User login
Tantrum-taming edibles, support gators, and chemo eggs
Chill out, kid
What do you do when your child has constant tantrums? A simple edible could do the trick, according to a Hollywood physician.

The natural medicine physician is in hot (bong) water after recommending marijuana cookies as treatment for a 4-year-old child’s ADHD and bipolar disorder. The icing on the cake (or cookie) is that both diagnoses weren’t even accurate. Perhaps the doctor was sampling his own treatments before the office visit?
The progressive physician has had his license revoked for the “grossly negligent” diagnosis, which he made in 30 minutes without consulting the child’s teachers, his father, or a psychiatrist. Probably not the best way to handle it, said the state medical board. Perhaps he should have suggested some CBD-infused Coke, instead?
Gator saver
Dogs, cats, peacocks – these are the animals that many people with anxiety, depression, and other mental health issues use for emotional support. But now, enter Wally, the emotional support alligator.

Spanning 5 feet long and sporting way too many teeth, Wally is the constant companion of a 65-year-old Pennsylvania man with depression. Wally’s owner decided to forgo pharmacologic treatment for something decidedly more reptilian.
Wally, who was rescued from Florida, loves chicken wings, hugs, and his adopted gator brother, Scrappy. He also has the potential to reach 16 feet long, which is … concerning. Something tells me you can’t take Wally on a plane as a service gator.
But don’t worry, Wally has been approved by a doctor. Rumors that Wally had the doctor’s arm in his jaws before approval are unsubstantiated.
Synergy is not always a good thing
Since it is generally agreed that two heads are better than one, three heads must be even better than two, right? But what if we’re not talking about heads? Suppose, instead, that the subject is global pandemics. Would it be better if three of the greatest threats to humanity’s existence on the planet decided to join forces?

The Lancet Global Syndemic Commission, a group of more than 40 international experts, said that obesity, undernutrition, and climate change “constitute a syndemic, or synergy of epidemics, because they co-occur in time and place, interact with each other to produce complex sequelae, and share common underlying societal drivers” (Lancet. 2019 Jan. 27. doi: 10.1016/S0140-6736[18]32822-8).
It gets better: The commission suggested that the “three interconnected health pandemics [have been] effectively orchestrated by the shadowy manipulations and influence of vested commercial interests – an entity collectively defined as ‘Big Food,’ ” according to Science Alert.
This all seems like a lot to overcome, but we here at LOTME have faith in science, and in the scientists who are working to solve these problems. After all, it’s not like anyone’s out there disregarding the science and saying that this stuff isn’t really happening. … Wait, what? … Climate change deniers? … Really? … The president tweeted what? … We’re doomed.
I prefer my medication sunny side up
Here’s a hypothetical question for you: If you were to have cancer, how would you prefer to be treated? Would you rather go through the rigors of chemotherapy? Or would you rather eat an omelet?

Okay, it probably wouldn’t work quite like that, but a group of physicians from the University of Edinburgh have successfully modified chickens to lay eggs containing a pair of human proteins within the egg white.
One of these proteins has antiviral and anticancer effects, and the other can help damaged tissue repair itself. The researchers added that the protein in the egg white could be modified to make the key ingredients for other protein-based drugs such as Avastin and Herceptin, which are used for treating cancer.
We know what you’re thinking: It’ll probably take a thousand eggs to make one dose – but no, it only takes three. Over the course of a year, one chicken could produce a hundred doses, and do it for far cheaper than is currently possible. We hate jumping on the social media bandwagon here, but frankly, this is an egg worth giving millions of Instagram likes.

Chill out, kid
What do you do when your child has constant tantrums? A simple edible could do the trick, according to a Hollywood physician.
The natural medicine physician is in hot (bong) water after recommending marijuana cookies as treatment for a 4-year-old child’s ADHD and bipolar disorder. The icing on the cake (or cookie) is that both diagnoses weren’t even accurate. Perhaps the doctor was sampling his own treatments before the office visit?
The progressive physician has had his license revoked for the “grossly negligent” diagnosis, which he made in 30 minutes without consulting the child’s teachers, his father, or a psychiatrist. Probably not the best way to handle it, said the state medical board. Perhaps he should have suggested some CBD-infused Coke, instead?
Gator saver
Dogs, cats, peacocks – these are the animals that many people with anxiety, depression, and other mental health issues use for emotional support. But now, enter Wally, the emotional support alligator.
Spanning 5 feet long and sporting way too many teeth, Wally is the constant companion of a 65-year-old Pennsylvania man with depression. Wally’s owner decided to forgo pharmacologic treatment for something decidedly more reptilian.
Wally, who was rescued from Florida, loves chicken wings, hugs, and his adopted gator brother, Scrappy. He also has the potential to reach 16 feet long, which is … concerning. Something tells me you can’t take Wally on a plane as a service gator.
But don’t worry, Wally has been approved by a doctor. Rumors that Wally had the doctor’s arm in his jaws before approval are unsubstantiated.
Synergy is not always a good thing
Since it is generally agreed that two heads are better than one, three heads must be even better than two, right? But what if we’re not talking about heads? Suppose, instead, that the subject is global pandemics. Would it be better if three of the greatest threats to humanity’s existence on the planet decided to join forces?
The Lancet Global Syndemic Commission, a group of more than 40 international experts, said that obesity, undernutrition, and climate change “constitute a syndemic, or synergy of epidemics, because they co-occur in time and place, interact with each other to produce complex sequelae, and share common underlying societal drivers” (Lancet. 2019 Jan. 27. doi: 10.1016/S0140-6736[18]32822-8).
It gets better: The commission suggested that the “three interconnected health pandemics [have been] effectively orchestrated by the shadowy manipulations and influence of vested commercial interests – an entity collectively defined as ‘Big Food,’ ” according to Science Alert.
This all seems like a lot to overcome, but we here at LOTME have faith in science, and in the scientists who are working to solve these problems. After all, it’s not like anyone’s out there disregarding the science and saying that this stuff isn’t really happening. … Wait, what? … Climate change deniers? … Really? … The president tweeted what? … We’re doomed.
I prefer my medication sunny side up
Here’s a hypothetical question for you: If you were to have cancer, how would you prefer to be treated? Would you rather go through the rigors of chemotherapy? Or would you rather eat an omelet?
Okay, it probably wouldn’t work quite like that, but a group of physicians from the University of Edinburgh have successfully modified chickens to lay eggs containing a pair of human proteins within the egg white.
One of these proteins has antiviral and anticancer effects, and the other can help damaged tissue repair itself. The researchers added that the protein in the egg white could be modified to make the key ingredients for other protein-based drugs such as Avastin and Herceptin, which are used for treating cancer.
We know what you’re thinking: It’ll probably take a thousand eggs to make one dose – but no, it only takes three. Over the course of a year, one chicken could produce a hundred doses, and do it for far cheaper than is currently possible. We hate jumping on the social media bandwagon here, but frankly, this is an egg worth giving millions of Instagram likes.
Chill out, kid
What do you do when your child has constant tantrums? A simple edible could do the trick, according to a Hollywood physician.
The natural medicine physician is in hot (bong) water after recommending marijuana cookies as treatment for a 4-year-old child’s ADHD and bipolar disorder. The icing on the cake (or cookie) is that both diagnoses weren’t even accurate. Perhaps the doctor was sampling his own treatments before the office visit?
The progressive physician has had his license revoked for the “grossly negligent” diagnosis, which he made in 30 minutes without consulting the child’s teachers, his father, or a psychiatrist. Probably not the best way to handle it, said the state medical board. Perhaps he should have suggested some CBD-infused Coke, instead?
Gator saver
Dogs, cats, peacocks – these are the animals that many people with anxiety, depression, and other mental health issues use for emotional support. But now, enter Wally, the emotional support alligator.
Spanning 5 feet long and sporting way too many teeth, Wally is the constant companion of a 65-year-old Pennsylvania man with depression. Wally’s owner decided to forgo pharmacologic treatment for something decidedly more reptilian.
Wally, who was rescued from Florida, loves chicken wings, hugs, and his adopted gator brother, Scrappy. He also has the potential to reach 16 feet long, which is … concerning. Something tells me you can’t take Wally on a plane as a service gator.
But don’t worry, Wally has been approved by a doctor. Rumors that Wally had the doctor’s arm in his jaws before approval are unsubstantiated.
Synergy is not always a good thing
Since it is generally agreed that two heads are better than one, three heads must be even better than two, right? But what if we’re not talking about heads? Suppose, instead, that the subject is global pandemics. Would it be better if three of the greatest threats to humanity’s existence on the planet decided to join forces?
The Lancet Global Syndemic Commission, a group of more than 40 international experts, said that obesity, undernutrition, and climate change “constitute a syndemic, or synergy of epidemics, because they co-occur in time and place, interact with each other to produce complex sequelae, and share common underlying societal drivers” (Lancet. 2019 Jan. 27. doi: 10.1016/S0140-6736[18]32822-8).
It gets better: The commission suggested that the “three interconnected health pandemics [have been] effectively orchestrated by the shadowy manipulations and influence of vested commercial interests – an entity collectively defined as ‘Big Food,’ ” according to Science Alert.
This all seems like a lot to overcome, but we here at LOTME have faith in science, and in the scientists who are working to solve these problems. After all, it’s not like anyone’s out there disregarding the science and saying that this stuff isn’t really happening. … Wait, what? … Climate change deniers? … Really? … The president tweeted what? … We’re doomed.
I prefer my medication sunny side up
Here’s a hypothetical question for you: If you were to have cancer, how would you prefer to be treated? Would you rather go through the rigors of chemotherapy? Or would you rather eat an omelet?
