User login
Scaling Up Efforts to Bring Weight Down: An Update on Recommendations, Techniques, and Pharmacotherapies for Adult Weight Management

|
Obesity meets 3 standard defining criteria of a disease: it is associated with impairment of normal bodily function, has characteristic signs and symptoms, and results in bodily harm.1 Accordingly, authoritative organizations, including the American Medical Association, formally recognize obesity as a disease—more specifically, a chronic, relapsing, neurobehavioral disease.1-6
Click here to read the activity.
Click here to complete the posttest and evaluation.
|
|
Obesity meets 3 standard defining criteria of a disease: it is associated with impairment of normal bodily function, has characteristic signs and symptoms, and results in bodily harm.1 Accordingly, authoritative organizations, including the American Medical Association, formally recognize obesity as a disease—more specifically, a chronic, relapsing, neurobehavioral disease.1-6
Click here to read the activity.
Click here to complete the posttest and evaluation.
|
|
Obesity meets 3 standard defining criteria of a disease: it is associated with impairment of normal bodily function, has characteristic signs and symptoms, and results in bodily harm.1 Accordingly, authoritative organizations, including the American Medical Association, formally recognize obesity as a disease—more specifically, a chronic, relapsing, neurobehavioral disease.1-6
Click here to read the activity.
Click here to complete the posttest and evaluation.
Biosimilar version of etanercept gains FDA approval
A biosimilar of etanercept received clearance for marketing from the Food and Drug Administration on Aug. 30 for all of the inflammatory disease indications held by the reference originator etanercept product, Enbrel, according to an announcement from the agency.
Approval for all of Enbrel’s indications – rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, and polyarticular juvenile idiopathic arthritis – was initially met with skepticism by members of the agency’s Arthritis Advisory Committee at a meeting in July because the biosimilar was compared against Enbrel in patients with plaque psoriasis only, but eventually all panel members voted to recommend approval.
The approval allows the biosimilar etanercept, called etanercept-szzs, to be marketed as a biosimilar only, not as an interchangeable product. The FDA has not yet developed guidance for manufacturers to follow to get approval for interchangeability, which means that a biosimilar “may be substituted for the reference product by a pharmacist without the intervention of the health care provider who prescribed the reference product,” according to the agency.
“We carefully evaluate the structural and functional characteristics of these complex molecules. Patients and providers can have confidence that there are no clinically meaningful differences in safety and efficacy from the reference product,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency’s announcement.
Etanercept-szzs will be marketed by Sandoz as Erelzi. Erelzi’s prescribing information can be found here. The biosimilar is currently undergoing review with the European Medicines Agency.
A biosimilar of etanercept received clearance for marketing from the Food and Drug Administration on Aug. 30 for all of the inflammatory disease indications held by the reference originator etanercept product, Enbrel, according to an announcement from the agency.
Approval for all of Enbrel’s indications – rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, and polyarticular juvenile idiopathic arthritis – was initially met with skepticism by members of the agency’s Arthritis Advisory Committee at a meeting in July because the biosimilar was compared against Enbrel in patients with plaque psoriasis only, but eventually all panel members voted to recommend approval.
The approval allows the biosimilar etanercept, called etanercept-szzs, to be marketed as a biosimilar only, not as an interchangeable product. The FDA has not yet developed guidance for manufacturers to follow to get approval for interchangeability, which means that a biosimilar “may be substituted for the reference product by a pharmacist without the intervention of the health care provider who prescribed the reference product,” according to the agency.
“We carefully evaluate the structural and functional characteristics of these complex molecules. Patients and providers can have confidence that there are no clinically meaningful differences in safety and efficacy from the reference product,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency’s announcement.
Etanercept-szzs will be marketed by Sandoz as Erelzi. Erelzi’s prescribing information can be found here. The biosimilar is currently undergoing review with the European Medicines Agency.
A biosimilar of etanercept received clearance for marketing from the Food and Drug Administration on Aug. 30 for all of the inflammatory disease indications held by the reference originator etanercept product, Enbrel, according to an announcement from the agency.
Approval for all of Enbrel’s indications – rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, and polyarticular juvenile idiopathic arthritis – was initially met with skepticism by members of the agency’s Arthritis Advisory Committee at a meeting in July because the biosimilar was compared against Enbrel in patients with plaque psoriasis only, but eventually all panel members voted to recommend approval.
The approval allows the biosimilar etanercept, called etanercept-szzs, to be marketed as a biosimilar only, not as an interchangeable product. The FDA has not yet developed guidance for manufacturers to follow to get approval for interchangeability, which means that a biosimilar “may be substituted for the reference product by a pharmacist without the intervention of the health care provider who prescribed the reference product,” according to the agency.
“We carefully evaluate the structural and functional characteristics of these complex molecules. Patients and providers can have confidence that there are no clinically meaningful differences in safety and efficacy from the reference product,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency’s announcement.
Etanercept-szzs will be marketed by Sandoz as Erelzi. Erelzi’s prescribing information can be found here. The biosimilar is currently undergoing review with the European Medicines Agency.
Postpartum HIV treatment reduces key maternal illnesses
DURBAN, SOUTH AFRICA – HIV-infected women who remained on antiretroviral therapy throughout the postpartum period reduced their risk of clinical stage 2 or 3 HIV disease events by 53%, compared with those who stopped treatment postpartum in the PROMISE 1077HS trial, Judith Currier, MD, reported at the 21st International AIDS Conference.
PROMISE (Promoting Maternal and Infant Survival Everywhere) is an ongoing multinational, multicomponent series of clinical trials. PROMISE 1077HS was designed to assess the risks and benefits of continued antiretroviral therapy (ART), compared with stopping therapy among nonbreastfeeding women postpartum, explained Dr. Currier, professor of medicine and chief of infectious diseases at the University of California, Los Angeles.

PROMISE 1077HS included 1,653 HIV-positive women in the United States and seven low- or middle-income countries. All participants were relatively healthy as evidenced by their median CD4+ count of 550 cells/mm3 prior to starting ART in pregnancy. None were planning to breastfeed. Upon delivery, the women were randomized to continue ART – the chief regimen was ritonavir-boosted lopinavir (Kaletra) plus tenofovir/emtricitabine (Truvada) – or stop therapy, restarting only when their CD4+ count fell below 350 cells/mm3.
Participants were prospectively followed for a median of 2.3 years post delivery. At that point, in summer 2015, the results of the landmark START trial were released (N Engl J Med. 2015 Aug 27;373[9]:795-807), paving the way for the current global strategy of ART for life in HIV-infected patients regardless of their CD4+ cell count.
The primary efficacy endpoint in PROMISE 1077HS was a composite of an AIDS event, death, or a serious cardiovascular, renal, or hepatic event. In this relatively healthy population, too few of these events occurred to allow the researchers to draw conclusions (four in the continued-ART group and six in the controls who stopped ART post partum).
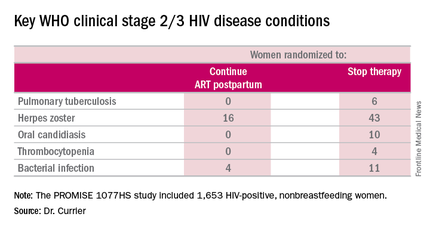
But the secondary endpoint of time to World Health Organization (WHO) clinical stage 2 or 3 HIV disease events was a different story. A total of 39 of these events occurred in the continued-ART group, for a rate of 2.02% per year, compared with 80 events and a rate of 4.36% per year in controls. That difference translated into a 53% relative risk reduction. Some key WHO clinical stage 2 and 3 HIV disease events include pulmonary tuberculosis, herpes zoster, oral candidiasis, thrombocytopenia, and bacterial infection.
Fully 23% of patients in the continued-ART group had laboratory-confirmed virologic failure as defined by a viral load of HIV RNA in excess of 1,000 copies/mL after at least 24 weeks of postpartum ART. Additionally, resistance testing indicated two-thirds of affected patients showed no evidence of resistance at the time of virologic failure, meaning their viremia was due to nonadherence to ART.
“This virologic failure rate highlights the importance of the challenge of adherence in this population,” Dr. Currier said. “Interventions to improve adherence as well as studies to examine newer regimens with a high genetic barrier to resistance are needed to ensure maximal long-term benefit from this strategy of continued ART postpartum.”
The PROMISE studies are funded by the National Institutes of Health. Dr. Currier reported having no financial disclosures.
DURBAN, SOUTH AFRICA – HIV-infected women who remained on antiretroviral therapy throughout the postpartum period reduced their risk of clinical stage 2 or 3 HIV disease events by 53%, compared with those who stopped treatment postpartum in the PROMISE 1077HS trial, Judith Currier, MD, reported at the 21st International AIDS Conference.
PROMISE (Promoting Maternal and Infant Survival Everywhere) is an ongoing multinational, multicomponent series of clinical trials. PROMISE 1077HS was designed to assess the risks and benefits of continued antiretroviral therapy (ART), compared with stopping therapy among nonbreastfeeding women postpartum, explained Dr. Currier, professor of medicine and chief of infectious diseases at the University of California, Los Angeles.
PROMISE 1077HS included 1,653 HIV-positive women in the United States and seven low- or middle-income countries. All participants were relatively healthy as evidenced by their median CD4+ count of 550 cells/mm3 prior to starting ART in pregnancy. None were planning to breastfeed. Upon delivery, the women were randomized to continue ART – the chief regimen was ritonavir-boosted lopinavir (Kaletra) plus tenofovir/emtricitabine (Truvada) – or stop therapy, restarting only when their CD4+ count fell below 350 cells/mm3.
Participants were prospectively followed for a median of 2.3 years post delivery. At that point, in summer 2015, the results of the landmark START trial were released (N Engl J Med. 2015 Aug 27;373[9]:795-807), paving the way for the current global strategy of ART for life in HIV-infected patients regardless of their CD4+ cell count.
The primary efficacy endpoint in PROMISE 1077HS was a composite of an AIDS event, death, or a serious cardiovascular, renal, or hepatic event. In this relatively healthy population, too few of these events occurred to allow the researchers to draw conclusions (four in the continued-ART group and six in the controls who stopped ART post partum).
But the secondary endpoint of time to World Health Organization (WHO) clinical stage 2 or 3 HIV disease events was a different story. A total of 39 of these events occurred in the continued-ART group, for a rate of 2.02% per year, compared with 80 events and a rate of 4.36% per year in controls. That difference translated into a 53% relative risk reduction. Some key WHO clinical stage 2 and 3 HIV disease events include pulmonary tuberculosis, herpes zoster, oral candidiasis, thrombocytopenia, and bacterial infection.
Fully 23% of patients in the continued-ART group had laboratory-confirmed virologic failure as defined by a viral load of HIV RNA in excess of 1,000 copies/mL after at least 24 weeks of postpartum ART. Additionally, resistance testing indicated two-thirds of affected patients showed no evidence of resistance at the time of virologic failure, meaning their viremia was due to nonadherence to ART.
“This virologic failure rate highlights the importance of the challenge of adherence in this population,” Dr. Currier said. “Interventions to improve adherence as well as studies to examine newer regimens with a high genetic barrier to resistance are needed to ensure maximal long-term benefit from this strategy of continued ART postpartum.”
The PROMISE studies are funded by the National Institutes of Health. Dr. Currier reported having no financial disclosures.
DURBAN, SOUTH AFRICA – HIV-infected women who remained on antiretroviral therapy throughout the postpartum period reduced their risk of clinical stage 2 or 3 HIV disease events by 53%, compared with those who stopped treatment postpartum in the PROMISE 1077HS trial, Judith Currier, MD, reported at the 21st International AIDS Conference.
PROMISE (Promoting Maternal and Infant Survival Everywhere) is an ongoing multinational, multicomponent series of clinical trials. PROMISE 1077HS was designed to assess the risks and benefits of continued antiretroviral therapy (ART), compared with stopping therapy among nonbreastfeeding women postpartum, explained Dr. Currier, professor of medicine and chief of infectious diseases at the University of California, Los Angeles.
PROMISE 1077HS included 1,653 HIV-positive women in the United States and seven low- or middle-income countries. All participants were relatively healthy as evidenced by their median CD4+ count of 550 cells/mm3 prior to starting ART in pregnancy. None were planning to breastfeed. Upon delivery, the women were randomized to continue ART – the chief regimen was ritonavir-boosted lopinavir (Kaletra) plus tenofovir/emtricitabine (Truvada) – or stop therapy, restarting only when their CD4+ count fell below 350 cells/mm3.
Participants were prospectively followed for a median of 2.3 years post delivery. At that point, in summer 2015, the results of the landmark START trial were released (N Engl J Med. 2015 Aug 27;373[9]:795-807), paving the way for the current global strategy of ART for life in HIV-infected patients regardless of their CD4+ cell count.
The primary efficacy endpoint in PROMISE 1077HS was a composite of an AIDS event, death, or a serious cardiovascular, renal, or hepatic event. In this relatively healthy population, too few of these events occurred to allow the researchers to draw conclusions (four in the continued-ART group and six in the controls who stopped ART post partum).
But the secondary endpoint of time to World Health Organization (WHO) clinical stage 2 or 3 HIV disease events was a different story. A total of 39 of these events occurred in the continued-ART group, for a rate of 2.02% per year, compared with 80 events and a rate of 4.36% per year in controls. That difference translated into a 53% relative risk reduction. Some key WHO clinical stage 2 and 3 HIV disease events include pulmonary tuberculosis, herpes zoster, oral candidiasis, thrombocytopenia, and bacterial infection.
Fully 23% of patients in the continued-ART group had laboratory-confirmed virologic failure as defined by a viral load of HIV RNA in excess of 1,000 copies/mL after at least 24 weeks of postpartum ART. Additionally, resistance testing indicated two-thirds of affected patients showed no evidence of resistance at the time of virologic failure, meaning their viremia was due to nonadherence to ART.
“This virologic failure rate highlights the importance of the challenge of adherence in this population,” Dr. Currier said. “Interventions to improve adherence as well as studies to examine newer regimens with a high genetic barrier to resistance are needed to ensure maximal long-term benefit from this strategy of continued ART postpartum.”
The PROMISE studies are funded by the National Institutes of Health. Dr. Currier reported having no financial disclosures.
AT AIDS 2016
Key clinical point: Women with HIV should continue antiretroviral therapy post partum.
Major finding: HIV-infected women who continued antiretroviral therapy post partum experienced 53% fewer WHO clinical stage 2 or 3 HIV disease events than women assigned to stop therapy after delivery.
Data source: The PROMISE 1077HS study included 1,653 HIV-positive, nonbreastfeeding women randomized to either continue or stop antiretroviral therapy post partum.
Disclosures: The PROMISE studies are funded by the National Institutes of Health. Dr. Currier reported having no financial disclosures.

QUIZ: Which Pain Medication to Use for Patients with ESRD on HD?
[WpProQuiz 13] [WpProQuiz_toplist 13]
[WpProQuiz 13] [WpProQuiz_toplist 13]
[WpProQuiz 13] [WpProQuiz_toplist 13]
Single dose bacterial vaginosis treatment performs well in phase III trial
ANNAPOLIS, MD. – Women with bacterial vaginosis could soon have an effective oral, single-dose treatment option, if results of a phase III study result in approval by the Food and Drug Administration.
In a modified intention-to-treat study of 189 women with bacterial vaginosis (BV) randomly assigned 2:1 to treatment or placebo, a single, granulated oral dose of secnidazole 2g was found to be statistically superior to placebo on all clinical endpoints.

