User login
Nivolumab + ipilimumab induced fulminant, fatal myocarditis
Two patients taking the immune checkpoint inhibitors nivolumab and ipilimumab for metastatic melanoma developed fulminant, fatal myocarditis, investigators reported in the New England Journal of Medicine.
Even though this adverse effect is rare, “clinicians should be vigilant for immune-mediated myocarditis, particularly because of its early onset, nonspecific symptomatology, and fulminant progression,” said Douglas B. Johnson, MD, of Vanderbilt University Medical Center, Nashville, and his associates.
The first case involved a 65-year-old woman with no cardiac risk factors who was admitted to the hospital with chest pain, dyspnea, and fatigue 12 days after she received her first dose of the combination therapy. She was found to have myocarditis and myositis with rhabdomylysis. Despite treatment with high-dose glucocorticoids, she developed intraventricular conduction delay within 24 hours, followed by complete heart block. She died from multisystem organ failure and refractory ventricular tachycardia.
The second case involved a 63-year-old man with no cardiac risk factors who was admitted with fatigue and myalgias 15 days after he received his first dose of the combination therapy. He showed profound ST-segment depression, an intraventricular conduction delay, myocarditis, and myositis. He also was treated with high-dose glucocorticoids but developed complete heart block and died from cardiac arrest.
Both patients had “strikingly elevated troponin levels and refractory conduction-system abnormalities with preserved cardiac function,” the investigators noted. Postmortem assessments showed intense lymphocytic infiltrates only in striated cardiac and skeletal muscle and in metastases; adjacent smooth muscle and other tissues were unaffected. Pathology results “were reminiscent of those observed in patients with acute allograft rejection after cardiac transplantation,” Dr. Johnson and his associates said (N Engl J Med. 2016 Nov 3. doi: 10.1056/NEJMoa1609214).
To assess the frequency of myocarditis and myositis in patients receiving immune checkpoint inhibitors for many different cancers, the investigators searched Bristol-Myers Squibb safety databases. They found 18 drug-related cases of severe myocarditis among 20,594 patients, for a frequency of 0.09%. Patients who received combined nivolumab and ipilimumab had more frequent and more severe myocarditis than those who took either agent alone.
“There are no known data regarding what monitoring strategy may be of value; in our practice, we are performing baseline ECG and weekly testing of troponin levels during weeks 1-3 for patients receiving combination immunotherapy,” the researchers noted.
This work was supported by the Bready Family Foundation, the National Cancer Institute, Vanderbilt-Ingram Cancer Center Ambassadors, the Breast Cancer Specialized Program of Research Excellence, the National Comprehensive Cancer Network, the National Institutes of Health, the Howard Hughes Medical Institute, and Gilead Life Sciences. Dr. Johnson reported receiving personal fees from Genoptix and Bristol-Myers Squibb, and his associates reported ties to numerous industry sources.
Two patients taking the immune checkpoint inhibitors nivolumab and ipilimumab for metastatic melanoma developed fulminant, fatal myocarditis, investigators reported in the New England Journal of Medicine.
Even though this adverse effect is rare, “clinicians should be vigilant for immune-mediated myocarditis, particularly because of its early onset, nonspecific symptomatology, and fulminant progression,” said Douglas B. Johnson, MD, of Vanderbilt University Medical Center, Nashville, and his associates.
The first case involved a 65-year-old woman with no cardiac risk factors who was admitted to the hospital with chest pain, dyspnea, and fatigue 12 days after she received her first dose of the combination therapy. She was found to have myocarditis and myositis with rhabdomylysis. Despite treatment with high-dose glucocorticoids, she developed intraventricular conduction delay within 24 hours, followed by complete heart block. She died from multisystem organ failure and refractory ventricular tachycardia.
The second case involved a 63-year-old man with no cardiac risk factors who was admitted with fatigue and myalgias 15 days after he received his first dose of the combination therapy. He showed profound ST-segment depression, an intraventricular conduction delay, myocarditis, and myositis. He also was treated with high-dose glucocorticoids but developed complete heart block and died from cardiac arrest.
Both patients had “strikingly elevated troponin levels and refractory conduction-system abnormalities with preserved cardiac function,” the investigators noted. Postmortem assessments showed intense lymphocytic infiltrates only in striated cardiac and skeletal muscle and in metastases; adjacent smooth muscle and other tissues were unaffected. Pathology results “were reminiscent of those observed in patients with acute allograft rejection after cardiac transplantation,” Dr. Johnson and his associates said (N Engl J Med. 2016 Nov 3. doi: 10.1056/NEJMoa1609214).
To assess the frequency of myocarditis and myositis in patients receiving immune checkpoint inhibitors for many different cancers, the investigators searched Bristol-Myers Squibb safety databases. They found 18 drug-related cases of severe myocarditis among 20,594 patients, for a frequency of 0.09%. Patients who received combined nivolumab and ipilimumab had more frequent and more severe myocarditis than those who took either agent alone.
“There are no known data regarding what monitoring strategy may be of value; in our practice, we are performing baseline ECG and weekly testing of troponin levels during weeks 1-3 for patients receiving combination immunotherapy,” the researchers noted.
This work was supported by the Bready Family Foundation, the National Cancer Institute, Vanderbilt-Ingram Cancer Center Ambassadors, the Breast Cancer Specialized Program of Research Excellence, the National Comprehensive Cancer Network, the National Institutes of Health, the Howard Hughes Medical Institute, and Gilead Life Sciences. Dr. Johnson reported receiving personal fees from Genoptix and Bristol-Myers Squibb, and his associates reported ties to numerous industry sources.
Two patients taking the immune checkpoint inhibitors nivolumab and ipilimumab for metastatic melanoma developed fulminant, fatal myocarditis, investigators reported in the New England Journal of Medicine.
Even though this adverse effect is rare, “clinicians should be vigilant for immune-mediated myocarditis, particularly because of its early onset, nonspecific symptomatology, and fulminant progression,” said Douglas B. Johnson, MD, of Vanderbilt University Medical Center, Nashville, and his associates.
The first case involved a 65-year-old woman with no cardiac risk factors who was admitted to the hospital with chest pain, dyspnea, and fatigue 12 days after she received her first dose of the combination therapy. She was found to have myocarditis and myositis with rhabdomylysis. Despite treatment with high-dose glucocorticoids, she developed intraventricular conduction delay within 24 hours, followed by complete heart block. She died from multisystem organ failure and refractory ventricular tachycardia.
The second case involved a 63-year-old man with no cardiac risk factors who was admitted with fatigue and myalgias 15 days after he received his first dose of the combination therapy. He showed profound ST-segment depression, an intraventricular conduction delay, myocarditis, and myositis. He also was treated with high-dose glucocorticoids but developed complete heart block and died from cardiac arrest.
Both patients had “strikingly elevated troponin levels and refractory conduction-system abnormalities with preserved cardiac function,” the investigators noted. Postmortem assessments showed intense lymphocytic infiltrates only in striated cardiac and skeletal muscle and in metastases; adjacent smooth muscle and other tissues were unaffected. Pathology results “were reminiscent of those observed in patients with acute allograft rejection after cardiac transplantation,” Dr. Johnson and his associates said (N Engl J Med. 2016 Nov 3. doi: 10.1056/NEJMoa1609214).
To assess the frequency of myocarditis and myositis in patients receiving immune checkpoint inhibitors for many different cancers, the investigators searched Bristol-Myers Squibb safety databases. They found 18 drug-related cases of severe myocarditis among 20,594 patients, for a frequency of 0.09%. Patients who received combined nivolumab and ipilimumab had more frequent and more severe myocarditis than those who took either agent alone.
“There are no known data regarding what monitoring strategy may be of value; in our practice, we are performing baseline ECG and weekly testing of troponin levels during weeks 1-3 for patients receiving combination immunotherapy,” the researchers noted.
This work was supported by the Bready Family Foundation, the National Cancer Institute, Vanderbilt-Ingram Cancer Center Ambassadors, the Breast Cancer Specialized Program of Research Excellence, the National Comprehensive Cancer Network, the National Institutes of Health, the Howard Hughes Medical Institute, and Gilead Life Sciences. Dr. Johnson reported receiving personal fees from Genoptix and Bristol-Myers Squibb, and his associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Two patients taking the immune checkpoint inhibitors nivolumab and ipilimumab for metastatic melanoma developed fulminant, fatal myocarditis.
Major finding: A search of Bristol-Myers Squibb safety databases found 18 drug-related cases of severe myocarditis among 20,594 patients, for a frequency of 0.09%.
Data source: Two case reports of a rare adverse effect of treatment with immune checkpoint inhibitors.
Disclosures: This work was supported by the Bready Family Foundation, the National Cancer Institute, Vanderbilt-Ingram Cancer Center Ambassadors, the Breast Cancer Specialized Program of Research Excellence, the National Comprehensive Cancer Network, the National Institutes of Health, the Howard Hughes Medical Institute, and Gilead Life Sciences. Dr. Johnson reported receiving personal fees from Genoptix and Bristol-Myers Squibb, and his associates reported ties to numerous industry sources.
Discussing screen time with parents
The American Academy of Pediatrics released a new set of recommendations for the appropriate amount of screen time for children and adolescents in October 2016.
Among other changes, the AAP now recommends no screen time (except for video chatting) for infants and children up to 18 months old. For 18- to 24-month-olds, the AAP discourages screen time, recommending that parents introduce only selected “high-quality” programming and cowatch with their children. Likewise, for children up to 5 years old, the AAP urges parents to limit all screen time to 1 hour/day, half of its previous recommendation, and again recommends that parents cowatch with their children and use only reliable providers of quality content, such as the Public Broadcasting Service (PBS). For older children, the AAP does not set specific time limits, but recommends that parents collaborate with the children on a media plan that limits screen time so that it does not interfere with other important activities, including homework, social time, exercise, and sleep.



There also is evidence that teenagers who spend substantial time engaged passively in social media (seeing what others are doing or saying via Facebook or Instagram) report higher levels of depression and anxiety, whereas those who use social media as a platform to stay connected (via two-way communication) report lower levels of these symptoms. While many young people use social sites as a forum to find peer support, share concerns, or develop their own “voice,” some young people might be vulnerable to exploitation, cyberbullying, or even online solicitation. The key here may be for parents, who have a sense of their child’s strengths and vulnerabilities, to be aware of where their children are spending their virtual time and to check in about the kinds of connections they have there. Of course, screen time can be equally seductive for parents. And when a parent is spending time reading texts or checking for Facebook updates, they are missing opportunities to be engaged with their children, helping them with homework or simply noticing that they seem stressed, or catching an opportunity to talk with them.
The pediatrician has the opportunity to educate parents about the potential risks that unchecked screen time can pose to their children’s healthy development. But it is critical that you approach these conversations with specificity and compassion. Customize the conversation to the age and personality of the child and family. A computer in the bedroom may make sense for an academically oriented 9th grader in a demanding school who is generally well-balanced in activities and friendships. A bedroom computer may be a poor choice for an isolated 9th grader almost addicted to video games with few friends or activities.
Simply reciting recommendations may heighten a parent’s feelings of isolation and shame, and not lead to meaningful change. Instead, start by asking about the details: Where are the screens in the home? Bedrooms? Who has a computer, tablet, or smartphone? How are these screens used in the context of the child’s overall psychosocial functioning? Depending on the circumstance, a smaller change, such as “no phones while doing homework,” can make a big difference. Simple, clear rules can be easier to explain and enforce, and protect parents from the perils of daily negotiations of screen terms with their children or teenagers. Perhaps they can have a “phone zone” where phones get parked and charged once kids get home from school. Perhaps there are limits on TV or video games on school nights (for the student performing below potential, rather than the driven student who would benefit from down time). Perhaps for preteens, computer-based homework can be done only on the desktop computer that is kept in a family study, rather than a laptop in a bedroom where kids are more likely to become distracted and surf the net. Pediatricians can help families think through the right approach to screen time that may range from restriction to shared use exploring shared interests to jointly watching a favorite TV show or sporting event.
You can help parents consider how they will talk about all this, acknowledging what is fun and rewarding about TV shows, social media, and the Internet alongside the problems of excessive use. Ask parents if it is hard for them to put down their own phones or tablets. They can acknowledge this explicitly with their children when establishing new media use rules. It is powerful for children, especially teenagers, to hear their parents acknowledge that “phones, tablets, and computers are powerful tools, but we all need to improve our skills at being in control of our use of them.” You might suggest that parents try this exercise: list all of the activities they wish they had time for in every day, and how much time they would spend in them. Then they should guess how much time they spend in screen-based entertainment. If they wish to protect time for screen-based entertainment, they can actively choose to do so. If you are able to help parents better understand the risks of excessive screen time and facilitate desired and appropriate use of media, you will have added to the quality of the family’s life.
The AAP has resources to help pediatricians partner with parents to create a Family Media Use Plan (www.healthychildren.org/MediaUsePlan).
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton Wellesley Hospital, also in Boston. Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email them at [email protected].
The American Academy of Pediatrics released a new set of recommendations for the appropriate amount of screen time for children and adolescents in October 2016.
Among other changes, the AAP now recommends no screen time (except for video chatting) for infants and children up to 18 months old. For 18- to 24-month-olds, the AAP discourages screen time, recommending that parents introduce only selected “high-quality” programming and cowatch with their children. Likewise, for children up to 5 years old, the AAP urges parents to limit all screen time to 1 hour/day, half of its previous recommendation, and again recommends that parents cowatch with their children and use only reliable providers of quality content, such as the Public Broadcasting Service (PBS). For older children, the AAP does not set specific time limits, but recommends that parents collaborate with the children on a media plan that limits screen time so that it does not interfere with other important activities, including homework, social time, exercise, and sleep.
