User login
FDA authorizes use of new Zika assay
The US Food and Drug Administration (FDA) has granted emergency use authorization (EUA) for a new assay designed to detect Zika virus infection.
The Aptima® Zika Virus assay is a molecular diagnostic tool that can be used to detect RNA from the Zika virus in human serum and plasma specimens.
The assay has not been FDA-cleared or approved. An EUA allows for the use of unapproved medical products or unapproved uses of approved medical products in an emergency.
The products must be used to diagnose, treat, or prevent serious or life-threatening conditions caused by chemical, biological, radiological, or nuclear threat agents, when there are no adequate alternatives.
This means the Aptima Zika Virus assay is only authorized as long as circumstances exist to justify the authorization of the emergency use of in vitro diagnostics for the detection of Zika virus under section 564(b)(1) of the Federal Food, Drug & Cosmetic Act, 21 U.S.C.§360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
About the assay
The Aptima Zika Virus assay was developed by Hologic, Inc. The assay runs on the Hologic Panther® system, an integrated platform that fully automates all aspects of nucleic acid amplification testing.
The Aptima Zika Virus assay will be available for use in all 50 states, Puerto Rico, and US territories.
The assay is designed to be used in individuals meeting Centers for Disease Control and Prevention (CDC) Zika virus clinical criteria (eg, clinical signs and symptoms associated with Zika virus infection) and/or CDC Zika virus epidemiological criteria (eg, history of residence in or travel to a geographic region with active Zika transmission at the time of travel, or other epidemiologic criteria for which Zika virus testing may be indicated), by laboratories in the US that are certified under the Clinical Laboratory Improvement Amendments of 1988, 42 U.S.C. § 263a, to perform high complexity tests, or by similarly qualified non-US laboratories.
For more information about the Aptima Zika Virus assay, visit www.hologic.com/zika.
About Zika testing
The FDA has granted EUAs for 4 other tests designed to detect Zika virus:
- RealStar® Zika Virus RT-PCR Kit U.S. (altona Diagnostics GmbH)
- Zika Virus RNA Qualitative Real-Time RT-PCR test (Focus Diagnostics)
- Trioplex Real-time RT-PCR Assay (CDC)
- Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA, CDC).
The FDA has also authorized use of the cobas® Zika test (Roche) to screen blood donations for Zika virus. The test may be used under an investigational new drug application for screening donated blood in areas with active mosquito-borne transmission of the virus. ![]()
The US Food and Drug Administration (FDA) has granted emergency use authorization (EUA) for a new assay designed to detect Zika virus infection.
The Aptima® Zika Virus assay is a molecular diagnostic tool that can be used to detect RNA from the Zika virus in human serum and plasma specimens.
The assay has not been FDA-cleared or approved. An EUA allows for the use of unapproved medical products or unapproved uses of approved medical products in an emergency.
The products must be used to diagnose, treat, or prevent serious or life-threatening conditions caused by chemical, biological, radiological, or nuclear threat agents, when there are no adequate alternatives.
This means the Aptima Zika Virus assay is only authorized as long as circumstances exist to justify the authorization of the emergency use of in vitro diagnostics for the detection of Zika virus under section 564(b)(1) of the Federal Food, Drug & Cosmetic Act, 21 U.S.C.§360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
About the assay
The Aptima Zika Virus assay was developed by Hologic, Inc. The assay runs on the Hologic Panther® system, an integrated platform that fully automates all aspects of nucleic acid amplification testing.
The Aptima Zika Virus assay will be available for use in all 50 states, Puerto Rico, and US territories.
The assay is designed to be used in individuals meeting Centers for Disease Control and Prevention (CDC) Zika virus clinical criteria (eg, clinical signs and symptoms associated with Zika virus infection) and/or CDC Zika virus epidemiological criteria (eg, history of residence in or travel to a geographic region with active Zika transmission at the time of travel, or other epidemiologic criteria for which Zika virus testing may be indicated), by laboratories in the US that are certified under the Clinical Laboratory Improvement Amendments of 1988, 42 U.S.C. § 263a, to perform high complexity tests, or by similarly qualified non-US laboratories.
For more information about the Aptima Zika Virus assay, visit www.hologic.com/zika.
About Zika testing
The FDA has granted EUAs for 4 other tests designed to detect Zika virus:
- RealStar® Zika Virus RT-PCR Kit U.S. (altona Diagnostics GmbH)
- Zika Virus RNA Qualitative Real-Time RT-PCR test (Focus Diagnostics)
- Trioplex Real-time RT-PCR Assay (CDC)
- Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA, CDC).
The FDA has also authorized use of the cobas® Zika test (Roche) to screen blood donations for Zika virus. The test may be used under an investigational new drug application for screening donated blood in areas with active mosquito-borne transmission of the virus. ![]()
The US Food and Drug Administration (FDA) has granted emergency use authorization (EUA) for a new assay designed to detect Zika virus infection.
The Aptima® Zika Virus assay is a molecular diagnostic tool that can be used to detect RNA from the Zika virus in human serum and plasma specimens.
The assay has not been FDA-cleared or approved. An EUA allows for the use of unapproved medical products or unapproved uses of approved medical products in an emergency.
The products must be used to diagnose, treat, or prevent serious or life-threatening conditions caused by chemical, biological, radiological, or nuclear threat agents, when there are no adequate alternatives.
This means the Aptima Zika Virus assay is only authorized as long as circumstances exist to justify the authorization of the emergency use of in vitro diagnostics for the detection of Zika virus under section 564(b)(1) of the Federal Food, Drug & Cosmetic Act, 21 U.S.C.§360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
About the assay
The Aptima Zika Virus assay was developed by Hologic, Inc. The assay runs on the Hologic Panther® system, an integrated platform that fully automates all aspects of nucleic acid amplification testing.
The Aptima Zika Virus assay will be available for use in all 50 states, Puerto Rico, and US territories.
The assay is designed to be used in individuals meeting Centers for Disease Control and Prevention (CDC) Zika virus clinical criteria (eg, clinical signs and symptoms associated with Zika virus infection) and/or CDC Zika virus epidemiological criteria (eg, history of residence in or travel to a geographic region with active Zika transmission at the time of travel, or other epidemiologic criteria for which Zika virus testing may be indicated), by laboratories in the US that are certified under the Clinical Laboratory Improvement Amendments of 1988, 42 U.S.C. § 263a, to perform high complexity tests, or by similarly qualified non-US laboratories.
For more information about the Aptima Zika Virus assay, visit www.hologic.com/zika.
About Zika testing
The FDA has granted EUAs for 4 other tests designed to detect Zika virus:
- RealStar® Zika Virus RT-PCR Kit U.S. (altona Diagnostics GmbH)
- Zika Virus RNA Qualitative Real-Time RT-PCR test (Focus Diagnostics)
- Trioplex Real-time RT-PCR Assay (CDC)
- Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA, CDC).
The FDA has also authorized use of the cobas® Zika test (Roche) to screen blood donations for Zika virus. The test may be used under an investigational new drug application for screening donated blood in areas with active mosquito-borne transmission of the virus. ![]()
Estimating the number of sports-related concussions in U.S. children
Between 1.1 million and 1.9 million sports- and recreation-related concussions (SRRCs) occur annually in U.S. children aged 18 years and under, according to a new study.
In the United States, more than 44 million children participate in sports annually, yet the number of SRRCs is unknown. “One challenge in calculating the incidence of SRRCs is that injured youth may not receive treatment, or may receive care from a variety of providers including certified athletic trainers, primary care, and emergency medicine physicians,” said Dr. Mersine A. Bryan of the University of Washington, Seattle, and her associates.

Three national databases were used in this study to conduct research for 2013. “Incidence rates were calculated on the basis of the number of claims divided by the number of enrollees,” they said.
Of children with SRRCs seen in health care settings, outpatient treatment had the highest number at 377,978 visits. The emergency department received between 115,479 and 166,929 sports-related visits, and there were 2,886-4,936 actual hospitalizations. Athletic trainer visits for all school sports totaled 85,885. The number of SRRCs not seen by any health care provider was estimated at 511,590-1,240,972.
The Centers for Disease Control and Prevention is currently developing a surveillance system for SRRCs that would include recreational sources of concussion.
Find the full study at the Journal of Pediatrics (2016 Jun 20. doi: 10.1542/peds.2015-4635).
Between 1.1 million and 1.9 million sports- and recreation-related concussions (SRRCs) occur annually in U.S. children aged 18 years and under, according to a new study.
In the United States, more than 44 million children participate in sports annually, yet the number of SRRCs is unknown. “One challenge in calculating the incidence of SRRCs is that injured youth may not receive treatment, or may receive care from a variety of providers including certified athletic trainers, primary care, and emergency medicine physicians,” said Dr. Mersine A. Bryan of the University of Washington, Seattle, and her associates.
Three national databases were used in this study to conduct research for 2013. “Incidence rates were calculated on the basis of the number of claims divided by the number of enrollees,” they said.
Of children with SRRCs seen in health care settings, outpatient treatment had the highest number at 377,978 visits. The emergency department received between 115,479 and 166,929 sports-related visits, and there were 2,886-4,936 actual hospitalizations. Athletic trainer visits for all school sports totaled 85,885. The number of SRRCs not seen by any health care provider was estimated at 511,590-1,240,972.
The Centers for Disease Control and Prevention is currently developing a surveillance system for SRRCs that would include recreational sources of concussion.
Find the full study at the Journal of Pediatrics (2016 Jun 20. doi: 10.1542/peds.2015-4635).
Between 1.1 million and 1.9 million sports- and recreation-related concussions (SRRCs) occur annually in U.S. children aged 18 years and under, according to a new study.
In the United States, more than 44 million children participate in sports annually, yet the number of SRRCs is unknown. “One challenge in calculating the incidence of SRRCs is that injured youth may not receive treatment, or may receive care from a variety of providers including certified athletic trainers, primary care, and emergency medicine physicians,” said Dr. Mersine A. Bryan of the University of Washington, Seattle, and her associates.
Three national databases were used in this study to conduct research for 2013. “Incidence rates were calculated on the basis of the number of claims divided by the number of enrollees,” they said.
Of children with SRRCs seen in health care settings, outpatient treatment had the highest number at 377,978 visits. The emergency department received between 115,479 and 166,929 sports-related visits, and there were 2,886-4,936 actual hospitalizations. Athletic trainer visits for all school sports totaled 85,885. The number of SRRCs not seen by any health care provider was estimated at 511,590-1,240,972.
The Centers for Disease Control and Prevention is currently developing a surveillance system for SRRCs that would include recreational sources of concussion.
Find the full study at the Journal of Pediatrics (2016 Jun 20. doi: 10.1542/peds.2015-4635).
FROM THE JOURNAL OF PEDIATRICS

Hospital-acquired respiratory viruses cause significant morbidity, mortality
BOSTON – Hospital-acquired respiratory viral infections may be a significant and underappreciated cause of morbidity and mortality among hospitalized patients.
According to a multisite, retrospective chart review of 44 patients with hospital-acquired respiratory viral illnesses (HA-RVIs), 17 patients (39%) died in-hospital. Further, of the 27 who survived, 18 (66.6%) were discharged to an advanced care setting rather than to home, though just 11/44 (25%) had been living in an advanced care setting before admission.

