User login
CNS lymphoma guidelines stress patient fitness, not age, in choosing treatment
for the diagnosis and management of primary central nervous system diffuse large B‐cell lymphoma.
PCNSL, implicated in some 3% of all brain tumors, is complex to diagnose and treat. People with suspected PCNSL must receive quick and coordinated attention from a multidisciplinary team of neurologists, hematologist-oncologists, and ocular specialists, according to the guidelines, published in the British Journal of Haematology.
Christopher P. Fox, MD, of the Nottingham (England) University Hospitals NHS Trust, and his colleagues, stress the importance of early multidisciplinary attention, aggressive induction treatment, helping patients into trials, universal screening for eye involvement, attaining histological diagnoses in addition to imaging findings, and avoidance or discontinuation of any corticosteroids before biopsy, as even a short course of steroids can impede diagnosis.
The guidelines incorporate findings from studies published since the society’s last comprehensive PCNSL guideline was issued more than a decade ago.
Dr. Fox and his colleagues say definitive treatment for PCNSL – induction of remission followed by consolidation – should start within 2 weeks of diagnosis and that a treatment regimen should be chosen according to a patient’s physiological fitness, not age. The fittest patients, who have better organ function and fewer comorbidities, should be eligible for intensive combination immunochemotherapy incorporating high-dose methotrexate (optimally four cycles of HD-MTX, cytarabine, thiotepa, and rituximab). Those deemed unfit for this regimen should be offered induction treatment with HD-MTX, rituximab and procarbazine, the guidelines’ authors say.
If patients cannot tolerate HD-MTX, oral chemotherapy and/or whole-brain radiotherapy may be offered. Response should be assessed with contrast-enhanced magnetic resonance imaging.
Consolidation therapy should be initiated after induction for all patients with nonprogressive disease, and high-dose thiotepa-based chemotherapy with autologous stem cell transplant is the recommended first-line option for consolidation. Response to consolidation, again measured with contrast-enhanced MRI, should be carried out at between 1 and 2 months after therapy is completed, and patients should be referred for neuropsychological testing to assess cognitive function.
Patients with relapsed or refractory disease should be approached with maximum urgency – the guidelines offer an algorithm for retreatment options – and offered clinical trial entry wherever possible.
The PCNSL guideline writing process was sponsored by the British Society for Haematology, and some coauthors, including the lead author, disclosed receiving fees from pharmaceutical manufacturers Adienne or F. Hoffman-La Roche.
SOURCE: Fox et al. Br J Haematol. 2018 Nov 23 doi: 10.1111/bjh.15661.
for the diagnosis and management of primary central nervous system diffuse large B‐cell lymphoma.
PCNSL, implicated in some 3% of all brain tumors, is complex to diagnose and treat. People with suspected PCNSL must receive quick and coordinated attention from a multidisciplinary team of neurologists, hematologist-oncologists, and ocular specialists, according to the guidelines, published in the British Journal of Haematology.
Christopher P. Fox, MD, of the Nottingham (England) University Hospitals NHS Trust, and his colleagues, stress the importance of early multidisciplinary attention, aggressive induction treatment, helping patients into trials, universal screening for eye involvement, attaining histological diagnoses in addition to imaging findings, and avoidance or discontinuation of any corticosteroids before biopsy, as even a short course of steroids can impede diagnosis.
The guidelines incorporate findings from studies published since the society’s last comprehensive PCNSL guideline was issued more than a decade ago.
Dr. Fox and his colleagues say definitive treatment for PCNSL – induction of remission followed by consolidation – should start within 2 weeks of diagnosis and that a treatment regimen should be chosen according to a patient’s physiological fitness, not age. The fittest patients, who have better organ function and fewer comorbidities, should be eligible for intensive combination immunochemotherapy incorporating high-dose methotrexate (optimally four cycles of HD-MTX, cytarabine, thiotepa, and rituximab). Those deemed unfit for this regimen should be offered induction treatment with HD-MTX, rituximab and procarbazine, the guidelines’ authors say.
If patients cannot tolerate HD-MTX, oral chemotherapy and/or whole-brain radiotherapy may be offered. Response should be assessed with contrast-enhanced magnetic resonance imaging.
Consolidation therapy should be initiated after induction for all patients with nonprogressive disease, and high-dose thiotepa-based chemotherapy with autologous stem cell transplant is the recommended first-line option for consolidation. Response to consolidation, again measured with contrast-enhanced MRI, should be carried out at between 1 and 2 months after therapy is completed, and patients should be referred for neuropsychological testing to assess cognitive function.
Patients with relapsed or refractory disease should be approached with maximum urgency – the guidelines offer an algorithm for retreatment options – and offered clinical trial entry wherever possible.
The PCNSL guideline writing process was sponsored by the British Society for Haematology, and some coauthors, including the lead author, disclosed receiving fees from pharmaceutical manufacturers Adienne or F. Hoffman-La Roche.
SOURCE: Fox et al. Br J Haematol. 2018 Nov 23 doi: 10.1111/bjh.15661.
for the diagnosis and management of primary central nervous system diffuse large B‐cell lymphoma.
PCNSL, implicated in some 3% of all brain tumors, is complex to diagnose and treat. People with suspected PCNSL must receive quick and coordinated attention from a multidisciplinary team of neurologists, hematologist-oncologists, and ocular specialists, according to the guidelines, published in the British Journal of Haematology.
Christopher P. Fox, MD, of the Nottingham (England) University Hospitals NHS Trust, and his colleagues, stress the importance of early multidisciplinary attention, aggressive induction treatment, helping patients into trials, universal screening for eye involvement, attaining histological diagnoses in addition to imaging findings, and avoidance or discontinuation of any corticosteroids before biopsy, as even a short course of steroids can impede diagnosis.
The guidelines incorporate findings from studies published since the society’s last comprehensive PCNSL guideline was issued more than a decade ago.
Dr. Fox and his colleagues say definitive treatment for PCNSL – induction of remission followed by consolidation – should start within 2 weeks of diagnosis and that a treatment regimen should be chosen according to a patient’s physiological fitness, not age. The fittest patients, who have better organ function and fewer comorbidities, should be eligible for intensive combination immunochemotherapy incorporating high-dose methotrexate (optimally four cycles of HD-MTX, cytarabine, thiotepa, and rituximab). Those deemed unfit for this regimen should be offered induction treatment with HD-MTX, rituximab and procarbazine, the guidelines’ authors say.
If patients cannot tolerate HD-MTX, oral chemotherapy and/or whole-brain radiotherapy may be offered. Response should be assessed with contrast-enhanced magnetic resonance imaging.
Consolidation therapy should be initiated after induction for all patients with nonprogressive disease, and high-dose thiotepa-based chemotherapy with autologous stem cell transplant is the recommended first-line option for consolidation. Response to consolidation, again measured with contrast-enhanced MRI, should be carried out at between 1 and 2 months after therapy is completed, and patients should be referred for neuropsychological testing to assess cognitive function.
Patients with relapsed or refractory disease should be approached with maximum urgency – the guidelines offer an algorithm for retreatment options – and offered clinical trial entry wherever possible.
The PCNSL guideline writing process was sponsored by the British Society for Haematology, and some coauthors, including the lead author, disclosed receiving fees from pharmaceutical manufacturers Adienne or F. Hoffman-La Roche.
SOURCE: Fox et al. Br J Haematol. 2018 Nov 23 doi: 10.1111/bjh.15661.
FROM THE BRITISH JOURNAL OF HAEMATOLOGY
Stereotactic radiotherapy highly effective for brain mets in RCC
Stereotactic radiosurgery (SRS) is being used increasingly for brain metastases to prevent central nervous system (CNS) toxicity, and new findings published in Clinical Genitourinary Cancer demonstrate that it is a highly effective procedure for brain metastases from renal cell carcinoma (RCC).
In this study, the 1-year local control rate was more than 90% and durability of control was up to 2 years. SRS appears to be the most efficacious for smaller metastases, while escalated dosing or fractionation are needed to effectively treat larger lesions.
In metastatic RCC, the incidence of brain metastases is as high as 15%, and while surgical resection is associated with an improved survival compared with whole brain radiotherapy alone, for patients who are not candidates for resection, SRS is an alternative option. For this study, the authors sought to determine outcomes after treatment with SRS for brain metastases in 268 patients with metastatic RCC who were treated between 2006 and 2015 at a single institution.
Of this group, 38 patients were identified with brain metastases and a total of 243 lesions were treated with SRS, reported Zabi Wardak, MD, and his associates.
The median lesion size was 0.6 cm (0.2-3.1) and patients received a median SRS treatment dose of 18 Gy (12-24). The median age of patients was 65 years and the most common histology was clear cell RCC. Three quarters (74%) of patients received only one GammaKnife session, with a median number of two lesions treated at each session. The most common intracranial location for metastases was the frontal lobe (39% of lesions) followed by the parietal and temporal lobes (15% of lesions). The median size of treated metastases was 0.6 cm (0.2-3.1), reported Dr. Wardak, of the University of Texas UT Southwestern Medical Center, Dallas, and his associates.
At 1 year, local control rates were 91.8% (95% confidence interval, 85.7-95.4) and at 2 years, 86.1% (77.1-91.7). The median overall survival after a diagnosis of brain metastases was 13.8 months (95% CI, 8.1-28.0), with a 1-year survival of 57.5% (95% CI, 40.2-71.4). For patients with a single brain metastasis, 1-year overall survival was 56.3% (95% CI, 29.5-76.2) and for those with multiple lesions, 59.1% (95% CI, 36.1-76.2).
“Patients with multiple brain metastases (greater than five lesions) from RCC did not have a worse survival and should be considered for SRS to achieve intracranial tumor control while avoiding neurological decline,” wrote Dr. Wardak and his associates.
Coauthor James Brugarolas is supported by a grant from the National Institutes of Health. No other study funding was disclosed. The other authors have no disclosures.
SOURCE: Wardak Z et al. Clin Genitourin Cancer. 2018 Nov 20. doi: 10.1016/j.clgc.2018.11.006.
Stereotactic radiosurgery (SRS) is being used increasingly for brain metastases to prevent central nervous system (CNS) toxicity, and new findings published in Clinical Genitourinary Cancer demonstrate that it is a highly effective procedure for brain metastases from renal cell carcinoma (RCC).
In this study, the 1-year local control rate was more than 90% and durability of control was up to 2 years. SRS appears to be the most efficacious for smaller metastases, while escalated dosing or fractionation are needed to effectively treat larger lesions.
In metastatic RCC, the incidence of brain metastases is as high as 15%, and while surgical resection is associated with an improved survival compared with whole brain radiotherapy alone, for patients who are not candidates for resection, SRS is an alternative option. For this study, the authors sought to determine outcomes after treatment with SRS for brain metastases in 268 patients with metastatic RCC who were treated between 2006 and 2015 at a single institution.
Of this group, 38 patients were identified with brain metastases and a total of 243 lesions were treated with SRS, reported Zabi Wardak, MD, and his associates.
The median lesion size was 0.6 cm (0.2-3.1) and patients received a median SRS treatment dose of 18 Gy (12-24). The median age of patients was 65 years and the most common histology was clear cell RCC. Three quarters (74%) of patients received only one GammaKnife session, with a median number of two lesions treated at each session. The most common intracranial location for metastases was the frontal lobe (39% of lesions) followed by the parietal and temporal lobes (15% of lesions). The median size of treated metastases was 0.6 cm (0.2-3.1), reported Dr. Wardak, of the University of Texas UT Southwestern Medical Center, Dallas, and his associates.
At 1 year, local control rates were 91.8% (95% confidence interval, 85.7-95.4) and at 2 years, 86.1% (77.1-91.7). The median overall survival after a diagnosis of brain metastases was 13.8 months (95% CI, 8.1-28.0), with a 1-year survival of 57.5% (95% CI, 40.2-71.4). For patients with a single brain metastasis, 1-year overall survival was 56.3% (95% CI, 29.5-76.2) and for those with multiple lesions, 59.1% (95% CI, 36.1-76.2).
“Patients with multiple brain metastases (greater than five lesions) from RCC did not have a worse survival and should be considered for SRS to achieve intracranial tumor control while avoiding neurological decline,” wrote Dr. Wardak and his associates.
Coauthor James Brugarolas is supported by a grant from the National Institutes of Health. No other study funding was disclosed. The other authors have no disclosures.
SOURCE: Wardak Z et al. Clin Genitourin Cancer. 2018 Nov 20. doi: 10.1016/j.clgc.2018.11.006.
Stereotactic radiosurgery (SRS) is being used increasingly for brain metastases to prevent central nervous system (CNS) toxicity, and new findings published in Clinical Genitourinary Cancer demonstrate that it is a highly effective procedure for brain metastases from renal cell carcinoma (RCC).
In this study, the 1-year local control rate was more than 90% and durability of control was up to 2 years. SRS appears to be the most efficacious for smaller metastases, while escalated dosing or fractionation are needed to effectively treat larger lesions.
In metastatic RCC, the incidence of brain metastases is as high as 15%, and while surgical resection is associated with an improved survival compared with whole brain radiotherapy alone, for patients who are not candidates for resection, SRS is an alternative option. For this study, the authors sought to determine outcomes after treatment with SRS for brain metastases in 268 patients with metastatic RCC who were treated between 2006 and 2015 at a single institution.
Of this group, 38 patients were identified with brain metastases and a total of 243 lesions were treated with SRS, reported Zabi Wardak, MD, and his associates.
The median lesion size was 0.6 cm (0.2-3.1) and patients received a median SRS treatment dose of 18 Gy (12-24). The median age of patients was 65 years and the most common histology was clear cell RCC. Three quarters (74%) of patients received only one GammaKnife session, with a median number of two lesions treated at each session. The most common intracranial location for metastases was the frontal lobe (39% of lesions) followed by the parietal and temporal lobes (15% of lesions). The median size of treated metastases was 0.6 cm (0.2-3.1), reported Dr. Wardak, of the University of Texas UT Southwestern Medical Center, Dallas, and his associates.
At 1 year, local control rates were 91.8% (95% confidence interval, 85.7-95.4) and at 2 years, 86.1% (77.1-91.7). The median overall survival after a diagnosis of brain metastases was 13.8 months (95% CI, 8.1-28.0), with a 1-year survival of 57.5% (95% CI, 40.2-71.4). For patients with a single brain metastasis, 1-year overall survival was 56.3% (95% CI, 29.5-76.2) and for those with multiple lesions, 59.1% (95% CI, 36.1-76.2).
“Patients with multiple brain metastases (greater than five lesions) from RCC did not have a worse survival and should be considered for SRS to achieve intracranial tumor control while avoiding neurological decline,” wrote Dr. Wardak and his associates.
Coauthor James Brugarolas is supported by a grant from the National Institutes of Health. No other study funding was disclosed. The other authors have no disclosures.
SOURCE: Wardak Z et al. Clin Genitourin Cancer. 2018 Nov 20. doi: 10.1016/j.clgc.2018.11.006.
FROM CLINICAL GENITOURINARY CANCER
Key clinical point: SRS can effectively control brain metastases in patients with RCC, with more than half of treated patients surviving more than a year.
Major finding:. Treated local control rates at 1 and 2 years were 91.8% (95% CI, 85.7-95.4) and 86.1% (77.1-91.7)
Study details: A single-institution study that reviewed the use of SRS to treat brain lesions in 38 patients with a total of 243 brain metastases.
Disclosures: Coauthor James Brugarolas is supported by a grant from the National Institutes of Health. No other study funding was disclosed. The other authors have no disclosures.
Source: Wardak Z et al. Clin Genitourin Cancer. 2018 Nov 20. doi: 10.1016/j.clgc.2018.11.006.
Viral tweet leads to physician backlash, #ThisISMyLane
When the National Rifle Association responded to an American College of Physicians position paper updating its policy on reducing firearm injuries by telling the physicians to “stay in their lane,” the group got an earful on Twitter.

“Many of the Tweet responses relayed heart-wrenching stories of doctors caring for patients who suffered and died from gun shot wounds,” writes Forbes contributor Bruce Y. Lee, MD, an associate professor of public health at Johns Hopkins University, Baltimore. “Some Tweets included pictures of blood-spattered scenes to emphasize what doctors have to regularly address.”
The NRA’s response to the ACP update led to the creation of the hashtags #ThisISMyLane and #ThisIsOurLane.
says Rebecca Cunningham, MD, an emergency physician at the University of Michigan, Ann Arbor, on the WBUR radio show, “On Point.” Talking to families about gun safety is “absolutely in our lane.” Meanwhile, Dr. Cunningham, principal investigator of Firearm Safety Among Children and Teens, tweeted that 50 women die per month “by gun by intimate partner.”
The twitter account @ThisIsOurLane, described as a group of “medical professionals who care for #GunViolence Victims,” currently has more than 26 million followers.
Japanese concepts offer perspective
Western culture is fueled by immediacy, and as a result, life can feel askew.
“We’re living in the busiest time of history of humanity, and we often do not have enough time to get everything done that we need to,” futurist and trends guru Daniel Levine says in an interview with NBC News. “The promise of technology was that it would handle our work for us and let us hang out more and relax, but the opposite has happened. Rather than helping us slow down, technology is forcing us to move even faster.”
In seeking another way, Mr. Levine cites “a countertrend against the barrage of tasks and technology that we are inundated with everyday. Patience is the other side of the coin of speed, and we’re looking more to [integrate] that into our lives.”
One step might be to take part in the Japanese tea ceremony of wabi-sabi. At the heart of the ceremony is the reality that things are not perfect but that the imperfections can be embraced to provide fulfillment. This attitude can extend to finding acceptance of personal imperfections.
