User login
Revisiting citizenship bonus and surge capacity
I devoted an entire column to the idea of a citizenship bonus in November 2011. At that time I expressed some ambivalence about its effectiveness. Since then I’ve become disenchanted and think it may do more harm than good.
SHM’s 2016 State of Hospital Medicine (SOHM) Report, based on 2015 data, shows that 46% of Hospital Medicine Groups (HMGs) connect some portion of bonus dollars to a provider’s citizenship.1 This is a relatively new phenomenon in the last 5 years or so. My anecdotal experience is that it isn’t limited to hospitalists; it is pretty common for doctors in any specialty who are employed by a hospital or other large organization.

HMGs vary in their definitions of what constitutes citizenship, but usually include things like committee participation, lectures, grand rounds presentations, community talks, research publications.
Our hospitalist group at my hospital has well-defined criteria that require attendance at more than 75% of meetings as a “light switch” (pays nothing itself, but “turns on” availability to citizenship bonus). Bonus dollars are paid for success in any one of several activities, such as making an in-person visit to two PCP offices or completing a meaningful project related to practice operations or clinical care.
I’ve been a supporter of a citizenship bonus for a long time, but two things have made me ambivalent or even opposed to it. The first is a book by Daniel Pink titled Drive: The Surprising Truth About What Motivates Us. It’s a short and very thought-provoking book summarizing research that suggests the effect of providing external rewards like compensation is to “…extinguish intrinsic motivation, diminish performance, crush creativity, and crowd out good behavior.”
The second reason for my ambivalence is my experience working with a lot of HMGs around the country. Those that have a citizenship bonus don’t seem to realize improved operations, more engaged doctors, or lower turnover, and so on. In fact, my experience is that the bonus tends to do exactly what Pink says – steer individuals and the group as a whole away from what is desired.
I’m not ready to say a citizenship bonus is always a bad idea. But it sure seems like it works out badly for many or most groups.
But if you do have a citizenship bonus, then don’t make the mistake of tying it to very basic expectations of the job, like attending group meetings or completing chart documentation on time. Doing those things should never be seen as a reason for a bonus.
Jeopardy (‘surge’) staffing: Not catching on?
As I write, influenza has swept through our region, and my hospital – like most along the west coast – is experiencing incredibly high volumes. I enter the building through a patient care unit that has been mothballed for several years, but today people from building maintenance were busy getting it ready for patients. The hospital is offering various incentives for patient care staff to work extra shifts to manage this volume surge, and our hospitalists have days with encounters near or at our highest-ever level. So surge capacity is once again on my mind.
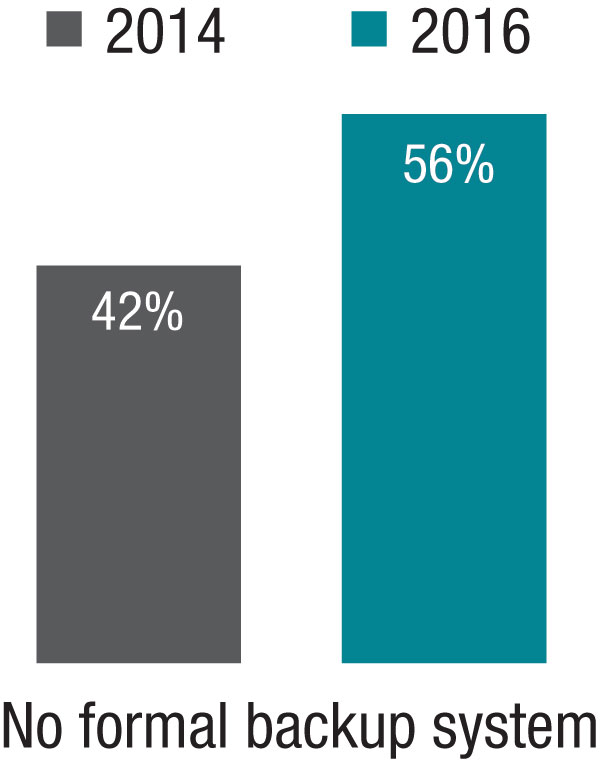
But if every hospitalist in the group went from, say, 156 to 190 shifts annually, the practice might be able to staff every day with an additional provider without adding staff or spending more money. And a doc’s average day would be less busy, which for some people (okay, not very many) would be a worthwhile trade-off. I realize this is a tough sell and to many people it sounds crazy.
The 2014 SOHM showed 42% of HMGs had “no formal backup system,” and this had climbed to 58% in the 2016 Report. I don’t know if jeopardy or surge backup systems are really becoming less common, but it seems pretty clear they aren’t becoming more common. So it’s worth thinking about whether there is a practical way to remove inhibitors of surge capacity.
Dr. Nelson has been a practicing hospitalist since 1988. He is cofounder and past president of SHM, and principal in Nelson Flores Hospital Medicine Consultants. He is course co-director for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. Write to him at [email protected].
Endnotes
1. Note that this is down from 2014, when 64% of groups reported having a citizenship element in their bonus. But I’m skeptical this is a real trend of decreasing popularity and suspect the drop is mostly explained by a much larger portion of respondents in this particular survey coming from hospitalist management companies which I think much less often have a citizenship bonus.
I devoted an entire column to the idea of a citizenship bonus in November 2011. At that time I expressed some ambivalence about its effectiveness. Since then I’ve become disenchanted and think it may do more harm than good.
SHM’s 2016 State of Hospital Medicine (SOHM) Report, based on 2015 data, shows that 46% of Hospital Medicine Groups (HMGs) connect some portion of bonus dollars to a provider’s citizenship.1 This is a relatively new phenomenon in the last 5 years or so. My anecdotal experience is that it isn’t limited to hospitalists; it is pretty common for doctors in any specialty who are employed by a hospital or other large organization.
HMGs vary in their definitions of what constitutes citizenship, but usually include things like committee participation, lectures, grand rounds presentations, community talks, research publications.
Our hospitalist group at my hospital has well-defined criteria that require attendance at more than 75% of meetings as a “light switch” (pays nothing itself, but “turns on” availability to citizenship bonus). Bonus dollars are paid for success in any one of several activities, such as making an in-person visit to two PCP offices or completing a meaningful project related to practice operations or clinical care.
I’ve been a supporter of a citizenship bonus for a long time, but two things have made me ambivalent or even opposed to it. The first is a book by Daniel Pink titled Drive: The Surprising Truth About What Motivates Us. It’s a short and very thought-provoking book summarizing research that suggests the effect of providing external rewards like compensation is to “…extinguish intrinsic motivation, diminish performance, crush creativity, and crowd out good behavior.”
The second reason for my ambivalence is my experience working with a lot of HMGs around the country. Those that have a citizenship bonus don’t seem to realize improved operations, more engaged doctors, or lower turnover, and so on. In fact, my experience is that the bonus tends to do exactly what Pink says – steer individuals and the group as a whole away from what is desired.
I’m not ready to say a citizenship bonus is always a bad idea. But it sure seems like it works out badly for many or most groups.
But if you do have a citizenship bonus, then don’t make the mistake of tying it to very basic expectations of the job, like attending group meetings or completing chart documentation on time. Doing those things should never be seen as a reason for a bonus.
Jeopardy (‘surge’) staffing: Not catching on?
As I write, influenza has swept through our region, and my hospital – like most along the west coast – is experiencing incredibly high volumes. I enter the building through a patient care unit that has been mothballed for several years, but today people from building maintenance were busy getting it ready for patients. The hospital is offering various incentives for patient care staff to work extra shifts to manage this volume surge, and our hospitalists have days with encounters near or at our highest-ever level. So surge capacity is once again on my mind.
But if every hospitalist in the group went from, say, 156 to 190 shifts annually, the practice might be able to staff every day with an additional provider without adding staff or spending more money. And a doc’s average day would be less busy, which for some people (okay, not very many) would be a worthwhile trade-off. I realize this is a tough sell and to many people it sounds crazy.
The 2014 SOHM showed 42% of HMGs had “no formal backup system,” and this had climbed to 58% in the 2016 Report. I don’t know if jeopardy or surge backup systems are really becoming less common, but it seems pretty clear they aren’t becoming more common. So it’s worth thinking about whether there is a practical way to remove inhibitors of surge capacity.
Dr. Nelson has been a practicing hospitalist since 1988. He is cofounder and past president of SHM, and principal in Nelson Flores Hospital Medicine Consultants. He is course co-director for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. Write to him at [email protected].
Endnotes
1. Note that this is down from 2014, when 64% of groups reported having a citizenship element in their bonus. But I’m skeptical this is a real trend of decreasing popularity and suspect the drop is mostly explained by a much larger portion of respondents in this particular survey coming from hospitalist management companies which I think much less often have a citizenship bonus.
I devoted an entire column to the idea of a citizenship bonus in November 2011. At that time I expressed some ambivalence about its effectiveness. Since then I’ve become disenchanted and think it may do more harm than good.
SHM’s 2016 State of Hospital Medicine (SOHM) Report, based on 2015 data, shows that 46% of Hospital Medicine Groups (HMGs) connect some portion of bonus dollars to a provider’s citizenship.1 This is a relatively new phenomenon in the last 5 years or so. My anecdotal experience is that it isn’t limited to hospitalists; it is pretty common for doctors in any specialty who are employed by a hospital or other large organization.
HMGs vary in their definitions of what constitutes citizenship, but usually include things like committee participation, lectures, grand rounds presentations, community talks, research publications.
Our hospitalist group at my hospital has well-defined criteria that require attendance at more than 75% of meetings as a “light switch” (pays nothing itself, but “turns on” availability to citizenship bonus). Bonus dollars are paid for success in any one of several activities, such as making an in-person visit to two PCP offices or completing a meaningful project related to practice operations or clinical care.
