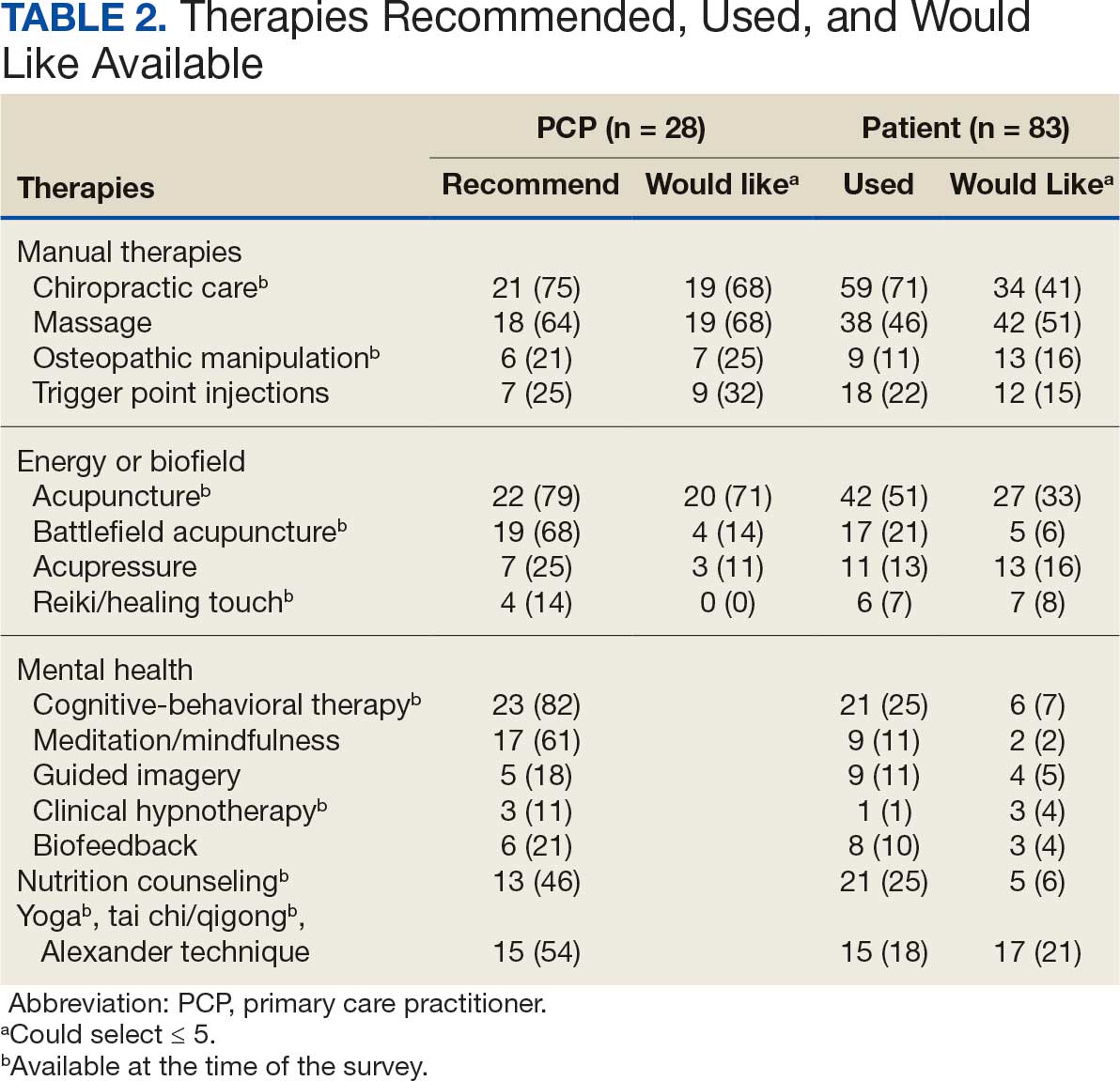
Integrative Therapies Desired
A majority of PCPs identified acupuncture (n = 20, 71%), chiropractic (n = 19, 68%), and massage (n = 19, 68%) as therapies they would most like to have available for patients with chronic pain (Figure 2). Similarly, patients identified massage (n = 42, 51%), chiropractic (n = 34, 41%), and acupuncture (n = 27, 33%) as most desired. Seventeen patients (21%) expressed interest in movement therapies.
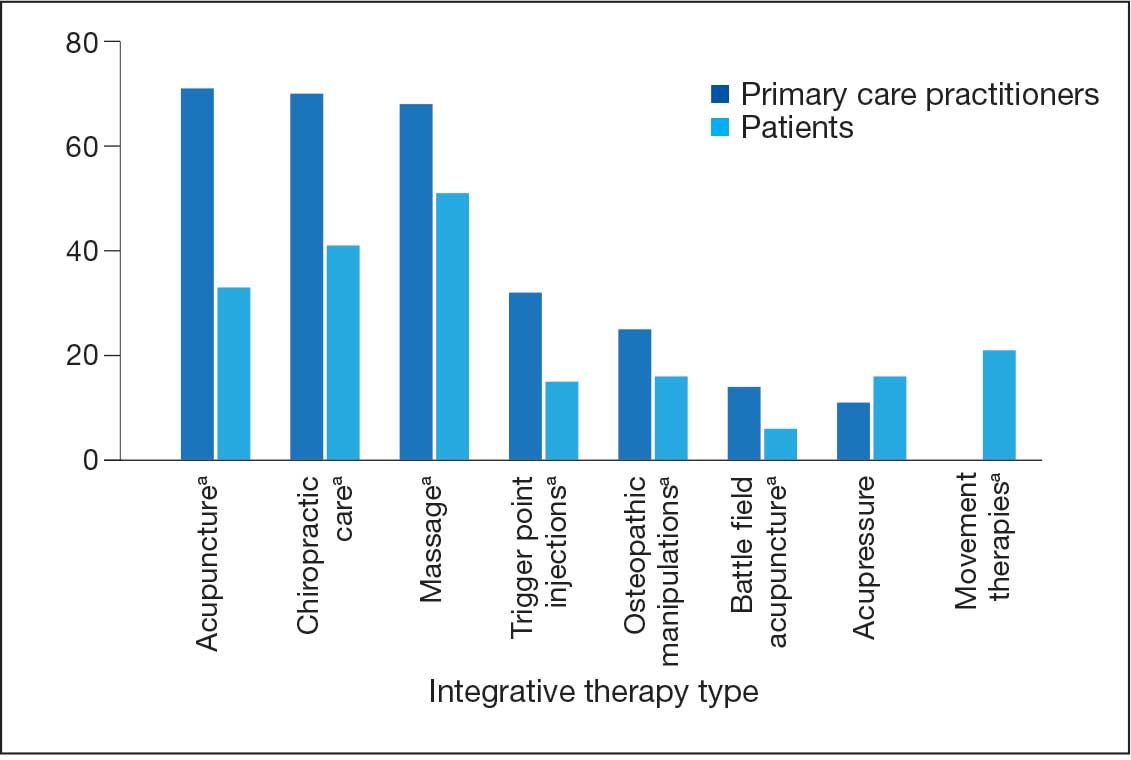
Barriers to Integrative Therapies Use
When asked about barriers to use, 26 PCPs (93%) identified access to services as a somewhat or extremely likely barrier, and 22 identified time constraints (79%) (Table 3). However, 17 PCPs (61%) noted lack of familiarity, and 18 (64%) noted a lack of scientific evidence as barriers to recommending integrative modalities. Among patients, 33 (40%) indicated not knowing what services were available at their facility as a barrier, 32 (39%) were not familiar with specific therapies, and 21 (25%) indicated a lack of clarity about the benefits of a specific therapy. Only 14 patients (17%) indicated that there were no obstacles to use.

DISCUSSION
Use of integrative therapies, including complementary treatments, is an increasingly important part of chronic pain management. This survey study suggests VA PCPs are willing to recommend integrative therapies and patients with chronic back pain both desire and use several therapies. Moreover, both groups expressed interest in greater availability of similar therapies. The results also highlight key barriers, such as knowledge gaps, that should be addressed to increase the uptake of integrative modalities for managing chronic pain.
An increasing number of US adults are using complementary health approaches, an important component of integrative therapy.12 This trend includes an increase in use for pain management, from 42.3% in 2002 to 49.2% in 2022; chiropractic care, acupuncture, and massage were most frequently used.12 Similarly, chiropractic, acupuncture and massage were most often used by this sample of veterans with chronic back pain and were identified by the highest percentages of PCPs and patients as the therapies they would most like available.
There were areas where the opinions of patients and clinicians differed. As has been seen previously reported, clinicians largely recommended cognitive-behavioral therapy while patients showed less interest.17 Additionally, while patients expressed interest in the availability of movement therapies, such as yoga, PCPs expressed more interest in other strategies, such as trigger point injections. These differences may reflect true preference or a tendency for clinicians and patients to select therapies with which they are more familiar. Additional research is needed to better understand the acceptability and potential use of integrative health treatments across a broad array of therapeutic options.
Despite VHA policy requiring facilities to provide certain complementary and integrative health modalities, almost all PCPs identified access to services as a major obstacle.15 Based on evidence and a rigorous vetting process, services currently required on-site, via telehealth, or through community partners include acupuncture and battlefield acupuncture (battlefield auricular acupuncture), biofeedback, clinical hypnosis, guided imagery, medical massage therapy, medication, tai chi/qigong, and yoga. Optional approaches, which may be made available to veterans, include chiropractic and healing touch. Outside the VHA, some states have introduced or enacted legislation mandating insurance coverage of nonpharmacological pain treatments.18 However, these requirements and mandates do not help address challenges such as the availability of trained/qualified practitioners.19,20 Ensuring access to complementary and integrative health treatments requires a more concerted effort to ensure that supply meets demand. It is also important to acknowledge the budgetary and physical space constraints that further limit access to services. Although expansion and integration of integrative medicine services remain a priority within the VA Whole Health program, implementation is contingent on available financial and infrastructure resources.
Time was also identified by PCPs as a barrier to recommending integrative therapies to patients. Developing and implementing time-efficient communication strategies for patient education such as concise talking points and informational handouts could help address this barrier. Furthermore, leveraging existing programs and engaging the entire health care team in patient education and referral could help increase integrative and complementary therapy uptake and use.
Although access and time were identified as major barriers, these findings also suggest that PCP and patient knowledge are another target area for enhancing the use of complementary and integrative therapies. Like prior research, most clinicians identified a lack of familiarity with certain services and a lack of scientific evidence as extremely or somewhat likely to affect their ability to offer integrative services to patients with chronic pain.21 Likewise, about 40% of patients identified being unfamiliar with a specific therapy as one of the major obstacles to receiving integrative therapies, with a similar number identifying PCPs as a source of information. The lack of familiarity may be due in part to the evolving nomenclature, with terms such as alternative, complementary, and integrative used to describe approaches outside what is often considered conventional medicine.10 On the other hand, there has also been considerable expansion in the number of therapies within this domain, along with an expanding evidence base. This suggests a need for targeted educational strategies for clinicians and patients, which can be rapidly deployed and continuously adapted as new therapies and evidence emerge.
Limitations
There are some inherent limitations with a survey-based approach, including sampling, non-response, and social desirability biases. In addition, this study only included PCPs and patients affiliated with a single VA medical center. Steps to mitigate these limitations included maintaining survey anonymity and reporting information about respondent characteristics to enhance transparency about the representativeness of the study findings.
CONCLUSIONS
Expanding the use of nonpharmacological pain treatments, including integrative modalities, is essential for safe and effective chronic pain management and reducing opioid use. Our findings show that VA PCPs and patients with chronic back pain are interested in and have some experience with certain integrative therapies. However, even within the context of a health care system that supports the use of integrative therapies for chronic pain as part of whole person care, increasing uptake will require addressing access and time-related constraints as well as ongoing clinician and patient education.
- Rikard SM, Strahan AE, Schmit KM, et al. Chronic pain among adults — United States, 2018-2021. MMWR Morb Mortal Wkly Rep. 2023;72:379-385. doi:10.15585/mmwr.mm7215a1
- Yong RJ, Mullins PM, Bhattacharyya N. Prevalence of chronic pain among adults in the United States. Pain. 2022;163:E328-E332. doi:10.1097/j.pain.0000000000002291
- Nahin RL, Feinberg T, Kapos FP, Terman GW. Estimated rates of incident and persistent chronic pain among US adults, 2019-2020. JAMA Netw Open. 2023;6:e2313563. doi:10.1001/jamanetworkopen.2023.13563
- Ferrari AJ, Santomauro DF, Aali A, et al. Global incidence, prevalence, years lived with disability (YLDs), disability-adjusted life-years (DALYs), and healthy life expectancy (HALE) for 371 diseases and injuries in 204 countries and territories and 811 subnational locations, 1990–2021: a systematic analysis for the Global Burden of Disease Study 2021. The Lancet. 2024;403:2133-2161. doi:10.1016/S0140-6736(24)00757-8 5.
- Qureshi AR, Patel M, Neumark S, et al. Prevalence of chronic non-cancer pain among military veterans: a systematic review and meta-analysis of observational studies. BMJ Mil Health. 2025;171:310-314. doi:10.1136/military-2023-002554
- Feldman DE, Nahin RL. Disability among persons with chronic severe back pain: results from a nationally representative population-based sample. J Pain. 2022;23:2144-2154. doi:10.1016/j.jpain.2022.07.016
- Qaseem A, Wilt TJ, McLean RM, Forciea MA. Noninvasive treatments for acute, subacute, and chronic low back pain: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:514-530. doi:10.7326/M16-2367
- van Erp RMA, Huijnen IPJ, Jakobs MLG, Kleijnen J, Smeets RJEM. Effectiveness of primary care interventions using a biopsychosocial approach in chronic low back pain: a systematic review. Pain Practice. 2019;19:224-241. doi:10.1111/papr.12735
- Chou R, Deyo R, Friedly J, et al. Nonpharmacologic therapies for low back pain: a systematic review for an American College of physicians clinical practice guideline. Ann Intern Med. 2017;166:493-505. doi:10.7326/M16-2459
- Complementary, alternative, or integrative health: what’s in a name? National Institutes of Health, National Center for Complementary and Integrative Health. Updated April 2021. Accessed December 15, 2025. https://www.nccih.nih.gov/health/complementary-alternative-or-integrative-health-whats-in-a-name.
- Taylor SL, Elwy AR. Complementary and alternative medicine for US veterans and active duty military personnel promising steps to improve their health. Med Care. 2014;52:S1-S4. doi:10.1097/MLR.0000000000000270.
- Nahin RL, Rhee A, Stussman B. Use of complementary health approaches overall and for pain management by US adults. JAMA. 2024;331:613-615. doi:10.1001/jama.2023.26775
- Gantt CJ, Donovan N, Khung M. Veterans Affairs’ Whole Health System of Care for transitioning service members and veterans. Mil Med. 2023;188:28-32. doi:10.1093/milmed/usad047
- Bokhour BG, Hyde J, Kligler B, et al. From patient outcomes to system change: evaluating the impact of VHA’s implementation of the Whole Health System of Care. Health Serv Res. 2022;57:53-65. doi:10.1111/1475-6773.13938
- Department of Veterans Affairs VHA. VHA Policy Directive 1137: Provision of Complementary and Integrative Health. December 2022. Accessed December 15, 2025. https://www.va.gov/VHApublications/ViewPublication.asp?pub_ID=10072
- Giannitrapani KF, Holliday JR, Miake-Lye IM, Hempel S, Taylor SL. Synthesizing the strength of the evidence of complementary and integrative health therapies for pain. Pain Med. 2019;20:1831-1840. doi:10.1093/pm/pnz068
- Belitskaya-Levy I, David Clark J, Shih MC, Bair MJ. Treatment preferences for chronic low back pain: views of veterans and their providers. J Pain Res. 2021;14:161-171. doi:10.2147/JPR.S290400
- Onstott TN, Hurst S, Kronick R, Tsou AC, Groessl E, McMenamin SB. Health insurance mandates for nonpharmacological pain treatments in 7 US states. JAMA Netw Open. 2024;7:E245737. doi:10.1001/jamanetworkopen.2024.5737
- Sullivan M, Leach M, Snow J, Moonaz S. The North American yoga therapy workforce survey. Complement Ther Med. 2017;31:39-48. doi:10.1016/j.ctim.2017.01.006
- Bolton R, Ritter G, Highland K, Larson MJ. The relationship between capacity and utilization of nonpharmacologic therapies in the US Military Health System. BMC Health Serv Res. 2022;22. doi:10.1186/s12913-022-07700-4
- Stussman BJ, Nahin RL, Barnes PM, Scott R, Feinberg T, Ward BW. Reasons office-based physicians in the United States recommend common complementary health approaches to patients: an exploratory study using a national survey. J Integr Complement Med. 2022;28:651-663. doi:10.1089/jicm.2022.0493
More than 50 million US adults report experiencing chronic pain, with nearly 7% experiencing high-impact chronic pain.1-3 Chronic pain negatively affects daily function, results in lost productivity, is a leading cause of disability, and is more prevalent among veterans compared with the general population.1,2,4-6 Estimates from 2021 suggest the prevalence of chronic pain among veterans exceeds 30%; > 11% experienced high-impact chronic pain.1
Primary care practitioners (PCPs) have a prominent role in chronic pain management. Pharmacologic options for treating pain, once a mainstay of therapy, present several challenges for patients and PCPs, including drug-drug interactions and adverse effects.7 The US opioid epidemic and shift to a biopsychosocial model of chronic pain care have increased emphasis on nonpharmacologic treatment options.8,9 These include integrative modalities, which incorporate conventional approaches with an array of complementary health approaches.10-12
Integrative therapy is a prominent feature in whole person care, which may be best exemplified by the US Department of Veterans Affairs (VA) Whole Health System of care.13-14 Whole health empowers an individual to take charge of their health and well-being so they can “live their life to the fullest.”14 As implemented in the Veterans Health Administration (VHA), whole health includes the use of evidence-based
METHODS
Using a cross-sectional survey design, PCPs and patients with chronic back pain affiliated with the VA Ann Arbor Healthcare System were invited to participate in separate but similar surveys to assess knowledge, interest, and use of nonpharmacologic integrative modalities for the treatment of chronic pain. In May, June, and July 2023, 78 PCPs received 3 email
Both survey instruments are available upon request, were developed by the study team, and included a mix of yes/no questions, “select all that apply” items, Likert scale response items, and open-ended questions. For one question about which modalities they would like available, the respondent was instructed to select up to 5 modalities. The instruments were extensively pretested by members of the study team, which included 2 PCPs and a nonveteran with chronic back pain.
The list of integrative modalities included in the survey was derived from the tier 1 and tier 2 complementary and integrative health modalities identified in a VHA Directive on complementary and integrative health.15,16 Tier 1 approaches are considered to have sufficient evidence and must be made available to veterans either within a VA medical facility or in the community. Tier 2 approaches are generally considered safe and may be made available but do not have sufficient evidence to mandate their provision. For participant ease, the integrative modalities were divided into 5 subgroups: manual therapies, energy/biofield therapies, mental health therapies, nutrition counseling, and movement therapies. The clinician survey assessed clinicians’ training and interest, clinical and personal use, and perceived barriers to providing integrative modalities for chronic pain. Professional and personal demographic data were also collected. Similarly, the patient survey assessed use of integrative therapies, perceptions of and interest in integrative modalities, and potential barriers to use. Demographic and health-related information was also collected.
Data analysis included descriptive statistics (eg, frequency counts, means, medians) and visual graphic displays. Separate analyses were conducted for clinicians and patients in addition to a comparative analysis of the use and potential interest in integrative modalities. Analysis were conducted using R software. This study was deemed nonresearch quality improvement by the VA Ann Arbor Healthcare System facility research oversight board and institutional review board approval was not solicited.
RESULTS
Twenty-eight clinicians completed the survey, yielding a participation rate of 36%. Participating clinicians had a median (IQR) age of 48 years (9.5), 15 self-identified as White (54%), 8 as Asian (29%), 15 as female (54%), 26 as non-Hispanic (93%), and 25 were medical doctors or doctors of osteopathy (89%). Nineteen (68%) worked at the main hospital outpatient clinic, and 9 practiced at community-based outpatient clinics (CBOCs). Thirteen respondents (46%) reported having no formal education or training in integrative approaches. Among those with prior training, 8 clinicians had nutrition counseling (29%) and 7 had psychologic therapy training (25%). Thirteen respondents (46%) also reported using integrative modalities for personal health needs: 8 used psychological therapies, 8 used movement therapies, 10 used integrative modalities for stress management or relaxation, and 8 used them for physical symptoms (Table 1).
Overall, 85 of 200 patients (43%) responded to the study survey. Two patients indicated they did not have chronic back pain and were excluded. Patients had a median (IQR) age of 66 (20) years, with 66 self-identifying as White (80%), 69 as male (83%), and 66 as non-Hispanic (80%). Forty-four patients (53%) received care at CBOCs. Forty-seven patients reported excellent, very good, or good overall health (57%), while 53 reported excellent, very good, or good mental health (64%). Fifty-nine patients reported back pain duration > 5 years (71%), and 67 (81%) indicated experiencing back pain flare-ups at least once per week over the previous 12 months. Sixty patients (72%) indicated they were somewhat or very interested in using integrative therapies as a back pain treatment; however, 40 patients (48%) indicated they had not received information about these therapies. Among those who indicated they had received information, the most frequently reported source was their PCP (41%). Most patients (72%) also reported feeling somewhat to very comfortable discussing integrative medicine therapies with their PCP.
Integrative Therapy Recommendations and Use
PCPs reported recommending multiple integrative modalities: 23 (82%) recommended cognitive-behavioral therapy, 22 (79%) recommended acupuncture, 21 (75%) recommended chiropractic, 19 (68%) recommended battlefield acupuncture, recommended massage 18 (64%), 17 (61%) recommended meditation or mindfulness, and 15 (54%) recommended movement therapies such as yoga or tai chi/qigong (Figure 1). The only therapies used by at least half of the patients were chiropractic used by 59 patients (71%) and acupuncture by 42 patients (51%). Thirty-eight patients (46%) reported massage use and 21 patients (25%) used cognitive-behavioral therapy (Table 2).
Integrative Therapies Desired
A majority of PCPs identified acupuncture (n = 20, 71%), chiropractic (n = 19, 68%), and massage (n = 19, 68%) as therapies they would most like to have available for patients with chronic pain (Figure 2). Similarly, patients identified massage (n = 42, 51%), chiropractic (n = 34, 41%), and acupuncture (n = 27, 33%) as most desired. Seventeen patients (21%) expressed interest in movement therapies.
Barriers to Integrative Therapies Use
When asked about barriers to use, 26 PCPs (93%) identified access to services as a somewhat or extremely likely barrier, and 22 identified time constraints (79%) (Table 3). However, 17 PCPs (61%) noted lack of familiarity, and 18 (64%) noted a lack of scientific evidence as barriers to recommending integrative modalities. Among patients, 33 (40%) indicated not knowing what services were available at their facility as a barrier, 32 (39%) were not familiar with specific therapies, and 21 (25%) indicated a lack of clarity about the benefits of a specific therapy. Only 14 patients (17%) indicated that there were no obstacles to use.
DISCUSSION
Use of integrative therapies, including complementary treatments, is an increasingly important part of chronic pain management. This survey study suggests VA PCPs are willing to recommend integrative therapies and patients with chronic back pain both desire and use several therapies. Moreover, both groups expressed interest in greater availability of similar therapies. The results also highlight key barriers, such as knowledge gaps, that should be addressed to increase the uptake of integrative modalities for managing chronic pain.
An increasing number of US adults are using complementary health approaches, an important component of integrative therapy.12 This trend includes an increase in use for pain management, from 42.3% in 2002 to 49.2% in 2022; chiropractic care, acupuncture, and massage were most frequently used.12 Similarly, chiropractic, acupuncture and massage were most often used by this sample of veterans with chronic back pain and were identified by the highest percentages of PCPs and patients as the therapies they would most like available.
There were areas where the opinions of patients and clinicians differed. As has been seen previously reported, clinicians largely recommended cognitive-behavioral therapy while patients showed less interest.17 Additionally, while patients expressed interest in the availability of movement therapies, such as yoga, PCPs expressed more interest in other strategies, such as trigger point injections. These differences may reflect true preference or a tendency for clinicians and patients to select therapies with which they are more familiar. Additional research is needed to better understand the acceptability and potential use of integrative health treatments across a broad array of therapeutic options.
Despite VHA policy requiring facilities to provide certain complementary and integrative health modalities, almost all PCPs identified access to services as a major obstacle.15 Based on evidence and a rigorous vetting process, services currently required on-site, via telehealth, or through community partners include acupuncture and battlefield acupuncture (battlefield auricular acupuncture), biofeedback, clinical hypnosis, guided imagery, medical massage therapy, medication, tai chi/qigong, and yoga. Optional approaches, which may be made available to veterans, include chiropractic and healing touch. Outside the VHA, some states have introduced or enacted legislation mandating insurance coverage of nonpharmacological pain treatments.18 However, these requirements and mandates do not help address challenges such as the availability of trained/qualified practitioners.19,20 Ensuring access to complementary and integrative health treatments requires a more concerted effort to ensure that supply meets demand. It is also important to acknowledge the budgetary and physical space constraints that further limit access to services. Although expansion and integration of integrative medicine services remain a priority within the VA Whole Health program, implementation is contingent on available financial and infrastructure resources.
Time was also identified by PCPs as a barrier to recommending integrative therapies to patients. Developing and implementing time-efficient communication strategies for patient education such as concise talking points and informational handouts could help address this barrier. Furthermore, leveraging existing programs and engaging the entire health care team in patient education and referral could help increase integrative and complementary therapy uptake and use.
Although access and time were identified as major barriers, these findings also suggest that PCP and patient knowledge are another target area for enhancing the use of complementary and integrative therapies. Like prior research, most clinicians identified a lack of familiarity with certain services and a lack of scientific evidence as extremely or somewhat likely to affect their ability to offer integrative services to patients with chronic pain.21 Likewise, about 40% of patients identified being unfamiliar with a specific therapy as one of the major obstacles to receiving integrative therapies, with a similar number identifying PCPs as a source of information. The lack of familiarity may be due in part to the evolving nomenclature, with terms such as alternative, complementary, and integrative used to describe approaches outside what is often considered conventional medicine.10 On the other hand, there has also been considerable expansion in the number of therapies within this domain, along with an expanding evidence base. This suggests a need for targeted educational strategies for clinicians and patients, which can be rapidly deployed and continuously adapted as new therapies and evidence emerge.
Limitations
There are some inherent limitations with a survey-based approach, including sampling, non-response, and social desirability biases. In addition, this study only included PCPs and patients affiliated with a single VA medical center. Steps to mitigate these limitations included maintaining survey anonymity and reporting information about respondent characteristics to enhance transparency about the representativeness of the study findings.
CONCLUSIONS
Expanding the use of nonpharmacological pain treatments, including integrative modalities, is essential for safe and effective chronic pain management and reducing opioid use. Our findings show that VA PCPs and patients with chronic back pain are interested in and have some experience with certain integrative therapies. However, even within the context of a health care system that supports the use of integrative therapies for chronic pain as part of whole person care, increasing uptake will require addressing access and time-related constraints as well as ongoing clinician and patient education.
More than 50 million US adults report experiencing chronic pain, with nearly 7% experiencing high-impact chronic pain.1-3 Chronic pain negatively affects daily function, results in lost productivity, is a leading cause of disability, and is more prevalent among veterans compared with the general population.1,2,4-6 Estimates from 2021 suggest the prevalence of chronic pain among veterans exceeds 30%; > 11% experienced high-impact chronic pain.1
Primary care practitioners (PCPs) have a prominent role in chronic pain management. Pharmacologic options for treating pain, once a mainstay of therapy, present several challenges for patients and PCPs, including drug-drug interactions and adverse effects.7 The US opioid epidemic and shift to a biopsychosocial model of chronic pain care have increased emphasis on nonpharmacologic treatment options.8,9 These include integrative modalities, which incorporate conventional approaches with an array of complementary health approaches.10-12
Integrative therapy is a prominent feature in whole person care, which may be best exemplified by the US Department of Veterans Affairs (VA) Whole Health System of care.13-14 Whole health empowers an individual to take charge of their health and well-being so they can “live their life to the fullest.”14 As implemented in the Veterans Health Administration (VHA), whole health includes the use of evidence-based
METHODS
Using a cross-sectional survey design, PCPs and patients with chronic back pain affiliated with the VA Ann Arbor Healthcare System were invited to participate in separate but similar surveys to assess knowledge, interest, and use of nonpharmacologic integrative modalities for the treatment of chronic pain. In May, June, and July 2023, 78 PCPs received 3 email
Both survey instruments are available upon request, were developed by the study team, and included a mix of yes/no questions, “select all that apply” items, Likert scale response items, and open-ended questions. For one question about which modalities they would like available, the respondent was instructed to select up to 5 modalities. The instruments were extensively pretested by members of the study team, which included 2 PCPs and a nonveteran with chronic back pain.
The list of integrative modalities included in the survey was derived from the tier 1 and tier 2 complementary and integrative health modalities identified in a VHA Directive on complementary and integrative health.15,16 Tier 1 approaches are considered to have sufficient evidence and must be made available to veterans either within a VA medical facility or in the community. Tier 2 approaches are generally considered safe and may be made available but do not have sufficient evidence to mandate their provision. For participant ease, the integrative modalities were divided into 5 subgroups: manual therapies, energy/biofield therapies, mental health therapies, nutrition counseling, and movement therapies. The clinician survey assessed clinicians’ training and interest, clinical and personal use, and perceived barriers to providing integrative modalities for chronic pain. Professional and personal demographic data were also collected. Similarly, the patient survey assessed use of integrative therapies, perceptions of and interest in integrative modalities, and potential barriers to use. Demographic and health-related information was also collected.
Data analysis included descriptive statistics (eg, frequency counts, means, medians) and visual graphic displays. Separate analyses were conducted for clinicians and patients in addition to a comparative analysis of the use and potential interest in integrative modalities. Analysis were conducted using R software. This study was deemed nonresearch quality improvement by the VA Ann Arbor Healthcare System facility research oversight board and institutional review board approval was not solicited.
RESULTS
Twenty-eight clinicians completed the survey, yielding a participation rate of 36%. Participating clinicians had a median (IQR) age of 48 years (9.5), 15 self-identified as White (54%), 8 as Asian (29%), 15 as female (54%), 26 as non-Hispanic (93%), and 25 were medical doctors or doctors of osteopathy (89%). Nineteen (68%) worked at the main hospital outpatient clinic, and 9 practiced at community-based outpatient clinics (CBOCs). Thirteen respondents (46%) reported having no formal education or training in integrative approaches. Among those with prior training, 8 clinicians had nutrition counseling (29%) and 7 had psychologic therapy training (25%). Thirteen respondents (46%) also reported using integrative modalities for personal health needs: 8 used psychological therapies, 8 used movement therapies, 10 used integrative modalities for stress management or relaxation, and 8 used them for physical symptoms (Table 1).
Overall, 85 of 200 patients (43%) responded to the study survey. Two patients indicated they did not have chronic back pain and were excluded. Patients had a median (IQR) age of 66 (20) years, with 66 self-identifying as White (80%), 69 as male (83%), and 66 as non-Hispanic (80%). Forty-four patients (53%) received care at CBOCs. Forty-seven patients reported excellent, very good, or good overall health (57%), while 53 reported excellent, very good, or good mental health (64%). Fifty-nine patients reported back pain duration > 5 years (71%), and 67 (81%) indicated experiencing back pain flare-ups at least once per week over the previous 12 months. Sixty patients (72%) indicated they were somewhat or very interested in using integrative therapies as a back pain treatment; however, 40 patients (48%) indicated they had not received information about these therapies. Among those who indicated they had received information, the most frequently reported source was their PCP (41%). Most patients (72%) also reported feeling somewhat to very comfortable discussing integrative medicine therapies with their PCP.
Integrative Therapy Recommendations and Use
PCPs reported recommending multiple integrative modalities: 23 (82%) recommended cognitive-behavioral therapy, 22 (79%) recommended acupuncture, 21 (75%) recommended chiropractic, 19 (68%) recommended battlefield acupuncture, recommended massage 18 (64%), 17 (61%) recommended meditation or mindfulness, and 15 (54%) recommended movement therapies such as yoga or tai chi/qigong (Figure 1). The only therapies used by at least half of the patients were chiropractic used by 59 patients (71%) and acupuncture by 42 patients (51%). Thirty-eight patients (46%) reported massage use and 21 patients (25%) used cognitive-behavioral therapy (Table 2).
Integrative Therapies Desired
A majority of PCPs identified acupuncture (n = 20, 71%), chiropractic (n = 19, 68%), and massage (n = 19, 68%) as therapies they would most like to have available for patients with chronic pain (Figure 2). Similarly, patients identified massage (n = 42, 51%), chiropractic (n = 34, 41%), and acupuncture (n = 27, 33%) as most desired. Seventeen patients (21%) expressed interest in movement therapies.
Barriers to Integrative Therapies Use
When asked about barriers to use, 26 PCPs (93%) identified access to services as a somewhat or extremely likely barrier, and 22 identified time constraints (79%) (Table 3). However, 17 PCPs (61%) noted lack of familiarity, and 18 (64%) noted a lack of scientific evidence as barriers to recommending integrative modalities. Among patients, 33 (40%) indicated not knowing what services were available at their facility as a barrier, 32 (39%) were not familiar with specific therapies, and 21 (25%) indicated a lack of clarity about the benefits of a specific therapy. Only 14 patients (17%) indicated that there were no obstacles to use.
DISCUSSION
Use of integrative therapies, including complementary treatments, is an increasingly important part of chronic pain management. This survey study suggests VA PCPs are willing to recommend integrative therapies and patients with chronic back pain both desire and use several therapies. Moreover, both groups expressed interest in greater availability of similar therapies. The results also highlight key barriers, such as knowledge gaps, that should be addressed to increase the uptake of integrative modalities for managing chronic pain.
An increasing number of US adults are using complementary health approaches, an important component of integrative therapy.12 This trend includes an increase in use for pain management, from 42.3% in 2002 to 49.2% in 2022; chiropractic care, acupuncture, and massage were most frequently used.12 Similarly, chiropractic, acupuncture and massage were most often used by this sample of veterans with chronic back pain and were identified by the highest percentages of PCPs and patients as the therapies they would most like available.
There were areas where the opinions of patients and clinicians differed. As has been seen previously reported, clinicians largely recommended cognitive-behavioral therapy while patients showed less interest.17 Additionally, while patients expressed interest in the availability of movement therapies, such as yoga, PCPs expressed more interest in other strategies, such as trigger point injections. These differences may reflect true preference or a tendency for clinicians and patients to select therapies with which they are more familiar. Additional research is needed to better understand the acceptability and potential use of integrative health treatments across a broad array of therapeutic options.
Despite VHA policy requiring facilities to provide certain complementary and integrative health modalities, almost all PCPs identified access to services as a major obstacle.15 Based on evidence and a rigorous vetting process, services currently required on-site, via telehealth, or through community partners include acupuncture and battlefield acupuncture (battlefield auricular acupuncture), biofeedback, clinical hypnosis, guided imagery, medical massage therapy, medication, tai chi/qigong, and yoga. Optional approaches, which may be made available to veterans, include chiropractic and healing touch. Outside the VHA, some states have introduced or enacted legislation mandating insurance coverage of nonpharmacological pain treatments.18 However, these requirements and mandates do not help address challenges such as the availability of trained/qualified practitioners.19,20 Ensuring access to complementary and integrative health treatments requires a more concerted effort to ensure that supply meets demand. It is also important to acknowledge the budgetary and physical space constraints that further limit access to services. Although expansion and integration of integrative medicine services remain a priority within the VA Whole Health program, implementation is contingent on available financial and infrastructure resources.
Time was also identified by PCPs as a barrier to recommending integrative therapies to patients. Developing and implementing time-efficient communication strategies for patient education such as concise talking points and informational handouts could help address this barrier. Furthermore, leveraging existing programs and engaging the entire health care team in patient education and referral could help increase integrative and complementary therapy uptake and use.
Although access and time were identified as major barriers, these findings also suggest that PCP and patient knowledge are another target area for enhancing the use of complementary and integrative therapies. Like prior research, most clinicians identified a lack of familiarity with certain services and a lack of scientific evidence as extremely or somewhat likely to affect their ability to offer integrative services to patients with chronic pain.21 Likewise, about 40% of patients identified being unfamiliar with a specific therapy as one of the major obstacles to receiving integrative therapies, with a similar number identifying PCPs as a source of information. The lack of familiarity may be due in part to the evolving nomenclature, with terms such as alternative, complementary, and integrative used to describe approaches outside what is often considered conventional medicine.10 On the other hand, there has also been considerable expansion in the number of therapies within this domain, along with an expanding evidence base. This suggests a need for targeted educational strategies for clinicians and patients, which can be rapidly deployed and continuously adapted as new therapies and evidence emerge.
Limitations
There are some inherent limitations with a survey-based approach, including sampling, non-response, and social desirability biases. In addition, this study only included PCPs and patients affiliated with a single VA medical center. Steps to mitigate these limitations included maintaining survey anonymity and reporting information about respondent characteristics to enhance transparency about the representativeness of the study findings.
CONCLUSIONS
Expanding the use of nonpharmacological pain treatments, including integrative modalities, is essential for safe and effective chronic pain management and reducing opioid use. Our findings show that VA PCPs and patients with chronic back pain are interested in and have some experience with certain integrative therapies. However, even within the context of a health care system that supports the use of integrative therapies for chronic pain as part of whole person care, increasing uptake will require addressing access and time-related constraints as well as ongoing clinician and patient education.