Okay, it probably wouldn’t work quite like that, but a group of physicians from the University of Edinburgh have successfully modified chickens to lay eggs containing a pair of human proteins within the egg white.
One of these proteins has antiviral and anticancer effects, and the other can help damaged tissue repair itself. The researchers added that the protein in the egg white could be modified to make the key ingredients for other protein-based drugs such as Avastin and Herceptin, which are used for treating cancer.
We know what you’re thinking: It’ll probably take a thousand eggs to make one dose – but no, it only takes three. Over the course of a year, one chicken could produce a hundred doses, and do it for far cheaper than is currently possible. We hate jumping on the social media bandwagon here, but frankly, this is an egg worth giving millions of Instagram likes.
The ongoing issue of gender disparities in interventional cardiology
As gender disparities persist in interventional cardiology, a new survey is shedding light on what is keeping women away from the field.
With women representing only 9% of interventional cardiologists in the United States as of 2017, researchers under the direction of the American College of Cardiology Women in Cardiology Leadership Council sought to asses the perspectives of fellows-in-training (FIT) regarding the factors influencing their cardiology subspecialty decisions.
A total of 574 FIT completed the survey, with 190 respondents anticipating pursuit of a career in interventional cardiology. The results of the survey were published online in JACC: Cardiovascular Interventions. According to the report, unlike other studies that looked at gender disparities in interventional cardiology that focused on the training (residency) or later (practicing cardiologists), this is the first to look at the time when the decision is made during general cardiology fellowship.
The goal of the survey was “to try to understand in the current realm of our millennials who are studying and are in fellowship and in training and in the trenches, what is dissuading them to be in the subspecialty of interventional cardiology,” Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, and coauthor of the study, said in an interview.
Lead author Celina Yong, MD, and her colleagues wrote in their report on the survey that women “were more likely to express interest in all other cardiovascular specialties (general/clinical cardiology, advanced imaging, heart failure/transplant, adult congenital, and other), with the exception of electrophysiology (13% women vs. 87% men, P = .001).”
Researchers analyzed the 504 remaining survey responses after excluding those considering electrophysiology to get a better understanding about the influencing factors related to the decision to pursue interventional cardiology.
“Logistic regression of all demographic characteristics revealed that male sex was the most significant predictor of a career choice in interventional cardiology [odds ratio, 3.98; P less than 0.001],” the authors noted.
All respondents who intended to pursue a career in interventional cardiology had a list of 15 options to select the reasons for choosing this path. The top five, in descending order, were the opportunity to pursue hands-on procedures, personal interest in the specialty subject area, the opportunity for immediate gratification or sense of accomplishment, the thrill of treating ill patients in critical situations, and having mentors or role models the respondent identified with.
“When disaggregated by gender, there were six attributes that were significantly different between men and women in terms of reasons for pursuing” interventional cardiology, the authors stated. “Men were more likely to be driven by innovation in the field, importance of being an expert, likelihood of employment after completion of training, financial advantages, and prestige. Women were more likely to be driven by having a female mentor or role model.”
For those not pursuing a career in interventional cardiology, the top five reasons, in descending order, were an uncontrollable or unpredictable lifestyle, concern over long work hours and poor work/life balance, greater interest in another field, a desire for different type of patient contact, and wanting to have children in the next 5 years.
“There were seven attributes identified that negatively influenced IC choice differently by sex,” noted Dr. Yong, of VA Palo Alto (Calif.) Medical Center, and her colleagues. “Women were more likely to be negatively influenced by all seven of these factors compared to men (in descending order)”:
1) Greater interest in another field.
2) Little flexibility in job prospects/opportunities over a lifetime.
3) Physically demanding nature of job (e.g., wearing heavy lead).
4) Radiation exposure concerns during childbearing.
5) “Old boys club” culture.
6) Lack of female role models.
7) Gender discrimination or harassment.
Dr. Mehran said that despite some limitations, the survey results were not surprising.
“Unfortunately, surveys are very subjective,” she said. Also, one can question how biased some of these questions are. “But nonetheless, I think the result is very similar to what we had expected and have been talking about.”
She noted that the subspecialty of interventional cardiology needs to be more family friendly.
“I think we are going to lose a lot of good men also who are not choosing interventional cardiology,” she said. “There is no question that we have to think about how we can enhance and improve and pave the way for men and women, but mostly women because there are hardly any women and that’s important. The family friendly environment is very, very important in interventional cardiology.”
The patriarchal culture is another area that needs to be addressed, she said.
“I feel that, hopefully, that’s a perception and not much of a reality,” Dr. Mehran said, though she did note that there are plenty of examples where female doctors do not get shown the same level of respect their male counterparts do. She noted, for example, at scientific meetings, when a woman is on a panel and speaking, audience members can be seen tuning out, using it as opportunity to look at phones. Sometimes the women on the panels are not even referred to as “doctor.”
“I think we have to have a standard that those kinds of things will not be tolerated, that people will be called out if they didn’t do the extra work to find the best women for those important panels and leadership roles. There has to be a code of conduct that is equal and gender neutral,” she said, adding: “I think we are trying to work very hard to equalize the playing field but we have to come up with solutions.”
To that end, Dr. Mehran created a not-for-profit organization, Women As One, to tackle these gender disparities.
“We are really looking for solutions,” she said. “We will hold several think tanks with key opinion leaders, men and women, to come up with how best can academic organizations make sure that there is gender equality, good representation, and no discrimination on the basis of sex. ... We have to come up with solutions. Otherwise we just keep showing the same statistics over and over again and its not improving.”
SOURCE: JACC: Cardiovasc Interven. 2019 Jan; doi: 10.1016/j.jcin.2018.09.036
As gender disparities persist in interventional cardiology, a new survey is shedding light on what is keeping women away from the field.
With women representing only 9% of interventional cardiologists in the United States as of 2017, researchers under the direction of the American College of Cardiology Women in Cardiology Leadership Council sought to asses the perspectives of fellows-in-training (FIT) regarding the factors influencing their cardiology subspecialty decisions.
A total of 574 FIT completed the survey, with 190 respondents anticipating pursuit of a career in interventional cardiology. The results of the survey were published online in JACC: Cardiovascular Interventions. According to the report, unlike other studies that looked at gender disparities in interventional cardiology that focused on the training (residency) or later (practicing cardiologists), this is the first to look at the time when the decision is made during general cardiology fellowship.
The goal of the survey was “to try to understand in the current realm of our millennials who are studying and are in fellowship and in training and in the trenches, what is dissuading them to be in the subspecialty of interventional cardiology,” Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, and coauthor of the study, said in an interview.
Lead author Celina Yong, MD, and her colleagues wrote in their report on the survey that women “were more likely to express interest in all other cardiovascular specialties (general/clinical cardiology, advanced imaging, heart failure/transplant, adult congenital, and other), with the exception of electrophysiology (13% women vs. 87% men, P = .001).”
Researchers analyzed the 504 remaining survey responses after excluding those considering electrophysiology to get a better understanding about the influencing factors related to the decision to pursue interventional cardiology.
“Logistic regression of all demographic characteristics revealed that male sex was the most significant predictor of a career choice in interventional cardiology [odds ratio, 3.98; P less than 0.001],” the authors noted.
All respondents who intended to pursue a career in interventional cardiology had a list of 15 options to select the reasons for choosing this path. The top five, in descending order, were the opportunity to pursue hands-on procedures, personal interest in the specialty subject area, the opportunity for immediate gratification or sense of accomplishment, the thrill of treating ill patients in critical situations, and having mentors or role models the respondent identified with.
“When disaggregated by gender, there were six attributes that were significantly different between men and women in terms of reasons for pursuing” interventional cardiology, the authors stated. “Men were more likely to be driven by innovation in the field, importance of being an expert, likelihood of employment after completion of training, financial advantages, and prestige. Women were more likely to be driven by having a female mentor or role model.”
For those not pursuing a career in interventional cardiology, the top five reasons, in descending order, were an uncontrollable or unpredictable lifestyle, concern over long work hours and poor work/life balance, greater interest in another field, a desire for different type of patient contact, and wanting to have children in the next 5 years.
“There were seven attributes identified that negatively influenced IC choice differently by sex,” noted Dr. Yong, of VA Palo Alto (Calif.) Medical Center, and her colleagues. “Women were more likely to be negatively influenced by all seven of these factors compared to men (in descending order)”:
1) Greater interest in another field.
2) Little flexibility in job prospects/opportunities over a lifetime.
3) Physically demanding nature of job (e.g., wearing heavy lead).
4) Radiation exposure concerns during childbearing.
5) “Old boys club” culture.
6) Lack of female role models.
7) Gender discrimination or harassment.
Dr. Mehran said that despite some limitations, the survey results were not surprising.
“Unfortunately, surveys are very subjective,” she said. Also, one can question how biased some of these questions are. “But nonetheless, I think the result is very similar to what we had expected and have been talking about.”
She noted that the subspecialty of interventional cardiology needs to be more family friendly.
“I think we are going to lose a lot of good men also who are not choosing interventional cardiology,” she said. “There is no question that we have to think about how we can enhance and improve and pave the way for men and women, but mostly women because there are hardly any women and that’s important. The family friendly environment is very, very important in interventional cardiology.”
The patriarchal culture is another area that needs to be addressed, she said.
“I feel that, hopefully, that’s a perception and not much of a reality,” Dr. Mehran said, though she did note that there are plenty of examples where female doctors do not get shown the same level of respect their male counterparts do. She noted, for example, at scientific meetings, when a woman is on a panel and speaking, audience members can be seen tuning out, using it as opportunity to look at phones. Sometimes the women on the panels are not even referred to as “doctor.”
