User login
Transitioning from your current medical practice: an abbreviated step-by-step guide
You have decided it is time to move on from your current hospital or medical group position and transition into a new role. While this decision is exciting and well-earned after years of hard work, it is critical that you make a plan and take specific steps to ensure that the transition is seamless.
The steps below are recommendations to make this process smoother.
Step 1: Determine how you are leaving the practice and your proposed timeline
Before anything else, you should decide how you are leaving your practice. Are you leaving the practice of medicine altogether, or are you simply leaving your current position for a different position elsewhere? This distinction will dictate what steps are necessary. Timing is also critical when leaving a practice, as it will dictate what steps should be taken and when. Having specific but realistic goals is imperative. Select a goal date for leaving the practice, but be aware that this goal may need to be adjusted.
Step 2: Create your team of advisers
Whether you are leaving your current practice or transitioning to a different position, it is extremely important to have the right individuals on your team. You should consider enlisting an attorney, a financial adviser, and an accountant to help facilitate the process. Enlisting lawyers with certain areas of expertise, such as in the areas of employment restrictive covenants, health care, or tax, may also be extremely beneficial and helpful throughout the process.

Step 3: Review your current employment agreement
It is quite likely that at the onset of your current employment arrangement, you signed an employment agreement with your hospital or group. You will want to carefully review this agreement, as it may contain provisions that can affect the steps you should take before you leave your current practice and work elsewhere. These provisions include the following:
a) Noncompetition provisions
It is critical to determine whether or not there are any restrictive covenants in your employment agreement that limit where you can work after you transition from your current practice into a new role. Restrictive covenants include noncompetition and nonsolicitation provisions, and prohibit employees from working at certain places or in certain geographic areas after they leave their current place of employment. Rules surrounding restrictive covenants vary from state to state. If there are restrictive covenants in your agreement, be sure to understand the scope of the covenant, including the geographic and temporal scope, as well as the types of medicine you are prohibited from practicing. If the covenants seem too broad or unnecessarily restrictive, consult with an attorney, as overly broad or unduly burdensome covenants are often unenforceable. However, a state-by-state analysis is required.
b) Notice and termination provisions
It is important to review whether or not there are any notice requirements in your employment agreement, which may require you to notify your employer in advance of a departure. Make sure to comply with the time requirements in the notice provision to avoid a breach of the agreement. It is also critical to determine whether terminating an agreement early will result in any termination penalties. At times, employers will impose a penalty if an employee prematurely terminates a working relationship. Understanding the penalties associated with terminating your agreement will allow you to decide whether you want to cancel the agreement and pay the penalty or push back your timeline until the end of the agreement’s term to avoid termination fees.
Step 4: Licensure obligations

Further, if your practice bills Medicare, you will want to file certain forms with Medicare to show that you are either changing your practice location or leaving medicine. For example, if you are leaving the hospital or group to practice elsewhere, you will need to fill out forms in order for your old group to submit claims and receive payments for Medicare services you provided while you were still part of that group. Furthermore, you will need to file reassignment forms to allow your new practice to bill on your behalf. Understanding which forms to complete can be confusing, so enlisting the help of a healthcare attorney may be worthwhile.
Step 5: Discuss your transition with your insurance representative
Even after you leave your current practice, you may be exposed to litigation for services you provided while you were employed or otherwise retained by such practice. To ensure that you are protected, discuss your insurance policy with your insurance representative. Review whether your insurance policy is “occurrence” or “claims-made.” If you have an occurrence policy, you are protected from covered incidents that occur during the policy period, regardless if your policy is still in existence. Claims-made policies only provide coverage for claims where both the incident and the claim occur during the policy period. For example, if you cancel your policy on March 1, and are sued on April 1 for an incident that allegedly occurred on Feb. 1, your claims-made insurance policy will not protect you. Therefore, it is important to analyze your policies to determine if tail insurance is needed.
There are a number of other issues you will want to address before you leave your practice, including financial responsibilities and medical record and privacy obligations. To ensure that you leave your practice properly, you should contact an experienced lawyer who can help you navigate this process.
Steven M. Harris is a nationally recognized health care attorney and a member of the law firm McDonald Hopkins LLC in Chicago. Write to him at [email protected].
You have decided it is time to move on from your current hospital or medical group position and transition into a new role. While this decision is exciting and well-earned after years of hard work, it is critical that you make a plan and take specific steps to ensure that the transition is seamless.
The steps below are recommendations to make this process smoother.
Step 1: Determine how you are leaving the practice and your proposed timeline
Before anything else, you should decide how you are leaving your practice. Are you leaving the practice of medicine altogether, or are you simply leaving your current position for a different position elsewhere? This distinction will dictate what steps are necessary. Timing is also critical when leaving a practice, as it will dictate what steps should be taken and when. Having specific but realistic goals is imperative. Select a goal date for leaving the practice, but be aware that this goal may need to be adjusted.
Step 2: Create your team of advisers
Whether you are leaving your current practice or transitioning to a different position, it is extremely important to have the right individuals on your team. You should consider enlisting an attorney, a financial adviser, and an accountant to help facilitate the process. Enlisting lawyers with certain areas of expertise, such as in the areas of employment restrictive covenants, health care, or tax, may also be extremely beneficial and helpful throughout the process.
Step 3: Review your current employment agreement
It is quite likely that at the onset of your current employment arrangement, you signed an employment agreement with your hospital or group. You will want to carefully review this agreement, as it may contain provisions that can affect the steps you should take before you leave your current practice and work elsewhere. These provisions include the following:
a) Noncompetition provisions
It is critical to determine whether or not there are any restrictive covenants in your employment agreement that limit where you can work after you transition from your current practice into a new role. Restrictive covenants include noncompetition and nonsolicitation provisions, and prohibit employees from working at certain places or in certain geographic areas after they leave their current place of employment. Rules surrounding restrictive covenants vary from state to state. If there are restrictive covenants in your agreement, be sure to understand the scope of the covenant, including the geographic and temporal scope, as well as the types of medicine you are prohibited from practicing. If the covenants seem too broad or unnecessarily restrictive, consult with an attorney, as overly broad or unduly burdensome covenants are often unenforceable. However, a state-by-state analysis is required.
b) Notice and termination provisions
It is important to review whether or not there are any notice requirements in your employment agreement, which may require you to notify your employer in advance of a departure. Make sure to comply with the time requirements in the notice provision to avoid a breach of the agreement. It is also critical to determine whether terminating an agreement early will result in any termination penalties. At times, employers will impose a penalty if an employee prematurely terminates a working relationship. Understanding the penalties associated with terminating your agreement will allow you to decide whether you want to cancel the agreement and pay the penalty or push back your timeline until the end of the agreement’s term to avoid termination fees.
Step 4: Licensure obligations
Further, if your practice bills Medicare, you will want to file certain forms with Medicare to show that you are either changing your practice location or leaving medicine. For example, if you are leaving the hospital or group to practice elsewhere, you will need to fill out forms in order for your old group to submit claims and receive payments for Medicare services you provided while you were still part of that group. Furthermore, you will need to file reassignment forms to allow your new practice to bill on your behalf. Understanding which forms to complete can be confusing, so enlisting the help of a healthcare attorney may be worthwhile.
Step 5: Discuss your transition with your insurance representative
Even after you leave your current practice, you may be exposed to litigation for services you provided while you were employed or otherwise retained by such practice. To ensure that you are protected, discuss your insurance policy with your insurance representative. Review whether your insurance policy is “occurrence” or “claims-made.” If you have an occurrence policy, you are protected from covered incidents that occur during the policy period, regardless if your policy is still in existence. Claims-made policies only provide coverage for claims where both the incident and the claim occur during the policy period. For example, if you cancel your policy on March 1, and are sued on April 1 for an incident that allegedly occurred on Feb. 1, your claims-made insurance policy will not protect you. Therefore, it is important to analyze your policies to determine if tail insurance is needed.
There are a number of other issues you will want to address before you leave your practice, including financial responsibilities and medical record and privacy obligations. To ensure that you leave your practice properly, you should contact an experienced lawyer who can help you navigate this process.
Steven M. Harris is a nationally recognized health care attorney and a member of the law firm McDonald Hopkins LLC in Chicago. Write to him at [email protected].
You have decided it is time to move on from your current hospital or medical group position and transition into a new role. While this decision is exciting and well-earned after years of hard work, it is critical that you make a plan and take specific steps to ensure that the transition is seamless.
The steps below are recommendations to make this process smoother.
Step 1: Determine how you are leaving the practice and your proposed timeline
Before anything else, you should decide how you are leaving your practice. Are you leaving the practice of medicine altogether, or are you simply leaving your current position for a different position elsewhere? This distinction will dictate what steps are necessary. Timing is also critical when leaving a practice, as it will dictate what steps should be taken and when. Having specific but realistic goals is imperative. Select a goal date for leaving the practice, but be aware that this goal may need to be adjusted.
Step 2: Create your team of advisers
Whether you are leaving your current practice or transitioning to a different position, it is extremely important to have the right individuals on your team. You should consider enlisting an attorney, a financial adviser, and an accountant to help facilitate the process. Enlisting lawyers with certain areas of expertise, such as in the areas of employment restrictive covenants, health care, or tax, may also be extremely beneficial and helpful throughout the process.
Step 3: Review your current employment agreement
It is quite likely that at the onset of your current employment arrangement, you signed an employment agreement with your hospital or group. You will want to carefully review this agreement, as it may contain provisions that can affect the steps you should take before you leave your current practice and work elsewhere. These provisions include the following:
a) Noncompetition provisions
It is critical to determine whether or not there are any restrictive covenants in your employment agreement that limit where you can work after you transition from your current practice into a new role. Restrictive covenants include noncompetition and nonsolicitation provisions, and prohibit employees from working at certain places or in certain geographic areas after they leave their current place of employment. Rules surrounding restrictive covenants vary from state to state. If there are restrictive covenants in your agreement, be sure to understand the scope of the covenant, including the geographic and temporal scope, as well as the types of medicine you are prohibited from practicing. If the covenants seem too broad or unnecessarily restrictive, consult with an attorney, as overly broad or unduly burdensome covenants are often unenforceable. However, a state-by-state analysis is required.
b) Notice and termination provisions
It is important to review whether or not there are any notice requirements in your employment agreement, which may require you to notify your employer in advance of a departure. Make sure to comply with the time requirements in the notice provision to avoid a breach of the agreement. It is also critical to determine whether terminating an agreement early will result in any termination penalties. At times, employers will impose a penalty if an employee prematurely terminates a working relationship. Understanding the penalties associated with terminating your agreement will allow you to decide whether you want to cancel the agreement and pay the penalty or push back your timeline until the end of the agreement’s term to avoid termination fees.
Step 4: Licensure obligations
Further, if your practice bills Medicare, you will want to file certain forms with Medicare to show that you are either changing your practice location or leaving medicine. For example, if you are leaving the hospital or group to practice elsewhere, you will need to fill out forms in order for your old group to submit claims and receive payments for Medicare services you provided while you were still part of that group. Furthermore, you will need to file reassignment forms to allow your new practice to bill on your behalf. Understanding which forms to complete can be confusing, so enlisting the help of a healthcare attorney may be worthwhile.
Step 5: Discuss your transition with your insurance representative
Even after you leave your current practice, you may be exposed to litigation for services you provided while you were employed or otherwise retained by such practice. To ensure that you are protected, discuss your insurance policy with your insurance representative. Review whether your insurance policy is “occurrence” or “claims-made.” If you have an occurrence policy, you are protected from covered incidents that occur during the policy period, regardless if your policy is still in existence. Claims-made policies only provide coverage for claims where both the incident and the claim occur during the policy period. For example, if you cancel your policy on March 1, and are sued on April 1 for an incident that allegedly occurred on Feb. 1, your claims-made insurance policy will not protect you. Therefore, it is important to analyze your policies to determine if tail insurance is needed.
There are a number of other issues you will want to address before you leave your practice, including financial responsibilities and medical record and privacy obligations. To ensure that you leave your practice properly, you should contact an experienced lawyer who can help you navigate this process.
Steven M. Harris is a nationally recognized health care attorney and a member of the law firm McDonald Hopkins LLC in Chicago. Write to him at [email protected].
Antibodies Part 3: In whose corner is genome editing’s best cut-man sitting?
We are in the midst of a revolution in genome editing. Science now exists in “AC,” or after CRISPR. Able to speedily and efficiently make genomic cuts with surgical precision, CRISPR/Cas9 is used almost ubiquitously now in the scientific community to study and alter DNA across fields ranging from medicine to agriculture to zoology. The possibilities of the biological and therapeutic implications are seemingly endless, as are the important ethical implications of their impact. Likely because of the latter, CRISPR technology has made its way from publications like Science and Nature into the lay public domains of Newsweek and NBC News.
In fact, CRISPR technology made its way into one of my favorite podcasts, WNYC’s “Radio Lab” in June 20151. The episode was entitled “Antibodies Part 1,” perhaps assuming that other technologies would also be discussed later although that has never happened. Actually, in an update early this year, the podcast jokingly addressed never moving on to “Part 2,” then followed with an update on how far CRISPR technology has progressed. Putting aside the technological advances and the early clinical applications, as well as the immense ethical considerations, CRISPR technology faces a new controversy, not one from a white coat but rather from a black robe.

In her CommonHealth blog2, Carey Goldberg of WBUR Boston Public Radio compared the case with the bout between undefeated Muhammad Ali and undefeated Joe Frazier at New York’s Madison Square Garden. Both men had legitimate claims to the title of World Heavyweight Champion. What transpired is now known as the “Fight of the Century.”
The analogy is apt. Boxing is about speed and control. Ali dominated the first three rounds with his jab, a punch that is both offensive with its attack and defensive in keeping one’s opponent at a distance. Dr. Doudna and her collaborator Emmanuelle Charpentier, PhD, published their work first (Science. 2012 Aug 17;337[6096]:816-21)3. UC Berkeley filed their patent first in May 2012.
Boxing is about timing and opportunity. Under the barrage of Ali’s jabs, Frazier found an inside position and caught Ali with a left hook. Dr. Zhang’s work followed closely after but had previously applied the technology in murine and human cells (Science. 2013 Feb 15; 339[6121]:819-23)4. The Broad Institute used this key difference to apply for its own patents under expedited review, which were granted in April 2014.
Boxing is about a punch and a counterpunch. Though fatigued, Ali continued to connect with combination punches. Frazier’s left hook pummeled Ali’s jaw. UC Berkeley filed an interference motion to invalidate the Broad Institute patent claim on the basis that the extension to eukaryotic cells was “obvious” based on the published work by Dr. Doudna’s group. In February, USPTO ruled that the Broad patent application may proceed, citing “patentably distinct subject matter.” Initial reports had indicated that Berkeley may appeal the decision, but no official filings have been made public.
Like the Fight of the Century, this case may go the distance and be decided by the judges. In a unanimous decision, Joe Frazier won the first of three epic bouts. The final scorecard is not known in the patent disputes; on March 28, the European Patent Office announced it will grant the patent application on behalf of Dr. Doudna and Dr. Charpentier.
Much like a prizefight, Wall Street has also been taking bets on who will prevail, with CRISPR-based biotech backing both sides mirroring the mid-bout odds, just as Ali dominated early with the jab, until Frazier evened the match with a left hook to the jaw. Ali fell to his knee on the canvas in the 11th round; will the European Patent Office decision prove to be a slip or a decisive knockdown? With so much at stake, the only assurance is that, as with the Ali-Frazier bout, there is likely more fighting to be done.
References:
1. Radio Lab
2. CommonHealth blog
3. Jinek M, et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21.
4. Le Cong et al., Multiplex genome engineering using CRISPR/Cas Systems. Science. 2013 Feb 15;339(6121):819-23. Multiplex genome engineering using CRISPR/Cas Systems.
Science. 2017 Feb 15: Round one of CRISPR patent legal battle goes to the Broad Institute.
StreetInsider.com: CRISPR Therapeutics (CRSP) says EPO to grant CRISPR/Cas gene editing patent.
