User login
Elevated liver function tests in a patient on palbociclib and fulvestrant
About 12.4% of women in the United States will be diagnosed with breast cancer at some point in their lifetime.1 A percentage of these women will develop metastatic disease and are estimated to have a 5-year survival rate of 22%.2 There have been meaningful improvements in su
However, endocrine resistance inevitably occurs, and a great deal of research has been focused on developing strategies to combat resistance. One mechanism of endocrine resistance is though the Cyclin-dependent kinases 4 and 6 (CDK4/6) complexes. Among the most promising of the strategies to prevent resistance are the CDK4/6 inhibitors. There are now 3 approved CDK4/6 inhibitor drugs that can be used in combination with endocrine therapy, 1 of which can also be used as a single agent. When used in combination with endocrine therapy, the use of CDK 4/6 inhibitors has significantly improved progression-free survival (PFS) in patients with hormone-sensitive HER2-negative metastatic breast cancer by inhibiting cellular division and growth.3 In postmenopausal women, endocrine therapy plus CDK4/6 inhibitors are the preferred first-line regimen for metastatic disease.
Since the approval of palbociclib by the US Food and Drug Administration in 2015, the most common hematologic lab abnormalities are anemia, leukopenia, neutropenia, and thrombocytopenia. The most common nonhematologic adverse events (AEs) are fatigue, infection, nausea, and stomatitis. Hepatic toxicity has not been commonly observed. We report here the case of a 57-year-old woman on palbociclib and fulvestrant who developed significant elevation of liver function tests after starting palbociclib, suggesting a possible drug-induced liver injury from palbociclib.
Case presentation and summary
A 57-year-old woman with history of hypothyroidism and hypertension presented in May 2016 with a lump in her right breast and back pain. The lump was biopsied and revealed invasive ductal carcinoma, moderately differentiated, estrogen receptor (ER) positive 100%, progesterone receptor (PR) positive 95%, and HER2 negative. A positron emission tomography (PET)–computed tomography (CT) scan and magnetic resonance imaging showed bone metastasis at several vertebral levels, and the results of a bone biopsy confirmed metastatic adenocarcinoma of breast origin, ER positive 60%, PR positive 40%, and HER2 negative. No liver lesions were seen on imaging, but there was suggestion of fatty liver. She was started on letrozole 2.5 mg daily in July 2016 while undergoing kyphoplasty and subsequent radiation. A restaging PET scan revealed progression of disease on letrozole, with possible new rib lesion and progression in the breast. No liver disease was noted. Therapy was changed to fulvestrant and palbociclib. Fulvestrant was started in March 2017 with standard dosing of 500 mg intramuscular on days 1, 15, and 29, and then once a month thereafter. Her first cycle of palbociclib was started on April 5, dosed at 125 mg by mouth daily for 21 days, followed by 7 days off, repeated every 28 days (all dates hereinafter fell within 2017, unless otherwise stipulated).
Labs checked on April 28 and May 26 were unremarkable. A restaging CT scan of the chest, abdomen, and pelvis was done on June 21 after completion of 3 cycles of fulvestrant and palbociclib. There was no evidence of liver metastases, only the fatty infiltration of the liver that had been seen previously. On June 23, 2017, lab results showed a transaminitis with an alanine aminotransferase (ALT) level of 446 IU/L (reference range 10-33 IU/L) and aspartate aminotransferase (AST) level of 183 IU/L (reference range 0-32 IU/L).
The patient’s liver enzyme levels continued to increase and peaked on July 3 at ALT >700 IU/L and AST at 421 IU/L. Her total bilirubin and alkaline phosphatase levels remained within normal limits. She had received her final dose of fulvestrant on May 31 and had taken her last dose of palbociclib on June 20, 2017. She had no history of elevated liver enzymes or liver disease, although the initial PET scan done at diagnosis had suggested hepatic steatosis. She said she had not recently used antibiotics, alcohol, or over-the-counter medications or supplements. There was no family history of liver problems, inflammatory bowel disease, or gastrointestinal malignancy. The only other medications she had taken recently were denosumab, levothyroxine for hypothyroidism, and amlodipine for hypertension. She was seen by hepatology for evaluation of acute hepatitis. Other etiologies for her elevated liver enzymes were ruled out, and she was diagnosed with a drug-induced liver injury from one of her anticancer medications. Her treatments with fulvestrant and palbociclib were held, and the results of her liver function tests normalized by September 2017.
Fulvestrant was restarted on August 24, and her lab results remained normal through November of that year, when restaging scans showed progression with new axillary adenopathy suspicious for metastasis. Imaging also showed a 1.6-cm hepatic lesion suggestive of a focal area of fat deposition or atypical hemangioma without definitive evidence of metastasis. Follow-up imaging was recommended. She was therefore rechallenged with palbociclib at a reduced dose of 100 mg by mouth daily and received the first dose on November 30. On December 8, repeat labs again showed elevated liver function tests (ALT, 285 IU/L; AST, 112 IU/L). Treatment with palbociclib was discontinued on December 10. Because the patient was not able to tolerate palbociclib, and fulvestrant alone was not controlling the disease, she was started on an alternate endocrine therapy with tamoxifen on December 26. The patient’s liver function tests normalized again by January 2018.
Discussion
The use of targeted therapies has changed the landscape of oncologic treatments. Several studies have evaluated the safety and efficacy of palbociclib in combination with endocrine therapy. The Palbociclib Ongoing Trials in the Management of Breast Cancer (PALOMA)-1 study, an open-label, randomized, phase-2 trial involving patients with newly diagnosed metastatic hormone sensitive HER2-negative breast cancer, demonstrated that palbociclib in combination with letrozole was associated with significantly longer PFS than letrozole alone.4 These results were later confirmed in the larger PALOMA-2 study, a randomized, double-blind, phase-3 trial that evaluated 666 postmenopausal patients with no prior systemic therapy. In that study, median PFS for the palbociclib–letrozole group was 24.8 months, compared with 14.5 months for the letrozole-alone group (hazard ratio [HR] for disease progression or death, 0.58 [0.46–0.72], P < .001).5 The most recent PALOMA-3 study, a phase-3 trial involving 521 patients with advanced hormone receptor–positive, HER2-negative breast cancer that had progressed during initial endocrine therapy, evaluated the efficacy of combined palbociclib and fulvestrant in a randomized, double-blind, placebo-controlled, parallel-group trial. The result was that the palbociclib–fulvestrant combination resulted in longer median PFS of 9.2 months, compared with 3.8 months with fulvestrant alone (P < .001).6
These trials also monitored the number of AEs as secondary aims. The most commonly reported AEs in the PALOMA trials for those patients in the palbociclib group were hematologic, with neutropenia being the most common, followed by leukopenia, anemia, and thrombocytopenia. The most common nonhematologic AEs reported in the palbociclib-fulvestrant group were fatigue, nausea, and headache. Elevated liver function tests were a rare but reported AE in 7.2% of the palbociclib-treated patients in the PALOMA-1 study.7 In the PALOMA-2 study, ALT and AST elevations were reported as AEs (all grades) in 9.9% and 9.7% of palbociclib-treated patients, respectively.5 In the PALOMA-3 study, there was 1 fatal serious AE of hepatic failure with grade 5 disease progression in the palbociclib group; however, the patient’s medical history included progressive liver metastasis and disease progression.6 A pooled safety analysis conducted across all PALOMA studies demonstrated that grade 3/4 AST and ALT elevations occurred in 3.3% and 2.3% of palbociclib-treated patients, respectively, again highlighting a reported but rare occurrence.8
The patient described in the present case report started on combination fulvestrant and palbociclib after her disease showed progression on letrozole. She developed an increase in transaminases after completing 3 cycles of palbociclib. Liver function tests increased nearly 12 weeks after beginning her first cycle of the CDK 4/6 inhibitor. Staging scans of the patient demonstrated fatty liver. It is not known if her fatty liver contributed to her transminitis; however, her baseline labs showed normal liver function tests, and they did not increase until after therapy with fulvestrant–palbociclib was started. It might have been that her fatty liver caused her to be at higher risk of transaminitis with administration of palbociclib, although we cannot be certain. Her lab results remained normal while she was on fulvestrant alone, and the liver function test results increased only after palbociclib was started, making this drug the more likely culprit.
Both events of increased liver enzymes occurred within a week of the last palbociclib dose; however, we note that hepatotoxicity developed at a faster rate when the patient was rechallenged with palbociclib at a lower dose, with elevated liver function tests increasing 1 week after restarting treatment as opposed to the first episode that occurred after 3 cycles of the palbociclib. After discontinuation of the medication, liver function tests again normalized, suggesting that palbociclib was most likely the causative agent. In addition, the degree of elevated liver enzymes was less severe on re-exposure at the lower dose of 100 mg, which raises the possibility that there could be a dose-dependent association between palbociclib and hepatotoxicity. There have been few case reports of increased liver enzymes associated with palbociclib, and it is only recently that this association has been more recognized. A meta-analysis by Zaw and colleagues has demonstrated that CDK 4/6 inhibitor–based regimens are associated with a higher risk of elevated AST and ALT; however, their relation with dose dependence was not described. In particular, they found that CDK 4/6 inhibitors increased the risk of high-grade, elevated ALT with a relative risk of 4.33 (95% confidence interval, 2.15-8.71; P < .0001). The meta-analysis also included other CDK 4/6 inhibitors such as abemaciclib and ribociclib, which have been more commonly associated with liver toxicity than palbociclib has.9 Our case report highlights the specific association between palbociclib and elevated liver enzymes.
In conclusion, this case report illustrates that our patient’s elevated liver enzymes were likely related to palbociclib. This is further supported by the fact that this AE occurred twice, both times after palbociclib exposure. In each instance, liver enzymes normalized after discontinuation of palbociclib. One cannot entirely rule out that fulvestrant might have been the culprit medication, but the patient’s normal hepatic panel for several months after starting fulvestrant suggests that is less likely. This case report is indicative of an uncommon complication in the treatment of metastatic breast cancer, one that is starting to gain more recognition, and we must think of palbociclib as a possible cause of drug-induced liver injury when targeted CDK 4/6–based regimens are used.
1. Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2014. Bethesda, MD: National Cancer Institute; 2017. https://seer.cancer.gov/csr/1975_2014/. Accessed April 3, 2018.
2. American Cancer Society. Breast cancer survival rates. https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-survival-rates.html. Accessed April 3, 2018.
3. Wolff AC. CDK4 and CDK6 inhibition in breast cancer - a new standard. N Engl J Med. 2016; 375(20):1993-1994.
4. Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomized phase 2 study. Lancet Oncol. 2015;16(1):25-35.
5. Finn RS, Martin M, Rugo, HS et. al. Palbociclib and letrozole in advanced breast cancer. New Engl J Med. 2016;375:1925-1936
6. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomized controlled trial. Lancet Oncol. 2016;17(4):425-439.
7. Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209-219.
8. Dieras V, Rugo HS, Schnell P, et al. Long-term pooled safety analysis of palbociclib in combination with endocrine therapy for HR+/HR- advanced breast cancer [published online July 18, 2018]. Natl Cancer Inst. 2018;111.
9. Zaw M, Thein KZ, Tun A, et al. A systematic review and meta-analysis of randomized controlled trials to evaluate the risk of gastrointestinal and hepatic toxicities in patients with hormone receptor positive HER2-negative breast cancer treated with CKD 4/6 inhibitors. J Clin Oncol. 2017;35(suppl 31):209.
About 12.4% of women in the United States will be diagnosed with breast cancer at some point in their lifetime.1 A percentage of these women will develop metastatic disease and are estimated to have a 5-year survival rate of 22%.2 There have been meaningful improvements in su
However, endocrine resistance inevitably occurs, and a great deal of research has been focused on developing strategies to combat resistance. One mechanism of endocrine resistance is though the Cyclin-dependent kinases 4 and 6 (CDK4/6) complexes. Among the most promising of the strategies to prevent resistance are the CDK4/6 inhibitors. There are now 3 approved CDK4/6 inhibitor drugs that can be used in combination with endocrine therapy, 1 of which can also be used as a single agent. When used in combination with endocrine therapy, the use of CDK 4/6 inhibitors has significantly improved progression-free survival (PFS) in patients with hormone-sensitive HER2-negative metastatic breast cancer by inhibiting cellular division and growth.3 In postmenopausal women, endocrine therapy plus CDK4/6 inhibitors are the preferred first-line regimen for metastatic disease.
Since the approval of palbociclib by the US Food and Drug Administration in 2015, the most common hematologic lab abnormalities are anemia, leukopenia, neutropenia, and thrombocytopenia. The most common nonhematologic adverse events (AEs) are fatigue, infection, nausea, and stomatitis. Hepatic toxicity has not been commonly observed. We report here the case of a 57-year-old woman on palbociclib and fulvestrant who developed significant elevation of liver function tests after starting palbociclib, suggesting a possible drug-induced liver injury from palbociclib.
Case presentation and summary
A 57-year-old woman with history of hypothyroidism and hypertension presented in May 2016 with a lump in her right breast and back pain. The lump was biopsied and revealed invasive ductal carcinoma, moderately differentiated, estrogen receptor (ER) positive 100%, progesterone receptor (PR) positive 95%, and HER2 negative. A positron emission tomography (PET)–computed tomography (CT) scan and magnetic resonance imaging showed bone metastasis at several vertebral levels, and the results of a bone biopsy confirmed metastatic adenocarcinoma of breast origin, ER positive 60%, PR positive 40%, and HER2 negative. No liver lesions were seen on imaging, but there was suggestion of fatty liver. She was started on letrozole 2.5 mg daily in July 2016 while undergoing kyphoplasty and subsequent radiation. A restaging PET scan revealed progression of disease on letrozole, with possible new rib lesion and progression in the breast. No liver disease was noted. Therapy was changed to fulvestrant and palbociclib. Fulvestrant was started in March 2017 with standard dosing of 500 mg intramuscular on days 1, 15, and 29, and then once a month thereafter. Her first cycle of palbociclib was started on April 5, dosed at 125 mg by mouth daily for 21 days, followed by 7 days off, repeated every 28 days (all dates hereinafter fell within 2017, unless otherwise stipulated).
Labs checked on April 28 and May 26 were unremarkable. A restaging CT scan of the chest, abdomen, and pelvis was done on June 21 after completion of 3 cycles of fulvestrant and palbociclib. There was no evidence of liver metastases, only the fatty infiltration of the liver that had been seen previously. On June 23, 2017, lab results showed a transaminitis with an alanine aminotransferase (ALT) level of 446 IU/L (reference range 10-33 IU/L) and aspartate aminotransferase (AST) level of 183 IU/L (reference range 0-32 IU/L).
The patient’s liver enzyme levels continued to increase and peaked on July 3 at ALT >700 IU/L and AST at 421 IU/L. Her total bilirubin and alkaline phosphatase levels remained within normal limits. She had received her final dose of fulvestrant on May 31 and had taken her last dose of palbociclib on June 20, 2017. She had no history of elevated liver enzymes or liver disease, although the initial PET scan done at diagnosis had suggested hepatic steatosis. She said she had not recently used antibiotics, alcohol, or over-the-counter medications or supplements. There was no family history of liver problems, inflammatory bowel disease, or gastrointestinal malignancy. The only other medications she had taken recently were denosumab, levothyroxine for hypothyroidism, and amlodipine for hypertension. She was seen by hepatology for evaluation of acute hepatitis. Other etiologies for her elevated liver enzymes were ruled out, and she was diagnosed with a drug-induced liver injury from one of her anticancer medications. Her treatments with fulvestrant and palbociclib were held, and the results of her liver function tests normalized by September 2017.
Fulvestrant was restarted on August 24, and her lab results remained normal through November of that year, when restaging scans showed progression with new axillary adenopathy suspicious for metastasis. Imaging also showed a 1.6-cm hepatic lesion suggestive of a focal area of fat deposition or atypical hemangioma without definitive evidence of metastasis. Follow-up imaging was recommended. She was therefore rechallenged with palbociclib at a reduced dose of 100 mg by mouth daily and received the first dose on November 30. On December 8, repeat labs again showed elevated liver function tests (ALT, 285 IU/L; AST, 112 IU/L). Treatment with palbociclib was discontinued on December 10. Because the patient was not able to tolerate palbociclib, and fulvestrant alone was not controlling the disease, she was started on an alternate endocrine therapy with tamoxifen on December 26. The patient’s liver function tests normalized again by January 2018.
Discussion
The use of targeted therapies has changed the landscape of oncologic treatments. Several studies have evaluated the safety and efficacy of palbociclib in combination with endocrine therapy. The Palbociclib Ongoing Trials in the Management of Breast Cancer (PALOMA)-1 study, an open-label, randomized, phase-2 trial involving patients with newly diagnosed metastatic hormone sensitive HER2-negative breast cancer, demonstrated that palbociclib in combination with letrozole was associated with significantly longer PFS than letrozole alone.4 These results were later confirmed in the larger PALOMA-2 study, a randomized, double-blind, phase-3 trial that evaluated 666 postmenopausal patients with no prior systemic therapy. In that study, median PFS for the palbociclib–letrozole group was 24.8 months, compared with 14.5 months for the letrozole-alone group (hazard ratio [HR] for disease progression or death, 0.58 [0.46–0.72], P < .001).5 The most recent PALOMA-3 study, a phase-3 trial involving 521 patients with advanced hormone receptor–positive, HER2-negative breast cancer that had progressed during initial endocrine therapy, evaluated the efficacy of combined palbociclib and fulvestrant in a randomized, double-blind, placebo-controlled, parallel-group trial. The result was that the palbociclib–fulvestrant combination resulted in longer median PFS of 9.2 months, compared with 3.8 months with fulvestrant alone (P < .001).6
These trials also monitored the number of AEs as secondary aims. The most commonly reported AEs in the PALOMA trials for those patients in the palbociclib group were hematologic, with neutropenia being the most common, followed by leukopenia, anemia, and thrombocytopenia. The most common nonhematologic AEs reported in the palbociclib-fulvestrant group were fatigue, nausea, and headache. Elevated liver function tests were a rare but reported AE in 7.2% of the palbociclib-treated patients in the PALOMA-1 study.7 In the PALOMA-2 study, ALT and AST elevations were reported as AEs (all grades) in 9.9% and 9.7% of palbociclib-treated patients, respectively.5 In the PALOMA-3 study, there was 1 fatal serious AE of hepatic failure with grade 5 disease progression in the palbociclib group; however, the patient’s medical history included progressive liver metastasis and disease progression.6 A pooled safety analysis conducted across all PALOMA studies demonstrated that grade 3/4 AST and ALT elevations occurred in 3.3% and 2.3% of palbociclib-treated patients, respectively, again highlighting a reported but rare occurrence.8
The patient described in the present case report started on combination fulvestrant and palbociclib after her disease showed progression on letrozole. She developed an increase in transaminases after completing 3 cycles of palbociclib. Liver function tests increased nearly 12 weeks after beginning her first cycle of the CDK 4/6 inhibitor. Staging scans of the patient demonstrated fatty liver. It is not known if her fatty liver contributed to her transminitis; however, her baseline labs showed normal liver function tests, and they did not increase until after therapy with fulvestrant–palbociclib was started. It might have been that her fatty liver caused her to be at higher risk of transaminitis with administration of palbociclib, although we cannot be certain. Her lab results remained normal while she was on fulvestrant alone, and the liver function test results increased only after palbociclib was started, making this drug the more likely culprit.
Both events of increased liver enzymes occurred within a week of the last palbociclib dose; however, we note that hepatotoxicity developed at a faster rate when the patient was rechallenged with palbociclib at a lower dose, with elevated liver function tests increasing 1 week after restarting treatment as opposed to the first episode that occurred after 3 cycles of the palbociclib. After discontinuation of the medication, liver function tests again normalized, suggesting that palbociclib was most likely the causative agent. In addition, the degree of elevated liver enzymes was less severe on re-exposure at the lower dose of 100 mg, which raises the possibility that there could be a dose-dependent association between palbociclib and hepatotoxicity. There have been few case reports of increased liver enzymes associated with palbociclib, and it is only recently that this association has been more recognized. A meta-analysis by Zaw and colleagues has demonstrated that CDK 4/6 inhibitor–based regimens are associated with a higher risk of elevated AST and ALT; however, their relation with dose dependence was not described. In particular, they found that CDK 4/6 inhibitors increased the risk of high-grade, elevated ALT with a relative risk of 4.33 (95% confidence interval, 2.15-8.71; P < .0001). The meta-analysis also included other CDK 4/6 inhibitors such as abemaciclib and ribociclib, which have been more commonly associated with liver toxicity than palbociclib has.9 Our case report highlights the specific association between palbociclib and elevated liver enzymes.
In conclusion, this case report illustrates that our patient’s elevated liver enzymes were likely related to palbociclib. This is further supported by the fact that this AE occurred twice, both times after palbociclib exposure. In each instance, liver enzymes normalized after discontinuation of palbociclib. One cannot entirely rule out that fulvestrant might have been the culprit medication, but the patient’s normal hepatic panel for several months after starting fulvestrant suggests that is less likely. This case report is indicative of an uncommon complication in the treatment of metastatic breast cancer, one that is starting to gain more recognition, and we must think of palbociclib as a possible cause of drug-induced liver injury when targeted CDK 4/6–based regimens are used.
About 12.4% of women in the United States will be diagnosed with breast cancer at some point in their lifetime.1 A percentage of these women will develop metastatic disease and are estimated to have a 5-year survival rate of 22%.2 There have been meaningful improvements in su
However, endocrine resistance inevitably occurs, and a great deal of research has been focused on developing strategies to combat resistance. One mechanism of endocrine resistance is though the Cyclin-dependent kinases 4 and 6 (CDK4/6) complexes. Among the most promising of the strategies to prevent resistance are the CDK4/6 inhibitors. There are now 3 approved CDK4/6 inhibitor drugs that can be used in combination with endocrine therapy, 1 of which can also be used as a single agent. When used in combination with endocrine therapy, the use of CDK 4/6 inhibitors has significantly improved progression-free survival (PFS) in patients with hormone-sensitive HER2-negative metastatic breast cancer by inhibiting cellular division and growth.3 In postmenopausal women, endocrine therapy plus CDK4/6 inhibitors are the preferred first-line regimen for metastatic disease.
Since the approval of palbociclib by the US Food and Drug Administration in 2015, the most common hematologic lab abnormalities are anemia, leukopenia, neutropenia, and thrombocytopenia. The most common nonhematologic adverse events (AEs) are fatigue, infection, nausea, and stomatitis. Hepatic toxicity has not been commonly observed. We report here the case of a 57-year-old woman on palbociclib and fulvestrant who developed significant elevation of liver function tests after starting palbociclib, suggesting a possible drug-induced liver injury from palbociclib.
Case presentation and summary
A 57-year-old woman with history of hypothyroidism and hypertension presented in May 2016 with a lump in her right breast and back pain. The lump was biopsied and revealed invasive ductal carcinoma, moderately differentiated, estrogen receptor (ER) positive 100%, progesterone receptor (PR) positive 95%, and HER2 negative. A positron emission tomography (PET)–computed tomography (CT) scan and magnetic resonance imaging showed bone metastasis at several vertebral levels, and the results of a bone biopsy confirmed metastatic adenocarcinoma of breast origin, ER positive 60%, PR positive 40%, and HER2 negative. No liver lesions were seen on imaging, but there was suggestion of fatty liver. She was started on letrozole 2.5 mg daily in July 2016 while undergoing kyphoplasty and subsequent radiation. A restaging PET scan revealed progression of disease on letrozole, with possible new rib lesion and progression in the breast. No liver disease was noted. Therapy was changed to fulvestrant and palbociclib. Fulvestrant was started in March 2017 with standard dosing of 500 mg intramuscular on days 1, 15, and 29, and then once a month thereafter. Her first cycle of palbociclib was started on April 5, dosed at 125 mg by mouth daily for 21 days, followed by 7 days off, repeated every 28 days (all dates hereinafter fell within 2017, unless otherwise stipulated).
