User login
Benign Pneumatosis Intestinalis: A Case Report and Review of the Literature
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
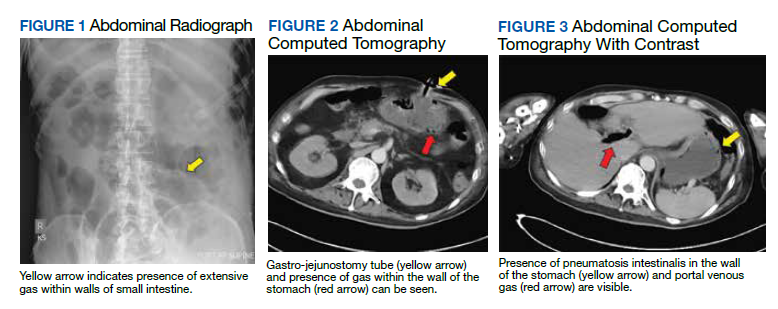
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011
62-year-old woman • dysuria • dyspareunia • urinary incontinence • Dx?
THE CASE
A 62-year-old postmenopausal woman presented to the clinic as a new patient for her annual physical examination. She reported a 9-year history of symptoms including dysuria, post-void dribbling, dyspareunia, and urinary incontinence on review of systems. Her physical examination revealed an anterior vaginal wall bulge (FIGURE). Results of a urinalysis were negative. The patient was referred to Urology for further evaluation.

THE DIAGNOSIS
A pelvic magnetic resonance imaging (MRI) scan revealed a large periurethral diverticulum with a horseshoe shape.
DISCUSSION
Women are more likely than men to develop urethral diverticulum, and it can manifest at any age, usually in the third through seventh decade.4,5 It was once thought to be more common in Black women, although the literature does not support this.6 Black women are 3 times more likely to be operated on than White women to treat urethral diverticula.7
Unknown origin. Most cases of urethral diverticulum are acquired; the etiology is uncertain.8,9 The assumption is that urethral diverticulum occurs as a result of repeated infection of the periurethral glands with subsequent obstruction, abscess formation, and chronic inflammation.1,2,4 Childbirth trauma, iatrogenic causes, and urethral instrumentation have also been implicated.3,4 In rare cases of congenital urethral diverticula, the diverticula are thought to be remnants of Gartner duct cysts, and yet, incidence in the pediatric population is low.8
Diagnosis is confirmed through physical exam and imaging
The urethral diverticulum manifests anteriorly and palpation of the anterior vaginal wall may reveal a painful mass.10 A split-speculum is used for careful inspection and palpation of the anterior vaginal wall.9 If the diverticulum is found to be firm on palpation, or there is bloody urethral drainage, malignancy (although rare) must be ruled out.4,5 Refer such patients to a urologist or urogynecologist.
Radiologic imaging (eg, ultrasound,
Continue to: Nonspecific symptoms may lead to misdiagnosis
Nonspecific symptoms may lead to misdiagnosis. The symptoms associated with urethral diverticulum are diverse and linked to several differential diagnoses (TABLE).3,4,12 The most common signs and symptoms are pelvic pain, urethral mass, dyspareunia, dysuria, urinary incontinence, and post-void dribbling—all of which are considered nonspecific.3,10,11 These nonspecific symptoms (or even an absence of symptoms), along with a physician’s lack of familiarity with urethral diverticulum, can result in a misdiagnosis or even a delayed diagnosis (up to 5.2 years).3,10

Managing symptoms vs preventing recurrence
Conservative management with antibiotics, anticholinergics, and/or observation is acceptable for patients with mild symptoms and those who are pregnant or who have a current infection or serious comorbidities that preclude surgery.3,9 Complete excision of the urethral diverticulum with reconstruction is considered the most effective surgical management for symptom relief and recurrence prevention.3,4,11,14
Our patient underwent a successful transvaginal suburethral diverticulectomy.
THE TAKEAWAY
The diagnosis of female urethral diverticulum is often delayed or misdiagnosed because symptoms are diverse and nonspecific. One should have a high degree of suspicion for urethral diverticulum in patients with dysuria, dyspareunia, pelvic pain, urinary incontinence, and irritative voiding symptoms who are not responding to conservative management. Ultrasound is an appropriate first-line imaging modality. However, a pelvic MRI is the most sensitive and specific in diagnosing urethral diverticulum.12
CORRESPONDENCE
Folashade Omole, MD, FAAFP, 720 Westview Drive, Atlanta, GA 30310; [email protected]
1. Billow M, James R, Resnick K, et al. An unusual presentation of a urethral diverticulum as a vaginal wall mass: a case report. J Med Case Rep. 2013;7:171. doi: 10.1186/1752-1947-7-171
2. El-Nashar SA, Bacon MM, Kim-Fine S, et al. Incidence of female urethral diverticulum: a population-based analysis and literature review. Int Urogynecol J. 2014;25:73-79. doi: 10.1007/s00192-013-2155-2
3. Cameron AP. Urethral diverticulum in the female: a meta-analysis of modern series. Minerva Ginecol. 2016;68:186-210.
4. Greiman AK, Rolef J, Rovner ES. Urethral diverticulum: a systematic review. Arab J Urol. 2019;17:49-57. doi: 10.1080/2090598X.2019.1589748
5. Allen D, Mishra V, Pepper W, et al. A single-center experience of symptomatic male urethral diverticula. Urology. 2007;70:650-653. doi: 10.1016/j.urology.2007.06.1111
6. O’Connor E, Iatropoulou D, Hashimoto S, et al. Urethral diverticulum carcinoma in females—a case series and review of the English and Japanese literature. Transl Androl Urol. 2018;7:703-729. doi: 10.21037/tau.2018.07.08
7. Burrows LJ, Howden NL, Meyn L, et al. Surgical procedures for urethral diverticula in women in the United States, 1979-1997. Int Urogynecol J Pelvic Floor Dysfunct. 2005;16:158-161. doi: 10.1007/s00192-004-1145-9
8. Riyach O, Ahsaini M, Tazi MF, et al. Female urethral diverticulum: cases report and literature. Ann Surg Innov Res. 2014;8:1. doi: 10.1186/1750-1164-8-1
9. Antosh DD, Gutman RE. Diagnosis and management of female urethral diverticulum. Female Pelvic Med Reconstr Surg. 2011;17:264-271. doi: 10.1097/SPV.0b013e318234a242
10. Romanzi LJ, Groutz A, Blaivas JG. Urethral diverticulum in women: diverse presentations resulting in diagnostic delay and mismanagement. J Urol. 2000;164:428-433.
11. Reeves FA, Inman RD, Chapple CR. Management of symptomatic urethral diverticula in women: a single-centre experience. Eur Urol. 2014;66:164-172. doi: 10.1016/j.eururo.2014.02.041
12. Dwarkasing RS, Dinkelaar W, Hop WCJ, et al. MRI evaluation of urethral diverticula and differential diagnosis in symptomatic women. AJR Am J Roentgenol. 2011;197:676-682. doi: 10.2214/AJR.10.6144
13. Porten S, Kielb S. Diagnosis of female diverticula using magnetic resonance imaging. Adv Urol. 2008;2008:213516. doi: 10.1155/2008/213516
14. Ockrim JL, Allen DJ, Shah PJ, et al. A tertiary experience of urethral diverticulectomy: diagnosis, imaging and surgical outcomes. BJU Int. 2009;103:1550-1554. doi: 10.1111/j.1464-410X.2009.08348.x
THE CASE
A 62-year-old postmenopausal woman presented to the clinic as a new patient for her annual physical examination. She reported a 9-year history of symptoms including dysuria, post-void dribbling, dyspareunia, and urinary incontinence on review of systems. Her physical examination revealed an anterior vaginal wall bulge (FIGURE). Results of a urinalysis were negative. The patient was referred to Urology for further evaluation.
THE DIAGNOSIS
A pelvic magnetic resonance imaging (MRI) scan revealed a large periurethral diverticulum with a horseshoe shape.
DISCUSSION
Women are more likely than men to develop urethral diverticulum, and it can manifest at any age, usually in the third through seventh decade.4,5 It was once thought to be more common in Black women, although the literature does not support this.6 Black women are 3 times more likely to be operated on than White women to treat urethral diverticula.7
Unknown origin. Most cases of urethral diverticulum are acquired; the etiology is uncertain.8,9 The assumption is that urethral diverticulum occurs as a result of repeated infection of the periurethral glands with subsequent obstruction, abscess formation, and chronic inflammation.1,2,4 Childbirth trauma, iatrogenic causes, and urethral instrumentation have also been implicated.3,4 In rare cases of congenital urethral diverticula, the diverticula are thought to be remnants of Gartner duct cysts, and yet, incidence in the pediatric population is low.8
Diagnosis is confirmed through physical exam and imaging
The urethral diverticulum manifests anteriorly and palpation of the anterior vaginal wall may reveal a painful mass.10 A split-speculum is used for careful inspection and palpation of the anterior vaginal wall.9 If the diverticulum is found to be firm on palpation, or there is bloody urethral drainage, malignancy (although rare) must be ruled out.4,5 Refer such patients to a urologist or urogynecologist.
Radiologic imaging (eg, ultrasound,
Continue to: Nonspecific symptoms may lead to misdiagnosis
Nonspecific symptoms may lead to misdiagnosis. The symptoms associated with urethral diverticulum are diverse and linked to several differential diagnoses (TABLE).3,4,12 The most common signs and symptoms are pelvic pain, urethral mass, dyspareunia, dysuria, urinary incontinence, and post-void dribbling—all of which are considered nonspecific.3,10,11 These nonspecific symptoms (or even an absence of symptoms), along with a physician’s lack of familiarity with urethral diverticulum, can result in a misdiagnosis or even a delayed diagnosis (up to 5.2 years).3,10
Managing symptoms vs preventing recurrence
Conservative management with antibiotics, anticholinergics, and/or observation is acceptable for patients with mild symptoms and those who are pregnant or who have a current infection or serious comorbidities that preclude surgery.3,9 Complete excision of the urethral diverticulum with reconstruction is considered the most effective surgical management for symptom relief and recurrence prevention.3,4,11,14
Our patient underwent a successful transvaginal suburethral diverticulectomy.
THE TAKEAWAY
The diagnosis of female urethral diverticulum is often delayed or misdiagnosed because symptoms are diverse and nonspecific. One should have a high degree of suspicion for urethral diverticulum in patients with dysuria, dyspareunia, pelvic pain, urinary incontinence, and irritative voiding symptoms who are not responding to conservative management. Ultrasound is an appropriate first-line imaging modality. However, a pelvic MRI is the most sensitive and specific in diagnosing urethral diverticulum.12
CORRESPONDENCE
Folashade Omole, MD, FAAFP, 720 Westview Drive, Atlanta, GA 30310; [email protected]
THE CASE
A 62-year-old postmenopausal woman presented to the clinic as a new patient for her annual physical examination. She reported a 9-year history of symptoms including dysuria, post-void dribbling, dyspareunia, and urinary incontinence on review of systems. Her physical examination revealed an anterior vaginal wall bulge (FIGURE). Results of a urinalysis were negative. The patient was referred to Urology for further evaluation.
THE DIAGNOSIS
A pelvic magnetic resonance imaging (MRI) scan revealed a large periurethral diverticulum with a horseshoe shape.
DISCUSSION
Women are more likely than men to develop urethral diverticulum, and it can manifest at any age, usually in the third through seventh decade.4,5 It was once thought to be more common in Black women, although the literature does not support this.6 Black women are 3 times more likely to be operated on than White women to treat urethral diverticula.7
Unknown origin. Most cases of urethral diverticulum are acquired; the etiology is uncertain.8,9 The assumption is that urethral diverticulum occurs as a result of repeated infection of the periurethral glands with subsequent obstruction, abscess formation, and chronic inflammation.1,2,4 Childbirth trauma, iatrogenic causes, and urethral instrumentation have also been implicated.3,4 In rare cases of congenital urethral diverticula, the diverticula are thought to be remnants of Gartner duct cysts, and yet, incidence in the pediatric population is low.8
Diagnosis is confirmed through physical exam and imaging
The urethral diverticulum manifests anteriorly and palpation of the anterior vaginal wall may reveal a painful mass.10 A split-speculum is used for careful inspection and palpation of the anterior vaginal wall.9 If the diverticulum is found to be firm on palpation, or there is bloody urethral drainage, malignancy (although rare) must be ruled out.4,5 Refer such patients to a urologist or urogynecologist.
Radiologic imaging (eg, ultrasound,
Continue to: Nonspecific symptoms may lead to misdiagnosis
Nonspecific symptoms may lead to misdiagnosis. The symptoms associated with urethral diverticulum are diverse and linked to several differential diagnoses (TABLE).3,4,12 The most common signs and symptoms are pelvic pain, urethral mass, dyspareunia, dysuria, urinary incontinence, and post-void dribbling—all of which are considered nonspecific.3,10,11 These nonspecific symptoms (or even an absence of symptoms), along with a physician’s lack of familiarity with urethral diverticulum, can result in a misdiagnosis or even a delayed diagnosis (up to 5.2 years).3,10
Managing symptoms vs preventing recurrence
Conservative management with antibiotics, anticholinergics, and/or observation is acceptable for patients with mild symptoms and those who are pregnant or who have a current infection or serious comorbidities that preclude surgery.3,9 Complete excision of the urethral diverticulum with reconstruction is considered the most effective surgical management for symptom relief and recurrence prevention.3,4,11,14
Our patient underwent a successful transvaginal suburethral diverticulectomy.
THE TAKEAWAY
The diagnosis of female urethral diverticulum is often delayed or misdiagnosed because symptoms are diverse and nonspecific. One should have a high degree of suspicion for urethral diverticulum in patients with dysuria, dyspareunia, pelvic pain, urinary incontinence, and irritative voiding symptoms who are not responding to conservative management. Ultrasound is an appropriate first-line imaging modality. However, a pelvic MRI is the most sensitive and specific in diagnosing urethral diverticulum.12
CORRESPONDENCE
Folashade Omole, MD, FAAFP, 720 Westview Drive, Atlanta, GA 30310; [email protected]
1. Billow M, James R, Resnick K, et al. An unusual presentation of a urethral diverticulum as a vaginal wall mass: a case report. J Med Case Rep. 2013;7:171. doi: 10.1186/1752-1947-7-171
2. El-Nashar SA, Bacon MM, Kim-Fine S, et al. Incidence of female urethral diverticulum: a population-based analysis and literature review. Int Urogynecol J. 2014;25:73-79. doi: 10.1007/s00192-013-2155-2
3. Cameron AP. Urethral diverticulum in the female: a meta-analysis of modern series. Minerva Ginecol. 2016;68:186-210.
4. Greiman AK, Rolef J, Rovner ES. Urethral diverticulum: a systematic review. Arab J Urol. 2019;17:49-57. doi: 10.1080/2090598X.2019.1589748
5. Allen D, Mishra V, Pepper W, et al. A single-center experience of symptomatic male urethral diverticula. Urology. 2007;70:650-653. doi: 10.1016/j.urology.2007.06.1111
6. O’Connor E, Iatropoulou D, Hashimoto S, et al. Urethral diverticulum carcinoma in females—a case series and review of the English and Japanese literature. Transl Androl Urol. 2018;7:703-729. doi: 10.21037/tau.2018.07.08
7. Burrows LJ, Howden NL, Meyn L, et al. Surgical procedures for urethral diverticula in women in the United States, 1979-1997. Int Urogynecol J Pelvic Floor Dysfunct. 2005;16:158-161. doi: 10.1007/s00192-004-1145-9
8. Riyach O, Ahsaini M, Tazi MF, et al. Female urethral diverticulum: cases report and literature. Ann Surg Innov Res. 2014;8:1. doi: 10.1186/1750-1164-8-1
9. Antosh DD, Gutman RE. Diagnosis and management of female urethral diverticulum. Female Pelvic Med Reconstr Surg. 2011;17:264-271. doi: 10.1097/SPV.0b013e318234a242
10. Romanzi LJ, Groutz A, Blaivas JG. Urethral diverticulum in women: diverse presentations resulting in diagnostic delay and mismanagement. J Urol. 2000;164:428-433.
11. Reeves FA, Inman RD, Chapple CR. Management of symptomatic urethral diverticula in women: a single-centre experience. Eur Urol. 2014;66:164-172. doi: 10.1016/j.eururo.2014.02.041
12. Dwarkasing RS, Dinkelaar W, Hop WCJ, et al. MRI evaluation of urethral diverticula and differential diagnosis in symptomatic women. AJR Am J Roentgenol. 2011;197:676-682. doi: 10.2214/AJR.10.6144
13. Porten S, Kielb S. Diagnosis of female diverticula using magnetic resonance imaging. Adv Urol. 2008;2008:213516. doi: 10.1155/2008/213516
14. Ockrim JL, Allen DJ, Shah PJ, et al. A tertiary experience of urethral diverticulectomy: diagnosis, imaging and surgical outcomes. BJU Int. 2009;103:1550-1554. doi: 10.1111/j.1464-410X.2009.08348.x
1. Billow M, James R, Resnick K, et al. An unusual presentation of a urethral diverticulum as a vaginal wall mass: a case report. J Med Case Rep. 2013;7:171. doi: 10.1186/1752-1947-7-171
2. El-Nashar SA, Bacon MM, Kim-Fine S, et al. Incidence of female urethral diverticulum: a population-based analysis and literature review. Int Urogynecol J. 2014;25:73-79. doi: 10.1007/s00192-013-2155-2
3. Cameron AP. Urethral diverticulum in the female: a meta-analysis of modern series. Minerva Ginecol. 2016;68:186-210.
4. Greiman AK, Rolef J, Rovner ES. Urethral diverticulum: a systematic review. Arab J Urol. 2019;17:49-57. doi: 10.1080/2090598X.2019.1589748
5. Allen D, Mishra V, Pepper W, et al. A single-center experience of symptomatic male urethral diverticula. Urology. 2007;70:650-653. doi: 10.1016/j.urology.2007.06.1111
6. O’Connor E, Iatropoulou D, Hashimoto S, et al. Urethral diverticulum carcinoma in females—a case series and review of the English and Japanese literature. Transl Androl Urol. 2018;7:703-729. doi: 10.21037/tau.2018.07.08
7. Burrows LJ, Howden NL, Meyn L, et al. Surgical procedures for urethral diverticula in women in the United States, 1979-1997. Int Urogynecol J Pelvic Floor Dysfunct. 2005;16:158-161. doi: 10.1007/s00192-004-1145-9
8. Riyach O, Ahsaini M, Tazi MF, et al. Female urethral diverticulum: cases report and literature. Ann Surg Innov Res. 2014;8:1. doi: 10.1186/1750-1164-8-1
9. Antosh DD, Gutman RE. Diagnosis and management of female urethral diverticulum. Female Pelvic Med Reconstr Surg. 2011;17:264-271. doi: 10.1097/SPV.0b013e318234a242
10. Romanzi LJ, Groutz A, Blaivas JG. Urethral diverticulum in women: diverse presentations resulting in diagnostic delay and mismanagement. J Urol. 2000;164:428-433.
11. Reeves FA, Inman RD, Chapple CR. Management of symptomatic urethral diverticula in women: a single-centre experience. Eur Urol. 2014;66:164-172. doi: 10.1016/j.eururo.2014.02.041
12. Dwarkasing RS, Dinkelaar W, Hop WCJ, et al. MRI evaluation of urethral diverticula and differential diagnosis in symptomatic women. AJR Am J Roentgenol. 2011;197:676-682. doi: 10.2214/AJR.10.6144
13. Porten S, Kielb S. Diagnosis of female diverticula using magnetic resonance imaging. Adv Urol. 2008;2008:213516. doi: 10.1155/2008/213516
14. Ockrim JL, Allen DJ, Shah PJ, et al. A tertiary experience of urethral diverticulectomy: diagnosis, imaging and surgical outcomes. BJU Int. 2009;103:1550-1554. doi: 10.1111/j.1464-410X.2009.08348.x

Deployed Airbag Causes Bullous Reaction Following a Motor Vehicle Accident
Airbags are lifesaving during motor vehicle accidents (MVAs), but their deployment has been associated with skin issues such as irritant dermatitis1; lacerations2; abrasions3; and thermal, friction, and chemical burns.4-6 Ocular issues such as alkaline chemical keratitis7 and ocular alkali injuries8 also have been described.
Airbag deployment is triggered by rapid deceleration and impact, which ignite a sodium azide cartridge, causing the woven nylon bag to inflate with hydrocarbon gases.8 This leads to release of sodium hydroxide, sodium bicarbonate, and metallic oxides in an aerosolized form. If a tear in the meshwork of the airbag occurs, exposure to an even larger amount of powder containing caustic alkali chemicals can occur.8
We describe a patient who developed a bullous reaction to airbag contents after he was involved in an MVA in which the airbag deployed.
Case Report
A 35-year-old man with a history of type 2 diabetes mellitus and chronic hepatitis B presented to the dermatology clinic for an evaluation of new-onset blisters. The rash occurred 1 day after the patient was involved in an MVA in which he was exposed to the airbag’s contents after it burst. He had been evaluated twice in the emergency department for the skin eruption before being referred to dermatology. He noted the lesions were pruritic and painful. Prior treatments included silver sulfadiazine cream 1% and clobetasol cream 0.05%, though he discontinued using the latter because of burning with application. Physical examination revealed tense vesicles and bullae on an erythematous base on the right lower flank, forearms, and legs, with the exception of the lower right leg where a cast had been from a prior injury (Figure 1).

Two punch biopsies of the left arm were performed and sent for hematoxylin and eosin staining and direct immunofluorescence to rule out bullous diseases, such as bullous pemphigoid, linear IgA, and bullous lupus. Hematoxylin and eosin staining revealed extensive spongiosis with blister formation and a dense perivascular infiltrate in the superficial and mid dermis composed of lymphocytes with numerous scattered eosinophils (Figures 2 and 3). Direct immunofluorescence studies were negative. Treatment with oral prednisone and oral antihistamines was initiated.


At 10-day follow-up, the patient had a few residual bullae; most lesions were demonstrating various stages of healing (Figure 4). The patient’s cast had been removed, and there were no lesions in this previously covered area. At 6-week follow-up he had continued healing of the bullae and erosions as well as postinflammatory hyperpigmentation (Figure 5).


Comment
With the advent of airbags for safety purposes, these potentially lifesaving devices also have been known to cause injury.9 In 1998, the most commonly reported airbag injuries were ocular injuries.10 Cutaneous manifestations of airbag injury are less well known.11
Two cases of airbag deployment with skin blistering have been reported in the literature based on a PubMed search of articles indexed for MEDLINE using the terms airbag blistering or airbag bullae12,13; however, the blistering was described in the context of a burn. One case of the effects of airbag deployment residue highlights a patient arriving to the emergency department with erythema and blisters on the hands within 48 hours of airbag deployment in an MVA, and the treatment was standard burn therapy.12 Another case report described a patient with a second-degree burn with a 12-cm blister occurring on the radial side of the hand and distal forearm following an MVA and airbag deployment, which was treated conservatively.13 Cases of thermal burns, chemical burns, and irritant contact dermatitis after airbag deployment have been described in the literature.4-6,11,12,14,15 Our patient’s distal right lower leg was covered with a cast for osteomyelitis, and no blisters had developed in this area. It is likely that the transfer of airbag contents occurred during the process of unbuckling his seatbelt, which could explain the bullae that developed on the right flank. Per the Centers for Disease Control and Prevention, individuals should quickly remove clothing and wash their body with large amounts of soap and water following exposure to sodium azide.16
In 1989, the Federal Motor Vehicle Safety Standard No. 208 (occupant crash protection) became effective, stating all cars must have vehicle crash protection.12 Prior to 1993, it was reported that there had been no associated chemical injuries with airbag deployment. Subsequently, a 6-month retrospective study in 1993 showed that dermal injuries were found in connection with the presence of sodium hydroxide in automobile airbags.12 By 2004, it was known that airbags could cause chemical and thermal burns in addition to traumatic injuries from deployment.1 Since 2007, all motor vehicles have been required to have advanced airbags, which are engineered to sense the presence of passengers and determine if the airbag will deploy, and if so, how much to deploy to minimize airbag-related injury.3
The brand of car that our patient drove during the MVA is one with known airbag recalls due to safety defects; however, the year and actual model of the vehicle are not known, so specific information about the airbag in question is not available. It has been noted that some defective airbag inflators that were exposed to excess moisture during the manufacturing process could explode during deployment, causing shrapnel and airbag rupture, which has been linked to nearly 300 injuries worldwide.17
Conclusion
It is evident that the use of airbag devices reduces morbidity and mortality due to MVAs.9 It also had been reported that up to 96% of airbag-related injuries are relatively minor, which many would argue justifies their use.18 Furthermore, it has been reported that 99.8% of skin injuries following airbag deployment are minor.19 In the United States, it is mandated that every vehicle have functional airbags installed.8
This case highlights the potential for substantial airbag-induced skin reactions, specifically a bullous reaction, following airbag deployment. The persistent pruritus and lasting postinflammatory hyperpigmentation seen in this case were certainly worrisome for our patient. We also present this case to remind dermatology providers of possible treatment approaches to these skin reactions. Immediate cleansing of the affected areas of skin may help avoid such reactions.
- Corazza M, Trincone S, Zampino MR, et al. Air bags and the skin. Skinmed. 2004;3:256-258.
- Corazza M, Trincone S, Virgili A. Effects of airbag deployment: lesions, epidemiology, and management. Am J Clin Dermatol. 2004;5:295-300.
- Kuska TC. Air bag safety: an update. J Emerg Nurs. 2016;42:438-441.
- Ulrich D, Noah EM, Fuchs P, et al. Burn injuries caused by air bag deployment. Burns. 2001;27:196-199.
- Erpenbeck SP, Roy E, Ziembicki JA, et al. A systematic review on airbag-induced burns. J Burn Care Res. 2021;42:481-487.
- Skibba KEH, Cleveland CN, Bell DE. Airbag burns: an unfortunate consequence of motor vehicle safety. J Burn Care Res. 2021;42:71-73.
- Smally AJ, Binzer A, Dolin S, et al. Alkaline chemical keratitis: eye injury from airbags. Ann Emerg Med. 1992;21:1400-1402.
- Barnes SS, Wong W Jr, Affeldt JC. A case of severe airbag related ocular alkali injury. Hawaii J Med Public Health. 2012;71:229-231.
- Wallis LA, Greaves I. Injuries associated with airbag deployment. Emerg Med J. 2002;19:490-493.
- Mohamed AA, Banerjee A. Patterns of injury associated with automobile airbag use. Postgrad Med J. 1998;74:455-458.
- Foley E, Helm TN. Air bag injury and the dermatologist. Cutis. 2000;66:251-252.
- Swanson-Biearman B, Mrvos R, Dean BS, et al. Air bags: lifesaving with toxic potential? Am J Emerg Med. 1993;11:38-39.
- Roth T, Meredith P. Traumatic lesions caused by the “air-bag” system [in French]. Z Unfallchir Versicherungsmed. 1993;86:189-193.
- Wu JJ, Sanchez-Palacios C, Brieva J, et al. A case of air bag dermatitis. Arch Dermatol. 2002;138:1383-1384.
- Vitello W, Kim M, Johnson RM, et al. Full-thickness burn to the hand from an automobile airbag. J Burn Care Rehabil. 1999;20:212-215.
- Centers for Disease Control and Prevention. Facts about sodium azide. Updated April 4, 2018. Accessed May 15, 2022. https://emergency.cdc.gov/agent/sodiumazide/basics/facts.asp
- Shepardson D. Honda to recall 1.2 million vehicles in North America to replace Takata airbags. March 12, 2019. Accessed March 22, 2022. https://www.reuters.com/article/us-honda-takata-recall/honda-to-recall-1-2-million-vehicles-in-north-america-to-replace-takata-airbags-idUSKBN1QT1C9
- Gabauer DJ, Gabler HC. The effects of airbags and seatbelts on occupant injury in longitudinal barrier crashes. J Safety Res. 2010;41:9-15.
- Rath AL, Jernigan MV, Stitzel JD, et al. The effects of depowered airbags on skin injuries in frontal automobile crashes. Plast Reconstr Surg. 2005;115:428-435.
Airbags are lifesaving during motor vehicle accidents (MVAs), but their deployment has been associated with skin issues such as irritant dermatitis1; lacerations2; abrasions3; and thermal, friction, and chemical burns.4-6 Ocular issues such as alkaline chemical keratitis7 and ocular alkali injuries8 also have been described.
Airbag deployment is triggered by rapid deceleration and impact, which ignite a sodium azide cartridge, causing the woven nylon bag to inflate with hydrocarbon gases.8 This leads to release of sodium hydroxide, sodium bicarbonate, and metallic oxides in an aerosolized form. If a tear in the meshwork of the airbag occurs, exposure to an even larger amount of powder containing caustic alkali chemicals can occur.8
We describe a patient who developed a bullous reaction to airbag contents after he was involved in an MVA in which the airbag deployed.
Case Report
A 35-year-old man with a history of type 2 diabetes mellitus and chronic hepatitis B presented to the dermatology clinic for an evaluation of new-onset blisters. The rash occurred 1 day after the patient was involved in an MVA in which he was exposed to the airbag’s contents after it burst. He had been evaluated twice in the emergency department for the skin eruption before being referred to dermatology. He noted the lesions were pruritic and painful. Prior treatments included silver sulfadiazine cream 1% and clobetasol cream 0.05%, though he discontinued using the latter because of burning with application. Physical examination revealed tense vesicles and bullae on an erythematous base on the right lower flank, forearms, and legs, with the exception of the lower right leg where a cast had been from a prior injury (Figure 1).
Two punch biopsies of the left arm were performed and sent for hematoxylin and eosin staining and direct immunofluorescence to rule out bullous diseases, such as bullous pemphigoid, linear IgA, and bullous lupus. Hematoxylin and eosin staining revealed extensive spongiosis with blister formation and a dense perivascular infiltrate in the superficial and mid dermis composed of lymphocytes with numerous scattered eosinophils (Figures 2 and 3). Direct immunofluorescence studies were negative. Treatment with oral prednisone and oral antihistamines was initiated.
At 10-day follow-up, the patient had a few residual bullae; most lesions were demonstrating various stages of healing (Figure 4). The patient’s cast had been removed, and there were no lesions in this previously covered area. At 6-week follow-up he had continued healing of the bullae and erosions as well as postinflammatory hyperpigmentation (Figure 5).
Comment
With the advent of airbags for safety purposes, these potentially lifesaving devices also have been known to cause injury.9 In 1998, the most commonly reported airbag injuries were ocular injuries.10 Cutaneous manifestations of airbag injury are less well known.11
Two cases of airbag deployment with skin blistering have been reported in the literature based on a PubMed search of articles indexed for MEDLINE using the terms airbag blistering or airbag bullae12,13; however, the blistering was described in the context of a burn. One case of the effects of airbag deployment residue highlights a patient arriving to the emergency department with erythema and blisters on the hands within 48 hours of airbag deployment in an MVA, and the treatment was standard burn therapy.12 Another case report described a patient with a second-degree burn with a 12-cm blister occurring on the radial side of the hand and distal forearm following an MVA and airbag deployment, which was treated conservatively.13 Cases of thermal burns, chemical burns, and irritant contact dermatitis after airbag deployment have been described in the literature.4-6,11,12,14,15 Our patient’s distal right lower leg was covered with a cast for osteomyelitis, and no blisters had developed in this area. It is likely that the transfer of airbag contents occurred during the process of unbuckling his seatbelt, which could explain the bullae that developed on the right flank. Per the Centers for Disease Control and Prevention, individuals should quickly remove clothing and wash their body with large amounts of soap and water following exposure to sodium azide.16
In 1989, the Federal Motor Vehicle Safety Standard No. 208 (occupant crash protection) became effective, stating all cars must have vehicle crash protection.12 Prior to 1993, it was reported that there had been no associated chemical injuries with airbag deployment. Subsequently, a 6-month retrospective study in 1993 showed that dermal injuries were found in connection with the presence of sodium hydroxide in automobile airbags.12 By 2004, it was known that airbags could cause chemical and thermal burns in addition to traumatic injuries from deployment.1 Since 2007, all motor vehicles have been required to have advanced airbags, which are engineered to sense the presence of passengers and determine if the airbag will deploy, and if so, how much to deploy to minimize airbag-related injury.3
The brand of car that our patient drove during the MVA is one with known airbag recalls due to safety defects; however, the year and actual model of the vehicle are not known, so specific information about the airbag in question is not available. It has been noted that some defective airbag inflators that were exposed to excess moisture during the manufacturing process could explode during deployment, causing shrapnel and airbag rupture, which has been linked to nearly 300 injuries worldwide.17
Conclusion
It is evident that the use of airbag devices reduces morbidity and mortality due to MVAs.9 It also had been reported that up to 96% of airbag-related injuries are relatively minor, which many would argue justifies their use.18 Furthermore, it has been reported that 99.8% of skin injuries following airbag deployment are minor.19 In the United States, it is mandated that every vehicle have functional airbags installed.8
This case highlights the potential for substantial airbag-induced skin reactions, specifically a bullous reaction, following airbag deployment. The persistent pruritus and lasting postinflammatory hyperpigmentation seen in this case were certainly worrisome for our patient. We also present this case to remind dermatology providers of possible treatment approaches to these skin reactions. Immediate cleansing of the affected areas of skin may help avoid such reactions.
Airbags are lifesaving during motor vehicle accidents (MVAs), but their deployment has been associated with skin issues such as irritant dermatitis1; lacerations2; abrasions3; and thermal, friction, and chemical burns.4-6 Ocular issues such as alkaline chemical keratitis7 and ocular alkali injuries8 also have been described.
Airbag deployment is triggered by rapid deceleration and impact, which ignite a sodium azide cartridge, causing the woven nylon bag to inflate with hydrocarbon gases.8 This leads to release of sodium hydroxide, sodium bicarbonate, and metallic oxides in an aerosolized form. If a tear in the meshwork of the airbag occurs, exposure to an even larger amount of powder containing caustic alkali chemicals can occur.8
We describe a patient who developed a bullous reaction to airbag contents after he was involved in an MVA in which the airbag deployed.
Case Report
A 35-year-old man with a history of type 2 diabetes mellitus and chronic hepatitis B presented to the dermatology clinic for an evaluation of new-onset blisters. The rash occurred 1 day after the patient was involved in an MVA in which he was exposed to the airbag’s contents after it burst. He had been evaluated twice in the emergency department for the skin eruption before being referred to dermatology. He noted the lesions were pruritic and painful. Prior treatments included silver sulfadiazine cream 1% and clobetasol cream 0.05%, though he discontinued using the latter because of burning with application. Physical examination revealed tense vesicles and bullae on an erythematous base on the right lower flank, forearms, and legs, with the exception of the lower right leg where a cast had been from a prior injury (Figure 1).
Two punch biopsies of the left arm were performed and sent for hematoxylin and eosin staining and direct immunofluorescence to rule out bullous diseases, such as bullous pemphigoid, linear IgA, and bullous lupus. Hematoxylin and eosin staining revealed extensive spongiosis with blister formation and a dense perivascular infiltrate in the superficial and mid dermis composed of lymphocytes with numerous scattered eosinophils (Figures 2 and 3). Direct immunofluorescence studies were negative. Treatment with oral prednisone and oral antihistamines was initiated.
At 10-day follow-up, the patient had a few residual bullae; most lesions were demonstrating various stages of healing (Figure 4). The patient’s cast had been removed, and there were no lesions in this previously covered area. At 6-week follow-up he had continued healing of the bullae and erosions as well as postinflammatory hyperpigmentation (Figure 5).
Comment
With the advent of airbags for safety purposes, these potentially lifesaving devices also have been known to cause injury.9 In 1998, the most commonly reported airbag injuries were ocular injuries.10 Cutaneous manifestations of airbag injury are less well known.11
Two cases of airbag deployment with skin blistering have been reported in the literature based on a PubMed search of articles indexed for MEDLINE using the terms airbag blistering or airbag bullae12,13; however, the blistering was described in the context of a burn. One case of the effects of airbag deployment residue highlights a patient arriving to the emergency department with erythema and blisters on the hands within 48 hours of airbag deployment in an MVA, and the treatment was standard burn therapy.12 Another case report described a patient with a second-degree burn with a 12-cm blister occurring on the radial side of the hand and distal forearm following an MVA and airbag deployment, which was treated conservatively.13 Cases of thermal burns, chemical burns, and irritant contact dermatitis after airbag deployment have been described in the literature.4-6,11,12,14,15 Our patient’s distal right lower leg was covered with a cast for osteomyelitis, and no blisters had developed in this area. It is likely that the transfer of airbag contents occurred during the process of unbuckling his seatbelt, which could explain the bullae that developed on the right flank. Per the Centers for Disease Control and Prevention, individuals should quickly remove clothing and wash their body with large amounts of soap and water following exposure to sodium azide.16
In 1989, the Federal Motor Vehicle Safety Standard No. 208 (occupant crash protection) became effective, stating all cars must have vehicle crash protection.12 Prior to 1993, it was reported that there had been no associated chemical injuries with airbag deployment. Subsequently, a 6-month retrospective study in 1993 showed that dermal injuries were found in connection with the presence of sodium hydroxide in automobile airbags.12 By 2004, it was known that airbags could cause chemical and thermal burns in addition to traumatic injuries from deployment.1 Since 2007, all motor vehicles have been required to have advanced airbags, which are engineered to sense the presence of passengers and determine if the airbag will deploy, and if so, how much to deploy to minimize airbag-related injury.3
The brand of car that our patient drove during the MVA is one with known airbag recalls due to safety defects; however, the year and actual model of the vehicle are not known, so specific information about the airbag in question is not available. It has been noted that some defective airbag inflators that were exposed to excess moisture during the manufacturing process could explode during deployment, causing shrapnel and airbag rupture, which has been linked to nearly 300 injuries worldwide.17
Conclusion
It is evident that the use of airbag devices reduces morbidity and mortality due to MVAs.9 It also had been reported that up to 96% of airbag-related injuries are relatively minor, which many would argue justifies their use.18 Furthermore, it has been reported that 99.8% of skin injuries following airbag deployment are minor.19 In the United States, it is mandated that every vehicle have functional airbags installed.8
This case highlights the potential for substantial airbag-induced skin reactions, specifically a bullous reaction, following airbag deployment. The persistent pruritus and lasting postinflammatory hyperpigmentation seen in this case were certainly worrisome for our patient. We also present this case to remind dermatology providers of possible treatment approaches to these skin reactions. Immediate cleansing of the affected areas of skin may help avoid such reactions.
- Corazza M, Trincone S, Zampino MR, et al. Air bags and the skin. Skinmed. 2004;3:256-258.
- Corazza M, Trincone S, Virgili A. Effects of airbag deployment: lesions, epidemiology, and management. Am J Clin Dermatol. 2004;5:295-300.
- Kuska TC. Air bag safety: an update. J Emerg Nurs. 2016;42:438-441.
- Ulrich D, Noah EM, Fuchs P, et al. Burn injuries caused by air bag deployment. Burns. 2001;27:196-199.
- Erpenbeck SP, Roy E, Ziembicki JA, et al. A systematic review on airbag-induced burns. J Burn Care Res. 2021;42:481-487.
- Skibba KEH, Cleveland CN, Bell DE. Airbag burns: an unfortunate consequence of motor vehicle safety. J Burn Care Res. 2021;42:71-73.
- Smally AJ, Binzer A, Dolin S, et al. Alkaline chemical keratitis: eye injury from airbags. Ann Emerg Med. 1992;21:1400-1402.
- Barnes SS, Wong W Jr, Affeldt JC. A case of severe airbag related ocular alkali injury. Hawaii J Med Public Health. 2012;71:229-231.
- Wallis LA, Greaves I. Injuries associated with airbag deployment. Emerg Med J. 2002;19:490-493.
- Mohamed AA, Banerjee A. Patterns of injury associated with automobile airbag use. Postgrad Med J. 1998;74:455-458.
- Foley E, Helm TN. Air bag injury and the dermatologist. Cutis. 2000;66:251-252.
- Swanson-Biearman B, Mrvos R, Dean BS, et al. Air bags: lifesaving with toxic potential? Am J Emerg Med. 1993;11:38-39.
- Roth T, Meredith P. Traumatic lesions caused by the “air-bag” system [in French]. Z Unfallchir Versicherungsmed. 1993;86:189-193.
- Wu JJ, Sanchez-Palacios C, Brieva J, et al. A case of air bag dermatitis. Arch Dermatol. 2002;138:1383-1384.
- Vitello W, Kim M, Johnson RM, et al. Full-thickness burn to the hand from an automobile airbag. J Burn Care Rehabil. 1999;20:212-215.
- Centers for Disease Control and Prevention. Facts about sodium azide. Updated April 4, 2018. Accessed May 15, 2022. https://emergency.cdc.gov/agent/sodiumazide/basics/facts.asp
- Shepardson D. Honda to recall 1.2 million vehicles in North America to replace Takata airbags. March 12, 2019. Accessed March 22, 2022. https://www.reuters.com/article/us-honda-takata-recall/honda-to-recall-1-2-million-vehicles-in-north-america-to-replace-takata-airbags-idUSKBN1QT1C9
- Gabauer DJ, Gabler HC. The effects of airbags and seatbelts on occupant injury in longitudinal barrier crashes. J Safety Res. 2010;41:9-15.
- Rath AL, Jernigan MV, Stitzel JD, et al. The effects of depowered airbags on skin injuries in frontal automobile crashes. Plast Reconstr Surg. 2005;115:428-435.
- Corazza M, Trincone S, Zampino MR, et al. Air bags and the skin. Skinmed. 2004;3:256-258.
