User login
Primary Cutaneous Epstein-Barr Virus–Positive Diffuse Large B-Cell Lymphoma: A Rare and Aggressive Cutaneous Lymphoma
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
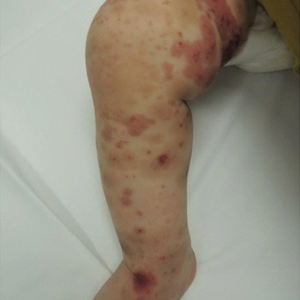
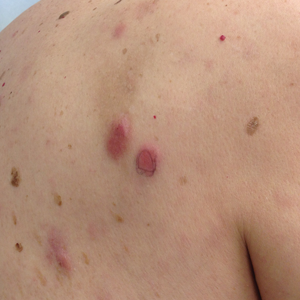
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
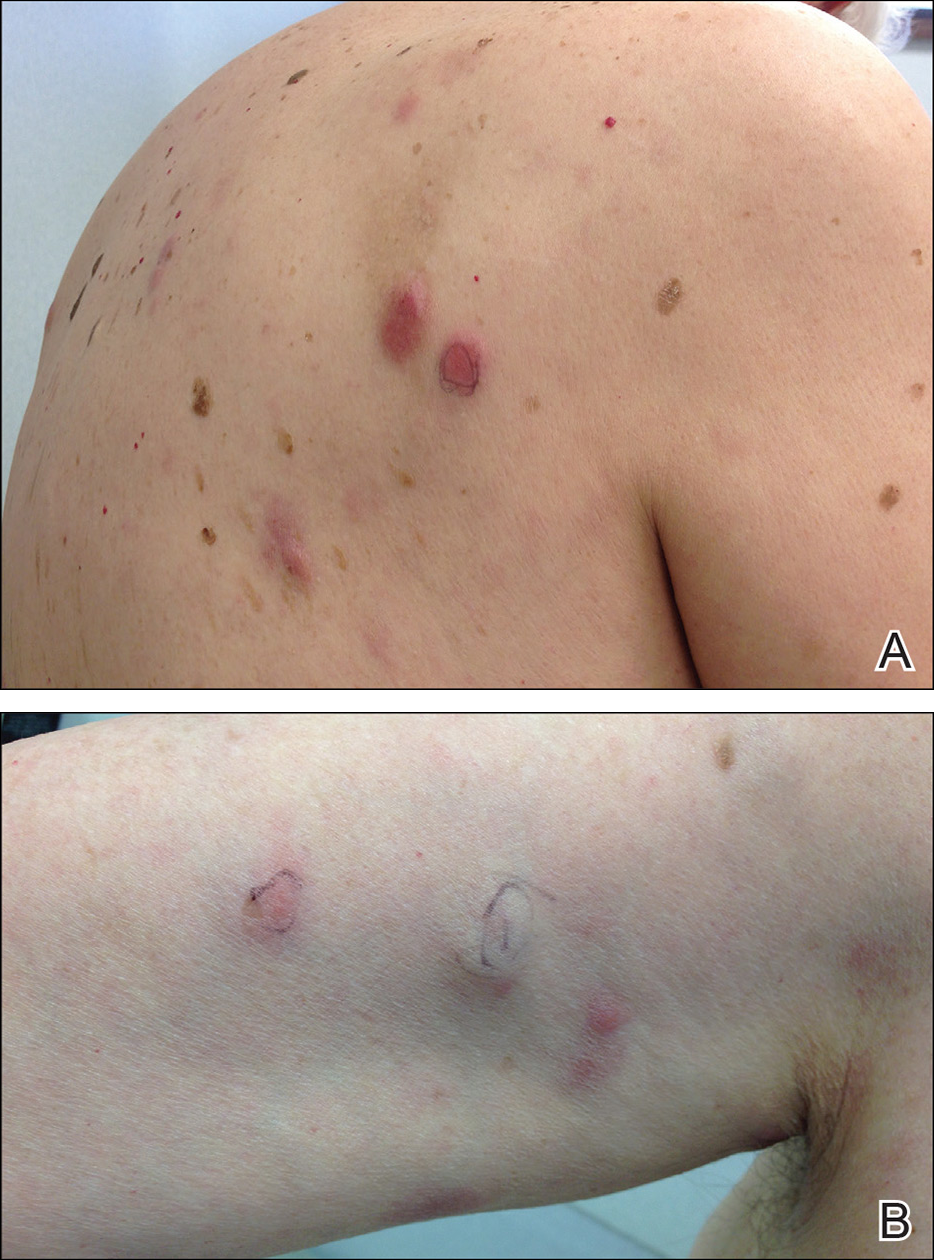
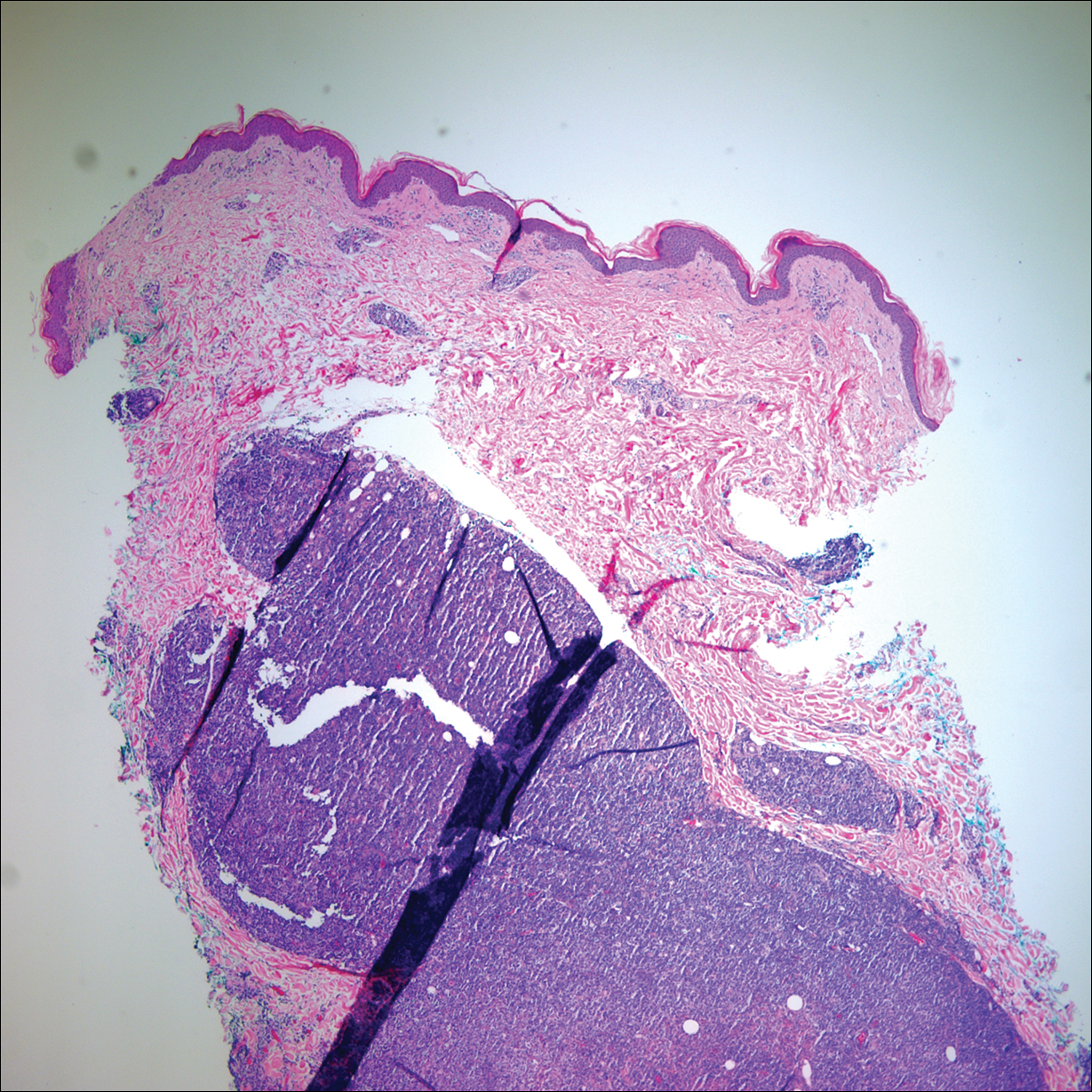
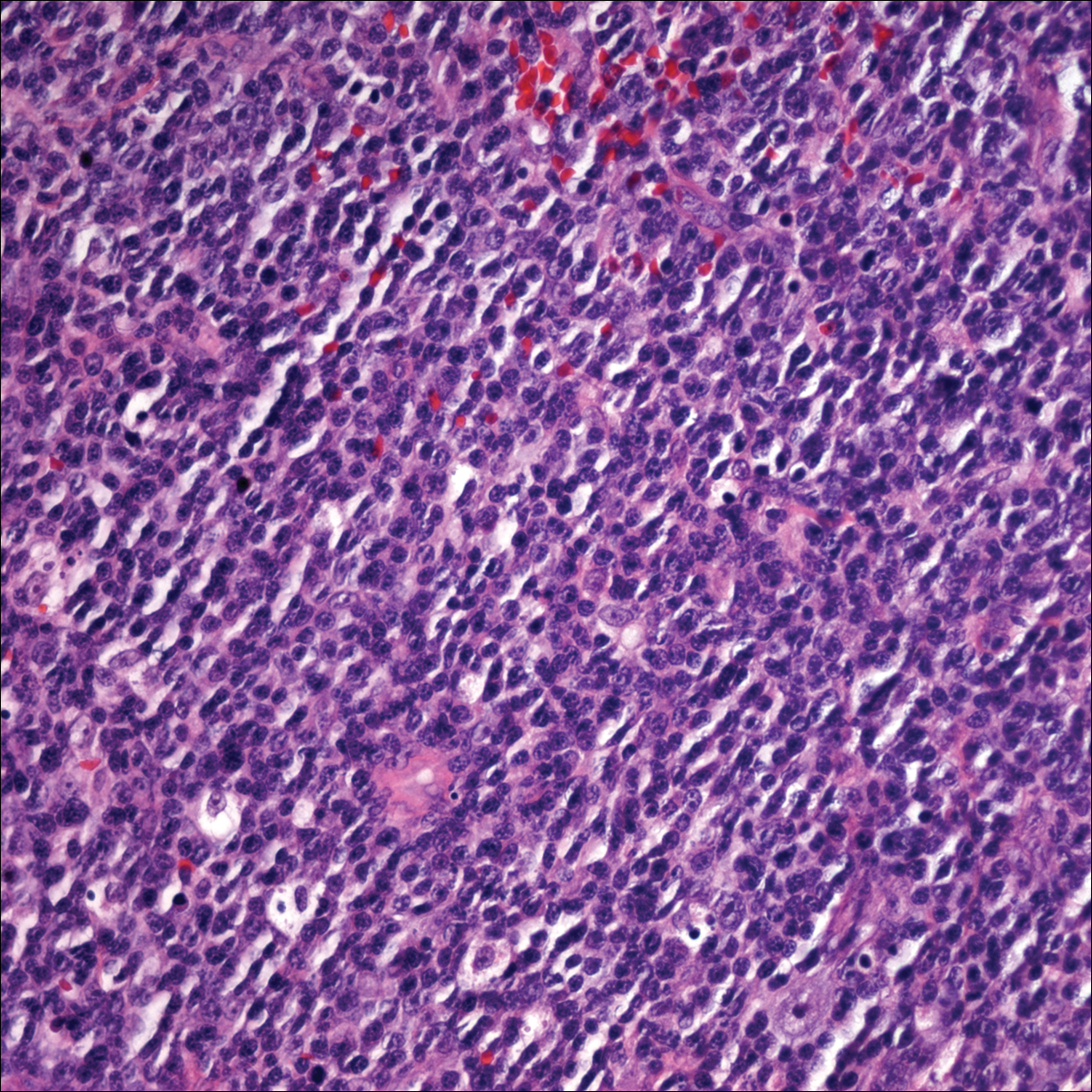
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Practice Points
- Primary cutaneous lymphomas are malignant lymphomas confined to the skin.
- Complete staging workup is necessary to rule out secondary involvement of the skin from a nodal lymphoma.
- Epstein-Barr virus-positive diffuse large B-cell lymphoma is a rare and aggressive primary cutaneous lymphoma.
Ichthyosiform Sarcoidosis and Systemic Involvement
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
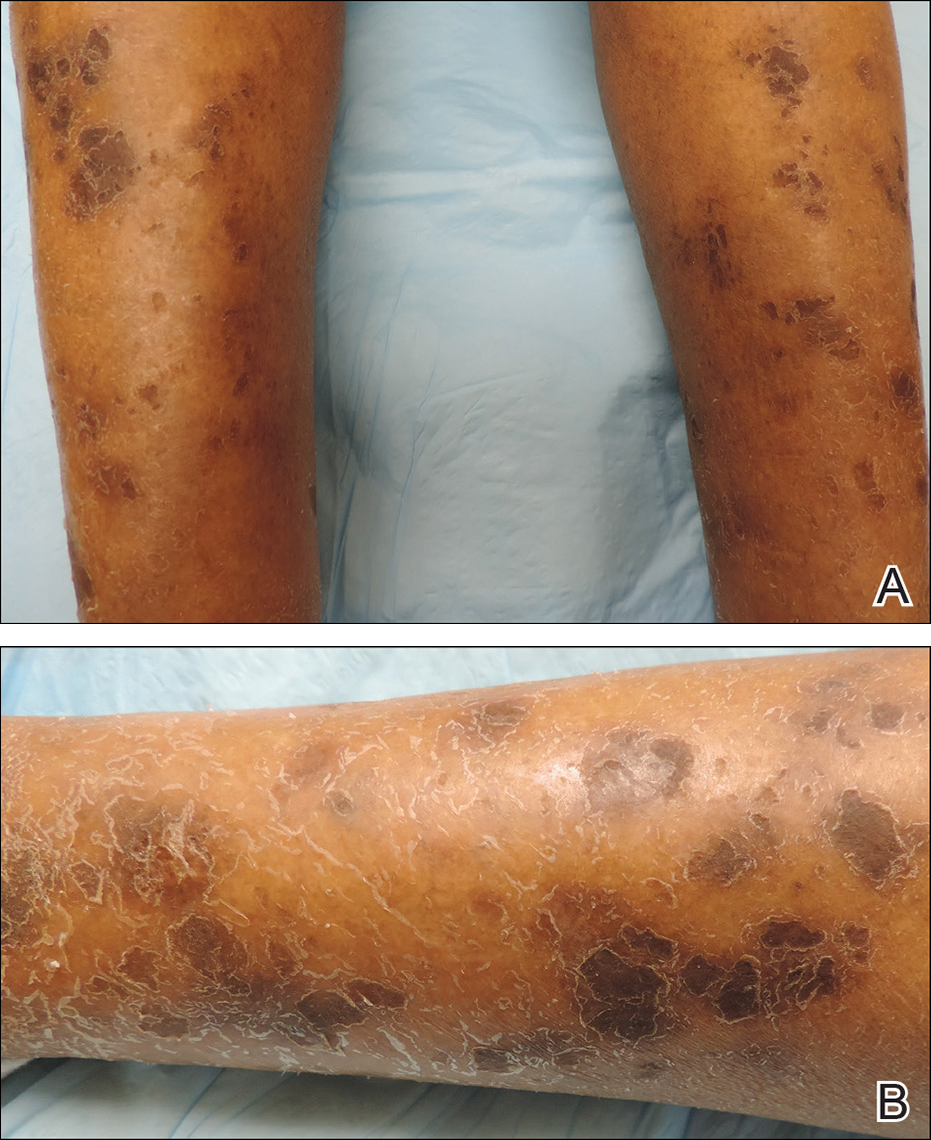
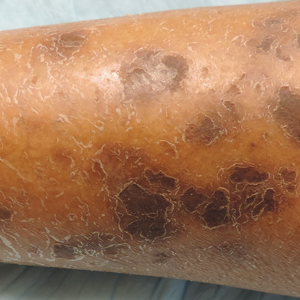
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
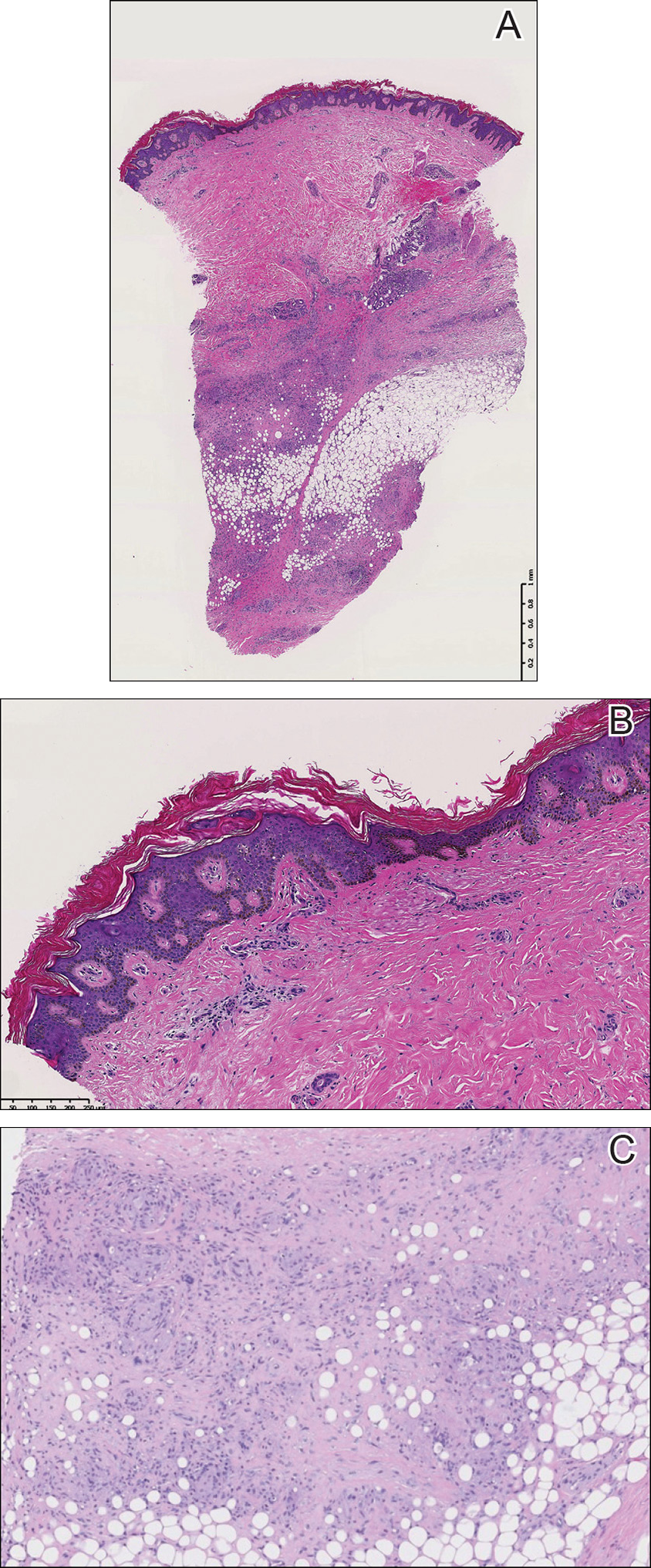
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
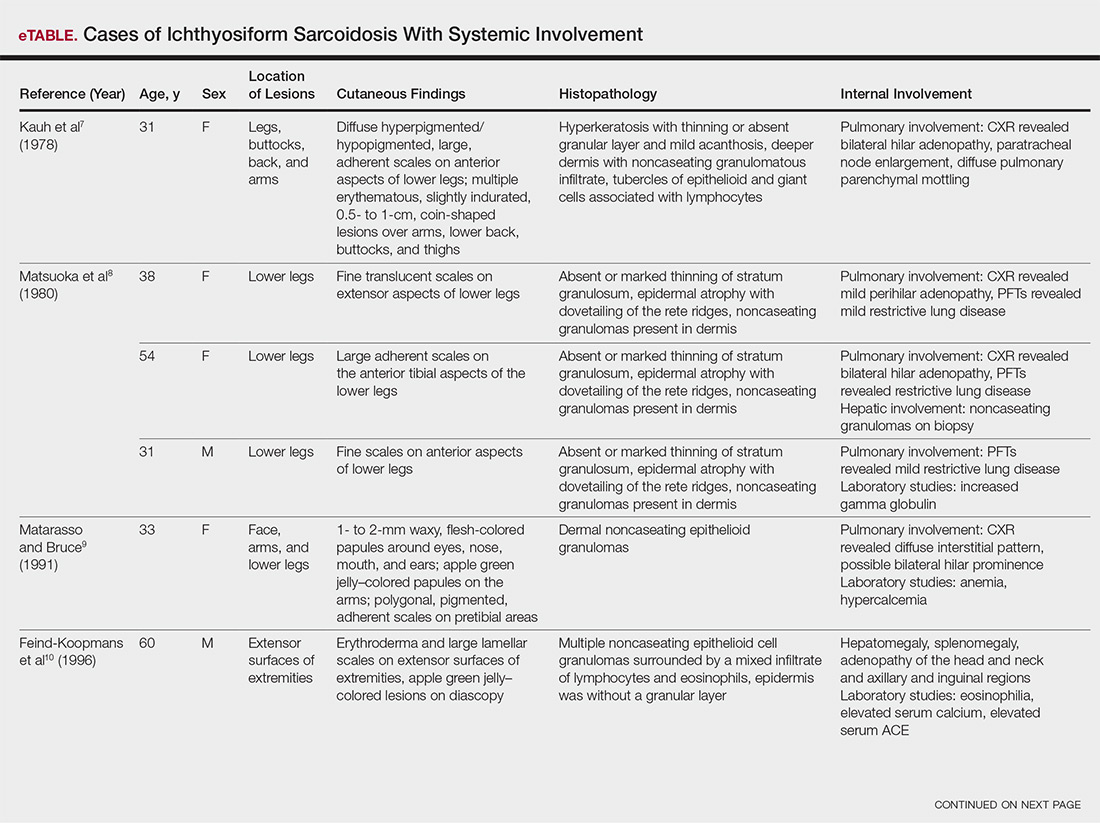
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
Practice Points
- Ichthyosiform sarcoidosis is a rare form of sarcoidosis that presents as polygonal adherent scales.