[email protected]
On Twitter @thedoctorisvin
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. Contact Dr. Viny at [email protected].
We are in the midst of a revolution in genome editing. Science now exists in “AC,” or after CRISPR. Able to speedily and efficiently make genomic cuts with surgical precision, CRISPR/Cas9 is used almost ubiquitously now in the scientific community to study and alter DNA across fields ranging from medicine to agriculture to zoology. The possibilities of the biological and therapeutic implications are seemingly endless, as are the important ethical implications of their impact. Likely because of the latter, CRISPR technology has made its way from publications like Science and Nature into the lay public domains of Newsweek and NBC News.
In fact, CRISPR technology made its way into one of my favorite podcasts, WNYC’s “Radio Lab” in June 20151. The episode was entitled “Antibodies Part 1,” perhaps assuming that other technologies would also be discussed later although that has never happened. Actually, in an update early this year, the podcast jokingly addressed never moving on to “Part 2,” then followed with an update on how far CRISPR technology has progressed. Putting aside the technological advances and the early clinical applications, as well as the immense ethical considerations, CRISPR technology faces a new controversy, not one from a white coat but rather from a black robe.
In her CommonHealth blog2, Carey Goldberg of WBUR Boston Public Radio compared the case with the bout between undefeated Muhammad Ali and undefeated Joe Frazier at New York’s Madison Square Garden. Both men had legitimate claims to the title of World Heavyweight Champion. What transpired is now known as the “Fight of the Century.”
The analogy is apt. Boxing is about speed and control. Ali dominated the first three rounds with his jab, a punch that is both offensive with its attack and defensive in keeping one’s opponent at a distance. Dr. Doudna and her collaborator Emmanuelle Charpentier, PhD, published their work first (Science. 2012 Aug 17;337[6096]:816-21)3. UC Berkeley filed their patent first in May 2012.
Boxing is about timing and opportunity. Under the barrage of Ali’s jabs, Frazier found an inside position and caught Ali with a left hook. Dr. Zhang’s work followed closely after but had previously applied the technology in murine and human cells (Science. 2013 Feb 15; 339[6121]:819-23)4. The Broad Institute used this key difference to apply for its own patents under expedited review, which were granted in April 2014.
Boxing is about a punch and a counterpunch. Though fatigued, Ali continued to connect with combination punches. Frazier’s left hook pummeled Ali’s jaw. UC Berkeley filed an interference motion to invalidate the Broad Institute patent claim on the basis that the extension to eukaryotic cells was “obvious” based on the published work by Dr. Doudna’s group. In February, USPTO ruled that the Broad patent application may proceed, citing “patentably distinct subject matter.” Initial reports had indicated that Berkeley may appeal the decision, but no official filings have been made public.
Like the Fight of the Century, this case may go the distance and be decided by the judges. In a unanimous decision, Joe Frazier won the first of three epic bouts. The final scorecard is not known in the patent disputes; on March 28, the European Patent Office announced it will grant the patent application on behalf of Dr. Doudna and Dr. Charpentier.
Much like a prizefight, Wall Street has also been taking bets on who will prevail, with CRISPR-based biotech backing both sides mirroring the mid-bout odds, just as Ali dominated early with the jab, until Frazier evened the match with a left hook to the jaw. Ali fell to his knee on the canvas in the 11th round; will the European Patent Office decision prove to be a slip or a decisive knockdown? With so much at stake, the only assurance is that, as with the Ali-Frazier bout, there is likely more fighting to be done.
References:
1. Radio Lab
2. CommonHealth blog
3. Jinek M, et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21.
4. Le Cong et al., Multiplex genome engineering using CRISPR/Cas Systems. Science. 2013 Feb 15;339(6121):819-23. Multiplex genome engineering using CRISPR/Cas Systems.
Science. 2017 Feb 15: Round one of CRISPR patent legal battle goes to the Broad Institute.
StreetInsider.com: CRISPR Therapeutics (CRSP) says EPO to grant CRISPR/Cas gene editing patent.
[email protected]
On Twitter @thedoctorisvin
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. Contact Dr. Viny at [email protected].
We are in the midst of a revolution in genome editing. Science now exists in “AC,” or after CRISPR. Able to speedily and efficiently make genomic cuts with surgical precision, CRISPR/Cas9 is used almost ubiquitously now in the scientific community to study and alter DNA across fields ranging from medicine to agriculture to zoology. The possibilities of the biological and therapeutic implications are seemingly endless, as are the important ethical implications of their impact. Likely because of the latter, CRISPR technology has made its way from publications like Science and Nature into the lay public domains of Newsweek and NBC News.
In fact, CRISPR technology made its way into one of my favorite podcasts, WNYC’s “Radio Lab” in June 20151. The episode was entitled “Antibodies Part 1,” perhaps assuming that other technologies would also be discussed later although that has never happened. Actually, in an update early this year, the podcast jokingly addressed never moving on to “Part 2,” then followed with an update on how far CRISPR technology has progressed. Putting aside the technological advances and the early clinical applications, as well as the immense ethical considerations, CRISPR technology faces a new controversy, not one from a white coat but rather from a black robe.
In her CommonHealth blog2, Carey Goldberg of WBUR Boston Public Radio compared the case with the bout between undefeated Muhammad Ali and undefeated Joe Frazier at New York’s Madison Square Garden. Both men had legitimate claims to the title of World Heavyweight Champion. What transpired is now known as the “Fight of the Century.”
The analogy is apt. Boxing is about speed and control. Ali dominated the first three rounds with his jab, a punch that is both offensive with its attack and defensive in keeping one’s opponent at a distance. Dr. Doudna and her collaborator Emmanuelle Charpentier, PhD, published their work first (Science. 2012 Aug 17;337[6096]:816-21)3. UC Berkeley filed their patent first in May 2012.
Boxing is about timing and opportunity. Under the barrage of Ali’s jabs, Frazier found an inside position and caught Ali with a left hook. Dr. Zhang’s work followed closely after but had previously applied the technology in murine and human cells (Science. 2013 Feb 15; 339[6121]:819-23)4. The Broad Institute used this key difference to apply for its own patents under expedited review, which were granted in April 2014.
Boxing is about a punch and a counterpunch. Though fatigued, Ali continued to connect with combination punches. Frazier’s left hook pummeled Ali’s jaw. UC Berkeley filed an interference motion to invalidate the Broad Institute patent claim on the basis that the extension to eukaryotic cells was “obvious” based on the published work by Dr. Doudna’s group. In February, USPTO ruled that the Broad patent application may proceed, citing “patentably distinct subject matter.” Initial reports had indicated that Berkeley may appeal the decision, but no official filings have been made public.
Like the Fight of the Century, this case may go the distance and be decided by the judges. In a unanimous decision, Joe Frazier won the first of three epic bouts. The final scorecard is not known in the patent disputes; on March 28, the European Patent Office announced it will grant the patent application on behalf of Dr. Doudna and Dr. Charpentier.
Much like a prizefight, Wall Street has also been taking bets on who will prevail, with CRISPR-based biotech backing both sides mirroring the mid-bout odds, just as Ali dominated early with the jab, until Frazier evened the match with a left hook to the jaw. Ali fell to his knee on the canvas in the 11th round; will the European Patent Office decision prove to be a slip or a decisive knockdown? With so much at stake, the only assurance is that, as with the Ali-Frazier bout, there is likely more fighting to be done.
References:
1. Radio Lab
2. CommonHealth blog
3. Jinek M, et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21.
4. Le Cong et al., Multiplex genome engineering using CRISPR/Cas Systems. Science. 2013 Feb 15;339(6121):819-23. Multiplex genome engineering using CRISPR/Cas Systems.
Science. 2017 Feb 15: Round one of CRISPR patent legal battle goes to the Broad Institute.
StreetInsider.com: CRISPR Therapeutics (CRSP) says EPO to grant CRISPR/Cas gene editing patent.
[email protected]
On Twitter @thedoctorisvin
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. Contact Dr. Viny at [email protected].
Improving Patient Satisfaction in Dermatology: A Prospective Study of an Urban Dermatology Clinic
The Patient Protection and Affordable Care Act was signed into law in 2010, aiming to expand access to and improve the quality of health care in the United States. In the states that expanded Medicaid eligibility, uninsurance among adults decreased from 15.8% in September 2013 to 7.3% in March 2016, a decline of 53.8%.1 On average, these newly insured individuals were younger and more likely to report fair to poor health than those previously insured. Approximately half of the newly insured have family incomes at or below 138% of the federal poverty level.1
Improvement in quality in medicine is not as easily quantified. Several programs have been implemented through the Centers for Medicare & Medicaid Services to measure and reimburse hospital systems and providers based on the quality and value of care being provided. Because of the complexity in defining quality in medicine, patient satisfaction has become a proxy measurement tool.2 With higher numbers of insured patients and an increased demand for services, dermatologists are being challenged to improve availability of services and respond to patients’ needs and desires as expressed through satisfaction surveys.
Few studies have assessed patient satisfaction in dermatology practices. As patient satisfaction surveys move to the forefront under the Patient Protection and Affordable Care Act, hospitals and providers will try to demonstrate the quality of their care through positive survey responses from patients. Importantly, patient satisfaction is a strong determinate if patients will comply with treatment and continue seeing their practitioner.3 A better understanding of patients’ perceptions regarding quality will allow for targeted interventions to be implemented. This study assesses and analyzes patient satisfaction, nonattendance rates, and cycle times in an outpatient dermatology clinic to provide a snapshot of patient satisfaction in an urban dermatology clinic.
Dr. Adam Sutton discusses the results of this study with Editor-in-Chief Vincent A. DeLeo, MD, in a "Peer to Peer" audiocast, "Measuring Patient Satisfaction: How Do Patients Perceive Quality of Care Delivered by Dermatologists?"
Methods
We conducted a prospective study that was approved by the University of Southern California Health Sciences (Los Angeles, California) institutional review board. A convenience sample of patients 18 years and older who spoke English or Spanish were recruited to participate in the study and agreed to complete the Patient Satisfaction Questionnaire Short Form (PSQ-18) and a demographic questionnaire, both in English or Spanish, at the conclusion of their visit.
Based on schedules and availability, medical students came to our clinic and obtained the surveys in the following manner: After patients checked in, the students approached the patients in the waiting area and asked if they would be willing to participate in the study. If patients agreed to participate, they provided written consent and the medical student handed them an envelope containing paper copies of the survey in English or Spanish, depending on the patient’s preference. Patients were asked to complete the surveys at the end of the visit and return them to the student in the envelope. The medical students did not otherwise participate in the patient’s visit.
Surveys were collected over an 8-month period at Los Angeles County+USC Medical Center dermatology clinics, which are part of a large safety-net health system. Among this population, it is common for patients to lack reliable Internet access or permanent home addresses; therefore, we elected to use point-of-care printed survey forms. Midway through the survey collection, we moved our clinic location; however, patients and physicians did not change. The comparison between clinics showed no substantive differences and did not change the conclusions of the study.
Patient Demographics
Demographic variables were age, sex, ethnicity, highest education level, annual household income, and primary language. Patients were grouped into 4 age categories: 18 to 29 years, 30 to 49 years, 50 to 64 years, and 65 years and older. Ethnicity was classified as Hispanic/Latino or other. Highest education level was classified as high school diploma or lower, and some college or higher. Annual household income was grouped into 3 categories: less than $15,000, $15,000 to $35,000, and more than $35,000.
Patient Satisfaction Questionnaire
The PSQ-18 survey was developed by the RAND Corporation (Santa Monica, California) and has been validated.4 The survey asks patients to rate aspects of their care experience on a 5-point Likert scale (strongly agree, agree, uncertain, disagree, strongly disagree), with 5 representing highest satisfaction. The survey contains 18 questions and is scored on 7 subscales: general satisfaction, technical quality, interpersonal manner, communication, financial aspects, time spent with doctor, and accessibility and convenience. The survey typically takes less than 5 minutes to complete.
Cycle Times and Nonattendance Rates
Cycle time is defined as the total amount of time that a patient spends in a clinic from check in to checkout, which was collected from our scheduling system for each patient who agreed to participate in the study. Cycle times were grouped into 4 categories: 0 to 60 minutes, 61 to 90 minutes, 91 to 120 minutes, and 121 minutes or more. During the study period, data also were collected from the electronic health record system regarding the number of patients with appointments scheduled and the number of patients who attended each clinic. From these figures, the rate of nonattendance for each clinic was calculated.
Statistical Analysis
Demographic results were calculated using arithmetic means. The PSQ-18 subscale scores were compared among demographic subgroups using a generalized linear model. Covariates included age, sex, ethnicity, highest education level, annual household income, and primary language. All statistical analyses were conducted using SAS software version 9.2.
Results
Of the 298 participants surveyed, the average age was 49 years, 51% were male, 73% self-identified as Hispanic/Latino, 64% spoke Spanish, 58% had a high school diploma or lower, and 68% reported an annual household income of less than $15,000 (Table 1).
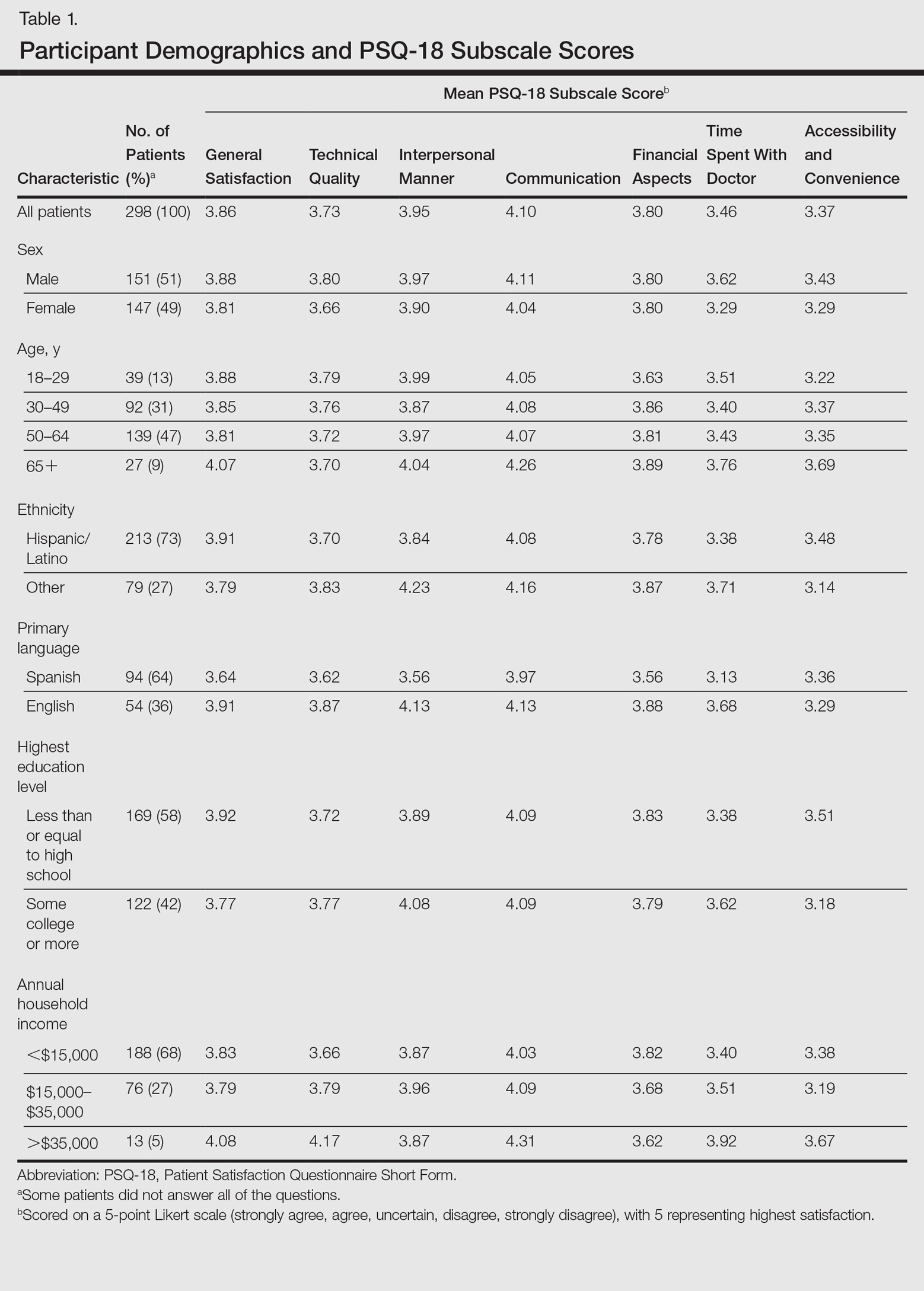
Table 1 shows PSQ-18 scores for all patients stratified by demographics. Notably, patients with some college or more were significantly more satisfied on the interpersonal manner (P<.03) and time spent with doctor (P<.007) subscales when compared to those who were less educated, but they had lower general satisfaction scores (P<.001). Patients with a reported annual household income of greater than $35,000 were more satisfied on the technical quality (P<.07) and time spent with doctor (P<.04) subscales when compared to those making less than $15,000. The patients with a household income greater than $35,000 also were more satisfied with accessibility and convenience (P<.05) than those making $15,000 to $35,000. When stratified by sex, the time spent with doctor subscale was significantly higher in males than females (P<.001). (Statistically significant differences when stratifying by age, ethnicity, and language are noted in the “Comment” section.)
