User login
Wearing White—Right or Wrong
Physicians wear white coats. In many medical centers, the length of one's coat grows with seniority; medical students, interns, and residents wear short white coats, while attending staff graduate to long white coats with their names embroidered on the front. This traditional uniform serves a similar role to the stripes on a military sleeve. That is, by examining the length of a person's coat, a nurse or other hospital employee can rapidly determine the seniority, and theoretically the increased medical knowledge, of the person inside.
Of course, it isn't quite this simple. In many places, all residents wear long coats (with embroidered names). In other hospitals, attending staff wear short coats. The problem of distinguishing between a medical student, resident, and attending physician can be quite vexing, particularly after the summer months when the anxiety visible on the faces of new house staff and third‐year medical students fades away into the more seasoned look of fatigue and malaise. As hospital‐based practitioners, what are we to do?
To be certain there are clues one can use to identify one's rank in the medical profession. Often medical students wear a medical school pin bestowed upon them at a robing ceremony or other coming‐of‐age festivity to mark the transition to the clinical training years.1 (Little do they know that the primary utility of such adornments is to mark them, as though with a scarlet letter, for easy identification when one wishes to pimp the medical team.)2 Similarly, one can look for an arm patch. Just as a mother hen has the need to identify her own chicks, so too is it helpful for the residency director to have each of his or her own residents easily identifiable by the residency patch emblazoned upon the arm of their (long) white coat. The patch test can fail, though, for too often the residency emblem is merely a simple modification of the hospital or medical center logo, and thus not easily distinguishable.
When all else fails, of course, you can look to the pockets. As one's medical knowledge and training rank increases, the number of papers, pens, and instruments in the pockets of the white coat decreases. This inverse relationship provides some increased ability to identify medical students and junior house staff. For example, a short white coat, busting with oversized index cards held together by a large metal ring, several tattered and folded journal articles, and a worn spiral‐bound text suggests a medical student or intern. If they have an electrocardiogram (ECG), calipers, and tuning fork visible, safe bet is you've found a medical student. Conversely, if the white coat sports several free pens (with medication logos embossed on their stems) and has more than a few stains, chances are you've spotted an intern.
Unfortunately, none of this is of much utility in identifying a physician, as the initial premise that physicians wear white coats may be false. The problem is 2‐fold. First of all, not all physicians wear white coats (or any coat for that matter), and, many nonphysicians wear white coats. Commonly, phlebotomists, pharmacists, and respiratory technicians wear white coats; long white coats, in fact. So do nutritionists, speech pathologists, the clerk at the radiology file room, and even the cashiers in the cafeteria. And why not? Each of these persons has an important role to play in the operation of a modern medical center. The long white coat serves as professional uniform to identify the wearer as contributing to the mission of the medical center. It engenders confidence and suggests cleanliness and purity.
The problem remains, however, what should I, as an attending physician wear? You see, patients encounter so many persons in their visits to the hospital that it is not clear who their doctor is. (It's not statistically likely to be the man in a long white coat.) Being able to identify the attending physician is important. The attending physician is ultimately responsible for the patient's care.
There are many guidelines for how to dress. For example, don't wear white after Labor Day. The pleats on a cummerbund point so that the folds open pointing up (apparently it was originally meant to serve as a ticket holder, perhaps in the days before pockets?). Match your belt to your shoes. The list goes on. However, physicians are not commonly associated with natty attire, so fashion help for the MD is hard to find, though nonetheless necessary.
Even wearing white is questionable. White robes have been associated with purity and sanctity for centuries. Religious leaders have donned white for spiritual cleansing of their communities on the most holy of days. I like the idea of appearing pure and holy. Of course, I don't want to mislead anybody, either.
Brides have traditionally worn white dresses. But, while many believe this is to convey a sense of premarital purity, more likely the tradition stems from an ancient Roman practice of wearing white as a symbol of joyous celebration. Interestingly, in some parts of Asia, people at a funeral wear white while the deceased is dressed in red.
In some environments, the culture of the medical center prescribes appropriate apparel. The Mayo Clinic in Minnesota, for example, has had a tradition of having their physician staff dress in business attire. By this, they mean conservative sport coats or suits with ties for men, and similar appropriate women's business clothing. As noted by Leonard L. Berry and Neeli Bendapudi in their article Why Docs Don't Wear White Coats or Polo Shirts at the Mayo Clinic3 in the Harvard Business School publication Working Knowledge for Business Leaders, while some may consider this semiformal business dress pretentious, it should be considered no more pretentious than, say, the dress code for airline pilots. Airline passengers don't want to see their pilot in a polo shirt, and patients feel the same way about their doctors. The business attire is a uniform; it's a visible clue that communicates respect to patients and their families. But, isn't a white coat a uniform that conveys respect? Perhaps a white coat isn't safe if the physician wants to stand out to oncoming traffic in his snowy Minnesota environs.
Besides, is it true that patients don't want their physician to wear a polo shirt? Perhaps casual dress will break down 1 of the patient‐doctor barriers to communication and allow for improved comfort with greater honesty in patientphysician interactions? Prior to designing a prospective controlled randomized trial to answer this question myself, I reviewed the available literature.
It turns out, that an evaluation of polo‐shirt‐wearing physicians has been carried out. Drs. Barrett and Booth4 from the Birmingham Maternity Hospital in England questioned 203 groups of parents and children (406 individuals) about various levels of physician dress. Seventy percent of participants thought that how the doctor dressed was important. Among children, a male physician in a polo shirt was considered more friendly and gentle than the male physician in a white coat (who did get points for being more competent and more concerned). Women physicians in T‐shirts were also though to be friendlier than if in a white coat, but similarly less competent. Parents were more likely to prefer casually‐dressed physicians and were poor at predicting what their children would want.
So, a polo shirt makes me look friendly, gentle, and less competent? What about a polo shirt under a white coatis that the whole package? What about business attire, as per the Mayo Clinic requirements? These questions remain unanswered. So, back to the literature.
In the Journal of the American Medical Association, in 1987, Dunn et al.5 evaluated 200 medical patients in Boston and San Francisco regarding their preference for physician dress. Sixty‐five percent believed physicians should wear a white coat, 27% said no tennis shoes, one‐half said no blue jeans, and about one‐third thought male physicians should wear ties and female physicians should be in a skirt or dress. I suppose the conclusion here is to wear a white coat with or without jeans and most of the time without a tie, though almost always with proper shoes, or at least without tennis shoes. (I practice in Boston and trained partly in San Francisco so this advice seems particularly valid for me.)
It may be more obvious what to do in Japan. Ikusaka et al.6 evaluated the experience of patients at a university clinic seen by a consulting physician in either a white coat, or private clothes. To me, private clothes imply pajamas and a bathrobe, but we must assume this means some form of professional dress lacking a white coat. It turns out that 71% of the Japanese patients seeing a doctor in a white coat preferred a white coat, though more patients seeing a physician in a white coat (vs. private clothes) felt tense during their consultation. The researchers stress that the presence of a white coat did not increase satisfaction with the consultation. They conclude, that while patients may say they prefer a white coat, maybe it would be better not to wear one since it makes patients feel tense.
In addition to feeling tense, white coats may cause hypertension. The phenomenon of elevated blood pressure when in the presence of the physician (or other hospital staff in a white coat) has been long documented. This white coat hypertension can be found in more than 15% of the population who have a measured blood pressure in the office of over 140/90 mmHg with normal daytime mean ambulatory blood pressure readings (when not around a white‐coat‐wearing stress‐inducing medical worker).7 Older adults, females, and nonsmokers were more likely to have white coat hypertension than other persons.
And yet, older adults prefer white coats. The Japanese study (Ikusaka et al.6) concluded that elderly patients prefer a white coat to other attire. Similarly, a study from the Royal Free Hospital, London, showed that white coats were twice as popular with patients as they were with physicians.8 Specifically, patients found the white coats made doctors easier to identify. In an article by the British Broadcasting Corporation (BBC) on the subject, it was noted that the elderly largely preferred physicians in white coats, while children preferred physicians without white coats. British children must prefer a friendly doctor to a competent one.
The article further suggests that only 1 in 8 physicians wears a white coat, complaining that they are too hot and uncomfortable, and may carry the risk of transmitting infections. The white coat, the symbol of cleanliness and purity, a source of infection? To add hypocrisy to the equation, one‐half of physicians who thought white coats should be worn admitted to never actually wearing a white coat. In fact, only 7 of 86 physicians surveyed wore their white coat on a daily basis. The BBC goes on to note that in Australia, the white coat is gaining momentum as there seems to be a movement towards rediscovering the white coat as a symbol of purpose and pride as a profession.8
Really? Let's consult the literature! According to Dr. D.A. Watson,9 White coats have largely disappeared from Australian teaching hospitals and the majority of junior doctors in Australia oppose the wearing of white coats. In a survey of 337 junior medical officers, only 16% preferred to wear a white coat. Peer pressure seems to have something to do with this, as 70% say they don't wear a white coat because nobody else wears a white coat. This is indeed a compelling argument.
Of course, a better argument against wearing a white coat may be that it causes tension (at least in Japanese patients) and may cause white‐coat‐hypertension, resulting in the inappropriate diagnosis and treatment of elevated blood pressure. In Australia, however, it seems that wearing a white coat may make patients too relaxed. Wing et al.10 noted that in 21% to 45% of elderly patients, blood pressure was atypically low when checked in the physician's office as compared to mean ambulatory blood pressure. This reverse‐white‐coat‐hypertension could result in the omission of necessary blood pressure treatment.
In the United States, residentsour own version of the Australian junior medical officercommonly wear scrubs covered by a white coat. Scrub clothes are typically available without charge from the hospital, limit the amount of laundry a busy resident needs to do, and can be put on with little concern as to pressed pleats or matching colors. The overlying white coat adds a moderate degree of formality to what could otherwise be mistaken as pajamas, while providing convenient pockets for the aforementioned papers and miscellaneous equipment and souvenirs. This outfit is likely one of practicality rather than a desire to be most appealing to patients. But, what do you know? It seems that patients prefer their resident physicians to dress this way. Dr. A. Cha et al.11 at the Northeastern Ohio College of Medicine found that patients in an obstetrics and gynecology clinic overall felt that resident physicians dressed in surgical scrubs with a white coat made them feel more comfortable and confident than if dressed otherwise. On the question of a white coat specifically, the majority had no preference that their physician wear one.
So, it's very much unclear whether a white coat is a tension‐causing, blood‐pressure‐elevating, infection risk or a competence‐implying blood‐pressure‐lowering way to identify a physician. As a result, the jury is still out on whether physicians should wear white.
One thing I do know, however, is that patients shouldn't wear white. But they do. As an OtolaryngologistHead and Neck Surgeon, my clinical practice is split between surgical procedures and office visits. Commonly, patients with sinus or nasal surgery will require some form of cotton gauze or foam material within the nose in order to tamponade bleeding. Similarly, patients presenting to the emergency department or urgent care center with epistaxis may have their noses packed and then be told to see an otolaryngologist (eg, me) in 3 or 4 days to have the packing removed. I also commonly remove facial skin lesions, biopsy tongue masses, reduce nasal fractures, and otherwise engage in activities with an above average propensity to result in a mess. More often than happenstance will allow, patients come to see me for such visits in their white Sunday best. I truly care for my patients. I respect them as individuals and desire to do no harm. This includes not staining their shirts, ties, or pants. However, there is no amount of blue towels or gauze pads than can keep a white shirt clean when you have that first sneeze after removal of your nasal packing.12
So what is it that makes so many patients come to the office in a white shirt? Perhaps patients subconsciously associate healthcare with the color white since their doctors wear white coats and the nurses wear starched white dresses with tight white folded caps on top of their head. I've never seen a nurse in a white dress and hat, but believe television programs have shown this in the past.
Maybe the answer lies in an adaptation of data from another Australian study. In a survey of 180 oncology patients about white coats on physicians, the most common argument against wearing the white coat was that it represented a barrier between the physician and the patient.13 However, it is indeed the sane patient who desires to have a barrier between their physician and the removal of nasal packing, a skin lesion, or a tongue mass. Another possibility is that as fewer and fewer physicians wear white, patients are gravitating toward this color as a way of distinguishing themselves from the medical staff. Perhaps a person dressed in white is less likely to be grabbed in the hallway with a Doctor come quick! and more likely to be allowed to just sit peacefully in the waiting room with a 1997 issue of Ladies Home Journal or Senior Fisherman magazine.
Of course, perhaps patients believe the fashion experts who expound that white goes with everything. If so, they soon learn that white doesn't go so well with blotchy splattered red.
The truth is, I don't want my patients to wear white. Between making certain I project myself as approachable and easy to speak with, remembering to cover all the appropriate irrelevant parts of the history and physical to comply with billing requirements, entering data in our easy‐to‐navigate electronic medical record, and attempting to both diagnose the problem and discern an acceptable and effective treatment, the last thing I need is to worry about staining patient shirts! I believe that this phenomenon is widespread among medical practitioners and should be called white‐shirt‐hypertension.
Conclusion
After reviewing the available literature, I've determined that patients should not wear white. However, I'm still not certain how I should dress. For now, I'll stick with whatever is clean and professional and make sure my belt and shoes match. I may or may not wear a tie, since another study showed that 30% of patients believed their physician wore a tie even if they didn't.14 Of course, I'll put on the white coat for the semiannual meeting with the chairman, or if I'm having something messy for lunch and wore a tie that day.
I'll also wear my name badge. It says I'm an attending physician and not a resident. It opens doors around the hospital. Literally. It's got a magnetic stripe.
- .Deconstructing the white coat.Ann Intern Med.1998;129:740–742.
- .The art of pimping.JAMA.1989;262(1):89–90.
- ,. “Why Docs don't wear white coats or Polo Shirts at the Mayo Clinic” working knowledge for business leaders. Available at:http://hbswk.hbs.edu/pubitem.jhtml?id=3380309(6970):1710–1712.
- ,,,,.Patient and house officer attitudes on physician attire and etiquette.JAMA.1987;257(1):65–68.
- ,,, et al.Patients' attitude toward consultations by a physician without a white coat in Japan.Intern Med.1999;38(7):533–536.
- ,,, et al.Determinants of white‐coat hypertension.Blood Press Monit.2004;9(6):307–309.
- Doctors ‘should wear white coats.’ BBC news, Thursday, 13 May, 2004. Available at:http://news.bbc.co.uk/2/hi/health/3706783.stm. Accessed May2009.
- .What do Australian junior doctors think of white coats?Med Educ.2002;36(12):1209–1213.
- ,,, ,;ANBP2 Management Committee and Investigators.Second Australian National Blood Pressure Study. ‘Reverse white‐coat hypertension’ in older hypertensives.J Hypertens.2002;20(4):639–644.
- ,,,.Resident physician attire: does it make a difference to our patients?Am J Obstet Gynecol.2004;190(5):1484–1488.
- . Personal communication.1994–2008.
- .Should doctors wear white coats?Med J Aust.2001;174:343–344.
- ,,,,.Does wearing a necktie influence patient perceptions of emergency department care?J Emerg Med.1998;16(4):541–543.
Physicians wear white coats. In many medical centers, the length of one's coat grows with seniority; medical students, interns, and residents wear short white coats, while attending staff graduate to long white coats with their names embroidered on the front. This traditional uniform serves a similar role to the stripes on a military sleeve. That is, by examining the length of a person's coat, a nurse or other hospital employee can rapidly determine the seniority, and theoretically the increased medical knowledge, of the person inside.
Of course, it isn't quite this simple. In many places, all residents wear long coats (with embroidered names). In other hospitals, attending staff wear short coats. The problem of distinguishing between a medical student, resident, and attending physician can be quite vexing, particularly after the summer months when the anxiety visible on the faces of new house staff and third‐year medical students fades away into the more seasoned look of fatigue and malaise. As hospital‐based practitioners, what are we to do?
To be certain there are clues one can use to identify one's rank in the medical profession. Often medical students wear a medical school pin bestowed upon them at a robing ceremony or other coming‐of‐age festivity to mark the transition to the clinical training years.1 (Little do they know that the primary utility of such adornments is to mark them, as though with a scarlet letter, for easy identification when one wishes to pimp the medical team.)2 Similarly, one can look for an arm patch. Just as a mother hen has the need to identify her own chicks, so too is it helpful for the residency director to have each of his or her own residents easily identifiable by the residency patch emblazoned upon the arm of their (long) white coat. The patch test can fail, though, for too often the residency emblem is merely a simple modification of the hospital or medical center logo, and thus not easily distinguishable.
When all else fails, of course, you can look to the pockets. As one's medical knowledge and training rank increases, the number of papers, pens, and instruments in the pockets of the white coat decreases. This inverse relationship provides some increased ability to identify medical students and junior house staff. For example, a short white coat, busting with oversized index cards held together by a large metal ring, several tattered and folded journal articles, and a worn spiral‐bound text suggests a medical student or intern. If they have an electrocardiogram (ECG), calipers, and tuning fork visible, safe bet is you've found a medical student. Conversely, if the white coat sports several free pens (with medication logos embossed on their stems) and has more than a few stains, chances are you've spotted an intern.
Unfortunately, none of this is of much utility in identifying a physician, as the initial premise that physicians wear white coats may be false. The problem is 2‐fold. First of all, not all physicians wear white coats (or any coat for that matter), and, many nonphysicians wear white coats. Commonly, phlebotomists, pharmacists, and respiratory technicians wear white coats; long white coats, in fact. So do nutritionists, speech pathologists, the clerk at the radiology file room, and even the cashiers in the cafeteria. And why not? Each of these persons has an important role to play in the operation of a modern medical center. The long white coat serves as professional uniform to identify the wearer as contributing to the mission of the medical center. It engenders confidence and suggests cleanliness and purity.
The problem remains, however, what should I, as an attending physician wear? You see, patients encounter so many persons in their visits to the hospital that it is not clear who their doctor is. (It's not statistically likely to be the man in a long white coat.) Being able to identify the attending physician is important. The attending physician is ultimately responsible for the patient's care.
There are many guidelines for how to dress. For example, don't wear white after Labor Day. The pleats on a cummerbund point so that the folds open pointing up (apparently it was originally meant to serve as a ticket holder, perhaps in the days before pockets?). Match your belt to your shoes. The list goes on. However, physicians are not commonly associated with natty attire, so fashion help for the MD is hard to find, though nonetheless necessary.
Even wearing white is questionable. White robes have been associated with purity and sanctity for centuries. Religious leaders have donned white for spiritual cleansing of their communities on the most holy of days. I like the idea of appearing pure and holy. Of course, I don't want to mislead anybody, either.
Brides have traditionally worn white dresses. But, while many believe this is to convey a sense of premarital purity, more likely the tradition stems from an ancient Roman practice of wearing white as a symbol of joyous celebration. Interestingly, in some parts of Asia, people at a funeral wear white while the deceased is dressed in red.
In some environments, the culture of the medical center prescribes appropriate apparel. The Mayo Clinic in Minnesota, for example, has had a tradition of having their physician staff dress in business attire. By this, they mean conservative sport coats or suits with ties for men, and similar appropriate women's business clothing. As noted by Leonard L. Berry and Neeli Bendapudi in their article Why Docs Don't Wear White Coats or Polo Shirts at the Mayo Clinic3 in the Harvard Business School publication Working Knowledge for Business Leaders, while some may consider this semiformal business dress pretentious, it should be considered no more pretentious than, say, the dress code for airline pilots. Airline passengers don't want to see their pilot in a polo shirt, and patients feel the same way about their doctors. The business attire is a uniform; it's a visible clue that communicates respect to patients and their families. But, isn't a white coat a uniform that conveys respect? Perhaps a white coat isn't safe if the physician wants to stand out to oncoming traffic in his snowy Minnesota environs.
Besides, is it true that patients don't want their physician to wear a polo shirt? Perhaps casual dress will break down 1 of the patient‐doctor barriers to communication and allow for improved comfort with greater honesty in patientphysician interactions? Prior to designing a prospective controlled randomized trial to answer this question myself, I reviewed the available literature.
It turns out, that an evaluation of polo‐shirt‐wearing physicians has been carried out. Drs. Barrett and Booth4 from the Birmingham Maternity Hospital in England questioned 203 groups of parents and children (406 individuals) about various levels of physician dress. Seventy percent of participants thought that how the doctor dressed was important. Among children, a male physician in a polo shirt was considered more friendly and gentle than the male physician in a white coat (who did get points for being more competent and more concerned). Women physicians in T‐shirts were also though to be friendlier than if in a white coat, but similarly less competent. Parents were more likely to prefer casually‐dressed physicians and were poor at predicting what their children would want.
So, a polo shirt makes me look friendly, gentle, and less competent? What about a polo shirt under a white coatis that the whole package? What about business attire, as per the Mayo Clinic requirements? These questions remain unanswered. So, back to the literature.
In the Journal of the American Medical Association, in 1987, Dunn et al.5 evaluated 200 medical patients in Boston and San Francisco regarding their preference for physician dress. Sixty‐five percent believed physicians should wear a white coat, 27% said no tennis shoes, one‐half said no blue jeans, and about one‐third thought male physicians should wear ties and female physicians should be in a skirt or dress. I suppose the conclusion here is to wear a white coat with or without jeans and most of the time without a tie, though almost always with proper shoes, or at least without tennis shoes. (I practice in Boston and trained partly in San Francisco so this advice seems particularly valid for me.)
It may be more obvious what to do in Japan. Ikusaka et al.6 evaluated the experience of patients at a university clinic seen by a consulting physician in either a white coat, or private clothes. To me, private clothes imply pajamas and a bathrobe, but we must assume this means some form of professional dress lacking a white coat. It turns out that 71% of the Japanese patients seeing a doctor in a white coat preferred a white coat, though more patients seeing a physician in a white coat (vs. private clothes) felt tense during their consultation. The researchers stress that the presence of a white coat did not increase satisfaction with the consultation. They conclude, that while patients may say they prefer a white coat, maybe it would be better not to wear one since it makes patients feel tense.
In addition to feeling tense, white coats may cause hypertension. The phenomenon of elevated blood pressure when in the presence of the physician (or other hospital staff in a white coat) has been long documented. This white coat hypertension can be found in more than 15% of the population who have a measured blood pressure in the office of over 140/90 mmHg with normal daytime mean ambulatory blood pressure readings (when not around a white‐coat‐wearing stress‐inducing medical worker).7 Older adults, females, and nonsmokers were more likely to have white coat hypertension than other persons.
And yet, older adults prefer white coats. The Japanese study (Ikusaka et al.6) concluded that elderly patients prefer a white coat to other attire. Similarly, a study from the Royal Free Hospital, London, showed that white coats were twice as popular with patients as they were with physicians.8 Specifically, patients found the white coats made doctors easier to identify. In an article by the British Broadcasting Corporation (BBC) on the subject, it was noted that the elderly largely preferred physicians in white coats, while children preferred physicians without white coats. British children must prefer a friendly doctor to a competent one.
The article further suggests that only 1 in 8 physicians wears a white coat, complaining that they are too hot and uncomfortable, and may carry the risk of transmitting infections. The white coat, the symbol of cleanliness and purity, a source of infection? To add hypocrisy to the equation, one‐half of physicians who thought white coats should be worn admitted to never actually wearing a white coat. In fact, only 7 of 86 physicians surveyed wore their white coat on a daily basis. The BBC goes on to note that in Australia, the white coat is gaining momentum as there seems to be a movement towards rediscovering the white coat as a symbol of purpose and pride as a profession.8
Really? Let's consult the literature! According to Dr. D.A. Watson,9 White coats have largely disappeared from Australian teaching hospitals and the majority of junior doctors in Australia oppose the wearing of white coats. In a survey of 337 junior medical officers, only 16% preferred to wear a white coat. Peer pressure seems to have something to do with this, as 70% say they don't wear a white coat because nobody else wears a white coat. This is indeed a compelling argument.
Of course, a better argument against wearing a white coat may be that it causes tension (at least in Japanese patients) and may cause white‐coat‐hypertension, resulting in the inappropriate diagnosis and treatment of elevated blood pressure. In Australia, however, it seems that wearing a white coat may make patients too relaxed. Wing et al.10 noted that in 21% to 45% of elderly patients, blood pressure was atypically low when checked in the physician's office as compared to mean ambulatory blood pressure. This reverse‐white‐coat‐hypertension could result in the omission of necessary blood pressure treatment.
In the United States, residentsour own version of the Australian junior medical officercommonly wear scrubs covered by a white coat. Scrub clothes are typically available without charge from the hospital, limit the amount of laundry a busy resident needs to do, and can be put on with little concern as to pressed pleats or matching colors. The overlying white coat adds a moderate degree of formality to what could otherwise be mistaken as pajamas, while providing convenient pockets for the aforementioned papers and miscellaneous equipment and souvenirs. This outfit is likely one of practicality rather than a desire to be most appealing to patients. But, what do you know? It seems that patients prefer their resident physicians to dress this way. Dr. A. Cha et al.11 at the Northeastern Ohio College of Medicine found that patients in an obstetrics and gynecology clinic overall felt that resident physicians dressed in surgical scrubs with a white coat made them feel more comfortable and confident than if dressed otherwise. On the question of a white coat specifically, the majority had no preference that their physician wear one.
So, it's very much unclear whether a white coat is a tension‐causing, blood‐pressure‐elevating, infection risk or a competence‐implying blood‐pressure‐lowering way to identify a physician. As a result, the jury is still out on whether physicians should wear white.
One thing I do know, however, is that patients shouldn't wear white. But they do. As an OtolaryngologistHead and Neck Surgeon, my clinical practice is split between surgical procedures and office visits. Commonly, patients with sinus or nasal surgery will require some form of cotton gauze or foam material within the nose in order to tamponade bleeding. Similarly, patients presenting to the emergency department or urgent care center with epistaxis may have their noses packed and then be told to see an otolaryngologist (eg, me) in 3 or 4 days to have the packing removed. I also commonly remove facial skin lesions, biopsy tongue masses, reduce nasal fractures, and otherwise engage in activities with an above average propensity to result in a mess. More often than happenstance will allow, patients come to see me for such visits in their white Sunday best. I truly care for my patients. I respect them as individuals and desire to do no harm. This includes not staining their shirts, ties, or pants. However, there is no amount of blue towels or gauze pads than can keep a white shirt clean when you have that first sneeze after removal of your nasal packing.12
So what is it that makes so many patients come to the office in a white shirt? Perhaps patients subconsciously associate healthcare with the color white since their doctors wear white coats and the nurses wear starched white dresses with tight white folded caps on top of their head. I've never seen a nurse in a white dress and hat, but believe television programs have shown this in the past.
Maybe the answer lies in an adaptation of data from another Australian study. In a survey of 180 oncology patients about white coats on physicians, the most common argument against wearing the white coat was that it represented a barrier between the physician and the patient.13 However, it is indeed the sane patient who desires to have a barrier between their physician and the removal of nasal packing, a skin lesion, or a tongue mass. Another possibility is that as fewer and fewer physicians wear white, patients are gravitating toward this color as a way of distinguishing themselves from the medical staff. Perhaps a person dressed in white is less likely to be grabbed in the hallway with a Doctor come quick! and more likely to be allowed to just sit peacefully in the waiting room with a 1997 issue of Ladies Home Journal or Senior Fisherman magazine.
Of course, perhaps patients believe the fashion experts who expound that white goes with everything. If so, they soon learn that white doesn't go so well with blotchy splattered red.
The truth is, I don't want my patients to wear white. Between making certain I project myself as approachable and easy to speak with, remembering to cover all the appropriate irrelevant parts of the history and physical to comply with billing requirements, entering data in our easy‐to‐navigate electronic medical record, and attempting to both diagnose the problem and discern an acceptable and effective treatment, the last thing I need is to worry about staining patient shirts! I believe that this phenomenon is widespread among medical practitioners and should be called white‐shirt‐hypertension.
Conclusion
After reviewing the available literature, I've determined that patients should not wear white. However, I'm still not certain how I should dress. For now, I'll stick with whatever is clean and professional and make sure my belt and shoes match. I may or may not wear a tie, since another study showed that 30% of patients believed their physician wore a tie even if they didn't.14 Of course, I'll put on the white coat for the semiannual meeting with the chairman, or if I'm having something messy for lunch and wore a tie that day.
I'll also wear my name badge. It says I'm an attending physician and not a resident. It opens doors around the hospital. Literally. It's got a magnetic stripe.
Physicians wear white coats. In many medical centers, the length of one's coat grows with seniority; medical students, interns, and residents wear short white coats, while attending staff graduate to long white coats with their names embroidered on the front. This traditional uniform serves a similar role to the stripes on a military sleeve. That is, by examining the length of a person's coat, a nurse or other hospital employee can rapidly determine the seniority, and theoretically the increased medical knowledge, of the person inside.
Of course, it isn't quite this simple. In many places, all residents wear long coats (with embroidered names). In other hospitals, attending staff wear short coats. The problem of distinguishing between a medical student, resident, and attending physician can be quite vexing, particularly after the summer months when the anxiety visible on the faces of new house staff and third‐year medical students fades away into the more seasoned look of fatigue and malaise. As hospital‐based practitioners, what are we to do?
To be certain there are clues one can use to identify one's rank in the medical profession. Often medical students wear a medical school pin bestowed upon them at a robing ceremony or other coming‐of‐age festivity to mark the transition to the clinical training years.1 (Little do they know that the primary utility of such adornments is to mark them, as though with a scarlet letter, for easy identification when one wishes to pimp the medical team.)2 Similarly, one can look for an arm patch. Just as a mother hen has the need to identify her own chicks, so too is it helpful for the residency director to have each of his or her own residents easily identifiable by the residency patch emblazoned upon the arm of their (long) white coat. The patch test can fail, though, for too often the residency emblem is merely a simple modification of the hospital or medical center logo, and thus not easily distinguishable.
When all else fails, of course, you can look to the pockets. As one's medical knowledge and training rank increases, the number of papers, pens, and instruments in the pockets of the white coat decreases. This inverse relationship provides some increased ability to identify medical students and junior house staff. For example, a short white coat, busting with oversized index cards held together by a large metal ring, several tattered and folded journal articles, and a worn spiral‐bound text suggests a medical student or intern. If they have an electrocardiogram (ECG), calipers, and tuning fork visible, safe bet is you've found a medical student. Conversely, if the white coat sports several free pens (with medication logos embossed on their stems) and has more than a few stains, chances are you've spotted an intern.
Unfortunately, none of this is of much utility in identifying a physician, as the initial premise that physicians wear white coats may be false. The problem is 2‐fold. First of all, not all physicians wear white coats (or any coat for that matter), and, many nonphysicians wear white coats. Commonly, phlebotomists, pharmacists, and respiratory technicians wear white coats; long white coats, in fact. So do nutritionists, speech pathologists, the clerk at the radiology file room, and even the cashiers in the cafeteria. And why not? Each of these persons has an important role to play in the operation of a modern medical center. The long white coat serves as professional uniform to identify the wearer as contributing to the mission of the medical center. It engenders confidence and suggests cleanliness and purity.
The problem remains, however, what should I, as an attending physician wear? You see, patients encounter so many persons in their visits to the hospital that it is not clear who their doctor is. (It's not statistically likely to be the man in a long white coat.) Being able to identify the attending physician is important. The attending physician is ultimately responsible for the patient's care.
There are many guidelines for how to dress. For example, don't wear white after Labor Day. The pleats on a cummerbund point so that the folds open pointing up (apparently it was originally meant to serve as a ticket holder, perhaps in the days before pockets?). Match your belt to your shoes. The list goes on. However, physicians are not commonly associated with natty attire, so fashion help for the MD is hard to find, though nonetheless necessary.
Even wearing white is questionable. White robes have been associated with purity and sanctity for centuries. Religious leaders have donned white for spiritual cleansing of their communities on the most holy of days. I like the idea of appearing pure and holy. Of course, I don't want to mislead anybody, either.
Brides have traditionally worn white dresses. But, while many believe this is to convey a sense of premarital purity, more likely the tradition stems from an ancient Roman practice of wearing white as a symbol of joyous celebration. Interestingly, in some parts of Asia, people at a funeral wear white while the deceased is dressed in red.
In some environments, the culture of the medical center prescribes appropriate apparel. The Mayo Clinic in Minnesota, for example, has had a tradition of having their physician staff dress in business attire. By this, they mean conservative sport coats or suits with ties for men, and similar appropriate women's business clothing. As noted by Leonard L. Berry and Neeli Bendapudi in their article Why Docs Don't Wear White Coats or Polo Shirts at the Mayo Clinic3 in the Harvard Business School publication Working Knowledge for Business Leaders, while some may consider this semiformal business dress pretentious, it should be considered no more pretentious than, say, the dress code for airline pilots. Airline passengers don't want to see their pilot in a polo shirt, and patients feel the same way about their doctors. The business attire is a uniform; it's a visible clue that communicates respect to patients and their families. But, isn't a white coat a uniform that conveys respect? Perhaps a white coat isn't safe if the physician wants to stand out to oncoming traffic in his snowy Minnesota environs.
Besides, is it true that patients don't want their physician to wear a polo shirt? Perhaps casual dress will break down 1 of the patient‐doctor barriers to communication and allow for improved comfort with greater honesty in patientphysician interactions? Prior to designing a prospective controlled randomized trial to answer this question myself, I reviewed the available literature.
It turns out, that an evaluation of polo‐shirt‐wearing physicians has been carried out. Drs. Barrett and Booth4 from the Birmingham Maternity Hospital in England questioned 203 groups of parents and children (406 individuals) about various levels of physician dress. Seventy percent of participants thought that how the doctor dressed was important. Among children, a male physician in a polo shirt was considered more friendly and gentle than the male physician in a white coat (who did get points for being more competent and more concerned). Women physicians in T‐shirts were also though to be friendlier than if in a white coat, but similarly less competent. Parents were more likely to prefer casually‐dressed physicians and were poor at predicting what their children would want.
So, a polo shirt makes me look friendly, gentle, and less competent? What about a polo shirt under a white coatis that the whole package? What about business attire, as per the Mayo Clinic requirements? These questions remain unanswered. So, back to the literature.
In the Journal of the American Medical Association, in 1987, Dunn et al.5 evaluated 200 medical patients in Boston and San Francisco regarding their preference for physician dress. Sixty‐five percent believed physicians should wear a white coat, 27% said no tennis shoes, one‐half said no blue jeans, and about one‐third thought male physicians should wear ties and female physicians should be in a skirt or dress. I suppose the conclusion here is to wear a white coat with or without jeans and most of the time without a tie, though almost always with proper shoes, or at least without tennis shoes. (I practice in Boston and trained partly in San Francisco so this advice seems particularly valid for me.)
It may be more obvious what to do in Japan. Ikusaka et al.6 evaluated the experience of patients at a university clinic seen by a consulting physician in either a white coat, or private clothes. To me, private clothes imply pajamas and a bathrobe, but we must assume this means some form of professional dress lacking a white coat. It turns out that 71% of the Japanese patients seeing a doctor in a white coat preferred a white coat, though more patients seeing a physician in a white coat (vs. private clothes) felt tense during their consultation. The researchers stress that the presence of a white coat did not increase satisfaction with the consultation. They conclude, that while patients may say they prefer a white coat, maybe it would be better not to wear one since it makes patients feel tense.
In addition to feeling tense, white coats may cause hypertension. The phenomenon of elevated blood pressure when in the presence of the physician (or other hospital staff in a white coat) has been long documented. This white coat hypertension can be found in more than 15% of the population who have a measured blood pressure in the office of over 140/90 mmHg with normal daytime mean ambulatory blood pressure readings (when not around a white‐coat‐wearing stress‐inducing medical worker).7 Older adults, females, and nonsmokers were more likely to have white coat hypertension than other persons.
And yet, older adults prefer white coats. The Japanese study (Ikusaka et al.6) concluded that elderly patients prefer a white coat to other attire. Similarly, a study from the Royal Free Hospital, London, showed that white coats were twice as popular with patients as they were with physicians.8 Specifically, patients found the white coats made doctors easier to identify. In an article by the British Broadcasting Corporation (BBC) on the subject, it was noted that the elderly largely preferred physicians in white coats, while children preferred physicians without white coats. British children must prefer a friendly doctor to a competent one.
The article further suggests that only 1 in 8 physicians wears a white coat, complaining that they are too hot and uncomfortable, and may carry the risk of transmitting infections. The white coat, the symbol of cleanliness and purity, a source of infection? To add hypocrisy to the equation, one‐half of physicians who thought white coats should be worn admitted to never actually wearing a white coat. In fact, only 7 of 86 physicians surveyed wore their white coat on a daily basis. The BBC goes on to note that in Australia, the white coat is gaining momentum as there seems to be a movement towards rediscovering the white coat as a symbol of purpose and pride as a profession.8
Really? Let's consult the literature! According to Dr. D.A. Watson,9 White coats have largely disappeared from Australian teaching hospitals and the majority of junior doctors in Australia oppose the wearing of white coats. In a survey of 337 junior medical officers, only 16% preferred to wear a white coat. Peer pressure seems to have something to do with this, as 70% say they don't wear a white coat because nobody else wears a white coat. This is indeed a compelling argument.
Of course, a better argument against wearing a white coat may be that it causes tension (at least in Japanese patients) and may cause white‐coat‐hypertension, resulting in the inappropriate diagnosis and treatment of elevated blood pressure. In Australia, however, it seems that wearing a white coat may make patients too relaxed. Wing et al.10 noted that in 21% to 45% of elderly patients, blood pressure was atypically low when checked in the physician's office as compared to mean ambulatory blood pressure. This reverse‐white‐coat‐hypertension could result in the omission of necessary blood pressure treatment.
In the United States, residentsour own version of the Australian junior medical officercommonly wear scrubs covered by a white coat. Scrub clothes are typically available without charge from the hospital, limit the amount of laundry a busy resident needs to do, and can be put on with little concern as to pressed pleats or matching colors. The overlying white coat adds a moderate degree of formality to what could otherwise be mistaken as pajamas, while providing convenient pockets for the aforementioned papers and miscellaneous equipment and souvenirs. This outfit is likely one of practicality rather than a desire to be most appealing to patients. But, what do you know? It seems that patients prefer their resident physicians to dress this way. Dr. A. Cha et al.11 at the Northeastern Ohio College of Medicine found that patients in an obstetrics and gynecology clinic overall felt that resident physicians dressed in surgical scrubs with a white coat made them feel more comfortable and confident than if dressed otherwise. On the question of a white coat specifically, the majority had no preference that their physician wear one.
So, it's very much unclear whether a white coat is a tension‐causing, blood‐pressure‐elevating, infection risk or a competence‐implying blood‐pressure‐lowering way to identify a physician. As a result, the jury is still out on whether physicians should wear white.
One thing I do know, however, is that patients shouldn't wear white. But they do. As an OtolaryngologistHead and Neck Surgeon, my clinical practice is split between surgical procedures and office visits. Commonly, patients with sinus or nasal surgery will require some form of cotton gauze or foam material within the nose in order to tamponade bleeding. Similarly, patients presenting to the emergency department or urgent care center with epistaxis may have their noses packed and then be told to see an otolaryngologist (eg, me) in 3 or 4 days to have the packing removed. I also commonly remove facial skin lesions, biopsy tongue masses, reduce nasal fractures, and otherwise engage in activities with an above average propensity to result in a mess. More often than happenstance will allow, patients come to see me for such visits in their white Sunday best. I truly care for my patients. I respect them as individuals and desire to do no harm. This includes not staining their shirts, ties, or pants. However, there is no amount of blue towels or gauze pads than can keep a white shirt clean when you have that first sneeze after removal of your nasal packing.12
So what is it that makes so many patients come to the office in a white shirt? Perhaps patients subconsciously associate healthcare with the color white since their doctors wear white coats and the nurses wear starched white dresses with tight white folded caps on top of their head. I've never seen a nurse in a white dress and hat, but believe television programs have shown this in the past.
Maybe the answer lies in an adaptation of data from another Australian study. In a survey of 180 oncology patients about white coats on physicians, the most common argument against wearing the white coat was that it represented a barrier between the physician and the patient.13 However, it is indeed the sane patient who desires to have a barrier between their physician and the removal of nasal packing, a skin lesion, or a tongue mass. Another possibility is that as fewer and fewer physicians wear white, patients are gravitating toward this color as a way of distinguishing themselves from the medical staff. Perhaps a person dressed in white is less likely to be grabbed in the hallway with a Doctor come quick! and more likely to be allowed to just sit peacefully in the waiting room with a 1997 issue of Ladies Home Journal or Senior Fisherman magazine.
Of course, perhaps patients believe the fashion experts who expound that white goes with everything. If so, they soon learn that white doesn't go so well with blotchy splattered red.
The truth is, I don't want my patients to wear white. Between making certain I project myself as approachable and easy to speak with, remembering to cover all the appropriate irrelevant parts of the history and physical to comply with billing requirements, entering data in our easy‐to‐navigate electronic medical record, and attempting to both diagnose the problem and discern an acceptable and effective treatment, the last thing I need is to worry about staining patient shirts! I believe that this phenomenon is widespread among medical practitioners and should be called white‐shirt‐hypertension.
Conclusion
After reviewing the available literature, I've determined that patients should not wear white. However, I'm still not certain how I should dress. For now, I'll stick with whatever is clean and professional and make sure my belt and shoes match. I may or may not wear a tie, since another study showed that 30% of patients believed their physician wore a tie even if they didn't.14 Of course, I'll put on the white coat for the semiannual meeting with the chairman, or if I'm having something messy for lunch and wore a tie that day.
I'll also wear my name badge. It says I'm an attending physician and not a resident. It opens doors around the hospital. Literally. It's got a magnetic stripe.
- .Deconstructing the white coat.Ann Intern Med.1998;129:740–742.
- .The art of pimping.JAMA.1989;262(1):89–90.
- ,. “Why Docs don't wear white coats or Polo Shirts at the Mayo Clinic” working knowledge for business leaders. Available at:http://hbswk.hbs.edu/pubitem.jhtml?id=3380309(6970):1710–1712.
- ,,,,.Patient and house officer attitudes on physician attire and etiquette.JAMA.1987;257(1):65–68.
- ,,, et al.Patients' attitude toward consultations by a physician without a white coat in Japan.Intern Med.1999;38(7):533–536.
- ,,, et al.Determinants of white‐coat hypertension.Blood Press Monit.2004;9(6):307–309.
- Doctors ‘should wear white coats.’ BBC news, Thursday, 13 May, 2004. Available at:http://news.bbc.co.uk/2/hi/health/3706783.stm. Accessed May2009.
- .What do Australian junior doctors think of white coats?Med Educ.2002;36(12):1209–1213.
- ,,, ,;ANBP2 Management Committee and Investigators.Second Australian National Blood Pressure Study. ‘Reverse white‐coat hypertension’ in older hypertensives.J Hypertens.2002;20(4):639–644.
- ,,,.Resident physician attire: does it make a difference to our patients?Am J Obstet Gynecol.2004;190(5):1484–1488.
- . Personal communication.1994–2008.
- .Should doctors wear white coats?Med J Aust.2001;174:343–344.
- ,,,,.Does wearing a necktie influence patient perceptions of emergency department care?J Emerg Med.1998;16(4):541–543.
- .Deconstructing the white coat.Ann Intern Med.1998;129:740–742.
- .The art of pimping.JAMA.1989;262(1):89–90.
- ,. “Why Docs don't wear white coats or Polo Shirts at the Mayo Clinic” working knowledge for business leaders. Available at:http://hbswk.hbs.edu/pubitem.jhtml?id=3380309(6970):1710–1712.
- ,,,,.Patient and house officer attitudes on physician attire and etiquette.JAMA.1987;257(1):65–68.
- ,,, et al.Patients' attitude toward consultations by a physician without a white coat in Japan.Intern Med.1999;38(7):533–536.
- ,,, et al.Determinants of white‐coat hypertension.Blood Press Monit.2004;9(6):307–309.
- Doctors ‘should wear white coats.’ BBC news, Thursday, 13 May, 2004. Available at:http://news.bbc.co.uk/2/hi/health/3706783.stm. Accessed May2009.
- .What do Australian junior doctors think of white coats?Med Educ.2002;36(12):1209–1213.
- ,,, ,;ANBP2 Management Committee and Investigators.Second Australian National Blood Pressure Study. ‘Reverse white‐coat hypertension’ in older hypertensives.J Hypertens.2002;20(4):639–644.
- ,,,.Resident physician attire: does it make a difference to our patients?Am J Obstet Gynecol.2004;190(5):1484–1488.
- . Personal communication.1994–2008.
- .Should doctors wear white coats?Med J Aust.2001;174:343–344.
- ,,,,.Does wearing a necktie influence patient perceptions of emergency department care?J Emerg Med.1998;16(4):541–543.
Port‐A‐Cath Embolization
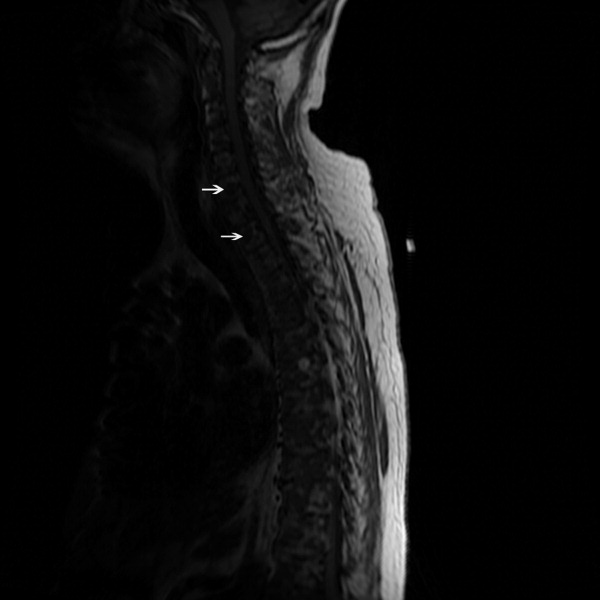
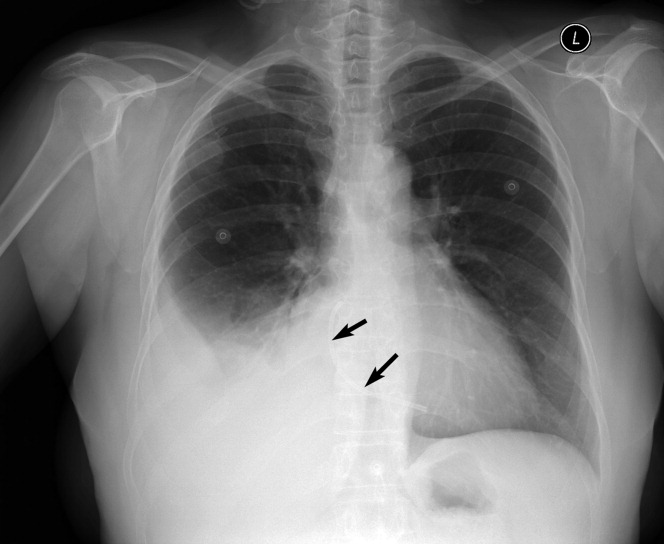
A 59‐year‐old white female, with a 3‐year history of Port‐A‐Cath (PAC) placement for abdominal mesothelioma, presented with 2 episodes of cardiac palpitations. The onset of palpitations occurred 2 days prior to admission, following 15 minutes of vigorous but failed attempts to access the PAC with normal saline and tissue plasminogen activator at her oncologist's office. Although asymptomatic at the time of manipulation, each episode was triggered by subsequent exertion, lasting for about 1 minute, and not associated with chest pain. Electrocardiogram showed normal sinus rhythm and occasional premature ventricular contractions. A chest x‐ray showed a catheter fragment spanning the right atrium and ventricle (Figure 1). Computed tomography (CT) scan confirmed a 10‐cm dislodged catheter (Figure 2). Following emergent catheter retrieval via right‐sided heart catheterization, the patient's symptoms resolved. At least 42 cases of catheter embolization have been reported in the recent literature.1 Of these cases, only 7% had palpitations. Although rare, catheter fracture should be considered in patients with palpitations and history of indwelling venous catheter.

- ,.Ventricular tachycardia secondary to Port‐A‐Cath fracture and embolization.J Emerg Med.2003;24:29–34.
A 59‐year‐old white female, with a 3‐year history of Port‐A‐Cath (PAC) placement for abdominal mesothelioma, presented with 2 episodes of cardiac palpitations. The onset of palpitations occurred 2 days prior to admission, following 15 minutes of vigorous but failed attempts to access the PAC with normal saline and tissue plasminogen activator at her oncologist's office. Although asymptomatic at the time of manipulation, each episode was triggered by subsequent exertion, lasting for about 1 minute, and not associated with chest pain. Electrocardiogram showed normal sinus rhythm and occasional premature ventricular contractions. A chest x‐ray showed a catheter fragment spanning the right atrium and ventricle (Figure 1). Computed tomography (CT) scan confirmed a 10‐cm dislodged catheter (Figure 2). Following emergent catheter retrieval via right‐sided heart catheterization, the patient's symptoms resolved. At least 42 cases of catheter embolization have been reported in the recent literature.1 Of these cases, only 7% had palpitations. Although rare, catheter fracture should be considered in patients with palpitations and history of indwelling venous catheter.
A 59‐year‐old white female, with a 3‐year history of Port‐A‐Cath (PAC) placement for abdominal mesothelioma, presented with 2 episodes of cardiac palpitations. The onset of palpitations occurred 2 days prior to admission, following 15 minutes of vigorous but failed attempts to access the PAC with normal saline and tissue plasminogen activator at her oncologist's office. Although asymptomatic at the time of manipulation, each episode was triggered by subsequent exertion, lasting for about 1 minute, and not associated with chest pain. Electrocardiogram showed normal sinus rhythm and occasional premature ventricular contractions. A chest x‐ray showed a catheter fragment spanning the right atrium and ventricle (Figure 1). Computed tomography (CT) scan confirmed a 10‐cm dislodged catheter (Figure 2). Following emergent catheter retrieval via right‐sided heart catheterization, the patient's symptoms resolved. At least 42 cases of catheter embolization have been reported in the recent literature.1 Of these cases, only 7% had palpitations. Although rare, catheter fracture should be considered in patients with palpitations and history of indwelling venous catheter.
- ,.Ventricular tachycardia secondary to Port‐A‐Cath fracture and embolization.J Emerg Med.2003;24:29–34.
- ,.Ventricular tachycardia secondary to Port‐A‐Cath fracture and embolization.J Emerg Med.2003;24:29–34.
CSI Risk in Infants with Focal Infection
Focal bacterial infections (FBIs), including otitis media (OM), cellulitis, and lymphadenitis, can occur at any age and are usually treated with oral antibiotics. When patients with focal infections present as a young infant, healthcare providers often worry about the risk of coexisting or concomitant systemic infections (CSIs), often termed serious bacterial infections (SBIs) in the published literature.1, 2 Risk of CSI becomes more worrisome when young infants have fever as part of their presentation, especially within the traditional rule‐out sepsis age range of less than 2 months. Often, these patients are well‐appearing and lack prenatal and neonatal risk factors for systemic infections, but are assumed to be at high risk for CSI based on the presence of focal bacterial infection.1, 3, 4
FBIs have been the subject of multiple studies, especially OM and cellulitis. However, few investigations have looked specifically at infants less than 60 days of age, lending uncertainty to medical decision‐making for both community and emergency physicians. Therefore, no standardized evidence‐based practice exists for treating young infants with FBIs. While some clinicians treat these infections using systemic antibiotics without performing laboratory tests, others opt for a full diagnostic evaluation for CSI; including serum blood counts, blood cultures, lumbar punctures (LPs), and bladder catheterizations. Also, some clinicians opt for treatment with oral antibiotics at home, while others choose intravenous (IV) antibiotics and hospitalization.
These decisions are likely due to studies of febrile infants under 60 days of age who present to an emergency department (ED), appear well, and have no focal source of infection on exam, yet are known to have a risk of systemic or occult bacterial infection of roughly 10%.310 There are no such studies documenting the risk of CSI in well‐appearing, afebrile patients, as fever is regarded as the main risk factor for CSI in this age.
Based on our clinical experience and review of the literature, we felt that afebrile, well‐appearing infants less than 60 days of age with an FBI on exam would have a very low risk of CSI. The primary objectives of this study were to determine the risk of CSI when presented with a well‐appearing infant who has an FBI on exam and to examine that risk in relation to the presence of fever. Other objectives were to describe the clinical presentation of FBIs in well‐appearing infants in this age group and to describe the current management and resource utilization of such infections in the ED.
Patients and Methods
Study Population
Cincinnati Children's Hospital Medical Center (CCHMC) is a 423‐bed tertiary‐care hospital located in southwestern Ohio. The medical center serves as the sole pediatric hospital in a 70‐mile radius, serving a metropolitan population of roughly 1 million. In addition, CCHMC cares for patients from a wide demographic and socioeconomic spectrum, including the urban, suburban, and rural areas of Cincinnati, southwestern Ohio, northern Kentucky, and southeastern Indiana. The ED sees an average of 90,000 patients annually, with an average of 15,000 admissions.
Patient Selection
Data were retrospectively collected from a consecutive series of infants age 0 to 59 days who presented to the CCHMC ED between January 1, 2000 and December 31, 2005. Patients met inclusion criteria if they were documented to be well‐appearing on exam; had normal‐for‐age heart rate, respiratory rate, blood pressure, and oxygen saturation (if measured); were discharged from their hospital of birth to home before presentation to the ED; and received a discharge diagnosis consistent with an FBI. Specifically, FBIs included the following diagnoses: soft tissue infection, cellulitis, mastoiditis, abscess, OM, omphalitis, mastitis, mammitis, paronychia, balanitis, posthitis, impetigo, or lymphadenitis. We excluded patients with a history of immunodeficiency, central venous catheter, tracheostomy, gastrostomy tube, chronic lung disease, previous admission for a documented bacterial infection, or if they had been taking systemic antibiotics at the time of evaluation. We also excluded infants noted to be toxic or ill‐appearing on examination.
Definitions
Fever was defined as a temperature greater than or equal to 38C (100.4F), recorded by the ED or by parental report. All infants seen in the CCHMC ED had rectal temperatures measured per protocol. A nontoxic infant was defined by chart review as documentation of: nontoxic, well‐appearing, or alert infant without signs of respiratory distress or hemodynamic instability. Patients were excluded from the nontoxic category with the following parameters: heart rate >180 beats per minute, respiratory rate >60 breaths per minute, oxygen saturation <90% on pulse oximetry, or systolic blood pressure <60 mmHg on 2 or more measurements. CSIs were defined as any of the following: bacteremia, urinary tract infection (UTI) identified by catheterized urine specimen, meningitis, septic arthritis, or osteomyelitis. Additionally, pneumonia was included as a CSI if confirmed by radiographs, based on final reading by an attending radiologist. Pneumonia is included as an SBI in a number of prior publications in this age group; therefore, we felt it was important to include it in our definition of CSI.7, 1115
Procedures
The chart review consisted of both electronic and paper medical records. We searched the electronic database for all the infants in this age group receiving a discharge diagnosis of an FBI as defined above. Information from the medical record was entered onto a standardized data sheet by one of the investigators or a research assistant. Data gathered included baseline demographic information (age at ED evaluation, gestational age at birth, gender, race), presence or absence of fever, vital signs on ED presentation, use of diagnostic evaluation (complete blood count [CBC], blood culture, C‐reactive protein, erythrocyte sedimentation rate, urinalysis, urine culture, LP, wound culture, radiographic studies), laboratory test results, therapeutic interventions (including antibiotics and subspecialty consultation), initial disposition (home, admission to hospital), and clinical outcome of both admitted and discharged patients. The results of all bacterial cultures were obtained from computerized microbiology records. Bacterial isolates from blood cultures that are considered skin flora, such as Staphylococcus epidermidis or Viridans group Streptococcus, were considered contaminants unless they were positive for growth within 24 hours of initial culture.1618 Radiology results were based on final interpretation by an attending radiologist.
Statistical Analysis
The primary objectives of this study were to determine the risk of CSI when presented with a well‐appearing infant who has an FBI on exam and to examine that risk in relation to the presence of fever. Thus, this study was powered to investigate the difference in CSI between febrile and afebrile patients. Again, we hypothesized that in afebrile, well‐appearing infants less than 60 days of age with an FBI on exam there is a very low risk of CSI. For statistical purposes and power calculations we used <1% risk as very low. Using the approximate risk of 10% CSI in well‐appearing, febrile infants this age without CSI, we assumed there would be approximately 10% less CSIs in the afebrile patients compared to the febrile patients. As per consultation with our statistician, for a significance level of = 0.05 and = 0.2 a 2‐sided t test required a total of 188 subjects to be enrolled.
Additional outcome measures (such as use of laboratory testing, IV antibiotics, subspecialty consultation, and admission) were reported as descriptive statistics with frequencies. Fisher's Exact test was used for these nominal variables to test significance of relationship between febrile and afebrile patients. The Exact method was used to calculate 95% confidence intervals (CIs) around the primary outcome point estimates. A P value < 0.05 was considered significant. All data were entered into a Microsoft Excel (Microsoft, Redmond, WA) database.
Results
Study Population
We identified 246 patients less than 60 days of age who were diagnosed in the ED with an FBI during the study period. Thirty‐seven patients met the exclusion criteria, leaving 209 patients for potential enrollment. Twelve (5.7%) charts were not located, resulting in a study population of 197 infants. Of these, 158 patients were afebrile and 39 were febrile. To put these numbers into context, over the study period our ED evaluated 2341 infants less than 60 days of age whose diagnosis included the word fever, febrile, or sepsis. The febrile infants included in our study group were part of these 2341 found in the retrospective search, but the afebrile infants included were not.
The mean age of these 197 infants was 29.6 days, with a standard deviation (SD) of 15.9 days. On average, the febrile patients (mean 40.7 days, SD 15.3 days) were older than the afebrile patients (mean 26.8 days, SD 16.1 days). No statistical differences were found between febrile and afebrile patients in respect to gender, race, and gestational age.
Primary Study OutcomeRisk of SBI
Four patients had a documented CSI, for an overall risk of CSI in our study population of 2.0% (Table 1). Statistically, febrile infants had a significantly higher risk of CSI (odds ratio [OR], 13.08; 95% CI, 1.32‐129.46) than afebrile infants. The febrile infants with a coexisting CSI included the following: a 32‐day‐old male with a buttock abscess and E. coli UTI; a 47‐day‐old male with periorbital cellulitis and Streptococcus pneumoniae bacteremia; and a 21‐day‐old female with trunk cellulitis and E. coli UTI. The afebrile infant with a CSI was an 11‐day‐old male with acute OM (AOM) and E. coli UTI. No cases of bacterial meningitis, septic arthritis, osteomyelitis, radiographic pneumonia, or death occurred.
| CSI | No CSI | Risk (%) | 95% CI (%) | |
|---|---|---|---|---|
| ||||
| Febrile (n = 39) | 3 | 36 | 7.69 | 1.62‐20.87 |
| Afebrile (n = 158) | 1 | 157 | 0.63 | 0.02‐3.48 |
| Total (n = 197) | 4 | 193 | 2.03 | 0.56‐5.12 |
Eight other patients had positive culture results, all of which were considered contaminants. Four patients had peripheral blood cultures that were deemed contaminants: 2 cases of Viridans group streptococcus and 2 cases of coagulase‐negative staphylococcus. Four urine cultures were considered contaminants: 1 staphylococcus species; 1 mixed flora specimen; 1 lactobacillus species, and 1 E. coli.
This positive culture for E. coli occurred in an 11‐day‐old female who presented without fever, was diagnosed clinically and by ultrasound with mastitis, and was admitted to the hospital. She underwent a full diagnostic workup for CSI. Initial screening laboratory test results were normal, including a negative urinalysis and 1 to 2 white blood cells (WBCs)/high‐powered field on the microscopic exam. The catheterized urine specimen grew the following: (1) 10,000 to 100,000 colony forming units (CFU)/mL of E. coli; and (2) normal flora 1000 to 9000 CFU/mL. Her blood and cerebrospinal fluid (CSF) culture were negative. The primary team did not diagnose or treat this patient as a UTI despite the positive culture. According to their documentation, it was a suspected contaminant because of negative chemical and microscopic urinalysis. If this were considered a true positive culture, which seem practical clinically, clinically the overall risk of CSI would be 2.5% (5/197 infants) and the risk of coexisting CSI in afebrile patients would be 1.3% (2/158 infants); however, febrile infants would still had a significantly higher risk of CSI (OR, 6.5; 95% CI, 1.05‐40.34) than afebrile infants.
