User login
Diet in the pathophysiology and management of irritable bowel syndrome
Diet plays an important role in the pathophysiology of irritable bowel syndrome (IBS) and is an effective tool in managing this disorder. This includes a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs).
These indigestible and poorly absorbed short-chain carbohydrates trigger IBS symptoms and are thought to exert their effects by increasing osmotic pressure in the lumen of the intestine and by providing a substrate for bacterial fermentation with consequent gas production.1 The gas causes abdominal distention, and the change in pressure in the lumen of the large intestine affects the release of serotonin, causing abdominal pain and discomfort.
THE MECHANISMS ARE COMPLICATED
Recent studies have shown that the mechanisms by which FODMAPs exert their effects are more complicated than originally thought.1
All segments of the gastrointestinal tract contain endocrine cells scattered between the mucosal epithelial cells facing the intestinal lumen.1,2 There are at least 10 types of endocrine cell, and they regulate gastrointestinal motility, secretion, absorption, visceral sensitivity, local immune defense, cell proliferation, and appetite.2–4 Abnormal densities of gastrointestinal endocrine cells have been reported in patients with IBS, which may explain the dysmotility, visceral hypersensitivity, and abnormal intestinal secretion seen in these patients.5
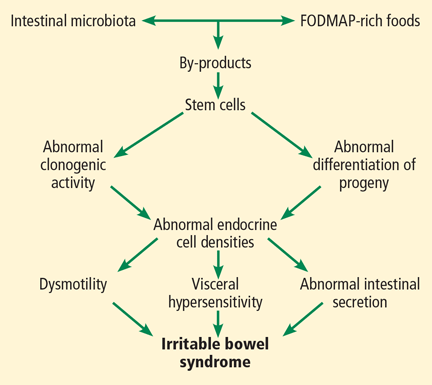
But other factors such as diet, intestinal microbiota, genetics, and low-grade inflammation also play pivotal roles in the pathophysiology of IBS by exerting effects on gastrointestinal endocrine cells. The abnormalities in the gastrointestinal endocrine cells in IBS are thought to be brought about by aberrant differentiation of stem cells into endocrine cells (Figure 1).6
A diet low in FODMAPs appears to induce changes in the intestinal microbiota and gastrointestinal endocrine cells and to reduce IBS symptoms.6
GLUTEN IS IMPLICATED
Another dietary factor in IBS is gluten. Symptoms of IBS and celiac disease overlap: most studies have found that fewer than 5% of patients with celiac disease are misdiagnosed as having IBS based on the symptom criteria for IBS, but some studies report a rate as high as 32%.7 In addition, 38% of patients with celiac disease who consume a gluten-free diet fulfill the symptom-based Rome criteria for IBS.7
The contribution of gluten to IBS does not end with the coexistence of IBS and celiac disease, but also includes the newly debated diagnosis of nonceliac gluten sensitivity, characterized by gastrointestinal symptoms (abdominal pain, diarrhea, constipation, nausea, and vomiting) and other symptoms (headache, musculoskeletal pain, “brain fog,” fatigue, and depression) that are similar to those of IBS. Symptoms are triggered by the ingestion of wheat products, are improved after wheat products are removed from the diet, and relapse after a wheat challenge.7
Nonceliac gluten sensitivity is often perceivable by patients, resulting in self-diagnosis and self-treatment.4 However, it is not clear whether it is gluten or the fructans and galactans in wheat that are responsible for triggering their symptoms.7
DIETARY GUIDANCE IS NEEDED
A low-FODMAP diet with small amounts of insoluble dietary fiber improves symptoms and quality of life in patients with IBS, but dietary guidance is critical and should be personalized because patients differ in how they tolerate foods rich in FODMAPs, probably owing to differing intestinal microbiota among individuals.3,8 Intake of probiotics increases tolerance of FODMAP-rich foods and should also be recommended.3,8,9 Because of the rigorous restrictions of the low-FODMAP diet, patients who receive personalized guidance are more inclined to adhere to the diet and to avoid vitamin and mineral deficiencies.9
- El-Salhy M, Gundersen D. Diet in irritable bowel syndrome. Nutr J 2015; 14:36–46.
- El-Salhy M, Seim I, Chopin L, Gundersen D, Hatlebakk JG, Hausken T. Irritable bowel syndrome: the role of gut neuroendocrine peptides. Front Biosci (Elite Ed) 2012; 4:2783–2800.
- El-Salhy M, Gundersen D, Hatlebakk JG, Hausken T. Irritable bowel syndrome: diagnosis, pathogenesis and treatment options. 1st ed. New York, NY: Nova Science Publishers, Inc.; 2014.
- El-Salhy M, Ostgaard H, Gundersen D, Hatlebakk JG, Hausken T. The role of diet in the pathogenesis and management of irritable bowel syndrome (review). Int J Mol Med 2012; 29:723–731.
- El-Salhy M, Gundersen D, Gilja OH, Hatlebakk JG, Hausken T. Is irritable bowel syndrome an organic disorder? World J Gastroenterol 2014; 20:384–400.
- El-Salhy M. Recent developments in the pathophysiology of irritable bowel syndrome. World J Gastroenterol 2015; 21:7621–7636.
- El-Salhy M, Hatlebakk JG, Gilja OH, Hausken T. The relation between celiac disease, nonceliac gluten sensitivity and irritable bowel syndrome. Nutr J 2015; 14:92–99.
- El-Salhy M, Lillebo E, Reinemo A, Salmelid L, Hausken T. Effects of a health program comprising reassurance, diet management, probiotic administration and regular exercise on symptoms and quality of life in patients with irritable bowel syndrome. Gastroenterology Insights 2010; 2:21–26. doi: http://dx.doi.org/10.4081/gi.2010.e6.
- Ostgaard H, Hausken T, Gundersen D, El-Salhy M. Diet and effects of diet management on quality of life and symptoms in patients with irritable bowel syndrome. Mol Med Rep 2012; 5:1382–1390.
Diet plays an important role in the pathophysiology of irritable bowel syndrome (IBS) and is an effective tool in managing this disorder. This includes a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs).
These indigestible and poorly absorbed short-chain carbohydrates trigger IBS symptoms and are thought to exert their effects by increasing osmotic pressure in the lumen of the intestine and by providing a substrate for bacterial fermentation with consequent gas production.1 The gas causes abdominal distention, and the change in pressure in the lumen of the large intestine affects the release of serotonin, causing abdominal pain and discomfort.
THE MECHANISMS ARE COMPLICATED
Recent studies have shown that the mechanisms by which FODMAPs exert their effects are more complicated than originally thought.1
All segments of the gastrointestinal tract contain endocrine cells scattered between the mucosal epithelial cells facing the intestinal lumen.1,2 There are at least 10 types of endocrine cell, and they regulate gastrointestinal motility, secretion, absorption, visceral sensitivity, local immune defense, cell proliferation, and appetite.2–4 Abnormal densities of gastrointestinal endocrine cells have been reported in patients with IBS, which may explain the dysmotility, visceral hypersensitivity, and abnormal intestinal secretion seen in these patients.5
But other factors such as diet, intestinal microbiota, genetics, and low-grade inflammation also play pivotal roles in the pathophysiology of IBS by exerting effects on gastrointestinal endocrine cells. The abnormalities in the gastrointestinal endocrine cells in IBS are thought to be brought about by aberrant differentiation of stem cells into endocrine cells (Figure 1).6
A diet low in FODMAPs appears to induce changes in the intestinal microbiota and gastrointestinal endocrine cells and to reduce IBS symptoms.6
GLUTEN IS IMPLICATED
Another dietary factor in IBS is gluten. Symptoms of IBS and celiac disease overlap: most studies have found that fewer than 5% of patients with celiac disease are misdiagnosed as having IBS based on the symptom criteria for IBS, but some studies report a rate as high as 32%.7 In addition, 38% of patients with celiac disease who consume a gluten-free diet fulfill the symptom-based Rome criteria for IBS.7
The contribution of gluten to IBS does not end with the coexistence of IBS and celiac disease, but also includes the newly debated diagnosis of nonceliac gluten sensitivity, characterized by gastrointestinal symptoms (abdominal pain, diarrhea, constipation, nausea, and vomiting) and other symptoms (headache, musculoskeletal pain, “brain fog,” fatigue, and depression) that are similar to those of IBS. Symptoms are triggered by the ingestion of wheat products, are improved after wheat products are removed from the diet, and relapse after a wheat challenge.7
Nonceliac gluten sensitivity is often perceivable by patients, resulting in self-diagnosis and self-treatment.4 However, it is not clear whether it is gluten or the fructans and galactans in wheat that are responsible for triggering their symptoms.7
DIETARY GUIDANCE IS NEEDED
A low-FODMAP diet with small amounts of insoluble dietary fiber improves symptoms and quality of life in patients with IBS, but dietary guidance is critical and should be personalized because patients differ in how they tolerate foods rich in FODMAPs, probably owing to differing intestinal microbiota among individuals.3,8 Intake of probiotics increases tolerance of FODMAP-rich foods and should also be recommended.3,8,9 Because of the rigorous restrictions of the low-FODMAP diet, patients who receive personalized guidance are more inclined to adhere to the diet and to avoid vitamin and mineral deficiencies.9
Diet plays an important role in the pathophysiology of irritable bowel syndrome (IBS) and is an effective tool in managing this disorder. This includes a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs).
These indigestible and poorly absorbed short-chain carbohydrates trigger IBS symptoms and are thought to exert their effects by increasing osmotic pressure in the lumen of the intestine and by providing a substrate for bacterial fermentation with consequent gas production.1 The gas causes abdominal distention, and the change in pressure in the lumen of the large intestine affects the release of serotonin, causing abdominal pain and discomfort.
THE MECHANISMS ARE COMPLICATED
Recent studies have shown that the mechanisms by which FODMAPs exert their effects are more complicated than originally thought.1
All segments of the gastrointestinal tract contain endocrine cells scattered between the mucosal epithelial cells facing the intestinal lumen.1,2 There are at least 10 types of endocrine cell, and they regulate gastrointestinal motility, secretion, absorption, visceral sensitivity, local immune defense, cell proliferation, and appetite.2–4 Abnormal densities of gastrointestinal endocrine cells have been reported in patients with IBS, which may explain the dysmotility, visceral hypersensitivity, and abnormal intestinal secretion seen in these patients.5
But other factors such as diet, intestinal microbiota, genetics, and low-grade inflammation also play pivotal roles in the pathophysiology of IBS by exerting effects on gastrointestinal endocrine cells. The abnormalities in the gastrointestinal endocrine cells in IBS are thought to be brought about by aberrant differentiation of stem cells into endocrine cells (Figure 1).6
A diet low in FODMAPs appears to induce changes in the intestinal microbiota and gastrointestinal endocrine cells and to reduce IBS symptoms.6
GLUTEN IS IMPLICATED
Another dietary factor in IBS is gluten. Symptoms of IBS and celiac disease overlap: most studies have found that fewer than 5% of patients with celiac disease are misdiagnosed as having IBS based on the symptom criteria for IBS, but some studies report a rate as high as 32%.7 In addition, 38% of patients with celiac disease who consume a gluten-free diet fulfill the symptom-based Rome criteria for IBS.7
The contribution of gluten to IBS does not end with the coexistence of IBS and celiac disease, but also includes the newly debated diagnosis of nonceliac gluten sensitivity, characterized by gastrointestinal symptoms (abdominal pain, diarrhea, constipation, nausea, and vomiting) and other symptoms (headache, musculoskeletal pain, “brain fog,” fatigue, and depression) that are similar to those of IBS. Symptoms are triggered by the ingestion of wheat products, are improved after wheat products are removed from the diet, and relapse after a wheat challenge.7
Nonceliac gluten sensitivity is often perceivable by patients, resulting in self-diagnosis and self-treatment.4 However, it is not clear whether it is gluten or the fructans and galactans in wheat that are responsible for triggering their symptoms.7
DIETARY GUIDANCE IS NEEDED
A low-FODMAP diet with small amounts of insoluble dietary fiber improves symptoms and quality of life in patients with IBS, but dietary guidance is critical and should be personalized because patients differ in how they tolerate foods rich in FODMAPs, probably owing to differing intestinal microbiota among individuals.3,8 Intake of probiotics increases tolerance of FODMAP-rich foods and should also be recommended.3,8,9 Because of the rigorous restrictions of the low-FODMAP diet, patients who receive personalized guidance are more inclined to adhere to the diet and to avoid vitamin and mineral deficiencies.9
- El-Salhy M, Gundersen D. Diet in irritable bowel syndrome. Nutr J 2015; 14:36–46.
