User login
Infiltrating Wound Vacuum-Assisted Closure With Topical Amphotericin for Mucormycosis Infection of the Achilles Tendon
Vacuum-assisted closure (VAC) of wounds has become a foundational tool in the armamentarium of wound care specialists. Using a system consisting of a sponge, semi-occlusive barrier, and fluid collection device, VAC systems apply constant negative pressure resulting in macro and micro deformation to a wound, stabilization of the wound environment, and removal of inflammatory factors in wound fluid.1 These conditions allow for the removal of drainage and fluid from a wound bed, reduced edema and inflammation, reduced bacterial load, recruitment of healing factors, approximation of wound edges, and increased blood flow to the wound.2
In complex, infected wounds, a variation of negative pressure wound therapy (NPWT) via the instillation of topical antibiotics (instillation VAC) has been used.3 This variation has been advantageous even in soft tissue fungal infections. Early and aggressive treatment of such infections is critical to prevent dissemination, particularly in aggressive infections, such as mucormycosis.4 We present a case of a patient with a mucormycosis infection of his left Achilles tendon and overlying skin who was successfully treated with surgical debridement and wound care with instillation NPWT with topical amphotericin B.
Case Presentation
A 53-year-old man underwent left Achilles tendon reconstruction with allograft after a complete tear during exercise. He had no relevant medical history and was otherwise healthy, which he attributed to working out daily. About a week after the operation, he began having incisional breakdown, prompting presentation to an emergency department. There, he received IV antibiotics along with multiple debridements. After the wound failed to improve and intra-operative cultures grew mucormycosis, he was transferred to our facility for a higher level of care. On admission, he was immediately given IV amphotericin B and scheduled for repeat debridement.
After 1 prior debridement and 10 total days of IV amphotericin, a repeat debridement was performed. After the debridement, the installation VAC was applied to the patient’s left lower extremity wound with an instilling fluid of amphotericin B and the settings as follows: smart phase instill volume, 110 mL; soak time, 3.5 hours; target pressure, 125 mm Hg; intensity, low; and VAC therapy mode, continuous. After 5 days, the wound bed appeared clean without overt signs of infection. However, due to some toxicity to healthy surrounding soft tissue, the instillation VAC was discontinued and standard NPWT was started. The patient underwent 2 additional rounds of debridement with partial delayed closure. Four weeks after discontinuation of the instillation VAC, the wound appeared healthy and granulated so the patient underwent split-thickness skin grafting to the left posterior ankle. He subsequently completed a course of oral antifungal medication as an outpatient.
The patient was seen in the outpatient clinic for 14 months from the initial mucormycosis infection (Figure). 
Discussion
Mucormycosis is an infection caused by fungi in the class Zygomycetes and of the order Mucorales that typically occurs in immunocompromised patients, especially those with diabetic ketoacidosis and neutropenia. Given that this patient had no relevant medical history and was otherwise healthy, he was at extremely low risk of this type of infection. In this patient’s case, the spores of this nonseptate hyphae wide-branching species were most likely introduced at the time of left Achilles tendon repair. Mucormycosis is progressive and can be fatal unless treated, with a mortality rate approaching 70%.5 The rarity and heterogeneity of mucormycosis make treatment variable.6 No prospective or randomized clinical trials exist in plastic surgery literature.
The use of wound VAC in combination with the instillation of amphotericin B to treat cutaneous mucormycosis is not well documented. Mucormycosis infections are traditionally addressed with surgical debridement and antifungal therapy, specifically IV amphotericin B.7,8 As previously noted, NPWT has become the gold standard in treating complex wounds.3 Additionally, wound VAC therapy with instillation has been noted in the literature as a reliable method to treat bacteria-infected wounds, providing a shorter treatment period and earlier wound closure.9 Instillation VAC therapy has proven particularly useful in complex, infected wounds, such as aggressive fungal infections.
Mucormycosis treatment is challenging particularly in the extremities as management must balance both mortality and limb salvage. In this case, the use of NPWT with wound VAC and intervals of instilling amphotericin B facilitated infection control in this lower extremity mucormycosis infection. The significant adverse effect profile of amphotericin B, particularly the nephrotoxicity, should be seriously considered when deciding the treatment regimen for patients affected by mucormycosis. Locally, topical amphotericin B has been reported to cause blistering, itchiness, redness, peeling, and dryness. However, topical preparations of amphotericin B are nontoxic unlike their IV counterpart, able to cross the physiological barriers of the skin while simultaneously targeting macrophages in the dermis and epidermis.10
Conclusions
Although the mainstay of treatment for systemic mucormycosis is radical debridement and IV amphotericin B, a more localized infection may benefit from an adjunct like an instillation wound VAC with topical amphotericin B, as presented in this case study. Swift treatment with wound VAC was beneficial in the overall recovery and tissue healing of this patient and may be beneficial in similar cases.
1. Normandin S, Safran T, Winocour S, et al. negative pressure wound therapy: mechanism of action and clinical applications. Semin Plast Surg. 2021;35(3):164-170. doi:10.1055/s-0041-1731792
2. Agarwal P, Kukrele R, Sharma D. Vacuum assisted closure (VAC)/negative pressure wound therapy (NPWT) for difficult wounds: a review. J Clin Orthop Trauma. 2019;10(5):845-848. doi:10.1016/j.jcot.2019.06.015
3. Gabriel A, Shores J, Bernstein B, et al. A clinical review of infected wound treatment with Vacuum Assisted Closure (V.A.C.) therapy: experience and case series. Int Wound J. 2009;6(suppl 2):1-25. doi:10.1111/j.1742-481X.2009.00628.x
4. Guégan S, Lanternier F, Rouzaud C, Dupin N, Lortholary O. Fungal skin and soft tissue infections. Curr Opin Infect Dis. 2016;29(2):124-130. doi:10.1097/QCO.0000000000000252
5. Ibrahim AS, Spellberg B, Walsh TJ, Kontoyiannis DP. Pathogenesis of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S16-S22. doi:10.1093/cid/cir865
6. Sipsas NV, Gamaletsou MN, Anastasopoulou A, Kontoyiannis DP. Therapy of mucormycosis. J Fungi (Basel). 2018;4(3):90. Published 2018 Jul 31. doi:10.3390/jof4030090
7. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18(3):556-569. doi:10.1128/CMR.18.3.556-569.2005
8. Losee JE, Selber J, Vega S, Hall C, Scott G, Serletti JM. Primary cutaneous mucormycosis: guide to surgical management. Ann Plast Surg. 2002;49(4):385-390. doi:10.1097/00000637-200210000-00009
9. Webb LX. New techniques in wound management: vacuum-assisted wound closure. J Am Acad Orthop Surg. 2002;10(5):303-311. doi:10.5435/00124635-200209000-00002
10. Varikuti S, Oghumu S, Saljoughian N, et al. Topical treatment with nanoliposomal Amphotericin B reduces early lesion growth but fails to induce cure in an experimental model of cutaneous leishmaniasis caused by Leishmania mexicana. Acta Trop. 2017;173:102-108. doi:10.1016/j.actatropica.2017.06.004
Vacuum-assisted closure (VAC) of wounds has become a foundational tool in the armamentarium of wound care specialists. Using a system consisting of a sponge, semi-occlusive barrier, and fluid collection device, VAC systems apply constant negative pressure resulting in macro and micro deformation to a wound, stabilization of the wound environment, and removal of inflammatory factors in wound fluid.1 These conditions allow for the removal of drainage and fluid from a wound bed, reduced edema and inflammation, reduced bacterial load, recruitment of healing factors, approximation of wound edges, and increased blood flow to the wound.2
In complex, infected wounds, a variation of negative pressure wound therapy (NPWT) via the instillation of topical antibiotics (instillation VAC) has been used.3 This variation has been advantageous even in soft tissue fungal infections. Early and aggressive treatment of such infections is critical to prevent dissemination, particularly in aggressive infections, such as mucormycosis.4 We present a case of a patient with a mucormycosis infection of his left Achilles tendon and overlying skin who was successfully treated with surgical debridement and wound care with instillation NPWT with topical amphotericin B.
Case Presentation
A 53-year-old man underwent left Achilles tendon reconstruction with allograft after a complete tear during exercise. He had no relevant medical history and was otherwise healthy, which he attributed to working out daily. About a week after the operation, he began having incisional breakdown, prompting presentation to an emergency department. There, he received IV antibiotics along with multiple debridements. After the wound failed to improve and intra-operative cultures grew mucormycosis, he was transferred to our facility for a higher level of care. On admission, he was immediately given IV amphotericin B and scheduled for repeat debridement.
After 1 prior debridement and 10 total days of IV amphotericin, a repeat debridement was performed. After the debridement, the installation VAC was applied to the patient’s left lower extremity wound with an instilling fluid of amphotericin B and the settings as follows: smart phase instill volume, 110 mL; soak time, 3.5 hours; target pressure, 125 mm Hg; intensity, low; and VAC therapy mode, continuous. After 5 days, the wound bed appeared clean without overt signs of infection. However, due to some toxicity to healthy surrounding soft tissue, the instillation VAC was discontinued and standard NPWT was started. The patient underwent 2 additional rounds of debridement with partial delayed closure. Four weeks after discontinuation of the instillation VAC, the wound appeared healthy and granulated so the patient underwent split-thickness skin grafting to the left posterior ankle. He subsequently completed a course of oral antifungal medication as an outpatient.
The patient was seen in the outpatient clinic for 14 months from the initial mucormycosis infection (Figure).
Discussion
Mucormycosis is an infection caused by fungi in the class Zygomycetes and of the order Mucorales that typically occurs in immunocompromised patients, especially those with diabetic ketoacidosis and neutropenia. Given that this patient had no relevant medical history and was otherwise healthy, he was at extremely low risk of this type of infection. In this patient’s case, the spores of this nonseptate hyphae wide-branching species were most likely introduced at the time of left Achilles tendon repair. Mucormycosis is progressive and can be fatal unless treated, with a mortality rate approaching 70%.5 The rarity and heterogeneity of mucormycosis make treatment variable.6 No prospective or randomized clinical trials exist in plastic surgery literature.
The use of wound VAC in combination with the instillation of amphotericin B to treat cutaneous mucormycosis is not well documented. Mucormycosis infections are traditionally addressed with surgical debridement and antifungal therapy, specifically IV amphotericin B.7,8 As previously noted, NPWT has become the gold standard in treating complex wounds.3 Additionally, wound VAC therapy with instillation has been noted in the literature as a reliable method to treat bacteria-infected wounds, providing a shorter treatment period and earlier wound closure.9 Instillation VAC therapy has proven particularly useful in complex, infected wounds, such as aggressive fungal infections.
Mucormycosis treatment is challenging particularly in the extremities as management must balance both mortality and limb salvage. In this case, the use of NPWT with wound VAC and intervals of instilling amphotericin B facilitated infection control in this lower extremity mucormycosis infection. The significant adverse effect profile of amphotericin B, particularly the nephrotoxicity, should be seriously considered when deciding the treatment regimen for patients affected by mucormycosis. Locally, topical amphotericin B has been reported to cause blistering, itchiness, redness, peeling, and dryness. However, topical preparations of amphotericin B are nontoxic unlike their IV counterpart, able to cross the physiological barriers of the skin while simultaneously targeting macrophages in the dermis and epidermis.10
Conclusions
Although the mainstay of treatment for systemic mucormycosis is radical debridement and IV amphotericin B, a more localized infection may benefit from an adjunct like an instillation wound VAC with topical amphotericin B, as presented in this case study. Swift treatment with wound VAC was beneficial in the overall recovery and tissue healing of this patient and may be beneficial in similar cases.
Vacuum-assisted closure (VAC) of wounds has become a foundational tool in the armamentarium of wound care specialists. Using a system consisting of a sponge, semi-occlusive barrier, and fluid collection device, VAC systems apply constant negative pressure resulting in macro and micro deformation to a wound, stabilization of the wound environment, and removal of inflammatory factors in wound fluid.1 These conditions allow for the removal of drainage and fluid from a wound bed, reduced edema and inflammation, reduced bacterial load, recruitment of healing factors, approximation of wound edges, and increased blood flow to the wound.2
In complex, infected wounds, a variation of negative pressure wound therapy (NPWT) via the instillation of topical antibiotics (instillation VAC) has been used.3 This variation has been advantageous even in soft tissue fungal infections. Early and aggressive treatment of such infections is critical to prevent dissemination, particularly in aggressive infections, such as mucormycosis.4 We present a case of a patient with a mucormycosis infection of his left Achilles tendon and overlying skin who was successfully treated with surgical debridement and wound care with instillation NPWT with topical amphotericin B.
Case Presentation
A 53-year-old man underwent left Achilles tendon reconstruction with allograft after a complete tear during exercise. He had no relevant medical history and was otherwise healthy, which he attributed to working out daily. About a week after the operation, he began having incisional breakdown, prompting presentation to an emergency department. There, he received IV antibiotics along with multiple debridements. After the wound failed to improve and intra-operative cultures grew mucormycosis, he was transferred to our facility for a higher level of care. On admission, he was immediately given IV amphotericin B and scheduled for repeat debridement.
After 1 prior debridement and 10 total days of IV amphotericin, a repeat debridement was performed. After the debridement, the installation VAC was applied to the patient’s left lower extremity wound with an instilling fluid of amphotericin B and the settings as follows: smart phase instill volume, 110 mL; soak time, 3.5 hours; target pressure, 125 mm Hg; intensity, low; and VAC therapy mode, continuous. After 5 days, the wound bed appeared clean without overt signs of infection. However, due to some toxicity to healthy surrounding soft tissue, the instillation VAC was discontinued and standard NPWT was started. The patient underwent 2 additional rounds of debridement with partial delayed closure. Four weeks after discontinuation of the instillation VAC, the wound appeared healthy and granulated so the patient underwent split-thickness skin grafting to the left posterior ankle. He subsequently completed a course of oral antifungal medication as an outpatient.
The patient was seen in the outpatient clinic for 14 months from the initial mucormycosis infection (Figure).
Discussion
Mucormycosis is an infection caused by fungi in the class Zygomycetes and of the order Mucorales that typically occurs in immunocompromised patients, especially those with diabetic ketoacidosis and neutropenia. Given that this patient had no relevant medical history and was otherwise healthy, he was at extremely low risk of this type of infection. In this patient’s case, the spores of this nonseptate hyphae wide-branching species were most likely introduced at the time of left Achilles tendon repair. Mucormycosis is progressive and can be fatal unless treated, with a mortality rate approaching 70%.5 The rarity and heterogeneity of mucormycosis make treatment variable.6 No prospective or randomized clinical trials exist in plastic surgery literature.
The use of wound VAC in combination with the instillation of amphotericin B to treat cutaneous mucormycosis is not well documented. Mucormycosis infections are traditionally addressed with surgical debridement and antifungal therapy, specifically IV amphotericin B.7,8 As previously noted, NPWT has become the gold standard in treating complex wounds.3 Additionally, wound VAC therapy with instillation has been noted in the literature as a reliable method to treat bacteria-infected wounds, providing a shorter treatment period and earlier wound closure.9 Instillation VAC therapy has proven particularly useful in complex, infected wounds, such as aggressive fungal infections.
Mucormycosis treatment is challenging particularly in the extremities as management must balance both mortality and limb salvage. In this case, the use of NPWT with wound VAC and intervals of instilling amphotericin B facilitated infection control in this lower extremity mucormycosis infection. The significant adverse effect profile of amphotericin B, particularly the nephrotoxicity, should be seriously considered when deciding the treatment regimen for patients affected by mucormycosis. Locally, topical amphotericin B has been reported to cause blistering, itchiness, redness, peeling, and dryness. However, topical preparations of amphotericin B are nontoxic unlike their IV counterpart, able to cross the physiological barriers of the skin while simultaneously targeting macrophages in the dermis and epidermis.10
Conclusions
Although the mainstay of treatment for systemic mucormycosis is radical debridement and IV amphotericin B, a more localized infection may benefit from an adjunct like an instillation wound VAC with topical amphotericin B, as presented in this case study. Swift treatment with wound VAC was beneficial in the overall recovery and tissue healing of this patient and may be beneficial in similar cases.
1. Normandin S, Safran T, Winocour S, et al. negative pressure wound therapy: mechanism of action and clinical applications. Semin Plast Surg. 2021;35(3):164-170. doi:10.1055/s-0041-1731792
2. Agarwal P, Kukrele R, Sharma D. Vacuum assisted closure (VAC)/negative pressure wound therapy (NPWT) for difficult wounds: a review. J Clin Orthop Trauma. 2019;10(5):845-848. doi:10.1016/j.jcot.2019.06.015
3. Gabriel A, Shores J, Bernstein B, et al. A clinical review of infected wound treatment with Vacuum Assisted Closure (V.A.C.) therapy: experience and case series. Int Wound J. 2009;6(suppl 2):1-25. doi:10.1111/j.1742-481X.2009.00628.x
4. Guégan S, Lanternier F, Rouzaud C, Dupin N, Lortholary O. Fungal skin and soft tissue infections. Curr Opin Infect Dis. 2016;29(2):124-130. doi:10.1097/QCO.0000000000000252
5. Ibrahim AS, Spellberg B, Walsh TJ, Kontoyiannis DP. Pathogenesis of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S16-S22. doi:10.1093/cid/cir865
6. Sipsas NV, Gamaletsou MN, Anastasopoulou A, Kontoyiannis DP. Therapy of mucormycosis. J Fungi (Basel). 2018;4(3):90. Published 2018 Jul 31. doi:10.3390/jof4030090
7. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18(3):556-569. doi:10.1128/CMR.18.3.556-569.2005
8. Losee JE, Selber J, Vega S, Hall C, Scott G, Serletti JM. Primary cutaneous mucormycosis: guide to surgical management. Ann Plast Surg. 2002;49(4):385-390. doi:10.1097/00000637-200210000-00009
9. Webb LX. New techniques in wound management: vacuum-assisted wound closure. J Am Acad Orthop Surg. 2002;10(5):303-311. doi:10.5435/00124635-200209000-00002
10. Varikuti S, Oghumu S, Saljoughian N, et al. Topical treatment with nanoliposomal Amphotericin B reduces early lesion growth but fails to induce cure in an experimental model of cutaneous leishmaniasis caused by Leishmania mexicana. Acta Trop. 2017;173:102-108. doi:10.1016/j.actatropica.2017.06.004
1. Normandin S, Safran T, Winocour S, et al. negative pressure wound therapy: mechanism of action and clinical applications. Semin Plast Surg. 2021;35(3):164-170. doi:10.1055/s-0041-1731792
2. Agarwal P, Kukrele R, Sharma D. Vacuum assisted closure (VAC)/negative pressure wound therapy (NPWT) for difficult wounds: a review. J Clin Orthop Trauma. 2019;10(5):845-848. doi:10.1016/j.jcot.2019.06.015
3. Gabriel A, Shores J, Bernstein B, et al. A clinical review of infected wound treatment with Vacuum Assisted Closure (V.A.C.) therapy: experience and case series. Int Wound J. 2009;6(suppl 2):1-25. doi:10.1111/j.1742-481X.2009.00628.x
4. Guégan S, Lanternier F, Rouzaud C, Dupin N, Lortholary O. Fungal skin and soft tissue infections. Curr Opin Infect Dis. 2016;29(2):124-130. doi:10.1097/QCO.0000000000000252
5. Ibrahim AS, Spellberg B, Walsh TJ, Kontoyiannis DP. Pathogenesis of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S16-S22. doi:10.1093/cid/cir865
6. Sipsas NV, Gamaletsou MN, Anastasopoulou A, Kontoyiannis DP. Therapy of mucormycosis. J Fungi (Basel). 2018;4(3):90. Published 2018 Jul 31. doi:10.3390/jof4030090
7. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18(3):556-569. doi:10.1128/CMR.18.3.556-569.2005
8. Losee JE, Selber J, Vega S, Hall C, Scott G, Serletti JM. Primary cutaneous mucormycosis: guide to surgical management. Ann Plast Surg. 2002;49(4):385-390. doi:10.1097/00000637-200210000-00009
9. Webb LX. New techniques in wound management: vacuum-assisted wound closure. J Am Acad Orthop Surg. 2002;10(5):303-311. doi:10.5435/00124635-200209000-00002
10. Varikuti S, Oghumu S, Saljoughian N, et al. Topical treatment with nanoliposomal Amphotericin B reduces early lesion growth but fails to induce cure in an experimental model of cutaneous leishmaniasis caused by Leishmania mexicana. Acta Trop. 2017;173:102-108. doi:10.1016/j.actatropica.2017.06.004

14-year-old boy • aching midsternal pain following a basketball injury • worsening pain with direct pressure and when the patient sneezed • Dx?
THE CASE
A 14-year-old boy sought care at our clinic for persistent chest pain after being hit in the chest with a teammate’s shoulder during a basketball game 3 weeks earlier. He had aching midsternal chest pain that worsened with direct pressure and when he sneezed, twisted, or bent forward. There was no bruising or swelling.
On examination, the patient demonstrated normal perfusion and normal work of breathing. He had focal tenderness with palpation at the manubrium with no noticeable step-off, and mild tenderness at the adjacent costochondral junctions and over his pectoral muscles. His sternal pain along the proximal sternum was reproducible with a weighted wall push-up. Although the patient maintained full range of motion in his upper extremities, he did have sternal pain with flexion, abduction, and external rotation of the bilateral upper extremities against resistance. Anteroposterior (AP) and lateral chest radiographs were unremarkable.
THE DIAGNOSIS
The unremarkable chest radiographs prompted further investigation with a diagnostic ultrasound, which revealed a small cortical defect with overlying anechoic fluid collection in the area of focal tenderness. T2-weighted magnetic resonance imaging (MRI) of the chest was performed; it revealed a transverse, nondisplaced fracture of the superior body of the sternum with surrounding bone marrow edema (FIGURE).

DISCUSSION
Fractures of the sternum comprise < 1% of traumatic fractures and have a low mortality rate (0.7%).1,2 The rarity of these fractures is attributed to the ribs’ elastic recoil, which protects the chest wall from anterior forces.1,3 These fractures are even more unusual in children due to the increased elasticity of their chest walls.4-6 Thus, it takes a significant amount of force for a child’s sternum to fracture.
While isolated sternum fractures can occur, two-thirds of sternum fractures are nonisolated and are associated with injuries to surrounding structures (including the heart, lungs, and vasculature) or fractures of the ribs and spine.2,3 Most often, these injuries are caused by significant blunt trauma to the anterior chest, rapid deceleration, or flexion-compression injury.2,3 They are typically transverse and localized, with 70% of fractures occurring in the mid-body and 17.6% at the manubriosternal joint.1,3,6
Athletes with a sternal fracture typically present as our patient did, with a history of blunt force trauma to the chest and with pain and tenderness over the anterior midline of the chest that increases with respiration or movement.1 A physical examination that includes chest palpation and auscultation of the heart and lungs must be performed to rule out damage to intrathoracic structures and assess the patient’s cardiac and pulmonary stability. An electrocardiogram should be performed to confirm that there are no cardiovascular complications.3,4
Initial imaging should include AP and lateral chest radiographs because any displacement will occur in the sagittal plane.1,2,4-6 If the radiograph shows no clear pathology, follow up with computed tomography, ultrasound, MRI, or technetium bone scans to gain additional information.1 Diagnosis of sternal fractures is especially difficult in children due to the presence of ossification centers for bone growth, which may be misinterpreted as a sternal fracture in the absence of a proper understanding of sternal development.5,6 On ultrasound, sternal fractures appear as a sharp step-off in the cortex, whereas in the absence of fracture, there is no cortical step-off and the cartilaginous plate between ossification centers appears in line with the cortex.7
Continue to: A self-limiting injury that requires proper pain control
A self-limiting injury that requires proper pain control
Isolated sternal fractures are typically self-limiting with a good prognosis.2 These injuries are managed supportively with rest, ice, and analgesics1; proper pain control is crucial to prevent respiratory compromise.8
Complete recovery for most patients occurs in 10 to 12 weeks.9 Recovery periods longer than 12 weeks are associated with nonisolated sternal fractures that are complicated by soft-tissue injury, injuries to the chest wall (such as sternoclavicular joint dislocation, usually from a fall on the shoulder), or fracture nonunion.1,2,5
Anterior sternoclavicular joint dislocations and stable posterior dislocations are managed with closed reduction and immobilization in a figure-of-eight brace.1 Operative management is reserved for patients with displaced fractures, sternal deformity, chest wall instability, respiratory insufficiency, uncontrolled pain, or fracture nonunion.1,3,8
A return-to-play protocol can begin once the patient is asymptomatic.1 The timeframe for a full return to play can vary from 6 weeks to 6 months, depending on the severity of the fracture.1 This process is guided by how quickly the symptoms resolve and by radiographic stability.9
Our patient was followed every 3 to 4 weeks and started physical therapy 6 weeks after his injury occurred. He was held from play for 10 weeks and gradually returned to play; he returned to full-contact activity after tolerating a practice without pain.
THE TAKEAWAY
Children typically have greater chest wall elasticity, and thus, it is unusual for them to sustain a sternal fracture. Diagnosis in children is complicated by the presence of ossification centers for bone growth on imaging. In this case, the fracture was first noticed on ultrasound and confirmed with MRI. Since these fractures can be associated with damage to surrounding structures, additional injuries should be considered when evaluating a patient with a sternum fracture.
CORRESPONDENCE
Catherine Romaine, East Carolina University, Brody School of Medicine, 600 Moye Boulevard, Greenville, NC 27834; [email protected]
1. Alent J, Narducci DM, Moran B, et al. Sternal injuries in sport: a review of the literature. Sports Med. 2018;48:2715-2724. doi: 10.1007/s40279-018-0990-5
2. Khoriati A-A, Rajakulasingam R, Shah R. Sternal fractures and their management. J Emerg Trauma Shock. 2013;6:113-116. doi: 10.4103/0974-2700.110763
3. Athanassiadi K, Gerazounis M, Moustardas M, et al. Sternal fractures: retrospective analysis of 100 cases. World J Surg. 2002;26:1243-1246. doi: 10.1007/s00268-002-6511-5
4. Ferguson LP, Wilkinson AG, Beattie TF. Fracture of the sternum in children. Emerg Med J. 2003;20:518-520. doi: 10.1136/emj.20.6.518
5. Ramgopal S, Shaffiey SA, Conti KA. Pediatric sternal fractures from a Level 1 trauma center. J Pediatr Surg. 2019;54:1628-1631. doi: 10.1016/j.jpedsurg.2018.08.040
6. Sesia SB, Prüfer F, Mayr J. Sternal fracture in children: diagnosis by ultrasonography. European J Pediatr Surg Rep. 2017;5:e39-e42. doi: 10.1055/s-0037-1606197
7. Nickson C, Rippey J. Ultrasonography of sternal fractures. Australas J Ultrasound Med. 2011;14:6-11. doi: 10.1002/j.2205-0140.2011.tb00131.x
8. Bauman ZM, Yanala U, Waibel BH, et al. Sternal fixation for isolated traumatic sternal fractures improves pain and upper extremity range of motion. Eur J Trauma Emerg Surg. 2022;48:225-230. doi: 10.1007/s00068-020-01568-x
9. Culp B, Hurbanek JG, Novak J, et al. Acute traumatic sternum fracture in a female college hockey player. Orthopedics. 2010;33:683. doi: 10.3928/01477447-20100722-17
THE CASE
A 14-year-old boy sought care at our clinic for persistent chest pain after being hit in the chest with a teammate’s shoulder during a basketball game 3 weeks earlier. He had aching midsternal chest pain that worsened with direct pressure and when he sneezed, twisted, or bent forward. There was no bruising or swelling.
On examination, the patient demonstrated normal perfusion and normal work of breathing. He had focal tenderness with palpation at the manubrium with no noticeable step-off, and mild tenderness at the adjacent costochondral junctions and over his pectoral muscles. His sternal pain along the proximal sternum was reproducible with a weighted wall push-up. Although the patient maintained full range of motion in his upper extremities, he did have sternal pain with flexion, abduction, and external rotation of the bilateral upper extremities against resistance. Anteroposterior (AP) and lateral chest radiographs were unremarkable.
THE DIAGNOSIS
The unremarkable chest radiographs prompted further investigation with a diagnostic ultrasound, which revealed a small cortical defect with overlying anechoic fluid collection in the area of focal tenderness. T2-weighted magnetic resonance imaging (MRI) of the chest was performed; it revealed a transverse, nondisplaced fracture of the superior body of the sternum with surrounding bone marrow edema (FIGURE).
DISCUSSION
Fractures of the sternum comprise < 1% of traumatic fractures and have a low mortality rate (0.7%).1,2 The rarity of these fractures is attributed to the ribs’ elastic recoil, which protects the chest wall from anterior forces.1,3 These fractures are even more unusual in children due to the increased elasticity of their chest walls.4-6 Thus, it takes a significant amount of force for a child’s sternum to fracture.
While isolated sternum fractures can occur, two-thirds of sternum fractures are nonisolated and are associated with injuries to surrounding structures (including the heart, lungs, and vasculature) or fractures of the ribs and spine.2,3 Most often, these injuries are caused by significant blunt trauma to the anterior chest, rapid deceleration, or flexion-compression injury.2,3 They are typically transverse and localized, with 70% of fractures occurring in the mid-body and 17.6% at the manubriosternal joint.1,3,6
Athletes with a sternal fracture typically present as our patient did, with a history of blunt force trauma to the chest and with pain and tenderness over the anterior midline of the chest that increases with respiration or movement.1 A physical examination that includes chest palpation and auscultation of the heart and lungs must be performed to rule out damage to intrathoracic structures and assess the patient’s cardiac and pulmonary stability. An electrocardiogram should be performed to confirm that there are no cardiovascular complications.3,4
Initial imaging should include AP and lateral chest radiographs because any displacement will occur in the sagittal plane.1,2,4-6 If the radiograph shows no clear pathology, follow up with computed tomography, ultrasound, MRI, or technetium bone scans to gain additional information.1 Diagnosis of sternal fractures is especially difficult in children due to the presence of ossification centers for bone growth, which may be misinterpreted as a sternal fracture in the absence of a proper understanding of sternal development.5,6 On ultrasound, sternal fractures appear as a sharp step-off in the cortex, whereas in the absence of fracture, there is no cortical step-off and the cartilaginous plate between ossification centers appears in line with the cortex.7
Continue to: A self-limiting injury that requires proper pain control
A self-limiting injury that requires proper pain control
Isolated sternal fractures are typically self-limiting with a good prognosis.2 These injuries are managed supportively with rest, ice, and analgesics1; proper pain control is crucial to prevent respiratory compromise.8
Complete recovery for most patients occurs in 10 to 12 weeks.9 Recovery periods longer than 12 weeks are associated with nonisolated sternal fractures that are complicated by soft-tissue injury, injuries to the chest wall (such as sternoclavicular joint dislocation, usually from a fall on the shoulder), or fracture nonunion.1,2,5
Anterior sternoclavicular joint dislocations and stable posterior dislocations are managed with closed reduction and immobilization in a figure-of-eight brace.1 Operative management is reserved for patients with displaced fractures, sternal deformity, chest wall instability, respiratory insufficiency, uncontrolled pain, or fracture nonunion.1,3,8
A return-to-play protocol can begin once the patient is asymptomatic.1 The timeframe for a full return to play can vary from 6 weeks to 6 months, depending on the severity of the fracture.1 This process is guided by how quickly the symptoms resolve and by radiographic stability.9
Our patient was followed every 3 to 4 weeks and started physical therapy 6 weeks after his injury occurred. He was held from play for 10 weeks and gradually returned to play; he returned to full-contact activity after tolerating a practice without pain.
