User login
Database may help predict cancer patients’ survival

Photo by Darren Baker
A newly developed database may help physicians predict survival outcomes in patients with hematologic and solid tumor malignancies, according to a paper published in Nature Medicine.
The database, known as PRECOG, integrates gene expression patterns of 39 types of cancer from nearly 18,000 patients with data about how long those patients lived.
By combining these data, researchers were able to see broad patterns that correlate with survival. They also believe this information could help them pinpoint potential therapeutic targets for a range of cancers.
“We were able to identify key pathways that can dramatically stratify survival across diverse cancer types,” said Ash Alizadeh, MD, PhD, of Stanford University in California.
“The patterns were very striking, especially because few such examples are currently available for the use of genes or immune cells for cancer prognosis.”
In addition to identifying potentially useful gene expression patterns, the researchers used an analytical tool called CIBERSORT to determine the composition of leukocytes that flock to a tumor.
“We were able to infer which immune cells are present or absent in individual solid tumors, to estimate their prevalence, and to correlate that information with patient survival,” said Aaron Newman, PhD, of Stanford University.
“We found you can even broadly distinguish cancer types just based on what kind of immune cells have infiltrated the tumor.”
Compiling the data
Researchers have tried for years to identify specific patterns of gene expression in cancerous tumors that differ from those in normal tissue. But the extreme variability among individual patients and tumors has made the process difficult, even when focused on particular cancer types.
“There are many more genes in a cell than there are patients with any one type of cancer, and this makes discovering the important genes for cancer outcomes a tough problem,” said Andrew Gentles, PhD, of Stanford University.
“Because it’s easy to find spurious associations that don’t hold up in follow-up studies, we combined information from a vast array of cancer types to better see meaningful correlations.”
The researchers first collected publicly available data on gene expression patterns of many types of cancers.
They then matched the gene expression profiles with clinical information about the patients, including their age, disease status, and how long they survived after diagnosis. Finally, the team combined the studies in a database.
“We wanted to be able to connect gene expression data with patient outcome for thousands of people at once,” Dr Alizadeh said. “Then, we could ask what we could learn more broadly.”
Surprising findings
The researchers were surprised to find that prognostic genes were often shared among distinct cancer types, suggesting that similar biological programs impact survival across cancers.
They were able to identify the top 10 genes that seemed to confer adverse outcomes—FOXM1, BIRC5, TOP2A, TPX2, NME1, CCNB1, CEP55, TYMS, CENPF, and CDKN3—and the top 10 genes associated with more positive outcomes—KLRB1, ITM2B, CBX7, CD2, CREBL2, SATB1, NR3C1, TMEM66, KLRK1, and FUCA1.
Many of these genes are involved in aspects of cell division or are associated with distinct leukocytes that flood a tumor.
The researchers were also able to identify combinations of leukocytes that appear to be correlated with outcomes.
In particular, elevated numbers of plasma cells and certain types of T cells correlated with better patient survival rates across many different solid tumors. But a high proportion of granulocytes was associated with adverse outcomes.
The researchers hope that PRECOG and CIBERSORT will increase our understanding of cancer biology and aid the development of new therapies for cancer patients. The team is applying these tools to better predict which patients will respond to new and emerging anticancer therapies.
Dr Alizadeh said this is especially important given recent advances in the development of drugs that engage immune responses but work well only for a subset of cancer patients. ![]()
Photo by Darren Baker
A newly developed database may help physicians predict survival outcomes in patients with hematologic and solid tumor malignancies, according to a paper published in Nature Medicine.
The database, known as PRECOG, integrates gene expression patterns of 39 types of cancer from nearly 18,000 patients with data about how long those patients lived.
By combining these data, researchers were able to see broad patterns that correlate with survival. They also believe this information could help them pinpoint potential therapeutic targets for a range of cancers.
“We were able to identify key pathways that can dramatically stratify survival across diverse cancer types,” said Ash Alizadeh, MD, PhD, of Stanford University in California.
“The patterns were very striking, especially because few such examples are currently available for the use of genes or immune cells for cancer prognosis.”
In addition to identifying potentially useful gene expression patterns, the researchers used an analytical tool called CIBERSORT to determine the composition of leukocytes that flock to a tumor.
“We were able to infer which immune cells are present or absent in individual solid tumors, to estimate their prevalence, and to correlate that information with patient survival,” said Aaron Newman, PhD, of Stanford University.
“We found you can even broadly distinguish cancer types just based on what kind of immune cells have infiltrated the tumor.”
Compiling the data
Researchers have tried for years to identify specific patterns of gene expression in cancerous tumors that differ from those in normal tissue. But the extreme variability among individual patients and tumors has made the process difficult, even when focused on particular cancer types.
“There are many more genes in a cell than there are patients with any one type of cancer, and this makes discovering the important genes for cancer outcomes a tough problem,” said Andrew Gentles, PhD, of Stanford University.
“Because it’s easy to find spurious associations that don’t hold up in follow-up studies, we combined information from a vast array of cancer types to better see meaningful correlations.”
The researchers first collected publicly available data on gene expression patterns of many types of cancers.
They then matched the gene expression profiles with clinical information about the patients, including their age, disease status, and how long they survived after diagnosis. Finally, the team combined the studies in a database.
“We wanted to be able to connect gene expression data with patient outcome for thousands of people at once,” Dr Alizadeh said. “Then, we could ask what we could learn more broadly.”
Surprising findings
The researchers were surprised to find that prognostic genes were often shared among distinct cancer types, suggesting that similar biological programs impact survival across cancers.
They were able to identify the top 10 genes that seemed to confer adverse outcomes—FOXM1, BIRC5, TOP2A, TPX2, NME1, CCNB1, CEP55, TYMS, CENPF, and CDKN3—and the top 10 genes associated with more positive outcomes—KLRB1, ITM2B, CBX7, CD2, CREBL2, SATB1, NR3C1, TMEM66, KLRK1, and FUCA1.
Many of these genes are involved in aspects of cell division or are associated with distinct leukocytes that flood a tumor.
The researchers were also able to identify combinations of leukocytes that appear to be correlated with outcomes.
In particular, elevated numbers of plasma cells and certain types of T cells correlated with better patient survival rates across many different solid tumors. But a high proportion of granulocytes was associated with adverse outcomes.
The researchers hope that PRECOG and CIBERSORT will increase our understanding of cancer biology and aid the development of new therapies for cancer patients. The team is applying these tools to better predict which patients will respond to new and emerging anticancer therapies.
Dr Alizadeh said this is especially important given recent advances in the development of drugs that engage immune responses but work well only for a subset of cancer patients. ![]()
Photo by Darren Baker
A newly developed database may help physicians predict survival outcomes in patients with hematologic and solid tumor malignancies, according to a paper published in Nature Medicine.
The database, known as PRECOG, integrates gene expression patterns of 39 types of cancer from nearly 18,000 patients with data about how long those patients lived.
By combining these data, researchers were able to see broad patterns that correlate with survival. They also believe this information could help them pinpoint potential therapeutic targets for a range of cancers.
“We were able to identify key pathways that can dramatically stratify survival across diverse cancer types,” said Ash Alizadeh, MD, PhD, of Stanford University in California.
“The patterns were very striking, especially because few such examples are currently available for the use of genes or immune cells for cancer prognosis.”
In addition to identifying potentially useful gene expression patterns, the researchers used an analytical tool called CIBERSORT to determine the composition of leukocytes that flock to a tumor.
“We were able to infer which immune cells are present or absent in individual solid tumors, to estimate their prevalence, and to correlate that information with patient survival,” said Aaron Newman, PhD, of Stanford University.
“We found you can even broadly distinguish cancer types just based on what kind of immune cells have infiltrated the tumor.”
Compiling the data
Researchers have tried for years to identify specific patterns of gene expression in cancerous tumors that differ from those in normal tissue. But the extreme variability among individual patients and tumors has made the process difficult, even when focused on particular cancer types.
“There are many more genes in a cell than there are patients with any one type of cancer, and this makes discovering the important genes for cancer outcomes a tough problem,” said Andrew Gentles, PhD, of Stanford University.
“Because it’s easy to find spurious associations that don’t hold up in follow-up studies, we combined information from a vast array of cancer types to better see meaningful correlations.”
The researchers first collected publicly available data on gene expression patterns of many types of cancers.
They then matched the gene expression profiles with clinical information about the patients, including their age, disease status, and how long they survived after diagnosis. Finally, the team combined the studies in a database.
“We wanted to be able to connect gene expression data with patient outcome for thousands of people at once,” Dr Alizadeh said. “Then, we could ask what we could learn more broadly.”
Surprising findings
The researchers were surprised to find that prognostic genes were often shared among distinct cancer types, suggesting that similar biological programs impact survival across cancers.
They were able to identify the top 10 genes that seemed to confer adverse outcomes—FOXM1, BIRC5, TOP2A, TPX2, NME1, CCNB1, CEP55, TYMS, CENPF, and CDKN3—and the top 10 genes associated with more positive outcomes—KLRB1, ITM2B, CBX7, CD2, CREBL2, SATB1, NR3C1, TMEM66, KLRK1, and FUCA1.
Many of these genes are involved in aspects of cell division or are associated with distinct leukocytes that flood a tumor.
The researchers were also able to identify combinations of leukocytes that appear to be correlated with outcomes.
In particular, elevated numbers of plasma cells and certain types of T cells correlated with better patient survival rates across many different solid tumors. But a high proportion of granulocytes was associated with adverse outcomes.
The researchers hope that PRECOG and CIBERSORT will increase our understanding of cancer biology and aid the development of new therapies for cancer patients. The team is applying these tools to better predict which patients will respond to new and emerging anticancer therapies.
Dr Alizadeh said this is especially important given recent advances in the development of drugs that engage immune responses but work well only for a subset of cancer patients. ![]()
Antibiotic can affect INR levels

Treatment with the antibiotic dicloxacillin may cause a significant decrease in international normalized ratio (INR) levels among patients taking vitamin K antagonists (VKAs), according to research published in JAMA.
Researchers studied 7400 patients on VKA therapy and found that 61% of patients taking warfarin and dicloxacillin experienced a decrease in INR after dicloxacillin exposure.
Forty-one percent of patients taking phenprocoumon had a decrease in INR after exposure to the antibiotic.
Anton Pottegard, PhD, of the University of Southern Denmark, Odense, and his colleagues conducted this research using Thrombobase, a clinical database of all VKA-treated patients followed up by 3 outpatient clinics and 50 general practitioners in Funen, Denmark.
The researchers included all patients who filled a prescription for dicloxacillin while receiving warfarin or phenprocoumon between March 1998 and November 2012.
INR results were grouped by the week relative to dicloxacillin exposure. The last INR measurement before dicloxacillin exposure was compared with the first measurement within weeks 2 to 4 after dicloxacillin exposure.
Of the 519 patients taking warfarin and initiating treatment with dicloxacillin, 236 met inclusion criteria. The average INR level was 2.6 prior to dicloxacillin exposure and 2 at two to four weeks after dicloxacillin exposure.
In total, 61% of patients (n=144) experienced sub-therapeutic INR levels (<2.0) within 2 to 4 weeks of dicloxacillin exposure.
Among patients taking phenprocoumon (n=64), average INR levels were 2.6 before exposure to dicloxacillin and 2.3 at two to four weeks after exposure. The proportion of patients with sub-therapeutic INR levels after dicloxacillin exposure was 41% (n=26).
The researchers said these results suggest treatment with dicloxacillin can cause a significant decrease in INR levels among patients taking VKAs. ![]()
Treatment with the antibiotic dicloxacillin may cause a significant decrease in international normalized ratio (INR) levels among patients taking vitamin K antagonists (VKAs), according to research published in JAMA.
Researchers studied 7400 patients on VKA therapy and found that 61% of patients taking warfarin and dicloxacillin experienced a decrease in INR after dicloxacillin exposure.
Forty-one percent of patients taking phenprocoumon had a decrease in INR after exposure to the antibiotic.
Anton Pottegard, PhD, of the University of Southern Denmark, Odense, and his colleagues conducted this research using Thrombobase, a clinical database of all VKA-treated patients followed up by 3 outpatient clinics and 50 general practitioners in Funen, Denmark.
The researchers included all patients who filled a prescription for dicloxacillin while receiving warfarin or phenprocoumon between March 1998 and November 2012.
INR results were grouped by the week relative to dicloxacillin exposure. The last INR measurement before dicloxacillin exposure was compared with the first measurement within weeks 2 to 4 after dicloxacillin exposure.
Of the 519 patients taking warfarin and initiating treatment with dicloxacillin, 236 met inclusion criteria. The average INR level was 2.6 prior to dicloxacillin exposure and 2 at two to four weeks after dicloxacillin exposure.
In total, 61% of patients (n=144) experienced sub-therapeutic INR levels (<2.0) within 2 to 4 weeks of dicloxacillin exposure.
Among patients taking phenprocoumon (n=64), average INR levels were 2.6 before exposure to dicloxacillin and 2.3 at two to four weeks after exposure. The proportion of patients with sub-therapeutic INR levels after dicloxacillin exposure was 41% (n=26).
The researchers said these results suggest treatment with dicloxacillin can cause a significant decrease in INR levels among patients taking VKAs. ![]()
Treatment with the antibiotic dicloxacillin may cause a significant decrease in international normalized ratio (INR) levels among patients taking vitamin K antagonists (VKAs), according to research published in JAMA.
Researchers studied 7400 patients on VKA therapy and found that 61% of patients taking warfarin and dicloxacillin experienced a decrease in INR after dicloxacillin exposure.
Forty-one percent of patients taking phenprocoumon had a decrease in INR after exposure to the antibiotic.
Anton Pottegard, PhD, of the University of Southern Denmark, Odense, and his colleagues conducted this research using Thrombobase, a clinical database of all VKA-treated patients followed up by 3 outpatient clinics and 50 general practitioners in Funen, Denmark.
The researchers included all patients who filled a prescription for dicloxacillin while receiving warfarin or phenprocoumon between March 1998 and November 2012.
INR results were grouped by the week relative to dicloxacillin exposure. The last INR measurement before dicloxacillin exposure was compared with the first measurement within weeks 2 to 4 after dicloxacillin exposure.
Of the 519 patients taking warfarin and initiating treatment with dicloxacillin, 236 met inclusion criteria. The average INR level was 2.6 prior to dicloxacillin exposure and 2 at two to four weeks after dicloxacillin exposure.
In total, 61% of patients (n=144) experienced sub-therapeutic INR levels (<2.0) within 2 to 4 weeks of dicloxacillin exposure.
Among patients taking phenprocoumon (n=64), average INR levels were 2.6 before exposure to dicloxacillin and 2.3 at two to four weeks after exposure. The proportion of patients with sub-therapeutic INR levels after dicloxacillin exposure was 41% (n=26).
The researchers said these results suggest treatment with dicloxacillin can cause a significant decrease in INR levels among patients taking VKAs. ![]()
Common chemicals may increase cancer risk

Common environmental chemicals assumed to be safe at low doses may act separately or together to induce cancer development, according to research published in Carcinogenesis.
Investigators studied low-dose effects of 85 common chemicals not considered to be carcinogenic to humans, reviewing the actions of these chemicals against a long list of mechanisms that are important for cancer development.
The team compared the chemicals’ biological activity patterns to 11 known hallmarks of cancer—distinctive patterns of cellular and genetic disruption associated with early cancer development.
The chemicals included the pain reliever acetaminophen; bisphenol A (BPA), which is used in plastic food and beverage containers; rotenone, a broad-spectrum insecticide; paraquat, an agricultural herbicide; and triclosan, an antibacterial agent used in soaps and cosmetics.
The investigators learned that 50 of the 85 chemicals they analyzed can disrupt cell function in ways that correlate with known early patterns of cancer, even at the low, presumably benign levels at which most people are exposed.
For 13 of the chemicals, the team found evidence of a dose-response threshold—a level of exposure at which a chemical is considered toxic by regulators. For 22 chemicals, there was no toxicity information at all.
“Our findings also suggest these molecules may be acting in synergy to increase cancer activity,” said William Bisson, PhD, of Oregon State University in Corvallis.
“For example, EDTA, a metal-ion-binding compound used in manufacturing and medicine, interferes with the body’s repair of damaged genes. EDTA doesn’t cause genetic mutations itself, but if you’re exposed to it along with some substance that is mutagenic, it enhances the effect because it disrupts DNA repair, a key layer of cancer defense.”
Dr Bisson said the main purpose of this study was to highlight gaps in knowledge of environmentally influenced cancers and to set forth a research agenda for the next few years. He added that more research is still necessary to assess early exposure and to understand early stages of cancer development.
Traditional risk assessment, Dr Bisson said, has historically focused on a quest for single chemicals and single modes of action—approaches that may underestimate cancer risk. With this study, investigators took a different tack, examining the interplay over time of independent molecular processes triggered by low-dose exposures to chemicals.
“Cancer is a disease of diseases,” Dr Bisson said. “It follows multi-step development patterns, and, in most cases, it has a long latency period. It has to be tackled from an angle that considers the complexity of these patterns. A better understanding of what’s driving things to the point where they get uncontrollable will be key for the development of effective strategies for prevention and early detection.” ![]()
Common environmental chemicals assumed to be safe at low doses may act separately or together to induce cancer development, according to research published in Carcinogenesis.
Investigators studied low-dose effects of 85 common chemicals not considered to be carcinogenic to humans, reviewing the actions of these chemicals against a long list of mechanisms that are important for cancer development.
The team compared the chemicals’ biological activity patterns to 11 known hallmarks of cancer—distinctive patterns of cellular and genetic disruption associated with early cancer development.
The chemicals included the pain reliever acetaminophen; bisphenol A (BPA), which is used in plastic food and beverage containers; rotenone, a broad-spectrum insecticide; paraquat, an agricultural herbicide; and triclosan, an antibacterial agent used in soaps and cosmetics.
The investigators learned that 50 of the 85 chemicals they analyzed can disrupt cell function in ways that correlate with known early patterns of cancer, even at the low, presumably benign levels at which most people are exposed.
For 13 of the chemicals, the team found evidence of a dose-response threshold—a level of exposure at which a chemical is considered toxic by regulators. For 22 chemicals, there was no toxicity information at all.
“Our findings also suggest these molecules may be acting in synergy to increase cancer activity,” said William Bisson, PhD, of Oregon State University in Corvallis.