I’ve been a supporter of a citizenship bonus for a long time, but two things have made me ambivalent or even opposed to it. The first is a book by Daniel Pink titled Drive: The Surprising Truth About What Motivates Us. It’s a short and very thought-provoking book summarizing research that suggests the effect of providing external rewards like compensation is to “…extinguish intrinsic motivation, diminish performance, crush creativity, and crowd out good behavior.”
The second reason for my ambivalence is my experience working with a lot of HMGs around the country. Those that have a citizenship bonus don’t seem to realize improved operations, more engaged doctors, or lower turnover, and so on. In fact, my experience is that the bonus tends to do exactly what Pink says – steer individuals and the group as a whole away from what is desired.
I’m not ready to say a citizenship bonus is always a bad idea. But it sure seems like it works out badly for many or most groups.
But if you do have a citizenship bonus, then don’t make the mistake of tying it to very basic expectations of the job, like attending group meetings or completing chart documentation on time. Doing those things should never be seen as a reason for a bonus.
Jeopardy (‘surge’) staffing: Not catching on?
As I write, influenza has swept through our region, and my hospital – like most along the west coast – is experiencing incredibly high volumes. I enter the building through a patient care unit that has been mothballed for several years, but today people from building maintenance were busy getting it ready for patients. The hospital is offering various incentives for patient care staff to work extra shifts to manage this volume surge, and our hospitalists have days with encounters near or at our highest-ever level. So surge capacity is once again on my mind.
But if every hospitalist in the group went from, say, 156 to 190 shifts annually, the practice might be able to staff every day with an additional provider without adding staff or spending more money. And a doc’s average day would be less busy, which for some people (okay, not very many) would be a worthwhile trade-off. I realize this is a tough sell and to many people it sounds crazy.
The 2014 SOHM showed 42% of HMGs had “no formal backup system,” and this had climbed to 58% in the 2016 Report. I don’t know if jeopardy or surge backup systems are really becoming less common, but it seems pretty clear they aren’t becoming more common. So it’s worth thinking about whether there is a practical way to remove inhibitors of surge capacity.
Dr. Nelson has been a practicing hospitalist since 1988. He is cofounder and past president of SHM, and principal in Nelson Flores Hospital Medicine Consultants. He is course co-director for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. Write to him at [email protected].
Endnotes
1. Note that this is down from 2014, when 64% of groups reported having a citizenship element in their bonus. But I’m skeptical this is a real trend of decreasing popularity and suspect the drop is mostly explained by a much larger portion of respondents in this particular survey coming from hospitalist management companies which I think much less often have a citizenship bonus.
Wired to win
In 1929, an industrialist in Philadelphia whose factories had been plagued by vandalism sought to curtail the problem by organizing the boys in the community into athletic teams. Within a few years, his effort became Pop Warner Football. A few years later, a group of parents in Williamsport, Pa., started what was to become Little League Baseball.
Prior to the development of these two programs, kids organized their own games using shared equipment, if any at all. They drew foul lines and cobbled together goals in the bare dirt and the stubbly weeds of vacant lots and backyards. Kids shared equipment with each other. They picked teams in a manner that reflected the sometimes painful reality that some kids were proven winners and others were not. Rules were adjusted to fit the situation. Disagreements were settled without referees, or the game dissolved and a lesson was learned.
From its start in the 1930’s, the model of adult-organized and miniaturized versions of professional sports has spread from baseball and football to almost every team sport, including soccer, hockey, and lacrosse. Children may have been deprived of some self-organizing and negotiating skills, but, when one considers the electronically dominated sedentary alternatives, for the most part, adult-organized team youth sports have been a positive.

Of course, there have been some growing pains because an adult sport that has simply been miniaturized doesn’t necessarily fit well with young minds and bodies that are still developing. In some sports, adult/parent coaches now are required to undergo rigorous training in hopes of making the sport more child appropriate. However, the truth remains that, when teams compete, there are going to be winners and losers.
I recently read a newspaper article that included references to a few recent studies that suggest humans are hard wired to win (Sapolsky, Robert. “The Grim Truth Behind the ‘Winner Effect.’ ”The Wall Street Journal. Feb. 24, 2017). Well, not to win exactly but to be more likely to win again once they have been victorious, a phenomenon known as the “winner effect.”
A mouse that has been allowed to win a fixed fight with another mouse is more likely to win his next fight. Other studies on a variety of species, including humans, have found that winning can elevate testosterone levels and suppress stress-mediating hormones – winning boosts confidence and risk taking. More recent studies on zebra fish have demonstrated that a region of the habenula, a portion of the brain, seems to be critical for controlling these behaviors and chemical mediators.
Of course, the problem is that, when there are winners, there have to be losers. From time to time, the adult organizers have struggled with how to compensate for this unfortunate reality in the structure of their youth sports programs. One response has been to give every participant a trophy. Except when the children are so young that they don’t know which goal is theirs, however, awarding trophies to all is a transparent and foolish charade. The winners know who they are and so do the losers. Skillful and compassionate coaches of both winning and losing teams can cooperate to soften the cutting edge of competition, but it will never disappear. It should be fun to play, but it is always going to be more fun to win.
If there is a solution, it falls on the shoulders of parents, educators, and sometimes pediatricians to help the losers find environments and activities in which their skills and aptitudes will give them the greatest chance of enjoying the benefits of the “winner effect.” Winning isn’t everything, but it feels a lot better than losing. If we can help a child to win once – whether it is on the athletic field or in a classroom – it is more likely he or she will do it again.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
In 1929, an industrialist in Philadelphia whose factories had been plagued by vandalism sought to curtail the problem by organizing the boys in the community into athletic teams. Within a few years, his effort became Pop Warner Football. A few years later, a group of parents in Williamsport, Pa., started what was to become Little League Baseball.
Prior to the development of these two programs, kids organized their own games using shared equipment, if any at all. They drew foul lines and cobbled together goals in the bare dirt and the stubbly weeds of vacant lots and backyards. Kids shared equipment with each other. They picked teams in a manner that reflected the sometimes painful reality that some kids were proven winners and others were not. Rules were adjusted to fit the situation. Disagreements were settled without referees, or the game dissolved and a lesson was learned.
From its start in the 1930’s, the model of adult-organized and miniaturized versions of professional sports has spread from baseball and football to almost every team sport, including soccer, hockey, and lacrosse. Children may have been deprived of some self-organizing and negotiating skills, but, when one considers the electronically dominated sedentary alternatives, for the most part, adult-organized team youth sports have been a positive.
Of course, there have been some growing pains because an adult sport that has simply been miniaturized doesn’t necessarily fit well with young minds and bodies that are still developing. In some sports, adult/parent coaches now are required to undergo rigorous training in hopes of making the sport more child appropriate. However, the truth remains that, when teams compete, there are going to be winners and losers.
I recently read a newspaper article that included references to a few recent studies that suggest humans are hard wired to win (Sapolsky, Robert. “The Grim Truth Behind the ‘Winner Effect.’ ”The Wall Street Journal. Feb. 24, 2017). Well, not to win exactly but to be more likely to win again once they have been victorious, a phenomenon known as the “winner effect.”
A mouse that has been allowed to win a fixed fight with another mouse is more likely to win his next fight. Other studies on a variety of species, including humans, have found that winning can elevate testosterone levels and suppress stress-mediating hormones – winning boosts confidence and risk taking. More recent studies on zebra fish have demonstrated that a region of the habenula, a portion of the brain, seems to be critical for controlling these behaviors and chemical mediators.
Of course, the problem is that, when there are winners, there have to be losers. From time to time, the adult organizers have struggled with how to compensate for this unfortunate reality in the structure of their youth sports programs. One response has been to give every participant a trophy. Except when the children are so young that they don’t know which goal is theirs, however, awarding trophies to all is a transparent and foolish charade. The winners know who they are and so do the losers. Skillful and compassionate coaches of both winning and losing teams can cooperate to soften the cutting edge of competition, but it will never disappear. It should be fun to play, but it is always going to be more fun to win.
If there is a solution, it falls on the shoulders of parents, educators, and sometimes pediatricians to help the losers find environments and activities in which their skills and aptitudes will give them the greatest chance of enjoying the benefits of the “winner effect.” Winning isn’t everything, but it feels a lot better than losing. If we can help a child to win once – whether it is on the athletic field or in a classroom – it is more likely he or she will do it again.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
In 1929, an industrialist in Philadelphia whose factories had been plagued by vandalism sought to curtail the problem by organizing the boys in the community into athletic teams. Within a few years, his effort became Pop Warner Football. A few years later, a group of parents in Williamsport, Pa., started what was to become Little League Baseball.
Prior to the development of these two programs, kids organized their own games using shared equipment, if any at all. They drew foul lines and cobbled together goals in the bare dirt and the stubbly weeds of vacant lots and backyards. Kids shared equipment with each other. They picked teams in a manner that reflected the sometimes painful reality that some kids were proven winners and others were not. Rules were adjusted to fit the situation. Disagreements were settled without referees, or the game dissolved and a lesson was learned.
From its start in the 1930’s, the model of adult-organized and miniaturized versions of professional sports has spread from baseball and football to almost every team sport, including soccer, hockey, and lacrosse. Children may have been deprived of some self-organizing and negotiating skills, but, when one considers the electronically dominated sedentary alternatives, for the most part, adult-organized team youth sports have been a positive.
Of course, there have been some growing pains because an adult sport that has simply been miniaturized doesn’t necessarily fit well with young minds and bodies that are still developing. In some sports, adult/parent coaches now are required to undergo rigorous training in hopes of making the sport more child appropriate. However, the truth remains that, when teams compete, there are going to be winners and losers.