- Corazza M, Trincone S, Virgili A. Effects of airbag deployment: lesions, epidemiology, and management. Am J Clin Dermatol. 2004;5:295-300.
- Kuska TC. Air bag safety: an update. J Emerg Nurs. 2016;42:438-441.
- Ulrich D, Noah EM, Fuchs P, et al. Burn injuries caused by air bag deployment. Burns. 2001;27:196-199.
- Erpenbeck SP, Roy E, Ziembicki JA, et al. A systematic review on airbag-induced burns. J Burn Care Res. 2021;42:481-487.
- Skibba KEH, Cleveland CN, Bell DE. Airbag burns: an unfortunate consequence of motor vehicle safety. J Burn Care Res. 2021;42:71-73.
- Smally AJ, Binzer A, Dolin S, et al. Alkaline chemical keratitis: eye injury from airbags. Ann Emerg Med. 1992;21:1400-1402.
- Barnes SS, Wong W Jr, Affeldt JC. A case of severe airbag related ocular alkali injury. Hawaii J Med Public Health. 2012;71:229-231.
- Wallis LA, Greaves I. Injuries associated with airbag deployment. Emerg Med J. 2002;19:490-493.
- Mohamed AA, Banerjee A. Patterns of injury associated with automobile airbag use. Postgrad Med J. 1998;74:455-458.
- Foley E, Helm TN. Air bag injury and the dermatologist. Cutis. 2000;66:251-252.
- Swanson-Biearman B, Mrvos R, Dean BS, et al. Air bags: lifesaving with toxic potential? Am J Emerg Med. 1993;11:38-39.
- Roth T, Meredith P. Traumatic lesions caused by the “air-bag” system [in French]. Z Unfallchir Versicherungsmed. 1993;86:189-193.
- Wu JJ, Sanchez-Palacios C, Brieva J, et al. A case of air bag dermatitis. Arch Dermatol. 2002;138:1383-1384.
- Vitello W, Kim M, Johnson RM, et al. Full-thickness burn to the hand from an automobile airbag. J Burn Care Rehabil. 1999;20:212-215.
- Centers for Disease Control and Prevention. Facts about sodium azide. Updated April 4, 2018. Accessed May 15, 2022. https://emergency.cdc.gov/agent/sodiumazide/basics/facts.asp
- Shepardson D. Honda to recall 1.2 million vehicles in North America to replace Takata airbags. March 12, 2019. Accessed March 22, 2022. https://www.reuters.com/article/us-honda-takata-recall/honda-to-recall-1-2-million-vehicles-in-north-america-to-replace-takata-airbags-idUSKBN1QT1C9
- Gabauer DJ, Gabler HC. The effects of airbags and seatbelts on occupant injury in longitudinal barrier crashes. J Safety Res. 2010;41:9-15.
- Rath AL, Jernigan MV, Stitzel JD, et al. The effects of depowered airbags on skin injuries in frontal automobile crashes. Plast Reconstr Surg. 2005;115:428-435.
Practice Points
- This case highlights the potential for a bullous reaction following airbag deployment.
- After airbag deployment, it is important to immediately cleanse the affected areas of skin with soap and water.

Pemphigus Vulgaris Aggravated: Rifampicin Found at the Scene of the Crime
Case Report
A 60-year-old man presented with eroded areas in the mouth and blistering eruptions on the scalp, face, trunk, arms, and legs. He initially presented to an outside hospital 4 years prior and was treated with oral prednisone 50 mg daily, to which the eruptions responded rapidly; however, following a nearly 5-mg reduction of the dose per week by the patient and irregular oral administration, he experienced several episodes of recurrence, but he could not remember the exact dosage of prednisone he had taken during that period. Subsequently, he was admitted to our hospital because of large areas of erythema and erosions on the scalp, trunk, arms, and legs.
Since starting the prednisone regimen 4 years prior, the patient had experienced onset of hypertension, diabetes, glaucoma, cataracts, optic nerve atrophy, aseptic necrosis of the femoral head, and osteoporosis. Biopsy of a new skin lesion

The patient initially was started again prednisone 50 mg daily, to which the skin eruptions responded, and 2 weeks later, the disease was considered controlled. The prednisone dosage was tapered to 20 mg daily 3 months later with no new blister formation. However, 2 weeks later, the patient was diagnosed by a tuberculosis specialist with pulmonary tuberculosis, and a daily regimen of isoniazid, rifampicin, ethambutol, and levofloxacin was instituted.
Ten days after starting antituberculosis therapy, the patient developed new erythematous blisters that could not be controlled and self-adjusted the prednisone dose to 50 mg daily. Two months later, blister formation continued.
Six months after the initial presentation, the patient returned to our hospital because of uncontrollable rashes (Figure 2). On admission, he had a Pemphigus Disease Area Index (PDAI) score of 32 with disease involving 30% of the body surface area. Laboratory testing showed a desmoglein 1 level of 233 U/mL and desmoglein 3 level of 228 U/mL. A tuberculosis specialist from an outside hospital was consulted to evaluate the patient’s condition and assist in treatment. Based on findings from a pulmonary computed tomography scan, which showed the inflammation was considerably absorbed, treatment was adjusted to stop using ethambutol and levofloxacin and continue rifampicin and isoniazid. For the PV, prednisone was titrated upward to 75 mg daily, mycophenolate mofetil (MMF) 1 g twice daily was added, and IVIG 400 mg/kg daily was administered for 7 days. After 3 weeks, the rash still expanded.

In considering possible interactions between the drugs, we consulted the literature and found reports1-3 that rifampicin accelerated glucocorticoid metabolism, of which the tuberculosis specialist that we consulted was not aware. Therefore, rifampicin was stopped, and the antituberculosis therapy was adjusted to levofloxacin and isoniazid. Meanwhile, the steroid was changed to methylprednisolone 120 mg daily for 3 days, then to 80 mg daily for 2 days.
After 5 days, the rash was controlled with no new development and the patient was discharged. He continued on prednisone 80 mg daily and MMF 1 g twice daily.
At 2-month follow-up, no new rash had developed. The patient had already self-discontinued the MMF for 1 month because it was difficult to obtain at local hospitals. The prednisone was reduced to 40 mg daily. Pulmonary computed tomography showed no signs of reactivation of tuberculosis.
Comment
Drugs that depend on these enzymes for their metabolism are prone to
Rifampicin causes a marked reduction in dose-corrected mycophenolic acid exposure when administered simultaneously with MMF through induction of glucuronidation activity and inhibition of enterohepatic recirculation.5,10In in vitro studies, rifampin and other cytochrome P450 inducers have been identified as potentially useful for increasing the rate of cyclophosphamide and ifosfamide (an isomeric analogue of cyclophosphamide) 4-hydroxylation in the human liver in a manner that could have a favorable impact on the clinical pharmacokinetics of these anticancer prodrugs.11 However, clinical analysis of 16 patients indicated that co-administration of ifosfamide with rifampin did not result in changes in the pharmacokinetics of the parent drug or its metabolites.12
The steroids and
Conclusion
In our patient, the use of rifapentine resulted in a recurrence of previously controlled PV and resistance to treatment. The patient’s disease was quickly controlled after discontinuation of rifampicin and with a short-term course of high-dose methylprednisolone and remained stable when the dosages of MMF and prednisone were reduced.
- Miyagawa S, Yamashina Y, Okuchi T, et al. Exacerbation of pemphigus by rifampicin. Br J Dermatol. 1986;114:729-732. doi:10.1111/j.1365-2133.1986.tb04882.x
- Gange RW, Rhodes EL, Edwards CO, et al. Pemphigus induced by rifampicin. Br J Dermatol. 1976;95:445-448. doi:10.1111/j.1365-2133.1976.tb00849.x
- Bergrem H, Refvem OK. Altered prednisolone pharmacokinetics in patients treated with rifampicin. Acta Med Scand. 1983;213:339-343. doi:10.1111/j.0954-6820.1983.tb03748.x
- McAllister WA, Thompson PJ, Al-Habet SM, et al. Rifampicin reduces effectiveness and bioavailability of prednisolone. Br Med J (Clin Res Ed). 1983;286:923-925. doi:10.1136/bmj.286.6369.923
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106. doi:10.1007/s00403-017-1790-8
- Barman H, Dass R, Duwarah SG. Use of high-dose prednisolone to overcome rifampicin-induced corticosteroid non-responsiveness in childhood nephrotic syndrome. Saudi J Kidney Dis Transpl. 2016;27:157-160. doi:10.4103/1319-2442.174198
- Okey AB, Roberts EA, Harper PA, et al. Induction of drug-metabolizing enzymes: mechanisms and consequences. Clin Biochem. 1986;19:132-141. doi:10.1016/s0009-9120(86)80060-1
- Venkatesan K. Pharmacokinetic interactions with rifampicin. Clin Pharmacokinet. 1992;22:47-65. doi:10.2165/00003088-199222010-00005
- Naesens M, Kuypers DRJ, Streit F, et al. Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther. 2006;80:509-521. doi:10.1016/j.clpt.2006.08.002
- Kuypers DRJ, Verleden G, Naesens M, et al. Drug interaction between mycophenolate mofetil and rifampin: possible induction of uridine diphosphate–glucuronosyltransferase. Clin Pharmacol Ther. 2005;78:81-88. doi:10.1016/j.clpt.2005.03.004
- Chenhsu RY, Loong CC, Chou MH, et al. Renal allograft dysfunction associated with rifampin–tacrolimus interaction. Ann Pharmacother. 2000;34:27-31. doi:10.1345/aph.19069
- Douglas JG, McLeod MJ. Pharmacokinetic factors in the modern drug treatment of tuberculosis. Clin Pharmacokinet. 1999;37:127-146. doi:10.2165/00003088-199937020-00003
Case Report
A 60-year-old man presented with eroded areas in the mouth and blistering eruptions on the scalp, face, trunk, arms, and legs. He initially presented to an outside hospital 4 years prior and was treated with oral prednisone 50 mg daily, to which the eruptions responded rapidly; however, following a nearly 5-mg reduction of the dose per week by the patient and irregular oral administration, he experienced several episodes of recurrence, but he could not remember the exact dosage of prednisone he had taken during that period. Subsequently, he was admitted to our hospital because of large areas of erythema and erosions on the scalp, trunk, arms, and legs.
Since starting the prednisone regimen 4 years prior, the patient had experienced onset of hypertension, diabetes, glaucoma, cataracts, optic nerve atrophy, aseptic necrosis of the femoral head, and osteoporosis. Biopsy of a new skin lesion
The patient initially was started again prednisone 50 mg daily, to which the skin eruptions responded, and 2 weeks later, the disease was considered controlled. The prednisone dosage was tapered to 20 mg daily 3 months later with no new blister formation. However, 2 weeks later, the patient was diagnosed by a tuberculosis specialist with pulmonary tuberculosis, and a daily regimen of isoniazid, rifampicin, ethambutol, and levofloxacin was instituted.
Ten days after starting antituberculosis therapy, the patient developed new erythematous blisters that could not be controlled and self-adjusted the prednisone dose to 50 mg daily. Two months later, blister formation continued.
Six months after the initial presentation, the patient returned to our hospital because of uncontrollable rashes (Figure 2). On admission, he had a Pemphigus Disease Area Index (PDAI) score of 32 with disease involving 30% of the body surface area. Laboratory testing showed a desmoglein 1 level of 233 U/mL and desmoglein 3 level of 228 U/mL. A tuberculosis specialist from an outside hospital was consulted to evaluate the patient’s condition and assist in treatment. Based on findings from a pulmonary computed tomography scan, which showed the inflammation was considerably absorbed, treatment was adjusted to stop using ethambutol and levofloxacin and continue rifampicin and isoniazid. For the PV, prednisone was titrated upward to 75 mg daily, mycophenolate mofetil (MMF) 1 g twice daily was added, and IVIG 400 mg/kg daily was administered for 7 days. After 3 weeks, the rash still expanded.
In considering possible interactions between the drugs, we consulted the literature and found reports1-3 that rifampicin accelerated glucocorticoid metabolism, of which the tuberculosis specialist that we consulted was not aware. Therefore, rifampicin was stopped, and the antituberculosis therapy was adjusted to levofloxacin and isoniazid. Meanwhile, the steroid was changed to methylprednisolone 120 mg daily for 3 days, then to 80 mg daily for 2 days.
After 5 days, the rash was controlled with no new development and the patient was discharged. He continued on prednisone 80 mg daily and MMF 1 g twice daily.
At 2-month follow-up, no new rash had developed. The patient had already self-discontinued the MMF for 1 month because it was difficult to obtain at local hospitals. The prednisone was reduced to 40 mg daily. Pulmonary computed tomography showed no signs of reactivation of tuberculosis.
Comment
Drugs that depend on these enzymes for their metabolism are prone to
Rifampicin causes a marked reduction in dose-corrected mycophenolic acid exposure when administered simultaneously with MMF through induction of glucuronidation activity and inhibition of enterohepatic recirculation.5,10In in vitro studies, rifampin and other cytochrome P450 inducers have been identified as potentially useful for increasing the rate of cyclophosphamide and ifosfamide (an isomeric analogue of cyclophosphamide) 4-hydroxylation in the human liver in a manner that could have a favorable impact on the clinical pharmacokinetics of these anticancer prodrugs.11 However, clinical analysis of 16 patients indicated that co-administration of ifosfamide with rifampin did not result in changes in the pharmacokinetics of the parent drug or its metabolites.12
The steroids and
Conclusion
In our patient, the use of rifapentine resulted in a recurrence of previously controlled PV and resistance to treatment. The patient’s disease was quickly controlled after discontinuation of rifampicin and with a short-term course of high-dose methylprednisolone and remained stable when the dosages of MMF and prednisone were reduced.
Case Report
A 60-year-old man presented with eroded areas in the mouth and blistering eruptions on the scalp, face, trunk, arms, and legs. He initially presented to an outside hospital 4 years prior and was treated with oral prednisone 50 mg daily, to which the eruptions responded rapidly; however, following a nearly 5-mg reduction of the dose per week by the patient and irregular oral administration, he experienced several episodes of recurrence, but he could not remember the exact dosage of prednisone he had taken during that period. Subsequently, he was admitted to our hospital because of large areas of erythema and erosions on the scalp, trunk, arms, and legs.
Since starting the prednisone regimen 4 years prior, the patient had experienced onset of hypertension, diabetes, glaucoma, cataracts, optic nerve atrophy, aseptic necrosis of the femoral head, and osteoporosis. Biopsy of a new skin lesion
The patient initially was started again prednisone 50 mg daily, to which the skin eruptions responded, and 2 weeks later, the disease was considered controlled. The prednisone dosage was tapered to 20 mg daily 3 months later with no new blister formation. However, 2 weeks later, the patient was diagnosed by a tuberculosis specialist with pulmonary tuberculosis, and a daily regimen of isoniazid, rifampicin, ethambutol, and levofloxacin was instituted.
Ten days after starting antituberculosis therapy, the patient developed new erythematous blisters that could not be controlled and self-adjusted the prednisone dose to 50 mg daily. Two months later, blister formation continued.
Six months after the initial presentation, the patient returned to our hospital because of uncontrollable rashes (Figure 2). On admission, he had a Pemphigus Disease Area Index (PDAI) score of 32 with disease involving 30% of the body surface area. Laboratory testing showed a desmoglein 1 level of 233 U/mL and desmoglein 3 level of 228 U/mL. A tuberculosis specialist from an outside hospital was consulted to evaluate the patient’s condition and assist in treatment. Based on findings from a pulmonary computed tomography scan, which showed the inflammation was considerably absorbed, treatment was adjusted to stop using ethambutol and levofloxacin and continue rifampicin and isoniazid. For the PV, prednisone was titrated upward to 75 mg daily, mycophenolate mofetil (MMF) 1 g twice daily was added, and IVIG 400 mg/kg daily was administered for 7 days. After 3 weeks, the rash still expanded.
In considering possible interactions between the drugs, we consulted the literature and found reports1-3 that rifampicin accelerated glucocorticoid metabolism, of which the tuberculosis specialist that we consulted was not aware. Therefore, rifampicin was stopped, and the antituberculosis therapy was adjusted to levofloxacin and isoniazid. Meanwhile, the steroid was changed to methylprednisolone 120 mg daily for 3 days, then to 80 mg daily for 2 days.
After 5 days, the rash was controlled with no new development and the patient was discharged. He continued on prednisone 80 mg daily and MMF 1 g twice daily.
At 2-month follow-up, no new rash had developed. The patient had already self-discontinued the MMF for 1 month because it was difficult to obtain at local hospitals. The prednisone was reduced to 40 mg daily. Pulmonary computed tomography showed no signs of reactivation of tuberculosis.
Comment
Drugs that depend on these enzymes for their metabolism are prone to
Rifampicin causes a marked reduction in dose-corrected mycophenolic acid exposure when administered simultaneously with MMF through induction of glucuronidation activity and inhibition of enterohepatic recirculation.5,10In in vitro studies, rifampin and other cytochrome P450 inducers have been identified as potentially useful for increasing the rate of cyclophosphamide and ifosfamide (an isomeric analogue of cyclophosphamide) 4-hydroxylation in the human liver in a manner that could have a favorable impact on the clinical pharmacokinetics of these anticancer prodrugs.11 However, clinical analysis of 16 patients indicated that co-administration of ifosfamide with rifampin did not result in changes in the pharmacokinetics of the parent drug or its metabolites.12
The steroids and
Conclusion
In our patient, the use of rifapentine resulted in a recurrence of previously controlled PV and resistance to treatment. The patient’s disease was quickly controlled after discontinuation of rifampicin and with a short-term course of high-dose methylprednisolone and remained stable when the dosages of MMF and prednisone were reduced.
- Miyagawa S, Yamashina Y, Okuchi T, et al. Exacerbation of pemphigus by rifampicin. Br J Dermatol. 1986;114:729-732. doi:10.1111/j.1365-2133.1986.tb04882.x
- Gange RW, Rhodes EL, Edwards CO, et al. Pemphigus induced by rifampicin. Br J Dermatol. 1976;95:445-448. doi:10.1111/j.1365-2133.1976.tb00849.x
- Bergrem H, Refvem OK. Altered prednisolone pharmacokinetics in patients treated with rifampicin. Acta Med Scand. 1983;213:339-343. doi:10.1111/j.0954-6820.1983.tb03748.x
- McAllister WA, Thompson PJ, Al-Habet SM, et al. Rifampicin reduces effectiveness and bioavailability of prednisolone. Br Med J (Clin Res Ed). 1983;286:923-925. doi:10.1136/bmj.286.6369.923
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106. doi:10.1007/s00403-017-1790-8
- Barman H, Dass R, Duwarah SG. Use of high-dose prednisolone to overcome rifampicin-induced corticosteroid non-responsiveness in childhood nephrotic syndrome. Saudi J Kidney Dis Transpl. 2016;27:157-160. doi:10.4103/1319-2442.174198
- Okey AB, Roberts EA, Harper PA, et al. Induction of drug-metabolizing enzymes: mechanisms and consequences. Clin Biochem. 1986;19:132-141. doi:10.1016/s0009-9120(86)80060-1
- Venkatesan K. Pharmacokinetic interactions with rifampicin. Clin Pharmacokinet. 1992;22:47-65. doi:10.2165/00003088-199222010-00005
- Naesens M, Kuypers DRJ, Streit F, et al. Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther. 2006;80:509-521. doi:10.1016/j.clpt.2006.08.002
- Kuypers DRJ, Verleden G, Naesens M, et al. Drug interaction between mycophenolate mofetil and rifampin: possible induction of uridine diphosphate–glucuronosyltransferase. Clin Pharmacol Ther. 2005;78:81-88. doi:10.1016/j.clpt.2005.03.004
- Chenhsu RY, Loong CC, Chou MH, et al. Renal allograft dysfunction associated with rifampin–tacrolimus interaction. Ann Pharmacother. 2000;34:27-31. doi:10.1345/aph.19069
- Douglas JG, McLeod MJ. Pharmacokinetic factors in the modern drug treatment of tuberculosis. Clin Pharmacokinet. 1999;37:127-146. doi:10.2165/00003088-199937020-00003
- Miyagawa S, Yamashina Y, Okuchi T, et al. Exacerbation of pemphigus by rifampicin. Br J Dermatol. 1986;114:729-732. doi:10.1111/j.1365-2133.1986.tb04882.x
- Gange RW, Rhodes EL, Edwards CO, et al. Pemphigus induced by rifampicin. Br J Dermatol. 1976;95:445-448. doi:10.1111/j.1365-2133.1976.tb00849.x
- Bergrem H, Refvem OK. Altered prednisolone pharmacokinetics in patients treated with rifampicin. Acta Med Scand. 1983;213:339-343. doi:10.1111/j.0954-6820.1983.tb03748.x
- McAllister WA, Thompson PJ, Al-Habet SM, et al. Rifampicin reduces effectiveness and bioavailability of prednisolone. Br Med J (Clin Res Ed). 1983;286:923-925. doi:10.1136/bmj.286.6369.923
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106. doi:10.1007/s00403-017-1790-8
- Barman H, Dass R, Duwarah SG. Use of high-dose prednisolone to overcome rifampicin-induced corticosteroid non-responsiveness in childhood nephrotic syndrome. Saudi J Kidney Dis Transpl. 2016;27:157-160. doi:10.4103/1319-2442.174198
- Okey AB, Roberts EA, Harper PA, et al. Induction of drug-metabolizing enzymes: mechanisms and consequences. Clin Biochem. 1986;19:132-141. doi:10.1016/s0009-9120(86)80060-1
- Venkatesan K. Pharmacokinetic interactions with rifampicin. Clin Pharmacokinet. 1992;22:47-65. doi:10.2165/00003088-199222010-00005
- Naesens M, Kuypers DRJ, Streit F, et al. Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther. 2006;80:509-521. doi:10.1016/j.clpt.2006.08.002
- Kuypers DRJ, Verleden G, Naesens M, et al. Drug interaction between mycophenolate mofetil and rifampin: possible induction of uridine diphosphate–glucuronosyltransferase. Clin Pharmacol Ther. 2005;78:81-88. doi:10.1016/j.clpt.2005.03.004
- Chenhsu RY, Loong CC, Chou MH, et al. Renal allograft dysfunction associated with rifampin–tacrolimus interaction. Ann Pharmacother. 2000;34:27-31. doi:10.1345/aph.19069
- Douglas JG, McLeod MJ. Pharmacokinetic factors in the modern drug treatment of tuberculosis. Clin Pharmacokinet. 1999;37:127-146. doi:10.2165/00003088-199937020-00003
Practice Points
- Long-term use of immunosuppressants requires constant attention for infections, especially latent infections in the body.
- Clinicians should carefully inquire with patients about concomitant diseases and medications used, and be vigilant about drug interactions.

Leiomyosarcoma of the Penis: A Case Report and Re-Appraisal
Penile cancer is rare with a worldwide incidence of 0.8 cases per 100,000 men.1 The most common type is squamous cell carcinoma (SCC) followed by soft tissue sarcoma (STS) and Kaposi sarcoma.2 Leiomyosarcoma (LMS) is the second most common STS subtype at this location.3 Approximately 50 cases of penile LMS have been reported in the English literature, most as isolated case reports while Fetsch and colleagues reported 14 cases from a single institute.4 We present a case of penile LMS with a review of 31 cases. We also describe presentation, treatment options, and recurrence pattern of this rare malignancy.
Case Presentation
A patient aged 70 years presented to the urology clinic with 1-year history of a slowly enlarging penile mass associated with phimosis. He reported no pain, dysuria, or hesitancy. On examination a 2 × 2-cm smooth, mobile, nonulcerating mass was seen on the tip of his left glans without inguinal lymphadenopathy. He underwent circumcision and excision biopsy that revealed an encapsulated tan-white mass measuring 3 × 2.2 × 1.5 cm under the surface of the foreskin. Histology showed a spindle cell tumor with areas of increased cellularity, prominent atypia, and pleomorphism, focal necrosis, and scattered mitoses, including atypical forms. The tumor stained positive for smooth muscle actin and desmin. Ki-67 staining showed foci with a very high proliferation index (Figure). Resection margins were negative. Final Fédération Nationale des Centres de Lutte Contre Le Cancer score was grade 2 (differentiation, 1; mitotic, 3; necrosis, 1). Computed tomography of the chest, abdomen, and pelvis did not show evidence of metastasis. The tumor was classified as superficial, stage IIA (pT1cN0cM0). Local excision with negative margins was deemed adequate treatment.
Discussion
Penile LMS is rare and arises from smooth muscles, which in the penis can be from dartos fascia, erector pili in the skin covering the shaft, or from tunica media of the superficial vessels and cavernosa.5 It commonly presents as a nodule or ulcer that might be accompanied by paraphimosis, phimosis, erectile dysfunction, and lower urinary tract symptoms depending on the extent of local tissue involvement. In our review of 31 cases, the age at presentation ranged from 38 to 85 years, with 1 case report of LMS in a 6-year-old. The highest incidence was in the 6th decade. Tumor behavior can be indolent or aggressive. Most patients in our review had asymptomatic, slow-growing lesions for 6 to 24 months before presentation—including our patient—while others had an aggressive tumor with symptoms for a few weeks followed by rapid metastatic spread.6,7
Histology and Staging
Diagnosis requires biopsy followed by histologic examination and immunohistochemistry of the lesion. Typically, LMS shows fascicles of spindle cells with varying degrees of nuclear atypia, pleomorphisms, and necrotic regions. Mitotic rate is variable and usually > 5 per high power field. Cells stain positive for smooth muscle actin, desmin, and h-caldesmon.8 TNM (tumor, nodes, metastasis) stage is determined by the American Joint Committee on Cancer guidelines for STS.
Pratt and colleagues were the first to categorize penile LMS as superficial or deep.9 The former includes all lesions superficial to tunica albuginea while the latter run deep to this layer. Anatomical distinction is an important factor in tumor behavior, treatment selection, and prognosis. In our review, we found 14 cases of superficial and 17 cases of deep LMS.
Treatment
There are no established guidelines on optimum treatment of penile LMS. However, we can extrapolate principles from current guidelines on penile cancer, cutaneous leiomyosarcoma, and limb sarcomas. At present, the first-line treatment for superficial penile LMS is wide local excision to achieve negative margins. Circumcision alone might be sufficient for tumors of the distal prepuce, as in our case.10 Radical resection generally is not required for these early-stage tumors. In our review, no patient in this category developed recurrence or metastasis regardless of initial surgery type (Table 1).6,11,12
For deep lesions, partial—if functional penile stump and negative margins can be achieved—or total penectomy is required.10 In our review, more conservative approaches to deep tumors were associated with local recurrences.7,13,14 Lymphatic spread is rare for LMS. Additionally, involvement of local lymph nodes usually coincides with distant spread. Inguinal lymph node dissection is not indicated if initial negative surgical margins are achieved.
For STS at other sites in the body, radiation therapy is recommended postoperatively for high-grade lesions, which can be extrapolated to penile LMS as well. The benefit of preoperative radiation therapy is less certain. In limb sarcomas, radiation is associated with better local control for large-sized tumors and is used for patients with initial unresectable tumors.15 Similar recommendation could be extended to penile LMS with local spread to inguinal lymph nodes, scrotum, or abdominal wall. In our review, postoperative radiation therapy was used in 3 patients with deep tumors.16-18 Of these, short-term relapse occurred in 1 patient.
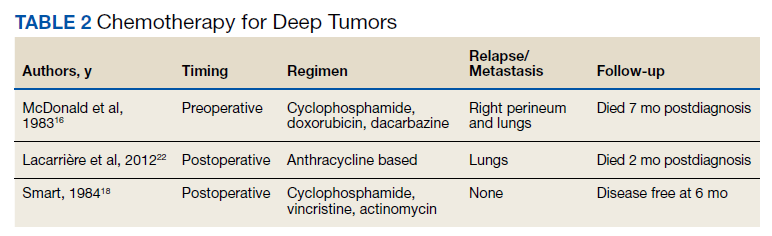
Chemotherapy for LMS remains controversial. The tumor generally is resistant to chemotherapy and systemic therapy, if employed, is for palliative purpose. The most promising results for adjuvant chemotherapy for resectable STS is seen in limb and uterine sarcomas with high-grade, metastatic, or relapsed tumors but improvement in overall survival has been marginal.19,20Single and multidrug regimens based on doxorubicin, ifosfamide, and gemcitabine have been studied with results showing no efficacy or a slight benefit.8,21 Immunotherapy and targeted therapy for penile STS have not been studied. In our review, postoperative chemotherapy was used for 2 patients with deep tumors and 1 patient with a superficial tumor while preoperative chemotherapy was used for 1 patient.16,18,22 Short-term relapse was seen in 2 of 4 of these patients (Table 2).
Metastatic Disease
LMS tends to metastasize hematogenously and lymphatic spread is uncommon. In our review, 7 patients developed metastasis. These patients had deep tumors at presentation with tumor size > 3 cm. Five of 7 patients had involvement of corpora cavernosa at presentation. The lung was the most common site of metastasis, followed by local extension to lower abdominal wall and scrotum. Of the 7 patients, 3 were treated with initial limited excision or partial penectomy and then experienced local recurrence or distant metastasis.7,13,14,23 This supports the use of radical surgery in large, deep tumors. In an additional 4 cases, metastasis occurred despite initial treatment with total penectomy and use of adjuvant chemoradiation therapy.
In most cases penile LMS is a de novo tumor, however, on occasion it could be accompanied by another epithelial malignancy. Similarly, penile LMS might be a site of recurrence for a primary LMS at another site, as seen in 3 of the reviewed cases. In the first, a patient presented with a nodule on the glans suspicious for SCC, second with synchronous SCC and LMS, and a third case where a patient presented with penile LMS 9 years after being treated for similar tumor in the epididymis.17,24,25
Prognosis
Penile LMS prognosis is difficult to ascertain because reported cases are rare. In our review, the longest documented disease-free survival was 3.5 years for a patient with superficial LMS treated with local excision.26 In cases of distant metastasis, average survival was 4.6 months, while the longest survival since initial presentation and last documented local recurrence was 16 years.14 Five-year survival has not been reported.
Conclusions
LMS of the penis is a rare and potentially aggressive malignancy. It can be classified as superficial or deep based on tumor relation to the tunica albuginea. Deep tumors, those > 3 cm, high-grade lesions, and tumors with involvement of corpora cavernosa, tend to spread locally, metastasize to distant areas, and require more radical surgery with or without postoperative radiation therapy. In comparison, superficial lesions can be treated with local excision only. Both superficial and deep tumors require close follow-up.
1. Montes Cardona CE, García-Perdomo HA. Incidence of penile cancer worldwide: systematic review and meta-analysis. Rev Panam Salud Publica. 2017;41:e117. Published 2017 Nov 30. doi:10.26633/RPSP.2017.117
2. Volker HU, Zettl A, Haralambieva E, et al. Leiomyosarcoma of the larynx as a local relapse of squamous cell carcinoma—report of an unusual case. Head Neck. 2010;32(5):679-683. doi:10.1002/hed.21127
3. Wollina U, Steinbach F, Verma S, et al. Penile tumours: a review. J Eur Acad Dermatol Venereol. 2014;28(10):1267-1276. doi:10.1111/jdv.12491
4. Fetsch JF, Davis CJ Jr, Miettinen M, Sesterhenn IA. Leiomyosarcoma of the penis: a clinicopathologic study of 14 cases with review of the literature and discussion of the differential diagnosis. Am J Surg Pathol. 2004;28(1):115-125. doi:10.1097/00000478-200401000-00014
5. Sundersingh S, Majhi U, Narayanaswamy K, Balasubramanian S. Primary leiomyosarcoma of the penis. Indian J Pathol Microbiol. 2009;52(3):447-448. doi:10.4103/0377-4929.55028
6. Mendis D, Bott SR, Davies JH. Subcutaneous leiomyosarcoma of the frenulum. Scientific World J. 2005;5:571-575. doi:10.1100/tsw.2005.76
7. Elem B, Nieslanik J. Leiomyosarcoma of the penis. Br J Urol. 1979;51(1):46. doi:10.1111/j.1464-410x.1979.tb04244.x
8. Serrano C, George S. Leiomyosarcoma. Hematol Oncol Clin North Am. 2013;27(5):957-974. doi:10.1016/j.hoc.2013.07.002
9. Pratt RM, Ross RT. Leiomyosarcoma of the penis. A report of a case. Br J Surg. 1969;56(11):870-872. doi:10.1002/bjs.1800561122
10. National Comprehensive Cancer Network. Penile cancer. NCCN evidence blocks. Version 2.2022 Updated January 26, 2022. Accessed March 16, 2022. https://www.nccn.org/professionals/physician_gls/pdf/penile_blocks.pdf
11. Ashley DJ, Edwards EC. Sarcoma of the penis; leiomyosarcoma of the penis: report of a case with a review of the literature on sarcoma of the penis. Br J Surg. 1957;45(190):170-179. doi:10.1002/bjs.18004519011
12. Pow-Sang MR, Orihuela E. Leiomyosarcoma of the penis. J Urol. 1994;151(6):1643-1645. doi:10.1016/s0022-5347(17)35328-413. Isa SS, Almaraz R, Magovern J. Leiomyosarcoma of the penis. Case report and review of the literature. Cancer. 1984;54(5):939-942. doi:10.1002/1097-0142(19840901)54:5<939::aid-cncr2820540533>3.0.co;2-y
14. Hutcheson JB, Wittaker WW, Fronstin MH. Leiomyosarcoma of the penis: case report and review of literature. J Urol. 1969;101(6):874-875. doi:10.1016/s0022-5347(17)62446-7
15. Grimer R, Judson I, Peake D, et al. Guidelines for the management of soft tissue sarcomas. Sarcoma. 2010;2010:506182. doi:10.1155/2010/506182
16. McDonald MW, O’Connell JR, Manning JT, Benjamin RS. Leiomyosarcoma of the penis. J Urol. 1983;130(4):788-789. doi:10.1016/s0022-5347(17)51464-0
17. Planz B, Brunner K, Kalem T, Schlick RW, Kind M. Primary leiomyosarcoma of the epididymis and late recurrence on the penis. J Urol. 1998;159(2):508. doi:10.1016/s0022-5347(01)63966-1
18. Smart RH. Leiomyosarcoma of the penis. J Urol. 1984;132(2):356-357. doi:10.1016/s0022-5347(17)49624-8
19. Patrikidou A, Domont J, Cioffi A, Le Cesne A. Treating soft tissue sarcomas with adjuvant chemotherapy. Curr Treat Options Oncol. 2011;12(1):21-31. doi:10.1007/s11864-011-0145-5
20. Italiano A, Delva F, Mathoulin-Pelissier S, et al. Effect of adjuvant chemotherapy on survival in FNCLCC grade 3 soft tissue sarcomas: a multivariate analysis of the French Sarcoma Group Database. Ann Oncol. 2010;21(12):2436-2441. doi:10.1093/annonc/mdq238
21. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008;113(3):573-581. doi:10.1002/cncr.23592
22. Lacarrière E, Galliot I, Gobet F, Sibert L. Leiomyosarcoma of the corpus cavernosum mimicking a Peyronie’s plaque. Urology. 2012;79(4):e53-e54. doi:10.1016/j.urology.2011.07.1410
23. Hamal PB. Leiomyosarcoma of penis—case report and review of the literature. Br J Urol. 1975;47(3):319-324. doi:10.1111/j.1464-410x.1975.tb03974.x
24. Greenwood N, Fox H, Edwards EC. Leiomyosarcoma of the penis. Cancer. 1972;29(2):481-483. doi:10.1002/1097-0142(197202)29:2<481::aid -cncr2820290237>3.0.co;2-q
25. Koizumi H, Nagano K, Kosaka S. A case of penile tumor: combination of leiomyosarcoma and squamous cell carcinoma. Hinyokika Kiyo. 1987;33(9):1489-1491.
26. Romero Gonzalez EJ, Marenco Jimenez JL, Mayorga Pineda MP, Martínez Morán A, Castiñeiras Fernández J. Leiomyosarcoma of the penis, an exceptional entity. Urol Case Rep. 2015;3(3):63-64. doi:10.1016/j.eucr.2014.12.007
Penile cancer is rare with a worldwide incidence of 0.8 cases per 100,000 men.1 The most common type is squamous cell carcinoma (SCC) followed by soft tissue sarcoma (STS) and Kaposi sarcoma.2 Leiomyosarcoma (LMS) is the second most common STS subtype at this location.3 Approximately 50 cases of penile LMS have been reported in the English literature, most as isolated case reports while Fetsch and colleagues reported 14 cases from a single institute.4 We present a case of penile LMS with a review of 31 cases. We also describe presentation, treatment options, and recurrence pattern of this rare malignancy.
Case Presentation
A patient aged 70 years presented to the urology clinic with 1-year history of a slowly enlarging penile mass associated with phimosis. He reported no pain, dysuria, or hesitancy. On examination a 2 × 2-cm smooth, mobile, nonulcerating mass was seen on the tip of his left glans without inguinal lymphadenopathy. He underwent circumcision and excision biopsy that revealed an encapsulated tan-white mass measuring 3 × 2.2 × 1.5 cm under the surface of the foreskin. Histology showed a spindle cell tumor with areas of increased cellularity, prominent atypia, and pleomorphism, focal necrosis, and scattered mitoses, including atypical forms. The tumor stained positive for smooth muscle actin and desmin. Ki-67 staining showed foci with a very high proliferation index (Figure). Resection margins were negative. Final Fédération Nationale des Centres de Lutte Contre Le Cancer score was grade 2 (differentiation, 1; mitotic, 3; necrosis, 1). Computed tomography of the chest, abdomen, and pelvis did not show evidence of metastasis. The tumor was classified as superficial, stage IIA (pT1cN0cM0). Local excision with negative margins was deemed adequate treatment.
Discussion
Penile LMS is rare and arises from smooth muscles, which in the penis can be from dartos fascia, erector pili in the skin covering the shaft, or from tunica media of the superficial vessels and cavernosa.5 It commonly presents as a nodule or ulcer that might be accompanied by paraphimosis, phimosis, erectile dysfunction, and lower urinary tract symptoms depending on the extent of local tissue involvement. In our review of 31 cases, the age at presentation ranged from 38 to 85 years, with 1 case report of LMS in a 6-year-old. The highest incidence was in the 6th decade. Tumor behavior can be indolent or aggressive. Most patients in our review had asymptomatic, slow-growing lesions for 6 to 24 months before presentation—including our patient—while others had an aggressive tumor with symptoms for a few weeks followed by rapid metastatic spread.6,7
Histology and Staging
Diagnosis requires biopsy followed by histologic examination and immunohistochemistry of the lesion. Typically, LMS shows fascicles of spindle cells with varying degrees of nuclear atypia, pleomorphisms, and necrotic regions. Mitotic rate is variable and usually > 5 per high power field. Cells stain positive for smooth muscle actin, desmin, and h-caldesmon.8 TNM (tumor, nodes, metastasis) stage is determined by the American Joint Committee on Cancer guidelines for STS.
Pratt and colleagues were the first to categorize penile LMS as superficial or deep.9 The former includes all lesions superficial to tunica albuginea while the latter run deep to this layer. Anatomical distinction is an important factor in tumor behavior, treatment selection, and prognosis. In our review, we found 14 cases of superficial and 17 cases of deep LMS.
Treatment
There are no established guidelines on optimum treatment of penile LMS. However, we can extrapolate principles from current guidelines on penile cancer, cutaneous leiomyosarcoma, and limb sarcomas. At present, the first-line treatment for superficial penile LMS is wide local excision to achieve negative margins. Circumcision alone might be sufficient for tumors of the distal prepuce, as in our case.10 Radical resection generally is not required for these early-stage tumors. In our review, no patient in this category developed recurrence or metastasis regardless of initial surgery type (Table 1).6,11,12
For deep lesions, partial—if functional penile stump and negative margins can be achieved—or total penectomy is required.10 In our review, more conservative approaches to deep tumors were associated with local recurrences.7,13,14 Lymphatic spread is rare for LMS. Additionally, involvement of local lymph nodes usually coincides with distant spread. Inguinal lymph node dissection is not indicated if initial negative surgical margins are achieved.
For STS at other sites in the body, radiation therapy is recommended postoperatively for high-grade lesions, which can be extrapolated to penile LMS as well. The benefit of preoperative radiation therapy is less certain. In limb sarcomas, radiation is associated with better local control for large-sized tumors and is used for patients with initial unresectable tumors.15 Similar recommendation could be extended to penile LMS with local spread to inguinal lymph nodes, scrotum, or abdominal wall. In our review, postoperative radiation therapy was used in 3 patients with deep tumors.16-18 Of these, short-term relapse occurred in 1 patient.
Chemotherapy for LMS remains controversial. The tumor generally is resistant to chemotherapy and systemic therapy, if employed, is for palliative purpose. The most promising results for adjuvant chemotherapy for resectable STS is seen in limb and uterine sarcomas with high-grade, metastatic, or relapsed tumors but improvement in overall survival has been marginal.19,20Single and multidrug regimens based on doxorubicin, ifosfamide, and gemcitabine have been studied with results showing no efficacy or a slight benefit.8,21 Immunotherapy and targeted therapy for penile STS have not been studied. In our review, postoperative chemotherapy was used for 2 patients with deep tumors and 1 patient with a superficial tumor while preoperative chemotherapy was used for 1 patient.16,18,22 Short-term relapse was seen in 2 of 4 of these patients (Table 2).
Metastatic Disease
LMS tends to metastasize hematogenously and lymphatic spread is uncommon. In our review, 7 patients developed metastasis. These patients had deep tumors at presentation with tumor size > 3 cm. Five of 7 patients had involvement of corpora cavernosa at presentation. The lung was the most common site of metastasis, followed by local extension to lower abdominal wall and scrotum. Of the 7 patients, 3 were treated with initial limited excision or partial penectomy and then experienced local recurrence or distant metastasis.7,13,14,23 This supports the use of radical surgery in large, deep tumors. In an additional 4 cases, metastasis occurred despite initial treatment with total penectomy and use of adjuvant chemoradiation therapy.
