User login
Drug receives priority review for FL

The US Food and Drug Administration (FDA) has granted priority review to a supplemental biologics license application (sBLA) for obinutuzumab (Gazyva®).
With this sBLA, Genentech is seeking approval for obinutuzumab to be used, first in combination with chemotherapy and then alone as maintenance, in patients with previously untreated follicular lymphoma (FL).
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA plans to make a decision on the sBLA for obinutuzumab by December 23, 2017.
The sBLA is supported by results of the GALLIUM study, which were presented at the 2016 ASH Annual Meeting.
About obinutuzumab
Obinutuzumab is a glycoengineered, humanized, monoclonal antibody that selectively binds to the extracellular domain of the CD20 antigen on B cells.
The drug is FDA-approved for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
Obinutuzumab is also approved to treat FL patients who relapse after, or are refractory to, a rituximab-containing regimen. In these patients, obinutuzumab is given first in combination with bendamustine and then alone as maintenance.
The full prescribing information for obinutuzumab is available at http://www.Gazyva.com.
About the GALLIUM study
GALLIUM enrolled 1401 patients with previously untreated, indolent non-Hodgkin lymphoma, including 1202 with FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
Patients who received obinutuzumab had significantly better progression-free survival than patients who received rituximab. The 3-year progression-free survival rate was 73.3% in the rituximab arm and 80% in the obinutuzumab arm (hazard ratio [HR]=0.66, P=0.0012).
There was no significant difference between the treatment arms with regard to overall survival. The 3-year overall survival was 92.1% in the rituximab arm and 94% in the obinutuzumab arm (HR=0.75, P=0.21).
The overall incidence of adverse events (AEs) was 98.3% in the rituximab arm and 99.5% in the obinutuzumab arm. The incidence of serious AEs was 39.9% and 46.1%, respectively.
The incidence of grade 3 or higher AEs was higher among patients who received obinutuzumab.
Grade 3 or higher AEs occurring in at least 5% of patients in either arm (rituximab and obinutuzumab, respectively) included neutropenia (67.8% and 74.6%), leukopenia (37.9% and 43.9%), febrile neutropenia (4.9% and 6.9%), infections and infestations (3.7% and 6.7%), and thrombocytopenia (2.7% and 6.1%). ![]()
The US Food and Drug Administration (FDA) has granted priority review to a supplemental biologics license application (sBLA) for obinutuzumab (Gazyva®).
With this sBLA, Genentech is seeking approval for obinutuzumab to be used, first in combination with chemotherapy and then alone as maintenance, in patients with previously untreated follicular lymphoma (FL).
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA plans to make a decision on the sBLA for obinutuzumab by December 23, 2017.
The sBLA is supported by results of the GALLIUM study, which were presented at the 2016 ASH Annual Meeting.
About obinutuzumab
Obinutuzumab is a glycoengineered, humanized, monoclonal antibody that selectively binds to the extracellular domain of the CD20 antigen on B cells.
The drug is FDA-approved for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
Obinutuzumab is also approved to treat FL patients who relapse after, or are refractory to, a rituximab-containing regimen. In these patients, obinutuzumab is given first in combination with bendamustine and then alone as maintenance.
The full prescribing information for obinutuzumab is available at http://www.Gazyva.com.
About the GALLIUM study
GALLIUM enrolled 1401 patients with previously untreated, indolent non-Hodgkin lymphoma, including 1202 with FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
Patients who received obinutuzumab had significantly better progression-free survival than patients who received rituximab. The 3-year progression-free survival rate was 73.3% in the rituximab arm and 80% in the obinutuzumab arm (hazard ratio [HR]=0.66, P=0.0012).
There was no significant difference between the treatment arms with regard to overall survival. The 3-year overall survival was 92.1% in the rituximab arm and 94% in the obinutuzumab arm (HR=0.75, P=0.21).
The overall incidence of adverse events (AEs) was 98.3% in the rituximab arm and 99.5% in the obinutuzumab arm. The incidence of serious AEs was 39.9% and 46.1%, respectively.
The incidence of grade 3 or higher AEs was higher among patients who received obinutuzumab.
Grade 3 or higher AEs occurring in at least 5% of patients in either arm (rituximab and obinutuzumab, respectively) included neutropenia (67.8% and 74.6%), leukopenia (37.9% and 43.9%), febrile neutropenia (4.9% and 6.9%), infections and infestations (3.7% and 6.7%), and thrombocytopenia (2.7% and 6.1%). ![]()
The US Food and Drug Administration (FDA) has granted priority review to a supplemental biologics license application (sBLA) for obinutuzumab (Gazyva®).
With this sBLA, Genentech is seeking approval for obinutuzumab to be used, first in combination with chemotherapy and then alone as maintenance, in patients with previously untreated follicular lymphoma (FL).
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA plans to make a decision on the sBLA for obinutuzumab by December 23, 2017.
The sBLA is supported by results of the GALLIUM study, which were presented at the 2016 ASH Annual Meeting.
About obinutuzumab
Obinutuzumab is a glycoengineered, humanized, monoclonal antibody that selectively binds to the extracellular domain of the CD20 antigen on B cells.
The drug is FDA-approved for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
Obinutuzumab is also approved to treat FL patients who relapse after, or are refractory to, a rituximab-containing regimen. In these patients, obinutuzumab is given first in combination with bendamustine and then alone as maintenance.
The full prescribing information for obinutuzumab is available at http://www.Gazyva.com.
About the GALLIUM study
GALLIUM enrolled 1401 patients with previously untreated, indolent non-Hodgkin lymphoma, including 1202 with FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
Patients who received obinutuzumab had significantly better progression-free survival than patients who received rituximab. The 3-year progression-free survival rate was 73.3% in the rituximab arm and 80% in the obinutuzumab arm (hazard ratio [HR]=0.66, P=0.0012).
There was no significant difference between the treatment arms with regard to overall survival. The 3-year overall survival was 92.1% in the rituximab arm and 94% in the obinutuzumab arm (HR=0.75, P=0.21).
The overall incidence of adverse events (AEs) was 98.3% in the rituximab arm and 99.5% in the obinutuzumab arm. The incidence of serious AEs was 39.9% and 46.1%, respectively.
The incidence of grade 3 or higher AEs was higher among patients who received obinutuzumab.
Grade 3 or higher AEs occurring in at least 5% of patients in either arm (rituximab and obinutuzumab, respectively) included neutropenia (67.8% and 74.6%), leukopenia (37.9% and 43.9%), febrile neutropenia (4.9% and 6.9%), infections and infestations (3.7% and 6.7%), and thrombocytopenia (2.7% and 6.1%). ![]()
Bacterial infection inhibits hematopoiesis

New research suggests that bacterial infection activates hematopoietic stem cells (HSCs) and significantly reduces their ability to produce blood by forcibly inducing proliferation.
These findings indicate that bacterial infections might cause dysregulated hematopoiesis like that which occurs in patients with anemias and leukemias.
Hitoshi Takizawa, PhD, of Kumamoto University’s International Research Center for Medical Sciences in Kumamoto, Japan, and his colleagues reported these findings in Cell Stem Cell.
The researchers gave lipopolysaccharide to mice to generate a bacterial infection model and analyzed the role of Toll-like receptors (TLRs) in HSCs.
The team found that lipopolysaccharides spread throughout the body, with some eventually reaching the bone marrow. This stimulated TLR4 and induced HSCs to proliferate.
However, the stimulus also induced stress on the HSCs.
So although HSCs proliferate temporarily upon TLR4 stimulation, their ability to successfully self-replicate decreases, resulting in diminished blood production.
The researchers observed similar results after infecting mice with Salmonella Typhimurium.
“Fortunately, we were able to confirm that this molecular reaction can be inhibited by drugs,” Dr Takizawa said.
Specifically, the researchers found that activation of reactive oxygen species and p38 through TLR4 ligation is key to HSC dysfunction.
And small-molecule inhibitors of reactive oxygen species and p38 were able to prevent HSC dysfunction.
“The medication maintains the production of blood and immune cells without weakening the immune reaction against pathogenic bacteria,” Dr Takizawa said. “It might be possible to simultaneously prevent blood diseases and many bacterial infections in the future.” ![]()
New research suggests that bacterial infection activates hematopoietic stem cells (HSCs) and significantly reduces their ability to produce blood by forcibly inducing proliferation.
These findings indicate that bacterial infections might cause dysregulated hematopoiesis like that which occurs in patients with anemias and leukemias.
Hitoshi Takizawa, PhD, of Kumamoto University’s International Research Center for Medical Sciences in Kumamoto, Japan, and his colleagues reported these findings in Cell Stem Cell.
The researchers gave lipopolysaccharide to mice to generate a bacterial infection model and analyzed the role of Toll-like receptors (TLRs) in HSCs.
The team found that lipopolysaccharides spread throughout the body, with some eventually reaching the bone marrow. This stimulated TLR4 and induced HSCs to proliferate.
However, the stimulus also induced stress on the HSCs.
So although HSCs proliferate temporarily upon TLR4 stimulation, their ability to successfully self-replicate decreases, resulting in diminished blood production.
The researchers observed similar results after infecting mice with Salmonella Typhimurium.
“Fortunately, we were able to confirm that this molecular reaction can be inhibited by drugs,” Dr Takizawa said.
Specifically, the researchers found that activation of reactive oxygen species and p38 through TLR4 ligation is key to HSC dysfunction.
And small-molecule inhibitors of reactive oxygen species and p38 were able to prevent HSC dysfunction.
“The medication maintains the production of blood and immune cells without weakening the immune reaction against pathogenic bacteria,” Dr Takizawa said. “It might be possible to simultaneously prevent blood diseases and many bacterial infections in the future.” ![]()
New research suggests that bacterial infection activates hematopoietic stem cells (HSCs) and significantly reduces their ability to produce blood by forcibly inducing proliferation.
These findings indicate that bacterial infections might cause dysregulated hematopoiesis like that which occurs in patients with anemias and leukemias.
Hitoshi Takizawa, PhD, of Kumamoto University’s International Research Center for Medical Sciences in Kumamoto, Japan, and his colleagues reported these findings in Cell Stem Cell.
The researchers gave lipopolysaccharide to mice to generate a bacterial infection model and analyzed the role of Toll-like receptors (TLRs) in HSCs.
The team found that lipopolysaccharides spread throughout the body, with some eventually reaching the bone marrow. This stimulated TLR4 and induced HSCs to proliferate.
However, the stimulus also induced stress on the HSCs.
So although HSCs proliferate temporarily upon TLR4 stimulation, their ability to successfully self-replicate decreases, resulting in diminished blood production.
The researchers observed similar results after infecting mice with Salmonella Typhimurium.
“Fortunately, we were able to confirm that this molecular reaction can be inhibited by drugs,” Dr Takizawa said.