There also is evidence that teenagers who spend substantial time engaged passively in social media (seeing what others are doing or saying via Facebook or Instagram) report higher levels of depression and anxiety, whereas those who use social media as a platform to stay connected (via two-way communication) report lower levels of these symptoms. While many young people use social sites as a forum to find peer support, share concerns, or develop their own “voice,” some young people might be vulnerable to exploitation, cyberbullying, or even online solicitation. The key here may be for parents, who have a sense of their child’s strengths and vulnerabilities, to be aware of where their children are spending their virtual time and to check in about the kinds of connections they have there. Of course, screen time can be equally seductive for parents. And when a parent is spending time reading texts or checking for Facebook updates, they are missing opportunities to be engaged with their children, helping them with homework or simply noticing that they seem stressed, or catching an opportunity to talk with them.
The pediatrician has the opportunity to educate parents about the potential risks that unchecked screen time can pose to their children’s healthy development. But it is critical that you approach these conversations with specificity and compassion. Customize the conversation to the age and personality of the child and family. A computer in the bedroom may make sense for an academically oriented 9th grader in a demanding school who is generally well-balanced in activities and friendships. A bedroom computer may be a poor choice for an isolated 9th grader almost addicted to video games with few friends or activities.
Simply reciting recommendations may heighten a parent’s feelings of isolation and shame, and not lead to meaningful change. Instead, start by asking about the details: Where are the screens in the home? Bedrooms? Who has a computer, tablet, or smartphone? How are these screens used in the context of the child’s overall psychosocial functioning? Depending on the circumstance, a smaller change, such as “no phones while doing homework,” can make a big difference. Simple, clear rules can be easier to explain and enforce, and protect parents from the perils of daily negotiations of screen terms with their children or teenagers. Perhaps they can have a “phone zone” where phones get parked and charged once kids get home from school. Perhaps there are limits on TV or video games on school nights (for the student performing below potential, rather than the driven student who would benefit from down time). Perhaps for preteens, computer-based homework can be done only on the desktop computer that is kept in a family study, rather than a laptop in a bedroom where kids are more likely to become distracted and surf the net. Pediatricians can help families think through the right approach to screen time that may range from restriction to shared use exploring shared interests to jointly watching a favorite TV show or sporting event.
You can help parents consider how they will talk about all this, acknowledging what is fun and rewarding about TV shows, social media, and the Internet alongside the problems of excessive use. Ask parents if it is hard for them to put down their own phones or tablets. They can acknowledge this explicitly with their children when establishing new media use rules. It is powerful for children, especially teenagers, to hear their parents acknowledge that “phones, tablets, and computers are powerful tools, but we all need to improve our skills at being in control of our use of them.” You might suggest that parents try this exercise: list all of the activities they wish they had time for in every day, and how much time they would spend in them. Then they should guess how much time they spend in screen-based entertainment. If they wish to protect time for screen-based entertainment, they can actively choose to do so. If you are able to help parents better understand the risks of excessive screen time and facilitate desired and appropriate use of media, you will have added to the quality of the family’s life.
The AAP has resources to help pediatricians partner with parents to create a Family Media Use Plan (www.healthychildren.org/MediaUsePlan).
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton Wellesley Hospital, also in Boston. Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email them at [email protected].
The American Academy of Pediatrics released a new set of recommendations for the appropriate amount of screen time for children and adolescents in October 2016.
Among other changes, the AAP now recommends no screen time (except for video chatting) for infants and children up to 18 months old. For 18- to 24-month-olds, the AAP discourages screen time, recommending that parents introduce only selected “high-quality” programming and cowatch with their children. Likewise, for children up to 5 years old, the AAP urges parents to limit all screen time to 1 hour/day, half of its previous recommendation, and again recommends that parents cowatch with their children and use only reliable providers of quality content, such as the Public Broadcasting Service (PBS). For older children, the AAP does not set specific time limits, but recommends that parents collaborate with the children on a media plan that limits screen time so that it does not interfere with other important activities, including homework, social time, exercise, and sleep.
There also is evidence that teenagers who spend substantial time engaged passively in social media (seeing what others are doing or saying via Facebook or Instagram) report higher levels of depression and anxiety, whereas those who use social media as a platform to stay connected (via two-way communication) report lower levels of these symptoms. While many young people use social sites as a forum to find peer support, share concerns, or develop their own “voice,” some young people might be vulnerable to exploitation, cyberbullying, or even online solicitation. The key here may be for parents, who have a sense of their child’s strengths and vulnerabilities, to be aware of where their children are spending their virtual time and to check in about the kinds of connections they have there. Of course, screen time can be equally seductive for parents. And when a parent is spending time reading texts or checking for Facebook updates, they are missing opportunities to be engaged with their children, helping them with homework or simply noticing that they seem stressed, or catching an opportunity to talk with them.
The pediatrician has the opportunity to educate parents about the potential risks that unchecked screen time can pose to their children’s healthy development. But it is critical that you approach these conversations with specificity and compassion. Customize the conversation to the age and personality of the child and family. A computer in the bedroom may make sense for an academically oriented 9th grader in a demanding school who is generally well-balanced in activities and friendships. A bedroom computer may be a poor choice for an isolated 9th grader almost addicted to video games with few friends or activities.
Simply reciting recommendations may heighten a parent’s feelings of isolation and shame, and not lead to meaningful change. Instead, start by asking about the details: Where are the screens in the home? Bedrooms? Who has a computer, tablet, or smartphone? How are these screens used in the context of the child’s overall psychosocial functioning? Depending on the circumstance, a smaller change, such as “no phones while doing homework,” can make a big difference. Simple, clear rules can be easier to explain and enforce, and protect parents from the perils of daily negotiations of screen terms with their children or teenagers. Perhaps they can have a “phone zone” where phones get parked and charged once kids get home from school. Perhaps there are limits on TV or video games on school nights (for the student performing below potential, rather than the driven student who would benefit from down time). Perhaps for preteens, computer-based homework can be done only on the desktop computer that is kept in a family study, rather than a laptop in a bedroom where kids are more likely to become distracted and surf the net. Pediatricians can help families think through the right approach to screen time that may range from restriction to shared use exploring shared interests to jointly watching a favorite TV show or sporting event.
You can help parents consider how they will talk about all this, acknowledging what is fun and rewarding about TV shows, social media, and the Internet alongside the problems of excessive use. Ask parents if it is hard for them to put down their own phones or tablets. They can acknowledge this explicitly with their children when establishing new media use rules. It is powerful for children, especially teenagers, to hear their parents acknowledge that “phones, tablets, and computers are powerful tools, but we all need to improve our skills at being in control of our use of them.” You might suggest that parents try this exercise: list all of the activities they wish they had time for in every day, and how much time they would spend in them. Then they should guess how much time they spend in screen-based entertainment. If they wish to protect time for screen-based entertainment, they can actively choose to do so. If you are able to help parents better understand the risks of excessive screen time and facilitate desired and appropriate use of media, you will have added to the quality of the family’s life.
The AAP has resources to help pediatricians partner with parents to create a Family Media Use Plan (www.healthychildren.org/MediaUsePlan).
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton Wellesley Hospital, also in Boston. Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email them at [email protected].
Cosmetic Corner: Dermatologists Weigh in on OTC Rosacea Treatments
To improve patient care and outcomes, leading dermatologists offered their recommendations on OTC rosacea treatments. Consideration must be given to:
- Eucerin Redness Relief
Beiersdorf Inc
Recommended by Gary Goldenberg, MD, New York, New York
- Rosaliac CC Cream
La Roche-Posay Laboratoire Dermatologique
“This product provides hydration and an even tint to correct and reduce the erythema of rosacea. It is made with both titanium dioxide and organic UV filters to give broad-spectrum coverage with SPF 30.”—Cherise Mizrahi-Levi, DO, New York, New York
- Vanicream
Pharmaceutical Specialties, Inc.
“I like Vanicream products since they are mild and hypoallergenic.”—Gary Goldenberg, MD, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, skin-lightening agents, and self-tanners will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on OTC rosacea treatments. Consideration must be given to:
- Eucerin Redness Relief
Beiersdorf Inc
Recommended by Gary Goldenberg, MD, New York, New York
- Rosaliac CC Cream
La Roche-Posay Laboratoire Dermatologique
“This product provides hydration and an even tint to correct and reduce the erythema of rosacea. It is made with both titanium dioxide and organic UV filters to give broad-spectrum coverage with SPF 30.”—Cherise Mizrahi-Levi, DO, New York, New York
- Vanicream
Pharmaceutical Specialties, Inc.
“I like Vanicream products since they are mild and hypoallergenic.”—Gary Goldenberg, MD, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, skin-lightening agents, and self-tanners will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on OTC rosacea treatments. Consideration must be given to:
- Eucerin Redness Relief
Beiersdorf Inc
Recommended by Gary Goldenberg, MD, New York, New York
- Rosaliac CC Cream
La Roche-Posay Laboratoire Dermatologique
“This product provides hydration and an even tint to correct and reduce the erythema of rosacea. It is made with both titanium dioxide and organic UV filters to give broad-spectrum coverage with SPF 30.”—Cherise Mizrahi-Levi, DO, New York, New York
- Vanicream
Pharmaceutical Specialties, Inc.
“I like Vanicream products since they are mild and hypoallergenic.”—Gary Goldenberg, MD, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, skin-lightening agents, and self-tanners will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Discharging Nodule on the Jaw
The Diagnosis: Dental Sinus Secondary to Osteonecrosis of the Jaw
Cone beam computed tomography revealed an area of lucency measuring 40×20 mm in the body of the right mandible (Figure). The patient subsequently underwent curettage of the wound with sequestrectomy of the involved area.
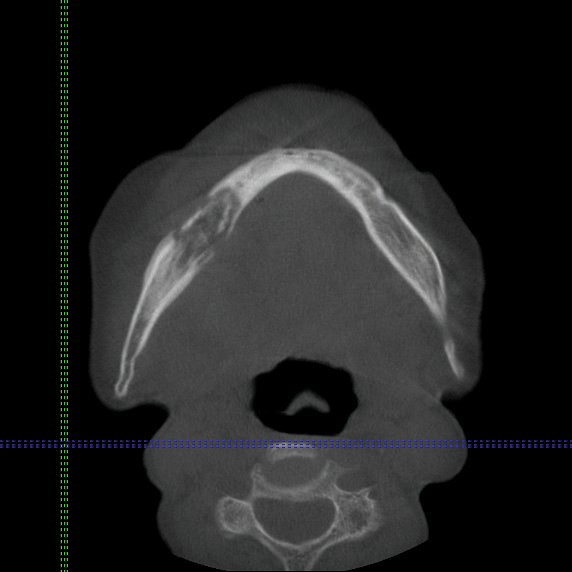
Osteonecrosis of the jaw is a form of avascular necrosis. It is an uncommon but potentially serious side effect of bisphosphonate use.1 Bisphosphonates commonly are used as first-line therapy for osteoporosis, with proven efficacy to reduce fracture risk by exerting an antiresorptive effect on bones.2 Bisphosphonate-related osteonecrosis of the jaw (BRONJ) is defined by the American Association of Oral and Maxillofacial Surgeons as the presence of exposed necrotic bone in the maxillofacial region that has persisted for more than 8 weeks, with current or prior treatment with a bisphosphonate and absence of prior radiation therapy to the jaw.3 Bisphosphonate-related osteonecrosis of the jaw can be associated with infections, pathologic fractures, extraoral fistulae, or osteolysis extending to the inferior border.
Our patient had a dental sinus that resulted from the underlying BRONJ. The jawbones, unlike the long bones, are in a special environment in that both acute and chronic infections occur often within the bone, and surgical procedures as well as masticatory trauma expose the bone to a bacteria-laden environment.4 Infection around the root apex of a tooth results in a dental abscess and a sinus tract can develop from the abscess, draining either intraorally or extraorally.5 Facial sinus tracts can be either odontogenic or nonodontogenic, and sometimes the lesions of dental origin may be confused with dermatological lesions.
Bisphosphonates inhibit osteoclasts, which are responsible for bone resorption. Antiangiogenetic effects also have been reported in bisphosphonates, resulting in devitalized bone.6 The potent and prolonged inhibition of bone remodeling likely plays an important role in BRONJ. The more frequently occurring microdamage inflicted on the lower jawbone with mastication also may represent a contributory factor.7
Bisphosphonate-related osteonecrosis of the jaw more often is associated with the use of high-dose intravenous (IV) bisphosphonate in cancer-related hypercalcemia and less so with oral bisphosphonates, which are generally used to treat osteoporosis.3 In a Swedish study conducted from 2003 to 2010, 55 cases of BRONJ were documented in a population of 1.2 million individuals. The prevalence of BRONJ in patients on oral bisphosphonates and IV bisphosphonates was estimated to be 0.024% and 2.8%, respectively.8
Bisphosphonates are widely used worldwide as the main treatment of osteoporosis. The association between osteonecrosis of the jaw and oral bisphosphonates is contentious among the osteoporosis population, as most studies focus on IV bisphosphonate use in cancer patients.9 Bisphosphonate-related osteonecrosis of the jaw adversely affects the patient's quality of life, producing notable morbidity in afflicted patients. Thus, a complete dental assessment and treatment is recommended before the initiation of bisphosphonate treatment. The risk for developing BRONJ associated with oral bisphosphonates increases when the duration of therapy exceeds 3 years.3 It has been reported that antifracture efficacy would persist for 1 to 2 years following discontinuation of alendronate or risedronate that had been taken for 3 to 5 years, but patients with low bone mineral density at the femoral neck (T-score below -2.5) after 3 to 5 years of treatment of bisphosphonates are at the highest risk for vertebral fractures and therefore appear to benefit most from continuation of therapy.10 For dental procedures, the American Association of Oral and Maxillofacial Surgeons suggests that if systemic conditions persist, the clinician might consider discontinuation of oral bisphosphonates for a 3-month period before and after elective invasive dental surgery to lower the risk for BRONJ.3 When possible, invasive dentoalveolar procedures such as extractions should be avoided; conservative endodontic treatment is preferable.
Bisphosphonate-related osteonecrosis of the jaw is a devastating condition that is difficult to treat and manage, thus the focus should be on prevention through dental clearance prior to starting bisphosphonates. It also is crucial to have a high index of suspicion for BRONJ in patients presenting with orofacial lesions so that they can be treated expediently.