I recently read a newspaper article that included references to a few recent studies that suggest humans are hard wired to win (Sapolsky, Robert. “The Grim Truth Behind the ‘Winner Effect.’ ”The Wall Street Journal. Feb. 24, 2017). Well, not to win exactly but to be more likely to win again once they have been victorious, a phenomenon known as the “winner effect.”
A mouse that has been allowed to win a fixed fight with another mouse is more likely to win his next fight. Other studies on a variety of species, including humans, have found that winning can elevate testosterone levels and suppress stress-mediating hormones – winning boosts confidence and risk taking. More recent studies on zebra fish have demonstrated that a region of the habenula, a portion of the brain, seems to be critical for controlling these behaviors and chemical mediators.
Of course, the problem is that, when there are winners, there have to be losers. From time to time, the adult organizers have struggled with how to compensate for this unfortunate reality in the structure of their youth sports programs. One response has been to give every participant a trophy. Except when the children are so young that they don’t know which goal is theirs, however, awarding trophies to all is a transparent and foolish charade. The winners know who they are and so do the losers. Skillful and compassionate coaches of both winning and losing teams can cooperate to soften the cutting edge of competition, but it will never disappear. It should be fun to play, but it is always going to be more fun to win.
If there is a solution, it falls on the shoulders of parents, educators, and sometimes pediatricians to help the losers find environments and activities in which their skills and aptitudes will give them the greatest chance of enjoying the benefits of the “winner effect.” Winning isn’t everything, but it feels a lot better than losing. If we can help a child to win once – whether it is on the athletic field or in a classroom – it is more likely he or she will do it again.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
New 52-week EASI, pruritus data strengthen case for dupilumab in adult atopic dermatitis
ORLANDO – Treatment with dupilumab was associated with significantly improved measures of disease severity, including in quality of life and pruritus symptoms, at 16 and 52 weeks in adults with moderate to severe atopic dermatitis (AD) in the phase III CHRONOS trial.
In the CHRONOS study of adults with uncontrolled, moderate to severe AD, patients were treated with the investigational biologic dupilumab (Dupixent), an interleukin-4 and interleukin-13 pathway blocker administered in subcutaneous injections, in combination with topical corticosteroids. At 52 weeks, they had achieved significantly improved measures of overall disease severity, compared with those who received corticosteroids alone, according to Andrew Blauvelt, MD, MBA, president of Oregon Medical Research Center, Portland, who presented the new data from the study in a late-breaking clinical session at the annual meeting of the American Academy of Dermatology.

The new 52-week data presented at AAD show that the mean improvement in the EASI score from baseline was 80% in the 300 mg dupilumab every week plus corticosteroid group (group 1) and 78% in the group treated every 2 weeks (group 2), compared with 46% in the placebo plus corticosteroids group (control) (P less than .0001).
The mean improvement in self-reported itch from baseline, as measured by the Pruritus Numerical Rating Scale, was 54% in the first group, 56% in the second group, compared with 27% in controls (P less than .0001).
In the first group, 65% achieved a 4-point or greater improvement in their Patient Oriented Eczema Measure scores, as did 76% of the second group, compared with 26% of controls (P less than .0001).
At least a 4-point improvement over baseline in Dermatology Life Quality Index scores was seen in 63% of group 1, 80% of group 2, and 30% of controls (P less than .0001).
Adverse events across the study were similar, although the treatment groups had higher incidences of injection site reactions: 19% in group 1 and 15% in group 2, compared with 8% in controls. The treatment groups also had higher rates of conjunctivitis: 19% in group 1 and 14% in group 2, compared with 8% in controls.
Dr. Blauvelt said that patients who were “exited from the trial were continued for follow-up” and that rescue therapies such as cyclosporine, and other systemic agents, were also available. The rate of rescue therapy was about 15% in the first two groups, while half of controls needed rescue therapy. “We considered those patients who needed rescue nonresponders,” he noted.
The dropout rate at week 52 was about 15% across the treatment groups, compared with twice that in controls.
“Atopic dermatitis is the new psoriasis. We’re in an exciting area now, and we’ll be seeing more biologic therapies for moderate to severe atopic dermatitis. We have a tremendous need for this,” Dr. Blauvelt commented.
The Food and Drug Administration is expected to make a decision on approval of dupilumab by March 29, 2017. Dupilumab was designated by the FDA as a breakthrough therapy for uncontrolled, moderate to severe AD in 2014.
Dr. Blauvelt disclosed many pharmaceutical industry relationships, including with Regeneron Pharmaceuticals and Sanofi, which are developing dupilumab. (If approved, Regeneron and Sanofi Genzyme, part of Sanofi, will commercialize dupilumab).
CORRECTION 3/10/17: An earlier version of this article misstated the rates of clear or nearly clear skin.
[email protected]
On Twitter @whitneymcknight
ORLANDO – Treatment with dupilumab was associated with significantly improved measures of disease severity, including in quality of life and pruritus symptoms, at 16 and 52 weeks in adults with moderate to severe atopic dermatitis (AD) in the phase III CHRONOS trial.
In the CHRONOS study of adults with uncontrolled, moderate to severe AD, patients were treated with the investigational biologic dupilumab (Dupixent), an interleukin-4 and interleukin-13 pathway blocker administered in subcutaneous injections, in combination with topical corticosteroids. At 52 weeks, they had achieved significantly improved measures of overall disease severity, compared with those who received corticosteroids alone, according to Andrew Blauvelt, MD, MBA, president of Oregon Medical Research Center, Portland, who presented the new data from the study in a late-breaking clinical session at the annual meeting of the American Academy of Dermatology.
The new 52-week data presented at AAD show that the mean improvement in the EASI score from baseline was 80% in the 300 mg dupilumab every week plus corticosteroid group (group 1) and 78% in the group treated every 2 weeks (group 2), compared with 46% in the placebo plus corticosteroids group (control) (P less than .0001).
The mean improvement in self-reported itch from baseline, as measured by the Pruritus Numerical Rating Scale, was 54% in the first group, 56% in the second group, compared with 27% in controls (P less than .0001).
In the first group, 65% achieved a 4-point or greater improvement in their Patient Oriented Eczema Measure scores, as did 76% of the second group, compared with 26% of controls (P less than .0001).
At least a 4-point improvement over baseline in Dermatology Life Quality Index scores was seen in 63% of group 1, 80% of group 2, and 30% of controls (P less than .0001).
Adverse events across the study were similar, although the treatment groups had higher incidences of injection site reactions: 19% in group 1 and 15% in group 2, compared with 8% in controls. The treatment groups also had higher rates of conjunctivitis: 19% in group 1 and 14% in group 2, compared with 8% in controls.
Dr. Blauvelt said that patients who were “exited from the trial were continued for follow-up” and that rescue therapies such as cyclosporine, and other systemic agents, were also available. The rate of rescue therapy was about 15% in the first two groups, while half of controls needed rescue therapy. “We considered those patients who needed rescue nonresponders,” he noted.
The dropout rate at week 52 was about 15% across the treatment groups, compared with twice that in controls.
“Atopic dermatitis is the new psoriasis. We’re in an exciting area now, and we’ll be seeing more biologic therapies for moderate to severe atopic dermatitis. We have a tremendous need for this,” Dr. Blauvelt commented.
The Food and Drug Administration is expected to make a decision on approval of dupilumab by March 29, 2017. Dupilumab was designated by the FDA as a breakthrough therapy for uncontrolled, moderate to severe AD in 2014.
Dr. Blauvelt disclosed many pharmaceutical industry relationships, including with Regeneron Pharmaceuticals and Sanofi, which are developing dupilumab. (If approved, Regeneron and Sanofi Genzyme, part of Sanofi, will commercialize dupilumab).
CORRECTION 3/10/17: An earlier version of this article misstated the rates of clear or nearly clear skin.
[email protected]
On Twitter @whitneymcknight
ORLANDO – Treatment with dupilumab was associated with significantly improved measures of disease severity, including in quality of life and pruritus symptoms, at 16 and 52 weeks in adults with moderate to severe atopic dermatitis (AD) in the phase III CHRONOS trial.
In the CHRONOS study of adults with uncontrolled, moderate to severe AD, patients were treated with the investigational biologic dupilumab (Dupixent), an interleukin-4 and interleukin-13 pathway blocker administered in subcutaneous injections, in combination with topical corticosteroids. At 52 weeks, they had achieved significantly improved measures of overall disease severity, compared with those who received corticosteroids alone, according to Andrew Blauvelt, MD, MBA, president of Oregon Medical Research Center, Portland, who presented the new data from the study in a late-breaking clinical session at the annual meeting of the American Academy of Dermatology.
The new 52-week data presented at AAD show that the mean improvement in the EASI score from baseline was 80% in the 300 mg dupilumab every week plus corticosteroid group (group 1) and 78% in the group treated every 2 weeks (group 2), compared with 46% in the placebo plus corticosteroids group (control) (P less than .0001).
The mean improvement in self-reported itch from baseline, as measured by the Pruritus Numerical Rating Scale, was 54% in the first group, 56% in the second group, compared with 27% in controls (P less than .0001).
In the first group, 65% achieved a 4-point or greater improvement in their Patient Oriented Eczema Measure scores, as did 76% of the second group, compared with 26% of controls (P less than .0001).
At least a 4-point improvement over baseline in Dermatology Life Quality Index scores was seen in 63% of group 1, 80% of group 2, and 30% of controls (P less than .0001).
Adverse events across the study were similar, although the treatment groups had higher incidences of injection site reactions: 19% in group 1 and 15% in group 2, compared with 8% in controls. The treatment groups also had higher rates of conjunctivitis: 19% in group 1 and 14% in group 2, compared with 8% in controls.