Labs checked on April 28 and May 26 were unremarkable. A restaging CT scan of the chest, abdomen, and pelvis was done on June 21 after completion of 3 cycles of fulvestrant and palbociclib. There was no evidence of liver metastases, only the fatty infiltration of the liver that had been seen previously. On June 23, 2017, lab results showed a transaminitis with an alanine aminotransferase (ALT) level of 446 IU/L (reference range 10-33 IU/L) and aspartate aminotransferase (AST) level of 183 IU/L (reference range 0-32 IU/L).
The patient’s liver enzyme levels continued to increase and peaked on July 3 at ALT >700 IU/L and AST at 421 IU/L. Her total bilirubin and alkaline phosphatase levels remained within normal limits. She had received her final dose of fulvestrant on May 31 and had taken her last dose of palbociclib on June 20, 2017. She had no history of elevated liver enzymes or liver disease, although the initial PET scan done at diagnosis had suggested hepatic steatosis. She said she had not recently used antibiotics, alcohol, or over-the-counter medications or supplements. There was no family history of liver problems, inflammatory bowel disease, or gastrointestinal malignancy. The only other medications she had taken recently were denosumab, levothyroxine for hypothyroidism, and amlodipine for hypertension. She was seen by hepatology for evaluation of acute hepatitis. Other etiologies for her elevated liver enzymes were ruled out, and she was diagnosed with a drug-induced liver injury from one of her anticancer medications. Her treatments with fulvestrant and palbociclib were held, and the results of her liver function tests normalized by September 2017.
Fulvestrant was restarted on August 24, and her lab results remained normal through November of that year, when restaging scans showed progression with new axillary adenopathy suspicious for metastasis. Imaging also showed a 1.6-cm hepatic lesion suggestive of a focal area of fat deposition or atypical hemangioma without definitive evidence of metastasis. Follow-up imaging was recommended. She was therefore rechallenged with palbociclib at a reduced dose of 100 mg by mouth daily and received the first dose on November 30. On December 8, repeat labs again showed elevated liver function tests (ALT, 285 IU/L; AST, 112 IU/L). Treatment with palbociclib was discontinued on December 10. Because the patient was not able to tolerate palbociclib, and fulvestrant alone was not controlling the disease, she was started on an alternate endocrine therapy with tamoxifen on December 26. The patient’s liver function tests normalized again by January 2018.
Discussion
The use of targeted therapies has changed the landscape of oncologic treatments. Several studies have evaluated the safety and efficacy of palbociclib in combination with endocrine therapy. The Palbociclib Ongoing Trials in the Management of Breast Cancer (PALOMA)-1 study, an open-label, randomized, phase-2 trial involving patients with newly diagnosed metastatic hormone sensitive HER2-negative breast cancer, demonstrated that palbociclib in combination with letrozole was associated with significantly longer PFS than letrozole alone.4 These results were later confirmed in the larger PALOMA-2 study, a randomized, double-blind, phase-3 trial that evaluated 666 postmenopausal patients with no prior systemic therapy. In that study, median PFS for the palbociclib–letrozole group was 24.8 months, compared with 14.5 months for the letrozole-alone group (hazard ratio [HR] for disease progression or death, 0.58 [0.46–0.72], P < .001).5 The most recent PALOMA-3 study, a phase-3 trial involving 521 patients with advanced hormone receptor–positive, HER2-negative breast cancer that had progressed during initial endocrine therapy, evaluated the efficacy of combined palbociclib and fulvestrant in a randomized, double-blind, placebo-controlled, parallel-group trial. The result was that the palbociclib–fulvestrant combination resulted in longer median PFS of 9.2 months, compared with 3.8 months with fulvestrant alone (P < .001).6
These trials also monitored the number of AEs as secondary aims. The most commonly reported AEs in the PALOMA trials for those patients in the palbociclib group were hematologic, with neutropenia being the most common, followed by leukopenia, anemia, and thrombocytopenia. The most common nonhematologic AEs reported in the palbociclib-fulvestrant group were fatigue, nausea, and headache. Elevated liver function tests were a rare but reported AE in 7.2% of the palbociclib-treated patients in the PALOMA-1 study.7 In the PALOMA-2 study, ALT and AST elevations were reported as AEs (all grades) in 9.9% and 9.7% of palbociclib-treated patients, respectively.5 In the PALOMA-3 study, there was 1 fatal serious AE of hepatic failure with grade 5 disease progression in the palbociclib group; however, the patient’s medical history included progressive liver metastasis and disease progression.6 A pooled safety analysis conducted across all PALOMA studies demonstrated that grade 3/4 AST and ALT elevations occurred in 3.3% and 2.3% of palbociclib-treated patients, respectively, again highlighting a reported but rare occurrence.8
The patient described in the present case report started on combination fulvestrant and palbociclib after her disease showed progression on letrozole. She developed an increase in transaminases after completing 3 cycles of palbociclib. Liver function tests increased nearly 12 weeks after beginning her first cycle of the CDK 4/6 inhibitor. Staging scans of the patient demonstrated fatty liver. It is not known if her fatty liver contributed to her transminitis; however, her baseline labs showed normal liver function tests, and they did not increase until after therapy with fulvestrant–palbociclib was started. It might have been that her fatty liver caused her to be at higher risk of transaminitis with administration of palbociclib, although we cannot be certain. Her lab results remained normal while she was on fulvestrant alone, and the liver function test results increased only after palbociclib was started, making this drug the more likely culprit.
Both events of increased liver enzymes occurred within a week of the last palbociclib dose; however, we note that hepatotoxicity developed at a faster rate when the patient was rechallenged with palbociclib at a lower dose, with elevated liver function tests increasing 1 week after restarting treatment as opposed to the first episode that occurred after 3 cycles of the palbociclib. After discontinuation of the medication, liver function tests again normalized, suggesting that palbociclib was most likely the causative agent. In addition, the degree of elevated liver enzymes was less severe on re-exposure at the lower dose of 100 mg, which raises the possibility that there could be a dose-dependent association between palbociclib and hepatotoxicity. There have been few case reports of increased liver enzymes associated with palbociclib, and it is only recently that this association has been more recognized. A meta-analysis by Zaw and colleagues has demonstrated that CDK 4/6 inhibitor–based regimens are associated with a higher risk of elevated AST and ALT; however, their relation with dose dependence was not described. In particular, they found that CDK 4/6 inhibitors increased the risk of high-grade, elevated ALT with a relative risk of 4.33 (95% confidence interval, 2.15-8.71; P < .0001). The meta-analysis also included other CDK 4/6 inhibitors such as abemaciclib and ribociclib, which have been more commonly associated with liver toxicity than palbociclib has.9 Our case report highlights the specific association between palbociclib and elevated liver enzymes.
In conclusion, this case report illustrates that our patient’s elevated liver enzymes were likely related to palbociclib. This is further supported by the fact that this AE occurred twice, both times after palbociclib exposure. In each instance, liver enzymes normalized after discontinuation of palbociclib. One cannot entirely rule out that fulvestrant might have been the culprit medication, but the patient’s normal hepatic panel for several months after starting fulvestrant suggests that is less likely. This case report is indicative of an uncommon complication in the treatment of metastatic breast cancer, one that is starting to gain more recognition, and we must think of palbociclib as a possible cause of drug-induced liver injury when targeted CDK 4/6–based regimens are used.
1. Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2014. Bethesda, MD: National Cancer Institute; 2017. https://seer.cancer.gov/csr/1975_2014/. Accessed April 3, 2018.
2. American Cancer Society. Breast cancer survival rates. https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-survival-rates.html. Accessed April 3, 2018.
3. Wolff AC. CDK4 and CDK6 inhibition in breast cancer - a new standard. N Engl J Med. 2016; 375(20):1993-1994.
4. Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomized phase 2 study. Lancet Oncol. 2015;16(1):25-35.
5. Finn RS, Martin M, Rugo, HS et. al. Palbociclib and letrozole in advanced breast cancer. New Engl J Med. 2016;375:1925-1936
6. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomized controlled trial. Lancet Oncol. 2016;17(4):425-439.
7. Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209-219.
8. Dieras V, Rugo HS, Schnell P, et al. Long-term pooled safety analysis of palbociclib in combination with endocrine therapy for HR+/HR- advanced breast cancer [published online July 18, 2018]. Natl Cancer Inst. 2018;111.
9. Zaw M, Thein KZ, Tun A, et al. A systematic review and meta-analysis of randomized controlled trials to evaluate the risk of gastrointestinal and hepatic toxicities in patients with hormone receptor positive HER2-negative breast cancer treated with CKD 4/6 inhibitors. J Clin Oncol. 2017;35(suppl 31):209.
1. Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2014. Bethesda, MD: National Cancer Institute; 2017. https://seer.cancer.gov/csr/1975_2014/. Accessed April 3, 2018.
2. American Cancer Society. Breast cancer survival rates. https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-survival-rates.html. Accessed April 3, 2018.
3. Wolff AC. CDK4 and CDK6 inhibition in breast cancer - a new standard. N Engl J Med. 2016; 375(20):1993-1994.
4. Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomized phase 2 study. Lancet Oncol. 2015;16(1):25-35.
5. Finn RS, Martin M, Rugo, HS et. al. Palbociclib and letrozole in advanced breast cancer. New Engl J Med. 2016;375:1925-1936
6. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomized controlled trial. Lancet Oncol. 2016;17(4):425-439.
7. Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209-219.
8. Dieras V, Rugo HS, Schnell P, et al. Long-term pooled safety analysis of palbociclib in combination with endocrine therapy for HR+/HR- advanced breast cancer [published online July 18, 2018]. Natl Cancer Inst. 2018;111.
9. Zaw M, Thein KZ, Tun A, et al. A systematic review and meta-analysis of randomized controlled trials to evaluate the risk of gastrointestinal and hepatic toxicities in patients with hormone receptor positive HER2-negative breast cancer treated with CKD 4/6 inhibitors. J Clin Oncol. 2017;35(suppl 31):209.
The challenge of managing a cetuximab rash
Epidermal growth factor receptor antibodies (EGFR) such as cetuximab have been approved for use as first-line management as well as salvage therapy for head and neck and colorectal cancers. Among the most common expected toxicity is a cutaneous eruption described as acneiform. The presence of a rash has been postulated to predict a more favorable treatment outcome for cancers of the head and neck1 but not for colorectum.2 With more severe drug reactions, patients may require a treatment break, which has been shown to reduce locoregional control and survival, particularly in patients with head and neck cancer.3 This has prompted clinicians to affect rapid therapy to reverse the drug eruption. Given the controversy around rapid and effective reversal of this drug reaction, this report aims to address the current status of clinical management using an actual patient vignette.
Case presentation and summary
The patient was a 57-year-old white man who had been diagnosed with stage 4 T4N0M1 grade 3 cutaneous squamous cell carcinoma (SCC) of the right postauricular soft tissues, with erosion into the right mastoid and biopsy-proven metastatic disease involving the contralateral left supraclavicular fossa and bilateral lungs. His disease became chemotherapy-refractory, and he was referred for palliative local therapy to the base of skull. Because of the size of the tumor (4 cm × 5 cm), he was considered for sensitizing chemotherapy, but cisplatin was not appropriate because of chronic hearing loss.4 The patient was recommended sensitizing doses of cetuximab. This EGFR antibody has been shown to offer similar benefits to those seen with cisplatin in the definitive management of head and neck SCC.5
The standard loading dose of cetuximab was given at 400 mg/m2 intravenously (IV). The following week, the sensitizing dose of 250 mg/m2 IV was given along with daily radiotherapy to the target volumes. The weekly dose of cetuximab continued at 250 mg/m2. The radiotherapy prescription was for 6,000 cGy in 200 cGy daily fractions, encompassing the gross tumor volume as identified on a computed-tomographic scan with 3-mm cuts. We used a noncoplanar arc radiotherapy beam arrangement because it inherently spreads the dose over a larger volume of normal tissue while conformally delivering its largest dose to the gross tumor volume. As such, a volume of the patient’s oropharynx and oral cavity was included within the radiotherapy dose penumbra. After receiving 3 weekly doses of cetuximab (1 loading dose and 2 weekly sensitizing doses) and 2,000 cGy of radiotherapy, the patient developed a robust grade 2 cutaneous eruption delimited to the face, with few scattered lesions on the upper anterior chest. He was seen in the medical oncology department and was prescribed doxycycline 100 mg orally twice daily and topical clindamycin 2% ointment twice daily.
In the radiation oncology clinic, his drug therapy was manipulated. His cetuximab cutaneous reaction was a grade 2, manifested by moderate erythema with nonconfluent moist desquamation. Because of concern that the patient would develop oral candida, which would further delay his therapy, the oral and topical antibiotics were discontinued, as was the oral prednisone. He was prescribed triamcinolone cream 0.1% to be applied to the facial and few chest wall areas twice daily and an oncology mouth rinse to address early nonconfluent mucositis. The accompanying images show the extent of the patient’s cetuximab cutaneous reaction at baseline before treatment initiation (Figure 1), at 4 days after the intervention (Figure 2), and again at 6 days after the intervention (Figure 3). The patient consented to having his photographs taken and understood that they would be used for educational and research publication purposes.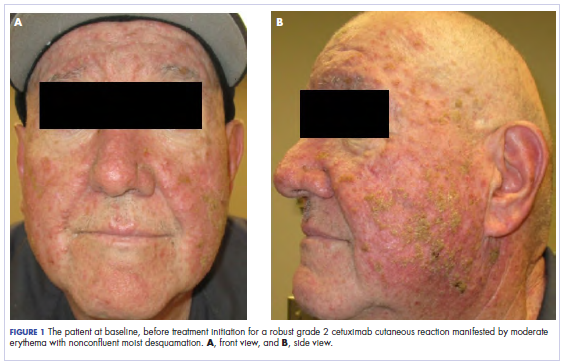
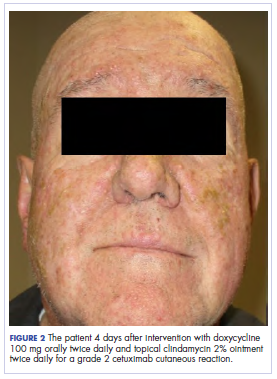
As can be seen from the photographs, the patient’s rash began to dry and peel by day 4 after the intervention, and there were no new eruptions. The pruritus that accompanied the rash had entirely resolved. By day 6, the rash had completely subsided. Because of the response to the topical steroid, the patient continued cetuximab without a dose modification. He was recommended to continue with the triamcinolone cream until the chemoradiotherapy course concluded.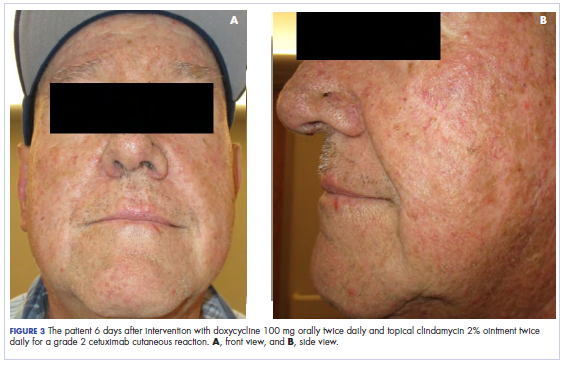
Discussion
A cetuximab-induced rash is common. In a 2011 meta-analysis quantifying grades 1 to 4 in severity, about 75% of patients treated with an EGFR inhibitor experienced a rash. Most of the rashes were lower than grade 3, and the drug was either dose-reduced or temporarily held, but it was not generally discontinued.6 Of note is that in a nonselected survey of medical oncologists who were prescribing cetuximab, 76% reported holding the drug owing to rash severity, 60% reported dose reductions for a drug rash, and 32% reported changing the drug because of rash severity.7
In the initial pharmaceutical registration trial, 76% to 88% of patients who received cetuximab developed a rash, 17% of which were at least grade 3. The pharma recommendations for managing the drug rash include a drug delay for up to 2 weeks for a rash of grade 3 or less and to terminate use of the drug if there is no clinical improvement after 2 weeks.8 Biopsies of the rash confirm a suppurative inflammatory reaction separate from an infectious acne reaction,9 resulting in a recommendation to treat with topical steroid therapy. In some circumstances, the drug reaction can become infected or involve the paronychia, often related to Staphylococcus aureus.10 Despite what would otherwise be a problem addressed by anti-inflammatory medical therapy, the clinical appearance of the rash marked by pustules, coupled with the relative immunosuppressed state of a cancer patient, has prompted medical oncologists to prescribe antibiotic therapy.
To address the many single-institutional reports on management of the EGFR rash, several guidelines have been published. The earliest guideline – after a report that concurrent cetuximab and radiotherapy was superior to radiotherapy alone in locally advanced head and neck cancer, which documented a 23% incidence of at least grade 3 cutaneous toxicity in the cetuximab arm1 – attempted to score the severity of the rash according to the National Cancer Institute’s (NCI) Common Terminology Criteria for Adverse Events (CTCAE). Under those criteria, the authors defined grade 2 toxicity as moderate to brisk erythema with patchy moist desquamation, mostly confined to skin folds and creases. Grade 3 toxicity was described as moist desquamation other than skin folds and creases with bleeding induced by minor trauma, and grade 4 skin toxicity was defined as skin necrosis or ulceration of full thickness dermis with spontaneous bleeding from the involved site. The authors went on to describe a grade-related treatment algorithm that included gently washing the skin, keeping it dry, and using topical anti-inflammatory agents, including steroids. Antibiotics should be used in the presence of a suspected infection after culturing the area, and grade 4 toxicity should be referred to a wound care center.11
In a consensus statement from the National Comprehensive Cancer Network, the authors noted that most management recommendations were anecdotal. They recommended against the use of astringents and other drying agents because they exacerbate pain. The ultimate choice of topical steroids or antibiotics was based entirely on subjective judgement given the absence of prospective data.12
A Spanish consensus conference report argued against any prophylaxis against a skin reaction, other than keeping the skin clean and dry.13 The authors of the report recommended against washing the affected skin more than twice a day to avoid excess drying, and they advocated for moisturizers and debridement of skin crusting with hydrogels to reduce superinfection and bleeding.13 The authors also noted that some guidelines have suggested that topical steroids might exacerbate a skin rash,14 but they concluded that topical steroids are beneficial as long as they are used for less than 2 weeks. Any use of antibiotics should be based on clear evidence of an infection.13
In the first modification of the NCI’s CTCAE rash grading scale, an international panel addressed the increasing number of reports in the literature suggesting that the previous toxicity scale was possibly inadequate in its recommendations for appropriate treatment. The initial scale had defined only the skin reaction and not what therapy should be administered; therefore, in the update, the descriptions for grades 1 and 2 toxicity remained unchanged, but oral antibiotics were recommended for grade 3 lesion, and parenteral antibiotics with skin grafting were required with grade 4 toxicity.15
An Asian expert panel suggested modifying the bioradiation dermatitis scale, defining a grade 3 dermatitis as >50% moist desquamation of the involved field with formation of confluent lesions because of treatment. They recommended both topical and oral therapy, wound care, and possible hospitalization in severe cases. The panel suggested topical and systemic steroids and antibiotics.16
Finally, in an Italian consensus report, the members again modified the skin toxicity grading and were notably more aggressive in terms of their management recommendations. They defined grade 2 toxicity as pustules or papules covering 10% to 30% of the body surface area, with potential pruritus or tenderness. They also noted the psychosocial impact of skin toxicities on patients and the limits to their activities of daily living. They recommended vitamin K1 (menadione) cream, topical antibiotics, topical intermediate potency steroids, and oral antibiotic therapy for up to 4 weeks for grade 2 toxicity. Despite this aggressive treatment course, the authors admitted that the utility of topical steroids and antibiotics was unknown. They defined grade 3 toxicity as pustules or papules covering more than 30% of the body surface area, with signs of possible pruritus and tenderness. Activities of daily living and self-care were affected, and there was evidence of a superinfection. The panel suggested use of antibiotics pending culture results, oral prednisone, antihistamines, and oral analgesics. Topical therapy was not included.17 It is noteworthy that only the Italian panel recommended the use of vitamin K1 cream. In a prospective randomized, double-blinded, placebo-controlled phase 2 trial of 30 patients, menadione exhibited no clinical benefit in terms of reducing the severity of cetuximab skin lesions.18
Figure 4 illustrates our institutional approach to treating cetuximab rash based on a combination of the Spanish and NCI approaches.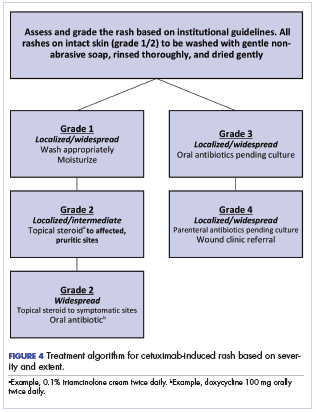
The ultimate choice of therapy to manage a cetuximab rash must be patient and treatment specific. Our institutional approach, like that of the Spanish series,13 is to avoid chemoprophylaxis against a rash; rather, we recommend daily washing of the skin with a gentle soap followed by thorough rinsing and adequate, nonaggressive drying. Moisturizing the intact skin has been shown to reduce exfoliation, and we have incorporated that approach into our regimen.19
In our patient, whose head and neck radiotherapy tumor volume included a portion of the oral cavity and oropharynx, systemic antibiotic and steroid therapy would likely lead to further complications with the development of oral candidiasis. Therefore, while the severity of the reaction remained a grade 2, it seemed appropriate to treat with topical intermediate potency steroids and skin cleansing only. If the reaction had become more severe, then cultures would have been obtained to guide our decision on antibiotic therapy. Our patient’s response to topical steroids was predictable and effective, and he was able to proceed with his course of cancer therapy.
1. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21-28.
2. Sommeijer DW, Karapetis CS, Zalcberg JR, et al. The relationship between rash, tumor mutation KRAS status and clinical and quality of life outcomes in patients with advanced colorectal cancer treated with cetuximab in the NCIC CTG/AGITG CO.17. Acta Oncol. 2014;53(7):877-884.
3. Vahabzadeh-Hagh AM, Rwigema JM, Nabili V, Wang MB, Lorentz WC. Predictors of prolongation in radiation treatment time in a veteran population treated with chemoradiation for oropharyngeal cancer. Acta Otolaryngol. 2018;138(1):80-84.
4. Waissbluth S, Peleva E, Daniel SJ. Platinum-induced ototoxicity: a prevailing ototoxicity criteria. Eur Arch Otorhinlaryngol. 2017;274(3):1187-1196.
5. Huang J, Zhang J, Shi C, Liu L, Wei Y. Survival, recurrence and toxicity of HNSCC in comparison of a radiotherapy combination with cisplatin versus cetuximab: a meta-analysis. BMC cancer. 2016;16(1):689-713.
6. Mittman N, Seung SJ. Rash rates with EGFR inhibitors: meta-analysis. Curr Oncol. 2011;18(2):e54-e63.
7. Boone SL, Rademaker A, Liu D, Pfeiffer C, Mauro DJ, Lacouture ME. Impact and management of skin toxicity associated with anti-epidermal growth factor receptor therapy: survey results. Oncology. 2007;72(3-4):152-159.
8. Erbitux (cetuximab). Ask Lilly website. www.erbitux.com/hcp/index.html. Updated July 3, 2018. Accessed November 27.
9. Busam KJ, Capodieci P, Motzer R, Kiehn T, Phelan D, Halpern AC. Cutaneous side-effects in cancer patients treated with antiepidermal growth factor receptor antibody C225. Br J Dermatol. 2001;144(6):1169-1176.
10. Agero AL, Dusza SW, Benvenuto-Andrade C, Busam KJ, Myskowski P, Halpern AC. Dermatologic side effects associated with the epidermal growth factor receptor inhibitors. J Am Acad Dermatol. 2006;55:657-670, 2006.
11. Bernier J, Bonner J, Vermorken JB, et al. Consensus guidelines for the management of radiation dermatitis and coexisting acne-like rash in patients receiving radiotherapy plus EGFR inhibitors for the treatment of squamous cell carcinoma of the head and neck. Ann Oncol. 2008;19(1):142-149.