In most cases penile LMS is a de novo tumor, however, on occasion it could be accompanied by another epithelial malignancy. Similarly, penile LMS might be a site of recurrence for a primary LMS at another site, as seen in 3 of the reviewed cases. In the first, a patient presented with a nodule on the glans suspicious for SCC, second with synchronous SCC and LMS, and a third case where a patient presented with penile LMS 9 years after being treated for similar tumor in the epididymis.17,24,25
Prognosis
Penile LMS prognosis is difficult to ascertain because reported cases are rare. In our review, the longest documented disease-free survival was 3.5 years for a patient with superficial LMS treated with local excision.26 In cases of distant metastasis, average survival was 4.6 months, while the longest survival since initial presentation and last documented local recurrence was 16 years.14 Five-year survival has not been reported.
Conclusions
LMS of the penis is a rare and potentially aggressive malignancy. It can be classified as superficial or deep based on tumor relation to the tunica albuginea. Deep tumors, those > 3 cm, high-grade lesions, and tumors with involvement of corpora cavernosa, tend to spread locally, metastasize to distant areas, and require more radical surgery with or without postoperative radiation therapy. In comparison, superficial lesions can be treated with local excision only. Both superficial and deep tumors require close follow-up.
Penile cancer is rare with a worldwide incidence of 0.8 cases per 100,000 men.1 The most common type is squamous cell carcinoma (SCC) followed by soft tissue sarcoma (STS) and Kaposi sarcoma.2 Leiomyosarcoma (LMS) is the second most common STS subtype at this location.3 Approximately 50 cases of penile LMS have been reported in the English literature, most as isolated case reports while Fetsch and colleagues reported 14 cases from a single institute.4 We present a case of penile LMS with a review of 31 cases. We also describe presentation, treatment options, and recurrence pattern of this rare malignancy.
Case Presentation
A patient aged 70 years presented to the urology clinic with 1-year history of a slowly enlarging penile mass associated with phimosis. He reported no pain, dysuria, or hesitancy. On examination a 2 × 2-cm smooth, mobile, nonulcerating mass was seen on the tip of his left glans without inguinal lymphadenopathy. He underwent circumcision and excision biopsy that revealed an encapsulated tan-white mass measuring 3 × 2.2 × 1.5 cm under the surface of the foreskin. Histology showed a spindle cell tumor with areas of increased cellularity, prominent atypia, and pleomorphism, focal necrosis, and scattered mitoses, including atypical forms. The tumor stained positive for smooth muscle actin and desmin. Ki-67 staining showed foci with a very high proliferation index (Figure). Resection margins were negative. Final Fédération Nationale des Centres de Lutte Contre Le Cancer score was grade 2 (differentiation, 1; mitotic, 3; necrosis, 1). Computed tomography of the chest, abdomen, and pelvis did not show evidence of metastasis. The tumor was classified as superficial, stage IIA (pT1cN0cM0). Local excision with negative margins was deemed adequate treatment.
Discussion
Penile LMS is rare and arises from smooth muscles, which in the penis can be from dartos fascia, erector pili in the skin covering the shaft, or from tunica media of the superficial vessels and cavernosa.5 It commonly presents as a nodule or ulcer that might be accompanied by paraphimosis, phimosis, erectile dysfunction, and lower urinary tract symptoms depending on the extent of local tissue involvement. In our review of 31 cases, the age at presentation ranged from 38 to 85 years, with 1 case report of LMS in a 6-year-old. The highest incidence was in the 6th decade. Tumor behavior can be indolent or aggressive. Most patients in our review had asymptomatic, slow-growing lesions for 6 to 24 months before presentation—including our patient—while others had an aggressive tumor with symptoms for a few weeks followed by rapid metastatic spread.6,7
Histology and Staging
Diagnosis requires biopsy followed by histologic examination and immunohistochemistry of the lesion. Typically, LMS shows fascicles of spindle cells with varying degrees of nuclear atypia, pleomorphisms, and necrotic regions. Mitotic rate is variable and usually > 5 per high power field. Cells stain positive for smooth muscle actin, desmin, and h-caldesmon.8 TNM (tumor, nodes, metastasis) stage is determined by the American Joint Committee on Cancer guidelines for STS.
Pratt and colleagues were the first to categorize penile LMS as superficial or deep.9 The former includes all lesions superficial to tunica albuginea while the latter run deep to this layer. Anatomical distinction is an important factor in tumor behavior, treatment selection, and prognosis. In our review, we found 14 cases of superficial and 17 cases of deep LMS.
Treatment
There are no established guidelines on optimum treatment of penile LMS. However, we can extrapolate principles from current guidelines on penile cancer, cutaneous leiomyosarcoma, and limb sarcomas. At present, the first-line treatment for superficial penile LMS is wide local excision to achieve negative margins. Circumcision alone might be sufficient for tumors of the distal prepuce, as in our case.10 Radical resection generally is not required for these early-stage tumors. In our review, no patient in this category developed recurrence or metastasis regardless of initial surgery type (Table 1).6,11,12
For deep lesions, partial—if functional penile stump and negative margins can be achieved—or total penectomy is required.10 In our review, more conservative approaches to deep tumors were associated with local recurrences.7,13,14 Lymphatic spread is rare for LMS. Additionally, involvement of local lymph nodes usually coincides with distant spread. Inguinal lymph node dissection is not indicated if initial negative surgical margins are achieved.
For STS at other sites in the body, radiation therapy is recommended postoperatively for high-grade lesions, which can be extrapolated to penile LMS as well. The benefit of preoperative radiation therapy is less certain. In limb sarcomas, radiation is associated with better local control for large-sized tumors and is used for patients with initial unresectable tumors.15 Similar recommendation could be extended to penile LMS with local spread to inguinal lymph nodes, scrotum, or abdominal wall. In our review, postoperative radiation therapy was used in 3 patients with deep tumors.16-18 Of these, short-term relapse occurred in 1 patient.
Chemotherapy for LMS remains controversial. The tumor generally is resistant to chemotherapy and systemic therapy, if employed, is for palliative purpose. The most promising results for adjuvant chemotherapy for resectable STS is seen in limb and uterine sarcomas with high-grade, metastatic, or relapsed tumors but improvement in overall survival has been marginal.19,20Single and multidrug regimens based on doxorubicin, ifosfamide, and gemcitabine have been studied with results showing no efficacy or a slight benefit.8,21 Immunotherapy and targeted therapy for penile STS have not been studied. In our review, postoperative chemotherapy was used for 2 patients with deep tumors and 1 patient with a superficial tumor while preoperative chemotherapy was used for 1 patient.16,18,22 Short-term relapse was seen in 2 of 4 of these patients (Table 2).
Metastatic Disease
LMS tends to metastasize hematogenously and lymphatic spread is uncommon. In our review, 7 patients developed metastasis. These patients had deep tumors at presentation with tumor size > 3 cm. Five of 7 patients had involvement of corpora cavernosa at presentation. The lung was the most common site of metastasis, followed by local extension to lower abdominal wall and scrotum. Of the 7 patients, 3 were treated with initial limited excision or partial penectomy and then experienced local recurrence or distant metastasis.7,13,14,23 This supports the use of radical surgery in large, deep tumors. In an additional 4 cases, metastasis occurred despite initial treatment with total penectomy and use of adjuvant chemoradiation therapy.
In most cases penile LMS is a de novo tumor, however, on occasion it could be accompanied by another epithelial malignancy. Similarly, penile LMS might be a site of recurrence for a primary LMS at another site, as seen in 3 of the reviewed cases. In the first, a patient presented with a nodule on the glans suspicious for SCC, second with synchronous SCC and LMS, and a third case where a patient presented with penile LMS 9 years after being treated for similar tumor in the epididymis.17,24,25
Prognosis
Penile LMS prognosis is difficult to ascertain because reported cases are rare. In our review, the longest documented disease-free survival was 3.5 years for a patient with superficial LMS treated with local excision.26 In cases of distant metastasis, average survival was 4.6 months, while the longest survival since initial presentation and last documented local recurrence was 16 years.14 Five-year survival has not been reported.
Conclusions
LMS of the penis is a rare and potentially aggressive malignancy. It can be classified as superficial or deep based on tumor relation to the tunica albuginea. Deep tumors, those > 3 cm, high-grade lesions, and tumors with involvement of corpora cavernosa, tend to spread locally, metastasize to distant areas, and require more radical surgery with or without postoperative radiation therapy. In comparison, superficial lesions can be treated with local excision only. Both superficial and deep tumors require close follow-up.
1. Montes Cardona CE, García-Perdomo HA. Incidence of penile cancer worldwide: systematic review and meta-analysis. Rev Panam Salud Publica. 2017;41:e117. Published 2017 Nov 30. doi:10.26633/RPSP.2017.117
2. Volker HU, Zettl A, Haralambieva E, et al. Leiomyosarcoma of the larynx as a local relapse of squamous cell carcinoma—report of an unusual case. Head Neck. 2010;32(5):679-683. doi:10.1002/hed.21127
3. Wollina U, Steinbach F, Verma S, et al. Penile tumours: a review. J Eur Acad Dermatol Venereol. 2014;28(10):1267-1276. doi:10.1111/jdv.12491
4. Fetsch JF, Davis CJ Jr, Miettinen M, Sesterhenn IA. Leiomyosarcoma of the penis: a clinicopathologic study of 14 cases with review of the literature and discussion of the differential diagnosis. Am J Surg Pathol. 2004;28(1):115-125. doi:10.1097/00000478-200401000-00014
5. Sundersingh S, Majhi U, Narayanaswamy K, Balasubramanian S. Primary leiomyosarcoma of the penis. Indian J Pathol Microbiol. 2009;52(3):447-448. doi:10.4103/0377-4929.55028
6. Mendis D, Bott SR, Davies JH. Subcutaneous leiomyosarcoma of the frenulum. Scientific World J. 2005;5:571-575. doi:10.1100/tsw.2005.76
7. Elem B, Nieslanik J. Leiomyosarcoma of the penis. Br J Urol. 1979;51(1):46. doi:10.1111/j.1464-410x.1979.tb04244.x
8. Serrano C, George S. Leiomyosarcoma. Hematol Oncol Clin North Am. 2013;27(5):957-974. doi:10.1016/j.hoc.2013.07.002
9. Pratt RM, Ross RT. Leiomyosarcoma of the penis. A report of a case. Br J Surg. 1969;56(11):870-872. doi:10.1002/bjs.1800561122
10. National Comprehensive Cancer Network. Penile cancer. NCCN evidence blocks. Version 2.2022 Updated January 26, 2022. Accessed March 16, 2022. https://www.nccn.org/professionals/physician_gls/pdf/penile_blocks.pdf
11. Ashley DJ, Edwards EC. Sarcoma of the penis; leiomyosarcoma of the penis: report of a case with a review of the literature on sarcoma of the penis. Br J Surg. 1957;45(190):170-179. doi:10.1002/bjs.18004519011
12. Pow-Sang MR, Orihuela E. Leiomyosarcoma of the penis. J Urol. 1994;151(6):1643-1645. doi:10.1016/s0022-5347(17)35328-413. Isa SS, Almaraz R, Magovern J. Leiomyosarcoma of the penis. Case report and review of the literature. Cancer. 1984;54(5):939-942. doi:10.1002/1097-0142(19840901)54:5<939::aid-cncr2820540533>3.0.co;2-y
14. Hutcheson JB, Wittaker WW, Fronstin MH. Leiomyosarcoma of the penis: case report and review of literature. J Urol. 1969;101(6):874-875. doi:10.1016/s0022-5347(17)62446-7
15. Grimer R, Judson I, Peake D, et al. Guidelines for the management of soft tissue sarcomas. Sarcoma. 2010;2010:506182. doi:10.1155/2010/506182
16. McDonald MW, O’Connell JR, Manning JT, Benjamin RS. Leiomyosarcoma of the penis. J Urol. 1983;130(4):788-789. doi:10.1016/s0022-5347(17)51464-0
17. Planz B, Brunner K, Kalem T, Schlick RW, Kind M. Primary leiomyosarcoma of the epididymis and late recurrence on the penis. J Urol. 1998;159(2):508. doi:10.1016/s0022-5347(01)63966-1
18. Smart RH. Leiomyosarcoma of the penis. J Urol. 1984;132(2):356-357. doi:10.1016/s0022-5347(17)49624-8
19. Patrikidou A, Domont J, Cioffi A, Le Cesne A. Treating soft tissue sarcomas with adjuvant chemotherapy. Curr Treat Options Oncol. 2011;12(1):21-31. doi:10.1007/s11864-011-0145-5
20. Italiano A, Delva F, Mathoulin-Pelissier S, et al. Effect of adjuvant chemotherapy on survival in FNCLCC grade 3 soft tissue sarcomas: a multivariate analysis of the French Sarcoma Group Database. Ann Oncol. 2010;21(12):2436-2441. doi:10.1093/annonc/mdq238
21. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008;113(3):573-581. doi:10.1002/cncr.23592
22. Lacarrière E, Galliot I, Gobet F, Sibert L. Leiomyosarcoma of the corpus cavernosum mimicking a Peyronie’s plaque. Urology. 2012;79(4):e53-e54. doi:10.1016/j.urology.2011.07.1410
23. Hamal PB. Leiomyosarcoma of penis—case report and review of the literature. Br J Urol. 1975;47(3):319-324. doi:10.1111/j.1464-410x.1975.tb03974.x
24. Greenwood N, Fox H, Edwards EC. Leiomyosarcoma of the penis. Cancer. 1972;29(2):481-483. doi:10.1002/1097-0142(197202)29:2<481::aid -cncr2820290237>3.0.co;2-q
25. Koizumi H, Nagano K, Kosaka S. A case of penile tumor: combination of leiomyosarcoma and squamous cell carcinoma. Hinyokika Kiyo. 1987;33(9):1489-1491.
26. Romero Gonzalez EJ, Marenco Jimenez JL, Mayorga Pineda MP, Martínez Morán A, Castiñeiras Fernández J. Leiomyosarcoma of the penis, an exceptional entity. Urol Case Rep. 2015;3(3):63-64. doi:10.1016/j.eucr.2014.12.007
1. Montes Cardona CE, García-Perdomo HA. Incidence of penile cancer worldwide: systematic review and meta-analysis. Rev Panam Salud Publica. 2017;41:e117. Published 2017 Nov 30. doi:10.26633/RPSP.2017.117
2. Volker HU, Zettl A, Haralambieva E, et al. Leiomyosarcoma of the larynx as a local relapse of squamous cell carcinoma—report of an unusual case. Head Neck. 2010;32(5):679-683. doi:10.1002/hed.21127
3. Wollina U, Steinbach F, Verma S, et al. Penile tumours: a review. J Eur Acad Dermatol Venereol. 2014;28(10):1267-1276. doi:10.1111/jdv.12491
4. Fetsch JF, Davis CJ Jr, Miettinen M, Sesterhenn IA. Leiomyosarcoma of the penis: a clinicopathologic study of 14 cases with review of the literature and discussion of the differential diagnosis. Am J Surg Pathol. 2004;28(1):115-125. doi:10.1097/00000478-200401000-00014
5. Sundersingh S, Majhi U, Narayanaswamy K, Balasubramanian S. Primary leiomyosarcoma of the penis. Indian J Pathol Microbiol. 2009;52(3):447-448. doi:10.4103/0377-4929.55028
6. Mendis D, Bott SR, Davies JH. Subcutaneous leiomyosarcoma of the frenulum. Scientific World J. 2005;5:571-575. doi:10.1100/tsw.2005.76
7. Elem B, Nieslanik J. Leiomyosarcoma of the penis. Br J Urol. 1979;51(1):46. doi:10.1111/j.1464-410x.1979.tb04244.x
8. Serrano C, George S. Leiomyosarcoma. Hematol Oncol Clin North Am. 2013;27(5):957-974. doi:10.1016/j.hoc.2013.07.002
9. Pratt RM, Ross RT. Leiomyosarcoma of the penis. A report of a case. Br J Surg. 1969;56(11):870-872. doi:10.1002/bjs.1800561122
10. National Comprehensive Cancer Network. Penile cancer. NCCN evidence blocks. Version 2.2022 Updated January 26, 2022. Accessed March 16, 2022. https://www.nccn.org/professionals/physician_gls/pdf/penile_blocks.pdf
11. Ashley DJ, Edwards EC. Sarcoma of the penis; leiomyosarcoma of the penis: report of a case with a review of the literature on sarcoma of the penis. Br J Surg. 1957;45(190):170-179. doi:10.1002/bjs.18004519011
12. Pow-Sang MR, Orihuela E. Leiomyosarcoma of the penis. J Urol. 1994;151(6):1643-1645. doi:10.1016/s0022-5347(17)35328-413. Isa SS, Almaraz R, Magovern J. Leiomyosarcoma of the penis. Case report and review of the literature. Cancer. 1984;54(5):939-942. doi:10.1002/1097-0142(19840901)54:5<939::aid-cncr2820540533>3.0.co;2-y
14. Hutcheson JB, Wittaker WW, Fronstin MH. Leiomyosarcoma of the penis: case report and review of literature. J Urol. 1969;101(6):874-875. doi:10.1016/s0022-5347(17)62446-7
15. Grimer R, Judson I, Peake D, et al. Guidelines for the management of soft tissue sarcomas. Sarcoma. 2010;2010:506182. doi:10.1155/2010/506182
16. McDonald MW, O’Connell JR, Manning JT, Benjamin RS. Leiomyosarcoma of the penis. J Urol. 1983;130(4):788-789. doi:10.1016/s0022-5347(17)51464-0
17. Planz B, Brunner K, Kalem T, Schlick RW, Kind M. Primary leiomyosarcoma of the epididymis and late recurrence on the penis. J Urol. 1998;159(2):508. doi:10.1016/s0022-5347(01)63966-1
18. Smart RH. Leiomyosarcoma of the penis. J Urol. 1984;132(2):356-357. doi:10.1016/s0022-5347(17)49624-8
19. Patrikidou A, Domont J, Cioffi A, Le Cesne A. Treating soft tissue sarcomas with adjuvant chemotherapy. Curr Treat Options Oncol. 2011;12(1):21-31. doi:10.1007/s11864-011-0145-5
20. Italiano A, Delva F, Mathoulin-Pelissier S, et al. Effect of adjuvant chemotherapy on survival in FNCLCC grade 3 soft tissue sarcomas: a multivariate analysis of the French Sarcoma Group Database. Ann Oncol. 2010;21(12):2436-2441. doi:10.1093/annonc/mdq238
21. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008;113(3):573-581. doi:10.1002/cncr.23592
22. Lacarrière E, Galliot I, Gobet F, Sibert L. Leiomyosarcoma of the corpus cavernosum mimicking a Peyronie’s plaque. Urology. 2012;79(4):e53-e54. doi:10.1016/j.urology.2011.07.1410
23. Hamal PB. Leiomyosarcoma of penis—case report and review of the literature. Br J Urol. 1975;47(3):319-324. doi:10.1111/j.1464-410x.1975.tb03974.x
24. Greenwood N, Fox H, Edwards EC. Leiomyosarcoma of the penis. Cancer. 1972;29(2):481-483. doi:10.1002/1097-0142(197202)29:2<481::aid -cncr2820290237>3.0.co;2-q
25. Koizumi H, Nagano K, Kosaka S. A case of penile tumor: combination of leiomyosarcoma and squamous cell carcinoma. Hinyokika Kiyo. 1987;33(9):1489-1491.
26. Romero Gonzalez EJ, Marenco Jimenez JL, Mayorga Pineda MP, Martínez Morán A, Castiñeiras Fernández J. Leiomyosarcoma of the penis, an exceptional entity. Urol Case Rep. 2015;3(3):63-64. doi:10.1016/j.eucr.2014.12.007
Inverted Appendix in a Patient With Weakness and Occult Bleeding
Appendiceal mucinous neoplasms (AMNs) are rare tumors of the appendix that can be asymptomatic or present with acute right lower quadrant (RLQ) pain mimicking appendicitis. Due to their potential to cause either no symptoms or nonspecific symptoms, such as abdominal pain, nausea, or vomiting, AMNs are often found incidentally during appendectomies or, even more rarely, colonoscopies. Most AMNs grow slowly and have little metastatic potential. However, due to potential complications, such as bowel obstruction and rupture, timely detection and removal of AMN is essential. We describe the case of a patient who appeared to have acute appendicitis complicated by rupture on imaging who was found instead to have a perforated low-grade AMN during surgery.
Case Presentation
A male patient aged 72 years with a history of type 2 diabetes mellitus, hypertension, and aortic stenosis, but no prior abdominal surgery, presented with a chief concern of generalized weakness. As part of the workup for his weakness, a computed tomography (CT) scan of the abdomen was performed which showed an RLQ phlegmon and mild fat stranding in the area. Imaging also revealed an asymptomatic gallstone measuring 1.5 cm with no evidence of cholecystitis. The patient had no fever and reported no abdominal pain, nausea, vomiting, or change in bowel habits. On physical examination, the patient’s abdomen was soft, nontender, and nondistended with normoactive bowel sounds and no rebound or guarding.
To manage the appendicitis, the patient started a 2-week course of amoxicillin clavulanate 875 mg twice daily and was instructed to schedule an interval appendectomy in the coming months. Four days later, during a follow-up with his primary care physician, he was found to be asymptomatic. However, at this visit his stool was found to be positive for occult blood. Given this finding and the lack of a previous colonoscopy, the patient underwent a colonoscopy, which revealed bulging at the appendiceal orifice, consistent with an inverted appendix. Portions of the appendix were biopsied (Figure 1). Histologic analysis of the appendiceal biopsies revealed no dysplasia or malignancy. The colonoscopy also revealed an 8-mm sessile polyp in the ascending colon which was resected, and histologic analysis of this polyp revealed a low-grade tubular adenoma. Additionally, a large angiodysplastic lesion was found in the ascending colon as well as external and medium-sized internal hemorrhoids.
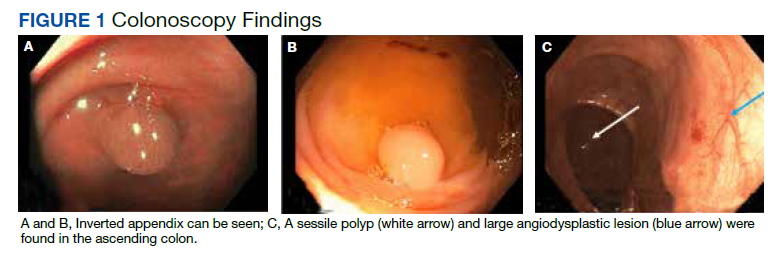
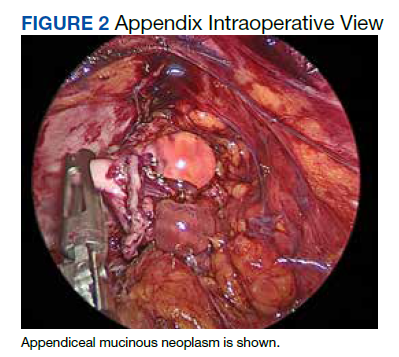
Six weeks after the colonoscopy, the patient was taken to the operating room for a laparoscopic appendectomy. Upon entry of the abdomen, extensive adhesions throughout the RLQ were found which required adhesiolysis. A calcified fecalith adherent to the mesentery of the small intestine in the RLQ was also found and resected. After lysis of the adhesions, the appendix and fibrotic tissue surrounding it could be seen (Figure 2). The appendix was dilated and the tip showed perforation. During dissection of the appendix, clear gelatinous material was found coming from the appendiceal lumen as well as from the fibrotic tissue around the appendix. On postoperative day 1 the appendix was resected and the patient was discharged.
Histologic specimens of the appendix were notable for evidence of perforation and neoplasia leading to a diagnosis of low-grade AMN. The presence of atypical mucinous epithelial cells on the serosal surface of the appendix, confirmed with a positive pancytokeratin stain, provided histologic evidence of appendiceal perforation (Figure 3). The presence of nuclear atypia demonstrated that the appendix was involved by a neoplastic process. Additionally, attenuation of the normal appendiceal epithelium, evidence of a chronic process, further helped to differentiate the AMN from complicated appendicitis. The presence of mucin involving the serosa of the appendix led to the classification of this patient’s neoplasm as grade pT4a. Of note, histologic examination demonstrated that the surgical margins contained tumor cells.
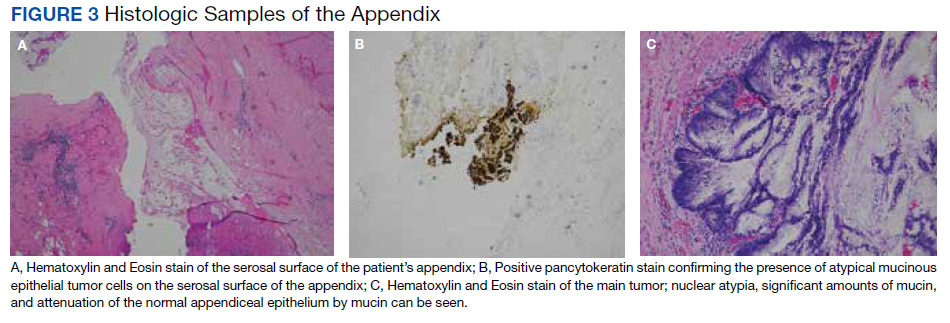
Given the positive margins of the resected AMN and the relatively large size of the neoplasm, a laparoscopic right hemicolectomy was performed 2 months later. Although multiple adhesions were found in the terminal ileum, cecum, and ascending colon during the hemicolectomy, no mucinous lesions were observed grossly. Histologic analysis showed no residual neoplasm as well as no lymph node involvement. On postoperative day 3 the patient was discharged and had an uneventful recovery. At his first surveillance visit 6 months after his hemicolectomy, the patient appeared to be doing well and reported no abdominal pain, nausea, vomiting, change in bowel habits, or any blood in the stool.
Discussion
AMNs are rare tumors with an annual age-adjusted incidence of approximately 0.12 per 1,000,000 people.1 These neoplasms can present as acute or chronic abdominal pain, gastrointestinal bleeding, intestinal obstruction, or acute abdomen.2-4 Most AMNs, however, are asymptomatic and are usually found incidentally during appendectomies for appendicitis, and can even be found during colonoscopies,such as in this case.5,6
Low-grade AMNs are distinguished from appendiceal mucinous adenocarcinomas by their lack of wall invasion.7 Additionally, low-grade AMNs have a very good prognosis as even neoplasms that have spread outside of the appendix have a 5-year overall survival rate of 79 to 86%.8 These low-grade neoplasms also have extremely low rates of recurrence after resection.9 In contrast, appendiceal mucinous adenocarcinomas have a much worse prognosis with a 5-year overall survival rate of 53.6%.10
Treatment of AMNs depends on the extent of their spread. Neoplasms that are confined to the appendix can typically be treated with appendectomy alone, while those that have spread beyond the appendix may require cytoreductive surgery and chemotherapy, namely, hyperthermic intraperitoneal chemotherapy (HIPEC), in addition to appendectomy.11 Cases in which neoplasms are not confined to the appendix also require more frequent surveillance for recurrence as compared to appendix-restricted neoplasms.11
Appendiceal inversion is a rare finding in adults with an estimated prevalence of 0.01%.6 Not only is appendiceal inversion rare in and of itself, it is even more rarely found in combination with appendiceal neoplasms.6 Other causes of appendiceal inversion include intussusception, acute appendicitis, appendiceal nodule, or even iatrogenic due to appendectomy.12-14 While appendiceal inversion can be completely benign, because these morphological changes of the appendix can resemble a polyp, these lesions are often biopsied and/or resected.15 However, lesion resection may be quite problematic due to high risk of bleeding and perforation.15 In order to avoid the risks associated with resection of a potentially benign finding, biopsy should be performed prior to any attempted resection of inverted appendices.15
Another interesting aspect of this case is the finding of fecal occult blood. The differential for fecal occult blood is quite broad and the patient had multiple conditions that could have led to the finding of occult blood in his stool. Hemorrhoids can cause a positive result on a fecal occult blood test (FOBT) although this is relatively uncommon, and hemorrhoids are more likely to cause frank blood to be seen.16 The sessile polyp found in the patient’s colon may also have caused the FOBT to be positive. This patient was also found to have an angiodysplasia (a finding that is associated with aortic stenosis, which this patient has a history of) which can also cause gastrointestinal bleeding.17 Lastly, AMNs may also cause gastrointestinal bleeding and thus a positive FOBT, although bleeding is a relatively uncommon presentation of AMNs, especially those that are low-grade as in this case.18
This case also highlights the association between appendiceal neoplasms and colonic neoplastic lesions. Patients with appendiceal neoplasms are more likely to have colonic neoplastic lesions than patients without appendiceal neoplasms.19 Studies have found that approximately 13 to 42% of patients with appendiceal neoplasms also have colonic neoplastic lesions.19 The majority of these lesions in the colon were right-sided and this finding was also seen in this case as the patient’s polyp was located in the ascending colon.19 Due to this association between appendiceal and colorectal neoplasia, the American Society of Colon and Rectal Surgeons strongly recommends that patients with appendiceal neoplasms or who are suspected of having them receive a colonoscopy.19
Additionally, perforation of an AMN, as was seen in this case, is a finding that should raise significant concern. Perforation of an AMN allows for the spread of malignant mucinous epithelial cells throughout the abdomen. The finding of extensive adhesions throughout the patient’s RLQ was unexpected as abdominal adhesions are most often seen in patients with a history of abdominal surgeries. Considering the lack of any prior abdominal surgeries in this patient, these adhesions were most likely the result of the spread and proliferation of malignant mucinous epithelial cells from the perforated AMN in the RLQ.20 The adhesiolysis performed in this case was thus not only important in order to visualize the appendix, but also for preventing future complications of abdominal adhesions such as bowel obstruction.20 Perforated AMN is also so concerning because it can potentially lead to pseudomyxoma peritonei—a condition in which malignant mucinous epithelial cells accumulate in the abdomen.21 Pseudomyxoma peritonei is extremely rare with an incidence of approximately 1 to 2 cases per million per year.22 Early recognition of AMNs and surgical referral are critically important as pseudomyxoma peritonei is difficult to treat, has a high rate of recurrence, and can be fatal.23
Lastly, this case highlights how findings of a ruptured appendix and/or mucin surrounding the appendix on imaging should warrant laparoscopy because only pathologic analysis of the appendix can definitively rule out AMNs. The utility of laparoscopic evaluation of the appendix is especially apparent as nonsurgical treatment of appendicitis using antibiotics is gaining favor for treating even complicated appendicitis.24 Appendicitis is much more common than AMNs. However, had the patient in this case only been given antibiotics for his suspected complicated appendicitis without any colonoscopy or appendectomy, the neoplasm in his appendix would have gone undetected and continued to grow, causing significant complications. The patient’s age at presentation in this case also necessitated laparoscopic evaluation of the appendix as the incidence of AMNs is highest among patients aged > 60 years.25 Additionally, because appendiceal inversion may be seen with AMNs,the patient’s inverted appendix seen during his colonoscopy was another compelling reason for laparoscopic evaluation of his appendix.6
Conclusions
AMNs can present with nonspecific symptoms or can be completely asymptomatic and are often found incidentally during colonoscopies or appendectomies for acute appendicitis. While it is true that AMNs have low metastatic potential and grow slowly, AMNs can rupture leading to pseudomyxoma peritonei or even cause bowel obstruction warranting timely identification and removal of these neoplasms. Laparoscopic evaluation in cases of ruptured appendices is critical not only for treatment, but also for determining the presence of a potential underlying appendiceal malignancy. Although AMNs are a rare pathology, physicians should still consider the possibility of these neoplasms even when imaging findings suggest appendicitis. Having AMNs as part of the differential diagnosis is especially necessary in cases, such as this one, in which the patient has appendiceal inversion, is aged > 50 years, and has concurrent colorectal neoplasms.
1. Shaib WL, Goodman M, Chen Z, et al. Incidence and survival of appendiceal mucinous neoplasms: a SEER analysis. Am J Clin Oncol. 2017;40(6):569-573. doi:10.1097/COC.0000000000000210
2. Kehagias I, Zygomalas A, Markopoulos G, Papandreou T, Kraniotis P. Diagnosis and treatment of mucinous appendiceal neoplasm presented as acute appendicitis. Case Rep Oncol Med. 2016;3:1-6. doi:10.1155/2016/2161952
3. Karatas M, Simsek C, Gunay S, et al. Acute lower gastrointestinal bleeding due to low-grade mucinous neoplasm of appendix. Acta Chir Belg. 2020;120(4):1-4. doi:10.1080/00015458.2020.1860397
4. Mourad FH, Hussein M, Bahlawan M, Haddad M, Tawil A. Intestinal obstruction secondary to appendiceal mucocele. Dig Dis Sci. 1999;44(8):1594-1599. doi:10.1023/a:1026615010989
5. Benabe SH, Leeman R, Brady AC, Hirzel A, Langshaw AH. Low-grade appendiceal mucinous neoplasm in an adolescent patient with untreated Crohn’s disease. ACG Case Reports J. 2020;7(3). doi:10.14309/crj.0000000000000338
6. Liu X, Liu G, Liu Y, et al. Complete appendiceal inversion with local high-grade intraepithelial neoplasia in an adult female: A case report. BMC Surg. 2019;19(1). doi:10.1186/s12893-019-0632-3
7. Gündog˘ar ÖS, Kımılog˘lu ES, Komut NS, et al. The evaluation of appendiceal mucinous neoplasms with a new classification system. Turk J Gastroenterol. 2018;29(5):532-542. doi:10.5152/tjg.2018.17605
8. Misdraji J, Yantiss RK, Graeme-Cook FM, Balis UJ, Young RH. Appendiceal mucinous neoplasms: a clinicopathologic analysis of 107 cases. Am J Surg Pathol. 2003;27(8):1089-1103. doi:10.1097/00000478-200308000-00006
9. Pai RK, Beck AH, Norton JA, Longacre TA. Appendiceal mucinous neoplasms: clinicopathologic study of 116 cases with analysis of factors predicting recurrence. Am J Surg Pathol. 2009;33(10):1425-1439. doi:10.1097/PAS.0b013e3181af6067
10. Asare EA, Compton CC, Hanna NN, et al. The impact of stage, grade, and mucinous histology on the efficacy of systemic chemotherapy in adenocarcinomas of the appendix: analysis of the National Cancer Data Base. Cancer. 2015;122(2):213-221. doi:10.1002/cncr.29744
11. Shaib WL, Assi R, Shamseddine A, et al. Appendiceal mucinous neoplasms: diagnosis and management. Oncologist. 2018;23(1):137. doi:10.1634/theoncologist.2017-0081erratum
12. Tran C, Sakioka J, Nguyen E, Beutler BD, Hsu J. An inverted appendix found on routine colonoscopy: a case report with discussion of imaging findings. Radiol Case Rep. 2019;14(8):952-955. doi:10.1016/j.radcr.2019.05.022
13. Shafi A, Azab M. A case of everted appendix with benign appendiceal nodule masquerading as appendiceal mucocele: a case report. Am J Gastroenterol. 2018;113:S1436. doi:10.14309/00000434-201810001-02585
14. Pokhrel B, Chang M, Anand G, Savides T, Fehmi S. Appendiceal mucinous neoplasm in an inverted appendix found on prior colonoscopy. VideoGIE. 2020;5(1):34-36. doi:10.1016/j.vgie.2019.09.013
15. Johnson EK, Arcila ME, Steele SR. Appendiceal inversion: a diagnostic and therapeutic dilemma. JSLS. 2009;13(1):92-95.
16. van Turenhout ST, Oort FA, sive Droste JST, et al. Hemorrhoids detected at colonoscopy: an infrequent cause of false-positive fecal immunochemical test results. Gastrointest Endosc. 2012;76(1):136-143. doi:10.1016/j.gie.2012.03.169
17. Hudzik B, Wilczek K, Gasior M. Heyde syndrome: Gastrointestinal bleeding and aortic stenosis. CMAJ. 2016;188(2):135-138. doi:10.1503/cmaj.150194
18. Leonards LM, Pahwa A, Patel MK, Petersen J, Nguyen MJ, Jude CM. Neoplasms of the appendix: pictorial review with clinical and pathologic correlation. RadioGraphics. 2017;37(4):1059-1083. doi:10.1148/rg.2017160150
19. Glasgow SC, Gaertner W, Stewart D, et al. The American Society of Colon and Rectal Surgeons, clinical practice guidelines for the management of appendiceal neoplasms. Dis Colon Rectum. 2019;62(12):1425-1438. doi:10.1097/DCR.0000000000001530
20. Panagopoulos P, Tsokaki T, Misiakos E, et al. Low-grade appendiceal mucinous neoplasm presenting as an adnexal mass. Case Reports in Obstetrics and Gynecology. 2017;2017:1-3. doi:10.1155/2017/7165321
21. Ramaswamy V. Pathology of mucinous appendiceal tumors and pseudomyxoma peritonei. Indian J Surg Oncol. 2016;7(2):258-267. doi:10.1007/s13193-016-0516-2.
22. Bevan KE, Mohamed F, Moran BJ. Pseudomyxoma peritonei. World J Gastrointest Oncol. 2010;2(1):44-50. doi:10.4251/wjgo.v2.i1.44
23. Mercier F, Dagbert F, Pocard M, et al. Recurrence of pseudomyxoma peritonei after cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. BJS Open. 2018;3(2):195-202. doi:10.1002/bjs5.97
24. David A, Dodgion C, Eddine SBZ, Davila D, Webb TP, Trevino CM. Perforated appendicitis: Short duration antibiotics are noninferior to traditional long duration antibiotics. Surgery. 2020;167(2):475-477. doi:10.1016/j.surg.2019.08.007
25. Raijman I, Leong S, Hassaram S, Marcon NE. Appendiceal mucocele: Endoscopic appearance. Endoscopy. 1994;26(3):326-328. doi:10.1055/s-2007-1008979
Appendiceal mucinous neoplasms (AMNs) are rare tumors of the appendix that can be asymptomatic or present with acute right lower quadrant (RLQ) pain mimicking appendicitis. Due to their potential to cause either no symptoms or nonspecific symptoms, such as abdominal pain, nausea, or vomiting, AMNs are often found incidentally during appendectomies or, even more rarely, colonoscopies. Most AMNs grow slowly and have little metastatic potential. However, due to potential complications, such as bowel obstruction and rupture, timely detection and removal of AMN is essential. We describe the case of a patient who appeared to have acute appendicitis complicated by rupture on imaging who was found instead to have a perforated low-grade AMN during surgery.
Case Presentation
A male patient aged 72 years with a history of type 2 diabetes mellitus, hypertension, and aortic stenosis, but no prior abdominal surgery, presented with a chief concern of generalized weakness. As part of the workup for his weakness, a computed tomography (CT) scan of the abdomen was performed which showed an RLQ phlegmon and mild fat stranding in the area. Imaging also revealed an asymptomatic gallstone measuring 1.5 cm with no evidence of cholecystitis. The patient had no fever and reported no abdominal pain, nausea, vomiting, or change in bowel habits. On physical examination, the patient’s abdomen was soft, nontender, and nondistended with normoactive bowel sounds and no rebound or guarding.
To manage the appendicitis, the patient started a 2-week course of amoxicillin clavulanate 875 mg twice daily and was instructed to schedule an interval appendectomy in the coming months. Four days later, during a follow-up with his primary care physician, he was found to be asymptomatic. However, at this visit his stool was found to be positive for occult blood. Given this finding and the lack of a previous colonoscopy, the patient underwent a colonoscopy, which revealed bulging at the appendiceal orifice, consistent with an inverted appendix. Portions of the appendix were biopsied (Figure 1). Histologic analysis of the appendiceal biopsies revealed no dysplasia or malignancy. The colonoscopy also revealed an 8-mm sessile polyp in the ascending colon which was resected, and histologic analysis of this polyp revealed a low-grade tubular adenoma. Additionally, a large angiodysplastic lesion was found in the ascending colon as well as external and medium-sized internal hemorrhoids.
Six weeks after the colonoscopy, the patient was taken to the operating room for a laparoscopic appendectomy. Upon entry of the abdomen, extensive adhesions throughout the RLQ were found which required adhesiolysis. A calcified fecalith adherent to the mesentery of the small intestine in the RLQ was also found and resected. After lysis of the adhesions, the appendix and fibrotic tissue surrounding it could be seen (Figure 2). The appendix was dilated and the tip showed perforation. During dissection of the appendix, clear gelatinous material was found coming from the appendiceal lumen as well as from the fibrotic tissue around the appendix. On postoperative day 1 the appendix was resected and the patient was discharged.
Histologic specimens of the appendix were notable for evidence of perforation and neoplasia leading to a diagnosis of low-grade AMN. The presence of atypical mucinous epithelial cells on the serosal surface of the appendix, confirmed with a positive pancytokeratin stain, provided histologic evidence of appendiceal perforation (Figure 3). The presence of nuclear atypia demonstrated that the appendix was involved by a neoplastic process. Additionally, attenuation of the normal appendiceal epithelium, evidence of a chronic process, further helped to differentiate the AMN from complicated appendicitis. The presence of mucin involving the serosa of the appendix led to the classification of this patient’s neoplasm as grade pT4a. Of note, histologic examination demonstrated that the surgical margins contained tumor cells.