Secondary Study OutcomeClinical Presentation of FBI in Well‐appearing Infants in This Age Group
Among the 197 study infants, there were multiple types of FBIs found on examination (Table 2). Soft tissue infections were the most common cause in afebrile patients, whereas AOM was the most common cause in the febrile patients. Overall, cellulitis and abscess were the leading causes of focal infections in this age group. A total of 46 patients were diagnosed with abscesses, the buttock being the site in 24 (52%) cases. Abscesses were drained and sent for culture in 13 patients. Methicillin‐resistant Staphylococcus aureus (MRSA) was found in 4 of these 13 cultures. Of the 55 patients diagnosed with cellulitis, 20 (36%) had cultures drawn from the cellulitis site, of which 5 cultures grew MRSA.
| Focal Infection | Afebrile (n = 158) [n (%)] | Febrile (n = 39) [n (%)] | Totals (n = 197) [n (%)] |
|---|---|---|---|
| |||
| AOM | 19 (12) | 22 (56) | 41 (21) |
| Cellulitis | 49 (31) | 6 (15) | 55 (28) |
| Abscess | 40 (25) | 6 (15) | 46 (23) |
| Impetigo | 20 (13) | 1 (3) | 21 (11) |
| Mastitis | 11 (7) | 2 (5) | 13 (7) |
| Lymphadenitis | 4 (3) | 1 (3) | 5 (3) |
| Omphalitis | 2 (1) | 0 (0) | 2 (1) |
| SSSS | 2 (1) | 0 (0) | 2 (1) |
| Paronychia | 1 (1) | 0 (0) | 1 (0) |
| Other | 10 (6) | 1 (3) | 11 (6) |
Of note, a 13‐day‐old male presenting without fever but with a scalp lesion had herpes simplex virus grown from his wound culture. His diagnostic workup was negative for CSI. Omphalitis, uncommon in the United States, was diagnosed in 2 patients in our study and neither of these patients developed necrotizing fasciitis. Mastitis was not a significant focal infection in either group; and no patient was diagnosed with mastoiditis, balanitis, or posthitis.
Secondary Study OutcomeCurrent Management of FBI in the ED: Resource Utilization
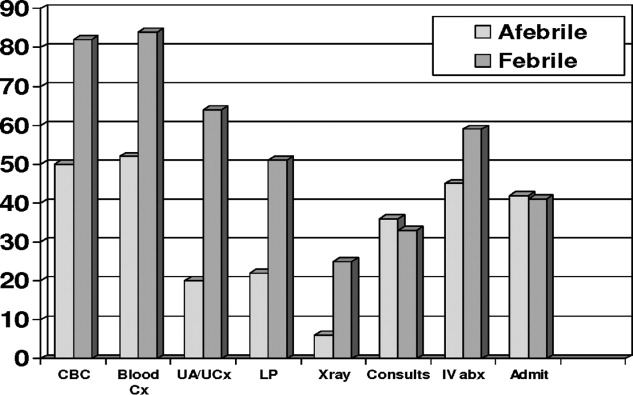
Febrile patients underwent significantly more diagnostic testing procedures than afebrile infants. Specifically, CBCs, peripheral blood cultures, urine analyses with cultures, LPs, and radiographic imaging were performed more often in infants with fever (Figure 1). Fifty‐seven infants had urine cultures performed: 32 of 158 afebrile infants and 25 of 39 febrile infants. Among these selected infants, again 3 UTIs occurred: 1 of 32 tested (3.1%) in the afebrile group and 2 of 25 tested (8%) in the febrile group. A total of 115 infants had blood cultures performed: 82 of the 158 afebrile infants and 33 of the 39 febrile infants. Among these selected infants, again 1 positive blood culture occurred: 0 of 82 tested (0%) in the afebrile group and 1 of 33 tested (3%) in the febrile group. Fifty‐five infants had LP performed: 35 of 158 afebrile infants and 30 of 39 febrile infants. No case of meningitis occurred in these selected infants. Twenty infants had diagnostic radiographs performed, 10 in the febrile group and 10 in the afebrile group. None of the radiographs showed signs of pneumonia on final radiology attending reading.
Overall, 33 of 39 febrile infants had cultures drawn and/or radiographs performed, with 3 CSIs discovered, and 87 of 158 afebrile infants had cultures drawn and/or radiographs performed, with 1 CSI discovered (Table 3). This means that 77 infants diagnosed with FBIs did not have any diagnostic testing for CSI performed while in the ED. The overall risk of CSI in patients who were tested was 3.3%. Comparing febrile and afebrile patients who had diagnostic workups performed, febrile infants had a trend toward increased risk of CSI (OR, 8.6; 95% CI, 0.8613‐85.8686).
| CSI | No CSI | Risk (%) | |
|---|---|---|---|
| |||
| Febrile (n = 33) | 3 | 30 | 9.1 |
| Afebrile (n = 87) | 1 | 86 | 1.1 |
| Totals (n = 120) | 4 | 116 | 3.3 |
No significant differences existed in the use of consultants, use of IV antibiotics, or admission rate. Sixty‐six (42%) of the 158 afebrile infants were admitted. The average length of stay for these infants was 2.59 days (range, 1‐22 days). This included a 22‐day‐old female with mastitis who required multiple surgeries and a protracted hospital course. Sixteen (41%) of the febrile infants were admitted and had an average stay of 2.87 days (range, 1‐6 days).
Patient Outcomes
Ninety‐two (58%) of the afebrile infants were discharged home from the ED, of which 5 (5.4%) returned to the ED within 72 hours after discharge. Three of these were planned ED returns as follow‐up with a primary care physician could not be guaranteed. The fourth was a 47‐day‐old with a buttock abscess who returned for reevaluation because of parental concern. The fifth was the 11‐day‐old male initially diagnosed with AOM who was called back to the ED when his urine culture grew E. coli, at which point he was admitted to the hospital. Twenty‐three (59%) of the febrile infants were discharged home from the ED. Two (8.7%) of these 23 returned to the ED within 72 hours; both were initially diagnosed with AOM and had planned follow‐ups as no primary care was available. No admitted patients required transfer to the pediatric intensive care unit and no deaths occurred.
Discussion
This study is the first to investigate the risk of CSI specifically in well‐appearing infants under 60 days of age presenting with FBI. Inclusion criteria were not limited to 1 specific infection, ie, OM, but included all FBI. This study demonstrated that the risk of CSI in afebrile infants is small (0.6%), which is similar to what has been shown in the OM literature.19, 20 However, the risk of CSI among febrile infants is not negligible (7.7%), and is similar to the risk of SBIs among young infants presenting with a fever of unknown source.57
AOM
A total of 41 infants were diagnoses with AOM. Indeed, AOM was the most common discharge diagnosis among the febrile cohort. No febrile infants with AOM were diagnosed with a CSI. However, as noted above, an 11‐day‐old afebrile infant with AOM did have a UTI diagnosed. This male underwent blood, urine, and CSF cultures on initial presentation. He was discharged home from the ED, and called back for further evaluation once the urine culture grew E. coli.
We relied on the diagnostic skills of the ED attending physician for determination of OM, and no rigorous diagnostic criteria were established for an AOM in this young age group. However, on chart review of those patients with AOM who were admitted, otolaryngology was often consulted and confirmed the diagnosis of OM.
AOM has been the most commonly studied focal infection studied most in this age group.1, 2, 11, 2123 More recent studies have investigated the risk of CSI with AOM among febrile and afebrile cohorts. These authors found that in infants under 2 months with AOM, febrile infants had a 6% to 13% risk of CSI, whereas no cases of CSI were found in afebrile patients.19, 20 These results suggest that afebrile, nontoxic infants with AOM could be treated as outpatients without need for further systemic evaluation. Our study strengthens this argument if close follow‐up can be assured.
Cellulitis and Abscess
A total of 106 infants were diagnosed with either abscess and/or cellulitis, with the vast majority of them presenting without fever. Moreover, cellulitis and abscess comprised more than one‐half of all infants in the afebrile cohort. The buttocks were the most common site for abscess (23 cases), followed next by the scalp (8 cases). In our study population, there were 29 positive abscess, wound, lesion, or skin aspirate cultures for Staphylococcus aureus, with 13 (45%) being MRSA. None of these patients had a CSI.
Community‐acquired staphylococcal infections have been studied in the newborn population, with MRSA accounting for up to 69% of soft‐tissue infections in one recent study involving 89 neonates.24 The authors found 4 cases of bacteremia, 3 cases of UTI, and 2 cases of CSF pleocytosis. Application of these results to our patients is difficult, as there was no mention of fever or well‐appearance stratification and there is no specific mention of any of the 77 skin and soft‐tissue infections having a CSI.
The frequency of bacteremia in pediatric patients with periorbital cellulitis has been studied, with some authors suggested more selective use of LP in patients with these localized soft‐tissue infections in the older infant or child.25, 26 Unfortunately, there were not enough patients under 60 days of age in either study from which to draw conclusions. In our study, a total of 5 patients presented with periorbital cellulitis: 2 afebrile and 3 febrile, including the 47‐day‐old febrile infant with coexisting pneumococcal bacteremia.
Group B streptococcus (GBS) cellulitis‐adenitis syndrome has also been identified in neonates, with meningitis occurring in up to 24% of infants in 1 review of 32 cases.27 These studies only included patients with positive blood cultures, providing no denominator for assessing a true CSI risk in young infants with cellulitis. No infants in our study had GBS infections.
Impetigo was diagnosed in 21 patients, with 17 patients being discharged home from the ED. In addition, 2 infants with staphylococcal scalded skin syndrome were identified. None of these infants had a CSI.
Other Infections
Mastitis in the young infant has been studied, with the risk of CSI being 3% in 1 review.2830 In our study, there were 18 patients diagnosed with either mastitis or breast abscess, of which 4 (22%) had fever and none had positive blood or CSF cultures. Multiple studies, either from older literature in the United States or more contemporary from abroad, have investigated omphalitis and necrotizing fasciitis in the newborn.31, 32 These show that infants presenting with omphalitis have a lower mortality and morbidity than those with necrotizing fasciitis. In our study, we had 2 patients with omphalitis, both were afebrile and neither had a coexisting CSI. We had no patients with necrotizing fasciitis. We also found no patients with mastoiditis, balanitis, or posthitis as the listed diagnosis in our study.
UTI
Of the 4 cases of CSI in our study, UTI was the most common, occurring in 3 of the 4 cases. This predominance of UTI as the cause of CSI is well‐recognized in young febrile infants with OM and infants with fever of unknown origin; however, it has not been reported with other FBIs in this age group.1, 5, 9, 11, 19, 33 Specifically, there has not been a reported relationship between UTI and skin or soft tissue infections. However, 2 of our 4 infants with a CSI had a UTI in addition to their skin or soft tissue infection: the 32‐day‐old male with a buttock abscess and E. coli UTI; and the 21‐day‐old female with truncal cellulitis and E. coli UTI.
Resource Utilization
Young infants with FBI and fever underwent significantly more resource utilization than afebrile infants in our ED (Figure 1). Physicians of febrile infants ordered more serum blood counts and blood cultures, urinalysis and urine cultures, and performed more LPs and radiographic studies. This is not surprising given that fever is a risk factor for CSI in this age group; however, resource utilization is not as well‐described in previous reports on infants with FBIs. In 1 retrospective study of 137 infants less than 8 weeks of age with AOM, 88% had diagnostic blood cultures, 66% had LP, and 81% had urine cultures drawn.19 Additionally, in our study the decision to hospitalize infants, use of IV antibiotics, and hospital length of stay was not statistically different between febrile and afebrile groups. This potentially suggests that diagnostic testing for infants with FBIs did not affect management and outcome. Another study showed hospital admission is costly and not without safety risks, with a complication rate as high as 19% in infants.34 Given these results, in addition to the presumption that all FBIs would be treated with antibiotics, one could infer that a number of well‐appearing, afebrile infants underwent unwarranted testing.
Limitations
The main limitation of our study was the retrospective design. A research assistant utilizing a computerized database program of ED patients performed the initial query based upon search criteria. A local expert using an established list of International Statistical Classification of Diseases and Related Health Problems (ICD) codes for any and all of the FBIs performed the chart review. Secondary diagnoses may have been omitted as a result of limited physician charting, which could have resulted in missed patients. CCHMC is currently transitioning from paper records to electronic medical records, which may explain why 12 charts were not found, despite exhaustive searches for them. Ultimately, 94% of all the eligible patients were found. Yet, with so few CSI diagnosed, 1 or 2 could conceivably change our results. As noted above, focal infections in infants under 2 months of age seem to be uncommon, not only in the ED setting, but also in a large cohort of primary care patients.35 With only a small number FBIs present over 6 years and so few having CSIs, we were limited to a univariate analysis, which limited our potential results and conclusions. Performing this study prospectively would have addressed these limitations, but would have required years of recruitment or multicenter collaboration.
Another potential limitation was the search strategy to identify eligible infants. Specifically, we did not search the records for infants diagnosed with UTI, bacteremia, or meningitis. Infants with these more serious conditions may have been coded as the primary or only problem, even if other problems such as OM were also present. Therefore, we may have underestimated the occurrence of systemic bacterial complications in a systematic way. This form of spectrum bias was a potential limitation. We approached this clinical question from the eye of the emergency department practitioner and thus used a search strategy based on the discharge diagnosis from the ED. If we were to look back to see if any patients discharged from the hospital with CSI also had FBI, we doubt our retrospective design would have allowed identification of patients with FBIs.
Other limitations may be our definition of CSI and the potential for missed CSIs. We defined meningitis, bacteremia, and UTI as a positive CSF, blood, or urine culture, respectively. Many studies are now defining culture‐negative meningitis or UTI based on WBC findings on a screening cell count or urine microscopy. However, because no patients were pretreated with antibiotics, we felt that a positive culture from a sterile site was a reasonable definition. Also, not all of the enrolled patients received full diagnostic workups to screen for CSI. In the smaller cohort of infants who received diagnostic testing, febrile infants still seemed to be at higher risk for CSI. To capture all possible CSIs, one would have needed to perform blood culture, suprapubic bladder aspiration, or catheterization for urine culture, LP, and chest radiograph on all patients with an FBI. Given the low risk of CSI in the United States, the previous literature's hinting at low risk for CSI in afebrile patients, and the presence of multiple clinical trials defining low risk characteristics for CSI, such a protocol would not receive support as being standard clinical practice and have difficulty being permitted by an institutional review board.
Another limitation of our study was follow‐up. Ideally, every patient would have been contacted to ensure reliable outcomes measurement. Patients may have been seen after their initial evaluation at their primary care provider's office or at another healthcare facility and had more diagnostic studies performed. However, because our institution is the sole admitting pediatric center in the area, particularly in infants, and most CSIs in this age group require admission; we feel that a reasonable degree of reliability for the outcomes of interest was maintained.
Last, we could not establish the rationale of clinical decision making, as our study was retrospective. For example, staff physicians may have foregone diagnostic testing if they had committed to a therapeutic course of antibiotics and felt diagnostic cultures would not change management. Also, we cannot ensure that interpretation of culture results was consistent with national standards. As described previously, 1 afebrile infant in our study potentially had a CSI that was clinically felt to be a contaminant by the inpatient healthcare team. This 11‐day‐old female was admitted with mastitis and had a negative urinalysis, but grew E. coli on urine culture. This was treated as a contaminant by the ward team, despite her age.
Conclusions
CSI is very uncommon in afebrile, well‐appearing infants under 2 months of age with an FBI. However, febrile infants have a greater risk of CSI. UTI is the most common CSI in well‐appearing infants less than 2 months of age with an FBI found on examination. Prospective, multicentered studies need to be performed to determine whether diagnostic evaluation should change the management of well‐appearing infants with an FBI found on examination.
- ,,, et al.Febrile infants at low risk for serious bacterial infection—an appraisal of the Rochester Criteria and implications for management.Pediatrics.1994;94:390–396.
- ,,, et al.Identification of infants unlikely to have serious bacterial infection although hospitalized for suspected sepsis.J Pediatr.1985;107:855–860.
- ,,.Outpatient management without antibiotics of fever in selected infants.N Engl J Med.1993;329:1437–1441.
- ,,.Outpatient treatment of febrile infants 28 to 89 days of age with intramuscular administration of ceftriaxone.J Pediatr.1992;120:22–27.
- ,.Predictive model for serious bacterial infections among infants younger than 3 months of age.Pediatrics.2001;108:311–316.
- ,,, et al.Applying outpatient protocols to febrile infants 1–28 days of age: can the threshold be lowered?Clin Pediatr.2000;39:81–88.
- ,,.The efficacy of routine outpatient management without antibiotics of fever in selected infants.Pediatrics.1999;103:627–631.
- ,.Evaluation of the infant with fever without source: an evidence based approach.Emerg Med Clin North Am.1999;17:97–126.
- ,,, et al.Risk of serious bacterial infection in young febrile infants with respiratory syncytial virus infections.Pediatrics.2004;113:1728–1734.
- ,,, et al.Relationship of fever magnitude to rate of serious bacterial infections in infants 4–8 weeks.Clin Pediatr (Phila).1991;30:478–480.
- ,,, et al.Enhanced urinalysis improved identification of febrile infants ages 60 days and younger at low risk for serious bacterial illness.Pediatrics.2001;108:866–871.
- ,,.C‐reactive protein in febrile children 1 to 36 months of age with clinically undetectable serious bacterial infection.Pediatrics.2001;108:1275–1279.
- ,.Utility of the serum C‐reactive protein for detection of occult bacterial infection in children.Arch Pediatr Adolesc Med.2002;156:905–909.
- ,,, et al.Procalcitonin and C‐reactive protein as diagnostic markers of severe bacterial infections in febrile infants and children in the emergency department.Pediatr Infect Dis J.2007;26:672–677.
- ,,.Neonatal fever: utility of the Rochester criteria in determining low risk for serious bacterial infections.Am J Emerg Med.1997;15:299–302.
- ,,.Evaluation of false positive blood cultures: guidelines for early detection of contaminated cultures in febrile children.Pediatr Emerg Care.1994;10:20–22.
- ,,.Outpatient pediatric blood cultures: time to positivity.Pediatrics.2000;106:251–255.
- ,,, et al.Occult bacteremia from a pediatric emergency department: current prevalence, time to detection and outcome.Pediatrics.2000;106:505–511.
- ,,, et al.Acute otitis media in infants younger than two months of age: microbiology, clinical presentation and therapeutic approach.Pediatr Infect Dis J.2002;21:669–674.
- ,,, et al.Otitis media in infants aged 0–8 weeks: frequency of associated serious bacterial disease.Pediatr Emerg Care.1999;15:252–254.
- ,,.Otitis media in children less than 12 weeks of age.Pediatrics.1977;59:827–832.
- ,,.Are well‐appearing febrile infants with otitis media at risk for serious bacterial illness? [Abstract].Am J Dis Child.1992;146:468.
- ,,, et al.Streptococcus pneumoniae infections in the neonate.Pediatrics.2003;112:1095–1102.
- ,,, et al.Community‐acquired Staphylococcus aureus infections in term and near‐term previously health neonates.Pediatrics.2007;118:874–881.
- ,.Blood cultures in the evaluation of children with cellulitis.Pediatrics.1997;101:e4.
- ,.Lumbar puncture in children with periorbital and orbital cellulitis.J Pediatr.1993;122:355–359.
- ,.Is lumbar puncture necessary to exclude meningitis in neonates and young infants: lessons from the Group B streptococcus cellulitis‐adenitis syndrome.Pediatrics.1998;102:985–986.
- ,.Neonatal mastitis.Clin Pediatr.1986;25:395–399.
- ,.Pseudomonas aeruginosa mastitis in a neonate.Pediatr Infect Dis J.1993;12:104.
- ,,.Group D streptococcal neonatal mastitis.Pediatr Infect Dis J.1998;7:362.
- ,,, et al.Early recognition of neonatal abdominal wall necrotizing fasciitis.Am J Surg.1994;167:481–484.
- ,,, et al.Necrotizing fasciitis: a serious complication of omphalitis in neonates.J Pediatr Surg.1994;29:1414–1416.
- ,,, et al.Acute otitis media in infants less than three months of age: clinical presentation, etiology and concomitant diseases.Int J Pediatr Otorhinolaryngol.2006;70:613–617.
- ,,, et al.Iatrogenic risks and financial costs of hospitalizing febrile infants.Am J Dis Child.1983;137:1146–1149.
- ,,, et al.Management and outcomes of care of fever in early infancy.JAMA.2004;291:1203–1212.
Focal bacterial infections (FBIs), including otitis media (OM), cellulitis, and lymphadenitis, can occur at any age and are usually treated with oral antibiotics. When patients with focal infections present as a young infant, healthcare providers often worry about the risk of coexisting or concomitant systemic infections (CSIs), often termed serious bacterial infections (SBIs) in the published literature.1, 2 Risk of CSI becomes more worrisome when young infants have fever as part of their presentation, especially within the traditional rule‐out sepsis age range of less than 2 months. Often, these patients are well‐appearing and lack prenatal and neonatal risk factors for systemic infections, but are assumed to be at high risk for CSI based on the presence of focal bacterial infection.1, 3, 4
FBIs have been the subject of multiple studies, especially OM and cellulitis. However, few investigations have looked specifically at infants less than 60 days of age, lending uncertainty to medical decision‐making for both community and emergency physicians. Therefore, no standardized evidence‐based practice exists for treating young infants with FBIs. While some clinicians treat these infections using systemic antibiotics without performing laboratory tests, others opt for a full diagnostic evaluation for CSI; including serum blood counts, blood cultures, lumbar punctures (LPs), and bladder catheterizations. Also, some clinicians opt for treatment with oral antibiotics at home, while others choose intravenous (IV) antibiotics and hospitalization.
These decisions are likely due to studies of febrile infants under 60 days of age who present to an emergency department (ED), appear well, and have no focal source of infection on exam, yet are known to have a risk of systemic or occult bacterial infection of roughly 10%.310 There are no such studies documenting the risk of CSI in well‐appearing, afebrile patients, as fever is regarded as the main risk factor for CSI in this age.
Based on our clinical experience and review of the literature, we felt that afebrile, well‐appearing infants less than 60 days of age with an FBI on exam would have a very low risk of CSI. The primary objectives of this study were to determine the risk of CSI when presented with a well‐appearing infant who has an FBI on exam and to examine that risk in relation to the presence of fever. Other objectives were to describe the clinical presentation of FBIs in well‐appearing infants in this age group and to describe the current management and resource utilization of such infections in the ED.
Patients and Methods
Study Population
Cincinnati Children's Hospital Medical Center (CCHMC) is a 423‐bed tertiary‐care hospital located in southwestern Ohio. The medical center serves as the sole pediatric hospital in a 70‐mile radius, serving a metropolitan population of roughly 1 million. In addition, CCHMC cares for patients from a wide demographic and socioeconomic spectrum, including the urban, suburban, and rural areas of Cincinnati, southwestern Ohio, northern Kentucky, and southeastern Indiana. The ED sees an average of 90,000 patients annually, with an average of 15,000 admissions.
Patient Selection
Data were retrospectively collected from a consecutive series of infants age 0 to 59 days who presented to the CCHMC ED between January 1, 2000 and December 31, 2005. Patients met inclusion criteria if they were documented to be well‐appearing on exam; had normal‐for‐age heart rate, respiratory rate, blood pressure, and oxygen saturation (if measured); were discharged from their hospital of birth to home before presentation to the ED; and received a discharge diagnosis consistent with an FBI. Specifically, FBIs included the following diagnoses: soft tissue infection, cellulitis, mastoiditis, abscess, OM, omphalitis, mastitis, mammitis, paronychia, balanitis, posthitis, impetigo, or lymphadenitis. We excluded patients with a history of immunodeficiency, central venous catheter, tracheostomy, gastrostomy tube, chronic lung disease, previous admission for a documented bacterial infection, or if they had been taking systemic antibiotics at the time of evaluation. We also excluded infants noted to be toxic or ill‐appearing on examination.
Definitions
Fever was defined as a temperature greater than or equal to 38C (100.4F), recorded by the ED or by parental report. All infants seen in the CCHMC ED had rectal temperatures measured per protocol. A nontoxic infant was defined by chart review as documentation of: nontoxic, well‐appearing, or alert infant without signs of respiratory distress or hemodynamic instability. Patients were excluded from the nontoxic category with the following parameters: heart rate >180 beats per minute, respiratory rate >60 breaths per minute, oxygen saturation <90% on pulse oximetry, or systolic blood pressure <60 mmHg on 2 or more measurements. CSIs were defined as any of the following: bacteremia, urinary tract infection (UTI) identified by catheterized urine specimen, meningitis, septic arthritis, or osteomyelitis. Additionally, pneumonia was included as a CSI if confirmed by radiographs, based on final reading by an attending radiologist. Pneumonia is included as an SBI in a number of prior publications in this age group; therefore, we felt it was important to include it in our definition of CSI.7, 1115
Procedures
The chart review consisted of both electronic and paper medical records. We searched the electronic database for all the infants in this age group receiving a discharge diagnosis of an FBI as defined above. Information from the medical record was entered onto a standardized data sheet by one of the investigators or a research assistant. Data gathered included baseline demographic information (age at ED evaluation, gestational age at birth, gender, race), presence or absence of fever, vital signs on ED presentation, use of diagnostic evaluation (complete blood count [CBC], blood culture, C‐reactive protein, erythrocyte sedimentation rate, urinalysis, urine culture, LP, wound culture, radiographic studies), laboratory test results, therapeutic interventions (including antibiotics and subspecialty consultation), initial disposition (home, admission to hospital), and clinical outcome of both admitted and discharged patients. The results of all bacterial cultures were obtained from computerized microbiology records. Bacterial isolates from blood cultures that are considered skin flora, such as Staphylococcus epidermidis or Viridans group Streptococcus, were considered contaminants unless they were positive for growth within 24 hours of initial culture.1618 Radiology results were based on final interpretation by an attending radiologist.
Statistical Analysis
The primary objectives of this study were to determine the risk of CSI when presented with a well‐appearing infant who has an FBI on exam and to examine that risk in relation to the presence of fever. Thus, this study was powered to investigate the difference in CSI between febrile and afebrile patients. Again, we hypothesized that in afebrile, well‐appearing infants less than 60 days of age with an FBI on exam there is a very low risk of CSI. For statistical purposes and power calculations we used <1% risk as very low. Using the approximate risk of 10% CSI in well‐appearing, febrile infants this age without CSI, we assumed there would be approximately 10% less CSIs in the afebrile patients compared to the febrile patients. As per consultation with our statistician, for a significance level of = 0.05 and = 0.2 a 2‐sided t test required a total of 188 subjects to be enrolled.
Additional outcome measures (such as use of laboratory testing, IV antibiotics, subspecialty consultation, and admission) were reported as descriptive statistics with frequencies. Fisher's Exact test was used for these nominal variables to test significance of relationship between febrile and afebrile patients. The Exact method was used to calculate 95% confidence intervals (CIs) around the primary outcome point estimates. A P value < 0.05 was considered significant. All data were entered into a Microsoft Excel (Microsoft, Redmond, WA) database.
Results
Study Population
We identified 246 patients less than 60 days of age who were diagnosed in the ED with an FBI during the study period. Thirty‐seven patients met the exclusion criteria, leaving 209 patients for potential enrollment. Twelve (5.7%) charts were not located, resulting in a study population of 197 infants. Of these, 158 patients were afebrile and 39 were febrile. To put these numbers into context, over the study period our ED evaluated 2341 infants less than 60 days of age whose diagnosis included the word fever, febrile, or sepsis. The febrile infants included in our study group were part of these 2341 found in the retrospective search, but the afebrile infants included were not.
The mean age of these 197 infants was 29.6 days, with a standard deviation (SD) of 15.9 days. On average, the febrile patients (mean 40.7 days, SD 15.3 days) were older than the afebrile patients (mean 26.8 days, SD 16.1 days). No statistical differences were found between febrile and afebrile patients in respect to gender, race, and gestational age.
Primary Study OutcomeRisk of SBI
Four patients had a documented CSI, for an overall risk of CSI in our study population of 2.0% (Table 1). Statistically, febrile infants had a significantly higher risk of CSI (odds ratio [OR], 13.08; 95% CI, 1.32‐129.46) than afebrile infants. The febrile infants with a coexisting CSI included the following: a 32‐day‐old male with a buttock abscess and E. coli UTI; a 47‐day‐old male with periorbital cellulitis and Streptococcus pneumoniae bacteremia; and a 21‐day‐old female with trunk cellulitis and E. coli UTI. The afebrile infant with a CSI was an 11‐day‐old male with acute OM (AOM) and E. coli UTI. No cases of bacterial meningitis, septic arthritis, osteomyelitis, radiographic pneumonia, or death occurred.
| CSI | No CSI | Risk (%) | 95% CI (%) | |
|---|---|---|---|---|
| ||||
| Febrile (n = 39) | 3 | 36 | 7.69 | 1.62‐20.87 |
| Afebrile (n = 158) | 1 | 157 | 0.63 | 0.02‐3.48 |
| Total (n = 197) | 4 | 193 | 2.03 | 0.56‐5.12 |
Eight other patients had positive culture results, all of which were considered contaminants. Four patients had peripheral blood cultures that were deemed contaminants: 2 cases of Viridans group streptococcus and 2 cases of coagulase‐negative staphylococcus. Four urine cultures were considered contaminants: 1 staphylococcus species; 1 mixed flora specimen; 1 lactobacillus species, and 1 E. coli.
This positive culture for E. coli occurred in an 11‐day‐old female who presented without fever, was diagnosed clinically and by ultrasound with mastitis, and was admitted to the hospital. She underwent a full diagnostic workup for CSI. Initial screening laboratory test results were normal, including a negative urinalysis and 1 to 2 white blood cells (WBCs)/high‐powered field on the microscopic exam. The catheterized urine specimen grew the following: (1) 10,000 to 100,000 colony forming units (CFU)/mL of E. coli; and (2) normal flora 1000 to 9000 CFU/mL. Her blood and cerebrospinal fluid (CSF) culture were negative. The primary team did not diagnose or treat this patient as a UTI despite the positive culture. According to their documentation, it was a suspected contaminant because of negative chemical and microscopic urinalysis. If this were considered a true positive culture, which seem practical clinically, clinically the overall risk of CSI would be 2.5% (5/197 infants) and the risk of coexisting CSI in afebrile patients would be 1.3% (2/158 infants); however, febrile infants would still had a significantly higher risk of CSI (OR, 6.5; 95% CI, 1.05‐40.34) than afebrile infants.
Secondary Study OutcomeClinical Presentation of FBI in Well‐appearing Infants in This Age Group
Among the 197 study infants, there were multiple types of FBIs found on examination (Table 2). Soft tissue infections were the most common cause in afebrile patients, whereas AOM was the most common cause in the febrile patients. Overall, cellulitis and abscess were the leading causes of focal infections in this age group. A total of 46 patients were diagnosed with abscesses, the buttock being the site in 24 (52%) cases. Abscesses were drained and sent for culture in 13 patients. Methicillin‐resistant Staphylococcus aureus (MRSA) was found in 4 of these 13 cultures. Of the 55 patients diagnosed with cellulitis, 20 (36%) had cultures drawn from the cellulitis site, of which 5 cultures grew MRSA.
| Focal Infection | Afebrile (n = 158) [n (%)] | Febrile (n = 39) [n (%)] | Totals (n = 197) [n (%)] |
|---|---|---|---|
| |||
| AOM | 19 (12) | 22 (56) | 41 (21) |
| Cellulitis | 49 (31) | 6 (15) | 55 (28) |
| Abscess | 40 (25) | 6 (15) | 46 (23) |
| Impetigo | 20 (13) | 1 (3) | 21 (11) |
| Mastitis | 11 (7) | 2 (5) | 13 (7) |
| Lymphadenitis | 4 (3) | 1 (3) | 5 (3) |
| Omphalitis | 2 (1) | 0 (0) | 2 (1) |
| SSSS | 2 (1) | 0 (0) | 2 (1) |
| Paronychia | 1 (1) | 0 (0) | 1 (0) |
| Other | 10 (6) | 1 (3) | 11 (6) |
Of note, a 13‐day‐old male presenting without fever but with a scalp lesion had herpes simplex virus grown from his wound culture. His diagnostic workup was negative for CSI. Omphalitis, uncommon in the United States, was diagnosed in 2 patients in our study and neither of these patients developed necrotizing fasciitis. Mastitis was not a significant focal infection in either group; and no patient was diagnosed with mastoiditis, balanitis, or posthitis.
Secondary Study OutcomeCurrent Management of FBI in the ED: Resource Utilization
Febrile patients underwent significantly more diagnostic testing procedures than afebrile infants. Specifically, CBCs, peripheral blood cultures, urine analyses with cultures, LPs, and radiographic imaging were performed more often in infants with fever (Figure 1). Fifty‐seven infants had urine cultures performed: 32 of 158 afebrile infants and 25 of 39 febrile infants. Among these selected infants, again 3 UTIs occurred: 1 of 32 tested (3.1%) in the afebrile group and 2 of 25 tested (8%) in the febrile group. A total of 115 infants had blood cultures performed: 82 of the 158 afebrile infants and 33 of the 39 febrile infants. Among these selected infants, again 1 positive blood culture occurred: 0 of 82 tested (0%) in the afebrile group and 1 of 33 tested (3%) in the febrile group. Fifty‐five infants had LP performed: 35 of 158 afebrile infants and 30 of 39 febrile infants. No case of meningitis occurred in these selected infants. Twenty infants had diagnostic radiographs performed, 10 in the febrile group and 10 in the afebrile group. None of the radiographs showed signs of pneumonia on final radiology attending reading.
Overall, 33 of 39 febrile infants had cultures drawn and/or radiographs performed, with 3 CSIs discovered, and 87 of 158 afebrile infants had cultures drawn and/or radiographs performed, with 1 CSI discovered (Table 3). This means that 77 infants diagnosed with FBIs did not have any diagnostic testing for CSI performed while in the ED. The overall risk of CSI in patients who were tested was 3.3%. Comparing febrile and afebrile patients who had diagnostic workups performed, febrile infants had a trend toward increased risk of CSI (OR, 8.6; 95% CI, 0.8613‐85.8686).
| CSI | No CSI | Risk (%) | |
|---|---|---|---|
| |||
| Febrile (n = 33) | 3 | 30 | 9.1 |
| Afebrile (n = 87) | 1 | 86 | 1.1 |
| Totals (n = 120) | 4 | 116 | 3.3 |
No significant differences existed in the use of consultants, use of IV antibiotics, or admission rate. Sixty‐six (42%) of the 158 afebrile infants were admitted. The average length of stay for these infants was 2.59 days (range, 1‐22 days). This included a 22‐day‐old female with mastitis who required multiple surgeries and a protracted hospital course. Sixteen (41%) of the febrile infants were admitted and had an average stay of 2.87 days (range, 1‐6 days).
Patient Outcomes
Ninety‐two (58%) of the afebrile infants were discharged home from the ED, of which 5 (5.4%) returned to the ED within 72 hours after discharge. Three of these were planned ED returns as follow‐up with a primary care physician could not be guaranteed. The fourth was a 47‐day‐old with a buttock abscess who returned for reevaluation because of parental concern. The fifth was the 11‐day‐old male initially diagnosed with AOM who was called back to the ED when his urine culture grew E. coli, at which point he was admitted to the hospital. Twenty‐three (59%) of the febrile infants were discharged home from the ED. Two (8.7%) of these 23 returned to the ED within 72 hours; both were initially diagnosed with AOM and had planned follow‐ups as no primary care was available. No admitted patients required transfer to the pediatric intensive care unit and no deaths occurred.
Discussion
This study is the first to investigate the risk of CSI specifically in well‐appearing infants under 60 days of age presenting with FBI. Inclusion criteria were not limited to 1 specific infection, ie, OM, but included all FBI. This study demonstrated that the risk of CSI in afebrile infants is small (0.6%), which is similar to what has been shown in the OM literature.19, 20 However, the risk of CSI among febrile infants is not negligible (7.7%), and is similar to the risk of SBIs among young infants presenting with a fever of unknown source.57
AOM
A total of 41 infants were diagnoses with AOM. Indeed, AOM was the most common discharge diagnosis among the febrile cohort. No febrile infants with AOM were diagnosed with a CSI. However, as noted above, an 11‐day‐old afebrile infant with AOM did have a UTI diagnosed. This male underwent blood, urine, and CSF cultures on initial presentation. He was discharged home from the ED, and called back for further evaluation once the urine culture grew E. coli.
We relied on the diagnostic skills of the ED attending physician for determination of OM, and no rigorous diagnostic criteria were established for an AOM in this young age group. However, on chart review of those patients with AOM who were admitted, otolaryngology was often consulted and confirmed the diagnosis of OM.
AOM has been the most commonly studied focal infection studied most in this age group.1, 2, 11, 2123 More recent studies have investigated the risk of CSI with AOM among febrile and afebrile cohorts. These authors found that in infants under 2 months with AOM, febrile infants had a 6% to 13% risk of CSI, whereas no cases of CSI were found in afebrile patients.19, 20 These results suggest that afebrile, nontoxic infants with AOM could be treated as outpatients without need for further systemic evaluation. Our study strengthens this argument if close follow‐up can be assured.
Cellulitis and Abscess
A total of 106 infants were diagnosed with either abscess and/or cellulitis, with the vast majority of them presenting without fever. Moreover, cellulitis and abscess comprised more than one‐half of all infants in the afebrile cohort. The buttocks were the most common site for abscess (23 cases), followed next by the scalp (8 cases). In our study population, there were 29 positive abscess, wound, lesion, or skin aspirate cultures for Staphylococcus aureus, with 13 (45%) being MRSA. None of these patients had a CSI.
Community‐acquired staphylococcal infections have been studied in the newborn population, with MRSA accounting for up to 69% of soft‐tissue infections in one recent study involving 89 neonates.24 The authors found 4 cases of bacteremia, 3 cases of UTI, and 2 cases of CSF pleocytosis. Application of these results to our patients is difficult, as there was no mention of fever or well‐appearance stratification and there is no specific mention of any of the 77 skin and soft‐tissue infections having a CSI.
The frequency of bacteremia in pediatric patients with periorbital cellulitis has been studied, with some authors suggested more selective use of LP in patients with these localized soft‐tissue infections in the older infant or child.25, 26 Unfortunately, there were not enough patients under 60 days of age in either study from which to draw conclusions. In our study, a total of 5 patients presented with periorbital cellulitis: 2 afebrile and 3 febrile, including the 47‐day‐old febrile infant with coexisting pneumococcal bacteremia.
Group B streptococcus (GBS) cellulitis‐adenitis syndrome has also been identified in neonates, with meningitis occurring in up to 24% of infants in 1 review of 32 cases.27 These studies only included patients with positive blood cultures, providing no denominator for assessing a true CSI risk in young infants with cellulitis. No infants in our study had GBS infections.
Impetigo was diagnosed in 21 patients, with 17 patients being discharged home from the ED. In addition, 2 infants with staphylococcal scalded skin syndrome were identified. None of these infants had a CSI.
Other Infections
Mastitis in the young infant has been studied, with the risk of CSI being 3% in 1 review.2830 In our study, there were 18 patients diagnosed with either mastitis or breast abscess, of which 4 (22%) had fever and none had positive blood or CSF cultures. Multiple studies, either from older literature in the United States or more contemporary from abroad, have investigated omphalitis and necrotizing fasciitis in the newborn.31, 32 These show that infants presenting with omphalitis have a lower mortality and morbidity than those with necrotizing fasciitis. In our study, we had 2 patients with omphalitis, both were afebrile and neither had a coexisting CSI. We had no patients with necrotizing fasciitis. We also found no patients with mastoiditis, balanitis, or posthitis as the listed diagnosis in our study.
UTI
Of the 4 cases of CSI in our study, UTI was the most common, occurring in 3 of the 4 cases. This predominance of UTI as the cause of CSI is well‐recognized in young febrile infants with OM and infants with fever of unknown origin; however, it has not been reported with other FBIs in this age group.1, 5, 9, 11, 19, 33 Specifically, there has not been a reported relationship between UTI and skin or soft tissue infections. However, 2 of our 4 infants with a CSI had a UTI in addition to their skin or soft tissue infection: the 32‐day‐old male with a buttock abscess and E. coli UTI; and the 21‐day‐old female with truncal cellulitis and E. coli UTI.
Resource Utilization
Young infants with FBI and fever underwent significantly more resource utilization than afebrile infants in our ED (Figure 1). Physicians of febrile infants ordered more serum blood counts and blood cultures, urinalysis and urine cultures, and performed more LPs and radiographic studies. This is not surprising given that fever is a risk factor for CSI in this age group; however, resource utilization is not as well‐described in previous reports on infants with FBIs. In 1 retrospective study of 137 infants less than 8 weeks of age with AOM, 88% had diagnostic blood cultures, 66% had LP, and 81% had urine cultures drawn.19 Additionally, in our study the decision to hospitalize infants, use of IV antibiotics, and hospital length of stay was not statistically different between febrile and afebrile groups. This potentially suggests that diagnostic testing for infants with FBIs did not affect management and outcome. Another study showed hospital admission is costly and not without safety risks, with a complication rate as high as 19% in infants.34 Given these results, in addition to the presumption that all FBIs would be treated with antibiotics, one could infer that a number of well‐appearing, afebrile infants underwent unwarranted testing.
Limitations
The main limitation of our study was the retrospective design. A research assistant utilizing a computerized database program of ED patients performed the initial query based upon search criteria. A local expert using an established list of International Statistical Classification of Diseases and Related Health Problems (ICD) codes for any and all of the FBIs performed the chart review. Secondary diagnoses may have been omitted as a result of limited physician charting, which could have resulted in missed patients. CCHMC is currently transitioning from paper records to electronic medical records, which may explain why 12 charts were not found, despite exhaustive searches for them. Ultimately, 94% of all the eligible patients were found. Yet, with so few CSI diagnosed, 1 or 2 could conceivably change our results. As noted above, focal infections in infants under 2 months of age seem to be uncommon, not only in the ED setting, but also in a large cohort of primary care patients.35 With only a small number FBIs present over 6 years and so few having CSIs, we were limited to a univariate analysis, which limited our potential results and conclusions. Performing this study prospectively would have addressed these limitations, but would have required years of recruitment or multicenter collaboration.
Another potential limitation was the search strategy to identify eligible infants. Specifically, we did not search the records for infants diagnosed with UTI, bacteremia, or meningitis. Infants with these more serious conditions may have been coded as the primary or only problem, even if other problems such as OM were also present. Therefore, we may have underestimated the occurrence of systemic bacterial complications in a systematic way. This form of spectrum bias was a potential limitation. We approached this clinical question from the eye of the emergency department practitioner and thus used a search strategy based on the discharge diagnosis from the ED. If we were to look back to see if any patients discharged from the hospital with CSI also had FBI, we doubt our retrospective design would have allowed identification of patients with FBIs.
Other limitations may be our definition of CSI and the potential for missed CSIs. We defined meningitis, bacteremia, and UTI as a positive CSF, blood, or urine culture, respectively. Many studies are now defining culture‐negative meningitis or UTI based on WBC findings on a screening cell count or urine microscopy. However, because no patients were pretreated with antibiotics, we felt that a positive culture from a sterile site was a reasonable definition. Also, not all of the enrolled patients received full diagnostic workups to screen for CSI. In the smaller cohort of infants who received diagnostic testing, febrile infants still seemed to be at higher risk for CSI. To capture all possible CSIs, one would have needed to perform blood culture, suprapubic bladder aspiration, or catheterization for urine culture, LP, and chest radiograph on all patients with an FBI. Given the low risk of CSI in the United States, the previous literature's hinting at low risk for CSI in afebrile patients, and the presence of multiple clinical trials defining low risk characteristics for CSI, such a protocol would not receive support as being standard clinical practice and have difficulty being permitted by an institutional review board.
Another limitation of our study was follow‐up. Ideally, every patient would have been contacted to ensure reliable outcomes measurement. Patients may have been seen after their initial evaluation at their primary care provider's office or at another healthcare facility and had more diagnostic studies performed. However, because our institution is the sole admitting pediatric center in the area, particularly in infants, and most CSIs in this age group require admission; we feel that a reasonable degree of reliability for the outcomes of interest was maintained.
Last, we could not establish the rationale of clinical decision making, as our study was retrospective. For example, staff physicians may have foregone diagnostic testing if they had committed to a therapeutic course of antibiotics and felt diagnostic cultures would not change management. Also, we cannot ensure that interpretation of culture results was consistent with national standards. As described previously, 1 afebrile infant in our study potentially had a CSI that was clinically felt to be a contaminant by the inpatient healthcare team. This 11‐day‐old female was admitted with mastitis and had a negative urinalysis, but grew E. coli on urine culture. This was treated as a contaminant by the ward team, despite her age.
Conclusions
CSI is very uncommon in afebrile, well‐appearing infants under 2 months of age with an FBI. However, febrile infants have a greater risk of CSI. UTI is the most common CSI in well‐appearing infants less than 2 months of age with an FBI found on examination. Prospective, multicentered studies need to be performed to determine whether diagnostic evaluation should change the management of well‐appearing infants with an FBI found on examination.
Focal bacterial infections (FBIs), including otitis media (OM), cellulitis, and lymphadenitis, can occur at any age and are usually treated with oral antibiotics. When patients with focal infections present as a young infant, healthcare providers often worry about the risk of coexisting or concomitant systemic infections (CSIs), often termed serious bacterial infections (SBIs) in the published literature.1, 2 Risk of CSI becomes more worrisome when young infants have fever as part of their presentation, especially within the traditional rule‐out sepsis age range of less than 2 months. Often, these patients are well‐appearing and lack prenatal and neonatal risk factors for systemic infections, but are assumed to be at high risk for CSI based on the presence of focal bacterial infection.1, 3, 4
FBIs have been the subject of multiple studies, especially OM and cellulitis. However, few investigations have looked specifically at infants less than 60 days of age, lending uncertainty to medical decision‐making for both community and emergency physicians. Therefore, no standardized evidence‐based practice exists for treating young infants with FBIs. While some clinicians treat these infections using systemic antibiotics without performing laboratory tests, others opt for a full diagnostic evaluation for CSI; including serum blood counts, blood cultures, lumbar punctures (LPs), and bladder catheterizations. Also, some clinicians opt for treatment with oral antibiotics at home, while others choose intravenous (IV) antibiotics and hospitalization.
These decisions are likely due to studies of febrile infants under 60 days of age who present to an emergency department (ED), appear well, and have no focal source of infection on exam, yet are known to have a risk of systemic or occult bacterial infection of roughly 10%.310 There are no such studies documenting the risk of CSI in well‐appearing, afebrile patients, as fever is regarded as the main risk factor for CSI in this age.
Based on our clinical experience and review of the literature, we felt that afebrile, well‐appearing infants less than 60 days of age with an FBI on exam would have a very low risk of CSI. The primary objectives of this study were to determine the risk of CSI when presented with a well‐appearing infant who has an FBI on exam and to examine that risk in relation to the presence of fever. Other objectives were to describe the clinical presentation of FBIs in well‐appearing infants in this age group and to describe the current management and resource utilization of such infections in the ED.
Patients and Methods
Study Population
Cincinnati Children's Hospital Medical Center (CCHMC) is a 423‐bed tertiary‐care hospital located in southwestern Ohio. The medical center serves as the sole pediatric hospital in a 70‐mile radius, serving a metropolitan population of roughly 1 million. In addition, CCHMC cares for patients from a wide demographic and socioeconomic spectrum, including the urban, suburban, and rural areas of Cincinnati, southwestern Ohio, northern Kentucky, and southeastern Indiana. The ED sees an average of 90,000 patients annually, with an average of 15,000 admissions.
Patient Selection
Data were retrospectively collected from a consecutive series of infants age 0 to 59 days who presented to the CCHMC ED between January 1, 2000 and December 31, 2005. Patients met inclusion criteria if they were documented to be well‐appearing on exam; had normal‐for‐age heart rate, respiratory rate, blood pressure, and oxygen saturation (if measured); were discharged from their hospital of birth to home before presentation to the ED; and received a discharge diagnosis consistent with an FBI. Specifically, FBIs included the following diagnoses: soft tissue infection, cellulitis, mastoiditis, abscess, OM, omphalitis, mastitis, mammitis, paronychia, balanitis, posthitis, impetigo, or lymphadenitis. We excluded patients with a history of immunodeficiency, central venous catheter, tracheostomy, gastrostomy tube, chronic lung disease, previous admission for a documented bacterial infection, or if they had been taking systemic antibiotics at the time of evaluation. We also excluded infants noted to be toxic or ill‐appearing on examination.
Definitions
Fever was defined as a temperature greater than or equal to 38C (100.4F), recorded by the ED or by parental report. All infants seen in the CCHMC ED had rectal temperatures measured per protocol. A nontoxic infant was defined by chart review as documentation of: nontoxic, well‐appearing, or alert infant without signs of respiratory distress or hemodynamic instability. Patients were excluded from the nontoxic category with the following parameters: heart rate >180 beats per minute, respiratory rate >60 breaths per minute, oxygen saturation <90% on pulse oximetry, or systolic blood pressure <60 mmHg on 2 or more measurements. CSIs were defined as any of the following: bacteremia, urinary tract infection (UTI) identified by catheterized urine specimen, meningitis, septic arthritis, or osteomyelitis. Additionally, pneumonia was included as a CSI if confirmed by radiographs, based on final reading by an attending radiologist. Pneumonia is included as an SBI in a number of prior publications in this age group; therefore, we felt it was important to include it in our definition of CSI.7, 1115
Procedures
The chart review consisted of both electronic and paper medical records. We searched the electronic database for all the infants in this age group receiving a discharge diagnosis of an FBI as defined above. Information from the medical record was entered onto a standardized data sheet by one of the investigators or a research assistant. Data gathered included baseline demographic information (age at ED evaluation, gestational age at birth, gender, race), presence or absence of fever, vital signs on ED presentation, use of diagnostic evaluation (complete blood count [CBC], blood culture, C‐reactive protein, erythrocyte sedimentation rate, urinalysis, urine culture, LP, wound culture, radiographic studies), laboratory test results, therapeutic interventions (including antibiotics and subspecialty consultation), initial disposition (home, admission to hospital), and clinical outcome of both admitted and discharged patients. The results of all bacterial cultures were obtained from computerized microbiology records. Bacterial isolates from blood cultures that are considered skin flora, such as Staphylococcus epidermidis or Viridans group Streptococcus, were considered contaminants unless they were positive for growth within 24 hours of initial culture.1618 Radiology results were based on final interpretation by an attending radiologist.
Statistical Analysis
The primary objectives of this study were to determine the risk of CSI when presented with a well‐appearing infant who has an FBI on exam and to examine that risk in relation to the presence of fever. Thus, this study was powered to investigate the difference in CSI between febrile and afebrile patients. Again, we hypothesized that in afebrile, well‐appearing infants less than 60 days of age with an FBI on exam there is a very low risk of CSI. For statistical purposes and power calculations we used <1% risk as very low. Using the approximate risk of 10% CSI in well‐appearing, febrile infants this age without CSI, we assumed there would be approximately 10% less CSIs in the afebrile patients compared to the febrile patients. As per consultation with our statistician, for a significance level of = 0.05 and = 0.2 a 2‐sided t test required a total of 188 subjects to be enrolled.
Additional outcome measures (such as use of laboratory testing, IV antibiotics, subspecialty consultation, and admission) were reported as descriptive statistics with frequencies. Fisher's Exact test was used for these nominal variables to test significance of relationship between febrile and afebrile patients. The Exact method was used to calculate 95% confidence intervals (CIs) around the primary outcome point estimates. A P value < 0.05 was considered significant. All data were entered into a Microsoft Excel (Microsoft, Redmond, WA) database.
Results
Study Population
We identified 246 patients less than 60 days of age who were diagnosed in the ED with an FBI during the study period. Thirty‐seven patients met the exclusion criteria, leaving 209 patients for potential enrollment. Twelve (5.7%) charts were not located, resulting in a study population of 197 infants. Of these, 158 patients were afebrile and 39 were febrile. To put these numbers into context, over the study period our ED evaluated 2341 infants less than 60 days of age whose diagnosis included the word fever, febrile, or sepsis. The febrile infants included in our study group were part of these 2341 found in the retrospective search, but the afebrile infants included were not.
The mean age of these 197 infants was 29.6 days, with a standard deviation (SD) of 15.9 days. On average, the febrile patients (mean 40.7 days, SD 15.3 days) were older than the afebrile patients (mean 26.8 days, SD 16.1 days). No statistical differences were found between febrile and afebrile patients in respect to gender, race, and gestational age.
Primary Study OutcomeRisk of SBI
Four patients had a documented CSI, for an overall risk of CSI in our study population of 2.0% (Table 1). Statistically, febrile infants had a significantly higher risk of CSI (odds ratio [OR], 13.08; 95% CI, 1.32‐129.46) than afebrile infants. The febrile infants with a coexisting CSI included the following: a 32‐day‐old male with a buttock abscess and E. coli UTI; a 47‐day‐old male with periorbital cellulitis and Streptococcus pneumoniae bacteremia; and a 21‐day‐old female with trunk cellulitis and E. coli UTI. The afebrile infant with a CSI was an 11‐day‐old male with acute OM (AOM) and E. coli UTI. No cases of bacterial meningitis, septic arthritis, osteomyelitis, radiographic pneumonia, or death occurred.
| CSI | No CSI | Risk (%) | 95% CI (%) | |
|---|---|---|---|---|
| ||||
| Febrile (n = 39) | 3 | 36 | 7.69 | 1.62‐20.87 |
| Afebrile (n = 158) | 1 | 157 | 0.63 | 0.02‐3.48 |
| Total (n = 197) | 4 | 193 | 2.03 | 0.56‐5.12 |
Eight other patients had positive culture results, all of which were considered contaminants. Four patients had peripheral blood cultures that were deemed contaminants: 2 cases of Viridans group streptococcus and 2 cases of coagulase‐negative staphylococcus. Four urine cultures were considered contaminants: 1 staphylococcus species; 1 mixed flora specimen; 1 lactobacillus species, and 1 E. coli.
This positive culture for E. coli occurred in an 11‐day‐old female who presented without fever, was diagnosed clinically and by ultrasound with mastitis, and was admitted to the hospital. She underwent a full diagnostic workup for CSI. Initial screening laboratory test results were normal, including a negative urinalysis and 1 to 2 white blood cells (WBCs)/high‐powered field on the microscopic exam. The catheterized urine specimen grew the following: (1) 10,000 to 100,000 colony forming units (CFU)/mL of E. coli; and (2) normal flora 1000 to 9000 CFU/mL. Her blood and cerebrospinal fluid (CSF) culture were negative. The primary team did not diagnose or treat this patient as a UTI despite the positive culture. According to their documentation, it was a suspected contaminant because of negative chemical and microscopic urinalysis. If this were considered a true positive culture, which seem practical clinically, clinically the overall risk of CSI would be 2.5% (5/197 infants) and the risk of coexisting CSI in afebrile patients would be 1.3% (2/158 infants); however, febrile infants would still had a significantly higher risk of CSI (OR, 6.5; 95% CI, 1.05‐40.34) than afebrile infants.
Secondary Study OutcomeClinical Presentation of FBI in Well‐appearing Infants in This Age Group
Among the 197 study infants, there were multiple types of FBIs found on examination (Table 2). Soft tissue infections were the most common cause in afebrile patients, whereas AOM was the most common cause in the febrile patients. Overall, cellulitis and abscess were the leading causes of focal infections in this age group. A total of 46 patients were diagnosed with abscesses, the buttock being the site in 24 (52%) cases. Abscesses were drained and sent for culture in 13 patients. Methicillin‐resistant Staphylococcus aureus (MRSA) was found in 4 of these 13 cultures. Of the 55 patients diagnosed with cellulitis, 20 (36%) had cultures drawn from the cellulitis site, of which 5 cultures grew MRSA.
| Focal Infection | Afebrile (n = 158) [n (%)] | Febrile (n = 39) [n (%)] | Totals (n = 197) [n (%)] |
|---|---|---|---|
| |||
| AOM | 19 (12) | 22 (56) | 41 (21) |
| Cellulitis | 49 (31) | 6 (15) | 55 (28) |
| Abscess | 40 (25) | 6 (15) | 46 (23) |
| Impetigo | 20 (13) | 1 (3) | 21 (11) |
| Mastitis | 11 (7) | 2 (5) | 13 (7) |
| Lymphadenitis | 4 (3) | 1 (3) | 5 (3) |
| Omphalitis | 2 (1) | 0 (0) | 2 (1) |
| SSSS | 2 (1) | 0 (0) | 2 (1) |
| Paronychia | 1 (1) | 0 (0) | 1 (0) |
| Other | 10 (6) | 1 (3) | 11 (6) |
Of note, a 13‐day‐old male presenting without fever but with a scalp lesion had herpes simplex virus grown from his wound culture. His diagnostic workup was negative for CSI. Omphalitis, uncommon in the United States, was diagnosed in 2 patients in our study and neither of these patients developed necrotizing fasciitis. Mastitis was not a significant focal infection in either group; and no patient was diagnosed with mastoiditis, balanitis, or posthitis.
Secondary Study OutcomeCurrent Management of FBI in the ED: Resource Utilization
Febrile patients underwent significantly more diagnostic testing procedures than afebrile infants. Specifically, CBCs, peripheral blood cultures, urine analyses with cultures, LPs, and radiographic imaging were performed more often in infants with fever (Figure 1). Fifty‐seven infants had urine cultures performed: 32 of 158 afebrile infants and 25 of 39 febrile infants. Among these selected infants, again 3 UTIs occurred: 1 of 32 tested (3.1%) in the afebrile group and 2 of 25 tested (8%) in the febrile group. A total of 115 infants had blood cultures performed: 82 of the 158 afebrile infants and 33 of the 39 febrile infants. Among these selected infants, again 1 positive blood culture occurred: 0 of 82 tested (0%) in the afebrile group and 1 of 33 tested (3%) in the febrile group. Fifty‐five infants had LP performed: 35 of 158 afebrile infants and 30 of 39 febrile infants. No case of meningitis occurred in these selected infants. Twenty infants had diagnostic radiographs performed, 10 in the febrile group and 10 in the afebrile group. None of the radiographs showed signs of pneumonia on final radiology attending reading.
Overall, 33 of 39 febrile infants had cultures drawn and/or radiographs performed, with 3 CSIs discovered, and 87 of 158 afebrile infants had cultures drawn and/or radiographs performed, with 1 CSI discovered (Table 3). This means that 77 infants diagnosed with FBIs did not have any diagnostic testing for CSI performed while in the ED. The overall risk of CSI in patients who were tested was 3.3%. Comparing febrile and afebrile patients who had diagnostic workups performed, febrile infants had a trend toward increased risk of CSI (OR, 8.6; 95% CI, 0.8613‐85.8686).
| CSI | No CSI | Risk (%) | |
|---|---|---|---|
| |||
| Febrile (n = 33) | 3 | 30 | 9.1 |
| Afebrile (n = 87) | 1 | 86 | 1.1 |
| Totals (n = 120) | 4 | 116 | 3.3 |
No significant differences existed in the use of consultants, use of IV antibiotics, or admission rate. Sixty‐six (42%) of the 158 afebrile infants were admitted. The average length of stay for these infants was 2.59 days (range, 1‐22 days). This included a 22‐day‐old female with mastitis who required multiple surgeries and a protracted hospital course. Sixteen (41%) of the febrile infants were admitted and had an average stay of 2.87 days (range, 1‐6 days).
Patient Outcomes
Ninety‐two (58%) of the afebrile infants were discharged home from the ED, of which 5 (5.4%) returned to the ED within 72 hours after discharge. Three of these were planned ED returns as follow‐up with a primary care physician could not be guaranteed. The fourth was a 47‐day‐old with a buttock abscess who returned for reevaluation because of parental concern. The fifth was the 11‐day‐old male initially diagnosed with AOM who was called back to the ED when his urine culture grew E. coli, at which point he was admitted to the hospital. Twenty‐three (59%) of the febrile infants were discharged home from the ED. Two (8.7%) of these 23 returned to the ED within 72 hours; both were initially diagnosed with AOM and had planned follow‐ups as no primary care was available. No admitted patients required transfer to the pediatric intensive care unit and no deaths occurred.