For the hospitalizations complicated by HA-RVI, the average length of stay was 30.4 days, with a positive respiratory virus panel (RVP) result occurring at a mean 18 days after admission.
“HA-RVIs are an underappreciated event and appear to target the sickest patients in the hospital,” said coauthor Dr. Matthew Sims, director of infectious diseases research at Beaumont Hospital, Rochester, Mich., at a poster session of the annual meeting of the American Society of Microbiology.
First author Dr. Adam K. Skrzynski, also of Beaumont Health, and his coauthors performed the analysis of 4,065 patients with a positive RVP result during hospitalization at a regional hospital system in the September 2011-May 2015 study period; the 1.1% of patients with positive results who formed the study cohort had to have symptoms of a respiratory infection occurring after more than 5 days of hospitalization. Mortality data were collected for the first 33 days of hospitalization.
Positive RVP results for those included in the study came primarily from nasopharyngeal swab (n = 32), with the remainder from bronchoalveolar lavage (n = 11) and sputum (n = 1). Most patients were female (29/44, 66%), and elderly, with an average age of 73.8 years. In an interview, Dr. Sims said that many patients were smokers, and that chronic obstructive pulmonary disease and obesity were common comorbidities.
The prognosis was particularly grim for the 12 patients (27.3%) who were admitted to the ICU: 10 (83.3%) died after an average 9.6 days in the ICU. Advanced interventions did not seem to make a difference, either. “Intubation didn’t help these patients,” said Dr. Sims. Nine patients (20.5%) were intubated within 7 days of their positive RVP results. Intubation lasted an average 7.6 days, and all nine of these patients died.
The RVP came into use in 2011 and made it possible to identify whether a respiratory virus was causing symptoms – and which virus was the culprit – said Dr. Sims. For the studied population, 13 of 44 patients had influenza; 11 of those had influenza A and 2 had influenza B. The next most common pathogen was parainfluenza, with 10 positive RVP results.
Dr. Sims said he and his coinvestigators were surprised to find that, although influenza A was the most common pathogen, only 18.8% of the patients with influenza A died during the study period. “While it is possible that the high frequency of influenza infection in our study may be due to poor vaccine-strain matching for the years in question, the lower mortality rate seen in influenza A infection may be due to our hospital’s mandatory influenza vaccination policy and subsequent protection against mortality,” Dr. Skrzynski and his coauthors wrote.
There were seasonal trends in mortality, with 70.6% of mortality occurring in the spring (April-June) and an additional 23.3% happening in the winter (January-March). Parainfluenza infection peaked in the spring, and influenza peaked in the winter months.
Dr. Sims said the study underlines the importance of encouraging ill hospital staff members to stay home, and family members with respiratory symptoms should not be visiting fragile patients. Dr. Skrzynski and his coauthors also wrote that “immunization of healthcare personnel against influenza should be mandatory.”
Still to be answered, said Dr. Sims, is the association between comorbidities and the potentially lethal effects of HA-RVIs. They are currently performing a matched case-control study to tease out these relationships.
Dr. Skrzynski reported no outside funding source, and the study authors had no financial disclosures.
On Twitter @karioakes
BOSTON – Hospital-acquired respiratory viral infections may be a significant and underappreciated cause of morbidity and mortality among hospitalized patients.
According to a multisite, retrospective chart review of 44 patients with hospital-acquired respiratory viral illnesses (HA-RVIs), 17 patients (39%) died in-hospital. Further, of the 27 who survived, 18 (66.6%) were discharged to an advanced care setting rather than to home, though just 11/44 (25%) had been living in an advanced care setting before admission.
For the hospitalizations complicated by HA-RVI, the average length of stay was 30.4 days, with a positive respiratory virus panel (RVP) result occurring at a mean 18 days after admission.
“HA-RVIs are an underappreciated event and appear to target the sickest patients in the hospital,” said coauthor Dr. Matthew Sims, director of infectious diseases research at Beaumont Hospital, Rochester, Mich., at a poster session of the annual meeting of the American Society of Microbiology.
First author Dr. Adam K. Skrzynski, also of Beaumont Health, and his coauthors performed the analysis of 4,065 patients with a positive RVP result during hospitalization at a regional hospital system in the September 2011-May 2015 study period; the 1.1% of patients with positive results who formed the study cohort had to have symptoms of a respiratory infection occurring after more than 5 days of hospitalization. Mortality data were collected for the first 33 days of hospitalization.
Positive RVP results for those included in the study came primarily from nasopharyngeal swab (n = 32), with the remainder from bronchoalveolar lavage (n = 11) and sputum (n = 1). Most patients were female (29/44, 66%), and elderly, with an average age of 73.8 years. In an interview, Dr. Sims said that many patients were smokers, and that chronic obstructive pulmonary disease and obesity were common comorbidities.
The prognosis was particularly grim for the 12 patients (27.3%) who were admitted to the ICU: 10 (83.3%) died after an average 9.6 days in the ICU. Advanced interventions did not seem to make a difference, either. “Intubation didn’t help these patients,” said Dr. Sims. Nine patients (20.5%) were intubated within 7 days of their positive RVP results. Intubation lasted an average 7.6 days, and all nine of these patients died.
The RVP came into use in 2011 and made it possible to identify whether a respiratory virus was causing symptoms – and which virus was the culprit – said Dr. Sims. For the studied population, 13 of 44 patients had influenza; 11 of those had influenza A and 2 had influenza B. The next most common pathogen was parainfluenza, with 10 positive RVP results.
Dr. Sims said he and his coinvestigators were surprised to find that, although influenza A was the most common pathogen, only 18.8% of the patients with influenza A died during the study period. “While it is possible that the high frequency of influenza infection in our study may be due to poor vaccine-strain matching for the years in question, the lower mortality rate seen in influenza A infection may be due to our hospital’s mandatory influenza vaccination policy and subsequent protection against mortality,” Dr. Skrzynski and his coauthors wrote.
There were seasonal trends in mortality, with 70.6% of mortality occurring in the spring (April-June) and an additional 23.3% happening in the winter (January-March). Parainfluenza infection peaked in the spring, and influenza peaked in the winter months.
Dr. Sims said the study underlines the importance of encouraging ill hospital staff members to stay home, and family members with respiratory symptoms should not be visiting fragile patients. Dr. Skrzynski and his coauthors also wrote that “immunization of healthcare personnel against influenza should be mandatory.”
Still to be answered, said Dr. Sims, is the association between comorbidities and the potentially lethal effects of HA-RVIs. They are currently performing a matched case-control study to tease out these relationships.
Dr. Skrzynski reported no outside funding source, and the study authors had no financial disclosures.
On Twitter @karioakes
BOSTON – Hospital-acquired respiratory viral infections may be a significant and underappreciated cause of morbidity and mortality among hospitalized patients.
According to a multisite, retrospective chart review of 44 patients with hospital-acquired respiratory viral illnesses (HA-RVIs), 17 patients (39%) died in-hospital. Further, of the 27 who survived, 18 (66.6%) were discharged to an advanced care setting rather than to home, though just 11/44 (25%) had been living in an advanced care setting before admission.
For the hospitalizations complicated by HA-RVI, the average length of stay was 30.4 days, with a positive respiratory virus panel (RVP) result occurring at a mean 18 days after admission.
“HA-RVIs are an underappreciated event and appear to target the sickest patients in the hospital,” said coauthor Dr. Matthew Sims, director of infectious diseases research at Beaumont Hospital, Rochester, Mich., at a poster session of the annual meeting of the American Society of Microbiology.
First author Dr. Adam K. Skrzynski, also of Beaumont Health, and his coauthors performed the analysis of 4,065 patients with a positive RVP result during hospitalization at a regional hospital system in the September 2011-May 2015 study period; the 1.1% of patients with positive results who formed the study cohort had to have symptoms of a respiratory infection occurring after more than 5 days of hospitalization. Mortality data were collected for the first 33 days of hospitalization.
Positive RVP results for those included in the study came primarily from nasopharyngeal swab (n = 32), with the remainder from bronchoalveolar lavage (n = 11) and sputum (n = 1). Most patients were female (29/44, 66%), and elderly, with an average age of 73.8 years. In an interview, Dr. Sims said that many patients were smokers, and that chronic obstructive pulmonary disease and obesity were common comorbidities.
The prognosis was particularly grim for the 12 patients (27.3%) who were admitted to the ICU: 10 (83.3%) died after an average 9.6 days in the ICU. Advanced interventions did not seem to make a difference, either. “Intubation didn’t help these patients,” said Dr. Sims. Nine patients (20.5%) were intubated within 7 days of their positive RVP results. Intubation lasted an average 7.6 days, and all nine of these patients died.
The RVP came into use in 2011 and made it possible to identify whether a respiratory virus was causing symptoms – and which virus was the culprit – said Dr. Sims. For the studied population, 13 of 44 patients had influenza; 11 of those had influenza A and 2 had influenza B. The next most common pathogen was parainfluenza, with 10 positive RVP results.
Dr. Sims said he and his coinvestigators were surprised to find that, although influenza A was the most common pathogen, only 18.8% of the patients with influenza A died during the study period. “While it is possible that the high frequency of influenza infection in our study may be due to poor vaccine-strain matching for the years in question, the lower mortality rate seen in influenza A infection may be due to our hospital’s mandatory influenza vaccination policy and subsequent protection against mortality,” Dr. Skrzynski and his coauthors wrote.
There were seasonal trends in mortality, with 70.6% of mortality occurring in the spring (April-June) and an additional 23.3% happening in the winter (January-March). Parainfluenza infection peaked in the spring, and influenza peaked in the winter months.
Dr. Sims said the study underlines the importance of encouraging ill hospital staff members to stay home, and family members with respiratory symptoms should not be visiting fragile patients. Dr. Skrzynski and his coauthors also wrote that “immunization of healthcare personnel against influenza should be mandatory.”
Still to be answered, said Dr. Sims, is the association between comorbidities and the potentially lethal effects of HA-RVIs. They are currently performing a matched case-control study to tease out these relationships.
Dr. Skrzynski reported no outside funding source, and the study authors had no financial disclosures.
On Twitter @karioakes
AT ASM MICROBE 2016
Key clinical point: Hospital-acquired respiratory viral illnesses had a 39% mortality rate.
Major finding: Of 44 symptomatic patients with positive respiratory virus panel screens, 17 died and 2/3 of the survivors went to advanced care settings on discharge.
Data source: Retrospective multisite chart review of 44 patients with HA-RVIs and positive RVP screens.
Disclosures: No external funding source was reported, and the study authors had no disclosures.

HCV patients had distinct mucosal microbiome
SAN DIEGO – Patients with hepatitis C virus (HCV) infections had distinct duodenal mucosal microbiomes and greater intestinal permeability, compared with healthy controls and patients with other chronic liver diseases, Dr. Ashok Raj reported.
The findings might one day lead to therapies that aim to restore or normalize the microbiomes of patients with HCV, Dr. Raj said in an interview at the annual Digestive Disease Week.