Secnidazole (SYM-1219) has a longer half-life, compared with metronidazole, the current treatment standard, according to Jane R. Schwebke, MD, the study investigator and a professor of medicine in the infectious disease department of the University of Alabama, Birmingham. Dr. Schwebke credits the study drug’s high bioavailablility and rapid absorption for its efficacy.
“You get a very high peak with SYM-1219 initially, and I think that might be the reason for the single-dose therapy’s efficacy. It’s due to the pharmacokinetics of the drug itself,” she reported at the annual scientific meeting of the Infectious Diseases Society for Obstetrics and Gynecology.
If the drug is approved, it would likely mean better adherence when compared with current standards of treatment, according to Sharon Hillier, PhD, director of reproductive infectious disease research at the Magee-Women’s Hospital of the University of Pittsburgh.
“It will absolutely improve compliance,” Dr. Hillier said in an interview. “Obviously, it’s much easier than taking [metronidazole] twice a day for 7 days.”
Treatment with metronidazole also requires a week of abstinence from alcohol, compared with what Dr. Hillier anticipates would be 2 or 3 days of alcohol abstinence with secnidazole.

The initial study enrollment was 189 women who were randomized 2:1 to secnidazole or placebo and treated at 21 sites nationally. After assessment for common sexually transmitted diseases, Nugent scores of 4 or greater, and all Amsel criteria (including a vaginal pH of 4.7 or greater, clue cells at or greater than 20%, and a positive KOH whiff test), 164 women remained in the modified intention-to-treat (ITT) analysis. A quarter of all women across the modified ITT group were recurrent BV sufferers, having had at least four episodes of BV in the previous year, and 87% had Nugent scores of 7 or greater.
“These were true BV cases; none were in the intermediate or mild zone,” Dr. Schwebke said.
Responders were women who, between days 21 and 30, had “normal” discharge, less than 20% clue cells, and a negative KOH whiff test. In the study arm, 53.3% of women had “normal” discharge and less than 20% clue cells at their 21-30 day visit, compared with 19.3% in the placebo group (P less than .001). The secondary endpoint – Nugent scores of 3 or less at days 7-14 – was achieved by 43.9% in the study group, compared with 5.3% of controls (P less than .001).
Just over a third of women in the study arm experienced one or more adverse events, compared with 21.9% of controls. Yeast infections were the most common adverse event. Less than 5% of the study group experienced nausea, headache, or diarrhea, compared with up to 3% of controls.
“What’s exciting about this new product is that it will be a single dose oral [that women] can take with a meal and with none of the adverse effects, and it relieves symptoms as well as other treatments,” Dr. Hillier said.
How treatment efficacy should be defined was a matter of debate during the presentation’s question and answer period. The FDA did not issue BV treatment guidance until 1998, despite prior approval of BV treatments clindamycin and metronidazole. The rigorous definition of clinical cure rate put forward in the FDA guidance document caused the cure rates that had been generally accepted by physicians to drop from as high as 80% to around 40%, according to Dr. Hilliard.
“I personally would like to see some head-to-head comparisons of the various treatments we have to know whether some are better than others,” Dr. Hillier said in the interview.
The ideal BV treatment should also provoke a microbiological cure, according to Dr. Schwebke. “What I would do is combine a drug like this with a biofilm inhibitor. Right now, this is great, because it’s single dose oral, and it’s as good as anything out there, but, I don’t think we’re taking the next step necessarily with efficacy.”
The study was supported by Symbiomix. Dr. Schwebke is a consultant for Symbiomix and receives funding from Alfa Wassermann and Starpharma, among others. Dr. Hillier is coprincipal investigator of the Microbicide Trials Network, sponsored by the National Institutes of Health.
On Twitter @whitneymcknight
ANNAPOLIS, MD. – Women with bacterial vaginosis could soon have an effective oral, single-dose treatment option, if results of a phase III study result in approval by the Food and Drug Administration.
In a modified intention-to-treat study of 189 women with bacterial vaginosis (BV) randomly assigned 2:1 to treatment or placebo, a single, granulated oral dose of secnidazole 2g was found to be statistically superior to placebo on all clinical endpoints.
Secnidazole (SYM-1219) has a longer half-life, compared with metronidazole, the current treatment standard, according to Jane R. Schwebke, MD, the study investigator and a professor of medicine in the infectious disease department of the University of Alabama, Birmingham. Dr. Schwebke credits the study drug’s high bioavailablility and rapid absorption for its efficacy.
“You get a very high peak with SYM-1219 initially, and I think that might be the reason for the single-dose therapy’s efficacy. It’s due to the pharmacokinetics of the drug itself,” she reported at the annual scientific meeting of the Infectious Diseases Society for Obstetrics and Gynecology.
If the drug is approved, it would likely mean better adherence when compared with current standards of treatment, according to Sharon Hillier, PhD, director of reproductive infectious disease research at the Magee-Women’s Hospital of the University of Pittsburgh.
“It will absolutely improve compliance,” Dr. Hillier said in an interview. “Obviously, it’s much easier than taking [metronidazole] twice a day for 7 days.”
Treatment with metronidazole also requires a week of abstinence from alcohol, compared with what Dr. Hillier anticipates would be 2 or 3 days of alcohol abstinence with secnidazole.
The initial study enrollment was 189 women who were randomized 2:1 to secnidazole or placebo and treated at 21 sites nationally. After assessment for common sexually transmitted diseases, Nugent scores of 4 or greater, and all Amsel criteria (including a vaginal pH of 4.7 or greater, clue cells at or greater than 20%, and a positive KOH whiff test), 164 women remained in the modified intention-to-treat (ITT) analysis. A quarter of all women across the modified ITT group were recurrent BV sufferers, having had at least four episodes of BV in the previous year, and 87% had Nugent scores of 7 or greater.
“These were true BV cases; none were in the intermediate or mild zone,” Dr. Schwebke said.
Responders were women who, between days 21 and 30, had “normal” discharge, less than 20% clue cells, and a negative KOH whiff test. In the study arm, 53.3% of women had “normal” discharge and less than 20% clue cells at their 21-30 day visit, compared with 19.3% in the placebo group (P less than .001). The secondary endpoint – Nugent scores of 3 or less at days 7-14 – was achieved by 43.9% in the study group, compared with 5.3% of controls (P less than .001).
Just over a third of women in the study arm experienced one or more adverse events, compared with 21.9% of controls. Yeast infections were the most common adverse event. Less than 5% of the study group experienced nausea, headache, or diarrhea, compared with up to 3% of controls.
“What’s exciting about this new product is that it will be a single dose oral [that women] can take with a meal and with none of the adverse effects, and it relieves symptoms as well as other treatments,” Dr. Hillier said.
How treatment efficacy should be defined was a matter of debate during the presentation’s question and answer period. The FDA did not issue BV treatment guidance until 1998, despite prior approval of BV treatments clindamycin and metronidazole. The rigorous definition of clinical cure rate put forward in the FDA guidance document caused the cure rates that had been generally accepted by physicians to drop from as high as 80% to around 40%, according to Dr. Hilliard.
“I personally would like to see some head-to-head comparisons of the various treatments we have to know whether some are better than others,” Dr. Hillier said in the interview.
The ideal BV treatment should also provoke a microbiological cure, according to Dr. Schwebke. “What I would do is combine a drug like this with a biofilm inhibitor. Right now, this is great, because it’s single dose oral, and it’s as good as anything out there, but, I don’t think we’re taking the next step necessarily with efficacy.”
The study was supported by Symbiomix. Dr. Schwebke is a consultant for Symbiomix and receives funding from Alfa Wassermann and Starpharma, among others. Dr. Hillier is coprincipal investigator of the Microbicide Trials Network, sponsored by the National Institutes of Health.
On Twitter @whitneymcknight
ANNAPOLIS, MD. – Women with bacterial vaginosis could soon have an effective oral, single-dose treatment option, if results of a phase III study result in approval by the Food and Drug Administration.
In a modified intention-to-treat study of 189 women with bacterial vaginosis (BV) randomly assigned 2:1 to treatment or placebo, a single, granulated oral dose of secnidazole 2g was found to be statistically superior to placebo on all clinical endpoints.
Secnidazole (SYM-1219) has a longer half-life, compared with metronidazole, the current treatment standard, according to Jane R. Schwebke, MD, the study investigator and a professor of medicine in the infectious disease department of the University of Alabama, Birmingham. Dr. Schwebke credits the study drug’s high bioavailablility and rapid absorption for its efficacy.
“You get a very high peak with SYM-1219 initially, and I think that might be the reason for the single-dose therapy’s efficacy. It’s due to the pharmacokinetics of the drug itself,” she reported at the annual scientific meeting of the Infectious Diseases Society for Obstetrics and Gynecology.
If the drug is approved, it would likely mean better adherence when compared with current standards of treatment, according to Sharon Hillier, PhD, director of reproductive infectious disease research at the Magee-Women’s Hospital of the University of Pittsburgh.
“It will absolutely improve compliance,” Dr. Hillier said in an interview. “Obviously, it’s much easier than taking [metronidazole] twice a day for 7 days.”
Treatment with metronidazole also requires a week of abstinence from alcohol, compared with what Dr. Hillier anticipates would be 2 or 3 days of alcohol abstinence with secnidazole.
The initial study enrollment was 189 women who were randomized 2:1 to secnidazole or placebo and treated at 21 sites nationally. After assessment for common sexually transmitted diseases, Nugent scores of 4 or greater, and all Amsel criteria (including a vaginal pH of 4.7 or greater, clue cells at or greater than 20%, and a positive KOH whiff test), 164 women remained in the modified intention-to-treat (ITT) analysis. A quarter of all women across the modified ITT group were recurrent BV sufferers, having had at least four episodes of BV in the previous year, and 87% had Nugent scores of 7 or greater.
“These were true BV cases; none were in the intermediate or mild zone,” Dr. Schwebke said.
Responders were women who, between days 21 and 30, had “normal” discharge, less than 20% clue cells, and a negative KOH whiff test. In the study arm, 53.3% of women had “normal” discharge and less than 20% clue cells at their 21-30 day visit, compared with 19.3% in the placebo group (P less than .001). The secondary endpoint – Nugent scores of 3 or less at days 7-14 – was achieved by 43.9% in the study group, compared with 5.3% of controls (P less than .001).
Just over a third of women in the study arm experienced one or more adverse events, compared with 21.9% of controls. Yeast infections were the most common adverse event. Less than 5% of the study group experienced nausea, headache, or diarrhea, compared with up to 3% of controls.
“What’s exciting about this new product is that it will be a single dose oral [that women] can take with a meal and with none of the adverse effects, and it relieves symptoms as well as other treatments,” Dr. Hillier said.
How treatment efficacy should be defined was a matter of debate during the presentation’s question and answer period. The FDA did not issue BV treatment guidance until 1998, despite prior approval of BV treatments clindamycin and metronidazole. The rigorous definition of clinical cure rate put forward in the FDA guidance document caused the cure rates that had been generally accepted by physicians to drop from as high as 80% to around 40%, according to Dr. Hilliard.
“I personally would like to see some head-to-head comparisons of the various treatments we have to know whether some are better than others,” Dr. Hillier said in the interview.
The ideal BV treatment should also provoke a microbiological cure, according to Dr. Schwebke. “What I would do is combine a drug like this with a biofilm inhibitor. Right now, this is great, because it’s single dose oral, and it’s as good as anything out there, but, I don’t think we’re taking the next step necessarily with efficacy.”
The study was supported by Symbiomix. Dr. Schwebke is a consultant for Symbiomix and receives funding from Alfa Wassermann and Starpharma, among others. Dr. Hillier is coprincipal investigator of the Microbicide Trials Network, sponsored by the National Institutes of Health.
On Twitter @whitneymcknight
AT IDSOG
Key clinical point: Granulated, single-dose oral secnidazole was statistically superior to placebo in treating bacterial vaginosis.
Major finding: In the study arm, 53.3% of women had “normal” discharge and less than 20% clue cells at their 21-30 day visit, compared with 19.3% in the placebo group (P less than .001).
Data source: A randomized, controlled phase III study of 189 women with bacterial vaginosis.
Disclosures: The study was supported by Symbiomix. Dr. Schwebke is a consultant for Symbiomix and receives funding from Alfa Wassermann and Starpharma, among others. Dr. Hillier is coprincipal investigator of the Microbicide Trials Network, sponsored by the National Institutes of Health.

FDG-PET gives early indication of response to therapy for Ewing sarcoma
Among patients with Ewing sarcoma, functional imaging with 18fluorodeoxyglucose (FDG)–PET was a better predictor of response 9 days after the start of therapy than anatomic imaging modalities were at 6 weeks, results of a retrospective analysis suggest.
A study comparing the predictive ability of functional imaging modalities such as FDG-PET with that of anatomic imaging modalities such as CT or MRI showed that an early signal with FDG-PET was superior to anatomic imaging, and that newly defined tumor volume criteria were better at predicting response and clinical benefit than either World Health Organization (WHO) criteria or Response Evaluation Criteria in Solid Tumors (RECIST), reported Vadim S. Koshkin, MD, of the Cleveland Clinic and colleagues.
“The time needed to assess tumors volumetrically is slightly greater than to do so unidimensionally or bidimensionally, but the process is now automated. The analysis presented here suggests that assessment of tumor volume is superior to predict response in clinical trials, compared with the currently widely used RECIST and WHO criteria. This requires additional validation with prospective clinical trials,” they wrote (J Clin Oncol. 2016 Aug 29. doi: 10.1200/JCO.2016.68.1858].
To get a better idea of the relative benefits of FDG-PET and anatomic imaging for assessing clinical outcomes, the investigators took a retrospective look at patients with Ewing sarcoma who were enrolled in the SARC 011 trial, a single-arm, multicenter, multicohort phase II study of patients with recurrent Ewing sarcoma treated with the investigational insulinlike growth factor–1 receptor antibody R1507.
Of the 115 patients enrolled, 76 had available anatomic imaging at baseline and at 6 weeks after the start of treatment. The imaging studies were assessed by the treating oncologist according to WHO criteria and by a central, external group of radiologists blinded to clinical outcomes of individual patients. FDG-PET studies were performed at baseline and on day 9 and were assessed by central reviewers using PET Response Criteria in Solid Tumors (PERCIST).
The authors compared PERCIST 1.0 criteria for functional imaging with FDG-PET, and for anatomic imaging, WHO and RECIST criteria were assessed independently, and newly defined volumetric criteria were based on measurements done by the central radiology group using semi-automated solid tumor segmentation software.
As noted before, the investigators found results of day 9 FDG-PET scans were associated with reduced overall survival (OS) in patients with disease progression, compared with those without progression (P = .001), and with improved OS among patients with responses to the antibody, compared with those without responses.
“There was no significance in median survival between patients who responded to treatment and patients with stable disease for any of the imaging criteria. However, for all of the criteria, there was a trend toward longer survival for patients in the response group, compared with the stable disease group,” the authors wrote.
They found that the anatomic imaging criteria (WHO local and centralized assessments, RECIST, and volume) identified fewer patients in the response group than PERCIST (FDG-PET) criteria did.
When they looked at the subgroup of 66 patients who had both interpretable functional and anatomic imaging, they found that PERCIST identified 43.9% of patients as responders, and 90.9% as nonprogressors.
The authors acknowledged that their study was hampered by the retrospective design, small sample size relative to the trial population, and lack of FDG-PET standardization to common criteria across the treatment centers.
Among patients with Ewing sarcoma, functional imaging with 18fluorodeoxyglucose (FDG)–PET was a better predictor of response 9 days after the start of therapy than anatomic imaging modalities were at 6 weeks, results of a retrospective analysis suggest.