Discussion
This study is the first to investigate the risk of CSI specifically in well‐appearing infants under 60 days of age presenting with FBI. Inclusion criteria were not limited to 1 specific infection, ie, OM, but included all FBI. This study demonstrated that the risk of CSI in afebrile infants is small (0.6%), which is similar to what has been shown in the OM literature.19, 20 However, the risk of CSI among febrile infants is not negligible (7.7%), and is similar to the risk of SBIs among young infants presenting with a fever of unknown source.57
AOM
A total of 41 infants were diagnoses with AOM. Indeed, AOM was the most common discharge diagnosis among the febrile cohort. No febrile infants with AOM were diagnosed with a CSI. However, as noted above, an 11‐day‐old afebrile infant with AOM did have a UTI diagnosed. This male underwent blood, urine, and CSF cultures on initial presentation. He was discharged home from the ED, and called back for further evaluation once the urine culture grew E. coli.
We relied on the diagnostic skills of the ED attending physician for determination of OM, and no rigorous diagnostic criteria were established for an AOM in this young age group. However, on chart review of those patients with AOM who were admitted, otolaryngology was often consulted and confirmed the diagnosis of OM.
AOM has been the most commonly studied focal infection studied most in this age group.1, 2, 11, 2123 More recent studies have investigated the risk of CSI with AOM among febrile and afebrile cohorts. These authors found that in infants under 2 months with AOM, febrile infants had a 6% to 13% risk of CSI, whereas no cases of CSI were found in afebrile patients.19, 20 These results suggest that afebrile, nontoxic infants with AOM could be treated as outpatients without need for further systemic evaluation. Our study strengthens this argument if close follow‐up can be assured.
Cellulitis and Abscess
A total of 106 infants were diagnosed with either abscess and/or cellulitis, with the vast majority of them presenting without fever. Moreover, cellulitis and abscess comprised more than one‐half of all infants in the afebrile cohort. The buttocks were the most common site for abscess (23 cases), followed next by the scalp (8 cases). In our study population, there were 29 positive abscess, wound, lesion, or skin aspirate cultures for Staphylococcus aureus, with 13 (45%) being MRSA. None of these patients had a CSI.
Community‐acquired staphylococcal infections have been studied in the newborn population, with MRSA accounting for up to 69% of soft‐tissue infections in one recent study involving 89 neonates.24 The authors found 4 cases of bacteremia, 3 cases of UTI, and 2 cases of CSF pleocytosis. Application of these results to our patients is difficult, as there was no mention of fever or well‐appearance stratification and there is no specific mention of any of the 77 skin and soft‐tissue infections having a CSI.
The frequency of bacteremia in pediatric patients with periorbital cellulitis has been studied, with some authors suggested more selective use of LP in patients with these localized soft‐tissue infections in the older infant or child.25, 26 Unfortunately, there were not enough patients under 60 days of age in either study from which to draw conclusions. In our study, a total of 5 patients presented with periorbital cellulitis: 2 afebrile and 3 febrile, including the 47‐day‐old febrile infant with coexisting pneumococcal bacteremia.
Group B streptococcus (GBS) cellulitis‐adenitis syndrome has also been identified in neonates, with meningitis occurring in up to 24% of infants in 1 review of 32 cases.27 These studies only included patients with positive blood cultures, providing no denominator for assessing a true CSI risk in young infants with cellulitis. No infants in our study had GBS infections.
Impetigo was diagnosed in 21 patients, with 17 patients being discharged home from the ED. In addition, 2 infants with staphylococcal scalded skin syndrome were identified. None of these infants had a CSI.
Other Infections
Mastitis in the young infant has been studied, with the risk of CSI being 3% in 1 review.2830 In our study, there were 18 patients diagnosed with either mastitis or breast abscess, of which 4 (22%) had fever and none had positive blood or CSF cultures. Multiple studies, either from older literature in the United States or more contemporary from abroad, have investigated omphalitis and necrotizing fasciitis in the newborn.31, 32 These show that infants presenting with omphalitis have a lower mortality and morbidity than those with necrotizing fasciitis. In our study, we had 2 patients with omphalitis, both were afebrile and neither had a coexisting CSI. We had no patients with necrotizing fasciitis. We also found no patients with mastoiditis, balanitis, or posthitis as the listed diagnosis in our study.
UTI
Of the 4 cases of CSI in our study, UTI was the most common, occurring in 3 of the 4 cases. This predominance of UTI as the cause of CSI is well‐recognized in young febrile infants with OM and infants with fever of unknown origin; however, it has not been reported with other FBIs in this age group.1, 5, 9, 11, 19, 33 Specifically, there has not been a reported relationship between UTI and skin or soft tissue infections. However, 2 of our 4 infants with a CSI had a UTI in addition to their skin or soft tissue infection: the 32‐day‐old male with a buttock abscess and E. coli UTI; and the 21‐day‐old female with truncal cellulitis and E. coli UTI.
Resource Utilization
Young infants with FBI and fever underwent significantly more resource utilization than afebrile infants in our ED (Figure 1). Physicians of febrile infants ordered more serum blood counts and blood cultures, urinalysis and urine cultures, and performed more LPs and radiographic studies. This is not surprising given that fever is a risk factor for CSI in this age group; however, resource utilization is not as well‐described in previous reports on infants with FBIs. In 1 retrospective study of 137 infants less than 8 weeks of age with AOM, 88% had diagnostic blood cultures, 66% had LP, and 81% had urine cultures drawn.19 Additionally, in our study the decision to hospitalize infants, use of IV antibiotics, and hospital length of stay was not statistically different between febrile and afebrile groups. This potentially suggests that diagnostic testing for infants with FBIs did not affect management and outcome. Another study showed hospital admission is costly and not without safety risks, with a complication rate as high as 19% in infants.34 Given these results, in addition to the presumption that all FBIs would be treated with antibiotics, one could infer that a number of well‐appearing, afebrile infants underwent unwarranted testing.
Limitations
The main limitation of our study was the retrospective design. A research assistant utilizing a computerized database program of ED patients performed the initial query based upon search criteria. A local expert using an established list of International Statistical Classification of Diseases and Related Health Problems (ICD) codes for any and all of the FBIs performed the chart review. Secondary diagnoses may have been omitted as a result of limited physician charting, which could have resulted in missed patients. CCHMC is currently transitioning from paper records to electronic medical records, which may explain why 12 charts were not found, despite exhaustive searches for them. Ultimately, 94% of all the eligible patients were found. Yet, with so few CSI diagnosed, 1 or 2 could conceivably change our results. As noted above, focal infections in infants under 2 months of age seem to be uncommon, not only in the ED setting, but also in a large cohort of primary care patients.35 With only a small number FBIs present over 6 years and so few having CSIs, we were limited to a univariate analysis, which limited our potential results and conclusions. Performing this study prospectively would have addressed these limitations, but would have required years of recruitment or multicenter collaboration.
Another potential limitation was the search strategy to identify eligible infants. Specifically, we did not search the records for infants diagnosed with UTI, bacteremia, or meningitis. Infants with these more serious conditions may have been coded as the primary or only problem, even if other problems such as OM were also present. Therefore, we may have underestimated the occurrence of systemic bacterial complications in a systematic way. This form of spectrum bias was a potential limitation. We approached this clinical question from the eye of the emergency department practitioner and thus used a search strategy based on the discharge diagnosis from the ED. If we were to look back to see if any patients discharged from the hospital with CSI also had FBI, we doubt our retrospective design would have allowed identification of patients with FBIs.
Other limitations may be our definition of CSI and the potential for missed CSIs. We defined meningitis, bacteremia, and UTI as a positive CSF, blood, or urine culture, respectively. Many studies are now defining culture‐negative meningitis or UTI based on WBC findings on a screening cell count or urine microscopy. However, because no patients were pretreated with antibiotics, we felt that a positive culture from a sterile site was a reasonable definition. Also, not all of the enrolled patients received full diagnostic workups to screen for CSI. In the smaller cohort of infants who received diagnostic testing, febrile infants still seemed to be at higher risk for CSI. To capture all possible CSIs, one would have needed to perform blood culture, suprapubic bladder aspiration, or catheterization for urine culture, LP, and chest radiograph on all patients with an FBI. Given the low risk of CSI in the United States, the previous literature's hinting at low risk for CSI in afebrile patients, and the presence of multiple clinical trials defining low risk characteristics for CSI, such a protocol would not receive support as being standard clinical practice and have difficulty being permitted by an institutional review board.
Another limitation of our study was follow‐up. Ideally, every patient would have been contacted to ensure reliable outcomes measurement. Patients may have been seen after their initial evaluation at their primary care provider's office or at another healthcare facility and had more diagnostic studies performed. However, because our institution is the sole admitting pediatric center in the area, particularly in infants, and most CSIs in this age group require admission; we feel that a reasonable degree of reliability for the outcomes of interest was maintained.
Last, we could not establish the rationale of clinical decision making, as our study was retrospective. For example, staff physicians may have foregone diagnostic testing if they had committed to a therapeutic course of antibiotics and felt diagnostic cultures would not change management. Also, we cannot ensure that interpretation of culture results was consistent with national standards. As described previously, 1 afebrile infant in our study potentially had a CSI that was clinically felt to be a contaminant by the inpatient healthcare team. This 11‐day‐old female was admitted with mastitis and had a negative urinalysis, but grew E. coli on urine culture. This was treated as a contaminant by the ward team, despite her age.
Conclusions
CSI is very uncommon in afebrile, well‐appearing infants under 2 months of age with an FBI. However, febrile infants have a greater risk of CSI. UTI is the most common CSI in well‐appearing infants less than 2 months of age with an FBI found on examination. Prospective, multicentered studies need to be performed to determine whether diagnostic evaluation should change the management of well‐appearing infants with an FBI found on examination.
- ,,, et al.Febrile infants at low risk for serious bacterial infection—an appraisal of the Rochester Criteria and implications for management.Pediatrics.1994;94:390–396.
- ,,, et al.Identification of infants unlikely to have serious bacterial infection although hospitalized for suspected sepsis.J Pediatr.1985;107:855–860.
- ,,.Outpatient management without antibiotics of fever in selected infants.N Engl J Med.1993;329:1437–1441.
- ,,.Outpatient treatment of febrile infants 28 to 89 days of age with intramuscular administration of ceftriaxone.J Pediatr.1992;120:22–27.
- ,.Predictive model for serious bacterial infections among infants younger than 3 months of age.Pediatrics.2001;108:311–316.
- ,,, et al.Applying outpatient protocols to febrile infants 1–28 days of age: can the threshold be lowered?Clin Pediatr.2000;39:81–88.
- ,,.The efficacy of routine outpatient management without antibiotics of fever in selected infants.Pediatrics.1999;103:627–631.
- ,.Evaluation of the infant with fever without source: an evidence based approach.Emerg Med Clin North Am.1999;17:97–126.
- ,,, et al.Risk of serious bacterial infection in young febrile infants with respiratory syncytial virus infections.Pediatrics.2004;113:1728–1734.
- ,,, et al.Relationship of fever magnitude to rate of serious bacterial infections in infants 4–8 weeks.Clin Pediatr (Phila).1991;30:478–480.
- ,,, et al.Enhanced urinalysis improved identification of febrile infants ages 60 days and younger at low risk for serious bacterial illness.Pediatrics.2001;108:866–871.
- ,,.C‐reactive protein in febrile children 1 to 36 months of age with clinically undetectable serious bacterial infection.Pediatrics.2001;108:1275–1279.
- ,.Utility of the serum C‐reactive protein for detection of occult bacterial infection in children.Arch Pediatr Adolesc Med.2002;156:905–909.
- ,,, et al.Procalcitonin and C‐reactive protein as diagnostic markers of severe bacterial infections in febrile infants and children in the emergency department.Pediatr Infect Dis J.2007;26:672–677.
- ,,.Neonatal fever: utility of the Rochester criteria in determining low risk for serious bacterial infections.Am J Emerg Med.1997;15:299–302.
- ,,.Evaluation of false positive blood cultures: guidelines for early detection of contaminated cultures in febrile children.Pediatr Emerg Care.1994;10:20–22.
- ,,.Outpatient pediatric blood cultures: time to positivity.Pediatrics.2000;106:251–255.
- ,,, et al.Occult bacteremia from a pediatric emergency department: current prevalence, time to detection and outcome.Pediatrics.2000;106:505–511.
- ,,, et al.Acute otitis media in infants younger than two months of age: microbiology, clinical presentation and therapeutic approach.Pediatr Infect Dis J.2002;21:669–674.
- ,,, et al.Otitis media in infants aged 0–8 weeks: frequency of associated serious bacterial disease.Pediatr Emerg Care.1999;15:252–254.
- ,,.Otitis media in children less than 12 weeks of age.Pediatrics.1977;59:827–832.
- ,,.Are well‐appearing febrile infants with otitis media at risk for serious bacterial illness? [Abstract].Am J Dis Child.1992;146:468.
- ,,, et al.Streptococcus pneumoniae infections in the neonate.Pediatrics.2003;112:1095–1102.
- ,,, et al.Community‐acquired Staphylococcus aureus infections in term and near‐term previously health neonates.Pediatrics.2007;118:874–881.
- ,.Blood cultures in the evaluation of children with cellulitis.Pediatrics.1997;101:e4.
- ,.Lumbar puncture in children with periorbital and orbital cellulitis.J Pediatr.1993;122:355–359.
- ,.Is lumbar puncture necessary to exclude meningitis in neonates and young infants: lessons from the Group B streptococcus cellulitis‐adenitis syndrome.Pediatrics.1998;102:985–986.
- ,.Neonatal mastitis.Clin Pediatr.1986;25:395–399.
- ,.Pseudomonas aeruginosa mastitis in a neonate.Pediatr Infect Dis J.1993;12:104.
- ,,.Group D streptococcal neonatal mastitis.Pediatr Infect Dis J.1998;7:362.
- ,,, et al.Early recognition of neonatal abdominal wall necrotizing fasciitis.Am J Surg.1994;167:481–484.
- ,,, et al.Necrotizing fasciitis: a serious complication of omphalitis in neonates.J Pediatr Surg.1994;29:1414–1416.
- ,,, et al.Acute otitis media in infants less than three months of age: clinical presentation, etiology and concomitant diseases.Int J Pediatr Otorhinolaryngol.2006;70:613–617.
- ,,, et al.Iatrogenic risks and financial costs of hospitalizing febrile infants.Am J Dis Child.1983;137:1146–1149.
- ,,, et al.Management and outcomes of care of fever in early infancy.JAMA.2004;291:1203–1212.
- ,,, et al.Febrile infants at low risk for serious bacterial infection—an appraisal of the Rochester Criteria and implications for management.Pediatrics.1994;94:390–396.
- ,,, et al.Identification of infants unlikely to have serious bacterial infection although hospitalized for suspected sepsis.J Pediatr.1985;107:855–860.
- ,,.Outpatient management without antibiotics of fever in selected infants.N Engl J Med.1993;329:1437–1441.
- ,,.Outpatient treatment of febrile infants 28 to 89 days of age with intramuscular administration of ceftriaxone.J Pediatr.1992;120:22–27.
- ,.Predictive model for serious bacterial infections among infants younger than 3 months of age.Pediatrics.2001;108:311–316.
- ,,, et al.Applying outpatient protocols to febrile infants 1–28 days of age: can the threshold be lowered?Clin Pediatr.2000;39:81–88.
- ,,.The efficacy of routine outpatient management without antibiotics of fever in selected infants.Pediatrics.1999;103:627–631.
- ,.Evaluation of the infant with fever without source: an evidence based approach.Emerg Med Clin North Am.1999;17:97–126.
- ,,, et al.Risk of serious bacterial infection in young febrile infants with respiratory syncytial virus infections.Pediatrics.2004;113:1728–1734.
- ,,, et al.Relationship of fever magnitude to rate of serious bacterial infections in infants 4–8 weeks.Clin Pediatr (Phila).1991;30:478–480.
- ,,, et al.Enhanced urinalysis improved identification of febrile infants ages 60 days and younger at low risk for serious bacterial illness.Pediatrics.2001;108:866–871.
- ,,.C‐reactive protein in febrile children 1 to 36 months of age with clinically undetectable serious bacterial infection.Pediatrics.2001;108:1275–1279.
- ,.Utility of the serum C‐reactive protein for detection of occult bacterial infection in children.Arch Pediatr Adolesc Med.2002;156:905–909.
- ,,, et al.Procalcitonin and C‐reactive protein as diagnostic markers of severe bacterial infections in febrile infants and children in the emergency department.Pediatr Infect Dis J.2007;26:672–677.
- ,,.Neonatal fever: utility of the Rochester criteria in determining low risk for serious bacterial infections.Am J Emerg Med.1997;15:299–302.
- ,,.Evaluation of false positive blood cultures: guidelines for early detection of contaminated cultures in febrile children.Pediatr Emerg Care.1994;10:20–22.
- ,,.Outpatient pediatric blood cultures: time to positivity.Pediatrics.2000;106:251–255.
- ,,, et al.Occult bacteremia from a pediatric emergency department: current prevalence, time to detection and outcome.Pediatrics.2000;106:505–511.
- ,,, et al.Acute otitis media in infants younger than two months of age: microbiology, clinical presentation and therapeutic approach.Pediatr Infect Dis J.2002;21:669–674.
- ,,, et al.Otitis media in infants aged 0–8 weeks: frequency of associated serious bacterial disease.Pediatr Emerg Care.1999;15:252–254.
- ,,.Otitis media in children less than 12 weeks of age.Pediatrics.1977;59:827–832.
- ,,.Are well‐appearing febrile infants with otitis media at risk for serious bacterial illness? [Abstract].Am J Dis Child.1992;146:468.
- ,,, et al.Streptococcus pneumoniae infections in the neonate.Pediatrics.2003;112:1095–1102.
- ,,, et al.Community‐acquired Staphylococcus aureus infections in term and near‐term previously health neonates.Pediatrics.2007;118:874–881.
- ,.Blood cultures in the evaluation of children with cellulitis.Pediatrics.1997;101:e4.
- ,.Lumbar puncture in children with periorbital and orbital cellulitis.J Pediatr.1993;122:355–359.
- ,.Is lumbar puncture necessary to exclude meningitis in neonates and young infants: lessons from the Group B streptococcus cellulitis‐adenitis syndrome.Pediatrics.1998;102:985–986.
- ,.Neonatal mastitis.Clin Pediatr.1986;25:395–399.
- ,.Pseudomonas aeruginosa mastitis in a neonate.Pediatr Infect Dis J.1993;12:104.
- ,,.Group D streptococcal neonatal mastitis.Pediatr Infect Dis J.1998;7:362.
- ,,, et al.Early recognition of neonatal abdominal wall necrotizing fasciitis.Am J Surg.1994;167:481–484.
- ,,, et al.Necrotizing fasciitis: a serious complication of omphalitis in neonates.J Pediatr Surg.1994;29:1414–1416.
- ,,, et al.Acute otitis media in infants less than three months of age: clinical presentation, etiology and concomitant diseases.Int J Pediatr Otorhinolaryngol.2006;70:613–617.
- ,,, et al.Iatrogenic risks and financial costs of hospitalizing febrile infants.Am J Dis Child.1983;137:1146–1149.
- ,,, et al.Management and outcomes of care of fever in early infancy.JAMA.2004;291:1203–1212.
Copyright © 2010 Society of Hospital Medicine
Gary L. Geis, Division of Emergency Medicine, Assistant Medical Director, Center for Simulation and Research, Cincinnati Childrens Hospital Medical Center, University of Cincinnati, College of Medicine, 333 Burnet Avenue, MLC 12000 Cincinnati, Ohio 45229‐3039
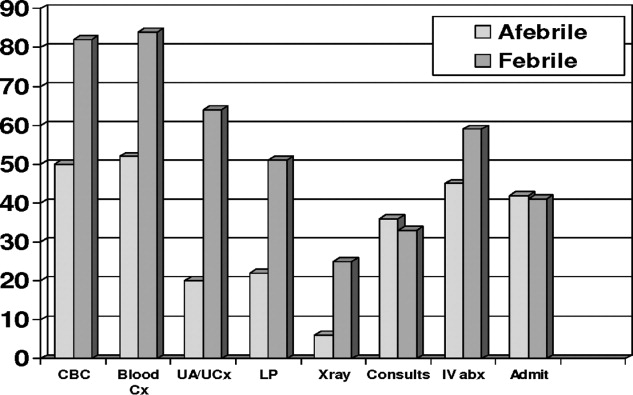
Continuing Medical Education Program in
If you wish to receive credit for this activity, which beginson the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to www.blackwellpublishing.com/cme.
-
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
If you wish to receive credit for this activity, which beginson the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to www.blackwellpublishing.com/cme.
-
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
If you wish to receive credit for this activity, which beginson the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to www.blackwellpublishing.com/cme.
-
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
Hypercalcemia and Milk‐Alkali Syndrome
Historically, the milk‐alkali syndrome developed as an adverse reaction to the Sippy regimen of frequent feedings of milk, cream, and alkaline powders as treatment for peptic ulcer disease.1 The classic description includes hypercalcemia, metabolic alkalosis, and renal failure. This syndrome seemingly disappeared when modern acid suppression therapies such as histamine‐2 blockers and proton pump inhibitors improved dyspepsia treatment. Over the past 20 years, milk‐alkali syndrome has had a resurgence, as consumption of supplements containing calcium has increased.2 Calcium carbonate supplements are a popular over‐the‐counter treatment for osteoporosis, dyspepsia, hypocalcemia, and hyperphosphatemia; these supplements provide both the calcium and alkali required for the development of milk‐alkali syndrome.
A 46‐year‐old man presented to the emergency department after his physician ordered outpatient laboratory tests to evaluate his fatigue. The patient was found to have acute renal failure and hypercalcemia. His serum creatinine was 3.6 mg/dL, increased from his baseline of 1.1 mg/dL several months prior, and his serum calcium was 14.9 mg/dL. Ten days prior to admission he developed increasing fatigue, decreased appetite, and decreased urine output, which he attributed to recent manual labor during summer. He reported taking an occasional calcium carbonate (Tums) for dyspepsia. He did not report pain or other complaints.
His medical history included hypertension and hyperlipidemia. He had a colonoscopy 1 year prior to presentation that was significant for a high‐grade dysplastic polyp and was currently due for repeat colonoscopy. His medications included clonidine, lisinopril, and aspirin. He had no recent medication changes. He had a 30 pack/year history of cigarette smoking and drank occasionally.
On physical exam, his temperature was 99.8F, blood pressure 97/48 mmHg, heart rate 89 beats per minute, respirations 20 breaths per minute, with a room air saturation of 97%. He had dry mucus membranes and the remainder of the physical exam was unremarkable.
In the emergency department laboratory testing revealed a creatinine of 4.6 mg/dL, serum total calcium of 15.9 mg/dL, serum bicarbonate level of 26 mmol/L, phosphate of 3.9 mg/dL, albumin of 4.4 gm/dL, and alkaline phosphatase of 92 IU/L. The urine specific gravity was 1.019 gm/mL (see Table 1 for the patient's complete admission laboratory values).
| Result (Normal Range) | |
|---|---|
| |
| At admission | |
| Sodium (mmol/L) | 135 (135‐145) |
| Potassium (mmol/L) | 4.4 (3.8‐5.0) |
| Chloride (mmol/L) | 97 (98‐107) |
| Bicarbonate (mmol/L) | 25 (22‐31) |
| BUN (mg/dL) | 64 (9‐20) |
| Creatinine (mg/dL) | 4.6 (0.5‐1.5) |
| Calcium (mg/dL) | 15.9 (8.4‐10.2) |
| Albumin (g/dL) | 4.4 (3.5‐5.0) |
| Alkaline phosphatase (U/L) | 92 (38‐126) |
| ALT (U/L) | 24 (11‐66) |
| AST (U/L) | 17 (15‐46) |
| Bilirubin, total (mg/dL) | 0.3 (0.3‐1.2) |
| Ionized calcium (mmol/L) | 1.58 (1.13‐1.32) |
| Creatinine kinase (U/L) | 83 (35‐232) |
| WBC (thousands/cm2) | 12.1 (4‐11) |
| Hemoglobin (g/dL) | 16.5 (14‐18) |
| Hematocrit (%) | 48.6 (40‐54) |
| Platelets (thousands/cm2) | 224 (150‐350) |
| Intact PTH (pg/mL) | 18.78 (15‐65) |
| PTHrP (pmol/L) | 2.5 (5) |
| 25‐OH‐Vitamin D (ng/mL) | 23 (16‐74) |
| H. pylori antibody | Negative |
| SPEP | No M‐spike |
| UPEP | No M‐spike |
| Hospital Day 4 (date of discharge) | |
| BUN (mg/dL) | 21 (9‐20) |
| Creatinine (mg/dL) | 1.6 (0.5‐1.5) |
| Calcium(mg/dL) | 8.4 (8.4‐10.2) |
| WBC (thousands/cm2) | 6.5 (4‐11) |
| Hemoglobin (g/dL) | 14.1 (14‐18) |
| Hematocrit (%) | 40.8 (40‐54) |
| Day 10 (at follow‐up) | |
| BUN (mg/dL) | 16 (9‐20) |
| Creatinine (mg/dL) | 0.9 (0.5‐1.5) |
| Calcium (mg/dL) | 8.1 (8.4‐10.2) |
| Intact PTH (pg/mL) | 240 (15‐65) |
His intact parathyroid (PTH) hormone was 18.8 pg/mL (normal, 15‐65). PTH hormone‐related peptide (PTHrP) was 2.5 pmol/L. After reviewing these laboratory test results, we proceeded with further questioning, during which he admitted to taking approximately 15 to 20 Tums (>7.5 gm of calcium carbonate) daily for dyspepsia rather than the occasional Tums he had originally reported. Over the next 3 days, his calcium decreased to 8.4 mg/dL and his creatinine decreased to 1.6 mg/dL with intravenous hydration. His fatigue improved. His 25‐hydroxy vitamin D (25‐OH‐Vitamin D) level was 23 ng/mL (normal, 16‐74 ng/mL).
At his 1 week follow‐up, his calcium was 8.1 mg/dL, and his creatinine had returned to normal at 0.9 mg/dL. His intact PTH level was elevated at 240 pg/mL.
Discussion
Milk‐alkali syndrome is now believed to be the third most common reason for hypercalcemia hospital admission.2, 3 Malignancy and primary hyperparathyroidism are the only 2 causes of hypercalcemia more common than milk‐alkali syndrome in hospitalized patients; these must be excluded before making a definitive diagnosis of milk‐alkali syndrome. The differential diagnosis for hypercalcemia also includes other less common etiologies such as medications (hydrochlorothiazide and lithium), as well as familial hypocalciuric hypercalcemia, hyperthyroidism, Addison's disease, acromegaly, tertiary hyperparathyroidism, and vitamin D intoxication. Physicians often discover hypercalcemia incidentally on routine laboratory tests, and diagnostic workup should include a thorough history and physical examination, as well as further laboratory evaluation.
The diagnosis of milk‐alkali syndrome requires a history of increased calcium and alkali intake, but is otherwise a diagnosis of exclusion. Given the increasing consumption of nonprescribed calcium supplements, one should have a high index of suspicion for the diagnosis of milk‐alkali syndrome, as patients may not consider calcium carbonate to be hazardous or even a medication and thus may not report calcium carbonate consumption. Patients often view calcium carbonate as a benign treatment for dyspepsia. Its over‐the‐counter availability and economical price make it a common self‐treatment for minor dyspepsia or as prevention of osteoporosis. Calcium supplementation is increasingly added to many products, making it easy for patients to consume large quantities of calcium unknowingly. Of note, without the absorbable alkali supplied by the carbonate in calcium carbonate (Tums), milk‐alkali syndrome does not occur. The amount of calcium carbonate necessary to cause milk‐alkali syndrome is not well known, though it is speculated to be as little as 5 to 10 g of calcium in the form of calcium carbonate, especially in those with other risk factors for hypercalcemia such as chronic renal insufficiency or vomiting.2 Workup of hypercalcemia should entail careful questioning about medications, as well as over‐the‐counter supplements, vitamins, and foods.
Manifestations of the milk‐alkali syndrome include renal failure, metabolic alkalosis, and volume contraction. Normally, the kidneys prevent hypercalcemia by excretion of excess calcium. Hypercalcemia can cause tubular damage and vasoconstriction of the renal afferent arteriole leading to acute renal failure.2, 4 Hypercalcemia can also cause nephrogenic diabetes insipidus, causing impaired renal concentrating ability, leading to increased sodium excretion and volume contraction.2 In addition, alkalosis further impairs calciuresis.2 Laboratory values usually reveal suppressed PTH and vitamin D levels due to hypercalcemia caused by exogenous intake of calcium.5 Hypercalcemia causes suppression of PTH, which can lead to hyperphosphatemia, as well as decreased conversion of vitamin D to the active 1,25‐dihyroxyvitamin‐D form.
The management of hypercalcemia due to milk‐alkali syndrome is supportive and includes saline hydration as well as withholding calcium carbonate. Management of hypercalcemia due to malignancy and hyperparathyroidism includes bisphosphonates with the addition of calcitonin if symptoms are severe.6, 7 There is no evidence that supports the use of bisphosphonates in the treatment of milk‐alkali syndrome. Loop diuretics are sometimes used to promote calciuresis, though evidence is lacking to support this, and it may worsen renal failure.6
In this case, a middle‐aged man took greater than the recommended dose of calcium carbonate for dyspepsia, which led to the development of acute renal failure and hypercalcemia. At first, the patient did not provide an accurate history of the extent of his calcium carbonate ingestion, leading us to focus on hyperparathyroidism or malignancy. With aggressive hydration and cessation of calcium carbonate, his renal function and serum calcium returned to baseline. Because we initially assumed occult malignancy as the most likely diagnosis, we gave the patient pamidronate. The patient did not have a significant alkalemia (serum bicarbonate level was normal). This was thought to be due to the patient's degree of renal failure causing a concomitant metabolic acidosis. The patient's follow‐up elevated PTH level may be explained by bisphosphonate administration or underlying primary hyperparathyroidism. Of note, decreasing calcium levels have also been speculated to be a cause of high PTH levels.8
In conclusion, physicians should have a high index of suspicion for milk‐alkali syndrome in patients with hypercalcemia. Calcium carbonate is responsible for most cases of milk‐alkali syndrome, and clinicians should inquire about the use of this supplement in all patients with hypercalcemia. Milk‐alkali syndrome is no longer a merely a historical curiosity; it is currently the third most common cause of hospital admissions for hypercalcemia.
- .Landmark article May 15, 1915: Gastric and duodenal ulcer. Medical cure by an efficient removal of gastric juice corrosion. By Bertram W. Sippy.JAMA.1983;250(16):2192–2197.
- ,,,.Milk‐alkali syndrome: a historical review and description of the modern version of the syndrome.Am J Med Sci.2006;331(5):233–242.
- ,,.Milk‐alkali syndrome is a major cause of hypercalcaemia among non‐end‐stage renal disease (non‐ESRD) inpatients.Clin Endocrinol (Oxf).2005;63(5):566–576.
- ,.Milk‐alkali syndrome associated with calcium carbonate consumption. Report of 7 patients with parathyroid hormone levels and an estimate of prevalence among patients hospitalized with hypercalcemia.Medicine (Baltimore).1995;74(2):89–96.
- ,,,,,.The milk‐alkali syndrome. A reversible form of acute renal failure.Arch Intern Med.1993;153(8):1005–1010.
- ,,.Narrative review: furosemide for hypercalcemia: an unproven yet common practice.Ann Intern Med.2008;149(4):259–263.
- .Salmon calcitonin in the acute management of hypercalcemia.Calcif Tissue Int.1990;46(suppl):S26–S30.
- ,.Milk alkali syndrome. Does it exist and can it be differentiated from primary hyperparathyroidism?Ann Surg.1983;197(4):427–433.
Historically, the milk‐alkali syndrome developed as an adverse reaction to the Sippy regimen of frequent feedings of milk, cream, and alkaline powders as treatment for peptic ulcer disease.1 The classic description includes hypercalcemia, metabolic alkalosis, and renal failure. This syndrome seemingly disappeared when modern acid suppression therapies such as histamine‐2 blockers and proton pump inhibitors improved dyspepsia treatment. Over the past 20 years, milk‐alkali syndrome has had a resurgence, as consumption of supplements containing calcium has increased.2 Calcium carbonate supplements are a popular over‐the‐counter treatment for osteoporosis, dyspepsia, hypocalcemia, and hyperphosphatemia; these supplements provide both the calcium and alkali required for the development of milk‐alkali syndrome.
A 46‐year‐old man presented to the emergency department after his physician ordered outpatient laboratory tests to evaluate his fatigue. The patient was found to have acute renal failure and hypercalcemia. His serum creatinine was 3.6 mg/dL, increased from his baseline of 1.1 mg/dL several months prior, and his serum calcium was 14.9 mg/dL. Ten days prior to admission he developed increasing fatigue, decreased appetite, and decreased urine output, which he attributed to recent manual labor during summer. He reported taking an occasional calcium carbonate (Tums) for dyspepsia. He did not report pain or other complaints.
His medical history included hypertension and hyperlipidemia. He had a colonoscopy 1 year prior to presentation that was significant for a high‐grade dysplastic polyp and was currently due for repeat colonoscopy. His medications included clonidine, lisinopril, and aspirin. He had no recent medication changes. He had a 30 pack/year history of cigarette smoking and drank occasionally.
On physical exam, his temperature was 99.8F, blood pressure 97/48 mmHg, heart rate 89 beats per minute, respirations 20 breaths per minute, with a room air saturation of 97%. He had dry mucus membranes and the remainder of the physical exam was unremarkable.
In the emergency department laboratory testing revealed a creatinine of 4.6 mg/dL, serum total calcium of 15.9 mg/dL, serum bicarbonate level of 26 mmol/L, phosphate of 3.9 mg/dL, albumin of 4.4 gm/dL, and alkaline phosphatase of 92 IU/L. The urine specific gravity was 1.019 gm/mL (see Table 1 for the patient's complete admission laboratory values).
| Result (Normal Range) | |
|---|---|
| |
| At admission | |
| Sodium (mmol/L) | 135 (135‐145) |
| Potassium (mmol/L) | 4.4 (3.8‐5.0) |
| Chloride (mmol/L) | 97 (98‐107) |
| Bicarbonate (mmol/L) | 25 (22‐31) |
| BUN (mg/dL) | 64 (9‐20) |
| Creatinine (mg/dL) | 4.6 (0.5‐1.5) |
| Calcium (mg/dL) | 15.9 (8.4‐10.2) |
| Albumin (g/dL) | 4.4 (3.5‐5.0) |
| Alkaline phosphatase (U/L) | 92 (38‐126) |
| ALT (U/L) | 24 (11‐66) |
| AST (U/L) | 17 (15‐46) |
| Bilirubin, total (mg/dL) | 0.3 (0.3‐1.2) |
| Ionized calcium (mmol/L) | 1.58 (1.13‐1.32) |
| Creatinine kinase (U/L) | 83 (35‐232) |
| WBC (thousands/cm2) | 12.1 (4‐11) |
| Hemoglobin (g/dL) | 16.5 (14‐18) |
| Hematocrit (%) | 48.6 (40‐54) |
| Platelets (thousands/cm2) | 224 (150‐350) |
| Intact PTH (pg/mL) | 18.78 (15‐65) |
| PTHrP (pmol/L) | 2.5 (5) |
| 25‐OH‐Vitamin D (ng/mL) | 23 (16‐74) |
| H. pylori antibody | Negative |
| SPEP | No M‐spike |
| UPEP | No M‐spike |
| Hospital Day 4 (date of discharge) | |
| BUN (mg/dL) | 21 (9‐20) |
| Creatinine (mg/dL) | 1.6 (0.5‐1.5) |
| Calcium(mg/dL) | 8.4 (8.4‐10.2) |
| WBC (thousands/cm2) | 6.5 (4‐11) |
| Hemoglobin (g/dL) | 14.1 (14‐18) |
| Hematocrit (%) | 40.8 (40‐54) |
| Day 10 (at follow‐up) | |
| BUN (mg/dL) | 16 (9‐20) |
| Creatinine (mg/dL) | 0.9 (0.5‐1.5) |
| Calcium (mg/dL) | 8.1 (8.4‐10.2) |
| Intact PTH (pg/mL) | 240 (15‐65) |
His intact parathyroid (PTH) hormone was 18.8 pg/mL (normal, 15‐65). PTH hormone‐related peptide (PTHrP) was 2.5 pmol/L. After reviewing these laboratory test results, we proceeded with further questioning, during which he admitted to taking approximately 15 to 20 Tums (>7.5 gm of calcium carbonate) daily for dyspepsia rather than the occasional Tums he had originally reported. Over the next 3 days, his calcium decreased to 8.4 mg/dL and his creatinine decreased to 1.6 mg/dL with intravenous hydration. His fatigue improved. His 25‐hydroxy vitamin D (25‐OH‐Vitamin D) level was 23 ng/mL (normal, 16‐74 ng/mL).
At his 1 week follow‐up, his calcium was 8.1 mg/dL, and his creatinine had returned to normal at 0.9 mg/dL. His intact PTH level was elevated at 240 pg/mL.
Discussion
Milk‐alkali syndrome is now believed to be the third most common reason for hypercalcemia hospital admission.2, 3 Malignancy and primary hyperparathyroidism are the only 2 causes of hypercalcemia more common than milk‐alkali syndrome in hospitalized patients; these must be excluded before making a definitive diagnosis of milk‐alkali syndrome. The differential diagnosis for hypercalcemia also includes other less common etiologies such as medications (hydrochlorothiazide and lithium), as well as familial hypocalciuric hypercalcemia, hyperthyroidism, Addison's disease, acromegaly, tertiary hyperparathyroidism, and vitamin D intoxication. Physicians often discover hypercalcemia incidentally on routine laboratory tests, and diagnostic workup should include a thorough history and physical examination, as well as further laboratory evaluation.
The diagnosis of milk‐alkali syndrome requires a history of increased calcium and alkali intake, but is otherwise a diagnosis of exclusion. Given the increasing consumption of nonprescribed calcium supplements, one should have a high index of suspicion for the diagnosis of milk‐alkali syndrome, as patients may not consider calcium carbonate to be hazardous or even a medication and thus may not report calcium carbonate consumption. Patients often view calcium carbonate as a benign treatment for dyspepsia. Its over‐the‐counter availability and economical price make it a common self‐treatment for minor dyspepsia or as prevention of osteoporosis. Calcium supplementation is increasingly added to many products, making it easy for patients to consume large quantities of calcium unknowingly. Of note, without the absorbable alkali supplied by the carbonate in calcium carbonate (Tums), milk‐alkali syndrome does not occur. The amount of calcium carbonate necessary to cause milk‐alkali syndrome is not well known, though it is speculated to be as little as 5 to 10 g of calcium in the form of calcium carbonate, especially in those with other risk factors for hypercalcemia such as chronic renal insufficiency or vomiting.2 Workup of hypercalcemia should entail careful questioning about medications, as well as over‐the‐counter supplements, vitamins, and foods.
Manifestations of the milk‐alkali syndrome include renal failure, metabolic alkalosis, and volume contraction. Normally, the kidneys prevent hypercalcemia by excretion of excess calcium. Hypercalcemia can cause tubular damage and vasoconstriction of the renal afferent arteriole leading to acute renal failure.2, 4 Hypercalcemia can also cause nephrogenic diabetes insipidus, causing impaired renal concentrating ability, leading to increased sodium excretion and volume contraction.2 In addition, alkalosis further impairs calciuresis.2 Laboratory values usually reveal suppressed PTH and vitamin D levels due to hypercalcemia caused by exogenous intake of calcium.5 Hypercalcemia causes suppression of PTH, which can lead to hyperphosphatemia, as well as decreased conversion of vitamin D to the active 1,25‐dihyroxyvitamin‐D form.
The management of hypercalcemia due to milk‐alkali syndrome is supportive and includes saline hydration as well as withholding calcium carbonate. Management of hypercalcemia due to malignancy and hyperparathyroidism includes bisphosphonates with the addition of calcitonin if symptoms are severe.6, 7 There is no evidence that supports the use of bisphosphonates in the treatment of milk‐alkali syndrome. Loop diuretics are sometimes used to promote calciuresis, though evidence is lacking to support this, and it may worsen renal failure.6
In this case, a middle‐aged man took greater than the recommended dose of calcium carbonate for dyspepsia, which led to the development of acute renal failure and hypercalcemia. At first, the patient did not provide an accurate history of the extent of his calcium carbonate ingestion, leading us to focus on hyperparathyroidism or malignancy. With aggressive hydration and cessation of calcium carbonate, his renal function and serum calcium returned to baseline. Because we initially assumed occult malignancy as the most likely diagnosis, we gave the patient pamidronate. The patient did not have a significant alkalemia (serum bicarbonate level was normal). This was thought to be due to the patient's degree of renal failure causing a concomitant metabolic acidosis. The patient's follow‐up elevated PTH level may be explained by bisphosphonate administration or underlying primary hyperparathyroidism. Of note, decreasing calcium levels have also been speculated to be a cause of high PTH levels.8
In conclusion, physicians should have a high index of suspicion for milk‐alkali syndrome in patients with hypercalcemia. Calcium carbonate is responsible for most cases of milk‐alkali syndrome, and clinicians should inquire about the use of this supplement in all patients with hypercalcemia. Milk‐alkali syndrome is no longer a merely a historical curiosity; it is currently the third most common cause of hospital admissions for hypercalcemia.
Historically, the milk‐alkali syndrome developed as an adverse reaction to the Sippy regimen of frequent feedings of milk, cream, and alkaline powders as treatment for peptic ulcer disease.1 The classic description includes hypercalcemia, metabolic alkalosis, and renal failure. This syndrome seemingly disappeared when modern acid suppression therapies such as histamine‐2 blockers and proton pump inhibitors improved dyspepsia treatment. Over the past 20 years, milk‐alkali syndrome has had a resurgence, as consumption of supplements containing calcium has increased.2 Calcium carbonate supplements are a popular over‐the‐counter treatment for osteoporosis, dyspepsia, hypocalcemia, and hyperphosphatemia; these supplements provide both the calcium and alkali required for the development of milk‐alkali syndrome.
A 46‐year‐old man presented to the emergency department after his physician ordered outpatient laboratory tests to evaluate his fatigue. The patient was found to have acute renal failure and hypercalcemia. His serum creatinine was 3.6 mg/dL, increased from his baseline of 1.1 mg/dL several months prior, and his serum calcium was 14.9 mg/dL. Ten days prior to admission he developed increasing fatigue, decreased appetite, and decreased urine output, which he attributed to recent manual labor during summer. He reported taking an occasional calcium carbonate (Tums) for dyspepsia. He did not report pain or other complaints.
His medical history included hypertension and hyperlipidemia. He had a colonoscopy 1 year prior to presentation that was significant for a high‐grade dysplastic polyp and was currently due for repeat colonoscopy. His medications included clonidine, lisinopril, and aspirin. He had no recent medication changes. He had a 30 pack/year history of cigarette smoking and drank occasionally.
On physical exam, his temperature was 99.8F, blood pressure 97/48 mmHg, heart rate 89 beats per minute, respirations 20 breaths per minute, with a room air saturation of 97%. He had dry mucus membranes and the remainder of the physical exam was unremarkable.
In the emergency department laboratory testing revealed a creatinine of 4.6 mg/dL, serum total calcium of 15.9 mg/dL, serum bicarbonate level of 26 mmol/L, phosphate of 3.9 mg/dL, albumin of 4.4 gm/dL, and alkaline phosphatase of 92 IU/L. The urine specific gravity was 1.019 gm/mL (see Table 1 for the patient's complete admission laboratory values).
| Result (Normal Range) | |
|---|---|
| |
| At admission | |
| Sodium (mmol/L) | 135 (135‐145) |
| Potassium (mmol/L) | 4.4 (3.8‐5.0) |
| Chloride (mmol/L) | 97 (98‐107) |
| Bicarbonate (mmol/L) | 25 (22‐31) |
| BUN (mg/dL) | 64 (9‐20) |
| Creatinine (mg/dL) | 4.6 (0.5‐1.5) |
| Calcium (mg/dL) | 15.9 (8.4‐10.2) |
| Albumin (g/dL) | 4.4 (3.5‐5.0) |
| Alkaline phosphatase (U/L) | 92 (38‐126) |
| ALT (U/L) | 24 (11‐66) |
| AST (U/L) | 17 (15‐46) |
| Bilirubin, total (mg/dL) | 0.3 (0.3‐1.2) |
| Ionized calcium (mmol/L) | 1.58 (1.13‐1.32) |
| Creatinine kinase (U/L) | 83 (35‐232) |
| WBC (thousands/cm2) | 12.1 (4‐11) |
| Hemoglobin (g/dL) | 16.5 (14‐18) |
| Hematocrit (%) | 48.6 (40‐54) |
| Platelets (thousands/cm2) | 224 (150‐350) |
| Intact PTH (pg/mL) | 18.78 (15‐65) |
| PTHrP (pmol/L) | 2.5 (5) |
| 25‐OH‐Vitamin D (ng/mL) | 23 (16‐74) |
| H. pylori antibody | Negative |
| SPEP | No M‐spike |
| UPEP | No M‐spike |
| Hospital Day 4 (date of discharge) | |
| BUN (mg/dL) | 21 (9‐20) |
| Creatinine (mg/dL) | 1.6 (0.5‐1.5) |
| Calcium(mg/dL) | 8.4 (8.4‐10.2) |
| WBC (thousands/cm2) | 6.5 (4‐11) |
| Hemoglobin (g/dL) | 14.1 (14‐18) |
| Hematocrit (%) | 40.8 (40‐54) |
| Day 10 (at follow‐up) | |
| BUN (mg/dL) | 16 (9‐20) |
| Creatinine (mg/dL) | 0.9 (0.5‐1.5) |
| Calcium (mg/dL) | 8.1 (8.4‐10.2) |
| Intact PTH (pg/mL) | 240 (15‐65) |
His intact parathyroid (PTH) hormone was 18.8 pg/mL (normal, 15‐65). PTH hormone‐related peptide (PTHrP) was 2.5 pmol/L. After reviewing these laboratory test results, we proceeded with further questioning, during which he admitted to taking approximately 15 to 20 Tums (>7.5 gm of calcium carbonate) daily for dyspepsia rather than the occasional Tums he had originally reported. Over the next 3 days, his calcium decreased to 8.4 mg/dL and his creatinine decreased to 1.6 mg/dL with intravenous hydration. His fatigue improved. His 25‐hydroxy vitamin D (25‐OH‐Vitamin D) level was 23 ng/mL (normal, 16‐74 ng/mL).
At his 1 week follow‐up, his calcium was 8.1 mg/dL, and his creatinine had returned to normal at 0.9 mg/dL. His intact PTH level was elevated at 240 pg/mL.
Discussion
Milk‐alkali syndrome is now believed to be the third most common reason for hypercalcemia hospital admission.2, 3 Malignancy and primary hyperparathyroidism are the only 2 causes of hypercalcemia more common than milk‐alkali syndrome in hospitalized patients; these must be excluded before making a definitive diagnosis of milk‐alkali syndrome. The differential diagnosis for hypercalcemia also includes other less common etiologies such as medications (hydrochlorothiazide and lithium), as well as familial hypocalciuric hypercalcemia, hyperthyroidism, Addison's disease, acromegaly, tertiary hyperparathyroidism, and vitamin D intoxication. Physicians often discover hypercalcemia incidentally on routine laboratory tests, and diagnostic workup should include a thorough history and physical examination, as well as further laboratory evaluation.
The diagnosis of milk‐alkali syndrome requires a history of increased calcium and alkali intake, but is otherwise a diagnosis of exclusion. Given the increasing consumption of nonprescribed calcium supplements, one should have a high index of suspicion for the diagnosis of milk‐alkali syndrome, as patients may not consider calcium carbonate to be hazardous or even a medication and thus may not report calcium carbonate consumption. Patients often view calcium carbonate as a benign treatment for dyspepsia. Its over‐the‐counter availability and economical price make it a common self‐treatment for minor dyspepsia or as prevention of osteoporosis. Calcium supplementation is increasingly added to many products, making it easy for patients to consume large quantities of calcium unknowingly. Of note, without the absorbable alkali supplied by the carbonate in calcium carbonate (Tums), milk‐alkali syndrome does not occur. The amount of calcium carbonate necessary to cause milk‐alkali syndrome is not well known, though it is speculated to be as little as 5 to 10 g of calcium in the form of calcium carbonate, especially in those with other risk factors for hypercalcemia such as chronic renal insufficiency or vomiting.2 Workup of hypercalcemia should entail careful questioning about medications, as well as over‐the‐counter supplements, vitamins, and foods.
Manifestations of the milk‐alkali syndrome include renal failure, metabolic alkalosis, and volume contraction. Normally, the kidneys prevent hypercalcemia by excretion of excess calcium. Hypercalcemia can cause tubular damage and vasoconstriction of the renal afferent arteriole leading to acute renal failure.2, 4 Hypercalcemia can also cause nephrogenic diabetes insipidus, causing impaired renal concentrating ability, leading to increased sodium excretion and volume contraction.2 In addition, alkalosis further impairs calciuresis.2 Laboratory values usually reveal suppressed PTH and vitamin D levels due to hypercalcemia caused by exogenous intake of calcium.5 Hypercalcemia causes suppression of PTH, which can lead to hyperphosphatemia, as well as decreased conversion of vitamin D to the active 1,25‐dihyroxyvitamin‐D form.
The management of hypercalcemia due to milk‐alkali syndrome is supportive and includes saline hydration as well as withholding calcium carbonate. Management of hypercalcemia due to malignancy and hyperparathyroidism includes bisphosphonates with the addition of calcitonin if symptoms are severe.6, 7 There is no evidence that supports the use of bisphosphonates in the treatment of milk‐alkali syndrome. Loop diuretics are sometimes used to promote calciuresis, though evidence is lacking to support this, and it may worsen renal failure.6
In this case, a middle‐aged man took greater than the recommended dose of calcium carbonate for dyspepsia, which led to the development of acute renal failure and hypercalcemia. At first, the patient did not provide an accurate history of the extent of his calcium carbonate ingestion, leading us to focus on hyperparathyroidism or malignancy. With aggressive hydration and cessation of calcium carbonate, his renal function and serum calcium returned to baseline. Because we initially assumed occult malignancy as the most likely diagnosis, we gave the patient pamidronate. The patient did not have a significant alkalemia (serum bicarbonate level was normal). This was thought to be due to the patient's degree of renal failure causing a concomitant metabolic acidosis. The patient's follow‐up elevated PTH level may be explained by bisphosphonate administration or underlying primary hyperparathyroidism. Of note, decreasing calcium levels have also been speculated to be a cause of high PTH levels.8
In conclusion, physicians should have a high index of suspicion for milk‐alkali syndrome in patients with hypercalcemia. Calcium carbonate is responsible for most cases of milk‐alkali syndrome, and clinicians should inquire about the use of this supplement in all patients with hypercalcemia. Milk‐alkali syndrome is no longer a merely a historical curiosity; it is currently the third most common cause of hospital admissions for hypercalcemia.
- .Landmark article May 15, 1915: Gastric and duodenal ulcer. Medical cure by an efficient removal of gastric juice corrosion. By Bertram W. Sippy.JAMA.1983;250(16):2192–2197.
- ,,,.Milk‐alkali syndrome: a historical review and description of the modern version of the syndrome.Am J Med Sci.2006;331(5):233–242.
- ,,.Milk‐alkali syndrome is a major cause of hypercalcaemia among non‐end‐stage renal disease (non‐ESRD) inpatients.Clin Endocrinol (Oxf).2005;63(5):566–576.
- ,.Milk‐alkali syndrome associated with calcium carbonate consumption. Report of 7 patients with parathyroid hormone levels and an estimate of prevalence among patients hospitalized with hypercalcemia.Medicine (Baltimore).1995;74(2):89–96.
- ,,,,,.The milk‐alkali syndrome. A reversible form of acute renal failure.Arch Intern Med.1993;153(8):1005–1010.
- ,,.Narrative review: furosemide for hypercalcemia: an unproven yet common practice.Ann Intern Med.2008;149(4):259–263.
- .Salmon calcitonin in the acute management of hypercalcemia.Calcif Tissue Int.1990;46(suppl):S26–S30.
- ,.Milk alkali syndrome. Does it exist and can it be differentiated from primary hyperparathyroidism?Ann Surg.1983;197(4):427–433.
- .Landmark article May 15, 1915: Gastric and duodenal ulcer. Medical cure by an efficient removal of gastric juice corrosion. By Bertram W. Sippy.JAMA.1983;250(16):2192–2197.
- ,,,.Milk‐alkali syndrome: a historical review and description of the modern version of the syndrome.Am J Med Sci.2006;331(5):233–242.
- ,,.Milk‐alkali syndrome is a major cause of hypercalcaemia among non‐end‐stage renal disease (non‐ESRD) inpatients.Clin Endocrinol (Oxf).2005;63(5):566–576.
- ,.Milk‐alkali syndrome associated with calcium carbonate consumption. Report of 7 patients with parathyroid hormone levels and an estimate of prevalence among patients hospitalized with hypercalcemia.Medicine (Baltimore).1995;74(2):89–96.
- ,,,,,.The milk‐alkali syndrome. A reversible form of acute renal failure.Arch Intern Med.1993;153(8):1005–1010.
- ,,.Narrative review: furosemide for hypercalcemia: an unproven yet common practice.Ann Intern Med.2008;149(4):259–263.
- .Salmon calcitonin in the acute management of hypercalcemia.Calcif Tissue Int.1990;46(suppl):S26–S30.
- ,.Milk alkali syndrome. Does it exist and can it be differentiated from primary hyperparathyroidism?Ann Surg.1983;197(4):427–433.
Nonphysician Providers
The current state of our profession is that the US population is aging rapidly, requiring ever more healthcare, and there is a stagnant number of physicians to care for them. The question of who will care for our aging population has been raised over and over in the past decade but the question is worth repeating. As our country continues to deliver state‐of‐the‐art medical care, it is slow to embrace the notion that in order for it to continue, it will need to incorporate the professions of advanced practice nurses and physician assistants. Without these nonphysician providers our medical community will not be able to reach the patients we have sworn to treat.
The percent of the US population age >65 years is projected to increase from 12.4% in 2000 to 19.6% in 2030. The number of persons age >65 years is expected to increase from approximately 35 million in 2000 to an estimated 71 million in 2030, and the number of persons age >80 years is expected to increase from 9.3 million in 2000 to 19.5 million in 2030.1 Our aging America is also coupled with a growing physician shortage. In its report entitled Physician Workforce Policy Guidelines for the United States, 2000‐2020, the Council on Graduate Medical Education recommended increasing the number of medical school graduates by 3000 per year by the year 2015 to meet the increasing need.2 Given the current trend of decreasing physician reimbursement coupled with the average medical school debt of $139,517,3 it is doubtful that the extra 3000 physicians needed to graduate in 2015 will actually ever do so. Despite this possible additional physician workforce, there still stands to be enormous need for the nonphysician provider with our rapidly expanding senior population.
Our nation's hospitals are by no means spared from our aging population or physician shortage. In fact, they are likely to be the hardest hit. Hospitalists are already feeling the pressure of an overstressed workforce coupled with increasing patient volume.4 There is a growing body of evidence supporting the successful collaboration between hospitalists and nurse practitioners (NPs)/physician assistants (PAs) (collectively, nonphysician providers [NPPs]). No longer are NPPs only working in outpatient practices or in the operating room, but they are actively involved with inpatient medical units improving our Hospital Medicine (HM) specialty. According to Myers et al.,5 the hospitalist NP model improved program finances and increased physician and resident satisfaction. In order for Hospital Medicine to create increasing value for its parent hospital or to the community it serves, NPPs will need increased integration into our care model for improved overall efficiency. We focus herein on the advantages and potential benefits of NPPs relating to their varied roles within HM.
Scope of Practice
The scope of practice of NPPs is regulated by each individual state board of registration. However, differences from state to state are usually minor and general statements on the practice scope of PAs and NPs can be made.
PAs
PAs practice under the supervision of a physician. PAs are trained in programs affiliated with medical schools and according to the medical model of care that emphasizes diagnosis and treatment. Most PAs graduate with a masters of science degree. According to the American Association of Physician Assistants (AAPA), the scope of practice is guided by state law, facility policy, and delegatory decisions made by the supervising physician.6 Prior experience and training should be the framework for scope of practice decisions. All 50 states allow PAs to prescribe with some oversight and restriction of schedule 2 controlled substances or by using a state formulary. The AAPA embraces the concept of the physician as the captain of the healthcare team and sees the PA role as entirely complementary to the care provided by physicians.7 This means that PAs, under an individual supervision agreement, can prescribe medicines, order and interpret tests, diagnose, and treat patients just as a physician would.
Advanced Practice Nurses
Advanced practice nurses (APNs) are trained under the nursing model and generally have some years of nursing experience before they pursue an entry‐level masters of science degree to become an APN. APNs can be divided into two categories: Clinical nurse specialists, who generally focus on patient and institutional education and are considered experts in nursing practice, and NPs, who have a focus on diagnosis and treatment of medical conditions. A clinical nurse specialist does not have prescriptive training or authority. NP training can be general (adult or family) or specific (eg, acute care, geriatric, pediatric, psychiatric). The American Association of Colleges of Nursing (AACN) has recommended that the entry level of all new NPs should be a clinical doctorate of nursing practice. Although controversial, many colleges have embraced this recommendation and are opening clinical doctorate‐level programs.8 Although some states allow NPs to practice independently, most NPs have a practice agreement with a collaborating physician that delineates the degree of supervision. Generally, the NP's scope of practice is identical to PAs and includes the above‐mentioned activities as proscribed by state regulations and facility bylaws. As with PAs, their prior experience and training should be the most important determinant of their scope of practice in a new position.
Potential Benefits of NPPs
Continuity
If a nonacademic hospitalist program has high yearly turnover due to use of recent medical graduates who are planning to do fellowships, NPPs can provide much needed stability and facilitate orientation of new physicians to the hospital. NPPs who work in academic settings can also provide increased continuity for patients and hospital staff. Residents, fellows, and attendings have certain rotational cycles on each medical service. NPPs generally do not rotate and can be the anchor of a medical team for patients and ancillary staff. Utilizing NPPs as liaisons between the hospitalist team and other members of the care team (eg, nurses, case managers, therapists, and administration) provides continuity for these groups and a central person who can help to facilitate change.
Quality Measures
NPPs can play an important role in hospital compliance with internal hospital or insurance provider quality initiatives. Surveillance of patients and charts for compliance with core measures, infection control, and prevention of complications are within the scope of practice of NPPs and can be incorporated into job descriptions. NPs and PAs will have the added responsibility of not only leading these surveillance teams but also in the correction of outliers given their prescriptive abilities. This will become an increasingly important task as reimbursement for preventable complications is curtailed. Additionally, the development and implementation of clinical pathways can be a focus of the NPP role to standardize and enhance quality of care.
Multidisciplinary Team Approach
Multidisciplinary teams that consist of NPPs, physicians, nurses, and therapists have been shown to increase communication and collaboration between participants.9 Mary Naylor, a Professor of Nursing at the University of Pennsylvania, has authored multiple articles and studies which examine the benefit of a multidisciplinary team that includes APNs with hospitalized patients. She has found that involving APNs in patient care, discharges, and routine follow‐up after discharge led to longer time to readmissions and decreased healthcare costs.1012 Furthermore, a nonteaching group consisting of NPPs, fellows, and attendings at the Mayo Clinic found increased physician satisfaction, shorter length of stay (LOS), and increased efficiency for their patients.13 A study done at JFK Medical Center in Florida noted that a collaborative practice which included unit‐based NPs serving in the dual role of NP and clinical nurse specialist increased patient satisfaction and improved patient outcomes.14
Financial Advantages
Efficiency and quality care are the cornerstones of HM. The partnership of NPPs within the specialty is creating even better performance. Models incorporating NPPs in the Hospitalist team approach are continuing to drive efficiency. Cowan et al.15 demonstrated that a multidisciplinary team, including nurse practitioners, decreased LOS from 6.01 to 5.0 and a reduced cost by $1,591 per patient. It is this team approach that will lift our specialty to be the model of care for all future hospital practice.
