User login
Interventions for Frequently Hospitalized Patients and Their Effect on Outcomes: A Systematic Review
In recent years, hospitals and health systems have engaged in considerable efforts to reduce readmissions, in part due to financial incentives from the Medicare Hospital Readmission Reduction Program.1,2 Though efforts to improve transitions of care for all patients are laudable, risk for readmission is not distributed equally; a small subset of patients accounts for a disproportionate number of hospital readmissions.3 This phenomenon of frequently hospitalized patients is similar to that seen in other populations in which a small proportion of patients account for a majority of healthcare utilization.3,4
Recognizing that the current system of healthcare delivery does not meet the needs of this population, healthcare organizations have begun to implement interventions that supplement or redesign the system of care for frequently hospitalized patients.5-7 Descriptive reviews of ambulatory "high-need, high-cost" patients emphasize complex case management and interdisciplinary, team-based care.8,9 Prior systematic reviews of studies aimed at patients with high use of emergency care demonstrate improvements in social outcomes such as homelessness but mixed results in reducing emergency department (ED) use.10 However, we were unable to identify any prior reviews that evaluated interventions intended specifically for patients with frequent hospital admissions. Our objective in this systematic review was to characterize interventions for frequently admitted patients and determine whether these interventions decrease use of healthcare resources, improve health outcomes, and/or reduce costs.
METHODS
Literature Search
We registered our study protocol in the PROSPERO database. A librarian (L.O.) collaboratively developed the search strategies with other review authors (A.G., B.H., N.N.) and in January 2018 ran searches on "super users," "high utilizers," and similar terms in the following databases: PubMed MEDLINE, Embase (embase.com), and Cochrane Central Register of Controlled Trials (CENTRAL) on the Wiley platform. The complete search strategies used are available in Appendix A.
We attempted to discover additional studies by searching the reference lists of key publications and contacted authors of relevant abstracts to determine whether studies had been published or were planned for peer-reviewed publication. We also contacted authors of included studies to locate additional studies meeting inclusion criteria.
Data Collection Process
Studies were eligible for inclusion in our review if they were (1) published in a peer-reviewed source, (2) defined a study population of patients frequently admitted to inpatient medical services, (3) evaluated an intervention targeting frequently hospitalized patients, and (4) included patients who were >18 years old and (5) admitted as inpatients on medical services. Of note, studies with patients admitted to psychiatric, obstetric, or surgical wards were not included, as the authors did not define these as "general medicine" units. Studies focused solely on an ambulatory population were similarly excluded. Given the heterogeneity of how studies defined frequently hospitalized patients, we did not establish a prespecified number of admissions for inclusion to ensure that we did not exclude interventions not meeting a strict set of criteria. The goal was not to examine interventions to reduce all readmissions, but rather, to look at patients who were recurrently hospitalized. Thus, patients had to be repeatedly admitted, but we let the studies define that usage explicitly.
Two members of a four-physician team (A.G., B.H., K.O., and N.N.) screened all initial results for eligibility through title and abstract review; potentially relevant articles were retained for full-text review to assess each study's eligibility. If a study's abstract did not clearly indicate whether inclusion criteria were met, we retained the article for full-text review. Two team members (A.G. and B.H.) independently reviewed the full text of each selected article to determine final inclusion in the study. The previously described inclusion criteria were again applied, and a final set of articles was identified for data extraction. Disagreements regarding inclusion in the final review (such as whether a study measured medical or psychiatric hospitalizations) were resolved through discussion among the entire four-physician review team to achieve consensus or, when required, by contacting authors of individual studies.
Data Abstraction and Risk of Bias Assessment
After selecting the final set of articles, we abstracted data using a tool developed by the Cochrane Effective Practice and Organization of Care Group.11 We then compiled study-level data into a single database for reporting. Extracted elements included study design, setting, patient characteristics, inclusion and exclusion criteria, control group identification, outcome measures, results, and length of follow-up. We also extracted individual characteristics of each intervention, including common intervention elements such as intervention setting, use of health information technology resources, and whether programs developed interdisciplinary care plans. We assessed the risk of bias of each study and the quality of studies using the Downs and Black Scale.12,13 Two team members (A.G. and B.H.) independently assessed the risk of bias for all nine studies, and differences were resolved by consensus. Due to the variation in the outcomes used, we were unable to conduct a meta-analysis.
RESULTS
Search Results
We found a total of 4,762 references in the three databases. After de-duplication using the EndNote software, there were 3,314 references to screen. We identified 116 studies for full-text review. Of those, we selected nine studies that met the criteria for this study (Figure). The most common reason for exclusion of an article for full-text review was that the patients studied were not defined as high utilizers of inpatient resources and were instead high-utilizers of ambulatory or emergency care (32 studies). We identified five of the included studies through the primary search and four through review of the references of the included papers.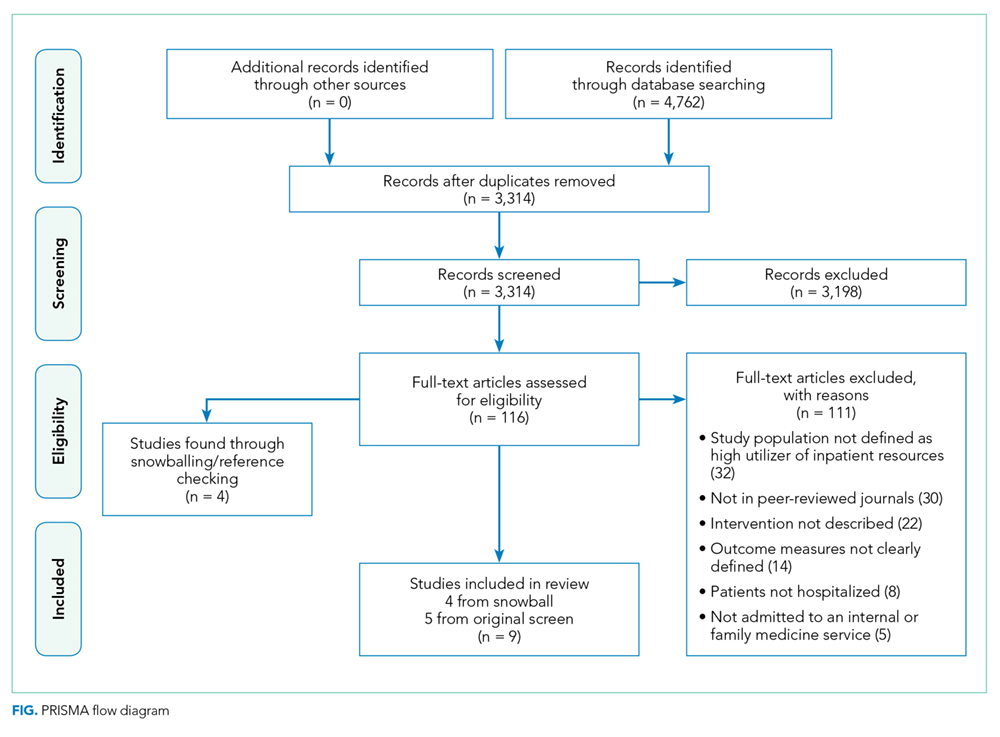
Study Designs and Included Studies
Of the nine included studies, three were randomized controlled trials, three were controlled retrospective cohort studies, and three were uncontrolled pre-post studies. The key characteristics of each study are described in Table 1.14-22 The included studies had different definitions for patients who were high utilizers of hospital care. Eight used a "threshold" model that predicted future admissions using past patterns; these studies included patients with at least two admissions over a period of 6 to 12 months, although many had higher thresholds. Zulman et al. used a prediction algorithm to identify patients at risk of future admission. Four studies also included some measure of medical complexity, such as a certain number of chronic medical conditions;14,17,18,22 in contrast, Sledge et al. excluded the most complex and high-cost patients.20
All studies measured hospital admissions as a primary or a secondary outcome (Table 1). Although all studies demonstrated a reduction in hospital admissions following implementation, those with the greatest reductions did not have a control group.14,15,17 All three randomized controlled trials showed equal reductions in admission rates between the intervention and control groups.18,20,22 Among those specifically examining readmissions to the hospital, similar trends emerged, although one study (Plant et al.) found a nonsignificant decrease in hospital readmissions (17% reduction in 24 months, P = .07).18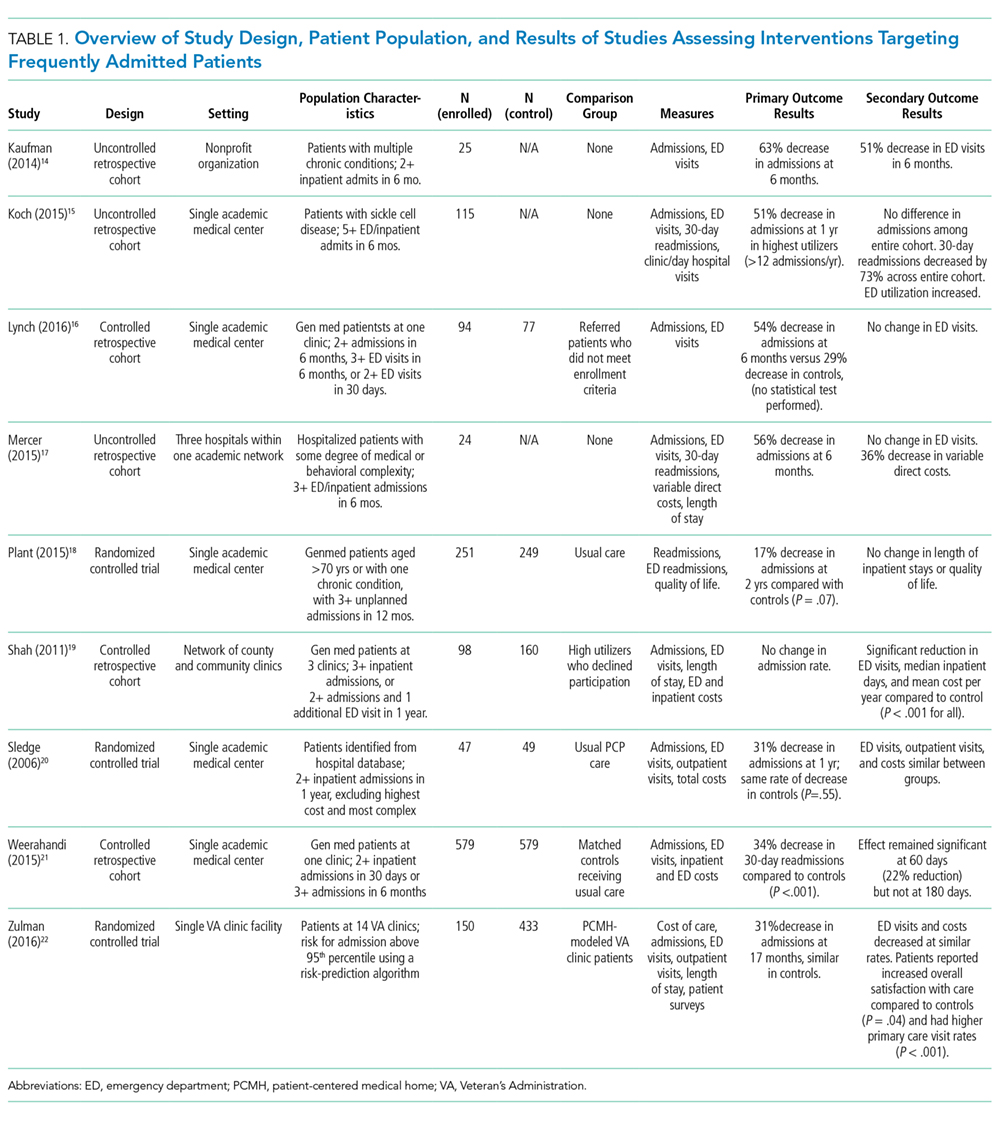
In the secondary outcome analysis, six of the nine studies found nonsignificant reductions in ED admissions (Table 1). Four studies measured costs to the hospital or the local hospital system, though none examined costs to patients or payors. Studies estimated cost differently, including the use of estimated hospital costs,17,20 "facility patient costs" at the VA,22 and a combination of inpatient and ED costs.19 The latter study (Shah et al., which implemented complex case management services) was the only one to find a statistically significant decrease in mean cost per year pre- and postintervention ($20,298 versus $7,053, P < .001).19
Only one study measured the quality of life, finding no significant change in summary scores after the intervention compared with controls (93.4 versus 92, P = .32).21 Another study conducted at a VA clinic network found no difference in a patient activation scale following the intervention but found significantly increased satisfaction with overall VA care (3.16 versus 2.90, P = .04).22
Intervention Characteristics
Intervention characteristics are summarized in Table 2. Although there was heterogeneity in study interventions, we identified common themes. Five of the nine interventions14-17,22 consisted of interdisciplinary teams that included community health workers, nurses, social workers, and physicians. Physicians were not included on every team; three interventions used them in direct care roles while two others contained physicians as advisors or in indirect roles. Intervention teams also had a variable level of involvement in a patient's care. Mercer et al. developed care plans for patients without physical interaction,17 whereas Zulman et al. recruited patients to a separate, intensive outpatient clinic outside the usual VA care team structure.22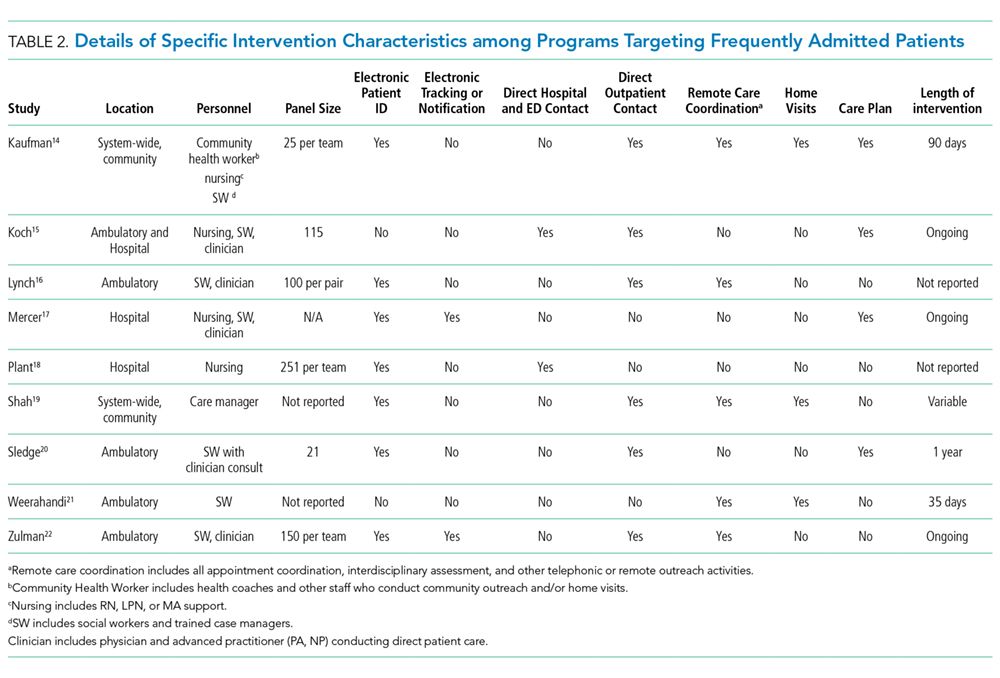
The majority of interventions added direct services or support - most commonly, a social worker - to usual care processes. Patient panel sizes were relatively small, with most of the teams recruiting fewer than 150 patients per interdisciplinary team (range, 25-251). There was variation in the length of intervention, from 35 days of case management following hospital discharge to one year of intensive social work support to others of an indefinite length.15,17,22
Additional common themes included caring for patients across settings and incorporating information technology (IT) into workflows. Four interventions reported either interacting with patients in multiple settings, such as the hospital, clinic, and day hospital, ED, at home, or in the community.14,19,21,22 Two others16,20 interacted with patients only in the clinic but expanded the scope of a "traditional" primary care practice to include open scheduling, flexible appointment times, interdisciplinary visits, or outreach. In addition, IT resources assisted seven of the nine interventions, most commonly by identifying eligible patients via an electronic data tracking system or by automated alerts when their patients arrived at affiliated care locations.
Risk of Study Bias
We systematically assessed the risk of bias of the nine included studies (Appendix B). Using the scale published by Downs and Black, a point-based scale in which a score of 18 denotes a high-quality study, the studies in this review scored 15.55 on average (range 6-22, standard deviation [SD] 5.0). Four of the nine studies met the benchmark for high quality.12,13,18-22 The risk of bias was highest for measures of internal validity and confounding (range 0-5, mean 2.83, SD 1.94). The risk of bias was lowest for reporting measures (range 0-13, mean 7.40, SD 3.43).
DISCUSSION
Overall, studies reported mixed results related to readmissions and hospital utilization. While low-quality studies found reductions in hospital use over time, higher quality studies found similar reductions in utilization between the intervention and control groups. Johnson et al. showed that frequent hospitalization rates in a cohort of high-utilizer patients declined naturally over the course of 1-2 years; only 10% of individuals in the initial cohort remained "chronically hospitalized."6 Thus, expanding on these findings, the decline in hospitalizations over time as observed in some of the studies included in this review may be due to study patients being identified during a "spike" in utilization, which naturally decreases as the underlying medical or social factors driving rehospitalization resolve. Alternatively, reduction in hospitalizations may represent patients choosing to pursue care at other neighboring hospitals.23 No study included in our review evaluated healthcare use at institutions other than their study hospital or health system.
A striking theme of this review was the heterogeneity in each study's patient population. Thresholds for "high utilizers" varied from two hospital admissions in six months to two to three admissions in 30 days, to a combination of ED and hospital admissions, and to the use of predictive algorithms. A standard "case definition" for this population could guide future research, enabling comparison of outcomes across settings. Thus, we propose that future studies use three or more hospital admissions within six months when evaluating interventions targeting "high utilizer" patients. Although patients with one prior hospitalization in the past year are at elevated risk of rehospitalization,2 we feel that a higher "threshold" for this population will identify those at the highest strata of risk. Although predictive models may be better than "threshold" models, more work in validating these tools needs to be done before these can be put to use across settings.
In contrast to interventions designed to reduce readmissions for heart failure, pneumonia, or other diagnoses, frequently admitted patients do not encompass one disease or pathology pattern. Rinehart et al., in a study characterizing frequently admitted patients across a health system, identified five "subgroups" of patients, including those with (1) unstable housing, (2) comorbid medical and psychiatric illness, (3) severe complex medical illness, (4) dual-diagnosis psychiatric illness and substance abuse, and (5) a combination of medical and psychosocial barriers.25 In light of this population's heterogeneity, interventions may need to be flexible and tailored to the needs of individual patients, while simultaneously accounting for the capabilities and priorities of the health system. More specific and standardized interventions, targeting more homogenous groups, may be appropriate for populations defined according to pathology (such as heart failure or sickle cell disease).27
The components of interventions used for frequently hospitalized patients were diverse. Although most of the studies used interdisciplinary teams, they focused their efforts in a variety of settings, often crossing modern "boundaries of care" by providing direct or indirect input on care across healthcare settings. Care fragmentation probably plays an important role in the risk for readmissions in this population;9 as such, interventions that address factors across the continuum of care may be more likely to succeed.21 Notably, six of nine studies were conducted at academic medical centers and an additional one at a VA facility affiliated with an academic center. Only two were located at community-based clinical networks, indicating a theoretical potential for publication bias as academic centers may be more likely to study and publish their work. There may be successful interventions that have not been formally studied or published in the peer-reviewed literature.
The breadth of the outcome measures in the included studies raises questions about what metrics should define success. Although all the studies looked at hospital utilization and readmission, measure definitions varied. Importantly, a minority of studies investigated quality of life and patient satisfaction, outcomes that may ultimately provide a more fertile ground for inquiry and intervention. Two studies looked at quality of life as an outcome,19,22 but only one found that patients reported increased satisfaction despite showing nonsignificant reductions in hospital use.22 As shown in multiple prior studies, patient engagement is associated with increased satisfaction and can be associated with lower healthcare costs.26,27 Hibbard et al. have demonstrated that patient activation is a specific component of patient engagement and inversely impacts healthcare cost, with lower levels of patient activation showing increased costs in comparison to those patients more engaged in their own care.27 By focusing on changing patients' perceptions about their own health and involvement in their own care team as a partner, programs may be able to make a greater impact.
Our systematic review has several limitations. Although we used a search strategy designed to identify all relevant studies, reviewed the references of included studies, and contacted the authors, we identified only nine studies meeting our inclusion criteria. Four of the nine studies were identified from a manual review of references of the included studies, suggesting the possibility of a suboptimal search strategy. Although the inclusion of articles that appear in a check of reference lists is a valid step in the systematic review article acquisition process, we conducted a post hoc investigation of alternate search strategies. We checked the titles, abstracts, and subject headings of the four articles found by reference review to determine whether the original search could have been improved. An analysis of the articles revealed that the terminology used was not consistent with the super user/utilizer terminology we were operating under, and that the four articles used terms such as "high risk" and "complex patients," which are more generic than our targeted terms. Only on a careful read of the abstracts and full-text did we find that these articles were useful to the study. Adjusting the original search to include these general terms would have resulted in an unwieldy set of results; hence, we felt it best to adhere to our original search strategy.
Additional limitations include that only four of the nine included studies were at low risk of bias. In addition to limitations based on study design and small sample sizes, the interventions were often limited to a short period. In light of the multiple factors that contribute to frequent hospitalizations, some of which cannot be addressed quickly, studies to evaluate interventions for longer durations are warranted.
CONCLUSIONS
We found mixed results for the effect of interventions on outcomes for frequently hospitalized patients. While low-quality studies found reductions in hospital use over time, higher quality studies generally found similar reductions in utilization between the intervention and control groups. The range of definitions, interventions, and outcomes used for frequently hospitalized patients is partly explained by the heterogeneity of the population. More rigorous studies using multifaceted interventions that adapt to patients' unique needs should be conducted to assess the effect on outcomes relevant to both providers and patients.
Acknowledgments
The authors would like the thank Dr. Luke Hansen, Dr. Margaret Chapman, and McKay Barra for their support and contributions to this paper and to Northwestern Memorial Hospital's CHAMP (Complex High Admission Management Program).
Disclosures
The authors have nothing to disclose.
Funding
The authors received no funding from external or internal sources for the completion of this project.
1. Center for Medicare and Medicaid Services. Readmissions Reduction Program (HRRP). https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Accessed March 23, 2018.
2. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. doi: 10.7326/0003-4819-155-8-201110180-00008. PubMed
3. Blumenthal D, Chernof B, Fulmer T, Lumpkin J, Selberg J. Caring for high-need, high-cost patients - an urgent priority. N Engl J Med. 2016;375(10):909-911. doi: 10.1056/NEJMp1608511. PubMed
4. Gawande A. The Hot Spotters. The New Yorker. 2011 Jan: 40-51.
5. Szekendi MK, Williams MV, Carrier D, Hensley L, Thomas S, Cerese J. The characteristics of patients frequently admitted to academic medical centers in the United States. J Hosp Med. 2015;10(9):563-568. doi: 10.1002/jhm.2375. PubMed
6. Johnson TL, Rinehart DJ, Durfee J, et al. For many patients who use large amounts of health care services, the need is intense yet temporary. Health Aff (Millwood). 2015;34(8):1312-1319. doi: 10.1377/hlthaff.2014.1186. PubMed
7. Tinetti ME, Reuben DB. The hospital-dependent patient. N Engl J Med. 2014;370:694-697. doi: 10.1056/NEJMp1315568. PubMed
8. Hong CS, Siegel AL, Ferris TG. Caring for high-need, high-cost patients: what makes for a successful care management program? Issue Brief (Commonw Fund). 2014;19:1-19. PubMed
9. Hochman M, Asch SM. Disruptive models in primary care: caring for high-needs, high-cost populations. J Gen Intern Med. 2017;32(4):392-397. doi: 10.1007/s11606-016-3945-2. PubMed
10. Althaus F1, Paroz S, Hugli O, et al. Effectiveness of interventions targeting frequent users of emergency departments: a systematic review. Ann Emerg Med. 2011 Jul;58(1):41-52.e42. doi: 10.1016/j.annemergmed.2011.03.007 PubMed
11. Cochrane Effective Practice and Organisation of Care (EPOC). What study designs should be included in an EPOC review? EPOC resources for review authors. Available at:http://epoc.cochrane.org/epoc-resources-review-authors. Accessed March 23, 2018.
12. Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377-384. doi: 10.1136/jech.52.6.377. PubMed
13. Goyal AA, Tur K, Mann J, Townsend W, Flanders SA, Chopra V. Do bedside visual tools improve patient and caregiver satisfaction? A systematic review of the literature. J Hosp Med 2017;12(11):930-936. doi: 10.12788/jhm.2871. PubMed
14. Kaufman S, Ali N, DeFiglio V, Craig K, Brenner J. Early efforts to target and enroll high-risk diabetic patients into urban community-based programs. Health Promot Pract. 2014;15(2 Suppl):62S-70S. doi: 10.1177/1524839914535776. PubMed
15. Koch KL, Karafin MS, Simpson P, Field JJ. Intensive management of high-utilizing adults with sickle cell disease lowers admissions. Am J Hematol. 2015;90(3):215-219. doi: 10.1002/ajh.23912. PubMed
16. Lynch CS, Wajnberg A, Jervis R, et al. Implementation science workshop: a novel multidisciplinary primary care program to improve care and outcomes for super-utilizers. J Gen Intern Med. 2016;31(7):797-802. doi: 10.1007/s11606-016-3598-1. PubMed
17. Mercer T, Bae J, Kipnes J, Velazquez M, Thomas S, Setji N. The highest utilizers of care: individualized care plans to coordinate care, improve healthcare service utilization, and reduce costs at an academic tertiary care center. J Hosp Med. 2015;10(7):419-424. doi: 10.1002/jhm.2351. PubMed
18. Plant NA, Kelly PJ, Leeder SR, et al. Coordinated care versus standard care in hospital admissions of people with chronic illness: a randomised controlled trial. Med J Aust. 2015;203(1):33-38. doi: 10.5694/mja14.01049. PubMed
19. Shah R, Chen C, O'Rourke S, Lee M, Mohanty SA, Abraham J. Evaluation of care management for the uninsured. Med Care. 2011;49(2):166-171. doi: 10.1097/MLR.0b013e3182028e81. PubMed
20. Sledge WH, Brown KE, Levine JM, et al. A randomized trial of primary intensive care to reduce hospital admissions in patients with high utilization of inpatient services. Dis Manag. 2006;9(6):328-338. doi: 10.1089/dis.2006.9.328. PubMed
21. Weerahandi H, Basso Lipani M, Kalman J, et al. Effects of a psychosocial transitional care model on hospitalizations and cost of care for high utilizers. Soc Work Health Care. 2015;54(6):485-498. doi: 10.1080/00981389.2015.1040141. PubMed
22. Zulman DM, Ezeji-Okoye SC, Shaw JG, et al. Partnered research in healthcare delivery redesign for high-need, high-cost patients: development and feasibility of an Intensive Management Patient-Aligned Care Team (ImPACT). J Gen Intern Med. 2014;29(4):861-869. doi: 10.1007/s11606-014-3022-7. PubMed
23. Mautner DB, Pang H, Brenner JC, et al. Generating hypotheses about care needs of high utilizers: lessons from patient interviews. Popul Health Manag. 2013;16 Suppl 1:S26-33. doi: 10.1089/pop.2013.0033. PubMed
24. Bodenheimer T. Strategies to reduce costs and improve care for high-utilizing Medicaid patients: Reflections on pioneering programs. Center for Health Care Strategies, Inc.;2013.
25. Rinehart DJ, Oronce C, Durfee MJ, et al. Identifying subgroups of adult superutilizers in an urban safety-net system using latent class analysis: implications for clinical practice. Med Care. 2018;56(1):e1-e9. doi: 10.1097/MLR.0000000000000628. PubMed
26. Boutwell A, Kunst E, Sorin J, Shniffer A, Logozzo J, Woodhouse D. DSRIP-Medicaid Accelerated eXchange (MAX) Series Program: Improving Care for Super Utilizers. January 2017. https://www.health.ny.gov/health_care/medicaid/redesign/dsrip/pps_workshops/docs/2017-01_imp_care.pdf. Accessed January 24, 2018.
27. Hibbard JH, Stockard J, Mahoney ER, Tusler M. Development of the Patient Activation Measure (PAM): conceptualizing and measuring activation in patients and consumers. Health Serv Res. 2004;39(4 Pt 1):1005-1026. doi: 10.1111/j.1475-6773.2004.00269.x PubMed
In recent years, hospitals and health systems have engaged in considerable efforts to reduce readmissions, in part due to financial incentives from the Medicare Hospital Readmission Reduction Program.1,2 Though efforts to improve transitions of care for all patients are laudable, risk for readmission is not distributed equally; a small subset of patients accounts for a disproportionate number of hospital readmissions.3 This phenomenon of frequently hospitalized patients is similar to that seen in other populations in which a small proportion of patients account for a majority of healthcare utilization.3,4
Recognizing that the current system of healthcare delivery does not meet the needs of this population, healthcare organizations have begun to implement interventions that supplement or redesign the system of care for frequently hospitalized patients.5-7 Descriptive reviews of ambulatory "high-need, high-cost" patients emphasize complex case management and interdisciplinary, team-based care.8,9 Prior systematic reviews of studies aimed at patients with high use of emergency care demonstrate improvements in social outcomes such as homelessness but mixed results in reducing emergency department (ED) use.10 However, we were unable to identify any prior reviews that evaluated interventions intended specifically for patients with frequent hospital admissions. Our objective in this systematic review was to characterize interventions for frequently admitted patients and determine whether these interventions decrease use of healthcare resources, improve health outcomes, and/or reduce costs.
METHODS
Literature Search
We registered our study protocol in the PROSPERO database. A librarian (L.O.) collaboratively developed the search strategies with other review authors (A.G., B.H., N.N.) and in January 2018 ran searches on "super users," "high utilizers," and similar terms in the following databases: PubMed MEDLINE, Embase (embase.com), and Cochrane Central Register of Controlled Trials (CENTRAL) on the Wiley platform. The complete search strategies used are available in Appendix A.
We attempted to discover additional studies by searching the reference lists of key publications and contacted authors of relevant abstracts to determine whether studies had been published or were planned for peer-reviewed publication. We also contacted authors of included studies to locate additional studies meeting inclusion criteria.
Data Collection Process
Studies were eligible for inclusion in our review if they were (1) published in a peer-reviewed source, (2) defined a study population of patients frequently admitted to inpatient medical services, (3) evaluated an intervention targeting frequently hospitalized patients, and (4) included patients who were >18 years old and (5) admitted as inpatients on medical services. Of note, studies with patients admitted to psychiatric, obstetric, or surgical wards were not included, as the authors did not define these as "general medicine" units. Studies focused solely on an ambulatory population were similarly excluded. Given the heterogeneity of how studies defined frequently hospitalized patients, we did not establish a prespecified number of admissions for inclusion to ensure that we did not exclude interventions not meeting a strict set of criteria. The goal was not to examine interventions to reduce all readmissions, but rather, to look at patients who were recurrently hospitalized. Thus, patients had to be repeatedly admitted, but we let the studies define that usage explicitly.
Two members of a four-physician team (A.G., B.H., K.O., and N.N.) screened all initial results for eligibility through title and abstract review; potentially relevant articles were retained for full-text review to assess each study's eligibility. If a study's abstract did not clearly indicate whether inclusion criteria were met, we retained the article for full-text review. Two team members (A.G. and B.H.) independently reviewed the full text of each selected article to determine final inclusion in the study. The previously described inclusion criteria were again applied, and a final set of articles was identified for data extraction. Disagreements regarding inclusion in the final review (such as whether a study measured medical or psychiatric hospitalizations) were resolved through discussion among the entire four-physician review team to achieve consensus or, when required, by contacting authors of individual studies.
Data Abstraction and Risk of Bias Assessment
After selecting the final set of articles, we abstracted data using a tool developed by the Cochrane Effective Practice and Organization of Care Group.11 We then compiled study-level data into a single database for reporting. Extracted elements included study design, setting, patient characteristics, inclusion and exclusion criteria, control group identification, outcome measures, results, and length of follow-up. We also extracted individual characteristics of each intervention, including common intervention elements such as intervention setting, use of health information technology resources, and whether programs developed interdisciplinary care plans. We assessed the risk of bias of each study and the quality of studies using the Downs and Black Scale.12,13 Two team members (A.G. and B.H.) independently assessed the risk of bias for all nine studies, and differences were resolved by consensus. Due to the variation in the outcomes used, we were unable to conduct a meta-analysis.
RESULTS
Search Results
We found a total of 4,762 references in the three databases. After de-duplication using the EndNote software, there were 3,314 references to screen. We identified 116 studies for full-text review. Of those, we selected nine studies that met the criteria for this study (Figure). The most common reason for exclusion of an article for full-text review was that the patients studied were not defined as high utilizers of inpatient resources and were instead high-utilizers of ambulatory or emergency care (32 studies). We identified five of the included studies through the primary search and four through review of the references of the included papers.
Study Designs and Included Studies
Of the nine included studies, three were randomized controlled trials, three were controlled retrospective cohort studies, and three were uncontrolled pre-post studies. The key characteristics of each study are described in Table 1.14-22 The included studies had different definitions for patients who were high utilizers of hospital care. Eight used a "threshold" model that predicted future admissions using past patterns; these studies included patients with at least two admissions over a period of 6 to 12 months, although many had higher thresholds. Zulman et al. used a prediction algorithm to identify patients at risk of future admission. Four studies also included some measure of medical complexity, such as a certain number of chronic medical conditions;14,17,18,22 in contrast, Sledge et al. excluded the most complex and high-cost patients.20
All studies measured hospital admissions as a primary or a secondary outcome (Table 1). Although all studies demonstrated a reduction in hospital admissions following implementation, those with the greatest reductions did not have a control group.14,15,17 All three randomized controlled trials showed equal reductions in admission rates between the intervention and control groups.18,20,22 Among those specifically examining readmissions to the hospital, similar trends emerged, although one study (Plant et al.) found a nonsignificant decrease in hospital readmissions (17% reduction in 24 months, P = .07).18
In the secondary outcome analysis, six of the nine studies found nonsignificant reductions in ED admissions (Table 1). Four studies measured costs to the hospital or the local hospital system, though none examined costs to patients or payors. Studies estimated cost differently, including the use of estimated hospital costs,17,20 "facility patient costs" at the VA,22 and a combination of inpatient and ED costs.19 The latter study (Shah et al., which implemented complex case management services) was the only one to find a statistically significant decrease in mean cost per year pre- and postintervention ($20,298 versus $7,053, P < .001).19
Only one study measured the quality of life, finding no significant change in summary scores after the intervention compared with controls (93.4 versus 92, P = .32).21 Another study conducted at a VA clinic network found no difference in a patient activation scale following the intervention but found significantly increased satisfaction with overall VA care (3.16 versus 2.90, P = .04).22
Intervention Characteristics
Intervention characteristics are summarized in Table 2. Although there was heterogeneity in study interventions, we identified common themes. Five of the nine interventions14-17,22 consisted of interdisciplinary teams that included community health workers, nurses, social workers, and physicians. Physicians were not included on every team; three interventions used them in direct care roles while two others contained physicians as advisors or in indirect roles. Intervention teams also had a variable level of involvement in a patient's care. Mercer et al. developed care plans for patients without physical interaction,17 whereas Zulman et al. recruited patients to a separate, intensive outpatient clinic outside the usual VA care team structure.22
The majority of interventions added direct services or support - most commonly, a social worker - to usual care processes. Patient panel sizes were relatively small, with most of the teams recruiting fewer than 150 patients per interdisciplinary team (range, 25-251). There was variation in the length of intervention, from 35 days of case management following hospital discharge to one year of intensive social work support to others of an indefinite length.15,17,22
Additional common themes included caring for patients across settings and incorporating information technology (IT) into workflows. Four interventions reported either interacting with patients in multiple settings, such as the hospital, clinic, and day hospital, ED, at home, or in the community.14,19,21,22 Two others16,20 interacted with patients only in the clinic but expanded the scope of a "traditional" primary care practice to include open scheduling, flexible appointment times, interdisciplinary visits, or outreach. In addition, IT resources assisted seven of the nine interventions, most commonly by identifying eligible patients via an electronic data tracking system or by automated alerts when their patients arrived at affiliated care locations.
Risk of Study Bias
We systematically assessed the risk of bias of the nine included studies (Appendix B). Using the scale published by Downs and Black, a point-based scale in which a score of 18 denotes a high-quality study, the studies in this review scored 15.55 on average (range 6-22, standard deviation [SD] 5.0). Four of the nine studies met the benchmark for high quality.12,13,18-22 The risk of bias was highest for measures of internal validity and confounding (range 0-5, mean 2.83, SD 1.94). The risk of bias was lowest for reporting measures (range 0-13, mean 7.40, SD 3.43).
DISCUSSION
Overall, studies reported mixed results related to readmissions and hospital utilization. While low-quality studies found reductions in hospital use over time, higher quality studies found similar reductions in utilization between the intervention and control groups. Johnson et al. showed that frequent hospitalization rates in a cohort of high-utilizer patients declined naturally over the course of 1-2 years; only 10% of individuals in the initial cohort remained "chronically hospitalized."6 Thus, expanding on these findings, the decline in hospitalizations over time as observed in some of the studies included in this review may be due to study patients being identified during a "spike" in utilization, which naturally decreases as the underlying medical or social factors driving rehospitalization resolve. Alternatively, reduction in hospitalizations may represent patients choosing to pursue care at other neighboring hospitals.23 No study included in our review evaluated healthcare use at institutions other than their study hospital or health system.
A striking theme of this review was the heterogeneity in each study's patient population. Thresholds for "high utilizers" varied from two hospital admissions in six months to two to three admissions in 30 days, to a combination of ED and hospital admissions, and to the use of predictive algorithms. A standard "case definition" for this population could guide future research, enabling comparison of outcomes across settings. Thus, we propose that future studies use three or more hospital admissions within six months when evaluating interventions targeting "high utilizer" patients. Although patients with one prior hospitalization in the past year are at elevated risk of rehospitalization,2 we feel that a higher "threshold" for this population will identify those at the highest strata of risk. Although predictive models may be better than "threshold" models, more work in validating these tools needs to be done before these can be put to use across settings.
In contrast to interventions designed to reduce readmissions for heart failure, pneumonia, or other diagnoses, frequently admitted patients do not encompass one disease or pathology pattern. Rinehart et al., in a study characterizing frequently admitted patients across a health system, identified five "subgroups" of patients, including those with (1) unstable housing, (2) comorbid medical and psychiatric illness, (3) severe complex medical illness, (4) dual-diagnosis psychiatric illness and substance abuse, and (5) a combination of medical and psychosocial barriers.25 In light of this population's heterogeneity, interventions may need to be flexible and tailored to the needs of individual patients, while simultaneously accounting for the capabilities and priorities of the health system. More specific and standardized interventions, targeting more homogenous groups, may be appropriate for populations defined according to pathology (such as heart failure or sickle cell disease).27
The components of interventions used for frequently hospitalized patients were diverse. Although most of the studies used interdisciplinary teams, they focused their efforts in a variety of settings, often crossing modern "boundaries of care" by providing direct or indirect input on care across healthcare settings. Care fragmentation probably plays an important role in the risk for readmissions in this population;9 as such, interventions that address factors across the continuum of care may be more likely to succeed.21 Notably, six of nine studies were conducted at academic medical centers and an additional one at a VA facility affiliated with an academic center. Only two were located at community-based clinical networks, indicating a theoretical potential for publication bias as academic centers may be more likely to study and publish their work. There may be successful interventions that have not been formally studied or published in the peer-reviewed literature.
The breadth of the outcome measures in the included studies raises questions about what metrics should define success. Although all the studies looked at hospital utilization and readmission, measure definitions varied. Importantly, a minority of studies investigated quality of life and patient satisfaction, outcomes that may ultimately provide a more fertile ground for inquiry and intervention. Two studies looked at quality of life as an outcome,19,22 but only one found that patients reported increased satisfaction despite showing nonsignificant reductions in hospital use.22 As shown in multiple prior studies, patient engagement is associated with increased satisfaction and can be associated with lower healthcare costs.26,27 Hibbard et al. have demonstrated that patient activation is a specific component of patient engagement and inversely impacts healthcare cost, with lower levels of patient activation showing increased costs in comparison to those patients more engaged in their own care.27 By focusing on changing patients' perceptions about their own health and involvement in their own care team as a partner, programs may be able to make a greater impact.