Dr. Blauvelt said that patients who were “exited from the trial were continued for follow-up” and that rescue therapies such as cyclosporine, and other systemic agents, were also available. The rate of rescue therapy was about 15% in the first two groups, while half of controls needed rescue therapy. “We considered those patients who needed rescue nonresponders,” he noted.
The dropout rate at week 52 was about 15% across the treatment groups, compared with twice that in controls.
“Atopic dermatitis is the new psoriasis. We’re in an exciting area now, and we’ll be seeing more biologic therapies for moderate to severe atopic dermatitis. We have a tremendous need for this,” Dr. Blauvelt commented.
The Food and Drug Administration is expected to make a decision on approval of dupilumab by March 29, 2017. Dupilumab was designated by the FDA as a breakthrough therapy for uncontrolled, moderate to severe AD in 2014.
Dr. Blauvelt disclosed many pharmaceutical industry relationships, including with Regeneron Pharmaceuticals and Sanofi, which are developing dupilumab. (If approved, Regeneron and Sanofi Genzyme, part of Sanofi, will commercialize dupilumab).
CORRECTION 3/10/17: An earlier version of this article misstated the rates of clear or nearly clear skin.
[email protected]
On Twitter @whitneymcknight
AT AAD 2017
Key clinical point:
Major finding: At 52 weeks, dupilumab 300 mg administered in a subcutaneous injection once a week or every two weeks plus topical corticosteroids resulted in significantly more clearing compared with topical corticosteroids alone (P less than .0001). Self-reported measures of itch and quality-of-life measures were also higher across treatment groups.
Data source: A phase III trial of 740 adults with moderate to severe AD, randomized to treatment with one of the two regimens or corticosteroids alone.
Disclosures: Dr. Blauvelt disclosed many pharmaceutical industry relationships, including with Regeneron Pharmaceuticals and Sanofi, which are developing dupilumab. (If approved, Regeneron and Sanofi Genzyme, part of Sanofi, will commercialize dupilumab).
Cannabis associated with increased risk of heart failure and stroke
Cannabis use was associated with an increased risk of cerebrovascular accidents and heart failure in a retrospective analysis of the Nationwide Inpatient Sample (NIS).
Aditi Kalla, MD, a cardiology fellow at Einstein Medical Center in Philadelphia, and her colleagues analyzed data from nearly 21 million adult patients aged 18-55 years from the NIS 2009-2010 database. Approximately 1.5% (316,397) were diagnosed as cannabis users.

Cannabis users also were more likely to report cardiac risk factors such as hypertension (19.9% vs. 15.7% of nonusers), tobacco use (47.2% vs. 11.4%), alcohol use (28.1% vs 3.8%), and obesity (7% vs. 6.5%). They were older, on average, with a mean age of 33 years, compared with 26 years, and were likely to be male (60%), Dr. Kalla noted during a press briefing held in advance of the annual meeting of the American College of Cardiology.
Using multivariate regression analysis to adjust for these traditional cardiovascular risk factors, the investigators found cannabis remained an independent predictor for heart failure, with an odds ratio of 1.1 (P less than .01) and cerebrovascular accident, with an OR of 1.24 (P less than .001).
“Even when we corrected for known risks, we still found a higher rate of both stroke and heart failure in these patients,” Dr. Kalla said. “That leads us to believe that there is something else going on besides just obesity or diet-related cardiovascular side effects.”
Dr. Kalla noted that an expert analysis published by the ACC in September 2016 linked cannabinoid receptor type 1 with atherogenesis.
Further research is needed on the topic of cannabis and cardiovascular effects, especially as the legalization of medical and recreational cannabis spreads across the country, Dr. Kalla said. “Decriminalization of cannabis has passed in several states, bringing the total count now up to 28 states, plus the District of Columbia. We now need to be more knowledgeable of the risks and benefits of cannabis, as patients in these states may inquire into the use of it, or even ask us for prescriptions for it.”
While the NIS provided a large and strong data set for this analysis, the number of cannabis users likely was underreported because cannabis was legal in just 14 states at the time, Dr. Kalla noted. The study also was limited by a lack of specific information regarding cannabis intake, method of intake (ingestion or smoking), quantity and frequency of use, and whether use was medical or recreational.
The information collected also excluded whether patients used marijuana for medical or recreational purpose and how it was taken, by smoking or ingestion.
[email protected]
On Twitter @EAZTweets
Cannabis use was associated with an increased risk of cerebrovascular accidents and heart failure in a retrospective analysis of the Nationwide Inpatient Sample (NIS).
Aditi Kalla, MD, a cardiology fellow at Einstein Medical Center in Philadelphia, and her colleagues analyzed data from nearly 21 million adult patients aged 18-55 years from the NIS 2009-2010 database. Approximately 1.5% (316,397) were diagnosed as cannabis users.
Cannabis users also were more likely to report cardiac risk factors such as hypertension (19.9% vs. 15.7% of nonusers), tobacco use (47.2% vs. 11.4%), alcohol use (28.1% vs 3.8%), and obesity (7% vs. 6.5%). They were older, on average, with a mean age of 33 years, compared with 26 years, and were likely to be male (60%), Dr. Kalla noted during a press briefing held in advance of the annual meeting of the American College of Cardiology.
Using multivariate regression analysis to adjust for these traditional cardiovascular risk factors, the investigators found cannabis remained an independent predictor for heart failure, with an odds ratio of 1.1 (P less than .01) and cerebrovascular accident, with an OR of 1.24 (P less than .001).
“Even when we corrected for known risks, we still found a higher rate of both stroke and heart failure in these patients,” Dr. Kalla said. “That leads us to believe that there is something else going on besides just obesity or diet-related cardiovascular side effects.”
Dr. Kalla noted that an expert analysis published by the ACC in September 2016 linked cannabinoid receptor type 1 with atherogenesis.
Further research is needed on the topic of cannabis and cardiovascular effects, especially as the legalization of medical and recreational cannabis spreads across the country, Dr. Kalla said. “Decriminalization of cannabis has passed in several states, bringing the total count now up to 28 states, plus the District of Columbia. We now need to be more knowledgeable of the risks and benefits of cannabis, as patients in these states may inquire into the use of it, or even ask us for prescriptions for it.”
While the NIS provided a large and strong data set for this analysis, the number of cannabis users likely was underreported because cannabis was legal in just 14 states at the time, Dr. Kalla noted. The study also was limited by a lack of specific information regarding cannabis intake, method of intake (ingestion or smoking), quantity and frequency of use, and whether use was medical or recreational.
The information collected also excluded whether patients used marijuana for medical or recreational purpose and how it was taken, by smoking or ingestion.
[email protected]
On Twitter @EAZTweets
Cannabis use was associated with an increased risk of cerebrovascular accidents and heart failure in a retrospective analysis of the Nationwide Inpatient Sample (NIS).
Aditi Kalla, MD, a cardiology fellow at Einstein Medical Center in Philadelphia, and her colleagues analyzed data from nearly 21 million adult patients aged 18-55 years from the NIS 2009-2010 database. Approximately 1.5% (316,397) were diagnosed as cannabis users.
Cannabis users also were more likely to report cardiac risk factors such as hypertension (19.9% vs. 15.7% of nonusers), tobacco use (47.2% vs. 11.4%), alcohol use (28.1% vs 3.8%), and obesity (7% vs. 6.5%). They were older, on average, with a mean age of 33 years, compared with 26 years, and were likely to be male (60%), Dr. Kalla noted during a press briefing held in advance of the annual meeting of the American College of Cardiology.
Using multivariate regression analysis to adjust for these traditional cardiovascular risk factors, the investigators found cannabis remained an independent predictor for heart failure, with an odds ratio of 1.1 (P less than .01) and cerebrovascular accident, with an OR of 1.24 (P less than .001).
“Even when we corrected for known risks, we still found a higher rate of both stroke and heart failure in these patients,” Dr. Kalla said. “That leads us to believe that there is something else going on besides just obesity or diet-related cardiovascular side effects.”
Dr. Kalla noted that an expert analysis published by the ACC in September 2016 linked cannabinoid receptor type 1 with atherogenesis.
Further research is needed on the topic of cannabis and cardiovascular effects, especially as the legalization of medical and recreational cannabis spreads across the country, Dr. Kalla said. “Decriminalization of cannabis has passed in several states, bringing the total count now up to 28 states, plus the District of Columbia. We now need to be more knowledgeable of the risks and benefits of cannabis, as patients in these states may inquire into the use of it, or even ask us for prescriptions for it.”
While the NIS provided a large and strong data set for this analysis, the number of cannabis users likely was underreported because cannabis was legal in just 14 states at the time, Dr. Kalla noted. The study also was limited by a lack of specific information regarding cannabis intake, method of intake (ingestion or smoking), quantity and frequency of use, and whether use was medical or recreational.
The information collected also excluded whether patients used marijuana for medical or recreational purpose and how it was taken, by smoking or ingestion.
[email protected]
On Twitter @EAZTweets
FROM ACC 17
Key clinical point:
Major finding: Cannabis users showed 26% increased risk (OR, 1.24) of stroke and 10% increased risk (OR, 1.1) of heart failure.
Data source: Retrospective study of over 20 million patients’ records aged 18-55 years gathered from the Nationwide Inpatient Sample 2009-2010 database.
Disclosures: Researchers reported no relevant conflicts of interest.
VIDEO: Tips, tricks, and pearls for keloid scar steroid injections
ORLANDO – What are the best ways to avoid atrophy when treating keloid scars with steroid injections? When should a keloid be treated with smaller amounts, but with a stronger steroid concentration? What is the most effective way to avoid precipitation in the syringe?
The answers to these questions – along with other tips, tricks, and pearls for treating keloids, both in patients with skin of color and those with white skin – are provided by Temitayo Ogunleye, MD, of the department of dermatology, University of Pennsylvania, Philadelphia, in a video interview at the annual meeting of the American Academy of Dermatology.