“I think we have to have a standard that those kinds of things will not be tolerated, that people will be called out if they didn’t do the extra work to find the best women for those important panels and leadership roles. There has to be a code of conduct that is equal and gender neutral,” she said, adding: “I think we are trying to work very hard to equalize the playing field but we have to come up with solutions.”
To that end, Dr. Mehran created a not-for-profit organization, Women As One, to tackle these gender disparities.
“We are really looking for solutions,” she said. “We will hold several think tanks with key opinion leaders, men and women, to come up with how best can academic organizations make sure that there is gender equality, good representation, and no discrimination on the basis of sex. ... We have to come up with solutions. Otherwise we just keep showing the same statistics over and over again and its not improving.”
SOURCE: JACC: Cardiovasc Interven. 2019 Jan; doi: 10.1016/j.jcin.2018.09.036
As gender disparities persist in interventional cardiology, a new survey is shedding light on what is keeping women away from the field.
With women representing only 9% of interventional cardiologists in the United States as of 2017, researchers under the direction of the American College of Cardiology Women in Cardiology Leadership Council sought to asses the perspectives of fellows-in-training (FIT) regarding the factors influencing their cardiology subspecialty decisions.
A total of 574 FIT completed the survey, with 190 respondents anticipating pursuit of a career in interventional cardiology. The results of the survey were published online in JACC: Cardiovascular Interventions. According to the report, unlike other studies that looked at gender disparities in interventional cardiology that focused on the training (residency) or later (practicing cardiologists), this is the first to look at the time when the decision is made during general cardiology fellowship.
The goal of the survey was “to try to understand in the current realm of our millennials who are studying and are in fellowship and in training and in the trenches, what is dissuading them to be in the subspecialty of interventional cardiology,” Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, and coauthor of the study, said in an interview.
Lead author Celina Yong, MD, and her colleagues wrote in their report on the survey that women “were more likely to express interest in all other cardiovascular specialties (general/clinical cardiology, advanced imaging, heart failure/transplant, adult congenital, and other), with the exception of electrophysiology (13% women vs. 87% men, P = .001).”
Researchers analyzed the 504 remaining survey responses after excluding those considering electrophysiology to get a better understanding about the influencing factors related to the decision to pursue interventional cardiology.
“Logistic regression of all demographic characteristics revealed that male sex was the most significant predictor of a career choice in interventional cardiology [odds ratio, 3.98; P less than 0.001],” the authors noted.
All respondents who intended to pursue a career in interventional cardiology had a list of 15 options to select the reasons for choosing this path. The top five, in descending order, were the opportunity to pursue hands-on procedures, personal interest in the specialty subject area, the opportunity for immediate gratification or sense of accomplishment, the thrill of treating ill patients in critical situations, and having mentors or role models the respondent identified with.
“When disaggregated by gender, there were six attributes that were significantly different between men and women in terms of reasons for pursuing” interventional cardiology, the authors stated. “Men were more likely to be driven by innovation in the field, importance of being an expert, likelihood of employment after completion of training, financial advantages, and prestige. Women were more likely to be driven by having a female mentor or role model.”
For those not pursuing a career in interventional cardiology, the top five reasons, in descending order, were an uncontrollable or unpredictable lifestyle, concern over long work hours and poor work/life balance, greater interest in another field, a desire for different type of patient contact, and wanting to have children in the next 5 years.
“There were seven attributes identified that negatively influenced IC choice differently by sex,” noted Dr. Yong, of VA Palo Alto (Calif.) Medical Center, and her colleagues. “Women were more likely to be negatively influenced by all seven of these factors compared to men (in descending order)”:
1) Greater interest in another field.
2) Little flexibility in job prospects/opportunities over a lifetime.
3) Physically demanding nature of job (e.g., wearing heavy lead).
4) Radiation exposure concerns during childbearing.
5) “Old boys club” culture.
6) Lack of female role models.
7) Gender discrimination or harassment.
Dr. Mehran said that despite some limitations, the survey results were not surprising.
“Unfortunately, surveys are very subjective,” she said. Also, one can question how biased some of these questions are. “But nonetheless, I think the result is very similar to what we had expected and have been talking about.”
She noted that the subspecialty of interventional cardiology needs to be more family friendly.
“I think we are going to lose a lot of good men also who are not choosing interventional cardiology,” she said. “There is no question that we have to think about how we can enhance and improve and pave the way for men and women, but mostly women because there are hardly any women and that’s important. The family friendly environment is very, very important in interventional cardiology.”
The patriarchal culture is another area that needs to be addressed, she said.
“I feel that, hopefully, that’s a perception and not much of a reality,” Dr. Mehran said, though she did note that there are plenty of examples where female doctors do not get shown the same level of respect their male counterparts do. She noted, for example, at scientific meetings, when a woman is on a panel and speaking, audience members can be seen tuning out, using it as opportunity to look at phones. Sometimes the women on the panels are not even referred to as “doctor.”
“I think we have to have a standard that those kinds of things will not be tolerated, that people will be called out if they didn’t do the extra work to find the best women for those important panels and leadership roles. There has to be a code of conduct that is equal and gender neutral,” she said, adding: “I think we are trying to work very hard to equalize the playing field but we have to come up with solutions.”
To that end, Dr. Mehran created a not-for-profit organization, Women As One, to tackle these gender disparities.
“We are really looking for solutions,” she said. “We will hold several think tanks with key opinion leaders, men and women, to come up with how best can academic organizations make sure that there is gender equality, good representation, and no discrimination on the basis of sex. ... We have to come up with solutions. Otherwise we just keep showing the same statistics over and over again and its not improving.”
SOURCE: JACC: Cardiovasc Interven. 2019 Jan; doi: 10.1016/j.jcin.2018.09.036
FROM JACC: CARDIOVASCULAR INTERVENTIONS
Key clinical point: Men are more likely to pursue a career in interventional cardiology than are women.
Major finding: Logistical regression of all demographic characteristics revealed being male was the most significant predictor of a career choice in IC .
Study details: Researchers analyzed survey responses from 574 fellows-in-training to determine the likelihood of pursuing a career in interventional cardiology.
Disclosures: The study was funded by the American College of Cardiology and the Women in Cardiology section of the ACC. The authors reported no financial disclosures.
Source: JACC: Cardiovasc Interven. 2019 Jan. doi: 10.1016/j.jcin.2018.09.036.
Thrombin generation looks promising as a hemophilia biomarker
SAN DIEGO – Thrombin generation may edge out baseline factor activity as a biomarker for predicting bleeding severity among patients with mild and moderate hemophilia, according to a study of 81 patients with nonsevere hemophilia.
Both baseline factor activity and thrombin generation had a similar correlation with bleeding severity, but thrombin generation had a higher sensitivity when differentiating between bleeding severities, Fadi Nossair, MD, of Children’s Hospital of King’s Daughters in Norfolk, Va., reported in a poster at the annual meeting of the American Society of Hematology.
Nonsevere cases of hemophilia A and B account for about half of all hemophilia cases in which factor level does not consistently correlate with bleeding phenotype. That makes it difficult to determine prophylaxis or surgery and highlights the need for a predictive biomarker, the investigators noted.
In the study, 81 patients had their bleeding assessed using standardized, self-administered and investigator-administered questionnaires. Bleeding phenotypes were also collected from EMRs.
One-time venous blood samples were collected after a washout period, when applicable. Additionally, platelet poor plasma was obtained to measure thrombin generation, phospholipid-dependent factor Xa initiated clotting time, factor VIII and IX activities, and von Willebrand factor.
Nearly three-quarters of patients in the study had a low bleeding score.
Both baseline factor level and thrombin generation values obtained with a regular reagent (5 pM of tissue factor) demonstrated a significant correlation with bleeding score (P less than .05). Values obtained with other reagents and biomarkers did not show a significant correlation, according to the researchers.
However, a sensitivity and specificity analysis that helped the researchers narrow down the optimal cutoff values for differentiating between bleeding severities also found that thrombin generation had superior sensitivity, compared with baseline factor level. All thrombin generation values had a higher sensitivity to predict bleeding severity, compared with baseline factor level (57%-62% versus 29%).
“Long-term prospective studies should evaluate the utility of this approach in predicting bleeding severity in this population,” the researchers said.
The study was supported by grants from Novo Nordisk. Dr. Nossair reported financial disclosures related to Novo Nordisk.
SOURCE: Nossair F et al. ASH 2018, Poster 3788.
SAN DIEGO – Thrombin generation may edge out baseline factor activity as a biomarker for predicting bleeding severity among patients with mild and moderate hemophilia, according to a study of 81 patients with nonsevere hemophilia.
Both baseline factor activity and thrombin generation had a similar correlation with bleeding severity, but thrombin generation had a higher sensitivity when differentiating between bleeding severities, Fadi Nossair, MD, of Children’s Hospital of King’s Daughters in Norfolk, Va., reported in a poster at the annual meeting of the American Society of Hematology.
Nonsevere cases of hemophilia A and B account for about half of all hemophilia cases in which factor level does not consistently correlate with bleeding phenotype. That makes it difficult to determine prophylaxis or surgery and highlights the need for a predictive biomarker, the investigators noted.
In the study, 81 patients had their bleeding assessed using standardized, self-administered and investigator-administered questionnaires. Bleeding phenotypes were also collected from EMRs.
