User login
Using CHIMPS for type A dissection in a high-risk patient
Traditional open repair for type A aortic dissection in patients with Marfan syndrome and a previous cardiovascular surgery carries a high risk of morbidity and mortality, but a team of surgeons from China have reported on a hybrid technique that combines open and endovascular approaches to repair type A dissection in a patient with Marfan syndrome.
In the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1191-3), Hong-wei Zhang, MD, and colleagues from West China Hospital of Sichuan University, explained their technique using chimney and sandwich grafts to repair a type A dissection in the patient late after Bentall surgery. “With great advancements in recent thoracic endovascular aortic repair technology, innovative hybrid operations combining open and endovascular techniques hold promising potential to expand treatment options,” Dr. Zhang and coauthors said.
They reported on a 33-year-old male with Marfan syndrome (MFS) who had elective aortic root and mechanical valve replacement 10 years earlier. Three days of persistent chest and back pain caused the patient to go to the emergency department, where computed tomography angiography (CTA) confirmed a type A aortic dissection from the distal ascending aorta to the iliac arteries and involving the proximal innominate artery and left common carotid artery (LCCA).
Because the patient refused another open surgery, Dr. Zhang and colleagues executed their hybrid approach, the first step of which was to create an LCCA-left axillar artery bypass with a 6-mm Gore-Tex graft (W.L. Gore & Associates). After they led the graft through the costoclavicular passage, they introduced the first (distal) thoracic stent (Valiant Captivia, Medtronic) from the right femoral artery and deployed it at the proximal descending aorta. They then inserted the second (proximal) thoracic stent graft into the previous ascending synthetic graft.
Next, they delivered the chimney grafts, two Fluency Plus covered stents (Bard Peripheral Vascular), from the right brachial and innominate artery into the ascending graft. Then they delivered two more Fluency grafts from the LCCA into the endolumen of the first (distal) thoracic stent graft.
After they deployed the second (proximal) thoracic stent graft, they deployed the precisely positioned stent grafts from the innominate artery and the LCCA, sandwiching the covered stents for the LCCA between the two thoracic stent grafts. They then occluded the left subclavian artery with a 10-mm double-disk vascular occlude.
Upon angiography at completion, Dr. Zhang and coauthors found an endoleak from the overlap zones between the two thoracic stent grafts.
However, the patient’s postoperative course was uneventful, and CTA 5 days after surgery showed complete sealing of the primary entry tear with patent chimney and sandwich grafts. The patient remained symptom-free at 30 days, when CTA again confirmed patency of the supra-arch grafts.
Dr. Zhang and coauthors acknowledge that carotid-to-carotid bypass could have been an alternative in order to use fewer stent grafts and to reduce the risk of endoleaks in this case, but they opted for this approach because of the dissection of the proximal innominate artery and LCCA and their concern of the long-term patency of a carotid-to-carotid bypass. “To our knowledge, this is the first reported case of a hybrid treatment for new-onset, type A aortic dissection in patients with MFS with a previous Bentall procedure,” Dr. Zhang and coauthors said. “Although further staged repairs are required in our case, this endovascular technique could be an effective and life-saving treatment option for the high-risk repeated surgical patients with MFS.”
Dr. Zhang and coauthors had no financial relationships to disclose.
In their invited commentary, Lars Svensson, MD, PhD, Matthew Eagleton, MD, and Eric Roselli, MD, of the Cleveland Clinic, said the approach Dr. Zhang and colleagues reported on is one of the “novel” endovascular CHIMPS methods for aortic arch repair – CHIMPS meaning chimneys, periscopes, snorkels, and sandwiches (J Thorac Cardiovasc Surg. 2016;152:958-9). But they noted that one of the ongoing challenges with these types of parallel grafts is the gutter leaks that occur between the sandwich grafts.
The commentators noted that CHIMPS procedures are easier alternatives to using spiral branch graft stents for the thoracoabdominal aorta or direct-connecting branch stems from an aortic stent in the arch, but they added, “An important caveat is that the blood supply maintenance and long-term durability may not be adequate.”
The patient Dr. Zhang and colleagues reported on “is young and will need a durable operation,” Dr. Svensson, Dr. Eagleton, and Dr. Roselli said. “Unfortunately, in our experience over time we have observed that these CHIMPS procedures tend to break down and leak into the arch, including the arch actually rupturing,” they said. These patients will need “intensive” monitoring. What’s more, patients with Marfan syndrome are prone to aneurysm formation “and are not good candidates for stenting,” the commentators said.
“Nevertheless, further engineering iterations of CHIMPS may address the problem with gutter leaks and become an alternative to the elephant trunk procedure for those patients who are at particularly high risk,” the commentators said.
Dr. Svensson disclosed he holds a patent with potential royalties for an aortic valve and aortic root stent graft with connecting branch grafts to the coronary ostia. Dr. Roselli is a consultant and investigator for Bolton, Gore, and Medtronic. Dr. Eagleton has no relationships to disclose.
In their invited commentary, Lars Svensson, MD, PhD, Matthew Eagleton, MD, and Eric Roselli, MD, of the Cleveland Clinic, said the approach Dr. Zhang and colleagues reported on is one of the “novel” endovascular CHIMPS methods for aortic arch repair – CHIMPS meaning chimneys, periscopes, snorkels, and sandwiches (J Thorac Cardiovasc Surg. 2016;152:958-9). But they noted that one of the ongoing challenges with these types of parallel grafts is the gutter leaks that occur between the sandwich grafts.
The commentators noted that CHIMPS procedures are easier alternatives to using spiral branch graft stents for the thoracoabdominal aorta or direct-connecting branch stems from an aortic stent in the arch, but they added, “An important caveat is that the blood supply maintenance and long-term durability may not be adequate.”
The patient Dr. Zhang and colleagues reported on “is young and will need a durable operation,” Dr. Svensson, Dr. Eagleton, and Dr. Roselli said. “Unfortunately, in our experience over time we have observed that these CHIMPS procedures tend to break down and leak into the arch, including the arch actually rupturing,” they said. These patients will need “intensive” monitoring. What’s more, patients with Marfan syndrome are prone to aneurysm formation “and are not good candidates for stenting,” the commentators said.
“Nevertheless, further engineering iterations of CHIMPS may address the problem with gutter leaks and become an alternative to the elephant trunk procedure for those patients who are at particularly high risk,” the commentators said.
Dr. Svensson disclosed he holds a patent with potential royalties for an aortic valve and aortic root stent graft with connecting branch grafts to the coronary ostia. Dr. Roselli is a consultant and investigator for Bolton, Gore, and Medtronic. Dr. Eagleton has no relationships to disclose.
In their invited commentary, Lars Svensson, MD, PhD, Matthew Eagleton, MD, and Eric Roselli, MD, of the Cleveland Clinic, said the approach Dr. Zhang and colleagues reported on is one of the “novel” endovascular CHIMPS methods for aortic arch repair – CHIMPS meaning chimneys, periscopes, snorkels, and sandwiches (J Thorac Cardiovasc Surg. 2016;152:958-9). But they noted that one of the ongoing challenges with these types of parallel grafts is the gutter leaks that occur between the sandwich grafts.
The commentators noted that CHIMPS procedures are easier alternatives to using spiral branch graft stents for the thoracoabdominal aorta or direct-connecting branch stems from an aortic stent in the arch, but they added, “An important caveat is that the blood supply maintenance and long-term durability may not be adequate.”
The patient Dr. Zhang and colleagues reported on “is young and will need a durable operation,” Dr. Svensson, Dr. Eagleton, and Dr. Roselli said. “Unfortunately, in our experience over time we have observed that these CHIMPS procedures tend to break down and leak into the arch, including the arch actually rupturing,” they said. These patients will need “intensive” monitoring. What’s more, patients with Marfan syndrome are prone to aneurysm formation “and are not good candidates for stenting,” the commentators said.
“Nevertheless, further engineering iterations of CHIMPS may address the problem with gutter leaks and become an alternative to the elephant trunk procedure for those patients who are at particularly high risk,” the commentators said.
Dr. Svensson disclosed he holds a patent with potential royalties for an aortic valve and aortic root stent graft with connecting branch grafts to the coronary ostia. Dr. Roselli is a consultant and investigator for Bolton, Gore, and Medtronic. Dr. Eagleton has no relationships to disclose.
Traditional open repair for type A aortic dissection in patients with Marfan syndrome and a previous cardiovascular surgery carries a high risk of morbidity and mortality, but a team of surgeons from China have reported on a hybrid technique that combines open and endovascular approaches to repair type A dissection in a patient with Marfan syndrome.
In the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1191-3), Hong-wei Zhang, MD, and colleagues from West China Hospital of Sichuan University, explained their technique using chimney and sandwich grafts to repair a type A dissection in the patient late after Bentall surgery. “With great advancements in recent thoracic endovascular aortic repair technology, innovative hybrid operations combining open and endovascular techniques hold promising potential to expand treatment options,” Dr. Zhang and coauthors said.
They reported on a 33-year-old male with Marfan syndrome (MFS) who had elective aortic root and mechanical valve replacement 10 years earlier. Three days of persistent chest and back pain caused the patient to go to the emergency department, where computed tomography angiography (CTA) confirmed a type A aortic dissection from the distal ascending aorta to the iliac arteries and involving the proximal innominate artery and left common carotid artery (LCCA).
Because the patient refused another open surgery, Dr. Zhang and colleagues executed their hybrid approach, the first step of which was to create an LCCA-left axillar artery bypass with a 6-mm Gore-Tex graft (W.L. Gore & Associates). After they led the graft through the costoclavicular passage, they introduced the first (distal) thoracic stent (Valiant Captivia, Medtronic) from the right femoral artery and deployed it at the proximal descending aorta. They then inserted the second (proximal) thoracic stent graft into the previous ascending synthetic graft.
Next, they delivered the chimney grafts, two Fluency Plus covered stents (Bard Peripheral Vascular), from the right brachial and innominate artery into the ascending graft. Then they delivered two more Fluency grafts from the LCCA into the endolumen of the first (distal) thoracic stent graft.
After they deployed the second (proximal) thoracic stent graft, they deployed the precisely positioned stent grafts from the innominate artery and the LCCA, sandwiching the covered stents for the LCCA between the two thoracic stent grafts. They then occluded the left subclavian artery with a 10-mm double-disk vascular occlude.
Upon angiography at completion, Dr. Zhang and coauthors found an endoleak from the overlap zones between the two thoracic stent grafts.
However, the patient’s postoperative course was uneventful, and CTA 5 days after surgery showed complete sealing of the primary entry tear with patent chimney and sandwich grafts. The patient remained symptom-free at 30 days, when CTA again confirmed patency of the supra-arch grafts.
Dr. Zhang and coauthors acknowledge that carotid-to-carotid bypass could have been an alternative in order to use fewer stent grafts and to reduce the risk of endoleaks in this case, but they opted for this approach because of the dissection of the proximal innominate artery and LCCA and their concern of the long-term patency of a carotid-to-carotid bypass. “To our knowledge, this is the first reported case of a hybrid treatment for new-onset, type A aortic dissection in patients with MFS with a previous Bentall procedure,” Dr. Zhang and coauthors said. “Although further staged repairs are required in our case, this endovascular technique could be an effective and life-saving treatment option for the high-risk repeated surgical patients with MFS.”
Dr. Zhang and coauthors had no financial relationships to disclose.
Traditional open repair for type A aortic dissection in patients with Marfan syndrome and a previous cardiovascular surgery carries a high risk of morbidity and mortality, but a team of surgeons from China have reported on a hybrid technique that combines open and endovascular approaches to repair type A dissection in a patient with Marfan syndrome.
In the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1191-3), Hong-wei Zhang, MD, and colleagues from West China Hospital of Sichuan University, explained their technique using chimney and sandwich grafts to repair a type A dissection in the patient late after Bentall surgery. “With great advancements in recent thoracic endovascular aortic repair technology, innovative hybrid operations combining open and endovascular techniques hold promising potential to expand treatment options,” Dr. Zhang and coauthors said.
