User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
div[contains(@class, 'medstat-accordion-set article-series')]
New coalition aims to revolutionize stalled lupus research
Clinical research into lupus has long been hampered by failures of medications that initially seemed promising. Now, a coalition of drugmakers, federal regulators, and activists has come together to forge a path toward better-designed studies and – potentially – groundbreaking new drugs.
“We have an opportunity to work collaboratively in lupus to address the challenges in drug development,” Teodora Staeva, PhD, vice president and chief scientific officer of the Lupus Research Alliance, said in an interview.
The alliance held a press conference on March 29 to announce the formation of the public-private Lupus Accelerating Breakthroughs Consortium. Coalition members include several major drugmakers, lupus organizations such as the LRA, the American College of Rheumatology, the Food and Drug Administration, and other federal agencies. Academic researchers, people living with lupus, caregivers and family members, and other members of the lupus community are also on board.
As Dr. Staeva explained, research into lupus has been marked by a high rate of failure. “Often, phase 2 trial successes have not translated into phase 3 successes,” she said.
But researchers, she said, don’t tend to think this is because the drugs themselves are useless.
Instead, it appears that “trial designs are not adequate to capture meaningful readouts of the drug effects, and that may have contributed to the multiple failures,” she said.
According to her, this may because the trials aren’t yet designed to fully detect whether drugs are useful. This is difficult to accomplish since patients have so many manifestations of the disease and trial participants already take a variety of existing drugs.
“Another major limitation has been the lack of integration of the patient’s voice and needs in the drug development process,” she said. It’s also challenging to recruit patients with the most severe lupus to participate in studies, especially since the trials often last 52 weeks.
The new coalition will not directly develop or favor specific drugs. Instead, it will focus on clinical research priorities. “It’s all open and collaborative,” Dr. Staeva explained, and a patient council will provide input. “We have a unique opportunity to bring the voice of people [living with lupus] to the table for the first time and be able to integrate their needs and priorities into the infrastructure.”
The new coalition was inspired by existing public-private partnerships such as the Kidney Health Initiative, she said. That initiative was founded in 2012 by the FDA and the American Society of Nephrology and has dozens of members, including multiple drugmakers and medical societies.
The leadership of the Lupus ABC coalition will include three nonvoting members from the FDA. They’ll offer guidance, Dr. Staeva said. At the press conference, Albert T. Roy, president and CEO of the LRA, said drug companies will appreciate the opportunity to speak with FDA representatives “in a space that is not competitive with respect to intellectual property or anything like that.”
The coalition will meet later in spring 2023, Dr. Staeva said. She hopes it will launch a couple of projects by the end of 2023 and be able to release preliminary results by the end of 2024.
One challenge will be figuring out how to stratify trial subjects so drug studies will more easily detect medications that may work in smaller populations of patients, Hoang Nguyen, PhD, director of scientific partnerships at the LRA, said in an interview. “Now we lump [patients] all together, and that’s not the optimal way to test drugs on patients who have a lot of differences.”
According to Dr. Staeva, the LRA funded the development of the coalition, and drugmakers will primarily provide financial support going forward. The pharmaceutical company members of the coalition are Biogen, Bristol-Myers Squibb, Eli Lilly, EMD Serono, Genentech, Gilead, GlaxoSmithKline, Merck, and Takeda.
Dr. Staeva, Dr. Nguyen, and Mr. Roy have no disclosures.
Clinical research into lupus has long been hampered by failures of medications that initially seemed promising. Now, a coalition of drugmakers, federal regulators, and activists has come together to forge a path toward better-designed studies and – potentially – groundbreaking new drugs.
“We have an opportunity to work collaboratively in lupus to address the challenges in drug development,” Teodora Staeva, PhD, vice president and chief scientific officer of the Lupus Research Alliance, said in an interview.
The alliance held a press conference on March 29 to announce the formation of the public-private Lupus Accelerating Breakthroughs Consortium. Coalition members include several major drugmakers, lupus organizations such as the LRA, the American College of Rheumatology, the Food and Drug Administration, and other federal agencies. Academic researchers, people living with lupus, caregivers and family members, and other members of the lupus community are also on board.
As Dr. Staeva explained, research into lupus has been marked by a high rate of failure. “Often, phase 2 trial successes have not translated into phase 3 successes,” she said.
But researchers, she said, don’t tend to think this is because the drugs themselves are useless.
Instead, it appears that “trial designs are not adequate to capture meaningful readouts of the drug effects, and that may have contributed to the multiple failures,” she said.
According to her, this may because the trials aren’t yet designed to fully detect whether drugs are useful. This is difficult to accomplish since patients have so many manifestations of the disease and trial participants already take a variety of existing drugs.
“Another major limitation has been the lack of integration of the patient’s voice and needs in the drug development process,” she said. It’s also challenging to recruit patients with the most severe lupus to participate in studies, especially since the trials often last 52 weeks.
The new coalition will not directly develop or favor specific drugs. Instead, it will focus on clinical research priorities. “It’s all open and collaborative,” Dr. Staeva explained, and a patient council will provide input. “We have a unique opportunity to bring the voice of people [living with lupus] to the table for the first time and be able to integrate their needs and priorities into the infrastructure.”
The new coalition was inspired by existing public-private partnerships such as the Kidney Health Initiative, she said. That initiative was founded in 2012 by the FDA and the American Society of Nephrology and has dozens of members, including multiple drugmakers and medical societies.
The leadership of the Lupus ABC coalition will include three nonvoting members from the FDA. They’ll offer guidance, Dr. Staeva said. At the press conference, Albert T. Roy, president and CEO of the LRA, said drug companies will appreciate the opportunity to speak with FDA representatives “in a space that is not competitive with respect to intellectual property or anything like that.”
The coalition will meet later in spring 2023, Dr. Staeva said. She hopes it will launch a couple of projects by the end of 2023 and be able to release preliminary results by the end of 2024.
One challenge will be figuring out how to stratify trial subjects so drug studies will more easily detect medications that may work in smaller populations of patients, Hoang Nguyen, PhD, director of scientific partnerships at the LRA, said in an interview. “Now we lump [patients] all together, and that’s not the optimal way to test drugs on patients who have a lot of differences.”
According to Dr. Staeva, the LRA funded the development of the coalition, and drugmakers will primarily provide financial support going forward. The pharmaceutical company members of the coalition are Biogen, Bristol-Myers Squibb, Eli Lilly, EMD Serono, Genentech, Gilead, GlaxoSmithKline, Merck, and Takeda.
Dr. Staeva, Dr. Nguyen, and Mr. Roy have no disclosures.
Clinical research into lupus has long been hampered by failures of medications that initially seemed promising. Now, a coalition of drugmakers, federal regulators, and activists has come together to forge a path toward better-designed studies and – potentially – groundbreaking new drugs.
“We have an opportunity to work collaboratively in lupus to address the challenges in drug development,” Teodora Staeva, PhD, vice president and chief scientific officer of the Lupus Research Alliance, said in an interview.
The alliance held a press conference on March 29 to announce the formation of the public-private Lupus Accelerating Breakthroughs Consortium. Coalition members include several major drugmakers, lupus organizations such as the LRA, the American College of Rheumatology, the Food and Drug Administration, and other federal agencies. Academic researchers, people living with lupus, caregivers and family members, and other members of the lupus community are also on board.
As Dr. Staeva explained, research into lupus has been marked by a high rate of failure. “Often, phase 2 trial successes have not translated into phase 3 successes,” she said.
But researchers, she said, don’t tend to think this is because the drugs themselves are useless.
Instead, it appears that “trial designs are not adequate to capture meaningful readouts of the drug effects, and that may have contributed to the multiple failures,” she said.
According to her, this may because the trials aren’t yet designed to fully detect whether drugs are useful. This is difficult to accomplish since patients have so many manifestations of the disease and trial participants already take a variety of existing drugs.
“Another major limitation has been the lack of integration of the patient’s voice and needs in the drug development process,” she said. It’s also challenging to recruit patients with the most severe lupus to participate in studies, especially since the trials often last 52 weeks.
The new coalition will not directly develop or favor specific drugs. Instead, it will focus on clinical research priorities. “It’s all open and collaborative,” Dr. Staeva explained, and a patient council will provide input. “We have a unique opportunity to bring the voice of people [living with lupus] to the table for the first time and be able to integrate their needs and priorities into the infrastructure.”
The new coalition was inspired by existing public-private partnerships such as the Kidney Health Initiative, she said. That initiative was founded in 2012 by the FDA and the American Society of Nephrology and has dozens of members, including multiple drugmakers and medical societies.
The leadership of the Lupus ABC coalition will include three nonvoting members from the FDA. They’ll offer guidance, Dr. Staeva said. At the press conference, Albert T. Roy, president and CEO of the LRA, said drug companies will appreciate the opportunity to speak with FDA representatives “in a space that is not competitive with respect to intellectual property or anything like that.”
The coalition will meet later in spring 2023, Dr. Staeva said. She hopes it will launch a couple of projects by the end of 2023 and be able to release preliminary results by the end of 2024.
One challenge will be figuring out how to stratify trial subjects so drug studies will more easily detect medications that may work in smaller populations of patients, Hoang Nguyen, PhD, director of scientific partnerships at the LRA, said in an interview. “Now we lump [patients] all together, and that’s not the optimal way to test drugs on patients who have a lot of differences.”
According to Dr. Staeva, the LRA funded the development of the coalition, and drugmakers will primarily provide financial support going forward. The pharmaceutical company members of the coalition are Biogen, Bristol-Myers Squibb, Eli Lilly, EMD Serono, Genentech, Gilead, GlaxoSmithKline, Merck, and Takeda.
Dr. Staeva, Dr. Nguyen, and Mr. Roy have no disclosures.
Biosimilars and patients: Discussions should address safety, cost, and anxiety about change
Rheumatologist Marcus Snow, MD, is comfortable with prescribing biosimilars as a first-line, first-time biologic, and discussing them with patients.
“If a biosimilar is on the market, it has gone through rigorous study proving its effectiveness and equivalence to a bio-originator,” said Dr. Snow, a rheumatologist with the University of Nebraska Medical Center, Omaha, and chair of the American College of Rheumatology’s Committee on Rheumatologic Care.

The formulary makes a big difference in the conversation about options, he said. “The formularies dictate what we can prescribe. It may not be appropriate, but it is reality. The cost of biologics for a patient without insurance coverage makes it impossible to afford.”
He will often tell patients that he’ll fight any changes or formulary restrictions he does not agree with. “However, when I see patients in follow-up, even if there is no known change on the horizon, I may bring up biosimilars when we have a moment to chat about them to familiarize them with what may happen in the future.”
The need for patient education on biosimilars presents a barrier to realizing their potential to save money and expand choice, noted Cardinal Health in its 2023 biosimilars report. Of 103 rheumatologists who responded to a Cardinal Health survey, 85% agreed that patient education was important. But those conversations can take an uncomfortable turn if the patient pushes back against taking a biosimilar owing to cost or safety concerns.
It’s not uncommon for a patient to express some anxiety about biosimilars, especially if they’re doing well on a current treatment plan. Most patients do not want any changes that may lead to worsening disease control, Dr. Snow said.

Patients and physicians alike often don’t understand the mechanics of biosimilars. “There’s a lot of misinformation about this,” said Sameer Awsare, MD, an associate executive director for The Permanente Medical Group in Campbell, Calif. Patients should know that a biosimilar will be as clinically efficacious as the medicine they’ve been on, with the same safety profiles, said Dr. Awsare, who works with Kaiser Permanente’s pharmacy partners on biosimilars.
Insurance often drives the conversation
The global anti-inflammatory biologics market is anticipated to reach $150 billion by 2027, according to a recent CVS report. As of March 2023, the Food and Drug Administration had approved 40 biosimilars to 11 different reference products. There are 28 on the U.S. market and 100 more in development. Projected to save more than $180 billion over the next 5 years, they are anticipated to expand choice and drive competition.
Rheumatologists, dermatologists, and gastroenterologists are frequent prescribers, although their choices for immune-mediated inflammatory diseases are limited to tumor necrosis factor inhibitors (infliximab [Remicade] originator and adalimumab [Humira] originator) and anti-CD20 agents, such as rituximab (Rituxan) originator.

Benefit design or formulary usually dictates what medicine a patient receives. “Because of significantly higher out-of-pocket cost or formulary positioning, patients may end up with a generic or a biosimilar instead of a brand-name medicine or branded biologic,” said Robert Popovian, PharmD, MS, chief science policy officer of the Global Healthy Living Foundation.
Insurers rarely offer both Remicade and biosimilar infliximab, allowing the doctor to choose, said Miguel Regueiro, MD, chair of the Cleveland Clinic’s Digestive Disease & Surgery Institute, who prescribes infliximab biosimilars. Most often, the payer will choose the lower-cost biosimilar. “I am fine with the biosimilar, either as a new start or a switch from the reference product.”

However, the patient might feel differently. They can form an attachment to the reference medication if it has prevented severe illness. “They do not want to change, as they feel they are going on a ‘new’ medication that will not work as well,” Dr. Regueiro said.
This is where the education comes in: to reassure patients that a biosimilar will work just as well as the reference product. “For patients who have done well for years on a biologic, more time needs to be spent reassuring them and answering questions,” compared with a patient just starting on a biosimilar, he advised.
But not all physicians are quick to prescribe biosimilars.

Especially with psoriasis, which has so many strong options for reference drugs, a switch may be hard to justify, said dermatologist Stephanie K. Fabbro, MD, assistant professor at Northeast Ohio Medical University, Rootstown. “If I have a preference, I would rather switch a patient to a drug from a different class without a biosimilar option to reduce the possibility of pushback.”
Dr. Fabbro, part of the core faculty in the Riverside Methodist Hospital Dermatology Residency Program in Columbus, will share data from clinical trials and postmarket surveillance with patients to support her decision.
Conversations about cost
Patients may also push back if they don’t save money when switching to a biosimilar. “This dilemma raises the question of who is profiting when a biosimilar is dispensed,” Dr. Popovian said. Insurers and pharmacy benefit managers (PBMs) that take additional concessions from biopharmaceutical manufacturers in the form of rebates and fees will often pocket this money as profit instead of passing savings back to the patient to help reduce their out-of-pocket requirement, he added.