Our systematic review has several limitations. Although we used a search strategy designed to identify all relevant studies, reviewed the references of included studies, and contacted the authors, we identified only nine studies meeting our inclusion criteria. Four of the nine studies were identified from a manual review of references of the included studies, suggesting the possibility of a suboptimal search strategy. Although the inclusion of articles that appear in a check of reference lists is a valid step in the systematic review article acquisition process, we conducted a post hoc investigation of alternate search strategies. We checked the titles, abstracts, and subject headings of the four articles found by reference review to determine whether the original search could have been improved. An analysis of the articles revealed that the terminology used was not consistent with the super user/utilizer terminology we were operating under, and that the four articles used terms such as "high risk" and "complex patients," which are more generic than our targeted terms. Only on a careful read of the abstracts and full-text did we find that these articles were useful to the study. Adjusting the original search to include these general terms would have resulted in an unwieldy set of results; hence, we felt it best to adhere to our original search strategy.
Additional limitations include that only four of the nine included studies were at low risk of bias. In addition to limitations based on study design and small sample sizes, the interventions were often limited to a short period. In light of the multiple factors that contribute to frequent hospitalizations, some of which cannot be addressed quickly, studies to evaluate interventions for longer durations are warranted.
CONCLUSIONS
We found mixed results for the effect of interventions on outcomes for frequently hospitalized patients. While low-quality studies found reductions in hospital use over time, higher quality studies generally found similar reductions in utilization between the intervention and control groups. The range of definitions, interventions, and outcomes used for frequently hospitalized patients is partly explained by the heterogeneity of the population. More rigorous studies using multifaceted interventions that adapt to patients' unique needs should be conducted to assess the effect on outcomes relevant to both providers and patients.
Acknowledgments
The authors would like the thank Dr. Luke Hansen, Dr. Margaret Chapman, and McKay Barra for their support and contributions to this paper and to Northwestern Memorial Hospital's CHAMP (Complex High Admission Management Program).
Disclosures
The authors have nothing to disclose.
Funding
The authors received no funding from external or internal sources for the completion of this project.
In recent years, hospitals and health systems have engaged in considerable efforts to reduce readmissions, in part due to financial incentives from the Medicare Hospital Readmission Reduction Program.1,2 Though efforts to improve transitions of care for all patients are laudable, risk for readmission is not distributed equally; a small subset of patients accounts for a disproportionate number of hospital readmissions.3 This phenomenon of frequently hospitalized patients is similar to that seen in other populations in which a small proportion of patients account for a majority of healthcare utilization.3,4
Recognizing that the current system of healthcare delivery does not meet the needs of this population, healthcare organizations have begun to implement interventions that supplement or redesign the system of care for frequently hospitalized patients.5-7 Descriptive reviews of ambulatory "high-need, high-cost" patients emphasize complex case management and interdisciplinary, team-based care.8,9 Prior systematic reviews of studies aimed at patients with high use of emergency care demonstrate improvements in social outcomes such as homelessness but mixed results in reducing emergency department (ED) use.10 However, we were unable to identify any prior reviews that evaluated interventions intended specifically for patients with frequent hospital admissions. Our objective in this systematic review was to characterize interventions for frequently admitted patients and determine whether these interventions decrease use of healthcare resources, improve health outcomes, and/or reduce costs.
METHODS
Literature Search
We registered our study protocol in the PROSPERO database. A librarian (L.O.) collaboratively developed the search strategies with other review authors (A.G., B.H., N.N.) and in January 2018 ran searches on "super users," "high utilizers," and similar terms in the following databases: PubMed MEDLINE, Embase (embase.com), and Cochrane Central Register of Controlled Trials (CENTRAL) on the Wiley platform. The complete search strategies used are available in Appendix A.
We attempted to discover additional studies by searching the reference lists of key publications and contacted authors of relevant abstracts to determine whether studies had been published or were planned for peer-reviewed publication. We also contacted authors of included studies to locate additional studies meeting inclusion criteria.
Data Collection Process
Studies were eligible for inclusion in our review if they were (1) published in a peer-reviewed source, (2) defined a study population of patients frequently admitted to inpatient medical services, (3) evaluated an intervention targeting frequently hospitalized patients, and (4) included patients who were >18 years old and (5) admitted as inpatients on medical services. Of note, studies with patients admitted to psychiatric, obstetric, or surgical wards were not included, as the authors did not define these as "general medicine" units. Studies focused solely on an ambulatory population were similarly excluded. Given the heterogeneity of how studies defined frequently hospitalized patients, we did not establish a prespecified number of admissions for inclusion to ensure that we did not exclude interventions not meeting a strict set of criteria. The goal was not to examine interventions to reduce all readmissions, but rather, to look at patients who were recurrently hospitalized. Thus, patients had to be repeatedly admitted, but we let the studies define that usage explicitly.
Two members of a four-physician team (A.G., B.H., K.O., and N.N.) screened all initial results for eligibility through title and abstract review; potentially relevant articles were retained for full-text review to assess each study's eligibility. If a study's abstract did not clearly indicate whether inclusion criteria were met, we retained the article for full-text review. Two team members (A.G. and B.H.) independently reviewed the full text of each selected article to determine final inclusion in the study. The previously described inclusion criteria were again applied, and a final set of articles was identified for data extraction. Disagreements regarding inclusion in the final review (such as whether a study measured medical or psychiatric hospitalizations) were resolved through discussion among the entire four-physician review team to achieve consensus or, when required, by contacting authors of individual studies.
Data Abstraction and Risk of Bias Assessment
After selecting the final set of articles, we abstracted data using a tool developed by the Cochrane Effective Practice and Organization of Care Group.11 We then compiled study-level data into a single database for reporting. Extracted elements included study design, setting, patient characteristics, inclusion and exclusion criteria, control group identification, outcome measures, results, and length of follow-up. We also extracted individual characteristics of each intervention, including common intervention elements such as intervention setting, use of health information technology resources, and whether programs developed interdisciplinary care plans. We assessed the risk of bias of each study and the quality of studies using the Downs and Black Scale.12,13 Two team members (A.G. and B.H.) independently assessed the risk of bias for all nine studies, and differences were resolved by consensus. Due to the variation in the outcomes used, we were unable to conduct a meta-analysis.
RESULTS
Search Results
We found a total of 4,762 references in the three databases. After de-duplication using the EndNote software, there were 3,314 references to screen. We identified 116 studies for full-text review. Of those, we selected nine studies that met the criteria for this study (Figure). The most common reason for exclusion of an article for full-text review was that the patients studied were not defined as high utilizers of inpatient resources and were instead high-utilizers of ambulatory or emergency care (32 studies). We identified five of the included studies through the primary search and four through review of the references of the included papers.
Study Designs and Included Studies
Of the nine included studies, three were randomized controlled trials, three were controlled retrospective cohort studies, and three were uncontrolled pre-post studies. The key characteristics of each study are described in Table 1.14-22 The included studies had different definitions for patients who were high utilizers of hospital care. Eight used a "threshold" model that predicted future admissions using past patterns; these studies included patients with at least two admissions over a period of 6 to 12 months, although many had higher thresholds. Zulman et al. used a prediction algorithm to identify patients at risk of future admission. Four studies also included some measure of medical complexity, such as a certain number of chronic medical conditions;14,17,18,22 in contrast, Sledge et al. excluded the most complex and high-cost patients.20
All studies measured hospital admissions as a primary or a secondary outcome (Table 1). Although all studies demonstrated a reduction in hospital admissions following implementation, those with the greatest reductions did not have a control group.14,15,17 All three randomized controlled trials showed equal reductions in admission rates between the intervention and control groups.18,20,22 Among those specifically examining readmissions to the hospital, similar trends emerged, although one study (Plant et al.) found a nonsignificant decrease in hospital readmissions (17% reduction in 24 months, P = .07).18
In the secondary outcome analysis, six of the nine studies found nonsignificant reductions in ED admissions (Table 1). Four studies measured costs to the hospital or the local hospital system, though none examined costs to patients or payors. Studies estimated cost differently, including the use of estimated hospital costs,17,20 "facility patient costs" at the VA,22 and a combination of inpatient and ED costs.19 The latter study (Shah et al., which implemented complex case management services) was the only one to find a statistically significant decrease in mean cost per year pre- and postintervention ($20,298 versus $7,053, P < .001).19
Only one study measured the quality of life, finding no significant change in summary scores after the intervention compared with controls (93.4 versus 92, P = .32).21 Another study conducted at a VA clinic network found no difference in a patient activation scale following the intervention but found significantly increased satisfaction with overall VA care (3.16 versus 2.90, P = .04).22
Intervention Characteristics
Intervention characteristics are summarized in Table 2. Although there was heterogeneity in study interventions, we identified common themes. Five of the nine interventions14-17,22 consisted of interdisciplinary teams that included community health workers, nurses, social workers, and physicians. Physicians were not included on every team; three interventions used them in direct care roles while two others contained physicians as advisors or in indirect roles. Intervention teams also had a variable level of involvement in a patient's care. Mercer et al. developed care plans for patients without physical interaction,17 whereas Zulman et al. recruited patients to a separate, intensive outpatient clinic outside the usual VA care team structure.22
The majority of interventions added direct services or support - most commonly, a social worker - to usual care processes. Patient panel sizes were relatively small, with most of the teams recruiting fewer than 150 patients per interdisciplinary team (range, 25-251). There was variation in the length of intervention, from 35 days of case management following hospital discharge to one year of intensive social work support to others of an indefinite length.15,17,22
Additional common themes included caring for patients across settings and incorporating information technology (IT) into workflows. Four interventions reported either interacting with patients in multiple settings, such as the hospital, clinic, and day hospital, ED, at home, or in the community.14,19,21,22 Two others16,20 interacted with patients only in the clinic but expanded the scope of a "traditional" primary care practice to include open scheduling, flexible appointment times, interdisciplinary visits, or outreach. In addition, IT resources assisted seven of the nine interventions, most commonly by identifying eligible patients via an electronic data tracking system or by automated alerts when their patients arrived at affiliated care locations.
Risk of Study Bias
We systematically assessed the risk of bias of the nine included studies (Appendix B). Using the scale published by Downs and Black, a point-based scale in which a score of 18 denotes a high-quality study, the studies in this review scored 15.55 on average (range 6-22, standard deviation [SD] 5.0). Four of the nine studies met the benchmark for high quality.12,13,18-22 The risk of bias was highest for measures of internal validity and confounding (range 0-5, mean 2.83, SD 1.94). The risk of bias was lowest for reporting measures (range 0-13, mean 7.40, SD 3.43).
DISCUSSION
Overall, studies reported mixed results related to readmissions and hospital utilization. While low-quality studies found reductions in hospital use over time, higher quality studies found similar reductions in utilization between the intervention and control groups. Johnson et al. showed that frequent hospitalization rates in a cohort of high-utilizer patients declined naturally over the course of 1-2 years; only 10% of individuals in the initial cohort remained "chronically hospitalized."6 Thus, expanding on these findings, the decline in hospitalizations over time as observed in some of the studies included in this review may be due to study patients being identified during a "spike" in utilization, which naturally decreases as the underlying medical or social factors driving rehospitalization resolve. Alternatively, reduction in hospitalizations may represent patients choosing to pursue care at other neighboring hospitals.23 No study included in our review evaluated healthcare use at institutions other than their study hospital or health system.
A striking theme of this review was the heterogeneity in each study's patient population. Thresholds for "high utilizers" varied from two hospital admissions in six months to two to three admissions in 30 days, to a combination of ED and hospital admissions, and to the use of predictive algorithms. A standard "case definition" for this population could guide future research, enabling comparison of outcomes across settings. Thus, we propose that future studies use three or more hospital admissions within six months when evaluating interventions targeting "high utilizer" patients. Although patients with one prior hospitalization in the past year are at elevated risk of rehospitalization,2 we feel that a higher "threshold" for this population will identify those at the highest strata of risk. Although predictive models may be better than "threshold" models, more work in validating these tools needs to be done before these can be put to use across settings.
In contrast to interventions designed to reduce readmissions for heart failure, pneumonia, or other diagnoses, frequently admitted patients do not encompass one disease or pathology pattern. Rinehart et al., in a study characterizing frequently admitted patients across a health system, identified five "subgroups" of patients, including those with (1) unstable housing, (2) comorbid medical and psychiatric illness, (3) severe complex medical illness, (4) dual-diagnosis psychiatric illness and substance abuse, and (5) a combination of medical and psychosocial barriers.25 In light of this population's heterogeneity, interventions may need to be flexible and tailored to the needs of individual patients, while simultaneously accounting for the capabilities and priorities of the health system. More specific and standardized interventions, targeting more homogenous groups, may be appropriate for populations defined according to pathology (such as heart failure or sickle cell disease).27
The components of interventions used for frequently hospitalized patients were diverse. Although most of the studies used interdisciplinary teams, they focused their efforts in a variety of settings, often crossing modern "boundaries of care" by providing direct or indirect input on care across healthcare settings. Care fragmentation probably plays an important role in the risk for readmissions in this population;9 as such, interventions that address factors across the continuum of care may be more likely to succeed.21 Notably, six of nine studies were conducted at academic medical centers and an additional one at a VA facility affiliated with an academic center. Only two were located at community-based clinical networks, indicating a theoretical potential for publication bias as academic centers may be more likely to study and publish their work. There may be successful interventions that have not been formally studied or published in the peer-reviewed literature.
The breadth of the outcome measures in the included studies raises questions about what metrics should define success. Although all the studies looked at hospital utilization and readmission, measure definitions varied. Importantly, a minority of studies investigated quality of life and patient satisfaction, outcomes that may ultimately provide a more fertile ground for inquiry and intervention. Two studies looked at quality of life as an outcome,19,22 but only one found that patients reported increased satisfaction despite showing nonsignificant reductions in hospital use.22 As shown in multiple prior studies, patient engagement is associated with increased satisfaction and can be associated with lower healthcare costs.26,27 Hibbard et al. have demonstrated that patient activation is a specific component of patient engagement and inversely impacts healthcare cost, with lower levels of patient activation showing increased costs in comparison to those patients more engaged in their own care.27 By focusing on changing patients' perceptions about their own health and involvement in their own care team as a partner, programs may be able to make a greater impact.
Our systematic review has several limitations. Although we used a search strategy designed to identify all relevant studies, reviewed the references of included studies, and contacted the authors, we identified only nine studies meeting our inclusion criteria. Four of the nine studies were identified from a manual review of references of the included studies, suggesting the possibility of a suboptimal search strategy. Although the inclusion of articles that appear in a check of reference lists is a valid step in the systematic review article acquisition process, we conducted a post hoc investigation of alternate search strategies. We checked the titles, abstracts, and subject headings of the four articles found by reference review to determine whether the original search could have been improved. An analysis of the articles revealed that the terminology used was not consistent with the super user/utilizer terminology we were operating under, and that the four articles used terms such as "high risk" and "complex patients," which are more generic than our targeted terms. Only on a careful read of the abstracts and full-text did we find that these articles were useful to the study. Adjusting the original search to include these general terms would have resulted in an unwieldy set of results; hence, we felt it best to adhere to our original search strategy.
Additional limitations include that only four of the nine included studies were at low risk of bias. In addition to limitations based on study design and small sample sizes, the interventions were often limited to a short period. In light of the multiple factors that contribute to frequent hospitalizations, some of which cannot be addressed quickly, studies to evaluate interventions for longer durations are warranted.
CONCLUSIONS
We found mixed results for the effect of interventions on outcomes for frequently hospitalized patients. While low-quality studies found reductions in hospital use over time, higher quality studies generally found similar reductions in utilization between the intervention and control groups. The range of definitions, interventions, and outcomes used for frequently hospitalized patients is partly explained by the heterogeneity of the population. More rigorous studies using multifaceted interventions that adapt to patients' unique needs should be conducted to assess the effect on outcomes relevant to both providers and patients.
Acknowledgments
The authors would like the thank Dr. Luke Hansen, Dr. Margaret Chapman, and McKay Barra for their support and contributions to this paper and to Northwestern Memorial Hospital's CHAMP (Complex High Admission Management Program).
Disclosures
The authors have nothing to disclose.
Funding
The authors received no funding from external or internal sources for the completion of this project.
1. Center for Medicare and Medicaid Services. Readmissions Reduction Program (HRRP). https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Accessed March 23, 2018.
2. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. doi: 10.7326/0003-4819-155-8-201110180-00008. PubMed
3. Blumenthal D, Chernof B, Fulmer T, Lumpkin J, Selberg J. Caring for high-need, high-cost patients - an urgent priority. N Engl J Med. 2016;375(10):909-911. doi: 10.1056/NEJMp1608511. PubMed
4. Gawande A. The Hot Spotters. The New Yorker. 2011 Jan: 40-51.
5. Szekendi MK, Williams MV, Carrier D, Hensley L, Thomas S, Cerese J. The characteristics of patients frequently admitted to academic medical centers in the United States. J Hosp Med. 2015;10(9):563-568. doi: 10.1002/jhm.2375. PubMed
6. Johnson TL, Rinehart DJ, Durfee J, et al. For many patients who use large amounts of health care services, the need is intense yet temporary. Health Aff (Millwood). 2015;34(8):1312-1319. doi: 10.1377/hlthaff.2014.1186. PubMed
7. Tinetti ME, Reuben DB. The hospital-dependent patient. N Engl J Med. 2014;370:694-697. doi: 10.1056/NEJMp1315568. PubMed
8. Hong CS, Siegel AL, Ferris TG. Caring for high-need, high-cost patients: what makes for a successful care management program? Issue Brief (Commonw Fund). 2014;19:1-19. PubMed
9. Hochman M, Asch SM. Disruptive models in primary care: caring for high-needs, high-cost populations. J Gen Intern Med. 2017;32(4):392-397. doi: 10.1007/s11606-016-3945-2. PubMed
10. Althaus F1, Paroz S, Hugli O, et al. Effectiveness of interventions targeting frequent users of emergency departments: a systematic review. Ann Emerg Med. 2011 Jul;58(1):41-52.e42. doi: 10.1016/j.annemergmed.2011.03.007 PubMed
11. Cochrane Effective Practice and Organisation of Care (EPOC). What study designs should be included in an EPOC review? EPOC resources for review authors. Available at:http://epoc.cochrane.org/epoc-resources-review-authors. Accessed March 23, 2018.
12. Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377-384. doi: 10.1136/jech.52.6.377. PubMed
13. Goyal AA, Tur K, Mann J, Townsend W, Flanders SA, Chopra V. Do bedside visual tools improve patient and caregiver satisfaction? A systematic review of the literature. J Hosp Med 2017;12(11):930-936. doi: 10.12788/jhm.2871. PubMed
14. Kaufman S, Ali N, DeFiglio V, Craig K, Brenner J. Early efforts to target and enroll high-risk diabetic patients into urban community-based programs. Health Promot Pract. 2014;15(2 Suppl):62S-70S. doi: 10.1177/1524839914535776. PubMed
15. Koch KL, Karafin MS, Simpson P, Field JJ. Intensive management of high-utilizing adults with sickle cell disease lowers admissions. Am J Hematol. 2015;90(3):215-219. doi: 10.1002/ajh.23912. PubMed
16. Lynch CS, Wajnberg A, Jervis R, et al. Implementation science workshop: a novel multidisciplinary primary care program to improve care and outcomes for super-utilizers. J Gen Intern Med. 2016;31(7):797-802. doi: 10.1007/s11606-016-3598-1. PubMed
17. Mercer T, Bae J, Kipnes J, Velazquez M, Thomas S, Setji N. The highest utilizers of care: individualized care plans to coordinate care, improve healthcare service utilization, and reduce costs at an academic tertiary care center. J Hosp Med. 2015;10(7):419-424. doi: 10.1002/jhm.2351. PubMed
18. Plant NA, Kelly PJ, Leeder SR, et al. Coordinated care versus standard care in hospital admissions of people with chronic illness: a randomised controlled trial. Med J Aust. 2015;203(1):33-38. doi: 10.5694/mja14.01049. PubMed
19. Shah R, Chen C, O'Rourke S, Lee M, Mohanty SA, Abraham J. Evaluation of care management for the uninsured. Med Care. 2011;49(2):166-171. doi: 10.1097/MLR.0b013e3182028e81. PubMed
20. Sledge WH, Brown KE, Levine JM, et al. A randomized trial of primary intensive care to reduce hospital admissions in patients with high utilization of inpatient services. Dis Manag. 2006;9(6):328-338. doi: 10.1089/dis.2006.9.328. PubMed
21. Weerahandi H, Basso Lipani M, Kalman J, et al. Effects of a psychosocial transitional care model on hospitalizations and cost of care for high utilizers. Soc Work Health Care. 2015;54(6):485-498. doi: 10.1080/00981389.2015.1040141. PubMed
22. Zulman DM, Ezeji-Okoye SC, Shaw JG, et al. Partnered research in healthcare delivery redesign for high-need, high-cost patients: development and feasibility of an Intensive Management Patient-Aligned Care Team (ImPACT). J Gen Intern Med. 2014;29(4):861-869. doi: 10.1007/s11606-014-3022-7. PubMed
23. Mautner DB, Pang H, Brenner JC, et al. Generating hypotheses about care needs of high utilizers: lessons from patient interviews. Popul Health Manag. 2013;16 Suppl 1:S26-33. doi: 10.1089/pop.2013.0033. PubMed
24. Bodenheimer T. Strategies to reduce costs and improve care for high-utilizing Medicaid patients: Reflections on pioneering programs. Center for Health Care Strategies, Inc.;2013.
25. Rinehart DJ, Oronce C, Durfee MJ, et al. Identifying subgroups of adult superutilizers in an urban safety-net system using latent class analysis: implications for clinical practice. Med Care. 2018;56(1):e1-e9. doi: 10.1097/MLR.0000000000000628. PubMed
26. Boutwell A, Kunst E, Sorin J, Shniffer A, Logozzo J, Woodhouse D. DSRIP-Medicaid Accelerated eXchange (MAX) Series Program: Improving Care for Super Utilizers. January 2017. https://www.health.ny.gov/health_care/medicaid/redesign/dsrip/pps_workshops/docs/2017-01_imp_care.pdf. Accessed January 24, 2018.
27. Hibbard JH, Stockard J, Mahoney ER, Tusler M. Development of the Patient Activation Measure (PAM): conceptualizing and measuring activation in patients and consumers. Health Serv Res. 2004;39(4 Pt 1):1005-1026. doi: 10.1111/j.1475-6773.2004.00269.x PubMed
1. Center for Medicare and Medicaid Services. Readmissions Reduction Program (HRRP). https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Accessed March 23, 2018.
2. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. doi: 10.7326/0003-4819-155-8-201110180-00008. PubMed
3. Blumenthal D, Chernof B, Fulmer T, Lumpkin J, Selberg J. Caring for high-need, high-cost patients - an urgent priority. N Engl J Med. 2016;375(10):909-911. doi: 10.1056/NEJMp1608511. PubMed
4. Gawande A. The Hot Spotters. The New Yorker. 2011 Jan: 40-51.
5. Szekendi MK, Williams MV, Carrier D, Hensley L, Thomas S, Cerese J. The characteristics of patients frequently admitted to academic medical centers in the United States. J Hosp Med. 2015;10(9):563-568. doi: 10.1002/jhm.2375. PubMed
6. Johnson TL, Rinehart DJ, Durfee J, et al. For many patients who use large amounts of health care services, the need is intense yet temporary. Health Aff (Millwood). 2015;34(8):1312-1319. doi: 10.1377/hlthaff.2014.1186. PubMed
7. Tinetti ME, Reuben DB. The hospital-dependent patient. N Engl J Med. 2014;370:694-697. doi: 10.1056/NEJMp1315568. PubMed
8. Hong CS, Siegel AL, Ferris TG. Caring for high-need, high-cost patients: what makes for a successful care management program? Issue Brief (Commonw Fund). 2014;19:1-19. PubMed
9. Hochman M, Asch SM. Disruptive models in primary care: caring for high-needs, high-cost populations. J Gen Intern Med. 2017;32(4):392-397. doi: 10.1007/s11606-016-3945-2. PubMed
10. Althaus F1, Paroz S, Hugli O, et al. Effectiveness of interventions targeting frequent users of emergency departments: a systematic review. Ann Emerg Med. 2011 Jul;58(1):41-52.e42. doi: 10.1016/j.annemergmed.2011.03.007 PubMed
11. Cochrane Effective Practice and Organisation of Care (EPOC). What study designs should be included in an EPOC review? EPOC resources for review authors. Available at:http://epoc.cochrane.org/epoc-resources-review-authors. Accessed March 23, 2018.
12. Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377-384. doi: 10.1136/jech.52.6.377. PubMed
13. Goyal AA, Tur K, Mann J, Townsend W, Flanders SA, Chopra V. Do bedside visual tools improve patient and caregiver satisfaction? A systematic review of the literature. J Hosp Med 2017;12(11):930-936. doi: 10.12788/jhm.2871. PubMed
14. Kaufman S, Ali N, DeFiglio V, Craig K, Brenner J. Early efforts to target and enroll high-risk diabetic patients into urban community-based programs. Health Promot Pract. 2014;15(2 Suppl):62S-70S. doi: 10.1177/1524839914535776. PubMed
15. Koch KL, Karafin MS, Simpson P, Field JJ. Intensive management of high-utilizing adults with sickle cell disease lowers admissions. Am J Hematol. 2015;90(3):215-219. doi: 10.1002/ajh.23912. PubMed
16. Lynch CS, Wajnberg A, Jervis R, et al. Implementation science workshop: a novel multidisciplinary primary care program to improve care and outcomes for super-utilizers. J Gen Intern Med. 2016;31(7):797-802. doi: 10.1007/s11606-016-3598-1. PubMed
17. Mercer T, Bae J, Kipnes J, Velazquez M, Thomas S, Setji N. The highest utilizers of care: individualized care plans to coordinate care, improve healthcare service utilization, and reduce costs at an academic tertiary care center. J Hosp Med. 2015;10(7):419-424. doi: 10.1002/jhm.2351. PubMed
18. Plant NA, Kelly PJ, Leeder SR, et al. Coordinated care versus standard care in hospital admissions of people with chronic illness: a randomised controlled trial. Med J Aust. 2015;203(1):33-38. doi: 10.5694/mja14.01049. PubMed
19. Shah R, Chen C, O'Rourke S, Lee M, Mohanty SA, Abraham J. Evaluation of care management for the uninsured. Med Care. 2011;49(2):166-171. doi: 10.1097/MLR.0b013e3182028e81. PubMed
20. Sledge WH, Brown KE, Levine JM, et al. A randomized trial of primary intensive care to reduce hospital admissions in patients with high utilization of inpatient services. Dis Manag. 2006;9(6):328-338. doi: 10.1089/dis.2006.9.328. PubMed
21. Weerahandi H, Basso Lipani M, Kalman J, et al. Effects of a psychosocial transitional care model on hospitalizations and cost of care for high utilizers. Soc Work Health Care. 2015;54(6):485-498. doi: 10.1080/00981389.2015.1040141. PubMed
22. Zulman DM, Ezeji-Okoye SC, Shaw JG, et al. Partnered research in healthcare delivery redesign for high-need, high-cost patients: development and feasibility of an Intensive Management Patient-Aligned Care Team (ImPACT). J Gen Intern Med. 2014;29(4):861-869. doi: 10.1007/s11606-014-3022-7. PubMed
23. Mautner DB, Pang H, Brenner JC, et al. Generating hypotheses about care needs of high utilizers: lessons from patient interviews. Popul Health Manag. 2013;16 Suppl 1:S26-33. doi: 10.1089/pop.2013.0033. PubMed
24. Bodenheimer T. Strategies to reduce costs and improve care for high-utilizing Medicaid patients: Reflections on pioneering programs. Center for Health Care Strategies, Inc.;2013.
25. Rinehart DJ, Oronce C, Durfee MJ, et al. Identifying subgroups of adult superutilizers in an urban safety-net system using latent class analysis: implications for clinical practice. Med Care. 2018;56(1):e1-e9. doi: 10.1097/MLR.0000000000000628. PubMed
26. Boutwell A, Kunst E, Sorin J, Shniffer A, Logozzo J, Woodhouse D. DSRIP-Medicaid Accelerated eXchange (MAX) Series Program: Improving Care for Super Utilizers. January 2017. https://www.health.ny.gov/health_care/medicaid/redesign/dsrip/pps_workshops/docs/2017-01_imp_care.pdf. Accessed January 24, 2018.
27. Hibbard JH, Stockard J, Mahoney ER, Tusler M. Development of the Patient Activation Measure (PAM): conceptualizing and measuring activation in patients and consumers. Health Serv Res. 2004;39(4 Pt 1):1005-1026. doi: 10.1111/j.1475-6773.2004.00269.x PubMed
© 2018 Society of Hospital Medicine
Acute Treatment of Hypertensive Urgency
The "Things We Do for No Reason" (TWDFNR) series reviews practices which have become common parts of hospital care but provide little value to our patients. Practices reviewed in the TWDFNR series do not represent "black and white" conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/

CLINICAL SCENARIO
A 67-year-old man is hospitalized with community-acquired pneumonia. He has a history of hypertension and is prescribed two antihypertensive medications (amlodipine and chlorthalidone) as an outpatient. On the evening of hospital day two, he is found to have a blood pressure of 192/95 on a scheduled vital signs check. He reports no symptoms other than cough, which is not new or worsening. The covering hospitalist reviews the documented blood pressures since admission and notes that many have been elevated despite continuation of his home regimen. The patient's nurse inquires about treating the patient with additional "as-needed" antihypertensive medications.
BACKGROUND
Hypertensive crises are common in hospitalized patients, with approximately one in seven patients experiencing an episode of hypertensive emergency and/or hypertensive urgency.1 Hypertensive emergency is typically defined as (1) a systolic blood pressure ≥180 mm Hg and/or a diastolic blood pressure ≥120 mm Hg with (2) evidence of new or worsening end-organ damage. The organs most commonly affected by severe hypertension are the brain (headache, confusion, stroke), heart (chest pain, myocardial infarction, pulmonary edema), large blood vessels (aortic dissection), and kidneys (acute hypertensive nephrosclerosis).2 With hypertensive urgency, patients experience similarly elevated blood pressure but have no symptoms or signs suggesting acute end-organ damage. Acute treatment with intravenous (IV) or immediate-acting oral medications is common; a single-center study showed that 7.4% of hospitalized patients had an order for "as needed" IV hydralazine or labetalol, with 60.3% receiving at least one dose.3 Among internal medicine and family medicine trainees in one survey, nearly half reported that they would use IV medications in a scenario where an inpatient had an asymptomatic blood pressure above 180 mm Hg.4
WHY YOU MIGHT THINK TREATING HYPERTENSIVE URGENCY IS NECESSARY
Treating patients with hypertensive urgency is based on an assumption: If one does not treat immediately, something bad (ie, end-organ damage) will occur over the next few hours. Data from the 1930s showed that patients with untreated hypertensive emergency had a one-year mortality rate >79% and a median survival of 10.4 months.5 More recent studies suggest that the in-hospital and one-year mortality for those with hypertensive emergency are 13% and 39%, respectively.6 These data demonstrate that patients with hypertensive emergency are at risk in both the short- and long-term.
Patients with hypertensive urgency are also at increased risk for long-term morbidity and mortality. The one-year mortality for those experiencing an episode of hypertensive urgency is approximately 9%.6 Given the concerns about poor outcomes, it remains a common practice in many facilities to acutely lower the blood pressure in patients with hypertensive urgency. This is highlighted by recommendations of a commonly used point-of-care medical resource, which suggests that "potential legal ramifications partially motivate lowering the blood pressure over several hours."7
WHY TREATING HYPERTENSIVE URGENCY IS UNNECESSARY AND POTENTIALLY HARMFUL
Concerns regarding overtreatment of hypertensive urgency relate to overestimated rates of hypertensive complications, the pathophysiology of hypertension itself, and the potential for adverse events related to treatment. Given that there are few trials examining hospitalized patients with hypertensive urgency, much of the data supporting a conservative approach are drawn from studies of outpatients or emergency department patients. In addition, there is little data suggesting that outcomes are different for patients presenting with a chief complaint of hypertensive urgency and those presenting with an alternate diagnosis but who are found to have blood pressures that meet the threshold for diagnosis of hypertensive urgency.
The landmark 1967 Veterans Affairs Cooperative Trial demonstrated the long-term benefits of treating patients with chronic hypertensive urgency.8 Importantly though, benefits accrued over a period of months to years, not hours. The time to the first adverse event in the placebo arm was two months, suggesting that even those with blood pressures chronically in the range of hypertensive urgency are unlikely to experience hyperacute (ie, within hours) events, even without treatment.
A more recent study, conducted by Patel et al., examined 58,836 patients seen in outpatient clinics and found to have blood pressures meeting the criteria for hypertensive urgency.9 This study included patients whose primary issue was hypertensive urgency and patients in whom the diagnosis was secondary. A total of 426 patients were referred to the hospital and only 100 (0.17%) were subsequently admitted. At seven days, the rates of the primary outcome (a composite of myocardial infarction, stroke, and/or transient ischemic attack) were 0.1% in those sent home and 0.5% in those sent to the hospital. In those patients with a systolic blood pressure ≥220 mm Hg, two out of 977 (0.2%) of those sent home and zero out of 81 of those sent to the hospital experienced the primary outcome. These data reinforce the message that, in patients with hypertensive urgency, rates of adverse events at seven days are low, even with extreme blood pressure elevation.
The human body has adapted to withstand wide variations in blood pressure.10 For example, through arteriolar constriction and reflex vasodilation, cerebral autoregulation maintains a constant cerebral blood flow within a wide range of perfusion pressures, ensuring that the brain is protected from higher mean arterial pressures.11 While this process is protective, over time the autoregulatory system becomes impaired, especially in patients with cerebrovascular disease. This places patients at risk for cerebral and/or cardiac ischemia with even slight drops in perfusion pressure.12,13 Indeed, in assessing treatment-related adverse events in a series of patients treated with intravenous nicardipine or nitroprusside for hypertensive emergency, Brooks and colleagues reported that 57% (27 of 47) of patients had overly large reductions in blood pressure (>25% reduction in mean arterial pressure) within the first 30 minutes of treatment.14 Two patients had acute ischemic events attributed to treatment with antihypertensive medications. Myocardial infarction and stroke have both been reported,12 and medication classes such as calcium channel blockers (sublingual nifedipine in particular), beta-blockers (eg, labetolol), angiotensin-converting-enzyme inhibitors (eg, captopril), and clonidine have all been implicated in treatment-related adverse events.12,15-17 Another potential issue derives from the observation that blood pressures obtained in the hospital setting are often inaccurate, owing to inappropriate patient preparation, faulty equipment, and inadequate training of staff obtaining the measurement.18
National guidelines support a cautious approach to the treatment of hypertensive urgency. The seventh Report of the Joint National Committee on Detection, Evaluation, and Treatment of Hypertension, published in 2003, noted that "patients with markedly elevated BP but without acute target-organ damage usually do not require hospitalization, but they should receive immediate combination oral antihypertensive therapy" and that "there is no evidence to suggest that failure to aggressively lower BP in the [emergency department] is associated with any increased short-term risk to the patient who presents with severe hypertension." JNC 7 also laments contemporary terminology: "Unfortunately, the term 'urgency' has led to overly aggressive management of many patients with severe, uncomplicated hypertension. Aggressive dosing with intravenous drugs or even oral agents, to rapidly lower BP is not without risk."19 The most recent JNC guideline does not comment on hypertensive urgency,20 and the 2017 American College of Cardiology/American Heart Association Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults argues that, "¬there is no indication for referral to the emergency department, immediate reduction in BP in the emergency department, or hospitalization for [patients with hypertensive urgency]."21
WHAT CLINICIANS SHOULD DO INSTEAD
After it is confirmed that a patient has no end-organ damage (ie, the patient has hypertensive urgency, not emergency), treatable causes of hypertension should be assessed. In hospitalized patients, these include missed or held doses of outpatient medications, pain, nausea, alcohol and/or benzodiazepine withdrawal, delirium, and obstructive sleep apnea.22 If no remediable cause is identified, patients should be allowed to rest for at least 30 minutes without the administration of additional antihypertensive medications, after which time the blood pressure should be measured using the correct technique.2 Clinical trials have shown that rest is effective at lowering blood pressure in patients with hypertensive urgency.23,24 One study initially treated 549 emergency department patients with a 30-minute rest period, after which time 32% of patients had responded (defined as a SBP <180 mm Hg and DBP <110 mm Hg, with at least a 20 mm Hg reduction in baseline SBP and/or a 10 mm Hg reduction in DBP).23 Another study randomized 138 patients with hypertensive urgency to either rest or active treatment with telmisartan. Blood pressures were checked every 30 minutes for four hours. The primary endpoint (reduction of MAP of 10%-35%) was similar in both groups (68.5% in the rest group and 69.1% in the telmisartan group).24 Even if rest is ineffective, the risk-benefit ratio of acutely lowering blood pressure will typically favor withholding acute treatment in asymptomatic patients. If blood pressure remains consistently elevated, augmentation of the home regimen (eg, increasing the dose of their next scheduled antihypertensive) of oral medications may be warranted. Though not all agree with management of antihypertensives in hospitalized patients,25 acute hospitalizations afford an opportunity to modify and observe chronic hypertension.26
RECOMMENDATIONS
- Ensure that patients do not have symptoms and/or signs of end-organ damage. This can be done with a brief review of systems and a physical examination. In select cases, an electrocardiogram and a chest x-ray may be warranted.
- Search for common causes of treatable hypertension in hospitalized patients; these include pain, nausea, withdrawal syndromes, and holding of usual antihypertensive medications.
- In those patients without symptoms and/or signs of end-organ damage, allow rest, followed by reassessment.
- Do not administer intravenous or immediate-acting oral antihypertensive medications to acutely lower blood pressure. Instead, address the issues raised in Recommendation #2 and consider modifying the chronic oral antihypertensive regimen in patients who are uncontrolled as outpatients or who are not treated as outpatients. Coordinate early postdischarge follow-up for repeat blood pressure evaluation and continued modification of a patient's chronic antihypertensive regimen.
CONCLUSION
Although patients with hypertensive urgency are often treated with medications to acutely lower their blood pressure, there is no evidence to support this practice, and a strong pathophysiologic basis suggests that harm may result. The patient in the case described above should be allowed to rest for at least 30 minutes, with reevaluation of his blood pressure. If it remains elevated and no treatable secondary causes are found, the treating hospitalist should consider altering his chronic antihypertensive regimen to promote long-term blood pressure control.
Do you think this is a low-value practice? Is this truly a "Thing We Do for No Reason?" Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other "Things We Do for No Reason" topics by emailing [email protected].
Disclosures
The authors have no conflicts of interest.