Given the positive margins of the resected AMN and the relatively large size of the neoplasm, a laparoscopic right hemicolectomy was performed 2 months later. Although multiple adhesions were found in the terminal ileum, cecum, and ascending colon during the hemicolectomy, no mucinous lesions were observed grossly. Histologic analysis showed no residual neoplasm as well as no lymph node involvement. On postoperative day 3 the patient was discharged and had an uneventful recovery. At his first surveillance visit 6 months after his hemicolectomy, the patient appeared to be doing well and reported no abdominal pain, nausea, vomiting, change in bowel habits, or any blood in the stool.
Discussion
AMNs are rare tumors with an annual age-adjusted incidence of approximately 0.12 per 1,000,000 people.1 These neoplasms can present as acute or chronic abdominal pain, gastrointestinal bleeding, intestinal obstruction, or acute abdomen.2-4 Most AMNs, however, are asymptomatic and are usually found incidentally during appendectomies for appendicitis, and can even be found during colonoscopies,such as in this case.5,6
Low-grade AMNs are distinguished from appendiceal mucinous adenocarcinomas by their lack of wall invasion.7 Additionally, low-grade AMNs have a very good prognosis as even neoplasms that have spread outside of the appendix have a 5-year overall survival rate of 79 to 86%.8 These low-grade neoplasms also have extremely low rates of recurrence after resection.9 In contrast, appendiceal mucinous adenocarcinomas have a much worse prognosis with a 5-year overall survival rate of 53.6%.10
Treatment of AMNs depends on the extent of their spread. Neoplasms that are confined to the appendix can typically be treated with appendectomy alone, while those that have spread beyond the appendix may require cytoreductive surgery and chemotherapy, namely, hyperthermic intraperitoneal chemotherapy (HIPEC), in addition to appendectomy.11 Cases in which neoplasms are not confined to the appendix also require more frequent surveillance for recurrence as compared to appendix-restricted neoplasms.11
Appendiceal inversion is a rare finding in adults with an estimated prevalence of 0.01%.6 Not only is appendiceal inversion rare in and of itself, it is even more rarely found in combination with appendiceal neoplasms.6 Other causes of appendiceal inversion include intussusception, acute appendicitis, appendiceal nodule, or even iatrogenic due to appendectomy.12-14 While appendiceal inversion can be completely benign, because these morphological changes of the appendix can resemble a polyp, these lesions are often biopsied and/or resected.15 However, lesion resection may be quite problematic due to high risk of bleeding and perforation.15 In order to avoid the risks associated with resection of a potentially benign finding, biopsy should be performed prior to any attempted resection of inverted appendices.15
Another interesting aspect of this case is the finding of fecal occult blood. The differential for fecal occult blood is quite broad and the patient had multiple conditions that could have led to the finding of occult blood in his stool. Hemorrhoids can cause a positive result on a fecal occult blood test (FOBT) although this is relatively uncommon, and hemorrhoids are more likely to cause frank blood to be seen.16 The sessile polyp found in the patient’s colon may also have caused the FOBT to be positive. This patient was also found to have an angiodysplasia (a finding that is associated with aortic stenosis, which this patient has a history of) which can also cause gastrointestinal bleeding.17 Lastly, AMNs may also cause gastrointestinal bleeding and thus a positive FOBT, although bleeding is a relatively uncommon presentation of AMNs, especially those that are low-grade as in this case.18
This case also highlights the association between appendiceal neoplasms and colonic neoplastic lesions. Patients with appendiceal neoplasms are more likely to have colonic neoplastic lesions than patients without appendiceal neoplasms.19 Studies have found that approximately 13 to 42% of patients with appendiceal neoplasms also have colonic neoplastic lesions.19 The majority of these lesions in the colon were right-sided and this finding was also seen in this case as the patient’s polyp was located in the ascending colon.19 Due to this association between appendiceal and colorectal neoplasia, the American Society of Colon and Rectal Surgeons strongly recommends that patients with appendiceal neoplasms or who are suspected of having them receive a colonoscopy.19
Additionally, perforation of an AMN, as was seen in this case, is a finding that should raise significant concern. Perforation of an AMN allows for the spread of malignant mucinous epithelial cells throughout the abdomen. The finding of extensive adhesions throughout the patient’s RLQ was unexpected as abdominal adhesions are most often seen in patients with a history of abdominal surgeries. Considering the lack of any prior abdominal surgeries in this patient, these adhesions were most likely the result of the spread and proliferation of malignant mucinous epithelial cells from the perforated AMN in the RLQ.20 The adhesiolysis performed in this case was thus not only important in order to visualize the appendix, but also for preventing future complications of abdominal adhesions such as bowel obstruction.20 Perforated AMN is also so concerning because it can potentially lead to pseudomyxoma peritonei—a condition in which malignant mucinous epithelial cells accumulate in the abdomen.21 Pseudomyxoma peritonei is extremely rare with an incidence of approximately 1 to 2 cases per million per year.22 Early recognition of AMNs and surgical referral are critically important as pseudomyxoma peritonei is difficult to treat, has a high rate of recurrence, and can be fatal.23
Lastly, this case highlights how findings of a ruptured appendix and/or mucin surrounding the appendix on imaging should warrant laparoscopy because only pathologic analysis of the appendix can definitively rule out AMNs. The utility of laparoscopic evaluation of the appendix is especially apparent as nonsurgical treatment of appendicitis using antibiotics is gaining favor for treating even complicated appendicitis.24 Appendicitis is much more common than AMNs. However, had the patient in this case only been given antibiotics for his suspected complicated appendicitis without any colonoscopy or appendectomy, the neoplasm in his appendix would have gone undetected and continued to grow, causing significant complications. The patient’s age at presentation in this case also necessitated laparoscopic evaluation of the appendix as the incidence of AMNs is highest among patients aged > 60 years.25 Additionally, because appendiceal inversion may be seen with AMNs,the patient’s inverted appendix seen during his colonoscopy was another compelling reason for laparoscopic evaluation of his appendix.6
Conclusions
AMNs can present with nonspecific symptoms or can be completely asymptomatic and are often found incidentally during colonoscopies or appendectomies for acute appendicitis. While it is true that AMNs have low metastatic potential and grow slowly, AMNs can rupture leading to pseudomyxoma peritonei or even cause bowel obstruction warranting timely identification and removal of these neoplasms. Laparoscopic evaluation in cases of ruptured appendices is critical not only for treatment, but also for determining the presence of a potential underlying appendiceal malignancy. Although AMNs are a rare pathology, physicians should still consider the possibility of these neoplasms even when imaging findings suggest appendicitis. Having AMNs as part of the differential diagnosis is especially necessary in cases, such as this one, in which the patient has appendiceal inversion, is aged > 50 years, and has concurrent colorectal neoplasms.
Appendiceal mucinous neoplasms (AMNs) are rare tumors of the appendix that can be asymptomatic or present with acute right lower quadrant (RLQ) pain mimicking appendicitis. Due to their potential to cause either no symptoms or nonspecific symptoms, such as abdominal pain, nausea, or vomiting, AMNs are often found incidentally during appendectomies or, even more rarely, colonoscopies. Most AMNs grow slowly and have little metastatic potential. However, due to potential complications, such as bowel obstruction and rupture, timely detection and removal of AMN is essential. We describe the case of a patient who appeared to have acute appendicitis complicated by rupture on imaging who was found instead to have a perforated low-grade AMN during surgery.
Case Presentation
A male patient aged 72 years with a history of type 2 diabetes mellitus, hypertension, and aortic stenosis, but no prior abdominal surgery, presented with a chief concern of generalized weakness. As part of the workup for his weakness, a computed tomography (CT) scan of the abdomen was performed which showed an RLQ phlegmon and mild fat stranding in the area. Imaging also revealed an asymptomatic gallstone measuring 1.5 cm with no evidence of cholecystitis. The patient had no fever and reported no abdominal pain, nausea, vomiting, or change in bowel habits. On physical examination, the patient’s abdomen was soft, nontender, and nondistended with normoactive bowel sounds and no rebound or guarding.
To manage the appendicitis, the patient started a 2-week course of amoxicillin clavulanate 875 mg twice daily and was instructed to schedule an interval appendectomy in the coming months. Four days later, during a follow-up with his primary care physician, he was found to be asymptomatic. However, at this visit his stool was found to be positive for occult blood. Given this finding and the lack of a previous colonoscopy, the patient underwent a colonoscopy, which revealed bulging at the appendiceal orifice, consistent with an inverted appendix. Portions of the appendix were biopsied (Figure 1). Histologic analysis of the appendiceal biopsies revealed no dysplasia or malignancy. The colonoscopy also revealed an 8-mm sessile polyp in the ascending colon which was resected, and histologic analysis of this polyp revealed a low-grade tubular adenoma. Additionally, a large angiodysplastic lesion was found in the ascending colon as well as external and medium-sized internal hemorrhoids.
Six weeks after the colonoscopy, the patient was taken to the operating room for a laparoscopic appendectomy. Upon entry of the abdomen, extensive adhesions throughout the RLQ were found which required adhesiolysis. A calcified fecalith adherent to the mesentery of the small intestine in the RLQ was also found and resected. After lysis of the adhesions, the appendix and fibrotic tissue surrounding it could be seen (Figure 2). The appendix was dilated and the tip showed perforation. During dissection of the appendix, clear gelatinous material was found coming from the appendiceal lumen as well as from the fibrotic tissue around the appendix. On postoperative day 1 the appendix was resected and the patient was discharged.
Histologic specimens of the appendix were notable for evidence of perforation and neoplasia leading to a diagnosis of low-grade AMN. The presence of atypical mucinous epithelial cells on the serosal surface of the appendix, confirmed with a positive pancytokeratin stain, provided histologic evidence of appendiceal perforation (Figure 3). The presence of nuclear atypia demonstrated that the appendix was involved by a neoplastic process. Additionally, attenuation of the normal appendiceal epithelium, evidence of a chronic process, further helped to differentiate the AMN from complicated appendicitis. The presence of mucin involving the serosa of the appendix led to the classification of this patient’s neoplasm as grade pT4a. Of note, histologic examination demonstrated that the surgical margins contained tumor cells.
Given the positive margins of the resected AMN and the relatively large size of the neoplasm, a laparoscopic right hemicolectomy was performed 2 months later. Although multiple adhesions were found in the terminal ileum, cecum, and ascending colon during the hemicolectomy, no mucinous lesions were observed grossly. Histologic analysis showed no residual neoplasm as well as no lymph node involvement. On postoperative day 3 the patient was discharged and had an uneventful recovery. At his first surveillance visit 6 months after his hemicolectomy, the patient appeared to be doing well and reported no abdominal pain, nausea, vomiting, change in bowel habits, or any blood in the stool.
Discussion
AMNs are rare tumors with an annual age-adjusted incidence of approximately 0.12 per 1,000,000 people.1 These neoplasms can present as acute or chronic abdominal pain, gastrointestinal bleeding, intestinal obstruction, or acute abdomen.2-4 Most AMNs, however, are asymptomatic and are usually found incidentally during appendectomies for appendicitis, and can even be found during colonoscopies,such as in this case.5,6
Low-grade AMNs are distinguished from appendiceal mucinous adenocarcinomas by their lack of wall invasion.7 Additionally, low-grade AMNs have a very good prognosis as even neoplasms that have spread outside of the appendix have a 5-year overall survival rate of 79 to 86%.8 These low-grade neoplasms also have extremely low rates of recurrence after resection.9 In contrast, appendiceal mucinous adenocarcinomas have a much worse prognosis with a 5-year overall survival rate of 53.6%.10
Treatment of AMNs depends on the extent of their spread. Neoplasms that are confined to the appendix can typically be treated with appendectomy alone, while those that have spread beyond the appendix may require cytoreductive surgery and chemotherapy, namely, hyperthermic intraperitoneal chemotherapy (HIPEC), in addition to appendectomy.11 Cases in which neoplasms are not confined to the appendix also require more frequent surveillance for recurrence as compared to appendix-restricted neoplasms.11
Appendiceal inversion is a rare finding in adults with an estimated prevalence of 0.01%.6 Not only is appendiceal inversion rare in and of itself, it is even more rarely found in combination with appendiceal neoplasms.6 Other causes of appendiceal inversion include intussusception, acute appendicitis, appendiceal nodule, or even iatrogenic due to appendectomy.12-14 While appendiceal inversion can be completely benign, because these morphological changes of the appendix can resemble a polyp, these lesions are often biopsied and/or resected.15 However, lesion resection may be quite problematic due to high risk of bleeding and perforation.15 In order to avoid the risks associated with resection of a potentially benign finding, biopsy should be performed prior to any attempted resection of inverted appendices.15
Another interesting aspect of this case is the finding of fecal occult blood. The differential for fecal occult blood is quite broad and the patient had multiple conditions that could have led to the finding of occult blood in his stool. Hemorrhoids can cause a positive result on a fecal occult blood test (FOBT) although this is relatively uncommon, and hemorrhoids are more likely to cause frank blood to be seen.16 The sessile polyp found in the patient’s colon may also have caused the FOBT to be positive. This patient was also found to have an angiodysplasia (a finding that is associated with aortic stenosis, which this patient has a history of) which can also cause gastrointestinal bleeding.17 Lastly, AMNs may also cause gastrointestinal bleeding and thus a positive FOBT, although bleeding is a relatively uncommon presentation of AMNs, especially those that are low-grade as in this case.18
This case also highlights the association between appendiceal neoplasms and colonic neoplastic lesions. Patients with appendiceal neoplasms are more likely to have colonic neoplastic lesions than patients without appendiceal neoplasms.19 Studies have found that approximately 13 to 42% of patients with appendiceal neoplasms also have colonic neoplastic lesions.19 The majority of these lesions in the colon were right-sided and this finding was also seen in this case as the patient’s polyp was located in the ascending colon.19 Due to this association between appendiceal and colorectal neoplasia, the American Society of Colon and Rectal Surgeons strongly recommends that patients with appendiceal neoplasms or who are suspected of having them receive a colonoscopy.19
Additionally, perforation of an AMN, as was seen in this case, is a finding that should raise significant concern. Perforation of an AMN allows for the spread of malignant mucinous epithelial cells throughout the abdomen. The finding of extensive adhesions throughout the patient’s RLQ was unexpected as abdominal adhesions are most often seen in patients with a history of abdominal surgeries. Considering the lack of any prior abdominal surgeries in this patient, these adhesions were most likely the result of the spread and proliferation of malignant mucinous epithelial cells from the perforated AMN in the RLQ.20 The adhesiolysis performed in this case was thus not only important in order to visualize the appendix, but also for preventing future complications of abdominal adhesions such as bowel obstruction.20 Perforated AMN is also so concerning because it can potentially lead to pseudomyxoma peritonei—a condition in which malignant mucinous epithelial cells accumulate in the abdomen.21 Pseudomyxoma peritonei is extremely rare with an incidence of approximately 1 to 2 cases per million per year.22 Early recognition of AMNs and surgical referral are critically important as pseudomyxoma peritonei is difficult to treat, has a high rate of recurrence, and can be fatal.23
Lastly, this case highlights how findings of a ruptured appendix and/or mucin surrounding the appendix on imaging should warrant laparoscopy because only pathologic analysis of the appendix can definitively rule out AMNs. The utility of laparoscopic evaluation of the appendix is especially apparent as nonsurgical treatment of appendicitis using antibiotics is gaining favor for treating even complicated appendicitis.24 Appendicitis is much more common than AMNs. However, had the patient in this case only been given antibiotics for his suspected complicated appendicitis without any colonoscopy or appendectomy, the neoplasm in his appendix would have gone undetected and continued to grow, causing significant complications. The patient’s age at presentation in this case also necessitated laparoscopic evaluation of the appendix as the incidence of AMNs is highest among patients aged > 60 years.25 Additionally, because appendiceal inversion may be seen with AMNs,the patient’s inverted appendix seen during his colonoscopy was another compelling reason for laparoscopic evaluation of his appendix.6
Conclusions
AMNs can present with nonspecific symptoms or can be completely asymptomatic and are often found incidentally during colonoscopies or appendectomies for acute appendicitis. While it is true that AMNs have low metastatic potential and grow slowly, AMNs can rupture leading to pseudomyxoma peritonei or even cause bowel obstruction warranting timely identification and removal of these neoplasms. Laparoscopic evaluation in cases of ruptured appendices is critical not only for treatment, but also for determining the presence of a potential underlying appendiceal malignancy. Although AMNs are a rare pathology, physicians should still consider the possibility of these neoplasms even when imaging findings suggest appendicitis. Having AMNs as part of the differential diagnosis is especially necessary in cases, such as this one, in which the patient has appendiceal inversion, is aged > 50 years, and has concurrent colorectal neoplasms.
1. Shaib WL, Goodman M, Chen Z, et al. Incidence and survival of appendiceal mucinous neoplasms: a SEER analysis. Am J Clin Oncol. 2017;40(6):569-573. doi:10.1097/COC.0000000000000210
2. Kehagias I, Zygomalas A, Markopoulos G, Papandreou T, Kraniotis P. Diagnosis and treatment of mucinous appendiceal neoplasm presented as acute appendicitis. Case Rep Oncol Med. 2016;3:1-6. doi:10.1155/2016/2161952
3. Karatas M, Simsek C, Gunay S, et al. Acute lower gastrointestinal bleeding due to low-grade mucinous neoplasm of appendix. Acta Chir Belg. 2020;120(4):1-4. doi:10.1080/00015458.2020.1860397
4. Mourad FH, Hussein M, Bahlawan M, Haddad M, Tawil A. Intestinal obstruction secondary to appendiceal mucocele. Dig Dis Sci. 1999;44(8):1594-1599. doi:10.1023/a:1026615010989
5. Benabe SH, Leeman R, Brady AC, Hirzel A, Langshaw AH. Low-grade appendiceal mucinous neoplasm in an adolescent patient with untreated Crohn’s disease. ACG Case Reports J. 2020;7(3). doi:10.14309/crj.0000000000000338
6. Liu X, Liu G, Liu Y, et al. Complete appendiceal inversion with local high-grade intraepithelial neoplasia in an adult female: A case report. BMC Surg. 2019;19(1). doi:10.1186/s12893-019-0632-3
7. Gündog˘ar ÖS, Kımılog˘lu ES, Komut NS, et al. The evaluation of appendiceal mucinous neoplasms with a new classification system. Turk J Gastroenterol. 2018;29(5):532-542. doi:10.5152/tjg.2018.17605
8. Misdraji J, Yantiss RK, Graeme-Cook FM, Balis UJ, Young RH. Appendiceal mucinous neoplasms: a clinicopathologic analysis of 107 cases. Am J Surg Pathol. 2003;27(8):1089-1103. doi:10.1097/00000478-200308000-00006
9. Pai RK, Beck AH, Norton JA, Longacre TA. Appendiceal mucinous neoplasms: clinicopathologic study of 116 cases with analysis of factors predicting recurrence. Am J Surg Pathol. 2009;33(10):1425-1439. doi:10.1097/PAS.0b013e3181af6067
10. Asare EA, Compton CC, Hanna NN, et al. The impact of stage, grade, and mucinous histology on the efficacy of systemic chemotherapy in adenocarcinomas of the appendix: analysis of the National Cancer Data Base. Cancer. 2015;122(2):213-221. doi:10.1002/cncr.29744
11. Shaib WL, Assi R, Shamseddine A, et al. Appendiceal mucinous neoplasms: diagnosis and management. Oncologist. 2018;23(1):137. doi:10.1634/theoncologist.2017-0081erratum
12. Tran C, Sakioka J, Nguyen E, Beutler BD, Hsu J. An inverted appendix found on routine colonoscopy: a case report with discussion of imaging findings. Radiol Case Rep. 2019;14(8):952-955. doi:10.1016/j.radcr.2019.05.022
13. Shafi A, Azab M. A case of everted appendix with benign appendiceal nodule masquerading as appendiceal mucocele: a case report. Am J Gastroenterol. 2018;113:S1436. doi:10.14309/00000434-201810001-02585
14. Pokhrel B, Chang M, Anand G, Savides T, Fehmi S. Appendiceal mucinous neoplasm in an inverted appendix found on prior colonoscopy. VideoGIE. 2020;5(1):34-36. doi:10.1016/j.vgie.2019.09.013
15. Johnson EK, Arcila ME, Steele SR. Appendiceal inversion: a diagnostic and therapeutic dilemma. JSLS. 2009;13(1):92-95.
16. van Turenhout ST, Oort FA, sive Droste JST, et al. Hemorrhoids detected at colonoscopy: an infrequent cause of false-positive fecal immunochemical test results. Gastrointest Endosc. 2012;76(1):136-143. doi:10.1016/j.gie.2012.03.169
17. Hudzik B, Wilczek K, Gasior M. Heyde syndrome: Gastrointestinal bleeding and aortic stenosis. CMAJ. 2016;188(2):135-138. doi:10.1503/cmaj.150194
18. Leonards LM, Pahwa A, Patel MK, Petersen J, Nguyen MJ, Jude CM. Neoplasms of the appendix: pictorial review with clinical and pathologic correlation. RadioGraphics. 2017;37(4):1059-1083. doi:10.1148/rg.2017160150
19. Glasgow SC, Gaertner W, Stewart D, et al. The American Society of Colon and Rectal Surgeons, clinical practice guidelines for the management of appendiceal neoplasms. Dis Colon Rectum. 2019;62(12):1425-1438. doi:10.1097/DCR.0000000000001530
20. Panagopoulos P, Tsokaki T, Misiakos E, et al. Low-grade appendiceal mucinous neoplasm presenting as an adnexal mass. Case Reports in Obstetrics and Gynecology. 2017;2017:1-3. doi:10.1155/2017/7165321
21. Ramaswamy V. Pathology of mucinous appendiceal tumors and pseudomyxoma peritonei. Indian J Surg Oncol. 2016;7(2):258-267. doi:10.1007/s13193-016-0516-2.
22. Bevan KE, Mohamed F, Moran BJ. Pseudomyxoma peritonei. World J Gastrointest Oncol. 2010;2(1):44-50. doi:10.4251/wjgo.v2.i1.44
23. Mercier F, Dagbert F, Pocard M, et al. Recurrence of pseudomyxoma peritonei after cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. BJS Open. 2018;3(2):195-202. doi:10.1002/bjs5.97
24. David A, Dodgion C, Eddine SBZ, Davila D, Webb TP, Trevino CM. Perforated appendicitis: Short duration antibiotics are noninferior to traditional long duration antibiotics. Surgery. 2020;167(2):475-477. doi:10.1016/j.surg.2019.08.007
25. Raijman I, Leong S, Hassaram S, Marcon NE. Appendiceal mucocele: Endoscopic appearance. Endoscopy. 1994;26(3):326-328. doi:10.1055/s-2007-1008979
1. Shaib WL, Goodman M, Chen Z, et al. Incidence and survival of appendiceal mucinous neoplasms: a SEER analysis. Am J Clin Oncol. 2017;40(6):569-573. doi:10.1097/COC.0000000000000210
2. Kehagias I, Zygomalas A, Markopoulos G, Papandreou T, Kraniotis P. Diagnosis and treatment of mucinous appendiceal neoplasm presented as acute appendicitis. Case Rep Oncol Med. 2016;3:1-6. doi:10.1155/2016/2161952
3. Karatas M, Simsek C, Gunay S, et al. Acute lower gastrointestinal bleeding due to low-grade mucinous neoplasm of appendix. Acta Chir Belg. 2020;120(4):1-4. doi:10.1080/00015458.2020.1860397
4. Mourad FH, Hussein M, Bahlawan M, Haddad M, Tawil A. Intestinal obstruction secondary to appendiceal mucocele. Dig Dis Sci. 1999;44(8):1594-1599. doi:10.1023/a:1026615010989
5. Benabe SH, Leeman R, Brady AC, Hirzel A, Langshaw AH. Low-grade appendiceal mucinous neoplasm in an adolescent patient with untreated Crohn’s disease. ACG Case Reports J. 2020;7(3). doi:10.14309/crj.0000000000000338
6. Liu X, Liu G, Liu Y, et al. Complete appendiceal inversion with local high-grade intraepithelial neoplasia in an adult female: A case report. BMC Surg. 2019;19(1). doi:10.1186/s12893-019-0632-3
7. Gündog˘ar ÖS, Kımılog˘lu ES, Komut NS, et al. The evaluation of appendiceal mucinous neoplasms with a new classification system. Turk J Gastroenterol. 2018;29(5):532-542. doi:10.5152/tjg.2018.17605
8. Misdraji J, Yantiss RK, Graeme-Cook FM, Balis UJ, Young RH. Appendiceal mucinous neoplasms: a clinicopathologic analysis of 107 cases. Am J Surg Pathol. 2003;27(8):1089-1103. doi:10.1097/00000478-200308000-00006
9. Pai RK, Beck AH, Norton JA, Longacre TA. Appendiceal mucinous neoplasms: clinicopathologic study of 116 cases with analysis of factors predicting recurrence. Am J Surg Pathol. 2009;33(10):1425-1439. doi:10.1097/PAS.0b013e3181af6067
10. Asare EA, Compton CC, Hanna NN, et al. The impact of stage, grade, and mucinous histology on the efficacy of systemic chemotherapy in adenocarcinomas of the appendix: analysis of the National Cancer Data Base. Cancer. 2015;122(2):213-221. doi:10.1002/cncr.29744
11. Shaib WL, Assi R, Shamseddine A, et al. Appendiceal mucinous neoplasms: diagnosis and management. Oncologist. 2018;23(1):137. doi:10.1634/theoncologist.2017-0081erratum
12. Tran C, Sakioka J, Nguyen E, Beutler BD, Hsu J. An inverted appendix found on routine colonoscopy: a case report with discussion of imaging findings. Radiol Case Rep. 2019;14(8):952-955. doi:10.1016/j.radcr.2019.05.022
13. Shafi A, Azab M. A case of everted appendix with benign appendiceal nodule masquerading as appendiceal mucocele: a case report. Am J Gastroenterol. 2018;113:S1436. doi:10.14309/00000434-201810001-02585
14. Pokhrel B, Chang M, Anand G, Savides T, Fehmi S. Appendiceal mucinous neoplasm in an inverted appendix found on prior colonoscopy. VideoGIE. 2020;5(1):34-36. doi:10.1016/j.vgie.2019.09.013
15. Johnson EK, Arcila ME, Steele SR. Appendiceal inversion: a diagnostic and therapeutic dilemma. JSLS. 2009;13(1):92-95.
16. van Turenhout ST, Oort FA, sive Droste JST, et al. Hemorrhoids detected at colonoscopy: an infrequent cause of false-positive fecal immunochemical test results. Gastrointest Endosc. 2012;76(1):136-143. doi:10.1016/j.gie.2012.03.169
17. Hudzik B, Wilczek K, Gasior M. Heyde syndrome: Gastrointestinal bleeding and aortic stenosis. CMAJ. 2016;188(2):135-138. doi:10.1503/cmaj.150194
18. Leonards LM, Pahwa A, Patel MK, Petersen J, Nguyen MJ, Jude CM. Neoplasms of the appendix: pictorial review with clinical and pathologic correlation. RadioGraphics. 2017;37(4):1059-1083. doi:10.1148/rg.2017160150
19. Glasgow SC, Gaertner W, Stewart D, et al. The American Society of Colon and Rectal Surgeons, clinical practice guidelines for the management of appendiceal neoplasms. Dis Colon Rectum. 2019;62(12):1425-1438. doi:10.1097/DCR.0000000000001530
20. Panagopoulos P, Tsokaki T, Misiakos E, et al. Low-grade appendiceal mucinous neoplasm presenting as an adnexal mass. Case Reports in Obstetrics and Gynecology. 2017;2017:1-3. doi:10.1155/2017/7165321
21. Ramaswamy V. Pathology of mucinous appendiceal tumors and pseudomyxoma peritonei. Indian J Surg Oncol. 2016;7(2):258-267. doi:10.1007/s13193-016-0516-2.
22. Bevan KE, Mohamed F, Moran BJ. Pseudomyxoma peritonei. World J Gastrointest Oncol. 2010;2(1):44-50. doi:10.4251/wjgo.v2.i1.44
23. Mercier F, Dagbert F, Pocard M, et al. Recurrence of pseudomyxoma peritonei after cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. BJS Open. 2018;3(2):195-202. doi:10.1002/bjs5.97
24. David A, Dodgion C, Eddine SBZ, Davila D, Webb TP, Trevino CM. Perforated appendicitis: Short duration antibiotics are noninferior to traditional long duration antibiotics. Surgery. 2020;167(2):475-477. doi:10.1016/j.surg.2019.08.007
25. Raijman I, Leong S, Hassaram S, Marcon NE. Appendiceal mucocele: Endoscopic appearance. Endoscopy. 1994;26(3):326-328. doi:10.1055/s-2007-1008979
Caregiver Support in a Case of Posttraumatic Stress Disorder and Lewy Body Dementia
Caregiving for a person with dementia in the community can be extremely difficult work. Much of this work falls on unpaid or informal caregivers. Sixty-three percent of older adults with dementia depend completely on unpaid caregivers, and an additional 26% receive some combination of paid and unpaid support, together comprising nearly 90% of the more than 3 million older Americans with dementia.1 In-home care is preferable for these patients. For veterans, the Caregiver Support Program (CSP) is the only US Department of Veterans Affairs (VA) program that exclusively supports caregivers. Although the CSP is not a nursing home diversion or cost savings program, successfully enabling at-home living in lieu of facility living also has the potential to reduce overall cost of care, and most importantly, to enable veterans who desire it to age at home.2,3
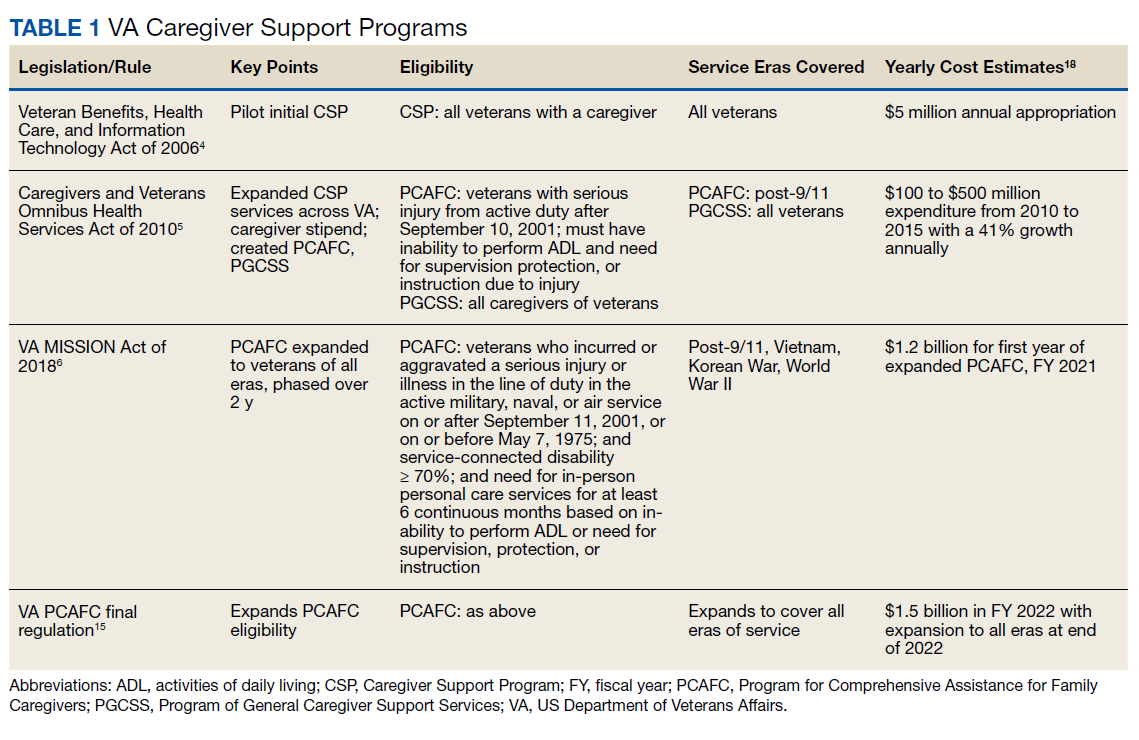
The CSP has 2 unique programs for caregivers of eligible veterans. The Program of General Caregiver Support Services (PGCSS) provides resources, education, and support to caregivers of all veterans enrolled in the Veterans Health Administration (VHA). The Program of Comprehensive Assistance for Family Caregivers (PCAFC) provides education and training, access to health care insurance if eligible, mental health counseling, access to a monthly caregiver stipend, enhanced respite care, wellness contacts, and travel compensation for VA health care appointments (Table 1).4,5
Patients undergo a rigorous assessment and highly specialized and individualized clinical decision-making process to confirm the service is appropriate for the patient. PCAFC was restructured and expanded on October 1, 2020.6 Currently, veterans who incurred or aggravated a serious injury (defined by a single or combined service-connection rating of ≥ 70%) in active military service before May 8, 1975, or after September 10, 2001, are eligible for PCAFC.6 Most notably, these changes opened eligibility in the PCAFC to caregivers of veterans from the Vietnam, Korean, and World War II eras of conflict and veterans with dependence in activities of daily living (ADL) due to a wider variety of illnesses, including dementia.6 The PCAFC is set to further expand to caregivers of otherwise eligible veterans of all eras of service on October 1, 2022, 2 years after the initial expansion, as laid out in the 2018 VA MISSION Act.6 Additional information on the history of the PGCSS and PCAFC and eligibility criteria for veterans and their family caregivers can be found in Tables 2 and 3.
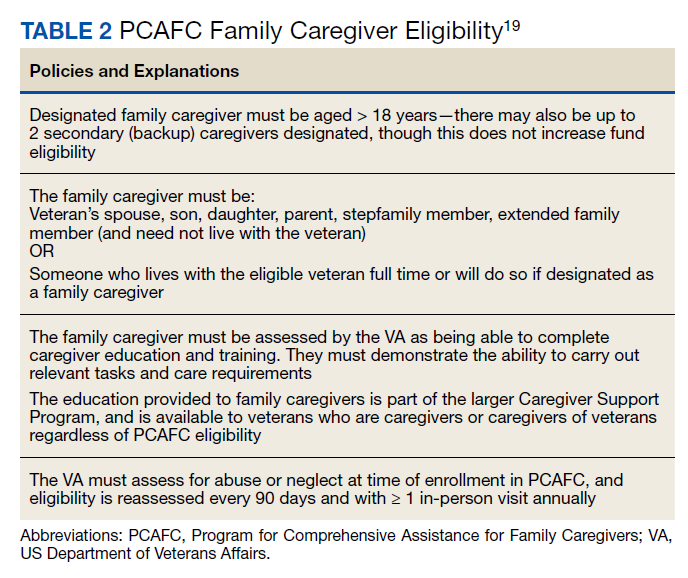

Posttraumatic stress disorder (PTSD) and cognitive impairment are 2 common causes of disability among veterans who receive VHA care. Among older veterans, rates of lifetime development of PTSD reach up to 30%.7 Dementia diagnosis is also more common in older veterans compared with age-matched civilians.8 Furthermore, a prior diagnosis of PTSD has been associated with nearly a 2-fold increase in risk of development of dementia in older age.7 These conditions are also linked to high degrees of service connection. PTSD is the third most prevalent service-connected disability for veterans receiving compensation and cognitive limitation is the third most prevalent category of service-connected disability among veterans.9
We present a case of a Vietnam-era veteran with a history of combat exposure and service-connected PTSD, and a later diagnosis of Lewy body dementia (LBD). Through combination of VHA geriatric services, the CSP, and the expanded PCAFC, the veteran’s primary family caregiver received the materials, support, and financial resources necessary to enable at-home living for the veteran, despite his illness and later complications.
Case Presentation
A male combat veteran presented to his primary care practitioner (PCP) with concerns of several years of progressive changes in gait, forgetfulness, and a gradual decline in the ability to live independently without assistance. At that time, his medical history was notable for PTSD (50% service connection), which had been diagnosed over a decade prior (but for which the veteran had refused medication or therapy on multiple occasions, stating he preferred to “breathe through” his intrusive symptom flare-ups), localized prostate cancer with a radical prostatectomy (100% service connection), multiple kidney stones with persistent left ureteral inflammation, and arteriosclerotic heart disease (10% service connection). A Saint Louis University Mental Status Exam (SLUMS) performed by the PCP was notable for a score of 9/30, in the dementia range. A computed tomography of the brain demonstrated scattered foci of hypoattenuation attributable to normal aging without any other pathology noted.
The veteran was referred to the Cognitive Care clinic, a local longitudinal multidisciplinary dementia care clinic, along with his spouse/caregiver. Cognitive testing was performed by a licensed clinical psychologist in the clinic and was notable for a Mini-Mental State Exam (MMSE) score of 18/30, also in the dementia range, and a more robust neuropsychiatric battery demonstrated borderline intact memory and language function but impairments in executive function and visuospatial skills. The patient’s clinical history included functional loss over time, with total dependence in instrumental activities of daily living (IADL), or tasks necessary to be fully independent or manage a household, including inability to manage finances, and some need for assistance in ADL, or personal care tasks such as dressing or grooming, including bathing. Physical examination was notable for bradykinesia, a shuffling gait, and rare episodes of speaking to someone who was not in the room, thought to be due to mild nondistressing hallucinations.
A diagnosis of LBD was made. At time of diagnosis, the patient met criteria for probable dementia with Lewy bodies, with 2 of 4 core clinical features (hallucinations and Parkinsonism), and multiple supportive features (gait disturbance, sensory disturbance, and altered mood).10,11 The veteran continued to develop more supportive features for diagnosis of LBD over time, including evidence of autonomic instability.
The veteran and his caregiver were educated on his diagnosis, and longitudinal support was offered. The veteran was no longer driving, and due to the severity of his symptoms, the importance of driving cessation was reinforced by the care team. Over the course of the next year, his illness progressed, with more frequent behaviors and psychological symptoms of dementia (BPSD). He began to exhibit nighttime wandering throughout the house and became more anxious and restless during the day. He lost the ability to make his own health care decisions, and his spouse became his activated health care power of attorney (HCPOA). His BPSD became more disruptive to daily life and was accompanied by a change in the character of his hallucinations, with prior nondistressing visions of other people being replaced with visions of war, burning bodies, and violence, much of it related to combat experiences in Vietnam. The BPSD began to include hiding behind furniture, running out of the house, and shouting and crying in response to hallucinations. At times, his BPSD became violent, lashing out in fear against his hallucinations and caregiver.
The veteran’s change in BPSD was concerning for a new baseline, rather than being clearly related to an underlying unmet physical need, such as pain, hunger, sleep, or discomfort. Multiple hospital admissions during that year involved IV hydration and treatment for urinary tract infections (UTI) for several days of inpatient stay at a time, but these behaviors persisted despite infection treatment and hydration. The patient’s changes in BPSD were thought to be secondary to uncovered and intensified PTSD in the setting of progressive dementia. Due to the clear danger the patient posed to himself and others, potential treatment options for these PTSD-related hallucinations were discussed with his caregiver. The caregiver shared that the patient’s BPSD and hallucinations were so distressing that “he would never want to live like this,” and that things had progressed to the point that “he has no quality of life.”
Oral aripiprazole 2 mg twice daily was prescribed after the risks of infection, cardiac complications, and exacerbation of movement disorder symptoms, such as increased stiffness and falls, were discussed with the caregiver. The caregiver was employed and relied on continued employment for income, but the patient could not be safely left alone. As the patient and his caregiver had reached a crisis point and living at home no longer appeared to be safe, the patient was referred to a VA-contracted skilled nursing facility (SNF) for long-term care. The patient’s caregiver was also referred to CSP for support during this transition. Due to the patient’s level of service connection and personal needs, as well as the patient and caregiver’s preference for the veteran to remain in his home, they were evaluated for the PCAFC for enhanced support to enable home as an alternative to facility living, should the patient respond to the antipsychotic therapy sufficiently, which was evaluated on a regular basis.
After several months, the patient’s BPSD had improved significantly, and he was no longer experiencing distressing hallucinations. However, his mobility also declined, and he became fully dependent in most ADL, including transfers, hygiene, and toileting. Due to the COVID-19 pandemic, visitation was limited, which was difficult for both the patient and his caregiver. The veteran and caregiver were approved for PCAFC due to the veteran’s combination of service-connected illnesses > 70%, dependence for most ADLs, and need for continuous supervision. A transfer home from the SNF was arranged.
The PCAFC allowed the veteran’s caregiver and family members to provide in-home full-time caregiving, as an alternative to facility placement. The caregiver received a variety of support, including access to peer support, instruction on ways to assist in his toileting, hygiene, and transfers, and a caregiving stipend. In addition to offsetting lost wages, the stipend also helped offset the cost of care supplies which were not provided or were not readily available from the VA, which at the time included the patient’s preferred nutritional supplement and some supplies for personal care.
The veteran’s care needs continued to escalate. A fall at home resulted in a hip fracture, which was treated with surgical pinning. Postfracture physical therapy in a facility was considered, but ultimately was provided at home. The patient also experienced multiple UTIs and resulting delirium, with accompanying agitation and hallucinations. These episodes improved with IV antibiotics and hydration during short hospital stays. Ultimately, a computed tomography demonstrated overflow incontinence likely related to urologic damage from prior kidney stones and stent placement was recommended.
Visiting skilled nurses for the patient’s area were difficult to coordinate but were eventually arranged. The patient continued residing in his home with the support of his caregiver, the PCAFC, and the local VA medical center geriatric and transitional care services. The patient was also referred to the palliative care outpatient specialty clinic for discussion of goals of care and assistance with advance care planning as his illness progressed. Mental health and geriatric psychiatry consult teams were considered for this case but not utilized.