Patients’ average cycle time from check in to checkout was 102 minutes (range, 24–177 minutes). There was no statistically significant difference in patient satisfaction subscale scores when stratifying patients by cycle time. During a period comparable to the time that surveys were collected, our mean (standard deviation [SD]) nonattendance rate was 30% (7%). Therefore, based on 2 SDs, there was a 95% chance that 16% to 44% of patients would not attend their scheduled appointments in each clinic.
Comment
Our dermatology clinic received an average general satisfaction subscale score of 3.86. Although the general impression of patients was positive, there were subscale scores in which the clinic performed below the general satisfaction score; the 2 lowest were time spent with doctor (3.46), and accessibility and convenience (3.37). One possible explanation for the lower time spent with doctor subscale score relates to visiting an academic medical center. Patients often are seen sequentially by a medical student, resident, and supervising physician. This educational model contributes to long cycle times; indeed, average patient visit length was more than 1.5 hours in our study. Meanwhile, patients may consider their “doctor” to be the last member of the medical team they see; thus, the percentage of the clinic visit time that a supervising physician spends with the patient may be perceived by patients as short compared to the overall time spent in the clinic.
Surprisingly, there was no statistically significant difference in patient satisfaction subscale scores, including time spent with doctor, for patients with longer cycle times compared to short cycle times (Table 2), which suggests that the length of clinic visits may have been longer than the threshold for further effect on satisfaction scores. To this point, prior research has shown that patient satisfaction notably drops after 15 minutes of waiting,5 defined as the time from check in to when the patient first sees the provider. Our data set did not allow us to analyze wait time by that definition. However, we used cycle time, which includes various periods of waiting during the patient’s visit. If we had more data points on cycle times less than 30 minutes, we might have detected a clearer relationship of cycle times to patient satisfaction scores.
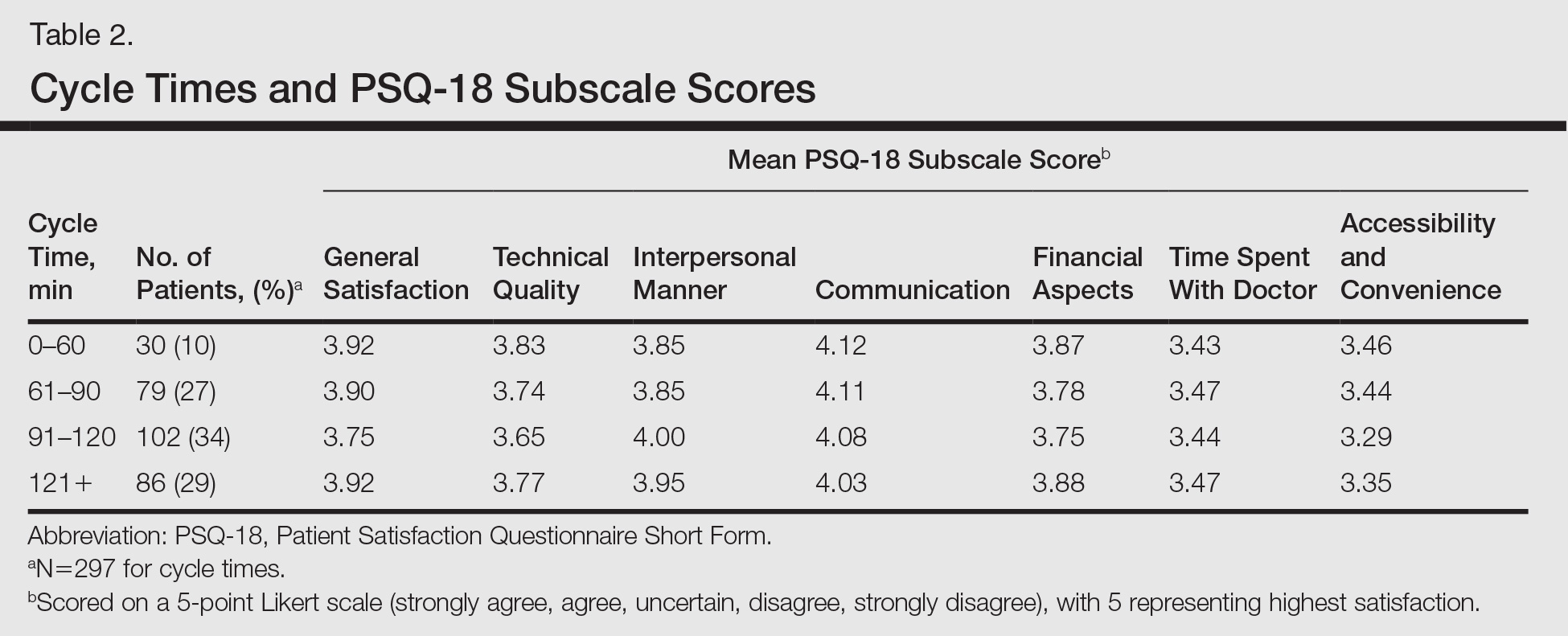
Satisfaction may not have varied with longer cycle times because differing perceptions might have balanced each other; in some cases, longer cycle times might reflect additional time spent with the provider, which could be perceived as valuable by the patient, and for others the long cycle time might be dissatisfying. Nevertheless, many of our patients were familiar with the county health system and expected to spend 90 minutes or more in clinic for each visit. Regardless, newly insured patients may have different expectations on how their health care should be delivered, an issue that could be investigated in the future.
The accessibility and convenience subscale scores reflected patients’ perception of timeliness and availability of medical care. The way that patients are scheduled at our clinic likely affected this subscale score, as patients must be referred through their primary care provider or the emergency department. We believe that many patients consider the wait for a primary care appointment as part of the overall wait for a dermatology appointment, which affects perception of accessibility and convenience for our clinic.
When we stratified by age, ethnicity, and language, other interesting trends occurred in satisfaction scores. Patients older than 65 years had a statistically significant higher accessibility and convenience subscale score when compared to the groups aged 18 to 29 years (P<.02) and 50 to 64 years (P<.05) as well as a higher but not statistically significant score compared to those aged 30 to 49 years (P<.07). Possible explanations include that older patients are familiar with the workings of our health system or that some of our patients older than 65 years may be retired and have fewer daily obligations. For the time spent with doctor subscale score, patients older than 65 years had higher scores when compared to those aged 30 to 49 years (P<.06) and 50 to 64 years (P<.07), perhaps because providers are spending more time with older individuals who may have more medical issues. A study involving a family medicine clinic also found that older patients were more satisfied with their overall care,6 which may be important given the changing demographics of Americans seeking medical care.
Differences in patient satisfaction when our patients were stratified by primary language and self-identified ethnicity also were noted. English-speaking patients were significantly more satisfied than Spanish-speaking patients in 4 subscales of satisfaction: technical quality (P<.01), interpersonal manner (P<.0001), financial aspects (P<.02), and time spent with doctor (P<.0006). For ethnicity, non-Hispanic/Latino patients had significantly higher subscale satisfaction scores for interpersonal manner (P<.0001) and time spent with doctor (P<.005). Variability in patient satisfaction based on primary language spoken and ethnicity has been described in other health care settings. Differences in satisfaction with care, understanding of potential side effects of a medication, compliance, and perceived rapport with physicians have been described.7-9
In addition to validating quality of care through patient satisfaction surveys, providers will be challenged to increase access to dermatologic services. Health systems that accept predominately Medicaid insurance, such as academic medical centers and safety-net hospitals, will be responsible for caring for millions of newly insured Medicaid patients. However, our high and variable nonattendance rates lead to inefficient use of our resources, often reducing the number of patients that are seen.
Canizares and Penneys10 studied an urban dermatology clinic over a 6-month period (N=508) and found that 17% of patients failed to keep their appointments; the subgroup of individuals with state-assisted insurance plans had the highest nonattendance rate (26%).10 In contrast, a group from Canada (N=5300) found that the nonattendance rate in a private dermatology practice was less than 8%.11 Our average nonattendance rate of 30% is within the range for urban clinics10,12; however, our SD of 7% leads to a high variability in patient volume each clinic day. As a result, on many days a reduced number of patients are seen resulting in a higher per-patient cost of delivering care.
Limitations
A potential bias is that the surveys were completed in the clinic and patients may have been concerned about possible repercussions for negative evaluations, which may have skewed results to be more positive than they otherwise would have been. We attempted to minimize this potential bias by having medical students who were not involved in the patients’ care administer the surveys. We also advised patients that their individual surveys would not be given to their providers and that any identifying information would be removed during data analysis. Our inferences could be affected by use of the terms satisfied and very satisfied in our patient satisfaction survey. Although we may interpret the results as patients reporting their degree of satisfaction, the patient may mean that there is room for improvement.13 Therefore, a survey that allows for more varied responses could potentially lead to different results.
Conclusion
Dermatology practitioners can support the specialty and validate the work they do by achieving high patient satisfaction scores. A study of online reviews compared patient ratings from 23 specialties and found that dermatology ranked second to last, ahead of only psychiatry.14 Our data has highlighted several opportunities to implement interventions that might improve patient satisfaction, though future studies would be required. Expanding or changing office hours, hiring more providers, or improving telephone access are potential interventions that might improve the accessibility and convenience subscale of patient satisfaction. Reducing the variability of nonattendance rates through the creation of resources to provide patients with clear directions and travel options, reminder calls, and instituting fees for missed appointments in some patient populations might allow for more predictable scheduling to optimize flow and the number of patients seen in each clinic.
Other approaches to improve satisfaction scores based on our results could include simple measures such as increasing the perception of time spent with the patient by having the physician sit down briefly in the examination room.15,16 It might be helpful to streamline translation assistance for patients who do not speak English as a primary language. It may be useful to recognize that younger patients have different expectations for clinic visits. For example, offering online scheduling to improve accessibility and convenience may improve satisfaction, particularly in patients who are accustomed to using technology.
It is our hope that while dermatologists continue to provide high quality care, they will work to demonstrate the value of their care by becoming leaders in patient satisfaction. Connecting their satisfaction with health care to patients’ quality of life has the potential to validate our specialty to insurers.
- Shatzer A, Long SK, Zuckerman S. Who are the newly insured as of early March 2014? Urban Institute Health Policy Center website. http://hrms.urban.org/briefs/Who-Are-the-Newly-Insured.html. Published May 22, 2014. Accessed March 17, 2017.
- Press I. Patient Satisfaction: Understanding and Measuring the Experience of Care. 2nd ed. Chicago, IL: Health Administration Press; 2006.
- Carr-Hill RA. The measurement of patient satisfaction. J Public Health Med. 1992;14:236-249.
- Thayparan A, Mahdi E. The Patient Satisfaction Questionnaire Short Form (PSQ-18) as an adaptable, reliable, and validated tool for use in various settings. Med Educ Online. 2013;18:21747.
- Garcia D, Kennedy C, Langager, J, et al. Pulse report 2009: outpatient: patient perspectives on American health care. South Bend, IN: Press Ganey Associates, Inc; 2009.
- Wetmore S, Boisvert L, Graham E, et al. Patient satisfaction with access and continuity of care in a multidisciplinary academic family medicine clinic. Can Fam Physician. 2014;60:E230-E236.
- Carrasquillo O, Orav EJ, Brennan TA, et al. Impact of language barriers on patient satisfaction in an emergency department. J Gen Intern Med. 1999;14:82-87.
- David RA, Rhee M. The impact of language as a barrier to effective health care in an underserved urban Hispanic community. Mt Sinai J Med. 1998;65:393-397.
- Ferguson WJ, Candib LM. Culture, language, and the doctor-patient relationship. Fam Med. 2002;34:353-361.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
- Pehr K. No show: incidence of nonattendance at a dermatology practice in a single universal payer model. J Cutan Med Surg. 2007;11:53-56.
- Penneys N, Glaser DA. The incidence of cancellation and non-attendance at a dermatology clinic. J Am Acad Dermatol. 1999;40:714-718.
- Collins K, O’Cathain A. The continuum of patient satisfaction—from satisfied to very satisfied. Soc Sci Med. 2003;57:2465-2470.
- Internet study: highest educated & trained doctors get poorest online reviews [news release]. Denver, CO: Vanguard Communications; April 22, 2015. https://vanguardcommunications.net/best-online-doctor-reviews/. Accessed November 28, 2016.
- Swayden KJ, Anderson KK, Connelly LM, et al. Effect of sitting vs. standing on perception of provider time at bedside: a pilot study. Patient Educ Couns. 2012;86:166-171.
- Sorenson E, Malakouti M, Brown G, et al. Enhancing patient satisfaction in dermatology. Am J Clin Dermatol. 2015;16:1-4.
The Patient Protection and Affordable Care Act was signed into law in 2010, aiming to expand access to and improve the quality of health care in the United States. In the states that expanded Medicaid eligibility, uninsurance among adults decreased from 15.8% in September 2013 to 7.3% in March 2016, a decline of 53.8%.1 On average, these newly insured individuals were younger and more likely to report fair to poor health than those previously insured. Approximately half of the newly insured have family incomes at or below 138% of the federal poverty level.1
Improvement in quality in medicine is not as easily quantified. Several programs have been implemented through the Centers for Medicare & Medicaid Services to measure and reimburse hospital systems and providers based on the quality and value of care being provided. Because of the complexity in defining quality in medicine, patient satisfaction has become a proxy measurement tool.2 With higher numbers of insured patients and an increased demand for services, dermatologists are being challenged to improve availability of services and respond to patients’ needs and desires as expressed through satisfaction surveys.
Few studies have assessed patient satisfaction in dermatology practices. As patient satisfaction surveys move to the forefront under the Patient Protection and Affordable Care Act, hospitals and providers will try to demonstrate the quality of their care through positive survey responses from patients. Importantly, patient satisfaction is a strong determinate if patients will comply with treatment and continue seeing their practitioner.3 A better understanding of patients’ perceptions regarding quality will allow for targeted interventions to be implemented. This study assesses and analyzes patient satisfaction, nonattendance rates, and cycle times in an outpatient dermatology clinic to provide a snapshot of patient satisfaction in an urban dermatology clinic.
Dr. Adam Sutton discusses the results of this study with Editor-in-Chief Vincent A. DeLeo, MD, in a "Peer to Peer" audiocast, "Measuring Patient Satisfaction: How Do Patients Perceive Quality of Care Delivered by Dermatologists?"
Methods
We conducted a prospective study that was approved by the University of Southern California Health Sciences (Los Angeles, California) institutional review board. A convenience sample of patients 18 years and older who spoke English or Spanish were recruited to participate in the study and agreed to complete the Patient Satisfaction Questionnaire Short Form (PSQ-18) and a demographic questionnaire, both in English or Spanish, at the conclusion of their visit.