1. Shorr AF, Zilberberg MD, Sun X, et al. Severe acute hypertension among inpatients admitted from the emergency department. J Hosp Med. 2012;7(3):203-210. doi: 10.1002/jhm.969. PubMed
2. Whelton PK, Carey RM, Aronow WS, et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the Prevention, detection, evaluation, and management of High blood pressure in adults: A report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Hypertension. 2017. PubMed
3. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: use of intravenous labetalol and hydralazine. J Clin Hypertens (Greenwich). 2010;12(1):29-33. doi: 10.1111/j.1751-7176.2009.00196.x. PubMed
4. Axon RN, Garrell R, Pfahl K, et al. Attitudes and practices of resident physicians regarding hypertension in the inpatient setting. J Clin Hypertens (Greenwich). 2010;12(9):698-705. doi: 10.1111/j.1751-7176.2010.00309.x. PubMed
5. Keith NM, Wagener HP, Barker NW. Some different types of essential hypertension: their course and prognosis. Am J Med Sci. 1974;268(6):336-345. doi: 10.1097/00000441-197412000-00004. PubMed
6. Guiga H, Decroux C, Michelet P, et al. Hospital and out-of-hospital mortality in 670 hypertensive emergencies and urgencies. J Clin Hypertens (Greenwich). 2017;19(11):1137-1142. doi: 10.1111/jch.13083. PubMed
7. Varon J, Williams EJ. Management of severe asymptomatic hypertension (hypertensive urgencies) in adults. In: Post T, ed. UpToDate, Waltham, MA. (Accessed February 13, 2018). PubMed
8. Effects of treatment on morbidity in hypertension. Results in patients with diastolic blood pressures averaging 115 through 129 mm Hg. JAMA. 1967;202(11):1028-1034. soi: 10.1001/jama.1967.03130240070013 PubMed
9. Patel KK, Young L, Howell EH, et al. Characteristics and outcomes of patients presenting with hypertensive urgency in the office setting. JAMA Intern Med. 2016;176(7):981-988. doi: 10.1001/jamainternmed.2016.1509. PubMed
10. MacDougall JD, Tuxen D, Sale DG, Moroz JR, Sutton JR. Arterial blood pressure response to heavy resistance exercise. J Appl Physiol. 1985;58(3):785-790. doi: 10.1152/jappl.1985.58.3.785. PubMed
11. Strandgaard S, Olesen J, Skinhoj E, Lassen NA. Autoregulation of brain circulation in severe arterial hypertension. Br Med J. 1973;1(5852):507-510. doi: 10.1136/bmj.1.5852.507. PubMed
12. Fischberg GM, Lozano E, Rajamani K, Ameriso S, Fisher MJ. Stroke precipitated by moderate blood pressure reduction. J Emerg Med. 2000;19(4):339-346. doi: 10.1016/S0736-4679(00)00267-5. PubMed
13. Ross RS. Pathophysiology of coronary circulation. Br Heart J. 1971;33(2):173-184. doi: 10.1136/hrt.33.2.173. PubMed
14. Brooks TW, Finch CK, Lobo BL, Deaton PR, Varner CF. Blood pressure management in acute hypertensive emergency. Am J Health Syst Pharm. 2007;64(24):2579-2582. doi: 10.2146/ajhp070105. PubMed
15. Grossman E, Messerli FH, Grodzicki T, Kowey P. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-1331. doi: 10.1001/jama.1996.03540160050032 PubMed
16. Hodsman GP, Isles CG, Murray GD et al. Factors related to first dose hypotensive effect of captopril: prediction and treatment. Br Med J (Clin Res Ed). 1983;286(6368):832-834. doi: 10.1136/bmj.286.6368.832. PubMed
17. Zeller KR, Von Kuhnert L, Matthews C. Rapid reduction of severe asymptomatic hypertension. A prospective, controlled trial. Arch Intern Med. 1989;149(10):2186-2189. doi: 10.1001/archinte.149.10.2186. PubMed
18. Pickering TG, Hall JE, Appel LJ, et al. Recommendations for blood pressure measurement in humans and experimental animals: Part 1: Blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation. 2005;111(5):697-716. doi: 10.1161/01.CIR.0000154900.76284.F6. PubMed
19. Chobanian AV, Bakris GL, Black HR, et al. The seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High blood pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2572. doi: 10.1001/jama.289.19.2560. PubMed
20. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. doi: 10.1001/jama.2013.284427 PubMed
21. Whelton PK, Carey RM, Aronow WS, et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the Prevention, detection, evaluation, and management of High blood pressure in adults: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2017. PubMed
22. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015;17(11):94. doi: 10.1007/s11886-015-0648-y. PubMed
23. Grassi D, O'Flaherty M, Pellizzari M, et al. Hypertensive urgencies in the emergency department: evaluating blood pressure response to rest and to antihypertensive drugs with different profiles. J Clin Hypertens (Greenwich). 2008;10(9):662-667. doi: 10.1111/j.1751-7176.2008.00001.x. PubMed
24. Park SK, Lee DY, Kim WJ, et al. Comparing the clinical efficacy of resting and antihypertensive medication in patients of hypertensive urgency: a randomized, control trial. J Hypertens. 2017;35(7):1474-1480. doi: 10.1097/HJH.0000000000001340. PubMed
25. Steinman MA, Auerbach AD. Managing chronic disease in hospitalized patients. JAMA Intern Med. 2013;173(20):1857-1858. doi: 10.1001/jamainternmed.2013.9511. PubMed
26. Breu AC, Allen-Dicker J, Mueller S et al. Hospitalist and primary care physician perspectives on medication management of chronic conditions for hospitalized patients. J Hosp Med. 2014;9(5):303-309. doi: 10.1002/jhm.2137. PubMed
The "Things We Do for No Reason" (TWDFNR) series reviews practices which have become common parts of hospital care but provide little value to our patients. Practices reviewed in the TWDFNR series do not represent "black and white" conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/
CLINICAL SCENARIO
A 67-year-old man is hospitalized with community-acquired pneumonia. He has a history of hypertension and is prescribed two antihypertensive medications (amlodipine and chlorthalidone) as an outpatient. On the evening of hospital day two, he is found to have a blood pressure of 192/95 on a scheduled vital signs check. He reports no symptoms other than cough, which is not new or worsening. The covering hospitalist reviews the documented blood pressures since admission and notes that many have been elevated despite continuation of his home regimen. The patient's nurse inquires about treating the patient with additional "as-needed" antihypertensive medications.
BACKGROUND
Hypertensive crises are common in hospitalized patients, with approximately one in seven patients experiencing an episode of hypertensive emergency and/or hypertensive urgency.1 Hypertensive emergency is typically defined as (1) a systolic blood pressure ≥180 mm Hg and/or a diastolic blood pressure ≥120 mm Hg with (2) evidence of new or worsening end-organ damage. The organs most commonly affected by severe hypertension are the brain (headache, confusion, stroke), heart (chest pain, myocardial infarction, pulmonary edema), large blood vessels (aortic dissection), and kidneys (acute hypertensive nephrosclerosis).2 With hypertensive urgency, patients experience similarly elevated blood pressure but have no symptoms or signs suggesting acute end-organ damage. Acute treatment with intravenous (IV) or immediate-acting oral medications is common; a single-center study showed that 7.4% of hospitalized patients had an order for "as needed" IV hydralazine or labetalol, with 60.3% receiving at least one dose.3 Among internal medicine and family medicine trainees in one survey, nearly half reported that they would use IV medications in a scenario where an inpatient had an asymptomatic blood pressure above 180 mm Hg.4
WHY YOU MIGHT THINK TREATING HYPERTENSIVE URGENCY IS NECESSARY
Treating patients with hypertensive urgency is based on an assumption: If one does not treat immediately, something bad (ie, end-organ damage) will occur over the next few hours. Data from the 1930s showed that patients with untreated hypertensive emergency had a one-year mortality rate >79% and a median survival of 10.4 months.5 More recent studies suggest that the in-hospital and one-year mortality for those with hypertensive emergency are 13% and 39%, respectively.6 These data demonstrate that patients with hypertensive emergency are at risk in both the short- and long-term.
Patients with hypertensive urgency are also at increased risk for long-term morbidity and mortality. The one-year mortality for those experiencing an episode of hypertensive urgency is approximately 9%.6 Given the concerns about poor outcomes, it remains a common practice in many facilities to acutely lower the blood pressure in patients with hypertensive urgency. This is highlighted by recommendations of a commonly used point-of-care medical resource, which suggests that "potential legal ramifications partially motivate lowering the blood pressure over several hours."7
WHY TREATING HYPERTENSIVE URGENCY IS UNNECESSARY AND POTENTIALLY HARMFUL
Concerns regarding overtreatment of hypertensive urgency relate to overestimated rates of hypertensive complications, the pathophysiology of hypertension itself, and the potential for adverse events related to treatment. Given that there are few trials examining hospitalized patients with hypertensive urgency, much of the data supporting a conservative approach are drawn from studies of outpatients or emergency department patients. In addition, there is little data suggesting that outcomes are different for patients presenting with a chief complaint of hypertensive urgency and those presenting with an alternate diagnosis but who are found to have blood pressures that meet the threshold for diagnosis of hypertensive urgency.
The landmark 1967 Veterans Affairs Cooperative Trial demonstrated the long-term benefits of treating patients with chronic hypertensive urgency.8 Importantly though, benefits accrued over a period of months to years, not hours. The time to the first adverse event in the placebo arm was two months, suggesting that even those with blood pressures chronically in the range of hypertensive urgency are unlikely to experience hyperacute (ie, within hours) events, even without treatment.
A more recent study, conducted by Patel et al., examined 58,836 patients seen in outpatient clinics and found to have blood pressures meeting the criteria for hypertensive urgency.9 This study included patients whose primary issue was hypertensive urgency and patients in whom the diagnosis was secondary. A total of 426 patients were referred to the hospital and only 100 (0.17%) were subsequently admitted. At seven days, the rates of the primary outcome (a composite of myocardial infarction, stroke, and/or transient ischemic attack) were 0.1% in those sent home and 0.5% in those sent to the hospital. In those patients with a systolic blood pressure ≥220 mm Hg, two out of 977 (0.2%) of those sent home and zero out of 81 of those sent to the hospital experienced the primary outcome. These data reinforce the message that, in patients with hypertensive urgency, rates of adverse events at seven days are low, even with extreme blood pressure elevation.
The human body has adapted to withstand wide variations in blood pressure.10 For example, through arteriolar constriction and reflex vasodilation, cerebral autoregulation maintains a constant cerebral blood flow within a wide range of perfusion pressures, ensuring that the brain is protected from higher mean arterial pressures.11 While this process is protective, over time the autoregulatory system becomes impaired, especially in patients with cerebrovascular disease. This places patients at risk for cerebral and/or cardiac ischemia with even slight drops in perfusion pressure.12,13 Indeed, in assessing treatment-related adverse events in a series of patients treated with intravenous nicardipine or nitroprusside for hypertensive emergency, Brooks and colleagues reported that 57% (27 of 47) of patients had overly large reductions in blood pressure (>25% reduction in mean arterial pressure) within the first 30 minutes of treatment.14 Two patients had acute ischemic events attributed to treatment with antihypertensive medications. Myocardial infarction and stroke have both been reported,12 and medication classes such as calcium channel blockers (sublingual nifedipine in particular), beta-blockers (eg, labetolol), angiotensin-converting-enzyme inhibitors (eg, captopril), and clonidine have all been implicated in treatment-related adverse events.12,15-17 Another potential issue derives from the observation that blood pressures obtained in the hospital setting are often inaccurate, owing to inappropriate patient preparation, faulty equipment, and inadequate training of staff obtaining the measurement.18
National guidelines support a cautious approach to the treatment of hypertensive urgency. The seventh Report of the Joint National Committee on Detection, Evaluation, and Treatment of Hypertension, published in 2003, noted that "patients with markedly elevated BP but without acute target-organ damage usually do not require hospitalization, but they should receive immediate combination oral antihypertensive therapy" and that "there is no evidence to suggest that failure to aggressively lower BP in the [emergency department] is associated with any increased short-term risk to the patient who presents with severe hypertension." JNC 7 also laments contemporary terminology: "Unfortunately, the term 'urgency' has led to overly aggressive management of many patients with severe, uncomplicated hypertension. Aggressive dosing with intravenous drugs or even oral agents, to rapidly lower BP is not without risk."19 The most recent JNC guideline does not comment on hypertensive urgency,20 and the 2017 American College of Cardiology/American Heart Association Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults argues that, "¬there is no indication for referral to the emergency department, immediate reduction in BP in the emergency department, or hospitalization for [patients with hypertensive urgency]."21
WHAT CLINICIANS SHOULD DO INSTEAD
After it is confirmed that a patient has no end-organ damage (ie, the patient has hypertensive urgency, not emergency), treatable causes of hypertension should be assessed. In hospitalized patients, these include missed or held doses of outpatient medications, pain, nausea, alcohol and/or benzodiazepine withdrawal, delirium, and obstructive sleep apnea.22 If no remediable cause is identified, patients should be allowed to rest for at least 30 minutes without the administration of additional antihypertensive medications, after which time the blood pressure should be measured using the correct technique.2 Clinical trials have shown that rest is effective at lowering blood pressure in patients with hypertensive urgency.23,24 One study initially treated 549 emergency department patients with a 30-minute rest period, after which time 32% of patients had responded (defined as a SBP <180 mm Hg and DBP <110 mm Hg, with at least a 20 mm Hg reduction in baseline SBP and/or a 10 mm Hg reduction in DBP).23 Another study randomized 138 patients with hypertensive urgency to either rest or active treatment with telmisartan. Blood pressures were checked every 30 minutes for four hours. The primary endpoint (reduction of MAP of 10%-35%) was similar in both groups (68.5% in the rest group and 69.1% in the telmisartan group).24 Even if rest is ineffective, the risk-benefit ratio of acutely lowering blood pressure will typically favor withholding acute treatment in asymptomatic patients. If blood pressure remains consistently elevated, augmentation of the home regimen (eg, increasing the dose of their next scheduled antihypertensive) of oral medications may be warranted. Though not all agree with management of antihypertensives in hospitalized patients,25 acute hospitalizations afford an opportunity to modify and observe chronic hypertension.26
RECOMMENDATIONS
- Ensure that patients do not have symptoms and/or signs of end-organ damage. This can be done with a brief review of systems and a physical examination. In select cases, an electrocardiogram and a chest x-ray may be warranted.
- Search for common causes of treatable hypertension in hospitalized patients; these include pain, nausea, withdrawal syndromes, and holding of usual antihypertensive medications.
- In those patients without symptoms and/or signs of end-organ damage, allow rest, followed by reassessment.
- Do not administer intravenous or immediate-acting oral antihypertensive medications to acutely lower blood pressure. Instead, address the issues raised in Recommendation #2 and consider modifying the chronic oral antihypertensive regimen in patients who are uncontrolled as outpatients or who are not treated as outpatients. Coordinate early postdischarge follow-up for repeat blood pressure evaluation and continued modification of a patient's chronic antihypertensive regimen.
CONCLUSION
Although patients with hypertensive urgency are often treated with medications to acutely lower their blood pressure, there is no evidence to support this practice, and a strong pathophysiologic basis suggests that harm may result. The patient in the case described above should be allowed to rest for at least 30 minutes, with reevaluation of his blood pressure. If it remains elevated and no treatable secondary causes are found, the treating hospitalist should consider altering his chronic antihypertensive regimen to promote long-term blood pressure control.
Do you think this is a low-value practice? Is this truly a "Thing We Do for No Reason?" Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other "Things We Do for No Reason" topics by emailing [email protected].
Disclosures
The authors have no conflicts of interest.
The "Things We Do for No Reason" (TWDFNR) series reviews practices which have become common parts of hospital care but provide little value to our patients. Practices reviewed in the TWDFNR series do not represent "black and white" conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/
CLINICAL SCENARIO
A 67-year-old man is hospitalized with community-acquired pneumonia. He has a history of hypertension and is prescribed two antihypertensive medications (amlodipine and chlorthalidone) as an outpatient. On the evening of hospital day two, he is found to have a blood pressure of 192/95 on a scheduled vital signs check. He reports no symptoms other than cough, which is not new or worsening. The covering hospitalist reviews the documented blood pressures since admission and notes that many have been elevated despite continuation of his home regimen. The patient's nurse inquires about treating the patient with additional "as-needed" antihypertensive medications.
BACKGROUND
Hypertensive crises are common in hospitalized patients, with approximately one in seven patients experiencing an episode of hypertensive emergency and/or hypertensive urgency.1 Hypertensive emergency is typically defined as (1) a systolic blood pressure ≥180 mm Hg and/or a diastolic blood pressure ≥120 mm Hg with (2) evidence of new or worsening end-organ damage. The organs most commonly affected by severe hypertension are the brain (headache, confusion, stroke), heart (chest pain, myocardial infarction, pulmonary edema), large blood vessels (aortic dissection), and kidneys (acute hypertensive nephrosclerosis).2 With hypertensive urgency, patients experience similarly elevated blood pressure but have no symptoms or signs suggesting acute end-organ damage. Acute treatment with intravenous (IV) or immediate-acting oral medications is common; a single-center study showed that 7.4% of hospitalized patients had an order for "as needed" IV hydralazine or labetalol, with 60.3% receiving at least one dose.3 Among internal medicine and family medicine trainees in one survey, nearly half reported that they would use IV medications in a scenario where an inpatient had an asymptomatic blood pressure above 180 mm Hg.4
WHY YOU MIGHT THINK TREATING HYPERTENSIVE URGENCY IS NECESSARY
Treating patients with hypertensive urgency is based on an assumption: If one does not treat immediately, something bad (ie, end-organ damage) will occur over the next few hours. Data from the 1930s showed that patients with untreated hypertensive emergency had a one-year mortality rate >79% and a median survival of 10.4 months.5 More recent studies suggest that the in-hospital and one-year mortality for those with hypertensive emergency are 13% and 39%, respectively.6 These data demonstrate that patients with hypertensive emergency are at risk in both the short- and long-term.
Patients with hypertensive urgency are also at increased risk for long-term morbidity and mortality. The one-year mortality for those experiencing an episode of hypertensive urgency is approximately 9%.6 Given the concerns about poor outcomes, it remains a common practice in many facilities to acutely lower the blood pressure in patients with hypertensive urgency. This is highlighted by recommendations of a commonly used point-of-care medical resource, which suggests that "potential legal ramifications partially motivate lowering the blood pressure over several hours."7
WHY TREATING HYPERTENSIVE URGENCY IS UNNECESSARY AND POTENTIALLY HARMFUL
Concerns regarding overtreatment of hypertensive urgency relate to overestimated rates of hypertensive complications, the pathophysiology of hypertension itself, and the potential for adverse events related to treatment. Given that there are few trials examining hospitalized patients with hypertensive urgency, much of the data supporting a conservative approach are drawn from studies of outpatients or emergency department patients. In addition, there is little data suggesting that outcomes are different for patients presenting with a chief complaint of hypertensive urgency and those presenting with an alternate diagnosis but who are found to have blood pressures that meet the threshold for diagnosis of hypertensive urgency.
The landmark 1967 Veterans Affairs Cooperative Trial demonstrated the long-term benefits of treating patients with chronic hypertensive urgency.8 Importantly though, benefits accrued over a period of months to years, not hours. The time to the first adverse event in the placebo arm was two months, suggesting that even those with blood pressures chronically in the range of hypertensive urgency are unlikely to experience hyperacute (ie, within hours) events, even without treatment.
A more recent study, conducted by Patel et al., examined 58,836 patients seen in outpatient clinics and found to have blood pressures meeting the criteria for hypertensive urgency.9 This study included patients whose primary issue was hypertensive urgency and patients in whom the diagnosis was secondary. A total of 426 patients were referred to the hospital and only 100 (0.17%) were subsequently admitted. At seven days, the rates of the primary outcome (a composite of myocardial infarction, stroke, and/or transient ischemic attack) were 0.1% in those sent home and 0.5% in those sent to the hospital. In those patients with a systolic blood pressure ≥220 mm Hg, two out of 977 (0.2%) of those sent home and zero out of 81 of those sent to the hospital experienced the primary outcome. These data reinforce the message that, in patients with hypertensive urgency, rates of adverse events at seven days are low, even with extreme blood pressure elevation.
The human body has adapted to withstand wide variations in blood pressure.10 For example, through arteriolar constriction and reflex vasodilation, cerebral autoregulation maintains a constant cerebral blood flow within a wide range of perfusion pressures, ensuring that the brain is protected from higher mean arterial pressures.11 While this process is protective, over time the autoregulatory system becomes impaired, especially in patients with cerebrovascular disease. This places patients at risk for cerebral and/or cardiac ischemia with even slight drops in perfusion pressure.12,13 Indeed, in assessing treatment-related adverse events in a series of patients treated with intravenous nicardipine or nitroprusside for hypertensive emergency, Brooks and colleagues reported that 57% (27 of 47) of patients had overly large reductions in blood pressure (>25% reduction in mean arterial pressure) within the first 30 minutes of treatment.14 Two patients had acute ischemic events attributed to treatment with antihypertensive medications. Myocardial infarction and stroke have both been reported,12 and medication classes such as calcium channel blockers (sublingual nifedipine in particular), beta-blockers (eg, labetolol), angiotensin-converting-enzyme inhibitors (eg, captopril), and clonidine have all been implicated in treatment-related adverse events.12,15-17 Another potential issue derives from the observation that blood pressures obtained in the hospital setting are often inaccurate, owing to inappropriate patient preparation, faulty equipment, and inadequate training of staff obtaining the measurement.18
National guidelines support a cautious approach to the treatment of hypertensive urgency. The seventh Report of the Joint National Committee on Detection, Evaluation, and Treatment of Hypertension, published in 2003, noted that "patients with markedly elevated BP but without acute target-organ damage usually do not require hospitalization, but they should receive immediate combination oral antihypertensive therapy" and that "there is no evidence to suggest that failure to aggressively lower BP in the [emergency department] is associated with any increased short-term risk to the patient who presents with severe hypertension." JNC 7 also laments contemporary terminology: "Unfortunately, the term 'urgency' has led to overly aggressive management of many patients with severe, uncomplicated hypertension. Aggressive dosing with intravenous drugs or even oral agents, to rapidly lower BP is not without risk."19 The most recent JNC guideline does not comment on hypertensive urgency,20 and the 2017 American College of Cardiology/American Heart Association Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults argues that, "¬there is no indication for referral to the emergency department, immediate reduction in BP in the emergency department, or hospitalization for [patients with hypertensive urgency]."21
WHAT CLINICIANS SHOULD DO INSTEAD
After it is confirmed that a patient has no end-organ damage (ie, the patient has hypertensive urgency, not emergency), treatable causes of hypertension should be assessed. In hospitalized patients, these include missed or held doses of outpatient medications, pain, nausea, alcohol and/or benzodiazepine withdrawal, delirium, and obstructive sleep apnea.22 If no remediable cause is identified, patients should be allowed to rest for at least 30 minutes without the administration of additional antihypertensive medications, after which time the blood pressure should be measured using the correct technique.2 Clinical trials have shown that rest is effective at lowering blood pressure in patients with hypertensive urgency.23,24 One study initially treated 549 emergency department patients with a 30-minute rest period, after which time 32% of patients had responded (defined as a SBP <180 mm Hg and DBP <110 mm Hg, with at least a 20 mm Hg reduction in baseline SBP and/or a 10 mm Hg reduction in DBP).23 Another study randomized 138 patients with hypertensive urgency to either rest or active treatment with telmisartan. Blood pressures were checked every 30 minutes for four hours. The primary endpoint (reduction of MAP of 10%-35%) was similar in both groups (68.5% in the rest group and 69.1% in the telmisartan group).24 Even if rest is ineffective, the risk-benefit ratio of acutely lowering blood pressure will typically favor withholding acute treatment in asymptomatic patients. If blood pressure remains consistently elevated, augmentation of the home regimen (eg, increasing the dose of their next scheduled antihypertensive) of oral medications may be warranted. Though not all agree with management of antihypertensives in hospitalized patients,25 acute hospitalizations afford an opportunity to modify and observe chronic hypertension.26
RECOMMENDATIONS
- Ensure that patients do not have symptoms and/or signs of end-organ damage. This can be done with a brief review of systems and a physical examination. In select cases, an electrocardiogram and a chest x-ray may be warranted.
- Search for common causes of treatable hypertension in hospitalized patients; these include pain, nausea, withdrawal syndromes, and holding of usual antihypertensive medications.
- In those patients without symptoms and/or signs of end-organ damage, allow rest, followed by reassessment.
- Do not administer intravenous or immediate-acting oral antihypertensive medications to acutely lower blood pressure. Instead, address the issues raised in Recommendation #2 and consider modifying the chronic oral antihypertensive regimen in patients who are uncontrolled as outpatients or who are not treated as outpatients. Coordinate early postdischarge follow-up for repeat blood pressure evaluation and continued modification of a patient's chronic antihypertensive regimen.
CONCLUSION
Although patients with hypertensive urgency are often treated with medications to acutely lower their blood pressure, there is no evidence to support this practice, and a strong pathophysiologic basis suggests that harm may result. The patient in the case described above should be allowed to rest for at least 30 minutes, with reevaluation of his blood pressure. If it remains elevated and no treatable secondary causes are found, the treating hospitalist should consider altering his chronic antihypertensive regimen to promote long-term blood pressure control.
Do you think this is a low-value practice? Is this truly a "Thing We Do for No Reason?" Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other "Things We Do for No Reason" topics by emailing [email protected].
Disclosures
The authors have no conflicts of interest.
1. Shorr AF, Zilberberg MD, Sun X, et al. Severe acute hypertension among inpatients admitted from the emergency department. J Hosp Med. 2012;7(3):203-210. doi: 10.1002/jhm.969. PubMed
2. Whelton PK, Carey RM, Aronow WS, et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the Prevention, detection, evaluation, and management of High blood pressure in adults: A report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Hypertension. 2017. PubMed
3. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: use of intravenous labetalol and hydralazine. J Clin Hypertens (Greenwich). 2010;12(1):29-33. doi: 10.1111/j.1751-7176.2009.00196.x. PubMed
4. Axon RN, Garrell R, Pfahl K, et al. Attitudes and practices of resident physicians regarding hypertension in the inpatient setting. J Clin Hypertens (Greenwich). 2010;12(9):698-705. doi: 10.1111/j.1751-7176.2010.00309.x. PubMed
5. Keith NM, Wagener HP, Barker NW. Some different types of essential hypertension: their course and prognosis. Am J Med Sci. 1974;268(6):336-345. doi: 10.1097/00000441-197412000-00004. PubMed
6. Guiga H, Decroux C, Michelet P, et al. Hospital and out-of-hospital mortality in 670 hypertensive emergencies and urgencies. J Clin Hypertens (Greenwich). 2017;19(11):1137-1142. doi: 10.1111/jch.13083. PubMed
7. Varon J, Williams EJ. Management of severe asymptomatic hypertension (hypertensive urgencies) in adults. In: Post T, ed. UpToDate, Waltham, MA. (Accessed February 13, 2018). PubMed
8. Effects of treatment on morbidity in hypertension. Results in patients with diastolic blood pressures averaging 115 through 129 mm Hg. JAMA. 1967;202(11):1028-1034. soi: 10.1001/jama.1967.03130240070013 PubMed
9. Patel KK, Young L, Howell EH, et al. Characteristics and outcomes of patients presenting with hypertensive urgency in the office setting. JAMA Intern Med. 2016;176(7):981-988. doi: 10.1001/jamainternmed.2016.1509. PubMed
10. MacDougall JD, Tuxen D, Sale DG, Moroz JR, Sutton JR. Arterial blood pressure response to heavy resistance exercise. J Appl Physiol. 1985;58(3):785-790. doi: 10.1152/jappl.1985.58.3.785. PubMed
11. Strandgaard S, Olesen J, Skinhoj E, Lassen NA. Autoregulation of brain circulation in severe arterial hypertension. Br Med J. 1973;1(5852):507-510. doi: 10.1136/bmj.1.5852.507. PubMed
12. Fischberg GM, Lozano E, Rajamani K, Ameriso S, Fisher MJ. Stroke precipitated by moderate blood pressure reduction. J Emerg Med. 2000;19(4):339-346. doi: 10.1016/S0736-4679(00)00267-5. PubMed
13. Ross RS. Pathophysiology of coronary circulation. Br Heart J. 1971;33(2):173-184. doi: 10.1136/hrt.33.2.173. PubMed
14. Brooks TW, Finch CK, Lobo BL, Deaton PR, Varner CF. Blood pressure management in acute hypertensive emergency. Am J Health Syst Pharm. 2007;64(24):2579-2582. doi: 10.2146/ajhp070105. PubMed
15. Grossman E, Messerli FH, Grodzicki T, Kowey P. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-1331. doi: 10.1001/jama.1996.03540160050032 PubMed
16. Hodsman GP, Isles CG, Murray GD et al. Factors related to first dose hypotensive effect of captopril: prediction and treatment. Br Med J (Clin Res Ed). 1983;286(6368):832-834. doi: 10.1136/bmj.286.6368.832. PubMed
17. Zeller KR, Von Kuhnert L, Matthews C. Rapid reduction of severe asymptomatic hypertension. A prospective, controlled trial. Arch Intern Med. 1989;149(10):2186-2189. doi: 10.1001/archinte.149.10.2186. PubMed
18. Pickering TG, Hall JE, Appel LJ, et al. Recommendations for blood pressure measurement in humans and experimental animals: Part 1: Blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation. 2005;111(5):697-716. doi: 10.1161/01.CIR.0000154900.76284.F6. PubMed
19. Chobanian AV, Bakris GL, Black HR, et al. The seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High blood pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2572. doi: 10.1001/jama.289.19.2560. PubMed
20. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. doi: 10.1001/jama.2013.284427 PubMed
21. Whelton PK, Carey RM, Aronow WS, et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the Prevention, detection, evaluation, and management of High blood pressure in adults: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2017. PubMed
22. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015;17(11):94. doi: 10.1007/s11886-015-0648-y. PubMed
23. Grassi D, O'Flaherty M, Pellizzari M, et al. Hypertensive urgencies in the emergency department: evaluating blood pressure response to rest and to antihypertensive drugs with different profiles. J Clin Hypertens (Greenwich). 2008;10(9):662-667. doi: 10.1111/j.1751-7176.2008.00001.x. PubMed
24. Park SK, Lee DY, Kim WJ, et al. Comparing the clinical efficacy of resting and antihypertensive medication in patients of hypertensive urgency: a randomized, control trial. J Hypertens. 2017;35(7):1474-1480. doi: 10.1097/HJH.0000000000001340. PubMed
25. Steinman MA, Auerbach AD. Managing chronic disease in hospitalized patients. JAMA Intern Med. 2013;173(20):1857-1858. doi: 10.1001/jamainternmed.2013.9511. PubMed
26. Breu AC, Allen-Dicker J, Mueller S et al. Hospitalist and primary care physician perspectives on medication management of chronic conditions for hospitalized patients. J Hosp Med. 2014;9(5):303-309. doi: 10.1002/jhm.2137. PubMed
1. Shorr AF, Zilberberg MD, Sun X, et al. Severe acute hypertension among inpatients admitted from the emergency department. J Hosp Med. 2012;7(3):203-210. doi: 10.1002/jhm.969. PubMed
2. Whelton PK, Carey RM, Aronow WS, et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the Prevention, detection, evaluation, and management of High blood pressure in adults: A report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Hypertension. 2017. PubMed
3. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: use of intravenous labetalol and hydralazine. J Clin Hypertens (Greenwich). 2010;12(1):29-33. doi: 10.1111/j.1751-7176.2009.00196.x. PubMed
4. Axon RN, Garrell R, Pfahl K, et al. Attitudes and practices of resident physicians regarding hypertension in the inpatient setting. J Clin Hypertens (Greenwich). 2010;12(9):698-705. doi: 10.1111/j.1751-7176.2010.00309.x. PubMed
5. Keith NM, Wagener HP, Barker NW. Some different types of essential hypertension: their course and prognosis. Am J Med Sci. 1974;268(6):336-345. doi: 10.1097/00000441-197412000-00004. PubMed
6. Guiga H, Decroux C, Michelet P, et al. Hospital and out-of-hospital mortality in 670 hypertensive emergencies and urgencies. J Clin Hypertens (Greenwich). 2017;19(11):1137-1142. doi: 10.1111/jch.13083. PubMed
7. Varon J, Williams EJ. Management of severe asymptomatic hypertension (hypertensive urgencies) in adults. In: Post T, ed. UpToDate, Waltham, MA. (Accessed February 13, 2018). PubMed
8. Effects of treatment on morbidity in hypertension. Results in patients with diastolic blood pressures averaging 115 through 129 mm Hg. JAMA. 1967;202(11):1028-1034. soi: 10.1001/jama.1967.03130240070013 PubMed
9. Patel KK, Young L, Howell EH, et al. Characteristics and outcomes of patients presenting with hypertensive urgency in the office setting. JAMA Intern Med. 2016;176(7):981-988. doi: 10.1001/jamainternmed.2016.1509. PubMed
10. MacDougall JD, Tuxen D, Sale DG, Moroz JR, Sutton JR. Arterial blood pressure response to heavy resistance exercise. J Appl Physiol. 1985;58(3):785-790. doi: 10.1152/jappl.1985.58.3.785. PubMed
11. Strandgaard S, Olesen J, Skinhoj E, Lassen NA. Autoregulation of brain circulation in severe arterial hypertension. Br Med J. 1973;1(5852):507-510. doi: 10.1136/bmj.1.5852.507. PubMed
12. Fischberg GM, Lozano E, Rajamani K, Ameriso S, Fisher MJ. Stroke precipitated by moderate blood pressure reduction. J Emerg Med. 2000;19(4):339-346. doi: 10.1016/S0736-4679(00)00267-5. PubMed
13. Ross RS. Pathophysiology of coronary circulation. Br Heart J. 1971;33(2):173-184. doi: 10.1136/hrt.33.2.173. PubMed
14. Brooks TW, Finch CK, Lobo BL, Deaton PR, Varner CF. Blood pressure management in acute hypertensive emergency. Am J Health Syst Pharm. 2007;64(24):2579-2582. doi: 10.2146/ajhp070105. PubMed
15. Grossman E, Messerli FH, Grodzicki T, Kowey P. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-1331. doi: 10.1001/jama.1996.03540160050032 PubMed
16. Hodsman GP, Isles CG, Murray GD et al. Factors related to first dose hypotensive effect of captopril: prediction and treatment. Br Med J (Clin Res Ed). 1983;286(6368):832-834. doi: 10.1136/bmj.286.6368.832. PubMed
17. Zeller KR, Von Kuhnert L, Matthews C. Rapid reduction of severe asymptomatic hypertension. A prospective, controlled trial. Arch Intern Med. 1989;149(10):2186-2189. doi: 10.1001/archinte.149.10.2186. PubMed
18. Pickering TG, Hall JE, Appel LJ, et al. Recommendations for blood pressure measurement in humans and experimental animals: Part 1: Blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation. 2005;111(5):697-716. doi: 10.1161/01.CIR.0000154900.76284.F6. PubMed
19. Chobanian AV, Bakris GL, Black HR, et al. The seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High blood pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2572. doi: 10.1001/jama.289.19.2560. PubMed
20. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. doi: 10.1001/jama.2013.284427 PubMed
21. Whelton PK, Carey RM, Aronow WS, et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the Prevention, detection, evaluation, and management of High blood pressure in adults: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2017. PubMed
22. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015;17(11):94. doi: 10.1007/s11886-015-0648-y. PubMed
23. Grassi D, O'Flaherty M, Pellizzari M, et al. Hypertensive urgencies in the emergency department: evaluating blood pressure response to rest and to antihypertensive drugs with different profiles. J Clin Hypertens (Greenwich). 2008;10(9):662-667. doi: 10.1111/j.1751-7176.2008.00001.x. PubMed
24. Park SK, Lee DY, Kim WJ, et al. Comparing the clinical efficacy of resting and antihypertensive medication in patients of hypertensive urgency: a randomized, control trial. J Hypertens. 2017;35(7):1474-1480. doi: 10.1097/HJH.0000000000001340. PubMed
25. Steinman MA, Auerbach AD. Managing chronic disease in hospitalized patients. JAMA Intern Med. 2013;173(20):1857-1858. doi: 10.1001/jamainternmed.2013.9511. PubMed
26. Breu AC, Allen-Dicker J, Mueller S et al. Hospitalist and primary care physician perspectives on medication management of chronic conditions for hospitalized patients. J Hosp Med. 2014;9(5):303-309. doi: 10.1002/jhm.2137. PubMed
© 2018 Society of Hospital Medicine
So Much More than Bald and Bloated
A 44-year-old previously healthy semiprofessional male athlete presented with five days of nausea, vomiting, and abdominal pain. He had also experienced several months of decreased energy and new episodes of constipation three weeks prior to presentation.
At this point, we do not have sufficient information to completely determine the cause of his abdominal symptoms. Common causes of abdominal pain and vomiting in adults of his age group include peptic ulcer disease, pancreatic or hepatobiliary track disorders, small or large bowel processes, appendicitis, or even renal pathology. Further characterization may be possible by describing the location and quality of pain and factors that might relieve or exacerbate his pain. Despite the ambiguity, multiple clues might allow us to narrow the broad differential diagnosis of abdominal pain. In a previously healthy, vigorous, middle-aged man with subacute abdominal pain associated with constipation, the differential diagnosis should include disease states that may cause a bowel obstruction; these states include inflammatory bowel disease (IBD), gastrointestinal malignancy, or peptic ulcer disease. Mechanical obstruction due to volvulus or intussusception would be less likely in his age group. Given his history of several months of fatigue and several weeks of constipation, he should be evaluated for metabolic causes of abdominal pain and constipation, such as hypothyroidism or hypercalcemia. In addition to basic laboratory and imaging studies, obtaining additional history regarding prior abdominal surgeries, medication use, alcohol intake, and family and travel history will be the key in directing the evaluation.
Six months prior to admission, the patient began to feel more fatigue and exercise intolerance, reduced sweating, increased cold intolerance, and increased presyncopal episodes. He was diagnosed with hypothyroidism (TSH 6.69 μIU/mL; free T4 not done) and initiated on levothyroxine. One month prior to presentation, he developed constipation, loss of taste, reduced appetite, and weight loss of 30 pounds. He developed blurry vision and photophobia. He also complained of erectile dysfunction, urinary hesitancy and straining, which were diagnosed as benign prostatic hypertrophy.
Given the addition of numerous historical features in a previously healthy man, it is important to strive for a parsimonious diagnosis to unify his seemingly disparate features. His fatigue, constipation, and cold intolerance are consistent with his diagnosis of hypothyroidism but are nonspecific. Whether the degree of hypothyroidism caused his symptoms or signs is doubtful. The constellation of symptoms and signs are more likely to be representative of a nonthyroidal illness. His abdominal pain, unexplained weight loss, and presyncopal episodes should raise consideration of adrenal insufficiency. The combination of hypothyroidism and adrenal insufficiency suggest the possibility of an autoimmune polyendocrine syndrome or other pituitary pathology. In this case, history of headache, dysgeusia, and visual disturbances might support the diagnosis of pituitary adenoma. A cosyntropin stimulation test could establish the diagnosis of adrenal insufficiency. A low ACTH level would establish a diagnosis of pituitary or hypothalamic hypofunction. If pituitary hypofunction is documented, then a brain MRI would be needed to confirm the diagnosis of pituitary adenoma.
His newly reported erectile dysfunction suggests the possibility of a psychiatric, neurologic, hormonal, or vascular process and should be explored further. Sexual dysfunction is also associated with adrenal insufficiency and hypopituitarism. However, the presence of suspected prostatic hypertrophy in a male competitive athlete in his forties also raises the question of exogenous androgen use.
His past medical history was notable for a two-year history of alopecia totalis, seasonal allergies, asthma, and a repaired congenital aortic web with known aortic insufficiency. He was married with two children, worked an office job, and had no history of injection drug use, blood transfusions, or multiple sexual partners. His family history was notable for hypothyroidism and asthma in several family members in addition to Crohn disease, celiac disease, diabetes, cardiovascular disease, and cancers of the breast and lung.
His past medical, surgical, and family history supports a diagnosis of autoimmune disease. Although there is a personal and family history of atopic disorders, including allergic rhinitis and asthma, no association is found between atopy and autoimmunity. His family history of hypothyroidism, Crohn disease, and diabetes suggests a familial autoimmune genetic predisposition. His history of alopecia totalis in the setting of hypothyroidism and possible autoimmune adrenal insufficiency or autoimmune hypophysitis raises suspicion for the previously suggested diagnosis of polyglandular autoimmune syndrome, also known as autoimmune polyendocrine syndrome. Type I polyglandular autoimmune syndrome is associated with hypoparathyroidism and mucocutaneous candidiasis. In the absence of these symptoms, the patient more likely has type II polyglandular autoimmune syndrome. Type II syndrome is more prevalent and can occur in the setting of other nonendocrine autoimmune disorders, such as vitiligo, myasthenia gravis, or rheumatoid arthritis. Adrenal insufficiency can be the initial and most prominent manifestation of type II syndrome.