Chronic liver disease (CLD) has been linked to dysbiosis, or abnormal shifts of the microbiome. But most studies have focused on fecal specimens, and “recent evidence suggests that the mucosal microbiota differ from fecal microbiota,” said Dr. Raj, a gastroenterologist and hepatologist at Princess Alexandra Hospital in Brisbane, Australia, and a PhD candidate at the University of Queensland at Brisbane.
“The small-intestinal mucosal microbiota are of particular interest to us,” Dr. Raj explained. “Anatomically, all the blood from this region of the gut drains into the portal vein and flows directly to the liver. Because of small-intestinal permeability, either bacteria or their products could travel to the liver and contribute to disease. But very little is known about this microbiota in CLD.”
Therefore, Dr. Raj and his associates sequenced bacterial DNA from mucosal biopsies of the second part of the duodenum from 38 prospectively recruited endoscopy patients with CLD and 13 healthy controls. The researchers also evaluated dietary habits, intestinal permeability, hepatic stiffness based on transient elastography, and the presence of metabolic syndrome, as measured by the International Diabetes Federation/American Heart Association/National Heart, Lung, and Blood Institute 2009 Consensus criteria. The CLD group included 28 men and 10 women aged 36-82 years, including 16 patients with HCV, 10 patients with nonalcoholic fatty liver disease, 7 patients with fatty liver disease, 3 patients with autoimmune hepatitis, and 2 patients with hepatitis B virus infection. The controls were between 24 and 73 years old, and 70% were women.
Sequencing of bacteria DNA revealed significant differences between patients and controls, particularly among patients with HCV, Dr. Raj said. The HCV patients not only had significantly less microbial diversity (P less than .02), but the overall changes in their microbiota were significant enough for them to cluster separately from controls and from patients with other types of CLD (P less than .01 for both comparisons). Furthermore, HCV patients had significantly greater small-intestinal permeability (mean ± SD log lactulose to rhamnose ratio, 1.57 ± 0.27) than controls (1.21 ± 0.25; P less than .01) or patients with other CLDs (1.24 ± 0.39; P = .01).
“Additionally, for the HCV patients, dietary fat intake showed a moderately strong positive correlation with intestinal permeability,” Dr. Raj said (r = 0.58; P = .03). “These findings are in keeping with animal models, which have shown that dietary fat can change the microbiota and also increase intestinal permeability.” However, the multivariate analysis found no links between microbial characteristics and hepatic stiffness or metabolic syndrome – perhaps because most patients were “at the cirrhotic end of the spectrum, reflecting their indication for endoscopy,” or because “these relationships are subtler and require larger sample numbers,” he said.
“Patients with HCV may have a unique small-intestinal microbiome,” Dr. Raj concluded. “These patients had higher intestinal permeability, and it is possible that the microbiota have a part to play in that.” Exactly how microbiota and gut permeability contribute to disease remains unclear, but pathology in the small intestine could help explain some features of the HCV trajectory, such as extrahepatic manifestations or variations in disease progression, he added. “Future studies may lead to targeting the small-intestinal gut microbiome to modulate and even treat HCV.”
The study was funded by a postgraduate award from the government of Australia and by the Princess Alexandra Hospital Research Foundation. Dr. Raj had no disclosures.
SAN DIEGO – Patients with hepatitis C virus (HCV) infections had distinct duodenal mucosal microbiomes and greater intestinal permeability, compared with healthy controls and patients with other chronic liver diseases, Dr. Ashok Raj reported.
The findings might one day lead to therapies that aim to restore or normalize the microbiomes of patients with HCV, Dr. Raj said in an interview at the annual Digestive Disease Week.
Chronic liver disease (CLD) has been linked to dysbiosis, or abnormal shifts of the microbiome. But most studies have focused on fecal specimens, and “recent evidence suggests that the mucosal microbiota differ from fecal microbiota,” said Dr. Raj, a gastroenterologist and hepatologist at Princess Alexandra Hospital in Brisbane, Australia, and a PhD candidate at the University of Queensland at Brisbane.
“The small-intestinal mucosal microbiota are of particular interest to us,” Dr. Raj explained. “Anatomically, all the blood from this region of the gut drains into the portal vein and flows directly to the liver. Because of small-intestinal permeability, either bacteria or their products could travel to the liver and contribute to disease. But very little is known about this microbiota in CLD.”
Therefore, Dr. Raj and his associates sequenced bacterial DNA from mucosal biopsies of the second part of the duodenum from 38 prospectively recruited endoscopy patients with CLD and 13 healthy controls. The researchers also evaluated dietary habits, intestinal permeability, hepatic stiffness based on transient elastography, and the presence of metabolic syndrome, as measured by the International Diabetes Federation/American Heart Association/National Heart, Lung, and Blood Institute 2009 Consensus criteria. The CLD group included 28 men and 10 women aged 36-82 years, including 16 patients with HCV, 10 patients with nonalcoholic fatty liver disease, 7 patients with fatty liver disease, 3 patients with autoimmune hepatitis, and 2 patients with hepatitis B virus infection. The controls were between 24 and 73 years old, and 70% were women.
Sequencing of bacteria DNA revealed significant differences between patients and controls, particularly among patients with HCV, Dr. Raj said. The HCV patients not only had significantly less microbial diversity (P less than .02), but the overall changes in their microbiota were significant enough for them to cluster separately from controls and from patients with other types of CLD (P less than .01 for both comparisons). Furthermore, HCV patients had significantly greater small-intestinal permeability (mean ± SD log lactulose to rhamnose ratio, 1.57 ± 0.27) than controls (1.21 ± 0.25; P less than .01) or patients with other CLDs (1.24 ± 0.39; P = .01).
“Additionally, for the HCV patients, dietary fat intake showed a moderately strong positive correlation with intestinal permeability,” Dr. Raj said (r = 0.58; P = .03). “These findings are in keeping with animal models, which have shown that dietary fat can change the microbiota and also increase intestinal permeability.” However, the multivariate analysis found no links between microbial characteristics and hepatic stiffness or metabolic syndrome – perhaps because most patients were “at the cirrhotic end of the spectrum, reflecting their indication for endoscopy,” or because “these relationships are subtler and require larger sample numbers,” he said.
“Patients with HCV may have a unique small-intestinal microbiome,” Dr. Raj concluded. “These patients had higher intestinal permeability, and it is possible that the microbiota have a part to play in that.” Exactly how microbiota and gut permeability contribute to disease remains unclear, but pathology in the small intestine could help explain some features of the HCV trajectory, such as extrahepatic manifestations or variations in disease progression, he added. “Future studies may lead to targeting the small-intestinal gut microbiome to modulate and even treat HCV.”
The study was funded by a postgraduate award from the government of Australia and by the Princess Alexandra Hospital Research Foundation. Dr. Raj had no disclosures.
SAN DIEGO – Patients with hepatitis C virus (HCV) infections had distinct duodenal mucosal microbiomes and greater intestinal permeability, compared with healthy controls and patients with other chronic liver diseases, Dr. Ashok Raj reported.
The findings might one day lead to therapies that aim to restore or normalize the microbiomes of patients with HCV, Dr. Raj said in an interview at the annual Digestive Disease Week.
Chronic liver disease (CLD) has been linked to dysbiosis, or abnormal shifts of the microbiome. But most studies have focused on fecal specimens, and “recent evidence suggests that the mucosal microbiota differ from fecal microbiota,” said Dr. Raj, a gastroenterologist and hepatologist at Princess Alexandra Hospital in Brisbane, Australia, and a PhD candidate at the University of Queensland at Brisbane.
“The small-intestinal mucosal microbiota are of particular interest to us,” Dr. Raj explained. “Anatomically, all the blood from this region of the gut drains into the portal vein and flows directly to the liver. Because of small-intestinal permeability, either bacteria or their products could travel to the liver and contribute to disease. But very little is known about this microbiota in CLD.”
Therefore, Dr. Raj and his associates sequenced bacterial DNA from mucosal biopsies of the second part of the duodenum from 38 prospectively recruited endoscopy patients with CLD and 13 healthy controls. The researchers also evaluated dietary habits, intestinal permeability, hepatic stiffness based on transient elastography, and the presence of metabolic syndrome, as measured by the International Diabetes Federation/American Heart Association/National Heart, Lung, and Blood Institute 2009 Consensus criteria. The CLD group included 28 men and 10 women aged 36-82 years, including 16 patients with HCV, 10 patients with nonalcoholic fatty liver disease, 7 patients with fatty liver disease, 3 patients with autoimmune hepatitis, and 2 patients with hepatitis B virus infection. The controls were between 24 and 73 years old, and 70% were women.
Sequencing of bacteria DNA revealed significant differences between patients and controls, particularly among patients with HCV, Dr. Raj said. The HCV patients not only had significantly less microbial diversity (P less than .02), but the overall changes in their microbiota were significant enough for them to cluster separately from controls and from patients with other types of CLD (P less than .01 for both comparisons). Furthermore, HCV patients had significantly greater small-intestinal permeability (mean ± SD log lactulose to rhamnose ratio, 1.57 ± 0.27) than controls (1.21 ± 0.25; P less than .01) or patients with other CLDs (1.24 ± 0.39; P = .01).
“Additionally, for the HCV patients, dietary fat intake showed a moderately strong positive correlation with intestinal permeability,” Dr. Raj said (r = 0.58; P = .03). “These findings are in keeping with animal models, which have shown that dietary fat can change the microbiota and also increase intestinal permeability.” However, the multivariate analysis found no links between microbial characteristics and hepatic stiffness or metabolic syndrome – perhaps because most patients were “at the cirrhotic end of the spectrum, reflecting their indication for endoscopy,” or because “these relationships are subtler and require larger sample numbers,” he said.
“Patients with HCV may have a unique small-intestinal microbiome,” Dr. Raj concluded. “These patients had higher intestinal permeability, and it is possible that the microbiota have a part to play in that.” Exactly how microbiota and gut permeability contribute to disease remains unclear, but pathology in the small intestine could help explain some features of the HCV trajectory, such as extrahepatic manifestations or variations in disease progression, he added. “Future studies may lead to targeting the small-intestinal gut microbiome to modulate and even treat HCV.”
The study was funded by a postgraduate award from the government of Australia and by the Princess Alexandra Hospital Research Foundation. Dr. Raj had no disclosures.
AT DDW® 2016
Key clinical point: Patients with hepatitis C virus infection had unique mucosal microbiomes, compared with controls or patients with other chronic liver diseases.
Major finding: The HCV patients not only had significantly less microbial diversity (P less than .02), but the overall changes in their microbiota were significant enough for them to cluster separately from controls and from patients with other types of chronic liver disease (P less than .01 for both comparisons).
Data source: Bacterial DNA sequencing of duodenal mucosal biopsies from 38 patients with chronic liver diseases and 10 controls.
Disclosures: The study was funded by a postgraduate award from the government of Australia and by the Princess Alexandra Hospital Research Foundation. Dr. Raj had no disclosures.

Recurrent C. diff infection much more costly to treat
The costs of treating patients with recurrent Clostridium difficile infection are approximately two to three times the treatment costs for patients with primary CDI, according to a study of hospitalized patients in a large tertiary care center.
Kevin W. Garey, Pharm.D., professor of pharmacy practice at the University of Houston College of Pharmacy, and his coauthors wrote in the Journal of Hospital Infection that the introduction of costly therapies, such as the narrow-spectrum macrocyclic antibiotic fidaxomicin and fecal microbiota transplantation, both of which reduce the risk of recurrent CDI, has increased interest in the economic burden of recurrent CDI (J Hosp Infect. 2016 Jul;93:286-9).