Developing patience also is important. Again, drawing on Japanese culture, the philosophical outlook of Shankankan espouses the beauty found in a slower pace.
“Patience is the understanding that this is a long journey and you can’t rush the process, particularly in the Zen meditation tradition of spiritual ripening,” says author and yoga teacher Kino MacGregor.
Ikigai – self-introspection as to one’s true purpose – is the another pearl of wisdom from Japanese culture. “I think the Western idea of purpose tends to be very focused on what your profession and livelihood are and how to make money,” Ms. MacGregor says. “Ikigai is quite different. It’s about finding what you love and what the world needs. That requires patience in the sense that it won’t be revealed to you in one moment. You’ll need space and time for those answers.”
Using animal-assisted therapy for children
A Canadian psychologist is putting her livestock to nontraditional use as part of a mental health therapy program for local children and youth in need.
Kali Eddy, who lives on a range in Saskatchewan, uses her critters to help treat anxiety, depression, and other mental health challenges, according to a report by Global News. “Really, it’s just a technique that I use in addition to traditional therapies,” she explains. “A lot of times in a traditional therapy setting, you’re sitting with a psychologist talking and looking at them in the eye – and sometimes this helps reduce some of that pressure if a client is petting an animal or interacting with an animal.”
As many pet owners can attest, having another living thing to focus on and care for can prove therapeutic. As part of a structured therapy, coming into contact with the animals can encourage conversations about personal struggles.
The tactile mental health program developed by Ms. Eddy has allowed her to use the animals that are part of her life to help her clients. And the need for mental health interventions is pressing: “10-20% of youth are affected by a mental illness or disorder, and I think those statistics are probably even higher because the number of youth who come to us who are diagnosed and struggling,” she says in the interview.
Advice for Alzheimer’s caregivers
Caring for a family member with Alzheimer’s disease can be a lonely responsibility, but advice from those who have made the journey can provide a roadmap.
Grieving for the patient while caring for them is important. “You have to learn how to grieve losing someone while they’re still alive,” Amy L. says in an interview with SELF. Amy cared for her father for 3 years until his death from Alzheimer’s in 2015.
“You always think about grief as something that happens once someone passes away, but this illness really changes who they are,” she adds. The knowledge that the disease is progressive and that cognitive and physical functions will spiral downward can be helpful, although very painful.
Trust in the ability to do what is needed for the affected person can prevent second-guessing and guilt later in life. “I wish I had known from the beginning to just listen to and trust myself because I am the only one who knows what it feels like to be in my own circumstance,” Linda G. says.
Having others to talk with is vital. “Connecting with others who know what [we’re] going through and who can offer support and suggestions for dealing with the disease’s various challenges has been very helpful,” explains Peggy M.
Global suicide rates down 29%
The number of suicides in the United States has increased since 2000, fueled by white, middle-aged men who have been hard hit by structural changes in the economy. But, according to an article in The Economist, compared with other countries around the world, the United States appears to be the exception. Globally, the suicide rate has dropped by 29% over the same period.
Notable declines have occurred among young women in China and India, middle-aged men in Russia, and elderly people in general. This might reflect increasing urbanization, with the accompanying access to health and mental health services, freedom from suffocating traditions that can spawn despair, and increased human interaction.
Spending on health services is another important factor. “Spending on health services, especially those that most benefit the old and sick, can make a big difference: Fear of chronic pain is one of the things that leads people to seek a quick way out. The remarkable recent fall in suicide among elderly Britons may have happened in part because Britain’s palliative-care system is the best in the world,” the authors write.
“For a few people – those who are terminally ill, in severe pain, and determined to die – suicide may be the least terrible option. In such circumstances, and with firm safeguards, doctors should be allowed to assist. But many of the 800,000 people who kill themselves each year act in haste, and more could be saved with better health services, labor-market policies, and curbs on booze, guns, pesticides, and pills.
“America, in particular, could spare much pain by learning from the progress elsewhere.”
When the National Rifle Association responded to an American College of Physicians position paper updating its policy on reducing firearm injuries by telling the physicians to “stay in their lane,” the group got an earful on Twitter.
“Many of the Tweet responses relayed heart-wrenching stories of doctors caring for patients who suffered and died from gun shot wounds,” writes Forbes contributor Bruce Y. Lee, MD, an associate professor of public health at Johns Hopkins University, Baltimore. “Some Tweets included pictures of blood-spattered scenes to emphasize what doctors have to regularly address.”
The NRA’s response to the ACP update led to the creation of the hashtags #ThisISMyLane and #ThisIsOurLane.
says Rebecca Cunningham, MD, an emergency physician at the University of Michigan, Ann Arbor, on the WBUR radio show, “On Point.” Talking to families about gun safety is “absolutely in our lane.” Meanwhile, Dr. Cunningham, principal investigator of Firearm Safety Among Children and Teens, tweeted that 50 women die per month “by gun by intimate partner.”
The twitter account @ThisIsOurLane, described as a group of “medical professionals who care for #GunViolence Victims,” currently has more than 26 million followers.
Japanese concepts offer perspective
Western culture is fueled by immediacy, and as a result, life can feel askew.
“We’re living in the busiest time of history of humanity, and we often do not have enough time to get everything done that we need to,” futurist and trends guru Daniel Levine says in an interview with NBC News. “The promise of technology was that it would handle our work for us and let us hang out more and relax, but the opposite has happened. Rather than helping us slow down, technology is forcing us to move even faster.”
In seeking another way, Mr. Levine cites “a countertrend against the barrage of tasks and technology that we are inundated with everyday. Patience is the other side of the coin of speed, and we’re looking more to [integrate] that into our lives.”
One step might be to take part in the Japanese tea ceremony of wabi-sabi. At the heart of the ceremony is the reality that things are not perfect but that the imperfections can be embraced to provide fulfillment. This attitude can extend to finding acceptance of personal imperfections.
Developing patience also is important. Again, drawing on Japanese culture, the philosophical outlook of Shankankan espouses the beauty found in a slower pace.
“Patience is the understanding that this is a long journey and you can’t rush the process, particularly in the Zen meditation tradition of spiritual ripening,” says author and yoga teacher Kino MacGregor.
Ikigai – self-introspection as to one’s true purpose – is the another pearl of wisdom from Japanese culture. “I think the Western idea of purpose tends to be very focused on what your profession and livelihood are and how to make money,” Ms. MacGregor says. “Ikigai is quite different. It’s about finding what you love and what the world needs. That requires patience in the sense that it won’t be revealed to you in one moment. You’ll need space and time for those answers.”
Using animal-assisted therapy for children
A Canadian psychologist is putting her livestock to nontraditional use as part of a mental health therapy program for local children and youth in need.
Kali Eddy, who lives on a range in Saskatchewan, uses her critters to help treat anxiety, depression, and other mental health challenges, according to a report by Global News. “Really, it’s just a technique that I use in addition to traditional therapies,” she explains. “A lot of times in a traditional therapy setting, you’re sitting with a psychologist talking and looking at them in the eye – and sometimes this helps reduce some of that pressure if a client is petting an animal or interacting with an animal.”
As many pet owners can attest, having another living thing to focus on and care for can prove therapeutic. As part of a structured therapy, coming into contact with the animals can encourage conversations about personal struggles.
The tactile mental health program developed by Ms. Eddy has allowed her to use the animals that are part of her life to help her clients. And the need for mental health interventions is pressing: “10-20% of youth are affected by a mental illness or disorder, and I think those statistics are probably even higher because the number of youth who come to us who are diagnosed and struggling,” she says in the interview.
Advice for Alzheimer’s caregivers
Caring for a family member with Alzheimer’s disease can be a lonely responsibility, but advice from those who have made the journey can provide a roadmap.
Grieving for the patient while caring for them is important. “You have to learn how to grieve losing someone while they’re still alive,” Amy L. says in an interview with SELF. Amy cared for her father for 3 years until his death from Alzheimer’s in 2015.
“You always think about grief as something that happens once someone passes away, but this illness really changes who they are,” she adds. The knowledge that the disease is progressive and that cognitive and physical functions will spiral downward can be helpful, although very painful.
Trust in the ability to do what is needed for the affected person can prevent second-guessing and guilt later in life. “I wish I had known from the beginning to just listen to and trust myself because I am the only one who knows what it feels like to be in my own circumstance,” Linda G. says.
Having others to talk with is vital. “Connecting with others who know what [we’re] going through and who can offer support and suggestions for dealing with the disease’s various challenges has been very helpful,” explains Peggy M.
Global suicide rates down 29%
The number of suicides in the United States has increased since 2000, fueled by white, middle-aged men who have been hard hit by structural changes in the economy. But, according to an article in The Economist, compared with other countries around the world, the United States appears to be the exception. Globally, the suicide rate has dropped by 29% over the same period.
Notable declines have occurred among young women in China and India, middle-aged men in Russia, and elderly people in general. This might reflect increasing urbanization, with the accompanying access to health and mental health services, freedom from suffocating traditions that can spawn despair, and increased human interaction.
Spending on health services is another important factor. “Spending on health services, especially those that most benefit the old and sick, can make a big difference: Fear of chronic pain is one of the things that leads people to seek a quick way out. The remarkable recent fall in suicide among elderly Britons may have happened in part because Britain’s palliative-care system is the best in the world,” the authors write.
“For a few people – those who are terminally ill, in severe pain, and determined to die – suicide may be the least terrible option. In such circumstances, and with firm safeguards, doctors should be allowed to assist. But many of the 800,000 people who kill themselves each year act in haste, and more could be saved with better health services, labor-market policies, and curbs on booze, guns, pesticides, and pills.
“America, in particular, could spare much pain by learning from the progress elsewhere.”
When the National Rifle Association responded to an American College of Physicians position paper updating its policy on reducing firearm injuries by telling the physicians to “stay in their lane,” the group got an earful on Twitter.
“Many of the Tweet responses relayed heart-wrenching stories of doctors caring for patients who suffered and died from gun shot wounds,” writes Forbes contributor Bruce Y. Lee, MD, an associate professor of public health at Johns Hopkins University, Baltimore. “Some Tweets included pictures of blood-spattered scenes to emphasize what doctors have to regularly address.”
The NRA’s response to the ACP update led to the creation of the hashtags #ThisISMyLane and #ThisIsOurLane.
says Rebecca Cunningham, MD, an emergency physician at the University of Michigan, Ann Arbor, on the WBUR radio show, “On Point.” Talking to families about gun safety is “absolutely in our lane.” Meanwhile, Dr. Cunningham, principal investigator of Firearm Safety Among Children and Teens, tweeted that 50 women die per month “by gun by intimate partner.”
The twitter account @ThisIsOurLane, described as a group of “medical professionals who care for #GunViolence Victims,” currently has more than 26 million followers.
Japanese concepts offer perspective
Western culture is fueled by immediacy, and as a result, life can feel askew.
“We’re living in the busiest time of history of humanity, and we often do not have enough time to get everything done that we need to,” futurist and trends guru Daniel Levine says in an interview with NBC News. “The promise of technology was that it would handle our work for us and let us hang out more and relax, but the opposite has happened. Rather than helping us slow down, technology is forcing us to move even faster.”
In seeking another way, Mr. Levine cites “a countertrend against the barrage of tasks and technology that we are inundated with everyday. Patience is the other side of the coin of speed, and we’re looking more to [integrate] that into our lives.”
One step might be to take part in the Japanese tea ceremony of wabi-sabi. At the heart of the ceremony is the reality that things are not perfect but that the imperfections can be embraced to provide fulfillment. This attitude can extend to finding acceptance of personal imperfections.
Developing patience also is important. Again, drawing on Japanese culture, the philosophical outlook of Shankankan espouses the beauty found in a slower pace.
“Patience is the understanding that this is a long journey and you can’t rush the process, particularly in the Zen meditation tradition of spiritual ripening,” says author and yoga teacher Kino MacGregor.
Ikigai – self-introspection as to one’s true purpose – is the another pearl of wisdom from Japanese culture. “I think the Western idea of purpose tends to be very focused on what your profession and livelihood are and how to make money,” Ms. MacGregor says. “Ikigai is quite different. It’s about finding what you love and what the world needs. That requires patience in the sense that it won’t be revealed to you in one moment. You’ll need space and time for those answers.”
Using animal-assisted therapy for children
A Canadian psychologist is putting her livestock to nontraditional use as part of a mental health therapy program for local children and youth in need.
Kali Eddy, who lives on a range in Saskatchewan, uses her critters to help treat anxiety, depression, and other mental health challenges, according to a report by Global News. “Really, it’s just a technique that I use in addition to traditional therapies,” she explains. “A lot of times in a traditional therapy setting, you’re sitting with a psychologist talking and looking at them in the eye – and sometimes this helps reduce some of that pressure if a client is petting an animal or interacting with an animal.”
As many pet owners can attest, having another living thing to focus on and care for can prove therapeutic. As part of a structured therapy, coming into contact with the animals can encourage conversations about personal struggles.
The tactile mental health program developed by Ms. Eddy has allowed her to use the animals that are part of her life to help her clients. And the need for mental health interventions is pressing: “10-20% of youth are affected by a mental illness or disorder, and I think those statistics are probably even higher because the number of youth who come to us who are diagnosed and struggling,” she says in the interview.
Advice for Alzheimer’s caregivers
Caring for a family member with Alzheimer’s disease can be a lonely responsibility, but advice from those who have made the journey can provide a roadmap.
Grieving for the patient while caring for them is important. “You have to learn how to grieve losing someone while they’re still alive,” Amy L. says in an interview with SELF. Amy cared for her father for 3 years until his death from Alzheimer’s in 2015.
“You always think about grief as something that happens once someone passes away, but this illness really changes who they are,” she adds. The knowledge that the disease is progressive and that cognitive and physical functions will spiral downward can be helpful, although very painful.
Trust in the ability to do what is needed for the affected person can prevent second-guessing and guilt later in life. “I wish I had known from the beginning to just listen to and trust myself because I am the only one who knows what it feels like to be in my own circumstance,” Linda G. says.
Having others to talk with is vital. “Connecting with others who know what [we’re] going through and who can offer support and suggestions for dealing with the disease’s various challenges has been very helpful,” explains Peggy M.
Global suicide rates down 29%
The number of suicides in the United States has increased since 2000, fueled by white, middle-aged men who have been hard hit by structural changes in the economy. But, according to an article in The Economist, compared with other countries around the world, the United States appears to be the exception. Globally, the suicide rate has dropped by 29% over the same period.
Notable declines have occurred among young women in China and India, middle-aged men in Russia, and elderly people in general. This might reflect increasing urbanization, with the accompanying access to health and mental health services, freedom from suffocating traditions that can spawn despair, and increased human interaction.
Spending on health services is another important factor. “Spending on health services, especially those that most benefit the old and sick, can make a big difference: Fear of chronic pain is one of the things that leads people to seek a quick way out. The remarkable recent fall in suicide among elderly Britons may have happened in part because Britain’s palliative-care system is the best in the world,” the authors write.
“For a few people – those who are terminally ill, in severe pain, and determined to die – suicide may be the least terrible option. In such circumstances, and with firm safeguards, doctors should be allowed to assist. But many of the 800,000 people who kill themselves each year act in haste, and more could be saved with better health services, labor-market policies, and curbs on booze, guns, pesticides, and pills.
“America, in particular, could spare much pain by learning from the progress elsewhere.”
FDA warns of serious side effect of AML treatment
The (Idhifa).

Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
A novel tracer shows promise for detecting CD8 T-cells in advanced solid tumors
WASHINGTON – and reference tissue in an open-label, phase 1, first-in-human study.
The findings demonstrate the ability of the tracer–an anti-CD8 zirconium-labeled minibody–to noninvasively detect CD8 distribution in patients with metastatic solid tumors, potentially providing more information – and more quickly – than is possible with a single biopsy, Michael S. Gordon, MD, reported during a late-breaking abstract session at the annual meeting of the Society for Immunotherapy of Cancer.
During a dose escalation period (stage 1) of the study, six patients received 3 mCi of 89Zr-IAB22M2C once intravenously followed by serial PET scans over a period of 5-7 days. The patients received increasing protein doses of 0.2 through 10 mg to establish safety and determine a “recommended protein dose and scanning parameters for subsequent trials,” explained Dr. Gordon of HonorHealth Research Institute, Scottsdale, Ariz.
Stage 1 was followed by a dose expansion period (stage 2) in which an additional nine subjects were scanned to better delineate the recommended phase 2 study dose, he said.
All patients were monitored for drug-related adverse events and evaluated with blood chemistry, hematology, cytokine assay, and anti-drug antibodies. Biodistribution, radiodosimetry and semi-quantitative evaluation of CD8-tracer uptake were performed in all patients.
“We saw rapid clearance with excretion through the hepatobiliary mechanism, uptake in T-cell rich tissues, and no uptake in background normal tissues – so no uptake in muscle, heart, brain, or lungs,” he said, adding that “tumor uptake was variable and was clearly seen in 10 out of 15 patients.
“The protein dose that was considered to have favorable biodistribution was the range between 0.5 and 1.5, and based upon the analysis, the most favorable imaging time point ... was deemed to be 24 hours,” he said, noting that changes could be seen in as early as 6 hours.
The estimated mean effective radiation dose was 2.4 rem/mCi, “which is consistent with other zirconium-labeled antibody or minibody technologies,” Dr. Gordon said.
Study subjects ranged in age from 31 to 82 years and included nine men and six women with solid tumor malignancies who were eligible to receive checkpoint inhibitor therapy. Their primary cancer types were melanoma (eight patients), non–small-cell lung cancer (six patients), and hepatocellular carcinoma (one patient).
