User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
nav[contains(@class, 'nav-ce-stack nav-ce-stack__large-screen')]
header[@id='header']
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
B6 a new approach for depression, anxiety?
Investigators compared supplementation with a 1-month course of vitamin B6 or B12 to supplementation with placebo in almost 500 adults. Results showed that vitamin B6 supplementation was associated with reductions in self-reported anxiety and a trend toward decreased depressive symptoms.
In addition, the vitamin B6 group showed increased levels of gamma-aminobutyric acid (GABA), as indicated by results on a visual test that was administered at the end of the trial. The test results demonstrated subtle changes in participants’ visual performance. The researchers considered this to be consistent with controlled levels of GABA-related brain activity.

However, “before practicing clinicians would recommend taking high doses of vitamin B6, a full-scale clinical trial would have to be carried out to verify the findings, assess any side effects, and find out which types of patients do or don’t benefit,” study investigator David Field, PhD, associate professor, School of Psychological and Clinical Language Sciences, University of Reading (England), told this news organization.
“My relatively small study can only be considered as an initial proof of concept,” Dr. Field said.
The findings were published online in the Journal of Human Psychopharmacology: Clinical and Experimental.
Eat Marmite?
“Recent research has connected mood disorders and some other neuropsychiatric conditions with disturbance in this balance, often in the direction of raised levels of brain activity,” Dr. Field noted.
Vitamin B6 is a coenzyme in the synthesis of GABA, an inhibitory neurotransmitter, from glutamate. Some previous research has suggested that vitamins B6 and B12 have a role in improving mood-related outcomes.
Dr. Field had reviewed a 2017 study of the effects on visual processing of eating Marmite, a type of food spread rich in vitamin B, every day for a few weeks.
“Remarkably, the results of that study suggested that eating Marmite had increased the level of the inhibitory neurotransmitter GABA in the visual part of the brain, damping down the level of neural activity slightly,” he said.
However, Marmite contains other B vitamins and other ingredients that might potentially account for this result, “plus, a lot of people don’t like the taste of Marmite,” Dr. Field noted.
Therefore, he wanted to “find out which individual ingredients were driving the effect, and B6 and B12 were the most plausible candidates.”
He decided to test these vitamins individually and to compare them to placebo. “I added the measures of anxiety and depression that were not in the Marmite study because I reasoned that if GABA levels were altered, this could improve those disorders, because we know that decreased levels of GABA in the brain occur in both of those conditions,” Dr. Field added.
Over the course of 5 years, investigators recruited 478 participants aged 18-58 years (mean age, 23 years; 381 women). Of these, 265 reported having anxiety, and 146 reported having depression.
The study participants were randomly assigned to receive either vitamin B6 (100 mg pyroxidine hydrochloride), vitamin B12 (1,000 mg methylcobalmin), or placebo tablets once daily for a month.
They also completed the Screen for Adult Anxiety Related Disorders (SCAARED) and the Mood and Feelings Questionnaire (MFQ) long version at baseline and following supplementation (“post test”), and they underwent three sensory tests that acted as assays of inhibitory function at post test.
In addition, 307 participants completed the Visual Contrast Sensitivity and Surround Suppression, which “measures the minimum percentage contrast between the lighter and darker regions of a striped pattern that can be detected (called the contrast threshold),” the investigators note.
The contrast threshold was measured with and without a suppressive surround mask that increases the threshold – an effect mediated by GABAergic connections in the visual cortex.
Participants (n = 172) also completed the Binocular Rivalry test and the Tactile Test Battery (n = 180). Both tests are designed to measure responses requiring GABAergic inhibitory activity.
‘Subtle changes’
ANOVA analyses revealed a “highly significant” reduction in anxiety at post test (F[1,173] = 10.03; P = .002; np 2 = .055), driven primarily by reduced anxiety in the B6 group (t[88] = 3.51; P < .001; d = .37). The placebo group also showed some reduction in anxiety, but it was not deemed significant, and the overall interaction itself did not reach significance.
A comparison of the B12 group with the group that received placebo revealed a significant reduction in anxiety at post test (F[1,175] = 4.08; P = .045; np 2 = .023), similarly driven by reduced anxiety in the B12 group (t[89] = 1.84; P = .069; d = .19) – but the interaction was not significant.
Among the B6 group, there was a highly significant reduction in scores on the generalized anxiety disorder and social anxiety subscales of the SCAARED, and there was a trend toward reductions on the other subscales. Among the B12 group, there was a significant reduction only on scores on the separation anxiety subscale. No significant changes were found in the placebo group.
The ANOVA test analysis of the B6 and placebo group data showed “no uniform direction of change” in depression at post test. The researchers found a “tendency” for depression scores to decrease between baseline and post test in the B6 group but to increase in the placebo group – an interaction that “approached” significance (F[1,96] = 3.08; P = .083; np 2 = .031), they report.
The ANOVA analysis of the B12 and placebo group data revealed no significant or trending effects, and the t-test comparing baseline and post-test scores in the B12 group was similarly nonsignificant.
B6 supplementation did change visual contrast thresholds, but only when a suppressive surround was present. There were “no clear effects” of B6 supplementation on other outcome measures, including binocular rivalry reversal rate and the tactile test battery, the investigators note.
“We found that supplementation with B6 produced subtle changes in tests of visual processing in a way that suggested an increase in the level of the inhibitory neurotransmitter GABA,” Dr. Field reported.
Vitamin B6 is a “cofactor for a metabolic pathway in the brain that converts the excitatory neurotransmitter glutamate into the inhibitory/calming GABA,” he said.
“By increasing the quantity of the cofactor, we slightly speed up the rate of this metabolic process, and so you end up with a bit more of the GABA neurotransmitter and a bit less glutamate. The net effect of this is to slightly reduce the amount of activity in the brain,” Dr. Field added.
Most common nutrient deficiency
Carol Johnston, PhD, professor and associate dean for faculty success, College of Health Solutions, Arizona State University, Phoenix, said vitamin B6 is “the most common nutrient deficiency in the United States;” 16% of men and 32% of women are reportedly B6 deficient.
“Young women on birth control are at higher risk for B6 deficiency due to effects of oral contraceptives on B6 metabolism,” whereas vitamin B12 deficiency is more common in older adults, said Dr. Johnston, who was not involved with the study.
The current study’s population mainly consisted of young women, and the interpretation of the data is “limited” because the researchers did not measure blood status for B6 and B12, Dr. Johnston noted. It is possible the sample was low in B6 and that the supplements “improved cognitive measures.”
Because the population was young – no one was older than 60 years – B12 status was likely “adequate in the sample, and supplementation did not have an impact,” she said.
Overall, Dr. Johnston cautioned that it is important to “alert clinicians and the general public about the concerns of overdosing B6.” For example, supplementation at high amounts can cause potentially irreversible sensory neuropathy, she noted.
“The safe upper limit defined by experts is 100 mg per day – the dosage used in this trial. Daily supplementation should not exceed this level,” Dr. Johnston said.
The vitamin tablets used in the study were supplied by Innopure. The investigators and Dr. Johnston have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Investigators compared supplementation with a 1-month course of vitamin B6 or B12 to supplementation with placebo in almost 500 adults. Results showed that vitamin B6 supplementation was associated with reductions in self-reported anxiety and a trend toward decreased depressive symptoms.
In addition, the vitamin B6 group showed increased levels of gamma-aminobutyric acid (GABA), as indicated by results on a visual test that was administered at the end of the trial. The test results demonstrated subtle changes in participants’ visual performance. The researchers considered this to be consistent with controlled levels of GABA-related brain activity.
However, “before practicing clinicians would recommend taking high doses of vitamin B6, a full-scale clinical trial would have to be carried out to verify the findings, assess any side effects, and find out which types of patients do or don’t benefit,” study investigator David Field, PhD, associate professor, School of Psychological and Clinical Language Sciences, University of Reading (England), told this news organization.
“My relatively small study can only be considered as an initial proof of concept,” Dr. Field said.
The findings were published online in the Journal of Human Psychopharmacology: Clinical and Experimental.
Eat Marmite?
“Recent research has connected mood disorders and some other neuropsychiatric conditions with disturbance in this balance, often in the direction of raised levels of brain activity,” Dr. Field noted.
Vitamin B6 is a coenzyme in the synthesis of GABA, an inhibitory neurotransmitter, from glutamate. Some previous research has suggested that vitamins B6 and B12 have a role in improving mood-related outcomes.
Dr. Field had reviewed a 2017 study of the effects on visual processing of eating Marmite, a type of food spread rich in vitamin B, every day for a few weeks.
“Remarkably, the results of that study suggested that eating Marmite had increased the level of the inhibitory neurotransmitter GABA in the visual part of the brain, damping down the level of neural activity slightly,” he said.
However, Marmite contains other B vitamins and other ingredients that might potentially account for this result, “plus, a lot of people don’t like the taste of Marmite,” Dr. Field noted.
Therefore, he wanted to “find out which individual ingredients were driving the effect, and B6 and B12 were the most plausible candidates.”
He decided to test these vitamins individually and to compare them to placebo. “I added the measures of anxiety and depression that were not in the Marmite study because I reasoned that if GABA levels were altered, this could improve those disorders, because we know that decreased levels of GABA in the brain occur in both of those conditions,” Dr. Field added.
Over the course of 5 years, investigators recruited 478 participants aged 18-58 years (mean age, 23 years; 381 women). Of these, 265 reported having anxiety, and 146 reported having depression.
The study participants were randomly assigned to receive either vitamin B6 (100 mg pyroxidine hydrochloride), vitamin B12 (1,000 mg methylcobalmin), or placebo tablets once daily for a month.
They also completed the Screen for Adult Anxiety Related Disorders (SCAARED) and the Mood and Feelings Questionnaire (MFQ) long version at baseline and following supplementation (“post test”), and they underwent three sensory tests that acted as assays of inhibitory function at post test.
In addition, 307 participants completed the Visual Contrast Sensitivity and Surround Suppression, which “measures the minimum percentage contrast between the lighter and darker regions of a striped pattern that can be detected (called the contrast threshold),” the investigators note.
The contrast threshold was measured with and without a suppressive surround mask that increases the threshold – an effect mediated by GABAergic connections in the visual cortex.
Participants (n = 172) also completed the Binocular Rivalry test and the Tactile Test Battery (n = 180). Both tests are designed to measure responses requiring GABAergic inhibitory activity.
‘Subtle changes’
ANOVA analyses revealed a “highly significant” reduction in anxiety at post test (F[1,173] = 10.03; P = .002; np 2 = .055), driven primarily by reduced anxiety in the B6 group (t[88] = 3.51; P < .001; d = .37). The placebo group also showed some reduction in anxiety, but it was not deemed significant, and the overall interaction itself did not reach significance.
A comparison of the B12 group with the group that received placebo revealed a significant reduction in anxiety at post test (F[1,175] = 4.08; P = .045; np 2 = .023), similarly driven by reduced anxiety in the B12 group (t[89] = 1.84; P = .069; d = .19) – but the interaction was not significant.
Among the B6 group, there was a highly significant reduction in scores on the generalized anxiety disorder and social anxiety subscales of the SCAARED, and there was a trend toward reductions on the other subscales. Among the B12 group, there was a significant reduction only on scores on the separation anxiety subscale. No significant changes were found in the placebo group.
The ANOVA test analysis of the B6 and placebo group data showed “no uniform direction of change” in depression at post test. The researchers found a “tendency” for depression scores to decrease between baseline and post test in the B6 group but to increase in the placebo group – an interaction that “approached” significance (F[1,96] = 3.08; P = .083; np 2 = .031), they report.
The ANOVA analysis of the B12 and placebo group data revealed no significant or trending effects, and the t-test comparing baseline and post-test scores in the B12 group was similarly nonsignificant.
B6 supplementation did change visual contrast thresholds, but only when a suppressive surround was present. There were “no clear effects” of B6 supplementation on other outcome measures, including binocular rivalry reversal rate and the tactile test battery, the investigators note.
“We found that supplementation with B6 produced subtle changes in tests of visual processing in a way that suggested an increase in the level of the inhibitory neurotransmitter GABA,” Dr. Field reported.
Vitamin B6 is a “cofactor for a metabolic pathway in the brain that converts the excitatory neurotransmitter glutamate into the inhibitory/calming GABA,” he said.
“By increasing the quantity of the cofactor, we slightly speed up the rate of this metabolic process, and so you end up with a bit more of the GABA neurotransmitter and a bit less glutamate. The net effect of this is to slightly reduce the amount of activity in the brain,” Dr. Field added.
Most common nutrient deficiency
Carol Johnston, PhD, professor and associate dean for faculty success, College of Health Solutions, Arizona State University, Phoenix, said vitamin B6 is “the most common nutrient deficiency in the United States;” 16% of men and 32% of women are reportedly B6 deficient.
“Young women on birth control are at higher risk for B6 deficiency due to effects of oral contraceptives on B6 metabolism,” whereas vitamin B12 deficiency is more common in older adults, said Dr. Johnston, who was not involved with the study.
The current study’s population mainly consisted of young women, and the interpretation of the data is “limited” because the researchers did not measure blood status for B6 and B12, Dr. Johnston noted. It is possible the sample was low in B6 and that the supplements “improved cognitive measures.”
Because the population was young – no one was older than 60 years – B12 status was likely “adequate in the sample, and supplementation did not have an impact,” she said.
Overall, Dr. Johnston cautioned that it is important to “alert clinicians and the general public about the concerns of overdosing B6.” For example, supplementation at high amounts can cause potentially irreversible sensory neuropathy, she noted.
“The safe upper limit defined by experts is 100 mg per day – the dosage used in this trial. Daily supplementation should not exceed this level,” Dr. Johnston said.
The vitamin tablets used in the study were supplied by Innopure. The investigators and Dr. Johnston have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Investigators compared supplementation with a 1-month course of vitamin B6 or B12 to supplementation with placebo in almost 500 adults. Results showed that vitamin B6 supplementation was associated with reductions in self-reported anxiety and a trend toward decreased depressive symptoms.
In addition, the vitamin B6 group showed increased levels of gamma-aminobutyric acid (GABA), as indicated by results on a visual test that was administered at the end of the trial. The test results demonstrated subtle changes in participants’ visual performance. The researchers considered this to be consistent with controlled levels of GABA-related brain activity.
However, “before practicing clinicians would recommend taking high doses of vitamin B6, a full-scale clinical trial would have to be carried out to verify the findings, assess any side effects, and find out which types of patients do or don’t benefit,” study investigator David Field, PhD, associate professor, School of Psychological and Clinical Language Sciences, University of Reading (England), told this news organization.
“My relatively small study can only be considered as an initial proof of concept,” Dr. Field said.
The findings were published online in the Journal of Human Psychopharmacology: Clinical and Experimental.
Eat Marmite?
“Recent research has connected mood disorders and some other neuropsychiatric conditions with disturbance in this balance, often in the direction of raised levels of brain activity,” Dr. Field noted.
Vitamin B6 is a coenzyme in the synthesis of GABA, an inhibitory neurotransmitter, from glutamate. Some previous research has suggested that vitamins B6 and B12 have a role in improving mood-related outcomes.
Dr. Field had reviewed a 2017 study of the effects on visual processing of eating Marmite, a type of food spread rich in vitamin B, every day for a few weeks.
“Remarkably, the results of that study suggested that eating Marmite had increased the level of the inhibitory neurotransmitter GABA in the visual part of the brain, damping down the level of neural activity slightly,” he said.
However, Marmite contains other B vitamins and other ingredients that might potentially account for this result, “plus, a lot of people don’t like the taste of Marmite,” Dr. Field noted.
Therefore, he wanted to “find out which individual ingredients were driving the effect, and B6 and B12 were the most plausible candidates.”
He decided to test these vitamins individually and to compare them to placebo. “I added the measures of anxiety and depression that were not in the Marmite study because I reasoned that if GABA levels were altered, this could improve those disorders, because we know that decreased levels of GABA in the brain occur in both of those conditions,” Dr. Field added.
Over the course of 5 years, investigators recruited 478 participants aged 18-58 years (mean age, 23 years; 381 women). Of these, 265 reported having anxiety, and 146 reported having depression.
The study participants were randomly assigned to receive either vitamin B6 (100 mg pyroxidine hydrochloride), vitamin B12 (1,000 mg methylcobalmin), or placebo tablets once daily for a month.
They also completed the Screen for Adult Anxiety Related Disorders (SCAARED) and the Mood and Feelings Questionnaire (MFQ) long version at baseline and following supplementation (“post test”), and they underwent three sensory tests that acted as assays of inhibitory function at post test.
In addition, 307 participants completed the Visual Contrast Sensitivity and Surround Suppression, which “measures the minimum percentage contrast between the lighter and darker regions of a striped pattern that can be detected (called the contrast threshold),” the investigators note.
The contrast threshold was measured with and without a suppressive surround mask that increases the threshold – an effect mediated by GABAergic connections in the visual cortex.
Participants (n = 172) also completed the Binocular Rivalry test and the Tactile Test Battery (n = 180). Both tests are designed to measure responses requiring GABAergic inhibitory activity.
‘Subtle changes’
ANOVA analyses revealed a “highly significant” reduction in anxiety at post test (F[1,173] = 10.03; P = .002; np 2 = .055), driven primarily by reduced anxiety in the B6 group (t[88] = 3.51; P < .001; d = .37). The placebo group also showed some reduction in anxiety, but it was not deemed significant, and the overall interaction itself did not reach significance.
A comparison of the B12 group with the group that received placebo revealed a significant reduction in anxiety at post test (F[1,175] = 4.08; P = .045; np 2 = .023), similarly driven by reduced anxiety in the B12 group (t[89] = 1.84; P = .069; d = .19) – but the interaction was not significant.
Among the B6 group, there was a highly significant reduction in scores on the generalized anxiety disorder and social anxiety subscales of the SCAARED, and there was a trend toward reductions on the other subscales. Among the B12 group, there was a significant reduction only on scores on the separation anxiety subscale. No significant changes were found in the placebo group.
The ANOVA test analysis of the B6 and placebo group data showed “no uniform direction of change” in depression at post test. The researchers found a “tendency” for depression scores to decrease between baseline and post test in the B6 group but to increase in the placebo group – an interaction that “approached” significance (F[1,96] = 3.08; P = .083; np 2 = .031), they report.