- El-Salhy M, Seim I, Chopin L, Gundersen D, Hatlebakk JG, Hausken T. Irritable bowel syndrome: the role of gut neuroendocrine peptides. Front Biosci (Elite Ed) 2012; 4:2783–2800.
- El-Salhy M, Gundersen D, Hatlebakk JG, Hausken T. Irritable bowel syndrome: diagnosis, pathogenesis and treatment options. 1st ed. New York, NY: Nova Science Publishers, Inc.; 2014.
- El-Salhy M, Ostgaard H, Gundersen D, Hatlebakk JG, Hausken T. The role of diet in the pathogenesis and management of irritable bowel syndrome (review). Int J Mol Med 2012; 29:723–731.
- El-Salhy M, Gundersen D, Gilja OH, Hatlebakk JG, Hausken T. Is irritable bowel syndrome an organic disorder? World J Gastroenterol 2014; 20:384–400.
- El-Salhy M. Recent developments in the pathophysiology of irritable bowel syndrome. World J Gastroenterol 2015; 21:7621–7636.
- El-Salhy M, Hatlebakk JG, Gilja OH, Hausken T. The relation between celiac disease, nonceliac gluten sensitivity and irritable bowel syndrome. Nutr J 2015; 14:92–99.
- El-Salhy M, Lillebo E, Reinemo A, Salmelid L, Hausken T. Effects of a health program comprising reassurance, diet management, probiotic administration and regular exercise on symptoms and quality of life in patients with irritable bowel syndrome. Gastroenterology Insights 2010; 2:21–26. doi: http://dx.doi.org/10.4081/gi.2010.e6.
- Ostgaard H, Hausken T, Gundersen D, El-Salhy M. Diet and effects of diet management on quality of life and symptoms in patients with irritable bowel syndrome. Mol Med Rep 2012; 5:1382–1390.
- El-Salhy M, Gundersen D. Diet in irritable bowel syndrome. Nutr J 2015; 14:36–46.
- El-Salhy M, Seim I, Chopin L, Gundersen D, Hatlebakk JG, Hausken T. Irritable bowel syndrome: the role of gut neuroendocrine peptides. Front Biosci (Elite Ed) 2012; 4:2783–2800.
- El-Salhy M, Gundersen D, Hatlebakk JG, Hausken T. Irritable bowel syndrome: diagnosis, pathogenesis and treatment options. 1st ed. New York, NY: Nova Science Publishers, Inc.; 2014.
- El-Salhy M, Ostgaard H, Gundersen D, Hatlebakk JG, Hausken T. The role of diet in the pathogenesis and management of irritable bowel syndrome (review). Int J Mol Med 2012; 29:723–731.
- El-Salhy M, Gundersen D, Gilja OH, Hatlebakk JG, Hausken T. Is irritable bowel syndrome an organic disorder? World J Gastroenterol 2014; 20:384–400.
- El-Salhy M. Recent developments in the pathophysiology of irritable bowel syndrome. World J Gastroenterol 2015; 21:7621–7636.
- El-Salhy M, Hatlebakk JG, Gilja OH, Hausken T. The relation between celiac disease, nonceliac gluten sensitivity and irritable bowel syndrome. Nutr J 2015; 14:92–99.
- El-Salhy M, Lillebo E, Reinemo A, Salmelid L, Hausken T. Effects of a health program comprising reassurance, diet management, probiotic administration and regular exercise on symptoms and quality of life in patients with irritable bowel syndrome. Gastroenterology Insights 2010; 2:21–26. doi: http://dx.doi.org/10.4081/gi.2010.e6.
- Ostgaard H, Hausken T, Gundersen D, El-Salhy M. Diet and effects of diet management on quality of life and symptoms in patients with irritable bowel syndrome. Mol Med Rep 2012; 5:1382–1390.
FDA: New labeling warns against combining opioids, benzodiazepines
Labeling for prescription opioid pain or cough medicines and benzodiazepines will now carry the strongest available warning regarding serious side effects and death associated with their combined use, according to the Food and Drug Administration.
The new boxed warnings urge health care professionals to limit prescribing opioid pain medicines with benzodiazepines or other central nervous system depressants only to patients for whom alternative treatment options are inadequate, and to limit dosages and treatment duration to the minimum possible while achieving the desired clinical effect.
![]()
“First, the FDA is requiring companies to update their product labeling for ... benzodiazepines and opioids to include possible harms when they are used together. Second, we are requiring new or updated medication guides for these drugs reflecting those same warnings,” said Doug Throckmorton, MD, deputy director of the FDA’s Center for Drug Evaluation and Research, during a telebriefing.
Opioids will include a warning regarding prescribing with benzodiazepines and other central nervous system depressants, including alcohol. Benzodiazepines will include a warning regarding prescribing with opioids.
In addition, the FDA has issued a safety communication to “warn the public about the serious risk of taking these products together to help make doctors more cautious and patients better informed,” Dr. Throckmorton said.
The action comes amid ongoing efforts to address an epidemic of opioid addiction across the United States, and in response to a first-of-its-kind “citizen petition” calling for the boxed warnings.
A coalition of health officials from multiple cities, states, and U.S. territories initiated that petition in February, and thousands of concerned community members started an additional online petition. Those petitions were in response to both the increasing combined use of opioids and benzodiazepines and a concomitant increase in the risk of serious side effects and deaths associated with their combined use, according to Baltimore City Health Commissioner Leana Wen, MD.
As an emergency physician, Dr. Wen said that she has seen firsthand the alarming trends; one in three unintentional overdose deaths from prescribed opioids also involve benzodiazepines, she noted.
“In my state of Maryland in 2014, benzodiazepines were associated with 19% of prescription opioid deaths, and 59% of benzodiazepine-associated deaths involved prescription opioids. We also noted the growing biological evidence that combining these medications caused sleepiness and slowed breathing, increasing the likelihood of a fatal overdose,” she said.
Dr. Throckmorton further noted that emergency department visits and deaths involving patients prescribed both opioids and benzodiazepines have increased significantly over time. From 2004 to 2011, the rate of nonmedical use–related emergency department visits increased significantly each year, and overdose deaths involving both drug classes during that period nearly tripled on an annual basis.

“Communities have been seeing this trend for some time, but ultimately we needed data in order to act today,” FDA Commissioner Robert Califf, MD, said during the telebriefing.
The current action is just “one part of a larger effort to address this epidemic.
“We remain focused and deeply committed to contributing to the comprehensive effort to address the opioid epidemic,” Dr. Califf said. The FDA “will continue to monitor these products carefully and take additional actions as needed, and will share updates with the public as necessary as we work to address this public health crisis.”
Dr. Califf noted that the current action is part of the FDA’s Opioids Action Plan, which is “importantly not meant just to cover illicit or abusive use of opioids.”
“So, you’ll be hearing a lot more from us, because this is a national crisis that is not going away. We’re making progress on the prescribing, and we’re seeing a reduction in the use of opioids now,” he noted. “But we’re still seeing many overdoses.
“This is a continuum, and we’ll continue to try to do everything we can to address the epidemic,” Dr. Califf concluded.
Labeling for prescription opioid pain or cough medicines and benzodiazepines will now carry the strongest available warning regarding serious side effects and death associated with their combined use, according to the Food and Drug Administration.
The new boxed warnings urge health care professionals to limit prescribing opioid pain medicines with benzodiazepines or other central nervous system depressants only to patients for whom alternative treatment options are inadequate, and to limit dosages and treatment duration to the minimum possible while achieving the desired clinical effect.
![]()
“First, the FDA is requiring companies to update their product labeling for ... benzodiazepines and opioids to include possible harms when they are used together. Second, we are requiring new or updated medication guides for these drugs reflecting those same warnings,” said Doug Throckmorton, MD, deputy director of the FDA’s Center for Drug Evaluation and Research, during a telebriefing.
Opioids will include a warning regarding prescribing with benzodiazepines and other central nervous system depressants, including alcohol. Benzodiazepines will include a warning regarding prescribing with opioids.
In addition, the FDA has issued a safety communication to “warn the public about the serious risk of taking these products together to help make doctors more cautious and patients better informed,” Dr. Throckmorton said.
The action comes amid ongoing efforts to address an epidemic of opioid addiction across the United States, and in response to a first-of-its-kind “citizen petition” calling for the boxed warnings.
A coalition of health officials from multiple cities, states, and U.S. territories initiated that petition in February, and thousands of concerned community members started an additional online petition. Those petitions were in response to both the increasing combined use of opioids and benzodiazepines and a concomitant increase in the risk of serious side effects and deaths associated with their combined use, according to Baltimore City Health Commissioner Leana Wen, MD.
As an emergency physician, Dr. Wen said that she has seen firsthand the alarming trends; one in three unintentional overdose deaths from prescribed opioids also involve benzodiazepines, she noted.
“In my state of Maryland in 2014, benzodiazepines were associated with 19% of prescription opioid deaths, and 59% of benzodiazepine-associated deaths involved prescription opioids. We also noted the growing biological evidence that combining these medications caused sleepiness and slowed breathing, increasing the likelihood of a fatal overdose,” she said.
Dr. Throckmorton further noted that emergency department visits and deaths involving patients prescribed both opioids and benzodiazepines have increased significantly over time. From 2004 to 2011, the rate of nonmedical use–related emergency department visits increased significantly each year, and overdose deaths involving both drug classes during that period nearly tripled on an annual basis.
“Communities have been seeing this trend for some time, but ultimately we needed data in order to act today,” FDA Commissioner Robert Califf, MD, said during the telebriefing.
The current action is just “one part of a larger effort to address this epidemic.
“We remain focused and deeply committed to contributing to the comprehensive effort to address the opioid epidemic,” Dr. Califf said. The FDA “will continue to monitor these products carefully and take additional actions as needed, and will share updates with the public as necessary as we work to address this public health crisis.”
Dr. Califf noted that the current action is part of the FDA’s Opioids Action Plan, which is “importantly not meant just to cover illicit or abusive use of opioids.”
“So, you’ll be hearing a lot more from us, because this is a national crisis that is not going away. We’re making progress on the prescribing, and we’re seeing a reduction in the use of opioids now,” he noted. “But we’re still seeing many overdoses.
“This is a continuum, and we’ll continue to try to do everything we can to address the epidemic,” Dr. Califf concluded.
Labeling for prescription opioid pain or cough medicines and benzodiazepines will now carry the strongest available warning regarding serious side effects and death associated with their combined use, according to the Food and Drug Administration.
The new boxed warnings urge health care professionals to limit prescribing opioid pain medicines with benzodiazepines or other central nervous system depressants only to patients for whom alternative treatment options are inadequate, and to limit dosages and treatment duration to the minimum possible while achieving the desired clinical effect.
![]()
“First, the FDA is requiring companies to update their product labeling for ... benzodiazepines and opioids to include possible harms when they are used together. Second, we are requiring new or updated medication guides for these drugs reflecting those same warnings,” said Doug Throckmorton, MD, deputy director of the FDA’s Center for Drug Evaluation and Research, during a telebriefing.