- Rikard SM, Strahan AE, Schmit KM, et al. Chronic pain among adults — United States, 2018-2021. MMWR Morb Mortal Wkly Rep. 2023;72:379-385. doi:10.15585/mmwr.mm7215a1
- Yong RJ, Mullins PM, Bhattacharyya N. Prevalence of chronic pain among adults in the United States. Pain. 2022;163:E328-E332. doi:10.1097/j.pain.0000000000002291
- Nahin RL, Feinberg T, Kapos FP, Terman GW. Estimated rates of incident and persistent chronic pain among US adults, 2019-2020. JAMA Netw Open. 2023;6:e2313563. doi:10.1001/jamanetworkopen.2023.13563
- Ferrari AJ, Santomauro DF, Aali A, et al. Global incidence, prevalence, years lived with disability (YLDs), disability-adjusted life-years (DALYs), and healthy life expectancy (HALE) for 371 diseases and injuries in 204 countries and territories and 811 subnational locations, 1990–2021: a systematic analysis for the Global Burden of Disease Study 2021. The Lancet. 2024;403:2133-2161. doi:10.1016/S0140-6736(24)00757-8 5.
- Qureshi AR, Patel M, Neumark S, et al. Prevalence of chronic non-cancer pain among military veterans: a systematic review and meta-analysis of observational studies. BMJ Mil Health. 2025;171:310-314. doi:10.1136/military-2023-002554
- Feldman DE, Nahin RL. Disability among persons with chronic severe back pain: results from a nationally representative population-based sample. J Pain. 2022;23:2144-2154. doi:10.1016/j.jpain.2022.07.016
- Qaseem A, Wilt TJ, McLean RM, Forciea MA. Noninvasive treatments for acute, subacute, and chronic low back pain: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:514-530. doi:10.7326/M16-2367
- van Erp RMA, Huijnen IPJ, Jakobs MLG, Kleijnen J, Smeets RJEM. Effectiveness of primary care interventions using a biopsychosocial approach in chronic low back pain: a systematic review. Pain Practice. 2019;19:224-241. doi:10.1111/papr.12735
- Chou R, Deyo R, Friedly J, et al. Nonpharmacologic therapies for low back pain: a systematic review for an American College of physicians clinical practice guideline. Ann Intern Med. 2017;166:493-505. doi:10.7326/M16-2459
- Complementary, alternative, or integrative health: what’s in a name? National Institutes of Health, National Center for Complementary and Integrative Health. Updated April 2021. Accessed December 15, 2025. https://www.nccih.nih.gov/health/complementary-alternative-or-integrative-health-whats-in-a-name.
- Taylor SL, Elwy AR. Complementary and alternative medicine for US veterans and active duty military personnel promising steps to improve their health. Med Care. 2014;52:S1-S4. doi:10.1097/MLR.0000000000000270.
- Nahin RL, Rhee A, Stussman B. Use of complementary health approaches overall and for pain management by US adults. JAMA. 2024;331:613-615. doi:10.1001/jama.2023.26775
- Gantt CJ, Donovan N, Khung M. Veterans Affairs’ Whole Health System of Care for transitioning service members and veterans. Mil Med. 2023;188:28-32. doi:10.1093/milmed/usad047
- Bokhour BG, Hyde J, Kligler B, et al. From patient outcomes to system change: evaluating the impact of VHA’s implementation of the Whole Health System of Care. Health Serv Res. 2022;57:53-65. doi:10.1111/1475-6773.13938
- Department of Veterans Affairs VHA. VHA Policy Directive 1137: Provision of Complementary and Integrative Health. December 2022. Accessed December 15, 2025. https://www.va.gov/VHApublications/ViewPublication.asp?pub_ID=10072
- Giannitrapani KF, Holliday JR, Miake-Lye IM, Hempel S, Taylor SL. Synthesizing the strength of the evidence of complementary and integrative health therapies for pain. Pain Med. 2019;20:1831-1840. doi:10.1093/pm/pnz068
- Belitskaya-Levy I, David Clark J, Shih MC, Bair MJ. Treatment preferences for chronic low back pain: views of veterans and their providers. J Pain Res. 2021;14:161-171. doi:10.2147/JPR.S290400
- Onstott TN, Hurst S, Kronick R, Tsou AC, Groessl E, McMenamin SB. Health insurance mandates for nonpharmacological pain treatments in 7 US states. JAMA Netw Open. 2024;7:E245737. doi:10.1001/jamanetworkopen.2024.5737
- Sullivan M, Leach M, Snow J, Moonaz S. The North American yoga therapy workforce survey. Complement Ther Med. 2017;31:39-48. doi:10.1016/j.ctim.2017.01.006
- Bolton R, Ritter G, Highland K, Larson MJ. The relationship between capacity and utilization of nonpharmacologic therapies in the US Military Health System. BMC Health Serv Res. 2022;22. doi:10.1186/s12913-022-07700-4
- Stussman BJ, Nahin RL, Barnes PM, Scott R, Feinberg T, Ward BW. Reasons office-based physicians in the United States recommend common complementary health approaches to patients: an exploratory study using a national survey. J Integr Complement Med. 2022;28:651-663. doi:10.1089/jicm.2022.0493
- Rikard SM, Strahan AE, Schmit KM, et al. Chronic pain among adults — United States, 2018-2021. MMWR Morb Mortal Wkly Rep. 2023;72:379-385. doi:10.15585/mmwr.mm7215a1
- Yong RJ, Mullins PM, Bhattacharyya N. Prevalence of chronic pain among adults in the United States. Pain. 2022;163:E328-E332. doi:10.1097/j.pain.0000000000002291
- Nahin RL, Feinberg T, Kapos FP, Terman GW. Estimated rates of incident and persistent chronic pain among US adults, 2019-2020. JAMA Netw Open. 2023;6:e2313563. doi:10.1001/jamanetworkopen.2023.13563
- Ferrari AJ, Santomauro DF, Aali A, et al. Global incidence, prevalence, years lived with disability (YLDs), disability-adjusted life-years (DALYs), and healthy life expectancy (HALE) for 371 diseases and injuries in 204 countries and territories and 811 subnational locations, 1990–2021: a systematic analysis for the Global Burden of Disease Study 2021. The Lancet. 2024;403:2133-2161. doi:10.1016/S0140-6736(24)00757-8 5.
- Qureshi AR, Patel M, Neumark S, et al. Prevalence of chronic non-cancer pain among military veterans: a systematic review and meta-analysis of observational studies. BMJ Mil Health. 2025;171:310-314. doi:10.1136/military-2023-002554
- Feldman DE, Nahin RL. Disability among persons with chronic severe back pain: results from a nationally representative population-based sample. J Pain. 2022;23:2144-2154. doi:10.1016/j.jpain.2022.07.016
- Qaseem A, Wilt TJ, McLean RM, Forciea MA. Noninvasive treatments for acute, subacute, and chronic low back pain: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:514-530. doi:10.7326/M16-2367
- van Erp RMA, Huijnen IPJ, Jakobs MLG, Kleijnen J, Smeets RJEM. Effectiveness of primary care interventions using a biopsychosocial approach in chronic low back pain: a systematic review. Pain Practice. 2019;19:224-241. doi:10.1111/papr.12735
- Chou R, Deyo R, Friedly J, et al. Nonpharmacologic therapies for low back pain: a systematic review for an American College of physicians clinical practice guideline. Ann Intern Med. 2017;166:493-505. doi:10.7326/M16-2459
- Complementary, alternative, or integrative health: what’s in a name? National Institutes of Health, National Center for Complementary and Integrative Health. Updated April 2021. Accessed December 15, 2025. https://www.nccih.nih.gov/health/complementary-alternative-or-integrative-health-whats-in-a-name.
- Taylor SL, Elwy AR. Complementary and alternative medicine for US veterans and active duty military personnel promising steps to improve their health. Med Care. 2014;52:S1-S4. doi:10.1097/MLR.0000000000000270.
- Nahin RL, Rhee A, Stussman B. Use of complementary health approaches overall and for pain management by US adults. JAMA. 2024;331:613-615. doi:10.1001/jama.2023.26775
- Gantt CJ, Donovan N, Khung M. Veterans Affairs’ Whole Health System of Care for transitioning service members and veterans. Mil Med. 2023;188:28-32. doi:10.1093/milmed/usad047
- Bokhour BG, Hyde J, Kligler B, et al. From patient outcomes to system change: evaluating the impact of VHA’s implementation of the Whole Health System of Care. Health Serv Res. 2022;57:53-65. doi:10.1111/1475-6773.13938
- Department of Veterans Affairs VHA. VHA Policy Directive 1137: Provision of Complementary and Integrative Health. December 2022. Accessed December 15, 2025. https://www.va.gov/VHApublications/ViewPublication.asp?pub_ID=10072
- Giannitrapani KF, Holliday JR, Miake-Lye IM, Hempel S, Taylor SL. Synthesizing the strength of the evidence of complementary and integrative health therapies for pain. Pain Med. 2019;20:1831-1840. doi:10.1093/pm/pnz068
- Belitskaya-Levy I, David Clark J, Shih MC, Bair MJ. Treatment preferences for chronic low back pain: views of veterans and their providers. J Pain Res. 2021;14:161-171. doi:10.2147/JPR.S290400
- Onstott TN, Hurst S, Kronick R, Tsou AC, Groessl E, McMenamin SB. Health insurance mandates for nonpharmacological pain treatments in 7 US states. JAMA Netw Open. 2024;7:E245737. doi:10.1001/jamanetworkopen.2024.5737
- Sullivan M, Leach M, Snow J, Moonaz S. The North American yoga therapy workforce survey. Complement Ther Med. 2017;31:39-48. doi:10.1016/j.ctim.2017.01.006
- Bolton R, Ritter G, Highland K, Larson MJ. The relationship between capacity and utilization of nonpharmacologic therapies in the US Military Health System. BMC Health Serv Res. 2022;22. doi:10.1186/s12913-022-07700-4
- Stussman BJ, Nahin RL, Barnes PM, Scott R, Feinberg T, Ward BW. Reasons office-based physicians in the United States recommend common complementary health approaches to patients: an exploratory study using a national survey. J Integr Complement Med. 2022;28:651-663. doi:10.1089/jicm.2022.0493
Primary Care Clinician and Patient Knowledge, Interest, and Use of Integrative Treatment Options for Chronic Low Back Pain Management
Primary Care Clinician and Patient Knowledge, Interest, and Use of Integrative Treatment Options for Chronic Low Back Pain Management

Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
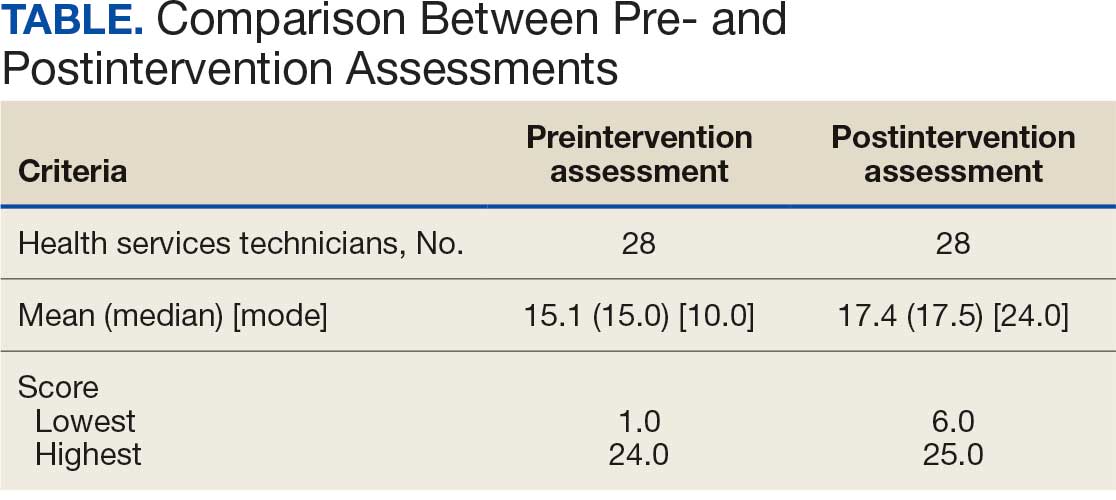
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Development and Validation of an Administrative Algorithm to Identify Veterans With Epilepsy
Development and Validation of an Administrative Algorithm to Identify Veterans With Epilepsy
Epilepsy affects about 4.5 million people in the United States and 150,000 new individuals are diagnosed each year.1,2 In 2019, epilepsy-attributable health care spending for noninstitutionalized people was around $5.4 billion and total epilepsy-attributable and epilepsy or seizure health care-related costs totaled $54 billion.3
Accurate surveillance of epilepsy in large health care systems can potentially improve health care delivery and resource allocation. A 2012 Institute of Medicine (IOM) report identified 13 recommendations to guide public health action on epilepsy, including validation of standard definitions for case ascertainment, identification of epilepsy through screening programs or protocols, and expansion of surveillance to better understand disease burden.4
A systematic review of validation studies concluded that it is reasonable to use administrative data to identify people with epilepsy in epidemiologic research. Combining The International Classification of Diseases (ICD) codes for epilepsy (ICD-10, G40-41; ICD-9, 345) with antiseizure medications (ASMs) could provide high positive predictive values (PPVs) and combining symptoms codes for convulsions (ICD-10, R56; ICD-9, 780.3, 780.39) with ASMs could lead to high sensitivity.5 However, identifying individuals with epilepsy from administrative data in large managed health care organizations is challenging.6 The IOM report noted that large managed health care organizations presented varying incidence and prevalence estimates due to differing methodology, geographic area, demographics, and definitions of epilepsy.
The Veterans Health Administration (VHA) is the largest integrated US health care system, providing care to > 9.1 million veterans.7 To improve the health and well-being of veterans with epilepsy (VWEs), a network of sites was established in 2008 called the US Department of Veterans Affairs (VA) Epilepsy Centers of Excellence (ECoE). Subsequent to the creation of the ECoE, efforts were made to identify VWEs within VHA databases.8,9 Prior to fiscal year (FY) 2016, the ECoE adopted a modified version of a well-established epilepsy diagnostic algorithm developed by Holden et al for large managed care organizations.10 The original algorithm identified patients by cross-matching ASMs with ICD-9 codes for an index year. But it failed to capture a considerable number of stable patients with epilepsy in the VHA due to incomplete documentation, and had false positives due to inclusion of patients identified from diagnostic clinics. The modified algorithm the ECoE used prior to FY 2016 considered additional prior years and excluded encounters from diagnostic clinics. The result was an improvement in the sensitivity and specificity of the algorithm. Researchers evaluating 500 patients with epilepsy estimated that the modified algorithm had a PPV of 82.0% (95% CI, 78.6%-85.4%).11
After implementation of ICD-10 codes in the VHA in FY 2016, the task of reliably and efficiently identifying VWE led to a 3-tier algorithm. This article presents a validation of the different tiers of this algorithm after the implementation of ICD-10 diagnosis codes and summarizes the surveillance data collected over the years within the VHA showing the trends of epilepsy.
Methods
The VHA National Neurology office commissioned a Neurology Cube dashboard in FY 2021 in collaboration with VHA Support Service Center (VSSC) for reporting and surveillance of VWEs as a quality improvement initiative. The Neurology Cube uses a 3-tier system for identifying VWE in the VHA databases. VSSC programmers extract data from the VHA Corporate Data Warehouse (CDW) and utilize Microsoft SQL Server and Microsoft Power BI for Neurology Cube reports. The 3-tier system identifies VWE and divides them into distinct groups. The first tier identifies VWE with the highest degree of confidence; Tiers 2 and 3 represent identification with successively lesser degrees of confidence (Figure 1).
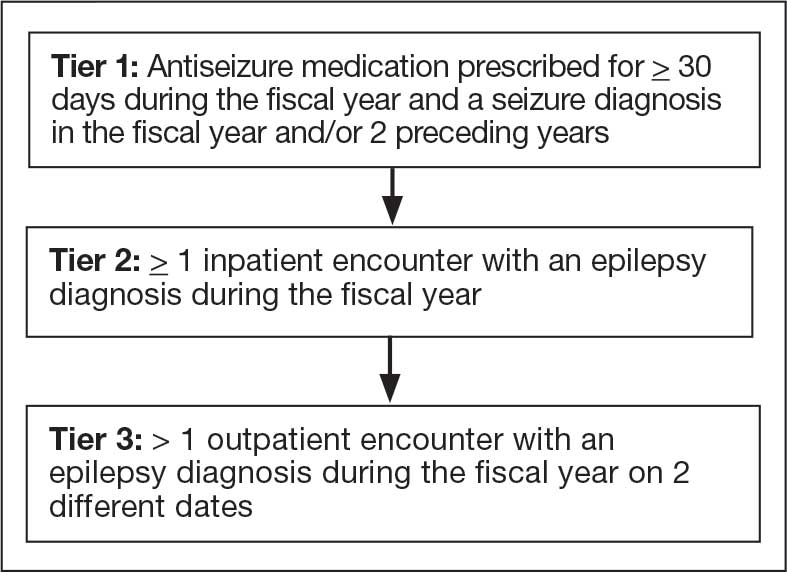
Tier 1
Definition. For a given index year and the preceding 2 years, any of following diagnosis codes on ≥ 1 clinical encounter are considered: 345.xx (epilepsy in ICD-9), 780.3x (other convulsions in ICD-9), G40.xxx (epilepsy in ICD-10), R40.4 (transient alteration of awareness), R56.1 (posttraumatic seizures), or R56.9 (unspecified convulsions). To reduce false positive rates, EEG clinic visits, which may include long-term monitoring, are excluded. Patients identified with ICD codes are then evaluated for an ASM prescription for ≥ 30 days during the index year. ASMs are listed in Appendix 1.
Validation. The development and validation of ICD-9 diagnosis codes crossmatched with an ASM prescription in the VHA has been published elsewhere.11 In FY 2017, after implementation of ICD-10 diagnostic codes, Tier 1 development and validation was performed in 2 phases. Even though Tier 1 study phases were conducted and completed during FY 2017, the patients for Tier 1 were identified from evaluation of FY 2016 data (October 1, 2015, to September 30, 2016). After the pilot analysis, the Tier 1 definition was implemented, and a chart review of 625 randomized patients was conducted at 5 sites for validation. Adequate preliminary data was not available to perform a sample size estimation for this study. Therefore, a practical target of 125 patients was set for Tier 1 from each site to obtain a final sample size of 625 patients. This second phase validated that the crossmatch of ICD-10 diagnosis codes with ASMs had a high PPV for identifying VWE.
Tiers 2 and 3
Definitions. For an index year, Tier 2 includes patients with ≥ 1 inpatient encounter documentation of either ICD-9 345.xx or ICD-10 G40.xxx, excluding EEG clinics. Tier 3 Includes patients who have had ≥ 2 outpatient encounters with diagnosis codes 345.xx or G40.xxx on 2 separate days, excluding EEG clinics. Tiers 2 and 3 do not require ASM prescriptions; this helps to identify VWEs who may be getting their medications outside of VHA or those who have received a new diagnosis.
Validations. Tiers 2 and 3 were included in the epilepsy identification algorithm in FY 2021 after validation was performed on a sample of 8 patients in each tier. Five patients were subsequently identified as having epilepsy in Tier 2 and 6 patients were identified in Tier 3. A more comprehensive validation of Tiers 2 and 3 was performed during FY 2022 that included patients at 5 sites seen during FY 2019 to FY 2022. Since yearly trends showed only about 8% of total patients were identified as having epilepsy through Tiers 2 and 3 we sought ≥ 20 patients per tier for the 5 sites for a total of 200 patients to ensure representation across the VHA. The final count was 126 patients for Tier 2 and 174 patients for Tier 3 (n = 300).
Gold Standard Criteria for Epilepsy Diagnosis
We used the International League Against Epilepsy (ILAE) definition of epilepsy for the validation of the 3 algorithm tiers. ILAE defines epilepsy as ≥ 2 unprovoked (or reflex) seizures occurring > 24 hours apart or 1 unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (≥ 60%) after 2 unprovoked seizures, occurring over the next 10 years.12
A standard protocol was provided to evaluators to identify patients using the VHA Computerized Patient Record System (Appendix 1). After review, evaluators categorized each patient in 1 of 4 ways: (1) Yes, definite: The patient’s health care practitioner (HCP) believes the patient has epilepsy and is treating with medication; (2) Yes, uncertain: The HCP has enough suspicion of epilepsy that a medication is prescribed, but uncertainty is expressed of the diagnosis; (3) No, definite: The HCP does not believe the patient has epilepsy and is therefore not treating with medication for seizure; (4) No, uncertain: The HCP is not treating with medication for epilepsy, because the diagnostic suspicion is not high enough, but there is suspicion for epilepsy.
As a quality improvement operational project, the Epilepsy National Program Office approved this validation project and determined that institutional review board approval was not required.
Statistical Analysis
Counts and percentages were computed for categories of epilepsy status. PPV of each tier was estimated with asymptotic 95% CIs.
Results
ICD-10 codes for 480 patients were evaluated in Tier 1 phase 1; 13.8% were documented with G40.xxx, 27.9% with R56.1, 34.4% with R56.9, and 24.0% with R40.4 (Appendix 2). In total, 68.1% fulfilled the criteria of epilepsy, 19.2% did not, and 12.7% were uncertain). From the validation of Tier 1 phase 2 (n = 625), the PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) was 85.1% (95% CI, 82.1%-87.8%) (Table).
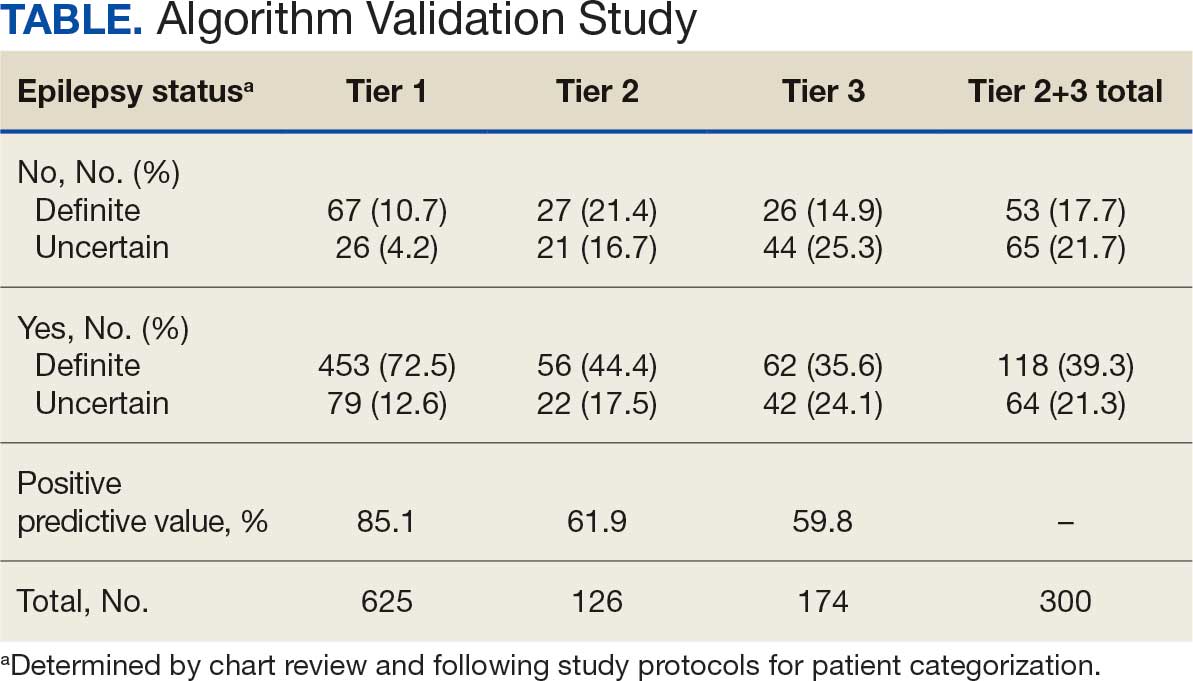
Of 300 patients evaluated, 126 (42.0%) were evaluated for Tier 2 with a PPV of 61.9% (95% CI, 53.4%-70.4%), and 174 (58.0%) patients were evaluated for Tier 3 with a PPV of 59.8% (95% CI, 52.5%-67.1%. The PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) were combined to calculate the PPV. Estimates of VHA VWE counts were computed for each tier from FY 2014 to FY 2023 using the VSSC Neurology Cube (Figure 2). For all years, > 92% patients were classified using the Tier 1 definition.
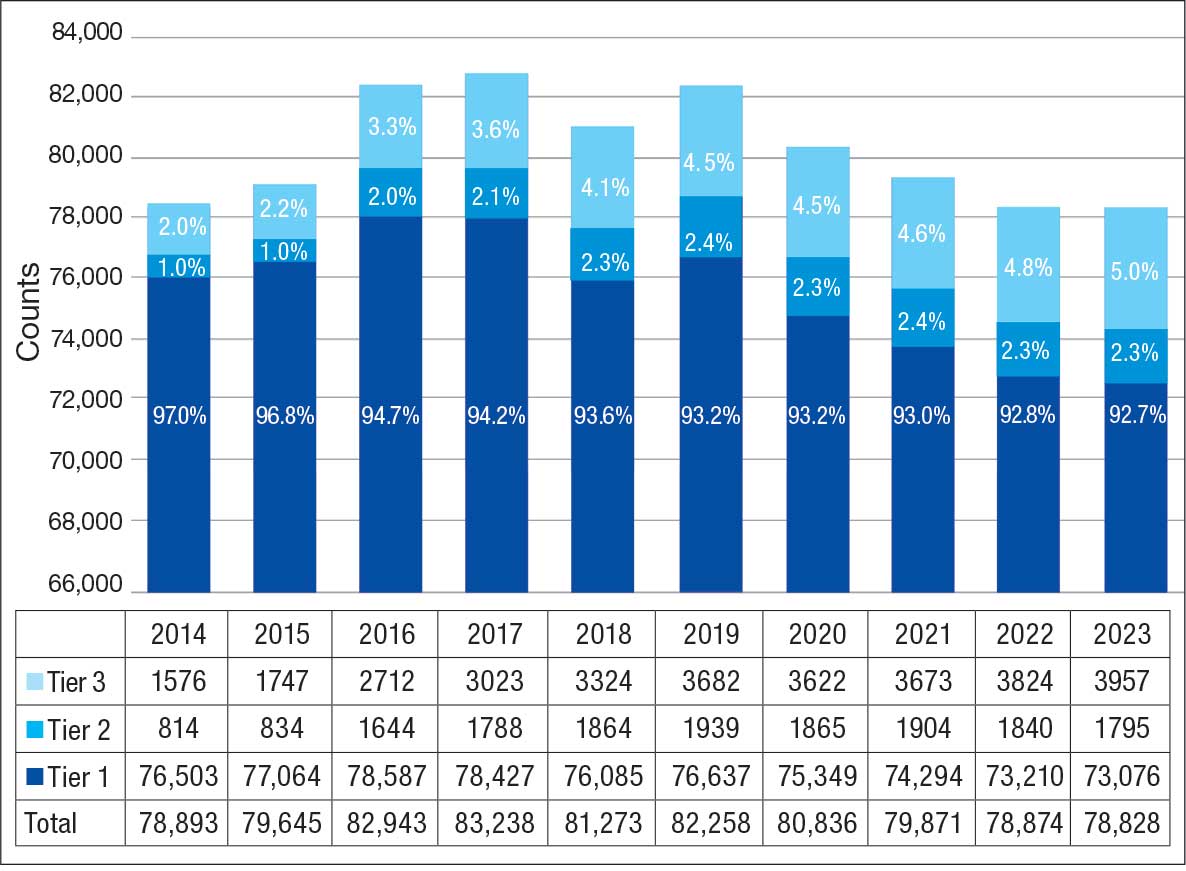
Discussion
The development and validation of the 3-tier diagnostic algorithm represents an important advancement in the surveillance and management of epilepsy among veterans within the VHA. The validation of this algorithm also demonstrates its practical utility in a large, integrated health care system.
Specific challenges were encountered when attempting to use pre-existing algorithms; these challenges included differences in the usage patterns of diagnostic codes and the patterns of ASM use within the VHA. These challenges prompted the need for a tailored approach, which led to the development of this algorithm. The inclusion of additional ICD-10 codes led to further revisions and subsequent validation. While many of the basic concepts of the algorithm, including ICD codes and ASMs, could work in other institutions, it would be wise for health care organizations to develop their own algorithms because of certain variables, including organizational size, patient demographics, common comorbidities, and the specific configurations of electronic health records and administrative data systems.
Studies have shown that ICD-10 codes for epilepsy (G40.* and/or R56.9) perform well in identifying epilepsy whether they are assigned by neurologists (sensitivity, 97.7%; specificity, 44.1%; PPV, 96.2%; negative predictive value, 57.7%), or in emergency department or hospital discharges (PPV, 75.5%).13,14 The pilot study of the algorithm’s Tier 1 development (phase 1) evaluated whether the selected ICD-10 diagnostic codes accurately included the VWE population within the VHA and revealed that while most codes (eg, epilepsy [G40.xxx]; posttraumatic seizures [R56.1]; and unspecified convulsions [R56.9]), had a low false positive rate (< 16%), the R40.4 code (transient alteration of awareness) had a higher false positivity of 42%. While this is not surprising given the broad spectrum of conditions that can manifest as transient alteration of awareness, it underscores the inherent challenges in diagnosing epilepsy using diagnosis codes.
In phase 2, the Tier 1 algorithm was validated as effective for identifying VWE in the VHA system, as its PPV was determined to be high (85%). In comparison, Tiers 2 and 3, whose criteria did not require data on VHA prescribed ASM use, had lower tiers of epilepsy predictability (PPV about 60% for both). This was thought to be acceptable because Tiers 2 and 3 represent a smaller population of the identified VWEs (about 8%). These VWEs may otherwise have been missed, partly because veterans are not required to get ASMs from the VHA.
Upon VHA implementation in FY 2021, this diagnostic algorithm exhibited significant clinical utility when integrated within the VSSC Neurology Cube. It facilitated an efficient approach to identifying VWEs using readily available databases. This led to better tracking of real-time epilepsy cases, which facilitated improving current resource allocation and targeted intervention strategies such as identification of drug-resistant epilepsy patients, optimizing strategies for telehealth and patient outreach for awareness of epilepsy care resources within VHA. Meanwhile, data acquired by the algorithm over the decade since its development (FY 2014 to FY 2023) contributed to more accurate epidemiologic information and identification of historic trends. Development of the algorithm represents one of the ways ECoEs have led to improved care for VWEs. ECoEs have been shown to improve health care for veterans in several metrics.15
A strength of this study is the rigorous multitiered validation process to confirm the diagnostic accuracy of ICD-10 codes against the gold standard ILAE definition of epilepsy to identify “definite” epilepsy cases within the VHA. The use of specific ICD codes further enhances the precision of epilepsy diagnoses. The inclusion of ASMs, which are sometimes prescribed for conditions other than epilepsy, could potentially inflate false positive rates.16
This study focused exclusively on the identification and validation of definite epilepsy cases within the VHA VSSC database, employing more stringent diagnostic criteria to ensure the highest level of certainty in ascertaining epilepsy. It is important to note there is a separate category of probable epilepsy, which involves a broader set of diagnostic criteria. While not covered in this study, probable epilepsy would be subject to future research and validation, which could provide insights into a wider spectrum of epilepsy diagnoses. Such future research could help refine the algorithm’s applicability and accuracy and potentially lead to more comprehensive surveillance and management strategies in clinical practice.
This study highlights the inherent challenges in leveraging administrative data for disease identification, particularly for conditions such as epilepsy, where diagnostic clarity can be complex. However, other conditions such as multiple sclerosis have noted similar success with the use of VHA administrative data for categorizing disease.17
Limitations
The algorithm discussed in this article is, in and of itself, generalizable. However, the validation process was unique to the VHA patient population, limiting the generalizability of the findings. Documentation practices and HCP attitudes within the VHA may differ from those in other health care settings. Identifying people with epilepsy can be challenging because of changing definitions of epilepsy over time. In addition to clinical evaluation, EEG and magnetic resonance imaging results, response to ASM treatment, and video-EEG monitoring of habitual events all can help establish the diagnosis. Therefore, studies may vary in how inclusive or exclusive the criteria are. ASMs such as gabapentin, pregabalin, carbamazepine, lamotrigine, topiramate, and valproate are used to treat other conditions, including headaches, generalized pain, and mood disorders. Consequently, including these ASMs in the Tier 1 definition may have increased the false positive rate. Additional research is needed to evaluate whether excluding these ASMs from the algorithm based on specific criteria (eg, dose of ASM used) can further refine the algorithm to identify patients with epilepsy.
Further refinement of this algorithm may also occur as technology changes. Future electronic health records may allow better tracking of different epilepsy factors, the integration of additional diagnostic criteria, and the use of natural language processing or other forms of artificial intelligence.
Conclusions
This study presents a significant step forward in epilepsy surveillance within the VHA. The algorithm offers a robust tool for identifying VWEs with good PPVs, facilitating better resource allocation and targeted care. Despite its limitations, this research lays a foundation for future advancements in the management and understanding of epilepsy within large health care systems. Since this VHA algorithm is based on ASMs and ICD diagnosis codes from patient records, other large managed health care systems also may be able to adapt this algorithm to their data specifications.

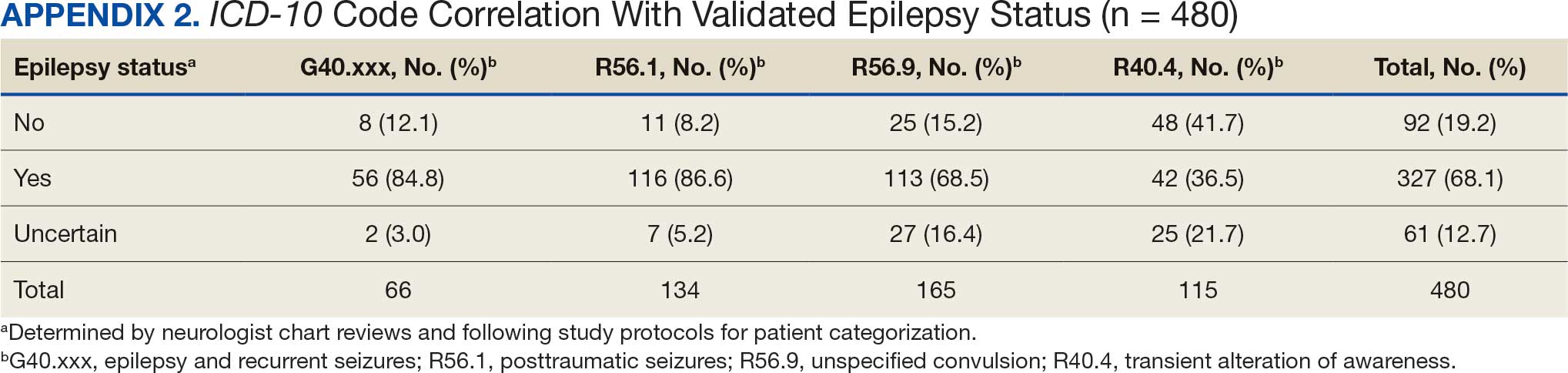
- Kobau R, Luncheon C, Greenlund K. Active epilepsy prevalence among U.S. adults is 1.1% and differs by educational level-National Health Interview Survey, United States, 2021. Epilepsy Behav. 2023;142:109180. doi:10.1016/j.yebeh.2023.109180
- GBD 2017 US Neurological Disorders Collaborators, Feigin VL, Vos T, et al. Burden of neurological disorders across the US from 1990-2017: a global burden of disease study. JAMA Neurol. 2021;78:165-176. doi:10.1001/jamaneurol.2020.4152
- Moura LMVR, Karakis I, Zack MM, et al. Drivers of US health care spending for persons with seizures and/or epilepsies, 2010-2018. Epilepsia. 2022;63:2144-2154. doi:10.1111/epi.17305
- Institute of Medicine. Epilepsy Across the Spectrum: Promoting Health and Understanding. The National Academies Press; 2012. Accessed November 11, 2025. www.nap.edu/catalog/13379
- Mbizvo GK, Bennett KH, Schnier C, Simpson CR, Duncan SE, Chin RFM. The accuracy of using administrative healthcare data to identify epilepsy cases: A systematic review of validation studies. Epilepsia. 2020;61:1319-1335. doi:10.1111/epi.16547
- Montouris GD. How will primary care physicians, specialists, and managed care treat epilepsy in the new millennium? Neurology. 2000;55:S42-S44.
- US Department of Veterans Affairs. Veterans Health Administration: About VHA. Accessed November 11, 2025. https://www.va.gov/health/aboutvha.asp
- Veterans’ Mental Health and Other Care Improvements Act of 2008, S 2162, 110th Cong (2008). Accessed November 11, 2025. https://www.congress.gov/bill/110th-congress/senate-bill/2162
- Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of Veterans diagnosed with seizures within Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762. doi:10.1682/JRRD.2014.10.0241
- Holden EW, Grossman E, Nguyen HT, et al. Developing a computer algorithm to identify epilepsy cases in managed care organizations. Dis Manag. 2005;8:1-14. doi:10.1089/dis.2005.8.1
- Rehman R, Everhart A, Frontera AT, et al. Implementation of an established algorithm and modifications for the identification of epilepsy patients in the Veterans Health Administration. Epilepsy Res. 2016;127:284-290. doi:10.1016/j.eplepsyres.2016.09.012
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475-482. doi:10.1111/epi.12550
- Smith JR, Jones FJS, Fureman BE, et al. Accuracy of ICD-10-CM claims-based definitions for epilepsy and seizure type. Epilepsy Res. 2020;166:106414. doi:10.1016/j.eplepsyres.2020.106414
- Jetté N, Reid AY, Quan H, et al. How accurate is ICD coding for epilepsy? Epilepsia. 2010;51:62-69. doi:10.1111/j.1528-1167.2009.02201.x
- Kelly P, Chinta R, Privitera G. Do centers of excellence reduce health care costs? Evidence from the US Veterans Health Administration Centers for Epilepsy. Glob Bus Organ Excell. 2015;34:18-29.
