User login
Current Thinking in OTC Analgesia: Patient Considerations and Practical Insights
December 2014
A supplement to Family Practice News® and Internal Medicine News®. This supplement is sponsored by McNeil Consumer Healthcare Division of McNEIL-PPC, Inc.

Click here to open the supplement
Topics
- Pain in the United States
- Critical Comorbid Illnesses
- Impact of Pain on Comorbidities and Patient Outcomes
- Pain Management for Patients Taking Concomitant Medications
- Benefits of OTC Analgesics
- Risks of OTC Analgesics
- A Nursing Perspective
- Not All Analgesics Are Appropriate for Every Patient
- Patients’ Safe Use of OTC Analgesics
- Conclusions
Faculty
Chairman
Charles P. Vega, Jr, MD, FAAFP
Clinical Professor, Department of Family Medicine
Director, Patient-Centered Advanced Clinical Education
Director, Program in Medical Education for the Latino Community
University of California, Irvine, School of Medicine
Irvine, California
Faculty
Christopher M. Chappel, MD
Medical Director
Chappel Health and Wellness
Kissimmee, Florida
Brett Badgley Snodgrass, MSN, APRN, FNP-BC
Family Nurse Practitioner
Saint Francis Hospital–Bartlett Interventional Pain Management Clinic
Chief Executive Officer and President
BBS Consultants, Inc.
Bartlett, Tennessee
Copyright © 2014 by Frontline Medical Communications Inc.
December 2014
A supplement to Family Practice News® and Internal Medicine News®. This supplement is sponsored by McNeil Consumer Healthcare Division of McNEIL-PPC, Inc.
Click here to open the supplement
Topics
- Pain in the United States
- Critical Comorbid Illnesses
- Impact of Pain on Comorbidities and Patient Outcomes
- Pain Management for Patients Taking Concomitant Medications
- Benefits of OTC Analgesics
- Risks of OTC Analgesics
- A Nursing Perspective
- Not All Analgesics Are Appropriate for Every Patient
- Patients’ Safe Use of OTC Analgesics
- Conclusions
Faculty
Chairman
Charles P. Vega, Jr, MD, FAAFP
Clinical Professor, Department of Family Medicine
Director, Patient-Centered Advanced Clinical Education
Director, Program in Medical Education for the Latino Community
University of California, Irvine, School of Medicine
Irvine, California
Faculty
Christopher M. Chappel, MD
Medical Director
Chappel Health and Wellness
Kissimmee, Florida
Brett Badgley Snodgrass, MSN, APRN, FNP-BC
Family Nurse Practitioner
Saint Francis Hospital–Bartlett Interventional Pain Management Clinic
Chief Executive Officer and President
BBS Consultants, Inc.
Bartlett, Tennessee
Copyright © 2014 by Frontline Medical Communications Inc.
December 2014
A supplement to Family Practice News® and Internal Medicine News®. This supplement is sponsored by McNeil Consumer Healthcare Division of McNEIL-PPC, Inc.
Click here to open the supplement
Topics
- Pain in the United States
- Critical Comorbid Illnesses
- Impact of Pain on Comorbidities and Patient Outcomes
- Pain Management for Patients Taking Concomitant Medications
- Benefits of OTC Analgesics
- Risks of OTC Analgesics
- A Nursing Perspective
- Not All Analgesics Are Appropriate for Every Patient
- Patients’ Safe Use of OTC Analgesics
- Conclusions
Faculty
Chairman
Charles P. Vega, Jr, MD, FAAFP
Clinical Professor, Department of Family Medicine
Director, Patient-Centered Advanced Clinical Education
Director, Program in Medical Education for the Latino Community
University of California, Irvine, School of Medicine
Irvine, California
Faculty
Christopher M. Chappel, MD
Medical Director
Chappel Health and Wellness
Kissimmee, Florida
Brett Badgley Snodgrass, MSN, APRN, FNP-BC
Family Nurse Practitioner
Saint Francis Hospital–Bartlett Interventional Pain Management Clinic
Chief Executive Officer and President
BBS Consultants, Inc.
Bartlett, Tennessee
Copyright © 2014 by Frontline Medical Communications Inc.

Product News: 12 2014
Heads Up! Education Program
The Skin Cancer Foundation is encouraging dermatologists to participate in Heads Up!, an education program that provides beauty professionals with tips on what to look for and how to speak to their clients if they spot a suspicious lesion. Dermatologists can host an educational event where hairstylists and aestheticians will learn about skin cancer and its warning signs. Because early detection is critical, the Heads Up! program ensures that this group of first responders will be prepared to give a “heads up” to their clients if they see something suspicious, encouraging the client to visit a dermatologist in a timely manner. For more information, visit www.skincancer.org/headsup.
Heliocare and Vitamin Angels
Ferndale Healthcare, Inc, announces support for Vitamin Angels and Walgreens with its Heliocare brand. Vitamin Angels helps at-risk populations in need, specifically pregnant women, new mothers, and children younger than 5 years, gain access to vitamins and minerals. Through 2017, a percentage of each Heliocare purchase at any Walgreens location will be donated to help Vitamin Angels. For more information, visit www.walgreens.com/vitaminangels.
Promiseb Topical Cream
Promius Pharma, LLC, introduces Promiseb Topical Cream in a 60-g box. Promiseb Topical Cream is a nonsteroidal cream for the management of seborrhea and seborrheic dermatitis that has demonstrated both anti-inflammatory and antifungal properties. The 60-g box provides an option for treatment of larger body surface areas, which may necessitate fewer refills. For more information, visit www.promiseb.com.
Total Relief Shampoo and Conditioner
Dr. Marder Skincare presents an over-the-counter scalp solution for itching, flaking, and scaling. This dermatologist-formulated hydrocortisone shampoo and conditioner can be used to relieve symptoms of psoriasis, seborrhea, and dandruff. Both products can be purchased online. For more information, visit www.drmarderskincare.com.
If you would like your product included in Product News, please e-mail a press release to the Editorial Office at [email protected]
Heads Up! Education Program
The Skin Cancer Foundation is encouraging dermatologists to participate in Heads Up!, an education program that provides beauty professionals with tips on what to look for and how to speak to their clients if they spot a suspicious lesion. Dermatologists can host an educational event where hairstylists and aestheticians will learn about skin cancer and its warning signs. Because early detection is critical, the Heads Up! program ensures that this group of first responders will be prepared to give a “heads up” to their clients if they see something suspicious, encouraging the client to visit a dermatologist in a timely manner. For more information, visit www.skincancer.org/headsup.
Heliocare and Vitamin Angels
Ferndale Healthcare, Inc, announces support for Vitamin Angels and Walgreens with its Heliocare brand. Vitamin Angels helps at-risk populations in need, specifically pregnant women, new mothers, and children younger than 5 years, gain access to vitamins and minerals. Through 2017, a percentage of each Heliocare purchase at any Walgreens location will be donated to help Vitamin Angels. For more information, visit www.walgreens.com/vitaminangels.
Promiseb Topical Cream
Promius Pharma, LLC, introduces Promiseb Topical Cream in a 60-g box. Promiseb Topical Cream is a nonsteroidal cream for the management of seborrhea and seborrheic dermatitis that has demonstrated both anti-inflammatory and antifungal properties. The 60-g box provides an option for treatment of larger body surface areas, which may necessitate fewer refills. For more information, visit www.promiseb.com.
Total Relief Shampoo and Conditioner
Dr. Marder Skincare presents an over-the-counter scalp solution for itching, flaking, and scaling. This dermatologist-formulated hydrocortisone shampoo and conditioner can be used to relieve symptoms of psoriasis, seborrhea, and dandruff. Both products can be purchased online. For more information, visit www.drmarderskincare.com.
If you would like your product included in Product News, please e-mail a press release to the Editorial Office at [email protected]
Heads Up! Education Program
The Skin Cancer Foundation is encouraging dermatologists to participate in Heads Up!, an education program that provides beauty professionals with tips on what to look for and how to speak to their clients if they spot a suspicious lesion. Dermatologists can host an educational event where hairstylists and aestheticians will learn about skin cancer and its warning signs. Because early detection is critical, the Heads Up! program ensures that this group of first responders will be prepared to give a “heads up” to their clients if they see something suspicious, encouraging the client to visit a dermatologist in a timely manner. For more information, visit www.skincancer.org/headsup.
Heliocare and Vitamin Angels
Ferndale Healthcare, Inc, announces support for Vitamin Angels and Walgreens with its Heliocare brand. Vitamin Angels helps at-risk populations in need, specifically pregnant women, new mothers, and children younger than 5 years, gain access to vitamins and minerals. Through 2017, a percentage of each Heliocare purchase at any Walgreens location will be donated to help Vitamin Angels. For more information, visit www.walgreens.com/vitaminangels.
Promiseb Topical Cream
Promius Pharma, LLC, introduces Promiseb Topical Cream in a 60-g box. Promiseb Topical Cream is a nonsteroidal cream for the management of seborrhea and seborrheic dermatitis that has demonstrated both anti-inflammatory and antifungal properties. The 60-g box provides an option for treatment of larger body surface areas, which may necessitate fewer refills. For more information, visit www.promiseb.com.
Total Relief Shampoo and Conditioner
Dr. Marder Skincare presents an over-the-counter scalp solution for itching, flaking, and scaling. This dermatologist-formulated hydrocortisone shampoo and conditioner can be used to relieve symptoms of psoriasis, seborrhea, and dandruff. Both products can be purchased online. For more information, visit www.drmarderskincare.com.
If you would like your product included in Product News, please e-mail a press release to the Editorial Office at [email protected]

Magnesium disappoints in sickle cell disease

sickle cell disease
Credit: St. Jude Hospital
SAN FRANCISCO—Magnesium does not improve outcomes in children hospitalized for sickle cell pain crises, results of the MAGiC study suggest.
Researchers hypothesized that magnesium—a known vasodilator, anti-inflammatory, and pain reliever—could alter the pathophysiology of pain crises.
However, when compared to normal saline, intravenous (IV) magnesium did not shorten hospital stays, lessen opioid use, or improve patients’ quality of life.
David C. Brousseau, MD, of the Medical College of Wisconsin and the Children’s Hospital of Wisconsin in Milwaukee, presented the results of this study at the 2014 ASH Annual Meeting (abstract 88).
Dr Brousseau noted that vasoocclusive crises are the most common acute complication of sickle cell disease and the most frequent cause of acute care or emergency department visits and hospitalizations. But recent changes in treatment have been minimal, with the judicious use of IV fluid and IV opioids being the mainstays of therapy.
“There have been few multicenter clinical trials evaluating new treatments, in part, due to a long history of difficulty with enrollment in interventional trials for sickle cell crises,” he continued. “These enrollment difficulties have been due to an inability to consent or to consent in a timely manner, leading to delayed initiation of study drug.”
With the MAGiC trial, Dr Brousseau and his colleagues sought to overcome this problem through a collaboration between pediatric emergency medicine physicians and pediatric hematologists.
In this randomized, double-blind trial, the researchers compared IV magnesium to normal saline. They enrolled children ages 4 to 21, with hemoglobin SS or hemoglobin SB° thalassemia, who were hospitalized after failing emergency department management for pain.
A total of 208 children were enrolled at 8 study sites over 3 years. Four children were excluded before receiving treatment, so 101 were randomized to receive magnesium and 103 to saline.
The children received 40 mg/kg of IV magnesium every 8 hours for a total of 6 doses or normal saline of an equivalent volume (1 mL/kg).
The treatment groups were well-balanced, with similar baseline age, sex, genotype, weight, history of acute chest syndrome or asthma, previous hospitalizations within the past 3 years, use of hydroxyurea, and days of pain prior to arrival.
The median time from the first emergency department opioid to the first study drug infusion was 7.3 hours in the magnesium group and 7.5 hours in the saline group.
For the study’s primary outcome, the researchers assessed patients’ length of stay from the first study drug infusion until 12 hours after the last IV opioid dose or the time of discharge, whichever came first.
“Approximately 50% of children [overall] met the study endpoint within 52 hours, and 25% met the study endpoint within 24 hours of the first drug infusion,” Dr Brousseau noted.
And there was no significant difference in the median length of stay between the treatment arms—56 hours in the magnesium arm and 47 hours in the placebo arm (P=0.264).
A secondary outcome was opioid use, recorded as morphine equivalents. There was no significant difference with this outcome, either. Patients in the magnesium arm received 1.46 mg/kg of morphine equivalents, compared to 1.28 mg/kg in the saline arm (P=0.12).
The researchers also assessed quality of life using the PedsQL sickle cell disease-specific module, fatigue module, and generic module. At 48 hours after the first infusion, there was no significant difference in quality of life scores between the treatment groups for any of the modules (P=0.17, 0.26, and 0.94, respectively). The same was true 1 week after discharge (P=0.55, 0.82, and 0.36, respectively).
As for safety, there was no significant difference between the treatment arms for most measures. However, patients in the magnesium arm were more likely to experience warmth upon infusion, at 26%, compared to 2% in the saline arm (P<0.01).
Acute chest syndrome occurred in 16% of patients in the magnesium arm and 14% in the saline arm (P=0.78). Hypotension occurred in 4% and 1%, respectively (P=0.39). And rehospitalization within 7 days occurred in 12% and 7%, respectively (P=0.11).
In closing, Dr Brousseau noted that, although the researchers did not prove their hypothesis correct, the MAGiC study was a success in one respect.
“Intravenous magnesium does not shorten length of stay, lessen opioid use, or improve quality of life in children hospitalized for sickle cell pain crises,” he said. “[However,] a collaboration between pediatric emergency department medicine physicians and pediatric hematologists allowed for successful enrollment in an acute intervention trial with a median time to first study drug of 7.5 hours.” ![]()
sickle cell disease
Credit: St. Jude Hospital
SAN FRANCISCO—Magnesium does not improve outcomes in children hospitalized for sickle cell pain crises, results of the MAGiC study suggest.
Researchers hypothesized that magnesium—a known vasodilator, anti-inflammatory, and pain reliever—could alter the pathophysiology of pain crises.
However, when compared to normal saline, intravenous (IV) magnesium did not shorten hospital stays, lessen opioid use, or improve patients’ quality of life.
David C. Brousseau, MD, of the Medical College of Wisconsin and the Children’s Hospital of Wisconsin in Milwaukee, presented the results of this study at the 2014 ASH Annual Meeting (abstract 88).
Dr Brousseau noted that vasoocclusive crises are the most common acute complication of sickle cell disease and the most frequent cause of acute care or emergency department visits and hospitalizations. But recent changes in treatment have been minimal, with the judicious use of IV fluid and IV opioids being the mainstays of therapy.
“There have been few multicenter clinical trials evaluating new treatments, in part, due to a long history of difficulty with enrollment in interventional trials for sickle cell crises,” he continued. “These enrollment difficulties have been due to an inability to consent or to consent in a timely manner, leading to delayed initiation of study drug.”
With the MAGiC trial, Dr Brousseau and his colleagues sought to overcome this problem through a collaboration between pediatric emergency medicine physicians and pediatric hematologists.
In this randomized, double-blind trial, the researchers compared IV magnesium to normal saline. They enrolled children ages 4 to 21, with hemoglobin SS or hemoglobin SB° thalassemia, who were hospitalized after failing emergency department management for pain.
A total of 208 children were enrolled at 8 study sites over 3 years. Four children were excluded before receiving treatment, so 101 were randomized to receive magnesium and 103 to saline.