Another factor in determining the fiscal advantage of NPPs is salary and medical liability comparison. According to the 2007 Society of Hospital Medicine (SHM) Survey, the average hospitalist salary is approaching $190,000, compared to an average NP earning $87,000 and PA earning $84,500.4 Furthermore, the average internal medicine malpractice payment for physicians ranges from $14,237 to $68,867.16 In comparison, the average malpractice insurance premium for NPPs varies from state to state but is approximately $800 to $2000 per year.17, 18 With increasing fiscal scrutiny from hospitals, HM groups (HMGs) will need to include NPPs to be fiscally stable.
Models of Care
There are many models for NPP roles in hospital medicine groups. Some groups use NPPs in the same role as physicians. They perform admissions, rounding, and discharges with varying degrees of oversight by physicians. Other groups use NPPs for a more limited role, such as exclusively performing histories and physicals in the emergency department or handling discharges on the wards. It is important to take into account the preferences and expectations of NPPs when designing job descriptions. While some NPPs may like the fast pace and quick turnover of admissions and discharges, others may prefer to follow patients throughout their hospital stay. The quality of handoffs is crucial if the former model is used, just as it is with physicians in this more truncated role. An NPP who works in a nonacademic model will likely have more autonomy and control over patient care decisions. An NPP role in the teaching service of an academic hospital is likely to be more collaborative and focus more on quality initiatives, patient teaching, and communication. It is crucial to design an NPP model that is sustainable with very strong support of management once the NPP is hired and orientated.
Registered Nurses And Hospital Medicine
Patient handoffs and communication are one of the most challenging aspects of an HMG. There is an increasing movement, throughout the country, to incorporate registered nurses (RNs) into daily workflow. The RN on the HM team can serve to augment the communication and workflow process. A highly motivated and organized registered nurse can help to improve overall provider's workflow efficiency. Communication to primary care physician and collecting ancillary medical information can allow the provider to treat more patients in a given shift and decrease the liability risk from lack of information. As HM organizations and hospitals become more financially bound, HMGs will need to become more efficient at time management and a dedicated RN can help smooth that process.
Potential Unintended Side Effects
Obviously, integration of NPPs can be a disaster for an HMG if not handled properly. Most hospitalists have heard of an integration of NPP into a group that was an unqualified failure. NPPs can feel unsupported, poorly oriented to the job, or thrown into a situation that is over their heads. Before an NPP is hired into an HMG, there needs to be a thorough examination of the rationale behind the decision and assessment of the hospital culture that will be the host of the new NPP. What does the HMG need for support? Are they looking for a short‐term fix for increased volume or a long‐term strategy to build a multidisciplinary team? Does the hospital culture see NPPs as poorly qualified to act as hospitalists or uniquely qualified to address shortcomings of the program? A clear job description should be the first step in determining what the NPP is expected to do. This can then be shared with the hospital leadership in advance to promote buy‐in. The second step is finding an NPP that fits the goals of the program. A new NPP, by virtue of the fact that they have less clinical hours in training than a physician hospitalist, will need more support and a longer orientation. NPPs who have experience in hospital medicine will have a much shorter orientation. A stepwise approach to orientation can be helpful in assessing skill level of new hires. These NPPs can be initially paired with an enthusiastic physician to provide support and assessment of existing skills. A gradual increase in independence can provide assurance that the NPP is qualified to provide care and gives many opportunities for reevaluation of the NPP. Clear expectations and constructive feedback should ultimately lead to a degree of comfort within the HMG, hospital, and the NPPs themselves.
Conclusions
It is clear that our healthcare system will need a very different approach to the economic problems it is facing. Standardization of care, integrated medical records, and expanded and universal resource utilization will drive the next generation of healthcare providers. The model of a private physician working alone under the direction of only his or her own medical knowledge is a thing of the past. Just as the HM specialty has grown from 300 in 1996 to more than 20,000 in 2008, so shall the integration of NPPs grow into our healthcare fabric.
- Centers for Disease Control and Prevention (CDC). Trends in aging—United States and worldwide. MMWR Morb Mortal Wkly Rep. 2003;52(6):101–104, 106.
- Council on Graduate Medical Education. Physician Workforce Policy Guidelines for the U.S. for 2000‐2020. Rockville, MD: U.S. Department of Health and Human Services;2005.
- American Medical Association. Medical Student Section. Advocacy and Policy. Medical Student Debt. Available at: http://www.ama‐assn.org/ama/pub/category/5349.html. Accessed June 2009.
- Society of Hospital Medicine (SHM). 2007‐2008 SHM Survey: State of the Hospital Medicine Movement. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Surveys2
The current state of our profession is that the US population is aging rapidly, requiring ever more healthcare, and there is a stagnant number of physicians to care for them. The question of who will care for our aging population has been raised over and over in the past decade but the question is worth repeating. As our country continues to deliver state‐of‐the‐art medical care, it is slow to embrace the notion that in order for it to continue, it will need to incorporate the professions of advanced practice nurses and physician assistants. Without these nonphysician providers our medical community will not be able to reach the patients we have sworn to treat.
The percent of the US population age >65 years is projected to increase from 12.4% in 2000 to 19.6% in 2030. The number of persons age >65 years is expected to increase from approximately 35 million in 2000 to an estimated 71 million in 2030, and the number of persons age >80 years is expected to increase from 9.3 million in 2000 to 19.5 million in 2030.1 Our aging America is also coupled with a growing physician shortage. In its report entitled Physician Workforce Policy Guidelines for the United States, 2000‐2020, the Council on Graduate Medical Education recommended increasing the number of medical school graduates by 3000 per year by the year 2015 to meet the increasing need.2 Given the current trend of decreasing physician reimbursement coupled with the average medical school debt of $139,517,3 it is doubtful that the extra 3000 physicians needed to graduate in 2015 will actually ever do so. Despite this possible additional physician workforce, there still stands to be enormous need for the nonphysician provider with our rapidly expanding senior population.
Our nation's hospitals are by no means spared from our aging population or physician shortage. In fact, they are likely to be the hardest hit. Hospitalists are already feeling the pressure of an overstressed workforce coupled with increasing patient volume.4 There is a growing body of evidence supporting the successful collaboration between hospitalists and nurse practitioners (NPs)/physician assistants (PAs) (collectively, nonphysician providers [NPPs]). No longer are NPPs only working in outpatient practices or in the operating room, but they are actively involved with inpatient medical units improving our Hospital Medicine (HM) specialty. According to Myers et al.,5 the hospitalist NP model improved program finances and increased physician and resident satisfaction. In order for Hospital Medicine to create increasing value for its parent hospital or to the community it serves, NPPs will need increased integration into our care model for improved overall efficiency. We focus herein on the advantages and potential benefits of NPPs relating to their varied roles within HM.
Scope of Practice
The scope of practice of NPPs is regulated by each individual state board of registration. However, differences from state to state are usually minor and general statements on the practice scope of PAs and NPs can be made.
PAs
PAs practice under the supervision of a physician. PAs are trained in programs affiliated with medical schools and according to the medical model of care that emphasizes diagnosis and treatment. Most PAs graduate with a masters of science degree. According to the American Association of Physician Assistants (AAPA), the scope of practice is guided by state law, facility policy, and delegatory decisions made by the supervising physician.6 Prior experience and training should be the framework for scope of practice decisions. All 50 states allow PAs to prescribe with some oversight and restriction of schedule 2 controlled substances or by using a state formulary. The AAPA embraces the concept of the physician as the captain of the healthcare team and sees the PA role as entirely complementary to the care provided by physicians.7 This means that PAs, under an individual supervision agreement, can prescribe medicines, order and interpret tests, diagnose, and treat patients just as a physician would.
Advanced Practice Nurses
Advanced practice nurses (APNs) are trained under the nursing model and generally have some years of nursing experience before they pursue an entry‐level masters of science degree to become an APN. APNs can be divided into two categories: Clinical nurse specialists, who generally focus on patient and institutional education and are considered experts in nursing practice, and NPs, who have a focus on diagnosis and treatment of medical conditions. A clinical nurse specialist does not have prescriptive training or authority. NP training can be general (adult or family) or specific (eg, acute care, geriatric, pediatric, psychiatric). The American Association of Colleges of Nursing (AACN) has recommended that the entry level of all new NPs should be a clinical doctorate of nursing practice. Although controversial, many colleges have embraced this recommendation and are opening clinical doctorate‐level programs.8 Although some states allow NPs to practice independently, most NPs have a practice agreement with a collaborating physician that delineates the degree of supervision. Generally, the NP's scope of practice is identical to PAs and includes the above‐mentioned activities as proscribed by state regulations and facility bylaws. As with PAs, their prior experience and training should be the most important determinant of their scope of practice in a new position.
Potential Benefits of NPPs
Continuity
If a nonacademic hospitalist program has high yearly turnover due to use of recent medical graduates who are planning to do fellowships, NPPs can provide much needed stability and facilitate orientation of new physicians to the hospital. NPPs who work in academic settings can also provide increased continuity for patients and hospital staff. Residents, fellows, and attendings have certain rotational cycles on each medical service. NPPs generally do not rotate and can be the anchor of a medical team for patients and ancillary staff. Utilizing NPPs as liaisons between the hospitalist team and other members of the care team (eg, nurses, case managers, therapists, and administration) provides continuity for these groups and a central person who can help to facilitate change.
Quality Measures
NPPs can play an important role in hospital compliance with internal hospital or insurance provider quality initiatives. Surveillance of patients and charts for compliance with core measures, infection control, and prevention of complications are within the scope of practice of NPPs and can be incorporated into job descriptions. NPs and PAs will have the added responsibility of not only leading these surveillance teams but also in the correction of outliers given their prescriptive abilities. This will become an increasingly important task as reimbursement for preventable complications is curtailed. Additionally, the development and implementation of clinical pathways can be a focus of the NPP role to standardize and enhance quality of care.
Multidisciplinary Team Approach
Multidisciplinary teams that consist of NPPs, physicians, nurses, and therapists have been shown to increase communication and collaboration between participants.9 Mary Naylor, a Professor of Nursing at the University of Pennsylvania, has authored multiple articles and studies which examine the benefit of a multidisciplinary team that includes APNs with hospitalized patients. She has found that involving APNs in patient care, discharges, and routine follow‐up after discharge led to longer time to readmissions and decreased healthcare costs.1012 Furthermore, a nonteaching group consisting of NPPs, fellows, and attendings at the Mayo Clinic found increased physician satisfaction, shorter length of stay (LOS), and increased efficiency for their patients.13 A study done at JFK Medical Center in Florida noted that a collaborative practice which included unit‐based NPs serving in the dual role of NP and clinical nurse specialist increased patient satisfaction and improved patient outcomes.14
Financial Advantages
Efficiency and quality care are the cornerstones of HM. The partnership of NPPs within the specialty is creating even better performance. Models incorporating NPPs in the Hospitalist team approach are continuing to drive efficiency. Cowan et al.15 demonstrated that a multidisciplinary team, including nurse practitioners, decreased LOS from 6.01 to 5.0 and a reduced cost by $1,591 per patient. It is this team approach that will lift our specialty to be the model of care for all future hospital practice.
Another factor in determining the fiscal advantage of NPPs is salary and medical liability comparison. According to the 2007 Society of Hospital Medicine (SHM) Survey, the average hospitalist salary is approaching $190,000, compared to an average NP earning $87,000 and PA earning $84,500.4 Furthermore, the average internal medicine malpractice payment for physicians ranges from $14,237 to $68,867.16 In comparison, the average malpractice insurance premium for NPPs varies from state to state but is approximately $800 to $2000 per year.17, 18 With increasing fiscal scrutiny from hospitals, HM groups (HMGs) will need to include NPPs to be fiscally stable.
Models of Care
There are many models for NPP roles in hospital medicine groups. Some groups use NPPs in the same role as physicians. They perform admissions, rounding, and discharges with varying degrees of oversight by physicians. Other groups use NPPs for a more limited role, such as exclusively performing histories and physicals in the emergency department or handling discharges on the wards. It is important to take into account the preferences and expectations of NPPs when designing job descriptions. While some NPPs may like the fast pace and quick turnover of admissions and discharges, others may prefer to follow patients throughout their hospital stay. The quality of handoffs is crucial if the former model is used, just as it is with physicians in this more truncated role. An NPP who works in a nonacademic model will likely have more autonomy and control over patient care decisions. An NPP role in the teaching service of an academic hospital is likely to be more collaborative and focus more on quality initiatives, patient teaching, and communication. It is crucial to design an NPP model that is sustainable with very strong support of management once the NPP is hired and orientated.
Registered Nurses And Hospital Medicine
Patient handoffs and communication are one of the most challenging aspects of an HMG. There is an increasing movement, throughout the country, to incorporate registered nurses (RNs) into daily workflow. The RN on the HM team can serve to augment the communication and workflow process. A highly motivated and organized registered nurse can help to improve overall provider's workflow efficiency. Communication to primary care physician and collecting ancillary medical information can allow the provider to treat more patients in a given shift and decrease the liability risk from lack of information. As HM organizations and hospitals become more financially bound, HMGs will need to become more efficient at time management and a dedicated RN can help smooth that process.
Potential Unintended Side Effects
Obviously, integration of NPPs can be a disaster for an HMG if not handled properly. Most hospitalists have heard of an integration of NPP into a group that was an unqualified failure. NPPs can feel unsupported, poorly oriented to the job, or thrown into a situation that is over their heads. Before an NPP is hired into an HMG, there needs to be a thorough examination of the rationale behind the decision and assessment of the hospital culture that will be the host of the new NPP. What does the HMG need for support? Are they looking for a short‐term fix for increased volume or a long‐term strategy to build a multidisciplinary team? Does the hospital culture see NPPs as poorly qualified to act as hospitalists or uniquely qualified to address shortcomings of the program? A clear job description should be the first step in determining what the NPP is expected to do. This can then be shared with the hospital leadership in advance to promote buy‐in. The second step is finding an NPP that fits the goals of the program. A new NPP, by virtue of the fact that they have less clinical hours in training than a physician hospitalist, will need more support and a longer orientation. NPPs who have experience in hospital medicine will have a much shorter orientation. A stepwise approach to orientation can be helpful in assessing skill level of new hires. These NPPs can be initially paired with an enthusiastic physician to provide support and assessment of existing skills. A gradual increase in independence can provide assurance that the NPP is qualified to provide care and gives many opportunities for reevaluation of the NPP. Clear expectations and constructive feedback should ultimately lead to a degree of comfort within the HMG, hospital, and the NPPs themselves.
Conclusions
It is clear that our healthcare system will need a very different approach to the economic problems it is facing. Standardization of care, integrated medical records, and expanded and universal resource utilization will drive the next generation of healthcare providers. The model of a private physician working alone under the direction of only his or her own medical knowledge is a thing of the past. Just as the HM specialty has grown from 300 in 1996 to more than 20,000 in 2008, so shall the integration of NPPs grow into our healthcare fabric.
The current state of our profession is that the US population is aging rapidly, requiring ever more healthcare, and there is a stagnant number of physicians to care for them. The question of who will care for our aging population has been raised over and over in the past decade but the question is worth repeating. As our country continues to deliver state‐of‐the‐art medical care, it is slow to embrace the notion that in order for it to continue, it will need to incorporate the professions of advanced practice nurses and physician assistants. Without these nonphysician providers our medical community will not be able to reach the patients we have sworn to treat.
The percent of the US population age >65 years is projected to increase from 12.4% in 2000 to 19.6% in 2030. The number of persons age >65 years is expected to increase from approximately 35 million in 2000 to an estimated 71 million in 2030, and the number of persons age >80 years is expected to increase from 9.3 million in 2000 to 19.5 million in 2030.1 Our aging America is also coupled with a growing physician shortage. In its report entitled Physician Workforce Policy Guidelines for the United States, 2000‐2020, the Council on Graduate Medical Education recommended increasing the number of medical school graduates by 3000 per year by the year 2015 to meet the increasing need.2 Given the current trend of decreasing physician reimbursement coupled with the average medical school debt of $139,517,3 it is doubtful that the extra 3000 physicians needed to graduate in 2015 will actually ever do so. Despite this possible additional physician workforce, there still stands to be enormous need for the nonphysician provider with our rapidly expanding senior population.
Our nation's hospitals are by no means spared from our aging population or physician shortage. In fact, they are likely to be the hardest hit. Hospitalists are already feeling the pressure of an overstressed workforce coupled with increasing patient volume.4 There is a growing body of evidence supporting the successful collaboration between hospitalists and nurse practitioners (NPs)/physician assistants (PAs) (collectively, nonphysician providers [NPPs]). No longer are NPPs only working in outpatient practices or in the operating room, but they are actively involved with inpatient medical units improving our Hospital Medicine (HM) specialty. According to Myers et al.,5 the hospitalist NP model improved program finances and increased physician and resident satisfaction. In order for Hospital Medicine to create increasing value for its parent hospital or to the community it serves, NPPs will need increased integration into our care model for improved overall efficiency. We focus herein on the advantages and potential benefits of NPPs relating to their varied roles within HM.
Scope of Practice
The scope of practice of NPPs is regulated by each individual state board of registration. However, differences from state to state are usually minor and general statements on the practice scope of PAs and NPs can be made.
PAs
PAs practice under the supervision of a physician. PAs are trained in programs affiliated with medical schools and according to the medical model of care that emphasizes diagnosis and treatment. Most PAs graduate with a masters of science degree. According to the American Association of Physician Assistants (AAPA), the scope of practice is guided by state law, facility policy, and delegatory decisions made by the supervising physician.6 Prior experience and training should be the framework for scope of practice decisions. All 50 states allow PAs to prescribe with some oversight and restriction of schedule 2 controlled substances or by using a state formulary. The AAPA embraces the concept of the physician as the captain of the healthcare team and sees the PA role as entirely complementary to the care provided by physicians.7 This means that PAs, under an individual supervision agreement, can prescribe medicines, order and interpret tests, diagnose, and treat patients just as a physician would.
Advanced Practice Nurses
Advanced practice nurses (APNs) are trained under the nursing model and generally have some years of nursing experience before they pursue an entry‐level masters of science degree to become an APN. APNs can be divided into two categories: Clinical nurse specialists, who generally focus on patient and institutional education and are considered experts in nursing practice, and NPs, who have a focus on diagnosis and treatment of medical conditions. A clinical nurse specialist does not have prescriptive training or authority. NP training can be general (adult or family) or specific (eg, acute care, geriatric, pediatric, psychiatric). The American Association of Colleges of Nursing (AACN) has recommended that the entry level of all new NPs should be a clinical doctorate of nursing practice. Although controversial, many colleges have embraced this recommendation and are opening clinical doctorate‐level programs.8 Although some states allow NPs to practice independently, most NPs have a practice agreement with a collaborating physician that delineates the degree of supervision. Generally, the NP's scope of practice is identical to PAs and includes the above‐mentioned activities as proscribed by state regulations and facility bylaws. As with PAs, their prior experience and training should be the most important determinant of their scope of practice in a new position.
Potential Benefits of NPPs
Continuity
If a nonacademic hospitalist program has high yearly turnover due to use of recent medical graduates who are planning to do fellowships, NPPs can provide much needed stability and facilitate orientation of new physicians to the hospital. NPPs who work in academic settings can also provide increased continuity for patients and hospital staff. Residents, fellows, and attendings have certain rotational cycles on each medical service. NPPs generally do not rotate and can be the anchor of a medical team for patients and ancillary staff. Utilizing NPPs as liaisons between the hospitalist team and other members of the care team (eg, nurses, case managers, therapists, and administration) provides continuity for these groups and a central person who can help to facilitate change.
Quality Measures
NPPs can play an important role in hospital compliance with internal hospital or insurance provider quality initiatives. Surveillance of patients and charts for compliance with core measures, infection control, and prevention of complications are within the scope of practice of NPPs and can be incorporated into job descriptions. NPs and PAs will have the added responsibility of not only leading these surveillance teams but also in the correction of outliers given their prescriptive abilities. This will become an increasingly important task as reimbursement for preventable complications is curtailed. Additionally, the development and implementation of clinical pathways can be a focus of the NPP role to standardize and enhance quality of care.
Multidisciplinary Team Approach
Multidisciplinary teams that consist of NPPs, physicians, nurses, and therapists have been shown to increase communication and collaboration between participants.9 Mary Naylor, a Professor of Nursing at the University of Pennsylvania, has authored multiple articles and studies which examine the benefit of a multidisciplinary team that includes APNs with hospitalized patients. She has found that involving APNs in patient care, discharges, and routine follow‐up after discharge led to longer time to readmissions and decreased healthcare costs.1012 Furthermore, a nonteaching group consisting of NPPs, fellows, and attendings at the Mayo Clinic found increased physician satisfaction, shorter length of stay (LOS), and increased efficiency for their patients.13 A study done at JFK Medical Center in Florida noted that a collaborative practice which included unit‐based NPs serving in the dual role of NP and clinical nurse specialist increased patient satisfaction and improved patient outcomes.14
Financial Advantages
Efficiency and quality care are the cornerstones of HM. The partnership of NPPs within the specialty is creating even better performance. Models incorporating NPPs in the Hospitalist team approach are continuing to drive efficiency. Cowan et al.15 demonstrated that a multidisciplinary team, including nurse practitioners, decreased LOS from 6.01 to 5.0 and a reduced cost by $1,591 per patient. It is this team approach that will lift our specialty to be the model of care for all future hospital practice.
Another factor in determining the fiscal advantage of NPPs is salary and medical liability comparison. According to the 2007 Society of Hospital Medicine (SHM) Survey, the average hospitalist salary is approaching $190,000, compared to an average NP earning $87,000 and PA earning $84,500.4 Furthermore, the average internal medicine malpractice payment for physicians ranges from $14,237 to $68,867.16 In comparison, the average malpractice insurance premium for NPPs varies from state to state but is approximately $800 to $2000 per year.17, 18 With increasing fiscal scrutiny from hospitals, HM groups (HMGs) will need to include NPPs to be fiscally stable.
Models of Care
There are many models for NPP roles in hospital medicine groups. Some groups use NPPs in the same role as physicians. They perform admissions, rounding, and discharges with varying degrees of oversight by physicians. Other groups use NPPs for a more limited role, such as exclusively performing histories and physicals in the emergency department or handling discharges on the wards. It is important to take into account the preferences and expectations of NPPs when designing job descriptions. While some NPPs may like the fast pace and quick turnover of admissions and discharges, others may prefer to follow patients throughout their hospital stay. The quality of handoffs is crucial if the former model is used, just as it is with physicians in this more truncated role. An NPP who works in a nonacademic model will likely have more autonomy and control over patient care decisions. An NPP role in the teaching service of an academic hospital is likely to be more collaborative and focus more on quality initiatives, patient teaching, and communication. It is crucial to design an NPP model that is sustainable with very strong support of management once the NPP is hired and orientated.
Registered Nurses And Hospital Medicine
Patient handoffs and communication are one of the most challenging aspects of an HMG. There is an increasing movement, throughout the country, to incorporate registered nurses (RNs) into daily workflow. The RN on the HM team can serve to augment the communication and workflow process. A highly motivated and organized registered nurse can help to improve overall provider's workflow efficiency. Communication to primary care physician and collecting ancillary medical information can allow the provider to treat more patients in a given shift and decrease the liability risk from lack of information. As HM organizations and hospitals become more financially bound, HMGs will need to become more efficient at time management and a dedicated RN can help smooth that process.
Potential Unintended Side Effects
Obviously, integration of NPPs can be a disaster for an HMG if not handled properly. Most hospitalists have heard of an integration of NPP into a group that was an unqualified failure. NPPs can feel unsupported, poorly oriented to the job, or thrown into a situation that is over their heads. Before an NPP is hired into an HMG, there needs to be a thorough examination of the rationale behind the decision and assessment of the hospital culture that will be the host of the new NPP. What does the HMG need for support? Are they looking for a short‐term fix for increased volume or a long‐term strategy to build a multidisciplinary team? Does the hospital culture see NPPs as poorly qualified to act as hospitalists or uniquely qualified to address shortcomings of the program? A clear job description should be the first step in determining what the NPP is expected to do. This can then be shared with the hospital leadership in advance to promote buy‐in. The second step is finding an NPP that fits the goals of the program. A new NPP, by virtue of the fact that they have less clinical hours in training than a physician hospitalist, will need more support and a longer orientation. NPPs who have experience in hospital medicine will have a much shorter orientation. A stepwise approach to orientation can be helpful in assessing skill level of new hires. These NPPs can be initially paired with an enthusiastic physician to provide support and assessment of existing skills. A gradual increase in independence can provide assurance that the NPP is qualified to provide care and gives many opportunities for reevaluation of the NPP. Clear expectations and constructive feedback should ultimately lead to a degree of comfort within the HMG, hospital, and the NPPs themselves.
Conclusions
It is clear that our healthcare system will need a very different approach to the economic problems it is facing. Standardization of care, integrated medical records, and expanded and universal resource utilization will drive the next generation of healthcare providers. The model of a private physician working alone under the direction of only his or her own medical knowledge is a thing of the past. Just as the HM specialty has grown from 300 in 1996 to more than 20,000 in 2008, so shall the integration of NPPs grow into our healthcare fabric.
- Centers for Disease Control and Prevention (CDC). Trends in aging—United States and worldwide. MMWR Morb Mortal Wkly Rep. 2003;52(6):101–104, 106.
- Council on Graduate Medical Education. Physician Workforce Policy Guidelines for the U.S. for 2000‐2020. Rockville, MD: U.S. Department of Health and Human Services;2005.
- American Medical Association. Medical Student Section. Advocacy and Policy. Medical Student Debt. Available at: http://www.ama‐assn.org/ama/pub/category/5349.html. Accessed June 2009.
- Society of Hospital Medicine (SHM). 2007‐2008 SHM Survey: State of the Hospital Medicine Movement. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Surveys2
- Centers for Disease Control and Prevention (CDC). Trends in aging—United States and worldwide. MMWR Morb Mortal Wkly Rep. 2003;52(6):101–104, 106.
- Council on Graduate Medical Education. Physician Workforce Policy Guidelines for the U.S. for 2000‐2020. Rockville, MD: U.S. Department of Health and Human Services;2005.
- American Medical Association. Medical Student Section. Advocacy and Policy. Medical Student Debt. Available at: http://www.ama‐assn.org/ama/pub/category/5349.html. Accessed June 2009.
- Society of Hospital Medicine (SHM). 2007‐2008 SHM Survey: State of the Hospital Medicine Movement. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Surveys2
Management of Hypertensive Urgencies
An association between hypertension and operative risk has been reported in small studies since the early 1970s. In two studies, Prys‐Roberts et al.1, 2 found that subjects with uncontrolled hypertension were more likely to have myocardial ischemic changes on electrocardiography with episodes of hypotension during induction of anesthesia. Subjects without hypertension or with hypertension controlled by medication were less likely to have episodes of hypotension, regardless of the type of anesthetic.
Hypertension increases the risk of developing perioperative heart failure (HF), renal failure, myocardial ischemia, or stroke. The level of risk is dependent upon the blood pressure (BP) level. It has been shown that a BP of 180/110 mm Hg without target‐organ damage (TOD) is not an independent risk factor for perioperative cardiovascular (CV) complications, suggesting this level of BP does not need to be reduced rapidly to normal.3, 4
The Joint National Committee defines hypertensive emergency as severe elevations in BP (usually >180/120 mm Hg) that produce evidence of TOD.5 Patients with this level of BP who are asymptomatic and have no signs of TOD are considered to have hypertensive urgency. As patients with this level of BP are at higher risk perioperatively, pharmacotherapy is indicated. When oral medications cannot be administered, hypertensive urgency can be managed with a parenteral medication. The agent should be easily and predictably titrated, safe, and convenient (Table 1). This article reviews the management of perioperative hypertensive urgency with parenteral medications. The management of hypertensive emergencies, aortic dissection, and hypertension of pregnancy is outside the scope of this review.
| Drug | Dose | Onset of Action | Duration | Use With Caution in | Adverse Reactions | Pregnancy Class* | Daily Cost |
|---|---|---|---|---|---|---|---|
| |||||||
| Hydralazine hydrochloride | 1020 mg IV q46h | 1020 minutes | 14 hours | Increased ICP; aortic dissection; myocardial ischemia | Reflex tachycardia; headache, flushing, vomiting | C | 20 mg q4h, $90 |
| Metoprolol | 1.255.0 mg IV q6h | 20 minutes | 58 hours | Heart block; bradycardia; acute heart failure | Bronchospasm | C (first trimester); D (second‐third trimesters) | 5 mg q6h, $10 |
| Enalaprilat | 1.255.0 mg IV q6h | 1530 minutes | 612 hours | Hyperkalemia; acute renal failure; hypovolemia | Hypotension; angioedema | C (first trimester); D (second‐third trimesters) | 5 mg q6h, $60 |
| Labetalol hydrochloride | 2080 mg IV q10min (max 300 mg daily) | 510 minutes | 36 hours | See metoprolol | Bronchospasm; nausea, vomitting; scalp tingling | C (first trimester); D (second‐third trimesters) | 300 mg, $15 |
| Transdermal clonidine | 0.10.3 mg once weekly | 23 days | 7 days | Abrupt withdrawal; elderly | Drowsiness, dizziness; local skin erythema; dry mouth | C | 0.3 mg/24‐hour patch, $10 |
Preoperative Considerations
In normotensive patients the induction of anesthesia can cause an acute elevation in BP (2030 mm Hg) and heart rate (HR) (1520 bpm).6 In patients with preexisting hypertension these changes are often greater, with elevations up to 90 mm Hg and 40 bpm. As anesthesia progresses systolic BP starts to fall (30 mm Hg), as a direct effect of both the anesthetic and the inhibition of the sympathetic nervous system (SNS). Patients with uncontrolled hypertension can have more severe reductions (60 mm Hg).6 This can result in intraoperative hypotension and shock. In a study of over 650 patients, marked intraoperative hypotension (50% of preoperative BP or a 33% reduction for more than 10 minutes) was an independent risk factor for perioperative CV complications (cardiac arrhythmia, ischemia, HF, or renal failure).7
Therefore, when BP is mildly elevated at the time of surgery (180/110 mm Hg), rapid reduction in BP is not necessary, and studies have been unable to demonstrate a benefit to delaying surgery.8 However, when BP is 180/110 mm Hg preoperatively, antihypertensive medications should be administered and intraoperative blood pressure monitored closely. There is a lack of data to support delay of surgery.9
Postoperative Considerations
The postoperative period is also associated with elevations in BP. In the immediate recovery phase from anesthesia, there is a mild elevation in BP within 10 to 15 mm Hg, but there are larger fluctuations in patients with preexisting hypertension.6 Otherwise postoperative hypertension can be seen from a variety of causes such as pain, excitement on emergence from anesthesia, and hypercarbia.10 Less common causes include agitation, hypoxemia, and hypervolemia. These secondary causes should be identified and treated before any antihypertensive medications are administered.
Drug Therapy
When evaluating a patient with a BP of 180/110 mm Hg, the physician must first classify the patient as having a hypertensive emergency or urgency. Hypertensive emergencies require immediate reduction in BP to prevent or limit hypertensive encephalopathy, intracerebral hemorrhage, acute myocardial infarction (MI), HF and aortic dissection.11 This is often accomplished by using continuous infusions of medications such as nitroprusside, nicardipine, or fenoldopam, and requires monitoring in an intensive care unit (ICU) with an intraarterial catheter.
As patients with hypertensive urgency are not at great risk for TOD, continuous infusions of the above medications that require ICU monitoring and intraarterial catheters seem to be unnecessary, and a possible misuse of resources. Treating hypertensive urgency in this manner could also be potentially dangerous.12, 13 Patients with chronic hypertension often have autoregulation of organ perfusion shifted to a higher range of mean arterial pressure, so excessive pressure reductions to normal BP values may induce organ hypoperfusion.14 Therefore, BP in hypertensive urgency can be lowered to 160/100 mm Hg over time.5 When oral medications cannot be used, there are several parenteral agents.
Diltiazem Hydrochloride and Verapamil
Diltiazem hydrochloride and verapamil are non‐dihydropyridine calcium‐channel blockers that produce vasodilation by decreasing calcium entry into vascular smooth muscle. In a study of 18 hypertensive patients, administration of intravenous diltiazem resulted in significant BP reductions within 5 minutes, however a variety of rhythm disturbances and heart block (HB) were observed.15 Verapamil has also been shown to successfully lower BP.16 However, when given at antihypertensive doses, verapamil has been shown to cause prolongation of the PR interval (30%), second‐degree block (0.7%), and complete HB (1.7%).17
Therefore, although oral diltiazem and verapamil may be appropriate for treating hypertension, the intravenous formulations are indicated only for the treatment of atrial fibrillation or flutter, and paroxysmal supraventricular tachycardia.18
Clonidine
Clonidine stimulates alpha2‐adrenoreceptors in the brain stem. This action results in reduced sympathetic outflow from the central nervous system, and decreases in peripheral resistance, renal vascular resistance, HR, and BP. Renal blood flow and glomerular filtration rate remain essentially unchanged. Normal postural reflexes are intact; therefore, orthostatic symptoms are mild and infrequent. Sudden cessation of treatment with clonidine has been associated with dangerous rebound hypertension.
Catapres‐TTS (clonidine) transdermal releases clonidine at a constant rate for 7 days. Therapeutic levels are achieved 2 to 3 days after initial application. After removal, therapeutic levels persist for about 8 hours and decline slowly over several days.19
Perioperatively, beneficial effects of clonidine include decreased anesthetic and opioid requirements, reduced hemodynamic responses to intubation and other stimuli, and improved postoperative renal function.20 Alpha2 agonists have also been shown to have significant antiischemic properties.21, 22
Beta‐adrenoreceptor () Blockers
Beta blockers are of particular interest in the management of perioperative hypertension. Several studies in the 1980s demonstrated that preoperative use of ‐blockers attenuated the severe BP fluctuations in the perioperative period; there was also a reduction in myocardial ischemia.2124 In addition, the preoperative ‐blockers in select at‐risk populations has been shown to decrease the rate of CV events (MI, unstable angina, need for coronary‐artery bypass, HF) and death.25, 26
Given these findings, the American College of Cardiology/American Heart Association (ACC/AHA) guidelines on the perioperative CV evaluation and care for noncardiac surgery recommended ‐blockers in patients receiving ‐blockers for angina, symptomatic arrhythmias, or hypertension; those undergoing vascular surgery with coronary artery disease or a revised cardiac risk index (RCRI) score >1; and those undergoing intermediate risk surgery with a RCRI of >1.27, 28 However, the recently published Perioperative Ischemic Evaluation Study (POISE) trial demonstrated that while ‐blockers reduced the risk of perioperative MI, there was an overall increase in net mortality.29 Given that most of the patients had an RCRI of 1 to 2, the ACC/AHA plans to revise this guideline.
If a ‐blocker is selected to manage perioperative hypertension, there are two available for parenteral use.
Metoprolol Tartrate
Metoprolol is a ‐1 selective adrenoreceptor antagonist available in both oral and intravenous formulations. Acutely, it decreases cardiac output by reducing both HR and contractility, therefore resulting in a decrease in BP. Over the course of a week it antagonizes ‐receptors in the juxtaglomerular complex, suppressing renin release and therefore production of angiotensin II.30 Metoprolol may lower BP by other mechanisms, including alteration of the sympathetic nervous system (SNS) and altered baroreceptor sensitivity.
The oral formulation is most commonly used to treat hypertension, MI, angina, atrial fibrillation, and HF. The intravenous form is only approved for the treatment of acute MI and supraventricular tachycardia. However, intravenous administration does induce its maximal hypotensive response within 20 minutes, generally lasting 3 to 4 hours. In a study investigating metoprolol and perioperative hypertension during extubation, the administration of intravenous metoprolol safely blunted the expected rise in BP.31 Similar findings were demonstrated in neurosurgical patients.32
Even though intravenous metoprolol can effectively lower BP, it does so mainly by reducing cardiac output. Therefore, caution must be taken in patients with a low cardiac index, and it should be avoided in acute HF, bradycardia or greater than first‐degree HB, or bronchospasm.
As metoprolol is a far more commonly used substitute for atenolol, we have deferred its specific discussion.
Labetalol Hydrochloride
Labetalol antagonizes both alpha1‐ and nonselective ‐adrenoreceptors. When given intravenously the onset of action is 5 minutes, but the duration can vary from 20 minutes to 23 hours, with an average of generally 6 hours. An initial dosage of 10 to 20 mg administered over 2 minutes can be followed by repeat doses every 10 minutes until the desired BP goal is achieved (maximum 300 mg daily). It decreases systemic vascular resistance and typically has no significant effect on cardiac index. In a multicenter study, bolus doses produced a rapid, smooth reduction in BP without reflex tachycardia or serious side effects.33 It has been shown to have similar efficacy and safety in cardiac surgery and other surgery requiring anesthesia.34, 35 Furthermore, it does not increase intracranial pressure,36 and is safe in patients with renal insufficiency or pregnancy. Contraindications to labetalol are hypotension, bradycardia, high‐degree HB, and severe asthma or chronic obstructive pulmonary disease.
Hydralazine Hydrochloride
Hydralazine reduces BP by increasing cyclic‐guanosine monophosphate in vascular smooth muscle, therefore leading to direct arterial vasodilation with little effect on venous circulation.37 It causes rapid reductions in BP, sometimes resulting in reflex tachycardia. When given intravenously, it has an onset of action of 5 minutes and duration of 3 to 8 hours, dependent mostly on hepatic clearance. This variability in hepatic acetylation and inactivation leads to some difficulty in drug titration.38 The starting dose is usually 10 mg, and it is administered every 4 to 6 hours. As stated, intravenous administration results in an increase in HR, cardiac output, myocardial contractility, and an overall increase in sympathetic activity.39
Although hydralazine has been used for the management of perioperative hypertension for several decades,40 its overall efficacy and safety have not been adequately defined for this setting. It has proven to be most successful during hypertension in pregnancy41 or hypertensive emergency.42 However, hydralazine is still widely used and is considered by some experts as an acceptable antihypertensive drug in the perioperative setting, as it can be administered in divided doses, routinely at 4 to 6 hour intervals, making it suitable for the treatment of hypertension in subjects unable to take medications by mouth or when a continuous infusion is unnecessary.
Hydralazine should be used with extreme caution in patients with evidence of cardiac ischemia, and it should be avoided in patients with aortic dissection or an increased intracranial pressure. The activation of the SNS and arterial vasodilation could have a potential benefit for patients with renal dysfunction.
Enalaprilat
Enalaprilat is the intravenous preparation of the active form of the angiotensin converting enzyme (ACE) inhibitor enalapril. By ACE inhibition, enalaprilat leads to a reduction in the production of angiotensin II, thereby reducing mean arterial pressure. The usual dose is 1.25 mg, and as much as 5 mg may be given every 6 hours as necessary,43 making it suitable for the treatment of hypertension in subjects unable to take medications by mouth.
Enalaprilat has demonstrated efficacy and safety when used in both CV surgery and neurosurgery. In a study of 14 patients with chronic HF, the administration of enalaprilat resulted in significant reductions in both mean arterial pressure (21%) and pulmonary capillary wedge pressure (33%).44 There was also an increase in the stroke volume index (20%) without a change in coronary blood flow or myocardial oxygen consumption, indicating an improvement in left ventricular function. As ACE inhibitors do not impair cerebral blood flow, enalaprilat may also be used safely in neurosurgery.45 Additionally, enalaprilat has been studied in the treatment of hypertensive urgencies. In a study of patients who had a diastolic BP between 100 and 114 mm Hg, the administration of 1.25 mg of enalaprilat lead to a significant reduction in systolic and diastolic BP within 60 minutes without any major adverse events.46
Even though enalaprilat has demonstrated safety and efficacy in several perioperative trials, its actions may be variable and not always predictable. When investigating the appropriate dose of enalaprilat, Hirschl et al.43 randomized 65 patients to receive different doses of enalaprilat. Response to treatment was defined as a stable reduction in BP to 180/95 mm Hg within 45 minutes. The goal was reached in only 63%, and surprisingly the response rates did not differ across differing dosages: 0.625 mg (67%), 1.25 mg (65%), 2.5 mg (59%), and 5 mg (62%).
Continuing chronic ACE inhibitor therapy within 12 to 24 hours preoperatively has been associated with severe hypotension at or shortly after induction of anesthesia. In a recent meta‐analysis, Rosenman et al.47 assessed the clinical consequences of preoperatively continuing vs. withholding ACE inhibitors or a angiotensin II receptor blocker (ARB) in patients treated chronically with these agents. Patients receiving an immediate preoperative ACE inhibitor or ARB were significantly more likely to develop hypotension requiring vasopressors. Although this observation cannot be directly translated, caution should be advised when selecting intravenous enalaprilat for the acute lowering of BP preoperatively.
Enalaprilat is contraindicated in pregnancy and patients with bilateral renal artery stenosis. It must also be used carefully in patients with hyperkalemia, acute renal failure, or hypovolemia.48 There should also be a dose adjustment when given to patients with severe chronic kidney disease.49 In addition, its use 12 to 24 hours prior to the induction of anesthesia should be discussed with the anesthesiologist.
Discussion
Nitroprusside, nitroglycerin, nicardipine, and fenoldopam are all effective antihypertensive medications. However, their availability only as continuous infusions requires ICU monitoring and an intraarterial catheter, and they are therefore unnecessary in the management of hypertensive urgency. The parenteral medications that do not require a continuous infusion are diltiazem, verapamil, metoprolol, labetalol, enalaprilat, hydralazine, and transdermal clonidine.
As stated, the intravenous formulations of diltiazem and verapamil are indicated only for certain arrhythmias. Because the onset of action of transdermal clonidine is about 2 days and the offset is 8 hours, it has limited usefulness in the treatment of perioperative hypertension. Therefore, only metoprolol, labetalol, enalaprilat, and hydralazine have a major role in the treatment of hypertensive urgency when oral medications cannot be used.
When given intravenously, enalaprilat and hydralazine are safe, effective, widely available, and inexpensive. When deciding between these 2 agents, a few other considerations may be of importance. Even though ACE inhibitors have well‐recognized benefits in the management of HF50 and diabetic nephropathy,51 these characteristics are not relevant in the short‐term use of enalaprilat to treat perioperative hypertension. However, enalaprilat may be preferred over hydralazine when activation of the SNS and reflex tachycardia is to be avoided (cardiac ischemia, aortic dissection, increased intracranial pressure). Hydralazine may be preferred in the setting of hyperkalemia and acute renal failure. It must be preferred in pregnancy or bilateral renal artery stenosis.
Although the weight of the evidence of perioperative ‐blocker use to reduce CV events in noncardiac surgery suggests a benefit, there are significant limitations. Few studies have compared different ‐blockers. Studies to determine the ideal target population, duration of therapy, and route of administration are lacking. Additionally, using perioperative ‐blockers may cause harm in low‐risk patients.52 Care should be taken when using labetalol and metoprolol in combination as they can induce a dangerous reduction in HR. The role of acute administration of intravenous ‐blockers in the setting of myocardial ischemia is debatable, and probably dangerous in the setting of hypotension, bradycardia, HB, pulmonary edema, or bronchospasm.53
Therefore, generalizing the perioperative ‐blocker data to all patients with perioperative hypertension seems unlikely to have significant benefit, and may possibly pose harm.29 However, it seems reasonable to use ‐blockers in those in whom it would be indicated otherwise, and to continue parenteral therapy in those already taking a ‐blocker preoperatively in order to avoid withdrawal.54
When deciding between metoprolol and labetalol, a few considerations may be of importance. First, there is much more evidence documenting the safety and efficacy of labetalol in perioperative hypertension. Second, even though metoprolol has proven benefit in patients with chronic HF, coronary artery disease (CAD), and MI, these long‐term studies investigated oral metoprolol, not the intravenous formulation.55 Most importantly, labetalol is more effective at lowering BP due to its additional blockade of alpha1 adrenoreceptors. Neither drug should be used in acute HF, bradycardia or greater than first‐degree HB, or bronchospasm. In conclusion, intravenous labetalol should be preferred over intravenous metoprolol for the management of perioperative hypertension.
Conclusions
Perioperative hypertension ideally should be evaluated well before the operative time period, when there is adequate time to initiate medications. Secondary causes such as pain, agitation, hypercarbia, hypoxemia, and hypervolemia should be treated directly prior to the administration of antihypertensive medications. It is uncertain whether patients with a BP of 180/110 mm Hg benefit from any specific parenteral medication, as there is little evidence from several studies that this level of BP without TOD leads to an increase in perioperative morbidity or mortality.3, 4, 7, 56 However, patients with hypertensive urgency are at higher risk for perioperative complications; therefore, their BP should be managed gradually to 160/110 mm Hg with the outlined recommended parenteral regimen (Figure 1).
When selecting a parenteral medication, we suggest first to exclude any contraindications, or see if an indication exists for a specific agent. Hydralazine, enalaprilat, metoprolol, or labetalol can be used as first‐line agents. Due to the scarcity of comparative trials looking at clinically significant outcomes (length of hospital stay, morbidity, mortality), decisions for the management of perioperative hypertension should be made based on comorbidity, efficacy, toxicity, and cost (Table 1).
Acknowledgements
The authors Henry R. Black, M.D. for his contribution.
- ,,.Studies of anaesthesia in relation to hypertension. I. Cardiovascular responses of treated and untreated patients.Br J Anaesth.1971;43(2):122–137.
- ,,, et al.Studies of anaesthesia in relation to hypertension. II. Haemodynamic consequences of induction and endotracheal intubation.Br J Anaesth.1971;43(6):531–547.
- ,,, et al.Multifactorial index of cardiac risk in noncardiac surgical procedures.N Engl J Med.1977;297(16):845–850.
- ,,, et al.Cardiac assessment for patients undergoing noncardiac surgery. A multifactorial clinical risk index.Arch Intern Med.1986;146(11):2131–2134.
- ,,, et al.The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report.JAMA.2003;289(19):2560–2572.
- ,.Management of anesthesia in patients with hypertension or ischemic heart disease.Int Anesthesiol Clin.1980;18(4):181–217.
- ,.Risks of general anesthesia and elective operation in the hypertensive patient.Anesthesiology.1979;50(4):285–292.
- ,,, et al.The dilemma of immediate preoperative hypertension: to treat and operate, or to postpone surgery?J Clin Anesth.2003;15(3):179–183.
- .Is blood pressure control necessary before surgery?Med Clin North Am.1993;77(2):349–363.
- ,.Hypertension in the immediate postoperative period.Br J Anaesth.1975;47(1):70–74.
- .Management of hypertensive emergencies.Lancet. 121994;344(8933):1335–1338.
- ,.Severely increased blood pressure in the emergency department.Ann Emerg Med.2003;41(4):513–529.
- ,,.Rapid reduction of severe asymptomatic hypertension. A prospective, controlled trial.Arch Intern Med.1989;149(10):2186–2189.
- ,.Cerebral blood flow and its pathophysiology in hypertension.Am J Hypertens.1989;2:(6 pt 1):486–492.
- ,,, et al.Efficacy, electrocardiographic and renal effects of intravenous diltiazem for essential hypertension.Am J Cardiol.1987;60(17):78I–84I.
- ,,, et al.Calcium entry blockers for the treatment of severe hypertension and hypertensive crisis.Am J Med.1984;77:(2B):35–45.
- ,.Rapid‐acting parenteral antihypertensive agents.J Clin Pharmacol.1990;30(3):195–209.
- ,.The role of calcium entry blockers in hypertensive emergencies.Circulation.1987;75(6 pt 2):V174–180.
- ,,, et al.Transdermal clonidine therapy in hypertensive patients. Effects on office and ambulatory recorded blood pressure values.JAMA.1985;253(2):233–235.
- ,,.Anesthesia and hypertension: the effect of clonidine on perioperative hemodynamics and isoflurane requirements.Anesthesiology.1987;67(1):3–10.
- ,,, et al.Small, oral dose of clonidine reduces the incidence of intraoperative myocardial ischemia in patients having vascular surgery.Anesthesiology.1996;85(4):706–712.
- ,,, et al.Effect of clonidine on cardiovascular morbidity and mortality after noncardiac surgery.Anesthesiology.2004;101(2):284–293.
- ,,, et al.Beta blockade to decrease silent myocardial ischemia during peripheral vascular surgery.Am J Surg.1989;158(2):113–116.
- ,,, et al.Haemodynamic effects of pretreatment with metoprolol in hypertensive patients undergoing surgery.Br J Anaesth.1986;58(3):251–260.
- ,,, et al.Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group.N Engl J Med.1996;335(23):1713–1720.
- ,,, et al.The effect of bisoprolol on perioperative mortality and myocardial infarction in high‐risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group.N Engl J Med.1999;341(24):1789–1794.
- ,,, et al.ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, and Society for Vascular Surgery.J Am Coll Cardiol.2007;50(17):e159–e241.
- ,,, et al.Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery.Circulation.1999;100(10):1043–1049.
- ,,, et al.Effects of extended‐release metoprolol succinate in patients undergoing non‐cardiac surgery (POISE trial): a randomised controlled trial.Lancet.2008;371(9627):1839–1847.
- ,.Inhibition of angiotensin II potentiation of sympathetic nerve activity by beta‐adrenergic antagonists.Hypertension.1980;2(1):90–96.
- ,,, et al.Comparison of intravenous metoprolol, verapamil and diltiazem on the attenuation of haemodynamic changes associated with tracheal extubation.Eur J Anaesthesiol.1999;16(7):462–467.
- ,,, et al.The effect of metoprolol upon blood pressure, cerebral blood flow and oxygen consumption in patients subjected to craniotomy for cerebral tumours.Acta Anaesthesiol Scand.1994;38(3):271–275.
- ,,, et al.Intravenous labetalol in the treatment of severe hypertension and hypertensive emergencies.Am J Med.1983;75:(4A):95–102.
- ,,.I.V. labetalol in the treatment of hypertension following coronary‐artery surgery.Br J Anaesth.1982;54(11):1191–1196.
- ,,, et al.Intravenous labetalol for treatment of postoperative hypertension.Anesthesiology.1987;67(3):413–416.
- ,,, et al.Labetalol to control blood pressure after cerebrovascular surgery.Crit Care Med.1988;16(8):765–768.
- ,,, et al.Cyclic guanosine 3′,5′ monophosphate concentrations in pre‐eclampsia: effects of hydralazine.Br J Obstet Gynaecol.1996;103(1):33–38.
- ,,, et al.Effect of intravenous dose on hydralazine kinetics after administration.Clin Pharmacol Ther.1983;34(2):148–152.
- ,,.Adverse effects of direct‐acting vasodilators.Drug Saf.1994;11(2):80–85.
- ,,, et al.Incidence and mechanism of post‐carotid endarterectomy hypertension.Arch Surg.1987;122(10):1153–1155.
- ,,, et al.Hydralazine for treatment of severe hypertension in pregnancy: meta‐analysis.Bmj.2003;327(7421):955–960.
- ,.Current diagnosis and management of hypertensive emergency.Semin Dial.2006;19(6):502–512.
- ,,, et al.Clinical evaluation of different doses of intravenous enalaprilat in patients with hypertensive crises.Arch Intern Med.1995;155(20):2217–2223.
- ,,, et al.Enalaprilat, a new parenteral angiotensin‐converting enzyme inhibitor: rapid changes in systemic and coronary hemodynamics and humoral profile in chronic heart failure.J Am Coll Cardiol.1987;9(5):1131–1138.
- ,,, et al.Cerebral blood flow in patients with congestive heart failure treated with captopril.Am J Med.1984;76:(5B):91–95.
- ,,, et al.The effect of intravenous enalaprilat (MK‐422) administration in patients with mild to moderate essential hypertension.J Clin Pharmacol.1987;27(5):415–418.
- ,,, et al.Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period.J Hosp Med.2008;3(4):319–325.
- ,.Toxic effects of drugs used in the ICU. Nitroprusside, nitroglycerin, and angiotensin‐converting enzyme inhibitors.Crit Care Clin.1991;7(3):555–581.
- ,,, et al.Disposition of enalapril and enalaprilat in renal insufficiency.Kidney Int Suppl.1987;20:S117–122.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group.N Engl J Med.1987;316(23):1429–1435.
- ,,, et al.Angiotensin‐receptor blockade versus converting‐enzyme inhibition in type 2 diabetes and nephropathy.N Engl J Med.2004;351(19):1952–1961.
- ,,, et al.Perioperative beta‐blocker therapy and mortality after major noncardiac surgery.N Engl J Med. 282005;353(4):349–361.
- ,,, et al.Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo‐controlled trial.Lancet.2005;366(9497):1622–1632.
- ,,, et al.Perioperative beta‐blocker withdrawal and mortality in vascular surgical patients.Am Heart J.2001;141(1):148–153.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT‐HF).Lancet.1999;353(9169):2001–2007. [No authors listed]
- ,,, et al.Preoperative and long‐term cardiac risk assessment. Predictive value of 23 clinical descriptors, 7 multivariate scoring systems, and quantitative dipyridamole imaging in 360 patients.Ann Surg.1992;216(2):192–204.
An association between hypertension and operative risk has been reported in small studies since the early 1970s. In two studies, Prys‐Roberts et al.1, 2 found that subjects with uncontrolled hypertension were more likely to have myocardial ischemic changes on electrocardiography with episodes of hypotension during induction of anesthesia. Subjects without hypertension or with hypertension controlled by medication were less likely to have episodes of hypotension, regardless of the type of anesthetic.
Hypertension increases the risk of developing perioperative heart failure (HF), renal failure, myocardial ischemia, or stroke. The level of risk is dependent upon the blood pressure (BP) level. It has been shown that a BP of 180/110 mm Hg without target‐organ damage (TOD) is not an independent risk factor for perioperative cardiovascular (CV) complications, suggesting this level of BP does not need to be reduced rapidly to normal.3, 4
The Joint National Committee defines hypertensive emergency as severe elevations in BP (usually >180/120 mm Hg) that produce evidence of TOD.5 Patients with this level of BP who are asymptomatic and have no signs of TOD are considered to have hypertensive urgency. As patients with this level of BP are at higher risk perioperatively, pharmacotherapy is indicated. When oral medications cannot be administered, hypertensive urgency can be managed with a parenteral medication. The agent should be easily and predictably titrated, safe, and convenient (Table 1). This article reviews the management of perioperative hypertensive urgency with parenteral medications. The management of hypertensive emergencies, aortic dissection, and hypertension of pregnancy is outside the scope of this review.
| Drug | Dose | Onset of Action | Duration | Use With Caution in | Adverse Reactions | Pregnancy Class* | Daily Cost |
|---|---|---|---|---|---|---|---|
| |||||||
| Hydralazine hydrochloride | 1020 mg IV q46h | 1020 minutes | 14 hours | Increased ICP; aortic dissection; myocardial ischemia | Reflex tachycardia; headache, flushing, vomiting | C | 20 mg q4h, $90 |
| Metoprolol | 1.255.0 mg IV q6h | 20 minutes | 58 hours | Heart block; bradycardia; acute heart failure | Bronchospasm | C (first trimester); D (second‐third trimesters) | 5 mg q6h, $10 |
| Enalaprilat | 1.255.0 mg IV q6h | 1530 minutes | 612 hours | Hyperkalemia; acute renal failure; hypovolemia | Hypotension; angioedema | C (first trimester); D (second‐third trimesters) | 5 mg q6h, $60 |
| Labetalol hydrochloride | 2080 mg IV q10min (max 300 mg daily) | 510 minutes | 36 hours | See metoprolol | Bronchospasm; nausea, vomitting; scalp tingling | C (first trimester); D (second‐third trimesters) | 300 mg, $15 |
| Transdermal clonidine | 0.10.3 mg once weekly | 23 days | 7 days | Abrupt withdrawal; elderly | Drowsiness, dizziness; local skin erythema; dry mouth | C | 0.3 mg/24‐hour patch, $10 |
Preoperative Considerations
In normotensive patients the induction of anesthesia can cause an acute elevation in BP (2030 mm Hg) and heart rate (HR) (1520 bpm).6 In patients with preexisting hypertension these changes are often greater, with elevations up to 90 mm Hg and 40 bpm. As anesthesia progresses systolic BP starts to fall (30 mm Hg), as a direct effect of both the anesthetic and the inhibition of the sympathetic nervous system (SNS). Patients with uncontrolled hypertension can have more severe reductions (60 mm Hg).6 This can result in intraoperative hypotension and shock. In a study of over 650 patients, marked intraoperative hypotension (50% of preoperative BP or a 33% reduction for more than 10 minutes) was an independent risk factor for perioperative CV complications (cardiac arrhythmia, ischemia, HF, or renal failure).7
Therefore, when BP is mildly elevated at the time of surgery (180/110 mm Hg), rapid reduction in BP is not necessary, and studies have been unable to demonstrate a benefit to delaying surgery.8 However, when BP is 180/110 mm Hg preoperatively, antihypertensive medications should be administered and intraoperative blood pressure monitored closely. There is a lack of data to support delay of surgery.9
Postoperative Considerations
The postoperative period is also associated with elevations in BP. In the immediate recovery phase from anesthesia, there is a mild elevation in BP within 10 to 15 mm Hg, but there are larger fluctuations in patients with preexisting hypertension.6 Otherwise postoperative hypertension can be seen from a variety of causes such as pain, excitement on emergence from anesthesia, and hypercarbia.10 Less common causes include agitation, hypoxemia, and hypervolemia. These secondary causes should be identified and treated before any antihypertensive medications are administered.
Drug Therapy
When evaluating a patient with a BP of 180/110 mm Hg, the physician must first classify the patient as having a hypertensive emergency or urgency. Hypertensive emergencies require immediate reduction in BP to prevent or limit hypertensive encephalopathy, intracerebral hemorrhage, acute myocardial infarction (MI), HF and aortic dissection.11 This is often accomplished by using continuous infusions of medications such as nitroprusside, nicardipine, or fenoldopam, and requires monitoring in an intensive care unit (ICU) with an intraarterial catheter.
As patients with hypertensive urgency are not at great risk for TOD, continuous infusions of the above medications that require ICU monitoring and intraarterial catheters seem to be unnecessary, and a possible misuse of resources. Treating hypertensive urgency in this manner could also be potentially dangerous.12, 13 Patients with chronic hypertension often have autoregulation of organ perfusion shifted to a higher range of mean arterial pressure, so excessive pressure reductions to normal BP values may induce organ hypoperfusion.14 Therefore, BP in hypertensive urgency can be lowered to 160/100 mm Hg over time.5 When oral medications cannot be used, there are several parenteral agents.
Diltiazem Hydrochloride and Verapamil
Diltiazem hydrochloride and verapamil are non‐dihydropyridine calcium‐channel blockers that produce vasodilation by decreasing calcium entry into vascular smooth muscle. In a study of 18 hypertensive patients, administration of intravenous diltiazem resulted in significant BP reductions within 5 minutes, however a variety of rhythm disturbances and heart block (HB) were observed.15 Verapamil has also been shown to successfully lower BP.16 However, when given at antihypertensive doses, verapamil has been shown to cause prolongation of the PR interval (30%), second‐degree block (0.7%), and complete HB (1.7%).17
Therefore, although oral diltiazem and verapamil may be appropriate for treating hypertension, the intravenous formulations are indicated only for the treatment of atrial fibrillation or flutter, and paroxysmal supraventricular tachycardia.18
Clonidine
Clonidine stimulates alpha2‐adrenoreceptors in the brain stem. This action results in reduced sympathetic outflow from the central nervous system, and decreases in peripheral resistance, renal vascular resistance, HR, and BP. Renal blood flow and glomerular filtration rate remain essentially unchanged. Normal postural reflexes are intact; therefore, orthostatic symptoms are mild and infrequent. Sudden cessation of treatment with clonidine has been associated with dangerous rebound hypertension.