On physical exam, he was afebrile, with a heart rate of 68 beats per minute, respiratory rate of 16 breaths per minute, and normal oxygen saturation. His supine blood pressure and heart rate were 116/72 mm Hg and 66 beats per minute, respectively, and his standing blood pressure and heart rates were 80/48 mm Hg and 68 beats per minute respectively. He was thin, had diffuse scalp and body alopecia, and was ill-appearing with dry skin and dry mucous membranes. No evidence of Osler nodes, Janeway lesions, or splinter hemorrhages were found on cutaneous examination. No Roth spots or conjunctival hemorrhages were noted on ophthalmologic examination. He had both a 3/6 crescendo–decrescendo systolic murmur best heard at the right clavicle and radiated to the carotids and a 3/6 early diastolic decrescendo murmur best heard at the left sternal border. His abdomen was slightly protuberant, with reduced bowel sounds, hyperresonant to tympanitic on percussion, and a diffusely, moderately tender without peritoneal signs. Neurologic examination revealed 8 mm pupils with minimal response to light and accommodation. The remaining portions of his cranial nerve and complete neurologic examination were normal.
The presence of postural hypotension supports the previous suspicion of adrenal insufficiency, and the possibility of a pituitary or hypothalamic process remains. However, his dilated and minimally responsive pupils and potentially adynamic bowel are inconsistent with these diagnoses. Mydriasis and adynamic bowel in combination with orthostatic hypotension, dysgeusia, urinary retention, and erectile dysfunction are strongly suggestive of an autonomic process. Endocarditis is worth considering given his multisystem involvement, subacute decline, and known valve pathology. The absence of fever or stigmata of endocarditis make it difficult to explain his clinical syndrome. An echocardiogram would be reasonable for further assessment. At this point, it is prudent to explore his adrenal and pituitary function; if unrevealing, embark on an evaluation of his autonomic dysfunction.
Initial laboratory investigations were notable for mild normocytic anemia and hypoalbuminemia. His cosyntropin stimulation test was normal at 60 minutes. An abdominal CT scan demonstrated marked dilation in the small bowel loops (6 cm in caliber) with associated small bowel wall thickening and hyperemia. The echocardiogram was unrevealing and only confirmed the ongoing, progression of his known valve pathology without evidence of vegetation.
The above testing rules out primary adrenal insufficiency, but an appropriate response to the cosyntropin stimulation test does not rule out secondary, or pituitary, adrenal insufficiency. The echocardiogram and lack of other features make infective endocarditis unlikely. Thus, as mentioned, it is important now to commence a complete work-up of his probable dysautonomia to explain the majority of his features. Additionally, his hypothyroidism (if more than sick euthyroid syndrome), family history of autoimmune processes, and alopecia totalis all suggest the possibility of an immune-related syndrome. His CT scan revealed some thickened hyperemic bowel, which could suggest an IBD, such as Crohn disease; however, the absence of other signs, such as fever, diarrhea, or bloody stools, argues against this diagnosis. A syndrome that could unify his presentation is autoimmune autonomic ganglionopathy (AAG), a rare genetic autonomic system disorder that presents with pandysautonomia. The spectrum of autoimmunity was considered early in this case, but the differential diagnosis included more common conditions, such as adrenal insufficiency. Similarly, IBD remains a consideration. The serologic studies for IBD can be useful but they lack definitive diagnostic accuracy. Given that treatment for AAG differs from that for IBD, additional information will help guide the therapeutic approach. Anti-α3gnAChR antibodies, which are associated with AAG, should be checked.
His history of presyncope, anhidrosis, urinary retention, and ileus raised suspicion for pandysautonomia, as characterized by signs of sympathetic and parasympathetic dysfunction. The suspicion for pandysautonomia was confirmed via specialized autonomic testing, which included reduced heart rate variation on Valsalva and deep breathing maneuvers, orthostatic hypotension consistent autonomic insufficiency on Tilt table testing, and reduced sweat response to acetylcholine application (QSART test). The patient underwent further diagnostic serologic testing to differentiate causes of autonomic failure (Table 1). His personal and family history of autoimmunity led to the working diagnosis of AAG. Ultimate testing revealed high titers of autoantibodies, specifically anti-α3gnAChR (3.29 nmol/L, normal <0.02 nmol/L), directed against the ganglionic nicotinic acetylcholine receptor. This finding strongly supported the diagnosis of AAG.1,4-7 
He was initially treated empirically with intravenous immunoglobulin (IVIG) with minimal improvement. He received additional immunomodulating therapies including methylprednisolone, plasmapheresis, and rituximab but did not tolerate a trial of mycophenolate. Six weeks after therapy initiation, his antibody titers decreased to 0.89 nmol/L with associated clinical improvement. Ultimately, he was discharged from the hospital on day 73 with a feeding tube and supplemental total parenteral nutrition. Four months postdischarge, he had returned to his prediagnosis weight, had eased back into his prior activities, and was off supplemental nutrition. Over a year later, he completed a 10-month prednisone taper and continued to receive monthly IVIG infusion
DISCUSSION
The clinical approach to dysautonomia is based on different etiologies: (1) those associated with neurodegenerative disorders; (2) those associated with peripheral neuropathies, and (3) isolated autonomic failure.2 Thus, clinical history and physical examination can assist greatly in guiding the evaluation of patients. Neurodegenerative disorders (such as Parkinson disease), combined disorders (such as multiple-system atrophy), and acquired or familial processes were considered. Our patient had neither a personal or family history nor physical examination supporting a neurodegenerative disorder. Disorders of the peripheral nerves were considered and can broadly be categorized as chronic sensorimotor neuropathies, sensory ganglionopathies, distal painful neuropathies, and acute or subacute motor polyradiculopathies. During evaluation, no historical, physical examination, or laboratory findings supported diabetes, amyloidosis, heavy metals, Sjögren syndrome, paraneoplastic neuropathy, sodium channel disorders, infectious etiologies, or porphyria (Table 1). Thus, in the absence of supportive evidence for primary neurodegenerative disorders or peripheral neuropathies, his syndrome appeared most compatible with an isolated autonomic failure syndrome. The principal differential for this syndrome is pure autonomic failure versus an immune-mediated autonomic disorder, including paraneoplastic autoimmune neuropathy and AAG. The diagnosis of pure autonomic failure is made after there is no clear unifying syndrome after more than five years of investigation. After exploration, no evidence of malignancy was discovered on body cross sectional imaging, PET scanning, bone marrow biopsy, colonoscopy, or laboratory testing. Thus, positive serologic testing in the absence of an underlying malignancy suggests a diagnosis of AAG.
AAG was first described in 1969 and is a rare, acquired disorder characterized by combined failure of the parasympathetic, sympathetic, and enteric nervous systems. This disorder typically presents in young-to-middle aged patients but has been described in all age groups. It is more commonly seen in patients with coexistent autoimmune diseases and/or a history of familial autoimmunity. The onset of clinical AAG may be subacute (less than three months) or insidious (more than three months). Patients present with signs or symptoms of pandysautonomia, such as severe orthostatic hypotension, syncope, constipation and gastrointestinal dysmotility, urinary retention, fixed and dilated pupils, and dry mouth and eyes (Table 2). Up to 40% of patients with AAG may also have significant cognitive impairment.3,4 Diagnosis relies on a combination of typical clinical features as discussed above and the exclusion of other diagnostic considerations. Diagnosis of AAG is aided by the presence of autoantibodies to ganglionic nicotinic acetylcholine receptors (gnAChR), particularly antiganglionic acetylcholine receptor α3 (anti-α3gAChR).1 Anti-gnAChR antibodies are only present in about half of patients with AAG. Antibody titers are highest in subacute AAG (40%-50%)3 compared with chronic AAG (30%-40%) or paraneoplastic AAG (10%-20%).5 Anti-gnAChR antibodies are not specific to AAG and have been identified in low levels in up to 20% of patients with thymomas, postural orthostatic tachycardia syndrome, chronic idiopathic anhidrosis, idiopathic gastrointestinal dysmotility, Lambert–Eaton syndrome, and myasthenia gravis without thymoma.1,5-7 These associations raise the question of shared pathology and perhaps a syndrome overlap. Individuals with seropositive AAG may also have other paraneoplastic antibodies, making it clinically indistinguishable from paraneoplastic autonomic neuropathy.5,8 Although the autoantibody lacks sensitivity and is imperfectly specific, its presence supports a diagnosis of AAG. Anti-gnAChR antibodies have been shown to be pathological in rabbit and mouse models.4 In patients with AAG, higher autoantibody titers correlate with increased disease severity.1,6,7 A decrease in autoantibody titers correlates with decreased disease severity.6 Case report series also described a distinct entity of seronegative AAG.2,3 Maintaining a high clinical suspicion for AAG even with negative antibodies is important.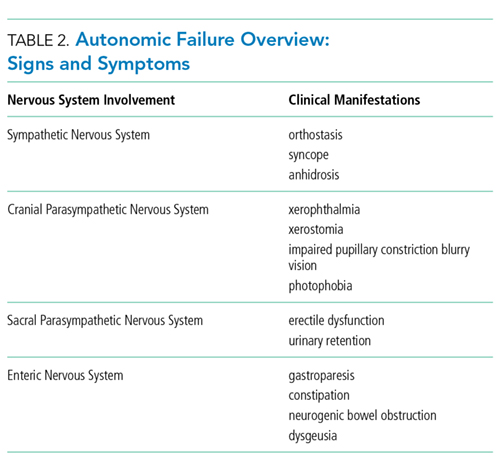
Given the rarity of the disease, no standard therapeutic regimens are available. About one-third of individuals improve on their own, while other individuals require extensive immunomodulation and symptom management. Case series and observational trials currently make up the vast array of treatment data. Therapies include glucocorticoids, plasmapheresis, IVIG, and other immunosuppressive agents, such as rituximab.9-12 Patients with and without identified anti-gnAChRs antibodies may respond to therapy.12 The overall long-term prognosis of the disease is poorly characterized.9,10,13
Despite the rarity of the syndrome discussed, this case represents how diagnostic reasoning strategies, such as law of parsimony, shift how the case is framed. For example, a middle-aged man with several new, distinctly unrelated diagnoses versus a middle-aged man with signs and symptoms of autonomic failure alters the subsequent clinical reasoning and diagnostic approach. Many diseases, both common and rare, are associated with dysautonomia. Therefore, clinicians should have an approach to autonomic failure
TEACHING POINTS
- Recognize the following signs and symptoms suggesting a dysautonomic syndrome: orthostasis, syncope, anhidrosis, xerophthalmia, xerostomia, impaired pupillary constriction, blurry vision, photophobia, erectile dysfunction, urinary retention, gastroparesis, constipation, neurogenic bowel obstruction, and dysgeusia.
- Recognize the clinical features, diagnostic approach, and management of autoimmune autonomic ganglionopathy.
- When faced with a complex clinical presentation, early application of the “law of parsimony” may help identify a unifying syndrome.
Acknowledgments
The authors wish to thank our Blinded Expert, Anthony Montanaro, MD, for his expertise and guidance during this process.
Disclosures
There are no known conflicts of interest.
1. Gibbons C, Freeman R. Antibody titers predict clinical features of autoimmune autonomic ganglionopathy. Auton Neurosci. 2009;146(1-2):8-12. doi: 10.1016/j.autneu.2008.11.013. PubMed
2. Golden E, Bryarly M, Vernino S. Seronegative autoimmune autonomic neuropathy: a distinct clinical entity. Clin Auton Res. 2018;28(1):115-123. doi: 10.1007/s10286-017-0493-8. PubMed
3. Sandroni P, Vernino S, Klein CM, et al. Idiopathic autonomic neuropathy: comparison of cases seropositive and seronegative for ganglionic acetylcholine receptor antibody. Arch Neurol. 2004;61(1):44-48. doi: 10.1001/archneur.61.1.44. PubMed
4. Vernino S, Ermilov L, Sha L, Szurszewski J, Low P, Lennon V. Passive transfer of autoimmune autonomic neuropathy to mice. J Neurosci. 2004;24(32):7037-7042. doi: 10.1523/JNEUROSCI.1485-04.2004. PubMed
5. Vernino S, Hopkins S, Wang Z. Autonomic ganglia, acetylcholine receptor antibodies, and autoimmune ganglionopathy. Auton Neurosci. 2009;146(1-2):3-7. doi: 10.1016/j.autneu.2008.09.005. PubMed
6. Vernino S, Low P, Fealey R, Stewart J, Farrugia G, Lennon V. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343(12):847-855. doi: 10.1056/NEJM200009213431204. PubMed
7. Gibbons C, Vernino S, Freeman R. Autoimmune autonomic ganglionopathy – Symptom antibody correlations. Auton Neurosci. 2015;192:130. doi: 10.1016/j.autneu.2015.07.241 .
8. Benarroch E. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol. 2014;10(7):396-407. doi: 10.1038/nrneurol.2014.88. PubMed
9. Baker SK, Morillo C, Vernino S. Autoimmune autonomic ganglionopathy with late-onset encephalopathy. Auton Neurosci. 2009;146(1-2):29-32. doi: 10.1016/j.autneu.2008.10.016. PubMed
10. Gibbons C, Centi J, Vernino S. Autoimmune autonomic ganglionoapthy with reversible cognitive impairment. Arch Neurol. 2012;69(4):461-466. doi: 10.1001/archneurol.2011.2372. PubMed
11. Boydston E, Muppidi S, Vernino S. Long-term outcomes in autoimmune autonomic ganglionopathy (P05.210). Neurology. 2012;78(1):P05.210. doi: 10.1212/WNL.78.1_MeetingAbstracts.P05.210.
12. Gehrking T, Sletten D, Fealey R, Low P, Singer W. 11-year follow-up of a case of autoimmune autonomic ganglionopathy (P03.024). Neurology. 2013;80(7):P03.024.
13. Imrich R, Vernino S, Eldadah BA, Holmes C, Goldstein DS. Autoimmune autonomic ganglionopathy: treatment by plasma exchanges and rituximab. Clin Auton Res. 2009;19(4):259-262. doi: 10.1007/s10286-009-0012-7. PubMed
14. Iodice V, Kimpinski K, Vernino S, Sandroni P, Fealey RD, Low PA. Efficacy of immunotherapy in seropositive and seronegative putative autoimmune autonomic ganglionopathy. Neurology. 2009;72(23):2002-8. doi: 10.1212/WNL.0b013e3181a92b52. PubMed
15. Hayashi M, Ishii Y. A Japanese case of autoimmune autonomic ganglionopathy (AAG) and a review of AAG cases in Japan. Auton Neurosci. 2009;146(1-2):26-8. doi: 10.1016/j.autneu.2008.12.013. PubMed
16. Baker, A. Simplicity. In: Baker A, Zalta E, eds. The Stanford Encyclopedia of Philosophy. Winter 2016 Edition. https://plato.stanford.edu/archives/win2016/entries/simplicity/. Accessed October 26, 2017.
A 44-year-old previously healthy semiprofessional male athlete presented with five days of nausea, vomiting, and abdominal pain. He had also experienced several months of decreased energy and new episodes of constipation three weeks prior to presentation.
At this point, we do not have sufficient information to completely determine the cause of his abdominal symptoms. Common causes of abdominal pain and vomiting in adults of his age group include peptic ulcer disease, pancreatic or hepatobiliary track disorders, small or large bowel processes, appendicitis, or even renal pathology. Further characterization may be possible by describing the location and quality of pain and factors that might relieve or exacerbate his pain. Despite the ambiguity, multiple clues might allow us to narrow the broad differential diagnosis of abdominal pain. In a previously healthy, vigorous, middle-aged man with subacute abdominal pain associated with constipation, the differential diagnosis should include disease states that may cause a bowel obstruction; these states include inflammatory bowel disease (IBD), gastrointestinal malignancy, or peptic ulcer disease. Mechanical obstruction due to volvulus or intussusception would be less likely in his age group. Given his history of several months of fatigue and several weeks of constipation, he should be evaluated for metabolic causes of abdominal pain and constipation, such as hypothyroidism or hypercalcemia. In addition to basic laboratory and imaging studies, obtaining additional history regarding prior abdominal surgeries, medication use, alcohol intake, and family and travel history will be the key in directing the evaluation.
Six months prior to admission, the patient began to feel more fatigue and exercise intolerance, reduced sweating, increased cold intolerance, and increased presyncopal episodes. He was diagnosed with hypothyroidism (TSH 6.69 μIU/mL; free T4 not done) and initiated on levothyroxine. One month prior to presentation, he developed constipation, loss of taste, reduced appetite, and weight loss of 30 pounds. He developed blurry vision and photophobia. He also complained of erectile dysfunction, urinary hesitancy and straining, which were diagnosed as benign prostatic hypertrophy.
Given the addition of numerous historical features in a previously healthy man, it is important to strive for a parsimonious diagnosis to unify his seemingly disparate features. His fatigue, constipation, and cold intolerance are consistent with his diagnosis of hypothyroidism but are nonspecific. Whether the degree of hypothyroidism caused his symptoms or signs is doubtful. The constellation of symptoms and signs are more likely to be representative of a nonthyroidal illness. His abdominal pain, unexplained weight loss, and presyncopal episodes should raise consideration of adrenal insufficiency. The combination of hypothyroidism and adrenal insufficiency suggest the possibility of an autoimmune polyendocrine syndrome or other pituitary pathology. In this case, history of headache, dysgeusia, and visual disturbances might support the diagnosis of pituitary adenoma. A cosyntropin stimulation test could establish the diagnosis of adrenal insufficiency. A low ACTH level would establish a diagnosis of pituitary or hypothalamic hypofunction. If pituitary hypofunction is documented, then a brain MRI would be needed to confirm the diagnosis of pituitary adenoma.
His newly reported erectile dysfunction suggests the possibility of a psychiatric, neurologic, hormonal, or vascular process and should be explored further. Sexual dysfunction is also associated with adrenal insufficiency and hypopituitarism. However, the presence of suspected prostatic hypertrophy in a male competitive athlete in his forties also raises the question of exogenous androgen use.
His past medical history was notable for a two-year history of alopecia totalis, seasonal allergies, asthma, and a repaired congenital aortic web with known aortic insufficiency. He was married with two children, worked an office job, and had no history of injection drug use, blood transfusions, or multiple sexual partners. His family history was notable for hypothyroidism and asthma in several family members in addition to Crohn disease, celiac disease, diabetes, cardiovascular disease, and cancers of the breast and lung.
His past medical, surgical, and family history supports a diagnosis of autoimmune disease. Although there is a personal and family history of atopic disorders, including allergic rhinitis and asthma, no association is found between atopy and autoimmunity. His family history of hypothyroidism, Crohn disease, and diabetes suggests a familial autoimmune genetic predisposition. His history of alopecia totalis in the setting of hypothyroidism and possible autoimmune adrenal insufficiency or autoimmune hypophysitis raises suspicion for the previously suggested diagnosis of polyglandular autoimmune syndrome, also known as autoimmune polyendocrine syndrome. Type I polyglandular autoimmune syndrome is associated with hypoparathyroidism and mucocutaneous candidiasis. In the absence of these symptoms, the patient more likely has type II polyglandular autoimmune syndrome. Type II syndrome is more prevalent and can occur in the setting of other nonendocrine autoimmune disorders, such as vitiligo, myasthenia gravis, or rheumatoid arthritis. Adrenal insufficiency can be the initial and most prominent manifestation of type II syndrome.
On physical exam, he was afebrile, with a heart rate of 68 beats per minute, respiratory rate of 16 breaths per minute, and normal oxygen saturation. His supine blood pressure and heart rate were 116/72 mm Hg and 66 beats per minute, respectively, and his standing blood pressure and heart rates were 80/48 mm Hg and 68 beats per minute respectively. He was thin, had diffuse scalp and body alopecia, and was ill-appearing with dry skin and dry mucous membranes. No evidence of Osler nodes, Janeway lesions, or splinter hemorrhages were found on cutaneous examination. No Roth spots or conjunctival hemorrhages were noted on ophthalmologic examination. He had both a 3/6 crescendo–decrescendo systolic murmur best heard at the right clavicle and radiated to the carotids and a 3/6 early diastolic decrescendo murmur best heard at the left sternal border. His abdomen was slightly protuberant, with reduced bowel sounds, hyperresonant to tympanitic on percussion, and a diffusely, moderately tender without peritoneal signs. Neurologic examination revealed 8 mm pupils with minimal response to light and accommodation. The remaining portions of his cranial nerve and complete neurologic examination were normal.
The presence of postural hypotension supports the previous suspicion of adrenal insufficiency, and the possibility of a pituitary or hypothalamic process remains. However, his dilated and minimally responsive pupils and potentially adynamic bowel are inconsistent with these diagnoses. Mydriasis and adynamic bowel in combination with orthostatic hypotension, dysgeusia, urinary retention, and erectile dysfunction are strongly suggestive of an autonomic process. Endocarditis is worth considering given his multisystem involvement, subacute decline, and known valve pathology. The absence of fever or stigmata of endocarditis make it difficult to explain his clinical syndrome. An echocardiogram would be reasonable for further assessment. At this point, it is prudent to explore his adrenal and pituitary function; if unrevealing, embark on an evaluation of his autonomic dysfunction.
Initial laboratory investigations were notable for mild normocytic anemia and hypoalbuminemia. His cosyntropin stimulation test was normal at 60 minutes. An abdominal CT scan demonstrated marked dilation in the small bowel loops (6 cm in caliber) with associated small bowel wall thickening and hyperemia. The echocardiogram was unrevealing and only confirmed the ongoing, progression of his known valve pathology without evidence of vegetation.
The above testing rules out primary adrenal insufficiency, but an appropriate response to the cosyntropin stimulation test does not rule out secondary, or pituitary, adrenal insufficiency. The echocardiogram and lack of other features make infective endocarditis unlikely. Thus, as mentioned, it is important now to commence a complete work-up of his probable dysautonomia to explain the majority of his features. Additionally, his hypothyroidism (if more than sick euthyroid syndrome), family history of autoimmune processes, and alopecia totalis all suggest the possibility of an immune-related syndrome. His CT scan revealed some thickened hyperemic bowel, which could suggest an IBD, such as Crohn disease; however, the absence of other signs, such as fever, diarrhea, or bloody stools, argues against this diagnosis. A syndrome that could unify his presentation is autoimmune autonomic ganglionopathy (AAG), a rare genetic autonomic system disorder that presents with pandysautonomia. The spectrum of autoimmunity was considered early in this case, but the differential diagnosis included more common conditions, such as adrenal insufficiency. Similarly, IBD remains a consideration. The serologic studies for IBD can be useful but they lack definitive diagnostic accuracy. Given that treatment for AAG differs from that for IBD, additional information will help guide the therapeutic approach. Anti-α3gnAChR antibodies, which are associated with AAG, should be checked.
His history of presyncope, anhidrosis, urinary retention, and ileus raised suspicion for pandysautonomia, as characterized by signs of sympathetic and parasympathetic dysfunction. The suspicion for pandysautonomia was confirmed via specialized autonomic testing, which included reduced heart rate variation on Valsalva and deep breathing maneuvers, orthostatic hypotension consistent autonomic insufficiency on Tilt table testing, and reduced sweat response to acetylcholine application (QSART test). The patient underwent further diagnostic serologic testing to differentiate causes of autonomic failure (Table 1). His personal and family history of autoimmunity led to the working diagnosis of AAG. Ultimate testing revealed high titers of autoantibodies, specifically anti-α3gnAChR (3.29 nmol/L, normal <0.02 nmol/L), directed against the ganglionic nicotinic acetylcholine receptor. This finding strongly supported the diagnosis of AAG.1,4-7
He was initially treated empirically with intravenous immunoglobulin (IVIG) with minimal improvement. He received additional immunomodulating therapies including methylprednisolone, plasmapheresis, and rituximab but did not tolerate a trial of mycophenolate. Six weeks after therapy initiation, his antibody titers decreased to 0.89 nmol/L with associated clinical improvement. Ultimately, he was discharged from the hospital on day 73 with a feeding tube and supplemental total parenteral nutrition. Four months postdischarge, he had returned to his prediagnosis weight, had eased back into his prior activities, and was off supplemental nutrition. Over a year later, he completed a 10-month prednisone taper and continued to receive monthly IVIG infusion
DISCUSSION
The clinical approach to dysautonomia is based on different etiologies: (1) those associated with neurodegenerative disorders; (2) those associated with peripheral neuropathies, and (3) isolated autonomic failure.2 Thus, clinical history and physical examination can assist greatly in guiding the evaluation of patients. Neurodegenerative disorders (such as Parkinson disease), combined disorders (such as multiple-system atrophy), and acquired or familial processes were considered. Our patient had neither a personal or family history nor physical examination supporting a neurodegenerative disorder. Disorders of the peripheral nerves were considered and can broadly be categorized as chronic sensorimotor neuropathies, sensory ganglionopathies, distal painful neuropathies, and acute or subacute motor polyradiculopathies. During evaluation, no historical, physical examination, or laboratory findings supported diabetes, amyloidosis, heavy metals, Sjögren syndrome, paraneoplastic neuropathy, sodium channel disorders, infectious etiologies, or porphyria (Table 1). Thus, in the absence of supportive evidence for primary neurodegenerative disorders or peripheral neuropathies, his syndrome appeared most compatible with an isolated autonomic failure syndrome. The principal differential for this syndrome is pure autonomic failure versus an immune-mediated autonomic disorder, including paraneoplastic autoimmune neuropathy and AAG. The diagnosis of pure autonomic failure is made after there is no clear unifying syndrome after more than five years of investigation. After exploration, no evidence of malignancy was discovered on body cross sectional imaging, PET scanning, bone marrow biopsy, colonoscopy, or laboratory testing. Thus, positive serologic testing in the absence of an underlying malignancy suggests a diagnosis of AAG.
AAG was first described in 1969 and is a rare, acquired disorder characterized by combined failure of the parasympathetic, sympathetic, and enteric nervous systems. This disorder typically presents in young-to-middle aged patients but has been described in all age groups. It is more commonly seen in patients with coexistent autoimmune diseases and/or a history of familial autoimmunity. The onset of clinical AAG may be subacute (less than three months) or insidious (more than three months). Patients present with signs or symptoms of pandysautonomia, such as severe orthostatic hypotension, syncope, constipation and gastrointestinal dysmotility, urinary retention, fixed and dilated pupils, and dry mouth and eyes (Table 2). Up to 40% of patients with AAG may also have significant cognitive impairment.3,4 Diagnosis relies on a combination of typical clinical features as discussed above and the exclusion of other diagnostic considerations. Diagnosis of AAG is aided by the presence of autoantibodies to ganglionic nicotinic acetylcholine receptors (gnAChR), particularly antiganglionic acetylcholine receptor α3 (anti-α3gAChR).1 Anti-gnAChR antibodies are only present in about half of patients with AAG. Antibody titers are highest in subacute AAG (40%-50%)3 compared with chronic AAG (30%-40%) or paraneoplastic AAG (10%-20%).5 Anti-gnAChR antibodies are not specific to AAG and have been identified in low levels in up to 20% of patients with thymomas, postural orthostatic tachycardia syndrome, chronic idiopathic anhidrosis, idiopathic gastrointestinal dysmotility, Lambert–Eaton syndrome, and myasthenia gravis without thymoma.1,5-7 These associations raise the question of shared pathology and perhaps a syndrome overlap. Individuals with seropositive AAG may also have other paraneoplastic antibodies, making it clinically indistinguishable from paraneoplastic autonomic neuropathy.5,8 Although the autoantibody lacks sensitivity and is imperfectly specific, its presence supports a diagnosis of AAG. Anti-gnAChR antibodies have been shown to be pathological in rabbit and mouse models.4 In patients with AAG, higher autoantibody titers correlate with increased disease severity.1,6,7 A decrease in autoantibody titers correlates with decreased disease severity.6 Case report series also described a distinct entity of seronegative AAG.2,3 Maintaining a high clinical suspicion for AAG even with negative antibodies is important.
Given the rarity of the disease, no standard therapeutic regimens are available. About one-third of individuals improve on their own, while other individuals require extensive immunomodulation and symptom management. Case series and observational trials currently make up the vast array of treatment data. Therapies include glucocorticoids, plasmapheresis, IVIG, and other immunosuppressive agents, such as rituximab.9-12 Patients with and without identified anti-gnAChRs antibodies may respond to therapy.12 The overall long-term prognosis of the disease is poorly characterized.9,10,13
Despite the rarity of the syndrome discussed, this case represents how diagnostic reasoning strategies, such as law of parsimony, shift how the case is framed. For example, a middle-aged man with several new, distinctly unrelated diagnoses versus a middle-aged man with signs and symptoms of autonomic failure alters the subsequent clinical reasoning and diagnostic approach. Many diseases, both common and rare, are associated with dysautonomia. Therefore, clinicians should have an approach to autonomic failure
TEACHING POINTS
- Recognize the following signs and symptoms suggesting a dysautonomic syndrome: orthostasis, syncope, anhidrosis, xerophthalmia, xerostomia, impaired pupillary constriction, blurry vision, photophobia, erectile dysfunction, urinary retention, gastroparesis, constipation, neurogenic bowel obstruction, and dysgeusia.
- Recognize the clinical features, diagnostic approach, and management of autoimmune autonomic ganglionopathy.
- When faced with a complex clinical presentation, early application of the “law of parsimony” may help identify a unifying syndrome.
Acknowledgments
The authors wish to thank our Blinded Expert, Anthony Montanaro, MD, for his expertise and guidance during this process.
Disclosures
There are no known conflicts of interest.
A 44-year-old previously healthy semiprofessional male athlete presented with five days of nausea, vomiting, and abdominal pain. He had also experienced several months of decreased energy and new episodes of constipation three weeks prior to presentation.
At this point, we do not have sufficient information to completely determine the cause of his abdominal symptoms. Common causes of abdominal pain and vomiting in adults of his age group include peptic ulcer disease, pancreatic or hepatobiliary track disorders, small or large bowel processes, appendicitis, or even renal pathology. Further characterization may be possible by describing the location and quality of pain and factors that might relieve or exacerbate his pain. Despite the ambiguity, multiple clues might allow us to narrow the broad differential diagnosis of abdominal pain. In a previously healthy, vigorous, middle-aged man with subacute abdominal pain associated with constipation, the differential diagnosis should include disease states that may cause a bowel obstruction; these states include inflammatory bowel disease (IBD), gastrointestinal malignancy, or peptic ulcer disease. Mechanical obstruction due to volvulus or intussusception would be less likely in his age group. Given his history of several months of fatigue and several weeks of constipation, he should be evaluated for metabolic causes of abdominal pain and constipation, such as hypothyroidism or hypercalcemia. In addition to basic laboratory and imaging studies, obtaining additional history regarding prior abdominal surgeries, medication use, alcohol intake, and family and travel history will be the key in directing the evaluation.
Six months prior to admission, the patient began to feel more fatigue and exercise intolerance, reduced sweating, increased cold intolerance, and increased presyncopal episodes. He was diagnosed with hypothyroidism (TSH 6.69 μIU/mL; free T4 not done) and initiated on levothyroxine. One month prior to presentation, he developed constipation, loss of taste, reduced appetite, and weight loss of 30 pounds. He developed blurry vision and photophobia. He also complained of erectile dysfunction, urinary hesitancy and straining, which were diagnosed as benign prostatic hypertrophy.
Given the addition of numerous historical features in a previously healthy man, it is important to strive for a parsimonious diagnosis to unify his seemingly disparate features. His fatigue, constipation, and cold intolerance are consistent with his diagnosis of hypothyroidism but are nonspecific. Whether the degree of hypothyroidism caused his symptoms or signs is doubtful. The constellation of symptoms and signs are more likely to be representative of a nonthyroidal illness. His abdominal pain, unexplained weight loss, and presyncopal episodes should raise consideration of adrenal insufficiency. The combination of hypothyroidism and adrenal insufficiency suggest the possibility of an autoimmune polyendocrine syndrome or other pituitary pathology. In this case, history of headache, dysgeusia, and visual disturbances might support the diagnosis of pituitary adenoma. A cosyntropin stimulation test could establish the diagnosis of adrenal insufficiency. A low ACTH level would establish a diagnosis of pituitary or hypothalamic hypofunction. If pituitary hypofunction is documented, then a brain MRI would be needed to confirm the diagnosis of pituitary adenoma.
His newly reported erectile dysfunction suggests the possibility of a psychiatric, neurologic, hormonal, or vascular process and should be explored further. Sexual dysfunction is also associated with adrenal insufficiency and hypopituitarism. However, the presence of suspected prostatic hypertrophy in a male competitive athlete in his forties also raises the question of exogenous androgen use.
His past medical history was notable for a two-year history of alopecia totalis, seasonal allergies, asthma, and a repaired congenital aortic web with known aortic insufficiency. He was married with two children, worked an office job, and had no history of injection drug use, blood transfusions, or multiple sexual partners. His family history was notable for hypothyroidism and asthma in several family members in addition to Crohn disease, celiac disease, diabetes, cardiovascular disease, and cancers of the breast and lung.
His past medical, surgical, and family history supports a diagnosis of autoimmune disease. Although there is a personal and family history of atopic disorders, including allergic rhinitis and asthma, no association is found between atopy and autoimmunity. His family history of hypothyroidism, Crohn disease, and diabetes suggests a familial autoimmune genetic predisposition. His history of alopecia totalis in the setting of hypothyroidism and possible autoimmune adrenal insufficiency or autoimmune hypophysitis raises suspicion for the previously suggested diagnosis of polyglandular autoimmune syndrome, also known as autoimmune polyendocrine syndrome. Type I polyglandular autoimmune syndrome is associated with hypoparathyroidism and mucocutaneous candidiasis. In the absence of these symptoms, the patient more likely has type II polyglandular autoimmune syndrome. Type II syndrome is more prevalent and can occur in the setting of other nonendocrine autoimmune disorders, such as vitiligo, myasthenia gravis, or rheumatoid arthritis. Adrenal insufficiency can be the initial and most prominent manifestation of type II syndrome.
On physical exam, he was afebrile, with a heart rate of 68 beats per minute, respiratory rate of 16 breaths per minute, and normal oxygen saturation. His supine blood pressure and heart rate were 116/72 mm Hg and 66 beats per minute, respectively, and his standing blood pressure and heart rates were 80/48 mm Hg and 68 beats per minute respectively. He was thin, had diffuse scalp and body alopecia, and was ill-appearing with dry skin and dry mucous membranes. No evidence of Osler nodes, Janeway lesions, or splinter hemorrhages were found on cutaneous examination. No Roth spots or conjunctival hemorrhages were noted on ophthalmologic examination. He had both a 3/6 crescendo–decrescendo systolic murmur best heard at the right clavicle and radiated to the carotids and a 3/6 early diastolic decrescendo murmur best heard at the left sternal border. His abdomen was slightly protuberant, with reduced bowel sounds, hyperresonant to tympanitic on percussion, and a diffusely, moderately tender without peritoneal signs. Neurologic examination revealed 8 mm pupils with minimal response to light and accommodation. The remaining portions of his cranial nerve and complete neurologic examination were normal.
The presence of postural hypotension supports the previous suspicion of adrenal insufficiency, and the possibility of a pituitary or hypothalamic process remains. However, his dilated and minimally responsive pupils and potentially adynamic bowel are inconsistent with these diagnoses. Mydriasis and adynamic bowel in combination with orthostatic hypotension, dysgeusia, urinary retention, and erectile dysfunction are strongly suggestive of an autonomic process. Endocarditis is worth considering given his multisystem involvement, subacute decline, and known valve pathology. The absence of fever or stigmata of endocarditis make it difficult to explain his clinical syndrome. An echocardiogram would be reasonable for further assessment. At this point, it is prudent to explore his adrenal and pituitary function; if unrevealing, embark on an evaluation of his autonomic dysfunction.
Initial laboratory investigations were notable for mild normocytic anemia and hypoalbuminemia. His cosyntropin stimulation test was normal at 60 minutes. An abdominal CT scan demonstrated marked dilation in the small bowel loops (6 cm in caliber) with associated small bowel wall thickening and hyperemia. The echocardiogram was unrevealing and only confirmed the ongoing, progression of his known valve pathology without evidence of vegetation.
The above testing rules out primary adrenal insufficiency, but an appropriate response to the cosyntropin stimulation test does not rule out secondary, or pituitary, adrenal insufficiency. The echocardiogram and lack of other features make infective endocarditis unlikely. Thus, as mentioned, it is important now to commence a complete work-up of his probable dysautonomia to explain the majority of his features. Additionally, his hypothyroidism (if more than sick euthyroid syndrome), family history of autoimmune processes, and alopecia totalis all suggest the possibility of an immune-related syndrome. His CT scan revealed some thickened hyperemic bowel, which could suggest an IBD, such as Crohn disease; however, the absence of other signs, such as fever, diarrhea, or bloody stools, argues against this diagnosis. A syndrome that could unify his presentation is autoimmune autonomic ganglionopathy (AAG), a rare genetic autonomic system disorder that presents with pandysautonomia. The spectrum of autoimmunity was considered early in this case, but the differential diagnosis included more common conditions, such as adrenal insufficiency. Similarly, IBD remains a consideration. The serologic studies for IBD can be useful but they lack definitive diagnostic accuracy. Given that treatment for AAG differs from that for IBD, additional information will help guide the therapeutic approach. Anti-α3gnAChR antibodies, which are associated with AAG, should be checked.
His history of presyncope, anhidrosis, urinary retention, and ileus raised suspicion for pandysautonomia, as characterized by signs of sympathetic and parasympathetic dysfunction. The suspicion for pandysautonomia was confirmed via specialized autonomic testing, which included reduced heart rate variation on Valsalva and deep breathing maneuvers, orthostatic hypotension consistent autonomic insufficiency on Tilt table testing, and reduced sweat response to acetylcholine application (QSART test). The patient underwent further diagnostic serologic testing to differentiate causes of autonomic failure (Table 1). His personal and family history of autoimmunity led to the working diagnosis of AAG. Ultimate testing revealed high titers of autoantibodies, specifically anti-α3gnAChR (3.29 nmol/L, normal <0.02 nmol/L), directed against the ganglionic nicotinic acetylcholine receptor. This finding strongly supported the diagnosis of AAG.1,4-7
He was initially treated empirically with intravenous immunoglobulin (IVIG) with minimal improvement. He received additional immunomodulating therapies including methylprednisolone, plasmapheresis, and rituximab but did not tolerate a trial of mycophenolate. Six weeks after therapy initiation, his antibody titers decreased to 0.89 nmol/L with associated clinical improvement. Ultimately, he was discharged from the hospital on day 73 with a feeding tube and supplemental total parenteral nutrition. Four months postdischarge, he had returned to his prediagnosis weight, had eased back into his prior activities, and was off supplemental nutrition. Over a year later, he completed a 10-month prednisone taper and continued to receive monthly IVIG infusion
DISCUSSION
The clinical approach to dysautonomia is based on different etiologies: (1) those associated with neurodegenerative disorders; (2) those associated with peripheral neuropathies, and (3) isolated autonomic failure.2 Thus, clinical history and physical examination can assist greatly in guiding the evaluation of patients. Neurodegenerative disorders (such as Parkinson disease), combined disorders (such as multiple-system atrophy), and acquired or familial processes were considered. Our patient had neither a personal or family history nor physical examination supporting a neurodegenerative disorder. Disorders of the peripheral nerves were considered and can broadly be categorized as chronic sensorimotor neuropathies, sensory ganglionopathies, distal painful neuropathies, and acute or subacute motor polyradiculopathies. During evaluation, no historical, physical examination, or laboratory findings supported diabetes, amyloidosis, heavy metals, Sjögren syndrome, paraneoplastic neuropathy, sodium channel disorders, infectious etiologies, or porphyria (Table 1). Thus, in the absence of supportive evidence for primary neurodegenerative disorders or peripheral neuropathies, his syndrome appeared most compatible with an isolated autonomic failure syndrome. The principal differential for this syndrome is pure autonomic failure versus an immune-mediated autonomic disorder, including paraneoplastic autoimmune neuropathy and AAG. The diagnosis of pure autonomic failure is made after there is no clear unifying syndrome after more than five years of investigation. After exploration, no evidence of malignancy was discovered on body cross sectional imaging, PET scanning, bone marrow biopsy, colonoscopy, or laboratory testing. Thus, positive serologic testing in the absence of an underlying malignancy suggests a diagnosis of AAG.