If an originator biologic and a biosimilar are available, “as a pharmacist, I will choose the medicine that will incur the lowest out-of-pocket cost for the patient,” Dr. Popovian said.

Discussing cost – and who dictates which biosimilar is on the formulary – is an important conversation to have with patients, said Vivek Kaul, MD, Segal-Watson Professor of Medicine at the University of Rochester (N.Y.) Medical Center.
Providing equivalent clinical efficacy while saving costs is the economic reality of biosimilars, Dr. Kaul said. Third-party payers regularly evaluate how to provide the same quality of care while saving money. Physicians and patients alike “must be mindful that as time goes on, if the science on biosimilars stays robust, if the adoption is more widespread and the cost-saving proposition turns out to be true, more formularies will be attracted to replacing the reference product with the biosimilar counterpart.”
Providers and patients can weigh the options if a formulary suddenly switches to a biosimilar, Dr. Kaul continued. “You can accept the novel product on the formulary or may have to face out-of-pocket expenses as a patient.” If providers and patients have concerns about the biosimilar, they can always appeal if there’s solid scientific evidence that supports reverting back to the reference product.
“If you think the biosimilar is equally efficacious, comes at a lower cost, and is right for the patient, then the providers should tell the patient that,” he added.
Some studies have questioned whether the biosimilars will save money, compared with the reference drug, Dr. Fabbro noted. Medicare, for example, may pay only for a certain percentage of an approved biosimilar, saddling the patient with a monthly copay costing thousands of dollars. “It is unclear whether biosimilar manufacturers will have the same level of patient support programs as the reference drug companies.”
For that reason, physicians should also inform patients about the robust patient assistance and copay assistance programs many reference drug manufacturers offer, she said.
Biosimilars 101: Familiarizing patients
Safety and ease of use are other common concerns about biosimilars. Patients may ask if the application is different, or why it’s advantageous to switch to a biosimilar, Dr. Awsare said.
Sometimes the syringe or injector for a biosimilar might look different from that of the originator drug, he said.
Anecdotally, Dr. Fabbro has heard stories of patients having injection reactions that they did not experience with the reference drug or having a disease flare-up after starting a biosimilar. 
As is the case with reference products, in their conversations with patients, clinicians should address the adverse event profile of biosimilars, offering data points from published studies and clinical guidelines that support the use of these products. “There should be an emphasis on patient education around efficacy and any side effects, and how the profile of the reference product compares with a proposed biosimilar,” Dr. Kaul suggested.
When Dr. Snow discusses biosimilars and generics, “I make sure to share this in an understandable way based on the patient’s scientific background, or lack thereof,” he said. If there is enough time, he also discusses how European- and U.S.-sourced biologics are slightly different.
Pharmacists should tell patients to expect the same clinical outcomes from a biosimilar, Dr. Popovian said. However, if they have any reduction in efficacy or potential safety concerns, they should communicate with their physician or pharmacist immediately.
In Dr. Regueiro’s practice, a pharmacist specializing in inflammatory bowel disease often has a one-on-one meeting with patients to educate and answer questions. “Additionally, we provide them the Crohn’s and Colitis Foundation web link on biosimilars,” said Dr. Regueiro.
A village approach to education
When biosimilars first came out, there were no formal education materials, Dr. Awsare said. Kaiser Permanente decided to create its own educational materials, not just for patients but also to help educate its primary care doctors; the rheumatologists, dermatologists, and gastroenterologists using the biosimilars; the nurses infusing patients; and the pharmacists preparing the biosimilars.
The health system also has a different approach to choosing medication. Instead of having an insurance company or PBM decide what’s in the formulary, clinicians work with the pharmacists at Kaiser to look at clinical evidence and decide which biosimilar to use. Most of its plans also provide lower copays to patients when they use the biosimilar.
This was the approach for Humira biosimilars, Dr. Awsare said. Eight will be on the market in 2023. “Our rheumatologists, dermatologists, and gastroenterologists looked at the data from Europe, looked at some real-world evidence, and then said: ‘We think this one’s going to be the best one for our patients.’ ”
Having clinicians choose the biosimilar instead of a health plan makes it a lot easier to have conversations with patients, he said. “Once we’ve moved that market share to that particular biosimilar, we give our physicians the time to have those discussions.”
Clinical pharmacists also provide educational support, offering guidance on issues such as side effects, as patients transition to the biosimilar. “We like to use the word ‘transition’ because it’s essentially the same biologic. So, you’re not actually switching,” Dr. Awsare said.
No consensus on interchangeability
Whether the conversation on interchangeability will affect patient conversations with physicians depends on who you ask.
If a biosimilar has an interchangeability designation, it means that the pharmacist can substitute it without the intervention of the clinician who prescribed the reference product. It does not relate to the quality, safety, or effectiveness of biosimilars or interchangeable biosimilar products, Dr. Popovian said.
The United States is the only country that has this designation. Even though it’s not identical to the originator drug, a biosimilar has the same clinical efficacy and safety profile. “So clinically, interchangeability is meaningless,” Dr. Awsare said.
In its report on biosimilars in the autoimmune category, CVS acknowledged that interchangeability was important but would not be a significant factor in driving adoption of biosimilars. However, in a Cardinal Health survey of 72 gastroenterologists, 38% cited the interchangeability of biosimilars as a top concern for adalimumab biosimilars, along with transitioning patients from Humira to a biosimilar (44%).
“Patient education regarding biosimilar safety, efficacy, and interchangeability appears paramount to the acceptance of these products, particularly for patients who are switched from a reference product,” Dr. Kaul noted in the Cardinal Health report.
Wherever supported by data, Dr. Kaul recommends incorporating biosimilar use and interchangeability into best practice guidelines going forward. “That will go a long way in disseminating the latest information on this topic and position this paradigm for increased adoption among providers.”
Some physicians like Dr. Snow aren’t that concerned with interchangeability. This hasn’t affected conversations with patients, he said. Multiple studies demonstrating the lack of antibody formation with multiple switches from different biosimilar drugs has eased his concern about multiple switches causing problems.
“Initially, there was a gap in demonstrating the long-term effect of multiple switches on antibody production and drug effectiveness. That gap has started to close as more data from Europe’s experience with biosimilars becomes available,” Dr. Snow said.
Resources for physicians, patients
The federal government has taken steps to advance biosimilars education and adoption. In 2021, President Biden signed the Advancing Education on Biosimilars Act into law, which directs the FDA to develop or improve continuing education programs that address prescribing of biosimilars and biological products.
The FDA provides educational materials on its website, including a comprehensive curriculum toolkit. The Accreditation Council for Medical Affairs has also created an online 40-hour curriculum for health care professionals called the Board-Certified Biologics and Biosimilars Specialist Program.
Dr. Fabbro recommended patients use the FDA page Biosimilar Basics for Patients to educate themselves on biosimilars. The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars, is another free resource for patients.
“While much has changed, the continued need for multistakeholder education, awareness, and dedicated research remains even more important as we expand into newer therapeutic areas and classes,” wrote the authors of the Cardinal Health report.
Help patients understand biologics and biosimilars by using AGA resources for providers and patients available at gastro.org/biosimilars.
Dr. Regueiro is on advisory boards and consults for AbbVie, Janssen, UCB, Takeda, Pfizer, Bristol-Myers Squibb, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET PharmaSolutions, Trellis, and Boehringer Ingelheim. Dr. Fabbro is a principal investigator for Castle Biosciences, on the speakers bureau for Valchlor, and on the advisory boards of Janssen and Bristol-Myers Squibb. Dr. Popovian, Dr. Snow, Dr. Awsare, and Dr. Kaul had no disclosures.
A version of this article originally appeared on Medscape.com.
Rheumatologist Marcus Snow, MD, is comfortable with prescribing biosimilars as a first-line, first-time biologic, and discussing them with patients.
“If a biosimilar is on the market, it has gone through rigorous study proving its effectiveness and equivalence to a bio-originator,” said Dr. Snow, a rheumatologist with the University of Nebraska Medical Center, Omaha, and chair of the American College of Rheumatology’s Committee on Rheumatologic Care.
The formulary makes a big difference in the conversation about options, he said. “The formularies dictate what we can prescribe. It may not be appropriate, but it is reality. The cost of biologics for a patient without insurance coverage makes it impossible to afford.”
He will often tell patients that he’ll fight any changes or formulary restrictions he does not agree with. “However, when I see patients in follow-up, even if there is no known change on the horizon, I may bring up biosimilars when we have a moment to chat about them to familiarize them with what may happen in the future.”
The need for patient education on biosimilars presents a barrier to realizing their potential to save money and expand choice, noted Cardinal Health in its 2023 biosimilars report. Of 103 rheumatologists who responded to a Cardinal Health survey, 85% agreed that patient education was important. But those conversations can take an uncomfortable turn if the patient pushes back against taking a biosimilar owing to cost or safety concerns.
It’s not uncommon for a patient to express some anxiety about biosimilars, especially if they’re doing well on a current treatment plan. Most patients do not want any changes that may lead to worsening disease control, Dr. Snow said.
Patients and physicians alike often don’t understand the mechanics of biosimilars. “There’s a lot of misinformation about this,” said Sameer Awsare, MD, an associate executive director for The Permanente Medical Group in Campbell, Calif. Patients should know that a biosimilar will be as clinically efficacious as the medicine they’ve been on, with the same safety profiles, said Dr. Awsare, who works with Kaiser Permanente’s pharmacy partners on biosimilars.
Insurance often drives the conversation
The global anti-inflammatory biologics market is anticipated to reach $150 billion by 2027, according to a recent CVS report. As of March 2023, the Food and Drug Administration had approved 40 biosimilars to 11 different reference products. There are 28 on the U.S. market and 100 more in development. Projected to save more than $180 billion over the next 5 years, they are anticipated to expand choice and drive competition.
Rheumatologists, dermatologists, and gastroenterologists are frequent prescribers, although their choices for immune-mediated inflammatory diseases are limited to tumor necrosis factor inhibitors (infliximab [Remicade] originator and adalimumab [Humira] originator) and anti-CD20 agents, such as rituximab (Rituxan) originator.
Benefit design or formulary usually dictates what medicine a patient receives. “Because of significantly higher out-of-pocket cost or formulary positioning, patients may end up with a generic or a biosimilar instead of a brand-name medicine or branded biologic,” said Robert Popovian, PharmD, MS, chief science policy officer of the Global Healthy Living Foundation.
Insurers rarely offer both Remicade and biosimilar infliximab, allowing the doctor to choose, said Miguel Regueiro, MD, chair of the Cleveland Clinic’s Digestive Disease & Surgery Institute, who prescribes infliximab biosimilars. Most often, the payer will choose the lower-cost biosimilar. “I am fine with the biosimilar, either as a new start or a switch from the reference product.”
However, the patient might feel differently. They can form an attachment to the reference medication if it has prevented severe illness. “They do not want to change, as they feel they are going on a ‘new’ medication that will not work as well,” Dr. Regueiro said.
This is where the education comes in: to reassure patients that a biosimilar will work just as well as the reference product. “For patients who have done well for years on a biologic, more time needs to be spent reassuring them and answering questions,” compared with a patient just starting on a biosimilar, he advised.
But not all physicians are quick to prescribe biosimilars.
Especially with psoriasis, which has so many strong options for reference drugs, a switch may be hard to justify, said dermatologist Stephanie K. Fabbro, MD, assistant professor at Northeast Ohio Medical University, Rootstown. “If I have a preference, I would rather switch a patient to a drug from a different class without a biosimilar option to reduce the possibility of pushback.”
Dr. Fabbro, part of the core faculty in the Riverside Methodist Hospital Dermatology Residency Program in Columbus, will share data from clinical trials and postmarket surveillance with patients to support her decision.
Conversations about cost
Patients may also push back if they don’t save money when switching to a biosimilar. “This dilemma raises the question of who is profiting when a biosimilar is dispensed,” Dr. Popovian said. Insurers and pharmacy benefit managers (PBMs) that take additional concessions from biopharmaceutical manufacturers in the form of rebates and fees will often pocket this money as profit instead of passing savings back to the patient to help reduce their out-of-pocket requirement, he added.
If an originator biologic and a biosimilar are available, “as a pharmacist, I will choose the medicine that will incur the lowest out-of-pocket cost for the patient,” Dr. Popovian said.
Discussing cost – and who dictates which biosimilar is on the formulary – is an important conversation to have with patients, said Vivek Kaul, MD, Segal-Watson Professor of Medicine at the University of Rochester (N.Y.) Medical Center.
Providing equivalent clinical efficacy while saving costs is the economic reality of biosimilars, Dr. Kaul said. Third-party payers regularly evaluate how to provide the same quality of care while saving money. Physicians and patients alike “must be mindful that as time goes on, if the science on biosimilars stays robust, if the adoption is more widespread and the cost-saving proposition turns out to be true, more formularies will be attracted to replacing the reference product with the biosimilar counterpart.”
Providers and patients can weigh the options if a formulary suddenly switches to a biosimilar, Dr. Kaul continued. “You can accept the novel product on the formulary or may have to face out-of-pocket expenses as a patient.” If providers and patients have concerns about the biosimilar, they can always appeal if there’s solid scientific evidence that supports reverting back to the reference product.
“If you think the biosimilar is equally efficacious, comes at a lower cost, and is right for the patient, then the providers should tell the patient that,” he added.
Some studies have questioned whether the biosimilars will save money, compared with the reference drug, Dr. Fabbro noted. Medicare, for example, may pay only for a certain percentage of an approved biosimilar, saddling the patient with a monthly copay costing thousands of dollars. “It is unclear whether biosimilar manufacturers will have the same level of patient support programs as the reference drug companies.”
For that reason, physicians should also inform patients about the robust patient assistance and copay assistance programs many reference drug manufacturers offer, she said.
Biosimilars 101: Familiarizing patients
Safety and ease of use are other common concerns about biosimilars. Patients may ask if the application is different, or why it’s advantageous to switch to a biosimilar, Dr. Awsare said.