Discussion
Older adult Americans are at high risk of poor financial wellbeing, with nearly one quarter of Americans aged > 62 years experiencing financial insecurity.12 Even in this case with health care provided by the VA, successful in-home care was challenging and required a dedicated live-in caregiver, care coordination resources, and financial support. As part of its mission of caring for veterans, the VA has instituted CSP, whose mission is to promote the health and well-being of family caregivers through education, support, and services.
PCAFC offers enhanced clinical support for caregivers of eligible veterans who are seriously injured. This includes resources, education, support, financial stipends, health insurance (if eligible), and beneficiary travel (if eligible) to primary caregivers of eligible veterans. PCAFC was originally reserved for veterans who had onset of service-related disability after September 11, 2001, with an associated personal care need. In this population, PCAFC demonstrated an increased usage of clinical resources, likely related to increased ease in accessing care.13
The cohort of post-9/11 veterans is very different from the cohort of veterans and their caregivers who may now qualify for the PCAFC after its October 2020 expansion. Veterans from the Vietnam, Korean, and World War II eras of conflict have rates of service-connected disability 2 to 3 times higher than those of post-9/11 era veterans and are at greater risk for dementia.9 Veterans aged ≥ 75 years who have service connection also report higher rates of difficulty with independent living and self-care compared with their younger peers.9 Since dementia and PTSD are common causes of service connection and disability it is likely that a significant proportion of older veterans will be eligible to apply for the newly expanded PCAFC.
To be eligible for PCAFC, a veteran must have a service-connected disability rating of ≥ 70% and must need in-person care services for ≥ 6 continuous months, based on either an inability to perform an ADL, or a need for supervision, protection, or instruction.
It is not clear how CSP use or increased access to PCAFC will impact costs. However, the PCAFC monthly stipend is scaled to the median wage of a home health aide and to the location of the caregiver, which is considerably less than the cost of recurrent hospitalization or a year of facility-level care.15 The CSP may eventually be a successful long-term investment in cost savings. In order to ensure the process for PCAFC approval is uniform and prompt as the program expands, CSP has restructured, increasing the number of employees, improving the patient review process, and expanding staff training.16 The VA plans to continually re-assess CSP using the infrastructure of the Caregiver Record Management Application as it continues to expand.17
Conclusions
Dementia and PTSD commonly coexist and are a significant source of disability in the service-connected veteran population. This case brings attention to the recent expansion of PCAFC, which now has the potential to support eligible veterans from the World War II, Korean, and Vietnam-era conflicts, in whom these illnesses are more common. In this case, in-home care was preferred by the veteran and primary caregiver but would not have been possible without a complex intervention. There is still more research to be done on the best way to meet the needs of older adults with dementia, the impact of in-home care, and the system-wide implications of PCAFC, especially as the program grows. However, in-home care is preferable to SNF living for many veterans and caregivers, and CSP will continue to be an essential element of providing care for this population.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin. The authors would also like to thank the members of the Veterans Affairs Central Office and National Caregiver Support Program Office, including Elyse Kaplan, Melinda Hogue, Beth Wolfsohn, Colleen Richardson, and Timothy Jobin, for their thorough review of the work and edits to ensure accurate program description.
1. Chi W, Graf E, Hughes L, et al. Community-dwelling older adults with dementia and their caregivers: key indicators from the National Health and Aging Trends study. Published January 29, 2019. Accessed February 16, 2022. https://aspe.hhs.gov/sites/default/files/migrated_legacy_files//186501/DemChartbook.pdf
2. Rapaport P, Burton A, Leverton M, et al. “I just keep thinking that I don’t want to rely on people.” A qualitative study of how people living with dementia achieve and maintain independence at home: stakeholder perspectives. BMC Geriatr. 2020;20(1):1-11. doi:10.1186/s12877-019-1406-6
3. Miller EA, Gidmark S, Gadbois E, Rudolph JL, Intrator O. Nursing home referral within the Veterans Health Administration: practice variation by payment source and facility type. Res Aging. 2018;40(7):687-711. doi:10.1177/0164027517730383
4. Veterans Benefits, Health Care, and Information Technology Act of 2006, Pub L No. 109-461, 120 Stat. 3403.
5. Caregivers and Veterans Omnibus Health Services Act of 2010, Pub L No. 111-163, 115 Stat 552.
6. VA MISSION Act of 2018. 38 CFR § 17.
7. Yaffe K, Vittinghoff E, Lindquist K, et al. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry. 2010;67(6):608-613. doi:10.1001/archgenpsychiatry.2010.61
8. Krishnan LL, Petersen NJ, Snow AL, et al. Prevalence of dementia among Veterans Affairs medical care system users. Dement Geriatr Cogn Disord. 2005;20(4):245-253. doi:10.1159/000087345
9. Holder, KA. The Disability of Veterans. Social, Economic, and Housing Statistics Division, US Census Bureau; 2014. Accessed February 9, 2022. https://www.census.gov/content/dam/Census/library/working-papers/2016/demo/Holder-2016-01.pdf
10. Sanford AM. Lewy body dementia. Clin Geriatr Med. 2018;34(4):603-615. doi:10.1016/j.cger.2018.06.007
11. Armstrong MJ. Lewy body dementias. Continuum (Minneap Minn). 2019;25(1):128-146. doi:10.1212/CON.0000000000000685
12. Bureau of Consumer Financial Protection. Financial well-being of older Americans. Published December 2018. Accessed February 17, 2022. https://files.consumerfinance.gov/f/documents/bcfp_financial-well-being-older-americans_report.pdf
13. Van Houtven CH, Smith VA, Stechuchak KM, et al. Comprehensive support for family caregivers: impact on veteran health care utilization and costs. Med Care Res Rev. 2019;76(1):89-114. doi:10.1177/1077558717697015
14. Boland L, Légaré F, Perez MMB, et al. Impact of home care versus alternative locations of care on elder health outcomes: an overview of systematic reviews. BMC Geriatr. 2017;17(1):20. doi:10.1186/s12877-016-0395-y
15. Program of Comprehensive Assistance for Family Caregivers Improvements and Amendments Under the VA MISSION Act of 2018. 85 FR § 13356.
16. Extension of Program of Comprehensive Assistance for Family Caregivers Eligibility for Legacy Participants and Legacy Applicants. 86 FR § 52614.
17. US Department of Veterans Affairs, 2020. Certification of the Implementation of the Caregiver Records Management Application (CARMA). 85 FR § 63358.
18. Sussman, JS. Department of Veterans Affairs: Caregiver Support. Congressional Research Service Report No. R46282. Published March 24, 2020. Accessed February 16, 2022. https://www.everycrsreport.com/files/20200324_R46282_656f1e8338af12a2a676c471be3b3c13b2fcb0bb.pdf
19. US Department of Veterans Affairs. Veterans Affairs Program of Comprehensive Assistance for Family Caregivers Eligibility Criteria Fact Sheet. Washington, DC: U.S. Department of Veterans Affairs; 2020. Accessed February 9, 2022. https://www.caregiver.va.gov/pdfs/MissionAct/EligibilityCriteriaFactsheet_Chapter2_Launch_Approved_Final_100120.pdf
Caregiving for a person with dementia in the community can be extremely difficult work. Much of this work falls on unpaid or informal caregivers. Sixty-three percent of older adults with dementia depend completely on unpaid caregivers, and an additional 26% receive some combination of paid and unpaid support, together comprising nearly 90% of the more than 3 million older Americans with dementia.1 In-home care is preferable for these patients. For veterans, the Caregiver Support Program (CSP) is the only US Department of Veterans Affairs (VA) program that exclusively supports caregivers. Although the CSP is not a nursing home diversion or cost savings program, successfully enabling at-home living in lieu of facility living also has the potential to reduce overall cost of care, and most importantly, to enable veterans who desire it to age at home.2,3
The CSP has 2 unique programs for caregivers of eligible veterans. The Program of General Caregiver Support Services (PGCSS) provides resources, education, and support to caregivers of all veterans enrolled in the Veterans Health Administration (VHA). The Program of Comprehensive Assistance for Family Caregivers (PCAFC) provides education and training, access to health care insurance if eligible, mental health counseling, access to a monthly caregiver stipend, enhanced respite care, wellness contacts, and travel compensation for VA health care appointments (Table 1).4,5
Patients undergo a rigorous assessment and highly specialized and individualized clinical decision-making process to confirm the service is appropriate for the patient. PCAFC was restructured and expanded on October 1, 2020.6 Currently, veterans who incurred or aggravated a serious injury (defined by a single or combined service-connection rating of ≥ 70%) in active military service before May 8, 1975, or after September 10, 2001, are eligible for PCAFC.6 Most notably, these changes opened eligibility in the PCAFC to caregivers of veterans from the Vietnam, Korean, and World War II eras of conflict and veterans with dependence in activities of daily living (ADL) due to a wider variety of illnesses, including dementia.6 The PCAFC is set to further expand to caregivers of otherwise eligible veterans of all eras of service on October 1, 2022, 2 years after the initial expansion, as laid out in the 2018 VA MISSION Act.6 Additional information on the history of the PGCSS and PCAFC and eligibility criteria for veterans and their family caregivers can be found in Tables 2 and 3.
Posttraumatic stress disorder (PTSD) and cognitive impairment are 2 common causes of disability among veterans who receive VHA care. Among older veterans, rates of lifetime development of PTSD reach up to 30%.7 Dementia diagnosis is also more common in older veterans compared with age-matched civilians.8 Furthermore, a prior diagnosis of PTSD has been associated with nearly a 2-fold increase in risk of development of dementia in older age.7 These conditions are also linked to high degrees of service connection. PTSD is the third most prevalent service-connected disability for veterans receiving compensation and cognitive limitation is the third most prevalent category of service-connected disability among veterans.9
We present a case of a Vietnam-era veteran with a history of combat exposure and service-connected PTSD, and a later diagnosis of Lewy body dementia (LBD). Through combination of VHA geriatric services, the CSP, and the expanded PCAFC, the veteran’s primary family caregiver received the materials, support, and financial resources necessary to enable at-home living for the veteran, despite his illness and later complications.
Case Presentation
A male combat veteran presented to his primary care practitioner (PCP) with concerns of several years of progressive changes in gait, forgetfulness, and a gradual decline in the ability to live independently without assistance. At that time, his medical history was notable for PTSD (50% service connection), which had been diagnosed over a decade prior (but for which the veteran had refused medication or therapy on multiple occasions, stating he preferred to “breathe through” his intrusive symptom flare-ups), localized prostate cancer with a radical prostatectomy (100% service connection), multiple kidney stones with persistent left ureteral inflammation, and arteriosclerotic heart disease (10% service connection). A Saint Louis University Mental Status Exam (SLUMS) performed by the PCP was notable for a score of 9/30, in the dementia range. A computed tomography of the brain demonstrated scattered foci of hypoattenuation attributable to normal aging without any other pathology noted.
The veteran was referred to the Cognitive Care clinic, a local longitudinal multidisciplinary dementia care clinic, along with his spouse/caregiver. Cognitive testing was performed by a licensed clinical psychologist in the clinic and was notable for a Mini-Mental State Exam (MMSE) score of 18/30, also in the dementia range, and a more robust neuropsychiatric battery demonstrated borderline intact memory and language function but impairments in executive function and visuospatial skills. The patient’s clinical history included functional loss over time, with total dependence in instrumental activities of daily living (IADL), or tasks necessary to be fully independent or manage a household, including inability to manage finances, and some need for assistance in ADL, or personal care tasks such as dressing or grooming, including bathing. Physical examination was notable for bradykinesia, a shuffling gait, and rare episodes of speaking to someone who was not in the room, thought to be due to mild nondistressing hallucinations.
A diagnosis of LBD was made. At time of diagnosis, the patient met criteria for probable dementia with Lewy bodies, with 2 of 4 core clinical features (hallucinations and Parkinsonism), and multiple supportive features (gait disturbance, sensory disturbance, and altered mood).10,11 The veteran continued to develop more supportive features for diagnosis of LBD over time, including evidence of autonomic instability.
The veteran and his caregiver were educated on his diagnosis, and longitudinal support was offered. The veteran was no longer driving, and due to the severity of his symptoms, the importance of driving cessation was reinforced by the care team. Over the course of the next year, his illness progressed, with more frequent behaviors and psychological symptoms of dementia (BPSD). He began to exhibit nighttime wandering throughout the house and became more anxious and restless during the day. He lost the ability to make his own health care decisions, and his spouse became his activated health care power of attorney (HCPOA). His BPSD became more disruptive to daily life and was accompanied by a change in the character of his hallucinations, with prior nondistressing visions of other people being replaced with visions of war, burning bodies, and violence, much of it related to combat experiences in Vietnam. The BPSD began to include hiding behind furniture, running out of the house, and shouting and crying in response to hallucinations. At times, his BPSD became violent, lashing out in fear against his hallucinations and caregiver.
The veteran’s change in BPSD was concerning for a new baseline, rather than being clearly related to an underlying unmet physical need, such as pain, hunger, sleep, or discomfort. Multiple hospital admissions during that year involved IV hydration and treatment for urinary tract infections (UTI) for several days of inpatient stay at a time, but these behaviors persisted despite infection treatment and hydration. The patient’s changes in BPSD were thought to be secondary to uncovered and intensified PTSD in the setting of progressive dementia. Due to the clear danger the patient posed to himself and others, potential treatment options for these PTSD-related hallucinations were discussed with his caregiver. The caregiver shared that the patient’s BPSD and hallucinations were so distressing that “he would never want to live like this,” and that things had progressed to the point that “he has no quality of life.”
Oral aripiprazole 2 mg twice daily was prescribed after the risks of infection, cardiac complications, and exacerbation of movement disorder symptoms, such as increased stiffness and falls, were discussed with the caregiver. The caregiver was employed and relied on continued employment for income, but the patient could not be safely left alone. As the patient and his caregiver had reached a crisis point and living at home no longer appeared to be safe, the patient was referred to a VA-contracted skilled nursing facility (SNF) for long-term care. The patient’s caregiver was also referred to CSP for support during this transition. Due to the patient’s level of service connection and personal needs, as well as the patient and caregiver’s preference for the veteran to remain in his home, they were evaluated for the PCAFC for enhanced support to enable home as an alternative to facility living, should the patient respond to the antipsychotic therapy sufficiently, which was evaluated on a regular basis.
After several months, the patient’s BPSD had improved significantly, and he was no longer experiencing distressing hallucinations. However, his mobility also declined, and he became fully dependent in most ADL, including transfers, hygiene, and toileting. Due to the COVID-19 pandemic, visitation was limited, which was difficult for both the patient and his caregiver. The veteran and caregiver were approved for PCAFC due to the veteran’s combination of service-connected illnesses > 70%, dependence for most ADLs, and need for continuous supervision. A transfer home from the SNF was arranged.
The PCAFC allowed the veteran’s caregiver and family members to provide in-home full-time caregiving, as an alternative to facility placement. The caregiver received a variety of support, including access to peer support, instruction on ways to assist in his toileting, hygiene, and transfers, and a caregiving stipend. In addition to offsetting lost wages, the stipend also helped offset the cost of care supplies which were not provided or were not readily available from the VA, which at the time included the patient’s preferred nutritional supplement and some supplies for personal care.
The veteran’s care needs continued to escalate. A fall at home resulted in a hip fracture, which was treated with surgical pinning. Postfracture physical therapy in a facility was considered, but ultimately was provided at home. The patient also experienced multiple UTIs and resulting delirium, with accompanying agitation and hallucinations. These episodes improved with IV antibiotics and hydration during short hospital stays. Ultimately, a computed tomography demonstrated overflow incontinence likely related to urologic damage from prior kidney stones and stent placement was recommended.
Visiting skilled nurses for the patient’s area were difficult to coordinate but were eventually arranged. The patient continued residing in his home with the support of his caregiver, the PCAFC, and the local VA medical center geriatric and transitional care services. The patient was also referred to the palliative care outpatient specialty clinic for discussion of goals of care and assistance with advance care planning as his illness progressed. Mental health and geriatric psychiatry consult teams were considered for this case but not utilized.
Discussion
Older adult Americans are at high risk of poor financial wellbeing, with nearly one quarter of Americans aged > 62 years experiencing financial insecurity.12 Even in this case with health care provided by the VA, successful in-home care was challenging and required a dedicated live-in caregiver, care coordination resources, and financial support. As part of its mission of caring for veterans, the VA has instituted CSP, whose mission is to promote the health and well-being of family caregivers through education, support, and services.
PCAFC offers enhanced clinical support for caregivers of eligible veterans who are seriously injured. This includes resources, education, support, financial stipends, health insurance (if eligible), and beneficiary travel (if eligible) to primary caregivers of eligible veterans. PCAFC was originally reserved for veterans who had onset of service-related disability after September 11, 2001, with an associated personal care need. In this population, PCAFC demonstrated an increased usage of clinical resources, likely related to increased ease in accessing care.13
The cohort of post-9/11 veterans is very different from the cohort of veterans and their caregivers who may now qualify for the PCAFC after its October 2020 expansion. Veterans from the Vietnam, Korean, and World War II eras of conflict have rates of service-connected disability 2 to 3 times higher than those of post-9/11 era veterans and are at greater risk for dementia.9 Veterans aged ≥ 75 years who have service connection also report higher rates of difficulty with independent living and self-care compared with their younger peers.9 Since dementia and PTSD are common causes of service connection and disability it is likely that a significant proportion of older veterans will be eligible to apply for the newly expanded PCAFC.
To be eligible for PCAFC, a veteran must have a service-connected disability rating of ≥ 70% and must need in-person care services for ≥ 6 continuous months, based on either an inability to perform an ADL, or a need for supervision, protection, or instruction.
It is not clear how CSP use or increased access to PCAFC will impact costs. However, the PCAFC monthly stipend is scaled to the median wage of a home health aide and to the location of the caregiver, which is considerably less than the cost of recurrent hospitalization or a year of facility-level care.15 The CSP may eventually be a successful long-term investment in cost savings. In order to ensure the process for PCAFC approval is uniform and prompt as the program expands, CSP has restructured, increasing the number of employees, improving the patient review process, and expanding staff training.16 The VA plans to continually re-assess CSP using the infrastructure of the Caregiver Record Management Application as it continues to expand.17
Conclusions
Dementia and PTSD commonly coexist and are a significant source of disability in the service-connected veteran population. This case brings attention to the recent expansion of PCAFC, which now has the potential to support eligible veterans from the World War II, Korean, and Vietnam-era conflicts, in whom these illnesses are more common. In this case, in-home care was preferred by the veteran and primary caregiver but would not have been possible without a complex intervention. There is still more research to be done on the best way to meet the needs of older adults with dementia, the impact of in-home care, and the system-wide implications of PCAFC, especially as the program grows. However, in-home care is preferable to SNF living for many veterans and caregivers, and CSP will continue to be an essential element of providing care for this population.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin. The authors would also like to thank the members of the Veterans Affairs Central Office and National Caregiver Support Program Office, including Elyse Kaplan, Melinda Hogue, Beth Wolfsohn, Colleen Richardson, and Timothy Jobin, for their thorough review of the work and edits to ensure accurate program description.
Caregiving for a person with dementia in the community can be extremely difficult work. Much of this work falls on unpaid or informal caregivers. Sixty-three percent of older adults with dementia depend completely on unpaid caregivers, and an additional 26% receive some combination of paid and unpaid support, together comprising nearly 90% of the more than 3 million older Americans with dementia.1 In-home care is preferable for these patients. For veterans, the Caregiver Support Program (CSP) is the only US Department of Veterans Affairs (VA) program that exclusively supports caregivers. Although the CSP is not a nursing home diversion or cost savings program, successfully enabling at-home living in lieu of facility living also has the potential to reduce overall cost of care, and most importantly, to enable veterans who desire it to age at home.2,3
The CSP has 2 unique programs for caregivers of eligible veterans. The Program of General Caregiver Support Services (PGCSS) provides resources, education, and support to caregivers of all veterans enrolled in the Veterans Health Administration (VHA). The Program of Comprehensive Assistance for Family Caregivers (PCAFC) provides education and training, access to health care insurance if eligible, mental health counseling, access to a monthly caregiver stipend, enhanced respite care, wellness contacts, and travel compensation for VA health care appointments (Table 1).4,5
Patients undergo a rigorous assessment and highly specialized and individualized clinical decision-making process to confirm the service is appropriate for the patient. PCAFC was restructured and expanded on October 1, 2020.6 Currently, veterans who incurred or aggravated a serious injury (defined by a single or combined service-connection rating of ≥ 70%) in active military service before May 8, 1975, or after September 10, 2001, are eligible for PCAFC.6 Most notably, these changes opened eligibility in the PCAFC to caregivers of veterans from the Vietnam, Korean, and World War II eras of conflict and veterans with dependence in activities of daily living (ADL) due to a wider variety of illnesses, including dementia.6 The PCAFC is set to further expand to caregivers of otherwise eligible veterans of all eras of service on October 1, 2022, 2 years after the initial expansion, as laid out in the 2018 VA MISSION Act.6 Additional information on the history of the PGCSS and PCAFC and eligibility criteria for veterans and their family caregivers can be found in Tables 2 and 3.
Posttraumatic stress disorder (PTSD) and cognitive impairment are 2 common causes of disability among veterans who receive VHA care. Among older veterans, rates of lifetime development of PTSD reach up to 30%.7 Dementia diagnosis is also more common in older veterans compared with age-matched civilians.8 Furthermore, a prior diagnosis of PTSD has been associated with nearly a 2-fold increase in risk of development of dementia in older age.7 These conditions are also linked to high degrees of service connection. PTSD is the third most prevalent service-connected disability for veterans receiving compensation and cognitive limitation is the third most prevalent category of service-connected disability among veterans.9
We present a case of a Vietnam-era veteran with a history of combat exposure and service-connected PTSD, and a later diagnosis of Lewy body dementia (LBD). Through combination of VHA geriatric services, the CSP, and the expanded PCAFC, the veteran’s primary family caregiver received the materials, support, and financial resources necessary to enable at-home living for the veteran, despite his illness and later complications.
Case Presentation
A male combat veteran presented to his primary care practitioner (PCP) with concerns of several years of progressive changes in gait, forgetfulness, and a gradual decline in the ability to live independently without assistance. At that time, his medical history was notable for PTSD (50% service connection), which had been diagnosed over a decade prior (but for which the veteran had refused medication or therapy on multiple occasions, stating he preferred to “breathe through” his intrusive symptom flare-ups), localized prostate cancer with a radical prostatectomy (100% service connection), multiple kidney stones with persistent left ureteral inflammation, and arteriosclerotic heart disease (10% service connection). A Saint Louis University Mental Status Exam (SLUMS) performed by the PCP was notable for a score of 9/30, in the dementia range. A computed tomography of the brain demonstrated scattered foci of hypoattenuation attributable to normal aging without any other pathology noted.
The veteran was referred to the Cognitive Care clinic, a local longitudinal multidisciplinary dementia care clinic, along with his spouse/caregiver. Cognitive testing was performed by a licensed clinical psychologist in the clinic and was notable for a Mini-Mental State Exam (MMSE) score of 18/30, also in the dementia range, and a more robust neuropsychiatric battery demonstrated borderline intact memory and language function but impairments in executive function and visuospatial skills. The patient’s clinical history included functional loss over time, with total dependence in instrumental activities of daily living (IADL), or tasks necessary to be fully independent or manage a household, including inability to manage finances, and some need for assistance in ADL, or personal care tasks such as dressing or grooming, including bathing. Physical examination was notable for bradykinesia, a shuffling gait, and rare episodes of speaking to someone who was not in the room, thought to be due to mild nondistressing hallucinations.
A diagnosis of LBD was made. At time of diagnosis, the patient met criteria for probable dementia with Lewy bodies, with 2 of 4 core clinical features (hallucinations and Parkinsonism), and multiple supportive features (gait disturbance, sensory disturbance, and altered mood).10,11 The veteran continued to develop more supportive features for diagnosis of LBD over time, including evidence of autonomic instability.
The veteran and his caregiver were educated on his diagnosis, and longitudinal support was offered. The veteran was no longer driving, and due to the severity of his symptoms, the importance of driving cessation was reinforced by the care team. Over the course of the next year, his illness progressed, with more frequent behaviors and psychological symptoms of dementia (BPSD). He began to exhibit nighttime wandering throughout the house and became more anxious and restless during the day. He lost the ability to make his own health care decisions, and his spouse became his activated health care power of attorney (HCPOA). His BPSD became more disruptive to daily life and was accompanied by a change in the character of his hallucinations, with prior nondistressing visions of other people being replaced with visions of war, burning bodies, and violence, much of it related to combat experiences in Vietnam. The BPSD began to include hiding behind furniture, running out of the house, and shouting and crying in response to hallucinations. At times, his BPSD became violent, lashing out in fear against his hallucinations and caregiver.
The veteran’s change in BPSD was concerning for a new baseline, rather than being clearly related to an underlying unmet physical need, such as pain, hunger, sleep, or discomfort. Multiple hospital admissions during that year involved IV hydration and treatment for urinary tract infections (UTI) for several days of inpatient stay at a time, but these behaviors persisted despite infection treatment and hydration. The patient’s changes in BPSD were thought to be secondary to uncovered and intensified PTSD in the setting of progressive dementia. Due to the clear danger the patient posed to himself and others, potential treatment options for these PTSD-related hallucinations were discussed with his caregiver. The caregiver shared that the patient’s BPSD and hallucinations were so distressing that “he would never want to live like this,” and that things had progressed to the point that “he has no quality of life.”
Oral aripiprazole 2 mg twice daily was prescribed after the risks of infection, cardiac complications, and exacerbation of movement disorder symptoms, such as increased stiffness and falls, were discussed with the caregiver. The caregiver was employed and relied on continued employment for income, but the patient could not be safely left alone. As the patient and his caregiver had reached a crisis point and living at home no longer appeared to be safe, the patient was referred to a VA-contracted skilled nursing facility (SNF) for long-term care. The patient’s caregiver was also referred to CSP for support during this transition. Due to the patient’s level of service connection and personal needs, as well as the patient and caregiver’s preference for the veteran to remain in his home, they were evaluated for the PCAFC for enhanced support to enable home as an alternative to facility living, should the patient respond to the antipsychotic therapy sufficiently, which was evaluated on a regular basis.
After several months, the patient’s BPSD had improved significantly, and he was no longer experiencing distressing hallucinations. However, his mobility also declined, and he became fully dependent in most ADL, including transfers, hygiene, and toileting. Due to the COVID-19 pandemic, visitation was limited, which was difficult for both the patient and his caregiver. The veteran and caregiver were approved for PCAFC due to the veteran’s combination of service-connected illnesses > 70%, dependence for most ADLs, and need for continuous supervision. A transfer home from the SNF was arranged.
The PCAFC allowed the veteran’s caregiver and family members to provide in-home full-time caregiving, as an alternative to facility placement. The caregiver received a variety of support, including access to peer support, instruction on ways to assist in his toileting, hygiene, and transfers, and a caregiving stipend. In addition to offsetting lost wages, the stipend also helped offset the cost of care supplies which were not provided or were not readily available from the VA, which at the time included the patient’s preferred nutritional supplement and some supplies for personal care.
The veteran’s care needs continued to escalate. A fall at home resulted in a hip fracture, which was treated with surgical pinning. Postfracture physical therapy in a facility was considered, but ultimately was provided at home. The patient also experienced multiple UTIs and resulting delirium, with accompanying agitation and hallucinations. These episodes improved with IV antibiotics and hydration during short hospital stays. Ultimately, a computed tomography demonstrated overflow incontinence likely related to urologic damage from prior kidney stones and stent placement was recommended.
Visiting skilled nurses for the patient’s area were difficult to coordinate but were eventually arranged. The patient continued residing in his home with the support of his caregiver, the PCAFC, and the local VA medical center geriatric and transitional care services. The patient was also referred to the palliative care outpatient specialty clinic for discussion of goals of care and assistance with advance care planning as his illness progressed. Mental health and geriatric psychiatry consult teams were considered for this case but not utilized.
Discussion
Older adult Americans are at high risk of poor financial wellbeing, with nearly one quarter of Americans aged > 62 years experiencing financial insecurity.12 Even in this case with health care provided by the VA, successful in-home care was challenging and required a dedicated live-in caregiver, care coordination resources, and financial support. As part of its mission of caring for veterans, the VA has instituted CSP, whose mission is to promote the health and well-being of family caregivers through education, support, and services.
PCAFC offers enhanced clinical support for caregivers of eligible veterans who are seriously injured. This includes resources, education, support, financial stipends, health insurance (if eligible), and beneficiary travel (if eligible) to primary caregivers of eligible veterans. PCAFC was originally reserved for veterans who had onset of service-related disability after September 11, 2001, with an associated personal care need. In this population, PCAFC demonstrated an increased usage of clinical resources, likely related to increased ease in accessing care.13
The cohort of post-9/11 veterans is very different from the cohort of veterans and their caregivers who may now qualify for the PCAFC after its October 2020 expansion. Veterans from the Vietnam, Korean, and World War II eras of conflict have rates of service-connected disability 2 to 3 times higher than those of post-9/11 era veterans and are at greater risk for dementia.9 Veterans aged ≥ 75 years who have service connection also report higher rates of difficulty with independent living and self-care compared with their younger peers.9 Since dementia and PTSD are common causes of service connection and disability it is likely that a significant proportion of older veterans will be eligible to apply for the newly expanded PCAFC.
To be eligible for PCAFC, a veteran must have a service-connected disability rating of ≥ 70% and must need in-person care services for ≥ 6 continuous months, based on either an inability to perform an ADL, or a need for supervision, protection, or instruction.
It is not clear how CSP use or increased access to PCAFC will impact costs. However, the PCAFC monthly stipend is scaled to the median wage of a home health aide and to the location of the caregiver, which is considerably less than the cost of recurrent hospitalization or a year of facility-level care.15 The CSP may eventually be a successful long-term investment in cost savings. In order to ensure the process for PCAFC approval is uniform and prompt as the program expands, CSP has restructured, increasing the number of employees, improving the patient review process, and expanding staff training.16 The VA plans to continually re-assess CSP using the infrastructure of the Caregiver Record Management Application as it continues to expand.17
Conclusions
Dementia and PTSD commonly coexist and are a significant source of disability in the service-connected veteran population. This case brings attention to the recent expansion of PCAFC, which now has the potential to support eligible veterans from the World War II, Korean, and Vietnam-era conflicts, in whom these illnesses are more common. In this case, in-home care was preferred by the veteran and primary caregiver but would not have been possible without a complex intervention. There is still more research to be done on the best way to meet the needs of older adults with dementia, the impact of in-home care, and the system-wide implications of PCAFC, especially as the program grows. However, in-home care is preferable to SNF living for many veterans and caregivers, and CSP will continue to be an essential element of providing care for this population.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin. The authors would also like to thank the members of the Veterans Affairs Central Office and National Caregiver Support Program Office, including Elyse Kaplan, Melinda Hogue, Beth Wolfsohn, Colleen Richardson, and Timothy Jobin, for their thorough review of the work and edits to ensure accurate program description.
1. Chi W, Graf E, Hughes L, et al. Community-dwelling older adults with dementia and their caregivers: key indicators from the National Health and Aging Trends study. Published January 29, 2019. Accessed February 16, 2022. https://aspe.hhs.gov/sites/default/files/migrated_legacy_files//186501/DemChartbook.pdf
2. Rapaport P, Burton A, Leverton M, et al. “I just keep thinking that I don’t want to rely on people.” A qualitative study of how people living with dementia achieve and maintain independence at home: stakeholder perspectives. BMC Geriatr. 2020;20(1):1-11. doi:10.1186/s12877-019-1406-6
3. Miller EA, Gidmark S, Gadbois E, Rudolph JL, Intrator O. Nursing home referral within the Veterans Health Administration: practice variation by payment source and facility type. Res Aging. 2018;40(7):687-711. doi:10.1177/0164027517730383
4. Veterans Benefits, Health Care, and Information Technology Act of 2006, Pub L No. 109-461, 120 Stat. 3403.
5. Caregivers and Veterans Omnibus Health Services Act of 2010, Pub L No. 111-163, 115 Stat 552.
6. VA MISSION Act of 2018. 38 CFR § 17.
7. Yaffe K, Vittinghoff E, Lindquist K, et al. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry. 2010;67(6):608-613. doi:10.1001/archgenpsychiatry.2010.61
8. Krishnan LL, Petersen NJ, Snow AL, et al. Prevalence of dementia among Veterans Affairs medical care system users. Dement Geriatr Cogn Disord. 2005;20(4):245-253. doi:10.1159/000087345
9. Holder, KA. The Disability of Veterans. Social, Economic, and Housing Statistics Division, US Census Bureau; 2014. Accessed February 9, 2022. https://www.census.gov/content/dam/Census/library/working-papers/2016/demo/Holder-2016-01.pdf
10. Sanford AM. Lewy body dementia. Clin Geriatr Med. 2018;34(4):603-615. doi:10.1016/j.cger.2018.06.007
11. Armstrong MJ. Lewy body dementias. Continuum (Minneap Minn). 2019;25(1):128-146. doi:10.1212/CON.0000000000000685
12. Bureau of Consumer Financial Protection. Financial well-being of older Americans. Published December 2018. Accessed February 17, 2022. https://files.consumerfinance.gov/f/documents/bcfp_financial-well-being-older-americans_report.pdf
13. Van Houtven CH, Smith VA, Stechuchak KM, et al. Comprehensive support for family caregivers: impact on veteran health care utilization and costs. Med Care Res Rev. 2019;76(1):89-114. doi:10.1177/1077558717697015
14. Boland L, Légaré F, Perez MMB, et al. Impact of home care versus alternative locations of care on elder health outcomes: an overview of systematic reviews. BMC Geriatr. 2017;17(1):20. doi:10.1186/s12877-016-0395-y
15. Program of Comprehensive Assistance for Family Caregivers Improvements and Amendments Under the VA MISSION Act of 2018. 85 FR § 13356.
16. Extension of Program of Comprehensive Assistance for Family Caregivers Eligibility for Legacy Participants and Legacy Applicants. 86 FR § 52614.
17. US Department of Veterans Affairs, 2020. Certification of the Implementation of the Caregiver Records Management Application (CARMA). 85 FR § 63358.
18. Sussman, JS. Department of Veterans Affairs: Caregiver Support. Congressional Research Service Report No. R46282. Published March 24, 2020. Accessed February 16, 2022. https://www.everycrsreport.com/files/20200324_R46282_656f1e8338af12a2a676c471be3b3c13b2fcb0bb.pdf
19. US Department of Veterans Affairs. Veterans Affairs Program of Comprehensive Assistance for Family Caregivers Eligibility Criteria Fact Sheet. Washington, DC: U.S. Department of Veterans Affairs; 2020. Accessed February 9, 2022. https://www.caregiver.va.gov/pdfs/MissionAct/EligibilityCriteriaFactsheet_Chapter2_Launch_Approved_Final_100120.pdf
1. Chi W, Graf E, Hughes L, et al. Community-dwelling older adults with dementia and their caregivers: key indicators from the National Health and Aging Trends study. Published January 29, 2019. Accessed February 16, 2022. https://aspe.hhs.gov/sites/default/files/migrated_legacy_files//186501/DemChartbook.pdf
2. Rapaport P, Burton A, Leverton M, et al. “I just keep thinking that I don’t want to rely on people.” A qualitative study of how people living with dementia achieve and maintain independence at home: stakeholder perspectives. BMC Geriatr. 2020;20(1):1-11. doi:10.1186/s12877-019-1406-6
3. Miller EA, Gidmark S, Gadbois E, Rudolph JL, Intrator O. Nursing home referral within the Veterans Health Administration: practice variation by payment source and facility type. Res Aging. 2018;40(7):687-711. doi:10.1177/0164027517730383
4. Veterans Benefits, Health Care, and Information Technology Act of 2006, Pub L No. 109-461, 120 Stat. 3403.
5. Caregivers and Veterans Omnibus Health Services Act of 2010, Pub L No. 111-163, 115 Stat 552.
6. VA MISSION Act of 2018. 38 CFR § 17.
7. Yaffe K, Vittinghoff E, Lindquist K, et al. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry. 2010;67(6):608-613. doi:10.1001/archgenpsychiatry.2010.61
8. Krishnan LL, Petersen NJ, Snow AL, et al. Prevalence of dementia among Veterans Affairs medical care system users. Dement Geriatr Cogn Disord. 2005;20(4):245-253. doi:10.1159/000087345
9. Holder, KA. The Disability of Veterans. Social, Economic, and Housing Statistics Division, US Census Bureau; 2014. Accessed February 9, 2022. https://www.census.gov/content/dam/Census/library/working-papers/2016/demo/Holder-2016-01.pdf
10. Sanford AM. Lewy body dementia. Clin Geriatr Med. 2018;34(4):603-615. doi:10.1016/j.cger.2018.06.007
11. Armstrong MJ. Lewy body dementias. Continuum (Minneap Minn). 2019;25(1):128-146. doi:10.1212/CON.0000000000000685
12. Bureau of Consumer Financial Protection. Financial well-being of older Americans. Published December 2018. Accessed February 17, 2022. https://files.consumerfinance.gov/f/documents/bcfp_financial-well-being-older-americans_report.pdf
13. Van Houtven CH, Smith VA, Stechuchak KM, et al. Comprehensive support for family caregivers: impact on veteran health care utilization and costs. Med Care Res Rev. 2019;76(1):89-114. doi:10.1177/1077558717697015
14. Boland L, Légaré F, Perez MMB, et al. Impact of home care versus alternative locations of care on elder health outcomes: an overview of systematic reviews. BMC Geriatr. 2017;17(1):20. doi:10.1186/s12877-016-0395-y
15. Program of Comprehensive Assistance for Family Caregivers Improvements and Amendments Under the VA MISSION Act of 2018. 85 FR § 13356.
16. Extension of Program of Comprehensive Assistance for Family Caregivers Eligibility for Legacy Participants and Legacy Applicants. 86 FR § 52614.
17. US Department of Veterans Affairs, 2020. Certification of the Implementation of the Caregiver Records Management Application (CARMA). 85 FR § 63358.
18. Sussman, JS. Department of Veterans Affairs: Caregiver Support. Congressional Research Service Report No. R46282. Published March 24, 2020. Accessed February 16, 2022. https://www.everycrsreport.com/files/20200324_R46282_656f1e8338af12a2a676c471be3b3c13b2fcb0bb.pdf
19. US Department of Veterans Affairs. Veterans Affairs Program of Comprehensive Assistance for Family Caregivers Eligibility Criteria Fact Sheet. Washington, DC: U.S. Department of Veterans Affairs; 2020. Accessed February 9, 2022. https://www.caregiver.va.gov/pdfs/MissionAct/EligibilityCriteriaFactsheet_Chapter2_Launch_Approved_Final_100120.pdf
Gadolinium Deposition Disease: A Case Report and the Prevalence of Enhanced MRI Procedures Within the Veterans Health Administration
Gadolinium (Gd)-based contrast agents are frequently used in health care for enhancing magnetic resonance image (MRI) signals at low concentrations. Contrary to popular opinion, this widely used heavy metal is not biologically inert. Once notable for its safety profile, there is mounting evidence for Gd deposition in various organ systems of the body, even in those with normal renal function. A large knowledge gap remains concerning the potential harms of Gd deposition and the factors determining its elimination from the body. However, the findings of deposited Gd throughout various organs and their intracellular compartments even years after the initial exposure have been established. Here, we describe a case of a Vietnam-era veteran whose presentation, clinical, and laboratory findings were consistent within the spectrum of Gd deposition disease.
Case Presentation
A Vietnam-era veteran aged > 70 years presented for evaluation of Gd-based contrast agent–induced chronic multisymptomatic illness His medical history was significant for chronic low back pain, chronic hypertension, type 2 diabetes mellitus, and hypogonadism. Surgical history was notable for back surgery (24 years prior), laminectomy (2 years prior), shoulder replacement (2 years prior), and an epidural complicated by a hematoma (1 year prior). His presenting concerns included a painful and pruritic rash that worsened with showering, pain originating at the right Achilles tendon with migration to the knee, and shoulder pain. His symptoms started shortly after receiving multiple exposures to Gd-based contrast agents to enhance MRIs during his clinical care (Omniscan 20 mL, Omniscan 20 mL, and Gadovist 10 mL, administered 578, 565, and 496 days prior to the clinic visit, respectively). New onset headaches coincided with the timeline of symptom onset, in addition to hoarseness and liberation of an “oily substance” from the skin. More than one year prior to this clinic visit, he was considered for having polymyalgia rheumatica given the ambiguity of symptoms. Functional status remained impaired despite treatment with prednisone and methotrexate.
The patient’s military service was in the mid-1960s. He was deployed to Japan and had no knowledge of an Agent Orange exposure. His tobacco history was distant, and he reported no tattoos, prior transfusions, or occupational metal exposure
Clinical Findings
The patient was afebrile, normotensive (146/88 mmHg), and normocardic. His weight was 100 kg. He was well nourished and in no acute distress. The thought process was attentive, and his affect pleasant. Ocular examination was notable for arcus senilus. The fundoscopic examination was limited on the left, but there was no neovascularization on the right. Jugular venous pulsation was normal at 8 cm. Right ventricular impulse was slightly hyperdynamic, the rhythm was regular, and there was no abnormal splitting of S2. A soft-grade I/VI crescendo/decrescendo murmur was auscultated along the apex. Radial pulses were 2/2. He was not in respiratory distress, with equally resonant fields bilaterally. Lung sounds were clear bilaterally. A papular, erythematous rash was present in a general distribution over the chest, with few telangiectasias and some varicosity along his left arm. The skin had normal elasticity, although the skin of the hands and legs was papyraceous.