- Ichthyosiform sarcoidosis is commonly associated with pulmonary, neurologic, and hepatic involvement.
- Acquired ichthyosis should warrant further investigation for systemic disease.
Crizotinib-Induced Lichenoid Drug Eruption in a Patient With Lung Cancer
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
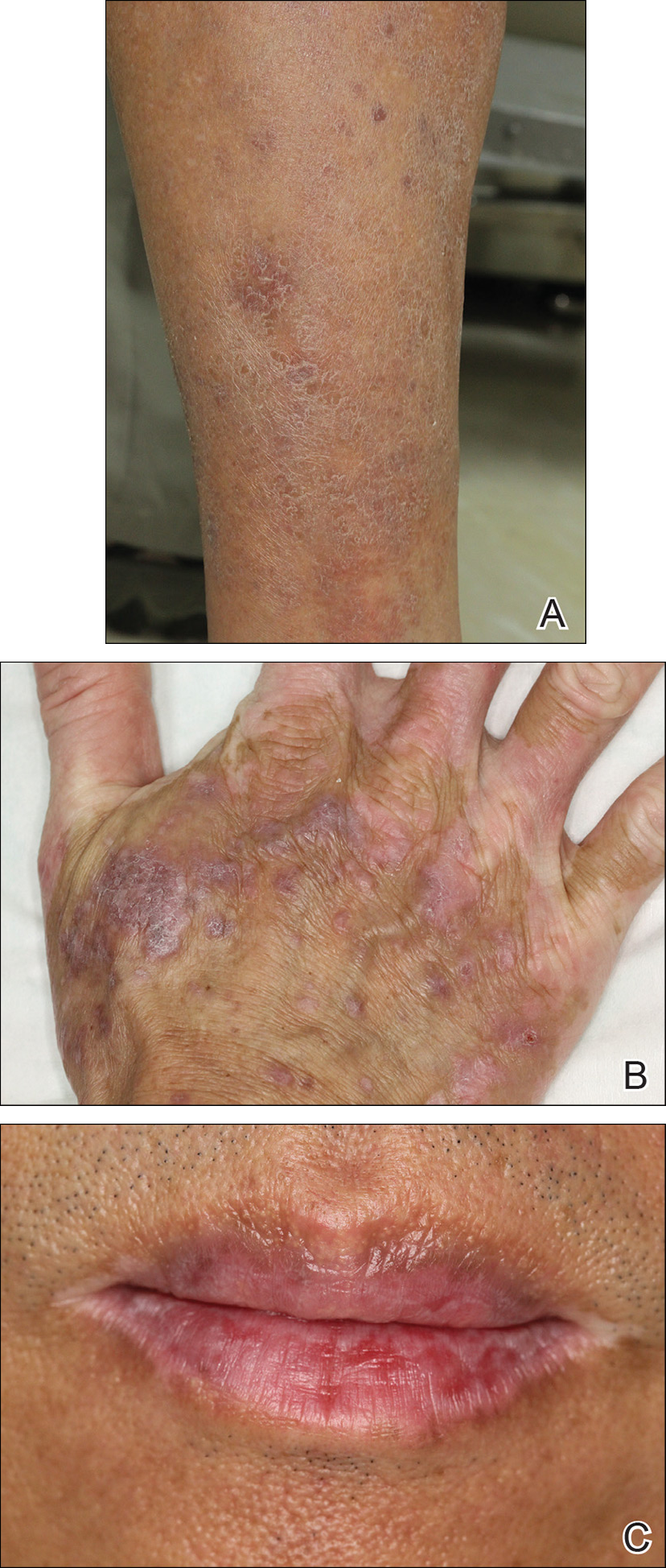
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
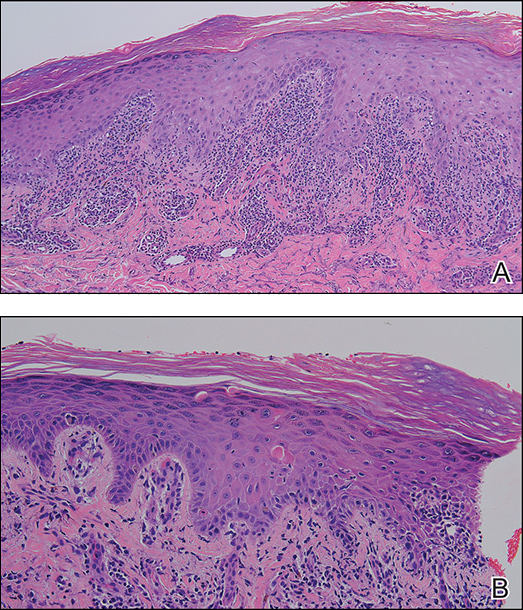
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
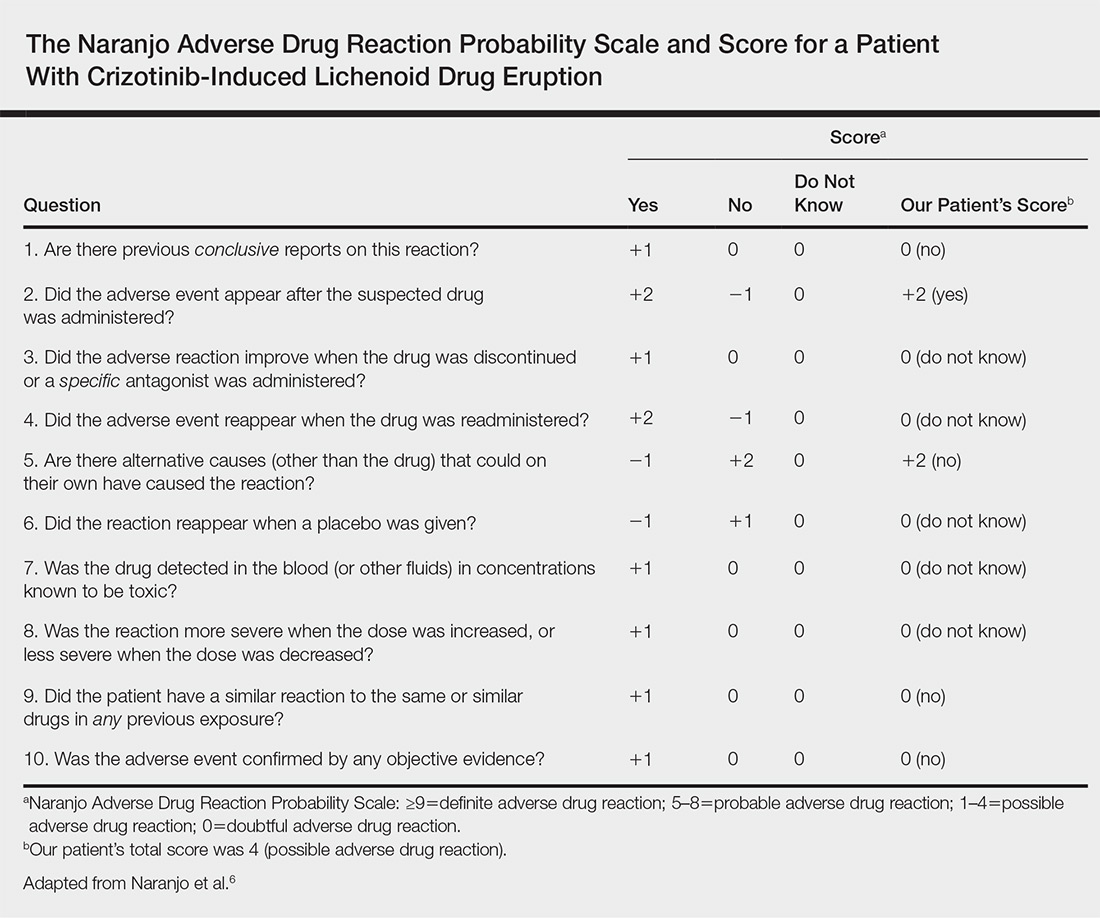
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
Practice Points
- Cutaneous lichenoid drug eruptions (LDEs) and photosensitive rash may be caused by crizotinib.
- The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.
A ‘double-hit’ bone marrow rare co-occurrence of 2 different pathologies
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Skin-Colored Papules on the Chest
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
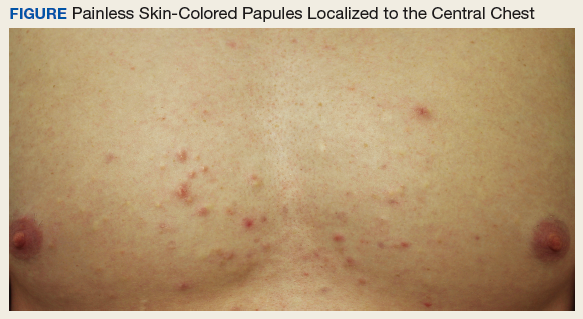
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
Concomitant Fibrofolliculoma and Trichodiscoma on the Abdomen
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
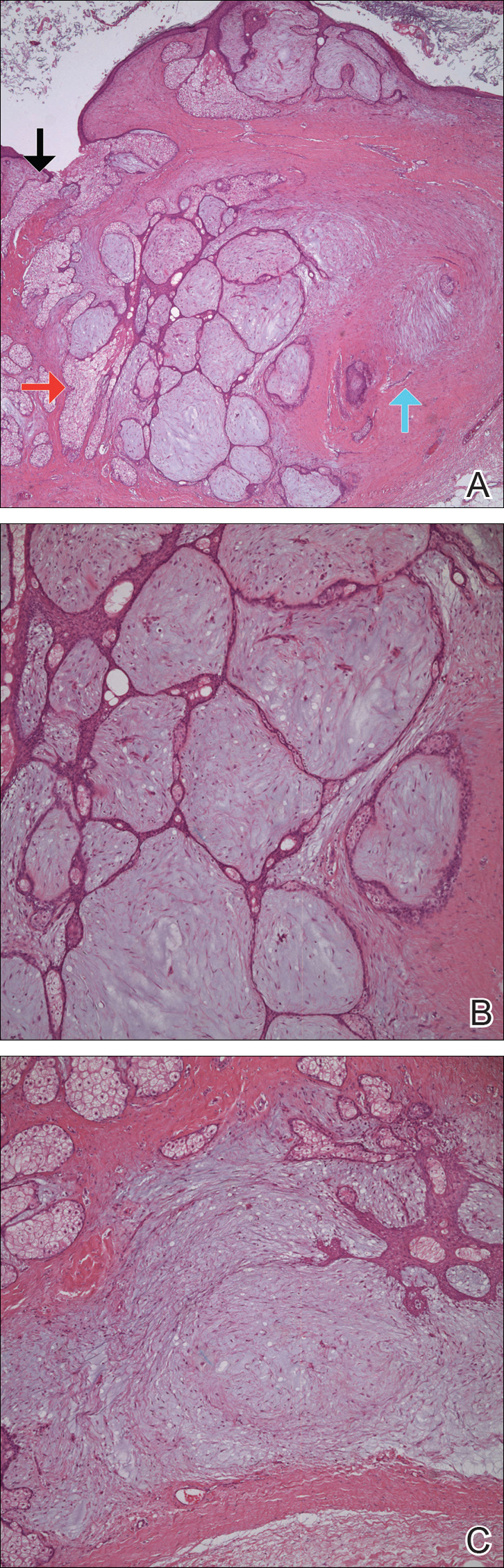
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.

Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Practice Points
- Fibrofolliculoma and trichodiscoma are flesh-colored adnexal tumors that arise from or around hair follicles.