THE TAKEAWAY
Children typically have greater chest wall elasticity, and thus, it is unusual for them to sustain a sternal fracture. Diagnosis in children is complicated by the presence of ossification centers for bone growth on imaging. In this case, the fracture was first noticed on ultrasound and confirmed with MRI. Since these fractures can be associated with damage to surrounding structures, additional injuries should be considered when evaluating a patient with a sternum fracture.
CORRESPONDENCE
Catherine Romaine, East Carolina University, Brody School of Medicine, 600 Moye Boulevard, Greenville, NC 27834; [email protected]
THE CASE
A 14-year-old boy sought care at our clinic for persistent chest pain after being hit in the chest with a teammate’s shoulder during a basketball game 3 weeks earlier. He had aching midsternal chest pain that worsened with direct pressure and when he sneezed, twisted, or bent forward. There was no bruising or swelling.
On examination, the patient demonstrated normal perfusion and normal work of breathing. He had focal tenderness with palpation at the manubrium with no noticeable step-off, and mild tenderness at the adjacent costochondral junctions and over his pectoral muscles. His sternal pain along the proximal sternum was reproducible with a weighted wall push-up. Although the patient maintained full range of motion in his upper extremities, he did have sternal pain with flexion, abduction, and external rotation of the bilateral upper extremities against resistance. Anteroposterior (AP) and lateral chest radiographs were unremarkable.
THE DIAGNOSIS
The unremarkable chest radiographs prompted further investigation with a diagnostic ultrasound, which revealed a small cortical defect with overlying anechoic fluid collection in the area of focal tenderness. T2-weighted magnetic resonance imaging (MRI) of the chest was performed; it revealed a transverse, nondisplaced fracture of the superior body of the sternum with surrounding bone marrow edema (FIGURE).
DISCUSSION
Fractures of the sternum comprise < 1% of traumatic fractures and have a low mortality rate (0.7%).1,2 The rarity of these fractures is attributed to the ribs’ elastic recoil, which protects the chest wall from anterior forces.1,3 These fractures are even more unusual in children due to the increased elasticity of their chest walls.4-6 Thus, it takes a significant amount of force for a child’s sternum to fracture.
While isolated sternum fractures can occur, two-thirds of sternum fractures are nonisolated and are associated with injuries to surrounding structures (including the heart, lungs, and vasculature) or fractures of the ribs and spine.2,3 Most often, these injuries are caused by significant blunt trauma to the anterior chest, rapid deceleration, or flexion-compression injury.2,3 They are typically transverse and localized, with 70% of fractures occurring in the mid-body and 17.6% at the manubriosternal joint.1,3,6
Athletes with a sternal fracture typically present as our patient did, with a history of blunt force trauma to the chest and with pain and tenderness over the anterior midline of the chest that increases with respiration or movement.1 A physical examination that includes chest palpation and auscultation of the heart and lungs must be performed to rule out damage to intrathoracic structures and assess the patient’s cardiac and pulmonary stability. An electrocardiogram should be performed to confirm that there are no cardiovascular complications.3,4
Initial imaging should include AP and lateral chest radiographs because any displacement will occur in the sagittal plane.1,2,4-6 If the radiograph shows no clear pathology, follow up with computed tomography, ultrasound, MRI, or technetium bone scans to gain additional information.1 Diagnosis of sternal fractures is especially difficult in children due to the presence of ossification centers for bone growth, which may be misinterpreted as a sternal fracture in the absence of a proper understanding of sternal development.5,6 On ultrasound, sternal fractures appear as a sharp step-off in the cortex, whereas in the absence of fracture, there is no cortical step-off and the cartilaginous plate between ossification centers appears in line with the cortex.7
Continue to: A self-limiting injury that requires proper pain control
A self-limiting injury that requires proper pain control
Isolated sternal fractures are typically self-limiting with a good prognosis.2 These injuries are managed supportively with rest, ice, and analgesics1; proper pain control is crucial to prevent respiratory compromise.8
Complete recovery for most patients occurs in 10 to 12 weeks.9 Recovery periods longer than 12 weeks are associated with nonisolated sternal fractures that are complicated by soft-tissue injury, injuries to the chest wall (such as sternoclavicular joint dislocation, usually from a fall on the shoulder), or fracture nonunion.1,2,5
Anterior sternoclavicular joint dislocations and stable posterior dislocations are managed with closed reduction and immobilization in a figure-of-eight brace.1 Operative management is reserved for patients with displaced fractures, sternal deformity, chest wall instability, respiratory insufficiency, uncontrolled pain, or fracture nonunion.1,3,8
A return-to-play protocol can begin once the patient is asymptomatic.1 The timeframe for a full return to play can vary from 6 weeks to 6 months, depending on the severity of the fracture.1 This process is guided by how quickly the symptoms resolve and by radiographic stability.9
Our patient was followed every 3 to 4 weeks and started physical therapy 6 weeks after his injury occurred. He was held from play for 10 weeks and gradually returned to play; he returned to full-contact activity after tolerating a practice without pain.
THE TAKEAWAY
Children typically have greater chest wall elasticity, and thus, it is unusual for them to sustain a sternal fracture. Diagnosis in children is complicated by the presence of ossification centers for bone growth on imaging. In this case, the fracture was first noticed on ultrasound and confirmed with MRI. Since these fractures can be associated with damage to surrounding structures, additional injuries should be considered when evaluating a patient with a sternum fracture.
CORRESPONDENCE
Catherine Romaine, East Carolina University, Brody School of Medicine, 600 Moye Boulevard, Greenville, NC 27834; [email protected]
1. Alent J, Narducci DM, Moran B, et al. Sternal injuries in sport: a review of the literature. Sports Med. 2018;48:2715-2724. doi: 10.1007/s40279-018-0990-5
2. Khoriati A-A, Rajakulasingam R, Shah R. Sternal fractures and their management. J Emerg Trauma Shock. 2013;6:113-116. doi: 10.4103/0974-2700.110763
3. Athanassiadi K, Gerazounis M, Moustardas M, et al. Sternal fractures: retrospective analysis of 100 cases. World J Surg. 2002;26:1243-1246. doi: 10.1007/s00268-002-6511-5
4. Ferguson LP, Wilkinson AG, Beattie TF. Fracture of the sternum in children. Emerg Med J. 2003;20:518-520. doi: 10.1136/emj.20.6.518
5. Ramgopal S, Shaffiey SA, Conti KA. Pediatric sternal fractures from a Level 1 trauma center. J Pediatr Surg. 2019;54:1628-1631. doi: 10.1016/j.jpedsurg.2018.08.040
6. Sesia SB, Prüfer F, Mayr J. Sternal fracture in children: diagnosis by ultrasonography. European J Pediatr Surg Rep. 2017;5:e39-e42. doi: 10.1055/s-0037-1606197
7. Nickson C, Rippey J. Ultrasonography of sternal fractures. Australas J Ultrasound Med. 2011;14:6-11. doi: 10.1002/j.2205-0140.2011.tb00131.x
8. Bauman ZM, Yanala U, Waibel BH, et al. Sternal fixation for isolated traumatic sternal fractures improves pain and upper extremity range of motion. Eur J Trauma Emerg Surg. 2022;48:225-230. doi: 10.1007/s00068-020-01568-x
9. Culp B, Hurbanek JG, Novak J, et al. Acute traumatic sternum fracture in a female college hockey player. Orthopedics. 2010;33:683. doi: 10.3928/01477447-20100722-17
1. Alent J, Narducci DM, Moran B, et al. Sternal injuries in sport: a review of the literature. Sports Med. 2018;48:2715-2724. doi: 10.1007/s40279-018-0990-5
2. Khoriati A-A, Rajakulasingam R, Shah R. Sternal fractures and their management. J Emerg Trauma Shock. 2013;6:113-116. doi: 10.4103/0974-2700.110763
3. Athanassiadi K, Gerazounis M, Moustardas M, et al. Sternal fractures: retrospective analysis of 100 cases. World J Surg. 2002;26:1243-1246. doi: 10.1007/s00268-002-6511-5
4. Ferguson LP, Wilkinson AG, Beattie TF. Fracture of the sternum in children. Emerg Med J. 2003;20:518-520. doi: 10.1136/emj.20.6.518
5. Ramgopal S, Shaffiey SA, Conti KA. Pediatric sternal fractures from a Level 1 trauma center. J Pediatr Surg. 2019;54:1628-1631. doi: 10.1016/j.jpedsurg.2018.08.040
6. Sesia SB, Prüfer F, Mayr J. Sternal fracture in children: diagnosis by ultrasonography. European J Pediatr Surg Rep. 2017;5:e39-e42. doi: 10.1055/s-0037-1606197
7. Nickson C, Rippey J. Ultrasonography of sternal fractures. Australas J Ultrasound Med. 2011;14:6-11. doi: 10.1002/j.2205-0140.2011.tb00131.x
8. Bauman ZM, Yanala U, Waibel BH, et al. Sternal fixation for isolated traumatic sternal fractures improves pain and upper extremity range of motion. Eur J Trauma Emerg Surg. 2022;48:225-230. doi: 10.1007/s00068-020-01568-x
9. Culp B, Hurbanek JG, Novak J, et al. Acute traumatic sternum fracture in a female college hockey player. Orthopedics. 2010;33:683. doi: 10.3928/01477447-20100722-17

42-year-old man • altered mental status • vomiting • agitation • Dx?
THE CASE
A 42-year-old man with a history of bipolar disorder with psychotic features, asthma, and chronic pain was brought to the emergency department (ED) by his father due to altered mental status, coughing, and vomiting. The patient was unable to recall events earlier in the day in detail but stated that he remembered using his inhaler for his cough, which seemed to precipitate his vomiting. The patient’s home medications were listed as albuterol 90 mcg, methadone 90 mg/d, and quetiapine 100 mg.
While in the ED, the patient was tachycardic (heart rate, 102 bpm), but all other vital signs were normal. He was agitated and at one point required restraints. On exam, he had epigastric tenderness to palpation, and his lungs were clear to auscultation bilaterally.
Blood work was notable for an elevated lipase level of 729 U/L (normal range, 0-160 U/L). Complete blood count, comprehensive metabolic panel, urinalysis, chest x-ray, and alcohol levels were unremarkable. Computed tomography of the abdomen/pelvis and ultrasound of the abdomen showed excess stool and gallbladder sludge without cholecystitis.
The patient was treated symptomatically with intravenous fluids, ondansetron, and lorazepam. He was admitted with a working diagnosis of acute pancreatitis and possible acute psychosis in the setting of schizophrenia.
A few hours after presentation, the patient returned to his baseline mental status. Over the next 24 hours, his lipase level trended down to normal.
THE DIAGNOSIS
After the patient’s discharge, the pharmacist from his primary care provider’s office called as part of the routine post-hospital follow-up and a medication reconciliation was performed. During this call, the patient stated he had used 2 different nasal sprays prior to his ED presentation.
The pharmacist asked him to read the names of each medication. He related the first was naloxone and the second, fluticasone (neither of which was included on his medication list). Upon further questioning, the pharmacist elicited clarification from the patient that he had, in fact, taken 2 doses of naloxone, shortly after which his vomiting began.
Continue to: This additional history...
This additional history suggested the patient’s true diagnosis was acute opioid withdrawal precipitated by his accidental self-administration of naloxone.
DISCUSSION
Naloxone is a pure mu-opioid receptor antagonist that is used for opioid overdose.1 In the past decade, in response to the opioid epidemic, naloxone has become increasingly available in the community as a way of decreasing opioid-related deaths.1,2 The US Food and Drug Administration recommends that all patients who are prescribed opioids for pain or opioid use disorder, as well as those who are at increased risk for opioid overdose, should be prescribed naloxone and educated on its use. Patients who received a naloxone prescription from their primary care provider have been found to have 47% fewer opioid-related ED visits.3
Quick effects, potential for complications. Use of naloxone can rapidly induce opioid withdrawal symptoms, including gastrointestinal effects, tachycardia, and agitation, as well as diaphoresis, shivering, lacrimation, tremor, anxiety, mydriasis, and hypertension. Naloxone use can also lead to severe complications, such as violent behaviors, ventricular tachycardia or fibrillation, asystole, or pulmonary edema, in the period immediately following administration.4 These effects most often subside within 20 to 60 minutes after administration of naloxone, as the antagonist effect wears off.
The treatment of naloxone toxicity is supportive, with particular attention paid to the patient’s mental and respiratory status.
Our patient was advised by his primary care physician on the proper use of all of his medications, including nasal sprays. The clinic pharmacist also met with him for an additional educational session on the proper use of naloxone.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Given the widespread use of naloxone, proper education and counselling regarding this medication is crucial. Patients should be advised of what to expect after its use. In addition, physicians should always maintain updated patient medication lists, ensuring that they include naloxone if it has been prescribed for use as needed for opioid reversal, to assist in the emergency treatment of affected patients.5
CORRESPONDENCE
Erik Weitz, DO, Troy Beaumont Family Medicine Residency, 44250 Dequindre Road, Sterling Heights, MI 48314; [email protected]
1. Parkin S, Neale J, Brown C, et al. Opioid overdose reversals using naloxone in New York City by people who use opioids: implications for public health and overdose harm reduction approaches from a qualitative study. Int J Drug Policy. 2020;79:102751. doi: 10.1016/j.drugpo.2020.102751
2. Rzasa Lynn R, Galinkin JL. Naloxone dosage for opioid reversal: current evidence and clinical implications. Ther Adv Drug Saf. 2018;9:63-88. doi: 10.1177/2042098617744161
3. Coffin PO, Behar E, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165:245-52. doi: 10.7326/M15-2771
4. Osterwalder JJ. Naloxone—for intoxications with intravenous heroin and heroin mixtures—harmless or hazardous? A prospective clinical study. J Toxicol Clin Toxicol. 1996;34:409-416. doi: 10.3109/15563659609013811
5. Kwan JL, Lo L, Sampson M, et al. Medication reconciliation during transitions of care as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5 pt 2):397-403. doi: 10.7326/0003-4819-158-5-201303051-00006
THE CASE
A 42-year-old man with a history of bipolar disorder with psychotic features, asthma, and chronic pain was brought to the emergency department (ED) by his father due to altered mental status, coughing, and vomiting. The patient was unable to recall events earlier in the day in detail but stated that he remembered using his inhaler for his cough, which seemed to precipitate his vomiting. The patient’s home medications were listed as albuterol 90 mcg, methadone 90 mg/d, and quetiapine 100 mg.
While in the ED, the patient was tachycardic (heart rate, 102 bpm), but all other vital signs were normal. He was agitated and at one point required restraints. On exam, he had epigastric tenderness to palpation, and his lungs were clear to auscultation bilaterally.
Blood work was notable for an elevated lipase level of 729 U/L (normal range, 0-160 U/L). Complete blood count, comprehensive metabolic panel, urinalysis, chest x-ray, and alcohol levels were unremarkable. Computed tomography of the abdomen/pelvis and ultrasound of the abdomen showed excess stool and gallbladder sludge without cholecystitis.
The patient was treated symptomatically with intravenous fluids, ondansetron, and lorazepam. He was admitted with a working diagnosis of acute pancreatitis and possible acute psychosis in the setting of schizophrenia.
A few hours after presentation, the patient returned to his baseline mental status. Over the next 24 hours, his lipase level trended down to normal.
THE DIAGNOSIS
After the patient’s discharge, the pharmacist from his primary care provider’s office called as part of the routine post-hospital follow-up and a medication reconciliation was performed. During this call, the patient stated he had used 2 different nasal sprays prior to his ED presentation.
The pharmacist asked him to read the names of each medication. He related the first was naloxone and the second, fluticasone (neither of which was included on his medication list). Upon further questioning, the pharmacist elicited clarification from the patient that he had, in fact, taken 2 doses of naloxone, shortly after which his vomiting began.
Continue to: This additional history...
This additional history suggested the patient’s true diagnosis was acute opioid withdrawal precipitated by his accidental self-administration of naloxone.
DISCUSSION
Naloxone is a pure mu-opioid receptor antagonist that is used for opioid overdose.1 In the past decade, in response to the opioid epidemic, naloxone has become increasingly available in the community as a way of decreasing opioid-related deaths.1,2 The US Food and Drug Administration recommends that all patients who are prescribed opioids for pain or opioid use disorder, as well as those who are at increased risk for opioid overdose, should be prescribed naloxone and educated on its use. Patients who received a naloxone prescription from their primary care provider have been found to have 47% fewer opioid-related ED visits.3
Quick effects, potential for complications. Use of naloxone can rapidly induce opioid withdrawal symptoms, including gastrointestinal effects, tachycardia, and agitation, as well as diaphoresis, shivering, lacrimation, tremor, anxiety, mydriasis, and hypertension. Naloxone use can also lead to severe complications, such as violent behaviors, ventricular tachycardia or fibrillation, asystole, or pulmonary edema, in the period immediately following administration.4 These effects most often subside within 20 to 60 minutes after administration of naloxone, as the antagonist effect wears off.
The treatment of naloxone toxicity is supportive, with particular attention paid to the patient’s mental and respiratory status.
Our patient was advised by his primary care physician on the proper use of all of his medications, including nasal sprays. The clinic pharmacist also met with him for an additional educational session on the proper use of naloxone.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Given the widespread use of naloxone, proper education and counselling regarding this medication is crucial. Patients should be advised of what to expect after its use. In addition, physicians should always maintain updated patient medication lists, ensuring that they include naloxone if it has been prescribed for use as needed for opioid reversal, to assist in the emergency treatment of affected patients.5
CORRESPONDENCE
Erik Weitz, DO, Troy Beaumont Family Medicine Residency, 44250 Dequindre Road, Sterling Heights, MI 48314; [email protected]
THE CASE
A 42-year-old man with a history of bipolar disorder with psychotic features, asthma, and chronic pain was brought to the emergency department (ED) by his father due to altered mental status, coughing, and vomiting. The patient was unable to recall events earlier in the day in detail but stated that he remembered using his inhaler for his cough, which seemed to precipitate his vomiting. The patient’s home medications were listed as albuterol 90 mcg, methadone 90 mg/d, and quetiapine 100 mg.
While in the ED, the patient was tachycardic (heart rate, 102 bpm), but all other vital signs were normal. He was agitated and at one point required restraints. On exam, he had epigastric tenderness to palpation, and his lungs were clear to auscultation bilaterally.
Blood work was notable for an elevated lipase level of 729 U/L (normal range, 0-160 U/L). Complete blood count, comprehensive metabolic panel, urinalysis, chest x-ray, and alcohol levels were unremarkable. Computed tomography of the abdomen/pelvis and ultrasound of the abdomen showed excess stool and gallbladder sludge without cholecystitis.
The patient was treated symptomatically with intravenous fluids, ondansetron, and lorazepam. He was admitted with a working diagnosis of acute pancreatitis and possible acute psychosis in the setting of schizophrenia.
A few hours after presentation, the patient returned to his baseline mental status. Over the next 24 hours, his lipase level trended down to normal.
THE DIAGNOSIS
After the patient’s discharge, the pharmacist from his primary care provider’s office called as part of the routine post-hospital follow-up and a medication reconciliation was performed. During this call, the patient stated he had used 2 different nasal sprays prior to his ED presentation.
The pharmacist asked him to read the names of each medication. He related the first was naloxone and the second, fluticasone (neither of which was included on his medication list). Upon further questioning, the pharmacist elicited clarification from the patient that he had, in fact, taken 2 doses of naloxone, shortly after which his vomiting began.
Continue to: This additional history...
This additional history suggested the patient’s true diagnosis was acute opioid withdrawal precipitated by his accidental self-administration of naloxone.
DISCUSSION
Naloxone is a pure mu-opioid receptor antagonist that is used for opioid overdose.1 In the past decade, in response to the opioid epidemic, naloxone has become increasingly available in the community as a way of decreasing opioid-related deaths.1,2 The US Food and Drug Administration recommends that all patients who are prescribed opioids for pain or opioid use disorder, as well as those who are at increased risk for opioid overdose, should be prescribed naloxone and educated on its use. Patients who received a naloxone prescription from their primary care provider have been found to have 47% fewer opioid-related ED visits.3
Quick effects, potential for complications. Use of naloxone can rapidly induce opioid withdrawal symptoms, including gastrointestinal effects, tachycardia, and agitation, as well as diaphoresis, shivering, lacrimation, tremor, anxiety, mydriasis, and hypertension. Naloxone use can also lead to severe complications, such as violent behaviors, ventricular tachycardia or fibrillation, asystole, or pulmonary edema, in the period immediately following administration.4 These effects most often subside within 20 to 60 minutes after administration of naloxone, as the antagonist effect wears off.
The treatment of naloxone toxicity is supportive, with particular attention paid to the patient’s mental and respiratory status.
Our patient was advised by his primary care physician on the proper use of all of his medications, including nasal sprays. The clinic pharmacist also met with him for an additional educational session on the proper use of naloxone.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Given the widespread use of naloxone, proper education and counselling regarding this medication is crucial. Patients should be advised of what to expect after its use. In addition, physicians should always maintain updated patient medication lists, ensuring that they include naloxone if it has been prescribed for use as needed for opioid reversal, to assist in the emergency treatment of affected patients.5
CORRESPONDENCE
Erik Weitz, DO, Troy Beaumont Family Medicine Residency, 44250 Dequindre Road, Sterling Heights, MI 48314; [email protected]
1. Parkin S, Neale J, Brown C, et al. Opioid overdose reversals using naloxone in New York City by people who use opioids: implications for public health and overdose harm reduction approaches from a qualitative study. Int J Drug Policy. 2020;79:102751. doi: 10.1016/j.drugpo.2020.102751
2. Rzasa Lynn R, Galinkin JL. Naloxone dosage for opioid reversal: current evidence and clinical implications. Ther Adv Drug Saf. 2018;9:63-88. doi: 10.1177/2042098617744161
3. Coffin PO, Behar E, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165:245-52. doi: 10.7326/M15-2771
4. Osterwalder JJ. Naloxone—for intoxications with intravenous heroin and heroin mixtures—harmless or hazardous? A prospective clinical study. J Toxicol Clin Toxicol. 1996;34:409-416. doi: 10.3109/15563659609013811
5. Kwan JL, Lo L, Sampson M, et al. Medication reconciliation during transitions of care as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5 pt 2):397-403. doi: 10.7326/0003-4819-158-5-201303051-00006
1. Parkin S, Neale J, Brown C, et al. Opioid overdose reversals using naloxone in New York City by people who use opioids: implications for public health and overdose harm reduction approaches from a qualitative study. Int J Drug Policy. 2020;79:102751. doi: 10.1016/j.drugpo.2020.102751
2. Rzasa Lynn R, Galinkin JL. Naloxone dosage for opioid reversal: current evidence and clinical implications. Ther Adv Drug Saf. 2018;9:63-88. doi: 10.1177/2042098617744161
3. Coffin PO, Behar E, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165:245-52. doi: 10.7326/M15-2771
4. Osterwalder JJ. Naloxone—for intoxications with intravenous heroin and heroin mixtures—harmless or hazardous? A prospective clinical study. J Toxicol Clin Toxicol. 1996;34:409-416. doi: 10.3109/15563659609013811
5. Kwan JL, Lo L, Sampson M, et al. Medication reconciliation during transitions of care as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5 pt 2):397-403. doi: 10.7326/0003-4819-158-5-201303051-00006

Kikuchi-Fujimoto Disease: A Case Report of Fever and Lymphadenopathy in a Young White Man
Kikuchi-Fujimoto disease (KFD) is a rare, usually self-limited cause of cervical lymphadenitis that is more prevalent among patients of Asian descent.1 The pathogenesis of KFD remains unknown. Clinically, KFD may mimic malignant lymphoproliferative disorders, autoimmune diseases such as systemic lupus erythematosus (SLE) lymphadenitis, and infectious conditions such as HIV and tuberculous lymphadenitis. The most common presentation of KFD involves fever and rapidly evolving cervical lymphadenopathy.2,3 Due to its rarity, KFD is not always considered in the differential diagnosis for fever with tender lymphadenopathy, and up to one-third of cases are initially misdiagnosed.2
Definitive diagnosis requires lymph node biopsy. It is critical to achieving a timely diagnosis of KFD to exclude more serious conditions, initiate appropriate treatment, and minimize undue stress for patients. We describe a case of KFD in a patient who was met with delays in obtaining a definitive diagnosis for his symptoms.
Case Presentation
A 27-year-old previously healthy White man presented to the emergency department with subacute, progressive right-sided neck pain and swelling. In the week leading up to presentation, he also noted intermittent fevers, night sweats, and abdominal pain. His symptoms were unrelieved with acetaminophen and aspirin. He reported no sick contacts, recent travel, or animal exposures. He had no known history of autoimmune disease, malignancy, or immunocompromising conditions. Vital signs at the time of presentation were notable for a temperature of 39.0 °C. On examination, he had several firm, mobile, and exquisitely tender lymph nodes in the right upper anterior cervical chain. Abdominal examination was notable for left upper quadrant tenderness with palpable splenomegaly. Due to initial concern that his symptoms represented bacterial lymphadenitis, he was started on broad-spectrum antibiotics and admitted to the hospital for an expedited infectious workup.
Initial laboratory studies were notable for a white blood cell count of 3.7 × 109/L with 57.5% neutrophils and 27.0% lymphocytes on differential.

Computed tomography (CT) of the neck revealed multiple heterogeneously enlarged lymph nodes along the right anterior cervical chain with necrotic changes (Figure 1).
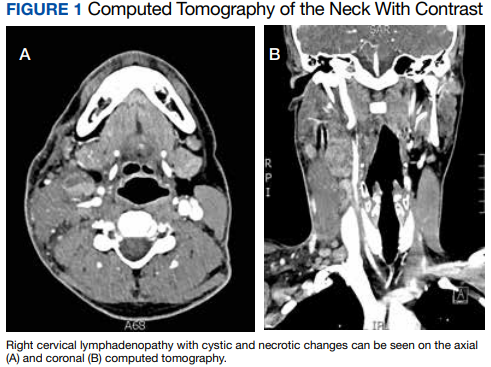
A core needle biopsy of a right-sided cervical lymph node was initially pursued, demonstrating necrotic tissue with minimal residual lymphoid tissue and no definitive evidence of lymphoma. Because these results were nondiagnostic, an excisional biopsy of the right-sided cervical lymph node was pursued 10 days later. Due to the stress of his 2-week hospitalization without a unifying diagnosis, the patient then elected to discharge home with close outpatient follow-up while awaiting his biopsy results. Antibiotics were not continued at the time of discharge as our broad infectious workup failed to yield a causative organism.
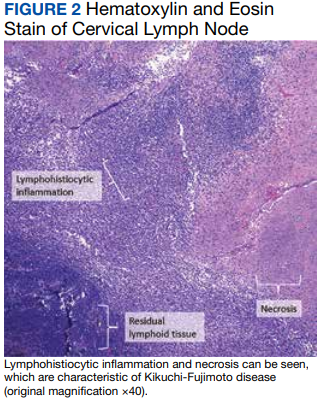
Two weeks postdischarge, the patient’s excisional lymph node biopsy returned demonstrating lymphohistiocytic inflammation with plasmacytoid dendritic cells, areas of necrosis, and scattered karyorrhectic nuclear debris, consistent with a diagnosis of KFD (Figure 2).
After 4 months of hydroxychloroquine therapy, the patient’s KFD symptoms resolved, prompting his dose to be reduced and eventually tapered. Repeat testing of his ANA and anti-dsDNA were performed at 1 and 6 months posthospitalization and returned within normal limits. A repeat PET-CT was performed 6 months posthospitalization showing resolution of his hypermetabolic right neck and right supraclavicular lymphadenopathy as well as his splenomegaly. It has now been more than a year since the patient’s initial presentation to the hospital, and he remains symptom-free and off prednisone and hydroxychloroquine.
Discussion
KFD is a rare cause of cervical lymphadenitis that was first described in 1972. Although cases have been reported worldwide, it is seen with higher prevalence in Asian countries. KFD was previously thought to have a female predominance, but recent reviews suggest a female to male ratio close to 1:1.1 The pathogenesis of KFD remains unknown, though some studies have suggested Epstein-Barr virus infection as a potential trigger.4,5 Human herpesvirus (HHV) 6, HHV 7, HHV 8, HSV, HIV, and parvovirus B19 also have been implicated as potential triggers, though no causative relationship has been established.2,5,6 Autoimmunity may also play a role in the pathogenesis of KFD given its histopathologic overlap with SLE lymphadenitis.1,7
The most common presenting symptoms of KFD include fever and tender cervical lymphadenopathy. Many patients also experience constitutional symptoms such as weight loss, night sweats, and fatigue.2 KFD is characterized by enlarged cervical lymph nodes, typically > 2 cm in diameter.3 Cutaneous manifestations of KFD are common and may manifest as nonspecific papules, plaques, nodules, or facial malar erythema.1,2 Case reports also have described KFD manifesting with ataxia, arthritis, parotitis, or ocular pathologies such as conjunctivitis and uveitis.1,2,8,9 Hepatosplenomegaly is a relatively rare manifestation of KFD seen in approximately 3% of cases.10 When present, hepatosplenomegaly may make the diagnosis of KFD especially difficult to distinguish from lymphoproliferative disorders such as lymphoma. Laboratory findings in KFD are nonspecific and include elevated levels of lactate dehydrogenase, erythrocyte sedimentation rate, C-reactive protein, and liver enzymes.3 Both lymphocytosis and lymphopenia have been described.3Definitive diagnosis of KFD is achieved through lymph node biopsy and histologic examination. Histopathologic findings of KFD include areas of coagulative necrosis and histiocytic proliferation within the cortical and paracortical regions of the lymph node. Scattered nuclear debris also may be seen, though this histologic finding also is seen with lymphoma. The absence of neutrophils is characteristic of KFD.2 In our patient, a core needle biopsy was initially pursued but returned nondiagnostic. A PET-CT also was obtained, though KFD may mimic lymphoma on PET as was seen in this patient’s case as well as in prior case reports.11 An excisional lymph node biopsy was ultimately performed and secured the diagnosis of KFD.
Although ultrasound-guided core needle biopsy was unable to yield the diagnosis for our patient, its diagnostic accuracy is still superior to that of fine needle aspiration and is therefore suggested as the primary diagnostic modality when KFD is suspected.12 Core needle biopsy also is less invasive, less time consuming, and perhaps more cost-effective than an open excisional biopsy, which often requires the use of an operating room and monitored anesthesia care.12 Understandably, our patient experienced significant stress while awaiting a final diagnosis. Whenever possible, lymph node biopsy should be prioritized over other diagnostic modalities to achieve a timely and definitive diagnosis.
KFD has no established treatment guidelines. Supportive care with antipyretics and analgesics is the most common initial approach, as KFD is typically a self-limited disease that resolves in 1 to 4 months.2 Patients with severe, persistent symptoms have been successfully treated with corticosteroids and hydroxychloroquine, with monotherapy typically trialed before concomitant use.2,13 After 2 courses of prednisone, our patient was prescribed single-agent hydroxychloroquine due to his recurrent symptoms and debilitating AEs from the steroids. Other case reports have described hydroxychloroquine as a treatment option when steroids fail to provide symptom relief or when there are recurrences of KFD.14-19 Retinopathy can occur as a result of long-term hydroxychloroquine use. As such, patients anticipated to require long-term hydroxychloroquine therapy should receive a baseline eye examination within months of drug initiation and repeat examination after 5 years of therapy.20
After symptom resolution, continued follow-up with a health care professional is recommended due to the potential for KFD recurrence or the development of a new autoimmune disease. The rate of KFD recurrence was previously described as 3%, but a more recent review found the rate of recurrence to be approximately 15% at > 6 months follow-up.1,3 Recurrence is often described during or shortly after the tapering of steroids.13,16,21,22 Recurrent KFD can be diagnosed with repeat lymph node biopsy, which also serves to exclude other disease processes.13,16 However, recurrence also has been diagnosed clinically based on the patient’s symptoms and laboratory investigations.21,22Continued surveillance of patients with KFD is also necessary to monitor for the development of new autoimmune diseases, especially SLE. SLE lymphadenitis shares many histopathologic characteristics with KFD. Case reports have described the development of SLE in patients with a history of KFD.2,7 Other autoimmune conditions described in patients with prior KFD include Sjögren syndrome, Hashimoto thyroiditis, Graves disease, mixed connective tissue disease, and antiphospholipid syndrome.3,23 Among patients with KFD, female sex, painful adenopathy, and cytopenias are significantly associated with the later development of autoimmune disease.23
Patient Perspective
This began for me in September 2020 out of the blue. I woke up one day with a random lymph node in my neck but otherwise felt completely healthy, and within 2 to 3 weeks I had never been more sick in my entire life. It came with bouts of fevers, neck pain from the swelling, stomach pain (I later learned an enlarged spleen was the source), terrible night sweats, violent chills where the shaking was uncontrollable for hours at a time, loss of appetite, and countless other symptoms that have come and gone over the past year.