One-time venous blood samples were collected after a washout period, when applicable. Additionally, platelet poor plasma was obtained to measure thrombin generation, phospholipid-dependent factor Xa initiated clotting time, factor VIII and IX activities, and von Willebrand factor.
Nearly three-quarters of patients in the study had a low bleeding score.
Both baseline factor level and thrombin generation values obtained with a regular reagent (5 pM of tissue factor) demonstrated a significant correlation with bleeding score (P less than .05). Values obtained with other reagents and biomarkers did not show a significant correlation, according to the researchers.
However, a sensitivity and specificity analysis that helped the researchers narrow down the optimal cutoff values for differentiating between bleeding severities also found that thrombin generation had superior sensitivity, compared with baseline factor level. All thrombin generation values had a higher sensitivity to predict bleeding severity, compared with baseline factor level (57%-62% versus 29%).
“Long-term prospective studies should evaluate the utility of this approach in predicting bleeding severity in this population,” the researchers said.
The study was supported by grants from Novo Nordisk. Dr. Nossair reported financial disclosures related to Novo Nordisk.
SOURCE: Nossair F et al. ASH 2018, Poster 3788.
SAN DIEGO – Thrombin generation may edge out baseline factor activity as a biomarker for predicting bleeding severity among patients with mild and moderate hemophilia, according to a study of 81 patients with nonsevere hemophilia.
Both baseline factor activity and thrombin generation had a similar correlation with bleeding severity, but thrombin generation had a higher sensitivity when differentiating between bleeding severities, Fadi Nossair, MD, of Children’s Hospital of King’s Daughters in Norfolk, Va., reported in a poster at the annual meeting of the American Society of Hematology.
Nonsevere cases of hemophilia A and B account for about half of all hemophilia cases in which factor level does not consistently correlate with bleeding phenotype. That makes it difficult to determine prophylaxis or surgery and highlights the need for a predictive biomarker, the investigators noted.
In the study, 81 patients had their bleeding assessed using standardized, self-administered and investigator-administered questionnaires. Bleeding phenotypes were also collected from EMRs.
One-time venous blood samples were collected after a washout period, when applicable. Additionally, platelet poor plasma was obtained to measure thrombin generation, phospholipid-dependent factor Xa initiated clotting time, factor VIII and IX activities, and von Willebrand factor.
Nearly three-quarters of patients in the study had a low bleeding score.
Both baseline factor level and thrombin generation values obtained with a regular reagent (5 pM of tissue factor) demonstrated a significant correlation with bleeding score (P less than .05). Values obtained with other reagents and biomarkers did not show a significant correlation, according to the researchers.
However, a sensitivity and specificity analysis that helped the researchers narrow down the optimal cutoff values for differentiating between bleeding severities also found that thrombin generation had superior sensitivity, compared with baseline factor level. All thrombin generation values had a higher sensitivity to predict bleeding severity, compared with baseline factor level (57%-62% versus 29%).
“Long-term prospective studies should evaluate the utility of this approach in predicting bleeding severity in this population,” the researchers said.
The study was supported by grants from Novo Nordisk. Dr. Nossair reported financial disclosures related to Novo Nordisk.
SOURCE: Nossair F et al. ASH 2018, Poster 3788.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: Compared with baseline factor level, all thrombin generation values had a higher sensitivity to predict bleeding severity (57%-62% versus 29%).
Study details: The study included 81 patients with mild or moderate hemophilia A or B and compared biomarkers for differentiating between bleeding phenotype severities.
Disclosures: The study was supported by grants from Novo Nordisk. Dr. Nossair reported financial disclosures related to Novo Nordisk.
Source: Nossair F et al. ASH 2018, Poster 3788.
Race/ethnicity, other factors predict PTSD and depression after mild TBI
Civilian patients with mild traumatic brain injury (TBI) who are black, have psychiatric history or lower education, or whose injury was caused by assault might be at greater risk of developing posttraumatic stress disorder or major depression, a longitudinal study suggests.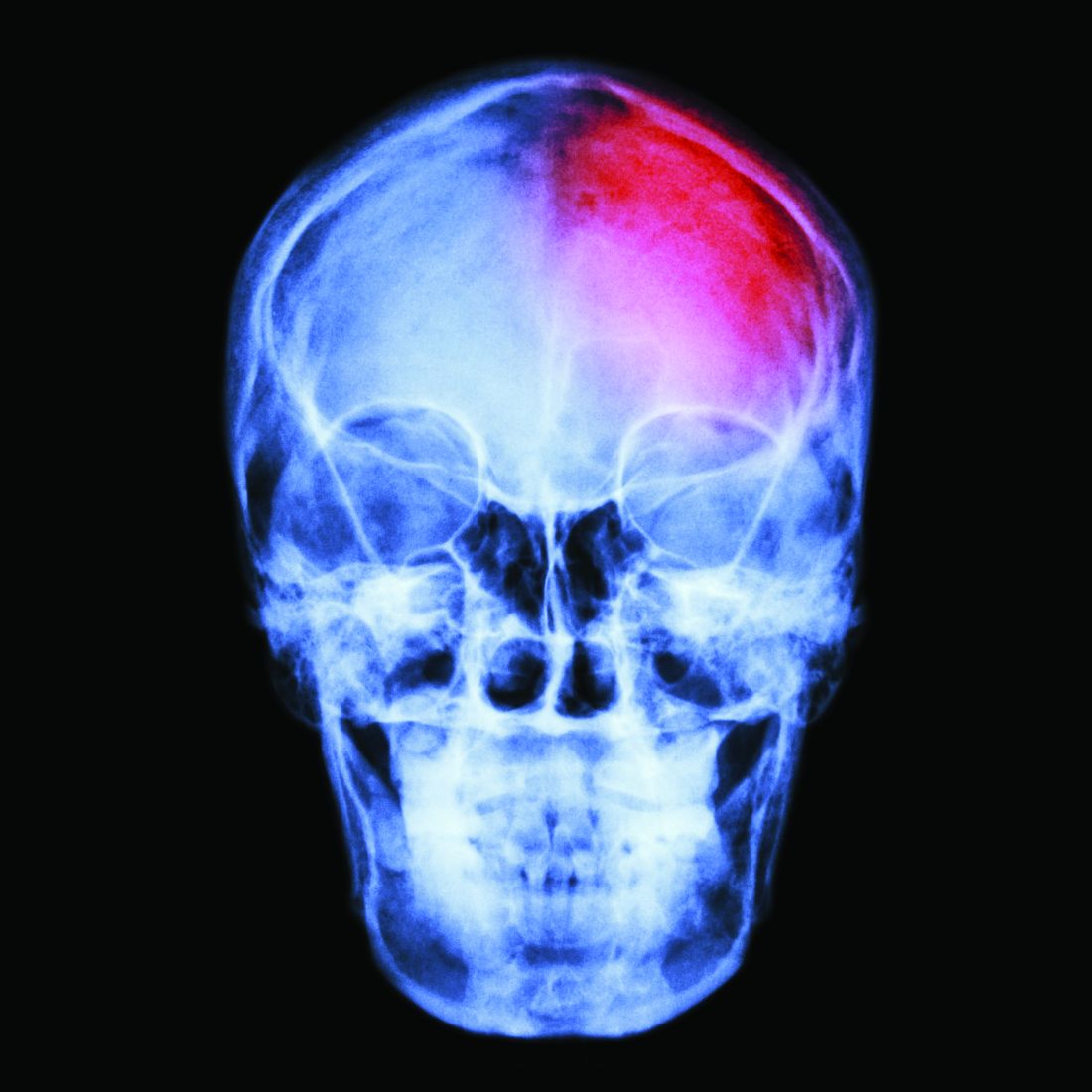
“Our findings may have implications for surveillance and treatment of mental disorders after TBI,” wrote Murray B. Stein, MD, MPH, and his associates. The study was published Jan. 30 in JAMA Psychiatry.
The researchers looked at the risk factors for and prevalence of posttraumatic stress disorder (PTSD) and major depressive disorder among 1,155 patients. The patients were enrolled at 11 level 1 trauma centers across the United States after they were evaluated for mild TBI in emergency departments as part of a prospective study called Transforming Research and Clinical Knowledge in Traumatic Brain Injury, or TRACK-TBI. The comparison group was 230 patients with nonhead orthopedic trauma injuries, wrote Dr. Stein, distinguished professor of psychiatry and family medicine and public health at the University of California, San Diego, and his associates.
They found that each additional year of education was associated with a significant 11% reduction in the risk of developing PTSD after mild TBI (P = .005). Also, black patients had a greater than fivefold higher risk of PTSD (P less than.001) than that of individuals who were not black.
Among patients with a history of mental illness and those who had experienced their injury as a result of assault or violence – as opposed to a motor vehicle accident or fall, for example – both had a greater than threefold higher risk of developing PTSD (odds ratio, 3.57 and 3.43 respectively). A prior TBI was nonsignificantly associated with an increased risk of developing PTSD.
Lower education duration, being black, or a history of mental illness also were all significantly associated with an increased risk of developing major depressive disorder after mild TBI.
However, duration of lost consciousness or posttraumatic amnesia, evidence of brain injury on CT, or hospitalization did not predict an increased risk of PTSD or major depression.
“Although MDD and PTSD are prevalent after TBI, little is known about which patients are at risk for developing them,” Dr. Stein and his associates wrote.
Noting that having a prior mental health problem was an “exceptionally strong” risk factor for PTSD and MDD after TBI, the authors said this could represent continuation or exacerbation of the prior mental health issue, or the triggering of a new episode in a person with a past history who had recovered.