The ANOVA analysis of the B12 and placebo group data revealed no significant or trending effects, and the t-test comparing baseline and post-test scores in the B12 group was similarly nonsignificant.
B6 supplementation did change visual contrast thresholds, but only when a suppressive surround was present. There were “no clear effects” of B6 supplementation on other outcome measures, including binocular rivalry reversal rate and the tactile test battery, the investigators note.
“We found that supplementation with B6 produced subtle changes in tests of visual processing in a way that suggested an increase in the level of the inhibitory neurotransmitter GABA,” Dr. Field reported.
Vitamin B6 is a “cofactor for a metabolic pathway in the brain that converts the excitatory neurotransmitter glutamate into the inhibitory/calming GABA,” he said.
“By increasing the quantity of the cofactor, we slightly speed up the rate of this metabolic process, and so you end up with a bit more of the GABA neurotransmitter and a bit less glutamate. The net effect of this is to slightly reduce the amount of activity in the brain,” Dr. Field added.
Most common nutrient deficiency
Carol Johnston, PhD, professor and associate dean for faculty success, College of Health Solutions, Arizona State University, Phoenix, said vitamin B6 is “the most common nutrient deficiency in the United States;” 16% of men and 32% of women are reportedly B6 deficient.
“Young women on birth control are at higher risk for B6 deficiency due to effects of oral contraceptives on B6 metabolism,” whereas vitamin B12 deficiency is more common in older adults, said Dr. Johnston, who was not involved with the study.
The current study’s population mainly consisted of young women, and the interpretation of the data is “limited” because the researchers did not measure blood status for B6 and B12, Dr. Johnston noted. It is possible the sample was low in B6 and that the supplements “improved cognitive measures.”
Because the population was young – no one was older than 60 years – B12 status was likely “adequate in the sample, and supplementation did not have an impact,” she said.
Overall, Dr. Johnston cautioned that it is important to “alert clinicians and the general public about the concerns of overdosing B6.” For example, supplementation at high amounts can cause potentially irreversible sensory neuropathy, she noted.
“The safe upper limit defined by experts is 100 mg per day – the dosage used in this trial. Daily supplementation should not exceed this level,” Dr. Johnston said.
The vitamin tablets used in the study were supplied by Innopure. The investigators and Dr. Johnston have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Routine weight counseling urged for women at midlife
Midlife women who are of normal weight or are overweight should routinely receive counseling aimed at limiting weight gain and preventing obesity and its associated health risks, a new clinical guideline states.
The recommendation, issued by the Women’s Preventive Services Initiative (WPSI) of the American College of Obstetricians and Gynecologists (ACOG), supports regular lifestyle counseling for women aged 40-60 years with normal or overweight body mass index of 18.5-29.9 kg/m2. Counseling could include individualized discussion of healthy eating and physical activity initiated by health professionals involved in preventive care.
Published online in Annals of Internal Medicine, the guideline addresses the prevalence and health burdens of obesity in U.S. women of middle age and seeks to reduce the known harms of obesity with an intervention of minimal anticipated harms. High BMI increases the risk for many chronic conditions including hypertension, dyslipidemia, type 2 diabetes, coronary artery disease, stroke, and all-cause mortality.
The best way to counsel, however, remains unclear. “Although the optimal approach could not be discerned from existing trials, a range of interventions of varying duration, frequency, and intensity showed benefit with potential clinical significance,” wrote the WPSI guideline panel, led by David P. Chelmow, MD, chair of the department of obstetrics and gynecology at Virginia Commonwealth University in Richmond.
The guideline rests on a systematic literature review led by family doctor Amy G. Cantor, MD, MPH, of the Pacific Northwest Evidence-based Practice Center, at Oregon Health & Science University in Portland, suggesting moderate reductions in weight could be achieved by offering advice to this age group.

The federally supported WPSI was launched by ACOG in 2016. The guideline fills a gap in current recommendations in that it targets a specific risk group and specifies individual counseling based on its effectiveness and applicability in primary care settings.
In another benefit of routine counseling, the panel stated, “Normalizing counseling about healthy diet and physical activity by providing it to all midlife women may also mitigate concerns about weight stigma resulting from only counseling women with obesity.”
The panelists noted that during 2017-2018, the prevalence of obesity (BMI ≥ 30.0 kg/m2) was 43.3% among U.S. women aged 40-59 years, while the prevalence of severe obesity (BMI ≥ 40.0 kg/m2) was highest in this age group at 11.5%. “Midlife women gain weight at an average of approximately 1.5 pounds per year, which increases their risk for transitioning from normal or overweight to obese BMI,” the panelists wrote.
The review
Dr. Cantor’s group analyzed seven randomized controlled trials (RCTs) published up to October 2021 from 12 publications involving 51,638 participants. Although the trials were largely small and heterogeneous, they suggested that counseling may result in modest differences in weight change without causing important harms.
Four RCTs showed significant favorable weight changes for counseling over no-counseling control groups, with a mean difference of 0.87 to 2.5 kg, whereas one trial of counseling and two trials of exercise showed no differences. One of two RCTs reported improved quality-of-life measures.
As for harms, while interventions did not increase measures of depression or stress in one trial, self-reported falls (37% vs. 29%, P < .001) and injuries (19% vs. 14%, P = .03) were more frequent with exercise counseling in one trial.
“More research is needed to determine optimal content, frequency, length, and number of sessions required and should include additional patient populations,” Dr. Cantor and associates wrote.
In terms of limitations, the authors acknowledged that trials of behavioral interventions in maintaining or reducing weight in midlife women demonstrate small magnitudes of effect.
Offering a nonparticipant’s perspective on the WPSI guideline for this news organization, JoAnn E. Manson, MD, DrPH, MACP, chief of the division of preventive medicine at Brigham and Women’s Hospital in Boston, said its message is of prime importance for women of middle age and it goes beyond concern about pounds lost or gained.

“Midlife and the transition to menopause are high-risk periods for women in terms of typical changes in body composition that increase the risk of adverse cardiometabolic outcomes,” said Dr. Manson, professor of women’s health at Harvard Medical School, Boston. “Counseling women should be a priority for physicians in clinical practice. And it’s not just whether weight gain is reflected on the scales or not but whether there’s an increase in central abdominal fat, a decrease in lean muscle mass, and an increase in adverse glucose tolerance.”
It is essential for women to be vigilant at this time, she added, and their exercise regimens should include strength and resistance training to preserve lean muscle mass and boost metabolic rate. Dr. Manson’s group has issued several statements stressing how important it is for clinicians to take decisive action on the counseling front and how they can do this in very little time during routine practice.
Also in full support of the guideline is Mary L. Rosser, MD, PhD, assistant professor of women’s health in obstetrics and gynecology at Columbia University Irving Medical Center in New York. “Midlife is a wonderful opportunity to encourage patients to assess their overall health status and make changes to impact their future health. Women in middle age tend to experience weight gain due to a variety of factors including aging and lifestyle,” said Dr. Rosser, who was not involved in the writing of the review or guideline.
While aging and genetics cannot be altered, behaviors can, and in her view, favorable behaviors would also include stress reduction and adequate sleep.
“The importance of reducing obesity with early intervention and prevention must focus on all women,” Dr. Rosser said. “We must narrow the inequities gap in care especially for high-risk minority groups and underserved populations. This will reduce disease and death and provide women the gift of active living and feeling better.”
The WPSI authors have made available a summary of the review and guideline for patients.
The systematic review and clinical guideline were funded by the federal Health Resources and Services Administration through ACOG. The authors of the guideline and the review authors disclosed no relevant financial conflicts of interest. Dr. Manson and Dr. Rosser disclosed no relevant competing interests with regard to their comments.
Midlife women who are of normal weight or are overweight should routinely receive counseling aimed at limiting weight gain and preventing obesity and its associated health risks, a new clinical guideline states.
The recommendation, issued by the Women’s Preventive Services Initiative (WPSI) of the American College of Obstetricians and Gynecologists (ACOG), supports regular lifestyle counseling for women aged 40-60 years with normal or overweight body mass index of 18.5-29.9 kg/m2. Counseling could include individualized discussion of healthy eating and physical activity initiated by health professionals involved in preventive care.
Published online in Annals of Internal Medicine, the guideline addresses the prevalence and health burdens of obesity in U.S. women of middle age and seeks to reduce the known harms of obesity with an intervention of minimal anticipated harms. High BMI increases the risk for many chronic conditions including hypertension, dyslipidemia, type 2 diabetes, coronary artery disease, stroke, and all-cause mortality.
The best way to counsel, however, remains unclear. “Although the optimal approach could not be discerned from existing trials, a range of interventions of varying duration, frequency, and intensity showed benefit with potential clinical significance,” wrote the WPSI guideline panel, led by David P. Chelmow, MD, chair of the department of obstetrics and gynecology at Virginia Commonwealth University in Richmond.
The guideline rests on a systematic literature review led by family doctor Amy G. Cantor, MD, MPH, of the Pacific Northwest Evidence-based Practice Center, at Oregon Health & Science University in Portland, suggesting moderate reductions in weight could be achieved by offering advice to this age group.
The federally supported WPSI was launched by ACOG in 2016. The guideline fills a gap in current recommendations in that it targets a specific risk group and specifies individual counseling based on its effectiveness and applicability in primary care settings.
In another benefit of routine counseling, the panel stated, “Normalizing counseling about healthy diet and physical activity by providing it to all midlife women may also mitigate concerns about weight stigma resulting from only counseling women with obesity.”
The panelists noted that during 2017-2018, the prevalence of obesity (BMI ≥ 30.0 kg/m2) was 43.3% among U.S. women aged 40-59 years, while the prevalence of severe obesity (BMI ≥ 40.0 kg/m2) was highest in this age group at 11.5%. “Midlife women gain weight at an average of approximately 1.5 pounds per year, which increases their risk for transitioning from normal or overweight to obese BMI,” the panelists wrote.
The review
Dr. Cantor’s group analyzed seven randomized controlled trials (RCTs) published up to October 2021 from 12 publications involving 51,638 participants. Although the trials were largely small and heterogeneous, they suggested that counseling may result in modest differences in weight change without causing important harms.
Four RCTs showed significant favorable weight changes for counseling over no-counseling control groups, with a mean difference of 0.87 to 2.5 kg, whereas one trial of counseling and two trials of exercise showed no differences. One of two RCTs reported improved quality-of-life measures.
As for harms, while interventions did not increase measures of depression or stress in one trial, self-reported falls (37% vs. 29%, P < .001) and injuries (19% vs. 14%, P = .03) were more frequent with exercise counseling in one trial.
“More research is needed to determine optimal content, frequency, length, and number of sessions required and should include additional patient populations,” Dr. Cantor and associates wrote.
In terms of limitations, the authors acknowledged that trials of behavioral interventions in maintaining or reducing weight in midlife women demonstrate small magnitudes of effect.
Offering a nonparticipant’s perspective on the WPSI guideline for this news organization, JoAnn E. Manson, MD, DrPH, MACP, chief of the division of preventive medicine at Brigham and Women’s Hospital in Boston, said its message is of prime importance for women of middle age and it goes beyond concern about pounds lost or gained.
“Midlife and the transition to menopause are high-risk periods for women in terms of typical changes in body composition that increase the risk of adverse cardiometabolic outcomes,” said Dr. Manson, professor of women’s health at Harvard Medical School, Boston. “Counseling women should be a priority for physicians in clinical practice. And it’s not just whether weight gain is reflected on the scales or not but whether there’s an increase in central abdominal fat, a decrease in lean muscle mass, and an increase in adverse glucose tolerance.”
It is essential for women to be vigilant at this time, she added, and their exercise regimens should include strength and resistance training to preserve lean muscle mass and boost metabolic rate. Dr. Manson’s group has issued several statements stressing how important it is for clinicians to take decisive action on the counseling front and how they can do this in very little time during routine practice.
Also in full support of the guideline is Mary L. Rosser, MD, PhD, assistant professor of women’s health in obstetrics and gynecology at Columbia University Irving Medical Center in New York. “Midlife is a wonderful opportunity to encourage patients to assess their overall health status and make changes to impact their future health. Women in middle age tend to experience weight gain due to a variety of factors including aging and lifestyle,” said Dr. Rosser, who was not involved in the writing of the review or guideline.
While aging and genetics cannot be altered, behaviors can, and in her view, favorable behaviors would also include stress reduction and adequate sleep.
“The importance of reducing obesity with early intervention and prevention must focus on all women,” Dr. Rosser said. “We must narrow the inequities gap in care especially for high-risk minority groups and underserved populations. This will reduce disease and death and provide women the gift of active living and feeling better.”
The WPSI authors have made available a summary of the review and guideline for patients.
The systematic review and clinical guideline were funded by the federal Health Resources and Services Administration through ACOG. The authors of the guideline and the review authors disclosed no relevant financial conflicts of interest. Dr. Manson and Dr. Rosser disclosed no relevant competing interests with regard to their comments.
Midlife women who are of normal weight or are overweight should routinely receive counseling aimed at limiting weight gain and preventing obesity and its associated health risks, a new clinical guideline states.
The recommendation, issued by the Women’s Preventive Services Initiative (WPSI) of the American College of Obstetricians and Gynecologists (ACOG), supports regular lifestyle counseling for women aged 40-60 years with normal or overweight body mass index of 18.5-29.9 kg/m2. Counseling could include individualized discussion of healthy eating and physical activity initiated by health professionals involved in preventive care.
Published online in Annals of Internal Medicine, the guideline addresses the prevalence and health burdens of obesity in U.S. women of middle age and seeks to reduce the known harms of obesity with an intervention of minimal anticipated harms. High BMI increases the risk for many chronic conditions including hypertension, dyslipidemia, type 2 diabetes, coronary artery disease, stroke, and all-cause mortality.
The best way to counsel, however, remains unclear. “Although the optimal approach could not be discerned from existing trials, a range of interventions of varying duration, frequency, and intensity showed benefit with potential clinical significance,” wrote the WPSI guideline panel, led by David P. Chelmow, MD, chair of the department of obstetrics and gynecology at Virginia Commonwealth University in Richmond.
The guideline rests on a systematic literature review led by family doctor Amy G. Cantor, MD, MPH, of the Pacific Northwest Evidence-based Practice Center, at Oregon Health & Science University in Portland, suggesting moderate reductions in weight could be achieved by offering advice to this age group.
The federally supported WPSI was launched by ACOG in 2016. The guideline fills a gap in current recommendations in that it targets a specific risk group and specifies individual counseling based on its effectiveness and applicability in primary care settings.
In another benefit of routine counseling, the panel stated, “Normalizing counseling about healthy diet and physical activity by providing it to all midlife women may also mitigate concerns about weight stigma resulting from only counseling women with obesity.”
The panelists noted that during 2017-2018, the prevalence of obesity (BMI ≥ 30.0 kg/m2) was 43.3% among U.S. women aged 40-59 years, while the prevalence of severe obesity (BMI ≥ 40.0 kg/m2) was highest in this age group at 11.5%. “Midlife women gain weight at an average of approximately 1.5 pounds per year, which increases their risk for transitioning from normal or overweight to obese BMI,” the panelists wrote.
The review
Dr. Cantor’s group analyzed seven randomized controlled trials (RCTs) published up to October 2021 from 12 publications involving 51,638 participants. Although the trials were largely small and heterogeneous, they suggested that counseling may result in modest differences in weight change without causing important harms.
Four RCTs showed significant favorable weight changes for counseling over no-counseling control groups, with a mean difference of 0.87 to 2.5 kg, whereas one trial of counseling and two trials of exercise showed no differences. One of two RCTs reported improved quality-of-life measures.
As for harms, while interventions did not increase measures of depression or stress in one trial, self-reported falls (37% vs. 29%, P < .001) and injuries (19% vs. 14%, P = .03) were more frequent with exercise counseling in one trial.
“More research is needed to determine optimal content, frequency, length, and number of sessions required and should include additional patient populations,” Dr. Cantor and associates wrote.
In terms of limitations, the authors acknowledged that trials of behavioral interventions in maintaining or reducing weight in midlife women demonstrate small magnitudes of effect.
Offering a nonparticipant’s perspective on the WPSI guideline for this news organization, JoAnn E. Manson, MD, DrPH, MACP, chief of the division of preventive medicine at Brigham and Women’s Hospital in Boston, said its message is of prime importance for women of middle age and it goes beyond concern about pounds lost or gained.
“Midlife and the transition to menopause are high-risk periods for women in terms of typical changes in body composition that increase the risk of adverse cardiometabolic outcomes,” said Dr. Manson, professor of women’s health at Harvard Medical School, Boston. “Counseling women should be a priority for physicians in clinical practice. And it’s not just whether weight gain is reflected on the scales or not but whether there’s an increase in central abdominal fat, a decrease in lean muscle mass, and an increase in adverse glucose tolerance.”
It is essential for women to be vigilant at this time, she added, and their exercise regimens should include strength and resistance training to preserve lean muscle mass and boost metabolic rate. Dr. Manson’s group has issued several statements stressing how important it is for clinicians to take decisive action on the counseling front and how they can do this in very little time during routine practice.
Also in full support of the guideline is Mary L. Rosser, MD, PhD, assistant professor of women’s health in obstetrics and gynecology at Columbia University Irving Medical Center in New York. “Midlife is a wonderful opportunity to encourage patients to assess their overall health status and make changes to impact their future health. Women in middle age tend to experience weight gain due to a variety of factors including aging and lifestyle,” said Dr. Rosser, who was not involved in the writing of the review or guideline.
While aging and genetics cannot be altered, behaviors can, and in her view, favorable behaviors would also include stress reduction and adequate sleep.
“The importance of reducing obesity with early intervention and prevention must focus on all women,” Dr. Rosser said. “We must narrow the inequities gap in care especially for high-risk minority groups and underserved populations. This will reduce disease and death and provide women the gift of active living and feeling better.”
The WPSI authors have made available a summary of the review and guideline for patients.
The systematic review and clinical guideline were funded by the federal Health Resources and Services Administration through ACOG. The authors of the guideline and the review authors disclosed no relevant financial conflicts of interest. Dr. Manson and Dr. Rosser disclosed no relevant competing interests with regard to their comments.
FROM ANNALS OF INTERNAL MEDICINE
Banana Boat recalls scalp sunscreen spray
.