- Edwards BJ, Gounder M, McKoy JM, et al. Pharmacovigilance and reporting oversight in US FDA fast-track process: bisphosphonates and osteonecrosis of the jaw. Lancet Oncol. 2008;9:1166-1172.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med. 2013;126:13-20.
- Ruggiero SL, Dodson TB, Assael LA, et al; American Association of Oral and Maxillofacial Surgeons. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws--2009 update. J Oral Maxillofac Surg. 2009;67(5 suppl):2-12.
- Mawardi H, Treister N, Richardson P, et al. Sinus tracts--an early sign of bisphosphonate-associated osteonecrosis of the jaws? J Oral Maxillofac Surg. 2009;67:593-601.
- Sammut S, Malden N, Lopes V. Facial cutaneous sinuses of dental origin--a diagnostic challenge. Br Dent J. 2013;215:555-558.
- Wood J, Bonjean K, Ruetz S, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055-1061.
- Hoefert S, Schmitz I, Tannapfel A, et al. Importance of microcracks in etiology of bisphosphonate-related osteonecrosis of the jaw: a possible pathogenetic model of symptomatic and non-symptomatic osteonecrosis of the jaw based on scanning electron microscopy findings. Clin Oral Investig. 2010;14:271-284.
- Hallmer F, Bjørnland T, Nicklasson A, et al. Osteonecrosis of the jaw in patients treated with oral and intravenous bisphosphonates: experience in Sweden. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:202-208.
- Lin TC, Yang CY, Kao Yang YH, et al. Incidence and risk of osteonecrosis of the jaw among the Taiwan osteoporosis population [published online February 11, 2014]. Osteoporos Int. 2014;25:1503-1511.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab. 2010;95:1555-1565.
The Diagnosis: Dental Sinus Secondary to Osteonecrosis of the Jaw
Cone beam computed tomography revealed an area of lucency measuring 40×20 mm in the body of the right mandible (Figure). The patient subsequently underwent curettage of the wound with sequestrectomy of the involved area.
Osteonecrosis of the jaw is a form of avascular necrosis. It is an uncommon but potentially serious side effect of bisphosphonate use.1 Bisphosphonates commonly are used as first-line therapy for osteoporosis, with proven efficacy to reduce fracture risk by exerting an antiresorptive effect on bones.2 Bisphosphonate-related osteonecrosis of the jaw (BRONJ) is defined by the American Association of Oral and Maxillofacial Surgeons as the presence of exposed necrotic bone in the maxillofacial region that has persisted for more than 8 weeks, with current or prior treatment with a bisphosphonate and absence of prior radiation therapy to the jaw.3 Bisphosphonate-related osteonecrosis of the jaw can be associated with infections, pathologic fractures, extraoral fistulae, or osteolysis extending to the inferior border.
Our patient had a dental sinus that resulted from the underlying BRONJ. The jawbones, unlike the long bones, are in a special environment in that both acute and chronic infections occur often within the bone, and surgical procedures as well as masticatory trauma expose the bone to a bacteria-laden environment.4 Infection around the root apex of a tooth results in a dental abscess and a sinus tract can develop from the abscess, draining either intraorally or extraorally.5 Facial sinus tracts can be either odontogenic or nonodontogenic, and sometimes the lesions of dental origin may be confused with dermatological lesions.
Bisphosphonates inhibit osteoclasts, which are responsible for bone resorption. Antiangiogenetic effects also have been reported in bisphosphonates, resulting in devitalized bone.6 The potent and prolonged inhibition of bone remodeling likely plays an important role in BRONJ. The more frequently occurring microdamage inflicted on the lower jawbone with mastication also may represent a contributory factor.7
Bisphosphonate-related osteonecrosis of the jaw more often is associated with the use of high-dose intravenous (IV) bisphosphonate in cancer-related hypercalcemia and less so with oral bisphosphonates, which are generally used to treat osteoporosis.3 In a Swedish study conducted from 2003 to 2010, 55 cases of BRONJ were documented in a population of 1.2 million individuals. The prevalence of BRONJ in patients on oral bisphosphonates and IV bisphosphonates was estimated to be 0.024% and 2.8%, respectively.8
Bisphosphonates are widely used worldwide as the main treatment of osteoporosis. The association between osteonecrosis of the jaw and oral bisphosphonates is contentious among the osteoporosis population, as most studies focus on IV bisphosphonate use in cancer patients.9 Bisphosphonate-related osteonecrosis of the jaw adversely affects the patient's quality of life, producing notable morbidity in afflicted patients. Thus, a complete dental assessment and treatment is recommended before the initiation of bisphosphonate treatment. The risk for developing BRONJ associated with oral bisphosphonates increases when the duration of therapy exceeds 3 years.3 It has been reported that antifracture efficacy would persist for 1 to 2 years following discontinuation of alendronate or risedronate that had been taken for 3 to 5 years, but patients with low bone mineral density at the femoral neck (T-score below -2.5) after 3 to 5 years of treatment of bisphosphonates are at the highest risk for vertebral fractures and therefore appear to benefit most from continuation of therapy.10 For dental procedures, the American Association of Oral and Maxillofacial Surgeons suggests that if systemic conditions persist, the clinician might consider discontinuation of oral bisphosphonates for a 3-month period before and after elective invasive dental surgery to lower the risk for BRONJ.3 When possible, invasive dentoalveolar procedures such as extractions should be avoided; conservative endodontic treatment is preferable.
Bisphosphonate-related osteonecrosis of the jaw is a devastating condition that is difficult to treat and manage, thus the focus should be on prevention through dental clearance prior to starting bisphosphonates. It also is crucial to have a high index of suspicion for BRONJ in patients presenting with orofacial lesions so that they can be treated expediently.
The Diagnosis: Dental Sinus Secondary to Osteonecrosis of the Jaw
Cone beam computed tomography revealed an area of lucency measuring 40×20 mm in the body of the right mandible (Figure). The patient subsequently underwent curettage of the wound with sequestrectomy of the involved area.
Osteonecrosis of the jaw is a form of avascular necrosis. It is an uncommon but potentially serious side effect of bisphosphonate use.1 Bisphosphonates commonly are used as first-line therapy for osteoporosis, with proven efficacy to reduce fracture risk by exerting an antiresorptive effect on bones.2 Bisphosphonate-related osteonecrosis of the jaw (BRONJ) is defined by the American Association of Oral and Maxillofacial Surgeons as the presence of exposed necrotic bone in the maxillofacial region that has persisted for more than 8 weeks, with current or prior treatment with a bisphosphonate and absence of prior radiation therapy to the jaw.3 Bisphosphonate-related osteonecrosis of the jaw can be associated with infections, pathologic fractures, extraoral fistulae, or osteolysis extending to the inferior border.
Our patient had a dental sinus that resulted from the underlying BRONJ. The jawbones, unlike the long bones, are in a special environment in that both acute and chronic infections occur often within the bone, and surgical procedures as well as masticatory trauma expose the bone to a bacteria-laden environment.4 Infection around the root apex of a tooth results in a dental abscess and a sinus tract can develop from the abscess, draining either intraorally or extraorally.5 Facial sinus tracts can be either odontogenic or nonodontogenic, and sometimes the lesions of dental origin may be confused with dermatological lesions.
Bisphosphonates inhibit osteoclasts, which are responsible for bone resorption. Antiangiogenetic effects also have been reported in bisphosphonates, resulting in devitalized bone.6 The potent and prolonged inhibition of bone remodeling likely plays an important role in BRONJ. The more frequently occurring microdamage inflicted on the lower jawbone with mastication also may represent a contributory factor.7
Bisphosphonate-related osteonecrosis of the jaw more often is associated with the use of high-dose intravenous (IV) bisphosphonate in cancer-related hypercalcemia and less so with oral bisphosphonates, which are generally used to treat osteoporosis.3 In a Swedish study conducted from 2003 to 2010, 55 cases of BRONJ were documented in a population of 1.2 million individuals. The prevalence of BRONJ in patients on oral bisphosphonates and IV bisphosphonates was estimated to be 0.024% and 2.8%, respectively.8
Bisphosphonates are widely used worldwide as the main treatment of osteoporosis. The association between osteonecrosis of the jaw and oral bisphosphonates is contentious among the osteoporosis population, as most studies focus on IV bisphosphonate use in cancer patients.9 Bisphosphonate-related osteonecrosis of the jaw adversely affects the patient's quality of life, producing notable morbidity in afflicted patients. Thus, a complete dental assessment and treatment is recommended before the initiation of bisphosphonate treatment. The risk for developing BRONJ associated with oral bisphosphonates increases when the duration of therapy exceeds 3 years.3 It has been reported that antifracture efficacy would persist for 1 to 2 years following discontinuation of alendronate or risedronate that had been taken for 3 to 5 years, but patients with low bone mineral density at the femoral neck (T-score below -2.5) after 3 to 5 years of treatment of bisphosphonates are at the highest risk for vertebral fractures and therefore appear to benefit most from continuation of therapy.10 For dental procedures, the American Association of Oral and Maxillofacial Surgeons suggests that if systemic conditions persist, the clinician might consider discontinuation of oral bisphosphonates for a 3-month period before and after elective invasive dental surgery to lower the risk for BRONJ.3 When possible, invasive dentoalveolar procedures such as extractions should be avoided; conservative endodontic treatment is preferable.
Bisphosphonate-related osteonecrosis of the jaw is a devastating condition that is difficult to treat and manage, thus the focus should be on prevention through dental clearance prior to starting bisphosphonates. It also is crucial to have a high index of suspicion for BRONJ in patients presenting with orofacial lesions so that they can be treated expediently.
- Edwards BJ, Gounder M, McKoy JM, et al. Pharmacovigilance and reporting oversight in US FDA fast-track process: bisphosphonates and osteonecrosis of the jaw. Lancet Oncol. 2008;9:1166-1172.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med. 2013;126:13-20.
- Ruggiero SL, Dodson TB, Assael LA, et al; American Association of Oral and Maxillofacial Surgeons. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws--2009 update. J Oral Maxillofac Surg. 2009;67(5 suppl):2-12.
- Mawardi H, Treister N, Richardson P, et al. Sinus tracts--an early sign of bisphosphonate-associated osteonecrosis of the jaws? J Oral Maxillofac Surg. 2009;67:593-601.
- Sammut S, Malden N, Lopes V. Facial cutaneous sinuses of dental origin--a diagnostic challenge. Br Dent J. 2013;215:555-558.
- Wood J, Bonjean K, Ruetz S, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055-1061.
- Hoefert S, Schmitz I, Tannapfel A, et al. Importance of microcracks in etiology of bisphosphonate-related osteonecrosis of the jaw: a possible pathogenetic model of symptomatic and non-symptomatic osteonecrosis of the jaw based on scanning electron microscopy findings. Clin Oral Investig. 2010;14:271-284.
- Hallmer F, Bjørnland T, Nicklasson A, et al. Osteonecrosis of the jaw in patients treated with oral and intravenous bisphosphonates: experience in Sweden. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:202-208.
- Lin TC, Yang CY, Kao Yang YH, et al. Incidence and risk of osteonecrosis of the jaw among the Taiwan osteoporosis population [published online February 11, 2014]. Osteoporos Int. 2014;25:1503-1511.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab. 2010;95:1555-1565.
- Edwards BJ, Gounder M, McKoy JM, et al. Pharmacovigilance and reporting oversight in US FDA fast-track process: bisphosphonates and osteonecrosis of the jaw. Lancet Oncol. 2008;9:1166-1172.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med. 2013;126:13-20.
- Ruggiero SL, Dodson TB, Assael LA, et al; American Association of Oral and Maxillofacial Surgeons. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws--2009 update. J Oral Maxillofac Surg. 2009;67(5 suppl):2-12.
- Mawardi H, Treister N, Richardson P, et al. Sinus tracts--an early sign of bisphosphonate-associated osteonecrosis of the jaws? J Oral Maxillofac Surg. 2009;67:593-601.
- Sammut S, Malden N, Lopes V. Facial cutaneous sinuses of dental origin--a diagnostic challenge. Br Dent J. 2013;215:555-558.
- Wood J, Bonjean K, Ruetz S, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055-1061.
- Hoefert S, Schmitz I, Tannapfel A, et al. Importance of microcracks in etiology of bisphosphonate-related osteonecrosis of the jaw: a possible pathogenetic model of symptomatic and non-symptomatic osteonecrosis of the jaw based on scanning electron microscopy findings. Clin Oral Investig. 2010;14:271-284.
- Hallmer F, Bjørnland T, Nicklasson A, et al. Osteonecrosis of the jaw in patients treated with oral and intravenous bisphosphonates: experience in Sweden. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:202-208.
- Lin TC, Yang CY, Kao Yang YH, et al. Incidence and risk of osteonecrosis of the jaw among the Taiwan osteoporosis population [published online February 11, 2014]. Osteoporos Int. 2014;25:1503-1511.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab. 2010;95:1555-1565.