Sometimes the syringe or injector for a biosimilar might look different from that of the originator drug, he said.
Anecdotally, Dr. Fabbro has heard stories of patients having injection reactions that they did not experience with the reference drug or having a disease flare-up after starting a biosimilar.
As is the case with reference products, in their conversations with patients, clinicians should address the adverse event profile of biosimilars, offering data points from published studies and clinical guidelines that support the use of these products. “There should be an emphasis on patient education around efficacy and any side effects, and how the profile of the reference product compares with a proposed biosimilar,” Dr. Kaul suggested.
When Dr. Snow discusses biosimilars and generics, “I make sure to share this in an understandable way based on the patient’s scientific background, or lack thereof,” he said. If there is enough time, he also discusses how European- and U.S.-sourced biologics are slightly different.
Pharmacists should tell patients to expect the same clinical outcomes from a biosimilar, Dr. Popovian said. However, if they have any reduction in efficacy or potential safety concerns, they should communicate with their physician or pharmacist immediately.
In Dr. Regueiro’s practice, a pharmacist specializing in inflammatory bowel disease often has a one-on-one meeting with patients to educate and answer questions. “Additionally, we provide them the Crohn’s and Colitis Foundation web link on biosimilars,” said Dr. Regueiro.
A village approach to education
When biosimilars first came out, there were no formal education materials, Dr. Awsare said. Kaiser Permanente decided to create its own educational materials, not just for patients but also to help educate its primary care doctors; the rheumatologists, dermatologists, and gastroenterologists using the biosimilars; the nurses infusing patients; and the pharmacists preparing the biosimilars.
The health system also has a different approach to choosing medication. Instead of having an insurance company or PBM decide what’s in the formulary, clinicians work with the pharmacists at Kaiser to look at clinical evidence and decide which biosimilar to use. Most of its plans also provide lower copays to patients when they use the biosimilar.
This was the approach for Humira biosimilars, Dr. Awsare said. Eight will be on the market in 2023. “Our rheumatologists, dermatologists, and gastroenterologists looked at the data from Europe, looked at some real-world evidence, and then said: ‘We think this one’s going to be the best one for our patients.’ ”
Having clinicians choose the biosimilar instead of a health plan makes it a lot easier to have conversations with patients, he said. “Once we’ve moved that market share to that particular biosimilar, we give our physicians the time to have those discussions.”
Clinical pharmacists also provide educational support, offering guidance on issues such as side effects, as patients transition to the biosimilar. “We like to use the word ‘transition’ because it’s essentially the same biologic. So, you’re not actually switching,” Dr. Awsare said.
No consensus on interchangeability
Whether the conversation on interchangeability will affect patient conversations with physicians depends on who you ask.
If a biosimilar has an interchangeability designation, it means that the pharmacist can substitute it without the intervention of the clinician who prescribed the reference product. It does not relate to the quality, safety, or effectiveness of biosimilars or interchangeable biosimilar products, Dr. Popovian said.
The United States is the only country that has this designation. Even though it’s not identical to the originator drug, a biosimilar has the same clinical efficacy and safety profile. “So clinically, interchangeability is meaningless,” Dr. Awsare said.
In its report on biosimilars in the autoimmune category, CVS acknowledged that interchangeability was important but would not be a significant factor in driving adoption of biosimilars. However, in a Cardinal Health survey of 72 gastroenterologists, 38% cited the interchangeability of biosimilars as a top concern for adalimumab biosimilars, along with transitioning patients from Humira to a biosimilar (44%).
“Patient education regarding biosimilar safety, efficacy, and interchangeability appears paramount to the acceptance of these products, particularly for patients who are switched from a reference product,” Dr. Kaul noted in the Cardinal Health report.
Wherever supported by data, Dr. Kaul recommends incorporating biosimilar use and interchangeability into best practice guidelines going forward. “That will go a long way in disseminating the latest information on this topic and position this paradigm for increased adoption among providers.”
Some physicians like Dr. Snow aren’t that concerned with interchangeability. This hasn’t affected conversations with patients, he said. Multiple studies demonstrating the lack of antibody formation with multiple switches from different biosimilar drugs has eased his concern about multiple switches causing problems.
“Initially, there was a gap in demonstrating the long-term effect of multiple switches on antibody production and drug effectiveness. That gap has started to close as more data from Europe’s experience with biosimilars becomes available,” Dr. Snow said.
Resources for physicians, patients
The federal government has taken steps to advance biosimilars education and adoption. In 2021, President Biden signed the Advancing Education on Biosimilars Act into law, which directs the FDA to develop or improve continuing education programs that address prescribing of biosimilars and biological products.
The FDA provides educational materials on its website, including a comprehensive curriculum toolkit. The Accreditation Council for Medical Affairs has also created an online 40-hour curriculum for health care professionals called the Board-Certified Biologics and Biosimilars Specialist Program.
Dr. Fabbro recommended patients use the FDA page Biosimilar Basics for Patients to educate themselves on biosimilars. The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars, is another free resource for patients.
“While much has changed, the continued need for multistakeholder education, awareness, and dedicated research remains even more important as we expand into newer therapeutic areas and classes,” wrote the authors of the Cardinal Health report.
Help patients understand biologics and biosimilars by using AGA resources for providers and patients available at gastro.org/biosimilars.
Dr. Regueiro is on advisory boards and consults for AbbVie, Janssen, UCB, Takeda, Pfizer, Bristol-Myers Squibb, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET PharmaSolutions, Trellis, and Boehringer Ingelheim. Dr. Fabbro is a principal investigator for Castle Biosciences, on the speakers bureau for Valchlor, and on the advisory boards of Janssen and Bristol-Myers Squibb. Dr. Popovian, Dr. Snow, Dr. Awsare, and Dr. Kaul had no disclosures.
A version of this article originally appeared on Medscape.com.
Rheumatologist Marcus Snow, MD, is comfortable with prescribing biosimilars as a first-line, first-time biologic, and discussing them with patients.
“If a biosimilar is on the market, it has gone through rigorous study proving its effectiveness and equivalence to a bio-originator,” said Dr. Snow, a rheumatologist with the University of Nebraska Medical Center, Omaha, and chair of the American College of Rheumatology’s Committee on Rheumatologic Care.
The formulary makes a big difference in the conversation about options, he said. “The formularies dictate what we can prescribe. It may not be appropriate, but it is reality. The cost of biologics for a patient without insurance coverage makes it impossible to afford.”
He will often tell patients that he’ll fight any changes or formulary restrictions he does not agree with. “However, when I see patients in follow-up, even if there is no known change on the horizon, I may bring up biosimilars when we have a moment to chat about them to familiarize them with what may happen in the future.”
The need for patient education on biosimilars presents a barrier to realizing their potential to save money and expand choice, noted Cardinal Health in its 2023 biosimilars report. Of 103 rheumatologists who responded to a Cardinal Health survey, 85% agreed that patient education was important. But those conversations can take an uncomfortable turn if the patient pushes back against taking a biosimilar owing to cost or safety concerns.
It’s not uncommon for a patient to express some anxiety about biosimilars, especially if they’re doing well on a current treatment plan. Most patients do not want any changes that may lead to worsening disease control, Dr. Snow said.
Patients and physicians alike often don’t understand the mechanics of biosimilars. “There’s a lot of misinformation about this,” said Sameer Awsare, MD, an associate executive director for The Permanente Medical Group in Campbell, Calif. Patients should know that a biosimilar will be as clinically efficacious as the medicine they’ve been on, with the same safety profiles, said Dr. Awsare, who works with Kaiser Permanente’s pharmacy partners on biosimilars.
Insurance often drives the conversation
The global anti-inflammatory biologics market is anticipated to reach $150 billion by 2027, according to a recent CVS report. As of March 2023, the Food and Drug Administration had approved 40 biosimilars to 11 different reference products. There are 28 on the U.S. market and 100 more in development. Projected to save more than $180 billion over the next 5 years, they are anticipated to expand choice and drive competition.
Rheumatologists, dermatologists, and gastroenterologists are frequent prescribers, although their choices for immune-mediated inflammatory diseases are limited to tumor necrosis factor inhibitors (infliximab [Remicade] originator and adalimumab [Humira] originator) and anti-CD20 agents, such as rituximab (Rituxan) originator.
Benefit design or formulary usually dictates what medicine a patient receives. “Because of significantly higher out-of-pocket cost or formulary positioning, patients may end up with a generic or a biosimilar instead of a brand-name medicine or branded biologic,” said Robert Popovian, PharmD, MS, chief science policy officer of the Global Healthy Living Foundation.
Insurers rarely offer both Remicade and biosimilar infliximab, allowing the doctor to choose, said Miguel Regueiro, MD, chair of the Cleveland Clinic’s Digestive Disease & Surgery Institute, who prescribes infliximab biosimilars. Most often, the payer will choose the lower-cost biosimilar. “I am fine with the biosimilar, either as a new start or a switch from the reference product.”
However, the patient might feel differently. They can form an attachment to the reference medication if it has prevented severe illness. “They do not want to change, as they feel they are going on a ‘new’ medication that will not work as well,” Dr. Regueiro said.
This is where the education comes in: to reassure patients that a biosimilar will work just as well as the reference product. “For patients who have done well for years on a biologic, more time needs to be spent reassuring them and answering questions,” compared with a patient just starting on a biosimilar, he advised.
But not all physicians are quick to prescribe biosimilars.
Especially with psoriasis, which has so many strong options for reference drugs, a switch may be hard to justify, said dermatologist Stephanie K. Fabbro, MD, assistant professor at Northeast Ohio Medical University, Rootstown. “If I have a preference, I would rather switch a patient to a drug from a different class without a biosimilar option to reduce the possibility of pushback.”
Dr. Fabbro, part of the core faculty in the Riverside Methodist Hospital Dermatology Residency Program in Columbus, will share data from clinical trials and postmarket surveillance with patients to support her decision.
Conversations about cost
Patients may also push back if they don’t save money when switching to a biosimilar. “This dilemma raises the question of who is profiting when a biosimilar is dispensed,” Dr. Popovian said. Insurers and pharmacy benefit managers (PBMs) that take additional concessions from biopharmaceutical manufacturers in the form of rebates and fees will often pocket this money as profit instead of passing savings back to the patient to help reduce their out-of-pocket requirement, he added.
If an originator biologic and a biosimilar are available, “as a pharmacist, I will choose the medicine that will incur the lowest out-of-pocket cost for the patient,” Dr. Popovian said.
Discussing cost – and who dictates which biosimilar is on the formulary – is an important conversation to have with patients, said Vivek Kaul, MD, Segal-Watson Professor of Medicine at the University of Rochester (N.Y.) Medical Center.
Providing equivalent clinical efficacy while saving costs is the economic reality of biosimilars, Dr. Kaul said. Third-party payers regularly evaluate how to provide the same quality of care while saving money. Physicians and patients alike “must be mindful that as time goes on, if the science on biosimilars stays robust, if the adoption is more widespread and the cost-saving proposition turns out to be true, more formularies will be attracted to replacing the reference product with the biosimilar counterpart.”
Providers and patients can weigh the options if a formulary suddenly switches to a biosimilar, Dr. Kaul continued. “You can accept the novel product on the formulary or may have to face out-of-pocket expenses as a patient.” If providers and patients have concerns about the biosimilar, they can always appeal if there’s solid scientific evidence that supports reverting back to the reference product.
“If you think the biosimilar is equally efficacious, comes at a lower cost, and is right for the patient, then the providers should tell the patient that,” he added.
Some studies have questioned whether the biosimilars will save money, compared with the reference drug, Dr. Fabbro noted. Medicare, for example, may pay only for a certain percentage of an approved biosimilar, saddling the patient with a monthly copay costing thousands of dollars. “It is unclear whether biosimilar manufacturers will have the same level of patient support programs as the reference drug companies.”
For that reason, physicians should also inform patients about the robust patient assistance and copay assistance programs many reference drug manufacturers offer, she said.
Biosimilars 101: Familiarizing patients
Safety and ease of use are other common concerns about biosimilars. Patients may ask if the application is different, or why it’s advantageous to switch to a biosimilar, Dr. Awsare said.
Sometimes the syringe or injector for a biosimilar might look different from that of the originator drug, he said.
Anecdotally, Dr. Fabbro has heard stories of patients having injection reactions that they did not experience with the reference drug or having a disease flare-up after starting a biosimilar.
As is the case with reference products, in their conversations with patients, clinicians should address the adverse event profile of biosimilars, offering data points from published studies and clinical guidelines that support the use of these products. “There should be an emphasis on patient education around efficacy and any side effects, and how the profile of the reference product compares with a proposed biosimilar,” Dr. Kaul suggested.
When Dr. Snow discusses biosimilars and generics, “I make sure to share this in an understandable way based on the patient’s scientific background, or lack thereof,” he said. If there is enough time, he also discusses how European- and U.S.-sourced biologics are slightly different.
Pharmacists should tell patients to expect the same clinical outcomes from a biosimilar, Dr. Popovian said. However, if they have any reduction in efficacy or potential safety concerns, they should communicate with their physician or pharmacist immediately.
In Dr. Regueiro’s practice, a pharmacist specializing in inflammatory bowel disease often has a one-on-one meeting with patients to educate and answer questions. “Additionally, we provide them the Crohn’s and Colitis Foundation web link on biosimilars,” said Dr. Regueiro.
A village approach to education
When biosimilars first came out, there were no formal education materials, Dr. Awsare said. Kaiser Permanente decided to create its own educational materials, not just for patients but also to help educate its primary care doctors; the rheumatologists, dermatologists, and gastroenterologists using the biosimilars; the nurses infusing patients; and the pharmacists preparing the biosimilars.
The health system also has a different approach to choosing medication. Instead of having an insurance company or PBM decide what’s in the formulary, clinicians work with the pharmacists at Kaiser to look at clinical evidence and decide which biosimilar to use. Most of its plans also provide lower copays to patients when they use the biosimilar.
