User login
Post-Discharge Phone Calls Prevent Hospital Readmissions
Two RIV posters presented at HM13 from University of California at San Francisco (UCSF) hospitalists analyzed outcomes from post-discharge phone calls to patients and found that those who were reached and interviewed by a call nurse had a 33% lower all-cause readmission rate.
UCSF joined SHM’s Project BOOST quality initiative in 2009 and adopted its recommendation to call patients within 72 hours of their hospital discharge, according to co-author Michelle Mourad, MD, assistant professor of clinical medicine and a UCSF hospitalist. “We reached out to about 60% to 70% of our patients with a standard script to address issues associated with readmissions,” Dr. Mourad explains. “We were also lucky enough to build a computer program with quantifiable outcomes in the database.”1
Researchers broke the data down into three categories: those called and interviewed by the nurse; those called who didn’t answer the phone or had a wrong number; and those who were never called due to errors in the administrative list of discharged patients. Interpreting the results is complicated, Dr. Mourad says, because of the challenges of separating factors leading to patients answering the survey from those that affect their readmission risk.
“These phone calls weren’t done in isolation and were part of our overall bridging interventions for patients going home from the hospital,” she says. “We designed the intervention to help people, and we found that 43% of those reached had at least one issue identified in the call for which the nurse tried to help.”
However, whether patients reported post-discharge issues and their responses to specific questions within the interview were not associated with readmission rates. “Does that mean the nurses’ calls are not helping? It either means the nurses are effectively managing these issues to prevent readmissions or that the factors affecting readmissions are more complicated than we currently understand,” Dr. Mourad says.
Larry Beresford is a freelance writer in San Francisco.
References
- Harrison J, Quinn K, Mourad M. Is anyone home? The association between being reached for a post-discharge telephone call and 30-day hospital readmission. Harrison J, Quinn K, Mourad M. Any questions? The relationship between responses to post-discharge call questions and 30-day hospital readmissions [abstracts]. Journal of Hospital Medicine, 2013, 8 Suppl 1.
- Institute of Medicine. Crossing the quality chasm: a new health system for the 21st century. Institute of Medicine website. Available at: http://www.iom.edu/~/media/Files/Report%20Files/2001/Crossing-the-Quality-Chasm/Quality%20Chasm%202001%20%20report%20brief.pdf. Accessed Sept. 9, 2013.
- Shlaes DM, Sahm D, Opiela C, Spellberg B. Commentary: the FDA reboot of antibiotic development. Antimicrob Agents Chemother. 29 Jul 2013 [Epub ahead of print].
- Alliance for Aging Research. HAIs growing problem, group says. Alliance for Aging Research website. Available at: http://www.agingresearch.org/content/article/detail/33504. Accessed Sept. 9, 2013.
- Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368:2255-2265.
- Hospitals in Pursuit of Excellence. Eliminating catheter-associated urinary tract infections. Hospitals in Pursuit of Excellence website. Available at: http://www.hpoe.org/Reports-HPOE/eliminating_catheter_associated_urinary_tract_infection.pdf. Accessed Sept. 9, 2013.
- Center to Advance Palliative Care. Growth of palliative care in U.S. hospitals 2013 snapshot. Center to Advance Palliative Care website. Available at: http://www.capc.org/capc-growth-analysis-snapshot-2013.pdf. Accessed Sept. 9, 2013.
Two RIV posters presented at HM13 from University of California at San Francisco (UCSF) hospitalists analyzed outcomes from post-discharge phone calls to patients and found that those who were reached and interviewed by a call nurse had a 33% lower all-cause readmission rate.
UCSF joined SHM’s Project BOOST quality initiative in 2009 and adopted its recommendation to call patients within 72 hours of their hospital discharge, according to co-author Michelle Mourad, MD, assistant professor of clinical medicine and a UCSF hospitalist. “We reached out to about 60% to 70% of our patients with a standard script to address issues associated with readmissions,” Dr. Mourad explains. “We were also lucky enough to build a computer program with quantifiable outcomes in the database.”1
Researchers broke the data down into three categories: those called and interviewed by the nurse; those called who didn’t answer the phone or had a wrong number; and those who were never called due to errors in the administrative list of discharged patients. Interpreting the results is complicated, Dr. Mourad says, because of the challenges of separating factors leading to patients answering the survey from those that affect their readmission risk.
“These phone calls weren’t done in isolation and were part of our overall bridging interventions for patients going home from the hospital,” she says. “We designed the intervention to help people, and we found that 43% of those reached had at least one issue identified in the call for which the nurse tried to help.”
However, whether patients reported post-discharge issues and their responses to specific questions within the interview were not associated with readmission rates. “Does that mean the nurses’ calls are not helping? It either means the nurses are effectively managing these issues to prevent readmissions or that the factors affecting readmissions are more complicated than we currently understand,” Dr. Mourad says.
Larry Beresford is a freelance writer in San Francisco.
References
- Harrison J, Quinn K, Mourad M. Is anyone home? The association between being reached for a post-discharge telephone call and 30-day hospital readmission. Harrison J, Quinn K, Mourad M. Any questions? The relationship between responses to post-discharge call questions and 30-day hospital readmissions [abstracts]. Journal of Hospital Medicine, 2013, 8 Suppl 1.
- Institute of Medicine. Crossing the quality chasm: a new health system for the 21st century. Institute of Medicine website. Available at: http://www.iom.edu/~/media/Files/Report%20Files/2001/Crossing-the-Quality-Chasm/Quality%20Chasm%202001%20%20report%20brief.pdf. Accessed Sept. 9, 2013.
- Shlaes DM, Sahm D, Opiela C, Spellberg B. Commentary: the FDA reboot of antibiotic development. Antimicrob Agents Chemother. 29 Jul 2013 [Epub ahead of print].
- Alliance for Aging Research. HAIs growing problem, group says. Alliance for Aging Research website. Available at: http://www.agingresearch.org/content/article/detail/33504. Accessed Sept. 9, 2013.
- Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368:2255-2265.
- Hospitals in Pursuit of Excellence. Eliminating catheter-associated urinary tract infections. Hospitals in Pursuit of Excellence website. Available at: http://www.hpoe.org/Reports-HPOE/eliminating_catheter_associated_urinary_tract_infection.pdf. Accessed Sept. 9, 2013.
- Center to Advance Palliative Care. Growth of palliative care in U.S. hospitals 2013 snapshot. Center to Advance Palliative Care website. Available at: http://www.capc.org/capc-growth-analysis-snapshot-2013.pdf. Accessed Sept. 9, 2013.
Two RIV posters presented at HM13 from University of California at San Francisco (UCSF) hospitalists analyzed outcomes from post-discharge phone calls to patients and found that those who were reached and interviewed by a call nurse had a 33% lower all-cause readmission rate.
UCSF joined SHM’s Project BOOST quality initiative in 2009 and adopted its recommendation to call patients within 72 hours of their hospital discharge, according to co-author Michelle Mourad, MD, assistant professor of clinical medicine and a UCSF hospitalist. “We reached out to about 60% to 70% of our patients with a standard script to address issues associated with readmissions,” Dr. Mourad explains. “We were also lucky enough to build a computer program with quantifiable outcomes in the database.”1
Researchers broke the data down into three categories: those called and interviewed by the nurse; those called who didn’t answer the phone or had a wrong number; and those who were never called due to errors in the administrative list of discharged patients. Interpreting the results is complicated, Dr. Mourad says, because of the challenges of separating factors leading to patients answering the survey from those that affect their readmission risk.
“These phone calls weren’t done in isolation and were part of our overall bridging interventions for patients going home from the hospital,” she says. “We designed the intervention to help people, and we found that 43% of those reached had at least one issue identified in the call for which the nurse tried to help.”
However, whether patients reported post-discharge issues and their responses to specific questions within the interview were not associated with readmission rates. “Does that mean the nurses’ calls are not helping? It either means the nurses are effectively managing these issues to prevent readmissions or that the factors affecting readmissions are more complicated than we currently understand,” Dr. Mourad says.
Larry Beresford is a freelance writer in San Francisco.
References
- Harrison J, Quinn K, Mourad M. Is anyone home? The association between being reached for a post-discharge telephone call and 30-day hospital readmission. Harrison J, Quinn K, Mourad M. Any questions? The relationship between responses to post-discharge call questions and 30-day hospital readmissions [abstracts]. Journal of Hospital Medicine, 2013, 8 Suppl 1.
- Institute of Medicine. Crossing the quality chasm: a new health system for the 21st century. Institute of Medicine website. Available at: http://www.iom.edu/~/media/Files/Report%20Files/2001/Crossing-the-Quality-Chasm/Quality%20Chasm%202001%20%20report%20brief.pdf. Accessed Sept. 9, 2013.
- Shlaes DM, Sahm D, Opiela C, Spellberg B. Commentary: the FDA reboot of antibiotic development. Antimicrob Agents Chemother. 29 Jul 2013 [Epub ahead of print].
- Alliance for Aging Research. HAIs growing problem, group says. Alliance for Aging Research website. Available at: http://www.agingresearch.org/content/article/detail/33504. Accessed Sept. 9, 2013.
- Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368:2255-2265.
- Hospitals in Pursuit of Excellence. Eliminating catheter-associated urinary tract infections. Hospitals in Pursuit of Excellence website. Available at: http://www.hpoe.org/Reports-HPOE/eliminating_catheter_associated_urinary_tract_infection.pdf. Accessed Sept. 9, 2013.
- Center to Advance Palliative Care. Growth of palliative care in U.S. hospitals 2013 snapshot. Center to Advance Palliative Care website. Available at: http://www.capc.org/capc-growth-analysis-snapshot-2013.pdf. Accessed Sept. 9, 2013.
What Is the Best Empiric Therapy for Community-Acquired Cellulitis?
Editor’s note: This month’s KCQ first appeared in July 2009, and since that time it has been one of our website’s most-read articles, generating 23,000-plus pageviews.
Case
A previously healthy 55-year-old white female presents to the ED with a three-day history of pain and erythema in her right hand. Examination reveals fluctuance as well. She is diagnosed with an abscess with surrounding cellulitis. The abscess is incised, drained, and cultured, and she is sent home on oral trimethoprim/sulfamethoxazole. The following day, her cellulitis has worsened. She is hospitalized and commenced on intravenous vancomycin. What is the best empiric therapy for community-acquired cellulitis?
Background
Cellulitis is defined as a skin and soft-tissue infection (SSTI), which develops as a result of bacterial entry via breaches in the skin barrier. Typically, it involves the dermis and subcutaneous tissue and is associated with local tenderness, erythema, swelling and fever. Cellulitis usually affects the lower extremities, but it can affect other locations, resulting in diagnoses such as periorbital, abdominal wall, buccal, and perianal cellulitis.1,2
Gram-positive organisms, especially Staphylococcus aureus and beta hemolytic streptococci, are the most common causes of cellulitis. Although it is less common, cellulitis can be caused by gram-negative organisms. The recent significant increase in the prevalence of SSTIs due to community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) has led to changes in the selection of antibiotics that were most commonly utilized to empirically treat cellulitis.
The diagnosis of cellulitis is based primarily on clinical manifestations. Due to low diagnostic yields, blood cultures, needle aspiration, or punch biopsy specimens usually are not helpful in the setting of simple cellulitis.3 Therefore, antibiotic therapy is almost universally started empirically. Starting appropriate initial antibiotic therapy improves patient outcomes by reducing mortality rates, length of stay, and inpatient costs.4
Cellulitis incidence is about two cases per 1,000 patient-years.5 This rather high incidence, coupled with escalating rates of SSTIs due to CA-MRSA, demands reliable and cost-effective treatment strategies for the management of community-acquired cellulitis.
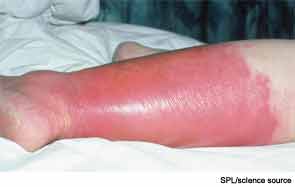
Review of the Data
The treatment of community-acquired cellulitis was straightforward until the past decade, as physicians saw a significant increase in CA-MRSA incidence.6 MRSA was reported initially in 1961, only two years after methicillin was introduced into clinical practice.7,8 Subsequently, MRSA prevalence increased dramatically, and by the beginning of this decade, more than 50% of the Staphylococcus aureus hospital strains were resistant to methicillin.8 Furthermore, 60% to 80% of community-acquired Staphylococcus aureus strains in the U.S. are methicillin-resistant.8
The two major types of MRSA infections are healthcare-acquired (HA-MRSA) and community-acquired (CA-MRSA). The HA-MRSA infection group is further subdivided into those strains that develop during a period of hospitalization and those that develop following contact with healthcare facilities (e.g. hospitalization or surgery within the previous year). This subgroup includes HA-MRSA infections in hemodialysis patients, residents of long-term-care facilities, and individuals who have a vascular catheter or other indwelling device.9,10
CA-MRSA infections, on the other hand, occur in individuals who have not had any contact with healthcare facilities. Higher rates of CA-MRSA infection are observed in settings where individuals have close contact with each other, including military trainees, athletes involved in contact sports, patients age 65 and older, men who have sex with other men, and parenteral substance abusers.8,11-13 However, in view of the high prevalence of CA-MRSA in the U.S., most patients, including those without any apparent risk factors, are at risk.8
HA-MRSA has the ability to survive on inanimate objects for extended time periods, increasing the likelihood of transmission to persons who come into contact with those objects. Although evidence has not confirmed that CA-MRSA has a similar capacity, it seems plausible that such spread does contribute to the propagation of CA-MRSA.12
The increasing importance of CA-MRSA also is evident in hospital settings, where it is replacing HA-MRSA as the most common type of Staphylococcus aureus. Because CA-MRSA tends to be susceptible to a larger number of antibiotics than HA-MRSA is, this has led to a reduced incidence of multidrug resistance. Fortunately, unlike HA-MRSA, CA-MRSA is susceptible to non-beta-lactam antibiotics, including tetracyclines, sulfonamides, and clindamycin.9
CA-MRSA most often causes SSTIs, and a tender abscess is a typical presentation.8 Patients commonly misinterpret early skin lesions as an insect or spider bite.12,14 When cutaneous CA-MRSA presents as an abscess, an incision and drainage procedure is essential for adequate treatment of the infection. For some CA-MRSA infections, particularly those characterized by the presence of a relatively small abscess, it might be adequate to do only an incision and drainage procedure, and not administer antibiotics.8,15 However, in most instances, especially when there is an area of cellulitis around the abscess, the initiation of antibiotic therapy improves patients’ clinical outcomes.9,16
When there is no apparent drainable purulent fluid collection, which often occurs with cellulitis, antibiotics should be the mainstay of therapy. The decision about which antibiotic to start can present some challenges, because the organism causing the cellulitis usually is not identified. This is because blood cultures are positive in less than 5% of cases. Also, positive culture results from needle aspiration are only helpful 5% to 40% of the time. Meanwhile, culture of punch biopsy specimens yields a pathogen in only 20% to 30% of cases.3,17-19
Due to increased CA-MRSA incidence, cephalexin should not be prescribed to treat cellulitis in the outpatient setting because it does not provide coverage for the pathogen.13 Instead, oral antibiotics (e.g. clindamycin or trimethoprim/sulfamethoxazole) should be prescribed. Doxycycline, minocycline, rifampin (usually prescribed in combination with fusidic acid to prevent resistance development), and linezolid are additional therapeutic options.
Trimethoprim/sulfamethoxazole and clindamycin have several advantages: good oral bioavailability, familiarity to physicians, and general affordability. A disadvantage to using both trimethoprim/sulfamethoxazole and doxycycline is that they provide inadequate coverage for group A streptococci, which are a common cause of cellulitis. Therefore, the simultaneous use of a beta-lactam antibiotic with either of these medications may improve outcomes for “nonpurulent” cellulitis.13,15 Linezolid has proven effective for SSTIs caused by MRSA, even though it is not bactericidal.
Excellent oral bioavailability of this drug is an attractive characteristic, as it facilitates the transition from the use of intravenous to oral antibiotic therapy later in a patient’s hospital course. Although oral linezolid has been studied in clinical trials and provides good coverage for MRSA, its use in the outpatient setting is relatively limited, largely due to its significant cost.20 In 2008, the cost of 10 days of treatment with oral linezolid was $1,286.80. In comparison, the generic trimethoprim/sulfamethoxazole cost $9.40, and generic clindamycin cost $95.10.8 The lack of routine availability in many outpatient pharmacies also hinders the widespread use of linezolid.13
To date, with the exception of linezolid, no randomized prospective clinical trials clearly demonstrate the efficacy of the oral agents that are commonly used for the outpatient treatment of cellulitis.20
When patients require hospitalization for the optimal treatment of cellulitis, it is important to select a parenteral antibiotic that provides coverage for MRSA.8 Vancomycin, daptomycin, linezolid, and tigecycline are the most commonly used agents.6
In the inpatient setting, failure to initiate appropriate medical therapy can result in longer hospital admissions, which significantly increase inpatient costs. Inadequate antibiotic therapy creates a significant financial burden and has been associated with increased mortality.4 Historically, vancomycin is used whenever a MRSA infection is suspected. However, there is concern about the declining efficacy of vancomycin related to a gradual increase in the rate of relative resistance—a minimal inhibitory concentration (MIC) increase—in MRSA strains. This MIC creep is noted in some medical centers and can lead to a failure to respond to vancomycin.13,20
Daptomycin is rapidly bactericidal against MRSA; in some institutions, its use may be preferred over vancomycin because the former antibiotic is associated with a significantly more rapid clinical response, which may shorten the required length of hospitalization.21 The once-daily dosing requirement for daptomycin allows for ease of use in both hospital and outpatient settings, and therefore may facilitate early hospital discharge or prevent the need for hospitalization altogether. Clinical experience also suggests potential economic advantages with the use of daptomycin.22
Tigecycline is a bacteriostatic antibiotic that achieves low serum concentrations. However, it penetrates the skin well and has a similar effectiveness to combination therapy with vancomycin and aztreonam. Thus far, tigecycline is not widely used for the treatment of MRSA infections, and it has been suggested that it may be preferred for polymicrobial infections or for patients who exhibit allergies to more commonly used agents.8
When selecting an antibiotic therapy, cost considerations play an important role in the decision-making process. For intravenous agents commonly used to treat CA-MRSA infections, the 2008 cost for 10 days of treatment with generic vancomycin was $182.80; daptomycin cost $1,660.80. For tigecycline and linezolid, the same duration of treatment cost $1,362 and $1,560, respectively.8
Back to the Case
Our patient, an otherwise healthy female, presented with no apparent risk factors for developing a CA-MRSA SSTI. However, more detailed history revealed that she regularly used sports equipment at her local fitness center. Based on her clinical presentation and concerns about the high local prevalence of CA-MRSA, an incision and drainage procedure was performed, and she was started empirically on IV vancomycin. She had a positive clinical response to this treatment.
Wound culture results confirmed CA-MRSA abscess and cellulitis, susceptible to trimethoprim/sulfamethoxazole. She was discharged on the oral formulation of this antibiotic to complete a 10-day course of treatment, including the days she received intravenous antibiotics.
Few well-designed trials have compared different lengths of cellulitis therapy. Most authorities recommend five to 10 days of treatment; however, longer courses might be required for more severe or complicated diseases.
Bottom line
Because of the high prevalence of CA-MRSA, initial antibiotic therapy for the treatment of community-acquired cellulitis must provide coverage for this organism.
Dr. Clarke is a hospitalist and assistant professor of medicine at Emory University School of Medicine, Atlanta. Dr. Dressler is a professor of medicine, hospital medicine associate division director for education, and associate program director for the J. Willis Hurst Internal Medicine Residency Program. Dr. Purohit, formerly an instructor in clinical medicine at Emory, is a hospitalist at WakeMed Health and Hospitals in Raleigh, N.C.
References
- Barzilai A, Choen HA. Isolation of group A streptococci from children with perianal cellulitis and from their siblings. Pediatr Infect Dis J. 1998;17(4):358-360.
- Thorsteinsdottir B, Tleyjeh IM, Baddour LM. Abdominal wall cellulitis in the morbidly obese. Scand J Infect Dis. 2005;37(8):605-608.
- Swartz MN. Clinical practice. Cellulitis. N Engl J Med. 2004;350(9):904-912.
- Edelsberg J, Berger A, Weber DJ, et al. Clinical and economic consequences of failure of initial antibiotic therapy for hospitalized patients with complicated skin and skin-structure infections. Infect Control Hosp Epidemiol. 2008;29(2):160-169.
- McNamara DR, Tleyjeh IM, Berbari EF, et al. Incidence of lower extremity cellulitis: a population-based study in Olmsted County, Minnesota. Mayo Clin Proc. 2007;82(7):817-821.
- Moellering RC. Current treatment options for community-acquired methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46(7):1032-1037.
- Chambers HF. The changing epidemiology of Staphylococcus aureus. Emerg Infect Dis. 2001;7(2):178-182.
- Moellering RC. A 39-year-old man with a skin infection. JAMA. 2008;299(1):79-87.
- Ruhe J, Smith N, Bradsher RW, Menon A. Community-onset methicillin-resistant Staphylococcus aureus skin and soft tissue infections: impact of antimicrobial therapy on outcome. Clin Infect Dis. 2007;44(6):777-784.
- David MZ, Glikman D, Crawford SE, et al. What is community-associated methicillin-resistant Staphylococcus aureus? J Infect Dis. 2008;197(9):1235-1243.
- Iyer S, Jones DH. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: a retrospective analysis of clinical presentation and treatment of a local outbreak. J Am Acad Dermatol. 2004;50(6):854-858.
- Centers for Disease Control and Prevention. Methicillin-resistant Staphylococcus aureus skin or soft tissue infections in a state prison—Mississippi, 2000. MMWR Morb Mortal Wkly Rep. 2001;50(42):919-922.
- Daum RS. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med. 2007;357(4):380-390.
- Dominguez TJ. It’s not a spider bite, it’s community-acquired methicillin-resistant Staphylococcus aureus. J Am Board Fam Pract. 2004;17(3):220-226.