- Haneef Z, Rehman R, Husain AM. Association between standardized mortality ratio and utilization of care in US veterans with drug-resistant epilepsy compared with all US veterans and the US general population. JAMA Neurol. 2022;79:879-887. doi:10.1001/jamaneurol.2022.2290
- Culpepper WJ, Marrie RA, Langer-Gould A, et al. Validation of an algorithm for identifying MS cases in administrative health claims datasets. Neurology. 2019;92:e1016-e1028 doi:10.1212/WNL.0000000000007043
Epilepsy affects about 4.5 million people in the United States and 150,000 new individuals are diagnosed each year.1,2 In 2019, epilepsy-attributable health care spending for noninstitutionalized people was around $5.4 billion and total epilepsy-attributable and epilepsy or seizure health care-related costs totaled $54 billion.3
Accurate surveillance of epilepsy in large health care systems can potentially improve health care delivery and resource allocation. A 2012 Institute of Medicine (IOM) report identified 13 recommendations to guide public health action on epilepsy, including validation of standard definitions for case ascertainment, identification of epilepsy through screening programs or protocols, and expansion of surveillance to better understand disease burden.4
A systematic review of validation studies concluded that it is reasonable to use administrative data to identify people with epilepsy in epidemiologic research. Combining The International Classification of Diseases (ICD) codes for epilepsy (ICD-10, G40-41; ICD-9, 345) with antiseizure medications (ASMs) could provide high positive predictive values (PPVs) and combining symptoms codes for convulsions (ICD-10, R56; ICD-9, 780.3, 780.39) with ASMs could lead to high sensitivity.5 However, identifying individuals with epilepsy from administrative data in large managed health care organizations is challenging.6 The IOM report noted that large managed health care organizations presented varying incidence and prevalence estimates due to differing methodology, geographic area, demographics, and definitions of epilepsy.
The Veterans Health Administration (VHA) is the largest integrated US health care system, providing care to > 9.1 million veterans.7 To improve the health and well-being of veterans with epilepsy (VWEs), a network of sites was established in 2008 called the US Department of Veterans Affairs (VA) Epilepsy Centers of Excellence (ECoE). Subsequent to the creation of the ECoE, efforts were made to identify VWEs within VHA databases.8,9 Prior to fiscal year (FY) 2016, the ECoE adopted a modified version of a well-established epilepsy diagnostic algorithm developed by Holden et al for large managed care organizations.10 The original algorithm identified patients by cross-matching ASMs with ICD-9 codes for an index year. But it failed to capture a considerable number of stable patients with epilepsy in the VHA due to incomplete documentation, and had false positives due to inclusion of patients identified from diagnostic clinics. The modified algorithm the ECoE used prior to FY 2016 considered additional prior years and excluded encounters from diagnostic clinics. The result was an improvement in the sensitivity and specificity of the algorithm. Researchers evaluating 500 patients with epilepsy estimated that the modified algorithm had a PPV of 82.0% (95% CI, 78.6%-85.4%).11
After implementation of ICD-10 codes in the VHA in FY 2016, the task of reliably and efficiently identifying VWE led to a 3-tier algorithm. This article presents a validation of the different tiers of this algorithm after the implementation of ICD-10 diagnosis codes and summarizes the surveillance data collected over the years within the VHA showing the trends of epilepsy.
Methods
The VHA National Neurology office commissioned a Neurology Cube dashboard in FY 2021 in collaboration with VHA Support Service Center (VSSC) for reporting and surveillance of VWEs as a quality improvement initiative. The Neurology Cube uses a 3-tier system for identifying VWE in the VHA databases. VSSC programmers extract data from the VHA Corporate Data Warehouse (CDW) and utilize Microsoft SQL Server and Microsoft Power BI for Neurology Cube reports. The 3-tier system identifies VWE and divides them into distinct groups. The first tier identifies VWE with the highest degree of confidence; Tiers 2 and 3 represent identification with successively lesser degrees of confidence (Figure 1).
Tier 1
Definition. For a given index year and the preceding 2 years, any of following diagnosis codes on ≥ 1 clinical encounter are considered: 345.xx (epilepsy in ICD-9), 780.3x (other convulsions in ICD-9), G40.xxx (epilepsy in ICD-10), R40.4 (transient alteration of awareness), R56.1 (posttraumatic seizures), or R56.9 (unspecified convulsions). To reduce false positive rates, EEG clinic visits, which may include long-term monitoring, are excluded. Patients identified with ICD codes are then evaluated for an ASM prescription for ≥ 30 days during the index year. ASMs are listed in Appendix 1.
Validation. The development and validation of ICD-9 diagnosis codes crossmatched with an ASM prescription in the VHA has been published elsewhere.11 In FY 2017, after implementation of ICD-10 diagnostic codes, Tier 1 development and validation was performed in 2 phases. Even though Tier 1 study phases were conducted and completed during FY 2017, the patients for Tier 1 were identified from evaluation of FY 2016 data (October 1, 2015, to September 30, 2016). After the pilot analysis, the Tier 1 definition was implemented, and a chart review of 625 randomized patients was conducted at 5 sites for validation. Adequate preliminary data was not available to perform a sample size estimation for this study. Therefore, a practical target of 125 patients was set for Tier 1 from each site to obtain a final sample size of 625 patients. This second phase validated that the crossmatch of ICD-10 diagnosis codes with ASMs had a high PPV for identifying VWE.
Tiers 2 and 3
Definitions. For an index year, Tier 2 includes patients with ≥ 1 inpatient encounter documentation of either ICD-9 345.xx or ICD-10 G40.xxx, excluding EEG clinics. Tier 3 Includes patients who have had ≥ 2 outpatient encounters with diagnosis codes 345.xx or G40.xxx on 2 separate days, excluding EEG clinics. Tiers 2 and 3 do not require ASM prescriptions; this helps to identify VWEs who may be getting their medications outside of VHA or those who have received a new diagnosis.
Validations. Tiers 2 and 3 were included in the epilepsy identification algorithm in FY 2021 after validation was performed on a sample of 8 patients in each tier. Five patients were subsequently identified as having epilepsy in Tier 2 and 6 patients were identified in Tier 3. A more comprehensive validation of Tiers 2 and 3 was performed during FY 2022 that included patients at 5 sites seen during FY 2019 to FY 2022. Since yearly trends showed only about 8% of total patients were identified as having epilepsy through Tiers 2 and 3 we sought ≥ 20 patients per tier for the 5 sites for a total of 200 patients to ensure representation across the VHA. The final count was 126 patients for Tier 2 and 174 patients for Tier 3 (n = 300).
Gold Standard Criteria for Epilepsy Diagnosis
We used the International League Against Epilepsy (ILAE) definition of epilepsy for the validation of the 3 algorithm tiers. ILAE defines epilepsy as ≥ 2 unprovoked (or reflex) seizures occurring > 24 hours apart or 1 unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (≥ 60%) after 2 unprovoked seizures, occurring over the next 10 years.12
A standard protocol was provided to evaluators to identify patients using the VHA Computerized Patient Record System (Appendix 1). After review, evaluators categorized each patient in 1 of 4 ways: (1) Yes, definite: The patient’s health care practitioner (HCP) believes the patient has epilepsy and is treating with medication; (2) Yes, uncertain: The HCP has enough suspicion of epilepsy that a medication is prescribed, but uncertainty is expressed of the diagnosis; (3) No, definite: The HCP does not believe the patient has epilepsy and is therefore not treating with medication for seizure; (4) No, uncertain: The HCP is not treating with medication for epilepsy, because the diagnostic suspicion is not high enough, but there is suspicion for epilepsy.
As a quality improvement operational project, the Epilepsy National Program Office approved this validation project and determined that institutional review board approval was not required.
Statistical Analysis
Counts and percentages were computed for categories of epilepsy status. PPV of each tier was estimated with asymptotic 95% CIs.
Results
ICD-10 codes for 480 patients were evaluated in Tier 1 phase 1; 13.8% were documented with G40.xxx, 27.9% with R56.1, 34.4% with R56.9, and 24.0% with R40.4 (Appendix 2). In total, 68.1% fulfilled the criteria of epilepsy, 19.2% did not, and 12.7% were uncertain). From the validation of Tier 1 phase 2 (n = 625), the PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) was 85.1% (95% CI, 82.1%-87.8%) (Table).
Of 300 patients evaluated, 126 (42.0%) were evaluated for Tier 2 with a PPV of 61.9% (95% CI, 53.4%-70.4%), and 174 (58.0%) patients were evaluated for Tier 3 with a PPV of 59.8% (95% CI, 52.5%-67.1%. The PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) were combined to calculate the PPV. Estimates of VHA VWE counts were computed for each tier from FY 2014 to FY 2023 using the VSSC Neurology Cube (Figure 2). For all years, > 92% patients were classified using the Tier 1 definition.
Discussion
The development and validation of the 3-tier diagnostic algorithm represents an important advancement in the surveillance and management of epilepsy among veterans within the VHA. The validation of this algorithm also demonstrates its practical utility in a large, integrated health care system.
Specific challenges were encountered when attempting to use pre-existing algorithms; these challenges included differences in the usage patterns of diagnostic codes and the patterns of ASM use within the VHA. These challenges prompted the need for a tailored approach, which led to the development of this algorithm. The inclusion of additional ICD-10 codes led to further revisions and subsequent validation. While many of the basic concepts of the algorithm, including ICD codes and ASMs, could work in other institutions, it would be wise for health care organizations to develop their own algorithms because of certain variables, including organizational size, patient demographics, common comorbidities, and the specific configurations of electronic health records and administrative data systems.
Studies have shown that ICD-10 codes for epilepsy (G40.* and/or R56.9) perform well in identifying epilepsy whether they are assigned by neurologists (sensitivity, 97.7%; specificity, 44.1%; PPV, 96.2%; negative predictive value, 57.7%), or in emergency department or hospital discharges (PPV, 75.5%).13,14 The pilot study of the algorithm’s Tier 1 development (phase 1) evaluated whether the selected ICD-10 diagnostic codes accurately included the VWE population within the VHA and revealed that while most codes (eg, epilepsy [G40.xxx]; posttraumatic seizures [R56.1]; and unspecified convulsions [R56.9]), had a low false positive rate (< 16%), the R40.4 code (transient alteration of awareness) had a higher false positivity of 42%. While this is not surprising given the broad spectrum of conditions that can manifest as transient alteration of awareness, it underscores the inherent challenges in diagnosing epilepsy using diagnosis codes.
In phase 2, the Tier 1 algorithm was validated as effective for identifying VWE in the VHA system, as its PPV was determined to be high (85%). In comparison, Tiers 2 and 3, whose criteria did not require data on VHA prescribed ASM use, had lower tiers of epilepsy predictability (PPV about 60% for both). This was thought to be acceptable because Tiers 2 and 3 represent a smaller population of the identified VWEs (about 8%). These VWEs may otherwise have been missed, partly because veterans are not required to get ASMs from the VHA.
Upon VHA implementation in FY 2021, this diagnostic algorithm exhibited significant clinical utility when integrated within the VSSC Neurology Cube. It facilitated an efficient approach to identifying VWEs using readily available databases. This led to better tracking of real-time epilepsy cases, which facilitated improving current resource allocation and targeted intervention strategies such as identification of drug-resistant epilepsy patients, optimizing strategies for telehealth and patient outreach for awareness of epilepsy care resources within VHA. Meanwhile, data acquired by the algorithm over the decade since its development (FY 2014 to FY 2023) contributed to more accurate epidemiologic information and identification of historic trends. Development of the algorithm represents one of the ways ECoEs have led to improved care for VWEs. ECoEs have been shown to improve health care for veterans in several metrics.15
A strength of this study is the rigorous multitiered validation process to confirm the diagnostic accuracy of ICD-10 codes against the gold standard ILAE definition of epilepsy to identify “definite” epilepsy cases within the VHA. The use of specific ICD codes further enhances the precision of epilepsy diagnoses. The inclusion of ASMs, which are sometimes prescribed for conditions other than epilepsy, could potentially inflate false positive rates.16
This study focused exclusively on the identification and validation of definite epilepsy cases within the VHA VSSC database, employing more stringent diagnostic criteria to ensure the highest level of certainty in ascertaining epilepsy. It is important to note there is a separate category of probable epilepsy, which involves a broader set of diagnostic criteria. While not covered in this study, probable epilepsy would be subject to future research and validation, which could provide insights into a wider spectrum of epilepsy diagnoses. Such future research could help refine the algorithm’s applicability and accuracy and potentially lead to more comprehensive surveillance and management strategies in clinical practice.
This study highlights the inherent challenges in leveraging administrative data for disease identification, particularly for conditions such as epilepsy, where diagnostic clarity can be complex. However, other conditions such as multiple sclerosis have noted similar success with the use of VHA administrative data for categorizing disease.17
Limitations
The algorithm discussed in this article is, in and of itself, generalizable. However, the validation process was unique to the VHA patient population, limiting the generalizability of the findings. Documentation practices and HCP attitudes within the VHA may differ from those in other health care settings. Identifying people with epilepsy can be challenging because of changing definitions of epilepsy over time. In addition to clinical evaluation, EEG and magnetic resonance imaging results, response to ASM treatment, and video-EEG monitoring of habitual events all can help establish the diagnosis. Therefore, studies may vary in how inclusive or exclusive the criteria are. ASMs such as gabapentin, pregabalin, carbamazepine, lamotrigine, topiramate, and valproate are used to treat other conditions, including headaches, generalized pain, and mood disorders. Consequently, including these ASMs in the Tier 1 definition may have increased the false positive rate. Additional research is needed to evaluate whether excluding these ASMs from the algorithm based on specific criteria (eg, dose of ASM used) can further refine the algorithm to identify patients with epilepsy.
Further refinement of this algorithm may also occur as technology changes. Future electronic health records may allow better tracking of different epilepsy factors, the integration of additional diagnostic criteria, and the use of natural language processing or other forms of artificial intelligence.
Conclusions
This study presents a significant step forward in epilepsy surveillance within the VHA. The algorithm offers a robust tool for identifying VWEs with good PPVs, facilitating better resource allocation and targeted care. Despite its limitations, this research lays a foundation for future advancements in the management and understanding of epilepsy within large health care systems. Since this VHA algorithm is based on ASMs and ICD diagnosis codes from patient records, other large managed health care systems also may be able to adapt this algorithm to their data specifications.
Epilepsy affects about 4.5 million people in the United States and 150,000 new individuals are diagnosed each year.1,2 In 2019, epilepsy-attributable health care spending for noninstitutionalized people was around $5.4 billion and total epilepsy-attributable and epilepsy or seizure health care-related costs totaled $54 billion.3
Accurate surveillance of epilepsy in large health care systems can potentially improve health care delivery and resource allocation. A 2012 Institute of Medicine (IOM) report identified 13 recommendations to guide public health action on epilepsy, including validation of standard definitions for case ascertainment, identification of epilepsy through screening programs or protocols, and expansion of surveillance to better understand disease burden.4
A systematic review of validation studies concluded that it is reasonable to use administrative data to identify people with epilepsy in epidemiologic research. Combining The International Classification of Diseases (ICD) codes for epilepsy (ICD-10, G40-41; ICD-9, 345) with antiseizure medications (ASMs) could provide high positive predictive values (PPVs) and combining symptoms codes for convulsions (ICD-10, R56; ICD-9, 780.3, 780.39) with ASMs could lead to high sensitivity.5 However, identifying individuals with epilepsy from administrative data in large managed health care organizations is challenging.6 The IOM report noted that large managed health care organizations presented varying incidence and prevalence estimates due to differing methodology, geographic area, demographics, and definitions of epilepsy.
The Veterans Health Administration (VHA) is the largest integrated US health care system, providing care to > 9.1 million veterans.7 To improve the health and well-being of veterans with epilepsy (VWEs), a network of sites was established in 2008 called the US Department of Veterans Affairs (VA) Epilepsy Centers of Excellence (ECoE). Subsequent to the creation of the ECoE, efforts were made to identify VWEs within VHA databases.8,9 Prior to fiscal year (FY) 2016, the ECoE adopted a modified version of a well-established epilepsy diagnostic algorithm developed by Holden et al for large managed care organizations.10 The original algorithm identified patients by cross-matching ASMs with ICD-9 codes for an index year. But it failed to capture a considerable number of stable patients with epilepsy in the VHA due to incomplete documentation, and had false positives due to inclusion of patients identified from diagnostic clinics. The modified algorithm the ECoE used prior to FY 2016 considered additional prior years and excluded encounters from diagnostic clinics. The result was an improvement in the sensitivity and specificity of the algorithm. Researchers evaluating 500 patients with epilepsy estimated that the modified algorithm had a PPV of 82.0% (95% CI, 78.6%-85.4%).11
After implementation of ICD-10 codes in the VHA in FY 2016, the task of reliably and efficiently identifying VWE led to a 3-tier algorithm. This article presents a validation of the different tiers of this algorithm after the implementation of ICD-10 diagnosis codes and summarizes the surveillance data collected over the years within the VHA showing the trends of epilepsy.
Methods
The VHA National Neurology office commissioned a Neurology Cube dashboard in FY 2021 in collaboration with VHA Support Service Center (VSSC) for reporting and surveillance of VWEs as a quality improvement initiative. The Neurology Cube uses a 3-tier system for identifying VWE in the VHA databases. VSSC programmers extract data from the VHA Corporate Data Warehouse (CDW) and utilize Microsoft SQL Server and Microsoft Power BI for Neurology Cube reports. The 3-tier system identifies VWE and divides them into distinct groups. The first tier identifies VWE with the highest degree of confidence; Tiers 2 and 3 represent identification with successively lesser degrees of confidence (Figure 1).
Tier 1
Definition. For a given index year and the preceding 2 years, any of following diagnosis codes on ≥ 1 clinical encounter are considered: 345.xx (epilepsy in ICD-9), 780.3x (other convulsions in ICD-9), G40.xxx (epilepsy in ICD-10), R40.4 (transient alteration of awareness), R56.1 (posttraumatic seizures), or R56.9 (unspecified convulsions). To reduce false positive rates, EEG clinic visits, which may include long-term monitoring, are excluded. Patients identified with ICD codes are then evaluated for an ASM prescription for ≥ 30 days during the index year. ASMs are listed in Appendix 1.
Validation. The development and validation of ICD-9 diagnosis codes crossmatched with an ASM prescription in the VHA has been published elsewhere.11 In FY 2017, after implementation of ICD-10 diagnostic codes, Tier 1 development and validation was performed in 2 phases. Even though Tier 1 study phases were conducted and completed during FY 2017, the patients for Tier 1 were identified from evaluation of FY 2016 data (October 1, 2015, to September 30, 2016). After the pilot analysis, the Tier 1 definition was implemented, and a chart review of 625 randomized patients was conducted at 5 sites for validation. Adequate preliminary data was not available to perform a sample size estimation for this study. Therefore, a practical target of 125 patients was set for Tier 1 from each site to obtain a final sample size of 625 patients. This second phase validated that the crossmatch of ICD-10 diagnosis codes with ASMs had a high PPV for identifying VWE.
Tiers 2 and 3
Definitions. For an index year, Tier 2 includes patients with ≥ 1 inpatient encounter documentation of either ICD-9 345.xx or ICD-10 G40.xxx, excluding EEG clinics. Tier 3 Includes patients who have had ≥ 2 outpatient encounters with diagnosis codes 345.xx or G40.xxx on 2 separate days, excluding EEG clinics. Tiers 2 and 3 do not require ASM prescriptions; this helps to identify VWEs who may be getting their medications outside of VHA or those who have received a new diagnosis.
Validations. Tiers 2 and 3 were included in the epilepsy identification algorithm in FY 2021 after validation was performed on a sample of 8 patients in each tier. Five patients were subsequently identified as having epilepsy in Tier 2 and 6 patients were identified in Tier 3. A more comprehensive validation of Tiers 2 and 3 was performed during FY 2022 that included patients at 5 sites seen during FY 2019 to FY 2022. Since yearly trends showed only about 8% of total patients were identified as having epilepsy through Tiers 2 and 3 we sought ≥ 20 patients per tier for the 5 sites for a total of 200 patients to ensure representation across the VHA. The final count was 126 patients for Tier 2 and 174 patients for Tier 3 (n = 300).
Gold Standard Criteria for Epilepsy Diagnosis
We used the International League Against Epilepsy (ILAE) definition of epilepsy for the validation of the 3 algorithm tiers. ILAE defines epilepsy as ≥ 2 unprovoked (or reflex) seizures occurring > 24 hours apart or 1 unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (≥ 60%) after 2 unprovoked seizures, occurring over the next 10 years.12
A standard protocol was provided to evaluators to identify patients using the VHA Computerized Patient Record System (Appendix 1). After review, evaluators categorized each patient in 1 of 4 ways: (1) Yes, definite: The patient’s health care practitioner (HCP) believes the patient has epilepsy and is treating with medication; (2) Yes, uncertain: The HCP has enough suspicion of epilepsy that a medication is prescribed, but uncertainty is expressed of the diagnosis; (3) No, definite: The HCP does not believe the patient has epilepsy and is therefore not treating with medication for seizure; (4) No, uncertain: The HCP is not treating with medication for epilepsy, because the diagnostic suspicion is not high enough, but there is suspicion for epilepsy.
As a quality improvement operational project, the Epilepsy National Program Office approved this validation project and determined that institutional review board approval was not required.
Statistical Analysis
Counts and percentages were computed for categories of epilepsy status. PPV of each tier was estimated with asymptotic 95% CIs.
Results
ICD-10 codes for 480 patients were evaluated in Tier 1 phase 1; 13.8% were documented with G40.xxx, 27.9% with R56.1, 34.4% with R56.9, and 24.0% with R40.4 (Appendix 2). In total, 68.1% fulfilled the criteria of epilepsy, 19.2% did not, and 12.7% were uncertain). From the validation of Tier 1 phase 2 (n = 625), the PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) was 85.1% (95% CI, 82.1%-87.8%) (Table).
Of 300 patients evaluated, 126 (42.0%) were evaluated for Tier 2 with a PPV of 61.9% (95% CI, 53.4%-70.4%), and 174 (58.0%) patients were evaluated for Tier 3 with a PPV of 59.8% (95% CI, 52.5%-67.1%. The PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) were combined to calculate the PPV. Estimates of VHA VWE counts were computed for each tier from FY 2014 to FY 2023 using the VSSC Neurology Cube (Figure 2). For all years, > 92% patients were classified using the Tier 1 definition.
Discussion
The development and validation of the 3-tier diagnostic algorithm represents an important advancement in the surveillance and management of epilepsy among veterans within the VHA. The validation of this algorithm also demonstrates its practical utility in a large, integrated health care system.
Specific challenges were encountered when attempting to use pre-existing algorithms; these challenges included differences in the usage patterns of diagnostic codes and the patterns of ASM use within the VHA. These challenges prompted the need for a tailored approach, which led to the development of this algorithm. The inclusion of additional ICD-10 codes led to further revisions and subsequent validation. While many of the basic concepts of the algorithm, including ICD codes and ASMs, could work in other institutions, it would be wise for health care organizations to develop their own algorithms because of certain variables, including organizational size, patient demographics, common comorbidities, and the specific configurations of electronic health records and administrative data systems.
Studies have shown that ICD-10 codes for epilepsy (G40.* and/or R56.9) perform well in identifying epilepsy whether they are assigned by neurologists (sensitivity, 97.7%; specificity, 44.1%; PPV, 96.2%; negative predictive value, 57.7%), or in emergency department or hospital discharges (PPV, 75.5%).13,14 The pilot study of the algorithm’s Tier 1 development (phase 1) evaluated whether the selected ICD-10 diagnostic codes accurately included the VWE population within the VHA and revealed that while most codes (eg, epilepsy [G40.xxx]; posttraumatic seizures [R56.1]; and unspecified convulsions [R56.9]), had a low false positive rate (< 16%), the R40.4 code (transient alteration of awareness) had a higher false positivity of 42%. While this is not surprising given the broad spectrum of conditions that can manifest as transient alteration of awareness, it underscores the inherent challenges in diagnosing epilepsy using diagnosis codes.
In phase 2, the Tier 1 algorithm was validated as effective for identifying VWE in the VHA system, as its PPV was determined to be high (85%). In comparison, Tiers 2 and 3, whose criteria did not require data on VHA prescribed ASM use, had lower tiers of epilepsy predictability (PPV about 60% for both). This was thought to be acceptable because Tiers 2 and 3 represent a smaller population of the identified VWEs (about 8%). These VWEs may otherwise have been missed, partly because veterans are not required to get ASMs from the VHA.
Upon VHA implementation in FY 2021, this diagnostic algorithm exhibited significant clinical utility when integrated within the VSSC Neurology Cube. It facilitated an efficient approach to identifying VWEs using readily available databases. This led to better tracking of real-time epilepsy cases, which facilitated improving current resource allocation and targeted intervention strategies such as identification of drug-resistant epilepsy patients, optimizing strategies for telehealth and patient outreach for awareness of epilepsy care resources within VHA. Meanwhile, data acquired by the algorithm over the decade since its development (FY 2014 to FY 2023) contributed to more accurate epidemiologic information and identification of historic trends. Development of the algorithm represents one of the ways ECoEs have led to improved care for VWEs. ECoEs have been shown to improve health care for veterans in several metrics.15
A strength of this study is the rigorous multitiered validation process to confirm the diagnostic accuracy of ICD-10 codes against the gold standard ILAE definition of epilepsy to identify “definite” epilepsy cases within the VHA. The use of specific ICD codes further enhances the precision of epilepsy diagnoses. The inclusion of ASMs, which are sometimes prescribed for conditions other than epilepsy, could potentially inflate false positive rates.16
This study focused exclusively on the identification and validation of definite epilepsy cases within the VHA VSSC database, employing more stringent diagnostic criteria to ensure the highest level of certainty in ascertaining epilepsy. It is important to note there is a separate category of probable epilepsy, which involves a broader set of diagnostic criteria. While not covered in this study, probable epilepsy would be subject to future research and validation, which could provide insights into a wider spectrum of epilepsy diagnoses. Such future research could help refine the algorithm’s applicability and accuracy and potentially lead to more comprehensive surveillance and management strategies in clinical practice.
This study highlights the inherent challenges in leveraging administrative data for disease identification, particularly for conditions such as epilepsy, where diagnostic clarity can be complex. However, other conditions such as multiple sclerosis have noted similar success with the use of VHA administrative data for categorizing disease.17
Limitations
The algorithm discussed in this article is, in and of itself, generalizable. However, the validation process was unique to the VHA patient population, limiting the generalizability of the findings. Documentation practices and HCP attitudes within the VHA may differ from those in other health care settings. Identifying people with epilepsy can be challenging because of changing definitions of epilepsy over time. In addition to clinical evaluation, EEG and magnetic resonance imaging results, response to ASM treatment, and video-EEG monitoring of habitual events all can help establish the diagnosis. Therefore, studies may vary in how inclusive or exclusive the criteria are. ASMs such as gabapentin, pregabalin, carbamazepine, lamotrigine, topiramate, and valproate are used to treat other conditions, including headaches, generalized pain, and mood disorders. Consequently, including these ASMs in the Tier 1 definition may have increased the false positive rate. Additional research is needed to evaluate whether excluding these ASMs from the algorithm based on specific criteria (eg, dose of ASM used) can further refine the algorithm to identify patients with epilepsy.
Further refinement of this algorithm may also occur as technology changes. Future electronic health records may allow better tracking of different epilepsy factors, the integration of additional diagnostic criteria, and the use of natural language processing or other forms of artificial intelligence.
Conclusions
This study presents a significant step forward in epilepsy surveillance within the VHA. The algorithm offers a robust tool for identifying VWEs with good PPVs, facilitating better resource allocation and targeted care. Despite its limitations, this research lays a foundation for future advancements in the management and understanding of epilepsy within large health care systems. Since this VHA algorithm is based on ASMs and ICD diagnosis codes from patient records, other large managed health care systems also may be able to adapt this algorithm to their data specifications.
- Kobau R, Luncheon C, Greenlund K. Active epilepsy prevalence among U.S. adults is 1.1% and differs by educational level-National Health Interview Survey, United States, 2021. Epilepsy Behav. 2023;142:109180. doi:10.1016/j.yebeh.2023.109180
- GBD 2017 US Neurological Disorders Collaborators, Feigin VL, Vos T, et al. Burden of neurological disorders across the US from 1990-2017: a global burden of disease study. JAMA Neurol. 2021;78:165-176. doi:10.1001/jamaneurol.2020.4152
- Moura LMVR, Karakis I, Zack MM, et al. Drivers of US health care spending for persons with seizures and/or epilepsies, 2010-2018. Epilepsia. 2022;63:2144-2154. doi:10.1111/epi.17305
- Institute of Medicine. Epilepsy Across the Spectrum: Promoting Health and Understanding. The National Academies Press; 2012. Accessed November 11, 2025. www.nap.edu/catalog/13379
- Mbizvo GK, Bennett KH, Schnier C, Simpson CR, Duncan SE, Chin RFM. The accuracy of using administrative healthcare data to identify epilepsy cases: A systematic review of validation studies. Epilepsia. 2020;61:1319-1335. doi:10.1111/epi.16547
- Montouris GD. How will primary care physicians, specialists, and managed care treat epilepsy in the new millennium? Neurology. 2000;55:S42-S44.
- US Department of Veterans Affairs. Veterans Health Administration: About VHA. Accessed November 11, 2025. https://www.va.gov/health/aboutvha.asp
- Veterans’ Mental Health and Other Care Improvements Act of 2008, S 2162, 110th Cong (2008). Accessed November 11, 2025. https://www.congress.gov/bill/110th-congress/senate-bill/2162
- Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of Veterans diagnosed with seizures within Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762. doi:10.1682/JRRD.2014.10.0241
- Holden EW, Grossman E, Nguyen HT, et al. Developing a computer algorithm to identify epilepsy cases in managed care organizations. Dis Manag. 2005;8:1-14. doi:10.1089/dis.2005.8.1
- Rehman R, Everhart A, Frontera AT, et al. Implementation of an established algorithm and modifications for the identification of epilepsy patients in the Veterans Health Administration. Epilepsy Res. 2016;127:284-290. doi:10.1016/j.eplepsyres.2016.09.012
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475-482. doi:10.1111/epi.12550
- Smith JR, Jones FJS, Fureman BE, et al. Accuracy of ICD-10-CM claims-based definitions for epilepsy and seizure type. Epilepsy Res. 2020;166:106414. doi:10.1016/j.eplepsyres.2020.106414
- Jetté N, Reid AY, Quan H, et al. How accurate is ICD coding for epilepsy? Epilepsia. 2010;51:62-69. doi:10.1111/j.1528-1167.2009.02201.x
- Kelly P, Chinta R, Privitera G. Do centers of excellence reduce health care costs? Evidence from the US Veterans Health Administration Centers for Epilepsy. Glob Bus Organ Excell. 2015;34:18-29.
- Haneef Z, Rehman R, Husain AM. Association between standardized mortality ratio and utilization of care in US veterans with drug-resistant epilepsy compared with all US veterans and the US general population. JAMA Neurol. 2022;79:879-887. doi:10.1001/jamaneurol.2022.2290
- Culpepper WJ, Marrie RA, Langer-Gould A, et al. Validation of an algorithm for identifying MS cases in administrative health claims datasets. Neurology. 2019;92:e1016-e1028 doi:10.1212/WNL.0000000000007043
- Kobau R, Luncheon C, Greenlund K. Active epilepsy prevalence among U.S. adults is 1.1% and differs by educational level-National Health Interview Survey, United States, 2021. Epilepsy Behav. 2023;142:109180. doi:10.1016/j.yebeh.2023.109180
- GBD 2017 US Neurological Disorders Collaborators, Feigin VL, Vos T, et al. Burden of neurological disorders across the US from 1990-2017: a global burden of disease study. JAMA Neurol. 2021;78:165-176. doi:10.1001/jamaneurol.2020.4152
- Moura LMVR, Karakis I, Zack MM, et al. Drivers of US health care spending for persons with seizures and/or epilepsies, 2010-2018. Epilepsia. 2022;63:2144-2154. doi:10.1111/epi.17305
- Institute of Medicine. Epilepsy Across the Spectrum: Promoting Health and Understanding. The National Academies Press; 2012. Accessed November 11, 2025. www.nap.edu/catalog/13379
- Mbizvo GK, Bennett KH, Schnier C, Simpson CR, Duncan SE, Chin RFM. The accuracy of using administrative healthcare data to identify epilepsy cases: A systematic review of validation studies. Epilepsia. 2020;61:1319-1335. doi:10.1111/epi.16547
- Montouris GD. How will primary care physicians, specialists, and managed care treat epilepsy in the new millennium? Neurology. 2000;55:S42-S44.
- US Department of Veterans Affairs. Veterans Health Administration: About VHA. Accessed November 11, 2025. https://www.va.gov/health/aboutvha.asp
- Veterans’ Mental Health and Other Care Improvements Act of 2008, S 2162, 110th Cong (2008). Accessed November 11, 2025. https://www.congress.gov/bill/110th-congress/senate-bill/2162
- Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of Veterans diagnosed with seizures within Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762. doi:10.1682/JRRD.2014.10.0241
- Holden EW, Grossman E, Nguyen HT, et al. Developing a computer algorithm to identify epilepsy cases in managed care organizations. Dis Manag. 2005;8:1-14. doi:10.1089/dis.2005.8.1
- Rehman R, Everhart A, Frontera AT, et al. Implementation of an established algorithm and modifications for the identification of epilepsy patients in the Veterans Health Administration. Epilepsy Res. 2016;127:284-290. doi:10.1016/j.eplepsyres.2016.09.012
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475-482. doi:10.1111/epi.12550
- Smith JR, Jones FJS, Fureman BE, et al. Accuracy of ICD-10-CM claims-based definitions for epilepsy and seizure type. Epilepsy Res. 2020;166:106414. doi:10.1016/j.eplepsyres.2020.106414
- Jetté N, Reid AY, Quan H, et al. How accurate is ICD coding for epilepsy? Epilepsia. 2010;51:62-69. doi:10.1111/j.1528-1167.2009.02201.x
- Kelly P, Chinta R, Privitera G. Do centers of excellence reduce health care costs? Evidence from the US Veterans Health Administration Centers for Epilepsy. Glob Bus Organ Excell. 2015;34:18-29.
- Haneef Z, Rehman R, Husain AM. Association between standardized mortality ratio and utilization of care in US veterans with drug-resistant epilepsy compared with all US veterans and the US general population. JAMA Neurol. 2022;79:879-887. doi:10.1001/jamaneurol.2022.2290
- Culpepper WJ, Marrie RA, Langer-Gould A, et al. Validation of an algorithm for identifying MS cases in administrative health claims datasets. Neurology. 2019;92:e1016-e1028 doi:10.1212/WNL.0000000000007043
Development and Validation of an Administrative Algorithm to Identify Veterans With Epilepsy
Development and Validation of an Administrative Algorithm to Identify Veterans With Epilepsy

Confronting Uncertainty and Addressing Urgency for Action Through the Establishment of a VA Long COVID Practice-Based Research Network
Confronting Uncertainty and Addressing Urgency for Action Through the Establishment of a VA Long COVID Practice-Based Research Network
Learning health systems (LHS) promote a continuous process that can assist in making sense of uncertainty when confronting emerging complex conditions such as Long COVID. Long COVID is an infection-associated chronic condition that detrimentally impacts veterans, their families, and the communities in which they live. This complex condition is defined by ongoing, new, or returning symptoms following COVID-19 infection that negatively affect return to meaningful participation in social, recreational, and vocational activities.1,2 The clinical uncertainty surrounding Long COVID is amplified by unclear etiology, prognosis, and expected course of symptoms.3,4 Uncertainty surrounding best clinical practices, processes, and policies for Long COVID care has resulted in practice variation despite the emerging evidence base for Long COVID care.4 Failure to address gaps in clinical evidence and care implementation threatens to perpetuate fragmented and unnecessary care.
The context surrounding Long COVID created an urgency to rapidly address clinically relevant questions and make sense of any uncertainty. Thus, the Veterans Health Administration (VHA) funded a Long COVID Practice-Based Research Network (LC-PBRN) to build an infrastructure that supports Long COVID research nationally and promotes interdisciplinary collaboration. The LC-PBRN vision is to centralize Long COVID clinical, research, and operational activities. The research infrastructure of the LC-PBRN is designed with an LHS lens to facilitate feedback loops and integrate knowledge learned while making progress towards this vision.5 This article describes the phases of infrastructure development and network building, as well as associated lessons learned.