The children received 40 mg/kg of IV magnesium every 8 hours for a total of 6 doses or normal saline of an equivalent volume (1 mL/kg).
The treatment groups were well-balanced, with similar baseline age, sex, genotype, weight, history of acute chest syndrome or asthma, previous hospitalizations within the past 3 years, use of hydroxyurea, and days of pain prior to arrival.
The median time from the first emergency department opioid to the first study drug infusion was 7.3 hours in the magnesium group and 7.5 hours in the saline group.
For the study’s primary outcome, the researchers assessed patients’ length of stay from the first study drug infusion until 12 hours after the last IV opioid dose or the time of discharge, whichever came first.
“Approximately 50% of children [overall] met the study endpoint within 52 hours, and 25% met the study endpoint within 24 hours of the first drug infusion,” Dr Brousseau noted.
And there was no significant difference in the median length of stay between the treatment arms—56 hours in the magnesium arm and 47 hours in the placebo arm (P=0.264).
A secondary outcome was opioid use, recorded as morphine equivalents. There was no significant difference with this outcome, either. Patients in the magnesium arm received 1.46 mg/kg of morphine equivalents, compared to 1.28 mg/kg in the saline arm (P=0.12).
The researchers also assessed quality of life using the PedsQL sickle cell disease-specific module, fatigue module, and generic module. At 48 hours after the first infusion, there was no significant difference in quality of life scores between the treatment groups for any of the modules (P=0.17, 0.26, and 0.94, respectively). The same was true 1 week after discharge (P=0.55, 0.82, and 0.36, respectively).
As for safety, there was no significant difference between the treatment arms for most measures. However, patients in the magnesium arm were more likely to experience warmth upon infusion, at 26%, compared to 2% in the saline arm (P<0.01).
Acute chest syndrome occurred in 16% of patients in the magnesium arm and 14% in the saline arm (P=0.78). Hypotension occurred in 4% and 1%, respectively (P=0.39). And rehospitalization within 7 days occurred in 12% and 7%, respectively (P=0.11).
In closing, Dr Brousseau noted that, although the researchers did not prove their hypothesis correct, the MAGiC study was a success in one respect.
“Intravenous magnesium does not shorten length of stay, lessen opioid use, or improve quality of life in children hospitalized for sickle cell pain crises,” he said. “[However,] a collaboration between pediatric emergency department medicine physicians and pediatric hematologists allowed for successful enrollment in an acute intervention trial with a median time to first study drug of 7.5 hours.” ![]()
sickle cell disease
Credit: St. Jude Hospital
SAN FRANCISCO—Magnesium does not improve outcomes in children hospitalized for sickle cell pain crises, results of the MAGiC study suggest.
Researchers hypothesized that magnesium—a known vasodilator, anti-inflammatory, and pain reliever—could alter the pathophysiology of pain crises.
However, when compared to normal saline, intravenous (IV) magnesium did not shorten hospital stays, lessen opioid use, or improve patients’ quality of life.
David C. Brousseau, MD, of the Medical College of Wisconsin and the Children’s Hospital of Wisconsin in Milwaukee, presented the results of this study at the 2014 ASH Annual Meeting (abstract 88).
Dr Brousseau noted that vasoocclusive crises are the most common acute complication of sickle cell disease and the most frequent cause of acute care or emergency department visits and hospitalizations. But recent changes in treatment have been minimal, with the judicious use of IV fluid and IV opioids being the mainstays of therapy.
“There have been few multicenter clinical trials evaluating new treatments, in part, due to a long history of difficulty with enrollment in interventional trials for sickle cell crises,” he continued. “These enrollment difficulties have been due to an inability to consent or to consent in a timely manner, leading to delayed initiation of study drug.”
With the MAGiC trial, Dr Brousseau and his colleagues sought to overcome this problem through a collaboration between pediatric emergency medicine physicians and pediatric hematologists.
In this randomized, double-blind trial, the researchers compared IV magnesium to normal saline. They enrolled children ages 4 to 21, with hemoglobin SS or hemoglobin SB° thalassemia, who were hospitalized after failing emergency department management for pain.
A total of 208 children were enrolled at 8 study sites over 3 years. Four children were excluded before receiving treatment, so 101 were randomized to receive magnesium and 103 to saline.
The children received 40 mg/kg of IV magnesium every 8 hours for a total of 6 doses or normal saline of an equivalent volume (1 mL/kg).
The treatment groups were well-balanced, with similar baseline age, sex, genotype, weight, history of acute chest syndrome or asthma, previous hospitalizations within the past 3 years, use of hydroxyurea, and days of pain prior to arrival.
The median time from the first emergency department opioid to the first study drug infusion was 7.3 hours in the magnesium group and 7.5 hours in the saline group.
For the study’s primary outcome, the researchers assessed patients’ length of stay from the first study drug infusion until 12 hours after the last IV opioid dose or the time of discharge, whichever came first.
“Approximately 50% of children [overall] met the study endpoint within 52 hours, and 25% met the study endpoint within 24 hours of the first drug infusion,” Dr Brousseau noted.
And there was no significant difference in the median length of stay between the treatment arms—56 hours in the magnesium arm and 47 hours in the placebo arm (P=0.264).
A secondary outcome was opioid use, recorded as morphine equivalents. There was no significant difference with this outcome, either. Patients in the magnesium arm received 1.46 mg/kg of morphine equivalents, compared to 1.28 mg/kg in the saline arm (P=0.12).
The researchers also assessed quality of life using the PedsQL sickle cell disease-specific module, fatigue module, and generic module. At 48 hours after the first infusion, there was no significant difference in quality of life scores between the treatment groups for any of the modules (P=0.17, 0.26, and 0.94, respectively). The same was true 1 week after discharge (P=0.55, 0.82, and 0.36, respectively).
As for safety, there was no significant difference between the treatment arms for most measures. However, patients in the magnesium arm were more likely to experience warmth upon infusion, at 26%, compared to 2% in the saline arm (P<0.01).
Acute chest syndrome occurred in 16% of patients in the magnesium arm and 14% in the saline arm (P=0.78). Hypotension occurred in 4% and 1%, respectively (P=0.39). And rehospitalization within 7 days occurred in 12% and 7%, respectively (P=0.11).
In closing, Dr Brousseau noted that, although the researchers did not prove their hypothesis correct, the MAGiC study was a success in one respect.
“Intravenous magnesium does not shorten length of stay, lessen opioid use, or improve quality of life in children hospitalized for sickle cell pain crises,” he said. “[However,] a collaboration between pediatric emergency department medicine physicians and pediatric hematologists allowed for successful enrollment in an acute intervention trial with a median time to first study drug of 7.5 hours.” ![]()
A Team Approach to Nonmelanotic Skin Cancer Procedures
For many decades, the treatment of choice for nonmetastatic but locally invasive nonmelanotic basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) has been complete surgical excision that ensures minimal tissue waste, yet retains adequate tumor-free resection margins. From early on, the primary challenge has been assessing the appropriateness of those margins at the time of the initial surgical procedure, rather than having to recall the patient later for an additional surgery to excise involved margins.
In 1953, Steven Mohs, MD, envisioned the use of a vital dye to distinguish benign from malignant skin tissue at the time of surgery.1-3 At that point intraoperative consultation with a pathologist and the process of examining frozen sections (FS) for diagnosis were not standards of care in oncologic surgery. This process allowed Mohs, with limited success, to excise tumors with negative margins. Mohs repeatedly revised and improved his procedure, including the utilization of intraoperative FS to examine the entire specimen margin, a process that is at the core of the Mohs micrographic surgery.1-3
Currently, the Mohs procedure is one of the most popular approaches to definitive skin cancer surgery, especially in the head and neck region where tissue preservation can be critical. It is usually performed as an outpatient or clinic procedure by a specially trained dermatologist who acts both as a surgeon and a pathologist, excising the lesion and processing it for FS diagnosis.4-6 In a hospital setting, other practitioners (surgeons and pathologists) often use the standard approach of limited sampling of resection margins for FS by serially sectioning a specimen that had already been inked or marked for the appropriate margins and freeze-sectioning representative portions of those margins. Reports published by experienced operators using these different approaches indicate variable cancer recurrence rates of 1% to 6%.7-9
At the VA it is a priority to deliver the same quality health care at a much lower price. In this setting it is prudent to periodically reexamine alternative approaches to patient care delivery that utilize existing resources or excess capacity to achieve comparable, if not superior, outcomes to the usually more costly private sector outsourcing contractual arrangements.
With that goal in mind, a few years ago Robley Rex VAMC (RRVAMC) embarked on a new team approach for resectable nonmelanotic skin cancer cases. The team consisted of a plastic surgeon and a pathologist with the appropriate technical and nursing support (histotechnicians, surgical nurse practitioners, and/or nurse anesthsesists) staff. None of the team members were exclusively dedicated to the procedure but were afforded adequate time and material resources to handle all such cases. In this report, the authors describe their experience and the impact of their approach on the affected patients.
Methods
At RRVAMC, primary care providers were encouraged to refer patients suspected of nonmelanotic skin cancer directly to a hospital-based plastic surgeon, who schedules them for a FS-controlled surgical excision of the suspected lesion. The plastic surgeon also plans to cover the resulting wound, if too large for primary closure, with a micrograft during the same procedure. The procedure is usually performed under local anesthesia. A general surgeon or surgical fellow with basic training in plastic surgery may substitute for the plastic surgeon. When not performing this procedure, the surgeon carries on other routine surgical duties.
A dedicated FS room was set up next to an operating room (OR), which was designated for this specialized skin cancer surgery, among other surgeries. The pathologist could walk into the OR anytime to assess the lesion, its location, and the surgeon’s plan of resection, and both physicians could discuss the best strategy for the initial resection or any subsequent margin reexcision. Both could also discuss whether a permanent section would be more appropriate under the conditions.
A small window separated the FS room from the OR, allowing two-way communication and the delivery of specimens. If the specimen was more complex in terms of margin definition, the pathologist could personally take the specimen after its excision directly from the surgeon who could offer further explanation of the special attributes of the specimen. The specimen was usually placed on a topographic drawing of the body region with one or more permanent marks that denoted specific landmarks for orientation.
Once the specimen was in the FS room, the pathologist proceeded with standard gross description followed by color inking of the margins and sampling, according to the following rules:
- Small specimen (< 0.5 cm): Embed as is; FSs may be cut parallel to epidermal surface and examined until no more tumor is seen.
- Medium specimen (0.5-3.0 cm): Serially cross-section and embed all in ≥ 1 blocks; ≥ 6 FSs (cuts) examined from each block.
- Large specimen (> 3.0 cm): Peripheral margins shaved; few central sections taken through deep margin.
For the very small specimens excised from cosmetically or biologically critical areas, such as the head and neck region, the pathologist could use the classic Mohs sampling technique of freezing the entire specimen as is and sectioning parallel to the skin surface until free margins were reached or the entire specimen was exhausted. The pathologist could use serial cross-sectioning at 2 mm intervals in medium-sized excisions, or limited sampling of peripheral and deep margins in very large specimens. In these latter sampling approaches, at least 6 sections are cut from each slice (block), each 5 µm to 10 µm thick. The sections were mounted on glass slides, stained with hematoxylin-eosin (H&E), and examined thoroughly under a microscope before rendering a diagnosis (assessment of the resection margin).
The diagnosis was communicated directly to the surgeon by the pathologist who walked into the OR or while viewing the slides with the surgeon at a double-headed microscope located in the FS room. Remnants of any frozen or unprocessed tissue were submitted for permanent section, and the findings of both the FS and permanent diagnosis were compared the following day. Similar to the main laboratory procedures, 10% of cases were subjected to retroactive peer review for quality assurance.
Freeze section duty was handled by a pathologist and a histotechnician. Once the FS case was completed, the pathologist and histotechnician returned to the main laboratory to attend to other routine duties.
The patient’s state of comfort and satisfaction was assessed informally but routinely by the surgical team before discharge and at the follow-up visit. The patient was asked about the overall experience and invited to submit written comments to the RRVAMC patient representative. A generic mailback card was also available for feedback.
For the cost analysis, budgeting for the recurrent annual cost of labor and supplies was based on a presumed maximum workload of 300 cases/year (3-4 cases/day; 2 days/week or 0.4 full-time equivalent employee [FTEE] for each member of the team) and estimated additional OR and histology laboratory supplies of about $500/case. At the end of the fiscal year, the budgeted estimates were reconciled with the actual expenses or the added financial burden that was associated with the program to calculate the expense per case, which then was compared with the average CMS (Centers for Medicare and Medicaid Services) reimbursement rate for Mohs procedures as usually billed by private practitioners.
Results
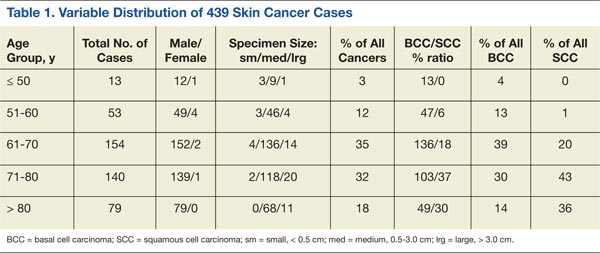
From 2006 to 2007, 439 procedures were performed at the RRVAMC program. Patients were followed up for recurrence or other complications through the end of 2012. No serious complications were encountered during any of these procedures. Patients’ comments after each procedure indicated complete satisfaction with the process, and no negative feedback or complaint was received. More than 5 years of follow-up on the initial 439 procedures yielded a rate of cancer recurrence of about 0.5% (2 patients, a 30-year-old woman and a 77-year-old man, both with basal cell carcinoma [BCC] of the nose), which is comparable or slightly better than that reported in relevant literature for the various methods, including the classic Mohs.10,11
Table 1 shows the distribution of the cases by age, gender, specimen size, and type of cancer. Most patients were white men (98.5%), and almost all (99%) cancers were from the head and neck region. Basal cell carcinoma was the diagnosis in 80% of the cases; the remainder were squamous cell carcinomas (SCCs). Both types of cancer were prevalent in the older age groups (> 50 years). Basal cell carcinoma was more prevalent in the group aged 51 to 70 years, whereas SCC predominated in patients aged > 70 years. The patients ranged in age from 30 to 89 years. The majority of specimens were medium sized (86%); 11% were large and the remaining 3% were small specimens. These demographics of patient’s age, cancer location, and prevalent diagnosis, were comparable to those of most VAMCs.
All acrediatation standards of the Clinical Laboratory Improvement Amendments of 1988 (CLIA 88) and College of American Pathologists (CAP) were observed in the RRVAMC FS laboratory, including monitoring frozen vs permanent tissue diagnosis and 10% retroactive peer review. Those indicators were always well below established thresholds or reasonable pathology practice community standards. The RRVAMC laboratory overall error (major discrepancy) rate has been < 0.2%. The FS laboratory has also been in compliance with the technical quality CAP accreditation standards, such as those for equipment, reagents, personnel, and environment controls.