They reported on a 33-year-old male with Marfan syndrome (MFS) who had elective aortic root and mechanical valve replacement 10 years earlier. Three days of persistent chest and back pain caused the patient to go to the emergency department, where computed tomography angiography (CTA) confirmed a type A aortic dissection from the distal ascending aorta to the iliac arteries and involving the proximal innominate artery and left common carotid artery (LCCA).
Because the patient refused another open surgery, Dr. Zhang and colleagues executed their hybrid approach, the first step of which was to create an LCCA-left axillar artery bypass with a 6-mm Gore-Tex graft (W.L. Gore & Associates). After they led the graft through the costoclavicular passage, they introduced the first (distal) thoracic stent (Valiant Captivia, Medtronic) from the right femoral artery and deployed it at the proximal descending aorta. They then inserted the second (proximal) thoracic stent graft into the previous ascending synthetic graft.
Next, they delivered the chimney grafts, two Fluency Plus covered stents (Bard Peripheral Vascular), from the right brachial and innominate artery into the ascending graft. Then they delivered two more Fluency grafts from the LCCA into the endolumen of the first (distal) thoracic stent graft.
After they deployed the second (proximal) thoracic stent graft, they deployed the precisely positioned stent grafts from the innominate artery and the LCCA, sandwiching the covered stents for the LCCA between the two thoracic stent grafts. They then occluded the left subclavian artery with a 10-mm double-disk vascular occlude.
Upon angiography at completion, Dr. Zhang and coauthors found an endoleak from the overlap zones between the two thoracic stent grafts.
However, the patient’s postoperative course was uneventful, and CTA 5 days after surgery showed complete sealing of the primary entry tear with patent chimney and sandwich grafts. The patient remained symptom-free at 30 days, when CTA again confirmed patency of the supra-arch grafts.
Dr. Zhang and coauthors acknowledge that carotid-to-carotid bypass could have been an alternative in order to use fewer stent grafts and to reduce the risk of endoleaks in this case, but they opted for this approach because of the dissection of the proximal innominate artery and LCCA and their concern of the long-term patency of a carotid-to-carotid bypass. “To our knowledge, this is the first reported case of a hybrid treatment for new-onset, type A aortic dissection in patients with MFS with a previous Bentall procedure,” Dr. Zhang and coauthors said. “Although further staged repairs are required in our case, this endovascular technique could be an effective and life-saving treatment option for the high-risk repeated surgical patients with MFS.”
Dr. Zhang and coauthors had no financial relationships to disclose.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Chimney and sandwich grafts facilitate hybrid repair of type A aortic dissection for a Marfan syndrome patient after Bentall surgery.
Major finding: A 33-year-old male with Marfan syndrome and a history of cardiac surgery was asymptomatic 30 days after hybrid repair for type A aortic dissection.
Data source: Case report of single patient at an academic medical center.
Disclosures: Dr. Zhang and coauthors reported having no financial disclosures.
Newborns with CHD have reduced cerebral oxygen delivery
Using a newer form of MRI to investigate oxygen levels in newborns with congenital heart disease, researchers in Canada reported that these patients may have impaired brain growth and development in the first weeks of life because of significantly lower cerebral oxygen delivery levels.
These findings suggest that oxygen delivery may impact brain growth, particularly in newborns with single-ventricle physiology, reported Jessie Mei Lim, BSc, of the University of Toronto, and her colleagues from McGill University, Montreal, and the Hospital for Sick Children, Toronto. The findings were published in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1095-103). Ms. Lim and her colleagues used cine phase-contrast (PC) MRI to measure cerebral blood flow in newborns with congenital heard disease (CHD). Previous studies used optical measures of tissue oxygenation and MRI arterial spin labeling to suggests that newborns with severe CHD have impaired CBF and cerebral oxygen delivery (CDO2) and CBF.
This single-center study involved 63 newborns from June 2013 to April 2015 at the Hospital for Sick Children. These subjects received an MRI of the head before surgery at an average of age 7.5 days. The scans were done without sedation or contrast while the infants were asleep. The study compared 31 age-matched controls with 32 subjects with various forms of CHD – 12 were managed surgically along a single-ventricle pathway (SVP), 4 had coarctation of the aorta, 13 had transposition of the great arteries (TGA), and 3 had other forms of CHD.
The researchers validated their method by reporting similarities between flows in the basilar and vertebral arteries in 14 controls, “suggesting good consistency and accuracy of our method for measuring CBF,” Ms. Lim and her coauthors noted. A comparison of CBF measured with an unpaired Student t test revealed no significant differences between the CHD group and controls. The average net CBF in CHD patients was 103.5 mL/min vs. 119.7 mL/min in controls.
However, when evaluating CDO2 using a Student t test, the researchers found significantly lower levels in the CHD group – an average of 1,1881 mLO2/min. vs. 2,712 mL O2/min in controls (P less than .0001). And when the researchers indexed CDO2 to brain volume yielding indexed oxygen delivery, the difference between the two groups was still significant: an average of 523.1 mL O2/min-1 .100 g-1 in the CHD group and 685.6 mL O2/min-1.100 g-1 in controls (P = .0006).
Among the CHD group, those with SVP and TGA had significantly lower CDO2 than that of controls. Brain volumes were also lower in those with CHD (mean of 338.5 mL vs. 377.7 mL in controls, P = .002).
The MRI findings were telling in the study population, Ms. Lim and her coauthors said. Five subjects in the CHD group had a combination of diffuse excessive high-signal intensity (DEHSI) and white-matter injury (WMI), 10 had an isolated finding of DEHSI, two had WMI alone and five others had other minor brain abnormalities. But the control group had no abnormal findings on conventional brain MRI.
The researchers acknowledged that, while the impact of reduced cerebral oxygen delivery is unknown, “theoretical reasons for thinking it might adversely impact ongoing brain growth and development during this period of rapid brain growth are considered.”
Cardiovascular surgeons should consider these findings when deciding on when to operate on newborns with CHD, the researchers said. “Further support for the concept that such a mechanism could lead to irreversible deficits in brain growth and development might result in attempts to expedite surgical repair of congenital cardiac lesions, which have conventionally not been addressed in the neonatal period,” they wrote.
Ms. Lim and her coauthors had no financial relationships to disclose.
Congenital heart disease (CHD) is heterogeneous and different types of lesions may cause different hemodynamics, Caitlin K. Rollins, MD, of Boston Children’s Hospital and Harvard Medical School said in her invited commentary (J Thorac Cardiovasc Surg. 2016;152-960-1).
Ms. Lim and her colleagues in this study confirmed that premise with their finding that newborns with CHD and controls had similar cerebral blood flow, but that those with CHD had reduced oxygen delivery. “These differences were most apparent in the neonates with single-ventricle physiology and transposition of the great arteries,” Dr. Rollins said. The study authors’ finding of an association between reduced oxygen delivery and impaired brain development, along with this group’s previous reports (Circulation 2015;131:1313-23) suggesting preserved cerebral blood flow in the late prenatal period, differ from other studies using traditional methods to show reduced cerebral blood flow in obstructive left-sided lesions, Dr. Rollins said. “Although technical differences may in part account for the discrepancy, the contrasting results also reflect that the relative contributions of abnormal cerebral blood flow and oxygenation differ among forms of CHD,” Dr. Rollins said.
Congenital heart disease (CHD) is heterogeneous and different types of lesions may cause different hemodynamics, Caitlin K. Rollins, MD, of Boston Children’s Hospital and Harvard Medical School said in her invited commentary (J Thorac Cardiovasc Surg. 2016;152-960-1).
Ms. Lim and her colleagues in this study confirmed that premise with their finding that newborns with CHD and controls had similar cerebral blood flow, but that those with CHD had reduced oxygen delivery. “These differences were most apparent in the neonates with single-ventricle physiology and transposition of the great arteries,” Dr. Rollins said. The study authors’ finding of an association between reduced oxygen delivery and impaired brain development, along with this group’s previous reports (Circulation 2015;131:1313-23) suggesting preserved cerebral blood flow in the late prenatal period, differ from other studies using traditional methods to show reduced cerebral blood flow in obstructive left-sided lesions, Dr. Rollins said. “Although technical differences may in part account for the discrepancy, the contrasting results also reflect that the relative contributions of abnormal cerebral blood flow and oxygenation differ among forms of CHD,” Dr. Rollins said.
Congenital heart disease (CHD) is heterogeneous and different types of lesions may cause different hemodynamics, Caitlin K. Rollins, MD, of Boston Children’s Hospital and Harvard Medical School said in her invited commentary (J Thorac Cardiovasc Surg. 2016;152-960-1).
Ms. Lim and her colleagues in this study confirmed that premise with their finding that newborns with CHD and controls had similar cerebral blood flow, but that those with CHD had reduced oxygen delivery. “These differences were most apparent in the neonates with single-ventricle physiology and transposition of the great arteries,” Dr. Rollins said. The study authors’ finding of an association between reduced oxygen delivery and impaired brain development, along with this group’s previous reports (Circulation 2015;131:1313-23) suggesting preserved cerebral blood flow in the late prenatal period, differ from other studies using traditional methods to show reduced cerebral blood flow in obstructive left-sided lesions, Dr. Rollins said. “Although technical differences may in part account for the discrepancy, the contrasting results also reflect that the relative contributions of abnormal cerebral blood flow and oxygenation differ among forms of CHD,” Dr. Rollins said.
Using a newer form of MRI to investigate oxygen levels in newborns with congenital heart disease, researchers in Canada reported that these patients may have impaired brain growth and development in the first weeks of life because of significantly lower cerebral oxygen delivery levels.
These findings suggest that oxygen delivery may impact brain growth, particularly in newborns with single-ventricle physiology, reported Jessie Mei Lim, BSc, of the University of Toronto, and her colleagues from McGill University, Montreal, and the Hospital for Sick Children, Toronto. The findings were published in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1095-103). Ms. Lim and her colleagues used cine phase-contrast (PC) MRI to measure cerebral blood flow in newborns with congenital heard disease (CHD). Previous studies used optical measures of tissue oxygenation and MRI arterial spin labeling to suggests that newborns with severe CHD have impaired CBF and cerebral oxygen delivery (CDO2) and CBF.
This single-center study involved 63 newborns from June 2013 to April 2015 at the Hospital for Sick Children. These subjects received an MRI of the head before surgery at an average of age 7.5 days. The scans were done without sedation or contrast while the infants were asleep. The study compared 31 age-matched controls with 32 subjects with various forms of CHD – 12 were managed surgically along a single-ventricle pathway (SVP), 4 had coarctation of the aorta, 13 had transposition of the great arteries (TGA), and 3 had other forms of CHD.
The researchers validated their method by reporting similarities between flows in the basilar and vertebral arteries in 14 controls, “suggesting good consistency and accuracy of our method for measuring CBF,” Ms. Lim and her coauthors noted. A comparison of CBF measured with an unpaired Student t test revealed no significant differences between the CHD group and controls. The average net CBF in CHD patients was 103.5 mL/min vs. 119.7 mL/min in controls.
However, when evaluating CDO2 using a Student t test, the researchers found significantly lower levels in the CHD group – an average of 1,1881 mLO2/min. vs. 2,712 mL O2/min in controls (P less than .0001). And when the researchers indexed CDO2 to brain volume yielding indexed oxygen delivery, the difference between the two groups was still significant: an average of 523.1 mL O2/min-1 .100 g-1 in the CHD group and 685.6 mL O2/min-1.100 g-1 in controls (P = .0006).
Among the CHD group, those with SVP and TGA had significantly lower CDO2 than that of controls. Brain volumes were also lower in those with CHD (mean of 338.5 mL vs. 377.7 mL in controls, P = .002).