Based on schedules and availability, medical students came to our clinic and obtained the surveys in the following manner: After patients checked in, the students approached the patients in the waiting area and asked if they would be willing to participate in the study. If patients agreed to participate, they provided written consent and the medical student handed them an envelope containing paper copies of the survey in English or Spanish, depending on the patient’s preference. Patients were asked to complete the surveys at the end of the visit and return them to the student in the envelope. The medical students did not otherwise participate in the patient’s visit.
Surveys were collected over an 8-month period at Los Angeles County+USC Medical Center dermatology clinics, which are part of a large safety-net health system. Among this population, it is common for patients to lack reliable Internet access or permanent home addresses; therefore, we elected to use point-of-care printed survey forms. Midway through the survey collection, we moved our clinic location; however, patients and physicians did not change. The comparison between clinics showed no substantive differences and did not change the conclusions of the study.
Patient Demographics
Demographic variables were age, sex, ethnicity, highest education level, annual household income, and primary language. Patients were grouped into 4 age categories: 18 to 29 years, 30 to 49 years, 50 to 64 years, and 65 years and older. Ethnicity was classified as Hispanic/Latino or other. Highest education level was classified as high school diploma or lower, and some college or higher. Annual household income was grouped into 3 categories: less than $15,000, $15,000 to $35,000, and more than $35,000.
Patient Satisfaction Questionnaire
The PSQ-18 survey was developed by the RAND Corporation (Santa Monica, California) and has been validated.4 The survey asks patients to rate aspects of their care experience on a 5-point Likert scale (strongly agree, agree, uncertain, disagree, strongly disagree), with 5 representing highest satisfaction. The survey contains 18 questions and is scored on 7 subscales: general satisfaction, technical quality, interpersonal manner, communication, financial aspects, time spent with doctor, and accessibility and convenience. The survey typically takes less than 5 minutes to complete.
Cycle Times and Nonattendance Rates
Cycle time is defined as the total amount of time that a patient spends in a clinic from check in to checkout, which was collected from our scheduling system for each patient who agreed to participate in the study. Cycle times were grouped into 4 categories: 0 to 60 minutes, 61 to 90 minutes, 91 to 120 minutes, and 121 minutes or more. During the study period, data also were collected from the electronic health record system regarding the number of patients with appointments scheduled and the number of patients who attended each clinic. From these figures, the rate of nonattendance for each clinic was calculated.
Statistical Analysis
Demographic results were calculated using arithmetic means. The PSQ-18 subscale scores were compared among demographic subgroups using a generalized linear model. Covariates included age, sex, ethnicity, highest education level, annual household income, and primary language. All statistical analyses were conducted using SAS software version 9.2.
Results
Of the 298 participants surveyed, the average age was 49 years, 51% were male, 73% self-identified as Hispanic/Latino, 64% spoke Spanish, 58% had a high school diploma or lower, and 68% reported an annual household income of less than $15,000 (Table 1).
Table 1 shows PSQ-18 scores for all patients stratified by demographics. Notably, patients with some college or more were significantly more satisfied on the interpersonal manner (P<.03) and time spent with doctor (P<.007) subscales when compared to those who were less educated, but they had lower general satisfaction scores (P<.001). Patients with a reported annual household income of greater than $35,000 were more satisfied on the technical quality (P<.07) and time spent with doctor (P<.04) subscales when compared to those making less than $15,000. The patients with a household income greater than $35,000 also were more satisfied with accessibility and convenience (P<.05) than those making $15,000 to $35,000. When stratified by sex, the time spent with doctor subscale was significantly higher in males than females (P<.001). (Statistically significant differences when stratifying by age, ethnicity, and language are noted in the “Comment” section.)
Patients’ average cycle time from check in to checkout was 102 minutes (range, 24–177 minutes). There was no statistically significant difference in patient satisfaction subscale scores when stratifying patients by cycle time. During a period comparable to the time that surveys were collected, our mean (standard deviation [SD]) nonattendance rate was 30% (7%). Therefore, based on 2 SDs, there was a 95% chance that 16% to 44% of patients would not attend their scheduled appointments in each clinic.
Comment
Our dermatology clinic received an average general satisfaction subscale score of 3.86. Although the general impression of patients was positive, there were subscale scores in which the clinic performed below the general satisfaction score; the 2 lowest were time spent with doctor (3.46), and accessibility and convenience (3.37). One possible explanation for the lower time spent with doctor subscale score relates to visiting an academic medical center. Patients often are seen sequentially by a medical student, resident, and supervising physician. This educational model contributes to long cycle times; indeed, average patient visit length was more than 1.5 hours in our study. Meanwhile, patients may consider their “doctor” to be the last member of the medical team they see; thus, the percentage of the clinic visit time that a supervising physician spends with the patient may be perceived by patients as short compared to the overall time spent in the clinic.
Surprisingly, there was no statistically significant difference in patient satisfaction subscale scores, including time spent with doctor, for patients with longer cycle times compared to short cycle times (Table 2), which suggests that the length of clinic visits may have been longer than the threshold for further effect on satisfaction scores. To this point, prior research has shown that patient satisfaction notably drops after 15 minutes of waiting,5 defined as the time from check in to when the patient first sees the provider. Our data set did not allow us to analyze wait time by that definition. However, we used cycle time, which includes various periods of waiting during the patient’s visit. If we had more data points on cycle times less than 30 minutes, we might have detected a clearer relationship of cycle times to patient satisfaction scores.
Satisfaction may not have varied with longer cycle times because differing perceptions might have balanced each other; in some cases, longer cycle times might reflect additional time spent with the provider, which could be perceived as valuable by the patient, and for others the long cycle time might be dissatisfying. Nevertheless, many of our patients were familiar with the county health system and expected to spend 90 minutes or more in clinic for each visit. Regardless, newly insured patients may have different expectations on how their health care should be delivered, an issue that could be investigated in the future.
The accessibility and convenience subscale scores reflected patients’ perception of timeliness and availability of medical care. The way that patients are scheduled at our clinic likely affected this subscale score, as patients must be referred through their primary care provider or the emergency department. We believe that many patients consider the wait for a primary care appointment as part of the overall wait for a dermatology appointment, which affects perception of accessibility and convenience for our clinic.
When we stratified by age, ethnicity, and language, other interesting trends occurred in satisfaction scores. Patients older than 65 years had a statistically significant higher accessibility and convenience subscale score when compared to the groups aged 18 to 29 years (P<.02) and 50 to 64 years (P<.05) as well as a higher but not statistically significant score compared to those aged 30 to 49 years (P<.07). Possible explanations include that older patients are familiar with the workings of our health system or that some of our patients older than 65 years may be retired and have fewer daily obligations. For the time spent with doctor subscale score, patients older than 65 years had higher scores when compared to those aged 30 to 49 years (P<.06) and 50 to 64 years (P<.07), perhaps because providers are spending more time with older individuals who may have more medical issues. A study involving a family medicine clinic also found that older patients were more satisfied with their overall care,6 which may be important given the changing demographics of Americans seeking medical care.
Differences in patient satisfaction when our patients were stratified by primary language and self-identified ethnicity also were noted. English-speaking patients were significantly more satisfied than Spanish-speaking patients in 4 subscales of satisfaction: technical quality (P<.01), interpersonal manner (P<.0001), financial aspects (P<.02), and time spent with doctor (P<.0006). For ethnicity, non-Hispanic/Latino patients had significantly higher subscale satisfaction scores for interpersonal manner (P<.0001) and time spent with doctor (P<.005). Variability in patient satisfaction based on primary language spoken and ethnicity has been described in other health care settings. Differences in satisfaction with care, understanding of potential side effects of a medication, compliance, and perceived rapport with physicians have been described.7-9
In addition to validating quality of care through patient satisfaction surveys, providers will be challenged to increase access to dermatologic services. Health systems that accept predominately Medicaid insurance, such as academic medical centers and safety-net hospitals, will be responsible for caring for millions of newly insured Medicaid patients. However, our high and variable nonattendance rates lead to inefficient use of our resources, often reducing the number of patients that are seen.
Canizares and Penneys10 studied an urban dermatology clinic over a 6-month period (N=508) and found that 17% of patients failed to keep their appointments; the subgroup of individuals with state-assisted insurance plans had the highest nonattendance rate (26%).10 In contrast, a group from Canada (N=5300) found that the nonattendance rate in a private dermatology practice was less than 8%.11 Our average nonattendance rate of 30% is within the range for urban clinics10,12; however, our SD of 7% leads to a high variability in patient volume each clinic day. As a result, on many days a reduced number of patients are seen resulting in a higher per-patient cost of delivering care.
Limitations
A potential bias is that the surveys were completed in the clinic and patients may have been concerned about possible repercussions for negative evaluations, which may have skewed results to be more positive than they otherwise would have been. We attempted to minimize this potential bias by having medical students who were not involved in the patients’ care administer the surveys. We also advised patients that their individual surveys would not be given to their providers and that any identifying information would be removed during data analysis. Our inferences could be affected by use of the terms satisfied and very satisfied in our patient satisfaction survey. Although we may interpret the results as patients reporting their degree of satisfaction, the patient may mean that there is room for improvement.13 Therefore, a survey that allows for more varied responses could potentially lead to different results.
Conclusion
Dermatology practitioners can support the specialty and validate the work they do by achieving high patient satisfaction scores. A study of online reviews compared patient ratings from 23 specialties and found that dermatology ranked second to last, ahead of only psychiatry.14 Our data has highlighted several opportunities to implement interventions that might improve patient satisfaction, though future studies would be required. Expanding or changing office hours, hiring more providers, or improving telephone access are potential interventions that might improve the accessibility and convenience subscale of patient satisfaction. Reducing the variability of nonattendance rates through the creation of resources to provide patients with clear directions and travel options, reminder calls, and instituting fees for missed appointments in some patient populations might allow for more predictable scheduling to optimize flow and the number of patients seen in each clinic.
Other approaches to improve satisfaction scores based on our results could include simple measures such as increasing the perception of time spent with the patient by having the physician sit down briefly in the examination room.15,16 It might be helpful to streamline translation assistance for patients who do not speak English as a primary language. It may be useful to recognize that younger patients have different expectations for clinic visits. For example, offering online scheduling to improve accessibility and convenience may improve satisfaction, particularly in patients who are accustomed to using technology.
It is our hope that while dermatologists continue to provide high quality care, they will work to demonstrate the value of their care by becoming leaders in patient satisfaction. Connecting their satisfaction with health care to patients’ quality of life has the potential to validate our specialty to insurers.
The Patient Protection and Affordable Care Act was signed into law in 2010, aiming to expand access to and improve the quality of health care in the United States. In the states that expanded Medicaid eligibility, uninsurance among adults decreased from 15.8% in September 2013 to 7.3% in March 2016, a decline of 53.8%.1 On average, these newly insured individuals were younger and more likely to report fair to poor health than those previously insured. Approximately half of the newly insured have family incomes at or below 138% of the federal poverty level.1
Improvement in quality in medicine is not as easily quantified. Several programs have been implemented through the Centers for Medicare & Medicaid Services to measure and reimburse hospital systems and providers based on the quality and value of care being provided. Because of the complexity in defining quality in medicine, patient satisfaction has become a proxy measurement tool.2 With higher numbers of insured patients and an increased demand for services, dermatologists are being challenged to improve availability of services and respond to patients’ needs and desires as expressed through satisfaction surveys.
Few studies have assessed patient satisfaction in dermatology practices. As patient satisfaction surveys move to the forefront under the Patient Protection and Affordable Care Act, hospitals and providers will try to demonstrate the quality of their care through positive survey responses from patients. Importantly, patient satisfaction is a strong determinate if patients will comply with treatment and continue seeing their practitioner.3 A better understanding of patients’ perceptions regarding quality will allow for targeted interventions to be implemented. This study assesses and analyzes patient satisfaction, nonattendance rates, and cycle times in an outpatient dermatology clinic to provide a snapshot of patient satisfaction in an urban dermatology clinic.
Dr. Adam Sutton discusses the results of this study with Editor-in-Chief Vincent A. DeLeo, MD, in a "Peer to Peer" audiocast, "Measuring Patient Satisfaction: How Do Patients Perceive Quality of Care Delivered by Dermatologists?"
Methods
We conducted a prospective study that was approved by the University of Southern California Health Sciences (Los Angeles, California) institutional review board. A convenience sample of patients 18 years and older who spoke English or Spanish were recruited to participate in the study and agreed to complete the Patient Satisfaction Questionnaire Short Form (PSQ-18) and a demographic questionnaire, both in English or Spanish, at the conclusion of their visit.
Based on schedules and availability, medical students came to our clinic and obtained the surveys in the following manner: After patients checked in, the students approached the patients in the waiting area and asked if they would be willing to participate in the study. If patients agreed to participate, they provided written consent and the medical student handed them an envelope containing paper copies of the survey in English or Spanish, depending on the patient’s preference. Patients were asked to complete the surveys at the end of the visit and return them to the student in the envelope. The medical students did not otherwise participate in the patient’s visit.
Surveys were collected over an 8-month period at Los Angeles County+USC Medical Center dermatology clinics, which are part of a large safety-net health system. Among this population, it is common for patients to lack reliable Internet access or permanent home addresses; therefore, we elected to use point-of-care printed survey forms. Midway through the survey collection, we moved our clinic location; however, patients and physicians did not change. The comparison between clinics showed no substantive differences and did not change the conclusions of the study.
Patient Demographics
Demographic variables were age, sex, ethnicity, highest education level, annual household income, and primary language. Patients were grouped into 4 age categories: 18 to 29 years, 30 to 49 years, 50 to 64 years, and 65 years and older. Ethnicity was classified as Hispanic/Latino or other. Highest education level was classified as high school diploma or lower, and some college or higher. Annual household income was grouped into 3 categories: less than $15,000, $15,000 to $35,000, and more than $35,000.
Patient Satisfaction Questionnaire
The PSQ-18 survey was developed by the RAND Corporation (Santa Monica, California) and has been validated.4 The survey asks patients to rate aspects of their care experience on a 5-point Likert scale (strongly agree, agree, uncertain, disagree, strongly disagree), with 5 representing highest satisfaction. The survey contains 18 questions and is scored on 7 subscales: general satisfaction, technical quality, interpersonal manner, communication, financial aspects, time spent with doctor, and accessibility and convenience. The survey typically takes less than 5 minutes to complete.
Cycle Times and Nonattendance Rates
Cycle time is defined as the total amount of time that a patient spends in a clinic from check in to checkout, which was collected from our scheduling system for each patient who agreed to participate in the study. Cycle times were grouped into 4 categories: 0 to 60 minutes, 61 to 90 minutes, 91 to 120 minutes, and 121 minutes or more. During the study period, data also were collected from the electronic health record system regarding the number of patients with appointments scheduled and the number of patients who attended each clinic. From these figures, the rate of nonattendance for each clinic was calculated.
Statistical Analysis
Demographic results were calculated using arithmetic means. The PSQ-18 subscale scores were compared among demographic subgroups using a generalized linear model. Covariates included age, sex, ethnicity, highest education level, annual household income, and primary language. All statistical analyses were conducted using SAS software version 9.2.
Results
Of the 298 participants surveyed, the average age was 49 years, 51% were male, 73% self-identified as Hispanic/Latino, 64% spoke Spanish, 58% had a high school diploma or lower, and 68% reported an annual household income of less than $15,000 (Table 1).
Table 1 shows PSQ-18 scores for all patients stratified by demographics. Notably, patients with some college or more were significantly more satisfied on the interpersonal manner (P<.03) and time spent with doctor (P<.007) subscales when compared to those who were less educated, but they had lower general satisfaction scores (P<.001). Patients with a reported annual household income of greater than $35,000 were more satisfied on the technical quality (P<.07) and time spent with doctor (P<.04) subscales when compared to those making less than $15,000. The patients with a household income greater than $35,000 also were more satisfied with accessibility and convenience (P<.05) than those making $15,000 to $35,000. When stratified by sex, the time spent with doctor subscale was significantly higher in males than females (P<.001). (Statistically significant differences when stratifying by age, ethnicity, and language are noted in the “Comment” section.)