“For example, EDTA, a metal-ion-binding compound used in manufacturing and medicine, interferes with the body’s repair of damaged genes. EDTA doesn’t cause genetic mutations itself, but if you’re exposed to it along with some substance that is mutagenic, it enhances the effect because it disrupts DNA repair, a key layer of cancer defense.”
Dr Bisson said the main purpose of this study was to highlight gaps in knowledge of environmentally influenced cancers and to set forth a research agenda for the next few years. He added that more research is still necessary to assess early exposure and to understand early stages of cancer development.
Traditional risk assessment, Dr Bisson said, has historically focused on a quest for single chemicals and single modes of action—approaches that may underestimate cancer risk. With this study, investigators took a different tack, examining the interplay over time of independent molecular processes triggered by low-dose exposures to chemicals.
“Cancer is a disease of diseases,” Dr Bisson said. “It follows multi-step development patterns, and, in most cases, it has a long latency period. It has to be tackled from an angle that considers the complexity of these patterns. A better understanding of what’s driving things to the point where they get uncontrollable will be key for the development of effective strategies for prevention and early detection.” ![]()
Common environmental chemicals assumed to be safe at low doses may act separately or together to induce cancer development, according to research published in Carcinogenesis.
Investigators studied low-dose effects of 85 common chemicals not considered to be carcinogenic to humans, reviewing the actions of these chemicals against a long list of mechanisms that are important for cancer development.
The team compared the chemicals’ biological activity patterns to 11 known hallmarks of cancer—distinctive patterns of cellular and genetic disruption associated with early cancer development.
The chemicals included the pain reliever acetaminophen; bisphenol A (BPA), which is used in plastic food and beverage containers; rotenone, a broad-spectrum insecticide; paraquat, an agricultural herbicide; and triclosan, an antibacterial agent used in soaps and cosmetics.
The investigators learned that 50 of the 85 chemicals they analyzed can disrupt cell function in ways that correlate with known early patterns of cancer, even at the low, presumably benign levels at which most people are exposed.
For 13 of the chemicals, the team found evidence of a dose-response threshold—a level of exposure at which a chemical is considered toxic by regulators. For 22 chemicals, there was no toxicity information at all.
“Our findings also suggest these molecules may be acting in synergy to increase cancer activity,” said William Bisson, PhD, of Oregon State University in Corvallis.
“For example, EDTA, a metal-ion-binding compound used in manufacturing and medicine, interferes with the body’s repair of damaged genes. EDTA doesn’t cause genetic mutations itself, but if you’re exposed to it along with some substance that is mutagenic, it enhances the effect because it disrupts DNA repair, a key layer of cancer defense.”
Dr Bisson said the main purpose of this study was to highlight gaps in knowledge of environmentally influenced cancers and to set forth a research agenda for the next few years. He added that more research is still necessary to assess early exposure and to understand early stages of cancer development.
Traditional risk assessment, Dr Bisson said, has historically focused on a quest for single chemicals and single modes of action—approaches that may underestimate cancer risk. With this study, investigators took a different tack, examining the interplay over time of independent molecular processes triggered by low-dose exposures to chemicals.
“Cancer is a disease of diseases,” Dr Bisson said. “It follows multi-step development patterns, and, in most cases, it has a long latency period. It has to be tackled from an angle that considers the complexity of these patterns. A better understanding of what’s driving things to the point where they get uncontrollable will be key for the development of effective strategies for prevention and early detection.” ![]()
DLBCL tied to metabolic disruption

Researchers say they have found evidence linking disrupted metabolism and diffuse large B-cell lymphoma (DLBCL).
“The link between metabolism and cancer has been proposed or inferred to exist for a long time, but what is more scarce is evidence for a direct connection—genetic mutations in metabolic enzymes,” said Ricardo C.T. Aguiar, MD, PhD, of the University of Texas Health Science Center at San Antonio.
“We have discovered a metabolic imbalance that is oncogenic or pro-cancer.”
Dr Aguiar and his colleagues described this discovery in Nature Communications.
The team found that the gene encoding the enzyme D2-hydroxyglutarate dehydrogenase (D2HGDH) is mutated in DLBCL.
The mutated lymphoma cell displays a deficiency of a metabolite called alpha-ketoglutarate (α-KG), which is needed in steady levels for cells to be healthy.
“When the levels of α-KG are abnormally low, another class of enzymes called dioxygenases don‘t function properly, resulting in a host of additional disturbances,” Dr Aguiar said.
He added that α-KG has been identified as a critical regulator of aging and stem cell maintenance. So the implications of his group’s findings are not limited to cancer biology. ![]()
Researchers say they have found evidence linking disrupted metabolism and diffuse large B-cell lymphoma (DLBCL).
“The link between metabolism and cancer has been proposed or inferred to exist for a long time, but what is more scarce is evidence for a direct connection—genetic mutations in metabolic enzymes,” said Ricardo C.T. Aguiar, MD, PhD, of the University of Texas Health Science Center at San Antonio.
“We have discovered a metabolic imbalance that is oncogenic or pro-cancer.”
Dr Aguiar and his colleagues described this discovery in Nature Communications.
The team found that the gene encoding the enzyme D2-hydroxyglutarate dehydrogenase (D2HGDH) is mutated in DLBCL.
The mutated lymphoma cell displays a deficiency of a metabolite called alpha-ketoglutarate (α-KG), which is needed in steady levels for cells to be healthy.
“When the levels of α-KG are abnormally low, another class of enzymes called dioxygenases don‘t function properly, resulting in a host of additional disturbances,” Dr Aguiar said.
He added that α-KG has been identified as a critical regulator of aging and stem cell maintenance. So the implications of his group’s findings are not limited to cancer biology. ![]()
Researchers say they have found evidence linking disrupted metabolism and diffuse large B-cell lymphoma (DLBCL).
“The link between metabolism and cancer has been proposed or inferred to exist for a long time, but what is more scarce is evidence for a direct connection—genetic mutations in metabolic enzymes,” said Ricardo C.T. Aguiar, MD, PhD, of the University of Texas Health Science Center at San Antonio.
“We have discovered a metabolic imbalance that is oncogenic or pro-cancer.”
Dr Aguiar and his colleagues described this discovery in Nature Communications.
The team found that the gene encoding the enzyme D2-hydroxyglutarate dehydrogenase (D2HGDH) is mutated in DLBCL.
The mutated lymphoma cell displays a deficiency of a metabolite called alpha-ketoglutarate (α-KG), which is needed in steady levels for cells to be healthy.
“When the levels of α-KG are abnormally low, another class of enzymes called dioxygenases don‘t function properly, resulting in a host of additional disturbances,” Dr Aguiar said.
He added that α-KG has been identified as a critical regulator of aging and stem cell maintenance. So the implications of his group’s findings are not limited to cancer biology. ![]()
Of Mice and Men
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
Handoffs From ED to Inpatient Care
Handoffs are the exchange of information between health professionals that accompany the transfer of patient‐care responsibility.[1] Poor handoff practices are associated with unsafe and inefficient care.[2, 3, 4] Teaching hospitals are especially at risk, as resident work‐hour restrictions have increased the number of handoffs.[5] Accreditation agencies now require that hospitals and residency programs have structured handoff processes[6, 7] and that medical students[8] and residents[9, 10, 11, 12] demonstrate competency in handoffs.
Physician handoff research has primarily focused on handoffs within a service or discipline. These within‐unit handoffs should be differentiated from interunit handoffs.[13, 14] Interunit handoffs, such as the transition from the emergency department (ED) to inpatient setting, are subject to unique challenges. The ED admission process involves changes in personnel, provider specialty, and location.[15] The transition occurs when the patient's clinical trajectory is uncertain, treatments are being initiated, and test results are pending. Other barriers include interdisciplinary cultural differences, interphysician conflict, unstructured communication, environmental factors, and complex care coordination.[13, 14, 15, 16, 17, 18] Despite these challenges, there is relatively little research specifically examining ED to inpatient handoffs, and most of what is available has focused on individual services within an institution.[13, 14, 15, 18, 19, 20, 21, 22, 23, 24, 25]
As part of an institutional effort to improve our ED admission handoff practices, we conducted a cross‐sectional, survey‐based needs‐assessment involving emergency medicine (EM) and 5 inpatient medical services. The objective of this study was to determine physicians' perceptions of the ED admission handoff process and to identify potential barriers to safe patient care.
METHODS
Survey Design
A study group comprised of resident and faculty physicians in internal medicine (IM) and EM, as well as a healthcare communication expert, designed analogous cross‐sectional surveys to determine the perceptions of admitting (see Supporting Information, Appendix 1, in the online version of this article) and EM (see Supporting Information, Appendix 2, in the online version of this article) physicians toward the admission handoff process. Using an iterative process to ensure content validity, we created questions in 6 domains based on the expert opinion of the authors and emergent themes identified in the literature.[15, 19, 22, 24] These themes were general communication quality, clinical information, interpersonal perceptions, responsibilities, organizational factors, and patient safety. We asked respondents to report their answers using 5‐point Likert and Likert‐like scales. Questions regarding frequency were assigned semiquantitative values: rarely=0% to 24%, sometimes=25% to 49%, often=50% to 74%, very often 75% to 99%, and always=100%. We also asked an open‐ended question, asking respondents to describe any handoff‐related adverse events (defined as patient harm or near miss) they encountered in the past 3 months. We pilot tested the survey for clarity and relevance prior to distribution on a group of 5 physicians from the participating services. The institutional internal review board approved the protocol (#046‐13‐EX).
Setting, Participants, and Recruitment
We conducted the study at a 627‐bed tertiary care academic medical center. Eligible participants included all resident, fellow, and faculty physicians directly involved in admission handoffs from EM and 5 medical inpatient services (university‐based IM, university‐based family medicine [FM], community‐based FM, cardiology, and critical care medicine). The admitting services accounted for two‐thirds of the institution's 10,000 annual adult, nonobstetric ED admissions. Physicians who had not participated in admission handoffs in the past 3 months were excluded.
At the time of the study, there was no standardized institutional process for admission handoff communication, nor was there policy delineating when patient‐care responsibility transferred from the EM to admitting physician. The admission handoff process generally relied on verbal handoff via telephone between EM and admitting physicians. All services used the same electronic health record, but there was no written handoff note, and EM physician documentation generally was not available at the time of handoff. To determine patient assignment schemes following admission handoff, we questioned leadership from the participating admitting services.
We distributed and collected anonymous hard‐copy surveys at educational conferences in March 2013. We emailed a link to an online survey to eligible participants who could not be reached in person. Subjects voluntarily participated and provided consent via cover letter.
Data Analysis
We compiled survey data and performed descriptive analysis. We assessed the internal consistency of the survey domains that were made up of at least 3 questions using Cronbach's . To compare the distribution of aggregate admitting service responses to EM responses, we used the Mann‐Whitney test. We used the Fisher exact test to examine the associations of dichotomized responses (<50% vs 50%) to the level of training (intern vs resident vs fellow/faculty) and to the admitting service affiliation (university‐based IM vs university‐based FM vs aggregate of other services). When indicated, we made pairwise comparisons using the Bonferroni method to compute adjusted P values. We analyzed data independently using both SPSS version 20 (IBM Corp., Armonk, NY) and SAS version 9.3 (SAS Inc., Cary, NC) software and considered a P value <0.05 to be significant. Three researchers independently categorized descriptions of adverse events based on a previously published qualitative analysis,[15] with disagreements settled by consensus.
RESULTS
After applying exclusion criteria, the survey response rate was 63% for admitting physicians (94/150) and 86% for EM physicians (32/37). Participants' service affiliation and level of training are shown in the Table 1. Table 2 provides the distribution of survey responses for EM and admitting physicians.
| Service Affiliation | Level of Training | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PGY1 | PGY2 | PGY3 | Fellow | Staff | |||||||
| No. | % | No. | % | No. | % | No. | % | No. | % | No. | |
| |||||||||||
| Admitting services | |||||||||||
| University‐based IM | 12 | 32.4 | 7 | 18.9 | 5 | 13.5 | 1 | 2.7 | 12 | 32.4 | 37 |
| University‐based FM | 15 | 44.1 | 13 | 38.2 | 5 | 14.7 | 1 | 2.9 | 0 | 0 | 34 |
| Community‐based FM | 5 | 50.0 | 1 | 10.0 | 3 | 30.0 | 0 | 0 | 1 | 10.0 | 10 |
| Critical care medicine | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 100.0 | 0 | 0 | 6 |
| Cardiology | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 100.0 | 0 | 0 | 7 |
| Admitting services total | 32 | 34.0 | 21 | 22.3 | 13 | 13.8 | 15 | 16.0 | 13 | 13.8 | 94 |
| Emergency medicine | 6 | 18.8 | 8 | 25.0 | 5 | 15.6 | 0 | 0 | 13 | 40.6 | 32 |
| Question | Service | Very Poor | Poor | Fair | Good | Very Good | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | No. | % | No. | % | ||
| Rarely (0%24%) | Sometimes (25%49%) | Often (50%74%) | Very Often (75%99%) | Always (100%) | |||||||
| |||||||||||
| Generally, the quality of communication between EM and admitting physicians is: | Admitting | 0 | 0 | 8 | 8.6% | 37 | 39.7% | 46 | 49.4% | 2 | 2.1% |
| EM | 0 | 0 | 2 | 6.2% | 4 | 12.5% | 20 | 62.5% | 6 | 18.7% | |
| The current handoff system's ability to ensure patient safety is generally: | Admitting | 1 | 1.0% | 10 | 10.7% | 43 | 46.2% | 37 | 39.7% | 2 | 2.1% |
| EM | 1 | 3.1% | 1 | 3.1% | 11 | 34.3% | 15 | 46.8% | 4 | 12.5% | |
| The current handoff system's ability to ensure efficient patient care is generally: | Admitting | 3 | 3.2% | 20 | 21.7% | 31 | 33.6% | 36 | 39.1% | 2 | 2.1% |
| EM | 2 | 6.2% | 5 | 15.6% | 15 | 46.8% | 10 | 31.2% | 0 | ||
| During handoff, how often does the EM physician provide the following information to the admitting service? | |||||||||||
| The working diagnosis of the EM physician | Admitting | 5 | 5.4% | 19 | 20.6% | 30 | 32.6% | 30 | 32.6% | 8 | 8.6% |
| EM | 0 | 4 | 12.5% | 0 | 12 | 37.5% | 16 | 50.0% | |||
| Relevant past medical/surgical history | Admitting | 5 | 5.4% | 25 | 27.1% | 40 | 43.4% | 18 | 19.5% | 4 | 4.3% |
| EM | 1 | 3.1% | 2 | 6.2% | 5 | 15.6% | 17 | 53.1% | 7 | 21.8% | |
| Relevant physical exam findings (including abnormal vital signs) | Admitting | 3 | 3.2% | 25 | 27.1% | 41 | 44.5% | 21 | 22.8% | 2 | 2.1% |
| EM | 0 | 5 | 15.6% | 2 | 6.2% | 15 | 46.8% | 10 | 31.2% | ||
| Results of relevant diagnostic studies (labs, imaging) | Admitting | 2 | 2.1% | 10 | 10.8% | 39 | 42.3% | 37 | 40.2% | 4 | 4.3% |
| EM | 1 | 3.1% | 0 | 3 | 9.3% | 14 | 43.7% | 14 | 43.7% | ||
| Procedures and therapeutic interventions initiated while in the ED | Admitting | 3 | 3.2% | 20 | 21.7% | 34 | 36.9% | 29 | 31.5% | 6 | 6.5% |
| EM | 1 | 3.1% | 0 | 3 | 9.3% | 18 | 56.2% | 10 | 31.2% | ||
| Trend in the patient's clinical condition while in the ED | Admitting | 12 | 13.1% | 27 | 29.6% | 33 | 36.2% | 17 | 18.6% | 2 | 2.1% |
| EM | 4 | 12.5% | 1 | 3.1% | 5 | 15.6% | 13 | 40.6% | 9 | 28.1% | |
| Current clinical condition of the patient (at time of handoff) | Admitting | 3 | 3.2% | 24 | 26.0% | 41 | 44.5% | 18 | 19.5% | 6 | 6.5% |
| EM | 1 | 3.1% | 1 | 3.1% | 3 | 9.3% | 13 | 40.6% | 14 | 43.7% | |
| Pending diagnostic studies (labs, imaging), if ordered | Admitting | 12 | 13.0% | 32 | 34.7% | 29 | 31.5% | 17 | 18.4% | 2 | 2.1% |
| EM | 0 | 5 | 15.6% | 6 | 18.7% | 14 | 43.7% | 7 | 21.8% | ||
| During handoff, how often are clinical questions asked about the patient being admitted? | Admitting | 2 | 2.1% | 1 | 1.0% | 13 | 14.1% | 29 | 31.5% | 47 | 51.0% |
| EM | 0 | 0 | 5 | 15.6% | 8 | 25.0% | 13 | 40.6% | 6 | 18.7% | |
| In general, how often do you agree with the clinical decisions made by the EM physician? | Admitting | 1 | 1.0% | 26 | 27.9% | 56 | 60.2% | 10 | 10.7% | 0 | 0 |
| Generally, how often do you feel you have to defend your clinical decisions to the admitting service? | EM | 2 | 6.2% | 15 | 46.8% | 5 | 15.6% | 10 | 31.2% | 0 | 0 |
| How often do you have clinically meaningful face‐to‐face communication with the EM/admitting physician about the patient being admitted? | Admitting | 24 | 25.8% | 38 | 40.8% | 22 | 23.6% | 8 | 8.6% | 1 | 1.0% |
| EM | 14 | 43.7% | 13 | 40.6% | 4 | 12.5% | 1 | 3.1% | 0 | ||
| On average, how often do competing clinical responsibilities distract you during handoff? | Admitting | 6 | 6.5% | 34 | 36.9% | 29 | 31.5% | 20 | 21.7% | 3 | 3.2% |
| EM | 7 | 21.8% | 8 | 25.0% | 9 | 28.1% | 8 | 25.0% | 0 | 0 | |
| On average, how often do environmental factors distract you during handoff? | Admitting | 44 | 48.3% | 31 | 34.0% | 10 | 10.9% | 6 | 6.5% | 0 | 0 |
| EM | 7 | 21.8% | 11 | 34.3% | 8 | 25.0% | 4 | 12.5% | 2 | 6.2% | |
The processes for assigning responsibilities following the initial handoff differed between admitting services, and within a service the process was often dynamic. For example, within the university‐based IM and community‐based FM services, the assignment process varied depending on timing (day vs night, weekday vs weekend). For the critical care medicine and cardiology services, fellows accepted admission handoff calls, and depending on competing clinical responsibilities and the patient's stability, either evaluated the patient independently or sent a resident to perform a preliminary evaluation. We reviewed and classified these varied admission assignment strategies into 4 general schemes (Figure 1). All 5 admitting services relied partly or entirely on housestaff for receiving admission handoffs, as did the EM service.