The MRI findings were telling in the study population, Ms. Lim and her coauthors said. Five subjects in the CHD group had a combination of diffuse excessive high-signal intensity (DEHSI) and white-matter injury (WMI), 10 had an isolated finding of DEHSI, two had WMI alone and five others had other minor brain abnormalities. But the control group had no abnormal findings on conventional brain MRI.
The researchers acknowledged that, while the impact of reduced cerebral oxygen delivery is unknown, “theoretical reasons for thinking it might adversely impact ongoing brain growth and development during this period of rapid brain growth are considered.”
Cardiovascular surgeons should consider these findings when deciding on when to operate on newborns with CHD, the researchers said. “Further support for the concept that such a mechanism could lead to irreversible deficits in brain growth and development might result in attempts to expedite surgical repair of congenital cardiac lesions, which have conventionally not been addressed in the neonatal period,” they wrote.
Ms. Lim and her coauthors had no financial relationships to disclose.
Using a newer form of MRI to investigate oxygen levels in newborns with congenital heart disease, researchers in Canada reported that these patients may have impaired brain growth and development in the first weeks of life because of significantly lower cerebral oxygen delivery levels.
These findings suggest that oxygen delivery may impact brain growth, particularly in newborns with single-ventricle physiology, reported Jessie Mei Lim, BSc, of the University of Toronto, and her colleagues from McGill University, Montreal, and the Hospital for Sick Children, Toronto. The findings were published in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:1095-103). Ms. Lim and her colleagues used cine phase-contrast (PC) MRI to measure cerebral blood flow in newborns with congenital heard disease (CHD). Previous studies used optical measures of tissue oxygenation and MRI arterial spin labeling to suggests that newborns with severe CHD have impaired CBF and cerebral oxygen delivery (CDO2) and CBF.
This single-center study involved 63 newborns from June 2013 to April 2015 at the Hospital for Sick Children. These subjects received an MRI of the head before surgery at an average of age 7.5 days. The scans were done without sedation or contrast while the infants were asleep. The study compared 31 age-matched controls with 32 subjects with various forms of CHD – 12 were managed surgically along a single-ventricle pathway (SVP), 4 had coarctation of the aorta, 13 had transposition of the great arteries (TGA), and 3 had other forms of CHD.
The researchers validated their method by reporting similarities between flows in the basilar and vertebral arteries in 14 controls, “suggesting good consistency and accuracy of our method for measuring CBF,” Ms. Lim and her coauthors noted. A comparison of CBF measured with an unpaired Student t test revealed no significant differences between the CHD group and controls. The average net CBF in CHD patients was 103.5 mL/min vs. 119.7 mL/min in controls.
However, when evaluating CDO2 using a Student t test, the researchers found significantly lower levels in the CHD group – an average of 1,1881 mLO2/min. vs. 2,712 mL O2/min in controls (P less than .0001). And when the researchers indexed CDO2 to brain volume yielding indexed oxygen delivery, the difference between the two groups was still significant: an average of 523.1 mL O2/min-1 .100 g-1 in the CHD group and 685.6 mL O2/min-1.100 g-1 in controls (P = .0006).
Among the CHD group, those with SVP and TGA had significantly lower CDO2 than that of controls. Brain volumes were also lower in those with CHD (mean of 338.5 mL vs. 377.7 mL in controls, P = .002).
The MRI findings were telling in the study population, Ms. Lim and her coauthors said. Five subjects in the CHD group had a combination of diffuse excessive high-signal intensity (DEHSI) and white-matter injury (WMI), 10 had an isolated finding of DEHSI, two had WMI alone and five others had other minor brain abnormalities. But the control group had no abnormal findings on conventional brain MRI.
The researchers acknowledged that, while the impact of reduced cerebral oxygen delivery is unknown, “theoretical reasons for thinking it might adversely impact ongoing brain growth and development during this period of rapid brain growth are considered.”
Cardiovascular surgeons should consider these findings when deciding on when to operate on newborns with CHD, the researchers said. “Further support for the concept that such a mechanism could lead to irreversible deficits in brain growth and development might result in attempts to expedite surgical repair of congenital cardiac lesions, which have conventionally not been addressed in the neonatal period,” they wrote.
Ms. Lim and her coauthors had no financial relationships to disclose.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Cerebral blood flow is maintained but cerebral oxygen delivery is decreased in preoperative newborns with cyanotic congenital heart disease (CHD).
Major finding: Average cerebral oxygen delivery measured 1,1881 mLO2/min in the CHD group when measured with Student t testing vs. 2,712 mLO2/min in controls (P less than .0001).
Data source: Single-center study of 32 neonates with various forms of CHD 31 age-matched controls.
Disclosures: Ms. Lim and coauthors have no financial relationships to disclose.
Time to rethink bioprosthetic valve guidelines?
Recent findings on the incidence and pathophysiology of bioprosthetic valve thrombosis require revisiting existing guidelines against routine echocardiography in the first 5 years after bioprosthetic valve replacement and a longer course of anticoagulation therapy than the current standard of 3 months, investigators from the Mayo Clinic said in an expert opinion article in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152;975-8).
In the expert commentary, Alexander C. Egbe, MBBS, of the departments of cardiovascular diseases and cardiovascular surgery at Mayo Clinic in Rochester, Minn., and coauthors explored the implications of their previous research, published in the Journal of the American College of Cardiology, that reported that bioprosthetic valve thrombosis (BPVT) is “not an uncommon cause of prosthetic valve dysfunction.” They identified BPVT in 46 of 397 (11%) bioprosthetic valves explanted at Mayo Clinic, and estimated the incidence of BPVT at 1% (J Am Coll Cardiol. 2015;66:2285-94), although Dr. Egbe and colleagues acknowledged the true incidence of BPVT is unknown, as is the time to occurrence. They noted that a different study design would be needed to determine that, along with the incidence of BPVT.
“The occurrence of BPVT is not restricted to surgically implanted bioprosthetic valves, but has also been observed after transcatheter aortic valve replacement (TAVR),” Dr. Egbe and colleagues said. They noted an association between BPVT and a lack of anticoagulation therapy in two earlier reports (N Engl J Med. 2015;373:2015-24; J Am Coll Cardiol. 2016;67:644-55). In their own study, 14 of 15 patients (93%) with diagnosed BPVT responded to anticoagulation therapy and avoided reoperation.
Dr. Egbe and coauthors did somewhat define the extent of the problem of misdiagnosis of BPVT. The diagnosis was considered in only 6 of 45 patients (13%) who had transesophageal echocardiography. “A significant proportion of the patients with BPVT were misdiagnosed as having structural failure and referred for reoperation,” Dr. Egbe and coauthors said. “This attests to the low level of awareness of the existence of BPVT and the lack of well-defined diagnostic criteria.”
They proposed a diagnostic model based on the echocardiography characteristics of three findings: a 50% increase in gradient within 5 years of implantation; increased cusp thickness; and abnormal cusp mobility. “The presence of all three echocardiographic features reliably diagnosed BPVT with a sensitivity of 72% and a specificity of 90%,” they said.
Their finding that 85% of BPVT cases occurred within 5 years of implantation flies in the face of clinical guidelines that state routine annual echocardiography is not recommended in that time frame (J Am So Echocardiogr. 2009;22;975-1014). But abnormal physical examination findings as a prerequisite for echocardiography may not be an effective method to diagnose BPVT. “In addition to transthoracic and transesophageal echocardiography, the use of other complementary imaging modalities, such as computed tomography, could be very effective in identifying subtle BPVT,” Dr. Egbe and colleagues said,
But preventing BPVT is more complicated. Clinical guidelines recommend anticoagulation of bioprosthetic valves for 3 months after implantation, but adhering to that guideline showed no protective effect against BPVT in their study, Dr. Egbe and coauthors said. Nor did antiplatelet therapy prove effective in preventing BPVT. However, a Danish study showed stopping anticoagulation within 6 months of surgical aortic valve replacement increased risk of thromboembolic complications and cardiovascular death (JAMA. 2012;308:2118-25). And the role of prosthesis type in BPVT “remains unclear.”
Dr. Egbe and coauthors acknowledged a number of questions persist with regard to BPVT in bioprosthetic valve dysfunction, including the true incidence, best screening method, risk factors, and the duration of anticoagulation, as well as the role of novel oral anticoagulants. “Answers to these questions will come from population-based prospective studies,” Dr. Egbe and colleagues said.
Dr. Egbe and his coauthors had no relationships to disclose.
Dr. Egbe and colleagues make a “provocative” case that it is the presence of thrombus on bioprosthetic valves, and not degeneration, that causes valve dysfunction, Clifford W. Barlow, MBBCh, DPhil, FRCS, of University Hospital Southampton (England) said in his invited commentary (J Thorac Cardiovasc Surg. 2016;152:978-80).
“This Expert Opinion is of particular interest because it relates to something commonly performed: conventional valve replacement,” Dr. Barlow said. Moreover, “BPVT is an under-recognized problem for which Dr. Egbe and colleagues concisely direct how future research should ascertain which diagnostic, preventive, and treatment strategies would improve long-term outcomes and avoid redo surgery.”
Dr. Egbe’s and colleagues’ recommendation of prolonged anticoagulation after bioprosthetic valve implantation complicates the selection of bioprosthetic valves – because cardiovascular surgeons frequently choose them to avoid anticoagulation, while accepting a higher risk of a reoperation because of valve degeneration, Dr. Barlow said.
And while Dr. Barlow noted this study found that porcine valves are not a predictor for BPVT, another Mayo Clinic study reported eight cases of BPVT, all in porcine valves (J Thorac Cardiovasc Surg. 2012;144:108-11). Nonetheless, the expert opinion by Dr. Egbe and colleagues is “relevant to much that is important – not only to improving outcomes with conventional valve replacement but also to these developing technologies,” Dr. Barlow said.
Dr. Egbe and colleagues make a “provocative” case that it is the presence of thrombus on bioprosthetic valves, and not degeneration, that causes valve dysfunction, Clifford W. Barlow, MBBCh, DPhil, FRCS, of University Hospital Southampton (England) said in his invited commentary (J Thorac Cardiovasc Surg. 2016;152:978-80).
“This Expert Opinion is of particular interest because it relates to something commonly performed: conventional valve replacement,” Dr. Barlow said. Moreover, “BPVT is an under-recognized problem for which Dr. Egbe and colleagues concisely direct how future research should ascertain which diagnostic, preventive, and treatment strategies would improve long-term outcomes and avoid redo surgery.”
Dr. Egbe’s and colleagues’ recommendation of prolonged anticoagulation after bioprosthetic valve implantation complicates the selection of bioprosthetic valves – because cardiovascular surgeons frequently choose them to avoid anticoagulation, while accepting a higher risk of a reoperation because of valve degeneration, Dr. Barlow said.
And while Dr. Barlow noted this study found that porcine valves are not a predictor for BPVT, another Mayo Clinic study reported eight cases of BPVT, all in porcine valves (J Thorac Cardiovasc Surg. 2012;144:108-11). Nonetheless, the expert opinion by Dr. Egbe and colleagues is “relevant to much that is important – not only to improving outcomes with conventional valve replacement but also to these developing technologies,” Dr. Barlow said.
Dr. Egbe and colleagues make a “provocative” case that it is the presence of thrombus on bioprosthetic valves, and not degeneration, that causes valve dysfunction, Clifford W. Barlow, MBBCh, DPhil, FRCS, of University Hospital Southampton (England) said in his invited commentary (J Thorac Cardiovasc Surg. 2016;152:978-80).
“This Expert Opinion is of particular interest because it relates to something commonly performed: conventional valve replacement,” Dr. Barlow said. Moreover, “BPVT is an under-recognized problem for which Dr. Egbe and colleagues concisely direct how future research should ascertain which diagnostic, preventive, and treatment strategies would improve long-term outcomes and avoid redo surgery.”
Dr. Egbe’s and colleagues’ recommendation of prolonged anticoagulation after bioprosthetic valve implantation complicates the selection of bioprosthetic valves – because cardiovascular surgeons frequently choose them to avoid anticoagulation, while accepting a higher risk of a reoperation because of valve degeneration, Dr. Barlow said.