“However, in either case this finding underscores the importance of clinicians being aware of the mental health history of their patients with [mild TBI], as this information is central to expectations regarding both short-term and long-term outcome,” they wrote.
Dr. Stein and his associates cited as a limitation their reliance on patient or family report. In addition, they said, the elevated risk for mental disorders among black individuals after mild TBI, which was independent of socioeconomic status or cause of injury, was not understood. “Unmeasured covariates may be part of the explanation; this is a topic needing further study,” they wrote.
The study was supported by the National Institutes of Health, the U.S. Department of Defense, Abbott Laboratories, and One Mind. Four authors declared consultancies, advisory board positions, speaking fees, and shares or stock options with the pharmaceutical and private industry. Two authors declared grants from the study sponsors.
SOURCE: Stein MB et al. JAMA Psychiatry. 2019. Jan 30. doi: 10.1001/jamapsychiatry.2018.4288.
Civilian patients with mild traumatic brain injury (TBI) who are black, have psychiatric history or lower education, or whose injury was caused by assault might be at greater risk of developing posttraumatic stress disorder or major depression, a longitudinal study suggests.
“Our findings may have implications for surveillance and treatment of mental disorders after TBI,” wrote Murray B. Stein, MD, MPH, and his associates. The study was published Jan. 30 in JAMA Psychiatry.
The researchers looked at the risk factors for and prevalence of posttraumatic stress disorder (PTSD) and major depressive disorder among 1,155 patients. The patients were enrolled at 11 level 1 trauma centers across the United States after they were evaluated for mild TBI in emergency departments as part of a prospective study called Transforming Research and Clinical Knowledge in Traumatic Brain Injury, or TRACK-TBI. The comparison group was 230 patients with nonhead orthopedic trauma injuries, wrote Dr. Stein, distinguished professor of psychiatry and family medicine and public health at the University of California, San Diego, and his associates.
They found that each additional year of education was associated with a significant 11% reduction in the risk of developing PTSD after mild TBI (P = .005). Also, black patients had a greater than fivefold higher risk of PTSD (P less than.001) than that of individuals who were not black.
Among patients with a history of mental illness and those who had experienced their injury as a result of assault or violence – as opposed to a motor vehicle accident or fall, for example – both had a greater than threefold higher risk of developing PTSD (odds ratio, 3.57 and 3.43 respectively). A prior TBI was nonsignificantly associated with an increased risk of developing PTSD.
Lower education duration, being black, or a history of mental illness also were all significantly associated with an increased risk of developing major depressive disorder after mild TBI.
However, duration of lost consciousness or posttraumatic amnesia, evidence of brain injury on CT, or hospitalization did not predict an increased risk of PTSD or major depression.
“Although MDD and PTSD are prevalent after TBI, little is known about which patients are at risk for developing them,” Dr. Stein and his associates wrote.
Noting that having a prior mental health problem was an “exceptionally strong” risk factor for PTSD and MDD after TBI, the authors said this could represent continuation or exacerbation of the prior mental health issue, or the triggering of a new episode in a person with a past history who had recovered.
“However, in either case this finding underscores the importance of clinicians being aware of the mental health history of their patients with [mild TBI], as this information is central to expectations regarding both short-term and long-term outcome,” they wrote.
Dr. Stein and his associates cited as a limitation their reliance on patient or family report. In addition, they said, the elevated risk for mental disorders among black individuals after mild TBI, which was independent of socioeconomic status or cause of injury, was not understood. “Unmeasured covariates may be part of the explanation; this is a topic needing further study,” they wrote.
The study was supported by the National Institutes of Health, the U.S. Department of Defense, Abbott Laboratories, and One Mind. Four authors declared consultancies, advisory board positions, speaking fees, and shares or stock options with the pharmaceutical and private industry. Two authors declared grants from the study sponsors.
SOURCE: Stein MB et al. JAMA Psychiatry. 2019. Jan 30. doi: 10.1001/jamapsychiatry.2018.4288.
Civilian patients with mild traumatic brain injury (TBI) who are black, have psychiatric history or lower education, or whose injury was caused by assault might be at greater risk of developing posttraumatic stress disorder or major depression, a longitudinal study suggests.
“Our findings may have implications for surveillance and treatment of mental disorders after TBI,” wrote Murray B. Stein, MD, MPH, and his associates. The study was published Jan. 30 in JAMA Psychiatry.
The researchers looked at the risk factors for and prevalence of posttraumatic stress disorder (PTSD) and major depressive disorder among 1,155 patients. The patients were enrolled at 11 level 1 trauma centers across the United States after they were evaluated for mild TBI in emergency departments as part of a prospective study called Transforming Research and Clinical Knowledge in Traumatic Brain Injury, or TRACK-TBI. The comparison group was 230 patients with nonhead orthopedic trauma injuries, wrote Dr. Stein, distinguished professor of psychiatry and family medicine and public health at the University of California, San Diego, and his associates.
They found that each additional year of education was associated with a significant 11% reduction in the risk of developing PTSD after mild TBI (P = .005). Also, black patients had a greater than fivefold higher risk of PTSD (P less than.001) than that of individuals who were not black.
Among patients with a history of mental illness and those who had experienced their injury as a result of assault or violence – as opposed to a motor vehicle accident or fall, for example – both had a greater than threefold higher risk of developing PTSD (odds ratio, 3.57 and 3.43 respectively). A prior TBI was nonsignificantly associated with an increased risk of developing PTSD.
Lower education duration, being black, or a history of mental illness also were all significantly associated with an increased risk of developing major depressive disorder after mild TBI.
However, duration of lost consciousness or posttraumatic amnesia, evidence of brain injury on CT, or hospitalization did not predict an increased risk of PTSD or major depression.
“Although MDD and PTSD are prevalent after TBI, little is known about which patients are at risk for developing them,” Dr. Stein and his associates wrote.
Noting that having a prior mental health problem was an “exceptionally strong” risk factor for PTSD and MDD after TBI, the authors said this could represent continuation or exacerbation of the prior mental health issue, or the triggering of a new episode in a person with a past history who had recovered.
“However, in either case this finding underscores the importance of clinicians being aware of the mental health history of their patients with [mild TBI], as this information is central to expectations regarding both short-term and long-term outcome,” they wrote.
Dr. Stein and his associates cited as a limitation their reliance on patient or family report. In addition, they said, the elevated risk for mental disorders among black individuals after mild TBI, which was independent of socioeconomic status or cause of injury, was not understood. “Unmeasured covariates may be part of the explanation; this is a topic needing further study,” they wrote.
The study was supported by the National Institutes of Health, the U.S. Department of Defense, Abbott Laboratories, and One Mind. Four authors declared consultancies, advisory board positions, speaking fees, and shares or stock options with the pharmaceutical and private industry. Two authors declared grants from the study sponsors.
SOURCE: Stein MB et al. JAMA Psychiatry. 2019. Jan 30. doi: 10.1001/jamapsychiatry.2018.4288.
FROM JAMA PSYCHIATRY
Key clinical point: The findings underscore “the importance of clinicians being aware of the mental health history of their patients with [mild TBI], as this information is central to expectations regarding both short-term and long-term outcome.”
Major finding: Black patients have fivefold higher risk of PTSD after brain injury.
Study details: Longitudinal cohort study of 1,155 patients with mild traumatic brain injury.
Disclosures: The study was supported by the National Institutes of Health, the U.S. Department of Defense, Abbott Laboratories, and One Mind. Four authors declared consultancies, advisory board positions, and speaking fees, shares, or stock options with the pharmaceutical and private industry. Two authors declared grants from the study sponsors.
Source: Stein MB et al. JAMA Psychiatry 2019. Jan 30. doi: 10.1001/jamapsychiatry.2018.4288.
Gastrectomy does not alter benefit of new oral chemo in gastric cancer
SAN FRANCISCO – suggests a preplanned subgroup analysis of the global phase 3 randomized controlled TAGS trial. Results were reported at the 2019 GI Cancers Symposium.

“The standard of care for early-stage gastric cancer is surgery, which is the only potentially curative treatment,” noted lead investigator David H. Ilson, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York. “Forty percent of patients with metastatic disease have had a history of previous gastrectomy.”
The TAGS trial assessed efficacy of combined trifluridine and tipiracil (Lonsurf) among 507 patients with metastatic gastric or gastroesophageal junction cancer who had received at least two prior chemotherapy regimens. (This combination chemotherapy is currently approved in the United States as later-line therapy for metastatic colorectal cancer.)
Compared with placebo, trifluridine/tipiracil prolonged overall survival by 2.6 months in the subgroup who had previously undergone gastrectomy, a benefit slightly greater than the 2.1 months previously reported for the entire trial population (Lancet Oncol. 2018;19:1437-48). And although the gastrectomy subgroup experienced more grade 3 or 4 adverse events, they were not more likely to stop treatment because of toxicity.
“The data from this analysis reinforce the benefit for trifluridine/tipiracil as prolonging survival versus placebo, and this is regardless of prior gastrectomy,” Dr. Ilson summarized. “Hematologic adverse events, such as neutropenia and leukopenia, may have been somewhat more frequent among the trifluridine/tipiracil–treated patients with gastrectomy than in the overall population, but this did not result in more treatment discontinuations. Exposure to the drug was similar between patients with gastrectomy and those in the overall population.”
“Trifluridine/tipiracil is an effective treatment option with a manageable toxicity profile for patients with metastatic gastric cancer, regardless of prior gastrectomy status,” he concluded.