It did take a little while to get a diagnosis, but I understand the autoimmune field is tricky. For about 4 to 5 weeks, I was told to prepare for a lymphoma diagnosis. I ended up doing 2 rounds of prednisone, one for 3 weeks at the end of 2020 and one for 2 months from March to May. The initial round helped quite a bit, but the second round did not have any effect on the lingering symptoms. In my opinion, prednisone is miserable to be on long term and I do not recommend it. The daily AEs that came with it included mood swings, insomnia, weight gain, and more. I have been on hydroxychloroquine now for almost 2 months and although it has some AEs of its own, it is nowhere near as rough as the prednisone and has helped manage my remaining symptoms quite a bit.
This certainly has not been a fun experience, but I was under great care during my time in the hospital and continue to be under good care through the rheumatology clinic. The one thing that could have made a huge difference would have been the issues involved in getting my surgery scheduled while I was still inpatient, which took quite a while. The pain during that time was so intense and unlike anything I have ever experienced before, and it was only the surgery that finally brought me some relief. To paint you a picture, I have broken bones, split my leg open, and have roughly 40 to 50 hours of tattoo work on me, and I have never experienced the level of pain like I felt in my neck and stomach. I remember feeling like someone had wound up and hit me with a baseball bat. The surgery brought me immense relief and if it had occurred when it was originally supposed to, I would have been spared 3 or so days of this type of pain.
It has been almost 10 months since my surgery and diagnosis, and life has mostly returned to normal for me. I am still on long-term medication as I mentioned, and I still deal with fatigue, spleen pain, and several other symptoms, but it is much more under control these days. I feel very fortunate to have been under and continue to be under such great care.
Conclusions
This case report highlights the importance of recognizing KFD as a rare but possible cause of fever and necrotizing cervical lymphadenopathy. KFD often mimics malignant lymphoproliferative disorders, autoimmune diseases such as SLE lymphadenitis, and infectious conditions such as HIV and tuberculous lymphadenitis. While KFD is seen with higher prevalence in Asian countries and was previously thought to be more predominant in females, the diagnosis should still be considered irrespective of geographic location or patient sex. Lymph node biopsy is the preferred diagnostic approach for patients with suspected KFD. Treatment is typically supportive but may consist of glucocorticoids in severe cases. Hydroxychloroquine may be used in refractory cases or as a steroid-sparing regimen when steroid AEs are poorly tolerated. Long-term follow-up is critical for patients with KFD to monitor for both disease recurrence and the development of autoimmune disease, especially SLE.
Acknowledgments
The authors thank Dr. Jacob Pilley for his detailed review of the patient’s pathology results. The authors also extend their gratitude to the patient, who deepened our understanding of this condition and what it is like to live with it.
1. Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol. 2004;122(1):141-152. doi:10.1309/YF08-1L4T-KYWV-YVPQ
2. Deaver D, Horna P, Cualing H, Sokol L. Pathogenesis, diagnosis, and management of Kikuchi-Fujimoto disease. Cancer Control. 2014;21(4):313-321. doi:10.1177/107327481402100407
3. Cheng CY, Sheng WH, Lo YC, Chung CS, Chen YC, Chang SC. Clinical presentations, laboratory results and outcomes of patients with Kikuchi’s disease: emphasis on the association between recurrent Kikuchi’s disease and autoimmune diseases. J Microbiol Immunol Infect. 2010;43(5):366-371. doi:10.1016/S1684-1182(10)60058-8
4. Stéphan JL, Jeannoël P, Chanoz J, Gentil-Përret A. Epstein-Barr virus-associated Kikuchi disease in two children. J Pediatr Hematol Oncol. 2001;23(4):240-243. doi:10.1097/00043426-200105000-00012
5. Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, Chen LM. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. Detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113(6):774-781. doi:10.1309/1A6Y-YCKP-5AVF-QTYR
6. Rosado FG, Tang YW, Hasserjian RP, McClain CM, Wang B, Mosse CA. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44(2):255-259. doi:10.1016/j.humpath.2012.05.016
7. Gordon JK, Magro C, Lu T, et al. Overlap between systemic lupus erythematosus and Kikuchi Fujimoto disease: a clinical pathology conference held by the Department of Rheumatology at Hospital for Special Surgery. HSS J. 2009;5(2):169-177. doi:10.1007/s11420-009-9123-x
8. Lo KB, Papazoglou A, Chua L, Candelario N. Case Report: Kikuchi: The great mimicker. F1000Res. 2018;7:520. Published 2018 Apr 30. doi:10.12688/f1000research.14758.1
9. Galor A, Georgy M, Leder HA, Dunn JP, Peters GB 3rd. Papillary conjunctivitis associated with Kikuchi disease. Cornea. 2008;27(8):944-946. doi:10.1097/ICO.0b013e31816bf488
10. Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26(1):50-54. doi:10.1007/s10067-006-0230-5
11. Lee DH, Lee JH, Shim EJ, et al. Disseminated Kikuchi-Fujimoto disease mimicking malignant lymphoma on positron emission tomography in a child. J Pediatr Hematol Oncol. 2009;31(9):687-689. doi:10.1097/MPH.0b013e31819a5d77
12. Park SG, Koo HR, Jang K, et al. Efficacy of ultrasound-guided needle biopsy in the diagnosis of Kikuchi-Fujimoto disease. Laryngoscope. 2021;131(5):E1519-E1523. doi:10.1002/lary.29160
13. Honda F, Tsuboi H, Toko H, et al. Recurrent Kikuchi-Fujimoto disease successfully treated by the concomitant use of hydroxychloroquine and corticosteroids. Intern Med. 2017;56(24):3373-3377. doi:10.2169/internalmedicine.9205-17
14. Rezayat T, Carroll MB, Ramsey BC, Smith A. A case of relapsing Kikuchi-Fujimoto disease. Case Rep Otolaryngol. 2013;2013:364795. doi:10.1155/2013/364795
15. Rezai K, Kuchipudi S, Chundi V, Ariga R, Loew J, Sha BE. Kikuchi-Fujimoto disease: hydroxychloroquine as a treatment. Clin Infect Dis. 2004;39(12):e124-e126. doi:10.1086/426144
16. Hyun M, So IT, Kim HA, Jung H, Ryu SY. Recurrent Kikuchi’s disease treated by hydroxychloroquine. Infect Chemother. 2016;48(2):127-131. doi:10.3947/ic.2016.48.2.127
17. Lin YC, Huang HH, Nong BR, et al. Pediatric Kikuchi-Fujimoto disease: A clinicopathologic study and the therapeutic effects of hydroxychloroquine. J Microbiol Immunol Infect. 2019;52(3):395-401. doi:10.1016/j.jmii.2017.08.023
18. Lin DY, Villegas MS, Tan PL, Wang S, Shek LP. Severe Kikuchi’s disease responsive to immune modulation. Singapore Med J. 2010;51(1):e18-e21.
19. Quintás-Cardama A, Fraga M, Cozzi SN, Caparrini A, Maceiras F, Forteza J. Fatal Kikuchi-Fujimoto disease: the lupus connection. Ann Hematol. 2003;82(3):186-188. doi:10.1007/s00277-003-0611-7
20. American Academy of Ophthalmology. ACR, AAD, RDS, and AAO 2020 Joint Statement on Hydroxychloroquine Use with Respect to Retinal Toxicity. Updated February 2021. Accessed November 28, 2022. https://www.aao.org/clinical-statement/acr-aad-rds-aao-2020-joint-statement-on-hydroxychl-2
21. Gerwig U, Weidmann RG, Lindner G. Relapsing Kikuchi-Fujimoto disease requiring prolonged steroid therapy. Case Rep Emerg Med. 2019;2019:6405687. Published 2019 Mar 7. doi:10.1155/2019/6405687
22. Faheem B, Kumar V, Ashkar H, Komal F, Sultana Y. Recurrent Kikuchi-Fujimoto disease masquerading as lymphoma successfully treated by anakinra. Cureus. 2020;12(11):e11655. Published 2020 Nov 23. doi:10.7759/cureus.11655
23. Sopeña B, Rivera A, Vázquez-Triñanes C, et al. Autoimmune manifestations of Kikuchi disease. Semin Arthritis Rheum. 2012;41(6):900-906. doi:10.1016/j.semarthrit.2011.11.001
Kikuchi-Fujimoto disease (KFD) is a rare, usually self-limited cause of cervical lymphadenitis that is more prevalent among patients of Asian descent.1 The pathogenesis of KFD remains unknown. Clinically, KFD may mimic malignant lymphoproliferative disorders, autoimmune diseases such as systemic lupus erythematosus (SLE) lymphadenitis, and infectious conditions such as HIV and tuberculous lymphadenitis. The most common presentation of KFD involves fever and rapidly evolving cervical lymphadenopathy.2,3 Due to its rarity, KFD is not always considered in the differential diagnosis for fever with tender lymphadenopathy, and up to one-third of cases are initially misdiagnosed.2
Definitive diagnosis requires lymph node biopsy. It is critical to achieving a timely diagnosis of KFD to exclude more serious conditions, initiate appropriate treatment, and minimize undue stress for patients. We describe a case of KFD in a patient who was met with delays in obtaining a definitive diagnosis for his symptoms.
Case Presentation
A 27-year-old previously healthy White man presented to the emergency department with subacute, progressive right-sided neck pain and swelling. In the week leading up to presentation, he also noted intermittent fevers, night sweats, and abdominal pain. His symptoms were unrelieved with acetaminophen and aspirin. He reported no sick contacts, recent travel, or animal exposures. He had no known history of autoimmune disease, malignancy, or immunocompromising conditions. Vital signs at the time of presentation were notable for a temperature of 39.0 °C. On examination, he had several firm, mobile, and exquisitely tender lymph nodes in the right upper anterior cervical chain. Abdominal examination was notable for left upper quadrant tenderness with palpable splenomegaly. Due to initial concern that his symptoms represented bacterial lymphadenitis, he was started on broad-spectrum antibiotics and admitted to the hospital for an expedited infectious workup.
Initial laboratory studies were notable for a white blood cell count of 3.7 × 109/L with 57.5% neutrophils and 27.0% lymphocytes on differential.
Computed tomography (CT) of the neck revealed multiple heterogeneously enlarged lymph nodes along the right anterior cervical chain with necrotic changes (Figure 1).
A core needle biopsy of a right-sided cervical lymph node was initially pursued, demonstrating necrotic tissue with minimal residual lymphoid tissue and no definitive evidence of lymphoma. Because these results were nondiagnostic, an excisional biopsy of the right-sided cervical lymph node was pursued 10 days later. Due to the stress of his 2-week hospitalization without a unifying diagnosis, the patient then elected to discharge home with close outpatient follow-up while awaiting his biopsy results. Antibiotics were not continued at the time of discharge as our broad infectious workup failed to yield a causative organism.
Two weeks postdischarge, the patient’s excisional lymph node biopsy returned demonstrating lymphohistiocytic inflammation with plasmacytoid dendritic cells, areas of necrosis, and scattered karyorrhectic nuclear debris, consistent with a diagnosis of KFD (Figure 2).
After 4 months of hydroxychloroquine therapy, the patient’s KFD symptoms resolved, prompting his dose to be reduced and eventually tapered. Repeat testing of his ANA and anti-dsDNA were performed at 1 and 6 months posthospitalization and returned within normal limits. A repeat PET-CT was performed 6 months posthospitalization showing resolution of his hypermetabolic right neck and right supraclavicular lymphadenopathy as well as his splenomegaly. It has now been more than a year since the patient’s initial presentation to the hospital, and he remains symptom-free and off prednisone and hydroxychloroquine.
Discussion
KFD is a rare cause of cervical lymphadenitis that was first described in 1972. Although cases have been reported worldwide, it is seen with higher prevalence in Asian countries. KFD was previously thought to have a female predominance, but recent reviews suggest a female to male ratio close to 1:1.1 The pathogenesis of KFD remains unknown, though some studies have suggested Epstein-Barr virus infection as a potential trigger.4,5 Human herpesvirus (HHV) 6, HHV 7, HHV 8, HSV, HIV, and parvovirus B19 also have been implicated as potential triggers, though no causative relationship has been established.2,5,6 Autoimmunity may also play a role in the pathogenesis of KFD given its histopathologic overlap with SLE lymphadenitis.1,7
The most common presenting symptoms of KFD include fever and tender cervical lymphadenopathy. Many patients also experience constitutional symptoms such as weight loss, night sweats, and fatigue.2 KFD is characterized by enlarged cervical lymph nodes, typically > 2 cm in diameter.3 Cutaneous manifestations of KFD are common and may manifest as nonspecific papules, plaques, nodules, or facial malar erythema.1,2 Case reports also have described KFD manifesting with ataxia, arthritis, parotitis, or ocular pathologies such as conjunctivitis and uveitis.1,2,8,9 Hepatosplenomegaly is a relatively rare manifestation of KFD seen in approximately 3% of cases.10 When present, hepatosplenomegaly may make the diagnosis of KFD especially difficult to distinguish from lymphoproliferative disorders such as lymphoma. Laboratory findings in KFD are nonspecific and include elevated levels of lactate dehydrogenase, erythrocyte sedimentation rate, C-reactive protein, and liver enzymes.3 Both lymphocytosis and lymphopenia have been described.3Definitive diagnosis of KFD is achieved through lymph node biopsy and histologic examination. Histopathologic findings of KFD include areas of coagulative necrosis and histiocytic proliferation within the cortical and paracortical regions of the lymph node. Scattered nuclear debris also may be seen, though this histologic finding also is seen with lymphoma. The absence of neutrophils is characteristic of KFD.2 In our patient, a core needle biopsy was initially pursued but returned nondiagnostic. A PET-CT also was obtained, though KFD may mimic lymphoma on PET as was seen in this patient’s case as well as in prior case reports.11 An excisional lymph node biopsy was ultimately performed and secured the diagnosis of KFD.
Although ultrasound-guided core needle biopsy was unable to yield the diagnosis for our patient, its diagnostic accuracy is still superior to that of fine needle aspiration and is therefore suggested as the primary diagnostic modality when KFD is suspected.12 Core needle biopsy also is less invasive, less time consuming, and perhaps more cost-effective than an open excisional biopsy, which often requires the use of an operating room and monitored anesthesia care.12 Understandably, our patient experienced significant stress while awaiting a final diagnosis. Whenever possible, lymph node biopsy should be prioritized over other diagnostic modalities to achieve a timely and definitive diagnosis.
KFD has no established treatment guidelines. Supportive care with antipyretics and analgesics is the most common initial approach, as KFD is typically a self-limited disease that resolves in 1 to 4 months.2 Patients with severe, persistent symptoms have been successfully treated with corticosteroids and hydroxychloroquine, with monotherapy typically trialed before concomitant use.2,13 After 2 courses of prednisone, our patient was prescribed single-agent hydroxychloroquine due to his recurrent symptoms and debilitating AEs from the steroids. Other case reports have described hydroxychloroquine as a treatment option when steroids fail to provide symptom relief or when there are recurrences of KFD.14-19 Retinopathy can occur as a result of long-term hydroxychloroquine use. As such, patients anticipated to require long-term hydroxychloroquine therapy should receive a baseline eye examination within months of drug initiation and repeat examination after 5 years of therapy.20
After symptom resolution, continued follow-up with a health care professional is recommended due to the potential for KFD recurrence or the development of a new autoimmune disease. The rate of KFD recurrence was previously described as 3%, but a more recent review found the rate of recurrence to be approximately 15% at > 6 months follow-up.1,3 Recurrence is often described during or shortly after the tapering of steroids.13,16,21,22 Recurrent KFD can be diagnosed with repeat lymph node biopsy, which also serves to exclude other disease processes.13,16 However, recurrence also has been diagnosed clinically based on the patient’s symptoms and laboratory investigations.21,22Continued surveillance of patients with KFD is also necessary to monitor for the development of new autoimmune diseases, especially SLE. SLE lymphadenitis shares many histopathologic characteristics with KFD. Case reports have described the development of SLE in patients with a history of KFD.2,7 Other autoimmune conditions described in patients with prior KFD include Sjögren syndrome, Hashimoto thyroiditis, Graves disease, mixed connective tissue disease, and antiphospholipid syndrome.3,23 Among patients with KFD, female sex, painful adenopathy, and cytopenias are significantly associated with the later development of autoimmune disease.23
Patient Perspective
This began for me in September 2020 out of the blue. I woke up one day with a random lymph node in my neck but otherwise felt completely healthy, and within 2 to 3 weeks I had never been more sick in my entire life. It came with bouts of fevers, neck pain from the swelling, stomach pain (I later learned an enlarged spleen was the source), terrible night sweats, violent chills where the shaking was uncontrollable for hours at a time, loss of appetite, and countless other symptoms that have come and gone over the past year.
It did take a little while to get a diagnosis, but I understand the autoimmune field is tricky. For about 4 to 5 weeks, I was told to prepare for a lymphoma diagnosis. I ended up doing 2 rounds of prednisone, one for 3 weeks at the end of 2020 and one for 2 months from March to May. The initial round helped quite a bit, but the second round did not have any effect on the lingering symptoms. In my opinion, prednisone is miserable to be on long term and I do not recommend it. The daily AEs that came with it included mood swings, insomnia, weight gain, and more. I have been on hydroxychloroquine now for almost 2 months and although it has some AEs of its own, it is nowhere near as rough as the prednisone and has helped manage my remaining symptoms quite a bit.
This certainly has not been a fun experience, but I was under great care during my time in the hospital and continue to be under good care through the rheumatology clinic. The one thing that could have made a huge difference would have been the issues involved in getting my surgery scheduled while I was still inpatient, which took quite a while. The pain during that time was so intense and unlike anything I have ever experienced before, and it was only the surgery that finally brought me some relief. To paint you a picture, I have broken bones, split my leg open, and have roughly 40 to 50 hours of tattoo work on me, and I have never experienced the level of pain like I felt in my neck and stomach. I remember feeling like someone had wound up and hit me with a baseball bat. The surgery brought me immense relief and if it had occurred when it was originally supposed to, I would have been spared 3 or so days of this type of pain.
It has been almost 10 months since my surgery and diagnosis, and life has mostly returned to normal for me. I am still on long-term medication as I mentioned, and I still deal with fatigue, spleen pain, and several other symptoms, but it is much more under control these days. I feel very fortunate to have been under and continue to be under such great care.
Conclusions
This case report highlights the importance of recognizing KFD as a rare but possible cause of fever and necrotizing cervical lymphadenopathy. KFD often mimics malignant lymphoproliferative disorders, autoimmune diseases such as SLE lymphadenitis, and infectious conditions such as HIV and tuberculous lymphadenitis. While KFD is seen with higher prevalence in Asian countries and was previously thought to be more predominant in females, the diagnosis should still be considered irrespective of geographic location or patient sex. Lymph node biopsy is the preferred diagnostic approach for patients with suspected KFD. Treatment is typically supportive but may consist of glucocorticoids in severe cases. Hydroxychloroquine may be used in refractory cases or as a steroid-sparing regimen when steroid AEs are poorly tolerated. Long-term follow-up is critical for patients with KFD to monitor for both disease recurrence and the development of autoimmune disease, especially SLE.
Acknowledgments
The authors thank Dr. Jacob Pilley for his detailed review of the patient’s pathology results. The authors also extend their gratitude to the patient, who deepened our understanding of this condition and what it is like to live with it.
Kikuchi-Fujimoto disease (KFD) is a rare, usually self-limited cause of cervical lymphadenitis that is more prevalent among patients of Asian descent.1 The pathogenesis of KFD remains unknown. Clinically, KFD may mimic malignant lymphoproliferative disorders, autoimmune diseases such as systemic lupus erythematosus (SLE) lymphadenitis, and infectious conditions such as HIV and tuberculous lymphadenitis. The most common presentation of KFD involves fever and rapidly evolving cervical lymphadenopathy.2,3 Due to its rarity, KFD is not always considered in the differential diagnosis for fever with tender lymphadenopathy, and up to one-third of cases are initially misdiagnosed.2
Definitive diagnosis requires lymph node biopsy. It is critical to achieving a timely diagnosis of KFD to exclude more serious conditions, initiate appropriate treatment, and minimize undue stress for patients. We describe a case of KFD in a patient who was met with delays in obtaining a definitive diagnosis for his symptoms.
Case Presentation
A 27-year-old previously healthy White man presented to the emergency department with subacute, progressive right-sided neck pain and swelling. In the week leading up to presentation, he also noted intermittent fevers, night sweats, and abdominal pain. His symptoms were unrelieved with acetaminophen and aspirin. He reported no sick contacts, recent travel, or animal exposures. He had no known history of autoimmune disease, malignancy, or immunocompromising conditions. Vital signs at the time of presentation were notable for a temperature of 39.0 °C. On examination, he had several firm, mobile, and exquisitely tender lymph nodes in the right upper anterior cervical chain. Abdominal examination was notable for left upper quadrant tenderness with palpable splenomegaly. Due to initial concern that his symptoms represented bacterial lymphadenitis, he was started on broad-spectrum antibiotics and admitted to the hospital for an expedited infectious workup.
Initial laboratory studies were notable for a white blood cell count of 3.7 × 109/L with 57.5% neutrophils and 27.0% lymphocytes on differential.
Computed tomography (CT) of the neck revealed multiple heterogeneously enlarged lymph nodes along the right anterior cervical chain with necrotic changes (Figure 1).
A core needle biopsy of a right-sided cervical lymph node was initially pursued, demonstrating necrotic tissue with minimal residual lymphoid tissue and no definitive evidence of lymphoma. Because these results were nondiagnostic, an excisional biopsy of the right-sided cervical lymph node was pursued 10 days later. Due to the stress of his 2-week hospitalization without a unifying diagnosis, the patient then elected to discharge home with close outpatient follow-up while awaiting his biopsy results. Antibiotics were not continued at the time of discharge as our broad infectious workup failed to yield a causative organism.
Two weeks postdischarge, the patient’s excisional lymph node biopsy returned demonstrating lymphohistiocytic inflammation with plasmacytoid dendritic cells, areas of necrosis, and scattered karyorrhectic nuclear debris, consistent with a diagnosis of KFD (Figure 2).
After 4 months of hydroxychloroquine therapy, the patient’s KFD symptoms resolved, prompting his dose to be reduced and eventually tapered. Repeat testing of his ANA and anti-dsDNA were performed at 1 and 6 months posthospitalization and returned within normal limits. A repeat PET-CT was performed 6 months posthospitalization showing resolution of his hypermetabolic right neck and right supraclavicular lymphadenopathy as well as his splenomegaly. It has now been more than a year since the patient’s initial presentation to the hospital, and he remains symptom-free and off prednisone and hydroxychloroquine.
Discussion
KFD is a rare cause of cervical lymphadenitis that was first described in 1972. Although cases have been reported worldwide, it is seen with higher prevalence in Asian countries. KFD was previously thought to have a female predominance, but recent reviews suggest a female to male ratio close to 1:1.1 The pathogenesis of KFD remains unknown, though some studies have suggested Epstein-Barr virus infection as a potential trigger.4,5 Human herpesvirus (HHV) 6, HHV 7, HHV 8, HSV, HIV, and parvovirus B19 also have been implicated as potential triggers, though no causative relationship has been established.2,5,6 Autoimmunity may also play a role in the pathogenesis of KFD given its histopathologic overlap with SLE lymphadenitis.1,7
The most common presenting symptoms of KFD include fever and tender cervical lymphadenopathy. Many patients also experience constitutional symptoms such as weight loss, night sweats, and fatigue.2 KFD is characterized by enlarged cervical lymph nodes, typically > 2 cm in diameter.3 Cutaneous manifestations of KFD are common and may manifest as nonspecific papules, plaques, nodules, or facial malar erythema.1,2 Case reports also have described KFD manifesting with ataxia, arthritis, parotitis, or ocular pathologies such as conjunctivitis and uveitis.1,2,8,9 Hepatosplenomegaly is a relatively rare manifestation of KFD seen in approximately 3% of cases.10 When present, hepatosplenomegaly may make the diagnosis of KFD especially difficult to distinguish from lymphoproliferative disorders such as lymphoma. Laboratory findings in KFD are nonspecific and include elevated levels of lactate dehydrogenase, erythrocyte sedimentation rate, C-reactive protein, and liver enzymes.3 Both lymphocytosis and lymphopenia have been described.3Definitive diagnosis of KFD is achieved through lymph node biopsy and histologic examination. Histopathologic findings of KFD include areas of coagulative necrosis and histiocytic proliferation within the cortical and paracortical regions of the lymph node. Scattered nuclear debris also may be seen, though this histologic finding also is seen with lymphoma. The absence of neutrophils is characteristic of KFD.2 In our patient, a core needle biopsy was initially pursued but returned nondiagnostic. A PET-CT also was obtained, though KFD may mimic lymphoma on PET as was seen in this patient’s case as well as in prior case reports.11 An excisional lymph node biopsy was ultimately performed and secured the diagnosis of KFD.
Although ultrasound-guided core needle biopsy was unable to yield the diagnosis for our patient, its diagnostic accuracy is still superior to that of fine needle aspiration and is therefore suggested as the primary diagnostic modality when KFD is suspected.12 Core needle biopsy also is less invasive, less time consuming, and perhaps more cost-effective than an open excisional biopsy, which often requires the use of an operating room and monitored anesthesia care.12 Understandably, our patient experienced significant stress while awaiting a final diagnosis. Whenever possible, lymph node biopsy should be prioritized over other diagnostic modalities to achieve a timely and definitive diagnosis.
KFD has no established treatment guidelines. Supportive care with antipyretics and analgesics is the most common initial approach, as KFD is typically a self-limited disease that resolves in 1 to 4 months.2 Patients with severe, persistent symptoms have been successfully treated with corticosteroids and hydroxychloroquine, with monotherapy typically trialed before concomitant use.2,13 After 2 courses of prednisone, our patient was prescribed single-agent hydroxychloroquine due to his recurrent symptoms and debilitating AEs from the steroids. Other case reports have described hydroxychloroquine as a treatment option when steroids fail to provide symptom relief or when there are recurrences of KFD.14-19 Retinopathy can occur as a result of long-term hydroxychloroquine use. As such, patients anticipated to require long-term hydroxychloroquine therapy should receive a baseline eye examination within months of drug initiation and repeat examination after 5 years of therapy.20
After symptom resolution, continued follow-up with a health care professional is recommended due to the potential for KFD recurrence or the development of a new autoimmune disease. The rate of KFD recurrence was previously described as 3%, but a more recent review found the rate of recurrence to be approximately 15% at > 6 months follow-up.1,3 Recurrence is often described during or shortly after the tapering of steroids.13,16,21,22 Recurrent KFD can be diagnosed with repeat lymph node biopsy, which also serves to exclude other disease processes.13,16 However, recurrence also has been diagnosed clinically based on the patient’s symptoms and laboratory investigations.21,22Continued surveillance of patients with KFD is also necessary to monitor for the development of new autoimmune diseases, especially SLE. SLE lymphadenitis shares many histopathologic characteristics with KFD. Case reports have described the development of SLE in patients with a history of KFD.2,7 Other autoimmune conditions described in patients with prior KFD include Sjögren syndrome, Hashimoto thyroiditis, Graves disease, mixed connective tissue disease, and antiphospholipid syndrome.3,23 Among patients with KFD, female sex, painful adenopathy, and cytopenias are significantly associated with the later development of autoimmune disease.23
Patient Perspective
This began for me in September 2020 out of the blue. I woke up one day with a random lymph node in my neck but otherwise felt completely healthy, and within 2 to 3 weeks I had never been more sick in my entire life. It came with bouts of fevers, neck pain from the swelling, stomach pain (I later learned an enlarged spleen was the source), terrible night sweats, violent chills where the shaking was uncontrollable for hours at a time, loss of appetite, and countless other symptoms that have come and gone over the past year.
It did take a little while to get a diagnosis, but I understand the autoimmune field is tricky. For about 4 to 5 weeks, I was told to prepare for a lymphoma diagnosis. I ended up doing 2 rounds of prednisone, one for 3 weeks at the end of 2020 and one for 2 months from March to May. The initial round helped quite a bit, but the second round did not have any effect on the lingering symptoms. In my opinion, prednisone is miserable to be on long term and I do not recommend it. The daily AEs that came with it included mood swings, insomnia, weight gain, and more. I have been on hydroxychloroquine now for almost 2 months and although it has some AEs of its own, it is nowhere near as rough as the prednisone and has helped manage my remaining symptoms quite a bit.
This certainly has not been a fun experience, but I was under great care during my time in the hospital and continue to be under good care through the rheumatology clinic. The one thing that could have made a huge difference would have been the issues involved in getting my surgery scheduled while I was still inpatient, which took quite a while. The pain during that time was so intense and unlike anything I have ever experienced before, and it was only the surgery that finally brought me some relief. To paint you a picture, I have broken bones, split my leg open, and have roughly 40 to 50 hours of tattoo work on me, and I have never experienced the level of pain like I felt in my neck and stomach. I remember feeling like someone had wound up and hit me with a baseball bat. The surgery brought me immense relief and if it had occurred when it was originally supposed to, I would have been spared 3 or so days of this type of pain.
It has been almost 10 months since my surgery and diagnosis, and life has mostly returned to normal for me. I am still on long-term medication as I mentioned, and I still deal with fatigue, spleen pain, and several other symptoms, but it is much more under control these days. I feel very fortunate to have been under and continue to be under such great care.
Conclusions
This case report highlights the importance of recognizing KFD as a rare but possible cause of fever and necrotizing cervical lymphadenopathy. KFD often mimics malignant lymphoproliferative disorders, autoimmune diseases such as SLE lymphadenitis, and infectious conditions such as HIV and tuberculous lymphadenitis. While KFD is seen with higher prevalence in Asian countries and was previously thought to be more predominant in females, the diagnosis should still be considered irrespective of geographic location or patient sex. Lymph node biopsy is the preferred diagnostic approach for patients with suspected KFD. Treatment is typically supportive but may consist of glucocorticoids in severe cases. Hydroxychloroquine may be used in refractory cases or as a steroid-sparing regimen when steroid AEs are poorly tolerated. Long-term follow-up is critical for patients with KFD to monitor for both disease recurrence and the development of autoimmune disease, especially SLE.
Acknowledgments
The authors thank Dr. Jacob Pilley for his detailed review of the patient’s pathology results. The authors also extend their gratitude to the patient, who deepened our understanding of this condition and what it is like to live with it.
1. Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol. 2004;122(1):141-152. doi:10.1309/YF08-1L4T-KYWV-YVPQ
2. Deaver D, Horna P, Cualing H, Sokol L. Pathogenesis, diagnosis, and management of Kikuchi-Fujimoto disease. Cancer Control. 2014;21(4):313-321. doi:10.1177/107327481402100407
3. Cheng CY, Sheng WH, Lo YC, Chung CS, Chen YC, Chang SC. Clinical presentations, laboratory results and outcomes of patients with Kikuchi’s disease: emphasis on the association between recurrent Kikuchi’s disease and autoimmune diseases. J Microbiol Immunol Infect. 2010;43(5):366-371. doi:10.1016/S1684-1182(10)60058-8
4. Stéphan JL, Jeannoël P, Chanoz J, Gentil-Përret A. Epstein-Barr virus-associated Kikuchi disease in two children. J Pediatr Hematol Oncol. 2001;23(4):240-243. doi:10.1097/00043426-200105000-00012
5. Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, Chen LM. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. Detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113(6):774-781. doi:10.1309/1A6Y-YCKP-5AVF-QTYR
6. Rosado FG, Tang YW, Hasserjian RP, McClain CM, Wang B, Mosse CA. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44(2):255-259. doi:10.1016/j.humpath.2012.05.016
7. Gordon JK, Magro C, Lu T, et al. Overlap between systemic lupus erythematosus and Kikuchi Fujimoto disease: a clinical pathology conference held by the Department of Rheumatology at Hospital for Special Surgery. HSS J. 2009;5(2):169-177. doi:10.1007/s11420-009-9123-x
8. Lo KB, Papazoglou A, Chua L, Candelario N. Case Report: Kikuchi: The great mimicker. F1000Res. 2018;7:520. Published 2018 Apr 30. doi:10.12688/f1000research.14758.1
9. Galor A, Georgy M, Leder HA, Dunn JP, Peters GB 3rd. Papillary conjunctivitis associated with Kikuchi disease. Cornea. 2008;27(8):944-946. doi:10.1097/ICO.0b013e31816bf488
10. Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26(1):50-54. doi:10.1007/s10067-006-0230-5
11. Lee DH, Lee JH, Shim EJ, et al. Disseminated Kikuchi-Fujimoto disease mimicking malignant lymphoma on positron emission tomography in a child. J Pediatr Hematol Oncol. 2009;31(9):687-689. doi:10.1097/MPH.0b013e31819a5d77
12. Park SG, Koo HR, Jang K, et al. Efficacy of ultrasound-guided needle biopsy in the diagnosis of Kikuchi-Fujimoto disease. Laryngoscope. 2021;131(5):E1519-E1523. doi:10.1002/lary.29160
13. Honda F, Tsuboi H, Toko H, et al. Recurrent Kikuchi-Fujimoto disease successfully treated by the concomitant use of hydroxychloroquine and corticosteroids. Intern Med. 2017;56(24):3373-3377. doi:10.2169/internalmedicine.9205-17
14. Rezayat T, Carroll MB, Ramsey BC, Smith A. A case of relapsing Kikuchi-Fujimoto disease. Case Rep Otolaryngol. 2013;2013:364795. doi:10.1155/2013/364795
15. Rezai K, Kuchipudi S, Chundi V, Ariga R, Loew J, Sha BE. Kikuchi-Fujimoto disease: hydroxychloroquine as a treatment. Clin Infect Dis. 2004;39(12):e124-e126. doi:10.1086/426144
16. Hyun M, So IT, Kim HA, Jung H, Ryu SY. Recurrent Kikuchi’s disease treated by hydroxychloroquine. Infect Chemother. 2016;48(2):127-131. doi:10.3947/ic.2016.48.2.127
17. Lin YC, Huang HH, Nong BR, et al. Pediatric Kikuchi-Fujimoto disease: A clinicopathologic study and the therapeutic effects of hydroxychloroquine. J Microbiol Immunol Infect. 2019;52(3):395-401. doi:10.1016/j.jmii.2017.08.023
18. Lin DY, Villegas MS, Tan PL, Wang S, Shek LP. Severe Kikuchi’s disease responsive to immune modulation. Singapore Med J. 2010;51(1):e18-e21.
19. Quintás-Cardama A, Fraga M, Cozzi SN, Caparrini A, Maceiras F, Forteza J. Fatal Kikuchi-Fujimoto disease: the lupus connection. Ann Hematol. 2003;82(3):186-188. doi:10.1007/s00277-003-0611-7
20. American Academy of Ophthalmology. ACR, AAD, RDS, and AAO 2020 Joint Statement on Hydroxychloroquine Use with Respect to Retinal Toxicity. Updated February 2021. Accessed November 28, 2022. https://www.aao.org/clinical-statement/acr-aad-rds-aao-2020-joint-statement-on-hydroxychl-2
21. Gerwig U, Weidmann RG, Lindner G. Relapsing Kikuchi-Fujimoto disease requiring prolonged steroid therapy. Case Rep Emerg Med. 2019;2019:6405687. Published 2019 Mar 7. doi:10.1155/2019/6405687
22. Faheem B, Kumar V, Ashkar H, Komal F, Sultana Y. Recurrent Kikuchi-Fujimoto disease masquerading as lymphoma successfully treated by anakinra. Cureus. 2020;12(11):e11655. Published 2020 Nov 23. doi:10.7759/cureus.11655
23. Sopeña B, Rivera A, Vázquez-Triñanes C, et al. Autoimmune manifestations of Kikuchi disease. Semin Arthritis Rheum. 2012;41(6):900-906. doi:10.1016/j.semarthrit.2011.11.001
1. Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol. 2004;122(1):141-152. doi:10.1309/YF08-1L4T-KYWV-YVPQ
2. Deaver D, Horna P, Cualing H, Sokol L. Pathogenesis, diagnosis, and management of Kikuchi-Fujimoto disease. Cancer Control. 2014;21(4):313-321. doi:10.1177/107327481402100407
3. Cheng CY, Sheng WH, Lo YC, Chung CS, Chen YC, Chang SC. Clinical presentations, laboratory results and outcomes of patients with Kikuchi’s disease: emphasis on the association between recurrent Kikuchi’s disease and autoimmune diseases. J Microbiol Immunol Infect. 2010;43(5):366-371. doi:10.1016/S1684-1182(10)60058-8
4. Stéphan JL, Jeannoël P, Chanoz J, Gentil-Përret A. Epstein-Barr virus-associated Kikuchi disease in two children. J Pediatr Hematol Oncol. 2001;23(4):240-243. doi:10.1097/00043426-200105000-00012
5. Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, Chen LM. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. Detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113(6):774-781. doi:10.1309/1A6Y-YCKP-5AVF-QTYR
6. Rosado FG, Tang YW, Hasserjian RP, McClain CM, Wang B, Mosse CA. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44(2):255-259. doi:10.1016/j.humpath.2012.05.016
7. Gordon JK, Magro C, Lu T, et al. Overlap between systemic lupus erythematosus and Kikuchi Fujimoto disease: a clinical pathology conference held by the Department of Rheumatology at Hospital for Special Surgery. HSS J. 2009;5(2):169-177. doi:10.1007/s11420-009-9123-x
8. Lo KB, Papazoglou A, Chua L, Candelario N. Case Report: Kikuchi: The great mimicker. F1000Res. 2018;7:520. Published 2018 Apr 30. doi:10.12688/f1000research.14758.1
9. Galor A, Georgy M, Leder HA, Dunn JP, Peters GB 3rd. Papillary conjunctivitis associated with Kikuchi disease. Cornea. 2008;27(8):944-946. doi:10.1097/ICO.0b013e31816bf488
10. Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26(1):50-54. doi:10.1007/s10067-006-0230-5
11. Lee DH, Lee JH, Shim EJ, et al. Disseminated Kikuchi-Fujimoto disease mimicking malignant lymphoma on positron emission tomography in a child. J Pediatr Hematol Oncol. 2009;31(9):687-689. doi:10.1097/MPH.0b013e31819a5d77
12. Park SG, Koo HR, Jang K, et al. Efficacy of ultrasound-guided needle biopsy in the diagnosis of Kikuchi-Fujimoto disease. Laryngoscope. 2021;131(5):E1519-E1523. doi:10.1002/lary.29160
13. Honda F, Tsuboi H, Toko H, et al. Recurrent Kikuchi-Fujimoto disease successfully treated by the concomitant use of hydroxychloroquine and corticosteroids. Intern Med. 2017;56(24):3373-3377. doi:10.2169/internalmedicine.9205-17
14. Rezayat T, Carroll MB, Ramsey BC, Smith A. A case of relapsing Kikuchi-Fujimoto disease. Case Rep Otolaryngol. 2013;2013:364795. doi:10.1155/2013/364795
15. Rezai K, Kuchipudi S, Chundi V, Ariga R, Loew J, Sha BE. Kikuchi-Fujimoto disease: hydroxychloroquine as a treatment. Clin Infect Dis. 2004;39(12):e124-e126. doi:10.1086/426144
16. Hyun M, So IT, Kim HA, Jung H, Ryu SY. Recurrent Kikuchi’s disease treated by hydroxychloroquine. Infect Chemother. 2016;48(2):127-131. doi:10.3947/ic.2016.48.2.127
17. Lin YC, Huang HH, Nong BR, et al. Pediatric Kikuchi-Fujimoto disease: A clinicopathologic study and the therapeutic effects of hydroxychloroquine. J Microbiol Immunol Infect. 2019;52(3):395-401. doi:10.1016/j.jmii.2017.08.023
18. Lin DY, Villegas MS, Tan PL, Wang S, Shek LP. Severe Kikuchi’s disease responsive to immune modulation. Singapore Med J. 2010;51(1):e18-e21.
19. Quintás-Cardama A, Fraga M, Cozzi SN, Caparrini A, Maceiras F, Forteza J. Fatal Kikuchi-Fujimoto disease: the lupus connection. Ann Hematol. 2003;82(3):186-188. doi:10.1007/s00277-003-0611-7
20. American Academy of Ophthalmology. ACR, AAD, RDS, and AAO 2020 Joint Statement on Hydroxychloroquine Use with Respect to Retinal Toxicity. Updated February 2021. Accessed November 28, 2022. https://www.aao.org/clinical-statement/acr-aad-rds-aao-2020-joint-statement-on-hydroxychl-2
21. Gerwig U, Weidmann RG, Lindner G. Relapsing Kikuchi-Fujimoto disease requiring prolonged steroid therapy. Case Rep Emerg Med. 2019;2019:6405687. Published 2019 Mar 7. doi:10.1155/2019/6405687
22. Faheem B, Kumar V, Ashkar H, Komal F, Sultana Y. Recurrent Kikuchi-Fujimoto disease masquerading as lymphoma successfully treated by anakinra. Cureus. 2020;12(11):e11655. Published 2020 Nov 23. doi:10.7759/cureus.11655
23. Sopeña B, Rivera A, Vázquez-Triñanes C, et al. Autoimmune manifestations of Kikuchi disease. Semin Arthritis Rheum. 2012;41(6):900-906. doi:10.1016/j.semarthrit.2011.11.001
Pyostomatitis Vegetans With Orofacial and Vulvar Granulomatosis in a Pediatric Patient
Case Report
A 7-year-old girl who was otherwise healthy was referred by pediatric gastroenterology for evaluation of cutaneous Crohn disease (CD). The patient had a 4-year history of persistent lip swelling and a 3-year history of asymmetric erythematous labial swelling and perianal erythema with skin tags. She had been applying the calcineurin inhibitor tacrolimus ointment 0.03% 1 or 2 times daily to her lesions with minimal improvement. She did not have a medical history of recurrent or unusual infectious diseases. There was no family history of autoimmune disease.
The patient and her guardian reported intermittent perianal pain but denied constipation, diarrhea, abdominal pain, and blood in the stool. She denied throat and tongue swelling, dysphagia, dyspnea, drooling, facial paralysis, and eyelid edema. She was a well-nourished child whose height and weight percentiles tracked at 30% and 25%, respectively. Physical examination revealed confluent symmetric lip swelling with mild angular cheilitis. Multiple 1- to 2-mm white pustules with pinpoint erosions covered the upper and lower labial mucosa and extended onto the buccal mucosa (Figure 1). She had symmetric erythema and swelling of the left labia majora extending to and involving the left perianal mucosa. Three perianal erythematous skin tags and a perianal fissure were identified.

The patient had been assessed 2 years earlier by pediatric dermatology and gastroenterology with an extensive evaluation that favored a diagnosis of cutaneous CD because the combination of orofacial granulomatosis (OFG), vulvar edema, and perianal skin tags is strongly associated.1-3 Contact dermatitis affecting the mouth was considered; however, allergen testing did not demonstrate a trigger.
A trial of a benzoate- and cinnamon-free diet, which has been reported to improve OFG,4 did not provide symptomatic improvement. Topical corticosteroids and tacrolimus reduced the perioral erythema, but the swelling persisted. An infectious cause was considered; however, topical mupirocin had no effect, and amoxicillin resulted in oral candidiasis.
A perianal biopsy revealed a granulomatous dermatitis. Fungal and bacterial cultures were negative. Upper and lower gastrointestinal (GI) endoscopy and a fecal calprotectin assay were not suggestive of inflammatory bowel disease (IBD). A complete blood cell count and QuantiFERON-TB Gold test measuring the immune response to tuberculosis antigens were normal. Chronic granulomatous disease, RAG1/RAG2 deficiency, common variable immunodeficiency, and NOD2 defects were ruled out with normal tests of dihydrorhodamine, quantitative immunoglobulins, and toll-like receptors.
Because of the discomfort associated with the patient’s lesions, she was offered treatment with tumor necrosis factor α inhibitors, including infliximab and adalimumab. These agents had been offered since the onset of symptoms; however, her parents declined systemic medication unless she developed GI involvement. Instead, the tacrolimus concentration was increased to 0.1% applied to the lips, labia, and perianal area, and fluocinonide gel 0.05% applied nightly to the oral pustules was added.
Two months later the patient had notably fewer oral pustules and diminished erythema but only slightly reduced oral, vulvar, and perianal swelling. A trial of oral metronidazole, which has been reported to clear a patient with cutaneous CD,5 was discontinued by her parents after 6 weeks because of a lack of interval improvement.
One year later, a pre-existing perianal skin tag doubled in size and became exquisitely tender. The calprotectin level—previously within reference range at less than 16 μg/g—was now elevated at 149 μg/g (reference range, 1–120 μg/g) and increased to 336 μg/g 3 weeks later. Testing for C-reactive protein, zinc, and stool occult blood; a comprehensive metabolic panel; and a complete blood cell count were unremarkable.
Repeat upper and lower GI endoscopy did not suggest CD. A biopsy using direct immunofluorescence (DIF) was obtained to evaluate for pyostomatitis vegetans (PSV) and rule out
The captured biopsy did not demonstrate the intended pustule; instead, it included less-affected mucosa and was obtained during topical treatment when few pustules and erosions persisted. Pathologic analysis revealed noncaseating granulomas without an increase in microabscesses, neutrophils, or eosinophils (Figure 2). Direct immunofluorescence staining for IgG, IgA, and C3 and indirect immunofluorescence staining for desmoglein-1 and desmoglein-3 antibodies were negative. Although the biopsy did not capture the intended pustule, diagnosis of PV was made based on clinical features and the constellation of cutaneous findings associated with IBD.
. B, Mild inflammation, predominantly lymphocytic, and irregular acanthosis were noted")
Intralesional triamcinolone, which has been of benefit for pediatric patients with orofacial granulomatosis,1,6,7 was instituted and normalized the vulva and perianal mucosa; however, lip swelling improved only minimally.
Comment
Pyostomatitis vegetans is characterized by multiple white or yellow, friable, miliary pustules that rupture, leaving behind ulcerations and erosions that cause a varying degree of oral pain.8 The disorder can involve any area of the oral mucosa—most often the labia-attached gingiva, soft and hard palates, buccal mucosa, vestibule, and tonsillar areas—but often spares the floor of the mouth and tongue.8-11 The term pyostomatitis vegetans was proposed in 1949 by McCarthy12 when he noted in a patient who presented with the characteristic appearance of the oral mucosa, though cases of vaginal, nasal, and periocular involvement have been reported.8,13,14
Histopathology—Pyostomatitis vegetans displays pseudoepithelial hyperplasia with acanthosis, hyperkeratosis, and intraepithelial or subepithelial microabscesses (or both) with neutrophils and eosinophils.8,9,15 There are a few possible explanations for this patient’s lack of tissue eosinophilia. It has been theorized that the presence of granulomas could mask concurrent PSV16 or that tissue in PSV contains fewer eosinophils as the disorder progresses.11 The oral biopsy obtained from our patient did not capture a pustule, and the condition had noticeably improved with topical tacrolimus at the time of biopsy; therefore, neither neutrophils nor eosinophils were identified. Peripheral eosinophilia, which is present in 42% to 90% of cases of PSV,9,17 can be a diagnostic clue.18 However, PE is associated with IBD,24 which usually occurs with PSV, so the absence of peripheral eosinophilia in our patient may be explained by her lack of bowel disease.
Pathogenesis—The pathogenesis of PSV is unknown. A proposed etiology includes cross-reacting antigens in the bowel and skin secondary to IBD as well as an aberrant immune response to an unidentified factor.8 Pyostomatitis vegetans is considered by many to be the mucosal variant of pyodermatitis vegetans,9,15,19 a neutrophilic dermatosis characterized by asymmetric, crusted, erythematous papulopustules that extend peripherally and coalesce to form large vegetating plaques. These lesions commonly manifest in the axillary folds, groin, and scalp and can involve the face, trunk, and distal extremities.9,18 Infection has been suggested as a cause of PSV, though cultures for pathogenic bacteria, viruses, and fungi consistently show only normal flora.20 Zinc deficiency attributed to malabsorption from CD was reported in an adult with PSV.21 The PSV resolved after 6 weeks of zinc supplementation.
Differential Diagnosis—The main entity in the clinical differential diagnosis for PSV is PVH, which is considered a variant of pemphigus vulgaris. Pemphigus vegetans of Hallopeau presents with pustules and progresses to hyperpigmented vegetative plaques with peripheral hypertrophic granulation tissue.22 The clinical and histological presentation of PVH can be similar to PSV; in PVH, however, DIF demonstrates intercellular IgG and C3 due to circulating IgG autoantibodies specific for desmoglein 3, a cell adhesion molecule.22-24 In PSV, DIF typically is negative for IgG, IgA, and C3.8 Immunohistochemical findings of PSV may overlap with IgA pemphigus, IgG/IgA pemphigus, and IgG pemphigus, which has sparked debate if PSV is an autoimmune blistering disorder or a secondary finding of epithelial injury.9,18,24
Pyostomatitis vegetans is most prevalent in patients aged 20 to 59 years25 but can occur at any age.8,19 Overall, extraintestinal symptoms, including mucocutaneous findings, are common in pediatric patients—in 30% to 71% of children with CD and 21% to 22% of children with ulcerative colitis26—and can predate onset of GI symptoms in 6% of pediatric patients.27
Oral disease is common in CD; manifestations are listed in the Table.28,29 In a prospective study of 48 children with CD, 42% (20/48) had oral manifestations identified at diagnosis28; in a similar study of 25 children, researchers noted that 48% (12/25) had disease-specific oral lesions.29 None of these children recognized the oral findings prior to the onset of systemic symptoms.28 Pyostomatitis vegetans was the least common oral manifestation, reported in 1 of 73 patients in the 2 studies combined.28,29

Two recent articles that looked at PSV in pediatric and adolescent populations identified only 9 patients with PSV.24,30 Only 2 patients (siblings) had documented onset of PSV before 12 years of age,31 which suggests an underlying genetic predisposition in young children.
It has been reported that active or subclinical (ie, asymptomatic with positive endoscopic findings) IBD in adults precedes onset of PSV, which may be considered a sign of relapse.9,30 However, PSV is incredibly rare in children and adolescents and can be an early finding of IBD in children.16,31,32
Our patient has not developed GI involvement since her initial presentation 5 years prior, though another pediatric patient developed symptomatic CD 9 years after onset of OFG.5 A retrospective review of pediatric OFG without CD met criteria for CD at a median of 3.1 years (range, 0.4–6.9 years).33 Regrettably, the early presence of PSV has been associated with future progression to CD and a complicated disease course.12,34
Management—Pyoderma stomatitis vegetans is treated with management of underlying IBD,8 with scarce literature available regarding pediatric patients. Oral lesions have been treated with antiseptics and topical corticosteroids, though these have limited benefit.8 In an adult with IBD, topical tacrolimus initially cleared PSV; however, lesions recurred until mesalamine was initiated.35 Systemic steroids were effective in a 16-year-old patient with CD and PSV,12 but recurrence is common after corticosteroids are stopped.34
Some patients benefit from steroid-sparing medications, such as dapsone, azathioprine, sulfamethoxypyridazine, methotrexate, mycophenolate mofetil, and tumor necrosis factor α inhibitors such as infliximab and adalimumab.8,9,15,23,34,36 A 12-year-old patient with pyodermatitis–PSV without intestinal disease was treated with prednisone, dapsone, and azathioprine with improvement but not complete resolution of oral erosions after 18 weeks of treatment.32 A 15-year-old patient with CD and pyodermatitis–PSV did not show improvement on prednisone, dapsone, and azathioprine but rapidly responded to infliximab.23 Infliximab led to complete clearance of oral lesions in an adult with severe fistulizing CD who developed PSV.11 However, 2 adolescent patients with CD developed PSV while on adalimumab,6,34 though 1 did improve after increasing adalimumab from once to twice weekly.6
Conclusion
The case described here—PSV in a prepubertal 7-year-old with multiple cutaneous findings suggestive of CD, including OFG, perianal and vulvar edema with biopsy-proven noncaseating granulomas, anal skin tags, and an elevated calprotectin level, noted during a cutaneous flare without clinical or endoscopically identified underlying bowel involvement—is an extremely rare presentation. Literature regarding management of PSV primarily is found in the form of case reports and focuses on treating underlying IBD. In patients with intestinal disease, treatment with biologic therapy appears most effective.6,23
ADDENDUM
Interestingly, 3 years after the patient’s original presentation to our clinic, chromosomal sequencing analysis to assess for copy number variants and whole exome gene sequencing identified a variant of unknown significance in the heat shock protein family A member 1-like gene, HSPA1L, which has an unknown mode of inheritance, but the literature suggests that both truncating and missense variants could be associated with individuals with ulcerative colitis, CD, and IBD.37,38 Although we cannot use this information to render a molecular diagnosis, it is highly suspicious that this is the cause of her clinical findings. Additionally, the patient currently is aged 10 years with unchanged cutaneous findings and has not developed gastrointestinal findings of IBD.
- Tuxen AJ, Orchard D. Childhood and adolescent orofacial granulomatosis is strongly associated with Crohn’s disease and responds to intralesional corticosteroids. Australas J Dermatol. 2010;51:124-127. doi:10.1111/j.1440-0960.2010.00627.x
- Vaid RM, Cohen BA. Cutaneous Crohn’s disease in the pediatric population. Pediatr Dermatol. 2010;27:279-281. doi:10.1111/j.1525-1470.2010.01138.x
- van de Scheur MR, van der Waal RIF, van der Waal I, et al. Ano-genital granulomatosis: the counterpart of oro-facial granulomatosis. J Eur Acad Dermatol Venereol. 2003;17:184-189. doi:10.1046/j.1468-3083.2003.00573.x
- Campbell HE, Escudier MP, Patel P, et al. Review article: cinnamon- and benzoate-free diet as a primary treatment for orofacial granulomatosis. Aliment Pharmacol Ther. 2011;34:687-701. doi:10.1111/j.1365-2036.2011.04792.x
- Duhra P, Paul CJ. Metastatic Crohn’s disease responding to metronidazole. Br J Dermatol. 1988;119:87-91. doi:10.1111/j.1365-2133.1988.tb07107.x
- Katsanos KH, Torres J, Roda G, et al. Review article: non-malignant oral manifestations in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015;42:40-60. doi:10.1111/apt.13217
- Schmitz BA, Unkel JH. Symptomatic oral Crohn’s disease in an adolescent. J Dent Child (Chic). 2018;85:66-69.
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Clark LG, Tolkachjov SN, Bridges AG, et al. Pyostomatitis vegetans (PSV)–pyodermatitis vegetans (PDV): a clinicopathologic study of 7 cases at a tertiary referral center. J Am Acad Dermatol. 2016;75:578-584. doi:10.1016/j.jaad.2016.03.047
- Hansen LS, Silverman S Jr, Daniels TE. The differential diagnosis of pyostomatitis vegetans and its relation to bowel disease. Oral Surg Oral Med Oral Pathol. 1983;55:363-373. doi:10.1016/0030-4220(83)90191-3
- Cataldo E, Covino MC, Tesone PE. Pyostomatitis vegetans. Oral Surg Oral Med Oral Pathol. 1981;52:172-177. doi:10.1016/0030-4220(81)90316-9
- McCarthy FP. Pyostomatitis vegetans; report of three cases. Arch Derm Syphilol. 1949;60:750-764.
- Bens G, Laharie D, Beylot-Barry M, et al. Successful treatment with infliximab and methotrexate of pyostomatitis vegetans associated with Crohn’s disease. Br J Dermatol. 2003;149:181-184. doi:10.1046/j.1365-2133.2003.05385.x
- Leibovitch I, Ooi C, Huilgol SC, et al. Pyodermatitis–pyostomatitis vegetans of the eyelids: case report and review of the literature. Ophthalmology. 2005;112:1809-1813. doi:10.1016/j.ophtha.2005.04.027
- Ruiz-Roca JA, Berini-Aytés L, Gay-Escoda C. Pyostomatitis vegetans. report of two cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:447-454. doi:10.1016/j.tripleo.2003.08.022
- Molnár T, Farkas K, Nagy F, et al. Third case: another pediatric patient with pyostomatitis vegetans and oral granuloma as one of the initial symptoms of Crohn’s disease. Inflamm Bowel Dis. 2011;17:E122-E123. doi:10.1002/ibd.21791
- Leydhecker W, Lund OE. Eye involvement in pyostomatitis vegetans. Klin Monbl Augenheilkd Augenarztl Fortbild. 1962;141:595-602.
- Thornhill MH, Zakrzewska JM, Gilkes JJ. Pyostomatitis vegetans: report of three cases and review of the literature. J Oral Pathol Med. 1992;21:128-133. doi:10.1111/j.1600-0714.1992.tb00996.x
- Chaudhry SI, Philpot NS, Odell EW, et al. Pyostomatitis vegetans associated with asymptomatic ulcerative colitis: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:327-330. doi:10.1016/s1079-2104(99)70217-9
- Konstantopoulou M, O’Dwyer EM, Steele JC, et al. Pyodermatitis–pyostomatitis vegetans complicated by methicillin-resistant Staphylococcus aureus infection. Clin Exp Dermatol. 2005;30:666-668. doi:10.1111/j.1365-2230.2005.01906.x
- Ficarra G, Cicchi P, Amorosi A, et al. Oral Crohn’s disease and pyostomatitis vegetans. an unusual association. Oral Surg Oral Med Oral Pathol. 1993;75:220-224. doi:10.1016/0030-4220(93)90097-n
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428. doi:10.1111/j.1365-4632.2004.02480.x
- Nico MMS, Hussein TP, Aoki V, et al. Pyostomatitis vegetans and its relation to inflammatory bowel disease, pyoderma gangrenosum, pyodermatitis vegetans, and pemphigus. J Oral Pathol Med. 2012;41:584-588. doi:10.1111/j.1600-0714.2012.01152.x
- Berzin D, Lahad A, Weiss B, et al. Inflammatory bowel disease presenting with pyodermatitis–pyostomatitis vegetans in a pediatric patient: a case report and review of the literature. Pediatr Dermatol. 2021;38:868-871. doi:10.1111/pde.14625
- Ballo FS, Camisa C, Allen CM. Pyostomatitis vegetans. report of a case and review of the literature. J Am Acad Dermatol. 1989;21:381-387.
- Greuter T, Bertoldo F, Rechner R, et al; Swiss IBD Cohort Study Group. Extraintestinal manifestations of pediatric inflammatory bowel disease: prevalence, presentation, and anti-TNF treatment. J Pediatr Gastroenterol Nutr. 2017;65:200-206. doi:10.1097/MPG.0000000000001455
- Jose FA, Garnett EA, Vittinghoff E, et al. Development of extraintestinal manifestations in pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:63-68. doi:10.1002/ibd.20604
- Harty S, Fleming P, Rowland M, et al. A prospective study of the oral manifestations of Crohn’s disease. Clin Gastroenterol Hepatol. 2005;3:886-891. doi:10.1016/s1542-3565(05)00424-6
- Pittock S, Drumm B, Fleming P, et al. The oral cavity in Crohn’s disease. J Pediatr. 2001;138:767-771. doi:10.1067/mpd.2001.113008
- Bardasi G, Romagnoli A, Foschini MP, et al. Pyostomatitis vegetans in a pediatric patient with ulcerative colitis: case report of a rare pediatric inflammatory bowel disease extraintestinal manifestation and review of the literature. Eur J Gastroenterol Hepatol. 2020;32:889-892. doi:10.1097/MEG.0000000000001723
- Mesquita Kde C, Costa IM. Case for diagnosis. An Bras Dermatol. 2012;87:929-931. doi:10.1590/s0365-05962012000600022
- Al-Rimawi HS, Hammad MM, Raweily EA, et al. Pyostomatitis vegetans in childhood. Eur J Pediatr. 1998;157:402-405. doi:10.1007/s004310050838
- Chen KL, Diiorio DA, Chiu YE, et al. Pediatric patients with orofacial granulomatosis likely to subsequently develop intestinal Crohn’s disease: brief report. Pediatr Dermatol. 2020;37:1162-1164. doi:10.1111/pde.14390
- Pazheri F, Alkhouri N, Radhakrishnan K. Pyostomatitis vegetans as an oral manifestation of Crohn’s disease in a pediatric patient. Inflamm Bowel Dis. 2010;16:2007. doi:10.1002/ibd.21245.
- Werchniak AE, Storm CA, Plunkett RW, et al. Treatment of pyostomatitis vegetans with topical tacrolimus. J Am Acad Dermatol. 2005;52:722-723. doi:10.1016/j.jaad.2004.11.041
- Stingeni L, Tramontana M, Bassotti G, et al. Pyodermatitis–pyostomatitis vegetans and antibullous pemphigoid antigen 180 autoantibodies: a casual association? Br J Dermatol. 2015;172:811-813. doi:10.1111/bjd.13297
- Takahashi S, Andreoletti G, Chen R, et al. De novo and rare mutations in the HSPA1L heat shock gene associated with inflammatory bowel disease. Genome Med. 2017;9:8. doi:10.1186/s13073-016-0394-9
- Crowley E, Warner N, Pan J, et al. Prevalence and clinical features of inflammatory bowel diseases associated with monogenic variants, identified by whole-exome sequencing in 1000 children at a single center. Gastroenterology. 2020;158:2208-2220. doi:10.1053/j .gastro.2020.02.023
Case Report
A 7-year-old girl who was otherwise healthy was referred by pediatric gastroenterology for evaluation of cutaneous Crohn disease (CD). The patient had a 4-year history of persistent lip swelling and a 3-year history of asymmetric erythematous labial swelling and perianal erythema with skin tags. She had been applying the calcineurin inhibitor tacrolimus ointment 0.03% 1 or 2 times daily to her lesions with minimal improvement. She did not have a medical history of recurrent or unusual infectious diseases. There was no family history of autoimmune disease.