And while Dr. Barlow noted this study found that porcine valves are not a predictor for BPVT, another Mayo Clinic study reported eight cases of BPVT, all in porcine valves (J Thorac Cardiovasc Surg. 2012;144:108-11). Nonetheless, the expert opinion by Dr. Egbe and colleagues is “relevant to much that is important – not only to improving outcomes with conventional valve replacement but also to these developing technologies,” Dr. Barlow said.
Recent findings on the incidence and pathophysiology of bioprosthetic valve thrombosis require revisiting existing guidelines against routine echocardiography in the first 5 years after bioprosthetic valve replacement and a longer course of anticoagulation therapy than the current standard of 3 months, investigators from the Mayo Clinic said in an expert opinion article in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152;975-8).
In the expert commentary, Alexander C. Egbe, MBBS, of the departments of cardiovascular diseases and cardiovascular surgery at Mayo Clinic in Rochester, Minn., and coauthors explored the implications of their previous research, published in the Journal of the American College of Cardiology, that reported that bioprosthetic valve thrombosis (BPVT) is “not an uncommon cause of prosthetic valve dysfunction.” They identified BPVT in 46 of 397 (11%) bioprosthetic valves explanted at Mayo Clinic, and estimated the incidence of BPVT at 1% (J Am Coll Cardiol. 2015;66:2285-94), although Dr. Egbe and colleagues acknowledged the true incidence of BPVT is unknown, as is the time to occurrence. They noted that a different study design would be needed to determine that, along with the incidence of BPVT.
“The occurrence of BPVT is not restricted to surgically implanted bioprosthetic valves, but has also been observed after transcatheter aortic valve replacement (TAVR),” Dr. Egbe and colleagues said. They noted an association between BPVT and a lack of anticoagulation therapy in two earlier reports (N Engl J Med. 2015;373:2015-24; J Am Coll Cardiol. 2016;67:644-55). In their own study, 14 of 15 patients (93%) with diagnosed BPVT responded to anticoagulation therapy and avoided reoperation.
Dr. Egbe and coauthors did somewhat define the extent of the problem of misdiagnosis of BPVT. The diagnosis was considered in only 6 of 45 patients (13%) who had transesophageal echocardiography. “A significant proportion of the patients with BPVT were misdiagnosed as having structural failure and referred for reoperation,” Dr. Egbe and coauthors said. “This attests to the low level of awareness of the existence of BPVT and the lack of well-defined diagnostic criteria.”
They proposed a diagnostic model based on the echocardiography characteristics of three findings: a 50% increase in gradient within 5 years of implantation; increased cusp thickness; and abnormal cusp mobility. “The presence of all three echocardiographic features reliably diagnosed BPVT with a sensitivity of 72% and a specificity of 90%,” they said.
Their finding that 85% of BPVT cases occurred within 5 years of implantation flies in the face of clinical guidelines that state routine annual echocardiography is not recommended in that time frame (J Am So Echocardiogr. 2009;22;975-1014). But abnormal physical examination findings as a prerequisite for echocardiography may not be an effective method to diagnose BPVT. “In addition to transthoracic and transesophageal echocardiography, the use of other complementary imaging modalities, such as computed tomography, could be very effective in identifying subtle BPVT,” Dr. Egbe and colleagues said,
But preventing BPVT is more complicated. Clinical guidelines recommend anticoagulation of bioprosthetic valves for 3 months after implantation, but adhering to that guideline showed no protective effect against BPVT in their study, Dr. Egbe and coauthors said. Nor did antiplatelet therapy prove effective in preventing BPVT. However, a Danish study showed stopping anticoagulation within 6 months of surgical aortic valve replacement increased risk of thromboembolic complications and cardiovascular death (JAMA. 2012;308:2118-25). And the role of prosthesis type in BPVT “remains unclear.”
Dr. Egbe and coauthors acknowledged a number of questions persist with regard to BPVT in bioprosthetic valve dysfunction, including the true incidence, best screening method, risk factors, and the duration of anticoagulation, as well as the role of novel oral anticoagulants. “Answers to these questions will come from population-based prospective studies,” Dr. Egbe and colleagues said.
Dr. Egbe and his coauthors had no relationships to disclose.
Recent findings on the incidence and pathophysiology of bioprosthetic valve thrombosis require revisiting existing guidelines against routine echocardiography in the first 5 years after bioprosthetic valve replacement and a longer course of anticoagulation therapy than the current standard of 3 months, investigators from the Mayo Clinic said in an expert opinion article in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152;975-8).
In the expert commentary, Alexander C. Egbe, MBBS, of the departments of cardiovascular diseases and cardiovascular surgery at Mayo Clinic in Rochester, Minn., and coauthors explored the implications of their previous research, published in the Journal of the American College of Cardiology, that reported that bioprosthetic valve thrombosis (BPVT) is “not an uncommon cause of prosthetic valve dysfunction.” They identified BPVT in 46 of 397 (11%) bioprosthetic valves explanted at Mayo Clinic, and estimated the incidence of BPVT at 1% (J Am Coll Cardiol. 2015;66:2285-94), although Dr. Egbe and colleagues acknowledged the true incidence of BPVT is unknown, as is the time to occurrence. They noted that a different study design would be needed to determine that, along with the incidence of BPVT.
“The occurrence of BPVT is not restricted to surgically implanted bioprosthetic valves, but has also been observed after transcatheter aortic valve replacement (TAVR),” Dr. Egbe and colleagues said. They noted an association between BPVT and a lack of anticoagulation therapy in two earlier reports (N Engl J Med. 2015;373:2015-24; J Am Coll Cardiol. 2016;67:644-55). In their own study, 14 of 15 patients (93%) with diagnosed BPVT responded to anticoagulation therapy and avoided reoperation.
Dr. Egbe and coauthors did somewhat define the extent of the problem of misdiagnosis of BPVT. The diagnosis was considered in only 6 of 45 patients (13%) who had transesophageal echocardiography. “A significant proportion of the patients with BPVT were misdiagnosed as having structural failure and referred for reoperation,” Dr. Egbe and coauthors said. “This attests to the low level of awareness of the existence of BPVT and the lack of well-defined diagnostic criteria.”
They proposed a diagnostic model based on the echocardiography characteristics of three findings: a 50% increase in gradient within 5 years of implantation; increased cusp thickness; and abnormal cusp mobility. “The presence of all three echocardiographic features reliably diagnosed BPVT with a sensitivity of 72% and a specificity of 90%,” they said.
Their finding that 85% of BPVT cases occurred within 5 years of implantation flies in the face of clinical guidelines that state routine annual echocardiography is not recommended in that time frame (J Am So Echocardiogr. 2009;22;975-1014). But abnormal physical examination findings as a prerequisite for echocardiography may not be an effective method to diagnose BPVT. “In addition to transthoracic and transesophageal echocardiography, the use of other complementary imaging modalities, such as computed tomography, could be very effective in identifying subtle BPVT,” Dr. Egbe and colleagues said,
But preventing BPVT is more complicated. Clinical guidelines recommend anticoagulation of bioprosthetic valves for 3 months after implantation, but adhering to that guideline showed no protective effect against BPVT in their study, Dr. Egbe and coauthors said. Nor did antiplatelet therapy prove effective in preventing BPVT. However, a Danish study showed stopping anticoagulation within 6 months of surgical aortic valve replacement increased risk of thromboembolic complications and cardiovascular death (JAMA. 2012;308:2118-25). And the role of prosthesis type in BPVT “remains unclear.”
Dr. Egbe and coauthors acknowledged a number of questions persist with regard to BPVT in bioprosthetic valve dysfunction, including the true incidence, best screening method, risk factors, and the duration of anticoagulation, as well as the role of novel oral anticoagulants. “Answers to these questions will come from population-based prospective studies,” Dr. Egbe and colleagues said.
Dr. Egbe and his coauthors had no relationships to disclose.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Preoperative echocardiography can aid in the diagnosis of BPVT.
Major finding: Sixty-five percent of all reoperations for BPVT occurred more than a year after implantation and up to 15% of these reoperations occurred more than 5 years after the initial implantation.
Data source: Single-center retrospective study of 397 valve explants.
Disclosures: Dr. Egbe and his coauthors reported having no financial disclosures.
Disease severity, QOL outcome measures need standardizing in atopic dermatitis research
The number of outcome measures used to assess disease severity and quality of life (QOL) in randomized controlled trials of patients with atopic dermatitis (AD) has risen in recent years, according to a systematic review reports.
An overall lack of standardization of these outcome measures, however, is hindering the synthesis and translation of research into clinical practice, reported Mary K. Hill of the University of Colorado, Aurora, and her associates.
“Standardization of disease severity and QOL outcome instruments is essential for comparability among studies and improved quality of research evidence,” they wrote (J Am Acad Dermatol. 2016 Nov;75[5]:906-17. doi: 10.1016/j.jaad.2016.07.002).
Their systematic review of 135 randomized controlled trials (RCTs) identified 62 disease-severity and 28 quality-of-life instruments used in studies of patients with AD between July 2010 and July 2015.
This was a drastic increase from the 20 disease severity scales and 14 QOL indices identified in a previous systematic review of 382 RCTs of AD therapies conducted between 1985 and 2010, they noted.
In their review, the most frequently used disease severity scale was the Scoring Atopic Dermatitis (SCORAD) index, which was used in 79 studies. That was followed by the visual analogue scale (VAS) for pruritus, used in 30 studies; the Investigator’s Global Assessment (IGA) tool, used in 29 studies; and the Eczema Area and Severity Index (EASI), used in 28 studies.
But despite the well-documented burden of AD, the researchers noted that only 33% of the RCTs they reviewed assessed QOL. “This is up from the 18% of RCTs on AD that reported QOL outcomes between 1985 and July 2010, perhaps signifying gradually increased attention to patient emotional well-being,” Ms. Hill and her associates wrote.
A trend described by the authors, however, as “perhaps the most disconcerting” was that 75% of identified QOL instruments were used only once. “Continued increases in the reporting of QOL outcomes will be of limited benefit for interstudy comparisons if the diversity of measures used also continues to rise,” they said.
Adding to the confusion, the researchers found frequent overlap in the naming and content of instruments used in studies, making it even more challenging to identify meaningful comparisons.
There was no funding source, and the authors had no conflicts of interest to declare.
The number of outcome measures used to assess disease severity and quality of life (QOL) in randomized controlled trials of patients with atopic dermatitis (AD) has risen in recent years, according to a systematic review reports.
An overall lack of standardization of these outcome measures, however, is hindering the synthesis and translation of research into clinical practice, reported Mary K. Hill of the University of Colorado, Aurora, and her associates.
“Standardization of disease severity and QOL outcome instruments is essential for comparability among studies and improved quality of research evidence,” they wrote (J Am Acad Dermatol. 2016 Nov;75[5]:906-17. doi: 10.1016/j.jaad.2016.07.002).
Their systematic review of 135 randomized controlled trials (RCTs) identified 62 disease-severity and 28 quality-of-life instruments used in studies of patients with AD between July 2010 and July 2015.
This was a drastic increase from the 20 disease severity scales and 14 QOL indices identified in a previous systematic review of 382 RCTs of AD therapies conducted between 1985 and 2010, they noted.
In their review, the most frequently used disease severity scale was the Scoring Atopic Dermatitis (SCORAD) index, which was used in 79 studies. That was followed by the visual analogue scale (VAS) for pruritus, used in 30 studies; the Investigator’s Global Assessment (IGA) tool, used in 29 studies; and the Eczema Area and Severity Index (EASI), used in 28 studies.
But despite the well-documented burden of AD, the researchers noted that only 33% of the RCTs they reviewed assessed QOL. “This is up from the 18% of RCTs on AD that reported QOL outcomes between 1985 and July 2010, perhaps signifying gradually increased attention to patient emotional well-being,” Ms. Hill and her associates wrote.