The company announced a voluntary recall for three batches of the Banana Boat Hair & Scalp Spray SPF 30, which came in 6-ounce bottles and was sold across the U.S. through various retailers and online, according to a recall alert by the Food and Drug Administration.
The three batches have a UPC label of 0-79656-04041-8 and fall under the lot codes 20016AF, 20084BF, and 21139AF, with the expiration dates of December 2022, February 2023, and April 2024, respectively.
“An internal review found that some samples of the product contained trace levels of benzene. While benzene is not an ingredient in any Banana Boat products, the review showed the unexpected levels of benzene came from the propellant that sprays the product out of the can,” according to the recall notice.
“Importantly, no other batches of Hair & Scalp (either before or after these batch codes) and no other Banana Boat products are in the scope of this recall and may continue to be used by consumers safely and as intended,” the company wrote.
Benzene is classified as a human carcinogen, the FDA wrote. Exposure to benzene can occur through the nose, mouth, and skin, and it can result in serious conditions such as leukemia, bone marrow cancer, and blood disorders.
“Benzene is ubiquitous in the environment. Humans around the world have daily exposures to it indoors and outdoors from multiple sources,” the company said. “Daily exposure to benzene in the recalled products would not be expected to cause adverse health consequences according to an independent health assessment using established exposure modeling guidelines.”
Edgewell said it hasn’t received any reports of bad events related to the recall. The company has told retailers to remove the affected batches from shelves.
Banana Boat will reimburse consumers who purchased a product with one of the affected lot codes, which are on the bottom of the can. In the meantime, consumers should stop using the affected product right away and discard it.
The recall comes a little over a year after Johnson & Johnson recalled five sunscreens due to low levels of benzene, according to The Associated Press. That recall included Aveeno and Neutrogena products in spray cans.
Consumers with questions about the recall can contact Edgewell Personal Care at 888-686-3988 Monday through Friday, 9 a.m. to 6 p.m. ET. People can also read more at the Banana Boat FAQ page or file for a refund directly on the Banana Boat Recall page.
A version of this article first appeared on WebMD.com.
.
The company announced a voluntary recall for three batches of the Banana Boat Hair & Scalp Spray SPF 30, which came in 6-ounce bottles and was sold across the U.S. through various retailers and online, according to a recall alert by the Food and Drug Administration.
The three batches have a UPC label of 0-79656-04041-8 and fall under the lot codes 20016AF, 20084BF, and 21139AF, with the expiration dates of December 2022, February 2023, and April 2024, respectively.
“An internal review found that some samples of the product contained trace levels of benzene. While benzene is not an ingredient in any Banana Boat products, the review showed the unexpected levels of benzene came from the propellant that sprays the product out of the can,” according to the recall notice.
“Importantly, no other batches of Hair & Scalp (either before or after these batch codes) and no other Banana Boat products are in the scope of this recall and may continue to be used by consumers safely and as intended,” the company wrote.
Benzene is classified as a human carcinogen, the FDA wrote. Exposure to benzene can occur through the nose, mouth, and skin, and it can result in serious conditions such as leukemia, bone marrow cancer, and blood disorders.
“Benzene is ubiquitous in the environment. Humans around the world have daily exposures to it indoors and outdoors from multiple sources,” the company said. “Daily exposure to benzene in the recalled products would not be expected to cause adverse health consequences according to an independent health assessment using established exposure modeling guidelines.”
Edgewell said it hasn’t received any reports of bad events related to the recall. The company has told retailers to remove the affected batches from shelves.
Banana Boat will reimburse consumers who purchased a product with one of the affected lot codes, which are on the bottom of the can. In the meantime, consumers should stop using the affected product right away and discard it.
The recall comes a little over a year after Johnson & Johnson recalled five sunscreens due to low levels of benzene, according to The Associated Press. That recall included Aveeno and Neutrogena products in spray cans.
Consumers with questions about the recall can contact Edgewell Personal Care at 888-686-3988 Monday through Friday, 9 a.m. to 6 p.m. ET. People can also read more at the Banana Boat FAQ page or file for a refund directly on the Banana Boat Recall page.
A version of this article first appeared on WebMD.com.
.
The company announced a voluntary recall for three batches of the Banana Boat Hair & Scalp Spray SPF 30, which came in 6-ounce bottles and was sold across the U.S. through various retailers and online, according to a recall alert by the Food and Drug Administration.
The three batches have a UPC label of 0-79656-04041-8 and fall under the lot codes 20016AF, 20084BF, and 21139AF, with the expiration dates of December 2022, February 2023, and April 2024, respectively.
“An internal review found that some samples of the product contained trace levels of benzene. While benzene is not an ingredient in any Banana Boat products, the review showed the unexpected levels of benzene came from the propellant that sprays the product out of the can,” according to the recall notice.
“Importantly, no other batches of Hair & Scalp (either before or after these batch codes) and no other Banana Boat products are in the scope of this recall and may continue to be used by consumers safely and as intended,” the company wrote.
Benzene is classified as a human carcinogen, the FDA wrote. Exposure to benzene can occur through the nose, mouth, and skin, and it can result in serious conditions such as leukemia, bone marrow cancer, and blood disorders.
“Benzene is ubiquitous in the environment. Humans around the world have daily exposures to it indoors and outdoors from multiple sources,” the company said. “Daily exposure to benzene in the recalled products would not be expected to cause adverse health consequences according to an independent health assessment using established exposure modeling guidelines.”
Edgewell said it hasn’t received any reports of bad events related to the recall. The company has told retailers to remove the affected batches from shelves.
Banana Boat will reimburse consumers who purchased a product with one of the affected lot codes, which are on the bottom of the can. In the meantime, consumers should stop using the affected product right away and discard it.
The recall comes a little over a year after Johnson & Johnson recalled five sunscreens due to low levels of benzene, according to The Associated Press. That recall included Aveeno and Neutrogena products in spray cans.
Consumers with questions about the recall can contact Edgewell Personal Care at 888-686-3988 Monday through Friday, 9 a.m. to 6 p.m. ET. People can also read more at the Banana Boat FAQ page or file for a refund directly on the Banana Boat Recall page.
A version of this article first appeared on WebMD.com.
Evusheld for COVID-19: Lifesaving and free, but still few takers
Evusheld (AstraZeneca), a medication used to prevent SARS-CoV-2 infection in patients at high risk, has problems: Namely, that supplies of the potentially lifesaving drug outweigh demand.
At least 7 million people who are immunocompromised could benefit from it, as could many others who are undergoing cancer treatment, have received a transplant, or who are allergic to the COVID-19 vaccines. The medication has laboratory-produced antibodies against SARS-CoV-2 and helps the body protect itself. It can slash the chances of becoming infected by 77%, according to the U.S. Food and Drug Administration.
And it’s free to eligible patients (although there may be an out-of-pocket administrative fee in some cases).
To meet demand, the Biden administration secured 1.7 million doses of the medicine, which was granted emergency use authorization by the FDA in December 2021. As of July 25, however, 793,348 doses have been ordered by the administration sites, and only 398,181 doses have been reported as used, a spokesperson for the Department of Health & Human Services tells this news organization.
Each week, a certain amount of doses from the 1.7 million dose stockpile is made available to state and territorial health departments. States have not been asking for their full allotment, the spokesperson said July 28.
Now, HHS and AstraZeneca have taken a number of steps to increase awareness of the medication and access to it.
- On July 27, HHS announced that individual providers and smaller sites of care that don’t currently receive Evusheld through the federal distribution process via the HHS Health Partner Order Portal can now order up to three patient courses of the medicine. These can be
- Health care providers can use the HHS’s COVID-19 Therapeutics Locator to find Evusheld in their area.
- AstraZeneca has launched a new website with educational materials and says it is working closely with patient and professional groups to inform patients and health care providers.
- A direct-to-consumer ad launched on June 22 and will run in the United States online and on TV (Yahoo, Fox, CBS Sports, MSN, ESPN) and be amplified on social and digital channels through year’s end, an AstraZeneca spokesperson said in an interview.
- AstraZeneca set up a toll-free number for providers: 1-833-EVUSHLD.
Evusheld includes two monoclonal antibodies, tixagevimab and cilgavimab. The medication is given as two consecutive intramuscular injections during a single visit to a doctor’s office, infusion center, or other health care facility. The antibodies bind to the SARS-CoV-2 spike protein and prevent the virus from getting into human cells and infecting them. It’s authorized for use in children and adults aged 12 years and older who weigh at least 88 pounds.
Studies have found that the medication decreases the risk of getting COVID-19 for up to 6 months after it is given. The FDA recommends repeat dosing every 6 months with the doses of 300 mg of each monoclonal antibody. In clinical trials, Evusheld reduced the incidence of COVID-19 symptomatic illness by 77%, compared with placebo.
Physicians monitor patients for an hour after administering Evusheld for allergic reactions. Other possible side effects include cardiac events, but they are not common.
Doctors and patients weigh in
Physicians – and patients – from the United States to the United Kingdom and beyond are questioning why the medication is underused while lauding the recent efforts to expand access and increase awareness.
The U.S. federal government may have underestimated the amount of communication needed to increase awareness of the medication and its applications, said infectious disease specialist William Schaffner, MD, professor of preventive medicine at Vanderbilt University School of Medicine, Nashville, Tenn.
“HHS hasn’t made a major educational effort to promote it,” he said in an interview.
Many physicians who need to know about it, such as transplant doctors and rheumatologists, are outside the typical public health communications loop, he said.
Eric Topol, MD, director of the Scripps Research Transational Institute and editor-in-chief of Medscape, has taken to social media to bemoan the lack of awareness.
Another infectious disease expert agrees. “In my experience, the awareness of Evusheld is low amongst many patients as well as many providers,” said Amesh Adalja, MD, a senior scholar at the Johns Hopkins Center for Health Security, Baltimore.
“Initially, there were scarce supplies of the drug, and certain hospital systems tiered eligibility based on degrees of immunosuppression, and only the most immunosuppressed were proactively approached for treatment.”
“Also, many community hospitals never initially ordered Evusheld – they may have been crowded out by academic centers who treat many more immunosuppressed patients and may not currently see it as a priority,” Dr. Adalja said in an interview. “As such, many immunosuppressed patients would have to seek treatment at academic medical centers, where the drug is more likely to be available.”
A version of this article first appeared on Medscape.com.
Evusheld (AstraZeneca), a medication used to prevent SARS-CoV-2 infection in patients at high risk, has problems: Namely, that supplies of the potentially lifesaving drug outweigh demand.
At least 7 million people who are immunocompromised could benefit from it, as could many others who are undergoing cancer treatment, have received a transplant, or who are allergic to the COVID-19 vaccines. The medication has laboratory-produced antibodies against SARS-CoV-2 and helps the body protect itself. It can slash the chances of becoming infected by 77%, according to the U.S. Food and Drug Administration.
And it’s free to eligible patients (although there may be an out-of-pocket administrative fee in some cases).
To meet demand, the Biden administration secured 1.7 million doses of the medicine, which was granted emergency use authorization by the FDA in December 2021. As of July 25, however, 793,348 doses have been ordered by the administration sites, and only 398,181 doses have been reported as used, a spokesperson for the Department of Health & Human Services tells this news organization.
Each week, a certain amount of doses from the 1.7 million dose stockpile is made available to state and territorial health departments. States have not been asking for their full allotment, the spokesperson said July 28.
Now, HHS and AstraZeneca have taken a number of steps to increase awareness of the medication and access to it.
- On July 27, HHS announced that individual providers and smaller sites of care that don’t currently receive Evusheld through the federal distribution process via the HHS Health Partner Order Portal can now order up to three patient courses of the medicine. These can be
- Health care providers can use the HHS’s COVID-19 Therapeutics Locator to find Evusheld in their area.
- AstraZeneca has launched a new website with educational materials and says it is working closely with patient and professional groups to inform patients and health care providers.
- A direct-to-consumer ad launched on June 22 and will run in the United States online and on TV (Yahoo, Fox, CBS Sports, MSN, ESPN) and be amplified on social and digital channels through year’s end, an AstraZeneca spokesperson said in an interview.
- AstraZeneca set up a toll-free number for providers: 1-833-EVUSHLD.
Evusheld includes two monoclonal antibodies, tixagevimab and cilgavimab. The medication is given as two consecutive intramuscular injections during a single visit to a doctor’s office, infusion center, or other health care facility. The antibodies bind to the SARS-CoV-2 spike protein and prevent the virus from getting into human cells and infecting them. It’s authorized for use in children and adults aged 12 years and older who weigh at least 88 pounds.
Studies have found that the medication decreases the risk of getting COVID-19 for up to 6 months after it is given. The FDA recommends repeat dosing every 6 months with the doses of 300 mg of each monoclonal antibody. In clinical trials, Evusheld reduced the incidence of COVID-19 symptomatic illness by 77%, compared with placebo.
Physicians monitor patients for an hour after administering Evusheld for allergic reactions. Other possible side effects include cardiac events, but they are not common.
Doctors and patients weigh in
Physicians – and patients – from the United States to the United Kingdom and beyond are questioning why the medication is underused while lauding the recent efforts to expand access and increase awareness.
The U.S. federal government may have underestimated the amount of communication needed to increase awareness of the medication and its applications, said infectious disease specialist William Schaffner, MD, professor of preventive medicine at Vanderbilt University School of Medicine, Nashville, Tenn.
“HHS hasn’t made a major educational effort to promote it,” he said in an interview.
Many physicians who need to know about it, such as transplant doctors and rheumatologists, are outside the typical public health communications loop, he said.
Eric Topol, MD, director of the Scripps Research Transational Institute and editor-in-chief of Medscape, has taken to social media to bemoan the lack of awareness.
Another infectious disease expert agrees. “In my experience, the awareness of Evusheld is low amongst many patients as well as many providers,” said Amesh Adalja, MD, a senior scholar at the Johns Hopkins Center for Health Security, Baltimore.
“Initially, there were scarce supplies of the drug, and certain hospital systems tiered eligibility based on degrees of immunosuppression, and only the most immunosuppressed were proactively approached for treatment.”
“Also, many community hospitals never initially ordered Evusheld – they may have been crowded out by academic centers who treat many more immunosuppressed patients and may not currently see it as a priority,” Dr. Adalja said in an interview. “As such, many immunosuppressed patients would have to seek treatment at academic medical centers, where the drug is more likely to be available.”
A version of this article first appeared on Medscape.com.
Evusheld (AstraZeneca), a medication used to prevent SARS-CoV-2 infection in patients at high risk, has problems: Namely, that supplies of the potentially lifesaving drug outweigh demand.
At least 7 million people who are immunocompromised could benefit from it, as could many others who are undergoing cancer treatment, have received a transplant, or who are allergic to the COVID-19 vaccines. The medication has laboratory-produced antibodies against SARS-CoV-2 and helps the body protect itself. It can slash the chances of becoming infected by 77%, according to the U.S. Food and Drug Administration.
And it’s free to eligible patients (although there may be an out-of-pocket administrative fee in some cases).
To meet demand, the Biden administration secured 1.7 million doses of the medicine, which was granted emergency use authorization by the FDA in December 2021. As of July 25, however, 793,348 doses have been ordered by the administration sites, and only 398,181 doses have been reported as used, a spokesperson for the Department of Health & Human Services tells this news organization.
Each week, a certain amount of doses from the 1.7 million dose stockpile is made available to state and territorial health departments. States have not been asking for their full allotment, the spokesperson said July 28.
Now, HHS and AstraZeneca have taken a number of steps to increase awareness of the medication and access to it.
- On July 27, HHS announced that individual providers and smaller sites of care that don’t currently receive Evusheld through the federal distribution process via the HHS Health Partner Order Portal can now order up to three patient courses of the medicine. These can be
- Health care providers can use the HHS’s COVID-19 Therapeutics Locator to find Evusheld in their area.
- AstraZeneca has launched a new website with educational materials and says it is working closely with patient and professional groups to inform patients and health care providers.
- A direct-to-consumer ad launched on June 22 and will run in the United States online and on TV (Yahoo, Fox, CBS Sports, MSN, ESPN) and be amplified on social and digital channels through year’s end, an AstraZeneca spokesperson said in an interview.
- AstraZeneca set up a toll-free number for providers: 1-833-EVUSHLD.
Evusheld includes two monoclonal antibodies, tixagevimab and cilgavimab. The medication is given as two consecutive intramuscular injections during a single visit to a doctor’s office, infusion center, or other health care facility. The antibodies bind to the SARS-CoV-2 spike protein and prevent the virus from getting into human cells and infecting them. It’s authorized for use in children and adults aged 12 years and older who weigh at least 88 pounds.
Studies have found that the medication decreases the risk of getting COVID-19 for up to 6 months after it is given. The FDA recommends repeat dosing every 6 months with the doses of 300 mg of each monoclonal antibody. In clinical trials, Evusheld reduced the incidence of COVID-19 symptomatic illness by 77%, compared with placebo.
Physicians monitor patients for an hour after administering Evusheld for allergic reactions. Other possible side effects include cardiac events, but they are not common.
Doctors and patients weigh in
Physicians – and patients – from the United States to the United Kingdom and beyond are questioning why the medication is underused while lauding the recent efforts to expand access and increase awareness.
The U.S. federal government may have underestimated the amount of communication needed to increase awareness of the medication and its applications, said infectious disease specialist William Schaffner, MD, professor of preventive medicine at Vanderbilt University School of Medicine, Nashville, Tenn.
“HHS hasn’t made a major educational effort to promote it,” he said in an interview.
Many physicians who need to know about it, such as transplant doctors and rheumatologists, are outside the typical public health communications loop, he said.
Eric Topol, MD, director of the Scripps Research Transational Institute and editor-in-chief of Medscape, has taken to social media to bemoan the lack of awareness.
Another infectious disease expert agrees. “In my experience, the awareness of Evusheld is low amongst many patients as well as many providers,” said Amesh Adalja, MD, a senior scholar at the Johns Hopkins Center for Health Security, Baltimore.
“Initially, there were scarce supplies of the drug, and certain hospital systems tiered eligibility based on degrees of immunosuppression, and only the most immunosuppressed were proactively approached for treatment.”
“Also, many community hospitals never initially ordered Evusheld – they may have been crowded out by academic centers who treat many more immunosuppressed patients and may not currently see it as a priority,” Dr. Adalja said in an interview. “As such, many immunosuppressed patients would have to seek treatment at academic medical centers, where the drug is more likely to be available.”