Still fit for treatment
The disconnect between toxicity and treatment discontinuation seen in the TAGS trial is not new, according to invited discussant Martine Extermann, MD, PhD, leader of the Senior Adult Oncology Program at the Moffitt Cancer Center in Tampa. Previous data among geriatric cancer patients have similarly shown that, despite substantial chemotherapy toxicity, by and large, there are only modest effects on health-related quality of life, performance status, and instrumental activities of daily living, she noted.
“The CTCAE [Common Terminology Criteria for Adverse Events] toxicity differences do not always translate into functional impact and treatment cessation. So this is only part of the picture. It’s a convenient part. It’s easily measurable. It’s well acknowledged as a measurement of side effects. But it does not tell the whole story. Quality of life and functional status add to the picture,” Dr. Extermann elaborated. “What the TAGS study is telling us is, despite a gastrectomy, these patients can be treated as a third-line treatment population for gastric cancer, which is not necessarily obvious to every oncologist.”
At the same time, she added that it would be helpful to have nutritional data on the study patients – and on all patients in similar trials, for that matter – because nutritional status is one of the components of the CRASH (Chemotherapy Risk Assessment Scale for High-Age Patients) score used to predict chemotherapy toxicity in older adults.
“I would support that [trifluridine/tipiracil] is an effective third-line chemotherapy in gastric cancer patients with or without prior gastrectomy, and this can be given safely,” Dr. Extermann concluded.
Study details
Patients in TAGS were randomized 2:1 to trifluridine/tipiracil (formerly TAS-102) or placebo, each added to best supportive care. (Trifluridine is a novel oral thymidine analogue, and tipiracil prevents trifluridine degradation.) Overall, 44% had undergone gastrectomy before entering the trial.
In the entire trial population, overall survival was 5.7 months with trifluridine/tipiracil and 3.6 months with placebo (hazard ratio, 0.69; 95% confidence interval, 0.56-0.85; P = .0006), as previously reported.
Among the subgroup who had undergone prior gastrectomy, overall survival was 6.0 months with trifluridine/tipiracil and 3.4 months with placebo (HR, 0.57; 95% CI, 0.41-0.79), Dr. Ilson reported at the symposium, which is sponsored by the American Gastroenterological Association, the American Society for Clinical Oncology, the American Society for Radiation Oncology, and the Society of Surgical Oncology. The combination also netted better progression-free survival (2.2 vs. 1.8 months; HR, 0.48; 95% CI, 0.35-0.65). “These data mirror the data seen in the overall treatment population,” he commented.
In a multivariate analysis including all prespecified factors, prior gastrectomy was neither prognostic nor predictive. Moreover, the treatment effect size remained the same after adjustment for potential prognostic factors.
Exposure to trifluridine/tipiracil was similar for the gastrectomy subgroup and the entire trial population in terms of relative dose intensity, median number of cycles, and treatment duration.
The rate of grade 3 or 4 treatment-related adverse events with trifluridine/tipiracil in the gastrectomy subgroup, 64%, was higher than that in the entire trial population, 53%, but the rate of discontinuation because of any-grade adverse events was similar, at 10% and 13%, respectively.
The difference in grade 3 or 4 adverse events between the gastrectomy subgroup and the entire trial population was mainly driven by higher rates of neutropenia (44% vs. 34%) and leukopenia (14% vs. 9%) in the former.
Dr. Ilson disclosed that he has a consulting role with Amgen, Astellas, AstraZeneca, Bayer, Bristol-Myers Squibb, Lilly, Merck, Pieris, Roche/Genentech, and Taiho and that he receives research support from Taiho. The study was funded by Taiho Oncology and Taiho Pharmaceutical.
SOURCE: Ilson DH et al. 2019 GI Cancers Symposium, Abstract 3.
SAN FRANCISCO – suggests a preplanned subgroup analysis of the global phase 3 randomized controlled TAGS trial. Results were reported at the 2019 GI Cancers Symposium.
“The standard of care for early-stage gastric cancer is surgery, which is the only potentially curative treatment,” noted lead investigator David H. Ilson, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York. “Forty percent of patients with metastatic disease have had a history of previous gastrectomy.”
The TAGS trial assessed efficacy of combined trifluridine and tipiracil (Lonsurf) among 507 patients with metastatic gastric or gastroesophageal junction cancer who had received at least two prior chemotherapy regimens. (This combination chemotherapy is currently approved in the United States as later-line therapy for metastatic colorectal cancer.)
Compared with placebo, trifluridine/tipiracil prolonged overall survival by 2.6 months in the subgroup who had previously undergone gastrectomy, a benefit slightly greater than the 2.1 months previously reported for the entire trial population (Lancet Oncol. 2018;19:1437-48). And although the gastrectomy subgroup experienced more grade 3 or 4 adverse events, they were not more likely to stop treatment because of toxicity.
“The data from this analysis reinforce the benefit for trifluridine/tipiracil as prolonging survival versus placebo, and this is regardless of prior gastrectomy,” Dr. Ilson summarized. “Hematologic adverse events, such as neutropenia and leukopenia, may have been somewhat more frequent among the trifluridine/tipiracil–treated patients with gastrectomy than in the overall population, but this did not result in more treatment discontinuations. Exposure to the drug was similar between patients with gastrectomy and those in the overall population.”
“Trifluridine/tipiracil is an effective treatment option with a manageable toxicity profile for patients with metastatic gastric cancer, regardless of prior gastrectomy status,” he concluded.
Still fit for treatment
The disconnect between toxicity and treatment discontinuation seen in the TAGS trial is not new, according to invited discussant Martine Extermann, MD, PhD, leader of the Senior Adult Oncology Program at the Moffitt Cancer Center in Tampa. Previous data among geriatric cancer patients have similarly shown that, despite substantial chemotherapy toxicity, by and large, there are only modest effects on health-related quality of life, performance status, and instrumental activities of daily living, she noted.
“The CTCAE [Common Terminology Criteria for Adverse Events] toxicity differences do not always translate into functional impact and treatment cessation. So this is only part of the picture. It’s a convenient part. It’s easily measurable. It’s well acknowledged as a measurement of side effects. But it does not tell the whole story. Quality of life and functional status add to the picture,” Dr. Extermann elaborated. “What the TAGS study is telling us is, despite a gastrectomy, these patients can be treated as a third-line treatment population for gastric cancer, which is not necessarily obvious to every oncologist.”
At the same time, she added that it would be helpful to have nutritional data on the study patients – and on all patients in similar trials, for that matter – because nutritional status is one of the components of the CRASH (Chemotherapy Risk Assessment Scale for High-Age Patients) score used to predict chemotherapy toxicity in older adults.
“I would support that [trifluridine/tipiracil] is an effective third-line chemotherapy in gastric cancer patients with or without prior gastrectomy, and this can be given safely,” Dr. Extermann concluded.
Study details
Patients in TAGS were randomized 2:1 to trifluridine/tipiracil (formerly TAS-102) or placebo, each added to best supportive care. (Trifluridine is a novel oral thymidine analogue, and tipiracil prevents trifluridine degradation.) Overall, 44% had undergone gastrectomy before entering the trial.
In the entire trial population, overall survival was 5.7 months with trifluridine/tipiracil and 3.6 months with placebo (hazard ratio, 0.69; 95% confidence interval, 0.56-0.85; P = .0006), as previously reported.
Among the subgroup who had undergone prior gastrectomy, overall survival was 6.0 months with trifluridine/tipiracil and 3.4 months with placebo (HR, 0.57; 95% CI, 0.41-0.79), Dr. Ilson reported at the symposium, which is sponsored by the American Gastroenterological Association, the American Society for Clinical Oncology, the American Society for Radiation Oncology, and the Society of Surgical Oncology. The combination also netted better progression-free survival (2.2 vs. 1.8 months; HR, 0.48; 95% CI, 0.35-0.65). “These data mirror the data seen in the overall treatment population,” he commented.
In a multivariate analysis including all prespecified factors, prior gastrectomy was neither prognostic nor predictive. Moreover, the treatment effect size remained the same after adjustment for potential prognostic factors.
Exposure to trifluridine/tipiracil was similar for the gastrectomy subgroup and the entire trial population in terms of relative dose intensity, median number of cycles, and treatment duration.
The rate of grade 3 or 4 treatment-related adverse events with trifluridine/tipiracil in the gastrectomy subgroup, 64%, was higher than that in the entire trial population, 53%, but the rate of discontinuation because of any-grade adverse events was similar, at 10% and 13%, respectively.
The difference in grade 3 or 4 adverse events between the gastrectomy subgroup and the entire trial population was mainly driven by higher rates of neutropenia (44% vs. 34%) and leukopenia (14% vs. 9%) in the former.
Dr. Ilson disclosed that he has a consulting role with Amgen, Astellas, AstraZeneca, Bayer, Bristol-Myers Squibb, Lilly, Merck, Pieris, Roche/Genentech, and Taiho and that he receives research support from Taiho. The study was funded by Taiho Oncology and Taiho Pharmaceutical.
SOURCE: Ilson DH et al. 2019 GI Cancers Symposium, Abstract 3.
SAN FRANCISCO – suggests a preplanned subgroup analysis of the global phase 3 randomized controlled TAGS trial. Results were reported at the 2019 GI Cancers Symposium.
“The standard of care for early-stage gastric cancer is surgery, which is the only potentially curative treatment,” noted lead investigator David H. Ilson, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York. “Forty percent of patients with metastatic disease have had a history of previous gastrectomy.”
The TAGS trial assessed efficacy of combined trifluridine and tipiracil (Lonsurf) among 507 patients with metastatic gastric or gastroesophageal junction cancer who had received at least two prior chemotherapy regimens. (This combination chemotherapy is currently approved in the United States as later-line therapy for metastatic colorectal cancer.)