12. Burtness B, Anadkat M, Basti S, et al. NCCN task force report: management of dermatologic and other toxicities associated with EGFR inhibition in patients with cancer. J Natl Compr Canc Netw. 2009;7(suppl 1):S5-S21.
13.
14. Li T, Perez-Soler R. Skin toxicity associated with epidermal growth factor receptor inhibitors. Target Oncol. 2009;4(2):107-119.
15. Bernier J, Russi EG, Homey B, et al. Management of radiation dermatitis in patients receiving cetuximab and radiotherapy for locally advanced squamous cell carcinoma of the head and neck: proposals for a revised grading system and consensus management guidelines. Ann Oncol. 2011;22(10):2191-2200.
16. Zhu G, Lin JC, Kim SB, Bernier J, et al. Asian expert recommendation on management of skin and mucosal effects of radiation, with or without the addition of cetuximab or chemotherapy, in treatment of head and neck squamous cell carcinoma. BMC Cancer. 2016;16:42-62.
17. Pinto C, Barone CA, Girolomoni G, et al. Management of skin reactions during cetuximab treatment in association with chemotherapy or radiotherapy: update of the Italian expert recommendations. Am J Clin Oncol. 2016;39(4):407-415.
18. Eriksen JG, Kaalund I, Clemmensen O, Overgaard J, Pfeiffer P. Placebo-controlled phase II study of vitamin K3 cream for the treatment of cetuximab-induced rash. Support Care Cancer. 2017;25(7):2179-2185.
19. Watanabe S, Nakamura M, Takahashi H, et al. Dermopathy associated with cetuximab and panitumumab: investigation of the usefulness of moisturizers in its management. Clin Cosmet Investig Dermatol. 2017;10:353-361.
Epidermal growth factor receptor antibodies (EGFR) such as cetuximab have been approved for use as first-line management as well as salvage therapy for head and neck and colorectal cancers. Among the most common expected toxicity is a cutaneous eruption described as acneiform. The presence of a rash has been postulated to predict a more favorable treatment outcome for cancers of the head and neck1 but not for colorectum.2 With more severe drug reactions, patients may require a treatment break, which has been shown to reduce locoregional control and survival, particularly in patients with head and neck cancer.3 This has prompted clinicians to affect rapid therapy to reverse the drug eruption. Given the controversy around rapid and effective reversal of this drug reaction, this report aims to address the current status of clinical management using an actual patient vignette.
Case presentation and summary
The patient was a 57-year-old white man who had been diagnosed with stage 4 T4N0M1 grade 3 cutaneous squamous cell carcinoma (SCC) of the right postauricular soft tissues, with erosion into the right mastoid and biopsy-proven metastatic disease involving the contralateral left supraclavicular fossa and bilateral lungs. His disease became chemotherapy-refractory, and he was referred for palliative local therapy to the base of skull. Because of the size of the tumor (4 cm × 5 cm), he was considered for sensitizing chemotherapy, but cisplatin was not appropriate because of chronic hearing loss.4 The patient was recommended sensitizing doses of cetuximab. This EGFR antibody has been shown to offer similar benefits to those seen with cisplatin in the definitive management of head and neck SCC.5
The standard loading dose of cetuximab was given at 400 mg/m2 intravenously (IV). The following week, the sensitizing dose of 250 mg/m2 IV was given along with daily radiotherapy to the target volumes. The weekly dose of cetuximab continued at 250 mg/m2. The radiotherapy prescription was for 6,000 cGy in 200 cGy daily fractions, encompassing the gross tumor volume as identified on a computed-tomographic scan with 3-mm cuts. We used a noncoplanar arc radiotherapy beam arrangement because it inherently spreads the dose over a larger volume of normal tissue while conformally delivering its largest dose to the gross tumor volume. As such, a volume of the patient’s oropharynx and oral cavity was included within the radiotherapy dose penumbra. After receiving 3 weekly doses of cetuximab (1 loading dose and 2 weekly sensitizing doses) and 2,000 cGy of radiotherapy, the patient developed a robust grade 2 cutaneous eruption delimited to the face, with few scattered lesions on the upper anterior chest. He was seen in the medical oncology department and was prescribed doxycycline 100 mg orally twice daily and topical clindamycin 2% ointment twice daily.
In the radiation oncology clinic, his drug therapy was manipulated. His cetuximab cutaneous reaction was a grade 2, manifested by moderate erythema with nonconfluent moist desquamation. Because of concern that the patient would develop oral candida, which would further delay his therapy, the oral and topical antibiotics were discontinued, as was the oral prednisone. He was prescribed triamcinolone cream 0.1% to be applied to the facial and few chest wall areas twice daily and an oncology mouth rinse to address early nonconfluent mucositis. The accompanying images show the extent of the patient’s cetuximab cutaneous reaction at baseline before treatment initiation (Figure 1), at 4 days after the intervention (Figure 2), and again at 6 days after the intervention (Figure 3). The patient consented to having his photographs taken and understood that they would be used for educational and research publication purposes.
As can be seen from the photographs, the patient’s rash began to dry and peel by day 4 after the intervention, and there were no new eruptions. The pruritus that accompanied the rash had entirely resolved. By day 6, the rash had completely subsided. Because of the response to the topical steroid, the patient continued cetuximab without a dose modification. He was recommended to continue with the triamcinolone cream until the chemoradiotherapy course concluded.
Discussion
A cetuximab-induced rash is common. In a 2011 meta-analysis quantifying grades 1 to 4 in severity, about 75% of patients treated with an EGFR inhibitor experienced a rash. Most of the rashes were lower than grade 3, and the drug was either dose-reduced or temporarily held, but it was not generally discontinued.6 Of note is that in a nonselected survey of medical oncologists who were prescribing cetuximab, 76% reported holding the drug owing to rash severity, 60% reported dose reductions for a drug rash, and 32% reported changing the drug because of rash severity.7
In the initial pharmaceutical registration trial, 76% to 88% of patients who received cetuximab developed a rash, 17% of which were at least grade 3. The pharma recommendations for managing the drug rash include a drug delay for up to 2 weeks for a rash of grade 3 or less and to terminate use of the drug if there is no clinical improvement after 2 weeks.8 Biopsies of the rash confirm a suppurative inflammatory reaction separate from an infectious acne reaction,9 resulting in a recommendation to treat with topical steroid therapy. In some circumstances, the drug reaction can become infected or involve the paronychia, often related to Staphylococcus aureus.10 Despite what would otherwise be a problem addressed by anti-inflammatory medical therapy, the clinical appearance of the rash marked by pustules, coupled with the relative immunosuppressed state of a cancer patient, has prompted medical oncologists to prescribe antibiotic therapy.
To address the many single-institutional reports on management of the EGFR rash, several guidelines have been published. The earliest guideline – after a report that concurrent cetuximab and radiotherapy was superior to radiotherapy alone in locally advanced head and neck cancer, which documented a 23% incidence of at least grade 3 cutaneous toxicity in the cetuximab arm1 – attempted to score the severity of the rash according to the National Cancer Institute’s (NCI) Common Terminology Criteria for Adverse Events (CTCAE). Under those criteria, the authors defined grade 2 toxicity as moderate to brisk erythema with patchy moist desquamation, mostly confined to skin folds and creases. Grade 3 toxicity was described as moist desquamation other than skin folds and creases with bleeding induced by minor trauma, and grade 4 skin toxicity was defined as skin necrosis or ulceration of full thickness dermis with spontaneous bleeding from the involved site. The authors went on to describe a grade-related treatment algorithm that included gently washing the skin, keeping it dry, and using topical anti-inflammatory agents, including steroids. Antibiotics should be used in the presence of a suspected infection after culturing the area, and grade 4 toxicity should be referred to a wound care center.11
In a consensus statement from the National Comprehensive Cancer Network, the authors noted that most management recommendations were anecdotal. They recommended against the use of astringents and other drying agents because they exacerbate pain. The ultimate choice of topical steroids or antibiotics was based entirely on subjective judgement given the absence of prospective data.12
A Spanish consensus conference report argued against any prophylaxis against a skin reaction, other than keeping the skin clean and dry.13 The authors of the report recommended against washing the affected skin more than twice a day to avoid excess drying, and they advocated for moisturizers and debridement of skin crusting with hydrogels to reduce superinfection and bleeding.13 The authors also noted that some guidelines have suggested that topical steroids might exacerbate a skin rash,14 but they concluded that topical steroids are beneficial as long as they are used for less than 2 weeks. Any use of antibiotics should be based on clear evidence of an infection.13
In the first modification of the NCI’s CTCAE rash grading scale, an international panel addressed the increasing number of reports in the literature suggesting that the previous toxicity scale was possibly inadequate in its recommendations for appropriate treatment. The initial scale had defined only the skin reaction and not what therapy should be administered; therefore, in the update, the descriptions for grades 1 and 2 toxicity remained unchanged, but oral antibiotics were recommended for grade 3 lesion, and parenteral antibiotics with skin grafting were required with grade 4 toxicity.15
An Asian expert panel suggested modifying the bioradiation dermatitis scale, defining a grade 3 dermatitis as >50% moist desquamation of the involved field with formation of confluent lesions because of treatment. They recommended both topical and oral therapy, wound care, and possible hospitalization in severe cases. The panel suggested topical and systemic steroids and antibiotics.16
Finally, in an Italian consensus report, the members again modified the skin toxicity grading and were notably more aggressive in terms of their management recommendations. They defined grade 2 toxicity as pustules or papules covering 10% to 30% of the body surface area, with potential pruritus or tenderness. They also noted the psychosocial impact of skin toxicities on patients and the limits to their activities of daily living. They recommended vitamin K1 (menadione) cream, topical antibiotics, topical intermediate potency steroids, and oral antibiotic therapy for up to 4 weeks for grade 2 toxicity. Despite this aggressive treatment course, the authors admitted that the utility of topical steroids and antibiotics was unknown. They defined grade 3 toxicity as pustules or papules covering more than 30% of the body surface area, with signs of possible pruritus and tenderness. Activities of daily living and self-care were affected, and there was evidence of a superinfection. The panel suggested use of antibiotics pending culture results, oral prednisone, antihistamines, and oral analgesics. Topical therapy was not included.17 It is noteworthy that only the Italian panel recommended the use of vitamin K1 cream. In a prospective randomized, double-blinded, placebo-controlled phase 2 trial of 30 patients, menadione exhibited no clinical benefit in terms of reducing the severity of cetuximab skin lesions.18
Figure 4 illustrates our institutional approach to treating cetuximab rash based on a combination of the Spanish and NCI approaches.
The ultimate choice of therapy to manage a cetuximab rash must be patient and treatment specific. Our institutional approach, like that of the Spanish series,13 is to avoid chemoprophylaxis against a rash; rather, we recommend daily washing of the skin with a gentle soap followed by thorough rinsing and adequate, nonaggressive drying. Moisturizing the intact skin has been shown to reduce exfoliation, and we have incorporated that approach into our regimen.19
In our patient, whose head and neck radiotherapy tumor volume included a portion of the oral cavity and oropharynx, systemic antibiotic and steroid therapy would likely lead to further complications with the development of oral candidiasis. Therefore, while the severity of the reaction remained a grade 2, it seemed appropriate to treat with topical intermediate potency steroids and skin cleansing only. If the reaction had become more severe, then cultures would have been obtained to guide our decision on antibiotic therapy. Our patient’s response to topical steroids was predictable and effective, and he was able to proceed with his course of cancer therapy.
Epidermal growth factor receptor antibodies (EGFR) such as cetuximab have been approved for use as first-line management as well as salvage therapy for head and neck and colorectal cancers. Among the most common expected toxicity is a cutaneous eruption described as acneiform. The presence of a rash has been postulated to predict a more favorable treatment outcome for cancers of the head and neck1 but not for colorectum.2 With more severe drug reactions, patients may require a treatment break, which has been shown to reduce locoregional control and survival, particularly in patients with head and neck cancer.3 This has prompted clinicians to affect rapid therapy to reverse the drug eruption. Given the controversy around rapid and effective reversal of this drug reaction, this report aims to address the current status of clinical management using an actual patient vignette.
Case presentation and summary
The patient was a 57-year-old white man who had been diagnosed with stage 4 T4N0M1 grade 3 cutaneous squamous cell carcinoma (SCC) of the right postauricular soft tissues, with erosion into the right mastoid and biopsy-proven metastatic disease involving the contralateral left supraclavicular fossa and bilateral lungs. His disease became chemotherapy-refractory, and he was referred for palliative local therapy to the base of skull. Because of the size of the tumor (4 cm × 5 cm), he was considered for sensitizing chemotherapy, but cisplatin was not appropriate because of chronic hearing loss.4 The patient was recommended sensitizing doses of cetuximab. This EGFR antibody has been shown to offer similar benefits to those seen with cisplatin in the definitive management of head and neck SCC.5
The standard loading dose of cetuximab was given at 400 mg/m2 intravenously (IV). The following week, the sensitizing dose of 250 mg/m2 IV was given along with daily radiotherapy to the target volumes. The weekly dose of cetuximab continued at 250 mg/m2. The radiotherapy prescription was for 6,000 cGy in 200 cGy daily fractions, encompassing the gross tumor volume as identified on a computed-tomographic scan with 3-mm cuts. We used a noncoplanar arc radiotherapy beam arrangement because it inherently spreads the dose over a larger volume of normal tissue while conformally delivering its largest dose to the gross tumor volume. As such, a volume of the patient’s oropharynx and oral cavity was included within the radiotherapy dose penumbra. After receiving 3 weekly doses of cetuximab (1 loading dose and 2 weekly sensitizing doses) and 2,000 cGy of radiotherapy, the patient developed a robust grade 2 cutaneous eruption delimited to the face, with few scattered lesions on the upper anterior chest. He was seen in the medical oncology department and was prescribed doxycycline 100 mg orally twice daily and topical clindamycin 2% ointment twice daily.
In the radiation oncology clinic, his drug therapy was manipulated. His cetuximab cutaneous reaction was a grade 2, manifested by moderate erythema with nonconfluent moist desquamation. Because of concern that the patient would develop oral candida, which would further delay his therapy, the oral and topical antibiotics were discontinued, as was the oral prednisone. He was prescribed triamcinolone cream 0.1% to be applied to the facial and few chest wall areas twice daily and an oncology mouth rinse to address early nonconfluent mucositis. The accompanying images show the extent of the patient’s cetuximab cutaneous reaction at baseline before treatment initiation (Figure 1), at 4 days after the intervention (Figure 2), and again at 6 days after the intervention (Figure 3). The patient consented to having his photographs taken and understood that they would be used for educational and research publication purposes.
As can be seen from the photographs, the patient’s rash began to dry and peel by day 4 after the intervention, and there were no new eruptions. The pruritus that accompanied the rash had entirely resolved. By day 6, the rash had completely subsided. Because of the response to the topical steroid, the patient continued cetuximab without a dose modification. He was recommended to continue with the triamcinolone cream until the chemoradiotherapy course concluded.
Discussion
A cetuximab-induced rash is common. In a 2011 meta-analysis quantifying grades 1 to 4 in severity, about 75% of patients treated with an EGFR inhibitor experienced a rash. Most of the rashes were lower than grade 3, and the drug was either dose-reduced or temporarily held, but it was not generally discontinued.6 Of note is that in a nonselected survey of medical oncologists who were prescribing cetuximab, 76% reported holding the drug owing to rash severity, 60% reported dose reductions for a drug rash, and 32% reported changing the drug because of rash severity.7
In the initial pharmaceutical registration trial, 76% to 88% of patients who received cetuximab developed a rash, 17% of which were at least grade 3. The pharma recommendations for managing the drug rash include a drug delay for up to 2 weeks for a rash of grade 3 or less and to terminate use of the drug if there is no clinical improvement after 2 weeks.8 Biopsies of the rash confirm a suppurative inflammatory reaction separate from an infectious acne reaction,9 resulting in a recommendation to treat with topical steroid therapy. In some circumstances, the drug reaction can become infected or involve the paronychia, often related to Staphylococcus aureus.10 Despite what would otherwise be a problem addressed by anti-inflammatory medical therapy, the clinical appearance of the rash marked by pustules, coupled with the relative immunosuppressed state of a cancer patient, has prompted medical oncologists to prescribe antibiotic therapy.
To address the many single-institutional reports on management of the EGFR rash, several guidelines have been published. The earliest guideline – after a report that concurrent cetuximab and radiotherapy was superior to radiotherapy alone in locally advanced head and neck cancer, which documented a 23% incidence of at least grade 3 cutaneous toxicity in the cetuximab arm1 – attempted to score the severity of the rash according to the National Cancer Institute’s (NCI) Common Terminology Criteria for Adverse Events (CTCAE). Under those criteria, the authors defined grade 2 toxicity as moderate to brisk erythema with patchy moist desquamation, mostly confined to skin folds and creases. Grade 3 toxicity was described as moist desquamation other than skin folds and creases with bleeding induced by minor trauma, and grade 4 skin toxicity was defined as skin necrosis or ulceration of full thickness dermis with spontaneous bleeding from the involved site. The authors went on to describe a grade-related treatment algorithm that included gently washing the skin, keeping it dry, and using topical anti-inflammatory agents, including steroids. Antibiotics should be used in the presence of a suspected infection after culturing the area, and grade 4 toxicity should be referred to a wound care center.11
In a consensus statement from the National Comprehensive Cancer Network, the authors noted that most management recommendations were anecdotal. They recommended against the use of astringents and other drying agents because they exacerbate pain. The ultimate choice of topical steroids or antibiotics was based entirely on subjective judgement given the absence of prospective data.12
A Spanish consensus conference report argued against any prophylaxis against a skin reaction, other than keeping the skin clean and dry.13 The authors of the report recommended against washing the affected skin more than twice a day to avoid excess drying, and they advocated for moisturizers and debridement of skin crusting with hydrogels to reduce superinfection and bleeding.13 The authors also noted that some guidelines have suggested that topical steroids might exacerbate a skin rash,14 but they concluded that topical steroids are beneficial as long as they are used for less than 2 weeks. Any use of antibiotics should be based on clear evidence of an infection.13
In the first modification of the NCI’s CTCAE rash grading scale, an international panel addressed the increasing number of reports in the literature suggesting that the previous toxicity scale was possibly inadequate in its recommendations for appropriate treatment. The initial scale had defined only the skin reaction and not what therapy should be administered; therefore, in the update, the descriptions for grades 1 and 2 toxicity remained unchanged, but oral antibiotics were recommended for grade 3 lesion, and parenteral antibiotics with skin grafting were required with grade 4 toxicity.15
An Asian expert panel suggested modifying the bioradiation dermatitis scale, defining a grade 3 dermatitis as >50% moist desquamation of the involved field with formation of confluent lesions because of treatment. They recommended both topical and oral therapy, wound care, and possible hospitalization in severe cases. The panel suggested topical and systemic steroids and antibiotics.16
Finally, in an Italian consensus report, the members again modified the skin toxicity grading and were notably more aggressive in terms of their management recommendations. They defined grade 2 toxicity as pustules or papules covering 10% to 30% of the body surface area, with potential pruritus or tenderness. They also noted the psychosocial impact of skin toxicities on patients and the limits to their activities of daily living. They recommended vitamin K1 (menadione) cream, topical antibiotics, topical intermediate potency steroids, and oral antibiotic therapy for up to 4 weeks for grade 2 toxicity. Despite this aggressive treatment course, the authors admitted that the utility of topical steroids and antibiotics was unknown. They defined grade 3 toxicity as pustules or papules covering more than 30% of the body surface area, with signs of possible pruritus and tenderness. Activities of daily living and self-care were affected, and there was evidence of a superinfection. The panel suggested use of antibiotics pending culture results, oral prednisone, antihistamines, and oral analgesics. Topical therapy was not included.17 It is noteworthy that only the Italian panel recommended the use of vitamin K1 cream. In a prospective randomized, double-blinded, placebo-controlled phase 2 trial of 30 patients, menadione exhibited no clinical benefit in terms of reducing the severity of cetuximab skin lesions.18
Figure 4 illustrates our institutional approach to treating cetuximab rash based on a combination of the Spanish and NCI approaches.
The ultimate choice of therapy to manage a cetuximab rash must be patient and treatment specific. Our institutional approach, like that of the Spanish series,13 is to avoid chemoprophylaxis against a rash; rather, we recommend daily washing of the skin with a gentle soap followed by thorough rinsing and adequate, nonaggressive drying. Moisturizing the intact skin has been shown to reduce exfoliation, and we have incorporated that approach into our regimen.19
In our patient, whose head and neck radiotherapy tumor volume included a portion of the oral cavity and oropharynx, systemic antibiotic and steroid therapy would likely lead to further complications with the development of oral candidiasis. Therefore, while the severity of the reaction remained a grade 2, it seemed appropriate to treat with topical intermediate potency steroids and skin cleansing only. If the reaction had become more severe, then cultures would have been obtained to guide our decision on antibiotic therapy. Our patient’s response to topical steroids was predictable and effective, and he was able to proceed with his course of cancer therapy.
1. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21-28.
2. Sommeijer DW, Karapetis CS, Zalcberg JR, et al. The relationship between rash, tumor mutation KRAS status and clinical and quality of life outcomes in patients with advanced colorectal cancer treated with cetuximab in the NCIC CTG/AGITG CO.17. Acta Oncol. 2014;53(7):877-884.
3. Vahabzadeh-Hagh AM, Rwigema JM, Nabili V, Wang MB, Lorentz WC. Predictors of prolongation in radiation treatment time in a veteran population treated with chemoradiation for oropharyngeal cancer. Acta Otolaryngol. 2018;138(1):80-84.
4. Waissbluth S, Peleva E, Daniel SJ. Platinum-induced ototoxicity: a prevailing ototoxicity criteria. Eur Arch Otorhinlaryngol. 2017;274(3):1187-1196.
5. Huang J, Zhang J, Shi C, Liu L, Wei Y. Survival, recurrence and toxicity of HNSCC in comparison of a radiotherapy combination with cisplatin versus cetuximab: a meta-analysis. BMC cancer. 2016;16(1):689-713.
6. Mittman N, Seung SJ. Rash rates with EGFR inhibitors: meta-analysis. Curr Oncol. 2011;18(2):e54-e63.
7. Boone SL, Rademaker A, Liu D, Pfeiffer C, Mauro DJ, Lacouture ME. Impact and management of skin toxicity associated with anti-epidermal growth factor receptor therapy: survey results. Oncology. 2007;72(3-4):152-159.
8. Erbitux (cetuximab). Ask Lilly website. www.erbitux.com/hcp/index.html. Updated July 3, 2018. Accessed November 27.
9. Busam KJ, Capodieci P, Motzer R, Kiehn T, Phelan D, Halpern AC. Cutaneous side-effects in cancer patients treated with antiepidermal growth factor receptor antibody C225. Br J Dermatol. 2001;144(6):1169-1176.
10. Agero AL, Dusza SW, Benvenuto-Andrade C, Busam KJ, Myskowski P, Halpern AC. Dermatologic side effects associated with the epidermal growth factor receptor inhibitors. J Am Acad Dermatol. 2006;55:657-670, 2006.
11. Bernier J, Bonner J, Vermorken JB, et al. Consensus guidelines for the management of radiation dermatitis and coexisting acne-like rash in patients receiving radiotherapy plus EGFR inhibitors for the treatment of squamous cell carcinoma of the head and neck. Ann Oncol. 2008;19(1):142-149.
12. Burtness B, Anadkat M, Basti S, et al. NCCN task force report: management of dermatologic and other toxicities associated with EGFR inhibition in patients with cancer. J Natl Compr Canc Netw. 2009;7(suppl 1):S5-S21.
13.
14. Li T, Perez-Soler R. Skin toxicity associated with epidermal growth factor receptor inhibitors. Target Oncol. 2009;4(2):107-119.
