CCJM delivers practical clinical articles relevant to internists, cardiologists, endocrinologists, and other specialists, all written by known experts.
Birds are among the most popular pets in the United States, ranking fourth behind dogs, cats, and fish. According to a commercial survey,1 6.4 million US households own at least one pet bird—and are therefore at risk of a number of bacterial, protozoal, fungal, viral, or parasitic zoonoses (infectious diseases of animals that are communicable to humans).2
This review focuses on the most common pet bird-associated diseases, with implications for bird-keepers’ health. Unless specifically stated otherwise, these diseases are not routinely transmissible from human to human.
NO NEED TO AVOID OWNING A BIRD
Although one can indeed acquire an infection from a pet bird, this possibility need not discourage people from owning birds.3,4
The ability of a microorganism to make a person sick varies with the virulence of the organism, the dose to which a person is exposed, and the route of infection. For a bird owner, prevention often involves simple hygiene, handwashing, sanitation, and regular veterinary care for the bird.3 Many of these diseases are transmitted by ingestion of food contaminated by avian fecal matter or inhalation of contaminated dust. Therefore, bird owners should take steps to minimize dander in the environment. Furthermore, wearing a mask during cage cleaning is suggested.5 Obtaining pet birds from reputable domestic sources decreases the risk of acquiring a bird with an infectious disease.4,6
These hygienic measures are especially important for people who are more susceptible to infection, eg, the very young or very old, those in poor health to begin with, and those with compromised immunity.
Most of the illnesses one can acquire from birds are asymptomatic or self-limited in humans, but they should be considered if a bird-keeper has persistent or unusual symptoms of infection or if the pet bird was recently acquired or has been ill or died.
DISEASES PRESENTING WITH FLU-LIKE OR PULMONARY SYMPTOMS
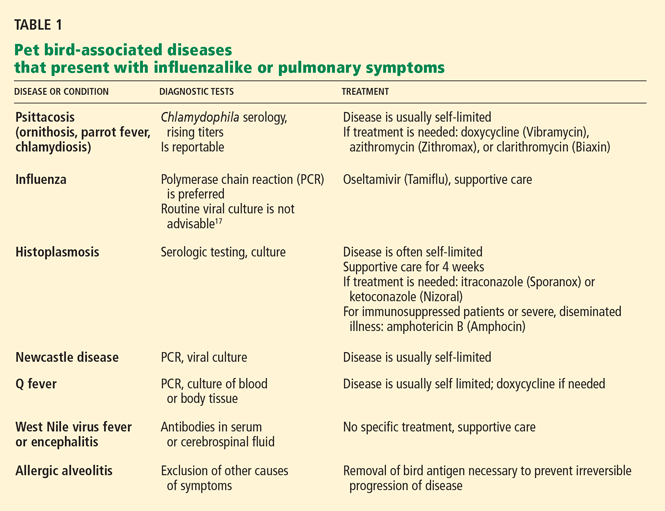
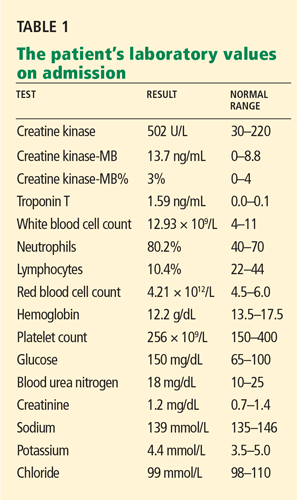
Bird-associated diseases that present with influenza-like or pulmonary symptoms are summarized in Table 1.
Psittacosis is caused by Chlamydophila (formerly Chlamydia) psittaci. The organism has been reported to be present in 40% of birds.4,7 Humans are considered incidental hosts.
Transmission of C psittaci is usually via inhalation of aerosolized particles contaminated with infected bird excreta, nasal secretions, tissue, or feathers. Beak-to-mouth transmission and transmission via bird bites have been reported.8 Unlike Chlamydophila pneumoniae, C psittaci has not conclusively been reported to be transmitted from human to human.
Many infected pet birds show no signs of the disease, but others may have conjunctivitis, liver disease, or generalized signs of severe infection such as ruffled feathers, loss of appetite, diarrhea, or lime-green urates (R. Stevenson, personal communication, 2007).2,5,9,10
Recognizing that the patient has been exposed to birds is clinically useful, as in one study 90% of patients with psittacosis had been exposed to birds as pets or in their occupations.11
Atypical pneumonia is the most common presentation of psittacosis in humans. The symptoms typically begin 7 to 14 days after exposure, with fever, chills, prominent headache, photophobia, and cough. Hepatosplenomegaly is clinically detectable in 10% to 70% of patients. Serious but uncommon presentations include pericarditis, myocarditis, bacterial culture-negative endocarditis, mental status changes, and thrombophlebitis. The combination of pneumonitis and hepatosplenomegaly should prompt consideration of psittacosis.11
Influenza
All known subtypes of influenza A virus can infect birds (influenza B virus cannot). However, there are substantial genetic differences between the subtypes of influenza A that typically infect both people and birds.12
Avian influenza viruses affecting birds and humans (H5N1, H7N7, H9N2, and others), commonly called “bird flu,” emerged in 1997 in association with poultry.13,14 Highly pathogenic H5N1 viral infections have attracted particular attention, as they have killed millions of birds and infected several hundred humans, half of whom have died.13 Various avian influenza viruses that can affect humans have been isolated from captive birds such as parrots, canaries, and poultry and from wild waterfowl and migrating birds.15 To our knowledge, pet birds have not been implicated in transmission, but they are a potential source of infection if they have been in contact with infected birds.
Transmission from bird to human has usually been via inhaled droplets and, less likely, from contaminated environmental sources. According to the US Centers for Disease Control and Prevention, family clusters of H5N1 infection have been observed,16 but person-to-person transmission has been very rare, limited, and unsustained.13
Signs of influenza in birds can range from none to respiratory tract infection, decreased egg production, systemic illness, and death.15
Avian influenza should be suspected in people with flu symptoms who have been exposed to sick poultry or wild birds, have travelled to an endemic area, or have had direct contact with a person known or suspected to be infected with avian influenza.17 Information about areas of the world in which avian influenza is endemic or is breaking out is available at the World Health Organization Epidemic and Pandemic Alert and Response Web page (www.who.int/csr/disease/avian_influenza/updates/en/index.html).
Signs and symptoms of avian influenza in humans include fever, upper respiratory tract infection, cough, and gastrointestinal symptoms. With highly pathogenic viruses such as H5N1, the disease can progress very rapidly from onset to death. Less-pathogenic viruses such as H9N2 cause milder symptoms.16
Histoplasmosis
Histoplasma capsulatum is a fungus that colonizes the gastrointestinal tract of birds and contaminates the soil via bird and bat droppings. The most highly endemic regions of the world are the Ohio and Mississippi River valleys. Typical pet birds such as canaries and parrots are not susceptible to symptomatic infection, but doves and pigeons (often treated as pets by bird-lovers) may become colonized if they contact contaminated birds or excreta (R. Stevenson, personal communication, 2007).5,18
Humans commonly acquire the organisms when they inhale disrupted soil contaminated with the organism. Human-to-human transmission has not been reported.
The degree of human illness depends on the inoculum size and the immunity of the person infected. In more than 90% of cases, the primary infection is minimally symptomatic or goes unnoticed. The typical incubation time is 7 to 21 days. In patients who become ill, symptoms include fever, chills, headache, nonproductive cough, and adenopathy-mediated chest pain. Disseminated disease generally occurs in immunocompromised patients and presents with fever, weight loss, hepatosplenomegaly, and pancytopenia.18
Newcastle disease (avian pneumoencephalitis)
The virus that causes Newcastle disease, avian paramyxovirus 1, can affect animals, reptiles, birds, and people. It is most common in wild birds, but parrots are also highly susceptible and can be reservoirs that continue to shed the virus for up to 12 months after the acute illness has subsided. Illegally imported Amazon parrots are the most likely source of infection for US households. The virus is spread through an infected bird’s feces and secretions from the nose, mouth, and eyes and can be carried on a person’s clothing, footwear, and equipment.
Some infected birds show no signs of it; others have respiratory signs, green diarrhea, muscle tremors, circling, paralysis, or swelling of tissues around the eyes and neck. The mortality rate in infected birds can be up to 100%.19
Human infection most often results in conjunctivitis. Chills, fever, and lethargy are exceptionally rare.15 Because the virus is prevalent in poultry, poultry workers are at greatest risk of infection. Rapid recovery in humans is common. People with conjunctivitis due to Newcastle disease virus should avoid contact with birds.2,20
Q fever
Q fever is caused by Coxiella burnetii, a gram-negative pleomorphic bacillus. Ticks and vertebrates (goats, sheep, and, less commonly, birds) are natural reservoirs for the organism. Human infection results from contact with infected animals or inhalation of dust contaminated with infected excreta or placental tissue.2 Birds may harbor the infection in experimental and natural settings.21,22
Symptoms in humans typically include fever, pneumonitis, severe headache, and photophobia. Meningitis, hepatitis, and thromboses are seen in more-severe disease. Infection acquired in pregnancy may lead to prematurity, abortion, or stillbirth.18,21
West Nile fever and West Nile encephalitis
Wild birds such as corvids and raptors harbor the West Nile virus; pet songbirds (passerines) can harbor it as well.23,24 The principal means of transmission of West Nile infection from birds to humans is via a mosquito biting an infected bird and then biting a human. Direct bird-to-human transmission has not been described.18 Infected birds may be asymptomatic or appear ill or reluctant to fly and die of disseminated viral infection.23
The incubation period in humans is generally 3 to 14 days, followed by the sudden onset of fever, malaise, nausea, vomiting, rash, lymphadenopathy, and retro-orbital pain. Neurologic presentations—ataxia, extrapyramidal signs, cranial nerve abnormalities, myelitis, optical neuritis, and seizures—are quite rare and generally occur in the elderly or immunocompromised. Fewer than 1% of affected people develop more-severe disease, such as acute encephalitis, aseptic meningitis, or Guillain-Barré syndrome.25
Allergic alveolitis
Allergic alveolitis (hypersensitivity pneumonitis, parakeet dander pneumoconiosis, pigeon lung disease, bird-breeder’s lung, bird-fancier’s disease) is not a zoonosis; the term describes diffuse parenchymal lung disease caused by repeated exposure to an inhaled allergen.26 However, it should be considered in patients with pulmonary symptoms and bird exposure. Avian proteins are a known trigger.27
Acute allergic alveolitis is clinically indistinguishable from a respiratory infection. It is characterized by the abrupt onset of an intense nonproductive cough, chest tightness, dyspnea, chills, fever, myalgia, and malaise. Symptoms gradually improve over 24 to 48 hours without antigen exposure but recur with repeated exposure.
Subacute illness presents with similar symptoms, which gradually worsen over weeks to months, and it can be indistinguishable from interstitial lung disease. Many patients with chronic allergic alveolitis are suspected of having tuberculosis or fungal pneumonia or receive a misdiagnosis of idiopathic pulmonary fibrosis.
Patients usually have a favorable outcome if the allergen is removed. If exposure continues, irreversible pulmonary fibrosis may develop.28
DISEASES PRESENTING WITH GASTROINTESTINAL SYMPTOMS
Bird-associated diseases that present with gastrointestinal symptoms are summarized in Table 2.
Salmonellosis
Nontyphoidal Salmonella species colonize the gastrointestinal tract of many animals, including birds. Up to 80% of chicken eggs are contaminated with this gram-negative bacterium.
Spread of nontyphoidal Salmonella to humans is much more common from poultry, poultry products, and pet reptiles than from pet birds, although ducks and baby chicks have transmitted infection to humans. Hand-to-mouth spread occurs after contact with pets or pet excreta.29,30 Infected birds may be healthy carriers, may develop enteritis or hemorrhagic hepatosplenic disease, or may even die.2,9
Gastroenteritis due to nontyphoidal Salmonella in humans begins with nausea, vomiting, fever, and loose, nonbloody diarrhea about 48 hours after ingestion. Most gastroenteritis infections are self-limited, with resolution of fever within 48 to 72 hours and resolution of diarrhea within 4 to 10 days.31
Systemic or severe infection warranting “preemptive” therapy is more likely in immunosuppressed patients, in patients with reduced gastric acid or impaired gastrointestinal mucosal integrity, in infants less than 3 months of age, and in patients with chronic gastrointestinal tract disease, malignant neoplasms, hemoglobinopathies, or infection with the human immunodeficiency virus.31 Systemic nontyphoidal Salmonella infection may settle in structurally abnormal sites such as in existing fractures, severe degenerative joint disease, organs affected by stones, or abnormal lung tissue. Large-vessel arteritis due to non-typhoidal Salmonella should be suspected in a person at risk (particularly if the person is elderly) who presents with back, chest, or abdominal pain preceded by gastroenteritis.31,32
Campylobacteriosis
The main reservoirs for Campylobacter jejuni are wild birds and poultry, although this bacterium can also affect other animals and pet birds.2 The most commonly affected pet birds are psittaciforms (parrots) and passeriforms (finches and canaries).
The organism colonizes the small intestine and colon of birds and can be spread to humans through contact with feces or carcasses of infected animals.33–35 Birds with campylobacteriosis develop hepatitis, lethargy, loss of appetite, weight loss, and yellow diarrhea and often die of the illness (R. Stevenson, personal communication, 2007).
The most important mode of transmission to humans is through handling or consuming chicken, milk, or other products contaminated with feces of carrier animals. However, in up to 24% of cases, the source of infection is unknown.33,34
Human infection with C jejuni most commonly leads to an acute, self-limited gastrointestinal illness characterized by fever, diarrhea, and abdominal cramps. The diarrhea is typically watery or bloody and occurs 8 to 10 times a day at peak illness. Fever can persist for up to a week. Most cases resolve within 7 days, but some patients may have a relapsing diarrheal illness lasting several weeks. Between 20% and 40% of cases of Guillain-Barré syndrome are preceded by infection with C jejuni.35
Giardiasis
Giardiasis is an intestinal protozoal infection caused by Giardia species (primarily G lamblia) that affect humans and other mammals. The parasite is found in bird droppings, but the role of birds in transmission to humans is unclear. Most infections are transmitted via contaminated surface water supplies, although person-to-person transmission has been documented.36 Infected pet birds have signs of gastroenteritis and can be treated, but reinfection often occurs.37
Giardia infections in humans are often asymptomatic, but about 50% of patients have diarrhea, abdominal pain, bloating, belching, nausea, and vomiting 3 days to 3 weeks after ingesting the parasite. A clinical clue may be new-onset lactose intolerance. Symptoms usually resolve after a week. Prolonged infection occurs in up to 20% of patients. People with hypochlorhydria or hypogammaglobulinemia, children, and travelers to endemic areas are at higher risk of infection.37
DISEASES PRESENTING WITH SKIN SYMPTOMS
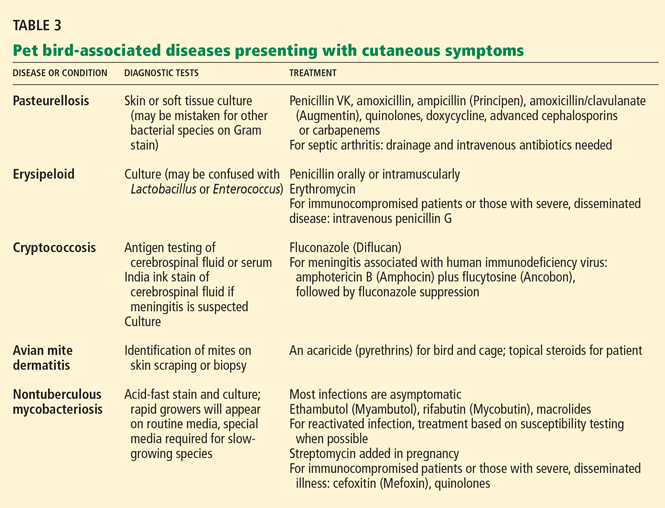
Bird-associated diseases that present with cutaneous symptoms are summarized in Table 3.
Pasteurellosis
Pasteurellosis is caused by Pasteurella multocida, an inhabitant of the healthy nasopharynx of some birds and also the causative agent of avian cholera.38 Many pet birds that acquire systemic Pasteurella infection from a cat bite die of avian cholera (Stevenson R, personal communication, 2007).
Pasteurella organisms are transmissible to humans via bites or scratches from pet birds. Infected wounds in humans are usually red and painful, but the physical findings may lead one to underestimate the severity of infection. Transmission via respiratory droplets is rare but may cause acute or subacute bronchitis, pneumonia, or septicemia.38
Erysipeloid
Erysipeloid, caused by the bacterium Erysipelothrix rhusiopathiae, is transmissible to humans via contact with domestic or wild fowl. Infection in pet birds can cause sepsis but is rarely seen in veterinary practice (R. Stevenson, personal communication, 2007).38,39
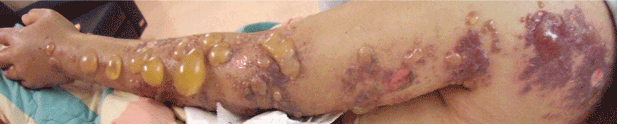
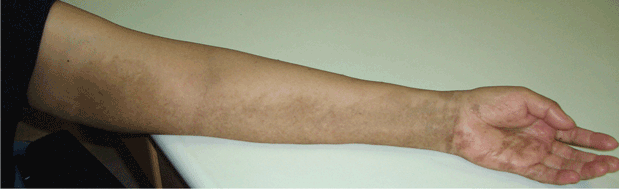
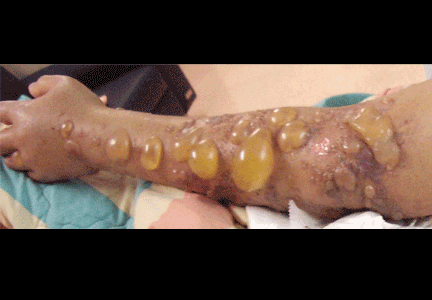
Human infection typically affects broken skin, causing a dramatic, localized skin infection that is painful and pruritic; at first it is livid-red, then blue-red. The infection can spread to nearby joints. Septicemia and endocarditis in humans are rare complications.40
Cryptococcosis
Cryptococcosis, caused by the encapsulated yeast Cryptococcus neoformans, can be harbored and transmitted by asymptomatic pet birds such as cockatoos via colonization of the gastrointestinal tract.38,41 The organism is found in soil contaminated by feces of colonized birds.
Pulmonary symptoms and meningitis are more typical of cryptococcal disease in general, although when contracted from a pet bird via a break in the skin, cutaneous cryptococcosis usually presents with skin lesions resembling cellulitis, molluscum, herpes, and Kaposi sarcomalike papulonodules.42 Infection beyond the skin in immunocompromised patients may involve the lungs and the central nervous system.41 Prostate and eye infections have also been reported.38
Avian mite dermatitis
Birds carry several kinds of mites: feather or “red” mites (which do not affect humans) and mites that can affect humans such as Ornithonyssus sylviarum (the northern fowl mite) and Dermanyssus gallinae (the poultry mite or chicken mite) (R. Stevenson, personal communication, 2007). O sylviarum and D gallinae are found in the commercial poultry industry, but uncommonly, pet birds can harbor them (R. Stevenson, personal communication, 2007).42–44
In humans, mites can cause an intensely pruritic, papular-papulovesicular eruption.42
Nontuberculous mycobacteriosis
Nontuberculous mycobacteria (Mycobacterium species chelonae, abscessus, fortuitum, avium, kansasii, ulcerans, and marinum) are ubiquitous in the environment and can colonize animals. M avium subsp avium causes avian tuberculosis.38 Birds may carry mycobacterial organisms on beaks, claws, and talons, facilitating passage to humans (J.M. Gaskin, personal communication, 2006). In one reported case, M chelonae skin infection was probably transmitted in this manner to a bird-keeper via a bird bite.45
In symptomatic nontuberculous mycobacterial infections, the site of inoculation usually determines the presenting signs. Mycobacteriosis should be suspected when skin or lung infections fail to improve with empiric treatment. Patients with structurally abnormal lungs or immunosuppression may be at higher risk of pulmonary or disseminated disease from infected birds.46,47
Acknowledgment: The authors extend special thanks to Jack M. Gaskin, DVM, PhD, Department of Infectious Disease and Pathology, University of Florida, Gainesville, FL; Rhoda Stevenson, DVM, Exotic Bird Hospital, Jacksonville, FL; and Stephanie L. Hines, MD, Section of General Internal Medicine, Mayo Clinic, Jacksonville, FL, for their review of this paper. Editing, proofreading, and reference verification were provided by the Mayo Clinic Section of Scientific Publications.
References
American Pet Product Manufacturer’s Association. 2005/2006 National Pet Owners Survey. In: Greenwich CT, American Pet Products Manufacturers Association 2005. www.appma.org.
Krauss H, Weber A, Appel M, et al. Zoonoses: Infectious Diseases Transmissible From Animals to Humans, 3rd ed. Washington, DC: American Society for Microbiology Press, 2003.
Hemsworth S, Pizer B. Pet ownership in immunocompromised children—a review of the literature and survey of existing guidelines. Eur J Oncol Nurs2006; 10:117–127.
Smith KA, Bradley KK, Stobierski MG, Tengelsen LA; National Association of State Public Health Veterinarians Psittacosis Compendium Committee. Compendium of measures to control Chlamydophila psittaci (formerly Chlamydia psittaci) infection among humans (psittacosis) and pet birds, 2005. J Am Vet Med Assoc2005; 226:532–539.
Jacob JP, Gaskin JM, Wilson HR, Mather FB. Avian diseases transmissible to humans. University of Florida Institute of Food and Agricultural Sciences Extension. http://edis.ifas.ufl.edu/PS019. Accessed 1/31/2009.
PAWS (Pets Are Wonderful Support). Safe Pet Guidelines: A Comprehensive Guide for Immunocompromised Animal Guardians. www.pawssf.org/SafePetGuide/SPG8.pdf(pp10–13). Accessed 1/31/2009.
Moroney JF, Guevara R, Iverson C, et al. Detection of chlamydiosis in a shipment of pet birds, leading to recognition of an outbreak of clinically mild psittacosis in humans. Clin Infect Dis1998; 26:1425–1429.
Glaser C, Lewis P, Wong S. Pet-, animal-, and vector-borne infections. Pediatr Rev2000; 21:219–232.
Grimes JE. Zoonoses acquired from pet birds. Vet Clin North Am Small Anim Pract1987; 17:209–218.
Spenser EL. Common infectious diseases of psittacine birds seen in practice. Vet Clin North Am Small Anim Pract1991; 21:1213–1230.
Richards MJ. Psittacosis. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
US Centers for Disease Control and Prevention. Avian influenza A viruses. www.cdc.gov/flu/avian/gen-info/avian-influenza.htm. Accessed 1/31/2009.
US Centers for Disease Control and Prevention. Key facts about avian influenza (bird flu) and avian influenza A (H5N1) virus. www.cdc.gov/flu/avian/gen-info/facts.htm. Accessed 1/31/2009.
Swayne DE, King DJ. Avian influenza and Newcastle disease. J Am Vet Med Assoc2003; 222:1534–1540.
Capua I, Alexander DJ. Human health implications of avian influenza viruses and paramyxoviruses. Eur J Clin Microbiol Infect Dis2004; 23:1–6.
US Centers for Disease Control and Prevention. Avian influenza A virus infections of humans. www.cdc.gov/flu/avian/gen-info/avian-flu-humans.htm. Accessed 1/31/2009.
US Centers for Disease Control and Prevention. Updated Interim Guidance for Laboratory Testing of Persons with Suspected Infection with Avian Influenza A (H5N1) Virus in the United States. www2a.cdc.gov/han/ArchiveSys/ViewMsgV.asp?AlertNum=00246. Accessed 11/2008.
Deepe GS. Histoplasma capsulatum. In: Mandell GL, Douglas RG, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases, 6th ed. New York: Churchill Livingstone, 2005:3012–3025.
Cross GM. Newcastle disease. Vet Clin North Am Small Anim Pract1991; 21:1231–1239.
Beard C. Velogenic Newcastle disease. In: Committee on Foreign Animal Diseases of the United States. Foreign Animal Diseases. Richmond, VA: Animal Health Association, 1998:370–376.
Behymer D, Riemann HP. Coxiella burnetii infection (Q fever). J Am Vet Med Assoc1989; 194:764–767.
To H, Sakai R, Shirota K, et al. Coxiellosis in domestic and wild birds from Japan. J Wild Dis1998; 34:310–316.
Weingartl HM, Neufeld JL, Copps J, Marszal P. Experimental West Nile virus infection in blue jays (Cyanocitta cristata) and crows (Corvus brachyrhynchos). Vet Pathol2004; 41:362–370.
Komar N, Panella NA, Langevin SA, et al. Avian hosts for West Nile virus in St. Tammany Parish, Louisiana, 2002. Am J Trop Med Hyg2005; 73:1031–1037.
Petersen LR. Clinical manifestations, diagnosis, and treatment of West Nile virus infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
King TE. Classification and clinical manifestations of hypersensitivity pneumonitis (extrinsic allergic alveolitis). In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
du Marchie Sarvaas GJ, Merkus PJ, de Jongste JC. A family with extrinsic allergic alveolitis caused by wild city pigeons: a case report. Pediatrics2000; 105:E62.
King TE. Treatment and prognosis of hypersensitivity pneumonitis (extrinsic allergic alveolitis). In :Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Woodward DL, Khakhria R, Johnson WM. Human salmonellosis associated with exotic pets. J Clin Microbiol1997; 35:2786–2790.
Hohmann EL. Microbiology and epidemiology of Salmonellosis. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Hohmann EL. Approach to the patient with nontyphoidal Salmonella in a stool culture. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Pegues DA, Miller SI. Salmonellosis. In: Kasper DL, Fauci AS, Longo DL, et al, editors. Harrison's Principles of Internal Medicine, 17th ed. New York: McGraw-Hill Medical, 2008:956–957.
Allos BM. Microbiology, pathogenesis, and epidemiology of Campylobacter infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Padungton P, Kaneene JB. Campylobacter spp in human, chickens, pigs and their antimicrobial resistance. J Vet Med Sci2003; 65:161–170.
Allos BM. Clinical features and treatment of Campylobacter infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Harris JM. Zoonotic diseases of birds. Vet Clin North Am Small Anim Pract1991; 21:1289–1298.
Leder K, Weller PF. Giardiasis in adults. In: Rose B, editor: UpToDate. Waltham, MA, 2008.
Hubálek Z. An annotated checklist of pathogenic microorganisms associated with migratory birds. J Wild Dis2004; 40:639–659.
Gartrell BD, Alley MR, Mack H, Donald J, McInnes K, Jansen P. Erysipelas in the critically endangered kakapo (Strigops habroptilus). Avian Pathol2005; 34:383–387.
Hand WL, Ho H. Erysipelothrix infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med2000; 132:205–208.
Rosen T, Jablon J. Infectious threats from exotic pets: dermatological implications. Dermatol Clin2003; 21:229–236.
Larson J, Gerlach S, Thompson K, et al. Mycobacterium chelonae/abcessus infection caused by a bird bite. Infect Dis Clin Pract2008; 16:60–61.
Griffith DE, Wallace JR. Clinical manifestations of non-tuberculous mycobacterial pulmonary infections in HIV-negative patients. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Griffith DE, Wallace JR. Epidemiology of nontuberculous mycobacterial infections. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2006.
Keels S. Jorn, MD Instructor, Mayo Clinic College of Medicine, Division of Community Internal Medicine, Mayo Clinic, Jacksonville, FL
Kristine M. Thompson, MD Assistant Professor, Mayo Clinic College of Medicine, Department of Emergency Medicine, Mayo Clinic, Jacksonville, FL
Jan M. Larson, MD Assistant Professor, Mayo Clinic College of Medicine, Department of Family Medicine, Mayo Clinic, Jacksonville, FL
Janis E. Blair, MD Professor, Mayo Clinic College of Medicine, Division of Infectious Diseases, Mayo Clinic, Scottsdale, AZ
Address: Keels S. Jorn, MD, Division of Community Internal Medicine, Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL 32224; e-mail [email protected]
Keels S. Jorn, MD Instructor, Mayo Clinic College of Medicine, Division of Community Internal Medicine, Mayo Clinic, Jacksonville, FL
Kristine M. Thompson, MD Assistant Professor, Mayo Clinic College of Medicine, Department of Emergency Medicine, Mayo Clinic, Jacksonville, FL
Jan M. Larson, MD Assistant Professor, Mayo Clinic College of Medicine, Department of Family Medicine, Mayo Clinic, Jacksonville, FL
Janis E. Blair, MD Professor, Mayo Clinic College of Medicine, Division of Infectious Diseases, Mayo Clinic, Scottsdale, AZ
Address: Keels S. Jorn, MD, Division of Community Internal Medicine, Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL 32224; e-mail [email protected]
Author and Disclosure Information
Keels S. Jorn, MD Instructor, Mayo Clinic College of Medicine, Division of Community Internal Medicine, Mayo Clinic, Jacksonville, FL
Kristine M. Thompson, MD Assistant Professor, Mayo Clinic College of Medicine, Department of Emergency Medicine, Mayo Clinic, Jacksonville, FL
Jan M. Larson, MD Assistant Professor, Mayo Clinic College of Medicine, Department of Family Medicine, Mayo Clinic, Jacksonville, FL
Janis E. Blair, MD Professor, Mayo Clinic College of Medicine, Division of Infectious Diseases, Mayo Clinic, Scottsdale, AZ
Address: Keels S. Jorn, MD, Division of Community Internal Medicine, Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL 32224; e-mail [email protected]
Birds are among the most popular pets in the United States, ranking fourth behind dogs, cats, and fish. According to a commercial survey,1 6.4 million US households own at least one pet bird—and are therefore at risk of a number of bacterial, protozoal, fungal, viral, or parasitic zoonoses (infectious diseases of animals that are communicable to humans).2
This review focuses on the most common pet bird-associated diseases, with implications for bird-keepers’ health. Unless specifically stated otherwise, these diseases are not routinely transmissible from human to human.
NO NEED TO AVOID OWNING A BIRD
Although one can indeed acquire an infection from a pet bird, this possibility need not discourage people from owning birds.3,4
The ability of a microorganism to make a person sick varies with the virulence of the organism, the dose to which a person is exposed, and the route of infection. For a bird owner, prevention often involves simple hygiene, handwashing, sanitation, and regular veterinary care for the bird.3 Many of these diseases are transmitted by ingestion of food contaminated by avian fecal matter or inhalation of contaminated dust. Therefore, bird owners should take steps to minimize dander in the environment. Furthermore, wearing a mask during cage cleaning is suggested.5 Obtaining pet birds from reputable domestic sources decreases the risk of acquiring a bird with an infectious disease.4,6
These hygienic measures are especially important for people who are more susceptible to infection, eg, the very young or very old, those in poor health to begin with, and those with compromised immunity.
Most of the illnesses one can acquire from birds are asymptomatic or self-limited in humans, but they should be considered if a bird-keeper has persistent or unusual symptoms of infection or if the pet bird was recently acquired or has been ill or died.
DISEASES PRESENTING WITH FLU-LIKE OR PULMONARY SYMPTOMS
Bird-associated diseases that present with influenza-like or pulmonary symptoms are summarized in Table 1.
Psittacosis is caused by Chlamydophila (formerly Chlamydia) psittaci. The organism has been reported to be present in 40% of birds.4,7 Humans are considered incidental hosts.
Transmission of C psittaci is usually via inhalation of aerosolized particles contaminated with infected bird excreta, nasal secretions, tissue, or feathers. Beak-to-mouth transmission and transmission via bird bites have been reported.8 Unlike Chlamydophila pneumoniae, C psittaci has not conclusively been reported to be transmitted from human to human.