The patient and her guardian reported intermittent perianal pain but denied constipation, diarrhea, abdominal pain, and blood in the stool. She denied throat and tongue swelling, dysphagia, dyspnea, drooling, facial paralysis, and eyelid edema. She was a well-nourished child whose height and weight percentiles tracked at 30% and 25%, respectively. Physical examination revealed confluent symmetric lip swelling with mild angular cheilitis. Multiple 1- to 2-mm white pustules with pinpoint erosions covered the upper and lower labial mucosa and extended onto the buccal mucosa (Figure 1). She had symmetric erythema and swelling of the left labia majora extending to and involving the left perianal mucosa. Three perianal erythematous skin tags and a perianal fissure were identified.
The patient had been assessed 2 years earlier by pediatric dermatology and gastroenterology with an extensive evaluation that favored a diagnosis of cutaneous CD because the combination of orofacial granulomatosis (OFG), vulvar edema, and perianal skin tags is strongly associated.1-3 Contact dermatitis affecting the mouth was considered; however, allergen testing did not demonstrate a trigger.
A trial of a benzoate- and cinnamon-free diet, which has been reported to improve OFG,4 did not provide symptomatic improvement. Topical corticosteroids and tacrolimus reduced the perioral erythema, but the swelling persisted. An infectious cause was considered; however, topical mupirocin had no effect, and amoxicillin resulted in oral candidiasis.
A perianal biopsy revealed a granulomatous dermatitis. Fungal and bacterial cultures were negative. Upper and lower gastrointestinal (GI) endoscopy and a fecal calprotectin assay were not suggestive of inflammatory bowel disease (IBD). A complete blood cell count and QuantiFERON-TB Gold test measuring the immune response to tuberculosis antigens were normal. Chronic granulomatous disease, RAG1/RAG2 deficiency, common variable immunodeficiency, and NOD2 defects were ruled out with normal tests of dihydrorhodamine, quantitative immunoglobulins, and toll-like receptors.
Because of the discomfort associated with the patient’s lesions, she was offered treatment with tumor necrosis factor α inhibitors, including infliximab and adalimumab. These agents had been offered since the onset of symptoms; however, her parents declined systemic medication unless she developed GI involvement. Instead, the tacrolimus concentration was increased to 0.1% applied to the lips, labia, and perianal area, and fluocinonide gel 0.05% applied nightly to the oral pustules was added.
Two months later the patient had notably fewer oral pustules and diminished erythema but only slightly reduced oral, vulvar, and perianal swelling. A trial of oral metronidazole, which has been reported to clear a patient with cutaneous CD,5 was discontinued by her parents after 6 weeks because of a lack of interval improvement.
One year later, a pre-existing perianal skin tag doubled in size and became exquisitely tender. The calprotectin level—previously within reference range at less than 16 μg/g—was now elevated at 149 μg/g (reference range, 1–120 μg/g) and increased to 336 μg/g 3 weeks later. Testing for C-reactive protein, zinc, and stool occult blood; a comprehensive metabolic panel; and a complete blood cell count were unremarkable.
Repeat upper and lower GI endoscopy did not suggest CD. A biopsy using direct immunofluorescence (DIF) was obtained to evaluate for pyostomatitis vegetans (PSV) and rule out
The captured biopsy did not demonstrate the intended pustule; instead, it included less-affected mucosa and was obtained during topical treatment when few pustules and erosions persisted. Pathologic analysis revealed noncaseating granulomas without an increase in microabscesses, neutrophils, or eosinophils (Figure 2). Direct immunofluorescence staining for IgG, IgA, and C3 and indirect immunofluorescence staining for desmoglein-1 and desmoglein-3 antibodies were negative. Although the biopsy did not capture the intended pustule, diagnosis of PV was made based on clinical features and the constellation of cutaneous findings associated with IBD.
Intralesional triamcinolone, which has been of benefit for pediatric patients with orofacial granulomatosis,1,6,7 was instituted and normalized the vulva and perianal mucosa; however, lip swelling improved only minimally.
Comment
Pyostomatitis vegetans is characterized by multiple white or yellow, friable, miliary pustules that rupture, leaving behind ulcerations and erosions that cause a varying degree of oral pain.8 The disorder can involve any area of the oral mucosa—most often the labia-attached gingiva, soft and hard palates, buccal mucosa, vestibule, and tonsillar areas—but often spares the floor of the mouth and tongue.8-11 The term pyostomatitis vegetans was proposed in 1949 by McCarthy12 when he noted in a patient who presented with the characteristic appearance of the oral mucosa, though cases of vaginal, nasal, and periocular involvement have been reported.8,13,14
Histopathology—Pyostomatitis vegetans displays pseudoepithelial hyperplasia with acanthosis, hyperkeratosis, and intraepithelial or subepithelial microabscesses (or both) with neutrophils and eosinophils.8,9,15 There are a few possible explanations for this patient’s lack of tissue eosinophilia. It has been theorized that the presence of granulomas could mask concurrent PSV16 or that tissue in PSV contains fewer eosinophils as the disorder progresses.11 The oral biopsy obtained from our patient did not capture a pustule, and the condition had noticeably improved with topical tacrolimus at the time of biopsy; therefore, neither neutrophils nor eosinophils were identified. Peripheral eosinophilia, which is present in 42% to 90% of cases of PSV,9,17 can be a diagnostic clue.18 However, PE is associated with IBD,24 which usually occurs with PSV, so the absence of peripheral eosinophilia in our patient may be explained by her lack of bowel disease.
Pathogenesis—The pathogenesis of PSV is unknown. A proposed etiology includes cross-reacting antigens in the bowel and skin secondary to IBD as well as an aberrant immune response to an unidentified factor.8 Pyostomatitis vegetans is considered by many to be the mucosal variant of pyodermatitis vegetans,9,15,19 a neutrophilic dermatosis characterized by asymmetric, crusted, erythematous papulopustules that extend peripherally and coalesce to form large vegetating plaques. These lesions commonly manifest in the axillary folds, groin, and scalp and can involve the face, trunk, and distal extremities.9,18 Infection has been suggested as a cause of PSV, though cultures for pathogenic bacteria, viruses, and fungi consistently show only normal flora.20 Zinc deficiency attributed to malabsorption from CD was reported in an adult with PSV.21 The PSV resolved after 6 weeks of zinc supplementation.
Differential Diagnosis—The main entity in the clinical differential diagnosis for PSV is PVH, which is considered a variant of pemphigus vulgaris. Pemphigus vegetans of Hallopeau presents with pustules and progresses to hyperpigmented vegetative plaques with peripheral hypertrophic granulation tissue.22 The clinical and histological presentation of PVH can be similar to PSV; in PVH, however, DIF demonstrates intercellular IgG and C3 due to circulating IgG autoantibodies specific for desmoglein 3, a cell adhesion molecule.22-24 In PSV, DIF typically is negative for IgG, IgA, and C3.8 Immunohistochemical findings of PSV may overlap with IgA pemphigus, IgG/IgA pemphigus, and IgG pemphigus, which has sparked debate if PSV is an autoimmune blistering disorder or a secondary finding of epithelial injury.9,18,24
Pyostomatitis vegetans is most prevalent in patients aged 20 to 59 years25 but can occur at any age.8,19 Overall, extraintestinal symptoms, including mucocutaneous findings, are common in pediatric patients—in 30% to 71% of children with CD and 21% to 22% of children with ulcerative colitis26—and can predate onset of GI symptoms in 6% of pediatric patients.27
Oral disease is common in CD; manifestations are listed in the Table.28,29 In a prospective study of 48 children with CD, 42% (20/48) had oral manifestations identified at diagnosis28; in a similar study of 25 children, researchers noted that 48% (12/25) had disease-specific oral lesions.29 None of these children recognized the oral findings prior to the onset of systemic symptoms.28 Pyostomatitis vegetans was the least common oral manifestation, reported in 1 of 73 patients in the 2 studies combined.28,29
Two recent articles that looked at PSV in pediatric and adolescent populations identified only 9 patients with PSV.24,30 Only 2 patients (siblings) had documented onset of PSV before 12 years of age,31 which suggests an underlying genetic predisposition in young children.
It has been reported that active or subclinical (ie, asymptomatic with positive endoscopic findings) IBD in adults precedes onset of PSV, which may be considered a sign of relapse.9,30 However, PSV is incredibly rare in children and adolescents and can be an early finding of IBD in children.16,31,32
Our patient has not developed GI involvement since her initial presentation 5 years prior, though another pediatric patient developed symptomatic CD 9 years after onset of OFG.5 A retrospective review of pediatric OFG without CD met criteria for CD at a median of 3.1 years (range, 0.4–6.9 years).33 Regrettably, the early presence of PSV has been associated with future progression to CD and a complicated disease course.12,34
Management—Pyoderma stomatitis vegetans is treated with management of underlying IBD,8 with scarce literature available regarding pediatric patients. Oral lesions have been treated with antiseptics and topical corticosteroids, though these have limited benefit.8 In an adult with IBD, topical tacrolimus initially cleared PSV; however, lesions recurred until mesalamine was initiated.35 Systemic steroids were effective in a 16-year-old patient with CD and PSV,12 but recurrence is common after corticosteroids are stopped.34
Some patients benefit from steroid-sparing medications, such as dapsone, azathioprine, sulfamethoxypyridazine, methotrexate, mycophenolate mofetil, and tumor necrosis factor α inhibitors such as infliximab and adalimumab.8,9,15,23,34,36 A 12-year-old patient with pyodermatitis–PSV without intestinal disease was treated with prednisone, dapsone, and azathioprine with improvement but not complete resolution of oral erosions after 18 weeks of treatment.32 A 15-year-old patient with CD and pyodermatitis–PSV did not show improvement on prednisone, dapsone, and azathioprine but rapidly responded to infliximab.23 Infliximab led to complete clearance of oral lesions in an adult with severe fistulizing CD who developed PSV.11 However, 2 adolescent patients with CD developed PSV while on adalimumab,6,34 though 1 did improve after increasing adalimumab from once to twice weekly.6
Conclusion
The case described here—PSV in a prepubertal 7-year-old with multiple cutaneous findings suggestive of CD, including OFG, perianal and vulvar edema with biopsy-proven noncaseating granulomas, anal skin tags, and an elevated calprotectin level, noted during a cutaneous flare without clinical or endoscopically identified underlying bowel involvement—is an extremely rare presentation. Literature regarding management of PSV primarily is found in the form of case reports and focuses on treating underlying IBD. In patients with intestinal disease, treatment with biologic therapy appears most effective.6,23
ADDENDUM
Interestingly, 3 years after the patient’s original presentation to our clinic, chromosomal sequencing analysis to assess for copy number variants and whole exome gene sequencing identified a variant of unknown significance in the heat shock protein family A member 1-like gene, HSPA1L, which has an unknown mode of inheritance, but the literature suggests that both truncating and missense variants could be associated with individuals with ulcerative colitis, CD, and IBD.37,38 Although we cannot use this information to render a molecular diagnosis, it is highly suspicious that this is the cause of her clinical findings. Additionally, the patient currently is aged 10 years with unchanged cutaneous findings and has not developed gastrointestinal findings of IBD.
Case Report
A 7-year-old girl who was otherwise healthy was referred by pediatric gastroenterology for evaluation of cutaneous Crohn disease (CD). The patient had a 4-year history of persistent lip swelling and a 3-year history of asymmetric erythematous labial swelling and perianal erythema with skin tags. She had been applying the calcineurin inhibitor tacrolimus ointment 0.03% 1 or 2 times daily to her lesions with minimal improvement. She did not have a medical history of recurrent or unusual infectious diseases. There was no family history of autoimmune disease.
The patient and her guardian reported intermittent perianal pain but denied constipation, diarrhea, abdominal pain, and blood in the stool. She denied throat and tongue swelling, dysphagia, dyspnea, drooling, facial paralysis, and eyelid edema. She was a well-nourished child whose height and weight percentiles tracked at 30% and 25%, respectively. Physical examination revealed confluent symmetric lip swelling with mild angular cheilitis. Multiple 1- to 2-mm white pustules with pinpoint erosions covered the upper and lower labial mucosa and extended onto the buccal mucosa (Figure 1). She had symmetric erythema and swelling of the left labia majora extending to and involving the left perianal mucosa. Three perianal erythematous skin tags and a perianal fissure were identified.
The patient had been assessed 2 years earlier by pediatric dermatology and gastroenterology with an extensive evaluation that favored a diagnosis of cutaneous CD because the combination of orofacial granulomatosis (OFG), vulvar edema, and perianal skin tags is strongly associated.1-3 Contact dermatitis affecting the mouth was considered; however, allergen testing did not demonstrate a trigger.
A trial of a benzoate- and cinnamon-free diet, which has been reported to improve OFG,4 did not provide symptomatic improvement. Topical corticosteroids and tacrolimus reduced the perioral erythema, but the swelling persisted. An infectious cause was considered; however, topical mupirocin had no effect, and amoxicillin resulted in oral candidiasis.
A perianal biopsy revealed a granulomatous dermatitis. Fungal and bacterial cultures were negative. Upper and lower gastrointestinal (GI) endoscopy and a fecal calprotectin assay were not suggestive of inflammatory bowel disease (IBD). A complete blood cell count and QuantiFERON-TB Gold test measuring the immune response to tuberculosis antigens were normal. Chronic granulomatous disease, RAG1/RAG2 deficiency, common variable immunodeficiency, and NOD2 defects were ruled out with normal tests of dihydrorhodamine, quantitative immunoglobulins, and toll-like receptors.
Because of the discomfort associated with the patient’s lesions, she was offered treatment with tumor necrosis factor α inhibitors, including infliximab and adalimumab. These agents had been offered since the onset of symptoms; however, her parents declined systemic medication unless she developed GI involvement. Instead, the tacrolimus concentration was increased to 0.1% applied to the lips, labia, and perianal area, and fluocinonide gel 0.05% applied nightly to the oral pustules was added.
Two months later the patient had notably fewer oral pustules and diminished erythema but only slightly reduced oral, vulvar, and perianal swelling. A trial of oral metronidazole, which has been reported to clear a patient with cutaneous CD,5 was discontinued by her parents after 6 weeks because of a lack of interval improvement.
One year later, a pre-existing perianal skin tag doubled in size and became exquisitely tender. The calprotectin level—previously within reference range at less than 16 μg/g—was now elevated at 149 μg/g (reference range, 1–120 μg/g) and increased to 336 μg/g 3 weeks later. Testing for C-reactive protein, zinc, and stool occult blood; a comprehensive metabolic panel; and a complete blood cell count were unremarkable.
Repeat upper and lower GI endoscopy did not suggest CD. A biopsy using direct immunofluorescence (DIF) was obtained to evaluate for pyostomatitis vegetans (PSV) and rule out
The captured biopsy did not demonstrate the intended pustule; instead, it included less-affected mucosa and was obtained during topical treatment when few pustules and erosions persisted. Pathologic analysis revealed noncaseating granulomas without an increase in microabscesses, neutrophils, or eosinophils (Figure 2). Direct immunofluorescence staining for IgG, IgA, and C3 and indirect immunofluorescence staining for desmoglein-1 and desmoglein-3 antibodies were negative. Although the biopsy did not capture the intended pustule, diagnosis of PV was made based on clinical features and the constellation of cutaneous findings associated with IBD.
Intralesional triamcinolone, which has been of benefit for pediatric patients with orofacial granulomatosis,1,6,7 was instituted and normalized the vulva and perianal mucosa; however, lip swelling improved only minimally.
Comment
Pyostomatitis vegetans is characterized by multiple white or yellow, friable, miliary pustules that rupture, leaving behind ulcerations and erosions that cause a varying degree of oral pain.8 The disorder can involve any area of the oral mucosa—most often the labia-attached gingiva, soft and hard palates, buccal mucosa, vestibule, and tonsillar areas—but often spares the floor of the mouth and tongue.8-11 The term pyostomatitis vegetans was proposed in 1949 by McCarthy12 when he noted in a patient who presented with the characteristic appearance of the oral mucosa, though cases of vaginal, nasal, and periocular involvement have been reported.8,13,14
Histopathology—Pyostomatitis vegetans displays pseudoepithelial hyperplasia with acanthosis, hyperkeratosis, and intraepithelial or subepithelial microabscesses (or both) with neutrophils and eosinophils.8,9,15 There are a few possible explanations for this patient’s lack of tissue eosinophilia. It has been theorized that the presence of granulomas could mask concurrent PSV16 or that tissue in PSV contains fewer eosinophils as the disorder progresses.11 The oral biopsy obtained from our patient did not capture a pustule, and the condition had noticeably improved with topical tacrolimus at the time of biopsy; therefore, neither neutrophils nor eosinophils were identified. Peripheral eosinophilia, which is present in 42% to 90% of cases of PSV,9,17 can be a diagnostic clue.18 However, PE is associated with IBD,24 which usually occurs with PSV, so the absence of peripheral eosinophilia in our patient may be explained by her lack of bowel disease.
Pathogenesis—The pathogenesis of PSV is unknown. A proposed etiology includes cross-reacting antigens in the bowel and skin secondary to IBD as well as an aberrant immune response to an unidentified factor.8 Pyostomatitis vegetans is considered by many to be the mucosal variant of pyodermatitis vegetans,9,15,19 a neutrophilic dermatosis characterized by asymmetric, crusted, erythematous papulopustules that extend peripherally and coalesce to form large vegetating plaques. These lesions commonly manifest in the axillary folds, groin, and scalp and can involve the face, trunk, and distal extremities.9,18 Infection has been suggested as a cause of PSV, though cultures for pathogenic bacteria, viruses, and fungi consistently show only normal flora.20 Zinc deficiency attributed to malabsorption from CD was reported in an adult with PSV.21 The PSV resolved after 6 weeks of zinc supplementation.
Differential Diagnosis—The main entity in the clinical differential diagnosis for PSV is PVH, which is considered a variant of pemphigus vulgaris. Pemphigus vegetans of Hallopeau presents with pustules and progresses to hyperpigmented vegetative plaques with peripheral hypertrophic granulation tissue.22 The clinical and histological presentation of PVH can be similar to PSV; in PVH, however, DIF demonstrates intercellular IgG and C3 due to circulating IgG autoantibodies specific for desmoglein 3, a cell adhesion molecule.22-24 In PSV, DIF typically is negative for IgG, IgA, and C3.8 Immunohistochemical findings of PSV may overlap with IgA pemphigus, IgG/IgA pemphigus, and IgG pemphigus, which has sparked debate if PSV is an autoimmune blistering disorder or a secondary finding of epithelial injury.9,18,24
Pyostomatitis vegetans is most prevalent in patients aged 20 to 59 years25 but can occur at any age.8,19 Overall, extraintestinal symptoms, including mucocutaneous findings, are common in pediatric patients—in 30% to 71% of children with CD and 21% to 22% of children with ulcerative colitis26—and can predate onset of GI symptoms in 6% of pediatric patients.27
Oral disease is common in CD; manifestations are listed in the Table.28,29 In a prospective study of 48 children with CD, 42% (20/48) had oral manifestations identified at diagnosis28; in a similar study of 25 children, researchers noted that 48% (12/25) had disease-specific oral lesions.29 None of these children recognized the oral findings prior to the onset of systemic symptoms.28 Pyostomatitis vegetans was the least common oral manifestation, reported in 1 of 73 patients in the 2 studies combined.28,29
Two recent articles that looked at PSV in pediatric and adolescent populations identified only 9 patients with PSV.24,30 Only 2 patients (siblings) had documented onset of PSV before 12 years of age,31 which suggests an underlying genetic predisposition in young children.
It has been reported that active or subclinical (ie, asymptomatic with positive endoscopic findings) IBD in adults precedes onset of PSV, which may be considered a sign of relapse.9,30 However, PSV is incredibly rare in children and adolescents and can be an early finding of IBD in children.16,31,32
Our patient has not developed GI involvement since her initial presentation 5 years prior, though another pediatric patient developed symptomatic CD 9 years after onset of OFG.5 A retrospective review of pediatric OFG without CD met criteria for CD at a median of 3.1 years (range, 0.4–6.9 years).33 Regrettably, the early presence of PSV has been associated with future progression to CD and a complicated disease course.12,34
Management—Pyoderma stomatitis vegetans is treated with management of underlying IBD,8 with scarce literature available regarding pediatric patients. Oral lesions have been treated with antiseptics and topical corticosteroids, though these have limited benefit.8 In an adult with IBD, topical tacrolimus initially cleared PSV; however, lesions recurred until mesalamine was initiated.35 Systemic steroids were effective in a 16-year-old patient with CD and PSV,12 but recurrence is common after corticosteroids are stopped.34
Some patients benefit from steroid-sparing medications, such as dapsone, azathioprine, sulfamethoxypyridazine, methotrexate, mycophenolate mofetil, and tumor necrosis factor α inhibitors such as infliximab and adalimumab.8,9,15,23,34,36 A 12-year-old patient with pyodermatitis–PSV without intestinal disease was treated with prednisone, dapsone, and azathioprine with improvement but not complete resolution of oral erosions after 18 weeks of treatment.32 A 15-year-old patient with CD and pyodermatitis–PSV did not show improvement on prednisone, dapsone, and azathioprine but rapidly responded to infliximab.23 Infliximab led to complete clearance of oral lesions in an adult with severe fistulizing CD who developed PSV.11 However, 2 adolescent patients with CD developed PSV while on adalimumab,6,34 though 1 did improve after increasing adalimumab from once to twice weekly.6
Conclusion
The case described here—PSV in a prepubertal 7-year-old with multiple cutaneous findings suggestive of CD, including OFG, perianal and vulvar edema with biopsy-proven noncaseating granulomas, anal skin tags, and an elevated calprotectin level, noted during a cutaneous flare without clinical or endoscopically identified underlying bowel involvement—is an extremely rare presentation. Literature regarding management of PSV primarily is found in the form of case reports and focuses on treating underlying IBD. In patients with intestinal disease, treatment with biologic therapy appears most effective.6,23
ADDENDUM
Interestingly, 3 years after the patient’s original presentation to our clinic, chromosomal sequencing analysis to assess for copy number variants and whole exome gene sequencing identified a variant of unknown significance in the heat shock protein family A member 1-like gene, HSPA1L, which has an unknown mode of inheritance, but the literature suggests that both truncating and missense variants could be associated with individuals with ulcerative colitis, CD, and IBD.37,38 Although we cannot use this information to render a molecular diagnosis, it is highly suspicious that this is the cause of her clinical findings. Additionally, the patient currently is aged 10 years with unchanged cutaneous findings and has not developed gastrointestinal findings of IBD.
- Tuxen AJ, Orchard D. Childhood and adolescent orofacial granulomatosis is strongly associated with Crohn’s disease and responds to intralesional corticosteroids. Australas J Dermatol. 2010;51:124-127. doi:10.1111/j.1440-0960.2010.00627.x
- Vaid RM, Cohen BA. Cutaneous Crohn’s disease in the pediatric population. Pediatr Dermatol. 2010;27:279-281. doi:10.1111/j.1525-1470.2010.01138.x
- van de Scheur MR, van der Waal RIF, van der Waal I, et al. Ano-genital granulomatosis: the counterpart of oro-facial granulomatosis. J Eur Acad Dermatol Venereol. 2003;17:184-189. doi:10.1046/j.1468-3083.2003.00573.x
- Campbell HE, Escudier MP, Patel P, et al. Review article: cinnamon- and benzoate-free diet as a primary treatment for orofacial granulomatosis. Aliment Pharmacol Ther. 2011;34:687-701. doi:10.1111/j.1365-2036.2011.04792.x
- Duhra P, Paul CJ. Metastatic Crohn’s disease responding to metronidazole. Br J Dermatol. 1988;119:87-91. doi:10.1111/j.1365-2133.1988.tb07107.x
- Katsanos KH, Torres J, Roda G, et al. Review article: non-malignant oral manifestations in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015;42:40-60. doi:10.1111/apt.13217
- Schmitz BA, Unkel JH. Symptomatic oral Crohn’s disease in an adolescent. J Dent Child (Chic). 2018;85:66-69.
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Clark LG, Tolkachjov SN, Bridges AG, et al. Pyostomatitis vegetans (PSV)–pyodermatitis vegetans (PDV): a clinicopathologic study of 7 cases at a tertiary referral center. J Am Acad Dermatol. 2016;75:578-584. doi:10.1016/j.jaad.2016.03.047
- Hansen LS, Silverman S Jr, Daniels TE. The differential diagnosis of pyostomatitis vegetans and its relation to bowel disease. Oral Surg Oral Med Oral Pathol. 1983;55:363-373. doi:10.1016/0030-4220(83)90191-3
- Cataldo E, Covino MC, Tesone PE. Pyostomatitis vegetans. Oral Surg Oral Med Oral Pathol. 1981;52:172-177. doi:10.1016/0030-4220(81)90316-9
- McCarthy FP. Pyostomatitis vegetans; report of three cases. Arch Derm Syphilol. 1949;60:750-764.
- Bens G, Laharie D, Beylot-Barry M, et al. Successful treatment with infliximab and methotrexate of pyostomatitis vegetans associated with Crohn’s disease. Br J Dermatol. 2003;149:181-184. doi:10.1046/j.1365-2133.2003.05385.x
- Leibovitch I, Ooi C, Huilgol SC, et al. Pyodermatitis–pyostomatitis vegetans of the eyelids: case report and review of the literature. Ophthalmology. 2005;112:1809-1813. doi:10.1016/j.ophtha.2005.04.027
- Ruiz-Roca JA, Berini-Aytés L, Gay-Escoda C. Pyostomatitis vegetans. report of two cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:447-454. doi:10.1016/j.tripleo.2003.08.022
- Molnár T, Farkas K, Nagy F, et al. Third case: another pediatric patient with pyostomatitis vegetans and oral granuloma as one of the initial symptoms of Crohn’s disease. Inflamm Bowel Dis. 2011;17:E122-E123. doi:10.1002/ibd.21791
- Leydhecker W, Lund OE. Eye involvement in pyostomatitis vegetans. Klin Monbl Augenheilkd Augenarztl Fortbild. 1962;141:595-602.
- Thornhill MH, Zakrzewska JM, Gilkes JJ. Pyostomatitis vegetans: report of three cases and review of the literature. J Oral Pathol Med. 1992;21:128-133. doi:10.1111/j.1600-0714.1992.tb00996.x
- Chaudhry SI, Philpot NS, Odell EW, et al. Pyostomatitis vegetans associated with asymptomatic ulcerative colitis: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:327-330. doi:10.1016/s1079-2104(99)70217-9
- Konstantopoulou M, O’Dwyer EM, Steele JC, et al. Pyodermatitis–pyostomatitis vegetans complicated by methicillin-resistant Staphylococcus aureus infection. Clin Exp Dermatol. 2005;30:666-668. doi:10.1111/j.1365-2230.2005.01906.x
- Ficarra G, Cicchi P, Amorosi A, et al. Oral Crohn’s disease and pyostomatitis vegetans. an unusual association. Oral Surg Oral Med Oral Pathol. 1993;75:220-224. doi:10.1016/0030-4220(93)90097-n
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428. doi:10.1111/j.1365-4632.2004.02480.x
- Nico MMS, Hussein TP, Aoki V, et al. Pyostomatitis vegetans and its relation to inflammatory bowel disease, pyoderma gangrenosum, pyodermatitis vegetans, and pemphigus. J Oral Pathol Med. 2012;41:584-588. doi:10.1111/j.1600-0714.2012.01152.x
- Berzin D, Lahad A, Weiss B, et al. Inflammatory bowel disease presenting with pyodermatitis–pyostomatitis vegetans in a pediatric patient: a case report and review of the literature. Pediatr Dermatol. 2021;38:868-871. doi:10.1111/pde.14625
- Ballo FS, Camisa C, Allen CM. Pyostomatitis vegetans. report of a case and review of the literature. J Am Acad Dermatol. 1989;21:381-387.
- Greuter T, Bertoldo F, Rechner R, et al; Swiss IBD Cohort Study Group. Extraintestinal manifestations of pediatric inflammatory bowel disease: prevalence, presentation, and anti-TNF treatment. J Pediatr Gastroenterol Nutr. 2017;65:200-206. doi:10.1097/MPG.0000000000001455
- Jose FA, Garnett EA, Vittinghoff E, et al. Development of extraintestinal manifestations in pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:63-68. doi:10.1002/ibd.20604
- Harty S, Fleming P, Rowland M, et al. A prospective study of the oral manifestations of Crohn’s disease. Clin Gastroenterol Hepatol. 2005;3:886-891. doi:10.1016/s1542-3565(05)00424-6
- Pittock S, Drumm B, Fleming P, et al. The oral cavity in Crohn’s disease. J Pediatr. 2001;138:767-771. doi:10.1067/mpd.2001.113008
- Bardasi G, Romagnoli A, Foschini MP, et al. Pyostomatitis vegetans in a pediatric patient with ulcerative colitis: case report of a rare pediatric inflammatory bowel disease extraintestinal manifestation and review of the literature. Eur J Gastroenterol Hepatol. 2020;32:889-892. doi:10.1097/MEG.0000000000001723
- Mesquita Kde C, Costa IM. Case for diagnosis. An Bras Dermatol. 2012;87:929-931. doi:10.1590/s0365-05962012000600022
- Al-Rimawi HS, Hammad MM, Raweily EA, et al. Pyostomatitis vegetans in childhood. Eur J Pediatr. 1998;157:402-405. doi:10.1007/s004310050838
- Chen KL, Diiorio DA, Chiu YE, et al. Pediatric patients with orofacial granulomatosis likely to subsequently develop intestinal Crohn’s disease: brief report. Pediatr Dermatol. 2020;37:1162-1164. doi:10.1111/pde.14390
- Pazheri F, Alkhouri N, Radhakrishnan K. Pyostomatitis vegetans as an oral manifestation of Crohn’s disease in a pediatric patient. Inflamm Bowel Dis. 2010;16:2007. doi:10.1002/ibd.21245.
- Werchniak AE, Storm CA, Plunkett RW, et al. Treatment of pyostomatitis vegetans with topical tacrolimus. J Am Acad Dermatol. 2005;52:722-723. doi:10.1016/j.jaad.2004.11.041
- Stingeni L, Tramontana M, Bassotti G, et al. Pyodermatitis–pyostomatitis vegetans and antibullous pemphigoid antigen 180 autoantibodies: a casual association? Br J Dermatol. 2015;172:811-813. doi:10.1111/bjd.13297
- Takahashi S, Andreoletti G, Chen R, et al. De novo and rare mutations in the HSPA1L heat shock gene associated with inflammatory bowel disease. Genome Med. 2017;9:8. doi:10.1186/s13073-016-0394-9
- Crowley E, Warner N, Pan J, et al. Prevalence and clinical features of inflammatory bowel diseases associated with monogenic variants, identified by whole-exome sequencing in 1000 children at a single center. Gastroenterology. 2020;158:2208-2220. doi:10.1053/j .gastro.2020.02.023
- Tuxen AJ, Orchard D. Childhood and adolescent orofacial granulomatosis is strongly associated with Crohn’s disease and responds to intralesional corticosteroids. Australas J Dermatol. 2010;51:124-127. doi:10.1111/j.1440-0960.2010.00627.x
- Vaid RM, Cohen BA. Cutaneous Crohn’s disease in the pediatric population. Pediatr Dermatol. 2010;27:279-281. doi:10.1111/j.1525-1470.2010.01138.x
- van de Scheur MR, van der Waal RIF, van der Waal I, et al. Ano-genital granulomatosis: the counterpart of oro-facial granulomatosis. J Eur Acad Dermatol Venereol. 2003;17:184-189. doi:10.1046/j.1468-3083.2003.00573.x
- Campbell HE, Escudier MP, Patel P, et al. Review article: cinnamon- and benzoate-free diet as a primary treatment for orofacial granulomatosis. Aliment Pharmacol Ther. 2011;34:687-701. doi:10.1111/j.1365-2036.2011.04792.x
- Duhra P, Paul CJ. Metastatic Crohn’s disease responding to metronidazole. Br J Dermatol. 1988;119:87-91. doi:10.1111/j.1365-2133.1988.tb07107.x
- Katsanos KH, Torres J, Roda G, et al. Review article: non-malignant oral manifestations in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015;42:40-60. doi:10.1111/apt.13217
- Schmitz BA, Unkel JH. Symptomatic oral Crohn’s disease in an adolescent. J Dent Child (Chic). 2018;85:66-69.