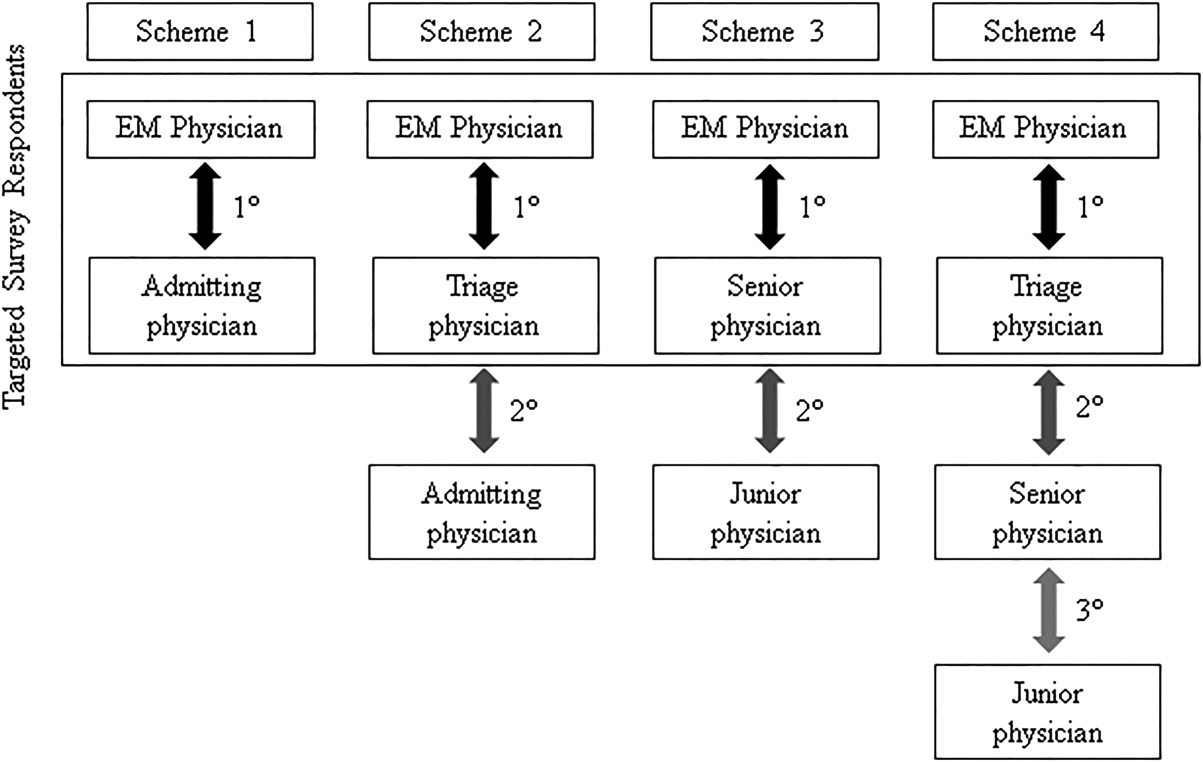
Communication Quality and Content
Cronbach's was 0.72 for general handoff questions and 0.89 for clinical information questions. Compared with EM respondents, admitting physicians reported worse quality of communication (P < 0.001) and less confidence in the handoff system's ability to ensure patient safety (P=0.04). Admitting physicians reported communication of clinical information occurred less frequently than EM physicians for all 8 content areas (P < 0.001 for all). There were no significant differences in responses between various levels of training and service affiliations.
Interpersonal Perceptions
EM respondents reported admitting physicians asked clinical questions less frequently than did admitting respondents (P < 0.001). Ninety‐four percent of EM physicians (n=30) felt they had to defend their clinical decisions at least sometimes. EM interns (P=0.009) and faculty (P=0.01) were more likely than residents to report feeling defensive. Most admitting physicians (60%, n=56) often agreed with decisions made by the EM provider, but 29% (n=27) agreed less than half the time. One‐third of admitting (n=31) and 16% of EM physicians (n=5) reported routine (ie, >50% of admissions) meaningful face‐to‐face communication with one another at the time of admission.
Responsibilities
When asked who was primarily responsible for patients boarding in the ED, defined as nonemergent patient care that occurs after handoff, but before a patient is physically transferred from the ED, 37.6% (n=47) of respondents answered the admitting physician, 21.6% (n=27) answered the EM physician, 34.4% (n=43) answered both, and 6.4% (n=8) answered don't know. Responses were similar for EM and admitting physicians.
Organizational Factors
Fifty‐six percent of all respondents (n=69) reported they were distracted during handoffs by competing clinical duties 50% of the time. Environmental factors, such as noise, more commonly distracted EM physicians (P=0.001). Approximately 60% (n=56) of admitting physicians reported using a triage system to distribute admissions, with a resultant 57% (n=32) reporting sequential handoffs (ie, handoffs of handoffs) occurred at least sometimes. About 80% of EM physicians (n=23) reported that shift change led to sequential handoffs at least sometimes. Seventy‐eight percent (n=67) of physicians felt sequential handoffs had a negative impact on patient care.
Patient Safety
Thirty‐four percent of admitting (n=30) and 19% of EM physicians (n=6) reported a patient was harmed or suffered a near miss in the past 3 months because of an ineffective handoff, with 58% (n=21) reporting 2 examples. Twenty‐four respondents described 29 adverse events. Respondents described perceived mistakes in diagnosis (n=11), treatment (n=16), and disposition (n=12), with some examples falling into more than 1 category. Absent or ineffective communication contributed to 27 of 29 examples. Other commonly cited areas of vulnerability included uncertain assignment of responsibility, sequential handoffs, and patient boarding.
DISCUSSION
Based upon physician self‐reporting, we identified perceived barriers to safe ED admission handoff across several domains. This study adds to the literature, as it provides a cross‐section of multiple inpatient services with varying admission schemes to underscore the complexities facing hospitals in safely transitioning patients between units. As noted in previous studies, one‐third of physicians reported a handoff‐related adverse event,[15] and there was significant disagreement between handoff participants about communication of critical information.[21, 26] These differences in perceptions suggest a failure of physicians to accurately transfer information to create a shared understanding of patient care,[21] which is the central function of handoffs.
EM physicians frequently felt that admitting physicians did not trust their clinical decisions, a perception supported by the fact that over 25% of admitting respondents' usually disagreed with decisions in the ED. Interdisciplinary trust is central in negotiating a shared plan of care[13] and mitigating conflict to ensure a safe transition of patient care.[16] Handoffs are complex social interactions, and feelings of defensiveness and mistrust are likely exacerbated by in‐group/out‐group biases,[15] conflicting information expectations,[19] and discordant ways of interpreting and framing handoff interactions.[13] Interestingly, EM residents were less likely than interns or faculty to report feeling defensive. This may be in part because residents from EM and admitting services develop relationships during interdisciplinary rotations, which may help facilitate future handoff interactions.[27] The fact that EM respondents felt defensive, despite reporting less‐frequent questioning than admitting physicians, suggests that tone and content of questions played an important role. These findings support the importance of interdisciplinary education and standardization of handoff communication between ED and admitting physicians.[23] Beach and colleagues have recommended a conceptual framework for interunit handoffs between EM and hospital physicians, but further research is needed to measure its impact in real‐world settings.[14]
We also found great variability in admitting services' processes for assigning patient‐care responsibility following the initial handoff. Even within an individual service, these processes were often dynamic and relied on physicians at different levels of training. This has several potential consequences. First, it may be difficult for physicians engaged in a handoff to know the level of experience and expertise of one another. These contextual variables play an important role in how handoff information is conveyed, as less experienced clinicians may require explicit information that a more experienced provider may infer.[1, 21] Second, the variability in admission assignment processes may further exacerbate uncertainty regarding responsibility for patients boarding in the ED, making it increasingly difficult for nurses and ancillary staff to know which physician is ultimately responsible for patient care. Finally, the diversity of admission schemes may complicate the development of standardized interunit handoff protocols, policy, and education.
A related finding was that sequential handoffs were common within both EM and admitting services. EM shift handoffs have their own set of barriers,[28] which can lead to ineffective communication.[29] Likewise, about two‐thirds of admitting respondents reported using an admission triage system. The goal of such systems is to simplify complex call schedules and diverse patient assignment schemes within admitting services, thus streamlining the admission process. These systems may also allow for more consistency in the quality of handoff communication through the creation of triage specialists. These potential advantages need to be weighed against the increased risk of communication breakdown. The introduction of sequential handoffs creates a game of telephone, in which there is no direct communication between the first and final caregivers (Figure 1), allowing misinformation to be propagated forward.[30] Sequential handoffs contributed to several reported adverse events, and the majority of surveyed physicians felt they negatively impacted patient care. Further research is necessary to determine the impact of centralized triage systems and to explore strategies to mitigate information decay that results from sequential handoffs, as quality‐improvement interventions may be of limited benefit if downstream communication remains ineffective. Potential strategies may include standardizing sequential handoff communication, leveraging centralized handoff notes within electronic health records, or developing handoff systems that ensure direct communication between the EM physician and the ultimate admitting provider.
Limitations
This was a single‐institution study, so results may not be generalizable, as handoff processes vary among hospitals.[24] Our study relied on a novel survey instrument, for which validity and reliability are uncertain, although internal consistency was good for domains that could be tested (Cronbach's 0.720.89). As with other survey‐based studies, participant selection, hindsight, recall, and response biases may have influenced the results. We attempted to minimize these risks by pilot testing the survey, targeting a relatively large number of respondents across multiple services, and by making efforts to maximize the response rate by contacting eligible participants both in person and via email. Because results reflect self‐reported perceptions, we cannot prove that the factors studied are actually associated with adverse outcomes, nor can we quantify their relative importance. Nevertheless, the reported perceptions raise concerns that warrant further study.
FUTURE DIRECTIONS
Further research is needed to examine interventions that may improve clinically relevant outcomes. Development of structured admission handoff protocols should be collaborative[31] and focus on clinical judgment, rather than rote recitation of data.[14] Based on our study findings, we are pilot testing a standardized approach for ED‐to‐hospital handoffs, and portions of this survey will be repeated in the postintervention assessment.
At our institution, housestaff at all levels of training regularly participated in the handoff process. The Accreditation Council for Graduate Medical Education requires that residents demonstrate competence in performing handoffs,[7] yet handoff training and assessment are inconsistent,[23, 32, 33] and published interventions have focused primarily on within‐unit handoffs.[34, 35, 36] Additional training should focus on the unique aspects of interunit handoffs. Approaches could include interprofessional communication training, simulation training, and enhanced assessment methods. Additionally, increasing face‐to‐face communication, perhaps as part of bedside handoffs, could improve relationships and the development of a shared mental model of patient care. More direct involvement by attending physicians will also be important, as there is evidence that such oversight may improve training[36] and safety,[37] as more experienced physicians better integrate handoff information.[21]
CONCLUSION
We identified several perceived barriers to safe interunit handoff from the ED to the inpatient setting. Handoff‐related adverse events, a pattern of conflicting physician perceptions, and frequent sequential handoffs were of particular concern. Our findings support the need for collaborative efforts to improve interdisciplinary communication.
Disclosure
Nothing to report.
- , . Handoffs in hospitals: a review of the literature on information exchange while transferring patient responsibility or control. Available at: http://deepblue.lib.umich.edu/handle/2027.42/61498. Updated 2009. Accessed May 15, 2014.
- . Handoffs causing patient harm: a survey of medical and surgical house staff. Jt Comm J Qual Patient Saf. 2008;34(10):563–570.
- . Consequences of inadequate sign‐out for patient care. Arch Intern Med. 2008;168(16):1755–1760.
- , . A systematic review of failures in handoff communication during intrahospital transfers. Jt Comm J Qual Patient Saf. 2011;37(6):274–284.
- , , , , . Managing discontinuity in academic medical centers: strategies for a safe and effective resident sign‐out. J Hosp Med. 2006;1(4):257–266.
- , . A model for building a standardized hand‐off protocol. Jt Comm J Qual Patient Saf. 2006;32(11):646–655.
- Accreditation Council for Graduate Medical Education. ACGME common program requirements. Available at: https://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/CPRs2013.pdf. Updated 2013. Accessed May 7, 2014.
- Association of American Medical Colleges. Core entrustable professional activities for entering residency. Available at: https://members.aamc.org/eweb/upload/Core%20EPA%20Faculty%20and%20Learner%20Guide.pdf. Updated 2014. Accessed July 7, 2014.
- Accreditation Council for Graduate Medical Education and American Board of Internal Medicine. The internal medicine milestones. Available at: http://www.acgme.org/acgmeweb/portals/0/pdfs/milestones/internalmedicinemilestones.pdf. Updated 2012. Accessed December 23, 2013.
- Accreditation Council for Graduate Medical Education and American Board of Emergency Medicine. The emergency medicine milestones. Available at: https://www.abem.org/public/docs/default‐source/migrated‐documents‐and‐files/em‐milestones.pdf?sfvrsn=4. Updated 2012. Accessed December 23, 2013.
- Accreditation Council for Graduate Medical Education and American Board of Family Medicine. The family medicine milestone project. Available at: http://www.acgme.org/acgmeweb/Portals/0/PDFs/Milestones/FamilyMedicineMilestones.pdf. Updated 2013. Accessed October 31, 2014.
- Accreditation Council for Graduate Medical Education and American Board of Pediatrics. The pediatrics milestone project. Available at: http://acgme.org/acgmeweb/Portals/0/PDFs/Milestones/PediatricsMilestones.pdf. Updated 2013. Accessed October, 31, 2014.
- , . The unappreciated challenges of between‐unit handoffs: negotiating and coordinating across boundaries. Ann Emerg Med. 2013;61(2):155–160.
- , , , et al. Improving interunit transitions of care between emergency physicians and hospital medicine physicians: a conceptual approach. Acad Emerg Med. 2012;19(10):1188–1195.
- , , , , , . Dropping the baton: a qualitative analysis of failures during the transition from emergency department to inpatient care. Ann Emerg Med. 2009;53(6):701–710.e4.
- , , , . Conflict prevention, conflict mitigation, and manifestations of conflict during emergency department consultations. Acad Emerg Med. 2014;21(3):308–313.
- , , , . I'm clear, you're clear, we're all clear: improving consultation communication skills in undergraduate medical education. Acad Med. 2013;88(6):753–758.
- , , , . Emergency physician to admitting physician handovers: an exploratory study. Proc Hum Factors Ergon Soc Annu Meet. 2002;46(16):1511–1515.
- , , . Communicating in the “gray zone”: perceptions about emergency physician hospitalist handoffs and patient safety. Acad Emerg Med. 2007;14(10):884–894.
- , . Chart biopsy: an emerging medical practice enabled by electronic health records and its impacts on emergency department‐inpatient admission handoffs. J Am Med Inform Assoc. 2013;20(2):260–267.
- , , , , . Admission handoff communications: clinician's shared understanding of patient severity of illness and problems. J Patient Saf. 2009;5(4):237–242.
- , , , et al. Exploring emergency physician‐hospitalist handoff interactions: development of the handoff communication assessment. Ann Emerg Med. 2010;55(2):161–170.
- , , , , , . Interunit handoffs of patients and transfers of information: a survey of current practices. Ann Emerg Med. 2014;64(4):343–349.e5.
- , , , et al. A conceptual framework for studying the safety of transitions in emergency care. In: Henriksen K, Battles JB, Marks ES, Lewin DI, eds. Advances in Patient Safety: From Research to Implementation. Vol. 2: Concepts and Methodology. Rockville, MD: Agency for Healthcare Research and Quality; 2005:309–321.
- , , , , , . Patient care transitions from the emergency department to the medicine ward: evaluation of a standardized electronic signout tool. Int J Qual Health Care. 2014;26(4):337–347.
- , , , , . Interns overestimate the effectiveness of their hand‐off communication. Pediatrics. 2010;125(3):491–496.
- , , , . Understanding the impact of residents' interpersonal relationships during emergency department referrals and consultations. J Grad Med Educ. 2013;5(4):576–581.
- , , , et al. Improving handoffs in the emergency department. Ann Emerg Med. 2010;55(2):171–180.
- , , . ED handoffs: observed practices and communication errors. Am J Emerg Med. 2011;29(5):502–511.
- , , , , . Characterizing information decay in patient handoffs. J Surg Educ. 2014;71(4):480–485.
- , , . Emergency medicine and hospital medicine: a call for collaboration. J Emerg Med. 2012;43(2):328–334.
- , , , et al. A survey of handoff practices in emergency medicine. Am J Med Qual. 2014;29(5):408–414.
- . Transfers of patient care between house staff on internal medicine wards: a national survey. Arch Intern Med. 2006;166(11):1173–1177.
- , , , , , . Effect of a systems intervention on the quality and safety of patient handoffs in an internal medicine residency program. J Gen Intern Med. 2013;28(8):986–993.
- , , , et al. Rates of medical errors and preventable adverse events among hospitalized children following implementation of a resident handoff bundle. JAMA. 2013;310(21):2262–2270.
- , , , et al. A structured handoff program for interns. Acad Med. 2009;84(3):347–352.
- , , , et al. Experience with faculty supervision of an electronic resident sign‐out system. Am J Med. 2010;123(4):376–381.
Handoffs are the exchange of information between health professionals that accompany the transfer of patient‐care responsibility.[1] Poor handoff practices are associated with unsafe and inefficient care.[2, 3, 4] Teaching hospitals are especially at risk, as resident work‐hour restrictions have increased the number of handoffs.[5] Accreditation agencies now require that hospitals and residency programs have structured handoff processes[6, 7] and that medical students[8] and residents[9, 10, 11, 12] demonstrate competency in handoffs.