Many infected pet birds show no signs of the disease, but others may have conjunctivitis, liver disease, or generalized signs of severe infection such as ruffled feathers, loss of appetite, diarrhea, or lime-green urates (R. Stevenson, personal communication, 2007).2,5,9,10
Recognizing that the patient has been exposed to birds is clinically useful, as in one study 90% of patients with psittacosis had been exposed to birds as pets or in their occupations.11
Atypical pneumonia is the most common presentation of psittacosis in humans. The symptoms typically begin 7 to 14 days after exposure, with fever, chills, prominent headache, photophobia, and cough. Hepatosplenomegaly is clinically detectable in 10% to 70% of patients. Serious but uncommon presentations include pericarditis, myocarditis, bacterial culture-negative endocarditis, mental status changes, and thrombophlebitis. The combination of pneumonitis and hepatosplenomegaly should prompt consideration of psittacosis.11
Influenza
All known subtypes of influenza A virus can infect birds (influenza B virus cannot). However, there are substantial genetic differences between the subtypes of influenza A that typically infect both people and birds.12
Avian influenza viruses affecting birds and humans (H5N1, H7N7, H9N2, and others), commonly called “bird flu,” emerged in 1997 in association with poultry.13,14 Highly pathogenic H5N1 viral infections have attracted particular attention, as they have killed millions of birds and infected several hundred humans, half of whom have died.13 Various avian influenza viruses that can affect humans have been isolated from captive birds such as parrots, canaries, and poultry and from wild waterfowl and migrating birds.15 To our knowledge, pet birds have not been implicated in transmission, but they are a potential source of infection if they have been in contact with infected birds.
Transmission from bird to human has usually been via inhaled droplets and, less likely, from contaminated environmental sources. According to the US Centers for Disease Control and Prevention, family clusters of H5N1 infection have been observed,16 but person-to-person transmission has been very rare, limited, and unsustained.13
Signs of influenza in birds can range from none to respiratory tract infection, decreased egg production, systemic illness, and death.15
Avian influenza should be suspected in people with flu symptoms who have been exposed to sick poultry or wild birds, have travelled to an endemic area, or have had direct contact with a person known or suspected to be infected with avian influenza.17 Information about areas of the world in which avian influenza is endemic or is breaking out is available at the World Health Organization Epidemic and Pandemic Alert and Response Web page (www.who.int/csr/disease/avian_influenza/updates/en/index.html).
Signs and symptoms of avian influenza in humans include fever, upper respiratory tract infection, cough, and gastrointestinal symptoms. With highly pathogenic viruses such as H5N1, the disease can progress very rapidly from onset to death. Less-pathogenic viruses such as H9N2 cause milder symptoms.16
Histoplasmosis
Histoplasma capsulatum is a fungus that colonizes the gastrointestinal tract of birds and contaminates the soil via bird and bat droppings. The most highly endemic regions of the world are the Ohio and Mississippi River valleys. Typical pet birds such as canaries and parrots are not susceptible to symptomatic infection, but doves and pigeons (often treated as pets by bird-lovers) may become colonized if they contact contaminated birds or excreta (R. Stevenson, personal communication, 2007).5,18
Humans commonly acquire the organisms when they inhale disrupted soil contaminated with the organism. Human-to-human transmission has not been reported.
The degree of human illness depends on the inoculum size and the immunity of the person infected. In more than 90% of cases, the primary infection is minimally symptomatic or goes unnoticed. The typical incubation time is 7 to 21 days. In patients who become ill, symptoms include fever, chills, headache, nonproductive cough, and adenopathy-mediated chest pain. Disseminated disease generally occurs in immunocompromised patients and presents with fever, weight loss, hepatosplenomegaly, and pancytopenia.18
Newcastle disease (avian pneumoencephalitis)
The virus that causes Newcastle disease, avian paramyxovirus 1, can affect animals, reptiles, birds, and people. It is most common in wild birds, but parrots are also highly susceptible and can be reservoirs that continue to shed the virus for up to 12 months after the acute illness has subsided. Illegally imported Amazon parrots are the most likely source of infection for US households. The virus is spread through an infected bird’s feces and secretions from the nose, mouth, and eyes and can be carried on a person’s clothing, footwear, and equipment.
Some infected birds show no signs of it; others have respiratory signs, green diarrhea, muscle tremors, circling, paralysis, or swelling of tissues around the eyes and neck. The mortality rate in infected birds can be up to 100%.19
Human infection most often results in conjunctivitis. Chills, fever, and lethargy are exceptionally rare.15 Because the virus is prevalent in poultry, poultry workers are at greatest risk of infection. Rapid recovery in humans is common. People with conjunctivitis due to Newcastle disease virus should avoid contact with birds.2,20
Q fever
Q fever is caused by Coxiella burnetii, a gram-negative pleomorphic bacillus. Ticks and vertebrates (goats, sheep, and, less commonly, birds) are natural reservoirs for the organism. Human infection results from contact with infected animals or inhalation of dust contaminated with infected excreta or placental tissue.2 Birds may harbor the infection in experimental and natural settings.21,22
Symptoms in humans typically include fever, pneumonitis, severe headache, and photophobia. Meningitis, hepatitis, and thromboses are seen in more-severe disease. Infection acquired in pregnancy may lead to prematurity, abortion, or stillbirth.18,21
West Nile fever and West Nile encephalitis
Wild birds such as corvids and raptors harbor the West Nile virus; pet songbirds (passerines) can harbor it as well.23,24 The principal means of transmission of West Nile infection from birds to humans is via a mosquito biting an infected bird and then biting a human. Direct bird-to-human transmission has not been described.18 Infected birds may be asymptomatic or appear ill or reluctant to fly and die of disseminated viral infection.23
The incubation period in humans is generally 3 to 14 days, followed by the sudden onset of fever, malaise, nausea, vomiting, rash, lymphadenopathy, and retro-orbital pain. Neurologic presentations—ataxia, extrapyramidal signs, cranial nerve abnormalities, myelitis, optical neuritis, and seizures—are quite rare and generally occur in the elderly or immunocompromised. Fewer than 1% of affected people develop more-severe disease, such as acute encephalitis, aseptic meningitis, or Guillain-Barré syndrome.25
Allergic alveolitis
Allergic alveolitis (hypersensitivity pneumonitis, parakeet dander pneumoconiosis, pigeon lung disease, bird-breeder’s lung, bird-fancier’s disease) is not a zoonosis; the term describes diffuse parenchymal lung disease caused by repeated exposure to an inhaled allergen.26 However, it should be considered in patients with pulmonary symptoms and bird exposure. Avian proteins are a known trigger.27
Acute allergic alveolitis is clinically indistinguishable from a respiratory infection. It is characterized by the abrupt onset of an intense nonproductive cough, chest tightness, dyspnea, chills, fever, myalgia, and malaise. Symptoms gradually improve over 24 to 48 hours without antigen exposure but recur with repeated exposure.
Subacute illness presents with similar symptoms, which gradually worsen over weeks to months, and it can be indistinguishable from interstitial lung disease. Many patients with chronic allergic alveolitis are suspected of having tuberculosis or fungal pneumonia or receive a misdiagnosis of idiopathic pulmonary fibrosis.
Patients usually have a favorable outcome if the allergen is removed. If exposure continues, irreversible pulmonary fibrosis may develop.28
DISEASES PRESENTING WITH GASTROINTESTINAL SYMPTOMS
Bird-associated diseases that present with gastrointestinal symptoms are summarized in Table 2.
Salmonellosis
Nontyphoidal Salmonella species colonize the gastrointestinal tract of many animals, including birds. Up to 80% of chicken eggs are contaminated with this gram-negative bacterium.
Spread of nontyphoidal Salmonella to humans is much more common from poultry, poultry products, and pet reptiles than from pet birds, although ducks and baby chicks have transmitted infection to humans. Hand-to-mouth spread occurs after contact with pets or pet excreta.29,30 Infected birds may be healthy carriers, may develop enteritis or hemorrhagic hepatosplenic disease, or may even die.2,9
Gastroenteritis due to nontyphoidal Salmonella in humans begins with nausea, vomiting, fever, and loose, nonbloody diarrhea about 48 hours after ingestion. Most gastroenteritis infections are self-limited, with resolution of fever within 48 to 72 hours and resolution of diarrhea within 4 to 10 days.31
Systemic or severe infection warranting “preemptive” therapy is more likely in immunosuppressed patients, in patients with reduced gastric acid or impaired gastrointestinal mucosal integrity, in infants less than 3 months of age, and in patients with chronic gastrointestinal tract disease, malignant neoplasms, hemoglobinopathies, or infection with the human immunodeficiency virus.31 Systemic nontyphoidal Salmonella infection may settle in structurally abnormal sites such as in existing fractures, severe degenerative joint disease, organs affected by stones, or abnormal lung tissue. Large-vessel arteritis due to non-typhoidal Salmonella should be suspected in a person at risk (particularly if the person is elderly) who presents with back, chest, or abdominal pain preceded by gastroenteritis.31,32
Campylobacteriosis
The main reservoirs for Campylobacter jejuni are wild birds and poultry, although this bacterium can also affect other animals and pet birds.2 The most commonly affected pet birds are psittaciforms (parrots) and passeriforms (finches and canaries).
The organism colonizes the small intestine and colon of birds and can be spread to humans through contact with feces or carcasses of infected animals.33–35 Birds with campylobacteriosis develop hepatitis, lethargy, loss of appetite, weight loss, and yellow diarrhea and often die of the illness (R. Stevenson, personal communication, 2007).
The most important mode of transmission to humans is through handling or consuming chicken, milk, or other products contaminated with feces of carrier animals. However, in up to 24% of cases, the source of infection is unknown.33,34
Human infection with C jejuni most commonly leads to an acute, self-limited gastrointestinal illness characterized by fever, diarrhea, and abdominal cramps. The diarrhea is typically watery or bloody and occurs 8 to 10 times a day at peak illness. Fever can persist for up to a week. Most cases resolve within 7 days, but some patients may have a relapsing diarrheal illness lasting several weeks. Between 20% and 40% of cases of Guillain-Barré syndrome are preceded by infection with C jejuni.35
Giardiasis
Giardiasis is an intestinal protozoal infection caused by Giardia species (primarily G lamblia) that affect humans and other mammals. The parasite is found in bird droppings, but the role of birds in transmission to humans is unclear. Most infections are transmitted via contaminated surface water supplies, although person-to-person transmission has been documented.36 Infected pet birds have signs of gastroenteritis and can be treated, but reinfection often occurs.37
Giardia infections in humans are often asymptomatic, but about 50% of patients have diarrhea, abdominal pain, bloating, belching, nausea, and vomiting 3 days to 3 weeks after ingesting the parasite. A clinical clue may be new-onset lactose intolerance. Symptoms usually resolve after a week. Prolonged infection occurs in up to 20% of patients. People with hypochlorhydria or hypogammaglobulinemia, children, and travelers to endemic areas are at higher risk of infection.37
DISEASES PRESENTING WITH SKIN SYMPTOMS
Bird-associated diseases that present with cutaneous symptoms are summarized in Table 3.
Pasteurellosis
Pasteurellosis is caused by Pasteurella multocida, an inhabitant of the healthy nasopharynx of some birds and also the causative agent of avian cholera.38 Many pet birds that acquire systemic Pasteurella infection from a cat bite die of avian cholera (Stevenson R, personal communication, 2007).
Pasteurella organisms are transmissible to humans via bites or scratches from pet birds. Infected wounds in humans are usually red and painful, but the physical findings may lead one to underestimate the severity of infection. Transmission via respiratory droplets is rare but may cause acute or subacute bronchitis, pneumonia, or septicemia.38
Erysipeloid
Erysipeloid, caused by the bacterium Erysipelothrix rhusiopathiae, is transmissible to humans via contact with domestic or wild fowl. Infection in pet birds can cause sepsis but is rarely seen in veterinary practice (R. Stevenson, personal communication, 2007).38,39
Human infection typically affects broken skin, causing a dramatic, localized skin infection that is painful and pruritic; at first it is livid-red, then blue-red. The infection can spread to nearby joints. Septicemia and endocarditis in humans are rare complications.40
Cryptococcosis
Cryptococcosis, caused by the encapsulated yeast Cryptococcus neoformans, can be harbored and transmitted by asymptomatic pet birds such as cockatoos via colonization of the gastrointestinal tract.38,41 The organism is found in soil contaminated by feces of colonized birds.
Pulmonary symptoms and meningitis are more typical of cryptococcal disease in general, although when contracted from a pet bird via a break in the skin, cutaneous cryptococcosis usually presents with skin lesions resembling cellulitis, molluscum, herpes, and Kaposi sarcomalike papulonodules.42 Infection beyond the skin in immunocompromised patients may involve the lungs and the central nervous system.41 Prostate and eye infections have also been reported.38
Avian mite dermatitis
Birds carry several kinds of mites: feather or “red” mites (which do not affect humans) and mites that can affect humans such as Ornithonyssus sylviarum (the northern fowl mite) and Dermanyssus gallinae (the poultry mite or chicken mite) (R. Stevenson, personal communication, 2007). O sylviarum and D gallinae are found in the commercial poultry industry, but uncommonly, pet birds can harbor them (R. Stevenson, personal communication, 2007).42–44
In humans, mites can cause an intensely pruritic, papular-papulovesicular eruption.42
Nontuberculous mycobacteriosis
Nontuberculous mycobacteria (Mycobacterium species chelonae, abscessus, fortuitum, avium, kansasii, ulcerans, and marinum) are ubiquitous in the environment and can colonize animals. M avium subsp avium causes avian tuberculosis.38 Birds may carry mycobacterial organisms on beaks, claws, and talons, facilitating passage to humans (J.M. Gaskin, personal communication, 2006). In one reported case, M chelonae skin infection was probably transmitted in this manner to a bird-keeper via a bird bite.45
In symptomatic nontuberculous mycobacterial infections, the site of inoculation usually determines the presenting signs. Mycobacteriosis should be suspected when skin or lung infections fail to improve with empiric treatment. Patients with structurally abnormal lungs or immunosuppression may be at higher risk of pulmonary or disseminated disease from infected birds.46,47
Acknowledgment: The authors extend special thanks to Jack M. Gaskin, DVM, PhD, Department of Infectious Disease and Pathology, University of Florida, Gainesville, FL; Rhoda Stevenson, DVM, Exotic Bird Hospital, Jacksonville, FL; and Stephanie L. Hines, MD, Section of General Internal Medicine, Mayo Clinic, Jacksonville, FL, for their review of this paper. Editing, proofreading, and reference verification were provided by the Mayo Clinic Section of Scientific Publications.
Birds are among the most popular pets in the United States, ranking fourth behind dogs, cats, and fish. According to a commercial survey,1 6.4 million US households own at least one pet bird—and are therefore at risk of a number of bacterial, protozoal, fungal, viral, or parasitic zoonoses (infectious diseases of animals that are communicable to humans).2
This review focuses on the most common pet bird-associated diseases, with implications for bird-keepers’ health. Unless specifically stated otherwise, these diseases are not routinely transmissible from human to human.
NO NEED TO AVOID OWNING A BIRD
Although one can indeed acquire an infection from a pet bird, this possibility need not discourage people from owning birds.3,4
The ability of a microorganism to make a person sick varies with the virulence of the organism, the dose to which a person is exposed, and the route of infection. For a bird owner, prevention often involves simple hygiene, handwashing, sanitation, and regular veterinary care for the bird.3 Many of these diseases are transmitted by ingestion of food contaminated by avian fecal matter or inhalation of contaminated dust. Therefore, bird owners should take steps to minimize dander in the environment. Furthermore, wearing a mask during cage cleaning is suggested.5 Obtaining pet birds from reputable domestic sources decreases the risk of acquiring a bird with an infectious disease.4,6
These hygienic measures are especially important for people who are more susceptible to infection, eg, the very young or very old, those in poor health to begin with, and those with compromised immunity.
Most of the illnesses one can acquire from birds are asymptomatic or self-limited in humans, but they should be considered if a bird-keeper has persistent or unusual symptoms of infection or if the pet bird was recently acquired or has been ill or died.
DISEASES PRESENTING WITH FLU-LIKE OR PULMONARY SYMPTOMS
Bird-associated diseases that present with influenza-like or pulmonary symptoms are summarized in Table 1.
Psittacosis is caused by Chlamydophila (formerly Chlamydia) psittaci. The organism has been reported to be present in 40% of birds.4,7 Humans are considered incidental hosts.
Transmission of C psittaci is usually via inhalation of aerosolized particles contaminated with infected bird excreta, nasal secretions, tissue, or feathers. Beak-to-mouth transmission and transmission via bird bites have been reported.8 Unlike Chlamydophila pneumoniae, C psittaci has not conclusively been reported to be transmitted from human to human.
Many infected pet birds show no signs of the disease, but others may have conjunctivitis, liver disease, or generalized signs of severe infection such as ruffled feathers, loss of appetite, diarrhea, or lime-green urates (R. Stevenson, personal communication, 2007).2,5,9,10
Recognizing that the patient has been exposed to birds is clinically useful, as in one study 90% of patients with psittacosis had been exposed to birds as pets or in their occupations.11
Atypical pneumonia is the most common presentation of psittacosis in humans. The symptoms typically begin 7 to 14 days after exposure, with fever, chills, prominent headache, photophobia, and cough. Hepatosplenomegaly is clinically detectable in 10% to 70% of patients. Serious but uncommon presentations include pericarditis, myocarditis, bacterial culture-negative endocarditis, mental status changes, and thrombophlebitis. The combination of pneumonitis and hepatosplenomegaly should prompt consideration of psittacosis.11
Influenza
All known subtypes of influenza A virus can infect birds (influenza B virus cannot). However, there are substantial genetic differences between the subtypes of influenza A that typically infect both people and birds.12
Avian influenza viruses affecting birds and humans (H5N1, H7N7, H9N2, and others), commonly called “bird flu,” emerged in 1997 in association with poultry.13,14 Highly pathogenic H5N1 viral infections have attracted particular attention, as they have killed millions of birds and infected several hundred humans, half of whom have died.13 Various avian influenza viruses that can affect humans have been isolated from captive birds such as parrots, canaries, and poultry and from wild waterfowl and migrating birds.15 To our knowledge, pet birds have not been implicated in transmission, but they are a potential source of infection if they have been in contact with infected birds.
Transmission from bird to human has usually been via inhaled droplets and, less likely, from contaminated environmental sources. According to the US Centers for Disease Control and Prevention, family clusters of H5N1 infection have been observed,16 but person-to-person transmission has been very rare, limited, and unsustained.13
Signs of influenza in birds can range from none to respiratory tract infection, decreased egg production, systemic illness, and death.15
Avian influenza should be suspected in people with flu symptoms who have been exposed to sick poultry or wild birds, have travelled to an endemic area, or have had direct contact with a person known or suspected to be infected with avian influenza.17 Information about areas of the world in which avian influenza is endemic or is breaking out is available at the World Health Organization Epidemic and Pandemic Alert and Response Web page (www.who.int/csr/disease/avian_influenza/updates/en/index.html).
Signs and symptoms of avian influenza in humans include fever, upper respiratory tract infection, cough, and gastrointestinal symptoms. With highly pathogenic viruses such as H5N1, the disease can progress very rapidly from onset to death. Less-pathogenic viruses such as H9N2 cause milder symptoms.16
Histoplasmosis
Histoplasma capsulatum is a fungus that colonizes the gastrointestinal tract of birds and contaminates the soil via bird and bat droppings. The most highly endemic regions of the world are the Ohio and Mississippi River valleys. Typical pet birds such as canaries and parrots are not susceptible to symptomatic infection, but doves and pigeons (often treated as pets by bird-lovers) may become colonized if they contact contaminated birds or excreta (R. Stevenson, personal communication, 2007).5,18
Humans commonly acquire the organisms when they inhale disrupted soil contaminated with the organism. Human-to-human transmission has not been reported.
The degree of human illness depends on the inoculum size and the immunity of the person infected. In more than 90% of cases, the primary infection is minimally symptomatic or goes unnoticed. The typical incubation time is 7 to 21 days. In patients who become ill, symptoms include fever, chills, headache, nonproductive cough, and adenopathy-mediated chest pain. Disseminated disease generally occurs in immunocompromised patients and presents with fever, weight loss, hepatosplenomegaly, and pancytopenia.18
Newcastle disease (avian pneumoencephalitis)
The virus that causes Newcastle disease, avian paramyxovirus 1, can affect animals, reptiles, birds, and people. It is most common in wild birds, but parrots are also highly susceptible and can be reservoirs that continue to shed the virus for up to 12 months after the acute illness has subsided. Illegally imported Amazon parrots are the most likely source of infection for US households. The virus is spread through an infected bird’s feces and secretions from the nose, mouth, and eyes and can be carried on a person’s clothing, footwear, and equipment.
Some infected birds show no signs of it; others have respiratory signs, green diarrhea, muscle tremors, circling, paralysis, or swelling of tissues around the eyes and neck. The mortality rate in infected birds can be up to 100%.19
Human infection most often results in conjunctivitis. Chills, fever, and lethargy are exceptionally rare.15 Because the virus is prevalent in poultry, poultry workers are at greatest risk of infection. Rapid recovery in humans is common. People with conjunctivitis due to Newcastle disease virus should avoid contact with birds.2,20
Q fever
Q fever is caused by Coxiella burnetii, a gram-negative pleomorphic bacillus. Ticks and vertebrates (goats, sheep, and, less commonly, birds) are natural reservoirs for the organism. Human infection results from contact with infected animals or inhalation of dust contaminated with infected excreta or placental tissue.2 Birds may harbor the infection in experimental and natural settings.21,22
Symptoms in humans typically include fever, pneumonitis, severe headache, and photophobia. Meningitis, hepatitis, and thromboses are seen in more-severe disease. Infection acquired in pregnancy may lead to prematurity, abortion, or stillbirth.18,21
West Nile fever and West Nile encephalitis
Wild birds such as corvids and raptors harbor the West Nile virus; pet songbirds (passerines) can harbor it as well.23,24 The principal means of transmission of West Nile infection from birds to humans is via a mosquito biting an infected bird and then biting a human. Direct bird-to-human transmission has not been described.18 Infected birds may be asymptomatic or appear ill or reluctant to fly and die of disseminated viral infection.23
The incubation period in humans is generally 3 to 14 days, followed by the sudden onset of fever, malaise, nausea, vomiting, rash, lymphadenopathy, and retro-orbital pain. Neurologic presentations—ataxia, extrapyramidal signs, cranial nerve abnormalities, myelitis, optical neuritis, and seizures—are quite rare and generally occur in the elderly or immunocompromised. Fewer than 1% of affected people develop more-severe disease, such as acute encephalitis, aseptic meningitis, or Guillain-Barré syndrome.25
Allergic alveolitis
Allergic alveolitis (hypersensitivity pneumonitis, parakeet dander pneumoconiosis, pigeon lung disease, bird-breeder’s lung, bird-fancier’s disease) is not a zoonosis; the term describes diffuse parenchymal lung disease caused by repeated exposure to an inhaled allergen.26 However, it should be considered in patients with pulmonary symptoms and bird exposure. Avian proteins are a known trigger.27
Acute allergic alveolitis is clinically indistinguishable from a respiratory infection. It is characterized by the abrupt onset of an intense nonproductive cough, chest tightness, dyspnea, chills, fever, myalgia, and malaise. Symptoms gradually improve over 24 to 48 hours without antigen exposure but recur with repeated exposure.
Subacute illness presents with similar symptoms, which gradually worsen over weeks to months, and it can be indistinguishable from interstitial lung disease. Many patients with chronic allergic alveolitis are suspected of having tuberculosis or fungal pneumonia or receive a misdiagnosis of idiopathic pulmonary fibrosis.
Patients usually have a favorable outcome if the allergen is removed. If exposure continues, irreversible pulmonary fibrosis may develop.28
DISEASES PRESENTING WITH GASTROINTESTINAL SYMPTOMS
Bird-associated diseases that present with gastrointestinal symptoms are summarized in Table 2.
Salmonellosis
Nontyphoidal Salmonella species colonize the gastrointestinal tract of many animals, including birds. Up to 80% of chicken eggs are contaminated with this gram-negative bacterium.
Spread of nontyphoidal Salmonella to humans is much more common from poultry, poultry products, and pet reptiles than from pet birds, although ducks and baby chicks have transmitted infection to humans. Hand-to-mouth spread occurs after contact with pets or pet excreta.29,30 Infected birds may be healthy carriers, may develop enteritis or hemorrhagic hepatosplenic disease, or may even die.2,9
Gastroenteritis due to nontyphoidal Salmonella in humans begins with nausea, vomiting, fever, and loose, nonbloody diarrhea about 48 hours after ingestion. Most gastroenteritis infections are self-limited, with resolution of fever within 48 to 72 hours and resolution of diarrhea within 4 to 10 days.31
Systemic or severe infection warranting “preemptive” therapy is more likely in immunosuppressed patients, in patients with reduced gastric acid or impaired gastrointestinal mucosal integrity, in infants less than 3 months of age, and in patients with chronic gastrointestinal tract disease, malignant neoplasms, hemoglobinopathies, or infection with the human immunodeficiency virus.31 Systemic nontyphoidal Salmonella infection may settle in structurally abnormal sites such as in existing fractures, severe degenerative joint disease, organs affected by stones, or abnormal lung tissue. Large-vessel arteritis due to non-typhoidal Salmonella should be suspected in a person at risk (particularly if the person is elderly) who presents with back, chest, or abdominal pain preceded by gastroenteritis.31,32
Campylobacteriosis
The main reservoirs for Campylobacter jejuni are wild birds and poultry, although this bacterium can also affect other animals and pet birds.2 The most commonly affected pet birds are psittaciforms (parrots) and passeriforms (finches and canaries).
The organism colonizes the small intestine and colon of birds and can be spread to humans through contact with feces or carcasses of infected animals.33–35 Birds with campylobacteriosis develop hepatitis, lethargy, loss of appetite, weight loss, and yellow diarrhea and often die of the illness (R. Stevenson, personal communication, 2007).
The most important mode of transmission to humans is through handling or consuming chicken, milk, or other products contaminated with feces of carrier animals. However, in up to 24% of cases, the source of infection is unknown.33,34
Human infection with C jejuni most commonly leads to an acute, self-limited gastrointestinal illness characterized by fever, diarrhea, and abdominal cramps. The diarrhea is typically watery or bloody and occurs 8 to 10 times a day at peak illness. Fever can persist for up to a week. Most cases resolve within 7 days, but some patients may have a relapsing diarrheal illness lasting several weeks. Between 20% and 40% of cases of Guillain-Barré syndrome are preceded by infection with C jejuni.35
Giardiasis
Giardiasis is an intestinal protozoal infection caused by Giardia species (primarily G lamblia) that affect humans and other mammals. The parasite is found in bird droppings, but the role of birds in transmission to humans is unclear. Most infections are transmitted via contaminated surface water supplies, although person-to-person transmission has been documented.36 Infected pet birds have signs of gastroenteritis and can be treated, but reinfection often occurs.37
Giardia infections in humans are often asymptomatic, but about 50% of patients have diarrhea, abdominal pain, bloating, belching, nausea, and vomiting 3 days to 3 weeks after ingesting the parasite. A clinical clue may be new-onset lactose intolerance. Symptoms usually resolve after a week. Prolonged infection occurs in up to 20% of patients. People with hypochlorhydria or hypogammaglobulinemia, children, and travelers to endemic areas are at higher risk of infection.37
DISEASES PRESENTING WITH SKIN SYMPTOMS
Bird-associated diseases that present with cutaneous symptoms are summarized in Table 3.
Pasteurellosis
Pasteurellosis is caused by Pasteurella multocida, an inhabitant of the healthy nasopharynx of some birds and also the causative agent of avian cholera.38 Many pet birds that acquire systemic Pasteurella infection from a cat bite die of avian cholera (Stevenson R, personal communication, 2007).
Pasteurella organisms are transmissible to humans via bites or scratches from pet birds. Infected wounds in humans are usually red and painful, but the physical findings may lead one to underestimate the severity of infection. Transmission via respiratory droplets is rare but may cause acute or subacute bronchitis, pneumonia, or septicemia.38
Erysipeloid
Erysipeloid, caused by the bacterium Erysipelothrix rhusiopathiae, is transmissible to humans via contact with domestic or wild fowl. Infection in pet birds can cause sepsis but is rarely seen in veterinary practice (R. Stevenson, personal communication, 2007).38,39
Human infection typically affects broken skin, causing a dramatic, localized skin infection that is painful and pruritic; at first it is livid-red, then blue-red. The infection can spread to nearby joints. Septicemia and endocarditis in humans are rare complications.40
Cryptococcosis
Cryptococcosis, caused by the encapsulated yeast Cryptococcus neoformans, can be harbored and transmitted by asymptomatic pet birds such as cockatoos via colonization of the gastrointestinal tract.38,41 The organism is found in soil contaminated by feces of colonized birds.
Pulmonary symptoms and meningitis are more typical of cryptococcal disease in general, although when contracted from a pet bird via a break in the skin, cutaneous cryptococcosis usually presents with skin lesions resembling cellulitis, molluscum, herpes, and Kaposi sarcomalike papulonodules.42 Infection beyond the skin in immunocompromised patients may involve the lungs and the central nervous system.41 Prostate and eye infections have also been reported.38
Avian mite dermatitis
Birds carry several kinds of mites: feather or “red” mites (which do not affect humans) and mites that can affect humans such as Ornithonyssus sylviarum (the northern fowl mite) and Dermanyssus gallinae (the poultry mite or chicken mite) (R. Stevenson, personal communication, 2007). O sylviarum and D gallinae are found in the commercial poultry industry, but uncommonly, pet birds can harbor them (R. Stevenson, personal communication, 2007).42–44
In humans, mites can cause an intensely pruritic, papular-papulovesicular eruption.42
Nontuberculous mycobacteriosis
Nontuberculous mycobacteria (Mycobacterium species chelonae, abscessus, fortuitum, avium, kansasii, ulcerans, and marinum) are ubiquitous in the environment and can colonize animals. M avium subsp avium causes avian tuberculosis.38 Birds may carry mycobacterial organisms on beaks, claws, and talons, facilitating passage to humans (J.M. Gaskin, personal communication, 2006). In one reported case, M chelonae skin infection was probably transmitted in this manner to a bird-keeper via a bird bite.45
In symptomatic nontuberculous mycobacterial infections, the site of inoculation usually determines the presenting signs. Mycobacteriosis should be suspected when skin or lung infections fail to improve with empiric treatment. Patients with structurally abnormal lungs or immunosuppression may be at higher risk of pulmonary or disseminated disease from infected birds.46,47
Acknowledgment: The authors extend special thanks to Jack M. Gaskin, DVM, PhD, Department of Infectious Disease and Pathology, University of Florida, Gainesville, FL; Rhoda Stevenson, DVM, Exotic Bird Hospital, Jacksonville, FL; and Stephanie L. Hines, MD, Section of General Internal Medicine, Mayo Clinic, Jacksonville, FL, for their review of this paper. Editing, proofreading, and reference verification were provided by the Mayo Clinic Section of Scientific Publications.
References
American Pet Product Manufacturer’s Association. 2005/2006 National Pet Owners Survey. In: Greenwich CT, American Pet Products Manufacturers Association 2005. www.appma.org.
Krauss H, Weber A, Appel M, et al. Zoonoses: Infectious Diseases Transmissible From Animals to Humans, 3rd ed. Washington, DC: American Society for Microbiology Press, 2003.
Hemsworth S, Pizer B. Pet ownership in immunocompromised children—a review of the literature and survey of existing guidelines. Eur J Oncol Nurs2006; 10:117–127.