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Clark LG, Tolkachjov SN, Bridges AG, et al. Pyostomatitis vegetans (PSV)–pyodermatitis vegetans (PDV): a clinicopathologic study of 7 cases at a tertiary referral center. J Am Acad Dermatol. 2016;75:578-584. doi:10.1016/j.jaad.2016.03.047
- Hansen LS, Silverman S Jr, Daniels TE. The differential diagnosis of pyostomatitis vegetans and its relation to bowel disease. Oral Surg Oral Med Oral Pathol. 1983;55:363-373. doi:10.1016/0030-4220(83)90191-3
- Cataldo E, Covino MC, Tesone PE. Pyostomatitis vegetans. Oral Surg Oral Med Oral Pathol. 1981;52:172-177. doi:10.1016/0030-4220(81)90316-9
- McCarthy FP. Pyostomatitis vegetans; report of three cases. Arch Derm Syphilol. 1949;60:750-764.
- Bens G, Laharie D, Beylot-Barry M, et al. Successful treatment with infliximab and methotrexate of pyostomatitis vegetans associated with Crohn’s disease. Br J Dermatol. 2003;149:181-184. doi:10.1046/j.1365-2133.2003.05385.x
- Leibovitch I, Ooi C, Huilgol SC, et al. Pyodermatitis–pyostomatitis vegetans of the eyelids: case report and review of the literature. Ophthalmology. 2005;112:1809-1813. doi:10.1016/j.ophtha.2005.04.027
- Ruiz-Roca JA, Berini-Aytés L, Gay-Escoda C. Pyostomatitis vegetans. report of two cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:447-454. doi:10.1016/j.tripleo.2003.08.022
- Molnár T, Farkas K, Nagy F, et al. Third case: another pediatric patient with pyostomatitis vegetans and oral granuloma as one of the initial symptoms of Crohn’s disease. Inflamm Bowel Dis. 2011;17:E122-E123. doi:10.1002/ibd.21791
- Leydhecker W, Lund OE. Eye involvement in pyostomatitis vegetans. Klin Monbl Augenheilkd Augenarztl Fortbild. 1962;141:595-602.
- Thornhill MH, Zakrzewska JM, Gilkes JJ. Pyostomatitis vegetans: report of three cases and review of the literature. J Oral Pathol Med. 1992;21:128-133. doi:10.1111/j.1600-0714.1992.tb00996.x
- Chaudhry SI, Philpot NS, Odell EW, et al. Pyostomatitis vegetans associated with asymptomatic ulcerative colitis: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:327-330. doi:10.1016/s1079-2104(99)70217-9
- Konstantopoulou M, O’Dwyer EM, Steele JC, et al. Pyodermatitis–pyostomatitis vegetans complicated by methicillin-resistant Staphylococcus aureus infection. Clin Exp Dermatol. 2005;30:666-668. doi:10.1111/j.1365-2230.2005.01906.x
- Ficarra G, Cicchi P, Amorosi A, et al. Oral Crohn’s disease and pyostomatitis vegetans. an unusual association. Oral Surg Oral Med Oral Pathol. 1993;75:220-224. doi:10.1016/0030-4220(93)90097-n
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428. doi:10.1111/j.1365-4632.2004.02480.x
- Nico MMS, Hussein TP, Aoki V, et al. Pyostomatitis vegetans and its relation to inflammatory bowel disease, pyoderma gangrenosum, pyodermatitis vegetans, and pemphigus. J Oral Pathol Med. 2012;41:584-588. doi:10.1111/j.1600-0714.2012.01152.x
- Berzin D, Lahad A, Weiss B, et al. Inflammatory bowel disease presenting with pyodermatitis–pyostomatitis vegetans in a pediatric patient: a case report and review of the literature. Pediatr Dermatol. 2021;38:868-871. doi:10.1111/pde.14625
- Ballo FS, Camisa C, Allen CM. Pyostomatitis vegetans. report of a case and review of the literature. J Am Acad Dermatol. 1989;21:381-387.
- Greuter T, Bertoldo F, Rechner R, et al; Swiss IBD Cohort Study Group. Extraintestinal manifestations of pediatric inflammatory bowel disease: prevalence, presentation, and anti-TNF treatment. J Pediatr Gastroenterol Nutr. 2017;65:200-206. doi:10.1097/MPG.0000000000001455
- Jose FA, Garnett EA, Vittinghoff E, et al. Development of extraintestinal manifestations in pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:63-68. doi:10.1002/ibd.20604
- Harty S, Fleming P, Rowland M, et al. A prospective study of the oral manifestations of Crohn’s disease. Clin Gastroenterol Hepatol. 2005;3:886-891. doi:10.1016/s1542-3565(05)00424-6
- Pittock S, Drumm B, Fleming P, et al. The oral cavity in Crohn’s disease. J Pediatr. 2001;138:767-771. doi:10.1067/mpd.2001.113008
- Bardasi G, Romagnoli A, Foschini MP, et al. Pyostomatitis vegetans in a pediatric patient with ulcerative colitis: case report of a rare pediatric inflammatory bowel disease extraintestinal manifestation and review of the literature. Eur J Gastroenterol Hepatol. 2020;32:889-892. doi:10.1097/MEG.0000000000001723
- Mesquita Kde C, Costa IM. Case for diagnosis. An Bras Dermatol. 2012;87:929-931. doi:10.1590/s0365-05962012000600022
- Al-Rimawi HS, Hammad MM, Raweily EA, et al. Pyostomatitis vegetans in childhood. Eur J Pediatr. 1998;157:402-405. doi:10.1007/s004310050838
- Chen KL, Diiorio DA, Chiu YE, et al. Pediatric patients with orofacial granulomatosis likely to subsequently develop intestinal Crohn’s disease: brief report. Pediatr Dermatol. 2020;37:1162-1164. doi:10.1111/pde.14390
- Pazheri F, Alkhouri N, Radhakrishnan K. Pyostomatitis vegetans as an oral manifestation of Crohn’s disease in a pediatric patient. Inflamm Bowel Dis. 2010;16:2007. doi:10.1002/ibd.21245.
- Werchniak AE, Storm CA, Plunkett RW, et al. Treatment of pyostomatitis vegetans with topical tacrolimus. J Am Acad Dermatol. 2005;52:722-723. doi:10.1016/j.jaad.2004.11.041
- Stingeni L, Tramontana M, Bassotti G, et al. Pyodermatitis–pyostomatitis vegetans and antibullous pemphigoid antigen 180 autoantibodies: a casual association? Br J Dermatol. 2015;172:811-813. doi:10.1111/bjd.13297
- Takahashi S, Andreoletti G, Chen R, et al. De novo and rare mutations in the HSPA1L heat shock gene associated with inflammatory bowel disease. Genome Med. 2017;9:8. doi:10.1186/s13073-016-0394-9
- Crowley E, Warner N, Pan J, et al. Prevalence and clinical features of inflammatory bowel diseases associated with monogenic variants, identified by whole-exome sequencing in 1000 children at a single center. Gastroenterology. 2020;158:2208-2220. doi:10.1053/j .gastro.2020.02.023
Practice Points
- Pyostomatitis vegetans (PSV) is a rare manifestation of cutaneous Crohn disease in children and can precede the onset of bowel pathology.
- Although topical and intralesional corticosteroids were beneficial in our patient, systemic corticosteroids and tumor necrosis factor α inhibitors, including infliximab and adalimumab, used to treat underlying inflammatory bowel disease appear to be the most efficacious option for treating PSV.

40-year-old woman • fever • rash • arthralgia • Dx?
THE CASE
A 40-year-old woman with no significant medical history sought care at the emergency department for a fever, rash, and arthralgia. On admission, she had worsening bilateral ankle pain and was having difficulty walking. During the previous 3 months, she’d had 3 episodes of tonsillitis, all of which were presumed to be caused by Streptococcus, although no swabs were obtained. Her primary care physician treated her with antibiotics each time: 1 round of amoxicillin 500 mg twice daily for 10 days and 2 rounds of amoxicillin/clavulanate 875 mg twice daily for 7 to 10 days. During the previous month, she’d experienced intermittent fevers ranging from 100.2 °F to 100.8

The patient said that 2 weeks prior to her admission to the hospital, she’d developed a rash on her right arm, which was papular, nondraining, nonpruritic, and not painful (FIGURE 1). Six days later, the rash spread to her left arm, chest, and back, with a few lesions on her legs (FIGURE 2). A few days later, she developed arthralgias in her hips, knees, and ankles. These were associated with the appearance of large, flat, erythematous lesions on her anterior lower extremities (FIGURE 2). About 5 days before she was admitted to our hospital, the patient was seen at another hospital and treated for possible cellulitis with cephalexin (500 mg 4 times daily for 5-7 days), but her symptoms persisted.

At this point, she sought care at our hospital for her worsening lower extremity arthralgia, difficulty walking, and the persistent rash. An initial lab report showed a white blood cell (WBC) count of 12.6 × 103/µL (normal range, 4.0-10.0 × 103/µL) with an absolute neutrophil count of 9.7 × 103/µL (normal, 1.7-7.0 × 103/µL). Her C-reactive protein (CRP) level was elevated (194.7 mg/L; normal, 0.0-5.0 mg/L), as was her erythrocyte sedimentation rate (ESR) (102.0 mm/h; normal, 0.0-20.0 mm/h). A rapid pharyngeal strep test was negative. Her anti-streptolysin O (ASO) titer was elevated (2092.0 IU/mL; normal, < 250.0 IU/mL), and her rheumatic factor was mildly elevated (19.0 IU/mL; normal, 0.0-14.0 IU/mL). An antinuclear antibody panel was positive at 1:80. Further testing was performed, and the patient was found to be negative for Sjögren syndrome A, Sjögren syndrome B, anti-Smith, scleroderma-70, double-stranded DNA, and chromatin AB—making an autoimmune disease unlikely.
THE DIAGNOSIS
The patient met the American Heart Association’s revised Jones criteria for the diagnosis of rheumatic fever: She had a positive ASO titer; polyarthritis and subcutaneous nodules (2 major criteria); and ESR > 60 mm/h and CRP > 3 mg/L (1 minor criterion).1 She started taking naproxen 500 mg twice per day and was given a penicillin G 1.5-million-unit injection. A transthoracic echocardiogram also was performed during her admission to rule out endocarditis; no abnormalities were found.
A few days after starting treatment for rheumatic fever, the patient’s WBC count returned to within normal limits and her joint swelling and pain improved; however, her rash did not go away, leading us to wonder if there was a second disease at work. Dermatology was consulted, and a punch biopsy was obtained. The results showed acute febrile neutrophilic dermatosis, or Sweet syndrome.
DISCUSSION
Sweet syndrome is considered rare, and incidence numbers are elusive.2 It has a worldwide distribution and no racial bias.3 Sweet syndrome usually occurs in women ages 30 to 50 years, although it may also occur in younger adults and children.
Three subtypes have been defined based on etiology: (1) classical (or idiopathic) Sweet syndrome; (2) malignancy-associated Sweet syndrome, which is most often related to acute myelogenous leukemia; and (3) drug-induced Sweet syndrome, which is usually associated with granulocyte colony–stimulating factor treatment.4 Our patient had the most common subtype: classical Sweet syndrome.
Continue to: What you'll see
What you’ll see.
Corticosteroid therapy is the gold standard for treatment of classical Sweet syndrome. Dosing usually starts with prednisone 1 mg/kg/d, which can be tapered to 10 mg/d within 4 to 6 weeks.5 If steroid treatment is contraindicated in the patient, alternative treatments are colchicine 0.5 mg 3 times daily for 10 to 21 days or enteric-coated potassium iodide 300 mg 3 times daily until the rash subsides.5 Without treatment, symptoms may resolve within weeks to months; with treatment, the rash usually resolves within 2 to 5 days. Some resistant forms may require 2 to 3 months of treatment.
There is a risk of recurrence in approximately one-third of patients after successful treatment of classical Sweet syndrome.5 Recurrence can be caused by another inciting factor (ie, irritable bowel disease, upper respiratory tract infection, malignancy, or a new medication), making a new investigation necessary. However, treatment would entail the same medications.5
The patient was placed on penicillin V 250 mg twice daily for 5 years due to the significant risk of carditis in the setting of rheumatic fever. She started an oral steroid regimen of a prednisone weekly taper, starting with 60 mg/d, for 4 to 6 weeks. Her papular rash improved soon after initiation of steroid therapy.
THE TAKEAWAY
On presentation, this patient’s symptoms met the Jones criteria for rheumatic fever, but she did not respond to treatment. This led us to revisit her case, order additional tests, and identify a second diagnosis—Sweet syndrome—that responded positively to treatment. This case is a reminder that sometimes the signs and symptoms we are looking at are the result of 2 underlying illnesses, with 1 possibly triggering the other. That was likely what occurred in this case.
Farah Leclercq, DO, Department of Family Medicine, University of Florida, 12041 Southwest 1 Lane, Gainesville, FL 32607; [email protected]
1. Gewitz MH, Baltimore SR, Tani LY, et al. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of doppler echocardiography: a scientific statement from the American Heart Association. Circulation. 2015;131:1806-1818. doi: 10.1161/CIR.0000000000000205
2. Joshi TP, Friske SK, Hsiou DA, Duvic M. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi: 10.1007/s40257-022-00673-4
3. Cohen PR, Kurzrock R. Sweets syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778. doi: 10.1046/j.1365-4362.2003.01891.x
4. Merola JF. Sweet syndrome (acute febrile neutrophilic dermatosis): pathogenesis, clinical manifestations, and diagnosis. UpToDate. August 9, 2020. Accessed October 27, 2022. www.uptodate.com/contents/sweet-syndrome-acute-febrile-neutrophilic-dermatosis-pathogenesis-clinical-manifestations-and-diagnosis
5. Cohen PR. Sweets syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi: 10.1186/1750-1172-2-34
THE CASE
A 40-year-old woman with no significant medical history sought care at the emergency department for a fever, rash, and arthralgia. On admission, she had worsening bilateral ankle pain and was having difficulty walking. During the previous 3 months, she’d had 3 episodes of tonsillitis, all of which were presumed to be caused by Streptococcus, although no swabs were obtained. Her primary care physician treated her with antibiotics each time: 1 round of amoxicillin 500 mg twice daily for 10 days and 2 rounds of amoxicillin/clavulanate 875 mg twice daily for 7 to 10 days. During the previous month, she’d experienced intermittent fevers ranging from 100.2 °F to 100.8
The patient said that 2 weeks prior to her admission to the hospital, she’d developed a rash on her right arm, which was papular, nondraining, nonpruritic, and not painful (FIGURE 1). Six days later, the rash spread to her left arm, chest, and back, with a few lesions on her legs (FIGURE 2). A few days later, she developed arthralgias in her hips, knees, and ankles. These were associated with the appearance of large, flat, erythematous lesions on her anterior lower extremities (FIGURE 2). About 5 days before she was admitted to our hospital, the patient was seen at another hospital and treated for possible cellulitis with cephalexin (500 mg 4 times daily for 5-7 days), but her symptoms persisted.
At this point, she sought care at our hospital for her worsening lower extremity arthralgia, difficulty walking, and the persistent rash. An initial lab report showed a white blood cell (WBC) count of 12.6 × 103/µL (normal range, 4.0-10.0 × 103/µL) with an absolute neutrophil count of 9.7 × 103/µL (normal, 1.7-7.0 × 103/µL). Her C-reactive protein (CRP) level was elevated (194.7 mg/L; normal, 0.0-5.0 mg/L), as was her erythrocyte sedimentation rate (ESR) (102.0 mm/h; normal, 0.0-20.0 mm/h). A rapid pharyngeal strep test was negative. Her anti-streptolysin O (ASO) titer was elevated (2092.0 IU/mL; normal, < 250.0 IU/mL), and her rheumatic factor was mildly elevated (19.0 IU/mL; normal, 0.0-14.0 IU/mL). An antinuclear antibody panel was positive at 1:80. Further testing was performed, and the patient was found to be negative for Sjögren syndrome A, Sjögren syndrome B, anti-Smith, scleroderma-70, double-stranded DNA, and chromatin AB—making an autoimmune disease unlikely.
THE DIAGNOSIS
The patient met the American Heart Association’s revised Jones criteria for the diagnosis of rheumatic fever: She had a positive ASO titer; polyarthritis and subcutaneous nodules (2 major criteria); and ESR > 60 mm/h and CRP > 3 mg/L (1 minor criterion).1 She started taking naproxen 500 mg twice per day and was given a penicillin G 1.5-million-unit injection. A transthoracic echocardiogram also was performed during her admission to rule out endocarditis; no abnormalities were found.
A few days after starting treatment for rheumatic fever, the patient’s WBC count returned to within normal limits and her joint swelling and pain improved; however, her rash did not go away, leading us to wonder if there was a second disease at work. Dermatology was consulted, and a punch biopsy was obtained. The results showed acute febrile neutrophilic dermatosis, or Sweet syndrome.
DISCUSSION
Sweet syndrome is considered rare, and incidence numbers are elusive.2 It has a worldwide distribution and no racial bias.3 Sweet syndrome usually occurs in women ages 30 to 50 years, although it may also occur in younger adults and children.
Three subtypes have been defined based on etiology: (1) classical (or idiopathic) Sweet syndrome; (2) malignancy-associated Sweet syndrome, which is most often related to acute myelogenous leukemia; and (3) drug-induced Sweet syndrome, which is usually associated with granulocyte colony–stimulating factor treatment.4 Our patient had the most common subtype: classical Sweet syndrome.
Continue to: What you'll see
What you’ll see.
Corticosteroid therapy is the gold standard for treatment of classical Sweet syndrome. Dosing usually starts with prednisone 1 mg/kg/d, which can be tapered to 10 mg/d within 4 to 6 weeks.5 If steroid treatment is contraindicated in the patient, alternative treatments are colchicine 0.5 mg 3 times daily for 10 to 21 days or enteric-coated potassium iodide 300 mg 3 times daily until the rash subsides.5 Without treatment, symptoms may resolve within weeks to months; with treatment, the rash usually resolves within 2 to 5 days. Some resistant forms may require 2 to 3 months of treatment.
There is a risk of recurrence in approximately one-third of patients after successful treatment of classical Sweet syndrome.5 Recurrence can be caused by another inciting factor (ie, irritable bowel disease, upper respiratory tract infection, malignancy, or a new medication), making a new investigation necessary. However, treatment would entail the same medications.5
The patient was placed on penicillin V 250 mg twice daily for 5 years due to the significant risk of carditis in the setting of rheumatic fever. She started an oral steroid regimen of a prednisone weekly taper, starting with 60 mg/d, for 4 to 6 weeks. Her papular rash improved soon after initiation of steroid therapy.
THE TAKEAWAY
On presentation, this patient’s symptoms met the Jones criteria for rheumatic fever, but she did not respond to treatment. This led us to revisit her case, order additional tests, and identify a second diagnosis—Sweet syndrome—that responded positively to treatment. This case is a reminder that sometimes the signs and symptoms we are looking at are the result of 2 underlying illnesses, with 1 possibly triggering the other. That was likely what occurred in this case.
Farah Leclercq, DO, Department of Family Medicine, University of Florida, 12041 Southwest 1 Lane, Gainesville, FL 32607; [email protected]
THE CASE
A 40-year-old woman with no significant medical history sought care at the emergency department for a fever, rash, and arthralgia. On admission, she had worsening bilateral ankle pain and was having difficulty walking. During the previous 3 months, she’d had 3 episodes of tonsillitis, all of which were presumed to be caused by Streptococcus, although no swabs were obtained. Her primary care physician treated her with antibiotics each time: 1 round of amoxicillin 500 mg twice daily for 10 days and 2 rounds of amoxicillin/clavulanate 875 mg twice daily for 7 to 10 days. During the previous month, she’d experienced intermittent fevers ranging from 100.2 °F to 100.8
The patient said that 2 weeks prior to her admission to the hospital, she’d developed a rash on her right arm, which was papular, nondraining, nonpruritic, and not painful (FIGURE 1). Six days later, the rash spread to her left arm, chest, and back, with a few lesions on her legs (FIGURE 2). A few days later, she developed arthralgias in her hips, knees, and ankles. These were associated with the appearance of large, flat, erythematous lesions on her anterior lower extremities (FIGURE 2). About 5 days before she was admitted to our hospital, the patient was seen at another hospital and treated for possible cellulitis with cephalexin (500 mg 4 times daily for 5-7 days), but her symptoms persisted.
At this point, she sought care at our hospital for her worsening lower extremity arthralgia, difficulty walking, and the persistent rash. An initial lab report showed a white blood cell (WBC) count of 12.6 × 103/µL (normal range, 4.0-10.0 × 103/µL) with an absolute neutrophil count of 9.7 × 103/µL (normal, 1.7-7.0 × 103/µL). Her C-reactive protein (CRP) level was elevated (194.7 mg/L; normal, 0.0-5.0 mg/L), as was her erythrocyte sedimentation rate (ESR) (102.0 mm/h; normal, 0.0-20.0 mm/h). A rapid pharyngeal strep test was negative. Her anti-streptolysin O (ASO) titer was elevated (2092.0 IU/mL; normal, < 250.0 IU/mL), and her rheumatic factor was mildly elevated (19.0 IU/mL; normal, 0.0-14.0 IU/mL). An antinuclear antibody panel was positive at 1:80. Further testing was performed, and the patient was found to be negative for Sjögren syndrome A, Sjögren syndrome B, anti-Smith, scleroderma-70, double-stranded DNA, and chromatin AB—making an autoimmune disease unlikely.
THE DIAGNOSIS
The patient met the American Heart Association’s revised Jones criteria for the diagnosis of rheumatic fever: She had a positive ASO titer; polyarthritis and subcutaneous nodules (2 major criteria); and ESR > 60 mm/h and CRP > 3 mg/L (1 minor criterion).1 She started taking naproxen 500 mg twice per day and was given a penicillin G 1.5-million-unit injection. A transthoracic echocardiogram also was performed during her admission to rule out endocarditis; no abnormalities were found.
A few days after starting treatment for rheumatic fever, the patient’s WBC count returned to within normal limits and her joint swelling and pain improved; however, her rash did not go away, leading us to wonder if there was a second disease at work. Dermatology was consulted, and a punch biopsy was obtained. The results showed acute febrile neutrophilic dermatosis, or Sweet syndrome.
DISCUSSION
Sweet syndrome is considered rare, and incidence numbers are elusive.2 It has a worldwide distribution and no racial bias.3 Sweet syndrome usually occurs in women ages 30 to 50 years, although it may also occur in younger adults and children.
Three subtypes have been defined based on etiology: (1) classical (or idiopathic) Sweet syndrome; (2) malignancy-associated Sweet syndrome, which is most often related to acute myelogenous leukemia; and (3) drug-induced Sweet syndrome, which is usually associated with granulocyte colony–stimulating factor treatment.4 Our patient had the most common subtype: classical Sweet syndrome.
Continue to: What you'll see
What you’ll see.
Corticosteroid therapy is the gold standard for treatment of classical Sweet syndrome. Dosing usually starts with prednisone 1 mg/kg/d, which can be tapered to 10 mg/d within 4 to 6 weeks.5 If steroid treatment is contraindicated in the patient, alternative treatments are colchicine 0.5 mg 3 times daily for 10 to 21 days or enteric-coated potassium iodide 300 mg 3 times daily until the rash subsides.5 Without treatment, symptoms may resolve within weeks to months; with treatment, the rash usually resolves within 2 to 5 days. Some resistant forms may require 2 to 3 months of treatment.
There is a risk of recurrence in approximately one-third of patients after successful treatment of classical Sweet syndrome.5 Recurrence can be caused by another inciting factor (ie, irritable bowel disease, upper respiratory tract infection, malignancy, or a new medication), making a new investigation necessary. However, treatment would entail the same medications.5
The patient was placed on penicillin V 250 mg twice daily for 5 years due to the significant risk of carditis in the setting of rheumatic fever. She started an oral steroid regimen of a prednisone weekly taper, starting with 60 mg/d, for 4 to 6 weeks. Her papular rash improved soon after initiation of steroid therapy.
THE TAKEAWAY
On presentation, this patient’s symptoms met the Jones criteria for rheumatic fever, but she did not respond to treatment. This led us to revisit her case, order additional tests, and identify a second diagnosis—Sweet syndrome—that responded positively to treatment. This case is a reminder that sometimes the signs and symptoms we are looking at are the result of 2 underlying illnesses, with 1 possibly triggering the other. That was likely what occurred in this case.
Farah Leclercq, DO, Department of Family Medicine, University of Florida, 12041 Southwest 1 Lane, Gainesville, FL 32607; [email protected]
1. Gewitz MH, Baltimore SR, Tani LY, et al. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of doppler echocardiography: a scientific statement from the American Heart Association. Circulation. 2015;131:1806-1818. doi: 10.1161/CIR.0000000000000205
2. Joshi TP, Friske SK, Hsiou DA, Duvic M. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi: 10.1007/s40257-022-00673-4
3. Cohen PR, Kurzrock R. Sweets syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778. doi: 10.1046/j.1365-4362.2003.01891.x
4. Merola JF. Sweet syndrome (acute febrile neutrophilic dermatosis): pathogenesis, clinical manifestations, and diagnosis. UpToDate. August 9, 2020. Accessed October 27, 2022. www.uptodate.com/contents/sweet-syndrome-acute-febrile-neutrophilic-dermatosis-pathogenesis-clinical-manifestations-and-diagnosis
5. Cohen PR. Sweets syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi: 10.1186/1750-1172-2-34
1. Gewitz MH, Baltimore SR, Tani LY, et al. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of doppler echocardiography: a scientific statement from the American Heart Association. Circulation. 2015;131:1806-1818. doi: 10.1161/CIR.0000000000000205
2. Joshi TP, Friske SK, Hsiou DA, Duvic M. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi: 10.1007/s40257-022-00673-4
3. Cohen PR, Kurzrock R. Sweets syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778. doi: 10.1046/j.1365-4362.2003.01891.x
4. Merola JF. Sweet syndrome (acute febrile neutrophilic dermatosis): pathogenesis, clinical manifestations, and diagnosis. UpToDate. August 9, 2020. Accessed October 27, 2022. www.uptodate.com/contents/sweet-syndrome-acute-febrile-neutrophilic-dermatosis-pathogenesis-clinical-manifestations-and-diagnosis
5. Cohen PR. Sweets syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi: 10.1186/1750-1172-2-34

Patient With Severe Headache After IV Immunoglobulin
A 35-year-old woman with a history of hypothyroidism and idiopathic small fiber autonomic and sensory neuropathy presented to the emergency department (ED) 48 hours after IV immunoglobulin (IG) infusion with a severe headache, nausea, neck stiffness, photophobia, and episodes of intense positional eye pressure. The patient reported previous episodes of headaches post-IVIG infusion but not nearly as severe. On ED arrival, the patient was afebrile with vital signs within normal limits. Initial laboratory results were notable for levels within reference range parameters: 5.9 × 109/L white blood cell (WBC) count, 13.3 g/dL hemoglobin, 38.7% hematocrit, and 279 × 109/L platelet count; there were no abnormal urinalysis findings, and she was negative for human chorionic gonadotropin.
Due to the patient’s symptoms concerning for an acute intracranial process, a brain computed tomography (CT) without contrast was ordered. The CT demonstrated no intracranial abnormalities, but the patient’s symptoms continued to worsen. The patient was started on IV fluids and 1 g IV acetaminophen and underwent a lumbar puncture (LP). Her opening pressure was elevated at 29 cm H2O (reference range, 6-20 cm), and the fluid was notably clear. During the LP, 25 mL of cerebrospinal fluid (CSF) was collected for laboratory analysis to include a polymerase chain reaction (PCR) panel and cultures, and a closing pressure of 12 cm H2O was recorded at the end of the procedure with the patient reporting some relief of pressure. The patient was admitted to the medicine ward for further workup and observations.The patient’s meningitis/encephalitis PCR panel detected no pathogens in the CSF, but her WBC count was 84 × 109/L (reference range, 4-11) with 30 segmented neutrophils (reference range, 0-6) and red blood cell count of 24 (reference range, 0-1); her normal glucose at 60 mg/dL (reference range, 40-70) and protein of 33 mg/dL (reference range, 15-45) were within normal parameters. Brain magnetic resonance images with and without contrast was inconsistent with any acute intracranial pathology to include subarachnoid hemorrhage or central nervous system neoplasm (Figure 1). Bacterial and fungal cultures were negative.
- What is your diagnosis?
- How would you treat this patient?
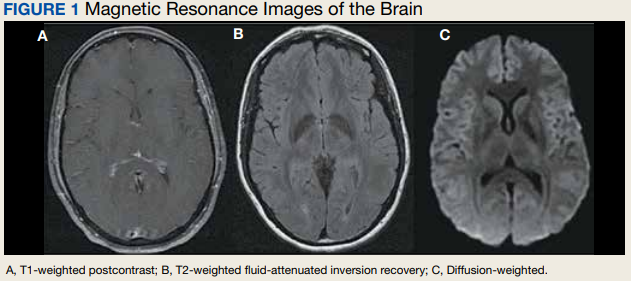
Discussion
Aseptic meningitis presents with a typical clinical picture of meningitis to include headache, stiffened neck, and photophobia. In the event of negative CSF bacterial and fungal cultures and negative viral PCR, a diagnosis of aseptic meningitis is considered.1 Though the differential for aseptic meningitis is broad, in the immunocompetent patient, the most common etiology of aseptic meningitis in the United States is by far viral, and specifically, enterovirus (50.9%). It is less commonly caused by herpes simplex virus (8.3%), varicella zoster virus, and finally, the mosquito-borne St. Louis encephalitis and West Nile viruses typically acquired in the summer or early fall months. Other infectious agents that can present with aseptic meningitis are spirochetes (Lyme disease and syphilis), tuberculous meningitis, fungal infections (cryptococcal meningitis), and other bacterial infections that have a negative culture.
The patient’s history, physical examination, vital signs, imaging, and lumbar puncture findings were most concerning for drug-induced aseptic meningitis (DIAM) secondary to her recent IVIG infusion. An algorithm can be used to work through the diagnostic approach (Figure 2).3,4 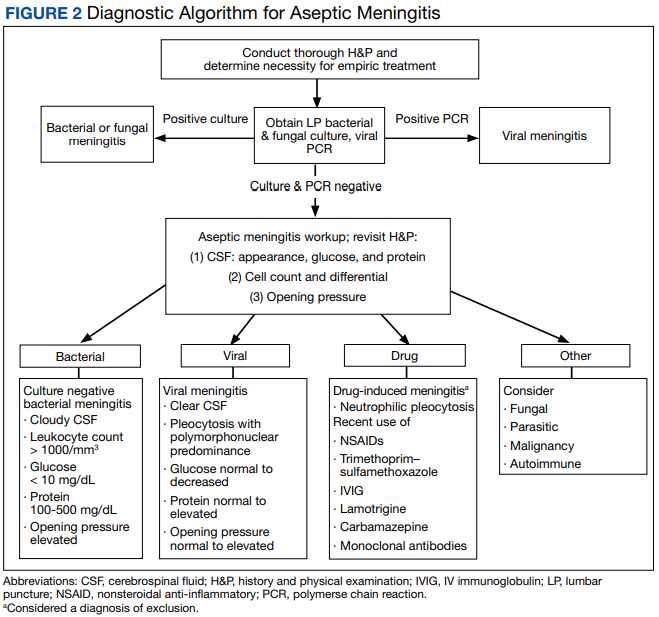
Immediate and delayed adverse reactions to IVIG are known risks for IVIG therapy. About 1% to 15% of patients who receive IVIG will experience mild immediate reactions to the infusion.6 These immediate reactions include fever (78.6%), acrocyanosis (71.4%), rash (64.3%), headache (57.1%), shortness of breath (42.8%), hypotension (35.7%), and chest pain (21.4%).