Specifically, the researchers found that activation of reactive oxygen species and p38 through TLR4 ligation is key to HSC dysfunction.
And small-molecule inhibitors of reactive oxygen species and p38 were able to prevent HSC dysfunction.
“The medication maintains the production of blood and immune cells without weakening the immune reaction against pathogenic bacteria,” Dr Takizawa said. “It might be possible to simultaneously prevent blood diseases and many bacterial infections in the future.” ![]()
Simplified HOSPITAL score predicts 30-day readmissions
Clinical Question: Will a simplified HOSPITAL score accurately predict 30-day readmissions?
Background: Hospital readmissions stress patients and health care systems. Interventions to prevent avoidable readmissions are complex and expensive. The HOSPITAL score predicts 30-day readmissions which may help direct resources toward high risk patients.

Setting: Nine hospitals in four countries.
Synopsis: The HOSPITAL score was simplified by removing the procedure variable, expanding the oncology criteria to include a diagnosis of cancer, and dividing patients into high and low risk groups. The simplified HOSPITAL score was used to predict avoidable readmissions of 117,065 patients from nine hospitals. Readmission rates predicted by the simplified HOSPITAL score matched observed outcomes with a sensitivity of 94% and specificity of 73%. Its discriminatory power was comparable with the original HOSPITAL score.
This was a robust study of medical patients but may not be generalizable to surgical patients. The score does not include patients’ socioeconomic status or support systems. It also cannot indicate what type of intervention may prevent readmissions.
Bottom Line: The simplified HOSPITAL score accurately predicts avoidable 30-day readmission rates.
Citation: Aubert CE, Schnipper JL, Williams MV, et al. Simplification of the HOSPITAL score for predicting 30-day readmissions. BMJ Qual Saf. Published online first. 17 Apr 2017. doi: 10.1136/bmjqs-2016-006239.
Dr. Helfrich is an assistant professor in the University of Kentucky division of hospital medicine.
Clinical Question: Will a simplified HOSPITAL score accurately predict 30-day readmissions?
Background: Hospital readmissions stress patients and health care systems. Interventions to prevent avoidable readmissions are complex and expensive. The HOSPITAL score predicts 30-day readmissions which may help direct resources toward high risk patients.
Setting: Nine hospitals in four countries.
Synopsis: The HOSPITAL score was simplified by removing the procedure variable, expanding the oncology criteria to include a diagnosis of cancer, and dividing patients into high and low risk groups. The simplified HOSPITAL score was used to predict avoidable readmissions of 117,065 patients from nine hospitals. Readmission rates predicted by the simplified HOSPITAL score matched observed outcomes with a sensitivity of 94% and specificity of 73%. Its discriminatory power was comparable with the original HOSPITAL score.
This was a robust study of medical patients but may not be generalizable to surgical patients. The score does not include patients’ socioeconomic status or support systems. It also cannot indicate what type of intervention may prevent readmissions.
Bottom Line: The simplified HOSPITAL score accurately predicts avoidable 30-day readmission rates.
Citation: Aubert CE, Schnipper JL, Williams MV, et al. Simplification of the HOSPITAL score for predicting 30-day readmissions. BMJ Qual Saf. Published online first. 17 Apr 2017. doi: 10.1136/bmjqs-2016-006239.
Dr. Helfrich is an assistant professor in the University of Kentucky division of hospital medicine.
Clinical Question: Will a simplified HOSPITAL score accurately predict 30-day readmissions?
Background: Hospital readmissions stress patients and health care systems. Interventions to prevent avoidable readmissions are complex and expensive. The HOSPITAL score predicts 30-day readmissions which may help direct resources toward high risk patients.
Setting: Nine hospitals in four countries.
Synopsis: The HOSPITAL score was simplified by removing the procedure variable, expanding the oncology criteria to include a diagnosis of cancer, and dividing patients into high and low risk groups. The simplified HOSPITAL score was used to predict avoidable readmissions of 117,065 patients from nine hospitals. Readmission rates predicted by the simplified HOSPITAL score matched observed outcomes with a sensitivity of 94% and specificity of 73%. Its discriminatory power was comparable with the original HOSPITAL score.
This was a robust study of medical patients but may not be generalizable to surgical patients. The score does not include patients’ socioeconomic status or support systems. It also cannot indicate what type of intervention may prevent readmissions.
Bottom Line: The simplified HOSPITAL score accurately predicts avoidable 30-day readmission rates.
Citation: Aubert CE, Schnipper JL, Williams MV, et al. Simplification of the HOSPITAL score for predicting 30-day readmissions. BMJ Qual Saf. Published online first. 17 Apr 2017. doi: 10.1136/bmjqs-2016-006239.
Dr. Helfrich is an assistant professor in the University of Kentucky division of hospital medicine.

Patient Preference Before and After Arthroscopic Rotator Cuff Repair: Which Is More Important, Pain Relief or Strength Return?
Take-Home Points
- Pain relief and return of strength are important satisfaction variables for patients undergoing ARCR.
- Pain relief and strength return are equally desirable in the majority (50%) of the patients before and after ARCR.
- Overall, patient preference for strength return dominates pain relief in long-term.
- Increasing age is associated with a stronger preference for pain relief.
- Improved understanding of patient expectations after ARCR will promote meaningful changes in patient satisfaction.
A rotator cuff tear (RCT) can cause significant pain, weakness, stiffness, and loss of function in the shoulder. In most patients, arthroscopic rotator cuff repair (ARCR) provides significant and reproducible pain relief and variable return of shoulder strength and function.1-4 ARCR outcomes are well described and well represented by validated outcome measures.5-9 However, these outcomes do not always correlate with patient satisfaction. For example, after ARCR, 2 patients with similar outcome scores may have different satisfaction levels.
Patient satisfaction involves multiple factors and varies with the patient’s preoperative expectations and the degree to which the surgery matches the patient’s desired outcomes.10-15 In clinical studies, Tashjian and colleagues,10 Henn and colleagues,11 and O’Holleran and colleagues12 found patient satisfaction correlated most highly with postoperative shoulder pain, shoulder function, general health status, and outcome scores. However, our understanding of patients’ desired outcomes and expectations of ARCR is limited, particularly regarding the importance of pain relief and strength return relative to each other. We believe patients’ preoperative expectations are influenced by their self-assessments of symptom severity and by their understanding of the outcomes of surgical procedures and of the information they receive from their surgeons during preoperative evaluation.
We conducted an observational study to determine patients’ preoperative preferences and the importance of post-ARCR pain relief and strength return relative to each other. After surgery, preferences and ratings of pain relief and strength return were reevaluated to determine if they were altered by outcomes. We also studied the influence of multiple factors, including severity of preoperative symptoms (pain, weakness), age, sex, occupation, and active sports involvement, on patients’ preoperative ratings of the importance of post-ARCR improvements in pain relief and strength return. We hypothesized that patients would vary in how they preoperatively value and desire post-ARCR pain relief and strength return.
Materials and Methods
The simple shoulder questionnaire (Figure) designed for this study had 12 items. Patients subjectively assessed the severity of their symptoms (pain level, shoulder weakness) and rated the importance of both pain relief and strength return to their occupational and personal life.

Before patients underwent surgery for symptomatic suspected RCTs, they were approached to participate in this prospective study. Sixty-five patients provided informed consent on forms approved by an Institutional Review Board. Inclusion criteria were suspected unilateral rotator cuff pathology and willingness to participate. Of the 65 patients, 60 underwent ARCR without another procedure, such as shoulder instability repair, SLAP (superior labrum anterior-to-posterior) repair, or distal clavicle excision; the other 5 patients elected nonoperative treatment and were excluded from review. At a mean (SD) follow-up of 5.2 (0.2) years, the 60 patients who had surgery completed the questionnaire again and rated the importance of pain relief and strength return relative to each other.
Patients with RCTs were divided according to age, sex, shoulder dominance, occupation type, and active sports involvement. Standard definitions for occupation types were used: blue-collar, manual labor jobs; white-collar, salaried/educated positions; and retired.
Matched-pairs t tests were used to compare preoperative and postoperative continuous variables (strength return preference, pain relief preference, SPD). One-way analysis of variance (ANOVA) was used to compare categorical variables (sex, shoulder dominance, active sports involvement) with continuous variables (SPD), and bivariate regression was used to compare groups with continuous data (age, SPD). In cases involving more than 2 groups (occupation types), the Tukey honestly significant difference (HSD) test was used to evaluate intergroup differences. P < .05 was used for statistical significance.
Results
ARCR Outcomes
After ARCR, there was significant improvement in patient-reported pain and subjective strength scores. Mean (SD) pain score improved from 5.9 (2.3) to 1.3 (2.3) after ARCR (P < .001), and mean (SD) strength improved from 46% (22%) of normal to 84% (17%) of normal (P < .001).
Importance of Post-ARCR Pain Relief and Strength Return
Analysis of preoperative questionnaire responses
revealed that, of 60 patients, 29 (48.3%) considered pain relief and strength return equally important, 20 (33.3%) valued postoperative strength return was more important, and 11 patients (18.3%) rated pain relief was more important than strength return. After a mean (SD) follow-up of 5.2 (0.2) years, 33 patients (55 %) valued pain relief and strength return as equally important, 17 patients (28.3%) preferred a strength recovery, and 10 patients (16.7%) preferred pain relief.
Overall patient ratings were significantly higher for strength return compared to pain relief before surgery, mean (SD), 9.2 (2.1) and 8.6 (2.3) (P = .02), and afterward, 8.9 (1.9) and 8.2 (3.1) (P = .03) (Table 1).

Subgroup Analyses
Sex and Age. Of the 60 patients, 43 were male and 17 female. Mean (SD) preoperative SPD was 1.0 (2.7) for males and 0.7 (2.3) females; the difference was not significant (P = .61). After surgery, females emphasized strength return over pain relief more than males did: Mean (SD) SPD was significantly higher (P = .04) for females, 1.7 (3.0), than for males, 0.4 (2.5). There were no preoperative–postoperative differences (P = .33) for males or females (Table 2).

Hand Dominance. RCT was found in the dominant shoulder of 31 patients (52%). Shoulder dominance did not affect SPD: Mean (SD) preoperative SPD was 1.3 (2.3) for dominant shoulders and 0.5 (2.7) for nondominant shoulders (P = .21), and postoperative SPD was 0.7 (2.6) for dominant and 0.9 (2.8) for nondominant (P = .79). SPD did not change from before surgery to after surgery for dominant (P = .14) or nondominant (P = .28) shoulders (Table 2).
Active Sports Participation. Thirty-two patients (53%) reported preoperative involvement in sports; 35 (58%) reported postoperative involvement (P = .37). Mean (SD) preoperative SPD was 1.4 (3.0) for involved patients and 0.3 (1.7) for uninvolved patients (P = .09), and postoperative SPD was 0.6 (2.8) for involved patients and 1.0 (2.6) for uninvolved patients (P = .53). SPD did not change from before surgery to after surgery for involved (P = .17) or uninvolved (P = .26) patients (Table 2).