Opioids will include a warning regarding prescribing with benzodiazepines and other central nervous system depressants, including alcohol. Benzodiazepines will include a warning regarding prescribing with opioids.
In addition, the FDA has issued a safety communication to “warn the public about the serious risk of taking these products together to help make doctors more cautious and patients better informed,” Dr. Throckmorton said.
The action comes amid ongoing efforts to address an epidemic of opioid addiction across the United States, and in response to a first-of-its-kind “citizen petition” calling for the boxed warnings.
A coalition of health officials from multiple cities, states, and U.S. territories initiated that petition in February, and thousands of concerned community members started an additional online petition. Those petitions were in response to both the increasing combined use of opioids and benzodiazepines and a concomitant increase in the risk of serious side effects and deaths associated with their combined use, according to Baltimore City Health Commissioner Leana Wen, MD.
As an emergency physician, Dr. Wen said that she has seen firsthand the alarming trends; one in three unintentional overdose deaths from prescribed opioids also involve benzodiazepines, she noted.
“In my state of Maryland in 2014, benzodiazepines were associated with 19% of prescription opioid deaths, and 59% of benzodiazepine-associated deaths involved prescription opioids. We also noted the growing biological evidence that combining these medications caused sleepiness and slowed breathing, increasing the likelihood of a fatal overdose,” she said.
Dr. Throckmorton further noted that emergency department visits and deaths involving patients prescribed both opioids and benzodiazepines have increased significantly over time. From 2004 to 2011, the rate of nonmedical use–related emergency department visits increased significantly each year, and overdose deaths involving both drug classes during that period nearly tripled on an annual basis.
“Communities have been seeing this trend for some time, but ultimately we needed data in order to act today,” FDA Commissioner Robert Califf, MD, said during the telebriefing.
The current action is just “one part of a larger effort to address this epidemic.
“We remain focused and deeply committed to contributing to the comprehensive effort to address the opioid epidemic,” Dr. Califf said. The FDA “will continue to monitor these products carefully and take additional actions as needed, and will share updates with the public as necessary as we work to address this public health crisis.”
Dr. Califf noted that the current action is part of the FDA’s Opioids Action Plan, which is “importantly not meant just to cover illicit or abusive use of opioids.”
“So, you’ll be hearing a lot more from us, because this is a national crisis that is not going away. We’re making progress on the prescribing, and we’re seeing a reduction in the use of opioids now,” he noted. “But we’re still seeing many overdoses.
“This is a continuum, and we’ll continue to try to do everything we can to address the epidemic,” Dr. Califf concluded.
Drugs in the pipeline hold promise for atopic dermatitis
NEWPORT BEACH, CALIF. – In the clinical opinion of Kelly M. Cordoro, MD, anyone who cares for patients with severe atopic dermatitis understands the sense of misery that can ensue.
“Atopic dermatitis patients don’t sleep well; they have poor school and work performance,” she said at the annual meeting of the Pacific Dermatologic Association. “They have absences. They’re unable to play; they can’t exercise. This leads to social disability; isolation from peers, and it goes on and on. The patients are miserable, the whole family is miserable, and we as physicians trying to sort out how to optimally treat them are miserable trying to figure out what the next best step is.”

The good news is, several drugs in the pipeline hold promise for atopic dermatitis patients, thanks largely to emerging data on its pathophysiology. In addition, mechanisms of itch, which are not yet fully understood, are also being unraveled. “It’s exciting to read the literature about the interaction of the skin, the immune system, and the nervous system,” said Dr. Cordoro, a pediatric dermatologist at the University of California, San Francisco. “Many of the mediators of itch are being identified. That has allowed for the development of targeted therapies against many of them.”
One of the promising treatments on the horizon for atopic dermatitis patients is phosphodiesterase-4 (PDE4) inhibitors. PDE-4 is a predominant cAMP-degrading enzyme in keratinocytes and inflammatory cells. “It’s really a candidate for not only atopic dermatitis but for psoriasis,” she said.
Oral PDE-4 inhibitors are already approved for psoriasis. Apremilast (Otezla) was approved by the Food and Drug Administration in 2014 for psoriasis and psoriatic arthritis, and a phase II trial of topical apremilast in adults with AD has been completed and the results are pending. “I look forward to seeing if this can help our patients,” Dr. Cordoro said.
Another promising agent for atopic dermatitis is 2% crisaborole topical ointment, a boron-based PDE-4 inhibitor developed by Anacor Pharmaceuticals. Dr. Cordoro described this compound as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways.
“Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said (J Am Acad Dermatol. 2016 Sept;75[3]:494-503.e). The FDA review of crisaborole for the treatment of mild to moderate atopic dermatitis in children and adults is currently underway, and is expected to be completed by early January 2017.
An especially favorable drug in development for atopic dermatitis is dupilumab, a fully human monoclonal antibody that targets the interleukin (IL)–4 receptor, and inhibits IL-4 and IL-13 signaling. A published trial of its use in adults with moderate to severe atopic dermatitis showed rapid improvements in all atopic dermatitis clinical indices (N Engl J Med. 2014;371[2]:130-9). The most common side effects were headache and pharyngitis, and skin infections and flares were more common in the placebo group, compared with the treatment group.
Dupilumab “has the potential to shift the treatment landscape of atopic dermatitis, because it can actually change the molecular signature of dermatitic skin, reducing inflammatory and proliferative markers,” Dr. Cordoro said. There are ongoing trials in adult and pediatric populations and FDA approval is anticipated in early 2017.
Published reports also suggest a role for the IL-12/23 pathway inhibitor ustekinumab in severe refractory adult atopic dermatitis (Int J Dermatol. 2012;51[1]:115-6 and JAAD Case Reports 2015;1:25-6). Additional studies are ongoing.
Therapies for itch that have completed phase II trials include the anti-IL31R monoclonal antibody nemolizumab (CIM331); the neurokinin-1R antagonist VLY-686; and the neurokinin-1R antagonist aprepitant gel.
Dr. Cordoro disclosed that she is a consultant for Celgene Corporation, Valeant, and Anacor Pharmaceuticals.
NEWPORT BEACH, CALIF. – In the clinical opinion of Kelly M. Cordoro, MD, anyone who cares for patients with severe atopic dermatitis understands the sense of misery that can ensue.
“Atopic dermatitis patients don’t sleep well; they have poor school and work performance,” she said at the annual meeting of the Pacific Dermatologic Association. “They have absences. They’re unable to play; they can’t exercise. This leads to social disability; isolation from peers, and it goes on and on. The patients are miserable, the whole family is miserable, and we as physicians trying to sort out how to optimally treat them are miserable trying to figure out what the next best step is.”
The good news is, several drugs in the pipeline hold promise for atopic dermatitis patients, thanks largely to emerging data on its pathophysiology. In addition, mechanisms of itch, which are not yet fully understood, are also being unraveled. “It’s exciting to read the literature about the interaction of the skin, the immune system, and the nervous system,” said Dr. Cordoro, a pediatric dermatologist at the University of California, San Francisco. “Many of the mediators of itch are being identified. That has allowed for the development of targeted therapies against many of them.”
One of the promising treatments on the horizon for atopic dermatitis patients is phosphodiesterase-4 (PDE4) inhibitors. PDE-4 is a predominant cAMP-degrading enzyme in keratinocytes and inflammatory cells. “It’s really a candidate for not only atopic dermatitis but for psoriasis,” she said.
Oral PDE-4 inhibitors are already approved for psoriasis. Apremilast (Otezla) was approved by the Food and Drug Administration in 2014 for psoriasis and psoriatic arthritis, and a phase II trial of topical apremilast in adults with AD has been completed and the results are pending. “I look forward to seeing if this can help our patients,” Dr. Cordoro said.
Another promising agent for atopic dermatitis is 2% crisaborole topical ointment, a boron-based PDE-4 inhibitor developed by Anacor Pharmaceuticals. Dr. Cordoro described this compound as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways.
“Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said (J Am Acad Dermatol. 2016 Sept;75[3]:494-503.e). The FDA review of crisaborole for the treatment of mild to moderate atopic dermatitis in children and adults is currently underway, and is expected to be completed by early January 2017.
An especially favorable drug in development for atopic dermatitis is dupilumab, a fully human monoclonal antibody that targets the interleukin (IL)–4 receptor, and inhibits IL-4 and IL-13 signaling. A published trial of its use in adults with moderate to severe atopic dermatitis showed rapid improvements in all atopic dermatitis clinical indices (N Engl J Med. 2014;371[2]:130-9). The most common side effects were headache and pharyngitis, and skin infections and flares were more common in the placebo group, compared with the treatment group.
Dupilumab “has the potential to shift the treatment landscape of atopic dermatitis, because it can actually change the molecular signature of dermatitic skin, reducing inflammatory and proliferative markers,” Dr. Cordoro said. There are ongoing trials in adult and pediatric populations and FDA approval is anticipated in early 2017.
Published reports also suggest a role for the IL-12/23 pathway inhibitor ustekinumab in severe refractory adult atopic dermatitis (Int J Dermatol. 2012;51[1]:115-6 and JAAD Case Reports 2015;1:25-6). Additional studies are ongoing.
Therapies for itch that have completed phase II trials include the anti-IL31R monoclonal antibody nemolizumab (CIM331); the neurokinin-1R antagonist VLY-686; and the neurokinin-1R antagonist aprepitant gel.
Dr. Cordoro disclosed that she is a consultant for Celgene Corporation, Valeant, and Anacor Pharmaceuticals.
NEWPORT BEACH, CALIF. – In the clinical opinion of Kelly M. Cordoro, MD, anyone who cares for patients with severe atopic dermatitis understands the sense of misery that can ensue.
“Atopic dermatitis patients don’t sleep well; they have poor school and work performance,” she said at the annual meeting of the Pacific Dermatologic Association. “They have absences. They’re unable to play; they can’t exercise. This leads to social disability; isolation from peers, and it goes on and on. The patients are miserable, the whole family is miserable, and we as physicians trying to sort out how to optimally treat them are miserable trying to figure out what the next best step is.”
The good news is, several drugs in the pipeline hold promise for atopic dermatitis patients, thanks largely to emerging data on its pathophysiology. In addition, mechanisms of itch, which are not yet fully understood, are also being unraveled. “It’s exciting to read the literature about the interaction of the skin, the immune system, and the nervous system,” said Dr. Cordoro, a pediatric dermatologist at the University of California, San Francisco. “Many of the mediators of itch are being identified. That has allowed for the development of targeted therapies against many of them.”
One of the promising treatments on the horizon for atopic dermatitis patients is phosphodiesterase-4 (PDE4) inhibitors. PDE-4 is a predominant cAMP-degrading enzyme in keratinocytes and inflammatory cells. “It’s really a candidate for not only atopic dermatitis but for psoriasis,” she said.
Oral PDE-4 inhibitors are already approved for psoriasis. Apremilast (Otezla) was approved by the Food and Drug Administration in 2014 for psoriasis and psoriatic arthritis, and a phase II trial of topical apremilast in adults with AD has been completed and the results are pending. “I look forward to seeing if this can help our patients,” Dr. Cordoro said.
Another promising agent for atopic dermatitis is 2% crisaborole topical ointment, a boron-based PDE-4 inhibitor developed by Anacor Pharmaceuticals. Dr. Cordoro described this compound as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways.
“Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said (J Am Acad Dermatol. 2016 Sept;75[3]:494-503.e). The FDA review of crisaborole for the treatment of mild to moderate atopic dermatitis in children and adults is currently underway, and is expected to be completed by early January 2017.
An especially favorable drug in development for atopic dermatitis is dupilumab, a fully human monoclonal antibody that targets the interleukin (IL)–4 receptor, and inhibits IL-4 and IL-13 signaling. A published trial of its use in adults with moderate to severe atopic dermatitis showed rapid improvements in all atopic dermatitis clinical indices (N Engl J Med. 2014;371[2]:130-9). The most common side effects were headache and pharyngitis, and skin infections and flares were more common in the placebo group, compared with the treatment group.