This was the approach for Humira biosimilars, Dr. Awsare said. Eight will be on the market in 2023. “Our rheumatologists, dermatologists, and gastroenterologists looked at the data from Europe, looked at some real-world evidence, and then said: ‘We think this one’s going to be the best one for our patients.’ ”
Having clinicians choose the biosimilar instead of a health plan makes it a lot easier to have conversations with patients, he said. “Once we’ve moved that market share to that particular biosimilar, we give our physicians the time to have those discussions.”
Clinical pharmacists also provide educational support, offering guidance on issues such as side effects, as patients transition to the biosimilar. “We like to use the word ‘transition’ because it’s essentially the same biologic. So, you’re not actually switching,” Dr. Awsare said.
No consensus on interchangeability
Whether the conversation on interchangeability will affect patient conversations with physicians depends on who you ask.
If a biosimilar has an interchangeability designation, it means that the pharmacist can substitute it without the intervention of the clinician who prescribed the reference product. It does not relate to the quality, safety, or effectiveness of biosimilars or interchangeable biosimilar products, Dr. Popovian said.
The United States is the only country that has this designation. Even though it’s not identical to the originator drug, a biosimilar has the same clinical efficacy and safety profile. “So clinically, interchangeability is meaningless,” Dr. Awsare said.
In its report on biosimilars in the autoimmune category, CVS acknowledged that interchangeability was important but would not be a significant factor in driving adoption of biosimilars. However, in a Cardinal Health survey of 72 gastroenterologists, 38% cited the interchangeability of biosimilars as a top concern for adalimumab biosimilars, along with transitioning patients from Humira to a biosimilar (44%).
“Patient education regarding biosimilar safety, efficacy, and interchangeability appears paramount to the acceptance of these products, particularly for patients who are switched from a reference product,” Dr. Kaul noted in the Cardinal Health report.
Wherever supported by data, Dr. Kaul recommends incorporating biosimilar use and interchangeability into best practice guidelines going forward. “That will go a long way in disseminating the latest information on this topic and position this paradigm for increased adoption among providers.”
Some physicians like Dr. Snow aren’t that concerned with interchangeability. This hasn’t affected conversations with patients, he said. Multiple studies demonstrating the lack of antibody formation with multiple switches from different biosimilar drugs has eased his concern about multiple switches causing problems.
“Initially, there was a gap in demonstrating the long-term effect of multiple switches on antibody production and drug effectiveness. That gap has started to close as more data from Europe’s experience with biosimilars becomes available,” Dr. Snow said.
Resources for physicians, patients
The federal government has taken steps to advance biosimilars education and adoption. In 2021, President Biden signed the Advancing Education on Biosimilars Act into law, which directs the FDA to develop or improve continuing education programs that address prescribing of biosimilars and biological products.
The FDA provides educational materials on its website, including a comprehensive curriculum toolkit. The Accreditation Council for Medical Affairs has also created an online 40-hour curriculum for health care professionals called the Board-Certified Biologics and Biosimilars Specialist Program.
Dr. Fabbro recommended patients use the FDA page Biosimilar Basics for Patients to educate themselves on biosimilars. The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars, is another free resource for patients.
“While much has changed, the continued need for multistakeholder education, awareness, and dedicated research remains even more important as we expand into newer therapeutic areas and classes,” wrote the authors of the Cardinal Health report.
Help patients understand biologics and biosimilars by using AGA resources for providers and patients available at gastro.org/biosimilars.
Dr. Regueiro is on advisory boards and consults for AbbVie, Janssen, UCB, Takeda, Pfizer, Bristol-Myers Squibb, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET PharmaSolutions, Trellis, and Boehringer Ingelheim. Dr. Fabbro is a principal investigator for Castle Biosciences, on the speakers bureau for Valchlor, and on the advisory boards of Janssen and Bristol-Myers Squibb. Dr. Popovian, Dr. Snow, Dr. Awsare, and Dr. Kaul had no disclosures.
A version of this article originally appeared on Medscape.com.
Sweaty treatment for social anxiety could pass the sniff test
Getting sweet on sweat
Are you the sort of person who struggles in social situations? Have the past 3 years been a secret respite from the terror and exhaustion of meeting new people? We understand your plight. People kind of suck. And you don’t have to look far to be reminded of it.
Unfortunately, on occasion we all have to interact with other human beings. If you suffer from social anxiety, this is not a fun thing to do. But new research indicates that there may be a way to alleviate the stress for those with social anxiety: armpits.
Specifically, sweat from the armpits of other people. Yes, this means a group of scientists gathered up some volunteers and collected their armpit sweat while the volunteers watched a variety of movies (horror, comedy, romance, etc.). Our condolences to the poor unpaid interns tasked with gathering the sweat.
Once they had their precious new medicine, the researchers took a group of women and administered a round of mindfulness therapy. Some of the participants then received the various sweats, while the rest were forced to smell only clean air. (The horror!) Lo and behold, the sweat groups had their anxiety scores reduced by about 40% after their therapy, compared with just 17% in the control group.
The researchers also found that the source of the sweat didn’t matter. Their study subjects responded the same to sweat excreted during a scary movie as they did to sweat from a comedy, a result that surprised the researchers. They suggested chemosignals in the sweat may affect the treatment response and advised further research. Which means more sweat collection! They plan on testing emotionally neutral movies next time, and if we can make a humble suggestion, they also should try the sweatiest movies.
Before the Food and Drug Administration can approve armpit sweat as a treatment for social anxiety, we have some advice for those shut-in introverts out there. Next time you have to interact with rabid extroverts, instead of shaking their hands, walk up to them and take a deep whiff of their armpits. Establish dominance. Someone will feel awkward, and science has proved it won’t be you.
The puff that vaccinates
Ever been shot with a Nerf gun or hit with a foam pool tube? More annoying than painful, right? If we asked if you’d rather get pelted with one of those than receive a traditional vaccine injection, you would choose the former. Maybe someday you actually will.

During the boredom of the early pandemic lockdown, Jeremiah Gassensmith, PhD, of the department of chemistry and biochemistry at the University of Texas, Dallas, ordered a compressed gas–powered jet injection system to fool around with at home. Hey, who didn’t? Anyway, when it was time to go back to the lab he handed it over to one of his grad students, Yalini Wijesundara, and asked her to see what could be done with it.
In her tinkering she found that the jet injector could deliver metal-organic frameworks (MOFs) that can hold a bunch of different materials, like proteins and nucleic acids, through the skin.
Thus the “MOF-Jet” was born!
Jet injectors are nothing new, but they hurt. The MOF-Jet, however, is practically painless and cheaper than the gene guns that veterinarians use to inject biological cargo attached to the surface of a metal microparticle.
Changing the carrier gas also changes the time needed to break down the MOF and thus alters delivery of the drug inside. “If you shoot it with carbon dioxide, it will release its cargo faster within cells; if you use regular air, it will take 4 or 5 days,” Ms. Wijesundara explained in a written statement. That means the same drug could be released over different timescales without changing its formulation.
While testing on onion cells and mice, Ms. Wijesundara noted that it was as easy as “pointing and shooting” to distribute the puff of gas into the cells. A saving grace to those with needle anxiety. Not that we would know anything about needle anxiety.
More testing needs to be done before bringing this technology to human use, obviously, but we’re looking forward to saying goodbye to that dreaded prick and hello to a puff.
Your hippocampus is showing
Brain anatomy is one of the many, many things that’s not really our thing, but we do know a cool picture when we see one. Case in point: The image just below, which happens to be a full-scale, single-cell resolution model of the CA1 region of the hippocampus that “replicates the structure and architecture of the area, along with the position and relative connectivity of the neurons,” according to a statement from the Human Brain Project.
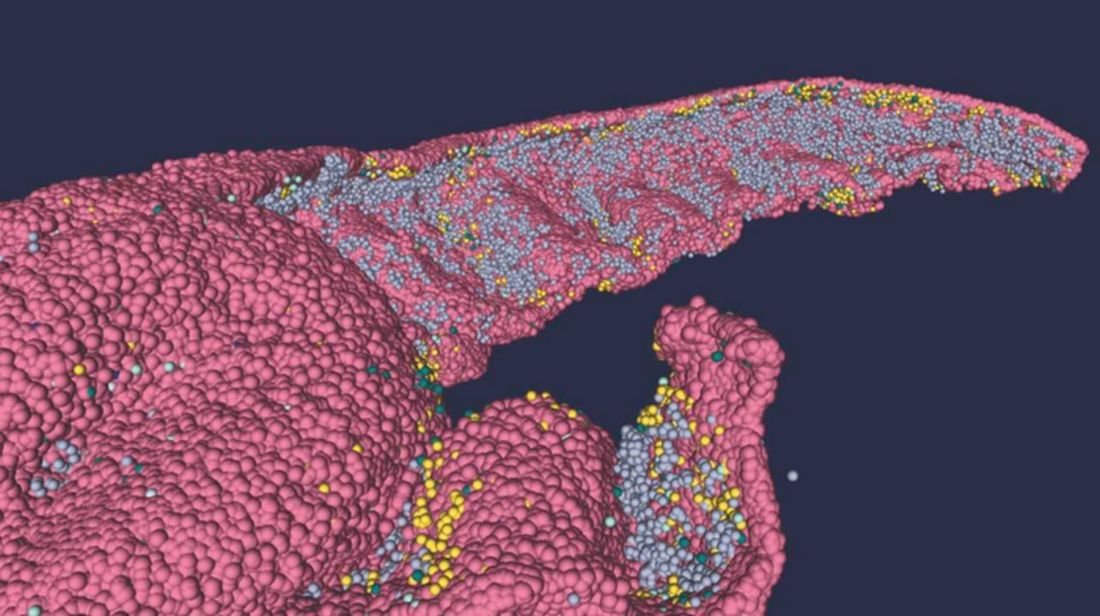
“We have performed a data mining operation on high resolution images of the human hippocampus, obtained from the BigBrain database. The position of individual neurons has been derived from a detailed analysis of these images,” said senior author Michele Migliore, PhD, of the Italian National Research Council’s Institute of Biophysics in Palermo.
Yes, he did say BigBrain database. BigBrain is – we checked and it’s definitely not this – a 3D model of a brain that was sectioned into 7,404 slices just 20 micrometers thick and then scanned by MRI. Digital reconstruction of those slices was done by supercomputer and the results are now available for analysis.
Dr. Migliore and his associates developed an image-processing algorithm to obtain neuronal positioning distribution and an algorithm to generate neuronal connectivity by approximating the shapes of dendrites and axons. (Our brains are starting to hurt just trying to write this.) “Some fit into narrow cones, others have a broad complex extension that can be approximated by dedicated geometrical volumes, and the connectivity to nearby neurons changes accordingly,” explained lead author Daniela Gandolfi of the University of Modena (Italy) and Reggio Emilia.
The investigators have made their dataset and the extraction methodology available on the EBRAINS platform and through the Human Brain Project and are moving on to other brain regions. And then, once everyone can find their way in and around the old gray matter, it should bring an end to conversations like this, which no doubt occur between male and female neuroscientists every day:
“Arnold, I think we’re lost.”
“Don’t worry, Bev, I know where I’m going.”
“Stop and ask this lady for directions.”
“I said I can find it.”
“Just ask her.”
“Fine. Excuse me, ma’am, can you tell us how to get to the corpora quadrigemina from here?
Getting sweet on sweat
Are you the sort of person who struggles in social situations? Have the past 3 years been a secret respite from the terror and exhaustion of meeting new people? We understand your plight. People kind of suck. And you don’t have to look far to be reminded of it.
Unfortunately, on occasion we all have to interact with other human beings. If you suffer from social anxiety, this is not a fun thing to do. But new research indicates that there may be a way to alleviate the stress for those with social anxiety: armpits.
Specifically, sweat from the armpits of other people. Yes, this means a group of scientists gathered up some volunteers and collected their armpit sweat while the volunteers watched a variety of movies (horror, comedy, romance, etc.). Our condolences to the poor unpaid interns tasked with gathering the sweat.
Once they had their precious new medicine, the researchers took a group of women and administered a round of mindfulness therapy. Some of the participants then received the various sweats, while the rest were forced to smell only clean air. (The horror!) Lo and behold, the sweat groups had their anxiety scores reduced by about 40% after their therapy, compared with just 17% in the control group.
The researchers also found that the source of the sweat didn’t matter. Their study subjects responded the same to sweat excreted during a scary movie as they did to sweat from a comedy, a result that surprised the researchers. They suggested chemosignals in the sweat may affect the treatment response and advised further research. Which means more sweat collection! They plan on testing emotionally neutral movies next time, and if we can make a humble suggestion, they also should try the sweatiest movies.
Before the Food and Drug Administration can approve armpit sweat as a treatment for social anxiety, we have some advice for those shut-in introverts out there. Next time you have to interact with rabid extroverts, instead of shaking their hands, walk up to them and take a deep whiff of their armpits. Establish dominance. Someone will feel awkward, and science has proved it won’t be you.
The puff that vaccinates
Ever been shot with a Nerf gun or hit with a foam pool tube? More annoying than painful, right? If we asked if you’d rather get pelted with one of those than receive a traditional vaccine injection, you would choose the former. Maybe someday you actually will.
During the boredom of the early pandemic lockdown, Jeremiah Gassensmith, PhD, of the department of chemistry and biochemistry at the University of Texas, Dallas, ordered a compressed gas–powered jet injection system to fool around with at home. Hey, who didn’t? Anyway, when it was time to go back to the lab he handed it over to one of his grad students, Yalini Wijesundara, and asked her to see what could be done with it.
In her tinkering she found that the jet injector could deliver metal-organic frameworks (MOFs) that can hold a bunch of different materials, like proteins and nucleic acids, through the skin.
Thus the “MOF-Jet” was born!
Jet injectors are nothing new, but they hurt. The MOF-Jet, however, is practically painless and cheaper than the gene guns that veterinarians use to inject biological cargo attached to the surface of a metal microparticle.
Changing the carrier gas also changes the time needed to break down the MOF and thus alters delivery of the drug inside. “If you shoot it with carbon dioxide, it will release its cargo faster within cells; if you use regular air, it will take 4 or 5 days,” Ms. Wijesundara explained in a written statement. That means the same drug could be released over different timescales without changing its formulation.