Occupation Type. There were 9 blue-collar workers (15%), 32 white-collar workers (53%), and 19 retirees (32%). Mean (SD) preoperative SPD was 2.8 (4.2) for blue-collar workers, 1.2 (2.1) for white-collar workers, and –0.4 (0.4) for retirees. There were no significant differences in preoperative SPD between blue-collar and white-collar workers (P = .19) or between white-collar workers and retirees (P = .06), but there was a significant difference between blue-collar workers and retirees (P = .004). Mean (SD) postoperative SPD was 1.3 (2.7) for blue-collar workers, 1.2 (3.1) for white-collar workers, and –0.3 (1.6) for retirees. There were no significant differences between blue-collar and white-collar workers (P = .99), white-collar workers and retirees (P = .13), or blue-collar workers and retirees (P = .3).
Discussion
In this study, we wanted to determine patients’ pre- and postoperative preferences for pain relief and strength return after ARCR. Preoperative and postoperative preference analysis of the 60 patients who underwent ARCR revealed that the majority valued pain relief and strength return equally. However, overall, there was higher ratings for strength return in long term after ARCR, irrespective of age, sex, preoperative levels of shoulder pain and weakness, and preoperative and postoperative sports involvement.
Patients’ preoperative expectations are a function of their assessment of their symptoms, their perceptions of expected surgical outcomes, and their understanding of preoperative discussion with their surgeons. In this study, patients self-assessed their shoulder symptoms and their effect on their occupational and personal life. They also rated the importance of post-ARCR pain relief and strength return relative to each other. To assess whether surgical outcomes affected perceptions of pain relief and strength return, patients completed the questionnaire before and after surgery. Overall, patients rated postoperative strength return over pain relief on long-term (5 years).
Subgroup analysis revealed a weak positive correlation between patient-reported preoperative pain scores and ratings of the importance of pain relief after surgery, but there was no correlation between postoperative pain scores and ratings of the importance of pain relief after surgery. This finding was surprising because we thought pain relief would be more important than strength return for patients with higher pain scores.1-3,16-21 We would like to clarify a point about this study: That patients preferred strength return over pain relief does not mean they did not care about pain relief. A substantial subset of patients (~50%) valued pain relief and strength return equally. In rotator cuff pathology, pain and weakness are to an extent interrelated. Shoulder pain that limits a patient’s ability to perform a strenuous task can be perceived as shoulder weakness, which may explain why, despite having higher pain scores, patients preferred strength return over pain relief. Increasing age showed a positive correlation with preference for pain relief, which explains the finding that retirees preferred pain relief over strength return. We used SPD to express the preference for strength return over pain relief before and after ARCR. Unfortunately, SPD may not be used to quantitatively define the preference for strength return over pain relief.
Patient satisfaction after RCR involves multiple factors and has been well studied. In a retrospective analysis of 112 patients, Tashjian and colleagues10 found that patient satisfaction was affected by preoperative expectations, marital status, disability status, preoperative pain function, and general health status after RCR. They also found a positive but weak correlation between patient satisfaction and functional outcome scores, including visual analog scale (VAS), Simple Shoulder Test (SST), and Disabilities of the Arm, Shoulder, and Hand (DASH) scores. Henn and colleagues11 evaluated 125 patients who underwent primary RCR for a chronic RCT. Higher preoperative expectations correlated with better postoperative VAS, SST, DASH, and Short Form 36 performance, irrespective of worker compensation status, symptom duration, number of patient comorbidities, tear size, repair technique, and number of previous operations. In a prospective cohort analysis of 311 RCR patients, O’Holleran and colleagues12 found that decreased patient satisfaction was associated with postoperative pain and dysfunction. Furthermore, willingness to recommend surgery to another person was significantly related to patient satisfaction. In the present study, we did not correlate preoperative expectations with postoperative outcome scores or evaluate the effect of other known factors on RCR outcomes. Our main goal was to understand ARCR patients’ preoperative and postoperative evaluations of the importance of pain relief and strength return relative to each other. Improved understanding of patients’ expectations will allow us to identify disparities between expectations and outcomes.
Our study had several limitations. First, our questionnaire was not validated. However, we used it only as an assessment tool, to collect data, and do not propose using it to assess ARCR outcomes. Second, objective strength measurements were not performed, before or after surgery, and therefore patients’ perceptions of weakness were not tested. Third, we did not correlate preoperative or postoperative shoulder outcome scores with patients’ expectations. Our intention was to understand how ARCR patients rate the importance of pain relief and strength return relative to each other. Fourth, we did not correlate patients’ expectations of strength return and pain relief with preoperative tear size or postoperative retear status.
Our observational study results showed that, before undergoing ARCR, most patients valued postoperative pain relief and strength return equally. However, there was an overall preference for strength return over pain relief. Furthermore, this preference held up irrespective of age, sex, sports involvement, or preoperative symptom severity. These findings add to our understanding of patients’ preoperative expectations of ARCR.
Am J Orthop. 2017;46(4):E244-E250. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Cole BJ, McCarty LP 3rd, Kang RW, Alford W, Lewis PB, Hayden JK. Arthroscopic rotator cuff repair: prospective functional outcome and repair integrity at minimum 2-year follow-up. J Shoulder Elbow Surg. 2007;16(5):579-585.
2. Huijsmans PE, Pritchard MP, Berghs BM, van Rooyen KS, Wallace AL, de Beer JF. Arthroscopic rotator cuff repair with double-row fixation. J Bone Joint Surg Am. 2007;89(6):1248-1257.
3. Wilson F, Hinov V, Adams G. Arthroscopic repair of full-thickness tears of the rotator cuff: 2- to 14-year follow-up. Arthroscopy. 2002;18(2):136-144.
4. Denard PJ, Jiwani AZ, Lädermann A, Burkhart SS. Long-term outcome of a consecutive series of subscapularis tendon tears repaired arthroscopically. Arthroscopy. 2012;28(11):1587-1591.
5. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3(6):347-352.
6. Roach KE, Budiman-Mak E, Songsiridej N, Lertratanakul Y. Development of a shoulder pain and disability index. Arthritis Care Res. 1991;4(4):143-149.
7. Constant CR, Murley AH. A clinical method of functional assessment of the shoulder. Clin Orthop Relat Res. 1987;(214):160-164.
8. Michener LA, McClure PW, Sennett BJ. American Shoulder and Elbow Surgeons Standardized Shoulder Assessment Form, patient self-report section: reliability, validity, and responsiveness. J Shoulder Elbow Surg. 2002;11(6):587-594.
9. Romeo AA, Bach BR Jr, O’Halloran KL. Scoring systems for shoulder conditions. Am J Sports Med. 1996;24(4):472-476.
10. Tashjian RZ, Bradley MP, Tocci S, Rey J, Henn RF, Green A. Factors influencing patient satisfaction after rotator cuff repair. J Shoulder Elbow Surg. 2007;16(6):752-758.
11. Henn RF 3rd, Kang L, Tashjian RZ, Green A. Patients’ preoperative expectations predict the outcome of rotator cuff repair. J Bone Joint Surg Am. 2007;89(9):1913-1919.
12. O’Holleran JD, Kocher MS, Horan MP, Briggs KK, Hawkins RJ. Determinants of patient satisfaction with outcome after rotator cuff surgery. J Bone Joint Surg Am. 2005;87(1):121-126.
13. Namdari S, Donegan RP, Chamberlain AM, Galatz LM, Yamaguchi K, Keener JD. Factors affecting outcome after structural failure of repaired rotator cuff tears. J Bone Joint Surg Am. 2014;96(2):99-105.
14. Nho SJ, Brown BS, Lyman S, Adler RS, Altchek DW, MacGillivray JD. Prospective analysis of arthroscopic rotator cuff repair: prognostic factors affecting clinical and ultrasound outcome. J Shoulder Elbow Surg. 2009;18(1):13-20.
15. Sonnabend DH, Watson EM. Structural factors affecting the outcome of rotator cuff repair. J Shoulder Elbow Surg. 2002;11(3):212-218.
16. Boileau P, Brassart N, Watkinson DJ, Carles M, Hatzidakis AM, Krishnan SG. Arthroscopic repair of full-thickness tears of the supraspinatus: does the tendon really heal? J Bone Joint Surg Am. 2005;87(6):1229-1240.
17. Sugaya H, Maeda K, Matsuki K, Moriishi J. Repair integrity and functional outcome after arthroscopic double-row rotator cuff repair. A prospective outcome study. J Bone Joint Surg Am. 2007;89(5):953-960.
18. DeFranco MJ, Bershadsky B, Ciccone J, Yum JK, Iannotti JP. Functional outcome of arthroscopic rotator cuff repairs: a correlation of anatomic and clinical results. J Shoulder Elbow Surg. 2007;16(6):759-765.
19. Galatz LM, Ball CM, Teefey SA, Middleton WD, Yamaguchi K. The outcome and repair integrity of completely arthroscopically repaired large and massive rotator cuff tears. J Bone Joint Surg Am. 2004;86(2):219-224.
20. Harryman DT 2nd, Mack LA, Wang KY, Jackins SE, Richardson ML, Matsen FA 3rd. Repairs of the rotator cuff. Correlation of functional results with integrity of the cuff. J Bone Joint Surg Am. 1991;73(7):982-989.
21. Romeo AA, Hang DW, Bach BR Jr, Shott S. Repair of full thickness rotator cuff tears. Gender, age, and other factors affecting outcome. Clin Orthop Relat Res. 1999;(367):243-255.
Take-Home Points
- Pain relief and return of strength are important satisfaction variables for patients undergoing ARCR.
- Pain relief and strength return are equally desirable in the majority (50%) of the patients before and after ARCR.
- Overall, patient preference for strength return dominates pain relief in long-term.
- Increasing age is associated with a stronger preference for pain relief.
- Improved understanding of patient expectations after ARCR will promote meaningful changes in patient satisfaction.
A rotator cuff tear (RCT) can cause significant pain, weakness, stiffness, and loss of function in the shoulder. In most patients, arthroscopic rotator cuff repair (ARCR) provides significant and reproducible pain relief and variable return of shoulder strength and function.1-4 ARCR outcomes are well described and well represented by validated outcome measures.5-9 However, these outcomes do not always correlate with patient satisfaction. For example, after ARCR, 2 patients with similar outcome scores may have different satisfaction levels.
Patient satisfaction involves multiple factors and varies with the patient’s preoperative expectations and the degree to which the surgery matches the patient’s desired outcomes.10-15 In clinical studies, Tashjian and colleagues,10 Henn and colleagues,11 and O’Holleran and colleagues12 found patient satisfaction correlated most highly with postoperative shoulder pain, shoulder function, general health status, and outcome scores. However, our understanding of patients’ desired outcomes and expectations of ARCR is limited, particularly regarding the importance of pain relief and strength return relative to each other. We believe patients’ preoperative expectations are influenced by their self-assessments of symptom severity and by their understanding of the outcomes of surgical procedures and of the information they receive from their surgeons during preoperative evaluation.