Physician handoff research has primarily focused on handoffs within a service or discipline. These within‐unit handoffs should be differentiated from interunit handoffs.[13, 14] Interunit handoffs, such as the transition from the emergency department (ED) to inpatient setting, are subject to unique challenges. The ED admission process involves changes in personnel, provider specialty, and location.[15] The transition occurs when the patient's clinical trajectory is uncertain, treatments are being initiated, and test results are pending. Other barriers include interdisciplinary cultural differences, interphysician conflict, unstructured communication, environmental factors, and complex care coordination.[13, 14, 15, 16, 17, 18] Despite these challenges, there is relatively little research specifically examining ED to inpatient handoffs, and most of what is available has focused on individual services within an institution.[13, 14, 15, 18, 19, 20, 21, 22, 23, 24, 25]
As part of an institutional effort to improve our ED admission handoff practices, we conducted a cross‐sectional, survey‐based needs‐assessment involving emergency medicine (EM) and 5 inpatient medical services. The objective of this study was to determine physicians' perceptions of the ED admission handoff process and to identify potential barriers to safe patient care.
METHODS
Survey Design
A study group comprised of resident and faculty physicians in internal medicine (IM) and EM, as well as a healthcare communication expert, designed analogous cross‐sectional surveys to determine the perceptions of admitting (see Supporting Information, Appendix 1, in the online version of this article) and EM (see Supporting Information, Appendix 2, in the online version of this article) physicians toward the admission handoff process. Using an iterative process to ensure content validity, we created questions in 6 domains based on the expert opinion of the authors and emergent themes identified in the literature.[15, 19, 22, 24] These themes were general communication quality, clinical information, interpersonal perceptions, responsibilities, organizational factors, and patient safety. We asked respondents to report their answers using 5‐point Likert and Likert‐like scales. Questions regarding frequency were assigned semiquantitative values: rarely=0% to 24%, sometimes=25% to 49%, often=50% to 74%, very often 75% to 99%, and always=100%. We also asked an open‐ended question, asking respondents to describe any handoff‐related adverse events (defined as patient harm or near miss) they encountered in the past 3 months. We pilot tested the survey for clarity and relevance prior to distribution on a group of 5 physicians from the participating services. The institutional internal review board approved the protocol (#046‐13‐EX).
Setting, Participants, and Recruitment
We conducted the study at a 627‐bed tertiary care academic medical center. Eligible participants included all resident, fellow, and faculty physicians directly involved in admission handoffs from EM and 5 medical inpatient services (university‐based IM, university‐based family medicine [FM], community‐based FM, cardiology, and critical care medicine). The admitting services accounted for two‐thirds of the institution's 10,000 annual adult, nonobstetric ED admissions. Physicians who had not participated in admission handoffs in the past 3 months were excluded.
At the time of the study, there was no standardized institutional process for admission handoff communication, nor was there policy delineating when patient‐care responsibility transferred from the EM to admitting physician. The admission handoff process generally relied on verbal handoff via telephone between EM and admitting physicians. All services used the same electronic health record, but there was no written handoff note, and EM physician documentation generally was not available at the time of handoff. To determine patient assignment schemes following admission handoff, we questioned leadership from the participating admitting services.
We distributed and collected anonymous hard‐copy surveys at educational conferences in March 2013. We emailed a link to an online survey to eligible participants who could not be reached in person. Subjects voluntarily participated and provided consent via cover letter.
Data Analysis
We compiled survey data and performed descriptive analysis. We assessed the internal consistency of the survey domains that were made up of at least 3 questions using Cronbach's . To compare the distribution of aggregate admitting service responses to EM responses, we used the Mann‐Whitney test. We used the Fisher exact test to examine the associations of dichotomized responses (<50% vs 50%) to the level of training (intern vs resident vs fellow/faculty) and to the admitting service affiliation (university‐based IM vs university‐based FM vs aggregate of other services). When indicated, we made pairwise comparisons using the Bonferroni method to compute adjusted P values. We analyzed data independently using both SPSS version 20 (IBM Corp., Armonk, NY) and SAS version 9.3 (SAS Inc., Cary, NC) software and considered a P value <0.05 to be significant. Three researchers independently categorized descriptions of adverse events based on a previously published qualitative analysis,[15] with disagreements settled by consensus.
RESULTS
After applying exclusion criteria, the survey response rate was 63% for admitting physicians (94/150) and 86% for EM physicians (32/37). Participants' service affiliation and level of training are shown in the Table 1. Table 2 provides the distribution of survey responses for EM and admitting physicians.
| Service Affiliation | Level of Training | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PGY1 | PGY2 | PGY3 | Fellow | Staff | |||||||
| No. | % | No. | % | No. | % | No. | % | No. | % | No. | |
| |||||||||||
| Admitting services | |||||||||||
| University‐based IM | 12 | 32.4 | 7 | 18.9 | 5 | 13.5 | 1 | 2.7 | 12 | 32.4 | 37 |
| University‐based FM | 15 | 44.1 | 13 | 38.2 | 5 | 14.7 | 1 | 2.9 | 0 | 0 | 34 |
| Community‐based FM | 5 | 50.0 | 1 | 10.0 | 3 | 30.0 | 0 | 0 | 1 | 10.0 | 10 |
| Critical care medicine | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 100.0 | 0 | 0 | 6 |
| Cardiology | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 100.0 | 0 | 0 | 7 |
| Admitting services total | 32 | 34.0 | 21 | 22.3 | 13 | 13.8 | 15 | 16.0 | 13 | 13.8 | 94 |
| Emergency medicine | 6 | 18.8 | 8 | 25.0 | 5 | 15.6 | 0 | 0 | 13 | 40.6 | 32 |
| Question | Service | Very Poor | Poor | Fair | Good | Very Good | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | No. | % | No. | % | ||
| Rarely (0%24%) | Sometimes (25%49%) | Often (50%74%) | Very Often (75%99%) | Always (100%) | |||||||
| |||||||||||
| Generally, the quality of communication between EM and admitting physicians is: | Admitting | 0 | 0 | 8 | 8.6% | 37 | 39.7% | 46 | 49.4% | 2 | 2.1% |
| EM | 0 | 0 | 2 | 6.2% | 4 | 12.5% | 20 | 62.5% | 6 | 18.7% | |
| The current handoff system's ability to ensure patient safety is generally: | Admitting | 1 | 1.0% | 10 | 10.7% | 43 | 46.2% | 37 | 39.7% | 2 | 2.1% |
| EM | 1 | 3.1% | 1 | 3.1% | 11 | 34.3% | 15 | 46.8% | 4 | 12.5% | |
| The current handoff system's ability to ensure efficient patient care is generally: | Admitting | 3 | 3.2% | 20 | 21.7% | 31 | 33.6% | 36 | 39.1% | 2 | 2.1% |
| EM | 2 | 6.2% | 5 | 15.6% | 15 | 46.8% | 10 | 31.2% | 0 | ||
| During handoff, how often does the EM physician provide the following information to the admitting service? | |||||||||||
| The working diagnosis of the EM physician | Admitting | 5 | 5.4% | 19 | 20.6% | 30 | 32.6% | 30 | 32.6% | 8 | 8.6% |
| EM | 0 | 4 | 12.5% | 0 | 12 | 37.5% | 16 | 50.0% | |||
| Relevant past medical/surgical history | Admitting | 5 | 5.4% | 25 | 27.1% | 40 | 43.4% | 18 | 19.5% | 4 | 4.3% |
| EM | 1 | 3.1% | 2 | 6.2% | 5 | 15.6% | 17 | 53.1% | 7 | 21.8% | |
| Relevant physical exam findings (including abnormal vital signs) | Admitting | 3 | 3.2% | 25 | 27.1% | 41 | 44.5% | 21 | 22.8% | 2 | 2.1% |
| EM | 0 | 5 | 15.6% | 2 | 6.2% | 15 | 46.8% | 10 | 31.2% | ||
| Results of relevant diagnostic studies (labs, imaging) | Admitting | 2 | 2.1% | 10 | 10.8% | 39 | 42.3% | 37 | 40.2% | 4 | 4.3% |
| EM | 1 | 3.1% | 0 | 3 | 9.3% | 14 | 43.7% | 14 | 43.7% | ||
| Procedures and therapeutic interventions initiated while in the ED | Admitting | 3 | 3.2% | 20 | 21.7% | 34 | 36.9% | 29 | 31.5% | 6 | 6.5% |
| EM | 1 | 3.1% | 0 | 3 | 9.3% | 18 | 56.2% | 10 | 31.2% | ||
| Trend in the patient's clinical condition while in the ED | Admitting | 12 | 13.1% | 27 | 29.6% | 33 | 36.2% | 17 | 18.6% | 2 | 2.1% |
| EM | 4 | 12.5% | 1 | 3.1% | 5 | 15.6% | 13 | 40.6% | 9 | 28.1% | |
| Current clinical condition of the patient (at time of handoff) | Admitting | 3 | 3.2% | 24 | 26.0% | 41 | 44.5% | 18 | 19.5% | 6 | 6.5% |
| EM | 1 | 3.1% | 1 | 3.1% | 3 | 9.3% | 13 | 40.6% | 14 | 43.7% | |
| Pending diagnostic studies (labs, imaging), if ordered | Admitting | 12 | 13.0% | 32 | 34.7% | 29 | 31.5% | 17 | 18.4% | 2 | 2.1% |
| EM | 0 | 5 | 15.6% | 6 | 18.7% | 14 | 43.7% | 7 | 21.8% | ||
| During handoff, how often are clinical questions asked about the patient being admitted? | Admitting | 2 | 2.1% | 1 | 1.0% | 13 | 14.1% | 29 | 31.5% | 47 | 51.0% |
| EM | 0 | 0 | 5 | 15.6% | 8 | 25.0% | 13 | 40.6% | 6 | 18.7% | |
| In general, how often do you agree with the clinical decisions made by the EM physician? | Admitting | 1 | 1.0% | 26 | 27.9% | 56 | 60.2% | 10 | 10.7% | 0 | 0 |
| Generally, how often do you feel you have to defend your clinical decisions to the admitting service? | EM | 2 | 6.2% | 15 | 46.8% | 5 | 15.6% | 10 | 31.2% | 0 | 0 |
| How often do you have clinically meaningful face‐to‐face communication with the EM/admitting physician about the patient being admitted? | Admitting | 24 | 25.8% | 38 | 40.8% | 22 | 23.6% | 8 | 8.6% | 1 | 1.0% |
| EM | 14 | 43.7% | 13 | 40.6% | 4 | 12.5% | 1 | 3.1% | 0 | ||
| On average, how often do competing clinical responsibilities distract you during handoff? | Admitting | 6 | 6.5% | 34 | 36.9% | 29 | 31.5% | 20 | 21.7% | 3 | 3.2% |
| EM | 7 | 21.8% | 8 | 25.0% | 9 | 28.1% | 8 | 25.0% | 0 | 0 | |
| On average, how often do environmental factors distract you during handoff? | Admitting | 44 | 48.3% | 31 | 34.0% | 10 | 10.9% | 6 | 6.5% | 0 | 0 |
| EM | 7 | 21.8% | 11 | 34.3% | 8 | 25.0% | 4 | 12.5% | 2 | 6.2% | |
The processes for assigning responsibilities following the initial handoff differed between admitting services, and within a service the process was often dynamic. For example, within the university‐based IM and community‐based FM services, the assignment process varied depending on timing (day vs night, weekday vs weekend). For the critical care medicine and cardiology services, fellows accepted admission handoff calls, and depending on competing clinical responsibilities and the patient's stability, either evaluated the patient independently or sent a resident to perform a preliminary evaluation. We reviewed and classified these varied admission assignment strategies into 4 general schemes (Figure 1). All 5 admitting services relied partly or entirely on housestaff for receiving admission handoffs, as did the EM service.
Communication Quality and Content
Cronbach's was 0.72 for general handoff questions and 0.89 for clinical information questions. Compared with EM respondents, admitting physicians reported worse quality of communication (P < 0.001) and less confidence in the handoff system's ability to ensure patient safety (P=0.04). Admitting physicians reported communication of clinical information occurred less frequently than EM physicians for all 8 content areas (P < 0.001 for all). There were no significant differences in responses between various levels of training and service affiliations.
Interpersonal Perceptions
EM respondents reported admitting physicians asked clinical questions less frequently than did admitting respondents (P < 0.001). Ninety‐four percent of EM physicians (n=30) felt they had to defend their clinical decisions at least sometimes. EM interns (P=0.009) and faculty (P=0.01) were more likely than residents to report feeling defensive. Most admitting physicians (60%, n=56) often agreed with decisions made by the EM provider, but 29% (n=27) agreed less than half the time. One‐third of admitting (n=31) and 16% of EM physicians (n=5) reported routine (ie, >50% of admissions) meaningful face‐to‐face communication with one another at the time of admission.
Responsibilities
When asked who was primarily responsible for patients boarding in the ED, defined as nonemergent patient care that occurs after handoff, but before a patient is physically transferred from the ED, 37.6% (n=47) of respondents answered the admitting physician, 21.6% (n=27) answered the EM physician, 34.4% (n=43) answered both, and 6.4% (n=8) answered don't know. Responses were similar for EM and admitting physicians.
Organizational Factors
Fifty‐six percent of all respondents (n=69) reported they were distracted during handoffs by competing clinical duties 50% of the time. Environmental factors, such as noise, more commonly distracted EM physicians (P=0.001). Approximately 60% (n=56) of admitting physicians reported using a triage system to distribute admissions, with a resultant 57% (n=32) reporting sequential handoffs (ie, handoffs of handoffs) occurred at least sometimes. About 80% of EM physicians (n=23) reported that shift change led to sequential handoffs at least sometimes. Seventy‐eight percent (n=67) of physicians felt sequential handoffs had a negative impact on patient care.
Patient Safety
Thirty‐four percent of admitting (n=30) and 19% of EM physicians (n=6) reported a patient was harmed or suffered a near miss in the past 3 months because of an ineffective handoff, with 58% (n=21) reporting 2 examples. Twenty‐four respondents described 29 adverse events. Respondents described perceived mistakes in diagnosis (n=11), treatment (n=16), and disposition (n=12), with some examples falling into more than 1 category. Absent or ineffective communication contributed to 27 of 29 examples. Other commonly cited areas of vulnerability included uncertain assignment of responsibility, sequential handoffs, and patient boarding.
DISCUSSION
Based upon physician self‐reporting, we identified perceived barriers to safe ED admission handoff across several domains. This study adds to the literature, as it provides a cross‐section of multiple inpatient services with varying admission schemes to underscore the complexities facing hospitals in safely transitioning patients between units. As noted in previous studies, one‐third of physicians reported a handoff‐related adverse event,[15] and there was significant disagreement between handoff participants about communication of critical information.[21, 26] These differences in perceptions suggest a failure of physicians to accurately transfer information to create a shared understanding of patient care,[21] which is the central function of handoffs.
EM physicians frequently felt that admitting physicians did not trust their clinical decisions, a perception supported by the fact that over 25% of admitting respondents' usually disagreed with decisions in the ED. Interdisciplinary trust is central in negotiating a shared plan of care[13] and mitigating conflict to ensure a safe transition of patient care.[16] Handoffs are complex social interactions, and feelings of defensiveness and mistrust are likely exacerbated by in‐group/out‐group biases,[15] conflicting information expectations,[19] and discordant ways of interpreting and framing handoff interactions.[13] Interestingly, EM residents were less likely than interns or faculty to report feeling defensive. This may be in part because residents from EM and admitting services develop relationships during interdisciplinary rotations, which may help facilitate future handoff interactions.[27] The fact that EM respondents felt defensive, despite reporting less‐frequent questioning than admitting physicians, suggests that tone and content of questions played an important role. These findings support the importance of interdisciplinary education and standardization of handoff communication between ED and admitting physicians.[23] Beach and colleagues have recommended a conceptual framework for interunit handoffs between EM and hospital physicians, but further research is needed to measure its impact in real‐world settings.[14]
We also found great variability in admitting services' processes for assigning patient‐care responsibility following the initial handoff. Even within an individual service, these processes were often dynamic and relied on physicians at different levels of training. This has several potential consequences. First, it may be difficult for physicians engaged in a handoff to know the level of experience and expertise of one another. These contextual variables play an important role in how handoff information is conveyed, as less experienced clinicians may require explicit information that a more experienced provider may infer.[1, 21] Second, the variability in admission assignment processes may further exacerbate uncertainty regarding responsibility for patients boarding in the ED, making it increasingly difficult for nurses and ancillary staff to know which physician is ultimately responsible for patient care. Finally, the diversity of admission schemes may complicate the development of standardized interunit handoff protocols, policy, and education.
A related finding was that sequential handoffs were common within both EM and admitting services. EM shift handoffs have their own set of barriers,[28] which can lead to ineffective communication.[29] Likewise, about two‐thirds of admitting respondents reported using an admission triage system. The goal of such systems is to simplify complex call schedules and diverse patient assignment schemes within admitting services, thus streamlining the admission process. These systems may also allow for more consistency in the quality of handoff communication through the creation of triage specialists. These potential advantages need to be weighed against the increased risk of communication breakdown. The introduction of sequential handoffs creates a game of telephone, in which there is no direct communication between the first and final caregivers (Figure 1), allowing misinformation to be propagated forward.[30] Sequential handoffs contributed to several reported adverse events, and the majority of surveyed physicians felt they negatively impacted patient care. Further research is necessary to determine the impact of centralized triage systems and to explore strategies to mitigate information decay that results from sequential handoffs, as quality‐improvement interventions may be of limited benefit if downstream communication remains ineffective. Potential strategies may include standardizing sequential handoff communication, leveraging centralized handoff notes within electronic health records, or developing handoff systems that ensure direct communication between the EM physician and the ultimate admitting provider.
Limitations
This was a single‐institution study, so results may not be generalizable, as handoff processes vary among hospitals.[24] Our study relied on a novel survey instrument, for which validity and reliability are uncertain, although internal consistency was good for domains that could be tested (Cronbach's 0.720.89). As with other survey‐based studies, participant selection, hindsight, recall, and response biases may have influenced the results. We attempted to minimize these risks by pilot testing the survey, targeting a relatively large number of respondents across multiple services, and by making efforts to maximize the response rate by contacting eligible participants both in person and via email. Because results reflect self‐reported perceptions, we cannot prove that the factors studied are actually associated with adverse outcomes, nor can we quantify their relative importance. Nevertheless, the reported perceptions raise concerns that warrant further study.