AAG was first described in 1969 and is a rare, acquired disorder characterized by combined failure of the parasympathetic, sympathetic, and enteric nervous systems. This disorder typically presents in young-to-middle aged patients but has been described in all age groups. It is more commonly seen in patients with coexistent autoimmune diseases and/or a history of familial autoimmunity. The onset of clinical AAG may be subacute (less than three months) or insidious (more than three months). Patients present with signs or symptoms of pandysautonomia, such as severe orthostatic hypotension, syncope, constipation and gastrointestinal dysmotility, urinary retention, fixed and dilated pupils, and dry mouth and eyes (Table 2). Up to 40% of patients with AAG may also have significant cognitive impairment.3,4 Diagnosis relies on a combination of typical clinical features as discussed above and the exclusion of other diagnostic considerations. Diagnosis of AAG is aided by the presence of autoantibodies to ganglionic nicotinic acetylcholine receptors (gnAChR), particularly antiganglionic acetylcholine receptor α3 (anti-α3gAChR).1 Anti-gnAChR antibodies are only present in about half of patients with AAG. Antibody titers are highest in subacute AAG (40%-50%)3 compared with chronic AAG (30%-40%) or paraneoplastic AAG (10%-20%).5 Anti-gnAChR antibodies are not specific to AAG and have been identified in low levels in up to 20% of patients with thymomas, postural orthostatic tachycardia syndrome, chronic idiopathic anhidrosis, idiopathic gastrointestinal dysmotility, Lambert–Eaton syndrome, and myasthenia gravis without thymoma.1,5-7 These associations raise the question of shared pathology and perhaps a syndrome overlap. Individuals with seropositive AAG may also have other paraneoplastic antibodies, making it clinically indistinguishable from paraneoplastic autonomic neuropathy.5,8 Although the autoantibody lacks sensitivity and is imperfectly specific, its presence supports a diagnosis of AAG. Anti-gnAChR antibodies have been shown to be pathological in rabbit and mouse models.4 In patients with AAG, higher autoantibody titers correlate with increased disease severity.1,6,7 A decrease in autoantibody titers correlates with decreased disease severity.6 Case report series also described a distinct entity of seronegative AAG.2,3 Maintaining a high clinical suspicion for AAG even with negative antibodies is important.
Given the rarity of the disease, no standard therapeutic regimens are available. About one-third of individuals improve on their own, while other individuals require extensive immunomodulation and symptom management. Case series and observational trials currently make up the vast array of treatment data. Therapies include glucocorticoids, plasmapheresis, IVIG, and other immunosuppressive agents, such as rituximab.9-12 Patients with and without identified anti-gnAChRs antibodies may respond to therapy.12 The overall long-term prognosis of the disease is poorly characterized.9,10,13
Despite the rarity of the syndrome discussed, this case represents how diagnostic reasoning strategies, such as law of parsimony, shift how the case is framed. For example, a middle-aged man with several new, distinctly unrelated diagnoses versus a middle-aged man with signs and symptoms of autonomic failure alters the subsequent clinical reasoning and diagnostic approach. Many diseases, both common and rare, are associated with dysautonomia. Therefore, clinicians should have an approach to autonomic failure
TEACHING POINTS
- Recognize the following signs and symptoms suggesting a dysautonomic syndrome: orthostasis, syncope, anhidrosis, xerophthalmia, xerostomia, impaired pupillary constriction, blurry vision, photophobia, erectile dysfunction, urinary retention, gastroparesis, constipation, neurogenic bowel obstruction, and dysgeusia.
- Recognize the clinical features, diagnostic approach, and management of autoimmune autonomic ganglionopathy.
- When faced with a complex clinical presentation, early application of the “law of parsimony” may help identify a unifying syndrome.
Acknowledgments
The authors wish to thank our Blinded Expert, Anthony Montanaro, MD, for his expertise and guidance during this process.
Disclosures
There are no known conflicts of interest.
1. Gibbons C, Freeman R. Antibody titers predict clinical features of autoimmune autonomic ganglionopathy. Auton Neurosci. 2009;146(1-2):8-12. doi: 10.1016/j.autneu.2008.11.013. PubMed
2. Golden E, Bryarly M, Vernino S. Seronegative autoimmune autonomic neuropathy: a distinct clinical entity. Clin Auton Res. 2018;28(1):115-123. doi: 10.1007/s10286-017-0493-8. PubMed
3. Sandroni P, Vernino S, Klein CM, et al. Idiopathic autonomic neuropathy: comparison of cases seropositive and seronegative for ganglionic acetylcholine receptor antibody. Arch Neurol. 2004;61(1):44-48. doi: 10.1001/archneur.61.1.44. PubMed
4. Vernino S, Ermilov L, Sha L, Szurszewski J, Low P, Lennon V. Passive transfer of autoimmune autonomic neuropathy to mice. J Neurosci. 2004;24(32):7037-7042. doi: 10.1523/JNEUROSCI.1485-04.2004. PubMed
5. Vernino S, Hopkins S, Wang Z. Autonomic ganglia, acetylcholine receptor antibodies, and autoimmune ganglionopathy. Auton Neurosci. 2009;146(1-2):3-7. doi: 10.1016/j.autneu.2008.09.005. PubMed
6. Vernino S, Low P, Fealey R, Stewart J, Farrugia G, Lennon V. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343(12):847-855. doi: 10.1056/NEJM200009213431204. PubMed
7. Gibbons C, Vernino S, Freeman R. Autoimmune autonomic ganglionopathy – Symptom antibody correlations. Auton Neurosci. 2015;192:130. doi: 10.1016/j.autneu.2015.07.241 .
8. Benarroch E. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol. 2014;10(7):396-407. doi: 10.1038/nrneurol.2014.88. PubMed
9. Baker SK, Morillo C, Vernino S. Autoimmune autonomic ganglionopathy with late-onset encephalopathy. Auton Neurosci. 2009;146(1-2):29-32. doi: 10.1016/j.autneu.2008.10.016. PubMed
10. Gibbons C, Centi J, Vernino S. Autoimmune autonomic ganglionoapthy with reversible cognitive impairment. Arch Neurol. 2012;69(4):461-466. doi: 10.1001/archneurol.2011.2372. PubMed
11. Boydston E, Muppidi S, Vernino S. Long-term outcomes in autoimmune autonomic ganglionopathy (P05.210). Neurology. 2012;78(1):P05.210. doi: 10.1212/WNL.78.1_MeetingAbstracts.P05.210.
12. Gehrking T, Sletten D, Fealey R, Low P, Singer W. 11-year follow-up of a case of autoimmune autonomic ganglionopathy (P03.024). Neurology. 2013;80(7):P03.024.
13. Imrich R, Vernino S, Eldadah BA, Holmes C, Goldstein DS. Autoimmune autonomic ganglionopathy: treatment by plasma exchanges and rituximab. Clin Auton Res. 2009;19(4):259-262. doi: 10.1007/s10286-009-0012-7. PubMed
14. Iodice V, Kimpinski K, Vernino S, Sandroni P, Fealey RD, Low PA. Efficacy of immunotherapy in seropositive and seronegative putative autoimmune autonomic ganglionopathy. Neurology. 2009;72(23):2002-8. doi: 10.1212/WNL.0b013e3181a92b52. PubMed
15. Hayashi M, Ishii Y. A Japanese case of autoimmune autonomic ganglionopathy (AAG) and a review of AAG cases in Japan. Auton Neurosci. 2009;146(1-2):26-8. doi: 10.1016/j.autneu.2008.12.013. PubMed
16. Baker, A. Simplicity. In: Baker A, Zalta E, eds. The Stanford Encyclopedia of Philosophy. Winter 2016 Edition. https://plato.stanford.edu/archives/win2016/entries/simplicity/. Accessed October 26, 2017.
1. Gibbons C, Freeman R. Antibody titers predict clinical features of autoimmune autonomic ganglionopathy. Auton Neurosci. 2009;146(1-2):8-12. doi: 10.1016/j.autneu.2008.11.013. PubMed
2. Golden E, Bryarly M, Vernino S. Seronegative autoimmune autonomic neuropathy: a distinct clinical entity. Clin Auton Res. 2018;28(1):115-123. doi: 10.1007/s10286-017-0493-8. PubMed
3. Sandroni P, Vernino S, Klein CM, et al. Idiopathic autonomic neuropathy: comparison of cases seropositive and seronegative for ganglionic acetylcholine receptor antibody. Arch Neurol. 2004;61(1):44-48. doi: 10.1001/archneur.61.1.44. PubMed
4. Vernino S, Ermilov L, Sha L, Szurszewski J, Low P, Lennon V. Passive transfer of autoimmune autonomic neuropathy to mice. J Neurosci. 2004;24(32):7037-7042. doi: 10.1523/JNEUROSCI.1485-04.2004. PubMed
5. Vernino S, Hopkins S, Wang Z. Autonomic ganglia, acetylcholine receptor antibodies, and autoimmune ganglionopathy. Auton Neurosci. 2009;146(1-2):3-7. doi: 10.1016/j.autneu.2008.09.005. PubMed
6. Vernino S, Low P, Fealey R, Stewart J, Farrugia G, Lennon V. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343(12):847-855. doi: 10.1056/NEJM200009213431204. PubMed
7. Gibbons C, Vernino S, Freeman R. Autoimmune autonomic ganglionopathy – Symptom antibody correlations. Auton Neurosci. 2015;192:130. doi: 10.1016/j.autneu.2015.07.241 .
8. Benarroch E. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol. 2014;10(7):396-407. doi: 10.1038/nrneurol.2014.88. PubMed
9. Baker SK, Morillo C, Vernino S. Autoimmune autonomic ganglionopathy with late-onset encephalopathy. Auton Neurosci. 2009;146(1-2):29-32. doi: 10.1016/j.autneu.2008.10.016. PubMed
10. Gibbons C, Centi J, Vernino S. Autoimmune autonomic ganglionoapthy with reversible cognitive impairment. Arch Neurol. 2012;69(4):461-466. doi: 10.1001/archneurol.2011.2372. PubMed
11. Boydston E, Muppidi S, Vernino S. Long-term outcomes in autoimmune autonomic ganglionopathy (P05.210). Neurology. 2012;78(1):P05.210. doi: 10.1212/WNL.78.1_MeetingAbstracts.P05.210.
12. Gehrking T, Sletten D, Fealey R, Low P, Singer W. 11-year follow-up of a case of autoimmune autonomic ganglionopathy (P03.024). Neurology. 2013;80(7):P03.024.
13. Imrich R, Vernino S, Eldadah BA, Holmes C, Goldstein DS. Autoimmune autonomic ganglionopathy: treatment by plasma exchanges and rituximab. Clin Auton Res. 2009;19(4):259-262. doi: 10.1007/s10286-009-0012-7. PubMed
14. Iodice V, Kimpinski K, Vernino S, Sandroni P, Fealey RD, Low PA. Efficacy of immunotherapy in seropositive and seronegative putative autoimmune autonomic ganglionopathy. Neurology. 2009;72(23):2002-8. doi: 10.1212/WNL.0b013e3181a92b52. PubMed
15. Hayashi M, Ishii Y. A Japanese case of autoimmune autonomic ganglionopathy (AAG) and a review of AAG cases in Japan. Auton Neurosci. 2009;146(1-2):26-8. doi: 10.1016/j.autneu.2008.12.013. PubMed
16. Baker, A. Simplicity. In: Baker A, Zalta E, eds. The Stanford Encyclopedia of Philosophy. Winter 2016 Edition. https://plato.stanford.edu/archives/win2016/entries/simplicity/. Accessed October 26, 2017.
© 2018 Society of Hospital Medicine
A Model to Improve Hospital-Based Palliative Care: The Palliative Care Redistribution Integrated System Model (PRISM)
Palliative care is an essential component of inpatient medicine. At its core, it is an interdisciplinary philosophy of care aiming to achieve the best quality of life for patients and families in the physical, psychosocial, and spiritual domains. With the aging population and growing complexity of hospitalized patients, inpatient palliative care needs are only projected to rise. However, a mismatch exists between the number of palliative care–trained physicians and the demand for such physicians. Currently, only 6,600 US physicians are board certified in palliative care – just 37% of the projected need.1 These workforce shortages have serious implications. In fact, it is estimated that nearly 40% of all hospitalized patients who need palliative care go without it.2
Existing efforts to improve access to palliative care have largely focused on bolstering the palliative care workforce. One tactic particularly relevant to hospitalists centers on frontline physicians providing “primary” palliative care: basic symptom management, patient-centered communication, and goals of care assessment, regardless of the disease state.3 Such physicians constitute the base of today’s palliative care workforce model – a three-tiered pyramid built on clinician availability and skills. In this model, the second tier (“secondary” palliative care) includes physicians supported by a palliative care consultant or referral. The third level (“tertiary” palliative care) encompasses care provided directly by specialized palliative care teams, usually within academic medical centers (Figure).4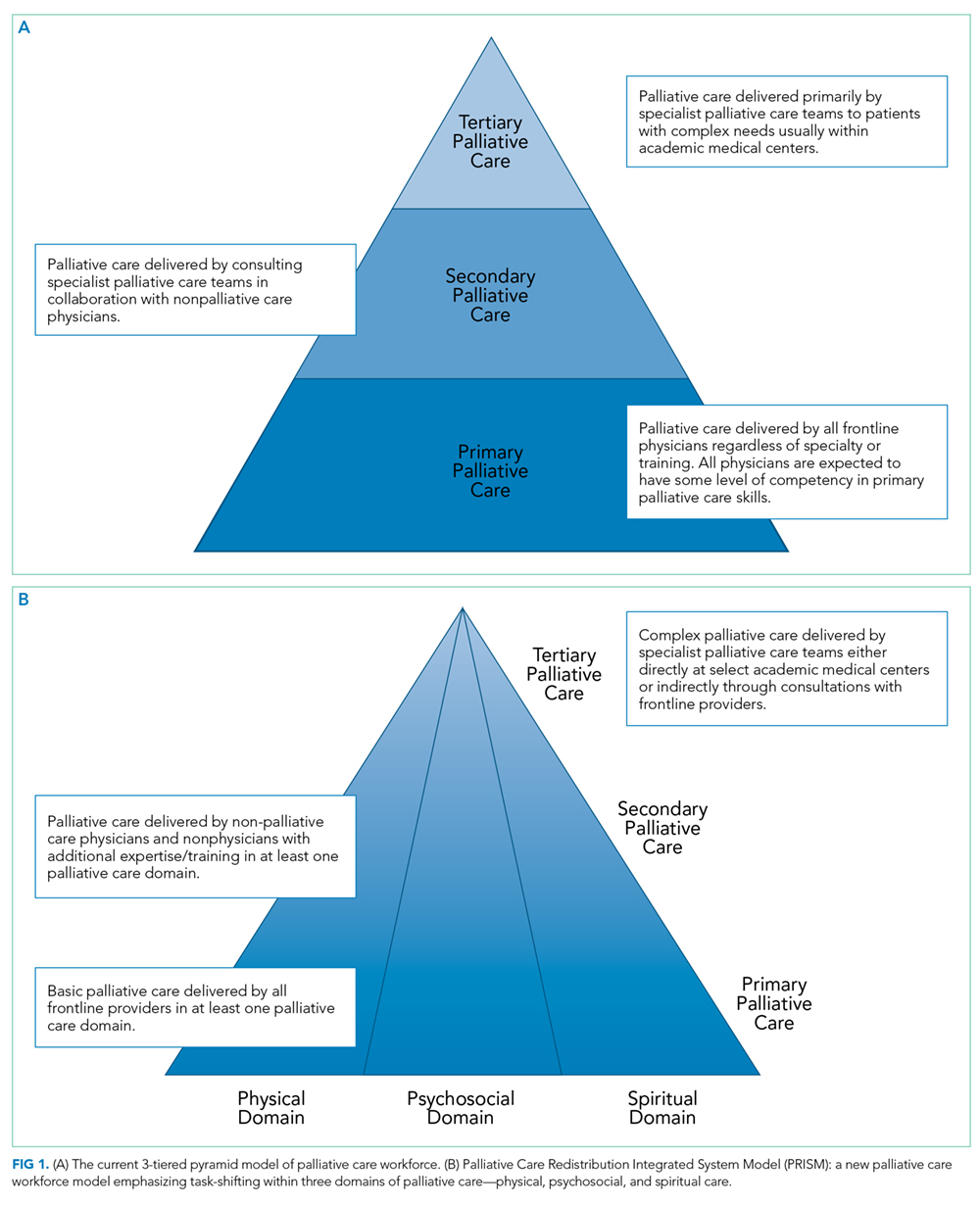
The practice of primary palliative care is central to the practice of hospital medicine.5,6 After all, hospitalists generate nearly half of all inpatient palliative care consultations7 and routinely interface with social workers, pharmacists, nurses, chaplains, and other consultants in their daily activities. Consequently, they are also well versed in serious illness communication and prognostication.8 In many ways, they are ideal purveyors of palliative care in the hospital.
Why then does the challenge to meet the demands of patients with palliative care needs persist? The truth may lie in at least three central shortcomings within the tiered palliative care workforce model. First, physicians comprising the base (where hospitalists typically fall) possess variable skills and knowledge in caring for seriously ill patients. While training opportunities exist for interested individuals,7 education alone can rarely achieve a systematic change. Second, some physicians may have the requisite skills but lack
The Palliative Care Redistribution Integrated Service Model (PRISM)
To better address the current palliative care access problem, we propose a new model: “The Palliative care Redistribution Integrated Service Model (PRISM; Figure 1).” Using the industrial engineering principle of “task shifting,” this approach leverages disciplinary diversity and shifts specific activities from more specialized to less specialized members.9 In this way, PRISM integrates hospital-based interdisciplinary teams across all tiers of palliative care delivery.
PRISM sheds a tier-based approach in favor of flexible, skill-based verticals that span all physician and nonphysician providers. By dividing the original pyramid into three domains – physical, psychosocial, and spiritual – providers with various spheres of expertise may serve patients on multiple tiers. For example, a bedside nurse may perform basic psychosocial assessment consistent with his or her training, while physicians may focus on code status or prescribe antiemetics or low-dose opiate monotherapy – skills they have refined during medical school. Analogously, secondary palliative care may be delivered by any provider with more advanced skills in communication or symptom management. In this way, we expand the pool of clinicians available to provide palliative care to include nurses, hospitalists, oncologists, intensivists, social workers, and chaplains and also recognize the diversity of skill sets within and between disciplines. Thus, a hospitalist may clarify the goals of care but may ask a social worker trained in psychosocial assessment for assistance with difficult family dynamics or a chaplain for spiritual needs. Interdisciplinary teamwork and cross-disciplinary communication – hallmarks of palliative care – are encouraged and valued. Furthermore, if providers feel uncomfortable providing a certain type of care, they can ask for assistance from more experienced providers within their discipline or outside of it. In rare cases, the most complex patients may be referred to specialist palliative care teams.
Inherent within PRISM is a recognition that all providers must have a basic palliative care skillset obtained through educational initiatives.7 Yet focusing solely on training the workforce as a strategy has and will continue to miss the mark. Rather, structural changes to the means of providing care are also needed. Within hospitals, these changes often rely heavily on hospitalists due to their central position in care delivery. In this way, hospitalists are well primed to be the agents of change in this model.
The Role of Technology
Since many hospitalized patients have unrecognized and underserved palliative care needs, a formal approach to assessment is needed. Lin et al. proposed criteria for a “sentinel hospitalization,” marking a major illness or transition in high-risk patients necessitating palliative interventions.10 Similar screening criteria have been validated among hospitalized oncology patients11 and in critical care.12 While checklists have been shown to help identify hospitalized patients with palliative care needs,13 their implementation has been slow, presumably because they are burdensome for busy providers to complete.
Technological automation may be a solution to the checklist conundrum. For example, if palliative care screening criteria could be automatically extracted from electronic health records, scoring systems could trigger hospitalists to consider the goals of care discussions or engage an interdisciplinary care team to fulfill a variety of needs. Frameworks for such scoring systems already exist and are familiar to most hospitalists. For example, admission order sets routinely calculate the Padua or Caprini score to facilitate decision-making for prophylaxis of deep vein thrombosis. An admission order set that screens and prompts decision-making around palliative care needs is thus feasible. One example is a hard stop for entering code status in the admission order set; in turn, this hard stop could also trigger providers to complete a “check-box” palliative care screening checklist. Automatic extraction of certain data from the record – such as age, prior code status, recent hospitalizations, or mobility scores – could auto-populate to facilitate decision-making. In turn, measuring the influence of such tools on access to palliative care, workflow, and capacity will be important, as most tools may not have quality or value intended.14
Streamlining Workflow
It is common for hospitalists to oversee care for 15-20 patients at a time. Thus, they may not have the time to meaningfully engage patients to assess palliative care needs. Creating designated hospitalist palliative care teams with enhanced interdisciplinary support for patients identified using sentinel hospitalization or checklist-based tools may help to solve this dilemma. These teams may also employ lower “caps,” freeing up time for critical discussions and planning around end of life. At the University of Michigan, we are planning just such an approach, a strategy which has the additional benefit of bypassing the binary “care versus no care” dilemma faced by patients choosing palliation. Rather, patients may continue to receive treatments congruent with the goals of care in such teams.
Making Palliative Care a Standard of Care
A call for health systems to develop and implement palliative care quality metrics has emerged. Given their role in quality improvement and health system reform, hospitalists are well positioned to shepherd this imperative. Creating incentives to screen inpatients for palliative care needs and develop new homes in which to care for these patients are but a few ways to help set the tone. Additionally, developing and sharing quality metrics and benchmarks currently captured in repositories such as the Palliative Care Quality Network, Global Palliative Care Quality Alliance, and Center to Advance Palliative Care can help to assess and continually improve care delivery. Creating and sharing dashboards from these metrics with all providers, regardless of discipline or training, will ensure accountability to deliver quality palliative care.
CONCLUSION
Many hospitalized patients do not receive appropriate attention to their palliative care needs. A new interdisciplinary workforce model that task shifts to physician and nonphysician providers and pairs system-level innovations and quality may solve this problem. Input and endorsement from a wide variety of disciplines (particularly our nonphysician colleagues) are needed to make PRISM operational. The proof of concept will lie in testing feasibility among key stakeholders and rigorously studying the proposed interventions. Through innovation in technology, workflow, and quality improvement, hospitalists are well poised to lead this change. After all, our patients deserve nothing less.
Disclosures
The authors have nothing to disclose.Funding: Dr. Abedini’s work is supported by the University of Michigan National Clinician Scholars Program at the Institute for Healthcare Policy and Innovation, as well as the Un
1. Lupu D. American Academy of Hospice and Palliative Medicine Task Force. Estimate of current hospice and palliative medicine physician workforce shortage. J Pain Symptom Manage. 2010;40(6):899-911. doi: 10.1016/j.jpainsymman.2010.07.004. PubMed
2. Chuang E, Hope AA, Allyn K, Szalkiewicz E, Gary B, Gong MN. Gaps in provision of primary and specialty palliative care in the acute care setting by race and ethnicity. J Pain Symptom Manage. 2017;54(5):645-653. doi: 10.1016/j.jpainsymman.2017.05.001 PubMed
3. Quill TE, Abernethy AP. Generalist plus specialist palliative care--creating a more sustainable model. N Engl J Med. 2013;368(13):1173-1175. doi: 10.1056/NEJMp1215620 PubMed
4. von Gunten CF. Secondary and tertiary palliative care in US hospitals. JAMA. 2002;287(7):875-881. doi: 10.1001/jama.287.7.875 PubMed
5. Pantilat SZ. Hope to reality: the future of hospitalists and palliative care. J Hosp Med. 2015;10(10):701-702. doi: 10.1002/jhm.2401 PubMed
6. Meier DE. Palliative care in hospitals. J Hosp Med. 2006;1(1):21-28. doi: 10.1016/j.cger.2004.07.006 PubMed
7. Fail RE, Meier DE. Improving quality of care for seriously ill patients: Opportunities for hospitalists. J Hosp Med. 2018;13(3):194-197. doi: 10.12788/jhm.2896. [Epub ahead of print] PubMed
8. Rosenberg LB, Greenwald J, Caponi B, et al. Confidence with and barriers to serious illness communication: A national survey of hospitalists. J Palliat Med. 2017;20(9):1013-1019. doi: 10.1089/jpm.2016.0515 PubMed
9. Carayon P, Gurses AP. Nursing workload and patient safety–a human factors engineering perspective. In: Hughes RG, ed.Patient Safety and Quality: An Evidence-Based Handbook for Nurses. Rockville, MD: Agency for Healthcare Research and Quality (US); 2008. PubMed
10. Lin RJ, Adelman RD, Diamond RR, Evans AT. The sentinel hospitalization and the role of palliative care. J Hosp Med. 2014;9(5):320-323. doi: 10.1002/jhm.2160 PubMed
11. Glare PA, Chow K. Validation of a simple screening tool for identifying unmet palliative care needs in patients with cancer. J Oncol Pract. 2015;11(1):e81-e86. doi: 10.1200/JOP.2014.001487. PubMed
12. Zalenski RJ, Jones SS, Courage C, et al. Impact of a palliative care screening and consultation in the ICU: A multihospital quality improvement project. J Pain Symptom Manage. 2017;53(1):5-12.e3. doi: 10.1016/j.jpainsymman.2016.08.003. PubMed
13. Weissman DE, Meier DE. Identifying patients in need of palliative care assessment in the hospital setting: a consensus report from the Center to Advance Palliative Care. J Palliat Med. 2011;14(1):17-23. doi: PubMed
14. MacLean CH, Kerr EA, Qaseem A. Time out-charting a path for improving performance measurement. N Engl J Med. 2018. Epub ahead of print. doi: 10.1056/NEJMp1802595 PubMed
Palliative care is an essential component of inpatient medicine. At its core, it is an interdisciplinary philosophy of care aiming to achieve the best quality of life for patients and families in the physical, psychosocial, and spiritual domains. With the aging population and growing complexity of hospitalized patients, inpatient palliative care needs are only projected to rise. However, a mismatch exists between the number of palliative care–trained physicians and the demand for such physicians. Currently, only 6,600 US physicians are board certified in palliative care – just 37% of the projected need.1 These workforce shortages have serious implications. In fact, it is estimated that nearly 40% of all hospitalized patients who need palliative care go without it.2
Existing efforts to improve access to palliative care have largely focused on bolstering the palliative care workforce. One tactic particularly relevant to hospitalists centers on frontline physicians providing “primary” palliative care: basic symptom management, patient-centered communication, and goals of care assessment, regardless of the disease state.3 Such physicians constitute the base of today’s palliative care workforce model – a three-tiered pyramid built on clinician availability and skills. In this model, the second tier (“secondary” palliative care) includes physicians supported by a palliative care consultant or referral. The third level (“tertiary” palliative care) encompasses care provided directly by specialized palliative care teams, usually within academic medical centers (Figure).4
The practice of primary palliative care is central to the practice of hospital medicine.5,6 After all, hospitalists generate nearly half of all inpatient palliative care consultations7 and routinely interface with social workers, pharmacists, nurses, chaplains, and other consultants in their daily activities. Consequently, they are also well versed in serious illness communication and prognostication.8 In many ways, they are ideal purveyors of palliative care in the hospital.
Why then does the challenge to meet the demands of patients with palliative care needs persist? The truth may lie in at least three central shortcomings within the tiered palliative care workforce model. First, physicians comprising the base (where hospitalists typically fall) possess variable skills and knowledge in caring for seriously ill patients. While training opportunities exist for interested individuals,7 education alone can rarely achieve a systematic change. Second, some physicians may have the requisite skills but lack
The Palliative Care Redistribution Integrated Service Model (PRISM)
To better address the current palliative care access problem, we propose a new model: “The Palliative care Redistribution Integrated Service Model (PRISM; Figure 1).” Using the industrial engineering principle of “task shifting,” this approach leverages disciplinary diversity and shifts specific activities from more specialized to less specialized members.9 In this way, PRISM integrates hospital-based interdisciplinary teams across all tiers of palliative care delivery.
PRISM sheds a tier-based approach in favor of flexible, skill-based verticals that span all physician and nonphysician providers. By dividing the original pyramid into three domains – physical, psychosocial, and spiritual – providers with various spheres of expertise may serve patients on multiple tiers. For example, a bedside nurse may perform basic psychosocial assessment consistent with his or her training, while physicians may focus on code status or prescribe antiemetics or low-dose opiate monotherapy – skills they have refined during medical school. Analogously, secondary palliative care may be delivered by any provider with more advanced skills in communication or symptom management. In this way, we expand the pool of clinicians available to provide palliative care to include nurses, hospitalists, oncologists, intensivists, social workers, and chaplains and also recognize the diversity of skill sets within and between disciplines. Thus, a hospitalist may clarify the goals of care but may ask a social worker trained in psychosocial assessment for assistance with difficult family dynamics or a chaplain for spiritual needs. Interdisciplinary teamwork and cross-disciplinary communication – hallmarks of palliative care – are encouraged and valued. Furthermore, if providers feel uncomfortable providing a certain type of care, they can ask for assistance from more experienced providers within their discipline or outside of it. In rare cases, the most complex patients may be referred to specialist palliative care teams.
Inherent within PRISM is a recognition that all providers must have a basic palliative care skillset obtained through educational initiatives.7 Yet focusing solely on training the workforce as a strategy has and will continue to miss the mark. Rather, structural changes to the means of providing care are also needed. Within hospitals, these changes often rely heavily on hospitalists due to their central position in care delivery. In this way, hospitalists are well primed to be the agents of change in this model.
The Role of Technology
Since many hospitalized patients have unrecognized and underserved palliative care needs, a formal approach to assessment is needed. Lin et al. proposed criteria for a “sentinel hospitalization,” marking a major illness or transition in high-risk patients necessitating palliative interventions.10 Similar screening criteria have been validated among hospitalized oncology patients11 and in critical care.12 While checklists have been shown to help identify hospitalized patients with palliative care needs,13 their implementation has been slow, presumably because they are burdensome for busy providers to complete.
Technological automation may be a solution to the checklist conundrum. For example, if palliative care screening criteria could be automatically extracted from electronic health records, scoring systems could trigger hospitalists to consider the goals of care discussions or engage an interdisciplinary care team to fulfill a variety of needs. Frameworks for such scoring systems already exist and are familiar to most hospitalists. For example, admission order sets routinely calculate the Padua or Caprini score to facilitate decision-making for prophylaxis of deep vein thrombosis. An admission order set that screens and prompts decision-making around palliative care needs is thus feasible. One example is a hard stop for entering code status in the admission order set; in turn, this hard stop could also trigger providers to complete a “check-box” palliative care screening checklist. Automatic extraction of certain data from the record – such as age, prior code status, recent hospitalizations, or mobility scores – could auto-populate to facilitate decision-making. In turn, measuring the influence of such tools on access to palliative care, workflow, and capacity will be important, as most tools may not have quality or value intended.14
Streamlining Workflow
It is common for hospitalists to oversee care for 15-20 patients at a time. Thus, they may not have the time to meaningfully engage patients to assess palliative care needs. Creating designated hospitalist palliative care teams with enhanced interdisciplinary support for patients identified using sentinel hospitalization or checklist-based tools may help to solve this dilemma. These teams may also employ lower “caps,” freeing up time for critical discussions and planning around end of life. At the University of Michigan, we are planning just such an approach, a strategy which has the additional benefit of bypassing the binary “care versus no care” dilemma faced by patients choosing palliation. Rather, patients may continue to receive treatments congruent with the goals of care in such teams.
Making Palliative Care a Standard of Care
A call for health systems to develop and implement palliative care quality metrics has emerged. Given their role in quality improvement and health system reform, hospitalists are well positioned to shepherd this imperative. Creating incentives to screen inpatients for palliative care needs and develop new homes in which to care for these patients are but a few ways to help set the tone. Additionally, developing and sharing quality metrics and benchmarks currently captured in repositories such as the Palliative Care Quality Network, Global Palliative Care Quality Alliance, and Center to Advance Palliative Care can help to assess and continually improve care delivery. Creating and sharing dashboards from these metrics with all providers, regardless of discipline or training, will ensure accountability to deliver quality palliative care.
CONCLUSION
Many hospitalized patients do not receive appropriate attention to their palliative care needs. A new interdisciplinary workforce model that task shifts to physician and nonphysician providers and pairs system-level innovations and quality may solve this problem. Input and endorsement from a wide variety of disciplines (particularly our nonphysician colleagues) are needed to make PRISM operational. The proof of concept will lie in testing feasibility among key stakeholders and rigorously studying the proposed interventions. Through innovation in technology, workflow, and quality improvement, hospitalists are well poised to lead this change. After all, our patients deserve nothing less.
Disclosures
The authors have nothing to disclose.Funding: Dr. Abedini’s work is supported by the University of Michigan National Clinician Scholars Program at the Institute for Healthcare Policy and Innovation, as well as the Un
Palliative care is an essential component of inpatient medicine. At its core, it is an interdisciplinary philosophy of care aiming to achieve the best quality of life for patients and families in the physical, psychosocial, and spiritual domains. With the aging population and growing complexity of hospitalized patients, inpatient palliative care needs are only projected to rise. However, a mismatch exists between the number of palliative care–trained physicians and the demand for such physicians. Currently, only 6,600 US physicians are board certified in palliative care – just 37% of the projected need.1 These workforce shortages have serious implications. In fact, it is estimated that nearly 40% of all hospitalized patients who need palliative care go without it.2
Existing efforts to improve access to palliative care have largely focused on bolstering the palliative care workforce. One tactic particularly relevant to hospitalists centers on frontline physicians providing “primary” palliative care: basic symptom management, patient-centered communication, and goals of care assessment, regardless of the disease state.3 Such physicians constitute the base of today’s palliative care workforce model – a three-tiered pyramid built on clinician availability and skills. In this model, the second tier (“secondary” palliative care) includes physicians supported by a palliative care consultant or referral. The third level (“tertiary” palliative care) encompasses care provided directly by specialized palliative care teams, usually within academic medical centers (Figure).4
The practice of primary palliative care is central to the practice of hospital medicine.5,6 After all, hospitalists generate nearly half of all inpatient palliative care consultations7 and routinely interface with social workers, pharmacists, nurses, chaplains, and other consultants in their daily activities. Consequently, they are also well versed in serious illness communication and prognostication.8 In many ways, they are ideal purveyors of palliative care in the hospital.
Why then does the challenge to meet the demands of patients with palliative care needs persist? The truth may lie in at least three central shortcomings within the tiered palliative care workforce model. First, physicians comprising the base (where hospitalists typically fall) possess variable skills and knowledge in caring for seriously ill patients. While training opportunities exist for interested individuals,7 education alone can rarely achieve a systematic change. Second, some physicians may have the requisite skills but lack
The Palliative Care Redistribution Integrated Service Model (PRISM)
To better address the current palliative care access problem, we propose a new model: “The Palliative care Redistribution Integrated Service Model (PRISM; Figure 1).” Using the industrial engineering principle of “task shifting,” this approach leverages disciplinary diversity and shifts specific activities from more specialized to less specialized members.9 In this way, PRISM integrates hospital-based interdisciplinary teams across all tiers of palliative care delivery.
PRISM sheds a tier-based approach in favor of flexible, skill-based verticals that span all physician and nonphysician providers. By dividing the original pyramid into three domains – physical, psychosocial, and spiritual – providers with various spheres of expertise may serve patients on multiple tiers. For example, a bedside nurse may perform basic psychosocial assessment consistent with his or her training, while physicians may focus on code status or prescribe antiemetics or low-dose opiate monotherapy – skills they have refined during medical school. Analogously, secondary palliative care may be delivered by any provider with more advanced skills in communication or symptom management. In this way, we expand the pool of clinicians available to provide palliative care to include nurses, hospitalists, oncologists, intensivists, social workers, and chaplains and also recognize the diversity of skill sets within and between disciplines. Thus, a hospitalist may clarify the goals of care but may ask a social worker trained in psychosocial assessment for assistance with difficult family dynamics or a chaplain for spiritual needs. Interdisciplinary teamwork and cross-disciplinary communication – hallmarks of palliative care – are encouraged and valued. Furthermore, if providers feel uncomfortable providing a certain type of care, they can ask for assistance from more experienced providers within their discipline or outside of it. In rare cases, the most complex patients may be referred to specialist palliative care teams.
Inherent within PRISM is a recognition that all providers must have a basic palliative care skillset obtained through educational initiatives.7 Yet focusing solely on training the workforce as a strategy has and will continue to miss the mark. Rather, structural changes to the means of providing care are also needed. Within hospitals, these changes often rely heavily on hospitalists due to their central position in care delivery. In this way, hospitalists are well primed to be the agents of change in this model.
The Role of Technology
Since many hospitalized patients have unrecognized and underserved palliative care needs, a formal approach to assessment is needed. Lin et al. proposed criteria for a “sentinel hospitalization,” marking a major illness or transition in high-risk patients necessitating palliative interventions.10 Similar screening criteria have been validated among hospitalized oncology patients11 and in critical care.12 While checklists have been shown to help identify hospitalized patients with palliative care needs,13 their implementation has been slow, presumably because they are burdensome for busy providers to complete.
Technological automation may be a solution to the checklist conundrum. For example, if palliative care screening criteria could be automatically extracted from electronic health records, scoring systems could trigger hospitalists to consider the goals of care discussions or engage an interdisciplinary care team to fulfill a variety of needs. Frameworks for such scoring systems already exist and are familiar to most hospitalists. For example, admission order sets routinely calculate the Padua or Caprini score to facilitate decision-making for prophylaxis of deep vein thrombosis. An admission order set that screens and prompts decision-making around palliative care needs is thus feasible. One example is a hard stop for entering code status in the admission order set; in turn, this hard stop could also trigger providers to complete a “check-box” palliative care screening checklist. Automatic extraction of certain data from the record – such as age, prior code status, recent hospitalizations, or mobility scores – could auto-populate to facilitate decision-making. In turn, measuring the influence of such tools on access to palliative care, workflow, and capacity will be important, as most tools may not have quality or value intended.14
Streamlining Workflow
It is common for hospitalists to oversee care for 15-20 patients at a time. Thus, they may not have the time to meaningfully engage patients to assess palliative care needs. Creating designated hospitalist palliative care teams with enhanced interdisciplinary support for patients identified using sentinel hospitalization or checklist-based tools may help to solve this dilemma. These teams may also employ lower “caps,” freeing up time for critical discussions and planning around end of life. At the University of Michigan, we are planning just such an approach, a strategy which has the additional benefit of bypassing the binary “care versus no care” dilemma faced by patients choosing palliation. Rather, patients may continue to receive treatments congruent with the goals of care in such teams.
Making Palliative Care a Standard of Care
A call for health systems to develop and implement palliative care quality metrics has emerged. Given their role in quality improvement and health system reform, hospitalists are well positioned to shepherd this imperative. Creating incentives to screen inpatients for palliative care needs and develop new homes in which to care for these patients are but a few ways to help set the tone. Additionally, developing and sharing quality metrics and benchmarks currently captured in repositories such as the Palliative Care Quality Network, Global Palliative Care Quality Alliance, and Center to Advance Palliative Care can help to assess and continually improve care delivery. Creating and sharing dashboards from these metrics with all providers, regardless of discipline or training, will ensure accountability to deliver quality palliative care.
CONCLUSION
Many hospitalized patients do not receive appropriate attention to their palliative care needs. A new interdisciplinary workforce model that task shifts to physician and nonphysician providers and pairs system-level innovations and quality may solve this problem. Input and endorsement from a wide variety of disciplines (particularly our nonphysician colleagues) are needed to make PRISM operational. The proof of concept will lie in testing feasibility among key stakeholders and rigorously studying the proposed interventions. Through innovation in technology, workflow, and quality improvement, hospitalists are well poised to lead this change. After all, our patients deserve nothing less.