We conducted an observational study to determine patients’ preoperative preferences and the importance of post-ARCR pain relief and strength return relative to each other. After surgery, preferences and ratings of pain relief and strength return were reevaluated to determine if they were altered by outcomes. We also studied the influence of multiple factors, including severity of preoperative symptoms (pain, weakness), age, sex, occupation, and active sports involvement, on patients’ preoperative ratings of the importance of post-ARCR improvements in pain relief and strength return. We hypothesized that patients would vary in how they preoperatively value and desire post-ARCR pain relief and strength return.
Materials and Methods
The simple shoulder questionnaire (Figure) designed for this study had 12 items. Patients subjectively assessed the severity of their symptoms (pain level, shoulder weakness) and rated the importance of both pain relief and strength return to their occupational and personal life.
Before patients underwent surgery for symptomatic suspected RCTs, they were approached to participate in this prospective study. Sixty-five patients provided informed consent on forms approved by an Institutional Review Board. Inclusion criteria were suspected unilateral rotator cuff pathology and willingness to participate. Of the 65 patients, 60 underwent ARCR without another procedure, such as shoulder instability repair, SLAP (superior labrum anterior-to-posterior) repair, or distal clavicle excision; the other 5 patients elected nonoperative treatment and were excluded from review. At a mean (SD) follow-up of 5.2 (0.2) years, the 60 patients who had surgery completed the questionnaire again and rated the importance of pain relief and strength return relative to each other.
Patients with RCTs were divided according to age, sex, shoulder dominance, occupation type, and active sports involvement. Standard definitions for occupation types were used: blue-collar, manual labor jobs; white-collar, salaried/educated positions; and retired.
Matched-pairs t tests were used to compare preoperative and postoperative continuous variables (strength return preference, pain relief preference, SPD). One-way analysis of variance (ANOVA) was used to compare categorical variables (sex, shoulder dominance, active sports involvement) with continuous variables (SPD), and bivariate regression was used to compare groups with continuous data (age, SPD). In cases involving more than 2 groups (occupation types), the Tukey honestly significant difference (HSD) test was used to evaluate intergroup differences. P < .05 was used for statistical significance.
Results
ARCR Outcomes
After ARCR, there was significant improvement in patient-reported pain and subjective strength scores. Mean (SD) pain score improved from 5.9 (2.3) to 1.3 (2.3) after ARCR (P < .001), and mean (SD) strength improved from 46% (22%) of normal to 84% (17%) of normal (P < .001).
Importance of Post-ARCR Pain Relief and Strength Return
Analysis of preoperative questionnaire responses
revealed that, of 60 patients, 29 (48.3%) considered pain relief and strength return equally important, 20 (33.3%) valued postoperative strength return was more important, and 11 patients (18.3%) rated pain relief was more important than strength return. After a mean (SD) follow-up of 5.2 (0.2) years, 33 patients (55 %) valued pain relief and strength return as equally important, 17 patients (28.3%) preferred a strength recovery, and 10 patients (16.7%) preferred pain relief.
Overall patient ratings were significantly higher for strength return compared to pain relief before surgery, mean (SD), 9.2 (2.1) and 8.6 (2.3) (P = .02), and afterward, 8.9 (1.9) and 8.2 (3.1) (P = .03) (Table 1).
Subgroup Analyses
Sex and Age. Of the 60 patients, 43 were male and 17 female. Mean (SD) preoperative SPD was 1.0 (2.7) for males and 0.7 (2.3) females; the difference was not significant (P = .61). After surgery, females emphasized strength return over pain relief more than males did: Mean (SD) SPD was significantly higher (P = .04) for females, 1.7 (3.0), than for males, 0.4 (2.5). There were no preoperative–postoperative differences (P = .33) for males or females (Table 2).
Hand Dominance. RCT was found in the dominant shoulder of 31 patients (52%). Shoulder dominance did not affect SPD: Mean (SD) preoperative SPD was 1.3 (2.3) for dominant shoulders and 0.5 (2.7) for nondominant shoulders (P = .21), and postoperative SPD was 0.7 (2.6) for dominant and 0.9 (2.8) for nondominant (P = .79). SPD did not change from before surgery to after surgery for dominant (P = .14) or nondominant (P = .28) shoulders (Table 2).
Active Sports Participation. Thirty-two patients (53%) reported preoperative involvement in sports; 35 (58%) reported postoperative involvement (P = .37). Mean (SD) preoperative SPD was 1.4 (3.0) for involved patients and 0.3 (1.7) for uninvolved patients (P = .09), and postoperative SPD was 0.6 (2.8) for involved patients and 1.0 (2.6) for uninvolved patients (P = .53). SPD did not change from before surgery to after surgery for involved (P = .17) or uninvolved (P = .26) patients (Table 2).
Occupation Type. There were 9 blue-collar workers (15%), 32 white-collar workers (53%), and 19 retirees (32%). Mean (SD) preoperative SPD was 2.8 (4.2) for blue-collar workers, 1.2 (2.1) for white-collar workers, and –0.4 (0.4) for retirees. There were no significant differences in preoperative SPD between blue-collar and white-collar workers (P = .19) or between white-collar workers and retirees (P = .06), but there was a significant difference between blue-collar workers and retirees (P = .004). Mean (SD) postoperative SPD was 1.3 (2.7) for blue-collar workers, 1.2 (3.1) for white-collar workers, and –0.3 (1.6) for retirees. There were no significant differences between blue-collar and white-collar workers (P = .99), white-collar workers and retirees (P = .13), or blue-collar workers and retirees (P = .3).
Discussion
In this study, we wanted to determine patients’ pre- and postoperative preferences for pain relief and strength return after ARCR. Preoperative and postoperative preference analysis of the 60 patients who underwent ARCR revealed that the majority valued pain relief and strength return equally. However, overall, there was higher ratings for strength return in long term after ARCR, irrespective of age, sex, preoperative levels of shoulder pain and weakness, and preoperative and postoperative sports involvement.
Patients’ preoperative expectations are a function of their assessment of their symptoms, their perceptions of expected surgical outcomes, and their understanding of preoperative discussion with their surgeons. In this study, patients self-assessed their shoulder symptoms and their effect on their occupational and personal life. They also rated the importance of post-ARCR pain relief and strength return relative to each other. To assess whether surgical outcomes affected perceptions of pain relief and strength return, patients completed the questionnaire before and after surgery. Overall, patients rated postoperative strength return over pain relief on long-term (5 years).
Subgroup analysis revealed a weak positive correlation between patient-reported preoperative pain scores and ratings of the importance of pain relief after surgery, but there was no correlation between postoperative pain scores and ratings of the importance of pain relief after surgery. This finding was surprising because we thought pain relief would be more important than strength return for patients with higher pain scores.1-3,16-21 We would like to clarify a point about this study: That patients preferred strength return over pain relief does not mean they did not care about pain relief. A substantial subset of patients (~50%) valued pain relief and strength return equally. In rotator cuff pathology, pain and weakness are to an extent interrelated. Shoulder pain that limits a patient’s ability to perform a strenuous task can be perceived as shoulder weakness, which may explain why, despite having higher pain scores, patients preferred strength return over pain relief. Increasing age showed a positive correlation with preference for pain relief, which explains the finding that retirees preferred pain relief over strength return. We used SPD to express the preference for strength return over pain relief before and after ARCR. Unfortunately, SPD may not be used to quantitatively define the preference for strength return over pain relief.
Patient satisfaction after RCR involves multiple factors and has been well studied. In a retrospective analysis of 112 patients, Tashjian and colleagues10 found that patient satisfaction was affected by preoperative expectations, marital status, disability status, preoperative pain function, and general health status after RCR. They also found a positive but weak correlation between patient satisfaction and functional outcome scores, including visual analog scale (VAS), Simple Shoulder Test (SST), and Disabilities of the Arm, Shoulder, and Hand (DASH) scores. Henn and colleagues11 evaluated 125 patients who underwent primary RCR for a chronic RCT. Higher preoperative expectations correlated with better postoperative VAS, SST, DASH, and Short Form 36 performance, irrespective of worker compensation status, symptom duration, number of patient comorbidities, tear size, repair technique, and number of previous operations. In a prospective cohort analysis of 311 RCR patients, O’Holleran and colleagues12 found that decreased patient satisfaction was associated with postoperative pain and dysfunction. Furthermore, willingness to recommend surgery to another person was significantly related to patient satisfaction. In the present study, we did not correlate preoperative expectations with postoperative outcome scores or evaluate the effect of other known factors on RCR outcomes. Our main goal was to understand ARCR patients’ preoperative and postoperative evaluations of the importance of pain relief and strength return relative to each other. Improved understanding of patients’ expectations will allow us to identify disparities between expectations and outcomes.
Our study had several limitations. First, our questionnaire was not validated. However, we used it only as an assessment tool, to collect data, and do not propose using it to assess ARCR outcomes. Second, objective strength measurements were not performed, before or after surgery, and therefore patients’ perceptions of weakness were not tested. Third, we did not correlate preoperative or postoperative shoulder outcome scores with patients’ expectations. Our intention was to understand how ARCR patients rate the importance of pain relief and strength return relative to each other. Fourth, we did not correlate patients’ expectations of strength return and pain relief with preoperative tear size or postoperative retear status.
Our observational study results showed that, before undergoing ARCR, most patients valued postoperative pain relief and strength return equally. However, there was an overall preference for strength return over pain relief. Furthermore, this preference held up irrespective of age, sex, sports involvement, or preoperative symptom severity. These findings add to our understanding of patients’ preoperative expectations of ARCR.
Am J Orthop. 2017;46(4):E244-E250. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Pain relief and return of strength are important satisfaction variables for patients undergoing ARCR.
- Pain relief and strength return are equally desirable in the majority (50%) of the patients before and after ARCR.
- Overall, patient preference for strength return dominates pain relief in long-term.
- Increasing age is associated with a stronger preference for pain relief.
- Improved understanding of patient expectations after ARCR will promote meaningful changes in patient satisfaction.
A rotator cuff tear (RCT) can cause significant pain, weakness, stiffness, and loss of function in the shoulder. In most patients, arthroscopic rotator cuff repair (ARCR) provides significant and reproducible pain relief and variable return of shoulder strength and function.1-4 ARCR outcomes are well described and well represented by validated outcome measures.5-9 However, these outcomes do not always correlate with patient satisfaction. For example, after ARCR, 2 patients with similar outcome scores may have different satisfaction levels.