15. Bernier J, Russi EG, Homey B, et al. Management of radiation dermatitis in patients receiving cetuximab and radiotherapy for locally advanced squamous cell carcinoma of the head and neck: proposals for a revised grading system and consensus management guidelines. Ann Oncol. 2011;22(10):2191-2200.
16. Zhu G, Lin JC, Kim SB, Bernier J, et al. Asian expert recommendation on management of skin and mucosal effects of radiation, with or without the addition of cetuximab or chemotherapy, in treatment of head and neck squamous cell carcinoma. BMC Cancer. 2016;16:42-62.
17. Pinto C, Barone CA, Girolomoni G, et al. Management of skin reactions during cetuximab treatment in association with chemotherapy or radiotherapy: update of the Italian expert recommendations. Am J Clin Oncol. 2016;39(4):407-415.
18. Eriksen JG, Kaalund I, Clemmensen O, Overgaard J, Pfeiffer P. Placebo-controlled phase II study of vitamin K3 cream for the treatment of cetuximab-induced rash. Support Care Cancer. 2017;25(7):2179-2185.
19. Watanabe S, Nakamura M, Takahashi H, et al. Dermopathy associated with cetuximab and panitumumab: investigation of the usefulness of moisturizers in its management. Clin Cosmet Investig Dermatol. 2017;10:353-361.
1. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21-28.
2. Sommeijer DW, Karapetis CS, Zalcberg JR, et al. The relationship between rash, tumor mutation KRAS status and clinical and quality of life outcomes in patients with advanced colorectal cancer treated with cetuximab in the NCIC CTG/AGITG CO.17. Acta Oncol. 2014;53(7):877-884.
3. Vahabzadeh-Hagh AM, Rwigema JM, Nabili V, Wang MB, Lorentz WC. Predictors of prolongation in radiation treatment time in a veteran population treated with chemoradiation for oropharyngeal cancer. Acta Otolaryngol. 2018;138(1):80-84.
4. Waissbluth S, Peleva E, Daniel SJ. Platinum-induced ototoxicity: a prevailing ototoxicity criteria. Eur Arch Otorhinlaryngol. 2017;274(3):1187-1196.
5. Huang J, Zhang J, Shi C, Liu L, Wei Y. Survival, recurrence and toxicity of HNSCC in comparison of a radiotherapy combination with cisplatin versus cetuximab: a meta-analysis. BMC cancer. 2016;16(1):689-713.
6. Mittman N, Seung SJ. Rash rates with EGFR inhibitors: meta-analysis. Curr Oncol. 2011;18(2):e54-e63.
7. Boone SL, Rademaker A, Liu D, Pfeiffer C, Mauro DJ, Lacouture ME. Impact and management of skin toxicity associated with anti-epidermal growth factor receptor therapy: survey results. Oncology. 2007;72(3-4):152-159.
8. Erbitux (cetuximab). Ask Lilly website. www.erbitux.com/hcp/index.html. Updated July 3, 2018. Accessed November 27.
9. Busam KJ, Capodieci P, Motzer R, Kiehn T, Phelan D, Halpern AC. Cutaneous side-effects in cancer patients treated with antiepidermal growth factor receptor antibody C225. Br J Dermatol. 2001;144(6):1169-1176.
10. Agero AL, Dusza SW, Benvenuto-Andrade C, Busam KJ, Myskowski P, Halpern AC. Dermatologic side effects associated with the epidermal growth factor receptor inhibitors. J Am Acad Dermatol. 2006;55:657-670, 2006.
11. Bernier J, Bonner J, Vermorken JB, et al. Consensus guidelines for the management of radiation dermatitis and coexisting acne-like rash in patients receiving radiotherapy plus EGFR inhibitors for the treatment of squamous cell carcinoma of the head and neck. Ann Oncol. 2008;19(1):142-149.
12. Burtness B, Anadkat M, Basti S, et al. NCCN task force report: management of dermatologic and other toxicities associated with EGFR inhibition in patients with cancer. J Natl Compr Canc Netw. 2009;7(suppl 1):S5-S21.
13.
14. Li T, Perez-Soler R. Skin toxicity associated with epidermal growth factor receptor inhibitors. Target Oncol. 2009;4(2):107-119.
15. Bernier J, Russi EG, Homey B, et al. Management of radiation dermatitis in patients receiving cetuximab and radiotherapy for locally advanced squamous cell carcinoma of the head and neck: proposals for a revised grading system and consensus management guidelines. Ann Oncol. 2011;22(10):2191-2200.
16. Zhu G, Lin JC, Kim SB, Bernier J, et al. Asian expert recommendation on management of skin and mucosal effects of radiation, with or without the addition of cetuximab or chemotherapy, in treatment of head and neck squamous cell carcinoma. BMC Cancer. 2016;16:42-62.
17. Pinto C, Barone CA, Girolomoni G, et al. Management of skin reactions during cetuximab treatment in association with chemotherapy or radiotherapy: update of the Italian expert recommendations. Am J Clin Oncol. 2016;39(4):407-415.
18. Eriksen JG, Kaalund I, Clemmensen O, Overgaard J, Pfeiffer P. Placebo-controlled phase II study of vitamin K3 cream for the treatment of cetuximab-induced rash. Support Care Cancer. 2017;25(7):2179-2185.
19. Watanabe S, Nakamura M, Takahashi H, et al. Dermopathy associated with cetuximab and panitumumab: investigation of the usefulness of moisturizers in its management. Clin Cosmet Investig Dermatol. 2017;10:353-361.
Incidental Asymptomatic Fibular Stress Fractures Presenting as Varus Knee Osteoarthritis: A Case Report
ABSTRACT
Stress fractures are often missed, especially in unusual clinical settings. We report on 2 patients who presented to our orthopedic surgery clinic with incidental findings of asymptomatic proximal fibular tension side stress fractures in severe longstanding varus osteoarthritic knees. Initial plain films demonstrated an expansile deformity of the proximal fibular shaft, and differential diagnosis included a healed or healing fracture versus possible neoplasm. Magnetic resonance imaging with and without gadolinium was utilized to rule out the latter prior to planned total knee arthroplasty.
Continue to: The proximal fibula...
The proximal fibula is a rare site for stress fractures, with most of these fractures occurring in military recruits.1 To the authors’ knowledge, there has been only 1 documented case of a proximal fibular stress fracture in patients with severe osteoarthritis (OA) and fixed varus deformity, which mimicked L5 radiculopathy.2 We are not aware of any reports of asymptomatic tension-side fibular stress fractures in varus knees. In our 2 cases, the patients were indicated for total knee arthroplasty (TKA) for varus degenerative joint disease after failing nonoperative treatment; however, further work-up was justified to rule out neoplasm after plain films revealed expansile deformities of the proximal fibular shaft. Each patient subsequently underwent magnetic resonance imaging (MRI) with and without gadolinium contrast, which demonstrated a healed and healing proximal fibular stress fracture. Magnetic resonance imaging is rarely indicated in the evaluation of degenerative joint disease, and stress fractures about a varus knee generally occur on the compression side of the tibia and are symptomatic.3-7 The patients provided informed written consent for print and electronic publication of this case report.

CASE REPORT
The first patient was a 77-year-old male who presented with longstanding knee pain, left greater than right, exacerbated by weight-bearing activities. The patient had no improvement with physical therapy or anti-inflammatory medication. He denied any history of trauma, weakness, paresthesias, or a recent increase in activity. The patient also denied any fevers, chills, night sweats, or other constitutional symptoms. On physical examination, the patient had an antalgic gait and limited range of motion bilaterally. Examination of his right lower extremity demonstrated a fixed 5° varus deformity. No distinct point tenderness was noted.

Radiographs of the right knee demonstrated varus deformity and tricompartmental degenerative changes with severe medial joint space narrowing. An expansile deformity of the proximal right fibular shaft was also noted (Figure 1), which was not present on the films 2 years earlier (Figure 2). The absence of this deformity on previous imaging raised the suspicion of a tumor. An MRI with and without gadolinium, which was obtained to rule out a neoplastic process, showed an old, healed proximal fibular shaft fracture with chronic periosteal reaction (Figure 3). There was no marrow edema to suggest acute injury and no neoplastic lesion. He was reassured regarding the benign findings and was scheduled for a left TKA, as his pain was more severe on the left knee. The patient’s stress fracture healed without complications, and he underwent a successful left TKA. He returned approximately 6 months after his procedure with worsening right knee pain and underwent a successful TKA on the right knee as well.

The second patient was a 67-year-old male with longstanding bilateral knee pain, right greater than left, with no antecedent trauma. He denied a history of increased activity, or weakness or paresthesias. He denied any fevers, chills, night sweats, or other constitutional symptoms. One year prior to presentation at our clinic, he had received corticosteroid injections and hyaluronic acid, without relief. The patient also had a history with another surgeon of arthroscopy 1 year earlier and subchondroplasty 3 years before presentation to our clinic. On physical examination, the patient’s right knee displayed a fixed 7° varus deformity with decreased range of motion, effusion, and diffuse crepitus. Further examination revealed tenderness to palpation of the proximal fibula.

Radiographs of the right knee showed degenerative joint disease with varus deformity and medial compartment joint space narrowing. They also demonstrated an expansile deformity of mixed lucency and sclerosis involving the proximal right fibular shaft (Figure 4). Although these findings appeared to be consistent with a stress fracture, their appearance was also suspicious for a neoplasm. To rule out malignancy, an MRI with and without gadolinium was obtained that revealed a healing stress fracture of the proximal fibula (Figure 5). The patient was reassured, and plans were made to proceed with a TKA. The patient’s stress fracture healed without complications, and he underwent successful right TKA. Radiographs from the patient’s 8-week follow-up showed a healed fibular stress fracture (Figure 6).


Continue to: DISCUSSION
DISCUSSION
To our knowledge, this is the first report of incidental tension-side stress fractures in varus osteoarthritic knees. Stress fractures have been classified into 2 groups, fatigue fractures and insufficiency fractures. Fatigue fractures occur when abnormal stress is applied to normal bones, and insufficiency fractures result when normal stress is applied to abnormal bones.8 Stress fractures can also be classified into risk categories based on which bone is involved and the loading of the bone.9 Sites loaded in tension have increased risk of nonunion, progression to complete fracture, and reoccurrence compared with sites loaded in compression.9 Stress fractures of the fibula occur rarely, and when present, they are more commonly observed in the distal fibula in athletes and military recruits.1 Stress fractures occur rarely in patients with primary OA, and when present in this setting, obesity and malalignment are the contributing factors.3 Neither patient was obese in our case (body mass index of 27 and 28, respectively), but significant varus deformity was present in both patients. Stress fractures occurring near the knee in the setting of a varus deformity generally occur on the compression side of the tibia and are symptomatic.3-7
Regarding malalignment, Cheung and colleagues10 reported about a case of an elderly female with OA of the knee with valgus deformity that initially developed a proximal fibular stress fracture followed by a proximal tibial stress fracture. However, both of our patients had varus deformities. Mullaji and Shetty3 documented stress fractures in 34 patients with OA, a majority with varus deformities, but did not report any isolated proximal fibular stress fractures. Manish and colleagues2 reported the only documented case of an isolated proximal fibular stress fracture in a patient with osteoarthritic varus deformity. The patient presented initially with pain and paresthesias of the lower thigh and leg consistent with an L5 radiculopathy. They believed that the varus deformity and the repetitive contraction of the lateral knee muscles put increased shear forces on the fibula leading to the stress fracture. Our patients did not present with any radicular symptoms, a history of acute worsening pain, or an increased activity concerning for a stress fracture. Instead, our patients presented with progressively worsening knee pain typical of severe OA and incidental findings on imaging of tension-side fibular stress fractures. An MRI with and without gadolinium confirmed the diagnosis of a healed fracture in our first patient and a healing fracture in our second patient.
CONCLUSION
Although exceedingly rare in osteoarthritic varus knees, we presented 2 cases of MRI-confirmed proximal fibular stress fractures in this report. As demonstrated, patients may present with symptoms of OA or radicular symptoms as described by Manish and colleagues.2 Presentation may also include an expansile lesion on imaging, prompting a differential diagnosis that includes a neoplasm. If present in the setting of an osteoarthritic varus knee, stress fractures of the proximal fibula should heal with conservative treatment and not affect the plan or outcome of TKA.
- Devas MB, Sweetnam R. Stress fractures of the fibula; a review of fifty cases in athletes. J Bone Joint Surg Br. 1956;38-B(4):818-829.
- Manish KK, Agnivesh T, Pramod PS, Samir SD. Isolated proximal fibular stress fracture in osteoarthritis knee presenting as L5 radiculopathy. J Orthop Case Reports. 2015;5(3):75-77. doi:10.13107/jocr.2250-0685.315.
- Mullaji A, Shetty G. Total knee arthroplasty for arthritic knees with tibiofibular stress fractures: classification and treatment guidelines. J Arthroplasty. 2010;25(2):295-301. doi:10.1016/j.arth.2008.11.012.
- Sourlas I, Papachristou G, Pilichou A, Giannoudis PV, Efstathopoulos N, Nikolaou VS. Proximal tibial stress fractures associated with primary degenerative knee osteoarthritis. Am J Orthop (Belle Mead NJ). 2009;38(3):120-124
- Demir B, Gursu S, Oke R, Ozturk K, Sahin V. Proximal tibia stress fracture caused by severe arthrosis of the knee with varus deformity. Am J Orthop (Belle Mead NJ). 2009;38(9):457-459.
- Satku K, Kumar VP, Pho RW. Stress fractures of the tibia in osteoarthritis of the knee. J Bone Joint Surg Br. 1987;69(2):309-311. doi:10.1302/0301-620X.69B2.3818767.
- Martin LM, Bourne RB, Rorabeck CH. Stress fractures associated with osteoarthritis of the knee. A report of three cases. J Bone Joint Surg Am. 1988;70(5):771-774.
- Hong SH, Chu IT. Stress fracture of the proximal fibula in military recruits. Clin Orthop Surg. 2009;1(3):161-164. doi:10.4055/cios.2009.1.3.161
- Knapik JJ, Reynolds K, Hoedebecke KL. Stress fractures: Etiology, epidemiology, diagnosis, treatment, and prevention. J Spec Oper Med. 17(2):120-130.
- Cheung MHS, Lee M-F, Lui TH. Insufficiency fracture of the proximal fibula and then tibia: A case report. J Orthop Surg. 2013;21(1):103-105. doi:10.1177/230949901302100126
ABSTRACT
Stress fractures are often missed, especially in unusual clinical settings. We report on 2 patients who presented to our orthopedic surgery clinic with incidental findings of asymptomatic proximal fibular tension side stress fractures in severe longstanding varus osteoarthritic knees. Initial plain films demonstrated an expansile deformity of the proximal fibular shaft, and differential diagnosis included a healed or healing fracture versus possible neoplasm. Magnetic resonance imaging with and without gadolinium was utilized to rule out the latter prior to planned total knee arthroplasty.
Continue to: The proximal fibula...
The proximal fibula is a rare site for stress fractures, with most of these fractures occurring in military recruits.1 To the authors’ knowledge, there has been only 1 documented case of a proximal fibular stress fracture in patients with severe osteoarthritis (OA) and fixed varus deformity, which mimicked L5 radiculopathy.2 We are not aware of any reports of asymptomatic tension-side fibular stress fractures in varus knees. In our 2 cases, the patients were indicated for total knee arthroplasty (TKA) for varus degenerative joint disease after failing nonoperative treatment; however, further work-up was justified to rule out neoplasm after plain films revealed expansile deformities of the proximal fibular shaft. Each patient subsequently underwent magnetic resonance imaging (MRI) with and without gadolinium contrast, which demonstrated a healed and healing proximal fibular stress fracture. Magnetic resonance imaging is rarely indicated in the evaluation of degenerative joint disease, and stress fractures about a varus knee generally occur on the compression side of the tibia and are symptomatic.3-7 The patients provided informed written consent for print and electronic publication of this case report.
CASE REPORT
The first patient was a 77-year-old male who presented with longstanding knee pain, left greater than right, exacerbated by weight-bearing activities. The patient had no improvement with physical therapy or anti-inflammatory medication. He denied any history of trauma, weakness, paresthesias, or a recent increase in activity. The patient also denied any fevers, chills, night sweats, or other constitutional symptoms. On physical examination, the patient had an antalgic gait and limited range of motion bilaterally. Examination of his right lower extremity demonstrated a fixed 5° varus deformity. No distinct point tenderness was noted.
Radiographs of the right knee demonstrated varus deformity and tricompartmental degenerative changes with severe medial joint space narrowing. An expansile deformity of the proximal right fibular shaft was also noted (Figure 1), which was not present on the films 2 years earlier (Figure 2). The absence of this deformity on previous imaging raised the suspicion of a tumor. An MRI with and without gadolinium, which was obtained to rule out a neoplastic process, showed an old, healed proximal fibular shaft fracture with chronic periosteal reaction (Figure 3). There was no marrow edema to suggest acute injury and no neoplastic lesion. He was reassured regarding the benign findings and was scheduled for a left TKA, as his pain was more severe on the left knee. The patient’s stress fracture healed without complications, and he underwent a successful left TKA. He returned approximately 6 months after his procedure with worsening right knee pain and underwent a successful TKA on the right knee as well.
The second patient was a 67-year-old male with longstanding bilateral knee pain, right greater than left, with no antecedent trauma. He denied a history of increased activity, or weakness or paresthesias. He denied any fevers, chills, night sweats, or other constitutional symptoms. One year prior to presentation at our clinic, he had received corticosteroid injections and hyaluronic acid, without relief. The patient also had a history with another surgeon of arthroscopy 1 year earlier and subchondroplasty 3 years before presentation to our clinic. On physical examination, the patient’s right knee displayed a fixed 7° varus deformity with decreased range of motion, effusion, and diffuse crepitus. Further examination revealed tenderness to palpation of the proximal fibula.
Radiographs of the right knee showed degenerative joint disease with varus deformity and medial compartment joint space narrowing. They also demonstrated an expansile deformity of mixed lucency and sclerosis involving the proximal right fibular shaft (Figure 4). Although these findings appeared to be consistent with a stress fracture, their appearance was also suspicious for a neoplasm. To rule out malignancy, an MRI with and without gadolinium was obtained that revealed a healing stress fracture of the proximal fibula (Figure 5). The patient was reassured, and plans were made to proceed with a TKA. The patient’s stress fracture healed without complications, and he underwent successful right TKA. Radiographs from the patient’s 8-week follow-up showed a healed fibular stress fracture (Figure 6).
Continue to: DISCUSSION
DISCUSSION
To our knowledge, this is the first report of incidental tension-side stress fractures in varus osteoarthritic knees. Stress fractures have been classified into 2 groups, fatigue fractures and insufficiency fractures. Fatigue fractures occur when abnormal stress is applied to normal bones, and insufficiency fractures result when normal stress is applied to abnormal bones.8 Stress fractures can also be classified into risk categories based on which bone is involved and the loading of the bone.9 Sites loaded in tension have increased risk of nonunion, progression to complete fracture, and reoccurrence compared with sites loaded in compression.9 Stress fractures of the fibula occur rarely, and when present, they are more commonly observed in the distal fibula in athletes and military recruits.1 Stress fractures occur rarely in patients with primary OA, and when present in this setting, obesity and malalignment are the contributing factors.3 Neither patient was obese in our case (body mass index of 27 and 28, respectively), but significant varus deformity was present in both patients. Stress fractures occurring near the knee in the setting of a varus deformity generally occur on the compression side of the tibia and are symptomatic.3-7
Regarding malalignment, Cheung and colleagues10 reported about a case of an elderly female with OA of the knee with valgus deformity that initially developed a proximal fibular stress fracture followed by a proximal tibial stress fracture. However, both of our patients had varus deformities. Mullaji and Shetty3 documented stress fractures in 34 patients with OA, a majority with varus deformities, but did not report any isolated proximal fibular stress fractures. Manish and colleagues2 reported the only documented case of an isolated proximal fibular stress fracture in a patient with osteoarthritic varus deformity. The patient presented initially with pain and paresthesias of the lower thigh and leg consistent with an L5 radiculopathy. They believed that the varus deformity and the repetitive contraction of the lateral knee muscles put increased shear forces on the fibula leading to the stress fracture. Our patients did not present with any radicular symptoms, a history of acute worsening pain, or an increased activity concerning for a stress fracture. Instead, our patients presented with progressively worsening knee pain typical of severe OA and incidental findings on imaging of tension-side fibular stress fractures. An MRI with and without gadolinium confirmed the diagnosis of a healed fracture in our first patient and a healing fracture in our second patient.
CONCLUSION
Although exceedingly rare in osteoarthritic varus knees, we presented 2 cases of MRI-confirmed proximal fibular stress fractures in this report. As demonstrated, patients may present with symptoms of OA or radicular symptoms as described by Manish and colleagues.2 Presentation may also include an expansile lesion on imaging, prompting a differential diagnosis that includes a neoplasm. If present in the setting of an osteoarthritic varus knee, stress fractures of the proximal fibula should heal with conservative treatment and not affect the plan or outcome of TKA.
ABSTRACT
Stress fractures are often missed, especially in unusual clinical settings. We report on 2 patients who presented to our orthopedic surgery clinic with incidental findings of asymptomatic proximal fibular tension side stress fractures in severe longstanding varus osteoarthritic knees. Initial plain films demonstrated an expansile deformity of the proximal fibular shaft, and differential diagnosis included a healed or healing fracture versus possible neoplasm. Magnetic resonance imaging with and without gadolinium was utilized to rule out the latter prior to planned total knee arthroplasty.
Continue to: The proximal fibula...
The proximal fibula is a rare site for stress fractures, with most of these fractures occurring in military recruits.1 To the authors’ knowledge, there has been only 1 documented case of a proximal fibular stress fracture in patients with severe osteoarthritis (OA) and fixed varus deformity, which mimicked L5 radiculopathy.2 We are not aware of any reports of asymptomatic tension-side fibular stress fractures in varus knees. In our 2 cases, the patients were indicated for total knee arthroplasty (TKA) for varus degenerative joint disease after failing nonoperative treatment; however, further work-up was justified to rule out neoplasm after plain films revealed expansile deformities of the proximal fibular shaft. Each patient subsequently underwent magnetic resonance imaging (MRI) with and without gadolinium contrast, which demonstrated a healed and healing proximal fibular stress fracture. Magnetic resonance imaging is rarely indicated in the evaluation of degenerative joint disease, and stress fractures about a varus knee generally occur on the compression side of the tibia and are symptomatic.3-7 The patients provided informed written consent for print and electronic publication of this case report.
CASE REPORT
The first patient was a 77-year-old male who presented with longstanding knee pain, left greater than right, exacerbated by weight-bearing activities. The patient had no improvement with physical therapy or anti-inflammatory medication. He denied any history of trauma, weakness, paresthesias, or a recent increase in activity. The patient also denied any fevers, chills, night sweats, or other constitutional symptoms. On physical examination, the patient had an antalgic gait and limited range of motion bilaterally. Examination of his right lower extremity demonstrated a fixed 5° varus deformity. No distinct point tenderness was noted.
Radiographs of the right knee demonstrated varus deformity and tricompartmental degenerative changes with severe medial joint space narrowing. An expansile deformity of the proximal right fibular shaft was also noted (Figure 1), which was not present on the films 2 years earlier (Figure 2). The absence of this deformity on previous imaging raised the suspicion of a tumor. An MRI with and without gadolinium, which was obtained to rule out a neoplastic process, showed an old, healed proximal fibular shaft fracture with chronic periosteal reaction (Figure 3). There was no marrow edema to suggest acute injury and no neoplastic lesion. He was reassured regarding the benign findings and was scheduled for a left TKA, as his pain was more severe on the left knee. The patient’s stress fracture healed without complications, and he underwent a successful left TKA. He returned approximately 6 months after his procedure with worsening right knee pain and underwent a successful TKA on the right knee as well.