Patients’ average cycle time from check in to checkout was 102 minutes (range, 24–177 minutes). There was no statistically significant difference in patient satisfaction subscale scores when stratifying patients by cycle time. During a period comparable to the time that surveys were collected, our mean (standard deviation [SD]) nonattendance rate was 30% (7%). Therefore, based on 2 SDs, there was a 95% chance that 16% to 44% of patients would not attend their scheduled appointments in each clinic.
Comment
Our dermatology clinic received an average general satisfaction subscale score of 3.86. Although the general impression of patients was positive, there were subscale scores in which the clinic performed below the general satisfaction score; the 2 lowest were time spent with doctor (3.46), and accessibility and convenience (3.37). One possible explanation for the lower time spent with doctor subscale score relates to visiting an academic medical center. Patients often are seen sequentially by a medical student, resident, and supervising physician. This educational model contributes to long cycle times; indeed, average patient visit length was more than 1.5 hours in our study. Meanwhile, patients may consider their “doctor” to be the last member of the medical team they see; thus, the percentage of the clinic visit time that a supervising physician spends with the patient may be perceived by patients as short compared to the overall time spent in the clinic.
Surprisingly, there was no statistically significant difference in patient satisfaction subscale scores, including time spent with doctor, for patients with longer cycle times compared to short cycle times (Table 2), which suggests that the length of clinic visits may have been longer than the threshold for further effect on satisfaction scores. To this point, prior research has shown that patient satisfaction notably drops after 15 minutes of waiting,5 defined as the time from check in to when the patient first sees the provider. Our data set did not allow us to analyze wait time by that definition. However, we used cycle time, which includes various periods of waiting during the patient’s visit. If we had more data points on cycle times less than 30 minutes, we might have detected a clearer relationship of cycle times to patient satisfaction scores.
Satisfaction may not have varied with longer cycle times because differing perceptions might have balanced each other; in some cases, longer cycle times might reflect additional time spent with the provider, which could be perceived as valuable by the patient, and for others the long cycle time might be dissatisfying. Nevertheless, many of our patients were familiar with the county health system and expected to spend 90 minutes or more in clinic for each visit. Regardless, newly insured patients may have different expectations on how their health care should be delivered, an issue that could be investigated in the future.
The accessibility and convenience subscale scores reflected patients’ perception of timeliness and availability of medical care. The way that patients are scheduled at our clinic likely affected this subscale score, as patients must be referred through their primary care provider or the emergency department. We believe that many patients consider the wait for a primary care appointment as part of the overall wait for a dermatology appointment, which affects perception of accessibility and convenience for our clinic.
When we stratified by age, ethnicity, and language, other interesting trends occurred in satisfaction scores. Patients older than 65 years had a statistically significant higher accessibility and convenience subscale score when compared to the groups aged 18 to 29 years (P<.02) and 50 to 64 years (P<.05) as well as a higher but not statistically significant score compared to those aged 30 to 49 years (P<.07). Possible explanations include that older patients are familiar with the workings of our health system or that some of our patients older than 65 years may be retired and have fewer daily obligations. For the time spent with doctor subscale score, patients older than 65 years had higher scores when compared to those aged 30 to 49 years (P<.06) and 50 to 64 years (P<.07), perhaps because providers are spending more time with older individuals who may have more medical issues. A study involving a family medicine clinic also found that older patients were more satisfied with their overall care,6 which may be important given the changing demographics of Americans seeking medical care.
Differences in patient satisfaction when our patients were stratified by primary language and self-identified ethnicity also were noted. English-speaking patients were significantly more satisfied than Spanish-speaking patients in 4 subscales of satisfaction: technical quality (P<.01), interpersonal manner (P<.0001), financial aspects (P<.02), and time spent with doctor (P<.0006). For ethnicity, non-Hispanic/Latino patients had significantly higher subscale satisfaction scores for interpersonal manner (P<.0001) and time spent with doctor (P<.005). Variability in patient satisfaction based on primary language spoken and ethnicity has been described in other health care settings. Differences in satisfaction with care, understanding of potential side effects of a medication, compliance, and perceived rapport with physicians have been described.7-9
In addition to validating quality of care through patient satisfaction surveys, providers will be challenged to increase access to dermatologic services. Health systems that accept predominately Medicaid insurance, such as academic medical centers and safety-net hospitals, will be responsible for caring for millions of newly insured Medicaid patients. However, our high and variable nonattendance rates lead to inefficient use of our resources, often reducing the number of patients that are seen.
Canizares and Penneys10 studied an urban dermatology clinic over a 6-month period (N=508) and found that 17% of patients failed to keep their appointments; the subgroup of individuals with state-assisted insurance plans had the highest nonattendance rate (26%).10 In contrast, a group from Canada (N=5300) found that the nonattendance rate in a private dermatology practice was less than 8%.11 Our average nonattendance rate of 30% is within the range for urban clinics10,12; however, our SD of 7% leads to a high variability in patient volume each clinic day. As a result, on many days a reduced number of patients are seen resulting in a higher per-patient cost of delivering care.
Limitations
A potential bias is that the surveys were completed in the clinic and patients may have been concerned about possible repercussions for negative evaluations, which may have skewed results to be more positive than they otherwise would have been. We attempted to minimize this potential bias by having medical students who were not involved in the patients’ care administer the surveys. We also advised patients that their individual surveys would not be given to their providers and that any identifying information would be removed during data analysis. Our inferences could be affected by use of the terms satisfied and very satisfied in our patient satisfaction survey. Although we may interpret the results as patients reporting their degree of satisfaction, the patient may mean that there is room for improvement.13 Therefore, a survey that allows for more varied responses could potentially lead to different results.
Conclusion
Dermatology practitioners can support the specialty and validate the work they do by achieving high patient satisfaction scores. A study of online reviews compared patient ratings from 23 specialties and found that dermatology ranked second to last, ahead of only psychiatry.14 Our data has highlighted several opportunities to implement interventions that might improve patient satisfaction, though future studies would be required. Expanding or changing office hours, hiring more providers, or improving telephone access are potential interventions that might improve the accessibility and convenience subscale of patient satisfaction. Reducing the variability of nonattendance rates through the creation of resources to provide patients with clear directions and travel options, reminder calls, and instituting fees for missed appointments in some patient populations might allow for more predictable scheduling to optimize flow and the number of patients seen in each clinic.
Other approaches to improve satisfaction scores based on our results could include simple measures such as increasing the perception of time spent with the patient by having the physician sit down briefly in the examination room.15,16 It might be helpful to streamline translation assistance for patients who do not speak English as a primary language. It may be useful to recognize that younger patients have different expectations for clinic visits. For example, offering online scheduling to improve accessibility and convenience may improve satisfaction, particularly in patients who are accustomed to using technology.
It is our hope that while dermatologists continue to provide high quality care, they will work to demonstrate the value of their care by becoming leaders in patient satisfaction. Connecting their satisfaction with health care to patients’ quality of life has the potential to validate our specialty to insurers.
- Shatzer A, Long SK, Zuckerman S. Who are the newly insured as of early March 2014? Urban Institute Health Policy Center website. http://hrms.urban.org/briefs/Who-Are-the-Newly-Insured.html. Published May 22, 2014. Accessed March 17, 2017.
- Press I. Patient Satisfaction: Understanding and Measuring the Experience of Care. 2nd ed. Chicago, IL: Health Administration Press; 2006.
- Carr-Hill RA. The measurement of patient satisfaction. J Public Health Med. 1992;14:236-249.
- Thayparan A, Mahdi E. The Patient Satisfaction Questionnaire Short Form (PSQ-18) as an adaptable, reliable, and validated tool for use in various settings. Med Educ Online. 2013;18:21747.
- Garcia D, Kennedy C, Langager, J, et al. Pulse report 2009: outpatient: patient perspectives on American health care. South Bend, IN: Press Ganey Associates, Inc; 2009.
- Wetmore S, Boisvert L, Graham E, et al. Patient satisfaction with access and continuity of care in a multidisciplinary academic family medicine clinic. Can Fam Physician. 2014;60:E230-E236.
- Carrasquillo O, Orav EJ, Brennan TA, et al. Impact of language barriers on patient satisfaction in an emergency department. J Gen Intern Med. 1999;14:82-87.
- David RA, Rhee M. The impact of language as a barrier to effective health care in an underserved urban Hispanic community. Mt Sinai J Med. 1998;65:393-397.
- Ferguson WJ, Candib LM. Culture, language, and the doctor-patient relationship. Fam Med. 2002;34:353-361.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
- Pehr K. No show: incidence of nonattendance at a dermatology practice in a single universal payer model. J Cutan Med Surg. 2007;11:53-56.
- Penneys N, Glaser DA. The incidence of cancellation and non-attendance at a dermatology clinic. J Am Acad Dermatol. 1999;40:714-718.
- Collins K, O’Cathain A. The continuum of patient satisfaction—from satisfied to very satisfied. Soc Sci Med. 2003;57:2465-2470.
- Internet study: highest educated & trained doctors get poorest online reviews [news release]. Denver, CO: Vanguard Communications; April 22, 2015. https://vanguardcommunications.net/best-online-doctor-reviews/. Accessed November 28, 2016.
- Swayden KJ, Anderson KK, Connelly LM, et al. Effect of sitting vs. standing on perception of provider time at bedside: a pilot study. Patient Educ Couns. 2012;86:166-171.
- Sorenson E, Malakouti M, Brown G, et al. Enhancing patient satisfaction in dermatology. Am J Clin Dermatol. 2015;16:1-4.
- Shatzer A, Long SK, Zuckerman S. Who are the newly insured as of early March 2014? Urban Institute Health Policy Center website. http://hrms.urban.org/briefs/Who-Are-the-Newly-Insured.html. Published May 22, 2014. Accessed March 17, 2017.
- Press I. Patient Satisfaction: Understanding and Measuring the Experience of Care. 2nd ed. Chicago, IL: Health Administration Press; 2006.
- Carr-Hill RA. The measurement of patient satisfaction. J Public Health Med. 1992;14:236-249.
- Thayparan A, Mahdi E. The Patient Satisfaction Questionnaire Short Form (PSQ-18) as an adaptable, reliable, and validated tool for use in various settings. Med Educ Online. 2013;18:21747.
- Garcia D, Kennedy C, Langager, J, et al. Pulse report 2009: outpatient: patient perspectives on American health care. South Bend, IN: Press Ganey Associates, Inc; 2009.
- Wetmore S, Boisvert L, Graham E, et al. Patient satisfaction with access and continuity of care in a multidisciplinary academic family medicine clinic. Can Fam Physician. 2014;60:E230-E236.
- Carrasquillo O, Orav EJ, Brennan TA, et al. Impact of language barriers on patient satisfaction in an emergency department. J Gen Intern Med. 1999;14:82-87.
- David RA, Rhee M. The impact of language as a barrier to effective health care in an underserved urban Hispanic community. Mt Sinai J Med. 1998;65:393-397.
- Ferguson WJ, Candib LM. Culture, language, and the doctor-patient relationship. Fam Med. 2002;34:353-361.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
- Pehr K. No show: incidence of nonattendance at a dermatology practice in a single universal payer model. J Cutan Med Surg. 2007;11:53-56.
- Penneys N, Glaser DA. The incidence of cancellation and non-attendance at a dermatology clinic. J Am Acad Dermatol. 1999;40:714-718.
- Collins K, O’Cathain A. The continuum of patient satisfaction—from satisfied to very satisfied. Soc Sci Med. 2003;57:2465-2470.
- Internet study: highest educated & trained doctors get poorest online reviews [news release]. Denver, CO: Vanguard Communications; April 22, 2015. https://vanguardcommunications.net/best-online-doctor-reviews/. Accessed November 28, 2016.
- Swayden KJ, Anderson KK, Connelly LM, et al. Effect of sitting vs. standing on perception of provider time at bedside: a pilot study. Patient Educ Couns. 2012;86:166-171.
- Sorenson E, Malakouti M, Brown G, et al. Enhancing patient satisfaction in dermatology. Am J Clin Dermatol. 2015;16:1-4.
Practice Points
- Patient experience can be measured through brief point-of-service patient satisfaction questionnaires.
- Stratifying and analyzing patient satisfaction allows for targeted interventions to be developed and implemented.
- Educational handouts in the patient's primary language may help increase satisfaction and improve compliance.

Birth defects found in 10% of confirmed U.S. Zika pregnancies
FROM MMWR
About 1 in 10 pregnant women with confirmed Zika infection in the United States had a fetus or baby with Zika-related birth defects in 2016, officials at the Centers for Disease Control and Prevention reported.
Among 972 completed pregnancies with laboratory evidence of possible recent Zika virus infection – 895 liveborn infants and 77 pregnancy losses – there were 51 fetuses/infants with Zika-related birth defects (5%). Among 250 pregnancies with laboratory-confirmed Zika infection, there were 24 cases of birth defects (10%). Among the 60 pregnancies in which Zika infection was confirmed and onset occurred during the first trimester, there were nine cases of birth defects (15%).![]()
“We are seeing about 30-40 new Zika cases in pregnant women each week in the United States. With the current tally of more than 1,600 pregnant women with evidence of Zika” reported in at least 44 states, mostly from travel to endemic areas, “this devastating outbreak is far from over,” CDC Acting Director Anne Schuchat, MD, said during an April 4 press conference.
But current birth defect estimates may not reflect the full impact of Zika virus in pregnancy, since some Zika-related developmental problems may not become apparent until months after birth. “We recommend babies receive close developmental monitoring and follow-up,” she said.
Despite CDC recommendations, one in three infants with possible congenital Zika infection had no report of Zika testing at birth, and only 1 in 4 had brain imaging (Morb Mortal Wkly Rep. 2017 Apr 4. doi: 10.15585/mmwr.mm6613e1).
“Because the full clinical spectrum of congenital Zika virus infection is not yet known, all infants born to women with laboratory evidence of possible recent Zika virus infection during pregnancy should receive postnatal neuroimaging and Zika virus testing in addition to a comprehensive newborn physical exam and hearing screen,” the CDC officials wrote.
Birth defects – most commonly microcephaly or brain abnormalities – were reported in similar proportions of fetuses/infants whose mothers did and did not report Zika symptoms.
Dr. Schuchat noted that Zika virus can cause vision problems, hearing problems, and seizures. Some infants have little or no control over their arms or legs, and cannot reach out to touch things because of their constricted joints. There can be problems reaching developmental milestones, such as sitting up, as well as problems with feeding, swallowing, and breathing. Some babies cry inconsolably. Others may be born with a normal head size but experience slow head growth later on and develop microcephaly.
The study confirms the need for pregnant women to continue taking steps to prevent Zika virus exposure through mosquito bites and sexual transmission.
“We encourage [health care providers] to ask about possible Zika exposure when caring for both pregnant women and their babies, and to follow CDC guidance for evaluation and care of infants with possible Zika infection,” said Peggy Honein, PhD, senior investigator on the review, in a statement.
The mothers of the 51 fetuses or infants with birth defects were exposed to Zika during trips to 16 countries, including Barbados, Belize, Brazil, Cape Verde, Colombia, Dominican Republic, El Salvador, Guatemala, Guyana, Haiti, Honduras, Jamaica, Mexico, Puerto Rico, Republic of Marshall Islands, and Venezuela.
FROM MMWR
About 1 in 10 pregnant women with confirmed Zika infection in the United States had a fetus or baby with Zika-related birth defects in 2016, officials at the Centers for Disease Control and Prevention reported.
Among 972 completed pregnancies with laboratory evidence of possible recent Zika virus infection – 895 liveborn infants and 77 pregnancy losses – there were 51 fetuses/infants with Zika-related birth defects (5%). Among 250 pregnancies with laboratory-confirmed Zika infection, there were 24 cases of birth defects (10%). Among the 60 pregnancies in which Zika infection was confirmed and onset occurred during the first trimester, there were nine cases of birth defects (15%).![]()
“We are seeing about 30-40 new Zika cases in pregnant women each week in the United States. With the current tally of more than 1,600 pregnant women with evidence of Zika” reported in at least 44 states, mostly from travel to endemic areas, “this devastating outbreak is far from over,” CDC Acting Director Anne Schuchat, MD, said during an April 4 press conference.