A version of this article first appeared on Medscape.com.
Higher ADR continues to show ‘strong, consistent’ link with lower interval CRC
Higher adenoma detection rates (ADR) during colonoscopies were associated with lower rates of interim colorectal cancer (CRC), and the relationship held true along a broad range of ADR values, according to a retrospective study.
The new study, published online in JAMA, examined ADRs and rates of interim colorectal cancer among patients in California and Washington State between 2011 and 2017. The authors found a 3% reduction in risk for each additional 1% value of ADR. The reduction in risk held true even at high ADRs.
“It basically reaffirms what we’ve believed for the longest time, and other research work has documented – that interim cancers are higher in association with lower adenoma detection rates. The higher you can get that adenoma detection rate, the more we’re going to be able to lower the [rate of] cancers that develop within 3 years of a colonoscopy,” said Lawrence Kosinski, MD, who was asked to comment on the study.
The study included 735,396 patients with a median age of 61.4 years. Among these patients, 852,624 negative colonoscopies were performed by 383 eligible physicians. Participating physicians had to perform at least 25 screening colonoscopies and 100 total colonoscopies per year. After 2.4 million person-years of follow-up, the researchers observed 619 postcolonoscopy colorectal cancers and 36 related deaths over a median follow-up of 3.25 years.
There was an association between each 1% increase in ADR and a reduced probability of postcolonoscopy CRC (hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.96-0.98) and mortality from postcolonoscopy CRC (HR, 0.95; 95% CI, 0.92-0.99).
The median ADR was 28.3%. There was an association between ADR above the median versus below the median and a reduced risk of postcolonoscopy CRC with 1.79 cases versus 3.10 cases per 10,000 person-years, respectively (absolute difference in 7-year risk, –12.2 per 10,000 negative colonoscopies; HR, 0.61; 95% CI, 0.52-0.73). There was a similar reduction in risk of postcolonoscopy CRC-related mortality (0.05 versus 0.22 per 10,000 person-years; absolute difference in 7-year risk, –1.2 per 10,000 negative colonoscopies; HR, 0.26; 95% CI, 0.11-0.65).
These findings may be limited in generalizability to physicians with lower procedure volumes or to populations with different adenoma prevalence.
“Given the strong, consistent associations of higher adenoma detection rates with colonoscopy effectiveness for reducing colorectal cancer incidence and mortality, the current results support more research to identify reliable and readily adoptable methods for increasing adenoma detection rates among physicians with lower values across diverse settings,” the researchers wrote.
The improvement over a broad range of ADRs, along with other recent findings, suggests that there may need to be updates to the use of ADRs as a quality metric, according to an accompanying editorial by Douglas K. Rex, MD, of the division of gastroenterology/hepatology at Indiana University, Indianapolis. For example, it’s possible that ADRs could be measured by averaging values from screening, diagnostic, and surveillance colonoscopy. The editorialist suggested that, if improvements in interim cancer rates continue as ADRs approach 50%, the current view of ADRs, as a minimally acceptable standard, may require reconsideration. Instead, it may be appropriate to continue with a minimum threshold, but add a much higher, aspirational target. Dr. Rex also suggested that highly-variable detection of sessile serrated lesions could be excluded from ADRs in order to reduce variability.
Factors to consider
The study is useful, but it doesn’t address the disparity in adenoma detection that exists between individual doctors, according to Dr. Kosinski, founder and chief medical officer of SonarMD and previously director of a large gastroenterology clinic. “Even if you look at doctors who do a minimum of 250 screening colonoscopies in a year, there’s still variability. There was even a study published in 2014 showing ADRs anywhere from 7.4% to 52.5%. The bell curve is broad,” he said.
As patients age, they have a higher frequency of polyps appearing on the right side of the colon, and those polyps are flatter and more easily missed than polyps on the left side. “The variation in ADR is higher on the right side of the colon than it is on the left. Doctors have to really do a very good job of examining that right side of the colon so that they don’t miss the flat polyps,” said Dr. Kosinski.
To improve ADRs, Dr. Kosinski emphasized the need to take the required time out to complete a procedure, despite the tight schedules often faced by ambulatory centers. “It’s the time you take coming out of the colon that’s critical. You owe it to the patient,” he said.
And if a patient hasn’t prepped well enough, it’s better to send the patient home without the procedure than to conduct a poor-quality screening. “If you can’t see the mucosal surface, you can’t tell the patient that they have a negative colonoscopy. If you have to do more cleaning during the procedure, then do more cleaning during the procedure. If you have to cancel the procedure and bring the patient back, it’s better to do that than it is to do an incomplete colonoscopy,” said Dr. Kosinski.
He also stressed the need to make sure that the patient is properly sedated and comfortable “so that you can do the job you’re supposed to do,” he said.
Some authors disclosed relationships with Amgen and the National Cancer Institute. Dr. Rex disclosed relationships with Olympus, Boston Scientific, Aries, and others, all outside the submitted work.
Higher adenoma detection rates (ADR) during colonoscopies were associated with lower rates of interim colorectal cancer (CRC), and the relationship held true along a broad range of ADR values, according to a retrospective study.
The new study, published online in JAMA, examined ADRs and rates of interim colorectal cancer among patients in California and Washington State between 2011 and 2017. The authors found a 3% reduction in risk for each additional 1% value of ADR. The reduction in risk held true even at high ADRs.
“It basically reaffirms what we’ve believed for the longest time, and other research work has documented – that interim cancers are higher in association with lower adenoma detection rates. The higher you can get that adenoma detection rate, the more we’re going to be able to lower the [rate of] cancers that develop within 3 years of a colonoscopy,” said Lawrence Kosinski, MD, who was asked to comment on the study.
The study included 735,396 patients with a median age of 61.4 years. Among these patients, 852,624 negative colonoscopies were performed by 383 eligible physicians. Participating physicians had to perform at least 25 screening colonoscopies and 100 total colonoscopies per year. After 2.4 million person-years of follow-up, the researchers observed 619 postcolonoscopy colorectal cancers and 36 related deaths over a median follow-up of 3.25 years.
There was an association between each 1% increase in ADR and a reduced probability of postcolonoscopy CRC (hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.96-0.98) and mortality from postcolonoscopy CRC (HR, 0.95; 95% CI, 0.92-0.99).
The median ADR was 28.3%. There was an association between ADR above the median versus below the median and a reduced risk of postcolonoscopy CRC with 1.79 cases versus 3.10 cases per 10,000 person-years, respectively (absolute difference in 7-year risk, –12.2 per 10,000 negative colonoscopies; HR, 0.61; 95% CI, 0.52-0.73). There was a similar reduction in risk of postcolonoscopy CRC-related mortality (0.05 versus 0.22 per 10,000 person-years; absolute difference in 7-year risk, –1.2 per 10,000 negative colonoscopies; HR, 0.26; 95% CI, 0.11-0.65).
These findings may be limited in generalizability to physicians with lower procedure volumes or to populations with different adenoma prevalence.
“Given the strong, consistent associations of higher adenoma detection rates with colonoscopy effectiveness for reducing colorectal cancer incidence and mortality, the current results support more research to identify reliable and readily adoptable methods for increasing adenoma detection rates among physicians with lower values across diverse settings,” the researchers wrote.
The improvement over a broad range of ADRs, along with other recent findings, suggests that there may need to be updates to the use of ADRs as a quality metric, according to an accompanying editorial by Douglas K. Rex, MD, of the division of gastroenterology/hepatology at Indiana University, Indianapolis. For example, it’s possible that ADRs could be measured by averaging values from screening, diagnostic, and surveillance colonoscopy. The editorialist suggested that, if improvements in interim cancer rates continue as ADRs approach 50%, the current view of ADRs, as a minimally acceptable standard, may require reconsideration. Instead, it may be appropriate to continue with a minimum threshold, but add a much higher, aspirational target. Dr. Rex also suggested that highly-variable detection of sessile serrated lesions could be excluded from ADRs in order to reduce variability.
Factors to consider
The study is useful, but it doesn’t address the disparity in adenoma detection that exists between individual doctors, according to Dr. Kosinski, founder and chief medical officer of SonarMD and previously director of a large gastroenterology clinic. “Even if you look at doctors who do a minimum of 250 screening colonoscopies in a year, there’s still variability. There was even a study published in 2014 showing ADRs anywhere from 7.4% to 52.5%. The bell curve is broad,” he said.
As patients age, they have a higher frequency of polyps appearing on the right side of the colon, and those polyps are flatter and more easily missed than polyps on the left side. “The variation in ADR is higher on the right side of the colon than it is on the left. Doctors have to really do a very good job of examining that right side of the colon so that they don’t miss the flat polyps,” said Dr. Kosinski.
To improve ADRs, Dr. Kosinski emphasized the need to take the required time out to complete a procedure, despite the tight schedules often faced by ambulatory centers. “It’s the time you take coming out of the colon that’s critical. You owe it to the patient,” he said.
And if a patient hasn’t prepped well enough, it’s better to send the patient home without the procedure than to conduct a poor-quality screening. “If you can’t see the mucosal surface, you can’t tell the patient that they have a negative colonoscopy. If you have to do more cleaning during the procedure, then do more cleaning during the procedure. If you have to cancel the procedure and bring the patient back, it’s better to do that than it is to do an incomplete colonoscopy,” said Dr. Kosinski.
He also stressed the need to make sure that the patient is properly sedated and comfortable “so that you can do the job you’re supposed to do,” he said.
Some authors disclosed relationships with Amgen and the National Cancer Institute. Dr. Rex disclosed relationships with Olympus, Boston Scientific, Aries, and others, all outside the submitted work.
Higher adenoma detection rates (ADR) during colonoscopies were associated with lower rates of interim colorectal cancer (CRC), and the relationship held true along a broad range of ADR values, according to a retrospective study.
The new study, published online in JAMA, examined ADRs and rates of interim colorectal cancer among patients in California and Washington State between 2011 and 2017. The authors found a 3% reduction in risk for each additional 1% value of ADR. The reduction in risk held true even at high ADRs.
“It basically reaffirms what we’ve believed for the longest time, and other research work has documented – that interim cancers are higher in association with lower adenoma detection rates. The higher you can get that adenoma detection rate, the more we’re going to be able to lower the [rate of] cancers that develop within 3 years of a colonoscopy,” said Lawrence Kosinski, MD, who was asked to comment on the study.
The study included 735,396 patients with a median age of 61.4 years. Among these patients, 852,624 negative colonoscopies were performed by 383 eligible physicians. Participating physicians had to perform at least 25 screening colonoscopies and 100 total colonoscopies per year. After 2.4 million person-years of follow-up, the researchers observed 619 postcolonoscopy colorectal cancers and 36 related deaths over a median follow-up of 3.25 years.
There was an association between each 1% increase in ADR and a reduced probability of postcolonoscopy CRC (hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.96-0.98) and mortality from postcolonoscopy CRC (HR, 0.95; 95% CI, 0.92-0.99).
The median ADR was 28.3%. There was an association between ADR above the median versus below the median and a reduced risk of postcolonoscopy CRC with 1.79 cases versus 3.10 cases per 10,000 person-years, respectively (absolute difference in 7-year risk, –12.2 per 10,000 negative colonoscopies; HR, 0.61; 95% CI, 0.52-0.73). There was a similar reduction in risk of postcolonoscopy CRC-related mortality (0.05 versus 0.22 per 10,000 person-years; absolute difference in 7-year risk, –1.2 per 10,000 negative colonoscopies; HR, 0.26; 95% CI, 0.11-0.65).
These findings may be limited in generalizability to physicians with lower procedure volumes or to populations with different adenoma prevalence.
“Given the strong, consistent associations of higher adenoma detection rates with colonoscopy effectiveness for reducing colorectal cancer incidence and mortality, the current results support more research to identify reliable and readily adoptable methods for increasing adenoma detection rates among physicians with lower values across diverse settings,” the researchers wrote.
The improvement over a broad range of ADRs, along with other recent findings, suggests that there may need to be updates to the use of ADRs as a quality metric, according to an accompanying editorial by Douglas K. Rex, MD, of the division of gastroenterology/hepatology at Indiana University, Indianapolis. For example, it’s possible that ADRs could be measured by averaging values from screening, diagnostic, and surveillance colonoscopy. The editorialist suggested that, if improvements in interim cancer rates continue as ADRs approach 50%, the current view of ADRs, as a minimally acceptable standard, may require reconsideration. Instead, it may be appropriate to continue with a minimum threshold, but add a much higher, aspirational target. Dr. Rex also suggested that highly-variable detection of sessile serrated lesions could be excluded from ADRs in order to reduce variability.
Factors to consider
The study is useful, but it doesn’t address the disparity in adenoma detection that exists between individual doctors, according to Dr. Kosinski, founder and chief medical officer of SonarMD and previously director of a large gastroenterology clinic. “Even if you look at doctors who do a minimum of 250 screening colonoscopies in a year, there’s still variability. There was even a study published in 2014 showing ADRs anywhere from 7.4% to 52.5%. The bell curve is broad,” he said.
As patients age, they have a higher frequency of polyps appearing on the right side of the colon, and those polyps are flatter and more easily missed than polyps on the left side. “The variation in ADR is higher on the right side of the colon than it is on the left. Doctors have to really do a very good job of examining that right side of the colon so that they don’t miss the flat polyps,” said Dr. Kosinski.
To improve ADRs, Dr. Kosinski emphasized the need to take the required time out to complete a procedure, despite the tight schedules often faced by ambulatory centers. “It’s the time you take coming out of the colon that’s critical. You owe it to the patient,” he said.
And if a patient hasn’t prepped well enough, it’s better to send the patient home without the procedure than to conduct a poor-quality screening. “If you can’t see the mucosal surface, you can’t tell the patient that they have a negative colonoscopy. If you have to do more cleaning during the procedure, then do more cleaning during the procedure. If you have to cancel the procedure and bring the patient back, it’s better to do that than it is to do an incomplete colonoscopy,” said Dr. Kosinski.
He also stressed the need to make sure that the patient is properly sedated and comfortable “so that you can do the job you’re supposed to do,” he said.
Some authors disclosed relationships with Amgen and the National Cancer Institute. Dr. Rex disclosed relationships with Olympus, Boston Scientific, Aries, and others, all outside the submitted work.
FROM JAMA
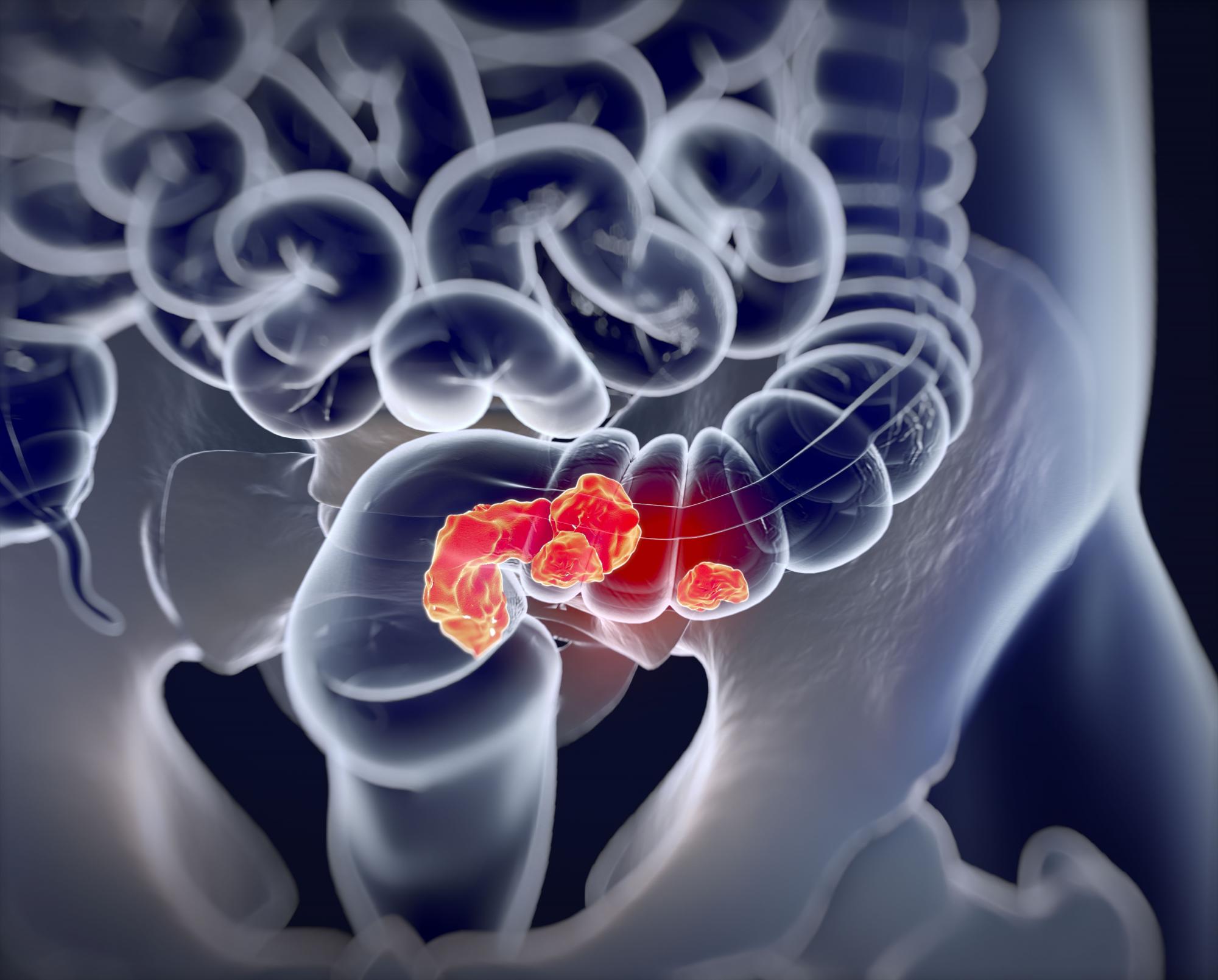
Ezetimibe plus statin: Attractive bypass to high-dose monotherapy
More patients with established atherosclerotic cardiovascular disease (ASCVD) achieved a low-density lipoprotein (LDL) cholesterol of less than 70 mg/dL, and fewer discontinued treatment with ezetimibe plus a moderate-dose statin, than did those on high-intensity statin monotherapy, a noninferiority trial shows.