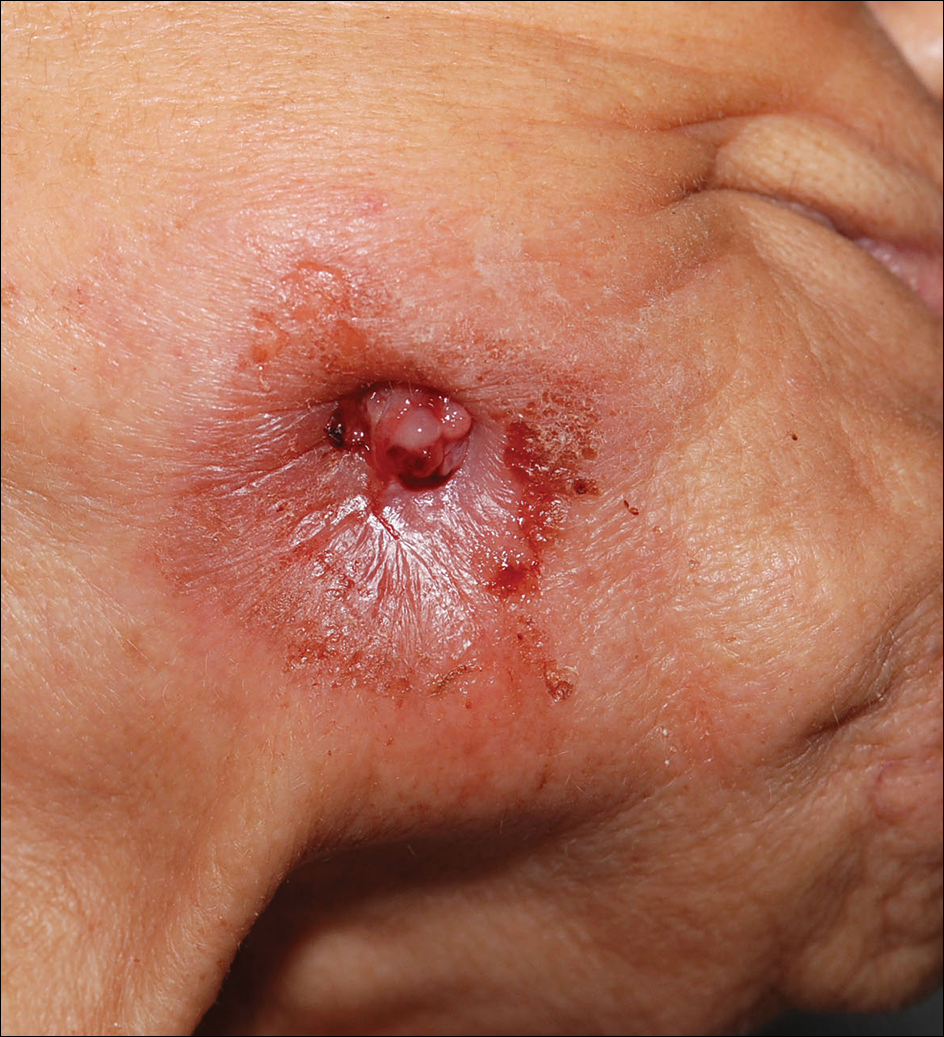
An 83-year-old woman presented with a painless, discharging, swollen nodule on the right side of the jaw of 6 months' duration. She had a history of osteoporosis diagnosed 3 years prior for which she was taking alendronate and cholecalciferol. Bone mineral density test scores were -3.93 (spine) and -2.81 (hip)(reference range, -2 and above). She also had hypertension that was treated with amlodipine. On examination there was fetor oris and a discharging sinus with purulent discharge at the jaw. The lower jaw was edentulous. A 5-mm area of red beefy granulation tissue was attached to underlying bone. An exposed sequestrum was seen intraorally with a 3-cm opening at the mandible. There also was submandibular lymphadenopathy.
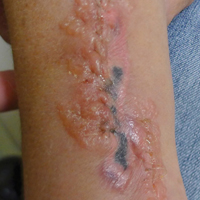
Contact Allergy to Poliglecaprone 25 Sutures
To the Editor:
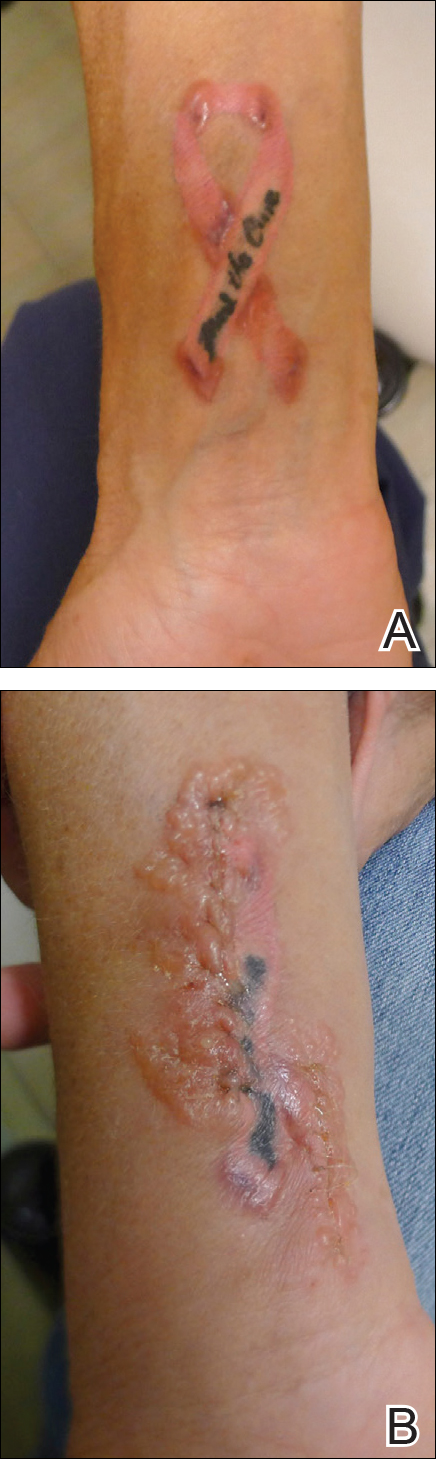
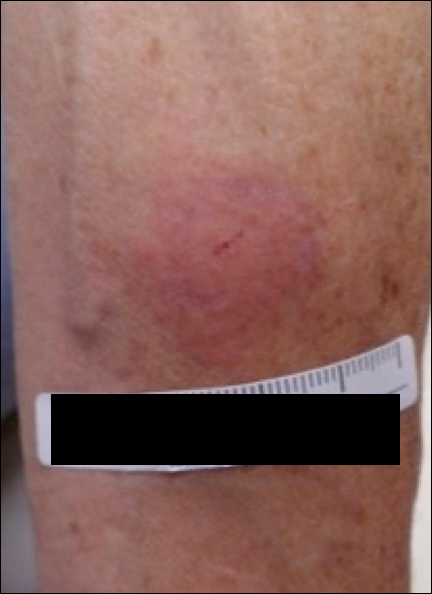
A 42-year-old woman who had a tattoo on the right wrist surgically removed 2 days prior developed severe erythema and swelling at the incision site (Figure 1). Exposure at the incision site was limited to bacitracin, poliglecaprone 25 suture, and plain cotton gauze. Patch testing of bacitracin was performed, which was ++ (moderately positive reaction) at the 96-hour reading, indicating that part of the reaction was due to the topical antibiotic. Testing of the suture was performed by tying the suture to the skin of the forearm and removing it at 48 hours. There was a ++ reaction to the suture prior to removal at 48 hours, which increased to +++ (severely positive reaction) after suture removal at 96 hours (Figure 2). Therefore, it appears that allergy to the suture also was partially responsible for the postsurgical reaction.
Poliglecaprone 25 suture is a monofilament synthetic absorbable material that is a copolymer of glycolide and ε-caprolactone. One case report of oral contact allergy to this suture material resulted in failure of an oral graft; however, no testing was performed to verify the contact allergy.1 Caprolactam ([CH2]5C[O]NH) is a related chemical that can be synthesized by treating caprolactone ([CH2]5CO2) with ammonia at elevated temperatures.2 Contact allergy has been reported to polyamide 6 suture, which is obtained by polymerizing ε-caprolactam. This report stated that contact allergy to ε-caprolactam also has been reported occupationally during manufacture and from its use in fishing nets, socks, gloves, and stockings.3
The package insert for the poliglecaprone 25 suture states that the material is “nonantigenic, nonpyrogenic and elicits only a slight tissue reaction during absorption.”4 We present a case of contact allergy to poliglecaprone 25 suture that was confirmed by allergy testing.
- Mawardi H. Oral contact allergy to suture material results in connective tissue graft failure: a case report. J Periodontol Online. 2014;4:155-160.
- Buntara T, Noel S, Phua PH, et al. Caprolactam from renewable resources: catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew Chem Int Ed Engl. 2011;50:7083-7087.
- Hausen BM. Allergic contact dermatitis from colored surgical suture material: contact allergy to epsilon-caprolactam and acid blue 158. Am J Contact Dermat. 2003;14:174-175.
- Monocryl [package insert]. Somerville, NJ: Ethicon, Inc; 1996.
To the Editor:
A 42-year-old woman who had a tattoo on the right wrist surgically removed 2 days prior developed severe erythema and swelling at the incision site (Figure 1). Exposure at the incision site was limited to bacitracin, poliglecaprone 25 suture, and plain cotton gauze. Patch testing of bacitracin was performed, which was ++ (moderately positive reaction) at the 96-hour reading, indicating that part of the reaction was due to the topical antibiotic. Testing of the suture was performed by tying the suture to the skin of the forearm and removing it at 48 hours. There was a ++ reaction to the suture prior to removal at 48 hours, which increased to +++ (severely positive reaction) after suture removal at 96 hours (Figure 2). Therefore, it appears that allergy to the suture also was partially responsible for the postsurgical reaction.
Poliglecaprone 25 suture is a monofilament synthetic absorbable material that is a copolymer of glycolide and ε-caprolactone. One case report of oral contact allergy to this suture material resulted in failure of an oral graft; however, no testing was performed to verify the contact allergy.1 Caprolactam ([CH2]5C[O]NH) is a related chemical that can be synthesized by treating caprolactone ([CH2]5CO2) with ammonia at elevated temperatures.2 Contact allergy has been reported to polyamide 6 suture, which is obtained by polymerizing ε-caprolactam. This report stated that contact allergy to ε-caprolactam also has been reported occupationally during manufacture and from its use in fishing nets, socks, gloves, and stockings.3
The package insert for the poliglecaprone 25 suture states that the material is “nonantigenic, nonpyrogenic and elicits only a slight tissue reaction during absorption.”4 We present a case of contact allergy to poliglecaprone 25 suture that was confirmed by allergy testing.
To the Editor:
A 42-year-old woman who had a tattoo on the right wrist surgically removed 2 days prior developed severe erythema and swelling at the incision site (Figure 1). Exposure at the incision site was limited to bacitracin, poliglecaprone 25 suture, and plain cotton gauze. Patch testing of bacitracin was performed, which was ++ (moderately positive reaction) at the 96-hour reading, indicating that part of the reaction was due to the topical antibiotic. Testing of the suture was performed by tying the suture to the skin of the forearm and removing it at 48 hours. There was a ++ reaction to the suture prior to removal at 48 hours, which increased to +++ (severely positive reaction) after suture removal at 96 hours (Figure 2). Therefore, it appears that allergy to the suture also was partially responsible for the postsurgical reaction.
Poliglecaprone 25 suture is a monofilament synthetic absorbable material that is a copolymer of glycolide and ε-caprolactone. One case report of oral contact allergy to this suture material resulted in failure of an oral graft; however, no testing was performed to verify the contact allergy.1 Caprolactam ([CH2]5C[O]NH) is a related chemical that can be synthesized by treating caprolactone ([CH2]5CO2) with ammonia at elevated temperatures.2 Contact allergy has been reported to polyamide 6 suture, which is obtained by polymerizing ε-caprolactam. This report stated that contact allergy to ε-caprolactam also has been reported occupationally during manufacture and from its use in fishing nets, socks, gloves, and stockings.3
The package insert for the poliglecaprone 25 suture states that the material is “nonantigenic, nonpyrogenic and elicits only a slight tissue reaction during absorption.”4 We present a case of contact allergy to poliglecaprone 25 suture that was confirmed by allergy testing.
- Mawardi H. Oral contact allergy to suture material results in connective tissue graft failure: a case report. J Periodontol Online. 2014;4:155-160.
- Buntara T, Noel S, Phua PH, et al. Caprolactam from renewable resources: catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew Chem Int Ed Engl. 2011;50:7083-7087.
- Hausen BM. Allergic contact dermatitis from colored surgical suture material: contact allergy to epsilon-caprolactam and acid blue 158. Am J Contact Dermat. 2003;14:174-175.
- Monocryl [package insert]. Somerville, NJ: Ethicon, Inc; 1996.
- Mawardi H. Oral contact allergy to suture material results in connective tissue graft failure: a case report. J Periodontol Online. 2014;4:155-160.
- Buntara T, Noel S, Phua PH, et al. Caprolactam from renewable resources: catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew Chem Int Ed Engl. 2011;50:7083-7087.
- Hausen BM. Allergic contact dermatitis from colored surgical suture material: contact allergy to epsilon-caprolactam and acid blue 158. Am J Contact Dermat. 2003;14:174-175.
- Monocryl [package insert]. Somerville, NJ: Ethicon, Inc; 1996.
Practice Point
- Physicians should be aware that rare contact reactions can occur with certain types of sutures.
The culture change of assessing parents for ACEs
Several years ago, pediatricians R.J. Gillespie, MD, MHPE, and Teri Pettersen, MD, piloted the use of a questionnaire about adverse childhood experiences (ACEs) and resilience at the 4-month well-child visit.
They and six other pediatricians at The Children’s Clinic in Portland, Ore., explained in a cover letter why they were posing the questions of parents, and they ended the survey by asking them about their interest in potential resources.
[[{"fid":"172157","view_mode":"medstat_image_flush_left","fields":{"format":"medstat_image_flush_left","field_file_image_alt_text[und][0][value]":"R.J. Gillespie, MD, MHPE","field_file_image_credit[und][0][value]":"Courtesy The Children's Clinic","field_file_image_caption[und][0][value]":"Dr. R.J. Gillespie "},"type":"media","attributes":{"class":"media-element file-medstat_image_flush_left"}}]]Today, all 28 of the pediatricians at the clinic screen for ACEs and resilience, and Dr. Pettersen, now retired from the practice, travels through the state conducting training for the Oregon Pediatric Society about the impact of ACEs in parents and their children, and how to go about identifying and addressing them.
