User login
Updates in the Management of Peripheral Arterial Disease: Focus on Reduction of Atherothrombotic Risk


This October 2021 online edition of the Hot Topics in Primary Care supplement provides updates on the management of peripheral arterial disease (PAD) and treatment approaches for reducing atherothrombotic risk.
Click here to Read More on PAD
Click here to read the Hot Topics in Primary Care 2021 Supplement
This October 2021 online edition of the Hot Topics in Primary Care supplement provides updates on the management of peripheral arterial disease (PAD) and treatment approaches for reducing atherothrombotic risk.
Click here to Read More on PAD
Click here to read the Hot Topics in Primary Care 2021 Supplement
This October 2021 online edition of the Hot Topics in Primary Care supplement provides updates on the management of peripheral arterial disease (PAD) and treatment approaches for reducing atherothrombotic risk.
Click here to Read More on PAD
Click here to read the Hot Topics in Primary Care 2021 Supplement
New FDA guidance aims to cut sodium in processed foods
The Food and Drug Administration has issued voluntary, short-term sodium reduction targets for food manufacturers, chain restaurants, and food service operators for processed, packaged, and prepared foods, with an eye toward reducing diet-related conditions such as heart disease and obesity.
The new targets seek to decrease average sodium intake from approximately 3,400 mg/day to 3,000 mg/day, about a 12% reduction, over the next 2.5 years, acting FDA Commissioner Janet Woodcock, MD, and Susan Mayne, PhD, director of the FDA’s Center for Food Safety and Applied Nutrition, said in joint statement.
Although this reduction keeps the average intake above the recommended limit of 2,300 mg/day for individuals 14 years and older as per the Dietary Guidelines for Americans, “we know that even these modest reductions made slowly over the next few years will substantially decrease diet-related diseases,” they added.
The FDA first proposed recommendations for reducing sodium content in draft guidance released in 2016.
Since, then a number of companies in the food industry have already made changes to sodium content in their products, “which is encouraging, but additional support across all types of foods to help consumers meet recommended sodium limits is needed,” Dr. Woodcock and Dr. Mayne said.
They emphasized that the new guidance represents short-term goals that the food industry should work to meet as soon as possible to help optimize public health.
“We will continue our discussions with the food industry as we monitor the sodium content of the food supply to evaluate progress. In the future, we plan to issue revised, subsequent targets to further lower the sodium content incrementally and continue to help reduce sodium intake,” Dr. Woodcock and Dr. Mayne said.
AHA: A good first step that does not go far enough
In a statement, the American Heart Association said the new targets will play “a critical role in helping people across the country achieve healthier levels of sodium and improved well-being overall. These targets will be an important driver to reduce sodium consumption, which can have significant health benefits and lead to lower medical costs.”
“Lowering sodium levels in the food supply would reduce risk of hypertension, heart disease, stroke, heart attack, and death in addition to saving billions of dollars in health care costs over the next decade,” the AHA said.
But the AHA also said lowering sodium intake to 3,000 mg/day is not enough.
“Lowering sodium further to 2,300 mg could prevent an estimated 450,000 cases of cardiovascular disease, gain 2 million quality-adjusted life-years, and save approximately $40 billion in health care costs over a 20-year period,” the AHA said.
The AHA is urging the FDA to “follow [this] action with additional targets to further lower the amount of sodium in the food supply and help people in America attain an appropriate sodium intake.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has issued voluntary, short-term sodium reduction targets for food manufacturers, chain restaurants, and food service operators for processed, packaged, and prepared foods, with an eye toward reducing diet-related conditions such as heart disease and obesity.
The new targets seek to decrease average sodium intake from approximately 3,400 mg/day to 3,000 mg/day, about a 12% reduction, over the next 2.5 years, acting FDA Commissioner Janet Woodcock, MD, and Susan Mayne, PhD, director of the FDA’s Center for Food Safety and Applied Nutrition, said in joint statement.
Although this reduction keeps the average intake above the recommended limit of 2,300 mg/day for individuals 14 years and older as per the Dietary Guidelines for Americans, “we know that even these modest reductions made slowly over the next few years will substantially decrease diet-related diseases,” they added.
The FDA first proposed recommendations for reducing sodium content in draft guidance released in 2016.
Since, then a number of companies in the food industry have already made changes to sodium content in their products, “which is encouraging, but additional support across all types of foods to help consumers meet recommended sodium limits is needed,” Dr. Woodcock and Dr. Mayne said.
They emphasized that the new guidance represents short-term goals that the food industry should work to meet as soon as possible to help optimize public health.
“We will continue our discussions with the food industry as we monitor the sodium content of the food supply to evaluate progress. In the future, we plan to issue revised, subsequent targets to further lower the sodium content incrementally and continue to help reduce sodium intake,” Dr. Woodcock and Dr. Mayne said.
AHA: A good first step that does not go far enough
In a statement, the American Heart Association said the new targets will play “a critical role in helping people across the country achieve healthier levels of sodium and improved well-being overall. These targets will be an important driver to reduce sodium consumption, which can have significant health benefits and lead to lower medical costs.”
“Lowering sodium levels in the food supply would reduce risk of hypertension, heart disease, stroke, heart attack, and death in addition to saving billions of dollars in health care costs over the next decade,” the AHA said.
But the AHA also said lowering sodium intake to 3,000 mg/day is not enough.
“Lowering sodium further to 2,300 mg could prevent an estimated 450,000 cases of cardiovascular disease, gain 2 million quality-adjusted life-years, and save approximately $40 billion in health care costs over a 20-year period,” the AHA said.
The AHA is urging the FDA to “follow [this] action with additional targets to further lower the amount of sodium in the food supply and help people in America attain an appropriate sodium intake.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has issued voluntary, short-term sodium reduction targets for food manufacturers, chain restaurants, and food service operators for processed, packaged, and prepared foods, with an eye toward reducing diet-related conditions such as heart disease and obesity.
The new targets seek to decrease average sodium intake from approximately 3,400 mg/day to 3,000 mg/day, about a 12% reduction, over the next 2.5 years, acting FDA Commissioner Janet Woodcock, MD, and Susan Mayne, PhD, director of the FDA’s Center for Food Safety and Applied Nutrition, said in joint statement.
Although this reduction keeps the average intake above the recommended limit of 2,300 mg/day for individuals 14 years and older as per the Dietary Guidelines for Americans, “we know that even these modest reductions made slowly over the next few years will substantially decrease diet-related diseases,” they added.
The FDA first proposed recommendations for reducing sodium content in draft guidance released in 2016.
Since, then a number of companies in the food industry have already made changes to sodium content in their products, “which is encouraging, but additional support across all types of foods to help consumers meet recommended sodium limits is needed,” Dr. Woodcock and Dr. Mayne said.
They emphasized that the new guidance represents short-term goals that the food industry should work to meet as soon as possible to help optimize public health.
“We will continue our discussions with the food industry as we monitor the sodium content of the food supply to evaluate progress. In the future, we plan to issue revised, subsequent targets to further lower the sodium content incrementally and continue to help reduce sodium intake,” Dr. Woodcock and Dr. Mayne said.
AHA: A good first step that does not go far enough
In a statement, the American Heart Association said the new targets will play “a critical role in helping people across the country achieve healthier levels of sodium and improved well-being overall. These targets will be an important driver to reduce sodium consumption, which can have significant health benefits and lead to lower medical costs.”
“Lowering sodium levels in the food supply would reduce risk of hypertension, heart disease, stroke, heart attack, and death in addition to saving billions of dollars in health care costs over the next decade,” the AHA said.
But the AHA also said lowering sodium intake to 3,000 mg/day is not enough.
“Lowering sodium further to 2,300 mg could prevent an estimated 450,000 cases of cardiovascular disease, gain 2 million quality-adjusted life-years, and save approximately $40 billion in health care costs over a 20-year period,” the AHA said.
The AHA is urging the FDA to “follow [this] action with additional targets to further lower the amount of sodium in the food supply and help people in America attain an appropriate sodium intake.”
A version of this article first appeared on Medscape.com.
HHS okays first U.S. pilot to mandate coverage of gender-affirming care
The approval means transgender-related care must be included as part of the essential benefits offered on the state’s Affordable Care Act marketplace, which includes private individual and small group insurance plans. The coverage will start Jan. 1, 2023. Colorado is the first state in the United States to require such coverage.
The HHS notes that gender-affirming treatments to be covered include eye and lid modifications, face tightening, facial bone remodeling for facial feminization, breast/chest construction and reductions, and laser hair removal.
“I am proud to stand with Colorado to remove barriers that have historically made it difficult for transgender people to access health coverage and medical care,” said HHS Secretary Xavier Becerra in a statement.
“Colorado’s expansion of their essential health benefits to include gender-affirming surgery and other treatments is a model for other states to follow, and we invite other states to follow suit,” said Centers for Medicare & Medicaid Services Administrator Chiquita Brooks-LaSure in the statement.
Medicaid already covers comprehensive transgender care in Colorado.
The LGBTQ+ advocacy group One Colorado estimated that, thanks to the Affordable Care Act, only 5% of the state’s LGBTQ+ community was uninsured in 2019, compared to 10% in 2011.
However, 34% of transgender respondents to a One Colorado poll in 2018 said they had been denied coverage for an LGBTQ-specific medical service, such as gender-affirming care. Sixty-two percent said that a lack of insurance or limited insurance was a barrier to care; 84% said another barrier was the lack of adequately trained mental and behavioral health professionals.
Mental health also covered
The Colorado plan requires individual and small group plans to cover an annual 45- to 60-minute mental health wellness exam with a qualified mental health care practitioner. The visit can include behavioral health screening, education and consultation about healthy lifestyle changes, referrals to mental health treatment, and discussion of potential medication options.
The plans also must cover an additional 15 medications as alternatives to opioids and up to six acupuncture visits annually.
“This plan expands access to mental health services for Coloradans while helping those fighting substance abuse to overcome their addiction,” said Governor Jared Polis in a statement.
“This improves care for Coloradans and ensures that even more Coloradans have access to help when they need it,” he said.
A version of this article first appeared on Medscape.com.
The approval means transgender-related care must be included as part of the essential benefits offered on the state’s Affordable Care Act marketplace, which includes private individual and small group insurance plans. The coverage will start Jan. 1, 2023. Colorado is the first state in the United States to require such coverage.
The HHS notes that gender-affirming treatments to be covered include eye and lid modifications, face tightening, facial bone remodeling for facial feminization, breast/chest construction and reductions, and laser hair removal.
“I am proud to stand with Colorado to remove barriers that have historically made it difficult for transgender people to access health coverage and medical care,” said HHS Secretary Xavier Becerra in a statement.
“Colorado’s expansion of their essential health benefits to include gender-affirming surgery and other treatments is a model for other states to follow, and we invite other states to follow suit,” said Centers for Medicare & Medicaid Services Administrator Chiquita Brooks-LaSure in the statement.
Medicaid already covers comprehensive transgender care in Colorado.
The LGBTQ+ advocacy group One Colorado estimated that, thanks to the Affordable Care Act, only 5% of the state’s LGBTQ+ community was uninsured in 2019, compared to 10% in 2011.
However, 34% of transgender respondents to a One Colorado poll in 2018 said they had been denied coverage for an LGBTQ-specific medical service, such as gender-affirming care. Sixty-two percent said that a lack of insurance or limited insurance was a barrier to care; 84% said another barrier was the lack of adequately trained mental and behavioral health professionals.
Mental health also covered
The Colorado plan requires individual and small group plans to cover an annual 45- to 60-minute mental health wellness exam with a qualified mental health care practitioner. The visit can include behavioral health screening, education and consultation about healthy lifestyle changes, referrals to mental health treatment, and discussion of potential medication options.
The plans also must cover an additional 15 medications as alternatives to opioids and up to six acupuncture visits annually.
“This plan expands access to mental health services for Coloradans while helping those fighting substance abuse to overcome their addiction,” said Governor Jared Polis in a statement.
“This improves care for Coloradans and ensures that even more Coloradans have access to help when they need it,” he said.
A version of this article first appeared on Medscape.com.
The approval means transgender-related care must be included as part of the essential benefits offered on the state’s Affordable Care Act marketplace, which includes private individual and small group insurance plans. The coverage will start Jan. 1, 2023. Colorado is the first state in the United States to require such coverage.
The HHS notes that gender-affirming treatments to be covered include eye and lid modifications, face tightening, facial bone remodeling for facial feminization, breast/chest construction and reductions, and laser hair removal.
“I am proud to stand with Colorado to remove barriers that have historically made it difficult for transgender people to access health coverage and medical care,” said HHS Secretary Xavier Becerra in a statement.
“Colorado’s expansion of their essential health benefits to include gender-affirming surgery and other treatments is a model for other states to follow, and we invite other states to follow suit,” said Centers for Medicare & Medicaid Services Administrator Chiquita Brooks-LaSure in the statement.
Medicaid already covers comprehensive transgender care in Colorado.
The LGBTQ+ advocacy group One Colorado estimated that, thanks to the Affordable Care Act, only 5% of the state’s LGBTQ+ community was uninsured in 2019, compared to 10% in 2011.
However, 34% of transgender respondents to a One Colorado poll in 2018 said they had been denied coverage for an LGBTQ-specific medical service, such as gender-affirming care. Sixty-two percent said that a lack of insurance or limited insurance was a barrier to care; 84% said another barrier was the lack of adequately trained mental and behavioral health professionals.