Between 2007-2013, Dr. Garey and his colleagues attempted to assess the additional costs of recurrent CDI by calculating total hospital length of stay (LOS), CDI-attributable LOS, and pharmacologic and hospitalization costs for 540 hospitalized adult patients (42% male) with a diagnosis of primary CDI. Of these patients, 95 (18%) experienced 101 recurrent CDI episodes. CDI-attributable hospital admissions occurred in 307 out of 540 (57%) primary CDI episodes, and 64 out of 101 (63%) recurrent CDI episodes.
The investigators estimated total and CDI-attributable hospitalization costs based on Healthcare Cost and Utilization Project data for average daily health care costs, multiplied by the total and CDI-attributable length of hospital stay. They then compared total hospital LOS, CDI-attributable LOS, and pharmacologic and hospitalization costs between patients with primary CDI only versus patients who experienced recurrent CDI.
Dr. Garey and his colleagues found that CDI-attributable median LOS and costs increased from 7 days and $13,168 for patients with primary CDI only, vs .15 days and $28,218 for patients with recurrent CDI (P less than .0001, each). Total hospital median LOS and costs increased from 11 days and $20,693 for patients with primary CDI only, vs. 24 days and $45,148 for patients with recurrent CDI (P less than .0001, each).
The median cost of pharmacologic treatment while hospitalized was $60 for patients with primary CDI only, and $140 for patients with recurrent CDI (P = .0013).
“Patients with CDI experience a significant health care economic burden that can be directly related to CDI,” the authors concluded.
The study was funded by a research grant from Merck. Dr. Garey reported receiving funding from Cubist, Summit, and Merck, while other coauthors reported research support from Merck, Cubist, and Actelion.
On Twitter @richpizzi
The costs of treating patients with recurrent Clostridium difficile infection are approximately two to three times the treatment costs for patients with primary CDI, according to a study of hospitalized patients in a large tertiary care center.
Kevin W. Garey, Pharm.D., professor of pharmacy practice at the University of Houston College of Pharmacy, and his coauthors wrote in the Journal of Hospital Infection that the introduction of costly therapies, such as the narrow-spectrum macrocyclic antibiotic fidaxomicin and fecal microbiota transplantation, both of which reduce the risk of recurrent CDI, has increased interest in the economic burden of recurrent CDI (J Hosp Infect. 2016 Jul;93:286-9).
Between 2007-2013, Dr. Garey and his colleagues attempted to assess the additional costs of recurrent CDI by calculating total hospital length of stay (LOS), CDI-attributable LOS, and pharmacologic and hospitalization costs for 540 hospitalized adult patients (42% male) with a diagnosis of primary CDI. Of these patients, 95 (18%) experienced 101 recurrent CDI episodes. CDI-attributable hospital admissions occurred in 307 out of 540 (57%) primary CDI episodes, and 64 out of 101 (63%) recurrent CDI episodes.
The investigators estimated total and CDI-attributable hospitalization costs based on Healthcare Cost and Utilization Project data for average daily health care costs, multiplied by the total and CDI-attributable length of hospital stay. They then compared total hospital LOS, CDI-attributable LOS, and pharmacologic and hospitalization costs between patients with primary CDI only versus patients who experienced recurrent CDI.
Dr. Garey and his colleagues found that CDI-attributable median LOS and costs increased from 7 days and $13,168 for patients with primary CDI only, vs .15 days and $28,218 for patients with recurrent CDI (P less than .0001, each). Total hospital median LOS and costs increased from 11 days and $20,693 for patients with primary CDI only, vs. 24 days and $45,148 for patients with recurrent CDI (P less than .0001, each).
The median cost of pharmacologic treatment while hospitalized was $60 for patients with primary CDI only, and $140 for patients with recurrent CDI (P = .0013).
“Patients with CDI experience a significant health care economic burden that can be directly related to CDI,” the authors concluded.
The study was funded by a research grant from Merck. Dr. Garey reported receiving funding from Cubist, Summit, and Merck, while other coauthors reported research support from Merck, Cubist, and Actelion.
On Twitter @richpizzi
The costs of treating patients with recurrent Clostridium difficile infection are approximately two to three times the treatment costs for patients with primary CDI, according to a study of hospitalized patients in a large tertiary care center.
Kevin W. Garey, Pharm.D., professor of pharmacy practice at the University of Houston College of Pharmacy, and his coauthors wrote in the Journal of Hospital Infection that the introduction of costly therapies, such as the narrow-spectrum macrocyclic antibiotic fidaxomicin and fecal microbiota transplantation, both of which reduce the risk of recurrent CDI, has increased interest in the economic burden of recurrent CDI (J Hosp Infect. 2016 Jul;93:286-9).
Between 2007-2013, Dr. Garey and his colleagues attempted to assess the additional costs of recurrent CDI by calculating total hospital length of stay (LOS), CDI-attributable LOS, and pharmacologic and hospitalization costs for 540 hospitalized adult patients (42% male) with a diagnosis of primary CDI. Of these patients, 95 (18%) experienced 101 recurrent CDI episodes. CDI-attributable hospital admissions occurred in 307 out of 540 (57%) primary CDI episodes, and 64 out of 101 (63%) recurrent CDI episodes.
The investigators estimated total and CDI-attributable hospitalization costs based on Healthcare Cost and Utilization Project data for average daily health care costs, multiplied by the total and CDI-attributable length of hospital stay. They then compared total hospital LOS, CDI-attributable LOS, and pharmacologic and hospitalization costs between patients with primary CDI only versus patients who experienced recurrent CDI.
Dr. Garey and his colleagues found that CDI-attributable median LOS and costs increased from 7 days and $13,168 for patients with primary CDI only, vs .15 days and $28,218 for patients with recurrent CDI (P less than .0001, each). Total hospital median LOS and costs increased from 11 days and $20,693 for patients with primary CDI only, vs. 24 days and $45,148 for patients with recurrent CDI (P less than .0001, each).
The median cost of pharmacologic treatment while hospitalized was $60 for patients with primary CDI only, and $140 for patients with recurrent CDI (P = .0013).
“Patients with CDI experience a significant health care economic burden that can be directly related to CDI,” the authors concluded.
The study was funded by a research grant from Merck. Dr. Garey reported receiving funding from Cubist, Summit, and Merck, while other coauthors reported research support from Merck, Cubist, and Actelion.
On Twitter @richpizzi
FROM THE JOURNAL OF HOSPITAL INFECTION
Key clinical point: The economic cost burden increased significantly for patients with recurrent C. difficile infections, compared with patients with primary CDI.
Major finding: Median hospital length of stay and costs attributable to C. difficile infections increased from 7 days and $13,168 for patients with primary CDI only, vs. 15 days and $28,218 for patients with recurrent CDI.
Data source: A prospective, observational cohort study of 540 hospitalized adult patients with primary CDI followed for 3 months to assess for recurrent CDI episodes.
Disclosures: The study was funded by a research grant from Merck. Dr. Garey reported receiving funding from Cubist, Summit, and Merck, while other coauthors reported research support from Merck, Cubist, and Actelion.

European initiative unveils pediatric care recommendations
LONDON – Recommendations on managing juvenile idiopathic arthritis and connective tissue disorders in children and young people across Europe were unveiled at the European Congress of Rheumatology.
The recommendations, which come from the SHARE (Single Hub and Access Point for Paediatric Rheumatology in Europe) project, cover best practices and provide guidance based on current evidence and expert opinion for the optimal diagnosis and treatment of these rare rheumatic diseases that affect the pediatric population.
It is hoped that the recommendations will be used to improve access to treatment and care within individual countries such that a child in one country will be able to receive the same standard of care as a child in another, Dr. Nico Wulffraat of University Medical Center Utrecht (the Netherlands) said in an interview.
Dr. Wulffraat, one of the driving forces behind the project, noted that the SHARE project was set up to look at making the management of rare pediatric rheumatic diseases more uniform across Europe. It addressed conditions such as juvenile idiopathic arthritis (JIA), childhood-onset systemic lupus erythematosus (cSLE), childhood antiphospholipid syndrome (APS), childhood vasculitis, juvenile dermatomyositis, and pediatric scleroderma. In addition, recommendations on diagnosis and treatment of periodic fever syndromes have been developed in collaboration with experts from the Eurofever Project.

“Our evidence- and consensus-based recommendations will hopefully drive access to uniform and optimal care throughout Europe, including off-label therapy when appropriate according to international consensus–derived expert advice,” Dr. Sebastiaan Vastert, SHARE project co-coordinator, said in an interview. He added: “The SHARE network will be invaluable for further international collaboration, both for optimization of care and for international collaboration in research as well.”
Dr. Wulffraat observed that while the recommendations are primarily directed at health care professionals, they also are of use for other stakeholders such as health authorities and insurance companies, and of course patients themselves to ensure the best level of care is being achieved throughout Europe.
The process for developing the guidelines was perhaps as important as the recommendations themselves, said Dr. Vastert, also of University Medical Center Utrecht. The process helped to build a network of international experts who could work together to develop future recommendations for improving patient care.
The recommendations for JIA and other pediatric rheumatic diseases included 51 “cross-cutting” statements, Dr. Vastert said. One of these statements was that a pediatric rheumatologist should manage children with signs of rheumatic disease. Another highlighted the members of a multidisciplinary team who should be involved as appropriate, such as a nurse specializing in pediatric rheumatic disease, a physiotherapist or occupational therapist, and a psychologist or psychosocial worker. Dr. Vastert also noted that good communication between team members is essential. In addition, there needs to be clear guidance on when to refer to a pediatric rheumatologist.
The SHARE project JIA recommendations include 10 evidence-based statements on diagnosis, 31 evidence-based statements on treatment, and 17 general statements on specific care for JIA, Dr. Vastert said. A few examples of the latter are that new patients should be seen in a specialist center within 4 weeks of referral; new patients and those starting a new therapy should be reviewed within 2-3 months to check on adherence, tolerance, and disease progression; and monitoring response to ongoing treatment should be every 3-6 months, preferably using existing standardized disease activity tools.