“So many of our visits are about behavioral problems or emotional disturbances, and so often at the root of these issues is some sort of trauma the child is experiencing,” Dr. Gillespie said in an interview. “What we’re seeing in many of these cases really are coping strategies for that child to deal with the toxic stress in his or her life.”
By assessing parents’ exposure to ACEs, briefly talking with them about how ACEs might impact their parenting, and tailoring their counseling and anticipatory guidance, the pediatricians hope to prevent ACEs and consequent toxic stress from developing in children.

The driving science
The term ACEs entered the medical lexicon after 1998, when a landmark study called the Adverse Childhood Experiences Study showed that traumatic experiences in childhood – abuse, neglect, and other severe dysfunctions in a household – not only are common among American adults but are associated with numerous poor health outcomes.
In the study and subsequent analyses, Dr. Vincent Felitti of Kaiser Permanente in San Diego and Dr. Robert Anda of the Centers for Disease Control and Prevention surveyed more than 17,000 patients about 10 types of ACEs and their current health status and behaviors. About two-thirds reported having at least one ACE, and one in eight reported four or more (Am J Prev Med. 1998;14[4]:245-58, www.cdc.gov/violenceprevention/acestudy/about.html).
Adults with four or more ACEs were not only significantly more likely to report health risk behaviors (smoking, substance abuse) and poor mental health outcomes (depression, suicide attempt); they were also significantly more likely to have poor physical health outcomes, with 2.2 times the risk of ischemic heart disease, 1.9 times the risk of cancer, and 3.9 times the risk of chronic bronchitis or emphysema, for instance. There was a strong dose-response relationship between ACEs and poor outcomes.
The Felitti study spawned dozens of analyses and additional research – in children as well as adults – on the associations between early-life adversity and the incidence of poor behavioral, mental, and physical outcomes, as well as on potential mechanisms.
Some research suggested a direct link between ACEs and negative outcomes, independent of whether individuals adopt risky behavior. Other studies suggested what experts in child development and mental health have long argued – that the more ACEs a parent has, the more ACEs their child will have.
And a growing body of biomedical literature linked the extreme, frequent, or prolonged activation of the body’s stress response in childhood – what has come to be known as “toxic stress” – with disruptions of the developing nervous, cardiovascular, immune, and metabolic systems.

While precise connections and mechanisms need to be clarified, “we now know that the repeated activation of the stress response leads to [negative] changes in the neuroendocrine immune pathways,” said Dr. Burke Harris, who coauthored a recent review of toxic stress in children and adolescents (Adv Pediatr. 2016;63[1]:403-28).
In January 2012, the American Academy of Pediatrics published a policy statement titled “Early Childhood Adversity, Toxic Stress, and the Role of the Pediatrician: Translating Developmental Science into Lifelong Health,” in which it urged pediatricians to consider actively screening for precipitants of toxic stress that are common in their communities (Pediatrics. 2011 Dec. doi: 10.1542/peds.2011-2662). But it stopped short of recommending particular tools or methods.
Dr. Gillespie and Dr. Pettersen did not want to wait for tools to be validated and approaches to be proven. “We’re building the plane as we fly,” Dr. Pettersen said.
The clinic’s roll-out
Dr. Pettersen learned about the ACE study and related research about 8 years ago while on a sabbatical to learn more about mental health issues. It “changed everything” about the way she viewed children and families and adversity. “I knew (we) didn’t have the infrastructure at the clinic, or the clinic’s support, to really start assessing children for what was happening to them,” she said, so she began thinking about ACE prevention and a focus on parenting.
Dr. Gillespie, in the meantime, was active in various quality improvement efforts at the state and national level, and had also become increasingly bothered by visits in which he saw children affected by maternal depression, abnormal attachment, and other problems. “I was seeing the consequences of ACEs, but I didn’t know specifically what was going on or how to talk about it,” he said.
The two pediatricians agreed to ask parents about ACEs at the 4-month well visit – a time when the families “knew us a little bit” and when “we could still influence parenting styles.”
In March 2013, they and their colleagues in the pilot group began giving parents a questionnaire that included the 10 ACE questions from Felitti’s study, questions about resilience from the Children’s Resilience Initiative, and a list of potential resources so they could understand parents’ needs.
They created a confidential field in their electronic medical record for documentation that appears during a visit, but does not print into notes and therefore will not be inadvertently released.
As they moved through the pilot phase, the pediatricians used various approaches to follow up on the assessment face-to-face. Eventually, they chose three particular questions as nonthreatening and helpful for conversation: Are there any experiences that still bother you? Of those experiences that don’t bother you, how did you get to the point where they don’t bother you? And how do these experiences affect your parenting now?
“It’s a motivational interviewing sort of style,” said Dr. Gillespie. “Parents can start identifying for themselves the solutions for the problems they’ve experienced, and they can start thinking about how their parenting might be impacted by things that have happened [or are still happening] to them.”
As the project rolled out, the physicians tweaked their process. They added four more ACE questions to address issues – community violence, extreme bullying, racism and prejudice, and foster care exposure – that they thought might lead to toxic stress in their population, for instance. And rather than ask on the written questionnaire for a “yes” or “no” to each of the ACE questions, they began asking the parent how many of the ACE questions applied to them. Moving away from the yes-no format to asking for a total count has led to more disclosures, Dr. Gillespie said.
To “keep the conversation going” in subsequent well-child visits, they developed a few questions to ask high-risk parents, like “How do you and your partner resolve conflict?” and “How did your parents resolve conflict in your household when you were a child?” And they provided training to all of the clinic’s staff on trauma-informed care and the need for support and compassion in their interactions with family members.
In the 3-plus years since incorporating ACEs assessments, the clinic’s pediatricians have made soft referrals to mental health professionals in only several cases – in each case, by suggesting that the parent contact their primary care physician. What most parents have wanted, says Dr. Gillespie, is recommendations for parenting classes and support groups. The clinic’s care manager assists the pediatricians in maintaining and providing links and handouts for various resources.
For Dr. Gillespie, the impact of the culture shift has been dramatic. “I’ve had 8-10 moms spontaneously reveal domestic violence to me in a subsequent visit, and say that they need a little help, because they’ve gotten the message that this is a safe place to talk about their experiences,” he said. “That had never happened to me in the previous 12 years of so of my career.”
Dr. Pettersen’s relationships with parents became “more intimate and more honest.” There was more trust. “If we can talk with parents [about ACEs] and not judge them for it,” she said, “then nothing is off the table.”
The ‘Two-Gen’ approach

“But I’d push back and say, parents know they have toxic stress but they don’t name it,” she said. “What we can do as trusted providers who want to advocate for families is to bear witness to their history by asking about it. Once they realize it’s not what’s wrong with [them], it’s what’s happened to [them], a shift occurs. That’s extremely validating for parents.”
That validation is part of a two-generation approach that she and Dr. Burke Harris see as part of a movement to break cycles of ACEs and toxic stress. At the California Pacific Medical Center’s Bayview Child Health Center in San Francisco, Dr. Burke Harris uses three ACE questionnaires – two of them ask parents (of children or teens) to report how many adverse experience types, or categories, apply to them and/or their child or teen, and one surveys adolescents themselves.
With the resources and clinical support of the Center for Youth Wellness, whose major funders include Google, Dr. Burke Harris can initiate a “warm hand-off” of patients with a high ACE score to a care coordinator or therapist. (The Center for Youth Wellness is beginning research to validate its ACE screening tools.) And in the meantime, the medical care she provides is trauma-informed.
“If a patient comes in for ADHD [attention-deficit/hyperactivity disorder] and has an ACE score of 6, my differential diagnosis and assessment will be different than if I see a patient sent by the school who has an ACE score of 0,” she said.
At the Portland Clinic, even though ACEs screening is now tied with the 4-month visit, pediatricians are much more attentive across the board to possible ACEs and toxic stress, and feel better able to converse with families, Dr. Gillespie said. One of his partners recently saw a 12-year-old boy who was failing in school and not making friends. Trauma-informed history-taking revealed at least several ACEs, and conversation turned to “all the resilience pieces… the connections he was missing and what he needed to cope,” he said.
References
• Resilience Project: This AAP project houses a “trauma toolkit” for primary care, case studies, and a variety of other tools.
• Center for Youth Wellness: The ACEs screening tools used by Dr. Burke Harris may be accessed at this website, along with a user guide containing sample scripts, and two white papers on ACEs and toxic stress.
• Resilience: The Biology of Stress and the Science of Hope: This documentary film, released in September 2016, is about ACEs and “a new movement” to treat and prevent toxic stress; it features the work of Dr. Burke Harris and others.
• Academy on Violence & Abuse: Various papers on ACEs screening and case finding in practice may be accessed here.
Several years ago, pediatricians R.J. Gillespie, MD, MHPE, and Teri Pettersen, MD, piloted the use of a questionnaire about adverse childhood experiences (ACEs) and resilience at the 4-month well-child visit.
They and six other pediatricians at The Children’s Clinic in Portland, Ore., explained in a cover letter why they were posing the questions of parents, and they ended the survey by asking them about their interest in potential resources.
[[{"fid":"172157","view_mode":"medstat_image_flush_left","fields":{"format":"medstat_image_flush_left","field_file_image_alt_text[und][0][value]":"R.J. Gillespie, MD, MHPE","field_file_image_credit[und][0][value]":"Courtesy The Children's Clinic","field_file_image_caption[und][0][value]":"Dr. R.J. Gillespie "},"type":"media","attributes":{"class":"media-element file-medstat_image_flush_left"}}]]Today, all 28 of the pediatricians at the clinic screen for ACEs and resilience, and Dr. Pettersen, now retired from the practice, travels through the state conducting training for the Oregon Pediatric Society about the impact of ACEs in parents and their children, and how to go about identifying and addressing them.
“So many of our visits are about behavioral problems or emotional disturbances, and so often at the root of these issues is some sort of trauma the child is experiencing,” Dr. Gillespie said in an interview. “What we’re seeing in many of these cases really are coping strategies for that child to deal with the toxic stress in his or her life.”
By assessing parents’ exposure to ACEs, briefly talking with them about how ACEs might impact their parenting, and tailoring their counseling and anticipatory guidance, the pediatricians hope to prevent ACEs and consequent toxic stress from developing in children.
The driving science
The term ACEs entered the medical lexicon after 1998, when a landmark study called the Adverse Childhood Experiences Study showed that traumatic experiences in childhood – abuse, neglect, and other severe dysfunctions in a household – not only are common among American adults but are associated with numerous poor health outcomes.
In the study and subsequent analyses, Dr. Vincent Felitti of Kaiser Permanente in San Diego and Dr. Robert Anda of the Centers for Disease Control and Prevention surveyed more than 17,000 patients about 10 types of ACEs and their current health status and behaviors. About two-thirds reported having at least one ACE, and one in eight reported four or more (Am J Prev Med. 1998;14[4]:245-58, www.cdc.gov/violenceprevention/acestudy/about.html).
Adults with four or more ACEs were not only significantly more likely to report health risk behaviors (smoking, substance abuse) and poor mental health outcomes (depression, suicide attempt); they were also significantly more likely to have poor physical health outcomes, with 2.2 times the risk of ischemic heart disease, 1.9 times the risk of cancer, and 3.9 times the risk of chronic bronchitis or emphysema, for instance. There was a strong dose-response relationship between ACEs and poor outcomes.
The Felitti study spawned dozens of analyses and additional research – in children as well as adults – on the associations between early-life adversity and the incidence of poor behavioral, mental, and physical outcomes, as well as on potential mechanisms.
Some research suggested a direct link between ACEs and negative outcomes, independent of whether individuals adopt risky behavior. Other studies suggested what experts in child development and mental health have long argued – that the more ACEs a parent has, the more ACEs their child will have.
And a growing body of biomedical literature linked the extreme, frequent, or prolonged activation of the body’s stress response in childhood – what has come to be known as “toxic stress” – with disruptions of the developing nervous, cardiovascular, immune, and metabolic systems.
While precise connections and mechanisms need to be clarified, “we now know that the repeated activation of the stress response leads to [negative] changes in the neuroendocrine immune pathways,” said Dr. Burke Harris, who coauthored a recent review of toxic stress in children and adolescents (Adv Pediatr. 2016;63[1]:403-28).
In January 2012, the American Academy of Pediatrics published a policy statement titled “Early Childhood Adversity, Toxic Stress, and the Role of the Pediatrician: Translating Developmental Science into Lifelong Health,” in which it urged pediatricians to consider actively screening for precipitants of toxic stress that are common in their communities (Pediatrics. 2011 Dec. doi: 10.1542/peds.2011-2662). But it stopped short of recommending particular tools or methods.
Dr. Gillespie and Dr. Pettersen did not want to wait for tools to be validated and approaches to be proven. “We’re building the plane as we fly,” Dr. Pettersen said.
The clinic’s roll-out
Dr. Pettersen learned about the ACE study and related research about 8 years ago while on a sabbatical to learn more about mental health issues. It “changed everything” about the way she viewed children and families and adversity. “I knew (we) didn’t have the infrastructure at the clinic, or the clinic’s support, to really start assessing children for what was happening to them,” she said, so she began thinking about ACE prevention and a focus on parenting.
Dr. Gillespie, in the meantime, was active in various quality improvement efforts at the state and national level, and had also become increasingly bothered by visits in which he saw children affected by maternal depression, abnormal attachment, and other problems. “I was seeing the consequences of ACEs, but I didn’t know specifically what was going on or how to talk about it,” he said.
The two pediatricians agreed to ask parents about ACEs at the 4-month well visit – a time when the families “knew us a little bit” and when “we could still influence parenting styles.”
In March 2013, they and their colleagues in the pilot group began giving parents a questionnaire that included the 10 ACE questions from Felitti’s study, questions about resilience from the Children’s Resilience Initiative, and a list of potential resources so they could understand parents’ needs.
They created a confidential field in their electronic medical record for documentation that appears during a visit, but does not print into notes and therefore will not be inadvertently released.