Dr. Ogunleye had no relevant disclosures.
[email protected]
On Twitter @whitneymcknight
ORLANDO – What are the best ways to avoid atrophy when treating keloid scars with steroid injections? When should a keloid be treated with smaller amounts, but with a stronger steroid concentration? What is the most effective way to avoid precipitation in the syringe?
The answers to these questions – along with other tips, tricks, and pearls for treating keloids, both in patients with skin of color and those with white skin – are provided by Temitayo Ogunleye, MD, of the department of dermatology, University of Pennsylvania, Philadelphia, in a video interview at the annual meeting of the American Academy of Dermatology.
Dr. Ogunleye had no relevant disclosures.
[email protected]
On Twitter @whitneymcknight
ORLANDO – What are the best ways to avoid atrophy when treating keloid scars with steroid injections? When should a keloid be treated with smaller amounts, but with a stronger steroid concentration? What is the most effective way to avoid precipitation in the syringe?
The answers to these questions – along with other tips, tricks, and pearls for treating keloids, both in patients with skin of color and those with white skin – are provided by Temitayo Ogunleye, MD, of the department of dermatology, University of Pennsylvania, Philadelphia, in a video interview at the annual meeting of the American Academy of Dermatology.
Dr. Ogunleye had no relevant disclosures.
[email protected]
On Twitter @whitneymcknight
AT AAD 17

Norovirus reporting tool yields real-time outbreak data
NoroSTAT, the Centers for Disease Control and Prevention’s new program with which states can report norovirus outbreaks, yields more timely and more complete epidemiologic and laboratory data, which allows a faster and better-informed public health response to such outbreaks, according to a report published in the Morbidity and Mortality Weekly Report.
The CDC launched NoroSTAT (Norovirus Sentinel Testing and Tracking) in 2012 to permit the health departments in selected states to report specific epidemiologic and laboratory data regarding norovirus outbreaks more rapidly than usual – within 7 business days, said Minesh P. Shah, MD, of the Epidemic Intelligence Service and the division of viral diseases, CDC, Atlanta, and his associates.
NoroSTAT significantly reduced the median interval in reporting epidemiologic data concerning norovirus from 22 days to 2 days and significantly reduced the median interval in reporting relevant laboratory data from 21 days to 3 days. The percentage of reports submitted within 7 business days increased from 26% to 95% among the states participating in NoroSTAT, while remaining low – only 12%-13% – in nonparticipating states. The number of complete reports also increased substantially, from 87% to 99.9%, among the participating states.
These improvements likely result from NoroSTAT’s stringent reporting requirements and from the program’s ability “to enhance communication between epidemiologists and laboratorians in both state health departments and at CDC,” Dr. Shah and his associates said (MMWR Morbidity and Mortality Weekly Report. 2017 Feb 24;66:185-9).
NoroSTAT represents a key advancement in norovirus outbreak surveillance and has proved valuable in early identification and better characterization of outbreaks. It was expanded to include nine states in August 2016, the investigators added.
NoroSTAT, the Centers for Disease Control and Prevention’s new program with which states can report norovirus outbreaks, yields more timely and more complete epidemiologic and laboratory data, which allows a faster and better-informed public health response to such outbreaks, according to a report published in the Morbidity and Mortality Weekly Report.
The CDC launched NoroSTAT (Norovirus Sentinel Testing and Tracking) in 2012 to permit the health departments in selected states to report specific epidemiologic and laboratory data regarding norovirus outbreaks more rapidly than usual – within 7 business days, said Minesh P. Shah, MD, of the Epidemic Intelligence Service and the division of viral diseases, CDC, Atlanta, and his associates.
NoroSTAT significantly reduced the median interval in reporting epidemiologic data concerning norovirus from 22 days to 2 days and significantly reduced the median interval in reporting relevant laboratory data from 21 days to 3 days. The percentage of reports submitted within 7 business days increased from 26% to 95% among the states participating in NoroSTAT, while remaining low – only 12%-13% – in nonparticipating states. The number of complete reports also increased substantially, from 87% to 99.9%, among the participating states.
These improvements likely result from NoroSTAT’s stringent reporting requirements and from the program’s ability “to enhance communication between epidemiologists and laboratorians in both state health departments and at CDC,” Dr. Shah and his associates said (MMWR Morbidity and Mortality Weekly Report. 2017 Feb 24;66:185-9).
NoroSTAT represents a key advancement in norovirus outbreak surveillance and has proved valuable in early identification and better characterization of outbreaks. It was expanded to include nine states in August 2016, the investigators added.
NoroSTAT, the Centers for Disease Control and Prevention’s new program with which states can report norovirus outbreaks, yields more timely and more complete epidemiologic and laboratory data, which allows a faster and better-informed public health response to such outbreaks, according to a report published in the Morbidity and Mortality Weekly Report.
The CDC launched NoroSTAT (Norovirus Sentinel Testing and Tracking) in 2012 to permit the health departments in selected states to report specific epidemiologic and laboratory data regarding norovirus outbreaks more rapidly than usual – within 7 business days, said Minesh P. Shah, MD, of the Epidemic Intelligence Service and the division of viral diseases, CDC, Atlanta, and his associates.
NoroSTAT significantly reduced the median interval in reporting epidemiologic data concerning norovirus from 22 days to 2 days and significantly reduced the median interval in reporting relevant laboratory data from 21 days to 3 days. The percentage of reports submitted within 7 business days increased from 26% to 95% among the states participating in NoroSTAT, while remaining low – only 12%-13% – in nonparticipating states. The number of complete reports also increased substantially, from 87% to 99.9%, among the participating states.
These improvements likely result from NoroSTAT’s stringent reporting requirements and from the program’s ability “to enhance communication between epidemiologists and laboratorians in both state health departments and at CDC,” Dr. Shah and his associates said (MMWR Morbidity and Mortality Weekly Report. 2017 Feb 24;66:185-9).
NoroSTAT represents a key advancement in norovirus outbreak surveillance and has proved valuable in early identification and better characterization of outbreaks. It was expanded to include nine states in August 2016, the investigators added.
Key clinical point: NoroSTAT, the CDC’s new program with which states can report norovirus outbreaks, yields more timely and complete epidemiologic data.
Major finding: NoroSTAT significantly reduced the median interval in reporting epidemiologic data concerning norovirus from 22 days to 2 days and significantly reduced the median interval in reporting relevant laboratory data from 21 days to 3 days.
Data source: A comparison of epidemiologic and laboratory data reported by all 50 states for the 3 years before and the 3 years after NoroSTAT was implemented in 5 states.
Disclosures: This study was sponsored by the Centers for Disease Control and Prevention. No financial disclosures were provided.
Sneak Peek: Journal of Hospital Medicine
Background: Communication among team members within hospitals is typically fragmented. Bedside interdisciplinary rounds (IDR) have the potential to improve communication and outcomes through enhanced structure and patient engagement.
Objective: To decrease length of stay (LOS) and complications through the transformation of daily IDR to a bedside model.
Setting: Two geographic areas of a medical unit using a clinical microsystem structure.
Patients: 2,005 hospitalizations over a 12-month period.
Interventions: A bedside model (mobile interdisciplinary care rounds [MICRO]) was developed. MICRO featured a defined structure, scripting, patient engagement, and a patient safety checklist.
Measurements: The primary outcomes were clinical deterioration (composite of death, transfer to a higher level of care, or development of a hospital-acquired complication) and length of stay (LOS). Patient safety culture and perceptions of bedside interdisciplinary rounding were assessed pre- and post-implementation.
Results: There was no difference in LOS (6.6 vs. 7.0 days, P = .17, for the MICRO and control groups, respectively) or clinical deterioration (7.7% vs. 9.3%, P = .46). LOS was reduced for patients transferred to the study unit (10.4 vs. 14.0 days, P = .02, for the MICRO and control groups, respectively). Nurses and hospitalists gave significantly higher scores for patient safety climate and the efficiency of rounds after implementation of the MICRO model.
Limitations: The trial was performed at a single hospital.
Conclusions: Bedside IDR did not reduce overall LOS or clinical deterioration. Future studies should examine whether comprehensive transformation of medical units, including co-leadership, geographic cohorting of teams, and bedside interdisciplinary rounding, improves clinical outcomes compared to units without these features.
Also in the Journal of Hospital Medicine …
Standardized Attending Rounds to Improve the Patient Experience: A Pragmatic Cluster Randomized Controlled Trial
Authors: Bradley Monash, MD, Nader Najafi, MD, Michelle Mourad, MD, Alvin Rajkomar, MD, Sumant R. Ranji, MD, Margaret C. Fang, MD, MPH, FHM, Marcia Glass, MD, Dimiter Milev, MPH, Yile Ding, MD, Andy Shen, BA, Bradley A. Sharpe, MD, FACP, SFHM, James D Harrison, MPH, PhD
All Together Now: Impact of a Regionalization and Bedside Rounding Initiative on the Efficiency and Inclusiveness of Clinical Rounds
Authors: Kristin T. L. Huang, MD, Jacquelyn Minahan, Patricia Brita-Rossi, RN, MSN, MBA, Patricia Aylward, RN, MSN, Joel T. Katz, MD, SFHM, Christopher Roy, MD, Jeffrey L. Schnipper, MD, MPH, FHM, Robert Boxer, MD, PhD
Family Report Compared to Clinician-Documented Diagnoses for Psychiatric Conditions Among Hospitalized Children
Authors: Stephanie K. Doupnik, MD, Chris Feudtner, MD, PhD, MPH, Steven C. Marcus, PhD
Perceived Safety and Value of Inpatient ‘Very Important Person’ Services
Authors: Joshua Allen-Dicker, MD, MPH, Andrew Auerbach, MD, MPH, SFHM, Shoshana J. Herzig, MD, MPH
A Time and Motion Study of Pharmacists and Pharmacy Technicians Obtaining Admission Medication Histories
Authors: Caroline B. Nguyen, PharmD, BCPS, Rita Shane, PharmD, FASHP, FCSHP, Douglas S. Bell, MD, PhD, Galen Cook-Wiens, MS, Joshua M. Pevnick, MD, MSHS
Background: Communication among team members within hospitals is typically fragmented. Bedside interdisciplinary rounds (IDR) have the potential to improve communication and outcomes through enhanced structure and patient engagement.