While it’s now established that drug combinations can achieve better efficacy with lower risks than high-dose monotherapy, the study is the first to show the benefits of the strategy for ASCVD in a randomized trial with long-term follow-up.
The primary endpoint – 3-year composite of cardiovascular death, major cardiovascular events, or nonfatal stroke – occurred in about 9% of patients in each group, showing non-inferiority.
Furthermore, the authors suggest that ezetimibe combination therapy be considered earlier in the treatment of those at high risk of adverse events, rather than doubling the statin dose.
The study was published online in The Lancet.
Less intolerance, less discontinuations
The open-label study, dubbed RACING, randomized 3,780 patients with ASCVD to receive moderate-intensity rosuvastatin 10 mg plus ezetimibe 10 mg or high-intensity 20 mg rosuvastatin monotherapy. Participants’ average age was 64 and 75% were men.
The primary endpoint occurred in 9.1% of patients in the combination therapy group and 9.9% in the high-intensity monotherapy group. The absolute between-group difference was −0.78% (90% confidence interval [CI], −2.39 to 0.83), well below the 2% noninferiority margin.
In the combination therapy group, LDL cholesterol concentrations of less than 70 mg/dL were achieved in 73% of patients at 1 year, 75% at 2 years, and 72% at 3 years. By contrast, in the monotherapy group, the lower concentrations were seen in 55% at 1 year, 60% at 2 years, and 58% at 3 years.
Further, a post hoc analysis showed LDL concentrations of less than 55 mg/dL at 1, 2, and 3 years in 42%, 45%, and 42% of patients in the combination therapy group versus 25%, 29%, and 25% of those in the high-intensity statin monotherapy group.
Eighty-eight patients (4.8%) on combination therapy discontinued medication or received a dose reduction, versus 150 patients (8.2%) on monotherapy.
Rates of myonecrosis were similar in the combination therapy and high-intensity statin groups (11 vs. 13), whereas myalgia was more common with high-intensity statins (29 vs. 17). The open-label design could have led to bias in reporting of patient symptoms, the authors noted. All clinical events, however, were adjudicated by an independent committee masked to treatment assignment.
There might be “some level of difference” when extending the findings to other populations because the trial involved only Koreans, coauthor Myeong-Ki Hong, MD, Yonsei University, Seoul, South Korea, acknowledged in response to a query from this news organization. He thinks the findings can be applied broadly nonetheless, and his team is currently investigating whether certain patients might benefit more than others from the combination.
Various options for patients
“The field of hypertension changed [its] guidelines almost 20 years ago to consider the initial use of combination therapy in hard-to-treat patients,” Christie Mitchell Ballantyne, MD, Baylor College of Medicine, Houston, said in an interview. He coauthored an accompanying editorial with Baylor colleague Layla A. Abushamat, MD.
“We now have enough evidence of the efficacy and safety of combination therapy to consider early initiation of this approach in patients with challenging lipid disorders who are at increased risk of ASCVD events,” affirmed Dr. Ballantyne.
“This study reinforces important principles in the management and secondary prevention of cardiovascular disease, namely that LDL reduction and associated risk reduction can be achieved in various ways,” said Daniel Muñoz, MD, MPA, executive medical director of the Vanderbilt Heart & Vascular Institute, Vanderbilt University Medical Center, Nashville, Tenn.
However, he noted, “The high-intensity statin dose used as a comparator in this study was rosuvastatin 20 mg. In clinical practice, we often target maximally aggressive reduction of LDL via higher doses – that is, rosuvastatin 40 mg or atorvastatin 80 mg.”
The bottom line, said Dr. Muñoz, who was not involved in the study: “There are different ways to achieve LDL-lowering and associated risk reduction in patients with CVD. For patients who warrant but might not tolerate high-intensity statin therapy, this study supports the use of a moderate-intensity statin in combination with ezetimibe.”
The study was funded by Hanmi Pharmaceutical, Seoul, South Korea. One study coauthor received an institutional research grant from the company. No other authors reported relevant financial relationships, nor did Dr. Ballantyne, Dr. Abushamat, or Dr. Muñoz.
A version of this article first appeared on Medscape.com.
More patients with established atherosclerotic cardiovascular disease (ASCVD) achieved a low-density lipoprotein (LDL) cholesterol of less than 70 mg/dL, and fewer discontinued treatment with ezetimibe plus a moderate-dose statin, than did those on high-intensity statin monotherapy, a noninferiority trial shows.
While it’s now established that drug combinations can achieve better efficacy with lower risks than high-dose monotherapy, the study is the first to show the benefits of the strategy for ASCVD in a randomized trial with long-term follow-up.
The primary endpoint – 3-year composite of cardiovascular death, major cardiovascular events, or nonfatal stroke – occurred in about 9% of patients in each group, showing non-inferiority.
Furthermore, the authors suggest that ezetimibe combination therapy be considered earlier in the treatment of those at high risk of adverse events, rather than doubling the statin dose.
The study was published online in The Lancet.
Less intolerance, less discontinuations
The open-label study, dubbed RACING, randomized 3,780 patients with ASCVD to receive moderate-intensity rosuvastatin 10 mg plus ezetimibe 10 mg or high-intensity 20 mg rosuvastatin monotherapy. Participants’ average age was 64 and 75% were men.
The primary endpoint occurred in 9.1% of patients in the combination therapy group and 9.9% in the high-intensity monotherapy group. The absolute between-group difference was −0.78% (90% confidence interval [CI], −2.39 to 0.83), well below the 2% noninferiority margin.
In the combination therapy group, LDL cholesterol concentrations of less than 70 mg/dL were achieved in 73% of patients at 1 year, 75% at 2 years, and 72% at 3 years. By contrast, in the monotherapy group, the lower concentrations were seen in 55% at 1 year, 60% at 2 years, and 58% at 3 years.
Further, a post hoc analysis showed LDL concentrations of less than 55 mg/dL at 1, 2, and 3 years in 42%, 45%, and 42% of patients in the combination therapy group versus 25%, 29%, and 25% of those in the high-intensity statin monotherapy group.
Eighty-eight patients (4.8%) on combination therapy discontinued medication or received a dose reduction, versus 150 patients (8.2%) on monotherapy.
Rates of myonecrosis were similar in the combination therapy and high-intensity statin groups (11 vs. 13), whereas myalgia was more common with high-intensity statins (29 vs. 17). The open-label design could have led to bias in reporting of patient symptoms, the authors noted. All clinical events, however, were adjudicated by an independent committee masked to treatment assignment.
There might be “some level of difference” when extending the findings to other populations because the trial involved only Koreans, coauthor Myeong-Ki Hong, MD, Yonsei University, Seoul, South Korea, acknowledged in response to a query from this news organization. He thinks the findings can be applied broadly nonetheless, and his team is currently investigating whether certain patients might benefit more than others from the combination.
Various options for patients
“The field of hypertension changed [its] guidelines almost 20 years ago to consider the initial use of combination therapy in hard-to-treat patients,” Christie Mitchell Ballantyne, MD, Baylor College of Medicine, Houston, said in an interview. He coauthored an accompanying editorial with Baylor colleague Layla A. Abushamat, MD.
“We now have enough evidence of the efficacy and safety of combination therapy to consider early initiation of this approach in patients with challenging lipid disorders who are at increased risk of ASCVD events,” affirmed Dr. Ballantyne.
“This study reinforces important principles in the management and secondary prevention of cardiovascular disease, namely that LDL reduction and associated risk reduction can be achieved in various ways,” said Daniel Muñoz, MD, MPA, executive medical director of the Vanderbilt Heart & Vascular Institute, Vanderbilt University Medical Center, Nashville, Tenn.
However, he noted, “The high-intensity statin dose used as a comparator in this study was rosuvastatin 20 mg. In clinical practice, we often target maximally aggressive reduction of LDL via higher doses – that is, rosuvastatin 40 mg or atorvastatin 80 mg.”
The bottom line, said Dr. Muñoz, who was not involved in the study: “There are different ways to achieve LDL-lowering and associated risk reduction in patients with CVD. For patients who warrant but might not tolerate high-intensity statin therapy, this study supports the use of a moderate-intensity statin in combination with ezetimibe.”
The study was funded by Hanmi Pharmaceutical, Seoul, South Korea. One study coauthor received an institutional research grant from the company. No other authors reported relevant financial relationships, nor did Dr. Ballantyne, Dr. Abushamat, or Dr. Muñoz.
A version of this article first appeared on Medscape.com.
More patients with established atherosclerotic cardiovascular disease (ASCVD) achieved a low-density lipoprotein (LDL) cholesterol of less than 70 mg/dL, and fewer discontinued treatment with ezetimibe plus a moderate-dose statin, than did those on high-intensity statin monotherapy, a noninferiority trial shows.
While it’s now established that drug combinations can achieve better efficacy with lower risks than high-dose monotherapy, the study is the first to show the benefits of the strategy for ASCVD in a randomized trial with long-term follow-up.
The primary endpoint – 3-year composite of cardiovascular death, major cardiovascular events, or nonfatal stroke – occurred in about 9% of patients in each group, showing non-inferiority.
Furthermore, the authors suggest that ezetimibe combination therapy be considered earlier in the treatment of those at high risk of adverse events, rather than doubling the statin dose.
The study was published online in The Lancet.
Less intolerance, less discontinuations
The open-label study, dubbed RACING, randomized 3,780 patients with ASCVD to receive moderate-intensity rosuvastatin 10 mg plus ezetimibe 10 mg or high-intensity 20 mg rosuvastatin monotherapy. Participants’ average age was 64 and 75% were men.
The primary endpoint occurred in 9.1% of patients in the combination therapy group and 9.9% in the high-intensity monotherapy group. The absolute between-group difference was −0.78% (90% confidence interval [CI], −2.39 to 0.83), well below the 2% noninferiority margin.
In the combination therapy group, LDL cholesterol concentrations of less than 70 mg/dL were achieved in 73% of patients at 1 year, 75% at 2 years, and 72% at 3 years. By contrast, in the monotherapy group, the lower concentrations were seen in 55% at 1 year, 60% at 2 years, and 58% at 3 years.
Further, a post hoc analysis showed LDL concentrations of less than 55 mg/dL at 1, 2, and 3 years in 42%, 45%, and 42% of patients in the combination therapy group versus 25%, 29%, and 25% of those in the high-intensity statin monotherapy group.
Eighty-eight patients (4.8%) on combination therapy discontinued medication or received a dose reduction, versus 150 patients (8.2%) on monotherapy.
Rates of myonecrosis were similar in the combination therapy and high-intensity statin groups (11 vs. 13), whereas myalgia was more common with high-intensity statins (29 vs. 17). The open-label design could have led to bias in reporting of patient symptoms, the authors noted. All clinical events, however, were adjudicated by an independent committee masked to treatment assignment.
There might be “some level of difference” when extending the findings to other populations because the trial involved only Koreans, coauthor Myeong-Ki Hong, MD, Yonsei University, Seoul, South Korea, acknowledged in response to a query from this news organization. He thinks the findings can be applied broadly nonetheless, and his team is currently investigating whether certain patients might benefit more than others from the combination.
Various options for patients
“The field of hypertension changed [its] guidelines almost 20 years ago to consider the initial use of combination therapy in hard-to-treat patients,” Christie Mitchell Ballantyne, MD, Baylor College of Medicine, Houston, said in an interview. He coauthored an accompanying editorial with Baylor colleague Layla A. Abushamat, MD.
“We now have enough evidence of the efficacy and safety of combination therapy to consider early initiation of this approach in patients with challenging lipid disorders who are at increased risk of ASCVD events,” affirmed Dr. Ballantyne.
“This study reinforces important principles in the management and secondary prevention of cardiovascular disease, namely that LDL reduction and associated risk reduction can be achieved in various ways,” said Daniel Muñoz, MD, MPA, executive medical director of the Vanderbilt Heart & Vascular Institute, Vanderbilt University Medical Center, Nashville, Tenn.
However, he noted, “The high-intensity statin dose used as a comparator in this study was rosuvastatin 20 mg. In clinical practice, we often target maximally aggressive reduction of LDL via higher doses – that is, rosuvastatin 40 mg or atorvastatin 80 mg.”
The bottom line, said Dr. Muñoz, who was not involved in the study: “There are different ways to achieve LDL-lowering and associated risk reduction in patients with CVD. For patients who warrant but might not tolerate high-intensity statin therapy, this study supports the use of a moderate-intensity statin in combination with ezetimibe.”
The study was funded by Hanmi Pharmaceutical, Seoul, South Korea. One study coauthor received an institutional research grant from the company. No other authors reported relevant financial relationships, nor did Dr. Ballantyne, Dr. Abushamat, or Dr. Muñoz.
A version of this article first appeared on Medscape.com.
Topical roflumilast approved for psoriasis in adults and adolescents
.
Roflumilast is a selective inhibitor of phosphodiesterase 4 (PDE4), the first approved for treating psoriasis, according to manufacturer Arcutis Biotherapeutics. The company announced the approval on July 29. Oral roflumilast (Daliresp ) was approved in 2011 for treating chronic obstructive pulmonary disease (COPD).“It’s a breakthrough topical therapy,” says Mark G. Lebwohl, MD, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York, and principal investigator in trials of topical roflumilast. In an interview, Dr. Lebwohl noted that the treatment significantly reduced psoriasis symptoms in both short- and long-term trials.

In addition, two features of this treatment set it apart from other topical psoriasis treatments, he said. Roflumilast is not a steroid, so does not have the risk for topical steroid-related side effects associated with chronic use, and, in clinical trials, topical roflumilast was effective in treating psoriasis in intertriginous areas, including the buttocks, underarms, and beneath the breasts, which are difficult to treat.
FDA approval is based on data from two phase 3 randomized, double-blind, vehicle-controlled trials, according to Arcutis. The primary endpoint was Investigator Global Assessment (IGA) success, defined as clear or almost clear with at least a two-grade improvement from baseline, and at least a two-grade IGA score improvement from baseline at 8 weeks.
At 8 weeks, 42.4% and 37.5% of the patients treated with topical roflumilast achieved an IGA success rate compared with 6.1% and 6.9% in the control groups, respectively (P < .0001 for both studies).
Treated patients also experienced significant improvements compared with those in the vehicle groups in secondary endpoints in the trials: Those included Intertriginous IGA (I-IGA) Success, Psoriasis Area Severity Index–75 (PASI-75), reductions in itch based on the Worst Itch–Numerical Rating Scale (WI-NRS), and self-reported psoriasis symptoms diary (PSD).
In the studies, 72% and 68% of patients treated with roflumilast met the I-IGA endpoint at 8 weeks versus 14% and 17%, respectively, of those on vehicle (P < .0001 for both studies).
In addition, by week 2, some participants treated with roflumilast had experienced reduced itchiness in both studies. At 8 weeks, among those with a WI-NRS score of 4 or more at baseline, 67% and 69% of the treated patients had at least a four-point reduction in the WI-NRS versus 26% and 33%, respectively, among those on vehicle (P < .0001 for both studies), according to the company.
In general, the cream was well tolerated. There were reports of diarrhea (3%), headache (2%), insomnia (1%), nausea (1%), application-site pain (1%), upper respiratory tract infections (1%), and urinary tract infections (1%). However, Dr. Lebwohl noted that these events were also observed in the control group.
“The study was unequivocal about the improvement in the intertriginous sites,” Dr. Lebwohl said. He contrasts that to the data from other nonsteroidal topicals, which he said can be associated with a rash or irritation in sensitive areas.
Dr. Lebwohl noted that PDE4 is an enzyme that increases inflammation and decreases anti-inflammatory mediators and that inhibiting PDE4 may interrupt some of the inflammation response responsible for psoriasis symptoms, as it has for other conditions such as atopic dermatitis. Data from the 8-week phase 3 trials and yearlong phase 2b, open-label studies support that hypothesis.
“I’m always excited for new psoriasis treatments to broaden our treatment armamentarium,” said Lauren E. Ploch, MD, a dermatologist who practices in Augusta, Ga., and Aiken, S.C., who was asked to comment on the approval.
Even a symptom that seems benign, like itching, Dr. Ploch added, can lead to reduced sleep and increased irritability. Referring to the data on the treatment in the sensitive, intertriginous areas, she noted that the skin in these areas is often thinner, so treatment with steroids can cause further thinning and damage to the skin. If roflumilast doesn’t cause burning, itching, or thinning, it will be a great option to treat these areas, she said in an interview. She was not involved in the trials.
Roflumilast cream will be marketed under the trade name Zoryve, and is expected to be available by mid-August, according to Arcutis.
Roflumilast cream is also under review in Canada for treatment of plaque psoriasis in adults and adolescents.
The studies were funded by Arcutis Biotherapeutics. Dr. Lebwohl reported receiving grant support and consulting fees from Arcutis. Dr. Ploch reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
.
Roflumilast is a selective inhibitor of phosphodiesterase 4 (PDE4), the first approved for treating psoriasis, according to manufacturer Arcutis Biotherapeutics. The company announced the approval on July 29. Oral roflumilast (Daliresp ) was approved in 2011 for treating chronic obstructive pulmonary disease (COPD).“It’s a breakthrough topical therapy,” says Mark G. Lebwohl, MD, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York, and principal investigator in trials of topical roflumilast. In an interview, Dr. Lebwohl noted that the treatment significantly reduced psoriasis symptoms in both short- and long-term trials.
In addition, two features of this treatment set it apart from other topical psoriasis treatments, he said. Roflumilast is not a steroid, so does not have the risk for topical steroid-related side effects associated with chronic use, and, in clinical trials, topical roflumilast was effective in treating psoriasis in intertriginous areas, including the buttocks, underarms, and beneath the breasts, which are difficult to treat.
FDA approval is based on data from two phase 3 randomized, double-blind, vehicle-controlled trials, according to Arcutis. The primary endpoint was Investigator Global Assessment (IGA) success, defined as clear or almost clear with at least a two-grade improvement from baseline, and at least a two-grade IGA score improvement from baseline at 8 weeks.
At 8 weeks, 42.4% and 37.5% of the patients treated with topical roflumilast achieved an IGA success rate compared with 6.1% and 6.9% in the control groups, respectively (P < .0001 for both studies).