- It is important to recognize these entities, as they can be related to Birt-Hogg-Dubé syndrome.
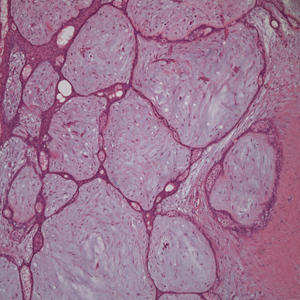
Roundtable Discussion: Anticoagulation Management
Case 1
Tracy Minichiello, MD. The first case we’ll discuss is a 75-year-old man with mild chronic kidney disease (CKD). His calculated creatinine clearance (CrCl) is about 52 mL/min, and he has a remote history of a gastrointestinal (GI) bleeding 3 years previously from a peptic ulcer. He presents with new onset nonvalvular atrial fibrillation (AF), and he’s already on aspirin for his stable coronary artery disease (CAD).
How do we think about anticoagulant selection in this patient? We have a number of new oral anticoagulants and we have warfarin. How do we decide between warfarin vs one of the direct-acting oral anticoagulants (DOACs)? If we choose a DOAC, which one would we select?
David Parra, PharmD. The first step for anticoagulation is to assess a patient’s thromboembolic risk utilizing the CHA2DS2-VASc and bleeding risk using a HAS-BLED score, or something similar. The next question is which oral anticoagulant to use. We have widespread experience with warfarin and can measure the anticoagulant effect easily. Warfarin has a long duration of action, so perhaps it’s more forgiving if you miss a dose. It also has an antidote. Lastly, organ dysfunction doesn’t preclude use of warfarin as you can still monitor the anticoagulant effect. So there still may be patients that may benefit significantly from warfarin vs a DOAC.
On the flip side, DOACs are easier to use and perform quite acceptably in comparison with warfarin in nonvalvular AF. There are some scenarios where a specific DOAC may be preferred over another, such as recent GI bleeding.
Dr. Minichiello. Do you consider renal function, bleeding history, or concomitant antiplatelet therapy?
Geoffrey Barnes, MD, MSc. A couple of factors are relevant. I think we should consider renal function for this gentleman. However, I look at some of the other features as Dr. Parra suggested. What’s the likelihood that this patient is going to take the medicine as prescribed? Is a twice-a-day regimen going to be something that’s particularly challenging? I also look at the real-world vs randomized trial experience.
This patient has a remote GI bleeding history. Some of the real-world data suggest there might be some more GI bleeding with rivaroxaban, but across the board, apixaban (in both the randomized trials and much of the real-world data) seem to have a favorable bleeding risk profile. For a patient who is open and reliable for taking medicine twice a day, apixaban might be a good option as long as we make sure that the dose is appropriate.