Catapres‐TTS (clonidine) transdermal releases clonidine at a constant rate for 7 days. Therapeutic levels are achieved 2 to 3 days after initial application. After removal, therapeutic levels persist for about 8 hours and decline slowly over several days.19
Perioperatively, beneficial effects of clonidine include decreased anesthetic and opioid requirements, reduced hemodynamic responses to intubation and other stimuli, and improved postoperative renal function.20 Alpha2 agonists have also been shown to have significant antiischemic properties.21, 22
Beta‐adrenoreceptor () Blockers
Beta blockers are of particular interest in the management of perioperative hypertension. Several studies in the 1980s demonstrated that preoperative use of ‐blockers attenuated the severe BP fluctuations in the perioperative period; there was also a reduction in myocardial ischemia.2124 In addition, the preoperative ‐blockers in select at‐risk populations has been shown to decrease the rate of CV events (MI, unstable angina, need for coronary‐artery bypass, HF) and death.25, 26
Given these findings, the American College of Cardiology/American Heart Association (ACC/AHA) guidelines on the perioperative CV evaluation and care for noncardiac surgery recommended ‐blockers in patients receiving ‐blockers for angina, symptomatic arrhythmias, or hypertension; those undergoing vascular surgery with coronary artery disease or a revised cardiac risk index (RCRI) score >1; and those undergoing intermediate risk surgery with a RCRI of >1.27, 28 However, the recently published Perioperative Ischemic Evaluation Study (POISE) trial demonstrated that while ‐blockers reduced the risk of perioperative MI, there was an overall increase in net mortality.29 Given that most of the patients had an RCRI of 1 to 2, the ACC/AHA plans to revise this guideline.
If a ‐blocker is selected to manage perioperative hypertension, there are two available for parenteral use.
Metoprolol Tartrate
Metoprolol is a ‐1 selective adrenoreceptor antagonist available in both oral and intravenous formulations. Acutely, it decreases cardiac output by reducing both HR and contractility, therefore resulting in a decrease in BP. Over the course of a week it antagonizes ‐receptors in the juxtaglomerular complex, suppressing renin release and therefore production of angiotensin II.30 Metoprolol may lower BP by other mechanisms, including alteration of the sympathetic nervous system (SNS) and altered baroreceptor sensitivity.
The oral formulation is most commonly used to treat hypertension, MI, angina, atrial fibrillation, and HF. The intravenous form is only approved for the treatment of acute MI and supraventricular tachycardia. However, intravenous administration does induce its maximal hypotensive response within 20 minutes, generally lasting 3 to 4 hours. In a study investigating metoprolol and perioperative hypertension during extubation, the administration of intravenous metoprolol safely blunted the expected rise in BP.31 Similar findings were demonstrated in neurosurgical patients.32
Even though intravenous metoprolol can effectively lower BP, it does so mainly by reducing cardiac output. Therefore, caution must be taken in patients with a low cardiac index, and it should be avoided in acute HF, bradycardia or greater than first‐degree HB, or bronchospasm.
As metoprolol is a far more commonly used substitute for atenolol, we have deferred its specific discussion.
Labetalol Hydrochloride
Labetalol antagonizes both alpha1‐ and nonselective ‐adrenoreceptors. When given intravenously the onset of action is 5 minutes, but the duration can vary from 20 minutes to 23 hours, with an average of generally 6 hours. An initial dosage of 10 to 20 mg administered over 2 minutes can be followed by repeat doses every 10 minutes until the desired BP goal is achieved (maximum 300 mg daily). It decreases systemic vascular resistance and typically has no significant effect on cardiac index. In a multicenter study, bolus doses produced a rapid, smooth reduction in BP without reflex tachycardia or serious side effects.33 It has been shown to have similar efficacy and safety in cardiac surgery and other surgery requiring anesthesia.34, 35 Furthermore, it does not increase intracranial pressure,36 and is safe in patients with renal insufficiency or pregnancy. Contraindications to labetalol are hypotension, bradycardia, high‐degree HB, and severe asthma or chronic obstructive pulmonary disease.
Hydralazine Hydrochloride
Hydralazine reduces BP by increasing cyclic‐guanosine monophosphate in vascular smooth muscle, therefore leading to direct arterial vasodilation with little effect on venous circulation.37 It causes rapid reductions in BP, sometimes resulting in reflex tachycardia. When given intravenously, it has an onset of action of 5 minutes and duration of 3 to 8 hours, dependent mostly on hepatic clearance. This variability in hepatic acetylation and inactivation leads to some difficulty in drug titration.38 The starting dose is usually 10 mg, and it is administered every 4 to 6 hours. As stated, intravenous administration results in an increase in HR, cardiac output, myocardial contractility, and an overall increase in sympathetic activity.39
Although hydralazine has been used for the management of perioperative hypertension for several decades,40 its overall efficacy and safety have not been adequately defined for this setting. It has proven to be most successful during hypertension in pregnancy41 or hypertensive emergency.42 However, hydralazine is still widely used and is considered by some experts as an acceptable antihypertensive drug in the perioperative setting, as it can be administered in divided doses, routinely at 4 to 6 hour intervals, making it suitable for the treatment of hypertension in subjects unable to take medications by mouth or when a continuous infusion is unnecessary.
Hydralazine should be used with extreme caution in patients with evidence of cardiac ischemia, and it should be avoided in patients with aortic dissection or an increased intracranial pressure. The activation of the SNS and arterial vasodilation could have a potential benefit for patients with renal dysfunction.
Enalaprilat
Enalaprilat is the intravenous preparation of the active form of the angiotensin converting enzyme (ACE) inhibitor enalapril. By ACE inhibition, enalaprilat leads to a reduction in the production of angiotensin II, thereby reducing mean arterial pressure. The usual dose is 1.25 mg, and as much as 5 mg may be given every 6 hours as necessary,43 making it suitable for the treatment of hypertension in subjects unable to take medications by mouth.
Enalaprilat has demonstrated efficacy and safety when used in both CV surgery and neurosurgery. In a study of 14 patients with chronic HF, the administration of enalaprilat resulted in significant reductions in both mean arterial pressure (21%) and pulmonary capillary wedge pressure (33%).44 There was also an increase in the stroke volume index (20%) without a change in coronary blood flow or myocardial oxygen consumption, indicating an improvement in left ventricular function. As ACE inhibitors do not impair cerebral blood flow, enalaprilat may also be used safely in neurosurgery.45 Additionally, enalaprilat has been studied in the treatment of hypertensive urgencies. In a study of patients who had a diastolic BP between 100 and 114 mm Hg, the administration of 1.25 mg of enalaprilat lead to a significant reduction in systolic and diastolic BP within 60 minutes without any major adverse events.46
Even though enalaprilat has demonstrated safety and efficacy in several perioperative trials, its actions may be variable and not always predictable. When investigating the appropriate dose of enalaprilat, Hirschl et al.43 randomized 65 patients to receive different doses of enalaprilat. Response to treatment was defined as a stable reduction in BP to 180/95 mm Hg within 45 minutes. The goal was reached in only 63%, and surprisingly the response rates did not differ across differing dosages: 0.625 mg (67%), 1.25 mg (65%), 2.5 mg (59%), and 5 mg (62%).
Continuing chronic ACE inhibitor therapy within 12 to 24 hours preoperatively has been associated with severe hypotension at or shortly after induction of anesthesia. In a recent meta‐analysis, Rosenman et al.47 assessed the clinical consequences of preoperatively continuing vs. withholding ACE inhibitors or a angiotensin II receptor blocker (ARB) in patients treated chronically with these agents. Patients receiving an immediate preoperative ACE inhibitor or ARB were significantly more likely to develop hypotension requiring vasopressors. Although this observation cannot be directly translated, caution should be advised when selecting intravenous enalaprilat for the acute lowering of BP preoperatively.
Enalaprilat is contraindicated in pregnancy and patients with bilateral renal artery stenosis. It must also be used carefully in patients with hyperkalemia, acute renal failure, or hypovolemia.48 There should also be a dose adjustment when given to patients with severe chronic kidney disease.49 In addition, its use 12 to 24 hours prior to the induction of anesthesia should be discussed with the anesthesiologist.
Discussion
Nitroprusside, nitroglycerin, nicardipine, and fenoldopam are all effective antihypertensive medications. However, their availability only as continuous infusions requires ICU monitoring and an intraarterial catheter, and they are therefore unnecessary in the management of hypertensive urgency. The parenteral medications that do not require a continuous infusion are diltiazem, verapamil, metoprolol, labetalol, enalaprilat, hydralazine, and transdermal clonidine.
As stated, the intravenous formulations of diltiazem and verapamil are indicated only for certain arrhythmias. Because the onset of action of transdermal clonidine is about 2 days and the offset is 8 hours, it has limited usefulness in the treatment of perioperative hypertension. Therefore, only metoprolol, labetalol, enalaprilat, and hydralazine have a major role in the treatment of hypertensive urgency when oral medications cannot be used.
When given intravenously, enalaprilat and hydralazine are safe, effective, widely available, and inexpensive. When deciding between these 2 agents, a few other considerations may be of importance. Even though ACE inhibitors have well‐recognized benefits in the management of HF50 and diabetic nephropathy,51 these characteristics are not relevant in the short‐term use of enalaprilat to treat perioperative hypertension. However, enalaprilat may be preferred over hydralazine when activation of the SNS and reflex tachycardia is to be avoided (cardiac ischemia, aortic dissection, increased intracranial pressure). Hydralazine may be preferred in the setting of hyperkalemia and acute renal failure. It must be preferred in pregnancy or bilateral renal artery stenosis.
Although the weight of the evidence of perioperative ‐blocker use to reduce CV events in noncardiac surgery suggests a benefit, there are significant limitations. Few studies have compared different ‐blockers. Studies to determine the ideal target population, duration of therapy, and route of administration are lacking. Additionally, using perioperative ‐blockers may cause harm in low‐risk patients.52 Care should be taken when using labetalol and metoprolol in combination as they can induce a dangerous reduction in HR. The role of acute administration of intravenous ‐blockers in the setting of myocardial ischemia is debatable, and probably dangerous in the setting of hypotension, bradycardia, HB, pulmonary edema, or bronchospasm.53
Therefore, generalizing the perioperative ‐blocker data to all patients with perioperative hypertension seems unlikely to have significant benefit, and may possibly pose harm.29 However, it seems reasonable to use ‐blockers in those in whom it would be indicated otherwise, and to continue parenteral therapy in those already taking a ‐blocker preoperatively in order to avoid withdrawal.54
When deciding between metoprolol and labetalol, a few considerations may be of importance. First, there is much more evidence documenting the safety and efficacy of labetalol in perioperative hypertension. Second, even though metoprolol has proven benefit in patients with chronic HF, coronary artery disease (CAD), and MI, these long‐term studies investigated oral metoprolol, not the intravenous formulation.55 Most importantly, labetalol is more effective at lowering BP due to its additional blockade of alpha1 adrenoreceptors. Neither drug should be used in acute HF, bradycardia or greater than first‐degree HB, or bronchospasm. In conclusion, intravenous labetalol should be preferred over intravenous metoprolol for the management of perioperative hypertension.
Conclusions
Perioperative hypertension ideally should be evaluated well before the operative time period, when there is adequate time to initiate medications. Secondary causes such as pain, agitation, hypercarbia, hypoxemia, and hypervolemia should be treated directly prior to the administration of antihypertensive medications. It is uncertain whether patients with a BP of 180/110 mm Hg benefit from any specific parenteral medication, as there is little evidence from several studies that this level of BP without TOD leads to an increase in perioperative morbidity or mortality.3, 4, 7, 56 However, patients with hypertensive urgency are at higher risk for perioperative complications; therefore, their BP should be managed gradually to 160/110 mm Hg with the outlined recommended parenteral regimen (Figure 1).
When selecting a parenteral medication, we suggest first to exclude any contraindications, or see if an indication exists for a specific agent. Hydralazine, enalaprilat, metoprolol, or labetalol can be used as first‐line agents. Due to the scarcity of comparative trials looking at clinically significant outcomes (length of hospital stay, morbidity, mortality), decisions for the management of perioperative hypertension should be made based on comorbidity, efficacy, toxicity, and cost (Table 1).
Acknowledgements
The authors Henry R. Black, M.D. for his contribution.
An association between hypertension and operative risk has been reported in small studies since the early 1970s. In two studies, Prys‐Roberts et al.1, 2 found that subjects with uncontrolled hypertension were more likely to have myocardial ischemic changes on electrocardiography with episodes of hypotension during induction of anesthesia. Subjects without hypertension or with hypertension controlled by medication were less likely to have episodes of hypotension, regardless of the type of anesthetic.
Hypertension increases the risk of developing perioperative heart failure (HF), renal failure, myocardial ischemia, or stroke. The level of risk is dependent upon the blood pressure (BP) level. It has been shown that a BP of 180/110 mm Hg without target‐organ damage (TOD) is not an independent risk factor for perioperative cardiovascular (CV) complications, suggesting this level of BP does not need to be reduced rapidly to normal.3, 4
The Joint National Committee defines hypertensive emergency as severe elevations in BP (usually >180/120 mm Hg) that produce evidence of TOD.5 Patients with this level of BP who are asymptomatic and have no signs of TOD are considered to have hypertensive urgency. As patients with this level of BP are at higher risk perioperatively, pharmacotherapy is indicated. When oral medications cannot be administered, hypertensive urgency can be managed with a parenteral medication. The agent should be easily and predictably titrated, safe, and convenient (Table 1). This article reviews the management of perioperative hypertensive urgency with parenteral medications. The management of hypertensive emergencies, aortic dissection, and hypertension of pregnancy is outside the scope of this review.
| Drug | Dose | Onset of Action | Duration | Use With Caution in | Adverse Reactions | Pregnancy Class* | Daily Cost |
|---|---|---|---|---|---|---|---|
| |||||||
| Hydralazine hydrochloride | 1020 mg IV q46h | 1020 minutes | 14 hours | Increased ICP; aortic dissection; myocardial ischemia | Reflex tachycardia; headache, flushing, vomiting | C | 20 mg q4h, $90 |
| Metoprolol | 1.255.0 mg IV q6h | 20 minutes | 58 hours | Heart block; bradycardia; acute heart failure | Bronchospasm | C (first trimester); D (second‐third trimesters) | 5 mg q6h, $10 |
| Enalaprilat | 1.255.0 mg IV q6h | 1530 minutes | 612 hours | Hyperkalemia; acute renal failure; hypovolemia | Hypotension; angioedema | C (first trimester); D (second‐third trimesters) | 5 mg q6h, $60 |
| Labetalol hydrochloride | 2080 mg IV q10min (max 300 mg daily) | 510 minutes | 36 hours | See metoprolol | Bronchospasm; nausea, vomitting; scalp tingling | C (first trimester); D (second‐third trimesters) | 300 mg, $15 |
| Transdermal clonidine | 0.10.3 mg once weekly | 23 days | 7 days | Abrupt withdrawal; elderly | Drowsiness, dizziness; local skin erythema; dry mouth | C | 0.3 mg/24‐hour patch, $10 |
Preoperative Considerations
In normotensive patients the induction of anesthesia can cause an acute elevation in BP (2030 mm Hg) and heart rate (HR) (1520 bpm).6 In patients with preexisting hypertension these changes are often greater, with elevations up to 90 mm Hg and 40 bpm. As anesthesia progresses systolic BP starts to fall (30 mm Hg), as a direct effect of both the anesthetic and the inhibition of the sympathetic nervous system (SNS). Patients with uncontrolled hypertension can have more severe reductions (60 mm Hg).6 This can result in intraoperative hypotension and shock. In a study of over 650 patients, marked intraoperative hypotension (50% of preoperative BP or a 33% reduction for more than 10 minutes) was an independent risk factor for perioperative CV complications (cardiac arrhythmia, ischemia, HF, or renal failure).7
Therefore, when BP is mildly elevated at the time of surgery (180/110 mm Hg), rapid reduction in BP is not necessary, and studies have been unable to demonstrate a benefit to delaying surgery.8 However, when BP is 180/110 mm Hg preoperatively, antihypertensive medications should be administered and intraoperative blood pressure monitored closely. There is a lack of data to support delay of surgery.9
Postoperative Considerations
The postoperative period is also associated with elevations in BP. In the immediate recovery phase from anesthesia, there is a mild elevation in BP within 10 to 15 mm Hg, but there are larger fluctuations in patients with preexisting hypertension.6 Otherwise postoperative hypertension can be seen from a variety of causes such as pain, excitement on emergence from anesthesia, and hypercarbia.10 Less common causes include agitation, hypoxemia, and hypervolemia. These secondary causes should be identified and treated before any antihypertensive medications are administered.
Drug Therapy
When evaluating a patient with a BP of 180/110 mm Hg, the physician must first classify the patient as having a hypertensive emergency or urgency. Hypertensive emergencies require immediate reduction in BP to prevent or limit hypertensive encephalopathy, intracerebral hemorrhage, acute myocardial infarction (MI), HF and aortic dissection.11 This is often accomplished by using continuous infusions of medications such as nitroprusside, nicardipine, or fenoldopam, and requires monitoring in an intensive care unit (ICU) with an intraarterial catheter.
As patients with hypertensive urgency are not at great risk for TOD, continuous infusions of the above medications that require ICU monitoring and intraarterial catheters seem to be unnecessary, and a possible misuse of resources. Treating hypertensive urgency in this manner could also be potentially dangerous.12, 13 Patients with chronic hypertension often have autoregulation of organ perfusion shifted to a higher range of mean arterial pressure, so excessive pressure reductions to normal BP values may induce organ hypoperfusion.14 Therefore, BP in hypertensive urgency can be lowered to 160/100 mm Hg over time.5 When oral medications cannot be used, there are several parenteral agents.
Diltiazem Hydrochloride and Verapamil
Diltiazem hydrochloride and verapamil are non‐dihydropyridine calcium‐channel blockers that produce vasodilation by decreasing calcium entry into vascular smooth muscle. In a study of 18 hypertensive patients, administration of intravenous diltiazem resulted in significant BP reductions within 5 minutes, however a variety of rhythm disturbances and heart block (HB) were observed.15 Verapamil has also been shown to successfully lower BP.16 However, when given at antihypertensive doses, verapamil has been shown to cause prolongation of the PR interval (30%), second‐degree block (0.7%), and complete HB (1.7%).17
Therefore, although oral diltiazem and verapamil may be appropriate for treating hypertension, the intravenous formulations are indicated only for the treatment of atrial fibrillation or flutter, and paroxysmal supraventricular tachycardia.18
Clonidine
Clonidine stimulates alpha2‐adrenoreceptors in the brain stem. This action results in reduced sympathetic outflow from the central nervous system, and decreases in peripheral resistance, renal vascular resistance, HR, and BP. Renal blood flow and glomerular filtration rate remain essentially unchanged. Normal postural reflexes are intact; therefore, orthostatic symptoms are mild and infrequent. Sudden cessation of treatment with clonidine has been associated with dangerous rebound hypertension.
Catapres‐TTS (clonidine) transdermal releases clonidine at a constant rate for 7 days. Therapeutic levels are achieved 2 to 3 days after initial application. After removal, therapeutic levels persist for about 8 hours and decline slowly over several days.19
Perioperatively, beneficial effects of clonidine include decreased anesthetic and opioid requirements, reduced hemodynamic responses to intubation and other stimuli, and improved postoperative renal function.20 Alpha2 agonists have also been shown to have significant antiischemic properties.21, 22
Beta‐adrenoreceptor () Blockers
Beta blockers are of particular interest in the management of perioperative hypertension. Several studies in the 1980s demonstrated that preoperative use of ‐blockers attenuated the severe BP fluctuations in the perioperative period; there was also a reduction in myocardial ischemia.2124 In addition, the preoperative ‐blockers in select at‐risk populations has been shown to decrease the rate of CV events (MI, unstable angina, need for coronary‐artery bypass, HF) and death.25, 26
Given these findings, the American College of Cardiology/American Heart Association (ACC/AHA) guidelines on the perioperative CV evaluation and care for noncardiac surgery recommended ‐blockers in patients receiving ‐blockers for angina, symptomatic arrhythmias, or hypertension; those undergoing vascular surgery with coronary artery disease or a revised cardiac risk index (RCRI) score >1; and those undergoing intermediate risk surgery with a RCRI of >1.27, 28 However, the recently published Perioperative Ischemic Evaluation Study (POISE) trial demonstrated that while ‐blockers reduced the risk of perioperative MI, there was an overall increase in net mortality.29 Given that most of the patients had an RCRI of 1 to 2, the ACC/AHA plans to revise this guideline.
If a ‐blocker is selected to manage perioperative hypertension, there are two available for parenteral use.
Metoprolol Tartrate
Metoprolol is a ‐1 selective adrenoreceptor antagonist available in both oral and intravenous formulations. Acutely, it decreases cardiac output by reducing both HR and contractility, therefore resulting in a decrease in BP. Over the course of a week it antagonizes ‐receptors in the juxtaglomerular complex, suppressing renin release and therefore production of angiotensin II.30 Metoprolol may lower BP by other mechanisms, including alteration of the sympathetic nervous system (SNS) and altered baroreceptor sensitivity.
The oral formulation is most commonly used to treat hypertension, MI, angina, atrial fibrillation, and HF. The intravenous form is only approved for the treatment of acute MI and supraventricular tachycardia. However, intravenous administration does induce its maximal hypotensive response within 20 minutes, generally lasting 3 to 4 hours. In a study investigating metoprolol and perioperative hypertension during extubation, the administration of intravenous metoprolol safely blunted the expected rise in BP.31 Similar findings were demonstrated in neurosurgical patients.32
Even though intravenous metoprolol can effectively lower BP, it does so mainly by reducing cardiac output. Therefore, caution must be taken in patients with a low cardiac index, and it should be avoided in acute HF, bradycardia or greater than first‐degree HB, or bronchospasm.
As metoprolol is a far more commonly used substitute for atenolol, we have deferred its specific discussion.
Labetalol Hydrochloride
Labetalol antagonizes both alpha1‐ and nonselective ‐adrenoreceptors. When given intravenously the onset of action is 5 minutes, but the duration can vary from 20 minutes to 23 hours, with an average of generally 6 hours. An initial dosage of 10 to 20 mg administered over 2 minutes can be followed by repeat doses every 10 minutes until the desired BP goal is achieved (maximum 300 mg daily). It decreases systemic vascular resistance and typically has no significant effect on cardiac index. In a multicenter study, bolus doses produced a rapid, smooth reduction in BP without reflex tachycardia or serious side effects.33 It has been shown to have similar efficacy and safety in cardiac surgery and other surgery requiring anesthesia.34, 35 Furthermore, it does not increase intracranial pressure,36 and is safe in patients with renal insufficiency or pregnancy. Contraindications to labetalol are hypotension, bradycardia, high‐degree HB, and severe asthma or chronic obstructive pulmonary disease.
Hydralazine Hydrochloride
Hydralazine reduces BP by increasing cyclic‐guanosine monophosphate in vascular smooth muscle, therefore leading to direct arterial vasodilation with little effect on venous circulation.37 It causes rapid reductions in BP, sometimes resulting in reflex tachycardia. When given intravenously, it has an onset of action of 5 minutes and duration of 3 to 8 hours, dependent mostly on hepatic clearance. This variability in hepatic acetylation and inactivation leads to some difficulty in drug titration.38 The starting dose is usually 10 mg, and it is administered every 4 to 6 hours. As stated, intravenous administration results in an increase in HR, cardiac output, myocardial contractility, and an overall increase in sympathetic activity.39
Although hydralazine has been used for the management of perioperative hypertension for several decades,40 its overall efficacy and safety have not been adequately defined for this setting. It has proven to be most successful during hypertension in pregnancy41 or hypertensive emergency.42 However, hydralazine is still widely used and is considered by some experts as an acceptable antihypertensive drug in the perioperative setting, as it can be administered in divided doses, routinely at 4 to 6 hour intervals, making it suitable for the treatment of hypertension in subjects unable to take medications by mouth or when a continuous infusion is unnecessary.
Hydralazine should be used with extreme caution in patients with evidence of cardiac ischemia, and it should be avoided in patients with aortic dissection or an increased intracranial pressure. The activation of the SNS and arterial vasodilation could have a potential benefit for patients with renal dysfunction.
Enalaprilat
Enalaprilat is the intravenous preparation of the active form of the angiotensin converting enzyme (ACE) inhibitor enalapril. By ACE inhibition, enalaprilat leads to a reduction in the production of angiotensin II, thereby reducing mean arterial pressure. The usual dose is 1.25 mg, and as much as 5 mg may be given every 6 hours as necessary,43 making it suitable for the treatment of hypertension in subjects unable to take medications by mouth.
Enalaprilat has demonstrated efficacy and safety when used in both CV surgery and neurosurgery. In a study of 14 patients with chronic HF, the administration of enalaprilat resulted in significant reductions in both mean arterial pressure (21%) and pulmonary capillary wedge pressure (33%).44 There was also an increase in the stroke volume index (20%) without a change in coronary blood flow or myocardial oxygen consumption, indicating an improvement in left ventricular function. As ACE inhibitors do not impair cerebral blood flow, enalaprilat may also be used safely in neurosurgery.45 Additionally, enalaprilat has been studied in the treatment of hypertensive urgencies. In a study of patients who had a diastolic BP between 100 and 114 mm Hg, the administration of 1.25 mg of enalaprilat lead to a significant reduction in systolic and diastolic BP within 60 minutes without any major adverse events.46
Even though enalaprilat has demonstrated safety and efficacy in several perioperative trials, its actions may be variable and not always predictable. When investigating the appropriate dose of enalaprilat, Hirschl et al.43 randomized 65 patients to receive different doses of enalaprilat. Response to treatment was defined as a stable reduction in BP to 180/95 mm Hg within 45 minutes. The goal was reached in only 63%, and surprisingly the response rates did not differ across differing dosages: 0.625 mg (67%), 1.25 mg (65%), 2.5 mg (59%), and 5 mg (62%).
Continuing chronic ACE inhibitor therapy within 12 to 24 hours preoperatively has been associated with severe hypotension at or shortly after induction of anesthesia. In a recent meta‐analysis, Rosenman et al.47 assessed the clinical consequences of preoperatively continuing vs. withholding ACE inhibitors or a angiotensin II receptor blocker (ARB) in patients treated chronically with these agents. Patients receiving an immediate preoperative ACE inhibitor or ARB were significantly more likely to develop hypotension requiring vasopressors. Although this observation cannot be directly translated, caution should be advised when selecting intravenous enalaprilat for the acute lowering of BP preoperatively.
Enalaprilat is contraindicated in pregnancy and patients with bilateral renal artery stenosis. It must also be used carefully in patients with hyperkalemia, acute renal failure, or hypovolemia.48 There should also be a dose adjustment when given to patients with severe chronic kidney disease.49 In addition, its use 12 to 24 hours prior to the induction of anesthesia should be discussed with the anesthesiologist.
Discussion
Nitroprusside, nitroglycerin, nicardipine, and fenoldopam are all effective antihypertensive medications. However, their availability only as continuous infusions requires ICU monitoring and an intraarterial catheter, and they are therefore unnecessary in the management of hypertensive urgency. The parenteral medications that do not require a continuous infusion are diltiazem, verapamil, metoprolol, labetalol, enalaprilat, hydralazine, and transdermal clonidine.
As stated, the intravenous formulations of diltiazem and verapamil are indicated only for certain arrhythmias. Because the onset of action of transdermal clonidine is about 2 days and the offset is 8 hours, it has limited usefulness in the treatment of perioperative hypertension. Therefore, only metoprolol, labetalol, enalaprilat, and hydralazine have a major role in the treatment of hypertensive urgency when oral medications cannot be used.
When given intravenously, enalaprilat and hydralazine are safe, effective, widely available, and inexpensive. When deciding between these 2 agents, a few other considerations may be of importance. Even though ACE inhibitors have well‐recognized benefits in the management of HF50 and diabetic nephropathy,51 these characteristics are not relevant in the short‐term use of enalaprilat to treat perioperative hypertension. However, enalaprilat may be preferred over hydralazine when activation of the SNS and reflex tachycardia is to be avoided (cardiac ischemia, aortic dissection, increased intracranial pressure). Hydralazine may be preferred in the setting of hyperkalemia and acute renal failure. It must be preferred in pregnancy or bilateral renal artery stenosis.
Although the weight of the evidence of perioperative ‐blocker use to reduce CV events in noncardiac surgery suggests a benefit, there are significant limitations. Few studies have compared different ‐blockers. Studies to determine the ideal target population, duration of therapy, and route of administration are lacking. Additionally, using perioperative ‐blockers may cause harm in low‐risk patients.52 Care should be taken when using labetalol and metoprolol in combination as they can induce a dangerous reduction in HR. The role of acute administration of intravenous ‐blockers in the setting of myocardial ischemia is debatable, and probably dangerous in the setting of hypotension, bradycardia, HB, pulmonary edema, or bronchospasm.53
Therefore, generalizing the perioperative ‐blocker data to all patients with perioperative hypertension seems unlikely to have significant benefit, and may possibly pose harm.29 However, it seems reasonable to use ‐blockers in those in whom it would be indicated otherwise, and to continue parenteral therapy in those already taking a ‐blocker preoperatively in order to avoid withdrawal.54
When deciding between metoprolol and labetalol, a few considerations may be of importance. First, there is much more evidence documenting the safety and efficacy of labetalol in perioperative hypertension. Second, even though metoprolol has proven benefit in patients with chronic HF, coronary artery disease (CAD), and MI, these long‐term studies investigated oral metoprolol, not the intravenous formulation.55 Most importantly, labetalol is more effective at lowering BP due to its additional blockade of alpha1 adrenoreceptors. Neither drug should be used in acute HF, bradycardia or greater than first‐degree HB, or bronchospasm. In conclusion, intravenous labetalol should be preferred over intravenous metoprolol for the management of perioperative hypertension.
Conclusions
Perioperative hypertension ideally should be evaluated well before the operative time period, when there is adequate time to initiate medications. Secondary causes such as pain, agitation, hypercarbia, hypoxemia, and hypervolemia should be treated directly prior to the administration of antihypertensive medications. It is uncertain whether patients with a BP of 180/110 mm Hg benefit from any specific parenteral medication, as there is little evidence from several studies that this level of BP without TOD leads to an increase in perioperative morbidity or mortality.3, 4, 7, 56 However, patients with hypertensive urgency are at higher risk for perioperative complications; therefore, their BP should be managed gradually to 160/110 mm Hg with the outlined recommended parenteral regimen (Figure 1).
When selecting a parenteral medication, we suggest first to exclude any contraindications, or see if an indication exists for a specific agent. Hydralazine, enalaprilat, metoprolol, or labetalol can be used as first‐line agents. Due to the scarcity of comparative trials looking at clinically significant outcomes (length of hospital stay, morbidity, mortality), decisions for the management of perioperative hypertension should be made based on comorbidity, efficacy, toxicity, and cost (Table 1).
Acknowledgements
The authors Henry R. Black, M.D. for his contribution.
- ,,.Studies of anaesthesia in relation to hypertension. I. Cardiovascular responses of treated and untreated patients.Br J Anaesth.1971;43(2):122–137.
- ,,, et al.Studies of anaesthesia in relation to hypertension. II. Haemodynamic consequences of induction and endotracheal intubation.Br J Anaesth.1971;43(6):531–547.
- ,,, et al.Multifactorial index of cardiac risk in noncardiac surgical procedures.N Engl J Med.1977;297(16):845–850.
- ,,, et al.Cardiac assessment for patients undergoing noncardiac surgery. A multifactorial clinical risk index.Arch Intern Med.1986;146(11):2131–2134.
- ,,, et al.The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report.JAMA.2003;289(19):2560–2572.
- ,.Management of anesthesia in patients with hypertension or ischemic heart disease.Int Anesthesiol Clin.1980;18(4):181–217.
- ,.Risks of general anesthesia and elective operation in the hypertensive patient.Anesthesiology.1979;50(4):285–292.
- ,,, et al.The dilemma of immediate preoperative hypertension: to treat and operate, or to postpone surgery?J Clin Anesth.2003;15(3):179–183.
- .Is blood pressure control necessary before surgery?Med Clin North Am.1993;77(2):349–363.
- ,.Hypertension in the immediate postoperative period.Br J Anaesth.1975;47(1):70–74.
- .Management of hypertensive emergencies.Lancet. 121994;344(8933):1335–1338.
- ,.Severely increased blood pressure in the emergency department.Ann Emerg Med.2003;41(4):513–529.
- ,,.Rapid reduction of severe asymptomatic hypertension. A prospective, controlled trial.Arch Intern Med.1989;149(10):2186–2189.
- ,.Cerebral blood flow and its pathophysiology in hypertension.Am J Hypertens.1989;2:(6 pt 1):486–492.
- ,,, et al.Efficacy, electrocardiographic and renal effects of intravenous diltiazem for essential hypertension.Am J Cardiol.1987;60(17):78I–84I.
- ,,, et al.Calcium entry blockers for the treatment of severe hypertension and hypertensive crisis.Am J Med.1984;77:(2B):35–45.
- ,.Rapid‐acting parenteral antihypertensive agents.J Clin Pharmacol.1990;30(3):195–209.
- ,.The role of calcium entry blockers in hypertensive emergencies.Circulation.1987;75(6 pt 2):V174–180.
- ,,, et al.Transdermal clonidine therapy in hypertensive patients. Effects on office and ambulatory recorded blood pressure values.JAMA.1985;253(2):233–235.
- ,,.Anesthesia and hypertension: the effect of clonidine on perioperative hemodynamics and isoflurane requirements.Anesthesiology.1987;67(1):3–10.
- ,,, et al.Small, oral dose of clonidine reduces the incidence of intraoperative myocardial ischemia in patients having vascular surgery.Anesthesiology.1996;85(4):706–712.
- ,,, et al.Effect of clonidine on cardiovascular morbidity and mortality after noncardiac surgery.Anesthesiology.2004;101(2):284–293.
- ,,, et al.Beta blockade to decrease silent myocardial ischemia during peripheral vascular surgery.Am J Surg.1989;158(2):113–116.
- ,,, et al.Haemodynamic effects of pretreatment with metoprolol in hypertensive patients undergoing surgery.Br J Anaesth.1986;58(3):251–260.
- ,,, et al.Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group.N Engl J Med.1996;335(23):1713–1720.
- ,,, et al.The effect of bisoprolol on perioperative mortality and myocardial infarction in high‐risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group.N Engl J Med.1999;341(24):1789–1794.
- ,,, et al.ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, and Society for Vascular Surgery.J Am Coll Cardiol.2007;50(17):e159–e241.
- ,,, et al.Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery.Circulation.1999;100(10):1043–1049.
- ,,, et al.Effects of extended‐release metoprolol succinate in patients undergoing non‐cardiac surgery (POISE trial): a randomised controlled trial.Lancet.2008;371(9627):1839–1847.
- ,.Inhibition of angiotensin II potentiation of sympathetic nerve activity by beta‐adrenergic antagonists.Hypertension.1980;2(1):90–96.
- ,,, et al.Comparison of intravenous metoprolol, verapamil and diltiazem on the attenuation of haemodynamic changes associated with tracheal extubation.Eur J Anaesthesiol.1999;16(7):462–467.
- ,,, et al.The effect of metoprolol upon blood pressure, cerebral blood flow and oxygen consumption in patients subjected to craniotomy for cerebral tumours.Acta Anaesthesiol Scand.1994;38(3):271–275.
- ,,, et al.Intravenous labetalol in the treatment of severe hypertension and hypertensive emergencies.Am J Med.1983;75:(4A):95–102.
- ,,.I.V. labetalol in the treatment of hypertension following coronary‐artery surgery.Br J Anaesth.1982;54(11):1191–1196.
- ,,, et al.Intravenous labetalol for treatment of postoperative hypertension.Anesthesiology.1987;67(3):413–416.
- ,,, et al.Labetalol to control blood pressure after cerebrovascular surgery.Crit Care Med.1988;16(8):765–768.
- ,,, et al.Cyclic guanosine 3′,5′ monophosphate concentrations in pre‐eclampsia: effects of hydralazine.Br J Obstet Gynaecol.1996;103(1):33–38.
- ,,, et al.Effect of intravenous dose on hydralazine kinetics after administration.Clin Pharmacol Ther.1983;34(2):148–152.
- ,,.Adverse effects of direct‐acting vasodilators.Drug Saf.1994;11(2):80–85.
- ,,, et al.Incidence and mechanism of post‐carotid endarterectomy hypertension.Arch Surg.1987;122(10):1153–1155.
- ,,, et al.Hydralazine for treatment of severe hypertension in pregnancy: meta‐analysis.Bmj.2003;327(7421):955–960.
- ,.Current diagnosis and management of hypertensive emergency.Semin Dial.2006;19(6):502–512.
- ,,, et al.Clinical evaluation of different doses of intravenous enalaprilat in patients with hypertensive crises.Arch Intern Med.1995;155(20):2217–2223.
- ,,, et al.Enalaprilat, a new parenteral angiotensin‐converting enzyme inhibitor: rapid changes in systemic and coronary hemodynamics and humoral profile in chronic heart failure.J Am Coll Cardiol.1987;9(5):1131–1138.
- ,,, et al.Cerebral blood flow in patients with congestive heart failure treated with captopril.Am J Med.1984;76:(5B):91–95.
- ,,, et al.The effect of intravenous enalaprilat (MK‐422) administration in patients with mild to moderate essential hypertension.J Clin Pharmacol.1987;27(5):415–418.
- ,,, et al.Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period.J Hosp Med.2008;3(4):319–325.
- ,.Toxic effects of drugs used in the ICU. Nitroprusside, nitroglycerin, and angiotensin‐converting enzyme inhibitors.Crit Care Clin.1991;7(3):555–581.
- ,,, et al.Disposition of enalapril and enalaprilat in renal insufficiency.Kidney Int Suppl.1987;20:S117–122.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group.N Engl J Med.1987;316(23):1429–1435.
- ,,, et al.Angiotensin‐receptor blockade versus converting‐enzyme inhibition in type 2 diabetes and nephropathy.N Engl J Med.2004;351(19):1952–1961.
- ,,, et al.Perioperative beta‐blocker therapy and mortality after major noncardiac surgery.N Engl J Med. 282005;353(4):349–361.
- ,,, et al.Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo‐controlled trial.Lancet.2005;366(9497):1622–1632.
- ,,, et al.Perioperative beta‐blocker withdrawal and mortality in vascular surgical patients.Am Heart J.2001;141(1):148–153.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT‐HF).Lancet.1999;353(9169):2001–2007. [No authors listed]
- ,,, et al.Preoperative and long‐term cardiac risk assessment. Predictive value of 23 clinical descriptors, 7 multivariate scoring systems, and quantitative dipyridamole imaging in 360 patients.Ann Surg.1992;216(2):192–204.
- ,,.Studies of anaesthesia in relation to hypertension. I. Cardiovascular responses of treated and untreated patients.Br J Anaesth.1971;43(2):122–137.
- ,,, et al.Studies of anaesthesia in relation to hypertension. II. Haemodynamic consequences of induction and endotracheal intubation.Br J Anaesth.1971;43(6):531–547.
- ,,, et al.Multifactorial index of cardiac risk in noncardiac surgical procedures.N Engl J Med.1977;297(16):845–850.
- ,,, et al.Cardiac assessment for patients undergoing noncardiac surgery. A multifactorial clinical risk index.Arch Intern Med.1986;146(11):2131–2134.
- ,,, et al.The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report.JAMA.2003;289(19):2560–2572.
- ,.Management of anesthesia in patients with hypertension or ischemic heart disease.Int Anesthesiol Clin.1980;18(4):181–217.
- ,.Risks of general anesthesia and elective operation in the hypertensive patient.Anesthesiology.1979;50(4):285–292.
- ,,, et al.The dilemma of immediate preoperative hypertension: to treat and operate, or to postpone surgery?J Clin Anesth.2003;15(3):179–183.
- .Is blood pressure control necessary before surgery?Med Clin North Am.1993;77(2):349–363.
- ,.Hypertension in the immediate postoperative period.Br J Anaesth.1975;47(1):70–74.
- .Management of hypertensive emergencies.Lancet. 121994;344(8933):1335–1338.
- ,.Severely increased blood pressure in the emergency department.Ann Emerg Med.2003;41(4):513–529.
- ,,.Rapid reduction of severe asymptomatic hypertension. A prospective, controlled trial.Arch Intern Med.1989;149(10):2186–2189.
- ,.Cerebral blood flow and its pathophysiology in hypertension.Am J Hypertens.1989;2:(6 pt 1):486–492.
- ,,, et al.Efficacy, electrocardiographic and renal effects of intravenous diltiazem for essential hypertension.Am J Cardiol.1987;60(17):78I–84I.
- ,,, et al.Calcium entry blockers for the treatment of severe hypertension and hypertensive crisis.Am J Med.1984;77:(2B):35–45.
- ,.Rapid‐acting parenteral antihypertensive agents.J Clin Pharmacol.1990;30(3):195–209.
- ,.The role of calcium entry blockers in hypertensive emergencies.Circulation.1987;75(6 pt 2):V174–180.
- ,,, et al.Transdermal clonidine therapy in hypertensive patients. Effects on office and ambulatory recorded blood pressure values.JAMA.1985;253(2):233–235.
- ,,.Anesthesia and hypertension: the effect of clonidine on perioperative hemodynamics and isoflurane requirements.Anesthesiology.1987;67(1):3–10.
- ,,, et al.Small, oral dose of clonidine reduces the incidence of intraoperative myocardial ischemia in patients having vascular surgery.Anesthesiology.1996;85(4):706–712.
- ,,, et al.Effect of clonidine on cardiovascular morbidity and mortality after noncardiac surgery.Anesthesiology.2004;101(2):284–293.
- ,,, et al.Beta blockade to decrease silent myocardial ischemia during peripheral vascular surgery.Am J Surg.1989;158(2):113–116.
- ,,, et al.Haemodynamic effects of pretreatment with metoprolol in hypertensive patients undergoing surgery.Br J Anaesth.1986;58(3):251–260.
- ,,, et al.Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group.N Engl J Med.1996;335(23):1713–1720.
- ,,, et al.The effect of bisoprolol on perioperative mortality and myocardial infarction in high‐risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group.N Engl J Med.1999;341(24):1789–1794.
- ,,, et al.ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, and Society for Vascular Surgery.J Am Coll Cardiol.2007;50(17):e159–e241.
- ,,, et al.Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery.Circulation.1999;100(10):1043–1049.
- ,,, et al.Effects of extended‐release metoprolol succinate in patients undergoing non‐cardiac surgery (POISE trial): a randomised controlled trial.Lancet.2008;371(9627):1839–1847.
- ,.Inhibition of angiotensin II potentiation of sympathetic nerve activity by beta‐adrenergic antagonists.Hypertension.1980;2(1):90–96.
- ,,, et al.Comparison of intravenous metoprolol, verapamil and diltiazem on the attenuation of haemodynamic changes associated with tracheal extubation.Eur J Anaesthesiol.1999;16(7):462–467.
- ,,, et al.The effect of metoprolol upon blood pressure, cerebral blood flow and oxygen consumption in patients subjected to craniotomy for cerebral tumours.Acta Anaesthesiol Scand.1994;38(3):271–275.
- ,,, et al.Intravenous labetalol in the treatment of severe hypertension and hypertensive emergencies.Am J Med.1983;75:(4A):95–102.
- ,,.I.V. labetalol in the treatment of hypertension following coronary‐artery surgery.Br J Anaesth.1982;54(11):1191–1196.
- ,,, et al.Intravenous labetalol for treatment of postoperative hypertension.Anesthesiology.1987;67(3):413–416.
- ,,, et al.Labetalol to control blood pressure after cerebrovascular surgery.Crit Care Med.1988;16(8):765–768.
- ,,, et al.Cyclic guanosine 3′,5′ monophosphate concentrations in pre‐eclampsia: effects of hydralazine.Br J Obstet Gynaecol.1996;103(1):33–38.
- ,,, et al.Effect of intravenous dose on hydralazine kinetics after administration.Clin Pharmacol Ther.1983;34(2):148–152.
- ,,.Adverse effects of direct‐acting vasodilators.Drug Saf.1994;11(2):80–85.
- ,,, et al.Incidence and mechanism of post‐carotid endarterectomy hypertension.Arch Surg.1987;122(10):1153–1155.
- ,,, et al.Hydralazine for treatment of severe hypertension in pregnancy: meta‐analysis.Bmj.2003;327(7421):955–960.
- ,.Current diagnosis and management of hypertensive emergency.Semin Dial.2006;19(6):502–512.
- ,,, et al.Clinical evaluation of different doses of intravenous enalaprilat in patients with hypertensive crises.Arch Intern Med.1995;155(20):2217–2223.
- ,,, et al.Enalaprilat, a new parenteral angiotensin‐converting enzyme inhibitor: rapid changes in systemic and coronary hemodynamics and humoral profile in chronic heart failure.J Am Coll Cardiol.1987;9(5):1131–1138.
- ,,, et al.Cerebral blood flow in patients with congestive heart failure treated with captopril.Am J Med.1984;76:(5B):91–95.
- ,,, et al.The effect of intravenous enalaprilat (MK‐422) administration in patients with mild to moderate essential hypertension.J Clin Pharmacol.1987;27(5):415–418.
- ,,, et al.Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period.J Hosp Med.2008;3(4):319–325.
- ,.Toxic effects of drugs used in the ICU. Nitroprusside, nitroglycerin, and angiotensin‐converting enzyme inhibitors.Crit Care Clin.1991;7(3):555–581.
- ,,, et al.Disposition of enalapril and enalaprilat in renal insufficiency.Kidney Int Suppl.1987;20:S117–122.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group.N Engl J Med.1987;316(23):1429–1435.
- ,,, et al.Angiotensin‐receptor blockade versus converting‐enzyme inhibition in type 2 diabetes and nephropathy.N Engl J Med.2004;351(19):1952–1961.
- ,,, et al.Perioperative beta‐blocker therapy and mortality after major noncardiac surgery.N Engl J Med. 282005;353(4):349–361.
- ,,, et al.Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo‐controlled trial.Lancet.2005;366(9497):1622–1632.
- ,,, et al.Perioperative beta‐blocker withdrawal and mortality in vascular surgical patients.Am Heart J.2001;141(1):148–153.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT‐HF).Lancet.1999;353(9169):2001–2007. [No authors listed]
- ,,, et al.Preoperative and long‐term cardiac risk assessment. Predictive value of 23 clinical descriptors, 7 multivariate scoring systems, and quantitative dipyridamole imaging in 360 patients.Ann Surg.1992;216(2):192–204.
Hospitalist Postgraduate PA Training Program
In recent years, the demand for hospitalists has outstripped the supply, creating a national shortage.1, 2 A recent Society of Hospital Medicine (SHM) survey found that in the last 2 years there has been a 31% mean growth increase in the number of hospitalist groups.3 As hospitalists are becoming more difficult to recruit, many practices are utilizing physician assistants (PAs) and nurse practitioners (NPs), collectively referred to as nonphysician providers (NPPs) to help offset the workload.4 The SHM survey also noted that the number of hospitalist groups utilizing NPPs increased from 29% to 38%.3 The exact number of NPPs working for hospitalist groups is unknown.
Hospitalist NPPs are in demand for reasons other than just physician shortages. NPPs have been utilized to fill the gap in many institutions where the workforce was impacted by the 2002 Accreditation Council for Graduate Medical Education (ACGME) ruling to restrict resident work hours. Several studies have documented NPPs' ability to assist with the compliance of ACCGME resident work‐hour restrictions while maintaining patient continuity of care, improving length of stays, and reducing health care costs on various hospital services.59 Dresselhaus et al.10 found that 56% of medical resident's time on service was delegated to tasks not related to direct patient care. They proposed that these tasks can be delegated to the NPPs, leaving more time for the residents to focus on direct patient care. In a recent study performed at a Pennsylvania hospital, patients presenting to the emergency department with low‐risk chest pain (based upon thrombolysis in myocardial infarction [TIMI] risk score) were admitted to a nonteaching service staffed with NPPs and attending physicians. Simultaneously, a similar group of low‐risk chest pain patients were admitted to a traditional internal medicine resident service. The results demonstrated lower median length of stay and hospital charges on the nonteaching service. This study suggested that NPPs can offset the workload volume for medical residents, allowing them to focus on patients with higher acuity and greater learning value.11
Barriers to Finding Experienced NPPs in Hospital Medicine
Although many hospitalist groups are interested in hiring NPPs, there can be significant obstacles to recruitment. For example, most experienced PAs and NPs have clinical backgrounds in either surgical or medical subspecialties and therefore typically need extensive on‐the‐job training in hospital medicine, which can often take at least 6 to 12 months to acquire the basic skill set.12 Hiring new graduates may require even longer training periods.
The inexperience of new graduates has become an even more pertinent issue due to recent changes in PA education. Traditionally, PA programs attracted older students with prior healthcare experience, who wished to return to school for additional training. However, in 2005 a major shift occurred in PA education: programs began transitioning from graduating trainees with a bachelor's degree to now requiring a master's level degree for completion of the PA program.13 The acquisition of more advanced degrees has changed the demographics of the students matriculating into PA programs, attracting younger students, straight from undergraduate institutions, with less prior healthcare experience.14 As a result, not only are new PA graduates less experienced overall, but they are particularly lacking in exposure to hospital medicine. After PA students complete their first 12 months of PA school in the basic sciences and didactic coursework, they embark on 12 to 15 months of clinical rotations, which are largely rooted in primary care. In fact, many PA programs find it difficult to offer hospital‐based rotations while fulfilling the required rotations in primary care. These factors have resulted in the need for more extensive on‐the‐job training particularly for those new graduates interested in hospital medicine. In light of these challenges, our institution created a 12‐month postgraduate PA fellowship program in Hospital Medicine.
Postgraduate PA Training Programs
Postgraduate PA fellowships, interchangeably called residencies, are voluntary 1‐year training programs that provide both didactic instruction and clinical experience in a medical or surgical subspecialty, thereby lessening the need for on‐the‐job training. These programs are recognized by the Association of Postgraduate Physician Assistant Programs.15 Currently, there are 44 postgraduate training programs in the United States, in a wide range of medical and surgical specialties. At the end of these 1‐year postgraduate PA programs, most graduates receive a certificate of completion. Until now, the only postgraduate education option for PAs interested in Hospital Medicine was a master's completion program only available to PAs who were already employed by a hospitalist group.15 This work reviews the first reported postgraduate hospitalist training program for PAs. Specifically, the program's background, curriculum, anticipated program outcomes, and future plans are discussed.
Background for A Hospitalist Postgraduate PA Fellowship
Mayo Clinic Arizona is a multispecialty private group comprised of both outpatient services and a tertiary care hospital medical center, located in the metropolitan Phoenix, AZ, area. The Mayo Clinic Hospital is a 7‐story facility with 244 licensed beds, 18 operating rooms, and a Level II emergency department. The Mayo Hospitalist group is composed of 15 full time hospitalists and 6 part‐time hospitalists, all of whom are salaried Mayo employees. The group provides 24‐hour in‐house staffing, covering both resident services (teams composed of interns and residents supervised by a staff hospitalist) and nonresident services (staff hospitalists). Over the years there has been steady growth in the number of nonresident services, in part due to resident work‐hour restrictions. To support the physicians working on these nonresident services, the first PA was hired in 2001. Since then, the number of NPPs in our Hospitalist group has increased to 9.35 full‐time equivalents (FTEs), including 1 nurse practitioner. However, one of the greatest challenges in expanding the NPP service was the difficulty finding candidates with experience in hospital internal medicine. This need inspired the creation of a PA fellowship in Hospital Medicine. At the time, there were 2 other postgraduate PA training programs at the Mayo Clinic Arizona in Hepatology and Otolaryngology/Ear, Nose, and Throat (ENT) Surgery.
Program Description
The Mayo Clinic Arizona PA fellowship in Hospital Medicine began in October 2007 and currently accepts 1 fellow per year. Applicants must be graduates of an Accreditation Review Commission in Education for the Physician Assistant (ARC‐PA)‐accredited PA program and be certified through the National Commission on Certification of Physician Assistants (NCCPA). Furthermore, they must be licensed to work as a PA in the state of Arizona. The program is 12 months in duration, and is comprised of both didactic and clinical components. Upon graduation, the fellow earns a certificate of completion from the Mayo Clinic College of Medicine. The program has received recognition with the Association of Postgraduate Physician Assistant Programs (APPAP).
Two physician assistants act as co‐program directors of the PA fellowship in hospital medicine. They are given 0.10 full‐time equivalent (FTE) for management of the program, which includes day‐to‐day operations, curriculum development, and candidate selection. The program also has 2 volunteer physician medical directors, both of whom have previous medical residency experience. The physicians and NPPs in our hospitalist group volunteer their time to serve as faculty for the program, assisting with much of the didactic and clinical education. The program receives a budget of $99,500 per year, which is funded by the organization's foundation through the department of education. This includes the fellow stipend of $44,000 per 12 months and institutional malpractice insurance coverage. The fellow also receives health and dental insurance, 2 weeks of paid vacation, and $500 stipend toward attendance of a continuing medical education (CME) conference.
CURRICULUM
The PA fellowship curriculum is designed in a diverse unique format that strives to accommodate all types of learners. It includes clinical rotations in various medicine/surgical subspecialties, didactic instruction, and teaching modules (Figure 1). The curriculum is based upon the SHM Core Competencies.15
Clinical Rotations
The PA fellow completes 12 to 14 general hospital medicine and medical specialty rotations, each 2 to 4 weeks in duration. The rotation calendar for the current fellow is given in Figure 2. These rotations are all inpatient‐based and are supervised by either the hospitalist or the respective inpatient subspecialists. The PA fellow's specific clinical responsibilities vary from rotation to rotation, and are designed to maximize the fellow's exposure to that particular specialty. Each rotation has specific written objectives created by the program directors and reviewed by the rotation's preceptor(s) (Figure 2). During the clinical rotations, complementary didactic lectures, coursework, and readings are provided to ensure the PA fellow receives a strong foundation. Didactic instruction is designed by the program directors, physician preceptors and staff NPPs, and is coordinated with the clinical rotation specialty. At the end of each rotation the fellow is evaluated by the preceptor and given direct feedback on their performance.

Didactic Instruction
The didactic instruction is organized in a system‐based manner and occurs on a weekly basis during the Hospital Internal Medicine service and Medicine Consults rotations. Hospitalist NPPs and physician faculty are responsible for most of the teaching. This formal didactic instruction is supplemented by journal club presentations given by the PA fellow to faculty in the division of hospital internal medicine. The fellow is also required to attend daily medical resident lunchtime educational lectures, weekly medical grand rounds, and any lectures provided by the medicine subspecialties while the PA is on that particular rotation.
Teaching Modules
One component of the Hospital Medicine PA fellowship curriculum that may be unique is the concept of teaching modules. While receiving regular didactic instruction and completing their clinical rotations, the PA is also expected to complete self‐directed teaching module assignments. These modules serve to educate the PA fellow on the hospital as a systemthe true essence of hospital medicine. The modules cover a variety of topics not directly addressed during their rotations. These topics are outlined in Figure 3. Each teaching module consists of a didactic component, clinical application, and assessment (Figure 4) and has its own specific objectives and goals. Teaching modules are often taught by the local expert in the hospital in that particular area. For example, for the infectious control teaching module, the PA fellow will rotate with the infection control nursing staff learning about the isolation and infection control policies of the institution.


Assessment Tools
There are several tools utilized to assess both the PA fellow and the fellowship program itself (Figure 5). The assessment tools used include both ongoing and summative assessments. To fulfill the ongoing assessment, each rotation and teaching module contains assessment tools provided by the preceptor, which are reviewed by the program directors. Additionally, during the clinical rotations, skills are assessed using competency checklists that require the preceptor to directly observe the PA fellow perform a specific task or skill‐set and sign off on its successful completion (Supplementary Figures 6, 7).

There are 2 forms of summative assessment for the PA fellow. First, to assess the PA fellow's knowledge, comprehensive mid‐year and end‐year examinations are utilized. These multiple‐choice examinations are comprised of questions which align with the didactic lectures/objectives provided by the Hospital Medicine faculty throughout the year. The second form of summative evaluation of the fellow is project‐based and divided into 2 parts. First, the fellow is expected to write a publication‐quality manuscript on a hospital medicine topic by the end of the year. Second, the PA fellow is expected to create a professional portfolio, which is comprised of a collection of all of the rotation/module assessments, the formal program assessments, and documentation of all of the skills obtained by the fellow throughout year (competency checklists). This portfolio can be used by the graduate to demonstrate to future employers what skills they possess and provide documentation of knowledge gained during the fellowship.
The program itself is evaluated by several measures. First, the fellow provides formal feedback during the mid‐year and end‐of‐the‐year assessments, which are used to enhance the experience of future fellows. Second, there is ongoing review by both the division of Hospital Medicine and the institution's Allied Health Education Committee, which ensures that the program maintains the appropriate standards and goals.
Future Goals for the PA Fellowship
The program graduated its first fellow at the end of October 2008 and has enjoyed early success. Integrating the PA fellow onto the hospitalist services augmented the present mid‐level and physician teams. There has been excellent institutional support for the program with extremely positive feedback from the rotation preceptors. There are several futures plans for the program. Our first goal is to seek accreditation from the Accreditation Review Commission for Physician Assistants (ARC‐PA), the organization that accredits entry level PA programs and which began formal, voluntary accreditation of postgraduate programs in early 2008. We plan to begin this process within the next academic year.
Our second long‐term goal for the program is to include NPs in the training program. Because of the desire to seek accreditation, the program directors felt temporarily limiting the fellowship to PAs would aide in the rigorous accreditation process, which can take approximately 1 year to complete. There is an NP on our faculty and the program has received interest from NPs. Once we obtain accreditation, expand the program enrollment, and develop an NP curriculum, we plan to open the fellowship to either PA or NP applicants.
Our third goal is to substantiate our PA Fellowship validity with outcome measures and ultimately publishable data. Thus far, the success of the PA fellowship is qualitative, and with small numbers of graduates it is difficult to quantify. After graduation of many subsequent PA fellows, our goal is to obtain quantifiable data that can be used to improve the quality of the PA fellowship and demonstrate the value of postgraduate training for physician assistants.
Perhaps the most important goal of the program is to eventually accept additional PA/NP fellows per year. While 1 program does not meet the demands of a national shortage of hospitalist providers, it may serve as a model that other institutions can adapt to their own needs. Since the program is based upon the SHM Core Competencies, the curriculum can be applied to a variety of hospitalist programs, and its relatively low operating cost makes it feasible for both academic‐based and community‐based institutions. Importantly, since recruitment and retention of employees is such a challenge for most hospitalist groups, this PA fellowship program may serve as a vehicle for recruitment and long‐term retention of well‐trained employees. This precedent has been set, as our division has hired our first PA fellow, whose transition from PA fellow to PA staff was seamless.
In conclusion, our PA fellowship in Hospital Medicine represents the first reported postgraduate PA program of this kind in the United States offering a certificate of completion. As the need for hospitalists increase so will the need for NPPs, particularly those with additional training in hospital medicine. This program serves as an example of 1 type of training tool for physician assistants looking to work in hospital medicine.
- ,,,.Health care market trends and the evolution of hospitalist use and roles.J Gen Intern Med.2004;20:101–107.
- .Innovations in the management of hospitalized patients. Nurse Pract Spring2006 (suppl):2–3.
- .Hospitalist pay up, productivity steady in SHM's latest survey.Hospitalist.2008;12(5):7,16.
- .Physician assistants: filling the gap in patient care in academic hospitals.Perspect Physician Assist Educ.2003;14(3):158–167.
- ,,, et al.The effect of a multidisciplinary hospitalist/physician and advanced practice nurse collaboration on hospital costs.J Nurs Adm.2006;36(2):79–85.
- ,,, et al.Physician extenders impact trauma systems.J Trauma.2005;58(5):917–920.
- ,,.The role of physician assistants in critical care units.Chest.1991;99:89–91.
- .Alliances: invaluable assistants.Hospitalist.2006;April:32–33.
- ,,.Resource use by physician assistant services versus teaching services.JAAPA.2002;15:33–42.
- ,,,,,.Analyzing the time and value of house staff inpatient work.J Intern Med.1998;13:534–540.
- ,,, et al.Improving resource utilization in a teaching hospital: Development of a nonteaching service for chest pain admissions.Acad Med.2006;81(5):432–435.
- .Midlevels make a rocky entrance into hospital medicine.Todays Hospitalist.2007;5(1):28–32.
- Accreditation Review Commission for Physician Assistant Education.3rd ed. 2005. Available at: http://www.arc‐pa.org/Standards/standards.html. Accessed September2009.