Patient satisfaction involves multiple factors and varies with the patient’s preoperative expectations and the degree to which the surgery matches the patient’s desired outcomes.10-15 In clinical studies, Tashjian and colleagues,10 Henn and colleagues,11 and O’Holleran and colleagues12 found patient satisfaction correlated most highly with postoperative shoulder pain, shoulder function, general health status, and outcome scores. However, our understanding of patients’ desired outcomes and expectations of ARCR is limited, particularly regarding the importance of pain relief and strength return relative to each other. We believe patients’ preoperative expectations are influenced by their self-assessments of symptom severity and by their understanding of the outcomes of surgical procedures and of the information they receive from their surgeons during preoperative evaluation.
We conducted an observational study to determine patients’ preoperative preferences and the importance of post-ARCR pain relief and strength return relative to each other. After surgery, preferences and ratings of pain relief and strength return were reevaluated to determine if they were altered by outcomes. We also studied the influence of multiple factors, including severity of preoperative symptoms (pain, weakness), age, sex, occupation, and active sports involvement, on patients’ preoperative ratings of the importance of post-ARCR improvements in pain relief and strength return. We hypothesized that patients would vary in how they preoperatively value and desire post-ARCR pain relief and strength return.
Materials and Methods
The simple shoulder questionnaire (Figure) designed for this study had 12 items. Patients subjectively assessed the severity of their symptoms (pain level, shoulder weakness) and rated the importance of both pain relief and strength return to their occupational and personal life.
Before patients underwent surgery for symptomatic suspected RCTs, they were approached to participate in this prospective study. Sixty-five patients provided informed consent on forms approved by an Institutional Review Board. Inclusion criteria were suspected unilateral rotator cuff pathology and willingness to participate. Of the 65 patients, 60 underwent ARCR without another procedure, such as shoulder instability repair, SLAP (superior labrum anterior-to-posterior) repair, or distal clavicle excision; the other 5 patients elected nonoperative treatment and were excluded from review. At a mean (SD) follow-up of 5.2 (0.2) years, the 60 patients who had surgery completed the questionnaire again and rated the importance of pain relief and strength return relative to each other.
Patients with RCTs were divided according to age, sex, shoulder dominance, occupation type, and active sports involvement. Standard definitions for occupation types were used: blue-collar, manual labor jobs; white-collar, salaried/educated positions; and retired.
Matched-pairs t tests were used to compare preoperative and postoperative continuous variables (strength return preference, pain relief preference, SPD). One-way analysis of variance (ANOVA) was used to compare categorical variables (sex, shoulder dominance, active sports involvement) with continuous variables (SPD), and bivariate regression was used to compare groups with continuous data (age, SPD). In cases involving more than 2 groups (occupation types), the Tukey honestly significant difference (HSD) test was used to evaluate intergroup differences. P < .05 was used for statistical significance.
Results
ARCR Outcomes
After ARCR, there was significant improvement in patient-reported pain and subjective strength scores. Mean (SD) pain score improved from 5.9 (2.3) to 1.3 (2.3) after ARCR (P < .001), and mean (SD) strength improved from 46% (22%) of normal to 84% (17%) of normal (P < .001).
Importance of Post-ARCR Pain Relief and Strength Return
Analysis of preoperative questionnaire responses
revealed that, of 60 patients, 29 (48.3%) considered pain relief and strength return equally important, 20 (33.3%) valued postoperative strength return was more important, and 11 patients (18.3%) rated pain relief was more important than strength return. After a mean (SD) follow-up of 5.2 (0.2) years, 33 patients (55 %) valued pain relief and strength return as equally important, 17 patients (28.3%) preferred a strength recovery, and 10 patients (16.7%) preferred pain relief.
Overall patient ratings were significantly higher for strength return compared to pain relief before surgery, mean (SD), 9.2 (2.1) and 8.6 (2.3) (P = .02), and afterward, 8.9 (1.9) and 8.2 (3.1) (P = .03) (Table 1).
Subgroup Analyses
Sex and Age. Of the 60 patients, 43 were male and 17 female. Mean (SD) preoperative SPD was 1.0 (2.7) for males and 0.7 (2.3) females; the difference was not significant (P = .61). After surgery, females emphasized strength return over pain relief more than males did: Mean (SD) SPD was significantly higher (P = .04) for females, 1.7 (3.0), than for males, 0.4 (2.5). There were no preoperative–postoperative differences (P = .33) for males or females (Table 2).
Hand Dominance. RCT was found in the dominant shoulder of 31 patients (52%). Shoulder dominance did not affect SPD: Mean (SD) preoperative SPD was 1.3 (2.3) for dominant shoulders and 0.5 (2.7) for nondominant shoulders (P = .21), and postoperative SPD was 0.7 (2.6) for dominant and 0.9 (2.8) for nondominant (P = .79). SPD did not change from before surgery to after surgery for dominant (P = .14) or nondominant (P = .28) shoulders (Table 2).
Active Sports Participation. Thirty-two patients (53%) reported preoperative involvement in sports; 35 (58%) reported postoperative involvement (P = .37). Mean (SD) preoperative SPD was 1.4 (3.0) for involved patients and 0.3 (1.7) for uninvolved patients (P = .09), and postoperative SPD was 0.6 (2.8) for involved patients and 1.0 (2.6) for uninvolved patients (P = .53). SPD did not change from before surgery to after surgery for involved (P = .17) or uninvolved (P = .26) patients (Table 2).
Occupation Type. There were 9 blue-collar workers (15%), 32 white-collar workers (53%), and 19 retirees (32%). Mean (SD) preoperative SPD was 2.8 (4.2) for blue-collar workers, 1.2 (2.1) for white-collar workers, and –0.4 (0.4) for retirees. There were no significant differences in preoperative SPD between blue-collar and white-collar workers (P = .19) or between white-collar workers and retirees (P = .06), but there was a significant difference between blue-collar workers and retirees (P = .004). Mean (SD) postoperative SPD was 1.3 (2.7) for blue-collar workers, 1.2 (3.1) for white-collar workers, and –0.3 (1.6) for retirees. There were no significant differences between blue-collar and white-collar workers (P = .99), white-collar workers and retirees (P = .13), or blue-collar workers and retirees (P = .3).
Discussion
In this study, we wanted to determine patients’ pre- and postoperative preferences for pain relief and strength return after ARCR. Preoperative and postoperative preference analysis of the 60 patients who underwent ARCR revealed that the majority valued pain relief and strength return equally. However, overall, there was higher ratings for strength return in long term after ARCR, irrespective of age, sex, preoperative levels of shoulder pain and weakness, and preoperative and postoperative sports involvement.
Patients’ preoperative expectations are a function of their assessment of their symptoms, their perceptions of expected surgical outcomes, and their understanding of preoperative discussion with their surgeons. In this study, patients self-assessed their shoulder symptoms and their effect on their occupational and personal life. They also rated the importance of post-ARCR pain relief and strength return relative to each other. To assess whether surgical outcomes affected perceptions of pain relief and strength return, patients completed the questionnaire before and after surgery. Overall, patients rated postoperative strength return over pain relief on long-term (5 years).
Subgroup analysis revealed a weak positive correlation between patient-reported preoperative pain scores and ratings of the importance of pain relief after surgery, but there was no correlation between postoperative pain scores and ratings of the importance of pain relief after surgery. This finding was surprising because we thought pain relief would be more important than strength return for patients with higher pain scores.1-3,16-21 We would like to clarify a point about this study: That patients preferred strength return over pain relief does not mean they did not care about pain relief. A substantial subset of patients (~50%) valued pain relief and strength return equally. In rotator cuff pathology, pain and weakness are to an extent interrelated. Shoulder pain that limits a patient’s ability to perform a strenuous task can be perceived as shoulder weakness, which may explain why, despite having higher pain scores, patients preferred strength return over pain relief. Increasing age showed a positive correlation with preference for pain relief, which explains the finding that retirees preferred pain relief over strength return. We used SPD to express the preference for strength return over pain relief before and after ARCR. Unfortunately, SPD may not be used to quantitatively define the preference for strength return over pain relief.
Patient satisfaction after RCR involves multiple factors and has been well studied. In a retrospective analysis of 112 patients, Tashjian and colleagues10 found that patient satisfaction was affected by preoperative expectations, marital status, disability status, preoperative pain function, and general health status after RCR. They also found a positive but weak correlation between patient satisfaction and functional outcome scores, including visual analog scale (VAS), Simple Shoulder Test (SST), and Disabilities of the Arm, Shoulder, and Hand (DASH) scores. Henn and colleagues11 evaluated 125 patients who underwent primary RCR for a chronic RCT. Higher preoperative expectations correlated with better postoperative VAS, SST, DASH, and Short Form 36 performance, irrespective of worker compensation status, symptom duration, number of patient comorbidities, tear size, repair technique, and number of previous operations. In a prospective cohort analysis of 311 RCR patients, O’Holleran and colleagues12 found that decreased patient satisfaction was associated with postoperative pain and dysfunction. Furthermore, willingness to recommend surgery to another person was significantly related to patient satisfaction. In the present study, we did not correlate preoperative expectations with postoperative outcome scores or evaluate the effect of other known factors on RCR outcomes. Our main goal was to understand ARCR patients’ preoperative and postoperative evaluations of the importance of pain relief and strength return relative to each other. Improved understanding of patients’ expectations will allow us to identify disparities between expectations and outcomes.
Our study had several limitations. First, our questionnaire was not validated. However, we used it only as an assessment tool, to collect data, and do not propose using it to assess ARCR outcomes. Second, objective strength measurements were not performed, before or after surgery, and therefore patients’ perceptions of weakness were not tested. Third, we did not correlate preoperative or postoperative shoulder outcome scores with patients’ expectations. Our intention was to understand how ARCR patients rate the importance of pain relief and strength return relative to each other. Fourth, we did not correlate patients’ expectations of strength return and pain relief with preoperative tear size or postoperative retear status.
Our observational study results showed that, before undergoing ARCR, most patients valued postoperative pain relief and strength return equally. However, there was an overall preference for strength return over pain relief. Furthermore, this preference held up irrespective of age, sex, sports involvement, or preoperative symptom severity. These findings add to our understanding of patients’ preoperative expectations of ARCR.
Am J Orthop. 2017;46(4):E244-E250. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Cole BJ, McCarty LP 3rd, Kang RW, Alford W, Lewis PB, Hayden JK. Arthroscopic rotator cuff repair: prospective functional outcome and repair integrity at minimum 2-year follow-up. J Shoulder Elbow Surg. 2007;16(5):579-585.
2. Huijsmans PE, Pritchard MP, Berghs BM, van Rooyen KS, Wallace AL, de Beer JF. Arthroscopic rotator cuff repair with double-row fixation. J Bone Joint Surg Am. 2007;89(6):1248-1257.
3. Wilson F, Hinov V, Adams G. Arthroscopic repair of full-thickness tears of the rotator cuff: 2- to 14-year follow-up. Arthroscopy. 2002;18(2):136-144.