IVIG is an increasingly used biologic pharmacologic agent used for a variety of medical conditions. This can be attributed to its multifaceted properties and ability to fight infection when given as replacement therapy and provide immunomodulation in conjunction with its more well-known anti-inflammatory properties.8 The number of conditions that can potentially benefit from IVIG is so vast that the American Academy of Allergy, Asthma and Immunology had to divide the indication for IVIG therapy into definitely beneficial, probably beneficial, may provide benefit, and unlikely to provide benefit categories.8
Conclusions
We encourage heightened clinical suspicion of DIAM in patients who have recently undergone IVIG infusion and present with meningeal signs (stiff neck, headache, photophobia, and ear/eye pressure) without any evidence of infection on physical examination or laboratory results. With such, we hope to improve clinician suspicion, detection, as well as patient education and outcomes in cases of DIAM.
1. Kareva L, Mironska K, Stavric K, Hasani A. Adverse reactions to intravenous immunoglobulins—our experience. Open Access Maced J Med Sci. 2018;6(12):2359-2362. doi:10.3889/oamjms.2018.513
2. Mount HR, Boyle SD. Aseptic and bacterial meningitis: evaluation, treatment, and prevention. Am Fam Physician. 2017;96(5):314-322.
3. Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician. 2003;68(6):1103-1108.
4. Connolly KJ, Hammer SM. The acute aseptic meningitis syndrome. Infect Dis Clin North Am. 1990;4(4):599-622.
5. Jolles S, Sewell WA, Leighton C. Drug-induced aseptic meningitis: diagnosis and management. Drug Saf. 2000;22(3):215-226. doi:10.2165/00002018-200022030-00005
6. Yelehe-Okouma M, Czmil-Garon J, Pape E, Petitpain N, Gillet P. Drug-induced aseptic meningitis: a mini-review. Fundam Clin Pharmacol. 2018;32(3):252-260. doi:10.1111/fcp.12349
7. Kepa L, Oczko-Grzesik B, Stolarz W, Sobala-Szczygiel B. Drug-induced aseptic meningitis in suspected central nervous system infections. J Clin Neurosci. 2005;12(5):562-564. doi:10.1016/j.jocn.2004.08.024
8. Perez EE, Orange JS, Bonilla F, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139(3S):S1-S46. doi:10.1016/j.jaci.2016.09.023
9. Kaarthigeyan K, Burli VV. Aseptic meningitis following intravenous immunoglobulin therapy of common variable immunodeficiency. J Pediatr Neurosci. 2011;6(2):160-161. doi:10.4103/1817-1745.92858
A 35-year-old woman with a history of hypothyroidism and idiopathic small fiber autonomic and sensory neuropathy presented to the emergency department (ED) 48 hours after IV immunoglobulin (IG) infusion with a severe headache, nausea, neck stiffness, photophobia, and episodes of intense positional eye pressure. The patient reported previous episodes of headaches post-IVIG infusion but not nearly as severe. On ED arrival, the patient was afebrile with vital signs within normal limits. Initial laboratory results were notable for levels within reference range parameters: 5.9 × 109/L white blood cell (WBC) count, 13.3 g/dL hemoglobin, 38.7% hematocrit, and 279 × 109/L platelet count; there were no abnormal urinalysis findings, and she was negative for human chorionic gonadotropin.
Due to the patient’s symptoms concerning for an acute intracranial process, a brain computed tomography (CT) without contrast was ordered. The CT demonstrated no intracranial abnormalities, but the patient’s symptoms continued to worsen. The patient was started on IV fluids and 1 g IV acetaminophen and underwent a lumbar puncture (LP). Her opening pressure was elevated at 29 cm H2O (reference range, 6-20 cm), and the fluid was notably clear. During the LP, 25 mL of cerebrospinal fluid (CSF) was collected for laboratory analysis to include a polymerase chain reaction (PCR) panel and cultures, and a closing pressure of 12 cm H2O was recorded at the end of the procedure with the patient reporting some relief of pressure. The patient was admitted to the medicine ward for further workup and observations.The patient’s meningitis/encephalitis PCR panel detected no pathogens in the CSF, but her WBC count was 84 × 109/L (reference range, 4-11) with 30 segmented neutrophils (reference range, 0-6) and red blood cell count of 24 (reference range, 0-1); her normal glucose at 60 mg/dL (reference range, 40-70) and protein of 33 mg/dL (reference range, 15-45) were within normal parameters. Brain magnetic resonance images with and without contrast was inconsistent with any acute intracranial pathology to include subarachnoid hemorrhage or central nervous system neoplasm (Figure 1). Bacterial and fungal cultures were negative.
- What is your diagnosis?
- How would you treat this patient?
Discussion
Aseptic meningitis presents with a typical clinical picture of meningitis to include headache, stiffened neck, and photophobia. In the event of negative CSF bacterial and fungal cultures and negative viral PCR, a diagnosis of aseptic meningitis is considered.1 Though the differential for aseptic meningitis is broad, in the immunocompetent patient, the most common etiology of aseptic meningitis in the United States is by far viral, and specifically, enterovirus (50.9%). It is less commonly caused by herpes simplex virus (8.3%), varicella zoster virus, and finally, the mosquito-borne St. Louis encephalitis and West Nile viruses typically acquired in the summer or early fall months. Other infectious agents that can present with aseptic meningitis are spirochetes (Lyme disease and syphilis), tuberculous meningitis, fungal infections (cryptococcal meningitis), and other bacterial infections that have a negative culture.
The patient’s history, physical examination, vital signs, imaging, and lumbar puncture findings were most concerning for drug-induced aseptic meningitis (DIAM) secondary to her recent IVIG infusion. An algorithm can be used to work through the diagnostic approach (Figure 2).3,4
Immediate and delayed adverse reactions to IVIG are known risks for IVIG therapy. About 1% to 15% of patients who receive IVIG will experience mild immediate reactions to the infusion.6 These immediate reactions include fever (78.6%), acrocyanosis (71.4%), rash (64.3%), headache (57.1%), shortness of breath (42.8%), hypotension (35.7%), and chest pain (21.4%).
IVIG is an increasingly used biologic pharmacologic agent used for a variety of medical conditions. This can be attributed to its multifaceted properties and ability to fight infection when given as replacement therapy and provide immunomodulation in conjunction with its more well-known anti-inflammatory properties.8 The number of conditions that can potentially benefit from IVIG is so vast that the American Academy of Allergy, Asthma and Immunology had to divide the indication for IVIG therapy into definitely beneficial, probably beneficial, may provide benefit, and unlikely to provide benefit categories.8
Conclusions
We encourage heightened clinical suspicion of DIAM in patients who have recently undergone IVIG infusion and present with meningeal signs (stiff neck, headache, photophobia, and ear/eye pressure) without any evidence of infection on physical examination or laboratory results. With such, we hope to improve clinician suspicion, detection, as well as patient education and outcomes in cases of DIAM.
A 35-year-old woman with a history of hypothyroidism and idiopathic small fiber autonomic and sensory neuropathy presented to the emergency department (ED) 48 hours after IV immunoglobulin (IG) infusion with a severe headache, nausea, neck stiffness, photophobia, and episodes of intense positional eye pressure. The patient reported previous episodes of headaches post-IVIG infusion but not nearly as severe. On ED arrival, the patient was afebrile with vital signs within normal limits. Initial laboratory results were notable for levels within reference range parameters: 5.9 × 109/L white blood cell (WBC) count, 13.3 g/dL hemoglobin, 38.7% hematocrit, and 279 × 109/L platelet count; there were no abnormal urinalysis findings, and she was negative for human chorionic gonadotropin.
Due to the patient’s symptoms concerning for an acute intracranial process, a brain computed tomography (CT) without contrast was ordered. The CT demonstrated no intracranial abnormalities, but the patient’s symptoms continued to worsen. The patient was started on IV fluids and 1 g IV acetaminophen and underwent a lumbar puncture (LP). Her opening pressure was elevated at 29 cm H2O (reference range, 6-20 cm), and the fluid was notably clear. During the LP, 25 mL of cerebrospinal fluid (CSF) was collected for laboratory analysis to include a polymerase chain reaction (PCR) panel and cultures, and a closing pressure of 12 cm H2O was recorded at the end of the procedure with the patient reporting some relief of pressure. The patient was admitted to the medicine ward for further workup and observations.The patient’s meningitis/encephalitis PCR panel detected no pathogens in the CSF, but her WBC count was 84 × 109/L (reference range, 4-11) with 30 segmented neutrophils (reference range, 0-6) and red blood cell count of 24 (reference range, 0-1); her normal glucose at 60 mg/dL (reference range, 40-70) and protein of 33 mg/dL (reference range, 15-45) were within normal parameters. Brain magnetic resonance images with and without contrast was inconsistent with any acute intracranial pathology to include subarachnoid hemorrhage or central nervous system neoplasm (Figure 1). Bacterial and fungal cultures were negative.
- What is your diagnosis?
- How would you treat this patient?
Discussion
Aseptic meningitis presents with a typical clinical picture of meningitis to include headache, stiffened neck, and photophobia. In the event of negative CSF bacterial and fungal cultures and negative viral PCR, a diagnosis of aseptic meningitis is considered.1 Though the differential for aseptic meningitis is broad, in the immunocompetent patient, the most common etiology of aseptic meningitis in the United States is by far viral, and specifically, enterovirus (50.9%). It is less commonly caused by herpes simplex virus (8.3%), varicella zoster virus, and finally, the mosquito-borne St. Louis encephalitis and West Nile viruses typically acquired in the summer or early fall months. Other infectious agents that can present with aseptic meningitis are spirochetes (Lyme disease and syphilis), tuberculous meningitis, fungal infections (cryptococcal meningitis), and other bacterial infections that have a negative culture.
The patient’s history, physical examination, vital signs, imaging, and lumbar puncture findings were most concerning for drug-induced aseptic meningitis (DIAM) secondary to her recent IVIG infusion. An algorithm can be used to work through the diagnostic approach (Figure 2).3,4
Immediate and delayed adverse reactions to IVIG are known risks for IVIG therapy. About 1% to 15% of patients who receive IVIG will experience mild immediate reactions to the infusion.6 These immediate reactions include fever (78.6%), acrocyanosis (71.4%), rash (64.3%), headache (57.1%), shortness of breath (42.8%), hypotension (35.7%), and chest pain (21.4%).
IVIG is an increasingly used biologic pharmacologic agent used for a variety of medical conditions. This can be attributed to its multifaceted properties and ability to fight infection when given as replacement therapy and provide immunomodulation in conjunction with its more well-known anti-inflammatory properties.8 The number of conditions that can potentially benefit from IVIG is so vast that the American Academy of Allergy, Asthma and Immunology had to divide the indication for IVIG therapy into definitely beneficial, probably beneficial, may provide benefit, and unlikely to provide benefit categories.8
Conclusions
We encourage heightened clinical suspicion of DIAM in patients who have recently undergone IVIG infusion and present with meningeal signs (stiff neck, headache, photophobia, and ear/eye pressure) without any evidence of infection on physical examination or laboratory results. With such, we hope to improve clinician suspicion, detection, as well as patient education and outcomes in cases of DIAM.
1. Kareva L, Mironska K, Stavric K, Hasani A. Adverse reactions to intravenous immunoglobulins—our experience. Open Access Maced J Med Sci. 2018;6(12):2359-2362. doi:10.3889/oamjms.2018.513
2. Mount HR, Boyle SD. Aseptic and bacterial meningitis: evaluation, treatment, and prevention. Am Fam Physician. 2017;96(5):314-322.
3. Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician. 2003;68(6):1103-1108.
4. Connolly KJ, Hammer SM. The acute aseptic meningitis syndrome. Infect Dis Clin North Am. 1990;4(4):599-622.
5. Jolles S, Sewell WA, Leighton C. Drug-induced aseptic meningitis: diagnosis and management. Drug Saf. 2000;22(3):215-226. doi:10.2165/00002018-200022030-00005
6. Yelehe-Okouma M, Czmil-Garon J, Pape E, Petitpain N, Gillet P. Drug-induced aseptic meningitis: a mini-review. Fundam Clin Pharmacol. 2018;32(3):252-260. doi:10.1111/fcp.12349
7. Kepa L, Oczko-Grzesik B, Stolarz W, Sobala-Szczygiel B. Drug-induced aseptic meningitis in suspected central nervous system infections. J Clin Neurosci. 2005;12(5):562-564. doi:10.1016/j.jocn.2004.08.024
8. Perez EE, Orange JS, Bonilla F, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139(3S):S1-S46. doi:10.1016/j.jaci.2016.09.023
9. Kaarthigeyan K, Burli VV. Aseptic meningitis following intravenous immunoglobulin therapy of common variable immunodeficiency. J Pediatr Neurosci. 2011;6(2):160-161. doi:10.4103/1817-1745.92858
1. Kareva L, Mironska K, Stavric K, Hasani A. Adverse reactions to intravenous immunoglobulins—our experience. Open Access Maced J Med Sci. 2018;6(12):2359-2362. doi:10.3889/oamjms.2018.513
2. Mount HR, Boyle SD. Aseptic and bacterial meningitis: evaluation, treatment, and prevention. Am Fam Physician. 2017;96(5):314-322.
3. Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician. 2003;68(6):1103-1108.
4. Connolly KJ, Hammer SM. The acute aseptic meningitis syndrome. Infect Dis Clin North Am. 1990;4(4):599-622.
5. Jolles S, Sewell WA, Leighton C. Drug-induced aseptic meningitis: diagnosis and management. Drug Saf. 2000;22(3):215-226. doi:10.2165/00002018-200022030-00005
6. Yelehe-Okouma M, Czmil-Garon J, Pape E, Petitpain N, Gillet P. Drug-induced aseptic meningitis: a mini-review. Fundam Clin Pharmacol. 2018;32(3):252-260. doi:10.1111/fcp.12349
7. Kepa L, Oczko-Grzesik B, Stolarz W, Sobala-Szczygiel B. Drug-induced aseptic meningitis in suspected central nervous system infections. J Clin Neurosci. 2005;12(5):562-564. doi:10.1016/j.jocn.2004.08.024
8. Perez EE, Orange JS, Bonilla F, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139(3S):S1-S46. doi:10.1016/j.jaci.2016.09.023
9. Kaarthigeyan K, Burli VV. Aseptic meningitis following intravenous immunoglobulin therapy of common variable immunodeficiency. J Pediatr Neurosci. 2011;6(2):160-161. doi:10.4103/1817-1745.92858
Oral Therapy for Aerococcus urinae Bacteremia and Thoracic Spondylodiscitis of Presumed Urinary Origin
Aerococcus urinae (A urinae), a gram-positive coccus readily mistaken for a Staphylococcus species, was first identified in 1992.1-3 It now reportedly accounts for 0.2% to 0.8% of clinical urine isolates.4-6 A urinae bacteriuria is typically asymptomatic and mainly occurs in women.7-9 Symptomatic A urinae urinary tract infection (UTI) occurs predominantly in older men with underlying urologic abnormalities.4-10
Serious A urinae infections are rare. The first 2 reported cases involved men with A urinae endocarditis, one of whom died.11,12 To date, only 8 cases of spondylodiscitis due to A urinae have been reported.13-20 Optimal treatment for invasive A urinae infection is undefined; however, the reported cases were treated successfully with diverse antibiotic regimen combinations; all including a β-lactam and beginning with at least 2 weeks of IV antibiotics.13-20 We describe a man with A urinae bacteremia and spondylodiscitis, presumably arising from a urinary source in the setting of bladder outlet obstruction, who was treated successfully.
Case Presentation
A 74-year-old man with morbid obesity, type 2 diabetes mellitus, stage 2 chronic kidney disease, and tobacco use presented to the emergency department after 2 weeks of progressive, nonradiating, midthoracic back pain, lower extremity weakness, gait imbalance, fatigue, anorexia, rigors, and subjective fevers. On presentation, he was afebrile and hemodynamically stable. A physical examination revealed point tenderness of the midthoracic vertebrae, nontender costovertebral angles, diffusely decreased strength, nonsustained clonus in both lower extremities, inguinal intertrigo, and a buried penis with purulent meatal discharge.
Laboratory results indicated a white blood cell (WBC) count of 13.5 K/μL (reference range, 4.0-11.0), absolute neutrophil count of 11.48 K/μL (reference range, 2.0-7.7), C-reactive protein (CRP) level of 225.3 mg/L (reference range, ≤ 5.0), erythrocyte sedimentation rate of 85 mm/h (reference range, 5-15), serum blood urea nitrogen of 76 mg/dL (reference range, 8-26), and serum creatinine (SCr) of 1.9 mg/dL (reference range, 1.1-1.4). A urinalysis showed positive leukocyte esterase, WBC clumps, and little bacteria. Abdominal/pelvic computed tomography showed spondylodiscitis-like changes at T7-T8, bilateral perinephric fat stranding, bladder distension, and bladder wall thickening.
The patient was presumed to have discitis secondary to a UTI, with possible pyelonephritis, and was given empiric vancomycin and ceftriaxone. Spinal magnetic resonance imaging with contrast supported spondylodiscitis at T7-T8, extending to T8-T9. Preliminary results from the admission blood and urine cultures showed gram-positive cocci in clusters, which were presumed initially to be Staphylococcus aureus (S aureus).
The final urine culture report listed multiple organisms, predominantly A urinae (Table 1); 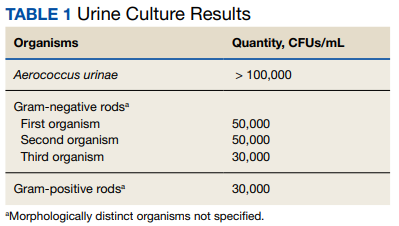
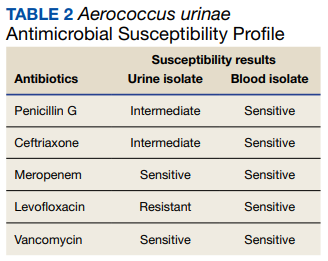
On hospital day 6, the patient’s back pain had resolved, micturition was normal, appetite had normalized, and SCr was minimally above baseline (1.4 mg/dL). He insisted on completing antibiotic treatment at home and had no other medical indication for continued hospitalization. Thus, antibiotic therapy was changed to an all-oral regimen of amoxicillin 1 g 3 times daily for 10 days and levofloxacin 750 mg daily for 6 weeks, and the patient was discharged to home.
The patient returned 5 days postdischarge due to anuria. Investigation showed severe acute kidney injury (SCr, 6.8 mg/dL) and bladder outlet obstruction due to phimosis and urethral meatal stenosis. Urinalysis was unremarkable. His CRP had declined from 225 mg/L (initial admission) to 154 mg/L. A urinae culture and 2 sets of blood cultures were finalized as no growth. He was diagnosed with postrenal acute kidney injury and underwent meatal dilation and Foley catheterization but declined surgical correction. When seen in the clinic 2 months postantimicrobial therapy, the patient had normal micturition, no symptoms or signs of infection, and steadily down-trending inflammatory markers.
Discussion
A urinae, historically considered a rare pathogen, has been identified with increasing frequency in urine cultures due to improved microbiologic diagnostic techniques. However, there are only 8 reported cases of A urinae spondylodiscitis. Urinary pathology is an accepted risk factor for A urinae infections; consequently, we suspect that our patient’s urinary outflow obstruction and poor genitourinary hygiene were related to his invasive A urinae infection.10,21,22 We surmise that he had a chronic urinary outflow obstruction contributing to his infection, as evidenced by imaging findings, while the phimosis and urethral meatal stenosis were most likely infectious sequelae considering his anuria and acute kidney injury 5 days postdischarge. Indeed, the correlation between A urinae and urinary tract pathology may justify an evaluation for urinary pathology in any man with A urinae infection, regardless of the presence of symptoms.
By contrast, the implications of A urinae bacteriuria remain unclear. From a public health perspective, A urinae bacteriuria is rare, but the infectious mechanism remains undetermined with a case report suggesting the possibility of sexual transmission.4-6,23 In our case, the patient was not sexually active and had no clear origin of infection. Considering the potential severity of infection, more studies are needed to determine the infectious mechanism of A urinae.
In terms of infectious morbidity, the results seem to vary by sex. In a retrospective study of about 30,000 clinical urine samples, 62 (58 from women, 4 from men) yielded A urinae. The 62 corresponding patients lacked systemic infectious complications, leading the authors to conclude that A urinae is a relatively avirulent organism.24 Although possibly true in women, we are wary of drawing conclusions, especially regarding men, from a study that included only 62 urine samples were A urinae–positive, with only 4 from men. More evidence is needed to define the prognostic implications of A urinae bacteriuria in men.
As illustrated by the present case and previous reports, severe A urinae infections can occur, and the contributory factors deserve consideration. In our patient, the actual mechanism for bacteremia remains unclear. The initial concern for acute pyelonephritis was prompted by a computed tomography finding of bilateral perinephric fat stranding. This finding was questioned because it is common in older patients without infection, hence, is highly nonspecific. A correlation with urinary outflow obstruction may be an important clue in cases like this one.25,26
Furthermore, whether the urinary tract truly was the source of the patient’s bacteremia is clouded by the differing antimicrobial susceptibility patterns of the A urinae blood and urine isolates. The simplest explanation for this discordance may be that all the isolates shared a common initial origin but adapted to different environments in the host (perhaps over time) or laboratory, producing phenotypic variation. Alternatively, the infection could have been polyclonal from the onset, with sampling error leading to the differing detected susceptibility patterns, or the blood and urine isolates may have represented independent acquisition events, involving distinct A urinae strains. Unfortunately (from an academic perspective), given patient preferences and recommendations from the infectious disease consultant, no bone biopsy was done for histology and culture to confirm infection and to allow comparative strain identification if A urinae was isolated.
Optimal treatment for A urinae spondylodiscitis has yet to be established. β-lactams have shown good clinical efficacy despite being bacteriostatic in vitro.27 Early in vitro studies showed synergistic bactericidal synergistic activity with penicillin plus aminoglycoside combination therapies.27-30 Cases of endocarditis have been successfully treated mainly with the combination of a β-lactam plus aminoglycoside combination therapy.30,31 Previous cases of spondylodiscitis have been treated successfully with diverse antimicrobial agents, including clindamycin, β-lactams, cephalosporins, fluoroquinolones, and aminoglycosides.14
Our patient improved rapidly while receiving empiric therapy with vancomycin and ceftriaxone and tolerated a rapid transition to oral amoxicillin and levofloxacin. This is the shortest IV treatment course for A urinae spondylodiscitis reported to date. We suspect that such rapid IV-to-oral transitions will suffice in most stable patients with A urinae spondylodiscitis or other invasive A urinae infections in line with the results of the OVIVA and POET trials.32,33
Conclusions
We believe A urinae UTI in the absence of obvious predisposing factors should prompt evaluation for urinary outflow obstruction. Despite improved laboratory diagnostic techniques, spondylodiscitis related to A urinae remains a rare entity and thus definitive treatment recommendations are difficult to make. However, we suspect that in many cases it is reasonable to extrapolate from the results of the POET and OVIVA trials and rapidly transition therapy of A urinae spondylodiscitis from IV to oral antibiotics. We suspect a review of the US Department of Veterans Affairs population might uncover a higher incidence of A urinae infection than previously estimated due to the population demographics and the epidemiology of A urinae.
1. Christensen JJ, Korner B, Kjaergaard H. Aerococcus-like organism—an unnoticed urinary tract pathogen. APMIS. 1989;97(6):539-546. doi:10.1111/j.1699-0463.1989.tb00828.x
2. Aguirre M, Collins MD. Phylogenetic analysis of some Aerococcus-like organisms from urinary tract infections: description of Aerococcus urinae sp. nov. J Gen Microbiol. 1992;138(2):401-405. doi:10.1099/00221287-138-2-401
3. Williams RE, Hirch A, Cowan ST. Aerococcus, a new bacterial genus. J Gen Microbiol. 1953;8(3):475-480. doi:10.1099/00221287-8-3-475
4. Kline KA, Lewis AL. Gram-positive uropathogens, polymicrobial urinary tract infection, and the emerging microbiota of the urinary tract. Microbiol Spectr. 2016;4(2). doi:10.1128/microbiolspec.UTI-0012-2012
5. Schuur PM, Kasteren ME, Sabbe L, Vos MC, Janssens MM, Buiting AG. Urinary tract infections with Aerococcus urinae in the south of The Netherlands. Eur J Clin Microbiol Infect Dis. 1997;16(12):871-875. doi:10.1007/BF01700552
6. Grude N, Tveten Y. Aerococcus urinae og urinveisinfeksjon [Aerococcus urinae and urinary tract infection]. Tidsskr Nor Laegeforen. 2002;122(2):174-175.
7. Narayanasamy S, King K, Dennison A, Spelman DW, Aung AK. Clinical characteristics and laboratory identification of Aerococcus infections: an Australian tertiary centre perspective. Int J Microbiol. 2017;2017. doi:10.1155/2017/5684614
8. Hilt EE, McKinley K, Pearce MM, et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol. 2014;52(3):871-876. doi:10.1128/JCM.02876-13
9. Pearce MM, Hilt EE, Rosenfeld AB, et al. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. mBio. 2014;5(4):e01283-14. doi:10.1128/mBio.01283-14
10. Sahu KK, Lal A, Mishra AK, Abraham GM. Aerococcus-related infections and their significance: a 9-year retrospective study. J Microsc Ultrastruct. 2021;9(1):18-25. doi:10.4103/JMAU.JMAU_61_19
11. Skov RL, Klarlund M, Thorsen S. Fatal endocarditis due to Aerococcus urinae. Diagn Microbiol Infect Dis. 1995;21(4):219-221. doi:10.1016/0732-8893(95)00037-b
12. Kristensen B, Nielsen G. Endocarditis caused by Aerococcus urinae, a newly recognized pathogen. Eur J Clin Microbiol Infect Dis. 1995;14(1):49-51. doi:10.1007/BF02112619
13. Astudillo L, Sailler L, Porte L, Lefevre JC, Massip P, Arlet-Suau E. Spondylodiscitis due to Aerococcus urinae: a first report. Scand J Infect Dis. 2003;35(11-12):890-891. doi:10.1080/00365540310016664
14. Lyagoubi A, Souffi C, Baroiller V, Vallee E. Spondylodiscitis: an increasingly described localization. EJIFCC. 2020;31(2):169-173.
15. Jerome M, Slim J, Sison R, Marton R. A case of Aerococcus urinae vertebral osteomyelitis. J Glob Infect Dis. 2015;7(2):85-86. doi:10.4103/0974-777X.157246
16. Tekin A, Tekin G, Turunç T, Demiroğlu Z, Kizilkiliç O. Infective endocarditis and spondylodiscitis in a patient due to Aerococcus urinae: first report. Int J Cardiol. 2007;115(3):402-403. doi:10.1016/j.ijcard.2006.01.046
17. Rougier E, Braud A, Argemi X, et al. Spondylodiscitis due to Aerococcus urinae and literature review. Infection. 2018;46(3):419-421. doi:10.1007/s15010-017-1106-0
18. Degroote E, Yildiz H, Lecouvet F, Verroken A, Belkhir L. Aerococcus urinae: an underestimated cause of spine infection? Case report and review of the literature. Acta Clin Belg. 2018;73(6):444-447. doi:10.1080/17843286.2018.1443003
19. Torres-Martos E, Pérez-Cortés S, Sánchez-Calvo JM, López-Prieto MD. Spondylodiscitis due to Aerococcus urinae infection in an elderly immunocompetent patient. Enferm Infecc Microbiol Clin. 2017;35(10):682-684. doi:10.1016/j.eimc.2017.02.005
20. Senneby E, Petersson AC, Rasmussen M. Clinical and microbiological features of bacteraemia with Aerococcus urinae. Clin Microbiol Infect. 2012;18(6):546-550. doi:10.1111/j.1469-0691.2011.03609.x
21. Sunnerhagen T, Nilson B, Olaison L, Rasmussen M. Clinical and microbiological features of infective endocarditis caused by aerococci. Infection. 2016;44(2):167-173. doi:10.1007/s15010-015-0812-8
22. de Jong MF, Soetekouw R, ten Kate RW, Veenendaal D. Aerococcus urinae: severe and fatal bloodstream infections and endocarditis. J Clin Microbiol. 2010;48(9):3445-3447. doi:10.1128/JCM.00835-10
23. Babaeer AA, Nader C, Iacoviello V, Tomera K. Necrotizing urethritis due to Aerococcus urinae. Case Rep Urol. 2015;2015:136147. doi:10.1155/2015/136147
24. Sierra-Hoffman M, Watkins K, Jinadatha C, Fader R, Carpenter JL. Clinical significance of Aerococcus urinae: a retrospective review. Diagn Microbiol Infect Dis. 2005;53(4):289-292. doi:10.1016/j.diagmicrobio.2005.06.021
25. Fukami H, Takeuchi Y, Kagaya S, et al. Perirenal fat stranding is not a powerful diagnostic tool for acute pyelonephritis. Int J Gen Med. 2017;10:137-144. doi:10.2147/IJGM.S133685
26. Han NY, Sung DJ, Kim MJ, Park BJ, Sim KC, Cho SB. Perirenal fat stranding on CT: is there an association with bladder outlet obstruction? Br J Radiol. 2016;89(1063):20160195. doi:10.1259/bjr.20160195
27. Hirzel C, Hirzberger L, Furrer H, Endimiani A. Bactericidal activity of penicillin, ceftriaxone, gentamicin and daptomycin alone and in combination against Aerococcus urinae. Int J Antimicrob Agents. 2016;48(3):271-276. doi:10.1016/j.ijantimicag.2016.05.007
28. Zbinden R, Santanam P, Hunziker L, Leuzinger B, von Graevenitz A. Endocarditis due to Aerococcus urinae: diagnostic tests, fatty acid composition and killing kinetics. Infection. 1999;27(2):122-124. doi:10.1007/BF02560511
29. Skov R, Christensen JJ, Korner B, Frimodt-Møller N, Espersen F. In vitro antimicrobial susceptibility of Aerococcus urinae to 14 antibiotics, and time-kill curves for penicillin, gentamicin and vancomycin. J Antimicrob Chemother. 2001;48(5):653-658. doi:10.1093/jac/48.5.653
30. Ebnöther C, Altwegg M, Gottschalk J, Seebach JD, Kronenberg A. Aerococcus urinae endocarditis: case report and review of the literature. Infection. 2002;30(5):310-313. doi:10.1007/s15010-002-3106-x
31. Tai DBG, Go JR, Fida M, Saleh OA. Management and treatment of Aerococcus bacteremia and endocarditis. Int J Infect Dis. 2021;102:584-589. doi:10.1016/j.ijid.2020.10.096
32. Li H-K, Rombach I, Zambellas R, et al; OVIVA Trial Collaborators. Oral versus intravenous antibiotics for bone and joint infection. N Engl J Med. 2019;380(5):425-436. doi:10.1056/NEJMoa1710926
33. Iversen K, Ihlemann N, Gill SU, et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Engl J Med. 2019;380(5):415-424. doi:10.1056/NEJMoa1808312
Aerococcus urinae (A urinae), a gram-positive coccus readily mistaken for a Staphylococcus species, was first identified in 1992.1-3 It now reportedly accounts for 0.2% to 0.8% of clinical urine isolates.4-6 A urinae bacteriuria is typically asymptomatic and mainly occurs in women.7-9 Symptomatic A urinae urinary tract infection (UTI) occurs predominantly in older men with underlying urologic abnormalities.4-10
Serious A urinae infections are rare. The first 2 reported cases involved men with A urinae endocarditis, one of whom died.11,12 To date, only 8 cases of spondylodiscitis due to A urinae have been reported.13-20 Optimal treatment for invasive A urinae infection is undefined; however, the reported cases were treated successfully with diverse antibiotic regimen combinations; all including a β-lactam and beginning with at least 2 weeks of IV antibiotics.13-20 We describe a man with A urinae bacteremia and spondylodiscitis, presumably arising from a urinary source in the setting of bladder outlet obstruction, who was treated successfully.