A study comparing the predictive ability of functional imaging modalities such as FDG-PET with that of anatomic imaging modalities such as CT or MRI showed that an early signal with FDG-PET was superior to anatomic imaging, and that newly defined tumor volume criteria were better at predicting response and clinical benefit than either World Health Organization (WHO) criteria or Response Evaluation Criteria in Solid Tumors (RECIST), reported Vadim S. Koshkin, MD, of the Cleveland Clinic and colleagues.
“The time needed to assess tumors volumetrically is slightly greater than to do so unidimensionally or bidimensionally, but the process is now automated. The analysis presented here suggests that assessment of tumor volume is superior to predict response in clinical trials, compared with the currently widely used RECIST and WHO criteria. This requires additional validation with prospective clinical trials,” they wrote (J Clin Oncol. 2016 Aug 29. doi: 10.1200/JCO.2016.68.1858].
To get a better idea of the relative benefits of FDG-PET and anatomic imaging for assessing clinical outcomes, the investigators took a retrospective look at patients with Ewing sarcoma who were enrolled in the SARC 011 trial, a single-arm, multicenter, multicohort phase II study of patients with recurrent Ewing sarcoma treated with the investigational insulinlike growth factor–1 receptor antibody R1507.
Of the 115 patients enrolled, 76 had available anatomic imaging at baseline and at 6 weeks after the start of treatment. The imaging studies were assessed by the treating oncologist according to WHO criteria and by a central, external group of radiologists blinded to clinical outcomes of individual patients. FDG-PET studies were performed at baseline and on day 9 and were assessed by central reviewers using PET Response Criteria in Solid Tumors (PERCIST).
The authors compared PERCIST 1.0 criteria for functional imaging with FDG-PET, and for anatomic imaging, WHO and RECIST criteria were assessed independently, and newly defined volumetric criteria were based on measurements done by the central radiology group using semi-automated solid tumor segmentation software.
As noted before, the investigators found results of day 9 FDG-PET scans were associated with reduced overall survival (OS) in patients with disease progression, compared with those without progression (P = .001), and with improved OS among patients with responses to the antibody, compared with those without responses.
“There was no significance in median survival between patients who responded to treatment and patients with stable disease for any of the imaging criteria. However, for all of the criteria, there was a trend toward longer survival for patients in the response group, compared with the stable disease group,” the authors wrote.
They found that the anatomic imaging criteria (WHO local and centralized assessments, RECIST, and volume) identified fewer patients in the response group than PERCIST (FDG-PET) criteria did.
When they looked at the subgroup of 66 patients who had both interpretable functional and anatomic imaging, they found that PERCIST identified 43.9% of patients as responders, and 90.9% as nonprogressors.
The authors acknowledged that their study was hampered by the retrospective design, small sample size relative to the trial population, and lack of FDG-PET standardization to common criteria across the treatment centers.
Among patients with Ewing sarcoma, functional imaging with 18fluorodeoxyglucose (FDG)–PET was a better predictor of response 9 days after the start of therapy than anatomic imaging modalities were at 6 weeks, results of a retrospective analysis suggest.
A study comparing the predictive ability of functional imaging modalities such as FDG-PET with that of anatomic imaging modalities such as CT or MRI showed that an early signal with FDG-PET was superior to anatomic imaging, and that newly defined tumor volume criteria were better at predicting response and clinical benefit than either World Health Organization (WHO) criteria or Response Evaluation Criteria in Solid Tumors (RECIST), reported Vadim S. Koshkin, MD, of the Cleveland Clinic and colleagues.
“The time needed to assess tumors volumetrically is slightly greater than to do so unidimensionally or bidimensionally, but the process is now automated. The analysis presented here suggests that assessment of tumor volume is superior to predict response in clinical trials, compared with the currently widely used RECIST and WHO criteria. This requires additional validation with prospective clinical trials,” they wrote (J Clin Oncol. 2016 Aug 29. doi: 10.1200/JCO.2016.68.1858].
To get a better idea of the relative benefits of FDG-PET and anatomic imaging for assessing clinical outcomes, the investigators took a retrospective look at patients with Ewing sarcoma who were enrolled in the SARC 011 trial, a single-arm, multicenter, multicohort phase II study of patients with recurrent Ewing sarcoma treated with the investigational insulinlike growth factor–1 receptor antibody R1507.
Of the 115 patients enrolled, 76 had available anatomic imaging at baseline and at 6 weeks after the start of treatment. The imaging studies were assessed by the treating oncologist according to WHO criteria and by a central, external group of radiologists blinded to clinical outcomes of individual patients. FDG-PET studies were performed at baseline and on day 9 and were assessed by central reviewers using PET Response Criteria in Solid Tumors (PERCIST).
The authors compared PERCIST 1.0 criteria for functional imaging with FDG-PET, and for anatomic imaging, WHO and RECIST criteria were assessed independently, and newly defined volumetric criteria were based on measurements done by the central radiology group using semi-automated solid tumor segmentation software.
As noted before, the investigators found results of day 9 FDG-PET scans were associated with reduced overall survival (OS) in patients with disease progression, compared with those without progression (P = .001), and with improved OS among patients with responses to the antibody, compared with those without responses.
“There was no significance in median survival between patients who responded to treatment and patients with stable disease for any of the imaging criteria. However, for all of the criteria, there was a trend toward longer survival for patients in the response group, compared with the stable disease group,” the authors wrote.
They found that the anatomic imaging criteria (WHO local and centralized assessments, RECIST, and volume) identified fewer patients in the response group than PERCIST (FDG-PET) criteria did.
When they looked at the subgroup of 66 patients who had both interpretable functional and anatomic imaging, they found that PERCIST identified 43.9% of patients as responders, and 90.9% as nonprogressors.
The authors acknowledged that their study was hampered by the retrospective design, small sample size relative to the trial population, and lack of FDG-PET standardization to common criteria across the treatment centers.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: FDG-PET predicted response to Ewing sarcoma therapy significantly earlier than MRI or CT.
Major finding: Day 9 FDG-PET scans were associated with reduced overall survival (OS) in patients with disease progression, compared with those without progression (P = .001).
Data source: Retrospective analysis of data on 76 patients enrolled in the SARC 011 trial.
Disclosures: The study was supported by the Radiological Society of North America; Quantitative Imaging Biomarker Alliance; the Sarcoma Alliance for Research Through Collaboration (SARC), and the National Institutes of Health. Vadim S. Koshkin and three coauthors reported no conflicts of interest. Seven coauthors reported financial relationships with various drug and/or device companies.
Vascular surgeons assisting nonvascular colleagues require depth/breadth of experience
Vascular surgeons called upon to provide intraoperative assistance should be prepared to undertake a wide range of repairs.
Nonvascular surgery patients who required any vascular repair had a higher incidence of the primary endpoint of death, myocardial infarction, or unplanned return to the operating room within 30 days post surgery. In addition, such cases accounted for almost 7% of the operative volume of the hospital’s vascular surgery service, according to the results of a retrospective record review of all 533 patients who underwent nonvascular surgery requiring intraoperative assistance by a vascular surgeon at Northwestern Memorial Hospital, Chicago, between January 2010 and June 2014.
After excluding 28 trauma patients and 226 who required placement of an inferior vena cava filter only, the remaining cohort of 299 patients were assessed. This cohort represented 6.9% of the entire operative output of the vascular surgery service at the hospital during the period assessed. The cohort comprised 49.5% men and a had mean patient age of 56.4 years, according to Tadaki M. Tomita, MD, and his colleagues at Northwestern University, Chicago.
Intraoperative assistance was requested by 12 different surgical subspecialties during the period studied, with the most common being neurosurgery (33.8%), orthopedic surgery (26.4%), urology (15.7%), and surgical oncology (6.7%). The major vascular surgeon participation by indications were spine exposure (52%), vascular reconstruction (19%), vascular control without hemorrhage (14.4%), and control of hemorrhage (14.4%), according to a report published online in JAMA Surgery (2016 Aug 3. doi: 10.1001/jamasurg.2016.2247).
For the entire cohort, 110 patients (36.8%) required vascular repairs, with 13 bypasses (4.4%), 18 patch angioplasties (6.0%), and 79 primary repairs (26.4%) performed; 64 cases were venous (21.4%) and 43 arterial (14.7%). The anatomic distribution in patients requiring vascular repair was 72.7% truncal and 27.4% peripheral.
Patients who required any vascular repair had a significantly higher incidence of the primary endpoint than did patients who did not require vascular repair (17.4% vs. 7.9%; P = .01), with five deaths, 16 MIs, and 20 unplanned returns to the OR.
“Vascular surgeons are often called on by nonvascular surgeons for assistance in the OR in a variety of clinical situations and anatomic locations,” the researchers stated. The vascular surgeon in all cases performed an open surgical exposure and open repair was performed in all cases that required vascular repair.
“While most consultations occurred preoperatively, a high proportion of emergent cases that are more likely to require vascular repair demonstrates the importance of having vascular surgeons immediately available at the hospital. To continue providing this valuable service, vascular trainees will need to continue to learn the full breadth of anatomic exposures and open vascular reconstructions,” the researchers concluded.
The authors reported that they had no disclosures.
Vascular surgeons called upon to provide intraoperative assistance should be prepared to undertake a wide range of repairs.
Nonvascular surgery patients who required any vascular repair had a higher incidence of the primary endpoint of death, myocardial infarction, or unplanned return to the operating room within 30 days post surgery. In addition, such cases accounted for almost 7% of the operative volume of the hospital’s vascular surgery service, according to the results of a retrospective record review of all 533 patients who underwent nonvascular surgery requiring intraoperative assistance by a vascular surgeon at Northwestern Memorial Hospital, Chicago, between January 2010 and June 2014.
After excluding 28 trauma patients and 226 who required placement of an inferior vena cava filter only, the remaining cohort of 299 patients were assessed. This cohort represented 6.9% of the entire operative output of the vascular surgery service at the hospital during the period assessed. The cohort comprised 49.5% men and a had mean patient age of 56.4 years, according to Tadaki M. Tomita, MD, and his colleagues at Northwestern University, Chicago.
Intraoperative assistance was requested by 12 different surgical subspecialties during the period studied, with the most common being neurosurgery (33.8%), orthopedic surgery (26.4%), urology (15.7%), and surgical oncology (6.7%). The major vascular surgeon participation by indications were spine exposure (52%), vascular reconstruction (19%), vascular control without hemorrhage (14.4%), and control of hemorrhage (14.4%), according to a report published online in JAMA Surgery (2016 Aug 3. doi: 10.1001/jamasurg.2016.2247).
For the entire cohort, 110 patients (36.8%) required vascular repairs, with 13 bypasses (4.4%), 18 patch angioplasties (6.0%), and 79 primary repairs (26.4%) performed; 64 cases were venous (21.4%) and 43 arterial (14.7%). The anatomic distribution in patients requiring vascular repair was 72.7% truncal and 27.4% peripheral.
Patients who required any vascular repair had a significantly higher incidence of the primary endpoint than did patients who did not require vascular repair (17.4% vs. 7.9%; P = .01), with five deaths, 16 MIs, and 20 unplanned returns to the OR.
“Vascular surgeons are often called on by nonvascular surgeons for assistance in the OR in a variety of clinical situations and anatomic locations,” the researchers stated. The vascular surgeon in all cases performed an open surgical exposure and open repair was performed in all cases that required vascular repair.
“While most consultations occurred preoperatively, a high proportion of emergent cases that are more likely to require vascular repair demonstrates the importance of having vascular surgeons immediately available at the hospital. To continue providing this valuable service, vascular trainees will need to continue to learn the full breadth of anatomic exposures and open vascular reconstructions,” the researchers concluded.
The authors reported that they had no disclosures.
Vascular surgeons called upon to provide intraoperative assistance should be prepared to undertake a wide range of repairs.
Nonvascular surgery patients who required any vascular repair had a higher incidence of the primary endpoint of death, myocardial infarction, or unplanned return to the operating room within 30 days post surgery. In addition, such cases accounted for almost 7% of the operative volume of the hospital’s vascular surgery service, according to the results of a retrospective record review of all 533 patients who underwent nonvascular surgery requiring intraoperative assistance by a vascular surgeon at Northwestern Memorial Hospital, Chicago, between January 2010 and June 2014.
After excluding 28 trauma patients and 226 who required placement of an inferior vena cava filter only, the remaining cohort of 299 patients were assessed. This cohort represented 6.9% of the entire operative output of the vascular surgery service at the hospital during the period assessed. The cohort comprised 49.5% men and a had mean patient age of 56.4 years, according to Tadaki M. Tomita, MD, and his colleagues at Northwestern University, Chicago.
Intraoperative assistance was requested by 12 different surgical subspecialties during the period studied, with the most common being neurosurgery (33.8%), orthopedic surgery (26.4%), urology (15.7%), and surgical oncology (6.7%). The major vascular surgeon participation by indications were spine exposure (52%), vascular reconstruction (19%), vascular control without hemorrhage (14.4%), and control of hemorrhage (14.4%), according to a report published online in JAMA Surgery (2016 Aug 3. doi: 10.1001/jamasurg.2016.2247).
For the entire cohort, 110 patients (36.8%) required vascular repairs, with 13 bypasses (4.4%), 18 patch angioplasties (6.0%), and 79 primary repairs (26.4%) performed; 64 cases were venous (21.4%) and 43 arterial (14.7%). The anatomic distribution in patients requiring vascular repair was 72.7% truncal and 27.4% peripheral.
Patients who required any vascular repair had a significantly higher incidence of the primary endpoint than did patients who did not require vascular repair (17.4% vs. 7.9%; P = .01), with five deaths, 16 MIs, and 20 unplanned returns to the OR.
“Vascular surgeons are often called on by nonvascular surgeons for assistance in the OR in a variety of clinical situations and anatomic locations,” the researchers stated. The vascular surgeon in all cases performed an open surgical exposure and open repair was performed in all cases that required vascular repair.
“While most consultations occurred preoperatively, a high proportion of emergent cases that are more likely to require vascular repair demonstrates the importance of having vascular surgeons immediately available at the hospital. To continue providing this valuable service, vascular trainees will need to continue to learn the full breadth of anatomic exposures and open vascular reconstructions,” the researchers concluded.
The authors reported that they had no disclosures.
FROM JAMA SURGERY
Key clinical point: Intraoperative assistance of vascular surgeons in nonvascular procedures accounted for nearly 7% of vascular work at a single institution and uniformly required open repair.
Major finding: Patients who required any intraoperative vascular repair had a higher incidence of the primary endpoint of death, myocardial infarction, or unplanned return to the operating room within 30 days post surgery.
Data source: The study was a retrospective review of all 299 patients undergoing nonvascular surgery who required intraoperative vascular surgery assistance at a single institution between January 2010 and June 2014.
Disclosures: The authors reported that they had no disclosures.
Serious infections in second trimester increase epilepsy risk
ANNAPOLIS, MD. – Febrile infections occurring in the second trimester appear to pose the greatest risk to the neurodevelopment of the fetus, a population based cohort study has shown.
In a review of 8,618,171 California births between January 1991 and December 2008, Ms. Hilary Haber, a third-year medical student at the University of California, Davis, and her coinvestigators found that maternal infections requiring hospitalizations during the second trimester were associated with a relative risk of 2.5 of having a child with epilepsy, a relative risk of 2.3 of having a child with an intellectual disability, and a relative risk of 1.2 of having a child with autism.