Mental health also covered
The Colorado plan requires individual and small group plans to cover an annual 45- to 60-minute mental health wellness exam with a qualified mental health care practitioner. The visit can include behavioral health screening, education and consultation about healthy lifestyle changes, referrals to mental health treatment, and discussion of potential medication options.
The plans also must cover an additional 15 medications as alternatives to opioids and up to six acupuncture visits annually.
“This plan expands access to mental health services for Coloradans while helping those fighting substance abuse to overcome their addiction,” said Governor Jared Polis in a statement.
“This improves care for Coloradans and ensures that even more Coloradans have access to help when they need it,” he said.
A version of this article first appeared on Medscape.com.
Stay tuned for CSI: Olive oil
Cracking down on food fraud
How do you know the olive oil in your pantry is from Greece? Or that the avocados on your toast are from Mexico? The label, right? Well, maybe not. False claims of origin are a huge problem in the food industry, costing over $30 billion in economic damage annually.

Fear not, citizens, because botanists are on the job, and they’ve found a cheaper and more efficient way to expose that non-Greek olive oil.
How? Florian Cueni, PhD, of the University of Basel, Switzerland, and associates developed a new model to simulate oxygen isotope ratios in plants from a specific region, based on the temperature, precipitation, growing season information, and humidity data. Previously, botanists had to collect reference data from the claimed origin country and from other regions to validate where the product actually came from.
“With minor adjustments to the parameters, our model can be used to determine all plant products,” said senior investigator Ansgar Kahmen. This can open up the door for even more plant forensics, including drug confiscations and illegal timber logging, with information that will hold up in court.
Why pay Greek-olive prices for olives from California?
Fear leads to anger, anger leads to unhelpful online reviews
And reading angry online reviews leads to hate and suffering. We may have co-opted Master Yoda’s wise words ever so slightly, but anyone who’s done any shopping online (so everyone) knows that the review section of any product can be downright villainous. Do these reviews affect what we buy?

The angry online product review was the subject of a recent study published in MIS Quarterly. In a series of experiments, participants were shown a series of realistic online reviews with varying amounts of anger but with similar amounts of information. After reading the reviews, participants rated helpfulness, their personal opinion of the product/retailer, and whether or not they would buy the product.
Participants overwhelmingly rated calmly written reviews as more helpful than angrily written ones. One would expect, then, that those unhelpful angry reviews would have little effect on the participant’s view or willingness to buy a product, but the study investigators found the opposite. Reading angry reviews made the participants more likely to reject the product, even though they didn’t think the angry review was useful. And when you think about it, it does make sense. Anger means drama, and we can’t resist a juicy bit of drama.
So while we should all aspire to be Yoda and rise above anger and hatred, in reality we seem to be channeling Emperor Palpatine. We let the hate flow through us, and in our anger, we ignore perfectly good products. On the plus side, now we can shoot lightning out of our hands, so that’s pretty cool.
Health care is heading to the hall of fame
We couldn’t be happier here at LOTME because it’s that time of year again.

No, we’re not talking about Healthcare Security and Safety Week or National Metric Week, although those are both kind of important. Hmm, maybe we should talk about health care security or the metric system. After all, in this country, medicine is one of the metric system’s biggest customers. And who doesn’t love picograms? They’re the unit-of-measurement equivalent of a koala.
So we’re doing the metric system, then? Nah.
We’re excited because the 2022 inductees to the National Inventors Hall of Fame were just announced, and, as usual, the world of health care is well represented.
First up is the surprisingly relevant (thanks to the party guest that won’t leave, SARS-CoV-2) pair of Katalin Karikó, PhD, and Drew Weissman, MD, who worked together in the early 2000s to modify mRNA “so it could avoid immediate immune detection, remain active longer and efficiently instruct cells to create antigens to protect against severe disease.” Their discoveries eventually led to the use of modified mRNA in the COVID-19 vaccines.
The second, albeit posthumous, physician-inductee is Patricia Bath, MD, who was the first Black female physician to receive a U.S. patent for a medical invention. The laserphaco device and technique to remove cataracts “performed all steps of cataract removal: making the incision, destroying the lens, and vacuuming out the fractured pieces.”
Two other inductees have somewhat tenuous connections to medical care. Lonnie Johnson invented the Super Soaker, a powerful squirt gun that has been criticized by psychologists for encouraging violence, and Carl Benz invented the automobile, which sort of means he invented the ambulance, so there you go.
The induction ceremony takes place on May 5, 2022, in Washington, DC. If you’re attending the black-tie dinner at The Anthem, let us know and we’ll split an Uber. It’s our only night to be fancy.
Cracking down on food fraud
How do you know the olive oil in your pantry is from Greece? Or that the avocados on your toast are from Mexico? The label, right? Well, maybe not. False claims of origin are a huge problem in the food industry, costing over $30 billion in economic damage annually.
Fear not, citizens, because botanists are on the job, and they’ve found a cheaper and more efficient way to expose that non-Greek olive oil.
How? Florian Cueni, PhD, of the University of Basel, Switzerland, and associates developed a new model to simulate oxygen isotope ratios in plants from a specific region, based on the temperature, precipitation, growing season information, and humidity data. Previously, botanists had to collect reference data from the claimed origin country and from other regions to validate where the product actually came from.
“With minor adjustments to the parameters, our model can be used to determine all plant products,” said senior investigator Ansgar Kahmen. This can open up the door for even more plant forensics, including drug confiscations and illegal timber logging, with information that will hold up in court.
Why pay Greek-olive prices for olives from California?
Fear leads to anger, anger leads to unhelpful online reviews
And reading angry online reviews leads to hate and suffering. We may have co-opted Master Yoda’s wise words ever so slightly, but anyone who’s done any shopping online (so everyone) knows that the review section of any product can be downright villainous. Do these reviews affect what we buy?
The angry online product review was the subject of a recent study published in MIS Quarterly. In a series of experiments, participants were shown a series of realistic online reviews with varying amounts of anger but with similar amounts of information. After reading the reviews, participants rated helpfulness, their personal opinion of the product/retailer, and whether or not they would buy the product.
Participants overwhelmingly rated calmly written reviews as more helpful than angrily written ones. One would expect, then, that those unhelpful angry reviews would have little effect on the participant’s view or willingness to buy a product, but the study investigators found the opposite. Reading angry reviews made the participants more likely to reject the product, even though they didn’t think the angry review was useful. And when you think about it, it does make sense. Anger means drama, and we can’t resist a juicy bit of drama.
So while we should all aspire to be Yoda and rise above anger and hatred, in reality we seem to be channeling Emperor Palpatine. We let the hate flow through us, and in our anger, we ignore perfectly good products. On the plus side, now we can shoot lightning out of our hands, so that’s pretty cool.
Health care is heading to the hall of fame
We couldn’t be happier here at LOTME because it’s that time of year again.
No, we’re not talking about Healthcare Security and Safety Week or National Metric Week, although those are both kind of important. Hmm, maybe we should talk about health care security or the metric system. After all, in this country, medicine is one of the metric system’s biggest customers. And who doesn’t love picograms? They’re the unit-of-measurement equivalent of a koala.
So we’re doing the metric system, then? Nah.
We’re excited because the 2022 inductees to the National Inventors Hall of Fame were just announced, and, as usual, the world of health care is well represented.
First up is the surprisingly relevant (thanks to the party guest that won’t leave, SARS-CoV-2) pair of Katalin Karikó, PhD, and Drew Weissman, MD, who worked together in the early 2000s to modify mRNA “so it could avoid immediate immune detection, remain active longer and efficiently instruct cells to create antigens to protect against severe disease.” Their discoveries eventually led to the use of modified mRNA in the COVID-19 vaccines.
The second, albeit posthumous, physician-inductee is Patricia Bath, MD, who was the first Black female physician to receive a U.S. patent for a medical invention. The laserphaco device and technique to remove cataracts “performed all steps of cataract removal: making the incision, destroying the lens, and vacuuming out the fractured pieces.”
Two other inductees have somewhat tenuous connections to medical care. Lonnie Johnson invented the Super Soaker, a powerful squirt gun that has been criticized by psychologists for encouraging violence, and Carl Benz invented the automobile, which sort of means he invented the ambulance, so there you go.
The induction ceremony takes place on May 5, 2022, in Washington, DC. If you’re attending the black-tie dinner at The Anthem, let us know and we’ll split an Uber. It’s our only night to be fancy.
Cracking down on food fraud
How do you know the olive oil in your pantry is from Greece? Or that the avocados on your toast are from Mexico? The label, right? Well, maybe not. False claims of origin are a huge problem in the food industry, costing over $30 billion in economic damage annually.
Fear not, citizens, because botanists are on the job, and they’ve found a cheaper and more efficient way to expose that non-Greek olive oil.
How? Florian Cueni, PhD, of the University of Basel, Switzerland, and associates developed a new model to simulate oxygen isotope ratios in plants from a specific region, based on the temperature, precipitation, growing season information, and humidity data. Previously, botanists had to collect reference data from the claimed origin country and from other regions to validate where the product actually came from.
“With minor adjustments to the parameters, our model can be used to determine all plant products,” said senior investigator Ansgar Kahmen. This can open up the door for even more plant forensics, including drug confiscations and illegal timber logging, with information that will hold up in court.
Why pay Greek-olive prices for olives from California?
Fear leads to anger, anger leads to unhelpful online reviews
And reading angry online reviews leads to hate and suffering. We may have co-opted Master Yoda’s wise words ever so slightly, but anyone who’s done any shopping online (so everyone) knows that the review section of any product can be downright villainous. Do these reviews affect what we buy?
The angry online product review was the subject of a recent study published in MIS Quarterly. In a series of experiments, participants were shown a series of realistic online reviews with varying amounts of anger but with similar amounts of information. After reading the reviews, participants rated helpfulness, their personal opinion of the product/retailer, and whether or not they would buy the product.
Participants overwhelmingly rated calmly written reviews as more helpful than angrily written ones. One would expect, then, that those unhelpful angry reviews would have little effect on the participant’s view or willingness to buy a product, but the study investigators found the opposite. Reading angry reviews made the participants more likely to reject the product, even though they didn’t think the angry review was useful. And when you think about it, it does make sense. Anger means drama, and we can’t resist a juicy bit of drama.
So while we should all aspire to be Yoda and rise above anger and hatred, in reality we seem to be channeling Emperor Palpatine. We let the hate flow through us, and in our anger, we ignore perfectly good products. On the plus side, now we can shoot lightning out of our hands, so that’s pretty cool.
Health care is heading to the hall of fame
We couldn’t be happier here at LOTME because it’s that time of year again.
No, we’re not talking about Healthcare Security and Safety Week or National Metric Week, although those are both kind of important. Hmm, maybe we should talk about health care security or the metric system. After all, in this country, medicine is one of the metric system’s biggest customers. And who doesn’t love picograms? They’re the unit-of-measurement equivalent of a koala.
So we’re doing the metric system, then? Nah.
We’re excited because the 2022 inductees to the National Inventors Hall of Fame were just announced, and, as usual, the world of health care is well represented.
First up is the surprisingly relevant (thanks to the party guest that won’t leave, SARS-CoV-2) pair of Katalin Karikó, PhD, and Drew Weissman, MD, who worked together in the early 2000s to modify mRNA “so it could avoid immediate immune detection, remain active longer and efficiently instruct cells to create antigens to protect against severe disease.” Their discoveries eventually led to the use of modified mRNA in the COVID-19 vaccines.
The second, albeit posthumous, physician-inductee is Patricia Bath, MD, who was the first Black female physician to receive a U.S. patent for a medical invention. The laserphaco device and technique to remove cataracts “performed all steps of cataract removal: making the incision, destroying the lens, and vacuuming out the fractured pieces.”
Two other inductees have somewhat tenuous connections to medical care. Lonnie Johnson invented the Super Soaker, a powerful squirt gun that has been criticized by psychologists for encouraging violence, and Carl Benz invented the automobile, which sort of means he invented the ambulance, so there you go.
The induction ceremony takes place on May 5, 2022, in Washington, DC. If you’re attending the black-tie dinner at The Anthem, let us know and we’ll split an Uber. It’s our only night to be fancy.
Upadacitinib meets primary endpoints for improvement in ankylosing spondylitis and nonradiographic disease
The selective and reversible Janus kinase inhibitor upadacitinib (Rinvoq) significantly improved symptoms in adults with either ankylosing spondylitis (AS) or nonradiographic axial spondyloarthritis (nr-axSpA) when compared with placebo in a pair of studies from the phase 3 SELECT-AXIS 2 clinical trial, according to press releases issued Oct. 7 by manufacturer AbbVie.
Upadacitinib is currently approved in the European Union for patients with active AS, as well as patients with moderate to severe active rheumatoid arthritis and active psoriatic arthritis. Upadacitinib is approved by the Food and Drug Administration for adults with moderately to severely active RA, but is not currently approved for active AS or nr-axSpA.
In study 1, significantly more patients with AS who were randomly assigned to upadacitinib 15 mg once daily met the primary endpoint of 40% improvement in Assessment in Spondyloarthritis International Society (ASAS 40) response criteria at week 14, compared with placebo (45% vs. 18%) after 14 weeks (P < .0001).
The study of 420 patients with an inadequate response to biologic disease-modifying antirheumatic drug therapy gave half upadacitinib for 104 weeks and the other half placebo for 14 weeks, followed by upadacitinib for 90 weeks.
Patients treated with upadacitinib showed significant improvements in secondary endpoints of back pain, inflammation, physical function, and disease activity at week 14, compared with placebo.