- Moran GJ, Krishnadasan A, Gorwitz RJ, et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355(7):666-674.
- Jetton L. Therapy for methicillin-resistant Staphylococcus aureus. N Engl J Med. 2006;355(20):2153-2155.
- Hook EW, Hooton TM, Horton CA, et al. Microbiologic evaluation of cutaneous cellulitis in adults. Arch Intern Med. 1986;146(2):295-297. Duvanel T, Auckenthaler R, Rohner P, Harms M,
- Saurat JH. Quantitative cultures of biopsy specimens from cutaneous cellulitis. Arch Intern Med. 1989;149(2):293-296.
- Newell PM, Norden CW. Value of needle aspiration in bacteriologic diagnosis of cellulitis in adults. J Clin Microbiol. 1988; 26(3):401-404.
- Loffler CA, Macdougall C. Update on prevalence and treatment of methicillin-resistant Staphylococcus aureus infections. Expert Rev Anti Infect Ther. 2007;5(6):961-981.
- Davis SL, McKinnon PS, Hall LM, et al. Daptomycin versus vancomycin for complicated skin and skin structure infections: clinical and economic outcomes. Pharmacotherapy. 2007;27(12):1611-1618.
- Seaton RA. Daptomycin: rationale and role in the management of skin and soft tissue infections. J Antimicrob Chemother. 2008;62(Suppl 3):iii15-23.
Editor’s note: This month’s KCQ first appeared in July 2009, and since that time it has been one of our website’s most-read articles, generating 23,000-plus pageviews.
Case
A previously healthy 55-year-old white female presents to the ED with a three-day history of pain and erythema in her right hand. Examination reveals fluctuance as well. She is diagnosed with an abscess with surrounding cellulitis. The abscess is incised, drained, and cultured, and she is sent home on oral trimethoprim/sulfamethoxazole. The following day, her cellulitis has worsened. She is hospitalized and commenced on intravenous vancomycin. What is the best empiric therapy for community-acquired cellulitis?
Background
Cellulitis is defined as a skin and soft-tissue infection (SSTI), which develops as a result of bacterial entry via breaches in the skin barrier. Typically, it involves the dermis and subcutaneous tissue and is associated with local tenderness, erythema, swelling and fever. Cellulitis usually affects the lower extremities, but it can affect other locations, resulting in diagnoses such as periorbital, abdominal wall, buccal, and perianal cellulitis.1,2
Gram-positive organisms, especially Staphylococcus aureus and beta hemolytic streptococci, are the most common causes of cellulitis. Although it is less common, cellulitis can be caused by gram-negative organisms. The recent significant increase in the prevalence of SSTIs due to community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) has led to changes in the selection of antibiotics that were most commonly utilized to empirically treat cellulitis.
The diagnosis of cellulitis is based primarily on clinical manifestations. Due to low diagnostic yields, blood cultures, needle aspiration, or punch biopsy specimens usually are not helpful in the setting of simple cellulitis.3 Therefore, antibiotic therapy is almost universally started empirically. Starting appropriate initial antibiotic therapy improves patient outcomes by reducing mortality rates, length of stay, and inpatient costs.4
Cellulitis incidence is about two cases per 1,000 patient-years.5 This rather high incidence, coupled with escalating rates of SSTIs due to CA-MRSA, demands reliable and cost-effective treatment strategies for the management of community-acquired cellulitis.
Review of the Data
The treatment of community-acquired cellulitis was straightforward until the past decade, as physicians saw a significant increase in CA-MRSA incidence.6 MRSA was reported initially in 1961, only two years after methicillin was introduced into clinical practice.7,8 Subsequently, MRSA prevalence increased dramatically, and by the beginning of this decade, more than 50% of the Staphylococcus aureus hospital strains were resistant to methicillin.8 Furthermore, 60% to 80% of community-acquired Staphylococcus aureus strains in the U.S. are methicillin-resistant.8
The two major types of MRSA infections are healthcare-acquired (HA-MRSA) and community-acquired (CA-MRSA). The HA-MRSA infection group is further subdivided into those strains that develop during a period of hospitalization and those that develop following contact with healthcare facilities (e.g. hospitalization or surgery within the previous year). This subgroup includes HA-MRSA infections in hemodialysis patients, residents of long-term-care facilities, and individuals who have a vascular catheter or other indwelling device.9,10
CA-MRSA infections, on the other hand, occur in individuals who have not had any contact with healthcare facilities. Higher rates of CA-MRSA infection are observed in settings where individuals have close contact with each other, including military trainees, athletes involved in contact sports, patients age 65 and older, men who have sex with other men, and parenteral substance abusers.8,11-13 However, in view of the high prevalence of CA-MRSA in the U.S., most patients, including those without any apparent risk factors, are at risk.8
HA-MRSA has the ability to survive on inanimate objects for extended time periods, increasing the likelihood of transmission to persons who come into contact with those objects. Although evidence has not confirmed that CA-MRSA has a similar capacity, it seems plausible that such spread does contribute to the propagation of CA-MRSA.12
The increasing importance of CA-MRSA also is evident in hospital settings, where it is replacing HA-MRSA as the most common type of Staphylococcus aureus. Because CA-MRSA tends to be susceptible to a larger number of antibiotics than HA-MRSA is, this has led to a reduced incidence of multidrug resistance. Fortunately, unlike HA-MRSA, CA-MRSA is susceptible to non-beta-lactam antibiotics, including tetracyclines, sulfonamides, and clindamycin.9
CA-MRSA most often causes SSTIs, and a tender abscess is a typical presentation.8 Patients commonly misinterpret early skin lesions as an insect or spider bite.12,14 When cutaneous CA-MRSA presents as an abscess, an incision and drainage procedure is essential for adequate treatment of the infection. For some CA-MRSA infections, particularly those characterized by the presence of a relatively small abscess, it might be adequate to do only an incision and drainage procedure, and not administer antibiotics.8,15 However, in most instances, especially when there is an area of cellulitis around the abscess, the initiation of antibiotic therapy improves patients’ clinical outcomes.9,16
When there is no apparent drainable purulent fluid collection, which often occurs with cellulitis, antibiotics should be the mainstay of therapy. The decision about which antibiotic to start can present some challenges, because the organism causing the cellulitis usually is not identified. This is because blood cultures are positive in less than 5% of cases. Also, positive culture results from needle aspiration are only helpful 5% to 40% of the time. Meanwhile, culture of punch biopsy specimens yields a pathogen in only 20% to 30% of cases.3,17-19
Due to increased CA-MRSA incidence, cephalexin should not be prescribed to treat cellulitis in the outpatient setting because it does not provide coverage for the pathogen.13 Instead, oral antibiotics (e.g. clindamycin or trimethoprim/sulfamethoxazole) should be prescribed. Doxycycline, minocycline, rifampin (usually prescribed in combination with fusidic acid to prevent resistance development), and linezolid are additional therapeutic options.
Trimethoprim/sulfamethoxazole and clindamycin have several advantages: good oral bioavailability, familiarity to physicians, and general affordability. A disadvantage to using both trimethoprim/sulfamethoxazole and doxycycline is that they provide inadequate coverage for group A streptococci, which are a common cause of cellulitis. Therefore, the simultaneous use of a beta-lactam antibiotic with either of these medications may improve outcomes for “nonpurulent” cellulitis.13,15 Linezolid has proven effective for SSTIs caused by MRSA, even though it is not bactericidal.
Excellent oral bioavailability of this drug is an attractive characteristic, as it facilitates the transition from the use of intravenous to oral antibiotic therapy later in a patient’s hospital course. Although oral linezolid has been studied in clinical trials and provides good coverage for MRSA, its use in the outpatient setting is relatively limited, largely due to its significant cost.20 In 2008, the cost of 10 days of treatment with oral linezolid was $1,286.80. In comparison, the generic trimethoprim/sulfamethoxazole cost $9.40, and generic clindamycin cost $95.10.8 The lack of routine availability in many outpatient pharmacies also hinders the widespread use of linezolid.13
To date, with the exception of linezolid, no randomized prospective clinical trials clearly demonstrate the efficacy of the oral agents that are commonly used for the outpatient treatment of cellulitis.20
When patients require hospitalization for the optimal treatment of cellulitis, it is important to select a parenteral antibiotic that provides coverage for MRSA.8 Vancomycin, daptomycin, linezolid, and tigecycline are the most commonly used agents.6
In the inpatient setting, failure to initiate appropriate medical therapy can result in longer hospital admissions, which significantly increase inpatient costs. Inadequate antibiotic therapy creates a significant financial burden and has been associated with increased mortality.4 Historically, vancomycin is used whenever a MRSA infection is suspected. However, there is concern about the declining efficacy of vancomycin related to a gradual increase in the rate of relative resistance—a minimal inhibitory concentration (MIC) increase—in MRSA strains. This MIC creep is noted in some medical centers and can lead to a failure to respond to vancomycin.13,20
Daptomycin is rapidly bactericidal against MRSA; in some institutions, its use may be preferred over vancomycin because the former antibiotic is associated with a significantly more rapid clinical response, which may shorten the required length of hospitalization.21 The once-daily dosing requirement for daptomycin allows for ease of use in both hospital and outpatient settings, and therefore may facilitate early hospital discharge or prevent the need for hospitalization altogether. Clinical experience also suggests potential economic advantages with the use of daptomycin.22
Tigecycline is a bacteriostatic antibiotic that achieves low serum concentrations. However, it penetrates the skin well and has a similar effectiveness to combination therapy with vancomycin and aztreonam. Thus far, tigecycline is not widely used for the treatment of MRSA infections, and it has been suggested that it may be preferred for polymicrobial infections or for patients who exhibit allergies to more commonly used agents.8
When selecting an antibiotic therapy, cost considerations play an important role in the decision-making process. For intravenous agents commonly used to treat CA-MRSA infections, the 2008 cost for 10 days of treatment with generic vancomycin was $182.80; daptomycin cost $1,660.80. For tigecycline and linezolid, the same duration of treatment cost $1,362 and $1,560, respectively.8
Back to the Case
Our patient, an otherwise healthy female, presented with no apparent risk factors for developing a CA-MRSA SSTI. However, more detailed history revealed that she regularly used sports equipment at her local fitness center. Based on her clinical presentation and concerns about the high local prevalence of CA-MRSA, an incision and drainage procedure was performed, and she was started empirically on IV vancomycin. She had a positive clinical response to this treatment.
Wound culture results confirmed CA-MRSA abscess and cellulitis, susceptible to trimethoprim/sulfamethoxazole. She was discharged on the oral formulation of this antibiotic to complete a 10-day course of treatment, including the days she received intravenous antibiotics.
Few well-designed trials have compared different lengths of cellulitis therapy. Most authorities recommend five to 10 days of treatment; however, longer courses might be required for more severe or complicated diseases.
Bottom line
Because of the high prevalence of CA-MRSA, initial antibiotic therapy for the treatment of community-acquired cellulitis must provide coverage for this organism.
Dr. Clarke is a hospitalist and assistant professor of medicine at Emory University School of Medicine, Atlanta. Dr. Dressler is a professor of medicine, hospital medicine associate division director for education, and associate program director for the J. Willis Hurst Internal Medicine Residency Program. Dr. Purohit, formerly an instructor in clinical medicine at Emory, is a hospitalist at WakeMed Health and Hospitals in Raleigh, N.C.
References
- Barzilai A, Choen HA. Isolation of group A streptococci from children with perianal cellulitis and from their siblings. Pediatr Infect Dis J. 1998;17(4):358-360.
- Thorsteinsdottir B, Tleyjeh IM, Baddour LM. Abdominal wall cellulitis in the morbidly obese. Scand J Infect Dis. 2005;37(8):605-608.
- Swartz MN. Clinical practice. Cellulitis. N Engl J Med. 2004;350(9):904-912.
- Edelsberg J, Berger A, Weber DJ, et al. Clinical and economic consequences of failure of initial antibiotic therapy for hospitalized patients with complicated skin and skin-structure infections. Infect Control Hosp Epidemiol. 2008;29(2):160-169.
- McNamara DR, Tleyjeh IM, Berbari EF, et al. Incidence of lower extremity cellulitis: a population-based study in Olmsted County, Minnesota. Mayo Clin Proc. 2007;82(7):817-821.
- Moellering RC. Current treatment options for community-acquired methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46(7):1032-1037.
- Chambers HF. The changing epidemiology of Staphylococcus aureus. Emerg Infect Dis. 2001;7(2):178-182.
- Moellering RC. A 39-year-old man with a skin infection. JAMA. 2008;299(1):79-87.
- Ruhe J, Smith N, Bradsher RW, Menon A. Community-onset methicillin-resistant Staphylococcus aureus skin and soft tissue infections: impact of antimicrobial therapy on outcome. Clin Infect Dis. 2007;44(6):777-784.
- David MZ, Glikman D, Crawford SE, et al. What is community-associated methicillin-resistant Staphylococcus aureus? J Infect Dis. 2008;197(9):1235-1243.
- Iyer S, Jones DH. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: a retrospective analysis of clinical presentation and treatment of a local outbreak. J Am Acad Dermatol. 2004;50(6):854-858.
- Centers for Disease Control and Prevention. Methicillin-resistant Staphylococcus aureus skin or soft tissue infections in a state prison—Mississippi, 2000. MMWR Morb Mortal Wkly Rep. 2001;50(42):919-922.
- Daum RS. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med. 2007;357(4):380-390.
- Dominguez TJ. It’s not a spider bite, it’s community-acquired methicillin-resistant Staphylococcus aureus. J Am Board Fam Pract. 2004;17(3):220-226.
- Moran GJ, Krishnadasan A, Gorwitz RJ, et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355(7):666-674.
- Jetton L. Therapy for methicillin-resistant Staphylococcus aureus. N Engl J Med. 2006;355(20):2153-2155.
- Hook EW, Hooton TM, Horton CA, et al. Microbiologic evaluation of cutaneous cellulitis in adults. Arch Intern Med. 1986;146(2):295-297. Duvanel T, Auckenthaler R, Rohner P, Harms M,
- Saurat JH. Quantitative cultures of biopsy specimens from cutaneous cellulitis. Arch Intern Med. 1989;149(2):293-296.
- Newell PM, Norden CW. Value of needle aspiration in bacteriologic diagnosis of cellulitis in adults. J Clin Microbiol. 1988; 26(3):401-404.
- Loffler CA, Macdougall C. Update on prevalence and treatment of methicillin-resistant Staphylococcus aureus infections. Expert Rev Anti Infect Ther. 2007;5(6):961-981.
- Davis SL, McKinnon PS, Hall LM, et al. Daptomycin versus vancomycin for complicated skin and skin structure infections: clinical and economic outcomes. Pharmacotherapy. 2007;27(12):1611-1618.
- Seaton RA. Daptomycin: rationale and role in the management of skin and soft tissue infections. J Antimicrob Chemother. 2008;62(Suppl 3):iii15-23.
Editor’s note: This month’s KCQ first appeared in July 2009, and since that time it has been one of our website’s most-read articles, generating 23,000-plus pageviews.
Case
A previously healthy 55-year-old white female presents to the ED with a three-day history of pain and erythema in her right hand. Examination reveals fluctuance as well. She is diagnosed with an abscess with surrounding cellulitis. The abscess is incised, drained, and cultured, and she is sent home on oral trimethoprim/sulfamethoxazole. The following day, her cellulitis has worsened. She is hospitalized and commenced on intravenous vancomycin. What is the best empiric therapy for community-acquired cellulitis?
Background
Cellulitis is defined as a skin and soft-tissue infection (SSTI), which develops as a result of bacterial entry via breaches in the skin barrier. Typically, it involves the dermis and subcutaneous tissue and is associated with local tenderness, erythema, swelling and fever. Cellulitis usually affects the lower extremities, but it can affect other locations, resulting in diagnoses such as periorbital, abdominal wall, buccal, and perianal cellulitis.1,2
Gram-positive organisms, especially Staphylococcus aureus and beta hemolytic streptococci, are the most common causes of cellulitis. Although it is less common, cellulitis can be caused by gram-negative organisms. The recent significant increase in the prevalence of SSTIs due to community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) has led to changes in the selection of antibiotics that were most commonly utilized to empirically treat cellulitis.
The diagnosis of cellulitis is based primarily on clinical manifestations. Due to low diagnostic yields, blood cultures, needle aspiration, or punch biopsy specimens usually are not helpful in the setting of simple cellulitis.3 Therefore, antibiotic therapy is almost universally started empirically. Starting appropriate initial antibiotic therapy improves patient outcomes by reducing mortality rates, length of stay, and inpatient costs.4
Cellulitis incidence is about two cases per 1,000 patient-years.5 This rather high incidence, coupled with escalating rates of SSTIs due to CA-MRSA, demands reliable and cost-effective treatment strategies for the management of community-acquired cellulitis.
Review of the Data
The treatment of community-acquired cellulitis was straightforward until the past decade, as physicians saw a significant increase in CA-MRSA incidence.6 MRSA was reported initially in 1961, only two years after methicillin was introduced into clinical practice.7,8 Subsequently, MRSA prevalence increased dramatically, and by the beginning of this decade, more than 50% of the Staphylococcus aureus hospital strains were resistant to methicillin.8 Furthermore, 60% to 80% of community-acquired Staphylococcus aureus strains in the U.S. are methicillin-resistant.8
The two major types of MRSA infections are healthcare-acquired (HA-MRSA) and community-acquired (CA-MRSA). The HA-MRSA infection group is further subdivided into those strains that develop during a period of hospitalization and those that develop following contact with healthcare facilities (e.g. hospitalization or surgery within the previous year). This subgroup includes HA-MRSA infections in hemodialysis patients, residents of long-term-care facilities, and individuals who have a vascular catheter or other indwelling device.9,10
CA-MRSA infections, on the other hand, occur in individuals who have not had any contact with healthcare facilities. Higher rates of CA-MRSA infection are observed in settings where individuals have close contact with each other, including military trainees, athletes involved in contact sports, patients age 65 and older, men who have sex with other men, and parenteral substance abusers.8,11-13 However, in view of the high prevalence of CA-MRSA in the U.S., most patients, including those without any apparent risk factors, are at risk.8
HA-MRSA has the ability to survive on inanimate objects for extended time periods, increasing the likelihood of transmission to persons who come into contact with those objects. Although evidence has not confirmed that CA-MRSA has a similar capacity, it seems plausible that such spread does contribute to the propagation of CA-MRSA.12
The increasing importance of CA-MRSA also is evident in hospital settings, where it is replacing HA-MRSA as the most common type of Staphylococcus aureus. Because CA-MRSA tends to be susceptible to a larger number of antibiotics than HA-MRSA is, this has led to a reduced incidence of multidrug resistance. Fortunately, unlike HA-MRSA, CA-MRSA is susceptible to non-beta-lactam antibiotics, including tetracyclines, sulfonamides, and clindamycin.9
CA-MRSA most often causes SSTIs, and a tender abscess is a typical presentation.8 Patients commonly misinterpret early skin lesions as an insect or spider bite.12,14 When cutaneous CA-MRSA presents as an abscess, an incision and drainage procedure is essential for adequate treatment of the infection. For some CA-MRSA infections, particularly those characterized by the presence of a relatively small abscess, it might be adequate to do only an incision and drainage procedure, and not administer antibiotics.8,15 However, in most instances, especially when there is an area of cellulitis around the abscess, the initiation of antibiotic therapy improves patients’ clinical outcomes.9,16
When there is no apparent drainable purulent fluid collection, which often occurs with cellulitis, antibiotics should be the mainstay of therapy. The decision about which antibiotic to start can present some challenges, because the organism causing the cellulitis usually is not identified. This is because blood cultures are positive in less than 5% of cases. Also, positive culture results from needle aspiration are only helpful 5% to 40% of the time. Meanwhile, culture of punch biopsy specimens yields a pathogen in only 20% to 30% of cases.3,17-19
Due to increased CA-MRSA incidence, cephalexin should not be prescribed to treat cellulitis in the outpatient setting because it does not provide coverage for the pathogen.13 Instead, oral antibiotics (e.g. clindamycin or trimethoprim/sulfamethoxazole) should be prescribed. Doxycycline, minocycline, rifampin (usually prescribed in combination with fusidic acid to prevent resistance development), and linezolid are additional therapeutic options.
Trimethoprim/sulfamethoxazole and clindamycin have several advantages: good oral bioavailability, familiarity to physicians, and general affordability. A disadvantage to using both trimethoprim/sulfamethoxazole and doxycycline is that they provide inadequate coverage for group A streptococci, which are a common cause of cellulitis. Therefore, the simultaneous use of a beta-lactam antibiotic with either of these medications may improve outcomes for “nonpurulent” cellulitis.13,15 Linezolid has proven effective for SSTIs caused by MRSA, even though it is not bactericidal.