- 22nd Annual Report on Physician Assistant Education in the U.S., 2005–2006. Available at: http://www.paeaonline.org. Accessed September2009.
- Association of Postgraduate Physician Assistant Programs. Available at: http://www.appap.org. Accessed September2009.
- ,,,,.The core competencies in hospital medicine: development and methodology.J Hosp Med.2006;1:48–56.
In recent years, the demand for hospitalists has outstripped the supply, creating a national shortage.1, 2 A recent Society of Hospital Medicine (SHM) survey found that in the last 2 years there has been a 31% mean growth increase in the number of hospitalist groups.3 As hospitalists are becoming more difficult to recruit, many practices are utilizing physician assistants (PAs) and nurse practitioners (NPs), collectively referred to as nonphysician providers (NPPs) to help offset the workload.4 The SHM survey also noted that the number of hospitalist groups utilizing NPPs increased from 29% to 38%.3 The exact number of NPPs working for hospitalist groups is unknown.
Hospitalist NPPs are in demand for reasons other than just physician shortages. NPPs have been utilized to fill the gap in many institutions where the workforce was impacted by the 2002 Accreditation Council for Graduate Medical Education (ACGME) ruling to restrict resident work hours. Several studies have documented NPPs' ability to assist with the compliance of ACCGME resident work‐hour restrictions while maintaining patient continuity of care, improving length of stays, and reducing health care costs on various hospital services.59 Dresselhaus et al.10 found that 56% of medical resident's time on service was delegated to tasks not related to direct patient care. They proposed that these tasks can be delegated to the NPPs, leaving more time for the residents to focus on direct patient care. In a recent study performed at a Pennsylvania hospital, patients presenting to the emergency department with low‐risk chest pain (based upon thrombolysis in myocardial infarction [TIMI] risk score) were admitted to a nonteaching service staffed with NPPs and attending physicians. Simultaneously, a similar group of low‐risk chest pain patients were admitted to a traditional internal medicine resident service. The results demonstrated lower median length of stay and hospital charges on the nonteaching service. This study suggested that NPPs can offset the workload volume for medical residents, allowing them to focus on patients with higher acuity and greater learning value.11
Barriers to Finding Experienced NPPs in Hospital Medicine
Although many hospitalist groups are interested in hiring NPPs, there can be significant obstacles to recruitment. For example, most experienced PAs and NPs have clinical backgrounds in either surgical or medical subspecialties and therefore typically need extensive on‐the‐job training in hospital medicine, which can often take at least 6 to 12 months to acquire the basic skill set.12 Hiring new graduates may require even longer training periods.
The inexperience of new graduates has become an even more pertinent issue due to recent changes in PA education. Traditionally, PA programs attracted older students with prior healthcare experience, who wished to return to school for additional training. However, in 2005 a major shift occurred in PA education: programs began transitioning from graduating trainees with a bachelor's degree to now requiring a master's level degree for completion of the PA program.13 The acquisition of more advanced degrees has changed the demographics of the students matriculating into PA programs, attracting younger students, straight from undergraduate institutions, with less prior healthcare experience.14 As a result, not only are new PA graduates less experienced overall, but they are particularly lacking in exposure to hospital medicine. After PA students complete their first 12 months of PA school in the basic sciences and didactic coursework, they embark on 12 to 15 months of clinical rotations, which are largely rooted in primary care. In fact, many PA programs find it difficult to offer hospital‐based rotations while fulfilling the required rotations in primary care. These factors have resulted in the need for more extensive on‐the‐job training particularly for those new graduates interested in hospital medicine. In light of these challenges, our institution created a 12‐month postgraduate PA fellowship program in Hospital Medicine.
Postgraduate PA Training Programs
Postgraduate PA fellowships, interchangeably called residencies, are voluntary 1‐year training programs that provide both didactic instruction and clinical experience in a medical or surgical subspecialty, thereby lessening the need for on‐the‐job training. These programs are recognized by the Association of Postgraduate Physician Assistant Programs.15 Currently, there are 44 postgraduate training programs in the United States, in a wide range of medical and surgical specialties. At the end of these 1‐year postgraduate PA programs, most graduates receive a certificate of completion. Until now, the only postgraduate education option for PAs interested in Hospital Medicine was a master's completion program only available to PAs who were already employed by a hospitalist group.15 This work reviews the first reported postgraduate hospitalist training program for PAs. Specifically, the program's background, curriculum, anticipated program outcomes, and future plans are discussed.
Background for A Hospitalist Postgraduate PA Fellowship
Mayo Clinic Arizona is a multispecialty private group comprised of both outpatient services and a tertiary care hospital medical center, located in the metropolitan Phoenix, AZ, area. The Mayo Clinic Hospital is a 7‐story facility with 244 licensed beds, 18 operating rooms, and a Level II emergency department. The Mayo Hospitalist group is composed of 15 full time hospitalists and 6 part‐time hospitalists, all of whom are salaried Mayo employees. The group provides 24‐hour in‐house staffing, covering both resident services (teams composed of interns and residents supervised by a staff hospitalist) and nonresident services (staff hospitalists). Over the years there has been steady growth in the number of nonresident services, in part due to resident work‐hour restrictions. To support the physicians working on these nonresident services, the first PA was hired in 2001. Since then, the number of NPPs in our Hospitalist group has increased to 9.35 full‐time equivalents (FTEs), including 1 nurse practitioner. However, one of the greatest challenges in expanding the NPP service was the difficulty finding candidates with experience in hospital internal medicine. This need inspired the creation of a PA fellowship in Hospital Medicine. At the time, there were 2 other postgraduate PA training programs at the Mayo Clinic Arizona in Hepatology and Otolaryngology/Ear, Nose, and Throat (ENT) Surgery.
Program Description
The Mayo Clinic Arizona PA fellowship in Hospital Medicine began in October 2007 and currently accepts 1 fellow per year. Applicants must be graduates of an Accreditation Review Commission in Education for the Physician Assistant (ARC‐PA)‐accredited PA program and be certified through the National Commission on Certification of Physician Assistants (NCCPA). Furthermore, they must be licensed to work as a PA in the state of Arizona. The program is 12 months in duration, and is comprised of both didactic and clinical components. Upon graduation, the fellow earns a certificate of completion from the Mayo Clinic College of Medicine. The program has received recognition with the Association of Postgraduate Physician Assistant Programs (APPAP).
Two physician assistants act as co‐program directors of the PA fellowship in hospital medicine. They are given 0.10 full‐time equivalent (FTE) for management of the program, which includes day‐to‐day operations, curriculum development, and candidate selection. The program also has 2 volunteer physician medical directors, both of whom have previous medical residency experience. The physicians and NPPs in our hospitalist group volunteer their time to serve as faculty for the program, assisting with much of the didactic and clinical education. The program receives a budget of $99,500 per year, which is funded by the organization's foundation through the department of education. This includes the fellow stipend of $44,000 per 12 months and institutional malpractice insurance coverage. The fellow also receives health and dental insurance, 2 weeks of paid vacation, and $500 stipend toward attendance of a continuing medical education (CME) conference.
CURRICULUM
The PA fellowship curriculum is designed in a diverse unique format that strives to accommodate all types of learners. It includes clinical rotations in various medicine/surgical subspecialties, didactic instruction, and teaching modules (Figure 1). The curriculum is based upon the SHM Core Competencies.15
Clinical Rotations
The PA fellow completes 12 to 14 general hospital medicine and medical specialty rotations, each 2 to 4 weeks in duration. The rotation calendar for the current fellow is given in Figure 2. These rotations are all inpatient‐based and are supervised by either the hospitalist or the respective inpatient subspecialists. The PA fellow's specific clinical responsibilities vary from rotation to rotation, and are designed to maximize the fellow's exposure to that particular specialty. Each rotation has specific written objectives created by the program directors and reviewed by the rotation's preceptor(s) (Figure 2). During the clinical rotations, complementary didactic lectures, coursework, and readings are provided to ensure the PA fellow receives a strong foundation. Didactic instruction is designed by the program directors, physician preceptors and staff NPPs, and is coordinated with the clinical rotation specialty. At the end of each rotation the fellow is evaluated by the preceptor and given direct feedback on their performance.
Didactic Instruction
The didactic instruction is organized in a system‐based manner and occurs on a weekly basis during the Hospital Internal Medicine service and Medicine Consults rotations. Hospitalist NPPs and physician faculty are responsible for most of the teaching. This formal didactic instruction is supplemented by journal club presentations given by the PA fellow to faculty in the division of hospital internal medicine. The fellow is also required to attend daily medical resident lunchtime educational lectures, weekly medical grand rounds, and any lectures provided by the medicine subspecialties while the PA is on that particular rotation.
Teaching Modules
One component of the Hospital Medicine PA fellowship curriculum that may be unique is the concept of teaching modules. While receiving regular didactic instruction and completing their clinical rotations, the PA is also expected to complete self‐directed teaching module assignments. These modules serve to educate the PA fellow on the hospital as a systemthe true essence of hospital medicine. The modules cover a variety of topics not directly addressed during their rotations. These topics are outlined in Figure 3. Each teaching module consists of a didactic component, clinical application, and assessment (Figure 4) and has its own specific objectives and goals. Teaching modules are often taught by the local expert in the hospital in that particular area. For example, for the infectious control teaching module, the PA fellow will rotate with the infection control nursing staff learning about the isolation and infection control policies of the institution.
Assessment Tools
There are several tools utilized to assess both the PA fellow and the fellowship program itself (Figure 5). The assessment tools used include both ongoing and summative assessments. To fulfill the ongoing assessment, each rotation and teaching module contains assessment tools provided by the preceptor, which are reviewed by the program directors. Additionally, during the clinical rotations, skills are assessed using competency checklists that require the preceptor to directly observe the PA fellow perform a specific task or skill‐set and sign off on its successful completion (Supplementary Figures 6, 7).
There are 2 forms of summative assessment for the PA fellow. First, to assess the PA fellow's knowledge, comprehensive mid‐year and end‐year examinations are utilized. These multiple‐choice examinations are comprised of questions which align with the didactic lectures/objectives provided by the Hospital Medicine faculty throughout the year. The second form of summative evaluation of the fellow is project‐based and divided into 2 parts. First, the fellow is expected to write a publication‐quality manuscript on a hospital medicine topic by the end of the year. Second, the PA fellow is expected to create a professional portfolio, which is comprised of a collection of all of the rotation/module assessments, the formal program assessments, and documentation of all of the skills obtained by the fellow throughout year (competency checklists). This portfolio can be used by the graduate to demonstrate to future employers what skills they possess and provide documentation of knowledge gained during the fellowship.
The program itself is evaluated by several measures. First, the fellow provides formal feedback during the mid‐year and end‐of‐the‐year assessments, which are used to enhance the experience of future fellows. Second, there is ongoing review by both the division of Hospital Medicine and the institution's Allied Health Education Committee, which ensures that the program maintains the appropriate standards and goals.
Future Goals for the PA Fellowship
The program graduated its first fellow at the end of October 2008 and has enjoyed early success. Integrating the PA fellow onto the hospitalist services augmented the present mid‐level and physician teams. There has been excellent institutional support for the program with extremely positive feedback from the rotation preceptors. There are several futures plans for the program. Our first goal is to seek accreditation from the Accreditation Review Commission for Physician Assistants (ARC‐PA), the organization that accredits entry level PA programs and which began formal, voluntary accreditation of postgraduate programs in early 2008. We plan to begin this process within the next academic year.
Our second long‐term goal for the program is to include NPs in the training program. Because of the desire to seek accreditation, the program directors felt temporarily limiting the fellowship to PAs would aide in the rigorous accreditation process, which can take approximately 1 year to complete. There is an NP on our faculty and the program has received interest from NPs. Once we obtain accreditation, expand the program enrollment, and develop an NP curriculum, we plan to open the fellowship to either PA or NP applicants.
Our third goal is to substantiate our PA Fellowship validity with outcome measures and ultimately publishable data. Thus far, the success of the PA fellowship is qualitative, and with small numbers of graduates it is difficult to quantify. After graduation of many subsequent PA fellows, our goal is to obtain quantifiable data that can be used to improve the quality of the PA fellowship and demonstrate the value of postgraduate training for physician assistants.
Perhaps the most important goal of the program is to eventually accept additional PA/NP fellows per year. While 1 program does not meet the demands of a national shortage of hospitalist providers, it may serve as a model that other institutions can adapt to their own needs. Since the program is based upon the SHM Core Competencies, the curriculum can be applied to a variety of hospitalist programs, and its relatively low operating cost makes it feasible for both academic‐based and community‐based institutions. Importantly, since recruitment and retention of employees is such a challenge for most hospitalist groups, this PA fellowship program may serve as a vehicle for recruitment and long‐term retention of well‐trained employees. This precedent has been set, as our division has hired our first PA fellow, whose transition from PA fellow to PA staff was seamless.
In conclusion, our PA fellowship in Hospital Medicine represents the first reported postgraduate PA program of this kind in the United States offering a certificate of completion. As the need for hospitalists increase so will the need for NPPs, particularly those with additional training in hospital medicine. This program serves as an example of 1 type of training tool for physician assistants looking to work in hospital medicine.
In recent years, the demand for hospitalists has outstripped the supply, creating a national shortage.1, 2 A recent Society of Hospital Medicine (SHM) survey found that in the last 2 years there has been a 31% mean growth increase in the number of hospitalist groups.3 As hospitalists are becoming more difficult to recruit, many practices are utilizing physician assistants (PAs) and nurse practitioners (NPs), collectively referred to as nonphysician providers (NPPs) to help offset the workload.4 The SHM survey also noted that the number of hospitalist groups utilizing NPPs increased from 29% to 38%.3 The exact number of NPPs working for hospitalist groups is unknown.
Hospitalist NPPs are in demand for reasons other than just physician shortages. NPPs have been utilized to fill the gap in many institutions where the workforce was impacted by the 2002 Accreditation Council for Graduate Medical Education (ACGME) ruling to restrict resident work hours. Several studies have documented NPPs' ability to assist with the compliance of ACCGME resident work‐hour restrictions while maintaining patient continuity of care, improving length of stays, and reducing health care costs on various hospital services.59 Dresselhaus et al.10 found that 56% of medical resident's time on service was delegated to tasks not related to direct patient care. They proposed that these tasks can be delegated to the NPPs, leaving more time for the residents to focus on direct patient care. In a recent study performed at a Pennsylvania hospital, patients presenting to the emergency department with low‐risk chest pain (based upon thrombolysis in myocardial infarction [TIMI] risk score) were admitted to a nonteaching service staffed with NPPs and attending physicians. Simultaneously, a similar group of low‐risk chest pain patients were admitted to a traditional internal medicine resident service. The results demonstrated lower median length of stay and hospital charges on the nonteaching service. This study suggested that NPPs can offset the workload volume for medical residents, allowing them to focus on patients with higher acuity and greater learning value.11
Barriers to Finding Experienced NPPs in Hospital Medicine
Although many hospitalist groups are interested in hiring NPPs, there can be significant obstacles to recruitment. For example, most experienced PAs and NPs have clinical backgrounds in either surgical or medical subspecialties and therefore typically need extensive on‐the‐job training in hospital medicine, which can often take at least 6 to 12 months to acquire the basic skill set.12 Hiring new graduates may require even longer training periods.
The inexperience of new graduates has become an even more pertinent issue due to recent changes in PA education. Traditionally, PA programs attracted older students with prior healthcare experience, who wished to return to school for additional training. However, in 2005 a major shift occurred in PA education: programs began transitioning from graduating trainees with a bachelor's degree to now requiring a master's level degree for completion of the PA program.13 The acquisition of more advanced degrees has changed the demographics of the students matriculating into PA programs, attracting younger students, straight from undergraduate institutions, with less prior healthcare experience.14 As a result, not only are new PA graduates less experienced overall, but they are particularly lacking in exposure to hospital medicine. After PA students complete their first 12 months of PA school in the basic sciences and didactic coursework, they embark on 12 to 15 months of clinical rotations, which are largely rooted in primary care. In fact, many PA programs find it difficult to offer hospital‐based rotations while fulfilling the required rotations in primary care. These factors have resulted in the need for more extensive on‐the‐job training particularly for those new graduates interested in hospital medicine. In light of these challenges, our institution created a 12‐month postgraduate PA fellowship program in Hospital Medicine.
Postgraduate PA Training Programs
Postgraduate PA fellowships, interchangeably called residencies, are voluntary 1‐year training programs that provide both didactic instruction and clinical experience in a medical or surgical subspecialty, thereby lessening the need for on‐the‐job training. These programs are recognized by the Association of Postgraduate Physician Assistant Programs.15 Currently, there are 44 postgraduate training programs in the United States, in a wide range of medical and surgical specialties. At the end of these 1‐year postgraduate PA programs, most graduates receive a certificate of completion. Until now, the only postgraduate education option for PAs interested in Hospital Medicine was a master's completion program only available to PAs who were already employed by a hospitalist group.15 This work reviews the first reported postgraduate hospitalist training program for PAs. Specifically, the program's background, curriculum, anticipated program outcomes, and future plans are discussed.
Background for A Hospitalist Postgraduate PA Fellowship
Mayo Clinic Arizona is a multispecialty private group comprised of both outpatient services and a tertiary care hospital medical center, located in the metropolitan Phoenix, AZ, area. The Mayo Clinic Hospital is a 7‐story facility with 244 licensed beds, 18 operating rooms, and a Level II emergency department. The Mayo Hospitalist group is composed of 15 full time hospitalists and 6 part‐time hospitalists, all of whom are salaried Mayo employees. The group provides 24‐hour in‐house staffing, covering both resident services (teams composed of interns and residents supervised by a staff hospitalist) and nonresident services (staff hospitalists). Over the years there has been steady growth in the number of nonresident services, in part due to resident work‐hour restrictions. To support the physicians working on these nonresident services, the first PA was hired in 2001. Since then, the number of NPPs in our Hospitalist group has increased to 9.35 full‐time equivalents (FTEs), including 1 nurse practitioner. However, one of the greatest challenges in expanding the NPP service was the difficulty finding candidates with experience in hospital internal medicine. This need inspired the creation of a PA fellowship in Hospital Medicine. At the time, there were 2 other postgraduate PA training programs at the Mayo Clinic Arizona in Hepatology and Otolaryngology/Ear, Nose, and Throat (ENT) Surgery.
Program Description
The Mayo Clinic Arizona PA fellowship in Hospital Medicine began in October 2007 and currently accepts 1 fellow per year. Applicants must be graduates of an Accreditation Review Commission in Education for the Physician Assistant (ARC‐PA)‐accredited PA program and be certified through the National Commission on Certification of Physician Assistants (NCCPA). Furthermore, they must be licensed to work as a PA in the state of Arizona. The program is 12 months in duration, and is comprised of both didactic and clinical components. Upon graduation, the fellow earns a certificate of completion from the Mayo Clinic College of Medicine. The program has received recognition with the Association of Postgraduate Physician Assistant Programs (APPAP).
Two physician assistants act as co‐program directors of the PA fellowship in hospital medicine. They are given 0.10 full‐time equivalent (FTE) for management of the program, which includes day‐to‐day operations, curriculum development, and candidate selection. The program also has 2 volunteer physician medical directors, both of whom have previous medical residency experience. The physicians and NPPs in our hospitalist group volunteer their time to serve as faculty for the program, assisting with much of the didactic and clinical education. The program receives a budget of $99,500 per year, which is funded by the organization's foundation through the department of education. This includes the fellow stipend of $44,000 per 12 months and institutional malpractice insurance coverage. The fellow also receives health and dental insurance, 2 weeks of paid vacation, and $500 stipend toward attendance of a continuing medical education (CME) conference.
CURRICULUM
The PA fellowship curriculum is designed in a diverse unique format that strives to accommodate all types of learners. It includes clinical rotations in various medicine/surgical subspecialties, didactic instruction, and teaching modules (Figure 1). The curriculum is based upon the SHM Core Competencies.15
Clinical Rotations
The PA fellow completes 12 to 14 general hospital medicine and medical specialty rotations, each 2 to 4 weeks in duration. The rotation calendar for the current fellow is given in Figure 2. These rotations are all inpatient‐based and are supervised by either the hospitalist or the respective inpatient subspecialists. The PA fellow's specific clinical responsibilities vary from rotation to rotation, and are designed to maximize the fellow's exposure to that particular specialty. Each rotation has specific written objectives created by the program directors and reviewed by the rotation's preceptor(s) (Figure 2). During the clinical rotations, complementary didactic lectures, coursework, and readings are provided to ensure the PA fellow receives a strong foundation. Didactic instruction is designed by the program directors, physician preceptors and staff NPPs, and is coordinated with the clinical rotation specialty. At the end of each rotation the fellow is evaluated by the preceptor and given direct feedback on their performance.
Didactic Instruction
The didactic instruction is organized in a system‐based manner and occurs on a weekly basis during the Hospital Internal Medicine service and Medicine Consults rotations. Hospitalist NPPs and physician faculty are responsible for most of the teaching. This formal didactic instruction is supplemented by journal club presentations given by the PA fellow to faculty in the division of hospital internal medicine. The fellow is also required to attend daily medical resident lunchtime educational lectures, weekly medical grand rounds, and any lectures provided by the medicine subspecialties while the PA is on that particular rotation.
Teaching Modules
One component of the Hospital Medicine PA fellowship curriculum that may be unique is the concept of teaching modules. While receiving regular didactic instruction and completing their clinical rotations, the PA is also expected to complete self‐directed teaching module assignments. These modules serve to educate the PA fellow on the hospital as a systemthe true essence of hospital medicine. The modules cover a variety of topics not directly addressed during their rotations. These topics are outlined in Figure 3. Each teaching module consists of a didactic component, clinical application, and assessment (Figure 4) and has its own specific objectives and goals. Teaching modules are often taught by the local expert in the hospital in that particular area. For example, for the infectious control teaching module, the PA fellow will rotate with the infection control nursing staff learning about the isolation and infection control policies of the institution.
Assessment Tools
There are several tools utilized to assess both the PA fellow and the fellowship program itself (Figure 5). The assessment tools used include both ongoing and summative assessments. To fulfill the ongoing assessment, each rotation and teaching module contains assessment tools provided by the preceptor, which are reviewed by the program directors. Additionally, during the clinical rotations, skills are assessed using competency checklists that require the preceptor to directly observe the PA fellow perform a specific task or skill‐set and sign off on its successful completion (Supplementary Figures 6, 7).
There are 2 forms of summative assessment for the PA fellow. First, to assess the PA fellow's knowledge, comprehensive mid‐year and end‐year examinations are utilized. These multiple‐choice examinations are comprised of questions which align with the didactic lectures/objectives provided by the Hospital Medicine faculty throughout the year. The second form of summative evaluation of the fellow is project‐based and divided into 2 parts. First, the fellow is expected to write a publication‐quality manuscript on a hospital medicine topic by the end of the year. Second, the PA fellow is expected to create a professional portfolio, which is comprised of a collection of all of the rotation/module assessments, the formal program assessments, and documentation of all of the skills obtained by the fellow throughout year (competency checklists). This portfolio can be used by the graduate to demonstrate to future employers what skills they possess and provide documentation of knowledge gained during the fellowship.
The program itself is evaluated by several measures. First, the fellow provides formal feedback during the mid‐year and end‐of‐the‐year assessments, which are used to enhance the experience of future fellows. Second, there is ongoing review by both the division of Hospital Medicine and the institution's Allied Health Education Committee, which ensures that the program maintains the appropriate standards and goals.
Future Goals for the PA Fellowship
The program graduated its first fellow at the end of October 2008 and has enjoyed early success. Integrating the PA fellow onto the hospitalist services augmented the present mid‐level and physician teams. There has been excellent institutional support for the program with extremely positive feedback from the rotation preceptors. There are several futures plans for the program. Our first goal is to seek accreditation from the Accreditation Review Commission for Physician Assistants (ARC‐PA), the organization that accredits entry level PA programs and which began formal, voluntary accreditation of postgraduate programs in early 2008. We plan to begin this process within the next academic year.
Our second long‐term goal for the program is to include NPs in the training program. Because of the desire to seek accreditation, the program directors felt temporarily limiting the fellowship to PAs would aide in the rigorous accreditation process, which can take approximately 1 year to complete. There is an NP on our faculty and the program has received interest from NPs. Once we obtain accreditation, expand the program enrollment, and develop an NP curriculum, we plan to open the fellowship to either PA or NP applicants.
Our third goal is to substantiate our PA Fellowship validity with outcome measures and ultimately publishable data. Thus far, the success of the PA fellowship is qualitative, and with small numbers of graduates it is difficult to quantify. After graduation of many subsequent PA fellows, our goal is to obtain quantifiable data that can be used to improve the quality of the PA fellowship and demonstrate the value of postgraduate training for physician assistants.
Perhaps the most important goal of the program is to eventually accept additional PA/NP fellows per year. While 1 program does not meet the demands of a national shortage of hospitalist providers, it may serve as a model that other institutions can adapt to their own needs. Since the program is based upon the SHM Core Competencies, the curriculum can be applied to a variety of hospitalist programs, and its relatively low operating cost makes it feasible for both academic‐based and community‐based institutions. Importantly, since recruitment and retention of employees is such a challenge for most hospitalist groups, this PA fellowship program may serve as a vehicle for recruitment and long‐term retention of well‐trained employees. This precedent has been set, as our division has hired our first PA fellow, whose transition from PA fellow to PA staff was seamless.
In conclusion, our PA fellowship in Hospital Medicine represents the first reported postgraduate PA program of this kind in the United States offering a certificate of completion. As the need for hospitalists increase so will the need for NPPs, particularly those with additional training in hospital medicine. This program serves as an example of 1 type of training tool for physician assistants looking to work in hospital medicine.
- ,,,.Health care market trends and the evolution of hospitalist use and roles.J Gen Intern Med.2004;20:101–107.
- .Innovations in the management of hospitalized patients. Nurse Pract Spring2006 (suppl):2–3.
- .Hospitalist pay up, productivity steady in SHM's latest survey.Hospitalist.2008;12(5):7,16.
- .Physician assistants: filling the gap in patient care in academic hospitals.Perspect Physician Assist Educ.2003;14(3):158–167.
- ,,, et al.The effect of a multidisciplinary hospitalist/physician and advanced practice nurse collaboration on hospital costs.J Nurs Adm.2006;36(2):79–85.
- ,,, et al.Physician extenders impact trauma systems.J Trauma.2005;58(5):917–920.
- ,,.The role of physician assistants in critical care units.Chest.1991;99:89–91.
- .Alliances: invaluable assistants.Hospitalist.2006;April:32–33.
- ,,.Resource use by physician assistant services versus teaching services.JAAPA.2002;15:33–42.
- ,,,,,.Analyzing the time and value of house staff inpatient work.J Intern Med.1998;13:534–540.
- ,,, et al.Improving resource utilization in a teaching hospital: Development of a nonteaching service for chest pain admissions.Acad Med.2006;81(5):432–435.
- .Midlevels make a rocky entrance into hospital medicine.Todays Hospitalist.2007;5(1):28–32.
- Accreditation Review Commission for Physician Assistant Education.3rd ed. 2005. Available at: http://www.arc‐pa.org/Standards/standards.html. Accessed September2009.
- 22nd Annual Report on Physician Assistant Education in the U.S., 2005–2006. Available at: http://www.paeaonline.org. Accessed September2009.
- Association of Postgraduate Physician Assistant Programs. Available at: http://www.appap.org. Accessed September2009.
- ,,,,.The core competencies in hospital medicine: development and methodology.J Hosp Med.2006;1:48–56.
- ,,,.Health care market trends and the evolution of hospitalist use and roles.J Gen Intern Med.2004;20:101–107.
- .Innovations in the management of hospitalized patients. Nurse Pract Spring2006 (suppl):2–3.
- .Hospitalist pay up, productivity steady in SHM's latest survey.Hospitalist.2008;12(5):7,16.
- .Physician assistants: filling the gap in patient care in academic hospitals.Perspect Physician Assist Educ.2003;14(3):158–167.
- ,,, et al.The effect of a multidisciplinary hospitalist/physician and advanced practice nurse collaboration on hospital costs.J Nurs Adm.2006;36(2):79–85.
- ,,, et al.Physician extenders impact trauma systems.J Trauma.2005;58(5):917–920.
- ,,.The role of physician assistants in critical care units.Chest.1991;99:89–91.
- .Alliances: invaluable assistants.Hospitalist.2006;April:32–33.
- ,,.Resource use by physician assistant services versus teaching services.JAAPA.2002;15:33–42.
- ,,,,,.Analyzing the time and value of house staff inpatient work.J Intern Med.1998;13:534–540.
- ,,, et al.Improving resource utilization in a teaching hospital: Development of a nonteaching service for chest pain admissions.Acad Med.2006;81(5):432–435.
- .Midlevels make a rocky entrance into hospital medicine.Todays Hospitalist.2007;5(1):28–32.
- Accreditation Review Commission for Physician Assistant Education.3rd ed. 2005. Available at: http://www.arc‐pa.org/Standards/standards.html. Accessed September2009.
- 22nd Annual Report on Physician Assistant Education in the U.S., 2005–2006. Available at: http://www.paeaonline.org. Accessed September2009.
- Association of Postgraduate Physician Assistant Programs. Available at: http://www.appap.org. Accessed September2009.
- ,,,,.The core competencies in hospital medicine: development and methodology.J Hosp Med.2006;1:48–56.
Copyright © 2010 Society of Hospital Medicine


Impact of CI Among Hospitalized Elders
In 2001, approximately 12.6 million individuals age 65 and older were discharged from American hospitals with an average length of stay of 5.8 days1 and up to 66% of them suffered from cognitive impairment (CI).220 CI in hospitalized older adults includes a variety of disorders ranging from mild cognitive deficit, delirium, to full‐blown dementia. Dementia is a syndrome of decline in memory plus at least 1 other cognitive domain, such as language, visuospatial, or executive function sufficient to interfere with social or occupational functioning in an alert person.21 Delirium is a disturbance of consciousness with reduced ability to focus, sustain, or shift attention that occurs over a short period of time and tends to fluctuate over the course of the day.22 Mild CI without dementia is defined as the presence of a cognitive deficit in the absence of delirium that does not affect functional performance.23
Hospitalized older adults with CI are vulnerable to hospital complications, including delirium, physical restraints, urinary catheters, and tethers.2, 3, 2435 The management of their medical or surgical illnesses requires avoiding certain medications with anticholinergic activities that might worsen cognition.36 Furthermore, CI may delay diagnostic and therapeutic procedures, demand more time for informed consentrelated issues, and result in difficulty in adherence to medical recommendations.37, 38 The special needs of hospitalized older adults with delirium and dementia has been shown to increase demands on nursing staff, risk of postdischarge institutionalization, length of stay, and health care costs.310, 27, 3948 We wanted to look specifically at CI because it often goes undetected4951 and can have a great impact on the hospital course of elders.
Screening for CI among hospitalized older adults has been considered to have potential benefit in hospital care of older adults.52 Screening may lead to early detection by uncovering subtle symptoms not yet apparent to families or other caregivers who know the patient well but do not notice small declines or changes in day‐to‐day functioning. Early recognition of CI may lead to early treatment and subsequently may delay progression of cognitive decline and improve health outcomes. Screening may enhance physician prescribing practices and reduce exposure to harmful medications among these vulnerable patients. Finally, delirium is an important prognostic indicator, and screening patients could provide invaluable information toward the overall clinical picture. Despite all of this, the current literature does not provide sufficient information to support the use of routine screening on admission.220, 41, 5254 Most of the published studies were conducted among elders who stayed in the hospital for more than 48 hours, missing data on the crucial first 48 hours of the hospital course.220, 41, 5254 These studies did not evaluate the impact of unrecognized CI on the hospital course and the majority of these studies were not conducted in the urban and lower socioeconomic status populations of elders that are the most vulnerable to bad health outcomes.220, 41, 5254 Finally, few studies evaluated the impact of delirium superimposed on CI on the hospital course and mortality of elders.220, 41, 5254
With these details in mind, we wanted to explore the impact of CI recognition among patients age 65 years and older admitted to the medical services of an urban, public hospital in Indianapolis to determine the prevalence and the impact of recognized and unrecognized CI on the hospital course of these elders. Furthermore, we examined the role of delirium superimposed on these hospitalized elders with CI.
Patients and Methods
The study was approved by the Indiana University Purdue University at Indianapolis Institutional Review Board (IRB).
Study Setting and Population
The study was conducted on the inpatient general medicine service of Wishard Memorial Hospital (WMH). WMH is a 450‐bed, university‐affiliated, urban, public hospital that is staffed by Indiana University School of Medicine faculty and house staff. It serves a population of approximately 750,000 in Marion County.
Inclusion and Exclusion Criteria
Patients were enrolled in the study based on the following criteria: (1) at least 65 years of age; (2) hospitalized on a medical ward; (3) able to speak English; and (4) have CI at the time of hospital admission (see below). Patients were excluded if they had previously enrolled in the study, were enrolled in another clinical study at the time of admission, or were aphasic or unresponsive at the time of screening.
Cognitive Screening
CI was determined by the Short Portable Mental Status Questionnaire (SPMSQ),55, 56 chosen for its accuracy56 and the fact that it is entirely verbal in administration. In most cases, patients were followed and reassessed daily. Patients having 2 or more errors, indicating a score of 8 or less on the SPMSQ after adjusting for race and education were considered to have cognitive impairment. The SPMSQ is a brief 10‐item screening test with a sensitivity of 86% and specificity 99.0% for dementia among medical inpatients.56 At the time of cognitive screening, delirium was assessed by using the Confusion Assessment Method (CAM).22 This was also done daily in most cases. The CAM22 is a structured instrument that evaluates the 10 symptoms of delirium specified in the Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III‐R: acute onset, fluctuating course, inattention, disorganized thinking, altered level of consciousness, disorientation, memory impairment, perceptual disturbances, psychomotor agitation or retardation, and sleep/wake disturbance. The CAM score is determined by examining the patient, investigating the chart and interviewing the nurse and/or a family member for: (1) acute and fluctuating changes in mental status, (2) inattention, (3) disorganized or incoherent thinking, and (4) altered level of consciousness. A CAM score is considered to be positive if the patient displays both (1) and (2) with at least one of (3) or (4). The CAM diagnosis of delirium was validated against the clinical judgment of a psychiatrist and found to have a sensitivity of 97% and a specificity of 92%.22 A research assistant (RA) was trained for a period of 9 months by a physician as a rater to interview the patient and administer both the SPMSQ and the CAM at the time of admission and then every weekday. When feasible, the RA administered both the SPMSQ and the CAM within the first few hours of hospitalization, and then followed up with our patients each day. More than 70% of our initial cognitive screening occurred in the first 48 hours of hospital admission, and was repeated on a daily basis. In addition to cognitive assessment, the RA reported the presence or absence of Foley catheterization, physical restraints, and tethers during the cognitive assessment. Agreement was obtained from the general internal medicine group practice physicians both to participate in the study and to request screening for CI as part of the recognized admission standard of care among their hospitalized patients aged 65 years and older. The study coordinator was notified of all admissions for patients aged 65 or older by the hospital intranet e‐mail and paging system. Admission notifications were sent by page and e‐mail on an hourly basis from Monday through Friday, 8:00 AM through 5:00 PM. Those admissions occurring between the hours of 5:00 PM and 8:00 AM were sent during the next normal batch notification. Pages and e‐mails for admissions occurring on Saturday and Sunday were sent on Monday morning at 8:00 AM.
Regenstrief Medical Record System at WMH
The computerized Regenstrief Medical Record System (RMRS) is the primary instrument for processing data and monitoring patient and physician activity for Wishard Health System.57, 58 The RMRS is a modular system, composed of Registration and Scheduling, Laboratory, and Pharmacy database modules. The Registration and Scheduling module is used to make all outpatient appointments for the office practices associated with Wishard Health System. The Laboratory module handles all data for all inpatient and outpatient laboratories. This module also produces all laboratory reports and data used for billing. In addition to laboratory data, this module stores coded results and full‐text interpretations of all imaging studies and special procedures. The Pharmacy module contains information on medication orders captured by the computerized physician order enter (CPOE). The Database module stores all the above data by date in a fully‐coded form. Thus, these data are readily retrievable for individual patients by healthcare providers using online terminals. Data for large numbers of patients are retrievable using a locally developed English‐like language called CARE. Patients can be identified either by a certain restriction list (eg, the list of subjects in a study) or by clinical criteria. The RMRS also maintains a number of other databases including diagnoses, vital signs, results of laboratory tests and diagnostic tests, full‐text discharge summaries, preventive health maneuvers, and detailed information on all inpatient and outpatient charges. It contains death certificate information from the Indiana State Board of Health for all registered patients who die in, or outside of, Indiana. Therefore, the RMRS collects and monitors a broad array of physician and patient activity, practice patterns, utilization, diagnostic test finding, and offers a wonderful array of outcome measures.
Other Data Collections
Patient demographics such as age, gender, race, and education level were determined by the RMRS and by information obtained during the time of cognitive screening. Length of hospital stay and 30‐day posthospitalization mortality were obtained from the RMRS. Comorbidity level was measured by reviewing the RMRS and determining each patient's Charlson comorbidity index total score.59, 60 This score was determined using International Statistical Classification of Diseases and Related Health Problems, 9th edition (ICD‐9) codes gathered from 1 year prior to admission until the patient was discharged from the hospital. Anticholinergic medications were determined by using the Anticholinergic Cognitive Burden Scale,61 an expert‐based practical index. The scale was developed based on a review of all published studies from 1996 to 2007 that measured the anticholinergic activities of a drug and its association with cognitive function in older adults. The list of drugs reviewed was presented to an expert interdisciplinary panel that included geriatricians, geriatric pharmacists, geriatric psychiatrists, general physicians, geriatric nurses, and aging brain researchers. The panel categorized each medication into a possible or definite anticholinergic category based on the severity of its cognitive anticholinergic effects.61 A patient who received at least 1 order of a possible or definite anticholinergic during their hospitalization was considered to be an anticholinergic user. Prior recognition of CI was determined by searching the RMRS for any ICD‐9 code (see Appendix) indicative of dementia, Alzheimer disease, or delirium reported at hospital admission, discharge, or during an 1‐year period prior to hospitalization for every patient enrolled in the study. Those patients with documented ICD‐9 codes were felt recognized as having some form of cognitive impairment. Those who had a positive screen but no prior documentation according to ICD‐9 coding, were said to have unrecognized CI.
Analysis
Descriptive statistics were calculated, including percentages for binary categorical variables, and means and standard deviations for continuous variables. Comparisons between groups were based upon Fisher's Exact Tests for binary categorical variables and t tests for continuous variables. When controlling for covariates such as age, gender, race, Charlson comorbidity index, and SPMSQ at screening, group comparisons were made by using logistic regression for binary categorical variables and multiple regression for continuous variables. Since the distributions of length of stay and Charlson comorbidity index were skewed, all statistical tests comparing them across groups were actually performed on their log‐transformed values.
Results
The Prevalence and Recognition of CI
Table 1 describes the demographic characteristic of our study population, which is a reflection of the public and urban nature of our target hospital. Our study assessed the cognitive status of 997 older adults usually (>70% of the time) within 48 hours of their admission to the medical ward of this urban hospital between July of 2006 and March 2008 (see Table 1) and found that 43% of these elders had evidence of CI as determined by a SPMSQ score of 8 points or less. However, 61% of the 424 cognitively impaired elders were not documented or recognized by the electronic medical record system to have cognitive deficit.
| Variable | n | %/Mean (SD) |
|---|---|---|
| ||
| Age (years), mean (SD) | 997 | 74.8 (7.5) |
| Age 85 (%) | 997 | 12.6 |
| Female (%) | 997 | 67.8 |
| African American (%) | 997 | 59.4 |
| Education (years), mean (SD) | 910 | 10.3 (2.8) |
| Education <12 years (%) | 910 | 59.1 |
| Screened within 48 hours of admission (%) | 997 | 73.2 |
| SPMSQ score at screening, mean (SD) | 997 | 7.7 (2.8) |
| Cognitive impairment based on the SPMSQ score 8 (%) | 997 | 42.5 |
The Impact of Unrecognized CI on the Hospital Course
As expected, hospitalized elders with documented CI were older (mean age 79.1 years vs. 76.1 years; P < 0.001) and had worse cognitive function upon screening than those with unrecognized CI (mean SPMSQ 3.4 points vs. 6.3; P < 0.001). Furthermore, CI recognition was influenced by the elders' race and comorbidity (Table 2); a higher percentage of elders with documented CI were African American (69% vs. 54%; P = 0.003) and had less comorbidity (mean Charlson index 1.9 vs. 2.3; P = 0.03). After adjusting for age, gender, race, comorbidity, and cognitive function at screening, our study found no differences between elders with previously recognized CI and those with unrecognized CI in regard to the length of hospital stay (6.7 days vs. 7.5 days; P = 0.59), 30‐day posthospital mortality (4.8% vs. 6.6%; P > 0.2), home discharge (32% vs. 45%; P > 0.7), hospital readmission (19.2% vs.18.8%; P > 0.6), delirium incidence (27% vs. 21%; P > 0.9), and physical restraints (1.8% vs. 1.5%; P > 0.4). We also found that elders with undocumented CI were not more likely to receive definite anticholinergics (33.2% vs. 32.7%; P > 0.9).
| CI Documented | CI Undocumented | P Value | P Value* | |
|---|---|---|---|---|
| ||||
| n (%) | 165 (39) | 259 (61) | n/a | |
| Age, mean (SD) | 79.1 (7.9) | 76.1 (8.0) | <0.001 | |
| Female (%) | 68.5 | 64.5 | 0.40 | |
| African American (%) | 68.5 | 53.7 | <0.01 | |
| SPMSQ at screen, mean (SD) | 3.4 (2.7) | 6.3 (2.1) | <0.001 | |
| Charlson comorbidity index, mean (SD) | 1.9 (1.9) | 2.3 (2.1) | 0.03 | |
| Length of hospital stay, mean (SD) | 6.7 (5.1) | 7.5 (7.1) | 0.49 | 0.59 |
| Survived at 30 days postdischarge (%) | 95.2 | 93.4 | 0.53 | 0.25 |
| Discharged home (%) | 31.5 | 45.2 | 0.01 | 0.74 |
| Readmission within 30 days after discharge home (%) | 19.2 | 18.8 | 0.99 | 0.66 |
| Incidence of delirium (%) | 26.7 | 20.6 | 0.52 | 0.99 |
| Observed with Foley catheter (%) | 43.6 | 27.4 | <0.001 | 0.61 |
| Observed with physical restraint (%) | 1.8 | 1.5 | 0.99 | 0.31 |
| Observed with tethers (%) | 81.8 | 73.8 | 0.06 | 0.58 |
| With at least 1 Ach (%) | 83.6 | 90.7 | 0.03 | 0.22 |
| Possible Ach (%) | 81.2 | 88.4 | 0.05 | 0.31 |
| Definite Ach (%) | 32.7 | 33.2 | 0.99 | 0.64 |
The Impact of Delirium on the Hospital Course of Elders with CI
Among the 424 hospitalized elders with CI, 163 (38%) had delirium at least once during their hospital course and 24% had delirium on the day of hospital discharge. In comparison to elders who had CI but not delirium during their hospitalization (Table 3), those with at least 1 day of delirium had a higher 30‐day posthospitalization mortality risk (8.6% vs. 4.2%; P = 0.09), stayed in the hospital 3.3 additional days (9.2 days vs. 5.9 days; P < 0.001), were less likely to be discharged home (25% vs. 49%; P < 0.001), were more likely to receive a Foley catheterization (52% vs. 23%; P < 0.001), more likely to be physically restrained (4% vs. 0%; P < 0.01), and more likely to receive tethers during their care (89% vs. 69%; P < 0.001). There was no statistically significant difference between the 2 groups in terms of 30‐day hospital readmission rates or in their use of definite anticholinergics (Table 3).
| Delirium+* | Delirium | P value | |
|---|---|---|---|
| |||
| n (%) | 163 (38) | 261 (62) | n/a |
| Age, mean (SD) | 78.4 (8.5) | 76.5 (7.8) | 0.02 |
| Female (%) | 60.1 | 69.7 | 0.05 |
| African American (%) | 64.4 | 56.3 | 0.10 |
| Charlson comorbidity index, mean (SD) | 1.8 (1.9) | 2.3 (2.1) | 0.01 |
| Length of hospital stay, mean (SD) | 9.2 (7.9) | 5.9 (4.9) | <0.001 |
| Survived at 30‐day postdischarge (%) | 91.4 | 95.8 | 0.09 |
| Discharged home (%) | 24.5 | 49.4 | <0.001 |
| Readmission within 30 days after discharge home (%) | 22.5 | 17.8 | 0.50 |
| Observed with Foley catheter (%) | 51.5 | 22.6 | <0.001 |
| Observed with physical restraint (%) | 4.3 | 0.0 | <0.01 |
| Observed with tethers (%) | 89.0 | 69.4 | <0.001 |
| With at least 1 anticholinergic (%) | 83.4 | 90.8 | 0.03 |
| Possible anticholinergic (%) | 80.4 | 88.9 | 0.02 |
| Definite anticholinergic (%) | 36.8 | 30.7 | 0.20 |
Discussion
Our study found that in an urban, public hospital, acute or preexisting CI affects more than one‐third of hospitalized elders admitted to general medical services. Unfortunately, our hospital system does not currently recognize the majority of these vulnerable patients. Our study also found that delirium affects more than one‐third of hospitalized elders with CI during their hospital course. Delirium complicates hospital care by prolonging length of stay and decreasing the probability of surviving and getting discharged home. It leads to high use of Foley catheterization, physical restraints, and tethers.
The high prevalence of CI with and without delirium in our cohort is within the rates reported previously in the literature. It is estimated that the prevalence of CI in hospitalized older adults ranges from 14% to 66%, depending on the method used to measure cognition, the definition of CI, and the type of hospital ward (surgical, medical, and geriatric units).220 One particular study that used a similar cognitive assessment method reported higher prevalence rates for both CI and delirium.11 The study randomly evaluated a sample of 201 patients age 65 and over who were hospitalized for a medical illness and found that 56% of the cohort suffered from CI and among those with CI, 47% had delirium.11 The difference between this finding and our study is most likely due to our sampling technique; more than 70% of our cognitive screening occurred in the first 48 hours of hospital admission whereas the Australian study, in similar enrollment criteria to all of the published studies in this area, excluded patients who were discharged within 48 hours of admission. We believe, however, that by including the first 48 hours of admission in our design, our study provides a more generalizable reflection of the actual acute care experience.
The impact of delirium on the course of hospital care found in our study supports some of the findings from previous studies conducted in the past 2 decades.5, 6, 11 Despite 2 decades of clinical research, delirium continues to increase mortality, hospital stays, and posthospital institutionalization.
We were surprised to find that patients suffering from delirium continue to receive at least 1 definite anticholinergic medication. Such medications are considered inappropriate among patients with any form of cognitive impairment.36, 62 Although the impact of anticholinergic medications on hospitalized outcomes is less well‐described, their use has been suspected to negatively impact long‐term outcomes of cognitive impairment.61, 63 Our study found no difference in the use of anticholinergic medications between those with CI who experienced delirium and those who did not; however, the total burden of anticholinergic medication was not assessed in a quantitative manner. It is still unknown if certain anticholinergic medications or a cumulative effect of anticholinergic medications may impact cognitive or health‐related outcomes in a vulnerable older population with CI.
Although our study reported for the first time in a systematic way the rate of undocumented CI among hospitalized elders found to have CI on admission, we found no impact of such underrecognition on the length of hospital stay, mortality, discharge location, and delirium occurrence. Although the use of anticholinergic medications is not recommended for patients with any form of CI, our results indicate that a significant number of patients with cognitive impairment continue to receive inappropriate medications. CI recognition in the elderly was not shown to have a statistically significant affect on length of stay, cost, or mortality.
Our study has some limitations. First of all, we did not determine the underlying types of CI such as Alzheimer disease, vascular dementia, mild cognitive impairment, or reversible etiology other than delirium. Such a categorization requires posthospital assessment, which was not included in our study design. Second, our delirium incidence rate and delirium impact on hospital outcomes might be very conservative and may underestimate its true prevalence and correlation due to our data collection methods. Despite the fluctuating nature of delirium, our study was not designed to assess the presence of delirium every shift and tried to assess cognitive function on a daily basis throughout the patient's hospitalization. Therefore, the severity and duration of delirium could not be accurately assessed. Our reported rates of use of Foley catheterization, physical restraints, and tethers are also very conservative and we could not determine the appropriateness of these procedures. Our study was conducted in 1 public hospital in an urban city with a higher percentage of African Americans. Thus, our sample is not a true representative sample. However, studies with significant representation of minority groups are not common in the research literature, especially in CI research; we hope to fulfill some of the gaps in the literature regarding the most vulnerable older American population. Finally, we were limited in our use of ICD‐9 coding to determine if patients had previously been recognized by other providers as having CI. ICD‐9 coding, while useful, is not perfect in identifying all if a patient's medical problems. Use of coding to determine whether a patient had been recognized as impaired also does not allow us to determine when the diagnosis was made.
In conclusion, our study evaluated cognitive impairment in hospitalized elders and found that in our cohort of 997 patients, 43% were cognitively impaired on admission. Of those with CI, 61% were not documented or recognized as impaired. We found no statistically significant difference between those with documented CI and those with undocumented CI in terms of length of stay, mortality, home discharge, readmission rates, incidence of delirium, or potential to receive anticholinergics or restraints. Among those with CI, 38% had delirium. Those with delirium experienced increased length of stay, decreased discharge to home, and increased use of Foley catheters and restraints.
- ,.National hospital discharge survey: annual summary, 1994.Vital Health Stat 13.1997;(128):i–v;1–50.
- .The dilemma of delirium: clinical and research controversies regarding diagnosis and evaluation of delirium in hospitalized elderly medical patients.Am J Med.1994;97:278–288.
- ,,.Dementia in medical wards.J Clin Epidemiol.1988;41:123–126.
- ,,,,,.Mental and behavioral disturbances in dementia: findings from the Cache County Study on Memory in Aging.Am J Psychiatry.2000;157:708–714.
- ,,, et al.Randomized, placebo‐controlled, double‐blind clinical trial of sertraline in the treatment of depression complicating Alzheimer's disease: initial results from the Depression in Alzheimer's Disease study.Am J Psychiatry.2000;157:1686–1689.
- ,,.Dementia in elderly persons in a general hospital.Am J Psychiatry.2000;157:704–707.
- ,,,,.A prospective study of the impact of psychiatric comorbidity on length of hospital stays of elderly medical‐surgical inpatients.Psychosomatics.1998;39:273–280.
- ,.Psychiatric comorbidity and length of stay in the general hospital. A critical review of outcome studies.Psychosomatics.1994;35:233–252.
- ,,,.Dementia among medical inpatients. Evaluation of 2000 consecutive admissions.Arch Intern Med.1986;146:1923–1926.
- ,,,.The consequences of non‐cognitive symptoms of dementia in medical hospital departments.Int J Psychiatry Med.2003;33:257–271.
- ,,.Cognitive impairment in medical inpatients. I: Screening for dementia—is history better than mental state?Age Ageing.1997;26:31–35.
- ,,, et al.Acute confusional states in elderly patients treated for femoral neck fracture.J Am Geriatr Soc.1988;36:525–530.
- ,.A prospective study of elderly general surgical patients: II. Post‐operative complications.Age Ageing.1989;18:316–326.
- ,,,.Dementia and depression in elderly medical inpatients.Int Psychogeriatr.2000;12:67–75.
- ,,.[Psychiatric disorders in elderly general hospital patients: incidence and long‐term prognosis].Nervenarzt.1993;64:53–61. [German]
- ,.Delirium and dementia in acute medical admissions of elderly patients in Iceland.Acta Psychiatr Scand.1993;87:123–127.
- ,,,,,.[Psychiatric morbidity in elderly patients admitted to a general hospital. A day‐prevalence study].Med Clin (Barc).1991;97:206–210. [Spanish]
- ,,,,,.Detection of psychiatric disorders in elderly medical inpatients.Age Ageing.1994;23:307–311.
- ,,,.Cognitive impairment, emotional disorder and length of stay of elderly patients in a district general hospital.Br J Med Psychol.1987;60(Pt 2):133–139.
- ,,.An investigation of the components of best nursing practice in the care of acutely ill hospitalized older patients with coincidental dementia: a multi‐method design.J Adv Nurs1999;30:1127–1136.
- American Psychiatric Association.Diagnostic and Statistical Manual of Mental Disorders.4th ed.Washington, DC:American Psychiatric Association;1994.
- ,,,,,.Clarifying confusion: the confusion assessment method. A new method for detection of delirium.Ann Intern Med.1990;113:941–948.
- ,,, et al.Prevalence of cognitive impairment: data from the Indianapolis Study of Health and Aging.Neurology.2001;57:1655–1662.
- ,,.Delirium: a symptom of how hospital care is failing older persons and a window to improve quality of hospital care.Am J Med.1999;106:565–573.
- ,,, et al.A multicomponent intervention to prevent delirium in hospitalized older patients.N Engl J Med.1999;340:669–676.
- ,,,.Iatrogenic causes of falls in hospitalised elderly patients: a case‐control study.Postgrad Med J.2002;78:487–489.
- ,,.A prospective study of delirium in hospitalized elderly.JAMA.1990;263:1097–1101.
- ,.The prognostic significance of delirium in older hospital patients.J Am Geriatr Soc.1997;45:174–178.
- ,.Prognosis of delirium in elderly hospital patients.CMAJ.1993;149:41–46.
- ,,,,.The detection of psychiatric morbidity and its effects on outcome in acute elderly medical admissions.Int J Ger Psych1991;6:861–866.
- ,,.Adverse consequences of hospitalization in the elderly.Soc Sci Med.1982;16:1033–1038.
- ,,, et al.Incidence of adverse events and negligence in hospitalized patients. Results of the Harvard Medical Practice Study I.N Engl J Med.1991;324:370–376.
- ,,,.Delirium in elderly patients: an overview of the state of the science.J Gerontol Nurs.2001;27:12–20.
- ,,,,.A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics.Ann Intern Med.1993;119:474–481.
- ,.Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability.JAMA.1996;275:852–857.
- ,,,,,.Updating the Beers criteria for potentially inappropriate medication use in older adults: results of a US consensus panel of experts.Arch Intern Med.2003;163:2716–2724.
- ,,, et al.A clinical prediction rule for delirium after elective noncardiac surgery.JAMA.1994;271:134–139.
- ,,, et al.The relationship of postoperative delirium with psychoactive medications.JAMA.1994;272:1518–1522.
- ,,, et al.How do delirium and dementia increase length of stay of elderly general medical inpatients?Psychosomatics.2004;45:235–242.
- ,,,,,.The relationship between a dementia diagnosis, chronic illness, Medicare expenditures, and hospital use.J Am Geriatr Soc.2004;52:187–194.
- ,,, et al.Risk factors for delirium in hospitalized elderly.JAMA.1992;267:827–831.
- ,,,,.Psychological comorbidity and length of stay in the general hospital.Am J Psychiatry.1991;148:324–329.
- ,,, et al.Delirium. The occurrence and persistence of symptoms among elderly hospitalized patients.Arch Intern Med.1992;152:334–340.
- ,,.What happens to medical patients with psychiatric disorder?J Psychosom Res1988;32:541–549.
- ,,,,.[Dementia syndromes and length of stay of elderly patients in internal medicine].Ann Med Interne (Paris).1997;148:424–426. [French]
- ,,, et al.[Hospital discharge planning and length of hospital stay in elderly patients admitted through the emergency department].Rev Epidemiol Sante Publique.1995;43:337–347. [French]
- ,,,,.The effect of dementia on acute care in a geriatric medical unit.Int Psychogeriatr.1992;4:231–239.
- ,,,.Cognitive impairment. Can it predict the course of hospitalized patients?J Am Geriatr Soc.1986;34:579–585.
- ,,,,;US Preventive Services Task Force. Screening for dementia in primary care: a summary of the evidence for the U.S. Preventive Services Task Force.Ann Intern Med.2003;138(11):927–937.
- ,,,.Cognitive impairment in the elderly medically ill: how often is it missed?Int J Geriatr Psychiatry.1993;8:929–937.
- .Recognition of cognitive impairment in elderly medical in‐patients.J R Soc Med.1995;88:183–184.
- ,.Quality indicators for dementia in vulnerable community‐dwelling and hospitalized elders.Ann Intern Med.2001;135:668–676.
- ,,,.Reducing delirium after hip fracture: a randomized trial.J Am Geriatr Soc.2001;49:516–522.
- ,.Prevalence of psychotic symptoms in delirium.Psychosomatics.2000;41:519–522.
- .A short portable mental status questionnaire for the assessment of organic brain deficit in elderly patients.J Am Geriatr Soc.1975;23:433–441.
- ,,,.Short Portable Mental Status Questionnaire as a screening test for dementia and delirium among the elderly.J Am Geriatr Soc.1987;35:412–416.
- ,,, et al.The Regenstrief Medical Record System: a quarter century experience.Int J Med Inform.1999;54:225–253.
- ,,,,,.Factors determining the decision to institutionalize dementing individuals: a prospective study.Gerontologist.1993;33:714–720.
- ,,,,,.Resuscitation: how do we decide? A prospective study of physicians' preferences and the clinical course of hospitalized patients.JAMA.1986;255:1316–1322.
- ,,,,,.Assessing illness severity: does clinical judgment work?J Chronic Dis.1986;39:439–452.
- ,,,,.Impact of anticholinergics on the aging brain: a review and practical application.Aging Health.2008;4(3):311–320.
- ,.Delirium in hospitalized older adults. In: Ham R, Sloane P, Warshaw G, eds.Primary Care Geriatrics: A Case‐Based Approach.5th ed.Philadelphia, PA:Mosby Elsevier;2007:210–218.
- ,,, et al.The association between cognition and histamine‐2 receptor antagonists in African Americans.J Am Geriatr Soc.2007;55(8):1248–1253.
In 2001, approximately 12.6 million individuals age 65 and older were discharged from American hospitals with an average length of stay of 5.8 days1 and up to 66% of them suffered from cognitive impairment (CI).220 CI in hospitalized older adults includes a variety of disorders ranging from mild cognitive deficit, delirium, to full‐blown dementia. Dementia is a syndrome of decline in memory plus at least 1 other cognitive domain, such as language, visuospatial, or executive function sufficient to interfere with social or occupational functioning in an alert person.21 Delirium is a disturbance of consciousness with reduced ability to focus, sustain, or shift attention that occurs over a short period of time and tends to fluctuate over the course of the day.22 Mild CI without dementia is defined as the presence of a cognitive deficit in the absence of delirium that does not affect functional performance.23
Hospitalized older adults with CI are vulnerable to hospital complications, including delirium, physical restraints, urinary catheters, and tethers.2, 3, 2435 The management of their medical or surgical illnesses requires avoiding certain medications with anticholinergic activities that might worsen cognition.36 Furthermore, CI may delay diagnostic and therapeutic procedures, demand more time for informed consentrelated issues, and result in difficulty in adherence to medical recommendations.37, 38 The special needs of hospitalized older adults with delirium and dementia has been shown to increase demands on nursing staff, risk of postdischarge institutionalization, length of stay, and health care costs.310, 27, 3948 We wanted to look specifically at CI because it often goes undetected4951 and can have a great impact on the hospital course of elders.
Screening for CI among hospitalized older adults has been considered to have potential benefit in hospital care of older adults.52 Screening may lead to early detection by uncovering subtle symptoms not yet apparent to families or other caregivers who know the patient well but do not notice small declines or changes in day‐to‐day functioning. Early recognition of CI may lead to early treatment and subsequently may delay progression of cognitive decline and improve health outcomes. Screening may enhance physician prescribing practices and reduce exposure to harmful medications among these vulnerable patients. Finally, delirium is an important prognostic indicator, and screening patients could provide invaluable information toward the overall clinical picture. Despite all of this, the current literature does not provide sufficient information to support the use of routine screening on admission.220, 41, 5254 Most of the published studies were conducted among elders who stayed in the hospital for more than 48 hours, missing data on the crucial first 48 hours of the hospital course.220, 41, 5254 These studies did not evaluate the impact of unrecognized CI on the hospital course and the majority of these studies were not conducted in the urban and lower socioeconomic status populations of elders that are the most vulnerable to bad health outcomes.220, 41, 5254 Finally, few studies evaluated the impact of delirium superimposed on CI on the hospital course and mortality of elders.220, 41, 5254
With these details in mind, we wanted to explore the impact of CI recognition among patients age 65 years and older admitted to the medical services of an urban, public hospital in Indianapolis to determine the prevalence and the impact of recognized and unrecognized CI on the hospital course of these elders. Furthermore, we examined the role of delirium superimposed on these hospitalized elders with CI.