EULAR standard operating procedures were followed when developing the various SHARE recommendations, said Dr. Michael Beresford of the University of Liverpool (England) and the lead for the recommendations on childhood connective tissue disorders. Dr. Beresford noted that the latter were a rare, and in some cases extremely rare, complex group of pediatric rheumatic diseases that could lead to significant morbidity and mortality.
“Evidence-based guidelines have been lacking, and management is based mainly on physician experience. Consequently, treatment regimens vary widely throughout Europe,” Dr. Beresford observed. “These [recommendations] provide evidence-based, internationally agreed-upon standards of optimal care for pediatric connective tissue disorders.”
Specifically, the connective tissue disorder recommendations cover when to refer and how to diagnose, treat, and monitor cSLE (including neuropsychiatric SLE), childhood APS, and juvenile vasculitides, including rare pediatric vasculitides such as Takayasu arteritis. The SHARE recommendations for the management of juvenile dermatomyositis are currently in press in Annals of the Rheumatic Diseases, Dr. Beresford said.
Giving a few examples of recommendations for cSLE, Dr. Beresford noted that one of the challenges is to try to prevent delay in diagnosis. The expert panel decided that the 2012 Systemic Lupus International Collaborating Clinics (SLICC) criteria could be used for diagnosis. A referral to a pediatric rheumatologist is warranted, they determined, when a child has a positive antinuclear antibody (ANA) test and meets two clinical SLICC criteria. Dr. Beresford conceded that antibody testing might not be available because of cost in all countries, but they “decided to draw a line in the sand” to say that it is important that it is routinely done in order to come closer to a definitive diagnosis.
The aim of treatment for cSLE, the recommendations advise, is to optimize control and prevent damage caused by both the disease and by its treatment. For example, all children should be on hydroxychloroquine, and if tapering of prednisone is not possible, a disease-modifying antirheumatic drug should be added. It’s also important to actively check compliance with therapy, Dr. Beresford said.
The SHARE project was initially funded by a grant from the European Agency for Health and Consumers between 2012 and 2015 and now continues under the auspices of the Paediatric Rheumatology European Society. All speakers reported having no relevant disclosures.
LONDON – Recommendations on managing juvenile idiopathic arthritis and connective tissue disorders in children and young people across Europe were unveiled at the European Congress of Rheumatology.
The recommendations, which come from the SHARE (Single Hub and Access Point for Paediatric Rheumatology in Europe) project, cover best practices and provide guidance based on current evidence and expert opinion for the optimal diagnosis and treatment of these rare rheumatic diseases that affect the pediatric population.
It is hoped that the recommendations will be used to improve access to treatment and care within individual countries such that a child in one country will be able to receive the same standard of care as a child in another, Dr. Nico Wulffraat of University Medical Center Utrecht (the Netherlands) said in an interview.
Dr. Wulffraat, one of the driving forces behind the project, noted that the SHARE project was set up to look at making the management of rare pediatric rheumatic diseases more uniform across Europe. It addressed conditions such as juvenile idiopathic arthritis (JIA), childhood-onset systemic lupus erythematosus (cSLE), childhood antiphospholipid syndrome (APS), childhood vasculitis, juvenile dermatomyositis, and pediatric scleroderma. In addition, recommendations on diagnosis and treatment of periodic fever syndromes have been developed in collaboration with experts from the Eurofever Project.
“Our evidence- and consensus-based recommendations will hopefully drive access to uniform and optimal care throughout Europe, including off-label therapy when appropriate according to international consensus–derived expert advice,” Dr. Sebastiaan Vastert, SHARE project co-coordinator, said in an interview. He added: “The SHARE network will be invaluable for further international collaboration, both for optimization of care and for international collaboration in research as well.”
Dr. Wulffraat observed that while the recommendations are primarily directed at health care professionals, they also are of use for other stakeholders such as health authorities and insurance companies, and of course patients themselves to ensure the best level of care is being achieved throughout Europe.
The process for developing the guidelines was perhaps as important as the recommendations themselves, said Dr. Vastert, also of University Medical Center Utrecht. The process helped to build a network of international experts who could work together to develop future recommendations for improving patient care.
The recommendations for JIA and other pediatric rheumatic diseases included 51 “cross-cutting” statements, Dr. Vastert said. One of these statements was that a pediatric rheumatologist should manage children with signs of rheumatic disease. Another highlighted the members of a multidisciplinary team who should be involved as appropriate, such as a nurse specializing in pediatric rheumatic disease, a physiotherapist or occupational therapist, and a psychologist or psychosocial worker. Dr. Vastert also noted that good communication between team members is essential. In addition, there needs to be clear guidance on when to refer to a pediatric rheumatologist.
The SHARE project JIA recommendations include 10 evidence-based statements on diagnosis, 31 evidence-based statements on treatment, and 17 general statements on specific care for JIA, Dr. Vastert said. A few examples of the latter are that new patients should be seen in a specialist center within 4 weeks of referral; new patients and those starting a new therapy should be reviewed within 2-3 months to check on adherence, tolerance, and disease progression; and monitoring response to ongoing treatment should be every 3-6 months, preferably using existing standardized disease activity tools.
EULAR standard operating procedures were followed when developing the various SHARE recommendations, said Dr. Michael Beresford of the University of Liverpool (England) and the lead for the recommendations on childhood connective tissue disorders. Dr. Beresford noted that the latter were a rare, and in some cases extremely rare, complex group of pediatric rheumatic diseases that could lead to significant morbidity and mortality.
“Evidence-based guidelines have been lacking, and management is based mainly on physician experience. Consequently, treatment regimens vary widely throughout Europe,” Dr. Beresford observed. “These [recommendations] provide evidence-based, internationally agreed-upon standards of optimal care for pediatric connective tissue disorders.”
Specifically, the connective tissue disorder recommendations cover when to refer and how to diagnose, treat, and monitor cSLE (including neuropsychiatric SLE), childhood APS, and juvenile vasculitides, including rare pediatric vasculitides such as Takayasu arteritis. The SHARE recommendations for the management of juvenile dermatomyositis are currently in press in Annals of the Rheumatic Diseases, Dr. Beresford said.
Giving a few examples of recommendations for cSLE, Dr. Beresford noted that one of the challenges is to try to prevent delay in diagnosis. The expert panel decided that the 2012 Systemic Lupus International Collaborating Clinics (SLICC) criteria could be used for diagnosis. A referral to a pediatric rheumatologist is warranted, they determined, when a child has a positive antinuclear antibody (ANA) test and meets two clinical SLICC criteria. Dr. Beresford conceded that antibody testing might not be available because of cost in all countries, but they “decided to draw a line in the sand” to say that it is important that it is routinely done in order to come closer to a definitive diagnosis.
The aim of treatment for cSLE, the recommendations advise, is to optimize control and prevent damage caused by both the disease and by its treatment. For example, all children should be on hydroxychloroquine, and if tapering of prednisone is not possible, a disease-modifying antirheumatic drug should be added. It’s also important to actively check compliance with therapy, Dr. Beresford said.
The SHARE project was initially funded by a grant from the European Agency for Health and Consumers between 2012 and 2015 and now continues under the auspices of the Paediatric Rheumatology European Society. All speakers reported having no relevant disclosures.
LONDON – Recommendations on managing juvenile idiopathic arthritis and connective tissue disorders in children and young people across Europe were unveiled at the European Congress of Rheumatology.
The recommendations, which come from the SHARE (Single Hub and Access Point for Paediatric Rheumatology in Europe) project, cover best practices and provide guidance based on current evidence and expert opinion for the optimal diagnosis and treatment of these rare rheumatic diseases that affect the pediatric population.
It is hoped that the recommendations will be used to improve access to treatment and care within individual countries such that a child in one country will be able to receive the same standard of care as a child in another, Dr. Nico Wulffraat of University Medical Center Utrecht (the Netherlands) said in an interview.
Dr. Wulffraat, one of the driving forces behind the project, noted that the SHARE project was set up to look at making the management of rare pediatric rheumatic diseases more uniform across Europe. It addressed conditions such as juvenile idiopathic arthritis (JIA), childhood-onset systemic lupus erythematosus (cSLE), childhood antiphospholipid syndrome (APS), childhood vasculitis, juvenile dermatomyositis, and pediatric scleroderma. In addition, recommendations on diagnosis and treatment of periodic fever syndromes have been developed in collaboration with experts from the Eurofever Project.
“Our evidence- and consensus-based recommendations will hopefully drive access to uniform and optimal care throughout Europe, including off-label therapy when appropriate according to international consensus–derived expert advice,” Dr. Sebastiaan Vastert, SHARE project co-coordinator, said in an interview. He added: “The SHARE network will be invaluable for further international collaboration, both for optimization of care and for international collaboration in research as well.”
Dr. Wulffraat observed that while the recommendations are primarily directed at health care professionals, they also are of use for other stakeholders such as health authorities and insurance companies, and of course patients themselves to ensure the best level of care is being achieved throughout Europe.
The process for developing the guidelines was perhaps as important as the recommendations themselves, said Dr. Vastert, also of University Medical Center Utrecht. The process helped to build a network of international experts who could work together to develop future recommendations for improving patient care.
The recommendations for JIA and other pediatric rheumatic diseases included 51 “cross-cutting” statements, Dr. Vastert said. One of these statements was that a pediatric rheumatologist should manage children with signs of rheumatic disease. Another highlighted the members of a multidisciplinary team who should be involved as appropriate, such as a nurse specializing in pediatric rheumatic disease, a physiotherapist or occupational therapist, and a psychologist or psychosocial worker. Dr. Vastert also noted that good communication between team members is essential. In addition, there needs to be clear guidance on when to refer to a pediatric rheumatologist.
The SHARE project JIA recommendations include 10 evidence-based statements on diagnosis, 31 evidence-based statements on treatment, and 17 general statements on specific care for JIA, Dr. Vastert said. A few examples of the latter are that new patients should be seen in a specialist center within 4 weeks of referral; new patients and those starting a new therapy should be reviewed within 2-3 months to check on adherence, tolerance, and disease progression; and monitoring response to ongoing treatment should be every 3-6 months, preferably using existing standardized disease activity tools.
EULAR standard operating procedures were followed when developing the various SHARE recommendations, said Dr. Michael Beresford of the University of Liverpool (England) and the lead for the recommendations on childhood connective tissue disorders. Dr. Beresford noted that the latter were a rare, and in some cases extremely rare, complex group of pediatric rheumatic diseases that could lead to significant morbidity and mortality.
“Evidence-based guidelines have been lacking, and management is based mainly on physician experience. Consequently, treatment regimens vary widely throughout Europe,” Dr. Beresford observed. “These [recommendations] provide evidence-based, internationally agreed-upon standards of optimal care for pediatric connective tissue disorders.”
Specifically, the connective tissue disorder recommendations cover when to refer and how to diagnose, treat, and monitor cSLE (including neuropsychiatric SLE), childhood APS, and juvenile vasculitides, including rare pediatric vasculitides such as Takayasu arteritis. The SHARE recommendations for the management of juvenile dermatomyositis are currently in press in Annals of the Rheumatic Diseases, Dr. Beresford said.
Giving a few examples of recommendations for cSLE, Dr. Beresford noted that one of the challenges is to try to prevent delay in diagnosis. The expert panel decided that the 2012 Systemic Lupus International Collaborating Clinics (SLICC) criteria could be used for diagnosis. A referral to a pediatric rheumatologist is warranted, they determined, when a child has a positive antinuclear antibody (ANA) test and meets two clinical SLICC criteria. Dr. Beresford conceded that antibody testing might not be available because of cost in all countries, but they “decided to draw a line in the sand” to say that it is important that it is routinely done in order to come closer to a definitive diagnosis.
The aim of treatment for cSLE, the recommendations advise, is to optimize control and prevent damage caused by both the disease and by its treatment. For example, all children should be on hydroxychloroquine, and if tapering of prednisone is not possible, a disease-modifying antirheumatic drug should be added. It’s also important to actively check compliance with therapy, Dr. Beresford said.
The SHARE project was initially funded by a grant from the European Agency for Health and Consumers between 2012 and 2015 and now continues under the auspices of the Paediatric Rheumatology European Society. All speakers reported having no relevant disclosures.
EXPERT ANALYSIS FROM THE EULAR 2016 CONGRESS

Early results promising for pembrolizumab in combination regimens for NSCLC
CHICAGO – Overall response rates ranged from 48% to 71% when pembrolizumab was added to chemotherapy combinations in patients with advanced non–small cell lung cancer (NSCLC), and close to 30% when added to an R2 inhibitor in patients with advanced NSCLC and other tumor types, according to two early phase clinical trials presented at the annual meeting of the American Society of Clinical Oncology.
Safety and efficacy of pembrolizumab in combination with either carboplatin and paclitaxel; carboplatin, paclitaxel, and bevacizumab; or carboplatin and pemetrexed were evaluated in the KEYNOTE-021 trial. Patients were split between two doses of pembrolizumab: 2 or 10 mg/kg every 3 weeks. The overall response rate for the entire cohort of 74 patients with advanced NSCLC was 57% (95% confidence interval, 45-68), reported Dr. Shirish Gadgeel of Karmanos Cancer Institute, Detroit.
For the 25 patients who received pembrolizumab, carboplatin, and paclitaxel (cohort A) the overall response was 52% (95% CI, 31-72). For the 25 patients who also received bevacizumab in addition to pembrolizumab, carboplatin, and paclitaxel (cohort B), the overall response rate was 48% (95% CI, 28-69).
Among 24 patients who received pemetrexed with pembrolizumab and carboplatin (cohort C), the overall response was the highest at 71% (95% CI, 49-87).
Grade 3 or 4 adverse events occurred in 36%, 46%, and 42% of patients in cohort A, B, and C, respectively. The most common adverse events were anemia and neutropenia including febrile neutropenia.
“Pembro in combination with standard chemotherapy regimens is feasible and yields substantial clinical efficacy regardless of pembro dose or PD-L1 status in treatment-naïve advanced NSCLC,” Dr Gadgeel said.
A randomized phase III study evaluating pemetrexed/platinum with or without pembrolizumab is currently recruiting, he said.
These combinations, while perhaps better than chemotherapy alone, may be no better than just immunotherapy alone, session moderator Dr. Scott Antonia of the Moffitt Cancer Center, Tampa, Fla., commented. “And the truth is that concurrent therapy is clearly more toxic.”