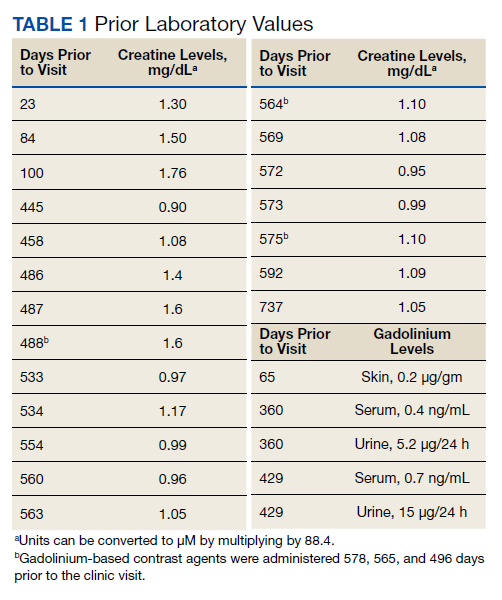
Gd levels were measured in the blood and urine (Table 1). Gd was detectable in the skin (0.2 µg/g) nearly 400 days after the last exposure. Gd was still detectable in the patient’s blood and urine (0.2 ng/mL and 0.5 µg/24 h, respectively) more than 3 years after his last exposure.
Discussion
In the United States, there are 40.44 MRI units per million people and 40 million MRIs are conducted annually. From 30 to 50% of these are enhanced with Gd-based contrast agents. In the past 30 years, there have been > 450 million contrast-enhanced MRI procedures.1
Gd is a rare earth metal. Among commercially available elements Gd has exceptional properties for enhancing MRI signals at low concentrations.1 The nonphysiologic metal is detoxified by chelation with proprietary multidentate formulations that enhance (primarily renal) elimination while retaining the paramagnetic and chemical properties for imaging. Gd exposure was found to be associated to iatrogenic nephrogenic systemic fibrosis in 2006 and later confirmed via multiple systematic reviews.2 Gd is retained in every vital organ after exposure.3 Gd-based contrast agents stimulate bone marrow–derived fibrocytes in mediating fibrosis, and bone marrow develop a memory of prior contrast exposure (Figure 1).4-6 Systemic fibrosis is mediated by the monocyte chemoattractant protein 1/C-C chemokine receptor 2.6,7 Even in the setting of normal renal function, Gd-based contrast induces the formation of Gd-rich nanoparticles in the skin and kidney.7,8 Far from being inert, Gd-based contrast agents induce systemic metabolic changes such as hypertriglyceridemia, elevations in low-density lipoprotein cholesterol, insulin resistance, and the Warburg effect (glycolytic/energy switching) in the renal cortex concomitant with profound mitochondrial abnormalities.8
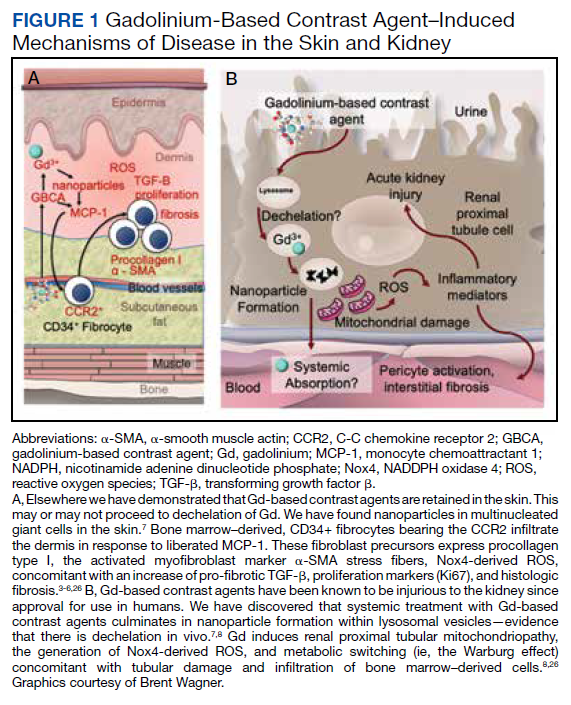
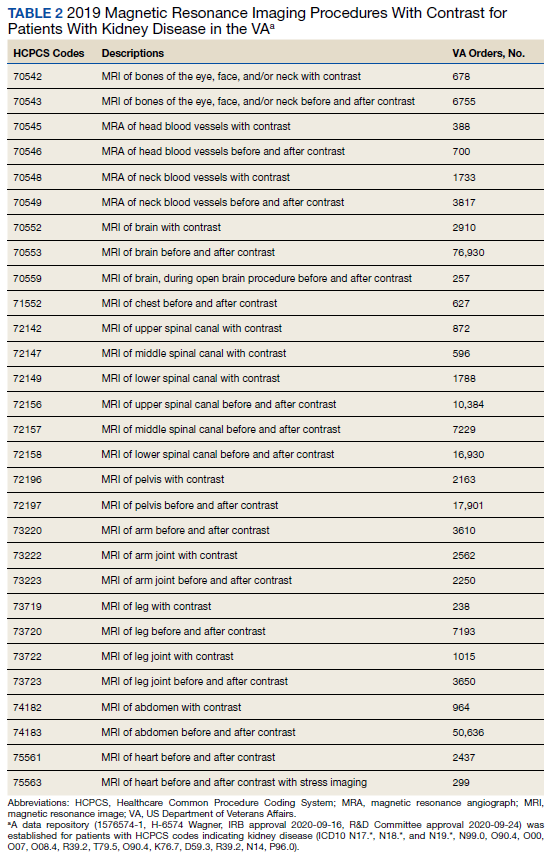
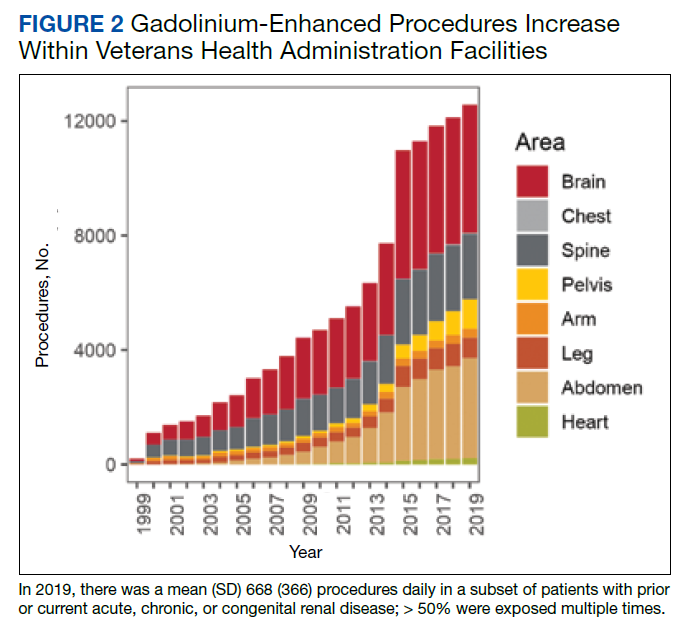
We have discovered that the rate of Gd-enhanced procedures has increased immensely within the Veterans Health Administration (VHA) system in a subset of patients with designated kidney disease (Table 2). Although a substantial number of procedures are dedicated to head and brain imaging within the VHA, the indications for Gd-enhanced diagnoses (eg, cardiac) are increasing (Figure 2).
Retention of Gd can be modeled as a function of time (t) by the half-lives of the fast, intermediate, and slow phases of elimination (Ta, Tb, and Tc, respectively):9
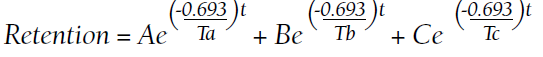
A, B, and C are the proportions (adding to 100%) that represent each of the compartments:
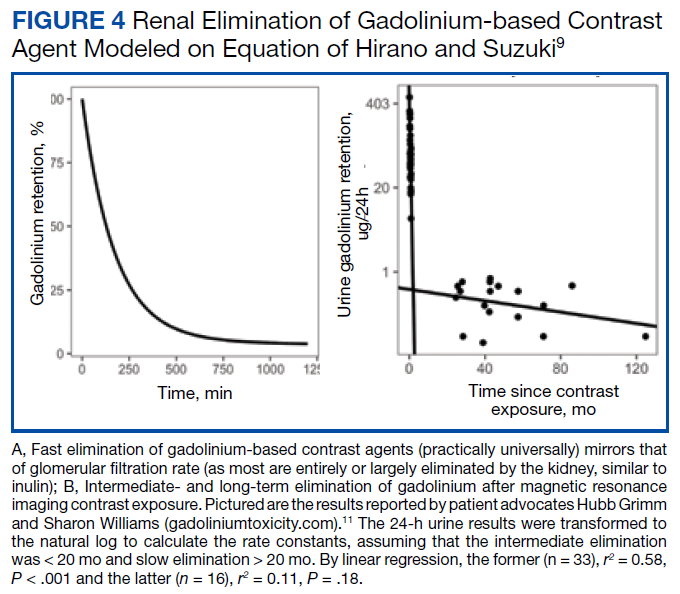
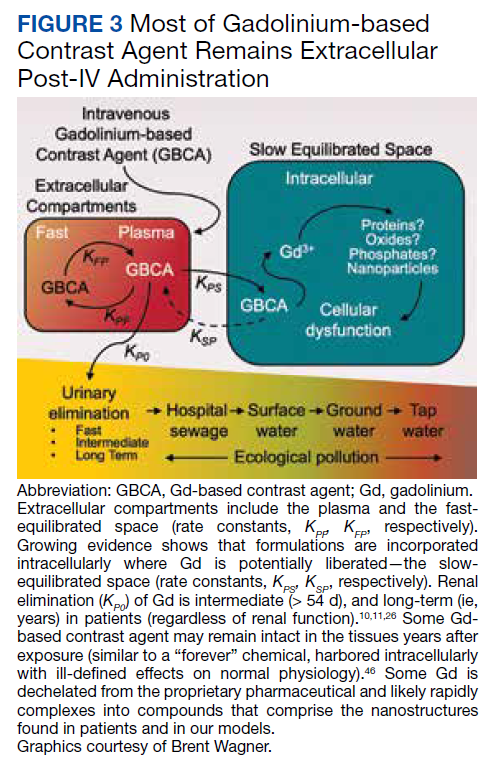
Numerous patients with normal renal function developed similar or novel symptoms that have been attributed to Gd concomitant with detectable urinary Gd years after exposure.11 Gd-based contrast agents are increasingly associated with cutaneous abnormalities even outside of nephrogenic systemic fibrosis. Gd-associated plaques develop in patients without kidney disease—these range from asymptomatic, pruritic, to burning.13 Histologic specimens reveal CD68 and factor XIIIa–positive spindle-shaped myeloid cells (the same mediators of iatrogenic systemic fibrosis) or CD34-positive cells. CD68 and factor XIIIa are distinctive for histologic specimens from patients with systemic fibrosis, and these markers have been detected in our preclinical models that demonstrated that bone marrow–derived cells are involved in mediating fibrosis.3,4,14-19 Similarly, CD34-positive cells have been historically associated with systemic fibrosis lesions.15,16,18-23 Plump osteocyte-appearing cells have also been noted (note that extraosseous metaplasia makes the histologic diagnosis of systemic fibrosis).14 Nephrogenic systemic fibrosis is an iatrogenic disease that can manifest years after exposure to Gd.5 Gd induces the recruitment of bone marrow–derived cells to the affected sites.4
The VA Health Service Research and Development Evidence Synthesis Program reviewed the safety of Gd-based contrast agents in patients with impaired kidney function.24,25 The group found only a single study of Gd and veterans. “Awareness and concern are growing about the long-term deposition of gadolinium in [the] brain and other tissues among patients with normal kidney function,” according to Lunyera and colleagues.25 The largest knowledge gap was that a comprehensive review “of all potential harms associated with gadolinium exposure” was not addressed. Furthermore, the group advised “caution in the use of [Gd-based contrast agents] in patients with severely impaired kidney function and acute kidney injury remains prudent, because the exact clinical factors contributing to [nephrogenic systemic fibrosis] risk in these subpopulations are still unknown.”25
Gd-based contrast agents—contrary to a widely held misconception—are not biologically inert.1 Gd-based contrast agents have a long history of association with acute renal injury. We have demonstrated that systemic treatment with MRI contrast agents leads to vacuolization of the proximal tubule and tubular injury.7,8 Kidney injury may be mediated by the generation of reactive oxygen species from NADPH oxidase 4 (Nox4).26
Gd retention, Gd-induced multisymptomatic illnesses, Gd-associated plaques, Gd-induced neurotoxicity, and nephrogenic systemic fibrosis are part of a continuum (with Gd as the common thread)—a theme of the September 8, 2017, US Food and Drug Administration (FDA) Medical Imaging Drugs Advisory Committee meeting.27 Patients, patient advocacy groups, and regulating agencies are concerned about long-term retention of a nonphysiologic rare earth element such as Gd.28-30 A patient advocacy group, The Lighthouse Project, collected information from patients linking the last date of Gd-based contrast agent exposure and urinary Gd.11 Data from their report suggest that the rate constants (valuable for the elimination equation above) are obtainable from 24-hour urine collections. Conceptually, Gd-induced diseases may represent a continuum that results from the retention of a nonphysiologic, toxic heavy rare earth metal.
As a heavy metal, Gd is not a natural physiologic trace element. Similar to numerous nonphysiologic metals, Gd is toxic. Inhaled Gd oxide (Gd2O3) dust leads to a number of time-dependent pathologies. Animal lung studies demonstrate reduced elasticity, enlarged cells, thickened lung walls, and recruitment of immune cells.31 Symptoms of acute IV Gd toxicity include decreased respiration, lethargy, abdominal cramps, and diarrhea.32 Pharmacologically, Gd concentrates in the liver and kidney and accumulates in the bone.32 Animals demonstrate intestinal depression and low blood pressure in response to Gd and, with higher doses, cardiovascular collapse.32 IV Gd chloride leads to metal deposition in the small blood vessels diffusely throughout the body, particularly in the lung and kidney and the metal is absorbed by the scavenging white blood cells.33 Gd chloride induces severe damage to the liver, spleen, and the digestive tract.33 Furthermore, this form of the toxicant metal markedly impacted functions associated with bleeding and clotting, ie, decreased platelet numbers and an increase in the laboratory-measured coagulation parameters.33 Semelka and colleagues have characterized chronic symptoms attributed to Gd-based contrast agents (not limited to chronic pain, headache, bone pain, skin thickening, and clouded mentation).34,35 Because Gd-induced conditions are underrecognized and ill-defined, disinherited patients often resort to untested (and potentially dangerous) chelation therapies.36
This patient presented with numerous symptoms that arose after Gd exposure. It is well established that Gd-based contrast agents (of any class) are retained in multiple organs (including the brain), for months to years. Gd-based contrast agents enter the cerebrospinal fluid within minutes of IV administration.37 Gd was found in the cerebrospinal fluid 9 months after administration in a case presented to the FDA Medical Imaging Drugs Advisory Committee.38 We know from intentional and accidental intrathecal administrations that Gd-based contrast agents are neurotoxic.39 Runge and colleagues demonstrated that Gd-based contrast agents exert mitochondrial toxicity in cultured neurons in vitro.40 McDonald and his team found Gd-rich nanoparticles within the brain neurons (cytoplasm and nuclei) from patients exposed to MRI contrast in the normal course of care.41 These nanoparticles are similar to what we have found in rodent models of Gd-induced disease.7,8,42
Prolonged elimination of Gd after MRI contrast administration (months to years) may be universal.10 Gd compartmentalizes into leukocytes and erythrocytes and into the cerebrospinal fluid within minutes.37,43 Patients with multisymptomatic illnesses attributed to Gd (Gd deposition disease) have perturbations in cytokine levels, many inflammatory.44,45 The results are concerning: Gd is retained intracellularly in vital organs, including brain neurons. It is inarguable that Gd is an alien, nonphysiologic element. With mounting evidence that Gd retention has clinical consequences, patients should be provided proper informed consent. Complications of renal insufficiency (ie, hyperkalemia, hyperphosphatemia, renal osteodystrophy, hyponatremia, anemia, immunosuppression, etc) follow a smooth, curvilinear slope as the true (not estimated) glomerular filtration declines; the worst iatrogenic complication from Gd—systemic fibrosis—is likely no different.
Patient Perspective
“Seems like it’s one thing after another. My family doctor said that once I had the gadolinium exposures, I have had problems ever since that I don’t recover from.” This includes chronic numbness from the rectum to the bilateral lower extremities and an indolent worsening kidney function; “I have already developed stage 3B chronic kidney disease.” Similar to many suffering with gadolinium retention, the patient was concerned about the long-term consequences. Gadolinium “is a toxic metal that is going through my body for 4 years. That has to be a problem. How come we don’t have that answer?” Clinician ignorance of Gd-induced complications and long-term retention is frustrating. “Not one of my doctors has taken gadolinium retention seriously. Where else are patients supposed to go?”
Conclusions
Health care professionals should be considering subclinical manifestations of nephrogenic systemic fibrosis or open to considering that intracellular neuronal retention of Gd may correlate with symptoms arising after MRI contrast exposures. The science concerning the mechanisms of how Gd exerts its pathologic effects is lagging behind the commercialization of enhancing Gd elimination (ie, chelation therapies) and other untested remedies. Practitioners need to acknowledge the unknown potential consequences of Gd and listen to patients who suspect chronic adverse effects.
1. Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28(2):154-162. doi:10.1097/MNH.0000000000000475
2. Grobner T. Gadolinium—a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis?. Nephrol Dial Transplant. 2006;21(4):1104-1108. doi:10.1093/ndt/gfk062
3. Do C, Barnes JL, Tan C, Wagner B. Type of MRI contrast, tissue gadolinium, and fibrosis. Am J Physiol Renal Physiol. 2014;307(7):F844-F855. doi:10.1152/ajprenal.00379.2014
4. Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
5. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311(1):F1-F11. doi:10.1152/ajprenal.00166.2016
6. Drel VR, Tan C, Barnes JL, Gorin Y, Lee DY, Wagner B. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
7. Do C, Drel V, Tan C, Lee D, Wagner B. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143.e2. doi:10.1016/j.jid.2019.03.1145
8. Do C, Ford B, Lee DY, Tan C, Escobar P, Wagner B. Gadolinium-based contrast agents: stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
9. Hirano S, Suzuki KT. Exposure, metabolism, and toxicity of rare earths and related compounds. Environ Health Perspect. 1996;104(suppl 1):85-95. doi:10.1289/ehp.96104s185
10. Alwasiyah D, Murphy C, Jannetto P, Hogg M, Beuhler MC. Urinary gadolinium levels after contrast-enhanced MRI in individuals with normal renal function: a pilot study. J Med Toxicol. 2019;15(2):121-127. doi:10.1007/s13181-018-0693-1
11. Williams S, Grimm H. gadolinium toxicity: shedding light on the effects of retained gadolinium from contrast MRI. Accessed April 11, 2022. https://gdtoxicity.files.wordpress.com/2018/12/gadolinium-clearance-times-for-135-contrast-mri-cases-final-v1-1.pdf
12. DeBevits JJ, Reshma M, Bageac D, et al. Gray matter nucleus hyperintensity after monthly triple-dose gadopentetate dimeglumine with long-term magnetic resonance imaging. Invest Radiol. 2020;55(10):629-635. doi:10.1097/RLI.0000000000000663
13. Gathings RM, Reddy R, Santa Cruz D, Brodell RT. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151(3):316-319. doi:10.1001/jamadermatol.2014.2660
14. Girardi M, Kay J, Elston DM, Leboit PE, Abu-Alfa A, Cowper SE. Nephrogenic systemic fibrosis: clinicopathological definition and workup recommendations. J Am Acad Dermatol. 2011;65(6):1095-1106 e7. doi:10.1016/j.jaad.2010.08.041
15. Daram SR, Cortese CM, Bastani B. Nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis: report of a new case with literature review. Am J Kidney Dis. 2005;46(4):754-759. doi:10.1053/j.ajkd.2005.06.024
16. Ortonne N, Lipsker D, Chantrel F, Boehm N, Grosshans E, Cribier B. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br J Dermatol. 2004;150(5):1050-1052. doi:10.1111/j.1365-2133.2004.05900.x
17. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-49. doi:10.1016/j.semarthrit.2005.08.002
18. Lewis KG, Lester BW, Pan TD, Robinson-Bostom L. Nephrogenic fibrosing dermopathyand calciphylaxis with pseudoxanthoma elasticum-like changes. J Cutan Pathol. 2006;33(10):695-700. doi:10.1111/j.1600-0560.2006.00490.x
19. Gibson SE, Farver CF, Prayson RA. Multiorgan involvement in nephrogenic fibrosing dermopathy: an autopsy case and review of the literature. Arch Pathol Lab Med. 2006;130(2):209-212. doi:10.5858/2006-130-209-MIINFD
20. Cassis TB, Jackson JM, Sonnier GB, Callen JP. Nephrogenic fibrosing dermopathy in a patient with acute renal failure never requiring dialysis. Int J Dermatol. 2006;45(1):56-59. doi:10.1111/j.1365-4632.2005.02701.x
21. Kucher C, Steere J, Elenitsas R, Siegel DL, Xu X. Nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis with diaphragmatic involvement in a patient with respiratory failure. J Am Acad Dermatol. 2006;54(suppl 2):S31-S34. doi:10.1016/j.jaad.2005.04.024
22. Sanyal S, Marckmann P, Scherer S, Abraham JL. Multiorgan gadolinium (Gd) deposition and fibrosis in a patient with nephrogenic systemic fibrosis—an autopsy-based review. Nephrol Dial Transplant. 2011;26(11):3616-3626. doi:10.1093/ndt/gfr085
23. Kucher C, Xu X, Pasha T, Elenitsas R. Histopathologic comparison of nephrogenic fibrosing dermopathy and scleromyxedema. J Cutan Pathol. 2005;32(7):484-490. doi:10.1111/j.0303-6987.2005.00365.x
24. Goldstein KM, Lunyera J, Mohottige D, et al. Risk of Nephrogenic Systemic Fibrosis after Exposure to Newer Gadolinium Agents. Washington (DC): Department of Veterans Affairs (US); October 2019. https://www.ncbi.nlm.nih.gov/books/NBK559376/25. Lunyera J, Mohottige D, Alexopoulos AS, et al. Risk for nephrogenic systemic fibrosis after exposure to newer gadolinium agents: a systematic review. Ann Intern Med. 2020;173(2):110-119. doi:10.7326/M20-0299
26. Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH Oxidase 4 (Nox4) for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
27. Wagner B. The pathophysiology and retention of gadolinium. United States Food & Drug Administration Medical Imaging Drugs Advisory Committee. 2017:1-23. https://www.fda.gov/advisory-committees/medical-imaging-drugs-advisory-committee/2017-meeting-materials-medical-imaging-drugs-advisory-committee?msclkid=6b5764ccbaa611ec95e35dddf8db57af
28. Runge VM. Critical questions regarding gadolinium deposition in the brain and body after injections of the gadolinium-based contrast agents, safety, and clinical recommendations in consideration of the EMA’s pharmacovigilance and risk assessment committee recommendation for suspension of the marketing authorizations for 4 linear agents. Invest Radiol. 2017;52(6):317-323. doi:10.1097/RLI.0000000000000374
29. Wagner B. Scared to the marrow: pitfalls and pearls in renal imaging. Adv Chronic Kidney Dis. 2017;24(3):136-137. doi:10.1053/j.ackd.2017.03.008
30. US Food and Drug Administration. Transcript for the September 8, 2017 Meeting of the Medical Imaging Drugs Advisory Committee (MIDAC). September 8, 2017. Accessed April 11, 2022. https://www.fda.gov/media/108935/download
31. Abel M, Talbot RB. Gadolinium oxide inhalation by guinea pigs: a correlative functional and histopathologic study. J Pharmacol Exp Ther. 1967;157(1):207-213.
32. Haley TJ, Raymond K, Komesu N, Upham HC. Toxicological and pharmacological effects of gadolinium and samarium chlorides. Br J Pharmacol Chemother. 1961;17(3):526-532. doi:10.1111/j.1476-5381.1961.tb01139.x

33. Spencer AJ, Wilson SA, Batchelor J, Reid A, Rees J, Harpur E. Gadolinium chloride toxicity in the rat. Toxicol Pathol. 1997;25(3):245-255. doi:10.1177/019262339702500301
34. Semelka RC, Ramalho M, AlObaidy M, Ramalho J. Gadolinium in humans: a family of disorders. AJR Am J Roentgenol. 2016;207(2):229-233. doi:10.2214/AJR.15.15842
35. Semelka RC, Ramalho M. Physicians with self-diagnosed gadolinium deposition disease: a case series. Radiol Bras. 2021;54(4):238-242. doi:10.1590/0100-3984.2020.0073
36. Layne KA, Wood DM, Dargan PI. Gadolinium-based contrast agents—what is the evidence for ‘gadolinium deposition disease’ and the use of chelation therapy? Clin Toxicol (Phila). 2020;58(3):151-160. doi:10.1080/15563650.2019.1681442
37. Nehra AK, McDonald RJ, Bluhm AM, et al. Accumulation of gadolinium in human cerebrospinal fluid after gadobutrol-enhanced MR imaging: a prospective observational cohort study. Radiology. 2018;288(2):416-423. doi:10.1148/radiol.2018171105
38. US Food and Drug Administration. Medical Imaging Drugs Advisory Committee Meeting. Gadolinium retention after gadolinium based contrast magnetic resonance imaging in patients with normal renal function. Briefing document. 2017. Accessed April 12, 2022. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/MedicalImagingDrugsAdvisoryCommittee/UCM572848.pdf
39. Calvo N, Jamil M, Feldman S, Shah A, Nauman F, Ferrara J. Neurotoxicity from intrathecal gadolinium administration: case presentation and brief review. Neurol Clin Pract. 2020;10(1):e7-e10. doi:10.1212/CPJ.0000000000000696
40. Bower DV, Richter JK, von Tengg-Kobligk H, Heverhagen JT, Runge VM. Gadolinium-based MRI contrast agents induce mitochondrial toxicity and cell death in human neurons, and toxicity increases with reduced kinetic stability of the agent. Invest Radiol. 2019;54(8):453-463. doi:10.1097/RLI.0000000000000567
41. McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
42. Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1(6):561-568. doi:10.34067/KID.0000272019
43. Di Gregorio E, Furlan C, Atlante S, Stefania R, Gianolio E, Aime S. Gadolinium retention in erythrocytes and leukocytes from human and murine blood upon treatment with gadolinium-based contrast agents for magnetic resonance imaging. Invest Radiol. 2020;55(1):30-37. doi:10.1097/RLI.0000000000000608
44. Maecker HT, Siebert JC, Rosenberg-Hasson Y, Koran LM, Ramalho M, Semelka RC. Acute chelation therapy-associated changes in urine gadolinium, self-reported flare severity, and serum cytokines in gadolinium deposition disease. Invest Radiol. 2021;56(6):374-384. doi:10.1097/RLI.0000000000000752
45. Maecker HT, Wang W, Rosenberg-Hasson Y, Semelka RC, Hickey J, Koran LM. An initial investigation of serum cytokine levels in patients with gadolinium retention. Radiol Bras. 2020;53(5):306-313. doi:10.1590/0100-3984.2019.0075
46. Birka M, Wentker KS, Lusmöller E, et al. Diagnosis of nephrogenic systemic fibrosis by means of elemental bioimaging and speciation analysis. Anal Chem. 2015;87(6):3321-3328. doi:10.1021/ac504488k
Gadolinium (Gd)-based contrast agents are frequently used in health care for enhancing magnetic resonance image (MRI) signals at low concentrations. Contrary to popular opinion, this widely used heavy metal is not biologically inert. Once notable for its safety profile, there is mounting evidence for Gd deposition in various organ systems of the body, even in those with normal renal function. A large knowledge gap remains concerning the potential harms of Gd deposition and the factors determining its elimination from the body. However, the findings of deposited Gd throughout various organs and their intracellular compartments even years after the initial exposure have been established. Here, we describe a case of a Vietnam-era veteran whose presentation, clinical, and laboratory findings were consistent within the spectrum of Gd deposition disease.
Case Presentation
A Vietnam-era veteran aged > 70 years presented for evaluation of Gd-based contrast agent–induced chronic multisymptomatic illness His medical history was significant for chronic low back pain, chronic hypertension, type 2 diabetes mellitus, and hypogonadism. Surgical history was notable for back surgery (24 years prior), laminectomy (2 years prior), shoulder replacement (2 years prior), and an epidural complicated by a hematoma (1 year prior). His presenting concerns included a painful and pruritic rash that worsened with showering, pain originating at the right Achilles tendon with migration to the knee, and shoulder pain. His symptoms started shortly after receiving multiple exposures to Gd-based contrast agents to enhance MRIs during his clinical care (Omniscan 20 mL, Omniscan 20 mL, and Gadovist 10 mL, administered 578, 565, and 496 days prior to the clinic visit, respectively). New onset headaches coincided with the timeline of symptom onset, in addition to hoarseness and liberation of an “oily substance” from the skin. More than one year prior to this clinic visit, he was considered for having polymyalgia rheumatica given the ambiguity of symptoms. Functional status remained impaired despite treatment with prednisone and methotrexate.
The patient’s military service was in the mid-1960s. He was deployed to Japan and had no knowledge of an Agent Orange exposure. His tobacco history was distant, and he reported no tattoos, prior transfusions, or occupational metal exposure
Clinical Findings
The patient was afebrile, normotensive (146/88 mmHg), and normocardic. His weight was 100 kg. He was well nourished and in no acute distress. The thought process was attentive, and his affect pleasant. Ocular examination was notable for arcus senilus. The fundoscopic examination was limited on the left, but there was no neovascularization on the right. Jugular venous pulsation was normal at 8 cm. Right ventricular impulse was slightly hyperdynamic, the rhythm was regular, and there was no abnormal splitting of S2. A soft-grade I/VI crescendo/decrescendo murmur was auscultated along the apex. Radial pulses were 2/2. He was not in respiratory distress, with equally resonant fields bilaterally. Lung sounds were clear bilaterally. A papular, erythematous rash was present in a general distribution over the chest, with few telangiectasias and some varicosity along his left arm. The skin had normal elasticity, although the skin of the hands and legs was papyraceous.
Gd levels were measured in the blood and urine (Table 1). Gd was detectable in the skin (0.2 µg/g) nearly 400 days after the last exposure. Gd was still detectable in the patient’s blood and urine (0.2 ng/mL and 0.5 µg/24 h, respectively) more than 3 years after his last exposure.
Discussion
In the United States, there are 40.44 MRI units per million people and 40 million MRIs are conducted annually. From 30 to 50% of these are enhanced with Gd-based contrast agents. In the past 30 years, there have been > 450 million contrast-enhanced MRI procedures.1
Gd is a rare earth metal. Among commercially available elements Gd has exceptional properties for enhancing MRI signals at low concentrations.1 The nonphysiologic metal is detoxified by chelation with proprietary multidentate formulations that enhance (primarily renal) elimination while retaining the paramagnetic and chemical properties for imaging. Gd exposure was found to be associated to iatrogenic nephrogenic systemic fibrosis in 2006 and later confirmed via multiple systematic reviews.2 Gd is retained in every vital organ after exposure.3 Gd-based contrast agents stimulate bone marrow–derived fibrocytes in mediating fibrosis, and bone marrow develop a memory of prior contrast exposure (Figure 1).4-6 Systemic fibrosis is mediated by the monocyte chemoattractant protein 1/C-C chemokine receptor 2.6,7 Even in the setting of normal renal function, Gd-based contrast induces the formation of Gd-rich nanoparticles in the skin and kidney.7,8 Far from being inert, Gd-based contrast agents induce systemic metabolic changes such as hypertriglyceridemia, elevations in low-density lipoprotein cholesterol, insulin resistance, and the Warburg effect (glycolytic/energy switching) in the renal cortex concomitant with profound mitochondrial abnormalities.8
We have discovered that the rate of Gd-enhanced procedures has increased immensely within the Veterans Health Administration (VHA) system in a subset of patients with designated kidney disease (Table 2). Although a substantial number of procedures are dedicated to head and brain imaging within the VHA, the indications for Gd-enhanced diagnoses (eg, cardiac) are increasing (Figure 2).
Retention of Gd can be modeled as a function of time (t) by the half-lives of the fast, intermediate, and slow phases of elimination (Ta, Tb, and Tc, respectively):9
A, B, and C are the proportions (adding to 100%) that represent each of the compartments:
Numerous patients with normal renal function developed similar or novel symptoms that have been attributed to Gd concomitant with detectable urinary Gd years after exposure.11 Gd-based contrast agents are increasingly associated with cutaneous abnormalities even outside of nephrogenic systemic fibrosis. Gd-associated plaques develop in patients without kidney disease—these range from asymptomatic, pruritic, to burning.13 Histologic specimens reveal CD68 and factor XIIIa–positive spindle-shaped myeloid cells (the same mediators of iatrogenic systemic fibrosis) or CD34-positive cells. CD68 and factor XIIIa are distinctive for histologic specimens from patients with systemic fibrosis, and these markers have been detected in our preclinical models that demonstrated that bone marrow–derived cells are involved in mediating fibrosis.3,4,14-19 Similarly, CD34-positive cells have been historically associated with systemic fibrosis lesions.15,16,18-23 Plump osteocyte-appearing cells have also been noted (note that extraosseous metaplasia makes the histologic diagnosis of systemic fibrosis).14 Nephrogenic systemic fibrosis is an iatrogenic disease that can manifest years after exposure to Gd.5 Gd induces the recruitment of bone marrow–derived cells to the affected sites.4
The VA Health Service Research and Development Evidence Synthesis Program reviewed the safety of Gd-based contrast agents in patients with impaired kidney function.24,25 The group found only a single study of Gd and veterans. “Awareness and concern are growing about the long-term deposition of gadolinium in [the] brain and other tissues among patients with normal kidney function,” according to Lunyera and colleagues.25 The largest knowledge gap was that a comprehensive review “of all potential harms associated with gadolinium exposure” was not addressed. Furthermore, the group advised “caution in the use of [Gd-based contrast agents] in patients with severely impaired kidney function and acute kidney injury remains prudent, because the exact clinical factors contributing to [nephrogenic systemic fibrosis] risk in these subpopulations are still unknown.”25
Gd-based contrast agents—contrary to a widely held misconception—are not biologically inert.1 Gd-based contrast agents have a long history of association with acute renal injury. We have demonstrated that systemic treatment with MRI contrast agents leads to vacuolization of the proximal tubule and tubular injury.7,8 Kidney injury may be mediated by the generation of reactive oxygen species from NADPH oxidase 4 (Nox4).26
Gd retention, Gd-induced multisymptomatic illnesses, Gd-associated plaques, Gd-induced neurotoxicity, and nephrogenic systemic fibrosis are part of a continuum (with Gd as the common thread)—a theme of the September 8, 2017, US Food and Drug Administration (FDA) Medical Imaging Drugs Advisory Committee meeting.27 Patients, patient advocacy groups, and regulating agencies are concerned about long-term retention of a nonphysiologic rare earth element such as Gd.28-30 A patient advocacy group, The Lighthouse Project, collected information from patients linking the last date of Gd-based contrast agent exposure and urinary Gd.11 Data from their report suggest that the rate constants (valuable for the elimination equation above) are obtainable from 24-hour urine collections. Conceptually, Gd-induced diseases may represent a continuum that results from the retention of a nonphysiologic, toxic heavy rare earth metal.
As a heavy metal, Gd is not a natural physiologic trace element. Similar to numerous nonphysiologic metals, Gd is toxic. Inhaled Gd oxide (Gd2O3) dust leads to a number of time-dependent pathologies. Animal lung studies demonstrate reduced elasticity, enlarged cells, thickened lung walls, and recruitment of immune cells.31 Symptoms of acute IV Gd toxicity include decreased respiration, lethargy, abdominal cramps, and diarrhea.32 Pharmacologically, Gd concentrates in the liver and kidney and accumulates in the bone.32 Animals demonstrate intestinal depression and low blood pressure in response to Gd and, with higher doses, cardiovascular collapse.32 IV Gd chloride leads to metal deposition in the small blood vessels diffusely throughout the body, particularly in the lung and kidney and the metal is absorbed by the scavenging white blood cells.33 Gd chloride induces severe damage to the liver, spleen, and the digestive tract.33 Furthermore, this form of the toxicant metal markedly impacted functions associated with bleeding and clotting, ie, decreased platelet numbers and an increase in the laboratory-measured coagulation parameters.33 Semelka and colleagues have characterized chronic symptoms attributed to Gd-based contrast agents (not limited to chronic pain, headache, bone pain, skin thickening, and clouded mentation).34,35 Because Gd-induced conditions are underrecognized and ill-defined, disinherited patients often resort to untested (and potentially dangerous) chelation therapies.36
This patient presented with numerous symptoms that arose after Gd exposure. It is well established that Gd-based contrast agents (of any class) are retained in multiple organs (including the brain), for months to years. Gd-based contrast agents enter the cerebrospinal fluid within minutes of IV administration.37 Gd was found in the cerebrospinal fluid 9 months after administration in a case presented to the FDA Medical Imaging Drugs Advisory Committee.38 We know from intentional and accidental intrathecal administrations that Gd-based contrast agents are neurotoxic.39 Runge and colleagues demonstrated that Gd-based contrast agents exert mitochondrial toxicity in cultured neurons in vitro.40 McDonald and his team found Gd-rich nanoparticles within the brain neurons (cytoplasm and nuclei) from patients exposed to MRI contrast in the normal course of care.41 These nanoparticles are similar to what we have found in rodent models of Gd-induced disease.7,8,42
Prolonged elimination of Gd after MRI contrast administration (months to years) may be universal.10 Gd compartmentalizes into leukocytes and erythrocytes and into the cerebrospinal fluid within minutes.37,43 Patients with multisymptomatic illnesses attributed to Gd (Gd deposition disease) have perturbations in cytokine levels, many inflammatory.44,45 The results are concerning: Gd is retained intracellularly in vital organs, including brain neurons. It is inarguable that Gd is an alien, nonphysiologic element. With mounting evidence that Gd retention has clinical consequences, patients should be provided proper informed consent. Complications of renal insufficiency (ie, hyperkalemia, hyperphosphatemia, renal osteodystrophy, hyponatremia, anemia, immunosuppression, etc) follow a smooth, curvilinear slope as the true (not estimated) glomerular filtration declines; the worst iatrogenic complication from Gd—systemic fibrosis—is likely no different.
Patient Perspective
“Seems like it’s one thing after another. My family doctor said that once I had the gadolinium exposures, I have had problems ever since that I don’t recover from.” This includes chronic numbness from the rectum to the bilateral lower extremities and an indolent worsening kidney function; “I have already developed stage 3B chronic kidney disease.” Similar to many suffering with gadolinium retention, the patient was concerned about the long-term consequences. Gadolinium “is a toxic metal that is going through my body for 4 years. That has to be a problem. How come we don’t have that answer?” Clinician ignorance of Gd-induced complications and long-term retention is frustrating. “Not one of my doctors has taken gadolinium retention seriously. Where else are patients supposed to go?”
Conclusions
Health care professionals should be considering subclinical manifestations of nephrogenic systemic fibrosis or open to considering that intracellular neuronal retention of Gd may correlate with symptoms arising after MRI contrast exposures. The science concerning the mechanisms of how Gd exerts its pathologic effects is lagging behind the commercialization of enhancing Gd elimination (ie, chelation therapies) and other untested remedies. Practitioners need to acknowledge the unknown potential consequences of Gd and listen to patients who suspect chronic adverse effects.
Gadolinium (Gd)-based contrast agents are frequently used in health care for enhancing magnetic resonance image (MRI) signals at low concentrations. Contrary to popular opinion, this widely used heavy metal is not biologically inert. Once notable for its safety profile, there is mounting evidence for Gd deposition in various organ systems of the body, even in those with normal renal function. A large knowledge gap remains concerning the potential harms of Gd deposition and the factors determining its elimination from the body. However, the findings of deposited Gd throughout various organs and their intracellular compartments even years after the initial exposure have been established. Here, we describe a case of a Vietnam-era veteran whose presentation, clinical, and laboratory findings were consistent within the spectrum of Gd deposition disease.
Case Presentation
A Vietnam-era veteran aged > 70 years presented for evaluation of Gd-based contrast agent–induced chronic multisymptomatic illness His medical history was significant for chronic low back pain, chronic hypertension, type 2 diabetes mellitus, and hypogonadism. Surgical history was notable for back surgery (24 years prior), laminectomy (2 years prior), shoulder replacement (2 years prior), and an epidural complicated by a hematoma (1 year prior). His presenting concerns included a painful and pruritic rash that worsened with showering, pain originating at the right Achilles tendon with migration to the knee, and shoulder pain. His symptoms started shortly after receiving multiple exposures to Gd-based contrast agents to enhance MRIs during his clinical care (Omniscan 20 mL, Omniscan 20 mL, and Gadovist 10 mL, administered 578, 565, and 496 days prior to the clinic visit, respectively). New onset headaches coincided with the timeline of symptom onset, in addition to hoarseness and liberation of an “oily substance” from the skin. More than one year prior to this clinic visit, he was considered for having polymyalgia rheumatica given the ambiguity of symptoms. Functional status remained impaired despite treatment with prednisone and methotrexate.