Arthur L. Allen, PharmD, CACP. In pivotal trial experience, dabigatran and rivaroxaban demonstrated an increased incidence of GI bleeding compared with warfarin. In some of the real-world studies, rivaroxaban mirrors warfarin with regard to bleeding, whereas dabigatran and apixaban have a lower incidence. In the pivotal trials, apixaban did not have a trigger of increased GI bleeding, but I would let the details of this patient’s GI bleeding history help me determine how important an issue this is at this point.
The other thing that is important to understand when considering choice of agents: As Dr. Parra mentioned, we do have quite a bit of experience with warfarin. But comparing the quality of evidence, the DOACs have been investigated in a far more rigorous fashion and in far more patients than warfarin ever was in its more than 60 years on the market. For example, the RE-LY trial alone enrolled more than 18,000 patients. Each of the DOACs have been studied in tens of thousands of patients for their approved indications. Further, we shouldn’t forget that the risk of intracranial hemorrhage is reduced by roughly 50% by choosing a DOAC over warfarin, which should be a consideration in this elderly gentleman.
Dr. Minichiello. In the veteran population, there is a sense of comfort with warfarin, and some concerns have been raised over a lack of reversibility for the newer agents. We have patients who have trepidation about starting one of the new anticoagulants. However, there is a marked reduction in the risk of the most devastating bleeding complication, namely intracranial hemorrhage, making the use of these agents most compelling. And when they did have bleeding complications, at least in the trials, their outcomes were no worse than they were with warfarin, where there is a reversal agent. In most cases the outcomes were actually better. 
Dr. Barnes. You often have to remind patients that there was no reversal agent in these huge trials where the DOACs showed similar or safer bleeding risk profiles, especially for the most serious bleeding, such as intracranial hemorrhage. I find patients often are reassured by knowing that.
Dr. Allen. I agree that there is concern about the lack of reversibility, but I think it has been completely overplayed. In the pivotal trials, patients who bled on DOAC therapy actually had better outcomes than those that bled on warfarin. This includes intracranial hemorrhage. There was a paper published in Stroke in 2012 that evaluated the subgroup of patients in the RE-LY trial that suffered intracranial hemorrhage. Patients on dabigatran actually fared better despite a lack of a specific reversal agent. When evaluating the available data about reversal of the DOACs, I’m not 100% convinced that we’re significantly impacting outcomes by reversing these agents. We’re certainly running up the bill, but are we treating the patient or treating the providers? As long as the renal function remains intact, the DOACs clear quickly, perhaps more quickly than warfarin can typically be reversed with standard reversal agents.
Dr. Minichiello. Remember that this patient has a history of a GI bleed. We are going to start him on full-dose anticoagulation for stroke prevention for his nonvalvular AF. He’s also on aspirin, and he has stable coronary disease. He does not have any stents in place but he did have a remote non-ST elevation myocardial infarction (MI) a number of years ago. Do we feel that the risk of dual therapy—anticoagulation combined with antiplatelet therapy—outweighs the risks? And how do we approach that risk?
Dr. Barnes. This is an important point to discuss. There has been a lot of discussion in the literature recently. When I start this type of patient on an oral anticoagulant, I try to discontinue the antiplatelet agent because I know how much bleeding risk that brings. The European guidelines (for example, Eur Heart J. 2014;35[45]:3155-3179) have been forward thinking with this for the last couple of years and have highlighted that if there’s an indication for anticoagulation for patients with stable coronary disease, meaning no MI and no stent within the past year, then we should stop the antiplatelet agents after a year in order to reduce the risk of MI. This is based on a lot of older literature where warfarin was compared with aspirin and shown to be protective in coronary patients, but at the risk of bleeding.
It’s important because there have been recent studies that have raised questions, including a recent Swedish article in (Circulation. 2017;136:1183-1192) that suggested discontinuing aspirin led to increased mortality. But it’s important to look at the details. While that was true for most patients, it was not true for the group of patients who were on an oral anticoagulant. Many colleagues ask me questions about that particular paper and its media coverage. I tell them that for our patients on chronic oral anticoagulants, the paper supports the notion that there is not increased mortality when aspirin use is stopped. We know that aspirin plus an anticoagulant leads to increased bleeding, so I try to stop it for patients who have stable CAD but are on long-term anticoagulation.
Dr. Allen. This isn’t a new thought. Back in early 2012, the 9th edition of the American College of Chest Physicians (CHEST) Antithrombotic Guidelines probably gave us the best guidance that we had ever seen to help us address this issue. Since that time the cardiology guidelines have caught up to recommend that we do not need additional antiplatelet therapy for stable CAD, and, in fact, it should be limited even in the setting of acute coronary syndromes and percutaneous coronary intervention (PCI).
Dr. Minichiello. That’s a good point because people are not necessarily clear about when there would be an indication to continue dual therapy and when it is safe to go to monotherapy. Scenarios where benefit of dual therapy may outweigh risk suggested in the CHEST 2012 guidelines include acute coronary syndrome or a recent stent, high-risk mechanical valves, and history of coronary artery bypass surgery.
I think the important thing is to consider each case individually and not to reflexively continue aspirin therapy. Often what we see is once on aspirin—always on aspirin. Being thoughtful about it, we should acknowledge that it likely results in a 2-fold increased risk of bleeding and make sure that we believe that the benefit outweighs the risk.
Dr. Allen. I agree. We probably have better evidence in the CAD population, but what do we do for patients with significant peripheral vascular disease, or those patients with symptomatic carotid stenosis or history of strokes? Some of the European guidance suggests taking a similar approach to CAD, but these are the patients for whom stopping aspirin makes me more nervous.
Dr. Parra. This is a perfect example of where less is more. All too often the reflex is to continue aspirin treatment indefinitely because the patient has a history of acute coronary syndrome or even peripheral arterial disease, when the best thing to do would be to drop the aspirin. It involves an individualized risk assessment and underscores the need to periodically do a risk/benefit assessment in all patients on anticoagulants, whether it’s warfarin or a DOAC.
I’d like to take a moment and step back to the case in the context of the GI bleeding. When we look at patients with a history of GI bleeding, it is important to understand the circumstances that surround it. This individual had a GI bleed 3 years previously and peptic ulcer disease. In these situations I ask whether the patient was taking over-the-counter nonsteroidal anti-inflamitory drugs at the time, had excessive alcohol use, or was successfully treated for Helicobacter pylori. All of these may influence whether or not I think the GI bleed is significant to influence the DOAC choice.
The other thing I consider is that the overall risk of major GI bleeding in those pivotal DOAC trials was quite small, < 1.5% per year with dabigatran 150 mg twice daily and < 1% per year with apixaban. The numbers needed to harm were quite high, over 200 patients per year with dabigatran 150 mg twice daily vs warfarin and over 350 patients per year with edoxaban 60 mg daily vs warfarin. There are no head-to-head comparisons with DOACs, but this small increased risk vs warfarin may still be an important consideration in some patients. In addition, it is important to remember that intracranial hemorrhage and fatal bleeding was less in all the pivotal NVAF trials with the DOACs when compared with warfarin. So that is something we need to reinforce with patients when we discuss treatment regardless of the DOAC selected.
Case 2
Dr. Minichiello. The next case is a 63-year-old man with hypertension, diabetes mellitus, nonvalvular AF, and he is taking dabigatran for stroke prevention. He presents in the emergency department with chest pain, and he is found to have a non-ST elevation MI. He goes to the cath lab and he is found to have a lesion in his left circumflex. The patient receives a newer generation drug-eluting stent. What are we going to do with his anticoagulation? We know he’s going to get some antiplatelet therapy, but what are our thoughts on this?
Dr. Parra. This is something that we run into all too often. I think the estimates are about 20% to 30% of patients who have indications for anticoagulation also end up having ischemic heart disease that requires PCI. The second thing is that we know combining an anticoagulant with antiplatelet therapy is associated with a 4% to 16% risk of fatal and nonfatal bleeding, and we have found out in patients with ischemic heart disease that when they bleed, they also have a higher mortality rate.
We’re trying to find the optimal balance between ischemic and thrombotic risk and bleeding risk. This is where some of the risk assessment tools that we have come into play. First, we need to establish the thrombotic risk by considering the CHA2DS2-VASc score, and the factors associated with increasing bleeding risk and stent thrombosis. You have time to work this out because, initially, all patients that are at sufficiently high thrombotic risk will receive dual antiplatelet therapy and anticoagulation therapy for a given period. This gives providers time to use some of those resources and figure out a long-term plan for the patient.
Dr. Allen. The fear and loathing that this brings up comes back to some historic things that should be considered. What we did for drug-eluting stents or bare-metal stents comes from older data where different stent technology was used. The stents used today are safer with a lower risk of in-stent thrombosis. Historically, we knew what to do for ACS and PCI and we knew what to do for AF, but we didn’t know what to do when the 2 crossed paths. We would put patients on warfarin and say, “Well, for now, target the INR between 2 and 2.5 and good luck with that.” That was all we had. The good news is now we have some evidence to move away from the use of dual antiplatelet plus anticoagulant therapies.
There were 2 DOAC studies recently published: The PIONEER-AF trial used rivaroxaban and more recently the RE-DUAL PCI trial used dabigatran. Each of the studies had some issues, but they were both studied in AF populations and aimed to address this issue of triple therapy. The PIONEER-AF trial looked at a number of different scenarios on different doses of rivaroxaban with either single or dual antiplatelet therapy compared to triple therapy with warfarin. The RE-DUAL PCI trial with dabigatran was less complex. Both studies were powered to look at safety, and they did show that with single antiplatelet plus oral anticoagulation regimens, the incidence of major bleeding complications was reduced.