Objective: To decrease length of stay (LOS) and complications through the transformation of daily IDR to a bedside model.
Setting: Two geographic areas of a medical unit using a clinical microsystem structure.
Patients: 2,005 hospitalizations over a 12-month period.
Interventions: A bedside model (mobile interdisciplinary care rounds [MICRO]) was developed. MICRO featured a defined structure, scripting, patient engagement, and a patient safety checklist.
Measurements: The primary outcomes were clinical deterioration (composite of death, transfer to a higher level of care, or development of a hospital-acquired complication) and length of stay (LOS). Patient safety culture and perceptions of bedside interdisciplinary rounding were assessed pre- and post-implementation.
Results: There was no difference in LOS (6.6 vs. 7.0 days, P = .17, for the MICRO and control groups, respectively) or clinical deterioration (7.7% vs. 9.3%, P = .46). LOS was reduced for patients transferred to the study unit (10.4 vs. 14.0 days, P = .02, for the MICRO and control groups, respectively). Nurses and hospitalists gave significantly higher scores for patient safety climate and the efficiency of rounds after implementation of the MICRO model.
Limitations: The trial was performed at a single hospital.
Conclusions: Bedside IDR did not reduce overall LOS or clinical deterioration. Future studies should examine whether comprehensive transformation of medical units, including co-leadership, geographic cohorting of teams, and bedside interdisciplinary rounding, improves clinical outcomes compared to units without these features.
Also in the Journal of Hospital Medicine …
Standardized Attending Rounds to Improve the Patient Experience: A Pragmatic Cluster Randomized Controlled Trial
Authors: Bradley Monash, MD, Nader Najafi, MD, Michelle Mourad, MD, Alvin Rajkomar, MD, Sumant R. Ranji, MD, Margaret C. Fang, MD, MPH, FHM, Marcia Glass, MD, Dimiter Milev, MPH, Yile Ding, MD, Andy Shen, BA, Bradley A. Sharpe, MD, FACP, SFHM, James D Harrison, MPH, PhD
All Together Now: Impact of a Regionalization and Bedside Rounding Initiative on the Efficiency and Inclusiveness of Clinical Rounds
Authors: Kristin T. L. Huang, MD, Jacquelyn Minahan, Patricia Brita-Rossi, RN, MSN, MBA, Patricia Aylward, RN, MSN, Joel T. Katz, MD, SFHM, Christopher Roy, MD, Jeffrey L. Schnipper, MD, MPH, FHM, Robert Boxer, MD, PhD
Family Report Compared to Clinician-Documented Diagnoses for Psychiatric Conditions Among Hospitalized Children
Authors: Stephanie K. Doupnik, MD, Chris Feudtner, MD, PhD, MPH, Steven C. Marcus, PhD
Perceived Safety and Value of Inpatient ‘Very Important Person’ Services
Authors: Joshua Allen-Dicker, MD, MPH, Andrew Auerbach, MD, MPH, SFHM, Shoshana J. Herzig, MD, MPH
A Time and Motion Study of Pharmacists and Pharmacy Technicians Obtaining Admission Medication Histories
Authors: Caroline B. Nguyen, PharmD, BCPS, Rita Shane, PharmD, FASHP, FCSHP, Douglas S. Bell, MD, PhD, Galen Cook-Wiens, MS, Joshua M. Pevnick, MD, MSHS
Background: Communication among team members within hospitals is typically fragmented. Bedside interdisciplinary rounds (IDR) have the potential to improve communication and outcomes through enhanced structure and patient engagement.
Objective: To decrease length of stay (LOS) and complications through the transformation of daily IDR to a bedside model.
Setting: Two geographic areas of a medical unit using a clinical microsystem structure.
Patients: 2,005 hospitalizations over a 12-month period.
Interventions: A bedside model (mobile interdisciplinary care rounds [MICRO]) was developed. MICRO featured a defined structure, scripting, patient engagement, and a patient safety checklist.
Measurements: The primary outcomes were clinical deterioration (composite of death, transfer to a higher level of care, or development of a hospital-acquired complication) and length of stay (LOS). Patient safety culture and perceptions of bedside interdisciplinary rounding were assessed pre- and post-implementation.
Results: There was no difference in LOS (6.6 vs. 7.0 days, P = .17, for the MICRO and control groups, respectively) or clinical deterioration (7.7% vs. 9.3%, P = .46). LOS was reduced for patients transferred to the study unit (10.4 vs. 14.0 days, P = .02, for the MICRO and control groups, respectively). Nurses and hospitalists gave significantly higher scores for patient safety climate and the efficiency of rounds after implementation of the MICRO model.
Limitations: The trial was performed at a single hospital.
Conclusions: Bedside IDR did not reduce overall LOS or clinical deterioration. Future studies should examine whether comprehensive transformation of medical units, including co-leadership, geographic cohorting of teams, and bedside interdisciplinary rounding, improves clinical outcomes compared to units without these features.
Also in the Journal of Hospital Medicine …
Standardized Attending Rounds to Improve the Patient Experience: A Pragmatic Cluster Randomized Controlled Trial
Authors: Bradley Monash, MD, Nader Najafi, MD, Michelle Mourad, MD, Alvin Rajkomar, MD, Sumant R. Ranji, MD, Margaret C. Fang, MD, MPH, FHM, Marcia Glass, MD, Dimiter Milev, MPH, Yile Ding, MD, Andy Shen, BA, Bradley A. Sharpe, MD, FACP, SFHM, James D Harrison, MPH, PhD
All Together Now: Impact of a Regionalization and Bedside Rounding Initiative on the Efficiency and Inclusiveness of Clinical Rounds
Authors: Kristin T. L. Huang, MD, Jacquelyn Minahan, Patricia Brita-Rossi, RN, MSN, MBA, Patricia Aylward, RN, MSN, Joel T. Katz, MD, SFHM, Christopher Roy, MD, Jeffrey L. Schnipper, MD, MPH, FHM, Robert Boxer, MD, PhD
Family Report Compared to Clinician-Documented Diagnoses for Psychiatric Conditions Among Hospitalized Children
Authors: Stephanie K. Doupnik, MD, Chris Feudtner, MD, PhD, MPH, Steven C. Marcus, PhD
Perceived Safety and Value of Inpatient ‘Very Important Person’ Services
Authors: Joshua Allen-Dicker, MD, MPH, Andrew Auerbach, MD, MPH, SFHM, Shoshana J. Herzig, MD, MPH
A Time and Motion Study of Pharmacists and Pharmacy Technicians Obtaining Admission Medication Histories
Authors: Caroline B. Nguyen, PharmD, BCPS, Rita Shane, PharmD, FASHP, FCSHP, Douglas S. Bell, MD, PhD, Galen Cook-Wiens, MS, Joshua M. Pevnick, MD, MSHS
Hospitalists trained in family medicine seek critical care training pathway
A nationwide shortage of intensivists has more hospitalists stepping into the critical care arena, but not all with the level of preparation and comfort of David Aymond, MD, a Louisiana-based hospitalist trained in family medicine (HTFM).
Dr. Aymond gained his ICU experience in a fellowship with the University of Alabama, where hospitalists also “were responsible for ICU patients,” he said. Years later, as an employee of both small and large hospitals with busy ICU services, and a faculty member for a family medicine residency with a busy ICU, Dr. Aymond moves seamlessly between roles.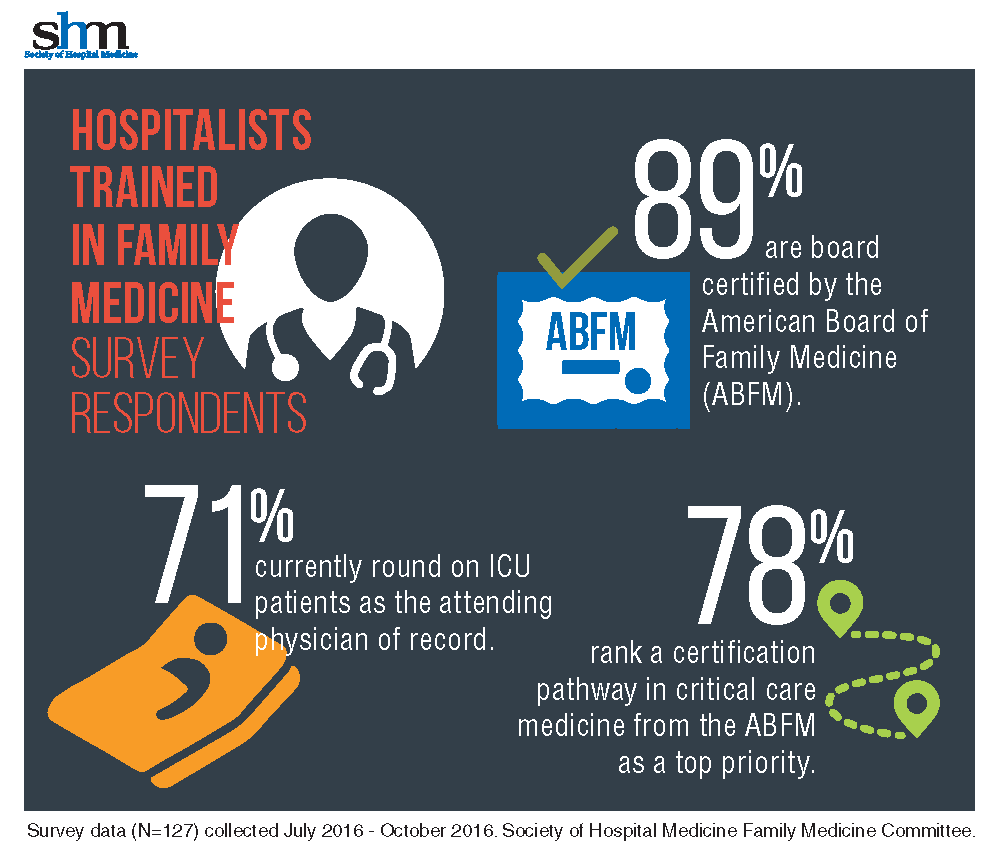
“It was eye-opening to learn how many [HTFM] are not only caring for patients in the ICU, but also are requesting additional training,” said Dr. Aymond, a member of the SHM Family Medicine Committee. “A critical care pathway would provide them with a level of expertise already available to physicians in internal medicine, emergency medicine, and surgery.”