Designing the LC-PBRN Infrastructure
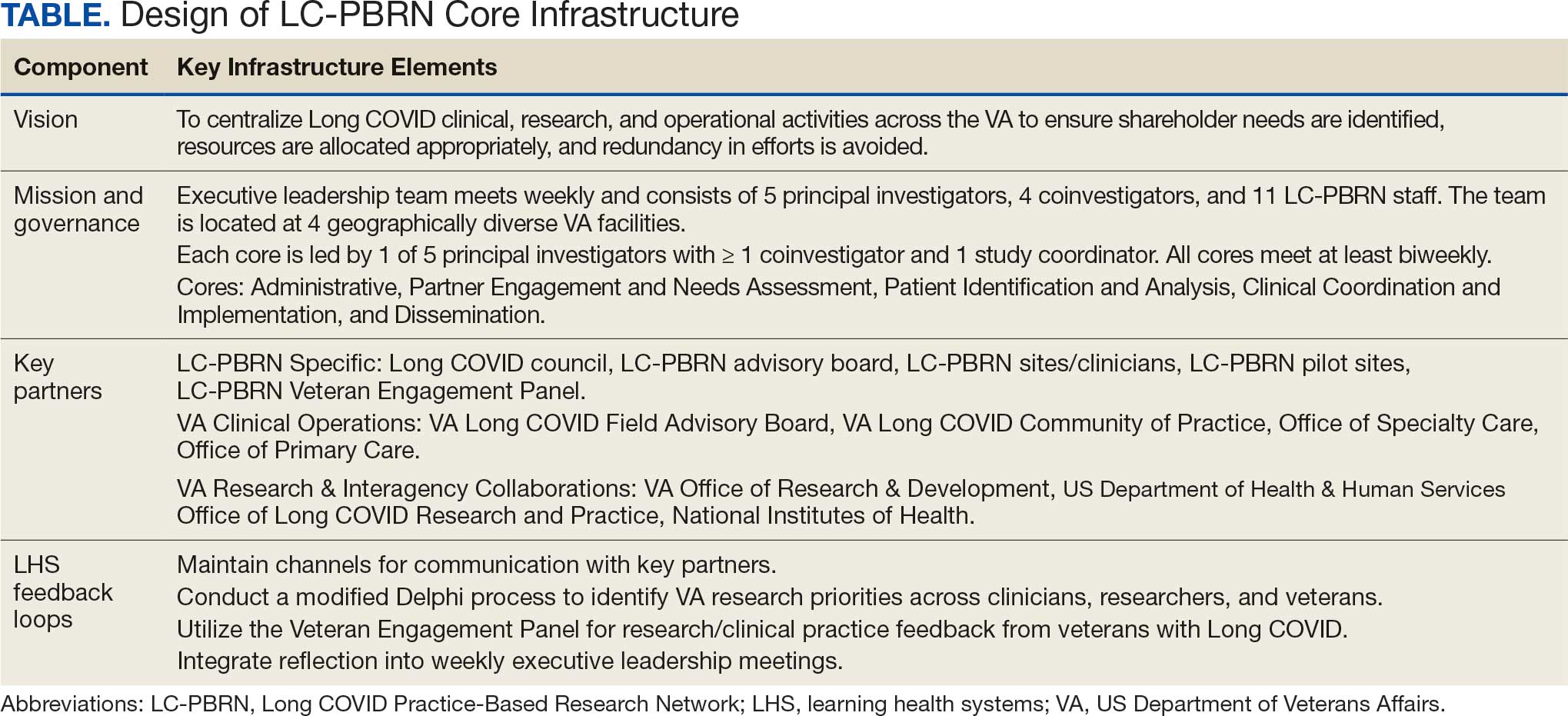
Vision
The LC-PBRN’s vision is to create an infrastructure that integrates an LHS framework by unifying the VA research approach to Long COVID to ensure veteran, clinician, operational, and researcher involvement (Figure 1).
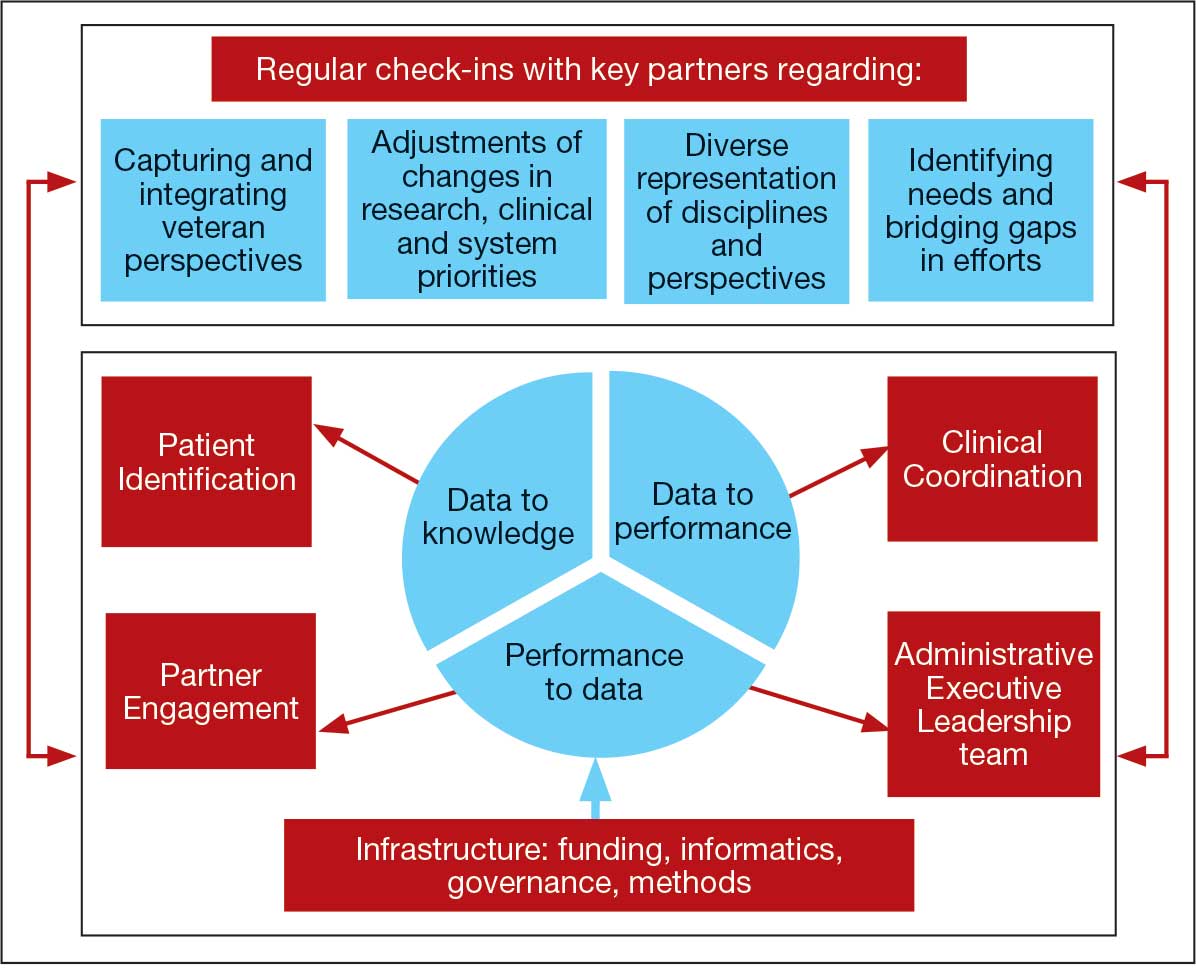
Mission and Governance
The LC-PBRN operates with an executive leadership team and 5 cores. The executive leadership team is responsible for overall LC-PBRN operations, management, and direction setting of the LC-PBRN. The executive leadership team meets weekly to provide oversight of each core, which specializes in different aspects. The cores include: Administrative, Partner Engagement and Needs Assessment, Patient Identification and Analysis, Clinical Coordination and Implementation, and Dissemination (Figure 2).
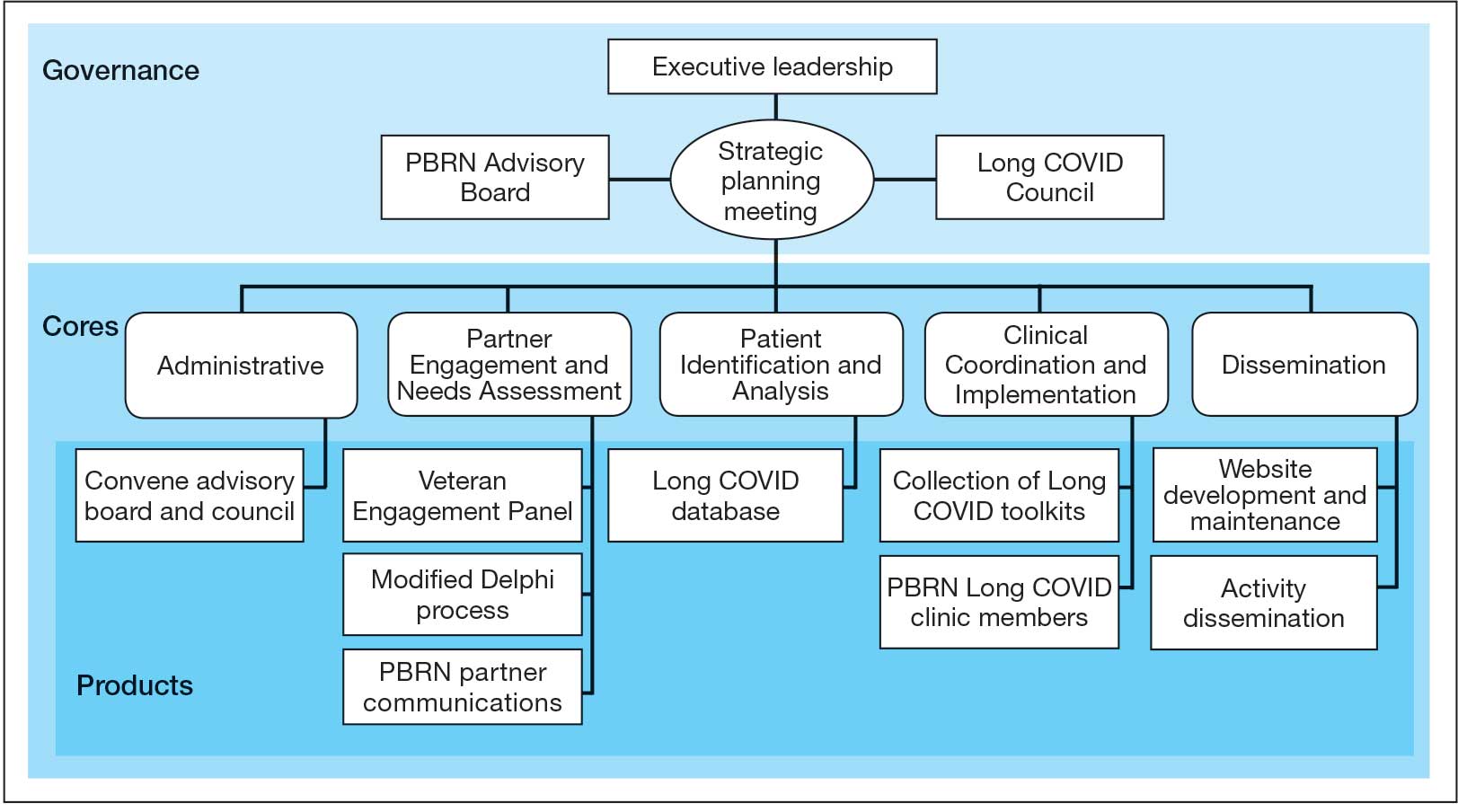
The Administrative core focuses on interagency collaboration to identify and network with key operational and agency leaders to allow for ongoing exploration of funding strategies for Long COVID research. The Administrative core manages 3 teams: an advisory board, Long COVID council, and the strategic planning team. The advisory board meets biannually to oversee achievement of LC-PBRN goals, deliverables, and tactics for meeting these goals. The advisory board includes the LC-PBRN executive leadership team and 13 interagency members from various shareholders (eg, Centers for Disease Control and Prevention, National Institutes of Health, and specialty departments within the VA).
The Long COVID council convenes quarterly to provide scientific input on important overarching issues in Long COVID research, practice, and policy. The council consists of 22 scientific representatives in VA and non-VA contexts, university affiliates, and veteran representatives. The strategic planning team convenes annually to identify how the LC-PBRN and its partners can meet the needs of the broader Long COVID ecosystem and conduct a strengths, opportunities, weaknesses, and threats analysis to identify strategic objectives and expected outcomes. The strategic planning team includes the executive leadership team and key Long COVID shareholders within VHA and affiliated partners. The Partner Engagement and Needs Assessment core aims to solicit feedback from veterans, clinicians, researchers, and operational leadership. Input is gathered through a Veteran Engagement Panel and a modified Delphi consensus process. The panel was formed using a Community Engagement Studio model to engage veterans as consultants on research.7 Currently, 10 members represent a range of ages, genders, racial and ethnic backgrounds, and military experience. All veterans have a history of Long COVID and are paid as consultants. Video conference panel meetings occur quarterly for 1 to 2 hours; the meeting length is shorter than typical engagement studios to accommodate for fatigue-related symptoms that may limit attention and ability to participate in longer meetings. Before each panel, the Partner Engagement and Needs Assessment core helps identify key questions and creates a structured agenda. Each panel begins with a presentation of a research study followed by a group discussion led by a trained facilitator. The modified Delphi consensus process focuses on identifying research priority areas for Long COVID within the VA. Veterans living with Long COVID, as well as clinicians and researchers who work closely with patients who have Long COVID, complete a series of progressive surveys to provide input on research priorities.
The Partner Engagement and Needs Assessment core also actively provides outreach to important partners in research, clinical care, and operational leadership to facilitate introductory meetings to (1) ask partners to describe their 5 largest pain points, (2) find pain points within the scope of LC-PBRN resources, and (3) discuss the strengths and capacity of the PBRN. During introductory meetings, communications preferences and a cadence for subsequent meetings are established. Subsequent engagement meetings aim to provide updates and codevelop solutions to emerging issues. This core maintains a living document to track engagement efforts, points of contact for identified and emerging partners, and ensure all communication is timely.
The Patient Identification and Analysis core develops a database of veterans with confirmed or suspected Long COVID. The goal is for researchers to use the database to identify potential participants for clinical trials and monitor clinical care outcomes. When possible, this core works with existing VA data to facilitate research that aligns with the LC-PBRN mission. The core can also use natural language processing and machine learning to work with researchers conducting clinical trials to help identify patients who may meet eligibility criteria.
The Clinical Coordination and Implementation core gathers information on the best practices for identifying and recruiting veterans for Long COVID research as well as compiles strategies for standardized clinical assessments that can both facilitate ongoing research and the successful implementation of evidence-based care. The Clinical Coordination and Implementation core provides support to pilot and multisite trials in 3 ways. First, it develops toolkits such as best practice strategies for recruiting participants for research, template examples of recruitment materials, and a library of patient-reported outcome measures, standardized clinical note titles and templates in use for Long COVID in the national electronic health record. Second, it partners with the Patient Identification and Analysis core to facilitate access to and use of algorithms that identify Long COVID cases based on electronic health records for recruitment. Finally, it compiles a detailed list of potential collaborating sites. The steps to facilitate patient identification and recruitment inform feasibility assessments and improve efficiency of launching pilot studies and multisite trials. The library of outcome measures, standardized clinical notes, and templates can aid and expedite data collection.
The Dissemination core focuses on developing a website, creating a dissemination plan, and actively disseminating products of the LC-PBRN and its partners. This core’s foundational framework is based on the Agency for Healthcare Research and Quality Quick-Start Guide to Dissemination for PBRNs.8,9 The core built an internal- and external-facing website to connect users with LC-PBRN products, potential outreach contacts, and promote timely updates on LC-PBRN activities. A manual of operating procedures will be drafted to include the development of training for practitioners involved in research projects to learn the processes involved in presenting clinical results for education and training initiatives, presentations, and manuscript preparation. A toolkit will also be developed to support dissemination activities designed to reach a variety of end-users, such as education materials, policy briefings, educational briefs, newsletters, and presentations at local, regional, and national levels.
Key Partners
Key partners exist specific to the LC-PBRN and within the broader VA ecosystem, including VA clinical operations, VA research, and intra-agency collaborations.
LC-PBRN Specific. In addition to the LC-PBRN council, advisory board, and Veteran Engagement Panel discussed earlier,
VA Clinical Operations. To support clinical operations, a Long COVID Field Advisory Board was formed through the VA Office of Specialty Care as an operational effort to develop clinical best practice. The LC-PBRN consults with this group on veteran engagement strategies for input on clinical guides and dissemination of practice guide materials. The LC-PBRN also partners with an existing Long COVID Community of Practice and the Office of Primary Care. The Community of Practice provides a learning space for VA staff interested in advancing Long COVID care and assists with disseminating LC-PBRN to the broader Long COVID clinical community. A member of the Office of Primary Care sits on the PBRN advisory board to provide input on engaging primary care practitioners and ensure their unique needs are considered in LC-PBRN initiatives.
VA Research & Interagency Collaborations. The LC-PBRN engages monthly with an interagency workgroup led by the US Department of Health and Human Services Office of Long COVID Research and Practice. These engagements support identification of research gaps that the VA may help address, monitor emerging funding opportunities, and foster collaborations. LC-PBRN representatives also meet with staff at the National Institutes of Health Researching COVID to Enhance Recovery initiative to identify pathways for veteran recruitment.
LHS Feedback Loops
The LC-PBRN was designed with an LHS approach in mind.10 Throughout development of the LC-PBRN, consideration was given to (1) capture data on new efforts within the Long COVID ecosystem (performance to data), (2) examine performance gaps and identify approaches for best practice (data to knowledge), and (3) implement best practices, develop toolkits, disseminate findings, and measure impacts (knowledge to performance). With this approach, the LC-PBRN is constantly evolving based on new information coming from the internal and external Long COVID ecosystem. Each element was deliberatively considered in relation to how data can be transformed into knowledge, knowledge into performance, and performance into data.
First, an important mechanism for feedback involves establishing clear channels of communication. Regular check-ins with key partners occur through virtual meetings to provide updates, assess needs and challenges, and codevelop action plans. For example, during a check-in with the Long COVID Field Advisory Board, members expressed a desire to incorporate veteran feedback into VA clinical practice recommendations. We provided expertise on different engagement modalities (eg, focus groups vs individual interviews), and collaboration occurred to identify key interview questions for veterans. This process resulted in a published clinician-facing Long COVID Nervous System Clinical Guide (available at [email protected]) that integrated critical feedback from veterans related to neurological symptoms.
Second, weekly executive leadership meetings include dedicated time for reflection on partner feedback, the current state of Long COVID, and contextual changes that impact deliverable priorities and timelines. Outcomes from these discussions are communicated with VHA Health Services Research and, when appropriate, to key partners to ensure alignment. For example, the Patient Identification and Analysis core was originally tasked with identifying a definition of Long COVID. However, as the broader community moved away from a singular definition, efforts were redirected toward higher-priority issues within the VA Long COVID ecosystem, including veteran enrollment in clinical trials.
Third, the Veteran Engagement Panel captures feedback from those with lived experience to inform Long COVID research and clinical efforts. The panel meetings are strategically designed to ask veterans living with Long COVID specific questions related to a given research or clinical topic of interest. For example, panel sessions with the Field Advisory Board focused on concerns articulated by veterans related to the mental health and gastroenterological symptoms associated with Long COVID. Insights from these discussions will inform development of Long COVID mental health and gastroenterological clinical care guides, with several PBRN investigators serving as subject matter experts. This collaborative approach ensures that veteran perspectives are represented in developing Long COVID clinical care processes.
Fourth, research priorities identified through the Delphi consensus process will inform development of VA Request for Funding Proposals related to Long COVID. The initial survey was developed in collaboration with veterans, clinicians, and researchers across the Veteran Engagement Panel, the Field Advisory Board, and the National Research Action Plan on Long COVID.11 The process was launched in October 2024 and concluded in June 2025. The team conducted 3 consensus rounds with veterans and VA clinicians and researchers. Top priority areas included the testing assessments for diagnosing Long COVID, studying subtypes of Long COVID and treatments for each, and finding biomarkers for Long COVID. A formal publication of the results and analysis is the focus of a future publication.
Fifth, ongoing engagement with the Field Advisory Board has supported adoption of a preliminary set of clinical outcome measures. If universally adopted, these instruments may contribute to the development of a standardized data collection process and serve as common data elements collected for epidemiologic, health services, or clinical trial research.
Lessons Learned and Practice Implications
Throughout the development of the LC-PBRN, several decisions were identified that have impacted infrastructure development and implementation.
Include veterans’ voices to ensure network efforts align with patient needs. Given the novelty of Long COVID, practitioners and researchers are learning as they go. It is important to listen to individuals who live with Long COVID. Throughout the development of the LC-PBRN, veteran perspective has proven how vital it is for them to be heard when it comes to their health care. Clinicians similarly highlighted the value of incorporating patient perspectives into the development of tools and treatment strategies. Develop an interdisciplinary leadership team to foster the diverse viewpoints needed to tackle multifaceted problems. It is important to consider as many clinical and research perspectives as possible because Long COVID is a complex condition with symptoms impacting major organ systems.12-15 Therefore, the team spans across a multitude of specialties and locations.
Set clear expectations and goals with partners to uphold timely deliverables and stay within the PBRN’s capacity. When including a multitude of partners, teams should consider each of those partners’ experiences and opinions in decision-making conversations. Expectation setting is important to ensure all partners are on the same page and understand the capacity of the LC-PBRN. This allows the team to focus its efforts, avoid being overwhelmed with requests, and provide quality deliverables.
Build engaging relationships to bridge gaps between internal and external partners. A substantial number of resources focus on building relationships with partners so they can trust the LC-PBRN has their best interests in mind. These relationships are important to ensure the VA avoids duplicate efforts. This includes prioritizing connecting partners who are working on similar efforts to promote collaboration across facilities.
Conclusions
PBRNs provide an important mechanism to use LHS approaches to successfully convene research around complex issues. PBRNs can support integration across the LHS cycle, allowing for multiple feedback loops, and coordinate activities that work to achieve a larger vision. PBRNs offer centralized mechanisms to collaboratively understand and address complex problems, such as Long COVID, where the uncertainty regarding how to treat occurs in tandem with the urgency to treat. The LC-PBRN model described in this article has the potential to transcend Long COVID by building infrastructure necessary to proactively address current or future clinical conditions or populations with a LHS lens. The infrastructure can require cross-system and sector collaborations, expediency, inclusivity, and patient- and family-centeredness. Future efforts will focus on building out a larger network of VHA sites, facilitating recruitment at site and veteran levels into Long COVID trials through case identification, and systematically support the standardization of clinical data for clinical utility and evaluation of quality and/or outcomes across the VHA.
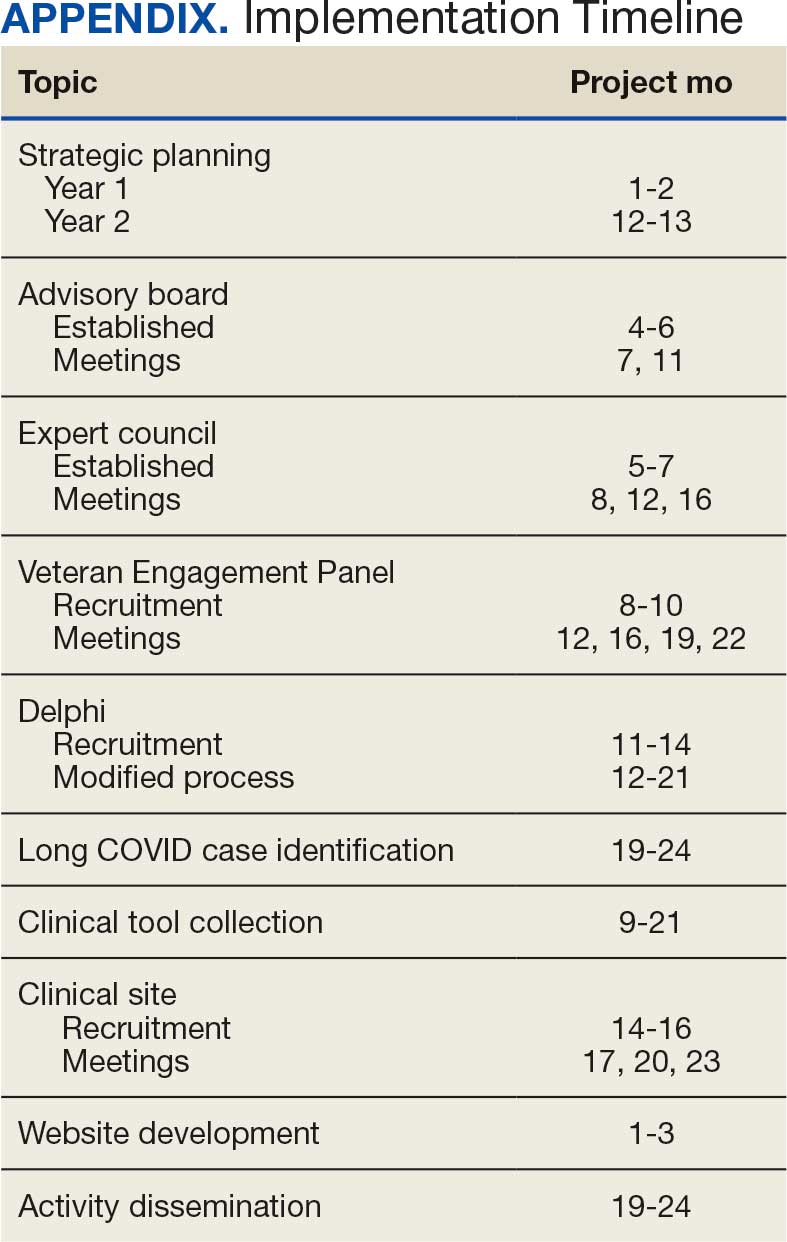
- Ottiger M, Poppele I, Sperling N, et al. Work ability and return-to-work of patients with post-COVID-19: a systematic review and meta-analysis. BMC Public Health. 2024;24:1811. doi:10.1186/s12889-024-19328-6
- Ziauddeen N, Gurdasani D, O’Hara ME, et al. Characteristics and impact of Long Covid: findings from an online survey. PLOS ONE. 2022;17:e0264331. doi:10.1371/journal.pone.0264331
- Graham F. Daily briefing: Answers emerge about long COVID recovery. Nature. Published online June 28, 2023. doi:10.1038/d41586-023-02190-8
- Al-Aly Z, Davis H, McCorkell L, et al. Long COVID science, research and policy. Nat Med. 2024;30:2148-2164. doi:10.1038/s41591-024-03173-6
- Atkins D, Kilbourne AM, Shulkin D. Moving from discovery to system-wide change: the role of research in a learning health care system: experience from three decades of health systems research in the Veterans Health Administration. Annu Rev Public Health. 2017;38:467-487. doi:10.1146/annurev-publhealth-031816-044255
- Ely EW, Brown LM, Fineberg HV. Long covid defined. N Engl J Med. 2024;391:1746-1753.doi:10.1056/NEJMsb2408466
- Joosten YA, Israel TL, Williams NA, et al. Community engagement studios: a structured approach to obtaining meaningful input from stakeholders to inform research. Acad Med. 2015;90:1646-1650. doi:10.1097/ACM.0000000000000794
- AHRQ. Quick-start guide to dissemination for practice-based research networks. Revised June 2014. Accessed December 2, 2025. https://www.ahrq.gov/sites/default/files/wysiwyg/ncepcr/resources/dissemination-quick-start-guide.pdf
- Gustavson AM, Morrow CD, Brown RJ, et al. Reimagining how we synthesize information to impact clinical care, policy, and research priorities in real time: examples and lessons learned from COVID-19. J Gen Intern Med. 2024;39:2554-2559. doi:10.1007/s11606-024-08855-y
- University of Minnesota. About the Center for Learning Health System Sciences. Updated December 11, 2025. Accessed December 12, 2025. https://med.umn.edu/clhss/about-us
- AHRQ. National Research Action Plan. Published online 2022. Accessed February 14, 2024. https://www.covid.gov/sites/default/files/documents/National-Research-Action-Plan-on-Long-COVID-08012022.pdf
- Gustavson AM, Eaton TL, Schapira RM, et al. Approaches to long COVID care: the Veterans Health Administration experience in 2021. BMJ Mil Health. 2024;170:179-180. doi:10.1136/military-2022-002185
- Gustavson AM. A learning health system approach to long COVID care. Fed Pract. 2022;39:7. doi:10.12788/fp.0288
- Palacio A, Bast E, Klimas N, et al. Lessons learned in implementing a multidisciplinary long COVID clinic. Am J Med. 2025;138:843-849.doi:10.1016/j.amjmed.2024.05.020
- Prusinski C, Yan D, Klasova J, et al. Multidisciplinary management strategies for long COVID: a narrative review. Cureus. 2024;16:e59478. doi:10.7759/cureus.59478
Learning health systems (LHS) promote a continuous process that can assist in making sense of uncertainty when confronting emerging complex conditions such as Long COVID. Long COVID is an infection-associated chronic condition that detrimentally impacts veterans, their families, and the communities in which they live. This complex condition is defined by ongoing, new, or returning symptoms following COVID-19 infection that negatively affect return to meaningful participation in social, recreational, and vocational activities.1,2 The clinical uncertainty surrounding Long COVID is amplified by unclear etiology, prognosis, and expected course of symptoms.3,4 Uncertainty surrounding best clinical practices, processes, and policies for Long COVID care has resulted in practice variation despite the emerging evidence base for Long COVID care.4 Failure to address gaps in clinical evidence and care implementation threatens to perpetuate fragmented and unnecessary care.
The context surrounding Long COVID created an urgency to rapidly address clinically relevant questions and make sense of any uncertainty. Thus, the Veterans Health Administration (VHA) funded a Long COVID Practice-Based Research Network (LC-PBRN) to build an infrastructure that supports Long COVID research nationally and promotes interdisciplinary collaboration. The LC-PBRN vision is to centralize Long COVID clinical, research, and operational activities. The research infrastructure of the LC-PBRN is designed with an LHS lens to facilitate feedback loops and integrate knowledge learned while making progress towards this vision.5 This article describes the phases of infrastructure development and network building, as well as associated lessons learned.
Designing the LC-PBRN Infrastructure
Vision
The LC-PBRN’s vision is to create an infrastructure that integrates an LHS framework by unifying the VA research approach to Long COVID to ensure veteran, clinician, operational, and researcher involvement (Figure 1).
Mission and Governance
The LC-PBRN operates with an executive leadership team and 5 cores. The executive leadership team is responsible for overall LC-PBRN operations, management, and direction setting of the LC-PBRN. The executive leadership team meets weekly to provide oversight of each core, which specializes in different aspects. The cores include: Administrative, Partner Engagement and Needs Assessment, Patient Identification and Analysis, Clinical Coordination and Implementation, and Dissemination (Figure 2).
The Administrative core focuses on interagency collaboration to identify and network with key operational and agency leaders to allow for ongoing exploration of funding strategies for Long COVID research. The Administrative core manages 3 teams: an advisory board, Long COVID council, and the strategic planning team. The advisory board meets biannually to oversee achievement of LC-PBRN goals, deliverables, and tactics for meeting these goals. The advisory board includes the LC-PBRN executive leadership team and 13 interagency members from various shareholders (eg, Centers for Disease Control and Prevention, National Institutes of Health, and specialty departments within the VA).
The Long COVID council convenes quarterly to provide scientific input on important overarching issues in Long COVID research, practice, and policy. The council consists of 22 scientific representatives in VA and non-VA contexts, university affiliates, and veteran representatives. The strategic planning team convenes annually to identify how the LC-PBRN and its partners can meet the needs of the broader Long COVID ecosystem and conduct a strengths, opportunities, weaknesses, and threats analysis to identify strategic objectives and expected outcomes. The strategic planning team includes the executive leadership team and key Long COVID shareholders within VHA and affiliated partners. The Partner Engagement and Needs Assessment core aims to solicit feedback from veterans, clinicians, researchers, and operational leadership. Input is gathered through a Veteran Engagement Panel and a modified Delphi consensus process. The panel was formed using a Community Engagement Studio model to engage veterans as consultants on research.7 Currently, 10 members represent a range of ages, genders, racial and ethnic backgrounds, and military experience. All veterans have a history of Long COVID and are paid as consultants. Video conference panel meetings occur quarterly for 1 to 2 hours; the meeting length is shorter than typical engagement studios to accommodate for fatigue-related symptoms that may limit attention and ability to participate in longer meetings. Before each panel, the Partner Engagement and Needs Assessment core helps identify key questions and creates a structured agenda. Each panel begins with a presentation of a research study followed by a group discussion led by a trained facilitator. The modified Delphi consensus process focuses on identifying research priority areas for Long COVID within the VA. Veterans living with Long COVID, as well as clinicians and researchers who work closely with patients who have Long COVID, complete a series of progressive surveys to provide input on research priorities.
The Partner Engagement and Needs Assessment core also actively provides outreach to important partners in research, clinical care, and operational leadership to facilitate introductory meetings to (1) ask partners to describe their 5 largest pain points, (2) find pain points within the scope of LC-PBRN resources, and (3) discuss the strengths and capacity of the PBRN. During introductory meetings, communications preferences and a cadence for subsequent meetings are established. Subsequent engagement meetings aim to provide updates and codevelop solutions to emerging issues. This core maintains a living document to track engagement efforts, points of contact for identified and emerging partners, and ensure all communication is timely.
The Patient Identification and Analysis core develops a database of veterans with confirmed or suspected Long COVID. The goal is for researchers to use the database to identify potential participants for clinical trials and monitor clinical care outcomes. When possible, this core works with existing VA data to facilitate research that aligns with the LC-PBRN mission. The core can also use natural language processing and machine learning to work with researchers conducting clinical trials to help identify patients who may meet eligibility criteria.
The Clinical Coordination and Implementation core gathers information on the best practices for identifying and recruiting veterans for Long COVID research as well as compiles strategies for standardized clinical assessments that can both facilitate ongoing research and the successful implementation of evidence-based care. The Clinical Coordination and Implementation core provides support to pilot and multisite trials in 3 ways. First, it develops toolkits such as best practice strategies for recruiting participants for research, template examples of recruitment materials, and a library of patient-reported outcome measures, standardized clinical note titles and templates in use for Long COVID in the national electronic health record. Second, it partners with the Patient Identification and Analysis core to facilitate access to and use of algorithms that identify Long COVID cases based on electronic health records for recruitment. Finally, it compiles a detailed list of potential collaborating sites. The steps to facilitate patient identification and recruitment inform feasibility assessments and improve efficiency of launching pilot studies and multisite trials. The library of outcome measures, standardized clinical notes, and templates can aid and expedite data collection.
The Dissemination core focuses on developing a website, creating a dissemination plan, and actively disseminating products of the LC-PBRN and its partners. This core’s foundational framework is based on the Agency for Healthcare Research and Quality Quick-Start Guide to Dissemination for PBRNs.8,9 The core built an internal- and external-facing website to connect users with LC-PBRN products, potential outreach contacts, and promote timely updates on LC-PBRN activities. A manual of operating procedures will be drafted to include the development of training for practitioners involved in research projects to learn the processes involved in presenting clinical results for education and training initiatives, presentations, and manuscript preparation. A toolkit will also be developed to support dissemination activities designed to reach a variety of end-users, such as education materials, policy briefings, educational briefs, newsletters, and presentations at local, regional, and national levels.
Key Partners
Key partners exist specific to the LC-PBRN and within the broader VA ecosystem, including VA clinical operations, VA research, and intra-agency collaborations.
LC-PBRN Specific. In addition to the LC-PBRN council, advisory board, and Veteran Engagement Panel discussed earlier,
VA Clinical Operations. To support clinical operations, a Long COVID Field Advisory Board was formed through the VA Office of Specialty Care as an operational effort to develop clinical best practice. The LC-PBRN consults with this group on veteran engagement strategies for input on clinical guides and dissemination of practice guide materials. The LC-PBRN also partners with an existing Long COVID Community of Practice and the Office of Primary Care. The Community of Practice provides a learning space for VA staff interested in advancing Long COVID care and assists with disseminating LC-PBRN to the broader Long COVID clinical community. A member of the Office of Primary Care sits on the PBRN advisory board to provide input on engaging primary care practitioners and ensure their unique needs are considered in LC-PBRN initiatives.
VA Research & Interagency Collaborations. The LC-PBRN engages monthly with an interagency workgroup led by the US Department of Health and Human Services Office of Long COVID Research and Practice. These engagements support identification of research gaps that the VA may help address, monitor emerging funding opportunities, and foster collaborations. LC-PBRN representatives also meet with staff at the National Institutes of Health Researching COVID to Enhance Recovery initiative to identify pathways for veteran recruitment.
LHS Feedback Loops
The LC-PBRN was designed with an LHS approach in mind.10 Throughout development of the LC-PBRN, consideration was given to (1) capture data on new efforts within the Long COVID ecosystem (performance to data), (2) examine performance gaps and identify approaches for best practice (data to knowledge), and (3) implement best practices, develop toolkits, disseminate findings, and measure impacts (knowledge to performance). With this approach, the LC-PBRN is constantly evolving based on new information coming from the internal and external Long COVID ecosystem. Each element was deliberatively considered in relation to how data can be transformed into knowledge, knowledge into performance, and performance into data.
First, an important mechanism for feedback involves establishing clear channels of communication. Regular check-ins with key partners occur through virtual meetings to provide updates, assess needs and challenges, and codevelop action plans. For example, during a check-in with the Long COVID Field Advisory Board, members expressed a desire to incorporate veteran feedback into VA clinical practice recommendations. We provided expertise on different engagement modalities (eg, focus groups vs individual interviews), and collaboration occurred to identify key interview questions for veterans. This process resulted in a published clinician-facing Long COVID Nervous System Clinical Guide (available at [email protected]) that integrated critical feedback from veterans related to neurological symptoms.
Second, weekly executive leadership meetings include dedicated time for reflection on partner feedback, the current state of Long COVID, and contextual changes that impact deliverable priorities and timelines. Outcomes from these discussions are communicated with VHA Health Services Research and, when appropriate, to key partners to ensure alignment. For example, the Patient Identification and Analysis core was originally tasked with identifying a definition of Long COVID. However, as the broader community moved away from a singular definition, efforts were redirected toward higher-priority issues within the VA Long COVID ecosystem, including veteran enrollment in clinical trials.