The second patient was a 67-year-old male with longstanding bilateral knee pain, right greater than left, with no antecedent trauma. He denied a history of increased activity, or weakness or paresthesias. He denied any fevers, chills, night sweats, or other constitutional symptoms. One year prior to presentation at our clinic, he had received corticosteroid injections and hyaluronic acid, without relief. The patient also had a history with another surgeon of arthroscopy 1 year earlier and subchondroplasty 3 years before presentation to our clinic. On physical examination, the patient’s right knee displayed a fixed 7° varus deformity with decreased range of motion, effusion, and diffuse crepitus. Further examination revealed tenderness to palpation of the proximal fibula.
Radiographs of the right knee showed degenerative joint disease with varus deformity and medial compartment joint space narrowing. They also demonstrated an expansile deformity of mixed lucency and sclerosis involving the proximal right fibular shaft (Figure 4). Although these findings appeared to be consistent with a stress fracture, their appearance was also suspicious for a neoplasm. To rule out malignancy, an MRI with and without gadolinium was obtained that revealed a healing stress fracture of the proximal fibula (Figure 5). The patient was reassured, and plans were made to proceed with a TKA. The patient’s stress fracture healed without complications, and he underwent successful right TKA. Radiographs from the patient’s 8-week follow-up showed a healed fibular stress fracture (Figure 6).
Continue to: DISCUSSION
DISCUSSION
To our knowledge, this is the first report of incidental tension-side stress fractures in varus osteoarthritic knees. Stress fractures have been classified into 2 groups, fatigue fractures and insufficiency fractures. Fatigue fractures occur when abnormal stress is applied to normal bones, and insufficiency fractures result when normal stress is applied to abnormal bones.8 Stress fractures can also be classified into risk categories based on which bone is involved and the loading of the bone.9 Sites loaded in tension have increased risk of nonunion, progression to complete fracture, and reoccurrence compared with sites loaded in compression.9 Stress fractures of the fibula occur rarely, and when present, they are more commonly observed in the distal fibula in athletes and military recruits.1 Stress fractures occur rarely in patients with primary OA, and when present in this setting, obesity and malalignment are the contributing factors.3 Neither patient was obese in our case (body mass index of 27 and 28, respectively), but significant varus deformity was present in both patients. Stress fractures occurring near the knee in the setting of a varus deformity generally occur on the compression side of the tibia and are symptomatic.3-7
Regarding malalignment, Cheung and colleagues10 reported about a case of an elderly female with OA of the knee with valgus deformity that initially developed a proximal fibular stress fracture followed by a proximal tibial stress fracture. However, both of our patients had varus deformities. Mullaji and Shetty3 documented stress fractures in 34 patients with OA, a majority with varus deformities, but did not report any isolated proximal fibular stress fractures. Manish and colleagues2 reported the only documented case of an isolated proximal fibular stress fracture in a patient with osteoarthritic varus deformity. The patient presented initially with pain and paresthesias of the lower thigh and leg consistent with an L5 radiculopathy. They believed that the varus deformity and the repetitive contraction of the lateral knee muscles put increased shear forces on the fibula leading to the stress fracture. Our patients did not present with any radicular symptoms, a history of acute worsening pain, or an increased activity concerning for a stress fracture. Instead, our patients presented with progressively worsening knee pain typical of severe OA and incidental findings on imaging of tension-side fibular stress fractures. An MRI with and without gadolinium confirmed the diagnosis of a healed fracture in our first patient and a healing fracture in our second patient.
CONCLUSION
Although exceedingly rare in osteoarthritic varus knees, we presented 2 cases of MRI-confirmed proximal fibular stress fractures in this report. As demonstrated, patients may present with symptoms of OA or radicular symptoms as described by Manish and colleagues.2 Presentation may also include an expansile lesion on imaging, prompting a differential diagnosis that includes a neoplasm. If present in the setting of an osteoarthritic varus knee, stress fractures of the proximal fibula should heal with conservative treatment and not affect the plan or outcome of TKA.
- Devas MB, Sweetnam R. Stress fractures of the fibula; a review of fifty cases in athletes. J Bone Joint Surg Br. 1956;38-B(4):818-829.
- Manish KK, Agnivesh T, Pramod PS, Samir SD. Isolated proximal fibular stress fracture in osteoarthritis knee presenting as L5 radiculopathy. J Orthop Case Reports. 2015;5(3):75-77. doi:10.13107/jocr.2250-0685.315.
- Mullaji A, Shetty G. Total knee arthroplasty for arthritic knees with tibiofibular stress fractures: classification and treatment guidelines. J Arthroplasty. 2010;25(2):295-301. doi:10.1016/j.arth.2008.11.012.
- Sourlas I, Papachristou G, Pilichou A, Giannoudis PV, Efstathopoulos N, Nikolaou VS. Proximal tibial stress fractures associated with primary degenerative knee osteoarthritis. Am J Orthop (Belle Mead NJ). 2009;38(3):120-124
- Demir B, Gursu S, Oke R, Ozturk K, Sahin V. Proximal tibia stress fracture caused by severe arthrosis of the knee with varus deformity. Am J Orthop (Belle Mead NJ). 2009;38(9):457-459.
- Satku K, Kumar VP, Pho RW. Stress fractures of the tibia in osteoarthritis of the knee. J Bone Joint Surg Br. 1987;69(2):309-311. doi:10.1302/0301-620X.69B2.3818767.
- Martin LM, Bourne RB, Rorabeck CH. Stress fractures associated with osteoarthritis of the knee. A report of three cases. J Bone Joint Surg Am. 1988;70(5):771-774.
- Hong SH, Chu IT. Stress fracture of the proximal fibula in military recruits. Clin Orthop Surg. 2009;1(3):161-164. doi:10.4055/cios.2009.1.3.161
- Knapik JJ, Reynolds K, Hoedebecke KL. Stress fractures: Etiology, epidemiology, diagnosis, treatment, and prevention. J Spec Oper Med. 17(2):120-130.
- Cheung MHS, Lee M-F, Lui TH. Insufficiency fracture of the proximal fibula and then tibia: A case report. J Orthop Surg. 2013;21(1):103-105. doi:10.1177/230949901302100126
- Devas MB, Sweetnam R. Stress fractures of the fibula; a review of fifty cases in athletes. J Bone Joint Surg Br. 1956;38-B(4):818-829.
- Manish KK, Agnivesh T, Pramod PS, Samir SD. Isolated proximal fibular stress fracture in osteoarthritis knee presenting as L5 radiculopathy. J Orthop Case Reports. 2015;5(3):75-77. doi:10.13107/jocr.2250-0685.315.
- Mullaji A, Shetty G. Total knee arthroplasty for arthritic knees with tibiofibular stress fractures: classification and treatment guidelines. J Arthroplasty. 2010;25(2):295-301. doi:10.1016/j.arth.2008.11.012.
- Sourlas I, Papachristou G, Pilichou A, Giannoudis PV, Efstathopoulos N, Nikolaou VS. Proximal tibial stress fractures associated with primary degenerative knee osteoarthritis. Am J Orthop (Belle Mead NJ). 2009;38(3):120-124
- Demir B, Gursu S, Oke R, Ozturk K, Sahin V. Proximal tibia stress fracture caused by severe arthrosis of the knee with varus deformity. Am J Orthop (Belle Mead NJ). 2009;38(9):457-459.
- Satku K, Kumar VP, Pho RW. Stress fractures of the tibia in osteoarthritis of the knee. J Bone Joint Surg Br. 1987;69(2):309-311. doi:10.1302/0301-620X.69B2.3818767.
- Martin LM, Bourne RB, Rorabeck CH. Stress fractures associated with osteoarthritis of the knee. A report of three cases. J Bone Joint Surg Am. 1988;70(5):771-774.
- Hong SH, Chu IT. Stress fracture of the proximal fibula in military recruits. Clin Orthop Surg. 2009;1(3):161-164. doi:10.4055/cios.2009.1.3.161
- Knapik JJ, Reynolds K, Hoedebecke KL. Stress fractures: Etiology, epidemiology, diagnosis, treatment, and prevention. J Spec Oper Med. 17(2):120-130.
- Cheung MHS, Lee M-F, Lui TH. Insufficiency fracture of the proximal fibula and then tibia: A case report. J Orthop Surg. 2013;21(1):103-105. doi:10.1177/230949901302100126
TAKE-HOME POINTS
- Proximal fibular stress fractures in patients with primary osteoarthritis and fixed varus deformity have rarely been reported.
- Stress fractures occurring near the knee in the setting of a varus deformity generally occur on the compression side of the tibia and are symptomatic.
- Proximal fibular stress fractures may present as an incidental finding of an expansile deformity on plain films in patients with varus osteoarthritic knees.
- Magnetic resonance imaging is rarely indicated in the evaluation of degenerative joint disease; however, it was justified in our case to rule out neoplasm.
- When present in the setting of an osteoarthritic varus knee, stress fractures of the proximal fibula should heal with conservative treatment and should not affect the plan or outcome of TKA.
Ice Pack–Induced Perniosis: A Rare and Underrecognized Association
Perniosis, or chilblain, is characterized by localized, tender, erythematous skin lesions that occur as an abnormal reaction to exposure to cold and damp conditions. Although the lesions favor the distal extremities, perniosis may present anywhere on the body. Lesions can develop within hours to days following exposure to temperature less than 10°C or damp environments with greater than 60% humidity.1 Acute cases may lead to pruritus and tenderness, whereas chronic cases may involve lesions that blister or ulcerate and can take weeks to heal. We report an unusual case of erythematous plaques arising on the buttocks of a 73-year-old woman using ice pack treatments for chronic low back pain.
Case Report
A 73-year-old woman presented with recurrent tender lesions on the buttocks of 5 years’ duration. Her medical history was remarkable for hypertension, hypothyroidism, and lumbar spinal fusion surgery 5 years prior. Physical examination revealed indurated erythematous plaques with areas of erosions on the left buttock with some involvement of the right buttock (Figure 1).
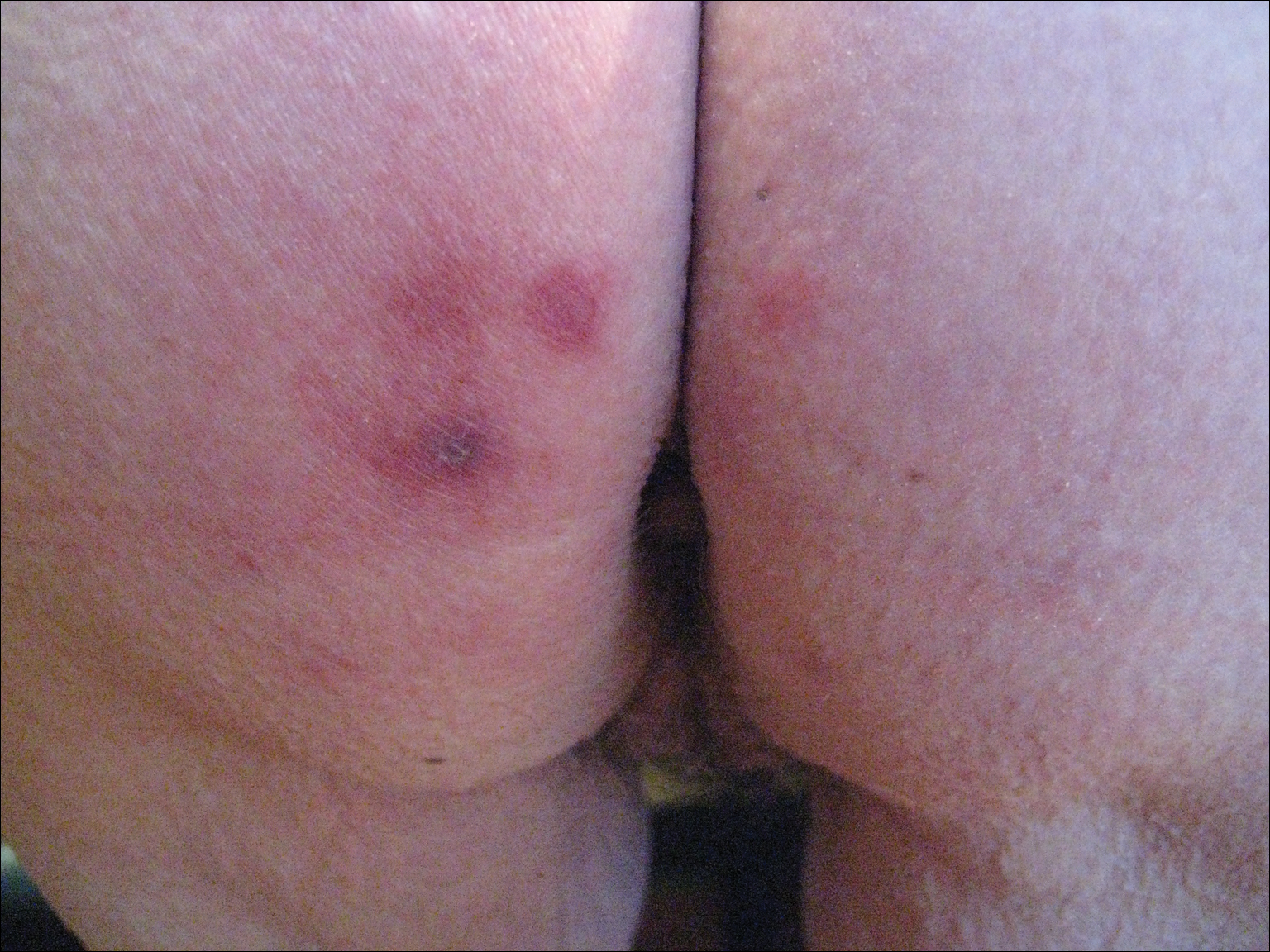
After a trial of oral valacyclovir for presumed herpes simplex infection provided no relief, a punch biopsy of the left buttock was performed, which revealed a cell-poor interface dermatitis with superficial and deep perivascular and periadnexal lymphocytic infiltrates (Figure 2). Perieccrine lymphocytes were present in a small portion of the reticular dermis (Figure 3). The patient revealed she had been sitting on ice packs for several hours daily since the lumbar spinal fusion surgery 5 years prior to alleviate chronic low back pain.
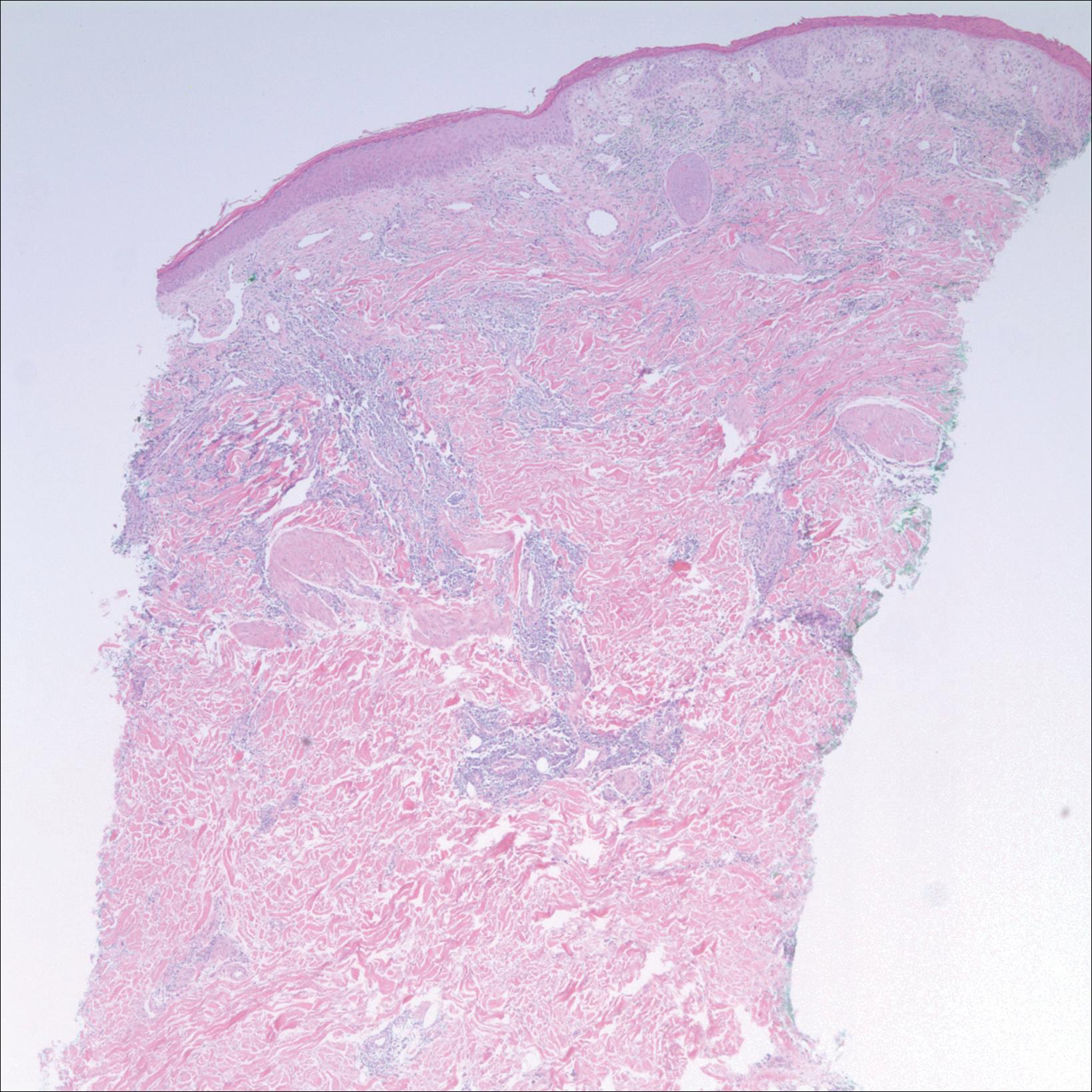
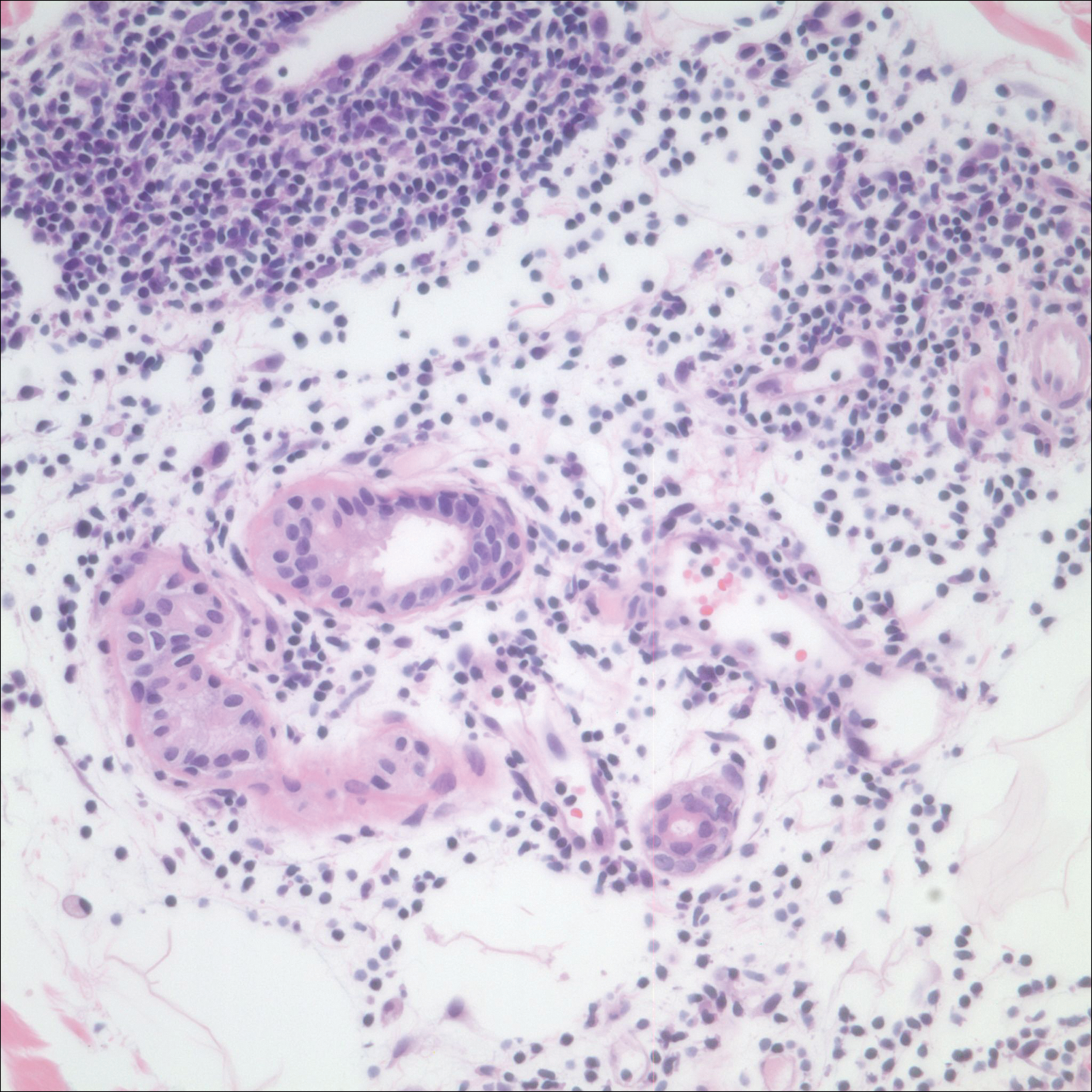
Based on the clinicopathologic correlation, a diagnosis of perniosis secondary to ice pack therapy was made. An evaluation for concomitant or underlying connective tissue disease (CTD) including a complete blood cell count with sedimentation rate, antinuclear antibodies (ANAs), serum protein electrophoresis, and serum levels of cryoglobulins and complement components was unremarkable. Our patient was treated with simple analgesia and was encouraged to avoid direct contact with ice packs for extended periods of time. Because of her low back pain, she continued to use ice packs but readjusted them sporadically and decreased frequency of use. She had complete resolution of the lesions at 6-month follow-up.
Comment
Perniosis is a self-limited condition, manifesting as erythematous plaques or nodules following exposure to cold and damp conditions. It was first reported in 1902 by Hochsinger2 as tender submental plaques occurring in children after exposure to cold weather. Since then, reports of perniosis have been described in equestrians and long-distance cyclists as well as in the context of other outdoor activities.3-5 In all cases, patients developed perniosis at sites of exposure to cold or damp conditions.
Perniosis arising in patients using ice pack therapy is a rare and recent phenomenon, with only 3 other known reported cases.6,7 In all cases, including ours, patients reported treating chronic low back pain with ice packs for more than 2 hours per day. Clinical presentations included erythematous to purpuric plaques with ulceration on the lower back or buttocks that reoccurred with subsequent use of ice packs. No concomitant CTD was reported.6
Much controversy exists as to whether idiopathic perniosis (IP) increases susceptibility to acquiring an autoimmune disease or if IP is a form of CTD that follows a more indolent course.8 In a prospective study of 33 patients with underlying IP, no patients developed lupus erythematosus (LE), with a median follow-up of 38 months.9 A study by Crowson and Magro8 revealed that 18 of 39 patients with perniotic lesions had an associated systemic disease including LE, human immunodeficiency virus, viral hepatitis, rheumatoid arthritis, cryofibrinogenemia, hypergammaglobulinemia, iritis, or Crohn disease. Of the 21 other patients who had no underlying CTD or systemic disease, 10 had a positive ANA test but no systemic symptoms; therefore, all 21 of these patients were classified as cases of IP.8
Cutaneous biopsy to distinguish between IP and autoimmune perniosis remains controversial; perniotic lesions and discoid LE share histopathologic features,9 as was evident with our case, which demonstrated overlapping findings of vacuolar change with superficial and deep perivascular and periadnexal lymphoid infiltrates. Typical features of IP include thrombosed capillaries in the papillary dermis and lymphocytic exocytosis localized to the acrosyringia, whereas secondary perniosis has superficial and deep perivascular and perieccrine lymphocytic infiltrates with vascular thrombosis in the reticular dermis. Vascular ectasia, dermal mucinosis, basement membrane zone thickening, and erythrocyte extravasation are not reliable and may be seen in both cases.8 One study revealed the only significant difference between both entities was the perieccrine distribution of lymphocytic infiltrate in cases of IP (P=.007), whereas an absence of perieccrine involvement was noted in autoimmune cases.9
Direct immunofluorescence (DIF) may help differentiate IP from autoimmune perniosis. In a prospective study by Viguier et al,9 6 of 9 patients with IP had negative DIF and 3 had slight nonspecific C3 immunoreactivity of dermal vessels. Conversely, in patients with autoimmune perniosis, positive DIF with the lupus band test was seen in 3 of 7 patients, all who had a positive ANA test9; however, positive ANA levels also were reported in patients with autoimmune perniosis but negative DIF, suggesting that DIF lacks specificity in diagnosing autoimmune perniosis.