While testing on onion cells and mice, Ms. Wijesundara noted that it was as easy as “pointing and shooting” to distribute the puff of gas into the cells. A saving grace to those with needle anxiety. Not that we would know anything about needle anxiety.
More testing needs to be done before bringing this technology to human use, obviously, but we’re looking forward to saying goodbye to that dreaded prick and hello to a puff.
Your hippocampus is showing
Brain anatomy is one of the many, many things that’s not really our thing, but we do know a cool picture when we see one. Case in point: The image just below, which happens to be a full-scale, single-cell resolution model of the CA1 region of the hippocampus that “replicates the structure and architecture of the area, along with the position and relative connectivity of the neurons,” according to a statement from the Human Brain Project.
“We have performed a data mining operation on high resolution images of the human hippocampus, obtained from the BigBrain database. The position of individual neurons has been derived from a detailed analysis of these images,” said senior author Michele Migliore, PhD, of the Italian National Research Council’s Institute of Biophysics in Palermo.
Yes, he did say BigBrain database. BigBrain is – we checked and it’s definitely not this – a 3D model of a brain that was sectioned into 7,404 slices just 20 micrometers thick and then scanned by MRI. Digital reconstruction of those slices was done by supercomputer and the results are now available for analysis.
Dr. Migliore and his associates developed an image-processing algorithm to obtain neuronal positioning distribution and an algorithm to generate neuronal connectivity by approximating the shapes of dendrites and axons. (Our brains are starting to hurt just trying to write this.) “Some fit into narrow cones, others have a broad complex extension that can be approximated by dedicated geometrical volumes, and the connectivity to nearby neurons changes accordingly,” explained lead author Daniela Gandolfi of the University of Modena (Italy) and Reggio Emilia.
The investigators have made their dataset and the extraction methodology available on the EBRAINS platform and through the Human Brain Project and are moving on to other brain regions. And then, once everyone can find their way in and around the old gray matter, it should bring an end to conversations like this, which no doubt occur between male and female neuroscientists every day:
“Arnold, I think we’re lost.”
“Don’t worry, Bev, I know where I’m going.”
“Stop and ask this lady for directions.”
“I said I can find it.”
“Just ask her.”
“Fine. Excuse me, ma’am, can you tell us how to get to the corpora quadrigemina from here?
Getting sweet on sweat
Are you the sort of person who struggles in social situations? Have the past 3 years been a secret respite from the terror and exhaustion of meeting new people? We understand your plight. People kind of suck. And you don’t have to look far to be reminded of it.
Unfortunately, on occasion we all have to interact with other human beings. If you suffer from social anxiety, this is not a fun thing to do. But new research indicates that there may be a way to alleviate the stress for those with social anxiety: armpits.
Specifically, sweat from the armpits of other people. Yes, this means a group of scientists gathered up some volunteers and collected their armpit sweat while the volunteers watched a variety of movies (horror, comedy, romance, etc.). Our condolences to the poor unpaid interns tasked with gathering the sweat.
Once they had their precious new medicine, the researchers took a group of women and administered a round of mindfulness therapy. Some of the participants then received the various sweats, while the rest were forced to smell only clean air. (The horror!) Lo and behold, the sweat groups had their anxiety scores reduced by about 40% after their therapy, compared with just 17% in the control group.
The researchers also found that the source of the sweat didn’t matter. Their study subjects responded the same to sweat excreted during a scary movie as they did to sweat from a comedy, a result that surprised the researchers. They suggested chemosignals in the sweat may affect the treatment response and advised further research. Which means more sweat collection! They plan on testing emotionally neutral movies next time, and if we can make a humble suggestion, they also should try the sweatiest movies.
Before the Food and Drug Administration can approve armpit sweat as a treatment for social anxiety, we have some advice for those shut-in introverts out there. Next time you have to interact with rabid extroverts, instead of shaking their hands, walk up to them and take a deep whiff of their armpits. Establish dominance. Someone will feel awkward, and science has proved it won’t be you.
The puff that vaccinates
Ever been shot with a Nerf gun or hit with a foam pool tube? More annoying than painful, right? If we asked if you’d rather get pelted with one of those than receive a traditional vaccine injection, you would choose the former. Maybe someday you actually will.
During the boredom of the early pandemic lockdown, Jeremiah Gassensmith, PhD, of the department of chemistry and biochemistry at the University of Texas, Dallas, ordered a compressed gas–powered jet injection system to fool around with at home. Hey, who didn’t? Anyway, when it was time to go back to the lab he handed it over to one of his grad students, Yalini Wijesundara, and asked her to see what could be done with it.
In her tinkering she found that the jet injector could deliver metal-organic frameworks (MOFs) that can hold a bunch of different materials, like proteins and nucleic acids, through the skin.
Thus the “MOF-Jet” was born!
Jet injectors are nothing new, but they hurt. The MOF-Jet, however, is practically painless and cheaper than the gene guns that veterinarians use to inject biological cargo attached to the surface of a metal microparticle.
Changing the carrier gas also changes the time needed to break down the MOF and thus alters delivery of the drug inside. “If you shoot it with carbon dioxide, it will release its cargo faster within cells; if you use regular air, it will take 4 or 5 days,” Ms. Wijesundara explained in a written statement. That means the same drug could be released over different timescales without changing its formulation.
While testing on onion cells and mice, Ms. Wijesundara noted that it was as easy as “pointing and shooting” to distribute the puff of gas into the cells. A saving grace to those with needle anxiety. Not that we would know anything about needle anxiety.
More testing needs to be done before bringing this technology to human use, obviously, but we’re looking forward to saying goodbye to that dreaded prick and hello to a puff.
Your hippocampus is showing
Brain anatomy is one of the many, many things that’s not really our thing, but we do know a cool picture when we see one. Case in point: The image just below, which happens to be a full-scale, single-cell resolution model of the CA1 region of the hippocampus that “replicates the structure and architecture of the area, along with the position and relative connectivity of the neurons,” according to a statement from the Human Brain Project.
“We have performed a data mining operation on high resolution images of the human hippocampus, obtained from the BigBrain database. The position of individual neurons has been derived from a detailed analysis of these images,” said senior author Michele Migliore, PhD, of the Italian National Research Council’s Institute of Biophysics in Palermo.
Yes, he did say BigBrain database. BigBrain is – we checked and it’s definitely not this – a 3D model of a brain that was sectioned into 7,404 slices just 20 micrometers thick and then scanned by MRI. Digital reconstruction of those slices was done by supercomputer and the results are now available for analysis.
Dr. Migliore and his associates developed an image-processing algorithm to obtain neuronal positioning distribution and an algorithm to generate neuronal connectivity by approximating the shapes of dendrites and axons. (Our brains are starting to hurt just trying to write this.) “Some fit into narrow cones, others have a broad complex extension that can be approximated by dedicated geometrical volumes, and the connectivity to nearby neurons changes accordingly,” explained lead author Daniela Gandolfi of the University of Modena (Italy) and Reggio Emilia.
The investigators have made their dataset and the extraction methodology available on the EBRAINS platform and through the Human Brain Project and are moving on to other brain regions. And then, once everyone can find their way in and around the old gray matter, it should bring an end to conversations like this, which no doubt occur between male and female neuroscientists every day:
“Arnold, I think we’re lost.”
“Don’t worry, Bev, I know where I’m going.”
“Stop and ask this lady for directions.”
“I said I can find it.”
“Just ask her.”
“Fine. Excuse me, ma’am, can you tell us how to get to the corpora quadrigemina from here?
Early treatment considerations in RA, April 2023
In evaluating the importance of early aggressive treatment of rheumatoid arthritis (RA), we often look at prognostic factors for severe disease, such as seropositivity, elevated inflammatory markers, and erosions. Eberhard and colleagues looked at the relationship between damage as seen on radiography (including erosions and joint space narrowing) and pain and disability in early RA using an inception cohort with <12 months of symptoms. Over 200 patients in Sweden were followed for 5 years with clinical, laboratory, and radiographic evaluations. Of interest, pain was associated with female sex, tender joint count, and inflammatory markers at various time points but not with radiographic damage. This may reflect that pain is related to current inflammation rather than past joint damage or that pain is related to other factors, such as central sensitization. Radiographic damage was, however, associated with disability and thus remains an important target and outcome measure for assessing treatment effectiveness.
Leon and colleagues also looked at early RA but instead, at the category of difficult-to-treat RA (D2T RA), meaning persistent RA symptoms after a trial of at least two biologic or targeted synthetic disease-modifying antirheumatic drugs. In order to gain better insight in preventing D2T RA, the authors examined its association with potentially modifiable risk factors early in the course of disease. Of the over 600 patients followed in this inception cohort, only about 6% were classified as having D2T RA. The study found that patients who had D2T RA tended to be younger, with a higher tender joint count, higher pain scores, and a higher initial level of disability. The Disease Activity Score (DAS28) itself was higher in patients with D2T RA, but the difference did not reach statistical significance. The small number of patients (35) in the D2T RA group may have affected the findings as well as their wider applicability. However, it is interesting to consider whether the associations may also reflect the impact of noninflammatory factors, as in the previous study, on the classification of D2T RA.
Park and colleagues evaluated the difference in clinical outcomes in postmenopausal patients with RA who underwent menopause at younger than 45 years or 45 years or older. Among over 2800 patients in Korea, those who underwent early menopause were more likely to be seronegative and have high disease activity and worse patient-reported outcome scores in fatigue, sleep, and health-related quality of life despite comparable treatments and prevalence of erosions. The authors suggest this may be related to lower cumulative estrogen exposure; whether this correlates to inflammatory cytokine signatures is not yet known. However, as with the prior studies, central sensitization and noninflammatory pain may also contribute and should be considered in interpreting response to therapy.
Finally, addressing the potential risk for cancer in patients with RA before or during treatment with immunosuppressive medications, Miyata and colleagues reported a study that screened nearly 2200 patients who underwent CT (from neck to pelvis) and compared them with those who underwent routine screening with physical exam plus radiography. The study found that CT screening enhanced cancer detection, with a large number of cancers detected at an earlier stage with CT screening compared with routine screening. The overall number of cancers detected was low (33), and thus routine screening with neck-to-pelvis CT for all patients with RA may not be a cost-effective practice. However, it bears further examination for potentially higher-risk populations or specific cancers.
In evaluating the importance of early aggressive treatment of rheumatoid arthritis (RA), we often look at prognostic factors for severe disease, such as seropositivity, elevated inflammatory markers, and erosions. Eberhard and colleagues looked at the relationship between damage as seen on radiography (including erosions and joint space narrowing) and pain and disability in early RA using an inception cohort with <12 months of symptoms. Over 200 patients in Sweden were followed for 5 years with clinical, laboratory, and radiographic evaluations. Of interest, pain was associated with female sex, tender joint count, and inflammatory markers at various time points but not with radiographic damage. This may reflect that pain is related to current inflammation rather than past joint damage or that pain is related to other factors, such as central sensitization. Radiographic damage was, however, associated with disability and thus remains an important target and outcome measure for assessing treatment effectiveness.
Leon and colleagues also looked at early RA but instead, at the category of difficult-to-treat RA (D2T RA), meaning persistent RA symptoms after a trial of at least two biologic or targeted synthetic disease-modifying antirheumatic drugs. In order to gain better insight in preventing D2T RA, the authors examined its association with potentially modifiable risk factors early in the course of disease. Of the over 600 patients followed in this inception cohort, only about 6% were classified as having D2T RA. The study found that patients who had D2T RA tended to be younger, with a higher tender joint count, higher pain scores, and a higher initial level of disability. The Disease Activity Score (DAS28) itself was higher in patients with D2T RA, but the difference did not reach statistical significance. The small number of patients (35) in the D2T RA group may have affected the findings as well as their wider applicability. However, it is interesting to consider whether the associations may also reflect the impact of noninflammatory factors, as in the previous study, on the classification of D2T RA.
Park and colleagues evaluated the difference in clinical outcomes in postmenopausal patients with RA who underwent menopause at younger than 45 years or 45 years or older. Among over 2800 patients in Korea, those who underwent early menopause were more likely to be seronegative and have high disease activity and worse patient-reported outcome scores in fatigue, sleep, and health-related quality of life despite comparable treatments and prevalence of erosions. The authors suggest this may be related to lower cumulative estrogen exposure; whether this correlates to inflammatory cytokine signatures is not yet known. However, as with the prior studies, central sensitization and noninflammatory pain may also contribute and should be considered in interpreting response to therapy.
Finally, addressing the potential risk for cancer in patients with RA before or during treatment with immunosuppressive medications, Miyata and colleagues reported a study that screened nearly 2200 patients who underwent CT (from neck to pelvis) and compared them with those who underwent routine screening with physical exam plus radiography. The study found that CT screening enhanced cancer detection, with a large number of cancers detected at an earlier stage with CT screening compared with routine screening. The overall number of cancers detected was low (33), and thus routine screening with neck-to-pelvis CT for all patients with RA may not be a cost-effective practice. However, it bears further examination for potentially higher-risk populations or specific cancers.
In evaluating the importance of early aggressive treatment of rheumatoid arthritis (RA), we often look at prognostic factors for severe disease, such as seropositivity, elevated inflammatory markers, and erosions. Eberhard and colleagues looked at the relationship between damage as seen on radiography (including erosions and joint space narrowing) and pain and disability in early RA using an inception cohort with <12 months of symptoms. Over 200 patients in Sweden were followed for 5 years with clinical, laboratory, and radiographic evaluations. Of interest, pain was associated with female sex, tender joint count, and inflammatory markers at various time points but not with radiographic damage. This may reflect that pain is related to current inflammation rather than past joint damage or that pain is related to other factors, such as central sensitization. Radiographic damage was, however, associated with disability and thus remains an important target and outcome measure for assessing treatment effectiveness.