As they moved through the pilot phase, the pediatricians used various approaches to follow up on the assessment face-to-face. Eventually, they chose three particular questions as nonthreatening and helpful for conversation: Are there any experiences that still bother you? Of those experiences that don’t bother you, how did you get to the point where they don’t bother you? And how do these experiences affect your parenting now?
“It’s a motivational interviewing sort of style,” said Dr. Gillespie. “Parents can start identifying for themselves the solutions for the problems they’ve experienced, and they can start thinking about how their parenting might be impacted by things that have happened [or are still happening] to them.”
As the project rolled out, the physicians tweaked their process. They added four more ACE questions to address issues – community violence, extreme bullying, racism and prejudice, and foster care exposure – that they thought might lead to toxic stress in their population, for instance. And rather than ask on the written questionnaire for a “yes” or “no” to each of the ACE questions, they began asking the parent how many of the ACE questions applied to them. Moving away from the yes-no format to asking for a total count has led to more disclosures, Dr. Gillespie said.
To “keep the conversation going” in subsequent well-child visits, they developed a few questions to ask high-risk parents, like “How do you and your partner resolve conflict?” and “How did your parents resolve conflict in your household when you were a child?” And they provided training to all of the clinic’s staff on trauma-informed care and the need for support and compassion in their interactions with family members.
In the 3-plus years since incorporating ACEs assessments, the clinic’s pediatricians have made soft referrals to mental health professionals in only several cases – in each case, by suggesting that the parent contact their primary care physician. What most parents have wanted, says Dr. Gillespie, is recommendations for parenting classes and support groups. The clinic’s care manager assists the pediatricians in maintaining and providing links and handouts for various resources.
For Dr. Gillespie, the impact of the culture shift has been dramatic. “I’ve had 8-10 moms spontaneously reveal domestic violence to me in a subsequent visit, and say that they need a little help, because they’ve gotten the message that this is a safe place to talk about their experiences,” he said. “That had never happened to me in the previous 12 years of so of my career.”
Dr. Pettersen’s relationships with parents became “more intimate and more honest.” There was more trust. “If we can talk with parents [about ACEs] and not judge them for it,” she said, “then nothing is off the table.”
The ‘Two-Gen’ approach
“But I’d push back and say, parents know they have toxic stress but they don’t name it,” she said. “What we can do as trusted providers who want to advocate for families is to bear witness to their history by asking about it. Once they realize it’s not what’s wrong with [them], it’s what’s happened to [them], a shift occurs. That’s extremely validating for parents.”
That validation is part of a two-generation approach that she and Dr. Burke Harris see as part of a movement to break cycles of ACEs and toxic stress. At the California Pacific Medical Center’s Bayview Child Health Center in San Francisco, Dr. Burke Harris uses three ACE questionnaires – two of them ask parents (of children or teens) to report how many adverse experience types, or categories, apply to them and/or their child or teen, and one surveys adolescents themselves.
With the resources and clinical support of the Center for Youth Wellness, whose major funders include Google, Dr. Burke Harris can initiate a “warm hand-off” of patients with a high ACE score to a care coordinator or therapist. (The Center for Youth Wellness is beginning research to validate its ACE screening tools.) And in the meantime, the medical care she provides is trauma-informed.
“If a patient comes in for ADHD [attention-deficit/hyperactivity disorder] and has an ACE score of 6, my differential diagnosis and assessment will be different than if I see a patient sent by the school who has an ACE score of 0,” she said.
At the Portland Clinic, even though ACEs screening is now tied with the 4-month visit, pediatricians are much more attentive across the board to possible ACEs and toxic stress, and feel better able to converse with families, Dr. Gillespie said. One of his partners recently saw a 12-year-old boy who was failing in school and not making friends. Trauma-informed history-taking revealed at least several ACEs, and conversation turned to “all the resilience pieces… the connections he was missing and what he needed to cope,” he said.
References
• Resilience Project: This AAP project houses a “trauma toolkit” for primary care, case studies, and a variety of other tools.
• Center for Youth Wellness: The ACEs screening tools used by Dr. Burke Harris may be accessed at this website, along with a user guide containing sample scripts, and two white papers on ACEs and toxic stress.
• Resilience: The Biology of Stress and the Science of Hope: This documentary film, released in September 2016, is about ACEs and “a new movement” to treat and prevent toxic stress; it features the work of Dr. Burke Harris and others.
• Academy on Violence & Abuse: Various papers on ACEs screening and case finding in practice may be accessed here.
Several years ago, pediatricians R.J. Gillespie, MD, MHPE, and Teri Pettersen, MD, piloted the use of a questionnaire about adverse childhood experiences (ACEs) and resilience at the 4-month well-child visit.
They and six other pediatricians at The Children’s Clinic in Portland, Ore., explained in a cover letter why they were posing the questions of parents, and they ended the survey by asking them about their interest in potential resources.
[[{"fid":"172157","view_mode":"medstat_image_flush_left","fields":{"format":"medstat_image_flush_left","field_file_image_alt_text[und][0][value]":"R.J. Gillespie, MD, MHPE","field_file_image_credit[und][0][value]":"Courtesy The Children's Clinic","field_file_image_caption[und][0][value]":"Dr. R.J. Gillespie "},"type":"media","attributes":{"class":"media-element file-medstat_image_flush_left"}}]]Today, all 28 of the pediatricians at the clinic screen for ACEs and resilience, and Dr. Pettersen, now retired from the practice, travels through the state conducting training for the Oregon Pediatric Society about the impact of ACEs in parents and their children, and how to go about identifying and addressing them.
“So many of our visits are about behavioral problems or emotional disturbances, and so often at the root of these issues is some sort of trauma the child is experiencing,” Dr. Gillespie said in an interview. “What we’re seeing in many of these cases really are coping strategies for that child to deal with the toxic stress in his or her life.”
By assessing parents’ exposure to ACEs, briefly talking with them about how ACEs might impact their parenting, and tailoring their counseling and anticipatory guidance, the pediatricians hope to prevent ACEs and consequent toxic stress from developing in children.
The driving science
The term ACEs entered the medical lexicon after 1998, when a landmark study called the Adverse Childhood Experiences Study showed that traumatic experiences in childhood – abuse, neglect, and other severe dysfunctions in a household – not only are common among American adults but are associated with numerous poor health outcomes.
In the study and subsequent analyses, Dr. Vincent Felitti of Kaiser Permanente in San Diego and Dr. Robert Anda of the Centers for Disease Control and Prevention surveyed more than 17,000 patients about 10 types of ACEs and their current health status and behaviors. About two-thirds reported having at least one ACE, and one in eight reported four or more (Am J Prev Med. 1998;14[4]:245-58, www.cdc.gov/violenceprevention/acestudy/about.html).
Adults with four or more ACEs were not only significantly more likely to report health risk behaviors (smoking, substance abuse) and poor mental health outcomes (depression, suicide attempt); they were also significantly more likely to have poor physical health outcomes, with 2.2 times the risk of ischemic heart disease, 1.9 times the risk of cancer, and 3.9 times the risk of chronic bronchitis or emphysema, for instance. There was a strong dose-response relationship between ACEs and poor outcomes.
The Felitti study spawned dozens of analyses and additional research – in children as well as adults – on the associations between early-life adversity and the incidence of poor behavioral, mental, and physical outcomes, as well as on potential mechanisms.
Some research suggested a direct link between ACEs and negative outcomes, independent of whether individuals adopt risky behavior. Other studies suggested what experts in child development and mental health have long argued – that the more ACEs a parent has, the more ACEs their child will have.
And a growing body of biomedical literature linked the extreme, frequent, or prolonged activation of the body’s stress response in childhood – what has come to be known as “toxic stress” – with disruptions of the developing nervous, cardiovascular, immune, and metabolic systems.
While precise connections and mechanisms need to be clarified, “we now know that the repeated activation of the stress response leads to [negative] changes in the neuroendocrine immune pathways,” said Dr. Burke Harris, who coauthored a recent review of toxic stress in children and adolescents (Adv Pediatr. 2016;63[1]:403-28).
In January 2012, the American Academy of Pediatrics published a policy statement titled “Early Childhood Adversity, Toxic Stress, and the Role of the Pediatrician: Translating Developmental Science into Lifelong Health,” in which it urged pediatricians to consider actively screening for precipitants of toxic stress that are common in their communities (Pediatrics. 2011 Dec. doi: 10.1542/peds.2011-2662). But it stopped short of recommending particular tools or methods.
Dr. Gillespie and Dr. Pettersen did not want to wait for tools to be validated and approaches to be proven. “We’re building the plane as we fly,” Dr. Pettersen said.
The clinic’s roll-out
Dr. Pettersen learned about the ACE study and related research about 8 years ago while on a sabbatical to learn more about mental health issues. It “changed everything” about the way she viewed children and families and adversity. “I knew (we) didn’t have the infrastructure at the clinic, or the clinic’s support, to really start assessing children for what was happening to them,” she said, so she began thinking about ACE prevention and a focus on parenting.
Dr. Gillespie, in the meantime, was active in various quality improvement efforts at the state and national level, and had also become increasingly bothered by visits in which he saw children affected by maternal depression, abnormal attachment, and other problems. “I was seeing the consequences of ACEs, but I didn’t know specifically what was going on or how to talk about it,” he said.
The two pediatricians agreed to ask parents about ACEs at the 4-month well visit – a time when the families “knew us a little bit” and when “we could still influence parenting styles.”
In March 2013, they and their colleagues in the pilot group began giving parents a questionnaire that included the 10 ACE questions from Felitti’s study, questions about resilience from the Children’s Resilience Initiative, and a list of potential resources so they could understand parents’ needs.
They created a confidential field in their electronic medical record for documentation that appears during a visit, but does not print into notes and therefore will not be inadvertently released.
As they moved through the pilot phase, the pediatricians used various approaches to follow up on the assessment face-to-face. Eventually, they chose three particular questions as nonthreatening and helpful for conversation: Are there any experiences that still bother you? Of those experiences that don’t bother you, how did you get to the point where they don’t bother you? And how do these experiences affect your parenting now?
“It’s a motivational interviewing sort of style,” said Dr. Gillespie. “Parents can start identifying for themselves the solutions for the problems they’ve experienced, and they can start thinking about how their parenting might be impacted by things that have happened [or are still happening] to them.”
As the project rolled out, the physicians tweaked their process. They added four more ACE questions to address issues – community violence, extreme bullying, racism and prejudice, and foster care exposure – that they thought might lead to toxic stress in their population, for instance. And rather than ask on the written questionnaire for a “yes” or “no” to each of the ACE questions, they began asking the parent how many of the ACE questions applied to them. Moving away from the yes-no format to asking for a total count has led to more disclosures, Dr. Gillespie said.
To “keep the conversation going” in subsequent well-child visits, they developed a few questions to ask high-risk parents, like “How do you and your partner resolve conflict?” and “How did your parents resolve conflict in your household when you were a child?” And they provided training to all of the clinic’s staff on trauma-informed care and the need for support and compassion in their interactions with family members.
In the 3-plus years since incorporating ACEs assessments, the clinic’s pediatricians have made soft referrals to mental health professionals in only several cases – in each case, by suggesting that the parent contact their primary care physician. What most parents have wanted, says Dr. Gillespie, is recommendations for parenting classes and support groups. The clinic’s care manager assists the pediatricians in maintaining and providing links and handouts for various resources.
For Dr. Gillespie, the impact of the culture shift has been dramatic. “I’ve had 8-10 moms spontaneously reveal domestic violence to me in a subsequent visit, and say that they need a little help, because they’ve gotten the message that this is a safe place to talk about their experiences,” he said. “That had never happened to me in the previous 12 years of so of my career.”
Dr. Pettersen’s relationships with parents became “more intimate and more honest.” There was more trust. “If we can talk with parents [about ACEs] and not judge them for it,” she said, “then nothing is off the table.”
The ‘Two-Gen’ approach
“But I’d push back and say, parents know they have toxic stress but they don’t name it,” she said. “What we can do as trusted providers who want to advocate for families is to bear witness to their history by asking about it. Once they realize it’s not what’s wrong with [them], it’s what’s happened to [them], a shift occurs. That’s extremely validating for parents.”
That validation is part of a two-generation approach that she and Dr. Burke Harris see as part of a movement to break cycles of ACEs and toxic stress. At the California Pacific Medical Center’s Bayview Child Health Center in San Francisco, Dr. Burke Harris uses three ACE questionnaires – two of them ask parents (of children or teens) to report how many adverse experience types, or categories, apply to them and/or their child or teen, and one surveys adolescents themselves.
With the resources and clinical support of the Center for Youth Wellness, whose major funders include Google, Dr. Burke Harris can initiate a “warm hand-off” of patients with a high ACE score to a care coordinator or therapist. (The Center for Youth Wellness is beginning research to validate its ACE screening tools.) And in the meantime, the medical care she provides is trauma-informed.
“If a patient comes in for ADHD [attention-deficit/hyperactivity disorder] and has an ACE score of 6, my differential diagnosis and assessment will be different than if I see a patient sent by the school who has an ACE score of 0,” she said.
At the Portland Clinic, even though ACEs screening is now tied with the 4-month visit, pediatricians are much more attentive across the board to possible ACEs and toxic stress, and feel better able to converse with families, Dr. Gillespie said. One of his partners recently saw a 12-year-old boy who was failing in school and not making friends. Trauma-informed history-taking revealed at least several ACEs, and conversation turned to “all the resilience pieces… the connections he was missing and what he needed to cope,” he said.
References
• Resilience Project: This AAP project houses a “trauma toolkit” for primary care, case studies, and a variety of other tools.
• Center for Youth Wellness: The ACEs screening tools used by Dr. Burke Harris may be accessed at this website, along with a user guide containing sample scripts, and two white papers on ACEs and toxic stress.
• Resilience: The Biology of Stress and the Science of Hope: This documentary film, released in September 2016, is about ACEs and “a new movement” to treat and prevent toxic stress; it features the work of Dr. Burke Harris and others.
• Academy on Violence & Abuse: Various papers on ACEs screening and case finding in practice may be accessed here.