Dupilumab “has the potential to shift the treatment landscape of atopic dermatitis, because it can actually change the molecular signature of dermatitic skin, reducing inflammatory and proliferative markers,” Dr. Cordoro said. There are ongoing trials in adult and pediatric populations and FDA approval is anticipated in early 2017.
Published reports also suggest a role for the IL-12/23 pathway inhibitor ustekinumab in severe refractory adult atopic dermatitis (Int J Dermatol. 2012;51[1]:115-6 and JAAD Case Reports 2015;1:25-6). Additional studies are ongoing.
Therapies for itch that have completed phase II trials include the anti-IL31R monoclonal antibody nemolizumab (CIM331); the neurokinin-1R antagonist VLY-686; and the neurokinin-1R antagonist aprepitant gel.
Dr. Cordoro disclosed that she is a consultant for Celgene Corporation, Valeant, and Anacor Pharmaceuticals.
EXPERT ANALYSIS FROM PDA 2016

Adding salmeterol to steroids didn’t boost kids’ serious asthma events
Adding the long-acting beta-agonist salmeterol to fluticasone in a fixed-dose combination didn’t increase serious asthma-related events among children aged 4-11 years, according to a report published online Sept. 1 in the New England Journal of Medicine.
After long-acting beta-agonists were introduced as add-on therapy for uncontrolled asthma, two large studies involving adults linked the treatment to an increase in asthma-related death. Other studies found no such association.
The FDA mandated that all four manufacturers of those agents in the United States perform large postmarketing safety trials to establish the noninferiority of the approach. In response, GlaxoSmithKline, the only maker of a long-acting beta-agonist with a pediatric indication (salmeterol), performed this international randomized, double-blind, controlled trial at 567 medical centers in 32 countries, said David A. Stempel, MD, of Respiratory Clinical Development, GSK, Research Triangle Park, N.C., and his associates.
The trial involved 6,208 children aged 4-11 years who had controlled or uncontrolled asthma with a history of exacerbations during the preceding year. The participants were randomly assigned to receive 26 weeks of a lower fixed-dose combination of salmeterol plus fluticasone, a higher fixed-dose combination, a lower dose of fluticasone alone, or a higher dose of fluticasone alone, delivered twice daily via a disk device.
The primary safety endpoint was a composite of death, endotracheal intubation, and hospitalization. No deaths or intubations occurred.
A total of 27 patients taking combined therapy and 21 taking fluticasone alone required hospitalization for asthma (hazard ratio, 1.28). The number of severe asthma exacerbations was 14% lower when salmeterol was added to fluticasone, a nonsignificant difference.
The results demonstrate the noninferiority of the combined therapy, Dr. Stempel and his associates said (N Engl J Med. 2016 Sep 1;375[9]:840-9).
The percentage of children who withdrew from the study because of asthma exacerbations was identical in the two groups (1.1% of each), and the percentage who had a serious adverse event was nearly identical (1.8% vs 1.7%, respectively). The mean percentage of rescue therapy–free days also was similar (83.0% vs 81.9%), as was the mean percentage of days in which asthma was controlled (74.8% vs. 73.4%).
At the conclusion of the study, 88.1% of the fluticasone-plus-salmeterol group had controlled asthma, as did 88.5% of the fluticasone-only group. Meaningful differences between the two treatments could not be identified among various subgroups of patients – defined by age, sex, and race – because the overall number of adverse events was so low, the investigators added.
They cautioned that the trial excluded children who had a history of multiple asthma-related hospitalizations and intubations. Therefore, the findings may not be applicable to patients with very severe asthma, the researchers cautioned.
GlaxoSmithKline sponsored the trial in response to a Food and Drug Administration mandate for large postmarketing safety studies from the marketers of long-acting beta agonist–containing products sold in the United States. Dr. Stempel is an employee of GSK; his associates reported ties to numerous industry sources.
These study findings provide reassuring evidence that combination inhalers are safe for the unusual child with asthma who needs more than inhaled glucocorticoids to control the disease or who has persistent, objectively documented variable airflow obstruction.

|
Dr. Andrew Bush |
But it’s important to emphasize that a combined inhaler is never indicated as first-line preventive therapy in children, because such use is increasingly creeping into practice. And monotherapy with a long-acting beta-agonist in a child should be considered medical negligence.
Andrew Bush, MD, is in the department of respiratory medicine at Royal Brompton Hospital, London. Urs Frey, MD, PhD, is in the department of pediatrics at the University of Basel (Switzerland) Children’s Hospital. They reported having no relevant financial disclosures. Dr. Bush and Dr. Frey made these remarks in an editorial accompanying Dr. Stempel’s report (N Engl J Med. 2016 Sep 1;375[9]:889-91).
These study findings provide reassuring evidence that combination inhalers are safe for the unusual child with asthma who needs more than inhaled glucocorticoids to control the disease or who has persistent, objectively documented variable airflow obstruction.
|
|
Dr. Andrew Bush |
But it’s important to emphasize that a combined inhaler is never indicated as first-line preventive therapy in children, because such use is increasingly creeping into practice. And monotherapy with a long-acting beta-agonist in a child should be considered medical negligence.
Andrew Bush, MD, is in the department of respiratory medicine at Royal Brompton Hospital, London. Urs Frey, MD, PhD, is in the department of pediatrics at the University of Basel (Switzerland) Children’s Hospital. They reported having no relevant financial disclosures. Dr. Bush and Dr. Frey made these remarks in an editorial accompanying Dr. Stempel’s report (N Engl J Med. 2016 Sep 1;375[9]:889-91).
These study findings provide reassuring evidence that combination inhalers are safe for the unusual child with asthma who needs more than inhaled glucocorticoids to control the disease or who has persistent, objectively documented variable airflow obstruction.
|
|
Dr. Andrew Bush |
But it’s important to emphasize that a combined inhaler is never indicated as first-line preventive therapy in children, because such use is increasingly creeping into practice. And monotherapy with a long-acting beta-agonist in a child should be considered medical negligence.
Andrew Bush, MD, is in the department of respiratory medicine at Royal Brompton Hospital, London. Urs Frey, MD, PhD, is in the department of pediatrics at the University of Basel (Switzerland) Children’s Hospital. They reported having no relevant financial disclosures. Dr. Bush and Dr. Frey made these remarks in an editorial accompanying Dr. Stempel’s report (N Engl J Med. 2016 Sep 1;375[9]:889-91).
Adding the long-acting beta-agonist salmeterol to fluticasone in a fixed-dose combination didn’t increase serious asthma-related events among children aged 4-11 years, according to a report published online Sept. 1 in the New England Journal of Medicine.
After long-acting beta-agonists were introduced as add-on therapy for uncontrolled asthma, two large studies involving adults linked the treatment to an increase in asthma-related death. Other studies found no such association.
The FDA mandated that all four manufacturers of those agents in the United States perform large postmarketing safety trials to establish the noninferiority of the approach. In response, GlaxoSmithKline, the only maker of a long-acting beta-agonist with a pediatric indication (salmeterol), performed this international randomized, double-blind, controlled trial at 567 medical centers in 32 countries, said David A. Stempel, MD, of Respiratory Clinical Development, GSK, Research Triangle Park, N.C., and his associates.
The trial involved 6,208 children aged 4-11 years who had controlled or uncontrolled asthma with a history of exacerbations during the preceding year. The participants were randomly assigned to receive 26 weeks of a lower fixed-dose combination of salmeterol plus fluticasone, a higher fixed-dose combination, a lower dose of fluticasone alone, or a higher dose of fluticasone alone, delivered twice daily via a disk device.
The primary safety endpoint was a composite of death, endotracheal intubation, and hospitalization. No deaths or intubations occurred.
A total of 27 patients taking combined therapy and 21 taking fluticasone alone required hospitalization for asthma (hazard ratio, 1.28). The number of severe asthma exacerbations was 14% lower when salmeterol was added to fluticasone, a nonsignificant difference.
The results demonstrate the noninferiority of the combined therapy, Dr. Stempel and his associates said (N Engl J Med. 2016 Sep 1;375[9]:840-9).
The percentage of children who withdrew from the study because of asthma exacerbations was identical in the two groups (1.1% of each), and the percentage who had a serious adverse event was nearly identical (1.8% vs 1.7%, respectively). The mean percentage of rescue therapy–free days also was similar (83.0% vs 81.9%), as was the mean percentage of days in which asthma was controlled (74.8% vs. 73.4%).
At the conclusion of the study, 88.1% of the fluticasone-plus-salmeterol group had controlled asthma, as did 88.5% of the fluticasone-only group. Meaningful differences between the two treatments could not be identified among various subgroups of patients – defined by age, sex, and race – because the overall number of adverse events was so low, the investigators added.
They cautioned that the trial excluded children who had a history of multiple asthma-related hospitalizations and intubations. Therefore, the findings may not be applicable to patients with very severe asthma, the researchers cautioned.
GlaxoSmithKline sponsored the trial in response to a Food and Drug Administration mandate for large postmarketing safety studies from the marketers of long-acting beta agonist–containing products sold in the United States. Dr. Stempel is an employee of GSK; his associates reported ties to numerous industry sources.
Adding the long-acting beta-agonist salmeterol to fluticasone in a fixed-dose combination didn’t increase serious asthma-related events among children aged 4-11 years, according to a report published online Sept. 1 in the New England Journal of Medicine.
After long-acting beta-agonists were introduced as add-on therapy for uncontrolled asthma, two large studies involving adults linked the treatment to an increase in asthma-related death. Other studies found no such association.
The FDA mandated that all four manufacturers of those agents in the United States perform large postmarketing safety trials to establish the noninferiority of the approach. In response, GlaxoSmithKline, the only maker of a long-acting beta-agonist with a pediatric indication (salmeterol), performed this international randomized, double-blind, controlled trial at 567 medical centers in 32 countries, said David A. Stempel, MD, of Respiratory Clinical Development, GSK, Research Triangle Park, N.C., and his associates.
The trial involved 6,208 children aged 4-11 years who had controlled or uncontrolled asthma with a history of exacerbations during the preceding year. The participants were randomly assigned to receive 26 weeks of a lower fixed-dose combination of salmeterol plus fluticasone, a higher fixed-dose combination, a lower dose of fluticasone alone, or a higher dose of fluticasone alone, delivered twice daily via a disk device.
The primary safety endpoint was a composite of death, endotracheal intubation, and hospitalization. No deaths or intubations occurred.
A total of 27 patients taking combined therapy and 21 taking fluticasone alone required hospitalization for asthma (hazard ratio, 1.28). The number of severe asthma exacerbations was 14% lower when salmeterol was added to fluticasone, a nonsignificant difference.
The results demonstrate the noninferiority of the combined therapy, Dr. Stempel and his associates said (N Engl J Med. 2016 Sep 1;375[9]:840-9).
The percentage of children who withdrew from the study because of asthma exacerbations was identical in the two groups (1.1% of each), and the percentage who had a serious adverse event was nearly identical (1.8% vs 1.7%, respectively). The mean percentage of rescue therapy–free days also was similar (83.0% vs 81.9%), as was the mean percentage of days in which asthma was controlled (74.8% vs. 73.4%).
At the conclusion of the study, 88.1% of the fluticasone-plus-salmeterol group had controlled asthma, as did 88.5% of the fluticasone-only group. Meaningful differences between the two treatments could not be identified among various subgroups of patients – defined by age, sex, and race – because the overall number of adverse events was so low, the investigators added.
They cautioned that the trial excluded children who had a history of multiple asthma-related hospitalizations and intubations. Therefore, the findings may not be applicable to patients with very severe asthma, the researchers cautioned.