4. Denard PJ, Jiwani AZ, Lädermann A, Burkhart SS. Long-term outcome of a consecutive series of subscapularis tendon tears repaired arthroscopically. Arthroscopy. 2012;28(11):1587-1591.
5. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3(6):347-352.
6. Roach KE, Budiman-Mak E, Songsiridej N, Lertratanakul Y. Development of a shoulder pain and disability index. Arthritis Care Res. 1991;4(4):143-149.
7. Constant CR, Murley AH. A clinical method of functional assessment of the shoulder. Clin Orthop Relat Res. 1987;(214):160-164.
8. Michener LA, McClure PW, Sennett BJ. American Shoulder and Elbow Surgeons Standardized Shoulder Assessment Form, patient self-report section: reliability, validity, and responsiveness. J Shoulder Elbow Surg. 2002;11(6):587-594.
9. Romeo AA, Bach BR Jr, O’Halloran KL. Scoring systems for shoulder conditions. Am J Sports Med. 1996;24(4):472-476.
10. Tashjian RZ, Bradley MP, Tocci S, Rey J, Henn RF, Green A. Factors influencing patient satisfaction after rotator cuff repair. J Shoulder Elbow Surg. 2007;16(6):752-758.
11. Henn RF 3rd, Kang L, Tashjian RZ, Green A. Patients’ preoperative expectations predict the outcome of rotator cuff repair. J Bone Joint Surg Am. 2007;89(9):1913-1919.
12. O’Holleran JD, Kocher MS, Horan MP, Briggs KK, Hawkins RJ. Determinants of patient satisfaction with outcome after rotator cuff surgery. J Bone Joint Surg Am. 2005;87(1):121-126.
13. Namdari S, Donegan RP, Chamberlain AM, Galatz LM, Yamaguchi K, Keener JD. Factors affecting outcome after structural failure of repaired rotator cuff tears. J Bone Joint Surg Am. 2014;96(2):99-105.
14. Nho SJ, Brown BS, Lyman S, Adler RS, Altchek DW, MacGillivray JD. Prospective analysis of arthroscopic rotator cuff repair: prognostic factors affecting clinical and ultrasound outcome. J Shoulder Elbow Surg. 2009;18(1):13-20.
15. Sonnabend DH, Watson EM. Structural factors affecting the outcome of rotator cuff repair. J Shoulder Elbow Surg. 2002;11(3):212-218.
16. Boileau P, Brassart N, Watkinson DJ, Carles M, Hatzidakis AM, Krishnan SG. Arthroscopic repair of full-thickness tears of the supraspinatus: does the tendon really heal? J Bone Joint Surg Am. 2005;87(6):1229-1240.
17. Sugaya H, Maeda K, Matsuki K, Moriishi J. Repair integrity and functional outcome after arthroscopic double-row rotator cuff repair. A prospective outcome study. J Bone Joint Surg Am. 2007;89(5):953-960.
18. DeFranco MJ, Bershadsky B, Ciccone J, Yum JK, Iannotti JP. Functional outcome of arthroscopic rotator cuff repairs: a correlation of anatomic and clinical results. J Shoulder Elbow Surg. 2007;16(6):759-765.
19. Galatz LM, Ball CM, Teefey SA, Middleton WD, Yamaguchi K. The outcome and repair integrity of completely arthroscopically repaired large and massive rotator cuff tears. J Bone Joint Surg Am. 2004;86(2):219-224.
20. Harryman DT 2nd, Mack LA, Wang KY, Jackins SE, Richardson ML, Matsen FA 3rd. Repairs of the rotator cuff. Correlation of functional results with integrity of the cuff. J Bone Joint Surg Am. 1991;73(7):982-989.
21. Romeo AA, Hang DW, Bach BR Jr, Shott S. Repair of full thickness rotator cuff tears. Gender, age, and other factors affecting outcome. Clin Orthop Relat Res. 1999;(367):243-255.
1. Cole BJ, McCarty LP 3rd, Kang RW, Alford W, Lewis PB, Hayden JK. Arthroscopic rotator cuff repair: prospective functional outcome and repair integrity at minimum 2-year follow-up. J Shoulder Elbow Surg. 2007;16(5):579-585.
2. Huijsmans PE, Pritchard MP, Berghs BM, van Rooyen KS, Wallace AL, de Beer JF. Arthroscopic rotator cuff repair with double-row fixation. J Bone Joint Surg Am. 2007;89(6):1248-1257.
3. Wilson F, Hinov V, Adams G. Arthroscopic repair of full-thickness tears of the rotator cuff: 2- to 14-year follow-up. Arthroscopy. 2002;18(2):136-144.
4. Denard PJ, Jiwani AZ, Lädermann A, Burkhart SS. Long-term outcome of a consecutive series of subscapularis tendon tears repaired arthroscopically. Arthroscopy. 2012;28(11):1587-1591.
5. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3(6):347-352.
6. Roach KE, Budiman-Mak E, Songsiridej N, Lertratanakul Y. Development of a shoulder pain and disability index. Arthritis Care Res. 1991;4(4):143-149.
7. Constant CR, Murley AH. A clinical method of functional assessment of the shoulder. Clin Orthop Relat Res. 1987;(214):160-164.
8. Michener LA, McClure PW, Sennett BJ. American Shoulder and Elbow Surgeons Standardized Shoulder Assessment Form, patient self-report section: reliability, validity, and responsiveness. J Shoulder Elbow Surg. 2002;11(6):587-594.
9. Romeo AA, Bach BR Jr, O’Halloran KL. Scoring systems for shoulder conditions. Am J Sports Med. 1996;24(4):472-476.
10. Tashjian RZ, Bradley MP, Tocci S, Rey J, Henn RF, Green A. Factors influencing patient satisfaction after rotator cuff repair. J Shoulder Elbow Surg. 2007;16(6):752-758.
11. Henn RF 3rd, Kang L, Tashjian RZ, Green A. Patients’ preoperative expectations predict the outcome of rotator cuff repair. J Bone Joint Surg Am. 2007;89(9):1913-1919.
12. O’Holleran JD, Kocher MS, Horan MP, Briggs KK, Hawkins RJ. Determinants of patient satisfaction with outcome after rotator cuff surgery. J Bone Joint Surg Am. 2005;87(1):121-126.
13. Namdari S, Donegan RP, Chamberlain AM, Galatz LM, Yamaguchi K, Keener JD. Factors affecting outcome after structural failure of repaired rotator cuff tears. J Bone Joint Surg Am. 2014;96(2):99-105.
14. Nho SJ, Brown BS, Lyman S, Adler RS, Altchek DW, MacGillivray JD. Prospective analysis of arthroscopic rotator cuff repair: prognostic factors affecting clinical and ultrasound outcome. J Shoulder Elbow Surg. 2009;18(1):13-20.
15. Sonnabend DH, Watson EM. Structural factors affecting the outcome of rotator cuff repair. J Shoulder Elbow Surg. 2002;11(3):212-218.
16. Boileau P, Brassart N, Watkinson DJ, Carles M, Hatzidakis AM, Krishnan SG. Arthroscopic repair of full-thickness tears of the supraspinatus: does the tendon really heal? J Bone Joint Surg Am. 2005;87(6):1229-1240.
17. Sugaya H, Maeda K, Matsuki K, Moriishi J. Repair integrity and functional outcome after arthroscopic double-row rotator cuff repair. A prospective outcome study. J Bone Joint Surg Am. 2007;89(5):953-960.
18. DeFranco MJ, Bershadsky B, Ciccone J, Yum JK, Iannotti JP. Functional outcome of arthroscopic rotator cuff repairs: a correlation of anatomic and clinical results. J Shoulder Elbow Surg. 2007;16(6):759-765.
19. Galatz LM, Ball CM, Teefey SA, Middleton WD, Yamaguchi K. The outcome and repair integrity of completely arthroscopically repaired large and massive rotator cuff tears. J Bone Joint Surg Am. 2004;86(2):219-224.
20. Harryman DT 2nd, Mack LA, Wang KY, Jackins SE, Richardson ML, Matsen FA 3rd. Repairs of the rotator cuff. Correlation of functional results with integrity of the cuff. J Bone Joint Surg Am. 1991;73(7):982-989.
21. Romeo AA, Hang DW, Bach BR Jr, Shott S. Repair of full thickness rotator cuff tears. Gender, age, and other factors affecting outcome. Clin Orthop Relat Res. 1999;(367):243-255.
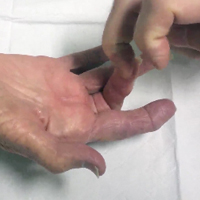
Percutaneous Trigger Finger Release
PD3 G1 SNA found to correlate with pentavalent rotavirus vaccine efficacy
(RV5), reported G. Frank Liu, PhD, and his associates at Merck in Kenilworth, N.J.
The researchers discovered this by analyzing data from five clinical trials of RV5 at both the individual and aggregated population level. Also, by looking at immunogenicity measures and case status of individuals from phase 2 and 3 trials of RV5, they found that “higher PD3 G1 SNA titers are associated with lower odds of contracting any [rotavirus gastroenteritis].”
Read more at (Hum Vaccin Immunother. 2017. doi: 10.1080/21645515.2017.1356522).
[email protected]
(RV5), reported G. Frank Liu, PhD, and his associates at Merck in Kenilworth, N.J.
The researchers discovered this by analyzing data from five clinical trials of RV5 at both the individual and aggregated population level. Also, by looking at immunogenicity measures and case status of individuals from phase 2 and 3 trials of RV5, they found that “higher PD3 G1 SNA titers are associated with lower odds of contracting any [rotavirus gastroenteritis].”
Read more at (Hum Vaccin Immunother. 2017. doi: 10.1080/21645515.2017.1356522).
[email protected]
(RV5), reported G. Frank Liu, PhD, and his associates at Merck in Kenilworth, N.J.
The researchers discovered this by analyzing data from five clinical trials of RV5 at both the individual and aggregated population level. Also, by looking at immunogenicity measures and case status of individuals from phase 2 and 3 trials of RV5, they found that “higher PD3 G1 SNA titers are associated with lower odds of contracting any [rotavirus gastroenteritis].”
Read more at (Hum Vaccin Immunother. 2017. doi: 10.1080/21645515.2017.1356522).
[email protected]
FROM HUMAN VACCINES & IMMUNOTHERAPEUTICS
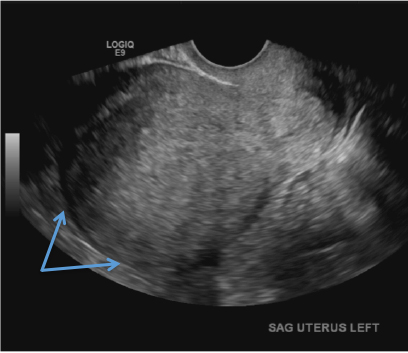
Adenomyosis in the spotlight, but which sign is featured?