Optimal management of GERD in IPF unknown
EXPERT ANALYSIS FROM CHEST 2016
LOS ANGELES – The optimal management of gastroesophageal reflux disease in patients with idiopathic pulmonary fibrosis has yet to be determined, according to Joyce S. Lee, MD.
“We need strong randomized clinical trial data to tell us whether or not medical or surgical treatment of GERD in IPF is indicated,” she said at the annual meeting of the American College of Chest Physicians.

Two proposed hypotheses explain the relationship between reflux and IPF. The first holds that reflux and microaspiration are involved in the pathogenesis of IPF. The second, favored by Dr. Lee, proposes that reflux and microaspiration impact the natural history, either through acute exacerbation, disease progression, or survival. Patients with IPF “have weakening of the lower esophageal sphincter, whether that’s due to the presence of a hiatal hernia, medications, or just aging of the tissue there,” she said. “We know how to diagnose reflux disease, but we don’t know how to diagnose microaspiration, which is defined as subclinical aspiration of small droplets of gastric contents. Reflux is a risk factor for the condition of microaspiration, but it is not a perfect surrogate. Not everybody with reflux will aspirate. There is a potential role for bronchoalveolar lavage pepsin and/or bile salt as a biomarker of microaspiration, but it is not validated or standardized in IPF yet.”
Reflux becomes pathologic when reflux of stomach contents causes troublesome symptoms and/or complications. “Troublesome” is defined as mild symptoms 2 or more days a week or moderate to severe symptoms more than 1 day a week. Dr. Lee said that chest physicians can diagnose GERD in their IPF patients the same way that gastroenterologists and primary care doctors do: with symptoms, barium swallow, 24-hour pH monitoring, impedance testing, and sometimes endoscopy. The 2015 IPF guidelines recommend that clinicians “use regular antacid treatment for patients with IPF (conditional recommendation, very low confidence in estimates of effect).” It does not extend to surgical treatment with fundoplication. (Am J Resp Crit Care Med. 2015 Jul 15;192[2]: e3-19).
In an effort to measure the relationship between antacid therapy and change in forced vital capacity, Dr. Lee and her associates evaluated IPF patients from placebo arms of the three Idiopathic Pulmonary Fibrosis Clinical Research Network randomized controlled trials. They found that, compared with patients who did not take antacid therapy at baseline, those who did experienced a slower decline in their forced vital capacity over time (Lancet Resp Med. 2013 Jul;1[5]:369-76). However, a more-recent analysis conducted by different investigators examined the placebo arms of three pirfenidone studies and found no significant effect of antacid therapy in IPF patients (Lancet Resp Med. 2016 May;4[5]:381-9). Dr. Lee said that both evaluations differed because they were secondary analyses of previously captured data. “There were also differences in the ways the trials obtained GERD history, medication indication, and dosing of the antacid therapy,” she said. “There were also differences in outcomes and different populations studied.”
Dr. Lee’s current approach to counseling IPF patients with GERD includes discussing lifestyle modifications and PPI therapy – either daily or twice a day dosing, depending on their symptoms. “Lifestyle modifications include weight loss, smoking cessation, raising the head of the bed 6-8 inches, and avoiding foods that cause acid reflux, including chocolate, alcohol, peppermint, and fatty or spicy foods, and avoiding large and late meals,” she said. “In terms of acid suppression therapy with H2 blockers and PPIs [proton pump inhibitors], symptom relief and healing of the esophagus occurs in 85%-90% of patients taking them correctly. This does not alter their risk of having microaspiration.” Laparoscopic antireflux therapy (fundoplication) is indicated only after the failure of medical therapy. “The goal is to correct any hernia and tighten the lower esophageal sphincter,” she said. “Efficacy and symptom relief is reported to be around 95%.”
An NIH-funded trial called Weighing Risks and Benefits of Laparoscopic Anti-Reflux Surgery in Patients with Idiopathic Fibrosis, is ongoing at six centers. Dr. Lee said that some results are expected within the next year. She reported having no financial disclosures.
EXPERT ANALYSIS FROM CHEST 2016
LOS ANGELES – The optimal management of gastroesophageal reflux disease in patients with idiopathic pulmonary fibrosis has yet to be determined, according to Joyce S. Lee, MD.
“We need strong randomized clinical trial data to tell us whether or not medical or surgical treatment of GERD in IPF is indicated,” she said at the annual meeting of the American College of Chest Physicians.
Two proposed hypotheses explain the relationship between reflux and IPF. The first holds that reflux and microaspiration are involved in the pathogenesis of IPF. The second, favored by Dr. Lee, proposes that reflux and microaspiration impact the natural history, either through acute exacerbation, disease progression, or survival. Patients with IPF “have weakening of the lower esophageal sphincter, whether that’s due to the presence of a hiatal hernia, medications, or just aging of the tissue there,” she said. “We know how to diagnose reflux disease, but we don’t know how to diagnose microaspiration, which is defined as subclinical aspiration of small droplets of gastric contents. Reflux is a risk factor for the condition of microaspiration, but it is not a perfect surrogate. Not everybody with reflux will aspirate. There is a potential role for bronchoalveolar lavage pepsin and/or bile salt as a biomarker of microaspiration, but it is not validated or standardized in IPF yet.”
Reflux becomes pathologic when reflux of stomach contents causes troublesome symptoms and/or complications. “Troublesome” is defined as mild symptoms 2 or more days a week or moderate to severe symptoms more than 1 day a week. Dr. Lee said that chest physicians can diagnose GERD in their IPF patients the same way that gastroenterologists and primary care doctors do: with symptoms, barium swallow, 24-hour pH monitoring, impedance testing, and sometimes endoscopy. The 2015 IPF guidelines recommend that clinicians “use regular antacid treatment for patients with IPF (conditional recommendation, very low confidence in estimates of effect).” It does not extend to surgical treatment with fundoplication. (Am J Resp Crit Care Med. 2015 Jul 15;192[2]: e3-19).
In an effort to measure the relationship between antacid therapy and change in forced vital capacity, Dr. Lee and her associates evaluated IPF patients from placebo arms of the three Idiopathic Pulmonary Fibrosis Clinical Research Network randomized controlled trials. They found that, compared with patients who did not take antacid therapy at baseline, those who did experienced a slower decline in their forced vital capacity over time (Lancet Resp Med. 2013 Jul;1[5]:369-76). However, a more-recent analysis conducted by different investigators examined the placebo arms of three pirfenidone studies and found no significant effect of antacid therapy in IPF patients (Lancet Resp Med. 2016 May;4[5]:381-9). Dr. Lee said that both evaluations differed because they were secondary analyses of previously captured data. “There were also differences in the ways the trials obtained GERD history, medication indication, and dosing of the antacid therapy,” she said. “There were also differences in outcomes and different populations studied.”
Dr. Lee’s current approach to counseling IPF patients with GERD includes discussing lifestyle modifications and PPI therapy – either daily or twice a day dosing, depending on their symptoms. “Lifestyle modifications include weight loss, smoking cessation, raising the head of the bed 6-8 inches, and avoiding foods that cause acid reflux, including chocolate, alcohol, peppermint, and fatty or spicy foods, and avoiding large and late meals,” she said. “In terms of acid suppression therapy with H2 blockers and PPIs [proton pump inhibitors], symptom relief and healing of the esophagus occurs in 85%-90% of patients taking them correctly. This does not alter their risk of having microaspiration.” Laparoscopic antireflux therapy (fundoplication) is indicated only after the failure of medical therapy. “The goal is to correct any hernia and tighten the lower esophageal sphincter,” she said. “Efficacy and symptom relief is reported to be around 95%.”
An NIH-funded trial called Weighing Risks and Benefits of Laparoscopic Anti-Reflux Surgery in Patients with Idiopathic Fibrosis, is ongoing at six centers. Dr. Lee said that some results are expected within the next year. She reported having no financial disclosures.
EXPERT ANALYSIS FROM CHEST 2016
LOS ANGELES – The optimal management of gastroesophageal reflux disease in patients with idiopathic pulmonary fibrosis has yet to be determined, according to Joyce S. Lee, MD.
“We need strong randomized clinical trial data to tell us whether or not medical or surgical treatment of GERD in IPF is indicated,” she said at the annual meeting of the American College of Chest Physicians.
Two proposed hypotheses explain the relationship between reflux and IPF. The first holds that reflux and microaspiration are involved in the pathogenesis of IPF. The second, favored by Dr. Lee, proposes that reflux and microaspiration impact the natural history, either through acute exacerbation, disease progression, or survival. Patients with IPF “have weakening of the lower esophageal sphincter, whether that’s due to the presence of a hiatal hernia, medications, or just aging of the tissue there,” she said. “We know how to diagnose reflux disease, but we don’t know how to diagnose microaspiration, which is defined as subclinical aspiration of small droplets of gastric contents. Reflux is a risk factor for the condition of microaspiration, but it is not a perfect surrogate. Not everybody with reflux will aspirate. There is a potential role for bronchoalveolar lavage pepsin and/or bile salt as a biomarker of microaspiration, but it is not validated or standardized in IPF yet.”
Reflux becomes pathologic when reflux of stomach contents causes troublesome symptoms and/or complications. “Troublesome” is defined as mild symptoms 2 or more days a week or moderate to severe symptoms more than 1 day a week. Dr. Lee said that chest physicians can diagnose GERD in their IPF patients the same way that gastroenterologists and primary care doctors do: with symptoms, barium swallow, 24-hour pH monitoring, impedance testing, and sometimes endoscopy. The 2015 IPF guidelines recommend that clinicians “use regular antacid treatment for patients with IPF (conditional recommendation, very low confidence in estimates of effect).” It does not extend to surgical treatment with fundoplication. (Am J Resp Crit Care Med. 2015 Jul 15;192[2]: e3-19).
In an effort to measure the relationship between antacid therapy and change in forced vital capacity, Dr. Lee and her associates evaluated IPF patients from placebo arms of the three Idiopathic Pulmonary Fibrosis Clinical Research Network randomized controlled trials. They found that, compared with patients who did not take antacid therapy at baseline, those who did experienced a slower decline in their forced vital capacity over time (Lancet Resp Med. 2013 Jul;1[5]:369-76). However, a more-recent analysis conducted by different investigators examined the placebo arms of three pirfenidone studies and found no significant effect of antacid therapy in IPF patients (Lancet Resp Med. 2016 May;4[5]:381-9). Dr. Lee said that both evaluations differed because they were secondary analyses of previously captured data. “There were also differences in the ways the trials obtained GERD history, medication indication, and dosing of the antacid therapy,” she said. “There were also differences in outcomes and different populations studied.”
Dr. Lee’s current approach to counseling IPF patients with GERD includes discussing lifestyle modifications and PPI therapy – either daily or twice a day dosing, depending on their symptoms. “Lifestyle modifications include weight loss, smoking cessation, raising the head of the bed 6-8 inches, and avoiding foods that cause acid reflux, including chocolate, alcohol, peppermint, and fatty or spicy foods, and avoiding large and late meals,” she said. “In terms of acid suppression therapy with H2 blockers and PPIs [proton pump inhibitors], symptom relief and healing of the esophagus occurs in 85%-90% of patients taking them correctly. This does not alter their risk of having microaspiration.” Laparoscopic antireflux therapy (fundoplication) is indicated only after the failure of medical therapy. “The goal is to correct any hernia and tighten the lower esophageal sphincter,” she said. “Efficacy and symptom relief is reported to be around 95%.”
An NIH-funded trial called Weighing Risks and Benefits of Laparoscopic Anti-Reflux Surgery in Patients with Idiopathic Fibrosis, is ongoing at six centers. Dr. Lee said that some results are expected within the next year. She reported having no financial disclosures.
Amantadine Hydrochloride Reduces Levodopa-Induced Dyskinesia in Patients With Parkinson’s Disease
PORTLAND, OR—Amantadine hydrochloride extended-release capsules, compared with placebo, result in a statistically significant and clinically meaningful improvement in levodopa-induced dyskinesia in patients with Parkinson’s disease, according to phase III trial results presented at the Fourth World Parkinson Congress. The drug also reduces daily off time and is generally well tolerated, researchers said.
Amantadine hydrochloride extended-release capsules, known as ADS-5102, are being developed by Adamas Pharmaceuticals with the aim of improving the pharmacokinetic profile of immediate-release amantadine.
Rajesh Pahwa, MD, Professor of Neurology at University of Kansas Medical Center in Kansas City, and colleagues reported efficacy and safety results from the EASE LID 3 trial, one of three phase III trials of the drug.

EASE LID 3 was a randomized, double-blind, placebo-controlled study conducted at 39 sites in the United States and Europe. Seventy-seven patients with Parkinson’s disease were randomized to receive 340 mg of amantadine hydrochloride or placebo once daily at bedtime for the treatment of levodopa-induced dyskinesia. Participants had an average age of 64.8, 52% were men, and they had been diagnosed with Parkinson’s disease for an average of 10.6 years. They had received levodopa treatment for an average of 8.1 years. Patients’ mean duration of levodopa-induced dyskinesia was 3.9 years.
Treatment Improved Outcomes
The primary efficacy analysis compared the least square mean change from baseline to week 12 in Unified Dyskinesia Rating Scale total score in the ADS-5102 group with that in the placebo group. Key secondary efficacy analyses compared the least square mean change from baseline to week 12 in on time without troublesome dyskinesia and off time, as recorded in participants’ home diaries.
Thirty-eight patients were randomized to receive ADS-5102 and 39 patients were randomized to receive placebo. In the ADS-5102 group, the observed mean Unified Dyskinesia Rating Scale total score from baseline to week 12 decreased by 46%, compared with a 16% reduction in the placebo group. At 12 weeks, daily on time without troublesome dyskinesia increased by 4.0 hours in the ADS-5102 group and by 2.1 hours in the placebo group. Daily off time decreased by 0.5 hours in the ADS-5102 group and increased by 0.6 hours in the placebo group.