GlaxoSmithKline sponsored the trial in response to a Food and Drug Administration mandate for large postmarketing safety studies from the marketers of long-acting beta agonist–containing products sold in the United States. Dr. Stempel is an employee of GSK; his associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Adding salmeterol to fluticasone therapy didn’t increase serious asthma-related events among children.
Major finding: 27 patients taking combined therapy and 21 taking fluticasone alone required hospitalization for asthma (HR, 1.28).
Data source: A 26-week international randomized, double-blind trial involving 6,208 patients aged 4-11 years.
Disclosures: GlaxoSmithKline sponsored the trial in response to a Food and Drug Administration mandate for large postmarketing safety studies from the marketers of long-acting beta agonist–containing products sold in the United States. Dr. Stempel is an employee of GSK; his associates reported ties to numerous industry sources.

Adding formoterol to budesonide does not increase serious asthma events
Adding formoterol to budesonide in a fixed-dose combination does not increase serous asthma-related events in adolescents and adults, according to a report published online Sept. 1 in the New England Journal of Medicine.
This finding from a multicenter randomized double-blind clinical trial involving 11,693 patients should allay safety concerns about adding long-acting beta-agonists to inhaled glucocorticoids in moderate to severe asthma. Previously, two large studies linked such additive therapy to increased asthma-related deaths and other serious outcomes, but other clinical trials and numerous meta-analyses found no such increase.
In 2009, the Food and Drug Administration mandated that the four manufacturers of long-acting beta-agonists available in the United States conduct postmarketing safety analyses of these agents. The current trial is AstraZeneca’s response to the mandate, said Stephen P. Peters, MD, PhD, of Wake Forest University, Winston-Salem N.C., and his associates.
They assessed patients aged 12 years and older who had taken daily asthma medication for at least 1 year before enrollment and had a history of at least one exacerbation during that year. These participants were enrolled at 534 medical centers in 25 countries during 2011-2015 and randomly assigned to receive either budesonide plus formoterol (5,846 patients) or budesonide alone (5,847 patients) through an inhaler twice daily for 26 weeks. The primary endpoint was a composite of asthma-related death, intubation, and hospitalization.
A total of 43 patients in the combined-therapy group had 49 serious asthma-related events, while 40 patients in the budesonide-only group had 45 such events. This is a nonsignificant difference and establishes the noninferiority of the combined treatment regarding this outcome, the investigators said (N Engl J Med. 2016 Sept 1. doi: 10.1056/NEJMoa1511190).
In addition, 539 (9.2%) of the patients in the combined-therapy group reported 637 asthma exacerbations, while 633 in the budesonide-only group had 762 exacerbations. Thus, the risk of having an asthma exacerbation was 16.5% lower with combined therapy (HR, 0.84).
Both study groups had a clinically relevant improvement in asthma control as measured by the ACQ-6, and the combined therapy yielded a significantly greater benefit. The percentage of patients who had a clinically relevant improvement in asthma control at the conclusion of treatment also favored budesonide plus formoterol (58.7% vs. 54.4%). And the combined-therapy group also had a greater mean number of symptom-free days, had fewer night-time awakenings, and used fewer doses of rescue medications, Dr. Peters and his associates said.
Given that asthma-related deaths are rare, none of the four individual manufacturer-sponsored postmarketing studies required by the FDA can be powered for a separate analysis of that endpoint. “Any between-group differences in asthma-related death will need to be evaluated in the context of pooled data from the four studies, once they are all completed,” the investigators added.
Dr. Peters and his associates reported ties to numerous industry sources.
Adding formoterol to budesonide in a fixed-dose combination does not increase serous asthma-related events in adolescents and adults, according to a report published online Sept. 1 in the New England Journal of Medicine.
This finding from a multicenter randomized double-blind clinical trial involving 11,693 patients should allay safety concerns about adding long-acting beta-agonists to inhaled glucocorticoids in moderate to severe asthma. Previously, two large studies linked such additive therapy to increased asthma-related deaths and other serious outcomes, but other clinical trials and numerous meta-analyses found no such increase.
In 2009, the Food and Drug Administration mandated that the four manufacturers of long-acting beta-agonists available in the United States conduct postmarketing safety analyses of these agents. The current trial is AstraZeneca’s response to the mandate, said Stephen P. Peters, MD, PhD, of Wake Forest University, Winston-Salem N.C., and his associates.
They assessed patients aged 12 years and older who had taken daily asthma medication for at least 1 year before enrollment and had a history of at least one exacerbation during that year. These participants were enrolled at 534 medical centers in 25 countries during 2011-2015 and randomly assigned to receive either budesonide plus formoterol (5,846 patients) or budesonide alone (5,847 patients) through an inhaler twice daily for 26 weeks. The primary endpoint was a composite of asthma-related death, intubation, and hospitalization.
A total of 43 patients in the combined-therapy group had 49 serious asthma-related events, while 40 patients in the budesonide-only group had 45 such events. This is a nonsignificant difference and establishes the noninferiority of the combined treatment regarding this outcome, the investigators said (N Engl J Med. 2016 Sept 1. doi: 10.1056/NEJMoa1511190).
In addition, 539 (9.2%) of the patients in the combined-therapy group reported 637 asthma exacerbations, while 633 in the budesonide-only group had 762 exacerbations. Thus, the risk of having an asthma exacerbation was 16.5% lower with combined therapy (HR, 0.84).
Both study groups had a clinically relevant improvement in asthma control as measured by the ACQ-6, and the combined therapy yielded a significantly greater benefit. The percentage of patients who had a clinically relevant improvement in asthma control at the conclusion of treatment also favored budesonide plus formoterol (58.7% vs. 54.4%). And the combined-therapy group also had a greater mean number of symptom-free days, had fewer night-time awakenings, and used fewer doses of rescue medications, Dr. Peters and his associates said.
Given that asthma-related deaths are rare, none of the four individual manufacturer-sponsored postmarketing studies required by the FDA can be powered for a separate analysis of that endpoint. “Any between-group differences in asthma-related death will need to be evaluated in the context of pooled data from the four studies, once they are all completed,” the investigators added.
Dr. Peters and his associates reported ties to numerous industry sources.
Adding formoterol to budesonide in a fixed-dose combination does not increase serous asthma-related events in adolescents and adults, according to a report published online Sept. 1 in the New England Journal of Medicine.
This finding from a multicenter randomized double-blind clinical trial involving 11,693 patients should allay safety concerns about adding long-acting beta-agonists to inhaled glucocorticoids in moderate to severe asthma. Previously, two large studies linked such additive therapy to increased asthma-related deaths and other serious outcomes, but other clinical trials and numerous meta-analyses found no such increase.
In 2009, the Food and Drug Administration mandated that the four manufacturers of long-acting beta-agonists available in the United States conduct postmarketing safety analyses of these agents. The current trial is AstraZeneca’s response to the mandate, said Stephen P. Peters, MD, PhD, of Wake Forest University, Winston-Salem N.C., and his associates.
They assessed patients aged 12 years and older who had taken daily asthma medication for at least 1 year before enrollment and had a history of at least one exacerbation during that year. These participants were enrolled at 534 medical centers in 25 countries during 2011-2015 and randomly assigned to receive either budesonide plus formoterol (5,846 patients) or budesonide alone (5,847 patients) through an inhaler twice daily for 26 weeks. The primary endpoint was a composite of asthma-related death, intubation, and hospitalization.
A total of 43 patients in the combined-therapy group had 49 serious asthma-related events, while 40 patients in the budesonide-only group had 45 such events. This is a nonsignificant difference and establishes the noninferiority of the combined treatment regarding this outcome, the investigators said (N Engl J Med. 2016 Sept 1. doi: 10.1056/NEJMoa1511190).
In addition, 539 (9.2%) of the patients in the combined-therapy group reported 637 asthma exacerbations, while 633 in the budesonide-only group had 762 exacerbations. Thus, the risk of having an asthma exacerbation was 16.5% lower with combined therapy (HR, 0.84).
Both study groups had a clinically relevant improvement in asthma control as measured by the ACQ-6, and the combined therapy yielded a significantly greater benefit. The percentage of patients who had a clinically relevant improvement in asthma control at the conclusion of treatment also favored budesonide plus formoterol (58.7% vs. 54.4%). And the combined-therapy group also had a greater mean number of symptom-free days, had fewer night-time awakenings, and used fewer doses of rescue medications, Dr. Peters and his associates said.
Given that asthma-related deaths are rare, none of the four individual manufacturer-sponsored postmarketing studies required by the FDA can be powered for a separate analysis of that endpoint. “Any between-group differences in asthma-related death will need to be evaluated in the context of pooled data from the four studies, once they are all completed,” the investigators added.
Dr. Peters and his associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Adding formoterol to budesonide in a fixed-dose combination does not increase serious asthma-related events in adolescents and adults.
Major finding: The risk of having an asthma exacerbation was 16.5% lower with combined therapy than with budesonide alone (HR, 0.84).
Data source: A 26-week multicenter randomized double-blind trial involving 11,693 asthma patients aged 12 and older.
Disclosures: This trial was sponsored by AstraZeneca in response to an FDA mandate for large postmarketing safety studies from the four marketers of long-acting beta-agonist-containing products sold in the United States. Dr. Peters and his associates reported ties to numerous industry sources.
AAP: MenB vaccines are safe for healthy adolescents, young adults
The serotype B meningococcal vaccines MenB-FHbp and MenB-4C are safe and can be administered to healthy people aged 10-25 years, according to a policy statement from the American Academy of Pediatrics Committee on Infectious Diseases.
The AAP recommends that people older than 10 years at increased risk for serogroup B meningococcal disease (category A) should receive MenB vaccines regularly. Category A includes people with persistent complement component deficiencies, people with anatomic or functional asplenia, and healthy people at increased risk because of a disease outbreak.

Young adults aged 16-23 years old may receive a vaccination, but it is not routinely recommended (category B), with a preferred vaccination age between 16 and 18 years.
Annual incidence of serogroup B meningococcal disease in people aged 11-24 years in the United States is about 50-60 cases per year, and a routine vaccination program would prevent 15-29 cases and 2-5 deaths per year, the researchers noted. The cost of routine vaccination in the general population would range from $3.7 million per quality-adjusted life year (QALY) to $9.4 million per QALY.
Both MenB-FHbp and MenB-4C have been safely administered in clinical trials, with no deaths related to either vaccine. Data on duration of immunogenicity and proportion of MenB strains covered by vaccines in different geographic regions remain incomplete, and both vaccine manufacturers must complete postmarketing studies to determine overall vaccine effectiveness.
“Pediatricians are encouraged to discuss the availability of the MenB vaccines with families. Discussion should include the low incidence of MenB disease and the unknown efficacy of the vaccines... The treating clinician should discuss the benefits, risks, and costs with patients and their families and then work with them to determine what is in their best interest,” the AAP committee noted.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-1890).
The serotype B meningococcal vaccines MenB-FHbp and MenB-4C are safe and can be administered to healthy people aged 10-25 years, according to a policy statement from the American Academy of Pediatrics Committee on Infectious Diseases.
The AAP recommends that people older than 10 years at increased risk for serogroup B meningococcal disease (category A) should receive MenB vaccines regularly. Category A includes people with persistent complement component deficiencies, people with anatomic or functional asplenia, and healthy people at increased risk because of a disease outbreak.
Young adults aged 16-23 years old may receive a vaccination, but it is not routinely recommended (category B), with a preferred vaccination age between 16 and 18 years.
Annual incidence of serogroup B meningococcal disease in people aged 11-24 years in the United States is about 50-60 cases per year, and a routine vaccination program would prevent 15-29 cases and 2-5 deaths per year, the researchers noted. The cost of routine vaccination in the general population would range from $3.7 million per quality-adjusted life year (QALY) to $9.4 million per QALY.