Significant associations were observed between subcategories of infection and intellectual disability and epilepsy, particularly those of a bacterial cause and from respiratory and genitourinary sites. Overall, any maternal infection during pregnancy was associated with a 43% increased risk of epilepsy, a 33% increased risk of intellectual disability, and an 8% increased risk of autism.
The exact mechanism of action between the maternal infection and adverse fetal neurodevelopmental outcomes is still unclear, Ms. Haber said at the annual scientific meeting of the Infectious Diseases Society for Obstetrics and Gynecology.
“Next, we are considering which specific [maternal] infections we should look at,” Ms. Haber said in an interview. “There is something about febrile infections, so we want to narrow that down and better characterize the outcomes from mild, moderate, severe infections.”
Ms. Haber reported having no relevant financial disclosures.
On Twitter @whitneymcknight
ANNAPOLIS, MD. – Febrile infections occurring in the second trimester appear to pose the greatest risk to the neurodevelopment of the fetus, a population based cohort study has shown.
In a review of 8,618,171 California births between January 1991 and December 2008, Ms. Hilary Haber, a third-year medical student at the University of California, Davis, and her coinvestigators found that maternal infections requiring hospitalizations during the second trimester were associated with a relative risk of 2.5 of having a child with epilepsy, a relative risk of 2.3 of having a child with an intellectual disability, and a relative risk of 1.2 of having a child with autism.
Significant associations were observed between subcategories of infection and intellectual disability and epilepsy, particularly those of a bacterial cause and from respiratory and genitourinary sites. Overall, any maternal infection during pregnancy was associated with a 43% increased risk of epilepsy, a 33% increased risk of intellectual disability, and an 8% increased risk of autism.
The exact mechanism of action between the maternal infection and adverse fetal neurodevelopmental outcomes is still unclear, Ms. Haber said at the annual scientific meeting of the Infectious Diseases Society for Obstetrics and Gynecology.
“Next, we are considering which specific [maternal] infections we should look at,” Ms. Haber said in an interview. “There is something about febrile infections, so we want to narrow that down and better characterize the outcomes from mild, moderate, severe infections.”
Ms. Haber reported having no relevant financial disclosures.
On Twitter @whitneymcknight
ANNAPOLIS, MD. – Febrile infections occurring in the second trimester appear to pose the greatest risk to the neurodevelopment of the fetus, a population based cohort study has shown.
In a review of 8,618,171 California births between January 1991 and December 2008, Ms. Hilary Haber, a third-year medical student at the University of California, Davis, and her coinvestigators found that maternal infections requiring hospitalizations during the second trimester were associated with a relative risk of 2.5 of having a child with epilepsy, a relative risk of 2.3 of having a child with an intellectual disability, and a relative risk of 1.2 of having a child with autism.
Significant associations were observed between subcategories of infection and intellectual disability and epilepsy, particularly those of a bacterial cause and from respiratory and genitourinary sites. Overall, any maternal infection during pregnancy was associated with a 43% increased risk of epilepsy, a 33% increased risk of intellectual disability, and an 8% increased risk of autism.
The exact mechanism of action between the maternal infection and adverse fetal neurodevelopmental outcomes is still unclear, Ms. Haber said at the annual scientific meeting of the Infectious Diseases Society for Obstetrics and Gynecology.
“Next, we are considering which specific [maternal] infections we should look at,” Ms. Haber said in an interview. “There is something about febrile infections, so we want to narrow that down and better characterize the outcomes from mild, moderate, severe infections.”
Ms. Haber reported having no relevant financial disclosures.
On Twitter @whitneymcknight
AT IDSOG
Key clinical point: Serious maternal infections in the second trimester pose an increased risk of having a child with epilepsy or intellectual disability.
Major finding: Maternal infections in the second trimester were associated with a relative risk of 2.5 of having a child with epilepsy.
Data source: Retrospective, population-based cohort study of more than 8 million births between 1991 and 2008.
Disclosures: Ms. Haber reported having no relevant financial disclosures.

Medication List Discrepancies and Therapeutic Duplications Among Dual Use Veterans
In the U.S., 4.5 million ambulatory care visits occur annually due to adverse drug reactions (ADRs) of prescription medications.1 Many ADRs are severe, and they result in more than 100,000 death per year.2 A significant number of these ADRs are preventable and are a result of inappropriate prescribing.3 It is well-documented that inappropriate prescribing is exacerbated by the number of patients who see multiple prescribers and use many different prescription medications.4 This situation results in many versions of a patient’s medication list and in discrepancies across data sources.5
Medication list discrepancies have been researched in the context of care transitions between the hospital and home.6,7 However, less attention has been given to community-dwelling adults who use multiple outpatient prescribers, a practice common among older adults with chronic conditions who see a primary care provider and several specialists.4 Also, veterans are a growing patient population who use providers from multiple health care systems.8 Up to 70% of veterans enrolled in VA health care use both VA and non-VA providers. These patients are referred to as dual users.9,10
There has been an increasing push for patients to be more actively engaged in their own health care, including maintenance of their medication list and other personal health information.11-13 Providers have realized that patients have important experiences and preferences to share about how they use medications at home.14,15 Research suggests that patient interest and ability to use patient portals is variable and dependent on age, technical abilities, health literacy, and endorsement by their providers.16 Greater patient engagement in the medication management process is potentially advantageous, especially because providers from different health care systems often lack the capability to share medication list information.12,17
My HealtheVet, the VA’s patient portal, offers veterans several features. For example, users can securely message providers, refill prescriptions, check appointments, self-enter information, and download their VA health record (including medication history) using the Blue Button (BB) feature. The BB is managed by the HHS to provide consistency across electronic health record platforms.18,19
This BB medication list gives VA patients the tool they need to inform their providers about the medications they take, particularly dual users. VA patients that use multiple prescribers are subject to medication list discrepancies because of the fragmentation of information.4,20
Objectives
The objectives of this study were to (1) describe discrepancies between VA medication lists and non-VA provider medication lists for a group of veteran dual users; (2) identify therapeutic duplications in these lists; and (3) contextualize discrepancies by interviewing non-VA providers about their medication reconciliation processes and management of dual use patients.
Methods
This analysis is based on data collected as part of a pilot randomized controlled trial by Turvey and colleagues.21 Veterans with a diagnosis of ≥ 1 chronic health condition (eg, diabetes, hypertension) were invited by letter to participate in a study about using online management of their health information. Interested patients were screened to meet additional inclusion criteria, such as taking ≥ 5 medications, receiving care from a non-VA provider, an appointment with a non-VA provider within the study time frame, and access to a computer, online access, and printer.
Eligible veterans were randomized to receive either (1) BB training (intervention group) instructing patients to download the Continuity of Care Document and bring it to their non-VA provider visit; or (2) a training evaluating medical information online (control group). Training information was mailed, including written materials and phone support, to both groups. The intervention group could also access an online training link.
One of the objectives was to test whether downloading and bringing the health information to a non-VA appointment decreased medication list discrepancies. The sample was small, and differences in discrepancy rates between groups were not significant. Therefore, groups were combined for the present analysis. Visits occurred between December 2013 and December 2014. Greater detail about study design and primary results are available in the study by Turvey and colleagues.21
Study procedures were approved by the University of Iowa Institutional Review Board and the Iowa City VA Health Care System Research and Development Committee. All participants provided consent.
Identifying Discrepancies
A 4-phase process was used to address medication discrepancies.22,23 The first phase defined medication discrepancy categories. The mutually exclusive categories were dose, frequency, and missing discrepancies. In cases where a medication was both dose and frequency discrepant, only dose discrepancy was applied. For missing medications, entities on only the VA list were marked as “non-VA missing” and medications appearing on only the non-VA list would be denoted as “VA missing.” Medications with no discrepancy were marked as such.
Phase 2 involved collecting medication data. Medication lists from the VA medical record were printed at the time of the non-VA provider appointment. Non-VA medication lists were obtained by sending a medical record request for the visit note, medication list, and any associated visit test results to the non-VA provider office within 2 to 3 weeks of the appointment. Patient names from both lists were replaced with unique patient identifiers.
In phase 3, a research assistant abstracted the hard copy medication lists into a database and identified discrepancies. Variables included medication name, dose, frequency, and administration route. Although administration routes were collected, discrepancies were not assessed because this information commonly was not specified. Medications also were coded as prescription or over-the-counter (OTC). Durable medical equipment often was present on VA lists (eg, syringes, test strips) and was excluded from all analyses. Medications also were not coded as discrepant if they were referenced in a visit note as being changed by the non-VA provider. These combined lists were evaluated by the research assistant based on the discrepancy categories specified in phase 1 and were verified by a pharmacist.
Phase 4 involved counting medication discrepancies. Medication discrepancy rates were calculated at the patient level, both descriptively (mean number of discrepancies per patient) and as a proportion of medications discrepant (number of discrepancies divided by total medications).
Identifying Duplications and High-Risk Medications
A pharmacist examined each combined medication list to identify therapeutic duplications, defined as a patient using ≥ 2 medications from the same medication class (eg, patient taking 2 statin drugs) but not 2 drugs for the same condition (eg, fish oil and atorvastatin for dyslipidemia). High-risk medications also were noted, including anticoagulants, certain nonsteroidal anti-inflammatory drugs, oral and injectable hypoglycemics, opioids, sedatives, and hypnotics.24-26 These medications received special focus because of their link to a high risk for ADRs.27
Descriptive statistics were calculated for patient characteristics and for each discrepancy type, both overall and according to prescription OTC, and high-risk medications. The proportion of discrepant medications was calculated for each category. Bivariate correlations were calculated for select variables to understand potential relationships.
Interviews With Non-VA Providers
All patients were instructed to bring a consent letter and the 1-page questionnaire to their non-VA provider appointment. The questionnaire contained an item asking whether non-VA providers could be contacted for a 15- to 30-minute follow-up interview. The semistructured, qualitative interviews assessed their experiences working with VA providers and VA patients, experiences with VA documents or records, preferences for receiving information from the VA, experience with personal health records, and sharing information with the VA. Eight interviews were conducted, audio-recorded, and transcribed. The goal of the interviews was to explore and understand provider perspectives on managing dual use veterans, including medication reconciliation processes to add context to the interpretation of medication list analysis. Because the data set was relatively small, summaries of each interview were created to highlight main points. These main points were sorted into topics, summarized, and representative quotes were selected.
Results
Fifty veterans were included in the analysis (Table 1). The mean age was 68.5 (SD 6.2); 90% were men. On average, they reported having 6 chronic health conditions and a fair-to-good health status. Based on the combined medication lists from VA and non-VA providers, veterans took an average of 15.8 (SD 7.0) unique medications (combined prescription and OTC/vitamins) and had an average of 10.0 (SD 6.1) all-type discrepancies (Table 2).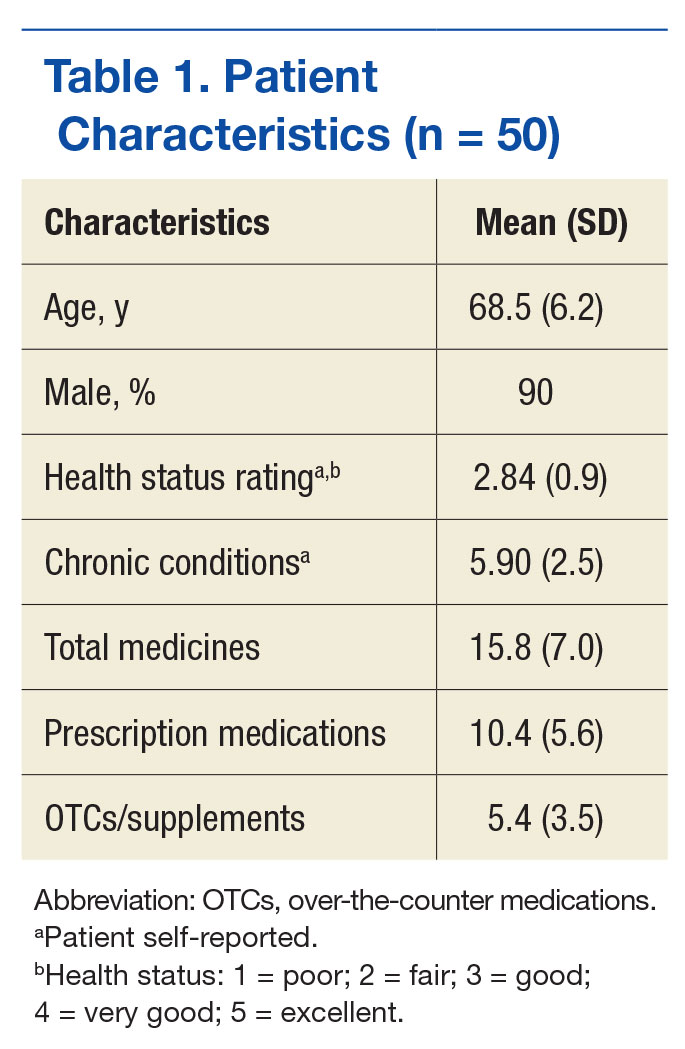
Overall, 58% of the prescription medications were discrepant: The most common discrepancy between the 2 lists was medication missing on one of the lists, which occurred 3.9 times per patient on average for prescription medications and 2.8 times per patient for OTCs. Frequency or dose discrepancies also were common between the lists at a rate of 1.9 discrepancies per patient for prescription medications and 1.2 discrepancies per patient for OTCs.
For high-risk medications, opiates and sedative medications had the most discrepancies between the lists because the VA practitioner may not have known that the patient was taking an opiate, although other discrepancies were present (Table 3). Anticoagulant discrepancies were the most consistent, most of these occurring with aspirin. Last, insulin commonly was dose discrepant between the 2 lists, although it also was missing from one list for a number of patients. Overall, high-risk medications shared a discrepancy rate (46.9%) similar to the overall rate.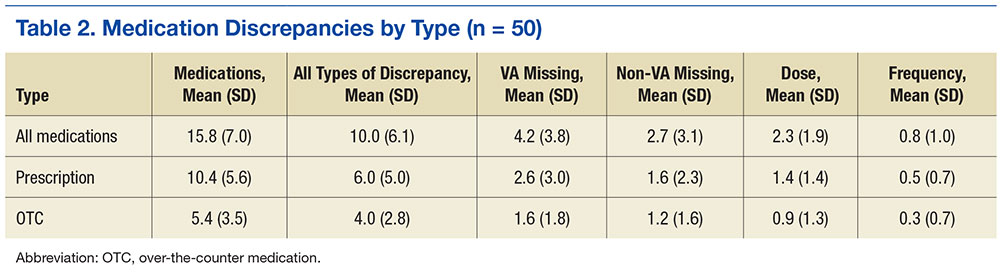
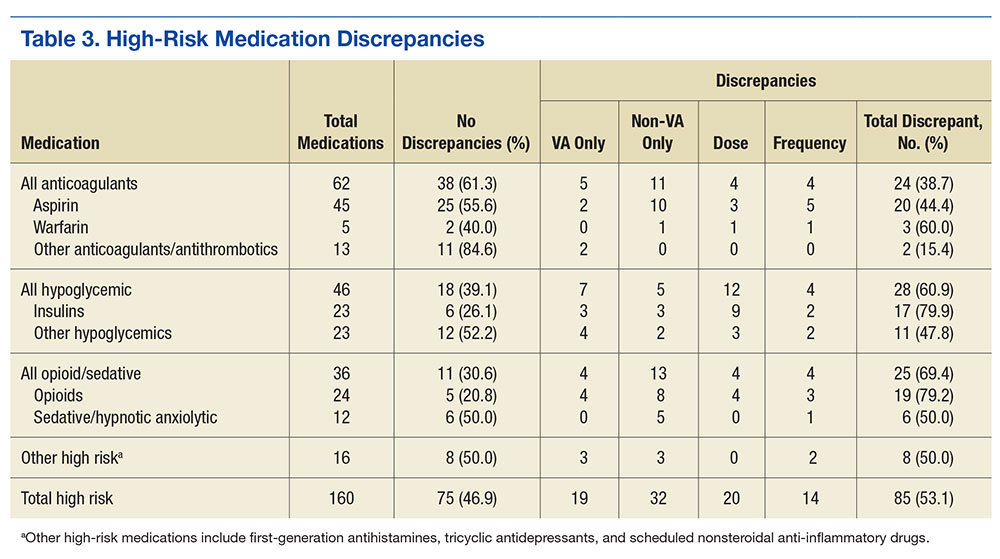
Twelve therapeutic duplications were identified in the sample.Ten were between-list duplications, that is, “provider A” thought the patient was on a particular medication and “provider B” thought that the patient was on a different medication (Table 4). In 6 instances, within-list duplications were identified (ie, a provider had 2 medications on the list that should not be taken together because they were in the same drug class). In 4 cases, both between- and within-list duplications were present.