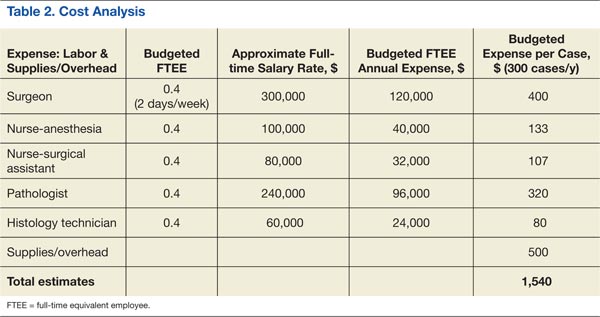
Cost analysis data are presented in Table 2. The data are based on realistic estimates in a hospital setting. The provided numbers for the FTEE salaries are average local estimates (based on VA-wide pay scale for employees according to their grades and within grade steps), though actual salary structure varied widely among institutions. Although budgeted estimates suggest an average expense of about $1,500 per case (including cases with multiple lesions that could be removed at the same session), the actual or realistic expense is far less, because some of the resources were preexisting or shared across the Surgical and Pathology Services, including FTEE time commitments. The RRVAMC planning strategy assumed 200 to 300 cases/year at $1,000 to $2,000/case.
Discussion
The RRVAMC approach of direct patient referral to the in-house plastic surgeon often spared the patient 2 additional clinical visits or procedures, which might otherwise have been required. Often, the primary care provider referred the patient to a dermatologist who would perform a shave or punch biopsy, awaiting a pathologist’s diagnosis before scheduling definitive (eg, Mohs) surgery with a separate provider. After that, the patient might be scheduled for reconstructive surgery, if necessary, by a plastic surgeon. With the RRVAMC approach, not only were the number of visits/procedures reduced, but the total time was shortened by several weeks, sparing the patient discomfort and uncertainty.
The RRVAMC cost analysis data show an average realistic cost at this setting (considering already available resources) of far less than $2,000 ($1,000-$1,500). This is substantially below the $2,000 to $10,000 cost per case (or lesion in patients with multiple lesions) that would have been required for a private sector referral, based on CMS reimbursement rates for Mohs procedures (CPT codes 17311-17315).
An important element in the cost-effectiveness, quality assurance, and time use in this approach is the flexibility of the key operators (surgery and pathology staff) and the sampling technique. For the latter, the pathologist can use the most efficient technique, depending on specimen source and size: The classic Mohs technique for very small (head and neck area) specimens, but serial cross-sectioning or limited sampling of peripheral and deep margins in other situations. All 3 sectioning approaches in the RRVAMC practice proved reliable in assessing the margins, as they were always verified either on permanent sections and/or through retroactive peer review. Furthermore, in a mostly elderly patient population, there is rarely a need for extremely conservative resection of the margins, as the skin often shows wrinkling or redundancy that allows for a more generous healthy rim around the lesion. In such cases, it may be indeed superfluous to apply the protracted and expensive Mohs procedural variant.
The quality assurance aspect of the RRVAMC approach is also important. Examining permanent sections as well as retroactive peer review can uncover diagnostic or processing errors even in the best of laboratories. That error rate in the surgical pathology community may reach more than 1% to 2%.12 In the RRVAMC practice, the major discrepancy rate is usually below 0.2%. There is a reason for concern in any FS laboratory where such monitoring is not done, considering that even BCC can be occasionally confused on FS with other small blue cell malignancies, such as lymphoma or Merkel cell carcinoma.
Conclusion
The authors offer the RRVAMC pathologist-plastic surgeon team approach to definitive skin cancer surgery as a reliable and less expensive in-house alternative to contractual outsourcing for those VA (or non-VA) medical centers that have a plastic surgeon (or trained equivalent) and a surgical pathologist on staff.
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Robley Rex VAMC in Louisville, Kentucky.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Robins P, Albom MJ. Mohs’ surgery—fresh tissue technique. J Dermatol Surg. 1975;1(2):37-41.
2. Mohs FE. Mohs micrographic surgery. A historical perspective. Dermatol Clin. 1989;7(4):609-611.
3. Mohs FE. Origin and progress of Mohs micrographic surgery. In: Mikhail GR, ed. Mohs Micrographic Surgery. Philadelphia, PA: WB Saunders; 1991:1-10.
4. Rowe DE, Carroll RJ, Day CL Jr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol. 1992;26(6):976-990.
5. Rowe DE. Comparison of treatment modalities for basal cell carcinoma. Clin Dermatol. 1995;13(6):617-620.
6. Smeets NW, Krekels GA, Ostertag JU, et al. Surgical excision vs Mohs’ micrographic surgery for basal-cell carcinoma of the face: Randomised controlled trial. Lancet. 2004;364(9447):1766-1772.
7. Bentkover SH, Grande DM, Soto H, Kozlicak BA, Guillaume D, Girouard S. Excision of head and neck basal cell carcinoma with a rapid, cross-sectional, frozen-section technique. Arch Facial Plast Surg. 2002;4(2):114-119.
8. Kimyai-Asadi A, Goldberg LH, Jih MH. Accuracy of serial transverse cross-sections in detecting residual basal cell carcinoma at the surgical margins of an elliptical excision specimen. J Am Acad Dermatol. 2004;53(3):469-474.
9. Dhingra N, Gajdasty A, Neal JW, Mukherjee AN, Lane CM. Confident complete excision of lid-margin BCCs using a marginal strip: An alternative to Mohs’ surgery. Brit J Ophthalmol. 2007;91(6):794-796.
10. Minton TJ. Contemporary Mohs surgery applications. Curr Opin Otolaryngol Head Neck Surg. 2008;16(4):376-380.
11. Mosterd K, Krekels GA, Nieman FH, et al. Surgical excision versus Mohs’ micrographic surgery for primary and recurrent basal-cell carcinoma of the face: A prospective randomised controlled trial with 5-years’ follow-up. Lancet Oncol. 2008;9(12):1149-1156.
12. Weiss MA. Analytic variables; diagnostic accuracy. In: Nakhleh RE, Fitzgibbons PL, eds. Quality Management in Anatomic Pathology. Northfield, IL: College of American Pathologists; 2005:50-76.
For many decades, the treatment of choice for nonmetastatic but locally invasive nonmelanotic basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) has been complete surgical excision that ensures minimal tissue waste, yet retains adequate tumor-free resection margins. From early on, the primary challenge has been assessing the appropriateness of those margins at the time of the initial surgical procedure, rather than having to recall the patient later for an additional surgery to excise involved margins.
In 1953, Steven Mohs, MD, envisioned the use of a vital dye to distinguish benign from malignant skin tissue at the time of surgery.1-3 At that point intraoperative consultation with a pathologist and the process of examining frozen sections (FS) for diagnosis were not standards of care in oncologic surgery. This process allowed Mohs, with limited success, to excise tumors with negative margins. Mohs repeatedly revised and improved his procedure, including the utilization of intraoperative FS to examine the entire specimen margin, a process that is at the core of the Mohs micrographic surgery.1-3
Currently, the Mohs procedure is one of the most popular approaches to definitive skin cancer surgery, especially in the head and neck region where tissue preservation can be critical. It is usually performed as an outpatient or clinic procedure by a specially trained dermatologist who acts both as a surgeon and a pathologist, excising the lesion and processing it for FS diagnosis.4-6 In a hospital setting, other practitioners (surgeons and pathologists) often use the standard approach of limited sampling of resection margins for FS by serially sectioning a specimen that had already been inked or marked for the appropriate margins and freeze-sectioning representative portions of those margins. Reports published by experienced operators using these different approaches indicate variable cancer recurrence rates of 1% to 6%.7-9
At the VA it is a priority to deliver the same quality health care at a much lower price. In this setting it is prudent to periodically reexamine alternative approaches to patient care delivery that utilize existing resources or excess capacity to achieve comparable, if not superior, outcomes to the usually more costly private sector outsourcing contractual arrangements.
With that goal in mind, a few years ago Robley Rex VAMC (RRVAMC) embarked on a new team approach for resectable nonmelanotic skin cancer cases. The team consisted of a plastic surgeon and a pathologist with the appropriate technical and nursing support (histotechnicians, surgical nurse practitioners, and/or nurse anesthsesists) staff. None of the team members were exclusively dedicated to the procedure but were afforded adequate time and material resources to handle all such cases. In this report, the authors describe their experience and the impact of their approach on the affected patients.
Methods
At RRVAMC, primary care providers were encouraged to refer patients suspected of nonmelanotic skin cancer directly to a hospital-based plastic surgeon, who schedules them for a FS-controlled surgical excision of the suspected lesion. The plastic surgeon also plans to cover the resulting wound, if too large for primary closure, with a micrograft during the same procedure. The procedure is usually performed under local anesthesia. A general surgeon or surgical fellow with basic training in plastic surgery may substitute for the plastic surgeon. When not performing this procedure, the surgeon carries on other routine surgical duties.
A dedicated FS room was set up next to an operating room (OR), which was designated for this specialized skin cancer surgery, among other surgeries. The pathologist could walk into the OR anytime to assess the lesion, its location, and the surgeon’s plan of resection, and both physicians could discuss the best strategy for the initial resection or any subsequent margin reexcision. Both could also discuss whether a permanent section would be more appropriate under the conditions.
A small window separated the FS room from the OR, allowing two-way communication and the delivery of specimens. If the specimen was more complex in terms of margin definition, the pathologist could personally take the specimen after its excision directly from the surgeon who could offer further explanation of the special attributes of the specimen. The specimen was usually placed on a topographic drawing of the body region with one or more permanent marks that denoted specific landmarks for orientation.
Once the specimen was in the FS room, the pathologist proceeded with standard gross description followed by color inking of the margins and sampling, according to the following rules:
- Small specimen (< 0.5 cm): Embed as is; FSs may be cut parallel to epidermal surface and examined until no more tumor is seen.
- Medium specimen (0.5-3.0 cm): Serially cross-section and embed all in ≥ 1 blocks; ≥ 6 FSs (cuts) examined from each block.
- Large specimen (> 3.0 cm): Peripheral margins shaved; few central sections taken through deep margin.
For the very small specimens excised from cosmetically or biologically critical areas, such as the head and neck region, the pathologist could use the classic Mohs sampling technique of freezing the entire specimen as is and sectioning parallel to the skin surface until free margins were reached or the entire specimen was exhausted. The pathologist could use serial cross-sectioning at 2 mm intervals in medium-sized excisions, or limited sampling of peripheral and deep margins in very large specimens. In these latter sampling approaches, at least 6 sections are cut from each slice (block), each 5 µm to 10 µm thick. The sections were mounted on glass slides, stained with hematoxylin-eosin (H&E), and examined thoroughly under a microscope before rendering a diagnosis (assessment of the resection margin).
The diagnosis was communicated directly to the surgeon by the pathologist who walked into the OR or while viewing the slides with the surgeon at a double-headed microscope located in the FS room. Remnants of any frozen or unprocessed tissue were submitted for permanent section, and the findings of both the FS and permanent diagnosis were compared the following day. Similar to the main laboratory procedures, 10% of cases were subjected to retroactive peer review for quality assurance.
Freeze section duty was handled by a pathologist and a histotechnician. Once the FS case was completed, the pathologist and histotechnician returned to the main laboratory to attend to other routine duties.
The patient’s state of comfort and satisfaction was assessed informally but routinely by the surgical team before discharge and at the follow-up visit. The patient was asked about the overall experience and invited to submit written comments to the RRVAMC patient representative. A generic mailback card was also available for feedback.
For the cost analysis, budgeting for the recurrent annual cost of labor and supplies was based on a presumed maximum workload of 300 cases/year (3-4 cases/day; 2 days/week or 0.4 full-time equivalent employee [FTEE] for each member of the team) and estimated additional OR and histology laboratory supplies of about $500/case. At the end of the fiscal year, the budgeted estimates were reconciled with the actual expenses or the added financial burden that was associated with the program to calculate the expense per case, which then was compared with the average CMS (Centers for Medicare and Medicaid Services) reimbursement rate for Mohs procedures as usually billed by private practitioners.
Results
From 2006 to 2007, 439 procedures were performed at the RRVAMC program. Patients were followed up for recurrence or other complications through the end of 2012. No serious complications were encountered during any of these procedures. Patients’ comments after each procedure indicated complete satisfaction with the process, and no negative feedback or complaint was received. More than 5 years of follow-up on the initial 439 procedures yielded a rate of cancer recurrence of about 0.5% (2 patients, a 30-year-old woman and a 77-year-old man, both with basal cell carcinoma [BCC] of the nose), which is comparable or slightly better than that reported in relevant literature for the various methods, including the classic Mohs.10,11
Table 1 shows the distribution of the cases by age, gender, specimen size, and type of cancer. Most patients were white men (98.5%), and almost all (99%) cancers were from the head and neck region. Basal cell carcinoma was the diagnosis in 80% of the cases; the remainder were squamous cell carcinomas (SCCs). Both types of cancer were prevalent in the older age groups (> 50 years). Basal cell carcinoma was more prevalent in the group aged 51 to 70 years, whereas SCC predominated in patients aged > 70 years. The patients ranged in age from 30 to 89 years. The majority of specimens were medium sized (86%); 11% were large and the remaining 3% were small specimens. These demographics of patient’s age, cancer location, and prevalent diagnosis, were comparable to those of most VAMCs.
All acrediatation standards of the Clinical Laboratory Improvement Amendments of 1988 (CLIA 88) and College of American Pathologists (CAP) were observed in the RRVAMC FS laboratory, including monitoring frozen vs permanent tissue diagnosis and 10% retroactive peer review. Those indicators were always well below established thresholds or reasonable pathology practice community standards. The RRVAMC laboratory overall error (major discrepancy) rate has been < 0.2%. The FS laboratory has also been in compliance with the technical quality CAP accreditation standards, such as those for equipment, reagents, personnel, and environment controls.
Cost analysis data are presented in Table 2. The data are based on realistic estimates in a hospital setting. The provided numbers for the FTEE salaries are average local estimates (based on VA-wide pay scale for employees according to their grades and within grade steps), though actual salary structure varied widely among institutions. Although budgeted estimates suggest an average expense of about $1,500 per case (including cases with multiple lesions that could be removed at the same session), the actual or realistic expense is far less, because some of the resources were preexisting or shared across the Surgical and Pathology Services, including FTEE time commitments. The RRVAMC planning strategy assumed 200 to 300 cases/year at $1,000 to $2,000/case.
Discussion
The RRVAMC approach of direct patient referral to the in-house plastic surgeon often spared the patient 2 additional clinical visits or procedures, which might otherwise have been required. Often, the primary care provider referred the patient to a dermatologist who would perform a shave or punch biopsy, awaiting a pathologist’s diagnosis before scheduling definitive (eg, Mohs) surgery with a separate provider. After that, the patient might be scheduled for reconstructive surgery, if necessary, by a plastic surgeon. With the RRVAMC approach, not only were the number of visits/procedures reduced, but the total time was shortened by several weeks, sparing the patient discomfort and uncertainty.
The RRVAMC cost analysis data show an average realistic cost at this setting (considering already available resources) of far less than $2,000 ($1,000-$1,500). This is substantially below the $2,000 to $10,000 cost per case (or lesion in patients with multiple lesions) that would have been required for a private sector referral, based on CMS reimbursement rates for Mohs procedures (CPT codes 17311-17315).
An important element in the cost-effectiveness, quality assurance, and time use in this approach is the flexibility of the key operators (surgery and pathology staff) and the sampling technique. For the latter, the pathologist can use the most efficient technique, depending on specimen source and size: The classic Mohs technique for very small (head and neck area) specimens, but serial cross-sectioning or limited sampling of peripheral and deep margins in other situations. All 3 sectioning approaches in the RRVAMC practice proved reliable in assessing the margins, as they were always verified either on permanent sections and/or through retroactive peer review. Furthermore, in a mostly elderly patient population, there is rarely a need for extremely conservative resection of the margins, as the skin often shows wrinkling or redundancy that allows for a more generous healthy rim around the lesion. In such cases, it may be indeed superfluous to apply the protracted and expensive Mohs procedural variant.