Both MenB-FHbp and MenB-4C have been safely administered in clinical trials, with no deaths related to either vaccine. Data on duration of immunogenicity and proportion of MenB strains covered by vaccines in different geographic regions remain incomplete, and both vaccine manufacturers must complete postmarketing studies to determine overall vaccine effectiveness.
“Pediatricians are encouraged to discuss the availability of the MenB vaccines with families. Discussion should include the low incidence of MenB disease and the unknown efficacy of the vaccines... The treating clinician should discuss the benefits, risks, and costs with patients and their families and then work with them to determine what is in their best interest,” the AAP committee noted.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-1890).
The serotype B meningococcal vaccines MenB-FHbp and MenB-4C are safe and can be administered to healthy people aged 10-25 years, according to a policy statement from the American Academy of Pediatrics Committee on Infectious Diseases.
The AAP recommends that people older than 10 years at increased risk for serogroup B meningococcal disease (category A) should receive MenB vaccines regularly. Category A includes people with persistent complement component deficiencies, people with anatomic or functional asplenia, and healthy people at increased risk because of a disease outbreak.
Young adults aged 16-23 years old may receive a vaccination, but it is not routinely recommended (category B), with a preferred vaccination age between 16 and 18 years.
Annual incidence of serogroup B meningococcal disease in people aged 11-24 years in the United States is about 50-60 cases per year, and a routine vaccination program would prevent 15-29 cases and 2-5 deaths per year, the researchers noted. The cost of routine vaccination in the general population would range from $3.7 million per quality-adjusted life year (QALY) to $9.4 million per QALY.
Both MenB-FHbp and MenB-4C have been safely administered in clinical trials, with no deaths related to either vaccine. Data on duration of immunogenicity and proportion of MenB strains covered by vaccines in different geographic regions remain incomplete, and both vaccine manufacturers must complete postmarketing studies to determine overall vaccine effectiveness.