FUTURE DIRECTIONS
Further research is needed to examine interventions that may improve clinically relevant outcomes. Development of structured admission handoff protocols should be collaborative[31] and focus on clinical judgment, rather than rote recitation of data.[14] Based on our study findings, we are pilot testing a standardized approach for ED‐to‐hospital handoffs, and portions of this survey will be repeated in the postintervention assessment.
At our institution, housestaff at all levels of training regularly participated in the handoff process. The Accreditation Council for Graduate Medical Education requires that residents demonstrate competence in performing handoffs,[7] yet handoff training and assessment are inconsistent,[23, 32, 33] and published interventions have focused primarily on within‐unit handoffs.[34, 35, 36] Additional training should focus on the unique aspects of interunit handoffs. Approaches could include interprofessional communication training, simulation training, and enhanced assessment methods. Additionally, increasing face‐to‐face communication, perhaps as part of bedside handoffs, could improve relationships and the development of a shared mental model of patient care. More direct involvement by attending physicians will also be important, as there is evidence that such oversight may improve training[36] and safety,[37] as more experienced physicians better integrate handoff information.[21]
CONCLUSION
We identified several perceived barriers to safe interunit handoff from the ED to the inpatient setting. Handoff‐related adverse events, a pattern of conflicting physician perceptions, and frequent sequential handoffs were of particular concern. Our findings support the need for collaborative efforts to improve interdisciplinary communication.
Disclosure
Nothing to report.
Handoffs are the exchange of information between health professionals that accompany the transfer of patient‐care responsibility.[1] Poor handoff practices are associated with unsafe and inefficient care.[2, 3, 4] Teaching hospitals are especially at risk, as resident work‐hour restrictions have increased the number of handoffs.[5] Accreditation agencies now require that hospitals and residency programs have structured handoff processes[6, 7] and that medical students[8] and residents[9, 10, 11, 12] demonstrate competency in handoffs.
Physician handoff research has primarily focused on handoffs within a service or discipline. These within‐unit handoffs should be differentiated from interunit handoffs.[13, 14] Interunit handoffs, such as the transition from the emergency department (ED) to inpatient setting, are subject to unique challenges. The ED admission process involves changes in personnel, provider specialty, and location.[15] The transition occurs when the patient's clinical trajectory is uncertain, treatments are being initiated, and test results are pending. Other barriers include interdisciplinary cultural differences, interphysician conflict, unstructured communication, environmental factors, and complex care coordination.[13, 14, 15, 16, 17, 18] Despite these challenges, there is relatively little research specifically examining ED to inpatient handoffs, and most of what is available has focused on individual services within an institution.[13, 14, 15, 18, 19, 20, 21, 22, 23, 24, 25]
As part of an institutional effort to improve our ED admission handoff practices, we conducted a cross‐sectional, survey‐based needs‐assessment involving emergency medicine (EM) and 5 inpatient medical services. The objective of this study was to determine physicians' perceptions of the ED admission handoff process and to identify potential barriers to safe patient care.
METHODS
Survey Design
A study group comprised of resident and faculty physicians in internal medicine (IM) and EM, as well as a healthcare communication expert, designed analogous cross‐sectional surveys to determine the perceptions of admitting (see Supporting Information, Appendix 1, in the online version of this article) and EM (see Supporting Information, Appendix 2, in the online version of this article) physicians toward the admission handoff process. Using an iterative process to ensure content validity, we created questions in 6 domains based on the expert opinion of the authors and emergent themes identified in the literature.[15, 19, 22, 24] These themes were general communication quality, clinical information, interpersonal perceptions, responsibilities, organizational factors, and patient safety. We asked respondents to report their answers using 5‐point Likert and Likert‐like scales. Questions regarding frequency were assigned semiquantitative values: rarely=0% to 24%, sometimes=25% to 49%, often=50% to 74%, very often 75% to 99%, and always=100%. We also asked an open‐ended question, asking respondents to describe any handoff‐related adverse events (defined as patient harm or near miss) they encountered in the past 3 months. We pilot tested the survey for clarity and relevance prior to distribution on a group of 5 physicians from the participating services. The institutional internal review board approved the protocol (#046‐13‐EX).
Setting, Participants, and Recruitment
We conducted the study at a 627‐bed tertiary care academic medical center. Eligible participants included all resident, fellow, and faculty physicians directly involved in admission handoffs from EM and 5 medical inpatient services (university‐based IM, university‐based family medicine [FM], community‐based FM, cardiology, and critical care medicine). The admitting services accounted for two‐thirds of the institution's 10,000 annual adult, nonobstetric ED admissions. Physicians who had not participated in admission handoffs in the past 3 months were excluded.
At the time of the study, there was no standardized institutional process for admission handoff communication, nor was there policy delineating when patient‐care responsibility transferred from the EM to admitting physician. The admission handoff process generally relied on verbal handoff via telephone between EM and admitting physicians. All services used the same electronic health record, but there was no written handoff note, and EM physician documentation generally was not available at the time of handoff. To determine patient assignment schemes following admission handoff, we questioned leadership from the participating admitting services.
We distributed and collected anonymous hard‐copy surveys at educational conferences in March 2013. We emailed a link to an online survey to eligible participants who could not be reached in person. Subjects voluntarily participated and provided consent via cover letter.
Data Analysis
We compiled survey data and performed descriptive analysis. We assessed the internal consistency of the survey domains that were made up of at least 3 questions using Cronbach's . To compare the distribution of aggregate admitting service responses to EM responses, we used the Mann‐Whitney test. We used the Fisher exact test to examine the associations of dichotomized responses (<50% vs 50%) to the level of training (intern vs resident vs fellow/faculty) and to the admitting service affiliation (university‐based IM vs university‐based FM vs aggregate of other services). When indicated, we made pairwise comparisons using the Bonferroni method to compute adjusted P values. We analyzed data independently using both SPSS version 20 (IBM Corp., Armonk, NY) and SAS version 9.3 (SAS Inc., Cary, NC) software and considered a P value <0.05 to be significant. Three researchers independently categorized descriptions of adverse events based on a previously published qualitative analysis,[15] with disagreements settled by consensus.
RESULTS
After applying exclusion criteria, the survey response rate was 63% for admitting physicians (94/150) and 86% for EM physicians (32/37). Participants' service affiliation and level of training are shown in the Table 1. Table 2 provides the distribution of survey responses for EM and admitting physicians.
| Service Affiliation | Level of Training | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PGY1 | PGY2 | PGY3 | Fellow | Staff | |||||||
| No. | % | No. | % | No. | % | No. | % | No. | % | No. | |
| |||||||||||
| Admitting services | |||||||||||
| University‐based IM | 12 | 32.4 | 7 | 18.9 | 5 | 13.5 | 1 | 2.7 | 12 | 32.4 | 37 |
| University‐based FM | 15 | 44.1 | 13 | 38.2 | 5 | 14.7 | 1 | 2.9 | 0 | 0 | 34 |
| Community‐based FM | 5 | 50.0 | 1 | 10.0 | 3 | 30.0 | 0 | 0 | 1 | 10.0 | 10 |
| Critical care medicine | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 100.0 | 0 | 0 | 6 |
| Cardiology | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 100.0 | 0 | 0 | 7 |
| Admitting services total | 32 | 34.0 | 21 | 22.3 | 13 | 13.8 | 15 | 16.0 | 13 | 13.8 | 94 |
| Emergency medicine | 6 | 18.8 | 8 | 25.0 | 5 | 15.6 | 0 | 0 | 13 | 40.6 | 32 |
| Question | Service | Very Poor | Poor | Fair | Good | Very Good | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | No. | % | No. | % | ||
| Rarely (0%24%) | Sometimes (25%49%) | Often (50%74%) | Very Often (75%99%) | Always (100%) | |||||||
| |||||||||||
| Generally, the quality of communication between EM and admitting physicians is: | Admitting | 0 | 0 | 8 | 8.6% | 37 | 39.7% | 46 | 49.4% | 2 | 2.1% |
| EM | 0 | 0 | 2 | 6.2% | 4 | 12.5% | 20 | 62.5% | 6 | 18.7% | |
| The current handoff system's ability to ensure patient safety is generally: | Admitting | 1 | 1.0% | 10 | 10.7% | 43 | 46.2% | 37 | 39.7% | 2 | 2.1% |
| EM | 1 | 3.1% | 1 | 3.1% | 11 | 34.3% | 15 | 46.8% | 4 | 12.5% | |
| The current handoff system's ability to ensure efficient patient care is generally: | Admitting | 3 | 3.2% | 20 | 21.7% | 31 | 33.6% | 36 | 39.1% | 2 | 2.1% |
| EM | 2 | 6.2% | 5 | 15.6% | 15 | 46.8% | 10 | 31.2% | 0 | ||
| During handoff, how often does the EM physician provide the following information to the admitting service? | |||||||||||
| The working diagnosis of the EM physician | Admitting | 5 | 5.4% | 19 | 20.6% | 30 | 32.6% | 30 | 32.6% | 8 | 8.6% |
| EM | 0 | 4 | 12.5% | 0 | 12 | 37.5% | 16 | 50.0% | |||
| Relevant past medical/surgical history | Admitting | 5 | 5.4% | 25 | 27.1% | 40 | 43.4% | 18 | 19.5% | 4 | 4.3% |
| EM | 1 | 3.1% | 2 | 6.2% | 5 | 15.6% | 17 | 53.1% | 7 | 21.8% | |
| Relevant physical exam findings (including abnormal vital signs) | Admitting | 3 | 3.2% | 25 | 27.1% | 41 | 44.5% | 21 | 22.8% | 2 | 2.1% |
| EM | 0 | 5 | 15.6% | 2 | 6.2% | 15 | 46.8% | 10 | 31.2% | ||
| Results of relevant diagnostic studies (labs, imaging) | Admitting | 2 | 2.1% | 10 | 10.8% | 39 | 42.3% | 37 | 40.2% | 4 | 4.3% |
| EM | 1 | 3.1% | 0 | 3 | 9.3% | 14 | 43.7% | 14 | 43.7% | ||
| Procedures and therapeutic interventions initiated while in the ED | Admitting | 3 | 3.2% | 20 | 21.7% | 34 | 36.9% | 29 | 31.5% | 6 | 6.5% |
| EM | 1 | 3.1% | 0 | 3 | 9.3% | 18 | 56.2% | 10 | 31.2% | ||
| Trend in the patient's clinical condition while in the ED | Admitting | 12 | 13.1% | 27 | 29.6% | 33 | 36.2% | 17 | 18.6% | 2 | 2.1% |
| EM | 4 | 12.5% | 1 | 3.1% | 5 | 15.6% | 13 | 40.6% | 9 | 28.1% | |
| Current clinical condition of the patient (at time of handoff) | Admitting | 3 | 3.2% | 24 | 26.0% | 41 | 44.5% | 18 | 19.5% | 6 | 6.5% |
| EM | 1 | 3.1% | 1 | 3.1% | 3 | 9.3% | 13 | 40.6% | 14 | 43.7% | |
| Pending diagnostic studies (labs, imaging), if ordered | Admitting | 12 | 13.0% | 32 | 34.7% | 29 | 31.5% | 17 | 18.4% | 2 | 2.1% |
| EM | 0 | 5 | 15.6% | 6 | 18.7% | 14 | 43.7% | 7 | 21.8% | ||
| During handoff, how often are clinical questions asked about the patient being admitted? | Admitting | 2 | 2.1% | 1 | 1.0% | 13 | 14.1% | 29 | 31.5% | 47 | 51.0% |
| EM | 0 | 0 | 5 | 15.6% | 8 | 25.0% | 13 | 40.6% | 6 | 18.7% | |
| In general, how often do you agree with the clinical decisions made by the EM physician? | Admitting | 1 | 1.0% | 26 | 27.9% | 56 | 60.2% | 10 | 10.7% | 0 | 0 |
| Generally, how often do you feel you have to defend your clinical decisions to the admitting service? | EM | 2 | 6.2% | 15 | 46.8% | 5 | 15.6% | 10 | 31.2% | 0 | 0 |
| How often do you have clinically meaningful face‐to‐face communication with the EM/admitting physician about the patient being admitted? | Admitting | 24 | 25.8% | 38 | 40.8% | 22 | 23.6% | 8 | 8.6% | 1 | 1.0% |
| EM | 14 | 43.7% | 13 | 40.6% | 4 | 12.5% | 1 | 3.1% | 0 | ||
| On average, how often do competing clinical responsibilities distract you during handoff? | Admitting | 6 | 6.5% | 34 | 36.9% | 29 | 31.5% | 20 | 21.7% | 3 | 3.2% |
| EM | 7 | 21.8% | 8 | 25.0% | 9 | 28.1% | 8 | 25.0% | 0 | 0 | |
| On average, how often do environmental factors distract you during handoff? | Admitting | 44 | 48.3% | 31 | 34.0% | 10 | 10.9% | 6 | 6.5% | 0 | 0 |
| EM | 7 | 21.8% | 11 | 34.3% | 8 | 25.0% | 4 | 12.5% | 2 | 6.2% | |
The processes for assigning responsibilities following the initial handoff differed between admitting services, and within a service the process was often dynamic. For example, within the university‐based IM and community‐based FM services, the assignment process varied depending on timing (day vs night, weekday vs weekend). For the critical care medicine and cardiology services, fellows accepted admission handoff calls, and depending on competing clinical responsibilities and the patient's stability, either evaluated the patient independently or sent a resident to perform a preliminary evaluation. We reviewed and classified these varied admission assignment strategies into 4 general schemes (Figure 1). All 5 admitting services relied partly or entirely on housestaff for receiving admission handoffs, as did the EM service.
Communication Quality and Content
Cronbach's was 0.72 for general handoff questions and 0.89 for clinical information questions. Compared with EM respondents, admitting physicians reported worse quality of communication (P < 0.001) and less confidence in the handoff system's ability to ensure patient safety (P=0.04). Admitting physicians reported communication of clinical information occurred less frequently than EM physicians for all 8 content areas (P < 0.001 for all). There were no significant differences in responses between various levels of training and service affiliations.
Interpersonal Perceptions
EM respondents reported admitting physicians asked clinical questions less frequently than did admitting respondents (P < 0.001). Ninety‐four percent of EM physicians (n=30) felt they had to defend their clinical decisions at least sometimes. EM interns (P=0.009) and faculty (P=0.01) were more likely than residents to report feeling defensive. Most admitting physicians (60%, n=56) often agreed with decisions made by the EM provider, but 29% (n=27) agreed less than half the time. One‐third of admitting (n=31) and 16% of EM physicians (n=5) reported routine (ie, >50% of admissions) meaningful face‐to‐face communication with one another at the time of admission.
Responsibilities
When asked who was primarily responsible for patients boarding in the ED, defined as nonemergent patient care that occurs after handoff, but before a patient is physically transferred from the ED, 37.6% (n=47) of respondents answered the admitting physician, 21.6% (n=27) answered the EM physician, 34.4% (n=43) answered both, and 6.4% (n=8) answered don't know. Responses were similar for EM and admitting physicians.
Organizational Factors
Fifty‐six percent of all respondents (n=69) reported they were distracted during handoffs by competing clinical duties 50% of the time. Environmental factors, such as noise, more commonly distracted EM physicians (P=0.001). Approximately 60% (n=56) of admitting physicians reported using a triage system to distribute admissions, with a resultant 57% (n=32) reporting sequential handoffs (ie, handoffs of handoffs) occurred at least sometimes. About 80% of EM physicians (n=23) reported that shift change led to sequential handoffs at least sometimes. Seventy‐eight percent (n=67) of physicians felt sequential handoffs had a negative impact on patient care.
Patient Safety
Thirty‐four percent of admitting (n=30) and 19% of EM physicians (n=6) reported a patient was harmed or suffered a near miss in the past 3 months because of an ineffective handoff, with 58% (n=21) reporting 2 examples. Twenty‐four respondents described 29 adverse events. Respondents described perceived mistakes in diagnosis (n=11), treatment (n=16), and disposition (n=12), with some examples falling into more than 1 category. Absent or ineffective communication contributed to 27 of 29 examples. Other commonly cited areas of vulnerability included uncertain assignment of responsibility, sequential handoffs, and patient boarding.
DISCUSSION
Based upon physician self‐reporting, we identified perceived barriers to safe ED admission handoff across several domains. This study adds to the literature, as it provides a cross‐section of multiple inpatient services with varying admission schemes to underscore the complexities facing hospitals in safely transitioning patients between units. As noted in previous studies, one‐third of physicians reported a handoff‐related adverse event,[15] and there was significant disagreement between handoff participants about communication of critical information.[21, 26] These differences in perceptions suggest a failure of physicians to accurately transfer information to create a shared understanding of patient care,[21] which is the central function of handoffs.
EM physicians frequently felt that admitting physicians did not trust their clinical decisions, a perception supported by the fact that over 25% of admitting respondents' usually disagreed with decisions in the ED. Interdisciplinary trust is central in negotiating a shared plan of care[13] and mitigating conflict to ensure a safe transition of patient care.[16] Handoffs are complex social interactions, and feelings of defensiveness and mistrust are likely exacerbated by in‐group/out‐group biases,[15] conflicting information expectations,[19] and discordant ways of interpreting and framing handoff interactions.[13] Interestingly, EM residents were less likely than interns or faculty to report feeling defensive. This may be in part because residents from EM and admitting services develop relationships during interdisciplinary rotations, which may help facilitate future handoff interactions.[27] The fact that EM respondents felt defensive, despite reporting less‐frequent questioning than admitting physicians, suggests that tone and content of questions played an important role. These findings support the importance of interdisciplinary education and standardization of handoff communication between ED and admitting physicians.[23] Beach and colleagues have recommended a conceptual framework for interunit handoffs between EM and hospital physicians, but further research is needed to measure its impact in real‐world settings.[14]
We also found great variability in admitting services' processes for assigning patient‐care responsibility following the initial handoff. Even within an individual service, these processes were often dynamic and relied on physicians at different levels of training. This has several potential consequences. First, it may be difficult for physicians engaged in a handoff to know the level of experience and expertise of one another. These contextual variables play an important role in how handoff information is conveyed, as less experienced clinicians may require explicit information that a more experienced provider may infer.[1, 21] Second, the variability in admission assignment processes may further exacerbate uncertainty regarding responsibility for patients boarding in the ED, making it increasingly difficult for nurses and ancillary staff to know which physician is ultimately responsible for patient care. Finally, the diversity of admission schemes may complicate the development of standardized interunit handoff protocols, policy, and education.