Third, the Veteran Engagement Panel captures feedback from those with lived experience to inform Long COVID research and clinical efforts. The panel meetings are strategically designed to ask veterans living with Long COVID specific questions related to a given research or clinical topic of interest. For example, panel sessions with the Field Advisory Board focused on concerns articulated by veterans related to the mental health and gastroenterological symptoms associated with Long COVID. Insights from these discussions will inform development of Long COVID mental health and gastroenterological clinical care guides, with several PBRN investigators serving as subject matter experts. This collaborative approach ensures that veteran perspectives are represented in developing Long COVID clinical care processes.
Fourth, research priorities identified through the Delphi consensus process will inform development of VA Request for Funding Proposals related to Long COVID. The initial survey was developed in collaboration with veterans, clinicians, and researchers across the Veteran Engagement Panel, the Field Advisory Board, and the National Research Action Plan on Long COVID.11 The process was launched in October 2024 and concluded in June 2025. The team conducted 3 consensus rounds with veterans and VA clinicians and researchers. Top priority areas included the testing assessments for diagnosing Long COVID, studying subtypes of Long COVID and treatments for each, and finding biomarkers for Long COVID. A formal publication of the results and analysis is the focus of a future publication.
Fifth, ongoing engagement with the Field Advisory Board has supported adoption of a preliminary set of clinical outcome measures. If universally adopted, these instruments may contribute to the development of a standardized data collection process and serve as common data elements collected for epidemiologic, health services, or clinical trial research.
Lessons Learned and Practice Implications
Throughout the development of the LC-PBRN, several decisions were identified that have impacted infrastructure development and implementation.
Include veterans’ voices to ensure network efforts align with patient needs. Given the novelty of Long COVID, practitioners and researchers are learning as they go. It is important to listen to individuals who live with Long COVID. Throughout the development of the LC-PBRN, veteran perspective has proven how vital it is for them to be heard when it comes to their health care. Clinicians similarly highlighted the value of incorporating patient perspectives into the development of tools and treatment strategies. Develop an interdisciplinary leadership team to foster the diverse viewpoints needed to tackle multifaceted problems. It is important to consider as many clinical and research perspectives as possible because Long COVID is a complex condition with symptoms impacting major organ systems.12-15 Therefore, the team spans across a multitude of specialties and locations.
Set clear expectations and goals with partners to uphold timely deliverables and stay within the PBRN’s capacity. When including a multitude of partners, teams should consider each of those partners’ experiences and opinions in decision-making conversations. Expectation setting is important to ensure all partners are on the same page and understand the capacity of the LC-PBRN. This allows the team to focus its efforts, avoid being overwhelmed with requests, and provide quality deliverables.
Build engaging relationships to bridge gaps between internal and external partners. A substantial number of resources focus on building relationships with partners so they can trust the LC-PBRN has their best interests in mind. These relationships are important to ensure the VA avoids duplicate efforts. This includes prioritizing connecting partners who are working on similar efforts to promote collaboration across facilities.
Conclusions
PBRNs provide an important mechanism to use LHS approaches to successfully convene research around complex issues. PBRNs can support integration across the LHS cycle, allowing for multiple feedback loops, and coordinate activities that work to achieve a larger vision. PBRNs offer centralized mechanisms to collaboratively understand and address complex problems, such as Long COVID, where the uncertainty regarding how to treat occurs in tandem with the urgency to treat. The LC-PBRN model described in this article has the potential to transcend Long COVID by building infrastructure necessary to proactively address current or future clinical conditions or populations with a LHS lens. The infrastructure can require cross-system and sector collaborations, expediency, inclusivity, and patient- and family-centeredness. Future efforts will focus on building out a larger network of VHA sites, facilitating recruitment at site and veteran levels into Long COVID trials through case identification, and systematically support the standardization of clinical data for clinical utility and evaluation of quality and/or outcomes across the VHA.
Learning health systems (LHS) promote a continuous process that can assist in making sense of uncertainty when confronting emerging complex conditions such as Long COVID. Long COVID is an infection-associated chronic condition that detrimentally impacts veterans, their families, and the communities in which they live. This complex condition is defined by ongoing, new, or returning symptoms following COVID-19 infection that negatively affect return to meaningful participation in social, recreational, and vocational activities.1,2 The clinical uncertainty surrounding Long COVID is amplified by unclear etiology, prognosis, and expected course of symptoms.3,4 Uncertainty surrounding best clinical practices, processes, and policies for Long COVID care has resulted in practice variation despite the emerging evidence base for Long COVID care.4 Failure to address gaps in clinical evidence and care implementation threatens to perpetuate fragmented and unnecessary care.
The context surrounding Long COVID created an urgency to rapidly address clinically relevant questions and make sense of any uncertainty. Thus, the Veterans Health Administration (VHA) funded a Long COVID Practice-Based Research Network (LC-PBRN) to build an infrastructure that supports Long COVID research nationally and promotes interdisciplinary collaboration. The LC-PBRN vision is to centralize Long COVID clinical, research, and operational activities. The research infrastructure of the LC-PBRN is designed with an LHS lens to facilitate feedback loops and integrate knowledge learned while making progress towards this vision.5 This article describes the phases of infrastructure development and network building, as well as associated lessons learned.
Designing the LC-PBRN Infrastructure
Vision
The LC-PBRN’s vision is to create an infrastructure that integrates an LHS framework by unifying the VA research approach to Long COVID to ensure veteran, clinician, operational, and researcher involvement (Figure 1).
Mission and Governance
The LC-PBRN operates with an executive leadership team and 5 cores. The executive leadership team is responsible for overall LC-PBRN operations, management, and direction setting of the LC-PBRN. The executive leadership team meets weekly to provide oversight of each core, which specializes in different aspects. The cores include: Administrative, Partner Engagement and Needs Assessment, Patient Identification and Analysis, Clinical Coordination and Implementation, and Dissemination (Figure 2).
The Administrative core focuses on interagency collaboration to identify and network with key operational and agency leaders to allow for ongoing exploration of funding strategies for Long COVID research. The Administrative core manages 3 teams: an advisory board, Long COVID council, and the strategic planning team. The advisory board meets biannually to oversee achievement of LC-PBRN goals, deliverables, and tactics for meeting these goals. The advisory board includes the LC-PBRN executive leadership team and 13 interagency members from various shareholders (eg, Centers for Disease Control and Prevention, National Institutes of Health, and specialty departments within the VA).
The Long COVID council convenes quarterly to provide scientific input on important overarching issues in Long COVID research, practice, and policy. The council consists of 22 scientific representatives in VA and non-VA contexts, university affiliates, and veteran representatives. The strategic planning team convenes annually to identify how the LC-PBRN and its partners can meet the needs of the broader Long COVID ecosystem and conduct a strengths, opportunities, weaknesses, and threats analysis to identify strategic objectives and expected outcomes. The strategic planning team includes the executive leadership team and key Long COVID shareholders within VHA and affiliated partners. The Partner Engagement and Needs Assessment core aims to solicit feedback from veterans, clinicians, researchers, and operational leadership. Input is gathered through a Veteran Engagement Panel and a modified Delphi consensus process. The panel was formed using a Community Engagement Studio model to engage veterans as consultants on research.7 Currently, 10 members represent a range of ages, genders, racial and ethnic backgrounds, and military experience. All veterans have a history of Long COVID and are paid as consultants. Video conference panel meetings occur quarterly for 1 to 2 hours; the meeting length is shorter than typical engagement studios to accommodate for fatigue-related symptoms that may limit attention and ability to participate in longer meetings. Before each panel, the Partner Engagement and Needs Assessment core helps identify key questions and creates a structured agenda. Each panel begins with a presentation of a research study followed by a group discussion led by a trained facilitator. The modified Delphi consensus process focuses on identifying research priority areas for Long COVID within the VA. Veterans living with Long COVID, as well as clinicians and researchers who work closely with patients who have Long COVID, complete a series of progressive surveys to provide input on research priorities.
The Partner Engagement and Needs Assessment core also actively provides outreach to important partners in research, clinical care, and operational leadership to facilitate introductory meetings to (1) ask partners to describe their 5 largest pain points, (2) find pain points within the scope of LC-PBRN resources, and (3) discuss the strengths and capacity of the PBRN. During introductory meetings, communications preferences and a cadence for subsequent meetings are established. Subsequent engagement meetings aim to provide updates and codevelop solutions to emerging issues. This core maintains a living document to track engagement efforts, points of contact for identified and emerging partners, and ensure all communication is timely.
The Patient Identification and Analysis core develops a database of veterans with confirmed or suspected Long COVID. The goal is for researchers to use the database to identify potential participants for clinical trials and monitor clinical care outcomes. When possible, this core works with existing VA data to facilitate research that aligns with the LC-PBRN mission. The core can also use natural language processing and machine learning to work with researchers conducting clinical trials to help identify patients who may meet eligibility criteria.
The Clinical Coordination and Implementation core gathers information on the best practices for identifying and recruiting veterans for Long COVID research as well as compiles strategies for standardized clinical assessments that can both facilitate ongoing research and the successful implementation of evidence-based care. The Clinical Coordination and Implementation core provides support to pilot and multisite trials in 3 ways. First, it develops toolkits such as best practice strategies for recruiting participants for research, template examples of recruitment materials, and a library of patient-reported outcome measures, standardized clinical note titles and templates in use for Long COVID in the national electronic health record. Second, it partners with the Patient Identification and Analysis core to facilitate access to and use of algorithms that identify Long COVID cases based on electronic health records for recruitment. Finally, it compiles a detailed list of potential collaborating sites. The steps to facilitate patient identification and recruitment inform feasibility assessments and improve efficiency of launching pilot studies and multisite trials. The library of outcome measures, standardized clinical notes, and templates can aid and expedite data collection.
The Dissemination core focuses on developing a website, creating a dissemination plan, and actively disseminating products of the LC-PBRN and its partners. This core’s foundational framework is based on the Agency for Healthcare Research and Quality Quick-Start Guide to Dissemination for PBRNs.8,9 The core built an internal- and external-facing website to connect users with LC-PBRN products, potential outreach contacts, and promote timely updates on LC-PBRN activities. A manual of operating procedures will be drafted to include the development of training for practitioners involved in research projects to learn the processes involved in presenting clinical results for education and training initiatives, presentations, and manuscript preparation. A toolkit will also be developed to support dissemination activities designed to reach a variety of end-users, such as education materials, policy briefings, educational briefs, newsletters, and presentations at local, regional, and national levels.
Key Partners
Key partners exist specific to the LC-PBRN and within the broader VA ecosystem, including VA clinical operations, VA research, and intra-agency collaborations.
LC-PBRN Specific. In addition to the LC-PBRN council, advisory board, and Veteran Engagement Panel discussed earlier,
VA Clinical Operations. To support clinical operations, a Long COVID Field Advisory Board was formed through the VA Office of Specialty Care as an operational effort to develop clinical best practice. The LC-PBRN consults with this group on veteran engagement strategies for input on clinical guides and dissemination of practice guide materials. The LC-PBRN also partners with an existing Long COVID Community of Practice and the Office of Primary Care. The Community of Practice provides a learning space for VA staff interested in advancing Long COVID care and assists with disseminating LC-PBRN to the broader Long COVID clinical community. A member of the Office of Primary Care sits on the PBRN advisory board to provide input on engaging primary care practitioners and ensure their unique needs are considered in LC-PBRN initiatives.
VA Research & Interagency Collaborations. The LC-PBRN engages monthly with an interagency workgroup led by the US Department of Health and Human Services Office of Long COVID Research and Practice. These engagements support identification of research gaps that the VA may help address, monitor emerging funding opportunities, and foster collaborations. LC-PBRN representatives also meet with staff at the National Institutes of Health Researching COVID to Enhance Recovery initiative to identify pathways for veteran recruitment.
LHS Feedback Loops
The LC-PBRN was designed with an LHS approach in mind.10 Throughout development of the LC-PBRN, consideration was given to (1) capture data on new efforts within the Long COVID ecosystem (performance to data), (2) examine performance gaps and identify approaches for best practice (data to knowledge), and (3) implement best practices, develop toolkits, disseminate findings, and measure impacts (knowledge to performance). With this approach, the LC-PBRN is constantly evolving based on new information coming from the internal and external Long COVID ecosystem. Each element was deliberatively considered in relation to how data can be transformed into knowledge, knowledge into performance, and performance into data.
First, an important mechanism for feedback involves establishing clear channels of communication. Regular check-ins with key partners occur through virtual meetings to provide updates, assess needs and challenges, and codevelop action plans. For example, during a check-in with the Long COVID Field Advisory Board, members expressed a desire to incorporate veteran feedback into VA clinical practice recommendations. We provided expertise on different engagement modalities (eg, focus groups vs individual interviews), and collaboration occurred to identify key interview questions for veterans. This process resulted in a published clinician-facing Long COVID Nervous System Clinical Guide (available at [email protected]) that integrated critical feedback from veterans related to neurological symptoms.
Second, weekly executive leadership meetings include dedicated time for reflection on partner feedback, the current state of Long COVID, and contextual changes that impact deliverable priorities and timelines. Outcomes from these discussions are communicated with VHA Health Services Research and, when appropriate, to key partners to ensure alignment. For example, the Patient Identification and Analysis core was originally tasked with identifying a definition of Long COVID. However, as the broader community moved away from a singular definition, efforts were redirected toward higher-priority issues within the VA Long COVID ecosystem, including veteran enrollment in clinical trials.
Third, the Veteran Engagement Panel captures feedback from those with lived experience to inform Long COVID research and clinical efforts. The panel meetings are strategically designed to ask veterans living with Long COVID specific questions related to a given research or clinical topic of interest. For example, panel sessions with the Field Advisory Board focused on concerns articulated by veterans related to the mental health and gastroenterological symptoms associated with Long COVID. Insights from these discussions will inform development of Long COVID mental health and gastroenterological clinical care guides, with several PBRN investigators serving as subject matter experts. This collaborative approach ensures that veteran perspectives are represented in developing Long COVID clinical care processes.
Fourth, research priorities identified through the Delphi consensus process will inform development of VA Request for Funding Proposals related to Long COVID. The initial survey was developed in collaboration with veterans, clinicians, and researchers across the Veteran Engagement Panel, the Field Advisory Board, and the National Research Action Plan on Long COVID.11 The process was launched in October 2024 and concluded in June 2025. The team conducted 3 consensus rounds with veterans and VA clinicians and researchers. Top priority areas included the testing assessments for diagnosing Long COVID, studying subtypes of Long COVID and treatments for each, and finding biomarkers for Long COVID. A formal publication of the results and analysis is the focus of a future publication.
Fifth, ongoing engagement with the Field Advisory Board has supported adoption of a preliminary set of clinical outcome measures. If universally adopted, these instruments may contribute to the development of a standardized data collection process and serve as common data elements collected for epidemiologic, health services, or clinical trial research.
Lessons Learned and Practice Implications
Throughout the development of the LC-PBRN, several decisions were identified that have impacted infrastructure development and implementation.
Include veterans’ voices to ensure network efforts align with patient needs. Given the novelty of Long COVID, practitioners and researchers are learning as they go. It is important to listen to individuals who live with Long COVID. Throughout the development of the LC-PBRN, veteran perspective has proven how vital it is for them to be heard when it comes to their health care. Clinicians similarly highlighted the value of incorporating patient perspectives into the development of tools and treatment strategies. Develop an interdisciplinary leadership team to foster the diverse viewpoints needed to tackle multifaceted problems. It is important to consider as many clinical and research perspectives as possible because Long COVID is a complex condition with symptoms impacting major organ systems.12-15 Therefore, the team spans across a multitude of specialties and locations.
Set clear expectations and goals with partners to uphold timely deliverables and stay within the PBRN’s capacity. When including a multitude of partners, teams should consider each of those partners’ experiences and opinions in decision-making conversations. Expectation setting is important to ensure all partners are on the same page and understand the capacity of the LC-PBRN. This allows the team to focus its efforts, avoid being overwhelmed with requests, and provide quality deliverables.
Build engaging relationships to bridge gaps between internal and external partners. A substantial number of resources focus on building relationships with partners so they can trust the LC-PBRN has their best interests in mind. These relationships are important to ensure the VA avoids duplicate efforts. This includes prioritizing connecting partners who are working on similar efforts to promote collaboration across facilities.
Conclusions
PBRNs provide an important mechanism to use LHS approaches to successfully convene research around complex issues. PBRNs can support integration across the LHS cycle, allowing for multiple feedback loops, and coordinate activities that work to achieve a larger vision. PBRNs offer centralized mechanisms to collaboratively understand and address complex problems, such as Long COVID, where the uncertainty regarding how to treat occurs in tandem with the urgency to treat. The LC-PBRN model described in this article has the potential to transcend Long COVID by building infrastructure necessary to proactively address current or future clinical conditions or populations with a LHS lens. The infrastructure can require cross-system and sector collaborations, expediency, inclusivity, and patient- and family-centeredness. Future efforts will focus on building out a larger network of VHA sites, facilitating recruitment at site and veteran levels into Long COVID trials through case identification, and systematically support the standardization of clinical data for clinical utility and evaluation of quality and/or outcomes across the VHA.
- Ottiger M, Poppele I, Sperling N, et al. Work ability and return-to-work of patients with post-COVID-19: a systematic review and meta-analysis. BMC Public Health. 2024;24:1811. doi:10.1186/s12889-024-19328-6
- Ziauddeen N, Gurdasani D, O’Hara ME, et al. Characteristics and impact of Long Covid: findings from an online survey. PLOS ONE. 2022;17:e0264331. doi:10.1371/journal.pone.0264331
- Graham F. Daily briefing: Answers emerge about long COVID recovery. Nature. Published online June 28, 2023. doi:10.1038/d41586-023-02190-8
- Al-Aly Z, Davis H, McCorkell L, et al. Long COVID science, research and policy. Nat Med. 2024;30:2148-2164. doi:10.1038/s41591-024-03173-6
- Atkins D, Kilbourne AM, Shulkin D. Moving from discovery to system-wide change: the role of research in a learning health care system: experience from three decades of health systems research in the Veterans Health Administration. Annu Rev Public Health. 2017;38:467-487. doi:10.1146/annurev-publhealth-031816-044255
- Ely EW, Brown LM, Fineberg HV. Long covid defined. N Engl J Med. 2024;391:1746-1753.doi:10.1056/NEJMsb2408466
- Joosten YA, Israel TL, Williams NA, et al. Community engagement studios: a structured approach to obtaining meaningful input from stakeholders to inform research. Acad Med. 2015;90:1646-1650. doi:10.1097/ACM.0000000000000794
- AHRQ. Quick-start guide to dissemination for practice-based research networks. Revised June 2014. Accessed December 2, 2025. https://www.ahrq.gov/sites/default/files/wysiwyg/ncepcr/resources/dissemination-quick-start-guide.pdf
- Gustavson AM, Morrow CD, Brown RJ, et al. Reimagining how we synthesize information to impact clinical care, policy, and research priorities in real time: examples and lessons learned from COVID-19. J Gen Intern Med. 2024;39:2554-2559. doi:10.1007/s11606-024-08855-y
- University of Minnesota. About the Center for Learning Health System Sciences. Updated December 11, 2025. Accessed December 12, 2025. https://med.umn.edu/clhss/about-us
- AHRQ. National Research Action Plan. Published online 2022. Accessed February 14, 2024. https://www.covid.gov/sites/default/files/documents/National-Research-Action-Plan-on-Long-COVID-08012022.pdf
- Gustavson AM, Eaton TL, Schapira RM, et al. Approaches to long COVID care: the Veterans Health Administration experience in 2021. BMJ Mil Health. 2024;170:179-180. doi:10.1136/military-2022-002185
- Gustavson AM. A learning health system approach to long COVID care. Fed Pract. 2022;39:7. doi:10.12788/fp.0288
- Palacio A, Bast E, Klimas N, et al. Lessons learned in implementing a multidisciplinary long COVID clinic. Am J Med. 2025;138:843-849.doi:10.1016/j.amjmed.2024.05.020
- Prusinski C, Yan D, Klasova J, et al. Multidisciplinary management strategies for long COVID: a narrative review. Cureus. 2024;16:e59478. doi:10.7759/cureus.59478
- Ottiger M, Poppele I, Sperling N, et al. Work ability and return-to-work of patients with post-COVID-19: a systematic review and meta-analysis. BMC Public Health. 2024;24:1811. doi:10.1186/s12889-024-19328-6
- Ziauddeen N, Gurdasani D, O’Hara ME, et al. Characteristics and impact of Long Covid: findings from an online survey. PLOS ONE. 2022;17:e0264331. doi:10.1371/journal.pone.0264331
- Graham F. Daily briefing: Answers emerge about long COVID recovery. Nature. Published online June 28, 2023. doi:10.1038/d41586-023-02190-8
- Al-Aly Z, Davis H, McCorkell L, et al. Long COVID science, research and policy. Nat Med. 2024;30:2148-2164. doi:10.1038/s41591-024-03173-6
- Atkins D, Kilbourne AM, Shulkin D. Moving from discovery to system-wide change: the role of research in a learning health care system: experience from three decades of health systems research in the Veterans Health Administration. Annu Rev Public Health. 2017;38:467-487. doi:10.1146/annurev-publhealth-031816-044255
- Ely EW, Brown LM, Fineberg HV. Long covid defined. N Engl J Med. 2024;391:1746-1753.doi:10.1056/NEJMsb2408466
- Joosten YA, Israel TL, Williams NA, et al. Community engagement studios: a structured approach to obtaining meaningful input from stakeholders to inform research. Acad Med. 2015;90:1646-1650. doi:10.1097/ACM.0000000000000794
- AHRQ. Quick-start guide to dissemination for practice-based research networks. Revised June 2014. Accessed December 2, 2025. https://www.ahrq.gov/sites/default/files/wysiwyg/ncepcr/resources/dissemination-quick-start-guide.pdf
- Gustavson AM, Morrow CD, Brown RJ, et al. Reimagining how we synthesize information to impact clinical care, policy, and research priorities in real time: examples and lessons learned from COVID-19. J Gen Intern Med. 2024;39:2554-2559. doi:10.1007/s11606-024-08855-y
- University of Minnesota. About the Center for Learning Health System Sciences. Updated December 11, 2025. Accessed December 12, 2025. https://med.umn.edu/clhss/about-us
- AHRQ. National Research Action Plan. Published online 2022. Accessed February 14, 2024. https://www.covid.gov/sites/default/files/documents/National-Research-Action-Plan-on-Long-COVID-08012022.pdf
- Gustavson AM, Eaton TL, Schapira RM, et al. Approaches to long COVID care: the Veterans Health Administration experience in 2021. BMJ Mil Health. 2024;170:179-180. doi:10.1136/military-2022-002185
- Gustavson AM. A learning health system approach to long COVID care. Fed Pract. 2022;39:7. doi:10.12788/fp.0288
- Palacio A, Bast E, Klimas N, et al. Lessons learned in implementing a multidisciplinary long COVID clinic. Am J Med. 2025;138:843-849.doi:10.1016/j.amjmed.2024.05.020
- Prusinski C, Yan D, Klasova J, et al. Multidisciplinary management strategies for long COVID: a narrative review. Cureus. 2024;16:e59478. doi:10.7759/cureus.59478
Confronting Uncertainty and Addressing Urgency for Action Through the Establishment of a VA Long COVID Practice-Based Research Network
Confronting Uncertainty and Addressing Urgency for Action Through the Establishment of a VA Long COVID Practice-Based Research Network

Effects of Lumbar Fusion and Dual-Mobility Liners on Dislocation Rates Following Total Hip Arthroplasty in a Veteran Population
Effects of Lumbar Fusion and Dual-Mobility Liners on Dislocation Rates Following Total Hip Arthroplasty in a Veteran Population
Total hip arthroplasty (THA) is among the most common elective orthopedic procedures performed annually in the United States, with an estimated 635,000 to 909,000 THAs expected each year by 2030.1 Consequently, complication rates and revision surgeries related to THA have been increasing, along with the financial burden on the health care system.2-4 Optimizing outcomes for patients undergoing THA and identifying risk factors for treatment failure have become areas of focus.
Over the last decade, there has been a renewed interest in the effect of previous lumbar spine fusion (LSF) surgery on THA outcomes. Studies have explored the rates of complications, postoperative mobility, and THA implant impingement.5-8 However, the outcome receiving the most attention in recent literature is the rate and effect of dislocation in patients with lumbar fusion surgery. Large Medicare database analyses have discovered an association with increased rates of dislocations in patients with lumbar fusion surgeries compared with those without.9,10 Prosthetic hip dislocation is an expensive complication of THA and is projected to have greater impact through 2035 due to a growing number of THA procedures.11 Identifying risk factors associated with hip dislocation is paramount to mitigating its effect on patients who have undergone THA.
Recent research has found increased rates of THA dislocation and revision surgery in patients with LSF, with some studies showing previous LSF as the strongest independent predictor.6-16 However, controversy surrounds this relationship, including the sequence of procedures (LSF before or after THA), the time between procedures, and involvement of the sacrum in LSF. One study found that patients had a 106% increased risk of dislocation when LSF was performed before THA compared with patients who underwent LSF 5 years after undergoing THA, while another study showed no significant difference in dislocations pre- vs post-LSF.16,17 An additional study showed no significant difference in the rate of dislocation in patients without sacral involvement in the LSF, while also showing significantly higher rates of dislocation in LSF with sacral involvement.12 The researchers also found a trend toward more dislocations in longer lumbosacral fusions. Recent studies have also examined dislocation rates with lumbar fusion in patients treated with dual-mobility liners.18-20 The consensus from these studies is that dual-mobility liners significantly decrease the rate of dislocation in primary THAs with lumbar fusion.
The present study sought to determine the rates of hip dislocations in a US Department of Veterans Affairs (VA) hospital setting. To the authors’ knowledge, no retrospective study focusing on THAs in the veteran population has been performed. This study benefits from controlling for various surgeon techniques and surgical preferences when compared to large Medicare database studies because the orthopedic surgeon (ABK) only performed the posterior approach for all patients during the study period.
The primary objective of this study was to determine whether the rates of hip dislocation would, in fact, be higher in patients with lumbar fusion surgery, as recent database studies suggest. Secondary objectives included determining whether patient characteristics, comorbidities, number of levels fused, or inclusion of the sacrum in the fusion construct influenced dislocation rates. Furthermore, VA Dayton Healthcare System (VADHS) began routine use of dual-mobility liners for lumbar fusion patients in 2018, allowing for examination of these patients.
Methods
The Wright State University and VADHS Institutional Review Board approved this study design. A retrospective review of all primary THAs at VADHS was performed to investigate the relationship between previous lumbar spine fusion and the incidence of THA revision. Manual chart review was performed for patients who underwent primary THA between January 2003, and December 2022. One surgeon performed all surgeries using only the posterior approach. Patients were not excluded if they had bilateral procedures and all eligible hips were included. Patients with a concomitant diagnosis of fracture of the femoral head or femoral neck at the time of surgery were excluded. Additionally, only patients with ≥ 12 months of follow-up data were included.
The primary outcome was dislocation within 12 months of THA; the primary independent variable was LSF prior to THA. Covariates included patient demographics (age, sex, body mass index [BMI]) and Charlson Comorbidity Index (CCI) score, with additional data collected on the number of levels fused, sacral spine involvement, revision rates, and use of dual-mobility liners. Year of surgery was also included in analyses to account for any changes that may have occurred during the study period.
Statistical Analysis
Statistical analyses were performed in SAS 9.4. Patients were grouped into 2 cohorts, depending on whether they had received LSF prior to THA. Analyses were adjusted for repeated measures to account for the small percentage of patients with bilateral procedures.
Univariate comparisons between cohorts for covariates, as well as rates of dislocation and revision, were performed using the independent samples t test for continuous variables and the Fisher exact test for dichotomous categorical variables. Significant comorbidities, as well as age, sex, BMI, liner type, LSF cohort, and surgery year, were included in a logistic regression model to determine what effect, if any, they had on the likelihood of dislocation. Variables were removed using a backward stepwise approach, starting with the nonsignificant variable effect with the lowest χ2 value, and continuing until reaching a final model where all remaining variable effects were significant. For the variables retained in the final model, odds ratios (ORs) with 95% CIs were derived, with dislocation designated as the event. Individual comorbidity subcomponents of the CCI were also analyzed for their effects on dislocation using backward stepwise logistic regression. A secondary analysis among patients with LSF tested for the influence of the number of vertebral levels fused, the presence or absence of sacral involvement in the fusion, and the use of dual-mobility liners on the likelihood of hip dislocation.
Results
The LSF cohort included 39 patients with THA and prior LSF, 3 of whom had bilateral procedures, for a total of 42 hips. The non-LSF cohort included 813 patients with THA, 112 of whom had bilateral procedures, for a total of 925 hips. The LSF and non-LSF cohorts did not differ significantly in age, sex, BMI, CCI, or revision rates (Table). The LSF cohort included a significantly higher percentage of hips receiving dual-mobility liners than did the non-LSF cohort (23.8% vs 0.6%; P < .001) and had more than twice the rate of dislocation (4 of 42 hips [9.5%] vs 35 of 925 hips [3.8%]), although this difference was not statistically significant (P = .08).
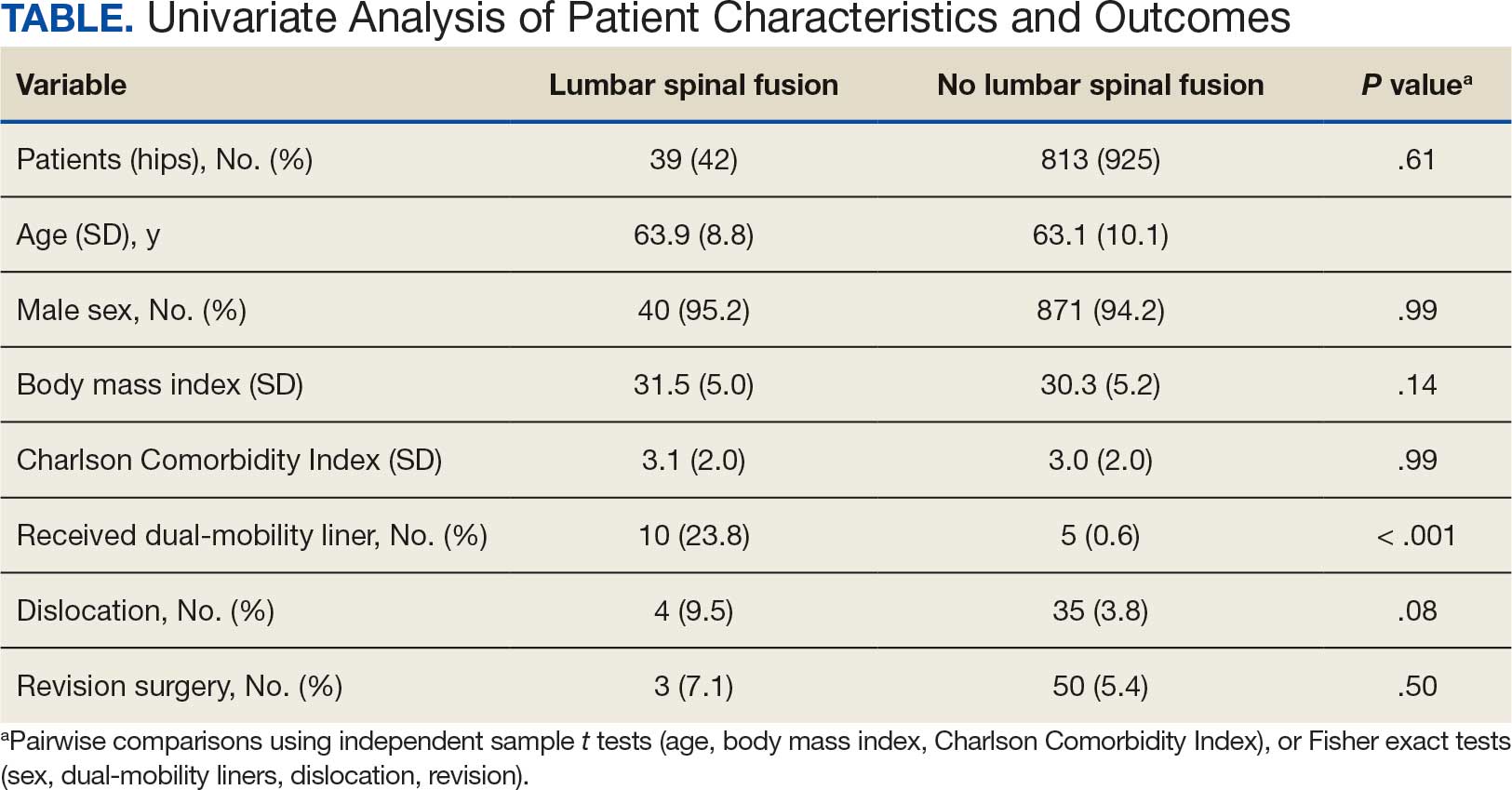
The final logistic regression model with dislocation as the outcome was statistically significant (χ2, 17.47; P < .001) and retained 2 significant predictor variables: LSF cohort (χ2, 4.63; P = .03), and sex (χ2, 18.27; P < .001). Females were more likely than males to experience dislocation (OR, 5.84; 95% CI, 2.60-13.13; P < .001) as were patients who had LSF prior to THA (OR, 3.42; 95% CI, 1.12-10.47; P = .03) (Figure). None of the CCI subcomponent comorbidities significantly affected the probability of dislocation (myocardial infarction, P = .46; congestive heart failure, P = .47; peripheral vascular disease, P = .97; stroke, P = .51; dementia, P = .99; chronic obstructive pulmonary disease, P = .95; connective tissue disease, P = .25; peptic ulcer, P = .41; liver disease, P = .30; diabetes, P = .06; hemiplegia, P = .99; chronic kidney disease, P = .82; solid tumor, P = .90; leukemia, P = .99; lymphoma, P = .99; AIDS, P = .99). Within the LSF cohort, neither the number of levels fused (P = .83) nor sacral involvement (P = .42), significantly affected the probability of hip dislocation. None of the patients in either cohort who received dual-mobility liners subsequently dislocated their hips, nor did any of them require revision surgery.
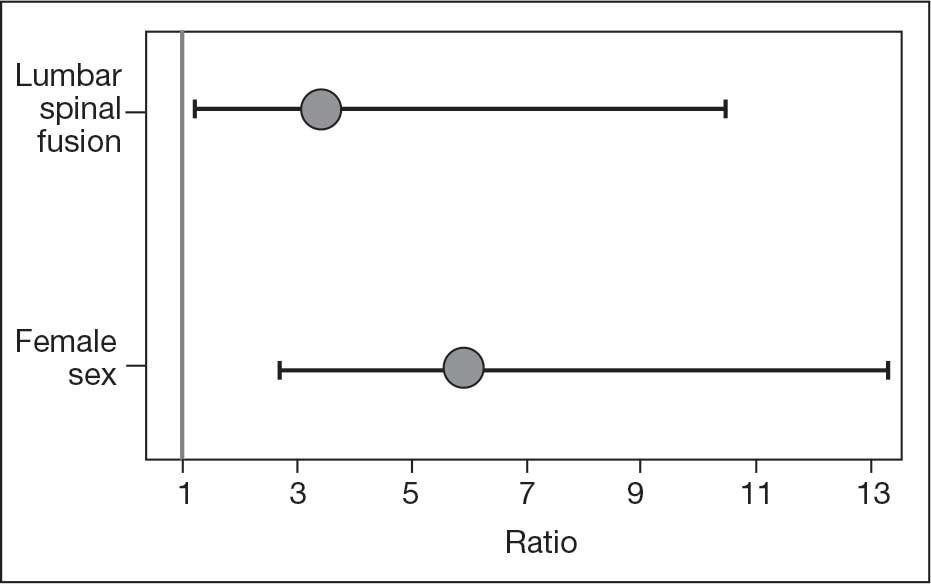
Discussion
Spinopelvic biomechanics have been an area of increasing interest and research. Spinal fusion has been shown to alter the mobility of the pelvis and has been associated with decreased stability of THA implants.21 For example, in the setting of a fused spine, the lack of compensatory changes in pelvic tilt or acetabular anteversion when adjusting to a seated or standing position may predispose patients to impingement because the acetabular component is not properly positioned. Dual-mobility constructs mitigate this risk by providing an additional articulation, which increases jump distance and range of motion prior to impingement, thereby enhancing stability.
The use of dual-mobility liners in patients with LSF has also been examined.18-20 These studies demonstrate a reduced risk of postoperative THA dislocation in patients with previous LSF. The rate of postoperative complications and revisions for LSF patients with dual-mobility liners was also found to be similar to that of THAs without dual-mobility in patients without prior LSF. This study focused on a veteran population to demonstrate the efficacy of dual-mobility liners in patients with LSF. The results indicate that LSF prior to THA and female sex were predictors for prosthetic hip dislocations in the 12-month postoperative period in this patient population, which aligns with the current literature.
The dislocation rate in the LSF-THA group (9.5%) was higher than the dislocation rate in the control group (3.8%). Although not statistically significant in the univariate analysis, LSF was shown to be a significant risk factor after controlling for patient sex. Other studies have found the dislocation rate to be 3% to 7%, which is lower than the dislocation rate observed in this study.8,10,16
The reasons for this higher rate of dislocation are not entirely clear. A veteran population has poorer overall health than the general population, which may contribute to the higher than previously reported dislocation rates.22 These results can be applied to the management of veterans seeking THA.
There have been conflicting reports regarding the impact a patient’s sex has on THA outcomes in the general population.23-26 This study found that female patients had higher rates of dislocation within 1 year of THA than male patients. This difference, which could be due to differences in baseline anatomic hip morphology between the sexes; females tend to have smaller femoral head sizes and less offset compared with males.27,28 However, this finding could have been confounded by the small number of female veterans in the study cohort.