A) Globular enlarged uterus INCORRECT
A homogeneously enlarged uterus in the absence of fibroids is characteristic of adenomyosis.1
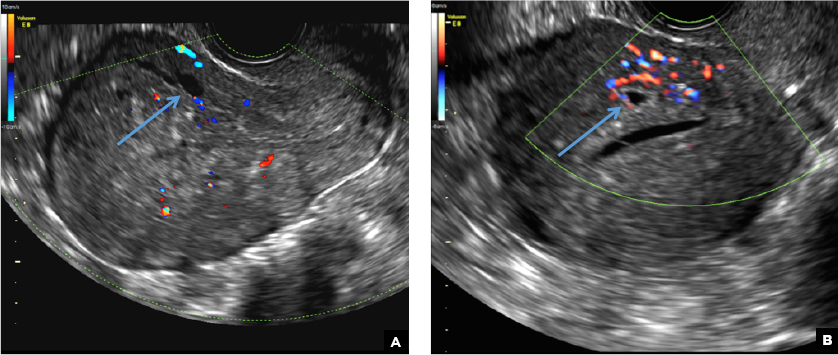
B) Cystic myometrial spaces INCORRECT
Myometrial cysts are dilated cystic glands or foci of hemorrhage within heterotopic endometrial tissue.2 They are often less than 5 mm in size but can be extensive and of variable sizes and can be differentiated from arcuate veins seen in the outer myometrium by the use of color Doppler.1,2
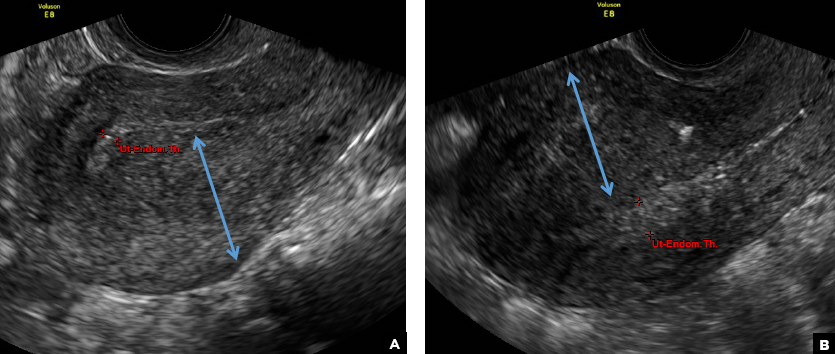
C) Asymmetric myometrial thickening INCORRECT
The finding of asymmetric uterine wall thickening in adenomyosis is usually seen when there is focal disease and presents with wall thickening that demonstrates anteroposterior asymmetry.1
D) Indistinct endomyometrial interface INCORRECT
The heterotopic endometrial tissue invading the myometrium obscures the normal endometrial myometrial border with pseudo-widening of the endometrial echo resulting in poor definition of the endomyometrial junction.1,2

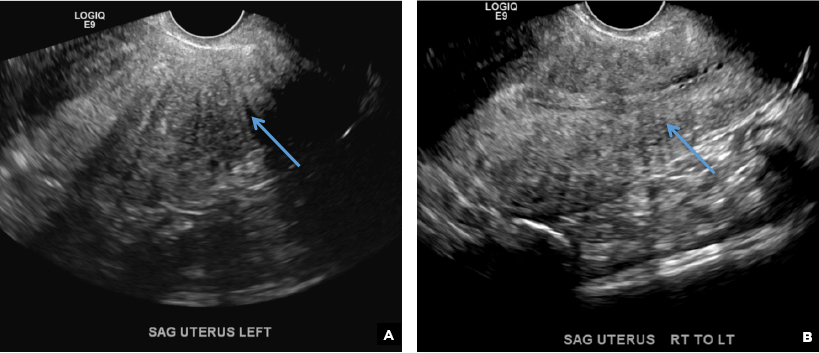
E) Myometrial linear striations CORRECT
A hyperplastic reaction to the infiltration of the heterotrophic endometrial glands into the myometrium results in radiating linear echogenic striations (sometimes referred to as the "venetian blinds" sign).1,2
- Sakhel A, Abuhamad A. Sonography of adenomyosis. J Ultrasound Med. 2012;31(5):805-808.
- Reinhold C, Tafazoli F, Mehio A, et al. Uterine adenomyosis: endovaginal US and MR imaging features with histopathologic correlation. Radiographics. 1999;19:S147-S160.

Dr. Kanmaniraja is Assistant Professor and Chief, Division of Abdominal Imaging, Department of Radiology, University of Florida College of Medicine-Jacksonville.

Dr. Kaunitz is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville. He is Medical Director and Director of Menopause and Gynecologic Ultrasound Services at UF Women's Health Specialists-Emerson. He also serves on the OBG Management Board of Editors.
The authors report no additional financial relationships relevant to this quiz.
Dr. Kanmaniraja is Assistant Professor and Chief, Division of Abdominal Imaging, Department of Radiology, University of Florida College of Medicine-Jacksonville.
Dr. Kaunitz is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville. He is Medical Director and Director of Menopause and Gynecologic Ultrasound Services at UF Women's Health Specialists-Emerson. He also serves on the OBG Management Board of Editors.
The authors report no additional financial relationships relevant to this quiz.
Dr. Kanmaniraja is Assistant Professor and Chief, Division of Abdominal Imaging, Department of Radiology, University of Florida College of Medicine-Jacksonville.
Dr. Kaunitz is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville. He is Medical Director and Director of Menopause and Gynecologic Ultrasound Services at UF Women's Health Specialists-Emerson. He also serves on the OBG Management Board of Editors.
The authors report no additional financial relationships relevant to this quiz.
A) Globular enlarged uterus INCORRECT
A homogeneously enlarged uterus in the absence of fibroids is characteristic of adenomyosis.1
B) Cystic myometrial spaces INCORRECT
Myometrial cysts are dilated cystic glands or foci of hemorrhage within heterotopic endometrial tissue.2 They are often less than 5 mm in size but can be extensive and of variable sizes and can be differentiated from arcuate veins seen in the outer myometrium by the use of color Doppler.1,2
C) Asymmetric myometrial thickening INCORRECT
The finding of asymmetric uterine wall thickening in adenomyosis is usually seen when there is focal disease and presents with wall thickening that demonstrates anteroposterior asymmetry.1
D) Indistinct endomyometrial interface INCORRECT
The heterotopic endometrial tissue invading the myometrium obscures the normal endometrial myometrial border with pseudo-widening of the endometrial echo resulting in poor definition of the endomyometrial junction.1,2
E) Myometrial linear striations CORRECT
A hyperplastic reaction to the infiltration of the heterotrophic endometrial glands into the myometrium results in radiating linear echogenic striations (sometimes referred to as the "venetian blinds" sign).1,2
A) Globular enlarged uterus INCORRECT
A homogeneously enlarged uterus in the absence of fibroids is characteristic of adenomyosis.1
B) Cystic myometrial spaces INCORRECT
Myometrial cysts are dilated cystic glands or foci of hemorrhage within heterotopic endometrial tissue.2 They are often less than 5 mm in size but can be extensive and of variable sizes and can be differentiated from arcuate veins seen in the outer myometrium by the use of color Doppler.1,2
C) Asymmetric myometrial thickening INCORRECT
The finding of asymmetric uterine wall thickening in adenomyosis is usually seen when there is focal disease and presents with wall thickening that demonstrates anteroposterior asymmetry.1
D) Indistinct endomyometrial interface INCORRECT
The heterotopic endometrial tissue invading the myometrium obscures the normal endometrial myometrial border with pseudo-widening of the endometrial echo resulting in poor definition of the endomyometrial junction.1,2
E) Myometrial linear striations CORRECT
A hyperplastic reaction to the infiltration of the heterotrophic endometrial glands into the myometrium results in radiating linear echogenic striations (sometimes referred to as the "venetian blinds" sign).1,2
- Sakhel A, Abuhamad A. Sonography of adenomyosis. J Ultrasound Med. 2012;31(5):805-808.
- Reinhold C, Tafazoli F, Mehio A, et al. Uterine adenomyosis: endovaginal US and MR imaging features with histopathologic correlation. Radiographics. 1999;19:S147-S160.
- Sakhel A, Abuhamad A. Sonography of adenomyosis. J Ultrasound Med. 2012;31(5):805-808.
- Reinhold C, Tafazoli F, Mehio A, et al. Uterine adenomyosis: endovaginal US and MR imaging features with histopathologic correlation. Radiographics. 1999;19:S147-S160.
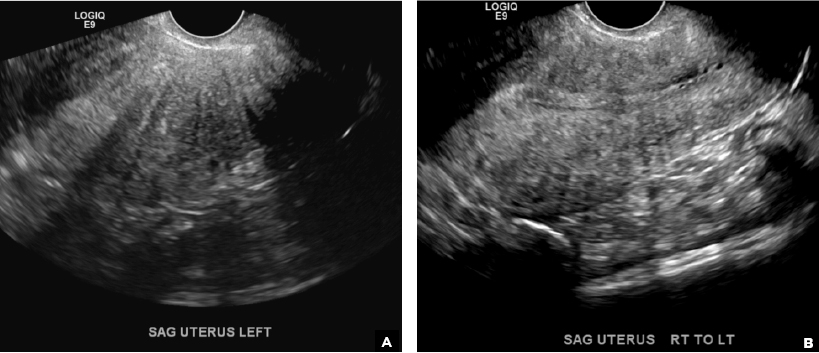
A 37-year-old woman with heavy menstrual bleeding and dysmenorrhea presents to her gynecologist. Physical exam suggests an enlarged uterus; the gynecologist orders pelvic ultrasonography.
Real-world adverse events reported after bivalent MenB vaccination in college students
, said Theresa M. Fiorito, MD, of Brown University, Providence, R.I., and her associates.
In February 2015, two unlinked, culture-confirmed cases of Neisseria meningitidis serogroup B (MenB) disease occurred at a local college in Rhode Island, and a bivalent MenB vaccine subsequently was administered. After the first-dose vaccination clinic, a 94% coverage rate was achieved, and there were no other cases of MenB disease. Injection site pain occurred in 78% of 1,736 students after dose one, in 64% of 1,395 after dose two, and in 71% of 609 students after dose three.
Serious adverse events, such as any allergic reaction; difficulty breathing; hives, welts, or a severe rash; and swelling of the face, mouth, or throat occurred in fewer than 5% of students after any of the three doses, the investigators reported.
“We found an overall lower rate of headache within our sample relative to the rates in reported clinical trials. The rates of injection site pain, fatigue, thermometer-confirmed fevers (100.4° F or higher), and chills were approximately similar in our study as in clinical trials. The rate of myalgia was higher for our sample than in clinical trials following the first and second doses of the vaccine, but similar after the third dose of vaccine,” Dr. Fiorito and her associates wrote.