Interview Summaries
Veterans and medication. Multiple non-VA providers said that the primary reason veteran patients were going to a VA provider was to obtain discounted medications. The use of the VA for medications also was a way for the non-VA provider to discover that the patient was a veteran. One non-VA provider was particularly concerned about the impact of new or different medications from VA prescribers on efforts to stabilize the patient’s chronic condition.
Several non-VA providers reported that veterans often brought a medication list to the appointment, and several providers recommended the practice to their patients. Non-VA providers preferred to have patients transfer information from VA, sometimes requesting that veterans bring in their records from recent appointments rather than the non-VA provider obtain the information directly from the VA.
Information sharing. Non-VA providers generally preferred hard copies of medication lists and other documents rather than scans because they were more likely to be included in decision making if the documents were presented during the visit. Also, document scans may be buried in the electronic medical record. Some providers mentioned their interest in electronic transfer of medical information like medication lists if the technology were more developed and better integrated.
“I think the long-term vision would be that it should be electronic… it wouldn’t necessarily be feasible at this time. Our system scans paper documents in to an e-version. … but when the patient comes to their encounter 10 days later, you don’t realize the stuff’s there… Having the patient bring them in is probably a more certain way to make sure that it’s actually included in your decision making as a provider.”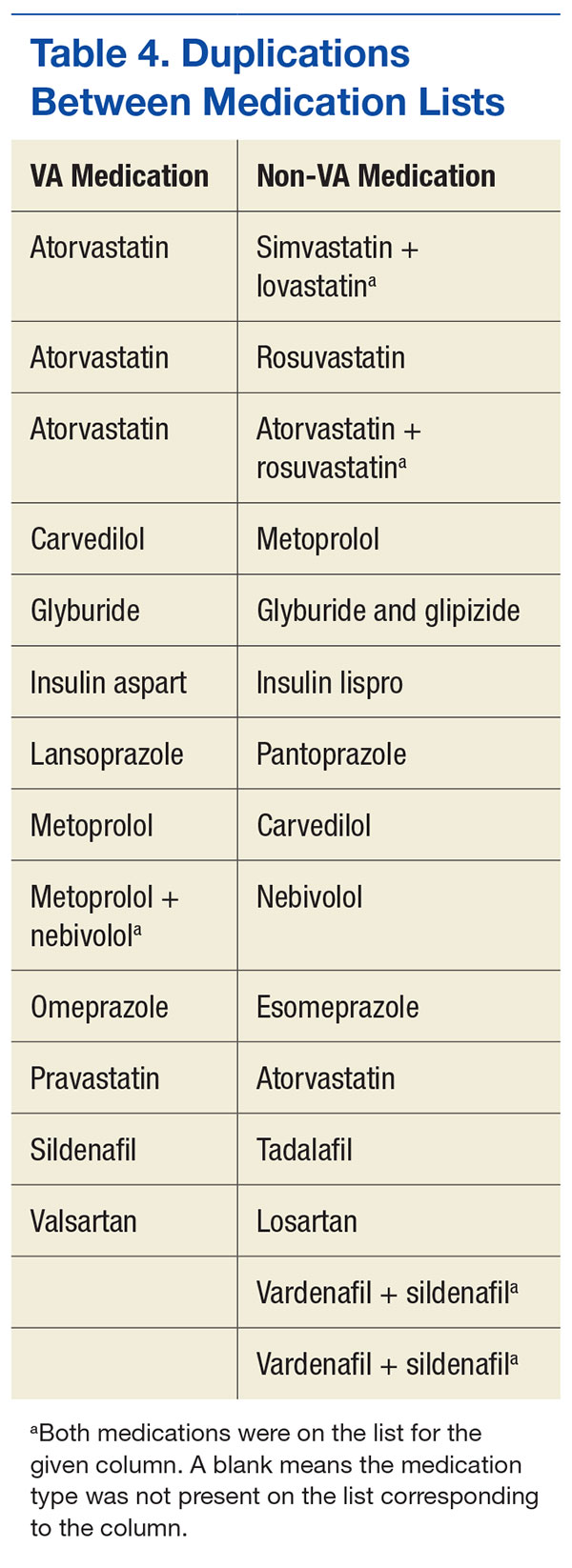
Most non-VA providers welcomed more information such as imaging studies because they reported rarely receiving this information from the VA system. Two mentioned the potential for too much information and wanted concise reports should the flow of information increase. Providers had little interest in logging in to a patient’s online health record portal as a delegate for reasons related to complexity, time, privacy, and lack of mechanism to document the information accessed.
Medication reconciliation. Non-VA providers generally reported that patients bringing their own or an outside medication list would prompt a process of medication reconciliation. The providers were interested in making changes to their records based on other lists, but outside data were verified against a patient self-report of actual use before adopting changes.
“I print out my med list of what I have in the computer and then I just check off my list against their list. And then whatever’s remaining, we talk about what the differences are, when they were changed, what they were changed for, if they were taken off of something, and if I don’t agree, then I’ll tell the patient, ‘look, there’s a disagreement here, they’ve told you not to be on this. I want you on this.”
Should a discrepancy arise, non-VA providers generally had a negative view of attempting to contact VA providers. Other mechanisms such as calling a local pharmacy would be done first.
Discussion
This study provided initial evidence that medication list discrepancies exist for dual use veterans. Other studies of medication list discrepancies have linked such inconsistencies to medication-related problems and negative outcomes for patients.27 Although efforts to increase access to care for veterans have advantages related to expediency, consequences to fragmenting care exist. More robust mechanisms for establishing and maintaining medication list consistency are needed, especially given the lack of a universally accepted medical record format or repository. A multifaceted approach, including patient engagement, seems necessary.
This study also showed that discrepancies of high-risk medications are common for veteran participants, placing them at risk for medication-related problems and harm. These risks included dose and frequency discrepancies that could result in over- or underdosing of medications and in medication omissions, which could cause duplicative therapies and unnecessary risks. For example, aspirin frequently was listed on non-VA lists but was omitted from VA medication lists. This could be problematic for patients who present to the VA for a procedure in which no information about aspirin could jeopardize their safety. Insulin doses also were commonly discrepant, which could impact glycemic control.
Many providers also had incomplete prescribing information for opiates. Those prescriptions are particularly relevant given the link between veterans, posttraumatic stress disorder, depression, and substance abuse.28-30 However, it was beyond the scope of this pilot study to link these discrepancies to ADRs, such as emergency department visits or hospital admissions. Other studies have demonstrated that discrepancies at hospital discharge can result in these types of negative outcomes.27,31 Subsequent research should determine the clinical significance of discrepancies that occur when veterans are dual users.
The qualitative interviews provided some initial context on prescriber perspectives about the role of veterans participating in the medication list sharing process and personal health records. It seemed that for the portion of patients who brought a list to their non-VA provider appointment, the information was welcomed but fell outside the usual visit workflow. Many provider visits are dominated by current patient symptoms, and issues of reconciling medications may be a lower priority.32 Also, some providers may delegate medication reconciliation functions to a nurse or other support staff. One physician offered that he delegated logging in to a patient’s online medication information to a health coach on staff. These findings were consistent with perspectives shared by non-VA family practice physicians about personal health records.33
The most common way to integrate outside medication lists into the non-VA provider’s medical record seemed to be scanning the document. Scanning had its limitations because the provider might be unaware of the scanned document, and there were no mechanisms to import medication names and doses. However, the process may improve only the non-VA providers’ records, as they reported that they had no easy or consistent way to transmit medication changes to notes to the VA.
In general, communicating with VA providers was seen as not feasible and not worth their time or effort. It may be beneficial to address this non-VA provider concern because it seems to inhibit the transfer of important health information and the maintenance of a concordant medication record. Information transfer is particularly relevant for veterans who are primarily cared for by non-VA providers and use the VA only to get prescription medications.
In the current approach, non-VA providers have no simple, direct way to update the VA medication list. Transmitting updates carries the risk of inappropriate changes and is concerning if neither or both prescribers consider themselves to be responsible for the patient’s medications. Also, the potential exists for all medication lists to be inaccurate if the lists do not reflect the medications patients take when left on their own. Patient nonadherence rates can exceed 50%, depending on the medication.34,35 Several interviewed non-VA physicians stressed the importance of asking patients to list the medications they were using during the medication reconciliation process.
This study offers several areas for additional inquiry, including understanding how providers make sense of medication lists from other sources and what technologies can be applied to increase list consistency without increasing the burden on providers.
Practice Implications
Although patient involvement in medication list sharing has the potential to improve information consistency, health systems, providers, and other stakeholders should be cautious in assuming that other prescribers will work to combat medication list entropy, especially if no systems exist to seamlessly incorporate this information into clinic workflow. Devising standardized procedures when patients bring in their records from other providers increases the likelihood that this information will be incorporated into clinical decision making and maintaining up-to-date medication information for patients who use multiple prescribers.
Limitations
These analyses are based on a small sample size (n = 50 for chart review) and (n = 8 for the semistructured interviews) from a single Midwestern state. These findings should be used as evidence for further inquiry.
Conclusion
This study illuminates the level of discrepancies between the medication lists of veteran dual users, including high rates of discrepancies for high-risk medications, such as anticoagulants and opiates. This study also provides evidence of deficiencies in the health care system to decrease medication list entropy that may place veterans at an elevated risk for adverse medication events.
1. Sarkar U, López A, Maselli JH, Gonzales R. Adverse drug events in US adult ambulatory medical care. Health Serv Res. 2011;46(5):1517-1533.
2. Kohn LT, Corrigan JM, Donaldson MS. To Err Is Human:Building a Safer Health System. Washington, DC: Institute of Medicine, National Academy Press; 1999.
3. Gandhi TK, Weingart SN, Borus J, et al. Adverse drug events in ambulatory care. N Eng J Med. 2003;348(16):1556-1564.
4. Tamblyn RM, McLeod PJ, Abrahamowicz M, Laprise R. Do too many cooks spoil the broth? Multiple physician involvement in medical management of elderly patients and potentially inappropriate drug combinations. CMAJ. 1996;154(8):1177-1184.
5. Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother. 2008;42(10):1373-1379.
6. Kripalani S, LeFevre F, Phillips CO, Williams MV, Basaviah P, Baker DW. Deficits in communication and information transfer between hospital-based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297(8):831-841.
7. McMillan A, Trompeter J, Havrda D, Fox J. Continuity of care between family practice physicians and hospitalist services. J Healthare Qual. 2013;35(1):41-49.
8. Liu CF, Manning WG, Burgess JF Jr, et al. Reliance on Veterans Affairs outpatient care by Medicare-eligible veterans. Med Care. 2011;49(10):911-917.
9. U.S. Department of Veterans Affairs, Veterans Health Administration. VHA Office of the ADUSH for Policy and Planning. 2011 survey of veteran enrollees’ health and reliance upon VA. http://www.va.gov/healthpolicyplanning/soe2011/soe2011_report.pdf. Published March 2012. Accessed August 2, 2016.
10. Nayar P, Apenteng B, Yu F, Woodbridge P, Fetrick A. Rural veterans’ perspectives of dual care. J Community Health. 2013;38(1):70-77.
11. Chae SY, Chae MH, Isaacson N, James TS. The patient medication list: can we get patients more involved in their medical care? J Am Board Fam Med. 2009;22(6):677-685.
12. Tang PC, Ash JS, Bates DW, Overhage JM, Sands DZ. Personal health records: definitions, benefits, and strategies for overcoming barriers to adoption. J Am Med Informatics Assoc. 2006;13(2):121-126.
13. Stroupe KT, Smith BM, Hogan TP, et al. Medication acquisition across systems of care and patient–provider communication among older veterans. Am J Health Syst Pharm. 2013;70(9):804-813.
14. Shoemaker SJ, Ramalho de Oliveira D, Alves M, Ekstrand M. The medication experience: preliminary evidence of its value for patient education and counseling on chronic medications. Patient Educ Couns. 2011;83(3):443-450.
15. Chewning B, Boh L, Wiederholt J, et al. Does the concordance concept serve patient medication management? Int J Pharm Pract. 2001;9(2):71-79.
16. Irizarry T, DeVito Dabbs A, Curran CR. Patient portals and patient engagement: a state of the science review. J Med Internet Res. 2015;17(6):e148.
17. Schnipper JL, Gandhi TK, Wald JS, et al. Effects of an online personal health record on medication accuracy and safety: a cluster-randomized trial. J Am Med Inform Assoc. 2012;19(5):728-734.
18. Turvey C, Klein D, Fix G, et al. Blue Button use by patients to access and share health record information using the Department of Veterans Affairs’ online patient portal. J Am Med Inform Assoc. 2014;21(4):657-663.
19. Hogan TP, Nazi KM, Luger TM, et al. Technology-assisted patient access to clinical information: an evaluation framework for Blue Button. JMIR Res Protoc. 2014;3(1):e18.
20. Steinman MA, Handler SM, Gurwitz JH, Schiff GD, Covinsky KE. Beyond the prescription: medication monitoring and adverse drug events in older adults. J Am Geriatr Soc. 2011;59(8):1520-1530.
21. Turvey CL, Klein DM, Witry M, et al. Patient education for consumer-mediated HIE. A pilot randomized controlled trial of the Department of Veterans Affairs Blue Button. Appl Clin Inform. 2016;7(3):765-776.
22. Polnaszek B, Gilmore-Bykovskyi A, Hovanes M, et al. Overcoming the challenges of unstructured data in multisite, electronic medical record-based abstraction [published online ahead of print June 25, 2014]. Med Care. doi: 10.1097/MLR.0000000000000108.
23. Kennelty K, Witry MJ, Gehring M, M D, Pulia N. A four-phase approach for systematically collecting data and measuring medication discrepancies when patients transition between health care settings. Res Social Adm Pharm. 2016;12(4):548-558.
24. American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60(4):616-631.
25. Gurwitz JH, Field TS, Harrold LR, et al. Incidence and preventability of adverse drug events among older persons in the ambulatory setting. JAMA. 2003;289(9):1107-1116.
26. Gurwitz JH, Field TS, Avorn J, et al. Incidence and preventability of adverse drug events in nursing homes. Am J Med. 2000;109(2):87-94.
27. Boockvar KS, Liu S, Goldstein N, Nebeker J, Siu A, Fried T. Prescribing discrepancies likely to cause adverse drug events after patient transfer. Qual Saf Health Care. 2009;18(1):32-36.