Significantly more upadacitinib- than placebo-treated patients reached low disease activity on the AS Disease Activity Score (ASDAS) (44% vs. 10%). Upadacitinib also led to significantly greater improvements from baseline than did placebo on MRI Spondyloarthritis Research Consortium of Canada (SPARCC) Score for Spine, Patient’s Assessment of Total Back Pain, and Bath AS Functional Index (BASFI) score (–2.26 vs. –1.09).
COVID-19 and headache were the most common adverse events that were seen with upadacitinib during the first 14 weeks of the study (occurring in 3% or more). No adverse events led to study discontinuation among patients taking upadacitinib, compared with 1.4% on placebo, and serious adverse events were reported in 2.8% taking upadacitinib and in 0.5% on placebo. Serious infections with upadacitinib included four cases of COVID-19 and one case of uveitis.
Study 2 in patients with nr-axSpA
Study 2, which included 313 adults with nr-axSpA, yielded results similar to those of study 1 on the primary endpoint of meeting ASAS40 response criteria at week 14 (45% with upadacitinib 15 mg once daily vs. 23% with placebo; P < .0001), as well as on a variety of secondary efficacy endpoints and safety data.
Significantly better responses were observed at week 14 with upadacitinib for rate of low disease activity according to ASDAS (42% vs. 18%), changes in MRI SPARCC Scores for SI joints (–2.49 vs. 0.57), Patient’s Assessment of Total Back Pain (-2.91 vs. -2.00), and physical function based on the BASFI (–2.61 vs. –1.47).
The most common adverse events at 14 weeks, occurring in at least 3% of patients taking upadacitinib, included headache, COVID-19, nasopharyngitis, and nausea. Adverse events leading to study discontinuation occurred in 2.6% with upadacitinib and 1.3% with placebo; serious adverse events occurred in 2.6% and 1.3%, respectively.
Serious infections included COVID-19-induced pneumonia and pyelonephritis in patients taking upadacitinib and one case of hemorrhagic fever with renal syndrome with placebo.
The full results of the SELECT-AXIS 2 trial will be presented at a future medical meeting and submitted for publication in a peer-reviewed journal, according to AbbVie.
The selective and reversible Janus kinase inhibitor upadacitinib (Rinvoq) significantly improved symptoms in adults with either ankylosing spondylitis (AS) or nonradiographic axial spondyloarthritis (nr-axSpA) when compared with placebo in a pair of studies from the phase 3 SELECT-AXIS 2 clinical trial, according to press releases issued Oct. 7 by manufacturer AbbVie.
Upadacitinib is currently approved in the European Union for patients with active AS, as well as patients with moderate to severe active rheumatoid arthritis and active psoriatic arthritis. Upadacitinib is approved by the Food and Drug Administration for adults with moderately to severely active RA, but is not currently approved for active AS or nr-axSpA.
In study 1, significantly more patients with AS who were randomly assigned to upadacitinib 15 mg once daily met the primary endpoint of 40% improvement in Assessment in Spondyloarthritis International Society (ASAS 40) response criteria at week 14, compared with placebo (45% vs. 18%) after 14 weeks (P < .0001).
The study of 420 patients with an inadequate response to biologic disease-modifying antirheumatic drug therapy gave half upadacitinib for 104 weeks and the other half placebo for 14 weeks, followed by upadacitinib for 90 weeks.
Patients treated with upadacitinib showed significant improvements in secondary endpoints of back pain, inflammation, physical function, and disease activity at week 14, compared with placebo.
Significantly more upadacitinib- than placebo-treated patients reached low disease activity on the AS Disease Activity Score (ASDAS) (44% vs. 10%). Upadacitinib also led to significantly greater improvements from baseline than did placebo on MRI Spondyloarthritis Research Consortium of Canada (SPARCC) Score for Spine, Patient’s Assessment of Total Back Pain, and Bath AS Functional Index (BASFI) score (–2.26 vs. –1.09).
COVID-19 and headache were the most common adverse events that were seen with upadacitinib during the first 14 weeks of the study (occurring in 3% or more). No adverse events led to study discontinuation among patients taking upadacitinib, compared with 1.4% on placebo, and serious adverse events were reported in 2.8% taking upadacitinib and in 0.5% on placebo. Serious infections with upadacitinib included four cases of COVID-19 and one case of uveitis.
Study 2 in patients with nr-axSpA
Study 2, which included 313 adults with nr-axSpA, yielded results similar to those of study 1 on the primary endpoint of meeting ASAS40 response criteria at week 14 (45% with upadacitinib 15 mg once daily vs. 23% with placebo; P < .0001), as well as on a variety of secondary efficacy endpoints and safety data.
Significantly better responses were observed at week 14 with upadacitinib for rate of low disease activity according to ASDAS (42% vs. 18%), changes in MRI SPARCC Scores for SI joints (–2.49 vs. 0.57), Patient’s Assessment of Total Back Pain (-2.91 vs. -2.00), and physical function based on the BASFI (–2.61 vs. –1.47).
The most common adverse events at 14 weeks, occurring in at least 3% of patients taking upadacitinib, included headache, COVID-19, nasopharyngitis, and nausea. Adverse events leading to study discontinuation occurred in 2.6% with upadacitinib and 1.3% with placebo; serious adverse events occurred in 2.6% and 1.3%, respectively.
Serious infections included COVID-19-induced pneumonia and pyelonephritis in patients taking upadacitinib and one case of hemorrhagic fever with renal syndrome with placebo.
The full results of the SELECT-AXIS 2 trial will be presented at a future medical meeting and submitted for publication in a peer-reviewed journal, according to AbbVie.
The selective and reversible Janus kinase inhibitor upadacitinib (Rinvoq) significantly improved symptoms in adults with either ankylosing spondylitis (AS) or nonradiographic axial spondyloarthritis (nr-axSpA) when compared with placebo in a pair of studies from the phase 3 SELECT-AXIS 2 clinical trial, according to press releases issued Oct. 7 by manufacturer AbbVie.
Upadacitinib is currently approved in the European Union for patients with active AS, as well as patients with moderate to severe active rheumatoid arthritis and active psoriatic arthritis. Upadacitinib is approved by the Food and Drug Administration for adults with moderately to severely active RA, but is not currently approved for active AS or nr-axSpA.
In study 1, significantly more patients with AS who were randomly assigned to upadacitinib 15 mg once daily met the primary endpoint of 40% improvement in Assessment in Spondyloarthritis International Society (ASAS 40) response criteria at week 14, compared with placebo (45% vs. 18%) after 14 weeks (P < .0001).
The study of 420 patients with an inadequate response to biologic disease-modifying antirheumatic drug therapy gave half upadacitinib for 104 weeks and the other half placebo for 14 weeks, followed by upadacitinib for 90 weeks.
Patients treated with upadacitinib showed significant improvements in secondary endpoints of back pain, inflammation, physical function, and disease activity at week 14, compared with placebo.
Significantly more upadacitinib- than placebo-treated patients reached low disease activity on the AS Disease Activity Score (ASDAS) (44% vs. 10%). Upadacitinib also led to significantly greater improvements from baseline than did placebo on MRI Spondyloarthritis Research Consortium of Canada (SPARCC) Score for Spine, Patient’s Assessment of Total Back Pain, and Bath AS Functional Index (BASFI) score (–2.26 vs. –1.09).
COVID-19 and headache were the most common adverse events that were seen with upadacitinib during the first 14 weeks of the study (occurring in 3% or more). No adverse events led to study discontinuation among patients taking upadacitinib, compared with 1.4% on placebo, and serious adverse events were reported in 2.8% taking upadacitinib and in 0.5% on placebo. Serious infections with upadacitinib included four cases of COVID-19 and one case of uveitis.
Study 2 in patients with nr-axSpA
Study 2, which included 313 adults with nr-axSpA, yielded results similar to those of study 1 on the primary endpoint of meeting ASAS40 response criteria at week 14 (45% with upadacitinib 15 mg once daily vs. 23% with placebo; P < .0001), as well as on a variety of secondary efficacy endpoints and safety data.
Significantly better responses were observed at week 14 with upadacitinib for rate of low disease activity according to ASDAS (42% vs. 18%), changes in MRI SPARCC Scores for SI joints (–2.49 vs. 0.57), Patient’s Assessment of Total Back Pain (-2.91 vs. -2.00), and physical function based on the BASFI (–2.61 vs. –1.47).
The most common adverse events at 14 weeks, occurring in at least 3% of patients taking upadacitinib, included headache, COVID-19, nasopharyngitis, and nausea. Adverse events leading to study discontinuation occurred in 2.6% with upadacitinib and 1.3% with placebo; serious adverse events occurred in 2.6% and 1.3%, respectively.
Serious infections included COVID-19-induced pneumonia and pyelonephritis in patients taking upadacitinib and one case of hemorrhagic fever with renal syndrome with placebo.
The full results of the SELECT-AXIS 2 trial will be presented at a future medical meeting and submitted for publication in a peer-reviewed journal, according to AbbVie.
Itchy vesicles
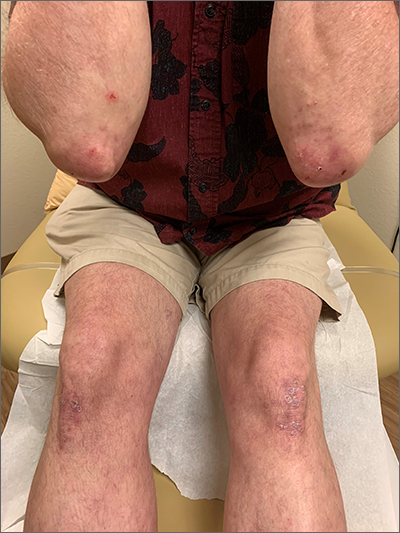
The patient’s history of celiac disease and the presence of vesicular lesions affecting primarily extensor surfaces pointed to a diagnosis of dermatitis herpetiformis (DH).
DH is an autoimmune skin condition that is associated with gluten sensitivity. It is more frequent in individuals of northern European heritage.1
The lesions associated with DH are intensely pruritic grouped papules, vesicles, and tense blisters that appear more commonly on extensor areas of the lower limbs, elbows, buttocks, and sacral region. Involvement of the oral mucosa is rare and not all patients with DH experience the intestinal symptoms of gluten sensitivity.
Direct immunofluorescence of perilesional skin is the gold standard to confirm the diagnosis. Histology of the lesions is also performed, but findings may vary depending on the age of the lesion. Additionally, serology can help to confirm the diagnosis. Both tissue and epidermal transglutaminase antibodies are often present in the serum but may be negative if the patient is following a gluten-free diet. In this case, biopsy was not ordered because the patient already had a biopsy-confirmed diagnosis of celiac disease and a classic presentation of lesions.
Treatment of DH consists of a strict gluten-free diet and dapsone as first-line pharmacologic therapy. Typically, dapsone is started at doses of 25 to 50 mg daily and increased, as needed and tolerated, to 100 to 200 mg per day. Improvement is usually seen within 2 days of treatment initiation. Dapsone has multiple potential adverse effects; the most common is hemolysis. Since individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency can develop severe hemolysis if treated with dapsone, it is necessary to screen for this condition prior to initiation of treatment. A complete blood count and liver and renal function testing are typically done before, and during, treatment with dapsone.1
This patient had normal levels of G6PD and his screening labs were also normal. He was started on dapsone orally 25 mg/d and follow-up was pending.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Marcella Colom, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
1. Mendes FBR, Hissa-Elian A, de Abreu MAMM, et al. Review: dermatitis herpetiformis. An Bras Dermatol. 2013;88:594-599. doi:10.1590/abd1806-4841.20131775
The patient’s history of celiac disease and the presence of vesicular lesions affecting primarily extensor surfaces pointed to a diagnosis of dermatitis herpetiformis (DH).
DH is an autoimmune skin condition that is associated with gluten sensitivity. It is more frequent in individuals of northern European heritage.1
The lesions associated with DH are intensely pruritic grouped papules, vesicles, and tense blisters that appear more commonly on extensor areas of the lower limbs, elbows, buttocks, and sacral region. Involvement of the oral mucosa is rare and not all patients with DH experience the intestinal symptoms of gluten sensitivity.
Direct immunofluorescence of perilesional skin is the gold standard to confirm the diagnosis. Histology of the lesions is also performed, but findings may vary depending on the age of the lesion. Additionally, serology can help to confirm the diagnosis. Both tissue and epidermal transglutaminase antibodies are often present in the serum but may be negative if the patient is following a gluten-free diet. In this case, biopsy was not ordered because the patient already had a biopsy-confirmed diagnosis of celiac disease and a classic presentation of lesions.
Treatment of DH consists of a strict gluten-free diet and dapsone as first-line pharmacologic therapy. Typically, dapsone is started at doses of 25 to 50 mg daily and increased, as needed and tolerated, to 100 to 200 mg per day. Improvement is usually seen within 2 days of treatment initiation. Dapsone has multiple potential adverse effects; the most common is hemolysis. Since individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency can develop severe hemolysis if treated with dapsone, it is necessary to screen for this condition prior to initiation of treatment. A complete blood count and liver and renal function testing are typically done before, and during, treatment with dapsone.1
This patient had normal levels of G6PD and his screening labs were also normal. He was started on dapsone orally 25 mg/d and follow-up was pending.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Marcella Colom, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
The patient’s history of celiac disease and the presence of vesicular lesions affecting primarily extensor surfaces pointed to a diagnosis of dermatitis herpetiformis (DH).