“Pediatricians are encouraged to discuss the availability of the MenB vaccines with families. Discussion should include the low incidence of MenB disease and the unknown efficacy of the vaccines... The treating clinician should discuss the benefits, risks, and costs with patients and their families and then work with them to determine what is in their best interest,” the AAP committee noted.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-1890).
FROM PEDIATRICS

Investigator-Reported Efficacy of Azelaic Acid Foam 15% in Patients With Papulopustular Rosacea: Secondary Efficacy Outcomes From a Randomized, Controlled, Double-blind, Phase 3 Trial
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
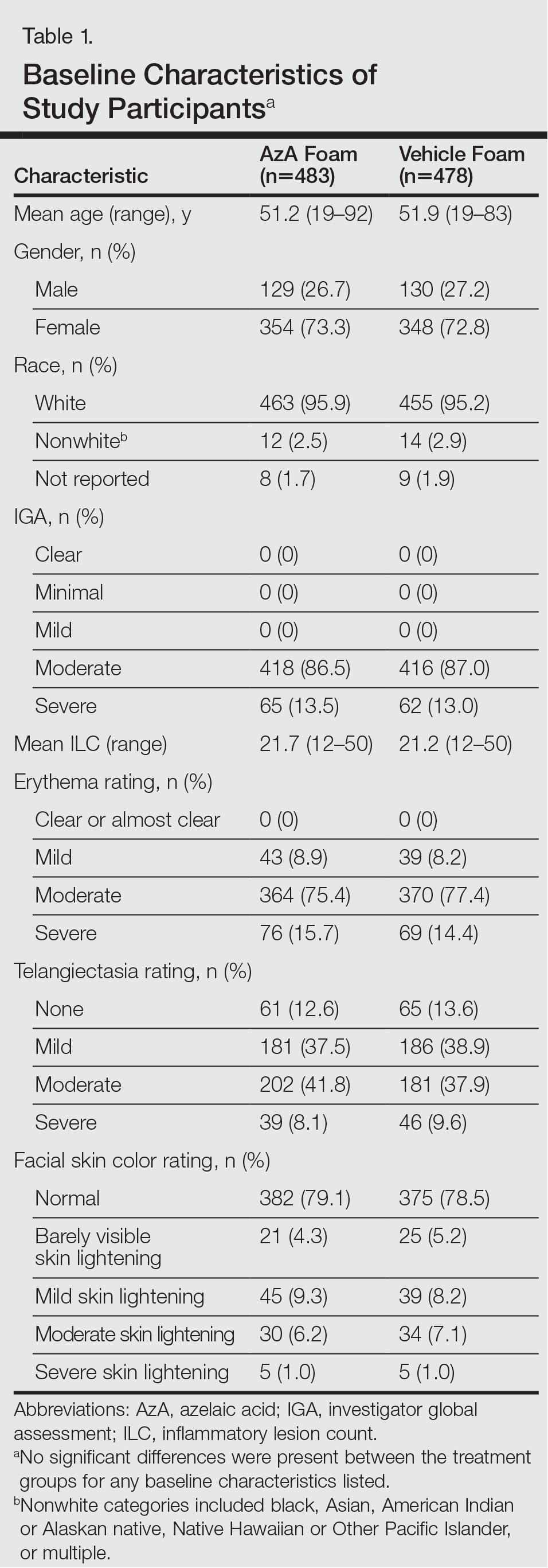
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
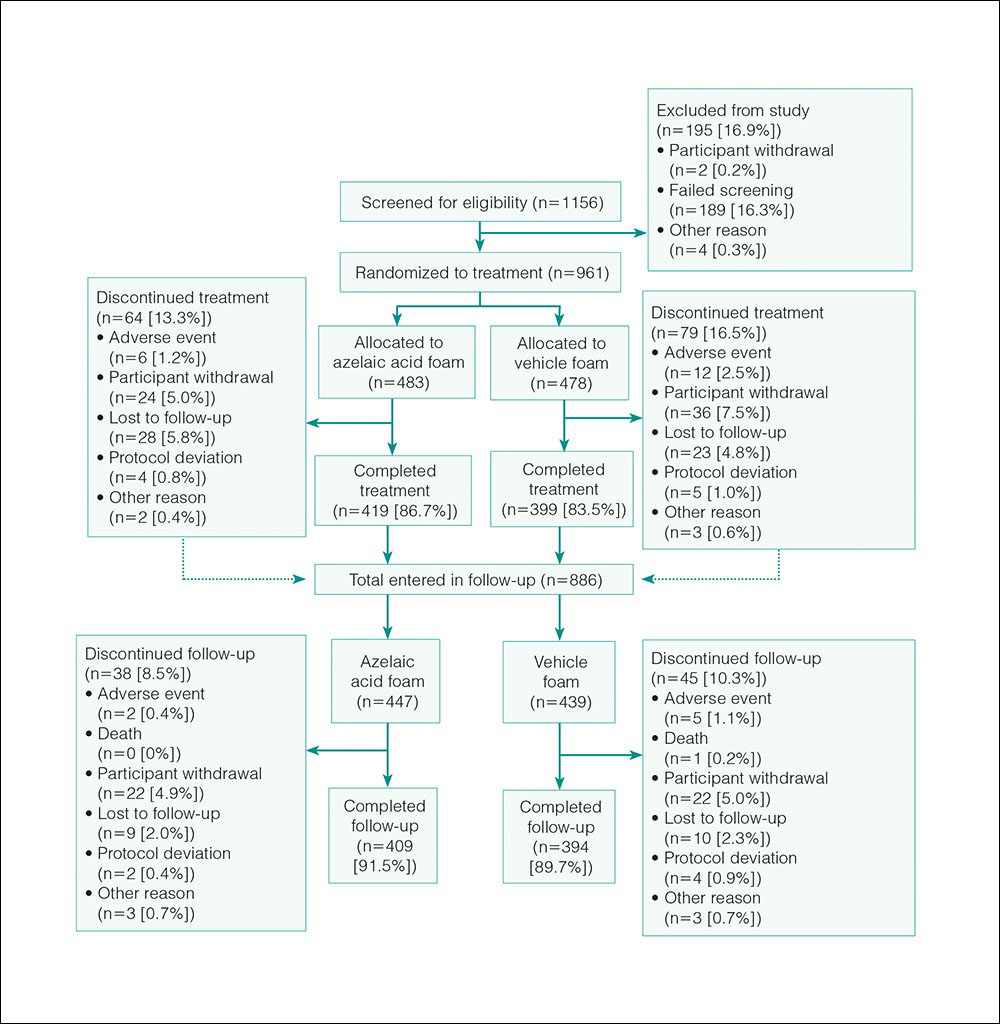
Study Participants
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
Efficacy
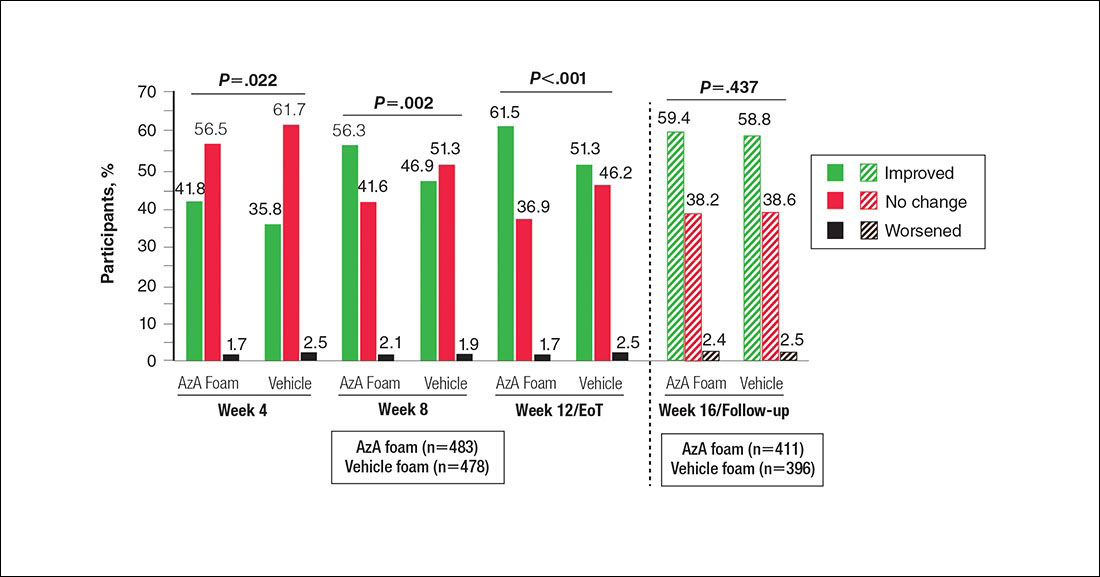
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
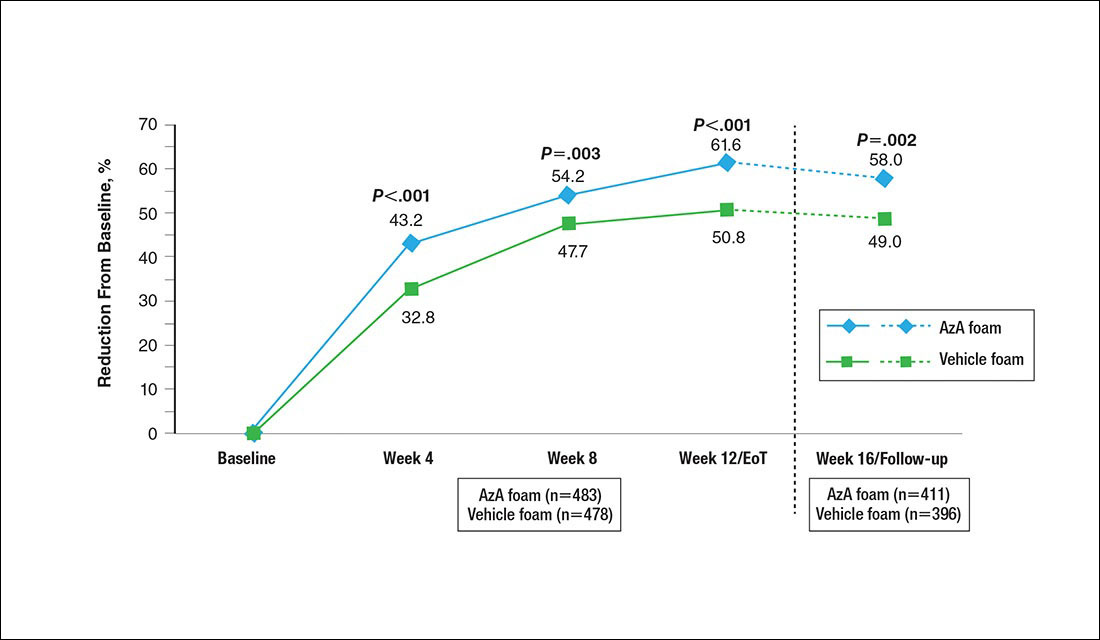
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
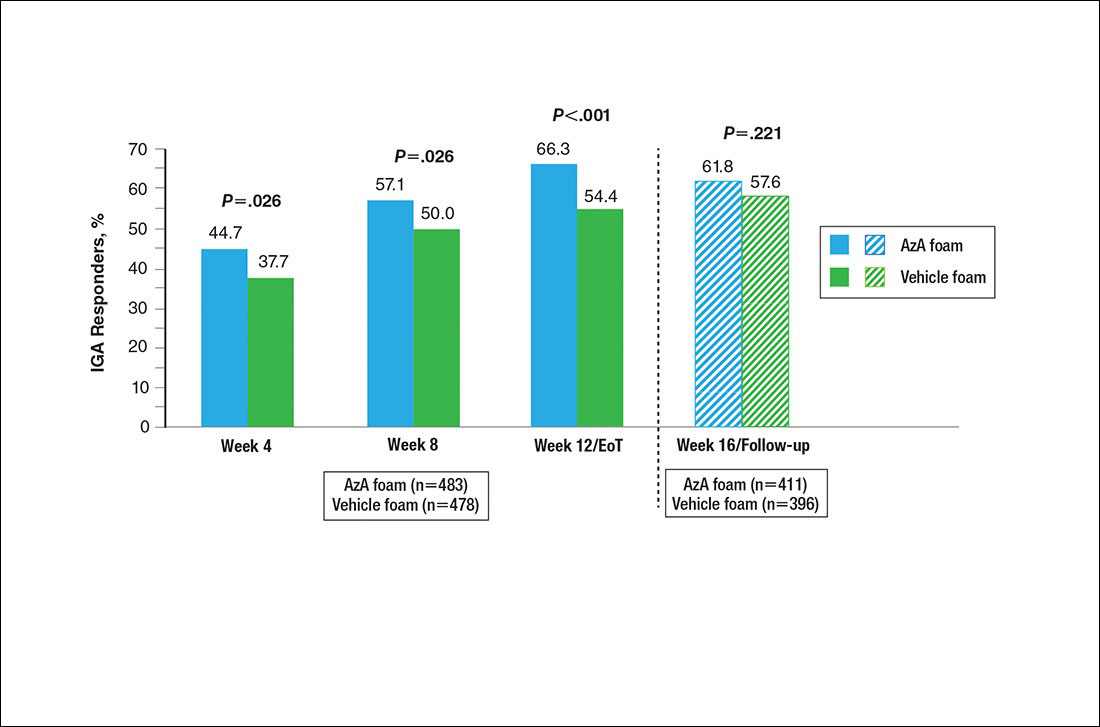
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
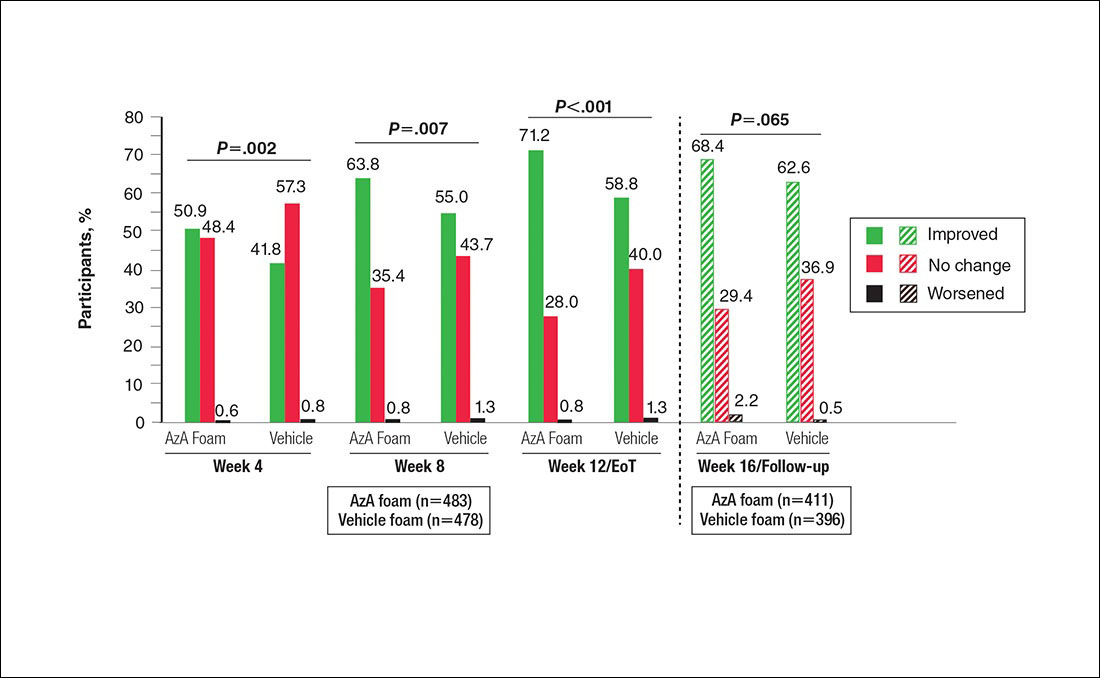
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
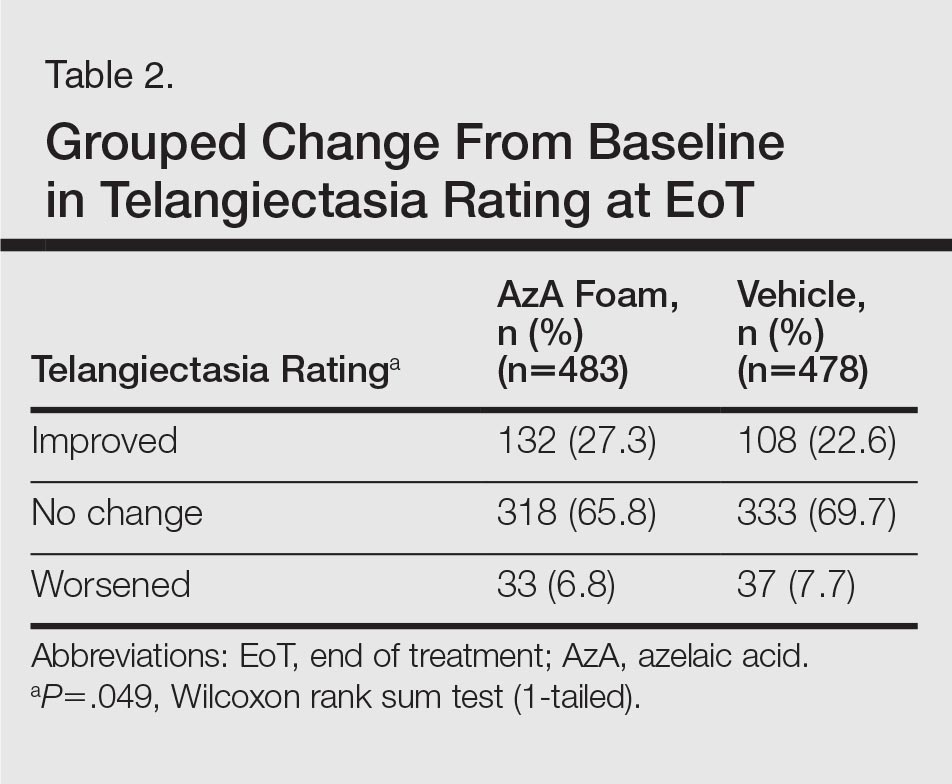
Safety
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
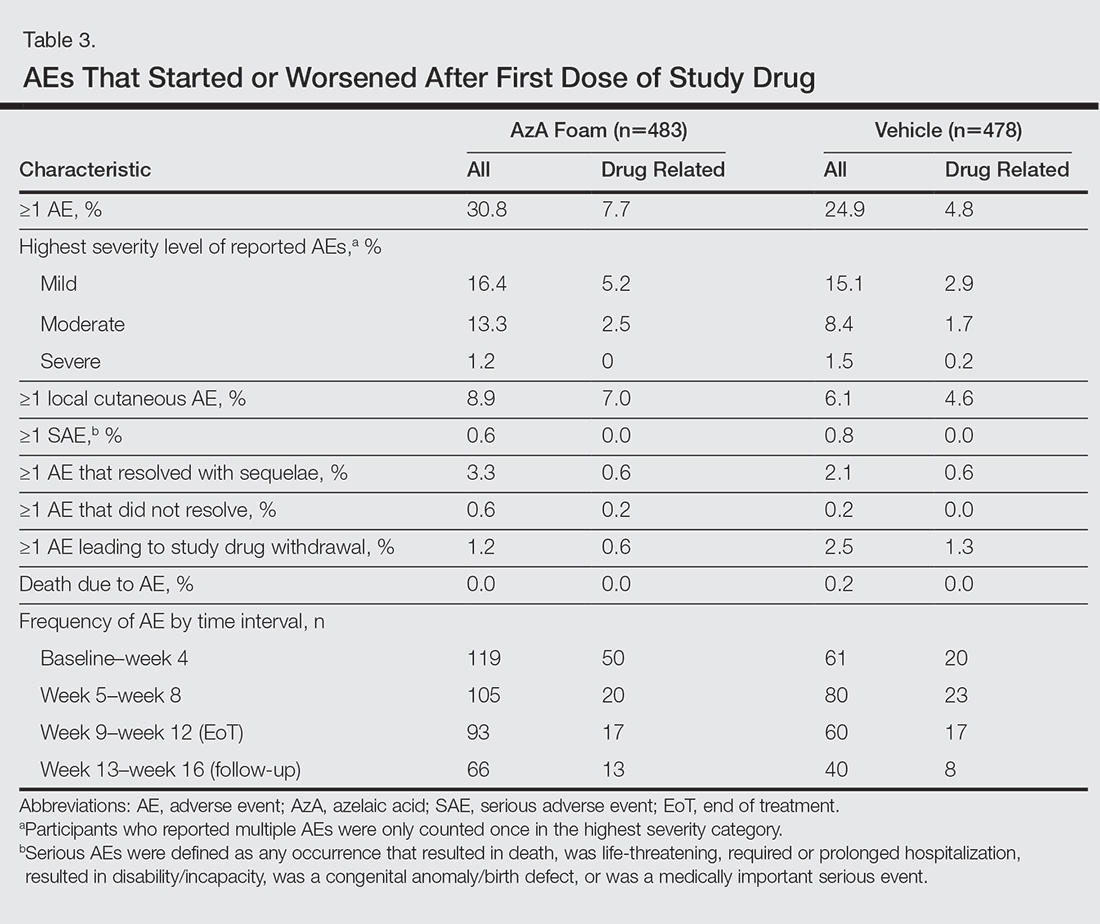
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
Study Participants
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
Efficacy
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
Safety
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
Study Participants
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
Efficacy
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
Safety
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
Practice Points
- Papulopustular rosacea (PPR) is a common chronic inflammatory dermatosis.