28. Shipherd JC, Stafford J, Tanner LR. Predicting alcohol and drug abuse in Persian Gulf War veterans: what role do PTSD symptoms play? Addict Behav. 2005;30(3):595-599.
29. Markou A, Kosten TR, Koob GF. Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology. 1998;18(3):135-174.
30. McFall ME, Mackay PW, Donovan DM. Combat-related posttraumatic stress disorder and severity of substance abuse in Vietnam veterans. J Stud Alcohol. 1992;53(4):357-363.
31. Kwan JL, Lo L, Sampson M, Shojania KG. Medication reconciliation during transitions of care as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5, pt 2):397-403.
32. Richard C, Lussier MT. Nature and frequency of exchanges on medications during primary care encounters. Patient Educ Couns. 2006;64(1-3):207-216.
33. Witry MJ, Doucette WR, Daly JM, Levy BT, Chrischilles EA. Family physician perceptions of personal health records. Perspect Health Inf Manag. 2010;7.
34. Horne R, Weinman J. Patients’ beliefs about prescribed medicines and their role in adherence to treatment in chronic physical illness. J Psychosom Res. 1999;47(6):555-567.
35. Osterberg L, Blaschke T. Adherence to medication. N Eng J Med. 2005;353(5):487-497.
In the U.S., 4.5 million ambulatory care visits occur annually due to adverse drug reactions (ADRs) of prescription medications.1 Many ADRs are severe, and they result in more than 100,000 death per year.2 A significant number of these ADRs are preventable and are a result of inappropriate prescribing.3 It is well-documented that inappropriate prescribing is exacerbated by the number of patients who see multiple prescribers and use many different prescription medications.4 This situation results in many versions of a patient’s medication list and in discrepancies across data sources.5
Medication list discrepancies have been researched in the context of care transitions between the hospital and home.6,7 However, less attention has been given to community-dwelling adults who use multiple outpatient prescribers, a practice common among older adults with chronic conditions who see a primary care provider and several specialists.4 Also, veterans are a growing patient population who use providers from multiple health care systems.8 Up to 70% of veterans enrolled in VA health care use both VA and non-VA providers. These patients are referred to as dual users.9,10
There has been an increasing push for patients to be more actively engaged in their own health care, including maintenance of their medication list and other personal health information.11-13 Providers have realized that patients have important experiences and preferences to share about how they use medications at home.14,15 Research suggests that patient interest and ability to use patient portals is variable and dependent on age, technical abilities, health literacy, and endorsement by their providers.16 Greater patient engagement in the medication management process is potentially advantageous, especially because providers from different health care systems often lack the capability to share medication list information.12,17
My HealtheVet, the VA’s patient portal, offers veterans several features. For example, users can securely message providers, refill prescriptions, check appointments, self-enter information, and download their VA health record (including medication history) using the Blue Button (BB) feature. The BB is managed by the HHS to provide consistency across electronic health record platforms.18,19
This BB medication list gives VA patients the tool they need to inform their providers about the medications they take, particularly dual users. VA patients that use multiple prescribers are subject to medication list discrepancies because of the fragmentation of information.4,20
Objectives
The objectives of this study were to (1) describe discrepancies between VA medication lists and non-VA provider medication lists for a group of veteran dual users; (2) identify therapeutic duplications in these lists; and (3) contextualize discrepancies by interviewing non-VA providers about their medication reconciliation processes and management of dual use patients.
Methods
This analysis is based on data collected as part of a pilot randomized controlled trial by Turvey and colleagues.21 Veterans with a diagnosis of ≥ 1 chronic health condition (eg, diabetes, hypertension) were invited by letter to participate in a study about using online management of their health information. Interested patients were screened to meet additional inclusion criteria, such as taking ≥ 5 medications, receiving care from a non-VA provider, an appointment with a non-VA provider within the study time frame, and access to a computer, online access, and printer.
Eligible veterans were randomized to receive either (1) BB training (intervention group) instructing patients to download the Continuity of Care Document and bring it to their non-VA provider visit; or (2) a training evaluating medical information online (control group). Training information was mailed, including written materials and phone support, to both groups. The intervention group could also access an online training link.
One of the objectives was to test whether downloading and bringing the health information to a non-VA appointment decreased medication list discrepancies. The sample was small, and differences in discrepancy rates between groups were not significant. Therefore, groups were combined for the present analysis. Visits occurred between December 2013 and December 2014. Greater detail about study design and primary results are available in the study by Turvey and colleagues.21
Study procedures were approved by the University of Iowa Institutional Review Board and the Iowa City VA Health Care System Research and Development Committee. All participants provided consent.
Identifying Discrepancies
A 4-phase process was used to address medication discrepancies.22,23 The first phase defined medication discrepancy categories. The mutually exclusive categories were dose, frequency, and missing discrepancies. In cases where a medication was both dose and frequency discrepant, only dose discrepancy was applied. For missing medications, entities on only the VA list were marked as “non-VA missing” and medications appearing on only the non-VA list would be denoted as “VA missing.” Medications with no discrepancy were marked as such.
Phase 2 involved collecting medication data. Medication lists from the VA medical record were printed at the time of the non-VA provider appointment. Non-VA medication lists were obtained by sending a medical record request for the visit note, medication list, and any associated visit test results to the non-VA provider office within 2 to 3 weeks of the appointment. Patient names from both lists were replaced with unique patient identifiers.
In phase 3, a research assistant abstracted the hard copy medication lists into a database and identified discrepancies. Variables included medication name, dose, frequency, and administration route. Although administration routes were collected, discrepancies were not assessed because this information commonly was not specified. Medications also were coded as prescription or over-the-counter (OTC). Durable medical equipment often was present on VA lists (eg, syringes, test strips) and was excluded from all analyses. Medications also were not coded as discrepant if they were referenced in a visit note as being changed by the non-VA provider. These combined lists were evaluated by the research assistant based on the discrepancy categories specified in phase 1 and were verified by a pharmacist.
Phase 4 involved counting medication discrepancies. Medication discrepancy rates were calculated at the patient level, both descriptively (mean number of discrepancies per patient) and as a proportion of medications discrepant (number of discrepancies divided by total medications).
Identifying Duplications and High-Risk Medications
A pharmacist examined each combined medication list to identify therapeutic duplications, defined as a patient using ≥ 2 medications from the same medication class (eg, patient taking 2 statin drugs) but not 2 drugs for the same condition (eg, fish oil and atorvastatin for dyslipidemia). High-risk medications also were noted, including anticoagulants, certain nonsteroidal anti-inflammatory drugs, oral and injectable hypoglycemics, opioids, sedatives, and hypnotics.24-26 These medications received special focus because of their link to a high risk for ADRs.27
Descriptive statistics were calculated for patient characteristics and for each discrepancy type, both overall and according to prescription OTC, and high-risk medications. The proportion of discrepant medications was calculated for each category. Bivariate correlations were calculated for select variables to understand potential relationships.
Interviews With Non-VA Providers
All patients were instructed to bring a consent letter and the 1-page questionnaire to their non-VA provider appointment. The questionnaire contained an item asking whether non-VA providers could be contacted for a 15- to 30-minute follow-up interview. The semistructured, qualitative interviews assessed their experiences working with VA providers and VA patients, experiences with VA documents or records, preferences for receiving information from the VA, experience with personal health records, and sharing information with the VA. Eight interviews were conducted, audio-recorded, and transcribed. The goal of the interviews was to explore and understand provider perspectives on managing dual use veterans, including medication reconciliation processes to add context to the interpretation of medication list analysis. Because the data set was relatively small, summaries of each interview were created to highlight main points. These main points were sorted into topics, summarized, and representative quotes were selected.
Results
Fifty veterans were included in the analysis (Table 1). The mean age was 68.5 (SD 6.2); 90% were men. On average, they reported having 6 chronic health conditions and a fair-to-good health status. Based on the combined medication lists from VA and non-VA providers, veterans took an average of 15.8 (SD 7.0) unique medications (combined prescription and OTC/vitamins) and had an average of 10.0 (SD 6.1) all-type discrepancies (Table 2).
Overall, 58% of the prescription medications were discrepant: The most common discrepancy between the 2 lists was medication missing on one of the lists, which occurred 3.9 times per patient on average for prescription medications and 2.8 times per patient for OTCs. Frequency or dose discrepancies also were common between the lists at a rate of 1.9 discrepancies per patient for prescription medications and 1.2 discrepancies per patient for OTCs.
For high-risk medications, opiates and sedative medications had the most discrepancies between the lists because the VA practitioner may not have known that the patient was taking an opiate, although other discrepancies were present (Table 3). Anticoagulant discrepancies were the most consistent, most of these occurring with aspirin. Last, insulin commonly was dose discrepant between the 2 lists, although it also was missing from one list for a number of patients. Overall, high-risk medications shared a discrepancy rate (46.9%) similar to the overall rate.
Twelve therapeutic duplications were identified in the sample.Ten were between-list duplications, that is, “provider A” thought the patient was on a particular medication and “provider B” thought that the patient was on a different medication (Table 4). In 6 instances, within-list duplications were identified (ie, a provider had 2 medications on the list that should not be taken together because they were in the same drug class). In 4 cases, both between- and within-list duplications were present.
Interview Summaries
Veterans and medication. Multiple non-VA providers said that the primary reason veteran patients were going to a VA provider was to obtain discounted medications. The use of the VA for medications also was a way for the non-VA provider to discover that the patient was a veteran. One non-VA provider was particularly concerned about the impact of new or different medications from VA prescribers on efforts to stabilize the patient’s chronic condition.
Several non-VA providers reported that veterans often brought a medication list to the appointment, and several providers recommended the practice to their patients. Non-VA providers preferred to have patients transfer information from VA, sometimes requesting that veterans bring in their records from recent appointments rather than the non-VA provider obtain the information directly from the VA.
Information sharing. Non-VA providers generally preferred hard copies of medication lists and other documents rather than scans because they were more likely to be included in decision making if the documents were presented during the visit. Also, document scans may be buried in the electronic medical record. Some providers mentioned their interest in electronic transfer of medical information like medication lists if the technology were more developed and better integrated.
“I think the long-term vision would be that it should be electronic… it wouldn’t necessarily be feasible at this time. Our system scans paper documents in to an e-version. … but when the patient comes to their encounter 10 days later, you don’t realize the stuff’s there… Having the patient bring them in is probably a more certain way to make sure that it’s actually included in your decision making as a provider.”
Most non-VA providers welcomed more information such as imaging studies because they reported rarely receiving this information from the VA system. Two mentioned the potential for too much information and wanted concise reports should the flow of information increase. Providers had little interest in logging in to a patient’s online health record portal as a delegate for reasons related to complexity, time, privacy, and lack of mechanism to document the information accessed.
Medication reconciliation. Non-VA providers generally reported that patients bringing their own or an outside medication list would prompt a process of medication reconciliation. The providers were interested in making changes to their records based on other lists, but outside data were verified against a patient self-report of actual use before adopting changes.
“I print out my med list of what I have in the computer and then I just check off my list against their list. And then whatever’s remaining, we talk about what the differences are, when they were changed, what they were changed for, if they were taken off of something, and if I don’t agree, then I’ll tell the patient, ‘look, there’s a disagreement here, they’ve told you not to be on this. I want you on this.”
Should a discrepancy arise, non-VA providers generally had a negative view of attempting to contact VA providers. Other mechanisms such as calling a local pharmacy would be done first.
Discussion
This study provided initial evidence that medication list discrepancies exist for dual use veterans. Other studies of medication list discrepancies have linked such inconsistencies to medication-related problems and negative outcomes for patients.27 Although efforts to increase access to care for veterans have advantages related to expediency, consequences to fragmenting care exist. More robust mechanisms for establishing and maintaining medication list consistency are needed, especially given the lack of a universally accepted medical record format or repository. A multifaceted approach, including patient engagement, seems necessary.
This study also showed that discrepancies of high-risk medications are common for veteran participants, placing them at risk for medication-related problems and harm. These risks included dose and frequency discrepancies that could result in over- or underdosing of medications and in medication omissions, which could cause duplicative therapies and unnecessary risks. For example, aspirin frequently was listed on non-VA lists but was omitted from VA medication lists. This could be problematic for patients who present to the VA for a procedure in which no information about aspirin could jeopardize their safety. Insulin doses also were commonly discrepant, which could impact glycemic control.
Many providers also had incomplete prescribing information for opiates. Those prescriptions are particularly relevant given the link between veterans, posttraumatic stress disorder, depression, and substance abuse.28-30 However, it was beyond the scope of this pilot study to link these discrepancies to ADRs, such as emergency department visits or hospital admissions. Other studies have demonstrated that discrepancies at hospital discharge can result in these types of negative outcomes.27,31 Subsequent research should determine the clinical significance of discrepancies that occur when veterans are dual users.
The qualitative interviews provided some initial context on prescriber perspectives about the role of veterans participating in the medication list sharing process and personal health records. It seemed that for the portion of patients who brought a list to their non-VA provider appointment, the information was welcomed but fell outside the usual visit workflow. Many provider visits are dominated by current patient symptoms, and issues of reconciling medications may be a lower priority.32 Also, some providers may delegate medication reconciliation functions to a nurse or other support staff. One physician offered that he delegated logging in to a patient’s online medication information to a health coach on staff. These findings were consistent with perspectives shared by non-VA family practice physicians about personal health records.33
The most common way to integrate outside medication lists into the non-VA provider’s medical record seemed to be scanning the document. Scanning had its limitations because the provider might be unaware of the scanned document, and there were no mechanisms to import medication names and doses. However, the process may improve only the non-VA providers’ records, as they reported that they had no easy or consistent way to transmit medication changes to notes to the VA.
In general, communicating with VA providers was seen as not feasible and not worth their time or effort. It may be beneficial to address this non-VA provider concern because it seems to inhibit the transfer of important health information and the maintenance of a concordant medication record. Information transfer is particularly relevant for veterans who are primarily cared for by non-VA providers and use the VA only to get prescription medications.
In the current approach, non-VA providers have no simple, direct way to update the VA medication list. Transmitting updates carries the risk of inappropriate changes and is concerning if neither or both prescribers consider themselves to be responsible for the patient’s medications. Also, the potential exists for all medication lists to be inaccurate if the lists do not reflect the medications patients take when left on their own. Patient nonadherence rates can exceed 50%, depending on the medication.34,35 Several interviewed non-VA physicians stressed the importance of asking patients to list the medications they were using during the medication reconciliation process.
This study offers several areas for additional inquiry, including understanding how providers make sense of medication lists from other sources and what technologies can be applied to increase list consistency without increasing the burden on providers.
Practice Implications
Although patient involvement in medication list sharing has the potential to improve information consistency, health systems, providers, and other stakeholders should be cautious in assuming that other prescribers will work to combat medication list entropy, especially if no systems exist to seamlessly incorporate this information into clinic workflow. Devising standardized procedures when patients bring in their records from other providers increases the likelihood that this information will be incorporated into clinical decision making and maintaining up-to-date medication information for patients who use multiple prescribers.
Limitations
These analyses are based on a small sample size (n = 50 for chart review) and (n = 8 for the semistructured interviews) from a single Midwestern state. These findings should be used as evidence for further inquiry.
Conclusion
This study illuminates the level of discrepancies between the medication lists of veteran dual users, including high rates of discrepancies for high-risk medications, such as anticoagulants and opiates. This study also provides evidence of deficiencies in the health care system to decrease medication list entropy that may place veterans at an elevated risk for adverse medication events.