Smith KA, Bradley KK, Stobierski MG, Tengelsen LA; National Association of State Public Health Veterinarians Psittacosis Compendium Committee. Compendium of measures to control Chlamydophila psittaci (formerly Chlamydia psittaci) infection among humans (psittacosis) and pet birds, 2005. J Am Vet Med Assoc2005; 226:532–539.
Jacob JP, Gaskin JM, Wilson HR, Mather FB. Avian diseases transmissible to humans. University of Florida Institute of Food and Agricultural Sciences Extension. http://edis.ifas.ufl.edu/PS019. Accessed 1/31/2009.
PAWS (Pets Are Wonderful Support). Safe Pet Guidelines: A Comprehensive Guide for Immunocompromised Animal Guardians. www.pawssf.org/SafePetGuide/SPG8.pdf(pp10–13). Accessed 1/31/2009.
Moroney JF, Guevara R, Iverson C, et al. Detection of chlamydiosis in a shipment of pet birds, leading to recognition of an outbreak of clinically mild psittacosis in humans. Clin Infect Dis1998; 26:1425–1429.
Glaser C, Lewis P, Wong S. Pet-, animal-, and vector-borne infections. Pediatr Rev2000; 21:219–232.
Grimes JE. Zoonoses acquired from pet birds. Vet Clin North Am Small Anim Pract1987; 17:209–218.
Spenser EL. Common infectious diseases of psittacine birds seen in practice. Vet Clin North Am Small Anim Pract1991; 21:1213–1230.
Richards MJ. Psittacosis. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
US Centers for Disease Control and Prevention. Avian influenza A viruses. www.cdc.gov/flu/avian/gen-info/avian-influenza.htm. Accessed 1/31/2009.
US Centers for Disease Control and Prevention. Key facts about avian influenza (bird flu) and avian influenza A (H5N1) virus. www.cdc.gov/flu/avian/gen-info/facts.htm. Accessed 1/31/2009.
Swayne DE, King DJ. Avian influenza and Newcastle disease. J Am Vet Med Assoc2003; 222:1534–1540.
Capua I, Alexander DJ. Human health implications of avian influenza viruses and paramyxoviruses. Eur J Clin Microbiol Infect Dis2004; 23:1–6.
US Centers for Disease Control and Prevention. Avian influenza A virus infections of humans. www.cdc.gov/flu/avian/gen-info/avian-flu-humans.htm. Accessed 1/31/2009.
US Centers for Disease Control and Prevention. Updated Interim Guidance for Laboratory Testing of Persons with Suspected Infection with Avian Influenza A (H5N1) Virus in the United States. www2a.cdc.gov/han/ArchiveSys/ViewMsgV.asp?AlertNum=00246. Accessed 11/2008.
Deepe GS. Histoplasma capsulatum. In: Mandell GL, Douglas RG, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases, 6th ed. New York: Churchill Livingstone, 2005:3012–3025.
Cross GM. Newcastle disease. Vet Clin North Am Small Anim Pract1991; 21:1231–1239.
Beard C. Velogenic Newcastle disease. In: Committee on Foreign Animal Diseases of the United States. Foreign Animal Diseases. Richmond, VA: Animal Health Association, 1998:370–376.
Behymer D, Riemann HP. Coxiella burnetii infection (Q fever). J Am Vet Med Assoc1989; 194:764–767.
To H, Sakai R, Shirota K, et al. Coxiellosis in domestic and wild birds from Japan. J Wild Dis1998; 34:310–316.
Weingartl HM, Neufeld JL, Copps J, Marszal P. Experimental West Nile virus infection in blue jays (Cyanocitta cristata) and crows (Corvus brachyrhynchos). Vet Pathol2004; 41:362–370.
Komar N, Panella NA, Langevin SA, et al. Avian hosts for West Nile virus in St. Tammany Parish, Louisiana, 2002. Am J Trop Med Hyg2005; 73:1031–1037.
Petersen LR. Clinical manifestations, diagnosis, and treatment of West Nile virus infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
King TE. Classification and clinical manifestations of hypersensitivity pneumonitis (extrinsic allergic alveolitis). In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
du Marchie Sarvaas GJ, Merkus PJ, de Jongste JC. A family with extrinsic allergic alveolitis caused by wild city pigeons: a case report. Pediatrics2000; 105:E62.
King TE. Treatment and prognosis of hypersensitivity pneumonitis (extrinsic allergic alveolitis). In :Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Woodward DL, Khakhria R, Johnson WM. Human salmonellosis associated with exotic pets. J Clin Microbiol1997; 35:2786–2790.
Hohmann EL. Microbiology and epidemiology of Salmonellosis. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Hohmann EL. Approach to the patient with nontyphoidal Salmonella in a stool culture. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Pegues DA, Miller SI. Salmonellosis. In: Kasper DL, Fauci AS, Longo DL, et al, editors. Harrison's Principles of Internal Medicine, 17th ed. New York: McGraw-Hill Medical, 2008:956–957.
Allos BM. Microbiology, pathogenesis, and epidemiology of Campylobacter infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Padungton P, Kaneene JB. Campylobacter spp in human, chickens, pigs and their antimicrobial resistance. J Vet Med Sci2003; 65:161–170.
Allos BM. Clinical features and treatment of Campylobacter infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Harris JM. Zoonotic diseases of birds. Vet Clin North Am Small Anim Pract1991; 21:1289–1298.
Leder K, Weller PF. Giardiasis in adults. In: Rose B, editor: UpToDate. Waltham, MA, 2008.
Hubálek Z. An annotated checklist of pathogenic microorganisms associated with migratory birds. J Wild Dis2004; 40:639–659.
Gartrell BD, Alley MR, Mack H, Donald J, McInnes K, Jansen P. Erysipelas in the critically endangered kakapo (Strigops habroptilus). Avian Pathol2005; 34:383–387.
Hand WL, Ho H. Erysipelothrix infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med2000; 132:205–208.
Rosen T, Jablon J. Infectious threats from exotic pets: dermatological implications. Dermatol Clin2003; 21:229–236.
Larson J, Gerlach S, Thompson K, et al. Mycobacterium chelonae/abcessus infection caused by a bird bite. Infect Dis Clin Pract2008; 16:60–61.
Griffith DE, Wallace JR. Clinical manifestations of non-tuberculous mycobacterial pulmonary infections in HIV-negative patients. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Griffith DE, Wallace JR. Epidemiology of nontuberculous mycobacterial infections. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2006.
References
American Pet Product Manufacturer’s Association. 2005/2006 National Pet Owners Survey. In: Greenwich CT, American Pet Products Manufacturers Association 2005. www.appma.org.
Krauss H, Weber A, Appel M, et al. Zoonoses: Infectious Diseases Transmissible From Animals to Humans, 3rd ed. Washington, DC: American Society for Microbiology Press, 2003.
Hemsworth S, Pizer B. Pet ownership in immunocompromised children—a review of the literature and survey of existing guidelines. Eur J Oncol Nurs2006; 10:117–127.
Smith KA, Bradley KK, Stobierski MG, Tengelsen LA; National Association of State Public Health Veterinarians Psittacosis Compendium Committee. Compendium of measures to control Chlamydophila psittaci (formerly Chlamydia psittaci) infection among humans (psittacosis) and pet birds, 2005. J Am Vet Med Assoc2005; 226:532–539.
Jacob JP, Gaskin JM, Wilson HR, Mather FB. Avian diseases transmissible to humans. University of Florida Institute of Food and Agricultural Sciences Extension. http://edis.ifas.ufl.edu/PS019. Accessed 1/31/2009.
PAWS (Pets Are Wonderful Support). Safe Pet Guidelines: A Comprehensive Guide for Immunocompromised Animal Guardians. www.pawssf.org/SafePetGuide/SPG8.pdf(pp10–13). Accessed 1/31/2009.
Moroney JF, Guevara R, Iverson C, et al. Detection of chlamydiosis in a shipment of pet birds, leading to recognition of an outbreak of clinically mild psittacosis in humans. Clin Infect Dis1998; 26:1425–1429.
Glaser C, Lewis P, Wong S. Pet-, animal-, and vector-borne infections. Pediatr Rev2000; 21:219–232.
Grimes JE. Zoonoses acquired from pet birds. Vet Clin North Am Small Anim Pract1987; 17:209–218.
Spenser EL. Common infectious diseases of psittacine birds seen in practice. Vet Clin North Am Small Anim Pract1991; 21:1213–1230.
Richards MJ. Psittacosis. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
US Centers for Disease Control and Prevention. Avian influenza A viruses. www.cdc.gov/flu/avian/gen-info/avian-influenza.htm. Accessed 1/31/2009.
US Centers for Disease Control and Prevention. Key facts about avian influenza (bird flu) and avian influenza A (H5N1) virus. www.cdc.gov/flu/avian/gen-info/facts.htm. Accessed 1/31/2009.
Swayne DE, King DJ. Avian influenza and Newcastle disease. J Am Vet Med Assoc2003; 222:1534–1540.
Capua I, Alexander DJ. Human health implications of avian influenza viruses and paramyxoviruses. Eur J Clin Microbiol Infect Dis2004; 23:1–6.
US Centers for Disease Control and Prevention. Avian influenza A virus infections of humans. www.cdc.gov/flu/avian/gen-info/avian-flu-humans.htm. Accessed 1/31/2009.
US Centers for Disease Control and Prevention. Updated Interim Guidance for Laboratory Testing of Persons with Suspected Infection with Avian Influenza A (H5N1) Virus in the United States. www2a.cdc.gov/han/ArchiveSys/ViewMsgV.asp?AlertNum=00246. Accessed 11/2008.
Deepe GS. Histoplasma capsulatum. In: Mandell GL, Douglas RG, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases, 6th ed. New York: Churchill Livingstone, 2005:3012–3025.
Cross GM. Newcastle disease. Vet Clin North Am Small Anim Pract1991; 21:1231–1239.
Beard C. Velogenic Newcastle disease. In: Committee on Foreign Animal Diseases of the United States. Foreign Animal Diseases. Richmond, VA: Animal Health Association, 1998:370–376.
Behymer D, Riemann HP. Coxiella burnetii infection (Q fever). J Am Vet Med Assoc1989; 194:764–767.
To H, Sakai R, Shirota K, et al. Coxiellosis in domestic and wild birds from Japan. J Wild Dis1998; 34:310–316.
Weingartl HM, Neufeld JL, Copps J, Marszal P. Experimental West Nile virus infection in blue jays (Cyanocitta cristata) and crows (Corvus brachyrhynchos). Vet Pathol2004; 41:362–370.
Komar N, Panella NA, Langevin SA, et al. Avian hosts for West Nile virus in St. Tammany Parish, Louisiana, 2002. Am J Trop Med Hyg2005; 73:1031–1037.
Petersen LR. Clinical manifestations, diagnosis, and treatment of West Nile virus infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
King TE. Classification and clinical manifestations of hypersensitivity pneumonitis (extrinsic allergic alveolitis). In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
du Marchie Sarvaas GJ, Merkus PJ, de Jongste JC. A family with extrinsic allergic alveolitis caused by wild city pigeons: a case report. Pediatrics2000; 105:E62.
King TE. Treatment and prognosis of hypersensitivity pneumonitis (extrinsic allergic alveolitis). In :Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Woodward DL, Khakhria R, Johnson WM. Human salmonellosis associated with exotic pets. J Clin Microbiol1997; 35:2786–2790.
Hohmann EL. Microbiology and epidemiology of Salmonellosis. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Hohmann EL. Approach to the patient with nontyphoidal Salmonella in a stool culture. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Pegues DA, Miller SI. Salmonellosis. In: Kasper DL, Fauci AS, Longo DL, et al, editors. Harrison's Principles of Internal Medicine, 17th ed. New York: McGraw-Hill Medical, 2008:956–957.
Allos BM. Microbiology, pathogenesis, and epidemiology of Campylobacter infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Padungton P, Kaneene JB. Campylobacter spp in human, chickens, pigs and their antimicrobial resistance. J Vet Med Sci2003; 65:161–170.
Allos BM. Clinical features and treatment of Campylobacter infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Harris JM. Zoonotic diseases of birds. Vet Clin North Am Small Anim Pract1991; 21:1289–1298.
Leder K, Weller PF. Giardiasis in adults. In: Rose B, editor: UpToDate. Waltham, MA, 2008.
Hubálek Z. An annotated checklist of pathogenic microorganisms associated with migratory birds. J Wild Dis2004; 40:639–659.
Gartrell BD, Alley MR, Mack H, Donald J, McInnes K, Jansen P. Erysipelas in the critically endangered kakapo (Strigops habroptilus). Avian Pathol2005; 34:383–387.
Hand WL, Ho H. Erysipelothrix infection. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med2000; 132:205–208.
Rosen T, Jablon J. Infectious threats from exotic pets: dermatological implications. Dermatol Clin2003; 21:229–236.
Larson J, Gerlach S, Thompson K, et al. Mycobacterium chelonae/abcessus infection caused by a bird bite. Infect Dis Clin Pract2008; 16:60–61.
Griffith DE, Wallace JR. Clinical manifestations of non-tuberculous mycobacterial pulmonary infections in HIV-negative patients. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Griffith DE, Wallace JR. Epidemiology of nontuberculous mycobacterial infections. In: Rose B, editor: UpToDate. Waltham, MA: UpToDate, 2006.
Most cases of pet bird-associated illness are asymptomatic or self-limited.
Transmission to humans occurs predominantly via inhalation or ingestion of infected or contaminated material. Prevention of human infection largely depends on proper hygiene and sanitation.
Bird-associated diseases that present with influenzalike or pulmonary symptoms include psittacosis, influenza, histoplasmosis, Newcastle disease, Q fever, West Nile virus fever or encephalitis, and allergic alveolitis.
Diseases presenting with gastrointestinal symptoms include salmonellosis, campylobacteriosis, and giardiasis.
Diseases presenting with cutaneous symptoms include pasteurellosis, erysipeloid, cryptococcosis, avian mite dermatitis, and nontuberculous mycobacteriosis.
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
Autoimmune disorders
Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
Structural or congenital abnormalities (pancreas divisum)
Trauma.
Figure 1.In this review, we focus on the causes of recurrent acute pancreatitis that can be managed by endoscopic therapy (Figure 1), ie:
Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
Sphincter of Oddi dysfunction
Pancreas divisum
Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis(Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
References
Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol2001; 96:2540–2555.
Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol2002; 97:1959–1962.
Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol2006; 41:681–685.
Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology2001; 120:708–717.
Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol2007; 102:1781–1788.
Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc2007; 66:213–218.
Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit?Hepatology1994; 20:523–525.
Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp1901; 12:182–188.
Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet1993; 341:1371–1373.
Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology1991; 101:1701–1709.
Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med1992; 326:589–593.
Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts?Gastrointest Endosc2002; 55:286–293.
Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology1986; 6:118–122.
Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology1986; 90:677–686.
Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol2000; 95:1702–1707.
Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther2006; 24:1059–1066.
Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas2003; 27:103–117.
Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am2003; 13:695–716.
Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc2005; 62:1–8.
Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy2003; 35:92.
Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg1977; 185:598–604.
Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol2008; 14:1023–1026.
Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med1996; 335:909–918.
Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology1996; 110:894–899.
Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology1990; 98:1008–1012.
Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up?Gastrointest Endosc2002; 55:523–526.
Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc2001; 54:42–48.
Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy2002; 34:273–279.
Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc2006; 64:60–65.
Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc2002; 55:157–162.
Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc1999; 49:599–604.
Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc2008; 67:AB102.
Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil2005; 17suppl 1:31–40.
McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol2007; 13:6333–6343.
Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci2007; 52:1211–1218.
Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep2002; 4:153–159.
Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas2005; 30:359–362.
Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP2004; 5:322–327.
Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc2004; 59:499–505.
Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility?Rev Gastroenterol Disord2004; 4:211–213.
Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy?Curr Gastroenterol Rep2002; 4:172–176.
Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol2007; 41:94–102.
Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther2006; 24:237–246.
Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci1989; 34:56–60.
Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med1989; 320:82–87.
Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy2002; 34:280–285.
Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol2007; 5:1354–1365.
Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford)2003; 5:142–145.
Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP2001; 2:382–400.
Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc2004; 60:419–425.
Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum?Pancreas2007; 34:21–45.
Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol2003; 17:381–383.
Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am1995; 5:145–170.
Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis?Pancreas1988; 3:108–110.
Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas2008; 37:151–153.
Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg1990; 159:59–64.
Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg1996; 183:65–70.
Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc2002; 55:376–381.
Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc1992; 38:430–434.
Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol2008; 14:1027–1033.
Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg1999; 134:311–315.
Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg2004; 67:309–310.
Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol2007; 13:2892–2894.
Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc1990; 36:63–65.
Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J2000; 41:528–532.
Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol2000; 31:172–174.
Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol2001; 16:1070,1075.
Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery2004; 136:909–916.
Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol2007; 5:75–79.
Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol2001; 96:705–709.
Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc2004; 60:673–678.
Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med2007; 356:676–684.
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
Autoimmune disorders
Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
Structural or congenital abnormalities (pancreas divisum)
Trauma.
Figure 1.In this review, we focus on the causes of recurrent acute pancreatitis that can be managed by endoscopic therapy (Figure 1), ie:
Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
Sphincter of Oddi dysfunction
Pancreas divisum
Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis(Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
Autoimmune disorders
Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
Structural or congenital abnormalities (pancreas divisum)
Trauma.
Figure 1.In this review, we focus on the causes of recurrent acute pancreatitis that can be managed by endoscopic therapy (Figure 1), ie:
Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
Sphincter of Oddi dysfunction
Pancreas divisum
Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis(Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
References
Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol2001; 96:2540–2555.
Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol2002; 97:1959–1962.
Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol2006; 41:681–685.
Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology2001; 120:708–717.
Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol2007; 102:1781–1788.
Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc2007; 66:213–218.
Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit?Hepatology1994; 20:523–525.
Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp1901; 12:182–188.
Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet1993; 341:1371–1373.
Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology1991; 101:1701–1709.
Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med1992; 326:589–593.
Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts?Gastrointest Endosc2002; 55:286–293.
Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology1986; 6:118–122.
Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology1986; 90:677–686.
Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol2000; 95:1702–1707.
Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther2006; 24:1059–1066.
Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas2003; 27:103–117.
Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am2003; 13:695–716.
Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc2005; 62:1–8.
Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy2003; 35:92.
Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg1977; 185:598–604.
Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol2008; 14:1023–1026.
Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med1996; 335:909–918.
Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology1996; 110:894–899.
Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology1990; 98:1008–1012.
Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up?Gastrointest Endosc2002; 55:523–526.
Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc2001; 54:42–48.
Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy2002; 34:273–279.
Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc2006; 64:60–65.
Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc2002; 55:157–162.
Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc1999; 49:599–604.
Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc2008; 67:AB102.
Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil2005; 17suppl 1:31–40.
McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol2007; 13:6333–6343.
Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci2007; 52:1211–1218.
Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep2002; 4:153–159.
Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas2005; 30:359–362.
Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP2004; 5:322–327.
Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc2004; 59:499–505.
Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility?Rev Gastroenterol Disord2004; 4:211–213.
Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy?Curr Gastroenterol Rep2002; 4:172–176.
Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol2007; 41:94–102.
Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther2006; 24:237–246.
Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci1989; 34:56–60.
Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med1989; 320:82–87.
Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy2002; 34:280–285.
Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol2007; 5:1354–1365.
Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford)2003; 5:142–145.
Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP2001; 2:382–400.
Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc2004; 60:419–425.
Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum?Pancreas2007; 34:21–45.
Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol2003; 17:381–383.
Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am1995; 5:145–170.
Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis?Pancreas1988; 3:108–110.
Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas2008; 37:151–153.
Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg1990; 159:59–64.
Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg1996; 183:65–70.
Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc2002; 55:376–381.
Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc1992; 38:430–434.
Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol2008; 14:1027–1033.
Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg1999; 134:311–315.
Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg2004; 67:309–310.
Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol2007; 13:2892–2894.
Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc1990; 36:63–65.
Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J2000; 41:528–532.
Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol2000; 31:172–174.
Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol2001; 16:1070,1075.
Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery2004; 136:909–916.
Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol2007; 5:75–79.
Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol2001; 96:705–709.
Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc2004; 60:673–678.
Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med2007; 356:676–684.
References
Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol2001; 96:2540–2555.
Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol2002; 97:1959–1962.
Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol2006; 41:681–685.
Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology2001; 120:708–717.
Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol2007; 102:1781–1788.
Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc2007; 66:213–218.
Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit?Hepatology1994; 20:523–525.
Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp1901; 12:182–188.
Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet1993; 341:1371–1373.
Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology1991; 101:1701–1709.
Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med1992; 326:589–593.
Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts?Gastrointest Endosc2002; 55:286–293.
Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology1986; 6:118–122.
Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology1986; 90:677–686.
Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol2000; 95:1702–1707.
Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther2006; 24:1059–1066.
Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas2003; 27:103–117.
Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am2003; 13:695–716.
Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc2005; 62:1–8.
Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy2003; 35:92.
Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg1977; 185:598–604.
Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol2008; 14:1023–1026.
Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med1996; 335:909–918.
Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology1996; 110:894–899.
Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology1990; 98:1008–1012.
Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up?Gastrointest Endosc2002; 55:523–526.
Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc2001; 54:42–48.
Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy2002; 34:273–279.
Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc2006; 64:60–65.
Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc2002; 55:157–162.
Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc1999; 49:599–604.
Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc2008; 67:AB102.
Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil2005; 17suppl 1:31–40.
McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol2007; 13:6333–6343.
Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci2007; 52:1211–1218.
Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep2002; 4:153–159.
Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas2005; 30:359–362.
Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP2004; 5:322–327.
Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc2004; 59:499–505.
Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility?Rev Gastroenterol Disord2004; 4:211–213.
Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy?Curr Gastroenterol Rep2002; 4:172–176.
Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol2007; 41:94–102.
Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther2006; 24:237–246.
Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci1989; 34:56–60.
Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med1989; 320:82–87.
Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy2002; 34:280–285.
Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol2007; 5:1354–1365.
Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford)2003; 5:142–145.
Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP2001; 2:382–400.
Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc2004; 60:419–425.
Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum?Pancreas2007; 34:21–45.
Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol2003; 17:381–383.
Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am1995; 5:145–170.
Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis?Pancreas1988; 3:108–110.
Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas2008; 37:151–153.
Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg1990; 159:59–64.
Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg1996; 183:65–70.
Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc2002; 55:376–381.
Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc1992; 38:430–434.
Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol2008; 14:1027–1033.
Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg1999; 134:311–315.
Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg2004; 67:309–310.
Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol2007; 13:2892–2894.
Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc1990; 36:63–65.
Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J2000; 41:528–532.
Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol2000; 31:172–174.
Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol2001; 16:1070,1075.
Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery2004; 136:909–916.
Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol2007; 5:75–79.
Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol2001; 96:705–709.
Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc2004; 60:673–678.
Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med2007; 356:676–684.
Recurrent attacks of acute pancreatitis can be prevented only by determining and treating the underlying cause.
Endoscopic procedures can cause anxiety and carry a risk of bleeding, perforation, and pancreatitis. The risks, benefits, and other treatment options should be discussed with the patient.
Endoscopic therapy is now the preferred treatment of sphincter of Oddi dysfunction at centers that have experience with this technique.
In patients with pancreas divisum and recurrent acute pancreatitis, surgical and endoscopic minor sphincterotomy are equally effective.
Figure 1.A 56-year-old woman presents with multiple huge bullae and crusted erosions in her left sixth to eighth cervical and first thoracic dermatomes (Figure 1), accompanied by severe, sharp, lancinating pain. She underwent renal transplantation 3 months ago for end-stage diabetic kidney disease and is now taking immunosuppressants, including tacrolimus (Prograf) (trough serum level 8–10 ng/dL), mycophenolate mofetil (CellCept) 500 mg twice a day, and prednisolone 5 mg per day.
Q: What is the most likely diagnosis?
Contact dermatitis
Herpes zoster
Herpes simplex
Pemphigus
Bullous pemphigoid
Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
Figure 2.This patient was admitted to the hospital and was put in a private room. The lesions were protected from further breakdown and secondary bacterial infection. We discontinued mycophenolate mofetil and prescribed acyclovir (Zovirax) 250 mg intravenously every 8 hours (dose adjusted according to her renal function) for 7 days. Antibiotics needed to be given later for cellulitis that developed as a complication. She had no sign of ophthalmic involvement, visceral involvement, or other complication. She was discharged with healing skin after 42 days of hospitalization (Figure 2) and is free from postherpetic neuralgia.
References
Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med2007; 74:489–504.
Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis2008; 10:260–268.
Ming-Ju Tsai, MD Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
Hung-Tien Kuo, MD Department of Internal Medicine, Kaohsiung Medical University Hospital and Faculty of Renal Care, Kaohsiung Medical University, Kaohsiung, Taiwan
Hung-Chun Chen, MD, PhD Department of Internal Medicine, Kaohsiung Medical University Hospital and Faculty of Renal Care, Kaohsiung Medical University, Kaohsiung, Taiwan
Address: Hung-Chun Chen, MD, PhD, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, No.100, Tzyou 1st Road, Kaohsiung 807, Taiwan; e-mail [email protected]
Ming-Ju Tsai, MD Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
Hung-Tien Kuo, MD Department of Internal Medicine, Kaohsiung Medical University Hospital and Faculty of Renal Care, Kaohsiung Medical University, Kaohsiung, Taiwan
Hung-Chun Chen, MD, PhD Department of Internal Medicine, Kaohsiung Medical University Hospital and Faculty of Renal Care, Kaohsiung Medical University, Kaohsiung, Taiwan
Address: Hung-Chun Chen, MD, PhD, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, No.100, Tzyou 1st Road, Kaohsiung 807, Taiwan; e-mail [email protected]
Author and Disclosure Information
Ming-Ju Tsai, MD Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
Hung-Tien Kuo, MD Department of Internal Medicine, Kaohsiung Medical University Hospital and Faculty of Renal Care, Kaohsiung Medical University, Kaohsiung, Taiwan
Hung-Chun Chen, MD, PhD Department of Internal Medicine, Kaohsiung Medical University Hospital and Faculty of Renal Care, Kaohsiung Medical University, Kaohsiung, Taiwan
Address: Hung-Chun Chen, MD, PhD, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, No.100, Tzyou 1st Road, Kaohsiung 807, Taiwan; e-mail [email protected]
Figure 1.A 56-year-old woman presents with multiple huge bullae and crusted erosions in her left sixth to eighth cervical and first thoracic dermatomes (Figure 1), accompanied by severe, sharp, lancinating pain. She underwent renal transplantation 3 months ago for end-stage diabetic kidney disease and is now taking immunosuppressants, including tacrolimus (Prograf) (trough serum level 8–10 ng/dL), mycophenolate mofetil (CellCept) 500 mg twice a day, and prednisolone 5 mg per day.
Q: What is the most likely diagnosis?
Contact dermatitis
Herpes zoster
Herpes simplex
Pemphigus
Bullous pemphigoid
Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
Figure 2.This patient was admitted to the hospital and was put in a private room. The lesions were protected from further breakdown and secondary bacterial infection. We discontinued mycophenolate mofetil and prescribed acyclovir (Zovirax) 250 mg intravenously every 8 hours (dose adjusted according to her renal function) for 7 days. Antibiotics needed to be given later for cellulitis that developed as a complication. She had no sign of ophthalmic involvement, visceral involvement, or other complication. She was discharged with healing skin after 42 days of hospitalization (Figure 2) and is free from postherpetic neuralgia.
Figure 1.A 56-year-old woman presents with multiple huge bullae and crusted erosions in her left sixth to eighth cervical and first thoracic dermatomes (Figure 1), accompanied by severe, sharp, lancinating pain. She underwent renal transplantation 3 months ago for end-stage diabetic kidney disease and is now taking immunosuppressants, including tacrolimus (Prograf) (trough serum level 8–10 ng/dL), mycophenolate mofetil (CellCept) 500 mg twice a day, and prednisolone 5 mg per day.
Q: What is the most likely diagnosis?
Contact dermatitis
Herpes zoster
Herpes simplex
Pemphigus
Bullous pemphigoid
Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
Figure 2.This patient was admitted to the hospital and was put in a private room. The lesions were protected from further breakdown and secondary bacterial infection. We discontinued mycophenolate mofetil and prescribed acyclovir (Zovirax) 250 mg intravenously every 8 hours (dose adjusted according to her renal function) for 7 days. Antibiotics needed to be given later for cellulitis that developed as a complication. She had no sign of ophthalmic involvement, visceral involvement, or other complication. She was discharged with healing skin after 42 days of hospitalization (Figure 2) and is free from postherpetic neuralgia.
References
Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med2007; 74:489–504.
Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis2008; 10:260–268.
References
Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med2007; 74:489–504.
Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis2008; 10:260–268.
With spring comes change and renewal at the Cleveland Clinic Journal of Medicine.
First, we welcome our new deputy editor Dr. Timothy Gilligan. Tim, a medical oncologist with a journalism degree, takes over the reins from Dr. David Rolston, who left last year to become Associate Director of General Internal Medicine at Geisinger Medical Center in Danville, PA. David continues to provide input to the Journal as a member of our editorial board.
Timothy Gilligan, MD, MSNew to our editorial board is geriatrician Dr. Theodore Suh, who will also oversee the independent peer review of the Journal’s supplements. We will further expand our editorial board in the coming months as part of our continuing effort to offer readers a balance of expertise across medical specialties.
Theodore Suh, MD, PhD, MHScExciting changes to our Web site, www.ccjm.org, will include links to activities offered by Cleveland Clinic’s Center for Continuing Medical Education and Center for Consumer Health Information. We will also continue to amplify the educational value of our online CME test and enhance the interactivity of this and other online offerings. Dr. Gilligan will play a prominent role in these upgrades.
Also, the Journal joins an expanding movement in medical journalism by screening all submitted manuscripts with a new online service called CrossCheck to detect plagiarism—whether self-plagiarism or traditional plagiarism. In this way, we will do our part to uphold the integrity of the medical literature.
And as always, I urge you to continue to submit your ideas for articles, particularly ideas for 1-Minute Consults. We review and value all of your suggestions.
With spring comes change and renewal at the Cleveland Clinic Journal of Medicine.
First, we welcome our new deputy editor Dr. Timothy Gilligan. Tim, a medical oncologist with a journalism degree, takes over the reins from Dr. David Rolston, who left last year to become Associate Director of General Internal Medicine at Geisinger Medical Center in Danville, PA. David continues to provide input to the Journal as a member of our editorial board.
Timothy Gilligan, MD, MSNew to our editorial board is geriatrician Dr. Theodore Suh, who will also oversee the independent peer review of the Journal’s supplements. We will further expand our editorial board in the coming months as part of our continuing effort to offer readers a balance of expertise across medical specialties.
Theodore Suh, MD, PhD, MHScExciting changes to our Web site, www.ccjm.org, will include links to activities offered by Cleveland Clinic’s Center for Continuing Medical Education and Center for Consumer Health Information. We will also continue to amplify the educational value of our online CME test and enhance the interactivity of this and other online offerings. Dr. Gilligan will play a prominent role in these upgrades.
Also, the Journal joins an expanding movement in medical journalism by screening all submitted manuscripts with a new online service called CrossCheck to detect plagiarism—whether self-plagiarism or traditional plagiarism. In this way, we will do our part to uphold the integrity of the medical literature.
And as always, I urge you to continue to submit your ideas for articles, particularly ideas for 1-Minute Consults. We review and value all of your suggestions.
With spring comes change and renewal at the Cleveland Clinic Journal of Medicine.
First, we welcome our new deputy editor Dr. Timothy Gilligan. Tim, a medical oncologist with a journalism degree, takes over the reins from Dr. David Rolston, who left last year to become Associate Director of General Internal Medicine at Geisinger Medical Center in Danville, PA. David continues to provide input to the Journal as a member of our editorial board.
Timothy Gilligan, MD, MSNew to our editorial board is geriatrician Dr. Theodore Suh, who will also oversee the independent peer review of the Journal’s supplements. We will further expand our editorial board in the coming months as part of our continuing effort to offer readers a balance of expertise across medical specialties.