Case Presentation
A 74-year-old man with morbid obesity, type 2 diabetes mellitus, stage 2 chronic kidney disease, and tobacco use presented to the emergency department after 2 weeks of progressive, nonradiating, midthoracic back pain, lower extremity weakness, gait imbalance, fatigue, anorexia, rigors, and subjective fevers. On presentation, he was afebrile and hemodynamically stable. A physical examination revealed point tenderness of the midthoracic vertebrae, nontender costovertebral angles, diffusely decreased strength, nonsustained clonus in both lower extremities, inguinal intertrigo, and a buried penis with purulent meatal discharge.
Laboratory results indicated a white blood cell (WBC) count of 13.5 K/μL (reference range, 4.0-11.0), absolute neutrophil count of 11.48 K/μL (reference range, 2.0-7.7), C-reactive protein (CRP) level of 225.3 mg/L (reference range, ≤ 5.0), erythrocyte sedimentation rate of 85 mm/h (reference range, 5-15), serum blood urea nitrogen of 76 mg/dL (reference range, 8-26), and serum creatinine (SCr) of 1.9 mg/dL (reference range, 1.1-1.4). A urinalysis showed positive leukocyte esterase, WBC clumps, and little bacteria. Abdominal/pelvic computed tomography showed spondylodiscitis-like changes at T7-T8, bilateral perinephric fat stranding, bladder distension, and bladder wall thickening.
The patient was presumed to have discitis secondary to a UTI, with possible pyelonephritis, and was given empiric vancomycin and ceftriaxone. Spinal magnetic resonance imaging with contrast supported spondylodiscitis at T7-T8, extending to T8-T9. Preliminary results from the admission blood and urine cultures showed gram-positive cocci in clusters, which were presumed initially to be Staphylococcus aureus (S aureus).
The final urine culture report listed multiple organisms, predominantly A urinae (Table 1);
On hospital day 6, the patient’s back pain had resolved, micturition was normal, appetite had normalized, and SCr was minimally above baseline (1.4 mg/dL). He insisted on completing antibiotic treatment at home and had no other medical indication for continued hospitalization. Thus, antibiotic therapy was changed to an all-oral regimen of amoxicillin 1 g 3 times daily for 10 days and levofloxacin 750 mg daily for 6 weeks, and the patient was discharged to home.
The patient returned 5 days postdischarge due to anuria. Investigation showed severe acute kidney injury (SCr, 6.8 mg/dL) and bladder outlet obstruction due to phimosis and urethral meatal stenosis. Urinalysis was unremarkable. His CRP had declined from 225 mg/L (initial admission) to 154 mg/L. A urinae culture and 2 sets of blood cultures were finalized as no growth. He was diagnosed with postrenal acute kidney injury and underwent meatal dilation and Foley catheterization but declined surgical correction. When seen in the clinic 2 months postantimicrobial therapy, the patient had normal micturition, no symptoms or signs of infection, and steadily down-trending inflammatory markers.
Discussion
A urinae, historically considered a rare pathogen, has been identified with increasing frequency in urine cultures due to improved microbiologic diagnostic techniques. However, there are only 8 reported cases of A urinae spondylodiscitis. Urinary pathology is an accepted risk factor for A urinae infections; consequently, we suspect that our patient’s urinary outflow obstruction and poor genitourinary hygiene were related to his invasive A urinae infection.10,21,22 We surmise that he had a chronic urinary outflow obstruction contributing to his infection, as evidenced by imaging findings, while the phimosis and urethral meatal stenosis were most likely infectious sequelae considering his anuria and acute kidney injury 5 days postdischarge. Indeed, the correlation between A urinae and urinary tract pathology may justify an evaluation for urinary pathology in any man with A urinae infection, regardless of the presence of symptoms.
By contrast, the implications of A urinae bacteriuria remain unclear. From a public health perspective, A urinae bacteriuria is rare, but the infectious mechanism remains undetermined with a case report suggesting the possibility of sexual transmission.4-6,23 In our case, the patient was not sexually active and had no clear origin of infection. Considering the potential severity of infection, more studies are needed to determine the infectious mechanism of A urinae.
In terms of infectious morbidity, the results seem to vary by sex. In a retrospective study of about 30,000 clinical urine samples, 62 (58 from women, 4 from men) yielded A urinae. The 62 corresponding patients lacked systemic infectious complications, leading the authors to conclude that A urinae is a relatively avirulent organism.24 Although possibly true in women, we are wary of drawing conclusions, especially regarding men, from a study that included only 62 urine samples were A urinae–positive, with only 4 from men. More evidence is needed to define the prognostic implications of A urinae bacteriuria in men.
As illustrated by the present case and previous reports, severe A urinae infections can occur, and the contributory factors deserve consideration. In our patient, the actual mechanism for bacteremia remains unclear. The initial concern for acute pyelonephritis was prompted by a computed tomography finding of bilateral perinephric fat stranding. This finding was questioned because it is common in older patients without infection, hence, is highly nonspecific. A correlation with urinary outflow obstruction may be an important clue in cases like this one.25,26
Furthermore, whether the urinary tract truly was the source of the patient’s bacteremia is clouded by the differing antimicrobial susceptibility patterns of the A urinae blood and urine isolates. The simplest explanation for this discordance may be that all the isolates shared a common initial origin but adapted to different environments in the host (perhaps over time) or laboratory, producing phenotypic variation. Alternatively, the infection could have been polyclonal from the onset, with sampling error leading to the differing detected susceptibility patterns, or the blood and urine isolates may have represented independent acquisition events, involving distinct A urinae strains. Unfortunately (from an academic perspective), given patient preferences and recommendations from the infectious disease consultant, no bone biopsy was done for histology and culture to confirm infection and to allow comparative strain identification if A urinae was isolated.
Optimal treatment for A urinae spondylodiscitis has yet to be established. β-lactams have shown good clinical efficacy despite being bacteriostatic in vitro.27 Early in vitro studies showed synergistic bactericidal synergistic activity with penicillin plus aminoglycoside combination therapies.27-30 Cases of endocarditis have been successfully treated mainly with the combination of a β-lactam plus aminoglycoside combination therapy.30,31 Previous cases of spondylodiscitis have been treated successfully with diverse antimicrobial agents, including clindamycin, β-lactams, cephalosporins, fluoroquinolones, and aminoglycosides.14
Our patient improved rapidly while receiving empiric therapy with vancomycin and ceftriaxone and tolerated a rapid transition to oral amoxicillin and levofloxacin. This is the shortest IV treatment course for A urinae spondylodiscitis reported to date. We suspect that such rapid IV-to-oral transitions will suffice in most stable patients with A urinae spondylodiscitis or other invasive A urinae infections in line with the results of the OVIVA and POET trials.32,33
Conclusions
We believe A urinae UTI in the absence of obvious predisposing factors should prompt evaluation for urinary outflow obstruction. Despite improved laboratory diagnostic techniques, spondylodiscitis related to A urinae remains a rare entity and thus definitive treatment recommendations are difficult to make. However, we suspect that in many cases it is reasonable to extrapolate from the results of the POET and OVIVA trials and rapidly transition therapy of A urinae spondylodiscitis from IV to oral antibiotics. We suspect a review of the US Department of Veterans Affairs population might uncover a higher incidence of A urinae infection than previously estimated due to the population demographics and the epidemiology of A urinae.
Aerococcus urinae (A urinae), a gram-positive coccus readily mistaken for a Staphylococcus species, was first identified in 1992.1-3 It now reportedly accounts for 0.2% to 0.8% of clinical urine isolates.4-6 A urinae bacteriuria is typically asymptomatic and mainly occurs in women.7-9 Symptomatic A urinae urinary tract infection (UTI) occurs predominantly in older men with underlying urologic abnormalities.4-10
Serious A urinae infections are rare. The first 2 reported cases involved men with A urinae endocarditis, one of whom died.11,12 To date, only 8 cases of spondylodiscitis due to A urinae have been reported.13-20 Optimal treatment for invasive A urinae infection is undefined; however, the reported cases were treated successfully with diverse antibiotic regimen combinations; all including a β-lactam and beginning with at least 2 weeks of IV antibiotics.13-20 We describe a man with A urinae bacteremia and spondylodiscitis, presumably arising from a urinary source in the setting of bladder outlet obstruction, who was treated successfully.
Case Presentation
A 74-year-old man with morbid obesity, type 2 diabetes mellitus, stage 2 chronic kidney disease, and tobacco use presented to the emergency department after 2 weeks of progressive, nonradiating, midthoracic back pain, lower extremity weakness, gait imbalance, fatigue, anorexia, rigors, and subjective fevers. On presentation, he was afebrile and hemodynamically stable. A physical examination revealed point tenderness of the midthoracic vertebrae, nontender costovertebral angles, diffusely decreased strength, nonsustained clonus in both lower extremities, inguinal intertrigo, and a buried penis with purulent meatal discharge.
Laboratory results indicated a white blood cell (WBC) count of 13.5 K/μL (reference range, 4.0-11.0), absolute neutrophil count of 11.48 K/μL (reference range, 2.0-7.7), C-reactive protein (CRP) level of 225.3 mg/L (reference range, ≤ 5.0), erythrocyte sedimentation rate of 85 mm/h (reference range, 5-15), serum blood urea nitrogen of 76 mg/dL (reference range, 8-26), and serum creatinine (SCr) of 1.9 mg/dL (reference range, 1.1-1.4). A urinalysis showed positive leukocyte esterase, WBC clumps, and little bacteria. Abdominal/pelvic computed tomography showed spondylodiscitis-like changes at T7-T8, bilateral perinephric fat stranding, bladder distension, and bladder wall thickening.
The patient was presumed to have discitis secondary to a UTI, with possible pyelonephritis, and was given empiric vancomycin and ceftriaxone. Spinal magnetic resonance imaging with contrast supported spondylodiscitis at T7-T8, extending to T8-T9. Preliminary results from the admission blood and urine cultures showed gram-positive cocci in clusters, which were presumed initially to be Staphylococcus aureus (S aureus).
The final urine culture report listed multiple organisms, predominantly A urinae (Table 1);
On hospital day 6, the patient’s back pain had resolved, micturition was normal, appetite had normalized, and SCr was minimally above baseline (1.4 mg/dL). He insisted on completing antibiotic treatment at home and had no other medical indication for continued hospitalization. Thus, antibiotic therapy was changed to an all-oral regimen of amoxicillin 1 g 3 times daily for 10 days and levofloxacin 750 mg daily for 6 weeks, and the patient was discharged to home.
The patient returned 5 days postdischarge due to anuria. Investigation showed severe acute kidney injury (SCr, 6.8 mg/dL) and bladder outlet obstruction due to phimosis and urethral meatal stenosis. Urinalysis was unremarkable. His CRP had declined from 225 mg/L (initial admission) to 154 mg/L. A urinae culture and 2 sets of blood cultures were finalized as no growth. He was diagnosed with postrenal acute kidney injury and underwent meatal dilation and Foley catheterization but declined surgical correction. When seen in the clinic 2 months postantimicrobial therapy, the patient had normal micturition, no symptoms or signs of infection, and steadily down-trending inflammatory markers.
Discussion
A urinae, historically considered a rare pathogen, has been identified with increasing frequency in urine cultures due to improved microbiologic diagnostic techniques. However, there are only 8 reported cases of A urinae spondylodiscitis. Urinary pathology is an accepted risk factor for A urinae infections; consequently, we suspect that our patient’s urinary outflow obstruction and poor genitourinary hygiene were related to his invasive A urinae infection.10,21,22 We surmise that he had a chronic urinary outflow obstruction contributing to his infection, as evidenced by imaging findings, while the phimosis and urethral meatal stenosis were most likely infectious sequelae considering his anuria and acute kidney injury 5 days postdischarge. Indeed, the correlation between A urinae and urinary tract pathology may justify an evaluation for urinary pathology in any man with A urinae infection, regardless of the presence of symptoms.
By contrast, the implications of A urinae bacteriuria remain unclear. From a public health perspective, A urinae bacteriuria is rare, but the infectious mechanism remains undetermined with a case report suggesting the possibility of sexual transmission.4-6,23 In our case, the patient was not sexually active and had no clear origin of infection. Considering the potential severity of infection, more studies are needed to determine the infectious mechanism of A urinae.
In terms of infectious morbidity, the results seem to vary by sex. In a retrospective study of about 30,000 clinical urine samples, 62 (58 from women, 4 from men) yielded A urinae. The 62 corresponding patients lacked systemic infectious complications, leading the authors to conclude that A urinae is a relatively avirulent organism.24 Although possibly true in women, we are wary of drawing conclusions, especially regarding men, from a study that included only 62 urine samples were A urinae–positive, with only 4 from men. More evidence is needed to define the prognostic implications of A urinae bacteriuria in men.
As illustrated by the present case and previous reports, severe A urinae infections can occur, and the contributory factors deserve consideration. In our patient, the actual mechanism for bacteremia remains unclear. The initial concern for acute pyelonephritis was prompted by a computed tomography finding of bilateral perinephric fat stranding. This finding was questioned because it is common in older patients without infection, hence, is highly nonspecific. A correlation with urinary outflow obstruction may be an important clue in cases like this one.25,26
Furthermore, whether the urinary tract truly was the source of the patient’s bacteremia is clouded by the differing antimicrobial susceptibility patterns of the A urinae blood and urine isolates. The simplest explanation for this discordance may be that all the isolates shared a common initial origin but adapted to different environments in the host (perhaps over time) or laboratory, producing phenotypic variation. Alternatively, the infection could have been polyclonal from the onset, with sampling error leading to the differing detected susceptibility patterns, or the blood and urine isolates may have represented independent acquisition events, involving distinct A urinae strains. Unfortunately (from an academic perspective), given patient preferences and recommendations from the infectious disease consultant, no bone biopsy was done for histology and culture to confirm infection and to allow comparative strain identification if A urinae was isolated.
Optimal treatment for A urinae spondylodiscitis has yet to be established. β-lactams have shown good clinical efficacy despite being bacteriostatic in vitro.27 Early in vitro studies showed synergistic bactericidal synergistic activity with penicillin plus aminoglycoside combination therapies.27-30 Cases of endocarditis have been successfully treated mainly with the combination of a β-lactam plus aminoglycoside combination therapy.30,31 Previous cases of spondylodiscitis have been treated successfully with diverse antimicrobial agents, including clindamycin, β-lactams, cephalosporins, fluoroquinolones, and aminoglycosides.14
Our patient improved rapidly while receiving empiric therapy with vancomycin and ceftriaxone and tolerated a rapid transition to oral amoxicillin and levofloxacin. This is the shortest IV treatment course for A urinae spondylodiscitis reported to date. We suspect that such rapid IV-to-oral transitions will suffice in most stable patients with A urinae spondylodiscitis or other invasive A urinae infections in line with the results of the OVIVA and POET trials.32,33
Conclusions
We believe A urinae UTI in the absence of obvious predisposing factors should prompt evaluation for urinary outflow obstruction. Despite improved laboratory diagnostic techniques, spondylodiscitis related to A urinae remains a rare entity and thus definitive treatment recommendations are difficult to make. However, we suspect that in many cases it is reasonable to extrapolate from the results of the POET and OVIVA trials and rapidly transition therapy of A urinae spondylodiscitis from IV to oral antibiotics. We suspect a review of the US Department of Veterans Affairs population might uncover a higher incidence of A urinae infection than previously estimated due to the population demographics and the epidemiology of A urinae.
1. Christensen JJ, Korner B, Kjaergaard H. Aerococcus-like organism—an unnoticed urinary tract pathogen. APMIS. 1989;97(6):539-546. doi:10.1111/j.1699-0463.1989.tb00828.x
2. Aguirre M, Collins MD. Phylogenetic analysis of some Aerococcus-like organisms from urinary tract infections: description of Aerococcus urinae sp. nov. J Gen Microbiol. 1992;138(2):401-405. doi:10.1099/00221287-138-2-401
3. Williams RE, Hirch A, Cowan ST. Aerococcus, a new bacterial genus. J Gen Microbiol. 1953;8(3):475-480. doi:10.1099/00221287-8-3-475
4. Kline KA, Lewis AL. Gram-positive uropathogens, polymicrobial urinary tract infection, and the emerging microbiota of the urinary tract. Microbiol Spectr. 2016;4(2). doi:10.1128/microbiolspec.UTI-0012-2012
5. Schuur PM, Kasteren ME, Sabbe L, Vos MC, Janssens MM, Buiting AG. Urinary tract infections with Aerococcus urinae in the south of The Netherlands. Eur J Clin Microbiol Infect Dis. 1997;16(12):871-875. doi:10.1007/BF01700552
6. Grude N, Tveten Y. Aerococcus urinae og urinveisinfeksjon [Aerococcus urinae and urinary tract infection]. Tidsskr Nor Laegeforen. 2002;122(2):174-175.
7. Narayanasamy S, King K, Dennison A, Spelman DW, Aung AK. Clinical characteristics and laboratory identification of Aerococcus infections: an Australian tertiary centre perspective. Int J Microbiol. 2017;2017. doi:10.1155/2017/5684614
8. Hilt EE, McKinley K, Pearce MM, et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol. 2014;52(3):871-876. doi:10.1128/JCM.02876-13
9. Pearce MM, Hilt EE, Rosenfeld AB, et al. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. mBio. 2014;5(4):e01283-14. doi:10.1128/mBio.01283-14
10. Sahu KK, Lal A, Mishra AK, Abraham GM. Aerococcus-related infections and their significance: a 9-year retrospective study. J Microsc Ultrastruct. 2021;9(1):18-25. doi:10.4103/JMAU.JMAU_61_19
11. Skov RL, Klarlund M, Thorsen S. Fatal endocarditis due to Aerococcus urinae. Diagn Microbiol Infect Dis. 1995;21(4):219-221. doi:10.1016/0732-8893(95)00037-b
12. Kristensen B, Nielsen G. Endocarditis caused by Aerococcus urinae, a newly recognized pathogen. Eur J Clin Microbiol Infect Dis. 1995;14(1):49-51. doi:10.1007/BF02112619
13. Astudillo L, Sailler L, Porte L, Lefevre JC, Massip P, Arlet-Suau E. Spondylodiscitis due to Aerococcus urinae: a first report. Scand J Infect Dis. 2003;35(11-12):890-891. doi:10.1080/00365540310016664
14. Lyagoubi A, Souffi C, Baroiller V, Vallee E. Spondylodiscitis: an increasingly described localization. EJIFCC. 2020;31(2):169-173.
15. Jerome M, Slim J, Sison R, Marton R. A case of Aerococcus urinae vertebral osteomyelitis. J Glob Infect Dis. 2015;7(2):85-86. doi:10.4103/0974-777X.157246
16. Tekin A, Tekin G, Turunç T, Demiroğlu Z, Kizilkiliç O. Infective endocarditis and spondylodiscitis in a patient due to Aerococcus urinae: first report. Int J Cardiol. 2007;115(3):402-403. doi:10.1016/j.ijcard.2006.01.046
17. Rougier E, Braud A, Argemi X, et al. Spondylodiscitis due to Aerococcus urinae and literature review. Infection. 2018;46(3):419-421. doi:10.1007/s15010-017-1106-0
18. Degroote E, Yildiz H, Lecouvet F, Verroken A, Belkhir L. Aerococcus urinae: an underestimated cause of spine infection? Case report and review of the literature. Acta Clin Belg. 2018;73(6):444-447. doi:10.1080/17843286.2018.1443003
19. Torres-Martos E, Pérez-Cortés S, Sánchez-Calvo JM, López-Prieto MD. Spondylodiscitis due to Aerococcus urinae infection in an elderly immunocompetent patient. Enferm Infecc Microbiol Clin. 2017;35(10):682-684. doi:10.1016/j.eimc.2017.02.005
20. Senneby E, Petersson AC, Rasmussen M. Clinical and microbiological features of bacteraemia with Aerococcus urinae. Clin Microbiol Infect. 2012;18(6):546-550. doi:10.1111/j.1469-0691.2011.03609.x
21. Sunnerhagen T, Nilson B, Olaison L, Rasmussen M. Clinical and microbiological features of infective endocarditis caused by aerococci. Infection. 2016;44(2):167-173. doi:10.1007/s15010-015-0812-8
22. de Jong MF, Soetekouw R, ten Kate RW, Veenendaal D. Aerococcus urinae: severe and fatal bloodstream infections and endocarditis. J Clin Microbiol. 2010;48(9):3445-3447. doi:10.1128/JCM.00835-10
23. Babaeer AA, Nader C, Iacoviello V, Tomera K. Necrotizing urethritis due to Aerococcus urinae. Case Rep Urol. 2015;2015:136147. doi:10.1155/2015/136147
24. Sierra-Hoffman M, Watkins K, Jinadatha C, Fader R, Carpenter JL. Clinical significance of Aerococcus urinae: a retrospective review. Diagn Microbiol Infect Dis. 2005;53(4):289-292. doi:10.1016/j.diagmicrobio.2005.06.021
25. Fukami H, Takeuchi Y, Kagaya S, et al. Perirenal fat stranding is not a powerful diagnostic tool for acute pyelonephritis. Int J Gen Med. 2017;10:137-144. doi:10.2147/IJGM.S133685
26. Han NY, Sung DJ, Kim MJ, Park BJ, Sim KC, Cho SB. Perirenal fat stranding on CT: is there an association with bladder outlet obstruction? Br J Radiol. 2016;89(1063):20160195. doi:10.1259/bjr.20160195
27. Hirzel C, Hirzberger L, Furrer H, Endimiani A. Bactericidal activity of penicillin, ceftriaxone, gentamicin and daptomycin alone and in combination against Aerococcus urinae. Int J Antimicrob Agents. 2016;48(3):271-276. doi:10.1016/j.ijantimicag.2016.05.007
28. Zbinden R, Santanam P, Hunziker L, Leuzinger B, von Graevenitz A. Endocarditis due to Aerococcus urinae: diagnostic tests, fatty acid composition and killing kinetics. Infection. 1999;27(2):122-124. doi:10.1007/BF02560511
29. Skov R, Christensen JJ, Korner B, Frimodt-Møller N, Espersen F. In vitro antimicrobial susceptibility of Aerococcus urinae to 14 antibiotics, and time-kill curves for penicillin, gentamicin and vancomycin. J Antimicrob Chemother. 2001;48(5):653-658. doi:10.1093/jac/48.5.653
30. Ebnöther C, Altwegg M, Gottschalk J, Seebach JD, Kronenberg A. Aerococcus urinae endocarditis: case report and review of the literature. Infection. 2002;30(5):310-313. doi:10.1007/s15010-002-3106-x
31. Tai DBG, Go JR, Fida M, Saleh OA. Management and treatment of Aerococcus bacteremia and endocarditis. Int J Infect Dis. 2021;102:584-589. doi:10.1016/j.ijid.2020.10.096
32. Li H-K, Rombach I, Zambellas R, et al; OVIVA Trial Collaborators. Oral versus intravenous antibiotics for bone and joint infection. N Engl J Med. 2019;380(5):425-436. doi:10.1056/NEJMoa1710926
33. Iversen K, Ihlemann N, Gill SU, et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Engl J Med. 2019;380(5):415-424. doi:10.1056/NEJMoa1808312
1. Christensen JJ, Korner B, Kjaergaard H. Aerococcus-like organism—an unnoticed urinary tract pathogen. APMIS. 1989;97(6):539-546. doi:10.1111/j.1699-0463.1989.tb00828.x
2. Aguirre M, Collins MD. Phylogenetic analysis of some Aerococcus-like organisms from urinary tract infections: description of Aerococcus urinae sp. nov. J Gen Microbiol. 1992;138(2):401-405. doi:10.1099/00221287-138-2-401
3. Williams RE, Hirch A, Cowan ST. Aerococcus, a new bacterial genus. J Gen Microbiol. 1953;8(3):475-480. doi:10.1099/00221287-8-3-475
4. Kline KA, Lewis AL. Gram-positive uropathogens, polymicrobial urinary tract infection, and the emerging microbiota of the urinary tract. Microbiol Spectr. 2016;4(2). doi:10.1128/microbiolspec.UTI-0012-2012
5. Schuur PM, Kasteren ME, Sabbe L, Vos MC, Janssens MM, Buiting AG. Urinary tract infections with Aerococcus urinae in the south of The Netherlands. Eur J Clin Microbiol Infect Dis. 1997;16(12):871-875. doi:10.1007/BF01700552
6. Grude N, Tveten Y. Aerococcus urinae og urinveisinfeksjon [Aerococcus urinae and urinary tract infection]. Tidsskr Nor Laegeforen. 2002;122(2):174-175.
7. Narayanasamy S, King K, Dennison A, Spelman DW, Aung AK. Clinical characteristics and laboratory identification of Aerococcus infections: an Australian tertiary centre perspective. Int J Microbiol. 2017;2017. doi:10.1155/2017/5684614
8. Hilt EE, McKinley K, Pearce MM, et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol. 2014;52(3):871-876. doi:10.1128/JCM.02876-13
9. Pearce MM, Hilt EE, Rosenfeld AB, et al. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. mBio. 2014;5(4):e01283-14. doi:10.1128/mBio.01283-14
10. Sahu KK, Lal A, Mishra AK, Abraham GM. Aerococcus-related infections and their significance: a 9-year retrospective study. J Microsc Ultrastruct. 2021;9(1):18-25. doi:10.4103/JMAU.JMAU_61_19
11. Skov RL, Klarlund M, Thorsen S. Fatal endocarditis due to Aerococcus urinae. Diagn Microbiol Infect Dis. 1995;21(4):219-221. doi:10.1016/0732-8893(95)00037-b
12. Kristensen B, Nielsen G. Endocarditis caused by Aerococcus urinae, a newly recognized pathogen. Eur J Clin Microbiol Infect Dis. 1995;14(1):49-51. doi:10.1007/BF02112619
13. Astudillo L, Sailler L, Porte L, Lefevre JC, Massip P, Arlet-Suau E. Spondylodiscitis due to Aerococcus urinae: a first report. Scand J Infect Dis. 2003;35(11-12):890-891. doi:10.1080/00365540310016664
14. Lyagoubi A, Souffi C, Baroiller V, Vallee E. Spondylodiscitis: an increasingly described localization. EJIFCC. 2020;31(2):169-173.
15. Jerome M, Slim J, Sison R, Marton R. A case of Aerococcus urinae vertebral osteomyelitis. J Glob Infect Dis. 2015;7(2):85-86. doi:10.4103/0974-777X.157246
16. Tekin A, Tekin G, Turunç T, Demiroğlu Z, Kizilkiliç O. Infective endocarditis and spondylodiscitis in a patient due to Aerococcus urinae: first report. Int J Cardiol. 2007;115(3):402-403. doi:10.1016/j.ijcard.2006.01.046
17. Rougier E, Braud A, Argemi X, et al. Spondylodiscitis due to Aerococcus urinae and literature review. Infection. 2018;46(3):419-421. doi:10.1007/s15010-017-1106-0
18. Degroote E, Yildiz H, Lecouvet F, Verroken A, Belkhir L. Aerococcus urinae: an underestimated cause of spine infection? Case report and review of the literature. Acta Clin Belg. 2018;73(6):444-447. doi:10.1080/17843286.2018.1443003
19. Torres-Martos E, Pérez-Cortés S, Sánchez-Calvo JM, López-Prieto MD. Spondylodiscitis due to Aerococcus urinae infection in an elderly immunocompetent patient. Enferm Infecc Microbiol Clin. 2017;35(10):682-684. doi:10.1016/j.eimc.2017.02.005
20. Senneby E, Petersson AC, Rasmussen M. Clinical and microbiological features of bacteraemia with Aerococcus urinae. Clin Microbiol Infect. 2012;18(6):546-550. doi:10.1111/j.1469-0691.2011.03609.x
21. Sunnerhagen T, Nilson B, Olaison L, Rasmussen M. Clinical and microbiological features of infective endocarditis caused by aerococci. Infection. 2016;44(2):167-173. doi:10.1007/s15010-015-0812-8
22. de Jong MF, Soetekouw R, ten Kate RW, Veenendaal D. Aerococcus urinae: severe and fatal bloodstream infections and endocarditis. J Clin Microbiol. 2010;48(9):3445-3447. doi:10.1128/JCM.00835-10
23. Babaeer AA, Nader C, Iacoviello V, Tomera K. Necrotizing urethritis due to Aerococcus urinae. Case Rep Urol. 2015;2015:136147. doi:10.1155/2015/136147
24. Sierra-Hoffman M, Watkins K, Jinadatha C, Fader R, Carpenter JL. Clinical significance of Aerococcus urinae: a retrospective review. Diagn Microbiol Infect Dis. 2005;53(4):289-292. doi:10.1016/j.diagmicrobio.2005.06.021
25. Fukami H, Takeuchi Y, Kagaya S, et al. Perirenal fat stranding is not a powerful diagnostic tool for acute pyelonephritis. Int J Gen Med. 2017;10:137-144. doi:10.2147/IJGM.S133685
26. Han NY, Sung DJ, Kim MJ, Park BJ, Sim KC, Cho SB. Perirenal fat stranding on CT: is there an association with bladder outlet obstruction? Br J Radiol. 2016;89(1063):20160195. doi:10.1259/bjr.20160195
27. Hirzel C, Hirzberger L, Furrer H, Endimiani A. Bactericidal activity of penicillin, ceftriaxone, gentamicin and daptomycin alone and in combination against Aerococcus urinae. Int J Antimicrob Agents. 2016;48(3):271-276. doi:10.1016/j.ijantimicag.2016.05.007
28. Zbinden R, Santanam P, Hunziker L, Leuzinger B, von Graevenitz A. Endocarditis due to Aerococcus urinae: diagnostic tests, fatty acid composition and killing kinetics. Infection. 1999;27(2):122-124. doi:10.1007/BF02560511
29. Skov R, Christensen JJ, Korner B, Frimodt-Møller N, Espersen F. In vitro antimicrobial susceptibility of Aerococcus urinae to 14 antibiotics, and time-kill curves for penicillin, gentamicin and vancomycin. J Antimicrob Chemother. 2001;48(5):653-658. doi:10.1093/jac/48.5.653
30. Ebnöther C, Altwegg M, Gottschalk J, Seebach JD, Kronenberg A. Aerococcus urinae endocarditis: case report and review of the literature. Infection. 2002;30(5):310-313. doi:10.1007/s15010-002-3106-x
31. Tai DBG, Go JR, Fida M, Saleh OA. Management and treatment of Aerococcus bacteremia and endocarditis. Int J Infect Dis. 2021;102:584-589. doi:10.1016/j.ijid.2020.10.096
32. Li H-K, Rombach I, Zambellas R, et al; OVIVA Trial Collaborators. Oral versus intravenous antibiotics for bone and joint infection. N Engl J Med. 2019;380(5):425-436. doi:10.1056/NEJMoa1710926
33. Iversen K, Ihlemann N, Gill SU, et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Engl J Med. 2019;380(5):415-424. doi:10.1056/NEJMoa1808312