With 71% of HTFM reporting that they round on ICU as the attending physician, the strong endorsement (78%) for critical care certification is not surprising.
“I am currently practicing as a full time intensivist and take consults from other providers, yet I only have a certificate from fellowship, no formal board certification in critical care,” noted a survey respondent.
Other participants stated, “it makes perfect sense to have a pathway to critical care if both family medicine and internal medicine coexist as hospitalists,” that certification is “imperative at rural and underserved hospitals,” and also “helpful for those …who work in larger hospitals and take care of critically ill patients.” More than half of those surveyed want the Family Medicine Committee to work with ABFM to create the pathway.
The majority (87%) of the HTFM survey respondents are certified by the ABFM, and 8% have attained Recognition of Focused Practice in Hospital Medicine. Common pathways for additional credentialing include SHM’s Fellow of Hospital Medicine program (38%), a fellowship in hospital medicine (19%), and certification in hospice and palliative care (15%). More than 38% reported “other qualifications,” such as years of work experience, certification by the American Osteopathic Board of Family Physicians, and prior training in internal medicine.
The survey also found that certification differences in internal medicine and family medicine hospitalists, which may have posed employment obstacles in the past for HTFM, are not as much of an issue.
“The critical care pathway is the bigger concern,” Dr. Aymond said.
SHM’s Family Medicine Committee will be working on a proposal to ABFM to create the training pathway in the coming months. Dr. Aymond wants intensivists to know that this not an attempt to encroach on their professional domain, “but an opportunity to fill the existing professional gap.
Family medicine physicians are already providing critical care services, so a pathway to obtain formal training makes sense,” he adds. “If a family medicine doc completes the fellowship and takes it back to a residency program [the residents] will be more prepared for their potential careers in hospital and ICU medicine and much more comfortable with high-acuity patients.”
Claudia Stahl is SHM’s content manager.
A nationwide shortage of intensivists has more hospitalists stepping into the critical care arena, but not all with the level of preparation and comfort of David Aymond, MD, a Louisiana-based hospitalist trained in family medicine (HTFM).
Dr. Aymond gained his ICU experience in a fellowship with the University of Alabama, where hospitalists also “were responsible for ICU patients,” he said. Years later, as an employee of both small and large hospitals with busy ICU services, and a faculty member for a family medicine residency with a busy ICU, Dr. Aymond moves seamlessly between roles.
“It was eye-opening to learn how many [HTFM] are not only caring for patients in the ICU, but also are requesting additional training,” said Dr. Aymond, a member of the SHM Family Medicine Committee. “A critical care pathway would provide them with a level of expertise already available to physicians in internal medicine, emergency medicine, and surgery.”
With 71% of HTFM reporting that they round on ICU as the attending physician, the strong endorsement (78%) for critical care certification is not surprising.
“I am currently practicing as a full time intensivist and take consults from other providers, yet I only have a certificate from fellowship, no formal board certification in critical care,” noted a survey respondent.
Other participants stated, “it makes perfect sense to have a pathway to critical care if both family medicine and internal medicine coexist as hospitalists,” that certification is “imperative at rural and underserved hospitals,” and also “helpful for those …who work in larger hospitals and take care of critically ill patients.” More than half of those surveyed want the Family Medicine Committee to work with ABFM to create the pathway.
The majority (87%) of the HTFM survey respondents are certified by the ABFM, and 8% have attained Recognition of Focused Practice in Hospital Medicine. Common pathways for additional credentialing include SHM’s Fellow of Hospital Medicine program (38%), a fellowship in hospital medicine (19%), and certification in hospice and palliative care (15%). More than 38% reported “other qualifications,” such as years of work experience, certification by the American Osteopathic Board of Family Physicians, and prior training in internal medicine.
The survey also found that certification differences in internal medicine and family medicine hospitalists, which may have posed employment obstacles in the past for HTFM, are not as much of an issue.
“The critical care pathway is the bigger concern,” Dr. Aymond said.
SHM’s Family Medicine Committee will be working on a proposal to ABFM to create the training pathway in the coming months. Dr. Aymond wants intensivists to know that this not an attempt to encroach on their professional domain, “but an opportunity to fill the existing professional gap.
Family medicine physicians are already providing critical care services, so a pathway to obtain formal training makes sense,” he adds. “If a family medicine doc completes the fellowship and takes it back to a residency program [the residents] will be more prepared for their potential careers in hospital and ICU medicine and much more comfortable with high-acuity patients.”
Claudia Stahl is SHM’s content manager.
A nationwide shortage of intensivists has more hospitalists stepping into the critical care arena, but not all with the level of preparation and comfort of David Aymond, MD, a Louisiana-based hospitalist trained in family medicine (HTFM).
Dr. Aymond gained his ICU experience in a fellowship with the University of Alabama, where hospitalists also “were responsible for ICU patients,” he said. Years later, as an employee of both small and large hospitals with busy ICU services, and a faculty member for a family medicine residency with a busy ICU, Dr. Aymond moves seamlessly between roles.
“It was eye-opening to learn how many [HTFM] are not only caring for patients in the ICU, but also are requesting additional training,” said Dr. Aymond, a member of the SHM Family Medicine Committee. “A critical care pathway would provide them with a level of expertise already available to physicians in internal medicine, emergency medicine, and surgery.”
With 71% of HTFM reporting that they round on ICU as the attending physician, the strong endorsement (78%) for critical care certification is not surprising.
“I am currently practicing as a full time intensivist and take consults from other providers, yet I only have a certificate from fellowship, no formal board certification in critical care,” noted a survey respondent.
Other participants stated, “it makes perfect sense to have a pathway to critical care if both family medicine and internal medicine coexist as hospitalists,” that certification is “imperative at rural and underserved hospitals,” and also “helpful for those …who work in larger hospitals and take care of critically ill patients.” More than half of those surveyed want the Family Medicine Committee to work with ABFM to create the pathway.
The majority (87%) of the HTFM survey respondents are certified by the ABFM, and 8% have attained Recognition of Focused Practice in Hospital Medicine. Common pathways for additional credentialing include SHM’s Fellow of Hospital Medicine program (38%), a fellowship in hospital medicine (19%), and certification in hospice and palliative care (15%). More than 38% reported “other qualifications,” such as years of work experience, certification by the American Osteopathic Board of Family Physicians, and prior training in internal medicine.
The survey also found that certification differences in internal medicine and family medicine hospitalists, which may have posed employment obstacles in the past for HTFM, are not as much of an issue.
“The critical care pathway is the bigger concern,” Dr. Aymond said.
SHM’s Family Medicine Committee will be working on a proposal to ABFM to create the training pathway in the coming months. Dr. Aymond wants intensivists to know that this not an attempt to encroach on their professional domain, “but an opportunity to fill the existing professional gap.
Family medicine physicians are already providing critical care services, so a pathway to obtain formal training makes sense,” he adds. “If a family medicine doc completes the fellowship and takes it back to a residency program [the residents] will be more prepared for their potential careers in hospital and ICU medicine and much more comfortable with high-acuity patients.”
Claudia Stahl is SHM’s content manager.
VIDEO: Working with alopecia patients’ insurers when using novel therapies
ORLANDO – Janus kinase inhibitors are “currently the most promising treatments” for alopecia areata, but they are expensive, are not approved for this indication, and so getting insurance coverage for these treatments can be difficult, Carolyn Goh, MD, said at the annual meeting of the American Academy of Dermatology.
In a video interview at the meeting, Dr. Goh of the department of dermatology, University of California, Los Angeles, shares the latest treatment algorithms that include these novel therapies, and thoughts on how to work with patients to increase their likelihood of getting insurance coverage for these treatments. Referring to the Janus kinase inhibitors, also known as JAK inhibitors, she said, “I think they would be very helpful for all patients with alopecia areata, but really given their side effect profile and risks involved, they should be reserved for more extensive disease.”
In the interview, Dr. Goh also discusses screening for thyroid disease in this patient population.
She had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @whitneymcknight
ORLANDO – Janus kinase inhibitors are “currently the most promising treatments” for alopecia areata, but they are expensive, are not approved for this indication, and so getting insurance coverage for these treatments can be difficult, Carolyn Goh, MD, said at the annual meeting of the American Academy of Dermatology.