The quality assurance aspect of the RRVAMC approach is also important. Examining permanent sections as well as retroactive peer review can uncover diagnostic or processing errors even in the best of laboratories. That error rate in the surgical pathology community may reach more than 1% to 2%.12 In the RRVAMC practice, the major discrepancy rate is usually below 0.2%. There is a reason for concern in any FS laboratory where such monitoring is not done, considering that even BCC can be occasionally confused on FS with other small blue cell malignancies, such as lymphoma or Merkel cell carcinoma.
Conclusion
The authors offer the RRVAMC pathologist-plastic surgeon team approach to definitive skin cancer surgery as a reliable and less expensive in-house alternative to contractual outsourcing for those VA (or non-VA) medical centers that have a plastic surgeon (or trained equivalent) and a surgical pathologist on staff.
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Robley Rex VAMC in Louisville, Kentucky.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
For many decades, the treatment of choice for nonmetastatic but locally invasive nonmelanotic basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) has been complete surgical excision that ensures minimal tissue waste, yet retains adequate tumor-free resection margins. From early on, the primary challenge has been assessing the appropriateness of those margins at the time of the initial surgical procedure, rather than having to recall the patient later for an additional surgery to excise involved margins.
In 1953, Steven Mohs, MD, envisioned the use of a vital dye to distinguish benign from malignant skin tissue at the time of surgery.1-3 At that point intraoperative consultation with a pathologist and the process of examining frozen sections (FS) for diagnosis were not standards of care in oncologic surgery. This process allowed Mohs, with limited success, to excise tumors with negative margins. Mohs repeatedly revised and improved his procedure, including the utilization of intraoperative FS to examine the entire specimen margin, a process that is at the core of the Mohs micrographic surgery.1-3
Currently, the Mohs procedure is one of the most popular approaches to definitive skin cancer surgery, especially in the head and neck region where tissue preservation can be critical. It is usually performed as an outpatient or clinic procedure by a specially trained dermatologist who acts both as a surgeon and a pathologist, excising the lesion and processing it for FS diagnosis.4-6 In a hospital setting, other practitioners (surgeons and pathologists) often use the standard approach of limited sampling of resection margins for FS by serially sectioning a specimen that had already been inked or marked for the appropriate margins and freeze-sectioning representative portions of those margins. Reports published by experienced operators using these different approaches indicate variable cancer recurrence rates of 1% to 6%.7-9
At the VA it is a priority to deliver the same quality health care at a much lower price. In this setting it is prudent to periodically reexamine alternative approaches to patient care delivery that utilize existing resources or excess capacity to achieve comparable, if not superior, outcomes to the usually more costly private sector outsourcing contractual arrangements.
With that goal in mind, a few years ago Robley Rex VAMC (RRVAMC) embarked on a new team approach for resectable nonmelanotic skin cancer cases. The team consisted of a plastic surgeon and a pathologist with the appropriate technical and nursing support (histotechnicians, surgical nurse practitioners, and/or nurse anesthsesists) staff. None of the team members were exclusively dedicated to the procedure but were afforded adequate time and material resources to handle all such cases. In this report, the authors describe their experience and the impact of their approach on the affected patients.
Methods
At RRVAMC, primary care providers were encouraged to refer patients suspected of nonmelanotic skin cancer directly to a hospital-based plastic surgeon, who schedules them for a FS-controlled surgical excision of the suspected lesion. The plastic surgeon also plans to cover the resulting wound, if too large for primary closure, with a micrograft during the same procedure. The procedure is usually performed under local anesthesia. A general surgeon or surgical fellow with basic training in plastic surgery may substitute for the plastic surgeon. When not performing this procedure, the surgeon carries on other routine surgical duties.
A dedicated FS room was set up next to an operating room (OR), which was designated for this specialized skin cancer surgery, among other surgeries. The pathologist could walk into the OR anytime to assess the lesion, its location, and the surgeon’s plan of resection, and both physicians could discuss the best strategy for the initial resection or any subsequent margin reexcision. Both could also discuss whether a permanent section would be more appropriate under the conditions.
A small window separated the FS room from the OR, allowing two-way communication and the delivery of specimens. If the specimen was more complex in terms of margin definition, the pathologist could personally take the specimen after its excision directly from the surgeon who could offer further explanation of the special attributes of the specimen. The specimen was usually placed on a topographic drawing of the body region with one or more permanent marks that denoted specific landmarks for orientation.
Once the specimen was in the FS room, the pathologist proceeded with standard gross description followed by color inking of the margins and sampling, according to the following rules:
- Small specimen (< 0.5 cm): Embed as is; FSs may be cut parallel to epidermal surface and examined until no more tumor is seen.
- Medium specimen (0.5-3.0 cm): Serially cross-section and embed all in ≥ 1 blocks; ≥ 6 FSs (cuts) examined from each block.
- Large specimen (> 3.0 cm): Peripheral margins shaved; few central sections taken through deep margin.
For the very small specimens excised from cosmetically or biologically critical areas, such as the head and neck region, the pathologist could use the classic Mohs sampling technique of freezing the entire specimen as is and sectioning parallel to the skin surface until free margins were reached or the entire specimen was exhausted. The pathologist could use serial cross-sectioning at 2 mm intervals in medium-sized excisions, or limited sampling of peripheral and deep margins in very large specimens. In these latter sampling approaches, at least 6 sections are cut from each slice (block), each 5 µm to 10 µm thick. The sections were mounted on glass slides, stained with hematoxylin-eosin (H&E), and examined thoroughly under a microscope before rendering a diagnosis (assessment of the resection margin).
The diagnosis was communicated directly to the surgeon by the pathologist who walked into the OR or while viewing the slides with the surgeon at a double-headed microscope located in the FS room. Remnants of any frozen or unprocessed tissue were submitted for permanent section, and the findings of both the FS and permanent diagnosis were compared the following day. Similar to the main laboratory procedures, 10% of cases were subjected to retroactive peer review for quality assurance.
Freeze section duty was handled by a pathologist and a histotechnician. Once the FS case was completed, the pathologist and histotechnician returned to the main laboratory to attend to other routine duties.
The patient’s state of comfort and satisfaction was assessed informally but routinely by the surgical team before discharge and at the follow-up visit. The patient was asked about the overall experience and invited to submit written comments to the RRVAMC patient representative. A generic mailback card was also available for feedback.
For the cost analysis, budgeting for the recurrent annual cost of labor and supplies was based on a presumed maximum workload of 300 cases/year (3-4 cases/day; 2 days/week or 0.4 full-time equivalent employee [FTEE] for each member of the team) and estimated additional OR and histology laboratory supplies of about $500/case. At the end of the fiscal year, the budgeted estimates were reconciled with the actual expenses or the added financial burden that was associated with the program to calculate the expense per case, which then was compared with the average CMS (Centers for Medicare and Medicaid Services) reimbursement rate for Mohs procedures as usually billed by private practitioners.
Results
From 2006 to 2007, 439 procedures were performed at the RRVAMC program. Patients were followed up for recurrence or other complications through the end of 2012. No serious complications were encountered during any of these procedures. Patients’ comments after each procedure indicated complete satisfaction with the process, and no negative feedback or complaint was received. More than 5 years of follow-up on the initial 439 procedures yielded a rate of cancer recurrence of about 0.5% (2 patients, a 30-year-old woman and a 77-year-old man, both with basal cell carcinoma [BCC] of the nose), which is comparable or slightly better than that reported in relevant literature for the various methods, including the classic Mohs.10,11
Table 1 shows the distribution of the cases by age, gender, specimen size, and type of cancer. Most patients were white men (98.5%), and almost all (99%) cancers were from the head and neck region. Basal cell carcinoma was the diagnosis in 80% of the cases; the remainder were squamous cell carcinomas (SCCs). Both types of cancer were prevalent in the older age groups (> 50 years). Basal cell carcinoma was more prevalent in the group aged 51 to 70 years, whereas SCC predominated in patients aged > 70 years. The patients ranged in age from 30 to 89 years. The majority of specimens were medium sized (86%); 11% were large and the remaining 3% were small specimens. These demographics of patient’s age, cancer location, and prevalent diagnosis, were comparable to those of most VAMCs.
All acrediatation standards of the Clinical Laboratory Improvement Amendments of 1988 (CLIA 88) and College of American Pathologists (CAP) were observed in the RRVAMC FS laboratory, including monitoring frozen vs permanent tissue diagnosis and 10% retroactive peer review. Those indicators were always well below established thresholds or reasonable pathology practice community standards. The RRVAMC laboratory overall error (major discrepancy) rate has been < 0.2%. The FS laboratory has also been in compliance with the technical quality CAP accreditation standards, such as those for equipment, reagents, personnel, and environment controls.
Cost analysis data are presented in Table 2. The data are based on realistic estimates in a hospital setting. The provided numbers for the FTEE salaries are average local estimates (based on VA-wide pay scale for employees according to their grades and within grade steps), though actual salary structure varied widely among institutions. Although budgeted estimates suggest an average expense of about $1,500 per case (including cases with multiple lesions that could be removed at the same session), the actual or realistic expense is far less, because some of the resources were preexisting or shared across the Surgical and Pathology Services, including FTEE time commitments. The RRVAMC planning strategy assumed 200 to 300 cases/year at $1,000 to $2,000/case.
Discussion
The RRVAMC approach of direct patient referral to the in-house plastic surgeon often spared the patient 2 additional clinical visits or procedures, which might otherwise have been required. Often, the primary care provider referred the patient to a dermatologist who would perform a shave or punch biopsy, awaiting a pathologist’s diagnosis before scheduling definitive (eg, Mohs) surgery with a separate provider. After that, the patient might be scheduled for reconstructive surgery, if necessary, by a plastic surgeon. With the RRVAMC approach, not only were the number of visits/procedures reduced, but the total time was shortened by several weeks, sparing the patient discomfort and uncertainty.
The RRVAMC cost analysis data show an average realistic cost at this setting (considering already available resources) of far less than $2,000 ($1,000-$1,500). This is substantially below the $2,000 to $10,000 cost per case (or lesion in patients with multiple lesions) that would have been required for a private sector referral, based on CMS reimbursement rates for Mohs procedures (CPT codes 17311-17315).
An important element in the cost-effectiveness, quality assurance, and time use in this approach is the flexibility of the key operators (surgery and pathology staff) and the sampling technique. For the latter, the pathologist can use the most efficient technique, depending on specimen source and size: The classic Mohs technique for very small (head and neck area) specimens, but serial cross-sectioning or limited sampling of peripheral and deep margins in other situations. All 3 sectioning approaches in the RRVAMC practice proved reliable in assessing the margins, as they were always verified either on permanent sections and/or through retroactive peer review. Furthermore, in a mostly elderly patient population, there is rarely a need for extremely conservative resection of the margins, as the skin often shows wrinkling or redundancy that allows for a more generous healthy rim around the lesion. In such cases, it may be indeed superfluous to apply the protracted and expensive Mohs procedural variant.
The quality assurance aspect of the RRVAMC approach is also important. Examining permanent sections as well as retroactive peer review can uncover diagnostic or processing errors even in the best of laboratories. That error rate in the surgical pathology community may reach more than 1% to 2%.12 In the RRVAMC practice, the major discrepancy rate is usually below 0.2%. There is a reason for concern in any FS laboratory where such monitoring is not done, considering that even BCC can be occasionally confused on FS with other small blue cell malignancies, such as lymphoma or Merkel cell carcinoma.
Conclusion
The authors offer the RRVAMC pathologist-plastic surgeon team approach to definitive skin cancer surgery as a reliable and less expensive in-house alternative to contractual outsourcing for those VA (or non-VA) medical centers that have a plastic surgeon (or trained equivalent) and a surgical pathologist on staff.
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Robley Rex VAMC in Louisville, Kentucky.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Robins P, Albom MJ. Mohs’ surgery—fresh tissue technique. J Dermatol Surg. 1975;1(2):37-41.
2. Mohs FE. Mohs micrographic surgery. A historical perspective. Dermatol Clin. 1989;7(4):609-611.
3. Mohs FE. Origin and progress of Mohs micrographic surgery. In: Mikhail GR, ed. Mohs Micrographic Surgery. Philadelphia, PA: WB Saunders; 1991:1-10.
4. Rowe DE, Carroll RJ, Day CL Jr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol. 1992;26(6):976-990.
5. Rowe DE. Comparison of treatment modalities for basal cell carcinoma. Clin Dermatol. 1995;13(6):617-620.
6. Smeets NW, Krekels GA, Ostertag JU, et al. Surgical excision vs Mohs’ micrographic surgery for basal-cell carcinoma of the face: Randomised controlled trial. Lancet. 2004;364(9447):1766-1772.
7. Bentkover SH, Grande DM, Soto H, Kozlicak BA, Guillaume D, Girouard S. Excision of head and neck basal cell carcinoma with a rapid, cross-sectional, frozen-section technique. Arch Facial Plast Surg. 2002;4(2):114-119.
8. Kimyai-Asadi A, Goldberg LH, Jih MH. Accuracy of serial transverse cross-sections in detecting residual basal cell carcinoma at the surgical margins of an elliptical excision specimen. J Am Acad Dermatol. 2004;53(3):469-474.
9. Dhingra N, Gajdasty A, Neal JW, Mukherjee AN, Lane CM. Confident complete excision of lid-margin BCCs using a marginal strip: An alternative to Mohs’ surgery. Brit J Ophthalmol. 2007;91(6):794-796.
10. Minton TJ. Contemporary Mohs surgery applications. Curr Opin Otolaryngol Head Neck Surg. 2008;16(4):376-380.
11. Mosterd K, Krekels GA, Nieman FH, et al. Surgical excision versus Mohs’ micrographic surgery for primary and recurrent basal-cell carcinoma of the face: A prospective randomised controlled trial with 5-years’ follow-up. Lancet Oncol. 2008;9(12):1149-1156.
12. Weiss MA. Analytic variables; diagnostic accuracy. In: Nakhleh RE, Fitzgibbons PL, eds. Quality Management in Anatomic Pathology. Northfield, IL: College of American Pathologists; 2005:50-76.
1. Robins P, Albom MJ. Mohs’ surgery—fresh tissue technique. J Dermatol Surg. 1975;1(2):37-41.
2. Mohs FE. Mohs micrographic surgery. A historical perspective. Dermatol Clin. 1989;7(4):609-611.
3. Mohs FE. Origin and progress of Mohs micrographic surgery. In: Mikhail GR, ed. Mohs Micrographic Surgery. Philadelphia, PA: WB Saunders; 1991:1-10.
4. Rowe DE, Carroll RJ, Day CL Jr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol. 1992;26(6):976-990.
5. Rowe DE. Comparison of treatment modalities for basal cell carcinoma. Clin Dermatol. 1995;13(6):617-620.
6. Smeets NW, Krekels GA, Ostertag JU, et al. Surgical excision vs Mohs’ micrographic surgery for basal-cell carcinoma of the face: Randomised controlled trial. Lancet. 2004;364(9447):1766-1772.