Leon and colleagues also looked at early RA but instead, at the category of difficult-to-treat RA (D2T RA), meaning persistent RA symptoms after a trial of at least two biologic or targeted synthetic disease-modifying antirheumatic drugs. In order to gain better insight in preventing D2T RA, the authors examined its association with potentially modifiable risk factors early in the course of disease. Of the over 600 patients followed in this inception cohort, only about 6% were classified as having D2T RA. The study found that patients who had D2T RA tended to be younger, with a higher tender joint count, higher pain scores, and a higher initial level of disability. The Disease Activity Score (DAS28) itself was higher in patients with D2T RA, but the difference did not reach statistical significance. The small number of patients (35) in the D2T RA group may have affected the findings as well as their wider applicability. However, it is interesting to consider whether the associations may also reflect the impact of noninflammatory factors, as in the previous study, on the classification of D2T RA.
Park and colleagues evaluated the difference in clinical outcomes in postmenopausal patients with RA who underwent menopause at younger than 45 years or 45 years or older. Among over 2800 patients in Korea, those who underwent early menopause were more likely to be seronegative and have high disease activity and worse patient-reported outcome scores in fatigue, sleep, and health-related quality of life despite comparable treatments and prevalence of erosions. The authors suggest this may be related to lower cumulative estrogen exposure; whether this correlates to inflammatory cytokine signatures is not yet known. However, as with the prior studies, central sensitization and noninflammatory pain may also contribute and should be considered in interpreting response to therapy.
Finally, addressing the potential risk for cancer in patients with RA before or during treatment with immunosuppressive medications, Miyata and colleagues reported a study that screened nearly 2200 patients who underwent CT (from neck to pelvis) and compared them with those who underwent routine screening with physical exam plus radiography. The study found that CT screening enhanced cancer detection, with a large number of cancers detected at an earlier stage with CT screening compared with routine screening. The overall number of cancers detected was low (33), and thus routine screening with neck-to-pelvis CT for all patients with RA may not be a cost-effective practice. However, it bears further examination for potentially higher-risk populations or specific cancers.
Osteoarthritis adjunctive therapies offer negligible added benefit to exercise
DENVER – Adding therapies such as acupuncture, electrophysical stimulation, or other interventions to standard exercise therapy does not appear to offer much benefit in pain relief or physical function for patients with knee osteoarthritis, according to a study presented at the OARSI 2023 World Congress. The findings were also published in the Cochrane Database of Systematic Reviews in October 2022.
“The results do not support the use of adjunctive therapies when we add them to exercise for pain, physical function, or quality of life, when compared against placebo, adjunctive therapy, and exercise,” Helen P. French, PhD, told attendees at the meeting sponsored by the Osteoarthritis Research Society International. The findings were similar for pain and physical function when comparing adjunctive therapies with exercise against exercise alone, said Dr. French, an associate professor in physiotherapy at the Royal College of Surgeons, Dublin, except that patients using adjunctive therapies reported feeling greater improvement in their global assessments.
Exercise is recommended as a core treatment for osteoarthritis, but some patients or clinicians may be interested in supplementing that therapy with acupuncture, heat therapy, electromagnetic fields, transcutaneous electrical nerve stimulation, braces/orthotics, and other interventions. Various Cochrane Reviews of the evidence exist for these interventions in treating chronic pain in general but not for their use as adjunctive therapies in addition to exercise for osteoarthritis pain.
Researchers therefore assessed the evidence for improvement in pain, physical function, and quality of life for two sets of comparisons: adjunctive therapies plus exercise versus exercise alone, and adjunctive therapies with exercise versus placebo adjunctive therapy with exercise. The review excluded studies looking at medications or supplements.
Pain was assessed with the Numeric Pain Rating Scale (NPRS, 0-10), with an improvement of at least 2 points (15% improvement) representing the minimum clinically important difference (MCID). Physical function was assessed with the Western Ontario and McMaster Universities Arthritis Index (WOMAC, 0-68), with 6 points (15%) considered the MCID, and quality of life was assessed with the SF-36 (0-100), with 6 points (12%) as the MCID.
The researchers identified trials on knee osteoarthritis that included an overall 6,508 participants with an average age ranging from 52 to 83 years. A total of 36 studies evaluated electrophysical agents. Another seven looked at manual therapies; four looked at acupuncture/dry needling or taping; three looked at psychological, dietary, or “whole body vibration” therapies; and two evaluated spa or peloid therapy. Only one trial evaluated foot insoles.
Nearly all the studies (98%) assessed pain, and most (87%) assessed physical function. Only about one in five (21%) assessed quality of life. The improvement in pain from adding adjunctive therapies to exercise, compared with placebo therapies plus exercise, was 0.77 points, or just under a 10% improvement, which fell short of the 15% MCID. Physical function improvement similarly fell short, with an average improvement of 5 points (12%).
In comparisons of exercise plus adjunctive therapies against exercise alone, the improvement from the additional interventions was even lower. Pain improvement was 0.41 points (7%), and physical function improvement was 2.8 points (9%). However, patients’ perceptions told a different story: 37% more patients who were using an adjunctive therapy reported feeling that the therapies were successful, compared with patients undergoing exercise therapy alone.
Adverse events were poorly reported in the trials, with only 10 trials reporting them at all, and the researchers found no significant difference in adverse events among the studies reporting them. The most common adverse events were increased pain in the joint with the osteoarthritis, pain elsewhere, or swelling and inflammation. It’s unclear, however, whether the pain, swelling, and inflammation were related to the interventions and how serious these effects might have been.
Michelle Hall, PhD, an associate professor in the department of physiotherapy at the University of Melbourne, comoderated the session with this presentation and found it interesting that more than one-third of patients perceived that they did better with the additional therapies even though improvement didn’t bear out in their pain or physical function assessments.
“But the other part of that was that the studies were of poor quality, so we can’t say with confidence, ‘Don’t do this therapy because it’s not going to work,’ ” Dr. Hall said in an interview. She said she personally would probably discourage patients from those therapies, “but I don’t think the evidence is there for everybody to do that,” she added.
Martin Van Der Esch, PhD, of Reade Centre of Rehabilitation and Rheumatology in Amsterdam, also comoderated the discussion and had more concerns about the use of adjunctive therapies in light of the study’s findings. He said in an interview that he tended to believe the patients’ overall self-reported improvement is likely a placebo effect, and he sees potential harm in that effect. If the pain is not truly decreasing as patients continue using those therapies, then the pain may become a more stable part of the nervous system, “so I think they need to do an intervention which really has evidence in reducing pain, an active approach that means exercising in the right way,” Dr. Van Der Esch said. If patients are undergoing therapy whose primary benefit is a placebo effect, “the pain will prolong and become more fixed in the nervous system,” shifting the patients toward greater risk of the pain becoming chronic, he said.
“I want to emphasize that we have an ethical role to our management, and it’s not ethical to give treatments which have no response and no pain relief except that the patient or the professional believes it will have an effect,” Dr. Van Der Esch said.
The research did not involve outside funding. Dr. French, Dr. Hall, and Dr. Van Der Esch reported having no relevant financial relationships.
DENVER – Adding therapies such as acupuncture, electrophysical stimulation, or other interventions to standard exercise therapy does not appear to offer much benefit in pain relief or physical function for patients with knee osteoarthritis, according to a study presented at the OARSI 2023 World Congress. The findings were also published in the Cochrane Database of Systematic Reviews in October 2022.
“The results do not support the use of adjunctive therapies when we add them to exercise for pain, physical function, or quality of life, when compared against placebo, adjunctive therapy, and exercise,” Helen P. French, PhD, told attendees at the meeting sponsored by the Osteoarthritis Research Society International. The findings were similar for pain and physical function when comparing adjunctive therapies with exercise against exercise alone, said Dr. French, an associate professor in physiotherapy at the Royal College of Surgeons, Dublin, except that patients using adjunctive therapies reported feeling greater improvement in their global assessments.
Exercise is recommended as a core treatment for osteoarthritis, but some patients or clinicians may be interested in supplementing that therapy with acupuncture, heat therapy, electromagnetic fields, transcutaneous electrical nerve stimulation, braces/orthotics, and other interventions. Various Cochrane Reviews of the evidence exist for these interventions in treating chronic pain in general but not for their use as adjunctive therapies in addition to exercise for osteoarthritis pain.
Researchers therefore assessed the evidence for improvement in pain, physical function, and quality of life for two sets of comparisons: adjunctive therapies plus exercise versus exercise alone, and adjunctive therapies with exercise versus placebo adjunctive therapy with exercise. The review excluded studies looking at medications or supplements.
Pain was assessed with the Numeric Pain Rating Scale (NPRS, 0-10), with an improvement of at least 2 points (15% improvement) representing the minimum clinically important difference (MCID). Physical function was assessed with the Western Ontario and McMaster Universities Arthritis Index (WOMAC, 0-68), with 6 points (15%) considered the MCID, and quality of life was assessed with the SF-36 (0-100), with 6 points (12%) as the MCID.
The researchers identified trials on knee osteoarthritis that included an overall 6,508 participants with an average age ranging from 52 to 83 years. A total of 36 studies evaluated electrophysical agents. Another seven looked at manual therapies; four looked at acupuncture/dry needling or taping; three looked at psychological, dietary, or “whole body vibration” therapies; and two evaluated spa or peloid therapy. Only one trial evaluated foot insoles.
Nearly all the studies (98%) assessed pain, and most (87%) assessed physical function. Only about one in five (21%) assessed quality of life. The improvement in pain from adding adjunctive therapies to exercise, compared with placebo therapies plus exercise, was 0.77 points, or just under a 10% improvement, which fell short of the 15% MCID. Physical function improvement similarly fell short, with an average improvement of 5 points (12%).
In comparisons of exercise plus adjunctive therapies against exercise alone, the improvement from the additional interventions was even lower. Pain improvement was 0.41 points (7%), and physical function improvement was 2.8 points (9%). However, patients’ perceptions told a different story: 37% more patients who were using an adjunctive therapy reported feeling that the therapies were successful, compared with patients undergoing exercise therapy alone.
Adverse events were poorly reported in the trials, with only 10 trials reporting them at all, and the researchers found no significant difference in adverse events among the studies reporting them. The most common adverse events were increased pain in the joint with the osteoarthritis, pain elsewhere, or swelling and inflammation. It’s unclear, however, whether the pain, swelling, and inflammation were related to the interventions and how serious these effects might have been.
Michelle Hall, PhD, an associate professor in the department of physiotherapy at the University of Melbourne, comoderated the session with this presentation and found it interesting that more than one-third of patients perceived that they did better with the additional therapies even though improvement didn’t bear out in their pain or physical function assessments.
“But the other part of that was that the studies were of poor quality, so we can’t say with confidence, ‘Don’t do this therapy because it’s not going to work,’ ” Dr. Hall said in an interview. She said she personally would probably discourage patients from those therapies, “but I don’t think the evidence is there for everybody to do that,” she added.
Martin Van Der Esch, PhD, of Reade Centre of Rehabilitation and Rheumatology in Amsterdam, also comoderated the discussion and had more concerns about the use of adjunctive therapies in light of the study’s findings. He said in an interview that he tended to believe the patients’ overall self-reported improvement is likely a placebo effect, and he sees potential harm in that effect. If the pain is not truly decreasing as patients continue using those therapies, then the pain may become a more stable part of the nervous system, “so I think they need to do an intervention which really has evidence in reducing pain, an active approach that means exercising in the right way,” Dr. Van Der Esch said. If patients are undergoing therapy whose primary benefit is a placebo effect, “the pain will prolong and become more fixed in the nervous system,” shifting the patients toward greater risk of the pain becoming chronic, he said.
“I want to emphasize that we have an ethical role to our management, and it’s not ethical to give treatments which have no response and no pain relief except that the patient or the professional believes it will have an effect,” Dr. Van Der Esch said.
The research did not involve outside funding. Dr. French, Dr. Hall, and Dr. Van Der Esch reported having no relevant financial relationships.
DENVER – Adding therapies such as acupuncture, electrophysical stimulation, or other interventions to standard exercise therapy does not appear to offer much benefit in pain relief or physical function for patients with knee osteoarthritis, according to a study presented at the OARSI 2023 World Congress. The findings were also published in the Cochrane Database of Systematic Reviews in October 2022.
“The results do not support the use of adjunctive therapies when we add them to exercise for pain, physical function, or quality of life, when compared against placebo, adjunctive therapy, and exercise,” Helen P. French, PhD, told attendees at the meeting sponsored by the Osteoarthritis Research Society International. The findings were similar for pain and physical function when comparing adjunctive therapies with exercise against exercise alone, said Dr. French, an associate professor in physiotherapy at the Royal College of Surgeons, Dublin, except that patients using adjunctive therapies reported feeling greater improvement in their global assessments.
Exercise is recommended as a core treatment for osteoarthritis, but some patients or clinicians may be interested in supplementing that therapy with acupuncture, heat therapy, electromagnetic fields, transcutaneous electrical nerve stimulation, braces/orthotics, and other interventions. Various Cochrane Reviews of the evidence exist for these interventions in treating chronic pain in general but not for their use as adjunctive therapies in addition to exercise for osteoarthritis pain.
Researchers therefore assessed the evidence for improvement in pain, physical function, and quality of life for two sets of comparisons: adjunctive therapies plus exercise versus exercise alone, and adjunctive therapies with exercise versus placebo adjunctive therapy with exercise. The review excluded studies looking at medications or supplements.
Pain was assessed with the Numeric Pain Rating Scale (NPRS, 0-10), with an improvement of at least 2 points (15% improvement) representing the minimum clinically important difference (MCID). Physical function was assessed with the Western Ontario and McMaster Universities Arthritis Index (WOMAC, 0-68), with 6 points (15%) considered the MCID, and quality of life was assessed with the SF-36 (0-100), with 6 points (12%) as the MCID.
The researchers identified trials on knee osteoarthritis that included an overall 6,508 participants with an average age ranging from 52 to 83 years. A total of 36 studies evaluated electrophysical agents. Another seven looked at manual therapies; four looked at acupuncture/dry needling or taping; three looked at psychological, dietary, or “whole body vibration” therapies; and two evaluated spa or peloid therapy. Only one trial evaluated foot insoles.
Nearly all the studies (98%) assessed pain, and most (87%) assessed physical function. Only about one in five (21%) assessed quality of life. The improvement in pain from adding adjunctive therapies to exercise, compared with placebo therapies plus exercise, was 0.77 points, or just under a 10% improvement, which fell short of the 15% MCID. Physical function improvement similarly fell short, with an average improvement of 5 points (12%).