Disclosures
The authors have nothing to disclose.Funding: Dr. Abedini’s work is supported by the University of Michigan National Clinician Scholars Program at the Institute for Healthcare Policy and Innovation, as well as the Un
1. Lupu D. American Academy of Hospice and Palliative Medicine Task Force. Estimate of current hospice and palliative medicine physician workforce shortage. J Pain Symptom Manage. 2010;40(6):899-911. doi: 10.1016/j.jpainsymman.2010.07.004. PubMed
2. Chuang E, Hope AA, Allyn K, Szalkiewicz E, Gary B, Gong MN. Gaps in provision of primary and specialty palliative care in the acute care setting by race and ethnicity. J Pain Symptom Manage. 2017;54(5):645-653. doi: 10.1016/j.jpainsymman.2017.05.001 PubMed
3. Quill TE, Abernethy AP. Generalist plus specialist palliative care--creating a more sustainable model. N Engl J Med. 2013;368(13):1173-1175. doi: 10.1056/NEJMp1215620 PubMed
4. von Gunten CF. Secondary and tertiary palliative care in US hospitals. JAMA. 2002;287(7):875-881. doi: 10.1001/jama.287.7.875 PubMed
5. Pantilat SZ. Hope to reality: the future of hospitalists and palliative care. J Hosp Med. 2015;10(10):701-702. doi: 10.1002/jhm.2401 PubMed
6. Meier DE. Palliative care in hospitals. J Hosp Med. 2006;1(1):21-28. doi: 10.1016/j.cger.2004.07.006 PubMed
7. Fail RE, Meier DE. Improving quality of care for seriously ill patients: Opportunities for hospitalists. J Hosp Med. 2018;13(3):194-197. doi: 10.12788/jhm.2896. [Epub ahead of print] PubMed
8. Rosenberg LB, Greenwald J, Caponi B, et al. Confidence with and barriers to serious illness communication: A national survey of hospitalists. J Palliat Med. 2017;20(9):1013-1019. doi: 10.1089/jpm.2016.0515 PubMed
9. Carayon P, Gurses AP. Nursing workload and patient safety–a human factors engineering perspective. In: Hughes RG, ed.Patient Safety and Quality: An Evidence-Based Handbook for Nurses. Rockville, MD: Agency for Healthcare Research and Quality (US); 2008. PubMed
10. Lin RJ, Adelman RD, Diamond RR, Evans AT. The sentinel hospitalization and the role of palliative care. J Hosp Med. 2014;9(5):320-323. doi: 10.1002/jhm.2160 PubMed
11. Glare PA, Chow K. Validation of a simple screening tool for identifying unmet palliative care needs in patients with cancer. J Oncol Pract. 2015;11(1):e81-e86. doi: 10.1200/JOP.2014.001487. PubMed
12. Zalenski RJ, Jones SS, Courage C, et al. Impact of a palliative care screening and consultation in the ICU: A multihospital quality improvement project. J Pain Symptom Manage. 2017;53(1):5-12.e3. doi: 10.1016/j.jpainsymman.2016.08.003. PubMed
13. Weissman DE, Meier DE. Identifying patients in need of palliative care assessment in the hospital setting: a consensus report from the Center to Advance Palliative Care. J Palliat Med. 2011;14(1):17-23. doi: PubMed
14. MacLean CH, Kerr EA, Qaseem A. Time out-charting a path for improving performance measurement. N Engl J Med. 2018. Epub ahead of print. doi: 10.1056/NEJMp1802595 PubMed
1. Lupu D. American Academy of Hospice and Palliative Medicine Task Force. Estimate of current hospice and palliative medicine physician workforce shortage. J Pain Symptom Manage. 2010;40(6):899-911. doi: 10.1016/j.jpainsymman.2010.07.004. PubMed
2. Chuang E, Hope AA, Allyn K, Szalkiewicz E, Gary B, Gong MN. Gaps in provision of primary and specialty palliative care in the acute care setting by race and ethnicity. J Pain Symptom Manage. 2017;54(5):645-653. doi: 10.1016/j.jpainsymman.2017.05.001 PubMed
3. Quill TE, Abernethy AP. Generalist plus specialist palliative care--creating a more sustainable model. N Engl J Med. 2013;368(13):1173-1175. doi: 10.1056/NEJMp1215620 PubMed
4. von Gunten CF. Secondary and tertiary palliative care in US hospitals. JAMA. 2002;287(7):875-881. doi: 10.1001/jama.287.7.875 PubMed
5. Pantilat SZ. Hope to reality: the future of hospitalists and palliative care. J Hosp Med. 2015;10(10):701-702. doi: 10.1002/jhm.2401 PubMed
6. Meier DE. Palliative care in hospitals. J Hosp Med. 2006;1(1):21-28. doi: 10.1016/j.cger.2004.07.006 PubMed
7. Fail RE, Meier DE. Improving quality of care for seriously ill patients: Opportunities for hospitalists. J Hosp Med. 2018;13(3):194-197. doi: 10.12788/jhm.2896. [Epub ahead of print] PubMed
8. Rosenberg LB, Greenwald J, Caponi B, et al. Confidence with and barriers to serious illness communication: A national survey of hospitalists. J Palliat Med. 2017;20(9):1013-1019. doi: 10.1089/jpm.2016.0515 PubMed
9. Carayon P, Gurses AP. Nursing workload and patient safety–a human factors engineering perspective. In: Hughes RG, ed.Patient Safety and Quality: An Evidence-Based Handbook for Nurses. Rockville, MD: Agency for Healthcare Research and Quality (US); 2008. PubMed
10. Lin RJ, Adelman RD, Diamond RR, Evans AT. The sentinel hospitalization and the role of palliative care. J Hosp Med. 2014;9(5):320-323. doi: 10.1002/jhm.2160 PubMed
11. Glare PA, Chow K. Validation of a simple screening tool for identifying unmet palliative care needs in patients with cancer. J Oncol Pract. 2015;11(1):e81-e86. doi: 10.1200/JOP.2014.001487. PubMed
12. Zalenski RJ, Jones SS, Courage C, et al. Impact of a palliative care screening and consultation in the ICU: A multihospital quality improvement project. J Pain Symptom Manage. 2017;53(1):5-12.e3. doi: 10.1016/j.jpainsymman.2016.08.003. PubMed
13. Weissman DE, Meier DE. Identifying patients in need of palliative care assessment in the hospital setting: a consensus report from the Center to Advance Palliative Care. J Palliat Med. 2011;14(1):17-23. doi: PubMed
14. MacLean CH, Kerr EA, Qaseem A. Time out-charting a path for improving performance measurement. N Engl J Med. 2018. Epub ahead of print. doi: 10.1056/NEJMp1802595 PubMed
© 2018 Society of Hospital Medicine
Barriers to Earlier Hospital Discharge: What Matters Most?
“Every system is perfectly designed to get the results it gets.”
—W. Edwards Deming inspired quote1
The timing of patient discharge represents a Gordian knot in hospital operations. Moving the time of discharge to earlier in the day is a complex challenge that defies replicable solutions and is often a barrier to optimal throughput and patient experience. In this issue of the Journal of Hospital Medicine, Zoucha et al. identify that discharge orders are frequently delayed due to physicians caring for other patients, heterogeneity in physician rounding styles, and other intrinsic factors such as census size, rounding style, and teaching versus nonteaching services.2 Some of these factors and their negative impact are consistent with the effect of higher hospitalist workload (census) when increasing length of stay that was identified by Elliott et al.3 Others, such as rounding style and balancing teaching and education, are a part of many hospitalist service operations. Other intrinsic factors identified by the authors include awaiting consultant recommendations, care completion by social workers, procedures, labs, radiology, therapy services, and home oxygen.
The authors, however, recognize hospitalist behaviors and hospital operations as intrinsic factors. This is significant because intrinsic factors are theoretically under the control of the hospital’s physicians, administration, and support services. They lend themselves to continuous improvement, re-engineering, and change management. They are a direct result of the people, processes, structure, and supporting information technology (IT).
The findings of this study contrast with the perceived dominance of extrinsic factors such as awaiting a ride, insurance authorization issues, or placement as the cause for discharge delays. Anecdotally, physicians and nurses in organizations often identify such extrinsic factors as causes of discharge delays before they call out intrinsic factors.
Frequently, the first reaction to managing complex intrinsic constraints is to add resources and complexity. Continuous improvement often reveals the culprit is poor design and waste found throughout the system. Zoucha et al. refer to LEAN successes by others4 as an example of how to approach these complex intrinsic issues. Increasing early discharge with improvement in length of stay and reducing or maintaining the readmission rate has been achieved using the Institute for Healthcare Improvement Model for Improvement,5 the Red/Yellow/Green Discharge Tool within the electronic medical record,6 and a comprehensive management plan.7 These examples were often accomplished through improving the deployment of existing resources and reducing wasted activity. New predictive tools using supervised machine learning can help identify appropriate patients for discharge earlier in the day.8 This approach is built on the concepts of “efficiency and communication as components of quality healthcare delivery.”6
Perhaps a practical reductionist approach is to start with the end in mind, and ask the question “what matters most?” Three key times occur in each discharge and the authors capture two of these: the discharge order time and discharge time. Not captured is the time the patient and family are told they are being discharged. It is against this backdrop that we can look at four perspectives: caregiver, organization, community, and the patient and family. “What matters most?” depends on the perspective of each one of the parties involved.
From the perspective of the caregivers (physicians and residents), the conclusions support prioritizing rounding on patients ready to discharge, lowering team census, and restructuring teaching rounds to drive earlier discharges. But only 7% of encounters prioritized patients ready for discharge first. Seventy-six percent prioritized sickest patients first (33%), room-by-room (27%), and newest patients (16%).2 The authors emphasize that such an approach needs to be balanced against the needs of the entire team census to ensure optimal care for all patients. Team and individual hospitalist census and processes must be optimized to improve the efficiency and effectiveness of the work. For teaching services, the goal is to accomplish effective teaching while maintaining or improving throughput. When
Healthcare is delivered by teams. As we look at supporting and structuring our hospitalist teams’ inpatient rounding we need to include the contributions of advanced practice professionals, pharmacists, nurses, care managers, social workers, and others. Achieving a team focus on a goal can be supported by number-by-time (n-by-T) target initiatives, which have been used successfully.10,11 Team-based solutions must be developed to address these complex issues and in recognition of the need to distribute this responsibility across the system, not just depending on physician changes to ensure optimal outcomes.
The perspectives of organization and community have the common goals of delivering healthcare value (outcomes, quality, safety, and sustainability) and ensuring access. To achieve these, it is important to separate the discharge curve (by shifting these patients’ time of discharge to the left) from the arrival curve, which is more fixed. The organization and community benefit from reduced cost of care, improved value delivery, and better access to services. For hospitals and health systems facing high occupancy, this becomes important for access and serving the community, especially during the peak hours for admissions and discharges.
Against this backdrop is the most important perspective, which is that of the patients and families. What matters most to them? When does their clock start? For patients and families, we believe that their expectations begin when the physician or APP says, “you are doing well and we can get you home today.” In the current study, the median time to discharge from the discharge order for four of the five hospitals was about three hours.2 It is reasonable to assume the time interval is on the order of four to six hours or more for many patients. Is this acceptable? We have little data to answer this question directly, and while the Hospital Consumers Assessment of Healthcare Providers and Systems (HCAHPS) survey asks select questions regarding the effectiveness of discharge information, it is silent on matters of discharge timeliness and expectations. While on the administrative side we often use readmission rates as a proxy for a safe and “effective” discharge, in reality, we lack meaningful patient-reported outcome measures to assess our effectiveness, which is a necessity for performance improvement.
The opportunities for improvement suggested by this study include restructuring rounding to prioritize discharges, managing census per provider, and rethinking resident education to accommodate both education and service. The authors’ approach includes identifying ways to improve the efficiency of the work through other team members (such as pharmacy techs for medication reconciliation) and balancing ancillary services support for all inpatient care and the outpatients they serve. Alternatively, tying incentives to the goal could be a convenient leadership response. The 2016 Society of Hospital Medicine State of Hospital Medicine Report notes that more than half (54%) of nonacademic hospitalist groups that treat adults have an incentive tied to early morning discharge orders or times. We believe that by keeping the patients and families at the center of this discussion, we are more likely to accomplish the goal of improved safety, efficiency, effectiveness, and patient experience.
The literature supports discharge delays as an international challenge with research on the topic in healthcare systems across the world.12 This may be related to an aging population, improvements, and access to advanced healthcare, and the challenges of occupancy and capacity mismatches in many healthcare systems worldwide. The authors have identified important intrinsic factors for these throughput and discharge delays. The results beg the question, are we willing to do the redesign and behavior change in our delivery of healthcare and healthcare education to achieve a more optimized system of care delivery?
A now-retired Cleveland Clinic performance improvement engineer frequently referenced W. Edwards Deming on “what makes the biggest difference in improving internal service quality?” He distilled this to two axioms based on Deming’s work: reducing cycle time and reducing defects. Both must be accomplished from the customer’s (patient’s) perspective without tradeoffs between the two. Cycle time is the time to accomplish a completed process or action, such as patient discharge or LOS. Defects are all the waste or “impossible” challenges that contribute to the feeling of resignation that lead to people dismissing the possibility of improvement, stating “it is what it is.” The challenge in the service of our patients and families, organizations, and communities is to move this dialog forward to “it is what we make it.”13
When we tell the patient and family they are being discharged it should happen safely, efficiently, predictably, and with empathy. From the perspective of clinicians, it should be as easy as possible to consistently do the right thing and do the work to which they have dedicated themselves. For communities and organizations struggling with access, improving throughput is vital.
Disclosures
Neither author has any conflicts to disclose. There are no external funding sources for this manuscript.
1. Institute for Healthcare Improvement. Available at: http://www.ihi.org/communities/blogs/origin-of-every-system-is-perfectly-designed-quote. Accessed August 2, 2018.
2. Zoucha J, Hull M, Keniston A, et al. Barriers to Early Hospital Discharge: A Cross Sectional Study at Five Academic Hospitals. J Hosp Med. 2018;13(12):816-822. doi: 10.12788/jhm.3074. PubMed
3. Elliott DJ, Young RS, Brice J, Aguiar R, Kolm P. Effect of Hospitalist Workload on the Quality and Efficiency of Care. JAMA Intern Med. 2014;174(5):786. doi: 10.1001/jamainternmed.2014.300. PubMed
4. Beck MJ, Okerblom D, Kumar A, Bandyopadhyay S, Scalzi LV. Lean intervention improves patient discharge times, improves emergency department throughput and reduces congestion. Hosp Pract. 2016;44(5):252-259. doi: 10.1080/21548331.2016.1254559. PubMed
5. Patel H, Morduchowicz S, Mourad M. Using a Systematic Framework of Interventions to Improve Early Discharges. Jt Comm J Qual Patient Saf. 2017;43(4):189-196. doi: 10.1016/j.jcjq.2016.12.003. PubMed
6. Mathews KS, Corso P, Bacon S, Jenq GY. Using the Red/Yellow/Green Discharge Tool to Improve the Timeliness of Hospital Discharges. Jt Comm J Qual Patient Saf. 2014;40(6). doi:10.1016/s1553-7250(14)40033-3. PubMed
7. Wertheimer B, Jacobs REA, Bailey M, et al. Discharge before noon: An achievable hospital goal. J Hosp Med. 2014;9(4):210-214. doi: 10.1002/jhm.2154. PubMed
8. Barnes S, Hamrock E, Toerper M, Siddiqui S, Levin S. Real-time prediction of inpatient length of stay for discharge prioritization. J Am Med Inform Assoc. 2015;23(e1). doi: 10.1093/jamia/ocv106. PubMed
9. Wachter RM. Hospitalist Workload. JAMA Intern Med. 2014;174(5):794. doi:1 0.1001/jamainternmed.2014.18. PubMed
10. Parikh PJ, Ballester N, Ramsey K, Kong N, Pook N. The n-by-T Target Discharge Strategy for Inpatient Units. Med Decis Making. 2017;37(5):534-543. doi:10.1177/0272989x17691735. PubMed
11. Kane M, Weinacker A, Arthofer R, et al. A Multidisciplinary Initiative to Increase Inpatient Discharges Before Noon. J Nurs Adm. 2016; 46(12):630-635.doi: 10.1097/NNA.0000000000000418 PubMed
12. Rojas-García A, Turner S, Pizzo E, Hudson E, Thomas J, Raine R. Impact and experiences of delayed discharge: A mixed-studies systematic review. Health Expect. 2017;21(1):41-56. doi: 10.1111/hex.12619. PubMed
13. Emmelhainz L. Achieving Excellence: Some Last Thoughts. Lecture presented: Health System Leadership at Cleveland Clinic Akron General; May 16, 2018; Akron, OH. PubMed
“Every system is perfectly designed to get the results it gets.”
—W. Edwards Deming inspired quote1
The timing of patient discharge represents a Gordian knot in hospital operations. Moving the time of discharge to earlier in the day is a complex challenge that defies replicable solutions and is often a barrier to optimal throughput and patient experience. In this issue of the Journal of Hospital Medicine, Zoucha et al. identify that discharge orders are frequently delayed due to physicians caring for other patients, heterogeneity in physician rounding styles, and other intrinsic factors such as census size, rounding style, and teaching versus nonteaching services.2 Some of these factors and their negative impact are consistent with the effect of higher hospitalist workload (census) when increasing length of stay that was identified by Elliott et al.3 Others, such as rounding style and balancing teaching and education, are a part of many hospitalist service operations. Other intrinsic factors identified by the authors include awaiting consultant recommendations, care completion by social workers, procedures, labs, radiology, therapy services, and home oxygen.
The authors, however, recognize hospitalist behaviors and hospital operations as intrinsic factors. This is significant because intrinsic factors are theoretically under the control of the hospital’s physicians, administration, and support services. They lend themselves to continuous improvement, re-engineering, and change management. They are a direct result of the people, processes, structure, and supporting information technology (IT).
The findings of this study contrast with the perceived dominance of extrinsic factors such as awaiting a ride, insurance authorization issues, or placement as the cause for discharge delays. Anecdotally, physicians and nurses in organizations often identify such extrinsic factors as causes of discharge delays before they call out intrinsic factors.
Frequently, the first reaction to managing complex intrinsic constraints is to add resources and complexity. Continuous improvement often reveals the culprit is poor design and waste found throughout the system. Zoucha et al. refer to LEAN successes by others4 as an example of how to approach these complex intrinsic issues. Increasing early discharge with improvement in length of stay and reducing or maintaining the readmission rate has been achieved using the Institute for Healthcare Improvement Model for Improvement,5 the Red/Yellow/Green Discharge Tool within the electronic medical record,6 and a comprehensive management plan.7 These examples were often accomplished through improving the deployment of existing resources and reducing wasted activity. New predictive tools using supervised machine learning can help identify appropriate patients for discharge earlier in the day.8 This approach is built on the concepts of “efficiency and communication as components of quality healthcare delivery.”6
Perhaps a practical reductionist approach is to start with the end in mind, and ask the question “what matters most?” Three key times occur in each discharge and the authors capture two of these: the discharge order time and discharge time. Not captured is the time the patient and family are told they are being discharged. It is against this backdrop that we can look at four perspectives: caregiver, organization, community, and the patient and family. “What matters most?” depends on the perspective of each one of the parties involved.
From the perspective of the caregivers (physicians and residents), the conclusions support prioritizing rounding on patients ready to discharge, lowering team census, and restructuring teaching rounds to drive earlier discharges. But only 7% of encounters prioritized patients ready for discharge first. Seventy-six percent prioritized sickest patients first (33%), room-by-room (27%), and newest patients (16%).2 The authors emphasize that such an approach needs to be balanced against the needs of the entire team census to ensure optimal care for all patients. Team and individual hospitalist census and processes must be optimized to improve the efficiency and effectiveness of the work. For teaching services, the goal is to accomplish effective teaching while maintaining or improving throughput. When
Healthcare is delivered by teams. As we look at supporting and structuring our hospitalist teams’ inpatient rounding we need to include the contributions of advanced practice professionals, pharmacists, nurses, care managers, social workers, and others. Achieving a team focus on a goal can be supported by number-by-time (n-by-T) target initiatives, which have been used successfully.10,11 Team-based solutions must be developed to address these complex issues and in recognition of the need to distribute this responsibility across the system, not just depending on physician changes to ensure optimal outcomes.
The perspectives of organization and community have the common goals of delivering healthcare value (outcomes, quality, safety, and sustainability) and ensuring access. To achieve these, it is important to separate the discharge curve (by shifting these patients’ time of discharge to the left) from the arrival curve, which is more fixed. The organization and community benefit from reduced cost of care, improved value delivery, and better access to services. For hospitals and health systems facing high occupancy, this becomes important for access and serving the community, especially during the peak hours for admissions and discharges.
Against this backdrop is the most important perspective, which is that of the patients and families. What matters most to them? When does their clock start? For patients and families, we believe that their expectations begin when the physician or APP says, “you are doing well and we can get you home today.” In the current study, the median time to discharge from the discharge order for four of the five hospitals was about three hours.2 It is reasonable to assume the time interval is on the order of four to six hours or more for many patients. Is this acceptable? We have little data to answer this question directly, and while the Hospital Consumers Assessment of Healthcare Providers and Systems (HCAHPS) survey asks select questions regarding the effectiveness of discharge information, it is silent on matters of discharge timeliness and expectations. While on the administrative side we often use readmission rates as a proxy for a safe and “effective” discharge, in reality, we lack meaningful patient-reported outcome measures to assess our effectiveness, which is a necessity for performance improvement.
The opportunities for improvement suggested by this study include restructuring rounding to prioritize discharges, managing census per provider, and rethinking resident education to accommodate both education and service. The authors’ approach includes identifying ways to improve the efficiency of the work through other team members (such as pharmacy techs for medication reconciliation) and balancing ancillary services support for all inpatient care and the outpatients they serve. Alternatively, tying incentives to the goal could be a convenient leadership response. The 2016 Society of Hospital Medicine State of Hospital Medicine Report notes that more than half (54%) of nonacademic hospitalist groups that treat adults have an incentive tied to early morning discharge orders or times. We believe that by keeping the patients and families at the center of this discussion, we are more likely to accomplish the goal of improved safety, efficiency, effectiveness, and patient experience.
The literature supports discharge delays as an international challenge with research on the topic in healthcare systems across the world.12 This may be related to an aging population, improvements, and access to advanced healthcare, and the challenges of occupancy and capacity mismatches in many healthcare systems worldwide. The authors have identified important intrinsic factors for these throughput and discharge delays. The results beg the question, are we willing to do the redesign and behavior change in our delivery of healthcare and healthcare education to achieve a more optimized system of care delivery?
A now-retired Cleveland Clinic performance improvement engineer frequently referenced W. Edwards Deming on “what makes the biggest difference in improving internal service quality?” He distilled this to two axioms based on Deming’s work: reducing cycle time and reducing defects. Both must be accomplished from the customer’s (patient’s) perspective without tradeoffs between the two. Cycle time is the time to accomplish a completed process or action, such as patient discharge or LOS. Defects are all the waste or “impossible” challenges that contribute to the feeling of resignation that lead to people dismissing the possibility of improvement, stating “it is what it is.” The challenge in the service of our patients and families, organizations, and communities is to move this dialog forward to “it is what we make it.”13
When we tell the patient and family they are being discharged it should happen safely, efficiently, predictably, and with empathy. From the perspective of clinicians, it should be as easy as possible to consistently do the right thing and do the work to which they have dedicated themselves. For communities and organizations struggling with access, improving throughput is vital.
Disclosures
Neither author has any conflicts to disclose. There are no external funding sources for this manuscript.
“Every system is perfectly designed to get the results it gets.”
—W. Edwards Deming inspired quote1
The timing of patient discharge represents a Gordian knot in hospital operations. Moving the time of discharge to earlier in the day is a complex challenge that defies replicable solutions and is often a barrier to optimal throughput and patient experience. In this issue of the Journal of Hospital Medicine, Zoucha et al. identify that discharge orders are frequently delayed due to physicians caring for other patients, heterogeneity in physician rounding styles, and other intrinsic factors such as census size, rounding style, and teaching versus nonteaching services.2 Some of these factors and their negative impact are consistent with the effect of higher hospitalist workload (census) when increasing length of stay that was identified by Elliott et al.3 Others, such as rounding style and balancing teaching and education, are a part of many hospitalist service operations. Other intrinsic factors identified by the authors include awaiting consultant recommendations, care completion by social workers, procedures, labs, radiology, therapy services, and home oxygen.
The authors, however, recognize hospitalist behaviors and hospital operations as intrinsic factors. This is significant because intrinsic factors are theoretically under the control of the hospital’s physicians, administration, and support services. They lend themselves to continuous improvement, re-engineering, and change management. They are a direct result of the people, processes, structure, and supporting information technology (IT).
The findings of this study contrast with the perceived dominance of extrinsic factors such as awaiting a ride, insurance authorization issues, or placement as the cause for discharge delays. Anecdotally, physicians and nurses in organizations often identify such extrinsic factors as causes of discharge delays before they call out intrinsic factors.
Frequently, the first reaction to managing complex intrinsic constraints is to add resources and complexity. Continuous improvement often reveals the culprit is poor design and waste found throughout the system. Zoucha et al. refer to LEAN successes by others4 as an example of how to approach these complex intrinsic issues. Increasing early discharge with improvement in length of stay and reducing or maintaining the readmission rate has been achieved using the Institute for Healthcare Improvement Model for Improvement,5 the Red/Yellow/Green Discharge Tool within the electronic medical record,6 and a comprehensive management plan.7 These examples were often accomplished through improving the deployment of existing resources and reducing wasted activity. New predictive tools using supervised machine learning can help identify appropriate patients for discharge earlier in the day.8 This approach is built on the concepts of “efficiency and communication as components of quality healthcare delivery.”6
Perhaps a practical reductionist approach is to start with the end in mind, and ask the question “what matters most?” Three key times occur in each discharge and the authors capture two of these: the discharge order time and discharge time. Not captured is the time the patient and family are told they are being discharged. It is against this backdrop that we can look at four perspectives: caregiver, organization, community, and the patient and family. “What matters most?” depends on the perspective of each one of the parties involved.
From the perspective of the caregivers (physicians and residents), the conclusions support prioritizing rounding on patients ready to discharge, lowering team census, and restructuring teaching rounds to drive earlier discharges. But only 7% of encounters prioritized patients ready for discharge first. Seventy-six percent prioritized sickest patients first (33%), room-by-room (27%), and newest patients (16%).2 The authors emphasize that such an approach needs to be balanced against the needs of the entire team census to ensure optimal care for all patients. Team and individual hospitalist census and processes must be optimized to improve the efficiency and effectiveness of the work. For teaching services, the goal is to accomplish effective teaching while maintaining or improving throughput. When
Healthcare is delivered by teams. As we look at supporting and structuring our hospitalist teams’ inpatient rounding we need to include the contributions of advanced practice professionals, pharmacists, nurses, care managers, social workers, and others. Achieving a team focus on a goal can be supported by number-by-time (n-by-T) target initiatives, which have been used successfully.10,11 Team-based solutions must be developed to address these complex issues and in recognition of the need to distribute this responsibility across the system, not just depending on physician changes to ensure optimal outcomes.
The perspectives of organization and community have the common goals of delivering healthcare value (outcomes, quality, safety, and sustainability) and ensuring access. To achieve these, it is important to separate the discharge curve (by shifting these patients’ time of discharge to the left) from the arrival curve, which is more fixed. The organization and community benefit from reduced cost of care, improved value delivery, and better access to services. For hospitals and health systems facing high occupancy, this becomes important for access and serving the community, especially during the peak hours for admissions and discharges.
Against this backdrop is the most important perspective, which is that of the patients and families. What matters most to them? When does their clock start? For patients and families, we believe that their expectations begin when the physician or APP says, “you are doing well and we can get you home today.” In the current study, the median time to discharge from the discharge order for four of the five hospitals was about three hours.2 It is reasonable to assume the time interval is on the order of four to six hours or more for many patients. Is this acceptable? We have little data to answer this question directly, and while the Hospital Consumers Assessment of Healthcare Providers and Systems (HCAHPS) survey asks select questions regarding the effectiveness of discharge information, it is silent on matters of discharge timeliness and expectations. While on the administrative side we often use readmission rates as a proxy for a safe and “effective” discharge, in reality, we lack meaningful patient-reported outcome measures to assess our effectiveness, which is a necessity for performance improvement.
The opportunities for improvement suggested by this study include restructuring rounding to prioritize discharges, managing census per provider, and rethinking resident education to accommodate both education and service. The authors’ approach includes identifying ways to improve the efficiency of the work through other team members (such as pharmacy techs for medication reconciliation) and balancing ancillary services support for all inpatient care and the outpatients they serve. Alternatively, tying incentives to the goal could be a convenient leadership response. The 2016 Society of Hospital Medicine State of Hospital Medicine Report notes that more than half (54%) of nonacademic hospitalist groups that treat adults have an incentive tied to early morning discharge orders or times. We believe that by keeping the patients and families at the center of this discussion, we are more likely to accomplish the goal of improved safety, efficiency, effectiveness, and patient experience.
The literature supports discharge delays as an international challenge with research on the topic in healthcare systems across the world.12 This may be related to an aging population, improvements, and access to advanced healthcare, and the challenges of occupancy and capacity mismatches in many healthcare systems worldwide. The authors have identified important intrinsic factors for these throughput and discharge delays. The results beg the question, are we willing to do the redesign and behavior change in our delivery of healthcare and healthcare education to achieve a more optimized system of care delivery?
A now-retired Cleveland Clinic performance improvement engineer frequently referenced W. Edwards Deming on “what makes the biggest difference in improving internal service quality?” He distilled this to two axioms based on Deming’s work: reducing cycle time and reducing defects. Both must be accomplished from the customer’s (patient’s) perspective without tradeoffs between the two. Cycle time is the time to accomplish a completed process or action, such as patient discharge or LOS. Defects are all the waste or “impossible” challenges that contribute to the feeling of resignation that lead to people dismissing the possibility of improvement, stating “it is what it is.” The challenge in the service of our patients and families, organizations, and communities is to move this dialog forward to “it is what we make it.”13
When we tell the patient and family they are being discharged it should happen safely, efficiently, predictably, and with empathy. From the perspective of clinicians, it should be as easy as possible to consistently do the right thing and do the work to which they have dedicated themselves. For communities and organizations struggling with access, improving throughput is vital.
Disclosures
Neither author has any conflicts to disclose. There are no external funding sources for this manuscript.
1. Institute for Healthcare Improvement. Available at: http://www.ihi.org/communities/blogs/origin-of-every-system-is-perfectly-designed-quote. Accessed August 2, 2018.
2. Zoucha J, Hull M, Keniston A, et al. Barriers to Early Hospital Discharge: A Cross Sectional Study at Five Academic Hospitals. J Hosp Med. 2018;13(12):816-822. doi: 10.12788/jhm.3074. PubMed
3. Elliott DJ, Young RS, Brice J, Aguiar R, Kolm P. Effect of Hospitalist Workload on the Quality and Efficiency of Care. JAMA Intern Med. 2014;174(5):786. doi: 10.1001/jamainternmed.2014.300. PubMed
4. Beck MJ, Okerblom D, Kumar A, Bandyopadhyay S, Scalzi LV. Lean intervention improves patient discharge times, improves emergency department throughput and reduces congestion. Hosp Pract. 2016;44(5):252-259. doi: 10.1080/21548331.2016.1254559. PubMed
5. Patel H, Morduchowicz S, Mourad M. Using a Systematic Framework of Interventions to Improve Early Discharges. Jt Comm J Qual Patient Saf. 2017;43(4):189-196. doi: 10.1016/j.jcjq.2016.12.003. PubMed
6. Mathews KS, Corso P, Bacon S, Jenq GY. Using the Red/Yellow/Green Discharge Tool to Improve the Timeliness of Hospital Discharges. Jt Comm J Qual Patient Saf. 2014;40(6). doi:10.1016/s1553-7250(14)40033-3. PubMed
7. Wertheimer B, Jacobs REA, Bailey M, et al. Discharge before noon: An achievable hospital goal. J Hosp Med. 2014;9(4):210-214. doi: 10.1002/jhm.2154. PubMed
8. Barnes S, Hamrock E, Toerper M, Siddiqui S, Levin S. Real-time prediction of inpatient length of stay for discharge prioritization. J Am Med Inform Assoc. 2015;23(e1). doi: 10.1093/jamia/ocv106. PubMed
9. Wachter RM. Hospitalist Workload. JAMA Intern Med. 2014;174(5):794. doi:1 0.1001/jamainternmed.2014.18. PubMed
10. Parikh PJ, Ballester N, Ramsey K, Kong N, Pook N. The n-by-T Target Discharge Strategy for Inpatient Units. Med Decis Making. 2017;37(5):534-543. doi:10.1177/0272989x17691735. PubMed
11. Kane M, Weinacker A, Arthofer R, et al. A Multidisciplinary Initiative to Increase Inpatient Discharges Before Noon. J Nurs Adm. 2016; 46(12):630-635.doi: 10.1097/NNA.0000000000000418 PubMed
12. Rojas-García A, Turner S, Pizzo E, Hudson E, Thomas J, Raine R. Impact and experiences of delayed discharge: A mixed-studies systematic review. Health Expect. 2017;21(1):41-56. doi: 10.1111/hex.12619. PubMed
13. Emmelhainz L. Achieving Excellence: Some Last Thoughts. Lecture presented: Health System Leadership at Cleveland Clinic Akron General; May 16, 2018; Akron, OH. PubMed
1. Institute for Healthcare Improvement. Available at: http://www.ihi.org/communities/blogs/origin-of-every-system-is-perfectly-designed-quote. Accessed August 2, 2018.
2. Zoucha J, Hull M, Keniston A, et al. Barriers to Early Hospital Discharge: A Cross Sectional Study at Five Academic Hospitals. J Hosp Med. 2018;13(12):816-822. doi: 10.12788/jhm.3074. PubMed
3. Elliott DJ, Young RS, Brice J, Aguiar R, Kolm P. Effect of Hospitalist Workload on the Quality and Efficiency of Care. JAMA Intern Med. 2014;174(5):786. doi: 10.1001/jamainternmed.2014.300. PubMed
4. Beck MJ, Okerblom D, Kumar A, Bandyopadhyay S, Scalzi LV. Lean intervention improves patient discharge times, improves emergency department throughput and reduces congestion. Hosp Pract. 2016;44(5):252-259. doi: 10.1080/21548331.2016.1254559. PubMed
5. Patel H, Morduchowicz S, Mourad M. Using a Systematic Framework of Interventions to Improve Early Discharges. Jt Comm J Qual Patient Saf. 2017;43(4):189-196. doi: 10.1016/j.jcjq.2016.12.003. PubMed
6. Mathews KS, Corso P, Bacon S, Jenq GY. Using the Red/Yellow/Green Discharge Tool to Improve the Timeliness of Hospital Discharges. Jt Comm J Qual Patient Saf. 2014;40(6). doi:10.1016/s1553-7250(14)40033-3. PubMed
7. Wertheimer B, Jacobs REA, Bailey M, et al. Discharge before noon: An achievable hospital goal. J Hosp Med. 2014;9(4):210-214. doi: 10.1002/jhm.2154. PubMed
8. Barnes S, Hamrock E, Toerper M, Siddiqui S, Levin S. Real-time prediction of inpatient length of stay for discharge prioritization. J Am Med Inform Assoc. 2015;23(e1). doi: 10.1093/jamia/ocv106. PubMed
9. Wachter RM. Hospitalist Workload. JAMA Intern Med. 2014;174(5):794. doi:1 0.1001/jamainternmed.2014.18. PubMed
10. Parikh PJ, Ballester N, Ramsey K, Kong N, Pook N. The n-by-T Target Discharge Strategy for Inpatient Units. Med Decis Making. 2017;37(5):534-543. doi:10.1177/0272989x17691735. PubMed
11. Kane M, Weinacker A, Arthofer R, et al. A Multidisciplinary Initiative to Increase Inpatient Discharges Before Noon. J Nurs Adm. 2016; 46(12):630-635.doi: 10.1097/NNA.0000000000000418 PubMed
12. Rojas-García A, Turner S, Pizzo E, Hudson E, Thomas J, Raine R. Impact and experiences of delayed discharge: A mixed-studies systematic review. Health Expect. 2017;21(1):41-56. doi: 10.1111/hex.12619. PubMed
13. Emmelhainz L. Achieving Excellence: Some Last Thoughts. Lecture presented: Health System Leadership at Cleveland Clinic Akron General; May 16, 2018; Akron, OH. PubMed
© 2018 Society of Hospital Medicine
Is Hospital Discharge the Rube Goldberg Machine of Academic Internal Medicine?
One of the least taught yet most complicated tasks confronting new trainees is the bewildering process of discharging a patient. On an internal medicine service, this process can often resemble a Rube Goldberg machine, in which a “simple” task is accomplished through a series of interconnected, almost comically convoluted, yet separate steps that are triggered one after another and must be executed perfectly in sequence for success. It seems easy at first; just tap out a few sentences in the discharge paperwork, do a quick medication reconciliation, and a click of a button later, voila! The patient magically falls off the list and is on their merry way home. In reality, it only takes one wrench thrown into the Rube Goldberg machine to take down the whole operation. Much to the chagrin of internal medicine interns across the country, residents quickly learn that discharge planning is usually far from straightforward and that a myriad of obstacles (often dynamic and frustratingly unpredictable) can stand in the way of a successful discharge.
While some surgical services can streamline discharge processes to target defined lengths of stay based on a particular diagnosis, general medicine patients tend to have greater numbers of comorbid conditions, complex hospital courses, and wider variation in access to posthospital healthcare. In addition, there is very little formal instruction in transitions of care, and most residents identify direct patient care (learning by doing) as the primary mode of education.1,2 Struggling through the process of finding an appropriate placement, ensuring adequate outpatient follow-up, and untangling a jumbled mess of a medication reconciliation is often the only way that housestaff learn the Sisyphean task of transitioning care out of the hospital. The unpredictability and intensity of patient care adds to the ever growing list of competing demands on the time and attention of residents. Attendings face pressure on all sides to not only provide exemplary patient care and an educational experience but also to optimize hospital throughput by discharging patients as soon as possible (and ideally before noon). No wonder that the discharge process can threaten to unravel at any time, with delays and complications in discharge metamorphosing into increased length of stay (LOS), poorer outcomes, and increased 30-day readmission rates. As on-the-ground providers, what realities do we face when challenging ourselves to discharge patients before noon, and what practical changes in our workflow can we make to reach this goal?
In this month’s Journal of Hospital Medicine, Zoucha et al. examine these questions in real time by identifying barriers preventing both “definite” and “possible” discharges at three representative time points over the course of randomly chosen weekdays. They surveyed both housestaff and attendings at five academic hospitals across the United States, and the majority of patients were cared for on teaching services.3 Reflecting the inherent differences in workflow between teaching and nonteaching services, delays in definite discharges on teaching services were most often hindered by completing rounds and the need to staff the patient with the attending, whereas nonresident services identified other patient-care-related (both urgent and nonurgent) issues to be the culprits. Late-afternoon discharges were delayed on teaching services due to outstanding paperwork and follow-up arrangements, both of which most senior residents are keenly aware of and make their best effort to complete ahead of time. Patients designated as “possible” discharges were awaiting clinical improvement and resolution of disposition issues dependent on social work and safe placement, which reasonably seemed independent of service type. These descriptive findings suggest that nonresident services are more efficient than resident teams, and we are keen to identify novel solutions, such as dedicated discharge coordinators,4 to facilitate the discharge process on resident teams without detracting from the educational value of the rotation.
Zoucha et al. also found that factors beyond our control (having a lower daily census, attending on a nonresident service) were significantly associated with both earlier discharge order entry times and the actual time of patient discharge.3 While it is tempting to foist the entirety of the blame on extrinsic factors such as discharge placement and insurance issues, the reality is there might be some workflow changes that could expedite the discharge process. The authors are correct to emphasize that rounding style, in which discharges are prioritized to be seen first, is a behavior modification worth targeting. The percentage of teams that routinely see discharges first is not well studied, as other factors, such as clinically unstable patients, new admissions from overnight, and even mundane characteristics such as geographic location in the hospital, can also compete for prioritization in rounding order. Given the authors’ findings, we are eager to see further work in this area as prioritization of discharges during rounds could conceivably be studied within the context of a randomized controlled trial. Other innovations in rounding styles such as rounding-in-flow5 (in which all tasks are completed for a single patient before rounding on the next patient) can also significantly reduce the time to discharge order placement.
With help from the Penn Medicine Center for Health Care Innovation, we are actively studying bottlenecks in the discharge process by developing an interactive platform focused on delivering real-time information to all members of the healthcare team. Rapid rounds are held every morning with the attending physician, floor nursing leadership, physical therapy, social worker, and case management to quickly identify pending tasks, anticipated disposition, and a target date of discharge. Efficiency is key, as each team is limited to approximately 5-10 minutes. Previous studies (mostly pre–post studies) have shown that this simple intervention significantly reduced LOS,6,7 increased rates of discharge before noon,8 and was improved by electronic tracking tools.9 Our multidisciplinary rounds are unique in that information is then entered into an intuitive, web-based platform, which allows consolidation and analysis that permits generation of real-time statistics. By standardizing the discharge planning process, we hope to streamline a previously fragmented process and maximize the efficiency of hospital resource utilization.