Two patients had received no prior treatment, three had discontinued prior checkpoint inhibitor therapy, and 10 were on immunotherapy.
No drug-related adverse events occurred during the course of the study, although one patient had a transient increase in anti-drug antibodies, Dr. Gordon said.
“Immunotherapy, and specifically checkpoint inhibitors (CPIs), have transformed the landscape of cancer care. Antitumor activity of CPIs is mediated by the CD8-positive T-cell cytotoxic effects, with preclinical and translational clinical studies demonstrating the importance of activated CD8-positive cells within the tumor microenvironment,” he explained, adding that currently available technology is limited in its ability to continually assess the presence of and change in the CD8 infiltrate; one biopsy may fail to capture the immunologic heterogeneity that exists among various tumors in an individual patient.
“As CPI therapy moves into front-line and earlier settings, the ability to have a noninvasive technology to assess whole body and intratumoral changes in CD8 trafficking or expansion in response to therapy is viewed as being crucial,” he said.
A phase 2 study to look closer at the potential for PET + 89Zr-IAB22M2C to fulfill that role will begin soon. The study will focus on correlating imaging with synchronous biopsies before and after primary immunotherapy to look for any predictive potential for this technology, he said.
This study was supported by ImaginAb and Parker Institute for Cancer Immunotherapy. Dr. Gordon reported having no disclosures.
SOURCE: Gordon M et al., SITC 2018: Abstract LB49.
WASHINGTON – and reference tissue in an open-label, phase 1, first-in-human study.
The findings demonstrate the ability of the tracer–an anti-CD8 zirconium-labeled minibody–to noninvasively detect CD8 distribution in patients with metastatic solid tumors, potentially providing more information – and more quickly – than is possible with a single biopsy, Michael S. Gordon, MD, reported during a late-breaking abstract session at the annual meeting of the Society for Immunotherapy of Cancer.
During a dose escalation period (stage 1) of the study, six patients received 3 mCi of 89Zr-IAB22M2C once intravenously followed by serial PET scans over a period of 5-7 days. The patients received increasing protein doses of 0.2 through 10 mg to establish safety and determine a “recommended protein dose and scanning parameters for subsequent trials,” explained Dr. Gordon of HonorHealth Research Institute, Scottsdale, Ariz.
Stage 1 was followed by a dose expansion period (stage 2) in which an additional nine subjects were scanned to better delineate the recommended phase 2 study dose, he said.
All patients were monitored for drug-related adverse events and evaluated with blood chemistry, hematology, cytokine assay, and anti-drug antibodies. Biodistribution, radiodosimetry and semi-quantitative evaluation of CD8-tracer uptake were performed in all patients.
“We saw rapid clearance with excretion through the hepatobiliary mechanism, uptake in T-cell rich tissues, and no uptake in background normal tissues – so no uptake in muscle, heart, brain, or lungs,” he said, adding that “tumor uptake was variable and was clearly seen in 10 out of 15 patients.
“The protein dose that was considered to have favorable biodistribution was the range between 0.5 and 1.5, and based upon the analysis, the most favorable imaging time point ... was deemed to be 24 hours,” he said, noting that changes could be seen in as early as 6 hours.
The estimated mean effective radiation dose was 2.4 rem/mCi, “which is consistent with other zirconium-labeled antibody or minibody technologies,” Dr. Gordon said.
Study subjects ranged in age from 31 to 82 years and included nine men and six women with solid tumor malignancies who were eligible to receive checkpoint inhibitor therapy. Their primary cancer types were melanoma (eight patients), non–small-cell lung cancer (six patients), and hepatocellular carcinoma (one patient).
Two patients had received no prior treatment, three had discontinued prior checkpoint inhibitor therapy, and 10 were on immunotherapy.
No drug-related adverse events occurred during the course of the study, although one patient had a transient increase in anti-drug antibodies, Dr. Gordon said.
“Immunotherapy, and specifically checkpoint inhibitors (CPIs), have transformed the landscape of cancer care. Antitumor activity of CPIs is mediated by the CD8-positive T-cell cytotoxic effects, with preclinical and translational clinical studies demonstrating the importance of activated CD8-positive cells within the tumor microenvironment,” he explained, adding that currently available technology is limited in its ability to continually assess the presence of and change in the CD8 infiltrate; one biopsy may fail to capture the immunologic heterogeneity that exists among various tumors in an individual patient.
“As CPI therapy moves into front-line and earlier settings, the ability to have a noninvasive technology to assess whole body and intratumoral changes in CD8 trafficking or expansion in response to therapy is viewed as being crucial,” he said.
A phase 2 study to look closer at the potential for PET + 89Zr-IAB22M2C to fulfill that role will begin soon. The study will focus on correlating imaging with synchronous biopsies before and after primary immunotherapy to look for any predictive potential for this technology, he said.
This study was supported by ImaginAb and Parker Institute for Cancer Immunotherapy. Dr. Gordon reported having no disclosures.
SOURCE: Gordon M et al., SITC 2018: Abstract LB49.
WASHINGTON – and reference tissue in an open-label, phase 1, first-in-human study.
The findings demonstrate the ability of the tracer–an anti-CD8 zirconium-labeled minibody–to noninvasively detect CD8 distribution in patients with metastatic solid tumors, potentially providing more information – and more quickly – than is possible with a single biopsy, Michael S. Gordon, MD, reported during a late-breaking abstract session at the annual meeting of the Society for Immunotherapy of Cancer.
During a dose escalation period (stage 1) of the study, six patients received 3 mCi of 89Zr-IAB22M2C once intravenously followed by serial PET scans over a period of 5-7 days. The patients received increasing protein doses of 0.2 through 10 mg to establish safety and determine a “recommended protein dose and scanning parameters for subsequent trials,” explained Dr. Gordon of HonorHealth Research Institute, Scottsdale, Ariz.
Stage 1 was followed by a dose expansion period (stage 2) in which an additional nine subjects were scanned to better delineate the recommended phase 2 study dose, he said.
All patients were monitored for drug-related adverse events and evaluated with blood chemistry, hematology, cytokine assay, and anti-drug antibodies. Biodistribution, radiodosimetry and semi-quantitative evaluation of CD8-tracer uptake were performed in all patients.
“We saw rapid clearance with excretion through the hepatobiliary mechanism, uptake in T-cell rich tissues, and no uptake in background normal tissues – so no uptake in muscle, heart, brain, or lungs,” he said, adding that “tumor uptake was variable and was clearly seen in 10 out of 15 patients.
“The protein dose that was considered to have favorable biodistribution was the range between 0.5 and 1.5, and based upon the analysis, the most favorable imaging time point ... was deemed to be 24 hours,” he said, noting that changes could be seen in as early as 6 hours.
The estimated mean effective radiation dose was 2.4 rem/mCi, “which is consistent with other zirconium-labeled antibody or minibody technologies,” Dr. Gordon said.
Study subjects ranged in age from 31 to 82 years and included nine men and six women with solid tumor malignancies who were eligible to receive checkpoint inhibitor therapy. Their primary cancer types were melanoma (eight patients), non–small-cell lung cancer (six patients), and hepatocellular carcinoma (one patient).
Two patients had received no prior treatment, three had discontinued prior checkpoint inhibitor therapy, and 10 were on immunotherapy.
No drug-related adverse events occurred during the course of the study, although one patient had a transient increase in anti-drug antibodies, Dr. Gordon said.
“Immunotherapy, and specifically checkpoint inhibitors (CPIs), have transformed the landscape of cancer care. Antitumor activity of CPIs is mediated by the CD8-positive T-cell cytotoxic effects, with preclinical and translational clinical studies demonstrating the importance of activated CD8-positive cells within the tumor microenvironment,” he explained, adding that currently available technology is limited in its ability to continually assess the presence of and change in the CD8 infiltrate; one biopsy may fail to capture the immunologic heterogeneity that exists among various tumors in an individual patient.
“As CPI therapy moves into front-line and earlier settings, the ability to have a noninvasive technology to assess whole body and intratumoral changes in CD8 trafficking or expansion in response to therapy is viewed as being crucial,” he said.
A phase 2 study to look closer at the potential for PET + 89Zr-IAB22M2C to fulfill that role will begin soon. The study will focus on correlating imaging with synchronous biopsies before and after primary immunotherapy to look for any predictive potential for this technology, he said.
This study was supported by ImaginAb and Parker Institute for Cancer Immunotherapy. Dr. Gordon reported having no disclosures.
SOURCE: Gordon M et al., SITC 2018: Abstract LB49.
REPORTING FROM SITC 2018
Key clinical point: PET with CD8-tracer 89Zr-IAB22M2C is safe, provides detailed CD8 T-cell information.
Major finding: Tumor uptake of the CD8-tracer was seen in 10 of 15 subjects.
Study details: An open-label phase 1 study of 15 patients.
Disclosures: This study was supported by ImaginAb and Parker Institute for Cancer Immunotherapy. Dr. Gordon reported having no disclosures.
Source: Gordon M et al. SITC 2018: Abstract LB49.
Hospitalist movers and shakers – Nov. 2018
George Kasarala, MD, recently was named the hospitalist medical director at Nash UNC Health Care in Rocky Mount, N.C. Dr. Kasarala will guide Nash UNC’s team of hospitalists, a program that has partnered with Sound Physicians.
Dr. Kasarala has a wealth of hospitalist experience, serving in a variety of positions since 2012. He comes to Nash UNC from Vidant Medical Center in Greenville, N.C. Prior to that, he was the associate hospitalist program director at the Apogee Hospitalist program in Elkhart, Ind.
In addition to his medical degree from Saint Louis University, Dr. Kasarala holds a master of business administration from the University of Findlay (Ohio).

Donald W. Woodburn, MD, has been selected as the new medical director at Carolinas Primary Care in Wadesboro, S.C. The longtime internist and hospitalist will stay in his role directing primary care for the facility, which is operated by Atrium Health.
A 35-year veteran in the medical field, Dr. Woodburn most recently was medical director for AnMed Hospitalist Services in Anderson, S.C. He has been a medical director in New York, Florida, and South Carolina since earning his medical degree from Howard University in Washington.
Rita Goyal, MD, has been hired as chief medical officer of ConcertCare, a health care technology company based in Birmingham, Ala. Dr. Goyal has expertise in both medicine and business was cited as the key to her appointment. She founded a Web-based medical consultation business in 2017, virtualMDvisit.net.
Dr. Goyal is an academic hospitalist at the University of Alabama, Birmingham, and will continue to serve as a hospitalist and in the University’s urgent care system.
Nirupma Sharma, MD, has been named chief of the newly minted division of pediatric hospital medicine at Augusta (Ga.) University Health. Dr. Sharma will oversee the pediatric hospitalist staff, including education, research, and clinical assistance.
Dr. Sharma has been the medical director of the 4C unit at Children’s Hospital of Georgia in Augusta. She also has served as associate director of the Medical College of Georgia’s department of pediatrics clerkship program.

Vineet Arora, MD, MHM, was recently named one of the top 10 doctors to follow on Twitter by Becker’s Hospital Review. Dr. Arora is an academic hospitalist at University of Chicago Medicine.
Using the hashtag #meded, Dr. Arora provides a wealth of medical knowledge on Twitter, currently boasting more than 29,000 followers on that social media platform. She also serves as the Journal of Hospital Medicine’s deputy social media editor, and blogs about topics trending in resident education.
BUSINESS MOVES
Aspirus Iron River (Mich.) Hospital has partnered with iNDIGO Health Partners to create a telehealth hospitalist program at night. iNDIGO, a private hospitalist group, will utilize two-way video to treat Aspirus patients during overnight hours.
The telehealth providers with iNDIGO are part of the staff at Aspirus Iron River and are familiar with the facility’s procedures. The remote physicians will be in contact with staff at the hospital, providing direction after meeting with patients via the video system.
The Hospitals of Providence Memorial Campus in El Paso, Tex., intends to have specialists on site at all times for expectant mothers after recently adopting an obstetric hospitalist program. The OB hospitalists will be available to treat patient concerns and medical emergencies that occur outside of normal hours for patients’ primary obstetricians.
All OB hospitalists will be board-certified OB physicians. The goal is to decrease wait times for expectant mothers, who can receive immediate assessments and treatment upon arrival in the emergency department.
George Kasarala, MD, recently was named the hospitalist medical director at Nash UNC Health Care in Rocky Mount, N.C. Dr. Kasarala will guide Nash UNC’s team of hospitalists, a program that has partnered with Sound Physicians.
Dr. Kasarala has a wealth of hospitalist experience, serving in a variety of positions since 2012. He comes to Nash UNC from Vidant Medical Center in Greenville, N.C. Prior to that, he was the associate hospitalist program director at the Apogee Hospitalist program in Elkhart, Ind.
In addition to his medical degree from Saint Louis University, Dr. Kasarala holds a master of business administration from the University of Findlay (Ohio).
Donald W. Woodburn, MD, has been selected as the new medical director at Carolinas Primary Care in Wadesboro, S.C. The longtime internist and hospitalist will stay in his role directing primary care for the facility, which is operated by Atrium Health.
A 35-year veteran in the medical field, Dr. Woodburn most recently was medical director for AnMed Hospitalist Services in Anderson, S.C. He has been a medical director in New York, Florida, and South Carolina since earning his medical degree from Howard University in Washington.
Rita Goyal, MD, has been hired as chief medical officer of ConcertCare, a health care technology company based in Birmingham, Ala. Dr. Goyal has expertise in both medicine and business was cited as the key to her appointment. She founded a Web-based medical consultation business in 2017, virtualMDvisit.net.
Dr. Goyal is an academic hospitalist at the University of Alabama, Birmingham, and will continue to serve as a hospitalist and in the University’s urgent care system.
Nirupma Sharma, MD, has been named chief of the newly minted division of pediatric hospital medicine at Augusta (Ga.) University Health. Dr. Sharma will oversee the pediatric hospitalist staff, including education, research, and clinical assistance.
Dr. Sharma has been the medical director of the 4C unit at Children’s Hospital of Georgia in Augusta. She also has served as associate director of the Medical College of Georgia’s department of pediatrics clerkship program.
Vineet Arora, MD, MHM, was recently named one of the top 10 doctors to follow on Twitter by Becker’s Hospital Review. Dr. Arora is an academic hospitalist at University of Chicago Medicine.
Using the hashtag #meded, Dr. Arora provides a wealth of medical knowledge on Twitter, currently boasting more than 29,000 followers on that social media platform. She also serves as the Journal of Hospital Medicine’s deputy social media editor, and blogs about topics trending in resident education.
BUSINESS MOVES
Aspirus Iron River (Mich.) Hospital has partnered with iNDIGO Health Partners to create a telehealth hospitalist program at night. iNDIGO, a private hospitalist group, will utilize two-way video to treat Aspirus patients during overnight hours.
The telehealth providers with iNDIGO are part of the staff at Aspirus Iron River and are familiar with the facility’s procedures. The remote physicians will be in contact with staff at the hospital, providing direction after meeting with patients via the video system.
The Hospitals of Providence Memorial Campus in El Paso, Tex., intends to have specialists on site at all times for expectant mothers after recently adopting an obstetric hospitalist program. The OB hospitalists will be available to treat patient concerns and medical emergencies that occur outside of normal hours for patients’ primary obstetricians.
All OB hospitalists will be board-certified OB physicians. The goal is to decrease wait times for expectant mothers, who can receive immediate assessments and treatment upon arrival in the emergency department.
George Kasarala, MD, recently was named the hospitalist medical director at Nash UNC Health Care in Rocky Mount, N.C. Dr. Kasarala will guide Nash UNC’s team of hospitalists, a program that has partnered with Sound Physicians.
Dr. Kasarala has a wealth of hospitalist experience, serving in a variety of positions since 2012. He comes to Nash UNC from Vidant Medical Center in Greenville, N.C. Prior to that, he was the associate hospitalist program director at the Apogee Hospitalist program in Elkhart, Ind.
In addition to his medical degree from Saint Louis University, Dr. Kasarala holds a master of business administration from the University of Findlay (Ohio).
Donald W. Woodburn, MD, has been selected as the new medical director at Carolinas Primary Care in Wadesboro, S.C. The longtime internist and hospitalist will stay in his role directing primary care for the facility, which is operated by Atrium Health.
A 35-year veteran in the medical field, Dr. Woodburn most recently was medical director for AnMed Hospitalist Services in Anderson, S.C. He has been a medical director in New York, Florida, and South Carolina since earning his medical degree from Howard University in Washington.
Rita Goyal, MD, has been hired as chief medical officer of ConcertCare, a health care technology company based in Birmingham, Ala. Dr. Goyal has expertise in both medicine and business was cited as the key to her appointment. She founded a Web-based medical consultation business in 2017, virtualMDvisit.net.
Dr. Goyal is an academic hospitalist at the University of Alabama, Birmingham, and will continue to serve as a hospitalist and in the University’s urgent care system.
Nirupma Sharma, MD, has been named chief of the newly minted division of pediatric hospital medicine at Augusta (Ga.) University Health. Dr. Sharma will oversee the pediatric hospitalist staff, including education, research, and clinical assistance.
Dr. Sharma has been the medical director of the 4C unit at Children’s Hospital of Georgia in Augusta. She also has served as associate director of the Medical College of Georgia’s department of pediatrics clerkship program.
Vineet Arora, MD, MHM, was recently named one of the top 10 doctors to follow on Twitter by Becker’s Hospital Review. Dr. Arora is an academic hospitalist at University of Chicago Medicine.