Excellent oral bioavailability of this drug is an attractive characteristic, as it facilitates the transition from the use of intravenous to oral antibiotic therapy later in a patient’s hospital course. Although oral linezolid has been studied in clinical trials and provides good coverage for MRSA, its use in the outpatient setting is relatively limited, largely due to its significant cost.20 In 2008, the cost of 10 days of treatment with oral linezolid was $1,286.80. In comparison, the generic trimethoprim/sulfamethoxazole cost $9.40, and generic clindamycin cost $95.10.8 The lack of routine availability in many outpatient pharmacies also hinders the widespread use of linezolid.13
To date, with the exception of linezolid, no randomized prospective clinical trials clearly demonstrate the efficacy of the oral agents that are commonly used for the outpatient treatment of cellulitis.20
When patients require hospitalization for the optimal treatment of cellulitis, it is important to select a parenteral antibiotic that provides coverage for MRSA.8 Vancomycin, daptomycin, linezolid, and tigecycline are the most commonly used agents.6
In the inpatient setting, failure to initiate appropriate medical therapy can result in longer hospital admissions, which significantly increase inpatient costs. Inadequate antibiotic therapy creates a significant financial burden and has been associated with increased mortality.4 Historically, vancomycin is used whenever a MRSA infection is suspected. However, there is concern about the declining efficacy of vancomycin related to a gradual increase in the rate of relative resistance—a minimal inhibitory concentration (MIC) increase—in MRSA strains. This MIC creep is noted in some medical centers and can lead to a failure to respond to vancomycin.13,20
Daptomycin is rapidly bactericidal against MRSA; in some institutions, its use may be preferred over vancomycin because the former antibiotic is associated with a significantly more rapid clinical response, which may shorten the required length of hospitalization.21 The once-daily dosing requirement for daptomycin allows for ease of use in both hospital and outpatient settings, and therefore may facilitate early hospital discharge or prevent the need for hospitalization altogether. Clinical experience also suggests potential economic advantages with the use of daptomycin.22
Tigecycline is a bacteriostatic antibiotic that achieves low serum concentrations. However, it penetrates the skin well and has a similar effectiveness to combination therapy with vancomycin and aztreonam. Thus far, tigecycline is not widely used for the treatment of MRSA infections, and it has been suggested that it may be preferred for polymicrobial infections or for patients who exhibit allergies to more commonly used agents.8
When selecting an antibiotic therapy, cost considerations play an important role in the decision-making process. For intravenous agents commonly used to treat CA-MRSA infections, the 2008 cost for 10 days of treatment with generic vancomycin was $182.80; daptomycin cost $1,660.80. For tigecycline and linezolid, the same duration of treatment cost $1,362 and $1,560, respectively.8
Back to the Case
Our patient, an otherwise healthy female, presented with no apparent risk factors for developing a CA-MRSA SSTI. However, more detailed history revealed that she regularly used sports equipment at her local fitness center. Based on her clinical presentation and concerns about the high local prevalence of CA-MRSA, an incision and drainage procedure was performed, and she was started empirically on IV vancomycin. She had a positive clinical response to this treatment.
Wound culture results confirmed CA-MRSA abscess and cellulitis, susceptible to trimethoprim/sulfamethoxazole. She was discharged on the oral formulation of this antibiotic to complete a 10-day course of treatment, including the days she received intravenous antibiotics.
Few well-designed trials have compared different lengths of cellulitis therapy. Most authorities recommend five to 10 days of treatment; however, longer courses might be required for more severe or complicated diseases.
Bottom line
Because of the high prevalence of CA-MRSA, initial antibiotic therapy for the treatment of community-acquired cellulitis must provide coverage for this organism.
Dr. Clarke is a hospitalist and assistant professor of medicine at Emory University School of Medicine, Atlanta. Dr. Dressler is a professor of medicine, hospital medicine associate division director for education, and associate program director for the J. Willis Hurst Internal Medicine Residency Program. Dr. Purohit, formerly an instructor in clinical medicine at Emory, is a hospitalist at WakeMed Health and Hospitals in Raleigh, N.C.
References
- Barzilai A, Choen HA. Isolation of group A streptococci from children with perianal cellulitis and from their siblings. Pediatr Infect Dis J. 1998;17(4):358-360.
- Thorsteinsdottir B, Tleyjeh IM, Baddour LM. Abdominal wall cellulitis in the morbidly obese. Scand J Infect Dis. 2005;37(8):605-608.
- Swartz MN. Clinical practice. Cellulitis. N Engl J Med. 2004;350(9):904-912.
- Edelsberg J, Berger A, Weber DJ, et al. Clinical and economic consequences of failure of initial antibiotic therapy for hospitalized patients with complicated skin and skin-structure infections. Infect Control Hosp Epidemiol. 2008;29(2):160-169.
- McNamara DR, Tleyjeh IM, Berbari EF, et al. Incidence of lower extremity cellulitis: a population-based study in Olmsted County, Minnesota. Mayo Clin Proc. 2007;82(7):817-821.
- Moellering RC. Current treatment options for community-acquired methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46(7):1032-1037.
- Chambers HF. The changing epidemiology of Staphylococcus aureus. Emerg Infect Dis. 2001;7(2):178-182.
- Moellering RC. A 39-year-old man with a skin infection. JAMA. 2008;299(1):79-87.
- Ruhe J, Smith N, Bradsher RW, Menon A. Community-onset methicillin-resistant Staphylococcus aureus skin and soft tissue infections: impact of antimicrobial therapy on outcome. Clin Infect Dis. 2007;44(6):777-784.
- David MZ, Glikman D, Crawford SE, et al. What is community-associated methicillin-resistant Staphylococcus aureus? J Infect Dis. 2008;197(9):1235-1243.
- Iyer S, Jones DH. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: a retrospective analysis of clinical presentation and treatment of a local outbreak. J Am Acad Dermatol. 2004;50(6):854-858.
- Centers for Disease Control and Prevention. Methicillin-resistant Staphylococcus aureus skin or soft tissue infections in a state prison—Mississippi, 2000. MMWR Morb Mortal Wkly Rep. 2001;50(42):919-922.
- Daum RS. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med. 2007;357(4):380-390.
- Dominguez TJ. It’s not a spider bite, it’s community-acquired methicillin-resistant Staphylococcus aureus. J Am Board Fam Pract. 2004;17(3):220-226.
- Moran GJ, Krishnadasan A, Gorwitz RJ, et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355(7):666-674.
- Jetton L. Therapy for methicillin-resistant Staphylococcus aureus. N Engl J Med. 2006;355(20):2153-2155.
- Hook EW, Hooton TM, Horton CA, et al. Microbiologic evaluation of cutaneous cellulitis in adults. Arch Intern Med. 1986;146(2):295-297. Duvanel T, Auckenthaler R, Rohner P, Harms M,
- Saurat JH. Quantitative cultures of biopsy specimens from cutaneous cellulitis. Arch Intern Med. 1989;149(2):293-296.
- Newell PM, Norden CW. Value of needle aspiration in bacteriologic diagnosis of cellulitis in adults. J Clin Microbiol. 1988; 26(3):401-404.
- Loffler CA, Macdougall C. Update on prevalence and treatment of methicillin-resistant Staphylococcus aureus infections. Expert Rev Anti Infect Ther. 2007;5(6):961-981.
- Davis SL, McKinnon PS, Hall LM, et al. Daptomycin versus vancomycin for complicated skin and skin structure infections: clinical and economic outcomes. Pharmacotherapy. 2007;27(12):1611-1618.
- Seaton RA. Daptomycin: rationale and role in the management of skin and soft tissue infections. J Antimicrob Chemother. 2008;62(Suppl 3):iii15-23.
ACCF/AHA 2013 Guidelines for Managing Heart Failure
Background
Heart failure (HF) is the No. 1 cause of both hospitalization and readmission among Americans 65 years and older.1 Hospitalizations in the HF population are associated with poor patient outcomes (30% mortality in the following year) and high costs, accounting for approximately 70% of the $32 billion spent on HF care annually in the United States.1 In 2009, the Centers for Medicare & Medicaid Services (CMS) introduced the 30-day, risk-standardized, all-cause readmission for HF as an indicator of quality and efficiency of care, which has since been incorporated into Medicare’s value-based purchasing program.2
HF is a complex syndrome that is associated with multiple comorbidities. Appropriate to these issues, management is multifaceted and involves care across the spectrum of disease:
- Diagnosing and treating underlying causes;
- Minimizing exacerbants;
- Optimizing management of comorbidities;
- Addressing psychosocial and environmental issues beyond the hospital; and
- Confronting end-of-life care.
In order to address this continuum of disease management, the American College of Cardiology Foundation (ACCF) and the American Heart Association (AHA) simultaneously released a new Guideline for the Management of HF in June in the Journal of the American College of Cardiology and Circulation.3,4 This update was developed in collaboration with the American Academy of Family Physicians, American College of Chest Physicians, Heart Rhythm Society, and the International Society for Heart and Lung Transplantation. The goals of the document are to improve quality of care, optimize patient outcomes, and advance the efficient use of healthcare resources. The guideline includes important recommendations for overall care, but particularly for hospital-based care and transitions of care that are largely the purview of hospitalists.
Guideline Update
The 2013 guideline is the third revision of the original guideline that was released in 2000. Despite being a complete rewrite of the 2009 HF guideline, the updated document contains relatively few changes to the recommendations that are Class I (should be performed) and III (no benefit or harm). The most significant randomized controlled trials in HF patients that have been published since the 2009 guideline include EMPHASIS-HF5 and MADIT-CRT/RAFT, which expand indications for aldosterone antagonists (AA) and cardiac resyncronization therapy (CRT), respectively, to patients with mild symptoms.5,6,7 Additionally, the WARCEF trial was published, which failed to demonstrate a significant difference in death, ischemic stroke, or intracerebral hemorrhage between treatment with warfarin or aspirin in patients with HF and reduced left ventricular ejection fraction (LVEF) in sinus rhythm.8
The most notable updates from the 2009 guideline include (* = Class I and III indications):
- The definition of HF has been revised to include: 1) HF with reduced ejection fraction (HFrEF; LVEF ≤40%), 2) HF failure with preserved ejection fraction (HFpEF; LVEF ≥50%), 3) HFpEF, borderline (LVEF 41-49%), and 4) HFpEF, improved (LVEF >40%).
- In the hospitalized patient, measurement of brain natriuretic peptide (BNP) or NT-proBNP is useful to support clinical judgment for the diagnosis of acutely decompensated HF, especially in the setting of uncertainty for the diagnosis.*
- AA should be used in patients with New York Heart Association (NYHA) functional Class II-IV and LVEF ≤35% unless contraindicated (creatinine ≤2.5 mg/dL in men, ≤2.0 mg/dL in women, and potassium <5.0 mEq/L).*
- CRT is indicated for patients who have LVEF of 35% or less, sinus rhythm, left bundle-branch block with a QRS duration of ³150 ms, and NYHA functional Class II-IV on guideline-directed medical therapy (GDMT).*
- Anticoagulation should not be used in patients with chronic HFrEF without atrial fibrillation, prior thromboembolic event, or cardioembolic source.*
- Transitions of care and GDMT can be improved by employing the following: 1) use of performance-improvement systems to identify HF patients, 2) development of multidisciplinary HF disease-management programs for patients at high risk of readmission, and 3) placing phone calls to the patient within three days of discharge and scheduling a follow-up visit within seven to 14 days.
Analysis
Overall, the new guideline provides a thorough reassessment and expert analysis on the diagnosis and management of HF for both inpatient and outpatient care. The authors introduce the phrase “guideline-directed medical therapy” (GDMT) to emphasize the smaller set of recommendations that constitute optimal medical therapy for HF patients. This designation, encompassing primarily Class I recommendations, helps providers rapidly determine the optimal treatment course for an individual patient. The mainstay of GDMT in HFrEF patients remains angiotensin-converting enzyme inhibitors (ACE-I), angiotensin receptor blockers (ARB) when ACE-I-intolerant, beta-blockers, and, in select patients, AA, hydralazine-nitrates, and diuretics.
A major shift in focus is seen in the new guideline with a greater emphasis on improved patient-centered outcomes across the spectrum of the disease. HF requires a continuum of care, from screening and genetic testing of family members of patients with idiopathic cardiomyopathy to conversations about palliative care and hospice. To this end, the authors highlight quality of life, shared decision-making, care coordination, transitions of care, and appropriateness of palliative care in a chronic disease state.
Further, the guideline expands upon previous recommendations for compliance with performance and quality metrics. Quality of care and adherence to performance measures of HF patients are becoming increasingly recognized, particularly in the hospital setting. The guideline offers recommendations for transitions of care in the hospitalized patient, which utilize systems of care coordination to ensure an evidence-based plan of care that includes the achievement of GDMT goals, effective management of comorbid conditions, timely follow-up, and appropriate dietary and physical activities.
HM Takeaways
HF is one of the most common, most challenging diseases managed by hospitalists. The 2013 ACCF/AHA Guideline for the Management of HF, while providing a comprehensive summary of evidence with recommendations for the totality of care for these patients throughout the course of the disease, places heavy emphasis on management during hospitalization and transitions. This includes repositioning of performance measures involving GDMT to better ensure optimal use of proven therapies in HFrEF, evidence-based steps to reduce readmissions, and greater recognition of the role of palliative care for patients with advanced disease.
Drs. McIlvennan and Allen are cardiologists in the Department of Medicine at the University of Colorado School of Medicine in Denver. Dr. Allen also works in the Colorado Health Outcomes Program.
References available at the-hospitalist.org.
Background
Heart failure (HF) is the No. 1 cause of both hospitalization and readmission among Americans 65 years and older.1 Hospitalizations in the HF population are associated with poor patient outcomes (30% mortality in the following year) and high costs, accounting for approximately 70% of the $32 billion spent on HF care annually in the United States.1 In 2009, the Centers for Medicare & Medicaid Services (CMS) introduced the 30-day, risk-standardized, all-cause readmission for HF as an indicator of quality and efficiency of care, which has since been incorporated into Medicare’s value-based purchasing program.2
HF is a complex syndrome that is associated with multiple comorbidities. Appropriate to these issues, management is multifaceted and involves care across the spectrum of disease:
- Diagnosing and treating underlying causes;
- Minimizing exacerbants;
- Optimizing management of comorbidities;
- Addressing psychosocial and environmental issues beyond the hospital; and
- Confronting end-of-life care.
In order to address this continuum of disease management, the American College of Cardiology Foundation (ACCF) and the American Heart Association (AHA) simultaneously released a new Guideline for the Management of HF in June in the Journal of the American College of Cardiology and Circulation.3,4 This update was developed in collaboration with the American Academy of Family Physicians, American College of Chest Physicians, Heart Rhythm Society, and the International Society for Heart and Lung Transplantation. The goals of the document are to improve quality of care, optimize patient outcomes, and advance the efficient use of healthcare resources. The guideline includes important recommendations for overall care, but particularly for hospital-based care and transitions of care that are largely the purview of hospitalists.
Guideline Update
The 2013 guideline is the third revision of the original guideline that was released in 2000. Despite being a complete rewrite of the 2009 HF guideline, the updated document contains relatively few changes to the recommendations that are Class I (should be performed) and III (no benefit or harm). The most significant randomized controlled trials in HF patients that have been published since the 2009 guideline include EMPHASIS-HF5 and MADIT-CRT/RAFT, which expand indications for aldosterone antagonists (AA) and cardiac resyncronization therapy (CRT), respectively, to patients with mild symptoms.5,6,7 Additionally, the WARCEF trial was published, which failed to demonstrate a significant difference in death, ischemic stroke, or intracerebral hemorrhage between treatment with warfarin or aspirin in patients with HF and reduced left ventricular ejection fraction (LVEF) in sinus rhythm.8
The most notable updates from the 2009 guideline include (* = Class I and III indications):
- The definition of HF has been revised to include: 1) HF with reduced ejection fraction (HFrEF; LVEF ≤40%), 2) HF failure with preserved ejection fraction (HFpEF; LVEF ≥50%), 3) HFpEF, borderline (LVEF 41-49%), and 4) HFpEF, improved (LVEF >40%).
- In the hospitalized patient, measurement of brain natriuretic peptide (BNP) or NT-proBNP is useful to support clinical judgment for the diagnosis of acutely decompensated HF, especially in the setting of uncertainty for the diagnosis.*
- AA should be used in patients with New York Heart Association (NYHA) functional Class II-IV and LVEF ≤35% unless contraindicated (creatinine ≤2.5 mg/dL in men, ≤2.0 mg/dL in women, and potassium <5.0 mEq/L).*
- CRT is indicated for patients who have LVEF of 35% or less, sinus rhythm, left bundle-branch block with a QRS duration of ³150 ms, and NYHA functional Class II-IV on guideline-directed medical therapy (GDMT).*
- Anticoagulation should not be used in patients with chronic HFrEF without atrial fibrillation, prior thromboembolic event, or cardioembolic source.*
- Transitions of care and GDMT can be improved by employing the following: 1) use of performance-improvement systems to identify HF patients, 2) development of multidisciplinary HF disease-management programs for patients at high risk of readmission, and 3) placing phone calls to the patient within three days of discharge and scheduling a follow-up visit within seven to 14 days.
Analysis
Overall, the new guideline provides a thorough reassessment and expert analysis on the diagnosis and management of HF for both inpatient and outpatient care. The authors introduce the phrase “guideline-directed medical therapy” (GDMT) to emphasize the smaller set of recommendations that constitute optimal medical therapy for HF patients. This designation, encompassing primarily Class I recommendations, helps providers rapidly determine the optimal treatment course for an individual patient. The mainstay of GDMT in HFrEF patients remains angiotensin-converting enzyme inhibitors (ACE-I), angiotensin receptor blockers (ARB) when ACE-I-intolerant, beta-blockers, and, in select patients, AA, hydralazine-nitrates, and diuretics.
A major shift in focus is seen in the new guideline with a greater emphasis on improved patient-centered outcomes across the spectrum of the disease. HF requires a continuum of care, from screening and genetic testing of family members of patients with idiopathic cardiomyopathy to conversations about palliative care and hospice. To this end, the authors highlight quality of life, shared decision-making, care coordination, transitions of care, and appropriateness of palliative care in a chronic disease state.
Further, the guideline expands upon previous recommendations for compliance with performance and quality metrics. Quality of care and adherence to performance measures of HF patients are becoming increasingly recognized, particularly in the hospital setting. The guideline offers recommendations for transitions of care in the hospitalized patient, which utilize systems of care coordination to ensure an evidence-based plan of care that includes the achievement of GDMT goals, effective management of comorbid conditions, timely follow-up, and appropriate dietary and physical activities.
HM Takeaways
HF is one of the most common, most challenging diseases managed by hospitalists. The 2013 ACCF/AHA Guideline for the Management of HF, while providing a comprehensive summary of evidence with recommendations for the totality of care for these patients throughout the course of the disease, places heavy emphasis on management during hospitalization and transitions. This includes repositioning of performance measures involving GDMT to better ensure optimal use of proven therapies in HFrEF, evidence-based steps to reduce readmissions, and greater recognition of the role of palliative care for patients with advanced disease.
Drs. McIlvennan and Allen are cardiologists in the Department of Medicine at the University of Colorado School of Medicine in Denver. Dr. Allen also works in the Colorado Health Outcomes Program.
References available at the-hospitalist.org.
Background
Heart failure (HF) is the No. 1 cause of both hospitalization and readmission among Americans 65 years and older.1 Hospitalizations in the HF population are associated with poor patient outcomes (30% mortality in the following year) and high costs, accounting for approximately 70% of the $32 billion spent on HF care annually in the United States.1 In 2009, the Centers for Medicare & Medicaid Services (CMS) introduced the 30-day, risk-standardized, all-cause readmission for HF as an indicator of quality and efficiency of care, which has since been incorporated into Medicare’s value-based purchasing program.2
HF is a complex syndrome that is associated with multiple comorbidities. Appropriate to these issues, management is multifaceted and involves care across the spectrum of disease:
- Diagnosing and treating underlying causes;
- Minimizing exacerbants;
- Optimizing management of comorbidities;
- Addressing psychosocial and environmental issues beyond the hospital; and
- Confronting end-of-life care.
In order to address this continuum of disease management, the American College of Cardiology Foundation (ACCF) and the American Heart Association (AHA) simultaneously released a new Guideline for the Management of HF in June in the Journal of the American College of Cardiology and Circulation.3,4 This update was developed in collaboration with the American Academy of Family Physicians, American College of Chest Physicians, Heart Rhythm Society, and the International Society for Heart and Lung Transplantation. The goals of the document are to improve quality of care, optimize patient outcomes, and advance the efficient use of healthcare resources. The guideline includes important recommendations for overall care, but particularly for hospital-based care and transitions of care that are largely the purview of hospitalists.
Guideline Update
The 2013 guideline is the third revision of the original guideline that was released in 2000. Despite being a complete rewrite of the 2009 HF guideline, the updated document contains relatively few changes to the recommendations that are Class I (should be performed) and III (no benefit or harm). The most significant randomized controlled trials in HF patients that have been published since the 2009 guideline include EMPHASIS-HF5 and MADIT-CRT/RAFT, which expand indications for aldosterone antagonists (AA) and cardiac resyncronization therapy (CRT), respectively, to patients with mild symptoms.5,6,7 Additionally, the WARCEF trial was published, which failed to demonstrate a significant difference in death, ischemic stroke, or intracerebral hemorrhage between treatment with warfarin or aspirin in patients with HF and reduced left ventricular ejection fraction (LVEF) in sinus rhythm.8
The most notable updates from the 2009 guideline include (* = Class I and III indications):
- The definition of HF has been revised to include: 1) HF with reduced ejection fraction (HFrEF; LVEF ≤40%), 2) HF failure with preserved ejection fraction (HFpEF; LVEF ≥50%), 3) HFpEF, borderline (LVEF 41-49%), and 4) HFpEF, improved (LVEF >40%).
- In the hospitalized patient, measurement of brain natriuretic peptide (BNP) or NT-proBNP is useful to support clinical judgment for the diagnosis of acutely decompensated HF, especially in the setting of uncertainty for the diagnosis.*
- AA should be used in patients with New York Heart Association (NYHA) functional Class II-IV and LVEF ≤35% unless contraindicated (creatinine ≤2.5 mg/dL in men, ≤2.0 mg/dL in women, and potassium <5.0 mEq/L).*
- CRT is indicated for patients who have LVEF of 35% or less, sinus rhythm, left bundle-branch block with a QRS duration of ³150 ms, and NYHA functional Class II-IV on guideline-directed medical therapy (GDMT).*
- Anticoagulation should not be used in patients with chronic HFrEF without atrial fibrillation, prior thromboembolic event, or cardioembolic source.*
- Transitions of care and GDMT can be improved by employing the following: 1) use of performance-improvement systems to identify HF patients, 2) development of multidisciplinary HF disease-management programs for patients at high risk of readmission, and 3) placing phone calls to the patient within three days of discharge and scheduling a follow-up visit within seven to 14 days.