Theodore Suh, MD, PhD, MHScExciting changes to our Web site, www.ccjm.org, will include links to activities offered by Cleveland Clinic’s Center for Continuing Medical Education and Center for Consumer Health Information. We will also continue to amplify the educational value of our online CME test and enhance the interactivity of this and other online offerings. Dr. Gilligan will play a prominent role in these upgrades.
Also, the Journal joins an expanding movement in medical journalism by screening all submitted manuscripts with a new online service called CrossCheck to detect plagiarism—whether self-plagiarism or traditional plagiarism. In this way, we will do our part to uphold the integrity of the medical literature.
And as always, I urge you to continue to submit your ideas for articles, particularly ideas for 1-Minute Consults. We review and value all of your suggestions.
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
Figure 1. The patient’s electrocardiogram on admission. See text for interpretation.On physical examination, he is a physically fit man in obvious distress. His jugular veins are not distended, and no lymph nodes are palpable in his neck. The heart sounds are muffled without murmurs, but a faint pericardial friction rub is heard that persists even when he holds his breath. His lungs are clear to auscultation, his abdomen is normal, and his lower extremities are warm, with normal pulses and no edema. Of note, neither a Kussmaul sign nor a paradoxical pulse is present. An electrocardiogram is ordered (Figure 1).
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
He goes on to add that his chest pain has recently increased in frequency, and it has occurred daily for the past 5 days. The pain is not related to exertion, occurs throughout the day, and is associated with significant shortness of breath. It worsens when he is taking a deep breath and improves when he leans forward. Although he is febrile, he says he has had no fevers or chills in the past. He gives no history of weight loss, cough, orthopnea, or paroxysmal nocturnal dyspnea, but has been experiencing malaise, weakness, and myalgia for the past month. His review of systems is otherwise negative.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
Acute myocardial infarction
Acute pericarditis
Myocarditis
Pulmonary embolism
Aortic dissection
Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
ST-segment elevation that is concave upward, occurring in all leads except aVR
T waves concordant with ST-segment deviation
PR-segment depression, sparing V1 and aVR
PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
Idiopathic
Neoplasm
Autoimmune
Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.
A more recent study in 453 patients revealed similar results: 377 (83.2%) of the cases were idiopathic, 23 (5.1%) were neoplastic, 17 (3.8%) were due to tuberculosis, 33 (7.3%) were autoimmune, and 3 (0.7%) were purulent. Of note, viral causes are categorized as idiopathic, since the diagnostic workup is usually unsuccessful and treatment is empiric6; therefore, most of the so-called idiopathic cases are likely viral. Table 2 summarizes the most common specific causes of acute pericarditis.
CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
Idiopathic
Infection
Malignancy
Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
Steroids
A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
Opioids
Colchicine
Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Figure 2. Magnetic resonance imaging shows multiple lesions in the liver.To our surprise, the MRI reveals innumerable hepatic lesions, some of which show radiographic features consistent with hemangiomas, while the remainder are atypical and appear to warrant a biopsy (Figure 2). An oncology consultation is obtained and the need for biopsy is confirmed.
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
Figure 3. Echocardiogram, four-chamber view, showing the tumor (crosshairs) in the right atrium (RA). LA = left atrium, LV = left ventricle, RV = right ventricle.To our astonishment, the echocardiogram reveals a mass in the right atrial free wall and right ventricle that appears to be invading the myocardial tissue (Figure 3).
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
Figure 4. Cardiac MRI; arrow points to the tumor. RA = right atrium, RV = right ventricle, LA = left atrium, LV = left ventricle.Cardiac MRI is performed, which confirms the finding and characterizes the mass as measuring 5.1 by 4.8 cm within the pericardial space adjacent to the right atrium and atrioventricular groove and adherent to the right atrium. There are small excrescences of soft tissue through the midportion of the right atrial wall, suggesting tissue invasion (Figure 4).
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
Myxoma
Papillary fibroelastoma
Sarcoma
Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
Figure 5. Liver biopsy shows cellular endothelial atypia with mitotic activity (black arrow), growth along the hepatic sinusoids, and papillary tufting (white arrow), all features of low-grade angiosarcoma.Fluorodeoxyglucose (FDG) positron-emission tomography is done, and shows a hypermetabolic right-sided pericardial tumor in addition to several suspicious hepatic lesions with heterogeneously increased FDG uptake. Biopsy with ultrasonographic guidance is performed again and reveals tissue consistent with hemangioma in addition to other areas with features strongly suggestive of a low-grade angiosarcoma (Figure 5). Pathology findings are unable to differentiate primary cardiac angiosarcoma from a metastatic cardiac tumor; however, given the multiple liver lesions and the presence of a solitary cardiac mass, this is most likely a primary cardiac tumor with metastasis to the liver.
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
References
Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev2007; 15:24–30.
LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J1986; 111:546–552.
Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol2001; 87:1326–1328.
Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol2006; 48:1–11.
Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol1995; 75:378–382.
Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation2007; 115:2739–2744.
Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore)2003; 82:385–391.
Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med2004; 351:2195–2202.
Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation2005; 112:2012–2016.
Heath D. Pathology of cardiac tumors. Am J Cardiol1968; 21:315–327.
Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol1980; 101:219–240.
Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J2003; 146:404–410.
Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol1968; 21:413–419.
Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer1997; 80:1497–1506.
Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest1987; 92:860–862.
Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer1998; 78:1624–1628.
Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg1991; 51:906–910.
Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J2002; 29:105–108.
Arash Aghel, MD Department of Internal Medicine, Cleveland Clinic
Richard A. Krasuski, MD Department of Cardiovascular Medicine, Section of Clinical Cardiology, Cleveland Clinic
Address: Richard Krasuski, MD, Department of Cardiovascular Medicine, J2-4, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Krasuski has disclosed that he has received honoraria from Pfizer for teaching and speaking and honoraria and consulting fees from Actelion for teaching, speaking, and consultation.
Arash Aghel, MD Department of Internal Medicine, Cleveland Clinic
Richard A. Krasuski, MD Department of Cardiovascular Medicine, Section of Clinical Cardiology, Cleveland Clinic
Address: Richard Krasuski, MD, Department of Cardiovascular Medicine, J2-4, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Krasuski has disclosed that he has received honoraria from Pfizer for teaching and speaking and honoraria and consulting fees from Actelion for teaching, speaking, and consultation.
Author and Disclosure Information
Arash Aghel, MD Department of Internal Medicine, Cleveland Clinic
Richard A. Krasuski, MD Department of Cardiovascular Medicine, Section of Clinical Cardiology, Cleveland Clinic
Address: Richard Krasuski, MD, Department of Cardiovascular Medicine, J2-4, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Krasuski has disclosed that he has received honoraria from Pfizer for teaching and speaking and honoraria and consulting fees from Actelion for teaching, speaking, and consultation.
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
Figure 1. The patient’s electrocardiogram on admission. See text for interpretation.On physical examination, he is a physically fit man in obvious distress. His jugular veins are not distended, and no lymph nodes are palpable in his neck. The heart sounds are muffled without murmurs, but a faint pericardial friction rub is heard that persists even when he holds his breath. His lungs are clear to auscultation, his abdomen is normal, and his lower extremities are warm, with normal pulses and no edema. Of note, neither a Kussmaul sign nor a paradoxical pulse is present. An electrocardiogram is ordered (Figure 1).
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
He goes on to add that his chest pain has recently increased in frequency, and it has occurred daily for the past 5 days. The pain is not related to exertion, occurs throughout the day, and is associated with significant shortness of breath. It worsens when he is taking a deep breath and improves when he leans forward. Although he is febrile, he says he has had no fevers or chills in the past. He gives no history of weight loss, cough, orthopnea, or paroxysmal nocturnal dyspnea, but has been experiencing malaise, weakness, and myalgia for the past month. His review of systems is otherwise negative.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
Acute myocardial infarction
Acute pericarditis
Myocarditis
Pulmonary embolism
Aortic dissection
Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
ST-segment elevation that is concave upward, occurring in all leads except aVR
T waves concordant with ST-segment deviation
PR-segment depression, sparing V1 and aVR
PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
Idiopathic
Neoplasm
Autoimmune
Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.
A more recent study in 453 patients revealed similar results: 377 (83.2%) of the cases were idiopathic, 23 (5.1%) were neoplastic, 17 (3.8%) were due to tuberculosis, 33 (7.3%) were autoimmune, and 3 (0.7%) were purulent. Of note, viral causes are categorized as idiopathic, since the diagnostic workup is usually unsuccessful and treatment is empiric6; therefore, most of the so-called idiopathic cases are likely viral. Table 2 summarizes the most common specific causes of acute pericarditis.
CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
Idiopathic
Infection
Malignancy
Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
Steroids
A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
Opioids
Colchicine
Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Figure 2. Magnetic resonance imaging shows multiple lesions in the liver.To our surprise, the MRI reveals innumerable hepatic lesions, some of which show radiographic features consistent with hemangiomas, while the remainder are atypical and appear to warrant a biopsy (Figure 2). An oncology consultation is obtained and the need for biopsy is confirmed.
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
Figure 3. Echocardiogram, four-chamber view, showing the tumor (crosshairs) in the right atrium (RA). LA = left atrium, LV = left ventricle, RV = right ventricle.To our astonishment, the echocardiogram reveals a mass in the right atrial free wall and right ventricle that appears to be invading the myocardial tissue (Figure 3).
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
Figure 4. Cardiac MRI; arrow points to the tumor. RA = right atrium, RV = right ventricle, LA = left atrium, LV = left ventricle.Cardiac MRI is performed, which confirms the finding and characterizes the mass as measuring 5.1 by 4.8 cm within the pericardial space adjacent to the right atrium and atrioventricular groove and adherent to the right atrium. There are small excrescences of soft tissue through the midportion of the right atrial wall, suggesting tissue invasion (Figure 4).
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
Myxoma
Papillary fibroelastoma
Sarcoma
Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
Figure 5. Liver biopsy shows cellular endothelial atypia with mitotic activity (black arrow), growth along the hepatic sinusoids, and papillary tufting (white arrow), all features of low-grade angiosarcoma.Fluorodeoxyglucose (FDG) positron-emission tomography is done, and shows a hypermetabolic right-sided pericardial tumor in addition to several suspicious hepatic lesions with heterogeneously increased FDG uptake. Biopsy with ultrasonographic guidance is performed again and reveals tissue consistent with hemangioma in addition to other areas with features strongly suggestive of a low-grade angiosarcoma (Figure 5). Pathology findings are unable to differentiate primary cardiac angiosarcoma from a metastatic cardiac tumor; however, given the multiple liver lesions and the presence of a solitary cardiac mass, this is most likely a primary cardiac tumor with metastasis to the liver.
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
Figure 1. The patient’s electrocardiogram on admission. See text for interpretation.On physical examination, he is a physically fit man in obvious distress. His jugular veins are not distended, and no lymph nodes are palpable in his neck. The heart sounds are muffled without murmurs, but a faint pericardial friction rub is heard that persists even when he holds his breath. His lungs are clear to auscultation, his abdomen is normal, and his lower extremities are warm, with normal pulses and no edema. Of note, neither a Kussmaul sign nor a paradoxical pulse is present. An electrocardiogram is ordered (Figure 1).
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
He goes on to add that his chest pain has recently increased in frequency, and it has occurred daily for the past 5 days. The pain is not related to exertion, occurs throughout the day, and is associated with significant shortness of breath. It worsens when he is taking a deep breath and improves when he leans forward. Although he is febrile, he says he has had no fevers or chills in the past. He gives no history of weight loss, cough, orthopnea, or paroxysmal nocturnal dyspnea, but has been experiencing malaise, weakness, and myalgia for the past month. His review of systems is otherwise negative.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
Acute myocardial infarction
Acute pericarditis
Myocarditis
Pulmonary embolism
Aortic dissection
Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
ST-segment elevation that is concave upward, occurring in all leads except aVR
T waves concordant with ST-segment deviation
PR-segment depression, sparing V1 and aVR
PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
Idiopathic
Neoplasm
Autoimmune
Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.
A more recent study in 453 patients revealed similar results: 377 (83.2%) of the cases were idiopathic, 23 (5.1%) were neoplastic, 17 (3.8%) were due to tuberculosis, 33 (7.3%) were autoimmune, and 3 (0.7%) were purulent. Of note, viral causes are categorized as idiopathic, since the diagnostic workup is usually unsuccessful and treatment is empiric6; therefore, most of the so-called idiopathic cases are likely viral. Table 2 summarizes the most common specific causes of acute pericarditis.
CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
Idiopathic
Infection
Malignancy
Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
Steroids
A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
Opioids
Colchicine
Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Figure 2. Magnetic resonance imaging shows multiple lesions in the liver.To our surprise, the MRI reveals innumerable hepatic lesions, some of which show radiographic features consistent with hemangiomas, while the remainder are atypical and appear to warrant a biopsy (Figure 2). An oncology consultation is obtained and the need for biopsy is confirmed.
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
Figure 3. Echocardiogram, four-chamber view, showing the tumor (crosshairs) in the right atrium (RA). LA = left atrium, LV = left ventricle, RV = right ventricle.To our astonishment, the echocardiogram reveals a mass in the right atrial free wall and right ventricle that appears to be invading the myocardial tissue (Figure 3).
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
Figure 4. Cardiac MRI; arrow points to the tumor. RA = right atrium, RV = right ventricle, LA = left atrium, LV = left ventricle.Cardiac MRI is performed, which confirms the finding and characterizes the mass as measuring 5.1 by 4.8 cm within the pericardial space adjacent to the right atrium and atrioventricular groove and adherent to the right atrium. There are small excrescences of soft tissue through the midportion of the right atrial wall, suggesting tissue invasion (Figure 4).
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
Myxoma
Papillary fibroelastoma
Sarcoma
Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
Figure 5. Liver biopsy shows cellular endothelial atypia with mitotic activity (black arrow), growth along the hepatic sinusoids, and papillary tufting (white arrow), all features of low-grade angiosarcoma.Fluorodeoxyglucose (FDG) positron-emission tomography is done, and shows a hypermetabolic right-sided pericardial tumor in addition to several suspicious hepatic lesions with heterogeneously increased FDG uptake. Biopsy with ultrasonographic guidance is performed again and reveals tissue consistent with hemangioma in addition to other areas with features strongly suggestive of a low-grade angiosarcoma (Figure 5). Pathology findings are unable to differentiate primary cardiac angiosarcoma from a metastatic cardiac tumor; however, given the multiple liver lesions and the presence of a solitary cardiac mass, this is most likely a primary cardiac tumor with metastasis to the liver.
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
References
Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev2007; 15:24–30.
LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J1986; 111:546–552.
Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol2001; 87:1326–1328.
Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol2006; 48:1–11.
Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol1995; 75:378–382.
Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation2007; 115:2739–2744.
Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore)2003; 82:385–391.
Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med2004; 351:2195–2202.
Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation2005; 112:2012–2016.
Heath D. Pathology of cardiac tumors. Am J Cardiol1968; 21:315–327.
Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol1980; 101:219–240.
Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J2003; 146:404–410.
Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol1968; 21:413–419.
Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer1997; 80:1497–1506.
Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest1987; 92:860–862.
Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer1998; 78:1624–1628.
Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg1991; 51:906–910.
Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J2002; 29:105–108.
References
Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev2007; 15:24–30.
LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J1986; 111:546–552.
Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol2001; 87:1326–1328.
Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol2006; 48:1–11.
Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol1995; 75:378–382.
Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation2007; 115:2739–2744.
Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore)2003; 82:385–391.
Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med2004; 351:2195–2202.
Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation2005; 112:2012–2016.
Heath D. Pathology of cardiac tumors. Am J Cardiol1968; 21:315–327.
Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol1980; 101:219–240.
Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J2003; 146:404–410.
Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol1968; 21:413–419.
Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer1997; 80:1497–1506.
Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest1987; 92:860–862.
Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer1998; 78:1624–1628.
Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg1991; 51:906–910.
Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J2002; 29:105–108.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
Hemoglobin 15.4 g/dL (12.0–16.0)
Platelet count 122 × 109/L (150–400)
International normalized ratio (INR) 1.1 (0.9–1.1)
Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
Atherosclerosis
Protein C deficiency
Use of oral contraceptive pills
The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
Yes
No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
Antithrombin levels
Protein C and S levels
Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
Antiphospholipid antibody syndrome
Heparin-induced thrombocytopenia
Malignancy
All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
The hypercoagulability associated with malignancy is also known as Trousseau syndrome. This term was originally used to describe migratory thrombophlebitis as a forewarning for occult visceral malignancy, and has grown over the years to describe malignancy-induced hypercoagulability.9
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
As stated above, the patient has a normal platelet count and negative results on cancer screening tests. Tests for antiphospholipid antibodies and lupus anticoagulant are repeated. Tests for the specific antiphospholipid antibodies against beta-2 glycoprotein I and cardiolipin are negative (Table 2). However, the test for lupus anticoagulant is positive by the criteria of the International Society on Thrombosis and Haemostasis: the patient has a prolonged clotting time screening test (hexagonal phase screen, DRVVT screen), positive mixing study (DRVVT 1:1 mix and circulating anticoagulant), positive phospholipid dependence (hexagonal phase screen, confirm, and delta; DRVVT confirm ratio; and platelet neutralization procedure), and no evidence of other factor-specific inhibitors (Table 3).14
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
Yes
No
Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
Lupus anticoagulant present
Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
Positron emission tomography and tumor marker levels
A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
True
False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
References
Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med2002; 346:752–763.
Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis1999; 42:91–138.
Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med2001; 135:367–373.
Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest2004; 126suppl 3:401S–428S.
Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract2003; 52:770–777.
Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol2001; 114:512–528.
Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood2007; 110:1723–1729.
Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci1974; 230:262–270.
Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res1983; 43:3963–3968.
Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry1985; 24:5558–5567.
Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res2001; 61:795–798.
Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost1995; 74:1597–1603.
Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost2006; 4:295–306.
Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med1996; 125:785–793.
Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost2004; 2:884–889.
Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol2007; 25:5490–5505.
Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med2003; 349:146–153.
Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med2006; 119:1062–1072.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
Hemoglobin 15.4 g/dL (12.0–16.0)
Platelet count 122 × 109/L (150–400)
International normalized ratio (INR) 1.1 (0.9–1.1)
Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
Atherosclerosis
Protein C deficiency
Use of oral contraceptive pills
The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
Yes
No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
Antithrombin levels
Protein C and S levels
Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
Antiphospholipid antibody syndrome
Heparin-induced thrombocytopenia
Malignancy
All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
The hypercoagulability associated with malignancy is also known as Trousseau syndrome. This term was originally used to describe migratory thrombophlebitis as a forewarning for occult visceral malignancy, and has grown over the years to describe malignancy-induced hypercoagulability.9
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
As stated above, the patient has a normal platelet count and negative results on cancer screening tests. Tests for antiphospholipid antibodies and lupus anticoagulant are repeated. Tests for the specific antiphospholipid antibodies against beta-2 glycoprotein I and cardiolipin are negative (Table 2). However, the test for lupus anticoagulant is positive by the criteria of the International Society on Thrombosis and Haemostasis: the patient has a prolonged clotting time screening test (hexagonal phase screen, DRVVT screen), positive mixing study (DRVVT 1:1 mix and circulating anticoagulant), positive phospholipid dependence (hexagonal phase screen, confirm, and delta; DRVVT confirm ratio; and platelet neutralization procedure), and no evidence of other factor-specific inhibitors (Table 3).14
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
Yes
No
Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
Lupus anticoagulant present
Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
Positron emission tomography and tumor marker levels
A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
True
False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
Hemoglobin 15.4 g/dL (12.0–16.0)
Platelet count 122 × 109/L (150–400)
International normalized ratio (INR) 1.1 (0.9–1.1)
Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
Atherosclerosis
Protein C deficiency
Use of oral contraceptive pills
The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
Yes
No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
Antithrombin levels
Protein C and S levels
Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
Antiphospholipid antibody syndrome
Heparin-induced thrombocytopenia
Malignancy
All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
The hypercoagulability associated with malignancy is also known as Trousseau syndrome. This term was originally used to describe migratory thrombophlebitis as a forewarning for occult visceral malignancy, and has grown over the years to describe malignancy-induced hypercoagulability.9
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
As stated above, the patient has a normal platelet count and negative results on cancer screening tests. Tests for antiphospholipid antibodies and lupus anticoagulant are repeated. Tests for the specific antiphospholipid antibodies against beta-2 glycoprotein I and cardiolipin are negative (Table 2). However, the test for lupus anticoagulant is positive by the criteria of the International Society on Thrombosis and Haemostasis: the patient has a prolonged clotting time screening test (hexagonal phase screen, DRVVT screen), positive mixing study (DRVVT 1:1 mix and circulating anticoagulant), positive phospholipid dependence (hexagonal phase screen, confirm, and delta; DRVVT confirm ratio; and platelet neutralization procedure), and no evidence of other factor-specific inhibitors (Table 3).14
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
Yes
No
Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
Lupus anticoagulant present
Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
Positron emission tomography and tumor marker levels
A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
True
False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
References
Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med2002; 346:752–763.
Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis1999; 42:91–138.
Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med2001; 135:367–373.
Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest2004; 126suppl 3:401S–428S.
Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract2003; 52:770–777.
Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol2001; 114:512–528.
Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood2007; 110:1723–1729.
Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci1974; 230:262–270.
Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res1983; 43:3963–3968.
Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry1985; 24:5558–5567.
Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res2001; 61:795–798.
Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost1995; 74:1597–1603.
Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost2006; 4:295–306.
Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med1996; 125:785–793.
Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost2004; 2:884–889.
Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol2007; 25:5490–5505.
Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med2003; 349:146–153.
Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med2006; 119:1062–1072.
References
Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med2002; 346:752–763.
Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis1999; 42:91–138.
Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med2001; 135:367–373.
Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest2004; 126suppl 3:401S–428S.
Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract2003; 52:770–777.
Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol2001; 114:512–528.
Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood2007; 110:1723–1729.
Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci1974; 230:262–270.
Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res1983; 43:3963–3968.
Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry1985; 24:5558–5567.
Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res2001; 61:795–798.
Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost1995; 74:1597–1603.
Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost2006; 4:295–306.
Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med1996; 125:785–793.
Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost2004; 2:884–889.
Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol2007; 25:5490–5505.
Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med2003; 349:146–153.
Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med2006; 119:1062–1072.
Two days ago, a 27-year-old woman noticed that her vision was blurry in her right eye. She has come to see her primary care physician for advice. This is the first time this has happened to her. She describes seeing a grayish blur over the center of her vision, but she has not noted any other symptoms except for some soreness around the right eye, which is worse with eye movements.
How should she be assessed and treated?
IMPORTANT TO RECOGNIZE
Sudden vision loss is one of the more common problems encountered in ophthalmology and neurology.
Optic neuritis, a demyelinating inflammatory condition that causes acute vision loss, is associated with multiple sclerosis (MS), and recognizing its classic clinical manifestations early is important so that appropriate diagnostic testing (magnetic resonance imaging [MRI]) and treatment (corticosteroids and immunomodulators) can be started.
Although a comprehensive review of all the optic neuropathies is beyond the scope of this paper, in the pages that follow we review some of the most common causes, which may be first seen by the general internist.
FOUR SUBTYPES OF OPTIC NEURITIS
There are four subtypes of optic neuritis:
Figure 1. In retrobulbar optic neuritis, the inflammation and demyelination occur behind the globe of the eye. The optic disc appears normal with no signs of swelling or pallor.Retrobulbar neuritis (Figure 1), or inflammation of the optic nerve behind the eye, is the form most commonly associated with MS.
Papillitis (Figure 2), or inflammation of the optic disc, can also be associated with MS.
Perineuritis is inflammation of the optic nerve sheath, sparing the optic nerve itself. Usually, patients are older, and vision loss is mild to moderate. Perineuritis is commonly due to infectious or inflammatory conditions, eg, syphilis or sarcoidosis.
Figure 2. In papillitis, mild swelling and elevation of the optic disc can be seen. The small splinter hemorrhage seen at 10 o’clock is not typical of optic neuritis associated with multiple sclerosis.Neuroretinitis may occur at any age. There is concomitant swelling of the optic nerve and macula. Exudates that form around the macula give the appearance of a star.
Perineuritis and neuroretinitis are not associated with MS, and if they are found they suggest another etiology. In the rest of this review, “optic neuritis” means retrobulbar optic neuritis, the form most commonly seen in clinical practice.
MOST COMMON IN YOUNG WOMEN
Acute demyelinating optic neuritis most often affects women in their 20s and 30s.1–3 Studies in the United States have estimated its annual incidence to be 5.1 to 6.4 per 100,000.4,5 The incidence is higher in populations at higher latitudes and lower near the equator. It is less common in blacks than in whites.6
In children, optic neuritis is not as strongly associated with MS, especially when there is optic disc swelling or bilateral involvement. Most children have a good visual outcome, although approximately 20% may be visually disabled.7–9
FEATURES: VISION LOSS AND EYE PAIN
Most of our current knowledge of the clinical features of optic neuritis comes from the Optic Neuritis Treatment Trial (ONTT),10 conducted in the 1990s. This trial enrolled 457 patients 18 to 46 years old who had acute unilateral optic neuritis. The patients had to have symptoms consistent with acute unilateral optic neuritis for 8 days or less. They could not have evidence of any systemic disease (except for MS) or have received prior treatment for MS. Therefore, this study was quite representative of the patients with optic neuritis that one might encounter in the clinic and is highly important in both characterizing optic neuritis and defining its treatment.
The study found that the two most common symptoms are vision loss and eye pain.
Vision loss in optic neuritis typically occurs over several hours to days, and vision reaches a nadir within 1 to 2 weeks. Typically, patients begin to recover 2 to 4 weeks after the onset of the vision loss. The optic nerve may take up to 6 to 12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.11 More than two-thirds of patients have at least 20/20 vision once they have fully recovered from the optic neuritis. Only 3% of patients become completely blind.
Eye pain is very common in optic neuritis (it affected 87% of patients in the ONTT) and typically worsens with eye movement. The eye is also sore to touch. The pain generally begins at the same time as the visual loss and improves along with it. Eye movements also may bring about photopsia (flickering or flashes of light), a symptom reported by 30% of the ONTT participants.
Loss of color vision out of proportion to the loss of visual acuity is characteristic of optic neuropathies. In the ONTT, 88% of the involved eyes had abnormal color vision as assessed by the Ishihara test (using pseudoisochromatic plates), and 94% as assessed by the Farnsworth-Munsell 100 hue test, which is more sensitive but cumbersome. The most common patterns of color vision loss in optic nerve disease are loss of red (protanopia) and green (deutranopia).
A good way to screen for loss of color vision is to test for color desaturation. First, ask the patient to fixate with the normal eye on a bright red object (for example, the cap from a bottle of ophthalmic mydriatic drops or a pen cap). Then ask the patient to compare the intensity of the redness between the good eye and the affected eye. The patient can quantify the color desaturation by rating what percentage of red is lost in the affected eye compared with the normal eye.
Temporary exacerbations of visual problems during fever (the Uhthoff phenomenon) can occur in patients who have had optic neuritis. These transient pseudoexacerbations are not new episodes of optic neuritis and should resolve after the body temperature returns to normal.
A relative afferent pupillary defect should be seen in the involved eye in all patients with optic neuritis if the other eye is uninvolved and healthy.12 The best way to elicit this sign is to perform the swinging light test in a dark room with the patient fixating at a distant target, which prevents miosis due to accommodation. When the light is swung to the involved eye, the pupil dilates because less light stimulus reaches the midbrain through the affected optic nerve. As the optic nerve heals and recovers, this sign may become subtle, but it persists in more than 90% of cases.12
Findings on funduscopy
Examination of the fundus is helpful in the clinical diagnosis of optic neuritis.
Two-thirds of the ONTT patients had retrobulbar neuritis, and one-third had papillitis. If optic nerve swelling is present, it is typically mild.
Peripapillary hemorrhages were exceedingly rare in the cases of papillitis (only 6%) and were associated with a very low to zero risk of developing MS. If peripapillary hemorrhages are found on examination, one should consider another diagnosis, such as anterior ischemic optic neuropathy.11
CASE CONTINUED
Our patient undergoes a neurologic examination, which reveals an afferent pupillary defect in the right eye and visual acuity of 20/100 in the right eye and 20/20 in the left. Visual fields are normal in the left eye as assessed by confrontation, but there is a central scotoma in the right.
OTHER TYPES OF NEUROPATHY
Optic neuritis is a clinical diagnosis based on the history and findings on examination. If the patient does not have its typical features (Table 1), then other diagnoses should be pursued with serologic and cerebrospinal fluid studies.
The following should be included in the differential diagnosis of optic neuritis:
Ischemic optic neuropathy
Ischemic optic neuropathy is more common in patients age 50 and older, whereas optic neuritis is more common in younger patients. Most patients with ischemic optic neuropathy have hypertension, hypercholesterolemia, diabetes mellitus, obstructive sleep apnea, or other vascular risk factors. The disease has several important subtypes, as discussed below.
Figure 3. Left, fundus photo several weeks after an attack of nonarteritic anterior ischemic optic neuropathy demonstrating pallor of the superior half of the disc. Middle, an associated inferior altitudinal defect. Right, sectoral swelling of the disc with flame or splinter hemorrhages is commonly seen. This is not typical of demyelinating optic neuritis.Nonarteritic anterior ischemic optic neuropathy is the most common form. Typically, there is acute onset of painless vision loss in one eye and an associated altitudinal field defect (Figure 3). For example, if the superior rim of the optic nerve acutely develops swelling and then becomes pale, a corresponding altitudinal cut would develop in the inferior visual field, respecting the horizontal meridian. Many patients first notice the vision loss upon waking up in the morning.13
Although patients with nonarteritic anterior ischemic optic neuropathy typically have vasculopathic risk factors such as hypertension, diabetes mellitus, peripheral vascular disease, or hypercholesterolemia, there is no proven causation between the two. The age of these patients ranges from 50 to 70, with an average age of 66.
The disc appears swollen and may have flame or splinter hemorrhages (Figure 3). The cup of the involved disc is typically small. The visual loss is believed to be the result of poor perfusion in the circulation of the posterior ciliary artery, which supplies the optic nerve head.1 If the other eye also has a small cup, it is considered to be at risk of future ischemic events. In one study,14 the opposite eye became involved within the next 5 years in 14.7% of all cases. The risk of recurrent disease in the same eye is low (6.4% in another study15).
Arteritic anterior ischemic optic neuropathy is more common in patients over age 70 and is usually due to giant cell arteritis, which has a significant association with polymyalgia rheumatica. Patients may have jaw claudication, proximal myalgia and arthralgia, scalp tenderness, headache, fatigue, and a significantly elevated erythrocyte sedimentation rate and C-reactive protein level. These features should be looked for in the review of systems, although patients may not have all of them.
The funduscopic examination may reveal a pale, swollen disc, peripapillary hemorrhages, branch or central retinal artery occlusions, or cotton-wool spots.
Temporal artery biopsy is the gold standard for diagnosis, but treatment with corticosteroids should not be delayed pending biopsy or other test results.1
Thrombocytosis has been associated with a higher risk of permanent vision loss in patients with giant cell arteritis.16
Posterior ischemic optic neuropathy is the least common form of ischemic optic neuropathy. This diagnosis should be entertained in older patients who report acute, painless vision loss but have a normal funduscopic examination. Giant cell arteritis must be considered first in this setting.
Bilateral posterior ischemic optic neuropathy can occur (although rarely) in patients undergoing cardiac or spinal surgery.17 Risk factors thought to be associated with perioperative disease include anemia, hypotension, substantial blood loss during the surgery, surgeries longer than 6.5 hours, carotid atherosclerosis, and diabetes.18
There are no effective treatments for most ischemic optic neuropathies with the crucial exception of giant cell arteritis.