Although histopathologic findings bear similarities to LE, there are no guidelines to suggest for or against laboratory testing for CTD in patients presenting with perniosis. Some investigators have suggested that any patient with clinical features suggestive of perniosis should undergo laboratory evaluation including a complete blood cell count and assessment for antibodies to Ro, ANA, rheumatoid factor, cryofibrinogens, and antiphospholipid antibodies.9 Serum protein electrophoresis and immunofixation electrophoresis may be done to exclude monoclonal gammopathy.
For idiopathic cases, treatment is aimed at limiting or removing cold exposure. Patients should be advised regarding the use of long-term ice pack use and the potential development of perniosis. For chronic perniosis lasting beyond several weeks, a combination of a slow taper of oral prednisone, hydroxychloroquine, and quinacrine has been successful in patients with persistent lesions despite making environmental modifications.3 Intralesional triamcinolone acetonide and nifedipine also have been effective in perniotic hand lesions.10
Conclusion
We report a rare case of perniosis on the buttocks that arose in a patient who utilized ice packs for treatment of chronic low back pain. Ice pack–induced perniosis may be an underreported entity. Histopathologic examination is nondescript, as overlapping features of perniosis and LE have been observed with no underlying CTD present. Correlation with patient history and clinical examination is paramount in diagnosis and management.
- Praminik T, Jha AK, Ghimire A. A retrospective study of cases with chilblains (perniosis) in Out Patient Department of Dermatology, Nepal Medical College and Teaching Hospital (NMCTH). Nepal Med Coll J. 2011;13:190-192.
- Hochsinger C. Acute perniosis in submental region of child [in German]. Monatsschr Kinderheilkd. 1902;1:323-327.
- Stewart CL, Adler DJ, Jacobson A, et al. Equestrian perniosis: a report of 2 cases and a review of the literature. Am J Dermatopathol. 2013;35:237-240.
- Neal AJ, Jarman AM, Bennett TG. Perniosis in a long-distance cyclist crossing Mongolia. J Travel Med. 2012;19:66-68.
- Price RD, Murdoch DR. Perniosis (chilblains) of the thigh: report of five cases including four following river crossings. High Alt Met Biol. 2001;2:535-538.
- West SA, McCalmont TH, North JP. Ice-pack dermatosis: a cold-induced dermatitis with similarities to cold panniculitis and perniosis that histopathologically resembles lupus. JAMA Dermatol. 2013;149:1314-1318.
- Haber JS, Ker KJ, Werth VP, et al. Ice‐pack dermatosis: a diagnositic pitfall for dermatopathologists that mimics lupus erythematosus. J Cutan Pathol. 2016;43:1-4.
- Crowson AN, Magro CM. Idiopathic perniosis and its mimics: a clinical and histological study of 38 cases. Hum Pathol. 1997;28:478-484.
- Viguier M, Pinguier L, Cavelier-Balloy B, et al. Clinical and histopathologic features and immunologic variables in patients with severe chilblains. a study of the relationship to lupus erythematosus. Medicine. 2001;80:180-188.
- Patra AK, Das AL, Ramadasan P. Diltiazem vs. nifedipine in chilblains: a clinical trial. Indian J Dermatol Venereol Leprol. 2003;69:209-211.
Perniosis, or chilblain, is characterized by localized, tender, erythematous skin lesions that occur as an abnormal reaction to exposure to cold and damp conditions. Although the lesions favor the distal extremities, perniosis may present anywhere on the body. Lesions can develop within hours to days following exposure to temperature less than 10°C or damp environments with greater than 60% humidity.1 Acute cases may lead to pruritus and tenderness, whereas chronic cases may involve lesions that blister or ulcerate and can take weeks to heal. We report an unusual case of erythematous plaques arising on the buttocks of a 73-year-old woman using ice pack treatments for chronic low back pain.
Case Report
A 73-year-old woman presented with recurrent tender lesions on the buttocks of 5 years’ duration. Her medical history was remarkable for hypertension, hypothyroidism, and lumbar spinal fusion surgery 5 years prior. Physical examination revealed indurated erythematous plaques with areas of erosions on the left buttock with some involvement of the right buttock (Figure 1).
After a trial of oral valacyclovir for presumed herpes simplex infection provided no relief, a punch biopsy of the left buttock was performed, which revealed a cell-poor interface dermatitis with superficial and deep perivascular and periadnexal lymphocytic infiltrates (Figure 2). Perieccrine lymphocytes were present in a small portion of the reticular dermis (Figure 3). The patient revealed she had been sitting on ice packs for several hours daily since the lumbar spinal fusion surgery 5 years prior to alleviate chronic low back pain.
Based on the clinicopathologic correlation, a diagnosis of perniosis secondary to ice pack therapy was made. An evaluation for concomitant or underlying connective tissue disease (CTD) including a complete blood cell count with sedimentation rate, antinuclear antibodies (ANAs), serum protein electrophoresis, and serum levels of cryoglobulins and complement components was unremarkable. Our patient was treated with simple analgesia and was encouraged to avoid direct contact with ice packs for extended periods of time. Because of her low back pain, she continued to use ice packs but readjusted them sporadically and decreased frequency of use. She had complete resolution of the lesions at 6-month follow-up.
Comment
Perniosis is a self-limited condition, manifesting as erythematous plaques or nodules following exposure to cold and damp conditions. It was first reported in 1902 by Hochsinger2 as tender submental plaques occurring in children after exposure to cold weather. Since then, reports of perniosis have been described in equestrians and long-distance cyclists as well as in the context of other outdoor activities.3-5 In all cases, patients developed perniosis at sites of exposure to cold or damp conditions.
Perniosis arising in patients using ice pack therapy is a rare and recent phenomenon, with only 3 other known reported cases.6,7 In all cases, including ours, patients reported treating chronic low back pain with ice packs for more than 2 hours per day. Clinical presentations included erythematous to purpuric plaques with ulceration on the lower back or buttocks that reoccurred with subsequent use of ice packs. No concomitant CTD was reported.6
Much controversy exists as to whether idiopathic perniosis (IP) increases susceptibility to acquiring an autoimmune disease or if IP is a form of CTD that follows a more indolent course.8 In a prospective study of 33 patients with underlying IP, no patients developed lupus erythematosus (LE), with a median follow-up of 38 months.9 A study by Crowson and Magro8 revealed that 18 of 39 patients with perniotic lesions had an associated systemic disease including LE, human immunodeficiency virus, viral hepatitis, rheumatoid arthritis, cryofibrinogenemia, hypergammaglobulinemia, iritis, or Crohn disease. Of the 21 other patients who had no underlying CTD or systemic disease, 10 had a positive ANA test but no systemic symptoms; therefore, all 21 of these patients were classified as cases of IP.8
Cutaneous biopsy to distinguish between IP and autoimmune perniosis remains controversial; perniotic lesions and discoid LE share histopathologic features,9 as was evident with our case, which demonstrated overlapping findings of vacuolar change with superficial and deep perivascular and periadnexal lymphoid infiltrates. Typical features of IP include thrombosed capillaries in the papillary dermis and lymphocytic exocytosis localized to the acrosyringia, whereas secondary perniosis has superficial and deep perivascular and perieccrine lymphocytic infiltrates with vascular thrombosis in the reticular dermis. Vascular ectasia, dermal mucinosis, basement membrane zone thickening, and erythrocyte extravasation are not reliable and may be seen in both cases.8 One study revealed the only significant difference between both entities was the perieccrine distribution of lymphocytic infiltrate in cases of IP (P=.007), whereas an absence of perieccrine involvement was noted in autoimmune cases.9
Direct immunofluorescence (DIF) may help differentiate IP from autoimmune perniosis. In a prospective study by Viguier et al,9 6 of 9 patients with IP had negative DIF and 3 had slight nonspecific C3 immunoreactivity of dermal vessels. Conversely, in patients with autoimmune perniosis, positive DIF with the lupus band test was seen in 3 of 7 patients, all who had a positive ANA test9; however, positive ANA levels also were reported in patients with autoimmune perniosis but negative DIF, suggesting that DIF lacks specificity in diagnosing autoimmune perniosis.
Although histopathologic findings bear similarities to LE, there are no guidelines to suggest for or against laboratory testing for CTD in patients presenting with perniosis. Some investigators have suggested that any patient with clinical features suggestive of perniosis should undergo laboratory evaluation including a complete blood cell count and assessment for antibodies to Ro, ANA, rheumatoid factor, cryofibrinogens, and antiphospholipid antibodies.9 Serum protein electrophoresis and immunofixation electrophoresis may be done to exclude monoclonal gammopathy.
For idiopathic cases, treatment is aimed at limiting or removing cold exposure. Patients should be advised regarding the use of long-term ice pack use and the potential development of perniosis. For chronic perniosis lasting beyond several weeks, a combination of a slow taper of oral prednisone, hydroxychloroquine, and quinacrine has been successful in patients with persistent lesions despite making environmental modifications.3 Intralesional triamcinolone acetonide and nifedipine also have been effective in perniotic hand lesions.10
Conclusion
We report a rare case of perniosis on the buttocks that arose in a patient who utilized ice packs for treatment of chronic low back pain. Ice pack–induced perniosis may be an underreported entity. Histopathologic examination is nondescript, as overlapping features of perniosis and LE have been observed with no underlying CTD present. Correlation with patient history and clinical examination is paramount in diagnosis and management.
Perniosis, or chilblain, is characterized by localized, tender, erythematous skin lesions that occur as an abnormal reaction to exposure to cold and damp conditions. Although the lesions favor the distal extremities, perniosis may present anywhere on the body. Lesions can develop within hours to days following exposure to temperature less than 10°C or damp environments with greater than 60% humidity.1 Acute cases may lead to pruritus and tenderness, whereas chronic cases may involve lesions that blister or ulcerate and can take weeks to heal. We report an unusual case of erythematous plaques arising on the buttocks of a 73-year-old woman using ice pack treatments for chronic low back pain.
Case Report
A 73-year-old woman presented with recurrent tender lesions on the buttocks of 5 years’ duration. Her medical history was remarkable for hypertension, hypothyroidism, and lumbar spinal fusion surgery 5 years prior. Physical examination revealed indurated erythematous plaques with areas of erosions on the left buttock with some involvement of the right buttock (Figure 1).
After a trial of oral valacyclovir for presumed herpes simplex infection provided no relief, a punch biopsy of the left buttock was performed, which revealed a cell-poor interface dermatitis with superficial and deep perivascular and periadnexal lymphocytic infiltrates (Figure 2). Perieccrine lymphocytes were present in a small portion of the reticular dermis (Figure 3). The patient revealed she had been sitting on ice packs for several hours daily since the lumbar spinal fusion surgery 5 years prior to alleviate chronic low back pain.
Based on the clinicopathologic correlation, a diagnosis of perniosis secondary to ice pack therapy was made. An evaluation for concomitant or underlying connective tissue disease (CTD) including a complete blood cell count with sedimentation rate, antinuclear antibodies (ANAs), serum protein electrophoresis, and serum levels of cryoglobulins and complement components was unremarkable. Our patient was treated with simple analgesia and was encouraged to avoid direct contact with ice packs for extended periods of time. Because of her low back pain, she continued to use ice packs but readjusted them sporadically and decreased frequency of use. She had complete resolution of the lesions at 6-month follow-up.
Comment
Perniosis is a self-limited condition, manifesting as erythematous plaques or nodules following exposure to cold and damp conditions. It was first reported in 1902 by Hochsinger2 as tender submental plaques occurring in children after exposure to cold weather. Since then, reports of perniosis have been described in equestrians and long-distance cyclists as well as in the context of other outdoor activities.3-5 In all cases, patients developed perniosis at sites of exposure to cold or damp conditions.
Perniosis arising in patients using ice pack therapy is a rare and recent phenomenon, with only 3 other known reported cases.6,7 In all cases, including ours, patients reported treating chronic low back pain with ice packs for more than 2 hours per day. Clinical presentations included erythematous to purpuric plaques with ulceration on the lower back or buttocks that reoccurred with subsequent use of ice packs. No concomitant CTD was reported.6
Much controversy exists as to whether idiopathic perniosis (IP) increases susceptibility to acquiring an autoimmune disease or if IP is a form of CTD that follows a more indolent course.8 In a prospective study of 33 patients with underlying IP, no patients developed lupus erythematosus (LE), with a median follow-up of 38 months.9 A study by Crowson and Magro8 revealed that 18 of 39 patients with perniotic lesions had an associated systemic disease including LE, human immunodeficiency virus, viral hepatitis, rheumatoid arthritis, cryofibrinogenemia, hypergammaglobulinemia, iritis, or Crohn disease. Of the 21 other patients who had no underlying CTD or systemic disease, 10 had a positive ANA test but no systemic symptoms; therefore, all 21 of these patients were classified as cases of IP.8
Cutaneous biopsy to distinguish between IP and autoimmune perniosis remains controversial; perniotic lesions and discoid LE share histopathologic features,9 as was evident with our case, which demonstrated overlapping findings of vacuolar change with superficial and deep perivascular and periadnexal lymphoid infiltrates. Typical features of IP include thrombosed capillaries in the papillary dermis and lymphocytic exocytosis localized to the acrosyringia, whereas secondary perniosis has superficial and deep perivascular and perieccrine lymphocytic infiltrates with vascular thrombosis in the reticular dermis. Vascular ectasia, dermal mucinosis, basement membrane zone thickening, and erythrocyte extravasation are not reliable and may be seen in both cases.8 One study revealed the only significant difference between both entities was the perieccrine distribution of lymphocytic infiltrate in cases of IP (P=.007), whereas an absence of perieccrine involvement was noted in autoimmune cases.9
Direct immunofluorescence (DIF) may help differentiate IP from autoimmune perniosis. In a prospective study by Viguier et al,9 6 of 9 patients with IP had negative DIF and 3 had slight nonspecific C3 immunoreactivity of dermal vessels. Conversely, in patients with autoimmune perniosis, positive DIF with the lupus band test was seen in 3 of 7 patients, all who had a positive ANA test9; however, positive ANA levels also were reported in patients with autoimmune perniosis but negative DIF, suggesting that DIF lacks specificity in diagnosing autoimmune perniosis.
Although histopathologic findings bear similarities to LE, there are no guidelines to suggest for or against laboratory testing for CTD in patients presenting with perniosis. Some investigators have suggested that any patient with clinical features suggestive of perniosis should undergo laboratory evaluation including a complete blood cell count and assessment for antibodies to Ro, ANA, rheumatoid factor, cryofibrinogens, and antiphospholipid antibodies.9 Serum protein electrophoresis and immunofixation electrophoresis may be done to exclude monoclonal gammopathy.
For idiopathic cases, treatment is aimed at limiting or removing cold exposure. Patients should be advised regarding the use of long-term ice pack use and the potential development of perniosis. For chronic perniosis lasting beyond several weeks, a combination of a slow taper of oral prednisone, hydroxychloroquine, and quinacrine has been successful in patients with persistent lesions despite making environmental modifications.3 Intralesional triamcinolone acetonide and nifedipine also have been effective in perniotic hand lesions.10
Conclusion
We report a rare case of perniosis on the buttocks that arose in a patient who utilized ice packs for treatment of chronic low back pain. Ice pack–induced perniosis may be an underreported entity. Histopathologic examination is nondescript, as overlapping features of perniosis and LE have been observed with no underlying CTD present. Correlation with patient history and clinical examination is paramount in diagnosis and management.
- Praminik T, Jha AK, Ghimire A. A retrospective study of cases with chilblains (perniosis) in Out Patient Department of Dermatology, Nepal Medical College and Teaching Hospital (NMCTH). Nepal Med Coll J. 2011;13:190-192.
- Hochsinger C. Acute perniosis in submental region of child [in German]. Monatsschr Kinderheilkd. 1902;1:323-327.
- Stewart CL, Adler DJ, Jacobson A, et al. Equestrian perniosis: a report of 2 cases and a review of the literature. Am J Dermatopathol. 2013;35:237-240.
- Neal AJ, Jarman AM, Bennett TG. Perniosis in a long-distance cyclist crossing Mongolia. J Travel Med. 2012;19:66-68.
- Price RD, Murdoch DR. Perniosis (chilblains) of the thigh: report of five cases including four following river crossings. High Alt Met Biol. 2001;2:535-538.
- West SA, McCalmont TH, North JP. Ice-pack dermatosis: a cold-induced dermatitis with similarities to cold panniculitis and perniosis that histopathologically resembles lupus. JAMA Dermatol. 2013;149:1314-1318.
- Haber JS, Ker KJ, Werth VP, et al. Ice‐pack dermatosis: a diagnositic pitfall for dermatopathologists that mimics lupus erythematosus. J Cutan Pathol. 2016;43:1-4.
- Crowson AN, Magro CM. Idiopathic perniosis and its mimics: a clinical and histological study of 38 cases. Hum Pathol. 1997;28:478-484.
- Viguier M, Pinguier L, Cavelier-Balloy B, et al. Clinical and histopathologic features and immunologic variables in patients with severe chilblains. a study of the relationship to lupus erythematosus. Medicine. 2001;80:180-188.
- Patra AK, Das AL, Ramadasan P. Diltiazem vs. nifedipine in chilblains: a clinical trial. Indian J Dermatol Venereol Leprol. 2003;69:209-211.
- Praminik T, Jha AK, Ghimire A. A retrospective study of cases with chilblains (perniosis) in Out Patient Department of Dermatology, Nepal Medical College and Teaching Hospital (NMCTH). Nepal Med Coll J. 2011;13:190-192.
- Hochsinger C. Acute perniosis in submental region of child [in German]. Monatsschr Kinderheilkd. 1902;1:323-327.
- Stewart CL, Adler DJ, Jacobson A, et al. Equestrian perniosis: a report of 2 cases and a review of the literature. Am J Dermatopathol. 2013;35:237-240.
- Neal AJ, Jarman AM, Bennett TG. Perniosis in a long-distance cyclist crossing Mongolia. J Travel Med. 2012;19:66-68.
- Price RD, Murdoch DR. Perniosis (chilblains) of the thigh: report of five cases including four following river crossings. High Alt Met Biol. 2001;2:535-538.
- West SA, McCalmont TH, North JP. Ice-pack dermatosis: a cold-induced dermatitis with similarities to cold panniculitis and perniosis that histopathologically resembles lupus. JAMA Dermatol. 2013;149:1314-1318.
- Haber JS, Ker KJ, Werth VP, et al. Ice‐pack dermatosis: a diagnositic pitfall for dermatopathologists that mimics lupus erythematosus. J Cutan Pathol. 2016;43:1-4.
- Crowson AN, Magro CM. Idiopathic perniosis and its mimics: a clinical and histological study of 38 cases. Hum Pathol. 1997;28:478-484.
- Viguier M, Pinguier L, Cavelier-Balloy B, et al. Clinical and histopathologic features and immunologic variables in patients with severe chilblains. a study of the relationship to lupus erythematosus. Medicine. 2001;80:180-188.
- Patra AK, Das AL, Ramadasan P. Diltiazem vs. nifedipine in chilblains: a clinical trial. Indian J Dermatol Venereol Leprol. 2003;69:209-211.
Practice Points
- Ice pack-induced perniosis is a rare condition that can occur in patients using long-term ice pack therapy.
- This entity histopathologically mimics cutaneous lupus erythematosus and can present a diagnostic challenge.
- A thorough clinical history and awareness of this diagnosis is essential for diagnostic accuracy.
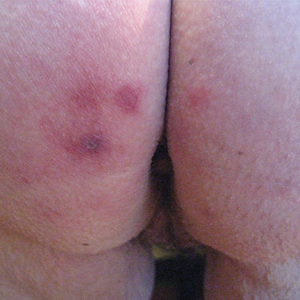
6-day history of fever • groin pain and swelling • recent hiking trip in Colorado • Dx?
THE CASE
A 33-year-old Caucasian woman presented to the emergency department with a 6-day history of fever (103°-104°F) and right groin pain and swelling. Associated symptoms included headache, diarrhea, malaise, weakness, nausea, cough, and anorexia. Upon presentation, she admitted to a recent hike on a bubonic plague–endemic trail in Colorado.
Her vital signs were unremarkable, and the physical examination demonstrated normal findings except for tender, erythematous, nonfluctuant right inguinal lymphadenopathy. The patient was admitted for intractable pain and fever and started on intravenous cefoxitin 2 g IV every 8 hours and oral doxycycline 100 mg every 12 hours for pelvic inflammatory disease vs tick- or flea-borne illness. Due to the patient’s recent trip to a plague-infested area, our suspicion for Yersinia pestis infection was high.
The patient’s work-up included a negative pregnancy test and urinalysis. A complete blood count demonstrated a white blood cell count of 8.6 (4.3-10.5) × 103/UL with a 3+ left shift and a platelet count of 112 (180-500) × 103/UL. A complete metabolic panel showed hypokalemia and hyponatremia (potassium 2.8 [3.5-5.1] mmol/L and sodium 134 [137-145] mmol/L). Blood cultures were negative for any bacterial or fungal growth after 48 hours; stool cultures were negative for Salmonella, Shigella, Campylobacter, Giardia, generalized Yersinia, and Escherichia coli O157:H7. Swabs for Gardnerella vaginalis, Trichomonas vaginalis, Candida, Chlamydia trachomatis, and Neisseria gonorrhea also were negative. Lyme, Bartonella henselae, and heterophile antibodies were also negative. Francisella tularensis was not cultured due to low suspicion.

Imaging included a normal chest x-ray and a computed tomography scan of the abdomen and pelvis that showed enlarged right inguinal lymph nodes with fatty stranding, a thicker distal right iliopsoas, hepatosplenomegaly, and an enlarged right adnexa (FIGURE 1). Initial ultrasound of the bubo showed 2 enlarged suprapubic lymph nodes, the largest measuring 3.5 × 1.4 × 2.4 cm3 (FIGURE 2), and 8 enlarged inguinal nodes.

The patient continued to have a low-grade fever, diarrhea, and inguinal lymphadenopathy throughout her first 2 hospitalized days. The cefoxitin was discontinued by Day 3, and the consulting infectious disease physician started oral metronidazole 500 mg every 12 hours due to the patient’s failure to improve. Later that night, the patient experienced increasing erythema and pain in her right inguinal region. A repeat ultrasound showed increased inguinal lymphadenopathy with the largest nodes measuring 2.9 × 1.5 × 2.5 cm3 and 2.7 × 1.3 × 2 cm3 (FIGURE 3).

Although doxycycline is considered an acceptable regimen for Y pestis infection, the infectious disease physician added oral ciprofloxacin 750 mg every 12 hours the following morning, as the patient had not improved.
THE DIAGNOSIS
Although the initial gram stain was negative for Yersinia, clinical suspicion pointed to a diagnosis of bubonic plague. Serology was considered; however, it was not available through the hospital. A definitive diagnosis required bubo aspiration and culture, which was performed but required 48 hours before results would be available.
Continue to: By Day 5, the patient was clinically improved and...