A related finding was that sequential handoffs were common within both EM and admitting services. EM shift handoffs have their own set of barriers,[28] which can lead to ineffective communication.[29] Likewise, about two‐thirds of admitting respondents reported using an admission triage system. The goal of such systems is to simplify complex call schedules and diverse patient assignment schemes within admitting services, thus streamlining the admission process. These systems may also allow for more consistency in the quality of handoff communication through the creation of triage specialists. These potential advantages need to be weighed against the increased risk of communication breakdown. The introduction of sequential handoffs creates a game of telephone, in which there is no direct communication between the first and final caregivers (Figure 1), allowing misinformation to be propagated forward.[30] Sequential handoffs contributed to several reported adverse events, and the majority of surveyed physicians felt they negatively impacted patient care. Further research is necessary to determine the impact of centralized triage systems and to explore strategies to mitigate information decay that results from sequential handoffs, as quality‐improvement interventions may be of limited benefit if downstream communication remains ineffective. Potential strategies may include standardizing sequential handoff communication, leveraging centralized handoff notes within electronic health records, or developing handoff systems that ensure direct communication between the EM physician and the ultimate admitting provider.
Limitations
This was a single‐institution study, so results may not be generalizable, as handoff processes vary among hospitals.[24] Our study relied on a novel survey instrument, for which validity and reliability are uncertain, although internal consistency was good for domains that could be tested (Cronbach's 0.720.89). As with other survey‐based studies, participant selection, hindsight, recall, and response biases may have influenced the results. We attempted to minimize these risks by pilot testing the survey, targeting a relatively large number of respondents across multiple services, and by making efforts to maximize the response rate by contacting eligible participants both in person and via email. Because results reflect self‐reported perceptions, we cannot prove that the factors studied are actually associated with adverse outcomes, nor can we quantify their relative importance. Nevertheless, the reported perceptions raise concerns that warrant further study.
FUTURE DIRECTIONS
Further research is needed to examine interventions that may improve clinically relevant outcomes. Development of structured admission handoff protocols should be collaborative[31] and focus on clinical judgment, rather than rote recitation of data.[14] Based on our study findings, we are pilot testing a standardized approach for ED‐to‐hospital handoffs, and portions of this survey will be repeated in the postintervention assessment.
At our institution, housestaff at all levels of training regularly participated in the handoff process. The Accreditation Council for Graduate Medical Education requires that residents demonstrate competence in performing handoffs,[7] yet handoff training and assessment are inconsistent,[23, 32, 33] and published interventions have focused primarily on within‐unit handoffs.[34, 35, 36] Additional training should focus on the unique aspects of interunit handoffs. Approaches could include interprofessional communication training, simulation training, and enhanced assessment methods. Additionally, increasing face‐to‐face communication, perhaps as part of bedside handoffs, could improve relationships and the development of a shared mental model of patient care. More direct involvement by attending physicians will also be important, as there is evidence that such oversight may improve training[36] and safety,[37] as more experienced physicians better integrate handoff information.[21]
CONCLUSION
We identified several perceived barriers to safe interunit handoff from the ED to the inpatient setting. Handoff‐related adverse events, a pattern of conflicting physician perceptions, and frequent sequential handoffs were of particular concern. Our findings support the need for collaborative efforts to improve interdisciplinary communication.
Disclosure
Nothing to report.
- , . Handoffs in hospitals: a review of the literature on information exchange while transferring patient responsibility or control. Available at: http://deepblue.lib.umich.edu/handle/2027.42/61498. Updated 2009. Accessed May 15, 2014.
- . Handoffs causing patient harm: a survey of medical and surgical house staff. Jt Comm J Qual Patient Saf. 2008;34(10):563–570.
- . Consequences of inadequate sign‐out for patient care. Arch Intern Med. 2008;168(16):1755–1760.
- , . A systematic review of failures in handoff communication during intrahospital transfers. Jt Comm J Qual Patient Saf. 2011;37(6):274–284.
- , , , , . Managing discontinuity in academic medical centers: strategies for a safe and effective resident sign‐out. J Hosp Med. 2006;1(4):257–266.
- , . A model for building a standardized hand‐off protocol. Jt Comm J Qual Patient Saf. 2006;32(11):646–655.
- Accreditation Council for Graduate Medical Education. ACGME common program requirements. Available at: https://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/CPRs2013.pdf. Updated 2013. Accessed May 7, 2014.
- Association of American Medical Colleges. Core entrustable professional activities for entering residency. Available at: https://members.aamc.org/eweb/upload/Core%20EPA%20Faculty%20and%20Learner%20Guide.pdf. Updated 2014. Accessed July 7, 2014.
- Accreditation Council for Graduate Medical Education and American Board of Internal Medicine. The internal medicine milestones. Available at: http://www.acgme.org/acgmeweb/portals/0/pdfs/milestones/internalmedicinemilestones.pdf. Updated 2012. Accessed December 23, 2013.
- Accreditation Council for Graduate Medical Education and American Board of Emergency Medicine. The emergency medicine milestones. Available at: https://www.abem.org/public/docs/default‐source/migrated‐documents‐and‐files/em‐milestones.pdf?sfvrsn=4. Updated 2012. Accessed December 23, 2013.
- Accreditation Council for Graduate Medical Education and American Board of Family Medicine. The family medicine milestone project. Available at: http://www.acgme.org/acgmeweb/Portals/0/PDFs/Milestones/FamilyMedicineMilestones.pdf. Updated 2013. Accessed October 31, 2014.
- Accreditation Council for Graduate Medical Education and American Board of Pediatrics. The pediatrics milestone project. Available at: http://acgme.org/acgmeweb/Portals/0/PDFs/Milestones/PediatricsMilestones.pdf. Updated 2013. Accessed October, 31, 2014.
- , . The unappreciated challenges of between‐unit handoffs: negotiating and coordinating across boundaries. Ann Emerg Med. 2013;61(2):155–160.
- , , , et al. Improving interunit transitions of care between emergency physicians and hospital medicine physicians: a conceptual approach. Acad Emerg Med. 2012;19(10):1188–1195.
- , , , , , . Dropping the baton: a qualitative analysis of failures during the transition from emergency department to inpatient care. Ann Emerg Med. 2009;53(6):701–710.e4.
- , , , . Conflict prevention, conflict mitigation, and manifestations of conflict during emergency department consultations. Acad Emerg Med. 2014;21(3):308–313.
- , , , . I'm clear, you're clear, we're all clear: improving consultation communication skills in undergraduate medical education. Acad Med. 2013;88(6):753–758.
- , , , . Emergency physician to admitting physician handovers: an exploratory study. Proc Hum Factors Ergon Soc Annu Meet. 2002;46(16):1511–1515.
- , , . Communicating in the “gray zone”: perceptions about emergency physician hospitalist handoffs and patient safety. Acad Emerg Med. 2007;14(10):884–894.
- , . Chart biopsy: an emerging medical practice enabled by electronic health records and its impacts on emergency department‐inpatient admission handoffs. J Am Med Inform Assoc. 2013;20(2):260–267.
- , , , , . Admission handoff communications: clinician's shared understanding of patient severity of illness and problems. J Patient Saf. 2009;5(4):237–242.
- , , , et al. Exploring emergency physician‐hospitalist handoff interactions: development of the handoff communication assessment. Ann Emerg Med. 2010;55(2):161–170.
- , , , , , . Interunit handoffs of patients and transfers of information: a survey of current practices. Ann Emerg Med. 2014;64(4):343–349.e5.
- , , , et al. A conceptual framework for studying the safety of transitions in emergency care. In: Henriksen K, Battles JB, Marks ES, Lewin DI, eds. Advances in Patient Safety: From Research to Implementation. Vol. 2: Concepts and Methodology. Rockville, MD: Agency for Healthcare Research and Quality; 2005:309–321.
- , , , , , . Patient care transitions from the emergency department to the medicine ward: evaluation of a standardized electronic signout tool. Int J Qual Health Care. 2014;26(4):337–347.
- , , , , . Interns overestimate the effectiveness of their hand‐off communication. Pediatrics. 2010;125(3):491–496.
- , , , . Understanding the impact of residents' interpersonal relationships during emergency department referrals and consultations. J Grad Med Educ. 2013;5(4):576–581.
- , , , et al. Improving handoffs in the emergency department. Ann Emerg Med. 2010;55(2):171–180.
- , , . ED handoffs: observed practices and communication errors. Am J Emerg Med. 2011;29(5):502–511.
- , , , , . Characterizing information decay in patient handoffs. J Surg Educ. 2014;71(4):480–485.
- , , . Emergency medicine and hospital medicine: a call for collaboration. J Emerg Med. 2012;43(2):328–334.
- , , , et al. A survey of handoff practices in emergency medicine. Am J Med Qual. 2014;29(5):408–414.
- . Transfers of patient care between house staff on internal medicine wards: a national survey. Arch Intern Med. 2006;166(11):1173–1177.
- , , , , , . Effect of a systems intervention on the quality and safety of patient handoffs in an internal medicine residency program. J Gen Intern Med. 2013;28(8):986–993.
- , , , et al. Rates of medical errors and preventable adverse events among hospitalized children following implementation of a resident handoff bundle. JAMA. 2013;310(21):2262–2270.
- , , , et al. A structured handoff program for interns. Acad Med. 2009;84(3):347–352.
- , , , et al. Experience with faculty supervision of an electronic resident sign‐out system. Am J Med. 2010;123(4):376–381.
- , . Handoffs in hospitals: a review of the literature on information exchange while transferring patient responsibility or control. Available at: http://deepblue.lib.umich.edu/handle/2027.42/61498. Updated 2009. Accessed May 15, 2014.
- . Handoffs causing patient harm: a survey of medical and surgical house staff. Jt Comm J Qual Patient Saf. 2008;34(10):563–570.
- . Consequences of inadequate sign‐out for patient care. Arch Intern Med. 2008;168(16):1755–1760.
- , . A systematic review of failures in handoff communication during intrahospital transfers. Jt Comm J Qual Patient Saf. 2011;37(6):274–284.
- , , , , . Managing discontinuity in academic medical centers: strategies for a safe and effective resident sign‐out. J Hosp Med. 2006;1(4):257–266.
- , . A model for building a standardized hand‐off protocol. Jt Comm J Qual Patient Saf. 2006;32(11):646–655.
- Accreditation Council for Graduate Medical Education. ACGME common program requirements. Available at: https://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/CPRs2013.pdf. Updated 2013. Accessed May 7, 2014.
- Association of American Medical Colleges. Core entrustable professional activities for entering residency. Available at: https://members.aamc.org/eweb/upload/Core%20EPA%20Faculty%20and%20Learner%20Guide.pdf. Updated 2014. Accessed July 7, 2014.
- Accreditation Council for Graduate Medical Education and American Board of Internal Medicine. The internal medicine milestones. Available at: http://www.acgme.org/acgmeweb/portals/0/pdfs/milestones/internalmedicinemilestones.pdf. Updated 2012. Accessed December 23, 2013.
- Accreditation Council for Graduate Medical Education and American Board of Emergency Medicine. The emergency medicine milestones. Available at: https://www.abem.org/public/docs/default‐source/migrated‐documents‐and‐files/em‐milestones.pdf?sfvrsn=4. Updated 2012. Accessed December 23, 2013.
- Accreditation Council for Graduate Medical Education and American Board of Family Medicine. The family medicine milestone project. Available at: http://www.acgme.org/acgmeweb/Portals/0/PDFs/Milestones/FamilyMedicineMilestones.pdf. Updated 2013. Accessed October 31, 2014.
- Accreditation Council for Graduate Medical Education and American Board of Pediatrics. The pediatrics milestone project. Available at: http://acgme.org/acgmeweb/Portals/0/PDFs/Milestones/PediatricsMilestones.pdf. Updated 2013. Accessed October, 31, 2014.
- , . The unappreciated challenges of between‐unit handoffs: negotiating and coordinating across boundaries. Ann Emerg Med. 2013;61(2):155–160.
- , , , et al. Improving interunit transitions of care between emergency physicians and hospital medicine physicians: a conceptual approach. Acad Emerg Med. 2012;19(10):1188–1195.
- , , , , , . Dropping the baton: a qualitative analysis of failures during the transition from emergency department to inpatient care. Ann Emerg Med. 2009;53(6):701–710.e4.
- , , , . Conflict prevention, conflict mitigation, and manifestations of conflict during emergency department consultations. Acad Emerg Med. 2014;21(3):308–313.
- , , , . I'm clear, you're clear, we're all clear: improving consultation communication skills in undergraduate medical education. Acad Med. 2013;88(6):753–758.
- , , , . Emergency physician to admitting physician handovers: an exploratory study. Proc Hum Factors Ergon Soc Annu Meet. 2002;46(16):1511–1515.
- , , . Communicating in the “gray zone”: perceptions about emergency physician hospitalist handoffs and patient safety. Acad Emerg Med. 2007;14(10):884–894.
- , . Chart biopsy: an emerging medical practice enabled by electronic health records and its impacts on emergency department‐inpatient admission handoffs. J Am Med Inform Assoc. 2013;20(2):260–267.
- , , , , . Admission handoff communications: clinician's shared understanding of patient severity of illness and problems. J Patient Saf. 2009;5(4):237–242.
- , , , et al. Exploring emergency physician‐hospitalist handoff interactions: development of the handoff communication assessment. Ann Emerg Med. 2010;55(2):161–170.
- , , , , , . Interunit handoffs of patients and transfers of information: a survey of current practices. Ann Emerg Med. 2014;64(4):343–349.e5.
- , , , et al. A conceptual framework for studying the safety of transitions in emergency care. In: Henriksen K, Battles JB, Marks ES, Lewin DI, eds. Advances in Patient Safety: From Research to Implementation. Vol. 2: Concepts and Methodology. Rockville, MD: Agency for Healthcare Research and Quality; 2005:309–321.
- , , , , , . Patient care transitions from the emergency department to the medicine ward: evaluation of a standardized electronic signout tool. Int J Qual Health Care. 2014;26(4):337–347.
- , , , , . Interns overestimate the effectiveness of their hand‐off communication. Pediatrics. 2010;125(3):491–496.
- , , , . Understanding the impact of residents' interpersonal relationships during emergency department referrals and consultations. J Grad Med Educ. 2013;5(4):576–581.
- , , , et al. Improving handoffs in the emergency department. Ann Emerg Med. 2010;55(2):171–180.
- , , . ED handoffs: observed practices and communication errors. Am J Emerg Med. 2011;29(5):502–511.
- , , , , . Characterizing information decay in patient handoffs. J Surg Educ. 2014;71(4):480–485.
- , , . Emergency medicine and hospital medicine: a call for collaboration. J Emerg Med. 2012;43(2):328–334.
- , , , et al. A survey of handoff practices in emergency medicine. Am J Med Qual. 2014;29(5):408–414.
- . Transfers of patient care between house staff on internal medicine wards: a national survey. Arch Intern Med. 2006;166(11):1173–1177.
- , , , , , . Effect of a systems intervention on the quality and safety of patient handoffs in an internal medicine residency program. J Gen Intern Med. 2013;28(8):986–993.
- , , , et al. Rates of medical errors and preventable adverse events among hospitalized children following implementation of a resident handoff bundle. JAMA. 2013;310(21):2262–2270.
- , , , et al. A structured handoff program for interns. Acad Med. 2009;84(3):347–352.
- , , , et al. Experience with faculty supervision of an electronic resident sign‐out system. Am J Med. 2010;123(4):376–381.
© 2015 Society of Hospital Medicine
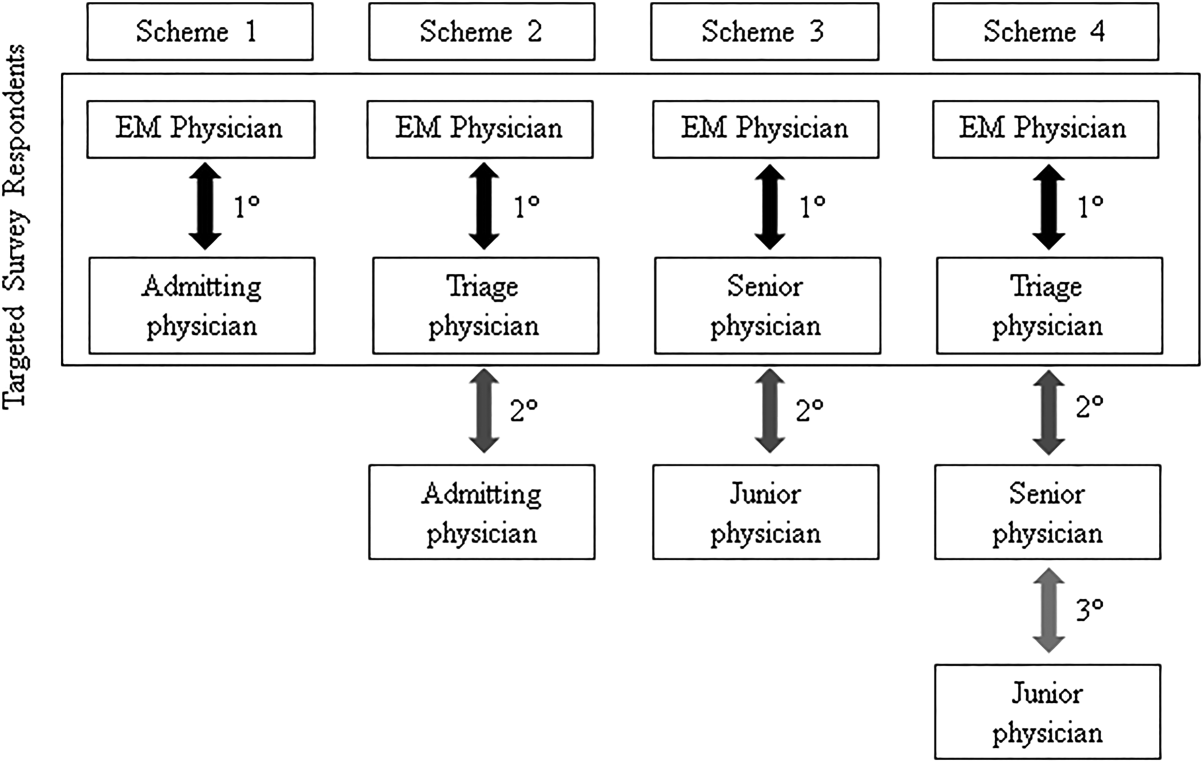
High-Flow Oxygen Therapy No Worse Than Noninvasive Ventilation, May Reduce Mortality
Clinical question: Does high-flow oxygen therapy result in a decreased rate of intubation for patients with nonhypercapnic acute hypoxemic respiratory failure?