Using the hashtag #meded, Dr. Arora provides a wealth of medical knowledge on Twitter, currently boasting more than 29,000 followers on that social media platform. She also serves as the Journal of Hospital Medicine’s deputy social media editor, and blogs about topics trending in resident education.
BUSINESS MOVES
Aspirus Iron River (Mich.) Hospital has partnered with iNDIGO Health Partners to create a telehealth hospitalist program at night. iNDIGO, a private hospitalist group, will utilize two-way video to treat Aspirus patients during overnight hours.
The telehealth providers with iNDIGO are part of the staff at Aspirus Iron River and are familiar with the facility’s procedures. The remote physicians will be in contact with staff at the hospital, providing direction after meeting with patients via the video system.
The Hospitals of Providence Memorial Campus in El Paso, Tex., intends to have specialists on site at all times for expectant mothers after recently adopting an obstetric hospitalist program. The OB hospitalists will be available to treat patient concerns and medical emergencies that occur outside of normal hours for patients’ primary obstetricians.
All OB hospitalists will be board-certified OB physicians. The goal is to decrease wait times for expectant mothers, who can receive immediate assessments and treatment upon arrival in the emergency department.
Model may predict prolonged status epilepticus outcomes
Two clinical parameters measurable at seizure onset appear to predict a return to baseline after prolonged status epilepticus (SE), based on a study of patients who presented to a single, tertiary academic medical center over a 12-year period.
Absence of nonconvulsive SE with coma and a decreasing Charlson Comorbidity Index were the only independent predictors for return to baseline in patients with SE duration greater than 48 hours, the researchers found. However, the research fell short of developing a model for identifying patients at risk for prolonged SE.
“These findings are of great clinical importance, as up to now, clinicians have had no reliable prediction tools to direct decisions regarding the level of care with progressive SE duration. Early and reliable identification of patients with potential favorable outcome despite having SE for several days is of utmost clinical importance, as this insight may urge clinicians to intensify treatment rather than consider care withdrawal as systemic and neurologic sequelae increase, and chances of SE termination decrease over time,” first author Raoul C. Sutter, MD, of University Hospital Basel (Switzerland), and his colleagues wrote about their findings in Epilepsia.
The researchers identified 467 adult patients with prolonged SE at University Hospital Basel during 2005-2016 – excluding those with SE as a consequence of hypoxic‐ischemic brain injury – who had a median age of 66.7 years and median SE duration of 1 day. While 11.8% of patients died in the hospital and 12.4% at 30 days after SE onset, 40.9% made a complete neurologic and functional recovery to their premorbid status.
There were significant differences in in-hospital outcomes between patients with different SE durations. For example, rates of returning to baseline differed significantly at 55.6% of those with a SE duration of 0-12 hours, 36.8% with 12-24 hours’ duration, 34.6% with 24-48 hours’ duration, and 25.5% with more than 48 hours.
A multivariable regression model identified absence of nonconvulsive SE with coma and a decreasing Charlson Comorbidity Index as the only independent predictors for return to baseline in patients with SE duration greater than 48 hours, and both remained significant predictors after adjustment for use of anesthetics and vasopressors. These predictors of a return to baseline after prolonged SE remained significant after excluding patients who died. This two-variable prediction model had an area under the receiver operating curve (AUROC) of 0.82, “indicating good discrimination,” and an AUROC of 0.76 following cross-validation.
The investigators also sought to develop a model to identify patients at risk for prolonged SE, but the model showed relatively poor discriminative ability with AUROCs of just 0.67-0.72 for predicting no termination of SE within 12, 24, or 48 hours. “Our attempt to generate a highly reliable prediction model for early recognition of patients at increased risk for developing prolonged SE failed, as demonstrated by the rather small AUROC and the fact that sensitivity analyses after exclusion of patients who died revealed inconsistent association of the identified predictors,” they wrote.
Prior reports identified younger age, absence of acute brain lesions at presentation, and the absence of multiple concomitant medical problems as factors associated with favorable outcome after prolonged SE, but “none of the studies performed multivariable regression models and generated or tested predictions models in this context,” they noted.
The authors cautioned that “although internal cross-validation of the final prediction model indicated adequate performance [based on an AUROC of 0.76], further external validation of our prediction model is warranted before our prediction model can be implemented and used for decision making in daily clinical practice.”
Some authors reported receiving research, travel, and/or personal grants or speaker fees from companies marketing antiepileptic drugs, such as UCB, Eisai, and GlaxoSmithKline.
SOURCE: Sutter RC et al. Epilepsia. 2018 Nov 22. doi: 10.1111/epi.14603
Two clinical parameters measurable at seizure onset appear to predict a return to baseline after prolonged status epilepticus (SE), based on a study of patients who presented to a single, tertiary academic medical center over a 12-year period.
Absence of nonconvulsive SE with coma and a decreasing Charlson Comorbidity Index were the only independent predictors for return to baseline in patients with SE duration greater than 48 hours, the researchers found. However, the research fell short of developing a model for identifying patients at risk for prolonged SE.
“These findings are of great clinical importance, as up to now, clinicians have had no reliable prediction tools to direct decisions regarding the level of care with progressive SE duration. Early and reliable identification of patients with potential favorable outcome despite having SE for several days is of utmost clinical importance, as this insight may urge clinicians to intensify treatment rather than consider care withdrawal as systemic and neurologic sequelae increase, and chances of SE termination decrease over time,” first author Raoul C. Sutter, MD, of University Hospital Basel (Switzerland), and his colleagues wrote about their findings in Epilepsia.
The researchers identified 467 adult patients with prolonged SE at University Hospital Basel during 2005-2016 – excluding those with SE as a consequence of hypoxic‐ischemic brain injury – who had a median age of 66.7 years and median SE duration of 1 day. While 11.8% of patients died in the hospital and 12.4% at 30 days after SE onset, 40.9% made a complete neurologic and functional recovery to their premorbid status.
There were significant differences in in-hospital outcomes between patients with different SE durations. For example, rates of returning to baseline differed significantly at 55.6% of those with a SE duration of 0-12 hours, 36.8% with 12-24 hours’ duration, 34.6% with 24-48 hours’ duration, and 25.5% with more than 48 hours.
A multivariable regression model identified absence of nonconvulsive SE with coma and a decreasing Charlson Comorbidity Index as the only independent predictors for return to baseline in patients with SE duration greater than 48 hours, and both remained significant predictors after adjustment for use of anesthetics and vasopressors. These predictors of a return to baseline after prolonged SE remained significant after excluding patients who died. This two-variable prediction model had an area under the receiver operating curve (AUROC) of 0.82, “indicating good discrimination,” and an AUROC of 0.76 following cross-validation.
The investigators also sought to develop a model to identify patients at risk for prolonged SE, but the model showed relatively poor discriminative ability with AUROCs of just 0.67-0.72 for predicting no termination of SE within 12, 24, or 48 hours. “Our attempt to generate a highly reliable prediction model for early recognition of patients at increased risk for developing prolonged SE failed, as demonstrated by the rather small AUROC and the fact that sensitivity analyses after exclusion of patients who died revealed inconsistent association of the identified predictors,” they wrote.
Prior reports identified younger age, absence of acute brain lesions at presentation, and the absence of multiple concomitant medical problems as factors associated with favorable outcome after prolonged SE, but “none of the studies performed multivariable regression models and generated or tested predictions models in this context,” they noted.
The authors cautioned that “although internal cross-validation of the final prediction model indicated adequate performance [based on an AUROC of 0.76], further external validation of our prediction model is warranted before our prediction model can be implemented and used for decision making in daily clinical practice.”
Some authors reported receiving research, travel, and/or personal grants or speaker fees from companies marketing antiepileptic drugs, such as UCB, Eisai, and GlaxoSmithKline.
SOURCE: Sutter RC et al. Epilepsia. 2018 Nov 22. doi: 10.1111/epi.14603
Two clinical parameters measurable at seizure onset appear to predict a return to baseline after prolonged status epilepticus (SE), based on a study of patients who presented to a single, tertiary academic medical center over a 12-year period.
Absence of nonconvulsive SE with coma and a decreasing Charlson Comorbidity Index were the only independent predictors for return to baseline in patients with SE duration greater than 48 hours, the researchers found. However, the research fell short of developing a model for identifying patients at risk for prolonged SE.
“These findings are of great clinical importance, as up to now, clinicians have had no reliable prediction tools to direct decisions regarding the level of care with progressive SE duration. Early and reliable identification of patients with potential favorable outcome despite having SE for several days is of utmost clinical importance, as this insight may urge clinicians to intensify treatment rather than consider care withdrawal as systemic and neurologic sequelae increase, and chances of SE termination decrease over time,” first author Raoul C. Sutter, MD, of University Hospital Basel (Switzerland), and his colleagues wrote about their findings in Epilepsia.
The researchers identified 467 adult patients with prolonged SE at University Hospital Basel during 2005-2016 – excluding those with SE as a consequence of hypoxic‐ischemic brain injury – who had a median age of 66.7 years and median SE duration of 1 day. While 11.8% of patients died in the hospital and 12.4% at 30 days after SE onset, 40.9% made a complete neurologic and functional recovery to their premorbid status.
There were significant differences in in-hospital outcomes between patients with different SE durations. For example, rates of returning to baseline differed significantly at 55.6% of those with a SE duration of 0-12 hours, 36.8% with 12-24 hours’ duration, 34.6% with 24-48 hours’ duration, and 25.5% with more than 48 hours.
A multivariable regression model identified absence of nonconvulsive SE with coma and a decreasing Charlson Comorbidity Index as the only independent predictors for return to baseline in patients with SE duration greater than 48 hours, and both remained significant predictors after adjustment for use of anesthetics and vasopressors. These predictors of a return to baseline after prolonged SE remained significant after excluding patients who died. This two-variable prediction model had an area under the receiver operating curve (AUROC) of 0.82, “indicating good discrimination,” and an AUROC of 0.76 following cross-validation.
The investigators also sought to develop a model to identify patients at risk for prolonged SE, but the model showed relatively poor discriminative ability with AUROCs of just 0.67-0.72 for predicting no termination of SE within 12, 24, or 48 hours. “Our attempt to generate a highly reliable prediction model for early recognition of patients at increased risk for developing prolonged SE failed, as demonstrated by the rather small AUROC and the fact that sensitivity analyses after exclusion of patients who died revealed inconsistent association of the identified predictors,” they wrote.
Prior reports identified younger age, absence of acute brain lesions at presentation, and the absence of multiple concomitant medical problems as factors associated with favorable outcome after prolonged SE, but “none of the studies performed multivariable regression models and generated or tested predictions models in this context,” they noted.
The authors cautioned that “although internal cross-validation of the final prediction model indicated adequate performance [based on an AUROC of 0.76], further external validation of our prediction model is warranted before our prediction model can be implemented and used for decision making in daily clinical practice.”
Some authors reported receiving research, travel, and/or personal grants or speaker fees from companies marketing antiepileptic drugs, such as UCB, Eisai, and GlaxoSmithKline.
SOURCE: Sutter RC et al. Epilepsia. 2018 Nov 22. doi: 10.1111/epi.14603
FROM Epilepsia
Key clinical point:
Major finding: A two-variable prediction model had an AUROC of 0.82.
Study details: A single-center study of 467 adult patients treated for status epilepticus during 2005-2016.
Disclosures: Some authors reported receiving research, travel, and/or personal grants or speaker fees from companies marketing antiepileptic drugs, such as UCB, Eisai, and GlaxoSmithKline.
Source: Sutter RC et al. Epilepsia. 2018 Nov 22. doi: 10.1111/epi.14603
Making a case for patient-reported outcomes in clinical inflammatory bowel disease practice
Patients seek medical care when they perceive a deterioration in their health. Gastroenterologists and health care providers are trained to seek out clinical, laboratory, radiologic, and endoscopic evidence of pathology. Conventional endpoints in inflammatory bowel disease (IBD) clinical trials and clinical care may fail to capture the full health status and disease experience from the patient perspective. The Food and Drug Administration has called for the development of coprimary endpoints in research trials to include an objective measure of inflammation in conjunction with patient-reported outcomes (PROs). The objective is to support labeling claims and improve safety and effectiveness in the drug approval process.1,2 There is also growing recognition that high-value care includes management of biologic and psychosocial factors to enable patients with chronic diseases to regain their health. Clinicians might follow suit by incorporating valid, reliable PRO measures to usual IBD care in order better to achieve patient-centered care, inform decision making, and improve the care provided.
What are patient-reported outcomes?
The FDA defines a PRO as “any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else.” Two PROs are used to measure various aspects of health including physical, emotional, or social domains. PROs have emerged as tools that may foster a better understanding of the patient’s condition, which may go beyond disease activity or symptoms. In effect, incorporating PROs into clinical practice enables a model of “coproduction” of health care, and may contribute to a more reciprocal patient-provider interaction where the needs of the patient may be more fully understood and incorporated into decision-making that may lead to improved patient satisfaction and outcomes.3,4

There are hundreds of available PROs in gastroenterology,5 ranging from simple (characterizing pain with a basic numeric rating scale) to complex multidomain, multi-item instruments. PROs may cover symptom assessment, health-related quality of life, and adherence to and satisfaction with treatment, and may be generic or disease specific. Numerous PROs have been developed for patients with IBD. Commonly used PROs in IBD include severity scales for pain, defecatory urgency, and bloody stool, and several disease-specific and generic instruments assessing different health-related quality-of-life domains have been used in research studies for patients with IBD.
The current approach to patient-centered care for IBD is limited
IBD is a difficult disease to manage – in part because there is no known biomarker that accurately reflects the full spectrum of disease activity. Numerous indices have been developed to better quantify disease activity and measure response to treatment. Among the most frequently used indices in clinical trials are the Crohn’s Disease Activity Index (CDAI) and (for ulcerative colitis [UC]) the Mayo Clinic Score. These endpoints incorporate signs and symptoms, laboratory findings (in the CDAI), and endoscopic assessments. The CDAI is a suboptimal instrument because of a lack of correlation with endoscopic inflammation and potential confounding with concomitant gastrointestinal illnesses, such as irritable bowel syndrome.6 The Mayo Clinic Score is difficult to interpret because of some subjective elements (what is considered a normal number of stools per day?); vagueness (mostly bloody stools more than half the time?); and need for a physician assessment, which often does not correspond with the patient’s perception of their disease.7 From a research perspective, this disconnect can compromise the quality of trial data. Clinically, it can negatively impact patients’ satisfaction and impair the patient-provider relationship.8
To that end, regulatory agencies, scientific bodies, and health care payors are shifting toward a more “patient-centered” approach with an emphasis on PROs. However, although the FDA is incorporating the patient perspective in its trials, measuring meaningful outcomes in day-to-day clinical care is challenging. In the absence of active inflammation, more than 30% of patients with IBD still suffer from gastrointestinal symptoms.9 Furthermore, physicians frequently underestimate the effect of depression, anxiety, fatigue, and sleep on patient health. Likewise, some patients with active small-bowel Crohn’s disease (CD) may experience few gastrointestinal symptoms but have profound fatigue, weight loss, and impaired quality of life. A focused assessment for disease activity may fail to identify aspects of health most relevant or important to individual patient well-being. There is a need for effective, efficient, and standardized strategies to better understand the concerns of the individual seeking help.

Although there are several PROs that measure disease activity primarily for clinical research trials,10 their prevalence in gastroenterology practices has not been assessed. Most likely, few clinical practices currently integrate standardized PROs in routine patient care. This may be because of several reasons, including lack of awareness of newly developed PROs, administrative burden including time and resources to collect PROs, potentially complex interpretation of results, and perhaps a reluctance among physicians to alter traditional patient interview methods of obtaining information about the health status of their patients. For effective use in clinical care, PROs require simple and relevant interpretation to add value to the clinician’s practice, and must minimally impact clinical flow and resources. The use of Internet-enabled tablets has been shown to be a feasible, efficient, and effective means of PRO assessment in gastroenterology practices, with good levels of patient satisfaction.11
Reaping potential benefits of patient-reported outcomes
The National Institutes of Health Patient-Reported Outcomes Measurement Information System (PROMIS) is an initiative developed to investigate and promote implementation of PRO measures among patients with chronic diseases. The collection of PROMIS measures has been shown to be feasible at a tertiary care IBD center, enabling a biopsychosocial model of care.12 Likewise, implementation of PROs in other clinical areas including oncology, orthopedics, and rheumatology has been robust.
In an innovative orthopedic study, PROMIS measures collected and linked to the electronic medical record predicted the likelihood of a clinically meaningful benefit from foot and ankle surgery.13 This facilitated tailored patient-specific preoperative discussions about the expected benefit of surgery. In a study at a rheumatology clinic patients with rheumatoid arthritis were asked to identify their highest priority treatment targets using PROMIS domains (fatigue, pain, depression, social function). The highest priority domain was tracked over time as a patient-centered marker of health, essentially personalizing measures of success for the individual patient.14
PROs have the unique potential to affect multiple levels of health care. At the patient level, PRO data can identify specific concerns, manage expectations of recovery, and tailor treatment decisions to personal preference. At the population level, PRO data can be used to standardize aspects of care to understand comparative health and disease among all patients in a practice or relative to outside practices, identify outliers, and drive improvement.