A trend described by the authors, however, as “perhaps the most disconcerting” was that 75% of identified QOL instruments were used only once. “Continued increases in the reporting of QOL outcomes will be of limited benefit for interstudy comparisons if the diversity of measures used also continues to rise,” they said.
Adding to the confusion, the researchers found frequent overlap in the naming and content of instruments used in studies, making it even more challenging to identify meaningful comparisons.
There was no funding source, and the authors had no conflicts of interest to declare.
The number of outcome measures used to assess disease severity and quality of life (QOL) in randomized controlled trials of patients with atopic dermatitis (AD) has risen in recent years, according to a systematic review reports.
An overall lack of standardization of these outcome measures, however, is hindering the synthesis and translation of research into clinical practice, reported Mary K. Hill of the University of Colorado, Aurora, and her associates.
“Standardization of disease severity and QOL outcome instruments is essential for comparability among studies and improved quality of research evidence,” they wrote (J Am Acad Dermatol. 2016 Nov;75[5]:906-17. doi: 10.1016/j.jaad.2016.07.002).
Their systematic review of 135 randomized controlled trials (RCTs) identified 62 disease-severity and 28 quality-of-life instruments used in studies of patients with AD between July 2010 and July 2015.
This was a drastic increase from the 20 disease severity scales and 14 QOL indices identified in a previous systematic review of 382 RCTs of AD therapies conducted between 1985 and 2010, they noted.
In their review, the most frequently used disease severity scale was the Scoring Atopic Dermatitis (SCORAD) index, which was used in 79 studies. That was followed by the visual analogue scale (VAS) for pruritus, used in 30 studies; the Investigator’s Global Assessment (IGA) tool, used in 29 studies; and the Eczema Area and Severity Index (EASI), used in 28 studies.
But despite the well-documented burden of AD, the researchers noted that only 33% of the RCTs they reviewed assessed QOL. “This is up from the 18% of RCTs on AD that reported QOL outcomes between 1985 and July 2010, perhaps signifying gradually increased attention to patient emotional well-being,” Ms. Hill and her associates wrote.
A trend described by the authors, however, as “perhaps the most disconcerting” was that 75% of identified QOL instruments were used only once. “Continued increases in the reporting of QOL outcomes will be of limited benefit for interstudy comparisons if the diversity of measures used also continues to rise,” they said.
Adding to the confusion, the researchers found frequent overlap in the naming and content of instruments used in studies, making it even more challenging to identify meaningful comparisons.
There was no funding source, and the authors had no conflicts of interest to declare.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: Disease severity and quality-of-life measures used in randomized controlled trials (RCTs) of atopic dermatitis (AD) need to be standardized so that study outcomes can be meaningfully compared and translated into clinical practice.
Major finding: The use of outcome measures used in atopic dermatitis RCTs has increased recently but still fall short in measuring quality-of-life outcomes.
Data source: A systematic review of 135 RCTs published between 2010-2015 that involved patients with AD.
Disclosures: There was no funding source, and the authors had no conflicts of interest to declare.
Vitamin D as an Add-On Therapy May Improve MRI Outcomes in MS
LONDON—High-dose vitamin D as an add-on therapy may improve MRI outcomes in patients with relapsing-remitting multiple sclerosis (MS), according to a study presented at the 32nd Congress of the European Committee for Treatment and Research in MS.
“Vitamin D is the precursor of a potent immunoregulatory molecule. However, whether supplementation of it improves outcomes is uncertain, since existing medical evidence is contradictory and involves small patient numbers,” said Raymond Hupperts, MD, PhD, Professor of Neurology at Maastricht University in the Netherlands.

Previous studies have found an association between low serum levels of vitamin D and a greater risk of developing MS and poor MS outcomes. As a result, Dr. Hupperts and colleagues conducted the SOLAR (Supplementation of VigantOL Oil Versus Placebo as Add-on in Patients With Relapsing Remitting MS Receiving Rebif Treatment) study to investigate the effects of vitamin D as add-on therapy in patients receiving subcutaneous interferon beta-1a.
The SOLAR study included 229 patients who were stratified by serum vitamin D level and randomized to vitamin D or placebo. Patients with serum vitamin D levels less than 150 nmol/L were randomized to one of two treatment groups. In treatment group one, patients were given 6,670 IU/day of vitamin D for four weeks, followed by 14,007 IU for 44 weeks or 92 weeks as an add-on to interferon beta-1a. In treatment group two, patients received matching placebo daily as an add-on therapy to interferon beta-1a.
The primary end point for the study was the proportion of patients with no evidence of disease activity (NEDA), which was defined as no relapses, no progression in Expanded Disability Status Scale (EDSS) score, and no gadolinium-enhancing T1 lesions or new or enlarging T2 MRI lesions, at Week 48. The secondary end points included annualized relapse rate (ARR) at Week 48, EDSS progression at Week 48, time to confirmed EDSS score progression, number of combined unique active (CUA) lesions per patient per scan at Week 48, number of T1-hypointense lesions at Week 48, and change from baseline in the total volume of T2 lesions at Week 48.
The ARR was lower in the vitamin D group, but the difference between groups was not statistically significant. However, the relapse rate in the vitamin D group was 0.28 versus 0.41 in the placebo group, a 30% difference at Week 48. In addition, vitamin D was associated with significantly better MRI outcomes. The number of CUA lesions at Week 48 was 1.09 in the vitamin D group and 1.49 in the placebo group. The change from baseline of the total volume of T2 lesions was 3.57% in the vitamin D group and 6.07% in the placebo group. Eighty-five percent of participants between ages 18 and 30 in the vitamin D group and 46.8% of this age group in the placebo arm had no new T1-hypointense lesions at Week 48.
“We conclude that SOLAR did not show a significant effect [of vitamin D] on the primary end point. However, because of ARR and MRI findings, we suggest a benefit of high-dose vitamin D,” said Dr. Hupperts. The researchers added that vitamin D supplementation may be more effective in early stages of MS, and they found no additional safety issues associated with high-dose vitamin D.
—Erica Tricarico
Suggested Reading
Ashtari F, Toghianifar N, Zarkesh-Esfahani SH, Mansourian M. High dose vitamin D intake and quality of life in relapsing-remitting multiple sclerosis: a randomized, double-blind, placebo-controlled clinical trial. Neurol Res. 2016;38(10):888-892.
LONDON—High-dose vitamin D as an add-on therapy may improve MRI outcomes in patients with relapsing-remitting multiple sclerosis (MS), according to a study presented at the 32nd Congress of the European Committee for Treatment and Research in MS.
“Vitamin D is the precursor of a potent immunoregulatory molecule. However, whether supplementation of it improves outcomes is uncertain, since existing medical evidence is contradictory and involves small patient numbers,” said Raymond Hupperts, MD, PhD, Professor of Neurology at Maastricht University in the Netherlands.
Previous studies have found an association between low serum levels of vitamin D and a greater risk of developing MS and poor MS outcomes. As a result, Dr. Hupperts and colleagues conducted the SOLAR (Supplementation of VigantOL Oil Versus Placebo as Add-on in Patients With Relapsing Remitting MS Receiving Rebif Treatment) study to investigate the effects of vitamin D as add-on therapy in patients receiving subcutaneous interferon beta-1a.
The SOLAR study included 229 patients who were stratified by serum vitamin D level and randomized to vitamin D or placebo. Patients with serum vitamin D levels less than 150 nmol/L were randomized to one of two treatment groups. In treatment group one, patients were given 6,670 IU/day of vitamin D for four weeks, followed by 14,007 IU for 44 weeks or 92 weeks as an add-on to interferon beta-1a. In treatment group two, patients received matching placebo daily as an add-on therapy to interferon beta-1a.
The primary end point for the study was the proportion of patients with no evidence of disease activity (NEDA), which was defined as no relapses, no progression in Expanded Disability Status Scale (EDSS) score, and no gadolinium-enhancing T1 lesions or new or enlarging T2 MRI lesions, at Week 48. The secondary end points included annualized relapse rate (ARR) at Week 48, EDSS progression at Week 48, time to confirmed EDSS score progression, number of combined unique active (CUA) lesions per patient per scan at Week 48, number of T1-hypointense lesions at Week 48, and change from baseline in the total volume of T2 lesions at Week 48.
The ARR was lower in the vitamin D group, but the difference between groups was not statistically significant. However, the relapse rate in the vitamin D group was 0.28 versus 0.41 in the placebo group, a 30% difference at Week 48. In addition, vitamin D was associated with significantly better MRI outcomes. The number of CUA lesions at Week 48 was 1.09 in the vitamin D group and 1.49 in the placebo group. The change from baseline of the total volume of T2 lesions was 3.57% in the vitamin D group and 6.07% in the placebo group. Eighty-five percent of participants between ages 18 and 30 in the vitamin D group and 46.8% of this age group in the placebo arm had no new T1-hypointense lesions at Week 48.
“We conclude that SOLAR did not show a significant effect [of vitamin D] on the primary end point. However, because of ARR and MRI findings, we suggest a benefit of high-dose vitamin D,” said Dr. Hupperts. The researchers added that vitamin D supplementation may be more effective in early stages of MS, and they found no additional safety issues associated with high-dose vitamin D.
—Erica Tricarico
Suggested Reading
Ashtari F, Toghianifar N, Zarkesh-Esfahani SH, Mansourian M. High dose vitamin D intake and quality of life in relapsing-remitting multiple sclerosis: a randomized, double-blind, placebo-controlled clinical trial. Neurol Res. 2016;38(10):888-892.
LONDON—High-dose vitamin D as an add-on therapy may improve MRI outcomes in patients with relapsing-remitting multiple sclerosis (MS), according to a study presented at the 32nd Congress of the European Committee for Treatment and Research in MS.
“Vitamin D is the precursor of a potent immunoregulatory molecule. However, whether supplementation of it improves outcomes is uncertain, since existing medical evidence is contradictory and involves small patient numbers,” said Raymond Hupperts, MD, PhD, Professor of Neurology at Maastricht University in the Netherlands.
Previous studies have found an association between low serum levels of vitamin D and a greater risk of developing MS and poor MS outcomes. As a result, Dr. Hupperts and colleagues conducted the SOLAR (Supplementation of VigantOL Oil Versus Placebo as Add-on in Patients With Relapsing Remitting MS Receiving Rebif Treatment) study to investigate the effects of vitamin D as add-on therapy in patients receiving subcutaneous interferon beta-1a.
The SOLAR study included 229 patients who were stratified by serum vitamin D level and randomized to vitamin D or placebo. Patients with serum vitamin D levels less than 150 nmol/L were randomized to one of two treatment groups. In treatment group one, patients were given 6,670 IU/day of vitamin D for four weeks, followed by 14,007 IU for 44 weeks or 92 weeks as an add-on to interferon beta-1a. In treatment group two, patients received matching placebo daily as an add-on therapy to interferon beta-1a.
The primary end point for the study was the proportion of patients with no evidence of disease activity (NEDA), which was defined as no relapses, no progression in Expanded Disability Status Scale (EDSS) score, and no gadolinium-enhancing T1 lesions or new or enlarging T2 MRI lesions, at Week 48. The secondary end points included annualized relapse rate (ARR) at Week 48, EDSS progression at Week 48, time to confirmed EDSS score progression, number of combined unique active (CUA) lesions per patient per scan at Week 48, number of T1-hypointense lesions at Week 48, and change from baseline in the total volume of T2 lesions at Week 48.
The ARR was lower in the vitamin D group, but the difference between groups was not statistically significant. However, the relapse rate in the vitamin D group was 0.28 versus 0.41 in the placebo group, a 30% difference at Week 48. In addition, vitamin D was associated with significantly better MRI outcomes. The number of CUA lesions at Week 48 was 1.09 in the vitamin D group and 1.49 in the placebo group. The change from baseline of the total volume of T2 lesions was 3.57% in the vitamin D group and 6.07% in the placebo group. Eighty-five percent of participants between ages 18 and 30 in the vitamin D group and 46.8% of this age group in the placebo arm had no new T1-hypointense lesions at Week 48.