Analysis
Overall, the new guideline provides a thorough reassessment and expert analysis on the diagnosis and management of HF for both inpatient and outpatient care. The authors introduce the phrase “guideline-directed medical therapy” (GDMT) to emphasize the smaller set of recommendations that constitute optimal medical therapy for HF patients. This designation, encompassing primarily Class I recommendations, helps providers rapidly determine the optimal treatment course for an individual patient. The mainstay of GDMT in HFrEF patients remains angiotensin-converting enzyme inhibitors (ACE-I), angiotensin receptor blockers (ARB) when ACE-I-intolerant, beta-blockers, and, in select patients, AA, hydralazine-nitrates, and diuretics.
A major shift in focus is seen in the new guideline with a greater emphasis on improved patient-centered outcomes across the spectrum of the disease. HF requires a continuum of care, from screening and genetic testing of family members of patients with idiopathic cardiomyopathy to conversations about palliative care and hospice. To this end, the authors highlight quality of life, shared decision-making, care coordination, transitions of care, and appropriateness of palliative care in a chronic disease state.
Further, the guideline expands upon previous recommendations for compliance with performance and quality metrics. Quality of care and adherence to performance measures of HF patients are becoming increasingly recognized, particularly in the hospital setting. The guideline offers recommendations for transitions of care in the hospitalized patient, which utilize systems of care coordination to ensure an evidence-based plan of care that includes the achievement of GDMT goals, effective management of comorbid conditions, timely follow-up, and appropriate dietary and physical activities.
HM Takeaways
HF is one of the most common, most challenging diseases managed by hospitalists. The 2013 ACCF/AHA Guideline for the Management of HF, while providing a comprehensive summary of evidence with recommendations for the totality of care for these patients throughout the course of the disease, places heavy emphasis on management during hospitalization and transitions. This includes repositioning of performance measures involving GDMT to better ensure optimal use of proven therapies in HFrEF, evidence-based steps to reduce readmissions, and greater recognition of the role of palliative care for patients with advanced disease.
Drs. McIlvennan and Allen are cardiologists in the Department of Medicine at the University of Colorado School of Medicine in Denver. Dr. Allen also works in the Colorado Health Outcomes Program.
References available at the-hospitalist.org.
Hospital-Based Palliative Care Reduces Length of Stay, Charges, Invasive Procedures, ICU Deaths

Clinical question: What are the characteristics of children who died in children’s hospitals while receiving palliative care (PC) compared to those who did not?
Background: Approximately 44,000 children die annually in hospitals in the U.S. Since the American Academy of Pediatrics (AAP) released a statement in August 2000 that presented an integrated model for providing PC to children with life-threatening conditions, pediatric PC programs have increased steadily in number. Children who receive PC services are commonly afflicted by genetic/congenital disorders, neuromuscular disorders, and cancer diagnoses. Although it is estimated that 6,320 people under the age of 24 received PC services in 2010, little data exist comparing pediatric inpatients receiving PC and those who do not.
Study design: Multicenter retrospective cohort study.
Setting: More than 40 freestanding children’s hospitals.
Synopsis: Using the Pediatric Health Information System (PHIS) database, which collects administrative and clinical data from more than 40 freestanding children’s hospitals belonging to the Children’s Hospital Association, researchers analyzed the characteristics of children under the age of 18 who died in the hospital more than five days after admission from 2001 to 2011. They extracted demographic data and categorized patients using major diagnostic categories (MDC) based on major organ system or etiology of disease. Identification of patients receiving PC services was by ICD-9 codes, and utilization of medications and procedures was identified by clinical transaction codes (CTC) and ICD-9 codes. The unit billing the last hospital day determined location of death.
Of the 24,342 children studied, only 3.8% received PC services based on coding. Patients less likely to receive PC services included black children (2.3%), those with circulatory diseases (2.8%), and those with neonatal diseases (1.9%). Children who did receive PC services had a significantly lower median length of stay (17 vs. 21 days), average daily charges ($9,348 vs. $11,806), received significantly fewer interventions (mechanical ventilation, invasive monitoring, surgical procedures), and died less frequently in an ICU setting (60% vs. 88%). PC services disproportionately altered the care of children with lymphatic/hematopoietic diseases, significantly decreasing use of mechanical ventilation (75% to 22%) and death in an ICU setting (66% to 21%).
Bottom line: Provision of PC services to children dying in children’s hospitals remains low. It is even lower for children with certain racial backgrounds and disease processes. When provided, PC services reduce length of stay, average daily charges, invasive procedures, and death in an ICU setting.
Citation: Keele L, Keenan HT, Sheetz J. Differences in characteristics of dying children who receive and do not receive palliative care. Pediatrics. 2013;132(1):72-78.
Reviewed by Pediatric Editor Weijen Chang, MD, SFHM, FAAP, associate clinical professor of medicine and pediatrics at the University of California at San Diego School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital.
Clinical question: What are the characteristics of children who died in children’s hospitals while receiving palliative care (PC) compared to those who did not?
Background: Approximately 44,000 children die annually in hospitals in the U.S. Since the American Academy of Pediatrics (AAP) released a statement in August 2000 that presented an integrated model for providing PC to children with life-threatening conditions, pediatric PC programs have increased steadily in number. Children who receive PC services are commonly afflicted by genetic/congenital disorders, neuromuscular disorders, and cancer diagnoses. Although it is estimated that 6,320 people under the age of 24 received PC services in 2010, little data exist comparing pediatric inpatients receiving PC and those who do not.
Study design: Multicenter retrospective cohort study.
Setting: More than 40 freestanding children’s hospitals.
Synopsis: Using the Pediatric Health Information System (PHIS) database, which collects administrative and clinical data from more than 40 freestanding children’s hospitals belonging to the Children’s Hospital Association, researchers analyzed the characteristics of children under the age of 18 who died in the hospital more than five days after admission from 2001 to 2011. They extracted demographic data and categorized patients using major diagnostic categories (MDC) based on major organ system or etiology of disease. Identification of patients receiving PC services was by ICD-9 codes, and utilization of medications and procedures was identified by clinical transaction codes (CTC) and ICD-9 codes. The unit billing the last hospital day determined location of death.
Of the 24,342 children studied, only 3.8% received PC services based on coding. Patients less likely to receive PC services included black children (2.3%), those with circulatory diseases (2.8%), and those with neonatal diseases (1.9%). Children who did receive PC services had a significantly lower median length of stay (17 vs. 21 days), average daily charges ($9,348 vs. $11,806), received significantly fewer interventions (mechanical ventilation, invasive monitoring, surgical procedures), and died less frequently in an ICU setting (60% vs. 88%). PC services disproportionately altered the care of children with lymphatic/hematopoietic diseases, significantly decreasing use of mechanical ventilation (75% to 22%) and death in an ICU setting (66% to 21%).
Bottom line: Provision of PC services to children dying in children’s hospitals remains low. It is even lower for children with certain racial backgrounds and disease processes. When provided, PC services reduce length of stay, average daily charges, invasive procedures, and death in an ICU setting.
Citation: Keele L, Keenan HT, Sheetz J. Differences in characteristics of dying children who receive and do not receive palliative care. Pediatrics. 2013;132(1):72-78.
Reviewed by Pediatric Editor Weijen Chang, MD, SFHM, FAAP, associate clinical professor of medicine and pediatrics at the University of California at San Diego School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital.
Clinical question: What are the characteristics of children who died in children’s hospitals while receiving palliative care (PC) compared to those who did not?
Background: Approximately 44,000 children die annually in hospitals in the U.S. Since the American Academy of Pediatrics (AAP) released a statement in August 2000 that presented an integrated model for providing PC to children with life-threatening conditions, pediatric PC programs have increased steadily in number. Children who receive PC services are commonly afflicted by genetic/congenital disorders, neuromuscular disorders, and cancer diagnoses. Although it is estimated that 6,320 people under the age of 24 received PC services in 2010, little data exist comparing pediatric inpatients receiving PC and those who do not.
Study design: Multicenter retrospective cohort study.
Setting: More than 40 freestanding children’s hospitals.
Synopsis: Using the Pediatric Health Information System (PHIS) database, which collects administrative and clinical data from more than 40 freestanding children’s hospitals belonging to the Children’s Hospital Association, researchers analyzed the characteristics of children under the age of 18 who died in the hospital more than five days after admission from 2001 to 2011. They extracted demographic data and categorized patients using major diagnostic categories (MDC) based on major organ system or etiology of disease. Identification of patients receiving PC services was by ICD-9 codes, and utilization of medications and procedures was identified by clinical transaction codes (CTC) and ICD-9 codes. The unit billing the last hospital day determined location of death.
Of the 24,342 children studied, only 3.8% received PC services based on coding. Patients less likely to receive PC services included black children (2.3%), those with circulatory diseases (2.8%), and those with neonatal diseases (1.9%). Children who did receive PC services had a significantly lower median length of stay (17 vs. 21 days), average daily charges ($9,348 vs. $11,806), received significantly fewer interventions (mechanical ventilation, invasive monitoring, surgical procedures), and died less frequently in an ICU setting (60% vs. 88%). PC services disproportionately altered the care of children with lymphatic/hematopoietic diseases, significantly decreasing use of mechanical ventilation (75% to 22%) and death in an ICU setting (66% to 21%).
Bottom line: Provision of PC services to children dying in children’s hospitals remains low. It is even lower for children with certain racial backgrounds and disease processes. When provided, PC services reduce length of stay, average daily charges, invasive procedures, and death in an ICU setting.
Citation: Keele L, Keenan HT, Sheetz J. Differences in characteristics of dying children who receive and do not receive palliative care. Pediatrics. 2013;132(1):72-78.
Reviewed by Pediatric Editor Weijen Chang, MD, SFHM, FAAP, associate clinical professor of medicine and pediatrics at the University of California at San Diego School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital.
Reviews of Reseach on Perioperative Morbidity, Capnography with Diabetic Ketoacidosis in the ED, Mortality Rate for Elective Surgeries
In This Edition
Literature At A Glance
A guide to this month’s studies
- Early treatment with intravenous tPA for acute stroke
- Perioperative morbidity, mortality for current smokers
- Statins associated with musculoskeletal conditions
- Antithrombotic medications in patients with history of stroke
- Extended prophylaxis with aspirin for patients after total hip arthroplasty
- Prognosis for symptomatic subsegmental pulmonary embolism
- Video-based educational workshops for academic hospitalists
- Increased mortality for elective surgeries on Fridays, weekends
- Basal plus correction insulin regimen and Type 2 diabetes
- Capnography to diagnose diabetic ketoacidosis in the ED
- How publicly reported mortality rates correlate with hospitals’ overall mortality
- Cost savings and preventable acute-care visits for Medicare patients
Early tPA in Acute Stroke Is Associated with Better Short-Term Outcomes in Routine Clinical Practice
Clinical question: Does early treatment with intravenous (IV) tissue plasminogen activator (tPA) result in better outcomes among patients with acute ischemic stroke in routine clinical practice?
Background: IV tPA for acute ischemic stroke is beneficial if given in the first 4.5 hours after symptom onset. However, pooled data from clinical trials have been limited in characterizing the extent to which onset-to-treatment (OTT) with IV tPA influences outcomes and how effective tPA is in routine clinical practice.
Study design: Data analysis from a stroke registry.
Setting: One thousand three hundred ninety-five U.S. hospitals participating in the Get with the Guidelines—Stroke Program.
Synopsis: Data were analyzed from 58,353 tPA-treated patients within 4.5 hours of symptom onset. Clinical outcomes were compared among patients treated in the 0-90-, 91-180-, and 181-270-minute OTT windows. Patient factors strongly associated with shorter OTT were greater stroke severity (odds ratio [OR] 2.8; 95% confidence interval [CI], 2.5-3.1 per five-point increase), arrival by ambulance (OR 5.9; 95% CI, 4.5-7.3), and arrival during regular hours (OR 4.6; 95% CI, 3.8-5.4). Faster OTT, in 15-minute increments, was associated with reduced in-hospital mortality (OR 0.96; 95% CI, 0.95-0.98; P<.001), reduced symptomatic intracranial hemorrhage (OR 0.96; 95% CI, 0.95-0.98; P<.001), increased achievement of independent ambulation at discharge (OR 1.04; 95% CI, 1.03-1.05; P<.001), and increased discharge to home (OR 1.03; 95% CI, 1.02-1.04; P<.001).
Data collected were dependent on the accuracy and completeness of the chart abstraction, and only short-term outcomes were reported. Although no post-discharge outcomes were reported, previous studies have shown that functional status at discharge strongly correlates with three-month disability outcomes.
Bottom line: In routine clinical practice, earlier tPA for acute ischemic strokes results in better short-term clinical outcomes.
Citation: Saver JL, Fonarow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA. 2013;309:2480-2488.
Current Smokers Have Higher Perioperative Morbidity and Mortality Compared to Past Smokers
Clinical question: Is there an association between current and past smoking on outcomes among patients having major surgery?
Background: Smoking is associated with adverse postoperative outcomes, but it is not known whether the associations are dose-dependent or limited to patients with smoking-related diseases. Smoking-related effects on postoperative events among patients having major surgery are also not well established.
Study design: Retrospective cohort study.
Setting: Four hundred forty-eight non-VA hospitals across the U.S., Canada, Lebanon, and the United Arab Emirates.
Synopsis: Data from 607,558 adult patients undergoing major surgery were obtained from the American College of Surgeons (ACS) National Surgical Quality Improvement Program (NSQIP) database. After adjusting for confounders (cardiopulmonary diseases and cancer), the effects of current and past smoking (quit >1 year prior) on 30-day post-operative outcomes were measured.
There were 125,192 (21%) current smokers and 78,763 (13%) past smokers. Increased odds of post-op mortality were noted in current smokers only (odds ratio [OR] 1.17; 95% CI, 1.10-1.24). The adjusted odds ratios were higher for arterial and respiratory events among current smokers compared with past smokers (OR 1.65; 95% CI, 1.51-1.81 vs. OR 1.20; CI, 1.09-1.31 for arterial events, respectively) and (OR, 1.45; CI, 1.40-1.51 vs. OR, 1.13; CI, 1.08-1.18, for respiratory events, respectively). No significant effects on venous events were observed.
There was an increased adjusted odds of mortality for current smokers with <10 pack-years, while the effects on arterial and respiratory events increased incrementally with increased pack-years. Smoking was associated with adverse post-op outcomes regardless of smoking-related diseases. Variability in hospital quality or surgical strategies may have confounded the results.
Bottom line: Among patients undergoing major surgery, current but not past smoking was associated with higher mortality; smoking cessation for at least a year prior to surgery may decrease post-operative adverse events.
Citation: Musallam KM, Rosendaal FR, Zaatari G, et al. Smoking and the risk of mortality and vascular and respiratory events in patients undergoing major surgery. JAMA Surg. 2013 Jun 19:1-8. doi: 10.1001/jamasurg.2013.2360 [Epub ahead of print].
Statins Associated with Several Musculoskeletal Conditions
Clinical question: Is statin use associated with musculoskeletal adverse events, including arthropathy and injury, in physically active individuals?
Background: Statin-induced musculoskeletal adverse events (AEs) include myalgias, muscle weakness, cramps, rhabdomyolysis, and tendinous disease. The full spectrum of AEs is unknown because randomized clinical trials have not been powered to detect uncommon AEs.
Study design: Retrospective cohort study with propensity score matching.
Setting: San Antonio military area.
Synopsis: A total of 46,249 patients aged 30 to 85 years who met study criteria were propensity-matched into 6,967 statin users and 6,967 nonusers. The occurrence of musculoskeletal conditions were categorized using ICD-9 codes: Msk1, all musculoskeletal diseases; Msk1a, arthropathies and related diseases; Msk1b, injury-related diseases; and Msk2, drug-associated musculoskeletal pain. Of these, statin users had a higher odds ratio (OR) for Msk1 (OR 1.19; 95% CI, 1.08-1.30), Msk1b (1.13; 1.05-1.21), and Msk2 (1.09; 1.02-1.18). Msk1b (arthropathies) had an OR of 1.07 (0.9-1.16, P=0.07). Simvastatin was used by 73.5% of patients, and years of simvastatin use was not a significant predictor of any of the outcome measures. Secondary and sensitivity analyses showed higher adjusted ORs for statin users in all groups. This study was limited by the use of ICD-9-CM codes for identification of baseline characteristics, and the musculoskeletal diagnosis groups used were not validated.
Bottom line: Statin use is associated with an increased likelihood of musculoskeletal conditions, arthropathies, injuries, and pain.
Citation: Mansi I, Frei CR, Pugh M, Makris U, Mortensen EM. Statins and musculoskeletal conditions, arthropathies, and injuries. JAMA Intern Med. 2013;173:1318-1326.
Evidence-Based Guidelines on Periprocedural Management of Antithrombotic Medications in Patients with History of Stroke
Clinical question: What is the evidence for the periprocedural management of antithrombotics in patients with ischemic cerebrovascular accidents (CVAs)?
Background: Evidence-based guidelines are needed to help clinicians determine the thromboembolic risk of temporary discontinuation of antithrombotic medications, the perioperative bleeding risks of continuing antithrombotic agents, whether bridging therapy should be used, and the appropriate timing of antithrombotic agent discontinuation.
Study design: Systematic literature review with practice recommendations.
Setting: American Academy of Neurology Guideline Development Subcommittee convened an expert panel to review and provide recommendations.
Synopsis: Researchers analyzed 133 literature reviews via MEDLINE and EMBASE. Aspirin in stroke patients:
- Should routinely be continued for dental procedures (Level A);
- Should probably be continued for invasive ocular anesthesia, cataract surgery, dermatologic procedures, transrectal ultrasound-guided prostate biopsy, spinal/epidural procedures, and carpal tunnel surgery (Level B); and
- Should possibly be continued for vitreoretinal surgery, electromyogram (EMG), transbronchial lung biopsy, colonoscopic polypectomy, upper endoscopy and biopsy/sphincterotomy, and abdominal ultrasound-guided biopsies (Level C).
Warfarin in stroke patients:
- Should routinely be continued for dental procedures (Level A); and
- Should possibly continued for dermatologic procedures (Level B) and EMG, prostate procedures, inguinal hemiorrhaphy, and endothermal ablation of great saphenous vein (Level C).
- There is a lack of evidence on warfarin for ophthalmologic procedures, with the exception of ocular anesthesia, where it probably does not increase clinically significant bleeding (Level B).
There was not enough evidence to support or refute a recommendation regarding heparin bridge therapy in reducing thromboembolism in chronically anticoagulated patients (Level B).
Bottom line: These are the most up-to-date guidelines for anticoagulant and antiplatelet agents in patients with transient ischemic attacks and strokes undergoing procedures, but further research is needed in many areas.
Citation: Armstrong MJ, Gronseth G Anderson DC, et al. Summary of evidence-based guideline: periprocedural management of antithrombotic medications in patients with ischemic cerebrovascular disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;80:2065-2069.
Extended Prophylaxis with Aspirin Was Noninferior to Extended Prophylaxis with Low-Molecular-Weight Heparin
Clinical question: Is aspirin as effective as low-molecular-weight heparin (LMWH) for the extended prophylaxis of venous thromboembolism (VTE) after total hip arthroplasty (THA)?
Background: Deep vein thrombosis (DVT) and pulmonary embolism (PE) are common complications after THA. After initial prophylaxis, LMWH given for up to 30 days has been shown to reduce VTE compared with placebo. However, LMWH is costly and may increase the risk of minor bleeding. Aspirin is a potentially simple, low-cost alternative.
Study design: Randomized, placebo-controlled trial.
Setting: Twelve university-affiliated orthopedic hospitals in Canada.
Synopsis: Patients undergoing elective THA without hip fracture, metastatic cancer, or bleeding precluding anticoagulants were eligible. All patients received dalteparin for 10 days and were then randomized to aspirin 81 mg daily or to continue dalteparin. The primary outcome was symptomatic proximal DVT or PE during 90 days’ follow-up. The study was terminated early due to slow enrollment. At that time, 2,364 patients had been enrolled, and an analysis by an independent data safety and monitoring board determined that continuing the study was unlikely to alter the main findings. Extended prophylaxis with aspirin was noninferior to LMWH for the primary outcome, which occurred in 0.3% vs. 1.3%, respectively (95% CI, -0.5% to 2.5%, P<.001 for noninferiority). There were no significant differences in major or minor bleeding.
Though the early termination is a concern, the sample size was large and the results do not suggest inadequate power as a reason for lack of superiority for LMWH. Also, all patients received 10 days of LMWH, which indicates a period of LMWH after discharge will still be needed for most patients prior to initiating aspirin.
Bottom line: After initial LMWH prophylaxis for 10 days, extended prophylaxis with aspirin can be considered, particularly for patients for whom LMWH may not be feasible.
Citation: Anderson DR, Dunbar MJ, Bohm ER, et al. Aspirin versus low-molecular-weight heparin for extended venous thromboembolism prophylaxis after total hip arthroplasty: a randomized trial. Ann Intern Med. 2013;158:800-806.
Symptomatic Subsegmental Pulmonary Embolism (PE) Has a Prognosis Similar to Proximal PE
Clinical question: Is the prognosis of a symptomatic subsegmental pulmonary embolism (PE) similar to that of a more proximal PE?
Background: The use of multidetector computed tomography angiography (CTA) has allowed for better assessment of the pulmonary vasculature and increased detection of distal emboli. Prior studies have raised questions on the clinical importance of subsegmental PE but have been limited by small size or retrospective design.
Study design: Combined data from two prospective trials of management of suspected PE.
Setting: Twelve hospitals in the Netherlands and four tertiary-care emergency departments in Canada.
Synopsis: The study cohort consisted of 3,769 patients with suspected PE, of which 2,688 underwent CTA. Of patients diagnosed with PE, 15.5% had isolated subsegmental emboli. All patients were treated with anticoagulation. During three months of follow-up, the incidence of symptomatic recurrence for subsegmental PE was similar to patients with proximal PE (3.6% vs. 2.5%, respectively). The mortality rates for patients with subsegmental and proximal PE were also similar (10.3% vs. 6.3%, respectively).
The study may have been underpowered to detect small differences in event rates; however, there was no trend suggesting that subsegmental PE had better outcomes than more proximal PE. Also, the study did not specifically investigate whether any management strategy is preferred based on thrombus location on CTA.