7. Bentkover SH, Grande DM, Soto H, Kozlicak BA, Guillaume D, Girouard S. Excision of head and neck basal cell carcinoma with a rapid, cross-sectional, frozen-section technique. Arch Facial Plast Surg. 2002;4(2):114-119.
8. Kimyai-Asadi A, Goldberg LH, Jih MH. Accuracy of serial transverse cross-sections in detecting residual basal cell carcinoma at the surgical margins of an elliptical excision specimen. J Am Acad Dermatol. 2004;53(3):469-474.
9. Dhingra N, Gajdasty A, Neal JW, Mukherjee AN, Lane CM. Confident complete excision of lid-margin BCCs using a marginal strip: An alternative to Mohs’ surgery. Brit J Ophthalmol. 2007;91(6):794-796.
10. Minton TJ. Contemporary Mohs surgery applications. Curr Opin Otolaryngol Head Neck Surg. 2008;16(4):376-380.
11. Mosterd K, Krekels GA, Nieman FH, et al. Surgical excision versus Mohs’ micrographic surgery for primary and recurrent basal-cell carcinoma of the face: A prospective randomised controlled trial with 5-years’ follow-up. Lancet Oncol. 2008;9(12):1149-1156.
12. Weiss MA. Analytic variables; diagnostic accuracy. In: Nakhleh RE, Fitzgibbons PL, eds. Quality Management in Anatomic Pathology. Northfield, IL: College of American Pathologists; 2005:50-76.
Reducing chemo drug’s cardiac side effects

Investigators have identified compounds that appear to prevent the cardiac damage caused by the chemotherapy drug doxorubicin.
The compounds target MDH2, an enzyme key to the generation of cellular energy in mitochondria.
And preclinical experiments showed that inhibiting MDH2 could prevent doxorubicin-induced damage to cardiac cells without reducing the drug’s antitumor effects.
The investigators detailed these experiments in Science Translational Medicine.
“Doxorubicin-induced cardiomyopathy limits the amount of the drug a patient can receive—which limits the ability to treat cancer—and even low, safer doses can lead to heart failure in up to 8% of patients,” explained study author Randall Peterson, PhD, of Massachusetts General Hospital in Charlestown.
“Finding an effective cardioprotective drug—essentially separating the good and bad effects of this form of chemotherapy—could increase the beneficial effects of doxorubicin against cancer while reducing the rate of heart failure in treated patients.”
To conduct a broad search for potential protective compounds, Dr Peterson and his colleagues developed a zebrafish model of doxorubicin-induced heart failure. They used this model to screen 3000 molecules from 2 chemical libraries for the ability to prevent the kind of cardiac damage caused by the drug.
Eight of the tested chemicals reduced damage to the hearts of zebrafish embryos, and two compounds—visnagin and diphenylurea—were the most potent in preventing both structural and functional damage.
Further in vitro and in vivo experiments revealed that either compound almost completely prevented the death of cardiac cells caused by doxorubicin. In mouse models of both high- and low-dose doxorubicin treatment, visnagin—a natural compound synthesized by the toothpick weed—was able to maintain cardiac function.
Investigation into the possible mechanism behind visnagin’s protective ability showed that the compound binds to and inhibits the action of MDH2, an enzyme essential to the generation of cellular energy by mitochondria.
Other agents that block MDH2 activity also protected zebrafish against doxorubicin-induced cardiac damage. And tests in both cellular and animal models of several types of cancer showed that neither visnagin nor diphenylurea reduced the antitumor action of doxorubicin.
“We are still trying to determine exactly how inhibition of MDH2 protects the heart, but one intriguing idea is that doxorubicin may kill cardiac and tumor cells in different ways,” Dr Peterson said. “Given the intense energy requirements of the beating heart, we speculate that cardiac cells may be especially susceptible to metabolic disturbance caused by doxorubicin and that inhibiting MDH2 may correct the metabolic imbalance and prevent the cells from dying.”
“It remains to be seen if visnagin’s protective effects are restricted to doxorubicin or if it can protect the heart from other kinds of damage. We are pursuing this question by testing its ability to protect heart muscle from oxygen deprivation during heart attacks and from the effects of other heart-damaging chemotherapy drugs.” ![]()
Investigators have identified compounds that appear to prevent the cardiac damage caused by the chemotherapy drug doxorubicin.
The compounds target MDH2, an enzyme key to the generation of cellular energy in mitochondria.
And preclinical experiments showed that inhibiting MDH2 could prevent doxorubicin-induced damage to cardiac cells without reducing the drug’s antitumor effects.
The investigators detailed these experiments in Science Translational Medicine.
“Doxorubicin-induced cardiomyopathy limits the amount of the drug a patient can receive—which limits the ability to treat cancer—and even low, safer doses can lead to heart failure in up to 8% of patients,” explained study author Randall Peterson, PhD, of Massachusetts General Hospital in Charlestown.
“Finding an effective cardioprotective drug—essentially separating the good and bad effects of this form of chemotherapy—could increase the beneficial effects of doxorubicin against cancer while reducing the rate of heart failure in treated patients.”
To conduct a broad search for potential protective compounds, Dr Peterson and his colleagues developed a zebrafish model of doxorubicin-induced heart failure. They used this model to screen 3000 molecules from 2 chemical libraries for the ability to prevent the kind of cardiac damage caused by the drug.
Eight of the tested chemicals reduced damage to the hearts of zebrafish embryos, and two compounds—visnagin and diphenylurea—were the most potent in preventing both structural and functional damage.
Further in vitro and in vivo experiments revealed that either compound almost completely prevented the death of cardiac cells caused by doxorubicin. In mouse models of both high- and low-dose doxorubicin treatment, visnagin—a natural compound synthesized by the toothpick weed—was able to maintain cardiac function.
Investigation into the possible mechanism behind visnagin’s protective ability showed that the compound binds to and inhibits the action of MDH2, an enzyme essential to the generation of cellular energy by mitochondria.
Other agents that block MDH2 activity also protected zebrafish against doxorubicin-induced cardiac damage. And tests in both cellular and animal models of several types of cancer showed that neither visnagin nor diphenylurea reduced the antitumor action of doxorubicin.
“We are still trying to determine exactly how inhibition of MDH2 protects the heart, but one intriguing idea is that doxorubicin may kill cardiac and tumor cells in different ways,” Dr Peterson said. “Given the intense energy requirements of the beating heart, we speculate that cardiac cells may be especially susceptible to metabolic disturbance caused by doxorubicin and that inhibiting MDH2 may correct the metabolic imbalance and prevent the cells from dying.”
“It remains to be seen if visnagin’s protective effects are restricted to doxorubicin or if it can protect the heart from other kinds of damage. We are pursuing this question by testing its ability to protect heart muscle from oxygen deprivation during heart attacks and from the effects of other heart-damaging chemotherapy drugs.” ![]()
Investigators have identified compounds that appear to prevent the cardiac damage caused by the chemotherapy drug doxorubicin.
The compounds target MDH2, an enzyme key to the generation of cellular energy in mitochondria.
And preclinical experiments showed that inhibiting MDH2 could prevent doxorubicin-induced damage to cardiac cells without reducing the drug’s antitumor effects.
The investigators detailed these experiments in Science Translational Medicine.
“Doxorubicin-induced cardiomyopathy limits the amount of the drug a patient can receive—which limits the ability to treat cancer—and even low, safer doses can lead to heart failure in up to 8% of patients,” explained study author Randall Peterson, PhD, of Massachusetts General Hospital in Charlestown.
“Finding an effective cardioprotective drug—essentially separating the good and bad effects of this form of chemotherapy—could increase the beneficial effects of doxorubicin against cancer while reducing the rate of heart failure in treated patients.”
To conduct a broad search for potential protective compounds, Dr Peterson and his colleagues developed a zebrafish model of doxorubicin-induced heart failure. They used this model to screen 3000 molecules from 2 chemical libraries for the ability to prevent the kind of cardiac damage caused by the drug.
Eight of the tested chemicals reduced damage to the hearts of zebrafish embryos, and two compounds—visnagin and diphenylurea—were the most potent in preventing both structural and functional damage.
Further in vitro and in vivo experiments revealed that either compound almost completely prevented the death of cardiac cells caused by doxorubicin. In mouse models of both high- and low-dose doxorubicin treatment, visnagin—a natural compound synthesized by the toothpick weed—was able to maintain cardiac function.
Investigation into the possible mechanism behind visnagin’s protective ability showed that the compound binds to and inhibits the action of MDH2, an enzyme essential to the generation of cellular energy by mitochondria.
Other agents that block MDH2 activity also protected zebrafish against doxorubicin-induced cardiac damage. And tests in both cellular and animal models of several types of cancer showed that neither visnagin nor diphenylurea reduced the antitumor action of doxorubicin.
“We are still trying to determine exactly how inhibition of MDH2 protects the heart, but one intriguing idea is that doxorubicin may kill cardiac and tumor cells in different ways,” Dr Peterson said. “Given the intense energy requirements of the beating heart, we speculate that cardiac cells may be especially susceptible to metabolic disturbance caused by doxorubicin and that inhibiting MDH2 may correct the metabolic imbalance and prevent the cells from dying.”
“It remains to be seen if visnagin’s protective effects are restricted to doxorubicin or if it can protect the heart from other kinds of damage. We are pursuing this question by testing its ability to protect heart muscle from oxygen deprivation during heart attacks and from the effects of other heart-damaging chemotherapy drugs.” ![]()
Home-Based Video Telehealth for Veterans With Dementia

For nearly 4 decades, the unifying focus of the 2-site New England Geriatric Research Education and Clinical Center (GRECC) has been on dementia and related disorders. Veterans with dementia are an extremely vulnerable population with high rates of health care use that is projected to total > $203 billion in the U.S. in 2013.1 Their caregivers are also among the most burdened, having provided about 17.5 billion hours of unpaid care in 2012, which is valued at more than $216 billion.1 Additionally, spouses, who are the most common caregivers of persons with dementia, often experience poor health outcomes related to the experience of living with the afflicted spouse.2
Currently > 200,000 VA patients have dementia, and that number is expected to increase.3 Dementia is largely a disease of the elderly; thus, many veterans with dementia also have other medical and orthopedic conditions that increase their frailty and decrease their mobility. Behavioral and psychological symptoms are present in > 75% of people with dementia, contributing to the relative isolation of both those with dementia and their families.4
Disruption in routine and removal from familiar surroundings can cause many patients, particularly in the moderate stages of dementia, to become disoriented and agitated. For these patients, VA clinics can be unsettling and may reveal behavior that is not the same as the veterans’ behavior at home. For these reasons, veterans with dementia likely may benefit from remote access to health care via telehealth. However, current telehealth applications for this population are vastly underdeveloped.
Video Dementia Management
Many GRECCs and other VA geriatric programs provide video-based dementia evaluations and management at community-based outpatient clinics (CBOCs) affiliated with their medical centers. At this point, close to a dozen GRECC and geriatric programs nationally have geriatric psychiatrists, geriatricians, or neurologists conducting such visits from their office or clinic space with video links to a telehealth-enabled room in the corresponding CBOC.
The visits usually entail having the veteran and, when appropriate, a family member check in at the local CBOC for the appointment and receiving assistance throughout the video visit from the telehealth technician at the CBOC.
The technician assists with the technical aspects of the encounter, including establishing and maintaining the video link to the VA medical center (VAMC), and often is trained to administer a brief standardized mental status assessment. The telehealth technician also helps pass along the physician’s written recommendations to the veteran and family once the recommendation summary has been sent by e-mail or printed on the CBOC printer. These VAMC-CBOC video telehealth programs have been very popular with veterans, particularly those in rural settings, since traveling to the CBOC is usually more convenient.
Home-based Video Program
While CBOC-based video telehealth programs expand the population of veterans able to benefit from specialty dementia care, any travel out of the home can be challenging or disruptive for many veterans and their families. In addition, the performance and demeanor of a veteran with dementia in a clinic setting is sometimes different from that which the family describes as their more typical behavior at home.
A new in-home video telehealth program developed by the GRECC at the Edith Nourse Rogers Memorial Veterans Hospital in Bedford is addressing these issues. The Bedford site of the New England GRECC offers in-home clinical video telehealth services to community-dwelling veterans and caregivers as an extension of their Interdisciplinary Memory Assessment Continuity Clinic (IMACC). Currently, the percentage of IMACC veterans/caregivers who have voluntarily signed up for the program is nearly 30%. About 70% of the families that have enrolled to date have their video visits with their spouse caregivers.
Veterans participating in the home video telehealth program have had at least 1 in-person visit at the Bedford IMACC before being invited to join. Veterans and their caregivers are invited to participate in the GRECC home telehealth program either at the time of an in-person IMACC visit or afterward via a telephone call from either a provider or a member of the telehealth staff.
Telehealth visits are offered as a supplement to regularly scheduled in-person visits, not as a substitute. The frequency of telehealth visits is individualized, depending on the medical status of the veteran and preferences of the caregiver. Some participating families, finding the telehealth format much more convenient, have asked whether they could postpone upcoming in-person visits at the VAMC. Many patients are particularly interested in minimizing medical visits in the winter months in New England.
Case Example
A male World War II veteran with moderate stage dementia lived with his wife in an apartment down the street from his adult son and daughter-in-law. His son and daughter-in-law visited and helped with the veteran’s care most days, but his wife was his primary caregiver. However, due to her own mobility issues, she was unable to attend the veteran’s in-person IMACC initial evaluation or subsequent follow-up visit.
This family enthusiastically embraced the opportunity to participate in the home video telehealth program and had multiple telehealth visits. During these video encounters, the veteran, his wife, and his son and daughter-in-law were present. The clinician, communicating via computer from the Bedford VAMC, was able to hear from all the caregivers, observe the veteran as he interacted with each person, and watch as he walked within the comfort and familiarity of his home.
Based on these observations, the veteran was clearly at risk for falls. The clinician ordered a home safety consultation as a result. Thus the home video telehealth program allowed this veteran’s mobility-impaired wife to participate directly in his dementia care. It gave the clinician an opportunity to spot potential fall risks within the veteran’s home before a disabling fall and provided the entire family with additional, convenient dementia-related care beyond the standard in-person VAMC visits.
Establishing Home Video Links
Veterans must already have broadband Internet access and a home computer or laptop to participate in the program. To assess the connectivity status and computer comfort level of the family, a Bedford VAMC telehealth technician calls the caregiver to assess their computer, operating system, presence of a webcam, and Internet service provider.
In addition to assessing the equipment necessary for the telehealth visit, the Bedford VAMC telehealth team also determines whether or not the caregiver has had experience with videoconferencing. Based on this information, the proper level of support is given to the family for both the initial software and, when necessary, VA-provided webcam installation.
On a few occasions, program staff have visited the veteran’s home to install the software and camera. Thus the telehealth program is fit to the family needs and resources to ensure a successful visit. The Bedford VAMC telehealth team provides enrolled families with live phone-based support for download and installation of the VA-approved videoconferencing software and webcam. For each scheduled video telehealth visit, a telehealth technician is available via phone to assist the caregiver with initiating the video call to the clinician.
Next Steps
GRECC neurologist Lauren Moo, MD, is leading this telehealth initiative as a clinical demonstration project and is studying implementation of the service. Dr. Moo is collecting data on whether IMACC veterans/caregivers accept or decline enrollment and their reasons for declining. The goal is to empirically determine the degree to which age, Internet access, and other variables are barriers to wider adoption.