In another study presented at the meeting, led by Dr. Roy Herbst of Yale Cancer Center, New Haven, Conn., pembrolizumab was combined with ramucirumab, a recently approved vascular endothelial growth factor receptor–2 inhibitor. “Hallmarks of tumor growth include angiogenesis and immunosuppression,” Dr. Herbst and his coauthors wrote in their abstract, noting that this was the first study to combine these agents to target both processes simultaneously.
“The idea is the R2 inhibitor might have an effect on the microimmune environment and drive T cells into the tumor,” Dr. Herbst said.
The study cohort included patients with advanced gastric or gastroesophageal junction adenocarcinoma, NSCLC, and urothelial carcinoma.
Overall, the response rate was close to 30% in the first 10 patients, Dr. Herbst reported. Furthermore, the preliminary data did not reveal any unexpected safety signals.
Although the data was from a small, ongoing phase I trial, Dr. Herbst noted the data is promising. Researchers are continuing to collect data on the safety profile of this drug combination, and are planning to enroll patients with additional tumor types.
The KEYNOTE-021 trial was funded by Merck Sharp and Dohme. Dr. Gadgeel reported having a consulting or advisory role and receiving research funding from multiple companies. The trial headed by Dr. Herbst was funded by Eli Lilly. Dr. Herbst reported having a consulting or advisory role and receiving honoraria and research funding from multiple companies including Eli Lilly.
On Twitter @jessnicolecraig
CHICAGO – Overall response rates ranged from 48% to 71% when pembrolizumab was added to chemotherapy combinations in patients with advanced non–small cell lung cancer (NSCLC), and close to 30% when added to an R2 inhibitor in patients with advanced NSCLC and other tumor types, according to two early phase clinical trials presented at the annual meeting of the American Society of Clinical Oncology.
Safety and efficacy of pembrolizumab in combination with either carboplatin and paclitaxel; carboplatin, paclitaxel, and bevacizumab; or carboplatin and pemetrexed were evaluated in the KEYNOTE-021 trial. Patients were split between two doses of pembrolizumab: 2 or 10 mg/kg every 3 weeks. The overall response rate for the entire cohort of 74 patients with advanced NSCLC was 57% (95% confidence interval, 45-68), reported Dr. Shirish Gadgeel of Karmanos Cancer Institute, Detroit.
For the 25 patients who received pembrolizumab, carboplatin, and paclitaxel (cohort A) the overall response was 52% (95% CI, 31-72). For the 25 patients who also received bevacizumab in addition to pembrolizumab, carboplatin, and paclitaxel (cohort B), the overall response rate was 48% (95% CI, 28-69).
Among 24 patients who received pemetrexed with pembrolizumab and carboplatin (cohort C), the overall response was the highest at 71% (95% CI, 49-87).
Grade 3 or 4 adverse events occurred in 36%, 46%, and 42% of patients in cohort A, B, and C, respectively. The most common adverse events were anemia and neutropenia including febrile neutropenia.
“Pembro in combination with standard chemotherapy regimens is feasible and yields substantial clinical efficacy regardless of pembro dose or PD-L1 status in treatment-naïve advanced NSCLC,” Dr Gadgeel said.
A randomized phase III study evaluating pemetrexed/platinum with or without pembrolizumab is currently recruiting, he said.
These combinations, while perhaps better than chemotherapy alone, may be no better than just immunotherapy alone, session moderator Dr. Scott Antonia of the Moffitt Cancer Center, Tampa, Fla., commented. “And the truth is that concurrent therapy is clearly more toxic.”
In another study presented at the meeting, led by Dr. Roy Herbst of Yale Cancer Center, New Haven, Conn., pembrolizumab was combined with ramucirumab, a recently approved vascular endothelial growth factor receptor–2 inhibitor. “Hallmarks of tumor growth include angiogenesis and immunosuppression,” Dr. Herbst and his coauthors wrote in their abstract, noting that this was the first study to combine these agents to target both processes simultaneously.
“The idea is the R2 inhibitor might have an effect on the microimmune environment and drive T cells into the tumor,” Dr. Herbst said.
The study cohort included patients with advanced gastric or gastroesophageal junction adenocarcinoma, NSCLC, and urothelial carcinoma.
Overall, the response rate was close to 30% in the first 10 patients, Dr. Herbst reported. Furthermore, the preliminary data did not reveal any unexpected safety signals.
Although the data was from a small, ongoing phase I trial, Dr. Herbst noted the data is promising. Researchers are continuing to collect data on the safety profile of this drug combination, and are planning to enroll patients with additional tumor types.
The KEYNOTE-021 trial was funded by Merck Sharp and Dohme. Dr. Gadgeel reported having a consulting or advisory role and receiving research funding from multiple companies. The trial headed by Dr. Herbst was funded by Eli Lilly. Dr. Herbst reported having a consulting or advisory role and receiving honoraria and research funding from multiple companies including Eli Lilly.
On Twitter @jessnicolecraig
CHICAGO – Overall response rates ranged from 48% to 71% when pembrolizumab was added to chemotherapy combinations in patients with advanced non–small cell lung cancer (NSCLC), and close to 30% when added to an R2 inhibitor in patients with advanced NSCLC and other tumor types, according to two early phase clinical trials presented at the annual meeting of the American Society of Clinical Oncology.
Safety and efficacy of pembrolizumab in combination with either carboplatin and paclitaxel; carboplatin, paclitaxel, and bevacizumab; or carboplatin and pemetrexed were evaluated in the KEYNOTE-021 trial. Patients were split between two doses of pembrolizumab: 2 or 10 mg/kg every 3 weeks. The overall response rate for the entire cohort of 74 patients with advanced NSCLC was 57% (95% confidence interval, 45-68), reported Dr. Shirish Gadgeel of Karmanos Cancer Institute, Detroit.
For the 25 patients who received pembrolizumab, carboplatin, and paclitaxel (cohort A) the overall response was 52% (95% CI, 31-72). For the 25 patients who also received bevacizumab in addition to pembrolizumab, carboplatin, and paclitaxel (cohort B), the overall response rate was 48% (95% CI, 28-69).
Among 24 patients who received pemetrexed with pembrolizumab and carboplatin (cohort C), the overall response was the highest at 71% (95% CI, 49-87).
Grade 3 or 4 adverse events occurred in 36%, 46%, and 42% of patients in cohort A, B, and C, respectively. The most common adverse events were anemia and neutropenia including febrile neutropenia.
“Pembro in combination with standard chemotherapy regimens is feasible and yields substantial clinical efficacy regardless of pembro dose or PD-L1 status in treatment-naïve advanced NSCLC,” Dr Gadgeel said.
A randomized phase III study evaluating pemetrexed/platinum with or without pembrolizumab is currently recruiting, he said.
These combinations, while perhaps better than chemotherapy alone, may be no better than just immunotherapy alone, session moderator Dr. Scott Antonia of the Moffitt Cancer Center, Tampa, Fla., commented. “And the truth is that concurrent therapy is clearly more toxic.”
In another study presented at the meeting, led by Dr. Roy Herbst of Yale Cancer Center, New Haven, Conn., pembrolizumab was combined with ramucirumab, a recently approved vascular endothelial growth factor receptor–2 inhibitor. “Hallmarks of tumor growth include angiogenesis and immunosuppression,” Dr. Herbst and his coauthors wrote in their abstract, noting that this was the first study to combine these agents to target both processes simultaneously.
“The idea is the R2 inhibitor might have an effect on the microimmune environment and drive T cells into the tumor,” Dr. Herbst said.
The study cohort included patients with advanced gastric or gastroesophageal junction adenocarcinoma, NSCLC, and urothelial carcinoma.
Overall, the response rate was close to 30% in the first 10 patients, Dr. Herbst reported. Furthermore, the preliminary data did not reveal any unexpected safety signals.
Although the data was from a small, ongoing phase I trial, Dr. Herbst noted the data is promising. Researchers are continuing to collect data on the safety profile of this drug combination, and are planning to enroll patients with additional tumor types.
The KEYNOTE-021 trial was funded by Merck Sharp and Dohme. Dr. Gadgeel reported having a consulting or advisory role and receiving research funding from multiple companies. The trial headed by Dr. Herbst was funded by Eli Lilly. Dr. Herbst reported having a consulting or advisory role and receiving honoraria and research funding from multiple companies including Eli Lilly.
On Twitter @jessnicolecraig
AT THE 2016 ASCO ANNUAL MEETING
Key clinical point: Overall response rates ranged from 30% to 71% when pembrolizumab was added to chemotherapy combinations or paired with an R2 inhibitor in patients with advanced NSCLC and other tumor types.
Major finding: Overall response rates ranged from 48% to 71% for NSCLC patients receiving pembrolizumab in combination with standard chemotherapy regimens, one with bevacizumab. In combinations with an R2-inhibitor, the overall response rate was nearly 30% for patients with advanced NSCLC or gastric or urothelial tumors.
Data source: The phase I/II KEYNOTE-021 trial of 74 patients with advanced NSCLC, and the phase I trial that included patients with advanced gastric or gastroesophageal junction adenocarcinoma, NSCLC, and urothelial carcinoma.
Disclosures: The KEYNOTE-021 trial was funded by Merck Sharp and Dohme. Dr. Gadgeel reported having a consulting or advisory role and receiving research funding from multiple companies. The trial headed by Dr. Herbst was funded by Eli Lilly. Dr. Herbst reported having a consulting or advisory role and receiving honoraria and research funding from multiple companies including Eli Lilly.

Symptoms linger after ‘successful’ gyn. cancer therapy
SAN DIEGO – Fully half of gynecologic cancer survivors who are done with treatment and cancer free nonetheless report having moderate to severe symptoms 2 and even 5 years after diagnosis, Dr. Michelle Rowland reported at the annual meeting of the Society of Gynecologic Oncology.
The most common symptoms in her cross-sectional study of 237 women with a history of uterine, ovarian, cervical, or vulvar cancer who were cancer free and at least 2 years post diagnosis were fatigue, insomnia, pain, memory impairment, and neuropathy, according to Dr. Rowland, a gynecologic oncology fellow at the University of Oklahoma in Oklahoma City.

She found the high prevalence of fatigue in gynecologic cancer survivors so distant from treatment particularly unexpected.
“The fact that 60% of women report some degree of fatigue 2 and 5 years out from their gynecologic cancer diagnosis was surprising to me,” she said in an interview. “Fatigue is something we usually think of as being related either to the treatment that they’re getting or to the cancer itself. But these women don’t have cancer anymore and are not being treated.”
The cross-sectional study included 237 patients who completed a self-assessment symptom questionnaire during a university outpatient gynecologic oncology clinic for ongoing routine disease surveillance. All were believed to be cancer free and none were receiving treatment 2 or more years post diagnosis. Seventy-seven of the women were 5 or more years out from diagnosis. The prevalence and self-rated severity of symptoms on a 0-10 scale were similar in the 2- and 5-year survivor groups.
One-quarter of the 2-year survivors reported having three or more moderate to severe symptoms as defined by a rating of 4-10. So did 29% of the 5-year survivors.
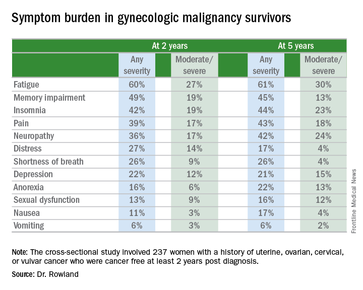
Three-quarters of women reported having one or more symptom.
In an effort to identify predictors of high symptom burden, Dr. Rowland and coinvestigators conducted a multivariate logistic regression analysis controlling for tumor stage, disease, site, race, and all forms of cancer therapy received. Prior chemotherapy proved to be an independent risk factor for high symptom burden at 2 years, while prior radiation therapy predicted high symptom burden at 5 or more years. Of note, cancer type was not predictive.
Roughly 40% of subjects reported currently being on medications for chronic pain, 11% were taking antianxiety drugs, and a similar proportion were using sleep aids.
Dr. Rowland concluded that long-term follow-up of gynecologic cancer survivors should include a symptom assessment. Survivor clinics, which are becoming increasingly common, offer a way to specifically address ongoing symptoms.
She reported having no financial conflicts regarding her study.
SAN DIEGO – Fully half of gynecologic cancer survivors who are done with treatment and cancer free nonetheless report having moderate to severe symptoms 2 and even 5 years after diagnosis, Dr. Michelle Rowland reported at the annual meeting of the Society of Gynecologic Oncology.
The most common symptoms in her cross-sectional study of 237 women with a history of uterine, ovarian, cervical, or vulvar cancer who were cancer free and at least 2 years post diagnosis were fatigue, insomnia, pain, memory impairment, and neuropathy, according to Dr. Rowland, a gynecologic oncology fellow at the University of Oklahoma in Oklahoma City.
She found the high prevalence of fatigue in gynecologic cancer survivors so distant from treatment particularly unexpected.
“The fact that 60% of women report some degree of fatigue 2 and 5 years out from their gynecologic cancer diagnosis was surprising to me,” she said in an interview. “Fatigue is something we usually think of as being related either to the treatment that they’re getting or to the cancer itself. But these women don’t have cancer anymore and are not being treated.”
The cross-sectional study included 237 patients who completed a self-assessment symptom questionnaire during a university outpatient gynecologic oncology clinic for ongoing routine disease surveillance. All were believed to be cancer free and none were receiving treatment 2 or more years post diagnosis. Seventy-seven of the women were 5 or more years out from diagnosis. The prevalence and self-rated severity of symptoms on a 0-10 scale were similar in the 2- and 5-year survivor groups.
One-quarter of the 2-year survivors reported having three or more moderate to severe symptoms as defined by a rating of 4-10. So did 29% of the 5-year survivors.
Three-quarters of women reported having one or more symptom.
In an effort to identify predictors of high symptom burden, Dr. Rowland and coinvestigators conducted a multivariate logistic regression analysis controlling for tumor stage, disease, site, race, and all forms of cancer therapy received. Prior chemotherapy proved to be an independent risk factor for high symptom burden at 2 years, while prior radiation therapy predicted high symptom burden at 5 or more years. Of note, cancer type was not predictive.
Roughly 40% of subjects reported currently being on medications for chronic pain, 11% were taking antianxiety drugs, and a similar proportion were using sleep aids.
Dr. Rowland concluded that long-term follow-up of gynecologic cancer survivors should include a symptom assessment. Survivor clinics, which are becoming increasingly common, offer a way to specifically address ongoing symptoms.
She reported having no financial conflicts regarding her study.
SAN DIEGO – Fully half of gynecologic cancer survivors who are done with treatment and cancer free nonetheless report having moderate to severe symptoms 2 and even 5 years after diagnosis, Dr. Michelle Rowland reported at the annual meeting of the Society of Gynecologic Oncology.
The most common symptoms in her cross-sectional study of 237 women with a history of uterine, ovarian, cervical, or vulvar cancer who were cancer free and at least 2 years post diagnosis were fatigue, insomnia, pain, memory impairment, and neuropathy, according to Dr. Rowland, a gynecologic oncology fellow at the University of Oklahoma in Oklahoma City.
She found the high prevalence of fatigue in gynecologic cancer survivors so distant from treatment particularly unexpected.
“The fact that 60% of women report some degree of fatigue 2 and 5 years out from their gynecologic cancer diagnosis was surprising to me,” she said in an interview. “Fatigue is something we usually think of as being related either to the treatment that they’re getting or to the cancer itself. But these women don’t have cancer anymore and are not being treated.”
The cross-sectional study included 237 patients who completed a self-assessment symptom questionnaire during a university outpatient gynecologic oncology clinic for ongoing routine disease surveillance. All were believed to be cancer free and none were receiving treatment 2 or more years post diagnosis. Seventy-seven of the women were 5 or more years out from diagnosis. The prevalence and self-rated severity of symptoms on a 0-10 scale were similar in the 2- and 5-year survivor groups.
One-quarter of the 2-year survivors reported having three or more moderate to severe symptoms as defined by a rating of 4-10. So did 29% of the 5-year survivors.
Three-quarters of women reported having one or more symptom.
In an effort to identify predictors of high symptom burden, Dr. Rowland and coinvestigators conducted a multivariate logistic regression analysis controlling for tumor stage, disease, site, race, and all forms of cancer therapy received. Prior chemotherapy proved to be an independent risk factor for high symptom burden at 2 years, while prior radiation therapy predicted high symptom burden at 5 or more years. Of note, cancer type was not predictive.
Roughly 40% of subjects reported currently being on medications for chronic pain, 11% were taking antianxiety drugs, and a similar proportion were using sleep aids.
Dr. Rowland concluded that long-term follow-up of gynecologic cancer survivors should include a symptom assessment. Survivor clinics, which are becoming increasingly common, offer a way to specifically address ongoing symptoms.
She reported having no financial conflicts regarding her study.
AT THE ANNUAL MEETING ON WOMEN’S CANCER
Key clinical point: Long-term follow-up of gynecologic cancer survivors should include assessment of cancer- or treatment-related symptoms.
Major finding: One-quarter of cancer-free patients reported three or more lingering symptoms of moderate to severe intensity 2 years after diagnosis.
Data source: This cross-sectional study utilized a structured questionnaire to evaluate the types and severity of symptoms present in 237 former gynecologic cancer patients who were off treatment and cancer free at least 2 years after diagnosis.
Disclosures: The study presenter reported having no financial conflicts of interest.