DH is an autoimmune skin condition that is associated with gluten sensitivity. It is more frequent in individuals of northern European heritage.1
The lesions associated with DH are intensely pruritic grouped papules, vesicles, and tense blisters that appear more commonly on extensor areas of the lower limbs, elbows, buttocks, and sacral region. Involvement of the oral mucosa is rare and not all patients with DH experience the intestinal symptoms of gluten sensitivity.
Direct immunofluorescence of perilesional skin is the gold standard to confirm the diagnosis. Histology of the lesions is also performed, but findings may vary depending on the age of the lesion. Additionally, serology can help to confirm the diagnosis. Both tissue and epidermal transglutaminase antibodies are often present in the serum but may be negative if the patient is following a gluten-free diet. In this case, biopsy was not ordered because the patient already had a biopsy-confirmed diagnosis of celiac disease and a classic presentation of lesions.
Treatment of DH consists of a strict gluten-free diet and dapsone as first-line pharmacologic therapy. Typically, dapsone is started at doses of 25 to 50 mg daily and increased, as needed and tolerated, to 100 to 200 mg per day. Improvement is usually seen within 2 days of treatment initiation. Dapsone has multiple potential adverse effects; the most common is hemolysis. Since individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency can develop severe hemolysis if treated with dapsone, it is necessary to screen for this condition prior to initiation of treatment. A complete blood count and liver and renal function testing are typically done before, and during, treatment with dapsone.1
This patient had normal levels of G6PD and his screening labs were also normal. He was started on dapsone orally 25 mg/d and follow-up was pending.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Marcella Colom, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
1. Mendes FBR, Hissa-Elian A, de Abreu MAMM, et al. Review: dermatitis herpetiformis. An Bras Dermatol. 2013;88:594-599. doi:10.1590/abd1806-4841.20131775
1. Mendes FBR, Hissa-Elian A, de Abreu MAMM, et al. Review: dermatitis herpetiformis. An Bras Dermatol. 2013;88:594-599. doi:10.1590/abd1806-4841.20131775
Lower thyroid hormone levels a red flag for elevated suicide risk?
Patients with comorbid anxiety and mood disorders who have reduced, albeit “normal” serum levels of thyroid-stimulating hormone (TSH) may be at increased risk for suicidal ideation, new research suggests.

In a cross-sectional study, clinical data on diagnosis, medication use, and symptom scores were gathered, along with assessments of blood levels of thyroid axis hormones, in patients with both anxiety and mood disorders.
After investigators accounted for age, gender, symptoms, medication use, and other potential confounders, patients with suicidal ideation were 54% less likely to have higher TSH levels. There was no association found with other thyroid hormones.
Based on the results, the assessment of thyroid hormone levels “may be important for suicide prevention and might allow clinicians to evaluate the potential of the suicidal ideation risk in individuals with [anxiety and mood disorders],” co-investigator Vilma Liaugaudaite, PhD student, Neuroscience Institute of the Lithuanian University of Health Sciences, Palanga, and colleagues note.
The findings were presented at the 34th European College of Neuropsychopharmacology (ECNP) Congress.
‘Complex mechanism’
Ms. Liaugaudaite told this news organization that thyroid hormones are known to have a “profound” effect on mood and behavior.
Recent studies show “various degrees of hypothalamic-pituitary-thyroid axis dysregulation are associated with suicidal behavior” in patients with depression, she added.
Noting that disturbances in the serotonin system “constitute the most common biochemical abnormality associated with suicidal behavior,” Ms. Liaugaudaite said it is thought thyroid hormones “are involved in a complex compensatory mechanism to correct reduced central 5-hydroxytryptamine activity” via lower TSH levels.
In addition, hypersecretion of thyrotropin-releasing hormone, which stimulates the release of TSH, “has been considered a compensatory mechanism to maintain normal thyroid hormone secretion and normalize serotonin activity in depressed patients,” she said.
To investigate associations between thyroid axis hormones and suicidality in individuals with comorbid anxiety and mood disorders, the researchers assessed consecutive patients attending a stress disorders clinic.
Sociodemographic and clinical information was gathered, and patients completed the Mini International Neuropsychiatric Interview, the Patient Health Questionnaire-9 (PHQ-9), and the General Anxiety Disorder-7 (GAD-7) scale.
Fasting blood samples were also tested for free thyroxine (FT4), free triiodothyronine (FT3), and TSH levels.
Significant association
Suicidal ideation was identified in 42 participants. Serum FT4, FT3, and TSH levels were within the normal range.

There were no significant differences between patients with and without suicidal ideation in terms of age, gender, education, obesity, smoking, and medication use.
Suicidal ideation was associated with higher scores on the PHQ-9 (15.5 vs. 13.3; P = .085), and with lower TSH levels (1.54 IU/L vs. 2.04 IU/L; P = .092).
The association between serum TSH levels and suicidal ideation was significant after multivariate logistic regression analysis accounted for age, gender, PHQ-9 and GAD-7 scores, education, body mass index, smoking, and use of antidepressants, tranquilizers, mood stabilizers, and neuroleptics.
Specifically, patients with suicidal ideation were significantly less likely to have higher TSH levels than those without, at an odds ratio of 0.46 (P = .027).
There were no significant associations between serum FT4 and FT3 levels and suicidal ideation.
Interesting, but preliminary
Commenting on the findings, Sanjeev Sockalingam, MD, vice chair and professor of psychiatry at the University of Toronto, said it is an “interesting study” because the literature on trying to identify individuals at risk for suicidal ideation or behaviors is “quite mixed, in terms of the results.”
However, it was a cross-sectional study with a relatively small sample size, and studies of this nature typically include patients with hypothyroidism “who end up having suicidal thoughts,” said Dr. Sockalingam, who was not involved with the research.
“I do wonder, given the sample size and patient population, if there may be other factors that may have been related to this,” he added.
Dr. Sockalingam noted that he would like to see more data on the medications the patients were taking, and he underlined that the thyroid levels were in the normal range, “so it’s a bit difficult to untangle what that means in terms of these subtle changes in thyroid levels.”
Robert Levitan, MD, Cameron Wilson Chair in Depression Research at the Centre for Addiction and Mental Health, Toronto, also emphasized that the thyroid levels were in the normal range.
He commented that it therefore “seems unlikely that there’s going to be some biological effect that’s going to affect the brain in a significant enough way” to influence suicidal ideation.
Dr. Levitan continued, “What’s probably happening is there’s some other clinical issue here that they just haven’t picked up on that’s leading in one direction to the suicidal ideation and perhaps affecting the TSH to some extent.”
Although the study is, therefore, “preliminary,” the findings are nevertheless “interesting,” he concluded.
The study received no funding. Ms. Liaugaudaite, Dr. Sockalingam, and Dr. Levitan have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Patients with comorbid anxiety and mood disorders who have reduced, albeit “normal” serum levels of thyroid-stimulating hormone (TSH) may be at increased risk for suicidal ideation, new research suggests.
In a cross-sectional study, clinical data on diagnosis, medication use, and symptom scores were gathered, along with assessments of blood levels of thyroid axis hormones, in patients with both anxiety and mood disorders.
After investigators accounted for age, gender, symptoms, medication use, and other potential confounders, patients with suicidal ideation were 54% less likely to have higher TSH levels. There was no association found with other thyroid hormones.
Based on the results, the assessment of thyroid hormone levels “may be important for suicide prevention and might allow clinicians to evaluate the potential of the suicidal ideation risk in individuals with [anxiety and mood disorders],” co-investigator Vilma Liaugaudaite, PhD student, Neuroscience Institute of the Lithuanian University of Health Sciences, Palanga, and colleagues note.
The findings were presented at the 34th European College of Neuropsychopharmacology (ECNP) Congress.
‘Complex mechanism’
Ms. Liaugaudaite told this news organization that thyroid hormones are known to have a “profound” effect on mood and behavior.
Recent studies show “various degrees of hypothalamic-pituitary-thyroid axis dysregulation are associated with suicidal behavior” in patients with depression, she added.
Noting that disturbances in the serotonin system “constitute the most common biochemical abnormality associated with suicidal behavior,” Ms. Liaugaudaite said it is thought thyroid hormones “are involved in a complex compensatory mechanism to correct reduced central 5-hydroxytryptamine activity” via lower TSH levels.
In addition, hypersecretion of thyrotropin-releasing hormone, which stimulates the release of TSH, “has been considered a compensatory mechanism to maintain normal thyroid hormone secretion and normalize serotonin activity in depressed patients,” she said.
To investigate associations between thyroid axis hormones and suicidality in individuals with comorbid anxiety and mood disorders, the researchers assessed consecutive patients attending a stress disorders clinic.
Sociodemographic and clinical information was gathered, and patients completed the Mini International Neuropsychiatric Interview, the Patient Health Questionnaire-9 (PHQ-9), and the General Anxiety Disorder-7 (GAD-7) scale.
Fasting blood samples were also tested for free thyroxine (FT4), free triiodothyronine (FT3), and TSH levels.
Significant association
Suicidal ideation was identified in 42 participants. Serum FT4, FT3, and TSH levels were within the normal range.
There were no significant differences between patients with and without suicidal ideation in terms of age, gender, education, obesity, smoking, and medication use.
Suicidal ideation was associated with higher scores on the PHQ-9 (15.5 vs. 13.3; P = .085), and with lower TSH levels (1.54 IU/L vs. 2.04 IU/L; P = .092).
The association between serum TSH levels and suicidal ideation was significant after multivariate logistic regression analysis accounted for age, gender, PHQ-9 and GAD-7 scores, education, body mass index, smoking, and use of antidepressants, tranquilizers, mood stabilizers, and neuroleptics.
Specifically, patients with suicidal ideation were significantly less likely to have higher TSH levels than those without, at an odds ratio of 0.46 (P = .027).
There were no significant associations between serum FT4 and FT3 levels and suicidal ideation.
Interesting, but preliminary
Commenting on the findings, Sanjeev Sockalingam, MD, vice chair and professor of psychiatry at the University of Toronto, said it is an “interesting study” because the literature on trying to identify individuals at risk for suicidal ideation or behaviors is “quite mixed, in terms of the results.”
However, it was a cross-sectional study with a relatively small sample size, and studies of this nature typically include patients with hypothyroidism “who end up having suicidal thoughts,” said Dr. Sockalingam, who was not involved with the research.
“I do wonder, given the sample size and patient population, if there may be other factors that may have been related to this,” he added.
Dr. Sockalingam noted that he would like to see more data on the medications the patients were taking, and he underlined that the thyroid levels were in the normal range, “so it’s a bit difficult to untangle what that means in terms of these subtle changes in thyroid levels.”
Robert Levitan, MD, Cameron Wilson Chair in Depression Research at the Centre for Addiction and Mental Health, Toronto, also emphasized that the thyroid levels were in the normal range.
He commented that it therefore “seems unlikely that there’s going to be some biological effect that’s going to affect the brain in a significant enough way” to influence suicidal ideation.
Dr. Levitan continued, “What’s probably happening is there’s some other clinical issue here that they just haven’t picked up on that’s leading in one direction to the suicidal ideation and perhaps affecting the TSH to some extent.”
Although the study is, therefore, “preliminary,” the findings are nevertheless “interesting,” he concluded.
The study received no funding. Ms. Liaugaudaite, Dr. Sockalingam, and Dr. Levitan have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Patients with comorbid anxiety and mood disorders who have reduced, albeit “normal” serum levels of thyroid-stimulating hormone (TSH) may be at increased risk for suicidal ideation, new research suggests.
In a cross-sectional study, clinical data on diagnosis, medication use, and symptom scores were gathered, along with assessments of blood levels of thyroid axis hormones, in patients with both anxiety and mood disorders.
After investigators accounted for age, gender, symptoms, medication use, and other potential confounders, patients with suicidal ideation were 54% less likely to have higher TSH levels. There was no association found with other thyroid hormones.
Based on the results, the assessment of thyroid hormone levels “may be important for suicide prevention and might allow clinicians to evaluate the potential of the suicidal ideation risk in individuals with [anxiety and mood disorders],” co-investigator Vilma Liaugaudaite, PhD student, Neuroscience Institute of the Lithuanian University of Health Sciences, Palanga, and colleagues note.
The findings were presented at the 34th European College of Neuropsychopharmacology (ECNP) Congress.
‘Complex mechanism’
Ms. Liaugaudaite told this news organization that thyroid hormones are known to have a “profound” effect on mood and behavior.
Recent studies show “various degrees of hypothalamic-pituitary-thyroid axis dysregulation are associated with suicidal behavior” in patients with depression, she added.
Noting that disturbances in the serotonin system “constitute the most common biochemical abnormality associated with suicidal behavior,” Ms. Liaugaudaite said it is thought thyroid hormones “are involved in a complex compensatory mechanism to correct reduced central 5-hydroxytryptamine activity” via lower TSH levels.
In addition, hypersecretion of thyrotropin-releasing hormone, which stimulates the release of TSH, “has been considered a compensatory mechanism to maintain normal thyroid hormone secretion and normalize serotonin activity in depressed patients,” she said.
To investigate associations between thyroid axis hormones and suicidality in individuals with comorbid anxiety and mood disorders, the researchers assessed consecutive patients attending a stress disorders clinic.
Sociodemographic and clinical information was gathered, and patients completed the Mini International Neuropsychiatric Interview, the Patient Health Questionnaire-9 (PHQ-9), and the General Anxiety Disorder-7 (GAD-7) scale.