In a video interview at the meeting, Dr. Goh of the department of dermatology, University of California, Los Angeles, shares the latest treatment algorithms that include these novel therapies, and thoughts on how to work with patients to increase their likelihood of getting insurance coverage for these treatments. Referring to the Janus kinase inhibitors, also known as JAK inhibitors, she said, “I think they would be very helpful for all patients with alopecia areata, but really given their side effect profile and risks involved, they should be reserved for more extensive disease.”
In the interview, Dr. Goh also discusses screening for thyroid disease in this patient population.
She had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @whitneymcknight
ORLANDO – Janus kinase inhibitors are “currently the most promising treatments” for alopecia areata, but they are expensive, are not approved for this indication, and so getting insurance coverage for these treatments can be difficult, Carolyn Goh, MD, said at the annual meeting of the American Academy of Dermatology.
In a video interview at the meeting, Dr. Goh of the department of dermatology, University of California, Los Angeles, shares the latest treatment algorithms that include these novel therapies, and thoughts on how to work with patients to increase their likelihood of getting insurance coverage for these treatments. Referring to the Janus kinase inhibitors, also known as JAK inhibitors, she said, “I think they would be very helpful for all patients with alopecia areata, but really given their side effect profile and risks involved, they should be reserved for more extensive disease.”
In the interview, Dr. Goh also discusses screening for thyroid disease in this patient population.
She had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @whitneymcknight
AT AAD 17

No benefit from adjuvant sunitinib or sorafenib for clear cell renal cancer
Adjuvant sunitinib or sorafenib show no significant advantages in disease-free or overall survival over placebo in patients with high-risk clear cell renal cancer, according to secondary analysis of data from the ASSURE trial.
The primary analysis of data from the ASSURE trial, which included patients with all types of renal cell carcinoma, had failed to show a benefit in disease-free survival.
“Given recently published results of a 750-patient randomized trial, S-TRAC, (sunitinib 50 mg daily [4/2 schedule] vs placebo in clear cell predominant pT3-4 or node-positive disease) that show improved [disease-free survival], the appropriate adjuvant strategy for high-risk patients is unclear,” Naomi B. Haas, MD, and coauthors wrote (JAMA Oncol. 2017 Mar 9. doi: 10.1001/jamaoncol.2017.0076).
Therefore, the investigators focused on a subset of patients from the ASSURE trial with high-risk clear cell renal cancer to determine if there might be a benefit in this group.
The secondary analysis involved 1,069 participants with pT3 and higher or node-positive renal cancer with clear cell histology who were randomized to receive 54 weeks of sunitinib (50mg, oral daily for 28 of 42 days per cycle), sorafenib (400 mg, oral twice daily continuously), or placebo.
The 5-year disease-free survival rate was 47.7% for patients in the sunitinib arm, 49.9% for those taking sorafenib, and 50% for placebo, with no statistically significant difference between the three groups. The 5-year overall survival rate was 75.2% for the sunitinib arm, 80.2% in the sorafenib arm, and 76.5% for those on placebo, with no statistically significant differences between the groups, reported Dr. Haas of the Abramson Cancer Center of the University of Pennsylvania, Philadelphia, and colleagues.
“This high-risk population had a better 5-year recurrence-free rate (around 50%) than expected (41.9% for high-risk disease and 36.0% for node-positive disease), possibly a result of better surgical technique, more accurate staging, or unknown biologic factors,” the authors wrote.
When the researchers analyzed disease-free survival according to quartiles of total dose per 6-week cycle, they also found no differences between each quartile of average dose per cycle.
There was, however, a significantly higher rate of grade 3 or higher adverse events in the sunitinib arm (66%) and sorafenib group (72%), compared with placebo (22%).
“Based on this analysis, a rationale for adjuvant therapy in this high-risk population is not elucidated,” Dr. Haas and colleagues said.
The study was coordinated by the ECOG-ACRIN Cancer Research Group and supported by Public Health Service grants, the National Cancer Institute, National Institutes of Health, and the Department of Health & Human Services. The drugs and placebos were provided by Bayer and Pfizer through the National Cancer Institute.
Adjuvant sunitinib or sorafenib show no significant advantages in disease-free or overall survival over placebo in patients with high-risk clear cell renal cancer, according to secondary analysis of data from the ASSURE trial.
The primary analysis of data from the ASSURE trial, which included patients with all types of renal cell carcinoma, had failed to show a benefit in disease-free survival.
“Given recently published results of a 750-patient randomized trial, S-TRAC, (sunitinib 50 mg daily [4/2 schedule] vs placebo in clear cell predominant pT3-4 or node-positive disease) that show improved [disease-free survival], the appropriate adjuvant strategy for high-risk patients is unclear,” Naomi B. Haas, MD, and coauthors wrote (JAMA Oncol. 2017 Mar 9. doi: 10.1001/jamaoncol.2017.0076).
Therefore, the investigators focused on a subset of patients from the ASSURE trial with high-risk clear cell renal cancer to determine if there might be a benefit in this group.
The secondary analysis involved 1,069 participants with pT3 and higher or node-positive renal cancer with clear cell histology who were randomized to receive 54 weeks of sunitinib (50mg, oral daily for 28 of 42 days per cycle), sorafenib (400 mg, oral twice daily continuously), or placebo.
The 5-year disease-free survival rate was 47.7% for patients in the sunitinib arm, 49.9% for those taking sorafenib, and 50% for placebo, with no statistically significant difference between the three groups. The 5-year overall survival rate was 75.2% for the sunitinib arm, 80.2% in the sorafenib arm, and 76.5% for those on placebo, with no statistically significant differences between the groups, reported Dr. Haas of the Abramson Cancer Center of the University of Pennsylvania, Philadelphia, and colleagues.
“This high-risk population had a better 5-year recurrence-free rate (around 50%) than expected (41.9% for high-risk disease and 36.0% for node-positive disease), possibly a result of better surgical technique, more accurate staging, or unknown biologic factors,” the authors wrote.
When the researchers analyzed disease-free survival according to quartiles of total dose per 6-week cycle, they also found no differences between each quartile of average dose per cycle.
There was, however, a significantly higher rate of grade 3 or higher adverse events in the sunitinib arm (66%) and sorafenib group (72%), compared with placebo (22%).
“Based on this analysis, a rationale for adjuvant therapy in this high-risk population is not elucidated,” Dr. Haas and colleagues said.
The study was coordinated by the ECOG-ACRIN Cancer Research Group and supported by Public Health Service grants, the National Cancer Institute, National Institutes of Health, and the Department of Health & Human Services. The drugs and placebos were provided by Bayer and Pfizer through the National Cancer Institute.
Adjuvant sunitinib or sorafenib show no significant advantages in disease-free or overall survival over placebo in patients with high-risk clear cell renal cancer, according to secondary analysis of data from the ASSURE trial.
The primary analysis of data from the ASSURE trial, which included patients with all types of renal cell carcinoma, had failed to show a benefit in disease-free survival.
“Given recently published results of a 750-patient randomized trial, S-TRAC, (sunitinib 50 mg daily [4/2 schedule] vs placebo in clear cell predominant pT3-4 or node-positive disease) that show improved [disease-free survival], the appropriate adjuvant strategy for high-risk patients is unclear,” Naomi B. Haas, MD, and coauthors wrote (JAMA Oncol. 2017 Mar 9. doi: 10.1001/jamaoncol.2017.0076).
Therefore, the investigators focused on a subset of patients from the ASSURE trial with high-risk clear cell renal cancer to determine if there might be a benefit in this group.
The secondary analysis involved 1,069 participants with pT3 and higher or node-positive renal cancer with clear cell histology who were randomized to receive 54 weeks of sunitinib (50mg, oral daily for 28 of 42 days per cycle), sorafenib (400 mg, oral twice daily continuously), or placebo.
The 5-year disease-free survival rate was 47.7% for patients in the sunitinib arm, 49.9% for those taking sorafenib, and 50% for placebo, with no statistically significant difference between the three groups. The 5-year overall survival rate was 75.2% for the sunitinib arm, 80.2% in the sorafenib arm, and 76.5% for those on placebo, with no statistically significant differences between the groups, reported Dr. Haas of the Abramson Cancer Center of the University of Pennsylvania, Philadelphia, and colleagues.
“This high-risk population had a better 5-year recurrence-free rate (around 50%) than expected (41.9% for high-risk disease and 36.0% for node-positive disease), possibly a result of better surgical technique, more accurate staging, or unknown biologic factors,” the authors wrote.
When the researchers analyzed disease-free survival according to quartiles of total dose per 6-week cycle, they also found no differences between each quartile of average dose per cycle.
There was, however, a significantly higher rate of grade 3 or higher adverse events in the sunitinib arm (66%) and sorafenib group (72%), compared with placebo (22%).
“Based on this analysis, a rationale for adjuvant therapy in this high-risk population is not elucidated,” Dr. Haas and colleagues said.
The study was coordinated by the ECOG-ACRIN Cancer Research Group and supported by Public Health Service grants, the National Cancer Institute, National Institutes of Health, and the Department of Health & Human Services. The drugs and placebos were provided by Bayer and Pfizer through the National Cancer Institute.
FROM JAMA ONCOLOGY
Key clinical point: Adjuvant sunitinib or sorafenib show no significant benefit in disease-free or overall survival in patients with high-risk clear cell renal cancer.
Major finding: The 5-year disease-free survival rate was 47.7% for patients treated with sunitinib, 49.9% for those treated with sorafenib, and 50% for those given placebo.
Data source: Secondary analysis of data from the ASSURE trial in 1,943 patients with pT3 and higher or node-positive renal cancer with clear cell histology.
Disclosures: The study was coordinated by the ECOG-ACRIN Cancer Research Group and supported by Public Health Service grants, the National Cancer Institute, National Institutes of Health, and the Department of Health & Human Services. The drugs and placebos were provided by Bayer and Pfizer through the National Cancer Institute.