Enhanced recovery protocol speeds discharge, decreases readmissions for ventral hernia repair
A postsurgical recovery program featuring early feeding and multimodal pain management hastened the return of bowel function and shortened hospital stay by 2 days for patients undergoing complex ventral hernia repair.
Despite leaving the hospital sooner, however, patients were 75% less likely to be readmitted within 90 days, Dr. Arnab Majumder and his colleagues wrote in the June issue of the Journal of the American College of Surgeons (2016 Jun;222:1106-15). Most of those who did return had wound complications, a stark contrast to readmissions among patients who didn’t experience the enhanced recovery program. Among that group, 75% of the readmissions were caused by bowel obstruction/ileus, deep venous thrombosis or pulmonary embolism, pneumonia, and urinary tract infections – all of them “problems [that] could be related to prolonged hospitalizations,” said Dr. Majumder of the University Hospitals Case Medical Center, Cleveland.

The investigators created an Enhanced Recovery After Surgery (ERAS) pathway specifically for patients undergoing complex ventral hernia repairs using transversus abdominis release and sublay synthetic mesh placement. The patients “present formidable challenges to the surgeon, not only in the operating room but also during perioperative management,” the authors noted.
The ERAS the team preemptively addressed patient issues in the preoperative, intra- and perioperative, and postoperative periods. They compared the outcomes of 100 patients treated with the protocol to those in a control group of 100 who underwent the same surgery before the protocol was implemented. The main outcomes measures were time to diet advancement, time to return of bowel function, time to oral narcotics, length of stay, and 90-day readmissions.
The ERAS begins with preoperative optimization. This consists of weight loss, smoking cessation, and managing diabetes and obstructive sleep apnea. No surgery occurs until the HbA1c is less than 8 and patients have been tobacco free for at least 1 month.
All patients receive an arginine and omega-3 supplement thrice daily for 5 days before surgery. The night before surgery, they have a nasal swab screen for methicillin-resistant Staphylococcus aureus (MRSA), and decolonization with mupirocin ointment and a chlorhexidine bath.
Preoperatively, they receive 5,000 units of unfractionated heparin, along with sequential compression devices for deep vein thrombosis protection. Use of both continue postoperatively, with heparin given every 8 hours until the patient is ambulatory.
Those without a history of narcotic use receive a preoperative dose of alvimopan. Everyone also receives oral gabapentin and preoperative antibiotics.
Intraoperatively, intravenous fluids are given judiciously to minimize bowel edema and decrease the risk of respiratory side effects. The surgeon also employs a transversus abdominis plane (TAP) block with liposomal bupivacaine.
There is a multimodal pain management program, which includes intravenous hydromorphone by patient-controlled anesthesia pump and oral gabapentin three times daily. Patients switch to oral analgesics on postoperative day 2. These consist of acetaminophen and NSAIDs on an alternating schedule. Intravenous diazepam can be added as an antispasmodic. Alvimopan is continued twice daily until first bowel movement or hospital discharge.
Clear liquids are limited to 250 mL/8 hours on postoperative day 1, along with a clear protein drink. By day 2, patients are advanced to unrestricted volumes of clear liquids, and on day 3, to a regular solid diet. At that point, the clear protein drink is switched for a regular protein drink (Boost Plus).
Patients who vomit or require a nasogastric tube for decompression, and those with severe persistent nausea are made NPO, but can resume diet when these symptoms improve. Those with mild-moderate nausea receive intravenous ondansetron and are allowed to self-limit oral intake.
Patients were a mean of 57 years old with a mean body mass index of about 33 kg/m2. The hernia was recurrent for about 60% of each group, with a mean area of about 300 cm2, and a mean width of 14 cm. The mean mesh size was about 1,000 cm2. The mean operative time was significantly shorter in the control group: 197 vs. 245 minutes.
Diet was advanced significantly more quickly in the ERAS group than the control group: 1 vs. 2 days on a liquid diet and 3 vs. 4.8 days to regular diet. Emesis after diet advancement was similar (4% for ERAS and 5% for control).
Compared with the control group, the ERAS group experienced significantly shorter average times to flatus (3.1 vs. 3.9 days), bowel movement (3.6 vs. 5.2 days), and reaching GI-3 status (3.4 vs. 4.8 days). Those in the ERAS group switched to oral narcotics sooner (2.2 vs. 3.6 days) also.
The average length of stay was significantly lower in ERAS group as well (4 vs. 6 days). About 18% of those in the ERAS group had a stay of less than 3 days, compared with 2% of the control group.
Readmissions within 90 days occurred in 16% of the control group and 4% of the ERAS group – a significant difference. There were four surgical site complications in the control group and three in the ERAS group. Bowel obstruction occurred in three control patients and one ERAS patient. All other complications requiring readmission occurred in the control group: two pulmonary embolisms, two deep vein thromboses, one pneumonia, one urinary tract infection and three other unspecified causes.
The authors noted that the shift to multimodal pain management and shorter-term use of IV opiates is a large contributor to the protocol’s good bowel outcomes. “Introduction of preoperative and postoperative gabapentin, intraoperative surgeon-delivered TAP block with long-acting liposomal bupivacaine, postoperative use of acetaminophen, and nonsteroidal agents have all appeared to contribute to better pain control and likely decrease in opioid consumption,” they said.
The use of diazepam as a pain medication is unusual, they said, but effective.
“We believe a large component of postoperative pain in hernia patients is due to muscle spasms after myofascial release, irritation from mesh placement, and transabdominal suture fixation. Therefore, in the context of our frequent use of myofascial releases for large incisional hernias, we believe the antispasmodic effects of diazepam potentially alleviate some of the postoperative discomfort caused by major abdominal wall reconstruction.”
None of the investigators reported financial conflicts.
A postsurgical recovery program featuring early feeding and multimodal pain management hastened the return of bowel function and shortened hospital stay by 2 days for patients undergoing complex ventral hernia repair.
Despite leaving the hospital sooner, however, patients were 75% less likely to be readmitted within 90 days, Dr. Arnab Majumder and his colleagues wrote in the June issue of the Journal of the American College of Surgeons (2016 Jun;222:1106-15). Most of those who did return had wound complications, a stark contrast to readmissions among patients who didn’t experience the enhanced recovery program. Among that group, 75% of the readmissions were caused by bowel obstruction/ileus, deep venous thrombosis or pulmonary embolism, pneumonia, and urinary tract infections – all of them “problems [that] could be related to prolonged hospitalizations,” said Dr. Majumder of the University Hospitals Case Medical Center, Cleveland.
The investigators created an Enhanced Recovery After Surgery (ERAS) pathway specifically for patients undergoing complex ventral hernia repairs using transversus abdominis release and sublay synthetic mesh placement. The patients “present formidable challenges to the surgeon, not only in the operating room but also during perioperative management,” the authors noted.
The ERAS the team preemptively addressed patient issues in the preoperative, intra- and perioperative, and postoperative periods. They compared the outcomes of 100 patients treated with the protocol to those in a control group of 100 who underwent the same surgery before the protocol was implemented. The main outcomes measures were time to diet advancement, time to return of bowel function, time to oral narcotics, length of stay, and 90-day readmissions.
The ERAS begins with preoperative optimization. This consists of weight loss, smoking cessation, and managing diabetes and obstructive sleep apnea. No surgery occurs until the HbA1c is less than 8 and patients have been tobacco free for at least 1 month.
All patients receive an arginine and omega-3 supplement thrice daily for 5 days before surgery. The night before surgery, they have a nasal swab screen for methicillin-resistant Staphylococcus aureus (MRSA), and decolonization with mupirocin ointment and a chlorhexidine bath.
Preoperatively, they receive 5,000 units of unfractionated heparin, along with sequential compression devices for deep vein thrombosis protection. Use of both continue postoperatively, with heparin given every 8 hours until the patient is ambulatory.
Those without a history of narcotic use receive a preoperative dose of alvimopan. Everyone also receives oral gabapentin and preoperative antibiotics.
Intraoperatively, intravenous fluids are given judiciously to minimize bowel edema and decrease the risk of respiratory side effects. The surgeon also employs a transversus abdominis plane (TAP) block with liposomal bupivacaine.
There is a multimodal pain management program, which includes intravenous hydromorphone by patient-controlled anesthesia pump and oral gabapentin three times daily. Patients switch to oral analgesics on postoperative day 2. These consist of acetaminophen and NSAIDs on an alternating schedule. Intravenous diazepam can be added as an antispasmodic. Alvimopan is continued twice daily until first bowel movement or hospital discharge.
Clear liquids are limited to 250 mL/8 hours on postoperative day 1, along with a clear protein drink. By day 2, patients are advanced to unrestricted volumes of clear liquids, and on day 3, to a regular solid diet. At that point, the clear protein drink is switched for a regular protein drink (Boost Plus).
Patients who vomit or require a nasogastric tube for decompression, and those with severe persistent nausea are made NPO, but can resume diet when these symptoms improve. Those with mild-moderate nausea receive intravenous ondansetron and are allowed to self-limit oral intake.
Patients were a mean of 57 years old with a mean body mass index of about 33 kg/m2. The hernia was recurrent for about 60% of each group, with a mean area of about 300 cm2, and a mean width of 14 cm. The mean mesh size was about 1,000 cm2. The mean operative time was significantly shorter in the control group: 197 vs. 245 minutes.
Diet was advanced significantly more quickly in the ERAS group than the control group: 1 vs. 2 days on a liquid diet and 3 vs. 4.8 days to regular diet. Emesis after diet advancement was similar (4% for ERAS and 5% for control).
Compared with the control group, the ERAS group experienced significantly shorter average times to flatus (3.1 vs. 3.9 days), bowel movement (3.6 vs. 5.2 days), and reaching GI-3 status (3.4 vs. 4.8 days). Those in the ERAS group switched to oral narcotics sooner (2.2 vs. 3.6 days) also.
The average length of stay was significantly lower in ERAS group as well (4 vs. 6 days). About 18% of those in the ERAS group had a stay of less than 3 days, compared with 2% of the control group.
Readmissions within 90 days occurred in 16% of the control group and 4% of the ERAS group – a significant difference. There were four surgical site complications in the control group and three in the ERAS group. Bowel obstruction occurred in three control patients and one ERAS patient. All other complications requiring readmission occurred in the control group: two pulmonary embolisms, two deep vein thromboses, one pneumonia, one urinary tract infection and three other unspecified causes.
The authors noted that the shift to multimodal pain management and shorter-term use of IV opiates is a large contributor to the protocol’s good bowel outcomes. “Introduction of preoperative and postoperative gabapentin, intraoperative surgeon-delivered TAP block with long-acting liposomal bupivacaine, postoperative use of acetaminophen, and nonsteroidal agents have all appeared to contribute to better pain control and likely decrease in opioid consumption,” they said.
The use of diazepam as a pain medication is unusual, they said, but effective.
“We believe a large component of postoperative pain in hernia patients is due to muscle spasms after myofascial release, irritation from mesh placement, and transabdominal suture fixation. Therefore, in the context of our frequent use of myofascial releases for large incisional hernias, we believe the antispasmodic effects of diazepam potentially alleviate some of the postoperative discomfort caused by major abdominal wall reconstruction.”
None of the investigators reported financial conflicts.
A postsurgical recovery program featuring early feeding and multimodal pain management hastened the return of bowel function and shortened hospital stay by 2 days for patients undergoing complex ventral hernia repair.
Despite leaving the hospital sooner, however, patients were 75% less likely to be readmitted within 90 days, Dr. Arnab Majumder and his colleagues wrote in the June issue of the Journal of the American College of Surgeons (2016 Jun;222:1106-15). Most of those who did return had wound complications, a stark contrast to readmissions among patients who didn’t experience the enhanced recovery program. Among that group, 75% of the readmissions were caused by bowel obstruction/ileus, deep venous thrombosis or pulmonary embolism, pneumonia, and urinary tract infections – all of them “problems [that] could be related to prolonged hospitalizations,” said Dr. Majumder of the University Hospitals Case Medical Center, Cleveland.
The investigators created an Enhanced Recovery After Surgery (ERAS) pathway specifically for patients undergoing complex ventral hernia repairs using transversus abdominis release and sublay synthetic mesh placement. The patients “present formidable challenges to the surgeon, not only in the operating room but also during perioperative management,” the authors noted.
The ERAS the team preemptively addressed patient issues in the preoperative, intra- and perioperative, and postoperative periods. They compared the outcomes of 100 patients treated with the protocol to those in a control group of 100 who underwent the same surgery before the protocol was implemented. The main outcomes measures were time to diet advancement, time to return of bowel function, time to oral narcotics, length of stay, and 90-day readmissions.
The ERAS begins with preoperative optimization. This consists of weight loss, smoking cessation, and managing diabetes and obstructive sleep apnea. No surgery occurs until the HbA1c is less than 8 and patients have been tobacco free for at least 1 month.
All patients receive an arginine and omega-3 supplement thrice daily for 5 days before surgery. The night before surgery, they have a nasal swab screen for methicillin-resistant Staphylococcus aureus (MRSA), and decolonization with mupirocin ointment and a chlorhexidine bath.
Preoperatively, they receive 5,000 units of unfractionated heparin, along with sequential compression devices for deep vein thrombosis protection. Use of both continue postoperatively, with heparin given every 8 hours until the patient is ambulatory.
Those without a history of narcotic use receive a preoperative dose of alvimopan. Everyone also receives oral gabapentin and preoperative antibiotics.
Intraoperatively, intravenous fluids are given judiciously to minimize bowel edema and decrease the risk of respiratory side effects. The surgeon also employs a transversus abdominis plane (TAP) block with liposomal bupivacaine.
There is a multimodal pain management program, which includes intravenous hydromorphone by patient-controlled anesthesia pump and oral gabapentin three times daily. Patients switch to oral analgesics on postoperative day 2. These consist of acetaminophen and NSAIDs on an alternating schedule. Intravenous diazepam can be added as an antispasmodic. Alvimopan is continued twice daily until first bowel movement or hospital discharge.
Clear liquids are limited to 250 mL/8 hours on postoperative day 1, along with a clear protein drink. By day 2, patients are advanced to unrestricted volumes of clear liquids, and on day 3, to a regular solid diet. At that point, the clear protein drink is switched for a regular protein drink (Boost Plus).
Patients who vomit or require a nasogastric tube for decompression, and those with severe persistent nausea are made NPO, but can resume diet when these symptoms improve. Those with mild-moderate nausea receive intravenous ondansetron and are allowed to self-limit oral intake.
Patients were a mean of 57 years old with a mean body mass index of about 33 kg/m2. The hernia was recurrent for about 60% of each group, with a mean area of about 300 cm2, and a mean width of 14 cm. The mean mesh size was about 1,000 cm2. The mean operative time was significantly shorter in the control group: 197 vs. 245 minutes.
Diet was advanced significantly more quickly in the ERAS group than the control group: 1 vs. 2 days on a liquid diet and 3 vs. 4.8 days to regular diet. Emesis after diet advancement was similar (4% for ERAS and 5% for control).
Compared with the control group, the ERAS group experienced significantly shorter average times to flatus (3.1 vs. 3.9 days), bowel movement (3.6 vs. 5.2 days), and reaching GI-3 status (3.4 vs. 4.8 days). Those in the ERAS group switched to oral narcotics sooner (2.2 vs. 3.6 days) also.
The average length of stay was significantly lower in ERAS group as well (4 vs. 6 days). About 18% of those in the ERAS group had a stay of less than 3 days, compared with 2% of the control group.
Readmissions within 90 days occurred in 16% of the control group and 4% of the ERAS group – a significant difference. There were four surgical site complications in the control group and three in the ERAS group. Bowel obstruction occurred in three control patients and one ERAS patient. All other complications requiring readmission occurred in the control group: two pulmonary embolisms, two deep vein thromboses, one pneumonia, one urinary tract infection and three other unspecified causes.
The authors noted that the shift to multimodal pain management and shorter-term use of IV opiates is a large contributor to the protocol’s good bowel outcomes. “Introduction of preoperative and postoperative gabapentin, intraoperative surgeon-delivered TAP block with long-acting liposomal bupivacaine, postoperative use of acetaminophen, and nonsteroidal agents have all appeared to contribute to better pain control and likely decrease in opioid consumption,” they said.
The use of diazepam as a pain medication is unusual, they said, but effective.
“We believe a large component of postoperative pain in hernia patients is due to muscle spasms after myofascial release, irritation from mesh placement, and transabdominal suture fixation. Therefore, in the context of our frequent use of myofascial releases for large incisional hernias, we believe the antispasmodic effects of diazepam potentially alleviate some of the postoperative discomfort caused by major abdominal wall reconstruction.”
None of the investigators reported financial conflicts.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF SURGEONS
Key clinical point: Early enteral feeding and multimodal pain management both contributed to early return of bowel function.
Major finding: Length of stay was 2 days shorter, and readmissions decreased by 75%, compared with a control group.
Data source: The prospective study comprised 200 patients.
Disclosures: Neither Dr. Majumder nor his colleagues had any financial disclosures.