Neuromyelitis optica (Devic disease)
Neuromyelitis optica (Devic disease) is a combination of optic neuritis and transverse myelitis (Table 2). Clinically, the disease spares the nervous system except for the optic nerves and spinal cord. The onset of the optic neuritis may precede or follow the onset of the transverse myelitis by up to 2 to 4 years.19 Usually, the optic neuritis is bilateral and retrobulbar and results in severe vision loss, worse than that seen in patients with MS.19,20
The transverse myelitis may cause paraplegia or quadriplegia, depending on the location of the lesion in the spinal cord (cervical vs thoracic). The transverse myelitis in neuromyelitis optica is distinct from that seen in MS. In neuromyelitis optica, the transverse myelitis is longitudinally extensive, spanning more than three vertebral bodies in length. In MS, spinal cord lesions usually are more discrete and involve one or two spinal cord segments.21
Recently, serum neuromyelitis optica immunoglobulin G (IgG) antibody has been shown to be a significant biomarker of this disease. Its sensitivity ranges from approximately 60% to 70% and its specificity is greater than 90%.22 This antibody binds to aquaporin-4, an important water-channel protein in the blood-brain barrier of the central nervous system, and evidence suggests that it is involved in the pathogenesis of the disease.23
Initially, it was proposed that MRI of the brain had to be normal for neuromyelitis optica to be diagnosed.21 However, the proposed 2006 criteria allow for some abnormal T2 and fluid-attenuated inversion recovery (FLAIR) hyperintensities in the periaqueductal gray matter and diencephalon.22
The spinal fluid in neuromyelitis optica may show a pleocytosis larger than that seen in MS (> 50 white blood cells per mm3) and may have a significant neutrophilic component.21 Oligoclonal bands are not typically present.
It is still debated whether neuromyelitis optica is a separate disease from MS or a subset of it. The implications of this debate may affect its management, as discussed below.
Inflammatory optic neuropathies
Inflammatory optic neuropathies can be caused by infections (eg, syphilis, cat scratch disease) or by noninfectious conditions (eg, sarcoidosis). Lyme disease is rarely a cause of retrobulbar optic neuritis but may cause papillitis.24 West Nile virus has also been reported to cause optic neuritis.25 Lupus may cause an optic neuropathy by inflammatory or ischemic mechanisms.26
Compressive optic neuropathies
Compressive optic neuropathies may be due to mass lesions, tumors, thyroid eye disease, or other orbital processes. MRI of the brain and orbits will confirm or rule out diagnoses associated with compressive optic neuropathy.
Genetic causes
Genetic causes of optic neuropathy include the Leber and Kjer hereditary optic neuropathies.
Leber optic neuropathy involves subacute and painless vision loss, with sequential involvement of both eyes over a period of weeks to months. This disorder predominantly affects men (80%–90% of patients) and is inherited from maternal mitochondrial DNA. The three most common mutations implicated in Leber optic neuropathy (located at base pairs 11,778, 3,460, and 14,484 in the mitochondrial DNA) are involved in more than 90% of cases. The prognosis for recovery varies depending on the genotype.27 These genes encode proteins that are part of complex I of the mitochondrial respiratory chain.28 Funduscopic examination most commonly shows circumpapillary telangiectasia, although up to one-third of patients can have a normal-appearing disc initially. Central vision is affected more severely than peripheral vision.29
Kjer autosomal-dominant optic atrophy is the most common hereditary optic neuropathy. This disease primarily affects children in the first decade of life with slowly progressive loss of vision. As with other optic neuropathies, there will eventually be pallor of the optic disc, a cecocentral scotoma, and loss of color perception. The OPA1 gene located on chromosome 3q28 has been implicated in most patients with dominant optic atrophy; a test is commercially available for diagnosis.30,31
Toxic and metabolic causes
Many agents can cause optic neuropathy. Toxins strongly associated with optic neuropathy include carbon monoxide, methanol, ethylene glycol, perchloroethylene, and tobacco. Drugs linked to optic neuropathy are ethambutol (Myambutol), clioquinol (Vioform), isoniazid (Nydrazid), amiodarone (Cordarone), linezolid (Zyvox), methotrexate, sildenafil (Viagra), oxymetazoline (contained in Afrin and other nasal sprays), and infliximab (Remicade).32–37 Additionally, several chemotherapeutic agents are known to cause optic atrophy, including vincristine (Oncovin), cisplatin (Platinol), carboplatin (Paraplatin), and paclitaxel (Abraxane, Onxol).
Nutritional deficiencies are presumed to have played a significant role in the endemics of optic neuropathy that have occurred in poor countries, such as in Cuba during the 1990s.38 Most nutritional optic neuropathies appear to be exacerbated by tobacco.39
MRI ASSESSES RISK OF MS
The diagnosis of optic neuritis is clinical, based on the history and physical findings.
However, MRI of the brain and orbits with gadolinium contrast has become the cornerstone of the evaluation in patients with optic neuritis. And MRI not only helps confirm the clinical diagnosis, but it also more importantly offers very strong prognostic information about the risk of future demyelinating events and MS.
Gadolinium-enhanced fat-saturated T1-weighted MRI of the orbits is the best sequence to show the inflammation of the optic nerve in optic neuritis (fat saturation is necessary to hide the bright signal of the orbital fat tissue).
Contrast-enhanced MRI can also help differentiate optic neuritis from nonarteritic anterior ischemic optic neuropathy. MRI of the orbits with gadolinium contrast shows enhancement of the affected optic nerve in approximately 95% of cases of optic neuritis, whereas optic nerve enhancement rarely occurs in nonarteritic anterior ischemic optic neuropathy.40
Brain MRI may show other white matter lesions (either hyperintensities on T2-weighted images or enhancement of T1-weighted images postcontrast), which denote a higher risk of developing MS. In 15-year follow-up data from the ONTT, monosymptomatic patients with no white matter lesions had a 25% risk of MS (defined at the time the ONTT was conducted as a second demyelinating event), while those with one lesion or more had a 72% risk.41
An earlier, prospective study in 102 Italian patients with optic neuritis found the risk of developing MS to be about 36% at 6 years and 42% at 8 years (using the Posner diagnostic criteria). When brain MRI data were analyzed, those with one or more lesions had a 52% risk of developing MS at 8 years, whereas those with no MRI lesions did not develop MS.42
Other studies have stratified the risk of MS in patients with clinically isolated syndromes (including not only optic neuritis, but also other neurologic symptoms such as brainstem, motor, or sensory deficits). At mean follow-ups ranging from 5 to 14 years, the risk of developing MS was 8% to 24% in patients with normal findings on brain MRI compared with 56% to 88% in those with abnormal MRI findings.43,44
Optic neuritis patients with atypical white matter lesions on brain MRI may benefit from lumbar puncture to look for oligoclonal bands, to measure the IgG index and the IgG synthesis rate, and to test for myelin basic protein in the cerebrospinal fluid. Of patients with acute optic neuritis, 79% have abnormalities in their cerebrospinal fluid. Oligoclonal bands are present in 69%, and for patients with oligoclonal bands, the 5-year probability of developing MS is estimated to be 65%, compared with 10% in those without bands. If the patient has no oligoclonal bands and has normal findings on brain MRI, he or she will not have MS 5 years later.45–47
Patients with optic neuritis who have no white matter lesions on brain MRI may be further risk-stratified on the basis of their clinical findings. In the ONTT 15-year follow-up, MS did not develop in any patient who had no brain lesions on baseline MRI, no prior optic neuritis in the contralateral eye, and no prior neurologic symptoms or signs, even if the patient had severe disc swelling (eg, peripapillary hemorrhage or retinal exudates) or if vision was reduced to no light perception.41
CASE CONTINUED: FINDINGS ON MRI
Figure 4. The patient’s magnetic resonance image. Top, an axial T2 image with contrast; bottom, sagittal T1 image with contrast. The white matter lesions indicate she is at risk of developing multiple sclerosis.Our patient undergoes MRI, which shows lesions on axial T2 and sagittal T1 imaging with contrast (Figure 4). Of note, there are significant lesions perpendicular to the corpus callosum (Dawson fingers), some of which enhance with contrast. The enhancement indicates breakdown of the blood-brain barrier and suggests that there is active inflammation in the white matter.
Patients in the ONTT were randomized to receive one of three treatments:
Oral prednisone 1 mg/kg/day for 14 days and then tapered over 4 days
Intravenous methylprednisolone (Solu-Medrol) 250 mg four times per day for 3 days followed by oral prednisone 1 mg/kg/day for 11 days and then tapered for 4 days
Oral placebo for 14 days.
The primary visual outcomes measured were visual acuity and contrast sensitivity.48
Patients who received intravenous methylprednisolone recovered their visual function faster, although the visual outcomes after 6 months were no better with methylprednisolone than with placebo or oral prednisone. Intravenous methylprednisolone also reduced the risk of MS within the first 2 years in patients with high-risk brain MRIs.
Surprisingly, patients in the oral prednisone group had a higher risk of recurrent optic neuritis in both eyes than did patients given intravenous methylprednisolone or placebo (30% at 2 years with oral prednisone vs 16% with placebo and 13% with intravenous methylprednisolone).48 At 10 years, the risk of recurrent optic neuritis was still higher in the oral prednisone group (44%) than in the intravenous methylprednisolone group (29%) (P = .03). However, the difference between the oral prednisone and placebo groups was no longer statistically significant (P = .07).49 Oral prednisone alone is therefore contraindicated in the treatment of typical unilateral demyelinating optic neuritis.
Many patients can now be treated with intravenous infusions of methylprednisolone at home for episodes of optic neuritis.
Risks vs benefits of corticosteroid therapy
When deciding whether to treat an individual patient who has optic neuritis with intravenous corticosteroids, one should consider all the benefits and risks.
Corticosteroids do not affect long-term visual outcome, although they do hasten recovery. Patients with mild vision loss (visual acuity better than 20/40), no significant visual field loss, and no enhancing lesions on brain MRI can forgo therapy with intravenous corticosteroids.
On the other hand, we strongly favor intravenous corticosteroid treatment in patients who have both acute optic neuritis and high signal abnormalities on brain MRI, since it may delay the onset of MS. In addition, patients with severe vision loss should receive intravenous corticosteroids to hasten their recovery. In the rare circumstance in which intravenous corticosteroids are not available, high-dose oral methyl-prednisolone (500 mg daily for 5 days and then tapered over 10 days) may be acceptable.50
The side effects of corticosteroids are minimal when they are given for a brief time in otherwise healthy patients. The most common side effects are mood changes, facial flushing, sleep perturbations, weight gain, and dyspepsia.48
IMMUNOGLOBUL IN: LITTLE BENEFIT
In a double-blind, randomized trial, patients were treated with intravenous immunoglobulin 0.4 g/kg or placebo on days 0, 1, 2, 30, and 60. No difference was found in the primary outcomes of contrast sensitivity, visual acuity, or color vision from 1 week up to 6 months. There was also no significant difference in MRI outcomes between the two groups. The number of relapses was similar between both groups after 6 months.51,52
PLASMA EXCHANGE: FEW DATA
Data on plasma exchange are too scarce for us to make any recommendations. In one trial in 10 patients with severe optic neuritis, 3 patients appeared to benefit from plasma exchange. All patients had already received two doses of intravenous steroids.53
IMMUNOMODULATORY THERAPY MAY PREVENT MULTIPLE SCLEROSIS IN SOME
The most important clinical decision to make in patients with optic neuritis is whether to begin immunomodulatory therapy. Patients who may benefit the most from immunomodulatory therapy are those with abnormal white matter lesions on brain MRI, as they are at higher risk of developing MS.
Collectively, data from three studies indicate that early treatment in patients with clinically isolated syndromes, such as optic neuritis, reduces the rate of MS to 35% (from 50% without treatment) and reduces the number of new active lesions on MRI by approximately 50%.54–56
In addition, the Betaferon/Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT) trial57 found that at 3 years the rate of disability was 40% lower in patients who started immunomodulatory therapy (interferon beta-1b; Betaseron) early vs later. (Early treatment meant starting within 60 days of the clinically isolated syndrome; late treatment began 2 years later.) This study suggests that early treatment may reduce future disability, although these results need to be confirmed in prospective trials.
Therefore, once the diagnosis is secure, patients with optic neuritis should be referred to a neurologist with experience in treating MS to begin treatment with immunomodulatory therapy, such as glatiramer acetate (Copaxone), interferon beta-1a (Avonex, Refib), or interferon beta-1b (Betaseron).
Patients who have a normal MRI of the brain may consider deferring therapy, since they are at low risk of developing MS. These patients should undergo surveillance MRI (at least annually at first) to look for the development of white matter lesions, as the ONTT showed even this cohort has a 22% risk of developing MS.
If neuromyelitis optica is suspected (ie, in patients with severe unilateral or bilateral vision loss, recurrent optic neuritis, paraplegia, or quadriplegia), the serum neuromyelitis optic antibody can be tested, keeping in mind that 30% to 40% of patients with neuromyelitis optica will be seronegative for this antibody. Other tests supporting the diagnosis of neuromyelitis optica may include an MRI of the spine showing longitudinally extensive transverse myelitis, a polymorphonuclear pleocytosis in the cerebrospinal fluid, absent oligoclonal bands in the cerebrospinal fluid, and normal MRI of the brain (with some possible signal abnormalities in the periaqueductal gray matter and around the diencephalon).
Because neuromyelitis optica is believed to be mediated primarily by the humoral immune system, immunomodulatory therapy is not a first-line treatment. Patients with neuromyelitis optica can be treated initially with corticosteroids, intravenous immunoglobulin therapy, plasma exchange, or immunosuppressive agents such as azathioprine (Imuran), rituximab (Rituxan), or cyclophosphamide (Cytoxan). The choice of medication should be deferred to a neurologist familiar with treatment of this disorder.
The risk of MS may be lower in children than in adults. One large, retrospective study found the cumulative risk of developing MS (the study predated the McDonald criteria) was 13% at 10 years and 19% by 20 years.58 More recently, a large series from Toronto reported a higher rate of MS development in children with optic neuritis (36% at two years).59 By comparison, studies of adults with unilateral optic neuritis found a 38% to 39% risk of converting to MS at 10 years.5,41 The use of immunomodulatory therapies to reduce the risk of MS has not been well studied in children, since the prevalence is low in this age group.
The most common side effects of the beta-interferons are flulike symptoms (fatigue, myalgia), injection site reactions, and elevations of aminotransferase levels. Most patients are able to tolerate the side effects if the beta-interferon is taken with acetaminophen (Tylenol) or with over-the-counter nonsteroidal anti-inflammatory drugs.
Glatiramer acetate does not cause flulike symptoms or elevate aminotransferases, but it does require more frequent injections. Rarely, it may cause an idiosyncratic panic-like attack.
CASE CONTINUED
The best therapeutic regimen for this patient would be intravenous methylprednisolone, and subsequently a disease-modifying, immunomodulatory agent (interferon beta or glatiramer acetate). Our patient chose to start therapy with interferon beta-1a 30 μg intramuscularly once a week. She has been tolerating the medication well and has had no new neurologic or visual symptoms for the past 2 years.
References
Liu GT. Visual loss: optic neuropathies. In: Liu GT, Volpe NJ, Galetta SL, editors. Neuro–Ophthalmology: Diagnosis and Management. Philadelphia, PA: WB Saunders, 2001:103–187.
Wray SH. Optic neuritis. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Philadelphia, PA: WB Saunders, 1994:2539–2568.
Optic Neuritis Study Group. The clinical profile of optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol1991; 109:1673–1678.
Percy AK, Nobrega FT, Kurland LT. Optic neuritis and multiple sclerosis: an epidemiologic study. Arch Ophthalmol1972; 87:135–139.
Rodriguez M, Siva A, Cross SA, O’Brien PC, Kurland LT. Optic neuritis: a population–based study in Olmsted County, Minnesota. Neurology1995; 45:244–250.
Phillips PH, Newman NJ, Lynn MJ. Optic neuritis in African Americans. Arch Neurol1998; 55:186–192.
Brady KM, Brar AS, Lee AG, Coats DK, Paysse EA, Steinkuller PG. Optic neuritis in children: clinical features and visual outcome. J AAPOS1999; 3:98–103.
Kriss A, Francis DA, Cuendet B, et al. Recovery after optic neuritis in childhood. J Neurol Neurosurg Psychiatry1988; 51:1253–1258.
Beck RW. The Optic Neuritis Treatment Trial. Arch Ophthalmol1988; 106:1051–1053.
Optic Neuritis Study Group. Visual function 15 years after optic neuritis. Ophthalmology2008; 115:1079–1082.
Cox TA, Thompson HS, Corbett JJ. Relative afferent pupillary defects in optic neuritis. Am J Ophthalmol1981; 92:685–690.
Arnold AC. Ischemic optic neuropathies. Ophthalmol Clin North Am2001; 14:83–98.
Newman NJ, Scherer R, Langenberg P, et al. The fellow eye in NAION: report from the ischemic optic neuropathy decompression trial follow–up study. Am J Ophthalmol2002; 134:317–328.
Hayreh SS, Podhajsky PA, Zimmerman B. Ipsilateral recurrence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol2001; 132:734–742.
Liozon E, Herrmann F, Ly K, et al. Risk factors for visual loss in giant cell (temporal) arteritis: a prospective study of 174 patients. Am J Med2001; 111:211–217.
Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol2005; 50:15–26.
American Society of Anesthesiologists Task Force on Perioperative Blindness. Practice advisory for perioperative visual loss associated with spine surgery: a report by the American Society of Anesthesiologists Task Force on Perioperative Blindness. Anesthesiology2006; 104:1319–1328.
Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology2007; 114:810–815.
Papais-Alvarenga RM, Carellos SC, Alvarenga MP, Holander C, Bichara RP, Thuler LC. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol2008; 126:12–16.
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology1999; 53:1107–1114.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology2006; 66:1485–1489.
Takahashi T, Fujihara K, Nakashima I, et al. Anti–aquaporin–4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain2007; 130:1235–1243.
Sibony P, Halperin J, Coyle PK, Patel K. Reactive Lyme serology in optic neuritis. J Neuroophthalmol2005; 25:71–82.
Anninger WV, Lomeo MD, Dingle J, Epstein AD, Lubow M. West Nile virus–associated optic neuritis and chorioretinitis. Am J Ophthalmol2003; 136:1183–1185.
Jabs DA, Miller NR, Newman SA, Johnson MA, Stevens MB. Optic neuropathy in systemic lupus erythematosus. Arch Ophthalmol1986; 104:564–568.
Howell N. LHON and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol2003; 37:94–108.
Newman NJ. Hereditary optic neuropathies. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. Philadelphia, PA: Lippincott Williams & Wilkins, 2005;465–501.
Smith JL, Hoyt WF, Susac JO. Ocular fundus in acute Leber optic neuropathy. Arch Ophthalmol1973; 90:349–354.
Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol2003; 87:48–53.
Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin– related GTPase, is mutated in autosomal dominant atrophy linked to chromosome 3q28. Nat Genet2000; 26:211–215.
McKinley SH, Foroozan R. Optic neuropathy associated with linezolid treatment. J Neuroophthalmol2005; 25:18–21.
Melamud A, Kosmorsky GS, Lee MS. Ocular ethambutol toxicity. Mayo Clin Proc2003; 78:1409–1411.
Kerrison JB. Optic neuropathies caused by toxins and adverse drug reactions. Ophthalmol Clin North Am2004; 17:481–488.
Pomeranz HD, Bhavsar AR. Nonarteritic ischemic optic neuropathy developing soon after use of sildenafil (Viagra): a report of seven new cases. J Neuroophthalmol2005; 25:9–13.
Fivgas GD, Newman NJ. Anterior ischemic optic neuropathy following the use of a nasal decongestant. Am J Ophthalmol1999; 127:104–106.
The Cuba Neuropathy Field Investigation Team. Epidemic optic neuropathy in Cuba—clinical characterization and risk factors. N Engl J Med1995; 333:1176–1182.
Solberg Y, Rosner M, Belkin M. The association between cigarette smoking and ocular diseases. Surv Ophthalmol1998; 42:535–547.
Rizzo JF, Andreoli CM, Rabinov JD. Use of magnetic resonance imaging to differentiate optic neuritis and nonarteritic anterior ischemic optic neuropathy. Ophthalmology2002; 109:1679–1684.
The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final Optic Neuritis Treatment Trial follow-up. Arch Neurol2008; 65:727–732.
Ghezzi A, Martinelli V, Torri V, et al. Long–term follow–up of isolated optic neuritis: the risk of developing multiple sclerosis, its outcome, and the prognostic role of paraclinical tests. J Neurol1999; 246:770– 775.
Brex PA, Ciccarelli O, O'Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med2002; 346:158–164.
Tintore M, Rovira A, Rio J, et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology2006; 67:968–972.
Söderström M, Ya–Ping J, Hillert J. Optic neuritis: prognosis for multiple sclerosis from MRI, CSF, and HLA findings. Neurology1998; 50:708–714.
Frederiksen JL, Madsen HO, Ryder LP, Larsson HB, Morling N, Svejgaard A. HLA typing in acute optic neuritis: relation to multiple sclerosis and magnetic resonance imaging findings. Arch Neurol1997; 54:76–80.
Frederiksen JL, Larsson HB, Oleson J. Correlation of magnetic resonance imaging and CSF findings in patients with acute monosymptomatic optic neuritis. Acta Neurol Scand1992; 86:317–322.
Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med1992; 326:581–588.
Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol2003; 121:944–949.
Sellebjerg F, Nielsen HS, Frederiksen JL, Olesen J. A randomized, controlled trial of oral high-dose methylprednisolone in acute optic neuritis. Neurology1999; 52:1479–1484.
Noseworthy JH, O’Brien PC, Peterson TM, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology2001; 56:1514–1522.
Roed HG, Langkilde A, Sellebjerg F, et al. A double–blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology2005; 64:804–810.
Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology2004; 63:1081–1083.
CHAMPS Study Group. Interferon beta-1a for optic neuritis patients at high risk for multiple sclerosis. Am J Ophthalmol2001; 132:463– 471.
Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet2001; 357:1576–1582.
Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology2006; 67:1242–1249.
Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet2007; 370:389–397.
Lucchinetti CF, Kiers L, O’Duffy A, et al. Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology1997; 49:1413–1418.
Wilejto M, Shroff M, Buncic JR, Kennedy J, Goia C, Banwell B. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology2006; 67:258–262.
Benjamin J. Osborne, MD Assistant Professor of Neurology and Ophthalmology, Departments of Neurology and Ophthalmology, Georgetown University Medical Center, Washington, DC
Nicholas J. Volpe, MD Niessen Professor of Ophthalmology and Neurology, Vice Chair for Clinical Practice and Residency Program Director, Division of Neuro-Ophthalmology, University of Pennsylvania School of Medicine, Scheie Eye Institute, Philadelphia, PA
Address: Nicholas J. Volpe, MD, Scheie Eye Institute; 39th and Market Streets, Philadelphia, PA 19104; e-mail [email protected]
Benjamin J. Osborne, MD Assistant Professor of Neurology and Ophthalmology, Departments of Neurology and Ophthalmology, Georgetown University Medical Center, Washington, DC
Nicholas J. Volpe, MD Niessen Professor of Ophthalmology and Neurology, Vice Chair for Clinical Practice and Residency Program Director, Division of Neuro-Ophthalmology, University of Pennsylvania School of Medicine, Scheie Eye Institute, Philadelphia, PA
Address: Nicholas J. Volpe, MD, Scheie Eye Institute; 39th and Market Streets, Philadelphia, PA 19104; e-mail [email protected]
Author and Disclosure Information
Benjamin J. Osborne, MD Assistant Professor of Neurology and Ophthalmology, Departments of Neurology and Ophthalmology, Georgetown University Medical Center, Washington, DC
Nicholas J. Volpe, MD Niessen Professor of Ophthalmology and Neurology, Vice Chair for Clinical Practice and Residency Program Director, Division of Neuro-Ophthalmology, University of Pennsylvania School of Medicine, Scheie Eye Institute, Philadelphia, PA
Address: Nicholas J. Volpe, MD, Scheie Eye Institute; 39th and Market Streets, Philadelphia, PA 19104; e-mail [email protected]
Two days ago, a 27-year-old woman noticed that her vision was blurry in her right eye. She has come to see her primary care physician for advice. This is the first time this has happened to her. She describes seeing a grayish blur over the center of her vision, but she has not noted any other symptoms except for some soreness around the right eye, which is worse with eye movements.
How should she be assessed and treated?
IMPORTANT TO RECOGNIZE
Sudden vision loss is one of the more common problems encountered in ophthalmology and neurology.
Optic neuritis, a demyelinating inflammatory condition that causes acute vision loss, is associated with multiple sclerosis (MS), and recognizing its classic clinical manifestations early is important so that appropriate diagnostic testing (magnetic resonance imaging [MRI]) and treatment (corticosteroids and immunomodulators) can be started.
Although a comprehensive review of all the optic neuropathies is beyond the scope of this paper, in the pages that follow we review some of the most common causes, which may be first seen by the general internist.
FOUR SUBTYPES OF OPTIC NEURITIS
There are four subtypes of optic neuritis:
Figure 1. In retrobulbar optic neuritis, the inflammation and demyelination occur behind the globe of the eye. The optic disc appears normal with no signs of swelling or pallor.Retrobulbar neuritis (Figure 1), or inflammation of the optic nerve behind the eye, is the form most commonly associated with MS.
Papillitis (Figure 2), or inflammation of the optic disc, can also be associated with MS.
Perineuritis is inflammation of the optic nerve sheath, sparing the optic nerve itself. Usually, patients are older, and vision loss is mild to moderate. Perineuritis is commonly due to infectious or inflammatory conditions, eg, syphilis or sarcoidosis.
Figure 2. In papillitis, mild swelling and elevation of the optic disc can be seen. The small splinter hemorrhage seen at 10 o’clock is not typical of optic neuritis associated with multiple sclerosis.Neuroretinitis may occur at any age. There is concomitant swelling of the optic nerve and macula. Exudates that form around the macula give the appearance of a star.
Perineuritis and neuroretinitis are not associated with MS, and if they are found they suggest another etiology. In the rest of this review, “optic neuritis” means retrobulbar optic neuritis, the form most commonly seen in clinical practice.
MOST COMMON IN YOUNG WOMEN
Acute demyelinating optic neuritis most often affects women in their 20s and 30s.1–3 Studies in the United States have estimated its annual incidence to be 5.1 to 6.4 per 100,000.4,5 The incidence is higher in populations at higher latitudes and lower near the equator. It is less common in blacks than in whites.6
In children, optic neuritis is not as strongly associated with MS, especially when there is optic disc swelling or bilateral involvement. Most children have a good visual outcome, although approximately 20% may be visually disabled.7–9
FEATURES: VISION LOSS AND EYE PAIN
Most of our current knowledge of the clinical features of optic neuritis comes from the Optic Neuritis Treatment Trial (ONTT),10 conducted in the 1990s. This trial enrolled 457 patients 18 to 46 years old who had acute unilateral optic neuritis. The patients had to have symptoms consistent with acute unilateral optic neuritis for 8 days or less. They could not have evidence of any systemic disease (except for MS) or have received prior treatment for MS. Therefore, this study was quite representative of the patients with optic neuritis that one might encounter in the clinic and is highly important in both characterizing optic neuritis and defining its treatment.
The study found that the two most common symptoms are vision loss and eye pain.
Vision loss in optic neuritis typically occurs over several hours to days, and vision reaches a nadir within 1 to 2 weeks. Typically, patients begin to recover 2 to 4 weeks after the onset of the vision loss. The optic nerve may take up to 6 to 12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.11 More than two-thirds of patients have at least 20/20 vision once they have fully recovered from the optic neuritis. Only 3% of patients become completely blind.
Eye pain is very common in optic neuritis (it affected 87% of patients in the ONTT) and typically worsens with eye movement. The eye is also sore to touch. The pain generally begins at the same time as the visual loss and improves along with it. Eye movements also may bring about photopsia (flickering or flashes of light), a symptom reported by 30% of the ONTT participants.
Loss of color vision out of proportion to the loss of visual acuity is characteristic of optic neuropathies. In the ONTT, 88% of the involved eyes had abnormal color vision as assessed by the Ishihara test (using pseudoisochromatic plates), and 94% as assessed by the Farnsworth-Munsell 100 hue test, which is more sensitive but cumbersome. The most common patterns of color vision loss in optic nerve disease are loss of red (protanopia) and green (deutranopia).
A good way to screen for loss of color vision is to test for color desaturation. First, ask the patient to fixate with the normal eye on a bright red object (for example, the cap from a bottle of ophthalmic mydriatic drops or a pen cap). Then ask the patient to compare the intensity of the redness between the good eye and the affected eye. The patient can quantify the color desaturation by rating what percentage of red is lost in the affected eye compared with the normal eye.
Temporary exacerbations of visual problems during fever (the Uhthoff phenomenon) can occur in patients who have had optic neuritis. These transient pseudoexacerbations are not new episodes of optic neuritis and should resolve after the body temperature returns to normal.
A relative afferent pupillary defect should be seen in the involved eye in all patients with optic neuritis if the other eye is uninvolved and healthy.12 The best way to elicit this sign is to perform the swinging light test in a dark room with the patient fixating at a distant target, which prevents miosis due to accommodation. When the light is swung to the involved eye, the pupil dilates because less light stimulus reaches the midbrain through the affected optic nerve. As the optic nerve heals and recovers, this sign may become subtle, but it persists in more than 90% of cases.12
Findings on funduscopy
Examination of the fundus is helpful in the clinical diagnosis of optic neuritis.
Two-thirds of the ONTT patients had retrobulbar neuritis, and one-third had papillitis. If optic nerve swelling is present, it is typically mild.
Peripapillary hemorrhages were exceedingly rare in the cases of papillitis (only 6%) and were associated with a very low to zero risk of developing MS. If peripapillary hemorrhages are found on examination, one should consider another diagnosis, such as anterior ischemic optic neuropathy.11
CASE CONTINUED
Our patient undergoes a neurologic examination, which reveals an afferent pupillary defect in the right eye and visual acuity of 20/100 in the right eye and 20/20 in the left. Visual fields are normal in the left eye as assessed by confrontation, but there is a central scotoma in the right.
OTHER TYPES OF NEUROPATHY
Optic neuritis is a clinical diagnosis based on the history and findings on examination. If the patient does not have its typical features (Table 1), then other diagnoses should be pursued with serologic and cerebrospinal fluid studies.
The following should be included in the differential diagnosis of optic neuritis:
Ischemic optic neuropathy
Ischemic optic neuropathy is more common in patients age 50 and older, whereas optic neuritis is more common in younger patients. Most patients with ischemic optic neuropathy have hypertension, hypercholesterolemia, diabetes mellitus, obstructive sleep apnea, or other vascular risk factors. The disease has several important subtypes, as discussed below.
Figure 3. Left, fundus photo several weeks after an attack of nonarteritic anterior ischemic optic neuropathy demonstrating pallor of the superior half of the disc. Middle, an associated inferior altitudinal defect. Right, sectoral swelling of the disc with flame or splinter hemorrhages is commonly seen. This is not typical of demyelinating optic neuritis.Nonarteritic anterior ischemic optic neuropathy is the most common form. Typically, there is acute onset of painless vision loss in one eye and an associated altitudinal field defect (Figure 3). For example, if the superior rim of the optic nerve acutely develops swelling and then becomes pale, a corresponding altitudinal cut would develop in the inferior visual field, respecting the horizontal meridian. Many patients first notice the vision loss upon waking up in the morning.13
Although patients with nonarteritic anterior ischemic optic neuropathy typically have vasculopathic risk factors such as hypertension, diabetes mellitus, peripheral vascular disease, or hypercholesterolemia, there is no proven causation between the two. The age of these patients ranges from 50 to 70, with an average age of 66.