Dr. Moo predicts that the improved access to clinical care offered by home video telehealth will translate into reduced hotline calls, emergency department visits, and delay in community living center placement. Easier access should facilitate earlier intervention for common dementia-related issues, such as fall risk, behavioral symptoms, and disruption of circadian rhythm, thereby improving quality of life and reducing overall health care utilization for this growing population of veterans.
There is the perception that geriatric veterans are not “wired” for Internet-based communications or lack the technical proficiency to use current and evolving technologies. However, a recent national survey suggests that while only 34% of those aged > 75 years use the Internet, there has been a significant jump in the percentage of Americans aged ≥ 65 years that use the Internet or e-mail: from 40% in 2010 to 53% in 2012.5
Once online, 70% of adults aged ≥ 65 years use the Internet on a typical day, suggesting that when given the necessary tools and training, seniors are enthusiastic technology adopters.5 Thus, it is anticipated that the number of geriatric veterans interested in and able to take advantage of the in-home video visit format will grow rapidly in the near future. The initial enrollment rate at Bedford of 30% is expected to grow as families and providers become more familiar with this modality.
The Bedford VAMC is in an urban/suburban region with multiple Internet service providers, a relatively educated population, and comparatively low levels of poverty. As such, the Bedford VAMC veterans with dementia and their caregivers are likely a best-case scenario population in which to pilot this dementia home telehealth program. If the preliminary success of this pilot program is sustained, expansion to a broader range of home telehealth services, such as social work and home safety assessments, to more rural settings would be the logical next steps.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Thies W, Bleiler L; Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9(2):208-245.
2. Kolanowski AM, Fick D, Waller JL, Shea D. Spouses of persons with dementia: Their healthcare problems, utilization, and costs. Res Nurs Health. 2004;27(5):296-306.
3. Veterans Health Administration Office of the Assistant Deputy Under Secretary for Health for Policy and Planning. Projections of the prevalence and incidence of dementias including Alzheimer’s Disease for the total, enrolled, and patient veteran populations age 65 or over. U.S. Department of Veterans Affairs Website. http://www4.va.gov/healthpolicyplanning/dementia/Dem022004.pdf. Published February 20, 2004. Accessed October 10, 2014.
4. Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: Results from the cardiovascular health study. JAMA. 2002;288(12):1475-1483.
5. Zickuhr K, Madden M. Older adults and Internet use. Pew Research Center’s Internet & American Life Project Website. http://pewinternet.org/Reports/2012/Older-adults-and-internet-use.aspx. Updated June 6, 2012. Accessed October 17, 2014.
For nearly 4 decades, the unifying focus of the 2-site New England Geriatric Research Education and Clinical Center (GRECC) has been on dementia and related disorders. Veterans with dementia are an extremely vulnerable population with high rates of health care use that is projected to total > $203 billion in the U.S. in 2013.1 Their caregivers are also among the most burdened, having provided about 17.5 billion hours of unpaid care in 2012, which is valued at more than $216 billion.1 Additionally, spouses, who are the most common caregivers of persons with dementia, often experience poor health outcomes related to the experience of living with the afflicted spouse.2
Currently > 200,000 VA patients have dementia, and that number is expected to increase.3 Dementia is largely a disease of the elderly; thus, many veterans with dementia also have other medical and orthopedic conditions that increase their frailty and decrease their mobility. Behavioral and psychological symptoms are present in > 75% of people with dementia, contributing to the relative isolation of both those with dementia and their families.4
Disruption in routine and removal from familiar surroundings can cause many patients, particularly in the moderate stages of dementia, to become disoriented and agitated. For these patients, VA clinics can be unsettling and may reveal behavior that is not the same as the veterans’ behavior at home. For these reasons, veterans with dementia likely may benefit from remote access to health care via telehealth. However, current telehealth applications for this population are vastly underdeveloped.
Video Dementia Management
Many GRECCs and other VA geriatric programs provide video-based dementia evaluations and management at community-based outpatient clinics (CBOCs) affiliated with their medical centers. At this point, close to a dozen GRECC and geriatric programs nationally have geriatric psychiatrists, geriatricians, or neurologists conducting such visits from their office or clinic space with video links to a telehealth-enabled room in the corresponding CBOC.
The visits usually entail having the veteran and, when appropriate, a family member check in at the local CBOC for the appointment and receiving assistance throughout the video visit from the telehealth technician at the CBOC.
The technician assists with the technical aspects of the encounter, including establishing and maintaining the video link to the VA medical center (VAMC), and often is trained to administer a brief standardized mental status assessment. The telehealth technician also helps pass along the physician’s written recommendations to the veteran and family once the recommendation summary has been sent by e-mail or printed on the CBOC printer. These VAMC-CBOC video telehealth programs have been very popular with veterans, particularly those in rural settings, since traveling to the CBOC is usually more convenient.
Home-based Video Program
While CBOC-based video telehealth programs expand the population of veterans able to benefit from specialty dementia care, any travel out of the home can be challenging or disruptive for many veterans and their families. In addition, the performance and demeanor of a veteran with dementia in a clinic setting is sometimes different from that which the family describes as their more typical behavior at home.
A new in-home video telehealth program developed by the GRECC at the Edith Nourse Rogers Memorial Veterans Hospital in Bedford is addressing these issues. The Bedford site of the New England GRECC offers in-home clinical video telehealth services to community-dwelling veterans and caregivers as an extension of their Interdisciplinary Memory Assessment Continuity Clinic (IMACC). Currently, the percentage of IMACC veterans/caregivers who have voluntarily signed up for the program is nearly 30%. About 70% of the families that have enrolled to date have their video visits with their spouse caregivers.
Veterans participating in the home video telehealth program have had at least 1 in-person visit at the Bedford IMACC before being invited to join. Veterans and their caregivers are invited to participate in the GRECC home telehealth program either at the time of an in-person IMACC visit or afterward via a telephone call from either a provider or a member of the telehealth staff.
Telehealth visits are offered as a supplement to regularly scheduled in-person visits, not as a substitute. The frequency of telehealth visits is individualized, depending on the medical status of the veteran and preferences of the caregiver. Some participating families, finding the telehealth format much more convenient, have asked whether they could postpone upcoming in-person visits at the VAMC. Many patients are particularly interested in minimizing medical visits in the winter months in New England.
Case Example
A male World War II veteran with moderate stage dementia lived with his wife in an apartment down the street from his adult son and daughter-in-law. His son and daughter-in-law visited and helped with the veteran’s care most days, but his wife was his primary caregiver. However, due to her own mobility issues, she was unable to attend the veteran’s in-person IMACC initial evaluation or subsequent follow-up visit.
This family enthusiastically embraced the opportunity to participate in the home video telehealth program and had multiple telehealth visits. During these video encounters, the veteran, his wife, and his son and daughter-in-law were present. The clinician, communicating via computer from the Bedford VAMC, was able to hear from all the caregivers, observe the veteran as he interacted with each person, and watch as he walked within the comfort and familiarity of his home.
Based on these observations, the veteran was clearly at risk for falls. The clinician ordered a home safety consultation as a result. Thus the home video telehealth program allowed this veteran’s mobility-impaired wife to participate directly in his dementia care. It gave the clinician an opportunity to spot potential fall risks within the veteran’s home before a disabling fall and provided the entire family with additional, convenient dementia-related care beyond the standard in-person VAMC visits.
Establishing Home Video Links
Veterans must already have broadband Internet access and a home computer or laptop to participate in the program. To assess the connectivity status and computer comfort level of the family, a Bedford VAMC telehealth technician calls the caregiver to assess their computer, operating system, presence of a webcam, and Internet service provider.
In addition to assessing the equipment necessary for the telehealth visit, the Bedford VAMC telehealth team also determines whether or not the caregiver has had experience with videoconferencing. Based on this information, the proper level of support is given to the family for both the initial software and, when necessary, VA-provided webcam installation.
On a few occasions, program staff have visited the veteran’s home to install the software and camera. Thus the telehealth program is fit to the family needs and resources to ensure a successful visit. The Bedford VAMC telehealth team provides enrolled families with live phone-based support for download and installation of the VA-approved videoconferencing software and webcam. For each scheduled video telehealth visit, a telehealth technician is available via phone to assist the caregiver with initiating the video call to the clinician.
Next Steps
GRECC neurologist Lauren Moo, MD, is leading this telehealth initiative as a clinical demonstration project and is studying implementation of the service. Dr. Moo is collecting data on whether IMACC veterans/caregivers accept or decline enrollment and their reasons for declining. The goal is to empirically determine the degree to which age, Internet access, and other variables are barriers to wider adoption.
Dr. Moo predicts that the improved access to clinical care offered by home video telehealth will translate into reduced hotline calls, emergency department visits, and delay in community living center placement. Easier access should facilitate earlier intervention for common dementia-related issues, such as fall risk, behavioral symptoms, and disruption of circadian rhythm, thereby improving quality of life and reducing overall health care utilization for this growing population of veterans.
There is the perception that geriatric veterans are not “wired” for Internet-based communications or lack the technical proficiency to use current and evolving technologies. However, a recent national survey suggests that while only 34% of those aged > 75 years use the Internet, there has been a significant jump in the percentage of Americans aged ≥ 65 years that use the Internet or e-mail: from 40% in 2010 to 53% in 2012.5
Once online, 70% of adults aged ≥ 65 years use the Internet on a typical day, suggesting that when given the necessary tools and training, seniors are enthusiastic technology adopters.5 Thus, it is anticipated that the number of geriatric veterans interested in and able to take advantage of the in-home video visit format will grow rapidly in the near future. The initial enrollment rate at Bedford of 30% is expected to grow as families and providers become more familiar with this modality.
The Bedford VAMC is in an urban/suburban region with multiple Internet service providers, a relatively educated population, and comparatively low levels of poverty. As such, the Bedford VAMC veterans with dementia and their caregivers are likely a best-case scenario population in which to pilot this dementia home telehealth program. If the preliminary success of this pilot program is sustained, expansion to a broader range of home telehealth services, such as social work and home safety assessments, to more rural settings would be the logical next steps.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
For nearly 4 decades, the unifying focus of the 2-site New England Geriatric Research Education and Clinical Center (GRECC) has been on dementia and related disorders. Veterans with dementia are an extremely vulnerable population with high rates of health care use that is projected to total > $203 billion in the U.S. in 2013.1 Their caregivers are also among the most burdened, having provided about 17.5 billion hours of unpaid care in 2012, which is valued at more than $216 billion.1 Additionally, spouses, who are the most common caregivers of persons with dementia, often experience poor health outcomes related to the experience of living with the afflicted spouse.2
Currently > 200,000 VA patients have dementia, and that number is expected to increase.3 Dementia is largely a disease of the elderly; thus, many veterans with dementia also have other medical and orthopedic conditions that increase their frailty and decrease their mobility. Behavioral and psychological symptoms are present in > 75% of people with dementia, contributing to the relative isolation of both those with dementia and their families.4
Disruption in routine and removal from familiar surroundings can cause many patients, particularly in the moderate stages of dementia, to become disoriented and agitated. For these patients, VA clinics can be unsettling and may reveal behavior that is not the same as the veterans’ behavior at home. For these reasons, veterans with dementia likely may benefit from remote access to health care via telehealth. However, current telehealth applications for this population are vastly underdeveloped.
Video Dementia Management
Many GRECCs and other VA geriatric programs provide video-based dementia evaluations and management at community-based outpatient clinics (CBOCs) affiliated with their medical centers. At this point, close to a dozen GRECC and geriatric programs nationally have geriatric psychiatrists, geriatricians, or neurologists conducting such visits from their office or clinic space with video links to a telehealth-enabled room in the corresponding CBOC.
The visits usually entail having the veteran and, when appropriate, a family member check in at the local CBOC for the appointment and receiving assistance throughout the video visit from the telehealth technician at the CBOC.
The technician assists with the technical aspects of the encounter, including establishing and maintaining the video link to the VA medical center (VAMC), and often is trained to administer a brief standardized mental status assessment. The telehealth technician also helps pass along the physician’s written recommendations to the veteran and family once the recommendation summary has been sent by e-mail or printed on the CBOC printer. These VAMC-CBOC video telehealth programs have been very popular with veterans, particularly those in rural settings, since traveling to the CBOC is usually more convenient.
Home-based Video Program
While CBOC-based video telehealth programs expand the population of veterans able to benefit from specialty dementia care, any travel out of the home can be challenging or disruptive for many veterans and their families. In addition, the performance and demeanor of a veteran with dementia in a clinic setting is sometimes different from that which the family describes as their more typical behavior at home.
A new in-home video telehealth program developed by the GRECC at the Edith Nourse Rogers Memorial Veterans Hospital in Bedford is addressing these issues. The Bedford site of the New England GRECC offers in-home clinical video telehealth services to community-dwelling veterans and caregivers as an extension of their Interdisciplinary Memory Assessment Continuity Clinic (IMACC). Currently, the percentage of IMACC veterans/caregivers who have voluntarily signed up for the program is nearly 30%. About 70% of the families that have enrolled to date have their video visits with their spouse caregivers.
Veterans participating in the home video telehealth program have had at least 1 in-person visit at the Bedford IMACC before being invited to join. Veterans and their caregivers are invited to participate in the GRECC home telehealth program either at the time of an in-person IMACC visit or afterward via a telephone call from either a provider or a member of the telehealth staff.
Telehealth visits are offered as a supplement to regularly scheduled in-person visits, not as a substitute. The frequency of telehealth visits is individualized, depending on the medical status of the veteran and preferences of the caregiver. Some participating families, finding the telehealth format much more convenient, have asked whether they could postpone upcoming in-person visits at the VAMC. Many patients are particularly interested in minimizing medical visits in the winter months in New England.
Case Example
A male World War II veteran with moderate stage dementia lived with his wife in an apartment down the street from his adult son and daughter-in-law. His son and daughter-in-law visited and helped with the veteran’s care most days, but his wife was his primary caregiver. However, due to her own mobility issues, she was unable to attend the veteran’s in-person IMACC initial evaluation or subsequent follow-up visit.
This family enthusiastically embraced the opportunity to participate in the home video telehealth program and had multiple telehealth visits. During these video encounters, the veteran, his wife, and his son and daughter-in-law were present. The clinician, communicating via computer from the Bedford VAMC, was able to hear from all the caregivers, observe the veteran as he interacted with each person, and watch as he walked within the comfort and familiarity of his home.
Based on these observations, the veteran was clearly at risk for falls. The clinician ordered a home safety consultation as a result. Thus the home video telehealth program allowed this veteran’s mobility-impaired wife to participate directly in his dementia care. It gave the clinician an opportunity to spot potential fall risks within the veteran’s home before a disabling fall and provided the entire family with additional, convenient dementia-related care beyond the standard in-person VAMC visits.
Establishing Home Video Links
Veterans must already have broadband Internet access and a home computer or laptop to participate in the program. To assess the connectivity status and computer comfort level of the family, a Bedford VAMC telehealth technician calls the caregiver to assess their computer, operating system, presence of a webcam, and Internet service provider.
In addition to assessing the equipment necessary for the telehealth visit, the Bedford VAMC telehealth team also determines whether or not the caregiver has had experience with videoconferencing. Based on this information, the proper level of support is given to the family for both the initial software and, when necessary, VA-provided webcam installation.
On a few occasions, program staff have visited the veteran’s home to install the software and camera. Thus the telehealth program is fit to the family needs and resources to ensure a successful visit. The Bedford VAMC telehealth team provides enrolled families with live phone-based support for download and installation of the VA-approved videoconferencing software and webcam. For each scheduled video telehealth visit, a telehealth technician is available via phone to assist the caregiver with initiating the video call to the clinician.