By Day 5, the patient was clinically improved and deemed safe for discharge on empiric treatment with ciprofloxacin 750 mg twice daily and doxycycline 100 mg twice daily to complete a 14-day course of antibiotic therapy for bubonic plague. The bubo culture subsequently grew Y pestis, confirming the diagnosis. The patient made a full recovery and was greatly improved when seen in the outpatient setting by the treating infectious disease physician. Outpatient ultrasound repeated 3 weeks after discharge showed borderline lymphadenopathy, no greater than 1 cm.
DISCUSSION
Between 2000 and 2009, there were 57 cases of Y pestis in the United States; in early 2015, 11 cases were found in 6 Western states.1 The plague presents in the bubonic form 80% to 95% of the time, and it has never been reported in Michigan (where we treated this patient); however, there was a laboratory case in Illinois. Although rats were traditionally the host for Y pestis, the prairie dog, Cynomys gunnisoni, is a host in the United States.2 Rodents are the most important hosts, but more than 200 mammalian species, including domestic pets, have had reported infections. Transmission is primarily via flea bites, but Y pestis also may be transmitted via respiratory secretion, inhalation, or direct handling of contaminated animal tissues. Due to the risk of respiratory spread, the Centers for Disease Control and Prevention must be notified of a diagnosis.3,4
Y pestis travels from the site of the flea bite to regional lymph nodes, where it reproduces, and the resultant inflammatory reaction creates buboes. The bacteria then circulate in the blood to other organs, although Y pestis bacteria are primarily removed by the liver and spleen. Patients often develop symptoms such as headache, fevers, chills, and gastrointestinal distress. Diagnosis is reached by bubo culture or rapid testing for the F1 antigen. Early intervention with antibiotics is crucial as untreated bubonic plague has a mortality rate of 50% to 90%.3,4
The differential diagnosis for unilateral inguinal lymphadenopathy with associated constitutional symptoms was broad, in this case, and included pelvic inflammatory disease, bubonic plague, iliopsoas abscess, lymphogranuloma venereum, bartonellosis, infectious mononucleosis, and tick-borne diseases, such as ehrlichiosis, tularemia, Lyme disease, Rocky Mountain spotted fever, and Colorado tick fever.
Treatment. Food and Drug Administration–approved treatments include streptomycin (gentamicin 5 mg/kg/day IM or IV for 14 days is more widely utilized), doxycycline 200 mg PO once daily for 10 to 14 days, and fluoroquinolones (ciprofloxacin 500-750 mg every 12 hours for 10-14 days). Trimethoprim-sulfamethoxazole may be used as an alternative, but limitations include potentially incomplete or slowed responses.
Continue to: THE TAKEAWAY
THE TAKEAWAY
This case points to the importance of a complete, systematic approach to each patient. While bubonic plague is not a diagnosis that would immediately come to mind in a patient visiting an emergency department in Michigan, a thorough history revealed a recent trip to a bubonic plague–endemic area. A thorough physical exam demonstrated unilateral painful inguinal adenopathy—which, when paired with the patient’s history—was consistent with the uncommon diagnosis of bubonic plague.
The authors thank Brian Waite, MD, and James Addison, MD, for critically revising this report for important intellectual content.
CORRESPONDENCE
Katherine Lazet, DO, 3838 N First Avenue, Evansville, IN 47710; [email protected]
1. Kwit N, Nelson C, Kugeler K, et al. Human Plague – United States, 2015. MMWR Morb Mortal Wkly Rep. 2015,64:918-919.
2. Friggens MM, Parmenter RR, Boyden M, et al. Flea abundance, diversity, and plague in Gunnison’s prairie dog (Cynomys gunnisoni) and their burrows in Montane grasslands in northern New Mexico. J Wildl Dis. 2010;46:356-367.
3. Mandell G, Bennett J, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2010:2943-2953.
4. Perry RD, Featherston JD. Yersinia pestis - etiologic agent of plague. Clin Microbiol Rev. 1997;10:35-66.
THE CASE
A 33-year-old Caucasian woman presented to the emergency department with a 6-day history of fever (103°-104°F) and right groin pain and swelling. Associated symptoms included headache, diarrhea, malaise, weakness, nausea, cough, and anorexia. Upon presentation, she admitted to a recent hike on a bubonic plague–endemic trail in Colorado.
Her vital signs were unremarkable, and the physical examination demonstrated normal findings except for tender, erythematous, nonfluctuant right inguinal lymphadenopathy. The patient was admitted for intractable pain and fever and started on intravenous cefoxitin 2 g IV every 8 hours and oral doxycycline 100 mg every 12 hours for pelvic inflammatory disease vs tick- or flea-borne illness. Due to the patient’s recent trip to a plague-infested area, our suspicion for Yersinia pestis infection was high.
The patient’s work-up included a negative pregnancy test and urinalysis. A complete blood count demonstrated a white blood cell count of 8.6 (4.3-10.5) × 103/UL with a 3+ left shift and a platelet count of 112 (180-500) × 103/UL. A complete metabolic panel showed hypokalemia and hyponatremia (potassium 2.8 [3.5-5.1] mmol/L and sodium 134 [137-145] mmol/L). Blood cultures were negative for any bacterial or fungal growth after 48 hours; stool cultures were negative for Salmonella, Shigella, Campylobacter, Giardia, generalized Yersinia, and Escherichia coli O157:H7. Swabs for Gardnerella vaginalis, Trichomonas vaginalis, Candida, Chlamydia trachomatis, and Neisseria gonorrhea also were negative. Lyme, Bartonella henselae, and heterophile antibodies were also negative. Francisella tularensis was not cultured due to low suspicion.
Imaging included a normal chest x-ray and a computed tomography scan of the abdomen and pelvis that showed enlarged right inguinal lymph nodes with fatty stranding, a thicker distal right iliopsoas, hepatosplenomegaly, and an enlarged right adnexa (FIGURE 1). Initial ultrasound of the bubo showed 2 enlarged suprapubic lymph nodes, the largest measuring 3.5 × 1.4 × 2.4 cm3 (FIGURE 2), and 8 enlarged inguinal nodes.
The patient continued to have a low-grade fever, diarrhea, and inguinal lymphadenopathy throughout her first 2 hospitalized days. The cefoxitin was discontinued by Day 3, and the consulting infectious disease physician started oral metronidazole 500 mg every 12 hours due to the patient’s failure to improve. Later that night, the patient experienced increasing erythema and pain in her right inguinal region. A repeat ultrasound showed increased inguinal lymphadenopathy with the largest nodes measuring 2.9 × 1.5 × 2.5 cm3 and 2.7 × 1.3 × 2 cm3 (FIGURE 3).
Although doxycycline is considered an acceptable regimen for Y pestis infection, the infectious disease physician added oral ciprofloxacin 750 mg every 12 hours the following morning, as the patient had not improved.
THE DIAGNOSIS
Although the initial gram stain was negative for Yersinia, clinical suspicion pointed to a diagnosis of bubonic plague. Serology was considered; however, it was not available through the hospital. A definitive diagnosis required bubo aspiration and culture, which was performed but required 48 hours before results would be available.
Continue to: By Day 5, the patient was clinically improved and...
By Day 5, the patient was clinically improved and deemed safe for discharge on empiric treatment with ciprofloxacin 750 mg twice daily and doxycycline 100 mg twice daily to complete a 14-day course of antibiotic therapy for bubonic plague. The bubo culture subsequently grew Y pestis, confirming the diagnosis. The patient made a full recovery and was greatly improved when seen in the outpatient setting by the treating infectious disease physician. Outpatient ultrasound repeated 3 weeks after discharge showed borderline lymphadenopathy, no greater than 1 cm.
DISCUSSION
Between 2000 and 2009, there were 57 cases of Y pestis in the United States; in early 2015, 11 cases were found in 6 Western states.1 The plague presents in the bubonic form 80% to 95% of the time, and it has never been reported in Michigan (where we treated this patient); however, there was a laboratory case in Illinois. Although rats were traditionally the host for Y pestis, the prairie dog, Cynomys gunnisoni, is a host in the United States.2 Rodents are the most important hosts, but more than 200 mammalian species, including domestic pets, have had reported infections. Transmission is primarily via flea bites, but Y pestis also may be transmitted via respiratory secretion, inhalation, or direct handling of contaminated animal tissues. Due to the risk of respiratory spread, the Centers for Disease Control and Prevention must be notified of a diagnosis.3,4
Y pestis travels from the site of the flea bite to regional lymph nodes, where it reproduces, and the resultant inflammatory reaction creates buboes. The bacteria then circulate in the blood to other organs, although Y pestis bacteria are primarily removed by the liver and spleen. Patients often develop symptoms such as headache, fevers, chills, and gastrointestinal distress. Diagnosis is reached by bubo culture or rapid testing for the F1 antigen. Early intervention with antibiotics is crucial as untreated bubonic plague has a mortality rate of 50% to 90%.3,4
The differential diagnosis for unilateral inguinal lymphadenopathy with associated constitutional symptoms was broad, in this case, and included pelvic inflammatory disease, bubonic plague, iliopsoas abscess, lymphogranuloma venereum, bartonellosis, infectious mononucleosis, and tick-borne diseases, such as ehrlichiosis, tularemia, Lyme disease, Rocky Mountain spotted fever, and Colorado tick fever.
Treatment. Food and Drug Administration–approved treatments include streptomycin (gentamicin 5 mg/kg/day IM or IV for 14 days is more widely utilized), doxycycline 200 mg PO once daily for 10 to 14 days, and fluoroquinolones (ciprofloxacin 500-750 mg every 12 hours for 10-14 days). Trimethoprim-sulfamethoxazole may be used as an alternative, but limitations include potentially incomplete or slowed responses.
Continue to: THE TAKEAWAY
THE TAKEAWAY
This case points to the importance of a complete, systematic approach to each patient. While bubonic plague is not a diagnosis that would immediately come to mind in a patient visiting an emergency department in Michigan, a thorough history revealed a recent trip to a bubonic plague–endemic area. A thorough physical exam demonstrated unilateral painful inguinal adenopathy—which, when paired with the patient’s history—was consistent with the uncommon diagnosis of bubonic plague.
The authors thank Brian Waite, MD, and James Addison, MD, for critically revising this report for important intellectual content.
CORRESPONDENCE
Katherine Lazet, DO, 3838 N First Avenue, Evansville, IN 47710; [email protected]
THE CASE
A 33-year-old Caucasian woman presented to the emergency department with a 6-day history of fever (103°-104°F) and right groin pain and swelling. Associated symptoms included headache, diarrhea, malaise, weakness, nausea, cough, and anorexia. Upon presentation, she admitted to a recent hike on a bubonic plague–endemic trail in Colorado.
Her vital signs were unremarkable, and the physical examination demonstrated normal findings except for tender, erythematous, nonfluctuant right inguinal lymphadenopathy. The patient was admitted for intractable pain and fever and started on intravenous cefoxitin 2 g IV every 8 hours and oral doxycycline 100 mg every 12 hours for pelvic inflammatory disease vs tick- or flea-borne illness. Due to the patient’s recent trip to a plague-infested area, our suspicion for Yersinia pestis infection was high.
The patient’s work-up included a negative pregnancy test and urinalysis. A complete blood count demonstrated a white blood cell count of 8.6 (4.3-10.5) × 103/UL with a 3+ left shift and a platelet count of 112 (180-500) × 103/UL. A complete metabolic panel showed hypokalemia and hyponatremia (potassium 2.8 [3.5-5.1] mmol/L and sodium 134 [137-145] mmol/L). Blood cultures were negative for any bacterial or fungal growth after 48 hours; stool cultures were negative for Salmonella, Shigella, Campylobacter, Giardia, generalized Yersinia, and Escherichia coli O157:H7. Swabs for Gardnerella vaginalis, Trichomonas vaginalis, Candida, Chlamydia trachomatis, and Neisseria gonorrhea also were negative. Lyme, Bartonella henselae, and heterophile antibodies were also negative. Francisella tularensis was not cultured due to low suspicion.
Imaging included a normal chest x-ray and a computed tomography scan of the abdomen and pelvis that showed enlarged right inguinal lymph nodes with fatty stranding, a thicker distal right iliopsoas, hepatosplenomegaly, and an enlarged right adnexa (FIGURE 1). Initial ultrasound of the bubo showed 2 enlarged suprapubic lymph nodes, the largest measuring 3.5 × 1.4 × 2.4 cm3 (FIGURE 2), and 8 enlarged inguinal nodes.
The patient continued to have a low-grade fever, diarrhea, and inguinal lymphadenopathy throughout her first 2 hospitalized days. The cefoxitin was discontinued by Day 3, and the consulting infectious disease physician started oral metronidazole 500 mg every 12 hours due to the patient’s failure to improve. Later that night, the patient experienced increasing erythema and pain in her right inguinal region. A repeat ultrasound showed increased inguinal lymphadenopathy with the largest nodes measuring 2.9 × 1.5 × 2.5 cm3 and 2.7 × 1.3 × 2 cm3 (FIGURE 3).
Although doxycycline is considered an acceptable regimen for Y pestis infection, the infectious disease physician added oral ciprofloxacin 750 mg every 12 hours the following morning, as the patient had not improved.
THE DIAGNOSIS
Although the initial gram stain was negative for Yersinia, clinical suspicion pointed to a diagnosis of bubonic plague. Serology was considered; however, it was not available through the hospital. A definitive diagnosis required bubo aspiration and culture, which was performed but required 48 hours before results would be available.
Continue to: By Day 5, the patient was clinically improved and...
By Day 5, the patient was clinically improved and deemed safe for discharge on empiric treatment with ciprofloxacin 750 mg twice daily and doxycycline 100 mg twice daily to complete a 14-day course of antibiotic therapy for bubonic plague. The bubo culture subsequently grew Y pestis, confirming the diagnosis. The patient made a full recovery and was greatly improved when seen in the outpatient setting by the treating infectious disease physician. Outpatient ultrasound repeated 3 weeks after discharge showed borderline lymphadenopathy, no greater than 1 cm.
DISCUSSION
Between 2000 and 2009, there were 57 cases of Y pestis in the United States; in early 2015, 11 cases were found in 6 Western states.1 The plague presents in the bubonic form 80% to 95% of the time, and it has never been reported in Michigan (where we treated this patient); however, there was a laboratory case in Illinois. Although rats were traditionally the host for Y pestis, the prairie dog, Cynomys gunnisoni, is a host in the United States.2 Rodents are the most important hosts, but more than 200 mammalian species, including domestic pets, have had reported infections. Transmission is primarily via flea bites, but Y pestis also may be transmitted via respiratory secretion, inhalation, or direct handling of contaminated animal tissues. Due to the risk of respiratory spread, the Centers for Disease Control and Prevention must be notified of a diagnosis.3,4
Y pestis travels from the site of the flea bite to regional lymph nodes, where it reproduces, and the resultant inflammatory reaction creates buboes. The bacteria then circulate in the blood to other organs, although Y pestis bacteria are primarily removed by the liver and spleen. Patients often develop symptoms such as headache, fevers, chills, and gastrointestinal distress. Diagnosis is reached by bubo culture or rapid testing for the F1 antigen. Early intervention with antibiotics is crucial as untreated bubonic plague has a mortality rate of 50% to 90%.3,4
The differential diagnosis for unilateral inguinal lymphadenopathy with associated constitutional symptoms was broad, in this case, and included pelvic inflammatory disease, bubonic plague, iliopsoas abscess, lymphogranuloma venereum, bartonellosis, infectious mononucleosis, and tick-borne diseases, such as ehrlichiosis, tularemia, Lyme disease, Rocky Mountain spotted fever, and Colorado tick fever.
Treatment. Food and Drug Administration–approved treatments include streptomycin (gentamicin 5 mg/kg/day IM or IV for 14 days is more widely utilized), doxycycline 200 mg PO once daily for 10 to 14 days, and fluoroquinolones (ciprofloxacin 500-750 mg every 12 hours for 10-14 days). Trimethoprim-sulfamethoxazole may be used as an alternative, but limitations include potentially incomplete or slowed responses.
Continue to: THE TAKEAWAY
THE TAKEAWAY
This case points to the importance of a complete, systematic approach to each patient. While bubonic plague is not a diagnosis that would immediately come to mind in a patient visiting an emergency department in Michigan, a thorough history revealed a recent trip to a bubonic plague–endemic area. A thorough physical exam demonstrated unilateral painful inguinal adenopathy—which, when paired with the patient’s history—was consistent with the uncommon diagnosis of bubonic plague.
The authors thank Brian Waite, MD, and James Addison, MD, for critically revising this report for important intellectual content.
CORRESPONDENCE
Katherine Lazet, DO, 3838 N First Avenue, Evansville, IN 47710; [email protected]
1. Kwit N, Nelson C, Kugeler K, et al. Human Plague – United States, 2015. MMWR Morb Mortal Wkly Rep. 2015,64:918-919.
2. Friggens MM, Parmenter RR, Boyden M, et al. Flea abundance, diversity, and plague in Gunnison’s prairie dog (Cynomys gunnisoni) and their burrows in Montane grasslands in northern New Mexico. J Wildl Dis. 2010;46:356-367.
3. Mandell G, Bennett J, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2010:2943-2953.
4. Perry RD, Featherston JD. Yersinia pestis - etiologic agent of plague. Clin Microbiol Rev. 1997;10:35-66.
1. Kwit N, Nelson C, Kugeler K, et al. Human Plague – United States, 2015. MMWR Morb Mortal Wkly Rep. 2015,64:918-919.
2. Friggens MM, Parmenter RR, Boyden M, et al. Flea abundance, diversity, and plague in Gunnison’s prairie dog (Cynomys gunnisoni) and their burrows in Montane grasslands in northern New Mexico. J Wildl Dis. 2010;46:356-367.
3. Mandell G, Bennett J, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2010:2943-2953.
4. Perry RD, Featherston JD. Yersinia pestis - etiologic agent of plague. Clin Microbiol Rev. 1997;10:35-66.

Point of Care Ultrasound (POCUS) for Small Bowel Obstruction in the ED
Small bowel obstruction (SBO) accounts for 2% of all cases of abdominal pain presenting to the ED and 15% of abdominal pain admissions to surgical units from the ED.1,2 SBO can be a difficult diagnosis; the most common symptoms include nausea, vomiting, abdominal pain, obstipation, and constipation. The symptomatology depends on multiple factors: the area of the blockage, length of obstruction, and degree of the obstruction (either partial or complete).3 An upper gastrointestinal (GI) blockage classically presents with nausea and vomiting, while a lower GI blockage often presents with abdominal pain, constipation, and obstipation. Complications of obstruction range from significant morbidity—such as bowel strangulation (23%) and sepsis (31%)—to mortality (9%).4 ED POCUS allows for rapid and accurate diagnosis of SBO.
CASE
A 60-year-old female with a past medical history of peptic ulcer disease and multiple abdominal surgeries, including umbilical hernia repair, appendectomy, and total abdominal hysterectomy, presented to the ED with an 8-hour history of nausea and vomiting. She reported that her abdomen felt bloated. She had experienced non-bloody, watery stools for the prior 3 weeks. She also reported three to four weeks of epigastric abdominal pain similar to her previous “ulcer pain.” Of note, she was evaluated in GI clinic one day prior to her ED visit for dysphagia, abdominal distention, and diarrhea and was scheduled for an outpatient upper endoscopy. Initial vitals were significant for a heart rate of 100 beats/min. Physical exam was significant for a mildly distended abdomen, tender to palpation at epigastrium without rebound or guarding. Labs showed a white blood cell count of 11.8 K/uL and otherwise unremarkable complete blood count, basic metabolic panel, liver function tests, and lactate measurement. Given the patient’s history of multiple abdominal surgeries and clinical presentation, POCUS was performed to evaluate for SBO. Dilated loops of small bowel were visualized in the lower abdomen gas, suggestive of SBO.
Since the small bowel encompasses a large portion of the abdomen, to fully evaluate for SBO, multiple views are necessary. These include the epigastrium, bilateral colic gutters, and suprapubic regions.5 Use the low-frequency curvilinear transducer to obtain these views, scanning in the transverse and sagittal planes (see Figures 1 and 2). Scan while moving the transducer in columns (ie, “mowing the lawn”), making sure to cover the entire abdomen. To assure that you are evaluating the small bowel, and not the large bowel, look for the characteristic plicae circularis of the small bowel (shown in Figure 3). In children and very slender adults, the high-frequency linear probe may provide enough depth to obtain adequate views.
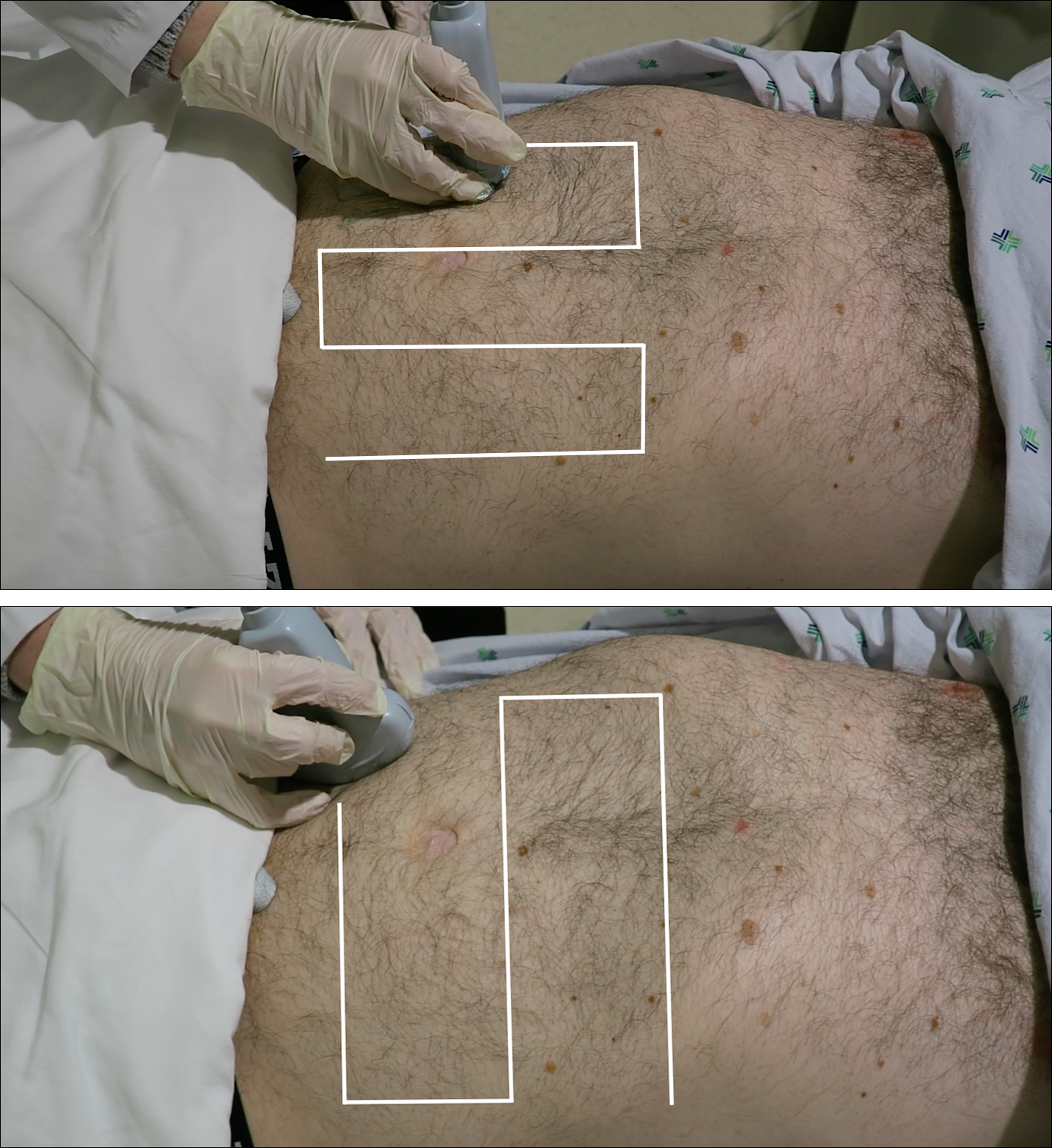
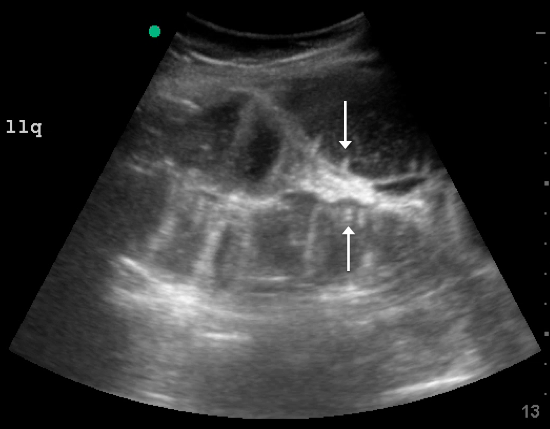
A fluid-filled small intestinal segment >2.5 cm is consistent with a diagnosis of SBO. Measuring the diameter of the small bowel is both the most sensitive and specific sign; a measurement of greater than 2.5 cm is diagnostic, with a sensitivity of 97% and specificity of 91% (see Figure 4).6 This can be somewhat difficult to visualize, as bowel loops are multidirectional and diameters can mistakenly be taken on an indirect cut; to avoid over- or underestimation of bowel diameter, you may want to measure in the short axis using a transverse cross-sectional view of the bowel.