In the U.S., 4.5 million ambulatory care visits occur annually due to adverse drug reactions (ADRs) of prescription medications.1 Many ADRs are severe, and they result in more than 100,000 death per year.2 A significant number of these ADRs are preventable and are a result of inappropriate prescribing.3 It is well-documented that inappropriate prescribing is exacerbated by the number of patients who see multiple prescribers and use many different prescription medications.4 This situation results in many versions of a patient’s medication list and in discrepancies across data sources.5
Medication list discrepancies have been researched in the context of care transitions between the hospital and home.6,7 However, less attention has been given to community-dwelling adults who use multiple outpatient prescribers, a practice common among older adults with chronic conditions who see a primary care provider and several specialists.4 Also, veterans are a growing patient population who use providers from multiple health care systems.8 Up to 70% of veterans enrolled in VA health care use both VA and non-VA providers. These patients are referred to as dual users.9,10
There has been an increasing push for patients to be more actively engaged in their own health care, including maintenance of their medication list and other personal health information.11-13 Providers have realized that patients have important experiences and preferences to share about how they use medications at home.14,15 Research suggests that patient interest and ability to use patient portals is variable and dependent on age, technical abilities, health literacy, and endorsement by their providers.16 Greater patient engagement in the medication management process is potentially advantageous, especially because providers from different health care systems often lack the capability to share medication list information.12,17
My HealtheVet, the VA’s patient portal, offers veterans several features. For example, users can securely message providers, refill prescriptions, check appointments, self-enter information, and download their VA health record (including medication history) using the Blue Button (BB) feature. The BB is managed by the HHS to provide consistency across electronic health record platforms.18,19
This BB medication list gives VA patients the tool they need to inform their providers about the medications they take, particularly dual users. VA patients that use multiple prescribers are subject to medication list discrepancies because of the fragmentation of information.4,20
Objectives
The objectives of this study were to (1) describe discrepancies between VA medication lists and non-VA provider medication lists for a group of veteran dual users; (2) identify therapeutic duplications in these lists; and (3) contextualize discrepancies by interviewing non-VA providers about their medication reconciliation processes and management of dual use patients.
Methods
This analysis is based on data collected as part of a pilot randomized controlled trial by Turvey and colleagues.21 Veterans with a diagnosis of ≥ 1 chronic health condition (eg, diabetes, hypertension) were invited by letter to participate in a study about using online management of their health information. Interested patients were screened to meet additional inclusion criteria, such as taking ≥ 5 medications, receiving care from a non-VA provider, an appointment with a non-VA provider within the study time frame, and access to a computer, online access, and printer.
Eligible veterans were randomized to receive either (1) BB training (intervention group) instructing patients to download the Continuity of Care Document and bring it to their non-VA provider visit; or (2) a training evaluating medical information online (control group). Training information was mailed, including written materials and phone support, to both groups. The intervention group could also access an online training link.
One of the objectives was to test whether downloading and bringing the health information to a non-VA appointment decreased medication list discrepancies. The sample was small, and differences in discrepancy rates between groups were not significant. Therefore, groups were combined for the present analysis. Visits occurred between December 2013 and December 2014. Greater detail about study design and primary results are available in the study by Turvey and colleagues.21
Study procedures were approved by the University of Iowa Institutional Review Board and the Iowa City VA Health Care System Research and Development Committee. All participants provided consent.
Identifying Discrepancies
A 4-phase process was used to address medication discrepancies.22,23 The first phase defined medication discrepancy categories. The mutually exclusive categories were dose, frequency, and missing discrepancies. In cases where a medication was both dose and frequency discrepant, only dose discrepancy was applied. For missing medications, entities on only the VA list were marked as “non-VA missing” and medications appearing on only the non-VA list would be denoted as “VA missing.” Medications with no discrepancy were marked as such.
Phase 2 involved collecting medication data. Medication lists from the VA medical record were printed at the time of the non-VA provider appointment. Non-VA medication lists were obtained by sending a medical record request for the visit note, medication list, and any associated visit test results to the non-VA provider office within 2 to 3 weeks of the appointment. Patient names from both lists were replaced with unique patient identifiers.
In phase 3, a research assistant abstracted the hard copy medication lists into a database and identified discrepancies. Variables included medication name, dose, frequency, and administration route. Although administration routes were collected, discrepancies were not assessed because this information commonly was not specified. Medications also were coded as prescription or over-the-counter (OTC). Durable medical equipment often was present on VA lists (eg, syringes, test strips) and was excluded from all analyses. Medications also were not coded as discrepant if they were referenced in a visit note as being changed by the non-VA provider. These combined lists were evaluated by the research assistant based on the discrepancy categories specified in phase 1 and were verified by a pharmacist.
Phase 4 involved counting medication discrepancies. Medication discrepancy rates were calculated at the patient level, both descriptively (mean number of discrepancies per patient) and as a proportion of medications discrepant (number of discrepancies divided by total medications).
Identifying Duplications and High-Risk Medications
A pharmacist examined each combined medication list to identify therapeutic duplications, defined as a patient using ≥ 2 medications from the same medication class (eg, patient taking 2 statin drugs) but not 2 drugs for the same condition (eg, fish oil and atorvastatin for dyslipidemia). High-risk medications also were noted, including anticoagulants, certain nonsteroidal anti-inflammatory drugs, oral and injectable hypoglycemics, opioids, sedatives, and hypnotics.24-26 These medications received special focus because of their link to a high risk for ADRs.27
Descriptive statistics were calculated for patient characteristics and for each discrepancy type, both overall and according to prescription OTC, and high-risk medications. The proportion of discrepant medications was calculated for each category. Bivariate correlations were calculated for select variables to understand potential relationships.
Interviews With Non-VA Providers
All patients were instructed to bring a consent letter and the 1-page questionnaire to their non-VA provider appointment. The questionnaire contained an item asking whether non-VA providers could be contacted for a 15- to 30-minute follow-up interview. The semistructured, qualitative interviews assessed their experiences working with VA providers and VA patients, experiences with VA documents or records, preferences for receiving information from the VA, experience with personal health records, and sharing information with the VA. Eight interviews were conducted, audio-recorded, and transcribed. The goal of the interviews was to explore and understand provider perspectives on managing dual use veterans, including medication reconciliation processes to add context to the interpretation of medication list analysis. Because the data set was relatively small, summaries of each interview were created to highlight main points. These main points were sorted into topics, summarized, and representative quotes were selected.
Results
Fifty veterans were included in the analysis (Table 1). The mean age was 68.5 (SD 6.2); 90% were men. On average, they reported having 6 chronic health conditions and a fair-to-good health status. Based on the combined medication lists from VA and non-VA providers, veterans took an average of 15.8 (SD 7.0) unique medications (combined prescription and OTC/vitamins) and had an average of 10.0 (SD 6.1) all-type discrepancies (Table 2).
Overall, 58% of the prescription medications were discrepant: The most common discrepancy between the 2 lists was medication missing on one of the lists, which occurred 3.9 times per patient on average for prescription medications and 2.8 times per patient for OTCs. Frequency or dose discrepancies also were common between the lists at a rate of 1.9 discrepancies per patient for prescription medications and 1.2 discrepancies per patient for OTCs.
For high-risk medications, opiates and sedative medications had the most discrepancies between the lists because the VA practitioner may not have known that the patient was taking an opiate, although other discrepancies were present (Table 3). Anticoagulant discrepancies were the most consistent, most of these occurring with aspirin. Last, insulin commonly was dose discrepant between the 2 lists, although it also was missing from one list for a number of patients. Overall, high-risk medications shared a discrepancy rate (46.9%) similar to the overall rate.
Twelve therapeutic duplications were identified in the sample.Ten were between-list duplications, that is, “provider A” thought the patient was on a particular medication and “provider B” thought that the patient was on a different medication (Table 4). In 6 instances, within-list duplications were identified (ie, a provider had 2 medications on the list that should not be taken together because they were in the same drug class). In 4 cases, both between- and within-list duplications were present.
Interview Summaries
Veterans and medication. Multiple non-VA providers said that the primary reason veteran patients were going to a VA provider was to obtain discounted medications. The use of the VA for medications also was a way for the non-VA provider to discover that the patient was a veteran. One non-VA provider was particularly concerned about the impact of new or different medications from VA prescribers on efforts to stabilize the patient’s chronic condition.
Several non-VA providers reported that veterans often brought a medication list to the appointment, and several providers recommended the practice to their patients. Non-VA providers preferred to have patients transfer information from VA, sometimes requesting that veterans bring in their records from recent appointments rather than the non-VA provider obtain the information directly from the VA.
Information sharing. Non-VA providers generally preferred hard copies of medication lists and other documents rather than scans because they were more likely to be included in decision making if the documents were presented during the visit. Also, document scans may be buried in the electronic medical record. Some providers mentioned their interest in electronic transfer of medical information like medication lists if the technology were more developed and better integrated.
“I think the long-term vision would be that it should be electronic… it wouldn’t necessarily be feasible at this time. Our system scans paper documents in to an e-version. … but when the patient comes to their encounter 10 days later, you don’t realize the stuff’s there… Having the patient bring them in is probably a more certain way to make sure that it’s actually included in your decision making as a provider.”
Most non-VA providers welcomed more information such as imaging studies because they reported rarely receiving this information from the VA system. Two mentioned the potential for too much information and wanted concise reports should the flow of information increase. Providers had little interest in logging in to a patient’s online health record portal as a delegate for reasons related to complexity, time, privacy, and lack of mechanism to document the information accessed.
Medication reconciliation. Non-VA providers generally reported that patients bringing their own or an outside medication list would prompt a process of medication reconciliation. The providers were interested in making changes to their records based on other lists, but outside data were verified against a patient self-report of actual use before adopting changes.
“I print out my med list of what I have in the computer and then I just check off my list against their list. And then whatever’s remaining, we talk about what the differences are, when they were changed, what they were changed for, if they were taken off of something, and if I don’t agree, then I’ll tell the patient, ‘look, there’s a disagreement here, they’ve told you not to be on this. I want you on this.”
Should a discrepancy arise, non-VA providers generally had a negative view of attempting to contact VA providers. Other mechanisms such as calling a local pharmacy would be done first.
Discussion
This study provided initial evidence that medication list discrepancies exist for dual use veterans. Other studies of medication list discrepancies have linked such inconsistencies to medication-related problems and negative outcomes for patients.27 Although efforts to increase access to care for veterans have advantages related to expediency, consequences to fragmenting care exist. More robust mechanisms for establishing and maintaining medication list consistency are needed, especially given the lack of a universally accepted medical record format or repository. A multifaceted approach, including patient engagement, seems necessary.
This study also showed that discrepancies of high-risk medications are common for veteran participants, placing them at risk for medication-related problems and harm. These risks included dose and frequency discrepancies that could result in over- or underdosing of medications and in medication omissions, which could cause duplicative therapies and unnecessary risks. For example, aspirin frequently was listed on non-VA lists but was omitted from VA medication lists. This could be problematic for patients who present to the VA for a procedure in which no information about aspirin could jeopardize their safety. Insulin doses also were commonly discrepant, which could impact glycemic control.
Many providers also had incomplete prescribing information for opiates. Those prescriptions are particularly relevant given the link between veterans, posttraumatic stress disorder, depression, and substance abuse.28-30 However, it was beyond the scope of this pilot study to link these discrepancies to ADRs, such as emergency department visits or hospital admissions. Other studies have demonstrated that discrepancies at hospital discharge can result in these types of negative outcomes.27,31 Subsequent research should determine the clinical significance of discrepancies that occur when veterans are dual users.
The qualitative interviews provided some initial context on prescriber perspectives about the role of veterans participating in the medication list sharing process and personal health records. It seemed that for the portion of patients who brought a list to their non-VA provider appointment, the information was welcomed but fell outside the usual visit workflow. Many provider visits are dominated by current patient symptoms, and issues of reconciling medications may be a lower priority.32 Also, some providers may delegate medication reconciliation functions to a nurse or other support staff. One physician offered that he delegated logging in to a patient’s online medication information to a health coach on staff. These findings were consistent with perspectives shared by non-VA family practice physicians about personal health records.33
The most common way to integrate outside medication lists into the non-VA provider’s medical record seemed to be scanning the document. Scanning had its limitations because the provider might be unaware of the scanned document, and there were no mechanisms to import medication names and doses. However, the process may improve only the non-VA providers’ records, as they reported that they had no easy or consistent way to transmit medication changes to notes to the VA.
In general, communicating with VA providers was seen as not feasible and not worth their time or effort. It may be beneficial to address this non-VA provider concern because it seems to inhibit the transfer of important health information and the maintenance of a concordant medication record. Information transfer is particularly relevant for veterans who are primarily cared for by non-VA providers and use the VA only to get prescription medications.
In the current approach, non-VA providers have no simple, direct way to update the VA medication list. Transmitting updates carries the risk of inappropriate changes and is concerning if neither or both prescribers consider themselves to be responsible for the patient’s medications. Also, the potential exists for all medication lists to be inaccurate if the lists do not reflect the medications patients take when left on their own. Patient nonadherence rates can exceed 50%, depending on the medication.34,35 Several interviewed non-VA physicians stressed the importance of asking patients to list the medications they were using during the medication reconciliation process.
This study offers several areas for additional inquiry, including understanding how providers make sense of medication lists from other sources and what technologies can be applied to increase list consistency without increasing the burden on providers.
Practice Implications
Although patient involvement in medication list sharing has the potential to improve information consistency, health systems, providers, and other stakeholders should be cautious in assuming that other prescribers will work to combat medication list entropy, especially if no systems exist to seamlessly incorporate this information into clinic workflow. Devising standardized procedures when patients bring in their records from other providers increases the likelihood that this information will be incorporated into clinical decision making and maintaining up-to-date medication information for patients who use multiple prescribers.
Limitations
These analyses are based on a small sample size (n = 50 for chart review) and (n = 8 for the semistructured interviews) from a single Midwestern state. These findings should be used as evidence for further inquiry.
Conclusion
This study illuminates the level of discrepancies between the medication lists of veteran dual users, including high rates of discrepancies for high-risk medications, such as anticoagulants and opiates. This study also provides evidence of deficiencies in the health care system to decrease medication list entropy that may place veterans at an elevated risk for adverse medication events.
1. Sarkar U, López A, Maselli JH, Gonzales R. Adverse drug events in US adult ambulatory medical care. Health Serv Res. 2011;46(5):1517-1533.
2. Kohn LT, Corrigan JM, Donaldson MS. To Err Is Human:Building a Safer Health System. Washington, DC: Institute of Medicine, National Academy Press; 1999.
3. Gandhi TK, Weingart SN, Borus J, et al. Adverse drug events in ambulatory care. N Eng J Med. 2003;348(16):1556-1564.
4. Tamblyn RM, McLeod PJ, Abrahamowicz M, Laprise R. Do too many cooks spoil the broth? Multiple physician involvement in medical management of elderly patients and potentially inappropriate drug combinations. CMAJ. 1996;154(8):1177-1184.
5. Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother. 2008;42(10):1373-1379.
6. Kripalani S, LeFevre F, Phillips CO, Williams MV, Basaviah P, Baker DW. Deficits in communication and information transfer between hospital-based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297(8):831-841.
7. McMillan A, Trompeter J, Havrda D, Fox J. Continuity of care between family practice physicians and hospitalist services. J Healthare Qual. 2013;35(1):41-49.
8. Liu CF, Manning WG, Burgess JF Jr, et al. Reliance on Veterans Affairs outpatient care by Medicare-eligible veterans. Med Care. 2011;49(10):911-917.