The disc appears swollen and may have flame or splinter hemorrhages (Figure 3). The cup of the involved disc is typically small. The visual loss is believed to be the result of poor perfusion in the circulation of the posterior ciliary artery, which supplies the optic nerve head.1 If the other eye also has a small cup, it is considered to be at risk of future ischemic events. In one study,14 the opposite eye became involved within the next 5 years in 14.7% of all cases. The risk of recurrent disease in the same eye is low (6.4% in another study15).
Arteritic anterior ischemic optic neuropathy is more common in patients over age 70 and is usually due to giant cell arteritis, which has a significant association with polymyalgia rheumatica. Patients may have jaw claudication, proximal myalgia and arthralgia, scalp tenderness, headache, fatigue, and a significantly elevated erythrocyte sedimentation rate and C-reactive protein level. These features should be looked for in the review of systems, although patients may not have all of them.
The funduscopic examination may reveal a pale, swollen disc, peripapillary hemorrhages, branch or central retinal artery occlusions, or cotton-wool spots.
Temporal artery biopsy is the gold standard for diagnosis, but treatment with corticosteroids should not be delayed pending biopsy or other test results.1
Thrombocytosis has been associated with a higher risk of permanent vision loss in patients with giant cell arteritis.16
Posterior ischemic optic neuropathy is the least common form of ischemic optic neuropathy. This diagnosis should be entertained in older patients who report acute, painless vision loss but have a normal funduscopic examination. Giant cell arteritis must be considered first in this setting.
Bilateral posterior ischemic optic neuropathy can occur (although rarely) in patients undergoing cardiac or spinal surgery.17 Risk factors thought to be associated with perioperative disease include anemia, hypotension, substantial blood loss during the surgery, surgeries longer than 6.5 hours, carotid atherosclerosis, and diabetes.18
There are no effective treatments for most ischemic optic neuropathies with the crucial exception of giant cell arteritis.
Neuromyelitis optica (Devic disease)
Neuromyelitis optica (Devic disease) is a combination of optic neuritis and transverse myelitis (Table 2). Clinically, the disease spares the nervous system except for the optic nerves and spinal cord. The onset of the optic neuritis may precede or follow the onset of the transverse myelitis by up to 2 to 4 years.19 Usually, the optic neuritis is bilateral and retrobulbar and results in severe vision loss, worse than that seen in patients with MS.19,20
The transverse myelitis may cause paraplegia or quadriplegia, depending on the location of the lesion in the spinal cord (cervical vs thoracic). The transverse myelitis in neuromyelitis optica is distinct from that seen in MS. In neuromyelitis optica, the transverse myelitis is longitudinally extensive, spanning more than three vertebral bodies in length. In MS, spinal cord lesions usually are more discrete and involve one or two spinal cord segments.21
Recently, serum neuromyelitis optica immunoglobulin G (IgG) antibody has been shown to be a significant biomarker of this disease. Its sensitivity ranges from approximately 60% to 70% and its specificity is greater than 90%.22 This antibody binds to aquaporin-4, an important water-channel protein in the blood-brain barrier of the central nervous system, and evidence suggests that it is involved in the pathogenesis of the disease.23
Initially, it was proposed that MRI of the brain had to be normal for neuromyelitis optica to be diagnosed.21 However, the proposed 2006 criteria allow for some abnormal T2 and fluid-attenuated inversion recovery (FLAIR) hyperintensities in the periaqueductal gray matter and diencephalon.22
The spinal fluid in neuromyelitis optica may show a pleocytosis larger than that seen in MS (> 50 white blood cells per mm3) and may have a significant neutrophilic component.21 Oligoclonal bands are not typically present.
It is still debated whether neuromyelitis optica is a separate disease from MS or a subset of it. The implications of this debate may affect its management, as discussed below.
Inflammatory optic neuropathies
Inflammatory optic neuropathies can be caused by infections (eg, syphilis, cat scratch disease) or by noninfectious conditions (eg, sarcoidosis). Lyme disease is rarely a cause of retrobulbar optic neuritis but may cause papillitis.24 West Nile virus has also been reported to cause optic neuritis.25 Lupus may cause an optic neuropathy by inflammatory or ischemic mechanisms.26
Compressive optic neuropathies
Compressive optic neuropathies may be due to mass lesions, tumors, thyroid eye disease, or other orbital processes. MRI of the brain and orbits will confirm or rule out diagnoses associated with compressive optic neuropathy.
Genetic causes
Genetic causes of optic neuropathy include the Leber and Kjer hereditary optic neuropathies.
Leber optic neuropathy involves subacute and painless vision loss, with sequential involvement of both eyes over a period of weeks to months. This disorder predominantly affects men (80%–90% of patients) and is inherited from maternal mitochondrial DNA. The three most common mutations implicated in Leber optic neuropathy (located at base pairs 11,778, 3,460, and 14,484 in the mitochondrial DNA) are involved in more than 90% of cases. The prognosis for recovery varies depending on the genotype.27 These genes encode proteins that are part of complex I of the mitochondrial respiratory chain.28 Funduscopic examination most commonly shows circumpapillary telangiectasia, although up to one-third of patients can have a normal-appearing disc initially. Central vision is affected more severely than peripheral vision.29
Kjer autosomal-dominant optic atrophy is the most common hereditary optic neuropathy. This disease primarily affects children in the first decade of life with slowly progressive loss of vision. As with other optic neuropathies, there will eventually be pallor of the optic disc, a cecocentral scotoma, and loss of color perception. The OPA1 gene located on chromosome 3q28 has been implicated in most patients with dominant optic atrophy; a test is commercially available for diagnosis.30,31
Toxic and metabolic causes
Many agents can cause optic neuropathy. Toxins strongly associated with optic neuropathy include carbon monoxide, methanol, ethylene glycol, perchloroethylene, and tobacco. Drugs linked to optic neuropathy are ethambutol (Myambutol), clioquinol (Vioform), isoniazid (Nydrazid), amiodarone (Cordarone), linezolid (Zyvox), methotrexate, sildenafil (Viagra), oxymetazoline (contained in Afrin and other nasal sprays), and infliximab (Remicade).32–37 Additionally, several chemotherapeutic agents are known to cause optic atrophy, including vincristine (Oncovin), cisplatin (Platinol), carboplatin (Paraplatin), and paclitaxel (Abraxane, Onxol).
Nutritional deficiencies are presumed to have played a significant role in the endemics of optic neuropathy that have occurred in poor countries, such as in Cuba during the 1990s.38 Most nutritional optic neuropathies appear to be exacerbated by tobacco.39
MRI ASSESSES RISK OF MS
The diagnosis of optic neuritis is clinical, based on the history and physical findings.
However, MRI of the brain and orbits with gadolinium contrast has become the cornerstone of the evaluation in patients with optic neuritis. And MRI not only helps confirm the clinical diagnosis, but it also more importantly offers very strong prognostic information about the risk of future demyelinating events and MS.
Gadolinium-enhanced fat-saturated T1-weighted MRI of the orbits is the best sequence to show the inflammation of the optic nerve in optic neuritis (fat saturation is necessary to hide the bright signal of the orbital fat tissue).
Contrast-enhanced MRI can also help differentiate optic neuritis from nonarteritic anterior ischemic optic neuropathy. MRI of the orbits with gadolinium contrast shows enhancement of the affected optic nerve in approximately 95% of cases of optic neuritis, whereas optic nerve enhancement rarely occurs in nonarteritic anterior ischemic optic neuropathy.40
Brain MRI may show other white matter lesions (either hyperintensities on T2-weighted images or enhancement of T1-weighted images postcontrast), which denote a higher risk of developing MS. In 15-year follow-up data from the ONTT, monosymptomatic patients with no white matter lesions had a 25% risk of MS (defined at the time the ONTT was conducted as a second demyelinating event), while those with one lesion or more had a 72% risk.41
An earlier, prospective study in 102 Italian patients with optic neuritis found the risk of developing MS to be about 36% at 6 years and 42% at 8 years (using the Posner diagnostic criteria). When brain MRI data were analyzed, those with one or more lesions had a 52% risk of developing MS at 8 years, whereas those with no MRI lesions did not develop MS.42
Other studies have stratified the risk of MS in patients with clinically isolated syndromes (including not only optic neuritis, but also other neurologic symptoms such as brainstem, motor, or sensory deficits). At mean follow-ups ranging from 5 to 14 years, the risk of developing MS was 8% to 24% in patients with normal findings on brain MRI compared with 56% to 88% in those with abnormal MRI findings.43,44
Optic neuritis patients with atypical white matter lesions on brain MRI may benefit from lumbar puncture to look for oligoclonal bands, to measure the IgG index and the IgG synthesis rate, and to test for myelin basic protein in the cerebrospinal fluid. Of patients with acute optic neuritis, 79% have abnormalities in their cerebrospinal fluid. Oligoclonal bands are present in 69%, and for patients with oligoclonal bands, the 5-year probability of developing MS is estimated to be 65%, compared with 10% in those without bands. If the patient has no oligoclonal bands and has normal findings on brain MRI, he or she will not have MS 5 years later.45–47
Patients with optic neuritis who have no white matter lesions on brain MRI may be further risk-stratified on the basis of their clinical findings. In the ONTT 15-year follow-up, MS did not develop in any patient who had no brain lesions on baseline MRI, no prior optic neuritis in the contralateral eye, and no prior neurologic symptoms or signs, even if the patient had severe disc swelling (eg, peripapillary hemorrhage or retinal exudates) or if vision was reduced to no light perception.41
CASE CONTINUED: FINDINGS ON MRI
Figure 4. The patient’s magnetic resonance image. Top, an axial T2 image with contrast; bottom, sagittal T1 image with contrast. The white matter lesions indicate she is at risk of developing multiple sclerosis.Our patient undergoes MRI, which shows lesions on axial T2 and sagittal T1 imaging with contrast (Figure 4). Of note, there are significant lesions perpendicular to the corpus callosum (Dawson fingers), some of which enhance with contrast. The enhancement indicates breakdown of the blood-brain barrier and suggests that there is active inflammation in the white matter.
Patients in the ONTT were randomized to receive one of three treatments:
Oral prednisone 1 mg/kg/day for 14 days and then tapered over 4 days
Intravenous methylprednisolone (Solu-Medrol) 250 mg four times per day for 3 days followed by oral prednisone 1 mg/kg/day for 11 days and then tapered for 4 days
Oral placebo for 14 days.
The primary visual outcomes measured were visual acuity and contrast sensitivity.48
Patients who received intravenous methylprednisolone recovered their visual function faster, although the visual outcomes after 6 months were no better with methylprednisolone than with placebo or oral prednisone. Intravenous methylprednisolone also reduced the risk of MS within the first 2 years in patients with high-risk brain MRIs.
Surprisingly, patients in the oral prednisone group had a higher risk of recurrent optic neuritis in both eyes than did patients given intravenous methylprednisolone or placebo (30% at 2 years with oral prednisone vs 16% with placebo and 13% with intravenous methylprednisolone).48 At 10 years, the risk of recurrent optic neuritis was still higher in the oral prednisone group (44%) than in the intravenous methylprednisolone group (29%) (P = .03). However, the difference between the oral prednisone and placebo groups was no longer statistically significant (P = .07).49 Oral prednisone alone is therefore contraindicated in the treatment of typical unilateral demyelinating optic neuritis.
Many patients can now be treated with intravenous infusions of methylprednisolone at home for episodes of optic neuritis.
Risks vs benefits of corticosteroid therapy
When deciding whether to treat an individual patient who has optic neuritis with intravenous corticosteroids, one should consider all the benefits and risks.
Corticosteroids do not affect long-term visual outcome, although they do hasten recovery. Patients with mild vision loss (visual acuity better than 20/40), no significant visual field loss, and no enhancing lesions on brain MRI can forgo therapy with intravenous corticosteroids.
On the other hand, we strongly favor intravenous corticosteroid treatment in patients who have both acute optic neuritis and high signal abnormalities on brain MRI, since it may delay the onset of MS. In addition, patients with severe vision loss should receive intravenous corticosteroids to hasten their recovery. In the rare circumstance in which intravenous corticosteroids are not available, high-dose oral methyl-prednisolone (500 mg daily for 5 days and then tapered over 10 days) may be acceptable.50
The side effects of corticosteroids are minimal when they are given for a brief time in otherwise healthy patients. The most common side effects are mood changes, facial flushing, sleep perturbations, weight gain, and dyspepsia.48
IMMUNOGLOBUL IN: LITTLE BENEFIT
In a double-blind, randomized trial, patients were treated with intravenous immunoglobulin 0.4 g/kg or placebo on days 0, 1, 2, 30, and 60. No difference was found in the primary outcomes of contrast sensitivity, visual acuity, or color vision from 1 week up to 6 months. There was also no significant difference in MRI outcomes between the two groups. The number of relapses was similar between both groups after 6 months.51,52
PLASMA EXCHANGE: FEW DATA
Data on plasma exchange are too scarce for us to make any recommendations. In one trial in 10 patients with severe optic neuritis, 3 patients appeared to benefit from plasma exchange. All patients had already received two doses of intravenous steroids.53
IMMUNOMODULATORY THERAPY MAY PREVENT MULTIPLE SCLEROSIS IN SOME
The most important clinical decision to make in patients with optic neuritis is whether to begin immunomodulatory therapy. Patients who may benefit the most from immunomodulatory therapy are those with abnormal white matter lesions on brain MRI, as they are at higher risk of developing MS.
Collectively, data from three studies indicate that early treatment in patients with clinically isolated syndromes, such as optic neuritis, reduces the rate of MS to 35% (from 50% without treatment) and reduces the number of new active lesions on MRI by approximately 50%.54–56
In addition, the Betaferon/Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT) trial57 found that at 3 years the rate of disability was 40% lower in patients who started immunomodulatory therapy (interferon beta-1b; Betaseron) early vs later. (Early treatment meant starting within 60 days of the clinically isolated syndrome; late treatment began 2 years later.) This study suggests that early treatment may reduce future disability, although these results need to be confirmed in prospective trials.
Therefore, once the diagnosis is secure, patients with optic neuritis should be referred to a neurologist with experience in treating MS to begin treatment with immunomodulatory therapy, such as glatiramer acetate (Copaxone), interferon beta-1a (Avonex, Refib), or interferon beta-1b (Betaseron).
Patients who have a normal MRI of the brain may consider deferring therapy, since they are at low risk of developing MS. These patients should undergo surveillance MRI (at least annually at first) to look for the development of white matter lesions, as the ONTT showed even this cohort has a 22% risk of developing MS.
If neuromyelitis optica is suspected (ie, in patients with severe unilateral or bilateral vision loss, recurrent optic neuritis, paraplegia, or quadriplegia), the serum neuromyelitis optic antibody can be tested, keeping in mind that 30% to 40% of patients with neuromyelitis optica will be seronegative for this antibody. Other tests supporting the diagnosis of neuromyelitis optica may include an MRI of the spine showing longitudinally extensive transverse myelitis, a polymorphonuclear pleocytosis in the cerebrospinal fluid, absent oligoclonal bands in the cerebrospinal fluid, and normal MRI of the brain (with some possible signal abnormalities in the periaqueductal gray matter and around the diencephalon).
Because neuromyelitis optica is believed to be mediated primarily by the humoral immune system, immunomodulatory therapy is not a first-line treatment. Patients with neuromyelitis optica can be treated initially with corticosteroids, intravenous immunoglobulin therapy, plasma exchange, or immunosuppressive agents such as azathioprine (Imuran), rituximab (Rituxan), or cyclophosphamide (Cytoxan). The choice of medication should be deferred to a neurologist familiar with treatment of this disorder.
The risk of MS may be lower in children than in adults. One large, retrospective study found the cumulative risk of developing MS (the study predated the McDonald criteria) was 13% at 10 years and 19% by 20 years.58 More recently, a large series from Toronto reported a higher rate of MS development in children with optic neuritis (36% at two years).59 By comparison, studies of adults with unilateral optic neuritis found a 38% to 39% risk of converting to MS at 10 years.5,41 The use of immunomodulatory therapies to reduce the risk of MS has not been well studied in children, since the prevalence is low in this age group.
The most common side effects of the beta-interferons are flulike symptoms (fatigue, myalgia), injection site reactions, and elevations of aminotransferase levels. Most patients are able to tolerate the side effects if the beta-interferon is taken with acetaminophen (Tylenol) or with over-the-counter nonsteroidal anti-inflammatory drugs.
Glatiramer acetate does not cause flulike symptoms or elevate aminotransferases, but it does require more frequent injections. Rarely, it may cause an idiosyncratic panic-like attack.
CASE CONTINUED
The best therapeutic regimen for this patient would be intravenous methylprednisolone, and subsequently a disease-modifying, immunomodulatory agent (interferon beta or glatiramer acetate). Our patient chose to start therapy with interferon beta-1a 30 μg intramuscularly once a week. She has been tolerating the medication well and has had no new neurologic or visual symptoms for the past 2 years.
Two days ago, a 27-year-old woman noticed that her vision was blurry in her right eye. She has come to see her primary care physician for advice. This is the first time this has happened to her. She describes seeing a grayish blur over the center of her vision, but she has not noted any other symptoms except for some soreness around the right eye, which is worse with eye movements.
How should she be assessed and treated?
IMPORTANT TO RECOGNIZE
Sudden vision loss is one of the more common problems encountered in ophthalmology and neurology.
Optic neuritis, a demyelinating inflammatory condition that causes acute vision loss, is associated with multiple sclerosis (MS), and recognizing its classic clinical manifestations early is important so that appropriate diagnostic testing (magnetic resonance imaging [MRI]) and treatment (corticosteroids and immunomodulators) can be started.
Although a comprehensive review of all the optic neuropathies is beyond the scope of this paper, in the pages that follow we review some of the most common causes, which may be first seen by the general internist.
FOUR SUBTYPES OF OPTIC NEURITIS
There are four subtypes of optic neuritis:
Figure 1. In retrobulbar optic neuritis, the inflammation and demyelination occur behind the globe of the eye. The optic disc appears normal with no signs of swelling or pallor.Retrobulbar neuritis (Figure 1), or inflammation of the optic nerve behind the eye, is the form most commonly associated with MS.
Papillitis (Figure 2), or inflammation of the optic disc, can also be associated with MS.
Perineuritis is inflammation of the optic nerve sheath, sparing the optic nerve itself. Usually, patients are older, and vision loss is mild to moderate. Perineuritis is commonly due to infectious or inflammatory conditions, eg, syphilis or sarcoidosis.
Figure 2. In papillitis, mild swelling and elevation of the optic disc can be seen. The small splinter hemorrhage seen at 10 o’clock is not typical of optic neuritis associated with multiple sclerosis.Neuroretinitis may occur at any age. There is concomitant swelling of the optic nerve and macula. Exudates that form around the macula give the appearance of a star.
Perineuritis and neuroretinitis are not associated with MS, and if they are found they suggest another etiology. In the rest of this review, “optic neuritis” means retrobulbar optic neuritis, the form most commonly seen in clinical practice.
MOST COMMON IN YOUNG WOMEN
Acute demyelinating optic neuritis most often affects women in their 20s and 30s.1–3 Studies in the United States have estimated its annual incidence to be 5.1 to 6.4 per 100,000.4,5 The incidence is higher in populations at higher latitudes and lower near the equator. It is less common in blacks than in whites.6
In children, optic neuritis is not as strongly associated with MS, especially when there is optic disc swelling or bilateral involvement. Most children have a good visual outcome, although approximately 20% may be visually disabled.7–9
FEATURES: VISION LOSS AND EYE PAIN
Most of our current knowledge of the clinical features of optic neuritis comes from the Optic Neuritis Treatment Trial (ONTT),10 conducted in the 1990s. This trial enrolled 457 patients 18 to 46 years old who had acute unilateral optic neuritis. The patients had to have symptoms consistent with acute unilateral optic neuritis for 8 days or less. They could not have evidence of any systemic disease (except for MS) or have received prior treatment for MS. Therefore, this study was quite representative of the patients with optic neuritis that one might encounter in the clinic and is highly important in both characterizing optic neuritis and defining its treatment.
The study found that the two most common symptoms are vision loss and eye pain.
Vision loss in optic neuritis typically occurs over several hours to days, and vision reaches a nadir within 1 to 2 weeks. Typically, patients begin to recover 2 to 4 weeks after the onset of the vision loss. The optic nerve may take up to 6 to 12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.11 More than two-thirds of patients have at least 20/20 vision once they have fully recovered from the optic neuritis. Only 3% of patients become completely blind.
Eye pain is very common in optic neuritis (it affected 87% of patients in the ONTT) and typically worsens with eye movement. The eye is also sore to touch. The pain generally begins at the same time as the visual loss and improves along with it. Eye movements also may bring about photopsia (flickering or flashes of light), a symptom reported by 30% of the ONTT participants.
Loss of color vision out of proportion to the loss of visual acuity is characteristic of optic neuropathies. In the ONTT, 88% of the involved eyes had abnormal color vision as assessed by the Ishihara test (using pseudoisochromatic plates), and 94% as assessed by the Farnsworth-Munsell 100 hue test, which is more sensitive but cumbersome. The most common patterns of color vision loss in optic nerve disease are loss of red (protanopia) and green (deutranopia).
A good way to screen for loss of color vision is to test for color desaturation. First, ask the patient to fixate with the normal eye on a bright red object (for example, the cap from a bottle of ophthalmic mydriatic drops or a pen cap). Then ask the patient to compare the intensity of the redness between the good eye and the affected eye. The patient can quantify the color desaturation by rating what percentage of red is lost in the affected eye compared with the normal eye.
Temporary exacerbations of visual problems during fever (the Uhthoff phenomenon) can occur in patients who have had optic neuritis. These transient pseudoexacerbations are not new episodes of optic neuritis and should resolve after the body temperature returns to normal.
A relative afferent pupillary defect should be seen in the involved eye in all patients with optic neuritis if the other eye is uninvolved and healthy.12 The best way to elicit this sign is to perform the swinging light test in a dark room with the patient fixating at a distant target, which prevents miosis due to accommodation. When the light is swung to the involved eye, the pupil dilates because less light stimulus reaches the midbrain through the affected optic nerve. As the optic nerve heals and recovers, this sign may become subtle, but it persists in more than 90% of cases.12
Findings on funduscopy
Examination of the fundus is helpful in the clinical diagnosis of optic neuritis.
Two-thirds of the ONTT patients had retrobulbar neuritis, and one-third had papillitis. If optic nerve swelling is present, it is typically mild.
Peripapillary hemorrhages were exceedingly rare in the cases of papillitis (only 6%) and were associated with a very low to zero risk of developing MS. If peripapillary hemorrhages are found on examination, one should consider another diagnosis, such as anterior ischemic optic neuropathy.11
CASE CONTINUED
Our patient undergoes a neurologic examination, which reveals an afferent pupillary defect in the right eye and visual acuity of 20/100 in the right eye and 20/20 in the left. Visual fields are normal in the left eye as assessed by confrontation, but there is a central scotoma in the right.
OTHER TYPES OF NEUROPATHY
Optic neuritis is a clinical diagnosis based on the history and findings on examination. If the patient does not have its typical features (Table 1), then other diagnoses should be pursued with serologic and cerebrospinal fluid studies.
The following should be included in the differential diagnosis of optic neuritis:
Ischemic optic neuropathy
Ischemic optic neuropathy is more common in patients age 50 and older, whereas optic neuritis is more common in younger patients. Most patients with ischemic optic neuropathy have hypertension, hypercholesterolemia, diabetes mellitus, obstructive sleep apnea, or other vascular risk factors. The disease has several important subtypes, as discussed below.
Figure 3. Left, fundus photo several weeks after an attack of nonarteritic anterior ischemic optic neuropathy demonstrating pallor of the superior half of the disc. Middle, an associated inferior altitudinal defect. Right, sectoral swelling of the disc with flame or splinter hemorrhages is commonly seen. This is not typical of demyelinating optic neuritis.Nonarteritic anterior ischemic optic neuropathy is the most common form. Typically, there is acute onset of painless vision loss in one eye and an associated altitudinal field defect (Figure 3). For example, if the superior rim of the optic nerve acutely develops swelling and then becomes pale, a corresponding altitudinal cut would develop in the inferior visual field, respecting the horizontal meridian. Many patients first notice the vision loss upon waking up in the morning.13
Although patients with nonarteritic anterior ischemic optic neuropathy typically have vasculopathic risk factors such as hypertension, diabetes mellitus, peripheral vascular disease, or hypercholesterolemia, there is no proven causation between the two. The age of these patients ranges from 50 to 70, with an average age of 66.
The disc appears swollen and may have flame or splinter hemorrhages (Figure 3). The cup of the involved disc is typically small. The visual loss is believed to be the result of poor perfusion in the circulation of the posterior ciliary artery, which supplies the optic nerve head.1 If the other eye also has a small cup, it is considered to be at risk of future ischemic events. In one study,14 the opposite eye became involved within the next 5 years in 14.7% of all cases. The risk of recurrent disease in the same eye is low (6.4% in another study15).
Arteritic anterior ischemic optic neuropathy is more common in patients over age 70 and is usually due to giant cell arteritis, which has a significant association with polymyalgia rheumatica. Patients may have jaw claudication, proximal myalgia and arthralgia, scalp tenderness, headache, fatigue, and a significantly elevated erythrocyte sedimentation rate and C-reactive protein level. These features should be looked for in the review of systems, although patients may not have all of them.
The funduscopic examination may reveal a pale, swollen disc, peripapillary hemorrhages, branch or central retinal artery occlusions, or cotton-wool spots.
Temporal artery biopsy is the gold standard for diagnosis, but treatment with corticosteroids should not be delayed pending biopsy or other test results.1
Thrombocytosis has been associated with a higher risk of permanent vision loss in patients with giant cell arteritis.16
Posterior ischemic optic neuropathy is the least common form of ischemic optic neuropathy. This diagnosis should be entertained in older patients who report acute, painless vision loss but have a normal funduscopic examination. Giant cell arteritis must be considered first in this setting.
Bilateral posterior ischemic optic neuropathy can occur (although rarely) in patients undergoing cardiac or spinal surgery.17 Risk factors thought to be associated with perioperative disease include anemia, hypotension, substantial blood loss during the surgery, surgeries longer than 6.5 hours, carotid atherosclerosis, and diabetes.18
There are no effective treatments for most ischemic optic neuropathies with the crucial exception of giant cell arteritis.
Neuromyelitis optica (Devic disease)
Neuromyelitis optica (Devic disease) is a combination of optic neuritis and transverse myelitis (Table 2). Clinically, the disease spares the nervous system except for the optic nerves and spinal cord. The onset of the optic neuritis may precede or follow the onset of the transverse myelitis by up to 2 to 4 years.19 Usually, the optic neuritis is bilateral and retrobulbar and results in severe vision loss, worse than that seen in patients with MS.19,20
The transverse myelitis may cause paraplegia or quadriplegia, depending on the location of the lesion in the spinal cord (cervical vs thoracic). The transverse myelitis in neuromyelitis optica is distinct from that seen in MS. In neuromyelitis optica, the transverse myelitis is longitudinally extensive, spanning more than three vertebral bodies in length. In MS, spinal cord lesions usually are more discrete and involve one or two spinal cord segments.21
Recently, serum neuromyelitis optica immunoglobulin G (IgG) antibody has been shown to be a significant biomarker of this disease. Its sensitivity ranges from approximately 60% to 70% and its specificity is greater than 90%.22 This antibody binds to aquaporin-4, an important water-channel protein in the blood-brain barrier of the central nervous system, and evidence suggests that it is involved in the pathogenesis of the disease.23
Initially, it was proposed that MRI of the brain had to be normal for neuromyelitis optica to be diagnosed.21 However, the proposed 2006 criteria allow for some abnormal T2 and fluid-attenuated inversion recovery (FLAIR) hyperintensities in the periaqueductal gray matter and diencephalon.22
The spinal fluid in neuromyelitis optica may show a pleocytosis larger than that seen in MS (> 50 white blood cells per mm3) and may have a significant neutrophilic component.21 Oligoclonal bands are not typically present.
It is still debated whether neuromyelitis optica is a separate disease from MS or a subset of it. The implications of this debate may affect its management, as discussed below.
Inflammatory optic neuropathies
Inflammatory optic neuropathies can be caused by infections (eg, syphilis, cat scratch disease) or by noninfectious conditions (eg, sarcoidosis). Lyme disease is rarely a cause of retrobulbar optic neuritis but may cause papillitis.24 West Nile virus has also been reported to cause optic neuritis.25 Lupus may cause an optic neuropathy by inflammatory or ischemic mechanisms.26
Compressive optic neuropathies
Compressive optic neuropathies may be due to mass lesions, tumors, thyroid eye disease, or other orbital processes. MRI of the brain and orbits will confirm or rule out diagnoses associated with compressive optic neuropathy.
Genetic causes
Genetic causes of optic neuropathy include the Leber and Kjer hereditary optic neuropathies.
Leber optic neuropathy involves subacute and painless vision loss, with sequential involvement of both eyes over a period of weeks to months. This disorder predominantly affects men (80%–90% of patients) and is inherited from maternal mitochondrial DNA. The three most common mutations implicated in Leber optic neuropathy (located at base pairs 11,778, 3,460, and 14,484 in the mitochondrial DNA) are involved in more than 90% of cases. The prognosis for recovery varies depending on the genotype.27 These genes encode proteins that are part of complex I of the mitochondrial respiratory chain.28 Funduscopic examination most commonly shows circumpapillary telangiectasia, although up to one-third of patients can have a normal-appearing disc initially. Central vision is affected more severely than peripheral vision.29
Kjer autosomal-dominant optic atrophy is the most common hereditary optic neuropathy. This disease primarily affects children in the first decade of life with slowly progressive loss of vision. As with other optic neuropathies, there will eventually be pallor of the optic disc, a cecocentral scotoma, and loss of color perception. The OPA1 gene located on chromosome 3q28 has been implicated in most patients with dominant optic atrophy; a test is commercially available for diagnosis.30,31
Toxic and metabolic causes
Many agents can cause optic neuropathy. Toxins strongly associated with optic neuropathy include carbon monoxide, methanol, ethylene glycol, perchloroethylene, and tobacco. Drugs linked to optic neuropathy are ethambutol (Myambutol), clioquinol (Vioform), isoniazid (Nydrazid), amiodarone (Cordarone), linezolid (Zyvox), methotrexate, sildenafil (Viagra), oxymetazoline (contained in Afrin and other nasal sprays), and infliximab (Remicade).32–37 Additionally, several chemotherapeutic agents are known to cause optic atrophy, including vincristine (Oncovin), cisplatin (Platinol), carboplatin (Paraplatin), and paclitaxel (Abraxane, Onxol).
Nutritional deficiencies are presumed to have played a significant role in the endemics of optic neuropathy that have occurred in poor countries, such as in Cuba during the 1990s.38 Most nutritional optic neuropathies appear to be exacerbated by tobacco.39
MRI ASSESSES RISK OF MS
The diagnosis of optic neuritis is clinical, based on the history and physical findings.
However, MRI of the brain and orbits with gadolinium contrast has become the cornerstone of the evaluation in patients with optic neuritis. And MRI not only helps confirm the clinical diagnosis, but it also more importantly offers very strong prognostic information about the risk of future demyelinating events and MS.
Gadolinium-enhanced fat-saturated T1-weighted MRI of the orbits is the best sequence to show the inflammation of the optic nerve in optic neuritis (fat saturation is necessary to hide the bright signal of the orbital fat tissue).