Treated patients also experienced significant improvements compared with those in the vehicle groups in secondary endpoints in the trials: Those included Intertriginous IGA (I-IGA) Success, Psoriasis Area Severity Index–75 (PASI-75), reductions in itch based on the Worst Itch–Numerical Rating Scale (WI-NRS), and self-reported psoriasis symptoms diary (PSD).
In the studies, 72% and 68% of patients treated with roflumilast met the I-IGA endpoint at 8 weeks versus 14% and 17%, respectively, of those on vehicle (P < .0001 for both studies).
In addition, by week 2, some participants treated with roflumilast had experienced reduced itchiness in both studies. At 8 weeks, among those with a WI-NRS score of 4 or more at baseline, 67% and 69% of the treated patients had at least a four-point reduction in the WI-NRS versus 26% and 33%, respectively, among those on vehicle (P < .0001 for both studies), according to the company.
In general, the cream was well tolerated. There were reports of diarrhea (3%), headache (2%), insomnia (1%), nausea (1%), application-site pain (1%), upper respiratory tract infections (1%), and urinary tract infections (1%). However, Dr. Lebwohl noted that these events were also observed in the control group.
“The study was unequivocal about the improvement in the intertriginous sites,” Dr. Lebwohl said. He contrasts that to the data from other nonsteroidal topicals, which he said can be associated with a rash or irritation in sensitive areas.
Dr. Lebwohl noted that PDE4 is an enzyme that increases inflammation and decreases anti-inflammatory mediators and that inhibiting PDE4 may interrupt some of the inflammation response responsible for psoriasis symptoms, as it has for other conditions such as atopic dermatitis. Data from the 8-week phase 3 trials and yearlong phase 2b, open-label studies support that hypothesis.
“I’m always excited for new psoriasis treatments to broaden our treatment armamentarium,” said Lauren E. Ploch, MD, a dermatologist who practices in Augusta, Ga., and Aiken, S.C., who was asked to comment on the approval.
Even a symptom that seems benign, like itching, Dr. Ploch added, can lead to reduced sleep and increased irritability. Referring to the data on the treatment in the sensitive, intertriginous areas, she noted that the skin in these areas is often thinner, so treatment with steroids can cause further thinning and damage to the skin. If roflumilast doesn’t cause burning, itching, or thinning, it will be a great option to treat these areas, she said in an interview. She was not involved in the trials.
Roflumilast cream will be marketed under the trade name Zoryve, and is expected to be available by mid-August, according to Arcutis.
Roflumilast cream is also under review in Canada for treatment of plaque psoriasis in adults and adolescents.
The studies were funded by Arcutis Biotherapeutics. Dr. Lebwohl reported receiving grant support and consulting fees from Arcutis. Dr. Ploch reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
.
Roflumilast is a selective inhibitor of phosphodiesterase 4 (PDE4), the first approved for treating psoriasis, according to manufacturer Arcutis Biotherapeutics. The company announced the approval on July 29. Oral roflumilast (Daliresp ) was approved in 2011 for treating chronic obstructive pulmonary disease (COPD).“It’s a breakthrough topical therapy,” says Mark G. Lebwohl, MD, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York, and principal investigator in trials of topical roflumilast. In an interview, Dr. Lebwohl noted that the treatment significantly reduced psoriasis symptoms in both short- and long-term trials.
In addition, two features of this treatment set it apart from other topical psoriasis treatments, he said. Roflumilast is not a steroid, so does not have the risk for topical steroid-related side effects associated with chronic use, and, in clinical trials, topical roflumilast was effective in treating psoriasis in intertriginous areas, including the buttocks, underarms, and beneath the breasts, which are difficult to treat.
FDA approval is based on data from two phase 3 randomized, double-blind, vehicle-controlled trials, according to Arcutis. The primary endpoint was Investigator Global Assessment (IGA) success, defined as clear or almost clear with at least a two-grade improvement from baseline, and at least a two-grade IGA score improvement from baseline at 8 weeks.
At 8 weeks, 42.4% and 37.5% of the patients treated with topical roflumilast achieved an IGA success rate compared with 6.1% and 6.9% in the control groups, respectively (P < .0001 for both studies).
Treated patients also experienced significant improvements compared with those in the vehicle groups in secondary endpoints in the trials: Those included Intertriginous IGA (I-IGA) Success, Psoriasis Area Severity Index–75 (PASI-75), reductions in itch based on the Worst Itch–Numerical Rating Scale (WI-NRS), and self-reported psoriasis symptoms diary (PSD).
In the studies, 72% and 68% of patients treated with roflumilast met the I-IGA endpoint at 8 weeks versus 14% and 17%, respectively, of those on vehicle (P < .0001 for both studies).
In addition, by week 2, some participants treated with roflumilast had experienced reduced itchiness in both studies. At 8 weeks, among those with a WI-NRS score of 4 or more at baseline, 67% and 69% of the treated patients had at least a four-point reduction in the WI-NRS versus 26% and 33%, respectively, among those on vehicle (P < .0001 for both studies), according to the company.
In general, the cream was well tolerated. There were reports of diarrhea (3%), headache (2%), insomnia (1%), nausea (1%), application-site pain (1%), upper respiratory tract infections (1%), and urinary tract infections (1%). However, Dr. Lebwohl noted that these events were also observed in the control group.
“The study was unequivocal about the improvement in the intertriginous sites,” Dr. Lebwohl said. He contrasts that to the data from other nonsteroidal topicals, which he said can be associated with a rash or irritation in sensitive areas.
Dr. Lebwohl noted that PDE4 is an enzyme that increases inflammation and decreases anti-inflammatory mediators and that inhibiting PDE4 may interrupt some of the inflammation response responsible for psoriasis symptoms, as it has for other conditions such as atopic dermatitis. Data from the 8-week phase 3 trials and yearlong phase 2b, open-label studies support that hypothesis.
“I’m always excited for new psoriasis treatments to broaden our treatment armamentarium,” said Lauren E. Ploch, MD, a dermatologist who practices in Augusta, Ga., and Aiken, S.C., who was asked to comment on the approval.
Even a symptom that seems benign, like itching, Dr. Ploch added, can lead to reduced sleep and increased irritability. Referring to the data on the treatment in the sensitive, intertriginous areas, she noted that the skin in these areas is often thinner, so treatment with steroids can cause further thinning and damage to the skin. If roflumilast doesn’t cause burning, itching, or thinning, it will be a great option to treat these areas, she said in an interview. She was not involved in the trials.
Roflumilast cream will be marketed under the trade name Zoryve, and is expected to be available by mid-August, according to Arcutis.
Roflumilast cream is also under review in Canada for treatment of plaque psoriasis in adults and adolescents.
The studies were funded by Arcutis Biotherapeutics. Dr. Lebwohl reported receiving grant support and consulting fees from Arcutis. Dr. Ploch reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.

FDA panel urges caution with skin cancer–detecting tools
A and how to address longstanding issues of racial equity in this field of medicine.
The Food and Drug Administration has scheduled two meetings to gather expert feedback about managing an expected expansion in the use of skin lesion apps and devices. Outside of the United States, there are apps promoted as being able to help spot skin lesions that should trigger a medical visit.
The general and plastic surgery devices panel of the FDA’s Medical Devices Advisory Committee began work on this topic on July 28, with a wide-ranging discussion about potential expanded use of computer-aided, skin lesion analyzer (SLA) devices. On Friday, the panel is considering an FDA proposal to shift the designation for an approved device for aiding dermatologists in skin cancer diagnoses from the most stringent regulatory category, class III, to the less restrictive class II.
The FDA called the meeting amid growing interest in using technology to aid in finding cancers, with some of these products already marketed to consumers outside of the United States. There are presently no legally marketed, FDA-cleared or FDA-approved SLA devices indicated for use by clinicians other than dermatologists or the lay public, the agency said in a briefing memo for the meeting. There are two devices with FDA approval, though, for aiding dermatologists. The FDA approved SciBase’s Nevisense in 2017 and Mela Sciences’ MelaFind, which has fallen out of use, in 2012. Both are class III devices.
But some companies intend to offer products for consumers in the United States. The company SkinVision, for example, has developed an app of the same name, which is intended to detect suspicious-looking skin spots via smartphone photos. SkinVision’s website says the product has been offered to consumers in Australia for remote skin checks since 2015. People in the Netherlands and United Kingdom also can use SkinVision, according to the company’s website. SkinVision says the company is working on providing the app for U.S. customers, “but we are not quite there yet.”
During the meeting, FDA panelists repeatedly emphasized the potential risks of these devices in terms of sensitivity (how often a test correctly generates a positive result) and of specificity (how often a test correctly generates a negative result).
New tools intended to aid in detection of skin cancer might produce too many false positives and thus trigger floods of worried patients seeking care and often facing unnecessary biopsies, the FDA panelists said. But more worrisome would be FDA clearance of tools that delivered too many false negative results, leaving people unaware of their cancers.

The standards would have to be set very high for new products, especially those intended for consumers, said FDA panelist Murad Alam, MD, a dermatologist and vice chair of the department of dermatology at Northwestern University, Chicago. Current technologies for analyzing skin lesions are not yet up to that task. Dr. Alam likened the situation to the hopes for self-driving cars.
“It sounds great in principle. If you read the predictions from 20 years ago, it should already have happened,” Dr. Alam said. “But we’re still struggling with that because there are serious points of failure.”
FDA panelist Veronica Rotemberg, MD, PhD, a dermatologist at Memorial Sloan Kettering Cancer Center, New York, also argued for well-designed studies to understand how consumers and clinicians would react to new tools.
“We have to define what prospective information, in the intended use setting, we need to feel comfortable saying that these tools could be in a layperson’s hand or a primary care person’s hand,” Dr. Rotemberg said.
The studies would not need to be large, especially in the case of nonmelanoma skin cancer, which is common, she added.
“There’s too much nuance here for us to be able to say: ‘This is what would happen,’ without testing it,” Dr. Rotemberg said. “I do not think these prospective studies would be very burdensome, but they would help us understand what the burden would be and what the costs would be and what the potential harms would be.”
Because of rules against disclosing corporate information, the FDA cannot tell the public about the kinds of inquiries it already may have fielded from companies interested in selling skin cancer detection tools.
But in response to a question during the FDA meeting, Binita Ashar, MD, a top official in the FDA’s Center for Devices and Radiological Health, said there is interest in having these kinds of products sold in the United States as well.
“I can tell you that this a very timely discussion and questions that we’re posing to you are the questions that we’re encountering or that we have been grappling with,” Dr. Ashar said.
FDA panelists noted that many patients cannot get access easily to dermatology visits.
Companies seeking to develop SLA devices likely will market their tools as attempts to fill a gap that now exists in medical care.
But there will be challenges ahead in explaining to patients how to interpret readings from these tools, the FDA panelists said. Consumers should know these tools are meant to assist in diagnosis, and not to make it.
“I’m not sure the layperson will hear that,” said FDA panelist Paula E. Bourelly, MD, a dermatologist from Olney, Md.
As a result, use of SLA tools could create tension between physicians and patients, with consumers demanding biopsies after seeing readings they don’t understand.
“I do have great concerns about the layperson feeling overly confident and reducing the provider to a technician,” she said.
The FDA panelists were not asked to cast formal votes on any issues discussed during the meeting They instead engaged in broad discussions around questions posed by the FDA in three key areas:
- What standards should be used to confirm lesion diagnosis in clinical testing of the accuracy of SLA devices?
- What would be acceptable true false-positive and false-negative results (sensitivity and specificity) for different diagnoses and users?
- How can the FDA address health equity considerations based on variable incidence of skin lesions?
Developing standards
The FDA asked the panel to consider several scenarios for SLA devices and to discuss how standards might vary depending on the user of the device, whether it would be dermatologists, other clinicians, or consumers.
The agency sought comments in particular about using histological diagnosis (core specimen processing with a consensus diagnosis from an expert dermatopathologist panel). In the briefing document for the meeting, the FDA argued that this approach provides the greatest certainty in the diagnosis.
“Device developers, however, cite concerns, both practical and ethical, in requiring biopsy of all lesions, particularly those that appear benign,” the FDA said. “They have proposed alternate means of defining ground truth, including consensus opinion of experts (of visual or dermoscopic examination of the lesion[s]), opinion of one expert (visual or dermoscopic examination), or other methods.”
In summarizing the discussion on this question, the FDA panel chairman, Hobart W. Harris, MD, MPH, a surgeon from the University of California, San Francisco, noted that there was broad support for histological data in clinical trials of SLA devices, with some allowance for cases where more hybrid approaches would be used.
There were also suggestions offered about designing trials and the need for biopsies of lesions that are clearly benign, as this would help gather data to help in developing algorithms.
Dr. Alam said care should be taken in explaining to study participants that they might have to undergo biopsies that they didn’t need, as part of the larger effort to gather data. This should be detailed in the consent form, he said.
“But I also think this is a relatively minor risk,” Dr. Alam said, comparing these biopsies to the blood samples that patients in many clinical studies routinely give.
“Are all of those blood draws necessary to track the change in whatever parameters that are being tracked? Probably not,” Dr. Alam said. “I think it would be possible to explain to a reasonable patient what this entails.”
Dr. Alam noted that companies might face extra hurdles in enrolling study participants and keeping them in the trials if the FDA seeks this kind of biopsy data. “But I don’t think inconvenience to the study sponsor is a good argument” for not seeking this kind of data, he added.
Leaving a loophole where certain kinds of clearly benign lesions don’t require a biopsy would eventually erode the quality of the research done on these devices. “That bar will be moved to accommodate the convenience of the sponsor, to make the study feasible,” Dr. Alam said. “And pretty soon, you’ll be missing a lot of patients that really should have biopsies.”
Acceptable rates of false positives, false negatives
The FDA panel chair noted that his colleagues had strongly urged review standards that would require that the devices improve on the rates of successful catches of suspicious lesions and lower false positives. But they did not endorse specific targets regarding the sensitivity and specificity rates.
“No one seems to be comfortable with providing or preordaining” these targets, Dr. Harris said.
Panelist Deneen Hesser, MSHSA, RN, urged a deep recognition of the power of a FDA clearance in the view of consumers.
“We need to be cognizant of what the term ‘FDA approved’ means to the lay individual,” said Ms. Hesser, who served as the patient representative on the panel. “A patient who sees that those tools are FDA approved will assume that each of those is the gold standard” in terms of expectations for delivering accurate results.
Like many of the panelists, Dr. Rotemberg urged the FDA to gather data about how patients would react to different messages encoded in consumer-oriented products.

“If the device says: ‘You should see a dermatologist for this’ and no other information, that’s very different from [saying]: ‘That lesion is suspicious for melanoma,’ ” Dr. Rotemberg said.
Despite the likely difficulties in conducting trials, the FDA needs to have the data to answer key questions about patient and physician reactions to readings from new tools, Dr. Rotemberg said.
“We don’t know how many additional biopsies we would cause with a specificity of 80%” for a new SLA tool, Dr. Rotemberg said, giving an example. “We don’t know how confident a dermatologist might be to say: ‘Actually, I’m not suspicious about that lesion and we can just fudge it or not biopsy it.’ We don’t know any of that until we study it in real life.”
The panelists also urged the FDA to seek to ensure that new tools used in analyzing skin lesions improve the quality of diagnosis.
Addressing equity
The FDA also asked the panel to weigh in on whether the agency should clear SLA tools in cases where the existing study data is drawn heavily from people considered to be at higher risk for skin cancer.

“To ensure generalizability across the entire U.S. population, should FDA require SLAs indicated for use beyond cancerous lesions be tested in a representative U.S. population?” the FDA asked.
The three most common skin cancers – melanoma, basal cell carcinoma, and squamous cell carcinoma – are more prevalent in people with Fitzpatrick I and II skin types, who tend to get sunburns, not tans. But people of color are more likely to develop melanoma in areas that are not sun exposed, such as the sole of the foot or under fingernails or toenails.
“Due in part to lower expected risk and screening, these melanomas are often detected late,” the FDA said in the briefing document.
There was broad consensus among panelists that the FDA should encourage companies to enroll people with all skin types and tones.
But they also looked for ways that the FDA could clear devices based on initial studies conducted largely with people considered to be at higher risk, with the agency then requiring follow-up trials to see how these products would work for the general U.S. population.
A version of this article first appeared on Medscape.com.
A and how to address longstanding issues of racial equity in this field of medicine.
The Food and Drug Administration has scheduled two meetings to gather expert feedback about managing an expected expansion in the use of skin lesion apps and devices. Outside of the United States, there are apps promoted as being able to help spot skin lesions that should trigger a medical visit.
The general and plastic surgery devices panel of the FDA’s Medical Devices Advisory Committee began work on this topic on July 28, with a wide-ranging discussion about potential expanded use of computer-aided, skin lesion analyzer (SLA) devices. On Friday, the panel is considering an FDA proposal to shift the designation for an approved device for aiding dermatologists in skin cancer diagnoses from the most stringent regulatory category, class III, to the less restrictive class II.
The FDA called the meeting amid growing interest in using technology to aid in finding cancers, with some of these products already marketed to consumers outside of the United States. There are presently no legally marketed, FDA-cleared or FDA-approved SLA devices indicated for use by clinicians other than dermatologists or the lay public, the agency said in a briefing memo for the meeting. There are two devices with FDA approval, though, for aiding dermatologists. The FDA approved SciBase’s Nevisense in 2017 and Mela Sciences’ MelaFind, which has fallen out of use, in 2012. Both are class III devices.
But some companies intend to offer products for consumers in the United States. The company SkinVision, for example, has developed an app of the same name, which is intended to detect suspicious-looking skin spots via smartphone photos. SkinVision’s website says the product has been offered to consumers in Australia for remote skin checks since 2015. People in the Netherlands and United Kingdom also can use SkinVision, according to the company’s website. SkinVision says the company is working on providing the app for U.S. customers, “but we are not quite there yet.”
During the meeting, FDA panelists repeatedly emphasized the potential risks of these devices in terms of sensitivity (how often a test correctly generates a positive result) and of specificity (how often a test correctly generates a negative result).
New tools intended to aid in detection of skin cancer might produce too many false positives and thus trigger floods of worried patients seeking care and often facing unnecessary biopsies, the FDA panelists said. But more worrisome would be FDA clearance of tools that delivered too many false negative results, leaving people unaware of their cancers.