Fasting blood samples were also tested for free thyroxine (FT4), free triiodothyronine (FT3), and TSH levels.
Significant association
Suicidal ideation was identified in 42 participants. Serum FT4, FT3, and TSH levels were within the normal range.
There were no significant differences between patients with and without suicidal ideation in terms of age, gender, education, obesity, smoking, and medication use.
Suicidal ideation was associated with higher scores on the PHQ-9 (15.5 vs. 13.3; P = .085), and with lower TSH levels (1.54 IU/L vs. 2.04 IU/L; P = .092).
The association between serum TSH levels and suicidal ideation was significant after multivariate logistic regression analysis accounted for age, gender, PHQ-9 and GAD-7 scores, education, body mass index, smoking, and use of antidepressants, tranquilizers, mood stabilizers, and neuroleptics.
Specifically, patients with suicidal ideation were significantly less likely to have higher TSH levels than those without, at an odds ratio of 0.46 (P = .027).
There were no significant associations between serum FT4 and FT3 levels and suicidal ideation.
Interesting, but preliminary
Commenting on the findings, Sanjeev Sockalingam, MD, vice chair and professor of psychiatry at the University of Toronto, said it is an “interesting study” because the literature on trying to identify individuals at risk for suicidal ideation or behaviors is “quite mixed, in terms of the results.”
However, it was a cross-sectional study with a relatively small sample size, and studies of this nature typically include patients with hypothyroidism “who end up having suicidal thoughts,” said Dr. Sockalingam, who was not involved with the research.
“I do wonder, given the sample size and patient population, if there may be other factors that may have been related to this,” he added.
Dr. Sockalingam noted that he would like to see more data on the medications the patients were taking, and he underlined that the thyroid levels were in the normal range, “so it’s a bit difficult to untangle what that means in terms of these subtle changes in thyroid levels.”
Robert Levitan, MD, Cameron Wilson Chair in Depression Research at the Centre for Addiction and Mental Health, Toronto, also emphasized that the thyroid levels were in the normal range.
He commented that it therefore “seems unlikely that there’s going to be some biological effect that’s going to affect the brain in a significant enough way” to influence suicidal ideation.
Dr. Levitan continued, “What’s probably happening is there’s some other clinical issue here that they just haven’t picked up on that’s leading in one direction to the suicidal ideation and perhaps affecting the TSH to some extent.”
Although the study is, therefore, “preliminary,” the findings are nevertheless “interesting,” he concluded.
The study received no funding. Ms. Liaugaudaite, Dr. Sockalingam, and Dr. Levitan have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ECNP 2021
WHO unveils global roadmap to defeat meningitis by 2030
The World Health Organization and its partners recently released an ambitious plan, Defeating meningitis by 2030: A global road map. The goal is to reduce deaths and disabilities from bacterial meningitis, which kills about 250,000 people annually of the 1.2 million estimated to be infected.
This type of infection around the brain and spinal cord also causes long-term disabilities – deafness, learning problems, seizures, loss of limbs – in about one-quarter of survivors.
The leading causes of bacterial meningitis are Neisseria meningitidis (meningococcus), Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, and group B streptococcus. As with malaria, about half of the cases are in children under age 5 years. The most severely affected area for both infections is sub-Saharan Africa.
The main goal of the roadmap is to reduce vaccine-preventable cases of bacterial meningitis by 50% and deaths by 70% by 2030. WHO’s partners included the Centers for Disease Control and Prevention, the London School of Hygiene and Tropical Medicine, Médecins Sans Frontières (Doctors Without Borders), the Meningitis Research Foundation, PATH, UNICEF, and numerous global consultants.
For primary prevention and epidemic control, a major goal is to achieve higher vaccine coverage. Another goal is developing and deploying rapid diagnostic tests to guide treatment and prevention activities and measure the impact of vaccination. The lack of laboratory capacity to confirm the bacteria is a significant challenge. Also, patients often receive antibiotics before appropriate tests are conducted, and lumbar punctures are frequently not done.
The commitment to this project emerged in 2017. It was followed by a baseline analysis in 2018 and a draft roadmap the following year. Consultations with experts and with more than 600 patient groups in more than 90 countries followed.
Prevention through greater vaccine uptake was the top priority. Vaccination is considered the first line of defense against antibiotic resistance among the targeted bacteria.
Another goal is to quantify the decrease in antibiotic use for invasive infections or prophylaxis and the subsequent reduction in antimicrobial resistance in relation to increased vaccination.
Surveillance is weak in many regions, limiting the ability to detect epidemics and to respond appropriately. Similarly, there are limited data on the burden of sequelae, such as deafness, on meningitis survivors.
There is an inadequate supply of affordable vaccines to respond to epidemics. Currently, routine vaccination against Neisseria meningitidis is occurring in 18 of 26 countries in the meningitis belt. Epidemics of meningococcus occur every few years in the driest time of the year and abate with the rains. Epidemics of pneumococcal meningitis are much rarer but follow a similar pattern; they have also been associated with crowding and alcohol use.
Care for those affected by meningitis is another focus, as is affirming the right to prevention and care. There’s a need for earlier recognition of the complications of meningitis and an increase in efforts to treat those complications.
WHO’s final goal in its roadmap is to boost awareness of meningitis and make it a priority for policymakers. Similarly, there is a need to educate communities about the disease, including how to access vaccines. If someone becomes ill, they need to be aware of the symptoms, the need for early treatment, and what aftercare is available.
Marie-Pierre Préziosi, MD, the core secretariat of WHO’s Technical Taskforce, told this news organization that while the roadmap looks aspirational, “it is feasible … you have strategic goals – each has milestones with time limits and who will do it.”
Regarding vaccinations, Dr. Préziosi said that “the strategy was a victim of its success. The mass campaign knocked down transmission completely.” Some countries are now waiting for multivalent vaccines. She said that vaccine hesitancy is not a significant problem in Africa “because the disease is so feared.”
Major obstacles to implementing the roadmap include the complacency of public health leaders and the COVID-19 lockdowns, which decreased vaccination coverage rates. “The second thing is also sufficient funding to do the research and innovation so that we get the affordable tools that we need globally,” Dr. Préziosi said.
Marilyn Felkner, DrPH, School of Human Ecology, University of Texas at Austin, said in an interview, “It’s very cliché, but we have often said that communicable diseases do not respect political boundaries. So to expect a country to be able to control that by themselves is a false hope.”
Regarding the roadmap, Dr. Felkner said, “I think that organizing ideas and having them in writing is always a good first step. And it can help people move forward if they’re feeling overwhelmed ... Having a written plan can certainly provide that fundamental basis. So, the important thing is not to say, ‘Oh, we have this great plan done; hope somebody picks up the plan.’ There’s got to be some momentum behind it, and hopefully some funding.”
Dr. Préziosi and Dr. Felkner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The World Health Organization and its partners recently released an ambitious plan, Defeating meningitis by 2030: A global road map. The goal is to reduce deaths and disabilities from bacterial meningitis, which kills about 250,000 people annually of the 1.2 million estimated to be infected.
This type of infection around the brain and spinal cord also causes long-term disabilities – deafness, learning problems, seizures, loss of limbs – in about one-quarter of survivors.
The leading causes of bacterial meningitis are Neisseria meningitidis (meningococcus), Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, and group B streptococcus. As with malaria, about half of the cases are in children under age 5 years. The most severely affected area for both infections is sub-Saharan Africa.
The main goal of the roadmap is to reduce vaccine-preventable cases of bacterial meningitis by 50% and deaths by 70% by 2030. WHO’s partners included the Centers for Disease Control and Prevention, the London School of Hygiene and Tropical Medicine, Médecins Sans Frontières (Doctors Without Borders), the Meningitis Research Foundation, PATH, UNICEF, and numerous global consultants.
For primary prevention and epidemic control, a major goal is to achieve higher vaccine coverage. Another goal is developing and deploying rapid diagnostic tests to guide treatment and prevention activities and measure the impact of vaccination. The lack of laboratory capacity to confirm the bacteria is a significant challenge. Also, patients often receive antibiotics before appropriate tests are conducted, and lumbar punctures are frequently not done.
The commitment to this project emerged in 2017. It was followed by a baseline analysis in 2018 and a draft roadmap the following year. Consultations with experts and with more than 600 patient groups in more than 90 countries followed.
Prevention through greater vaccine uptake was the top priority. Vaccination is considered the first line of defense against antibiotic resistance among the targeted bacteria.
Another goal is to quantify the decrease in antibiotic use for invasive infections or prophylaxis and the subsequent reduction in antimicrobial resistance in relation to increased vaccination.
Surveillance is weak in many regions, limiting the ability to detect epidemics and to respond appropriately. Similarly, there are limited data on the burden of sequelae, such as deafness, on meningitis survivors.
There is an inadequate supply of affordable vaccines to respond to epidemics. Currently, routine vaccination against Neisseria meningitidis is occurring in 18 of 26 countries in the meningitis belt. Epidemics of meningococcus occur every few years in the driest time of the year and abate with the rains. Epidemics of pneumococcal meningitis are much rarer but follow a similar pattern; they have also been associated with crowding and alcohol use.
Care for those affected by meningitis is another focus, as is affirming the right to prevention and care. There’s a need for earlier recognition of the complications of meningitis and an increase in efforts to treat those complications.
WHO’s final goal in its roadmap is to boost awareness of meningitis and make it a priority for policymakers. Similarly, there is a need to educate communities about the disease, including how to access vaccines. If someone becomes ill, they need to be aware of the symptoms, the need for early treatment, and what aftercare is available.
Marie-Pierre Préziosi, MD, the core secretariat of WHO’s Technical Taskforce, told this news organization that while the roadmap looks aspirational, “it is feasible … you have strategic goals – each has milestones with time limits and who will do it.”
Regarding vaccinations, Dr. Préziosi said that “the strategy was a victim of its success. The mass campaign knocked down transmission completely.” Some countries are now waiting for multivalent vaccines. She said that vaccine hesitancy is not a significant problem in Africa “because the disease is so feared.”
Major obstacles to implementing the roadmap include the complacency of public health leaders and the COVID-19 lockdowns, which decreased vaccination coverage rates. “The second thing is also sufficient funding to do the research and innovation so that we get the affordable tools that we need globally,” Dr. Préziosi said.
Marilyn Felkner, DrPH, School of Human Ecology, University of Texas at Austin, said in an interview, “It’s very cliché, but we have often said that communicable diseases do not respect political boundaries. So to expect a country to be able to control that by themselves is a false hope.”
Regarding the roadmap, Dr. Felkner said, “I think that organizing ideas and having them in writing is always a good first step. And it can help people move forward if they’re feeling overwhelmed ... Having a written plan can certainly provide that fundamental basis. So, the important thing is not to say, ‘Oh, we have this great plan done; hope somebody picks up the plan.’ There’s got to be some momentum behind it, and hopefully some funding.”
Dr. Préziosi and Dr. Felkner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The World Health Organization and its partners recently released an ambitious plan, Defeating meningitis by 2030: A global road map. The goal is to reduce deaths and disabilities from bacterial meningitis, which kills about 250,000 people annually of the 1.2 million estimated to be infected.
This type of infection around the brain and spinal cord also causes long-term disabilities – deafness, learning problems, seizures, loss of limbs – in about one-quarter of survivors.
The leading causes of bacterial meningitis are Neisseria meningitidis (meningococcus), Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, and group B streptococcus. As with malaria, about half of the cases are in children under age 5 years. The most severely affected area for both infections is sub-Saharan Africa.
The main goal of the roadmap is to reduce vaccine-preventable cases of bacterial meningitis by 50% and deaths by 70% by 2030. WHO’s partners included the Centers for Disease Control and Prevention, the London School of Hygiene and Tropical Medicine, Médecins Sans Frontières (Doctors Without Borders), the Meningitis Research Foundation, PATH, UNICEF, and numerous global consultants.
For primary prevention and epidemic control, a major goal is to achieve higher vaccine coverage. Another goal is developing and deploying rapid diagnostic tests to guide treatment and prevention activities and measure the impact of vaccination. The lack of laboratory capacity to confirm the bacteria is a significant challenge. Also, patients often receive antibiotics before appropriate tests are conducted, and lumbar punctures are frequently not done.
The commitment to this project emerged in 2017. It was followed by a baseline analysis in 2018 and a draft roadmap the following year. Consultations with experts and with more than 600 patient groups in more than 90 countries followed.
Prevention through greater vaccine uptake was the top priority. Vaccination is considered the first line of defense against antibiotic resistance among the targeted bacteria.
Another goal is to quantify the decrease in antibiotic use for invasive infections or prophylaxis and the subsequent reduction in antimicrobial resistance in relation to increased vaccination.
Surveillance is weak in many regions, limiting the ability to detect epidemics and to respond appropriately. Similarly, there are limited data on the burden of sequelae, such as deafness, on meningitis survivors.
There is an inadequate supply of affordable vaccines to respond to epidemics. Currently, routine vaccination against Neisseria meningitidis is occurring in 18 of 26 countries in the meningitis belt. Epidemics of meningococcus occur every few years in the driest time of the year and abate with the rains. Epidemics of pneumococcal meningitis are much rarer but follow a similar pattern; they have also been associated with crowding and alcohol use.
Care for those affected by meningitis is another focus, as is affirming the right to prevention and care. There’s a need for earlier recognition of the complications of meningitis and an increase in efforts to treat those complications.