- A novel hydrophilic foam formulation of azelaic acid (AzA) was approved for the treatment of PPR.
- In addition to effectively treating papules and pustules, AzA foam also may reduce rosacea-associated erythema.
- The unique AzA foam vehicle may improve epidermal barrier integrity and function, thereby offering patients a distinct topical approach to rosacea management.
Pediatric questionnaire sorts out psychosocial effects of skin conditions
MINNEAPOLIS – A new screening tool may help dermatologists address the psychosocial issues relating to appearance and body image in children and adolescents.
The Pediatric Dermatology Psychosocial Screen (PDPS), is being developed as a standardized tool to evaluate psychosocial stress related to birthmarks, skin diseases, and conditions affecting pigmentation or hair growth. Elizabeth Tocci, MD, and her colleagues, who have been involved with the development of the PDPS, envision it as a useful tool to provide support for pediatric dermatology patients and to help dermatologists decide when mental health consults are warranted in their pediatric patients.
Dr. Tocci, a resident in dermatology at Roger Williams Medical Center, Providence, R.I., and her colleagues described the tool and initial testing results in a poster session at the annual meeting of the Society for Pediatric Dermatology.
The PDPS is a refinement of a pilot survey, created by the coauthors in consultation with experts in neurodermatitis and body dysmorphic disorder (BDD). Following preliminary validity analysis of the pilot questionnaire, a revised PDPS was administered to 105 children, aged 8-19 years, who were patients at a pediatric dermatology clinic. In addition to completing the PDPS, they also filled out psychological questionnaires that assessed for depression, self-esteem, and social problems.
The PDPS asks general questions about the skin diagnosis and any treatments the patient may have used, such as over-the-counter products, prescription medications, and procedures, as well as the use of makeup. In addition, the PDPS asks what social and psychological supports or online resources the patient might have tried, including support groups and appointments with school counselors or mental health providers.
Psychosocial aspects of the skin condition are explored by asking how upset patients are about social sequelae of having a visible skin condition, and whether they are asked about the condition and whether they are made fun of, stared at, or avoided because of the condition. Other questions pertain to whether they notice others’ skin, are hyperobservant of their own skin condition, or feel their popularity and their willingness to date are affected by their skin condition.
Respondent resiliency as it relates to the skin condition is explored by asking whether the respondent found it difficult to move on after a negative social interaction related to the skin condition, and how long the feeling of upset persisted after a negative incident.
Of the 105 surveys, 87 were complete enough to allow analysis. The analysis showed that higher self-reported resiliency was associated with higher positive scores on other psychosocial factors, such as self-esteem, body image, fewer negative and more positive social supports, less self-consciousness, less negative affect, and less BDD. “Self-reported resilience was a significant predictor and determinant of all the psychosocial factors measured,” Dr. Tocci and her associates wrote.
Results indicate that the PDPS is useful to evaluate children and teens in a busy clinic setting, and is “an excellent self-reporting tool for measuring resilience versus psychosocial distress,” they added.
The test is not yet available; the next steps include refining the length and wording of the PDPS, with further validation and testing.
Dr. Tocci and her collaborators reported no conflicts of interest.
On Twitter @karioakes
MINNEAPOLIS – A new screening tool may help dermatologists address the psychosocial issues relating to appearance and body image in children and adolescents.
The Pediatric Dermatology Psychosocial Screen (PDPS), is being developed as a standardized tool to evaluate psychosocial stress related to birthmarks, skin diseases, and conditions affecting pigmentation or hair growth. Elizabeth Tocci, MD, and her colleagues, who have been involved with the development of the PDPS, envision it as a useful tool to provide support for pediatric dermatology patients and to help dermatologists decide when mental health consults are warranted in their pediatric patients.
Dr. Tocci, a resident in dermatology at Roger Williams Medical Center, Providence, R.I., and her colleagues described the tool and initial testing results in a poster session at the annual meeting of the Society for Pediatric Dermatology.
The PDPS is a refinement of a pilot survey, created by the coauthors in consultation with experts in neurodermatitis and body dysmorphic disorder (BDD). Following preliminary validity analysis of the pilot questionnaire, a revised PDPS was administered to 105 children, aged 8-19 years, who were patients at a pediatric dermatology clinic. In addition to completing the PDPS, they also filled out psychological questionnaires that assessed for depression, self-esteem, and social problems.
The PDPS asks general questions about the skin diagnosis and any treatments the patient may have used, such as over-the-counter products, prescription medications, and procedures, as well as the use of makeup. In addition, the PDPS asks what social and psychological supports or online resources the patient might have tried, including support groups and appointments with school counselors or mental health providers.
Psychosocial aspects of the skin condition are explored by asking how upset patients are about social sequelae of having a visible skin condition, and whether they are asked about the condition and whether they are made fun of, stared at, or avoided because of the condition. Other questions pertain to whether they notice others’ skin, are hyperobservant of their own skin condition, or feel their popularity and their willingness to date are affected by their skin condition.
Respondent resiliency as it relates to the skin condition is explored by asking whether the respondent found it difficult to move on after a negative social interaction related to the skin condition, and how long the feeling of upset persisted after a negative incident.
Of the 105 surveys, 87 were complete enough to allow analysis. The analysis showed that higher self-reported resiliency was associated with higher positive scores on other psychosocial factors, such as self-esteem, body image, fewer negative and more positive social supports, less self-consciousness, less negative affect, and less BDD. “Self-reported resilience was a significant predictor and determinant of all the psychosocial factors measured,” Dr. Tocci and her associates wrote.
Results indicate that the PDPS is useful to evaluate children and teens in a busy clinic setting, and is “an excellent self-reporting tool for measuring resilience versus psychosocial distress,” they added.
The test is not yet available; the next steps include refining the length and wording of the PDPS, with further validation and testing.
Dr. Tocci and her collaborators reported no conflicts of interest.
On Twitter @karioakes
MINNEAPOLIS – A new screening tool may help dermatologists address the psychosocial issues relating to appearance and body image in children and adolescents.
The Pediatric Dermatology Psychosocial Screen (PDPS), is being developed as a standardized tool to evaluate psychosocial stress related to birthmarks, skin diseases, and conditions affecting pigmentation or hair growth. Elizabeth Tocci, MD, and her colleagues, who have been involved with the development of the PDPS, envision it as a useful tool to provide support for pediatric dermatology patients and to help dermatologists decide when mental health consults are warranted in their pediatric patients.
Dr. Tocci, a resident in dermatology at Roger Williams Medical Center, Providence, R.I., and her colleagues described the tool and initial testing results in a poster session at the annual meeting of the Society for Pediatric Dermatology.
The PDPS is a refinement of a pilot survey, created by the coauthors in consultation with experts in neurodermatitis and body dysmorphic disorder (BDD). Following preliminary validity analysis of the pilot questionnaire, a revised PDPS was administered to 105 children, aged 8-19 years, who were patients at a pediatric dermatology clinic. In addition to completing the PDPS, they also filled out psychological questionnaires that assessed for depression, self-esteem, and social problems.
The PDPS asks general questions about the skin diagnosis and any treatments the patient may have used, such as over-the-counter products, prescription medications, and procedures, as well as the use of makeup. In addition, the PDPS asks what social and psychological supports or online resources the patient might have tried, including support groups and appointments with school counselors or mental health providers.
Psychosocial aspects of the skin condition are explored by asking how upset patients are about social sequelae of having a visible skin condition, and whether they are asked about the condition and whether they are made fun of, stared at, or avoided because of the condition. Other questions pertain to whether they notice others’ skin, are hyperobservant of their own skin condition, or feel their popularity and their willingness to date are affected by their skin condition.
Respondent resiliency as it relates to the skin condition is explored by asking whether the respondent found it difficult to move on after a negative social interaction related to the skin condition, and how long the feeling of upset persisted after a negative incident.
Of the 105 surveys, 87 were complete enough to allow analysis. The analysis showed that higher self-reported resiliency was associated with higher positive scores on other psychosocial factors, such as self-esteem, body image, fewer negative and more positive social supports, less self-consciousness, less negative affect, and less BDD. “Self-reported resilience was a significant predictor and determinant of all the psychosocial factors measured,” Dr. Tocci and her associates wrote.
Results indicate that the PDPS is useful to evaluate children and teens in a busy clinic setting, and is “an excellent self-reporting tool for measuring resilience versus psychosocial distress,” they added.
The test is not yet available; the next steps include refining the length and wording of the PDPS, with further validation and testing.
Dr. Tocci and her collaborators reported no conflicts of interest.
On Twitter @karioakes
EXPERT ANALYSIS FROM THE SPD ANNUAL MEETING
Some psoriasis patients benefit from switching anti-TNF agents
Psoriasis patients may have more success with a second tumor necrosis factor (TNF) antagonist after failure with a first, report Paul S. Yamauchi, MD, PhD, and coauthors.
Investigators analyzed 15 studies evaluating the efficacy of switching TNF antagonists after primary or secondary failure. Response rates at 24 weeks for a second antagonist were 30%-74% for a 75% improvement in Psoriasis Area and Severity Index score, and 20%-70% for achieving a Physician Global Assessment score of 0/1. Mean improvements in Dermatology Life Quality Index ranged from –3.5 to –13, Dr. Yamauchi and colleagues reported.

Patients who experienced secondary failure with initial treatment generally achieved better responses than those with primary failure, the authors said.
Though response rates to a second anti-TNF agent were lower than for a first, “a substantial proportion of patients in every study achieved treatment success,” they added.
Read the full article in the Journal of the American Academy of Dermatology.
Psoriasis patients may have more success with a second tumor necrosis factor (TNF) antagonist after failure with a first, report Paul S. Yamauchi, MD, PhD, and coauthors.
Investigators analyzed 15 studies evaluating the efficacy of switching TNF antagonists after primary or secondary failure. Response rates at 24 weeks for a second antagonist were 30%-74% for a 75% improvement in Psoriasis Area and Severity Index score, and 20%-70% for achieving a Physician Global Assessment score of 0/1. Mean improvements in Dermatology Life Quality Index ranged from –3.5 to –13, Dr. Yamauchi and colleagues reported.
Patients who experienced secondary failure with initial treatment generally achieved better responses than those with primary failure, the authors said.
Though response rates to a second anti-TNF agent were lower than for a first, “a substantial proportion of patients in every study achieved treatment success,” they added.
Read the full article in the Journal of the American Academy of Dermatology.
Psoriasis patients may have more success with a second tumor necrosis factor (TNF) antagonist after failure with a first, report Paul S. Yamauchi, MD, PhD, and coauthors.
Investigators analyzed 15 studies evaluating the efficacy of switching TNF antagonists after primary or secondary failure. Response rates at 24 weeks for a second antagonist were 30%-74% for a 75% improvement in Psoriasis Area and Severity Index score, and 20%-70% for achieving a Physician Global Assessment score of 0/1. Mean improvements in Dermatology Life Quality Index ranged from –3.5 to –13, Dr. Yamauchi and colleagues reported.
Patients who experienced secondary failure with initial treatment generally achieved better responses than those with primary failure, the authors said.
Though response rates to a second anti-TNF agent were lower than for a first, “a substantial proportion of patients in every study achieved treatment success,” they added.
Read the full article in the Journal of the American Academy of Dermatology.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY

An Update on Neurotoxin Products and Administration Methods
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
Practice Points
- Botulinum neurotoxin (BoNT) injection is the most popular nonsurgical aesthetic procedure available of which there are currently 4 products approved by the US Food and Drug Administration.
- A variety of new BoNT products with unique properties and formulations are currently being studied, some of which are already available for clinical use in foreign markets.
- Administration technique and novel product delivery methods also can be utilized to minimize pain and maximize aesthetic outcomes.