The standards would have to be set very high for new products, especially those intended for consumers, said FDA panelist Murad Alam, MD, a dermatologist and vice chair of the department of dermatology at Northwestern University, Chicago. Current technologies for analyzing skin lesions are not yet up to that task. Dr. Alam likened the situation to the hopes for self-driving cars.
“It sounds great in principle. If you read the predictions from 20 years ago, it should already have happened,” Dr. Alam said. “But we’re still struggling with that because there are serious points of failure.”
FDA panelist Veronica Rotemberg, MD, PhD, a dermatologist at Memorial Sloan Kettering Cancer Center, New York, also argued for well-designed studies to understand how consumers and clinicians would react to new tools.
“We have to define what prospective information, in the intended use setting, we need to feel comfortable saying that these tools could be in a layperson’s hand or a primary care person’s hand,” Dr. Rotemberg said.
The studies would not need to be large, especially in the case of nonmelanoma skin cancer, which is common, she added.
“There’s too much nuance here for us to be able to say: ‘This is what would happen,’ without testing it,” Dr. Rotemberg said. “I do not think these prospective studies would be very burdensome, but they would help us understand what the burden would be and what the costs would be and what the potential harms would be.”
Because of rules against disclosing corporate information, the FDA cannot tell the public about the kinds of inquiries it already may have fielded from companies interested in selling skin cancer detection tools.
But in response to a question during the FDA meeting, Binita Ashar, MD, a top official in the FDA’s Center for Devices and Radiological Health, said there is interest in having these kinds of products sold in the United States as well.
“I can tell you that this a very timely discussion and questions that we’re posing to you are the questions that we’re encountering or that we have been grappling with,” Dr. Ashar said.
FDA panelists noted that many patients cannot get access easily to dermatology visits.
Companies seeking to develop SLA devices likely will market their tools as attempts to fill a gap that now exists in medical care.
But there will be challenges ahead in explaining to patients how to interpret readings from these tools, the FDA panelists said. Consumers should know these tools are meant to assist in diagnosis, and not to make it.
“I’m not sure the layperson will hear that,” said FDA panelist Paula E. Bourelly, MD, a dermatologist from Olney, Md.
As a result, use of SLA tools could create tension between physicians and patients, with consumers demanding biopsies after seeing readings they don’t understand.
“I do have great concerns about the layperson feeling overly confident and reducing the provider to a technician,” she said.
The FDA panelists were not asked to cast formal votes on any issues discussed during the meeting They instead engaged in broad discussions around questions posed by the FDA in three key areas:
- What standards should be used to confirm lesion diagnosis in clinical testing of the accuracy of SLA devices?
- What would be acceptable true false-positive and false-negative results (sensitivity and specificity) for different diagnoses and users?
- How can the FDA address health equity considerations based on variable incidence of skin lesions?
Developing standards
The FDA asked the panel to consider several scenarios for SLA devices and to discuss how standards might vary depending on the user of the device, whether it would be dermatologists, other clinicians, or consumers.
The agency sought comments in particular about using histological diagnosis (core specimen processing with a consensus diagnosis from an expert dermatopathologist panel). In the briefing document for the meeting, the FDA argued that this approach provides the greatest certainty in the diagnosis.
“Device developers, however, cite concerns, both practical and ethical, in requiring biopsy of all lesions, particularly those that appear benign,” the FDA said. “They have proposed alternate means of defining ground truth, including consensus opinion of experts (of visual or dermoscopic examination of the lesion[s]), opinion of one expert (visual or dermoscopic examination), or other methods.”
In summarizing the discussion on this question, the FDA panel chairman, Hobart W. Harris, MD, MPH, a surgeon from the University of California, San Francisco, noted that there was broad support for histological data in clinical trials of SLA devices, with some allowance for cases where more hybrid approaches would be used.
There were also suggestions offered about designing trials and the need for biopsies of lesions that are clearly benign, as this would help gather data to help in developing algorithms.
Dr. Alam said care should be taken in explaining to study participants that they might have to undergo biopsies that they didn’t need, as part of the larger effort to gather data. This should be detailed in the consent form, he said.
“But I also think this is a relatively minor risk,” Dr. Alam said, comparing these biopsies to the blood samples that patients in many clinical studies routinely give.
“Are all of those blood draws necessary to track the change in whatever parameters that are being tracked? Probably not,” Dr. Alam said. “I think it would be possible to explain to a reasonable patient what this entails.”
Dr. Alam noted that companies might face extra hurdles in enrolling study participants and keeping them in the trials if the FDA seeks this kind of biopsy data. “But I don’t think inconvenience to the study sponsor is a good argument” for not seeking this kind of data, he added.
Leaving a loophole where certain kinds of clearly benign lesions don’t require a biopsy would eventually erode the quality of the research done on these devices. “That bar will be moved to accommodate the convenience of the sponsor, to make the study feasible,” Dr. Alam said. “And pretty soon, you’ll be missing a lot of patients that really should have biopsies.”
Acceptable rates of false positives, false negatives
The FDA panel chair noted that his colleagues had strongly urged review standards that would require that the devices improve on the rates of successful catches of suspicious lesions and lower false positives. But they did not endorse specific targets regarding the sensitivity and specificity rates.
“No one seems to be comfortable with providing or preordaining” these targets, Dr. Harris said.
Panelist Deneen Hesser, MSHSA, RN, urged a deep recognition of the power of a FDA clearance in the view of consumers.
“We need to be cognizant of what the term ‘FDA approved’ means to the lay individual,” said Ms. Hesser, who served as the patient representative on the panel. “A patient who sees that those tools are FDA approved will assume that each of those is the gold standard” in terms of expectations for delivering accurate results.
Like many of the panelists, Dr. Rotemberg urged the FDA to gather data about how patients would react to different messages encoded in consumer-oriented products.
“If the device says: ‘You should see a dermatologist for this’ and no other information, that’s very different from [saying]: ‘That lesion is suspicious for melanoma,’ ” Dr. Rotemberg said.
Despite the likely difficulties in conducting trials, the FDA needs to have the data to answer key questions about patient and physician reactions to readings from new tools, Dr. Rotemberg said.
“We don’t know how many additional biopsies we would cause with a specificity of 80%” for a new SLA tool, Dr. Rotemberg said, giving an example. “We don’t know how confident a dermatologist might be to say: ‘Actually, I’m not suspicious about that lesion and we can just fudge it or not biopsy it.’ We don’t know any of that until we study it in real life.”
The panelists also urged the FDA to seek to ensure that new tools used in analyzing skin lesions improve the quality of diagnosis.
Addressing equity
The FDA also asked the panel to weigh in on whether the agency should clear SLA tools in cases where the existing study data is drawn heavily from people considered to be at higher risk for skin cancer.
“To ensure generalizability across the entire U.S. population, should FDA require SLAs indicated for use beyond cancerous lesions be tested in a representative U.S. population?” the FDA asked.
The three most common skin cancers – melanoma, basal cell carcinoma, and squamous cell carcinoma – are more prevalent in people with Fitzpatrick I and II skin types, who tend to get sunburns, not tans. But people of color are more likely to develop melanoma in areas that are not sun exposed, such as the sole of the foot or under fingernails or toenails.
“Due in part to lower expected risk and screening, these melanomas are often detected late,” the FDA said in the briefing document.
There was broad consensus among panelists that the FDA should encourage companies to enroll people with all skin types and tones.
But they also looked for ways that the FDA could clear devices based on initial studies conducted largely with people considered to be at higher risk, with the agency then requiring follow-up trials to see how these products would work for the general U.S. population.
A version of this article first appeared on Medscape.com.
A and how to address longstanding issues of racial equity in this field of medicine.
The Food and Drug Administration has scheduled two meetings to gather expert feedback about managing an expected expansion in the use of skin lesion apps and devices. Outside of the United States, there are apps promoted as being able to help spot skin lesions that should trigger a medical visit.
The general and plastic surgery devices panel of the FDA’s Medical Devices Advisory Committee began work on this topic on July 28, with a wide-ranging discussion about potential expanded use of computer-aided, skin lesion analyzer (SLA) devices. On Friday, the panel is considering an FDA proposal to shift the designation for an approved device for aiding dermatologists in skin cancer diagnoses from the most stringent regulatory category, class III, to the less restrictive class II.
The FDA called the meeting amid growing interest in using technology to aid in finding cancers, with some of these products already marketed to consumers outside of the United States. There are presently no legally marketed, FDA-cleared or FDA-approved SLA devices indicated for use by clinicians other than dermatologists or the lay public, the agency said in a briefing memo for the meeting. There are two devices with FDA approval, though, for aiding dermatologists. The FDA approved SciBase’s Nevisense in 2017 and Mela Sciences’ MelaFind, which has fallen out of use, in 2012. Both are class III devices.
But some companies intend to offer products for consumers in the United States. The company SkinVision, for example, has developed an app of the same name, which is intended to detect suspicious-looking skin spots via smartphone photos. SkinVision’s website says the product has been offered to consumers in Australia for remote skin checks since 2015. People in the Netherlands and United Kingdom also can use SkinVision, according to the company’s website. SkinVision says the company is working on providing the app for U.S. customers, “but we are not quite there yet.”
During the meeting, FDA panelists repeatedly emphasized the potential risks of these devices in terms of sensitivity (how often a test correctly generates a positive result) and of specificity (how often a test correctly generates a negative result).
New tools intended to aid in detection of skin cancer might produce too many false positives and thus trigger floods of worried patients seeking care and often facing unnecessary biopsies, the FDA panelists said. But more worrisome would be FDA clearance of tools that delivered too many false negative results, leaving people unaware of their cancers.
The standards would have to be set very high for new products, especially those intended for consumers, said FDA panelist Murad Alam, MD, a dermatologist and vice chair of the department of dermatology at Northwestern University, Chicago. Current technologies for analyzing skin lesions are not yet up to that task. Dr. Alam likened the situation to the hopes for self-driving cars.
“It sounds great in principle. If you read the predictions from 20 years ago, it should already have happened,” Dr. Alam said. “But we’re still struggling with that because there are serious points of failure.”
FDA panelist Veronica Rotemberg, MD, PhD, a dermatologist at Memorial Sloan Kettering Cancer Center, New York, also argued for well-designed studies to understand how consumers and clinicians would react to new tools.
“We have to define what prospective information, in the intended use setting, we need to feel comfortable saying that these tools could be in a layperson’s hand or a primary care person’s hand,” Dr. Rotemberg said.
The studies would not need to be large, especially in the case of nonmelanoma skin cancer, which is common, she added.
“There’s too much nuance here for us to be able to say: ‘This is what would happen,’ without testing it,” Dr. Rotemberg said. “I do not think these prospective studies would be very burdensome, but they would help us understand what the burden would be and what the costs would be and what the potential harms would be.”
Because of rules against disclosing corporate information, the FDA cannot tell the public about the kinds of inquiries it already may have fielded from companies interested in selling skin cancer detection tools.
But in response to a question during the FDA meeting, Binita Ashar, MD, a top official in the FDA’s Center for Devices and Radiological Health, said there is interest in having these kinds of products sold in the United States as well.
“I can tell you that this a very timely discussion and questions that we’re posing to you are the questions that we’re encountering or that we have been grappling with,” Dr. Ashar said.
FDA panelists noted that many patients cannot get access easily to dermatology visits.
Companies seeking to develop SLA devices likely will market their tools as attempts to fill a gap that now exists in medical care.
But there will be challenges ahead in explaining to patients how to interpret readings from these tools, the FDA panelists said. Consumers should know these tools are meant to assist in diagnosis, and not to make it.
“I’m not sure the layperson will hear that,” said FDA panelist Paula E. Bourelly, MD, a dermatologist from Olney, Md.
As a result, use of SLA tools could create tension between physicians and patients, with consumers demanding biopsies after seeing readings they don’t understand.
“I do have great concerns about the layperson feeling overly confident and reducing the provider to a technician,” she said.
The FDA panelists were not asked to cast formal votes on any issues discussed during the meeting They instead engaged in broad discussions around questions posed by the FDA in three key areas:
- What standards should be used to confirm lesion diagnosis in clinical testing of the accuracy of SLA devices?
- What would be acceptable true false-positive and false-negative results (sensitivity and specificity) for different diagnoses and users?
- How can the FDA address health equity considerations based on variable incidence of skin lesions?
Developing standards
The FDA asked the panel to consider several scenarios for SLA devices and to discuss how standards might vary depending on the user of the device, whether it would be dermatologists, other clinicians, or consumers.
The agency sought comments in particular about using histological diagnosis (core specimen processing with a consensus diagnosis from an expert dermatopathologist panel). In the briefing document for the meeting, the FDA argued that this approach provides the greatest certainty in the diagnosis.
“Device developers, however, cite concerns, both practical and ethical, in requiring biopsy of all lesions, particularly those that appear benign,” the FDA said. “They have proposed alternate means of defining ground truth, including consensus opinion of experts (of visual or dermoscopic examination of the lesion[s]), opinion of one expert (visual or dermoscopic examination), or other methods.”
In summarizing the discussion on this question, the FDA panel chairman, Hobart W. Harris, MD, MPH, a surgeon from the University of California, San Francisco, noted that there was broad support for histological data in clinical trials of SLA devices, with some allowance for cases where more hybrid approaches would be used.
There were also suggestions offered about designing trials and the need for biopsies of lesions that are clearly benign, as this would help gather data to help in developing algorithms.
Dr. Alam said care should be taken in explaining to study participants that they might have to undergo biopsies that they didn’t need, as part of the larger effort to gather data. This should be detailed in the consent form, he said.
“But I also think this is a relatively minor risk,” Dr. Alam said, comparing these biopsies to the blood samples that patients in many clinical studies routinely give.
“Are all of those blood draws necessary to track the change in whatever parameters that are being tracked? Probably not,” Dr. Alam said. “I think it would be possible to explain to a reasonable patient what this entails.”
Dr. Alam noted that companies might face extra hurdles in enrolling study participants and keeping them in the trials if the FDA seeks this kind of biopsy data. “But I don’t think inconvenience to the study sponsor is a good argument” for not seeking this kind of data, he added.
Leaving a loophole where certain kinds of clearly benign lesions don’t require a biopsy would eventually erode the quality of the research done on these devices. “That bar will be moved to accommodate the convenience of the sponsor, to make the study feasible,” Dr. Alam said. “And pretty soon, you’ll be missing a lot of patients that really should have biopsies.”
Acceptable rates of false positives, false negatives
The FDA panel chair noted that his colleagues had strongly urged review standards that would require that the devices improve on the rates of successful catches of suspicious lesions and lower false positives. But they did not endorse specific targets regarding the sensitivity and specificity rates.
“No one seems to be comfortable with providing or preordaining” these targets, Dr. Harris said.
Panelist Deneen Hesser, MSHSA, RN, urged a deep recognition of the power of a FDA clearance in the view of consumers.
“We need to be cognizant of what the term ‘FDA approved’ means to the lay individual,” said Ms. Hesser, who served as the patient representative on the panel. “A patient who sees that those tools are FDA approved will assume that each of those is the gold standard” in terms of expectations for delivering accurate results.
Like many of the panelists, Dr. Rotemberg urged the FDA to gather data about how patients would react to different messages encoded in consumer-oriented products.
“If the device says: ‘You should see a dermatologist for this’ and no other information, that’s very different from [saying]: ‘That lesion is suspicious for melanoma,’ ” Dr. Rotemberg said.
Despite the likely difficulties in conducting trials, the FDA needs to have the data to answer key questions about patient and physician reactions to readings from new tools, Dr. Rotemberg said.
“We don’t know how many additional biopsies we would cause with a specificity of 80%” for a new SLA tool, Dr. Rotemberg said, giving an example. “We don’t know how confident a dermatologist might be to say: ‘Actually, I’m not suspicious about that lesion and we can just fudge it or not biopsy it.’ We don’t know any of that until we study it in real life.”
The panelists also urged the FDA to seek to ensure that new tools used in analyzing skin lesions improve the quality of diagnosis.
Addressing equity
The FDA also asked the panel to weigh in on whether the agency should clear SLA tools in cases where the existing study data is drawn heavily from people considered to be at higher risk for skin cancer.
“To ensure generalizability across the entire U.S. population, should FDA require SLAs indicated for use beyond cancerous lesions be tested in a representative U.S. population?” the FDA asked.
The three most common skin cancers – melanoma, basal cell carcinoma, and squamous cell carcinoma – are more prevalent in people with Fitzpatrick I and II skin types, who tend to get sunburns, not tans. But people of color are more likely to develop melanoma in areas that are not sun exposed, such as the sole of the foot or under fingernails or toenails.
“Due in part to lower expected risk and screening, these melanomas are often detected late,” the FDA said in the briefing document.
There was broad consensus among panelists that the FDA should encourage companies to enroll people with all skin types and tones.
But they also looked for ways that the FDA could clear devices based on initial studies conducted largely with people considered to be at higher risk, with the agency then requiring follow-up trials to see how these products would work for the general U.S. population.
A version of this article first appeared on Medscape.com.
Commentary: Treatments for IBS, August 2022

The study by Huang and colleagues investigates the long-term effects and potential mechanisms of action of transcutaneous electrical acustimulation (TEA) in patients with IBS with constipation (IBS-C) experiencing colonic transit issues and visceral hypersensitivity. Patients with IBS-C receiving TEA had increased frequency of spontaneous bowel movements and significant improvements in analog pain score compared with the placebo group. This supports the benefit of TEA for improving constipation and the concomitant symptoms by accelerating colonic transit and reducing rectal sensation. It can be inferred that the improvement of these symptoms will lead to enhanced quality of life in patients with IBS-C.
The study by Melchior and colleagues1 emphasizes the effect that IBS can have on a patient's life. Patients with IBS report reduced disease-specific quality of life. The cumulative impact of the demographic factors and the severity of psychological symptoms, somatic symptoms, and gastrointestinal symptoms are associated with decreased quality of life. The severity of this impact is determined by the combination of these factors and the level of severity at which they occur. This decrease in quality of life can lead to increased anxiety related to gastrointestinal function. It is important for providers to acknowledge the effect that gastrointestinal function has on the patient's life in a broader sense. The appreciation and understanding of the patient experience enhances the therapeutic relationship and shared decision-making between the patient and the healthcare provider.