Contrast-enhanced MRI can also help differentiate optic neuritis from nonarteritic anterior ischemic optic neuropathy. MRI of the orbits with gadolinium contrast shows enhancement of the affected optic nerve in approximately 95% of cases of optic neuritis, whereas optic nerve enhancement rarely occurs in nonarteritic anterior ischemic optic neuropathy.40
Brain MRI may show other white matter lesions (either hyperintensities on T2-weighted images or enhancement of T1-weighted images postcontrast), which denote a higher risk of developing MS. In 15-year follow-up data from the ONTT, monosymptomatic patients with no white matter lesions had a 25% risk of MS (defined at the time the ONTT was conducted as a second demyelinating event), while those with one lesion or more had a 72% risk.41
An earlier, prospective study in 102 Italian patients with optic neuritis found the risk of developing MS to be about 36% at 6 years and 42% at 8 years (using the Posner diagnostic criteria). When brain MRI data were analyzed, those with one or more lesions had a 52% risk of developing MS at 8 years, whereas those with no MRI lesions did not develop MS.42
Other studies have stratified the risk of MS in patients with clinically isolated syndromes (including not only optic neuritis, but also other neurologic symptoms such as brainstem, motor, or sensory deficits). At mean follow-ups ranging from 5 to 14 years, the risk of developing MS was 8% to 24% in patients with normal findings on brain MRI compared with 56% to 88% in those with abnormal MRI findings.43,44
Optic neuritis patients with atypical white matter lesions on brain MRI may benefit from lumbar puncture to look for oligoclonal bands, to measure the IgG index and the IgG synthesis rate, and to test for myelin basic protein in the cerebrospinal fluid. Of patients with acute optic neuritis, 79% have abnormalities in their cerebrospinal fluid. Oligoclonal bands are present in 69%, and for patients with oligoclonal bands, the 5-year probability of developing MS is estimated to be 65%, compared with 10% in those without bands. If the patient has no oligoclonal bands and has normal findings on brain MRI, he or she will not have MS 5 years later.45–47
Patients with optic neuritis who have no white matter lesions on brain MRI may be further risk-stratified on the basis of their clinical findings. In the ONTT 15-year follow-up, MS did not develop in any patient who had no brain lesions on baseline MRI, no prior optic neuritis in the contralateral eye, and no prior neurologic symptoms or signs, even if the patient had severe disc swelling (eg, peripapillary hemorrhage or retinal exudates) or if vision was reduced to no light perception.41
CASE CONTINUED: FINDINGS ON MRI
Figure 4. The patient’s magnetic resonance image. Top, an axial T2 image with contrast; bottom, sagittal T1 image with contrast. The white matter lesions indicate she is at risk of developing multiple sclerosis.Our patient undergoes MRI, which shows lesions on axial T2 and sagittal T1 imaging with contrast (Figure 4). Of note, there are significant lesions perpendicular to the corpus callosum (Dawson fingers), some of which enhance with contrast. The enhancement indicates breakdown of the blood-brain barrier and suggests that there is active inflammation in the white matter.
Patients in the ONTT were randomized to receive one of three treatments:
Oral prednisone 1 mg/kg/day for 14 days and then tapered over 4 days
Intravenous methylprednisolone (Solu-Medrol) 250 mg four times per day for 3 days followed by oral prednisone 1 mg/kg/day for 11 days and then tapered for 4 days
Oral placebo for 14 days.
The primary visual outcomes measured were visual acuity and contrast sensitivity.48
Patients who received intravenous methylprednisolone recovered their visual function faster, although the visual outcomes after 6 months were no better with methylprednisolone than with placebo or oral prednisone. Intravenous methylprednisolone also reduced the risk of MS within the first 2 years in patients with high-risk brain MRIs.
Surprisingly, patients in the oral prednisone group had a higher risk of recurrent optic neuritis in both eyes than did patients given intravenous methylprednisolone or placebo (30% at 2 years with oral prednisone vs 16% with placebo and 13% with intravenous methylprednisolone).48 At 10 years, the risk of recurrent optic neuritis was still higher in the oral prednisone group (44%) than in the intravenous methylprednisolone group (29%) (P = .03). However, the difference between the oral prednisone and placebo groups was no longer statistically significant (P = .07).49 Oral prednisone alone is therefore contraindicated in the treatment of typical unilateral demyelinating optic neuritis.
Many patients can now be treated with intravenous infusions of methylprednisolone at home for episodes of optic neuritis.
Risks vs benefits of corticosteroid therapy
When deciding whether to treat an individual patient who has optic neuritis with intravenous corticosteroids, one should consider all the benefits and risks.
Corticosteroids do not affect long-term visual outcome, although they do hasten recovery. Patients with mild vision loss (visual acuity better than 20/40), no significant visual field loss, and no enhancing lesions on brain MRI can forgo therapy with intravenous corticosteroids.
On the other hand, we strongly favor intravenous corticosteroid treatment in patients who have both acute optic neuritis and high signal abnormalities on brain MRI, since it may delay the onset of MS. In addition, patients with severe vision loss should receive intravenous corticosteroids to hasten their recovery. In the rare circumstance in which intravenous corticosteroids are not available, high-dose oral methyl-prednisolone (500 mg daily for 5 days and then tapered over 10 days) may be acceptable.50
The side effects of corticosteroids are minimal when they are given for a brief time in otherwise healthy patients. The most common side effects are mood changes, facial flushing, sleep perturbations, weight gain, and dyspepsia.48
IMMUNOGLOBUL IN: LITTLE BENEFIT
In a double-blind, randomized trial, patients were treated with intravenous immunoglobulin 0.4 g/kg or placebo on days 0, 1, 2, 30, and 60. No difference was found in the primary outcomes of contrast sensitivity, visual acuity, or color vision from 1 week up to 6 months. There was also no significant difference in MRI outcomes between the two groups. The number of relapses was similar between both groups after 6 months.51,52
PLASMA EXCHANGE: FEW DATA
Data on plasma exchange are too scarce for us to make any recommendations. In one trial in 10 patients with severe optic neuritis, 3 patients appeared to benefit from plasma exchange. All patients had already received two doses of intravenous steroids.53
IMMUNOMODULATORY THERAPY MAY PREVENT MULTIPLE SCLEROSIS IN SOME
The most important clinical decision to make in patients with optic neuritis is whether to begin immunomodulatory therapy. Patients who may benefit the most from immunomodulatory therapy are those with abnormal white matter lesions on brain MRI, as they are at higher risk of developing MS.
Collectively, data from three studies indicate that early treatment in patients with clinically isolated syndromes, such as optic neuritis, reduces the rate of MS to 35% (from 50% without treatment) and reduces the number of new active lesions on MRI by approximately 50%.54–56
In addition, the Betaferon/Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT) trial57 found that at 3 years the rate of disability was 40% lower in patients who started immunomodulatory therapy (interferon beta-1b; Betaseron) early vs later. (Early treatment meant starting within 60 days of the clinically isolated syndrome; late treatment began 2 years later.) This study suggests that early treatment may reduce future disability, although these results need to be confirmed in prospective trials.
Therefore, once the diagnosis is secure, patients with optic neuritis should be referred to a neurologist with experience in treating MS to begin treatment with immunomodulatory therapy, such as glatiramer acetate (Copaxone), interferon beta-1a (Avonex, Refib), or interferon beta-1b (Betaseron).
Patients who have a normal MRI of the brain may consider deferring therapy, since they are at low risk of developing MS. These patients should undergo surveillance MRI (at least annually at first) to look for the development of white matter lesions, as the ONTT showed even this cohort has a 22% risk of developing MS.
If neuromyelitis optica is suspected (ie, in patients with severe unilateral or bilateral vision loss, recurrent optic neuritis, paraplegia, or quadriplegia), the serum neuromyelitis optic antibody can be tested, keeping in mind that 30% to 40% of patients with neuromyelitis optica will be seronegative for this antibody. Other tests supporting the diagnosis of neuromyelitis optica may include an MRI of the spine showing longitudinally extensive transverse myelitis, a polymorphonuclear pleocytosis in the cerebrospinal fluid, absent oligoclonal bands in the cerebrospinal fluid, and normal MRI of the brain (with some possible signal abnormalities in the periaqueductal gray matter and around the diencephalon).
Because neuromyelitis optica is believed to be mediated primarily by the humoral immune system, immunomodulatory therapy is not a first-line treatment. Patients with neuromyelitis optica can be treated initially with corticosteroids, intravenous immunoglobulin therapy, plasma exchange, or immunosuppressive agents such as azathioprine (Imuran), rituximab (Rituxan), or cyclophosphamide (Cytoxan). The choice of medication should be deferred to a neurologist familiar with treatment of this disorder.
The risk of MS may be lower in children than in adults. One large, retrospective study found the cumulative risk of developing MS (the study predated the McDonald criteria) was 13% at 10 years and 19% by 20 years.58 More recently, a large series from Toronto reported a higher rate of MS development in children with optic neuritis (36% at two years).59 By comparison, studies of adults with unilateral optic neuritis found a 38% to 39% risk of converting to MS at 10 years.5,41 The use of immunomodulatory therapies to reduce the risk of MS has not been well studied in children, since the prevalence is low in this age group.
The most common side effects of the beta-interferons are flulike symptoms (fatigue, myalgia), injection site reactions, and elevations of aminotransferase levels. Most patients are able to tolerate the side effects if the beta-interferon is taken with acetaminophen (Tylenol) or with over-the-counter nonsteroidal anti-inflammatory drugs.
Glatiramer acetate does not cause flulike symptoms or elevate aminotransferases, but it does require more frequent injections. Rarely, it may cause an idiosyncratic panic-like attack.
CASE CONTINUED
The best therapeutic regimen for this patient would be intravenous methylprednisolone, and subsequently a disease-modifying, immunomodulatory agent (interferon beta or glatiramer acetate). Our patient chose to start therapy with interferon beta-1a 30 μg intramuscularly once a week. She has been tolerating the medication well and has had no new neurologic or visual symptoms for the past 2 years.
References
Liu GT. Visual loss: optic neuropathies. In: Liu GT, Volpe NJ, Galetta SL, editors. Neuro–Ophthalmology: Diagnosis and Management. Philadelphia, PA: WB Saunders, 2001:103–187.
Wray SH. Optic neuritis. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Philadelphia, PA: WB Saunders, 1994:2539–2568.
Optic Neuritis Study Group. The clinical profile of optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol1991; 109:1673–1678.
Percy AK, Nobrega FT, Kurland LT. Optic neuritis and multiple sclerosis: an epidemiologic study. Arch Ophthalmol1972; 87:135–139.
Rodriguez M, Siva A, Cross SA, O’Brien PC, Kurland LT. Optic neuritis: a population–based study in Olmsted County, Minnesota. Neurology1995; 45:244–250.
Phillips PH, Newman NJ, Lynn MJ. Optic neuritis in African Americans. Arch Neurol1998; 55:186–192.
Brady KM, Brar AS, Lee AG, Coats DK, Paysse EA, Steinkuller PG. Optic neuritis in children: clinical features and visual outcome. J AAPOS1999; 3:98–103.
Kriss A, Francis DA, Cuendet B, et al. Recovery after optic neuritis in childhood. J Neurol Neurosurg Psychiatry1988; 51:1253–1258.
Beck RW. The Optic Neuritis Treatment Trial. Arch Ophthalmol1988; 106:1051–1053.
Optic Neuritis Study Group. Visual function 15 years after optic neuritis. Ophthalmology2008; 115:1079–1082.
Cox TA, Thompson HS, Corbett JJ. Relative afferent pupillary defects in optic neuritis. Am J Ophthalmol1981; 92:685–690.
Arnold AC. Ischemic optic neuropathies. Ophthalmol Clin North Am2001; 14:83–98.
Newman NJ, Scherer R, Langenberg P, et al. The fellow eye in NAION: report from the ischemic optic neuropathy decompression trial follow–up study. Am J Ophthalmol2002; 134:317–328.
Hayreh SS, Podhajsky PA, Zimmerman B. Ipsilateral recurrence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol2001; 132:734–742.
Liozon E, Herrmann F, Ly K, et al. Risk factors for visual loss in giant cell (temporal) arteritis: a prospective study of 174 patients. Am J Med2001; 111:211–217.
Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol2005; 50:15–26.
American Society of Anesthesiologists Task Force on Perioperative Blindness. Practice advisory for perioperative visual loss associated with spine surgery: a report by the American Society of Anesthesiologists Task Force on Perioperative Blindness. Anesthesiology2006; 104:1319–1328.
Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology2007; 114:810–815.
Papais-Alvarenga RM, Carellos SC, Alvarenga MP, Holander C, Bichara RP, Thuler LC. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol2008; 126:12–16.
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology1999; 53:1107–1114.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology2006; 66:1485–1489.
Takahashi T, Fujihara K, Nakashima I, et al. Anti–aquaporin–4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain2007; 130:1235–1243.
Sibony P, Halperin J, Coyle PK, Patel K. Reactive Lyme serology in optic neuritis. J Neuroophthalmol2005; 25:71–82.
Anninger WV, Lomeo MD, Dingle J, Epstein AD, Lubow M. West Nile virus–associated optic neuritis and chorioretinitis. Am J Ophthalmol2003; 136:1183–1185.
Jabs DA, Miller NR, Newman SA, Johnson MA, Stevens MB. Optic neuropathy in systemic lupus erythematosus. Arch Ophthalmol1986; 104:564–568.
Howell N. LHON and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol2003; 37:94–108.
Newman NJ. Hereditary optic neuropathies. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. Philadelphia, PA: Lippincott Williams & Wilkins, 2005;465–501.
Smith JL, Hoyt WF, Susac JO. Ocular fundus in acute Leber optic neuropathy. Arch Ophthalmol1973; 90:349–354.
Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol2003; 87:48–53.
Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin– related GTPase, is mutated in autosomal dominant atrophy linked to chromosome 3q28. Nat Genet2000; 26:211–215.
McKinley SH, Foroozan R. Optic neuropathy associated with linezolid treatment. J Neuroophthalmol2005; 25:18–21.
Melamud A, Kosmorsky GS, Lee MS. Ocular ethambutol toxicity. Mayo Clin Proc2003; 78:1409–1411.
Kerrison JB. Optic neuropathies caused by toxins and adverse drug reactions. Ophthalmol Clin North Am2004; 17:481–488.
Pomeranz HD, Bhavsar AR. Nonarteritic ischemic optic neuropathy developing soon after use of sildenafil (Viagra): a report of seven new cases. J Neuroophthalmol2005; 25:9–13.
Fivgas GD, Newman NJ. Anterior ischemic optic neuropathy following the use of a nasal decongestant. Am J Ophthalmol1999; 127:104–106.
The Cuba Neuropathy Field Investigation Team. Epidemic optic neuropathy in Cuba—clinical characterization and risk factors. N Engl J Med1995; 333:1176–1182.
Solberg Y, Rosner M, Belkin M. The association between cigarette smoking and ocular diseases. Surv Ophthalmol1998; 42:535–547.
Rizzo JF, Andreoli CM, Rabinov JD. Use of magnetic resonance imaging to differentiate optic neuritis and nonarteritic anterior ischemic optic neuropathy. Ophthalmology2002; 109:1679–1684.
The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final Optic Neuritis Treatment Trial follow-up. Arch Neurol2008; 65:727–732.
Ghezzi A, Martinelli V, Torri V, et al. Long–term follow–up of isolated optic neuritis: the risk of developing multiple sclerosis, its outcome, and the prognostic role of paraclinical tests. J Neurol1999; 246:770– 775.
Brex PA, Ciccarelli O, O'Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med2002; 346:158–164.
Tintore M, Rovira A, Rio J, et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology2006; 67:968–972.
Söderström M, Ya–Ping J, Hillert J. Optic neuritis: prognosis for multiple sclerosis from MRI, CSF, and HLA findings. Neurology1998; 50:708–714.
Frederiksen JL, Madsen HO, Ryder LP, Larsson HB, Morling N, Svejgaard A. HLA typing in acute optic neuritis: relation to multiple sclerosis and magnetic resonance imaging findings. Arch Neurol1997; 54:76–80.
Frederiksen JL, Larsson HB, Oleson J. Correlation of magnetic resonance imaging and CSF findings in patients with acute monosymptomatic optic neuritis. Acta Neurol Scand1992; 86:317–322.
Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med1992; 326:581–588.
Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol2003; 121:944–949.
Sellebjerg F, Nielsen HS, Frederiksen JL, Olesen J. A randomized, controlled trial of oral high-dose methylprednisolone in acute optic neuritis. Neurology1999; 52:1479–1484.
Noseworthy JH, O’Brien PC, Peterson TM, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology2001; 56:1514–1522.
Roed HG, Langkilde A, Sellebjerg F, et al. A double–blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology2005; 64:804–810.
Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology2004; 63:1081–1083.
CHAMPS Study Group. Interferon beta-1a for optic neuritis patients at high risk for multiple sclerosis. Am J Ophthalmol2001; 132:463– 471.
Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet2001; 357:1576–1582.
Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology2006; 67:1242–1249.
Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet2007; 370:389–397.
Lucchinetti CF, Kiers L, O’Duffy A, et al. Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology1997; 49:1413–1418.
Wilejto M, Shroff M, Buncic JR, Kennedy J, Goia C, Banwell B. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology2006; 67:258–262.
References
Liu GT. Visual loss: optic neuropathies. In: Liu GT, Volpe NJ, Galetta SL, editors. Neuro–Ophthalmology: Diagnosis and Management. Philadelphia, PA: WB Saunders, 2001:103–187.
Wray SH. Optic neuritis. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Philadelphia, PA: WB Saunders, 1994:2539–2568.
Optic Neuritis Study Group. The clinical profile of optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol1991; 109:1673–1678.
Percy AK, Nobrega FT, Kurland LT. Optic neuritis and multiple sclerosis: an epidemiologic study. Arch Ophthalmol1972; 87:135–139.
Rodriguez M, Siva A, Cross SA, O’Brien PC, Kurland LT. Optic neuritis: a population–based study in Olmsted County, Minnesota. Neurology1995; 45:244–250.
Phillips PH, Newman NJ, Lynn MJ. Optic neuritis in African Americans. Arch Neurol1998; 55:186–192.
Brady KM, Brar AS, Lee AG, Coats DK, Paysse EA, Steinkuller PG. Optic neuritis in children: clinical features and visual outcome. J AAPOS1999; 3:98–103.
Kriss A, Francis DA, Cuendet B, et al. Recovery after optic neuritis in childhood. J Neurol Neurosurg Psychiatry1988; 51:1253–1258.
Beck RW. The Optic Neuritis Treatment Trial. Arch Ophthalmol1988; 106:1051–1053.
Optic Neuritis Study Group. Visual function 15 years after optic neuritis. Ophthalmology2008; 115:1079–1082.
Cox TA, Thompson HS, Corbett JJ. Relative afferent pupillary defects in optic neuritis. Am J Ophthalmol1981; 92:685–690.
Arnold AC. Ischemic optic neuropathies. Ophthalmol Clin North Am2001; 14:83–98.
Newman NJ, Scherer R, Langenberg P, et al. The fellow eye in NAION: report from the ischemic optic neuropathy decompression trial follow–up study. Am J Ophthalmol2002; 134:317–328.
Hayreh SS, Podhajsky PA, Zimmerman B. Ipsilateral recurrence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol2001; 132:734–742.
Liozon E, Herrmann F, Ly K, et al. Risk factors for visual loss in giant cell (temporal) arteritis: a prospective study of 174 patients. Am J Med2001; 111:211–217.
Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol2005; 50:15–26.
American Society of Anesthesiologists Task Force on Perioperative Blindness. Practice advisory for perioperative visual loss associated with spine surgery: a report by the American Society of Anesthesiologists Task Force on Perioperative Blindness. Anesthesiology2006; 104:1319–1328.
Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology2007; 114:810–815.
Papais-Alvarenga RM, Carellos SC, Alvarenga MP, Holander C, Bichara RP, Thuler LC. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol2008; 126:12–16.
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology1999; 53:1107–1114.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology2006; 66:1485–1489.
Takahashi T, Fujihara K, Nakashima I, et al. Anti–aquaporin–4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain2007; 130:1235–1243.
Sibony P, Halperin J, Coyle PK, Patel K. Reactive Lyme serology in optic neuritis. J Neuroophthalmol2005; 25:71–82.
Anninger WV, Lomeo MD, Dingle J, Epstein AD, Lubow M. West Nile virus–associated optic neuritis and chorioretinitis. Am J Ophthalmol2003; 136:1183–1185.
Jabs DA, Miller NR, Newman SA, Johnson MA, Stevens MB. Optic neuropathy in systemic lupus erythematosus. Arch Ophthalmol1986; 104:564–568.
Howell N. LHON and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol2003; 37:94–108.
Newman NJ. Hereditary optic neuropathies. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. Philadelphia, PA: Lippincott Williams & Wilkins, 2005;465–501.
Smith JL, Hoyt WF, Susac JO. Ocular fundus in acute Leber optic neuropathy. Arch Ophthalmol1973; 90:349–354.
Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol2003; 87:48–53.
Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin– related GTPase, is mutated in autosomal dominant atrophy linked to chromosome 3q28. Nat Genet2000; 26:211–215.
McKinley SH, Foroozan R. Optic neuropathy associated with linezolid treatment. J Neuroophthalmol2005; 25:18–21.
Melamud A, Kosmorsky GS, Lee MS. Ocular ethambutol toxicity. Mayo Clin Proc2003; 78:1409–1411.
Kerrison JB. Optic neuropathies caused by toxins and adverse drug reactions. Ophthalmol Clin North Am2004; 17:481–488.
Pomeranz HD, Bhavsar AR. Nonarteritic ischemic optic neuropathy developing soon after use of sildenafil (Viagra): a report of seven new cases. J Neuroophthalmol2005; 25:9–13.
Fivgas GD, Newman NJ. Anterior ischemic optic neuropathy following the use of a nasal decongestant. Am J Ophthalmol1999; 127:104–106.
The Cuba Neuropathy Field Investigation Team. Epidemic optic neuropathy in Cuba—clinical characterization and risk factors. N Engl J Med1995; 333:1176–1182.
Solberg Y, Rosner M, Belkin M. The association between cigarette smoking and ocular diseases. Surv Ophthalmol1998; 42:535–547.
Rizzo JF, Andreoli CM, Rabinov JD. Use of magnetic resonance imaging to differentiate optic neuritis and nonarteritic anterior ischemic optic neuropathy. Ophthalmology2002; 109:1679–1684.
The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final Optic Neuritis Treatment Trial follow-up. Arch Neurol2008; 65:727–732.
Ghezzi A, Martinelli V, Torri V, et al. Long–term follow–up of isolated optic neuritis: the risk of developing multiple sclerosis, its outcome, and the prognostic role of paraclinical tests. J Neurol1999; 246:770– 775.
Brex PA, Ciccarelli O, O'Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med2002; 346:158–164.
Tintore M, Rovira A, Rio J, et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology2006; 67:968–972.
Söderström M, Ya–Ping J, Hillert J. Optic neuritis: prognosis for multiple sclerosis from MRI, CSF, and HLA findings. Neurology1998; 50:708–714.
Frederiksen JL, Madsen HO, Ryder LP, Larsson HB, Morling N, Svejgaard A. HLA typing in acute optic neuritis: relation to multiple sclerosis and magnetic resonance imaging findings. Arch Neurol1997; 54:76–80.
Frederiksen JL, Larsson HB, Oleson J. Correlation of magnetic resonance imaging and CSF findings in patients with acute monosymptomatic optic neuritis. Acta Neurol Scand1992; 86:317–322.
Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med1992; 326:581–588.
Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol2003; 121:944–949.
Sellebjerg F, Nielsen HS, Frederiksen JL, Olesen J. A randomized, controlled trial of oral high-dose methylprednisolone in acute optic neuritis. Neurology1999; 52:1479–1484.
Noseworthy JH, O’Brien PC, Peterson TM, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology2001; 56:1514–1522.
Roed HG, Langkilde A, Sellebjerg F, et al. A double–blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology2005; 64:804–810.
Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology2004; 63:1081–1083.
CHAMPS Study Group. Interferon beta-1a for optic neuritis patients at high risk for multiple sclerosis. Am J Ophthalmol2001; 132:463– 471.
Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet2001; 357:1576–1582.
Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology2006; 67:1242–1249.
Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet2007; 370:389–397.
Lucchinetti CF, Kiers L, O’Duffy A, et al. Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology1997; 49:1413–1418.
Wilejto M, Shroff M, Buncic JR, Kennedy J, Goia C, Banwell B. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology2006; 67:258–262.
Optic neuritis is most common in women in their 20s and 30s, whereas ischemic optic neuropathy, which is more common, primarily affects older people.
The diagnosis of optic neuritis is primarily clinical, but magnetic resonance imaging confirms the diagnosis and, more importantly, assesses the risk of MS.
Intravenous methylprednisolone (Solu-Medrol) does not affect the long-term visual outcome, but it speeds visual recovery and reduces the risk of MS. Surprisingly, oral prednisone seems to increase the risk of recurrent optic neuritis and is therefore contraindicated.
Early treatment with interferon beta reduces the risk of MS and should be considered in patients at high risk.
A 28-year-old woman comes in for her annual checkup. Her physician notices a palpable, painless, 1-cm, well-demarcated mass in the left breast at the 3 o’clock position 2 cm from the nipple, with no associated skin changes, nipple retraction, or discharge. The patient has no personal or family history of breast cancer.
Given the patient’s age, physical findings, and medical history, the clinician believes it unlikely that the patient has cancer. How should she proceed with the workup of this patient?
PHYSICAL FINDINGS OF A BREAST MASS ARE NOT EXCLUSIVE
Figure 1. A simple cyst in the left breast. All three mammographic views—craniocaudal (A), mediolateral oblique (B), and spot-compression (C)—show a round, well-circumscribed mass in the mid-breast. Ultrasonography (D) shows a round, well-circumscribed anechoic lesion with a sharply defined posterior wall and posterior acoustic enhancement.Breast cancer is the most common female malignancy and the second-leading cause of cancer deaths in the United States.1 The incidence is low in young women and increases with advancing age. Benign breast disease is common in young women and less common in postmenopausal women.2,3 However, the discovery of a breast mass, whether by the woman herself or by a clinician, is a common occurrence and distressing for any woman.
Benign lesions tend to have discrete, well-defined margins and are typically mobile. Malignant lesions may be firm, may have indistinct borders, and are often immobile.2 Although most breast masses found by palpation are benign, imaging is the critical next step in the workup to help determine if the mass is benign or malignant.
Benign palpable masses include:
Figure 2. Fibroadenoma. On mammography, the craniocaudal (A) and mediolateral oblique (B) views with a bright metallic marker (arrows) show a round, well-circumscribed mass in the upper outer quadrant of the left breast. Ultrasonography (C) shows an oval, well-circumscribed, mildly heterogeneous, hypoechoic mass that is wider than tall, indicating a benign mass.Cysts (Figure 1)
Fibroadenomas (Figure 2)
Prominent fat lobules
Lymph nodes
Oil cysts
Lipomas
Hamartomas (Figure 3)
Hematomas
Fat necrosis
Galactoceles.
Malignant palpable masses include:
Figure 3. Hamartoma. Craniocaudal (A) and mediolateral oblique (B) mammographic views of the left breast show an apparently encapsulated, heterogeneous mass that contains fat mixed with fibroglandular tissue.Invasive ductal and lobular carcinoma (Figure 4)
Ductal carcinoma in situ (which rarely presents as a palpable mass.)
HISTORY AND PHYSICAL EXAMINATION
To ensure that imaging provides the most useful information about a palpable breast lump, it is important to first do a careful history and physical examination. Important aspects of the history include family history, personal history of breast cancer, and any previous breast biopsies. The onset and duration of the palpable mass, changes in its size, the relationship of these changes to the menstrual cycle, and the presence or lack of tenderness are additional important elements of the history.
Figure 4. Infiltrating ductal carcinoma. Craniocaudal (A) and mediolateral oblique (B) mammographic views of the right breast show an irregular, mildly spiculated, high-density lesion in the posterior, medial breast. Ultrasonography (C) shows an irregularly shaped hypoechoic mass which is taller than wide (a profile tending to indicate malignancy) and has mild posterior acoustic shadowing.On examination, it is important to note the clock-face location, size, texture, tenderness, and mobility of the lump. Accompanying nipple discharge and skin erythema or retraction are also important to report. In addition to conveying the location of the mass to the radiologist, it is equally important that the patient know what features the physician feels. This way, if the clinical information from the ordering physician is not available at the time of the radiologic evaluation, the patient will be able to guide the radiologist to the region of concern.
IMAGING TECHNIQUES
Mammography and ultrasonography are the primary imaging studies for evaluating palpable breast masses. Typically, in women under age 30, ultrasonography is the first or the only test ordered to evaluate the abnormality.4 In women age 30 or older, diagnostic mammography is typically the first test ordered. If mammography indicates that the palpable mass is not benign, then ultrasonography is the next study to be done.3 Although a powerful tool, magnetic resonance imaging of the breast does not currently have a role in the workup of a palpable abnormality and should not be used as a decision-delaying tactic or in place of biopsy.
Screening or diagnostic mammography?
Mammography is used in both screening and diagnosis. Screening mammography consists of two standard views of each breast—craniocaudal and mediolateral oblique—and is appropriate for asymptomatic women.
Women age 30 or older who present with a palpable breast mass require diagnostic mammography, in which standard mammographic views are obtained, as well as additional views (eg, tangential or spot-compression views) to better define the area of clinical concern. In a tangential view, a metallic skin marker is placed on the skin overlying the site of the palpable abnormality.
On mammography, a suspicious palpable mass has an irregular shape with spiculated margins. A benign mass typically has a round shape with well-circumscribed margins. If the palpable abnormality is not mammographically benign (eg, if it does not look like a lymph node, lipoma, or degenerating fibroadenoma), then ultrasonography is performed.
Mammography is less sensitive in younger women (ie, under age 30) because their breast tissue tends to be dense and glandular, whereas the tissue becomes more “fat-replaced” with age.3
Ultrasonography plays a complementary role
Ultrasonography complements diagnostic mammography and can be used as a first imaging study to evaluate a palpable breast mass in a young woman (ie, under age 30) with dense breast tissue. Ultrasonography is helpful in distinguishing cystic lesions from solid masses. It helps the radiologist delineate the shape, borders, and acoustic properties of the mass. It is also performed when a palpable mass is mammographically occult. When a mass appears suspicious on either mammography or ultrasonography, ultrasonography can be used to guide biopsy.
A suspicious mass on ultrasonography classically appears “taller than wide” and has posterior acoustic shadowing. Microlobulations and a spiculated margin also raise concern for malignancy. A benign sonographic appearance of a palpable mass includes a “wider than tall” (ellipsoid) shape, with homogeneous echogenicity, and four or fewer gentle lobulations. A thin, echogenic capsule also suggests the mass is benign.
Core-needle biopsy with ultrasonographic guidance
Core-needle biopsy is performed with a large-diameter (14-gauge to 18-gauge) needle to obtain tissue cores for histologic analysis. It has gained popularity over fine-needle aspiration because it includes surrounding tissue architecture, thus providing a more definitive histologic diagnosis.
Pathologic information obtained from core-needle biopsy allows the radiologist and surgeon to counsel the patient and determine the best surgical management or follow-up imaging study. If a clinician performs fine-needle biopsy in the office, it should be preceded by an imaging workup of the palpable finding.
WHAT IS APPROPRIATE FOR OUR 28-YEAR-OLD PATIENT?
Because she is under age 30, ultrasonography is the initial study of choice to evaluate the mass. If a simple cyst is detected, she can be reassured that the lesion is benign, and no subsequent follow-up is required. If the lesion is a solid mass with benign features, mammography may be considered, the patient may be followed with short-interval imaging (every 6 months) depending on patient-specific factors such as family history, or the mass can be biopsied. If the lesion is a solid mass with suspicious or malignant features, mammography with spot-compression views should be performed, and the patient should undergo core-needle biopsy with ultrasonographic guidance.