Ultimately, high-quality care of complex patients on internal medicine services from admission to discharge requires hard work, smart utilization of resources, and a little bit of luck. There may not be a secret ingredient that guarantees perfectly efficient discharges 100% of the time, but this study inspires us to ponder additional approaches to this longstanding problem. The authors are to be congratulated for a rigorous study that illuminates where we as healthcare providers are able to realistically intervene to expedite the discharge process. First, having a lower census cap may not be possible in this era of maximal hospital usage, but this work suggests that thoughtful management of time on rounds may be a way to address the underlying problem. Secondly, the superior efficiency of nonteaching services may merely reflect the increased experience of the providers, and a realistic solution could be to implement a formal curriculum to educate housestaff about the discharge process, which would simultaneously address residency competency standards for transitions of care. Finally, the role of innovative informatics tools will surely open further avenues of investigation, as we continually evolve in response to intensifying standards of modern, efficient healthcare delivery in the 21st century. It may not be possible to eliminate the complexity from this particular Rube Goldberg machine, but taking the steps above may allow us to implement as many fail-safes as we can.
Disclosures
The authors have nothing to disclose.
1. Young E, Stickrath C, McNulty M, et al. Residents’ exposure to educational experiences in facilitating hospital discharges. J Grad Med Educ. 2017;9(2):184-189. doi: 10.4300/JGME-D-16-00503.1. PubMed
2. Greysen SR, Schiliro D, Curry L, et al. “Learning by doing” - Resident perspectives on developing competency in high-quality discharge care. J Gen Intern Med. 2012;27(9):1188-1194. doi: 10.1007/s11606-012-2094-5. PubMed
3. Zoucha J, Hull M, Keniston A, et al. Barriers to Early Hospital Discharge: A Cross-Sectional Study at Five Academic Hospitals. J Hosp Med. 2018;13(12):816-822. doi: 10.12788/jhm.3074. PubMed
4. Finn KM, Heffner R, Chang Y, et al. Improving the discharge process by embedding a discharge facilitator in a resident team. J Hosp Med. 2011;6(9):494-500. doi: 10.1002/jhm.924. PubMed
5. Calderon AS, Blackmore CC, Williams BL, et al. Transforming ward rounds through rounding-in-flow. J Grad Med Educ. 2014;6(4):750-755. doi: 10.4300/JGME-D-13-00324.1. PubMed
6. Kane M, Rohatgi N, Heidenreich PA, et al. Lean-based redesign of multidisciplinary rounds on general medicine service. J Hosp Med. 2018;13(7):482-485. doi: 10.12788/jhm.2908. PubMed
7. Gonçalves-Bradley D, Lannin N, Clemson L, Cameron ID, Shepperd S. Discharge planning from hospital. Cochrane Database Syst Rev. 2016;1-3. doi: 10.1002/14651858.CD000313.pub5.www.cochranelibrary.com. PubMed
8. Wertheimer B, Jacobs REA, Bailey M, et al. Discharge before noon: an achievable hospital goal. J Hosp Med. 2014;9(4):210-214. doi: 10.1002/jhm.2154. PubMed
9. Meo N, Paul E, Wilson C, Powers J, Magbual M, Miles KM. Introducing an electronic tracking tool into daily multidisciplinary discharge rounds on a medicine service: a quality improvement project to reduce length of stay. BMJ Open Qual. 2018;7(3):e000174. doi: 10.1136/bmjoq-2017-000174. PubMed
One of the least taught yet most complicated tasks confronting new trainees is the bewildering process of discharging a patient. On an internal medicine service, this process can often resemble a Rube Goldberg machine, in which a “simple” task is accomplished through a series of interconnected, almost comically convoluted, yet separate steps that are triggered one after another and must be executed perfectly in sequence for success. It seems easy at first; just tap out a few sentences in the discharge paperwork, do a quick medication reconciliation, and a click of a button later, voila! The patient magically falls off the list and is on their merry way home. In reality, it only takes one wrench thrown into the Rube Goldberg machine to take down the whole operation. Much to the chagrin of internal medicine interns across the country, residents quickly learn that discharge planning is usually far from straightforward and that a myriad of obstacles (often dynamic and frustratingly unpredictable) can stand in the way of a successful discharge.
While some surgical services can streamline discharge processes to target defined lengths of stay based on a particular diagnosis, general medicine patients tend to have greater numbers of comorbid conditions, complex hospital courses, and wider variation in access to posthospital healthcare. In addition, there is very little formal instruction in transitions of care, and most residents identify direct patient care (learning by doing) as the primary mode of education.1,2 Struggling through the process of finding an appropriate placement, ensuring adequate outpatient follow-up, and untangling a jumbled mess of a medication reconciliation is often the only way that housestaff learn the Sisyphean task of transitioning care out of the hospital. The unpredictability and intensity of patient care adds to the ever growing list of competing demands on the time and attention of residents. Attendings face pressure on all sides to not only provide exemplary patient care and an educational experience but also to optimize hospital throughput by discharging patients as soon as possible (and ideally before noon). No wonder that the discharge process can threaten to unravel at any time, with delays and complications in discharge metamorphosing into increased length of stay (LOS), poorer outcomes, and increased 30-day readmission rates. As on-the-ground providers, what realities do we face when challenging ourselves to discharge patients before noon, and what practical changes in our workflow can we make to reach this goal?
In this month’s Journal of Hospital Medicine, Zoucha et al. examine these questions in real time by identifying barriers preventing both “definite” and “possible” discharges at three representative time points over the course of randomly chosen weekdays. They surveyed both housestaff and attendings at five academic hospitals across the United States, and the majority of patients were cared for on teaching services.3 Reflecting the inherent differences in workflow between teaching and nonteaching services, delays in definite discharges on teaching services were most often hindered by completing rounds and the need to staff the patient with the attending, whereas nonresident services identified other patient-care-related (both urgent and nonurgent) issues to be the culprits. Late-afternoon discharges were delayed on teaching services due to outstanding paperwork and follow-up arrangements, both of which most senior residents are keenly aware of and make their best effort to complete ahead of time. Patients designated as “possible” discharges were awaiting clinical improvement and resolution of disposition issues dependent on social work and safe placement, which reasonably seemed independent of service type. These descriptive findings suggest that nonresident services are more efficient than resident teams, and we are keen to identify novel solutions, such as dedicated discharge coordinators,4 to facilitate the discharge process on resident teams without detracting from the educational value of the rotation.
Zoucha et al. also found that factors beyond our control (having a lower daily census, attending on a nonresident service) were significantly associated with both earlier discharge order entry times and the actual time of patient discharge.3 While it is tempting to foist the entirety of the blame on extrinsic factors such as discharge placement and insurance issues, the reality is there might be some workflow changes that could expedite the discharge process. The authors are correct to emphasize that rounding style, in which discharges are prioritized to be seen first, is a behavior modification worth targeting. The percentage of teams that routinely see discharges first is not well studied, as other factors, such as clinically unstable patients, new admissions from overnight, and even mundane characteristics such as geographic location in the hospital, can also compete for prioritization in rounding order. Given the authors’ findings, we are eager to see further work in this area as prioritization of discharges during rounds could conceivably be studied within the context of a randomized controlled trial. Other innovations in rounding styles such as rounding-in-flow5 (in which all tasks are completed for a single patient before rounding on the next patient) can also significantly reduce the time to discharge order placement.
With help from the Penn Medicine Center for Health Care Innovation, we are actively studying bottlenecks in the discharge process by developing an interactive platform focused on delivering real-time information to all members of the healthcare team. Rapid rounds are held every morning with the attending physician, floor nursing leadership, physical therapy, social worker, and case management to quickly identify pending tasks, anticipated disposition, and a target date of discharge. Efficiency is key, as each team is limited to approximately 5-10 minutes. Previous studies (mostly pre–post studies) have shown that this simple intervention significantly reduced LOS,6,7 increased rates of discharge before noon,8 and was improved by electronic tracking tools.9 Our multidisciplinary rounds are unique in that information is then entered into an intuitive, web-based platform, which allows consolidation and analysis that permits generation of real-time statistics. By standardizing the discharge planning process, we hope to streamline a previously fragmented process and maximize the efficiency of hospital resource utilization.
Ultimately, high-quality care of complex patients on internal medicine services from admission to discharge requires hard work, smart utilization of resources, and a little bit of luck. There may not be a secret ingredient that guarantees perfectly efficient discharges 100% of the time, but this study inspires us to ponder additional approaches to this longstanding problem. The authors are to be congratulated for a rigorous study that illuminates where we as healthcare providers are able to realistically intervene to expedite the discharge process. First, having a lower census cap may not be possible in this era of maximal hospital usage, but this work suggests that thoughtful management of time on rounds may be a way to address the underlying problem. Secondly, the superior efficiency of nonteaching services may merely reflect the increased experience of the providers, and a realistic solution could be to implement a formal curriculum to educate housestaff about the discharge process, which would simultaneously address residency competency standards for transitions of care. Finally, the role of innovative informatics tools will surely open further avenues of investigation, as we continually evolve in response to intensifying standards of modern, efficient healthcare delivery in the 21st century. It may not be possible to eliminate the complexity from this particular Rube Goldberg machine, but taking the steps above may allow us to implement as many fail-safes as we can.
Disclosures
The authors have nothing to disclose.
One of the least taught yet most complicated tasks confronting new trainees is the bewildering process of discharging a patient. On an internal medicine service, this process can often resemble a Rube Goldberg machine, in which a “simple” task is accomplished through a series of interconnected, almost comically convoluted, yet separate steps that are triggered one after another and must be executed perfectly in sequence for success. It seems easy at first; just tap out a few sentences in the discharge paperwork, do a quick medication reconciliation, and a click of a button later, voila! The patient magically falls off the list and is on their merry way home. In reality, it only takes one wrench thrown into the Rube Goldberg machine to take down the whole operation. Much to the chagrin of internal medicine interns across the country, residents quickly learn that discharge planning is usually far from straightforward and that a myriad of obstacles (often dynamic and frustratingly unpredictable) can stand in the way of a successful discharge.
While some surgical services can streamline discharge processes to target defined lengths of stay based on a particular diagnosis, general medicine patients tend to have greater numbers of comorbid conditions, complex hospital courses, and wider variation in access to posthospital healthcare. In addition, there is very little formal instruction in transitions of care, and most residents identify direct patient care (learning by doing) as the primary mode of education.1,2 Struggling through the process of finding an appropriate placement, ensuring adequate outpatient follow-up, and untangling a jumbled mess of a medication reconciliation is often the only way that housestaff learn the Sisyphean task of transitioning care out of the hospital. The unpredictability and intensity of patient care adds to the ever growing list of competing demands on the time and attention of residents. Attendings face pressure on all sides to not only provide exemplary patient care and an educational experience but also to optimize hospital throughput by discharging patients as soon as possible (and ideally before noon). No wonder that the discharge process can threaten to unravel at any time, with delays and complications in discharge metamorphosing into increased length of stay (LOS), poorer outcomes, and increased 30-day readmission rates. As on-the-ground providers, what realities do we face when challenging ourselves to discharge patients before noon, and what practical changes in our workflow can we make to reach this goal?
In this month’s Journal of Hospital Medicine, Zoucha et al. examine these questions in real time by identifying barriers preventing both “definite” and “possible” discharges at three representative time points over the course of randomly chosen weekdays. They surveyed both housestaff and attendings at five academic hospitals across the United States, and the majority of patients were cared for on teaching services.3 Reflecting the inherent differences in workflow between teaching and nonteaching services, delays in definite discharges on teaching services were most often hindered by completing rounds and the need to staff the patient with the attending, whereas nonresident services identified other patient-care-related (both urgent and nonurgent) issues to be the culprits. Late-afternoon discharges were delayed on teaching services due to outstanding paperwork and follow-up arrangements, both of which most senior residents are keenly aware of and make their best effort to complete ahead of time. Patients designated as “possible” discharges were awaiting clinical improvement and resolution of disposition issues dependent on social work and safe placement, which reasonably seemed independent of service type. These descriptive findings suggest that nonresident services are more efficient than resident teams, and we are keen to identify novel solutions, such as dedicated discharge coordinators,4 to facilitate the discharge process on resident teams without detracting from the educational value of the rotation.
Zoucha et al. also found that factors beyond our control (having a lower daily census, attending on a nonresident service) were significantly associated with both earlier discharge order entry times and the actual time of patient discharge.3 While it is tempting to foist the entirety of the blame on extrinsic factors such as discharge placement and insurance issues, the reality is there might be some workflow changes that could expedite the discharge process. The authors are correct to emphasize that rounding style, in which discharges are prioritized to be seen first, is a behavior modification worth targeting. The percentage of teams that routinely see discharges first is not well studied, as other factors, such as clinically unstable patients, new admissions from overnight, and even mundane characteristics such as geographic location in the hospital, can also compete for prioritization in rounding order. Given the authors’ findings, we are eager to see further work in this area as prioritization of discharges during rounds could conceivably be studied within the context of a randomized controlled trial. Other innovations in rounding styles such as rounding-in-flow5 (in which all tasks are completed for a single patient before rounding on the next patient) can also significantly reduce the time to discharge order placement.
With help from the Penn Medicine Center for Health Care Innovation, we are actively studying bottlenecks in the discharge process by developing an interactive platform focused on delivering real-time information to all members of the healthcare team. Rapid rounds are held every morning with the attending physician, floor nursing leadership, physical therapy, social worker, and case management to quickly identify pending tasks, anticipated disposition, and a target date of discharge. Efficiency is key, as each team is limited to approximately 5-10 minutes. Previous studies (mostly pre–post studies) have shown that this simple intervention significantly reduced LOS,6,7 increased rates of discharge before noon,8 and was improved by electronic tracking tools.9 Our multidisciplinary rounds are unique in that information is then entered into an intuitive, web-based platform, which allows consolidation and analysis that permits generation of real-time statistics. By standardizing the discharge planning process, we hope to streamline a previously fragmented process and maximize the efficiency of hospital resource utilization.
Ultimately, high-quality care of complex patients on internal medicine services from admission to discharge requires hard work, smart utilization of resources, and a little bit of luck. There may not be a secret ingredient that guarantees perfectly efficient discharges 100% of the time, but this study inspires us to ponder additional approaches to this longstanding problem. The authors are to be congratulated for a rigorous study that illuminates where we as healthcare providers are able to realistically intervene to expedite the discharge process. First, having a lower census cap may not be possible in this era of maximal hospital usage, but this work suggests that thoughtful management of time on rounds may be a way to address the underlying problem. Secondly, the superior efficiency of nonteaching services may merely reflect the increased experience of the providers, and a realistic solution could be to implement a formal curriculum to educate housestaff about the discharge process, which would simultaneously address residency competency standards for transitions of care. Finally, the role of innovative informatics tools will surely open further avenues of investigation, as we continually evolve in response to intensifying standards of modern, efficient healthcare delivery in the 21st century. It may not be possible to eliminate the complexity from this particular Rube Goldberg machine, but taking the steps above may allow us to implement as many fail-safes as we can.
Disclosures
The authors have nothing to disclose.
1. Young E, Stickrath C, McNulty M, et al. Residents’ exposure to educational experiences in facilitating hospital discharges. J Grad Med Educ. 2017;9(2):184-189. doi: 10.4300/JGME-D-16-00503.1. PubMed
2. Greysen SR, Schiliro D, Curry L, et al. “Learning by doing” - Resident perspectives on developing competency in high-quality discharge care. J Gen Intern Med. 2012;27(9):1188-1194. doi: 10.1007/s11606-012-2094-5. PubMed
3. Zoucha J, Hull M, Keniston A, et al. Barriers to Early Hospital Discharge: A Cross-Sectional Study at Five Academic Hospitals. J Hosp Med. 2018;13(12):816-822. doi: 10.12788/jhm.3074. PubMed
4. Finn KM, Heffner R, Chang Y, et al. Improving the discharge process by embedding a discharge facilitator in a resident team. J Hosp Med. 2011;6(9):494-500. doi: 10.1002/jhm.924. PubMed
5. Calderon AS, Blackmore CC, Williams BL, et al. Transforming ward rounds through rounding-in-flow. J Grad Med Educ. 2014;6(4):750-755. doi: 10.4300/JGME-D-13-00324.1. PubMed
6. Kane M, Rohatgi N, Heidenreich PA, et al. Lean-based redesign of multidisciplinary rounds on general medicine service. J Hosp Med. 2018;13(7):482-485. doi: 10.12788/jhm.2908. PubMed
7. Gonçalves-Bradley D, Lannin N, Clemson L, Cameron ID, Shepperd S. Discharge planning from hospital. Cochrane Database Syst Rev. 2016;1-3. doi: 10.1002/14651858.CD000313.pub5.www.cochranelibrary.com. PubMed
8. Wertheimer B, Jacobs REA, Bailey M, et al. Discharge before noon: an achievable hospital goal. J Hosp Med. 2014;9(4):210-214. doi: 10.1002/jhm.2154. PubMed
9. Meo N, Paul E, Wilson C, Powers J, Magbual M, Miles KM. Introducing an electronic tracking tool into daily multidisciplinary discharge rounds on a medicine service: a quality improvement project to reduce length of stay. BMJ Open Qual. 2018;7(3):e000174. doi: 10.1136/bmjoq-2017-000174. PubMed
1. Young E, Stickrath C, McNulty M, et al. Residents’ exposure to educational experiences in facilitating hospital discharges. J Grad Med Educ. 2017;9(2):184-189. doi: 10.4300/JGME-D-16-00503.1. PubMed
2. Greysen SR, Schiliro D, Curry L, et al. “Learning by doing” - Resident perspectives on developing competency in high-quality discharge care. J Gen Intern Med. 2012;27(9):1188-1194. doi: 10.1007/s11606-012-2094-5. PubMed
3. Zoucha J, Hull M, Keniston A, et al. Barriers to Early Hospital Discharge: A Cross-Sectional Study at Five Academic Hospitals. J Hosp Med. 2018;13(12):816-822. doi: 10.12788/jhm.3074. PubMed
4. Finn KM, Heffner R, Chang Y, et al. Improving the discharge process by embedding a discharge facilitator in a resident team. J Hosp Med. 2011;6(9):494-500. doi: 10.1002/jhm.924. PubMed
5. Calderon AS, Blackmore CC, Williams BL, et al. Transforming ward rounds through rounding-in-flow. J Grad Med Educ. 2014;6(4):750-755. doi: 10.4300/JGME-D-13-00324.1. PubMed
6. Kane M, Rohatgi N, Heidenreich PA, et al. Lean-based redesign of multidisciplinary rounds on general medicine service. J Hosp Med. 2018;13(7):482-485. doi: 10.12788/jhm.2908. PubMed
7. Gonçalves-Bradley D, Lannin N, Clemson L, Cameron ID, Shepperd S. Discharge planning from hospital. Cochrane Database Syst Rev. 2016;1-3. doi: 10.1002/14651858.CD000313.pub5.www.cochranelibrary.com. PubMed
8. Wertheimer B, Jacobs REA, Bailey M, et al. Discharge before noon: an achievable hospital goal. J Hosp Med. 2014;9(4):210-214. doi: 10.1002/jhm.2154. PubMed
9. Meo N, Paul E, Wilson C, Powers J, Magbual M, Miles KM. Introducing an electronic tracking tool into daily multidisciplinary discharge rounds on a medicine service: a quality improvement project to reduce length of stay. BMJ Open Qual. 2018;7(3):e000174. doi: 10.1136/bmjoq-2017-000174. PubMed
© 2018 Society of Hospital Medicine
We May Not “Have It All,” But We Can Make It Better through Structural Changes
In this issue of the Journal of Hospital Medicine, the paper by Gottenborg et al. captures the experiences of female academic hospitalists navigating one of the most significant transitions they will face—becoming new mothers.1 This article gives an accessible voice to impersonal statistics about the barriers women physicians encounter within and across specialties in academia. The challenges and anecdotes shared by the study participants were eminently relatable and captured the all-too-familiar circumstances most of us with children have faced in our careers as physician mothers.
STUDY COMMENTARY AND DISCUSSION
This study uses qualitative research methods to illustrate the hurdles faced by mothers in hospital medicine beyond what is demonstrated by quantitative measures and provides the helpful step of proposing some solutions to the obstacles they have faced. While the sample size was very small, the women interviewed were diverse in their years in practice, geographic distribution, and percent clinical effort, providing evidence that the themes discussed prevail across demographic categories.
The snowball sampling via the Society of Hospital Medicine committees may not have yielded a representative sample of female hospitalists. It seems possible that women who are involved in this type of leadership may be better supported and/or have different work schedules than their peers who are not in leadership positions. We also wish there had been more emphasis on the systemic and structural factors that can improve the quality of life of physician mothers. These policies include paternity leave and other creative ways of acknowledging the useful skills and experience that motherhood brings to bear on clinical practice, such as increased empathy and compassion, as mentioned by one of the study participants.
Even with the aforementioned limitations, this study is important because it combines authentic quotes from practicing academic hospitalists with concrete and tangible suggestions for structural changes. The most striking element is that the majority of the study participants experienced uncertainty and a lack of transparency around parental leave policies. As nearly half of hospitalists are women and 80% are under age 40,2 it seems unimaginable that there would not be explicit policies in place for what is a common and largely anticipated life event. Medicine has made great strides toward gender equality, but we are unlikely to ever reach the goal of true parity without openly addressing the disproportionate effect of childbearing and child rearing on women physicians. Standardized, readily available, and equitable parental leave policies (for both birth parents and nonbirth parents) are the first and most critical step.
The absence of standard leave policies naturally puts physician mothers in the position of having to negotiate or “haggle” with various supervisors, the majority of whom are male division chiefs and department chairs,3 which places all parties in an uncomfortable position, further reinforcing inequities and sowing discord and resentment. Having formal policies around leave protects not only those who utilize parental leave but also the other members of a hospital medicine practice who take on the workload of the person on leave.
Uncertainty around how to address the increased clinical load and for how long, also creates anxiety among other group members and may lead to feelings of bitterness toward clinicians on leave, further contributing to the negative impact of new parenthood on female hospitalists. We can think of no other medical circumstance in which there is as much advance notice of the need for significant time away from work. Yet pregnancy, which is subject to complications and emergencies just like other medical conditions, is treated with so little concern that one may be asked to arrange for their own coverage during such an emergency, as one study subject reported.
We also empathize with the study participants’ reports of feeling that supervisors often mentally discounted their ability to participate in projects on return to work. These pernicious assumptions can compound a cycle of lost productivity, disengagement, and attrition from the workforce.
Female hospitalists returning from leave face additional challenges that place an undue burden on their professional activities, most notably related to breastfeeding. This is particularly relevant in the context of the intensity inherent in practicing hospital medicine, which includes long days of being the primary provider for acutely ill inpatients, as well as long stretches of many consecutive days when it may not be possible to return home before children’s bedtime. Even at our own institution, which has been recognized as a “Healthy Mothers Workplace,” breastfeeding accommodations are not set up to allow for ongoing clinical activities while taking time to express breastmilk, and the clinical schedule does not build in adjustments for this time-consuming and psychologically taxing commitment. Breastfeeding for at least one year is the medical recommendation of the American Academy of Pediatrics in line with a substantial body of evidence.4 One quote from the article poignantly notes, “Pumping every 3-4 hours: stopping what you’re doing, finding an empty room to pump, finding a place to store your milk, then going back to work, three times per shift, for the next 9 months of your life, was hell.” If we cannot enable our own medical providers to follow evidence-based recommendations, how can we possibly expect this of our patients?
CONCLUSIONS
The notion of women “having it all” is an impossible ideal—both work and life outside of work will inevitably require tradeoffs. However, there is an abundance of evidence and recommendations for concrete steps that can be taken to improve the experience of female physicians who have children. These include formal policies for childbearing and child rearing leave (the American Academy of Pediatrics recommends at least six to nine months5), convenient space and protected time for pumping milk during the first year, on-site childcare services and back-up child care, and flexible work schedules.6 It is time to stop treating childbirth among female physicians like an unexpected inconvenience and acknowledge the undeniable demographics of physicians in hospital medicine and the duty of healthcare systems and hospital medicine leaders to effectively plan for the needs of half of their workforce.
Disclosures
Neither of the authors have any financial conflicts of interest to disclose.
1. Gottenborg E, Maw A, Ngov LK, Burden M, Ponomaryova A, Jones CD. You can’t have it all: The experience of academic hospitalists during pregnancy, parental leave, and return to work. J Hosp Med. 2018;13(12):836-839. doi: 10.12788/jhm.3076. PubMed
2. Reid MB, Misky GJ, Harrison RA, Sharpe B, Auerbach A, Glasheen JJ. Mentorship, productivity, and promotion among academic hospitalists. J Gen Intern Med. 2012;27(1):23-27. doi: 10.1007/s11606-011-1892-5. PubMed
3. Association of American Medical Colleges. The state of women in academic medicine: The pipeline and pathways to leadership, 2015-2016. https://www.aamc.org/members/gwims/statistics/. Accessed October 1, 2018.
4. American Academy of Pediatrics. Breastfeeding and the use of human milk. Pediatrics. 2012;129(3):e827-e841. doi: 10.1542/peds.2011-3552. PubMed
5. National Public Radio. A Pediatrician’s View of Paid Parental Leave. https://www.npr.org/sections/health-shots/2016/10/10/497052014/a-pediatricians-view-of-paid-parental-leave. Accessed September 26, 2018.
6. Mangurian C, Linos E, Sarkar U, Rodriguez C, Jagsi R. What’s holding women in medicine back from leadership? (2018, June 19). Harvard Business Review. https://hbr.org/2018/06/whats-holding-women-in-medicine-back-from-leadership. Accessed October 1, 2018.
In this issue of the Journal of Hospital Medicine, the paper by Gottenborg et al. captures the experiences of female academic hospitalists navigating one of the most significant transitions they will face—becoming new mothers.1 This article gives an accessible voice to impersonal statistics about the barriers women physicians encounter within and across specialties in academia. The challenges and anecdotes shared by the study participants were eminently relatable and captured the all-too-familiar circumstances most of us with children have faced in our careers as physician mothers.
STUDY COMMENTARY AND DISCUSSION
This study uses qualitative research methods to illustrate the hurdles faced by mothers in hospital medicine beyond what is demonstrated by quantitative measures and provides the helpful step of proposing some solutions to the obstacles they have faced. While the sample size was very small, the women interviewed were diverse in their years in practice, geographic distribution, and percent clinical effort, providing evidence that the themes discussed prevail across demographic categories.
The snowball sampling via the Society of Hospital Medicine committees may not have yielded a representative sample of female hospitalists. It seems possible that women who are involved in this type of leadership may be better supported and/or have different work schedules than their peers who are not in leadership positions. We also wish there had been more emphasis on the systemic and structural factors that can improve the quality of life of physician mothers. These policies include paternity leave and other creative ways of acknowledging the useful skills and experience that motherhood brings to bear on clinical practice, such as increased empathy and compassion, as mentioned by one of the study participants.
Even with the aforementioned limitations, this study is important because it combines authentic quotes from practicing academic hospitalists with concrete and tangible suggestions for structural changes. The most striking element is that the majority of the study participants experienced uncertainty and a lack of transparency around parental leave policies. As nearly half of hospitalists are women and 80% are under age 40,2 it seems unimaginable that there would not be explicit policies in place for what is a common and largely anticipated life event. Medicine has made great strides toward gender equality, but we are unlikely to ever reach the goal of true parity without openly addressing the disproportionate effect of childbearing and child rearing on women physicians. Standardized, readily available, and equitable parental leave policies (for both birth parents and nonbirth parents) are the first and most critical step.
The absence of standard leave policies naturally puts physician mothers in the position of having to negotiate or “haggle” with various supervisors, the majority of whom are male division chiefs and department chairs,3 which places all parties in an uncomfortable position, further reinforcing inequities and sowing discord and resentment. Having formal policies around leave protects not only those who utilize parental leave but also the other members of a hospital medicine practice who take on the workload of the person on leave.
Uncertainty around how to address the increased clinical load and for how long, also creates anxiety among other group members and may lead to feelings of bitterness toward clinicians on leave, further contributing to the negative impact of new parenthood on female hospitalists. We can think of no other medical circumstance in which there is as much advance notice of the need for significant time away from work. Yet pregnancy, which is subject to complications and emergencies just like other medical conditions, is treated with so little concern that one may be asked to arrange for their own coverage during such an emergency, as one study subject reported.
We also empathize with the study participants’ reports of feeling that supervisors often mentally discounted their ability to participate in projects on return to work. These pernicious assumptions can compound a cycle of lost productivity, disengagement, and attrition from the workforce.
Female hospitalists returning from leave face additional challenges that place an undue burden on their professional activities, most notably related to breastfeeding. This is particularly relevant in the context of the intensity inherent in practicing hospital medicine, which includes long days of being the primary provider for acutely ill inpatients, as well as long stretches of many consecutive days when it may not be possible to return home before children’s bedtime. Even at our own institution, which has been recognized as a “Healthy Mothers Workplace,” breastfeeding accommodations are not set up to allow for ongoing clinical activities while taking time to express breastmilk, and the clinical schedule does not build in adjustments for this time-consuming and psychologically taxing commitment. Breastfeeding for at least one year is the medical recommendation of the American Academy of Pediatrics in line with a substantial body of evidence.4 One quote from the article poignantly notes, “Pumping every 3-4 hours: stopping what you’re doing, finding an empty room to pump, finding a place to store your milk, then going back to work, three times per shift, for the next 9 months of your life, was hell.” If we cannot enable our own medical providers to follow evidence-based recommendations, how can we possibly expect this of our patients?
CONCLUSIONS
The notion of women “having it all” is an impossible ideal—both work and life outside of work will inevitably require tradeoffs. However, there is an abundance of evidence and recommendations for concrete steps that can be taken to improve the experience of female physicians who have children. These include formal policies for childbearing and child rearing leave (the American Academy of Pediatrics recommends at least six to nine months5), convenient space and protected time for pumping milk during the first year, on-site childcare services and back-up child care, and flexible work schedules.6 It is time to stop treating childbirth among female physicians like an unexpected inconvenience and acknowledge the undeniable demographics of physicians in hospital medicine and the duty of healthcare systems and hospital medicine leaders to effectively plan for the needs of half of their workforce.
Disclosures
Neither of the authors have any financial conflicts of interest to disclose.
In this issue of the Journal of Hospital Medicine, the paper by Gottenborg et al. captures the experiences of female academic hospitalists navigating one of the most significant transitions they will face—becoming new mothers.1 This article gives an accessible voice to impersonal statistics about the barriers women physicians encounter within and across specialties in academia. The challenges and anecdotes shared by the study participants were eminently relatable and captured the all-too-familiar circumstances most of us with children have faced in our careers as physician mothers.
STUDY COMMENTARY AND DISCUSSION
This study uses qualitative research methods to illustrate the hurdles faced by mothers in hospital medicine beyond what is demonstrated by quantitative measures and provides the helpful step of proposing some solutions to the obstacles they have faced. While the sample size was very small, the women interviewed were diverse in their years in practice, geographic distribution, and percent clinical effort, providing evidence that the themes discussed prevail across demographic categories.
The snowball sampling via the Society of Hospital Medicine committees may not have yielded a representative sample of female hospitalists. It seems possible that women who are involved in this type of leadership may be better supported and/or have different work schedules than their peers who are not in leadership positions. We also wish there had been more emphasis on the systemic and structural factors that can improve the quality of life of physician mothers. These policies include paternity leave and other creative ways of acknowledging the useful skills and experience that motherhood brings to bear on clinical practice, such as increased empathy and compassion, as mentioned by one of the study participants.
Even with the aforementioned limitations, this study is important because it combines authentic quotes from practicing academic hospitalists with concrete and tangible suggestions for structural changes. The most striking element is that the majority of the study participants experienced uncertainty and a lack of transparency around parental leave policies. As nearly half of hospitalists are women and 80% are under age 40,2 it seems unimaginable that there would not be explicit policies in place for what is a common and largely anticipated life event. Medicine has made great strides toward gender equality, but we are unlikely to ever reach the goal of true parity without openly addressing the disproportionate effect of childbearing and child rearing on women physicians. Standardized, readily available, and equitable parental leave policies (for both birth parents and nonbirth parents) are the first and most critical step.
The absence of standard leave policies naturally puts physician mothers in the position of having to negotiate or “haggle” with various supervisors, the majority of whom are male division chiefs and department chairs,3 which places all parties in an uncomfortable position, further reinforcing inequities and sowing discord and resentment. Having formal policies around leave protects not only those who utilize parental leave but also the other members of a hospital medicine practice who take on the workload of the person on leave.
Uncertainty around how to address the increased clinical load and for how long, also creates anxiety among other group members and may lead to feelings of bitterness toward clinicians on leave, further contributing to the negative impact of new parenthood on female hospitalists. We can think of no other medical circumstance in which there is as much advance notice of the need for significant time away from work. Yet pregnancy, which is subject to complications and emergencies just like other medical conditions, is treated with so little concern that one may be asked to arrange for their own coverage during such an emergency, as one study subject reported.
We also empathize with the study participants’ reports of feeling that supervisors often mentally discounted their ability to participate in projects on return to work. These pernicious assumptions can compound a cycle of lost productivity, disengagement, and attrition from the workforce.
Female hospitalists returning from leave face additional challenges that place an undue burden on their professional activities, most notably related to breastfeeding. This is particularly relevant in the context of the intensity inherent in practicing hospital medicine, which includes long days of being the primary provider for acutely ill inpatients, as well as long stretches of many consecutive days when it may not be possible to return home before children’s bedtime. Even at our own institution, which has been recognized as a “Healthy Mothers Workplace,” breastfeeding accommodations are not set up to allow for ongoing clinical activities while taking time to express breastmilk, and the clinical schedule does not build in adjustments for this time-consuming and psychologically taxing commitment. Breastfeeding for at least one year is the medical recommendation of the American Academy of Pediatrics in line with a substantial body of evidence.4 One quote from the article poignantly notes, “Pumping every 3-4 hours: stopping what you’re doing, finding an empty room to pump, finding a place to store your milk, then going back to work, three times per shift, for the next 9 months of your life, was hell.” If we cannot enable our own medical providers to follow evidence-based recommendations, how can we possibly expect this of our patients?
CONCLUSIONS
The notion of women “having it all” is an impossible ideal—both work and life outside of work will inevitably require tradeoffs. However, there is an abundance of evidence and recommendations for concrete steps that can be taken to improve the experience of female physicians who have children. These include formal policies for childbearing and child rearing leave (the American Academy of Pediatrics recommends at least six to nine months5), convenient space and protected time for pumping milk during the first year, on-site childcare services and back-up child care, and flexible work schedules.6 It is time to stop treating childbirth among female physicians like an unexpected inconvenience and acknowledge the undeniable demographics of physicians in hospital medicine and the duty of healthcare systems and hospital medicine leaders to effectively plan for the needs of half of their workforce.
Disclosures
Neither of the authors have any financial conflicts of interest to disclose.
1. Gottenborg E, Maw A, Ngov LK, Burden M, Ponomaryova A, Jones CD. You can’t have it all: The experience of academic hospitalists during pregnancy, parental leave, and return to work. J Hosp Med. 2018;13(12):836-839. doi: 10.12788/jhm.3076. PubMed
2. Reid MB, Misky GJ, Harrison RA, Sharpe B, Auerbach A, Glasheen JJ. Mentorship, productivity, and promotion among academic hospitalists. J Gen Intern Med. 2012;27(1):23-27. doi: 10.1007/s11606-011-1892-5. PubMed
3. Association of American Medical Colleges. The state of women in academic medicine: The pipeline and pathways to leadership, 2015-2016. https://www.aamc.org/members/gwims/statistics/. Accessed October 1, 2018.
4. American Academy of Pediatrics. Breastfeeding and the use of human milk. Pediatrics. 2012;129(3):e827-e841. doi: 10.1542/peds.2011-3552. PubMed
5. National Public Radio. A Pediatrician’s View of Paid Parental Leave. https://www.npr.org/sections/health-shots/2016/10/10/497052014/a-pediatricians-view-of-paid-parental-leave. Accessed September 26, 2018.
6. Mangurian C, Linos E, Sarkar U, Rodriguez C, Jagsi R. What’s holding women in medicine back from leadership? (2018, June 19). Harvard Business Review. https://hbr.org/2018/06/whats-holding-women-in-medicine-back-from-leadership. Accessed October 1, 2018.
1. Gottenborg E, Maw A, Ngov LK, Burden M, Ponomaryova A, Jones CD. You can’t have it all: The experience of academic hospitalists during pregnancy, parental leave, and return to work. J Hosp Med. 2018;13(12):836-839. doi: 10.12788/jhm.3076. PubMed
2. Reid MB, Misky GJ, Harrison RA, Sharpe B, Auerbach A, Glasheen JJ. Mentorship, productivity, and promotion among academic hospitalists. J Gen Intern Med. 2012;27(1):23-27. doi: 10.1007/s11606-011-1892-5. PubMed
3. Association of American Medical Colleges. The state of women in academic medicine: The pipeline and pathways to leadership, 2015-2016. https://www.aamc.org/members/gwims/statistics/. Accessed October 1, 2018.
4. American Academy of Pediatrics. Breastfeeding and the use of human milk. Pediatrics. 2012;129(3):e827-e841. doi: 10.1542/peds.2011-3552. PubMed
5. National Public Radio. A Pediatrician’s View of Paid Parental Leave. https://www.npr.org/sections/health-shots/2016/10/10/497052014/a-pediatricians-view-of-paid-parental-leave. Accessed September 26, 2018.
6. Mangurian C, Linos E, Sarkar U, Rodriguez C, Jagsi R. What’s holding women in medicine back from leadership? (2018, June 19). Harvard Business Review. https://hbr.org/2018/06/whats-holding-women-in-medicine-back-from-leadership. Accessed October 1, 2018.
© 2018 Society of Hospital Medicine
On Decreasing Utilization: Models of Care for Frequently Hospitalized Patients and Their Effect on Outcomes
In this month’s edition of the Journal of Hospital Medicine, Goodwin and colleagues report their findings from their systematic review of models of care for frequently hospitalized patients. The authors reviewed the literature for interventions to reduce hospital admissions in frequently hospitalized patients with the goal of assessing the success of the interventions. This report contributes to the literature base of interventions to reduce healthcare utilization, particularly in the area of inpatient hospitalization.1
Goodwin et al. report that only nine studies met their criteria for review after a thorough search of the published literature. Of these nine studies, only four were determined to be high-quality studies. Interestingly, the low-quality studies found positive results in reducing hospital utilization, whereas the high-quality studies found decreases that were mirrored by their control groups. Impressive heterogeneity was found in the range of definitions, interventions, and outcome measures in the studies. These studies highlight the issue of “regression to the mean” for sicker individuals; however, they may not address readmission rates of specific medical systems or procedures that are also cost drivers, even if the patients are not considered critically ill. They also show where research partnerships can assist in increasing the number of members included in the studies for robust analyses.
From the perspective of a health plan, we applaud all efforts to improve patient outcomes and reduce cost. This report states that efforts to reduce chronic hospitalizations have not been unqualified successes. We must reflect upon how reducing utilization and improving outcomes align with our overall goals as a society. Recently, Federal Reserve Chairman Jay Powell summed up our nation’s particular issue, stating, “It is widely understood that the United States is on an unsustainable fiscal path, largely due to the interaction between an aging population and a healthcare system that delivers pretty average healthcare at a cost that is much higher than that of any other advanced economy.”2
A recent Kaiser Family Foundation analysis showed that 1% of patients accounted for 23% of all medical spending in the United States, and 97% of medical spending is attributed to the top 50% of patients.3 Pharmaceutical costs also play a role in this trend. Blue Cross and Blue Shield of Texas (BCBSTX) found that 2.5% of our population accounted for just under 50% of total medical spending. Conversely, when looking at patients with very high costs, only 0.4% had over $100,000 in spending exclusive of pharmacy. When including pharmacy, that number rises to 0.5%. As we consider annual medical and pharmacy trends year over year, we find that pharmacy spending may outpace hospital expenses in the near future.