Next Steps
GRECC neurologist Lauren Moo, MD, is leading this telehealth initiative as a clinical demonstration project and is studying implementation of the service. Dr. Moo is collecting data on whether IMACC veterans/caregivers accept or decline enrollment and their reasons for declining. The goal is to empirically determine the degree to which age, Internet access, and other variables are barriers to wider adoption.
Dr. Moo predicts that the improved access to clinical care offered by home video telehealth will translate into reduced hotline calls, emergency department visits, and delay in community living center placement. Easier access should facilitate earlier intervention for common dementia-related issues, such as fall risk, behavioral symptoms, and disruption of circadian rhythm, thereby improving quality of life and reducing overall health care utilization for this growing population of veterans.
There is the perception that geriatric veterans are not “wired” for Internet-based communications or lack the technical proficiency to use current and evolving technologies. However, a recent national survey suggests that while only 34% of those aged > 75 years use the Internet, there has been a significant jump in the percentage of Americans aged ≥ 65 years that use the Internet or e-mail: from 40% in 2010 to 53% in 2012.5
Once online, 70% of adults aged ≥ 65 years use the Internet on a typical day, suggesting that when given the necessary tools and training, seniors are enthusiastic technology adopters.5 Thus, it is anticipated that the number of geriatric veterans interested in and able to take advantage of the in-home video visit format will grow rapidly in the near future. The initial enrollment rate at Bedford of 30% is expected to grow as families and providers become more familiar with this modality.
The Bedford VAMC is in an urban/suburban region with multiple Internet service providers, a relatively educated population, and comparatively low levels of poverty. As such, the Bedford VAMC veterans with dementia and their caregivers are likely a best-case scenario population in which to pilot this dementia home telehealth program. If the preliminary success of this pilot program is sustained, expansion to a broader range of home telehealth services, such as social work and home safety assessments, to more rural settings would be the logical next steps.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Thies W, Bleiler L; Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9(2):208-245.
2. Kolanowski AM, Fick D, Waller JL, Shea D. Spouses of persons with dementia: Their healthcare problems, utilization, and costs. Res Nurs Health. 2004;27(5):296-306.
3. Veterans Health Administration Office of the Assistant Deputy Under Secretary for Health for Policy and Planning. Projections of the prevalence and incidence of dementias including Alzheimer’s Disease for the total, enrolled, and patient veteran populations age 65 or over. U.S. Department of Veterans Affairs Website. http://www4.va.gov/healthpolicyplanning/dementia/Dem022004.pdf. Published February 20, 2004. Accessed October 10, 2014.
4. Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: Results from the cardiovascular health study. JAMA. 2002;288(12):1475-1483.
5. Zickuhr K, Madden M. Older adults and Internet use. Pew Research Center’s Internet & American Life Project Website. http://pewinternet.org/Reports/2012/Older-adults-and-internet-use.aspx. Updated June 6, 2012. Accessed October 17, 2014.
1. Thies W, Bleiler L; Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9(2):208-245.
2. Kolanowski AM, Fick D, Waller JL, Shea D. Spouses of persons with dementia: Their healthcare problems, utilization, and costs. Res Nurs Health. 2004;27(5):296-306.
3. Veterans Health Administration Office of the Assistant Deputy Under Secretary for Health for Policy and Planning. Projections of the prevalence and incidence of dementias including Alzheimer’s Disease for the total, enrolled, and patient veteran populations age 65 or over. U.S. Department of Veterans Affairs Website. http://www4.va.gov/healthpolicyplanning/dementia/Dem022004.pdf. Published February 20, 2004. Accessed October 10, 2014.
4. Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: Results from the cardiovascular health study. JAMA. 2002;288(12):1475-1483.
5. Zickuhr K, Madden M. Older adults and Internet use. Pew Research Center’s Internet & American Life Project Website. http://pewinternet.org/Reports/2012/Older-adults-and-internet-use.aspx. Updated June 6, 2012. Accessed October 17, 2014.
Introducing a better bleeding risk score in atrial fib
CHICAGO – The ORBIT-AF bleeding risk score is a simple, user-friendly new tool for assessing the risk of major bleeding in patients with atrial fibrillation on oral anticoagulation, Emily C. O’Brien, Ph.D., announced at the American Heart Association scientific sessions.
This novel score offers significant advantages over existing bleeding risk scores, including HAS-BLED and ATRIA. Those scores were developed on the basis of small numbers of bleeding events, they show inconsistent performance, and their calculation requires data that’s often not readily accessible in busy daily practice, according to Dr. O’Brien of the Duke Clinical Research Institute in Durham, N.C.

The new score was derived from the ORBIT-AF registry, the largest prospective U.S. registry of patients with atrial fibrillation (AF).
The score was constructed using data on 7,411 AF patients in community practice settings at 173 U.S. sites. All subjects were on oral anticoagulant therapy at baseline. During 2 years of prospective follow-up, 581 patients (7.8%) experienced a major bleeding event as defined by International Society on Thrombosis and Haemostasis criteria.
After sifting through numerous potential candidate variables, Dr. O’Brien and coinvestigators settled upon five they identified as the most potent and practical baseline predictors of major bleeding risk while on oral anticoagulation. Then they packaged them in a convenient acronym: ORBIT, for Older than 74, Renal insufficiency with an estimated glomerular filtration rate below 60 mL/minute per/1.73 m2, Bleeding history, Insufficient hemoglobin/hematocrit or anemia, and Treatment with an antiplatelet agent. The two strongest predictors – renal insufficiency and bleeding history– were awarded two points each; the others are worth one point each.
The observed major bleeding rate among patients enrolled in the ORBIT-AF registry rose with an increasing risk score. The same was true upon application of the ORBIT bleeding score to an independent study sample comprised of participants in the ROCKET-AF randomized clinical trial.
Dr. O’Brien also compared the performance of the ORBIT bleeding score to that of two existing bleeding risk scores – HAS-BLED and ATRIA – in the ORBIT-AF and ROCKET-AF cohorts. The simpler, more user friendly ORBIT bleeding score had a C-statistic of 0.67, similar to the 0.64 for HAS-BLED and 0.66 for ATRIA.
Thus, the ORBIT bleeding score is a practical new tool for use alongside the CHA2DS2-VASc stroke risk score to support clinical decision making regarding whether or not to place an individual AF patient on oral anticoagulation, Dr. O’Brien concluded.
She reported having no financial conflicts regarding this study. The ORBIT-AF registry is sponsored by Janssen.
CHICAGO – The ORBIT-AF bleeding risk score is a simple, user-friendly new tool for assessing the risk of major bleeding in patients with atrial fibrillation on oral anticoagulation, Emily C. O’Brien, Ph.D., announced at the American Heart Association scientific sessions.
This novel score offers significant advantages over existing bleeding risk scores, including HAS-BLED and ATRIA. Those scores were developed on the basis of small numbers of bleeding events, they show inconsistent performance, and their calculation requires data that’s often not readily accessible in busy daily practice, according to Dr. O’Brien of the Duke Clinical Research Institute in Durham, N.C.
The new score was derived from the ORBIT-AF registry, the largest prospective U.S. registry of patients with atrial fibrillation (AF).
The score was constructed using data on 7,411 AF patients in community practice settings at 173 U.S. sites. All subjects were on oral anticoagulant therapy at baseline. During 2 years of prospective follow-up, 581 patients (7.8%) experienced a major bleeding event as defined by International Society on Thrombosis and Haemostasis criteria.
After sifting through numerous potential candidate variables, Dr. O’Brien and coinvestigators settled upon five they identified as the most potent and practical baseline predictors of major bleeding risk while on oral anticoagulation. Then they packaged them in a convenient acronym: ORBIT, for Older than 74, Renal insufficiency with an estimated glomerular filtration rate below 60 mL/minute per/1.73 m2, Bleeding history, Insufficient hemoglobin/hematocrit or anemia, and Treatment with an antiplatelet agent. The two strongest predictors – renal insufficiency and bleeding history– were awarded two points each; the others are worth one point each.
The observed major bleeding rate among patients enrolled in the ORBIT-AF registry rose with an increasing risk score. The same was true upon application of the ORBIT bleeding score to an independent study sample comprised of participants in the ROCKET-AF randomized clinical trial.
Dr. O’Brien also compared the performance of the ORBIT bleeding score to that of two existing bleeding risk scores – HAS-BLED and ATRIA – in the ORBIT-AF and ROCKET-AF cohorts. The simpler, more user friendly ORBIT bleeding score had a C-statistic of 0.67, similar to the 0.64 for HAS-BLED and 0.66 for ATRIA.
Thus, the ORBIT bleeding score is a practical new tool for use alongside the CHA2DS2-VASc stroke risk score to support clinical decision making regarding whether or not to place an individual AF patient on oral anticoagulation, Dr. O’Brien concluded.
She reported having no financial conflicts regarding this study. The ORBIT-AF registry is sponsored by Janssen.
CHICAGO – The ORBIT-AF bleeding risk score is a simple, user-friendly new tool for assessing the risk of major bleeding in patients with atrial fibrillation on oral anticoagulation, Emily C. O’Brien, Ph.D., announced at the American Heart Association scientific sessions.
This novel score offers significant advantages over existing bleeding risk scores, including HAS-BLED and ATRIA. Those scores were developed on the basis of small numbers of bleeding events, they show inconsistent performance, and their calculation requires data that’s often not readily accessible in busy daily practice, according to Dr. O’Brien of the Duke Clinical Research Institute in Durham, N.C.
The new score was derived from the ORBIT-AF registry, the largest prospective U.S. registry of patients with atrial fibrillation (AF).
The score was constructed using data on 7,411 AF patients in community practice settings at 173 U.S. sites. All subjects were on oral anticoagulant therapy at baseline. During 2 years of prospective follow-up, 581 patients (7.8%) experienced a major bleeding event as defined by International Society on Thrombosis and Haemostasis criteria.
After sifting through numerous potential candidate variables, Dr. O’Brien and coinvestigators settled upon five they identified as the most potent and practical baseline predictors of major bleeding risk while on oral anticoagulation. Then they packaged them in a convenient acronym: ORBIT, for Older than 74, Renal insufficiency with an estimated glomerular filtration rate below 60 mL/minute per/1.73 m2, Bleeding history, Insufficient hemoglobin/hematocrit or anemia, and Treatment with an antiplatelet agent. The two strongest predictors – renal insufficiency and bleeding history– were awarded two points each; the others are worth one point each.
The observed major bleeding rate among patients enrolled in the ORBIT-AF registry rose with an increasing risk score. The same was true upon application of the ORBIT bleeding score to an independent study sample comprised of participants in the ROCKET-AF randomized clinical trial.
Dr. O’Brien also compared the performance of the ORBIT bleeding score to that of two existing bleeding risk scores – HAS-BLED and ATRIA – in the ORBIT-AF and ROCKET-AF cohorts. The simpler, more user friendly ORBIT bleeding score had a C-statistic of 0.67, similar to the 0.64 for HAS-BLED and 0.66 for ATRIA.
Thus, the ORBIT bleeding score is a practical new tool for use alongside the CHA2DS2-VASc stroke risk score to support clinical decision making regarding whether or not to place an individual AF patient on oral anticoagulation, Dr. O’Brien concluded.
She reported having no financial conflicts regarding this study. The ORBIT-AF registry is sponsored by Janssen.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: A simple new score is available to assess major bleeding risk in patients with atrial fibrillation on oral anticoagulation.
Major finding: The major bleeding risk in patients with atrial fibrillation on oral anticoagulation ranged from 1.7 per 100 patient-years in those with an ORBIT risk score of 0% to 14.9% in those with a maximum score of 7.
Data source: The risk score was derived by analyzing prospective 2-year follow-up data on 7,411 U.S. patients with atrial fibrillation in a large registry.
Disclosures: The ORBIT-AF registry is sponsored by Janssen. The presenter reported having no financial conflicts.

Weak magnetic fields not responsible for leukemia, study suggests

Research first carried out in the 1970s revealed an association between living near overhead power lines and an increased risk of childhood leukemia.
Although some later studies failed to find such a link, the International Agency for Research on Cancer has categorized low-frequency magnetic fields as “possibly carcinogenic.”
However, a mechanism for this association has never been found, and, now, researchers have ruled out one of the prime candidates.
The team studied the effects of weak magnetic fields (WMFs) on key human proteins, including those
crucial for health, and found they have no detectable impact.
The researchers detailed this discovery in the Journal of the Royal Society Interface.
Alex Jones, PhD, of The University of Manchester in the UK, and his colleagues looked at how WMFs affect flavoproteins, which are key to processes vital for healthy human function, such as the nervous system, DNA repair, and the biological clock.
If these proteins malfunction, there are serious knock-on effects for human health. But after subjecting flavoproteins to WMFs in the lab, the researchers found that WMFs have no detectable impact on these proteins.
“There is still some concern among the public about this potential link, which has been found in some studies into cases of childhood leukemia, but without any clear mechanism for why,” Dr Jones said.
“Flavoproteins transfer electrons from one place to another. Along the path the electrons take, very short-lived chemical species known as radical pairs are often created. Biochemical reactions involving radical pairs are considered the most plausible candidates for sensitivity to WMFs, but for them to be so, the reaction conditions have to be right. This research suggests that the correct conditions for biochemical effects of WMFs are likely to be rare in human biology.”
“More work on other possible links will need to be done,” noted study author Nigel Scrutton, PhD, also of the University of Manchester.
“But this study definitely takes us nearer to the point where we can say that power lines, mobile phones, and other similar devices are likely to be safe for humans.” ![]()
Research first carried out in the 1970s revealed an association between living near overhead power lines and an increased risk of childhood leukemia.
Although some later studies failed to find such a link, the International Agency for Research on Cancer has categorized low-frequency magnetic fields as “possibly carcinogenic.”
However, a mechanism for this association has never been found, and, now, researchers have ruled out one of the prime candidates.
The team studied the effects of weak magnetic fields (WMFs) on key human proteins, including those
crucial for health, and found they have no detectable impact.
The researchers detailed this discovery in the Journal of the Royal Society Interface.
Alex Jones, PhD, of The University of Manchester in the UK, and his colleagues looked at how WMFs affect flavoproteins, which are key to processes vital for healthy human function, such as the nervous system, DNA repair, and the biological clock.
If these proteins malfunction, there are serious knock-on effects for human health. But after subjecting flavoproteins to WMFs in the lab, the researchers found that WMFs have no detectable impact on these proteins.
“There is still some concern among the public about this potential link, which has been found in some studies into cases of childhood leukemia, but without any clear mechanism for why,” Dr Jones said.
“Flavoproteins transfer electrons from one place to another. Along the path the electrons take, very short-lived chemical species known as radical pairs are often created. Biochemical reactions involving radical pairs are considered the most plausible candidates for sensitivity to WMFs, but for them to be so, the reaction conditions have to be right. This research suggests that the correct conditions for biochemical effects of WMFs are likely to be rare in human biology.”
“More work on other possible links will need to be done,” noted study author Nigel Scrutton, PhD, also of the University of Manchester.
“But this study definitely takes us nearer to the point where we can say that power lines, mobile phones, and other similar devices are likely to be safe for humans.” ![]()
Research first carried out in the 1970s revealed an association between living near overhead power lines and an increased risk of childhood leukemia.