Optimizing PROs for use in clinical trials: CD–PROs and UC–PROs
Developing standardized, validated instruments according to FDA guidance is a complex process. The lack of an FDA-approved PRO has resulted in substantial variability in the definitions of clinical response or remission in clinical trials to date.15 As a result, IBD-specific PROs (CD-PRO and UC-PRO) are being developed under FDA guidance for use in clinical trials.16 With achievement of prequalification for open use, UC-PRO and CD-PRO will cover five IBD-specific outcomes domains or modules: 1) bowel signs and symptoms, 2) systemic symptoms, 3) emotional impact, 4) coping behaviors, and 5) IBD impact on daily life. The bowel signs and symptoms module may also incorporate a functional impact assessment. Each module includes numerous pertinent items (e.g., “I feel worried,” “I feel scared,” “I feel alone” in the emotional impact module) and are currently being tailored and scored for practicality and relevance. It is hoped that UC-PRO and CD-PRO in final form will be relevant and applicable for clinical trials and gastroenterology practices alike.
Because the development of the UC-PRO and the CD-PRO is still underway, interim PROs are being used in ongoing clinical trials. These interim measures were extracted from existing components of the CDAI, Mayo Clinic Score, and UC Disease Activity Index. The CD PRO-2 consists of two items: abdominal pain and stool frequency. The UC PRO-2 is composed of rectal bleeding and stool frequency. The PRO-3 adds an item regarding general well-being. The sensitivity of these PROs was tested in studies for CD and UC. Both PROs performed similarly to their respective parent instrument. Important limitations include the lack of validation, and the fact that these interim measures were derived from parent measures with acknowledged limitations as previously discussed. Current clinical trials are coupling these interim measures with endoscopic data as coprimary endpoints.
PROs in routine clinical practice: Are we ready for prime time?
Few instruments developed to date have been widely implemented into routine IBD clinical practice. Table 1 highlights commonly available or recently developed PROs for IBD care. As clinicians strive to more effectively integrate PROs into clinical practice, we propose a three-step process to getting started: 1) select and administer a PRO instrument, 2) identify areas of impairment and create a targeted treatment strategy to focus on those areas, and 3) repeat the same PRO at follow-up to assess for improvement. The instrument can be administered before the visit or in the clinic waiting room. Focus a portion of the patient’s visit on discussing the results and identifying one or more domains to target for improvement. For example, the patient may indicate diarrhea as his/her most important area to target, triggering a symptom-specific investigation and therapeutic approach. The PRO may also highlight social or emotional impairment that may require an ancillary referral. The benefits of this PRO-driven approach to IBD care are twofold. First, the patient’s primary concerns are positioned at the forefront of the clinical visit. Second, aligning the clinician’s focus with the patient input may actually help to streamline each visit and improve overall visit efficiency and patient satisfaction.
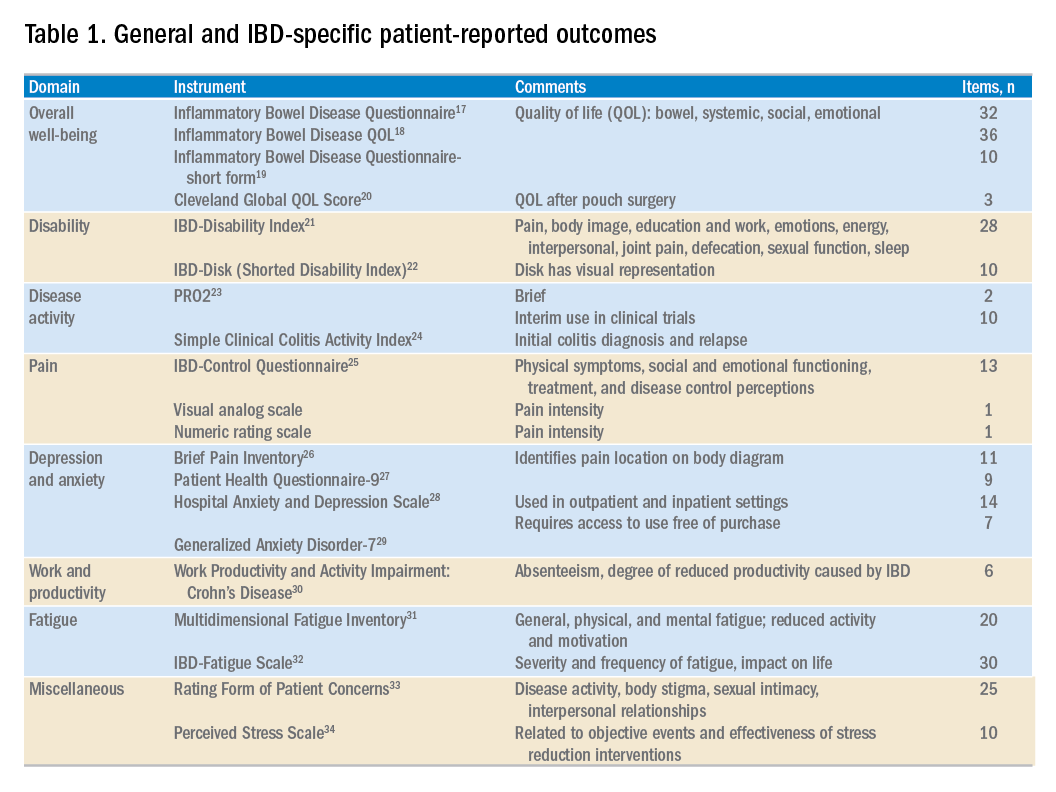
Conclusions
As therapies for IBD improve, so should standards of patient-centered care. Clinicians must actively seek and then listen to the concerns of patients and be able to address the multiple facets of living with a chronic disease. PROs empower patients, helping them identify important topics for discussion at the clinical visit. This affords clinicians a better understanding of primary patient concerns before the visit, and potentially improves the quality and value of care. At first, the process of incorporating PROs into a busy clinical practice may be challenging, but targeted treatment plans have the potential to foster a better patient – and physician – experience.
Content from this column was originally published in the “Practice Management: The Road Ahead” section of Clinical Gastroenterology and Hepatology (2018;16[5]:603-7).
References
1. Guidance for industry: patient-reported outcome measures: use in medical product development to support labeling claims: draft guidance. Health Qual Life Outcomes. 2006;4:79.
2. Burke, L.B., Kennedy, D.L., Miskala, P.H., et al. The use of patient-reported outcome measures in the evaluation of medical products for regulatory approval. Clin Pharmacol Ther. 2008;84:281-3.
3. Batalden, M., Baltalden, P., Margolis, P., et al. Coproduction of healthcare service. BMJ Qual Saf. 2016;25:509-17.
4. Johnson, L.C. Melmed, G.Y., Nelson, E.C., et al. Fostering collaboration through creation of an IBD learning health system. Am J Gastroenterol. 2017;112:406-8.
5. Khanna, P., Agarwal, N., Khanna, D., et al. Development of an online library of patient reported outcome measures in gastroenterology: the GI-PRO database. Am J Gastroenterol. 2014;109:234-48.
6. Bruining, D.H. Sandborn, W.J. Do not assume symptoms indicate failure of anti-tumor necrosis factor therapy in January 2015 Emerging Treatment Goals in IBD Trials and Practice 45 REVIEWS AND PERSPECTIVES Crohn’s disease. Clin Gastroenterol Hepatol. 2011;9:395-9.
7. Surti, B., Spiegel, B., Ippoliti, A., et al. Assessing health status in inflammatory bowel disease using a novel single-item numeric rating scale. Dig Dis Sci. 2013;58:1313-21.
8. Marshall, S., Haywood, K. Fitzpatrick R. Impact of patient-reported outcome measures on routine practice: a structured review. J Eval Clin Pract. 2006;12:559-68.
9. Simren, M., Axelsson, J., Gillberg, R., et al. Quality of life in inflammatory bowel disease in remission: the impact of IBD-like symptoms and associated psychological factors. Am J Gastroenterol. 2002;97:389-96.
10. De Jong, M.J., Huibregtse, R., Masclee, A.A.M., et al. Patient-reported outcome measures for use in clinical trials and clinical practice in inflammatory bowel diseases: a systematic review. Clin Gastroenterol Hepatol. 2018;16:648-63.
11. Atreja, A., Rizk, M. Capturing patient reported outcomes and quality of life in routine clinical practice: ready to prime time? Minerva Gastroenterol Dietol. 2012;58:19-24.
12. Ishak, W.W., Pan, D., Steiner, A.J., et al. Patient reported outcomes of quality of life, functioning, and GI/psychiatric symptom severity in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:798-803.
13. Ho, B., Houck, J.R., Flemister, A.S., et al. Preoperative PROMIS scores predict postoperative success in foot and ankle patients. Foot Ankle Int. 2016;37:911-8. 14. Bacalao, E., Greene, G.J., Beaumont, J.L., et al. Standardizing and personalizing the treat to target (T2T) approach for rheumatoid arthritis using the Patient-Reported Outcomes Measurement Information System (PROMIS): baseline findings on patient-centered treatment priorities. Clin Rheumatol. 2017;36:1729-36.
15. Ma, C., Panaccione, R., Fedorak, R.N., et al. Heterogeneity in definitions of endpoints for clinical trials of ulcerative colitis: a systematic review for development of a core outcome set. Clin Gastroenterol Hepatol. 2018;16:637-47.
16. Higgins P. Patient reported outcomes in IBD 2017. Available at: ibdctworkshop.files.wordpress.com/2017/01/patient-reported-outcomes-in-ibd___peter-higgins.pdf. Accessed Aug. 27, 2017.
17. Guyatt, G., Mitchell, A. Irvine, E.J., et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96:804-10.
18. Love, J.R., Irvine, E.J., Fedorak, R.N. Quality of life in inflammatory bowel disease. J Clin Gastroenterol. 1992;14:15-9.
19. Irvine, E.J., Zhou, Q., Thompson, A.K. The short inflammatory bowel disease questionnaire: a quality of life instrument for community physicians managing inflammatory bowel disease. CCRPT investigators. Canadian Crohn’s Relapse Prevention Trial. Am J Gastroenterol. 1996;91:1571-8.
20. Fazio, V.W., O’Riordain, M.G., Lavery, I.C., et al. Long-term functional outcome and quality of life after stapled restorative proctocolectomy. Ann Surg. 1999;230:575-84.
21. Gower-Rousseau, C., Sarter, H., Savoye, G., et al. Validation of the inflammatory bowel disease disability index in a population-based cohort. Gut. 2017;66:588-96.
22. Gosh, S., Louis, E., Beaugerie, L., et al. Development of the IBD-Disk: a visual self-administered tool assessing disability in inflammatory bowel diseases. Inflamm Bowel Dis. 2017;23:333-40.
23. Khanna, R., Zou, G., D’Haens, G., et al. A retrospective analysis: the development of patient reported outcome measures for the assessment of Crohn’s disease activity. Aliment Pharmacol Ther. 2015;41:77-86.
24. Walmsley, R.S., Ayres, R.C.S., Pounder, P.R., et al. A simple clinical colitis activity index. Gut. 1998;43:29-32.
25. Bodger, K., Ormerod, C., Shackcloth, D., et al. Development and validation of a rapid, general measure of disease control from the patient perspective: the IBD-Control questionnaire. Gut. 2014;63:1092-102.
26. Cleeland, C.S., Ryan, K.M. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singapore. 1994;23:129-38.
27. Kroenke, K., Spitzer, R.L., Williams, J.B.W. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-13.
28. Zigmond, A.S., Snaith, R.P. The Hospital Anxiety and Depression Scale. Acta Psychiatr Scand. 1983;67:361-70.
29. Spitzer, R.L., Korneke, K., Williams, J.B., et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092-7.
30. Reilly, M.C., Zbrozek, A.S. Dukes, E.M. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmachoeconomics. 1993;4:353-65.
31. Smets, E.M., Garssen, B. Bonke, B., et al. The Multidimensional Fatigue Inventory psychometric qualities of an instrument to assess fatigue. J Psychosom Res. 1995;39:315-25.
32. Czuber-Dochan, W., Norton, C., Bassettt, P., et al. Development and psychometric testing of inflammatory bowel disease fatigue (IBD-F) patient self-assessment scale. J Crohns Colitis. 2014;8:1398-406.
33. Drossman, D.A., Leserman, J., Li, Z.M., et al. The rating form of IBD patient concerns: a new measure of health status. Psychosom Med. 1991;53:701-12. 34. Cohen, S., Kamarck, T., Mermelstein, R. A global measure of perceived stress. J Health Soc Behav. 1983;24:385-96.
Dr. Cohen is in the division of digestive and liver diseases; Dr. Melmed is director, clinical inflammatory bowel disease, director, clinical research in the division of gastroenterology, and director, advanced inflammatory bowel disease fellowship program, Cedars-Sinai Medical Center, Los Angeles. Dr. Melmed is a consultant for AbbVie, Boehringer Ingelheim, Celgene, Genentech, Janssen, Pfizer, Samsung Bioepis, Takeda, and UCB; and received support for research from Prometheus Labs. The remaining author discloses no conflicts.
Patients seek medical care when they perceive a deterioration in their health. Gastroenterologists and health care providers are trained to seek out clinical, laboratory, radiologic, and endoscopic evidence of pathology. Conventional endpoints in inflammatory bowel disease (IBD) clinical trials and clinical care may fail to capture the full health status and disease experience from the patient perspective. The Food and Drug Administration has called for the development of coprimary endpoints in research trials to include an objective measure of inflammation in conjunction with patient-reported outcomes (PROs). The objective is to support labeling claims and improve safety and effectiveness in the drug approval process.1,2 There is also growing recognition that high-value care includes management of biologic and psychosocial factors to enable patients with chronic diseases to regain their health. Clinicians might follow suit by incorporating valid, reliable PRO measures to usual IBD care in order better to achieve patient-centered care, inform decision making, and improve the care provided.
What are patient-reported outcomes?
The FDA defines a PRO as “any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else.” Two PROs are used to measure various aspects of health including physical, emotional, or social domains. PROs have emerged as tools that may foster a better understanding of the patient’s condition, which may go beyond disease activity or symptoms. In effect, incorporating PROs into clinical practice enables a model of “coproduction” of health care, and may contribute to a more reciprocal patient-provider interaction where the needs of the patient may be more fully understood and incorporated into decision-making that may lead to improved patient satisfaction and outcomes.3,4
There are hundreds of available PROs in gastroenterology,5 ranging from simple (characterizing pain with a basic numeric rating scale) to complex multidomain, multi-item instruments. PROs may cover symptom assessment, health-related quality of life, and adherence to and satisfaction with treatment, and may be generic or disease specific. Numerous PROs have been developed for patients with IBD. Commonly used PROs in IBD include severity scales for pain, defecatory urgency, and bloody stool, and several disease-specific and generic instruments assessing different health-related quality-of-life domains have been used in research studies for patients with IBD.
The current approach to patient-centered care for IBD is limited
IBD is a difficult disease to manage – in part because there is no known biomarker that accurately reflects the full spectrum of disease activity. Numerous indices have been developed to better quantify disease activity and measure response to treatment. Among the most frequently used indices in clinical trials are the Crohn’s Disease Activity Index (CDAI) and (for ulcerative colitis [UC]) the Mayo Clinic Score. These endpoints incorporate signs and symptoms, laboratory findings (in the CDAI), and endoscopic assessments. The CDAI is a suboptimal instrument because of a lack of correlation with endoscopic inflammation and potential confounding with concomitant gastrointestinal illnesses, such as irritable bowel syndrome.6 The Mayo Clinic Score is difficult to interpret because of some subjective elements (what is considered a normal number of stools per day?); vagueness (mostly bloody stools more than half the time?); and need for a physician assessment, which often does not correspond with the patient’s perception of their disease.7 From a research perspective, this disconnect can compromise the quality of trial data. Clinically, it can negatively impact patients’ satisfaction and impair the patient-provider relationship.8
To that end, regulatory agencies, scientific bodies, and health care payors are shifting toward a more “patient-centered” approach with an emphasis on PROs. However, although the FDA is incorporating the patient perspective in its trials, measuring meaningful outcomes in day-to-day clinical care is challenging. In the absence of active inflammation, more than 30% of patients with IBD still suffer from gastrointestinal symptoms.9 Furthermore, physicians frequently underestimate the effect of depression, anxiety, fatigue, and sleep on patient health. Likewise, some patients with active small-bowel Crohn’s disease (CD) may experience few gastrointestinal symptoms but have profound fatigue, weight loss, and impaired quality of life. A focused assessment for disease activity may fail to identify aspects of health most relevant or important to individual patient well-being. There is a need for effective, efficient, and standardized strategies to better understand the concerns of the individual seeking help.
Although there are several PROs that measure disease activity primarily for clinical research trials,10 their prevalence in gastroenterology practices has not been assessed. Most likely, few clinical practices currently integrate standardized PROs in routine patient care. This may be because of several reasons, including lack of awareness of newly developed PROs, administrative burden including time and resources to collect PROs, potentially complex interpretation of results, and perhaps a reluctance among physicians to alter traditional patient interview methods of obtaining information about the health status of their patients. For effective use in clinical care, PROs require simple and relevant interpretation to add value to the clinician’s practice, and must minimally impact clinical flow and resources. The use of Internet-enabled tablets has been shown to be a feasible, efficient, and effective means of PRO assessment in gastroenterology practices, with good levels of patient satisfaction.11
Reaping potential benefits of patient-reported outcomes
The National Institutes of Health Patient-Reported Outcomes Measurement Information System (PROMIS) is an initiative developed to investigate and promote implementation of PRO measures among patients with chronic diseases. The collection of PROMIS measures has been shown to be feasible at a tertiary care IBD center, enabling a biopsychosocial model of care.12 Likewise, implementation of PROs in other clinical areas including oncology, orthopedics, and rheumatology has been robust.