Patients and Methods
The study was approved by the Indiana University Purdue University at Indianapolis Institutional Review Board (IRB).
Study Setting and Population
The study was conducted on the inpatient general medicine service of Wishard Memorial Hospital (WMH). WMH is a 450‐bed, university‐affiliated, urban, public hospital that is staffed by Indiana University School of Medicine faculty and house staff. It serves a population of approximately 750,000 in Marion County.
Inclusion and Exclusion Criteria
Patients were enrolled in the study based on the following criteria: (1) at least 65 years of age; (2) hospitalized on a medical ward; (3) able to speak English; and (4) have CI at the time of hospital admission (see below). Patients were excluded if they had previously enrolled in the study, were enrolled in another clinical study at the time of admission, or were aphasic or unresponsive at the time of screening.
Cognitive Screening
CI was determined by the Short Portable Mental Status Questionnaire (SPMSQ),55, 56 chosen for its accuracy56 and the fact that it is entirely verbal in administration. In most cases, patients were followed and reassessed daily. Patients having 2 or more errors, indicating a score of 8 or less on the SPMSQ after adjusting for race and education were considered to have cognitive impairment. The SPMSQ is a brief 10‐item screening test with a sensitivity of 86% and specificity 99.0% for dementia among medical inpatients.56 At the time of cognitive screening, delirium was assessed by using the Confusion Assessment Method (CAM).22 This was also done daily in most cases. The CAM22 is a structured instrument that evaluates the 10 symptoms of delirium specified in the Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III‐R: acute onset, fluctuating course, inattention, disorganized thinking, altered level of consciousness, disorientation, memory impairment, perceptual disturbances, psychomotor agitation or retardation, and sleep/wake disturbance. The CAM score is determined by examining the patient, investigating the chart and interviewing the nurse and/or a family member for: (1) acute and fluctuating changes in mental status, (2) inattention, (3) disorganized or incoherent thinking, and (4) altered level of consciousness. A CAM score is considered to be positive if the patient displays both (1) and (2) with at least one of (3) or (4). The CAM diagnosis of delirium was validated against the clinical judgment of a psychiatrist and found to have a sensitivity of 97% and a specificity of 92%.22 A research assistant (RA) was trained for a period of 9 months by a physician as a rater to interview the patient and administer both the SPMSQ and the CAM at the time of admission and then every weekday. When feasible, the RA administered both the SPMSQ and the CAM within the first few hours of hospitalization, and then followed up with our patients each day. More than 70% of our initial cognitive screening occurred in the first 48 hours of hospital admission, and was repeated on a daily basis. In addition to cognitive assessment, the RA reported the presence or absence of Foley catheterization, physical restraints, and tethers during the cognitive assessment. Agreement was obtained from the general internal medicine group practice physicians both to participate in the study and to request screening for CI as part of the recognized admission standard of care among their hospitalized patients aged 65 years and older. The study coordinator was notified of all admissions for patients aged 65 or older by the hospital intranet e‐mail and paging system. Admission notifications were sent by page and e‐mail on an hourly basis from Monday through Friday, 8:00 AM through 5:00 PM. Those admissions occurring between the hours of 5:00 PM and 8:00 AM were sent during the next normal batch notification. Pages and e‐mails for admissions occurring on Saturday and Sunday were sent on Monday morning at 8:00 AM.
Regenstrief Medical Record System at WMH
The computerized Regenstrief Medical Record System (RMRS) is the primary instrument for processing data and monitoring patient and physician activity for Wishard Health System.57, 58 The RMRS is a modular system, composed of Registration and Scheduling, Laboratory, and Pharmacy database modules. The Registration and Scheduling module is used to make all outpatient appointments for the office practices associated with Wishard Health System. The Laboratory module handles all data for all inpatient and outpatient laboratories. This module also produces all laboratory reports and data used for billing. In addition to laboratory data, this module stores coded results and full‐text interpretations of all imaging studies and special procedures. The Pharmacy module contains information on medication orders captured by the computerized physician order enter (CPOE). The Database module stores all the above data by date in a fully‐coded form. Thus, these data are readily retrievable for individual patients by healthcare providers using online terminals. Data for large numbers of patients are retrievable using a locally developed English‐like language called CARE. Patients can be identified either by a certain restriction list (eg, the list of subjects in a study) or by clinical criteria. The RMRS also maintains a number of other databases including diagnoses, vital signs, results of laboratory tests and diagnostic tests, full‐text discharge summaries, preventive health maneuvers, and detailed information on all inpatient and outpatient charges. It contains death certificate information from the Indiana State Board of Health for all registered patients who die in, or outside of, Indiana. Therefore, the RMRS collects and monitors a broad array of physician and patient activity, practice patterns, utilization, diagnostic test finding, and offers a wonderful array of outcome measures.
Other Data Collections
Patient demographics such as age, gender, race, and education level were determined by the RMRS and by information obtained during the time of cognitive screening. Length of hospital stay and 30‐day posthospitalization mortality were obtained from the RMRS. Comorbidity level was measured by reviewing the RMRS and determining each patient's Charlson comorbidity index total score.59, 60 This score was determined using International Statistical Classification of Diseases and Related Health Problems, 9th edition (ICD‐9) codes gathered from 1 year prior to admission until the patient was discharged from the hospital. Anticholinergic medications were determined by using the Anticholinergic Cognitive Burden Scale,61 an expert‐based practical index. The scale was developed based on a review of all published studies from 1996 to 2007 that measured the anticholinergic activities of a drug and its association with cognitive function in older adults. The list of drugs reviewed was presented to an expert interdisciplinary panel that included geriatricians, geriatric pharmacists, geriatric psychiatrists, general physicians, geriatric nurses, and aging brain researchers. The panel categorized each medication into a possible or definite anticholinergic category based on the severity of its cognitive anticholinergic effects.61 A patient who received at least 1 order of a possible or definite anticholinergic during their hospitalization was considered to be an anticholinergic user. Prior recognition of CI was determined by searching the RMRS for any ICD‐9 code (see Appendix) indicative of dementia, Alzheimer disease, or delirium reported at hospital admission, discharge, or during an 1‐year period prior to hospitalization for every patient enrolled in the study. Those patients with documented ICD‐9 codes were felt recognized as having some form of cognitive impairment. Those who had a positive screen but no prior documentation according to ICD‐9 coding, were said to have unrecognized CI.
Analysis
Descriptive statistics were calculated, including percentages for binary categorical variables, and means and standard deviations for continuous variables. Comparisons between groups were based upon Fisher's Exact Tests for binary categorical variables and t tests for continuous variables. When controlling for covariates such as age, gender, race, Charlson comorbidity index, and SPMSQ at screening, group comparisons were made by using logistic regression for binary categorical variables and multiple regression for continuous variables. Since the distributions of length of stay and Charlson comorbidity index were skewed, all statistical tests comparing them across groups were actually performed on their log‐transformed values.
Results
The Prevalence and Recognition of CI
Table 1 describes the demographic characteristic of our study population, which is a reflection of the public and urban nature of our target hospital. Our study assessed the cognitive status of 997 older adults usually (>70% of the time) within 48 hours of their admission to the medical ward of this urban hospital between July of 2006 and March 2008 (see Table 1) and found that 43% of these elders had evidence of CI as determined by a SPMSQ score of 8 points or less. However, 61% of the 424 cognitively impaired elders were not documented or recognized by the electronic medical record system to have cognitive deficit.
| Variable | n | %/Mean (SD) |
|---|---|---|
| ||
| Age (years), mean (SD) | 997 | 74.8 (7.5) |
| Age 85 (%) | 997 | 12.6 |
| Female (%) | 997 | 67.8 |
| African American (%) | 997 | 59.4 |
| Education (years), mean (SD) | 910 | 10.3 (2.8) |
| Education <12 years (%) | 910 | 59.1 |
| Screened within 48 hours of admission (%) | 997 | 73.2 |
| SPMSQ score at screening, mean (SD) | 997 | 7.7 (2.8) |
| Cognitive impairment based on the SPMSQ score 8 (%) | 997 | 42.5 |
The Impact of Unrecognized CI on the Hospital Course
As expected, hospitalized elders with documented CI were older (mean age 79.1 years vs. 76.1 years; P < 0.001) and had worse cognitive function upon screening than those with unrecognized CI (mean SPMSQ 3.4 points vs. 6.3; P < 0.001). Furthermore, CI recognition was influenced by the elders' race and comorbidity (Table 2); a higher percentage of elders with documented CI were African American (69% vs. 54%; P = 0.003) and had less comorbidity (mean Charlson index 1.9 vs. 2.3; P = 0.03). After adjusting for age, gender, race, comorbidity, and cognitive function at screening, our study found no differences between elders with previously recognized CI and those with unrecognized CI in regard to the length of hospital stay (6.7 days vs. 7.5 days; P = 0.59), 30‐day posthospital mortality (4.8% vs. 6.6%; P > 0.2), home discharge (32% vs. 45%; P > 0.7), hospital readmission (19.2% vs.18.8%; P > 0.6), delirium incidence (27% vs. 21%; P > 0.9), and physical restraints (1.8% vs. 1.5%; P > 0.4). We also found that elders with undocumented CI were not more likely to receive definite anticholinergics (33.2% vs. 32.7%; P > 0.9).
| CI Documented | CI Undocumented | P Value | P Value* | |
|---|---|---|---|---|
| ||||
| n (%) | 165 (39) | 259 (61) | n/a | |
| Age, mean (SD) | 79.1 (7.9) | 76.1 (8.0) | <0.001 | |
| Female (%) | 68.5 | 64.5 | 0.40 | |
| African American (%) | 68.5 | 53.7 | <0.01 | |
| SPMSQ at screen, mean (SD) | 3.4 (2.7) | 6.3 (2.1) | <0.001 | |
| Charlson comorbidity index, mean (SD) | 1.9 (1.9) | 2.3 (2.1) | 0.03 | |
| Length of hospital stay, mean (SD) | 6.7 (5.1) | 7.5 (7.1) | 0.49 | 0.59 |
| Survived at 30 days postdischarge (%) | 95.2 | 93.4 | 0.53 | 0.25 |
| Discharged home (%) | 31.5 | 45.2 | 0.01 | 0.74 |
| Readmission within 30 days after discharge home (%) | 19.2 | 18.8 | 0.99 | 0.66 |
| Incidence of delirium (%) | 26.7 | 20.6 | 0.52 | 0.99 |
| Observed with Foley catheter (%) | 43.6 | 27.4 | <0.001 | 0.61 |
| Observed with physical restraint (%) | 1.8 | 1.5 | 0.99 | 0.31 |
| Observed with tethers (%) | 81.8 | 73.8 | 0.06 | 0.58 |
| With at least 1 Ach (%) | 83.6 | 90.7 | 0.03 | 0.22 |
| Possible Ach (%) | 81.2 | 88.4 | 0.05 | 0.31 |
| Definite Ach (%) | 32.7 | 33.2 | 0.99 | 0.64 |
The Impact of Delirium on the Hospital Course of Elders with CI
Among the 424 hospitalized elders with CI, 163 (38%) had delirium at least once during their hospital course and 24% had delirium on the day of hospital discharge. In comparison to elders who had CI but not delirium during their hospitalization (Table 3), those with at least 1 day of delirium had a higher 30‐day posthospitalization mortality risk (8.6% vs. 4.2%; P = 0.09), stayed in the hospital 3.3 additional days (9.2 days vs. 5.9 days; P < 0.001), were less likely to be discharged home (25% vs. 49%; P < 0.001), were more likely to receive a Foley catheterization (52% vs. 23%; P < 0.001), more likely to be physically restrained (4% vs. 0%; P < 0.01), and more likely to receive tethers during their care (89% vs. 69%; P < 0.001). There was no statistically significant difference between the 2 groups in terms of 30‐day hospital readmission rates or in their use of definite anticholinergics (Table 3).
| Delirium+* | Delirium | P value | |
|---|---|---|---|
| |||
| n (%) | 163 (38) | 261 (62) | n/a |
| Age, mean (SD) | 78.4 (8.5) | 76.5 (7.8) | 0.02 |
| Female (%) | 60.1 | 69.7 | 0.05 |
| African American (%) | 64.4 | 56.3 | 0.10 |
| Charlson comorbidity index, mean (SD) | 1.8 (1.9) | 2.3 (2.1) | 0.01 |
| Length of hospital stay, mean (SD) | 9.2 (7.9) | 5.9 (4.9) | <0.001 |
| Survived at 30‐day postdischarge (%) | 91.4 | 95.8 | 0.09 |
| Discharged home (%) | 24.5 | 49.4 | <0.001 |
| Readmission within 30 days after discharge home (%) | 22.5 | 17.8 | 0.50 |
| Observed with Foley catheter (%) | 51.5 | 22.6 | <0.001 |
| Observed with physical restraint (%) | 4.3 | 0.0 | <0.01 |
| Observed with tethers (%) | 89.0 | 69.4 | <0.001 |
| With at least 1 anticholinergic (%) | 83.4 | 90.8 | 0.03 |
| Possible anticholinergic (%) | 80.4 | 88.9 | 0.02 |
| Definite anticholinergic (%) | 36.8 | 30.7 | 0.20 |
Discussion
Our study found that in an urban, public hospital, acute or preexisting CI affects more than one‐third of hospitalized elders admitted to general medical services. Unfortunately, our hospital system does not currently recognize the majority of these vulnerable patients. Our study also found that delirium affects more than one‐third of hospitalized elders with CI during their hospital course. Delirium complicates hospital care by prolonging length of stay and decreasing the probability of surviving and getting discharged home. It leads to high use of Foley catheterization, physical restraints, and tethers.
The high prevalence of CI with and without delirium in our cohort is within the rates reported previously in the literature. It is estimated that the prevalence of CI in hospitalized older adults ranges from 14% to 66%, depending on the method used to measure cognition, the definition of CI, and the type of hospital ward (surgical, medical, and geriatric units).220 One particular study that used a similar cognitive assessment method reported higher prevalence rates for both CI and delirium.11 The study randomly evaluated a sample of 201 patients age 65 and over who were hospitalized for a medical illness and found that 56% of the cohort suffered from CI and among those with CI, 47% had delirium.11 The difference between this finding and our study is most likely due to our sampling technique; more than 70% of our cognitive screening occurred in the first 48 hours of hospital admission whereas the Australian study, in similar enrollment criteria to all of the published studies in this area, excluded patients who were discharged within 48 hours of admission. We believe, however, that by including the first 48 hours of admission in our design, our study provides a more generalizable reflection of the actual acute care experience.
The impact of delirium on the course of hospital care found in our study supports some of the findings from previous studies conducted in the past 2 decades.5, 6, 11 Despite 2 decades of clinical research, delirium continues to increase mortality, hospital stays, and posthospital institutionalization.
We were surprised to find that patients suffering from delirium continue to receive at least 1 definite anticholinergic medication. Such medications are considered inappropriate among patients with any form of cognitive impairment.36, 62 Although the impact of anticholinergic medications on hospitalized outcomes is less well‐described, their use has been suspected to negatively impact long‐term outcomes of cognitive impairment.61, 63 Our study found no difference in the use of anticholinergic medications between those with CI who experienced delirium and those who did not; however, the total burden of anticholinergic medication was not assessed in a quantitative manner. It is still unknown if certain anticholinergic medications or a cumulative effect of anticholinergic medications may impact cognitive or health‐related outcomes in a vulnerable older population with CI.
Although our study reported for the first time in a systematic way the rate of undocumented CI among hospitalized elders found to have CI on admission, we found no impact of such underrecognition on the length of hospital stay, mortality, discharge location, and delirium occurrence. Although the use of anticholinergic medications is not recommended for patients with any form of CI, our results indicate that a significant number of patients with cognitive impairment continue to receive inappropriate medications. CI recognition in the elderly was not shown to have a statistically significant affect on length of stay, cost, or mortality.
Our study has some limitations. First of all, we did not determine the underlying types of CI such as Alzheimer disease, vascular dementia, mild cognitive impairment, or reversible etiology other than delirium. Such a categorization requires posthospital assessment, which was not included in our study design. Second, our delirium incidence rate and delirium impact on hospital outcomes might be very conservative and may underestimate its true prevalence and correlation due to our data collection methods. Despite the fluctuating nature of delirium, our study was not designed to assess the presence of delirium every shift and tried to assess cognitive function on a daily basis throughout the patient's hospitalization. Therefore, the severity and duration of delirium could not be accurately assessed. Our reported rates of use of Foley catheterization, physical restraints, and tethers are also very conservative and we could not determine the appropriateness of these procedures. Our study was conducted in 1 public hospital in an urban city with a higher percentage of African Americans. Thus, our sample is not a true representative sample. However, studies with significant representation of minority groups are not common in the research literature, especially in CI research; we hope to fulfill some of the gaps in the literature regarding the most vulnerable older American population. Finally, we were limited in our use of ICD‐9 coding to determine if patients had previously been recognized by other providers as having CI. ICD‐9 coding, while useful, is not perfect in identifying all if a patient's medical problems. Use of coding to determine whether a patient had been recognized as impaired also does not allow us to determine when the diagnosis was made.
In conclusion, our study evaluated cognitive impairment in hospitalized elders and found that in our cohort of 997 patients, 43% were cognitively impaired on admission. Of those with CI, 61% were not documented or recognized as impaired. We found no statistically significant difference between those with documented CI and those with undocumented CI in terms of length of stay, mortality, home discharge, readmission rates, incidence of delirium, or potential to receive anticholinergics or restraints. Among those with CI, 38% had delirium. Those with delirium experienced increased length of stay, decreased discharge to home, and increased use of Foley catheters and restraints.
In 2001, approximately 12.6 million individuals age 65 and older were discharged from American hospitals with an average length of stay of 5.8 days1 and up to 66% of them suffered from cognitive impairment (CI).220 CI in hospitalized older adults includes a variety of disorders ranging from mild cognitive deficit, delirium, to full‐blown dementia. Dementia is a syndrome of decline in memory plus at least 1 other cognitive domain, such as language, visuospatial, or executive function sufficient to interfere with social or occupational functioning in an alert person.21 Delirium is a disturbance of consciousness with reduced ability to focus, sustain, or shift attention that occurs over a short period of time and tends to fluctuate over the course of the day.22 Mild CI without dementia is defined as the presence of a cognitive deficit in the absence of delirium that does not affect functional performance.23
Hospitalized older adults with CI are vulnerable to hospital complications, including delirium, physical restraints, urinary catheters, and tethers.2, 3, 2435 The management of their medical or surgical illnesses requires avoiding certain medications with anticholinergic activities that might worsen cognition.36 Furthermore, CI may delay diagnostic and therapeutic procedures, demand more time for informed consentrelated issues, and result in difficulty in adherence to medical recommendations.37, 38 The special needs of hospitalized older adults with delirium and dementia has been shown to increase demands on nursing staff, risk of postdischarge institutionalization, length of stay, and health care costs.310, 27, 3948 We wanted to look specifically at CI because it often goes undetected4951 and can have a great impact on the hospital course of elders.
Screening for CI among hospitalized older adults has been considered to have potential benefit in hospital care of older adults.52 Screening may lead to early detection by uncovering subtle symptoms not yet apparent to families or other caregivers who know the patient well but do not notice small declines or changes in day‐to‐day functioning. Early recognition of CI may lead to early treatment and subsequently may delay progression of cognitive decline and improve health outcomes. Screening may enhance physician prescribing practices and reduce exposure to harmful medications among these vulnerable patients. Finally, delirium is an important prognostic indicator, and screening patients could provide invaluable information toward the overall clinical picture. Despite all of this, the current literature does not provide sufficient information to support the use of routine screening on admission.220, 41, 5254 Most of the published studies were conducted among elders who stayed in the hospital for more than 48 hours, missing data on the crucial first 48 hours of the hospital course.220, 41, 5254 These studies did not evaluate the impact of unrecognized CI on the hospital course and the majority of these studies were not conducted in the urban and lower socioeconomic status populations of elders that are the most vulnerable to bad health outcomes.220, 41, 5254 Finally, few studies evaluated the impact of delirium superimposed on CI on the hospital course and mortality of elders.220, 41, 5254
With these details in mind, we wanted to explore the impact of CI recognition among patients age 65 years and older admitted to the medical services of an urban, public hospital in Indianapolis to determine the prevalence and the impact of recognized and unrecognized CI on the hospital course of these elders. Furthermore, we examined the role of delirium superimposed on these hospitalized elders with CI.
Patients and Methods
The study was approved by the Indiana University Purdue University at Indianapolis Institutional Review Board (IRB).
Study Setting and Population
The study was conducted on the inpatient general medicine service of Wishard Memorial Hospital (WMH). WMH is a 450‐bed, university‐affiliated, urban, public hospital that is staffed by Indiana University School of Medicine faculty and house staff. It serves a population of approximately 750,000 in Marion County.
Inclusion and Exclusion Criteria
Patients were enrolled in the study based on the following criteria: (1) at least 65 years of age; (2) hospitalized on a medical ward; (3) able to speak English; and (4) have CI at the time of hospital admission (see below). Patients were excluded if they had previously enrolled in the study, were enrolled in another clinical study at the time of admission, or were aphasic or unresponsive at the time of screening.
Cognitive Screening
CI was determined by the Short Portable Mental Status Questionnaire (SPMSQ),55, 56 chosen for its accuracy56 and the fact that it is entirely verbal in administration. In most cases, patients were followed and reassessed daily. Patients having 2 or more errors, indicating a score of 8 or less on the SPMSQ after adjusting for race and education were considered to have cognitive impairment. The SPMSQ is a brief 10‐item screening test with a sensitivity of 86% and specificity 99.0% for dementia among medical inpatients.56 At the time of cognitive screening, delirium was assessed by using the Confusion Assessment Method (CAM).22 This was also done daily in most cases. The CAM22 is a structured instrument that evaluates the 10 symptoms of delirium specified in the Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III‐R: acute onset, fluctuating course, inattention, disorganized thinking, altered level of consciousness, disorientation, memory impairment, perceptual disturbances, psychomotor agitation or retardation, and sleep/wake disturbance. The CAM score is determined by examining the patient, investigating the chart and interviewing the nurse and/or a family member for: (1) acute and fluctuating changes in mental status, (2) inattention, (3) disorganized or incoherent thinking, and (4) altered level of consciousness. A CAM score is considered to be positive if the patient displays both (1) and (2) with at least one of (3) or (4). The CAM diagnosis of delirium was validated against the clinical judgment of a psychiatrist and found to have a sensitivity of 97% and a specificity of 92%.22 A research assistant (RA) was trained for a period of 9 months by a physician as a rater to interview the patient and administer both the SPMSQ and the CAM at the time of admission and then every weekday. When feasible, the RA administered both the SPMSQ and the CAM within the first few hours of hospitalization, and then followed up with our patients each day. More than 70% of our initial cognitive screening occurred in the first 48 hours of hospital admission, and was repeated on a daily basis. In addition to cognitive assessment, the RA reported the presence or absence of Foley catheterization, physical restraints, and tethers during the cognitive assessment. Agreement was obtained from the general internal medicine group practice physicians both to participate in the study and to request screening for CI as part of the recognized admission standard of care among their hospitalized patients aged 65 years and older. The study coordinator was notified of all admissions for patients aged 65 or older by the hospital intranet e‐mail and paging system. Admission notifications were sent by page and e‐mail on an hourly basis from Monday through Friday, 8:00 AM through 5:00 PM. Those admissions occurring between the hours of 5:00 PM and 8:00 AM were sent during the next normal batch notification. Pages and e‐mails for admissions occurring on Saturday and Sunday were sent on Monday morning at 8:00 AM.
Regenstrief Medical Record System at WMH
The computerized Regenstrief Medical Record System (RMRS) is the primary instrument for processing data and monitoring patient and physician activity for Wishard Health System.57, 58 The RMRS is a modular system, composed of Registration and Scheduling, Laboratory, and Pharmacy database modules. The Registration and Scheduling module is used to make all outpatient appointments for the office practices associated with Wishard Health System. The Laboratory module handles all data for all inpatient and outpatient laboratories. This module also produces all laboratory reports and data used for billing. In addition to laboratory data, this module stores coded results and full‐text interpretations of all imaging studies and special procedures. The Pharmacy module contains information on medication orders captured by the computerized physician order enter (CPOE). The Database module stores all the above data by date in a fully‐coded form. Thus, these data are readily retrievable for individual patients by healthcare providers using online terminals. Data for large numbers of patients are retrievable using a locally developed English‐like language called CARE. Patients can be identified either by a certain restriction list (eg, the list of subjects in a study) or by clinical criteria. The RMRS also maintains a number of other databases including diagnoses, vital signs, results of laboratory tests and diagnostic tests, full‐text discharge summaries, preventive health maneuvers, and detailed information on all inpatient and outpatient charges. It contains death certificate information from the Indiana State Board of Health for all registered patients who die in, or outside of, Indiana. Therefore, the RMRS collects and monitors a broad array of physician and patient activity, practice patterns, utilization, diagnostic test finding, and offers a wonderful array of outcome measures.
Other Data Collections
Patient demographics such as age, gender, race, and education level were determined by the RMRS and by information obtained during the time of cognitive screening. Length of hospital stay and 30‐day posthospitalization mortality were obtained from the RMRS. Comorbidity level was measured by reviewing the RMRS and determining each patient's Charlson comorbidity index total score.59, 60 This score was determined using International Statistical Classification of Diseases and Related Health Problems, 9th edition (ICD‐9) codes gathered from 1 year prior to admission until the patient was discharged from the hospital. Anticholinergic medications were determined by using the Anticholinergic Cognitive Burden Scale,61 an expert‐based practical index. The scale was developed based on a review of all published studies from 1996 to 2007 that measured the anticholinergic activities of a drug and its association with cognitive function in older adults. The list of drugs reviewed was presented to an expert interdisciplinary panel that included geriatricians, geriatric pharmacists, geriatric psychiatrists, general physicians, geriatric nurses, and aging brain researchers. The panel categorized each medication into a possible or definite anticholinergic category based on the severity of its cognitive anticholinergic effects.61 A patient who received at least 1 order of a possible or definite anticholinergic during their hospitalization was considered to be an anticholinergic user. Prior recognition of CI was determined by searching the RMRS for any ICD‐9 code (see Appendix) indicative of dementia, Alzheimer disease, or delirium reported at hospital admission, discharge, or during an 1‐year period prior to hospitalization for every patient enrolled in the study. Those patients with documented ICD‐9 codes were felt recognized as having some form of cognitive impairment. Those who had a positive screen but no prior documentation according to ICD‐9 coding, were said to have unrecognized CI.
Analysis
Descriptive statistics were calculated, including percentages for binary categorical variables, and means and standard deviations for continuous variables. Comparisons between groups were based upon Fisher's Exact Tests for binary categorical variables and t tests for continuous variables. When controlling for covariates such as age, gender, race, Charlson comorbidity index, and SPMSQ at screening, group comparisons were made by using logistic regression for binary categorical variables and multiple regression for continuous variables. Since the distributions of length of stay and Charlson comorbidity index were skewed, all statistical tests comparing them across groups were actually performed on their log‐transformed values.
Results
The Prevalence and Recognition of CI
Table 1 describes the demographic characteristic of our study population, which is a reflection of the public and urban nature of our target hospital. Our study assessed the cognitive status of 997 older adults usually (>70% of the time) within 48 hours of their admission to the medical ward of this urban hospital between July of 2006 and March 2008 (see Table 1) and found that 43% of these elders had evidence of CI as determined by a SPMSQ score of 8 points or less. However, 61% of the 424 cognitively impaired elders were not documented or recognized by the electronic medical record system to have cognitive deficit.
| Variable | n | %/Mean (SD) |
|---|---|---|
| ||
| Age (years), mean (SD) | 997 | 74.8 (7.5) |
| Age 85 (%) | 997 | 12.6 |
| Female (%) | 997 | 67.8 |
| African American (%) | 997 | 59.4 |
| Education (years), mean (SD) | 910 | 10.3 (2.8) |
| Education <12 years (%) | 910 | 59.1 |
| Screened within 48 hours of admission (%) | 997 | 73.2 |
| SPMSQ score at screening, mean (SD) | 997 | 7.7 (2.8) |
| Cognitive impairment based on the SPMSQ score 8 (%) | 997 | 42.5 |
The Impact of Unrecognized CI on the Hospital Course
As expected, hospitalized elders with documented CI were older (mean age 79.1 years vs. 76.1 years; P < 0.001) and had worse cognitive function upon screening than those with unrecognized CI (mean SPMSQ 3.4 points vs. 6.3; P < 0.001). Furthermore, CI recognition was influenced by the elders' race and comorbidity (Table 2); a higher percentage of elders with documented CI were African American (69% vs. 54%; P = 0.003) and had less comorbidity (mean Charlson index 1.9 vs. 2.3; P = 0.03). After adjusting for age, gender, race, comorbidity, and cognitive function at screening, our study found no differences between elders with previously recognized CI and those with unrecognized CI in regard to the length of hospital stay (6.7 days vs. 7.5 days; P = 0.59), 30‐day posthospital mortality (4.8% vs. 6.6%; P > 0.2), home discharge (32% vs. 45%; P > 0.7), hospital readmission (19.2% vs.18.8%; P > 0.6), delirium incidence (27% vs. 21%; P > 0.9), and physical restraints (1.8% vs. 1.5%; P > 0.4). We also found that elders with undocumented CI were not more likely to receive definite anticholinergics (33.2% vs. 32.7%; P > 0.9).
| CI Documented | CI Undocumented | P Value | P Value* | |
|---|---|---|---|---|
| ||||
| n (%) | 165 (39) | 259 (61) | n/a | |
| Age, mean (SD) | 79.1 (7.9) | 76.1 (8.0) | <0.001 | |
| Female (%) | 68.5 | 64.5 | 0.40 | |
| African American (%) | 68.5 | 53.7 | <0.01 | |
| SPMSQ at screen, mean (SD) | 3.4 (2.7) | 6.3 (2.1) | <0.001 | |
| Charlson comorbidity index, mean (SD) | 1.9 (1.9) | 2.3 (2.1) | 0.03 | |
| Length of hospital stay, mean (SD) | 6.7 (5.1) | 7.5 (7.1) | 0.49 | 0.59 |
| Survived at 30 days postdischarge (%) | 95.2 | 93.4 | 0.53 | 0.25 |
| Discharged home (%) | 31.5 | 45.2 | 0.01 | 0.74 |
| Readmission within 30 days after discharge home (%) | 19.2 | 18.8 | 0.99 | 0.66 |
| Incidence of delirium (%) | 26.7 | 20.6 | 0.52 | 0.99 |
| Observed with Foley catheter (%) | 43.6 | 27.4 | <0.001 | 0.61 |
| Observed with physical restraint (%) | 1.8 | 1.5 | 0.99 | 0.31 |
| Observed with tethers (%) | 81.8 | 73.8 | 0.06 | 0.58 |
| With at least 1 Ach (%) | 83.6 | 90.7 | 0.03 | 0.22 |
| Possible Ach (%) | 81.2 | 88.4 | 0.05 | 0.31 |
| Definite Ach (%) | 32.7 | 33.2 | 0.99 | 0.64 |
The Impact of Delirium on the Hospital Course of Elders with CI
Among the 424 hospitalized elders with CI, 163 (38%) had delirium at least once during their hospital course and 24% had delirium on the day of hospital discharge. In comparison to elders who had CI but not delirium during their hospitalization (Table 3), those with at least 1 day of delirium had a higher 30‐day posthospitalization mortality risk (8.6% vs. 4.2%; P = 0.09), stayed in the hospital 3.3 additional days (9.2 days vs. 5.9 days; P < 0.001), were less likely to be discharged home (25% vs. 49%; P < 0.001), were more likely to receive a Foley catheterization (52% vs. 23%; P < 0.001), more likely to be physically restrained (4% vs. 0%; P < 0.01), and more likely to receive tethers during their care (89% vs. 69%; P < 0.001). There was no statistically significant difference between the 2 groups in terms of 30‐day hospital readmission rates or in their use of definite anticholinergics (Table 3).
| Delirium+* | Delirium | P value | |
|---|---|---|---|
| |||
| n (%) | 163 (38) | 261 (62) | n/a |
| Age, mean (SD) | 78.4 (8.5) | 76.5 (7.8) | 0.02 |
| Female (%) | 60.1 | 69.7 | 0.05 |
| African American (%) | 64.4 | 56.3 | 0.10 |
| Charlson comorbidity index, mean (SD) | 1.8 (1.9) | 2.3 (2.1) | 0.01 |
| Length of hospital stay, mean (SD) | 9.2 (7.9) | 5.9 (4.9) | <0.001 |
| Survived at 30‐day postdischarge (%) | 91.4 | 95.8 | 0.09 |
| Discharged home (%) | 24.5 | 49.4 | <0.001 |
| Readmission within 30 days after discharge home (%) | 22.5 | 17.8 | 0.50 |
| Observed with Foley catheter (%) | 51.5 | 22.6 | <0.001 |
| Observed with physical restraint (%) | 4.3 | 0.0 | <0.01 |
| Observed with tethers (%) | 89.0 | 69.4 | <0.001 |
| With at least 1 anticholinergic (%) | 83.4 | 90.8 | 0.03 |
| Possible anticholinergic (%) | 80.4 | 88.9 | 0.02 |
| Definite anticholinergic (%) | 36.8 | 30.7 | 0.20 |
Discussion
Our study found that in an urban, public hospital, acute or preexisting CI affects more than one‐third of hospitalized elders admitted to general medical services. Unfortunately, our hospital system does not currently recognize the majority of these vulnerable patients. Our study also found that delirium affects more than one‐third of hospitalized elders with CI during their hospital course. Delirium complicates hospital care by prolonging length of stay and decreasing the probability of surviving and getting discharged home. It leads to high use of Foley catheterization, physical restraints, and tethers.
The high prevalence of CI with and without delirium in our cohort is within the rates reported previously in the literature. It is estimated that the prevalence of CI in hospitalized older adults ranges from 14% to 66%, depending on the method used to measure cognition, the definition of CI, and the type of hospital ward (surgical, medical, and geriatric units).220 One particular study that used a similar cognitive assessment method reported higher prevalence rates for both CI and delirium.11 The study randomly evaluated a sample of 201 patients age 65 and over who were hospitalized for a medical illness and found that 56% of the cohort suffered from CI and among those with CI, 47% had delirium.11 The difference between this finding and our study is most likely due to our sampling technique; more than 70% of our cognitive screening occurred in the first 48 hours of hospital admission whereas the Australian study, in similar enrollment criteria to all of the published studies in this area, excluded patients who were discharged within 48 hours of admission. We believe, however, that by including the first 48 hours of admission in our design, our study provides a more generalizable reflection of the actual acute care experience.
The impact of delirium on the course of hospital care found in our study supports some of the findings from previous studies conducted in the past 2 decades.5, 6, 11 Despite 2 decades of clinical research, delirium continues to increase mortality, hospital stays, and posthospital institutionalization.
We were surprised to find that patients suffering from delirium continue to receive at least 1 definite anticholinergic medication. Such medications are considered inappropriate among patients with any form of cognitive impairment.36, 62 Although the impact of anticholinergic medications on hospitalized outcomes is less well‐described, their use has been suspected to negatively impact long‐term outcomes of cognitive impairment.61, 63 Our study found no difference in the use of anticholinergic medications between those with CI who experienced delirium and those who did not; however, the total burden of anticholinergic medication was not assessed in a quantitative manner. It is still unknown if certain anticholinergic medications or a cumulative effect of anticholinergic medications may impact cognitive or health‐related outcomes in a vulnerable older population with CI.
Although our study reported for the first time in a systematic way the rate of undocumented CI among hospitalized elders found to have CI on admission, we found no impact of such underrecognition on the length of hospital stay, mortality, discharge location, and delirium occurrence. Although the use of anticholinergic medications is not recommended for patients with any form of CI, our results indicate that a significant number of patients with cognitive impairment continue to receive inappropriate medications. CI recognition in the elderly was not shown to have a statistically significant affect on length of stay, cost, or mortality.
Our study has some limitations. First of all, we did not determine the underlying types of CI such as Alzheimer disease, vascular dementia, mild cognitive impairment, or reversible etiology other than delirium. Such a categorization requires posthospital assessment, which was not included in our study design. Second, our delirium incidence rate and delirium impact on hospital outcomes might be very conservative and may underestimate its true prevalence and correlation due to our data collection methods. Despite the fluctuating nature of delirium, our study was not designed to assess the presence of delirium every shift and tried to assess cognitive function on a daily basis throughout the patient's hospitalization. Therefore, the severity and duration of delirium could not be accurately assessed. Our reported rates of use of Foley catheterization, physical restraints, and tethers are also very conservative and we could not determine the appropriateness of these procedures. Our study was conducted in 1 public hospital in an urban city with a higher percentage of African Americans. Thus, our sample is not a true representative sample. However, studies with significant representation of minority groups are not common in the research literature, especially in CI research; we hope to fulfill some of the gaps in the literature regarding the most vulnerable older American population. Finally, we were limited in our use of ICD‐9 coding to determine if patients had previously been recognized by other providers as having CI. ICD‐9 coding, while useful, is not perfect in identifying all if a patient's medical problems. Use of coding to determine whether a patient had been recognized as impaired also does not allow us to determine when the diagnosis was made.
In conclusion, our study evaluated cognitive impairment in hospitalized elders and found that in our cohort of 997 patients, 43% were cognitively impaired on admission. Of those with CI, 61% were not documented or recognized as impaired. We found no statistically significant difference between those with documented CI and those with undocumented CI in terms of length of stay, mortality, home discharge, readmission rates, incidence of delirium, or potential to receive anticholinergics or restraints. Among those with CI, 38% had delirium. Those with delirium experienced increased length of stay, decreased discharge to home, and increased use of Foley catheters and restraints.
- ,.National hospital discharge survey: annual summary, 1994.Vital Health Stat 13.1997;(128):i–v;1–50.
- .The dilemma of delirium: clinical and research controversies regarding diagnosis and evaluation of delirium in hospitalized elderly medical patients.Am J Med.1994;97:278–288.
- ,,.Dementia in medical wards.J Clin Epidemiol.1988;41:123–126.
- ,,,,,.Mental and behavioral disturbances in dementia: findings from the Cache County Study on Memory in Aging.Am J Psychiatry.2000;157:708–714.
- ,,, et al.Randomized, placebo‐controlled, double‐blind clinical trial of sertraline in the treatment of depression complicating Alzheimer's disease: initial results from the Depression in Alzheimer's Disease study.Am J Psychiatry.2000;157:1686–1689.
- ,,.Dementia in elderly persons in a general hospital.Am J Psychiatry.2000;157:704–707.
- ,,,,.A prospective study of the impact of psychiatric comorbidity on length of hospital stays of elderly medical‐surgical inpatients.Psychosomatics.1998;39:273–280.
- ,.Psychiatric comorbidity and length of stay in the general hospital. A critical review of outcome studies.Psychosomatics.1994;35:233–252.
- ,,,.Dementia among medical inpatients. Evaluation of 2000 consecutive admissions.Arch Intern Med.1986;146:1923–1926.
- ,,,.The consequences of non‐cognitive symptoms of dementia in medical hospital departments.Int J Psychiatry Med.2003;33:257–271.
- ,,.Cognitive impairment in medical inpatients. I: Screening for dementia—is history better than mental state?Age Ageing.1997;26:31–35.
- ,,, et al.Acute confusional states in elderly patients treated for femoral neck fracture.J Am Geriatr Soc.1988;36:525–530.
- ,.A prospective study of elderly general surgical patients: II. Post‐operative complications.Age Ageing.1989;18:316–326.
- ,,,.Dementia and depression in elderly medical inpatients.Int Psychogeriatr.2000;12:67–75.
- ,,.[Psychiatric disorders in elderly general hospital patients: incidence and long‐term prognosis].Nervenarzt.1993;64:53–61. [German]
- ,.Delirium and dementia in acute medical admissions of elderly patients in Iceland.Acta Psychiatr Scand.1993;87:123–127.
- ,,,,,.[Psychiatric morbidity in elderly patients admitted to a general hospital. A day‐prevalence study].Med Clin (Barc).1991;97:206–210. [Spanish]
- ,,,,,.Detection of psychiatric disorders in elderly medical inpatients.Age Ageing.1994;23:307–311.
- ,,,.Cognitive impairment, emotional disorder and length of stay of elderly patients in a district general hospital.Br J Med Psychol.1987;60(Pt 2):133–139.
- ,,.An investigation of the components of best nursing practice in the care of acutely ill hospitalized older patients with coincidental dementia: a multi‐method design.J Adv Nurs1999;30:1127–1136.
- American Psychiatric Association.Diagnostic and Statistical Manual of Mental Disorders.4th ed.Washington, DC:American Psychiatric Association;1994.
- ,,,,,.Clarifying confusion: the confusion assessment method. A new method for detection of delirium.Ann Intern Med.1990;113:941–948.
- ,,, et al.Prevalence of cognitive impairment: data from the Indianapolis Study of Health and Aging.Neurology.2001;57:1655–1662.
- ,,.Delirium: a symptom of how hospital care is failing older persons and a window to improve quality of hospital care.Am J Med.1999;106:565–573.
- ,,, et al.A multicomponent intervention to prevent delirium in hospitalized older patients.N Engl J Med.1999;340:669–676.
- ,,,.Iatrogenic causes of falls in hospitalised elderly patients: a case‐control study.Postgrad Med J.2002;78:487–489.
- ,,.A prospective study of delirium in hospitalized elderly.JAMA.1990;263:1097–1101.
- ,.The prognostic significance of delirium in older hospital patients.J Am Geriatr Soc.1997;45:174–178.
- ,.Prognosis of delirium in elderly hospital patients.CMAJ.1993;149:41–46.
- ,,,,.The detection of psychiatric morbidity and its effects on outcome in acute elderly medical admissions.Int J Ger Psych1991;6:861–866.
- ,,.Adverse consequences of hospitalization in the elderly.Soc Sci Med.1982;16:1033–1038.
- ,,, et al.Incidence of adverse events and negligence in hospitalized patients. Results of the Harvard Medical Practice Study I.N Engl J Med.1991;324:370–376.
- ,,,.Delirium in elderly patients: an overview of the state of the science.J Gerontol Nurs.2001;27:12–20.
- ,,,,.A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics.Ann Intern Med.1993;119:474–481.
- ,.Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability.JAMA.1996;275:852–857.
- ,,,,,.Updating the Beers criteria for potentially inappropriate medication use in older adults: results of a US consensus panel of experts.Arch Intern Med.2003;163:2716–2724.
- ,,, et al.A clinical prediction rule for delirium after elective noncardiac surgery.JAMA.1994;271:134–139.
- ,,, et al.The relationship of postoperative delirium with psychoactive medications.JAMA.1994;272:1518–1522.
- ,,, et al.How do delirium and dementia increase length of stay of elderly general medical inpatients?Psychosomatics.2004;45:235–242.
- ,,,,,.The relationship between a dementia diagnosis, chronic illness, Medicare expenditures, and hospital use.J Am Geriatr Soc.2004;52:187–194.
- ,,, et al.Risk factors for delirium in hospitalized elderly.JAMA.1992;267:827–831.
- ,,,,.Psychological comorbidity and length of stay in the general hospital.Am J Psychiatry.1991;148:324–329.
- ,,, et al.Delirium. The occurrence and persistence of symptoms among elderly hospitalized patients.Arch Intern Med.1992;152:334–340.
- ,,.What happens to medical patients with psychiatric disorder?J Psychosom Res1988;32:541–549.
- ,,,,.[Dementia syndromes and length of stay of elderly patients in internal medicine].Ann Med Interne (Paris).1997;148:424–426. [French]
- ,,, et al.[Hospital discharge planning and length of hospital stay in elderly patients admitted through the emergency department].Rev Epidemiol Sante Publique.1995;43:337–347. [French]
- ,,,,.The effect of dementia on acute care in a geriatric medical unit.Int Psychogeriatr.1992;4:231–239.
- ,,,.Cognitive impairment. Can it predict the course of hospitalized patients?J Am Geriatr Soc.1986;34:579–585.
- ,,,,;US Preventive Services Task Force. Screening for dementia in primary care: a summary of the evidence for the U.S. Preventive Services Task Force.Ann Intern Med.2003;138(11):927–937.
- ,,,.Cognitive impairment in the elderly medically ill: how often is it missed?Int J Geriatr Psychiatry.1993;8:929–937.
- .Recognition of cognitive impairment in elderly medical in‐patients.J R Soc Med.1995;88:183–184.
- ,.Quality indicators for dementia in vulnerable community‐dwelling and hospitalized elders.Ann Intern Med.2001;135:668–676.
- ,,,.Reducing delirium after hip fracture: a randomized trial.J Am Geriatr Soc.2001;49:516–522.
- ,.Prevalence of psychotic symptoms in delirium.Psychosomatics.2000;41:519–522.
- .A short portable mental status questionnaire for the assessment of organic brain deficit in elderly patients.J Am Geriatr Soc.1975;23:433–441.
- ,,,.Short Portable Mental Status Questionnaire as a screening test for dementia and delirium among the elderly.J Am Geriatr Soc.1987;35:412–416.
- ,,, et al.The Regenstrief Medical Record System: a quarter century experience.Int J Med Inform.1999;54:225–253.
- ,,,,,.Factors determining the decision to institutionalize dementing individuals: a prospective study.Gerontologist.1993;33:714–720.
- ,,,,,.Resuscitation: how do we decide? A prospective study of physicians' preferences and the clinical course of hospitalized patients.JAMA.1986;255:1316–1322.
- ,,,,,.Assessing illness severity: does clinical judgment work?J Chronic Dis.1986;39:439–452.
- ,,,,.Impact of anticholinergics on the aging brain: a review and practical application.Aging Health.2008;4(3):311–320.
- ,.Delirium in hospitalized older adults. In: Ham R, Sloane P, Warshaw G, eds.Primary Care Geriatrics: A Case‐Based Approach.5th ed.Philadelphia, PA:Mosby Elsevier;2007:210–218.
- ,,, et al.The association between cognition and histamine‐2 receptor antagonists in African Americans.J Am Geriatr Soc.2007;55(8):1248–1253.
- ,.National hospital discharge survey: annual summary, 1994.Vital Health Stat 13.1997;(128):i–v;1–50.
- .The dilemma of delirium: clinical and research controversies regarding diagnosis and evaluation of delirium in hospitalized elderly medical patients.Am J Med.1994;97:278–288.
- ,,.Dementia in medical wards.J Clin Epidemiol.1988;41:123–126.
- ,,,,,.Mental and behavioral disturbances in dementia: findings from the Cache County Study on Memory in Aging.Am J Psychiatry.2000;157:708–714.
- ,,, et al.Randomized, placebo‐controlled, double‐blind clinical trial of sertraline in the treatment of depression complicating Alzheimer's disease: initial results from the Depression in Alzheimer's Disease study.Am J Psychiatry.2000;157:1686–1689.
- ,,.Dementia in elderly persons in a general hospital.Am J Psychiatry.2000;157:704–707.
- ,,,,.A prospective study of the impact of psychiatric comorbidity on length of hospital stays of elderly medical‐surgical inpatients.Psychosomatics.1998;39:273–280.
- ,.Psychiatric comorbidity and length of stay in the general hospital. A critical review of outcome studies.Psychosomatics.1994;35:233–252.
- ,,,.Dementia among medical inpatients. Evaluation of 2000 consecutive admissions.Arch Intern Med.1986;146:1923–1926.
- ,,,.The consequences of non‐cognitive symptoms of dementia in medical hospital departments.Int J Psychiatry Med.2003;33:257–271.
- ,,.Cognitive impairment in medical inpatients. I: Screening for dementia—is history better than mental state?Age Ageing.1997;26:31–35.
- ,,, et al.Acute confusional states in elderly patients treated for femoral neck fracture.J Am Geriatr Soc.1988;36:525–530.
- ,.A prospective study of elderly general surgical patients: II. Post‐operative complications.Age Ageing.1989;18:316–326.
- ,,,.Dementia and depression in elderly medical inpatients.Int Psychogeriatr.2000;12:67–75.
- ,,.[Psychiatric disorders in elderly general hospital patients: incidence and long‐term prognosis].Nervenarzt.1993;64:53–61. [German]
- ,.Delirium and dementia in acute medical admissions of elderly patients in Iceland.Acta Psychiatr Scand.1993;87:123–127.
- ,,,,,.[Psychiatric morbidity in elderly patients admitted to a general hospital. A day‐prevalence study].Med Clin (Barc).1991;97:206–210. [Spanish]
- ,,,,,.Detection of psychiatric disorders in elderly medical inpatients.Age Ageing.1994;23:307–311.
- ,,,.Cognitive impairment, emotional disorder and length of stay of elderly patients in a district general hospital.Br J Med Psychol.1987;60(Pt 2):133–139.
- ,,.An investigation of the components of best nursing practice in the care of acutely ill hospitalized older patients with coincidental dementia: a multi‐method design.J Adv Nurs1999;30:1127–1136.
- American Psychiatric Association.Diagnostic and Statistical Manual of Mental Disorders.4th ed.Washington, DC:American Psychiatric Association;1994.
- ,,,,,.Clarifying confusion: the confusion assessment method. A new method for detection of delirium.Ann Intern Med.1990;113:941–948.
- ,,, et al.Prevalence of cognitive impairment: data from the Indianapolis Study of Health and Aging.Neurology.2001;57:1655–1662.
- ,,.Delirium: a symptom of how hospital care is failing older persons and a window to improve quality of hospital care.Am J Med.1999;106:565–573.
- ,,, et al.A multicomponent intervention to prevent delirium in hospitalized older patients.N Engl J Med.1999;340:669–676.
- ,,,.Iatrogenic causes of falls in hospitalised elderly patients: a case‐control study.Postgrad Med J.2002;78:487–489.
- ,,.A prospective study of delirium in hospitalized elderly.JAMA.1990;263:1097–1101.
- ,.The prognostic significance of delirium in older hospital patients.J Am Geriatr Soc.1997;45:174–178.
- ,.Prognosis of delirium in elderly hospital patients.CMAJ.1993;149:41–46.
- ,,,,.The detection of psychiatric morbidity and its effects on outcome in acute elderly medical admissions.Int J Ger Psych1991;6:861–866.
- ,,.Adverse consequences of hospitalization in the elderly.Soc Sci Med.1982;16:1033–1038.
- ,,, et al.Incidence of adverse events and negligence in hospitalized patients. Results of the Harvard Medical Practice Study I.N Engl J Med.1991;324:370–376.
- ,,,.Delirium in elderly patients: an overview of the state of the science.J Gerontol Nurs.2001;27:12–20.
- ,,,,.A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics.Ann Intern Med.1993;119:474–481.
- ,.Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability.JAMA.1996;275:852–857.
- ,,,,,.Updating the Beers criteria for potentially inappropriate medication use in older adults: results of a US consensus panel of experts.Arch Intern Med.2003;163:2716–2724.
- ,,, et al.A clinical prediction rule for delirium after elective noncardiac surgery.JAMA.1994;271:134–139.
- ,,, et al.The relationship of postoperative delirium with psychoactive medications.JAMA.1994;272:1518–1522.
- ,,, et al.How do delirium and dementia increase length of stay of elderly general medical inpatients?Psychosomatics.2004;45:235–242.
- ,,,,,.The relationship between a dementia diagnosis, chronic illness, Medicare expenditures, and hospital use.J Am Geriatr Soc.2004;52:187–194.
- ,,, et al.Risk factors for delirium in hospitalized elderly.JAMA.1992;267:827–831.
- ,,,,.Psychological comorbidity and length of stay in the general hospital.Am J Psychiatry.1991;148:324–329.
- ,,, et al.Delirium. The occurrence and persistence of symptoms among elderly hospitalized patients.Arch Intern Med.1992;152:334–340.
- ,,.What happens to medical patients with psychiatric disorder?J Psychosom Res1988;32:541–549.
- ,,,,.[Dementia syndromes and length of stay of elderly patients in internal medicine].Ann Med Interne (Paris).1997;148:424–426. [French]
- ,,, et al.[Hospital discharge planning and length of hospital stay in elderly patients admitted through the emergency department].Rev Epidemiol Sante Publique.1995;43:337–347. [French]
- ,,,,.The effect of dementia on acute care in a geriatric medical unit.Int Psychogeriatr.1992;4:231–239.
- ,,,.Cognitive impairment. Can it predict the course of hospitalized patients?J Am Geriatr Soc.1986;34:579–585.
- ,,,,;US Preventive Services Task Force. Screening for dementia in primary care: a summary of the evidence for the U.S. Preventive Services Task Force.Ann Intern Med.2003;138(11):927–937.
- ,,,.Cognitive impairment in the elderly medically ill: how often is it missed?Int J Geriatr Psychiatry.1993;8:929–937.
- .Recognition of cognitive impairment in elderly medical in‐patients.J R Soc Med.1995;88:183–184.
- ,.Quality indicators for dementia in vulnerable community‐dwelling and hospitalized elders.Ann Intern Med.2001;135:668–676.
- ,,,.Reducing delirium after hip fracture: a randomized trial.J Am Geriatr Soc.2001;49:516–522.
- ,.Prevalence of psychotic symptoms in delirium.Psychosomatics.2000;41:519–522.
- .A short portable mental status questionnaire for the assessment of organic brain deficit in elderly patients.J Am Geriatr Soc.1975;23:433–441.
- ,,,.Short Portable Mental Status Questionnaire as a screening test for dementia and delirium among the elderly.J Am Geriatr Soc.1987;35:412–416.
- ,,, et al.The Regenstrief Medical Record System: a quarter century experience.Int J Med Inform.1999;54:225–253.
- ,,,,,.Factors determining the decision to institutionalize dementing individuals: a prospective study.Gerontologist.1993;33:714–720.
- ,,,,,.Resuscitation: how do we decide? A prospective study of physicians' preferences and the clinical course of hospitalized patients.JAMA.1986;255:1316–1322.
- ,,,,,.Assessing illness severity: does clinical judgment work?J Chronic Dis.1986;39:439–452.
- ,,,,.Impact of anticholinergics on the aging brain: a review and practical application.Aging Health.2008;4(3):311–320.
- ,.Delirium in hospitalized older adults. In: Ham R, Sloane P, Warshaw G, eds.Primary Care Geriatrics: A Case‐Based Approach.5th ed.Philadelphia, PA:Mosby Elsevier;2007:210–218.
- ,,, et al.The association between cognition and histamine‐2 receptor antagonists in African Americans.J Am Geriatr Soc.2007;55(8):1248–1253.
Copyright © 2010 Society of Hospital Medicine
A Pain in the Bone
A 71‐year‐old man presented to a hospital with a one week history of fatigue, polyuria, and polydipsia. He also reported pain in his back, hips, and ribs, in addition to frequent falls, intermittent confusion, constipation, and a weight loss of 10 pounds over the last 2 weeks. He denied cough, shortness of breath, chest pain, fever, night sweats, headache, and focal weakness.
Polyuria, which is often associated with polydipsia, can be arbitrarily defined as a urine output exceeding 3 L per day. After excluding osmotic diuresis due to uncontrolled diabetes mellitus, the 3 major causes of polyuria are primary polydipsia, central diabetes insipidus, and nephrogenic diabetes insipidus. Approximately 30% to 50% of cases of central diabetes insipidus are idiopathic; however, primary or secondary brain tumors or infiltrative diseases involving the hypothalamic‐pituitary region need to be considered in this 71‐year‐old man. The most common causes of nephrogenic diabetes insipidus in adults are chronic lithium ingestion, hypokalemia, and hypercalcemia. The patient describes symptoms that can result from severe hypercalcemia, including fatigue, confusion, constipation, polyuria, and polydipsia.
The patient's past medical history included long‐standing, insulin‐requiring type 2 diabetes with associated complications including coronary artery disease, transient ischemic attacks, proliferative retinopathy, peripheral diabetic neuropathy, and nephropathy. Seven years prior to presentation, he received a cadaveric renal transplant that was complicated by BK virus (polyomavirus) nephropathy and secondary hyperparathyroidism. Three years after his transplant surgery, he developed squamous cell carcinoma of the skin, which was treated with local surgical resection. Two years after that, he developed stage I laryngeal cancer of the glottis and received laser surgery, and since then he had been considered disease‐free. He also had a history of hypertension, hypercholesterolemia, osteoporosis, and depression. His medications included aspirin, amlodipine, metoprolol succinate, valsartan, furosemide, simvastatin, insulin, prednisone, sirolimus, and sulfamethoxazole/trimethoprim. He was a married psychiatrist. He denied tobacco use and reported occasional alcohol use.
The prolonged immunosuppressive therapy that is required following organ transplantation carries a markedly increased risk of the subsequent development of malignant tumors, including cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma. Primary brain lymphoma resulting in central diabetes insipidus would be unlikely in the absence of headache or focal weakness. An increased risk of lung cancer occurs in recipients of heart and lung transplants, and to a much lesser degree, recipients of kidney transplants. However, metastatic lung cancer is less likely in the absence of respiratory symptoms and smoking history (present in approximately 90% of all lung cancers). Nephrogenic diabetes insipidus, in its mild form, is relatively common in elderly patients with acute or chronic renal insufficiency because of a reduction in maximum urinary concentrating ability. On the other hand, this alone does not explain his remaining symptoms. The instinctive diagnosis in this case is tertiary hyperparathyroidism due to progression of untreated secondary hyperparathyroidism. This causes hypercalcemia, nephrogenic diabetes insipidus, and significant bone pain related to renal osteodystrophy.
On physical exam, the patient appeared chronically ill, but was in no acute distress. He weighed 197.6 pounds and his height was 70.5 inches. He was afebrile with a blood pressure of 146/82 mm Hg, a heart rate of 76 beats per minute, a respiratory rate of 12 breaths per minute, and an oxygen saturation of 97% while breathing room air. He had no generalized lymphadenopathy. Thyroid examination was unremarkable. Examination of the lungs, heart, abdomen, and lower extremities was normal. The rectal examination revealed no masses or prostate nodules; a test for fecal occult blood was negative. He had loss of sensation to light touch and vibration in the feet with absent Achilles deep tendon reflexes. He had a poorly healing surgical wound on his forehead at the site of his prior skin cancer, but no rash or other lesions. There was no joint swelling or erythema. There were tender points over the cervical, thoracic, and lumbar spine; on multiple ribs; and on the pelvic rims.
Perhaps of greatest importance is the lack of lymphadenopathy, organomegaly, or other findings suggestive of diffuse lymphoproliferative disease. His multifocal bone tenderness is concerning for renal osteodystrophy, multiple myeloma, or primary or metastatic bone disease. Cancers in men that metastasize to the bone usually originate from the prostate, lung, kidney, or thyroid gland. In any case, his physical examination did not reveal an enlarged, asymmetric, or nodular prostate or thyroid gland. I recommend a chest film to rule out primary lung malignancy and a basic laboratory evaluation to narrow down the differential diagnosis.
A complete blood count showed a normocytic anemia with a hemoglobin of 8.7 g/dL and a hematocrit of 25%. Other laboratory tests revealed the following values: sodium, 139 mmol/L; potassium, 4.1 mmol/L; blood urea nitrogen, 70 mg/dL; creatinine, 3.5 mg/dL (most recent value 2 months ago was 1.9 mg/dL); total calcium, 13.2 mg/dL (normal range, 8.5‐10.5 mg/dL); phosphate, 5.3 mg/dL; magnesium, 2.5 mg/dL; total bilirubin, 0.5 mg/dL; alkaline phosphatase, 130 U/L; aspartate aminotransferase, 28 U/L; alanine aminotransferase, 19 U/L; albumin, 3.5 g/dL; and lactate dehydrogenase (LDH), 1258 IU/L (normal range, 105‐333 IU/L). A chest radiograph was normal.