Adverse Events
Four patients, all in the ADS-5102 group, experienced serious adverse events. In one patient, the serious adverse event was considered related to the study drug (ie, constipation and urinary retention). The patient completed study treatment and did not discontinue participation in the trial.
Seven patients in the ADS-5102 group (19%) versus three patients in the placebo group (8%) discontinued treatment due to adverse events. In the ADS-5102 group, the most frequent adverse events (ie, they occurred in more than two patients) were dry mouth, nausea, decreased appetite, insomnia, orthostatic hypotension, constipation, fall, and hallucination.
“ADS-5102 resulted in statistically significant and clinically meaningful improvement in levodopa-induced dyskinesia, confirming results from earlier controlled clinical trials,” the researchers concluded.
Investigators are conducting an open-label safety study of ADS-5102 that includes patients from the phase III trials as well as patients with levodopa-induced dyskinesia who have undergone deep brain stimulation. Adamas also is investigating ADS-5102 for use in patients with multiple sclerosis with walking impairment.
—Jake Remaly
PORTLAND, OR—Amantadine hydrochloride extended-release capsules, compared with placebo, result in a statistically significant and clinically meaningful improvement in levodopa-induced dyskinesia in patients with Parkinson’s disease, according to phase III trial results presented at the Fourth World Parkinson Congress. The drug also reduces daily off time and is generally well tolerated, researchers said.
Amantadine hydrochloride extended-release capsules, known as ADS-5102, are being developed by Adamas Pharmaceuticals with the aim of improving the pharmacokinetic profile of immediate-release amantadine.
Rajesh Pahwa, MD, Professor of Neurology at University of Kansas Medical Center in Kansas City, and colleagues reported efficacy and safety results from the EASE LID 3 trial, one of three phase III trials of the drug.
EASE LID 3 was a randomized, double-blind, placebo-controlled study conducted at 39 sites in the United States and Europe. Seventy-seven patients with Parkinson’s disease were randomized to receive 340 mg of amantadine hydrochloride or placebo once daily at bedtime for the treatment of levodopa-induced dyskinesia. Participants had an average age of 64.8, 52% were men, and they had been diagnosed with Parkinson’s disease for an average of 10.6 years. They had received levodopa treatment for an average of 8.1 years. Patients’ mean duration of levodopa-induced dyskinesia was 3.9 years.
Treatment Improved Outcomes
The primary efficacy analysis compared the least square mean change from baseline to week 12 in Unified Dyskinesia Rating Scale total score in the ADS-5102 group with that in the placebo group. Key secondary efficacy analyses compared the least square mean change from baseline to week 12 in on time without troublesome dyskinesia and off time, as recorded in participants’ home diaries.
Thirty-eight patients were randomized to receive ADS-5102 and 39 patients were randomized to receive placebo. In the ADS-5102 group, the observed mean Unified Dyskinesia Rating Scale total score from baseline to week 12 decreased by 46%, compared with a 16% reduction in the placebo group. At 12 weeks, daily on time without troublesome dyskinesia increased by 4.0 hours in the ADS-5102 group and by 2.1 hours in the placebo group. Daily off time decreased by 0.5 hours in the ADS-5102 group and increased by 0.6 hours in the placebo group.
Adverse Events
Four patients, all in the ADS-5102 group, experienced serious adverse events. In one patient, the serious adverse event was considered related to the study drug (ie, constipation and urinary retention). The patient completed study treatment and did not discontinue participation in the trial.
Seven patients in the ADS-5102 group (19%) versus three patients in the placebo group (8%) discontinued treatment due to adverse events. In the ADS-5102 group, the most frequent adverse events (ie, they occurred in more than two patients) were dry mouth, nausea, decreased appetite, insomnia, orthostatic hypotension, constipation, fall, and hallucination.
“ADS-5102 resulted in statistically significant and clinically meaningful improvement in levodopa-induced dyskinesia, confirming results from earlier controlled clinical trials,” the researchers concluded.
Investigators are conducting an open-label safety study of ADS-5102 that includes patients from the phase III trials as well as patients with levodopa-induced dyskinesia who have undergone deep brain stimulation. Adamas also is investigating ADS-5102 for use in patients with multiple sclerosis with walking impairment.
—Jake Remaly
PORTLAND, OR—Amantadine hydrochloride extended-release capsules, compared with placebo, result in a statistically significant and clinically meaningful improvement in levodopa-induced dyskinesia in patients with Parkinson’s disease, according to phase III trial results presented at the Fourth World Parkinson Congress. The drug also reduces daily off time and is generally well tolerated, researchers said.
Amantadine hydrochloride extended-release capsules, known as ADS-5102, are being developed by Adamas Pharmaceuticals with the aim of improving the pharmacokinetic profile of immediate-release amantadine.
Rajesh Pahwa, MD, Professor of Neurology at University of Kansas Medical Center in Kansas City, and colleagues reported efficacy and safety results from the EASE LID 3 trial, one of three phase III trials of the drug.
EASE LID 3 was a randomized, double-blind, placebo-controlled study conducted at 39 sites in the United States and Europe. Seventy-seven patients with Parkinson’s disease were randomized to receive 340 mg of amantadine hydrochloride or placebo once daily at bedtime for the treatment of levodopa-induced dyskinesia. Participants had an average age of 64.8, 52% were men, and they had been diagnosed with Parkinson’s disease for an average of 10.6 years. They had received levodopa treatment for an average of 8.1 years. Patients’ mean duration of levodopa-induced dyskinesia was 3.9 years.
Treatment Improved Outcomes
The primary efficacy analysis compared the least square mean change from baseline to week 12 in Unified Dyskinesia Rating Scale total score in the ADS-5102 group with that in the placebo group. Key secondary efficacy analyses compared the least square mean change from baseline to week 12 in on time without troublesome dyskinesia and off time, as recorded in participants’ home diaries.
Thirty-eight patients were randomized to receive ADS-5102 and 39 patients were randomized to receive placebo. In the ADS-5102 group, the observed mean Unified Dyskinesia Rating Scale total score from baseline to week 12 decreased by 46%, compared with a 16% reduction in the placebo group. At 12 weeks, daily on time without troublesome dyskinesia increased by 4.0 hours in the ADS-5102 group and by 2.1 hours in the placebo group. Daily off time decreased by 0.5 hours in the ADS-5102 group and increased by 0.6 hours in the placebo group.
Adverse Events
Four patients, all in the ADS-5102 group, experienced serious adverse events. In one patient, the serious adverse event was considered related to the study drug (ie, constipation and urinary retention). The patient completed study treatment and did not discontinue participation in the trial.
Seven patients in the ADS-5102 group (19%) versus three patients in the placebo group (8%) discontinued treatment due to adverse events. In the ADS-5102 group, the most frequent adverse events (ie, they occurred in more than two patients) were dry mouth, nausea, decreased appetite, insomnia, orthostatic hypotension, constipation, fall, and hallucination.
“ADS-5102 resulted in statistically significant and clinically meaningful improvement in levodopa-induced dyskinesia, confirming results from earlier controlled clinical trials,” the researchers concluded.
Investigators are conducting an open-label safety study of ADS-5102 that includes patients from the phase III trials as well as patients with levodopa-induced dyskinesia who have undergone deep brain stimulation. Adamas also is investigating ADS-5102 for use in patients with multiple sclerosis with walking impairment.
—Jake Remaly

Ricardo Jorge, MD

High-Intensity Exercise May Be Feasible in Patients With Parkinson’s Disease
PORTLAND, OR—High-intensity exercise is feasible and safe in patients with Parkinson's disease, according to trial results presented at the Fourth World Parkinson Congress. An eight-week high-intensity exercise and fall-prevention boot camp was well attended, and participants tolerated high-intensity aerobic, strength, and balance training exercises, researchers said.
Most contemporary exercise programs for people with Parkinson's disease are low to moderate in intensity. Recent studies, however, suggest that older adults can safely tolerate high-intensity exercise programs and attain more meaningful benefits. "The optimal parameters of exercise for Parkinson's disease have not been thoroughly explored," said Merrill R. Landers, PT, DPT, PhD, OCS, Professor of Physical Therapy, and James Navalta, PhD, Associate Professor of Kinesiology, both at the University of Nevada in Las Vegas.

To test the feasibility and safety of a high-intensity exercise and fall-prevention boot camp in people with Parkinson's disease, Drs. Landers and Navalta conducted a phase II, pragmatic, randomized clinical trial.
Twenty-seven participants were included in the study. Fourteen patients were randomized to the high-intensity exercise program, and 13 patients were randomized to a low-intensity control program (ie, Fitness Counts Exercise Program). Physiotherapists supervised the exercise programs at community exercise gyms.
Patients had an average age of about 64 and disease duration of nearly five years. Nineteen men and eight women enrolled in the study. Most patients were Hoehn and Yahr stage 2.
The high-intensity boot camp sessions consisted of 30 minutes of aerobic exercise (70% of heart rate maximum), 30 minutes of strengthening (50% to 80% of one-repetition maximum), 15 minutes of balance training, and 15 minutes of active rest and stretching. The control sessions consisted of 15 minutes of aerobic exercise (60% of heart rate maximum), 15 minutes of strengthening (less than 50% of one-repetition maximum), 10 minutes of balance training, 10 minutes of rest, and 10 minutes of stretching. There was not a significant difference in the rate of injuries, falls, or exercise side effects between groups. Adverse events were similar in the two arms. Compared with low-intensity exercise, high-intensity exercise produced greater improvements in balance, physical activity, parkinsonian symptoms, endurance, fatigue, and bone health.
"These results warrant further investigation in a large-scale comparative effectiveness trial," the researchers concluded.
—Jake Remaly
PORTLAND, OR—High-intensity exercise is feasible and safe in patients with Parkinson's disease, according to trial results presented at the Fourth World Parkinson Congress. An eight-week high-intensity exercise and fall-prevention boot camp was well attended, and participants tolerated high-intensity aerobic, strength, and balance training exercises, researchers said.
Most contemporary exercise programs for people with Parkinson's disease are low to moderate in intensity. Recent studies, however, suggest that older adults can safely tolerate high-intensity exercise programs and attain more meaningful benefits. "The optimal parameters of exercise for Parkinson's disease have not been thoroughly explored," said Merrill R. Landers, PT, DPT, PhD, OCS, Professor of Physical Therapy, and James Navalta, PhD, Associate Professor of Kinesiology, both at the University of Nevada in Las Vegas.
To test the feasibility and safety of a high-intensity exercise and fall-prevention boot camp in people with Parkinson's disease, Drs. Landers and Navalta conducted a phase II, pragmatic, randomized clinical trial.
Twenty-seven participants were included in the study. Fourteen patients were randomized to the high-intensity exercise program, and 13 patients were randomized to a low-intensity control program (ie, Fitness Counts Exercise Program). Physiotherapists supervised the exercise programs at community exercise gyms.
Patients had an average age of about 64 and disease duration of nearly five years. Nineteen men and eight women enrolled in the study. Most patients were Hoehn and Yahr stage 2.
The high-intensity boot camp sessions consisted of 30 minutes of aerobic exercise (70% of heart rate maximum), 30 minutes of strengthening (50% to 80% of one-repetition maximum), 15 minutes of balance training, and 15 minutes of active rest and stretching. The control sessions consisted of 15 minutes of aerobic exercise (60% of heart rate maximum), 15 minutes of strengthening (less than 50% of one-repetition maximum), 10 minutes of balance training, 10 minutes of rest, and 10 minutes of stretching. There was not a significant difference in the rate of injuries, falls, or exercise side effects between groups. Adverse events were similar in the two arms. Compared with low-intensity exercise, high-intensity exercise produced greater improvements in balance, physical activity, parkinsonian symptoms, endurance, fatigue, and bone health.
"These results warrant further investigation in a large-scale comparative effectiveness trial," the researchers concluded.
—Jake Remaly
PORTLAND, OR—High-intensity exercise is feasible and safe in patients with Parkinson's disease, according to trial results presented at the Fourth World Parkinson Congress. An eight-week high-intensity exercise and fall-prevention boot camp was well attended, and participants tolerated high-intensity aerobic, strength, and balance training exercises, researchers said.
Most contemporary exercise programs for people with Parkinson's disease are low to moderate in intensity. Recent studies, however, suggest that older adults can safely tolerate high-intensity exercise programs and attain more meaningful benefits. "The optimal parameters of exercise for Parkinson's disease have not been thoroughly explored," said Merrill R. Landers, PT, DPT, PhD, OCS, Professor of Physical Therapy, and James Navalta, PhD, Associate Professor of Kinesiology, both at the University of Nevada in Las Vegas.
To test the feasibility and safety of a high-intensity exercise and fall-prevention boot camp in people with Parkinson's disease, Drs. Landers and Navalta conducted a phase II, pragmatic, randomized clinical trial.
Twenty-seven participants were included in the study. Fourteen patients were randomized to the high-intensity exercise program, and 13 patients were randomized to a low-intensity control program (ie, Fitness Counts Exercise Program). Physiotherapists supervised the exercise programs at community exercise gyms.
Patients had an average age of about 64 and disease duration of nearly five years. Nineteen men and eight women enrolled in the study. Most patients were Hoehn and Yahr stage 2.
The high-intensity boot camp sessions consisted of 30 minutes of aerobic exercise (70% of heart rate maximum), 30 minutes of strengthening (50% to 80% of one-repetition maximum), 15 minutes of balance training, and 15 minutes of active rest and stretching. The control sessions consisted of 15 minutes of aerobic exercise (60% of heart rate maximum), 15 minutes of strengthening (less than 50% of one-repetition maximum), 10 minutes of balance training, 10 minutes of rest, and 10 minutes of stretching. There was not a significant difference in the rate of injuries, falls, or exercise side effects between groups. Adverse events were similar in the two arms. Compared with low-intensity exercise, high-intensity exercise produced greater improvements in balance, physical activity, parkinsonian symptoms, endurance, fatigue, and bone health.
"These results warrant further investigation in a large-scale comparative effectiveness trial," the researchers concluded.
—Jake Remaly