Read more at (Pediatr Infect Dis J. 2017. doi: 10.1097/INF.0000000000001742).
, said Theresa M. Fiorito, MD, of Brown University, Providence, R.I., and her associates.
In February 2015, two unlinked, culture-confirmed cases of Neisseria meningitidis serogroup B (MenB) disease occurred at a local college in Rhode Island, and a bivalent MenB vaccine subsequently was administered. After the first-dose vaccination clinic, a 94% coverage rate was achieved, and there were no other cases of MenB disease. Injection site pain occurred in 78% of 1,736 students after dose one, in 64% of 1,395 after dose two, and in 71% of 609 students after dose three.
Serious adverse events, such as any allergic reaction; difficulty breathing; hives, welts, or a severe rash; and swelling of the face, mouth, or throat occurred in fewer than 5% of students after any of the three doses, the investigators reported.
“We found an overall lower rate of headache within our sample relative to the rates in reported clinical trials. The rates of injection site pain, fatigue, thermometer-confirmed fevers (100.4° F or higher), and chills were approximately similar in our study as in clinical trials. The rate of myalgia was higher for our sample than in clinical trials following the first and second doses of the vaccine, but similar after the third dose of vaccine,” Dr. Fiorito and her associates wrote.
Read more at (Pediatr Infect Dis J. 2017. doi: 10.1097/INF.0000000000001742).
, said Theresa M. Fiorito, MD, of Brown University, Providence, R.I., and her associates.
In February 2015, two unlinked, culture-confirmed cases of Neisseria meningitidis serogroup B (MenB) disease occurred at a local college in Rhode Island, and a bivalent MenB vaccine subsequently was administered. After the first-dose vaccination clinic, a 94% coverage rate was achieved, and there were no other cases of MenB disease. Injection site pain occurred in 78% of 1,736 students after dose one, in 64% of 1,395 after dose two, and in 71% of 609 students after dose three.
Serious adverse events, such as any allergic reaction; difficulty breathing; hives, welts, or a severe rash; and swelling of the face, mouth, or throat occurred in fewer than 5% of students after any of the three doses, the investigators reported.
“We found an overall lower rate of headache within our sample relative to the rates in reported clinical trials. The rates of injection site pain, fatigue, thermometer-confirmed fevers (100.4° F or higher), and chills were approximately similar in our study as in clinical trials. The rate of myalgia was higher for our sample than in clinical trials following the first and second doses of the vaccine, but similar after the third dose of vaccine,” Dr. Fiorito and her associates wrote.
Read more at (Pediatr Infect Dis J. 2017. doi: 10.1097/INF.0000000000001742).
FROM THE PEDIATRIC INFECTIOUS DISEASE JOURNAL
Dr. Britt awarded NIH grant to develop strategies to address health care disparities
L. D. Britt, MD, MPH, DSc(Hon), FACS, FCCM, FRCSEng(Hon), FRCSEd(Hon), FWACS(Hon), FRCSI(Hon), FCS(SA)(Hon), FRCSGlasg(Hon), Henry Ford Professor and Edward Brickhouse Chairman, department of surgery, Eastern Virginia Medical School, Norfolk, and a Past-President of the American College of Surgeons (ACS), was recently awarded a multimillion-dollar National Institutes of Health (NIH) research grant. The grant will be used to develop strategies to address health care disparities in the various surgical specialties. Specifically, the emphasis of this research is “to determine the specific measures of health care disparities in the various surgical specialties in order to develop targeted interventions to mitigate such disparities,” said Dr. Britt, principal investigator of the research project.
The NIH R01 grants are among the most competitive awards in scientific research. Dr. Britt’s research team comprises experts in the field who work in medical organizations and academic institutions, such as the ACS; Harvard Medical School, Boston, MA; and the University of California, Los Angeles.
Dr. Britt has dedicated his career to patient care and addressing the multifaceted disparities in health care, and he believes that this research grant is a pivotal step toward countering one of the greatest challenges facing this country. He is particularly thankful for the unwavering support of David B. Hoyt, MD, FACS, ACS Executive Director; the Board of Regents; and the ACS Committee on Health Care Disparities, which he chairs. Adil Haider, MD, MPH, FACS, professor and director of the Center for Surgery and Public Health, Harvard Medical School, serves as Vice-Chair of the committee.
“This is a big step for the American College of Surgeons,” Dr. Britt said. “With its 100-plus year history of using data to address quality of care in surgery, if the College, in collaboration with the NIH, can’t solve this problem, no one can.”
Dr. Britt added that he anticipates that the College’s efforts to address disparities in health care with the help of the NIH will serve as a template for other professional organizations so that all patients have access to the services they need, from primary care to obstetrics-gynecology, and from cardiology to psychiatry. “Dr. Hoyt and I hope this is the start of movement to address health care disparities in all specialties, but it starts with the College.”
L. D. Britt, MD, MPH, DSc(Hon), FACS, FCCM, FRCSEng(Hon), FRCSEd(Hon), FWACS(Hon), FRCSI(Hon), FCS(SA)(Hon), FRCSGlasg(Hon), Henry Ford Professor and Edward Brickhouse Chairman, department of surgery, Eastern Virginia Medical School, Norfolk, and a Past-President of the American College of Surgeons (ACS), was recently awarded a multimillion-dollar National Institutes of Health (NIH) research grant. The grant will be used to develop strategies to address health care disparities in the various surgical specialties. Specifically, the emphasis of this research is “to determine the specific measures of health care disparities in the various surgical specialties in order to develop targeted interventions to mitigate such disparities,” said Dr. Britt, principal investigator of the research project.
The NIH R01 grants are among the most competitive awards in scientific research. Dr. Britt’s research team comprises experts in the field who work in medical organizations and academic institutions, such as the ACS; Harvard Medical School, Boston, MA; and the University of California, Los Angeles.
Dr. Britt has dedicated his career to patient care and addressing the multifaceted disparities in health care, and he believes that this research grant is a pivotal step toward countering one of the greatest challenges facing this country. He is particularly thankful for the unwavering support of David B. Hoyt, MD, FACS, ACS Executive Director; the Board of Regents; and the ACS Committee on Health Care Disparities, which he chairs. Adil Haider, MD, MPH, FACS, professor and director of the Center for Surgery and Public Health, Harvard Medical School, serves as Vice-Chair of the committee.
“This is a big step for the American College of Surgeons,” Dr. Britt said. “With its 100-plus year history of using data to address quality of care in surgery, if the College, in collaboration with the NIH, can’t solve this problem, no one can.”
Dr. Britt added that he anticipates that the College’s efforts to address disparities in health care with the help of the NIH will serve as a template for other professional organizations so that all patients have access to the services they need, from primary care to obstetrics-gynecology, and from cardiology to psychiatry. “Dr. Hoyt and I hope this is the start of movement to address health care disparities in all specialties, but it starts with the College.”
L. D. Britt, MD, MPH, DSc(Hon), FACS, FCCM, FRCSEng(Hon), FRCSEd(Hon), FWACS(Hon), FRCSI(Hon), FCS(SA)(Hon), FRCSGlasg(Hon), Henry Ford Professor and Edward Brickhouse Chairman, department of surgery, Eastern Virginia Medical School, Norfolk, and a Past-President of the American College of Surgeons (ACS), was recently awarded a multimillion-dollar National Institutes of Health (NIH) research grant. The grant will be used to develop strategies to address health care disparities in the various surgical specialties. Specifically, the emphasis of this research is “to determine the specific measures of health care disparities in the various surgical specialties in order to develop targeted interventions to mitigate such disparities,” said Dr. Britt, principal investigator of the research project.
The NIH R01 grants are among the most competitive awards in scientific research. Dr. Britt’s research team comprises experts in the field who work in medical organizations and academic institutions, such as the ACS; Harvard Medical School, Boston, MA; and the University of California, Los Angeles.
Dr. Britt has dedicated his career to patient care and addressing the multifaceted disparities in health care, and he believes that this research grant is a pivotal step toward countering one of the greatest challenges facing this country. He is particularly thankful for the unwavering support of David B. Hoyt, MD, FACS, ACS Executive Director; the Board of Regents; and the ACS Committee on Health Care Disparities, which he chairs. Adil Haider, MD, MPH, FACS, professor and director of the Center for Surgery and Public Health, Harvard Medical School, serves as Vice-Chair of the committee.
“This is a big step for the American College of Surgeons,” Dr. Britt said. “With its 100-plus year history of using data to address quality of care in surgery, if the College, in collaboration with the NIH, can’t solve this problem, no one can.”
Dr. Britt added that he anticipates that the College’s efforts to address disparities in health care with the help of the NIH will serve as a template for other professional organizations so that all patients have access to the services they need, from primary care to obstetrics-gynecology, and from cardiology to psychiatry. “Dr. Hoyt and I hope this is the start of movement to address health care disparities in all specialties, but it starts with the College.”
FDA approves second adalimumab biosimilar for multiple conditions
The Food and Drug Administration has approved Cyltezo (adalimumab-adbm) for multiple conditions.
Cyltezo is an injectable tumor necrosis factor blocker, and is a biosimilar to adalimumab (Humira). The drug is indicated to treat moderate to severe active rheumatoid arthritis, active psoriatic arthritis, active ankylosing spondylitis, moderate to severe active Crohn’s disease, moderate to severe active ulcerative colitis, moderately to severely active polyarticular juvenile idiopathic arthritis in patients 4 years of age and older, and moderate to severe plaque psoriasis.![]()
Find the Cyltezo labeling information here.
The Food and Drug Administration has approved Cyltezo (adalimumab-adbm) for multiple conditions.
Cyltezo is an injectable tumor necrosis factor blocker, and is a biosimilar to adalimumab (Humira). The drug is indicated to treat moderate to severe active rheumatoid arthritis, active psoriatic arthritis, active ankylosing spondylitis, moderate to severe active Crohn’s disease, moderate to severe active ulcerative colitis, moderately to severely active polyarticular juvenile idiopathic arthritis in patients 4 years of age and older, and moderate to severe plaque psoriasis.![]()
Find the Cyltezo labeling information here.
The Food and Drug Administration has approved Cyltezo (adalimumab-adbm) for multiple conditions.
Cyltezo is an injectable tumor necrosis factor blocker, and is a biosimilar to adalimumab (Humira). The drug is indicated to treat moderate to severe active rheumatoid arthritis, active psoriatic arthritis, active ankylosing spondylitis, moderate to severe active Crohn’s disease, moderate to severe active ulcerative colitis, moderately to severely active polyarticular juvenile idiopathic arthritis in patients 4 years of age and older, and moderate to severe plaque psoriasis.![]()
Find the Cyltezo labeling information here.