But current birth defect estimates may not reflect the full impact of Zika virus in pregnancy, since some Zika-related developmental problems may not become apparent until months after birth. “We recommend babies receive close developmental monitoring and follow-up,” she said.
Despite CDC recommendations, one in three infants with possible congenital Zika infection had no report of Zika testing at birth, and only 1 in 4 had brain imaging (Morb Mortal Wkly Rep. 2017 Apr 4. doi: 10.15585/mmwr.mm6613e1).
“Because the full clinical spectrum of congenital Zika virus infection is not yet known, all infants born to women with laboratory evidence of possible recent Zika virus infection during pregnancy should receive postnatal neuroimaging and Zika virus testing in addition to a comprehensive newborn physical exam and hearing screen,” the CDC officials wrote.
Birth defects – most commonly microcephaly or brain abnormalities – were reported in similar proportions of fetuses/infants whose mothers did and did not report Zika symptoms.
Dr. Schuchat noted that Zika virus can cause vision problems, hearing problems, and seizures. Some infants have little or no control over their arms or legs, and cannot reach out to touch things because of their constricted joints. There can be problems reaching developmental milestones, such as sitting up, as well as problems with feeding, swallowing, and breathing. Some babies cry inconsolably. Others may be born with a normal head size but experience slow head growth later on and develop microcephaly.
The study confirms the need for pregnant women to continue taking steps to prevent Zika virus exposure through mosquito bites and sexual transmission.
“We encourage [health care providers] to ask about possible Zika exposure when caring for both pregnant women and their babies, and to follow CDC guidance for evaluation and care of infants with possible Zika infection,” said Peggy Honein, PhD, senior investigator on the review, in a statement.
The mothers of the 51 fetuses or infants with birth defects were exposed to Zika during trips to 16 countries, including Barbados, Belize, Brazil, Cape Verde, Colombia, Dominican Republic, El Salvador, Guatemala, Guyana, Haiti, Honduras, Jamaica, Mexico, Puerto Rico, Republic of Marshall Islands, and Venezuela.
FROM MMWR
About 1 in 10 pregnant women with confirmed Zika infection in the United States had a fetus or baby with Zika-related birth defects in 2016, officials at the Centers for Disease Control and Prevention reported.
Among 972 completed pregnancies with laboratory evidence of possible recent Zika virus infection – 895 liveborn infants and 77 pregnancy losses – there were 51 fetuses/infants with Zika-related birth defects (5%). Among 250 pregnancies with laboratory-confirmed Zika infection, there were 24 cases of birth defects (10%). Among the 60 pregnancies in which Zika infection was confirmed and onset occurred during the first trimester, there were nine cases of birth defects (15%).![]()
“We are seeing about 30-40 new Zika cases in pregnant women each week in the United States. With the current tally of more than 1,600 pregnant women with evidence of Zika” reported in at least 44 states, mostly from travel to endemic areas, “this devastating outbreak is far from over,” CDC Acting Director Anne Schuchat, MD, said during an April 4 press conference.
But current birth defect estimates may not reflect the full impact of Zika virus in pregnancy, since some Zika-related developmental problems may not become apparent until months after birth. “We recommend babies receive close developmental monitoring and follow-up,” she said.
Despite CDC recommendations, one in three infants with possible congenital Zika infection had no report of Zika testing at birth, and only 1 in 4 had brain imaging (Morb Mortal Wkly Rep. 2017 Apr 4. doi: 10.15585/mmwr.mm6613e1).
“Because the full clinical spectrum of congenital Zika virus infection is not yet known, all infants born to women with laboratory evidence of possible recent Zika virus infection during pregnancy should receive postnatal neuroimaging and Zika virus testing in addition to a comprehensive newborn physical exam and hearing screen,” the CDC officials wrote.
Birth defects – most commonly microcephaly or brain abnormalities – were reported in similar proportions of fetuses/infants whose mothers did and did not report Zika symptoms.
Dr. Schuchat noted that Zika virus can cause vision problems, hearing problems, and seizures. Some infants have little or no control over their arms or legs, and cannot reach out to touch things because of their constricted joints. There can be problems reaching developmental milestones, such as sitting up, as well as problems with feeding, swallowing, and breathing. Some babies cry inconsolably. Others may be born with a normal head size but experience slow head growth later on and develop microcephaly.
The study confirms the need for pregnant women to continue taking steps to prevent Zika virus exposure through mosquito bites and sexual transmission.
“We encourage [health care providers] to ask about possible Zika exposure when caring for both pregnant women and their babies, and to follow CDC guidance for evaluation and care of infants with possible Zika infection,” said Peggy Honein, PhD, senior investigator on the review, in a statement.
The mothers of the 51 fetuses or infants with birth defects were exposed to Zika during trips to 16 countries, including Barbados, Belize, Brazil, Cape Verde, Colombia, Dominican Republic, El Salvador, Guatemala, Guyana, Haiti, Honduras, Jamaica, Mexico, Puerto Rico, Republic of Marshall Islands, and Venezuela.
Stopping TNF inhibitors for pregnancy may invite flares
Women with rheumatoid arthritis or axial spondyloarthritis who stop treatment with tumor necrosis factor inhibitors when they become pregnant may be inviting disease flares during the pregnancy, according to a report published in Arthritis Research & Therapy.
To examine the frequency of rheumatoid arthritis (RA) and axial spondyloarthritis (axSpA) flares during pregnancy, researchers prospectively followed 136 women treated at the Center for Pregnancy in Rheumatic Diseases at Inselspital Bern (Switzerland) during a 5-year period. These patients – 75 with RA and 61 with axSpA – were assessed before conception, during each trimester, and 6-8 weeks postpartum for disease activity and medication use, said Stephanie van den Brandt, MD, of the department of rheumatology, immunology, and allergology at the University of Bern, and her associates.
The relative risk of a disease flare was 3.33 among RA patients and 3.08 among axSpA patients who discontinued TNF inhibitors at the time of a positive pregnancy test. In comparison, rheumatic disease remained stable throughout pregnancy in most women who were not taking TNF inhibitors before pregnancy, the investigators said (Arthritis Res Ther. 2017 Mar 20. doi: 10.1186/s13075-017-1269-1).
Most disease flares occurred in the first trimester among women with RA and in the second half of pregnancy among women with axSpA. Most women with RA who resumed taking TNF inhibitors when their disease flared responded well to the treatment, with CRP levels dropping by 70% and remission being achieved rapidly. In contrast, most women with axSpA who resumed taking TNF inhibitors did not respond as well, with CRP levels dropping by only 35%. Their disease was ameliorated but not controlled by restarting the therapy.
No sponsor was cited for this study. Dr. van den Brandt and her associates reported having no relevant financial disclosures.
Women with rheumatoid arthritis or axial spondyloarthritis who stop treatment with tumor necrosis factor inhibitors when they become pregnant may be inviting disease flares during the pregnancy, according to a report published in Arthritis Research & Therapy.
To examine the frequency of rheumatoid arthritis (RA) and axial spondyloarthritis (axSpA) flares during pregnancy, researchers prospectively followed 136 women treated at the Center for Pregnancy in Rheumatic Diseases at Inselspital Bern (Switzerland) during a 5-year period. These patients – 75 with RA and 61 with axSpA – were assessed before conception, during each trimester, and 6-8 weeks postpartum for disease activity and medication use, said Stephanie van den Brandt, MD, of the department of rheumatology, immunology, and allergology at the University of Bern, and her associates.
The relative risk of a disease flare was 3.33 among RA patients and 3.08 among axSpA patients who discontinued TNF inhibitors at the time of a positive pregnancy test. In comparison, rheumatic disease remained stable throughout pregnancy in most women who were not taking TNF inhibitors before pregnancy, the investigators said (Arthritis Res Ther. 2017 Mar 20. doi: 10.1186/s13075-017-1269-1).
Most disease flares occurred in the first trimester among women with RA and in the second half of pregnancy among women with axSpA. Most women with RA who resumed taking TNF inhibitors when their disease flared responded well to the treatment, with CRP levels dropping by 70% and remission being achieved rapidly. In contrast, most women with axSpA who resumed taking TNF inhibitors did not respond as well, with CRP levels dropping by only 35%. Their disease was ameliorated but not controlled by restarting the therapy.
No sponsor was cited for this study. Dr. van den Brandt and her associates reported having no relevant financial disclosures.
Women with rheumatoid arthritis or axial spondyloarthritis who stop treatment with tumor necrosis factor inhibitors when they become pregnant may be inviting disease flares during the pregnancy, according to a report published in Arthritis Research & Therapy.
To examine the frequency of rheumatoid arthritis (RA) and axial spondyloarthritis (axSpA) flares during pregnancy, researchers prospectively followed 136 women treated at the Center for Pregnancy in Rheumatic Diseases at Inselspital Bern (Switzerland) during a 5-year period. These patients – 75 with RA and 61 with axSpA – were assessed before conception, during each trimester, and 6-8 weeks postpartum for disease activity and medication use, said Stephanie van den Brandt, MD, of the department of rheumatology, immunology, and allergology at the University of Bern, and her associates.
The relative risk of a disease flare was 3.33 among RA patients and 3.08 among axSpA patients who discontinued TNF inhibitors at the time of a positive pregnancy test. In comparison, rheumatic disease remained stable throughout pregnancy in most women who were not taking TNF inhibitors before pregnancy, the investigators said (Arthritis Res Ther. 2017 Mar 20. doi: 10.1186/s13075-017-1269-1).
Most disease flares occurred in the first trimester among women with RA and in the second half of pregnancy among women with axSpA. Most women with RA who resumed taking TNF inhibitors when their disease flared responded well to the treatment, with CRP levels dropping by 70% and remission being achieved rapidly. In contrast, most women with axSpA who resumed taking TNF inhibitors did not respond as well, with CRP levels dropping by only 35%. Their disease was ameliorated but not controlled by restarting the therapy.
No sponsor was cited for this study. Dr. van den Brandt and her associates reported having no relevant financial disclosures.
Key clinical point:
Major finding: The relative risk of a disease flare was 3.33 among RA patients and 3.08 among axSpA patients who discontinued TNF inhibitors at conception.
Data source: A prospective cohort study involving 75 pregnant women with RA and 61 with axial spondyloarthritis treated at one Swiss specialty center in 2000-2015.
Disclosures: No sponsor was cited for this study. Dr. van den Brandt and her associates reported having no relevant financial disclosures.
What’s old is new in topical psoriasis therapy
WAILEA, HAWAII – A fixed combination of halobetasol and tazarotene formulated as a once-daily lotion proved safe and effective for patients with moderate or severe plaque psoriasis in a phase II randomized clinical trial, Linda Stein Gold, MD, reported at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
The rationale for developing this product is that it should provide the efficacy of a class 1 superpotent topical corticosteroid, albeit with the topical retinoid lessening concern about the possibility of steroid-induced skin atrophy and other long-term safety issues, explained Dr. Stein Gold, director of dermatology research at Henry Ford Health System in Detroit.

The double-blind, multicenter, 8-week, phase II study included 212 patients randomized 2:2:1 to once-daily application of fixed-combination halobetasol propionate 0.01% and tazarotene 0.045% lotion, either topical agent as monotherapy, or vehicle. Ninety percent of participants had moderate plaque psoriasis as defined by a baseline Investigator’s Global Assessment (IGA) score of 3 and a median 5% body surface area involvement. The remaining patients were classified as having severe psoriasis, with a baseline IGA of 4, but no one with psoriasis over more than 12% of their body surface area was eligible for the trial.
The fixed-combination lotion, known for now as IDP-118, established superior efficacy over placebo by 2 weeks. At week 8, treatment success, as defined by at least a two-grade improvement from baseline in IGA score plus a rating of clear or almost clear, was documented in 53% of the IDP-118 lotion group, compared with 33% on halobetasol propionate monotherapy, 19% with tazarotene alone, and 10% with vehicle, Dr. Stein Gold reported.
Each patient had a target lesion 25-32 cm2 in size, which was designated for careful assessment of changes in the domains of lesional erythema, plaque elevation, and scaling. At week 8, the IDP-118 had a target lesion treatment success rate of 54% for erythema, 68% for plaque elevation, and 64% for scaling, she said.
The treatment discontinuation rate was 3.4% in the IDP-118 group, 12.1% with tazarotene, zero for halobetasol propionate, and 3.2% for vehicle. The most frequently reported treatment-emergent adverse events were mild or moderate application-site reactions. Skin atrophy was rare. No treatment-related serious adverse events occurred.
A 555-patient, phase III, open-label, 12-month safety study of IDP-118 is due to be completed later in 2017.
The phase II study was funded by Valeant Pharmaceuticals. Dr. Stein Gold reported serving as an investigator, advisor, and speaker for the company. SDEF and this news organization are owned by the same parent company.
WAILEA, HAWAII – A fixed combination of halobetasol and tazarotene formulated as a once-daily lotion proved safe and effective for patients with moderate or severe plaque psoriasis in a phase II randomized clinical trial, Linda Stein Gold, MD, reported at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
The rationale for developing this product is that it should provide the efficacy of a class 1 superpotent topical corticosteroid, albeit with the topical retinoid lessening concern about the possibility of steroid-induced skin atrophy and other long-term safety issues, explained Dr. Stein Gold, director of dermatology research at Henry Ford Health System in Detroit.
The double-blind, multicenter, 8-week, phase II study included 212 patients randomized 2:2:1 to once-daily application of fixed-combination halobetasol propionate 0.01% and tazarotene 0.045% lotion, either topical agent as monotherapy, or vehicle. Ninety percent of participants had moderate plaque psoriasis as defined by a baseline Investigator’s Global Assessment (IGA) score of 3 and a median 5% body surface area involvement. The remaining patients were classified as having severe psoriasis, with a baseline IGA of 4, but no one with psoriasis over more than 12% of their body surface area was eligible for the trial.
The fixed-combination lotion, known for now as IDP-118, established superior efficacy over placebo by 2 weeks. At week 8, treatment success, as defined by at least a two-grade improvement from baseline in IGA score plus a rating of clear or almost clear, was documented in 53% of the IDP-118 lotion group, compared with 33% on halobetasol propionate monotherapy, 19% with tazarotene alone, and 10% with vehicle, Dr. Stein Gold reported.
Each patient had a target lesion 25-32 cm2 in size, which was designated for careful assessment of changes in the domains of lesional erythema, plaque elevation, and scaling. At week 8, the IDP-118 had a target lesion treatment success rate of 54% for erythema, 68% for plaque elevation, and 64% for scaling, she said.
The treatment discontinuation rate was 3.4% in the IDP-118 group, 12.1% with tazarotene, zero for halobetasol propionate, and 3.2% for vehicle. The most frequently reported treatment-emergent adverse events were mild or moderate application-site reactions. Skin atrophy was rare. No treatment-related serious adverse events occurred.
A 555-patient, phase III, open-label, 12-month safety study of IDP-118 is due to be completed later in 2017.
The phase II study was funded by Valeant Pharmaceuticals. Dr. Stein Gold reported serving as an investigator, advisor, and speaker for the company. SDEF and this news organization are owned by the same parent company.
WAILEA, HAWAII – A fixed combination of halobetasol and tazarotene formulated as a once-daily lotion proved safe and effective for patients with moderate or severe plaque psoriasis in a phase II randomized clinical trial, Linda Stein Gold, MD, reported at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
The rationale for developing this product is that it should provide the efficacy of a class 1 superpotent topical corticosteroid, albeit with the topical retinoid lessening concern about the possibility of steroid-induced skin atrophy and other long-term safety issues, explained Dr. Stein Gold, director of dermatology research at Henry Ford Health System in Detroit.