New Framework for Quality Improvement
Improving healthcare means taking an efficacious intervention from one setting and effectively implementing it somewhere else.

“It is this key element of adapting what works to new settings that sets improvement in contrast to clinical research. The study of these complex systems will therefore require different methods of inquiry,” according to a recently published paper in the International Journal for Quality in Health Care titled “How Do We Learn about Improving Health Care: A Call for a New Epistemological Paradigm.”
“In biomedical sciences, we’re used to a golden standard that is the randomized controlled trial,” says lead author M. Rashad Massoud, MD, MPH, senior vice president, Quality & Performance Institute, University Research Co., LLC. “Of course, the nature of what we’re trying to do does not lend itself to that type of evaluation. It means that we can’t have an either/or situation where we either continue as we are or we go to flip side—which then inhibits the very nature of improvement from taking place, which is very contextual, very much adaptive in nature. There has to be a happy medium in between, where we can continue to do the improvements without inhibiting them and, at the same time, improve the rigor of the work.”
A new framework for how we learn about improvement could help in the design, implementation, and evaluation of QI by strengthening attribution and better understanding variations in effectiveness in different contexts, the authors assert.
“This will in turn allow us to understand which activities, under which conditions, are most effective at achieving sustained results in health outcomes,” the authors write.
In seeking a new framework for learning about QI, the authors suggest that the following questions must be considered:
- Did the improvements work?
- Why did they work?
- How do we know that the results can be attributed to the changes made?
- How can we replicate them?
“I think hospitalists would probably welcome the idea that not only can they measure improvements in the work that they’re doing but can actually do that in a more rigorous way and actually attribute the results they’re getting to the work that they’re doing,” Dr. Massoud says.
Reference
- Massoud MR, Barry D, Murphy A, Albrecht Y, Sax S, Parchman M. How do we learn about improving health care: a call for a new epistemological paradigm. Intl J Quality Health Care. doi:10.1093/intqhc/mzw039.
Improving healthcare means taking an efficacious intervention from one setting and effectively implementing it somewhere else.
“It is this key element of adapting what works to new settings that sets improvement in contrast to clinical research. The study of these complex systems will therefore require different methods of inquiry,” according to a recently published paper in the International Journal for Quality in Health Care titled “How Do We Learn about Improving Health Care: A Call for a New Epistemological Paradigm.”
“In biomedical sciences, we’re used to a golden standard that is the randomized controlled trial,” says lead author M. Rashad Massoud, MD, MPH, senior vice president, Quality & Performance Institute, University Research Co., LLC. “Of course, the nature of what we’re trying to do does not lend itself to that type of evaluation. It means that we can’t have an either/or situation where we either continue as we are or we go to flip side—which then inhibits the very nature of improvement from taking place, which is very contextual, very much adaptive in nature. There has to be a happy medium in between, where we can continue to do the improvements without inhibiting them and, at the same time, improve the rigor of the work.”
A new framework for how we learn about improvement could help in the design, implementation, and evaluation of QI by strengthening attribution and better understanding variations in effectiveness in different contexts, the authors assert.
“This will in turn allow us to understand which activities, under which conditions, are most effective at achieving sustained results in health outcomes,” the authors write.
In seeking a new framework for learning about QI, the authors suggest that the following questions must be considered:
- Did the improvements work?
- Why did they work?
- How do we know that the results can be attributed to the changes made?
- How can we replicate them?
“I think hospitalists would probably welcome the idea that not only can they measure improvements in the work that they’re doing but can actually do that in a more rigorous way and actually attribute the results they’re getting to the work that they’re doing,” Dr. Massoud says.
Reference
- Massoud MR, Barry D, Murphy A, Albrecht Y, Sax S, Parchman M. How do we learn about improving health care: a call for a new epistemological paradigm. Intl J Quality Health Care. doi:10.1093/intqhc/mzw039.
Improving healthcare means taking an efficacious intervention from one setting and effectively implementing it somewhere else.
“It is this key element of adapting what works to new settings that sets improvement in contrast to clinical research. The study of these complex systems will therefore require different methods of inquiry,” according to a recently published paper in the International Journal for Quality in Health Care titled “How Do We Learn about Improving Health Care: A Call for a New Epistemological Paradigm.”
“In biomedical sciences, we’re used to a golden standard that is the randomized controlled trial,” says lead author M. Rashad Massoud, MD, MPH, senior vice president, Quality & Performance Institute, University Research Co., LLC. “Of course, the nature of what we’re trying to do does not lend itself to that type of evaluation. It means that we can’t have an either/or situation where we either continue as we are or we go to flip side—which then inhibits the very nature of improvement from taking place, which is very contextual, very much adaptive in nature. There has to be a happy medium in between, where we can continue to do the improvements without inhibiting them and, at the same time, improve the rigor of the work.”
A new framework for how we learn about improvement could help in the design, implementation, and evaluation of QI by strengthening attribution and better understanding variations in effectiveness in different contexts, the authors assert.
“This will in turn allow us to understand which activities, under which conditions, are most effective at achieving sustained results in health outcomes,” the authors write.
In seeking a new framework for learning about QI, the authors suggest that the following questions must be considered:
- Did the improvements work?
- Why did they work?
- How do we know that the results can be attributed to the changes made?
- How can we replicate them?
“I think hospitalists would probably welcome the idea that not only can they measure improvements in the work that they’re doing but can actually do that in a more rigorous way and actually attribute the results they’re getting to the work that they’re doing,” Dr. Massoud says.
Reference
- Massoud MR, Barry D, Murphy A, Albrecht Y, Sax S, Parchman M. How do we learn about improving health care: a call for a new epistemological paradigm. Intl J Quality Health Care. doi:10.1093/intqhc/mzw039.