Bottom line: Clinicians should continue to anticoagulate patients with subsegmental PE as the prognosis is similar to those with proximal PE.
Citation: Den Exter PL, van Es J, Klok FA, et al. Risk profile and clinical outcome of symptomatic subsegmental pulmonary embolism. Blood. 2013;122:1144-1149.
Video-Based Educational Workshop for Academic Hospitalists and House Staff May Improve Professionalism
Clinical question: Can video-based education promote professionalism among academic hospitalists and house staff?
Background: Unprofessional behavior by academic hospitalists and residents can negatively impact the learning environment and patient safety. This behavior increases throughout training, and faculty behavior can be influential. There is a paucity of educational materials to train hospitalists and house staff to recognize and ameliorate unprofessional behaviors.
Study design: Educational survey study.
Setting: University of Chicago, Northwestern University, and NorthShore University Health System teaching hospitals.
Synopsis: Three videos were developed displaying three types of unprofessional behavior: disparaging other physicians, “blocking” admissions, and misrepresenting tests to expedite their completion. There were 44 hospitalists and 244 house staff who received a 60-minute workshop in which they watched the videos using a viewing tool and discussed the videos in small groups.
For all three videos, more than three-quarters of both hospitalists and house staff felt the behavior was unprofessional or somewhat unprofessional. Hospitalists and house staff found the workshop useful and effective (65.9% and 77.1%, respectively) and would change their behavior as a result of the workshop (65.9% and 67.2%, respectively). Those who perceived the videos as “very realistic” were more likely to report intent to change behavior (93% vs. 53%, P=0.01).
This study is limited by its small sample size and possible selection bias. Those interested or concerned about unprofessional behavior may have been more likely to attend the workshop.
Bottom line: Video-based professionalism education is a feasible and well-received way to educate hospitalists and residents about unprofessional behavior and may even affect their future behavior.
Citation: Farnan JM, O’Leary KJ, Didwania A, et al. Promoting professionalism via a video-based educational workshop for academic hospitalists and housestaff. J Hosp Med. 2013;8:386-389.
Friday and Weekend Elective Surgeries Have Increased Mortality
Clinical question: How can the association between mortality and the day of elective surgical procedures be assessed?
Background: Several studies have described the “weekend effect” for both surgical and medical patients, with higher mortality and length of stay in patients admitted on the weekend compared to weekdays. Two potential explanations are poorer quality of care being delivered on the weekend or more severely ill patients being operated on or admitted on the weekend.
Study design: Retrospective analysis of national hospital administrative data.
Setting: All acute-care and specialist hospitals in England from 2008 to 2011.
Synopsis: There were 4,133,346 elective, inpatient surgical procedures studied. Friday surgeries had an adjusted odds ratio of death within 30 days and within two days of 1.44 [95% CI, 1.39-1.50] and 1.42 [95% CI, 1.26-1.60], respectively, when compared with Monday. Weekend surgeries had an adjusted odds ratio of death within 30 days and within two days of 1.82 [95% CI, 1.71-1.94] and 2.67 [95% CI, 2.30-3.09], respectively, when compared with Monday. There were significant trends toward higher mortality at the end of the workweek and weekends for four high-risk procedures: esophagus and/or stomach excision, colon and/or rectum excisions, coronary artery bypass graft, and lung excision. For lower-risk procedures, there was a significant increase in mortality for Friday surgeries but not weekend surgeries. As with all studies using administrative data, inherent selection biases could not be adjusted for Friday or weekend procedures.
Bottom line: Elective surgeries that occur on the weekend and later in the week have an increased risk of mortality, implying that the weekend effect is due to poorer quality of care during weekends, rather than higher-acuity patients presenting on weekends.
Citation: Aylin P, Alexandrescu R, Jen MH, Mayer EK, Bottle A. Day of week of procedure and 30 day mortality for elective surgery: retrospective analysis of hospital episode statistics. BMJ. 2013;346:f2424.
Basal Plus Correction Insulin Regimen Is Effective in Hospitalized Patients with Type 2 Diabetes
Clinical question: Does a basal plus correction insulin regimen (as needed with meals) result in similar glycemic control and lower rates of hypoglycemia compared to a basal-bolus regimen?
Background: Basal bolus is the preferred insulin regimen for non-critically-ill hospitalized patients as per clinical guidelines. But use is limited due to the complexity of the regimen and the fear of inducing hypoglycemia. A less complex, easier-to-implement basal plus correction insulin regimen may be an effective alternative.
Study design: Multicenter, prospective, open-label, randomized study.
Setting: Six hospitals in the U.S.
Synopsis: A group of 375 medical and surgical patients with Type 2 diabetes treated with diet, oral anti-diabetic agents, or low-dose insulin (≤ 0.4 units/kg/day) were randomized to:
- Basal-bolus insulin regimen with glargine once daily and fixed doses of glusiline before meals;
- Basal plus correction insulin (“basal plus”) regimen with glargine once daily and glusiline sliding scale insulin (SSI) before meals; or
- Regular SSI alone.
After the first day of therapy, treatment with basal-bolus and basal-plus regimens resulted in similar improvements in daily blood glucose (BG) (P=0.16), and both were superior to SSI alone (P=0.04). Both regimens also resulted in less treatment failure (defined as mean daily BG of >240 mg/dl or >2 consecutive BG >240 mg/dl) than did treatment with SSI. Hypoglycemia (BG <70 mg/dl) occurred in 16%, 13%, and 3% of patients in the basal-bolus, basal-plus, and SSI groups, respectively (P=0.02). There were no between-group differences in the frequency of severe hypoglycemia (<40 mg/dl; P=0.76).
The study was not powered to evaluate hospital complications (infection, mortality, hospital stay, and readmissions) across groups.
Bottom line: The basal-plus regimen resulted in glycemic control similar to standard basal-bolus regimen and is an effective alternative for the initial management of hyperglycemia in general medical and surgical patients with Type 2 diabetes.
Citation: Umpierrez GE, Smiley D, Hermayer K, et al. Randomized study comparing a basal-bolus with a basal plus correction insulin regimen for the hospital management of medical and surgical patients with type 2 diabetes: Basal plus trial. Diabetes Care. 2013;36:2169-2174.
Capnography Can Help Diagnose Diabetic Ketoacidosis in the ED
Clinical question: Can capnography be used as a screening tool to identify patients with diabetic ketoacidosis (DKA)?
Background: Metabolic acidosis is a major criterion for diagnosing DKA. Previous studies have shown that end-tidal carbon dioxide (ETCO2) measurement by capnography can provide an accurate estimation of arterial carbon dioxide tension (PaCO2) and may be a noninvasive, fast, inexpensive measurement of acidosis in DKA. However, those studies were in pediatric patients and had small sample sizes.
Study design: Cross-sectional, prospective descriptive-analytic study.
Setting: The ED of Imam Reza Medical Research and Training Hospital, Tabriz, East Azarbaijan, Iran.
Synopsis: A total of 181 adult patients older than 18 with suspected DKA and blood sugar >250 mg/dl were included in the study. Simultaneous capnography and arterial blood gas (ABG) were obtained on all patients. Urine ketones, complete blood count, serum levels of potassium, urea, and creatinine were collected. Sixty-two patients were found to have DKA, while 119 had other conditions associated with metabolic acidosis. There was a significant linear relationship between pH and ETCO2 (P>0.0001, relative risk (R)=0.253), PaCO2 and ETCO2 (P>0.0001, R=0.572), and bicarbonate (HCO3) and ETCO2 (P>0.0001, R=0.730). ETCO2 values >24.5 mmHg had a sensitivity and specificity of 0.90 for ruling out DKA. No cutoff point could be determined for ruling in DKA.
The study was open to selection bias as patient collection was only done during the day, so eligible subjects may have been missed. Moreover, though the study suggests that capnography has a role in ruling out DKA, the exact cutoff value is unclear. Other studies found that higher values were needed to exclude diagnosis.
Bottom line: Using ETCO2 values >24.5 mmHg, capnography can help exclude the diagnosis of DKA in adult patients with elevated BG.
Citation: Soleimanour H, Taghizadieh A, Niafar M, Rahmani F, Golzari S, Esfanjani RM. Predictive value of capnography for diagnosis in patients with suspected diabetic ketoacidosis in the emergency department. West J Emerg Med. 2013. doi: 10.5811/westjem.2013.4.14296.
Publicly Reported Mortality Correlates with Overall Mortality
Clinical question: Are publicly reported mortality rates associated with a hospital’s overall medical and surgical mortality rate?
Background: Public reporting of mortality has become an important strategy in Medicare’s quality-improvement initiative. However, the mortality rate for only three conditions, acute myocardial infarction, congestive heart failure, and pneumonia are reported. It is unclear if these rates correlate to a hospital’s overall mortality rate.
Study design: Retrospective cohort.
Setting: National Medicare fee-for-service population.
Synopsis: Using 2008-2009 data from 2,322 acute-care hospitals with 6.7 million admissions, an aggregate mortality rate for the three publicly reported conditions, a standardized 30-day mortality rate for selected medical and surgical conditions, and an overall average composite mortality score was calculated for each hospital. Based on their mortality for the three publicly reported conditions, hospitals were grouped into quartiles from highest (top-performing hospitals) to lowest mortality (poor-performing hospitals).
Top-performing hospitals had a 3.6% (9.4%vs 13.0%; P<.001) lower mortality rate than poor-performing hospitals and an odds ratio >5 of being a top performer in overall mortality (OR 5.3; 95% CI, 4.3-6.5). They also had an 81% lower chance of being in the worst-performing quartile in overall mortality (OR 0.19; 95% CI, 0.14-0.27). Conversely, poor-performing hospitals had a 4.5 times higher risk of being in the lowest quartile in overall mortality. The study is limited by the use of administrative data, which limits the ability to adjust for severity of illness, overall health, and socioeconomic status of each hospital’s population.
Bottom line: A hospital’s mortality performance on the three publicly reported conditions may predict mortality rates across a wide range of medical and surgical conditions.
Citation: McCrum ML, Joynt KE, Orav EJ, Gawande AA, Jha AK. Mortality for publicly reported conditions and overall hospital mortality rates. JAMA Intern Med. 2013;173:1351-1357.
Cost Savings in Decreasing Preventable Acute-Care Visits Are Limited among High-Cost Medicare Utilizers
Clinical question: What role do preventable acute-care visits play in the overall costs of care for the highest Medicare utilizers?
Background: Some 10% of Medicare patients account for more than half the costs. Interventions targeted at decreasing acute-care costs (ED visits and inpatient hospitalizations) for this high-cost population are widespread, but it is unknown what impact they can have.
Study design: Retrospective cohort.
Setting: National Medicare fee-for-service population.
Synopsis: Standardized total costs were created for fee-for-service Medicare patients for 2009 and 2010 in order to identify high-cost and persistently high-cost patients. Algorithms were used to identify preventable ED visits and hospitalizations in both the high-cost and non-high-cost cohorts.
Of the more than 1 million patients in the sample Medicare population, as many as 113,341 were high-cost. As much as 73% of acute-care spending was attributable to this cohort. Overall, 10% of acute-care costs were felt to be preventable in the high-cost group, 13.5% in the persistently high-cost group, and 19% in the non-high-cost group for 2010. The most common reasons for preventable acute care in the high-cost cohort were heart failure, bacterial pneumonia, and chronic obstructive pulmonary disease. Catastrophic events (myocardial infarction, stroke, sepsis), cancer, and orthopedic procedures drove overall inpatient costs in the high-cost group.
Preventable costs were higher per capita in areas with higher numbers of primary-care and specialist physicians, but it’s unclear if this was a supply or demand issue. The study also used algorithms that possibly overestimate the amount of preventable acute care.
Bottom line: In the highest Medicare utilizers, cost savings aimed at preventable acute care may be limited and might be better targeted at efficiency during acute-care episodes.
Citation: Joynt KE, Gawande AA, Orav EJ, Jha AK. Contribution of preventable acute care spending to total spending for high-cost Medicare patients. JAMA. 2013;309:2572-2578.
In This Edition
Literature At A Glance
A guide to this month’s studies
- Early treatment with intravenous tPA for acute stroke
- Perioperative morbidity, mortality for current smokers
- Statins associated with musculoskeletal conditions
- Antithrombotic medications in patients with history of stroke
- Extended prophylaxis with aspirin for patients after total hip arthroplasty
- Prognosis for symptomatic subsegmental pulmonary embolism
- Video-based educational workshops for academic hospitalists
- Increased mortality for elective surgeries on Fridays, weekends
- Basal plus correction insulin regimen and Type 2 diabetes
- Capnography to diagnose diabetic ketoacidosis in the ED
- How publicly reported mortality rates correlate with hospitals’ overall mortality
- Cost savings and preventable acute-care visits for Medicare patients
Early tPA in Acute Stroke Is Associated with Better Short-Term Outcomes in Routine Clinical Practice
Clinical question: Does early treatment with intravenous (IV) tissue plasminogen activator (tPA) result in better outcomes among patients with acute ischemic stroke in routine clinical practice?
Background: IV tPA for acute ischemic stroke is beneficial if given in the first 4.5 hours after symptom onset. However, pooled data from clinical trials have been limited in characterizing the extent to which onset-to-treatment (OTT) with IV tPA influences outcomes and how effective tPA is in routine clinical practice.
Study design: Data analysis from a stroke registry.
Setting: One thousand three hundred ninety-five U.S. hospitals participating in the Get with the Guidelines—Stroke Program.
Synopsis: Data were analyzed from 58,353 tPA-treated patients within 4.5 hours of symptom onset. Clinical outcomes were compared among patients treated in the 0-90-, 91-180-, and 181-270-minute OTT windows. Patient factors strongly associated with shorter OTT were greater stroke severity (odds ratio [OR] 2.8; 95% confidence interval [CI], 2.5-3.1 per five-point increase), arrival by ambulance (OR 5.9; 95% CI, 4.5-7.3), and arrival during regular hours (OR 4.6; 95% CI, 3.8-5.4). Faster OTT, in 15-minute increments, was associated with reduced in-hospital mortality (OR 0.96; 95% CI, 0.95-0.98; P<.001), reduced symptomatic intracranial hemorrhage (OR 0.96; 95% CI, 0.95-0.98; P<.001), increased achievement of independent ambulation at discharge (OR 1.04; 95% CI, 1.03-1.05; P<.001), and increased discharge to home (OR 1.03; 95% CI, 1.02-1.04; P<.001).
Data collected were dependent on the accuracy and completeness of the chart abstraction, and only short-term outcomes were reported. Although no post-discharge outcomes were reported, previous studies have shown that functional status at discharge strongly correlates with three-month disability outcomes.
Bottom line: In routine clinical practice, earlier tPA for acute ischemic strokes results in better short-term clinical outcomes.
Citation: Saver JL, Fonarow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA. 2013;309:2480-2488.
Current Smokers Have Higher Perioperative Morbidity and Mortality Compared to Past Smokers
Clinical question: Is there an association between current and past smoking on outcomes among patients having major surgery?
Background: Smoking is associated with adverse postoperative outcomes, but it is not known whether the associations are dose-dependent or limited to patients with smoking-related diseases. Smoking-related effects on postoperative events among patients having major surgery are also not well established.
Study design: Retrospective cohort study.
Setting: Four hundred forty-eight non-VA hospitals across the U.S., Canada, Lebanon, and the United Arab Emirates.
Synopsis: Data from 607,558 adult patients undergoing major surgery were obtained from the American College of Surgeons (ACS) National Surgical Quality Improvement Program (NSQIP) database. After adjusting for confounders (cardiopulmonary diseases and cancer), the effects of current and past smoking (quit >1 year prior) on 30-day post-operative outcomes were measured.
There were 125,192 (21%) current smokers and 78,763 (13%) past smokers. Increased odds of post-op mortality were noted in current smokers only (odds ratio [OR] 1.17; 95% CI, 1.10-1.24). The adjusted odds ratios were higher for arterial and respiratory events among current smokers compared with past smokers (OR 1.65; 95% CI, 1.51-1.81 vs. OR 1.20; CI, 1.09-1.31 for arterial events, respectively) and (OR, 1.45; CI, 1.40-1.51 vs. OR, 1.13; CI, 1.08-1.18, for respiratory events, respectively). No significant effects on venous events were observed.
There was an increased adjusted odds of mortality for current smokers with <10 pack-years, while the effects on arterial and respiratory events increased incrementally with increased pack-years. Smoking was associated with adverse post-op outcomes regardless of smoking-related diseases. Variability in hospital quality or surgical strategies may have confounded the results.
Bottom line: Among patients undergoing major surgery, current but not past smoking was associated with higher mortality; smoking cessation for at least a year prior to surgery may decrease post-operative adverse events.
Citation: Musallam KM, Rosendaal FR, Zaatari G, et al. Smoking and the risk of mortality and vascular and respiratory events in patients undergoing major surgery. JAMA Surg. 2013 Jun 19:1-8. doi: 10.1001/jamasurg.2013.2360 [Epub ahead of print].
Statins Associated with Several Musculoskeletal Conditions
Clinical question: Is statin use associated with musculoskeletal adverse events, including arthropathy and injury, in physically active individuals?
Background: Statin-induced musculoskeletal adverse events (AEs) include myalgias, muscle weakness, cramps, rhabdomyolysis, and tendinous disease. The full spectrum of AEs is unknown because randomized clinical trials have not been powered to detect uncommon AEs.
Study design: Retrospective cohort study with propensity score matching.
Setting: San Antonio military area.
Synopsis: A total of 46,249 patients aged 30 to 85 years who met study criteria were propensity-matched into 6,967 statin users and 6,967 nonusers. The occurrence of musculoskeletal conditions were categorized using ICD-9 codes: Msk1, all musculoskeletal diseases; Msk1a, arthropathies and related diseases; Msk1b, injury-related diseases; and Msk2, drug-associated musculoskeletal pain. Of these, statin users had a higher odds ratio (OR) for Msk1 (OR 1.19; 95% CI, 1.08-1.30), Msk1b (1.13; 1.05-1.21), and Msk2 (1.09; 1.02-1.18). Msk1b (arthropathies) had an OR of 1.07 (0.9-1.16, P=0.07). Simvastatin was used by 73.5% of patients, and years of simvastatin use was not a significant predictor of any of the outcome measures. Secondary and sensitivity analyses showed higher adjusted ORs for statin users in all groups. This study was limited by the use of ICD-9-CM codes for identification of baseline characteristics, and the musculoskeletal diagnosis groups used were not validated.
Bottom line: Statin use is associated with an increased likelihood of musculoskeletal conditions, arthropathies, injuries, and pain.
Citation: Mansi I, Frei CR, Pugh M, Makris U, Mortensen EM. Statins and musculoskeletal conditions, arthropathies, and injuries. JAMA Intern Med. 2013;173:1318-1326.
Evidence-Based Guidelines on Periprocedural Management of Antithrombotic Medications in Patients with History of Stroke
Clinical question: What is the evidence for the periprocedural management of antithrombotics in patients with ischemic cerebrovascular accidents (CVAs)?
Background: Evidence-based guidelines are needed to help clinicians determine the thromboembolic risk of temporary discontinuation of antithrombotic medications, the perioperative bleeding risks of continuing antithrombotic agents, whether bridging therapy should be used, and the appropriate timing of antithrombotic agent discontinuation.
Study design: Systematic literature review with practice recommendations.
Setting: American Academy of Neurology Guideline Development Subcommittee convened an expert panel to review and provide recommendations.
Synopsis: Researchers analyzed 133 literature reviews via MEDLINE and EMBASE. Aspirin in stroke patients:
- Should routinely be continued for dental procedures (Level A);
- Should probably be continued for invasive ocular anesthesia, cataract surgery, dermatologic procedures, transrectal ultrasound-guided prostate biopsy, spinal/epidural procedures, and carpal tunnel surgery (Level B); and
- Should possibly be continued for vitreoretinal surgery, electromyogram (EMG), transbronchial lung biopsy, colonoscopic polypectomy, upper endoscopy and biopsy/sphincterotomy, and abdominal ultrasound-guided biopsies (Level C).
Warfarin in stroke patients:
- Should routinely be continued for dental procedures (Level A); and
- Should possibly continued for dermatologic procedures (Level B) and EMG, prostate procedures, inguinal hemiorrhaphy, and endothermal ablation of great saphenous vein (Level C).
- There is a lack of evidence on warfarin for ophthalmologic procedures, with the exception of ocular anesthesia, where it probably does not increase clinically significant bleeding (Level B).
There was not enough evidence to support or refute a recommendation regarding heparin bridge therapy in reducing thromboembolism in chronically anticoagulated patients (Level B).
Bottom line: These are the most up-to-date guidelines for anticoagulant and antiplatelet agents in patients with transient ischemic attacks and strokes undergoing procedures, but further research is needed in many areas.
Citation: Armstrong MJ, Gronseth G Anderson DC, et al. Summary of evidence-based guideline: periprocedural management of antithrombotic medications in patients with ischemic cerebrovascular disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;80:2065-2069.
Extended Prophylaxis with Aspirin Was Noninferior to Extended Prophylaxis with Low-Molecular-Weight Heparin
Clinical question: Is aspirin as effective as low-molecular-weight heparin (LMWH) for the extended prophylaxis of venous thromboembolism (VTE) after total hip arthroplasty (THA)?
Background: Deep vein thrombosis (DVT) and pulmonary embolism (PE) are common complications after THA. After initial prophylaxis, LMWH given for up to 30 days has been shown to reduce VTE compared with placebo. However, LMWH is costly and may increase the risk of minor bleeding. Aspirin is a potentially simple, low-cost alternative.
Study design: Randomized, placebo-controlled trial.
Setting: Twelve university-affiliated orthopedic hospitals in Canada.