Additional References
- Melchior C, Colomier E, Trindade IA, et al. Irritable bowel syndrome: Factors of importance for disease-specific quality of life. United European Gastroenterol J. 2022 Jul 13. Doi: 10.1002/ueg2.12277
The study by Huang and colleagues investigates the long-term effects and potential mechanisms of action of transcutaneous electrical acustimulation (TEA) in patients with IBS with constipation (IBS-C) experiencing colonic transit issues and visceral hypersensitivity. Patients with IBS-C receiving TEA had increased frequency of spontaneous bowel movements and significant improvements in analog pain score compared with the placebo group. This supports the benefit of TEA for improving constipation and the concomitant symptoms by accelerating colonic transit and reducing rectal sensation. It can be inferred that the improvement of these symptoms will lead to enhanced quality of life in patients with IBS-C.
The study by Melchior and colleagues1 emphasizes the effect that IBS can have on a patient's life. Patients with IBS report reduced disease-specific quality of life. The cumulative impact of the demographic factors and the severity of psychological symptoms, somatic symptoms, and gastrointestinal symptoms are associated with decreased quality of life. The severity of this impact is determined by the combination of these factors and the level of severity at which they occur. This decrease in quality of life can lead to increased anxiety related to gastrointestinal function. It is important for providers to acknowledge the effect that gastrointestinal function has on the patient's life in a broader sense. The appreciation and understanding of the patient experience enhances the therapeutic relationship and shared decision-making between the patient and the healthcare provider.
Additional References
- Melchior C, Colomier E, Trindade IA, et al. Irritable bowel syndrome: Factors of importance for disease-specific quality of life. United European Gastroenterol J. 2022 Jul 13. Doi: 10.1002/ueg2.12277
The study by Huang and colleagues investigates the long-term effects and potential mechanisms of action of transcutaneous electrical acustimulation (TEA) in patients with IBS with constipation (IBS-C) experiencing colonic transit issues and visceral hypersensitivity. Patients with IBS-C receiving TEA had increased frequency of spontaneous bowel movements and significant improvements in analog pain score compared with the placebo group. This supports the benefit of TEA for improving constipation and the concomitant symptoms by accelerating colonic transit and reducing rectal sensation. It can be inferred that the improvement of these symptoms will lead to enhanced quality of life in patients with IBS-C.
The study by Melchior and colleagues1 emphasizes the effect that IBS can have on a patient's life. Patients with IBS report reduced disease-specific quality of life. The cumulative impact of the demographic factors and the severity of psychological symptoms, somatic symptoms, and gastrointestinal symptoms are associated with decreased quality of life. The severity of this impact is determined by the combination of these factors and the level of severity at which they occur. This decrease in quality of life can lead to increased anxiety related to gastrointestinal function. It is important for providers to acknowledge the effect that gastrointestinal function has on the patient's life in a broader sense. The appreciation and understanding of the patient experience enhances the therapeutic relationship and shared decision-making between the patient and the healthcare provider.
Additional References
- Melchior C, Colomier E, Trindade IA, et al. Irritable bowel syndrome: Factors of importance for disease-specific quality of life. United European Gastroenterol J. 2022 Jul 13. Doi: 10.1002/ueg2.12277
High rate of mental health problems in transgender children
Transgender children, even those as young as 9 or 10 years old, already show increased susceptibility to mental health problems compared with their cisgender peers, new research suggests.
Investigators assessed a sample of more than 7000 children aged 9-10 years in the general population and found those who reported being transgender scored considerably higher on all six subscales of the DSM-5-oriented Child Behavior Checklist (CBCL).
Transgender children had almost sixfold higher odds of suicidality and over twice the odds of depressive and anxiety problems, compared with cisgender children. Moreover, transgender children displayed higher levels of mental health problems compared with previous studies of transgender children recruited from specialist gender clinics.
“Our findings emphasize the vulnerability of transgender children, including those who may not yet have accessed specialist support,” senior author Kenneth C. Pang, MBBS, BMedSc, PhD, associate professor, Murdoch Children’s Research Institute, University of Melbourne, Royal Children’s Hospital, Australia, told this news organization.
“Clinicians providing general health care to transgender children should keep this vulnerability in mind and proactively address any mental health problems that exist,” he said.
The findings were published online as a research letter in JAMA Network Open.
Higher levels of support?
“We felt this study was important to conduct because previous studies regarding the mental health of transgender children have been drawn from children receiving specialist gender-related care,” Dr. Pang said.
“Transgender children receiving such care are likely to enjoy higher levels of support than those unable to access such services, and this might create differences in mental health,” he added.
To investigate this issue, the researchers turned to participants (n = 7,169; mean age, 10.3 years) in the Adolescent Brain Cognitive Development (ABCD) study.
“The ABCD study is a longitudinal study of over 11,000 children who were recruited to reflect the sociodemographic variation of the U.S. population,” lead author Douglas H. Russell, MSc, a PhD candidate at the University of Melbourne, told this news organization.
To be included in the current study, children had to understand and respond to the question “Are you transgender?”
The researchers compared mental health outcomes between transgender and cisgender children (n = 58 and n = 7,111, respectively) using the CBCL, which study participants had completed at baseline.
Key protective factor
The transgender children recorded higher mean T scores for all six subscales of the CBCL, although all children scored in the references range; and the standardized mean difference was “small.”
Suicidality was measured by summing the two suicide-related items in the parent-report CBCL assessing suicidal ideation and attempts.
“For the CBCL, T scores are calculated for measures that are scored on a continuous scale,” Dr. Pang noted. “Responses to the suicidality questions on the CBCL were assessed in a categorical manner (at risk of suicide vs. not), as previously described by others. So T scores were therefore not able to be calculated.”
When the investigators determined the proportion of cisgender and transgender children who scored in the “borderline” or “clinical” range (T score, 65), they found increased odds of transgender children scoring in that range in all six subscales, as well as suicidality.
The researchers note the results for attention-deficit/hyperactivity disorder and oppositional defiant problems were not statistically significant.
Previous studies that used clinical samples of young transgender children (aged 5 -11 years) reported lower rates of depression and anxiety than what was found in the current study.
“Transgender children in the general population displayed higher levels of mental health problems compared to previous studies of transgender children recruited from specialist gender clinics,” Mr. Russell said.
One reason for that may be children in specialist clinics “are likely to have support from their families (a key protective factor for the mental health of transgender young people); in comparison, many transgender children in the general population lack parental support for their gender,” the investigators wrote.
“Our findings suggest that by 9 to 10 years of age transgender children already show increased susceptibility to mental health problems compared with their cisgender peers, which has important public health implications,” they added.
The researchers noted that whether this susceptibility “is due to stigma, minority stress, discrimination, or gender dysphoria is unclear, but providing appropriate mental health supports to this vulnerable group is paramount.”
“Pathologizing and damaging”
Commenting for this news organiztion, Jack L. Turban, MD, incoming assistant professor of child and adolescent psychiatry, University of California, San Francisco, said that “sadly” the findings are “largely in line with past studies that have shown dramatic mental health disparities” for transgender and gender diverse youth.
“The dramatically elevated odds of suicidality warrants particular public health concern,” said Dr. Turban, who was not involved with the study.
He noted these results “come at a time when transgender youth are under legislative attack in many states throughout the country, and the national rhetoric around them has been pathologizing and damaging.”
Dr. Turban said that he worries “if our national discourse around trans youth doesn’t change soon, that these disparities will worsen.”
Funding was provided to individual investigators by the Hugh Williamson Foundation, the Royal Children’s Hospital foundation, the National Health and Medical Research Council, and the Australian Government Research Training Program Scholarship. Mr. Russell and Dr. Pang reported being members of the Australian Professional Association for Trans Health. Dr. Pang is a member of the World Professional Association for Transgender Health and a member of the editorial board of the journal Transgender Health. Dr. Turban reported textbook royalties from Springer Nature, being on the scientific advisory board of Panorama Global (UpSwing Fund), and payments as an expert witness for the American Civil Liberties Union, Lambda Legal, and Cooley LLP. He has received a pilot research award from AACAP and pharmaceutical partners (Arbor and Pfizer), a research fellowship from the Sorensen Foundation, and freelance payments from the New York Times, the Washington Post, and the Los Angeles Times.
A version of this article first appeared on Medscape.com.
Transgender children, even those as young as 9 or 10 years old, already show increased susceptibility to mental health problems compared with their cisgender peers, new research suggests.
Investigators assessed a sample of more than 7000 children aged 9-10 years in the general population and found those who reported being transgender scored considerably higher on all six subscales of the DSM-5-oriented Child Behavior Checklist (CBCL).
Transgender children had almost sixfold higher odds of suicidality and over twice the odds of depressive and anxiety problems, compared with cisgender children. Moreover, transgender children displayed higher levels of mental health problems compared with previous studies of transgender children recruited from specialist gender clinics.
“Our findings emphasize the vulnerability of transgender children, including those who may not yet have accessed specialist support,” senior author Kenneth C. Pang, MBBS, BMedSc, PhD, associate professor, Murdoch Children’s Research Institute, University of Melbourne, Royal Children’s Hospital, Australia, told this news organization.
“Clinicians providing general health care to transgender children should keep this vulnerability in mind and proactively address any mental health problems that exist,” he said.
The findings were published online as a research letter in JAMA Network Open.
Higher levels of support?
“We felt this study was important to conduct because previous studies regarding the mental health of transgender children have been drawn from children receiving specialist gender-related care,” Dr. Pang said.
“Transgender children receiving such care are likely to enjoy higher levels of support than those unable to access such services, and this might create differences in mental health,” he added.
To investigate this issue, the researchers turned to participants (n = 7,169; mean age, 10.3 years) in the Adolescent Brain Cognitive Development (ABCD) study.
“The ABCD study is a longitudinal study of over 11,000 children who were recruited to reflect the sociodemographic variation of the U.S. population,” lead author Douglas H. Russell, MSc, a PhD candidate at the University of Melbourne, told this news organization.
To be included in the current study, children had to understand and respond to the question “Are you transgender?”
The researchers compared mental health outcomes between transgender and cisgender children (n = 58 and n = 7,111, respectively) using the CBCL, which study participants had completed at baseline.
Key protective factor
The transgender children recorded higher mean T scores for all six subscales of the CBCL, although all children scored in the references range; and the standardized mean difference was “small.”
Suicidality was measured by summing the two suicide-related items in the parent-report CBCL assessing suicidal ideation and attempts.
“For the CBCL, T scores are calculated for measures that are scored on a continuous scale,” Dr. Pang noted. “Responses to the suicidality questions on the CBCL were assessed in a categorical manner (at risk of suicide vs. not), as previously described by others. So T scores were therefore not able to be calculated.”
When the investigators determined the proportion of cisgender and transgender children who scored in the “borderline” or “clinical” range (T score, 65), they found increased odds of transgender children scoring in that range in all six subscales, as well as suicidality.
The researchers note the results for attention-deficit/hyperactivity disorder and oppositional defiant problems were not statistically significant.
Previous studies that used clinical samples of young transgender children (aged 5 -11 years) reported lower rates of depression and anxiety than what was found in the current study.
“Transgender children in the general population displayed higher levels of mental health problems compared to previous studies of transgender children recruited from specialist gender clinics,” Mr. Russell said.
One reason for that may be children in specialist clinics “are likely to have support from their families (a key protective factor for the mental health of transgender young people); in comparison, many transgender children in the general population lack parental support for their gender,” the investigators wrote.
“Our findings suggest that by 9 to 10 years of age transgender children already show increased susceptibility to mental health problems compared with their cisgender peers, which has important public health implications,” they added.
The researchers noted that whether this susceptibility “is due to stigma, minority stress, discrimination, or gender dysphoria is unclear, but providing appropriate mental health supports to this vulnerable group is paramount.”
“Pathologizing and damaging”
Commenting for this news organiztion, Jack L. Turban, MD, incoming assistant professor of child and adolescent psychiatry, University of California, San Francisco, said that “sadly” the findings are “largely in line with past studies that have shown dramatic mental health disparities” for transgender and gender diverse youth.
“The dramatically elevated odds of suicidality warrants particular public health concern,” said Dr. Turban, who was not involved with the study.
He noted these results “come at a time when transgender youth are under legislative attack in many states throughout the country, and the national rhetoric around them has been pathologizing and damaging.”
Dr. Turban said that he worries “if our national discourse around trans youth doesn’t change soon, that these disparities will worsen.”
Funding was provided to individual investigators by the Hugh Williamson Foundation, the Royal Children’s Hospital foundation, the National Health and Medical Research Council, and the Australian Government Research Training Program Scholarship. Mr. Russell and Dr. Pang reported being members of the Australian Professional Association for Trans Health. Dr. Pang is a member of the World Professional Association for Transgender Health and a member of the editorial board of the journal Transgender Health. Dr. Turban reported textbook royalties from Springer Nature, being on the scientific advisory board of Panorama Global (UpSwing Fund), and payments as an expert witness for the American Civil Liberties Union, Lambda Legal, and Cooley LLP. He has received a pilot research award from AACAP and pharmaceutical partners (Arbor and Pfizer), a research fellowship from the Sorensen Foundation, and freelance payments from the New York Times, the Washington Post, and the Los Angeles Times.
A version of this article first appeared on Medscape.com.
Transgender children, even those as young as 9 or 10 years old, already show increased susceptibility to mental health problems compared with their cisgender peers, new research suggests.
Investigators assessed a sample of more than 7000 children aged 9-10 years in the general population and found those who reported being transgender scored considerably higher on all six subscales of the DSM-5-oriented Child Behavior Checklist (CBCL).
Transgender children had almost sixfold higher odds of suicidality and over twice the odds of depressive and anxiety problems, compared with cisgender children. Moreover, transgender children displayed higher levels of mental health problems compared with previous studies of transgender children recruited from specialist gender clinics.
“Our findings emphasize the vulnerability of transgender children, including those who may not yet have accessed specialist support,” senior author Kenneth C. Pang, MBBS, BMedSc, PhD, associate professor, Murdoch Children’s Research Institute, University of Melbourne, Royal Children’s Hospital, Australia, told this news organization.
“Clinicians providing general health care to transgender children should keep this vulnerability in mind and proactively address any mental health problems that exist,” he said.
The findings were published online as a research letter in JAMA Network Open.
Higher levels of support?
“We felt this study was important to conduct because previous studies regarding the mental health of transgender children have been drawn from children receiving specialist gender-related care,” Dr. Pang said.
“Transgender children receiving such care are likely to enjoy higher levels of support than those unable to access such services, and this might create differences in mental health,” he added.
To investigate this issue, the researchers turned to participants (n = 7,169; mean age, 10.3 years) in the Adolescent Brain Cognitive Development (ABCD) study.
“The ABCD study is a longitudinal study of over 11,000 children who were recruited to reflect the sociodemographic variation of the U.S. population,” lead author Douglas H. Russell, MSc, a PhD candidate at the University of Melbourne, told this news organization.
To be included in the current study, children had to understand and respond to the question “Are you transgender?”
The researchers compared mental health outcomes between transgender and cisgender children (n = 58 and n = 7,111, respectively) using the CBCL, which study participants had completed at baseline.
Key protective factor
The transgender children recorded higher mean T scores for all six subscales of the CBCL, although all children scored in the references range; and the standardized mean difference was “small.”
Suicidality was measured by summing the two suicide-related items in the parent-report CBCL assessing suicidal ideation and attempts.
“For the CBCL, T scores are calculated for measures that are scored on a continuous scale,” Dr. Pang noted. “Responses to the suicidality questions on the CBCL were assessed in a categorical manner (at risk of suicide vs. not), as previously described by others. So T scores were therefore not able to be calculated.”
When the investigators determined the proportion of cisgender and transgender children who scored in the “borderline” or “clinical” range (T score, 65), they found increased odds of transgender children scoring in that range in all six subscales, as well as suicidality.
The researchers note the results for attention-deficit/hyperactivity disorder and oppositional defiant problems were not statistically significant.
Previous studies that used clinical samples of young transgender children (aged 5 -11 years) reported lower rates of depression and anxiety than what was found in the current study.
“Transgender children in the general population displayed higher levels of mental health problems compared to previous studies of transgender children recruited from specialist gender clinics,” Mr. Russell said.
One reason for that may be children in specialist clinics “are likely to have support from their families (a key protective factor for the mental health of transgender young people); in comparison, many transgender children in the general population lack parental support for their gender,” the investigators wrote.
“Our findings suggest that by 9 to 10 years of age transgender children already show increased susceptibility to mental health problems compared with their cisgender peers, which has important public health implications,” they added.
The researchers noted that whether this susceptibility “is due to stigma, minority stress, discrimination, or gender dysphoria is unclear, but providing appropriate mental health supports to this vulnerable group is paramount.”
“Pathologizing and damaging”
Commenting for this news organiztion, Jack L. Turban, MD, incoming assistant professor of child and adolescent psychiatry, University of California, San Francisco, said that “sadly” the findings are “largely in line with past studies that have shown dramatic mental health disparities” for transgender and gender diverse youth.
“The dramatically elevated odds of suicidality warrants particular public health concern,” said Dr. Turban, who was not involved with the study.
He noted these results “come at a time when transgender youth are under legislative attack in many states throughout the country, and the national rhetoric around them has been pathologizing and damaging.”
Dr. Turban said that he worries “if our national discourse around trans youth doesn’t change soon, that these disparities will worsen.”
Funding was provided to individual investigators by the Hugh Williamson Foundation, the Royal Children’s Hospital foundation, the National Health and Medical Research Council, and the Australian Government Research Training Program Scholarship. Mr. Russell and Dr. Pang reported being members of the Australian Professional Association for Trans Health. Dr. Pang is a member of the World Professional Association for Transgender Health and a member of the editorial board of the journal Transgender Health. Dr. Turban reported textbook royalties from Springer Nature, being on the scientific advisory board of Panorama Global (UpSwing Fund), and payments as an expert witness for the American Civil Liberties Union, Lambda Legal, and Cooley LLP. He has received a pilot research award from AACAP and pharmaceutical partners (Arbor and Pfizer), a research fellowship from the Sorensen Foundation, and freelance payments from the New York Times, the Washington Post, and the Los Angeles Times.
A version of this article first appeared on Medscape.com.
FROM JAMA NETWORK OPEN