Compared with placebo, trifluridine/tipiracil prolonged overall survival by 2.6 months in the subgroup who had previously undergone gastrectomy, a benefit slightly greater than the 2.1 months previously reported for the entire trial population (Lancet Oncol. 2018;19:1437-48). And although the gastrectomy subgroup experienced more grade 3 or 4 adverse events, they were not more likely to stop treatment because of toxicity.
“The data from this analysis reinforce the benefit for trifluridine/tipiracil as prolonging survival versus placebo, and this is regardless of prior gastrectomy,” Dr. Ilson summarized. “Hematologic adverse events, such as neutropenia and leukopenia, may have been somewhat more frequent among the trifluridine/tipiracil–treated patients with gastrectomy than in the overall population, but this did not result in more treatment discontinuations. Exposure to the drug was similar between patients with gastrectomy and those in the overall population.”
“Trifluridine/tipiracil is an effective treatment option with a manageable toxicity profile for patients with metastatic gastric cancer, regardless of prior gastrectomy status,” he concluded.
Still fit for treatment
The disconnect between toxicity and treatment discontinuation seen in the TAGS trial is not new, according to invited discussant Martine Extermann, MD, PhD, leader of the Senior Adult Oncology Program at the Moffitt Cancer Center in Tampa. Previous data among geriatric cancer patients have similarly shown that, despite substantial chemotherapy toxicity, by and large, there are only modest effects on health-related quality of life, performance status, and instrumental activities of daily living, she noted.
“The CTCAE [Common Terminology Criteria for Adverse Events] toxicity differences do not always translate into functional impact and treatment cessation. So this is only part of the picture. It’s a convenient part. It’s easily measurable. It’s well acknowledged as a measurement of side effects. But it does not tell the whole story. Quality of life and functional status add to the picture,” Dr. Extermann elaborated. “What the TAGS study is telling us is, despite a gastrectomy, these patients can be treated as a third-line treatment population for gastric cancer, which is not necessarily obvious to every oncologist.”
At the same time, she added that it would be helpful to have nutritional data on the study patients – and on all patients in similar trials, for that matter – because nutritional status is one of the components of the CRASH (Chemotherapy Risk Assessment Scale for High-Age Patients) score used to predict chemotherapy toxicity in older adults.
“I would support that [trifluridine/tipiracil] is an effective third-line chemotherapy in gastric cancer patients with or without prior gastrectomy, and this can be given safely,” Dr. Extermann concluded.
Study details
Patients in TAGS were randomized 2:1 to trifluridine/tipiracil (formerly TAS-102) or placebo, each added to best supportive care. (Trifluridine is a novel oral thymidine analogue, and tipiracil prevents trifluridine degradation.) Overall, 44% had undergone gastrectomy before entering the trial.
In the entire trial population, overall survival was 5.7 months with trifluridine/tipiracil and 3.6 months with placebo (hazard ratio, 0.69; 95% confidence interval, 0.56-0.85; P = .0006), as previously reported.
Among the subgroup who had undergone prior gastrectomy, overall survival was 6.0 months with trifluridine/tipiracil and 3.4 months with placebo (HR, 0.57; 95% CI, 0.41-0.79), Dr. Ilson reported at the symposium, which is sponsored by the American Gastroenterological Association, the American Society for Clinical Oncology, the American Society for Radiation Oncology, and the Society of Surgical Oncology. The combination also netted better progression-free survival (2.2 vs. 1.8 months; HR, 0.48; 95% CI, 0.35-0.65). “These data mirror the data seen in the overall treatment population,” he commented.
In a multivariate analysis including all prespecified factors, prior gastrectomy was neither prognostic nor predictive. Moreover, the treatment effect size remained the same after adjustment for potential prognostic factors.
Exposure to trifluridine/tipiracil was similar for the gastrectomy subgroup and the entire trial population in terms of relative dose intensity, median number of cycles, and treatment duration.
The rate of grade 3 or 4 treatment-related adverse events with trifluridine/tipiracil in the gastrectomy subgroup, 64%, was higher than that in the entire trial population, 53%, but the rate of discontinuation because of any-grade adverse events was similar, at 10% and 13%, respectively.
The difference in grade 3 or 4 adverse events between the gastrectomy subgroup and the entire trial population was mainly driven by higher rates of neutropenia (44% vs. 34%) and leukopenia (14% vs. 9%) in the former.
Dr. Ilson disclosed that he has a consulting role with Amgen, Astellas, AstraZeneca, Bayer, Bristol-Myers Squibb, Lilly, Merck, Pieris, Roche/Genentech, and Taiho and that he receives research support from Taiho. The study was funded by Taiho Oncology and Taiho Pharmaceutical.
SOURCE: Ilson DH et al. 2019 GI Cancers Symposium, Abstract 3.
REPORTING FROM THE 2019 GI CANCERS SYMPOSIUM
Key clinical point: Patients with metastatic gastric cancer experience largely similar efficacy and safety outcomes with oral trifluridine/tipiracil regardless of prior gastrectomy.
Major finding: Compared with placebo, trifluridine/tipiracil improved overall survival in the gastrectomy subgroup (hazard ratio, 0.57) with a higher rate of grade 3/4 adverse events in that subgroup (64% vs. 53%) but similar rate of discontinuation because of adverse events (10% vs. 13%).
Study details: Preplanned subgroup analysis of a phase 3 randomized controlled trial (TAGS trial) among 507 patients with metastatic gastric or gastroesophageal junction cancer who had received at least two prior chemotherapy regimens.
Disclosures: Dr. Ilson disclosed that he has a consulting role with Amgen, Astellas, AstraZeneca, Bayer, Bristol-Myers Squibb, Lilly, Merck, Pieris, Roche/Genentech, and Taiho and that he receives research support from Taiho. The study was funded by Taiho Oncology and Taiho Pharmaceutical.
Source: Ilson DH et al. 2019 GI Cancers Symposium, Abstract 3.
IPH4102 on fast track for Sézary syndrome
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
Suicide Prevention on the Job
Many adults spend a large part of their time at work—making the workplace an important but underused location for suicide prevention, say CDC researchers who analyzed data on 22,053 suicides in 17 states. The US suicide rate among working adults (aged 16-64 years) rose 34% between 2000 and 2016, from 12.9 to 17.3 per 100,000.
Suicide rates rose in many occupational groups between 2012 and 2015, but identifying the specific role that occupational factors might play in suicide risk is complicated, the researchers say: Both work (eg, little job control and job insecurity) and nonwork (eg, relationship conflict) factors are associated with psychological distress and suicide. And factors such as access to lethal means while on the job play a part as well.
The major occupational group with the highest male suicide rate was Construction and Extraction (from 43.6% in 2012 to 53.2% in 2015. The Arts, Design, Entertainment, Sports, and Media groups had the highest female suicide rate (15.6%, up from 11.7%).
Health Care Support, which ranked twelfth on the 2015 list, actually saw a drop in the numbers of male suicides (19.5/100,000 in 2015, vs 22.1 in 2012). But among women, the numbers rose 31%: from 8.4 per 100,000 to 11.0, making that category third among females.
With a 13% drop (from 10.3 to 9.0), Health Care Practitioners and Technical (female) moved from fourth to sixth place. For men, the category ranked eighth, with a rate change of 23%, from 20.8 to 25.6 suicide deaths per 100,000.
The researchers say better understanding of how suicides are distributed by occupational group might help inform prevention programs and policies. The CDC recommends a comprehensive approach, including strategies such as:
- Enhancing social connectedness;
- Strengthening state or local economic supports;
- Implementing practices that encourage help-seeking and reduce stigma;
- Providing referrals to mental health and other services; and
- Reducing access to lethal means among people at risk
The CDC also encourages decision makers, such as employers, to create a response plan, should someone in their organization commit suicide.
Many adults spend a large part of their time at work—making the workplace an important but underused location for suicide prevention, say CDC researchers who analyzed data on 22,053 suicides in 17 states. The US suicide rate among working adults (aged 16-64 years) rose 34% between 2000 and 2016, from 12.9 to 17.3 per 100,000.
Suicide rates rose in many occupational groups between 2012 and 2015, but identifying the specific role that occupational factors might play in suicide risk is complicated, the researchers say: Both work (eg, little job control and job insecurity) and nonwork (eg, relationship conflict) factors are associated with psychological distress and suicide. And factors such as access to lethal means while on the job play a part as well.
The major occupational group with the highest male suicide rate was Construction and Extraction (from 43.6% in 2012 to 53.2% in 2015. The Arts, Design, Entertainment, Sports, and Media groups had the highest female suicide rate (15.6%, up from 11.7%).
Health Care Support, which ranked twelfth on the 2015 list, actually saw a drop in the numbers of male suicides (19.5/100,000 in 2015, vs 22.1 in 2012). But among women, the numbers rose 31%: from 8.4 per 100,000 to 11.0, making that category third among females.
With a 13% drop (from 10.3 to 9.0), Health Care Practitioners and Technical (female) moved from fourth to sixth place. For men, the category ranked eighth, with a rate change of 23%, from 20.8 to 25.6 suicide deaths per 100,000.
The researchers say better understanding of how suicides are distributed by occupational group might help inform prevention programs and policies. The CDC recommends a comprehensive approach, including strategies such as:
- Enhancing social connectedness;
- Strengthening state or local economic supports;
- Implementing practices that encourage help-seeking and reduce stigma;
- Providing referrals to mental health and other services; and
- Reducing access to lethal means among people at risk
The CDC also encourages decision makers, such as employers, to create a response plan, should someone in their organization commit suicide.