A type 2 diabetes mellitus (T2DM) diagnosis, which is a component of CCI, trended toward increased risk of prosthetic hip dislocation. Multiple studies have also discussed the increased risk of postoperative infections and revisions following THA in patients with T2DM.29-31 One study found T2DM to be an independent risk factor for immediate in-hospital postoperative complications following hip arthroplasty.32
Another factor that may influence postoperative dislocation risk is surgical approach. The posterior approach has historically been associated with higher rates of instability when compared to anterior or lateral THA.33 Researchers have also looked at the role that surgical approach plays in patients with prior LSF. Huebschmann et al confirmed that not only is LSF a significant risk factor for dislocation following THA, but anterior and laterally based surgical approaches may mitigate this risk.34
Limitations
As a retrospective cohort study, the reliability of the data hinges on complete documentation. Documentation of all encounters for dislocations was obtained from the VA Computerized Patient Record System, which may have led to some dislocation events being missed. However, as long as there was adequate postoperative follow-up, it was assumed all events outside the VA were included. Another limitation of this study was that male patients greatly outnumbered female patients, and this fact could limit the generalizability of findings to the population as a whole.
Conclusions
This study in a veteran population found that prior LSF and female sex were significant predictors for postoperative dislocation within 1 year of THA surgery. Additionally, the use of a dual-mobility liner was found to be protective against postoperative dislocation events. These data allow clinicians to better counsel veterans on the risk factors associated with postoperative dislocation and strategies to mitigate this risk.
- Sloan M, Premkumar A, Sheth NP. Projected volume of primary total joint arthroplasty in the U.S., 2014 to 2030. J Bone Joint Surg Am. 2018;100:1455-1460. doi:10.2106/JBJS.17.01617
- Bozic KJ, Kurtz SM, Lau E, et al. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91:128-133. doi:10.2106/JBJS.H.00155
- Kurtz SM, Ong KL, Schmier J, et al. Future clinical and economic impact of revision total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89:144-151. doi:10.2106/JBJS.G.00587
- Kurtz SM, Ong KL, Schmier J, et al. Primary and revision arthroplasty surgery caseloads in the United States from 1990 to 2004. J Arthroplasty. 2009;24:195-203. doi:10.1016/j.arth.2007.11.015
- Yamato Y, Furuhashi H, Hasegawa T, et al. Simulation of implant impingement after spinal corrective fusion surgery in patients with previous total hip arthroplasty: a retrospective case series. Spine (Phila Pa 1976). 2021;46:512-519. doi:10.1097/BRS.0000000000003836
- Mudrick CA, Melvin JS, Springer BD. Late posterior hip instability after lumbar spinopelvic fusion. Arthroplast Today. 2015;1:25-29. doi:10.1016/j.artd.2015.05.002
- Diebo BG, Beyer GA, Grieco PW, et al. Complications in patients undergoing spinal fusion after THA. Clin Orthop Relat Res. 2018;476:412-417.doi:10.1007/s11999.0000000000000009 8.
- Sing DC, Barry JJ, Aguilar TU, et al. Prior lumbar spinal arthrodesis increases risk of prosthetic-related complication in total hip arthroplasty. J Arthroplasty. 2016;31:227-232.e1. doi:10.1016/j.arth.2016.02.069
- King CA, Landy DC, Martell JM, et al. Time to dislocation analysis of lumbar spine fusion following total hip arthroplasty: breaking up a happy home. J Arthroplasty. 2018;33:3768-3772. doi:10.1016/j.arth.2018.08.029
- Buckland AJ, Puvanesarajah V, Vigdorchik J, et al. Dislocation of a primary total hip arthroplasty is more common in patients with a lumbar spinal fusion. Bone Joint J. 2017;99-B:585-591.doi:10.1302/0301-620X.99B5.BJJ-2016-0657.R1
- Pirruccio K, Premkumar A, Sheth NP. The burden of prosthetic hip dislocations in the United States is projected to significantly increase by 2035. Hip Int. 2021;31:714-721. doi:10.1177/1120700020923619
- Salib CG, Reina N, Perry KI, et al. Lumbar fusion involving the sacrum increases dislocation risk in primary total hip arthroplasty. Bone Joint J. 2019;101-B:198-206. doi:10.1302/0301-620X.101B2.BJJ-2018-0754.R1
- An VVG, Phan K, Sivakumar BS, et al. Prior lumbar spinal fusion is associated with an increased risk of dislocation and revision in total hip arthroplasty: a meta-analysis. J Arthroplasty. 2018;33:297-300. doi:10.1016/j.arth.2017.08.040
- Klemt C, Padmanabha A, Tirumala V, et al. Lumbar spine fusion before revision total hip arthroplasty is associated with increased dislocation rates. J Am Acad Orthop Surg. 2021;29:e860-e868. doi:10.5435/JAAOS-D-20-00824
- Gausden EB, Parhar HS, Popper JE, et al. Risk factors for early dislocation following primary elective total hip arthroplasty. J Arthroplasty. 2018;33:1567-1571. doi:10.1016/j.arth.2017.12.034
- Malkani AL, Himschoot KJ, Ong KL, et al. Does timing of primary total hip arthroplasty prior to or after lumbar spine fusion have an effect on dislocation and revision rates?. J Arthroplasty. 2019;34:907-911. doi:10.1016/j.arth.2019.01.009
- Parilla FW, Shah RR, Gordon AC, et al. Does it matter: total hip arthroplasty or lumbar spinal fusion first? Preoperative sagittal spinopelvic measurements guide patient-specific surgical strategies in patients requiring both. J Arthroplasty. 2019;34:2652-2662. doi:10.1016/j.arth.2019.05.053
- Chalmers BP, Syku M, Sculco TP, et al. Dual-mobility constructs in primary total hip arthroplasty in high-risk patients with spinal fusions: our institutional experience. Arthroplast Today. 2020;6:749-754. doi:10.1016/j.artd.2020.07.024
- Nessler JM, Malkani AL, Sachdeva S, et al. Use of dual mobility cups in patients undergoing primary total hip arthroplasty with prior lumbar spine fusion. Int Orthop. 2020;44:857-862. doi:10.1007/s00264-020-04507-y
- Nessler JM, Malkani AL, Yep PJ, et al. Dislocation rates of primary total hip arthroplasty in patients with prior lumbar spine fusion and lumbar degenerative disk disease with and without utilization of dual mobility cups: an American Joint Replacement Registry study. J Am Acad Orthop Surg. 2023;31:e271-e277. doi:10.5435/JAAOS-D-22-00767
- Phan D, Bederman SS, Schwarzkopf R. The influence of sagittal spinal deformity on anteversion of the acetabular component in total hip arthroplasty. Bone Joint J. 2015;97-B:1017-1023. doi:10.1302/0301-620X.97B8.35700
- Agha Z, Lofgren RP, VanRuiswyk JV, et al. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160:3252-3257. doi:10.1001/archinte.160.21.325223.
- Basques BA, Bell JA, Fillingham YA, et al. Gender differences for hip and knee arthroplasty: complications and healthcare utilization. J Arthroplasty. 2019;34:1593-1597.e1. doi:10.1016/j.arth.2019.03.064
- Kim YH, Choi Y, Kim JS. Influence of patient-, design-, and surgery-related factors on rate of dislocation after primary cementless total hip arthroplasty. J Arthroplasty. 2009;24:1258-1263. doi:10.1016/j.arth.2009.03.017
- Chen A, Paxton L, Zheng X, et al. Association of sex with risk of 2-year revision among patients undergoing total hip arthroplasty. JAMA Netw Open. 2021;4:e2110687. doi:10.1001/jamanetworkopen.2021.10687
- Inacio MCS, Ake CF, Paxton EW, et al. Sex and risk of hip implant failure: assessing total hip arthroplasty outcomes in the United States. JAMA Intern Med. 2013;173:435-441. doi:10.1001/jamainternmed.2013.3271
- Karlson EW, Daltroy LH, Liang MH, et al. Gender differences in patient preferences may underlie differential utilization of elective surgery. Am J Med. 1997;102:524-530. doi:10.1016/s0002-9343(97)00050-8
- Kostamo T, Bourne RB, Whittaker JP, et al. No difference in gender-specific hip replacement outcomes. Clin Orthop Relat Res. 2009;467:135-140. doi:10.1007/s11999-008-0466-2
- Papagelopoulos PJ, Idusuyi OB, Wallrichs SL, et al. Long term outcome and survivorship analysis of primary total knee arthroplasty in patients with diabetes mellitus. Clin Orthop Relat Res. 1996;(330):124-132. doi:10.1097/00003086-199609000-00015
- Fitzgerald RH Jr, Nolan DR, Ilstrup DM, et al. Deep wound sepsis following total hip arthroplasty. J Bone Joint Surg Am. 1977;59:847-855.
- Blom AW, Brown J, Taylor AH, et al. Infection after total knee arthroplasty. J Bone Joint Surg Br. 2004;86:688-691. doi:10.1302/0301-620x.86b5.14887
- Jain NB, Guller U, Pietrobon R, et al. Comorbidities increase complication rates in patients having arthroplasty. Clin Orthop Relat Res. 2005;435:232-238. doi:10.1097/01.blo.0000156479.97488.a2
- Docter S, Philpott HT, Godkin L, et al. Comparison of intra and post-operative complication rates among surgical approaches in Total Hip Arthroplasty: A systematic review and meta-analysis. J Orthop. 2020;20:310-325. doi:10.1016/j.jor.2020.05.008
- Huebschmann NA, Lawrence KW, Robin JX, et al. Does surgical approach affect dislocation rate after total hip arthroplasty in patients who have prior lumbar spinal fusion? A retrospective analysis of 16,223 cases. J Arthroplasty. 2024;39:S306-S313. doi:10.1016/j.arth.2024.03.068
Total hip arthroplasty (THA) is among the most common elective orthopedic procedures performed annually in the United States, with an estimated 635,000 to 909,000 THAs expected each year by 2030.1 Consequently, complication rates and revision surgeries related to THA have been increasing, along with the financial burden on the health care system.2-4 Optimizing outcomes for patients undergoing THA and identifying risk factors for treatment failure have become areas of focus.
Over the last decade, there has been a renewed interest in the effect of previous lumbar spine fusion (LSF) surgery on THA outcomes. Studies have explored the rates of complications, postoperative mobility, and THA implant impingement.5-8 However, the outcome receiving the most attention in recent literature is the rate and effect of dislocation in patients with lumbar fusion surgery. Large Medicare database analyses have discovered an association with increased rates of dislocations in patients with lumbar fusion surgeries compared with those without.9,10 Prosthetic hip dislocation is an expensive complication of THA and is projected to have greater impact through 2035 due to a growing number of THA procedures.11 Identifying risk factors associated with hip dislocation is paramount to mitigating its effect on patients who have undergone THA.
Recent research has found increased rates of THA dislocation and revision surgery in patients with LSF, with some studies showing previous LSF as the strongest independent predictor.6-16 However, controversy surrounds this relationship, including the sequence of procedures (LSF before or after THA), the time between procedures, and involvement of the sacrum in LSF. One study found that patients had a 106% increased risk of dislocation when LSF was performed before THA compared with patients who underwent LSF 5 years after undergoing THA, while another study showed no significant difference in dislocations pre- vs post-LSF.16,17 An additional study showed no significant difference in the rate of dislocation in patients without sacral involvement in the LSF, while also showing significantly higher rates of dislocation in LSF with sacral involvement.12 The researchers also found a trend toward more dislocations in longer lumbosacral fusions. Recent studies have also examined dislocation rates with lumbar fusion in patients treated with dual-mobility liners.18-20 The consensus from these studies is that dual-mobility liners significantly decrease the rate of dislocation in primary THAs with lumbar fusion.
The present study sought to determine the rates of hip dislocations in a US Department of Veterans Affairs (VA) hospital setting. To the authors’ knowledge, no retrospective study focusing on THAs in the veteran population has been performed. This study benefits from controlling for various surgeon techniques and surgical preferences when compared to large Medicare database studies because the orthopedic surgeon (ABK) only performed the posterior approach for all patients during the study period.
The primary objective of this study was to determine whether the rates of hip dislocation would, in fact, be higher in patients with lumbar fusion surgery, as recent database studies suggest. Secondary objectives included determining whether patient characteristics, comorbidities, number of levels fused, or inclusion of the sacrum in the fusion construct influenced dislocation rates. Furthermore, VA Dayton Healthcare System (VADHS) began routine use of dual-mobility liners for lumbar fusion patients in 2018, allowing for examination of these patients.
Methods
The Wright State University and VADHS Institutional Review Board approved this study design. A retrospective review of all primary THAs at VADHS was performed to investigate the relationship between previous lumbar spine fusion and the incidence of THA revision. Manual chart review was performed for patients who underwent primary THA between January 2003, and December 2022. One surgeon performed all surgeries using only the posterior approach. Patients were not excluded if they had bilateral procedures and all eligible hips were included. Patients with a concomitant diagnosis of fracture of the femoral head or femoral neck at the time of surgery were excluded. Additionally, only patients with ≥ 12 months of follow-up data were included.
The primary outcome was dislocation within 12 months of THA; the primary independent variable was LSF prior to THA. Covariates included patient demographics (age, sex, body mass index [BMI]) and Charlson Comorbidity Index (CCI) score, with additional data collected on the number of levels fused, sacral spine involvement, revision rates, and use of dual-mobility liners. Year of surgery was also included in analyses to account for any changes that may have occurred during the study period.
Statistical Analysis
Statistical analyses were performed in SAS 9.4. Patients were grouped into 2 cohorts, depending on whether they had received LSF prior to THA. Analyses were adjusted for repeated measures to account for the small percentage of patients with bilateral procedures.
Univariate comparisons between cohorts for covariates, as well as rates of dislocation and revision, were performed using the independent samples t test for continuous variables and the Fisher exact test for dichotomous categorical variables. Significant comorbidities, as well as age, sex, BMI, liner type, LSF cohort, and surgery year, were included in a logistic regression model to determine what effect, if any, they had on the likelihood of dislocation. Variables were removed using a backward stepwise approach, starting with the nonsignificant variable effect with the lowest χ2 value, and continuing until reaching a final model where all remaining variable effects were significant. For the variables retained in the final model, odds ratios (ORs) with 95% CIs were derived, with dislocation designated as the event. Individual comorbidity subcomponents of the CCI were also analyzed for their effects on dislocation using backward stepwise logistic regression. A secondary analysis among patients with LSF tested for the influence of the number of vertebral levels fused, the presence or absence of sacral involvement in the fusion, and the use of dual-mobility liners on the likelihood of hip dislocation.
Results
The LSF cohort included 39 patients with THA and prior LSF, 3 of whom had bilateral procedures, for a total of 42 hips. The non-LSF cohort included 813 patients with THA, 112 of whom had bilateral procedures, for a total of 925 hips. The LSF and non-LSF cohorts did not differ significantly in age, sex, BMI, CCI, or revision rates (Table). The LSF cohort included a significantly higher percentage of hips receiving dual-mobility liners than did the non-LSF cohort (23.8% vs 0.6%; P < .001) and had more than twice the rate of dislocation (4 of 42 hips [9.5%] vs 35 of 925 hips [3.8%]), although this difference was not statistically significant (P = .08).
The final logistic regression model with dislocation as the outcome was statistically significant (χ2, 17.47; P < .001) and retained 2 significant predictor variables: LSF cohort (χ2, 4.63; P = .03), and sex (χ2, 18.27; P < .001). Females were more likely than males to experience dislocation (OR, 5.84; 95% CI, 2.60-13.13; P < .001) as were patients who had LSF prior to THA (OR, 3.42; 95% CI, 1.12-10.47; P = .03) (Figure). None of the CCI subcomponent comorbidities significantly affected the probability of dislocation (myocardial infarction, P = .46; congestive heart failure, P = .47; peripheral vascular disease, P = .97; stroke, P = .51; dementia, P = .99; chronic obstructive pulmonary disease, P = .95; connective tissue disease, P = .25; peptic ulcer, P = .41; liver disease, P = .30; diabetes, P = .06; hemiplegia, P = .99; chronic kidney disease, P = .82; solid tumor, P = .90; leukemia, P = .99; lymphoma, P = .99; AIDS, P = .99). Within the LSF cohort, neither the number of levels fused (P = .83) nor sacral involvement (P = .42), significantly affected the probability of hip dislocation. None of the patients in either cohort who received dual-mobility liners subsequently dislocated their hips, nor did any of them require revision surgery.
Discussion
Spinopelvic biomechanics have been an area of increasing interest and research. Spinal fusion has been shown to alter the mobility of the pelvis and has been associated with decreased stability of THA implants.21 For example, in the setting of a fused spine, the lack of compensatory changes in pelvic tilt or acetabular anteversion when adjusting to a seated or standing position may predispose patients to impingement because the acetabular component is not properly positioned. Dual-mobility constructs mitigate this risk by providing an additional articulation, which increases jump distance and range of motion prior to impingement, thereby enhancing stability.
The use of dual-mobility liners in patients with LSF has also been examined.18-20 These studies demonstrate a reduced risk of postoperative THA dislocation in patients with previous LSF. The rate of postoperative complications and revisions for LSF patients with dual-mobility liners was also found to be similar to that of THAs without dual-mobility in patients without prior LSF. This study focused on a veteran population to demonstrate the efficacy of dual-mobility liners in patients with LSF. The results indicate that LSF prior to THA and female sex were predictors for prosthetic hip dislocations in the 12-month postoperative period in this patient population, which aligns with the current literature.
The dislocation rate in the LSF-THA group (9.5%) was higher than the dislocation rate in the control group (3.8%). Although not statistically significant in the univariate analysis, LSF was shown to be a significant risk factor after controlling for patient sex. Other studies have found the dislocation rate to be 3% to 7%, which is lower than the dislocation rate observed in this study.8,10,16
The reasons for this higher rate of dislocation are not entirely clear. A veteran population has poorer overall health than the general population, which may contribute to the higher than previously reported dislocation rates.22 These results can be applied to the management of veterans seeking THA.
There have been conflicting reports regarding the impact a patient’s sex has on THA outcomes in the general population.23-26 This study found that female patients had higher rates of dislocation within 1 year of THA than male patients. This difference, which could be due to differences in baseline anatomic hip morphology between the sexes; females tend to have smaller femoral head sizes and less offset compared with males.27,28 However, this finding could have been confounded by the small number of female veterans in the study cohort.
A type 2 diabetes mellitus (T2DM) diagnosis, which is a component of CCI, trended toward increased risk of prosthetic hip dislocation. Multiple studies have also discussed the increased risk of postoperative infections and revisions following THA in patients with T2DM.29-31 One study found T2DM to be an independent risk factor for immediate in-hospital postoperative complications following hip arthroplasty.32
Another factor that may influence postoperative dislocation risk is surgical approach. The posterior approach has historically been associated with higher rates of instability when compared to anterior or lateral THA.33 Researchers have also looked at the role that surgical approach plays in patients with prior LSF. Huebschmann et al confirmed that not only is LSF a significant risk factor for dislocation following THA, but anterior and laterally based surgical approaches may mitigate this risk.34
Limitations
As a retrospective cohort study, the reliability of the data hinges on complete documentation. Documentation of all encounters for dislocations was obtained from the VA Computerized Patient Record System, which may have led to some dislocation events being missed. However, as long as there was adequate postoperative follow-up, it was assumed all events outside the VA were included. Another limitation of this study was that male patients greatly outnumbered female patients, and this fact could limit the generalizability of findings to the population as a whole.
Conclusions
This study in a veteran population found that prior LSF and female sex were significant predictors for postoperative dislocation within 1 year of THA surgery. Additionally, the use of a dual-mobility liner was found to be protective against postoperative dislocation events. These data allow clinicians to better counsel veterans on the risk factors associated with postoperative dislocation and strategies to mitigate this risk.
Total hip arthroplasty (THA) is among the most common elective orthopedic procedures performed annually in the United States, with an estimated 635,000 to 909,000 THAs expected each year by 2030.1 Consequently, complication rates and revision surgeries related to THA have been increasing, along with the financial burden on the health care system.2-4 Optimizing outcomes for patients undergoing THA and identifying risk factors for treatment failure have become areas of focus.
Over the last decade, there has been a renewed interest in the effect of previous lumbar spine fusion (LSF) surgery on THA outcomes. Studies have explored the rates of complications, postoperative mobility, and THA implant impingement.5-8 However, the outcome receiving the most attention in recent literature is the rate and effect of dislocation in patients with lumbar fusion surgery. Large Medicare database analyses have discovered an association with increased rates of dislocations in patients with lumbar fusion surgeries compared with those without.9,10 Prosthetic hip dislocation is an expensive complication of THA and is projected to have greater impact through 2035 due to a growing number of THA procedures.11 Identifying risk factors associated with hip dislocation is paramount to mitigating its effect on patients who have undergone THA.
Recent research has found increased rates of THA dislocation and revision surgery in patients with LSF, with some studies showing previous LSF as the strongest independent predictor.6-16 However, controversy surrounds this relationship, including the sequence of procedures (LSF before or after THA), the time between procedures, and involvement of the sacrum in LSF. One study found that patients had a 106% increased risk of dislocation when LSF was performed before THA compared with patients who underwent LSF 5 years after undergoing THA, while another study showed no significant difference in dislocations pre- vs post-LSF.16,17 An additional study showed no significant difference in the rate of dislocation in patients without sacral involvement in the LSF, while also showing significantly higher rates of dislocation in LSF with sacral involvement.12 The researchers also found a trend toward more dislocations in longer lumbosacral fusions. Recent studies have also examined dislocation rates with lumbar fusion in patients treated with dual-mobility liners.18-20 The consensus from these studies is that dual-mobility liners significantly decrease the rate of dislocation in primary THAs with lumbar fusion.
The present study sought to determine the rates of hip dislocations in a US Department of Veterans Affairs (VA) hospital setting. To the authors’ knowledge, no retrospective study focusing on THAs in the veteran population has been performed. This study benefits from controlling for various surgeon techniques and surgical preferences when compared to large Medicare database studies because the orthopedic surgeon (ABK) only performed the posterior approach for all patients during the study period.
The primary objective of this study was to determine whether the rates of hip dislocation would, in fact, be higher in patients with lumbar fusion surgery, as recent database studies suggest. Secondary objectives included determining whether patient characteristics, comorbidities, number of levels fused, or inclusion of the sacrum in the fusion construct influenced dislocation rates. Furthermore, VA Dayton Healthcare System (VADHS) began routine use of dual-mobility liners for lumbar fusion patients in 2018, allowing for examination of these patients.
Methods
The Wright State University and VADHS Institutional Review Board approved this study design. A retrospective review of all primary THAs at VADHS was performed to investigate the relationship between previous lumbar spine fusion and the incidence of THA revision. Manual chart review was performed for patients who underwent primary THA between January 2003, and December 2022. One surgeon performed all surgeries using only the posterior approach. Patients were not excluded if they had bilateral procedures and all eligible hips were included. Patients with a concomitant diagnosis of fracture of the femoral head or femoral neck at the time of surgery were excluded. Additionally, only patients with ≥ 12 months of follow-up data were included.
The primary outcome was dislocation within 12 months of THA; the primary independent variable was LSF prior to THA. Covariates included patient demographics (age, sex, body mass index [BMI]) and Charlson Comorbidity Index (CCI) score, with additional data collected on the number of levels fused, sacral spine involvement, revision rates, and use of dual-mobility liners. Year of surgery was also included in analyses to account for any changes that may have occurred during the study period.
Statistical Analysis
Statistical analyses were performed in SAS 9.4. Patients were grouped into 2 cohorts, depending on whether they had received LSF prior to THA. Analyses were adjusted for repeated measures to account for the small percentage of patients with bilateral procedures.
Univariate comparisons between cohorts for covariates, as well as rates of dislocation and revision, were performed using the independent samples t test for continuous variables and the Fisher exact test for dichotomous categorical variables. Significant comorbidities, as well as age, sex, BMI, liner type, LSF cohort, and surgery year, were included in a logistic regression model to determine what effect, if any, they had on the likelihood of dislocation. Variables were removed using a backward stepwise approach, starting with the nonsignificant variable effect with the lowest χ2 value, and continuing until reaching a final model where all remaining variable effects were significant. For the variables retained in the final model, odds ratios (ORs) with 95% CIs were derived, with dislocation designated as the event. Individual comorbidity subcomponents of the CCI were also analyzed for their effects on dislocation using backward stepwise logistic regression. A secondary analysis among patients with LSF tested for the influence of the number of vertebral levels fused, the presence or absence of sacral involvement in the fusion, and the use of dual-mobility liners on the likelihood of hip dislocation.
Results
The LSF cohort included 39 patients with THA and prior LSF, 3 of whom had bilateral procedures, for a total of 42 hips. The non-LSF cohort included 813 patients with THA, 112 of whom had bilateral procedures, for a total of 925 hips. The LSF and non-LSF cohorts did not differ significantly in age, sex, BMI, CCI, or revision rates (Table). The LSF cohort included a significantly higher percentage of hips receiving dual-mobility liners than did the non-LSF cohort (23.8% vs 0.6%; P < .001) and had more than twice the rate of dislocation (4 of 42 hips [9.5%] vs 35 of 925 hips [3.8%]), although this difference was not statistically significant (P = .08).
The final logistic regression model with dislocation as the outcome was statistically significant (χ2, 17.47; P < .001) and retained 2 significant predictor variables: LSF cohort (χ2, 4.63; P = .03), and sex (χ2, 18.27; P < .001). Females were more likely than males to experience dislocation (OR, 5.84; 95% CI, 2.60-13.13; P < .001) as were patients who had LSF prior to THA (OR, 3.42; 95% CI, 1.12-10.47; P = .03) (Figure). None of the CCI subcomponent comorbidities significantly affected the probability of dislocation (myocardial infarction, P = .46; congestive heart failure, P = .47; peripheral vascular disease, P = .97; stroke, P = .51; dementia, P = .99; chronic obstructive pulmonary disease, P = .95; connective tissue disease, P = .25; peptic ulcer, P = .41; liver disease, P = .30; diabetes, P = .06; hemiplegia, P = .99; chronic kidney disease, P = .82; solid tumor, P = .90; leukemia, P = .99; lymphoma, P = .99; AIDS, P = .99). Within the LSF cohort, neither the number of levels fused (P = .83) nor sacral involvement (P = .42), significantly affected the probability of hip dislocation. None of the patients in either cohort who received dual-mobility liners subsequently dislocated their hips, nor did any of them require revision surgery.
Discussion
Spinopelvic biomechanics have been an area of increasing interest and research. Spinal fusion has been shown to alter the mobility of the pelvis and has been associated with decreased stability of THA implants.21 For example, in the setting of a fused spine, the lack of compensatory changes in pelvic tilt or acetabular anteversion when adjusting to a seated or standing position may predispose patients to impingement because the acetabular component is not properly positioned. Dual-mobility constructs mitigate this risk by providing an additional articulation, which increases jump distance and range of motion prior to impingement, thereby enhancing stability.
The use of dual-mobility liners in patients with LSF has also been examined.18-20 These studies demonstrate a reduced risk of postoperative THA dislocation in patients with previous LSF. The rate of postoperative complications and revisions for LSF patients with dual-mobility liners was also found to be similar to that of THAs without dual-mobility in patients without prior LSF. This study focused on a veteran population to demonstrate the efficacy of dual-mobility liners in patients with LSF. The results indicate that LSF prior to THA and female sex were predictors for prosthetic hip dislocations in the 12-month postoperative period in this patient population, which aligns with the current literature.
The dislocation rate in the LSF-THA group (9.5%) was higher than the dislocation rate in the control group (3.8%). Although not statistically significant in the univariate analysis, LSF was shown to be a significant risk factor after controlling for patient sex. Other studies have found the dislocation rate to be 3% to 7%, which is lower than the dislocation rate observed in this study.8,10,16
The reasons for this higher rate of dislocation are not entirely clear. A veteran population has poorer overall health than the general population, which may contribute to the higher than previously reported dislocation rates.22 These results can be applied to the management of veterans seeking THA.
There have been conflicting reports regarding the impact a patient’s sex has on THA outcomes in the general population.23-26 This study found that female patients had higher rates of dislocation within 1 year of THA than male patients. This difference, which could be due to differences in baseline anatomic hip morphology between the sexes; females tend to have smaller femoral head sizes and less offset compared with males.27,28 However, this finding could have been confounded by the small number of female veterans in the study cohort.
A type 2 diabetes mellitus (T2DM) diagnosis, which is a component of CCI, trended toward increased risk of prosthetic hip dislocation. Multiple studies have also discussed the increased risk of postoperative infections and revisions following THA in patients with T2DM.29-31 One study found T2DM to be an independent risk factor for immediate in-hospital postoperative complications following hip arthroplasty.32
Another factor that may influence postoperative dislocation risk is surgical approach. The posterior approach has historically been associated with higher rates of instability when compared to anterior or lateral THA.33 Researchers have also looked at the role that surgical approach plays in patients with prior LSF. Huebschmann et al confirmed that not only is LSF a significant risk factor for dislocation following THA, but anterior and laterally based surgical approaches may mitigate this risk.34
Limitations
As a retrospective cohort study, the reliability of the data hinges on complete documentation. Documentation of all encounters for dislocations was obtained from the VA Computerized Patient Record System, which may have led to some dislocation events being missed. However, as long as there was adequate postoperative follow-up, it was assumed all events outside the VA were included. Another limitation of this study was that male patients greatly outnumbered female patients, and this fact could limit the generalizability of findings to the population as a whole.
Conclusions
This study in a veteran population found that prior LSF and female sex were significant predictors for postoperative dislocation within 1 year of THA surgery. Additionally, the use of a dual-mobility liner was found to be protective against postoperative dislocation events. These data allow clinicians to better counsel veterans on the risk factors associated with postoperative dislocation and strategies to mitigate this risk.
- Sloan M, Premkumar A, Sheth NP. Projected volume of primary total joint arthroplasty in the U.S., 2014 to 2030. J Bone Joint Surg Am. 2018;100:1455-1460. doi:10.2106/JBJS.17.01617
- Bozic KJ, Kurtz SM, Lau E, et al. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91:128-133. doi:10.2106/JBJS.H.00155
- Kurtz SM, Ong KL, Schmier J, et al. Future clinical and economic impact of revision total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89:144-151. doi:10.2106/JBJS.G.00587
- Kurtz SM, Ong KL, Schmier J, et al. Primary and revision arthroplasty surgery caseloads in the United States from 1990 to 2004. J Arthroplasty. 2009;24:195-203. doi:10.1016/j.arth.2007.11.015
- Yamato Y, Furuhashi H, Hasegawa T, et al. Simulation of implant impingement after spinal corrective fusion surgery in patients with previous total hip arthroplasty: a retrospective case series. Spine (Phila Pa 1976). 2021;46:512-519. doi:10.1097/BRS.0000000000003836
- Mudrick CA, Melvin JS, Springer BD. Late posterior hip instability after lumbar spinopelvic fusion. Arthroplast Today. 2015;1:25-29. doi:10.1016/j.artd.2015.05.002
- Diebo BG, Beyer GA, Grieco PW, et al. Complications in patients undergoing spinal fusion after THA. Clin Orthop Relat Res. 2018;476:412-417.doi:10.1007/s11999.0000000000000009 8.
- Sing DC, Barry JJ, Aguilar TU, et al. Prior lumbar spinal arthrodesis increases risk of prosthetic-related complication in total hip arthroplasty. J Arthroplasty. 2016;31:227-232.e1. doi:10.1016/j.arth.2016.02.069
- King CA, Landy DC, Martell JM, et al. Time to dislocation analysis of lumbar spine fusion following total hip arthroplasty: breaking up a happy home. J Arthroplasty. 2018;33:3768-3772. doi:10.1016/j.arth.2018.08.029
- Buckland AJ, Puvanesarajah V, Vigdorchik J, et al. Dislocation of a primary total hip arthroplasty is more common in patients with a lumbar spinal fusion. Bone Joint J. 2017;99-B:585-591.doi:10.1302/0301-620X.99B5.BJJ-2016-0657.R1
- Pirruccio K, Premkumar A, Sheth NP. The burden of prosthetic hip dislocations in the United States is projected to significantly increase by 2035. Hip Int. 2021;31:714-721. doi:10.1177/1120700020923619
- Salib CG, Reina N, Perry KI, et al. Lumbar fusion involving the sacrum increases dislocation risk in primary total hip arthroplasty. Bone Joint J. 2019;101-B:198-206. doi:10.1302/0301-620X.101B2.BJJ-2018-0754.R1
- An VVG, Phan K, Sivakumar BS, et al. Prior lumbar spinal fusion is associated with an increased risk of dislocation and revision in total hip arthroplasty: a meta-analysis. J Arthroplasty. 2018;33:297-300. doi:10.1016/j.arth.2017.08.040
- Klemt C, Padmanabha A, Tirumala V, et al. Lumbar spine fusion before revision total hip arthroplasty is associated with increased dislocation rates. J Am Acad Orthop Surg. 2021;29:e860-e868. doi:10.5435/JAAOS-D-20-00824
- Gausden EB, Parhar HS, Popper JE, et al. Risk factors for early dislocation following primary elective total hip arthroplasty. J Arthroplasty. 2018;33:1567-1571. doi:10.1016/j.arth.2017.12.034
- Malkani AL, Himschoot KJ, Ong KL, et al. Does timing of primary total hip arthroplasty prior to or after lumbar spine fusion have an effect on dislocation and revision rates?. J Arthroplasty. 2019;34:907-911. doi:10.1016/j.arth.2019.01.009
- Parilla FW, Shah RR, Gordon AC, et al. Does it matter: total hip arthroplasty or lumbar spinal fusion first? Preoperative sagittal spinopelvic measurements guide patient-specific surgical strategies in patients requiring both. J Arthroplasty. 2019;34:2652-2662. doi:10.1016/j.arth.2019.05.053
- Chalmers BP, Syku M, Sculco TP, et al. Dual-mobility constructs in primary total hip arthroplasty in high-risk patients with spinal fusions: our institutional experience. Arthroplast Today. 2020;6:749-754. doi:10.1016/j.artd.2020.07.024
- Nessler JM, Malkani AL, Sachdeva S, et al. Use of dual mobility cups in patients undergoing primary total hip arthroplasty with prior lumbar spine fusion. Int Orthop. 2020;44:857-862. doi:10.1007/s00264-020-04507-y
- Nessler JM, Malkani AL, Yep PJ, et al. Dislocation rates of primary total hip arthroplasty in patients with prior lumbar spine fusion and lumbar degenerative disk disease with and without utilization of dual mobility cups: an American Joint Replacement Registry study. J Am Acad Orthop Surg. 2023;31:e271-e277. doi:10.5435/JAAOS-D-22-00767
- Phan D, Bederman SS, Schwarzkopf R. The influence of sagittal spinal deformity on anteversion of the acetabular component in total hip arthroplasty. Bone Joint J. 2015;97-B:1017-1023. doi:10.1302/0301-620X.97B8.35700
- Agha Z, Lofgren RP, VanRuiswyk JV, et al. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160:3252-3257. doi:10.1001/archinte.160.21.325223.
- Basques BA, Bell JA, Fillingham YA, et al. Gender differences for hip and knee arthroplasty: complications and healthcare utilization. J Arthroplasty. 2019;34:1593-1597.e1. doi:10.1016/j.arth.2019.03.064
- Kim YH, Choi Y, Kim JS. Influence of patient-, design-, and surgery-related factors on rate of dislocation after primary cementless total hip arthroplasty. J Arthroplasty. 2009;24:1258-1263. doi:10.1016/j.arth.2009.03.017
- Chen A, Paxton L, Zheng X, et al. Association of sex with risk of 2-year revision among patients undergoing total hip arthroplasty. JAMA Netw Open. 2021;4:e2110687. doi:10.1001/jamanetworkopen.2021.10687
- Inacio MCS, Ake CF, Paxton EW, et al. Sex and risk of hip implant failure: assessing total hip arthroplasty outcomes in the United States. JAMA Intern Med. 2013;173:435-441. doi:10.1001/jamainternmed.2013.3271
- Karlson EW, Daltroy LH, Liang MH, et al. Gender differences in patient preferences may underlie differential utilization of elective surgery. Am J Med. 1997;102:524-530. doi:10.1016/s0002-9343(97)00050-8
- Kostamo T, Bourne RB, Whittaker JP, et al. No difference in gender-specific hip replacement outcomes. Clin Orthop Relat Res. 2009;467:135-140. doi:10.1007/s11999-008-0466-2
- Papagelopoulos PJ, Idusuyi OB, Wallrichs SL, et al. Long term outcome and survivorship analysis of primary total knee arthroplasty in patients with diabetes mellitus. Clin Orthop Relat Res. 1996;(330):124-132. doi:10.1097/00003086-199609000-00015
- Fitzgerald RH Jr, Nolan DR, Ilstrup DM, et al. Deep wound sepsis following total hip arthroplasty. J Bone Joint Surg Am. 1977;59:847-855.