“We conclude that SOLAR did not show a significant effect [of vitamin D] on the primary end point. However, because of ARR and MRI findings, we suggest a benefit of high-dose vitamin D,” said Dr. Hupperts. The researchers added that vitamin D supplementation may be more effective in early stages of MS, and they found no additional safety issues associated with high-dose vitamin D.
—Erica Tricarico
Suggested Reading
Ashtari F, Toghianifar N, Zarkesh-Esfahani SH, Mansourian M. High dose vitamin D intake and quality of life in relapsing-remitting multiple sclerosis: a randomized, double-blind, placebo-controlled clinical trial. Neurol Res. 2016;38(10):888-892.

Early Administration of Sertraline May Prevent Onset of Depression Following TBI
Sertraline may help to prevent the onset of depressive disorders after a traumatic brain injury (TBI), according to data published online ahead of print September 14 in JAMA Psychiatry.
“Our findings suggest that sertraline given at a low dosage early after TBI is an efficacious strategy to prevent depression after TBI,” said Ricardo E. Jorge, MD, Professor of Psychiatry and Behavioral Sciences at Baylor College of Medicine in Houston.

Every year, there are approximately 1.7 million cases of TBI in the United States. TBI contributes to 30% of all injury deaths and is a major cause of death and disability in the US, according to the Centers for Disease Control and Prevention.
Depressive disorders are common after TBI. In two studies, 58 of 157 patients developed a depressive disorder during the first year following TBI. Dr. Jorge and his colleagues conducted a double-blind, placebo-controlled study to assess the efficacy of sertraline in preventing depressive disorders following TBI. Their main outcome was time to onset of depressive disorder, as defined by the DSM-IV, associated with TBI.
“We hypothesized that the time from baseline to onset of depressive disorders would be greater in a group of patients randomized to receive sertraline treatment versus a group of patients randomized to receive placebo,” said Dr. Jorge. “We also hypothesized that, when compared with patients receiving placebo, patients receiving sertraline would show better performance in a set of neuropsychologic tests after six months of treatment.”
For the study, 94 patients were randomized to receive 100 mg/day of sertraline or placebo once daily for 24 weeks or until the development of a mood disorder. The age of participants ranged between 18 and 85, and patients had mild, moderate, or severe TBI. In addition, participants were required to have complete recovery of posttraumatic amnesia within four weeks of the traumatic episode. Patients with ongoing depression were excluded from the study. Furthermore, patients with mood disorders were required to have been in full remission for at least a year following discontinuation of treatment.
Researchers used the Mini-International Neuropsychiatric Interview and DSM-IV criteria to diagnose depressive disorders. In addition, participants were evaluated at baseline and at two, four, eight, 12, 16, 20, and 24 weeks. A Mini-International Neuropsychiatric Interview was administered via telephone on weeks six, 10, 14, 18, and 22.
The number of patients needed to treat to prevent development of depression after TBI at 24 weeks was 5.9. There were no incident cases of anxiety disorders, and one patient had suicidal ideation. Nearly all patients reported mild or moderate adverse events in the sertraline and placebo groups. Sexual adverse events were mild and did not significantly impact the quality of life of participants. Frequencies of dry mouth and diarrhea were higher among participants who received sertraline.
“The fact that small doses of sertraline are efficacious to prevent depression after TBI stands in sharp contrast to the lack of efficacy of antidepressants to treat depression in the chronic stage of TBI,” said Dr. Jorge.
Limitations of this study include its scarce representation of ethnic and racial minorities, small sample size, and limited follow-up following incident TBI.
—Erica Tricarico
Suggested Reading
Jorge RE, Acion L, Burin DI, Robinson RG. Sertraline for preventing mood disorders following traumatic brain injury. JAMA Psychiatry. 2016 Sep 14 [Epub ahead of print].
Sertraline may help to prevent the onset of depressive disorders after a traumatic brain injury (TBI), according to data published online ahead of print September 14 in JAMA Psychiatry.
“Our findings suggest that sertraline given at a low dosage early after TBI is an efficacious strategy to prevent depression after TBI,” said Ricardo E. Jorge, MD, Professor of Psychiatry and Behavioral Sciences at Baylor College of Medicine in Houston.
Every year, there are approximately 1.7 million cases of TBI in the United States. TBI contributes to 30% of all injury deaths and is a major cause of death and disability in the US, according to the Centers for Disease Control and Prevention.
Depressive disorders are common after TBI. In two studies, 58 of 157 patients developed a depressive disorder during the first year following TBI. Dr. Jorge and his colleagues conducted a double-blind, placebo-controlled study to assess the efficacy of sertraline in preventing depressive disorders following TBI. Their main outcome was time to onset of depressive disorder, as defined by the DSM-IV, associated with TBI.
“We hypothesized that the time from baseline to onset of depressive disorders would be greater in a group of patients randomized to receive sertraline treatment versus a group of patients randomized to receive placebo,” said Dr. Jorge. “We also hypothesized that, when compared with patients receiving placebo, patients receiving sertraline would show better performance in a set of neuropsychologic tests after six months of treatment.”
For the study, 94 patients were randomized to receive 100 mg/day of sertraline or placebo once daily for 24 weeks or until the development of a mood disorder. The age of participants ranged between 18 and 85, and patients had mild, moderate, or severe TBI. In addition, participants were required to have complete recovery of posttraumatic amnesia within four weeks of the traumatic episode. Patients with ongoing depression were excluded from the study. Furthermore, patients with mood disorders were required to have been in full remission for at least a year following discontinuation of treatment.
Researchers used the Mini-International Neuropsychiatric Interview and DSM-IV criteria to diagnose depressive disorders. In addition, participants were evaluated at baseline and at two, four, eight, 12, 16, 20, and 24 weeks. A Mini-International Neuropsychiatric Interview was administered via telephone on weeks six, 10, 14, 18, and 22.
The number of patients needed to treat to prevent development of depression after TBI at 24 weeks was 5.9. There were no incident cases of anxiety disorders, and one patient had suicidal ideation. Nearly all patients reported mild or moderate adverse events in the sertraline and placebo groups. Sexual adverse events were mild and did not significantly impact the quality of life of participants. Frequencies of dry mouth and diarrhea were higher among participants who received sertraline.
“The fact that small doses of sertraline are efficacious to prevent depression after TBI stands in sharp contrast to the lack of efficacy of antidepressants to treat depression in the chronic stage of TBI,” said Dr. Jorge.
Limitations of this study include its scarce representation of ethnic and racial minorities, small sample size, and limited follow-up following incident TBI.
—Erica Tricarico
Suggested Reading
Jorge RE, Acion L, Burin DI, Robinson RG. Sertraline for preventing mood disorders following traumatic brain injury. JAMA Psychiatry. 2016 Sep 14 [Epub ahead of print].
Sertraline may help to prevent the onset of depressive disorders after a traumatic brain injury (TBI), according to data published online ahead of print September 14 in JAMA Psychiatry.
“Our findings suggest that sertraline given at a low dosage early after TBI is an efficacious strategy to prevent depression after TBI,” said Ricardo E. Jorge, MD, Professor of Psychiatry and Behavioral Sciences at Baylor College of Medicine in Houston.
Every year, there are approximately 1.7 million cases of TBI in the United States. TBI contributes to 30% of all injury deaths and is a major cause of death and disability in the US, according to the Centers for Disease Control and Prevention.
Depressive disorders are common after TBI. In two studies, 58 of 157 patients developed a depressive disorder during the first year following TBI. Dr. Jorge and his colleagues conducted a double-blind, placebo-controlled study to assess the efficacy of sertraline in preventing depressive disorders following TBI. Their main outcome was time to onset of depressive disorder, as defined by the DSM-IV, associated with TBI.
“We hypothesized that the time from baseline to onset of depressive disorders would be greater in a group of patients randomized to receive sertraline treatment versus a group of patients randomized to receive placebo,” said Dr. Jorge. “We also hypothesized that, when compared with patients receiving placebo, patients receiving sertraline would show better performance in a set of neuropsychologic tests after six months of treatment.”
For the study, 94 patients were randomized to receive 100 mg/day of sertraline or placebo once daily for 24 weeks or until the development of a mood disorder. The age of participants ranged between 18 and 85, and patients had mild, moderate, or severe TBI. In addition, participants were required to have complete recovery of posttraumatic amnesia within four weeks of the traumatic episode. Patients with ongoing depression were excluded from the study. Furthermore, patients with mood disorders were required to have been in full remission for at least a year following discontinuation of treatment.
Researchers used the Mini-International Neuropsychiatric Interview and DSM-IV criteria to diagnose depressive disorders. In addition, participants were evaluated at baseline and at two, four, eight, 12, 16, 20, and 24 weeks. A Mini-International Neuropsychiatric Interview was administered via telephone on weeks six, 10, 14, 18, and 22.
The number of patients needed to treat to prevent development of depression after TBI at 24 weeks was 5.9. There were no incident cases of anxiety disorders, and one patient had suicidal ideation. Nearly all patients reported mild or moderate adverse events in the sertraline and placebo groups. Sexual adverse events were mild and did not significantly impact the quality of life of participants. Frequencies of dry mouth and diarrhea were higher among participants who received sertraline.
“The fact that small doses of sertraline are efficacious to prevent depression after TBI stands in sharp contrast to the lack of efficacy of antidepressants to treat depression in the chronic stage of TBI,” said Dr. Jorge.
Limitations of this study include its scarce representation of ethnic and racial minorities, small sample size, and limited follow-up following incident TBI.
—Erica Tricarico
Suggested Reading
Jorge RE, Acion L, Burin DI, Robinson RG. Sertraline for preventing mood disorders following traumatic brain injury. JAMA Psychiatry. 2016 Sep 14 [Epub ahead of print].

‘Stepping’ up to a better way to teach robotic lobectomy
Teaching minimally invasive robotic surgery to residents can be difficult in a health care environment obsessed with quality outcome measures and under scrutiny by hospital administrators and payers, but researchers at the University of Alabama at Birmingham may have devised a method to instruct residents in robotic lobectomy without compromising patient outcomes, according to a study published in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:991-7).
Robert J. Cerfolio, MD, MBA, and his coauthors divided the procedure into 19 sequential, teachable steps and allowed residents to perform selected steps during operations that Dr. Cerfolio directed. “We then applied simulation training, coaching techniques, and video review of each step to help improve the steps that residents could not complete,” Dr. Cerfolio and his coauthors said.

Surgeons in academic centers face the challenge of teaching “the art and science of surgery,” Dr. Cerfolio and his colleagues said, while maintaining quality outcomes. “Teaching minimally invasive surgery, especially robotic surgery, is challenging given the risks and the limited availability of the robot.”
The researchers acknowledged that other groups have taken a similar approach to training, but this is the first study that included video review, coaching, and instruction tied to time constraints, they said. “A major concern is that while teaching robotic surgery, patients can be injured, care is worse, and metrics that are increasingly used as surrogates for quality outcomes suffer,” they noted.
They allotted each step in the procedure a set amount of time in which the resident had to complete it, totaling 80 minutes for all 19 steps and ranging from 1 minute to inspect the pleura after placing ports (9 minutes) to 20 minutes to close the five incisions. If the resident completed the task in the allotted time, it was recorded as “performed.”
Between February 2010 and December 2010 Dr. Cerfolio performed 520 robotic lobectomies, and over time the percentage of successful steps per resident improved. For example, in the first year, 50% of thoracic surgery residents completed the first five steps (mark and place ports, inspect pleura, resect the inferior pulmonary ligament, and remove three lymph nodes), but by the last year of the study 90% of them successfully completed the five steps.