However, that brings up some issues about how the studies were conducted. Both studied AF populations and in some cases did not study doses approved for AF. Yet at the same time, the studies were not powered to look at stroke outcomes, which raises the question: Are we running the risk of giving up stroke efficacy for reduced bleeding? I don’t think that we’ve fully answered that, certainly not with the PIONEER AF trial.
Dr. Parra. I agree. When we look at those trials, 2 things come to mind. First, the doses of dabigatran used in the RE-DUAL PCI trial were doses that have been shown to be beneficial in the nonvalvular AF population. Second, a take-home point from those trials is the P2Y12 inhibitor that was utilized—close to 90% or more used clopidogrel in the PIONEER-AF trial. Clopidogrel remains the P2Y12 inhibitor of choice. One of the other findings is that the aspirin dosing should be low, < 100 mg daily, and that we need to consider routine use of proton pump inhibitors to protect against the bleeding that can be found with the antiplatelet agents.
Also of interest, the European Society of Cardiology (ESC) recently released a focused update with some excellent recommendations on dual antiplatelet therapy in coronary artery disease in which they incorporated the results from the PIONEER-AF-PCI trial. The RE-DUAL PCI trial had not been published when these came out. If you’re concerned about ischemic risk prevailing, ESC recommendations based upon risk stratification are triple therapy with aspirin, clopidogrel, and an oral anticoagulant for longer than 1 month and up to 6 months and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months; afterward just oral anticoagulation alone. If the concerns about bleeding prevail, then we have 2 different pathways: one limiting triple therapy to 1 month and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months. But the ESC also has a second recommendation for patients at high risk of bleeding, which is dual therapy with clopidogrel and an oral anticoagulant at the offset for up to 12 months. I found these guidelines to be particularly helpful in terms of how to put this into practice.
Dr. Allen. There’s still so much concern about in-stent thrombosis. Although a smaller trial, we knew from the WOEST trial that single antiplatelet therapy with warfarin was reasonable. We know from the PIONEER-AF and RE-DUAL PCI trials that we didn’t get significantly more in-stent thrombosis by giving up the second antiplatelet. Whether or not we answered the stroke question is another issue, but the cardiology societies are still hanging on to dual antiplatelet therapy. I question if that’s based on the older data and the older stent technologies.
Dr. Minichiello. This highlights again that often we have to consider these patients’ case-by-case analysis, and that these decisions require multidisciplinary input. It involves coming together and figuring out in this particular patient, which of those 3 options would be best. We have a lot more options than we did just a short year or year and a half ago with at least some data providing comfort that DOACs at effective doses for stroke prevention in nonvalvular AF look like they can be combined with single antiplatelet therapy for post-PCI patients.
Dr. Barnes. Speaking as a cardiologist, this is a question I encounter all the time. I think everything said here is really well taken. I’ll just summarize to say for patients who have acute coronary syndromes and AF. First, I’m okay with using warfarin and now I’m okay using the DOACs, but the anticoagulant needs to continue for stroke prevention. Second, the patient has to be on clopidogrel as a P2Y12 inhibitor because it has the lowest bleeding risk profile. Third, the patient doesn’t always need 1 year of dual antiplatelet therapy the way we used to think of it. With newer generation stents and ongoing anticoagulation, you can get away with shorter courses of your antiplatelets, albeit 3 or 6 months. Providers should have a conversation with patients and think hard about how to balance clotting vs that bleeding profile.
Case 3
Dr. Minichiello. This case involves a 66-year-old man who has nonvalvular AF and is on warfarin. He has CKD, and his CrCl is about 30. He has hypertension, diabetes mellitus, and he is going to go for a colonoscopy. The proceduralist lets you know about the date and wants to know whether he needs to be bridged. He also has a remote history of a transient ischemic attack.
Dr. Allen. Historically, we’ve had detailed guidance on how to risk assess patients with AF, venous thromboembolism (VT), and mechanical heart valves in the periprocedural period. From that risk assessment the guidance helped us determine whether or not we should offer periprocedural bridging with heparin or low-molecularweight heparin.
The issue is that the detailed guidance was always based on expert opinion not hard science and there was no great evidence that we were preventing thrombotic events or that there was a net clinical benefit to bridging. Some retrospective cohort studies started coming out around 2012 that demonstrated an increased incidence of major bleeding events associated with bridging with no reduction in thrombotic complications. Some might argue that this is because thrombotic complications are so rare that you would have to have tens of thousands of patients for adequate power. Nonetheless, these studies were adequately powered to show a significant increase in major bleeding.
The best prospective trial data we have for this population comes from the BRIDGE trial, which randomized AF patients to receive dalteparin bridge therapy vs placebo during periprocedural interruptions in warfarin therapy. It too demonstrated a significant increase in the risk of bleeding complications associated with bridging with no significant reduction in the risk of thromboembolic events. Critics point out that some of the higher risk patients were underrepresented, and the same could be said about some of the other retrospective studies in the VT and mechanical valve populations.
We have waited for quite a while for guidances to catch up with these data. The most recent guidance published would apply to this patient. It was guidance for the AF population published by the American College of Cardiology (ACC) in early 2017
To make a decision to bridge, you not only have to make an assumption that your patient is at such extraordinary thrombotic risk that you would find some reduction in thrombotic events associated with bridging, but also that the benefit is going to be so great that it would overcome this very clear increased risk of bleeding, resulting in a net clinical benefit. Only then would it make sense to bridge. Based on the available data, it is quite possible that no such patient population exists.
Dr. Parra. I agree. We found from the BRIDGE trial, with the caveat that you pointed out, that high or moderately high thrombotic risk individuals weren’t as well represented, that there is at least a 2.5-fold increase in major bleeding. We also know that higher thrombotic-risk patients tend to also have higher bleeding risk. So in high thrombotic-risk patients, while there is uncertainty about whether or not there’s going to be a thrombotic benefit from bridging, we can be confident there will be an increased risk of major bleeding perhaps even more than that seen in the BRIDGE trial.
One thing that really illustrates this is the 2017 American Heart Association/ACC-focused update on the 2014 guideline for the management of patients with valvular heart disease. The guideline has a section on bridging therapy for prosthetic valves. And that was recently downgraded from a Class I recommendation to Class IIA, recognizing that the data are limited in terms of even when to bridge prosthetic valves.
Dr. Allen. Bridging is an area where we continue to do something that we know causes harm because we hope it has some benefit. Despite what some societal guidance still says, I very rarely bridge patients. If I do, it’s because the patient and/or caregivers have heard the facts and have opted to do it.
Dr. Minichiello. Practically speaking, the data—the retrospective data, the observational data, the BRIDGE trial, etc—definitely make us step back and think about bridging and realize that it’s a HIGH RISK of intervention. We really need to be informing patients about that risk. We know that bridging increases the risk of bleeding. We do not have data showing a reduction in the risk of thromboembolic disease with bridge therapy. That’s all based on what we think would happen, what we hope would be the result of bridge therapy, but in truth we do not know if this is the case. The risks of bridge therapy must be weighed heavily each time we consider using it.
In my practice I reserve bridge therapy, which we know is associated with increased harm for patients in whom we think there is the very highest risk of thromboembolism, ie, those with very recent arterial or venous thrombosis, and by recently, I mean within the past 1 to 3 months; patients who have very severe thrombophilia like antitphospholipid antibody syndrome, some cancer patients, and those with mechanical valves in the mitral position or those with high-risk mechanical aortic valves. 
I don’t bridge most AF patients. In fact, I can’t remember the last time I bridged an AF patient. I do know that this is somewhat discordant with the ACC recommendations, but absent the data to support bridge therapy, I’m really concerned that the risk outweighs the benefit. This is particularly true in our VA population where the bleeding risk is high because of CKD or a history of bleeding or thrombocytopenia or concomitant aspirin or something else.
Dr. Barnes. We know from some studies that have been published that the biggest driver of a clinician deciding whether or not a patient should bridge is actually whether or not they’ve had a stroke. The truth is that the BRIDGE trial enrolled a sizeable population; it was about 15% of patients with a prior TIA or stroke. So it gave us some insight. Despite that, our event rates for thromboembolism, arterial thromboembolism, including stroke, were quite low.
I tend to look far more at the collection of risk factors and not just at a history of stroke. Now as you mentioned, Dr. Minichiello, a stroke within the past 1 to 3 months, I ask, “does this procedure even need to happen?” But outside of that, it’s not a history of stroke that’s going to make the decision for me: It’s all the risk factors together.
There’s an ongoing study, the PERIOP 2 study, that is enrolling patients at higher risk for stroke and patients with mechanical valves. This study may give us more insight into exactly what kind of risks these patients are at and whether they get benefit from bridging. But in the meantime, I’m really reserving bridging for my highest risk patients, those with multiple risk factors, CHADS2 scores of 5 and 6 or CHA2DS2-VASc of 7, 8, 9, and those with a recent VT or mechanical mitral valves.
Click here to read the digital edition.