The patient’s military service was in the mid-1960s. He was deployed to Japan and had no knowledge of an Agent Orange exposure. His tobacco history was distant, and he reported no tattoos, prior transfusions, or occupational metal exposure
Clinical Findings
The patient was afebrile, normotensive (146/88 mmHg), and normocardic. His weight was 100 kg. He was well nourished and in no acute distress. The thought process was attentive, and his affect pleasant. Ocular examination was notable for arcus senilus. The fundoscopic examination was limited on the left, but there was no neovascularization on the right. Jugular venous pulsation was normal at 8 cm. Right ventricular impulse was slightly hyperdynamic, the rhythm was regular, and there was no abnormal splitting of S2. A soft-grade I/VI crescendo/decrescendo murmur was auscultated along the apex. Radial pulses were 2/2. He was not in respiratory distress, with equally resonant fields bilaterally. Lung sounds were clear bilaterally. A papular, erythematous rash was present in a general distribution over the chest, with few telangiectasias and some varicosity along his left arm. The skin had normal elasticity, although the skin of the hands and legs was papyraceous.
Gd levels were measured in the blood and urine (Table 1). Gd was detectable in the skin (0.2 µg/g) nearly 400 days after the last exposure. Gd was still detectable in the patient’s blood and urine (0.2 ng/mL and 0.5 µg/24 h, respectively) more than 3 years after his last exposure.
Discussion
In the United States, there are 40.44 MRI units per million people and 40 million MRIs are conducted annually. From 30 to 50% of these are enhanced with Gd-based contrast agents. In the past 30 years, there have been > 450 million contrast-enhanced MRI procedures.1
Gd is a rare earth metal. Among commercially available elements Gd has exceptional properties for enhancing MRI signals at low concentrations.1 The nonphysiologic metal is detoxified by chelation with proprietary multidentate formulations that enhance (primarily renal) elimination while retaining the paramagnetic and chemical properties for imaging. Gd exposure was found to be associated to iatrogenic nephrogenic systemic fibrosis in 2006 and later confirmed via multiple systematic reviews.2 Gd is retained in every vital organ after exposure.3 Gd-based contrast agents stimulate bone marrow–derived fibrocytes in mediating fibrosis, and bone marrow develop a memory of prior contrast exposure (Figure 1).4-6 Systemic fibrosis is mediated by the monocyte chemoattractant protein 1/C-C chemokine receptor 2.6,7 Even in the setting of normal renal function, Gd-based contrast induces the formation of Gd-rich nanoparticles in the skin and kidney.7,8 Far from being inert, Gd-based contrast agents induce systemic metabolic changes such as hypertriglyceridemia, elevations in low-density lipoprotein cholesterol, insulin resistance, and the Warburg effect (glycolytic/energy switching) in the renal cortex concomitant with profound mitochondrial abnormalities.8
We have discovered that the rate of Gd-enhanced procedures has increased immensely within the Veterans Health Administration (VHA) system in a subset of patients with designated kidney disease (Table 2). Although a substantial number of procedures are dedicated to head and brain imaging within the VHA, the indications for Gd-enhanced diagnoses (eg, cardiac) are increasing (Figure 2).
Retention of Gd can be modeled as a function of time (t) by the half-lives of the fast, intermediate, and slow phases of elimination (Ta, Tb, and Tc, respectively):9
A, B, and C are the proportions (adding to 100%) that represent each of the compartments:
Numerous patients with normal renal function developed similar or novel symptoms that have been attributed to Gd concomitant with detectable urinary Gd years after exposure.11 Gd-based contrast agents are increasingly associated with cutaneous abnormalities even outside of nephrogenic systemic fibrosis. Gd-associated plaques develop in patients without kidney disease—these range from asymptomatic, pruritic, to burning.13 Histologic specimens reveal CD68 and factor XIIIa–positive spindle-shaped myeloid cells (the same mediators of iatrogenic systemic fibrosis) or CD34-positive cells. CD68 and factor XIIIa are distinctive for histologic specimens from patients with systemic fibrosis, and these markers have been detected in our preclinical models that demonstrated that bone marrow–derived cells are involved in mediating fibrosis.3,4,14-19 Similarly, CD34-positive cells have been historically associated with systemic fibrosis lesions.15,16,18-23 Plump osteocyte-appearing cells have also been noted (note that extraosseous metaplasia makes the histologic diagnosis of systemic fibrosis).14 Nephrogenic systemic fibrosis is an iatrogenic disease that can manifest years after exposure to Gd.5 Gd induces the recruitment of bone marrow–derived cells to the affected sites.4
The VA Health Service Research and Development Evidence Synthesis Program reviewed the safety of Gd-based contrast agents in patients with impaired kidney function.24,25 The group found only a single study of Gd and veterans. “Awareness and concern are growing about the long-term deposition of gadolinium in [the] brain and other tissues among patients with normal kidney function,” according to Lunyera and colleagues.25 The largest knowledge gap was that a comprehensive review “of all potential harms associated with gadolinium exposure” was not addressed. Furthermore, the group advised “caution in the use of [Gd-based contrast agents] in patients with severely impaired kidney function and acute kidney injury remains prudent, because the exact clinical factors contributing to [nephrogenic systemic fibrosis] risk in these subpopulations are still unknown.”25
Gd-based contrast agents—contrary to a widely held misconception—are not biologically inert.1 Gd-based contrast agents have a long history of association with acute renal injury. We have demonstrated that systemic treatment with MRI contrast agents leads to vacuolization of the proximal tubule and tubular injury.7,8 Kidney injury may be mediated by the generation of reactive oxygen species from NADPH oxidase 4 (Nox4).26
Gd retention, Gd-induced multisymptomatic illnesses, Gd-associated plaques, Gd-induced neurotoxicity, and nephrogenic systemic fibrosis are part of a continuum (with Gd as the common thread)—a theme of the September 8, 2017, US Food and Drug Administration (FDA) Medical Imaging Drugs Advisory Committee meeting.27 Patients, patient advocacy groups, and regulating agencies are concerned about long-term retention of a nonphysiologic rare earth element such as Gd.28-30 A patient advocacy group, The Lighthouse Project, collected information from patients linking the last date of Gd-based contrast agent exposure and urinary Gd.11 Data from their report suggest that the rate constants (valuable for the elimination equation above) are obtainable from 24-hour urine collections. Conceptually, Gd-induced diseases may represent a continuum that results from the retention of a nonphysiologic, toxic heavy rare earth metal.
As a heavy metal, Gd is not a natural physiologic trace element. Similar to numerous nonphysiologic metals, Gd is toxic. Inhaled Gd oxide (Gd2O3) dust leads to a number of time-dependent pathologies. Animal lung studies demonstrate reduced elasticity, enlarged cells, thickened lung walls, and recruitment of immune cells.31 Symptoms of acute IV Gd toxicity include decreased respiration, lethargy, abdominal cramps, and diarrhea.32 Pharmacologically, Gd concentrates in the liver and kidney and accumulates in the bone.32 Animals demonstrate intestinal depression and low blood pressure in response to Gd and, with higher doses, cardiovascular collapse.32 IV Gd chloride leads to metal deposition in the small blood vessels diffusely throughout the body, particularly in the lung and kidney and the metal is absorbed by the scavenging white blood cells.33 Gd chloride induces severe damage to the liver, spleen, and the digestive tract.33 Furthermore, this form of the toxicant metal markedly impacted functions associated with bleeding and clotting, ie, decreased platelet numbers and an increase in the laboratory-measured coagulation parameters.33 Semelka and colleagues have characterized chronic symptoms attributed to Gd-based contrast agents (not limited to chronic pain, headache, bone pain, skin thickening, and clouded mentation).34,35 Because Gd-induced conditions are underrecognized and ill-defined, disinherited patients often resort to untested (and potentially dangerous) chelation therapies.36
This patient presented with numerous symptoms that arose after Gd exposure. It is well established that Gd-based contrast agents (of any class) are retained in multiple organs (including the brain), for months to years. Gd-based contrast agents enter the cerebrospinal fluid within minutes of IV administration.37 Gd was found in the cerebrospinal fluid 9 months after administration in a case presented to the FDA Medical Imaging Drugs Advisory Committee.38 We know from intentional and accidental intrathecal administrations that Gd-based contrast agents are neurotoxic.39 Runge and colleagues demonstrated that Gd-based contrast agents exert mitochondrial toxicity in cultured neurons in vitro.40 McDonald and his team found Gd-rich nanoparticles within the brain neurons (cytoplasm and nuclei) from patients exposed to MRI contrast in the normal course of care.41 These nanoparticles are similar to what we have found in rodent models of Gd-induced disease.7,8,42
Prolonged elimination of Gd after MRI contrast administration (months to years) may be universal.10 Gd compartmentalizes into leukocytes and erythrocytes and into the cerebrospinal fluid within minutes.37,43 Patients with multisymptomatic illnesses attributed to Gd (Gd deposition disease) have perturbations in cytokine levels, many inflammatory.44,45 The results are concerning: Gd is retained intracellularly in vital organs, including brain neurons. It is inarguable that Gd is an alien, nonphysiologic element. With mounting evidence that Gd retention has clinical consequences, patients should be provided proper informed consent. Complications of renal insufficiency (ie, hyperkalemia, hyperphosphatemia, renal osteodystrophy, hyponatremia, anemia, immunosuppression, etc) follow a smooth, curvilinear slope as the true (not estimated) glomerular filtration declines; the worst iatrogenic complication from Gd—systemic fibrosis—is likely no different.
Patient Perspective
“Seems like it’s one thing after another. My family doctor said that once I had the gadolinium exposures, I have had problems ever since that I don’t recover from.” This includes chronic numbness from the rectum to the bilateral lower extremities and an indolent worsening kidney function; “I have already developed stage 3B chronic kidney disease.” Similar to many suffering with gadolinium retention, the patient was concerned about the long-term consequences. Gadolinium “is a toxic metal that is going through my body for 4 years. That has to be a problem. How come we don’t have that answer?” Clinician ignorance of Gd-induced complications and long-term retention is frustrating. “Not one of my doctors has taken gadolinium retention seriously. Where else are patients supposed to go?”
Conclusions
Health care professionals should be considering subclinical manifestations of nephrogenic systemic fibrosis or open to considering that intracellular neuronal retention of Gd may correlate with symptoms arising after MRI contrast exposures. The science concerning the mechanisms of how Gd exerts its pathologic effects is lagging behind the commercialization of enhancing Gd elimination (ie, chelation therapies) and other untested remedies. Practitioners need to acknowledge the unknown potential consequences of Gd and listen to patients who suspect chronic adverse effects.
1. Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28(2):154-162. doi:10.1097/MNH.0000000000000475
2. Grobner T. Gadolinium—a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis?. Nephrol Dial Transplant. 2006;21(4):1104-1108. doi:10.1093/ndt/gfk062
3. Do C, Barnes JL, Tan C, Wagner B. Type of MRI contrast, tissue gadolinium, and fibrosis. Am J Physiol Renal Physiol. 2014;307(7):F844-F855. doi:10.1152/ajprenal.00379.2014
4. Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
5. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311(1):F1-F11. doi:10.1152/ajprenal.00166.2016
6. Drel VR, Tan C, Barnes JL, Gorin Y, Lee DY, Wagner B. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
7. Do C, Drel V, Tan C, Lee D, Wagner B. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143.e2. doi:10.1016/j.jid.2019.03.1145
8. Do C, Ford B, Lee DY, Tan C, Escobar P, Wagner B. Gadolinium-based contrast agents: stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
9. Hirano S, Suzuki KT. Exposure, metabolism, and toxicity of rare earths and related compounds. Environ Health Perspect. 1996;104(suppl 1):85-95. doi:10.1289/ehp.96104s185
10. Alwasiyah D, Murphy C, Jannetto P, Hogg M, Beuhler MC. Urinary gadolinium levels after contrast-enhanced MRI in individuals with normal renal function: a pilot study. J Med Toxicol. 2019;15(2):121-127. doi:10.1007/s13181-018-0693-1
11. Williams S, Grimm H. gadolinium toxicity: shedding light on the effects of retained gadolinium from contrast MRI. Accessed April 11, 2022. https://gdtoxicity.files.wordpress.com/2018/12/gadolinium-clearance-times-for-135-contrast-mri-cases-final-v1-1.pdf
12. DeBevits JJ, Reshma M, Bageac D, et al. Gray matter nucleus hyperintensity after monthly triple-dose gadopentetate dimeglumine with long-term magnetic resonance imaging. Invest Radiol. 2020;55(10):629-635. doi:10.1097/RLI.0000000000000663
13. Gathings RM, Reddy R, Santa Cruz D, Brodell RT. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151(3):316-319. doi:10.1001/jamadermatol.2014.2660
14. Girardi M, Kay J, Elston DM, Leboit PE, Abu-Alfa A, Cowper SE. Nephrogenic systemic fibrosis: clinicopathological definition and workup recommendations. J Am Acad Dermatol. 2011;65(6):1095-1106 e7. doi:10.1016/j.jaad.2010.08.041
15. Daram SR, Cortese CM, Bastani B. Nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis: report of a new case with literature review. Am J Kidney Dis. 2005;46(4):754-759. doi:10.1053/j.ajkd.2005.06.024
16. Ortonne N, Lipsker D, Chantrel F, Boehm N, Grosshans E, Cribier B. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br J Dermatol. 2004;150(5):1050-1052. doi:10.1111/j.1365-2133.2004.05900.x
17. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-49. doi:10.1016/j.semarthrit.2005.08.002
18. Lewis KG, Lester BW, Pan TD, Robinson-Bostom L. Nephrogenic fibrosing dermopathyand calciphylaxis with pseudoxanthoma elasticum-like changes. J Cutan Pathol. 2006;33(10):695-700. doi:10.1111/j.1600-0560.2006.00490.x
19. Gibson SE, Farver CF, Prayson RA. Multiorgan involvement in nephrogenic fibrosing dermopathy: an autopsy case and review of the literature. Arch Pathol Lab Med. 2006;130(2):209-212. doi:10.5858/2006-130-209-MIINFD
20. Cassis TB, Jackson JM, Sonnier GB, Callen JP. Nephrogenic fibrosing dermopathy in a patient with acute renal failure never requiring dialysis. Int J Dermatol. 2006;45(1):56-59. doi:10.1111/j.1365-4632.2005.02701.x
21. Kucher C, Steere J, Elenitsas R, Siegel DL, Xu X. Nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis with diaphragmatic involvement in a patient with respiratory failure. J Am Acad Dermatol. 2006;54(suppl 2):S31-S34. doi:10.1016/j.jaad.2005.04.024
22. Sanyal S, Marckmann P, Scherer S, Abraham JL. Multiorgan gadolinium (Gd) deposition and fibrosis in a patient with nephrogenic systemic fibrosis—an autopsy-based review. Nephrol Dial Transplant. 2011;26(11):3616-3626. doi:10.1093/ndt/gfr085
23. Kucher C, Xu X, Pasha T, Elenitsas R. Histopathologic comparison of nephrogenic fibrosing dermopathy and scleromyxedema. J Cutan Pathol. 2005;32(7):484-490. doi:10.1111/j.0303-6987.2005.00365.x
24. Goldstein KM, Lunyera J, Mohottige D, et al. Risk of Nephrogenic Systemic Fibrosis after Exposure to Newer Gadolinium Agents. Washington (DC): Department of Veterans Affairs (US); October 2019. https://www.ncbi.nlm.nih.gov/books/NBK559376/25. Lunyera J, Mohottige D, Alexopoulos AS, et al. Risk for nephrogenic systemic fibrosis after exposure to newer gadolinium agents: a systematic review. Ann Intern Med. 2020;173(2):110-119. doi:10.7326/M20-0299
26. Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH Oxidase 4 (Nox4) for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
27. Wagner B. The pathophysiology and retention of gadolinium. United States Food & Drug Administration Medical Imaging Drugs Advisory Committee. 2017:1-23. https://www.fda.gov/advisory-committees/medical-imaging-drugs-advisory-committee/2017-meeting-materials-medical-imaging-drugs-advisory-committee?msclkid=6b5764ccbaa611ec95e35dddf8db57af
28. Runge VM. Critical questions regarding gadolinium deposition in the brain and body after injections of the gadolinium-based contrast agents, safety, and clinical recommendations in consideration of the EMA’s pharmacovigilance and risk assessment committee recommendation for suspension of the marketing authorizations for 4 linear agents. Invest Radiol. 2017;52(6):317-323. doi:10.1097/RLI.0000000000000374
29. Wagner B. Scared to the marrow: pitfalls and pearls in renal imaging. Adv Chronic Kidney Dis. 2017;24(3):136-137. doi:10.1053/j.ackd.2017.03.008
30. US Food and Drug Administration. Transcript for the September 8, 2017 Meeting of the Medical Imaging Drugs Advisory Committee (MIDAC). September 8, 2017. Accessed April 11, 2022. https://www.fda.gov/media/108935/download
31. Abel M, Talbot RB. Gadolinium oxide inhalation by guinea pigs: a correlative functional and histopathologic study. J Pharmacol Exp Ther. 1967;157(1):207-213.
32. Haley TJ, Raymond K, Komesu N, Upham HC. Toxicological and pharmacological effects of gadolinium and samarium chlorides. Br J Pharmacol Chemother. 1961;17(3):526-532. doi:10.1111/j.1476-5381.1961.tb01139.x
33. Spencer AJ, Wilson SA, Batchelor J, Reid A, Rees J, Harpur E. Gadolinium chloride toxicity in the rat. Toxicol Pathol. 1997;25(3):245-255. doi:10.1177/019262339702500301
34. Semelka RC, Ramalho M, AlObaidy M, Ramalho J. Gadolinium in humans: a family of disorders. AJR Am J Roentgenol. 2016;207(2):229-233. doi:10.2214/AJR.15.15842
35. Semelka RC, Ramalho M. Physicians with self-diagnosed gadolinium deposition disease: a case series. Radiol Bras. 2021;54(4):238-242. doi:10.1590/0100-3984.2020.0073
36. Layne KA, Wood DM, Dargan PI. Gadolinium-based contrast agents—what is the evidence for ‘gadolinium deposition disease’ and the use of chelation therapy? Clin Toxicol (Phila). 2020;58(3):151-160. doi:10.1080/15563650.2019.1681442
37. Nehra AK, McDonald RJ, Bluhm AM, et al. Accumulation of gadolinium in human cerebrospinal fluid after gadobutrol-enhanced MR imaging: a prospective observational cohort study. Radiology. 2018;288(2):416-423. doi:10.1148/radiol.2018171105
38. US Food and Drug Administration. Medical Imaging Drugs Advisory Committee Meeting. Gadolinium retention after gadolinium based contrast magnetic resonance imaging in patients with normal renal function. Briefing document. 2017. Accessed April 12, 2022. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/MedicalImagingDrugsAdvisoryCommittee/UCM572848.pdf
39. Calvo N, Jamil M, Feldman S, Shah A, Nauman F, Ferrara J. Neurotoxicity from intrathecal gadolinium administration: case presentation and brief review. Neurol Clin Pract. 2020;10(1):e7-e10. doi:10.1212/CPJ.0000000000000696
40. Bower DV, Richter JK, von Tengg-Kobligk H, Heverhagen JT, Runge VM. Gadolinium-based MRI contrast agents induce mitochondrial toxicity and cell death in human neurons, and toxicity increases with reduced kinetic stability of the agent. Invest Radiol. 2019;54(8):453-463. doi:10.1097/RLI.0000000000000567
41. McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
42. Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1(6):561-568. doi:10.34067/KID.0000272019
43. Di Gregorio E, Furlan C, Atlante S, Stefania R, Gianolio E, Aime S. Gadolinium retention in erythrocytes and leukocytes from human and murine blood upon treatment with gadolinium-based contrast agents for magnetic resonance imaging. Invest Radiol. 2020;55(1):30-37. doi:10.1097/RLI.0000000000000608
44. Maecker HT, Siebert JC, Rosenberg-Hasson Y, Koran LM, Ramalho M, Semelka RC. Acute chelation therapy-associated changes in urine gadolinium, self-reported flare severity, and serum cytokines in gadolinium deposition disease. Invest Radiol. 2021;56(6):374-384. doi:10.1097/RLI.0000000000000752
45. Maecker HT, Wang W, Rosenberg-Hasson Y, Semelka RC, Hickey J, Koran LM. An initial investigation of serum cytokine levels in patients with gadolinium retention. Radiol Bras. 2020;53(5):306-313. doi:10.1590/0100-3984.2019.0075
46. Birka M, Wentker KS, Lusmöller E, et al. Diagnosis of nephrogenic systemic fibrosis by means of elemental bioimaging and speciation analysis. Anal Chem. 2015;87(6):3321-3328. doi:10.1021/ac504488k
1. Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28(2):154-162. doi:10.1097/MNH.0000000000000475
2. Grobner T. Gadolinium—a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis?. Nephrol Dial Transplant. 2006;21(4):1104-1108. doi:10.1093/ndt/gfk062
3. Do C, Barnes JL, Tan C, Wagner B. Type of MRI contrast, tissue gadolinium, and fibrosis. Am J Physiol Renal Physiol. 2014;307(7):F844-F855. doi:10.1152/ajprenal.00379.2014
4. Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
5. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311(1):F1-F11. doi:10.1152/ajprenal.00166.2016
6. Drel VR, Tan C, Barnes JL, Gorin Y, Lee DY, Wagner B. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
7. Do C, Drel V, Tan C, Lee D, Wagner B. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143.e2. doi:10.1016/j.jid.2019.03.1145
8. Do C, Ford B, Lee DY, Tan C, Escobar P, Wagner B. Gadolinium-based contrast agents: stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
9. Hirano S, Suzuki KT. Exposure, metabolism, and toxicity of rare earths and related compounds. Environ Health Perspect. 1996;104(suppl 1):85-95. doi:10.1289/ehp.96104s185
10. Alwasiyah D, Murphy C, Jannetto P, Hogg M, Beuhler MC. Urinary gadolinium levels after contrast-enhanced MRI in individuals with normal renal function: a pilot study. J Med Toxicol. 2019;15(2):121-127. doi:10.1007/s13181-018-0693-1
11. Williams S, Grimm H. gadolinium toxicity: shedding light on the effects of retained gadolinium from contrast MRI. Accessed April 11, 2022. https://gdtoxicity.files.wordpress.com/2018/12/gadolinium-clearance-times-for-135-contrast-mri-cases-final-v1-1.pdf
12. DeBevits JJ, Reshma M, Bageac D, et al. Gray matter nucleus hyperintensity after monthly triple-dose gadopentetate dimeglumine with long-term magnetic resonance imaging. Invest Radiol. 2020;55(10):629-635. doi:10.1097/RLI.0000000000000663
13. Gathings RM, Reddy R, Santa Cruz D, Brodell RT. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151(3):316-319. doi:10.1001/jamadermatol.2014.2660
14. Girardi M, Kay J, Elston DM, Leboit PE, Abu-Alfa A, Cowper SE. Nephrogenic systemic fibrosis: clinicopathological definition and workup recommendations. J Am Acad Dermatol. 2011;65(6):1095-1106 e7. doi:10.1016/j.jaad.2010.08.041
15. Daram SR, Cortese CM, Bastani B. Nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis: report of a new case with literature review. Am J Kidney Dis. 2005;46(4):754-759. doi:10.1053/j.ajkd.2005.06.024
16. Ortonne N, Lipsker D, Chantrel F, Boehm N, Grosshans E, Cribier B. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br J Dermatol. 2004;150(5):1050-1052. doi:10.1111/j.1365-2133.2004.05900.x
17. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-49. doi:10.1016/j.semarthrit.2005.08.002
18. Lewis KG, Lester BW, Pan TD, Robinson-Bostom L. Nephrogenic fibrosing dermopathyand calciphylaxis with pseudoxanthoma elasticum-like changes. J Cutan Pathol. 2006;33(10):695-700. doi:10.1111/j.1600-0560.2006.00490.x
19. Gibson SE, Farver CF, Prayson RA. Multiorgan involvement in nephrogenic fibrosing dermopathy: an autopsy case and review of the literature. Arch Pathol Lab Med. 2006;130(2):209-212. doi:10.5858/2006-130-209-MIINFD
20. Cassis TB, Jackson JM, Sonnier GB, Callen JP. Nephrogenic fibrosing dermopathy in a patient with acute renal failure never requiring dialysis. Int J Dermatol. 2006;45(1):56-59. doi:10.1111/j.1365-4632.2005.02701.x
21. Kucher C, Steere J, Elenitsas R, Siegel DL, Xu X. Nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis with diaphragmatic involvement in a patient with respiratory failure. J Am Acad Dermatol. 2006;54(suppl 2):S31-S34. doi:10.1016/j.jaad.2005.04.024
22. Sanyal S, Marckmann P, Scherer S, Abraham JL. Multiorgan gadolinium (Gd) deposition and fibrosis in a patient with nephrogenic systemic fibrosis—an autopsy-based review. Nephrol Dial Transplant. 2011;26(11):3616-3626. doi:10.1093/ndt/gfr085
23. Kucher C, Xu X, Pasha T, Elenitsas R. Histopathologic comparison of nephrogenic fibrosing dermopathy and scleromyxedema. J Cutan Pathol. 2005;32(7):484-490. doi:10.1111/j.0303-6987.2005.00365.x
24. Goldstein KM, Lunyera J, Mohottige D, et al. Risk of Nephrogenic Systemic Fibrosis after Exposure to Newer Gadolinium Agents. Washington (DC): Department of Veterans Affairs (US); October 2019. https://www.ncbi.nlm.nih.gov/books/NBK559376/25. Lunyera J, Mohottige D, Alexopoulos AS, et al. Risk for nephrogenic systemic fibrosis after exposure to newer gadolinium agents: a systematic review. Ann Intern Med. 2020;173(2):110-119. doi:10.7326/M20-0299
26. Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH Oxidase 4 (Nox4) for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
27. Wagner B. The pathophysiology and retention of gadolinium. United States Food & Drug Administration Medical Imaging Drugs Advisory Committee. 2017:1-23. https://www.fda.gov/advisory-committees/medical-imaging-drugs-advisory-committee/2017-meeting-materials-medical-imaging-drugs-advisory-committee?msclkid=6b5764ccbaa611ec95e35dddf8db57af
28. Runge VM. Critical questions regarding gadolinium deposition in the brain and body after injections of the gadolinium-based contrast agents, safety, and clinical recommendations in consideration of the EMA’s pharmacovigilance and risk assessment committee recommendation for suspension of the marketing authorizations for 4 linear agents. Invest Radiol. 2017;52(6):317-323. doi:10.1097/RLI.0000000000000374
29. Wagner B. Scared to the marrow: pitfalls and pearls in renal imaging. Adv Chronic Kidney Dis. 2017;24(3):136-137. doi:10.1053/j.ackd.2017.03.008
30. US Food and Drug Administration. Transcript for the September 8, 2017 Meeting of the Medical Imaging Drugs Advisory Committee (MIDAC). September 8, 2017. Accessed April 11, 2022. https://www.fda.gov/media/108935/download
31. Abel M, Talbot RB. Gadolinium oxide inhalation by guinea pigs: a correlative functional and histopathologic study. J Pharmacol Exp Ther. 1967;157(1):207-213.
32. Haley TJ, Raymond K, Komesu N, Upham HC. Toxicological and pharmacological effects of gadolinium and samarium chlorides. Br J Pharmacol Chemother. 1961;17(3):526-532. doi:10.1111/j.1476-5381.1961.tb01139.x
33. Spencer AJ, Wilson SA, Batchelor J, Reid A, Rees J, Harpur E. Gadolinium chloride toxicity in the rat. Toxicol Pathol. 1997;25(3):245-255. doi:10.1177/019262339702500301
34. Semelka RC, Ramalho M, AlObaidy M, Ramalho J. Gadolinium in humans: a family of disorders. AJR Am J Roentgenol. 2016;207(2):229-233. doi:10.2214/AJR.15.15842
35. Semelka RC, Ramalho M. Physicians with self-diagnosed gadolinium deposition disease: a case series. Radiol Bras. 2021;54(4):238-242. doi:10.1590/0100-3984.2020.0073
36. Layne KA, Wood DM, Dargan PI. Gadolinium-based contrast agents—what is the evidence for ‘gadolinium deposition disease’ and the use of chelation therapy? Clin Toxicol (Phila). 2020;58(3):151-160. doi:10.1080/15563650.2019.1681442
37. Nehra AK, McDonald RJ, Bluhm AM, et al. Accumulation of gadolinium in human cerebrospinal fluid after gadobutrol-enhanced MR imaging: a prospective observational cohort study. Radiology. 2018;288(2):416-423. doi:10.1148/radiol.2018171105
38. US Food and Drug Administration. Medical Imaging Drugs Advisory Committee Meeting. Gadolinium retention after gadolinium based contrast magnetic resonance imaging in patients with normal renal function. Briefing document. 2017. Accessed April 12, 2022. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/MedicalImagingDrugsAdvisoryCommittee/UCM572848.pdf
39. Calvo N, Jamil M, Feldman S, Shah A, Nauman F, Ferrara J. Neurotoxicity from intrathecal gadolinium administration: case presentation and brief review. Neurol Clin Pract. 2020;10(1):e7-e10. doi:10.1212/CPJ.0000000000000696
40. Bower DV, Richter JK, von Tengg-Kobligk H, Heverhagen JT, Runge VM. Gadolinium-based MRI contrast agents induce mitochondrial toxicity and cell death in human neurons, and toxicity increases with reduced kinetic stability of the agent. Invest Radiol. 2019;54(8):453-463. doi:10.1097/RLI.0000000000000567
41. McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
42. Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1(6):561-568. doi:10.34067/KID.0000272019
43. Di Gregorio E, Furlan C, Atlante S, Stefania R, Gianolio E, Aime S. Gadolinium retention in erythrocytes and leukocytes from human and murine blood upon treatment with gadolinium-based contrast agents for magnetic resonance imaging. Invest Radiol. 2020;55(1):30-37. doi:10.1097/RLI.0000000000000608
44. Maecker HT, Siebert JC, Rosenberg-Hasson Y, Koran LM, Ramalho M, Semelka RC. Acute chelation therapy-associated changes in urine gadolinium, self-reported flare severity, and serum cytokines in gadolinium deposition disease. Invest Radiol. 2021;56(6):374-384. doi:10.1097/RLI.0000000000000752
45. Maecker HT, Wang W, Rosenberg-Hasson Y, Semelka RC, Hickey J, Koran LM. An initial investigation of serum cytokine levels in patients with gadolinium retention. Radiol Bras. 2020;53(5):306-313. doi:10.1590/0100-3984.2019.0075
46. Birka M, Wentker KS, Lusmöller E, et al. Diagnosis of nephrogenic systemic fibrosis by means of elemental bioimaging and speciation analysis. Anal Chem. 2015;87(6):3321-3328. doi:10.1021/ac504488k
Skull Base Regeneration During Treatment With Chemoradiation for Nasopharyngeal Carcinoma: A Case Report
Nasopharyngeal carcinoma (NPC) differs from other head and neck (H&N) cancers in its epidemiology and treatment. Unlike other H&N cancers, NPC has a distinct geographical distribution with a much higher incidence in endemic areas, such as southern China, than in areas where it is relatively uncommon, such as the United States.1 The etiology of NPC varies based on the geographical distribution, with Epstein-Barr virus (EBV) thought to be the primary etiologic agent in endemic areas. On the other hand, in North America 2 additional subsets of NPC have been identified: human papillomavirus (HPV)–positive/EBV-negative and HPV-negative/EBV-negative.2,3 NPC arises from the epithelial lining of the nasopharynx, often in the fossa of Rosenmuller, and is the most seen tumor in the nasopharynx.4 NPC is less surgically accessible than other H&N cancers, and surgery to the nasopharynx poses more risks given the proximity of critical surrounding structures. NPC is radiosensitive, and therefore radiotherapy (RT), in combination with chemotherapy for locally advanced tumors, has become the mainstay of treatment for nonmetastatic NPC.4
NPC often presents with an asymptomatic neck mass or with symptoms of epistaxis, nasal obstruction, and otitis media.5 Advanced cases of NPC can present with direct extension into the skull base, paranasal sinuses, and orbit, as well as involvement of cranial nerves. Radiation planning for tumors of the nasopharynx is complicated by the need to deliver an adequate dose to the tumor while limiting dose and toxicity to nearby critical structures such as the brainstem, optic chiasm, eyes, spinal cord (SC), temporal lobes, and cochleae. Achieving an adequate dose to nasopharyngeal primary tumors is especially complicated for T4 tumors invading the skull base with intracranial extension, in direct contact with these critical structures (Table 1).
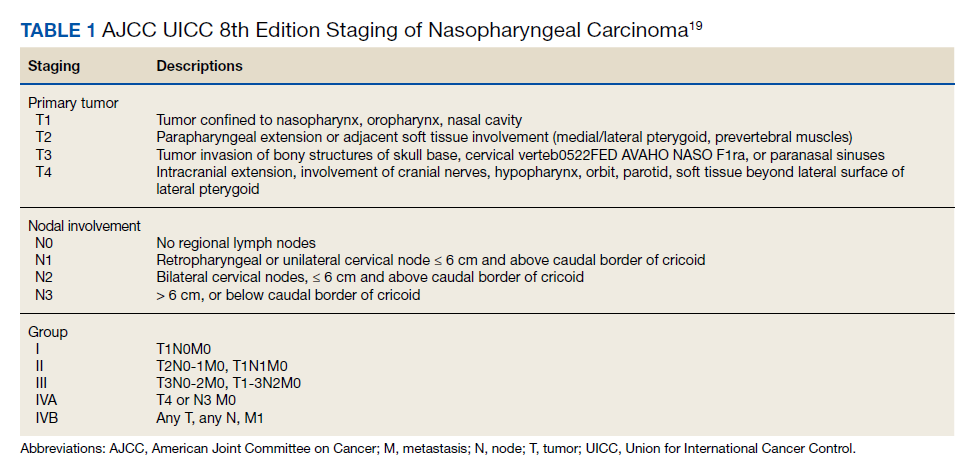
Skull base invasion is a poor prognostic factor, predicting for an increased risk of locoregional recurrence and worse overall survival. Furthermore, the extent of skull base invasion in NPC affects overall prognosis, with cranial nerve involvement and intracranial extension predictive for worse outcomes.5 Depending on the extent of destruction, a bony defect along the skull base could develop with tumor shrinkage during RT, resulting in complications such as cerebrospinal fluid leaks, herniation, and atlantoaxial instability.6
There is a paucity of literature on the ability of bone to regenerate during or after RT for cases of NPC with skull base destruction. To our knowledge, nothing has been published detailing the extent of bony regeneration that can occur during treatment itself, as the tumor regresses and poses a threat of a skull base defect. Here we present a case of T4 HPV-positive/EBV-negative NPC with intracranial extension and describe the RT planning methods leading to prolonged local control, limited toxicities, and bony regeneration of the skull base during treatment.
Case Presentation
A 34-year-old male patient with no previous medical history presented to the emergency department with worsening diplopia, nasal obstruction, facial pain, and neck stiffness. The patient reported a 3 pack-year smoking history with recent smoking cessation. His physical examination was notable for a right abducens nerve palsy and an ulcerated nasopharyngeal mass on endoscopy.
Computed tomography (CT) scan revealed a 7-cm mass in the nasopharynx, eroding through the skull base with destruction and replacement of the clivus by tumor. Also noted was erosion of the petrous apices, carotid canals, sella turcica, dens, and the bilateral occipital condyles. There was intracranial extension with replacement of portions of the cavernous sinuses as well as mass effect on the prepontine cistern. Additional brain imaging studies, including magnetic resonance imaging (MRI) and positron emission tomography (PET) scans, were obtained for completion of the staging workup. The MRI correlated with the findings noted on CT and demonstrated involvement of Meckel cave, foramen ovale, foramen rotundum, Dorello canal, and the hypoglossal canals. No cervical lymphadenopathy or distant metastases were noted on imaging. Pathology from biopsy revealed poorly differentiated squamous cell carcinoma, EBV-negative, strongly p16-positive, HPV-16 positive, and P53-negative.

The H&N multidisciplinary tumor board recommended concurrent chemoradiation for this stage IVA (T4N0M0) EBV-negative, HPV-positive, Word Health Organization type I NPC (Table 2). The patient underwent CT simulation for RT planning, and both tumor volumes and critical normal structures were contoured. The goal was to deliver 70 Gy to the gross tumor. However, given the inability to deliver this dose while meeting the SC dose tolerance of < 45 Gy, a 2-Gy fraction was removed. Therefore, 34 fractions of 2 Gy were delivered to the tumor volume for a total dose of 68 Gy. Weekly cisplatin, at a dose of 40 mg/m2, was administered concurrently with RT.
RT planning was complicated by the tumor’s contact with the brainstem and upper cervical SC, as well as proximity of the tumor to the optic apparatus. The patient underwent 2 replanning CT scans at 26 Gy and 44 Gy to evaluate for tumor shrinkage. These CT scans demonstrated shrinkage of the tumor away from critical neural structures, allowing the treatment volume to be reduced away from these structures in order to achieve required dose tolerances (brainstem < 54 Gy, optic nerves and chiasm < 50 Gy, SC < 45 Gy for this case). The replanning CT scan at 44 Gy, 5 weeks after treatment initiation, demonstrated that dramatic tumor shrinkage had occurred early in treatment, with separation of the remaining tumor from the area of the SC and brainstem with which it was initially in contact (Figure 1). This improvement allowed for shrinkage of the high-dose radiation field away from these critical neural structures.
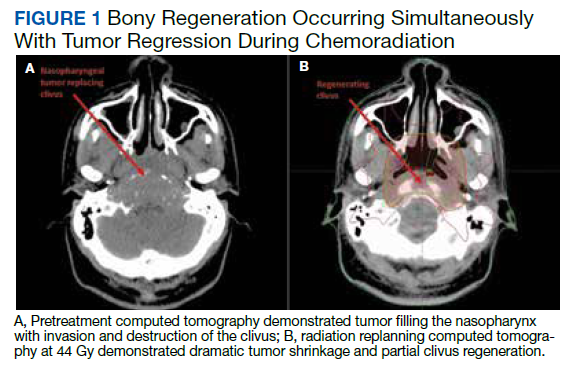
Baseline destruction of the skull base by tumor raised concern for craniospinal instability with tumor response. The patient was evaluated by neurosurgery before the start of RT, and the recommendation was for reimaging during treatment and close follow-up of the patient’s symptoms to determine whether surgical fixation would be indicated during or after treatment. The patient underwent a replanning CT scan at 44 Gy, 5 weeks after treatment initiation, that demonstrated impressive bony regeneration occurring during chemoradiation. New bone formation was noted in the region of the clivus and bilateral occipital condyles, which had been absent on CT prior to treatment initiation. Another CT at 54 Gy demonstrated further ossification of the clivus and bilateral occipital condyles, and bony regeneration occurring rapidly during chemoradiation. The posttreatment CT 3 months after completion of chemoradiation demonstrated complete skull base regeneration, maintaining stability of this area and precluding the need for neurosurgical intervention (Figure 2).
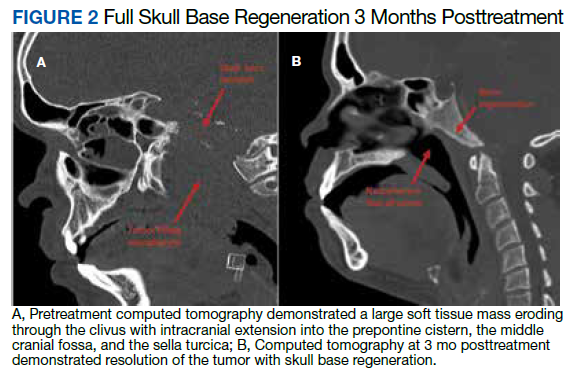
During RT,
The patient had no evidence of disease at 5 years posttreatment. After completing treatment, the patient experienced ongoing intermittent nasal congestion and occasional aural fullness. He experienced an early decay of several teeth starting 1 year after completion of RT, and he continues to visit his dentist for management. He experienced no other treatment-related toxicities. In particular, he has exhibited no signs of neurologic toxicity to date.
Discussion
RT for NPC is complicated by the proximity of these tumors to critical surrounding neural structures. It is challenging to achieve the required dose constraints to surrounding neural tissues while delivering the usual 70-Gy dose to the gross tumor, especially when the tumor comes into direct contact with these structures.
This case provides an example of response-adapted RT using imaging during treatment to shrink the high-dose target as the tumor shrinks away from critical surrounding structures.7 This strategy permits delivery of the maximum dose to the tumor while minimizing radiation dose, and therefore risk of toxicity, to normal surrounding structures. While it is typical to deliver 70 Gy to the full extent of tumor involvement for H&N tumors, this was not possible in this case as the tumor was in contact with the brainstem and upper cervical SC. Delivering the full 70 Gy to these areas of tumor would have placed this patient at substantial risk of brainstem and/or SC toxicity. This report demonstrates that response-adapted RT with shrinking fields can allow for tumor control while avoiding toxicity to critical neural structures for cases of locally advanced NPC in which tumor is abutting these structures.
Bony regeneration of the skull base following RT has been reported in the literature, but in limited reviews. Early reports used plain radiography to follow changes. Unger and colleagues demonstrated the regeneration of bone using skull radiographs 4 to 6 months after completion of RT for NPC.8 More recent literature details the ability of bone to regenerate after RT based on CT findings. Fang and colleagues reported on 90 cases of NPC with skull base destruction, with 63% having bony regeneration on posttreatment CT.9 Most of the patients in Fang’s report had bony regeneration within 1 year of treatment, and in general, bony regeneration became more evident on imaging with longer follow-up. Of note, local control was significantly greater in patients with regeneration vs persistent destruction (77% vs 21%, P < .001). On multivariate analysis, complete tumor response was significantly associated with bony regeneration; other factors such as age, sex, radiation dose, and chemotherapy were not significantly associated with the likelihood of bony regeneration.