Dr. Cerfolio and coauthors acknowledged “many flaws” in their study, but the study also had strengths: It involved only one operation and corroborated the database with each resident’s own surgical logs.
“Operations such as robotic lobectomy can be successfully taught by dividing them into a series of surgical maneuvers or steps,” the researchers noted. Recording what residents can and can’t do, reviewing video, and coaching contribute to the process to improve their skills. “Further studies that scientifically measure ‘ways to teach’ and ways to coach and mentor are needed,” they said.
Dr. Cerfolio disclosed relationships with Intuitive Surgical, Ethicon, Community Health Services, KCL, Bovie and C-SATS. Coauthor Douglas Minnich, MD, is a consultant to Medtronic. The other co-authors had no financial relationships to disclose.
Inderpal S. Sarkaria, MD, of the University of Pittsburgh acknowledged in his invited commentary how “metric-driven patient outcomes” have changed cardiothoracic surgical training (J Thorac Cardiovasc Surg. 2016;152:998).
But Dr. Sarkaria questioned the validity of using time performed as a metric in this study to evaluate a trainee’s competency. “Although ‘time’ is an important component, should not the primary focus be on ‘quality’ of the trainee’s work?” Dr. Sarkaria asked.

Despite these questions and the limitations of the study, he found the approach to surgical training “laudable.” Said Dr. Sarkaria: “It is arguable that the limitations of the study speak more to a common wisdom that certain aspects of surgical education remain an art to a greater or lesser extent, not easily amenable to our efforts to discretely compartmentalize and quantify the process.”
While the premise demands further study, Dr. Cerfolio and his coauthors “have laid a solid foundation on which further to build, explore, and potentially improve the science and art of teaching complex operations to our surgical residents,” Dr. Sarkaria said.
Dr. Sarkaria had no relationships to disclose.
Inderpal S. Sarkaria, MD, of the University of Pittsburgh acknowledged in his invited commentary how “metric-driven patient outcomes” have changed cardiothoracic surgical training (J Thorac Cardiovasc Surg. 2016;152:998).
But Dr. Sarkaria questioned the validity of using time performed as a metric in this study to evaluate a trainee’s competency. “Although ‘time’ is an important component, should not the primary focus be on ‘quality’ of the trainee’s work?” Dr. Sarkaria asked.
Despite these questions and the limitations of the study, he found the approach to surgical training “laudable.” Said Dr. Sarkaria: “It is arguable that the limitations of the study speak more to a common wisdom that certain aspects of surgical education remain an art to a greater or lesser extent, not easily amenable to our efforts to discretely compartmentalize and quantify the process.”
While the premise demands further study, Dr. Cerfolio and his coauthors “have laid a solid foundation on which further to build, explore, and potentially improve the science and art of teaching complex operations to our surgical residents,” Dr. Sarkaria said.
Dr. Sarkaria had no relationships to disclose.
Inderpal S. Sarkaria, MD, of the University of Pittsburgh acknowledged in his invited commentary how “metric-driven patient outcomes” have changed cardiothoracic surgical training (J Thorac Cardiovasc Surg. 2016;152:998).
But Dr. Sarkaria questioned the validity of using time performed as a metric in this study to evaluate a trainee’s competency. “Although ‘time’ is an important component, should not the primary focus be on ‘quality’ of the trainee’s work?” Dr. Sarkaria asked.
Despite these questions and the limitations of the study, he found the approach to surgical training “laudable.” Said Dr. Sarkaria: “It is arguable that the limitations of the study speak more to a common wisdom that certain aspects of surgical education remain an art to a greater or lesser extent, not easily amenable to our efforts to discretely compartmentalize and quantify the process.”
While the premise demands further study, Dr. Cerfolio and his coauthors “have laid a solid foundation on which further to build, explore, and potentially improve the science and art of teaching complex operations to our surgical residents,” Dr. Sarkaria said.
Dr. Sarkaria had no relationships to disclose.
Teaching minimally invasive robotic surgery to residents can be difficult in a health care environment obsessed with quality outcome measures and under scrutiny by hospital administrators and payers, but researchers at the University of Alabama at Birmingham may have devised a method to instruct residents in robotic lobectomy without compromising patient outcomes, according to a study published in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:991-7).
Robert J. Cerfolio, MD, MBA, and his coauthors divided the procedure into 19 sequential, teachable steps and allowed residents to perform selected steps during operations that Dr. Cerfolio directed. “We then applied simulation training, coaching techniques, and video review of each step to help improve the steps that residents could not complete,” Dr. Cerfolio and his coauthors said.
Surgeons in academic centers face the challenge of teaching “the art and science of surgery,” Dr. Cerfolio and his colleagues said, while maintaining quality outcomes. “Teaching minimally invasive surgery, especially robotic surgery, is challenging given the risks and the limited availability of the robot.”
The researchers acknowledged that other groups have taken a similar approach to training, but this is the first study that included video review, coaching, and instruction tied to time constraints, they said. “A major concern is that while teaching robotic surgery, patients can be injured, care is worse, and metrics that are increasingly used as surrogates for quality outcomes suffer,” they noted.
They allotted each step in the procedure a set amount of time in which the resident had to complete it, totaling 80 minutes for all 19 steps and ranging from 1 minute to inspect the pleura after placing ports (9 minutes) to 20 minutes to close the five incisions. If the resident completed the task in the allotted time, it was recorded as “performed.”
Between February 2010 and December 2010 Dr. Cerfolio performed 520 robotic lobectomies, and over time the percentage of successful steps per resident improved. For example, in the first year, 50% of thoracic surgery residents completed the first five steps (mark and place ports, inspect pleura, resect the inferior pulmonary ligament, and remove three lymph nodes), but by the last year of the study 90% of them successfully completed the five steps.
Dr. Cerfolio and coauthors acknowledged “many flaws” in their study, but the study also had strengths: It involved only one operation and corroborated the database with each resident’s own surgical logs.
“Operations such as robotic lobectomy can be successfully taught by dividing them into a series of surgical maneuvers or steps,” the researchers noted. Recording what residents can and can’t do, reviewing video, and coaching contribute to the process to improve their skills. “Further studies that scientifically measure ‘ways to teach’ and ways to coach and mentor are needed,” they said.
Dr. Cerfolio disclosed relationships with Intuitive Surgical, Ethicon, Community Health Services, KCL, Bovie and C-SATS. Coauthor Douglas Minnich, MD, is a consultant to Medtronic. The other co-authors had no financial relationships to disclose.
Teaching minimally invasive robotic surgery to residents can be difficult in a health care environment obsessed with quality outcome measures and under scrutiny by hospital administrators and payers, but researchers at the University of Alabama at Birmingham may have devised a method to instruct residents in robotic lobectomy without compromising patient outcomes, according to a study published in the October issue of the Journal of Thoracic and Cardiovascular Surgery (2016;152:991-7).
Robert J. Cerfolio, MD, MBA, and his coauthors divided the procedure into 19 sequential, teachable steps and allowed residents to perform selected steps during operations that Dr. Cerfolio directed. “We then applied simulation training, coaching techniques, and video review of each step to help improve the steps that residents could not complete,” Dr. Cerfolio and his coauthors said.
Surgeons in academic centers face the challenge of teaching “the art and science of surgery,” Dr. Cerfolio and his colleagues said, while maintaining quality outcomes. “Teaching minimally invasive surgery, especially robotic surgery, is challenging given the risks and the limited availability of the robot.”
The researchers acknowledged that other groups have taken a similar approach to training, but this is the first study that included video review, coaching, and instruction tied to time constraints, they said. “A major concern is that while teaching robotic surgery, patients can be injured, care is worse, and metrics that are increasingly used as surrogates for quality outcomes suffer,” they noted.
They allotted each step in the procedure a set amount of time in which the resident had to complete it, totaling 80 minutes for all 19 steps and ranging from 1 minute to inspect the pleura after placing ports (9 minutes) to 20 minutes to close the five incisions. If the resident completed the task in the allotted time, it was recorded as “performed.”
Between February 2010 and December 2010 Dr. Cerfolio performed 520 robotic lobectomies, and over time the percentage of successful steps per resident improved. For example, in the first year, 50% of thoracic surgery residents completed the first five steps (mark and place ports, inspect pleura, resect the inferior pulmonary ligament, and remove three lymph nodes), but by the last year of the study 90% of them successfully completed the five steps.
Dr. Cerfolio and coauthors acknowledged “many flaws” in their study, but the study also had strengths: It involved only one operation and corroborated the database with each resident’s own surgical logs.
“Operations such as robotic lobectomy can be successfully taught by dividing them into a series of surgical maneuvers or steps,” the researchers noted. Recording what residents can and can’t do, reviewing video, and coaching contribute to the process to improve their skills. “Further studies that scientifically measure ‘ways to teach’ and ways to coach and mentor are needed,” they said.
Dr. Cerfolio disclosed relationships with Intuitive Surgical, Ethicon, Community Health Services, KCL, Bovie and C-SATS. Coauthor Douglas Minnich, MD, is a consultant to Medtronic. The other co-authors had no financial relationships to disclose.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Surgical residents learn and safely perform robotic lobectomy by dividing the procedure into a series of surgical maneuvers.
Major finding: The percentage of thoracic surgery residents who completed the first 5 of 19 procedural steps of the operation improved from 50% in the first year to 90% in the fifth year.
Data source: Single-center study of 520 consecutive lobectomies over 5 years by 35 general surgery residents and 7 cardiothoracic residents from February 2010 to December 2015.
Disclosures: Dr. Cerfolio disclosed relationships with Intuitive Surgical, Ethicon, Community Health Services, KCL, Bovie and C-SATS. Coauthor Douglas Minnich, MD, is a consultant to Medtronic. The other coauthors had no financial relationships to disclose.
A tricky interplay, indeed
Bone disease in patients with kidney disease is indeed a tricky interplay, as the article by Nyman et al (J Fam Pract. 2016;65:606-612) aptly states in its title.
The author made incorrect statements on page 607 regarding hyperphosphatemia and hypocalcemia and the escalation of fracture risk. (Editor’s Note: See erratum.)
In addition, on page 610, the article mentions that 1,25-(OH)2 vitamin D may help prevent hypertension, myocardial infarction, and stroke in patients without chronic kidney disease. This is not supported by the literature and even the reference cited states that fact.
Roy N. Morcos, MD, FAAFP
Boardman, OH
Author’s response:
Thank you, Dr. Morcos, for your careful read of our article.
Regarding the discussion of 1,25-(OH)2 vitamin D, we are in agreement. In fact, the last sentence of our paragraph reads: “There are no data, however, confirming that 25(OH)D supplementation mitigates these outcomes.” We were simply calling attention to the fact that despite the lack of evidence, some providers are still prescribing native vitamin D for their patients with chronic kidney disease for reasons unrelated to parathyroid hormone suppression.
Karly Pippitt, MD,
on behalf of co-authors Heather Nyman, PharmD, BCPS;
Alisyn Hansen, PharmD, BCACP, CDE;
Karen Gunning, PharmD, BCPS, BCACP, FCCP
Salt Lake City, UT
Bone disease in patients with kidney disease is indeed a tricky interplay, as the article by Nyman et al (J Fam Pract. 2016;65:606-612) aptly states in its title.
The author made incorrect statements on page 607 regarding hyperphosphatemia and hypocalcemia and the escalation of fracture risk. (Editor’s Note: See erratum.)
In addition, on page 610, the article mentions that 1,25-(OH)2 vitamin D may help prevent hypertension, myocardial infarction, and stroke in patients without chronic kidney disease. This is not supported by the literature and even the reference cited states that fact.
Roy N. Morcos, MD, FAAFP
Boardman, OH
Author’s response:
Thank you, Dr. Morcos, for your careful read of our article.
Regarding the discussion of 1,25-(OH)2 vitamin D, we are in agreement. In fact, the last sentence of our paragraph reads: “There are no data, however, confirming that 25(OH)D supplementation mitigates these outcomes.” We were simply calling attention to the fact that despite the lack of evidence, some providers are still prescribing native vitamin D for their patients with chronic kidney disease for reasons unrelated to parathyroid hormone suppression.