- Blom AW, Brown J, Taylor AH, et al. Infection after total knee arthroplasty. J Bone Joint Surg Br. 2004;86:688-691. doi:10.1302/0301-620x.86b5.14887
- Jain NB, Guller U, Pietrobon R, et al. Comorbidities increase complication rates in patients having arthroplasty. Clin Orthop Relat Res. 2005;435:232-238. doi:10.1097/01.blo.0000156479.97488.a2
- Docter S, Philpott HT, Godkin L, et al. Comparison of intra and post-operative complication rates among surgical approaches in Total Hip Arthroplasty: A systematic review and meta-analysis. J Orthop. 2020;20:310-325. doi:10.1016/j.jor.2020.05.008
- Huebschmann NA, Lawrence KW, Robin JX, et al. Does surgical approach affect dislocation rate after total hip arthroplasty in patients who have prior lumbar spinal fusion? A retrospective analysis of 16,223 cases. J Arthroplasty. 2024;39:S306-S313. doi:10.1016/j.arth.2024.03.068
- Sloan M, Premkumar A, Sheth NP. Projected volume of primary total joint arthroplasty in the U.S., 2014 to 2030. J Bone Joint Surg Am. 2018;100:1455-1460. doi:10.2106/JBJS.17.01617
- Bozic KJ, Kurtz SM, Lau E, et al. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91:128-133. doi:10.2106/JBJS.H.00155
- Kurtz SM, Ong KL, Schmier J, et al. Future clinical and economic impact of revision total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89:144-151. doi:10.2106/JBJS.G.00587
- Kurtz SM, Ong KL, Schmier J, et al. Primary and revision arthroplasty surgery caseloads in the United States from 1990 to 2004. J Arthroplasty. 2009;24:195-203. doi:10.1016/j.arth.2007.11.015
- Yamato Y, Furuhashi H, Hasegawa T, et al. Simulation of implant impingement after spinal corrective fusion surgery in patients with previous total hip arthroplasty: a retrospective case series. Spine (Phila Pa 1976). 2021;46:512-519. doi:10.1097/BRS.0000000000003836
- Mudrick CA, Melvin JS, Springer BD. Late posterior hip instability after lumbar spinopelvic fusion. Arthroplast Today. 2015;1:25-29. doi:10.1016/j.artd.2015.05.002
- Diebo BG, Beyer GA, Grieco PW, et al. Complications in patients undergoing spinal fusion after THA. Clin Orthop Relat Res. 2018;476:412-417.doi:10.1007/s11999.0000000000000009 8.
- Sing DC, Barry JJ, Aguilar TU, et al. Prior lumbar spinal arthrodesis increases risk of prosthetic-related complication in total hip arthroplasty. J Arthroplasty. 2016;31:227-232.e1. doi:10.1016/j.arth.2016.02.069
- King CA, Landy DC, Martell JM, et al. Time to dislocation analysis of lumbar spine fusion following total hip arthroplasty: breaking up a happy home. J Arthroplasty. 2018;33:3768-3772. doi:10.1016/j.arth.2018.08.029
- Buckland AJ, Puvanesarajah V, Vigdorchik J, et al. Dislocation of a primary total hip arthroplasty is more common in patients with a lumbar spinal fusion. Bone Joint J. 2017;99-B:585-591.doi:10.1302/0301-620X.99B5.BJJ-2016-0657.R1
- Pirruccio K, Premkumar A, Sheth NP. The burden of prosthetic hip dislocations in the United States is projected to significantly increase by 2035. Hip Int. 2021;31:714-721. doi:10.1177/1120700020923619
- Salib CG, Reina N, Perry KI, et al. Lumbar fusion involving the sacrum increases dislocation risk in primary total hip arthroplasty. Bone Joint J. 2019;101-B:198-206. doi:10.1302/0301-620X.101B2.BJJ-2018-0754.R1
- An VVG, Phan K, Sivakumar BS, et al. Prior lumbar spinal fusion is associated with an increased risk of dislocation and revision in total hip arthroplasty: a meta-analysis. J Arthroplasty. 2018;33:297-300. doi:10.1016/j.arth.2017.08.040
- Klemt C, Padmanabha A, Tirumala V, et al. Lumbar spine fusion before revision total hip arthroplasty is associated with increased dislocation rates. J Am Acad Orthop Surg. 2021;29:e860-e868. doi:10.5435/JAAOS-D-20-00824
- Gausden EB, Parhar HS, Popper JE, et al. Risk factors for early dislocation following primary elective total hip arthroplasty. J Arthroplasty. 2018;33:1567-1571. doi:10.1016/j.arth.2017.12.034
- Malkani AL, Himschoot KJ, Ong KL, et al. Does timing of primary total hip arthroplasty prior to or after lumbar spine fusion have an effect on dislocation and revision rates?. J Arthroplasty. 2019;34:907-911. doi:10.1016/j.arth.2019.01.009
- Parilla FW, Shah RR, Gordon AC, et al. Does it matter: total hip arthroplasty or lumbar spinal fusion first? Preoperative sagittal spinopelvic measurements guide patient-specific surgical strategies in patients requiring both. J Arthroplasty. 2019;34:2652-2662. doi:10.1016/j.arth.2019.05.053
- Chalmers BP, Syku M, Sculco TP, et al. Dual-mobility constructs in primary total hip arthroplasty in high-risk patients with spinal fusions: our institutional experience. Arthroplast Today. 2020;6:749-754. doi:10.1016/j.artd.2020.07.024
- Nessler JM, Malkani AL, Sachdeva S, et al. Use of dual mobility cups in patients undergoing primary total hip arthroplasty with prior lumbar spine fusion. Int Orthop. 2020;44:857-862. doi:10.1007/s00264-020-04507-y
- Nessler JM, Malkani AL, Yep PJ, et al. Dislocation rates of primary total hip arthroplasty in patients with prior lumbar spine fusion and lumbar degenerative disk disease with and without utilization of dual mobility cups: an American Joint Replacement Registry study. J Am Acad Orthop Surg. 2023;31:e271-e277. doi:10.5435/JAAOS-D-22-00767
- Phan D, Bederman SS, Schwarzkopf R. The influence of sagittal spinal deformity on anteversion of the acetabular component in total hip arthroplasty. Bone Joint J. 2015;97-B:1017-1023. doi:10.1302/0301-620X.97B8.35700
- Agha Z, Lofgren RP, VanRuiswyk JV, et al. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160:3252-3257. doi:10.1001/archinte.160.21.325223.
- Basques BA, Bell JA, Fillingham YA, et al. Gender differences for hip and knee arthroplasty: complications and healthcare utilization. J Arthroplasty. 2019;34:1593-1597.e1. doi:10.1016/j.arth.2019.03.064
- Kim YH, Choi Y, Kim JS. Influence of patient-, design-, and surgery-related factors on rate of dislocation after primary cementless total hip arthroplasty. J Arthroplasty. 2009;24:1258-1263. doi:10.1016/j.arth.2009.03.017
- Chen A, Paxton L, Zheng X, et al. Association of sex with risk of 2-year revision among patients undergoing total hip arthroplasty. JAMA Netw Open. 2021;4:e2110687. doi:10.1001/jamanetworkopen.2021.10687
- Inacio MCS, Ake CF, Paxton EW, et al. Sex and risk of hip implant failure: assessing total hip arthroplasty outcomes in the United States. JAMA Intern Med. 2013;173:435-441. doi:10.1001/jamainternmed.2013.3271
- Karlson EW, Daltroy LH, Liang MH, et al. Gender differences in patient preferences may underlie differential utilization of elective surgery. Am J Med. 1997;102:524-530. doi:10.1016/s0002-9343(97)00050-8
- Kostamo T, Bourne RB, Whittaker JP, et al. No difference in gender-specific hip replacement outcomes. Clin Orthop Relat Res. 2009;467:135-140. doi:10.1007/s11999-008-0466-2
- Papagelopoulos PJ, Idusuyi OB, Wallrichs SL, et al. Long term outcome and survivorship analysis of primary total knee arthroplasty in patients with diabetes mellitus. Clin Orthop Relat Res. 1996;(330):124-132. doi:10.1097/00003086-199609000-00015
- Fitzgerald RH Jr, Nolan DR, Ilstrup DM, et al. Deep wound sepsis following total hip arthroplasty. J Bone Joint Surg Am. 1977;59:847-855.
- Blom AW, Brown J, Taylor AH, et al. Infection after total knee arthroplasty. J Bone Joint Surg Br. 2004;86:688-691. doi:10.1302/0301-620x.86b5.14887
- Jain NB, Guller U, Pietrobon R, et al. Comorbidities increase complication rates in patients having arthroplasty. Clin Orthop Relat Res. 2005;435:232-238. doi:10.1097/01.blo.0000156479.97488.a2
- Docter S, Philpott HT, Godkin L, et al. Comparison of intra and post-operative complication rates among surgical approaches in Total Hip Arthroplasty: A systematic review and meta-analysis. J Orthop. 2020;20:310-325. doi:10.1016/j.jor.2020.05.008
- Huebschmann NA, Lawrence KW, Robin JX, et al. Does surgical approach affect dislocation rate after total hip arthroplasty in patients who have prior lumbar spinal fusion? A retrospective analysis of 16,223 cases. J Arthroplasty. 2024;39:S306-S313. doi:10.1016/j.arth.2024.03.068
Effects of Lumbar Fusion and Dual-Mobility Liners on Dislocation Rates Following Total Hip Arthroplasty in a Veteran Population
Effects of Lumbar Fusion and Dual-Mobility Liners on Dislocation Rates Following Total Hip Arthroplasty in a Veteran Population
Progressive Dystrophy of the Fingernails and Toenails
Progressive Dystrophy of the Fingernails and Toenails
THE DIAGNOSIS: Nail Lichen Planus
The biopsy results showed features of hypergranulosis of the matricial epithelium, irregular acanthosis, apoptotic keratinocytes along the basal layer, and a lichenoid infiltrate consistent with nail lichen planus. The patient was started on topical clobetasol propionate 0.05% applied once daily under overnight occlusion. Additionally, intramatricial triamcinolone acetonide (2.5 mg/mL; 0.1 mL per injection) was administered into the affected nail matrix at 4-week intervals for a total of 2 sessions. At the 2-month follow-up visit, the patient reported improvement in longitudinal ridging; however, he subsequently was lost to follow-up.
Nail lichen planus is a chronic inflammatory disorder that occurs in 10% to 15% of patients with lichen planus worldwide and is more common in adults than children.1 It can manifest independently or concurrently with cutaneous and/or oral mucosal involvement. The fingernails are more commonly affected than the toenails.2 The clinical features of nail lichen planus can be classified based on involvement of the nail matrix (longitudinal ridging, red lunula, thinning of the nail plate, koilonychia, trachyonychia, pterygium, and anonychia) or nail bed (onycholysis, subungual hyperkeratosis, and splinter hemorrhages).1
In our patient, who presented with chronic progressive nail dystrophy affecting all 20 nails, onychomycosis, nail psoriasis, onychotillomania, and idiopathic trachyonychia were included in the differential.1
Onychomycosis manifests as white or yellow-brown discoloration of the nail, onycholysis, subungual hyperkeratosis, and thickening of the nail plate. Diagnosis is confirmed by the presence of septate hyphae (dermatophytes) or budding yeast cells (Candida species) on a potassium hydroxide mount. Other diagnostic modalities include dermoscopy, fungal culture, and histopathology of nail clippings, with demonstration of fungal elements identified on periodic acid-Schiff staining (eFigure 1).3
Nail psoriasis characteristically manifests as deep irregular pitting of the nails. Other features favoring psoriasis include involvement of the nail matrix manifesting as leukonychia, red lunula, and crumbling, as well as involvement of the nail bed manifesting as onycholysis, subungual hyperkeratosis, salmon patches/oil spots, and splinter hemorrhages (eFigure 2).4 Diagnosis primarily is clinical, supported by histopathology when uncertainty exists.
Onychotillomania is a behavioral disorder characterized by an irresistible urge or impulse in patients to either pick or pull at their fingernails and/or toenails. Clinicopathologic features of the involved nails are nonspecific and atypical, with possible involvement of periungual and digital skin. Diagnosis of onychotillomania is challenging.5 Dermoscopic features including anonychia with multiple obliquely arranged nail bed hemorrhages, gray pigmentation of the nail bed, and wavy lines, has been proposed to aid the diagnosis of onychotillomania.6
Idiopathic trachyonychia is isolated nail involvement characterized by rough, ridged, and thin nails affecting multiple or all of the fingernails and toenails without an underlying systemic or dermatologic condition (eFigure 3). The terms trachyonychia and 20-nail dystrophy have been used interchangeably in the literature; however, trachyonychia does not always involve all 20 nails. Other conditions causing widespread dystrophy of all 20 nails cannot be diagnosed as 20-nail dystrophy or trachyonychia without the distinct morphologic features of thin brittle nails with pronounced longitudinal ridging.7
Prompt diagnosis and early intervention in nail lichen planus is crucial due to the potential for irreversible scarring. First-line treatment options include intramatricial and intramuscular triamcinolone acetonide for 3 to 6 months.4 Second-line therapies include oral retinoids such as acitretin and alitretinoin and immunosuppressive agents such as azathioprine, mycophenolate mofetil, and cyclosporine. Other reported treatment options include clobetasol propionate, tacrolimus, dapsone, griseofulvin, etanercept, hydroxychloroquine, methotrexate, and UV therapy.4
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Leung AKC, Lam JM, Leong KF, et al. Onychomycosis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:32-45. doi:10.2174/1872213X13666191026090713
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Sidiropoulou P, Sgouros D, Theodoropoulos K, et al. Onychotillomania: a chameleon-like disorder: case report and review of literature. Skin Appendage Disord. 2019;5:104-107. doi:10.1159/000489941
- Maddy AJ, Tosti A. Dermoscopic features of onychotillomania: a study of 36 cases. J Am Acad Dermatol. 2018;79:702-705. doi:10.1016 /j.jaad.2018.04.015
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
THE DIAGNOSIS: Nail Lichen Planus
The biopsy results showed features of hypergranulosis of the matricial epithelium, irregular acanthosis, apoptotic keratinocytes along the basal layer, and a lichenoid infiltrate consistent with nail lichen planus. The patient was started on topical clobetasol propionate 0.05% applied once daily under overnight occlusion. Additionally, intramatricial triamcinolone acetonide (2.5 mg/mL; 0.1 mL per injection) was administered into the affected nail matrix at 4-week intervals for a total of 2 sessions. At the 2-month follow-up visit, the patient reported improvement in longitudinal ridging; however, he subsequently was lost to follow-up.
Nail lichen planus is a chronic inflammatory disorder that occurs in 10% to 15% of patients with lichen planus worldwide and is more common in adults than children.1 It can manifest independently or concurrently with cutaneous and/or oral mucosal involvement. The fingernails are more commonly affected than the toenails.2 The clinical features of nail lichen planus can be classified based on involvement of the nail matrix (longitudinal ridging, red lunula, thinning of the nail plate, koilonychia, trachyonychia, pterygium, and anonychia) or nail bed (onycholysis, subungual hyperkeratosis, and splinter hemorrhages).1
In our patient, who presented with chronic progressive nail dystrophy affecting all 20 nails, onychomycosis, nail psoriasis, onychotillomania, and idiopathic trachyonychia were included in the differential.1
Onychomycosis manifests as white or yellow-brown discoloration of the nail, onycholysis, subungual hyperkeratosis, and thickening of the nail plate. Diagnosis is confirmed by the presence of septate hyphae (dermatophytes) or budding yeast cells (Candida species) on a potassium hydroxide mount. Other diagnostic modalities include dermoscopy, fungal culture, and histopathology of nail clippings, with demonstration of fungal elements identified on periodic acid-Schiff staining (eFigure 1).3
Nail psoriasis characteristically manifests as deep irregular pitting of the nails. Other features favoring psoriasis include involvement of the nail matrix manifesting as leukonychia, red lunula, and crumbling, as well as involvement of the nail bed manifesting as onycholysis, subungual hyperkeratosis, salmon patches/oil spots, and splinter hemorrhages (eFigure 2).4 Diagnosis primarily is clinical, supported by histopathology when uncertainty exists.
Onychotillomania is a behavioral disorder characterized by an irresistible urge or impulse in patients to either pick or pull at their fingernails and/or toenails. Clinicopathologic features of the involved nails are nonspecific and atypical, with possible involvement of periungual and digital skin. Diagnosis of onychotillomania is challenging.5 Dermoscopic features including anonychia with multiple obliquely arranged nail bed hemorrhages, gray pigmentation of the nail bed, and wavy lines, has been proposed to aid the diagnosis of onychotillomania.6
Idiopathic trachyonychia is isolated nail involvement characterized by rough, ridged, and thin nails affecting multiple or all of the fingernails and toenails without an underlying systemic or dermatologic condition (eFigure 3). The terms trachyonychia and 20-nail dystrophy have been used interchangeably in the literature; however, trachyonychia does not always involve all 20 nails. Other conditions causing widespread dystrophy of all 20 nails cannot be diagnosed as 20-nail dystrophy or trachyonychia without the distinct morphologic features of thin brittle nails with pronounced longitudinal ridging.7
Prompt diagnosis and early intervention in nail lichen planus is crucial due to the potential for irreversible scarring. First-line treatment options include intramatricial and intramuscular triamcinolone acetonide for 3 to 6 months.4 Second-line therapies include oral retinoids such as acitretin and alitretinoin and immunosuppressive agents such as azathioprine, mycophenolate mofetil, and cyclosporine. Other reported treatment options include clobetasol propionate, tacrolimus, dapsone, griseofulvin, etanercept, hydroxychloroquine, methotrexate, and UV therapy.4
THE DIAGNOSIS: Nail Lichen Planus
The biopsy results showed features of hypergranulosis of the matricial epithelium, irregular acanthosis, apoptotic keratinocytes along the basal layer, and a lichenoid infiltrate consistent with nail lichen planus. The patient was started on topical clobetasol propionate 0.05% applied once daily under overnight occlusion. Additionally, intramatricial triamcinolone acetonide (2.5 mg/mL; 0.1 mL per injection) was administered into the affected nail matrix at 4-week intervals for a total of 2 sessions. At the 2-month follow-up visit, the patient reported improvement in longitudinal ridging; however, he subsequently was lost to follow-up.
Nail lichen planus is a chronic inflammatory disorder that occurs in 10% to 15% of patients with lichen planus worldwide and is more common in adults than children.1 It can manifest independently or concurrently with cutaneous and/or oral mucosal involvement. The fingernails are more commonly affected than the toenails.2 The clinical features of nail lichen planus can be classified based on involvement of the nail matrix (longitudinal ridging, red lunula, thinning of the nail plate, koilonychia, trachyonychia, pterygium, and anonychia) or nail bed (onycholysis, subungual hyperkeratosis, and splinter hemorrhages).1
In our patient, who presented with chronic progressive nail dystrophy affecting all 20 nails, onychomycosis, nail psoriasis, onychotillomania, and idiopathic trachyonychia were included in the differential.1
Onychomycosis manifests as white or yellow-brown discoloration of the nail, onycholysis, subungual hyperkeratosis, and thickening of the nail plate. Diagnosis is confirmed by the presence of septate hyphae (dermatophytes) or budding yeast cells (Candida species) on a potassium hydroxide mount. Other diagnostic modalities include dermoscopy, fungal culture, and histopathology of nail clippings, with demonstration of fungal elements identified on periodic acid-Schiff staining (eFigure 1).3
Nail psoriasis characteristically manifests as deep irregular pitting of the nails. Other features favoring psoriasis include involvement of the nail matrix manifesting as leukonychia, red lunula, and crumbling, as well as involvement of the nail bed manifesting as onycholysis, subungual hyperkeratosis, salmon patches/oil spots, and splinter hemorrhages (eFigure 2).4 Diagnosis primarily is clinical, supported by histopathology when uncertainty exists.
Onychotillomania is a behavioral disorder characterized by an irresistible urge or impulse in patients to either pick or pull at their fingernails and/or toenails. Clinicopathologic features of the involved nails are nonspecific and atypical, with possible involvement of periungual and digital skin. Diagnosis of onychotillomania is challenging.5 Dermoscopic features including anonychia with multiple obliquely arranged nail bed hemorrhages, gray pigmentation of the nail bed, and wavy lines, has been proposed to aid the diagnosis of onychotillomania.6
Idiopathic trachyonychia is isolated nail involvement characterized by rough, ridged, and thin nails affecting multiple or all of the fingernails and toenails without an underlying systemic or dermatologic condition (eFigure 3). The terms trachyonychia and 20-nail dystrophy have been used interchangeably in the literature; however, trachyonychia does not always involve all 20 nails. Other conditions causing widespread dystrophy of all 20 nails cannot be diagnosed as 20-nail dystrophy or trachyonychia without the distinct morphologic features of thin brittle nails with pronounced longitudinal ridging.7
Prompt diagnosis and early intervention in nail lichen planus is crucial due to the potential for irreversible scarring. First-line treatment options include intramatricial and intramuscular triamcinolone acetonide for 3 to 6 months.4 Second-line therapies include oral retinoids such as acitretin and alitretinoin and immunosuppressive agents such as azathioprine, mycophenolate mofetil, and cyclosporine. Other reported treatment options include clobetasol propionate, tacrolimus, dapsone, griseofulvin, etanercept, hydroxychloroquine, methotrexate, and UV therapy.4
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Leung AKC, Lam JM, Leong KF, et al. Onychomycosis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:32-45. doi:10.2174/1872213X13666191026090713
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Sidiropoulou P, Sgouros D, Theodoropoulos K, et al. Onychotillomania: a chameleon-like disorder: case report and review of literature. Skin Appendage Disord. 2019;5:104-107. doi:10.1159/000489941
- Maddy AJ, Tosti A. Dermoscopic features of onychotillomania: a study of 36 cases. J Am Acad Dermatol. 2018;79:702-705. doi:10.1016 /j.jaad.2018.04.015
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Leung AKC, Lam JM, Leong KF, et al. Onychomycosis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:32-45. doi:10.2174/1872213X13666191026090713
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Sidiropoulou P, Sgouros D, Theodoropoulos K, et al. Onychotillomania: a chameleon-like disorder: case report and review of literature. Skin Appendage Disord. 2019;5:104-107. doi:10.1159/000489941
- Maddy AJ, Tosti A. Dermoscopic features of onychotillomania: a study of 36 cases. J Am Acad Dermatol. 2018;79:702-705. doi:10.1016 /j.jaad.2018.04.015
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
Progressive Dystrophy of the Fingernails and Toenails
Progressive Dystrophy of the Fingernails and Toenails
A 35-year-old man presented to the dermatology department with gradually progressive dystrophy of the fingernails and toenails of 20 years’ duration. The patient reported no history of other dermatologic conditions. Physical examination revealed longitudinal ridging of all 20 nails and discoloration of the nail plates, as well as a few nails showing pterygium and anonychia; the skin and mucosal surfaces were otherwise normal, and nail plate thinning was not observed. A potassium hydroxide mount was negative. A biopsy of the nail matrix on the left thumbnail was performed.

Negotiating the VUCA World Through Tiered Huddles
Negotiating the VUCA World Through Tiered Huddles
To see what is in front of one’s nose needs a constant struggle.
George Orwell (1946)1
In 2019, the Veterans Health Administration (VHA) initiated a process to become a high reliability organization (HRO).2 The COVID-19 pandemic has been described in medical literature as a volatile, uncertain, complex, and ambiguous (VUCA) event, underscoring the necessity of resilient communication strategies.3 Challenges posed by 2024 Hurricanes Helene and Milton further highlighted the need for resilient communication strategies within HRO implementation.
Central to the HRO journey within the VHA has been the development of tiered huddles, an evolution of the safety huddle concept.4 Emerging organically as an effective communication mechanism across multiple facilities between 2019 and 2020, tiered huddles were, in part, spurred by the onset of COVID-19. Tiered huddles represent a proactive approach to identifying and addressing organizational threats in their early stages, thereby preventing their escalation to a VUCA-laden crisis.5 When conditions evolve beyond the horizon of tractability, where challenges are easily identified and resolved, tiered huddles serve as a resilient mechanism to restore dynamic equilibrium within the organization.6,7
This article describes how tiered huddles were integrated within Veterans Integrated Service Network (VISN) 4 and explores why these huddles are essential, particularly in the context of VUCA events. What began as a local-level tactic has now gained widespread acceptance and continues to evolve across the VHA with full support from the US Department of Veterans Affairs (VA) Under Secretary for Health.8
The VHA is divided into 18 VISNs. Nine VA Medical Centers (VAMCs) and 46 outpatient clinics across Pennsylvania, Delaware, and parts of Ohio, New York, and New Jersey make up VISN 4. Disseminating vital information across VISN 4, in addition to the 17 other VISNs—including 170 VAMCs and 1193 clinics—presents a formidable challenge. As the largest integrated system in the US, the VHA is realigning its workforce to address organizational inefficiencies. An enterprise of this scale, shaped by recurrent organizational change, faces ongoing challenges in sustaining clear communication across all levels. These transitions create uncertainty for staff as roles and resources shift, underscoring the need for dependable vertical and horizontal information flow. Tiered huddles offer a steady means to support coordinated communication and strengthen the system’s ability to adapt.9
ERIE VA MEDICAL CENTER HRO JOURNEY
In 2019, John Gennaro, the Erie VAMC executive director, attended a presentation that showcased the Cleveland Clinic’s tiered huddle process, with an opportunity to observe its 5-tiered system.10 Erie VAMC already had a 3-tiered huddle system, but the Cleveland Clinic’s more robust model inspired Gennaro to propose a VISN 4 pilot program. Tiered huddles were perceived as innovative, yet not fully embraced within the VHA; nonetheless, VISN 4, much like several other VISNs, moved forward and established a VISN-level (Tier 4) huddle.8 It is important to note that there was a notional fifth-tier capability as VISN and program office leaders already participated in daily VHA-wide meetings under the auspices of the Hospital Operations Center (HOC).
Expanding the Tiered Huddle Process
The Erie VAMC huddle process begins with the unit level Managers and Frontline Staff (Tier 1), then moves to Service Chiefs and Managers (Tier 2). Tier 3 involves facility executive leadership team and service chiefs, clinical directors and top VAMC administrators (these configurations may vary depending on context). The sequencing and flow of information is bidirectional across levels, reflecting the importance of closed-loop communication to ensure staff at all levels understand that issues raised are followed up on and/or closed out (Figure 1).2
Tier 4 composition may vary among VISNs depending on size and unique mission requirements.8,11 The VISN 4 Tier 4 huddle includes the VISN director, 9 VAMC directors, and key network administrators and clinical experts. The Tier 5 huddle includes 18 VISN 4 directors with the VHA HOC (Figure 2). The tiered huddle process emphasizes team-based culture and psychological safety.12-15 Staff at all levels are encouraged to identify and transparently resolve issues, fostering a proactive and problem-solving environment across the organization. A more nuanced and detailed process across tier levels is depicted in the Table.

The vetting and distillation of information can present challenges as vital information ascends and spreads across organization levels. Visual management systems (VMS), whether a whiteboard or a digital platform, are key to facilitate decision-making related to what needs to be prioritized and disseminated at each tier level.2,8 At Tier 5, the HOC uses a digital VMS to provide a structured, user-friendly format for categorizing issues and topics and enhances clarity and accessibility (Figure 3). The Tier 5 VMS also facilitates tracking and reciprocal information exchange, helping to close the loop on emerging issues by monitoring their progression and resolution up and across tiers.2,8 The Tier 5 huddle process and technology supporting continue to evolve offering increasing sophistication in organizational situational awareness and responsiveness.

VUCA: A Lens for Health Care Challenges
First introduced by social scientists at the US Army War College in 1995, VUCA describes complex and unpredictable conditions often encountered in military operations.16,17 Prompted by the COVID-19 pandemic, the acronym VUCA gained recognition in health care, as leaders acknowledged the challenge of navigating rapidly changing environments. van Stralen, Byrum and Inozu, recognized authorities in high reliability, cited VUCA as the rationale for implementing HRO principles and practices. They argued that “HRO solves the problem of operations and performance in a volatile, uncertain, complex, ambiguous environment.” 18 To fully appreciate the VUCA environment and its relevance to health care, it is essential to unpack the 4 components of the acronym: volatile, uncertain, complex, and ambiguous.
Volatile refers to the speed and unpredictability of change. Health care systems are interactively complex and tightly coupled, meaning that changes in 1 part of the system can rapidly impact others.6,18,19 This high degree of interdependence amplifies volatility, especially when unexpected events occur. The rapid spread of COVID- 19 and the evolving nature of its transmission challenged health care systems’ ability to respond swiftly and effectively. Volatility also may emerge in acute medical situations, such as the rapid deterioration of a patient’s condition.
Uncertain captures the lack of predictability inherent in complex systems. In health care, uncertainty arises when there is insufficient information or when an excess of data make it difficult to discern meaningful patterns. COVID-19 and recent natural disasters have introduced profound uncertainty, as the disease’s behavior, transmission, and impact were initially unknown. Health care practitioners struggled to make decisions in real time, lacking clear guidance or precedent.3,20 While health care planning and established protocols are grounded in predictability, the COVID-19 pandemic revealed that as complexity increases, predictability diminishes. Moreover, complexity can complicate protocol selection, as situations may arise in which multiple protocols conflict or compete. The cognitive challenge of operating in this environment is analogous to what military strategists call the fog of war, where situational awareness is low and decision-makers must navigate without clarity.21 Tiered huddles, a core practice in HROs, mitigate uncertainty by fostering real-time communication and shared situational awareness among teams.20
Complex refers to the intricate interplay of multiple, interconnected factors within a system.22 In health care, this complexity is heightened by the sociotechnical nature of the field—where human, technology, and organizational elements all converge.19 Systems designed to prevent failures, such as redundancies and safety protocols, can themselves contribute to increased complexity. HRO practices such as tiered huddles are implemented to mitigate the risk of catastrophic failure by fostering collaborative sensemaking, enhanced situational awareness, and rapid problem-solving.5,20,23
Ambiguous refers to situations in which multiple interpretations, causes, or outcomes are possible. It explains how, despite following protocols, failure can still occur, or how individuals may reach different conclusions from the same data. Ambiguity does not offer binary solutions; instead, it presents a murky, multifaceted reality that requires thoughtful interpretation and adaptive responses. In these moments, leaders must act decisively, even in the absence of complete information, making trade-offs that balance immediate needs with long-term consequences.
MANAGING VUCA ENVIRONMENTS WITH TIERED HUDDLES
The tiered huddle process provides several key benefits that enable real-time issue resolution. These include the rapid dissemination of vital information, enhanced agility and resilience, and improved sensemaking within a VUCA environment. Additionally, tiered huddles prevent organizational drift by fostering heightened situational awareness. The tiered huddle process also supports leadership development, as unit-level leaders gain valuable insights into strategic decision-making through active participation. Each component is outlined in the following section.
Spread: The Challenge of Communicating
“The hallmark of a great organization is how quickly bad news travels upward,” argued Jay Forrester, the father of system dynamics.24 Unfortunately, steep power gradients and siloed organizational structures inhibit the flow of unfavorable information from frontline staff to senior leadership. This suppression is not necessarily intentional but is often a byproduct of organizational culture. Tiered huddles address the weakness of top-down communication models by promoting a reciprocal, bidirectional information exchange, with an emphasis on closed-loop communication. Open communication can foster a culture of trust and transparency, allowing leaders to make more informed decisions and respond quickly to emerging risks.
Enhancing Agility and Resilience
Tiered huddles contribute to a mindful infrastructure, an important aspect of maintaining organizational awareness and agility.21,25 A mindful infrastructure enables an organization to detect early warning signs of potential disruptions and respond to them before they escalate. In this sense, tiered huddles serve as a signal-sensing mechanism, providing the agility needed to adapt to changing circumstances and prevent patient harm. Tiered huddles facilitate self-organization, a concept from chaos theory known as autopoiesis. 26 This self-organizing capability allows teams to develop novel solutions in response to unforeseen challenges, exemplifying the adaptability and resilience needed in a VUCA environment. The diverse backgrounds of tiered huddle participants—both cognitively and culturally—enable a broader range of perspectives, which is critical for making sound decisions in complex and uncertain situations. “HROs cultivate diversity not just because it helps them notice more in complex environments, but also because it helps them adapt to the complexities they do spot,” argues Weick et al.27 This diversity of thought and experience enhances the organization’s ability to respond to complexity, much like firefighters continually adapt to the VUCA conditions they face.
Sensemaking and Sensitivity to Operations
Leaders at all levels must be attuned to what is happening both within and outside their organization. This continual sensing of the environment—looking for weak signals, threats, and opportunities—is important for HROs. This signal detection capability allows organizations to address problems in their nascent emerging state within a tractable horizon to successfully manage fluctuations. The horizon of tractability reflects a zone where weak signals and evolving issues can be identified, addressed, and resolved early before they evolve and cascade outside of safe operations. 7 Tiered huddles facilitate this process by creating a platform for team members to engage in respectful, collaborative dialogue. The diversity inherent in tiered huddles also supports sensemaking, a process of interpreting and understanding complex situations.27 In a VUCA environment, this multiperspective approach helps filter out noise and identify the most important signals. Tiered huddles can help overcome the phenomenon of dysfunctional momentum associated with cognitive lockup, fixation error, and tunnel vision, in which individuals or teams fixate on a particular solution, thus missing important alternative views.21,28 By fostering a common operating picture of the fluctuating environment, tiered huddles can enable more accurate decision-making and improve organizational resilience.
Avoiding Organizational Drift
One of the most significant contributions of tiered huddles is the ability to detect early signs of organizational drift, or subtle deviations from standard practices that can accumulate over time and lead to serious failures. By continuously monitoring for precursor conditions and weak signals, tiered huddles allow organizations to intervene early and prevent drift from becoming catastrophic.29,30 This vigilance is essential in health care, where complacency can lead to patient harm. Tiered huddles foster a culture of mindfulness and accountability, ensuring that staff stay engaged and alert to potential risks. This proactive approach is a safeguard against human error and the gradual erosion of safety standards.
Leadership Development
Tiered huddles serve as a powerful tool for leadership development. Effective leaders must be able to anticipate potential risks and foresee system failures. Involving future leaders in tiered huddles can facilitate the transfer of these critical skills. When emerging leaders at lower tiers participate in ascending-tier huddles, they gain a unique opportunity to engage in a structured, collaborative setting. This environment provides a safe space to develop and practice strategic skills, enhancing their ability to think proactively and manage complexity. By integrating future leaders into tiered huddles, organizations offer essential, hands-on experience in real-time decision making. This experiential learning is invaluable for preparing leaders to navigate the demands of a VUCA environment.
CONCLUSIONS
Since implementing the tiered huddle process, the Erie VAMC and VISN 4 have emerged as early adopters of VUCA, thus contributing to the expansion of this innovative communication approach across the VHA. Tiered huddles strengthen organizational resilience and agility, facilitate critical information flow to manage risk, and support the cultivation of future leaders. The Erie VAMC director and the VISN 4 network director regard the expansion of tiered huddles, including Tiers 4 and 5, as an adaptable model for the VHA. While tiered huddles have not yet been mandated across the VHA, a pilot at the Tier 5 HOC level was initiated on May 20, 2024. In a complex world in which VUCA events will continue to be inevitable, implementation of robust tiered huddles within complex health care systems provides the opportunity for improved responses and delivery of care.
- Orwell S, Angus I, eds. In Front of Your Nose, 1945-1950. Godine; 2000. Orwell G. The Collected Essays, Journalism, and Letters of George Orwell; vol 4.
- Murray JS, Baghdadi A, Dannenberg W, Crews P, Walsh ND. The role of high reliability organization foundational practices in building a culture of safety. Fed Pract. 2024;41:214-221. doi:10.12788/fp.0486
- Goldenhar LM, Brady PW, Sutcliffe KM, Muething SE. Huddling for high reliability and situation awareness. BMJ Qual Saf. 2013;22:899-906. doi:10.1136/bmjqs-2012-001467
- Pandit M. Critical factors for successful management of VUCA times. BMJ Lead. 2021;5:121-123. doi:10.1136/leader-2020-000305
- Mihaljevic T. Tiered daily huddles: the power of teamwork in managing large healthcare organisations. BMJ Qual Saf. 2020;29:1050-1052. doi:10.1136/bmjqs-2019-010575
- van Stralen D, Mercer TA. High-reliability organizing (HRO) in the COVID-19 liminal zone: characteristics of workers and local leaders. Neonatology Today. 2021;16:90-101. http://www.neonatologytoday.net /newsletters/nt-apr21.pdf
- Nemeth C, Wears R, Woods D, Hollnagel E, Cook R. Minding the gaps: creating resilience in health care. In: Henriksen K, Battles JB, Keyes MA, Grady ML, eds. Advances in Patient Safety: New Directions and Alternative Approaches. Vol 3: Performance and Tools. Agency for Healthcare Research and Quality; 2008.
- Merchant NB, O’Neal J, Montoya A, Cox GR, Murray JS. Creating a process for the implementation of tiered huddles in a Veterans Affairs medical center. Mil Med. 2023;188:901-906. doi:10.1093/milmed/usac073
- Starbuck WH, Farjoun M, eds. Organization at the Limit: Lessons From the Columbia Disaster. 1st ed. Wiley-Blackwell; 2005.
- Mihaljevic T. Tiered daily huddles: the power of teamwork in managing large healthcare organisations. BMJ Qual Saf. 2020;29:1050-1052. doi:10.1136/bmjqs-2019-010575
- Donnelly LF, Cherian SS, Chua KB, et al. The Daily Readiness Huddle: a process to rapidly identify issues and foster improvement through problem-solving accountability. Pediatr Radiol. 2017;47:22-30. doi:10.1007/s00247-016-3712-x
- Clark TR. The 4 Stages of Psychological Safety: Defining the Path to Inclusion and Innovation. Berrett-Koehler Publishers, Inc.; 2020.
- Edmondson AC. The Fearless Organization: Creating Psychological Safety in the Workplace for Learning, Innovation, and Growth. John Wiley & Sons; 2018.
- Edmondson AC. The Right Kind of Wrong: The Science of Failing Well. Simon Element/Simon Acumen; 2023.
- Murray JS, Kelly S, Hanover C. Promoting psychological safety in healthcare organizations. Mil Med. 2022;187:808 -810. doi:10.1093/milmed/usac041
- Barber HF. Developing strategic leadership: the US Army War College experience. J Manag Dev. 1992;11:4-12. doi:10.1108/02621719210018208
- US Army Heritage & Education Center. Who first originated the term VUCA (volatility, uncertainty, complexity and ambiguity)? Accessed November 5, 2025. https://usawc .libanswers.com/ahec/faq/84869
- van Stralen D, Byrum SL, Inozu B. High Reliability for a Highly Unreliable World: Preparing for Code Blue Through Daily Operations in Healthcare. CreateSpace Independent Publishing Platform; 2018.
- Perrow C. Normal Accidents: Living With High-Risk Technologies. Princeton University Press; 2000.