9. U.S. Department of Veterans Affairs, Veterans Health Administration. VHA Office of the ADUSH for Policy and Planning. 2011 survey of veteran enrollees’ health and reliance upon VA. http://www.va.gov/healthpolicyplanning/soe2011/soe2011_report.pdf. Published March 2012. Accessed August 2, 2016.
10. Nayar P, Apenteng B, Yu F, Woodbridge P, Fetrick A. Rural veterans’ perspectives of dual care. J Community Health. 2013;38(1):70-77.
11. Chae SY, Chae MH, Isaacson N, James TS. The patient medication list: can we get patients more involved in their medical care? J Am Board Fam Med. 2009;22(6):677-685.
12. Tang PC, Ash JS, Bates DW, Overhage JM, Sands DZ. Personal health records: definitions, benefits, and strategies for overcoming barriers to adoption. J Am Med Informatics Assoc. 2006;13(2):121-126.
13. Stroupe KT, Smith BM, Hogan TP, et al. Medication acquisition across systems of care and patient–provider communication among older veterans. Am J Health Syst Pharm. 2013;70(9):804-813.
14. Shoemaker SJ, Ramalho de Oliveira D, Alves M, Ekstrand M. The medication experience: preliminary evidence of its value for patient education and counseling on chronic medications. Patient Educ Couns. 2011;83(3):443-450.
15. Chewning B, Boh L, Wiederholt J, et al. Does the concordance concept serve patient medication management? Int J Pharm Pract. 2001;9(2):71-79.
16. Irizarry T, DeVito Dabbs A, Curran CR. Patient portals and patient engagement: a state of the science review. J Med Internet Res. 2015;17(6):e148.
17. Schnipper JL, Gandhi TK, Wald JS, et al. Effects of an online personal health record on medication accuracy and safety: a cluster-randomized trial. J Am Med Inform Assoc. 2012;19(5):728-734.
18. Turvey C, Klein D, Fix G, et al. Blue Button use by patients to access and share health record information using the Department of Veterans Affairs’ online patient portal. J Am Med Inform Assoc. 2014;21(4):657-663.
19. Hogan TP, Nazi KM, Luger TM, et al. Technology-assisted patient access to clinical information: an evaluation framework for Blue Button. JMIR Res Protoc. 2014;3(1):e18.
20. Steinman MA, Handler SM, Gurwitz JH, Schiff GD, Covinsky KE. Beyond the prescription: medication monitoring and adverse drug events in older adults. J Am Geriatr Soc. 2011;59(8):1520-1530.
21. Turvey CL, Klein DM, Witry M, et al. Patient education for consumer-mediated HIE. A pilot randomized controlled trial of the Department of Veterans Affairs Blue Button. Appl Clin Inform. 2016;7(3):765-776.
22. Polnaszek B, Gilmore-Bykovskyi A, Hovanes M, et al. Overcoming the challenges of unstructured data in multisite, electronic medical record-based abstraction [published online ahead of print June 25, 2014]. Med Care. doi: 10.1097/MLR.0000000000000108.
23. Kennelty K, Witry MJ, Gehring M, M D, Pulia N. A four-phase approach for systematically collecting data and measuring medication discrepancies when patients transition between health care settings. Res Social Adm Pharm. 2016;12(4):548-558.
24. American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60(4):616-631.
25. Gurwitz JH, Field TS, Harrold LR, et al. Incidence and preventability of adverse drug events among older persons in the ambulatory setting. JAMA. 2003;289(9):1107-1116.
26. Gurwitz JH, Field TS, Avorn J, et al. Incidence and preventability of adverse drug events in nursing homes. Am J Med. 2000;109(2):87-94.
27. Boockvar KS, Liu S, Goldstein N, Nebeker J, Siu A, Fried T. Prescribing discrepancies likely to cause adverse drug events after patient transfer. Qual Saf Health Care. 2009;18(1):32-36.
28. Shipherd JC, Stafford J, Tanner LR. Predicting alcohol and drug abuse in Persian Gulf War veterans: what role do PTSD symptoms play? Addict Behav. 2005;30(3):595-599.
29. Markou A, Kosten TR, Koob GF. Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology. 1998;18(3):135-174.
30. McFall ME, Mackay PW, Donovan DM. Combat-related posttraumatic stress disorder and severity of substance abuse in Vietnam veterans. J Stud Alcohol. 1992;53(4):357-363.
31. Kwan JL, Lo L, Sampson M, Shojania KG. Medication reconciliation during transitions of care as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5, pt 2):397-403.
32. Richard C, Lussier MT. Nature and frequency of exchanges on medications during primary care encounters. Patient Educ Couns. 2006;64(1-3):207-216.
33. Witry MJ, Doucette WR, Daly JM, Levy BT, Chrischilles EA. Family physician perceptions of personal health records. Perspect Health Inf Manag. 2010;7.
34. Horne R, Weinman J. Patients’ beliefs about prescribed medicines and their role in adherence to treatment in chronic physical illness. J Psychosom Res. 1999;47(6):555-567.
35. Osterberg L, Blaschke T. Adherence to medication. N Eng J Med. 2005;353(5):487-497.
1. Sarkar U, López A, Maselli JH, Gonzales R. Adverse drug events in US adult ambulatory medical care. Health Serv Res. 2011;46(5):1517-1533.
2. Kohn LT, Corrigan JM, Donaldson MS. To Err Is Human:Building a Safer Health System. Washington, DC: Institute of Medicine, National Academy Press; 1999.
3. Gandhi TK, Weingart SN, Borus J, et al. Adverse drug events in ambulatory care. N Eng J Med. 2003;348(16):1556-1564.
4. Tamblyn RM, McLeod PJ, Abrahamowicz M, Laprise R. Do too many cooks spoil the broth? Multiple physician involvement in medical management of elderly patients and potentially inappropriate drug combinations. CMAJ. 1996;154(8):1177-1184.
5. Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother. 2008;42(10):1373-1379.
6. Kripalani S, LeFevre F, Phillips CO, Williams MV, Basaviah P, Baker DW. Deficits in communication and information transfer between hospital-based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297(8):831-841.
7. McMillan A, Trompeter J, Havrda D, Fox J. Continuity of care between family practice physicians and hospitalist services. J Healthare Qual. 2013;35(1):41-49.
8. Liu CF, Manning WG, Burgess JF Jr, et al. Reliance on Veterans Affairs outpatient care by Medicare-eligible veterans. Med Care. 2011;49(10):911-917.
9. U.S. Department of Veterans Affairs, Veterans Health Administration. VHA Office of the ADUSH for Policy and Planning. 2011 survey of veteran enrollees’ health and reliance upon VA. http://www.va.gov/healthpolicyplanning/soe2011/soe2011_report.pdf. Published March 2012. Accessed August 2, 2016.
10. Nayar P, Apenteng B, Yu F, Woodbridge P, Fetrick A. Rural veterans’ perspectives of dual care. J Community Health. 2013;38(1):70-77.
11. Chae SY, Chae MH, Isaacson N, James TS. The patient medication list: can we get patients more involved in their medical care? J Am Board Fam Med. 2009;22(6):677-685.
12. Tang PC, Ash JS, Bates DW, Overhage JM, Sands DZ. Personal health records: definitions, benefits, and strategies for overcoming barriers to adoption. J Am Med Informatics Assoc. 2006;13(2):121-126.
13. Stroupe KT, Smith BM, Hogan TP, et al. Medication acquisition across systems of care and patient–provider communication among older veterans. Am J Health Syst Pharm. 2013;70(9):804-813.
14. Shoemaker SJ, Ramalho de Oliveira D, Alves M, Ekstrand M. The medication experience: preliminary evidence of its value for patient education and counseling on chronic medications. Patient Educ Couns. 2011;83(3):443-450.
15. Chewning B, Boh L, Wiederholt J, et al. Does the concordance concept serve patient medication management? Int J Pharm Pract. 2001;9(2):71-79.
16. Irizarry T, DeVito Dabbs A, Curran CR. Patient portals and patient engagement: a state of the science review. J Med Internet Res. 2015;17(6):e148.
17. Schnipper JL, Gandhi TK, Wald JS, et al. Effects of an online personal health record on medication accuracy and safety: a cluster-randomized trial. J Am Med Inform Assoc. 2012;19(5):728-734.
18. Turvey C, Klein D, Fix G, et al. Blue Button use by patients to access and share health record information using the Department of Veterans Affairs’ online patient portal. J Am Med Inform Assoc. 2014;21(4):657-663.
19. Hogan TP, Nazi KM, Luger TM, et al. Technology-assisted patient access to clinical information: an evaluation framework for Blue Button. JMIR Res Protoc. 2014;3(1):e18.
20. Steinman MA, Handler SM, Gurwitz JH, Schiff GD, Covinsky KE. Beyond the prescription: medication monitoring and adverse drug events in older adults. J Am Geriatr Soc. 2011;59(8):1520-1530.
21. Turvey CL, Klein DM, Witry M, et al. Patient education for consumer-mediated HIE. A pilot randomized controlled trial of the Department of Veterans Affairs Blue Button. Appl Clin Inform. 2016;7(3):765-776.
22. Polnaszek B, Gilmore-Bykovskyi A, Hovanes M, et al. Overcoming the challenges of unstructured data in multisite, electronic medical record-based abstraction [published online ahead of print June 25, 2014]. Med Care. doi: 10.1097/MLR.0000000000000108.
23. Kennelty K, Witry MJ, Gehring M, M D, Pulia N. A four-phase approach for systematically collecting data and measuring medication discrepancies when patients transition between health care settings. Res Social Adm Pharm. 2016;12(4):548-558.
24. American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60(4):616-631.
25. Gurwitz JH, Field TS, Harrold LR, et al. Incidence and preventability of adverse drug events among older persons in the ambulatory setting. JAMA. 2003;289(9):1107-1116.
26. Gurwitz JH, Field TS, Avorn J, et al. Incidence and preventability of adverse drug events in nursing homes. Am J Med. 2000;109(2):87-94.
27. Boockvar KS, Liu S, Goldstein N, Nebeker J, Siu A, Fried T. Prescribing discrepancies likely to cause adverse drug events after patient transfer. Qual Saf Health Care. 2009;18(1):32-36.
28. Shipherd JC, Stafford J, Tanner LR. Predicting alcohol and drug abuse in Persian Gulf War veterans: what role do PTSD symptoms play? Addict Behav. 2005;30(3):595-599.
29. Markou A, Kosten TR, Koob GF. Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology. 1998;18(3):135-174.
30. McFall ME, Mackay PW, Donovan DM. Combat-related posttraumatic stress disorder and severity of substance abuse in Vietnam veterans. J Stud Alcohol. 1992;53(4):357-363.
31. Kwan JL, Lo L, Sampson M, Shojania KG. Medication reconciliation during transitions of care as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5, pt 2):397-403.
32. Richard C, Lussier MT. Nature and frequency of exchanges on medications during primary care encounters. Patient Educ Couns. 2006;64(1-3):207-216.
33. Witry MJ, Doucette WR, Daly JM, Levy BT, Chrischilles EA. Family physician perceptions of personal health records. Perspect Health Inf Manag. 2010;7.
34. Horne R, Weinman J. Patients’ beliefs about prescribed medicines and their role in adherence to treatment in chronic physical illness. J Psychosom Res. 1999;47(6):555-567.
35. Osterberg L, Blaschke T. Adherence to medication. N Eng J Med. 2005;353(5):487-497.
Rotavirus vaccination herd effect benefits newborns and infants
Unvaccinated newborns and infants 42 days old or younger had significantly fewer rotavirus infections after the introduction of a universal mass vaccination (UMV) program, based on a decade of surveillance data from 11 pediatric care facilities in Austria.
“The present study aimed to investigate the long-term effect of UMV on rotavirus (RV)–associated hospitalizations, with particular focus on neonates and infants less than 6 weeks of age, comparing surveillance data between the prevaccination and postvaccination periods,” wrote Martina Prelog, MD, of University Hospital Wuerzburg (Germany), and her colleagues.
The data included 10,960 laboratory-confirmed cases of RV covering the periods before and after the initiation of the mass vaccination program.
Overall, hospitalizations for community-acquired RV infections dropped by almost 90% across all age groups. Among young infants, nosocomial RV infection rates were 28% prior to the vaccination program and 19% afterwards. However, overall nosocomial RV infection rates increased from 6% before the vaccination program to 13% after the program, and 6% of the cases were breakthrough infections, generally after incomplete RV vaccination.
“High numbers of documented cases and similar trends in all centers bolster the conclusion that UMV with RV vaccination may be associated with lower rates of RV hospitalization in unvaccinated neonates and young infants, supporting the beneficial role of UMV,” Dr. Prelog and her associates wrote.
Find the full study here in the Journal of Infectious Diseases (2016. doi: 10.1093/infdis/jiw186).
Unvaccinated newborns and infants 42 days old or younger had significantly fewer rotavirus infections after the introduction of a universal mass vaccination (UMV) program, based on a decade of surveillance data from 11 pediatric care facilities in Austria.
“The present study aimed to investigate the long-term effect of UMV on rotavirus (RV)–associated hospitalizations, with particular focus on neonates and infants less than 6 weeks of age, comparing surveillance data between the prevaccination and postvaccination periods,” wrote Martina Prelog, MD, of University Hospital Wuerzburg (Germany), and her colleagues.
The data included 10,960 laboratory-confirmed cases of RV covering the periods before and after the initiation of the mass vaccination program.
Overall, hospitalizations for community-acquired RV infections dropped by almost 90% across all age groups. Among young infants, nosocomial RV infection rates were 28% prior to the vaccination program and 19% afterwards. However, overall nosocomial RV infection rates increased from 6% before the vaccination program to 13% after the program, and 6% of the cases were breakthrough infections, generally after incomplete RV vaccination.
“High numbers of documented cases and similar trends in all centers bolster the conclusion that UMV with RV vaccination may be associated with lower rates of RV hospitalization in unvaccinated neonates and young infants, supporting the beneficial role of UMV,” Dr. Prelog and her associates wrote.
Find the full study here in the Journal of Infectious Diseases (2016. doi: 10.1093/infdis/jiw186).
Unvaccinated newborns and infants 42 days old or younger had significantly fewer rotavirus infections after the introduction of a universal mass vaccination (UMV) program, based on a decade of surveillance data from 11 pediatric care facilities in Austria.
“The present study aimed to investigate the long-term effect of UMV on rotavirus (RV)–associated hospitalizations, with particular focus on neonates and infants less than 6 weeks of age, comparing surveillance data between the prevaccination and postvaccination periods,” wrote Martina Prelog, MD, of University Hospital Wuerzburg (Germany), and her colleagues.
The data included 10,960 laboratory-confirmed cases of RV covering the periods before and after the initiation of the mass vaccination program.
Overall, hospitalizations for community-acquired RV infections dropped by almost 90% across all age groups. Among young infants, nosocomial RV infection rates were 28% prior to the vaccination program and 19% afterwards. However, overall nosocomial RV infection rates increased from 6% before the vaccination program to 13% after the program, and 6% of the cases were breakthrough infections, generally after incomplete RV vaccination.
“High numbers of documented cases and similar trends in all centers bolster the conclusion that UMV with RV vaccination may be associated with lower rates of RV hospitalization in unvaccinated neonates and young infants, supporting the beneficial role of UMV,” Dr. Prelog and her associates wrote.
Find the full study here in the Journal of Infectious Diseases (2016. doi: 10.1093/infdis/jiw186).
FROM THE JOURNAL OF INFECTIOUS DISEASES