Synopsis: Patients undergoing elective THA without hip fracture, metastatic cancer, or bleeding precluding anticoagulants were eligible. All patients received dalteparin for 10 days and were then randomized to aspirin 81 mg daily or to continue dalteparin. The primary outcome was symptomatic proximal DVT or PE during 90 days’ follow-up. The study was terminated early due to slow enrollment. At that time, 2,364 patients had been enrolled, and an analysis by an independent data safety and monitoring board determined that continuing the study was unlikely to alter the main findings. Extended prophylaxis with aspirin was noninferior to LMWH for the primary outcome, which occurred in 0.3% vs. 1.3%, respectively (95% CI, -0.5% to 2.5%, P<.001 for noninferiority). There were no significant differences in major or minor bleeding.
Though the early termination is a concern, the sample size was large and the results do not suggest inadequate power as a reason for lack of superiority for LMWH. Also, all patients received 10 days of LMWH, which indicates a period of LMWH after discharge will still be needed for most patients prior to initiating aspirin.
Bottom line: After initial LMWH prophylaxis for 10 days, extended prophylaxis with aspirin can be considered, particularly for patients for whom LMWH may not be feasible.
Citation: Anderson DR, Dunbar MJ, Bohm ER, et al. Aspirin versus low-molecular-weight heparin for extended venous thromboembolism prophylaxis after total hip arthroplasty: a randomized trial. Ann Intern Med. 2013;158:800-806.
Symptomatic Subsegmental Pulmonary Embolism (PE) Has a Prognosis Similar to Proximal PE
Clinical question: Is the prognosis of a symptomatic subsegmental pulmonary embolism (PE) similar to that of a more proximal PE?
Background: The use of multidetector computed tomography angiography (CTA) has allowed for better assessment of the pulmonary vasculature and increased detection of distal emboli. Prior studies have raised questions on the clinical importance of subsegmental PE but have been limited by small size or retrospective design.
Study design: Combined data from two prospective trials of management of suspected PE.
Setting: Twelve hospitals in the Netherlands and four tertiary-care emergency departments in Canada.
Synopsis: The study cohort consisted of 3,769 patients with suspected PE, of which 2,688 underwent CTA. Of patients diagnosed with PE, 15.5% had isolated subsegmental emboli. All patients were treated with anticoagulation. During three months of follow-up, the incidence of symptomatic recurrence for subsegmental PE was similar to patients with proximal PE (3.6% vs. 2.5%, respectively). The mortality rates for patients with subsegmental and proximal PE were also similar (10.3% vs. 6.3%, respectively).
The study may have been underpowered to detect small differences in event rates; however, there was no trend suggesting that subsegmental PE had better outcomes than more proximal PE. Also, the study did not specifically investigate whether any management strategy is preferred based on thrombus location on CTA.
Bottom line: Clinicians should continue to anticoagulate patients with subsegmental PE as the prognosis is similar to those with proximal PE.
Citation: Den Exter PL, van Es J, Klok FA, et al. Risk profile and clinical outcome of symptomatic subsegmental pulmonary embolism. Blood. 2013;122:1144-1149.
Video-Based Educational Workshop for Academic Hospitalists and House Staff May Improve Professionalism
Clinical question: Can video-based education promote professionalism among academic hospitalists and house staff?
Background: Unprofessional behavior by academic hospitalists and residents can negatively impact the learning environment and patient safety. This behavior increases throughout training, and faculty behavior can be influential. There is a paucity of educational materials to train hospitalists and house staff to recognize and ameliorate unprofessional behaviors.
Study design: Educational survey study.
Setting: University of Chicago, Northwestern University, and NorthShore University Health System teaching hospitals.
Synopsis: Three videos were developed displaying three types of unprofessional behavior: disparaging other physicians, “blocking” admissions, and misrepresenting tests to expedite their completion. There were 44 hospitalists and 244 house staff who received a 60-minute workshop in which they watched the videos using a viewing tool and discussed the videos in small groups.
For all three videos, more than three-quarters of both hospitalists and house staff felt the behavior was unprofessional or somewhat unprofessional. Hospitalists and house staff found the workshop useful and effective (65.9% and 77.1%, respectively) and would change their behavior as a result of the workshop (65.9% and 67.2%, respectively). Those who perceived the videos as “very realistic” were more likely to report intent to change behavior (93% vs. 53%, P=0.01).
This study is limited by its small sample size and possible selection bias. Those interested or concerned about unprofessional behavior may have been more likely to attend the workshop.
Bottom line: Video-based professionalism education is a feasible and well-received way to educate hospitalists and residents about unprofessional behavior and may even affect their future behavior.
Citation: Farnan JM, O’Leary KJ, Didwania A, et al. Promoting professionalism via a video-based educational workshop for academic hospitalists and housestaff. J Hosp Med. 2013;8:386-389.
Friday and Weekend Elective Surgeries Have Increased Mortality
Clinical question: How can the association between mortality and the day of elective surgical procedures be assessed?
Background: Several studies have described the “weekend effect” for both surgical and medical patients, with higher mortality and length of stay in patients admitted on the weekend compared to weekdays. Two potential explanations are poorer quality of care being delivered on the weekend or more severely ill patients being operated on or admitted on the weekend.
Study design: Retrospective analysis of national hospital administrative data.
Setting: All acute-care and specialist hospitals in England from 2008 to 2011.
Synopsis: There were 4,133,346 elective, inpatient surgical procedures studied. Friday surgeries had an adjusted odds ratio of death within 30 days and within two days of 1.44 [95% CI, 1.39-1.50] and 1.42 [95% CI, 1.26-1.60], respectively, when compared with Monday. Weekend surgeries had an adjusted odds ratio of death within 30 days and within two days of 1.82 [95% CI, 1.71-1.94] and 2.67 [95% CI, 2.30-3.09], respectively, when compared with Monday. There were significant trends toward higher mortality at the end of the workweek and weekends for four high-risk procedures: esophagus and/or stomach excision, colon and/or rectum excisions, coronary artery bypass graft, and lung excision. For lower-risk procedures, there was a significant increase in mortality for Friday surgeries but not weekend surgeries. As with all studies using administrative data, inherent selection biases could not be adjusted for Friday or weekend procedures.
Bottom line: Elective surgeries that occur on the weekend and later in the week have an increased risk of mortality, implying that the weekend effect is due to poorer quality of care during weekends, rather than higher-acuity patients presenting on weekends.
Citation: Aylin P, Alexandrescu R, Jen MH, Mayer EK, Bottle A. Day of week of procedure and 30 day mortality for elective surgery: retrospective analysis of hospital episode statistics. BMJ. 2013;346:f2424.
Basal Plus Correction Insulin Regimen Is Effective in Hospitalized Patients with Type 2 Diabetes
Clinical question: Does a basal plus correction insulin regimen (as needed with meals) result in similar glycemic control and lower rates of hypoglycemia compared to a basal-bolus regimen?
Background: Basal bolus is the preferred insulin regimen for non-critically-ill hospitalized patients as per clinical guidelines. But use is limited due to the complexity of the regimen and the fear of inducing hypoglycemia. A less complex, easier-to-implement basal plus correction insulin regimen may be an effective alternative.
Study design: Multicenter, prospective, open-label, randomized study.
Setting: Six hospitals in the U.S.
Synopsis: A group of 375 medical and surgical patients with Type 2 diabetes treated with diet, oral anti-diabetic agents, or low-dose insulin (≤ 0.4 units/kg/day) were randomized to:
- Basal-bolus insulin regimen with glargine once daily and fixed doses of glusiline before meals;
- Basal plus correction insulin (“basal plus”) regimen with glargine once daily and glusiline sliding scale insulin (SSI) before meals; or
- Regular SSI alone.
After the first day of therapy, treatment with basal-bolus and basal-plus regimens resulted in similar improvements in daily blood glucose (BG) (P=0.16), and both were superior to SSI alone (P=0.04). Both regimens also resulted in less treatment failure (defined as mean daily BG of >240 mg/dl or >2 consecutive BG >240 mg/dl) than did treatment with SSI. Hypoglycemia (BG <70 mg/dl) occurred in 16%, 13%, and 3% of patients in the basal-bolus, basal-plus, and SSI groups, respectively (P=0.02). There were no between-group differences in the frequency of severe hypoglycemia (<40 mg/dl; P=0.76).
The study was not powered to evaluate hospital complications (infection, mortality, hospital stay, and readmissions) across groups.
Bottom line: The basal-plus regimen resulted in glycemic control similar to standard basal-bolus regimen and is an effective alternative for the initial management of hyperglycemia in general medical and surgical patients with Type 2 diabetes.
Citation: Umpierrez GE, Smiley D, Hermayer K, et al. Randomized study comparing a basal-bolus with a basal plus correction insulin regimen for the hospital management of medical and surgical patients with type 2 diabetes: Basal plus trial. Diabetes Care. 2013;36:2169-2174.
Capnography Can Help Diagnose Diabetic Ketoacidosis in the ED
Clinical question: Can capnography be used as a screening tool to identify patients with diabetic ketoacidosis (DKA)?
Background: Metabolic acidosis is a major criterion for diagnosing DKA. Previous studies have shown that end-tidal carbon dioxide (ETCO2) measurement by capnography can provide an accurate estimation of arterial carbon dioxide tension (PaCO2) and may be a noninvasive, fast, inexpensive measurement of acidosis in DKA. However, those studies were in pediatric patients and had small sample sizes.
Study design: Cross-sectional, prospective descriptive-analytic study.
Setting: The ED of Imam Reza Medical Research and Training Hospital, Tabriz, East Azarbaijan, Iran.
Synopsis: A total of 181 adult patients older than 18 with suspected DKA and blood sugar >250 mg/dl were included in the study. Simultaneous capnography and arterial blood gas (ABG) were obtained on all patients. Urine ketones, complete blood count, serum levels of potassium, urea, and creatinine were collected. Sixty-two patients were found to have DKA, while 119 had other conditions associated with metabolic acidosis. There was a significant linear relationship between pH and ETCO2 (P>0.0001, relative risk (R)=0.253), PaCO2 and ETCO2 (P>0.0001, R=0.572), and bicarbonate (HCO3) and ETCO2 (P>0.0001, R=0.730). ETCO2 values >24.5 mmHg had a sensitivity and specificity of 0.90 for ruling out DKA. No cutoff point could be determined for ruling in DKA.
The study was open to selection bias as patient collection was only done during the day, so eligible subjects may have been missed. Moreover, though the study suggests that capnography has a role in ruling out DKA, the exact cutoff value is unclear. Other studies found that higher values were needed to exclude diagnosis.
Bottom line: Using ETCO2 values >24.5 mmHg, capnography can help exclude the diagnosis of DKA in adult patients with elevated BG.
Citation: Soleimanour H, Taghizadieh A, Niafar M, Rahmani F, Golzari S, Esfanjani RM. Predictive value of capnography for diagnosis in patients with suspected diabetic ketoacidosis in the emergency department. West J Emerg Med. 2013. doi: 10.5811/westjem.2013.4.14296.
Publicly Reported Mortality Correlates with Overall Mortality
Clinical question: Are publicly reported mortality rates associated with a hospital’s overall medical and surgical mortality rate?
Background: Public reporting of mortality has become an important strategy in Medicare’s quality-improvement initiative. However, the mortality rate for only three conditions, acute myocardial infarction, congestive heart failure, and pneumonia are reported. It is unclear if these rates correlate to a hospital’s overall mortality rate.
Study design: Retrospective cohort.
Setting: National Medicare fee-for-service population.
Synopsis: Using 2008-2009 data from 2,322 acute-care hospitals with 6.7 million admissions, an aggregate mortality rate for the three publicly reported conditions, a standardized 30-day mortality rate for selected medical and surgical conditions, and an overall average composite mortality score was calculated for each hospital. Based on their mortality for the three publicly reported conditions, hospitals were grouped into quartiles from highest (top-performing hospitals) to lowest mortality (poor-performing hospitals).
Top-performing hospitals had a 3.6% (9.4%vs 13.0%; P<.001) lower mortality rate than poor-performing hospitals and an odds ratio >5 of being a top performer in overall mortality (OR 5.3; 95% CI, 4.3-6.5). They also had an 81% lower chance of being in the worst-performing quartile in overall mortality (OR 0.19; 95% CI, 0.14-0.27). Conversely, poor-performing hospitals had a 4.5 times higher risk of being in the lowest quartile in overall mortality. The study is limited by the use of administrative data, which limits the ability to adjust for severity of illness, overall health, and socioeconomic status of each hospital’s population.
Bottom line: A hospital’s mortality performance on the three publicly reported conditions may predict mortality rates across a wide range of medical and surgical conditions.
Citation: McCrum ML, Joynt KE, Orav EJ, Gawande AA, Jha AK. Mortality for publicly reported conditions and overall hospital mortality rates. JAMA Intern Med. 2013;173:1351-1357.
Cost Savings in Decreasing Preventable Acute-Care Visits Are Limited among High-Cost Medicare Utilizers
Clinical question: What role do preventable acute-care visits play in the overall costs of care for the highest Medicare utilizers?
Background: Some 10% of Medicare patients account for more than half the costs. Interventions targeted at decreasing acute-care costs (ED visits and inpatient hospitalizations) for this high-cost population are widespread, but it is unknown what impact they can have.
Study design: Retrospective cohort.
Setting: National Medicare fee-for-service population.
Synopsis: Standardized total costs were created for fee-for-service Medicare patients for 2009 and 2010 in order to identify high-cost and persistently high-cost patients. Algorithms were used to identify preventable ED visits and hospitalizations in both the high-cost and non-high-cost cohorts.
Of the more than 1 million patients in the sample Medicare population, as many as 113,341 were high-cost. As much as 73% of acute-care spending was attributable to this cohort. Overall, 10% of acute-care costs were felt to be preventable in the high-cost group, 13.5% in the persistently high-cost group, and 19% in the non-high-cost group for 2010. The most common reasons for preventable acute care in the high-cost cohort were heart failure, bacterial pneumonia, and chronic obstructive pulmonary disease. Catastrophic events (myocardial infarction, stroke, sepsis), cancer, and orthopedic procedures drove overall inpatient costs in the high-cost group.
Preventable costs were higher per capita in areas with higher numbers of primary-care and specialist physicians, but it’s unclear if this was a supply or demand issue. The study also used algorithms that possibly overestimate the amount of preventable acute care.
Bottom line: In the highest Medicare utilizers, cost savings aimed at preventable acute care may be limited and might be better targeted at efficiency during acute-care episodes.
Citation: Joynt KE, Gawande AA, Orav EJ, Jha AK. Contribution of preventable acute care spending to total spending for high-cost Medicare patients. JAMA. 2013;309:2572-2578.
In This Edition
Literature At A Glance
A guide to this month’s studies
- Early treatment with intravenous tPA for acute stroke
- Perioperative morbidity, mortality for current smokers
- Statins associated with musculoskeletal conditions
- Antithrombotic medications in patients with history of stroke
- Extended prophylaxis with aspirin for patients after total hip arthroplasty
- Prognosis for symptomatic subsegmental pulmonary embolism
- Video-based educational workshops for academic hospitalists
- Increased mortality for elective surgeries on Fridays, weekends
- Basal plus correction insulin regimen and Type 2 diabetes
- Capnography to diagnose diabetic ketoacidosis in the ED
- How publicly reported mortality rates correlate with hospitals’ overall mortality
- Cost savings and preventable acute-care visits for Medicare patients
Early tPA in Acute Stroke Is Associated with Better Short-Term Outcomes in Routine Clinical Practice
Clinical question: Does early treatment with intravenous (IV) tissue plasminogen activator (tPA) result in better outcomes among patients with acute ischemic stroke in routine clinical practice?
Background: IV tPA for acute ischemic stroke is beneficial if given in the first 4.5 hours after symptom onset. However, pooled data from clinical trials have been limited in characterizing the extent to which onset-to-treatment (OTT) with IV tPA influences outcomes and how effective tPA is in routine clinical practice.
Study design: Data analysis from a stroke registry.
Setting: One thousand three hundred ninety-five U.S. hospitals participating in the Get with the Guidelines—Stroke Program.
Synopsis: Data were analyzed from 58,353 tPA-treated patients within 4.5 hours of symptom onset. Clinical outcomes were compared among patients treated in the 0-90-, 91-180-, and 181-270-minute OTT windows. Patient factors strongly associated with shorter OTT were greater stroke severity (odds ratio [OR] 2.8; 95% confidence interval [CI], 2.5-3.1 per five-point increase), arrival by ambulance (OR 5.9; 95% CI, 4.5-7.3), and arrival during regular hours (OR 4.6; 95% CI, 3.8-5.4). Faster OTT, in 15-minute increments, was associated with reduced in-hospital mortality (OR 0.96; 95% CI, 0.95-0.98; P<.001), reduced symptomatic intracranial hemorrhage (OR 0.96; 95% CI, 0.95-0.98; P<.001), increased achievement of independent ambulation at discharge (OR 1.04; 95% CI, 1.03-1.05; P<.001), and increased discharge to home (OR 1.03; 95% CI, 1.02-1.04; P<.001).
Data collected were dependent on the accuracy and completeness of the chart abstraction, and only short-term outcomes were reported. Although no post-discharge outcomes were reported, previous studies have shown that functional status at discharge strongly correlates with three-month disability outcomes.
Bottom line: In routine clinical practice, earlier tPA for acute ischemic strokes results in better short-term clinical outcomes.
Citation: Saver JL, Fonarow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA. 2013;309:2480-2488.
Current Smokers Have Higher Perioperative Morbidity and Mortality Compared to Past Smokers
Clinical question: Is there an association between current and past smoking on outcomes among patients having major surgery?
Background: Smoking is associated with adverse postoperative outcomes, but it is not known whether the associations are dose-dependent or limited to patients with smoking-related diseases. Smoking-related effects on postoperative events among patients having major surgery are also not well established.
Study design: Retrospective cohort study.
Setting: Four hundred forty-eight non-VA hospitals across the U.S., Canada, Lebanon, and the United Arab Emirates.
Synopsis: Data from 607,558 adult patients undergoing major surgery were obtained from the American College of Surgeons (ACS) National Surgical Quality Improvement Program (NSQIP) database. After adjusting for confounders (cardiopulmonary diseases and cancer), the effects of current and past smoking (quit >1 year prior) on 30-day post-operative outcomes were measured.
There were 125,192 (21%) current smokers and 78,763 (13%) past smokers. Increased odds of post-op mortality were noted in current smokers only (odds ratio [OR] 1.17; 95% CI, 1.10-1.24). The adjusted odds ratios were higher for arterial and respiratory events among current smokers compared with past smokers (OR 1.65; 95% CI, 1.51-1.81 vs. OR 1.20; CI, 1.09-1.31 for arterial events, respectively) and (OR, 1.45; CI, 1.40-1.51 vs. OR, 1.13; CI, 1.08-1.18, for respiratory events, respectively). No significant effects on venous events were observed.
There was an increased adjusted odds of mortality for current smokers with <10 pack-years, while the effects on arterial and respiratory events increased incrementally with increased pack-years. Smoking was associated with adverse post-op outcomes regardless of smoking-related diseases. Variability in hospital quality or surgical strategies may have confounded the results.
Bottom line: Among patients undergoing major surgery, current but not past smoking was associated with higher mortality; smoking cessation for at least a year prior to surgery may decrease post-operative adverse events.
Citation: Musallam KM, Rosendaal FR, Zaatari G, et al. Smoking and the risk of mortality and vascular and respiratory events in patients undergoing major surgery. JAMA Surg. 2013 Jun 19:1-8. doi: 10.1001/jamasurg.2013.2360 [Epub ahead of print].
Statins Associated with Several Musculoskeletal Conditions
Clinical question: Is statin use associated with musculoskeletal adverse events, including arthropathy and injury, in physically active individuals?
Background: Statin-induced musculoskeletal adverse events (AEs) include myalgias, muscle weakness, cramps, rhabdomyolysis, and tendinous disease. The full spectrum of AEs is unknown because randomized clinical trials have not been powered to detect uncommon AEs.
Study design: Retrospective cohort study with propensity score matching.
Setting: San Antonio military area.
Synopsis: A total of 46,249 patients aged 30 to 85 years who met study criteria were propensity-matched into 6,967 statin users and 6,967 nonusers. The occurrence of musculoskeletal conditions were categorized using ICD-9 codes: Msk1, all musculoskeletal diseases; Msk1a, arthropathies and related diseases; Msk1b, injury-related diseases; and Msk2, drug-associated musculoskeletal pain. Of these, statin users had a higher odds ratio (OR) for Msk1 (OR 1.19; 95% CI, 1.08-1.30), Msk1b (1.13; 1.05-1.21), and Msk2 (1.09; 1.02-1.18). Msk1b (arthropathies) had an OR of 1.07 (0.9-1.16, P=0.07). Simvastatin was used by 73.5% of patients, and years of simvastatin use was not a significant predictor of any of the outcome measures. Secondary and sensitivity analyses showed higher adjusted ORs for statin users in all groups. This study was limited by the use of ICD-9-CM codes for identification of baseline characteristics, and the musculoskeletal diagnosis groups used were not validated.
Bottom line: Statin use is associated with an increased likelihood of musculoskeletal conditions, arthropathies, injuries, and pain.
Citation: Mansi I, Frei CR, Pugh M, Makris U, Mortensen EM. Statins and musculoskeletal conditions, arthropathies, and injuries. JAMA Intern Med. 2013;173:1318-1326.
Evidence-Based Guidelines on Periprocedural Management of Antithrombotic Medications in Patients with History of Stroke
Clinical question: What is the evidence for the periprocedural management of antithrombotics in patients with ischemic cerebrovascular accidents (CVAs)?
Background: Evidence-based guidelines are needed to help clinicians determine the thromboembolic risk of temporary discontinuation of antithrombotic medications, the perioperative bleeding risks of continuing antithrombotic agents, whether bridging therapy should be used, and the appropriate timing of antithrombotic agent discontinuation.
Study design: Systematic literature review with practice recommendations.
Setting: American Academy of Neurology Guideline Development Subcommittee convened an expert panel to review and provide recommendations.