Many adults spend a large part of their time at work—making the workplace an important but underused location for suicide prevention, say CDC researchers who analyzed data on 22,053 suicides in 17 states. The US suicide rate among working adults (aged 16-64 years) rose 34% between 2000 and 2016, from 12.9 to 17.3 per 100,000.
Suicide rates rose in many occupational groups between 2012 and 2015, but identifying the specific role that occupational factors might play in suicide risk is complicated, the researchers say: Both work (eg, little job control and job insecurity) and nonwork (eg, relationship conflict) factors are associated with psychological distress and suicide. And factors such as access to lethal means while on the job play a part as well.
The major occupational group with the highest male suicide rate was Construction and Extraction (from 43.6% in 2012 to 53.2% in 2015. The Arts, Design, Entertainment, Sports, and Media groups had the highest female suicide rate (15.6%, up from 11.7%).
Health Care Support, which ranked twelfth on the 2015 list, actually saw a drop in the numbers of male suicides (19.5/100,000 in 2015, vs 22.1 in 2012). But among women, the numbers rose 31%: from 8.4 per 100,000 to 11.0, making that category third among females.
With a 13% drop (from 10.3 to 9.0), Health Care Practitioners and Technical (female) moved from fourth to sixth place. For men, the category ranked eighth, with a rate change of 23%, from 20.8 to 25.6 suicide deaths per 100,000.
The researchers say better understanding of how suicides are distributed by occupational group might help inform prevention programs and policies. The CDC recommends a comprehensive approach, including strategies such as:
- Enhancing social connectedness;
- Strengthening state or local economic supports;
- Implementing practices that encourage help-seeking and reduce stigma;
- Providing referrals to mental health and other services; and
- Reducing access to lethal means among people at risk
The CDC also encourages decision makers, such as employers, to create a response plan, should someone in their organization commit suicide.
Medical ethics and economics
The balance between medical research and the pharmaceutical world has always been unsettling. The recent spate of articles in the press reporting the large payments by industry to a number of highly paid medical staff of the Memorial Sloan Kettering Cancer Institute in New York has raised again the continuing issue around that relationship.

When large sums of money are paid to medical leaders for serving on advisory boards, it is reasonable to question whom are they representing: industry or medical science. These relationships are not limited to cancer hospitals and can be presumed to pertain to cardiology and other specialties. One need only look at the disclosure statements of contemporary published articles to become aware of the entanglement of science and industry.
There is little question that both industry and science need to interact to focus direct resources to appropriate targets. No one is better able to do that than well informed scientists working in their disease fields. Industry needs scientific input and scientists need the financial resources of industry. I have been able to see that relationship play out to achieve major impacts on heart disease. But corporate decisions also can be driven by market forces and not altruism. Drug and device research has been redirected or stopped as a result of decisions made by sales forces. At other times, drugs that have great potential in the laboratory have been shelved because of a lack of scientific leadership.
So where is the moral and ethical balance? Published disclosures by authors is not much more than a catharsis in the process where action is required. Medical advisory boards are critical for successful drug and device development. That exchange is crucial to move medical science forward, but the large sums of money raise appropriate questions of what is driving the discussion.
At a more grass roots level, the financial role of investigators and hospitals in clinical trials has raised some concern. Traditionally, the institution and investigators have been reimbursed for their time and expense for recruiting and following patients. Patients, of course, are not reimbursed in clinical trials but are placed at considerable risk of an uncertain result. The reimbursements for marginal expenses seem to be appropriate. More recently, payments to physicians and hospitals have been made at current fee schedules for the implantation of a variety of new devices such as pacemakers and valves. In addition, both physicians and hospitals have invested in the financial success of these clinical trials clouding over their altruistic goals. It has been an incentive for recruiting patients for trials and has been a source of considerable revenue both for the physicians and the institution, without informing the patients of their financial relationship to industry. .
There is a lot of money sloshing around in the health care world that has the potential to lead to ethical uncertainty. It is the physician’s responsibility to build up ethical barriers to prevent us from slipping into that morass.
Dr. Goldstein is professor of medicine at Wayne State University and the division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit.
The balance between medical research and the pharmaceutical world has always been unsettling. The recent spate of articles in the press reporting the large payments by industry to a number of highly paid medical staff of the Memorial Sloan Kettering Cancer Institute in New York has raised again the continuing issue around that relationship.
When large sums of money are paid to medical leaders for serving on advisory boards, it is reasonable to question whom are they representing: industry or medical science. These relationships are not limited to cancer hospitals and can be presumed to pertain to cardiology and other specialties. One need only look at the disclosure statements of contemporary published articles to become aware of the entanglement of science and industry.
There is little question that both industry and science need to interact to focus direct resources to appropriate targets. No one is better able to do that than well informed scientists working in their disease fields. Industry needs scientific input and scientists need the financial resources of industry. I have been able to see that relationship play out to achieve major impacts on heart disease. But corporate decisions also can be driven by market forces and not altruism. Drug and device research has been redirected or stopped as a result of decisions made by sales forces. At other times, drugs that have great potential in the laboratory have been shelved because of a lack of scientific leadership.
So where is the moral and ethical balance? Published disclosures by authors is not much more than a catharsis in the process where action is required. Medical advisory boards are critical for successful drug and device development. That exchange is crucial to move medical science forward, but the large sums of money raise appropriate questions of what is driving the discussion.
At a more grass roots level, the financial role of investigators and hospitals in clinical trials has raised some concern. Traditionally, the institution and investigators have been reimbursed for their time and expense for recruiting and following patients. Patients, of course, are not reimbursed in clinical trials but are placed at considerable risk of an uncertain result. The reimbursements for marginal expenses seem to be appropriate. More recently, payments to physicians and hospitals have been made at current fee schedules for the implantation of a variety of new devices such as pacemakers and valves. In addition, both physicians and hospitals have invested in the financial success of these clinical trials clouding over their altruistic goals. It has been an incentive for recruiting patients for trials and has been a source of considerable revenue both for the physicians and the institution, without informing the patients of their financial relationship to industry. .
There is a lot of money sloshing around in the health care world that has the potential to lead to ethical uncertainty. It is the physician’s responsibility to build up ethical barriers to prevent us from slipping into that morass.
Dr. Goldstein is professor of medicine at Wayne State University and the division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit.
The balance between medical research and the pharmaceutical world has always been unsettling. The recent spate of articles in the press reporting the large payments by industry to a number of highly paid medical staff of the Memorial Sloan Kettering Cancer Institute in New York has raised again the continuing issue around that relationship.
When large sums of money are paid to medical leaders for serving on advisory boards, it is reasonable to question whom are they representing: industry or medical science. These relationships are not limited to cancer hospitals and can be presumed to pertain to cardiology and other specialties. One need only look at the disclosure statements of contemporary published articles to become aware of the entanglement of science and industry.
There is little question that both industry and science need to interact to focus direct resources to appropriate targets. No one is better able to do that than well informed scientists working in their disease fields. Industry needs scientific input and scientists need the financial resources of industry. I have been able to see that relationship play out to achieve major impacts on heart disease. But corporate decisions also can be driven by market forces and not altruism. Drug and device research has been redirected or stopped as a result of decisions made by sales forces. At other times, drugs that have great potential in the laboratory have been shelved because of a lack of scientific leadership.
So where is the moral and ethical balance? Published disclosures by authors is not much more than a catharsis in the process where action is required. Medical advisory boards are critical for successful drug and device development. That exchange is crucial to move medical science forward, but the large sums of money raise appropriate questions of what is driving the discussion.
At a more grass roots level, the financial role of investigators and hospitals in clinical trials has raised some concern. Traditionally, the institution and investigators have been reimbursed for their time and expense for recruiting and following patients. Patients, of course, are not reimbursed in clinical trials but are placed at considerable risk of an uncertain result. The reimbursements for marginal expenses seem to be appropriate. More recently, payments to physicians and hospitals have been made at current fee schedules for the implantation of a variety of new devices such as pacemakers and valves. In addition, both physicians and hospitals have invested in the financial success of these clinical trials clouding over their altruistic goals. It has been an incentive for recruiting patients for trials and has been a source of considerable revenue both for the physicians and the institution, without informing the patients of their financial relationship to industry. .
There is a lot of money sloshing around in the health care world that has the potential to lead to ethical uncertainty. It is the physician’s responsibility to build up ethical barriers to prevent us from slipping into that morass.
Dr. Goldstein is professor of medicine at Wayne State University and the division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit.
Tardive dyskinesia: Masterclass
In this Masterclass edition from the 2018 AACP Encore meeting in Las Vegas, Dr. Meyer talks about this movement disorder and analyzes how psychiatrists should approach treatment today.
In this Masterclass edition from the 2018 AACP Encore meeting in Las Vegas, Dr. Meyer talks about this movement disorder and analyzes how psychiatrists should approach treatment today.
In this Masterclass edition from the 2018 AACP Encore meeting in Las Vegas, Dr. Meyer talks about this movement disorder and analyzes how psychiatrists should approach treatment today.

Physician mothers caring for others at home
Also today, what one neurologist refuses to accept in his office, flu activity increases after 2 weeks of declines, and when sweaty palms are really primary hyperhidrosis.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
Also today, what one neurologist refuses to accept in his office, flu activity increases after 2 weeks of declines, and when sweaty palms are really primary hyperhidrosis.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
Also today, what one neurologist refuses to accept in his office, flu activity increases after 2 weeks of declines, and when sweaty palms are really primary hyperhidrosis.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