Although some later studies failed to find such a link, the International Agency for Research on Cancer has categorized low-frequency magnetic fields as “possibly carcinogenic.”
However, a mechanism for this association has never been found, and, now, researchers have ruled out one of the prime candidates.
The team studied the effects of weak magnetic fields (WMFs) on key human proteins, including those
crucial for health, and found they have no detectable impact.
The researchers detailed this discovery in the Journal of the Royal Society Interface.
Alex Jones, PhD, of The University of Manchester in the UK, and his colleagues looked at how WMFs affect flavoproteins, which are key to processes vital for healthy human function, such as the nervous system, DNA repair, and the biological clock.
If these proteins malfunction, there are serious knock-on effects for human health. But after subjecting flavoproteins to WMFs in the lab, the researchers found that WMFs have no detectable impact on these proteins.
“There is still some concern among the public about this potential link, which has been found in some studies into cases of childhood leukemia, but without any clear mechanism for why,” Dr Jones said.
“Flavoproteins transfer electrons from one place to another. Along the path the electrons take, very short-lived chemical species known as radical pairs are often created. Biochemical reactions involving radical pairs are considered the most plausible candidates for sensitivity to WMFs, but for them to be so, the reaction conditions have to be right. This research suggests that the correct conditions for biochemical effects of WMFs are likely to be rare in human biology.”
“More work on other possible links will need to be done,” noted study author Nigel Scrutton, PhD, also of the University of Manchester.
“But this study definitely takes us nearer to the point where we can say that power lines, mobile phones, and other similar devices are likely to be safe for humans.” ![]()
CDC offers pediatric health care providers resources on Ebola in children
Children’s needs differ significantly from the needs of adults, especially when it comes to handling dire situations like the current Ebola outbreak in West Africa that has resulted in a few cases in the United States.
Children may be at increased risk for developing the infection if they have recently traveled to one of the countries experiencing an outbreak. However, since they are very unlikely to be caregivers or participate in funeral activities that raise the risk of exposure, the chances of a child in the United States developing Ebola is very low, according to the Centers for Disease Control and Prevention.
Because information about Ebola can be scary and alarming for children, it is important for healthcare providers to recognize and address their developmental and psychological needs to help them better understand facts about the illness and their risk of exposure. It also helps to be prepared just in case the need arises to address a potential Ebola case.
The CDC recommends the following resources to guide health care providers who work with children:
1. Ebola Virus Disease and Children: What US Pediatricians Need to Know
2. What Obstetrician–Gynecologists Should Know About Ebola
3. Information for Healthcare Workers and Settings
4. Algorithm for Evaluation of the Returned Traveler
5. Checklist for Patients Being Evaluated for Ebola Virus Disease (EVD) in the United States
6. Interim Guidance for Environmental Infection Control in Hospitals for Ebola Virus
7. Ebola Preparedness Considerations for Outpatient/Ambulatory Care Centers
8. Ebola Screening Criteria for Outpatient/Ambulatory Care Centers
Children’s needs differ significantly from the needs of adults, especially when it comes to handling dire situations like the current Ebola outbreak in West Africa that has resulted in a few cases in the United States.
Children may be at increased risk for developing the infection if they have recently traveled to one of the countries experiencing an outbreak. However, since they are very unlikely to be caregivers or participate in funeral activities that raise the risk of exposure, the chances of a child in the United States developing Ebola is very low, according to the Centers for Disease Control and Prevention.
Because information about Ebola can be scary and alarming for children, it is important for healthcare providers to recognize and address their developmental and psychological needs to help them better understand facts about the illness and their risk of exposure. It also helps to be prepared just in case the need arises to address a potential Ebola case.
The CDC recommends the following resources to guide health care providers who work with children:
1. Ebola Virus Disease and Children: What US Pediatricians Need to Know
2. What Obstetrician–Gynecologists Should Know About Ebola
3. Information for Healthcare Workers and Settings
4. Algorithm for Evaluation of the Returned Traveler
5. Checklist for Patients Being Evaluated for Ebola Virus Disease (EVD) in the United States
6. Interim Guidance for Environmental Infection Control in Hospitals for Ebola Virus
7. Ebola Preparedness Considerations for Outpatient/Ambulatory Care Centers
8. Ebola Screening Criteria for Outpatient/Ambulatory Care Centers
Children’s needs differ significantly from the needs of adults, especially when it comes to handling dire situations like the current Ebola outbreak in West Africa that has resulted in a few cases in the United States.
Children may be at increased risk for developing the infection if they have recently traveled to one of the countries experiencing an outbreak. However, since they are very unlikely to be caregivers or participate in funeral activities that raise the risk of exposure, the chances of a child in the United States developing Ebola is very low, according to the Centers for Disease Control and Prevention.
Because information about Ebola can be scary and alarming for children, it is important for healthcare providers to recognize and address their developmental and psychological needs to help them better understand facts about the illness and their risk of exposure. It also helps to be prepared just in case the need arises to address a potential Ebola case.
The CDC recommends the following resources to guide health care providers who work with children:
1. Ebola Virus Disease and Children: What US Pediatricians Need to Know
2. What Obstetrician–Gynecologists Should Know About Ebola
3. Information for Healthcare Workers and Settings
4. Algorithm for Evaluation of the Returned Traveler
5. Checklist for Patients Being Evaluated for Ebola Virus Disease (EVD) in the United States
6. Interim Guidance for Environmental Infection Control in Hospitals for Ebola Virus
7. Ebola Preparedness Considerations for Outpatient/Ambulatory Care Centers
8. Ebola Screening Criteria for Outpatient/Ambulatory Care Centers
Product controls bleeding in kids with hemophilia A

the 2014 ASH Annual Meeting
SAN FRANCISCO—A recombinant factor VIII (FVIII) Fc fusion protein is effective for routine prophylaxis and control of bleeding in previously treated children with severe hemophilia A, according to the first phase 3 study of a long-acting FVIII in very young patients.
Prophylactic treatment of hemophilia A with recombinant FVIII requires frequent infusions, up to 3 to 4 per week.
Conventional FVIII replacement therapies have circulating half-lives of 8 to 12 hours.
And children exhibit faster clearance than adults, which may necessitate even more frequent infusions.
Recombinant FVIII Fc fusion protein (Eloctate) has been shown to have a 1.5-fold longer half-life when compared with recombinant FVIII (Advate) in a phase 3 study of adults and adolescents.
“In a pediatric population, we demonstrated similar pharmacokinetic safety and efficacy in terms of annualized bleeding rate, with no inhibitors and no adverse events related to the drug,” said Guy Young, MD, of the University of Southern California in Los Angeles.
At the 2014 ASH Annual Meeting, Dr Young and his colleagues reported results observed with recombinant FVIII Fc fusion protein in the KIDS A-LONG study (abstract 1494). This phase 3 trial was sponsored by Biogen Idec and Sobi, the companies developing recombinant FVIII Fc fusion protein.
The study enrolled 71 males under the age of 12 with severe hemophilia A (< 1 IU/dL endogenous FVIII activity), who had at least 50 prior exposure days to FVIII and no history of FVIII inhibitors.
The patients received twice-weekly prophylactic infusions of the drug, 25 IU/kg on day 1 and 50 IU/kg on day 4. Adjustments to dosing frequency up to once every 2 days and dose to ≤ 80 IU/kg were made as needed.
A subset of 25 patients under age 6 and 35 patients ages 6 to 11 underwent sequential pharmacokinetic evaluations with their prior FVIII therapy (50 IU/kg), followed by the recombinant FVIII Fc fusion protein (50 IU/kg).
“The recombinant factor VIII Fc fusion protein was effective for routine prophylaxis and for control of bleeding,” Dr Young said. “A low annualized bleeding rate was observed in both age cohorts.”
About three-quarters of the patients had a longer dosing interval with recombinant FVIII Fc fusion protein compared with their prior FVIII prophylactic dosing interval.
About 90% of the patients were on twice-weekly dosing at the end of the study compared with about 75% infusing at least 3 times a week pre-study, Dr Young said. Some 93% of bleeding episodes were controlled with 1 or 2 injections.
“The recombinant FVIII Fc fusion protein had a prolonged half-life and reduced clearance compared with conventional FVIII,” Dr Young noted.
Half-life extension was comparable to that observed in adults and adolescents.
Adverse events were generally similar to those expected for the pediatric hemophilia population.
Some 85.5% of subjects reported at least one adverse event, but no patient discontinued treatment due to an adverse event. Two non-serious events (myalgia and erythematous rash) were related to recombinant FVIII Fc fusion protein.
Five patients (7.2%) experienced a total of 7 serious adverse events, which were not related to treatment. There were no reports of anaphylaxis, vascular thrombotic events, or death.
Dr Young said the next step is to test the recombinant FVIII Fc fusion protein in previously untreated young hemophilia A patients to determine the rate of immunogenicity.
“We don’t expect to see antibodies in these patients,” he said. ![]()
the 2014 ASH Annual Meeting
SAN FRANCISCO—A recombinant factor VIII (FVIII) Fc fusion protein is effective for routine prophylaxis and control of bleeding in previously treated children with severe hemophilia A, according to the first phase 3 study of a long-acting FVIII in very young patients.
Prophylactic treatment of hemophilia A with recombinant FVIII requires frequent infusions, up to 3 to 4 per week.
Conventional FVIII replacement therapies have circulating half-lives of 8 to 12 hours.
And children exhibit faster clearance than adults, which may necessitate even more frequent infusions.
Recombinant FVIII Fc fusion protein (Eloctate) has been shown to have a 1.5-fold longer half-life when compared with recombinant FVIII (Advate) in a phase 3 study of adults and adolescents.
“In a pediatric population, we demonstrated similar pharmacokinetic safety and efficacy in terms of annualized bleeding rate, with no inhibitors and no adverse events related to the drug,” said Guy Young, MD, of the University of Southern California in Los Angeles.
At the 2014 ASH Annual Meeting, Dr Young and his colleagues reported results observed with recombinant FVIII Fc fusion protein in the KIDS A-LONG study (abstract 1494). This phase 3 trial was sponsored by Biogen Idec and Sobi, the companies developing recombinant FVIII Fc fusion protein.
The study enrolled 71 males under the age of 12 with severe hemophilia A (< 1 IU/dL endogenous FVIII activity), who had at least 50 prior exposure days to FVIII and no history of FVIII inhibitors.
The patients received twice-weekly prophylactic infusions of the drug, 25 IU/kg on day 1 and 50 IU/kg on day 4. Adjustments to dosing frequency up to once every 2 days and dose to ≤ 80 IU/kg were made as needed.
A subset of 25 patients under age 6 and 35 patients ages 6 to 11 underwent sequential pharmacokinetic evaluations with their prior FVIII therapy (50 IU/kg), followed by the recombinant FVIII Fc fusion protein (50 IU/kg).
“The recombinant factor VIII Fc fusion protein was effective for routine prophylaxis and for control of bleeding,” Dr Young said. “A low annualized bleeding rate was observed in both age cohorts.”
About three-quarters of the patients had a longer dosing interval with recombinant FVIII Fc fusion protein compared with their prior FVIII prophylactic dosing interval.
About 90% of the patients were on twice-weekly dosing at the end of the study compared with about 75% infusing at least 3 times a week pre-study, Dr Young said. Some 93% of bleeding episodes were controlled with 1 or 2 injections.
“The recombinant FVIII Fc fusion protein had a prolonged half-life and reduced clearance compared with conventional FVIII,” Dr Young noted.
Half-life extension was comparable to that observed in adults and adolescents.
Adverse events were generally similar to those expected for the pediatric hemophilia population.
Some 85.5% of subjects reported at least one adverse event, but no patient discontinued treatment due to an adverse event. Two non-serious events (myalgia and erythematous rash) were related to recombinant FVIII Fc fusion protein.
Five patients (7.2%) experienced a total of 7 serious adverse events, which were not related to treatment. There were no reports of anaphylaxis, vascular thrombotic events, or death.
Dr Young said the next step is to test the recombinant FVIII Fc fusion protein in previously untreated young hemophilia A patients to determine the rate of immunogenicity.
“We don’t expect to see antibodies in these patients,” he said. ![]()
the 2014 ASH Annual Meeting
SAN FRANCISCO—A recombinant factor VIII (FVIII) Fc fusion protein is effective for routine prophylaxis and control of bleeding in previously treated children with severe hemophilia A, according to the first phase 3 study of a long-acting FVIII in very young patients.
Prophylactic treatment of hemophilia A with recombinant FVIII requires frequent infusions, up to 3 to 4 per week.
Conventional FVIII replacement therapies have circulating half-lives of 8 to 12 hours.
And children exhibit faster clearance than adults, which may necessitate even more frequent infusions.
Recombinant FVIII Fc fusion protein (Eloctate) has been shown to have a 1.5-fold longer half-life when compared with recombinant FVIII (Advate) in a phase 3 study of adults and adolescents.
“In a pediatric population, we demonstrated similar pharmacokinetic safety and efficacy in terms of annualized bleeding rate, with no inhibitors and no adverse events related to the drug,” said Guy Young, MD, of the University of Southern California in Los Angeles.
At the 2014 ASH Annual Meeting, Dr Young and his colleagues reported results observed with recombinant FVIII Fc fusion protein in the KIDS A-LONG study (abstract 1494). This phase 3 trial was sponsored by Biogen Idec and Sobi, the companies developing recombinant FVIII Fc fusion protein.
The study enrolled 71 males under the age of 12 with severe hemophilia A (< 1 IU/dL endogenous FVIII activity), who had at least 50 prior exposure days to FVIII and no history of FVIII inhibitors.
The patients received twice-weekly prophylactic infusions of the drug, 25 IU/kg on day 1 and 50 IU/kg on day 4. Adjustments to dosing frequency up to once every 2 days and dose to ≤ 80 IU/kg were made as needed.
A subset of 25 patients under age 6 and 35 patients ages 6 to 11 underwent sequential pharmacokinetic evaluations with their prior FVIII therapy (50 IU/kg), followed by the recombinant FVIII Fc fusion protein (50 IU/kg).
“The recombinant factor VIII Fc fusion protein was effective for routine prophylaxis and for control of bleeding,” Dr Young said. “A low annualized bleeding rate was observed in both age cohorts.”
About three-quarters of the patients had a longer dosing interval with recombinant FVIII Fc fusion protein compared with their prior FVIII prophylactic dosing interval.
About 90% of the patients were on twice-weekly dosing at the end of the study compared with about 75% infusing at least 3 times a week pre-study, Dr Young said. Some 93% of bleeding episodes were controlled with 1 or 2 injections.
“The recombinant FVIII Fc fusion protein had a prolonged half-life and reduced clearance compared with conventional FVIII,” Dr Young noted.
Half-life extension was comparable to that observed in adults and adolescents.
Adverse events were generally similar to those expected for the pediatric hemophilia population.
Some 85.5% of subjects reported at least one adverse event, but no patient discontinued treatment due to an adverse event. Two non-serious events (myalgia and erythematous rash) were related to recombinant FVIII Fc fusion protein.
Five patients (7.2%) experienced a total of 7 serious adverse events, which were not related to treatment. There were no reports of anaphylaxis, vascular thrombotic events, or death.
Dr Young said the next step is to test the recombinant FVIII Fc fusion protein in previously untreated young hemophilia A patients to determine the rate of immunogenicity.
“We don’t expect to see antibodies in these patients,” he said. ![]()