WHO’s final goal in its roadmap is to boost awareness of meningitis and make it a priority for policymakers. Similarly, there is a need to educate communities about the disease, including how to access vaccines. If someone becomes ill, they need to be aware of the symptoms, the need for early treatment, and what aftercare is available.
Marie-Pierre Préziosi, MD, the core secretariat of WHO’s Technical Taskforce, told this news organization that while the roadmap looks aspirational, “it is feasible … you have strategic goals – each has milestones with time limits and who will do it.”
Regarding vaccinations, Dr. Préziosi said that “the strategy was a victim of its success. The mass campaign knocked down transmission completely.” Some countries are now waiting for multivalent vaccines. She said that vaccine hesitancy is not a significant problem in Africa “because the disease is so feared.”
Major obstacles to implementing the roadmap include the complacency of public health leaders and the COVID-19 lockdowns, which decreased vaccination coverage rates. “The second thing is also sufficient funding to do the research and innovation so that we get the affordable tools that we need globally,” Dr. Préziosi said.
Marilyn Felkner, DrPH, School of Human Ecology, University of Texas at Austin, said in an interview, “It’s very cliché, but we have often said that communicable diseases do not respect political boundaries. So to expect a country to be able to control that by themselves is a false hope.”
Regarding the roadmap, Dr. Felkner said, “I think that organizing ideas and having them in writing is always a good first step. And it can help people move forward if they’re feeling overwhelmed ... Having a written plan can certainly provide that fundamental basis. So, the important thing is not to say, ‘Oh, we have this great plan done; hope somebody picks up the plan.’ There’s got to be some momentum behind it, and hopefully some funding.”
Dr. Préziosi and Dr. Felkner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Fecal microbiota transplants may improve resistance to melanoma immunotherapy
In the fall of 2020, Hassane M. Zarour, MD, and colleagues began to pore over raw data from their

Preclinical mouse studies have demonstrated that the gut microbiota could influence the response of tumors to anti–PD-1 immunotherapy, but FMT had not been previously evaluated in human patients with malignant melanoma whose disease persisted or progressed after medical therapy. Only 30%-40% of melanoma patients respond to anti–PD-1 immunotherapy, so the researchers’ sense of anticipation was palpable. “It’s a high-risk, high-reward study, so you never know,” Dr. Zarour, a dermatologist and immunologist who is coleader of the melanoma program at the University of Pittsburgh Medical Center’s Hillman Cancer Center, said in an interview.
For the study, which was funded by the National Institutes of Health and published in Science, Dr. Zarour and a team of colleagues, including Diwakar Davar, MD, a medical oncologist/hematologist at UPMC and Giorgio Trinchieri, MD, head of the cancer immunology section at the National Cancer Institute, enrolled 16 patients with advanced melanoma whose disease had persisted or progressed with anti-PD-1 drugs; donors were 7 patients with advanced melanoma who had responded to pembrolizumab, 4 with a complete response and 3 with a partial response, with a median progression-free survival of 56 months.
After donors and patients underwent serial stool sampling and studies to stamp out the potential for transmitting infectious agents, the researchers administered the donor-derived FMT to patients via colonoscopy every 14 days for 3 weeks, followed by pembrolizumab. To their delight, 6 of the 15 evaluable recipients responded to treatment, with a reduction in tumor or long-term disease stabilization. Moreover, responders also showed increased abundance of taxa that were previously associated with response to immunotherapy, increased activation of CD8+ T cells, and decreased frequency of interleukin-8–expressing myeloid cells.
“This opens new doors for the future,” Dr. Zarour said. “It’s very encouraging, but I don’t want to overstate the data. It’s a small, nonrandomized trial, but one has to keep in mind that people were skeptical about this work; they didn’t think FMT would work. Now we see many people coming into the field to investigate the role of the microbiome as a therapeutic tool, which is great.”

Teri Greiling, MD, characterized the finding as a key development in understanding the microbiome’s potential to influence the course of melanoma and other diseases. “What’s emerging over the last decade of research is that our immune system has a close, back-and-forth relationship with our microbiota,” said Dr. Greiling, associate professor of dermatology at Oregon Health & Science University, Portland. “From day 1 of birth, we’re colonized by microbes that train our immune system how to function. In response, your immune system keeps those microbes in check and shapes which ones are allowed to colonize, and which ones are a target for attack. Thus, inflammatory responses are generated. Similarly, the goal of immunotherapy is to activate the immune system to fight cancer. This study shows that the immune system continues to need the colonizing microbes in our body to function optimally.”
Immunotherapy with checkpoint inhibitors was not an option for malignant melanoma patients until 2011, she noted, so the potential for FMT to further improve outcomes is welcome news for patients and their families. “We went from a less than 5% chance of survival with metastatic melanoma to now, with the right combination of checkpoint inhibitors, we’re up over 50%, which is amazing in a decade,” Dr. Greiling said. “Still, we’re losing half of our patients. If [FMT provides] a 30% improvement over that, that would be great, but it’s hard to extrapolate from such small numbers.”
Positive results in an Israeli study
Results from a similar, smaller phase 1 trial of 2 FMT donors and 10 recipients with metastatic melanoma who had progressed on anti-PD-1 therapy, from the Ella Lemelbaum Institute for Immuno-Oncology at Sheba Medical Center in Tel HaShomer, Israel, yielded similar results. The FMT protocol in this study included colonoscopy and oral stool capsules, followed by the reintroduction of anti–PD-1 therapy with nivolumab. The two FMT donors had previously been treated with anti–PD-1 monotherapy for metastatic melanoma and had achieved a clinical response for at least 1 year. Of the 10 FMT recipients, 1 had a complete response and 2 had a partial response.

“We expected changes in the immune system but did not expect that 3 out of the 10 patients in our study would be turned from nonresponders to responders,” the study’s lead author, Erez N. Baruch, MD, PhD, told this news organization. “Since this was a first-in-human study, we were aiming to assess safety and not clinical responses. [We found] that microbiota modulation can change the immune infiltration within melanoma tumors and by this affect response to immunotherapy.”
Dr. Baruch, an internal medicine resident in the physician-scientist track program at the University of Texas, Houston, said that the findings create a potential new therapeutic paradigm, or a new “playing ground” for drug development that can support existing immunotherapies. “It is important for dermatologists to understand that disruptions of the gut microbiota, mainly by antibiotics, may be harmful to melanoma patients,” he said. “Antibiotics in cancer patients should be used judiciously but of course should not be avoided when there’s an indication.”
As for next steps, Dr. Zarour and colleagues are recruiting more patients to boost their sample size and conducting sequential analysis of the microbiome of study participants “to better determine what the good and bad bugs are,” he said. “There are so many variables, including diet and geography. We need more data.” The hope is to develop a “microbiome signature” to identify patients likely to respond to FMT, and maybe one day, a probiotic capsule that patients take to optimize their response to immunotherapy.
“We don’t want to say that the microbiome is responsible for everything, but it’s responsible for some of the response and some of the resistance to treatment,” Dr. Zarour said. “So, we want to identify what candidate nonresponders are more likely to respond to FMT and be able to stick the right stool in the donor. This goes to better education of the microbiome signature. We are working hard on that.”
Dr. Baruch added that performing FMT for melanoma patients requires tight collaborations between oncologists, dermatologists, GI, and infectious disease experts. “These usually can be done in the setting of large cancer centers and will probably not be available in any hospital,” he said. “This is why understanding the mechanisms and developing an FMT-like drug is important. We are focusing on studying the mechanisms behind the clinical effect in order to develop a drug with an FMT-like effect without the safety and logistic issues related to FMTs.”

Tamia A. Harris-Tryon, MD, PhD, whose lab at the University of Texas Southwestern Medical Center at Dallas is studying how diet and the microbiota impact skin immunity, underscored the importance of evaluating the characteristics of the diet of patients as trials of FMT in melanoma patients carry on. “We know that the diet impacts the repertoire of microbes that colonize the gut,” said Dr. Harris-Tryon, assistant professor in the department of dermatology at the medical center. “The diet of the recipient likely has an impact” on the success of donor FMT.
She also noted that other skin conditions have been linked to a disrupted gut microbiome, such as psoriasis. “Given the safety of FMT in both of these studies, trials of FMT in psoriasis and other systemic skin conditions should be considered,” she said.
According to Dr. Zarour, mounting data from separate studies show that some gut microbiota play a role in adverse events experienced by melanoma patients on immunotherapy. “That is very important, especially with combination therapy,” he said. “There are also microbes involved in resistance to treatment, so the idea would be to identify these microbes.”
Studies raise more questions
In the opinion of Dr. Greiling, results from these two studies raise more questions than they answer. “The big question ... is why and how does FMT work, and how can we make the response better?” she said. “Is there one particular gene product from one microbe that is the key magic ingredient, and we can harness this as a drug? More likely it’s a complex interplay between multiple bacterial species needed to direct the immune response. Is there a group of microbes that is the same from person to person, or is it more complex?”
Then there are pending regulatory concerns. “We know that FMT works for [Clostridioides] difficile colitis but it’s not officially [Food and Drug Administration] approved,” Dr. Greiling said. “The FDA is really struggling with how to approve or regulate using bacteria as a drug. Where is that crossover? That inhibits things moving forward, for good reason. You want to balance safety with live microbes.”
The UPMC clinical trial was supported by Merck. Dr. Zarour disclosed that he is supported by grants from the National Cancer Institute and the James W. and Frances G. McGlothlin Chair in Melanoma Immunotherapy Research at UPMC. The Israeli study was funded by the Ella Lemelbaum Institute for Immuno-Oncology. Dr. Baruch was supported by the Allen Berg Fund for Excellence in Immuno-Oncology Research. Dr. Greiling and Dr. Harris-Tryon reported having no relevant financial disclosures.
In the fall of 2020, Hassane M. Zarour, MD, and colleagues began to pore over raw data from their
Preclinical mouse studies have demonstrated that the gut microbiota could influence the response of tumors to anti–PD-1 immunotherapy, but FMT had not been previously evaluated in human patients with malignant melanoma whose disease persisted or progressed after medical therapy. Only 30%-40% of melanoma patients respond to anti–PD-1 immunotherapy, so the researchers’ sense of anticipation was palpable. “It’s a high-risk, high-reward study, so you never know,” Dr. Zarour, a dermatologist and immunologist who is coleader of the melanoma program at the University of Pittsburgh Medical Center’s Hillman Cancer Center, said in an interview.
For the study, which was funded by the National Institutes of Health and published in Science, Dr. Zarour and a team of colleagues, including Diwakar Davar, MD, a medical oncologist/hematologist at UPMC and Giorgio Trinchieri, MD, head of the cancer immunology section at the National Cancer Institute, enrolled 16 patients with advanced melanoma whose disease had persisted or progressed with anti-PD-1 drugs; donors were 7 patients with advanced melanoma who had responded to pembrolizumab, 4 with a complete response and 3 with a partial response, with a median progression-free survival of 56 months.
After donors and patients underwent serial stool sampling and studies to stamp out the potential for transmitting infectious agents, the researchers administered the donor-derived FMT to patients via colonoscopy every 14 days for 3 weeks, followed by pembrolizumab. To their delight, 6 of the 15 evaluable recipients responded to treatment, with a reduction in tumor or long-term disease stabilization. Moreover, responders also showed increased abundance of taxa that were previously associated with response to immunotherapy, increased activation of CD8+ T cells, and decreased frequency of interleukin-8–expressing myeloid cells.
“This opens new doors for the future,” Dr. Zarour said. “It’s very encouraging, but I don’t want to overstate the data. It’s a small, nonrandomized trial, but one has to keep in mind that people were skeptical about this work; they didn’t think FMT would work. Now we see many people coming into the field to investigate the role of the microbiome as a therapeutic tool, which is great.”
Teri Greiling, MD, characterized the finding as a key development in understanding the microbiome’s potential to influence the course of melanoma and other diseases. “What’s emerging over the last decade of research is that our immune system has a close, back-and-forth relationship with our microbiota,” said Dr. Greiling, associate professor of dermatology at Oregon Health & Science University, Portland. “From day 1 of birth, we’re colonized by microbes that train our immune system how to function. In response, your immune system keeps those microbes in check and shapes which ones are allowed to colonize, and which ones are a target for attack. Thus, inflammatory responses are generated. Similarly, the goal of immunotherapy is to activate the immune system to fight cancer. This study shows that the immune system continues to need the colonizing microbes in our body to function optimally.”
Immunotherapy with checkpoint inhibitors was not an option for malignant melanoma patients until 2011, she noted, so the potential for FMT to further improve outcomes is welcome news for patients and their families. “We went from a less than 5% chance of survival with metastatic melanoma to now, with the right combination of checkpoint inhibitors, we’re up over 50%, which is amazing in a decade,” Dr. Greiling said. “Still, we’re losing half of our patients. If [FMT provides] a 30% improvement over that, that would be great, but it’s hard to extrapolate from such small numbers.”
Positive results in an Israeli study
Results from a similar, smaller phase 1 trial of 2 FMT donors and 10 recipients with metastatic melanoma who had progressed on anti-PD-1 therapy, from the Ella Lemelbaum Institute for Immuno-Oncology at Sheba Medical Center in Tel HaShomer, Israel, yielded similar results. The FMT protocol in this study included colonoscopy and oral stool capsules, followed by the reintroduction of anti–PD-1 therapy with nivolumab. The two FMT donors had previously been treated with anti–PD-1 monotherapy for metastatic melanoma and had achieved a clinical response for at least 1 year. Of the 10 FMT recipients, 1 had a complete response and 2 had a partial response.