Bottom line: In this underpowered study, the use of high-flow oxygen therapy did not significantly reduce the rate of intubation as compared with standard oxygen therapy or noninvasive positive pressure ventilation in patients with nonhypercapnic acute hypoxemic respiratory failure. However, patients in the high-flow oxygen group had decreased 90-day mortality, as well as an increased number of ventilator-free days. Patients in the high-flow oxygen group also reported less respiratory discomfort and dyspnea than patients in the other 2 groups. (LOE = 1b-)
Reference: Frat J, Thille AW, Mercat A, et al, for the FLORALI Study Group and the REVA Network. High-flow oxygen through nasal cannula in acute hypoxemic respiratory failure. N Engl J Med 2015;372(23):2185-2196.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (ICU only)
Synopsis: High-flow oxygen therapy uses oxygen delivered via nasal cannula at high flow rates to provide low-level positive pressure and reduce effective deadspace in the airways. Its effectiveness in the treatment of acute hypoxemic respiratory failure has not been established. In this study, investigators compared high-flow oxygen therapy with noninvasive positive pressure ventilation as well as with standard oxygen therapy in patients with nonhypercapnic acute hypoxemic respiratory failure.
Using concealed allocation, patients were randomized into 1 of 3 groups: (1) standard oxygen therapy using a nonrebreather face mask at a flow rate of 10 liters per minute or more; (2) high-flow oxygen therapy provided through a heated humidifier at a rate of 50 liters per minute for at least 2 days; or (3) noninvasive positive pressure ventilation for 8 hours per day for at least 2 days. With all 3 strategies, the goal was to maintain an oxygen saturation level of 92% or more. The 3 groups were similar at baseline with the majority of patients having community-acquired pneumonia as a cause of their acute respiratory failure. Analysis was by intention to treat.
For the primary outcome, the high-flow oxygen therapy group had a lower rate of intubation at 28 days than the other 2 groups, but this difference was not statistically significant (38% in high-flow group, 47% in standard group, 50% in noninvasive ventilation group; P = .18). Of note, the intubation rate in the standard oxygen therapy group was lower than the expected 60%, thus the study was underpowered to detect a difference if it truly exists. The high-flow therapy resulted in reduced 90-day mortality as compared with both standard therapy (hazard ratio [HR] = 2.01, 95% CI 1.01-3.99; P = .046) and noninvasive ventilation (HR = 2.50, 1.31-4.78; P = .006).
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: Does high-flow oxygen therapy result in a decreased rate of intubation for patients with nonhypercapnic acute hypoxemic respiratory failure?
Bottom line: In this underpowered study, the use of high-flow oxygen therapy did not significantly reduce the rate of intubation as compared with standard oxygen therapy or noninvasive positive pressure ventilation in patients with nonhypercapnic acute hypoxemic respiratory failure. However, patients in the high-flow oxygen group had decreased 90-day mortality, as well as an increased number of ventilator-free days. Patients in the high-flow oxygen group also reported less respiratory discomfort and dyspnea than patients in the other 2 groups. (LOE = 1b-)
Reference: Frat J, Thille AW, Mercat A, et al, for the FLORALI Study Group and the REVA Network. High-flow oxygen through nasal cannula in acute hypoxemic respiratory failure. N Engl J Med 2015;372(23):2185-2196.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (ICU only)
Synopsis: High-flow oxygen therapy uses oxygen delivered via nasal cannula at high flow rates to provide low-level positive pressure and reduce effective deadspace in the airways. Its effectiveness in the treatment of acute hypoxemic respiratory failure has not been established. In this study, investigators compared high-flow oxygen therapy with noninvasive positive pressure ventilation as well as with standard oxygen therapy in patients with nonhypercapnic acute hypoxemic respiratory failure.
Using concealed allocation, patients were randomized into 1 of 3 groups: (1) standard oxygen therapy using a nonrebreather face mask at a flow rate of 10 liters per minute or more; (2) high-flow oxygen therapy provided through a heated humidifier at a rate of 50 liters per minute for at least 2 days; or (3) noninvasive positive pressure ventilation for 8 hours per day for at least 2 days. With all 3 strategies, the goal was to maintain an oxygen saturation level of 92% or more. The 3 groups were similar at baseline with the majority of patients having community-acquired pneumonia as a cause of their acute respiratory failure. Analysis was by intention to treat.
For the primary outcome, the high-flow oxygen therapy group had a lower rate of intubation at 28 days than the other 2 groups, but this difference was not statistically significant (38% in high-flow group, 47% in standard group, 50% in noninvasive ventilation group; P = .18). Of note, the intubation rate in the standard oxygen therapy group was lower than the expected 60%, thus the study was underpowered to detect a difference if it truly exists. The high-flow therapy resulted in reduced 90-day mortality as compared with both standard therapy (hazard ratio [HR] = 2.01, 95% CI 1.01-3.99; P = .046) and noninvasive ventilation (HR = 2.50, 1.31-4.78; P = .006).
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: Does high-flow oxygen therapy result in a decreased rate of intubation for patients with nonhypercapnic acute hypoxemic respiratory failure?
Bottom line: In this underpowered study, the use of high-flow oxygen therapy did not significantly reduce the rate of intubation as compared with standard oxygen therapy or noninvasive positive pressure ventilation in patients with nonhypercapnic acute hypoxemic respiratory failure. However, patients in the high-flow oxygen group had decreased 90-day mortality, as well as an increased number of ventilator-free days. Patients in the high-flow oxygen group also reported less respiratory discomfort and dyspnea than patients in the other 2 groups. (LOE = 1b-)
Reference: Frat J, Thille AW, Mercat A, et al, for the FLORALI Study Group and the REVA Network. High-flow oxygen through nasal cannula in acute hypoxemic respiratory failure. N Engl J Med 2015;372(23):2185-2196.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (ICU only)
Synopsis: High-flow oxygen therapy uses oxygen delivered via nasal cannula at high flow rates to provide low-level positive pressure and reduce effective deadspace in the airways. Its effectiveness in the treatment of acute hypoxemic respiratory failure has not been established. In this study, investigators compared high-flow oxygen therapy with noninvasive positive pressure ventilation as well as with standard oxygen therapy in patients with nonhypercapnic acute hypoxemic respiratory failure.
Using concealed allocation, patients were randomized into 1 of 3 groups: (1) standard oxygen therapy using a nonrebreather face mask at a flow rate of 10 liters per minute or more; (2) high-flow oxygen therapy provided through a heated humidifier at a rate of 50 liters per minute for at least 2 days; or (3) noninvasive positive pressure ventilation for 8 hours per day for at least 2 days. With all 3 strategies, the goal was to maintain an oxygen saturation level of 92% or more. The 3 groups were similar at baseline with the majority of patients having community-acquired pneumonia as a cause of their acute respiratory failure. Analysis was by intention to treat.
For the primary outcome, the high-flow oxygen therapy group had a lower rate of intubation at 28 days than the other 2 groups, but this difference was not statistically significant (38% in high-flow group, 47% in standard group, 50% in noninvasive ventilation group; P = .18). Of note, the intubation rate in the standard oxygen therapy group was lower than the expected 60%, thus the study was underpowered to detect a difference if it truly exists. The high-flow therapy resulted in reduced 90-day mortality as compared with both standard therapy (hazard ratio [HR] = 2.01, 95% CI 1.01-3.99; P = .046) and noninvasive ventilation (HR = 2.50, 1.31-4.78; P = .006).
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Patient Navigators Reduce Readmission Rates for High-Risk Older Patients
Clinical question: Does the use of a patient navigator to guide hospitalized patients through the health care system reduce 30-day readmission rates?
Bottom line: The use of a community health worker acting as a patient navigator (PN), both during hospitalization and after discharge, to assist patients with coordination of care, follow-up appointments, provider communication, and medication compliance decreases the 30-day readmission rate in older high-risk patients, but increased admissions in younger patients, suggesting that the younger population may require different strategies to decrease their use of hospital-based care. (LOE = 1b-)
Reference: Balaban RB, Galbraith AA, Burns ME, Vialle-Valentin CE, Larochelle MR, Ross-Degnan D. A patient navigator intervention to reduce hospital readmissions among high-risk safety-net patients: a randomized controlled trial. J Gen Intern Med 2015;30(7):907-915.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (any location) with outpatient follow-up
Synopsis: This US study took place within a safety-net hospital system in Massachusetts that has a large underserved patient population. The authors enrolled more than 1500 hospitalized patients with at least 1 of 5 risk factors for readmission (older than 60 years, previous hospitalization within the last 6 months, length of stay of 3 days or more, or admission diagnoses of heart failure or chronic obstructive pulmonary disease). Patients in the intervention group were assigned to a hospital-based community health worker, or PN, while patients in the control group received usual care. The PN's primary responsibility was helping the patient navigate through the health care system, including assessing postdischarge needs, assisting with communication with inpatient providers and primary care physicians, confirming and rescheduling follow-up appointments, addressing barriers to taking medications, and assisting with transportation and insurance issues. These services were provided through a hospital visit and at least 3 weekly postdischarge phone calls. Patients in both groups were racially diverse and the majority carried public insurance. The patients older than 60 years were more medically complex, but younger patients had more psychiatric disorders and substance use disorders, as well as higher rates of previous hospitalizations and longer lengths of stay.
Overall, the 30-day readmission rate did not differ significantly between the control and intervention groups, but the study was underpowered given the limits to enrollment during the prespecified period. In an adjusted analysis of the 2 age subgroups, however, readmissions decreased by 4% in older intervention patients and they increased by 12% in younger intervention patients (both differences statistically significant).
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: Does the use of a patient navigator to guide hospitalized patients through the health care system reduce 30-day readmission rates?
Bottom line: The use of a community health worker acting as a patient navigator (PN), both during hospitalization and after discharge, to assist patients with coordination of care, follow-up appointments, provider communication, and medication compliance decreases the 30-day readmission rate in older high-risk patients, but increased admissions in younger patients, suggesting that the younger population may require different strategies to decrease their use of hospital-based care. (LOE = 1b-)
Reference: Balaban RB, Galbraith AA, Burns ME, Vialle-Valentin CE, Larochelle MR, Ross-Degnan D. A patient navigator intervention to reduce hospital readmissions among high-risk safety-net patients: a randomized controlled trial. J Gen Intern Med 2015;30(7):907-915.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (any location) with outpatient follow-up
Synopsis: This US study took place within a safety-net hospital system in Massachusetts that has a large underserved patient population. The authors enrolled more than 1500 hospitalized patients with at least 1 of 5 risk factors for readmission (older than 60 years, previous hospitalization within the last 6 months, length of stay of 3 days or more, or admission diagnoses of heart failure or chronic obstructive pulmonary disease). Patients in the intervention group were assigned to a hospital-based community health worker, or PN, while patients in the control group received usual care. The PN's primary responsibility was helping the patient navigate through the health care system, including assessing postdischarge needs, assisting with communication with inpatient providers and primary care physicians, confirming and rescheduling follow-up appointments, addressing barriers to taking medications, and assisting with transportation and insurance issues. These services were provided through a hospital visit and at least 3 weekly postdischarge phone calls. Patients in both groups were racially diverse and the majority carried public insurance. The patients older than 60 years were more medically complex, but younger patients had more psychiatric disorders and substance use disorders, as well as higher rates of previous hospitalizations and longer lengths of stay.
Overall, the 30-day readmission rate did not differ significantly between the control and intervention groups, but the study was underpowered given the limits to enrollment during the prespecified period. In an adjusted analysis of the 2 age subgroups, however, readmissions decreased by 4% in older intervention patients and they increased by 12% in younger intervention patients (both differences statistically significant).
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: Does the use of a patient navigator to guide hospitalized patients through the health care system reduce 30-day readmission rates?
Bottom line: The use of a community health worker acting as a patient navigator (PN), both during hospitalization and after discharge, to assist patients with coordination of care, follow-up appointments, provider communication, and medication compliance decreases the 30-day readmission rate in older high-risk patients, but increased admissions in younger patients, suggesting that the younger population may require different strategies to decrease their use of hospital-based care. (LOE = 1b-)
Reference: Balaban RB, Galbraith AA, Burns ME, Vialle-Valentin CE, Larochelle MR, Ross-Degnan D. A patient navigator intervention to reduce hospital readmissions among high-risk safety-net patients: a randomized controlled trial. J Gen Intern Med 2015;30(7):907-915.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (any location) with outpatient follow-up
Synopsis: This US study took place within a safety-net hospital system in Massachusetts that has a large underserved patient population. The authors enrolled more than 1500 hospitalized patients with at least 1 of 5 risk factors for readmission (older than 60 years, previous hospitalization within the last 6 months, length of stay of 3 days or more, or admission diagnoses of heart failure or chronic obstructive pulmonary disease). Patients in the intervention group were assigned to a hospital-based community health worker, or PN, while patients in the control group received usual care. The PN's primary responsibility was helping the patient navigate through the health care system, including assessing postdischarge needs, assisting with communication with inpatient providers and primary care physicians, confirming and rescheduling follow-up appointments, addressing barriers to taking medications, and assisting with transportation and insurance issues. These services were provided through a hospital visit and at least 3 weekly postdischarge phone calls. Patients in both groups were racially diverse and the majority carried public insurance. The patients older than 60 years were more medically complex, but younger patients had more psychiatric disorders and substance use disorders, as well as higher rates of previous hospitalizations and longer lengths of stay.
Overall, the 30-day readmission rate did not differ significantly between the control and intervention groups, but the study was underpowered given the limits to enrollment during the prespecified period. In an adjusted analysis of the 2 age subgroups, however, readmissions decreased by 4% in older intervention patients and they increased by 12% in younger intervention patients (both differences statistically significant).
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Is bread bad?
My 27-year-old patient with a body mass index of 64 kg/m2 presented to my clinic with back and leg pain. Unfortunately, I was not the least bit surprised.
When bending over, he developed acute onset of back pain, with radiating pain down the lateral side of his right leg to his foot. Exam and history were consistent with lumbar radiculopathy.

Over the next several months, the patient had intractable pain in the presence of escalating opioids. The neurosurgeons said that his weight created too high a risk for intraoperative and postoperative complications, with which I agreed.
And so began the work of titrating his pain-modulating agents, along with the significantly less glamorous and substantially more challenging task of helping him lose weight.
He was staunchly opposed to bariatric surgery and could not afford weight loss medications, presenting a bit of an impasse. To me, the fact that his insurance company would have covered his spinal surgery but not his FDA-approved weight loss medications embodies one of the great medical mysteries of modern times.
His mother accompanied him on one of his several visits and chimed in, “He needs to eat bread, doesn’t he?”
One of my common weight loss counseling mantras is “no whites for breakfast, lunch, or dinner.” These whites would include rice, bread, pasta, and potatoes. The airwaves have been crackling for a while with calls to decrease carbohydrate consumption to combat the obesity epidemic and to eat bread only if you’re a duck. As a result, bread consumption has declined worldwide.
So how bad is bread? Are all breads the same?
Luis Serra-Majem and Inmaculada Bautista-Castano of the University of Las Palmas de Gran Canaria, Spain, conducted a systematic review of the impact of bread consumption on obesity and abdominal adiposity (Br. J. Nutr. 2015;113:S29-S35). The authors concluded that white (refined grain) bread, but not whole-grain bread, may be associated with excess abdominal fat.
Proposed hypotheses for how breads impact adiposity differently are:
1. Whole-grain bread increases satiety more than white bread;
2. Whole-grain bread results in lower plasma glucose and insulin responses than white bread;
3. Higher fiber content of whole-grain bread limits glucose absorption more than white bread; and
4. Whole-grain bread may positively influence gut microbiota through a probiotic effect.
My advice to the patient was to restrict calories, avoid carbohydrates, and if bread must be consumed, then it must be whole grain. Baby steps.
But he found religion in this (and in walking) and lost 200 pounds over the next 5 years. Miracles are still possible.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.
My 27-year-old patient with a body mass index of 64 kg/m2 presented to my clinic with back and leg pain. Unfortunately, I was not the least bit surprised.
When bending over, he developed acute onset of back pain, with radiating pain down the lateral side of his right leg to his foot. Exam and history were consistent with lumbar radiculopathy.
Over the next several months, the patient had intractable pain in the presence of escalating opioids. The neurosurgeons said that his weight created too high a risk for intraoperative and postoperative complications, with which I agreed.
And so began the work of titrating his pain-modulating agents, along with the significantly less glamorous and substantially more challenging task of helping him lose weight.
He was staunchly opposed to bariatric surgery and could not afford weight loss medications, presenting a bit of an impasse. To me, the fact that his insurance company would have covered his spinal surgery but not his FDA-approved weight loss medications embodies one of the great medical mysteries of modern times.
His mother accompanied him on one of his several visits and chimed in, “He needs to eat bread, doesn’t he?”
One of my common weight loss counseling mantras is “no whites for breakfast, lunch, or dinner.” These whites would include rice, bread, pasta, and potatoes. The airwaves have been crackling for a while with calls to decrease carbohydrate consumption to combat the obesity epidemic and to eat bread only if you’re a duck. As a result, bread consumption has declined worldwide.
So how bad is bread? Are all breads the same?
Luis Serra-Majem and Inmaculada Bautista-Castano of the University of Las Palmas de Gran Canaria, Spain, conducted a systematic review of the impact of bread consumption on obesity and abdominal adiposity (Br. J. Nutr. 2015;113:S29-S35). The authors concluded that white (refined grain) bread, but not whole-grain bread, may be associated with excess abdominal fat.
Proposed hypotheses for how breads impact adiposity differently are:
1. Whole-grain bread increases satiety more than white bread;
2. Whole-grain bread results in lower plasma glucose and insulin responses than white bread;
3. Higher fiber content of whole-grain bread limits glucose absorption more than white bread; and
4. Whole-grain bread may positively influence gut microbiota through a probiotic effect.
My advice to the patient was to restrict calories, avoid carbohydrates, and if bread must be consumed, then it must be whole grain. Baby steps.