Case 1
Tracy Minichiello, MD. The first case we’ll discuss is a 75-year-old man with mild chronic kidney disease (CKD). His calculated creatinine clearance (CrCl) is about 52 mL/min, and he has a remote history of a gastrointestinal (GI) bleeding 3 years previously from a peptic ulcer. He presents with new onset nonvalvular atrial fibrillation (AF), and he’s already on aspirin for his stable coronary artery disease (CAD).
How do we think about anticoagulant selection in this patient? We have a number of new oral anticoagulants and we have warfarin. How do we decide between warfarin vs one of the direct-acting oral anticoagulants (DOACs)? If we choose a DOAC, which one would we select?
David Parra, PharmD. The first step for anticoagulation is to assess a patient’s thromboembolic risk utilizing the CHA2DS2-VASc and bleeding risk using a HAS-BLED score, or something similar. The next question is which oral anticoagulant to use. We have widespread experience with warfarin and can measure the anticoagulant effect easily. Warfarin has a long duration of action, so perhaps it’s more forgiving if you miss a dose. It also has an antidote. Lastly, organ dysfunction doesn’t preclude use of warfarin as you can still monitor the anticoagulant effect. So there still may be patients that may benefit significantly from warfarin vs a DOAC.
On the flip side, DOACs are easier to use and perform quite acceptably in comparison with warfarin in nonvalvular AF. There are some scenarios where a specific DOAC may be preferred over another, such as recent GI bleeding.
Dr. Minichiello. Do you consider renal function, bleeding history, or concomitant antiplatelet therapy?
Geoffrey Barnes, MD, MSc. A couple of factors are relevant. I think we should consider renal function for this gentleman. However, I look at some of the other features as Dr. Parra suggested. What’s the likelihood that this patient is going to take the medicine as prescribed? Is a twice-a-day regimen going to be something that’s particularly challenging? I also look at the real-world vs randomized trial experience.
This patient has a remote GI bleeding history. Some of the real-world data suggest there might be some more GI bleeding with rivaroxaban, but across the board, apixaban (in both the randomized trials and much of the real-world data) seem to have a favorable bleeding risk profile. For a patient who is open and reliable for taking medicine twice a day, apixaban might be a good option as long as we make sure that the dose is appropriate.
Arthur L. Allen, PharmD, CACP. In pivotal trial experience, dabigatran and rivaroxaban demonstrated an increased incidence of GI bleeding compared with warfarin. In some of the real-world studies, rivaroxaban mirrors warfarin with regard to bleeding, whereas dabigatran and apixaban have a lower incidence. In the pivotal trials, apixaban did not have a trigger of increased GI bleeding, but I would let the details of this patient’s GI bleeding history help me determine how important an issue this is at this point.
The other thing that is important to understand when considering choice of agents: As Dr. Parra mentioned, we do have quite a bit of experience with warfarin. But comparing the quality of evidence, the DOACs have been investigated in a far more rigorous fashion and in far more patients than warfarin ever was in its more than 60 years on the market. For example, the RE-LY trial alone enrolled more than 18,000 patients. Each of the DOACs have been studied in tens of thousands of patients for their approved indications. Further, we shouldn’t forget that the risk of intracranial hemorrhage is reduced by roughly 50% by choosing a DOAC over warfarin, which should be a consideration in this elderly gentleman.
Dr. Minichiello. In the veteran population, there is a sense of comfort with warfarin, and some concerns have been raised over a lack of reversibility for the newer agents. We have patients who have trepidation about starting one of the new anticoagulants. However, there is a marked reduction in the risk of the most devastating bleeding complication, namely intracranial hemorrhage, making the use of these agents most compelling. And when they did have bleeding complications, at least in the trials, their outcomes were no worse than they were with warfarin, where there is a reversal agent. In most cases the outcomes were actually better.
Dr. Barnes. You often have to remind patients that there was no reversal agent in these huge trials where the DOACs showed similar or safer bleeding risk profiles, especially for the most serious bleeding, such as intracranial hemorrhage. I find patients often are reassured by knowing that.
Dr. Allen. I agree that there is concern about the lack of reversibility, but I think it has been completely overplayed. In the pivotal trials, patients who bled on DOAC therapy actually had better outcomes than those that bled on warfarin. This includes intracranial hemorrhage. There was a paper published in Stroke in 2012 that evaluated the subgroup of patients in the RE-LY trial that suffered intracranial hemorrhage. Patients on dabigatran actually fared better despite a lack of a specific reversal agent. When evaluating the available data about reversal of the DOACs, I’m not 100% convinced that we’re significantly impacting outcomes by reversing these agents. We’re certainly running up the bill, but are we treating the patient or treating the providers? As long as the renal function remains intact, the DOACs clear quickly, perhaps more quickly than warfarin can typically be reversed with standard reversal agents.
Dr. Minichiello. Remember that this patient has a history of a GI bleed. We are going to start him on full-dose anticoagulation for stroke prevention for his nonvalvular AF. He’s also on aspirin, and he has stable coronary disease. He does not have any stents in place but he did have a remote non-ST elevation myocardial infarction (MI) a number of years ago. Do we feel that the risk of dual therapy—anticoagulation combined with antiplatelet therapy—outweighs the risks? And how do we approach that risk?
Dr. Barnes. This is an important point to discuss. There has been a lot of discussion in the literature recently. When I start this type of patient on an oral anticoagulant, I try to discontinue the antiplatelet agent because I know how much bleeding risk that brings. The European guidelines (for example, Eur Heart J. 2014;35[45]:3155-3179) have been forward thinking with this for the last couple of years and have highlighted that if there’s an indication for anticoagulation for patients with stable coronary disease, meaning no MI and no stent within the past year, then we should stop the antiplatelet agents after a year in order to reduce the risk of MI. This is based on a lot of older literature where warfarin was compared with aspirin and shown to be protective in coronary patients, but at the risk of bleeding.
It’s important because there have been recent studies that have raised questions, including a recent Swedish article in (Circulation. 2017;136:1183-1192) that suggested discontinuing aspirin led to increased mortality. But it’s important to look at the details. While that was true for most patients, it was not true for the group of patients who were on an oral anticoagulant. Many colleagues ask me questions about that particular paper and its media coverage. I tell them that for our patients on chronic oral anticoagulants, the paper supports the notion that there is not increased mortality when aspirin use is stopped. We know that aspirin plus an anticoagulant leads to increased bleeding, so I try to stop it for patients who have stable CAD but are on long-term anticoagulation.
Dr. Allen. This isn’t a new thought. Back in early 2012, the 9th edition of the American College of Chest Physicians (CHEST) Antithrombotic Guidelines probably gave us the best guidance that we had ever seen to help us address this issue. Since that time the cardiology guidelines have caught up to recommend that we do not need additional antiplatelet therapy for stable CAD, and, in fact, it should be limited even in the setting of acute coronary syndromes and percutaneous coronary intervention (PCI).
Dr. Minichiello. That’s a good point because people are not necessarily clear about when there would be an indication to continue dual therapy and when it is safe to go to monotherapy. Scenarios where benefit of dual therapy may outweigh risk suggested in the CHEST 2012 guidelines include acute coronary syndrome or a recent stent, high-risk mechanical valves, and history of coronary artery bypass surgery.
I think the important thing is to consider each case individually and not to reflexively continue aspirin therapy. Often what we see is once on aspirin—always on aspirin. Being thoughtful about it, we should acknowledge that it likely results in a 2-fold increased risk of bleeding and make sure that we believe that the benefit outweighs the risk.
Dr. Allen. I agree. We probably have better evidence in the CAD population, but what do we do for patients with significant peripheral vascular disease, or those patients with symptomatic carotid stenosis or history of strokes? Some of the European guidance suggests taking a similar approach to CAD, but these are the patients for whom stopping aspirin makes me more nervous.
Dr. Parra. This is a perfect example of where less is more. All too often the reflex is to continue aspirin treatment indefinitely because the patient has a history of acute coronary syndrome or even peripheral arterial disease, when the best thing to do would be to drop the aspirin. It involves an individualized risk assessment and underscores the need to periodically do a risk/benefit assessment in all patients on anticoagulants, whether it’s warfarin or a DOAC.
I’d like to take a moment and step back to the case in the context of the GI bleeding. When we look at patients with a history of GI bleeding, it is important to understand the circumstances that surround it. This individual had a GI bleed 3 years previously and peptic ulcer disease. In these situations I ask whether the patient was taking over-the-counter nonsteroidal anti-inflamitory drugs at the time, had excessive alcohol use, or was successfully treated for Helicobacter pylori. All of these may influence whether or not I think the GI bleed is significant to influence the DOAC choice.
The other thing I consider is that the overall risk of major GI bleeding in those pivotal DOAC trials was quite small, < 1.5% per year with dabigatran 150 mg twice daily and < 1% per year with apixaban. The numbers needed to harm were quite high, over 200 patients per year with dabigatran 150 mg twice daily vs warfarin and over 350 patients per year with edoxaban 60 mg daily vs warfarin. There are no head-to-head comparisons with DOACs, but this small increased risk vs warfarin may still be an important consideration in some patients. In addition, it is important to remember that intracranial hemorrhage and fatal bleeding was less in all the pivotal NVAF trials with the DOACs when compared with warfarin. So that is something we need to reinforce with patients when we discuss treatment regardless of the DOAC selected.