The double-blind, multicenter, 8-week, phase II study included 212 patients randomized 2:2:1 to once-daily application of fixed-combination halobetasol propionate 0.01% and tazarotene 0.045% lotion, either topical agent as monotherapy, or vehicle. Ninety percent of participants had moderate plaque psoriasis as defined by a baseline Investigator’s Global Assessment (IGA) score of 3 and a median 5% body surface area involvement. The remaining patients were classified as having severe psoriasis, with a baseline IGA of 4, but no one with psoriasis over more than 12% of their body surface area was eligible for the trial.
The fixed-combination lotion, known for now as IDP-118, established superior efficacy over placebo by 2 weeks. At week 8, treatment success, as defined by at least a two-grade improvement from baseline in IGA score plus a rating of clear or almost clear, was documented in 53% of the IDP-118 lotion group, compared with 33% on halobetasol propionate monotherapy, 19% with tazarotene alone, and 10% with vehicle, Dr. Stein Gold reported.
Each patient had a target lesion 25-32 cm2 in size, which was designated for careful assessment of changes in the domains of lesional erythema, plaque elevation, and scaling. At week 8, the IDP-118 had a target lesion treatment success rate of 54% for erythema, 68% for plaque elevation, and 64% for scaling, she said.
The treatment discontinuation rate was 3.4% in the IDP-118 group, 12.1% with tazarotene, zero for halobetasol propionate, and 3.2% for vehicle. The most frequently reported treatment-emergent adverse events were mild or moderate application-site reactions. Skin atrophy was rare. No treatment-related serious adverse events occurred.
A 555-patient, phase III, open-label, 12-month safety study of IDP-118 is due to be completed later in 2017.
The phase II study was funded by Valeant Pharmaceuticals. Dr. Stein Gold reported serving as an investigator, advisor, and speaker for the company. SDEF and this news organization are owned by the same parent company.
Key clinical point:
Major finding: An investigational fixed-combination lotion consisting of halobetasol propionate 0.01% and tazarotene 0.045% achieved scores of clear or almost clear in 53% of patients, significantly better than either component as monotherapy.
Data source: This phase II, double-blind, multicenter, vehicle-controlled, 8-week randomized clinical trial included 212 patients with moderate or severe plaque psoriasis.
Disclosures: The study was funded by Valeant Pharmaceuticals. The presenter reported serving as an investigator, advisor, and speaker for the company.
Alpha-blockers deemed safe in heart failure
WASHINGTON – Prescribing alpha-adrenergic blocking agents for treatment of benign prostatic hypertrophy in men with heart failure proved to be not only safe, but also was associated with significantly reduced rates of both heart failure readmission and all-cause mortality in a large retrospective cohort study.
“These findings are associative, not necessarily causative. But the novel association between alpha-blocker treatment and improved outcomes may warrant further study,” Alberta L. Warner, MD, said at the annual meeting of the American College of Cardiology.

Many men with heart failure also have benign prostatic hypertrophy (BPH), for which they receive treatment with an alpha-blocker. Yet little is known about the impact of this drug class on safety and outcomes in the setting of heart failure. Of note, the use of alpha-blockers for treatment of hypertension was associated with increased risk of new-onset heart failure in the ALLHAT study.
To shed light on the safety of alpha-blockers in patients with prevalent heart failure, Dr. Warner and her coinvestigators analyzed data on nearly 170,000 men with a primary diagnosis of heart failure discharged from Veterans Affairs hospitals in 2002-2015. They determined that fully 28% of them left the hospital on an alpha-blocker. From this pool of nearly 48,000 heart failure patients, they were able to propensity-score match 38,991 on the basis of 54 baseline clinical characteristics to an equal number of VA heart failure patients who weren’t on an alpha-blocker. The study outcomes were time to first readmission for heart failure or all-cause mortality during 2 years of follow-up.
Patients on an alpha-blocker had a 38% heart failure readmission rate at 2 years, compared with 40% in nonusers, for a modest, albeit statistically significant, 6% reduction in relative risk. The all-cause mortality rate also was significantly lower in patients on an alpha-blocker: 40% versus 44%, for a 9% relative risk reduction.
In a subanalysis restricted to the heart failure patients on an alpha-blocker, higher-dose therapy was associated with a significantly lower incidence of all-cause mortality than was a lower-dose alpha-blocker during 2 years of follow-up, with a 9% relative risk reduction. A high-dose alpha-blocker, however, wasn’t significantly more effective than lower-dose therapy in terms of reducing heart failure readmissions, said Dr. Warner, a cardiologist at the Veterans Affairs Greater Los Angeles Healthcare System and the University of California, Los Angeles.
Similarly, patients treated with a nonspecific, peripherally vasoactive alpha-blocker had a significantly lower mortality rate than those on an alpha-1a-adrenergic–specific agent, with a highly significant 15% reduction in relative risk. The type of alpha-blocker, however, didn’t make a significant difference in terms of heart failure readmissions, where both types of agents were similarly better than not being on an alpha-blocker at all, she said.
Eighty-five percent of study participants were on a beta-blocker. Heart failure readmissions and all-cause mortality were significantly lower with alpha-blocker therapy, regardless of whether patients were on background beta-blocker therapy.
One audience member, however, noting that the beta-blocker carvedilol possesses some alpha-blocking effects, asked if outcomes differed depending on whether a patient was on carvedilol or metoprolol.
Dr. Warner replied that those numbers are still being crunched, and that the final answer isn’t in yet, but her preliminary impression is that the benefits of alpha-blocker therapy in terms of both heart failure readmissions and all-cause mortality were attenuated in the patients on carvedilol, possibly due to that beta-blocker’s alpha-blocking properties.
She reported having no financial conflicts regarding the study.
WASHINGTON – Prescribing alpha-adrenergic blocking agents for treatment of benign prostatic hypertrophy in men with heart failure proved to be not only safe, but also was associated with significantly reduced rates of both heart failure readmission and all-cause mortality in a large retrospective cohort study.
“These findings are associative, not necessarily causative. But the novel association between alpha-blocker treatment and improved outcomes may warrant further study,” Alberta L. Warner, MD, said at the annual meeting of the American College of Cardiology.
Many men with heart failure also have benign prostatic hypertrophy (BPH), for which they receive treatment with an alpha-blocker. Yet little is known about the impact of this drug class on safety and outcomes in the setting of heart failure. Of note, the use of alpha-blockers for treatment of hypertension was associated with increased risk of new-onset heart failure in the ALLHAT study.
To shed light on the safety of alpha-blockers in patients with prevalent heart failure, Dr. Warner and her coinvestigators analyzed data on nearly 170,000 men with a primary diagnosis of heart failure discharged from Veterans Affairs hospitals in 2002-2015. They determined that fully 28% of them left the hospital on an alpha-blocker. From this pool of nearly 48,000 heart failure patients, they were able to propensity-score match 38,991 on the basis of 54 baseline clinical characteristics to an equal number of VA heart failure patients who weren’t on an alpha-blocker. The study outcomes were time to first readmission for heart failure or all-cause mortality during 2 years of follow-up.
Patients on an alpha-blocker had a 38% heart failure readmission rate at 2 years, compared with 40% in nonusers, for a modest, albeit statistically significant, 6% reduction in relative risk. The all-cause mortality rate also was significantly lower in patients on an alpha-blocker: 40% versus 44%, for a 9% relative risk reduction.
In a subanalysis restricted to the heart failure patients on an alpha-blocker, higher-dose therapy was associated with a significantly lower incidence of all-cause mortality than was a lower-dose alpha-blocker during 2 years of follow-up, with a 9% relative risk reduction. A high-dose alpha-blocker, however, wasn’t significantly more effective than lower-dose therapy in terms of reducing heart failure readmissions, said Dr. Warner, a cardiologist at the Veterans Affairs Greater Los Angeles Healthcare System and the University of California, Los Angeles.
Similarly, patients treated with a nonspecific, peripherally vasoactive alpha-blocker had a significantly lower mortality rate than those on an alpha-1a-adrenergic–specific agent, with a highly significant 15% reduction in relative risk. The type of alpha-blocker, however, didn’t make a significant difference in terms of heart failure readmissions, where both types of agents were similarly better than not being on an alpha-blocker at all, she said.
Eighty-five percent of study participants were on a beta-blocker. Heart failure readmissions and all-cause mortality were significantly lower with alpha-blocker therapy, regardless of whether patients were on background beta-blocker therapy.
One audience member, however, noting that the beta-blocker carvedilol possesses some alpha-blocking effects, asked if outcomes differed depending on whether a patient was on carvedilol or metoprolol.
Dr. Warner replied that those numbers are still being crunched, and that the final answer isn’t in yet, but her preliminary impression is that the benefits of alpha-blocker therapy in terms of both heart failure readmissions and all-cause mortality were attenuated in the patients on carvedilol, possibly due to that beta-blocker’s alpha-blocking properties.
She reported having no financial conflicts regarding the study.
WASHINGTON – Prescribing alpha-adrenergic blocking agents for treatment of benign prostatic hypertrophy in men with heart failure proved to be not only safe, but also was associated with significantly reduced rates of both heart failure readmission and all-cause mortality in a large retrospective cohort study.
“These findings are associative, not necessarily causative. But the novel association between alpha-blocker treatment and improved outcomes may warrant further study,” Alberta L. Warner, MD, said at the annual meeting of the American College of Cardiology.
Many men with heart failure also have benign prostatic hypertrophy (BPH), for which they receive treatment with an alpha-blocker. Yet little is known about the impact of this drug class on safety and outcomes in the setting of heart failure. Of note, the use of alpha-blockers for treatment of hypertension was associated with increased risk of new-onset heart failure in the ALLHAT study.
To shed light on the safety of alpha-blockers in patients with prevalent heart failure, Dr. Warner and her coinvestigators analyzed data on nearly 170,000 men with a primary diagnosis of heart failure discharged from Veterans Affairs hospitals in 2002-2015. They determined that fully 28% of them left the hospital on an alpha-blocker. From this pool of nearly 48,000 heart failure patients, they were able to propensity-score match 38,991 on the basis of 54 baseline clinical characteristics to an equal number of VA heart failure patients who weren’t on an alpha-blocker. The study outcomes were time to first readmission for heart failure or all-cause mortality during 2 years of follow-up.
Patients on an alpha-blocker had a 38% heart failure readmission rate at 2 years, compared with 40% in nonusers, for a modest, albeit statistically significant, 6% reduction in relative risk. The all-cause mortality rate also was significantly lower in patients on an alpha-blocker: 40% versus 44%, for a 9% relative risk reduction.
In a subanalysis restricted to the heart failure patients on an alpha-blocker, higher-dose therapy was associated with a significantly lower incidence of all-cause mortality than was a lower-dose alpha-blocker during 2 years of follow-up, with a 9% relative risk reduction. A high-dose alpha-blocker, however, wasn’t significantly more effective than lower-dose therapy in terms of reducing heart failure readmissions, said Dr. Warner, a cardiologist at the Veterans Affairs Greater Los Angeles Healthcare System and the University of California, Los Angeles.
Similarly, patients treated with a nonspecific, peripherally vasoactive alpha-blocker had a significantly lower mortality rate than those on an alpha-1a-adrenergic–specific agent, with a highly significant 15% reduction in relative risk. The type of alpha-blocker, however, didn’t make a significant difference in terms of heart failure readmissions, where both types of agents were similarly better than not being on an alpha-blocker at all, she said.
Eighty-five percent of study participants were on a beta-blocker. Heart failure readmissions and all-cause mortality were significantly lower with alpha-blocker therapy, regardless of whether patients were on background beta-blocker therapy.
One audience member, however, noting that the beta-blocker carvedilol possesses some alpha-blocking effects, asked if outcomes differed depending on whether a patient was on carvedilol or metoprolol.
Dr. Warner replied that those numbers are still being crunched, and that the final answer isn’t in yet, but her preliminary impression is that the benefits of alpha-blocker therapy in terms of both heart failure readmissions and all-cause mortality were attenuated in the patients on carvedilol, possibly due to that beta-blocker’s alpha-blocking properties.
She reported having no financial conflicts regarding the study.
Key clinical point:
Major finding: The risk of all-cause mortality in heart failure patients on an alpha-blocker was 9% lower than in extensively matched controls during 2 years of follow-up.
Data source: This retrospective cohort study included 38,991 heart failure patients taking an alpha-blocker for benign prostatic hypertrophy who were propensity matched on 54 clinical characteristics to an equal number of heart failure controls not on the medication.
Disclosures: The study presenter reported having no financial conflicts.
NCI to Study African-American Cancer Survivors
Studies have shown that African Americans have higher incidences of cancer than that of other racial/ethnic groups. They also are more likely to be diagnosed later and to die of the cancer. Compared with whites, African Americans have poorer survival rates for the 4 most common types of cancer (lung, breast, prostate, and colorectal). The ready-to-launch Detroit Research on Cancer Survivors study, funded by the National Cancer Institute (NCI), is “uniquely poised” to find out why, said Douglas Lowy, MD, acting director of NCI.
The largest such study to date will include 5,560 African American cancer survivors and 2,780 family members and will look at cancer progression, recurrence, mortality as well as quality of life for survivors and their families. The researchers will investigate the “myriad factors that may affect cancer survival,” including type of treatment, coexisting disease, genetics, social structure, support, neighborhood context, poverty, stress, racial discrimination, and literacy.
The participants are drawn from 3 counties around Detroit where about 21,000 people are diagnosed with cancer every year. The study also uses data from the Detroit area population-based cancer registry, part of NCI’s Surveillance, Epidemiology and End Results (SEER) Program. Joanne Elena, PhD, MPH, scientific program director for the grant funding the study, calls it a “great example of an efficient use of an existing structure to rapidly recruit cancer survivors into research studies.”
The grant is for $9 million over 5 years. “Investigating the complex factors that lead to disparities in cancer among underserved populations should lead to a greater understanding of the social and biologic causes of such differences,” said Robert Croyle, PhD, director of NCI’s Division of Cancer Control and Population Sciences. “And our hope is that this knowledge will lead to better outcomes.”
Studies have shown that African Americans have higher incidences of cancer than that of other racial/ethnic groups. They also are more likely to be diagnosed later and to die of the cancer. Compared with whites, African Americans have poorer survival rates for the 4 most common types of cancer (lung, breast, prostate, and colorectal). The ready-to-launch Detroit Research on Cancer Survivors study, funded by the National Cancer Institute (NCI), is “uniquely poised” to find out why, said Douglas Lowy, MD, acting director of NCI.
The largest such study to date will include 5,560 African American cancer survivors and 2,780 family members and will look at cancer progression, recurrence, mortality as well as quality of life for survivors and their families. The researchers will investigate the “myriad factors that may affect cancer survival,” including type of treatment, coexisting disease, genetics, social structure, support, neighborhood context, poverty, stress, racial discrimination, and literacy.
The participants are drawn from 3 counties around Detroit where about 21,000 people are diagnosed with cancer every year. The study also uses data from the Detroit area population-based cancer registry, part of NCI’s Surveillance, Epidemiology and End Results (SEER) Program. Joanne Elena, PhD, MPH, scientific program director for the grant funding the study, calls it a “great example of an efficient use of an existing structure to rapidly recruit cancer survivors into research studies.”
The grant is for $9 million over 5 years. “Investigating the complex factors that lead to disparities in cancer among underserved populations should lead to a greater understanding of the social and biologic causes of such differences,” said Robert Croyle, PhD, director of NCI’s Division of Cancer Control and Population Sciences. “And our hope is that this knowledge will lead to better outcomes.”
Studies have shown that African Americans have higher incidences of cancer than that of other racial/ethnic groups. They also are more likely to be diagnosed later and to die of the cancer. Compared with whites, African Americans have poorer survival rates for the 4 most common types of cancer (lung, breast, prostate, and colorectal). The ready-to-launch Detroit Research on Cancer Survivors study, funded by the National Cancer Institute (NCI), is “uniquely poised” to find out why, said Douglas Lowy, MD, acting director of NCI.
The largest such study to date will include 5,560 African American cancer survivors and 2,780 family members and will look at cancer progression, recurrence, mortality as well as quality of life for survivors and their families. The researchers will investigate the “myriad factors that may affect cancer survival,” including type of treatment, coexisting disease, genetics, social structure, support, neighborhood context, poverty, stress, racial discrimination, and literacy.