In an innovative orthopedic study, PROMIS measures collected and linked to the electronic medical record predicted the likelihood of a clinically meaningful benefit from foot and ankle surgery.13 This facilitated tailored patient-specific preoperative discussions about the expected benefit of surgery. In a study at a rheumatology clinic patients with rheumatoid arthritis were asked to identify their highest priority treatment targets using PROMIS domains (fatigue, pain, depression, social function). The highest priority domain was tracked over time as a patient-centered marker of health, essentially personalizing measures of success for the individual patient.14
PROs have the unique potential to affect multiple levels of health care. At the patient level, PRO data can identify specific concerns, manage expectations of recovery, and tailor treatment decisions to personal preference. At the population level, PRO data can be used to standardize aspects of care to understand comparative health and disease among all patients in a practice or relative to outside practices, identify outliers, and drive improvement.
Optimizing PROs for use in clinical trials: CD–PROs and UC–PROs
Developing standardized, validated instruments according to FDA guidance is a complex process. The lack of an FDA-approved PRO has resulted in substantial variability in the definitions of clinical response or remission in clinical trials to date.15 As a result, IBD-specific PROs (CD-PRO and UC-PRO) are being developed under FDA guidance for use in clinical trials.16 With achievement of prequalification for open use, UC-PRO and CD-PRO will cover five IBD-specific outcomes domains or modules: 1) bowel signs and symptoms, 2) systemic symptoms, 3) emotional impact, 4) coping behaviors, and 5) IBD impact on daily life. The bowel signs and symptoms module may also incorporate a functional impact assessment. Each module includes numerous pertinent items (e.g., “I feel worried,” “I feel scared,” “I feel alone” in the emotional impact module) and are currently being tailored and scored for practicality and relevance. It is hoped that UC-PRO and CD-PRO in final form will be relevant and applicable for clinical trials and gastroenterology practices alike.
Because the development of the UC-PRO and the CD-PRO is still underway, interim PROs are being used in ongoing clinical trials. These interim measures were extracted from existing components of the CDAI, Mayo Clinic Score, and UC Disease Activity Index. The CD PRO-2 consists of two items: abdominal pain and stool frequency. The UC PRO-2 is composed of rectal bleeding and stool frequency. The PRO-3 adds an item regarding general well-being. The sensitivity of these PROs was tested in studies for CD and UC. Both PROs performed similarly to their respective parent instrument. Important limitations include the lack of validation, and the fact that these interim measures were derived from parent measures with acknowledged limitations as previously discussed. Current clinical trials are coupling these interim measures with endoscopic data as coprimary endpoints.
PROs in routine clinical practice: Are we ready for prime time?
Few instruments developed to date have been widely implemented into routine IBD clinical practice. Table 1 highlights commonly available or recently developed PROs for IBD care. As clinicians strive to more effectively integrate PROs into clinical practice, we propose a three-step process to getting started: 1) select and administer a PRO instrument, 2) identify areas of impairment and create a targeted treatment strategy to focus on those areas, and 3) repeat the same PRO at follow-up to assess for improvement. The instrument can be administered before the visit or in the clinic waiting room. Focus a portion of the patient’s visit on discussing the results and identifying one or more domains to target for improvement. For example, the patient may indicate diarrhea as his/her most important area to target, triggering a symptom-specific investigation and therapeutic approach. The PRO may also highlight social or emotional impairment that may require an ancillary referral. The benefits of this PRO-driven approach to IBD care are twofold. First, the patient’s primary concerns are positioned at the forefront of the clinical visit. Second, aligning the clinician’s focus with the patient input may actually help to streamline each visit and improve overall visit efficiency and patient satisfaction.
Conclusions
As therapies for IBD improve, so should standards of patient-centered care. Clinicians must actively seek and then listen to the concerns of patients and be able to address the multiple facets of living with a chronic disease. PROs empower patients, helping them identify important topics for discussion at the clinical visit. This affords clinicians a better understanding of primary patient concerns before the visit, and potentially improves the quality and value of care. At first, the process of incorporating PROs into a busy clinical practice may be challenging, but targeted treatment plans have the potential to foster a better patient – and physician – experience.
Content from this column was originally published in the “Practice Management: The Road Ahead” section of Clinical Gastroenterology and Hepatology (2018;16[5]:603-7).
References
1. Guidance for industry: patient-reported outcome measures: use in medical product development to support labeling claims: draft guidance. Health Qual Life Outcomes. 2006;4:79.
2. Burke, L.B., Kennedy, D.L., Miskala, P.H., et al. The use of patient-reported outcome measures in the evaluation of medical products for regulatory approval. Clin Pharmacol Ther. 2008;84:281-3.
3. Batalden, M., Baltalden, P., Margolis, P., et al. Coproduction of healthcare service. BMJ Qual Saf. 2016;25:509-17.
4. Johnson, L.C. Melmed, G.Y., Nelson, E.C., et al. Fostering collaboration through creation of an IBD learning health system. Am J Gastroenterol. 2017;112:406-8.
5. Khanna, P., Agarwal, N., Khanna, D., et al. Development of an online library of patient reported outcome measures in gastroenterology: the GI-PRO database. Am J Gastroenterol. 2014;109:234-48.
6. Bruining, D.H. Sandborn, W.J. Do not assume symptoms indicate failure of anti-tumor necrosis factor therapy in January 2015 Emerging Treatment Goals in IBD Trials and Practice 45 REVIEWS AND PERSPECTIVES Crohn’s disease. Clin Gastroenterol Hepatol. 2011;9:395-9.
7. Surti, B., Spiegel, B., Ippoliti, A., et al. Assessing health status in inflammatory bowel disease using a novel single-item numeric rating scale. Dig Dis Sci. 2013;58:1313-21.
8. Marshall, S., Haywood, K. Fitzpatrick R. Impact of patient-reported outcome measures on routine practice: a structured review. J Eval Clin Pract. 2006;12:559-68.
9. Simren, M., Axelsson, J., Gillberg, R., et al. Quality of life in inflammatory bowel disease in remission: the impact of IBD-like symptoms and associated psychological factors. Am J Gastroenterol. 2002;97:389-96.
10. De Jong, M.J., Huibregtse, R., Masclee, A.A.M., et al. Patient-reported outcome measures for use in clinical trials and clinical practice in inflammatory bowel diseases: a systematic review. Clin Gastroenterol Hepatol. 2018;16:648-63.
11. Atreja, A., Rizk, M. Capturing patient reported outcomes and quality of life in routine clinical practice: ready to prime time? Minerva Gastroenterol Dietol. 2012;58:19-24.
12. Ishak, W.W., Pan, D., Steiner, A.J., et al. Patient reported outcomes of quality of life, functioning, and GI/psychiatric symptom severity in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:798-803.
13. Ho, B., Houck, J.R., Flemister, A.S., et al. Preoperative PROMIS scores predict postoperative success in foot and ankle patients. Foot Ankle Int. 2016;37:911-8. 14. Bacalao, E., Greene, G.J., Beaumont, J.L., et al. Standardizing and personalizing the treat to target (T2T) approach for rheumatoid arthritis using the Patient-Reported Outcomes Measurement Information System (PROMIS): baseline findings on patient-centered treatment priorities. Clin Rheumatol. 2017;36:1729-36.
15. Ma, C., Panaccione, R., Fedorak, R.N., et al. Heterogeneity in definitions of endpoints for clinical trials of ulcerative colitis: a systematic review for development of a core outcome set. Clin Gastroenterol Hepatol. 2018;16:637-47.
16. Higgins P. Patient reported outcomes in IBD 2017. Available at: ibdctworkshop.files.wordpress.com/2017/01/patient-reported-outcomes-in-ibd___peter-higgins.pdf. Accessed Aug. 27, 2017.
17. Guyatt, G., Mitchell, A. Irvine, E.J., et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96:804-10.
18. Love, J.R., Irvine, E.J., Fedorak, R.N. Quality of life in inflammatory bowel disease. J Clin Gastroenterol. 1992;14:15-9.
19. Irvine, E.J., Zhou, Q., Thompson, A.K. The short inflammatory bowel disease questionnaire: a quality of life instrument for community physicians managing inflammatory bowel disease. CCRPT investigators. Canadian Crohn’s Relapse Prevention Trial. Am J Gastroenterol. 1996;91:1571-8.
20. Fazio, V.W., O’Riordain, M.G., Lavery, I.C., et al. Long-term functional outcome and quality of life after stapled restorative proctocolectomy. Ann Surg. 1999;230:575-84.
21. Gower-Rousseau, C., Sarter, H., Savoye, G., et al. Validation of the inflammatory bowel disease disability index in a population-based cohort. Gut. 2017;66:588-96.
22. Gosh, S., Louis, E., Beaugerie, L., et al. Development of the IBD-Disk: a visual self-administered tool assessing disability in inflammatory bowel diseases. Inflamm Bowel Dis. 2017;23:333-40.
23. Khanna, R., Zou, G., D’Haens, G., et al. A retrospective analysis: the development of patient reported outcome measures for the assessment of Crohn’s disease activity. Aliment Pharmacol Ther. 2015;41:77-86.
24. Walmsley, R.S., Ayres, R.C.S., Pounder, P.R., et al. A simple clinical colitis activity index. Gut. 1998;43:29-32.
25. Bodger, K., Ormerod, C., Shackcloth, D., et al. Development and validation of a rapid, general measure of disease control from the patient perspective: the IBD-Control questionnaire. Gut. 2014;63:1092-102.
26. Cleeland, C.S., Ryan, K.M. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singapore. 1994;23:129-38.
27. Kroenke, K., Spitzer, R.L., Williams, J.B.W. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-13.
28. Zigmond, A.S., Snaith, R.P. The Hospital Anxiety and Depression Scale. Acta Psychiatr Scand. 1983;67:361-70.
29. Spitzer, R.L., Korneke, K., Williams, J.B., et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092-7.
30. Reilly, M.C., Zbrozek, A.S. Dukes, E.M. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmachoeconomics. 1993;4:353-65.
31. Smets, E.M., Garssen, B. Bonke, B., et al. The Multidimensional Fatigue Inventory psychometric qualities of an instrument to assess fatigue. J Psychosom Res. 1995;39:315-25.
32. Czuber-Dochan, W., Norton, C., Bassettt, P., et al. Development and psychometric testing of inflammatory bowel disease fatigue (IBD-F) patient self-assessment scale. J Crohns Colitis. 2014;8:1398-406.
33. Drossman, D.A., Leserman, J., Li, Z.M., et al. The rating form of IBD patient concerns: a new measure of health status. Psychosom Med. 1991;53:701-12. 34. Cohen, S., Kamarck, T., Mermelstein, R. A global measure of perceived stress. J Health Soc Behav. 1983;24:385-96.
Dr. Cohen is in the division of digestive and liver diseases; Dr. Melmed is director, clinical inflammatory bowel disease, director, clinical research in the division of gastroenterology, and director, advanced inflammatory bowel disease fellowship program, Cedars-Sinai Medical Center, Los Angeles. Dr. Melmed is a consultant for AbbVie, Boehringer Ingelheim, Celgene, Genentech, Janssen, Pfizer, Samsung Bioepis, Takeda, and UCB; and received support for research from Prometheus Labs. The remaining author discloses no conflicts.
Patients seek medical care when they perceive a deterioration in their health. Gastroenterologists and health care providers are trained to seek out clinical, laboratory, radiologic, and endoscopic evidence of pathology. Conventional endpoints in inflammatory bowel disease (IBD) clinical trials and clinical care may fail to capture the full health status and disease experience from the patient perspective. The Food and Drug Administration has called for the development of coprimary endpoints in research trials to include an objective measure of inflammation in conjunction with patient-reported outcomes (PROs). The objective is to support labeling claims and improve safety and effectiveness in the drug approval process.1,2 There is also growing recognition that high-value care includes management of biologic and psychosocial factors to enable patients with chronic diseases to regain their health. Clinicians might follow suit by incorporating valid, reliable PRO measures to usual IBD care in order better to achieve patient-centered care, inform decision making, and improve the care provided.
What are patient-reported outcomes?
The FDA defines a PRO as “any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else.” Two PROs are used to measure various aspects of health including physical, emotional, or social domains. PROs have emerged as tools that may foster a better understanding of the patient’s condition, which may go beyond disease activity or symptoms. In effect, incorporating PROs into clinical practice enables a model of “coproduction” of health care, and may contribute to a more reciprocal patient-provider interaction where the needs of the patient may be more fully understood and incorporated into decision-making that may lead to improved patient satisfaction and outcomes.3,4
There are hundreds of available PROs in gastroenterology,5 ranging from simple (characterizing pain with a basic numeric rating scale) to complex multidomain, multi-item instruments. PROs may cover symptom assessment, health-related quality of life, and adherence to and satisfaction with treatment, and may be generic or disease specific. Numerous PROs have been developed for patients with IBD. Commonly used PROs in IBD include severity scales for pain, defecatory urgency, and bloody stool, and several disease-specific and generic instruments assessing different health-related quality-of-life domains have been used in research studies for patients with IBD.
The current approach to patient-centered care for IBD is limited
IBD is a difficult disease to manage – in part because there is no known biomarker that accurately reflects the full spectrum of disease activity. Numerous indices have been developed to better quantify disease activity and measure response to treatment. Among the most frequently used indices in clinical trials are the Crohn’s Disease Activity Index (CDAI) and (for ulcerative colitis [UC]) the Mayo Clinic Score. These endpoints incorporate signs and symptoms, laboratory findings (in the CDAI), and endoscopic assessments. The CDAI is a suboptimal instrument because of a lack of correlation with endoscopic inflammation and potential confounding with concomitant gastrointestinal illnesses, such as irritable bowel syndrome.6 The Mayo Clinic Score is difficult to interpret because of some subjective elements (what is considered a normal number of stools per day?); vagueness (mostly bloody stools more than half the time?); and need for a physician assessment, which often does not correspond with the patient’s perception of their disease.7 From a research perspective, this disconnect can compromise the quality of trial data. Clinically, it can negatively impact patients’ satisfaction and impair the patient-provider relationship.8
To that end, regulatory agencies, scientific bodies, and health care payors are shifting toward a more “patient-centered” approach with an emphasis on PROs. However, although the FDA is incorporating the patient perspective in its trials, measuring meaningful outcomes in day-to-day clinical care is challenging. In the absence of active inflammation, more than 30% of patients with IBD still suffer from gastrointestinal symptoms.9 Furthermore, physicians frequently underestimate the effect of depression, anxiety, fatigue, and sleep on patient health. Likewise, some patients with active small-bowel Crohn’s disease (CD) may experience few gastrointestinal symptoms but have profound fatigue, weight loss, and impaired quality of life. A focused assessment for disease activity may fail to identify aspects of health most relevant or important to individual patient well-being. There is a need for effective, efficient, and standardized strategies to better understand the concerns of the individual seeking help.
Although there are several PROs that measure disease activity primarily for clinical research trials,10 their prevalence in gastroenterology practices has not been assessed. Most likely, few clinical practices currently integrate standardized PROs in routine patient care. This may be because of several reasons, including lack of awareness of newly developed PROs, administrative burden including time and resources to collect PROs, potentially complex interpretation of results, and perhaps a reluctance among physicians to alter traditional patient interview methods of obtaining information about the health status of their patients. For effective use in clinical care, PROs require simple and relevant interpretation to add value to the clinician’s practice, and must minimally impact clinical flow and resources. The use of Internet-enabled tablets has been shown to be a feasible, efficient, and effective means of PRO assessment in gastroenterology practices, with good levels of patient satisfaction.11
Reaping potential benefits of patient-reported outcomes
The National Institutes of Health Patient-Reported Outcomes Measurement Information System (PROMIS) is an initiative developed to investigate and promote implementation of PRO measures among patients with chronic diseases. The collection of PROMIS measures has been shown to be feasible at a tertiary care IBD center, enabling a biopsychosocial model of care.12 Likewise, implementation of PROs in other clinical areas including oncology, orthopedics, and rheumatology has been robust.
In an innovative orthopedic study, PROMIS measures collected and linked to the electronic medical record predicted the likelihood of a clinically meaningful benefit from foot and ankle surgery.13 This facilitated tailored patient-specific preoperative discussions about the expected benefit of surgery. In a study at a rheumatology clinic patients with rheumatoid arthritis were asked to identify their highest priority treatment targets using PROMIS domains (fatigue, pain, depression, social function). The highest priority domain was tracked over time as a patient-centered marker of health, essentially personalizing measures of success for the individual patient.14
PROs have the unique potential to affect multiple levels of health care. At the patient level, PRO data can identify specific concerns, manage expectations of recovery, and tailor treatment decisions to personal preference. At the population level, PRO data can be used to standardize aspects of care to understand comparative health and disease among all patients in a practice or relative to outside practices, identify outliers, and drive improvement.
Optimizing PROs for use in clinical trials: CD–PROs and UC–PROs
Developing standardized, validated instruments according to FDA guidance is a complex process. The lack of an FDA-approved PRO has resulted in substantial variability in the definitions of clinical response or remission in clinical trials to date.15 As a result, IBD-specific PROs (CD-PRO and UC-PRO) are being developed under FDA guidance for use in clinical trials.16 With achievement of prequalification for open use, UC-PRO and CD-PRO will cover five IBD-specific outcomes domains or modules: 1) bowel signs and symptoms, 2) systemic symptoms, 3) emotional impact, 4) coping behaviors, and 5) IBD impact on daily life. The bowel signs and symptoms module may also incorporate a functional impact assessment. Each module includes numerous pertinent items (e.g., “I feel worried,” “I feel scared,” “I feel alone” in the emotional impact module) and are currently being tailored and scored for practicality and relevance. It is hoped that UC-PRO and CD-PRO in final form will be relevant and applicable for clinical trials and gastroenterology practices alike.