“We expected changes in the immune system but did not expect that 3 out of the 10 patients in our study would be turned from nonresponders to responders,” the study’s lead author, Erez N. Baruch, MD, PhD, told this news organization. “Since this was a first-in-human study, we were aiming to assess safety and not clinical responses. [We found] that microbiota modulation can change the immune infiltration within melanoma tumors and by this affect response to immunotherapy.”
Dr. Baruch, an internal medicine resident in the physician-scientist track program at the University of Texas, Houston, said that the findings create a potential new therapeutic paradigm, or a new “playing ground” for drug development that can support existing immunotherapies. “It is important for dermatologists to understand that disruptions of the gut microbiota, mainly by antibiotics, may be harmful to melanoma patients,” he said. “Antibiotics in cancer patients should be used judiciously but of course should not be avoided when there’s an indication.”
As for next steps, Dr. Zarour and colleagues are recruiting more patients to boost their sample size and conducting sequential analysis of the microbiome of study participants “to better determine what the good and bad bugs are,” he said. “There are so many variables, including diet and geography. We need more data.” The hope is to develop a “microbiome signature” to identify patients likely to respond to FMT, and maybe one day, a probiotic capsule that patients take to optimize their response to immunotherapy.
“We don’t want to say that the microbiome is responsible for everything, but it’s responsible for some of the response and some of the resistance to treatment,” Dr. Zarour said. “So, we want to identify what candidate nonresponders are more likely to respond to FMT and be able to stick the right stool in the donor. This goes to better education of the microbiome signature. We are working hard on that.”
Dr. Baruch added that performing FMT for melanoma patients requires tight collaborations between oncologists, dermatologists, GI, and infectious disease experts. “These usually can be done in the setting of large cancer centers and will probably not be available in any hospital,” he said. “This is why understanding the mechanisms and developing an FMT-like drug is important. We are focusing on studying the mechanisms behind the clinical effect in order to develop a drug with an FMT-like effect without the safety and logistic issues related to FMTs.”
Tamia A. Harris-Tryon, MD, PhD, whose lab at the University of Texas Southwestern Medical Center at Dallas is studying how diet and the microbiota impact skin immunity, underscored the importance of evaluating the characteristics of the diet of patients as trials of FMT in melanoma patients carry on. “We know that the diet impacts the repertoire of microbes that colonize the gut,” said Dr. Harris-Tryon, assistant professor in the department of dermatology at the medical center. “The diet of the recipient likely has an impact” on the success of donor FMT.
She also noted that other skin conditions have been linked to a disrupted gut microbiome, such as psoriasis. “Given the safety of FMT in both of these studies, trials of FMT in psoriasis and other systemic skin conditions should be considered,” she said.
According to Dr. Zarour, mounting data from separate studies show that some gut microbiota play a role in adverse events experienced by melanoma patients on immunotherapy. “That is very important, especially with combination therapy,” he said. “There are also microbes involved in resistance to treatment, so the idea would be to identify these microbes.”
Studies raise more questions
In the opinion of Dr. Greiling, results from these two studies raise more questions than they answer. “The big question ... is why and how does FMT work, and how can we make the response better?” she said. “Is there one particular gene product from one microbe that is the key magic ingredient, and we can harness this as a drug? More likely it’s a complex interplay between multiple bacterial species needed to direct the immune response. Is there a group of microbes that is the same from person to person, or is it more complex?”
Then there are pending regulatory concerns. “We know that FMT works for [Clostridioides] difficile colitis but it’s not officially [Food and Drug Administration] approved,” Dr. Greiling said. “The FDA is really struggling with how to approve or regulate using bacteria as a drug. Where is that crossover? That inhibits things moving forward, for good reason. You want to balance safety with live microbes.”
The UPMC clinical trial was supported by Merck. Dr. Zarour disclosed that he is supported by grants from the National Cancer Institute and the James W. and Frances G. McGlothlin Chair in Melanoma Immunotherapy Research at UPMC. The Israeli study was funded by the Ella Lemelbaum Institute for Immuno-Oncology. Dr. Baruch was supported by the Allen Berg Fund for Excellence in Immuno-Oncology Research. Dr. Greiling and Dr. Harris-Tryon reported having no relevant financial disclosures.
In the fall of 2020, Hassane M. Zarour, MD, and colleagues began to pore over raw data from their
Preclinical mouse studies have demonstrated that the gut microbiota could influence the response of tumors to anti–PD-1 immunotherapy, but FMT had not been previously evaluated in human patients with malignant melanoma whose disease persisted or progressed after medical therapy. Only 30%-40% of melanoma patients respond to anti–PD-1 immunotherapy, so the researchers’ sense of anticipation was palpable. “It’s a high-risk, high-reward study, so you never know,” Dr. Zarour, a dermatologist and immunologist who is coleader of the melanoma program at the University of Pittsburgh Medical Center’s Hillman Cancer Center, said in an interview.
For the study, which was funded by the National Institutes of Health and published in Science, Dr. Zarour and a team of colleagues, including Diwakar Davar, MD, a medical oncologist/hematologist at UPMC and Giorgio Trinchieri, MD, head of the cancer immunology section at the National Cancer Institute, enrolled 16 patients with advanced melanoma whose disease had persisted or progressed with anti-PD-1 drugs; donors were 7 patients with advanced melanoma who had responded to pembrolizumab, 4 with a complete response and 3 with a partial response, with a median progression-free survival of 56 months.
After donors and patients underwent serial stool sampling and studies to stamp out the potential for transmitting infectious agents, the researchers administered the donor-derived FMT to patients via colonoscopy every 14 days for 3 weeks, followed by pembrolizumab. To their delight, 6 of the 15 evaluable recipients responded to treatment, with a reduction in tumor or long-term disease stabilization. Moreover, responders also showed increased abundance of taxa that were previously associated with response to immunotherapy, increased activation of CD8+ T cells, and decreased frequency of interleukin-8–expressing myeloid cells.
“This opens new doors for the future,” Dr. Zarour said. “It’s very encouraging, but I don’t want to overstate the data. It’s a small, nonrandomized trial, but one has to keep in mind that people were skeptical about this work; they didn’t think FMT would work. Now we see many people coming into the field to investigate the role of the microbiome as a therapeutic tool, which is great.”
Teri Greiling, MD, characterized the finding as a key development in understanding the microbiome’s potential to influence the course of melanoma and other diseases. “What’s emerging over the last decade of research is that our immune system has a close, back-and-forth relationship with our microbiota,” said Dr. Greiling, associate professor of dermatology at Oregon Health & Science University, Portland. “From day 1 of birth, we’re colonized by microbes that train our immune system how to function. In response, your immune system keeps those microbes in check and shapes which ones are allowed to colonize, and which ones are a target for attack. Thus, inflammatory responses are generated. Similarly, the goal of immunotherapy is to activate the immune system to fight cancer. This study shows that the immune system continues to need the colonizing microbes in our body to function optimally.”
Immunotherapy with checkpoint inhibitors was not an option for malignant melanoma patients until 2011, she noted, so the potential for FMT to further improve outcomes is welcome news for patients and their families. “We went from a less than 5% chance of survival with metastatic melanoma to now, with the right combination of checkpoint inhibitors, we’re up over 50%, which is amazing in a decade,” Dr. Greiling said. “Still, we’re losing half of our patients. If [FMT provides] a 30% improvement over that, that would be great, but it’s hard to extrapolate from such small numbers.”
Positive results in an Israeli study
Results from a similar, smaller phase 1 trial of 2 FMT donors and 10 recipients with metastatic melanoma who had progressed on anti-PD-1 therapy, from the Ella Lemelbaum Institute for Immuno-Oncology at Sheba Medical Center in Tel HaShomer, Israel, yielded similar results. The FMT protocol in this study included colonoscopy and oral stool capsules, followed by the reintroduction of anti–PD-1 therapy with nivolumab. The two FMT donors had previously been treated with anti–PD-1 monotherapy for metastatic melanoma and had achieved a clinical response for at least 1 year. Of the 10 FMT recipients, 1 had a complete response and 2 had a partial response.
“We expected changes in the immune system but did not expect that 3 out of the 10 patients in our study would be turned from nonresponders to responders,” the study’s lead author, Erez N. Baruch, MD, PhD, told this news organization. “Since this was a first-in-human study, we were aiming to assess safety and not clinical responses. [We found] that microbiota modulation can change the immune infiltration within melanoma tumors and by this affect response to immunotherapy.”
Dr. Baruch, an internal medicine resident in the physician-scientist track program at the University of Texas, Houston, said that the findings create a potential new therapeutic paradigm, or a new “playing ground” for drug development that can support existing immunotherapies. “It is important for dermatologists to understand that disruptions of the gut microbiota, mainly by antibiotics, may be harmful to melanoma patients,” he said. “Antibiotics in cancer patients should be used judiciously but of course should not be avoided when there’s an indication.”
As for next steps, Dr. Zarour and colleagues are recruiting more patients to boost their sample size and conducting sequential analysis of the microbiome of study participants “to better determine what the good and bad bugs are,” he said. “There are so many variables, including diet and geography. We need more data.” The hope is to develop a “microbiome signature” to identify patients likely to respond to FMT, and maybe one day, a probiotic capsule that patients take to optimize their response to immunotherapy.
“We don’t want to say that the microbiome is responsible for everything, but it’s responsible for some of the response and some of the resistance to treatment,” Dr. Zarour said. “So, we want to identify what candidate nonresponders are more likely to respond to FMT and be able to stick the right stool in the donor. This goes to better education of the microbiome signature. We are working hard on that.”
Dr. Baruch added that performing FMT for melanoma patients requires tight collaborations between oncologists, dermatologists, GI, and infectious disease experts. “These usually can be done in the setting of large cancer centers and will probably not be available in any hospital,” he said. “This is why understanding the mechanisms and developing an FMT-like drug is important. We are focusing on studying the mechanisms behind the clinical effect in order to develop a drug with an FMT-like effect without the safety and logistic issues related to FMTs.”
Tamia A. Harris-Tryon, MD, PhD, whose lab at the University of Texas Southwestern Medical Center at Dallas is studying how diet and the microbiota impact skin immunity, underscored the importance of evaluating the characteristics of the diet of patients as trials of FMT in melanoma patients carry on. “We know that the diet impacts the repertoire of microbes that colonize the gut,” said Dr. Harris-Tryon, assistant professor in the department of dermatology at the medical center. “The diet of the recipient likely has an impact” on the success of donor FMT.
She also noted that other skin conditions have been linked to a disrupted gut microbiome, such as psoriasis. “Given the safety of FMT in both of these studies, trials of FMT in psoriasis and other systemic skin conditions should be considered,” she said.
According to Dr. Zarour, mounting data from separate studies show that some gut microbiota play a role in adverse events experienced by melanoma patients on immunotherapy. “That is very important, especially with combination therapy,” he said. “There are also microbes involved in resistance to treatment, so the idea would be to identify these microbes.”
Studies raise more questions
In the opinion of Dr. Greiling, results from these two studies raise more questions than they answer. “The big question ... is why and how does FMT work, and how can we make the response better?” she said. “Is there one particular gene product from one microbe that is the key magic ingredient, and we can harness this as a drug? More likely it’s a complex interplay between multiple bacterial species needed to direct the immune response. Is there a group of microbes that is the same from person to person, or is it more complex?”
Then there are pending regulatory concerns. “We know that FMT works for [Clostridioides] difficile colitis but it’s not officially [Food and Drug Administration] approved,” Dr. Greiling said. “The FDA is really struggling with how to approve or regulate using bacteria as a drug. Where is that crossover? That inhibits things moving forward, for good reason. You want to balance safety with live microbes.”
The UPMC clinical trial was supported by Merck. Dr. Zarour disclosed that he is supported by grants from the National Cancer Institute and the James W. and Frances G. McGlothlin Chair in Melanoma Immunotherapy Research at UPMC. The Israeli study was funded by the Ella Lemelbaum Institute for Immuno-Oncology. Dr. Baruch was supported by the Allen Berg Fund for Excellence in Immuno-Oncology Research. Dr. Greiling and Dr. Harris-Tryon reported having no relevant financial disclosures.
COPD: Higher mortality with low baseline CO diffusing capacity
Patients with a baseline DLCO (diffusing capacity for carbon monoxide) of < 60% of predicted have more severe disease clinical expression with higher mortality risk, according to a long-term observational study of Global Initiative for Obstructive Lung Disease (GOLD) I chronic obstructive pulmonary disease (COPD) patients. Clarifying mechanisms of low DLCO may help clinicians direct interventions toward ameliorating the low capacity, Juan Pablo de Torres, MD, and colleagues wrote in the journal CHEST®.
Defining increased risk
“Can a DLCO threshold help define an increased risk of death and a different clinical presentation in GOLD I patients?” the researchers questioned. For evaluation of COPD, the GOLD does not currently promote the use of DLCO, and the clinical and prognostic utility of a low DLCO has not been studied, the authors noted.
Several COPD studies, however, have shown associations between low DLCO values and reduced exercise capacity, increased symptoms, risk of severe exacerbations, and mortality. The patients included in these studies, however, have generally had moderate to severe airflow limitation, and have not had postbronchodilator forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) < 0.70 and an FEV1 ≥ 80%, defined by GOLD as COPD spirometric stage I. These mild obstruction GOLD I patients, in large epidemiological studies, do have increased risk of death. But it is often assumed, Dr. de Torres and colleagues noted, that “mild” suggests a good prognosis. They propose that a simple DLCO measurement could help identify those GOLD I patients with “worse overall COPD compromise and an increased risk of death.” Importantly, GOLD I represents the largest percentage of patients with airflow limitation that epidemiological studies have identified.