The participants are drawn from 3 counties around Detroit where about 21,000 people are diagnosed with cancer every year. The study also uses data from the Detroit area population-based cancer registry, part of NCI’s Surveillance, Epidemiology and End Results (SEER) Program. Joanne Elena, PhD, MPH, scientific program director for the grant funding the study, calls it a “great example of an efficient use of an existing structure to rapidly recruit cancer survivors into research studies.”
The grant is for $9 million over 5 years. “Investigating the complex factors that lead to disparities in cancer among underserved populations should lead to a greater understanding of the social and biologic causes of such differences,” said Robert Croyle, PhD, director of NCI’s Division of Cancer Control and Population Sciences. “And our hope is that this knowledge will lead to better outcomes.”
Inhibitor produces durable responses in rel/ref iNHL

WASHINGTON, DC—An investigational drug can produce durable responses and has a manageable safety profile in patients with relapsed or refractory indolent non-Hodgkin lymphoma (iNHL), according to researchers.
The drug is copanlisib, an intravenous pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor.
In the phase 2 CHRONOS-1 trial, copanlisib produced an objective response rate (ORR) of 59.2%, with a complete response (CR) rate of 12%, in patients with relapsed/refractory iNHL.
The median duration of response exceeded 98 weeks.
There were 3 deaths considered related to copanlisib, and the most common treatment-related adverse events (AEs) were transient hyperglycemia and hypertension.
These results were presented at the AACR Annual Meeting 2017 (abstract CT149). The study is supported by Bayer, the company developing copanlisib.
CHRONOS-1 included 141 patients with iNHL. Most (n=104) had follicular lymphoma (FL), 23 had marginal zone lymphoma (MZL), 8 had small lymphocytic lymphoma, and 6 had lymphoplasmacytoid/Waldenstrӧm’s macroglobulinemia.
All patients had relapsed after or were refractory to at least 2 prior lines of therapy, which included both rituximab and an alkylating agent.
For this study, the patients received 60 mg of intravenous copanlisib intermittently on days 1, 8, and 15 of a 28-day cycle.
At the time of analysis, the median duration of treatment was 22 weeks (range, 1-105), and 46 patients were still receiving copanlisib.
Efficacy
For the entire cohort, the ORR was 59.2%. Twelve percent of patients achieved a CR, 47.2% had a partial response (PR), 29.6% had stable disease, and 2.1% had progressive disease.
Among patients with FL, the ORR was 58.7%, the CR rate was 14.4%, and the PR rate was 44.2%.
Among patients with MZL, the ORR was 69.6%, with 8.7% of patients achieving a CR and 60.9% achieving a PR.
For the entire cohort, the estimated median duration of response was 687 days (range, 0-687). For patients with FL, it was 370 days (range, 0-687).
The estimated median progression-free survival was 340 days (range, 0-736), and the median overall survival had not been reached at the time of analysis.
Safety
The most common treatment-related AEs were transient hyperglycemia (all grades, 49%/grade 3+, 40%) and hypertension (all grades, 29%/grade 3+, 23%).
The researchers said other AEs of interest were neutropenia (all grades, 25%/grade 3+, 19%), diarrhea (all grades, 18%/grade 3+, 4%), lung infection (all grades, 14%/grade 3+, 11%), pneumonitis (all grades, 7%/grade 3+, 1.4%), and colitis (0.7%, all grade 3+).
Laboratory AEs of interest were alanine aminotransferase increase (all grades, 23%/grade 1, 19%) and aspartate aminotransferase increase (all grades, 28%/grade 1, 25%).
There were 2 non-fatal opportunistic infections.
There were 6 deaths, and 3 of them were considered related to copanlisib. These 3 deaths were due to lung infection, respiratory failure, and a thromboembolic event.
“[I]nhibition of the PI3K pathway has been shown to be an effective therapeutic strategy in treating indolent lymphomas . . .,” said study investigator Martin Dreyling, MD, of Klinikum der Universität München-Grosshadern in Munich, Germany.
“However, concerns exist about the safety of available oral PI3K inhibitors . . . . The results of CHRONOS-1 demonstrate that intermittent intravenous administration of copanlisib achieved durable efficacy with a manageable safety profile in this difficult-to-treat patient population.” ![]()
WASHINGTON, DC—An investigational drug can produce durable responses and has a manageable safety profile in patients with relapsed or refractory indolent non-Hodgkin lymphoma (iNHL), according to researchers.
The drug is copanlisib, an intravenous pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor.
In the phase 2 CHRONOS-1 trial, copanlisib produced an objective response rate (ORR) of 59.2%, with a complete response (CR) rate of 12%, in patients with relapsed/refractory iNHL.
The median duration of response exceeded 98 weeks.
There were 3 deaths considered related to copanlisib, and the most common treatment-related adverse events (AEs) were transient hyperglycemia and hypertension.
These results were presented at the AACR Annual Meeting 2017 (abstract CT149). The study is supported by Bayer, the company developing copanlisib.
CHRONOS-1 included 141 patients with iNHL. Most (n=104) had follicular lymphoma (FL), 23 had marginal zone lymphoma (MZL), 8 had small lymphocytic lymphoma, and 6 had lymphoplasmacytoid/Waldenstrӧm’s macroglobulinemia.
All patients had relapsed after or were refractory to at least 2 prior lines of therapy, which included both rituximab and an alkylating agent.
For this study, the patients received 60 mg of intravenous copanlisib intermittently on days 1, 8, and 15 of a 28-day cycle.
At the time of analysis, the median duration of treatment was 22 weeks (range, 1-105), and 46 patients were still receiving copanlisib.
Efficacy
For the entire cohort, the ORR was 59.2%. Twelve percent of patients achieved a CR, 47.2% had a partial response (PR), 29.6% had stable disease, and 2.1% had progressive disease.
Among patients with FL, the ORR was 58.7%, the CR rate was 14.4%, and the PR rate was 44.2%.
Among patients with MZL, the ORR was 69.6%, with 8.7% of patients achieving a CR and 60.9% achieving a PR.
For the entire cohort, the estimated median duration of response was 687 days (range, 0-687). For patients with FL, it was 370 days (range, 0-687).
The estimated median progression-free survival was 340 days (range, 0-736), and the median overall survival had not been reached at the time of analysis.
Safety
The most common treatment-related AEs were transient hyperglycemia (all grades, 49%/grade 3+, 40%) and hypertension (all grades, 29%/grade 3+, 23%).
The researchers said other AEs of interest were neutropenia (all grades, 25%/grade 3+, 19%), diarrhea (all grades, 18%/grade 3+, 4%), lung infection (all grades, 14%/grade 3+, 11%), pneumonitis (all grades, 7%/grade 3+, 1.4%), and colitis (0.7%, all grade 3+).
Laboratory AEs of interest were alanine aminotransferase increase (all grades, 23%/grade 1, 19%) and aspartate aminotransferase increase (all grades, 28%/grade 1, 25%).
There were 2 non-fatal opportunistic infections.
There were 6 deaths, and 3 of them were considered related to copanlisib. These 3 deaths were due to lung infection, respiratory failure, and a thromboembolic event.
“[I]nhibition of the PI3K pathway has been shown to be an effective therapeutic strategy in treating indolent lymphomas . . .,” said study investigator Martin Dreyling, MD, of Klinikum der Universität München-Grosshadern in Munich, Germany.
“However, concerns exist about the safety of available oral PI3K inhibitors . . . . The results of CHRONOS-1 demonstrate that intermittent intravenous administration of copanlisib achieved durable efficacy with a manageable safety profile in this difficult-to-treat patient population.” ![]()
WASHINGTON, DC—An investigational drug can produce durable responses and has a manageable safety profile in patients with relapsed or refractory indolent non-Hodgkin lymphoma (iNHL), according to researchers.
The drug is copanlisib, an intravenous pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor.
In the phase 2 CHRONOS-1 trial, copanlisib produced an objective response rate (ORR) of 59.2%, with a complete response (CR) rate of 12%, in patients with relapsed/refractory iNHL.
The median duration of response exceeded 98 weeks.
There were 3 deaths considered related to copanlisib, and the most common treatment-related adverse events (AEs) were transient hyperglycemia and hypertension.
These results were presented at the AACR Annual Meeting 2017 (abstract CT149). The study is supported by Bayer, the company developing copanlisib.
CHRONOS-1 included 141 patients with iNHL. Most (n=104) had follicular lymphoma (FL), 23 had marginal zone lymphoma (MZL), 8 had small lymphocytic lymphoma, and 6 had lymphoplasmacytoid/Waldenstrӧm’s macroglobulinemia.
All patients had relapsed after or were refractory to at least 2 prior lines of therapy, which included both rituximab and an alkylating agent.
For this study, the patients received 60 mg of intravenous copanlisib intermittently on days 1, 8, and 15 of a 28-day cycle.
At the time of analysis, the median duration of treatment was 22 weeks (range, 1-105), and 46 patients were still receiving copanlisib.
Efficacy
For the entire cohort, the ORR was 59.2%. Twelve percent of patients achieved a CR, 47.2% had a partial response (PR), 29.6% had stable disease, and 2.1% had progressive disease.
Among patients with FL, the ORR was 58.7%, the CR rate was 14.4%, and the PR rate was 44.2%.
Among patients with MZL, the ORR was 69.6%, with 8.7% of patients achieving a CR and 60.9% achieving a PR.
For the entire cohort, the estimated median duration of response was 687 days (range, 0-687). For patients with FL, it was 370 days (range, 0-687).
The estimated median progression-free survival was 340 days (range, 0-736), and the median overall survival had not been reached at the time of analysis.
Safety
The most common treatment-related AEs were transient hyperglycemia (all grades, 49%/grade 3+, 40%) and hypertension (all grades, 29%/grade 3+, 23%).
The researchers said other AEs of interest were neutropenia (all grades, 25%/grade 3+, 19%), diarrhea (all grades, 18%/grade 3+, 4%), lung infection (all grades, 14%/grade 3+, 11%), pneumonitis (all grades, 7%/grade 3+, 1.4%), and colitis (0.7%, all grade 3+).
Laboratory AEs of interest were alanine aminotransferase increase (all grades, 23%/grade 1, 19%) and aspartate aminotransferase increase (all grades, 28%/grade 1, 25%).
There were 2 non-fatal opportunistic infections.
There were 6 deaths, and 3 of them were considered related to copanlisib. These 3 deaths were due to lung infection, respiratory failure, and a thromboembolic event.
“[I]nhibition of the PI3K pathway has been shown to be an effective therapeutic strategy in treating indolent lymphomas . . .,” said study investigator Martin Dreyling, MD, of Klinikum der Universität München-Grosshadern in Munich, Germany.
“However, concerns exist about the safety of available oral PI3K inhibitors . . . . The results of CHRONOS-1 demonstrate that intermittent intravenous administration of copanlisib achieved durable efficacy with a manageable safety profile in this difficult-to-treat patient population.” ![]()
First fossilized mammalian RBCs, Babesia-type pathogens found
An amber specimen found in the Dominican Republic contained the first fossilized mammalian red blood cells (RBCs) and intraerythrocytic hemoparasites, according to an article published in the Journal of Medical Entomology.
The fossil contained an engorged tick and RBCs infected with parasites that resemble existing members of the Babesiidae and Theileriidae families of the order Piroplasmida.
It appears that 2 small holes in the back of the tick allowed blood to ooze out just as the tick became stuck in tree sap that later fossilized into amber.
“These 2 tiny holes indicate that something picked a tick off the mammal it was feeding on, puncturing it in the process and dropping it immediately into tree sap,” said study author George Poinar, Jr, PhD, of Oregon State University in Corvallis.
“This would be consistent with the grooming behavior of monkeys that we know lived at that time in this region. The fossilized blood cells, infected with these parasites, are simply amazing in their detail. This discovery provides the only known fossils of Babesia-type pathogens.”
Dr Poinar said the amber specimen came from mines located in the Cordillera Septentrional of the Dominican Republic. It may have originated anywhere from 15 to 45 million years ago.
The fossil contained an engorged nymphal tick of the genus Ambylomma. The parasites found in the RBCs were also found in the gut epithelial cells and body cavity of the tick.
“The life forms we find in amber can reveal so much about the history and evolution of diseases we still struggle with today,” Dr Poinar said. “This parasite, for instance, was clearly around millions of years before humans and appears to have evolved alongside primates, among other hosts.”
Part of what makes this fossil unique, Dr Poinar said, is the way in which the parasites and RBCs were preserved, almost as if they had been stained and otherwise treated in a lab.
The parasites were different enough in texture and density to stand out clearly within the RBCs during the natural embalming process for which amber is famous. ![]()
An amber specimen found in the Dominican Republic contained the first fossilized mammalian red blood cells (RBCs) and intraerythrocytic hemoparasites, according to an article published in the Journal of Medical Entomology.
The fossil contained an engorged tick and RBCs infected with parasites that resemble existing members of the Babesiidae and Theileriidae families of the order Piroplasmida.
It appears that 2 small holes in the back of the tick allowed blood to ooze out just as the tick became stuck in tree sap that later fossilized into amber.
“These 2 tiny holes indicate that something picked a tick off the mammal it was feeding on, puncturing it in the process and dropping it immediately into tree sap,” said study author George Poinar, Jr, PhD, of Oregon State University in Corvallis.
“This would be consistent with the grooming behavior of monkeys that we know lived at that time in this region. The fossilized blood cells, infected with these parasites, are simply amazing in their detail. This discovery provides the only known fossils of Babesia-type pathogens.”
Dr Poinar said the amber specimen came from mines located in the Cordillera Septentrional of the Dominican Republic. It may have originated anywhere from 15 to 45 million years ago.
The fossil contained an engorged nymphal tick of the genus Ambylomma. The parasites found in the RBCs were also found in the gut epithelial cells and body cavity of the tick.
“The life forms we find in amber can reveal so much about the history and evolution of diseases we still struggle with today,” Dr Poinar said. “This parasite, for instance, was clearly around millions of years before humans and appears to have evolved alongside primates, among other hosts.”
Part of what makes this fossil unique, Dr Poinar said, is the way in which the parasites and RBCs were preserved, almost as if they had been stained and otherwise treated in a lab.
The parasites were different enough in texture and density to stand out clearly within the RBCs during the natural embalming process for which amber is famous. ![]()
An amber specimen found in the Dominican Republic contained the first fossilized mammalian red blood cells (RBCs) and intraerythrocytic hemoparasites, according to an article published in the Journal of Medical Entomology.
The fossil contained an engorged tick and RBCs infected with parasites that resemble existing members of the Babesiidae and Theileriidae families of the order Piroplasmida.
It appears that 2 small holes in the back of the tick allowed blood to ooze out just as the tick became stuck in tree sap that later fossilized into amber.
“These 2 tiny holes indicate that something picked a tick off the mammal it was feeding on, puncturing it in the process and dropping it immediately into tree sap,” said study author George Poinar, Jr, PhD, of Oregon State University in Corvallis.
“This would be consistent with the grooming behavior of monkeys that we know lived at that time in this region. The fossilized blood cells, infected with these parasites, are simply amazing in their detail. This discovery provides the only known fossils of Babesia-type pathogens.”
Dr Poinar said the amber specimen came from mines located in the Cordillera Septentrional of the Dominican Republic. It may have originated anywhere from 15 to 45 million years ago.
The fossil contained an engorged nymphal tick of the genus Ambylomma. The parasites found in the RBCs were also found in the gut epithelial cells and body cavity of the tick.
“The life forms we find in amber can reveal so much about the history and evolution of diseases we still struggle with today,” Dr Poinar said. “This parasite, for instance, was clearly around millions of years before humans and appears to have evolved alongside primates, among other hosts.”
Part of what makes this fossil unique, Dr Poinar said, is the way in which the parasites and RBCs were preserved, almost as if they had been stained and otherwise treated in a lab.
The parasites were different enough in texture and density to stand out clearly within the RBCs during the natural embalming process for which amber is famous. ![]()