But he found religion in this (and in walking) and lost 200 pounds over the next 5 years. Miracles are still possible.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.
My 27-year-old patient with a body mass index of 64 kg/m2 presented to my clinic with back and leg pain. Unfortunately, I was not the least bit surprised.
When bending over, he developed acute onset of back pain, with radiating pain down the lateral side of his right leg to his foot. Exam and history were consistent with lumbar radiculopathy.
Over the next several months, the patient had intractable pain in the presence of escalating opioids. The neurosurgeons said that his weight created too high a risk for intraoperative and postoperative complications, with which I agreed.
And so began the work of titrating his pain-modulating agents, along with the significantly less glamorous and substantially more challenging task of helping him lose weight.
He was staunchly opposed to bariatric surgery and could not afford weight loss medications, presenting a bit of an impasse. To me, the fact that his insurance company would have covered his spinal surgery but not his FDA-approved weight loss medications embodies one of the great medical mysteries of modern times.
His mother accompanied him on one of his several visits and chimed in, “He needs to eat bread, doesn’t he?”
One of my common weight loss counseling mantras is “no whites for breakfast, lunch, or dinner.” These whites would include rice, bread, pasta, and potatoes. The airwaves have been crackling for a while with calls to decrease carbohydrate consumption to combat the obesity epidemic and to eat bread only if you’re a duck. As a result, bread consumption has declined worldwide.
So how bad is bread? Are all breads the same?
Luis Serra-Majem and Inmaculada Bautista-Castano of the University of Las Palmas de Gran Canaria, Spain, conducted a systematic review of the impact of bread consumption on obesity and abdominal adiposity (Br. J. Nutr. 2015;113:S29-S35). The authors concluded that white (refined grain) bread, but not whole-grain bread, may be associated with excess abdominal fat.
Proposed hypotheses for how breads impact adiposity differently are:
1. Whole-grain bread increases satiety more than white bread;
2. Whole-grain bread results in lower plasma glucose and insulin responses than white bread;
3. Higher fiber content of whole-grain bread limits glucose absorption more than white bread; and
4. Whole-grain bread may positively influence gut microbiota through a probiotic effect.
My advice to the patient was to restrict calories, avoid carbohydrates, and if bread must be consumed, then it must be whole grain. Baby steps.
But he found religion in this (and in walking) and lost 200 pounds over the next 5 years. Miracles are still possible.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.

Why, oh why, won’t she go to bed?!
To weary parents, the fact that their child does not want to go to bed at night is both puzzling and exasperating. No matter what age, it is important to have a healthy bedtime for the child’s well-being as well as their caregiver’s!
Sleep, like the “canary in the mine,” is vulnerable to disruption by anything from minor illness to changes in schedule, things viewed on media, or emotions in the home, to life changes such as a new sibling, toilet training, or a new school year. In patients on stimulant medicine resisting bedtime, consider the need to finally eat. Asking about these specifically will help you plan an appropriate time to successfully address bedtime conflicts.

Knowing some basic principles about falling asleep can help your counseling. “Sleep drive” builds up over the day like a coiled spring, making falling asleep easier initially than for wakings later in the night. It also means that a nap too close to bedtime reduces the drive. Avoiding any naps (after age 4 years) and naps lasting past 4 p.m. for children under 4, even in car rides, is crucial. Beware of teens “having trouble falling asleep” who have sneaked in a nap after school!
To optimize sleep drive, calculate age-related sleep needs and, ideally, ask parents to keep a sleep diary for 1-2 weeks, especially checking on naps at daycare. Updated sleep duration standards (preschool 10-13 hours; school age 9-11 hours; teens 8-10 hours (see sleepfoundation.org) show that ranges of total sleep are remarkably stable, but may not meet parents’ ideals for time “off duty.”
If placed in bed when not yet tired, anyone will have trouble falling asleep (phase shift). For a child, lying awake in the dark alone is time for active imaginations to conjure fear of separation (> 4-6 months), monsters (for preschoolers), burglars (for anxious school-aged children), or the next day’s social or academic stresses (for school-aged children to teens). Children with anxiety disorders even worry that they may not get enough sleep! A soothing routine with a story in their bed (not media), a spritzing of “monster spray” or a “bedtime ticket” to cash in for “one more thing” (for those who beg) will usually suffice for preschoolers. A “magic flashlight” lends the child some control to check for monsters, but an “exorcism ritual” by the all-powerful parent can be added if needed. Teens are never too old for a chance to talk or even a story read by the parent (but they won’t ask for this).
A secret to managing bedtime struggles is to start the routine at the time the child is now falling asleep to avoid resistance, and keeping wake-up time appropriate to the new schedule. Once falling asleep within 10 minutes, move bedtime 15 minutes earlier each night to reach the schedule, then maintain 7 days per week (or within 1 hour) to prevent resetting the biological clock. Sorry, no movie nights until 2 a.m. or “sleeping in” on weekends! Teens who resist bedtime may be napping, socializing at the “only time” peers are up, addicted to media, or avoiding family. Their cooperation must be engaged to make a change by staying up all night once, then setting a new schedule, or staying up 1 hour later each night until the desired bedtime is reached.

Because sleep includes being paralyzed (REM stages), evolution encourages animals to sleep together to protect from predators. Sleeping alone requires a great deal of reassurance, such as from a friendly atmosphere, favorite stuffed toy, and familiar routine that implies safety. Children could use a toy, pet, or even a sibling to feel safe. While body contact is the most reassuring, children may not return to sleep from the many normal night wakings without it. Most can be weaned from this dependence by the parent sitting by the bed silently, moving one foot closer to the door each night. A promise to “check on you” in 5 minutes also helps.
Other factors making sleep easier include avoiding caffeine, stimulating medicines, or nicotine as well as exciting games, media, or exercise within 2 hours of bedtime. A quiet, dark, cool but not damp location used only for sleeping is helpful, but not always possible. A white noise generator, fan, or radio on static can help.
Many parents strangely expect their child to give up the pleasures of the day and take themselves to bed! As for other kinds of limit setting, parental company is typically needed for brushing teeth, pajamas, and a story. Ideally, it can be fun as a race or with songs, not a yelling match, which undermines the sense of safety. Setting rules about no electronics in the bedroom, even for charging, after a certain hour is often the only solution (even for adults) to the common struggle over ending media.
Often the bedtime complaint is “curtain calls” after being settled in bed. For children who call out, advise parents wait a few moments before responding, then reassure verbally without entering their room, waiting longer each time. For the child getting out of bed, one parent should lead them back without talking at all as many times as necessary. A reward for staying in bed without calling or coming out can be an extra story the next night and/or a morning reward. Alternatively, close the door and turn off the light if they come out or call out. After a few moments in the dark, give a “second chance” as long as they are quietly in bed. Gating the doorway works well for toddlers – sleeping on the floor is not dangerous!
Families often are ambivalent about asking for bedtime advice, thinking your solutions might be stressful, harmful to their relationship, or will take up their own precious sleep. For many, a prolonged bedtime is the best part of their day. Special Time earlier is often key to enforcing a healthy bedtime. Reassure them that these solutions usually take less than 3 weeks!
For the child, bedtime means giving up on fun but, more importantly, separation from the parent. This separation is harder if negative emotions are left from a day of behavior struggles or parents are even subtly angry at each other. For parents, bedtime means separating from their main pleasure in life. They also may be regretting their interactions during the day. Ambivalence about parting also may come from fears of being alone with their partner due to marital discord, intimate partner violence, chronic arguing, substance use, or simply depression. When simple advice fails, it is important to explore these meanings with families, encourage positive daytime behavior management methods, and avoid conflict in front of the children to resolve bedtime struggles.
Dr. Howard is assistant professor of pediatrics at the Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS (www.CHADIS.com). She has no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline. E-mail her at [email protected].
To weary parents, the fact that their child does not want to go to bed at night is both puzzling and exasperating. No matter what age, it is important to have a healthy bedtime for the child’s well-being as well as their caregiver’s!
Sleep, like the “canary in the mine,” is vulnerable to disruption by anything from minor illness to changes in schedule, things viewed on media, or emotions in the home, to life changes such as a new sibling, toilet training, or a new school year. In patients on stimulant medicine resisting bedtime, consider the need to finally eat. Asking about these specifically will help you plan an appropriate time to successfully address bedtime conflicts.
Knowing some basic principles about falling asleep can help your counseling. “Sleep drive” builds up over the day like a coiled spring, making falling asleep easier initially than for wakings later in the night. It also means that a nap too close to bedtime reduces the drive. Avoiding any naps (after age 4 years) and naps lasting past 4 p.m. for children under 4, even in car rides, is crucial. Beware of teens “having trouble falling asleep” who have sneaked in a nap after school!
To optimize sleep drive, calculate age-related sleep needs and, ideally, ask parents to keep a sleep diary for 1-2 weeks, especially checking on naps at daycare. Updated sleep duration standards (preschool 10-13 hours; school age 9-11 hours; teens 8-10 hours (see sleepfoundation.org) show that ranges of total sleep are remarkably stable, but may not meet parents’ ideals for time “off duty.”
If placed in bed when not yet tired, anyone will have trouble falling asleep (phase shift). For a child, lying awake in the dark alone is time for active imaginations to conjure fear of separation (> 4-6 months), monsters (for preschoolers), burglars (for anxious school-aged children), or the next day’s social or academic stresses (for school-aged children to teens). Children with anxiety disorders even worry that they may not get enough sleep! A soothing routine with a story in their bed (not media), a spritzing of “monster spray” or a “bedtime ticket” to cash in for “one more thing” (for those who beg) will usually suffice for preschoolers. A “magic flashlight” lends the child some control to check for monsters, but an “exorcism ritual” by the all-powerful parent can be added if needed. Teens are never too old for a chance to talk or even a story read by the parent (but they won’t ask for this).
A secret to managing bedtime struggles is to start the routine at the time the child is now falling asleep to avoid resistance, and keeping wake-up time appropriate to the new schedule. Once falling asleep within 10 minutes, move bedtime 15 minutes earlier each night to reach the schedule, then maintain 7 days per week (or within 1 hour) to prevent resetting the biological clock. Sorry, no movie nights until 2 a.m. or “sleeping in” on weekends! Teens who resist bedtime may be napping, socializing at the “only time” peers are up, addicted to media, or avoiding family. Their cooperation must be engaged to make a change by staying up all night once, then setting a new schedule, or staying up 1 hour later each night until the desired bedtime is reached.
Because sleep includes being paralyzed (REM stages), evolution encourages animals to sleep together to protect from predators. Sleeping alone requires a great deal of reassurance, such as from a friendly atmosphere, favorite stuffed toy, and familiar routine that implies safety. Children could use a toy, pet, or even a sibling to feel safe. While body contact is the most reassuring, children may not return to sleep from the many normal night wakings without it. Most can be weaned from this dependence by the parent sitting by the bed silently, moving one foot closer to the door each night. A promise to “check on you” in 5 minutes also helps.
Other factors making sleep easier include avoiding caffeine, stimulating medicines, or nicotine as well as exciting games, media, or exercise within 2 hours of bedtime. A quiet, dark, cool but not damp location used only for sleeping is helpful, but not always possible. A white noise generator, fan, or radio on static can help.
Many parents strangely expect their child to give up the pleasures of the day and take themselves to bed! As for other kinds of limit setting, parental company is typically needed for brushing teeth, pajamas, and a story. Ideally, it can be fun as a race or with songs, not a yelling match, which undermines the sense of safety. Setting rules about no electronics in the bedroom, even for charging, after a certain hour is often the only solution (even for adults) to the common struggle over ending media.
Often the bedtime complaint is “curtain calls” after being settled in bed. For children who call out, advise parents wait a few moments before responding, then reassure verbally without entering their room, waiting longer each time. For the child getting out of bed, one parent should lead them back without talking at all as many times as necessary. A reward for staying in bed without calling or coming out can be an extra story the next night and/or a morning reward. Alternatively, close the door and turn off the light if they come out or call out. After a few moments in the dark, give a “second chance” as long as they are quietly in bed. Gating the doorway works well for toddlers – sleeping on the floor is not dangerous!
Families often are ambivalent about asking for bedtime advice, thinking your solutions might be stressful, harmful to their relationship, or will take up their own precious sleep. For many, a prolonged bedtime is the best part of their day. Special Time earlier is often key to enforcing a healthy bedtime. Reassure them that these solutions usually take less than 3 weeks!
For the child, bedtime means giving up on fun but, more importantly, separation from the parent. This separation is harder if negative emotions are left from a day of behavior struggles or parents are even subtly angry at each other. For parents, bedtime means separating from their main pleasure in life. They also may be regretting their interactions during the day. Ambivalence about parting also may come from fears of being alone with their partner due to marital discord, intimate partner violence, chronic arguing, substance use, or simply depression. When simple advice fails, it is important to explore these meanings with families, encourage positive daytime behavior management methods, and avoid conflict in front of the children to resolve bedtime struggles.
Dr. Howard is assistant professor of pediatrics at the Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS (www.CHADIS.com). She has no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline. E-mail her at [email protected].
To weary parents, the fact that their child does not want to go to bed at night is both puzzling and exasperating. No matter what age, it is important to have a healthy bedtime for the child’s well-being as well as their caregiver’s!
Sleep, like the “canary in the mine,” is vulnerable to disruption by anything from minor illness to changes in schedule, things viewed on media, or emotions in the home, to life changes such as a new sibling, toilet training, or a new school year. In patients on stimulant medicine resisting bedtime, consider the need to finally eat. Asking about these specifically will help you plan an appropriate time to successfully address bedtime conflicts.
Knowing some basic principles about falling asleep can help your counseling. “Sleep drive” builds up over the day like a coiled spring, making falling asleep easier initially than for wakings later in the night. It also means that a nap too close to bedtime reduces the drive. Avoiding any naps (after age 4 years) and naps lasting past 4 p.m. for children under 4, even in car rides, is crucial. Beware of teens “having trouble falling asleep” who have sneaked in a nap after school!
To optimize sleep drive, calculate age-related sleep needs and, ideally, ask parents to keep a sleep diary for 1-2 weeks, especially checking on naps at daycare. Updated sleep duration standards (preschool 10-13 hours; school age 9-11 hours; teens 8-10 hours (see sleepfoundation.org) show that ranges of total sleep are remarkably stable, but may not meet parents’ ideals for time “off duty.”
If placed in bed when not yet tired, anyone will have trouble falling asleep (phase shift). For a child, lying awake in the dark alone is time for active imaginations to conjure fear of separation (> 4-6 months), monsters (for preschoolers), burglars (for anxious school-aged children), or the next day’s social or academic stresses (for school-aged children to teens). Children with anxiety disorders even worry that they may not get enough sleep! A soothing routine with a story in their bed (not media), a spritzing of “monster spray” or a “bedtime ticket” to cash in for “one more thing” (for those who beg) will usually suffice for preschoolers. A “magic flashlight” lends the child some control to check for monsters, but an “exorcism ritual” by the all-powerful parent can be added if needed. Teens are never too old for a chance to talk or even a story read by the parent (but they won’t ask for this).
A secret to managing bedtime struggles is to start the routine at the time the child is now falling asleep to avoid resistance, and keeping wake-up time appropriate to the new schedule. Once falling asleep within 10 minutes, move bedtime 15 minutes earlier each night to reach the schedule, then maintain 7 days per week (or within 1 hour) to prevent resetting the biological clock. Sorry, no movie nights until 2 a.m. or “sleeping in” on weekends! Teens who resist bedtime may be napping, socializing at the “only time” peers are up, addicted to media, or avoiding family. Their cooperation must be engaged to make a change by staying up all night once, then setting a new schedule, or staying up 1 hour later each night until the desired bedtime is reached.
Because sleep includes being paralyzed (REM stages), evolution encourages animals to sleep together to protect from predators. Sleeping alone requires a great deal of reassurance, such as from a friendly atmosphere, favorite stuffed toy, and familiar routine that implies safety. Children could use a toy, pet, or even a sibling to feel safe. While body contact is the most reassuring, children may not return to sleep from the many normal night wakings without it. Most can be weaned from this dependence by the parent sitting by the bed silently, moving one foot closer to the door each night. A promise to “check on you” in 5 minutes also helps.
Other factors making sleep easier include avoiding caffeine, stimulating medicines, or nicotine as well as exciting games, media, or exercise within 2 hours of bedtime. A quiet, dark, cool but not damp location used only for sleeping is helpful, but not always possible. A white noise generator, fan, or radio on static can help.
Many parents strangely expect their child to give up the pleasures of the day and take themselves to bed! As for other kinds of limit setting, parental company is typically needed for brushing teeth, pajamas, and a story. Ideally, it can be fun as a race or with songs, not a yelling match, which undermines the sense of safety. Setting rules about no electronics in the bedroom, even for charging, after a certain hour is often the only solution (even for adults) to the common struggle over ending media.
Often the bedtime complaint is “curtain calls” after being settled in bed. For children who call out, advise parents wait a few moments before responding, then reassure verbally without entering their room, waiting longer each time. For the child getting out of bed, one parent should lead them back without talking at all as many times as necessary. A reward for staying in bed without calling or coming out can be an extra story the next night and/or a morning reward. Alternatively, close the door and turn off the light if they come out or call out. After a few moments in the dark, give a “second chance” as long as they are quietly in bed. Gating the doorway works well for toddlers – sleeping on the floor is not dangerous!
Families often are ambivalent about asking for bedtime advice, thinking your solutions might be stressful, harmful to their relationship, or will take up their own precious sleep. For many, a prolonged bedtime is the best part of their day. Special Time earlier is often key to enforcing a healthy bedtime. Reassure them that these solutions usually take less than 3 weeks!
For the child, bedtime means giving up on fun but, more importantly, separation from the parent. This separation is harder if negative emotions are left from a day of behavior struggles or parents are even subtly angry at each other. For parents, bedtime means separating from their main pleasure in life. They also may be regretting their interactions during the day. Ambivalence about parting also may come from fears of being alone with their partner due to marital discord, intimate partner violence, chronic arguing, substance use, or simply depression. When simple advice fails, it is important to explore these meanings with families, encourage positive daytime behavior management methods, and avoid conflict in front of the children to resolve bedtime struggles.
Dr. Howard is assistant professor of pediatrics at the Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS (www.CHADIS.com). She has no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline. E-mail her at [email protected].