The researchers enrolled 360 GOLD stage I COPD patients, recording their age, sex, pack-years’ history, body mass index, dyspnea, lung function measurements, exercise capacity, BODE (body mass index, airflow obstruction, dyspnea, and exercise capacity) index, and history of exacerbations, and followed them for a mean of 109 months. They identified a cutoff DLCO value for all-cause mortality, compared the clinical and physiological characteristics of patients above and below the threshold, and explored the predictive power of that cutoff value.
All-cause mortality difference
The mean age in the overall population studied was 63 years (31% were women), with 43% active smokers, and pack-years history of 45. Overall mortality was 11% during the follow-up period. The predominantly male population was mildly overweight, had few comorbidities, normal FEV1 values, mild dyspnea, normal 6-minute walk distance, and very few exacerbations.
Analysis showed a DLCO cutoff value of < 60% was associated with a significant all-cause mortality differential (DLCO ≥ 60%: 9% vs. DLCO < 60%: 23%, P = .01). At a same FEV1% predicted and Charlson score, patients with DLCO < 60% had lower BMI, more dyspnea, lower inspiratory capacity (IC)/total lung capacity (TLC) ratio, lower 6-minute walk distance, and higher BODE index. Adjusted Cox multiple regression analysis confirmed that a DLCO < 60% was associated with an all-cause mortality hazard ratio [HR] of 3.37, (95% confidence interval, 1.35-8.39; P = .009).
Multiorgan loss of tissue
The researchers found that patients with baseline DLCO < 60% were more likely to be women (46% versus 28%), and to have a lower BMI (25 vs. 27), higher pack-year history (54 vs. 43), the same spirometric values but lower IC/TLC ratio (.37 vs. .40), a lower walk distance (443 vs. 485 meters), higher dyspnea (MRC score 1.1 vs. .7), similar exacerbation rate, higher BODE index (.5 vs. .2) and higher mortality than patients with higher DLCO % predicted values. This group, Dr. de Torres and colleagues suggest, represents a multiorgan loss of tissue, a phenotype associated with worse clinical outcomes and prognosis.
“Low DLCO in these patients,” Dr. de Torres said in an interview, “could mainly be secondary to coexistent emphysema, which is the most common cause of low DLCO in this population. Also possible, but less likely, is coexistent pulmonary hypertension.” He noted further that “This study opens the door to research specifically testing if such is the case, and if it is, for clinicians to use available therapies to prevent adverse outcomes.”
Comorbidity burden
Patients with GOLD I COPD die more often of cardiovascular disease instead of underlying lung disease, according to Richard H. Zou, MD, and Jessica Bon, MD, of the University of Pittsburgh, in an accompanying editorial in the journal CHEST.
Increased mortality rates, they suggest, may be related to higher comorbidity burden, particularly comorbidities associated with cardiovascular-related health. Subclinical cardiovascular disease is a common comorbidity in COPD, and concomitant endothelial dysfunction has been associated with both cardiovascular disease and early emphysema in smokers. They may have disproportionately reduced DLCO levels because of parenchymal destruction.
“This study suggests that DLCO can be used to identify patients with GOLD I COPD at increased death risk and that individuals with mild airflow obstruction with DLCO <60% predicted are a clinical phenotype distinct from those with higher DLCO levels,” Dr. Zhou and Dr. Bon concluded.
The researchers and the editorialists declared that they had no disclosures. One of the three cohorts assessed in the current study (CHAIN cohort in Spain) received funding from AstraZeneca.
Patients with a baseline DLCO (diffusing capacity for carbon monoxide) of < 60% of predicted have more severe disease clinical expression with higher mortality risk, according to a long-term observational study of Global Initiative for Obstructive Lung Disease (GOLD) I chronic obstructive pulmonary disease (COPD) patients. Clarifying mechanisms of low DLCO may help clinicians direct interventions toward ameliorating the low capacity, Juan Pablo de Torres, MD, and colleagues wrote in the journal CHEST®.
Defining increased risk
“Can a DLCO threshold help define an increased risk of death and a different clinical presentation in GOLD I patients?” the researchers questioned. For evaluation of COPD, the GOLD does not currently promote the use of DLCO, and the clinical and prognostic utility of a low DLCO has not been studied, the authors noted.
Several COPD studies, however, have shown associations between low DLCO values and reduced exercise capacity, increased symptoms, risk of severe exacerbations, and mortality. The patients included in these studies, however, have generally had moderate to severe airflow limitation, and have not had postbronchodilator forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) < 0.70 and an FEV1 ≥ 80%, defined by GOLD as COPD spirometric stage I. These mild obstruction GOLD I patients, in large epidemiological studies, do have increased risk of death. But it is often assumed, Dr. de Torres and colleagues noted, that “mild” suggests a good prognosis. They propose that a simple DLCO measurement could help identify those GOLD I patients with “worse overall COPD compromise and an increased risk of death.” Importantly, GOLD I represents the largest percentage of patients with airflow limitation that epidemiological studies have identified.
The researchers enrolled 360 GOLD stage I COPD patients, recording their age, sex, pack-years’ history, body mass index, dyspnea, lung function measurements, exercise capacity, BODE (body mass index, airflow obstruction, dyspnea, and exercise capacity) index, and history of exacerbations, and followed them for a mean of 109 months. They identified a cutoff DLCO value for all-cause mortality, compared the clinical and physiological characteristics of patients above and below the threshold, and explored the predictive power of that cutoff value.
All-cause mortality difference
The mean age in the overall population studied was 63 years (31% were women), with 43% active smokers, and pack-years history of 45. Overall mortality was 11% during the follow-up period. The predominantly male population was mildly overweight, had few comorbidities, normal FEV1 values, mild dyspnea, normal 6-minute walk distance, and very few exacerbations.
Analysis showed a DLCO cutoff value of < 60% was associated with a significant all-cause mortality differential (DLCO ≥ 60%: 9% vs. DLCO < 60%: 23%, P = .01). At a same FEV1% predicted and Charlson score, patients with DLCO < 60% had lower BMI, more dyspnea, lower inspiratory capacity (IC)/total lung capacity (TLC) ratio, lower 6-minute walk distance, and higher BODE index. Adjusted Cox multiple regression analysis confirmed that a DLCO < 60% was associated with an all-cause mortality hazard ratio [HR] of 3.37, (95% confidence interval, 1.35-8.39; P = .009).
Multiorgan loss of tissue
The researchers found that patients with baseline DLCO < 60% were more likely to be women (46% versus 28%), and to have a lower BMI (25 vs. 27), higher pack-year history (54 vs. 43), the same spirometric values but lower IC/TLC ratio (.37 vs. .40), a lower walk distance (443 vs. 485 meters), higher dyspnea (MRC score 1.1 vs. .7), similar exacerbation rate, higher BODE index (.5 vs. .2) and higher mortality than patients with higher DLCO % predicted values. This group, Dr. de Torres and colleagues suggest, represents a multiorgan loss of tissue, a phenotype associated with worse clinical outcomes and prognosis.
“Low DLCO in these patients,” Dr. de Torres said in an interview, “could mainly be secondary to coexistent emphysema, which is the most common cause of low DLCO in this population. Also possible, but less likely, is coexistent pulmonary hypertension.” He noted further that “This study opens the door to research specifically testing if such is the case, and if it is, for clinicians to use available therapies to prevent adverse outcomes.”
Comorbidity burden
Patients with GOLD I COPD die more often of cardiovascular disease instead of underlying lung disease, according to Richard H. Zou, MD, and Jessica Bon, MD, of the University of Pittsburgh, in an accompanying editorial in the journal CHEST.
Increased mortality rates, they suggest, may be related to higher comorbidity burden, particularly comorbidities associated with cardiovascular-related health. Subclinical cardiovascular disease is a common comorbidity in COPD, and concomitant endothelial dysfunction has been associated with both cardiovascular disease and early emphysema in smokers. They may have disproportionately reduced DLCO levels because of parenchymal destruction.
“This study suggests that DLCO can be used to identify patients with GOLD I COPD at increased death risk and that individuals with mild airflow obstruction with DLCO <60% predicted are a clinical phenotype distinct from those with higher DLCO levels,” Dr. Zhou and Dr. Bon concluded.
The researchers and the editorialists declared that they had no disclosures. One of the three cohorts assessed in the current study (CHAIN cohort in Spain) received funding from AstraZeneca.
Patients with a baseline DLCO (diffusing capacity for carbon monoxide) of < 60% of predicted have more severe disease clinical expression with higher mortality risk, according to a long-term observational study of Global Initiative for Obstructive Lung Disease (GOLD) I chronic obstructive pulmonary disease (COPD) patients. Clarifying mechanisms of low DLCO may help clinicians direct interventions toward ameliorating the low capacity, Juan Pablo de Torres, MD, and colleagues wrote in the journal CHEST®.
Defining increased risk
“Can a DLCO threshold help define an increased risk of death and a different clinical presentation in GOLD I patients?” the researchers questioned. For evaluation of COPD, the GOLD does not currently promote the use of DLCO, and the clinical and prognostic utility of a low DLCO has not been studied, the authors noted.
Several COPD studies, however, have shown associations between low DLCO values and reduced exercise capacity, increased symptoms, risk of severe exacerbations, and mortality. The patients included in these studies, however, have generally had moderate to severe airflow limitation, and have not had postbronchodilator forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) < 0.70 and an FEV1 ≥ 80%, defined by GOLD as COPD spirometric stage I. These mild obstruction GOLD I patients, in large epidemiological studies, do have increased risk of death. But it is often assumed, Dr. de Torres and colleagues noted, that “mild” suggests a good prognosis. They propose that a simple DLCO measurement could help identify those GOLD I patients with “worse overall COPD compromise and an increased risk of death.” Importantly, GOLD I represents the largest percentage of patients with airflow limitation that epidemiological studies have identified.
The researchers enrolled 360 GOLD stage I COPD patients, recording their age, sex, pack-years’ history, body mass index, dyspnea, lung function measurements, exercise capacity, BODE (body mass index, airflow obstruction, dyspnea, and exercise capacity) index, and history of exacerbations, and followed them for a mean of 109 months. They identified a cutoff DLCO value for all-cause mortality, compared the clinical and physiological characteristics of patients above and below the threshold, and explored the predictive power of that cutoff value.
All-cause mortality difference
The mean age in the overall population studied was 63 years (31% were women), with 43% active smokers, and pack-years history of 45. Overall mortality was 11% during the follow-up period. The predominantly male population was mildly overweight, had few comorbidities, normal FEV1 values, mild dyspnea, normal 6-minute walk distance, and very few exacerbations.
Analysis showed a DLCO cutoff value of < 60% was associated with a significant all-cause mortality differential (DLCO ≥ 60%: 9% vs. DLCO < 60%: 23%, P = .01). At a same FEV1% predicted and Charlson score, patients with DLCO < 60% had lower BMI, more dyspnea, lower inspiratory capacity (IC)/total lung capacity (TLC) ratio, lower 6-minute walk distance, and higher BODE index. Adjusted Cox multiple regression analysis confirmed that a DLCO < 60% was associated with an all-cause mortality hazard ratio [HR] of 3.37, (95% confidence interval, 1.35-8.39; P = .009).
Multiorgan loss of tissue
The researchers found that patients with baseline DLCO < 60% were more likely to be women (46% versus 28%), and to have a lower BMI (25 vs. 27), higher pack-year history (54 vs. 43), the same spirometric values but lower IC/TLC ratio (.37 vs. .40), a lower walk distance (443 vs. 485 meters), higher dyspnea (MRC score 1.1 vs. .7), similar exacerbation rate, higher BODE index (.5 vs. .2) and higher mortality than patients with higher DLCO % predicted values. This group, Dr. de Torres and colleagues suggest, represents a multiorgan loss of tissue, a phenotype associated with worse clinical outcomes and prognosis.
“Low DLCO in these patients,” Dr. de Torres said in an interview, “could mainly be secondary to coexistent emphysema, which is the most common cause of low DLCO in this population. Also possible, but less likely, is coexistent pulmonary hypertension.” He noted further that “This study opens the door to research specifically testing if such is the case, and if it is, for clinicians to use available therapies to prevent adverse outcomes.”
Comorbidity burden
Patients with GOLD I COPD die more often of cardiovascular disease instead of underlying lung disease, according to Richard H. Zou, MD, and Jessica Bon, MD, of the University of Pittsburgh, in an accompanying editorial in the journal CHEST.
Increased mortality rates, they suggest, may be related to higher comorbidity burden, particularly comorbidities associated with cardiovascular-related health. Subclinical cardiovascular disease is a common comorbidity in COPD, and concomitant endothelial dysfunction has been associated with both cardiovascular disease and early emphysema in smokers. They may have disproportionately reduced DLCO levels because of parenchymal destruction.
“This study suggests that DLCO can be used to identify patients with GOLD I COPD at increased death risk and that individuals with mild airflow obstruction with DLCO <60% predicted are a clinical phenotype distinct from those with higher DLCO levels,” Dr. Zhou and Dr. Bon concluded.
The researchers and the editorialists declared that they had no disclosures. One of the three cohorts assessed in the current study (CHAIN cohort in Spain) received funding from AstraZeneca.
FROM THE JOURNAL CHEST®