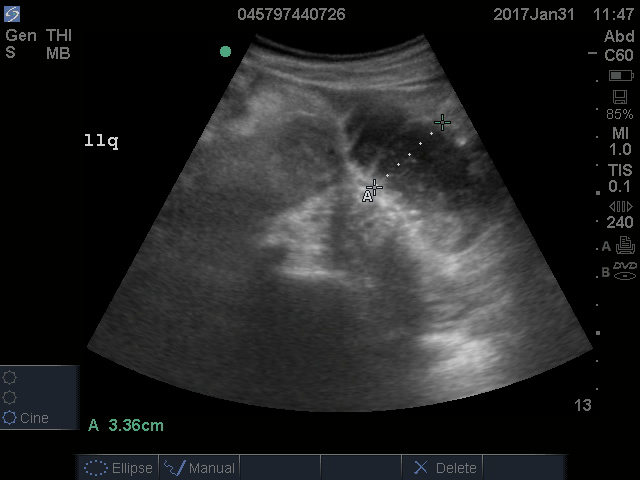
Lack of peristalsis is suggestive of a closed-loop obstruction. However, this finding may be more difficult to visualize, as it requires several continuous minutes of scanning or repeated exams to truly establish absent peristalsis. In prolonged courses of SBO, the bowel wall can measure >3 mm, which suggests necrosis, warranting accelerated surgical intervention. In addition, the detection of extraluminal peritoneal fluid can help determine the severity of the SBO, and small versus large fluid amounts can help determine whether medical or surgical management is warranted (see Figure 5).7
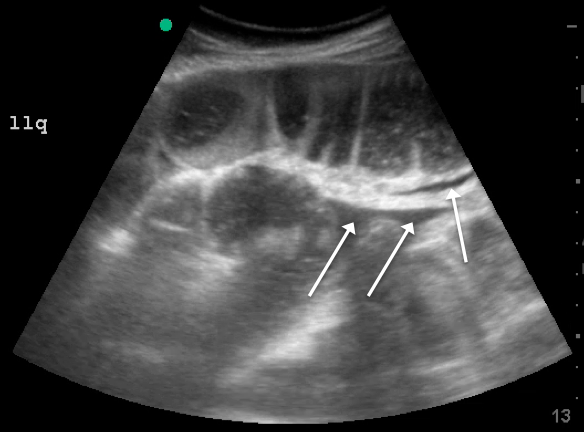
DISCUSSION
Increased time to diagnosis of SBO can lead to prolonged patient suffering and greater complication rates. The gold standard for diagnosing SBO—CT with intravenous and oral contrast—can take hours, requiring patients, who are often nauseated, to ingest and tolerate oral contrast. In the past, an “obstructive series” of x-rays would have been used early in the work-up of possible SBO.6
Recent literature suggests that POCUS is not only faster, more cost effective, and advantageous (involving no ionizing radiation), but also more accurate than x-rays. Specifically, a meta-analysis by Taylor et al showed pooled estimates for obstructive series x-rays have a sensitivity (Sn) of 75%, a specificity (Sp) of 66%, a positive likelihood ratio (+LR) of 1.6, and a negative likelihood ratio (-LR) of 0.43.1 On the other hand, pooled results from ED studies of emergency medicine (EM) residents performing POCUS in patients with signs and symptoms suspicious for SBO showed POCUS had a Sn of 97%, Sp of 90%, +LR of 9.5, and a -LR of 0.04.1,5,8 While detractors point to the operator-dependent nature of POCUS, literature suggests that with EM residents novice to POCUS for SBO (defined as less than 5 previous scans for SBO) were given a 10-minute didactic session and yielded Sn 94%, Sp 81%, +LR 5.0, -LR 0.07.5 Unluer et al trained novice EM residents for 6 hours and found them to yield Sn 98%, Sp 95%, +LR 19.5, and -LR 0.02.8 Thus, while it is no surprise that those with more training attain better results, both studies show it does not take much time for EM providers to surpass the accuracy of x-rays with POCUS.
CASE CONCLUSION
The findings on POCUS highly suggested the diagnosis of an SBO. A CT scan of the abdomen and pelvis with intravenous and oral contrast was ordered to further evaluate obstruction, transition point, and possible complications, including signs of ischemia per surgical request. CT demonstrated dilated loops of small bowel with transition point in the right lower quadrant, with a small amount of mesenteric fluid consistent with SBO with possible early bowel compromise due to ischemia. General surgery admitted the patient; conservative treatment with serial abdominal exams, nasogastric tube, NPO and bowel rest was ordered. The patient’s diet was gradually advanced, and she was discharged on the eleventh day of hospitalization.
SUMMARY
POCUS is a useful non-invasive tool that can accurately diagnose SBO. POCUS has increased sensitivity and specificity when compared to abdominal X-rays. This bedside imaging will not only give the ED provider rapid diagnostic information but also lead to expedited surgical intervention.
- Taylor MR, Lalani N. Adult small bowel obstruction. Acad Emerg Med. 2013;20(6):528-544.
- Hastings RS, Powers RD. Abdominal pain in the ED: a 35-year retrospective. Am J Emerg Med.2011;29:711-716.
- Markogiannakis H, Messaris E, Dardamanis D, et al. Acute mechanical bowel obstruction: clinical presentation, etiology, management and outcome. World J Gastroenterol. 2007;13:432.
- Bickell N, Federman A, Aufses A. Influence of time on risk of bowel resection in complete small bowel obstruction. J Am Coll Surg. 2005;201(6):847-854.
- Jang TB, Chandler D, Kaji AH. Bedside ultrasonography for the detection of small bowel obstruction in the emergency department. Emerg Med J. 2011;28:676-678.
- Carpenter CR, Pines JM. The end of X-rays for suspected small bowel obstruction? Using evidence-based diagnostics to inform best practices in emergency medicine. Acad Emerg Med. 2013;20:618-20.
- Grassi R, Romano S, D’Amario F, et al. The relevance of free fluid between intestinal loops detected by sonography in the clinical assessment of small bowel obstruction in adults. Eur J Radiol. 2004;50(1):5-14.
- Unlüer E, Yavaşi O, Eroğlu O, Yilmaz C, Akarca F. Ultrasonography by emergency medicine and radiology residents for the diagnosis of small bowel obstruction. Eur J Emerg Med. 2010;17(5):260-264.
Small bowel obstruction (SBO) accounts for 2% of all cases of abdominal pain presenting to the ED and 15% of abdominal pain admissions to surgical units from the ED.1,2 SBO can be a difficult diagnosis; the most common symptoms include nausea, vomiting, abdominal pain, obstipation, and constipation. The symptomatology depends on multiple factors: the area of the blockage, length of obstruction, and degree of the obstruction (either partial or complete).3 An upper gastrointestinal (GI) blockage classically presents with nausea and vomiting, while a lower GI blockage often presents with abdominal pain, constipation, and obstipation. Complications of obstruction range from significant morbidity—such as bowel strangulation (23%) and sepsis (31%)—to mortality (9%).4 ED POCUS allows for rapid and accurate diagnosis of SBO.
CASE
A 60-year-old female with a past medical history of peptic ulcer disease and multiple abdominal surgeries, including umbilical hernia repair, appendectomy, and total abdominal hysterectomy, presented to the ED with an 8-hour history of nausea and vomiting. She reported that her abdomen felt bloated. She had experienced non-bloody, watery stools for the prior 3 weeks. She also reported three to four weeks of epigastric abdominal pain similar to her previous “ulcer pain.” Of note, she was evaluated in GI clinic one day prior to her ED visit for dysphagia, abdominal distention, and diarrhea and was scheduled for an outpatient upper endoscopy. Initial vitals were significant for a heart rate of 100 beats/min. Physical exam was significant for a mildly distended abdomen, tender to palpation at epigastrium without rebound or guarding. Labs showed a white blood cell count of 11.8 K/uL and otherwise unremarkable complete blood count, basic metabolic panel, liver function tests, and lactate measurement. Given the patient’s history of multiple abdominal surgeries and clinical presentation, POCUS was performed to evaluate for SBO. Dilated loops of small bowel were visualized in the lower abdomen gas, suggestive of SBO.
Since the small bowel encompasses a large portion of the abdomen, to fully evaluate for SBO, multiple views are necessary. These include the epigastrium, bilateral colic gutters, and suprapubic regions.5 Use the low-frequency curvilinear transducer to obtain these views, scanning in the transverse and sagittal planes (see Figures 1 and 2). Scan while moving the transducer in columns (ie, “mowing the lawn”), making sure to cover the entire abdomen. To assure that you are evaluating the small bowel, and not the large bowel, look for the characteristic plicae circularis of the small bowel (shown in Figure 3). In children and very slender adults, the high-frequency linear probe may provide enough depth to obtain adequate views.
A fluid-filled small intestinal segment >2.5 cm is consistent with a diagnosis of SBO. Measuring the diameter of the small bowel is both the most sensitive and specific sign; a measurement of greater than 2.5 cm is diagnostic, with a sensitivity of 97% and specificity of 91% (see Figure 4).6 This can be somewhat difficult to visualize, as bowel loops are multidirectional and diameters can mistakenly be taken on an indirect cut; to avoid over- or underestimation of bowel diameter, you may want to measure in the short axis using a transverse cross-sectional view of the bowel.
Lack of peristalsis is suggestive of a closed-loop obstruction. However, this finding may be more difficult to visualize, as it requires several continuous minutes of scanning or repeated exams to truly establish absent peristalsis. In prolonged courses of SBO, the bowel wall can measure >3 mm, which suggests necrosis, warranting accelerated surgical intervention. In addition, the detection of extraluminal peritoneal fluid can help determine the severity of the SBO, and small versus large fluid amounts can help determine whether medical or surgical management is warranted (see Figure 5).7
DISCUSSION
Increased time to diagnosis of SBO can lead to prolonged patient suffering and greater complication rates. The gold standard for diagnosing SBO—CT with intravenous and oral contrast—can take hours, requiring patients, who are often nauseated, to ingest and tolerate oral contrast. In the past, an “obstructive series” of x-rays would have been used early in the work-up of possible SBO.6
Recent literature suggests that POCUS is not only faster, more cost effective, and advantageous (involving no ionizing radiation), but also more accurate than x-rays. Specifically, a meta-analysis by Taylor et al showed pooled estimates for obstructive series x-rays have a sensitivity (Sn) of 75%, a specificity (Sp) of 66%, a positive likelihood ratio (+LR) of 1.6, and a negative likelihood ratio (-LR) of 0.43.1 On the other hand, pooled results from ED studies of emergency medicine (EM) residents performing POCUS in patients with signs and symptoms suspicious for SBO showed POCUS had a Sn of 97%, Sp of 90%, +LR of 9.5, and a -LR of 0.04.1,5,8 While detractors point to the operator-dependent nature of POCUS, literature suggests that with EM residents novice to POCUS for SBO (defined as less than 5 previous scans for SBO) were given a 10-minute didactic session and yielded Sn 94%, Sp 81%, +LR 5.0, -LR 0.07.5 Unluer et al trained novice EM residents for 6 hours and found them to yield Sn 98%, Sp 95%, +LR 19.5, and -LR 0.02.8 Thus, while it is no surprise that those with more training attain better results, both studies show it does not take much time for EM providers to surpass the accuracy of x-rays with POCUS.
CASE CONCLUSION
The findings on POCUS highly suggested the diagnosis of an SBO. A CT scan of the abdomen and pelvis with intravenous and oral contrast was ordered to further evaluate obstruction, transition point, and possible complications, including signs of ischemia per surgical request. CT demonstrated dilated loops of small bowel with transition point in the right lower quadrant, with a small amount of mesenteric fluid consistent with SBO with possible early bowel compromise due to ischemia. General surgery admitted the patient; conservative treatment with serial abdominal exams, nasogastric tube, NPO and bowel rest was ordered. The patient’s diet was gradually advanced, and she was discharged on the eleventh day of hospitalization.
SUMMARY
POCUS is a useful non-invasive tool that can accurately diagnose SBO. POCUS has increased sensitivity and specificity when compared to abdominal X-rays. This bedside imaging will not only give the ED provider rapid diagnostic information but also lead to expedited surgical intervention.
Small bowel obstruction (SBO) accounts for 2% of all cases of abdominal pain presenting to the ED and 15% of abdominal pain admissions to surgical units from the ED.1,2 SBO can be a difficult diagnosis; the most common symptoms include nausea, vomiting, abdominal pain, obstipation, and constipation. The symptomatology depends on multiple factors: the area of the blockage, length of obstruction, and degree of the obstruction (either partial or complete).3 An upper gastrointestinal (GI) blockage classically presents with nausea and vomiting, while a lower GI blockage often presents with abdominal pain, constipation, and obstipation. Complications of obstruction range from significant morbidity—such as bowel strangulation (23%) and sepsis (31%)—to mortality (9%).4 ED POCUS allows for rapid and accurate diagnosis of SBO.
CASE
A 60-year-old female with a past medical history of peptic ulcer disease and multiple abdominal surgeries, including umbilical hernia repair, appendectomy, and total abdominal hysterectomy, presented to the ED with an 8-hour history of nausea and vomiting. She reported that her abdomen felt bloated. She had experienced non-bloody, watery stools for the prior 3 weeks. She also reported three to four weeks of epigastric abdominal pain similar to her previous “ulcer pain.” Of note, she was evaluated in GI clinic one day prior to her ED visit for dysphagia, abdominal distention, and diarrhea and was scheduled for an outpatient upper endoscopy. Initial vitals were significant for a heart rate of 100 beats/min. Physical exam was significant for a mildly distended abdomen, tender to palpation at epigastrium without rebound or guarding. Labs showed a white blood cell count of 11.8 K/uL and otherwise unremarkable complete blood count, basic metabolic panel, liver function tests, and lactate measurement. Given the patient’s history of multiple abdominal surgeries and clinical presentation, POCUS was performed to evaluate for SBO. Dilated loops of small bowel were visualized in the lower abdomen gas, suggestive of SBO.
Since the small bowel encompasses a large portion of the abdomen, to fully evaluate for SBO, multiple views are necessary. These include the epigastrium, bilateral colic gutters, and suprapubic regions.5 Use the low-frequency curvilinear transducer to obtain these views, scanning in the transverse and sagittal planes (see Figures 1 and 2). Scan while moving the transducer in columns (ie, “mowing the lawn”), making sure to cover the entire abdomen. To assure that you are evaluating the small bowel, and not the large bowel, look for the characteristic plicae circularis of the small bowel (shown in Figure 3). In children and very slender adults, the high-frequency linear probe may provide enough depth to obtain adequate views.
A fluid-filled small intestinal segment >2.5 cm is consistent with a diagnosis of SBO. Measuring the diameter of the small bowel is both the most sensitive and specific sign; a measurement of greater than 2.5 cm is diagnostic, with a sensitivity of 97% and specificity of 91% (see Figure 4).6 This can be somewhat difficult to visualize, as bowel loops are multidirectional and diameters can mistakenly be taken on an indirect cut; to avoid over- or underestimation of bowel diameter, you may want to measure in the short axis using a transverse cross-sectional view of the bowel.
Lack of peristalsis is suggestive of a closed-loop obstruction. However, this finding may be more difficult to visualize, as it requires several continuous minutes of scanning or repeated exams to truly establish absent peristalsis. In prolonged courses of SBO, the bowel wall can measure >3 mm, which suggests necrosis, warranting accelerated surgical intervention. In addition, the detection of extraluminal peritoneal fluid can help determine the severity of the SBO, and small versus large fluid amounts can help determine whether medical or surgical management is warranted (see Figure 5).7
DISCUSSION
Increased time to diagnosis of SBO can lead to prolonged patient suffering and greater complication rates. The gold standard for diagnosing SBO—CT with intravenous and oral contrast—can take hours, requiring patients, who are often nauseated, to ingest and tolerate oral contrast. In the past, an “obstructive series” of x-rays would have been used early in the work-up of possible SBO.6
Recent literature suggests that POCUS is not only faster, more cost effective, and advantageous (involving no ionizing radiation), but also more accurate than x-rays. Specifically, a meta-analysis by Taylor et al showed pooled estimates for obstructive series x-rays have a sensitivity (Sn) of 75%, a specificity (Sp) of 66%, a positive likelihood ratio (+LR) of 1.6, and a negative likelihood ratio (-LR) of 0.43.1 On the other hand, pooled results from ED studies of emergency medicine (EM) residents performing POCUS in patients with signs and symptoms suspicious for SBO showed POCUS had a Sn of 97%, Sp of 90%, +LR of 9.5, and a -LR of 0.04.1,5,8 While detractors point to the operator-dependent nature of POCUS, literature suggests that with EM residents novice to POCUS for SBO (defined as less than 5 previous scans for SBO) were given a 10-minute didactic session and yielded Sn 94%, Sp 81%, +LR 5.0, -LR 0.07.5 Unluer et al trained novice EM residents for 6 hours and found them to yield Sn 98%, Sp 95%, +LR 19.5, and -LR 0.02.8 Thus, while it is no surprise that those with more training attain better results, both studies show it does not take much time for EM providers to surpass the accuracy of x-rays with POCUS.
CASE CONCLUSION
The findings on POCUS highly suggested the diagnosis of an SBO. A CT scan of the abdomen and pelvis with intravenous and oral contrast was ordered to further evaluate obstruction, transition point, and possible complications, including signs of ischemia per surgical request. CT demonstrated dilated loops of small bowel with transition point in the right lower quadrant, with a small amount of mesenteric fluid consistent with SBO with possible early bowel compromise due to ischemia. General surgery admitted the patient; conservative treatment with serial abdominal exams, nasogastric tube, NPO and bowel rest was ordered. The patient’s diet was gradually advanced, and she was discharged on the eleventh day of hospitalization.
SUMMARY
POCUS is a useful non-invasive tool that can accurately diagnose SBO. POCUS has increased sensitivity and specificity when compared to abdominal X-rays. This bedside imaging will not only give the ED provider rapid diagnostic information but also lead to expedited surgical intervention.
- Taylor MR, Lalani N. Adult small bowel obstruction. Acad Emerg Med. 2013;20(6):528-544.
- Hastings RS, Powers RD. Abdominal pain in the ED: a 35-year retrospective. Am J Emerg Med.2011;29:711-716.
- Markogiannakis H, Messaris E, Dardamanis D, et al. Acute mechanical bowel obstruction: clinical presentation, etiology, management and outcome. World J Gastroenterol. 2007;13:432.
- Bickell N, Federman A, Aufses A. Influence of time on risk of bowel resection in complete small bowel obstruction. J Am Coll Surg. 2005;201(6):847-854.
- Jang TB, Chandler D, Kaji AH. Bedside ultrasonography for the detection of small bowel obstruction in the emergency department. Emerg Med J. 2011;28:676-678.
- Carpenter CR, Pines JM. The end of X-rays for suspected small bowel obstruction? Using evidence-based diagnostics to inform best practices in emergency medicine. Acad Emerg Med. 2013;20:618-20.
- Grassi R, Romano S, D’Amario F, et al. The relevance of free fluid between intestinal loops detected by sonography in the clinical assessment of small bowel obstruction in adults. Eur J Radiol. 2004;50(1):5-14.
- Unlüer E, Yavaşi O, Eroğlu O, Yilmaz C, Akarca F. Ultrasonography by emergency medicine and radiology residents for the diagnosis of small bowel obstruction. Eur J Emerg Med. 2010;17(5):260-264.
- Taylor MR, Lalani N. Adult small bowel obstruction. Acad Emerg Med. 2013;20(6):528-544.
- Hastings RS, Powers RD. Abdominal pain in the ED: a 35-year retrospective. Am J Emerg Med.2011;29:711-716.
- Markogiannakis H, Messaris E, Dardamanis D, et al. Acute mechanical bowel obstruction: clinical presentation, etiology, management and outcome. World J Gastroenterol. 2007;13:432.
- Bickell N, Federman A, Aufses A. Influence of time on risk of bowel resection in complete small bowel obstruction. J Am Coll Surg. 2005;201(6):847-854.
- Jang TB, Chandler D, Kaji AH. Bedside ultrasonography for the detection of small bowel obstruction in the emergency department. Emerg Med J. 2011;28:676-678.
- Carpenter CR, Pines JM. The end of X-rays for suspected small bowel obstruction? Using evidence-based diagnostics to inform best practices in emergency medicine. Acad Emerg Med. 2013;20:618-20.
- Grassi R, Romano S, D’Amario F, et al. The relevance of free fluid between intestinal loops detected by sonography in the clinical assessment of small bowel obstruction in adults. Eur J Radiol. 2004;50(1):5-14.
- Unlüer E, Yavaşi O, Eroğlu O, Yilmaz C, Akarca F. Ultrasonography by emergency medicine and radiology residents for the diagnosis of small bowel obstruction. Eur J Emerg Med. 2010;17(5):260-264.
Malignant olecranon bursitis in the setting of multiple myeloma relapse
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.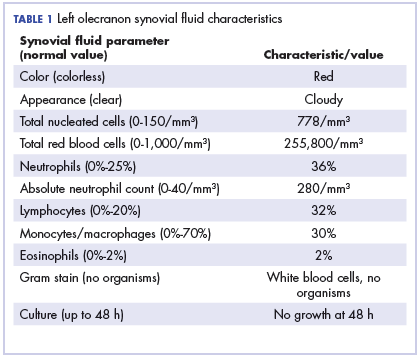
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).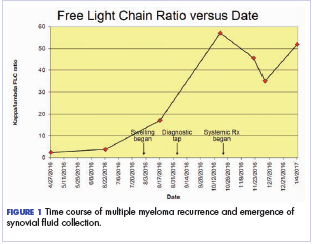
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.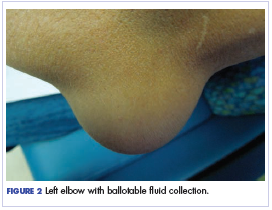
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).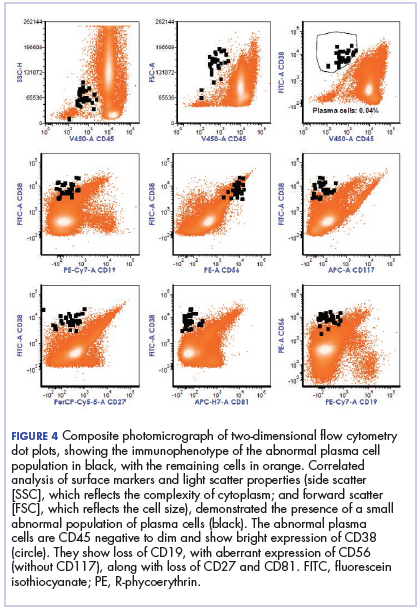
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.