Synopsis: Researchers analyzed 133 literature reviews via MEDLINE and EMBASE. Aspirin in stroke patients:
- Should routinely be continued for dental procedures (Level A);
- Should probably be continued for invasive ocular anesthesia, cataract surgery, dermatologic procedures, transrectal ultrasound-guided prostate biopsy, spinal/epidural procedures, and carpal tunnel surgery (Level B); and
- Should possibly be continued for vitreoretinal surgery, electromyogram (EMG), transbronchial lung biopsy, colonoscopic polypectomy, upper endoscopy and biopsy/sphincterotomy, and abdominal ultrasound-guided biopsies (Level C).
Warfarin in stroke patients:
- Should routinely be continued for dental procedures (Level A); and
- Should possibly continued for dermatologic procedures (Level B) and EMG, prostate procedures, inguinal hemiorrhaphy, and endothermal ablation of great saphenous vein (Level C).
- There is a lack of evidence on warfarin for ophthalmologic procedures, with the exception of ocular anesthesia, where it probably does not increase clinically significant bleeding (Level B).
There was not enough evidence to support or refute a recommendation regarding heparin bridge therapy in reducing thromboembolism in chronically anticoagulated patients (Level B).
Bottom line: These are the most up-to-date guidelines for anticoagulant and antiplatelet agents in patients with transient ischemic attacks and strokes undergoing procedures, but further research is needed in many areas.
Citation: Armstrong MJ, Gronseth G Anderson DC, et al. Summary of evidence-based guideline: periprocedural management of antithrombotic medications in patients with ischemic cerebrovascular disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;80:2065-2069.
Extended Prophylaxis with Aspirin Was Noninferior to Extended Prophylaxis with Low-Molecular-Weight Heparin
Clinical question: Is aspirin as effective as low-molecular-weight heparin (LMWH) for the extended prophylaxis of venous thromboembolism (VTE) after total hip arthroplasty (THA)?
Background: Deep vein thrombosis (DVT) and pulmonary embolism (PE) are common complications after THA. After initial prophylaxis, LMWH given for up to 30 days has been shown to reduce VTE compared with placebo. However, LMWH is costly and may increase the risk of minor bleeding. Aspirin is a potentially simple, low-cost alternative.
Study design: Randomized, placebo-controlled trial.
Setting: Twelve university-affiliated orthopedic hospitals in Canada.
Synopsis: Patients undergoing elective THA without hip fracture, metastatic cancer, or bleeding precluding anticoagulants were eligible. All patients received dalteparin for 10 days and were then randomized to aspirin 81 mg daily or to continue dalteparin. The primary outcome was symptomatic proximal DVT or PE during 90 days’ follow-up. The study was terminated early due to slow enrollment. At that time, 2,364 patients had been enrolled, and an analysis by an independent data safety and monitoring board determined that continuing the study was unlikely to alter the main findings. Extended prophylaxis with aspirin was noninferior to LMWH for the primary outcome, which occurred in 0.3% vs. 1.3%, respectively (95% CI, -0.5% to 2.5%, P<.001 for noninferiority). There were no significant differences in major or minor bleeding.
Though the early termination is a concern, the sample size was large and the results do not suggest inadequate power as a reason for lack of superiority for LMWH. Also, all patients received 10 days of LMWH, which indicates a period of LMWH after discharge will still be needed for most patients prior to initiating aspirin.
Bottom line: After initial LMWH prophylaxis for 10 days, extended prophylaxis with aspirin can be considered, particularly for patients for whom LMWH may not be feasible.
Citation: Anderson DR, Dunbar MJ, Bohm ER, et al. Aspirin versus low-molecular-weight heparin for extended venous thromboembolism prophylaxis after total hip arthroplasty: a randomized trial. Ann Intern Med. 2013;158:800-806.
Symptomatic Subsegmental Pulmonary Embolism (PE) Has a Prognosis Similar to Proximal PE
Clinical question: Is the prognosis of a symptomatic subsegmental pulmonary embolism (PE) similar to that of a more proximal PE?
Background: The use of multidetector computed tomography angiography (CTA) has allowed for better assessment of the pulmonary vasculature and increased detection of distal emboli. Prior studies have raised questions on the clinical importance of subsegmental PE but have been limited by small size or retrospective design.
Study design: Combined data from two prospective trials of management of suspected PE.
Setting: Twelve hospitals in the Netherlands and four tertiary-care emergency departments in Canada.
Synopsis: The study cohort consisted of 3,769 patients with suspected PE, of which 2,688 underwent CTA. Of patients diagnosed with PE, 15.5% had isolated subsegmental emboli. All patients were treated with anticoagulation. During three months of follow-up, the incidence of symptomatic recurrence for subsegmental PE was similar to patients with proximal PE (3.6% vs. 2.5%, respectively). The mortality rates for patients with subsegmental and proximal PE were also similar (10.3% vs. 6.3%, respectively).
The study may have been underpowered to detect small differences in event rates; however, there was no trend suggesting that subsegmental PE had better outcomes than more proximal PE. Also, the study did not specifically investigate whether any management strategy is preferred based on thrombus location on CTA.
Bottom line: Clinicians should continue to anticoagulate patients with subsegmental PE as the prognosis is similar to those with proximal PE.
Citation: Den Exter PL, van Es J, Klok FA, et al. Risk profile and clinical outcome of symptomatic subsegmental pulmonary embolism. Blood. 2013;122:1144-1149.
Video-Based Educational Workshop for Academic Hospitalists and House Staff May Improve Professionalism
Clinical question: Can video-based education promote professionalism among academic hospitalists and house staff?
Background: Unprofessional behavior by academic hospitalists and residents can negatively impact the learning environment and patient safety. This behavior increases throughout training, and faculty behavior can be influential. There is a paucity of educational materials to train hospitalists and house staff to recognize and ameliorate unprofessional behaviors.
Study design: Educational survey study.
Setting: University of Chicago, Northwestern University, and NorthShore University Health System teaching hospitals.
Synopsis: Three videos were developed displaying three types of unprofessional behavior: disparaging other physicians, “blocking” admissions, and misrepresenting tests to expedite their completion. There were 44 hospitalists and 244 house staff who received a 60-minute workshop in which they watched the videos using a viewing tool and discussed the videos in small groups.
For all three videos, more than three-quarters of both hospitalists and house staff felt the behavior was unprofessional or somewhat unprofessional. Hospitalists and house staff found the workshop useful and effective (65.9% and 77.1%, respectively) and would change their behavior as a result of the workshop (65.9% and 67.2%, respectively). Those who perceived the videos as “very realistic” were more likely to report intent to change behavior (93% vs. 53%, P=0.01).
This study is limited by its small sample size and possible selection bias. Those interested or concerned about unprofessional behavior may have been more likely to attend the workshop.
Bottom line: Video-based professionalism education is a feasible and well-received way to educate hospitalists and residents about unprofessional behavior and may even affect their future behavior.
Citation: Farnan JM, O’Leary KJ, Didwania A, et al. Promoting professionalism via a video-based educational workshop for academic hospitalists and housestaff. J Hosp Med. 2013;8:386-389.
Friday and Weekend Elective Surgeries Have Increased Mortality
Clinical question: How can the association between mortality and the day of elective surgical procedures be assessed?
Background: Several studies have described the “weekend effect” for both surgical and medical patients, with higher mortality and length of stay in patients admitted on the weekend compared to weekdays. Two potential explanations are poorer quality of care being delivered on the weekend or more severely ill patients being operated on or admitted on the weekend.
Study design: Retrospective analysis of national hospital administrative data.
Setting: All acute-care and specialist hospitals in England from 2008 to 2011.
Synopsis: There were 4,133,346 elective, inpatient surgical procedures studied. Friday surgeries had an adjusted odds ratio of death within 30 days and within two days of 1.44 [95% CI, 1.39-1.50] and 1.42 [95% CI, 1.26-1.60], respectively, when compared with Monday. Weekend surgeries had an adjusted odds ratio of death within 30 days and within two days of 1.82 [95% CI, 1.71-1.94] and 2.67 [95% CI, 2.30-3.09], respectively, when compared with Monday. There were significant trends toward higher mortality at the end of the workweek and weekends for four high-risk procedures: esophagus and/or stomach excision, colon and/or rectum excisions, coronary artery bypass graft, and lung excision. For lower-risk procedures, there was a significant increase in mortality for Friday surgeries but not weekend surgeries. As with all studies using administrative data, inherent selection biases could not be adjusted for Friday or weekend procedures.
Bottom line: Elective surgeries that occur on the weekend and later in the week have an increased risk of mortality, implying that the weekend effect is due to poorer quality of care during weekends, rather than higher-acuity patients presenting on weekends.
Citation: Aylin P, Alexandrescu R, Jen MH, Mayer EK, Bottle A. Day of week of procedure and 30 day mortality for elective surgery: retrospective analysis of hospital episode statistics. BMJ. 2013;346:f2424.
Basal Plus Correction Insulin Regimen Is Effective in Hospitalized Patients with Type 2 Diabetes
Clinical question: Does a basal plus correction insulin regimen (as needed with meals) result in similar glycemic control and lower rates of hypoglycemia compared to a basal-bolus regimen?
Background: Basal bolus is the preferred insulin regimen for non-critically-ill hospitalized patients as per clinical guidelines. But use is limited due to the complexity of the regimen and the fear of inducing hypoglycemia. A less complex, easier-to-implement basal plus correction insulin regimen may be an effective alternative.
Study design: Multicenter, prospective, open-label, randomized study.
Setting: Six hospitals in the U.S.
Synopsis: A group of 375 medical and surgical patients with Type 2 diabetes treated with diet, oral anti-diabetic agents, or low-dose insulin (≤ 0.4 units/kg/day) were randomized to:
- Basal-bolus insulin regimen with glargine once daily and fixed doses of glusiline before meals;
- Basal plus correction insulin (“basal plus”) regimen with glargine once daily and glusiline sliding scale insulin (SSI) before meals; or
- Regular SSI alone.
After the first day of therapy, treatment with basal-bolus and basal-plus regimens resulted in similar improvements in daily blood glucose (BG) (P=0.16), and both were superior to SSI alone (P=0.04). Both regimens also resulted in less treatment failure (defined as mean daily BG of >240 mg/dl or >2 consecutive BG >240 mg/dl) than did treatment with SSI. Hypoglycemia (BG <70 mg/dl) occurred in 16%, 13%, and 3% of patients in the basal-bolus, basal-plus, and SSI groups, respectively (P=0.02). There were no between-group differences in the frequency of severe hypoglycemia (<40 mg/dl; P=0.76).
The study was not powered to evaluate hospital complications (infection, mortality, hospital stay, and readmissions) across groups.
Bottom line: The basal-plus regimen resulted in glycemic control similar to standard basal-bolus regimen and is an effective alternative for the initial management of hyperglycemia in general medical and surgical patients with Type 2 diabetes.
Citation: Umpierrez GE, Smiley D, Hermayer K, et al. Randomized study comparing a basal-bolus with a basal plus correction insulin regimen for the hospital management of medical and surgical patients with type 2 diabetes: Basal plus trial. Diabetes Care. 2013;36:2169-2174.
Capnography Can Help Diagnose Diabetic Ketoacidosis in the ED
Clinical question: Can capnography be used as a screening tool to identify patients with diabetic ketoacidosis (DKA)?
Background: Metabolic acidosis is a major criterion for diagnosing DKA. Previous studies have shown that end-tidal carbon dioxide (ETCO2) measurement by capnography can provide an accurate estimation of arterial carbon dioxide tension (PaCO2) and may be a noninvasive, fast, inexpensive measurement of acidosis in DKA. However, those studies were in pediatric patients and had small sample sizes.
Study design: Cross-sectional, prospective descriptive-analytic study.
Setting: The ED of Imam Reza Medical Research and Training Hospital, Tabriz, East Azarbaijan, Iran.
Synopsis: A total of 181 adult patients older than 18 with suspected DKA and blood sugar >250 mg/dl were included in the study. Simultaneous capnography and arterial blood gas (ABG) were obtained on all patients. Urine ketones, complete blood count, serum levels of potassium, urea, and creatinine were collected. Sixty-two patients were found to have DKA, while 119 had other conditions associated with metabolic acidosis. There was a significant linear relationship between pH and ETCO2 (P>0.0001, relative risk (R)=0.253), PaCO2 and ETCO2 (P>0.0001, R=0.572), and bicarbonate (HCO3) and ETCO2 (P>0.0001, R=0.730). ETCO2 values >24.5 mmHg had a sensitivity and specificity of 0.90 for ruling out DKA. No cutoff point could be determined for ruling in DKA.
The study was open to selection bias as patient collection was only done during the day, so eligible subjects may have been missed. Moreover, though the study suggests that capnography has a role in ruling out DKA, the exact cutoff value is unclear. Other studies found that higher values were needed to exclude diagnosis.
Bottom line: Using ETCO2 values >24.5 mmHg, capnography can help exclude the diagnosis of DKA in adult patients with elevated BG.
Citation: Soleimanour H, Taghizadieh A, Niafar M, Rahmani F, Golzari S, Esfanjani RM. Predictive value of capnography for diagnosis in patients with suspected diabetic ketoacidosis in the emergency department. West J Emerg Med. 2013. doi: 10.5811/westjem.2013.4.14296.
Publicly Reported Mortality Correlates with Overall Mortality
Clinical question: Are publicly reported mortality rates associated with a hospital’s overall medical and surgical mortality rate?
Background: Public reporting of mortality has become an important strategy in Medicare’s quality-improvement initiative. However, the mortality rate for only three conditions, acute myocardial infarction, congestive heart failure, and pneumonia are reported. It is unclear if these rates correlate to a hospital’s overall mortality rate.
Study design: Retrospective cohort.
Setting: National Medicare fee-for-service population.
Synopsis: Using 2008-2009 data from 2,322 acute-care hospitals with 6.7 million admissions, an aggregate mortality rate for the three publicly reported conditions, a standardized 30-day mortality rate for selected medical and surgical conditions, and an overall average composite mortality score was calculated for each hospital. Based on their mortality for the three publicly reported conditions, hospitals were grouped into quartiles from highest (top-performing hospitals) to lowest mortality (poor-performing hospitals).
Top-performing hospitals had a 3.6% (9.4%vs 13.0%; P<.001) lower mortality rate than poor-performing hospitals and an odds ratio >5 of being a top performer in overall mortality (OR 5.3; 95% CI, 4.3-6.5). They also had an 81% lower chance of being in the worst-performing quartile in overall mortality (OR 0.19; 95% CI, 0.14-0.27). Conversely, poor-performing hospitals had a 4.5 times higher risk of being in the lowest quartile in overall mortality. The study is limited by the use of administrative data, which limits the ability to adjust for severity of illness, overall health, and socioeconomic status of each hospital’s population.
Bottom line: A hospital’s mortality performance on the three publicly reported conditions may predict mortality rates across a wide range of medical and surgical conditions.
Citation: McCrum ML, Joynt KE, Orav EJ, Gawande AA, Jha AK. Mortality for publicly reported conditions and overall hospital mortality rates. JAMA Intern Med. 2013;173:1351-1357.
Cost Savings in Decreasing Preventable Acute-Care Visits Are Limited among High-Cost Medicare Utilizers
Clinical question: What role do preventable acute-care visits play in the overall costs of care for the highest Medicare utilizers?
Background: Some 10% of Medicare patients account for more than half the costs. Interventions targeted at decreasing acute-care costs (ED visits and inpatient hospitalizations) for this high-cost population are widespread, but it is unknown what impact they can have.
Study design: Retrospective cohort.
Setting: National Medicare fee-for-service population.
Synopsis: Standardized total costs were created for fee-for-service Medicare patients for 2009 and 2010 in order to identify high-cost and persistently high-cost patients. Algorithms were used to identify preventable ED visits and hospitalizations in both the high-cost and non-high-cost cohorts.
Of the more than 1 million patients in the sample Medicare population, as many as 113,341 were high-cost. As much as 73% of acute-care spending was attributable to this cohort. Overall, 10% of acute-care costs were felt to be preventable in the high-cost group, 13.5% in the persistently high-cost group, and 19% in the non-high-cost group for 2010. The most common reasons for preventable acute care in the high-cost cohort were heart failure, bacterial pneumonia, and chronic obstructive pulmonary disease. Catastrophic events (myocardial infarction, stroke, sepsis), cancer, and orthopedic procedures drove overall inpatient costs in the high-cost group.
Preventable costs were higher per capita in areas with higher numbers of primary-care and specialist physicians, but it’s unclear if this was a supply or demand issue. The study also used algorithms that possibly overestimate the amount of preventable acute care.
Bottom line: In the highest Medicare utilizers, cost savings aimed at preventable acute care may be limited and might be better targeted at efficiency during acute-care episodes.
Citation: Joynt KE, Gawande AA, Orav EJ, Jha AK. Contribution of preventable acute care spending to total spending for high-cost Medicare patients. JAMA. 2013;309:2572-2578.
Hospitalist Compensation Models Evolve Toward Production, Performance-Based Variables
Hospitalists have long recognized that compensation varies significantly by geographic location and by the type of hospitalist medicine group (HMG) you work in: private vs. hospital-owned vs. national-management-owned. A review of SHM’s 2012 State of Hospital Medicine report suggests that hospitalist compensation is also evolving toward a model that more routinely includes both some production variable and performance-based pay (see Figure 1). Although the proportion of compensation paid as a base salary has been trending up over the last few years, so has the proportion paid as a performance incentive.
Source: 2012 State of Hospital Medicine report; www.hospitalmedicine.org/survey
The pay distribution of adult-medicine hospitalists employed by management companies is composed of a high base percentage (mean 88.3% by survey data) and relatively low production and performance variables (mean 6.8% and 4.9%, respectively) compared with other employment models. Contrast that with private hospitalist-only groups, where the mean base is 76.3% with an emphasis on a production component (19.4%) and slightly less on performance pay at 4.2%.
Of the three employment models, however, hospital-/health-system-employed groups have the highest proportion of compensation based on performance metrics with a mean of 7.8%. This makes sense given the financial penalties hospitals and health systems are facing from the Centers for Medicare & Medicaid Services (CMS) around pay-for-performance measures. Hospitals are looking for help from hospitalists in improving quality of care and patient satisfaction and avoiding incurring future penalties. Compensation models in these groups reflect the goals of aligning performance on these measures with financial incentives/risk for hospitalists working in these environments.
What are the top performance metrics hospitalists are being compensated for? CMS’ hospital value-based purchasing (HVBP) core measures and patient satisfaction scores are at the top of the list. More than 70% of all HMGs identify these two measures as part of their performance pay incentive, which is seen consistently by geographic location and by type of hospitalist group.
Beyond these top two metrics, management-company-employed groups also focus on ED throughput measures and early morning discharge times, with more than 70% of these groups having pay incentives aligned with these goals. They also have a higher proportion of their groups participating in several other measures, such as clinical protocols, medication reconciliation, EHR utilization, transitions of care, and readmission rates. In comparison, both hospital-employed and private groups have a wider variety of performance measures in which they participate. Differences are seen geographically, too, with hospitalists located in the Western region having a wider variety of performance measures than other regions.
How hospitalists are compensated for their work will likely continue to evolve. Overall, for nonacademic HMGs serving adults only, we are seeing an upward trend in percentage paid as base pay (from 76% in 2010 to 81% in 2012) and in performance (from 5% in 2010 to 7% in 2012). Hospitalists should anticipate that performance-based pay will continue to account for an increasingly larger percentage of their overall compensation, especially as CMS’ pay-for-performance measures for hospital systems really start to take effect.
Hospital CEOs and CFOs are looking to hospitalists to help deliver on quality, satisfaction, and other performance measures. Incentives will be put in place to reward those groups who do it well.
Dr. Sites is senior medical director of hospitalist programs at Providence Health and Services in Oregon. She is a member of SHM’s Practice Analysis Committee.
Hospitalists have long recognized that compensation varies significantly by geographic location and by the type of hospitalist medicine group (HMG) you work in: private vs. hospital-owned vs. national-management-owned. A review of SHM’s 2012 State of Hospital Medicine report suggests that hospitalist compensation is also evolving toward a model that more routinely includes both some production variable and performance-based pay (see Figure 1). Although the proportion of compensation paid as a base salary has been trending up over the last few years, so has the proportion paid as a performance incentive.
Source: 2012 State of Hospital Medicine report; www.hospitalmedicine.org/survey
The pay distribution of adult-medicine hospitalists employed by management companies is composed of a high base percentage (mean 88.3% by survey data) and relatively low production and performance variables (mean 6.8% and 4.9%, respectively) compared with other employment models. Contrast that with private hospitalist-only groups, where the mean base is 76.3% with an emphasis on a production component (19.4%) and slightly less on performance pay at 4.2%.
Of the three employment models, however, hospital-/health-system-employed groups have the highest proportion of compensation based on performance metrics with a mean of 7.8%. This makes sense given the financial penalties hospitals and health systems are facing from the Centers for Medicare & Medicaid Services (CMS) around pay-for-performance measures. Hospitals are looking for help from hospitalists in improving quality of care and patient satisfaction and avoiding incurring future penalties. Compensation models in these groups reflect the goals of aligning performance on these measures with financial incentives/risk for hospitalists working in these environments.
What are the top performance metrics hospitalists are being compensated for? CMS’ hospital value-based purchasing (HVBP) core measures and patient satisfaction scores are at the top of the list. More than 70% of all HMGs identify these two measures as part of their performance pay incentive, which is seen consistently by geographic location and by type of hospitalist group.
Beyond these top two metrics, management-company-employed groups also focus on ED throughput measures and early morning discharge times, with more than 70% of these groups having pay incentives aligned with these goals. They also have a higher proportion of their groups participating in several other measures, such as clinical protocols, medication reconciliation, EHR utilization, transitions of care, and readmission rates. In comparison, both hospital-employed and private groups have a wider variety of performance measures in which they participate. Differences are seen geographically, too, with hospitalists located in the Western region having a wider variety of performance measures than other regions.