The most important laboratory findings are severe hypercalcemia, acute on chronic renal failure, and anemia. Hypercalcemia most commonly results from malignancy or hyperparathyroidism. Less frequently, hypercalcemia may result from sarcoidosis, vitamin D intoxication, or hyperthyroidism. The degree of hypercalcemia is useful diagnostically as hyperparathyroidism commonly results in mild hypercalcemia (serum calcium concentration often below 11 mg/dL). Values above 13 mg/dL are unusual in hyperparathyroidism and are most often due to malignancy. Malignancy is often evident clinically by the time it causes hypercalcemia, and patients with hypercalcemia of malignancy are more often symptomatic than those with hyperparathyroidism. Additionally, localized bone pain and weight loss do not result from hypercalcemia itself and their presence also raises concern for malignancy.
Nonmelanoma skin cancer is the most common cancer occurring after transplantation but does not cause hypercalcemia. Squamous cancers of the head and neck can rarely cause hypercalcemia due to secretion of parathyroid hormone‐related peptide; however, his early‐stage laryngeal cancer and the expected high likelihood of cure argue against this possibility. Osteolytic metastases account for approximately 20% of cases of hypercalcemia of malignancy (Table 1). Prostate cancer rarely results in hypercalcemia since bone metastases are predominantly osteoblastic, whereas metastatic non‐small‐cell lung cancer, thyroid cancer, and kidney cancer more commonly cause hypercalcemia due to osteolytic bone lesions. The total alkaline phosphatase has been traditionally used to assess the osteoblastic component of bone remodeling. Its normal level tends to predict a negative bone scan and supports the likelihood of lytic lesions. Posttransplantation lymphoproliferative disorders, which include a wide range of syndromes, can rarely result in hypercalcemia. I am also worried about the possibility of multiple myeloma as he has the classic triad of hypercalcemia, bone pain, and subacute kidney injury.
|
| Osteolytic metastases |
| Breast cancer |
| Multiple myeloma |
| Lymphoma |
| Leukemia |
| Humoral hypercalcemia (PTH‐related protein) |
| Squamous cell carcinomas |
| Renal carcinomas |
| Bladder carcinoma |
| Breast cancer |
| Ovarian carcinoma |
| Leukemia |
| Lymphoma |
| 1,25‐Dihydroxyvitamin D secretion |
| Lymphoma |
| Ovarian dysgerminomas |
| Ectopic PTH secretion (rare) |
| Ovarian carcinoma |
| Lung carcinomas |
| Neuroectodermal tumor |
| Thyroid papillary carcinoma |
| Rhabdomyosarcoma |
| Pancreatic cancer |
The first purpose of the laboratory evaluation is to differentiate parathyroid hormone (PTH)‐mediated hypercalcemia (primary and tertiary hyperparathyroidism) from non‐PTH‐mediated hypercalcemia (primarily malignancy, hyperthyroidism, vitamin D intoxication, and granulomatous disease). The production of vitamin D metabolites, PTH‐related protein, or hypercalcemia from osteolysis in these latter cases results in suppressed PTH levels.
In severe elevations of calcium, the initial goals of treatment are directed toward fluid resuscitation with normal saline and, unless contraindicated, the immediate institution of bisphosphonate therapy. A loop diuretic such as furosemide is often used, but a recent review concluded that there is little evidence to support its use in this setting.
The patient was admitted and treated with intravenous saline and furosemide. Additional laboratory evaluation revealed normal levels of prostate‐specific antigen and thyroid‐stimulating hormone. PTH was 44 pg/mL (the most recent value was 906 pg/mL eight years ago; normal range, 15‐65 pg/mL) and beta‐2 microglobulin (B2M) was 8 mg/L (normal range, 0.8‐2.2 mg/L).
The normal PTH level makes tertiary hyperparathyroidism unlikely and points toward non‐PTH‐related hypercalcemia. An elevated B2M level may occur in patients with chronic graft rejection, renal tubular dysfunction, dialysis‐related amyloidosis, multiple myeloma, or lymphoma. LDH is often elevated in patients with multiple myeloma and lymphoma, but this is not a specific finding. The next laboratory test would be measurement of PTH‐related protein and vitamin D metabolites, as these tests can differentiate between the causes of non‐PTH‐mediated hypercalcemia.
Serum concentrations of the vitamin D metabolites, 25‐hydroxyvitamin D (calcidiol) and 1,25‐dihydroxyvitamin D (calcitriol), were low‐normal. PTH‐related protein was not detected.
The marked elevation of serum LDH and B2M, the relatively suppressed PTH level, combined with undetectable PTH‐related protein suggest multiple myeloma or lymphoma as the likely cause of the patient's clinical presentation. The combination of hypercalcemia and multifocal bone pain makes multiple myeloma the leading diagnosis as hypercalcemia is uncommon in patients with lymphoma, especially at the time of initial clinical presentation.
I would proceed with serum and urine protein electrophoresis (SPEP and UPEP, respectively) and a skeletal survey. If these tests do not confirm the diagnosis of multiple myeloma, I would order a noncontrast computed tomography (CT) of the chest and abdomen and a magnetic resonance imaging (MRI) of the spine. In addition, I would like to monitor his response to the intravenous saline and furosemide.
Forty‐eight hours after presentation, repeat serum calcium and creatinine levels were 11.3 mg/dL and 2.9 mg/dL, respectively. He received salmon calcitonin 4 U/kg every 12 hours. Pamidronate was avoided because of his kidney disease. His confusion resolved. He received intravenous morphine intermittently to alleviate his bone pain.
The SPEP revealed a monoclonal immunoglobulin G (IgG) lambda (light chain) spike representing roughly 3% (200 mg/dL) of total protein. His serum Ig levels were normal. The UPEP was negative for monoclonal immunoglobulin and Bence‐Jones protein. The skeletal survey revealed marked osteopenia, and the bone scan was normal. An MRI of the spine showed multiple round lesions in the cervical, thoracic, and lumbar spine (Figure 1). A CT of the chest showed similar bone lesions in the ribs and pelvis. A CT of the abdomen and chest did not suggest any primary malignancy nor did it show thoracic or abdominal lymphadenopathy.
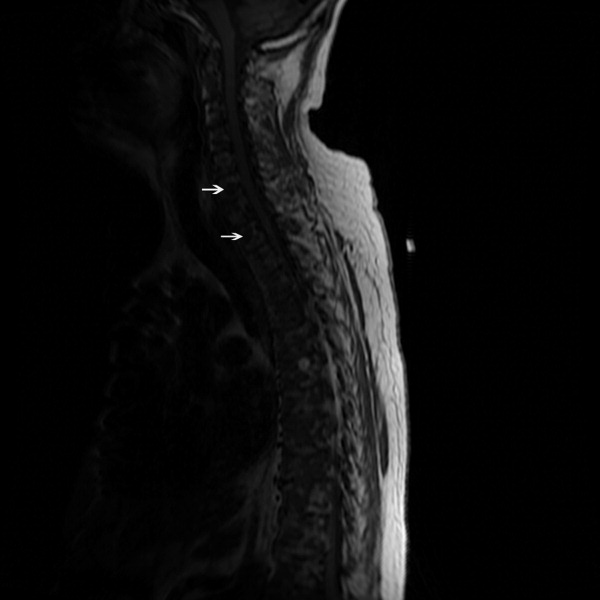
The lack of lymphadenopathy, splenomegaly, or a visceral mass by CT imaging and physical examination, along with the normal PSA level, exclude most common forms of non‐Hodgkin lymphoma and bone metastasis from solid tumors. In multiple myeloma, cytokines secreted by plasma cells suppress osteoblast activity; therefore, while discrete lytic bone lesions are apparent on skeletal survey, the bone scan is typically normal. The absence of lytic lesions, normal serum immunoglobulin levels, and unremarkable UPEP make multiple myeloma or light‐chain deposition disease a less likely diagnosis.
Typically, primary lymphoma of the bone produces increased uptake with bone scanning. However, because primary lymphoma of the bone is one of the least common primary skeletal malignancies and varies widely in appearance on imaging, confident diagnosis based on imaging alone usually is not possible.
Posttransplantation lymphoproliferative disorder (PTLD) refers to a syndrome that ranges from a self‐limited form of lymphoproliferation to an aggressive disseminated disease. Although the patient is at risk for PTLD, isolated bone involvement has only rarely been reported.
Primary lymphoma of the bone and PTLD are my leading diagnoses in this patient. At this point, I recommend a bone marrow biopsy and biopsy of an easily accessible representative bone lesion with special staining for Epstein‐Barr virus (EBV) (EBV‐encoded RNA [EBER] and latent membrane protein 1 [LMP1]). I expect this test to provide a definitive diagnosis. As 95% of PTLD cases are induced by infection with EBV, information regarding pretransplantation EBV status of the patient and the donor, current EBV status of the patient, and type and intensity of immunosuppression at the time of transplantation would be very helpful to determine their likelihood.
Seventy‐two hours after presentation, his serum calcium level normalized and most of his symptoms improved. Calcitonin was discontinued, and he was maintained on oral hydration. On hospital day number 5, he underwent CT‐guided bone biopsy of the L4 vertebral body, which showed large aggregates of atypical lymphoid cells (Figure 2). These cells were predominantly B‐cells interspersed with small reactive T‐cells. The cells did not express EBV LMP1 or EBER (Figure 3). On hospital day 7, he underwent a bone marrow biopsy, which revealed similar large atypical lymphoid cells that comprised the majority of marrow space (Figure 4). By immunohistochemistry, these cells brightly expressed the pan B cell marker, CD20, and coexpressed bcl‐2. EBER and LMP1 were also negative. A flow cytometry of the bone marrow demonstrated a lambda light chain restriction within the B lymphocytes.

The medical records indicated that the patient had positive pretransplantation EBV serologies. He received a regimen based on sirolimus, mycophenolate mofetil, and prednisone, and did not receive high doses of induction or maintenance immunosuppressive therapy.
The biopsy results establish a diagnosis of diffuse large B‐cell lymphoma of the bone. PTLD is unlikely given his positive pretransplantation EBV status, the late onset of his disease (6 years after transplantation), the isolated bone involvement, and the negative EBER and LMP1 tests.
The patient was discharged and was readmitted 1 week later for induction chemotherapy with etoposide, vincristine, doxorubicin, cyclophosphamide, and prednisone [EPOCH]Rituxan (rituximab). Over the next several months, he received 6 cycles of chemotherapy, his hypercalcemia resolved, and his back pain improved.
Commentary
Hypercalcemia is among the most common causes of nephrogenic diabetes insipidus in adults.1 A urinary concentrating defect usually becomes clinically apparent if the plasma calcium concentration is persistently above 11 mg/dL.1 This defect is generally reversible with correction of the hypercalcemia but may persist in patients in whom interstitial nephritis has induced permanent medullary damage. The mechanism by which the concentrating defect occurs is incompletely understood but may be related to impairments in sodium chloride reabsorption in the thick ascending limb and in the ability of antidiuretic hormone to increase water permeability in the collecting tubules.1
Although hypercalcemia in otherwise healthy outpatients is usually due to primary hyperparathyroidism, malignancy is more often responsible for hypercalcemia in hospitalized patients.2 While the signs and symptoms of hypercalcemia are similar regardless of the cause, several clinical features may help distinguish the etiology of hypercalcemia. For instance, the presence of tachycardia, warm skin, thinning of the hair, stare and lid lag, and widened pulse pressure points toward hypercalcemia related to hyperthyroidism. In addition, risk factors and comorbidities guide the diagnostic process. For example, low‐level hypercalcemia in an asymptomatic postmenopausal woman with a normal physical examination suggests primary hyperparathyroidism. In contrast, hypercalcemia in a transplant patient raises concern of malignancy including PTLDs.3, 4
PTLDs are uncommon causes of hypercalcemia but are among the most serious and potentially fatal complications of chronic immunosuppression in transplant recipients.5 They occur in 1.9% of patients after kidney transplantation. The lymphoproliferative disorders occurring after transplantation have different characteristics from those that occur in the general population. Non‐Hodgkin lymphoma accounts for 65% of lymphomas in the general population, compared to 93% in transplant recipients.5, 6 The pathogenesis of PTLD appears to be related to B cell proliferation induced by infection with EBV in the setting of chronic immunosuppression.6 Therefore, there is an increased frequency of PTLD among transplant recipients who are EBV seronegative at the time of operation. These patients, who have no preoperative immunity to EBV, usually acquire the infection from the donor. The level of immunosuppression (intensity and type) influences PTLD rates as well. The disease typically occurs within 12 months after transplantation and in two‐thirds of cases involves extranodal sites. Among these sites, the gastrointestinal tract is involved in about 26% of cases and central nervous system in about 27%. Isolated bone involvement is exceedingly rare.5, 6
Primary lymphoma of the bone is another rare cause of hypercalcemia and accounts for less than 5% of all primary bone tumors.7 The majority of cases are of the non‐Hodgkin's type, characterized as diffuse large B‐cell lymphomas, with peak occurrence in the sixth to seventh decades of life.8 The classic imaging findings of primary lymphoma of the bone are a solitary metadiaphyseal lesion with a layered periosteal reaction on plain radiographs, and corresponding surrounding soft‐tissue mass on MRI.9 Less commonly, primary lymphoma of the bone can be multifocal with diffuse osseous involvement and variable radiographic appearances, as in this case. Most series have reported that the long bones are affected most frequently (especially the femur), although a large series showed equal numbers of cases presenting in the long bones and the spine.712
In order to diagnose primary lymphoma of the bone, it is necessary to exclude nodal or disseminated disease by physical examination and imaging. As plain films are often normal, bone scan or MRI of clinically affected areas is necessary to establish disease extent.9 Distinguishing primary bone lymphomas (PLB) from other bone tumors is important because PLB has a better response to therapy and a better prognosis.10, 11
Randomized trials addressing treatment options for primary lymphoma of bone are not available. Historically, PLB was treated with radiotherapy alone with good local control. However, the rate of distant relapses was relatively high. Currently, chemotherapy with or without radiation therapy is preferred; 5‐year survival is approximately 70% after combined therapy.10, 11
In this case, symptomatic hypercalcemia, a history of transplantation, marked elevation of both LDH and B2M, and a normal PTH level all pointed toward the correct diagnosis of malignancy. Low or normal levels of vitamin D metabolites and PTH‐related protein occur in 20% of patients with hypercalcemia caused by malignancy.13, 14 Diffuse osteopenia on skeletal survey is a prominent feature of renal osteodystrophy or osteoporosis related to chronic corticosteroid use. However, in a patient with diffuse osteopenia and hypercalcemia, clinicians must consider multiple myeloma and other lymphoproliferative disorders; the absence of osteoblastic or osteolytic lesions and a normal alkaline phosphatase do not rule out these diagnoses. When the results of serum and urine protein electrophoresis exclude multiple myeloma, the next investigation should be a bone biopsy to exclude PLB, an uncommon cause of anemia, hypercalcemia, and osteopenic, painful bones.
Key Points for Hospitalists
-
Normal total alkaline phosphatase does not exclude primary or metastatic bone malignancy. While a normal level tends to predict a negative bone scan, further diagnostic tests are needed to exclude bone malignancy if high clinical suspicion exists.
-
The degree of hypercalcemia is useful diagnostically; values above 13 mg/dL are most often due to malignancy.
-
Hypercalcemia in transplant patients deserves special attention due to an increased risk of malignancy, including squamous cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma.
-
While rare, consider primary lymphoma of the bone in patients with hypercalcemia and bone pain, along with the more common diagnoses of multiple myeloma and metastatic bone disease.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
- ,.Hypercalcemia: clinical manifestations, pathogenesis, diagnosis, and management. In: Favus MJ, ed.Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism.5th ed.Washington, DC:American Society for Bone and Mineral Research;2003:225–230.
- ,,, et al.Malignancy after renal transplantation: analysis of incidence and risk factors in 1700 patients followed during a 25‐year period.Transplant Proc.1997;29:831–833.
- ,.Malignancy‐associated hypercalcemia. In: DeGroot L, Jameson LJ, eds.Endocrinology.4th ed.Philadelphia, PA:Saunders;2001:1093–1100.
- ,.Diagnosis and management of posttransplant lymphoproliferative disorder in solid‐organ transplant recipients.Clin Infect Dis.2001;33(suppl 1):S38–S46.
- ,,, et al.Epstein‐Barr virus‐induced posttransplant lymphoproliferative disorders: ASTS/ASTP EBV‐PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting.Transplantation.1999;68:1517–1525.
- ,,, et al.Primary bone lymphoma: a new and detailed characterization of 28 patients in a single‐institution study.Jpn J Clin Oncol.2007;37(3):216–223.
- ,,, et al.Diffuse large B‐cell lymphoma of bone. An analysis of differentiation‐associated antigens with clinical correlation.Am J Surg Pathol.2003;27:1269–1277.
- ,,,,,.Primary bone lymphoma: radiographic‐MR imaging correlation.Radiographics.2003;23:1371–1383.
- ,,, et al.Primary bone lymphoma in 24 patients treated between 1955 and 1999.Clin Orthop.2002;397:271–280.
- ,,, et al.A clinicopathological retrospective study of 131 patients with primary bone lymphoma: a population‐based study of successively treated cohorts from the British Columbia Cancer Agency.Ann Oncol.2007;18:129.
- ,,, et al.Malignant lymphoma of bone.Cancer.1986;58:2646–2655.
- .Hypercalcemia in malignant lymphoma and leukemia.Ann N Y Acad Sci.1974;230:240–246.
- .Incidence and prognostic significance of hypercalcemia in B‐cell non‐Hodgkin's lymphoma. [Letter]J Clin Pathol.2002;55:637–638.
A 71‐year‐old man presented to a hospital with a one week history of fatigue, polyuria, and polydipsia. He also reported pain in his back, hips, and ribs, in addition to frequent falls, intermittent confusion, constipation, and a weight loss of 10 pounds over the last 2 weeks. He denied cough, shortness of breath, chest pain, fever, night sweats, headache, and focal weakness.
Polyuria, which is often associated with polydipsia, can be arbitrarily defined as a urine output exceeding 3 L per day. After excluding osmotic diuresis due to uncontrolled diabetes mellitus, the 3 major causes of polyuria are primary polydipsia, central diabetes insipidus, and nephrogenic diabetes insipidus. Approximately 30% to 50% of cases of central diabetes insipidus are idiopathic; however, primary or secondary brain tumors or infiltrative diseases involving the hypothalamic‐pituitary region need to be considered in this 71‐year‐old man. The most common causes of nephrogenic diabetes insipidus in adults are chronic lithium ingestion, hypokalemia, and hypercalcemia. The patient describes symptoms that can result from severe hypercalcemia, including fatigue, confusion, constipation, polyuria, and polydipsia.
The patient's past medical history included long‐standing, insulin‐requiring type 2 diabetes with associated complications including coronary artery disease, transient ischemic attacks, proliferative retinopathy, peripheral diabetic neuropathy, and nephropathy. Seven years prior to presentation, he received a cadaveric renal transplant that was complicated by BK virus (polyomavirus) nephropathy and secondary hyperparathyroidism. Three years after his transplant surgery, he developed squamous cell carcinoma of the skin, which was treated with local surgical resection. Two years after that, he developed stage I laryngeal cancer of the glottis and received laser surgery, and since then he had been considered disease‐free. He also had a history of hypertension, hypercholesterolemia, osteoporosis, and depression. His medications included aspirin, amlodipine, metoprolol succinate, valsartan, furosemide, simvastatin, insulin, prednisone, sirolimus, and sulfamethoxazole/trimethoprim. He was a married psychiatrist. He denied tobacco use and reported occasional alcohol use.
The prolonged immunosuppressive therapy that is required following organ transplantation carries a markedly increased risk of the subsequent development of malignant tumors, including cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma. Primary brain lymphoma resulting in central diabetes insipidus would be unlikely in the absence of headache or focal weakness. An increased risk of lung cancer occurs in recipients of heart and lung transplants, and to a much lesser degree, recipients of kidney transplants. However, metastatic lung cancer is less likely in the absence of respiratory symptoms and smoking history (present in approximately 90% of all lung cancers). Nephrogenic diabetes insipidus, in its mild form, is relatively common in elderly patients with acute or chronic renal insufficiency because of a reduction in maximum urinary concentrating ability. On the other hand, this alone does not explain his remaining symptoms. The instinctive diagnosis in this case is tertiary hyperparathyroidism due to progression of untreated secondary hyperparathyroidism. This causes hypercalcemia, nephrogenic diabetes insipidus, and significant bone pain related to renal osteodystrophy.
On physical exam, the patient appeared chronically ill, but was in no acute distress. He weighed 197.6 pounds and his height was 70.5 inches. He was afebrile with a blood pressure of 146/82 mm Hg, a heart rate of 76 beats per minute, a respiratory rate of 12 breaths per minute, and an oxygen saturation of 97% while breathing room air. He had no generalized lymphadenopathy. Thyroid examination was unremarkable. Examination of the lungs, heart, abdomen, and lower extremities was normal. The rectal examination revealed no masses or prostate nodules; a test for fecal occult blood was negative. He had loss of sensation to light touch and vibration in the feet with absent Achilles deep tendon reflexes. He had a poorly healing surgical wound on his forehead at the site of his prior skin cancer, but no rash or other lesions. There was no joint swelling or erythema. There were tender points over the cervical, thoracic, and lumbar spine; on multiple ribs; and on the pelvic rims.
Perhaps of greatest importance is the lack of lymphadenopathy, organomegaly, or other findings suggestive of diffuse lymphoproliferative disease. His multifocal bone tenderness is concerning for renal osteodystrophy, multiple myeloma, or primary or metastatic bone disease. Cancers in men that metastasize to the bone usually originate from the prostate, lung, kidney, or thyroid gland. In any case, his physical examination did not reveal an enlarged, asymmetric, or nodular prostate or thyroid gland. I recommend a chest film to rule out primary lung malignancy and a basic laboratory evaluation to narrow down the differential diagnosis.
A complete blood count showed a normocytic anemia with a hemoglobin of 8.7 g/dL and a hematocrit of 25%. Other laboratory tests revealed the following values: sodium, 139 mmol/L; potassium, 4.1 mmol/L; blood urea nitrogen, 70 mg/dL; creatinine, 3.5 mg/dL (most recent value 2 months ago was 1.9 mg/dL); total calcium, 13.2 mg/dL (normal range, 8.5‐10.5 mg/dL); phosphate, 5.3 mg/dL; magnesium, 2.5 mg/dL; total bilirubin, 0.5 mg/dL; alkaline phosphatase, 130 U/L; aspartate aminotransferase, 28 U/L; alanine aminotransferase, 19 U/L; albumin, 3.5 g/dL; and lactate dehydrogenase (LDH), 1258 IU/L (normal range, 105‐333 IU/L). A chest radiograph was normal.
The most important laboratory findings are severe hypercalcemia, acute on chronic renal failure, and anemia. Hypercalcemia most commonly results from malignancy or hyperparathyroidism. Less frequently, hypercalcemia may result from sarcoidosis, vitamin D intoxication, or hyperthyroidism. The degree of hypercalcemia is useful diagnostically as hyperparathyroidism commonly results in mild hypercalcemia (serum calcium concentration often below 11 mg/dL). Values above 13 mg/dL are unusual in hyperparathyroidism and are most often due to malignancy. Malignancy is often evident clinically by the time it causes hypercalcemia, and patients with hypercalcemia of malignancy are more often symptomatic than those with hyperparathyroidism. Additionally, localized bone pain and weight loss do not result from hypercalcemia itself and their presence also raises concern for malignancy.
Nonmelanoma skin cancer is the most common cancer occurring after transplantation but does not cause hypercalcemia. Squamous cancers of the head and neck can rarely cause hypercalcemia due to secretion of parathyroid hormone‐related peptide; however, his early‐stage laryngeal cancer and the expected high likelihood of cure argue against this possibility. Osteolytic metastases account for approximately 20% of cases of hypercalcemia of malignancy (Table 1). Prostate cancer rarely results in hypercalcemia since bone metastases are predominantly osteoblastic, whereas metastatic non‐small‐cell lung cancer, thyroid cancer, and kidney cancer more commonly cause hypercalcemia due to osteolytic bone lesions. The total alkaline phosphatase has been traditionally used to assess the osteoblastic component of bone remodeling. Its normal level tends to predict a negative bone scan and supports the likelihood of lytic lesions. Posttransplantation lymphoproliferative disorders, which include a wide range of syndromes, can rarely result in hypercalcemia. I am also worried about the possibility of multiple myeloma as he has the classic triad of hypercalcemia, bone pain, and subacute kidney injury.
|
| Osteolytic metastases |
| Breast cancer |
| Multiple myeloma |
| Lymphoma |
| Leukemia |
| Humoral hypercalcemia (PTH‐related protein) |
| Squamous cell carcinomas |
| Renal carcinomas |
| Bladder carcinoma |
| Breast cancer |
| Ovarian carcinoma |
| Leukemia |
| Lymphoma |
| 1,25‐Dihydroxyvitamin D secretion |
| Lymphoma |
| Ovarian dysgerminomas |
| Ectopic PTH secretion (rare) |
| Ovarian carcinoma |
| Lung carcinomas |
| Neuroectodermal tumor |
| Thyroid papillary carcinoma |
| Rhabdomyosarcoma |
| Pancreatic cancer |
The first purpose of the laboratory evaluation is to differentiate parathyroid hormone (PTH)‐mediated hypercalcemia (primary and tertiary hyperparathyroidism) from non‐PTH‐mediated hypercalcemia (primarily malignancy, hyperthyroidism, vitamin D intoxication, and granulomatous disease). The production of vitamin D metabolites, PTH‐related protein, or hypercalcemia from osteolysis in these latter cases results in suppressed PTH levels.
In severe elevations of calcium, the initial goals of treatment are directed toward fluid resuscitation with normal saline and, unless contraindicated, the immediate institution of bisphosphonate therapy. A loop diuretic such as furosemide is often used, but a recent review concluded that there is little evidence to support its use in this setting.
The patient was admitted and treated with intravenous saline and furosemide. Additional laboratory evaluation revealed normal levels of prostate‐specific antigen and thyroid‐stimulating hormone. PTH was 44 pg/mL (the most recent value was 906 pg/mL eight years ago; normal range, 15‐65 pg/mL) and beta‐2 microglobulin (B2M) was 8 mg/L (normal range, 0.8‐2.2 mg/L).
The normal PTH level makes tertiary hyperparathyroidism unlikely and points toward non‐PTH‐related hypercalcemia. An elevated B2M level may occur in patients with chronic graft rejection, renal tubular dysfunction, dialysis‐related amyloidosis, multiple myeloma, or lymphoma. LDH is often elevated in patients with multiple myeloma and lymphoma, but this is not a specific finding. The next laboratory test would be measurement of PTH‐related protein and vitamin D metabolites, as these tests can differentiate between the causes of non‐PTH‐mediated hypercalcemia.
Serum concentrations of the vitamin D metabolites, 25‐hydroxyvitamin D (calcidiol) and 1,25‐dihydroxyvitamin D (calcitriol), were low‐normal. PTH‐related protein was not detected.
The marked elevation of serum LDH and B2M, the relatively suppressed PTH level, combined with undetectable PTH‐related protein suggest multiple myeloma or lymphoma as the likely cause of the patient's clinical presentation. The combination of hypercalcemia and multifocal bone pain makes multiple myeloma the leading diagnosis as hypercalcemia is uncommon in patients with lymphoma, especially at the time of initial clinical presentation.
I would proceed with serum and urine protein electrophoresis (SPEP and UPEP, respectively) and a skeletal survey. If these tests do not confirm the diagnosis of multiple myeloma, I would order a noncontrast computed tomography (CT) of the chest and abdomen and a magnetic resonance imaging (MRI) of the spine. In addition, I would like to monitor his response to the intravenous saline and furosemide.
Forty‐eight hours after presentation, repeat serum calcium and creatinine levels were 11.3 mg/dL and 2.9 mg/dL, respectively. He received salmon calcitonin 4 U/kg every 12 hours. Pamidronate was avoided because of his kidney disease. His confusion resolved. He received intravenous morphine intermittently to alleviate his bone pain.
The SPEP revealed a monoclonal immunoglobulin G (IgG) lambda (light chain) spike representing roughly 3% (200 mg/dL) of total protein. His serum Ig levels were normal. The UPEP was negative for monoclonal immunoglobulin and Bence‐Jones protein. The skeletal survey revealed marked osteopenia, and the bone scan was normal. An MRI of the spine showed multiple round lesions in the cervical, thoracic, and lumbar spine (Figure 1). A CT of the chest showed similar bone lesions in the ribs and pelvis. A CT of the abdomen and chest did not suggest any primary malignancy nor did it show thoracic or abdominal lymphadenopathy.
The lack of lymphadenopathy, splenomegaly, or a visceral mass by CT imaging and physical examination, along with the normal PSA level, exclude most common forms of non‐Hodgkin lymphoma and bone metastasis from solid tumors. In multiple myeloma, cytokines secreted by plasma cells suppress osteoblast activity; therefore, while discrete lytic bone lesions are apparent on skeletal survey, the bone scan is typically normal. The absence of lytic lesions, normal serum immunoglobulin levels, and unremarkable UPEP make multiple myeloma or light‐chain deposition disease a less likely diagnosis.
Typically, primary lymphoma of the bone produces increased uptake with bone scanning. However, because primary lymphoma of the bone is one of the least common primary skeletal malignancies and varies widely in appearance on imaging, confident diagnosis based on imaging alone usually is not possible.
Posttransplantation lymphoproliferative disorder (PTLD) refers to a syndrome that ranges from a self‐limited form of lymphoproliferation to an aggressive disseminated disease. Although the patient is at risk for PTLD, isolated bone involvement has only rarely been reported.
Primary lymphoma of the bone and PTLD are my leading diagnoses in this patient. At this point, I recommend a bone marrow biopsy and biopsy of an easily accessible representative bone lesion with special staining for Epstein‐Barr virus (EBV) (EBV‐encoded RNA [EBER] and latent membrane protein 1 [LMP1]). I expect this test to provide a definitive diagnosis. As 95% of PTLD cases are induced by infection with EBV, information regarding pretransplantation EBV status of the patient and the donor, current EBV status of the patient, and type and intensity of immunosuppression at the time of transplantation would be very helpful to determine their likelihood.
Seventy‐two hours after presentation, his serum calcium level normalized and most of his symptoms improved. Calcitonin was discontinued, and he was maintained on oral hydration. On hospital day number 5, he underwent CT‐guided bone biopsy of the L4 vertebral body, which showed large aggregates of atypical lymphoid cells (Figure 2). These cells were predominantly B‐cells interspersed with small reactive T‐cells. The cells did not express EBV LMP1 or EBER (Figure 3). On hospital day 7, he underwent a bone marrow biopsy, which revealed similar large atypical lymphoid cells that comprised the majority of marrow space (Figure 4). By immunohistochemistry, these cells brightly expressed the pan B cell marker, CD20, and coexpressed bcl‐2. EBER and LMP1 were also negative. A flow cytometry of the bone marrow demonstrated a lambda light chain restriction within the B lymphocytes.
The medical records indicated that the patient had positive pretransplantation EBV serologies. He received a regimen based on sirolimus, mycophenolate mofetil, and prednisone, and did not receive high doses of induction or maintenance immunosuppressive therapy.
The biopsy results establish a diagnosis of diffuse large B‐cell lymphoma of the bone. PTLD is unlikely given his positive pretransplantation EBV status, the late onset of his disease (6 years after transplantation), the isolated bone involvement, and the negative EBER and LMP1 tests.
The patient was discharged and was readmitted 1 week later for induction chemotherapy with etoposide, vincristine, doxorubicin, cyclophosphamide, and prednisone [EPOCH]Rituxan (rituximab). Over the next several months, he received 6 cycles of chemotherapy, his hypercalcemia resolved, and his back pain improved.
Commentary
Hypercalcemia is among the most common causes of nephrogenic diabetes insipidus in adults.1 A urinary concentrating defect usually becomes clinically apparent if the plasma calcium concentration is persistently above 11 mg/dL.1 This defect is generally reversible with correction of the hypercalcemia but may persist in patients in whom interstitial nephritis has induced permanent medullary damage. The mechanism by which the concentrating defect occurs is incompletely understood but may be related to impairments in sodium chloride reabsorption in the thick ascending limb and in the ability of antidiuretic hormone to increase water permeability in the collecting tubules.1
Although hypercalcemia in otherwise healthy outpatients is usually due to primary hyperparathyroidism, malignancy is more often responsible for hypercalcemia in hospitalized patients.2 While the signs and symptoms of hypercalcemia are similar regardless of the cause, several clinical features may help distinguish the etiology of hypercalcemia. For instance, the presence of tachycardia, warm skin, thinning of the hair, stare and lid lag, and widened pulse pressure points toward hypercalcemia related to hyperthyroidism. In addition, risk factors and comorbidities guide the diagnostic process. For example, low‐level hypercalcemia in an asymptomatic postmenopausal woman with a normal physical examination suggests primary hyperparathyroidism. In contrast, hypercalcemia in a transplant patient raises concern of malignancy including PTLDs.3, 4
PTLDs are uncommon causes of hypercalcemia but are among the most serious and potentially fatal complications of chronic immunosuppression in transplant recipients.5 They occur in 1.9% of patients after kidney transplantation. The lymphoproliferative disorders occurring after transplantation have different characteristics from those that occur in the general population. Non‐Hodgkin lymphoma accounts for 65% of lymphomas in the general population, compared to 93% in transplant recipients.5, 6 The pathogenesis of PTLD appears to be related to B cell proliferation induced by infection with EBV in the setting of chronic immunosuppression.6 Therefore, there is an increased frequency of PTLD among transplant recipients who are EBV seronegative at the time of operation. These patients, who have no preoperative immunity to EBV, usually acquire the infection from the donor. The level of immunosuppression (intensity and type) influences PTLD rates as well. The disease typically occurs within 12 months after transplantation and in two‐thirds of cases involves extranodal sites. Among these sites, the gastrointestinal tract is involved in about 26% of cases and central nervous system in about 27%. Isolated bone involvement is exceedingly rare.5, 6
Primary lymphoma of the bone is another rare cause of hypercalcemia and accounts for less than 5% of all primary bone tumors.7 The majority of cases are of the non‐Hodgkin's type, characterized as diffuse large B‐cell lymphomas, with peak occurrence in the sixth to seventh decades of life.8 The classic imaging findings of primary lymphoma of the bone are a solitary metadiaphyseal lesion with a layered periosteal reaction on plain radiographs, and corresponding surrounding soft‐tissue mass on MRI.9 Less commonly, primary lymphoma of the bone can be multifocal with diffuse osseous involvement and variable radiographic appearances, as in this case. Most series have reported that the long bones are affected most frequently (especially the femur), although a large series showed equal numbers of cases presenting in the long bones and the spine.712
In order to diagnose primary lymphoma of the bone, it is necessary to exclude nodal or disseminated disease by physical examination and imaging. As plain films are often normal, bone scan or MRI of clinically affected areas is necessary to establish disease extent.9 Distinguishing primary bone lymphomas (PLB) from other bone tumors is important because PLB has a better response to therapy and a better prognosis.10, 11
Randomized trials addressing treatment options for primary lymphoma of bone are not available. Historically, PLB was treated with radiotherapy alone with good local control. However, the rate of distant relapses was relatively high. Currently, chemotherapy with or without radiation therapy is preferred; 5‐year survival is approximately 70% after combined therapy.10, 11
In this case, symptomatic hypercalcemia, a history of transplantation, marked elevation of both LDH and B2M, and a normal PTH level all pointed toward the correct diagnosis of malignancy. Low or normal levels of vitamin D metabolites and PTH‐related protein occur in 20% of patients with hypercalcemia caused by malignancy.13, 14 Diffuse osteopenia on skeletal survey is a prominent feature of renal osteodystrophy or osteoporosis related to chronic corticosteroid use. However, in a patient with diffuse osteopenia and hypercalcemia, clinicians must consider multiple myeloma and other lymphoproliferative disorders; the absence of osteoblastic or osteolytic lesions and a normal alkaline phosphatase do not rule out these diagnoses. When the results of serum and urine protein electrophoresis exclude multiple myeloma, the next investigation should be a bone biopsy to exclude PLB, an uncommon cause of anemia, hypercalcemia, and osteopenic, painful bones.
Key Points for Hospitalists
-
Normal total alkaline phosphatase does not exclude primary or metastatic bone malignancy. While a normal level tends to predict a negative bone scan, further diagnostic tests are needed to exclude bone malignancy if high clinical suspicion exists.
-
The degree of hypercalcemia is useful diagnostically; values above 13 mg/dL are most often due to malignancy.
-
Hypercalcemia in transplant patients deserves special attention due to an increased risk of malignancy, including squamous cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma.
-
While rare, consider primary lymphoma of the bone in patients with hypercalcemia and bone pain, along with the more common diagnoses of multiple myeloma and metastatic bone disease.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
A 71‐year‐old man presented to a hospital with a one week history of fatigue, polyuria, and polydipsia. He also reported pain in his back, hips, and ribs, in addition to frequent falls, intermittent confusion, constipation, and a weight loss of 10 pounds over the last 2 weeks. He denied cough, shortness of breath, chest pain, fever, night sweats, headache, and focal weakness.
Polyuria, which is often associated with polydipsia, can be arbitrarily defined as a urine output exceeding 3 L per day. After excluding osmotic diuresis due to uncontrolled diabetes mellitus, the 3 major causes of polyuria are primary polydipsia, central diabetes insipidus, and nephrogenic diabetes insipidus. Approximately 30% to 50% of cases of central diabetes insipidus are idiopathic; however, primary or secondary brain tumors or infiltrative diseases involving the hypothalamic‐pituitary region need to be considered in this 71‐year‐old man. The most common causes of nephrogenic diabetes insipidus in adults are chronic lithium ingestion, hypokalemia, and hypercalcemia. The patient describes symptoms that can result from severe hypercalcemia, including fatigue, confusion, constipation, polyuria, and polydipsia.
The patient's past medical history included long‐standing, insulin‐requiring type 2 diabetes with associated complications including coronary artery disease, transient ischemic attacks, proliferative retinopathy, peripheral diabetic neuropathy, and nephropathy. Seven years prior to presentation, he received a cadaveric renal transplant that was complicated by BK virus (polyomavirus) nephropathy and secondary hyperparathyroidism. Three years after his transplant surgery, he developed squamous cell carcinoma of the skin, which was treated with local surgical resection. Two years after that, he developed stage I laryngeal cancer of the glottis and received laser surgery, and since then he had been considered disease‐free. He also had a history of hypertension, hypercholesterolemia, osteoporosis, and depression. His medications included aspirin, amlodipine, metoprolol succinate, valsartan, furosemide, simvastatin, insulin, prednisone, sirolimus, and sulfamethoxazole/trimethoprim. He was a married psychiatrist. He denied tobacco use and reported occasional alcohol use.
The prolonged immunosuppressive therapy that is required following organ transplantation carries a markedly increased risk of the subsequent development of malignant tumors, including cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma. Primary brain lymphoma resulting in central diabetes insipidus would be unlikely in the absence of headache or focal weakness. An increased risk of lung cancer occurs in recipients of heart and lung transplants, and to a much lesser degree, recipients of kidney transplants. However, metastatic lung cancer is less likely in the absence of respiratory symptoms and smoking history (present in approximately 90% of all lung cancers). Nephrogenic diabetes insipidus, in its mild form, is relatively common in elderly patients with acute or chronic renal insufficiency because of a reduction in maximum urinary concentrating ability. On the other hand, this alone does not explain his remaining symptoms. The instinctive diagnosis in this case is tertiary hyperparathyroidism due to progression of untreated secondary hyperparathyroidism. This causes hypercalcemia, nephrogenic diabetes insipidus, and significant bone pain related to renal osteodystrophy.
On physical exam, the patient appeared chronically ill, but was in no acute distress. He weighed 197.6 pounds and his height was 70.5 inches. He was afebrile with a blood pressure of 146/82 mm Hg, a heart rate of 76 beats per minute, a respiratory rate of 12 breaths per minute, and an oxygen saturation of 97% while breathing room air. He had no generalized lymphadenopathy. Thyroid examination was unremarkable. Examination of the lungs, heart, abdomen, and lower extremities was normal. The rectal examination revealed no masses or prostate nodules; a test for fecal occult blood was negative. He had loss of sensation to light touch and vibration in the feet with absent Achilles deep tendon reflexes. He had a poorly healing surgical wound on his forehead at the site of his prior skin cancer, but no rash or other lesions. There was no joint swelling or erythema. There were tender points over the cervical, thoracic, and lumbar spine; on multiple ribs; and on the pelvic rims.
Perhaps of greatest importance is the lack of lymphadenopathy, organomegaly, or other findings suggestive of diffuse lymphoproliferative disease. His multifocal bone tenderness is concerning for renal osteodystrophy, multiple myeloma, or primary or metastatic bone disease. Cancers in men that metastasize to the bone usually originate from the prostate, lung, kidney, or thyroid gland. In any case, his physical examination did not reveal an enlarged, asymmetric, or nodular prostate or thyroid gland. I recommend a chest film to rule out primary lung malignancy and a basic laboratory evaluation to narrow down the differential diagnosis.
A complete blood count showed a normocytic anemia with a hemoglobin of 8.7 g/dL and a hematocrit of 25%. Other laboratory tests revealed the following values: sodium, 139 mmol/L; potassium, 4.1 mmol/L; blood urea nitrogen, 70 mg/dL; creatinine, 3.5 mg/dL (most recent value 2 months ago was 1.9 mg/dL); total calcium, 13.2 mg/dL (normal range, 8.5‐10.5 mg/dL); phosphate, 5.3 mg/dL; magnesium, 2.5 mg/dL; total bilirubin, 0.5 mg/dL; alkaline phosphatase, 130 U/L; aspartate aminotransferase, 28 U/L; alanine aminotransferase, 19 U/L; albumin, 3.5 g/dL; and lactate dehydrogenase (LDH), 1258 IU/L (normal range, 105‐333 IU/L). A chest radiograph was normal.
The most important laboratory findings are severe hypercalcemia, acute on chronic renal failure, and anemia. Hypercalcemia most commonly results from malignancy or hyperparathyroidism. Less frequently, hypercalcemia may result from sarcoidosis, vitamin D intoxication, or hyperthyroidism. The degree of hypercalcemia is useful diagnostically as hyperparathyroidism commonly results in mild hypercalcemia (serum calcium concentration often below 11 mg/dL). Values above 13 mg/dL are unusual in hyperparathyroidism and are most often due to malignancy. Malignancy is often evident clinically by the time it causes hypercalcemia, and patients with hypercalcemia of malignancy are more often symptomatic than those with hyperparathyroidism. Additionally, localized bone pain and weight loss do not result from hypercalcemia itself and their presence also raises concern for malignancy.
Nonmelanoma skin cancer is the most common cancer occurring after transplantation but does not cause hypercalcemia. Squamous cancers of the head and neck can rarely cause hypercalcemia due to secretion of parathyroid hormone‐related peptide; however, his early‐stage laryngeal cancer and the expected high likelihood of cure argue against this possibility. Osteolytic metastases account for approximately 20% of cases of hypercalcemia of malignancy (Table 1). Prostate cancer rarely results in hypercalcemia since bone metastases are predominantly osteoblastic, whereas metastatic non‐small‐cell lung cancer, thyroid cancer, and kidney cancer more commonly cause hypercalcemia due to osteolytic bone lesions. The total alkaline phosphatase has been traditionally used to assess the osteoblastic component of bone remodeling. Its normal level tends to predict a negative bone scan and supports the likelihood of lytic lesions. Posttransplantation lymphoproliferative disorders, which include a wide range of syndromes, can rarely result in hypercalcemia. I am also worried about the possibility of multiple myeloma as he has the classic triad of hypercalcemia, bone pain, and subacute kidney injury.
|
| Osteolytic metastases |
| Breast cancer |
| Multiple myeloma |
| Lymphoma |
| Leukemia |
| Humoral hypercalcemia (PTH‐related protein) |
| Squamous cell carcinomas |
| Renal carcinomas |
| Bladder carcinoma |
| Breast cancer |
| Ovarian carcinoma |
| Leukemia |
| Lymphoma |
| 1,25‐Dihydroxyvitamin D secretion |
| Lymphoma |
| Ovarian dysgerminomas |
| Ectopic PTH secretion (rare) |
| Ovarian carcinoma |
| Lung carcinomas |
| Neuroectodermal tumor |
| Thyroid papillary carcinoma |
| Rhabdomyosarcoma |
| Pancreatic cancer |
The first purpose of the laboratory evaluation is to differentiate parathyroid hormone (PTH)‐mediated hypercalcemia (primary and tertiary hyperparathyroidism) from non‐PTH‐mediated hypercalcemia (primarily malignancy, hyperthyroidism, vitamin D intoxication, and granulomatous disease). The production of vitamin D metabolites, PTH‐related protein, or hypercalcemia from osteolysis in these latter cases results in suppressed PTH levels.
In severe elevations of calcium, the initial goals of treatment are directed toward fluid resuscitation with normal saline and, unless contraindicated, the immediate institution of bisphosphonate therapy. A loop diuretic such as furosemide is often used, but a recent review concluded that there is little evidence to support its use in this setting.
The patient was admitted and treated with intravenous saline and furosemide. Additional laboratory evaluation revealed normal levels of prostate‐specific antigen and thyroid‐stimulating hormone. PTH was 44 pg/mL (the most recent value was 906 pg/mL eight years ago; normal range, 15‐65 pg/mL) and beta‐2 microglobulin (B2M) was 8 mg/L (normal range, 0.8‐2.2 mg/L).
The normal PTH level makes tertiary hyperparathyroidism unlikely and points toward non‐PTH‐related hypercalcemia. An elevated B2M level may occur in patients with chronic graft rejection, renal tubular dysfunction, dialysis‐related amyloidosis, multiple myeloma, or lymphoma. LDH is often elevated in patients with multiple myeloma and lymphoma, but this is not a specific finding. The next laboratory test would be measurement of PTH‐related protein and vitamin D metabolites, as these tests can differentiate between the causes of non‐PTH‐mediated hypercalcemia.
Serum concentrations of the vitamin D metabolites, 25‐hydroxyvitamin D (calcidiol) and 1,25‐dihydroxyvitamin D (calcitriol), were low‐normal. PTH‐related protein was not detected.
The marked elevation of serum LDH and B2M, the relatively suppressed PTH level, combined with undetectable PTH‐related protein suggest multiple myeloma or lymphoma as the likely cause of the patient's clinical presentation. The combination of hypercalcemia and multifocal bone pain makes multiple myeloma the leading diagnosis as hypercalcemia is uncommon in patients with lymphoma, especially at the time of initial clinical presentation.
I would proceed with serum and urine protein electrophoresis (SPEP and UPEP, respectively) and a skeletal survey. If these tests do not confirm the diagnosis of multiple myeloma, I would order a noncontrast computed tomography (CT) of the chest and abdomen and a magnetic resonance imaging (MRI) of the spine. In addition, I would like to monitor his response to the intravenous saline and furosemide.
Forty‐eight hours after presentation, repeat serum calcium and creatinine levels were 11.3 mg/dL and 2.9 mg/dL, respectively. He received salmon calcitonin 4 U/kg every 12 hours. Pamidronate was avoided because of his kidney disease. His confusion resolved. He received intravenous morphine intermittently to alleviate his bone pain.
The SPEP revealed a monoclonal immunoglobulin G (IgG) lambda (light chain) spike representing roughly 3% (200 mg/dL) of total protein. His serum Ig levels were normal. The UPEP was negative for monoclonal immunoglobulin and Bence‐Jones protein. The skeletal survey revealed marked osteopenia, and the bone scan was normal. An MRI of the spine showed multiple round lesions in the cervical, thoracic, and lumbar spine (Figure 1). A CT of the chest showed similar bone lesions in the ribs and pelvis. A CT of the abdomen and chest did not suggest any primary malignancy nor did it show thoracic or abdominal lymphadenopathy.
The lack of lymphadenopathy, splenomegaly, or a visceral mass by CT imaging and physical examination, along with the normal PSA level, exclude most common forms of non‐Hodgkin lymphoma and bone metastasis from solid tumors. In multiple myeloma, cytokines secreted by plasma cells suppress osteoblast activity; therefore, while discrete lytic bone lesions are apparent on skeletal survey, the bone scan is typically normal. The absence of lytic lesions, normal serum immunoglobulin levels, and unremarkable UPEP make multiple myeloma or light‐chain deposition disease a less likely diagnosis.
Typically, primary lymphoma of the bone produces increased uptake with bone scanning. However, because primary lymphoma of the bone is one of the least common primary skeletal malignancies and varies widely in appearance on imaging, confident diagnosis based on imaging alone usually is not possible.
Posttransplantation lymphoproliferative disorder (PTLD) refers to a syndrome that ranges from a self‐limited form of lymphoproliferation to an aggressive disseminated disease. Although the patient is at risk for PTLD, isolated bone involvement has only rarely been reported.
Primary lymphoma of the bone and PTLD are my leading diagnoses in this patient. At this point, I recommend a bone marrow biopsy and biopsy of an easily accessible representative bone lesion with special staining for Epstein‐Barr virus (EBV) (EBV‐encoded RNA [EBER] and latent membrane protein 1 [LMP1]). I expect this test to provide a definitive diagnosis. As 95% of PTLD cases are induced by infection with EBV, information regarding pretransplantation EBV status of the patient and the donor, current EBV status of the patient, and type and intensity of immunosuppression at the time of transplantation would be very helpful to determine their likelihood.
Seventy‐two hours after presentation, his serum calcium level normalized and most of his symptoms improved. Calcitonin was discontinued, and he was maintained on oral hydration. On hospital day number 5, he underwent CT‐guided bone biopsy of the L4 vertebral body, which showed large aggregates of atypical lymphoid cells (Figure 2). These cells were predominantly B‐cells interspersed with small reactive T‐cells. The cells did not express EBV LMP1 or EBER (Figure 3). On hospital day 7, he underwent a bone marrow biopsy, which revealed similar large atypical lymphoid cells that comprised the majority of marrow space (Figure 4). By immunohistochemistry, these cells brightly expressed the pan B cell marker, CD20, and coexpressed bcl‐2. EBER and LMP1 were also negative. A flow cytometry of the bone marrow demonstrated a lambda light chain restriction within the B lymphocytes.
The medical records indicated that the patient had positive pretransplantation EBV serologies. He received a regimen based on sirolimus, mycophenolate mofetil, and prednisone, and did not receive high doses of induction or maintenance immunosuppressive therapy.
The biopsy results establish a diagnosis of diffuse large B‐cell lymphoma of the bone. PTLD is unlikely given his positive pretransplantation EBV status, the late onset of his disease (6 years after transplantation), the isolated bone involvement, and the negative EBER and LMP1 tests.
The patient was discharged and was readmitted 1 week later for induction chemotherapy with etoposide, vincristine, doxorubicin, cyclophosphamide, and prednisone [EPOCH]Rituxan (rituximab). Over the next several months, he received 6 cycles of chemotherapy, his hypercalcemia resolved, and his back pain improved.
Commentary
Hypercalcemia is among the most common causes of nephrogenic diabetes insipidus in adults.1 A urinary concentrating defect usually becomes clinically apparent if the plasma calcium concentration is persistently above 11 mg/dL.1 This defect is generally reversible with correction of the hypercalcemia but may persist in patients in whom interstitial nephritis has induced permanent medullary damage. The mechanism by which the concentrating defect occurs is incompletely understood but may be related to impairments in sodium chloride reabsorption in the thick ascending limb and in the ability of antidiuretic hormone to increase water permeability in the collecting tubules.1
Although hypercalcemia in otherwise healthy outpatients is usually due to primary hyperparathyroidism, malignancy is more often responsible for hypercalcemia in hospitalized patients.2 While the signs and symptoms of hypercalcemia are similar regardless of the cause, several clinical features may help distinguish the etiology of hypercalcemia. For instance, the presence of tachycardia, warm skin, thinning of the hair, stare and lid lag, and widened pulse pressure points toward hypercalcemia related to hyperthyroidism. In addition, risk factors and comorbidities guide the diagnostic process. For example, low‐level hypercalcemia in an asymptomatic postmenopausal woman with a normal physical examination suggests primary hyperparathyroidism. In contrast, hypercalcemia in a transplant patient raises concern of malignancy including PTLDs.3, 4
PTLDs are uncommon causes of hypercalcemia but are among the most serious and potentially fatal complications of chronic immunosuppression in transplant recipients.5 They occur in 1.9% of patients after kidney transplantation. The lymphoproliferative disorders occurring after transplantation have different characteristics from those that occur in the general population. Non‐Hodgkin lymphoma accounts for 65% of lymphomas in the general population, compared to 93% in transplant recipients.5, 6 The pathogenesis of PTLD appears to be related to B cell proliferation induced by infection with EBV in the setting of chronic immunosuppression.6 Therefore, there is an increased frequency of PTLD among transplant recipients who are EBV seronegative at the time of operation. These patients, who have no preoperative immunity to EBV, usually acquire the infection from the donor. The level of immunosuppression (intensity and type) influences PTLD rates as well. The disease typically occurs within 12 months after transplantation and in two‐thirds of cases involves extranodal sites. Among these sites, the gastrointestinal tract is involved in about 26% of cases and central nervous system in about 27%. Isolated bone involvement is exceedingly rare.5, 6
Primary lymphoma of the bone is another rare cause of hypercalcemia and accounts for less than 5% of all primary bone tumors.7 The majority of cases are of the non‐Hodgkin's type, characterized as diffuse large B‐cell lymphomas, with peak occurrence in the sixth to seventh decades of life.8 The classic imaging findings of primary lymphoma of the bone are a solitary metadiaphyseal lesion with a layered periosteal reaction on plain radiographs, and corresponding surrounding soft‐tissue mass on MRI.9 Less commonly, primary lymphoma of the bone can be multifocal with diffuse osseous involvement and variable radiographic appearances, as in this case. Most series have reported that the long bones are affected most frequently (especially the femur), although a large series showed equal numbers of cases presenting in the long bones and the spine.712
In order to diagnose primary lymphoma of the bone, it is necessary to exclude nodal or disseminated disease by physical examination and imaging. As plain films are often normal, bone scan or MRI of clinically affected areas is necessary to establish disease extent.9 Distinguishing primary bone lymphomas (PLB) from other bone tumors is important because PLB has a better response to therapy and a better prognosis.10, 11
Randomized trials addressing treatment options for primary lymphoma of bone are not available. Historically, PLB was treated with radiotherapy alone with good local control. However, the rate of distant relapses was relatively high. Currently, chemotherapy with or without radiation therapy is preferred; 5‐year survival is approximately 70% after combined therapy.10, 11
In this case, symptomatic hypercalcemia, a history of transplantation, marked elevation of both LDH and B2M, and a normal PTH level all pointed toward the correct diagnosis of malignancy. Low or normal levels of vitamin D metabolites and PTH‐related protein occur in 20% of patients with hypercalcemia caused by malignancy.13, 14 Diffuse osteopenia on skeletal survey is a prominent feature of renal osteodystrophy or osteoporosis related to chronic corticosteroid use. However, in a patient with diffuse osteopenia and hypercalcemia, clinicians must consider multiple myeloma and other lymphoproliferative disorders; the absence of osteoblastic or osteolytic lesions and a normal alkaline phosphatase do not rule out these diagnoses. When the results of serum and urine protein electrophoresis exclude multiple myeloma, the next investigation should be a bone biopsy to exclude PLB, an uncommon cause of anemia, hypercalcemia, and osteopenic, painful bones.
Key Points for Hospitalists
-
Normal total alkaline phosphatase does not exclude primary or metastatic bone malignancy. While a normal level tends to predict a negative bone scan, further diagnostic tests are needed to exclude bone malignancy if high clinical suspicion exists.
-
The degree of hypercalcemia is useful diagnostically; values above 13 mg/dL are most often due to malignancy.
-
Hypercalcemia in transplant patients deserves special attention due to an increased risk of malignancy, including squamous cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma.
-
While rare, consider primary lymphoma of the bone in patients with hypercalcemia and bone pain, along with the more common diagnoses of multiple myeloma and metastatic bone disease.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
- ,.Hypercalcemia: clinical manifestations, pathogenesis, diagnosis, and management. In: Favus MJ, ed.Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism.5th ed.Washington, DC:American Society for Bone and Mineral Research;2003:225–230.
- ,,, et al.Malignancy after renal transplantation: analysis of incidence and risk factors in 1700 patients followed during a 25‐year period.Transplant Proc.1997;29:831–833.
- ,.Malignancy‐associated hypercalcemia. In: DeGroot L, Jameson LJ, eds.Endocrinology.4th ed.Philadelphia, PA:Saunders;2001:1093–1100.
- ,.Diagnosis and management of posttransplant lymphoproliferative disorder in solid‐organ transplant recipients.Clin Infect Dis.2001;33(suppl 1):S38–S46.
- ,,, et al.Epstein‐Barr virus‐induced posttransplant lymphoproliferative disorders: ASTS/ASTP EBV‐PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting.Transplantation.1999;68:1517–1525.
- ,,, et al.Primary bone lymphoma: a new and detailed characterization of 28 patients in a single‐institution study.Jpn J Clin Oncol.2007;37(3):216–223.
- ,,, et al.Diffuse large B‐cell lymphoma of bone. An analysis of differentiation‐associated antigens with clinical correlation.Am J Surg Pathol.2003;27:1269–1277.
- ,,,,,.Primary bone lymphoma: radiographic‐MR imaging correlation.Radiographics.2003;23:1371–1383.
- ,,, et al.Primary bone lymphoma in 24 patients treated between 1955 and 1999.Clin Orthop.2002;397:271–280.
- ,,, et al.A clinicopathological retrospective study of 131 patients with primary bone lymphoma: a population‐based study of successively treated cohorts from the British Columbia Cancer Agency.Ann Oncol.2007;18:129.
- ,,, et al.Malignant lymphoma of bone.Cancer.1986;58:2646–2655.
- .Hypercalcemia in malignant lymphoma and leukemia.Ann N Y Acad Sci.1974;230:240–246.
- .Incidence and prognostic significance of hypercalcemia in B‐cell non‐Hodgkin's lymphoma. [Letter]J Clin Pathol.2002;55:637–638.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
- ,.Hypercalcemia: clinical manifestations, pathogenesis, diagnosis, and management. In: Favus MJ, ed.Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism.5th ed.Washington, DC:American Society for Bone and Mineral Research;2003:225–230.
- ,,, et al.Malignancy after renal transplantation: analysis of incidence and risk factors in 1700 patients followed during a 25‐year period.Transplant Proc.1997;29:831–833.
- ,.Malignancy‐associated hypercalcemia. In: DeGroot L, Jameson LJ, eds.Endocrinology.4th ed.Philadelphia, PA:Saunders;2001:1093–1100.
- ,.Diagnosis and management of posttransplant lymphoproliferative disorder in solid‐organ transplant recipients.Clin Infect Dis.2001;33(suppl 1):S38–S46.
- ,,, et al.Epstein‐Barr virus‐induced posttransplant lymphoproliferative disorders: ASTS/ASTP EBV‐PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting.Transplantation.1999;68:1517–1525.
- ,,, et al.Primary bone lymphoma: a new and detailed characterization of 28 patients in a single‐institution study.Jpn J Clin Oncol.2007;37(3):216–223.
- ,,, et al.Diffuse large B‐cell lymphoma of bone. An analysis of differentiation‐associated antigens with clinical correlation.Am J Surg Pathol.2003;27:1269–1277.
- ,,,,,.Primary bone lymphoma: radiographic‐MR imaging correlation.Radiographics.2003;23:1371–1383.
- ,,, et al.Primary bone lymphoma in 24 patients treated between 1955 and 1999.Clin Orthop.2002;397:271–280.
- ,,, et al.A clinicopathological retrospective study of 131 patients with primary bone lymphoma: a population‐based study of successively treated cohorts from the British Columbia Cancer Agency.Ann Oncol.2007;18:129.
- ,,, et al.Malignant lymphoma of bone.Cancer.1986;58:2646–2655.
- .Hypercalcemia in malignant lymphoma and leukemia.Ann N Y Acad Sci.1974;230:240–246.
- .Incidence and prognostic significance of hypercalcemia in B‐cell non‐Hodgkin's lymphoma. [Letter]J Clin Pathol.2002;55:637–638.