In comparisons of exercise plus adjunctive therapies against exercise alone, the improvement from the additional interventions was even lower. Pain improvement was 0.41 points (7%), and physical function improvement was 2.8 points (9%). However, patients’ perceptions told a different story: 37% more patients who were using an adjunctive therapy reported feeling that the therapies were successful, compared with patients undergoing exercise therapy alone.
Adverse events were poorly reported in the trials, with only 10 trials reporting them at all, and the researchers found no significant difference in adverse events among the studies reporting them. The most common adverse events were increased pain in the joint with the osteoarthritis, pain elsewhere, or swelling and inflammation. It’s unclear, however, whether the pain, swelling, and inflammation were related to the interventions and how serious these effects might have been.
Michelle Hall, PhD, an associate professor in the department of physiotherapy at the University of Melbourne, comoderated the session with this presentation and found it interesting that more than one-third of patients perceived that they did better with the additional therapies even though improvement didn’t bear out in their pain or physical function assessments.
“But the other part of that was that the studies were of poor quality, so we can’t say with confidence, ‘Don’t do this therapy because it’s not going to work,’ ” Dr. Hall said in an interview. She said she personally would probably discourage patients from those therapies, “but I don’t think the evidence is there for everybody to do that,” she added.
Martin Van Der Esch, PhD, of Reade Centre of Rehabilitation and Rheumatology in Amsterdam, also comoderated the discussion and had more concerns about the use of adjunctive therapies in light of the study’s findings. He said in an interview that he tended to believe the patients’ overall self-reported improvement is likely a placebo effect, and he sees potential harm in that effect. If the pain is not truly decreasing as patients continue using those therapies, then the pain may become a more stable part of the nervous system, “so I think they need to do an intervention which really has evidence in reducing pain, an active approach that means exercising in the right way,” Dr. Van Der Esch said. If patients are undergoing therapy whose primary benefit is a placebo effect, “the pain will prolong and become more fixed in the nervous system,” shifting the patients toward greater risk of the pain becoming chronic, he said.
“I want to emphasize that we have an ethical role to our management, and it’s not ethical to give treatments which have no response and no pain relief except that the patient or the professional believes it will have an effect,” Dr. Van Der Esch said.
The research did not involve outside funding. Dr. French, Dr. Hall, and Dr. Van Der Esch reported having no relevant financial relationships.
AT OARSI 2023
Antidepressants benefit some patients with osteoarthritis pain
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
FROM OARSI 2023
JAK inhibitor ivarmacitinib shows efficacy for atopic dermatitis in a pivotal trial
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
AT AAD 2023
DMARDs taper-to-discontinuation trial deemed inconclusive
The small size of a new study of the feasibility of tapering conventional synthetic disease-modifying antirheumatic drug (csDMARD) doses to half for patients with rheumatoid arthritis in remission, and then to zero, makes suspect the validity of its finding of no statistical difference between continuing half doses and stopping altogether, according to one rheumatologist’s analysis.
In the open-label, randomized trial of 56 patients, which was published as a research letter in JAMA, more patients in the group that discontinued csDMARDs experienced flares within 1 year than did the half-dose group, but this difference was not statistically significant.
Most patients in the drug-free group did not experience disease flares, the authors note.
“The results show that in this population, a majority of patients remained flare-free for at least a year after csDMARD discontinuation. This highlights a potential for drug-free remission in a subgroup of RA patients, and the data provide a basis for shared decision-making in this patient group. We know that tapering is a common question from patients and thus think that the data are especially clinically relevant,” first author Siri Lillegraven, MD, MPH, PhD, of the Center for Treatment of Rheumatic and Musculoskeletal Diseases at the Diakonhjemmet Hospital, Oslo, said in an interview.
While several studies have demonstrated that patients with RA can maintain remission on lower doses of medication, James O’Dell, MD, chief of the Division of Rheumatology at the University of Nebraska Medical Center, Omaha, urged caution in interpreting these results because the study was so small – just 56 patients. “Every analysis they did favored staying on treatment, but the confidence interval slightly crossed null, so they can’t say [that group] was superior,” Dr. O’Dell said in an interview. He was not involved in the research. “Had this study been double this size and they got the same results, they would have clearly shown that staying on medicines were superior,” he said.
Dr. Lillegraven acknowledged the impact of the trial’s small sample size. “This is a study with a limited study sample, and it is conceivable that a larger study might have shown a statistical difference between the groups,” she said.
In addition to the small number of patients in the study, Dr. O’Dell also noted that this study group was already a selected group of patients who had maintained remission on half-dose therapy for at least 1 year. Even then, “what they showed was that 39% of the patients who they discontinued [then] flared, compared with 17% when they didn’t taper [off medication],” he said. “That’s a pretty important clinical difference.”
While Dr. O’Dell thinks the study was too small to inform practice, he emphasized that tapering off full doses of medications can be beneficial for patients with RA that has been in remission for 6 months or longer. “It seems to take less medicines to keep somebody in remission than it did to get them there in the first place,” he said. “I come out strongly in favor of tapering medications in rheumatoid arthritis patients who are in remission, and that includes tapering and stopping biologics if patients are on conventional therapy,” he added, “but tapering patients off all of their conventional therapy is something that I think is a bridge too far.”
This trial was the second part of the ARCTIC REWIND study, which involved patients with RA that was in sustained remission, per their Disease Activity Score. In the first part of the trial, 160 participants from 10 hospitals in Norway were enrolled and were randomly assigned to either continue their standard csDMARD dosing or taper down to a half dose. Patients whose doses were tapered to a half dose and whose conditions were in remission for 1 year were eligible for the second half the study.
Of the 56 participants who were included, 26 discontinued csDMARD therapy, while 30 continued taking a half dose for 12 months of follow-up. Most patients in both groups had received methotrexate monotherapy (21 in the discontinuation group and 26 in the continued half-dosing group). Triple therapy (methotrexate, sulfasalazine, and hydroxychloroquine) was used by three patients in the discontinuation group and by two in the half-dose group. Two additional patients in the discontinuation group and two in the half-dose group took other mono/duo therapies. Clinic visits occurred every 4 months; visits were more frequent if there was an increase in disease activity. For patients who experienced a disease flare, full-dose csDMARD treatment was resumed.
Ten patients in the discontinuation group experienced flares during 1 year, compared with five patients in the half-dose group. The risk difference between the two groups was not statistically significant (RD, 21.5%; 95% CI, –3.4% to 49.7%). The median time to flare was 179 days in the discontinuation group and 133 days in the half-dose group.
Of those who experienced flares, 8 of 10 patients in the discontinuation group and 2 of 5 in the half-dose group regained remission when full-dose therapy was resumed.
The study was funded by the Research Council of Norway and the South-Eastern Norway Regional Health Authorities. Many of the authors disclosed financial ties to pharmaceutical companies. Dr. O’Dell disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The small size of a new study of the feasibility of tapering conventional synthetic disease-modifying antirheumatic drug (csDMARD) doses to half for patients with rheumatoid arthritis in remission, and then to zero, makes suspect the validity of its finding of no statistical difference between continuing half doses and stopping altogether, according to one rheumatologist’s analysis.
In the open-label, randomized trial of 56 patients, which was published as a research letter in JAMA, more patients in the group that discontinued csDMARDs experienced flares within 1 year than did the half-dose group, but this difference was not statistically significant.
Most patients in the drug-free group did not experience disease flares, the authors note.
“The results show that in this population, a majority of patients remained flare-free for at least a year after csDMARD discontinuation. This highlights a potential for drug-free remission in a subgroup of RA patients, and the data provide a basis for shared decision-making in this patient group. We know that tapering is a common question from patients and thus think that the data are especially clinically relevant,” first author Siri Lillegraven, MD, MPH, PhD, of the Center for Treatment of Rheumatic and Musculoskeletal Diseases at the Diakonhjemmet Hospital, Oslo, said in an interview.
While several studies have demonstrated that patients with RA can maintain remission on lower doses of medication, James O’Dell, MD, chief of the Division of Rheumatology at the University of Nebraska Medical Center, Omaha, urged caution in interpreting these results because the study was so small – just 56 patients. “Every analysis they did favored staying on treatment, but the confidence interval slightly crossed null, so they can’t say [that group] was superior,” Dr. O’Dell said in an interview. He was not involved in the research. “Had this study been double this size and they got the same results, they would have clearly shown that staying on medicines were superior,” he said.
Dr. Lillegraven acknowledged the impact of the trial’s small sample size. “This is a study with a limited study sample, and it is conceivable that a larger study might have shown a statistical difference between the groups,” she said.
In addition to the small number of patients in the study, Dr. O’Dell also noted that this study group was already a selected group of patients who had maintained remission on half-dose therapy for at least 1 year. Even then, “what they showed was that 39% of the patients who they discontinued [then] flared, compared with 17% when they didn’t taper [off medication],” he said. “That’s a pretty important clinical difference.”
While Dr. O’Dell thinks the study was too small to inform practice, he emphasized that tapering off full doses of medications can be beneficial for patients with RA that has been in remission for 6 months or longer. “It seems to take less medicines to keep somebody in remission than it did to get them there in the first place,” he said. “I come out strongly in favor of tapering medications in rheumatoid arthritis patients who are in remission, and that includes tapering and stopping biologics if patients are on conventional therapy,” he added, “but tapering patients off all of their conventional therapy is something that I think is a bridge too far.”
This trial was the second part of the ARCTIC REWIND study, which involved patients with RA that was in sustained remission, per their Disease Activity Score. In the first part of the trial, 160 participants from 10 hospitals in Norway were enrolled and were randomly assigned to either continue their standard csDMARD dosing or taper down to a half dose. Patients whose doses were tapered to a half dose and whose conditions were in remission for 1 year were eligible for the second half the study.
Of the 56 participants who were included, 26 discontinued csDMARD therapy, while 30 continued taking a half dose for 12 months of follow-up. Most patients in both groups had received methotrexate monotherapy (21 in the discontinuation group and 26 in the continued half-dosing group). Triple therapy (methotrexate, sulfasalazine, and hydroxychloroquine) was used by three patients in the discontinuation group and by two in the half-dose group. Two additional patients in the discontinuation group and two in the half-dose group took other mono/duo therapies. Clinic visits occurred every 4 months; visits were more frequent if there was an increase in disease activity. For patients who experienced a disease flare, full-dose csDMARD treatment was resumed.
Ten patients in the discontinuation group experienced flares during 1 year, compared with five patients in the half-dose group. The risk difference between the two groups was not statistically significant (RD, 21.5%; 95% CI, –3.4% to 49.7%). The median time to flare was 179 days in the discontinuation group and 133 days in the half-dose group.
Of those who experienced flares, 8 of 10 patients in the discontinuation group and 2 of 5 in the half-dose group regained remission when full-dose therapy was resumed.
The study was funded by the Research Council of Norway and the South-Eastern Norway Regional Health Authorities. Many of the authors disclosed financial ties to pharmaceutical companies. Dr. O’Dell disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The small size of a new study of the feasibility of tapering conventional synthetic disease-modifying antirheumatic drug (csDMARD) doses to half for patients with rheumatoid arthritis in remission, and then to zero, makes suspect the validity of its finding of no statistical difference between continuing half doses and stopping altogether, according to one rheumatologist’s analysis.
In the open-label, randomized trial of 56 patients, which was published as a research letter in JAMA, more patients in the group that discontinued csDMARDs experienced flares within 1 year than did the half-dose group, but this difference was not statistically significant.
Most patients in the drug-free group did not experience disease flares, the authors note.
“The results show that in this population, a majority of patients remained flare-free for at least a year after csDMARD discontinuation. This highlights a potential for drug-free remission in a subgroup of RA patients, and the data provide a basis for shared decision-making in this patient group. We know that tapering is a common question from patients and thus think that the data are especially clinically relevant,” first author Siri Lillegraven, MD, MPH, PhD, of the Center for Treatment of Rheumatic and Musculoskeletal Diseases at the Diakonhjemmet Hospital, Oslo, said in an interview.
While several studies have demonstrated that patients with RA can maintain remission on lower doses of medication, James O’Dell, MD, chief of the Division of Rheumatology at the University of Nebraska Medical Center, Omaha, urged caution in interpreting these results because the study was so small – just 56 patients. “Every analysis they did favored staying on treatment, but the confidence interval slightly crossed null, so they can’t say [that group] was superior,” Dr. O’Dell said in an interview. He was not involved in the research. “Had this study been double this size and they got the same results, they would have clearly shown that staying on medicines were superior,” he said.
Dr. Lillegraven acknowledged the impact of the trial’s small sample size. “This is a study with a limited study sample, and it is conceivable that a larger study might have shown a statistical difference between the groups,” she said.
In addition to the small number of patients in the study, Dr. O’Dell also noted that this study group was already a selected group of patients who had maintained remission on half-dose therapy for at least 1 year. Even then, “what they showed was that 39% of the patients who they discontinued [then] flared, compared with 17% when they didn’t taper [off medication],” he said. “That’s a pretty important clinical difference.”
While Dr. O’Dell thinks the study was too small to inform practice, he emphasized that tapering off full doses of medications can be beneficial for patients with RA that has been in remission for 6 months or longer. “It seems to take less medicines to keep somebody in remission than it did to get them there in the first place,” he said. “I come out strongly in favor of tapering medications in rheumatoid arthritis patients who are in remission, and that includes tapering and stopping biologics if patients are on conventional therapy,” he added, “but tapering patients off all of their conventional therapy is something that I think is a bridge too far.”
This trial was the second part of the ARCTIC REWIND study, which involved patients with RA that was in sustained remission, per their Disease Activity Score. In the first part of the trial, 160 participants from 10 hospitals in Norway were enrolled and were randomly assigned to either continue their standard csDMARD dosing or taper down to a half dose. Patients whose doses were tapered to a half dose and whose conditions were in remission for 1 year were eligible for the second half the study.