Because the development of the UC-PRO and the CD-PRO is still underway, interim PROs are being used in ongoing clinical trials. These interim measures were extracted from existing components of the CDAI, Mayo Clinic Score, and UC Disease Activity Index. The CD PRO-2 consists of two items: abdominal pain and stool frequency. The UC PRO-2 is composed of rectal bleeding and stool frequency. The PRO-3 adds an item regarding general well-being. The sensitivity of these PROs was tested in studies for CD and UC. Both PROs performed similarly to their respective parent instrument. Important limitations include the lack of validation, and the fact that these interim measures were derived from parent measures with acknowledged limitations as previously discussed. Current clinical trials are coupling these interim measures with endoscopic data as coprimary endpoints.
PROs in routine clinical practice: Are we ready for prime time?
Few instruments developed to date have been widely implemented into routine IBD clinical practice. Table 1 highlights commonly available or recently developed PROs for IBD care. As clinicians strive to more effectively integrate PROs into clinical practice, we propose a three-step process to getting started: 1) select and administer a PRO instrument, 2) identify areas of impairment and create a targeted treatment strategy to focus on those areas, and 3) repeat the same PRO at follow-up to assess for improvement. The instrument can be administered before the visit or in the clinic waiting room. Focus a portion of the patient’s visit on discussing the results and identifying one or more domains to target for improvement. For example, the patient may indicate diarrhea as his/her most important area to target, triggering a symptom-specific investigation and therapeutic approach. The PRO may also highlight social or emotional impairment that may require an ancillary referral. The benefits of this PRO-driven approach to IBD care are twofold. First, the patient’s primary concerns are positioned at the forefront of the clinical visit. Second, aligning the clinician’s focus with the patient input may actually help to streamline each visit and improve overall visit efficiency and patient satisfaction.
Conclusions
As therapies for IBD improve, so should standards of patient-centered care. Clinicians must actively seek and then listen to the concerns of patients and be able to address the multiple facets of living with a chronic disease. PROs empower patients, helping them identify important topics for discussion at the clinical visit. This affords clinicians a better understanding of primary patient concerns before the visit, and potentially improves the quality and value of care. At first, the process of incorporating PROs into a busy clinical practice may be challenging, but targeted treatment plans have the potential to foster a better patient – and physician – experience.
Content from this column was originally published in the “Practice Management: The Road Ahead” section of Clinical Gastroenterology and Hepatology (2018;16[5]:603-7).
References
1. Guidance for industry: patient-reported outcome measures: use in medical product development to support labeling claims: draft guidance. Health Qual Life Outcomes. 2006;4:79.
2. Burke, L.B., Kennedy, D.L., Miskala, P.H., et al. The use of patient-reported outcome measures in the evaluation of medical products for regulatory approval. Clin Pharmacol Ther. 2008;84:281-3.
3. Batalden, M., Baltalden, P., Margolis, P., et al. Coproduction of healthcare service. BMJ Qual Saf. 2016;25:509-17.
4. Johnson, L.C. Melmed, G.Y., Nelson, E.C., et al. Fostering collaboration through creation of an IBD learning health system. Am J Gastroenterol. 2017;112:406-8.
5. Khanna, P., Agarwal, N., Khanna, D., et al. Development of an online library of patient reported outcome measures in gastroenterology: the GI-PRO database. Am J Gastroenterol. 2014;109:234-48.
6. Bruining, D.H. Sandborn, W.J. Do not assume symptoms indicate failure of anti-tumor necrosis factor therapy in January 2015 Emerging Treatment Goals in IBD Trials and Practice 45 REVIEWS AND PERSPECTIVES Crohn’s disease. Clin Gastroenterol Hepatol. 2011;9:395-9.
7. Surti, B., Spiegel, B., Ippoliti, A., et al. Assessing health status in inflammatory bowel disease using a novel single-item numeric rating scale. Dig Dis Sci. 2013;58:1313-21.
8. Marshall, S., Haywood, K. Fitzpatrick R. Impact of patient-reported outcome measures on routine practice: a structured review. J Eval Clin Pract. 2006;12:559-68.
9. Simren, M., Axelsson, J., Gillberg, R., et al. Quality of life in inflammatory bowel disease in remission: the impact of IBD-like symptoms and associated psychological factors. Am J Gastroenterol. 2002;97:389-96.
10. De Jong, M.J., Huibregtse, R., Masclee, A.A.M., et al. Patient-reported outcome measures for use in clinical trials and clinical practice in inflammatory bowel diseases: a systematic review. Clin Gastroenterol Hepatol. 2018;16:648-63.
11. Atreja, A., Rizk, M. Capturing patient reported outcomes and quality of life in routine clinical practice: ready to prime time? Minerva Gastroenterol Dietol. 2012;58:19-24.
12. Ishak, W.W., Pan, D., Steiner, A.J., et al. Patient reported outcomes of quality of life, functioning, and GI/psychiatric symptom severity in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:798-803.
13. Ho, B., Houck, J.R., Flemister, A.S., et al. Preoperative PROMIS scores predict postoperative success in foot and ankle patients. Foot Ankle Int. 2016;37:911-8. 14. Bacalao, E., Greene, G.J., Beaumont, J.L., et al. Standardizing and personalizing the treat to target (T2T) approach for rheumatoid arthritis using the Patient-Reported Outcomes Measurement Information System (PROMIS): baseline findings on patient-centered treatment priorities. Clin Rheumatol. 2017;36:1729-36.
15. Ma, C., Panaccione, R., Fedorak, R.N., et al. Heterogeneity in definitions of endpoints for clinical trials of ulcerative colitis: a systematic review for development of a core outcome set. Clin Gastroenterol Hepatol. 2018;16:637-47.
16. Higgins P. Patient reported outcomes in IBD 2017. Available at: ibdctworkshop.files.wordpress.com/2017/01/patient-reported-outcomes-in-ibd___peter-higgins.pdf. Accessed Aug. 27, 2017.
17. Guyatt, G., Mitchell, A. Irvine, E.J., et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96:804-10.
18. Love, J.R., Irvine, E.J., Fedorak, R.N. Quality of life in inflammatory bowel disease. J Clin Gastroenterol. 1992;14:15-9.
19. Irvine, E.J., Zhou, Q., Thompson, A.K. The short inflammatory bowel disease questionnaire: a quality of life instrument for community physicians managing inflammatory bowel disease. CCRPT investigators. Canadian Crohn’s Relapse Prevention Trial. Am J Gastroenterol. 1996;91:1571-8.
20. Fazio, V.W., O’Riordain, M.G., Lavery, I.C., et al. Long-term functional outcome and quality of life after stapled restorative proctocolectomy. Ann Surg. 1999;230:575-84.
21. Gower-Rousseau, C., Sarter, H., Savoye, G., et al. Validation of the inflammatory bowel disease disability index in a population-based cohort. Gut. 2017;66:588-96.
22. Gosh, S., Louis, E., Beaugerie, L., et al. Development of the IBD-Disk: a visual self-administered tool assessing disability in inflammatory bowel diseases. Inflamm Bowel Dis. 2017;23:333-40.
23. Khanna, R., Zou, G., D’Haens, G., et al. A retrospective analysis: the development of patient reported outcome measures for the assessment of Crohn’s disease activity. Aliment Pharmacol Ther. 2015;41:77-86.
24. Walmsley, R.S., Ayres, R.C.S., Pounder, P.R., et al. A simple clinical colitis activity index. Gut. 1998;43:29-32.
25. Bodger, K., Ormerod, C., Shackcloth, D., et al. Development and validation of a rapid, general measure of disease control from the patient perspective: the IBD-Control questionnaire. Gut. 2014;63:1092-102.
26. Cleeland, C.S., Ryan, K.M. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singapore. 1994;23:129-38.
27. Kroenke, K., Spitzer, R.L., Williams, J.B.W. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-13.
28. Zigmond, A.S., Snaith, R.P. The Hospital Anxiety and Depression Scale. Acta Psychiatr Scand. 1983;67:361-70.
29. Spitzer, R.L., Korneke, K., Williams, J.B., et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092-7.
30. Reilly, M.C., Zbrozek, A.S. Dukes, E.M. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmachoeconomics. 1993;4:353-65.
31. Smets, E.M., Garssen, B. Bonke, B., et al. The Multidimensional Fatigue Inventory psychometric qualities of an instrument to assess fatigue. J Psychosom Res. 1995;39:315-25.
32. Czuber-Dochan, W., Norton, C., Bassettt, P., et al. Development and psychometric testing of inflammatory bowel disease fatigue (IBD-F) patient self-assessment scale. J Crohns Colitis. 2014;8:1398-406.
33. Drossman, D.A., Leserman, J., Li, Z.M., et al. The rating form of IBD patient concerns: a new measure of health status. Psychosom Med. 1991;53:701-12. 34. Cohen, S., Kamarck, T., Mermelstein, R. A global measure of perceived stress. J Health Soc Behav. 1983;24:385-96.
Dr. Cohen is in the division of digestive and liver diseases; Dr. Melmed is director, clinical inflammatory bowel disease, director, clinical research in the division of gastroenterology, and director, advanced inflammatory bowel disease fellowship program, Cedars-Sinai Medical Center, Los Angeles. Dr. Melmed is a consultant for AbbVie, Boehringer Ingelheim, Celgene, Genentech, Janssen, Pfizer, Samsung Bioepis, Takeda, and UCB; and received support for research from Prometheus Labs. The remaining author discloses no conflicts.
Trump scheme for Part B drugs raises red flags
A proposed Trump administration plan for paying for drugs under Medicare Part B has raised red flags for doctors.

The Centers for Medicare & Medicaid Services announced Oct. 25 that it will test paying for Part B drugs by more closely aligning those payments with international rates.
The so-called International Price Index (IPI) model “would test whether increasing competition for private-sector vendors to negotiate drug prices, and aligning Medicare payments for drugs with prices that are paid in foreign countries, improves beneficiary access and quality of care while reducing expenditures,” according to a government fact sheet.
Under the test, private vendors would “procure drugs, distribute them to physicians and hospitals, and take on the responsibility of billing Medicare. Vendors would aggregate purchasing, seek volume-based discounts, and compete for providers’ business, thereby creating competition where none exists today.”
Health care professionals and hospitals in certain geographic areas would receive their Part B drugs under this program, while the rest of the country would continue under the current buy-and-bill system. Eventually, over the 5-year phase-in period, half of the geographic regions would fall under this IPI model.
CMS officials note that the IPI model “would maintain beneficiaries’ choice of provider and treatments and would have meaningful beneficiary protections such as enhanced monitoring and Medicare Beneficiary Ombudsman supports.”
Initially, only single-source drugs and biologics with available international pricing data would be provided under the IPI model, which could be expanded over time to include drugs available via multiple sources.
Currently, Medicare typically pays average sales price (ASP) plus a 6% add-on for drugs under Part B. Under IPI, if the international price is determined to be lower than the ASP, the CMS would reimburse based on a target price derived from an international price index, with the hope that manufacturers would match the international price. The target price would be phased in over a 5-year period.
The plan also calls for an add-on price similar to the current buy-and-bill system; however, the CMS aims to bring the add-on back to 6% rather than the actual 4.3% under the budget sequestration.
Other add-ons are also under consideration, such as paying a fixed amount per encounter or per month as well as a unique payment based on drug class, physician specialty, or physician practice.
The American Gastroenterological Association also has concerns, noting that the proposed changes in policy are complex and certain details are lacking, which makes it difficult to assess fully the impact of the proposal.
While it’s true that the high cost of biologics, such as those used to treat inflammatory bowel disease, create barriers to patient access, efforts to address costs may create other patient access issues and penalize gastroenterologists for providing high-quality care to some of the most complex patients. The Competitive Acquisition Program previously abandoned created patient access issues. Moreover, utilization management strategies such as step therapy or “fail first” protocols have no place in the Medicare Part B program. Policy makers should be careful to not penalize Medicare patients who depend on timely access to needed therapies.
“The administration’s proposal for an International Pricing Index Model for Part B drugs raises a number of questions, and we need to have a greater understanding of the potential impact of the proposal on patients, physicians, and the health care system,” American Medical Association President Barbara McAneny, MD, said in a statement. “We look forward to working constructively with the Administration as it seeks feedback.”
Comments are due Dec. 24. The CMS plans to issue the proposed rule related to this model in the spring of 2019.
A proposed Trump administration plan for paying for drugs under Medicare Part B has raised red flags for doctors.
The Centers for Medicare & Medicaid Services announced Oct. 25 that it will test paying for Part B drugs by more closely aligning those payments with international rates.
The so-called International Price Index (IPI) model “would test whether increasing competition for private-sector vendors to negotiate drug prices, and aligning Medicare payments for drugs with prices that are paid in foreign countries, improves beneficiary access and quality of care while reducing expenditures,” according to a government fact sheet.
Under the test, private vendors would “procure drugs, distribute them to physicians and hospitals, and take on the responsibility of billing Medicare. Vendors would aggregate purchasing, seek volume-based discounts, and compete for providers’ business, thereby creating competition where none exists today.”
Health care professionals and hospitals in certain geographic areas would receive their Part B drugs under this program, while the rest of the country would continue under the current buy-and-bill system. Eventually, over the 5-year phase-in period, half of the geographic regions would fall under this IPI model.
CMS officials note that the IPI model “would maintain beneficiaries’ choice of provider and treatments and would have meaningful beneficiary protections such as enhanced monitoring and Medicare Beneficiary Ombudsman supports.”
Initially, only single-source drugs and biologics with available international pricing data would be provided under the IPI model, which could be expanded over time to include drugs available via multiple sources.
Currently, Medicare typically pays average sales price (ASP) plus a 6% add-on for drugs under Part B. Under IPI, if the international price is determined to be lower than the ASP, the CMS would reimburse based on a target price derived from an international price index, with the hope that manufacturers would match the international price. The target price would be phased in over a 5-year period.
The plan also calls for an add-on price similar to the current buy-and-bill system; however, the CMS aims to bring the add-on back to 6% rather than the actual 4.3% under the budget sequestration.
Other add-ons are also under consideration, such as paying a fixed amount per encounter or per month as well as a unique payment based on drug class, physician specialty, or physician practice.
The American Gastroenterological Association also has concerns, noting that the proposed changes in policy are complex and certain details are lacking, which makes it difficult to assess fully the impact of the proposal.
While it’s true that the high cost of biologics, such as those used to treat inflammatory bowel disease, create barriers to patient access, efforts to address costs may create other patient access issues and penalize gastroenterologists for providing high-quality care to some of the most complex patients. The Competitive Acquisition Program previously abandoned created patient access issues. Moreover, utilization management strategies such as step therapy or “fail first” protocols have no place in the Medicare Part B program. Policy makers should be careful to not penalize Medicare patients who depend on timely access to needed therapies.
“The administration’s proposal for an International Pricing Index Model for Part B drugs raises a number of questions, and we need to have a greater understanding of the potential impact of the proposal on patients, physicians, and the health care system,” American Medical Association President Barbara McAneny, MD, said in a statement. “We look forward to working constructively with the Administration as it seeks feedback.”
Comments are due Dec. 24. The CMS plans to issue the proposed rule related to this model in the spring of 2019.
A proposed Trump administration plan for paying for drugs under Medicare Part B has raised red flags for doctors.
The Centers for Medicare & Medicaid Services announced Oct. 25 that it will test paying for Part B drugs by more closely aligning those payments with international rates.
The so-called International Price Index (IPI) model “would test whether increasing competition for private-sector vendors to negotiate drug prices, and aligning Medicare payments for drugs with prices that are paid in foreign countries, improves beneficiary access and quality of care while reducing expenditures,” according to a government fact sheet.
Under the test, private vendors would “procure drugs, distribute them to physicians and hospitals, and take on the responsibility of billing Medicare. Vendors would aggregate purchasing, seek volume-based discounts, and compete for providers’ business, thereby creating competition where none exists today.”
Health care professionals and hospitals in certain geographic areas would receive their Part B drugs under this program, while the rest of the country would continue under the current buy-and-bill system. Eventually, over the 5-year phase-in period, half of the geographic regions would fall under this IPI model.
CMS officials note that the IPI model “would maintain beneficiaries’ choice of provider and treatments and would have meaningful beneficiary protections such as enhanced monitoring and Medicare Beneficiary Ombudsman supports.”
Initially, only single-source drugs and biologics with available international pricing data would be provided under the IPI model, which could be expanded over time to include drugs available via multiple sources.
Currently, Medicare typically pays average sales price (ASP) plus a 6% add-on for drugs under Part B. Under IPI, if the international price is determined to be lower than the ASP, the CMS would reimburse based on a target price derived from an international price index, with the hope that manufacturers would match the international price. The target price would be phased in over a 5-year period.
The plan also calls for an add-on price similar to the current buy-and-bill system; however, the CMS aims to bring the add-on back to 6% rather than the actual 4.3% under the budget sequestration.
Other add-ons are also under consideration, such as paying a fixed amount per encounter or per month as well as a unique payment based on drug class, physician specialty, or physician practice.
The American Gastroenterological Association also has concerns, noting that the proposed changes in policy are complex and certain details are lacking, which makes it difficult to assess fully the impact of the proposal.
While it’s true that the high cost of biologics, such as those used to treat inflammatory bowel disease, create barriers to patient access, efforts to address costs may create other patient access issues and penalize gastroenterologists for providing high-quality care to some of the most complex patients. The Competitive Acquisition Program previously abandoned created patient access issues. Moreover, utilization management strategies such as step therapy or “fail first” protocols have no place in the Medicare Part B program. Policy makers should be careful to not penalize Medicare patients who depend on timely access to needed therapies.