Our report details a nasopharyngeal tumor that destroyed the skull base with no intact bony barrier. In such cases, concern arises regarding craniospinal instability with tumor regression if there is not simultaneous bone regeneration. Tumor invasion of the skull base and C1-2 vertebral bodies and complications from treatment of such tumor extent can lead to symptoms of craniospinal instability, including pain, difficulty with neck range of motion, and loss of strength and sensation in the upper and lower extremities.10 A case report of a woman treated with chemoradiation for a plasmacytoma of the skull base detailed her posttreatment presentation with quadriparesis resulting from craniospinal instability after tumor regression.11 Such instability is generally treated surgically, and during this woman’s surgery, there was an injury to the right vertebral artery, although this did not cause any additional neurologic deficits.
RT leads to hypocellularity, hypovascularity, and hypoxia of treated tissues, resulting in a reduced ability for growth and healing. Studies demonstrate that irradiated bone contains fewer osteoblast cells and osteocytes than unirradiated bone, resulting in reduced regenerative capacity.12,13 Furthermore, the reconstruction of bony defects resulting after cancer treatment has been shown to be difficult and associated with a high risk of complications.14 Given the impaired ability of irradiated bone to regenerate, studies have evaluated the use of growth factors and gene therapy to promote bone formation after treatment.15 Bone marrow stem cells have been shown to reverse radiation-induced cellular depletion and to increase osteocyte counts in animal studies.12 Further, overexpression of miR-34a, a tumor suppressor involved in tissue development, has been shown to improve osteoblastic differentiation of irradiated bone marrow stem cells and promote bone regeneration in vitro and in animal studies.13 While several techniques are being studied in vitro and in animal studies to promote bony regeneration after RT, there is a lack of data on use of these techniques in humans with cancer.
With our case, there was great uncertainty related to the ability of bone to regenerate during treatment and concern regarding consequences of formation of a skull base defect during treatment. CT imaging revealed bony regeneration of the central skull base and clivus, as well as occipital condyles, that occurred throughout the RT course. There was clear evidence of bone regeneration on the replanning CT obtained 5 weeks after treatment initiation. To our knowledge, this is the first report to demonstrate rapid bony regeneration during RT, thereby maintaining the integrity of the skull base and precluding the need for neurosurgical intervention. Moving forward, imaging should be considered during treatment for patients with tumor-related destruction of the skull base and upper cervical spine to evaluate the extent of bony regeneration during treatment and estimate the potential risk of craniocervical instability. Further studies with imaging during treatment are needed for more information on the likelihood of bony regeneration and factors that correlate with bony regeneration during treatment. As in other reports, our case demonstrates that bony regeneration may predict complete response to RT.9
Our patient’s tumor was HPV-positive and EBV-negative. In the US, the rate of HPV-positive NPC is 35%.16 However, HPV-positive NPC is much less common in endemic areas. A recent study from China of 1,328 patients with NPC revealed a 6.4% rate of HPV-positive/EBV-negative cases.17 In that study, patients with HPV-positive/EBV-negative tumors had improved survival compared to patients whose tumors were HPV-negative/EBV-positive. Another study suggests that the impact of HPV in NPC varies according to race, with HPV-positivity predicting for improved outcomes in East Asian patients and worse outcomes in White patients.17 A study from the University of Michigan suggests that both HPV-positive/EBV-negative and HPV-negative/EBV-negative NPC are associated with worse overall survival and locoregional control than EBV-positive NPC.2 Overall, the prognostic role of HPV in NPC remains unclear given conflicting information in the literature and the lack of large population studies.18
Conclusions
There is a paucity of literature on bony regeneration in patients with skull base destruction from advanced NPC, and in particular, the ability of skull base regeneration to occur during treatment simultaneous with tumor regression. Our patient had HPV-positive/EBV-negative NPC, but it is unclear how this subtype affected his prognosis. Factors such as tumor histology, radiosensitivity with rapid tumor regression, and young age may have all contributed to the rapidity of bone regeneration in our patient. This case report demonstrates that an impressive tumor response to chemoradiation with simultaneous bony regeneration is possible among patients presenting with tumor destruction of the skull base, precluding the need for neurosurgical intervention.
1. Chang ET, Adami HO. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15(10):1765-1777. doi:10.1158/1055-9965.EPI-06-0353
2. Stenmark MH, McHugh JB, Schipper M, et al. Nonendemic HPV-positive nasopharyngeal carcinoma: association with poor prognosis. Int J Radiat Oncol Biol Phys. 2014;88(3):580-588. doi:10.1016/j.ijrobp.2013.11.246
3. Maxwell JH, Kumar B, Feng FY, et al. HPV-positive/p16-positive/EBV-negative nasopharyngeal carcinoma in white North Americans. Head Neck. 2010;32(5):562-567. doi:10.1002/hed.21216
4. Chen YP, Chan ATC, Le QT, Blanchard P, Sun Y, Ma J. Nasopharyngeal carcinoma. Lancet. 2019;394(10192):64-80. doi:10.1016/S0140-6736(19)30956-0
5. Roh JL, Sung MW, Kim KH, et al.. Nasopharyngeal carcinoma with skull base invasion: a necessity of staging subdivision. Am J Otolaryngol. 2004;25(1):26-32. doi:10.1016/j.amjoto.2003.09.011
6. Orr RD, Salo PT. Atlantoaxial instability complicating radiation therapy for recurrent nasopharyngeal carcinoma. A case report. Spine. 1998;23(11):1280-1282. doi:10.1097/00007632-199806010-00021
7. Morgan HE, Sher DJ. Adaptive radiotherapy for head and neck cancer. Cancers Head Neck. 2020;5:1. doi:10.1186/s41199-019-0046-z
8. Unger JD, Chiang LC, Unger GF. Apparent reformation of the base of the skull following radiotherapy for nasopharyngeal carcinoma. Radiology. 1978;126(3):779-782. doi:10.1148/126.3.779
9. Fang FM, Leung SW, Wang CJ, et al. Computed tomography findings of bony regeneration after radiotherapy for nasopharyngeal carcinoma with skull base destruction: implications for local control. Int J Radiat Oncol Biol Phys. 1999;44(2):305-309. doi:10.1016/s0360-3016(99)00004-8
10. Tiruchelvarayan R, Lee KA, Ng I. Surgery for atlanto-axial (C1-2) involvement or instability in nasopharyngeal carcinoma patients. Singapore Med J. 2012;53(6):416-421.
11. Samprón N, Arrazola M, Urculo E. Skull-base plasmacytoma with craniocervical instability [in Spanish]. Neurocirugia (Astur). 2009;20(5):478-483.
12. Zheutlin AR, Deshpande SS, Nelson NS, et al. Bone marrow stem cells assuage radiation-induced damage in a murine model of distraction osteogenesis: a histomorphometric evaluation. Cytotherapy. 2016;18(5):664-672. doi:10.1016/j.jcyt.2016.01.013
13. Liu H, Dong Y, Feng X, et al. miR-34a promotes bone regeneration in irradiated bone defects by enhancing osteoblast differentiation of mesenchymal stromal cells in rats. Stem Cell Res Ther. 2019;10(1):180. doi:10.1186/s13287-019-1285-y
14. Holzapfel BM, Wagner F, Martine LC, et al. Tissue engineering and regenerative medicine in musculoskeletal oncology. Cancer Metastasis Rev. 2016;35(3):475-487. doi:10.1007/s10555-016-9635-z
15. Hu WW, Ward BB, Wang Z, Krebsbach PH. Bone regeneration in defects compromised by radiotherapy. J Dent Res. 2010;89(1):77-81. doi:10.1177/0022034509352151
16. Wotman M, Oh EJ, Ahn S, Kraus D, Constantino P, Tham T. HPV status in patients with nasopharyngeal carcinoma in the United States: a SEER database study. Am J Otolaryngol. 2019;40(5):705-710. doi:10.1016/j.amjoto.2019.06.00717. Huang WB, Chan JYW, Liu DL. Human papillomavirus and World Health Organization type III nasopharyngeal carcinoma: multicenter study from an endemic area in Southern China. Cancer. 2018;124(3):530-536. doi:10.1002/cncr.31031.
18. Verma V, Simone CB 2nd, Lin C. Human papillomavirus and nasopharyngeal cancer. Head Neck. 2018;40(4):696-706. doi:10.1002/hed.24978
19. Lee AWM, Lydiatt WM, Colevas AD, et al. Nasopharynx. In: Amin MB, ed. AJCC Cancer Staging Manual. 8th ed. Springer; 2017:103.
20. Barnes L, Eveson JW, Reichart P, Sidransky D, eds. Pathology and genetics of head and neck tumors. In: World Health Organization Classification of Tumours. IARC Press; 2005.
Nasopharyngeal carcinoma (NPC) differs from other head and neck (H&N) cancers in its epidemiology and treatment. Unlike other H&N cancers, NPC has a distinct geographical distribution with a much higher incidence in endemic areas, such as southern China, than in areas where it is relatively uncommon, such as the United States.1 The etiology of NPC varies based on the geographical distribution, with Epstein-Barr virus (EBV) thought to be the primary etiologic agent in endemic areas. On the other hand, in North America 2 additional subsets of NPC have been identified: human papillomavirus (HPV)–positive/EBV-negative and HPV-negative/EBV-negative.2,3 NPC arises from the epithelial lining of the nasopharynx, often in the fossa of Rosenmuller, and is the most seen tumor in the nasopharynx.4 NPC is less surgically accessible than other H&N cancers, and surgery to the nasopharynx poses more risks given the proximity of critical surrounding structures. NPC is radiosensitive, and therefore radiotherapy (RT), in combination with chemotherapy for locally advanced tumors, has become the mainstay of treatment for nonmetastatic NPC.4
NPC often presents with an asymptomatic neck mass or with symptoms of epistaxis, nasal obstruction, and otitis media.5 Advanced cases of NPC can present with direct extension into the skull base, paranasal sinuses, and orbit, as well as involvement of cranial nerves. Radiation planning for tumors of the nasopharynx is complicated by the need to deliver an adequate dose to the tumor while limiting dose and toxicity to nearby critical structures such as the brainstem, optic chiasm, eyes, spinal cord (SC), temporal lobes, and cochleae. Achieving an adequate dose to nasopharyngeal primary tumors is especially complicated for T4 tumors invading the skull base with intracranial extension, in direct contact with these critical structures (Table 1).
Skull base invasion is a poor prognostic factor, predicting for an increased risk of locoregional recurrence and worse overall survival. Furthermore, the extent of skull base invasion in NPC affects overall prognosis, with cranial nerve involvement and intracranial extension predictive for worse outcomes.5 Depending on the extent of destruction, a bony defect along the skull base could develop with tumor shrinkage during RT, resulting in complications such as cerebrospinal fluid leaks, herniation, and atlantoaxial instability.6
There is a paucity of literature on the ability of bone to regenerate during or after RT for cases of NPC with skull base destruction. To our knowledge, nothing has been published detailing the extent of bony regeneration that can occur during treatment itself, as the tumor regresses and poses a threat of a skull base defect. Here we present a case of T4 HPV-positive/EBV-negative NPC with intracranial extension and describe the RT planning methods leading to prolonged local control, limited toxicities, and bony regeneration of the skull base during treatment.
Case Presentation
A 34-year-old male patient with no previous medical history presented to the emergency department with worsening diplopia, nasal obstruction, facial pain, and neck stiffness. The patient reported a 3 pack-year smoking history with recent smoking cessation. His physical examination was notable for a right abducens nerve palsy and an ulcerated nasopharyngeal mass on endoscopy.
Computed tomography (CT) scan revealed a 7-cm mass in the nasopharynx, eroding through the skull base with destruction and replacement of the clivus by tumor. Also noted was erosion of the petrous apices, carotid canals, sella turcica, dens, and the bilateral occipital condyles. There was intracranial extension with replacement of portions of the cavernous sinuses as well as mass effect on the prepontine cistern. Additional brain imaging studies, including magnetic resonance imaging (MRI) and positron emission tomography (PET) scans, were obtained for completion of the staging workup. The MRI correlated with the findings noted on CT and demonstrated involvement of Meckel cave, foramen ovale, foramen rotundum, Dorello canal, and the hypoglossal canals. No cervical lymphadenopathy or distant metastases were noted on imaging. Pathology from biopsy revealed poorly differentiated squamous cell carcinoma, EBV-negative, strongly p16-positive, HPV-16 positive, and P53-negative.
The H&N multidisciplinary tumor board recommended concurrent chemoradiation for this stage IVA (T4N0M0) EBV-negative, HPV-positive, Word Health Organization type I NPC (Table 2). The patient underwent CT simulation for RT planning, and both tumor volumes and critical normal structures were contoured. The goal was to deliver 70 Gy to the gross tumor. However, given the inability to deliver this dose while meeting the SC dose tolerance of < 45 Gy, a 2-Gy fraction was removed. Therefore, 34 fractions of 2 Gy were delivered to the tumor volume for a total dose of 68 Gy. Weekly cisplatin, at a dose of 40 mg/m2, was administered concurrently with RT.
RT planning was complicated by the tumor’s contact with the brainstem and upper cervical SC, as well as proximity of the tumor to the optic apparatus. The patient underwent 2 replanning CT scans at 26 Gy and 44 Gy to evaluate for tumor shrinkage. These CT scans demonstrated shrinkage of the tumor away from critical neural structures, allowing the treatment volume to be reduced away from these structures in order to achieve required dose tolerances (brainstem < 54 Gy, optic nerves and chiasm < 50 Gy, SC < 45 Gy for this case). The replanning CT scan at 44 Gy, 5 weeks after treatment initiation, demonstrated that dramatic tumor shrinkage had occurred early in treatment, with separation of the remaining tumor from the area of the SC and brainstem with which it was initially in contact (Figure 1). This improvement allowed for shrinkage of the high-dose radiation field away from these critical neural structures.
Baseline destruction of the skull base by tumor raised concern for craniospinal instability with tumor response. The patient was evaluated by neurosurgery before the start of RT, and the recommendation was for reimaging during treatment and close follow-up of the patient’s symptoms to determine whether surgical fixation would be indicated during or after treatment. The patient underwent a replanning CT scan at 44 Gy, 5 weeks after treatment initiation, that demonstrated impressive bony regeneration occurring during chemoradiation. New bone formation was noted in the region of the clivus and bilateral occipital condyles, which had been absent on CT prior to treatment initiation. Another CT at 54 Gy demonstrated further ossification of the clivus and bilateral occipital condyles, and bony regeneration occurring rapidly during chemoradiation. The posttreatment CT 3 months after completion of chemoradiation demonstrated complete skull base regeneration, maintaining stability of this area and precluding the need for neurosurgical intervention (Figure 2).
During RT,
The patient had no evidence of disease at 5 years posttreatment. After completing treatment, the patient experienced ongoing intermittent nasal congestion and occasional aural fullness. He experienced an early decay of several teeth starting 1 year after completion of RT, and he continues to visit his dentist for management. He experienced no other treatment-related toxicities. In particular, he has exhibited no signs of neurologic toxicity to date.
Discussion
RT for NPC is complicated by the proximity of these tumors to critical surrounding neural structures. It is challenging to achieve the required dose constraints to surrounding neural tissues while delivering the usual 70-Gy dose to the gross tumor, especially when the tumor comes into direct contact with these structures.
This case provides an example of response-adapted RT using imaging during treatment to shrink the high-dose target as the tumor shrinks away from critical surrounding structures.7 This strategy permits delivery of the maximum dose to the tumor while minimizing radiation dose, and therefore risk of toxicity, to normal surrounding structures. While it is typical to deliver 70 Gy to the full extent of tumor involvement for H&N tumors, this was not possible in this case as the tumor was in contact with the brainstem and upper cervical SC. Delivering the full 70 Gy to these areas of tumor would have placed this patient at substantial risk of brainstem and/or SC toxicity. This report demonstrates that response-adapted RT with shrinking fields can allow for tumor control while avoiding toxicity to critical neural structures for cases of locally advanced NPC in which tumor is abutting these structures.
Bony regeneration of the skull base following RT has been reported in the literature, but in limited reviews. Early reports used plain radiography to follow changes. Unger and colleagues demonstrated the regeneration of bone using skull radiographs 4 to 6 months after completion of RT for NPC.8 More recent literature details the ability of bone to regenerate after RT based on CT findings. Fang and colleagues reported on 90 cases of NPC with skull base destruction, with 63% having bony regeneration on posttreatment CT.9 Most of the patients in Fang’s report had bony regeneration within 1 year of treatment, and in general, bony regeneration became more evident on imaging with longer follow-up. Of note, local control was significantly greater in patients with regeneration vs persistent destruction (77% vs 21%, P < .001). On multivariate analysis, complete tumor response was significantly associated with bony regeneration; other factors such as age, sex, radiation dose, and chemotherapy were not significantly associated with the likelihood of bony regeneration.
Our report details a nasopharyngeal tumor that destroyed the skull base with no intact bony barrier. In such cases, concern arises regarding craniospinal instability with tumor regression if there is not simultaneous bone regeneration. Tumor invasion of the skull base and C1-2 vertebral bodies and complications from treatment of such tumor extent can lead to symptoms of craniospinal instability, including pain, difficulty with neck range of motion, and loss of strength and sensation in the upper and lower extremities.10 A case report of a woman treated with chemoradiation for a plasmacytoma of the skull base detailed her posttreatment presentation with quadriparesis resulting from craniospinal instability after tumor regression.11 Such instability is generally treated surgically, and during this woman’s surgery, there was an injury to the right vertebral artery, although this did not cause any additional neurologic deficits.
RT leads to hypocellularity, hypovascularity, and hypoxia of treated tissues, resulting in a reduced ability for growth and healing. Studies demonstrate that irradiated bone contains fewer osteoblast cells and osteocytes than unirradiated bone, resulting in reduced regenerative capacity.12,13 Furthermore, the reconstruction of bony defects resulting after cancer treatment has been shown to be difficult and associated with a high risk of complications.14 Given the impaired ability of irradiated bone to regenerate, studies have evaluated the use of growth factors and gene therapy to promote bone formation after treatment.15 Bone marrow stem cells have been shown to reverse radiation-induced cellular depletion and to increase osteocyte counts in animal studies.12 Further, overexpression of miR-34a, a tumor suppressor involved in tissue development, has been shown to improve osteoblastic differentiation of irradiated bone marrow stem cells and promote bone regeneration in vitro and in animal studies.13 While several techniques are being studied in vitro and in animal studies to promote bony regeneration after RT, there is a lack of data on use of these techniques in humans with cancer.
With our case, there was great uncertainty related to the ability of bone to regenerate during treatment and concern regarding consequences of formation of a skull base defect during treatment. CT imaging revealed bony regeneration of the central skull base and clivus, as well as occipital condyles, that occurred throughout the RT course. There was clear evidence of bone regeneration on the replanning CT obtained 5 weeks after treatment initiation. To our knowledge, this is the first report to demonstrate rapid bony regeneration during RT, thereby maintaining the integrity of the skull base and precluding the need for neurosurgical intervention. Moving forward, imaging should be considered during treatment for patients with tumor-related destruction of the skull base and upper cervical spine to evaluate the extent of bony regeneration during treatment and estimate the potential risk of craniocervical instability. Further studies with imaging during treatment are needed for more information on the likelihood of bony regeneration and factors that correlate with bony regeneration during treatment. As in other reports, our case demonstrates that bony regeneration may predict complete response to RT.9
Our patient’s tumor was HPV-positive and EBV-negative. In the US, the rate of HPV-positive NPC is 35%.16 However, HPV-positive NPC is much less common in endemic areas. A recent study from China of 1,328 patients with NPC revealed a 6.4% rate of HPV-positive/EBV-negative cases.17 In that study, patients with HPV-positive/EBV-negative tumors had improved survival compared to patients whose tumors were HPV-negative/EBV-positive. Another study suggests that the impact of HPV in NPC varies according to race, with HPV-positivity predicting for improved outcomes in East Asian patients and worse outcomes in White patients.17 A study from the University of Michigan suggests that both HPV-positive/EBV-negative and HPV-negative/EBV-negative NPC are associated with worse overall survival and locoregional control than EBV-positive NPC.2 Overall, the prognostic role of HPV in NPC remains unclear given conflicting information in the literature and the lack of large population studies.18
Conclusions
There is a paucity of literature on bony regeneration in patients with skull base destruction from advanced NPC, and in particular, the ability of skull base regeneration to occur during treatment simultaneous with tumor regression. Our patient had HPV-positive/EBV-negative NPC, but it is unclear how this subtype affected his prognosis. Factors such as tumor histology, radiosensitivity with rapid tumor regression, and young age may have all contributed to the rapidity of bone regeneration in our patient. This case report demonstrates that an impressive tumor response to chemoradiation with simultaneous bony regeneration is possible among patients presenting with tumor destruction of the skull base, precluding the need for neurosurgical intervention.
Nasopharyngeal carcinoma (NPC) differs from other head and neck (H&N) cancers in its epidemiology and treatment. Unlike other H&N cancers, NPC has a distinct geographical distribution with a much higher incidence in endemic areas, such as southern China, than in areas where it is relatively uncommon, such as the United States.1 The etiology of NPC varies based on the geographical distribution, with Epstein-Barr virus (EBV) thought to be the primary etiologic agent in endemic areas. On the other hand, in North America 2 additional subsets of NPC have been identified: human papillomavirus (HPV)–positive/EBV-negative and HPV-negative/EBV-negative.2,3 NPC arises from the epithelial lining of the nasopharynx, often in the fossa of Rosenmuller, and is the most seen tumor in the nasopharynx.4 NPC is less surgically accessible than other H&N cancers, and surgery to the nasopharynx poses more risks given the proximity of critical surrounding structures. NPC is radiosensitive, and therefore radiotherapy (RT), in combination with chemotherapy for locally advanced tumors, has become the mainstay of treatment for nonmetastatic NPC.4
NPC often presents with an asymptomatic neck mass or with symptoms of epistaxis, nasal obstruction, and otitis media.5 Advanced cases of NPC can present with direct extension into the skull base, paranasal sinuses, and orbit, as well as involvement of cranial nerves. Radiation planning for tumors of the nasopharynx is complicated by the need to deliver an adequate dose to the tumor while limiting dose and toxicity to nearby critical structures such as the brainstem, optic chiasm, eyes, spinal cord (SC), temporal lobes, and cochleae. Achieving an adequate dose to nasopharyngeal primary tumors is especially complicated for T4 tumors invading the skull base with intracranial extension, in direct contact with these critical structures (Table 1).
Skull base invasion is a poor prognostic factor, predicting for an increased risk of locoregional recurrence and worse overall survival. Furthermore, the extent of skull base invasion in NPC affects overall prognosis, with cranial nerve involvement and intracranial extension predictive for worse outcomes.5 Depending on the extent of destruction, a bony defect along the skull base could develop with tumor shrinkage during RT, resulting in complications such as cerebrospinal fluid leaks, herniation, and atlantoaxial instability.6
There is a paucity of literature on the ability of bone to regenerate during or after RT for cases of NPC with skull base destruction. To our knowledge, nothing has been published detailing the extent of bony regeneration that can occur during treatment itself, as the tumor regresses and poses a threat of a skull base defect. Here we present a case of T4 HPV-positive/EBV-negative NPC with intracranial extension and describe the RT planning methods leading to prolonged local control, limited toxicities, and bony regeneration of the skull base during treatment.
Case Presentation
A 34-year-old male patient with no previous medical history presented to the emergency department with worsening diplopia, nasal obstruction, facial pain, and neck stiffness. The patient reported a 3 pack-year smoking history with recent smoking cessation. His physical examination was notable for a right abducens nerve palsy and an ulcerated nasopharyngeal mass on endoscopy.
Computed tomography (CT) scan revealed a 7-cm mass in the nasopharynx, eroding through the skull base with destruction and replacement of the clivus by tumor. Also noted was erosion of the petrous apices, carotid canals, sella turcica, dens, and the bilateral occipital condyles. There was intracranial extension with replacement of portions of the cavernous sinuses as well as mass effect on the prepontine cistern. Additional brain imaging studies, including magnetic resonance imaging (MRI) and positron emission tomography (PET) scans, were obtained for completion of the staging workup. The MRI correlated with the findings noted on CT and demonstrated involvement of Meckel cave, foramen ovale, foramen rotundum, Dorello canal, and the hypoglossal canals. No cervical lymphadenopathy or distant metastases were noted on imaging. Pathology from biopsy revealed poorly differentiated squamous cell carcinoma, EBV-negative, strongly p16-positive, HPV-16 positive, and P53-negative.
The H&N multidisciplinary tumor board recommended concurrent chemoradiation for this stage IVA (T4N0M0) EBV-negative, HPV-positive, Word Health Organization type I NPC (Table 2). The patient underwent CT simulation for RT planning, and both tumor volumes and critical normal structures were contoured. The goal was to deliver 70 Gy to the gross tumor. However, given the inability to deliver this dose while meeting the SC dose tolerance of < 45 Gy, a 2-Gy fraction was removed. Therefore, 34 fractions of 2 Gy were delivered to the tumor volume for a total dose of 68 Gy. Weekly cisplatin, at a dose of 40 mg/m2, was administered concurrently with RT.
RT planning was complicated by the tumor’s contact with the brainstem and upper cervical SC, as well as proximity of the tumor to the optic apparatus. The patient underwent 2 replanning CT scans at 26 Gy and 44 Gy to evaluate for tumor shrinkage. These CT scans demonstrated shrinkage of the tumor away from critical neural structures, allowing the treatment volume to be reduced away from these structures in order to achieve required dose tolerances (brainstem < 54 Gy, optic nerves and chiasm < 50 Gy, SC < 45 Gy for this case). The replanning CT scan at 44 Gy, 5 weeks after treatment initiation, demonstrated that dramatic tumor shrinkage had occurred early in treatment, with separation of the remaining tumor from the area of the SC and brainstem with which it was initially in contact (Figure 1). This improvement allowed for shrinkage of the high-dose radiation field away from these critical neural structures.
Baseline destruction of the skull base by tumor raised concern for craniospinal instability with tumor response. The patient was evaluated by neurosurgery before the start of RT, and the recommendation was for reimaging during treatment and close follow-up of the patient’s symptoms to determine whether surgical fixation would be indicated during or after treatment. The patient underwent a replanning CT scan at 44 Gy, 5 weeks after treatment initiation, that demonstrated impressive bony regeneration occurring during chemoradiation. New bone formation was noted in the region of the clivus and bilateral occipital condyles, which had been absent on CT prior to treatment initiation. Another CT at 54 Gy demonstrated further ossification of the clivus and bilateral occipital condyles, and bony regeneration occurring rapidly during chemoradiation. The posttreatment CT 3 months after completion of chemoradiation demonstrated complete skull base regeneration, maintaining stability of this area and precluding the need for neurosurgical intervention (Figure 2).
During RT,
The patient had no evidence of disease at 5 years posttreatment. After completing treatment, the patient experienced ongoing intermittent nasal congestion and occasional aural fullness. He experienced an early decay of several teeth starting 1 year after completion of RT, and he continues to visit his dentist for management. He experienced no other treatment-related toxicities. In particular, he has exhibited no signs of neurologic toxicity to date.
Discussion
RT for NPC is complicated by the proximity of these tumors to critical surrounding neural structures. It is challenging to achieve the required dose constraints to surrounding neural tissues while delivering the usual 70-Gy dose to the gross tumor, especially when the tumor comes into direct contact with these structures.
This case provides an example of response-adapted RT using imaging during treatment to shrink the high-dose target as the tumor shrinks away from critical surrounding structures.7 This strategy permits delivery of the maximum dose to the tumor while minimizing radiation dose, and therefore risk of toxicity, to normal surrounding structures. While it is typical to deliver 70 Gy to the full extent of tumor involvement for H&N tumors, this was not possible in this case as the tumor was in contact with the brainstem and upper cervical SC. Delivering the full 70 Gy to these areas of tumor would have placed this patient at substantial risk of brainstem and/or SC toxicity. This report demonstrates that response-adapted RT with shrinking fields can allow for tumor control while avoiding toxicity to critical neural structures for cases of locally advanced NPC in which tumor is abutting these structures.
Bony regeneration of the skull base following RT has been reported in the literature, but in limited reviews. Early reports used plain radiography to follow changes. Unger and colleagues demonstrated the regeneration of bone using skull radiographs 4 to 6 months after completion of RT for NPC.8 More recent literature details the ability of bone to regenerate after RT based on CT findings. Fang and colleagues reported on 90 cases of NPC with skull base destruction, with 63% having bony regeneration on posttreatment CT.9 Most of the patients in Fang’s report had bony regeneration within 1 year of treatment, and in general, bony regeneration became more evident on imaging with longer follow-up. Of note, local control was significantly greater in patients with regeneration vs persistent destruction (77% vs 21%, P < .001). On multivariate analysis, complete tumor response was significantly associated with bony regeneration; other factors such as age, sex, radiation dose, and chemotherapy were not significantly associated with the likelihood of bony regeneration.
Our report details a nasopharyngeal tumor that destroyed the skull base with no intact bony barrier. In such cases, concern arises regarding craniospinal instability with tumor regression if there is not simultaneous bone regeneration. Tumor invasion of the skull base and C1-2 vertebral bodies and complications from treatment of such tumor extent can lead to symptoms of craniospinal instability, including pain, difficulty with neck range of motion, and loss of strength and sensation in the upper and lower extremities.10 A case report of a woman treated with chemoradiation for a plasmacytoma of the skull base detailed her posttreatment presentation with quadriparesis resulting from craniospinal instability after tumor regression.11 Such instability is generally treated surgically, and during this woman’s surgery, there was an injury to the right vertebral artery, although this did not cause any additional neurologic deficits.
RT leads to hypocellularity, hypovascularity, and hypoxia of treated tissues, resulting in a reduced ability for growth and healing. Studies demonstrate that irradiated bone contains fewer osteoblast cells and osteocytes than unirradiated bone, resulting in reduced regenerative capacity.12,13 Furthermore, the reconstruction of bony defects resulting after cancer treatment has been shown to be difficult and associated with a high risk of complications.14 Given the impaired ability of irradiated bone to regenerate, studies have evaluated the use of growth factors and gene therapy to promote bone formation after treatment.15 Bone marrow stem cells have been shown to reverse radiation-induced cellular depletion and to increase osteocyte counts in animal studies.12 Further, overexpression of miR-34a, a tumor suppressor involved in tissue development, has been shown to improve osteoblastic differentiation of irradiated bone marrow stem cells and promote bone regeneration in vitro and in animal studies.13 While several techniques are being studied in vitro and in animal studies to promote bony regeneration after RT, there is a lack of data on use of these techniques in humans with cancer.
With our case, there was great uncertainty related to the ability of bone to regenerate during treatment and concern regarding consequences of formation of a skull base defect during treatment. CT imaging revealed bony regeneration of the central skull base and clivus, as well as occipital condyles, that occurred throughout the RT course. There was clear evidence of bone regeneration on the replanning CT obtained 5 weeks after treatment initiation. To our knowledge, this is the first report to demonstrate rapid bony regeneration during RT, thereby maintaining the integrity of the skull base and precluding the need for neurosurgical intervention. Moving forward, imaging should be considered during treatment for patients with tumor-related destruction of the skull base and upper cervical spine to evaluate the extent of bony regeneration during treatment and estimate the potential risk of craniocervical instability. Further studies with imaging during treatment are needed for more information on the likelihood of bony regeneration and factors that correlate with bony regeneration during treatment. As in other reports, our case demonstrates that bony regeneration may predict complete response to RT.9
Our patient’s tumor was HPV-positive and EBV-negative. In the US, the rate of HPV-positive NPC is 35%.16 However, HPV-positive NPC is much less common in endemic areas. A recent study from China of 1,328 patients with NPC revealed a 6.4% rate of HPV-positive/EBV-negative cases.17 In that study, patients with HPV-positive/EBV-negative tumors had improved survival compared to patients whose tumors were HPV-negative/EBV-positive. Another study suggests that the impact of HPV in NPC varies according to race, with HPV-positivity predicting for improved outcomes in East Asian patients and worse outcomes in White patients.17 A study from the University of Michigan suggests that both HPV-positive/EBV-negative and HPV-negative/EBV-negative NPC are associated with worse overall survival and locoregional control than EBV-positive NPC.2 Overall, the prognostic role of HPV in NPC remains unclear given conflicting information in the literature and the lack of large population studies.18
Conclusions
There is a paucity of literature on bony regeneration in patients with skull base destruction from advanced NPC, and in particular, the ability of skull base regeneration to occur during treatment simultaneous with tumor regression. Our patient had HPV-positive/EBV-negative NPC, but it is unclear how this subtype affected his prognosis. Factors such as tumor histology, radiosensitivity with rapid tumor regression, and young age may have all contributed to the rapidity of bone regeneration in our patient. This case report demonstrates that an impressive tumor response to chemoradiation with simultaneous bony regeneration is possible among patients presenting with tumor destruction of the skull base, precluding the need for neurosurgical intervention.
1. Chang ET, Adami HO. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15(10):1765-1777. doi:10.1158/1055-9965.EPI-06-0353
2. Stenmark MH, McHugh JB, Schipper M, et al. Nonendemic HPV-positive nasopharyngeal carcinoma: association with poor prognosis. Int J Radiat Oncol Biol Phys. 2014;88(3):580-588. doi:10.1016/j.ijrobp.2013.11.246
3. Maxwell JH, Kumar B, Feng FY, et al. HPV-positive/p16-positive/EBV-negative nasopharyngeal carcinoma in white North Americans. Head Neck. 2010;32(5):562-567. doi:10.1002/hed.21216
4. Chen YP, Chan ATC, Le QT, Blanchard P, Sun Y, Ma J. Nasopharyngeal carcinoma. Lancet. 2019;394(10192):64-80. doi:10.1016/S0140-6736(19)30956-0
5. Roh JL, Sung MW, Kim KH, et al.. Nasopharyngeal carcinoma with skull base invasion: a necessity of staging subdivision. Am J Otolaryngol. 2004;25(1):26-32. doi:10.1016/j.amjoto.2003.09.011
6. Orr RD, Salo PT. Atlantoaxial instability complicating radiation therapy for recurrent nasopharyngeal carcinoma. A case report. Spine. 1998;23(11):1280-1282. doi:10.1097/00007632-199806010-00021
7. Morgan HE, Sher DJ. Adaptive radiotherapy for head and neck cancer. Cancers Head Neck. 2020;5:1. doi:10.1186/s41199-019-0046-z
8. Unger JD, Chiang LC, Unger GF. Apparent reformation of the base of the skull following radiotherapy for nasopharyngeal carcinoma. Radiology. 1978;126(3):779-782. doi:10.1148/126.3.779
9. Fang FM, Leung SW, Wang CJ, et al. Computed tomography findings of bony regeneration after radiotherapy for nasopharyngeal carcinoma with skull base destruction: implications for local control. Int J Radiat Oncol Biol Phys. 1999;44(2):305-309. doi:10.1016/s0360-3016(99)00004-8
10. Tiruchelvarayan R, Lee KA, Ng I. Surgery for atlanto-axial (C1-2) involvement or instability in nasopharyngeal carcinoma patients. Singapore Med J. 2012;53(6):416-421.
11. Samprón N, Arrazola M, Urculo E. Skull-base plasmacytoma with craniocervical instability [in Spanish]. Neurocirugia (Astur). 2009;20(5):478-483.
12. Zheutlin AR, Deshpande SS, Nelson NS, et al. Bone marrow stem cells assuage radiation-induced damage in a murine model of distraction osteogenesis: a histomorphometric evaluation. Cytotherapy. 2016;18(5):664-672. doi:10.1016/j.jcyt.2016.01.013
13. Liu H, Dong Y, Feng X, et al. miR-34a promotes bone regeneration in irradiated bone defects by enhancing osteoblast differentiation of mesenchymal stromal cells in rats. Stem Cell Res Ther. 2019;10(1):180. doi:10.1186/s13287-019-1285-y
14. Holzapfel BM, Wagner F, Martine LC, et al. Tissue engineering and regenerative medicine in musculoskeletal oncology. Cancer Metastasis Rev. 2016;35(3):475-487. doi:10.1007/s10555-016-9635-z
15. Hu WW, Ward BB, Wang Z, Krebsbach PH. Bone regeneration in defects compromised by radiotherapy. J Dent Res. 2010;89(1):77-81. doi:10.1177/0022034509352151
16. Wotman M, Oh EJ, Ahn S, Kraus D, Constantino P, Tham T. HPV status in patients with nasopharyngeal carcinoma in the United States: a SEER database study. Am J Otolaryngol. 2019;40(5):705-710. doi:10.1016/j.amjoto.2019.06.00717. Huang WB, Chan JYW, Liu DL. Human papillomavirus and World Health Organization type III nasopharyngeal carcinoma: multicenter study from an endemic area in Southern China. Cancer. 2018;124(3):530-536. doi:10.1002/cncr.31031.
18. Verma V, Simone CB 2nd, Lin C. Human papillomavirus and nasopharyngeal cancer. Head Neck. 2018;40(4):696-706. doi:10.1002/hed.24978
19. Lee AWM, Lydiatt WM, Colevas AD, et al. Nasopharynx. In: Amin MB, ed. AJCC Cancer Staging Manual. 8th ed. Springer; 2017:103.
20. Barnes L, Eveson JW, Reichart P, Sidransky D, eds. Pathology and genetics of head and neck tumors. In: World Health Organization Classification of Tumours. IARC Press; 2005.
1. Chang ET, Adami HO. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15(10):1765-1777. doi:10.1158/1055-9965.EPI-06-0353
2. Stenmark MH, McHugh JB, Schipper M, et al. Nonendemic HPV-positive nasopharyngeal carcinoma: association with poor prognosis. Int J Radiat Oncol Biol Phys. 2014;88(3):580-588. doi:10.1016/j.ijrobp.2013.11.246
3. Maxwell JH, Kumar B, Feng FY, et al. HPV-positive/p16-positive/EBV-negative nasopharyngeal carcinoma in white North Americans. Head Neck. 2010;32(5):562-567. doi:10.1002/hed.21216
4. Chen YP, Chan ATC, Le QT, Blanchard P, Sun Y, Ma J. Nasopharyngeal carcinoma. Lancet. 2019;394(10192):64-80. doi:10.1016/S0140-6736(19)30956-0
5. Roh JL, Sung MW, Kim KH, et al.. Nasopharyngeal carcinoma with skull base invasion: a necessity of staging subdivision. Am J Otolaryngol. 2004;25(1):26-32. doi:10.1016/j.amjoto.2003.09.011
6. Orr RD, Salo PT. Atlantoaxial instability complicating radiation therapy for recurrent nasopharyngeal carcinoma. A case report. Spine. 1998;23(11):1280-1282. doi:10.1097/00007632-199806010-00021
7. Morgan HE, Sher DJ. Adaptive radiotherapy for head and neck cancer. Cancers Head Neck. 2020;5:1. doi:10.1186/s41199-019-0046-z
8. Unger JD, Chiang LC, Unger GF. Apparent reformation of the base of the skull following radiotherapy for nasopharyngeal carcinoma. Radiology. 1978;126(3):779-782. doi:10.1148/126.3.779
9. Fang FM, Leung SW, Wang CJ, et al. Computed tomography findings of bony regeneration after radiotherapy for nasopharyngeal carcinoma with skull base destruction: implications for local control. Int J Radiat Oncol Biol Phys. 1999;44(2):305-309. doi:10.1016/s0360-3016(99)00004-8
10. Tiruchelvarayan R, Lee KA, Ng I. Surgery for atlanto-axial (C1-2) involvement or instability in nasopharyngeal carcinoma patients. Singapore Med J. 2012;53(6):416-421.
11. Samprón N, Arrazola M, Urculo E. Skull-base plasmacytoma with craniocervical instability [in Spanish]. Neurocirugia (Astur). 2009;20(5):478-483.
12. Zheutlin AR, Deshpande SS, Nelson NS, et al. Bone marrow stem cells assuage radiation-induced damage in a murine model of distraction osteogenesis: a histomorphometric evaluation. Cytotherapy. 2016;18(5):664-672. doi:10.1016/j.jcyt.2016.01.013
13. Liu H, Dong Y, Feng X, et al. miR-34a promotes bone regeneration in irradiated bone defects by enhancing osteoblast differentiation of mesenchymal stromal cells in rats. Stem Cell Res Ther. 2019;10(1):180. doi:10.1186/s13287-019-1285-y
14. Holzapfel BM, Wagner F, Martine LC, et al. Tissue engineering and regenerative medicine in musculoskeletal oncology. Cancer Metastasis Rev. 2016;35(3):475-487. doi:10.1007/s10555-016-9635-z
15. Hu WW, Ward BB, Wang Z, Krebsbach PH. Bone regeneration in defects compromised by radiotherapy. J Dent Res. 2010;89(1):77-81. doi:10.1177/0022034509352151
16. Wotman M, Oh EJ, Ahn S, Kraus D, Constantino P, Tham T. HPV status in patients with nasopharyngeal carcinoma in the United States: a SEER database study. Am J Otolaryngol. 2019;40(5):705-710. doi:10.1016/j.amjoto.2019.06.00717. Huang WB, Chan JYW, Liu DL. Human papillomavirus and World Health Organization type III nasopharyngeal carcinoma: multicenter study from an endemic area in Southern China. Cancer. 2018;124(3):530-536. doi:10.1002/cncr.31031.
18. Verma V, Simone CB 2nd, Lin C. Human papillomavirus and nasopharyngeal cancer. Head Neck. 2018;40(4):696-706. doi:10.1002/hed.24978
19. Lee AWM, Lydiatt WM, Colevas AD, et al. Nasopharynx. In: Amin MB, ed. AJCC Cancer Staging Manual. 8th ed. Springer; 2017:103.
20. Barnes L, Eveson JW, Reichart P, Sidransky D, eds. Pathology and genetics of head and neck tumors. In: World Health Organization Classification of Tumours. IARC Press; 2005.