How hospitalists are compensated for their work will likely continue to evolve. Overall, for nonacademic HMGs serving adults only, we are seeing an upward trend in percentage paid as base pay (from 76% in 2010 to 81% in 2012) and in performance (from 5% in 2010 to 7% in 2012). Hospitalists should anticipate that performance-based pay will continue to account for an increasingly larger percentage of their overall compensation, especially as CMS’ pay-for-performance measures for hospital systems really start to take effect.
Hospital CEOs and CFOs are looking to hospitalists to help deliver on quality, satisfaction, and other performance measures. Incentives will be put in place to reward those groups who do it well.
Dr. Sites is senior medical director of hospitalist programs at Providence Health and Services in Oregon. She is a member of SHM’s Practice Analysis Committee.
Hospitalists have long recognized that compensation varies significantly by geographic location and by the type of hospitalist medicine group (HMG) you work in: private vs. hospital-owned vs. national-management-owned. A review of SHM’s 2012 State of Hospital Medicine report suggests that hospitalist compensation is also evolving toward a model that more routinely includes both some production variable and performance-based pay (see Figure 1). Although the proportion of compensation paid as a base salary has been trending up over the last few years, so has the proportion paid as a performance incentive.
Source: 2012 State of Hospital Medicine report; www.hospitalmedicine.org/survey
The pay distribution of adult-medicine hospitalists employed by management companies is composed of a high base percentage (mean 88.3% by survey data) and relatively low production and performance variables (mean 6.8% and 4.9%, respectively) compared with other employment models. Contrast that with private hospitalist-only groups, where the mean base is 76.3% with an emphasis on a production component (19.4%) and slightly less on performance pay at 4.2%.
Of the three employment models, however, hospital-/health-system-employed groups have the highest proportion of compensation based on performance metrics with a mean of 7.8%. This makes sense given the financial penalties hospitals and health systems are facing from the Centers for Medicare & Medicaid Services (CMS) around pay-for-performance measures. Hospitals are looking for help from hospitalists in improving quality of care and patient satisfaction and avoiding incurring future penalties. Compensation models in these groups reflect the goals of aligning performance on these measures with financial incentives/risk for hospitalists working in these environments.
What are the top performance metrics hospitalists are being compensated for? CMS’ hospital value-based purchasing (HVBP) core measures and patient satisfaction scores are at the top of the list. More than 70% of all HMGs identify these two measures as part of their performance pay incentive, which is seen consistently by geographic location and by type of hospitalist group.
Beyond these top two metrics, management-company-employed groups also focus on ED throughput measures and early morning discharge times, with more than 70% of these groups having pay incentives aligned with these goals. They also have a higher proportion of their groups participating in several other measures, such as clinical protocols, medication reconciliation, EHR utilization, transitions of care, and readmission rates. In comparison, both hospital-employed and private groups have a wider variety of performance measures in which they participate. Differences are seen geographically, too, with hospitalists located in the Western region having a wider variety of performance measures than other regions.
How hospitalists are compensated for their work will likely continue to evolve. Overall, for nonacademic HMGs serving adults only, we are seeing an upward trend in percentage paid as base pay (from 76% in 2010 to 81% in 2012) and in performance (from 5% in 2010 to 7% in 2012). Hospitalists should anticipate that performance-based pay will continue to account for an increasingly larger percentage of their overall compensation, especially as CMS’ pay-for-performance measures for hospital systems really start to take effect.
Hospital CEOs and CFOs are looking to hospitalists to help deliver on quality, satisfaction, and other performance measures. Incentives will be put in place to reward those groups who do it well.
Dr. Sites is senior medical director of hospitalist programs at Providence Health and Services in Oregon. She is a member of SHM’s Practice Analysis Committee.
SHM Advocates for Medicare to Cover Skilled-Nursing Facilities
The Centers for Medicare & Medicaid Services (CMS) recently issued a Final Rule for the Inpatient Prospective Payment System, which guides payment and programs associated with inpatient hospitalizations. In this year’s rule, CMS adjusted the criteria for inpatient admissions in an attempt to simplify and clarify the decision-making process.
The policy would allow physicians to admit a patient if they reasonably expect and document in the medical record that a beneficiary will need to stay in the hospital for more than two midnights. Admissions based on this time-limited expectation will be presumed to be appropriate for Medicare Part A payment. CMS cited concerns about the growing trend of longer observation stays to support this change.
With observation stays, there are two major financial concerns for patients: whether the hospital stay is paid under Medicare Part A or Part B, and whether Medicare will pay for post-acute care in a skilled-nursing facility (SNF). Medicare Part A reimburses for inpatient admissions, with a one-time deductible for the benefit period. Outpatient services, such as observation care and physician services, are covered under Medicare Part B, which has copays and co-insurance that greatly increase the costs for beneficiaries. In addition, SNF coverage through Medicare Part A is determined by the three-day rule; a patient must be an inpatient for three days to qualify for coverage.
While the long-term impacts of this regulatory change to the admission criteria remain to be seen, SHM is concerned that the rule does not adequately address the broader problems associated with inpatient and observation status. As we note in our comments to CMS on the new rule:1
Even with these changes, the central tension created by the bifurcation in admission status still remains.…Other policies and programs, such as the attempts to reduce admissions, may inadvertently add pressure to the admission decision.
Indeed, for beneficiaries, the barrier to SNF coverage remains. CMS takes care to note that, while time under emergency care and observation care count toward the two-midnight presumption for inpatient admission, it does not count toward the three-day rule for SNF coverage. This is particularly problematic; as advances in medicine allow for the treatment of higher-acuity and -severity conditions with observation stays or shorter inpatient stays, patients might not be getting the follow-up care they need. This puts them at risk for additional complications and, ultimately, readmissions to the hospital.
In an era of seeking value in the healthcare system, it seems like an opportunity lost to streamline and coordinate care across settings and to ensure that patients are getting the follow-up care they require. It is for this reason that hospitalists continue to push for passage of the Improving Access to Medicare Coverage Act, a bill sponsored by Rep. Joe Courtney (D-Conn.), Rep. Tom Latham (R-Iowa), and Sen. Sherrod Brown (D-Ohio) that would count observation status as time toward the three-day requirement for SNF coverage.
A recent Office of Inspector General (OIG) report for the U.S. Department of Health and Human Services on observation status sums up the problem succinctly.2 The OIG states that “CMS should consider how to ensure that beneficiaries with similar post-hospital care needs have the same access and cost-sharing for SNF services.”2
SHM concurs.
Joshua Lapps is SHM’s government relations specialist.
References
- Society of Hospital Medicine. SHM submits comments in response to FY2014 inpatient prospective payment system proposed rule. Society of Hospital Medicine website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Letters_to_Congress_and_Regulatory_Agencies&Template=/CM/ContentDisplay.cfm&ContentID=34044. Accessed Sept. 9, 2013.
- Office of Inspector General. Memorandum report: Hospitals’ use of observations stays and short inpatient stays for Medicare beneficiaries, OEI-02-12-00040. U.S. Department of Health and Human Services website. Available at: http://oig.hhs.gov/oei/reports/oei-02-12-00040.pdf. Accessed Sept. 9, 2013.
The Centers for Medicare & Medicaid Services (CMS) recently issued a Final Rule for the Inpatient Prospective Payment System, which guides payment and programs associated with inpatient hospitalizations. In this year’s rule, CMS adjusted the criteria for inpatient admissions in an attempt to simplify and clarify the decision-making process.
The policy would allow physicians to admit a patient if they reasonably expect and document in the medical record that a beneficiary will need to stay in the hospital for more than two midnights. Admissions based on this time-limited expectation will be presumed to be appropriate for Medicare Part A payment. CMS cited concerns about the growing trend of longer observation stays to support this change.
With observation stays, there are two major financial concerns for patients: whether the hospital stay is paid under Medicare Part A or Part B, and whether Medicare will pay for post-acute care in a skilled-nursing facility (SNF). Medicare Part A reimburses for inpatient admissions, with a one-time deductible for the benefit period. Outpatient services, such as observation care and physician services, are covered under Medicare Part B, which has copays and co-insurance that greatly increase the costs for beneficiaries. In addition, SNF coverage through Medicare Part A is determined by the three-day rule; a patient must be an inpatient for three days to qualify for coverage.
While the long-term impacts of this regulatory change to the admission criteria remain to be seen, SHM is concerned that the rule does not adequately address the broader problems associated with inpatient and observation status. As we note in our comments to CMS on the new rule:1
Even with these changes, the central tension created by the bifurcation in admission status still remains.…Other policies and programs, such as the attempts to reduce admissions, may inadvertently add pressure to the admission decision.
Indeed, for beneficiaries, the barrier to SNF coverage remains. CMS takes care to note that, while time under emergency care and observation care count toward the two-midnight presumption for inpatient admission, it does not count toward the three-day rule for SNF coverage. This is particularly problematic; as advances in medicine allow for the treatment of higher-acuity and -severity conditions with observation stays or shorter inpatient stays, patients might not be getting the follow-up care they need. This puts them at risk for additional complications and, ultimately, readmissions to the hospital.
In an era of seeking value in the healthcare system, it seems like an opportunity lost to streamline and coordinate care across settings and to ensure that patients are getting the follow-up care they require. It is for this reason that hospitalists continue to push for passage of the Improving Access to Medicare Coverage Act, a bill sponsored by Rep. Joe Courtney (D-Conn.), Rep. Tom Latham (R-Iowa), and Sen. Sherrod Brown (D-Ohio) that would count observation status as time toward the three-day requirement for SNF coverage.
A recent Office of Inspector General (OIG) report for the U.S. Department of Health and Human Services on observation status sums up the problem succinctly.2 The OIG states that “CMS should consider how to ensure that beneficiaries with similar post-hospital care needs have the same access and cost-sharing for SNF services.”2
SHM concurs.
Joshua Lapps is SHM’s government relations specialist.
References
- Society of Hospital Medicine. SHM submits comments in response to FY2014 inpatient prospective payment system proposed rule. Society of Hospital Medicine website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Letters_to_Congress_and_Regulatory_Agencies&Template=/CM/ContentDisplay.cfm&ContentID=34044. Accessed Sept. 9, 2013.
- Office of Inspector General. Memorandum report: Hospitals’ use of observations stays and short inpatient stays for Medicare beneficiaries, OEI-02-12-00040. U.S. Department of Health and Human Services website. Available at: http://oig.hhs.gov/oei/reports/oei-02-12-00040.pdf. Accessed Sept. 9, 2013.
The Centers for Medicare & Medicaid Services (CMS) recently issued a Final Rule for the Inpatient Prospective Payment System, which guides payment and programs associated with inpatient hospitalizations. In this year’s rule, CMS adjusted the criteria for inpatient admissions in an attempt to simplify and clarify the decision-making process.
The policy would allow physicians to admit a patient if they reasonably expect and document in the medical record that a beneficiary will need to stay in the hospital for more than two midnights. Admissions based on this time-limited expectation will be presumed to be appropriate for Medicare Part A payment. CMS cited concerns about the growing trend of longer observation stays to support this change.
With observation stays, there are two major financial concerns for patients: whether the hospital stay is paid under Medicare Part A or Part B, and whether Medicare will pay for post-acute care in a skilled-nursing facility (SNF). Medicare Part A reimburses for inpatient admissions, with a one-time deductible for the benefit period. Outpatient services, such as observation care and physician services, are covered under Medicare Part B, which has copays and co-insurance that greatly increase the costs for beneficiaries. In addition, SNF coverage through Medicare Part A is determined by the three-day rule; a patient must be an inpatient for three days to qualify for coverage.
While the long-term impacts of this regulatory change to the admission criteria remain to be seen, SHM is concerned that the rule does not adequately address the broader problems associated with inpatient and observation status. As we note in our comments to CMS on the new rule:1
Even with these changes, the central tension created by the bifurcation in admission status still remains.…Other policies and programs, such as the attempts to reduce admissions, may inadvertently add pressure to the admission decision.
Indeed, for beneficiaries, the barrier to SNF coverage remains. CMS takes care to note that, while time under emergency care and observation care count toward the two-midnight presumption for inpatient admission, it does not count toward the three-day rule for SNF coverage. This is particularly problematic; as advances in medicine allow for the treatment of higher-acuity and -severity conditions with observation stays or shorter inpatient stays, patients might not be getting the follow-up care they need. This puts them at risk for additional complications and, ultimately, readmissions to the hospital.
In an era of seeking value in the healthcare system, it seems like an opportunity lost to streamline and coordinate care across settings and to ensure that patients are getting the follow-up care they require. It is for this reason that hospitalists continue to push for passage of the Improving Access to Medicare Coverage Act, a bill sponsored by Rep. Joe Courtney (D-Conn.), Rep. Tom Latham (R-Iowa), and Sen. Sherrod Brown (D-Ohio) that would count observation status as time toward the three-day requirement for SNF coverage.
A recent Office of Inspector General (OIG) report for the U.S. Department of Health and Human Services on observation status sums up the problem succinctly.2 The OIG states that “CMS should consider how to ensure that beneficiaries with similar post-hospital care needs have the same access and cost-sharing for SNF services.”2
SHM concurs.
Joshua Lapps is SHM’s government relations specialist.
References
- Society of Hospital Medicine. SHM submits comments in response to FY2014 inpatient prospective payment system proposed rule. Society of Hospital Medicine website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Letters_to_Congress_and_Regulatory_Agencies&Template=/CM/ContentDisplay.cfm&ContentID=34044. Accessed Sept. 9, 2013.
- Office of Inspector General. Memorandum report: Hospitals’ use of observations stays and short inpatient stays for Medicare beneficiaries, OEI-02-12-00040. U.S. Department of Health and Human Services website. Available at: http://oig.hhs.gov/oei/reports/oei-02-12-00040.pdf. Accessed Sept. 9, 2013.
Bleeding Risks, Anticoagulants, Hospital-Acquired Infections Among Can't Miss Topics at HM14
What breakout-session and pre-course topics are HM14 course director Daniel Brotman, MD, SFHM, and assistant course director Efren Manjarrez, MD, SFHM, looking forward to showcasing? Here is a sampling:
- Bleeding risks: a crucial yet misunderstood area.
- New anticoagulants: a quickly evolving area that will affect lots of hospitalists and patients.
- What keeps your CFO up at night: a financial perspective from a hospital president and hospitalist.
- Choosing Wisely: Learn how SHM turned the ABIM Foundation’s Choosing Wisely initiative into practical recommendations for hospitalists.
- Pediatric clinical conundrums.
- Updates in key specialty and content areas.
- Hospital-acquired infection control by Sanjay Saint.
- CMS’ meaningful use.
What breakout-session and pre-course topics are HM14 course director Daniel Brotman, MD, SFHM, and assistant course director Efren Manjarrez, MD, SFHM, looking forward to showcasing? Here is a sampling:
- Bleeding risks: a crucial yet misunderstood area.
- New anticoagulants: a quickly evolving area that will affect lots of hospitalists and patients.
- What keeps your CFO up at night: a financial perspective from a hospital president and hospitalist.
- Choosing Wisely: Learn how SHM turned the ABIM Foundation’s Choosing Wisely initiative into practical recommendations for hospitalists.
- Pediatric clinical conundrums.
- Updates in key specialty and content areas.
- Hospital-acquired infection control by Sanjay Saint.
- CMS’ meaningful use.
What breakout-session and pre-course topics are HM14 course director Daniel Brotman, MD, SFHM, and assistant course director Efren Manjarrez, MD, SFHM, looking forward to showcasing? Here is a sampling:
- Bleeding risks: a crucial yet misunderstood area.
- New anticoagulants: a quickly evolving area that will affect lots of hospitalists and patients.
- What keeps your CFO up at night: a financial perspective from a hospital president and hospitalist.
- Choosing Wisely: Learn how SHM turned the ABIM Foundation’s Choosing Wisely initiative into practical recommendations for hospitalists.
- Pediatric clinical conundrums.
- Updates in key specialty and content areas.
- Hospital-acquired infection control by Sanjay Saint.
- CMS’ meaningful use.
Make Plans Now for HM14
SHM’s next annual meeting, HM14, is only six months away. So today is the day to make scheduling requests and book a room. And, for the first time, the biggest annual event in hospital medicine will be in Las Vegas.
HM14 will be held March 24-27 at Mandalay Bay Resort and Casino in Las Vegas. Meeting registration is now open at www.hospitalmedicine2014.org. The early registration discount ends Jan. 26.
Who should attend HM14? Bring the whole team: hospitalists, pediatricians, academic hospitalists, general internists, family physicians, nurse practitioners, physician assistants, administrators, and providers practicing in acute-care settings.
SHM’s next annual meeting, HM14, is only six months away. So today is the day to make scheduling requests and book a room. And, for the first time, the biggest annual event in hospital medicine will be in Las Vegas.
HM14 will be held March 24-27 at Mandalay Bay Resort and Casino in Las Vegas. Meeting registration is now open at www.hospitalmedicine2014.org. The early registration discount ends Jan. 26.
Who should attend HM14? Bring the whole team: hospitalists, pediatricians, academic hospitalists, general internists, family physicians, nurse practitioners, physician assistants, administrators, and providers practicing in acute-care settings.
SHM’s next annual meeting, HM14, is only six months away. So today is the day to make scheduling requests and book a room. And, for the first time, the biggest annual event in hospital medicine will be in Las Vegas.
HM14 will be held March 24-27 at Mandalay Bay Resort and Casino in Las Vegas. Meeting registration is now open at www.hospitalmedicine2014.org. The early registration discount ends Jan. 26.
Who should attend HM14? Bring the whole team: hospitalists, pediatricians, academic hospitalists, general internists, family physicians, nurse practitioners, physician assistants, administrators, and providers practicing in acute-care settings.
SHM Fellow in Hospital Medicine Spotlight: Janet Nagamine, MD, BSN, SFHM

Dr. Nagamine is a hospitalist physician at Kaiser Permanente Medical Center in Santa Clara, Calif., where she previously was a quality chief and safety officer. She is a member of the National Quality Forum Patient Safety Complications Steering Committee, an SHM board member, and a member of SHM’s Hospital Quality and Patient Safety Committee, which she chaired for four years.
Undergraduate education: University of Hawaii, Honolulu.
Medical school: University of Hawaii.
Notable: In the early stages of the patient-safety movement, Dr. Nagamine worked with aviation-safety experts and Kaiser colleagues to develop innovative patient-safety programs. She has led numerous quality-improvement (QI) projects, including the development of a patient-safety curriculum for staff and anonymous reporting mechanisms that focus on identifying problems in the system rather than focusing simply on individuals. For her efforts, she was awarded SHM’s Clinical Award of Excellence in 2002.
After leading many other QI initiatives on a national level, she was named a Senior Fellow in Hospital Medicine in 2010. Dr. Nagamine has been with SHM’s Project BOOST since its inception, serving on the advisory board and as a mentor. She says the SFHM designation acknowledges that she has contributed to hospital medicine in a meaningful way.
FYI: A doctor who enjoys creative projects outside of the workplace, Dr. Nagamine is working on a documentary about her father’s struggles as a World War II soldier and a poor immigrant starting a new life in California.
Quotable: “Creating a culture of safety and transparency allows us to get real about the fact that we’re human and fallible. Ultimately, we get much more information about where the problems are and how to fix them.”
Dr. Nagamine is a hospitalist physician at Kaiser Permanente Medical Center in Santa Clara, Calif., where she previously was a quality chief and safety officer. She is a member of the National Quality Forum Patient Safety Complications Steering Committee, an SHM board member, and a member of SHM’s Hospital Quality and Patient Safety Committee, which she chaired for four years.
Undergraduate education: University of Hawaii, Honolulu.
Medical school: University of Hawaii.
Notable: In the early stages of the patient-safety movement, Dr. Nagamine worked with aviation-safety experts and Kaiser colleagues to develop innovative patient-safety programs. She has led numerous quality-improvement (QI) projects, including the development of a patient-safety curriculum for staff and anonymous reporting mechanisms that focus on identifying problems in the system rather than focusing simply on individuals. For her efforts, she was awarded SHM’s Clinical Award of Excellence in 2002.
After leading many other QI initiatives on a national level, she was named a Senior Fellow in Hospital Medicine in 2010. Dr. Nagamine has been with SHM’s Project BOOST since its inception, serving on the advisory board and as a mentor. She says the SFHM designation acknowledges that she has contributed to hospital medicine in a meaningful way.
FYI: A doctor who enjoys creative projects outside of the workplace, Dr. Nagamine is working on a documentary about her father’s struggles as a World War II soldier and a poor immigrant starting a new life in California.
Quotable: “Creating a culture of safety and transparency allows us to get real about the fact that we’re human and fallible. Ultimately, we get much more information about where the problems are and how to fix them.”
Dr. Nagamine is a hospitalist physician at Kaiser Permanente Medical Center in Santa Clara, Calif., where she previously was a quality chief and safety officer. She is a member of the National Quality Forum Patient Safety Complications Steering Committee, an SHM board member, and a member of SHM’s Hospital Quality and Patient Safety Committee, which she chaired for four years.
Undergraduate education: University of Hawaii, Honolulu.
Medical school: University of Hawaii.
Notable: In the early stages of the patient-safety movement, Dr. Nagamine worked with aviation-safety experts and Kaiser colleagues to develop innovative patient-safety programs. She has led numerous quality-improvement (QI) projects, including the development of a patient-safety curriculum for staff and anonymous reporting mechanisms that focus on identifying problems in the system rather than focusing simply on individuals. For her efforts, she was awarded SHM’s Clinical Award of Excellence in 2002.
After leading many other QI initiatives on a national level, she was named a Senior Fellow in Hospital Medicine in 2010. Dr. Nagamine has been with SHM’s Project BOOST since its inception, serving on the advisory board and as a mentor. She says the SFHM designation acknowledges that she has contributed to hospital medicine in a meaningful way.
FYI: A doctor who enjoys creative projects outside of the workplace, Dr. Nagamine is working on a documentary about her father’s struggles as a World War II soldier and a poor immigrant starting a new life in California.
Quotable: “Creating a culture of safety and transparency allows us to get real about the fact that we’re human and fallible. Ultimately, we get much more information about where the problems are and how to fix them.”