Karly Pippitt, MD,
on behalf of co-authors Heather Nyman, PharmD, BCPS;
Alisyn Hansen, PharmD, BCACP, CDE;
Karen Gunning, PharmD, BCPS, BCACP, FCCP
Salt Lake City, UT
Bone disease in patients with kidney disease is indeed a tricky interplay, as the article by Nyman et al (J Fam Pract. 2016;65:606-612) aptly states in its title.
The author made incorrect statements on page 607 regarding hyperphosphatemia and hypocalcemia and the escalation of fracture risk. (Editor’s Note: See erratum.)
In addition, on page 610, the article mentions that 1,25-(OH)2 vitamin D may help prevent hypertension, myocardial infarction, and stroke in patients without chronic kidney disease. This is not supported by the literature and even the reference cited states that fact.
Roy N. Morcos, MD, FAAFP
Boardman, OH
Author’s response:
Thank you, Dr. Morcos, for your careful read of our article.
Regarding the discussion of 1,25-(OH)2 vitamin D, we are in agreement. In fact, the last sentence of our paragraph reads: “There are no data, however, confirming that 25(OH)D supplementation mitigates these outcomes.” We were simply calling attention to the fact that despite the lack of evidence, some providers are still prescribing native vitamin D for their patients with chronic kidney disease for reasons unrelated to parathyroid hormone suppression.
Karly Pippitt, MD,
on behalf of co-authors Heather Nyman, PharmD, BCPS;
Alisyn Hansen, PharmD, BCACP, CDE;
Karen Gunning, PharmD, BCPS, BCACP, FCCP
Salt Lake City, UT
MS relapse predictors identified after patients stop treatment
Patients with multiple sclerosis treated with interferon-beta or glatiramer acetate who are aged 45 and over, and have no evidence of clinical disease activity for more than 4 years, have a high likelihood of remaining relapse free after stopping treatment, research showed.
The study, conducted by Gabriel Bsteh, MD, of the department of neurology at the Medical University of Innsbruck (Austria) and his colleagues, provides some evidence in the absence of randomized trials that may help guide discussions with patients – particularly those who have not had a relapse in a while – when they ask if they could discontinue disease-modifying treatment (DMT).
After a median follow-up period of 3.8 years, 98 patients (44.3%) had a relapse. Confirmed disability progression occurred in 46 patients (20.8%), and 15 patients (6.8%) converted to secondary progressive multiple sclerosis.
The independent predictors of absence of relapse after discontinuing treatment included age 45 years or older at discontinuation (HR = 0.47; confidence interval, 0.23–0.95; P = .038), absence of relapses for 4 or more years on DMT before discontinuation (HR = 0.29; CI, 0.10–0.82; P = .020), and absence of contrast-enhancing lesions (HR = 0.46; CI, 0.28–0.78; P =.004). A combination of age 45 years or older and absence of relapses after 4 or more years on DMT was associated with a very low risk of having a relapse after discontinuation, regardless of MRI results (HR = 0.06; CI, 0.01-0.44; P less than .001).
Higher Expanded Disability Status Scale (EDSS) scores at discontinuation, age older than 45 years at discontinuation, and longer disease duration were the only significant independent predictors of disability progression after discontinuation, irrespective of the presence of relapses on DMT or gadolinium-enhancing lesions.
“This underlines the concept of a window of opportunity in the treatment of MS, in the sense that once a certain extent of disability is reached, the impact of relapses, and therefore the effect of anti-inflammatory treatment, are drastically reduced,” the investigators wrote.
The study is limited because of its observational retrospective nature, but the research team said their results emphasized the importance of regular, thorough clinical evaluation of patients with RRMS. “While MRI may have a role in aiding decision making regarding DMT discontinuation, our data clearly show that demographic factors and clinical monitoring go a long way in risk stratification,” they wrote.
The authors received no financial support for the study, but several of the authors reported participating in meetings or receiving honoraria from several pharmaceutical companies.
Patients with multiple sclerosis treated with interferon-beta or glatiramer acetate who are aged 45 and over, and have no evidence of clinical disease activity for more than 4 years, have a high likelihood of remaining relapse free after stopping treatment, research showed.
The study, conducted by Gabriel Bsteh, MD, of the department of neurology at the Medical University of Innsbruck (Austria) and his colleagues, provides some evidence in the absence of randomized trials that may help guide discussions with patients – particularly those who have not had a relapse in a while – when they ask if they could discontinue disease-modifying treatment (DMT).
After a median follow-up period of 3.8 years, 98 patients (44.3%) had a relapse. Confirmed disability progression occurred in 46 patients (20.8%), and 15 patients (6.8%) converted to secondary progressive multiple sclerosis.
The independent predictors of absence of relapse after discontinuing treatment included age 45 years or older at discontinuation (HR = 0.47; confidence interval, 0.23–0.95; P = .038), absence of relapses for 4 or more years on DMT before discontinuation (HR = 0.29; CI, 0.10–0.82; P = .020), and absence of contrast-enhancing lesions (HR = 0.46; CI, 0.28–0.78; P =.004). A combination of age 45 years or older and absence of relapses after 4 or more years on DMT was associated with a very low risk of having a relapse after discontinuation, regardless of MRI results (HR = 0.06; CI, 0.01-0.44; P less than .001).
Higher Expanded Disability Status Scale (EDSS) scores at discontinuation, age older than 45 years at discontinuation, and longer disease duration were the only significant independent predictors of disability progression after discontinuation, irrespective of the presence of relapses on DMT or gadolinium-enhancing lesions.
“This underlines the concept of a window of opportunity in the treatment of MS, in the sense that once a certain extent of disability is reached, the impact of relapses, and therefore the effect of anti-inflammatory treatment, are drastically reduced,” the investigators wrote.
The study is limited because of its observational retrospective nature, but the research team said their results emphasized the importance of regular, thorough clinical evaluation of patients with RRMS. “While MRI may have a role in aiding decision making regarding DMT discontinuation, our data clearly show that demographic factors and clinical monitoring go a long way in risk stratification,” they wrote.
The authors received no financial support for the study, but several of the authors reported participating in meetings or receiving honoraria from several pharmaceutical companies.
Patients with multiple sclerosis treated with interferon-beta or glatiramer acetate who are aged 45 and over, and have no evidence of clinical disease activity for more than 4 years, have a high likelihood of remaining relapse free after stopping treatment, research showed.
The study, conducted by Gabriel Bsteh, MD, of the department of neurology at the Medical University of Innsbruck (Austria) and his colleagues, provides some evidence in the absence of randomized trials that may help guide discussions with patients – particularly those who have not had a relapse in a while – when they ask if they could discontinue disease-modifying treatment (DMT).
After a median follow-up period of 3.8 years, 98 patients (44.3%) had a relapse. Confirmed disability progression occurred in 46 patients (20.8%), and 15 patients (6.8%) converted to secondary progressive multiple sclerosis.
The independent predictors of absence of relapse after discontinuing treatment included age 45 years or older at discontinuation (HR = 0.47; confidence interval, 0.23–0.95; P = .038), absence of relapses for 4 or more years on DMT before discontinuation (HR = 0.29; CI, 0.10–0.82; P = .020), and absence of contrast-enhancing lesions (HR = 0.46; CI, 0.28–0.78; P =.004). A combination of age 45 years or older and absence of relapses after 4 or more years on DMT was associated with a very low risk of having a relapse after discontinuation, regardless of MRI results (HR = 0.06; CI, 0.01-0.44; P less than .001).
Higher Expanded Disability Status Scale (EDSS) scores at discontinuation, age older than 45 years at discontinuation, and longer disease duration were the only significant independent predictors of disability progression after discontinuation, irrespective of the presence of relapses on DMT or gadolinium-enhancing lesions.
“This underlines the concept of a window of opportunity in the treatment of MS, in the sense that once a certain extent of disability is reached, the impact of relapses, and therefore the effect of anti-inflammatory treatment, are drastically reduced,” the investigators wrote.
The study is limited because of its observational retrospective nature, but the research team said their results emphasized the importance of regular, thorough clinical evaluation of patients with RRMS. “While MRI may have a role in aiding decision making regarding DMT discontinuation, our data clearly show that demographic factors and clinical monitoring go a long way in risk stratification,” they wrote.
The authors received no financial support for the study, but several of the authors reported participating in meetings or receiving honoraria from several pharmaceutical companies.
FROM MULTIPLE SCLEROSIS JOURNAL
Key clinical point: Age and time since last relapse can predict relapsing-remitting multiple sclerosis patients with a lower risk of relapse after they discontinue interferon-beta or glatiramer acetate.
Main finding: A combination of age 45 years or older and absence of relapses after 4 or more years on disease-modifying treatment was associated with a very low risk of having a relapse after discontinuation, regardless of MRI results (HR = 0.06; CI, 0.01–0.44; P less than .001).
Data source: An observational retrospective analysis of 221 patients from the Innsbruck MS database with relapsing-remitting multiple sclerosis who discontinued disease-modifying treatment after more than 12 months and had documented follow-up at 2 years.
Disclosures: The authors received no financial support for the study, but several of the authors reported participating in meetings or receiving honoraria from several pharmaceutical companies.
Benefits and challenges of caring for international patients
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent years have seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
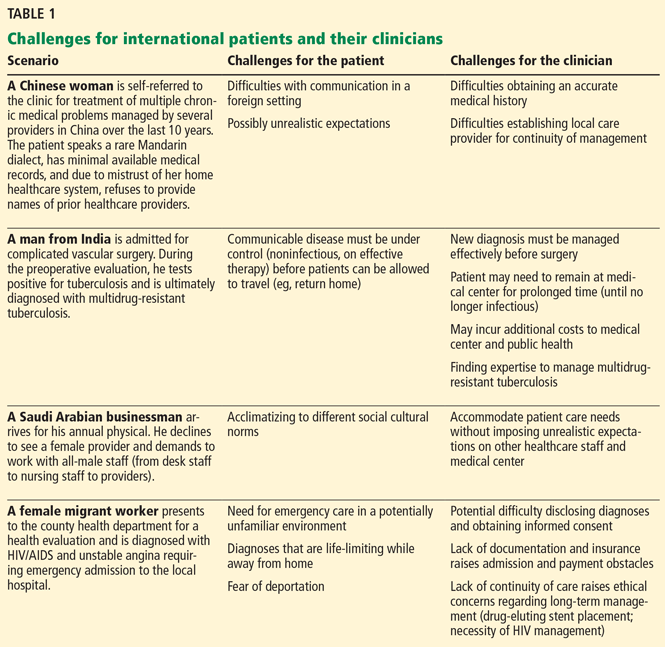
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitant costs of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.

Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement

Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
- William Osler. BrainyQuote.com, Xplore Inc, 2016. www.brainyquote.com/quotes/quotes/w/williamosl391388.html. Accessed September 21, 2016.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
- Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
- Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
- Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
- Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
- Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent years have seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitant costs of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.
Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement
Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent years have seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitant costs of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.
Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement
Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
- William Osler. BrainyQuote.com, Xplore Inc, 2016. www.brainyquote.com/quotes/quotes/w/williamosl391388.html. Accessed September 21, 2016.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
- Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
- Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
- Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
- Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
- Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
- William Osler. BrainyQuote.com, Xplore Inc, 2016. www.brainyquote.com/quotes/quotes/w/williamosl391388.html. Accessed September 21, 2016.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
- Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
- Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
- Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
- Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
- Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
KEY POINTS
- Challenges in caring for international patients include cultural differences, institutional barriers, communication difficulties, sparse medical records, and financial considerations.
- Understanding should be reached beforehand on potentially sensitive issues such as physical examinations, payment, tests, and treatment.
- Benefits to the provider and institution include enhanced medical skills, cultural competency, personal satisfaction, and institutional prestige.