“The administration’s proposal for an International Pricing Index Model for Part B drugs raises a number of questions, and we need to have a greater understanding of the potential impact of the proposal on patients, physicians, and the health care system,” American Medical Association President Barbara McAneny, MD, said in a statement. “We look forward to working constructively with the Administration as it seeks feedback.”
Comments are due Dec. 24. The CMS plans to issue the proposed rule related to this model in the spring of 2019.
Acute flaccid myelitis has unique MRI features
Acute flaccid myelitis appears to present most commonly as asymmetric weakness after respiratory viral infection and has distinctive MRI features that could help with early diagnosis.
In a paper published in JAMA Pediatrics, researchers presented the results of a retrospective case series of 45 children who were diagnosed between 2012 and 2016 with acute flaccid myelitis, or “pseudo polio,” using the Centers for Disease Control’s case definition.
Matthew J. Elrick, MD, PhD, of Johns Hopkins University, Baltimore, and his coauthors came up with a set of reproducible and distinctive features of acute flaccid myelitis. These were the presence of a prodromal fever or viral syndrome; weakness in a lower motor neuron pattern involving one or more limbs, neck, face, and/or bulbar muscles; supportive evidence either from MRI, nerve conduction studies, or cerebrospinal fluid; and the absence of objective sensory deficits, supratentorial white matter, cortical lesions greater than 1 cm in size, encephalopathy, elevated cerebrospinal fluid without pleocytosis, or any other alternative diagnosis.
The researchers commented that, while the CDC case definition has helped with epidemiologic surveillance of acute flaccid myelitis, it may also pick up children with acute weakness caused by other conditions such as transverse myelitis, Guillain-Barré syndrome, ischemic myelopathy, and other myelopathies.
To identify clinical features that might help differentiate patients with acute flaccid myelitis, the researchers attempted to see how many alternative diagnoses were captured in the CDC case definition.
The patients in their study all presented with acute flaccid paralysis in at least one limb and with either an MRI showing a spinal cord lesion spanning one or more spinal segments but largely restricted to gray matter or pleocytosis of the cerebrospinal fluid. The researchers divided the cases into those who also met a well-defined alternative diagnosis – who they categorized as “acute flaccid myelitis with possible alternative diagnosis” (AFM-ad) – and those who were categorized as “restrictively defined AFM” (rAFM). Overall, 34 patients were classified as rAFM and 11 as AFM-ad.
Those in the rAFD group nearly all had asymmetric onset of symptoms, while those in the AFM-ad group were more likely to experience bilateral onset in their lower extremities, “reflecting the pattern of symptoms often seen in other causes of myelopathy such as transverse myelitis and ischemic injury,” the authors noted.
While both groups often presented with decreased muscle tone and reflexes, this was more likely to evolve to increased tone or hyperreflexia in the AFM-ad group. Patients with AFM-ad were also more likely to experience impaired bowel or bladder function.
On MRI, lesions were mostly or completely restricted to the spinal cord gray matter in patients with rAFM or to involve the dorsal pons. These patients did not have any supratentorial brain lesions.
Patients in the rAFM category also had lower cerebrospinal fluid protein values than those in the AFM-ad category, but this was the only cerebrospinal fluid difference between the two groups.
All patients categorized as having rAFM had an infectious prodrome – such as viral syndrome, fever, congestion, and cough – compared with 63.6% of the patients categorized as AFM-ad. The pathogen was identified in only 13 of the rAFM patients, and included 5 patients with enterovirus D68, 2 with unspecified enterovirus, 2 with rhinovirus, 2 with adenovirus, and 2 with mycoplasma. Of the three patients in the AFM-ad group whose pathogen was identified, one had an untyped rhinovirus/enterovirus and mycoplasma, one had a rhinovirus B, and one had enterovirus D68.
“These results highlight that the CDC case definition, while appropriately sensitive for epidemiologic ascertainment of possible AFM cases, also encompasses other neurologic diseases that can cause acute weakness,” the authors wrote. However, they acknowledged that acute flaccid myelitis was still poorly understood and their own definition of the disease may change as more children are diagnosed.
“We propose that the definition of rAFM presented here be used as a starting point for developing inclusion and exclusion criteria for future research studies of AFM,” they wrote.
The study was supported by Johns Hopkins University, the Bart McLean Fund for Neuroimmunology Research, and Project Restore. Two authors reported funding from private industry outside the submitted work and five reported support from or involvement with research and funding bodies.
SOURCE: Elrick MJ et al. JAMA Pediatr. 2018 Nov 30. doi: 10.1001/jamapediatrics.2018.4890.
Acute flaccid myelitis (AFM) initially presents subtly, complicating its diagnosis. Children present with a rapid onset of weakness that is associated with a febrile illness, which can be respiratory, gastrointestinal, or with symptoms of hand-foot-and-mouth disease. Given the lack of effective treatments, early diagnosis and monitoring are essential for mitigating the risk of respiratory decline and long-term complications.
While patient history and physical examination can provide clues to the presence of AFM, confirming the diagnosis requires lumbar puncture and MRI of the spinal cord. On MRI, diagnostic confirmation will come from findings of longitudinal, butterfly-shaped, anterior horn–predominant T2 and fluid-attenuated inversion recovery hyperintensities of the central gray matter.
Patients with suspected AFM should be hospitalized because they can rapidly deteriorate to the point of respiratory compromise, particularly those with upper extremity and bulbar weakness.
Sarah E. Hopkins, MD, is from the division of neurology at the Children’s Hospital of Philadelphia; Matthew J. Elrick, MD, PhD, is from the department of neurology at Johns Hopkins University, Baltimore; and Kevin Messacar, MD, is from the department of pediatrics at the Children’s Hospital Colorado. These comments are taken from an accompanying viewpoint (JAMA Pediatr. 2018 Nov 30. doi: 10.1001/jamapediatrics.2018.4896). Dr. Messacar reported support from the National Institutes of Health/National Institute of Allergy and Infectious and Dr. Hopkins reported support from the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) initially presents subtly, complicating its diagnosis. Children present with a rapid onset of weakness that is associated with a febrile illness, which can be respiratory, gastrointestinal, or with symptoms of hand-foot-and-mouth disease. Given the lack of effective treatments, early diagnosis and monitoring are essential for mitigating the risk of respiratory decline and long-term complications.
While patient history and physical examination can provide clues to the presence of AFM, confirming the diagnosis requires lumbar puncture and MRI of the spinal cord. On MRI, diagnostic confirmation will come from findings of longitudinal, butterfly-shaped, anterior horn–predominant T2 and fluid-attenuated inversion recovery hyperintensities of the central gray matter.
Patients with suspected AFM should be hospitalized because they can rapidly deteriorate to the point of respiratory compromise, particularly those with upper extremity and bulbar weakness.
Sarah E. Hopkins, MD, is from the division of neurology at the Children’s Hospital of Philadelphia; Matthew J. Elrick, MD, PhD, is from the department of neurology at Johns Hopkins University, Baltimore; and Kevin Messacar, MD, is from the department of pediatrics at the Children’s Hospital Colorado. These comments are taken from an accompanying viewpoint (JAMA Pediatr. 2018 Nov 30. doi: 10.1001/jamapediatrics.2018.4896). Dr. Messacar reported support from the National Institutes of Health/National Institute of Allergy and Infectious and Dr. Hopkins reported support from the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) initially presents subtly, complicating its diagnosis. Children present with a rapid onset of weakness that is associated with a febrile illness, which can be respiratory, gastrointestinal, or with symptoms of hand-foot-and-mouth disease. Given the lack of effective treatments, early diagnosis and monitoring are essential for mitigating the risk of respiratory decline and long-term complications.
While patient history and physical examination can provide clues to the presence of AFM, confirming the diagnosis requires lumbar puncture and MRI of the spinal cord. On MRI, diagnostic confirmation will come from findings of longitudinal, butterfly-shaped, anterior horn–predominant T2 and fluid-attenuated inversion recovery hyperintensities of the central gray matter.
Patients with suspected AFM should be hospitalized because they can rapidly deteriorate to the point of respiratory compromise, particularly those with upper extremity and bulbar weakness.
Sarah E. Hopkins, MD, is from the division of neurology at the Children’s Hospital of Philadelphia; Matthew J. Elrick, MD, PhD, is from the department of neurology at Johns Hopkins University, Baltimore; and Kevin Messacar, MD, is from the department of pediatrics at the Children’s Hospital Colorado. These comments are taken from an accompanying viewpoint (JAMA Pediatr. 2018 Nov 30. doi: 10.1001/jamapediatrics.2018.4896). Dr. Messacar reported support from the National Institutes of Health/National Institute of Allergy and Infectious and Dr. Hopkins reported support from the Centers for Disease Control and Prevention.
Acute flaccid myelitis appears to present most commonly as asymmetric weakness after respiratory viral infection and has distinctive MRI features that could help with early diagnosis.
In a paper published in JAMA Pediatrics, researchers presented the results of a retrospective case series of 45 children who were diagnosed between 2012 and 2016 with acute flaccid myelitis, or “pseudo polio,” using the Centers for Disease Control’s case definition.
Matthew J. Elrick, MD, PhD, of Johns Hopkins University, Baltimore, and his coauthors came up with a set of reproducible and distinctive features of acute flaccid myelitis. These were the presence of a prodromal fever or viral syndrome; weakness in a lower motor neuron pattern involving one or more limbs, neck, face, and/or bulbar muscles; supportive evidence either from MRI, nerve conduction studies, or cerebrospinal fluid; and the absence of objective sensory deficits, supratentorial white matter, cortical lesions greater than 1 cm in size, encephalopathy, elevated cerebrospinal fluid without pleocytosis, or any other alternative diagnosis.
The researchers commented that, while the CDC case definition has helped with epidemiologic surveillance of acute flaccid myelitis, it may also pick up children with acute weakness caused by other conditions such as transverse myelitis, Guillain-Barré syndrome, ischemic myelopathy, and other myelopathies.
To identify clinical features that might help differentiate patients with acute flaccid myelitis, the researchers attempted to see how many alternative diagnoses were captured in the CDC case definition.
The patients in their study all presented with acute flaccid paralysis in at least one limb and with either an MRI showing a spinal cord lesion spanning one or more spinal segments but largely restricted to gray matter or pleocytosis of the cerebrospinal fluid. The researchers divided the cases into those who also met a well-defined alternative diagnosis – who they categorized as “acute flaccid myelitis with possible alternative diagnosis” (AFM-ad) – and those who were categorized as “restrictively defined AFM” (rAFM). Overall, 34 patients were classified as rAFM and 11 as AFM-ad.
Those in the rAFD group nearly all had asymmetric onset of symptoms, while those in the AFM-ad group were more likely to experience bilateral onset in their lower extremities, “reflecting the pattern of symptoms often seen in other causes of myelopathy such as transverse myelitis and ischemic injury,” the authors noted.
While both groups often presented with decreased muscle tone and reflexes, this was more likely to evolve to increased tone or hyperreflexia in the AFM-ad group. Patients with AFM-ad were also more likely to experience impaired bowel or bladder function.
On MRI, lesions were mostly or completely restricted to the spinal cord gray matter in patients with rAFM or to involve the dorsal pons. These patients did not have any supratentorial brain lesions.
Patients in the rAFM category also had lower cerebrospinal fluid protein values than those in the AFM-ad category, but this was the only cerebrospinal fluid difference between the two groups.
All patients categorized as having rAFM had an infectious prodrome – such as viral syndrome, fever, congestion, and cough – compared with 63.6% of the patients categorized as AFM-ad. The pathogen was identified in only 13 of the rAFM patients, and included 5 patients with enterovirus D68, 2 with unspecified enterovirus, 2 with rhinovirus, 2 with adenovirus, and 2 with mycoplasma. Of the three patients in the AFM-ad group whose pathogen was identified, one had an untyped rhinovirus/enterovirus and mycoplasma, one had a rhinovirus B, and one had enterovirus D68.
“These results highlight that the CDC case definition, while appropriately sensitive for epidemiologic ascertainment of possible AFM cases, also encompasses other neurologic diseases that can cause acute weakness,” the authors wrote. However, they acknowledged that acute flaccid myelitis was still poorly understood and their own definition of the disease may change as more children are diagnosed.
“We propose that the definition of rAFM presented here be used as a starting point for developing inclusion and exclusion criteria for future research studies of AFM,” they wrote.
The study was supported by Johns Hopkins University, the Bart McLean Fund for Neuroimmunology Research, and Project Restore. Two authors reported funding from private industry outside the submitted work and five reported support from or involvement with research and funding bodies.
SOURCE: Elrick MJ et al. JAMA Pediatr. 2018 Nov 30. doi: 10.1001/jamapediatrics.2018.4890.
Acute flaccid myelitis appears to present most commonly as asymmetric weakness after respiratory viral infection and has distinctive MRI features that could help with early diagnosis.
In a paper published in JAMA Pediatrics, researchers presented the results of a retrospective case series of 45 children who were diagnosed between 2012 and 2016 with acute flaccid myelitis, or “pseudo polio,” using the Centers for Disease Control’s case definition.
Matthew J. Elrick, MD, PhD, of Johns Hopkins University, Baltimore, and his coauthors came up with a set of reproducible and distinctive features of acute flaccid myelitis. These were the presence of a prodromal fever or viral syndrome; weakness in a lower motor neuron pattern involving one or more limbs, neck, face, and/or bulbar muscles; supportive evidence either from MRI, nerve conduction studies, or cerebrospinal fluid; and the absence of objective sensory deficits, supratentorial white matter, cortical lesions greater than 1 cm in size, encephalopathy, elevated cerebrospinal fluid without pleocytosis, or any other alternative diagnosis.
The researchers commented that, while the CDC case definition has helped with epidemiologic surveillance of acute flaccid myelitis, it may also pick up children with acute weakness caused by other conditions such as transverse myelitis, Guillain-Barré syndrome, ischemic myelopathy, and other myelopathies.
To identify clinical features that might help differentiate patients with acute flaccid myelitis, the researchers attempted to see how many alternative diagnoses were captured in the CDC case definition.
The patients in their study all presented with acute flaccid paralysis in at least one limb and with either an MRI showing a spinal cord lesion spanning one or more spinal segments but largely restricted to gray matter or pleocytosis of the cerebrospinal fluid. The researchers divided the cases into those who also met a well-defined alternative diagnosis – who they categorized as “acute flaccid myelitis with possible alternative diagnosis” (AFM-ad) – and those who were categorized as “restrictively defined AFM” (rAFM). Overall, 34 patients were classified as rAFM and 11 as AFM-ad.
Those in the rAFD group nearly all had asymmetric onset of symptoms, while those in the AFM-ad group were more likely to experience bilateral onset in their lower extremities, “reflecting the pattern of symptoms often seen in other causes of myelopathy such as transverse myelitis and ischemic injury,” the authors noted.
While both groups often presented with decreased muscle tone and reflexes, this was more likely to evolve to increased tone or hyperreflexia in the AFM-ad group. Patients with AFM-ad were also more likely to experience impaired bowel or bladder function.
On MRI, lesions were mostly or completely restricted to the spinal cord gray matter in patients with rAFM or to involve the dorsal pons. These patients did not have any supratentorial brain lesions.
Patients in the rAFM category also had lower cerebrospinal fluid protein values than those in the AFM-ad category, but this was the only cerebrospinal fluid difference between the two groups.
All patients categorized as having rAFM had an infectious prodrome – such as viral syndrome, fever, congestion, and cough – compared with 63.6% of the patients categorized as AFM-ad. The pathogen was identified in only 13 of the rAFM patients, and included 5 patients with enterovirus D68, 2 with unspecified enterovirus, 2 with rhinovirus, 2 with adenovirus, and 2 with mycoplasma. Of the three patients in the AFM-ad group whose pathogen was identified, one had an untyped rhinovirus/enterovirus and mycoplasma, one had a rhinovirus B, and one had enterovirus D68.
“These results highlight that the CDC case definition, while appropriately sensitive for epidemiologic ascertainment of possible AFM cases, also encompasses other neurologic diseases that can cause acute weakness,” the authors wrote. However, they acknowledged that acute flaccid myelitis was still poorly understood and their own definition of the disease may change as more children are diagnosed.
“We propose that the definition of rAFM presented here be used as a starting point for developing inclusion and exclusion criteria for future research studies of AFM,” they wrote.
The study was supported by Johns Hopkins University, the Bart McLean Fund for Neuroimmunology Research, and Project Restore. Two authors reported funding from private industry outside the submitted work and five reported support from or involvement with research and funding bodies.
SOURCE: Elrick MJ et al. JAMA Pediatr. 2018 Nov 30. doi: 10.1001/jamapediatrics.2018.4890.
FROM JAMA PEDIATRICS
Key clinical point: Acute flaccid myelitis has distinct features that can distinguish it from other similar conditions.
Major finding: Asymmetric onset of symptoms and MRI signature can help distinguish acute flaccid myelitis from alternative diagnoses.
Study details: A retrospective case series in 45 children diagnosed with acute flaccid myelitis.
Disclosures: The study was supported by Johns Hopkins University, the Bart McLean Fund for Neuroimmunology Research, and Project Restore. Two authors reported funding from private industry outside the submitted work and five reported support from or involvement with research and funding bodies.
Source: Elrick MJ et al. JAMA Pediatr. 2018 Nov 30. doi: 10.1001/jamapediatrics.2018.4890.