Of the 56 participants who were included, 26 discontinued csDMARD therapy, while 30 continued taking a half dose for 12 months of follow-up. Most patients in both groups had received methotrexate monotherapy (21 in the discontinuation group and 26 in the continued half-dosing group). Triple therapy (methotrexate, sulfasalazine, and hydroxychloroquine) was used by three patients in the discontinuation group and by two in the half-dose group. Two additional patients in the discontinuation group and two in the half-dose group took other mono/duo therapies. Clinic visits occurred every 4 months; visits were more frequent if there was an increase in disease activity. For patients who experienced a disease flare, full-dose csDMARD treatment was resumed.
Ten patients in the discontinuation group experienced flares during 1 year, compared with five patients in the half-dose group. The risk difference between the two groups was not statistically significant (RD, 21.5%; 95% CI, –3.4% to 49.7%). The median time to flare was 179 days in the discontinuation group and 133 days in the half-dose group.
Of those who experienced flares, 8 of 10 patients in the discontinuation group and 2 of 5 in the half-dose group regained remission when full-dose therapy was resumed.
The study was funded by the Research Council of Norway and the South-Eastern Norway Regional Health Authorities. Many of the authors disclosed financial ties to pharmaceutical companies. Dr. O’Dell disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM JAMA
Luxe vacations, private jets: Medical device maker, surgeon to pay $46 million penalty in kickback scheme
according to experts familiar with the federal Anti-Kickback Statute.
Historically, enforcement actions have primarily focused on the person or organization offering the perks – and not necessarily the physicians accepting it, Steven W. Ortquist, founder and principal of Arete Compliance Solutions, LLC, in Phoenix, told this news organization.
But that’s changing.
“In recent years, we are seeing a trend toward holding physicians and others on the receiving end of the inducement accountable as well,” said Mr. Ortquist, who is a past board member and president of the Health Care Compliance Association. He noted that authorities usually pursue the inducing company first before moving on to individual clinicians or practices.
The Department of Justice followed a similar pattern in a recently announced kickback settlement that ensnared an intraocular lens distributor, an ophthalmology equipment supplier, two CEOs, and a surgeon. Precision Lens must pay more than $43 million for offering high-end vacations and other expensive perks to surgeons who used its cataract products.
The verdict marks the end of a 6-week civil jury trial, where evidence emerged that Paul Ehlen, owner of Precision Lens and its parent company, Cameron-Ehlen Group, maintained a secret “slush fund” for paying kickbacks to ophthalmic surgeons. The inducement scheme netted the Minnesota-based company millions in sales and led to the submission of 64,575 false Medicare claims from 2006 to 2015, a violation of the Anti-Kickback Statute and the False Claims Act.
According to court documents, physicians received luxury travel and entertainment packages, including skiing, fishing, and golfing excursions at exclusive destinations, often traveling via private jet to attend Broadway musicals and major sporting events. Mr. Ehlen and company representatives also sold frequent flyer miles to physicians at a steep discount, allowing them to take personal and business trips below fair market value.
Federal authorities initially announced an investigation into the business practices of Precision Lens in 2017 after receiving a whistleblower complaint from Kipp Fesenmaier, a former executive at Sightpath Medical, an ophthalmology supplier and “corporate partner” of Precision Lens. Mr. Fesenmaier alleged that both companies were involved in an inducement scheme.
Sightpath Medical and its CEO, James Tiffany, agreed to a $12 million settlement to resolve the kickback allegations.
The Department of Justice subsequently investigated Jitendra Swarup, MD, an ophthalmologist and cataract surgeon who allegedly received “unlawful remuneration from Sightpath, Precision, and Ehlen” and filed false insurance claims. In addition to accepting expensive hunting and fishing trips from the medical device companies, Dr. Swarup was paid more than $100,000 per year for consulting services he did not fully render.
Dr. Swarup agreed to a nearly $3 million settlement and participation in a 3-year corporate integrity agreement with the Office of Inspector General. In exchange for compliance with such contracts, the OIG permits physicians to continue participating in Medicare, Medicaid, and other federal health care programs.
In a statement from attorneys, Precision Lens and Mr. Ehlen pledged to appeal the verdict and “defend ... our wholly appropriate actions” while remaining focused on their commitment to health care clinicians and manufacturers.
‘Endless’ opportunities for inducement
Unfortunately, opportunities for inducement are “endless,” experts say. Extravagant trips, dinners, and gifts can trigger a violation, but so can nearly anything of value.
Just last year, Biotronik reached a $12.95 million settlement amid allegations that company representatives wined and dined physicians to induce their use of its pacemakers and defibrillators. To date, no physicians have been charged.
But after a record-breaking number of whistleblower judgments last fiscal year totaling more than $2 billion, physicians should take note, Radha Bhatnagar, Esq, director of compliance at The CM Group, told the news organization.
“When manufacturers offer physicians kickbacks with the added element of fraudulent Medicare or Medicaid reimbursements, that is typically when manufacturers and individuals face civil and criminal liability,” said Ms. Bhatnagar, something the Department of Justice alluded to when announcing a settlement involving 15 Texas physicians last year.
In another case, Kingsley R. Chin, an orthopedic surgeon and designer of a spinal implant, was indicted in 2021 for paying millions of dollars in sham consulting fees to physicians who used his products. At least six surgeons who accepted money from Dr. Chin were later named in a civil case and ordered to pay $3.3 million in penalties.
Jason Montone, DO, an orthopedic surgeon who accepted the illicit payments, agreed to a plea deal with a reduced prison sentence, 1 year of supervised release, and a fine of $379,000.
Although Dr. Chin’s sentencing hasn’t been announced, violating kickback laws can result in a sentence of up to 10 years.
A version of this article originally appeared on Medscape.com.
according to experts familiar with the federal Anti-Kickback Statute.
Historically, enforcement actions have primarily focused on the person or organization offering the perks – and not necessarily the physicians accepting it, Steven W. Ortquist, founder and principal of Arete Compliance Solutions, LLC, in Phoenix, told this news organization.
But that’s changing.
“In recent years, we are seeing a trend toward holding physicians and others on the receiving end of the inducement accountable as well,” said Mr. Ortquist, who is a past board member and president of the Health Care Compliance Association. He noted that authorities usually pursue the inducing company first before moving on to individual clinicians or practices.
The Department of Justice followed a similar pattern in a recently announced kickback settlement that ensnared an intraocular lens distributor, an ophthalmology equipment supplier, two CEOs, and a surgeon. Precision Lens must pay more than $43 million for offering high-end vacations and other expensive perks to surgeons who used its cataract products.
The verdict marks the end of a 6-week civil jury trial, where evidence emerged that Paul Ehlen, owner of Precision Lens and its parent company, Cameron-Ehlen Group, maintained a secret “slush fund” for paying kickbacks to ophthalmic surgeons. The inducement scheme netted the Minnesota-based company millions in sales and led to the submission of 64,575 false Medicare claims from 2006 to 2015, a violation of the Anti-Kickback Statute and the False Claims Act.
According to court documents, physicians received luxury travel and entertainment packages, including skiing, fishing, and golfing excursions at exclusive destinations, often traveling via private jet to attend Broadway musicals and major sporting events. Mr. Ehlen and company representatives also sold frequent flyer miles to physicians at a steep discount, allowing them to take personal and business trips below fair market value.
Federal authorities initially announced an investigation into the business practices of Precision Lens in 2017 after receiving a whistleblower complaint from Kipp Fesenmaier, a former executive at Sightpath Medical, an ophthalmology supplier and “corporate partner” of Precision Lens. Mr. Fesenmaier alleged that both companies were involved in an inducement scheme.
Sightpath Medical and its CEO, James Tiffany, agreed to a $12 million settlement to resolve the kickback allegations.
The Department of Justice subsequently investigated Jitendra Swarup, MD, an ophthalmologist and cataract surgeon who allegedly received “unlawful remuneration from Sightpath, Precision, and Ehlen” and filed false insurance claims. In addition to accepting expensive hunting and fishing trips from the medical device companies, Dr. Swarup was paid more than $100,000 per year for consulting services he did not fully render.
Dr. Swarup agreed to a nearly $3 million settlement and participation in a 3-year corporate integrity agreement with the Office of Inspector General. In exchange for compliance with such contracts, the OIG permits physicians to continue participating in Medicare, Medicaid, and other federal health care programs.
In a statement from attorneys, Precision Lens and Mr. Ehlen pledged to appeal the verdict and “defend ... our wholly appropriate actions” while remaining focused on their commitment to health care clinicians and manufacturers.
‘Endless’ opportunities for inducement
Unfortunately, opportunities for inducement are “endless,” experts say. Extravagant trips, dinners, and gifts can trigger a violation, but so can nearly anything of value.
Just last year, Biotronik reached a $12.95 million settlement amid allegations that company representatives wined and dined physicians to induce their use of its pacemakers and defibrillators. To date, no physicians have been charged.
But after a record-breaking number of whistleblower judgments last fiscal year totaling more than $2 billion, physicians should take note, Radha Bhatnagar, Esq, director of compliance at The CM Group, told the news organization.
“When manufacturers offer physicians kickbacks with the added element of fraudulent Medicare or Medicaid reimbursements, that is typically when manufacturers and individuals face civil and criminal liability,” said Ms. Bhatnagar, something the Department of Justice alluded to when announcing a settlement involving 15 Texas physicians last year.
In another case, Kingsley R. Chin, an orthopedic surgeon and designer of a spinal implant, was indicted in 2021 for paying millions of dollars in sham consulting fees to physicians who used his products. At least six surgeons who accepted money from Dr. Chin were later named in a civil case and ordered to pay $3.3 million in penalties.
Jason Montone, DO, an orthopedic surgeon who accepted the illicit payments, agreed to a plea deal with a reduced prison sentence, 1 year of supervised release, and a fine of $379,000.
Although Dr. Chin’s sentencing hasn’t been announced, violating kickback laws can result in a sentence of up to 10 years.
A version of this article originally appeared on Medscape.com.
according to experts familiar with the federal Anti-Kickback Statute.
Historically, enforcement actions have primarily focused on the person or organization offering the perks – and not necessarily the physicians accepting it, Steven W. Ortquist, founder and principal of Arete Compliance Solutions, LLC, in Phoenix, told this news organization.
But that’s changing.
“In recent years, we are seeing a trend toward holding physicians and others on the receiving end of the inducement accountable as well,” said Mr. Ortquist, who is a past board member and president of the Health Care Compliance Association. He noted that authorities usually pursue the inducing company first before moving on to individual clinicians or practices.
The Department of Justice followed a similar pattern in a recently announced kickback settlement that ensnared an intraocular lens distributor, an ophthalmology equipment supplier, two CEOs, and a surgeon. Precision Lens must pay more than $43 million for offering high-end vacations and other expensive perks to surgeons who used its cataract products.
The verdict marks the end of a 6-week civil jury trial, where evidence emerged that Paul Ehlen, owner of Precision Lens and its parent company, Cameron-Ehlen Group, maintained a secret “slush fund” for paying kickbacks to ophthalmic surgeons. The inducement scheme netted the Minnesota-based company millions in sales and led to the submission of 64,575 false Medicare claims from 2006 to 2015, a violation of the Anti-Kickback Statute and the False Claims Act.
According to court documents, physicians received luxury travel and entertainment packages, including skiing, fishing, and golfing excursions at exclusive destinations, often traveling via private jet to attend Broadway musicals and major sporting events. Mr. Ehlen and company representatives also sold frequent flyer miles to physicians at a steep discount, allowing them to take personal and business trips below fair market value.
Federal authorities initially announced an investigation into the business practices of Precision Lens in 2017 after receiving a whistleblower complaint from Kipp Fesenmaier, a former executive at Sightpath Medical, an ophthalmology supplier and “corporate partner” of Precision Lens. Mr. Fesenmaier alleged that both companies were involved in an inducement scheme.
Sightpath Medical and its CEO, James Tiffany, agreed to a $12 million settlement to resolve the kickback allegations.
The Department of Justice subsequently investigated Jitendra Swarup, MD, an ophthalmologist and cataract surgeon who allegedly received “unlawful remuneration from Sightpath, Precision, and Ehlen” and filed false insurance claims. In addition to accepting expensive hunting and fishing trips from the medical device companies, Dr. Swarup was paid more than $100,000 per year for consulting services he did not fully render.
Dr. Swarup agreed to a nearly $3 million settlement and participation in a 3-year corporate integrity agreement with the Office of Inspector General. In exchange for compliance with such contracts, the OIG permits physicians to continue participating in Medicare, Medicaid, and other federal health care programs.
In a statement from attorneys, Precision Lens and Mr. Ehlen pledged to appeal the verdict and “defend ... our wholly appropriate actions” while remaining focused on their commitment to health care clinicians and manufacturers.
‘Endless’ opportunities for inducement
Unfortunately, opportunities for inducement are “endless,” experts say. Extravagant trips, dinners, and gifts can trigger a violation, but so can nearly anything of value.
Just last year, Biotronik reached a $12.95 million settlement amid allegations that company representatives wined and dined physicians to induce their use of its pacemakers and defibrillators. To date, no physicians have been charged.
But after a record-breaking number of whistleblower judgments last fiscal year totaling more than $2 billion, physicians should take note, Radha Bhatnagar, Esq, director of compliance at The CM Group, told the news organization.
“When manufacturers offer physicians kickbacks with the added element of fraudulent Medicare or Medicaid reimbursements, that is typically when manufacturers and individuals face civil and criminal liability,” said Ms. Bhatnagar, something the Department of Justice alluded to when announcing a settlement involving 15 Texas physicians last year.
In another case, Kingsley R. Chin, an orthopedic surgeon and designer of a spinal implant, was indicted in 2021 for paying millions of dollars in sham consulting fees to physicians who used his products. At least six surgeons who accepted money from Dr. Chin were later named in a civil case and ordered to pay $3.3 million in penalties.
Jason Montone, DO, an orthopedic surgeon who accepted the illicit payments, agreed to a plea deal with a reduced prison sentence, 1 year of supervised release, and a fine of $379,000.
Although Dr. Chin’s sentencing hasn’t been announced, violating kickback laws can result in a sentence of up to 10 years.
A version of this article originally appeared on Medscape.com.
FDA approves new formulation of Hyrimoz adalimumab biosimilar
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.