- Sculli G, Essen K. Soaring to Success: The Path to Developing High-Reliability Clinical Teams. HCPro; 2021. Accessed November 5, 2025. https://hcmarketplace.com /media/wysiwyg/CRM3_browse.pdf
- Barton MA, Sutcliffe KM, Vogus TJ, DeWitt T. Performing under uncertainty: contextualized engagement in wildland firefighting. J Contingencies Crisis Manag. 2015;23:74-83. doi:10.1111/1468-5973.12076
- Sutcliffe KM. Mindful organizing. In: Ramanujam R, Roberts KH, eds. Organizing for Reliability: A Guide for Research and Practice. Stanford University Press; 2018:61-89.
- Merchant NB, O’Neal J, Dealino-Perez C, Xiang J, Montoya A Jr, Murray JS. A high-reliability organization mindset. Am J Med Qual. 2022;37:504-510. doi:10.1097/jmq.0000000000000086
- Senge PM. The Fifth Discipline Fieldbook: Strategies and Tools for Building a Learning Organization. Crown Currency; 1994.
- Ramanujam R, Roberts KH, eds. Organizing for Reliability: A Guide for Research and Practice. Stanford University Press; 2018.
- Coveney PV. Self-organization and complexity: a new age for theory, computation and experiment. Philos Trans A Math Phys Eng Sci. 2003;361:1057-1079. doi:10.1098/rsta.2003.1191
- Weick KE, Sutcliffe KM. Managing the Unexpected: Sustained Performance in a Complex World. 3rd ed. Wiley; 2015.
- Barton M, Sutcliffe K. Overcoming dysfunctional momentum: organizational safety as a social achievement. Hum Relations. 2009;62:1327-1356. doi:10.1177/0018726709334491
- Dekker S. Drift Into Failure: From Hunting Broken Components to Understanding Complex Systems. Routledge; 2011.
- Price MR, Williams TC. When doing wrong feels so right: normalization of deviance. J Patient Saf. 2018;14:1-2. doi:10.1097/pts.0000000000000157
To see what is in front of one’s nose needs a constant struggle.
George Orwell (1946)1
In 2019, the Veterans Health Administration (VHA) initiated a process to become a high reliability organization (HRO).2 The COVID-19 pandemic has been described in medical literature as a volatile, uncertain, complex, and ambiguous (VUCA) event, underscoring the necessity of resilient communication strategies.3 Challenges posed by 2024 Hurricanes Helene and Milton further highlighted the need for resilient communication strategies within HRO implementation.
Central to the HRO journey within the VHA has been the development of tiered huddles, an evolution of the safety huddle concept.4 Emerging organically as an effective communication mechanism across multiple facilities between 2019 and 2020, tiered huddles were, in part, spurred by the onset of COVID-19. Tiered huddles represent a proactive approach to identifying and addressing organizational threats in their early stages, thereby preventing their escalation to a VUCA-laden crisis.5 When conditions evolve beyond the horizon of tractability, where challenges are easily identified and resolved, tiered huddles serve as a resilient mechanism to restore dynamic equilibrium within the organization.6,7
This article describes how tiered huddles were integrated within Veterans Integrated Service Network (VISN) 4 and explores why these huddles are essential, particularly in the context of VUCA events. What began as a local-level tactic has now gained widespread acceptance and continues to evolve across the VHA with full support from the US Department of Veterans Affairs (VA) Under Secretary for Health.8
The VHA is divided into 18 VISNs. Nine VA Medical Centers (VAMCs) and 46 outpatient clinics across Pennsylvania, Delaware, and parts of Ohio, New York, and New Jersey make up VISN 4. Disseminating vital information across VISN 4, in addition to the 17 other VISNs—including 170 VAMCs and 1193 clinics—presents a formidable challenge. As the largest integrated system in the US, the VHA is realigning its workforce to address organizational inefficiencies. An enterprise of this scale, shaped by recurrent organizational change, faces ongoing challenges in sustaining clear communication across all levels. These transitions create uncertainty for staff as roles and resources shift, underscoring the need for dependable vertical and horizontal information flow. Tiered huddles offer a steady means to support coordinated communication and strengthen the system’s ability to adapt.9
ERIE VA MEDICAL CENTER HRO JOURNEY
In 2019, John Gennaro, the Erie VAMC executive director, attended a presentation that showcased the Cleveland Clinic’s tiered huddle process, with an opportunity to observe its 5-tiered system.10 Erie VAMC already had a 3-tiered huddle system, but the Cleveland Clinic’s more robust model inspired Gennaro to propose a VISN 4 pilot program. Tiered huddles were perceived as innovative, yet not fully embraced within the VHA; nonetheless, VISN 4, much like several other VISNs, moved forward and established a VISN-level (Tier 4) huddle.8 It is important to note that there was a notional fifth-tier capability as VISN and program office leaders already participated in daily VHA-wide meetings under the auspices of the Hospital Operations Center (HOC).
Expanding the Tiered Huddle Process
The Erie VAMC huddle process begins with the unit level Managers and Frontline Staff (Tier 1), then moves to Service Chiefs and Managers (Tier 2). Tier 3 involves facility executive leadership team and service chiefs, clinical directors and top VAMC administrators (these configurations may vary depending on context). The sequencing and flow of information is bidirectional across levels, reflecting the importance of closed-loop communication to ensure staff at all levels understand that issues raised are followed up on and/or closed out (Figure 1).2
Tier 4 composition may vary among VISNs depending on size and unique mission requirements.8,11 The VISN 4 Tier 4 huddle includes the VISN director, 9 VAMC directors, and key network administrators and clinical experts. The Tier 5 huddle includes 18 VISN 4 directors with the VHA HOC (Figure 2). The tiered huddle process emphasizes team-based culture and psychological safety.12-15 Staff at all levels are encouraged to identify and transparently resolve issues, fostering a proactive and problem-solving environment across the organization. A more nuanced and detailed process across tier levels is depicted in the Table.
The vetting and distillation of information can present challenges as vital information ascends and spreads across organization levels. Visual management systems (VMS), whether a whiteboard or a digital platform, are key to facilitate decision-making related to what needs to be prioritized and disseminated at each tier level.2,8 At Tier 5, the HOC uses a digital VMS to provide a structured, user-friendly format for categorizing issues and topics and enhances clarity and accessibility (Figure 3). The Tier 5 VMS also facilitates tracking and reciprocal information exchange, helping to close the loop on emerging issues by monitoring their progression and resolution up and across tiers.2,8 The Tier 5 huddle process and technology supporting continue to evolve offering increasing sophistication in organizational situational awareness and responsiveness.
VUCA: A Lens for Health Care Challenges
First introduced by social scientists at the US Army War College in 1995, VUCA describes complex and unpredictable conditions often encountered in military operations.16,17 Prompted by the COVID-19 pandemic, the acronym VUCA gained recognition in health care, as leaders acknowledged the challenge of navigating rapidly changing environments. van Stralen, Byrum and Inozu, recognized authorities in high reliability, cited VUCA as the rationale for implementing HRO principles and practices. They argued that “HRO solves the problem of operations and performance in a volatile, uncertain, complex, ambiguous environment.” 18 To fully appreciate the VUCA environment and its relevance to health care, it is essential to unpack the 4 components of the acronym: volatile, uncertain, complex, and ambiguous.
Volatile refers to the speed and unpredictability of change. Health care systems are interactively complex and tightly coupled, meaning that changes in 1 part of the system can rapidly impact others.6,18,19 This high degree of interdependence amplifies volatility, especially when unexpected events occur. The rapid spread of COVID- 19 and the evolving nature of its transmission challenged health care systems’ ability to respond swiftly and effectively. Volatility also may emerge in acute medical situations, such as the rapid deterioration of a patient’s condition.
Uncertain captures the lack of predictability inherent in complex systems. In health care, uncertainty arises when there is insufficient information or when an excess of data make it difficult to discern meaningful patterns. COVID-19 and recent natural disasters have introduced profound uncertainty, as the disease’s behavior, transmission, and impact were initially unknown. Health care practitioners struggled to make decisions in real time, lacking clear guidance or precedent.3,20 While health care planning and established protocols are grounded in predictability, the COVID-19 pandemic revealed that as complexity increases, predictability diminishes. Moreover, complexity can complicate protocol selection, as situations may arise in which multiple protocols conflict or compete. The cognitive challenge of operating in this environment is analogous to what military strategists call the fog of war, where situational awareness is low and decision-makers must navigate without clarity.21 Tiered huddles, a core practice in HROs, mitigate uncertainty by fostering real-time communication and shared situational awareness among teams.20
Complex refers to the intricate interplay of multiple, interconnected factors within a system.22 In health care, this complexity is heightened by the sociotechnical nature of the field—where human, technology, and organizational elements all converge.19 Systems designed to prevent failures, such as redundancies and safety protocols, can themselves contribute to increased complexity. HRO practices such as tiered huddles are implemented to mitigate the risk of catastrophic failure by fostering collaborative sensemaking, enhanced situational awareness, and rapid problem-solving.5,20,23
Ambiguous refers to situations in which multiple interpretations, causes, or outcomes are possible. It explains how, despite following protocols, failure can still occur, or how individuals may reach different conclusions from the same data. Ambiguity does not offer binary solutions; instead, it presents a murky, multifaceted reality that requires thoughtful interpretation and adaptive responses. In these moments, leaders must act decisively, even in the absence of complete information, making trade-offs that balance immediate needs with long-term consequences.
MANAGING VUCA ENVIRONMENTS WITH TIERED HUDDLES
The tiered huddle process provides several key benefits that enable real-time issue resolution. These include the rapid dissemination of vital information, enhanced agility and resilience, and improved sensemaking within a VUCA environment. Additionally, tiered huddles prevent organizational drift by fostering heightened situational awareness. The tiered huddle process also supports leadership development, as unit-level leaders gain valuable insights into strategic decision-making through active participation. Each component is outlined in the following section.
Spread: The Challenge of Communicating
“The hallmark of a great organization is how quickly bad news travels upward,” argued Jay Forrester, the father of system dynamics.24 Unfortunately, steep power gradients and siloed organizational structures inhibit the flow of unfavorable information from frontline staff to senior leadership. This suppression is not necessarily intentional but is often a byproduct of organizational culture. Tiered huddles address the weakness of top-down communication models by promoting a reciprocal, bidirectional information exchange, with an emphasis on closed-loop communication. Open communication can foster a culture of trust and transparency, allowing leaders to make more informed decisions and respond quickly to emerging risks.
Enhancing Agility and Resilience
Tiered huddles contribute to a mindful infrastructure, an important aspect of maintaining organizational awareness and agility.21,25 A mindful infrastructure enables an organization to detect early warning signs of potential disruptions and respond to them before they escalate. In this sense, tiered huddles serve as a signal-sensing mechanism, providing the agility needed to adapt to changing circumstances and prevent patient harm. Tiered huddles facilitate self-organization, a concept from chaos theory known as autopoiesis. 26 This self-organizing capability allows teams to develop novel solutions in response to unforeseen challenges, exemplifying the adaptability and resilience needed in a VUCA environment. The diverse backgrounds of tiered huddle participants—both cognitively and culturally—enable a broader range of perspectives, which is critical for making sound decisions in complex and uncertain situations. “HROs cultivate diversity not just because it helps them notice more in complex environments, but also because it helps them adapt to the complexities they do spot,” argues Weick et al.27 This diversity of thought and experience enhances the organization’s ability to respond to complexity, much like firefighters continually adapt to the VUCA conditions they face.
Sensemaking and Sensitivity to Operations
Leaders at all levels must be attuned to what is happening both within and outside their organization. This continual sensing of the environment—looking for weak signals, threats, and opportunities—is important for HROs. This signal detection capability allows organizations to address problems in their nascent emerging state within a tractable horizon to successfully manage fluctuations. The horizon of tractability reflects a zone where weak signals and evolving issues can be identified, addressed, and resolved early before they evolve and cascade outside of safe operations. 7 Tiered huddles facilitate this process by creating a platform for team members to engage in respectful, collaborative dialogue. The diversity inherent in tiered huddles also supports sensemaking, a process of interpreting and understanding complex situations.27 In a VUCA environment, this multiperspective approach helps filter out noise and identify the most important signals. Tiered huddles can help overcome the phenomenon of dysfunctional momentum associated with cognitive lockup, fixation error, and tunnel vision, in which individuals or teams fixate on a particular solution, thus missing important alternative views.21,28 By fostering a common operating picture of the fluctuating environment, tiered huddles can enable more accurate decision-making and improve organizational resilience.
Avoiding Organizational Drift
One of the most significant contributions of tiered huddles is the ability to detect early signs of organizational drift, or subtle deviations from standard practices that can accumulate over time and lead to serious failures. By continuously monitoring for precursor conditions and weak signals, tiered huddles allow organizations to intervene early and prevent drift from becoming catastrophic.29,30 This vigilance is essential in health care, where complacency can lead to patient harm. Tiered huddles foster a culture of mindfulness and accountability, ensuring that staff stay engaged and alert to potential risks. This proactive approach is a safeguard against human error and the gradual erosion of safety standards.
Leadership Development
Tiered huddles serve as a powerful tool for leadership development. Effective leaders must be able to anticipate potential risks and foresee system failures. Involving future leaders in tiered huddles can facilitate the transfer of these critical skills. When emerging leaders at lower tiers participate in ascending-tier huddles, they gain a unique opportunity to engage in a structured, collaborative setting. This environment provides a safe space to develop and practice strategic skills, enhancing their ability to think proactively and manage complexity. By integrating future leaders into tiered huddles, organizations offer essential, hands-on experience in real-time decision making. This experiential learning is invaluable for preparing leaders to navigate the demands of a VUCA environment.
CONCLUSIONS
Since implementing the tiered huddle process, the Erie VAMC and VISN 4 have emerged as early adopters of VUCA, thus contributing to the expansion of this innovative communication approach across the VHA. Tiered huddles strengthen organizational resilience and agility, facilitate critical information flow to manage risk, and support the cultivation of future leaders. The Erie VAMC director and the VISN 4 network director regard the expansion of tiered huddles, including Tiers 4 and 5, as an adaptable model for the VHA. While tiered huddles have not yet been mandated across the VHA, a pilot at the Tier 5 HOC level was initiated on May 20, 2024. In a complex world in which VUCA events will continue to be inevitable, implementation of robust tiered huddles within complex health care systems provides the opportunity for improved responses and delivery of care.
To see what is in front of one’s nose needs a constant struggle.
George Orwell (1946)1
In 2019, the Veterans Health Administration (VHA) initiated a process to become a high reliability organization (HRO).2 The COVID-19 pandemic has been described in medical literature as a volatile, uncertain, complex, and ambiguous (VUCA) event, underscoring the necessity of resilient communication strategies.3 Challenges posed by 2024 Hurricanes Helene and Milton further highlighted the need for resilient communication strategies within HRO implementation.
Central to the HRO journey within the VHA has been the development of tiered huddles, an evolution of the safety huddle concept.4 Emerging organically as an effective communication mechanism across multiple facilities between 2019 and 2020, tiered huddles were, in part, spurred by the onset of COVID-19. Tiered huddles represent a proactive approach to identifying and addressing organizational threats in their early stages, thereby preventing their escalation to a VUCA-laden crisis.5 When conditions evolve beyond the horizon of tractability, where challenges are easily identified and resolved, tiered huddles serve as a resilient mechanism to restore dynamic equilibrium within the organization.6,7
This article describes how tiered huddles were integrated within Veterans Integrated Service Network (VISN) 4 and explores why these huddles are essential, particularly in the context of VUCA events. What began as a local-level tactic has now gained widespread acceptance and continues to evolve across the VHA with full support from the US Department of Veterans Affairs (VA) Under Secretary for Health.8
The VHA is divided into 18 VISNs. Nine VA Medical Centers (VAMCs) and 46 outpatient clinics across Pennsylvania, Delaware, and parts of Ohio, New York, and New Jersey make up VISN 4. Disseminating vital information across VISN 4, in addition to the 17 other VISNs—including 170 VAMCs and 1193 clinics—presents a formidable challenge. As the largest integrated system in the US, the VHA is realigning its workforce to address organizational inefficiencies. An enterprise of this scale, shaped by recurrent organizational change, faces ongoing challenges in sustaining clear communication across all levels. These transitions create uncertainty for staff as roles and resources shift, underscoring the need for dependable vertical and horizontal information flow. Tiered huddles offer a steady means to support coordinated communication and strengthen the system’s ability to adapt.9
ERIE VA MEDICAL CENTER HRO JOURNEY
In 2019, John Gennaro, the Erie VAMC executive director, attended a presentation that showcased the Cleveland Clinic’s tiered huddle process, with an opportunity to observe its 5-tiered system.10 Erie VAMC already had a 3-tiered huddle system, but the Cleveland Clinic’s more robust model inspired Gennaro to propose a VISN 4 pilot program. Tiered huddles were perceived as innovative, yet not fully embraced within the VHA; nonetheless, VISN 4, much like several other VISNs, moved forward and established a VISN-level (Tier 4) huddle.8 It is important to note that there was a notional fifth-tier capability as VISN and program office leaders already participated in daily VHA-wide meetings under the auspices of the Hospital Operations Center (HOC).
Expanding the Tiered Huddle Process
The Erie VAMC huddle process begins with the unit level Managers and Frontline Staff (Tier 1), then moves to Service Chiefs and Managers (Tier 2). Tier 3 involves facility executive leadership team and service chiefs, clinical directors and top VAMC administrators (these configurations may vary depending on context). The sequencing and flow of information is bidirectional across levels, reflecting the importance of closed-loop communication to ensure staff at all levels understand that issues raised are followed up on and/or closed out (Figure 1).2
Tier 4 composition may vary among VISNs depending on size and unique mission requirements.8,11 The VISN 4 Tier 4 huddle includes the VISN director, 9 VAMC directors, and key network administrators and clinical experts. The Tier 5 huddle includes 18 VISN 4 directors with the VHA HOC (Figure 2). The tiered huddle process emphasizes team-based culture and psychological safety.12-15 Staff at all levels are encouraged to identify and transparently resolve issues, fostering a proactive and problem-solving environment across the organization. A more nuanced and detailed process across tier levels is depicted in the Table.
The vetting and distillation of information can present challenges as vital information ascends and spreads across organization levels. Visual management systems (VMS), whether a whiteboard or a digital platform, are key to facilitate decision-making related to what needs to be prioritized and disseminated at each tier level.2,8 At Tier 5, the HOC uses a digital VMS to provide a structured, user-friendly format for categorizing issues and topics and enhances clarity and accessibility (Figure 3). The Tier 5 VMS also facilitates tracking and reciprocal information exchange, helping to close the loop on emerging issues by monitoring their progression and resolution up and across tiers.2,8 The Tier 5 huddle process and technology supporting continue to evolve offering increasing sophistication in organizational situational awareness and responsiveness.
VUCA: A Lens for Health Care Challenges
First introduced by social scientists at the US Army War College in 1995, VUCA describes complex and unpredictable conditions often encountered in military operations.16,17 Prompted by the COVID-19 pandemic, the acronym VUCA gained recognition in health care, as leaders acknowledged the challenge of navigating rapidly changing environments. van Stralen, Byrum and Inozu, recognized authorities in high reliability, cited VUCA as the rationale for implementing HRO principles and practices. They argued that “HRO solves the problem of operations and performance in a volatile, uncertain, complex, ambiguous environment.” 18 To fully appreciate the VUCA environment and its relevance to health care, it is essential to unpack the 4 components of the acronym: volatile, uncertain, complex, and ambiguous.
Volatile refers to the speed and unpredictability of change. Health care systems are interactively complex and tightly coupled, meaning that changes in 1 part of the system can rapidly impact others.6,18,19 This high degree of interdependence amplifies volatility, especially when unexpected events occur. The rapid spread of COVID- 19 and the evolving nature of its transmission challenged health care systems’ ability to respond swiftly and effectively. Volatility also may emerge in acute medical situations, such as the rapid deterioration of a patient’s condition.
Uncertain captures the lack of predictability inherent in complex systems. In health care, uncertainty arises when there is insufficient information or when an excess of data make it difficult to discern meaningful patterns. COVID-19 and recent natural disasters have introduced profound uncertainty, as the disease’s behavior, transmission, and impact were initially unknown. Health care practitioners struggled to make decisions in real time, lacking clear guidance or precedent.3,20 While health care planning and established protocols are grounded in predictability, the COVID-19 pandemic revealed that as complexity increases, predictability diminishes. Moreover, complexity can complicate protocol selection, as situations may arise in which multiple protocols conflict or compete. The cognitive challenge of operating in this environment is analogous to what military strategists call the fog of war, where situational awareness is low and decision-makers must navigate without clarity.21 Tiered huddles, a core practice in HROs, mitigate uncertainty by fostering real-time communication and shared situational awareness among teams.20
Complex refers to the intricate interplay of multiple, interconnected factors within a system.22 In health care, this complexity is heightened by the sociotechnical nature of the field—where human, technology, and organizational elements all converge.19 Systems designed to prevent failures, such as redundancies and safety protocols, can themselves contribute to increased complexity. HRO practices such as tiered huddles are implemented to mitigate the risk of catastrophic failure by fostering collaborative sensemaking, enhanced situational awareness, and rapid problem-solving.5,20,23
Ambiguous refers to situations in which multiple interpretations, causes, or outcomes are possible. It explains how, despite following protocols, failure can still occur, or how individuals may reach different conclusions from the same data. Ambiguity does not offer binary solutions; instead, it presents a murky, multifaceted reality that requires thoughtful interpretation and adaptive responses. In these moments, leaders must act decisively, even in the absence of complete information, making trade-offs that balance immediate needs with long-term consequences.
MANAGING VUCA ENVIRONMENTS WITH TIERED HUDDLES
The tiered huddle process provides several key benefits that enable real-time issue resolution. These include the rapid dissemination of vital information, enhanced agility and resilience, and improved sensemaking within a VUCA environment. Additionally, tiered huddles prevent organizational drift by fostering heightened situational awareness. The tiered huddle process also supports leadership development, as unit-level leaders gain valuable insights into strategic decision-making through active participation. Each component is outlined in the following section.
Spread: The Challenge of Communicating
“The hallmark of a great organization is how quickly bad news travels upward,” argued Jay Forrester, the father of system dynamics.24 Unfortunately, steep power gradients and siloed organizational structures inhibit the flow of unfavorable information from frontline staff to senior leadership. This suppression is not necessarily intentional but is often a byproduct of organizational culture. Tiered huddles address the weakness of top-down communication models by promoting a reciprocal, bidirectional information exchange, with an emphasis on closed-loop communication. Open communication can foster a culture of trust and transparency, allowing leaders to make more informed decisions and respond quickly to emerging risks.
Enhancing Agility and Resilience
Tiered huddles contribute to a mindful infrastructure, an important aspect of maintaining organizational awareness and agility.21,25 A mindful infrastructure enables an organization to detect early warning signs of potential disruptions and respond to them before they escalate. In this sense, tiered huddles serve as a signal-sensing mechanism, providing the agility needed to adapt to changing circumstances and prevent patient harm. Tiered huddles facilitate self-organization, a concept from chaos theory known as autopoiesis. 26 This self-organizing capability allows teams to develop novel solutions in response to unforeseen challenges, exemplifying the adaptability and resilience needed in a VUCA environment. The diverse backgrounds of tiered huddle participants—both cognitively and culturally—enable a broader range of perspectives, which is critical for making sound decisions in complex and uncertain situations. “HROs cultivate diversity not just because it helps them notice more in complex environments, but also because it helps them adapt to the complexities they do spot,” argues Weick et al.27 This diversity of thought and experience enhances the organization’s ability to respond to complexity, much like firefighters continually adapt to the VUCA conditions they face.
Sensemaking and Sensitivity to Operations
Leaders at all levels must be attuned to what is happening both within and outside their organization. This continual sensing of the environment—looking for weak signals, threats, and opportunities—is important for HROs. This signal detection capability allows organizations to address problems in their nascent emerging state within a tractable horizon to successfully manage fluctuations. The horizon of tractability reflects a zone where weak signals and evolving issues can be identified, addressed, and resolved early before they evolve and cascade outside of safe operations. 7 Tiered huddles facilitate this process by creating a platform for team members to engage in respectful, collaborative dialogue. The diversity inherent in tiered huddles also supports sensemaking, a process of interpreting and understanding complex situations.27 In a VUCA environment, this multiperspective approach helps filter out noise and identify the most important signals. Tiered huddles can help overcome the phenomenon of dysfunctional momentum associated with cognitive lockup, fixation error, and tunnel vision, in which individuals or teams fixate on a particular solution, thus missing important alternative views.21,28 By fostering a common operating picture of the fluctuating environment, tiered huddles can enable more accurate decision-making and improve organizational resilience.
Avoiding Organizational Drift
One of the most significant contributions of tiered huddles is the ability to detect early signs of organizational drift, or subtle deviations from standard practices that can accumulate over time and lead to serious failures. By continuously monitoring for precursor conditions and weak signals, tiered huddles allow organizations to intervene early and prevent drift from becoming catastrophic.29,30 This vigilance is essential in health care, where complacency can lead to patient harm. Tiered huddles foster a culture of mindfulness and accountability, ensuring that staff stay engaged and alert to potential risks. This proactive approach is a safeguard against human error and the gradual erosion of safety standards.
Leadership Development
Tiered huddles serve as a powerful tool for leadership development. Effective leaders must be able to anticipate potential risks and foresee system failures. Involving future leaders in tiered huddles can facilitate the transfer of these critical skills. When emerging leaders at lower tiers participate in ascending-tier huddles, they gain a unique opportunity to engage in a structured, collaborative setting. This environment provides a safe space to develop and practice strategic skills, enhancing their ability to think proactively and manage complexity. By integrating future leaders into tiered huddles, organizations offer essential, hands-on experience in real-time decision making. This experiential learning is invaluable for preparing leaders to navigate the demands of a VUCA environment.
CONCLUSIONS
Since implementing the tiered huddle process, the Erie VAMC and VISN 4 have emerged as early adopters of VUCA, thus contributing to the expansion of this innovative communication approach across the VHA. Tiered huddles strengthen organizational resilience and agility, facilitate critical information flow to manage risk, and support the cultivation of future leaders. The Erie VAMC director and the VISN 4 network director regard the expansion of tiered huddles, including Tiers 4 and 5, as an adaptable model for the VHA. While tiered huddles have not yet been mandated across the VHA, a pilot at the Tier 5 HOC level was initiated on May 20, 2024. In a complex world in which VUCA events will continue to be inevitable, implementation of robust tiered huddles within complex health care systems provides the opportunity for improved responses and delivery of care.
- Orwell S, Angus I, eds. In Front of Your Nose, 1945-1950. Godine; 2000. Orwell G. The Collected Essays, Journalism, and Letters of George Orwell; vol 4.
- Murray JS, Baghdadi A, Dannenberg W, Crews P, Walsh ND. The role of high reliability organization foundational practices in building a culture of safety. Fed Pract. 2024;41:214-221. doi:10.12788/fp.0486
- Goldenhar LM, Brady PW, Sutcliffe KM, Muething SE. Huddling for high reliability and situation awareness. BMJ Qual Saf. 2013;22:899-906. doi:10.1136/bmjqs-2012-001467
- Pandit M. Critical factors for successful management of VUCA times. BMJ Lead. 2021;5:121-123. doi:10.1136/leader-2020-000305
- Mihaljevic T. Tiered daily huddles: the power of teamwork in managing large healthcare organisations. BMJ Qual Saf. 2020;29:1050-1052. doi:10.1136/bmjqs-2019-010575
- van Stralen D, Mercer TA. High-reliability organizing (HRO) in the COVID-19 liminal zone: characteristics of workers and local leaders. Neonatology Today. 2021;16:90-101. http://www.neonatologytoday.net /newsletters/nt-apr21.pdf
- Nemeth C, Wears R, Woods D, Hollnagel E, Cook R. Minding the gaps: creating resilience in health care. In: Henriksen K, Battles JB, Keyes MA, Grady ML, eds. Advances in Patient Safety: New Directions and Alternative Approaches. Vol 3: Performance and Tools. Agency for Healthcare Research and Quality; 2008.
- Merchant NB, O’Neal J, Montoya A, Cox GR, Murray JS. Creating a process for the implementation of tiered huddles in a Veterans Affairs medical center. Mil Med. 2023;188:901-906. doi:10.1093/milmed/usac073
- Starbuck WH, Farjoun M, eds. Organization at the Limit: Lessons From the Columbia Disaster. 1st ed. Wiley-Blackwell; 2005.
- Mihaljevic T. Tiered daily huddles: the power of teamwork in managing large healthcare organisations. BMJ Qual Saf. 2020;29:1050-1052. doi:10.1136/bmjqs-2019-010575
- Donnelly LF, Cherian SS, Chua KB, et al. The Daily Readiness Huddle: a process to rapidly identify issues and foster improvement through problem-solving accountability. Pediatr Radiol. 2017;47:22-30. doi:10.1007/s00247-016-3712-x
- Clark TR. The 4 Stages of Psychological Safety: Defining the Path to Inclusion and Innovation. Berrett-Koehler Publishers, Inc.; 2020.
- Edmondson AC. The Fearless Organization: Creating Psychological Safety in the Workplace for Learning, Innovation, and Growth. John Wiley & Sons; 2018.
- Edmondson AC. The Right Kind of Wrong: The Science of Failing Well. Simon Element/Simon Acumen; 2023.
- Murray JS, Kelly S, Hanover C. Promoting psychological safety in healthcare organizations. Mil Med. 2022;187:808 -810. doi:10.1093/milmed/usac041
- Barber HF. Developing strategic leadership: the US Army War College experience. J Manag Dev. 1992;11:4-12. doi:10.1108/02621719210018208
- US Army Heritage & Education Center. Who first originated the term VUCA (volatility, uncertainty, complexity and ambiguity)? Accessed November 5, 2025. https://usawc .libanswers.com/ahec/faq/84869
- van Stralen D, Byrum SL, Inozu B. High Reliability for a Highly Unreliable World: Preparing for Code Blue Through Daily Operations in Healthcare. CreateSpace Independent Publishing Platform; 2018.
- Perrow C. Normal Accidents: Living With High-Risk Technologies. Princeton University Press; 2000.
- Sculli G, Essen K. Soaring to Success: The Path to Developing High-Reliability Clinical Teams. HCPro; 2021. Accessed November 5, 2025. https://hcmarketplace.com /media/wysiwyg/CRM3_browse.pdf
- Barton MA, Sutcliffe KM, Vogus TJ, DeWitt T. Performing under uncertainty: contextualized engagement in wildland firefighting. J Contingencies Crisis Manag. 2015;23:74-83. doi:10.1111/1468-5973.12076
- Sutcliffe KM. Mindful organizing. In: Ramanujam R, Roberts KH, eds. Organizing for Reliability: A Guide for Research and Practice. Stanford University Press; 2018:61-89.
- Merchant NB, O’Neal J, Dealino-Perez C, Xiang J, Montoya A Jr, Murray JS. A high-reliability organization mindset. Am J Med Qual. 2022;37:504-510. doi:10.1097/jmq.0000000000000086
- Senge PM. The Fifth Discipline Fieldbook: Strategies and Tools for Building a Learning Organization. Crown Currency; 1994.
- Ramanujam R, Roberts KH, eds. Organizing for Reliability: A Guide for Research and Practice. Stanford University Press; 2018.
- Coveney PV. Self-organization and complexity: a new age for theory, computation and experiment. Philos Trans A Math Phys Eng Sci. 2003;361:1057-1079. doi:10.1098/rsta.2003.1191
- Weick KE, Sutcliffe KM. Managing the Unexpected: Sustained Performance in a Complex World. 3rd ed. Wiley; 2015.
- Barton M, Sutcliffe K. Overcoming dysfunctional momentum: organizational safety as a social achievement. Hum Relations. 2009;62:1327-1356. doi:10.1177/0018726709334491
- Dekker S. Drift Into Failure: From Hunting Broken Components to Understanding Complex Systems. Routledge; 2011.
- Price MR, Williams TC. When doing wrong feels so right: normalization of deviance. J Patient Saf. 2018;14:1-2. doi:10.1097/pts.0000000000000157
- Orwell S, Angus I, eds. In Front of Your Nose, 1945-1950. Godine; 2000. Orwell G. The Collected Essays, Journalism, and Letters of George Orwell; vol 4.
- Murray JS, Baghdadi A, Dannenberg W, Crews P, Walsh ND. The role of high reliability organization foundational practices in building a culture of safety. Fed Pract. 2024;41:214-221. doi:10.12788/fp.0486
- Goldenhar LM, Brady PW, Sutcliffe KM, Muething SE. Huddling for high reliability and situation awareness. BMJ Qual Saf. 2013;22:899-906. doi:10.1136/bmjqs-2012-001467
- Pandit M. Critical factors for successful management of VUCA times. BMJ Lead. 2021;5:121-123. doi:10.1136/leader-2020-000305
- Mihaljevic T. Tiered daily huddles: the power of teamwork in managing large healthcare organisations. BMJ Qual Saf. 2020;29:1050-1052. doi:10.1136/bmjqs-2019-010575
- van Stralen D, Mercer TA. High-reliability organizing (HRO) in the COVID-19 liminal zone: characteristics of workers and local leaders. Neonatology Today. 2021;16:90-101. http://www.neonatologytoday.net /newsletters/nt-apr21.pdf
- Nemeth C, Wears R, Woods D, Hollnagel E, Cook R. Minding the gaps: creating resilience in health care. In: Henriksen K, Battles JB, Keyes MA, Grady ML, eds. Advances in Patient Safety: New Directions and Alternative Approaches. Vol 3: Performance and Tools. Agency for Healthcare Research and Quality; 2008.
- Merchant NB, O’Neal J, Montoya A, Cox GR, Murray JS. Creating a process for the implementation of tiered huddles in a Veterans Affairs medical center. Mil Med. 2023;188:901-906. doi:10.1093/milmed/usac073
- Starbuck WH, Farjoun M, eds. Organization at the Limit: Lessons From the Columbia Disaster. 1st ed. Wiley-Blackwell; 2005.
- Mihaljevic T. Tiered daily huddles: the power of teamwork in managing large healthcare organisations. BMJ Qual Saf. 2020;29:1050-1052. doi:10.1136/bmjqs-2019-010575
- Donnelly LF, Cherian SS, Chua KB, et al. The Daily Readiness Huddle: a process to rapidly identify issues and foster improvement through problem-solving accountability. Pediatr Radiol. 2017;47:22-30. doi:10.1007/s00247-016-3712-x
- Clark TR. The 4 Stages of Psychological Safety: Defining the Path to Inclusion and Innovation. Berrett-Koehler Publishers, Inc.; 2020.
- Edmondson AC. The Fearless Organization: Creating Psychological Safety in the Workplace for Learning, Innovation, and Growth. John Wiley & Sons; 2018.
- Edmondson AC. The Right Kind of Wrong: The Science of Failing Well. Simon Element/Simon Acumen; 2023.
- Murray JS, Kelly S, Hanover C. Promoting psychological safety in healthcare organizations. Mil Med. 2022;187:808 -810. doi:10.1093/milmed/usac041
- Barber HF. Developing strategic leadership: the US Army War College experience. J Manag Dev. 1992;11:4-12. doi:10.1108/02621719210018208
- US Army Heritage & Education Center. Who first originated the term VUCA (volatility, uncertainty, complexity and ambiguity)? Accessed November 5, 2025. https://usawc .libanswers.com/ahec/faq/84869
- van Stralen D, Byrum SL, Inozu B. High Reliability for a Highly Unreliable World: Preparing for Code Blue Through Daily Operations in Healthcare. CreateSpace Independent Publishing Platform; 2018.
- Perrow C. Normal Accidents: Living With High-Risk Technologies. Princeton University Press; 2000.
- Sculli G, Essen K. Soaring to Success: The Path to Developing High-Reliability Clinical Teams. HCPro; 2021. Accessed November 5, 2025. https://hcmarketplace.com /media/wysiwyg/CRM3_browse.pdf
- Barton MA, Sutcliffe KM, Vogus TJ, DeWitt T. Performing under uncertainty: contextualized engagement in wildland firefighting. J Contingencies Crisis Manag. 2015;23:74-83. doi:10.1111/1468-5973.12076
- Sutcliffe KM. Mindful organizing. In: Ramanujam R, Roberts KH, eds. Organizing for Reliability: A Guide for Research and Practice. Stanford University Press; 2018:61-89.
- Merchant NB, O’Neal J, Dealino-Perez C, Xiang J, Montoya A Jr, Murray JS. A high-reliability organization mindset. Am J Med Qual. 2022;37:504-510. doi:10.1097/jmq.0000000000000086
- Senge PM. The Fifth Discipline Fieldbook: Strategies and Tools for Building a Learning Organization. Crown Currency; 1994.
- Ramanujam R, Roberts KH, eds. Organizing for Reliability: A Guide for Research and Practice. Stanford University Press; 2018.
- Coveney PV. Self-organization and complexity: a new age for theory, computation and experiment. Philos Trans A Math Phys Eng Sci. 2003;361:1057-1079. doi:10.1098/rsta.2003.1191
- Weick KE, Sutcliffe KM. Managing the Unexpected: Sustained Performance in a Complex World. 3rd ed. Wiley; 2015.
- Barton M, Sutcliffe K. Overcoming dysfunctional momentum: organizational safety as a social achievement. Hum Relations. 2009;62:1327-1356. doi:10.1177/0018726709334491
- Dekker S. Drift Into Failure: From Hunting Broken Components to Understanding Complex Systems. Routledge; 2011.
- Price MR, Williams TC. When doing wrong feels so right: normalization of deviance. J Patient Saf. 2018;14:1-2. doi:10.1097/pts.0000000000000157
Negotiating the VUCA World Through Tiered Huddles
Negotiating the VUCA World Through Tiered Huddles