In a patient age 30 or older, diagnostic mammography is the imaging study of first choice.3 If the mass is clearly benign on mammography, no additional imaging would be necessary. If mammography fails to image the mass or shows it to have benign features such as fat, then the patient can undergo ultrasonography for further evaluation and confirmation of the clinical and mammographic findings. If the mass appears suspicious or malignant on mammography, ultrasonography is the next step, as it can help characterize the lesion and be used to guide core-needle biopsy.
IF A PREGNANT WOMAN HAS A PALPABLE BREAST MASS
Most publications on breast cancer in pregnancy report a prevalence of 3 per 10,000 pregnancies, accounting for 3% of all breast cancers diagnosed.5 Therefore, imaging evaluation of a palpable mass should not be postponed.
Hormonal changes throughout pregnancy may increase the nodularity of breast tissue, raising the concern of palpable masses. Additionally, there is a higher prevalence of galactoceles and lactating adenomas in these patients. Because contrasting fatty breast tissue is lost during pregnancy and because of the need to minimize radiation exposure, ultrasonography is often the imaging test of first choice. If mammography is required, the radiation dose is very low and the patient’s abdomen and pelvis can be shielded.6 In this situation, the patient can be reassured that the imaging test is not jeopardizing her fetus.
WHAT WORKUP IS REQUIRED IN MEN?
Breast cancer in men is rare, accounting for less than 0.5% of all cases.7 Most often, a palpable breast mass in a man presents as unilateral gynecomastia. Gynecomastia occurs in a bimodal age distribution (in the 2nd and 7th decades) and has a variety of hormonal and drug-related causes. Despite the low prevalence of breast cancer in men, the combination of mammography and ultrasonography is recommended for evaluation at all ages.
References
Klein S. Evaluation of palpable breast masses. Am Fam Physician2005; 71:1731–1738.
Pruthi S. Detection and evaluation of a palpable breast mass. Mayo Clin Proc2001; 76:641–648.
Harvey JA. Sonography of palpable breast masses. Semin Ultrasound CT MR2006; 27:284–297.
Mehta TS. Current uses of ultrasound in the evaluation of the breast. Radiol Clin North Am2003; 41:841–856.
Gallenberg MM, Lopines CL. Breast cancer and pregnancy. Semin Oncol1989; 16:369–376.
Barnavon Y, Wallack MK. Management of the pregnant patient with carcinoma of the breast. Surg Gynecol Obstet1990; 171:347–352.
Cardenosa G. The Core Curriculum: Breast Imaging. Philadelphia: Lippincott Williams and Wilkins, 2003;304.
Lauren Stein, MD Imaging Institute, Cleveland Clinic
Melanie Chellman-Jeffers, MD Center for Specialized Women’s Health and Section of Breast Imaging, Department of Diagnostic Radiology, Imaging Institute, Cleveland Clinic
Address: Melanie Chellman-Jeffers, MD, Imaging Institute, Section of Breast Imaging, A10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Lauren Stein, MD Imaging Institute, Cleveland Clinic
Melanie Chellman-Jeffers, MD Center for Specialized Women’s Health and Section of Breast Imaging, Department of Diagnostic Radiology, Imaging Institute, Cleveland Clinic
Address: Melanie Chellman-Jeffers, MD, Imaging Institute, Section of Breast Imaging, A10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Author and Disclosure Information
Lauren Stein, MD Imaging Institute, Cleveland Clinic
Melanie Chellman-Jeffers, MD Center for Specialized Women’s Health and Section of Breast Imaging, Department of Diagnostic Radiology, Imaging Institute, Cleveland Clinic
Address: Melanie Chellman-Jeffers, MD, Imaging Institute, Section of Breast Imaging, A10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
A 28-year-old woman comes in for her annual checkup. Her physician notices a palpable, painless, 1-cm, well-demarcated mass in the left breast at the 3 o’clock position 2 cm from the nipple, with no associated skin changes, nipple retraction, or discharge. The patient has no personal or family history of breast cancer.
Given the patient’s age, physical findings, and medical history, the clinician believes it unlikely that the patient has cancer. How should she proceed with the workup of this patient?
PHYSICAL FINDINGS OF A BREAST MASS ARE NOT EXCLUSIVE
Figure 1. A simple cyst in the left breast. All three mammographic views—craniocaudal (A), mediolateral oblique (B), and spot-compression (C)—show a round, well-circumscribed mass in the mid-breast. Ultrasonography (D) shows a round, well-circumscribed anechoic lesion with a sharply defined posterior wall and posterior acoustic enhancement.Breast cancer is the most common female malignancy and the second-leading cause of cancer deaths in the United States.1 The incidence is low in young women and increases with advancing age. Benign breast disease is common in young women and less common in postmenopausal women.2,3 However, the discovery of a breast mass, whether by the woman herself or by a clinician, is a common occurrence and distressing for any woman.
Benign lesions tend to have discrete, well-defined margins and are typically mobile. Malignant lesions may be firm, may have indistinct borders, and are often immobile.2 Although most breast masses found by palpation are benign, imaging is the critical next step in the workup to help determine if the mass is benign or malignant.
Benign palpable masses include:
Figure 2. Fibroadenoma. On mammography, the craniocaudal (A) and mediolateral oblique (B) views with a bright metallic marker (arrows) show a round, well-circumscribed mass in the upper outer quadrant of the left breast. Ultrasonography (C) shows an oval, well-circumscribed, mildly heterogeneous, hypoechoic mass that is wider than tall, indicating a benign mass.Cysts (Figure 1)
Fibroadenomas (Figure 2)
Prominent fat lobules
Lymph nodes
Oil cysts
Lipomas
Hamartomas (Figure 3)
Hematomas
Fat necrosis
Galactoceles.
Malignant palpable masses include:
Figure 3. Hamartoma. Craniocaudal (A) and mediolateral oblique (B) mammographic views of the left breast show an apparently encapsulated, heterogeneous mass that contains fat mixed with fibroglandular tissue.Invasive ductal and lobular carcinoma (Figure 4)
Ductal carcinoma in situ (which rarely presents as a palpable mass.)
HISTORY AND PHYSICAL EXAMINATION
To ensure that imaging provides the most useful information about a palpable breast lump, it is important to first do a careful history and physical examination. Important aspects of the history include family history, personal history of breast cancer, and any previous breast biopsies. The onset and duration of the palpable mass, changes in its size, the relationship of these changes to the menstrual cycle, and the presence or lack of tenderness are additional important elements of the history.
Figure 4. Infiltrating ductal carcinoma. Craniocaudal (A) and mediolateral oblique (B) mammographic views of the right breast show an irregular, mildly spiculated, high-density lesion in the posterior, medial breast. Ultrasonography (C) shows an irregularly shaped hypoechoic mass which is taller than wide (a profile tending to indicate malignancy) and has mild posterior acoustic shadowing.On examination, it is important to note the clock-face location, size, texture, tenderness, and mobility of the lump. Accompanying nipple discharge and skin erythema or retraction are also important to report. In addition to conveying the location of the mass to the radiologist, it is equally important that the patient know what features the physician feels. This way, if the clinical information from the ordering physician is not available at the time of the radiologic evaluation, the patient will be able to guide the radiologist to the region of concern.
IMAGING TECHNIQUES
Mammography and ultrasonography are the primary imaging studies for evaluating palpable breast masses. Typically, in women under age 30, ultrasonography is the first or the only test ordered to evaluate the abnormality.4 In women age 30 or older, diagnostic mammography is typically the first test ordered. If mammography indicates that the palpable mass is not benign, then ultrasonography is the next study to be done.3 Although a powerful tool, magnetic resonance imaging of the breast does not currently have a role in the workup of a palpable abnormality and should not be used as a decision-delaying tactic or in place of biopsy.
Screening or diagnostic mammography?
Mammography is used in both screening and diagnosis. Screening mammography consists of two standard views of each breast—craniocaudal and mediolateral oblique—and is appropriate for asymptomatic women.
Women age 30 or older who present with a palpable breast mass require diagnostic mammography, in which standard mammographic views are obtained, as well as additional views (eg, tangential or spot-compression views) to better define the area of clinical concern. In a tangential view, a metallic skin marker is placed on the skin overlying the site of the palpable abnormality.
On mammography, a suspicious palpable mass has an irregular shape with spiculated margins. A benign mass typically has a round shape with well-circumscribed margins. If the palpable abnormality is not mammographically benign (eg, if it does not look like a lymph node, lipoma, or degenerating fibroadenoma), then ultrasonography is performed.
Mammography is less sensitive in younger women (ie, under age 30) because their breast tissue tends to be dense and glandular, whereas the tissue becomes more “fat-replaced” with age.3
Ultrasonography plays a complementary role
Ultrasonography complements diagnostic mammography and can be used as a first imaging study to evaluate a palpable breast mass in a young woman (ie, under age 30) with dense breast tissue. Ultrasonography is helpful in distinguishing cystic lesions from solid masses. It helps the radiologist delineate the shape, borders, and acoustic properties of the mass. It is also performed when a palpable mass is mammographically occult. When a mass appears suspicious on either mammography or ultrasonography, ultrasonography can be used to guide biopsy.
A suspicious mass on ultrasonography classically appears “taller than wide” and has posterior acoustic shadowing. Microlobulations and a spiculated margin also raise concern for malignancy. A benign sonographic appearance of a palpable mass includes a “wider than tall” (ellipsoid) shape, with homogeneous echogenicity, and four or fewer gentle lobulations. A thin, echogenic capsule also suggests the mass is benign.
Core-needle biopsy with ultrasonographic guidance
Core-needle biopsy is performed with a large-diameter (14-gauge to 18-gauge) needle to obtain tissue cores for histologic analysis. It has gained popularity over fine-needle aspiration because it includes surrounding tissue architecture, thus providing a more definitive histologic diagnosis.
Pathologic information obtained from core-needle biopsy allows the radiologist and surgeon to counsel the patient and determine the best surgical management or follow-up imaging study. If a clinician performs fine-needle biopsy in the office, it should be preceded by an imaging workup of the palpable finding.
WHAT IS APPROPRIATE FOR OUR 28-YEAR-OLD PATIENT?
Because she is under age 30, ultrasonography is the initial study of choice to evaluate the mass. If a simple cyst is detected, she can be reassured that the lesion is benign, and no subsequent follow-up is required. If the lesion is a solid mass with benign features, mammography may be considered, the patient may be followed with short-interval imaging (every 6 months) depending on patient-specific factors such as family history, or the mass can be biopsied. If the lesion is a solid mass with suspicious or malignant features, mammography with spot-compression views should be performed, and the patient should undergo core-needle biopsy with ultrasonographic guidance.
In a patient age 30 or older, diagnostic mammography is the imaging study of first choice.3 If the mass is clearly benign on mammography, no additional imaging would be necessary. If mammography fails to image the mass or shows it to have benign features such as fat, then the patient can undergo ultrasonography for further evaluation and confirmation of the clinical and mammographic findings. If the mass appears suspicious or malignant on mammography, ultrasonography is the next step, as it can help characterize the lesion and be used to guide core-needle biopsy.
IF A PREGNANT WOMAN HAS A PALPABLE BREAST MASS
Most publications on breast cancer in pregnancy report a prevalence of 3 per 10,000 pregnancies, accounting for 3% of all breast cancers diagnosed.5 Therefore, imaging evaluation of a palpable mass should not be postponed.
Hormonal changes throughout pregnancy may increase the nodularity of breast tissue, raising the concern of palpable masses. Additionally, there is a higher prevalence of galactoceles and lactating adenomas in these patients. Because contrasting fatty breast tissue is lost during pregnancy and because of the need to minimize radiation exposure, ultrasonography is often the imaging test of first choice. If mammography is required, the radiation dose is very low and the patient’s abdomen and pelvis can be shielded.6 In this situation, the patient can be reassured that the imaging test is not jeopardizing her fetus.
WHAT WORKUP IS REQUIRED IN MEN?
Breast cancer in men is rare, accounting for less than 0.5% of all cases.7 Most often, a palpable breast mass in a man presents as unilateral gynecomastia. Gynecomastia occurs in a bimodal age distribution (in the 2nd and 7th decades) and has a variety of hormonal and drug-related causes. Despite the low prevalence of breast cancer in men, the combination of mammography and ultrasonography is recommended for evaluation at all ages.
A 28-year-old woman comes in for her annual checkup. Her physician notices a palpable, painless, 1-cm, well-demarcated mass in the left breast at the 3 o’clock position 2 cm from the nipple, with no associated skin changes, nipple retraction, or discharge. The patient has no personal or family history of breast cancer.
Given the patient’s age, physical findings, and medical history, the clinician believes it unlikely that the patient has cancer. How should she proceed with the workup of this patient?
PHYSICAL FINDINGS OF A BREAST MASS ARE NOT EXCLUSIVE
Figure 1. A simple cyst in the left breast. All three mammographic views—craniocaudal (A), mediolateral oblique (B), and spot-compression (C)—show a round, well-circumscribed mass in the mid-breast. Ultrasonography (D) shows a round, well-circumscribed anechoic lesion with a sharply defined posterior wall and posterior acoustic enhancement.Breast cancer is the most common female malignancy and the second-leading cause of cancer deaths in the United States.1 The incidence is low in young women and increases with advancing age. Benign breast disease is common in young women and less common in postmenopausal women.2,3 However, the discovery of a breast mass, whether by the woman herself or by a clinician, is a common occurrence and distressing for any woman.
Benign lesions tend to have discrete, well-defined margins and are typically mobile. Malignant lesions may be firm, may have indistinct borders, and are often immobile.2 Although most breast masses found by palpation are benign, imaging is the critical next step in the workup to help determine if the mass is benign or malignant.
Benign palpable masses include:
Figure 2. Fibroadenoma. On mammography, the craniocaudal (A) and mediolateral oblique (B) views with a bright metallic marker (arrows) show a round, well-circumscribed mass in the upper outer quadrant of the left breast. Ultrasonography (C) shows an oval, well-circumscribed, mildly heterogeneous, hypoechoic mass that is wider than tall, indicating a benign mass.Cysts (Figure 1)
Fibroadenomas (Figure 2)
Prominent fat lobules
Lymph nodes
Oil cysts
Lipomas
Hamartomas (Figure 3)
Hematomas
Fat necrosis
Galactoceles.
Malignant palpable masses include:
Figure 3. Hamartoma. Craniocaudal (A) and mediolateral oblique (B) mammographic views of the left breast show an apparently encapsulated, heterogeneous mass that contains fat mixed with fibroglandular tissue.Invasive ductal and lobular carcinoma (Figure 4)
Ductal carcinoma in situ (which rarely presents as a palpable mass.)
HISTORY AND PHYSICAL EXAMINATION
To ensure that imaging provides the most useful information about a palpable breast lump, it is important to first do a careful history and physical examination. Important aspects of the history include family history, personal history of breast cancer, and any previous breast biopsies. The onset and duration of the palpable mass, changes in its size, the relationship of these changes to the menstrual cycle, and the presence or lack of tenderness are additional important elements of the history.
Figure 4. Infiltrating ductal carcinoma. Craniocaudal (A) and mediolateral oblique (B) mammographic views of the right breast show an irregular, mildly spiculated, high-density lesion in the posterior, medial breast. Ultrasonography (C) shows an irregularly shaped hypoechoic mass which is taller than wide (a profile tending to indicate malignancy) and has mild posterior acoustic shadowing.On examination, it is important to note the clock-face location, size, texture, tenderness, and mobility of the lump. Accompanying nipple discharge and skin erythema or retraction are also important to report. In addition to conveying the location of the mass to the radiologist, it is equally important that the patient know what features the physician feels. This way, if the clinical information from the ordering physician is not available at the time of the radiologic evaluation, the patient will be able to guide the radiologist to the region of concern.
IMAGING TECHNIQUES
Mammography and ultrasonography are the primary imaging studies for evaluating palpable breast masses. Typically, in women under age 30, ultrasonography is the first or the only test ordered to evaluate the abnormality.4 In women age 30 or older, diagnostic mammography is typically the first test ordered. If mammography indicates that the palpable mass is not benign, then ultrasonography is the next study to be done.3 Although a powerful tool, magnetic resonance imaging of the breast does not currently have a role in the workup of a palpable abnormality and should not be used as a decision-delaying tactic or in place of biopsy.
Screening or diagnostic mammography?
Mammography is used in both screening and diagnosis. Screening mammography consists of two standard views of each breast—craniocaudal and mediolateral oblique—and is appropriate for asymptomatic women.
Women age 30 or older who present with a palpable breast mass require diagnostic mammography, in which standard mammographic views are obtained, as well as additional views (eg, tangential or spot-compression views) to better define the area of clinical concern. In a tangential view, a metallic skin marker is placed on the skin overlying the site of the palpable abnormality.
On mammography, a suspicious palpable mass has an irregular shape with spiculated margins. A benign mass typically has a round shape with well-circumscribed margins. If the palpable abnormality is not mammographically benign (eg, if it does not look like a lymph node, lipoma, or degenerating fibroadenoma), then ultrasonography is performed.
Mammography is less sensitive in younger women (ie, under age 30) because their breast tissue tends to be dense and glandular, whereas the tissue becomes more “fat-replaced” with age.3
Ultrasonography plays a complementary role
Ultrasonography complements diagnostic mammography and can be used as a first imaging study to evaluate a palpable breast mass in a young woman (ie, under age 30) with dense breast tissue. Ultrasonography is helpful in distinguishing cystic lesions from solid masses. It helps the radiologist delineate the shape, borders, and acoustic properties of the mass. It is also performed when a palpable mass is mammographically occult. When a mass appears suspicious on either mammography or ultrasonography, ultrasonography can be used to guide biopsy.
A suspicious mass on ultrasonography classically appears “taller than wide” and has posterior acoustic shadowing. Microlobulations and a spiculated margin also raise concern for malignancy. A benign sonographic appearance of a palpable mass includes a “wider than tall” (ellipsoid) shape, with homogeneous echogenicity, and four or fewer gentle lobulations. A thin, echogenic capsule also suggests the mass is benign.
Core-needle biopsy with ultrasonographic guidance
Core-needle biopsy is performed with a large-diameter (14-gauge to 18-gauge) needle to obtain tissue cores for histologic analysis. It has gained popularity over fine-needle aspiration because it includes surrounding tissue architecture, thus providing a more definitive histologic diagnosis.
Pathologic information obtained from core-needle biopsy allows the radiologist and surgeon to counsel the patient and determine the best surgical management or follow-up imaging study. If a clinician performs fine-needle biopsy in the office, it should be preceded by an imaging workup of the palpable finding.
WHAT IS APPROPRIATE FOR OUR 28-YEAR-OLD PATIENT?
Because she is under age 30, ultrasonography is the initial study of choice to evaluate the mass. If a simple cyst is detected, she can be reassured that the lesion is benign, and no subsequent follow-up is required. If the lesion is a solid mass with benign features, mammography may be considered, the patient may be followed with short-interval imaging (every 6 months) depending on patient-specific factors such as family history, or the mass can be biopsied. If the lesion is a solid mass with suspicious or malignant features, mammography with spot-compression views should be performed, and the patient should undergo core-needle biopsy with ultrasonographic guidance.
In a patient age 30 or older, diagnostic mammography is the imaging study of first choice.3 If the mass is clearly benign on mammography, no additional imaging would be necessary. If mammography fails to image the mass or shows it to have benign features such as fat, then the patient can undergo ultrasonography for further evaluation and confirmation of the clinical and mammographic findings. If the mass appears suspicious or malignant on mammography, ultrasonography is the next step, as it can help characterize the lesion and be used to guide core-needle biopsy.
IF A PREGNANT WOMAN HAS A PALPABLE BREAST MASS
Most publications on breast cancer in pregnancy report a prevalence of 3 per 10,000 pregnancies, accounting for 3% of all breast cancers diagnosed.5 Therefore, imaging evaluation of a palpable mass should not be postponed.
Hormonal changes throughout pregnancy may increase the nodularity of breast tissue, raising the concern of palpable masses. Additionally, there is a higher prevalence of galactoceles and lactating adenomas in these patients. Because contrasting fatty breast tissue is lost during pregnancy and because of the need to minimize radiation exposure, ultrasonography is often the imaging test of first choice. If mammography is required, the radiation dose is very low and the patient’s abdomen and pelvis can be shielded.6 In this situation, the patient can be reassured that the imaging test is not jeopardizing her fetus.
WHAT WORKUP IS REQUIRED IN MEN?
Breast cancer in men is rare, accounting for less than 0.5% of all cases.7 Most often, a palpable breast mass in a man presents as unilateral gynecomastia. Gynecomastia occurs in a bimodal age distribution (in the 2nd and 7th decades) and has a variety of hormonal and drug-related causes. Despite the low prevalence of breast cancer in men, the combination of mammography and ultrasonography is recommended for evaluation at all ages.
References
Klein S. Evaluation of palpable breast masses. Am Fam Physician2005; 71:1731–1738.
Pruthi S. Detection and evaluation of a palpable breast mass. Mayo Clin Proc2001; 76:641–648.
Harvey JA. Sonography of palpable breast masses. Semin Ultrasound CT MR2006; 27:284–297.
Mehta TS. Current uses of ultrasound in the evaluation of the breast. Radiol Clin North Am2003; 41:841–856.
Gallenberg MM, Lopines CL. Breast cancer and pregnancy. Semin Oncol1989; 16:369–376.
Barnavon Y, Wallack MK. Management of the pregnant patient with carcinoma of the breast. Surg Gynecol Obstet1990; 171:347–352.
Cardenosa G. The Core Curriculum: Breast Imaging. Philadelphia: Lippincott Williams and Wilkins, 2003;304.
References
Klein S. Evaluation of palpable breast masses. Am Fam Physician2005; 71:1731–1738.
Pruthi S. Detection and evaluation of a palpable breast mass. Mayo Clin Proc2001; 76:641–648.
Harvey JA. Sonography of palpable breast masses. Semin Ultrasound CT MR2006; 27:284–297.
Mehta TS. Current uses of ultrasound in the evaluation of the breast. Radiol Clin North Am2003; 41:841–856.
Gallenberg MM, Lopines CL. Breast cancer and pregnancy. Semin Oncol1989; 16:369–376.
Barnavon Y, Wallack MK. Management of the pregnant patient with carcinoma of the breast. Surg Gynecol Obstet1990; 171:347–352.
Cardenosa G. The Core Curriculum: Breast Imaging. Philadelphia: Lippincott Williams and Wilkins, 2003;304.
Typically, in women under age 30, ultrasonography is the first or the only test ordered to evaluate the abnormality. In women age 30 or older, diagnostic mammography is typically the first test ordered.
On mammography, a suspicious palpable mass has an irregular shape with spiculated margins. A benign mass typically has a round shape with well-circumscribed margins.
When mammography is required during pregnancy, the patient can be reassured that it will not jeopardize her fetus because the radiation dose is very low and the abdomen and pelvis can be shielded.
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
Figure 1. Multiple cotton wool spots in the peripapillary area in the left eye.Funduscopic examination of the left eye reveals multiple cotton wool spots in the peripapillary area (Figure 1). The visual acuity is 20/200. The right eye appears normal, with normal vision.
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
Hypertensive retinopathy
Diabetic retinopathy
Human immunodeficiency virus (HIV) retinopathy
Retinal involvement of systemic autoimmune disease
Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
References
Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update2007; 8:17–28.
Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol2001; 49:5–14.
Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol1988; 11:239–251.
Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol1989; 13:187–191.
Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci1996; 143:1–13.
Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med1998; 339:1415–1425.
Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated?J Neuroophthalmol2005; 25:299–302.
Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology2003; 110:1683–1689.
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
Figure 1. Multiple cotton wool spots in the peripapillary area in the left eye.Funduscopic examination of the left eye reveals multiple cotton wool spots in the peripapillary area (Figure 1). The visual acuity is 20/200. The right eye appears normal, with normal vision.
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
Hypertensive retinopathy
Diabetic retinopathy
Human immunodeficiency virus (HIV) retinopathy
Retinal involvement of systemic autoimmune disease
Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
Figure 1. Multiple cotton wool spots in the peripapillary area in the left eye.Funduscopic examination of the left eye reveals multiple cotton wool spots in the peripapillary area (Figure 1). The visual acuity is 20/200. The right eye appears normal, with normal vision.
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
Hypertensive retinopathy
Diabetic retinopathy
Human immunodeficiency virus (HIV) retinopathy
Retinal involvement of systemic autoimmune disease
Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
References
Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update2007; 8:17–28.
Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol2001; 49:5–14.
Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol1988; 11:239–251.
Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol1989; 13:187–191.
Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci1996; 143:1–13.
Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med1998; 339:1415–1425.
Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated?J Neuroophthalmol2005; 25:299–302.
Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology2003; 110:1683–1689.
References
Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update2007; 8:17–28.
Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol2001; 49:5–14.
Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol1988; 11:239–251.
Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol1989; 13:187–191.
Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci1996; 143:1–13.
Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med1998; 339:1415–1425.
Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated?J Neuroophthalmol2005; 25:299–302.
Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology2003; 110:1683–1689.
Long ago, an acute myocardial infarction (MI) was diagnosed by a combination of patient history, electrocardiographic (ECG) findings, elevation of aspartate aminotransferase and creatine kinase (CK), and the pattern of lactate dehydrogenase isozymes. The erythrocyte sedimentation rate was useful in distinguishing prolonged angina from an MI, and even the presence or absence of leukocytosis was sometimes a determining diagnostic test. The recognition that CK-MB was fairly specific for myocardial injury was a major step forward in the diagnosis of what would later be called the acute coronary syndrome.
But way back then, although the patient (and the physician) were often diaphoretic, the acute diagnosis was of limited significance to acute management. We rushed to put the patient to bed rest, started the lidocaine drip at the first sign of a few premature ventricular contractions, slapped on the oxygen prongs, got serial electrocardiograms to watch for conduction blocks—and a few forward thinkers began heparin drips.
As therapeutic options became more interventional, the need for rapid diagnostic tests and better biomarkers of prognosis became more critical. ST elevations took on new meaning, but the major diagnostic advance was the incorporation of cardiac troponin into our diagnostic algorithm.
In this issue of the Journal, Drs. Shaun Senter and Gary Francis discuss the power of these tests in the diagnosis of acute MI. They are not perfect tests. Acute pericarditis can still present diagnostic challenges, with sometimes confusing ECG findings, and almost a third of patients have elevated troponins (Bainey KR, Bhatt DL, Mayo Clin Proc 2009; 84:5–6; Imazio M, et al, J Am Coll Cardiol 2003; 42:2144–2148). Troponins may occasionally be elevated in acute severe heart failure and aortic dissection. In my practice, elevation of troponins may be difficult to interpret in the setting of chronic inflammatory muscle disease; it is not always easy to distinguish whether the leakage of these biomarkers is from injured regenerating skeletal muscle or from cardiac muscle.
When treating patients with a possible acute coronary syndrome, prompt diagnosis and intervention are often warranted, but the risks of using thrombolytic therapy inappropriately in the setting of pericarditis or an acute intracranial process with ECG changes are substantial.
Senter and Francis review for us the latest “precise definition” of acute myocardial infarction and provide a commentary on the utility of different diagnostic tests. They also highlight the value of using different diagnostic modalities to obtain the information we need for prognostication and treatment decisions.
Long ago, an acute myocardial infarction (MI) was diagnosed by a combination of patient history, electrocardiographic (ECG) findings, elevation of aspartate aminotransferase and creatine kinase (CK), and the pattern of lactate dehydrogenase isozymes. The erythrocyte sedimentation rate was useful in distinguishing prolonged angina from an MI, and even the presence or absence of leukocytosis was sometimes a determining diagnostic test. The recognition that CK-MB was fairly specific for myocardial injury was a major step forward in the diagnosis of what would later be called the acute coronary syndrome.
But way back then, although the patient (and the physician) were often diaphoretic, the acute diagnosis was of limited significance to acute management. We rushed to put the patient to bed rest, started the lidocaine drip at the first sign of a few premature ventricular contractions, slapped on the oxygen prongs, got serial electrocardiograms to watch for conduction blocks—and a few forward thinkers began heparin drips.
As therapeutic options became more interventional, the need for rapid diagnostic tests and better biomarkers of prognosis became more critical. ST elevations took on new meaning, but the major diagnostic advance was the incorporation of cardiac troponin into our diagnostic algorithm.
In this issue of the Journal, Drs. Shaun Senter and Gary Francis discuss the power of these tests in the diagnosis of acute MI. They are not perfect tests. Acute pericarditis can still present diagnostic challenges, with sometimes confusing ECG findings, and almost a third of patients have elevated troponins (Bainey KR, Bhatt DL, Mayo Clin Proc 2009; 84:5–6; Imazio M, et al, J Am Coll Cardiol 2003; 42:2144–2148). Troponins may occasionally be elevated in acute severe heart failure and aortic dissection. In my practice, elevation of troponins may be difficult to interpret in the setting of chronic inflammatory muscle disease; it is not always easy to distinguish whether the leakage of these biomarkers is from injured regenerating skeletal muscle or from cardiac muscle.
When treating patients with a possible acute coronary syndrome, prompt diagnosis and intervention are often warranted, but the risks of using thrombolytic therapy inappropriately in the setting of pericarditis or an acute intracranial process with ECG changes are substantial.
Senter and Francis review for us the latest “precise definition” of acute myocardial infarction and provide a commentary on the utility of different diagnostic tests. They also highlight the value of using different diagnostic modalities to obtain the information we need for prognostication and treatment decisions.
Long ago, an acute myocardial infarction (MI) was diagnosed by a combination of patient history, electrocardiographic (ECG) findings, elevation of aspartate aminotransferase and creatine kinase (CK), and the pattern of lactate dehydrogenase isozymes. The erythrocyte sedimentation rate was useful in distinguishing prolonged angina from an MI, and even the presence or absence of leukocytosis was sometimes a determining diagnostic test. The recognition that CK-MB was fairly specific for myocardial injury was a major step forward in the diagnosis of what would later be called the acute coronary syndrome.
But way back then, although the patient (and the physician) were often diaphoretic, the acute diagnosis was of limited significance to acute management. We rushed to put the patient to bed rest, started the lidocaine drip at the first sign of a few premature ventricular contractions, slapped on the oxygen prongs, got serial electrocardiograms to watch for conduction blocks—and a few forward thinkers began heparin drips.
As therapeutic options became more interventional, the need for rapid diagnostic tests and better biomarkers of prognosis became more critical. ST elevations took on new meaning, but the major diagnostic advance was the incorporation of cardiac troponin into our diagnostic algorithm.
In this issue of the Journal, Drs. Shaun Senter and Gary Francis discuss the power of these tests in the diagnosis of acute MI. They are not perfect tests. Acute pericarditis can still present diagnostic challenges, with sometimes confusing ECG findings, and almost a third of patients have elevated troponins (Bainey KR, Bhatt DL, Mayo Clin Proc 2009; 84:5–6; Imazio M, et al, J Am Coll Cardiol 2003; 42:2144–2148). Troponins may occasionally be elevated in acute severe heart failure and aortic dissection. In my practice, elevation of troponins may be difficult to interpret in the setting of chronic inflammatory muscle disease; it is not always easy to distinguish whether the leakage of these biomarkers is from injured regenerating skeletal muscle or from cardiac muscle.
When treating patients with a possible acute coronary syndrome, prompt diagnosis and intervention are often warranted, but the risks of using thrombolytic therapy inappropriately in the setting of pericarditis or an acute intracranial process with ECG changes are substantial.
Senter and Francis review for us the latest “precise definition” of acute myocardial infarction and provide a commentary on the utility of different diagnostic tests. They also highlight the value of using different diagnostic modalities to obtain the information we need for prognostication and treatment decisions.