Our internal data are also consistent with published reports that fewer than half of high-cost patients in one year continue to be high-cost cases the following year. Niall Brennan et al. reported that only 39% of the top 5% of spenders in a given year are also high spenders the following year.4 This finding not only coincides with the author’s statement around regression to the mean for the high admission utilizers, but it may be instructive to those looking to a Pareto method of attacking cost. If more than half of targeted patients will move out of the high cost category on their own, then demonstrating the effectiveness of interventions becomes challenging. Moreover, this regression finding speaks to the need to create effective programs to manage population health on a broad basis, which can address quality to all members and streamline costs for a large group that covers well more than 50% of medical spending.
BCBSTX emphasizes the creation of systems that let providers become responsible and accountable to outcomes and cost. Accountable Care Organizations (ACOs) and Intensive Medical Homes (IMHs) have played important roles in this journey, but physicians need to continue to invent and prioritize interventions that may achieve both goals. In particular, hospitalists have an important role to play. As ACOs flourish, hospitalists will need to join under the value-based umbrella and continue to intervene in patient care, policies, and procedures to reduce avoidable hospitalizations.
The development of value-based arrangements offers the healthcare system a unique opportunity to bring much-needed change. In our medical partnerships, direct communication with providers regarding their member experience and sharing of vital information about their patients’ health status have helped improve patient outcomes and decrease cost. Our IMH partnerships show a savings of up to $45,000 per member per year driven by decreases in admissions and ER visits, and in some cases, expensive medications. The hard work in these successes lies within the subtleties of fostering the relationship between payers and providers. Each pillar within the ecosystem plays a key role offering strengths, but the upside toward change comes in how we support each other’s weaknesses. This support is manifested in two ways: collaboration through communication and transparency through data sharing.
The road to change is one less traveled but not unpaved; advances in technology allow us to take experiences and build from them. At its core, technology has enhanced our collaboration and data capabilities. The ability to stay in touch with providers allows for almost real-time addressing of issues, promoting efficiency. The connection we have with providers has evolved from being solely paper contracts to a multichannel, multifunctional system. The ability to take claims experience, insert clinical acumen, and perform data analysis brings actionable solutions to be executed by our providers.
Those in the healthcare system will need to come together to continue to create interventions that improve quality while decreasing costs. The second part may require even more work than the first. The Health Care Cost Institute recently published data showing that inpatient utilization over a five-year period fell 12.9% in the commercially insured.5 However, over that same period, hospital prices for inpatient care rose 24.3%. The fundamental reason for the excess amount of money spent in US healthcare is that the prices are incredibly high.6 Currently, when diligence is exercised in reducing utilization, hospitals simply raise prices as a response to compensate for the lost income. Likewise, although prescription drug utilization only increased 1.8% during that period, the prices increased by 24.9%.
For the United States healthcare system to improve its quality and reduce its cost, we will need inventive partnerships to continue to create new systems to interact with patients in the most efficient and effective way possible. Readmissions and hospital utilization will be a large part of that improvement. Hospitals and hospitalists should ensure that they continue to focus on making healthcare more affordable by improving efficiency and outcomes and by resisting the tendencies of hospitals and pharmaceutical companies to raise prices in reaction to the improved efficiency.
Disclosures
The authors have nothing to disclose.
1. Goodwin A, Henschen BL, O’Dwyer LC, Nichols N, O’Leary KJ. Interventions for Frequently Hospitalized Patients and their Effect on Outcomes: A Systematic Review. J Hosp Med. 2018; 13(12):853-859. doi: 10.12788/jhm.3089. PubMed
2. Marketplace. Fed Chair Jay Powel. https://www.marketplace.org/2018/07/12/economy/powell-transcript. Accessed August 3, 2018.
3. Health System Tracker. https://www.healthsystemtracker.org/chart-collection/health-expenditures-vary-across-population/#item-start%2012/01/2017. Accessed August 3, 2018.
4. NEJM Catalyst. Consistently High Turnover in the Group of Top Health Care Spenders. https://catalyst.nejm.org/high-turnover-top-health-care-spenders/. Accessed August 3, 2018.
5. Health Care Cost Institute. 2016 Health Care Cost and Utilization Report. http://www.healthcostinstitute.org/report/2016-health-care-cost-utilization-report/. Accessed August 3, 2018.
6. Anderson GF, Reinhardt UE, Hussey PS, Peterosyan V. It’s the prices, stupid: why the United States is so different from other countries. Health Aff (Millwood). 2003;22(3):89-105. doi: 10.1377/hlthaff.22.3.89. PubMed
In this month’s edition of the Journal of Hospital Medicine, Goodwin and colleagues report their findings from their systematic review of models of care for frequently hospitalized patients. The authors reviewed the literature for interventions to reduce hospital admissions in frequently hospitalized patients with the goal of assessing the success of the interventions. This report contributes to the literature base of interventions to reduce healthcare utilization, particularly in the area of inpatient hospitalization.1
Goodwin et al. report that only nine studies met their criteria for review after a thorough search of the published literature. Of these nine studies, only four were determined to be high-quality studies. Interestingly, the low-quality studies found positive results in reducing hospital utilization, whereas the high-quality studies found decreases that were mirrored by their control groups. Impressive heterogeneity was found in the range of definitions, interventions, and outcome measures in the studies. These studies highlight the issue of “regression to the mean” for sicker individuals; however, they may not address readmission rates of specific medical systems or procedures that are also cost drivers, even if the patients are not considered critically ill. They also show where research partnerships can assist in increasing the number of members included in the studies for robust analyses.
From the perspective of a health plan, we applaud all efforts to improve patient outcomes and reduce cost. This report states that efforts to reduce chronic hospitalizations have not been unqualified successes. We must reflect upon how reducing utilization and improving outcomes align with our overall goals as a society. Recently, Federal Reserve Chairman Jay Powell summed up our nation’s particular issue, stating, “It is widely understood that the United States is on an unsustainable fiscal path, largely due to the interaction between an aging population and a healthcare system that delivers pretty average healthcare at a cost that is much higher than that of any other advanced economy.”2
A recent Kaiser Family Foundation analysis showed that 1% of patients accounted for 23% of all medical spending in the United States, and 97% of medical spending is attributed to the top 50% of patients.3 Pharmaceutical costs also play a role in this trend. Blue Cross and Blue Shield of Texas (BCBSTX) found that 2.5% of our population accounted for just under 50% of total medical spending. Conversely, when looking at patients with very high costs, only 0.4% had over $100,000 in spending exclusive of pharmacy. When including pharmacy, that number rises to 0.5%. As we consider annual medical and pharmacy trends year over year, we find that pharmacy spending may outpace hospital expenses in the near future.
Our internal data are also consistent with published reports that fewer than half of high-cost patients in one year continue to be high-cost cases the following year. Niall Brennan et al. reported that only 39% of the top 5% of spenders in a given year are also high spenders the following year.4 This finding not only coincides with the author’s statement around regression to the mean for the high admission utilizers, but it may be instructive to those looking to a Pareto method of attacking cost. If more than half of targeted patients will move out of the high cost category on their own, then demonstrating the effectiveness of interventions becomes challenging. Moreover, this regression finding speaks to the need to create effective programs to manage population health on a broad basis, which can address quality to all members and streamline costs for a large group that covers well more than 50% of medical spending.
BCBSTX emphasizes the creation of systems that let providers become responsible and accountable to outcomes and cost. Accountable Care Organizations (ACOs) and Intensive Medical Homes (IMHs) have played important roles in this journey, but physicians need to continue to invent and prioritize interventions that may achieve both goals. In particular, hospitalists have an important role to play. As ACOs flourish, hospitalists will need to join under the value-based umbrella and continue to intervene in patient care, policies, and procedures to reduce avoidable hospitalizations.
The development of value-based arrangements offers the healthcare system a unique opportunity to bring much-needed change. In our medical partnerships, direct communication with providers regarding their member experience and sharing of vital information about their patients’ health status have helped improve patient outcomes and decrease cost. Our IMH partnerships show a savings of up to $45,000 per member per year driven by decreases in admissions and ER visits, and in some cases, expensive medications. The hard work in these successes lies within the subtleties of fostering the relationship between payers and providers. Each pillar within the ecosystem plays a key role offering strengths, but the upside toward change comes in how we support each other’s weaknesses. This support is manifested in two ways: collaboration through communication and transparency through data sharing.
The road to change is one less traveled but not unpaved; advances in technology allow us to take experiences and build from them. At its core, technology has enhanced our collaboration and data capabilities. The ability to stay in touch with providers allows for almost real-time addressing of issues, promoting efficiency. The connection we have with providers has evolved from being solely paper contracts to a multichannel, multifunctional system. The ability to take claims experience, insert clinical acumen, and perform data analysis brings actionable solutions to be executed by our providers.
Those in the healthcare system will need to come together to continue to create interventions that improve quality while decreasing costs. The second part may require even more work than the first. The Health Care Cost Institute recently published data showing that inpatient utilization over a five-year period fell 12.9% in the commercially insured.5 However, over that same period, hospital prices for inpatient care rose 24.3%. The fundamental reason for the excess amount of money spent in US healthcare is that the prices are incredibly high.6 Currently, when diligence is exercised in reducing utilization, hospitals simply raise prices as a response to compensate for the lost income. Likewise, although prescription drug utilization only increased 1.8% during that period, the prices increased by 24.9%.
For the United States healthcare system to improve its quality and reduce its cost, we will need inventive partnerships to continue to create new systems to interact with patients in the most efficient and effective way possible. Readmissions and hospital utilization will be a large part of that improvement. Hospitals and hospitalists should ensure that they continue to focus on making healthcare more affordable by improving efficiency and outcomes and by resisting the tendencies of hospitals and pharmaceutical companies to raise prices in reaction to the improved efficiency.
Disclosures
The authors have nothing to disclose.
In this month’s edition of the Journal of Hospital Medicine, Goodwin and colleagues report their findings from their systematic review of models of care for frequently hospitalized patients. The authors reviewed the literature for interventions to reduce hospital admissions in frequently hospitalized patients with the goal of assessing the success of the interventions. This report contributes to the literature base of interventions to reduce healthcare utilization, particularly in the area of inpatient hospitalization.1
Goodwin et al. report that only nine studies met their criteria for review after a thorough search of the published literature. Of these nine studies, only four were determined to be high-quality studies. Interestingly, the low-quality studies found positive results in reducing hospital utilization, whereas the high-quality studies found decreases that were mirrored by their control groups. Impressive heterogeneity was found in the range of definitions, interventions, and outcome measures in the studies. These studies highlight the issue of “regression to the mean” for sicker individuals; however, they may not address readmission rates of specific medical systems or procedures that are also cost drivers, even if the patients are not considered critically ill. They also show where research partnerships can assist in increasing the number of members included in the studies for robust analyses.
From the perspective of a health plan, we applaud all efforts to improve patient outcomes and reduce cost. This report states that efforts to reduce chronic hospitalizations have not been unqualified successes. We must reflect upon how reducing utilization and improving outcomes align with our overall goals as a society. Recently, Federal Reserve Chairman Jay Powell summed up our nation’s particular issue, stating, “It is widely understood that the United States is on an unsustainable fiscal path, largely due to the interaction between an aging population and a healthcare system that delivers pretty average healthcare at a cost that is much higher than that of any other advanced economy.”2
A recent Kaiser Family Foundation analysis showed that 1% of patients accounted for 23% of all medical spending in the United States, and 97% of medical spending is attributed to the top 50% of patients.3 Pharmaceutical costs also play a role in this trend. Blue Cross and Blue Shield of Texas (BCBSTX) found that 2.5% of our population accounted for just under 50% of total medical spending. Conversely, when looking at patients with very high costs, only 0.4% had over $100,000 in spending exclusive of pharmacy. When including pharmacy, that number rises to 0.5%. As we consider annual medical and pharmacy trends year over year, we find that pharmacy spending may outpace hospital expenses in the near future.
Our internal data are also consistent with published reports that fewer than half of high-cost patients in one year continue to be high-cost cases the following year. Niall Brennan et al. reported that only 39% of the top 5% of spenders in a given year are also high spenders the following year.4 This finding not only coincides with the author’s statement around regression to the mean for the high admission utilizers, but it may be instructive to those looking to a Pareto method of attacking cost. If more than half of targeted patients will move out of the high cost category on their own, then demonstrating the effectiveness of interventions becomes challenging. Moreover, this regression finding speaks to the need to create effective programs to manage population health on a broad basis, which can address quality to all members and streamline costs for a large group that covers well more than 50% of medical spending.
BCBSTX emphasizes the creation of systems that let providers become responsible and accountable to outcomes and cost. Accountable Care Organizations (ACOs) and Intensive Medical Homes (IMHs) have played important roles in this journey, but physicians need to continue to invent and prioritize interventions that may achieve both goals. In particular, hospitalists have an important role to play. As ACOs flourish, hospitalists will need to join under the value-based umbrella and continue to intervene in patient care, policies, and procedures to reduce avoidable hospitalizations.
The development of value-based arrangements offers the healthcare system a unique opportunity to bring much-needed change. In our medical partnerships, direct communication with providers regarding their member experience and sharing of vital information about their patients’ health status have helped improve patient outcomes and decrease cost. Our IMH partnerships show a savings of up to $45,000 per member per year driven by decreases in admissions and ER visits, and in some cases, expensive medications. The hard work in these successes lies within the subtleties of fostering the relationship between payers and providers. Each pillar within the ecosystem plays a key role offering strengths, but the upside toward change comes in how we support each other’s weaknesses. This support is manifested in two ways: collaboration through communication and transparency through data sharing.
The road to change is one less traveled but not unpaved; advances in technology allow us to take experiences and build from them. At its core, technology has enhanced our collaboration and data capabilities. The ability to stay in touch with providers allows for almost real-time addressing of issues, promoting efficiency. The connection we have with providers has evolved from being solely paper contracts to a multichannel, multifunctional system. The ability to take claims experience, insert clinical acumen, and perform data analysis brings actionable solutions to be executed by our providers.
Those in the healthcare system will need to come together to continue to create interventions that improve quality while decreasing costs. The second part may require even more work than the first. The Health Care Cost Institute recently published data showing that inpatient utilization over a five-year period fell 12.9% in the commercially insured.5 However, over that same period, hospital prices for inpatient care rose 24.3%. The fundamental reason for the excess amount of money spent in US healthcare is that the prices are incredibly high.6 Currently, when diligence is exercised in reducing utilization, hospitals simply raise prices as a response to compensate for the lost income. Likewise, although prescription drug utilization only increased 1.8% during that period, the prices increased by 24.9%.
For the United States healthcare system to improve its quality and reduce its cost, we will need inventive partnerships to continue to create new systems to interact with patients in the most efficient and effective way possible. Readmissions and hospital utilization will be a large part of that improvement. Hospitals and hospitalists should ensure that they continue to focus on making healthcare more affordable by improving efficiency and outcomes and by resisting the tendencies of hospitals and pharmaceutical companies to raise prices in reaction to the improved efficiency.
Disclosures
The authors have nothing to disclose.
1. Goodwin A, Henschen BL, O’Dwyer LC, Nichols N, O’Leary KJ. Interventions for Frequently Hospitalized Patients and their Effect on Outcomes: A Systematic Review. J Hosp Med. 2018; 13(12):853-859. doi: 10.12788/jhm.3089. PubMed
2. Marketplace. Fed Chair Jay Powel. https://www.marketplace.org/2018/07/12/economy/powell-transcript. Accessed August 3, 2018.
3. Health System Tracker. https://www.healthsystemtracker.org/chart-collection/health-expenditures-vary-across-population/#item-start%2012/01/2017. Accessed August 3, 2018.
4. NEJM Catalyst. Consistently High Turnover in the Group of Top Health Care Spenders. https://catalyst.nejm.org/high-turnover-top-health-care-spenders/. Accessed August 3, 2018.
5. Health Care Cost Institute. 2016 Health Care Cost and Utilization Report. http://www.healthcostinstitute.org/report/2016-health-care-cost-utilization-report/. Accessed August 3, 2018.
6. Anderson GF, Reinhardt UE, Hussey PS, Peterosyan V. It’s the prices, stupid: why the United States is so different from other countries. Health Aff (Millwood). 2003;22(3):89-105. doi: 10.1377/hlthaff.22.3.89. PubMed
1. Goodwin A, Henschen BL, O’Dwyer LC, Nichols N, O’Leary KJ. Interventions for Frequently Hospitalized Patients and their Effect on Outcomes: A Systematic Review. J Hosp Med. 2018; 13(12):853-859. doi: 10.12788/jhm.3089. PubMed
2. Marketplace. Fed Chair Jay Powel. https://www.marketplace.org/2018/07/12/economy/powell-transcript. Accessed August 3, 2018.
3. Health System Tracker. https://www.healthsystemtracker.org/chart-collection/health-expenditures-vary-across-population/#item-start%2012/01/2017. Accessed August 3, 2018.
4. NEJM Catalyst. Consistently High Turnover in the Group of Top Health Care Spenders. https://catalyst.nejm.org/high-turnover-top-health-care-spenders/. Accessed August 3, 2018.
5. Health Care Cost Institute. 2016 Health Care Cost and Utilization Report. http://www.healthcostinstitute.org/report/2016-health-care-cost-utilization-report/. Accessed August 3, 2018.
6. Anderson GF, Reinhardt UE, Hussey PS, Peterosyan V. It’s the prices, stupid: why the United States is so different from other countries. Health Aff (Millwood). 2003;22(3):89-105. doi: 10.1377/hlthaff.22.3.89. PubMed
© 2018 Society of Hospital Medicine
Towards Scalable Hospital-Based Palliative Care: Challenges and Opportunities for Hospitalists
There is growing evidence that supports the ability of specialty palliative care to achieve the Triple Aim in healthcare: (1) improve patient and family experience of care, (2) improve health outcomes, and (3) reduce healthcare costs.1,2 However, the full realization of this value remains elusive due, in large part, to the increasing demand for specialty palliative care services outpacing the supply of specialists.3 Because expansion of the specialty palliative care workforce will never be sufficient to meet the needs of seriously ill patients, and nonspecialist physicians often fail to recognize palliative care needs in a timely manner,4 innovative and systematic solutions are needed to provide high-quality palliative care in a manner that is sustainable.5
To close the gap between workforce and patient needs, experts have largely advocated for two care delivery models that aim to improve the organization and allocation of limited palliative care resources: (1) a tier-based approach in which primary palliative care (basic skills for all clinicians) and specialty palliative care (advanced skills requiring additional training) have distinct but supportive roles, and (2) a need-based approach where different types of palliative care clinicians are deployed based on specific needs.5,6 In this issue, Abedini and Chopra propose a “Palliative Care Redistribution Integrated System Model” (PRISM) that combines these two approaches, with need-based care delivery that escalates through skill tiers to improve hospital-based palliative care.7
PRISM is attractive because it leverages the skill sets of clinicians across disciplines and is designed for the hospital, where the vast majority of specialty palliative care is provided in the United States. Moreover, it employs hospitalists who routinely care for a high volume of seriously ill patients, and are therefore well positioned to expand the palliative care workforce. The authors suggest several approaches to implement PRISM, such as designating certain hospitalist teams for palliative care, more interdisciplinary support, automated patient risk stratification or mandatory screening checklists, and strategic use of bedside nurses and social workers to facilitate early basic needs assessments. Although sound in principle, there are several foreseeable barriers to each of these approaches and potential unintended consequences of PRISM in the fields of hospital and palliative medicine.
Applying insights from behavioral economics will be essential for the successful implementation and dissemination of PRISM. Changing clinician behavior is not a challenge unique to palliative care interventions, but it may be particularly difficult due to misperceptions that palliative care is synonymous with end-of-life care and that such conversations are always time-intensive. Indeed, Abedini and Chopra acknowledge that all clinicians need to be well versed in basic palliative care skills for PRISM to succeed, yet most educational initiatives have shown modest results at best. The most promising clinician education programs, such as the Serious Illness Care Program and VitalTalk require intensive training simulations and are most effective when implemented on a system level to promote cultural change.8.9 Thus, training hospitalists in preparation for PRISM will require considerable upfront investment by hospitals. While policy efforts to improve palliative care training in medical education are progressing (Palliative Care and Hospice Education and Training Act, H.R.1676), any evidence of impact is nearly a generation away.
The authors also advocate for a technology-driven solution for systematic and early identification of palliative care needs. However, ideal clinical decision support would not rely on checklists to be completed by bedside clinicians or “hard stop” alerts in the electronic health record, as both of these approaches rely heavily upon consistent and accurate data entry by busy clinicians. Rather, innovative predictive analytics with machine learning and natural language processing methods hold great promise to support an electronic precision medicine approach for palliative care delivery. Even after such prediction models are developed, rigorous studies are needed to understand how they can change clinician behavior and impact the quality and cost of care.
Shifting palliative care tasks to nonspecialists has implications beyond quality and access. First, there are likely to be reimbursement implications as nonbillable clinicians such as social workers provide palliative care services that were previously provided by physicians and advance practice providers. As value-based payment models grow, healthcare systems may be wise to invest in innovative palliative care delivery models such as PRISM, but obtaining financial support will require rigorous evidence of value. Second, it will be important to monitor the already high rates of burnout and emotional exhaustion among palliative care clinicians10 when implementing care delivery models that select only the most complex patients for referral to specialty palliative care. Finally, new palliative care delivery models must fit within a larger national strategy to grow palliative care across the care continuum.11 This is of particular importance with hospital-focused solutions such as PRISM due to concerns about the growing split in care coordination between inpatient and outpatient care. Since seriously ill patients spend the majority of time outside the hospital and evidence for the value of palliative care is most robust in home and ambulatory settings,1 an important role for hospitalists could be to systematically identify and refer high-risk patients to community-based palliative care services after discharge from a sentinel hospitalization.
In conclusion, innovative palliative care delivery models such as PRISM are critical to ensuring that seriously ill patients have access to high-quality palliative care; however, more work is still needed to create the training programs, patient identification tools, scalable implementation, and evaluation processes necessary for success.
Disclosures
Dr. Courtright and Dr. O’Connor have nothing to disclose.
Funding
This work was funded in part by a career development award from the National Palliative Care Research Center (KRC). The views expressed herein solely represent those of the authors.
1. Kavalieratos D, Corbelli J, Zhang D, et al. Association between palliative care and patient and caregiver outcomes. Jama. 2016;316(20):2104. doi: 10.1001/jama.2016.16840. PubMed
2. May P, Normand C, Cassel JB, et al. Economics of palliative care for hospitalized adults with serious illness. JAMA Intern Med. 2018;178(6):820. doi: 10.1001/jamainternmed.2018.0750. PubMed
3. Dumanovsky T, Augustin R, Rogers M, Lettang K, Meier DE, Morrison RS. The growth of palliative care in U.S. hospitals: a status report. J Palliat Med. 2016;19(1):8-15. doi: 10.1089/jpm.2015.0351. PubMed
4. Heyland DK. Failure to Engage hospitalized elderly patients and their families in advance care planning. JAMA Intern Med. 2013;173(9):778. doi: 10.1001/jamainternmed.2013.180. PubMed
5. Courtright KR, Cassel JB, Halpern SD. A research agenda for high-value palliative care. Ann Intern Med. 2017;168(1):71. doi: 10.7326/m17-2164. PubMed
6. Billings JA, Bernacki R. Strategic targeting of advance care planning interventions. JAMA Intern Med. 2014;174(4):620. doi: 10.1001/jamainternmed.2013.14384. PubMed
7. Abedini NC, Chopra V. A Model to Improve Hospital-Based Palliative Care: The Palliative Care Redistribution Integrated System Model (PRISM). J Hosp Med. 2018;13(12):868-871. doi: 10.12788/jhm.3065 PubMed
8. Bernacki R, Hutchings M, Vick J, et al. Development of the Serious Illness Care Program: a randomized controlled trial of a palliative care communication intervention. BMJ Open. 2015;5(10):e009032. doi: 10.1136/bmjopen-2015-009032. PubMed
9. Clayton JM, Butow PN, Waters A, et al. Evaluation of a novel individualized communication-skills training intervention to improve doctors’ confidence and skills in end-of-life communication. Palliat Med. 2012;27(3):236-243. doi: 10.1177/0269216312449683. PubMed
10. Kamal AH, Bull JH, Wolf SP, et al. Prevalence and predictors of burnout among hospice and palliative care clinicians in the U.S. J Pain Symptom Manag. 2016;51(4):690-696. doi: 10.1016/j.jpainsymman.2015.10.020. PubMed
11. Meier DE, Back AL, Berman A, Block SD, Corrigan JM, Morrison RS. A national strategy for palliative care. Health Aff (Millwood). 2017;36(7):1265-1273. doi: 10.1377/hlthaff.2017.0164. PubMed
There is growing evidence that supports the ability of specialty palliative care to achieve the Triple Aim in healthcare: (1) improve patient and family experience of care, (2) improve health outcomes, and (3) reduce healthcare costs.1,2 However, the full realization of this value remains elusive due, in large part, to the increasing demand for specialty palliative care services outpacing the supply of specialists.3 Because expansion of the specialty palliative care workforce will never be sufficient to meet the needs of seriously ill patients, and nonspecialist physicians often fail to recognize palliative care needs in a timely manner,4 innovative and systematic solutions are needed to provide high-quality palliative care in a manner that is sustainable.5
To close the gap between workforce and patient needs, experts have largely advocated for two care delivery models that aim to improve the organization and allocation of limited palliative care resources: (1) a tier-based approach in which primary palliative care (basic skills for all clinicians) and specialty palliative care (advanced skills requiring additional training) have distinct but supportive roles, and (2) a need-based approach where different types of palliative care clinicians are deployed based on specific needs.5,6 In this issue, Abedini and Chopra propose a “Palliative Care Redistribution Integrated System Model” (PRISM) that combines these two approaches, with need-based care delivery that escalates through skill tiers to improve hospital-based palliative care.7
PRISM is attractive because it leverages the skill sets of clinicians across disciplines and is designed for the hospital, where the vast majority of specialty palliative care is provided in the United States. Moreover, it employs hospitalists who routinely care for a high volume of seriously ill patients, and are therefore well positioned to expand the palliative care workforce. The authors suggest several approaches to implement PRISM, such as designating certain hospitalist teams for palliative care, more interdisciplinary support, automated patient risk stratification or mandatory screening checklists, and strategic use of bedside nurses and social workers to facilitate early basic needs assessments. Although sound in principle, there are several foreseeable barriers to each of these approaches and potential unintended consequences of PRISM in the fields of hospital and palliative medicine.
Applying insights from behavioral economics will be essential for the successful implementation and dissemination of PRISM. Changing clinician behavior is not a challenge unique to palliative care interventions, but it may be particularly difficult due to misperceptions that palliative care is synonymous with end-of-life care and that such conversations are always time-intensive. Indeed, Abedini and Chopra acknowledge that all clinicians need to be well versed in basic palliative care skills for PRISM to succeed, yet most educational initiatives have shown modest results at best. The most promising clinician education programs, such as the Serious Illness Care Program and VitalTalk require intensive training simulations and are most effective when implemented on a system level to promote cultural change.8.9 Thus, training hospitalists in preparation for PRISM will require considerable upfront investment by hospitals. While policy efforts to improve palliative care training in medical education are progressing (Palliative Care and Hospice Education and Training Act, H.R.1676), any evidence of impact is nearly a generation away.
The authors also advocate for a technology-driven solution for systematic and early identification of palliative care needs. However, ideal clinical decision support would not rely on checklists to be completed by bedside clinicians or “hard stop” alerts in the electronic health record, as both of these approaches rely heavily upon consistent and accurate data entry by busy clinicians. Rather, innovative predictive analytics with machine learning and natural language processing methods hold great promise to support an electronic precision medicine approach for palliative care delivery. Even after such prediction models are developed, rigorous studies are needed to understand how they can change clinician behavior and impact the quality and cost of care.
Shifting palliative care tasks to nonspecialists has implications beyond quality and access. First, there are likely to be reimbursement implications as nonbillable clinicians such as social workers provide palliative care services that were previously provided by physicians and advance practice providers. As value-based payment models grow, healthcare systems may be wise to invest in innovative palliative care delivery models such as PRISM, but obtaining financial support will require rigorous evidence of value. Second, it will be important to monitor the already high rates of burnout and emotional exhaustion among palliative care clinicians10 when implementing care delivery models that select only the most complex patients for referral to specialty palliative care. Finally, new palliative care delivery models must fit within a larger national strategy to grow palliative care across the care continuum.11 This is of particular importance with hospital-focused solutions such as PRISM due to concerns about the growing split in care coordination between inpatient and outpatient care. Since seriously ill patients spend the majority of time outside the hospital and evidence for the value of palliative care is most robust in home and ambulatory settings,1 an important role for hospitalists could be to systematically identify and refer high-risk patients to community-based palliative care services after discharge from a sentinel hospitalization.
In conclusion, innovative palliative care delivery models such as PRISM are critical to ensuring that seriously ill patients have access to high-quality palliative care; however, more work is still needed to create the training programs, patient identification tools, scalable implementation, and evaluation processes necessary for success.
Disclosures
Dr. Courtright and Dr. O’Connor have nothing to disclose.
Funding
This work was funded in part by a career development award from the National Palliative Care Research Center (KRC). The views expressed herein solely represent those of the authors.
There is growing evidence that supports the ability of specialty palliative care to achieve the Triple Aim in healthcare: (1) improve patient and family experience of care, (2) improve health outcomes, and (3) reduce healthcare costs.1,2 However, the full realization of this value remains elusive due, in large part, to the increasing demand for specialty palliative care services outpacing the supply of specialists.3 Because expansion of the specialty palliative care workforce will never be sufficient to meet the needs of seriously ill patients, and nonspecialist physicians often fail to recognize palliative care needs in a timely manner,4 innovative and systematic solutions are needed to provide high-quality palliative care in a manner that is sustainable.5
To close the gap between workforce and patient needs, experts have largely advocated for two care delivery models that aim to improve the organization and allocation of limited palliative care resources: (1) a tier-based approach in which primary palliative care (basic skills for all clinicians) and specialty palliative care (advanced skills requiring additional training) have distinct but supportive roles, and (2) a need-based approach where different types of palliative care clinicians are deployed based on specific needs.5,6 In this issue, Abedini and Chopra propose a “Palliative Care Redistribution Integrated System Model” (PRISM) that combines these two approaches, with need-based care delivery that escalates through skill tiers to improve hospital-based palliative care.7
PRISM is attractive because it leverages the skill sets of clinicians across disciplines and is designed for the hospital, where the vast majority of specialty palliative care is provided in the United States. Moreover, it employs hospitalists who routinely care for a high volume of seriously ill patients, and are therefore well positioned to expand the palliative care workforce. The authors suggest several approaches to implement PRISM, such as designating certain hospitalist teams for palliative care, more interdisciplinary support, automated patient risk stratification or mandatory screening checklists, and strategic use of bedside nurses and social workers to facilitate early basic needs assessments. Although sound in principle, there are several foreseeable barriers to each of these approaches and potential unintended consequences of PRISM in the fields of hospital and palliative medicine.
Applying insights from behavioral economics will be essential for the successful implementation and dissemination of PRISM. Changing clinician behavior is not a challenge unique to palliative care interventions, but it may be particularly difficult due to misperceptions that palliative care is synonymous with end-of-life care and that such conversations are always time-intensive. Indeed, Abedini and Chopra acknowledge that all clinicians need to be well versed in basic palliative care skills for PRISM to succeed, yet most educational initiatives have shown modest results at best. The most promising clinician education programs, such as the Serious Illness Care Program and VitalTalk require intensive training simulations and are most effective when implemented on a system level to promote cultural change.8.9 Thus, training hospitalists in preparation for PRISM will require considerable upfront investment by hospitals. While policy efforts to improve palliative care training in medical education are progressing (Palliative Care and Hospice Education and Training Act, H.R.1676), any evidence of impact is nearly a generation away.
The authors also advocate for a technology-driven solution for systematic and early identification of palliative care needs. However, ideal clinical decision support would not rely on checklists to be completed by bedside clinicians or “hard stop” alerts in the electronic health record, as both of these approaches rely heavily upon consistent and accurate data entry by busy clinicians. Rather, innovative predictive analytics with machine learning and natural language processing methods hold great promise to support an electronic precision medicine approach for palliative care delivery. Even after such prediction models are developed, rigorous studies are needed to understand how they can change clinician behavior and impact the quality and cost of care.
Shifting palliative care tasks to nonspecialists has implications beyond quality and access. First, there are likely to be reimbursement implications as nonbillable clinicians such as social workers provide palliative care services that were previously provided by physicians and advance practice providers. As value-based payment models grow, healthcare systems may be wise to invest in innovative palliative care delivery models such as PRISM, but obtaining financial support will require rigorous evidence of value. Second, it will be important to monitor the already high rates of burnout and emotional exhaustion among palliative care clinicians10 when implementing care delivery models that select only the most complex patients for referral to specialty palliative care. Finally, new palliative care delivery models must fit within a larger national strategy to grow palliative care across the care continuum.11 This is of particular importance with hospital-focused solutions such as PRISM due to concerns about the growing split in care coordination between inpatient and outpatient care. Since seriously ill patients spend the majority of time outside the hospital and evidence for the value of palliative care is most robust in home and ambulatory settings,1 an important role for hospitalists could be to systematically identify and refer high-risk patients to community-based palliative care services after discharge from a sentinel hospitalization.
In conclusion, innovative palliative care delivery models such as PRISM are critical to ensuring that seriously ill patients have access to high-quality palliative care; however, more work is still needed to create the training programs, patient identification tools, scalable implementation, and evaluation processes necessary for success.
Disclosures
Dr. Courtright and Dr. O’Connor have nothing to disclose.
Funding
This work was funded in part by a career development award from the National Palliative Care Research Center (KRC). The views expressed herein solely represent those of the authors.
1. Kavalieratos D, Corbelli J, Zhang D, et al. Association between palliative care and patient and caregiver outcomes. Jama. 2016;316(20):2104. doi: 10.1001/jama.2016.16840. PubMed
2. May P, Normand C, Cassel JB, et al. Economics of palliative care for hospitalized adults with serious illness. JAMA Intern Med. 2018;178(6):820. doi: 10.1001/jamainternmed.2018.0750. PubMed
3. Dumanovsky T, Augustin R, Rogers M, Lettang K, Meier DE, Morrison RS. The growth of palliative care in U.S. hospitals: a status report. J Palliat Med. 2016;19(1):8-15. doi: 10.1089/jpm.2015.0351. PubMed
4. Heyland DK. Failure to Engage hospitalized elderly patients and their families in advance care planning. JAMA Intern Med. 2013;173(9):778. doi: 10.1001/jamainternmed.2013.180. PubMed
5. Courtright KR, Cassel JB, Halpern SD. A research agenda for high-value palliative care. Ann Intern Med. 2017;168(1):71. doi: 10.7326/m17-2164. PubMed
6. Billings JA, Bernacki R. Strategic targeting of advance care planning interventions. JAMA Intern Med. 2014;174(4):620. doi: 10.1001/jamainternmed.2013.14384. PubMed
7. Abedini NC, Chopra V. A Model to Improve Hospital-Based Palliative Care: The Palliative Care Redistribution Integrated System Model (PRISM). J Hosp Med. 2018;13(12):868-871. doi: 10.12788/jhm.3065 PubMed
8. Bernacki R, Hutchings M, Vick J, et al. Development of the Serious Illness Care Program: a randomized controlled trial of a palliative care communication intervention. BMJ Open. 2015;5(10):e009032. doi: 10.1136/bmjopen-2015-009032. PubMed
9. Clayton JM, Butow PN, Waters A, et al. Evaluation of a novel individualized communication-skills training intervention to improve doctors’ confidence and skills in end-of-life communication. Palliat Med. 2012;27(3):236-243. doi: 10.1177/0269216312449683. PubMed
10. Kamal AH, Bull JH, Wolf SP, et al. Prevalence and predictors of burnout among hospice and palliative care clinicians in the U.S. J Pain Symptom Manag. 2016;51(4):690-696. doi: 10.1016/j.jpainsymman.2015.10.020. PubMed
11. Meier DE, Back AL, Berman A, Block SD, Corrigan JM, Morrison RS. A national strategy for palliative care. Health Aff (Millwood). 2017;36(7):1265-1273. doi: 10.1377/hlthaff.2017.0164. PubMed
1. Kavalieratos D, Corbelli J, Zhang D, et al. Association between palliative care and patient and caregiver outcomes. Jama. 2016;316(20):2104. doi: 10.1001/jama.2016.16840. PubMed
2. May P, Normand C, Cassel JB, et al. Economics of palliative care for hospitalized adults with serious illness. JAMA Intern Med. 2018;178(6):820. doi: 10.1001/jamainternmed.2018.0750. PubMed
3. Dumanovsky T, Augustin R, Rogers M, Lettang K, Meier DE, Morrison RS. The growth of palliative care in U.S. hospitals: a status report. J Palliat Med. 2016;19(1):8-15. doi: 10.1089/jpm.2015.0351. PubMed
4. Heyland DK. Failure to Engage hospitalized elderly patients and their families in advance care planning. JAMA Intern Med. 2013;173(9):778. doi: 10.1001/jamainternmed.2013.180. PubMed
5. Courtright KR, Cassel JB, Halpern SD. A research agenda for high-value palliative care. Ann Intern Med. 2017;168(1):71. doi: 10.7326/m17-2164. PubMed
6. Billings JA, Bernacki R. Strategic targeting of advance care planning interventions. JAMA Intern Med. 2014;174(4):620. doi: 10.1001/jamainternmed.2013.14384. PubMed
7. Abedini NC, Chopra V. A Model to Improve Hospital-Based Palliative Care: The Palliative Care Redistribution Integrated System Model (PRISM). J Hosp Med. 2018;13(12):868-871. doi: 10.12788/jhm.3065 PubMed
8. Bernacki R, Hutchings M, Vick J, et al. Development of the Serious Illness Care Program: a randomized controlled trial of a palliative care communication intervention. BMJ Open. 2015;5(10):e009032. doi: 10.1136/bmjopen-2015-009032. PubMed
9. Clayton JM, Butow PN, Waters A, et al. Evaluation of a novel individualized communication-skills training intervention to improve doctors’ confidence and skills in end-of-life communication. Palliat Med. 2012;27(3):236-243. doi: 10.1177/0269216312449683. PubMed
10. Kamal AH, Bull JH, Wolf SP, et al. Prevalence and predictors of burnout among hospice and palliative care clinicians in the U.S. J Pain Symptom Manag. 2016;51(4):690-696. doi: 10.1016/j.jpainsymman.2015.10.020. PubMed
11. Meier DE, Back AL, Berman A, Block SD, Corrigan JM, Morrison RS. A national strategy for palliative care. Health Aff (Millwood). 2017;36(7):1265-1273. doi: 10.1377/hlthaff.2017.0164. PubMed
© 2018 Society of Hospital Medicine
December 2018 Digital Edition
Click here to access the December 2018 Digital Edition.
Table of Contents
- Anesthesia Care Practice Models in the Veterans Health Administration
- Role of Point-of-Care Ultrasonography in the Evaluation and Management of Kidney Disease
- PACT ICU Model: Interprofessional Case Conferences for High-Risk/High-Need Patients
- A Pharmacist-Led Transitional Care Program to Reduce Hospital Readmissions in Older Adults
- The Gift and the Thought Both Count
- Overemphasizing Communities in the National Suicide Strategy Could Undercut VA Successes

Click here to access the December 2018 Digital Edition.
Table of Contents
- Anesthesia Care Practice Models in the Veterans Health Administration
- Role of Point-of-Care Ultrasonography in the Evaluation and Management of Kidney Disease
- PACT ICU Model: Interprofessional Case Conferences for High-Risk/High-Need Patients
- A Pharmacist-Led Transitional Care Program to Reduce Hospital Readmissions in Older Adults
- The Gift and the Thought Both Count
- Overemphasizing Communities in the National Suicide Strategy Could Undercut VA Successes
Click here to access the December 2018 Digital Edition.
Table of Contents
- Anesthesia Care Practice Models in the Veterans Health Administration
- Role of Point-of-Care Ultrasonography in the Evaluation and Management of Kidney Disease
- PACT ICU Model: Interprofessional Case Conferences for High-Risk/High-Need Patients
- A Pharmacist-Led Transitional Care Program to Reduce Hospital Readmissions in Older Adults
- The Gift and the Thought Both Count
- Overemphasizing Communities in the National Suicide Strategy Could Undercut VA Successes