Case 2
Dr. Minichiello. The next case is a 63-year-old man with hypertension, diabetes mellitus, nonvalvular AF, and he is taking dabigatran for stroke prevention. He presents in the emergency department with chest pain, and he is found to have a non-ST elevation MI. He goes to the cath lab and he is found to have a lesion in his left circumflex. The patient receives a newer generation drug-eluting stent. What are we going to do with his anticoagulation? We know he’s going to get some antiplatelet therapy, but what are our thoughts on this?
Dr. Parra. This is something that we run into all too often. I think the estimates are about 20% to 30% of patients who have indications for anticoagulation also end up having ischemic heart disease that requires PCI. The second thing is that we know combining an anticoagulant with antiplatelet therapy is associated with a 4% to 16% risk of fatal and nonfatal bleeding, and we have found out in patients with ischemic heart disease that when they bleed, they also have a higher mortality rate.
We’re trying to find the optimal balance between ischemic and thrombotic risk and bleeding risk. This is where some of the risk assessment tools that we have come into play. First, we need to establish the thrombotic risk by considering the CHA2DS2-VASc score, and the factors associated with increasing bleeding risk and stent thrombosis. You have time to work this out because, initially, all patients that are at sufficiently high thrombotic risk will receive dual antiplatelet therapy and anticoagulation therapy for a given period. This gives providers time to use some of those resources and figure out a long-term plan for the patient.
Dr. Allen. The fear and loathing that this brings up comes back to some historic things that should be considered. What we did for drug-eluting stents or bare-metal stents comes from older data where different stent technology was used. The stents used today are safer with a lower risk of in-stent thrombosis. Historically, we knew what to do for ACS and PCI and we knew what to do for AF, but we didn’t know what to do when the 2 crossed paths. We would put patients on warfarin and say, “Well, for now, target the INR between 2 and 2.5 and good luck with that.” That was all we had. The good news is now we have some evidence to move away from the use of dual antiplatelet plus anticoagulant therapies.
There were 2 DOAC studies recently published: The PIONEER-AF trial used rivaroxaban and more recently the RE-DUAL PCI trial used dabigatran. Each of the studies had some issues, but they were both studied in AF populations and aimed to address this issue of triple therapy. The PIONEER-AF trial looked at a number of different scenarios on different doses of rivaroxaban with either single or dual antiplatelet therapy compared to triple therapy with warfarin. The RE-DUAL PCI trial with dabigatran was less complex. Both studies were powered to look at safety, and they did show that with single antiplatelet plus oral anticoagulation regimens, the incidence of major bleeding complications was reduced.
However, that brings up some issues about how the studies were conducted. Both studied AF populations and in some cases did not study doses approved for AF. Yet at the same time, the studies were not powered to look at stroke outcomes, which raises the question: Are we running the risk of giving up stroke efficacy for reduced bleeding? I don’t think that we’ve fully answered that, certainly not with the PIONEER AF trial.
Dr. Parra. I agree. When we look at those trials, 2 things come to mind. First, the doses of dabigatran used in the RE-DUAL PCI trial were doses that have been shown to be beneficial in the nonvalvular AF population. Second, a take-home point from those trials is the P2Y12 inhibitor that was utilized—close to 90% or more used clopidogrel in the PIONEER-AF trial. Clopidogrel remains the P2Y12 inhibitor of choice. One of the other findings is that the aspirin dosing should be low, < 100 mg daily, and that we need to consider routine use of proton pump inhibitors to protect against the bleeding that can be found with the antiplatelet agents.
Also of interest, the European Society of Cardiology (ESC) recently released a focused update with some excellent recommendations on dual antiplatelet therapy in coronary artery disease in which they incorporated the results from the PIONEER-AF-PCI trial. The RE-DUAL PCI trial had not been published when these came out. If you’re concerned about ischemic risk prevailing, ESC recommendations based upon risk stratification are triple therapy with aspirin, clopidogrel, and an oral anticoagulant for longer than 1 month and up to 6 months and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months; afterward just oral anticoagulation alone. If the concerns about bleeding prevail, then we have 2 different pathways: one limiting triple therapy to 1 month and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months. But the ESC also has a second recommendation for patients at high risk of bleeding, which is dual therapy with clopidogrel and an oral anticoagulant at the offset for up to 12 months. I found these guidelines to be particularly helpful in terms of how to put this into practice.
Dr. Allen. There’s still so much concern about in-stent thrombosis. Although a smaller trial, we knew from the WOEST trial that single antiplatelet therapy with warfarin was reasonable. We know from the PIONEER-AF and RE-DUAL PCI trials that we didn’t get significantly more in-stent thrombosis by giving up the second antiplatelet. Whether or not we answered the stroke question is another issue, but the cardiology societies are still hanging on to dual antiplatelet therapy. I question if that’s based on the older data and the older stent technologies.
Dr. Minichiello. This highlights again that often we have to consider these patients’ case-by-case analysis, and that these decisions require multidisciplinary input. It involves coming together and figuring out in this particular patient, which of those 3 options would be best. We have a lot more options than we did just a short year or year and a half ago with at least some data providing comfort that DOACs at effective doses for stroke prevention in nonvalvular AF look like they can be combined with single antiplatelet therapy for post-PCI patients.
Dr. Barnes. Speaking as a cardiologist, this is a question I encounter all the time. I think everything said here is really well taken. I’ll just summarize to say for patients who have acute coronary syndromes and AF. First, I’m okay with using warfarin and now I’m okay using the DOACs, but the anticoagulant needs to continue for stroke prevention. Second, the patient has to be on clopidogrel as a P2Y12 inhibitor because it has the lowest bleeding risk profile. Third, the patient doesn’t always need 1 year of dual antiplatelet therapy the way we used to think of it. With newer generation stents and ongoing anticoagulation, you can get away with shorter courses of your antiplatelets, albeit 3 or 6 months. Providers should have a conversation with patients and think hard about how to balance clotting vs that bleeding profile.
Case 3
Dr. Minichiello. This case involves a 66-year-old man who has nonvalvular AF and is on warfarin. He has CKD, and his CrCl is about 30. He has hypertension, diabetes mellitus, and he is going to go for a colonoscopy. The proceduralist lets you know about the date and wants to know whether he needs to be bridged. He also has a remote history of a transient ischemic attack.
Dr. Allen. Historically, we’ve had detailed guidance on how to risk assess patients with AF, venous thromboembolism (VT), and mechanical heart valves in the periprocedural period. From that risk assessment the guidance helped us determine whether or not we should offer periprocedural bridging with heparin or low-molecularweight heparin.
The issue is that the detailed guidance was always based on expert opinion not hard science and there was no great evidence that we were preventing thrombotic events or that there was a net clinical benefit to bridging. Some retrospective cohort studies started coming out around 2012 that demonstrated an increased incidence of major bleeding events associated with bridging with no reduction in thrombotic complications. Some might argue that this is because thrombotic complications are so rare that you would have to have tens of thousands of patients for adequate power. Nonetheless, these studies were adequately powered to show a significant increase in major bleeding.
The best prospective trial data we have for this population comes from the BRIDGE trial, which randomized AF patients to receive dalteparin bridge therapy vs placebo during periprocedural interruptions in warfarin therapy. It too demonstrated a significant increase in the risk of bleeding complications associated with bridging with no significant reduction in the risk of thromboembolic events. Critics point out that some of the higher risk patients were underrepresented, and the same could be said about some of the other retrospective studies in the VT and mechanical valve populations.
We have waited for quite a while for guidances to catch up with these data. The most recent guidance published would apply to this patient. It was guidance for the AF population published by the American College of Cardiology (ACC) in early 2017
To make a decision to bridge, you not only have to make an assumption that your patient is at such extraordinary thrombotic risk that you would find some reduction in thrombotic events associated with bridging, but also that the benefit is going to be so great that it would overcome this very clear increased risk of bleeding, resulting in a net clinical benefit. Only then would it make sense to bridge. Based on the available data, it is quite possible that no such patient population exists.
Dr. Parra. I agree. We found from the BRIDGE trial, with the caveat that you pointed out, that high or moderately high thrombotic risk individuals weren’t as well represented, that there is at least a 2.5-fold increase in major bleeding. We also know that higher thrombotic-risk patients tend to also have higher bleeding risk. So in high thrombotic-risk patients, while there is uncertainty about whether or not there’s going to be a thrombotic benefit from bridging, we can be confident there will be an increased risk of major bleeding perhaps even more than that seen in the BRIDGE trial.
One thing that really illustrates this is the 2017 American Heart Association/ACC-focused update on the 2014 guideline for the management of patients with valvular heart disease. The guideline has a section on bridging therapy for prosthetic valves. And that was recently downgraded from a Class I recommendation to Class IIA, recognizing that the data are limited in terms of even when to bridge prosthetic valves.
Dr. Allen. Bridging is an area where we continue to do something that we know causes harm because we hope it has some benefit. Despite what some societal guidance still says, I very rarely bridge patients. If I do, it’s because the patient and/or caregivers have heard the facts and have opted to do it.
Dr. Minichiello. Practically speaking, the data—the retrospective data, the observational data, the BRIDGE trial, etc—definitely make us step back and think about bridging and realize that it’s a HIGH RISK of intervention. We really need to be informing patients about that risk. We know that bridging increases the risk of bleeding. We do not have data showing a reduction in the risk of thromboembolic disease with bridge therapy. That’s all based on what we think would happen, what we hope would be the result of bridge therapy, but in truth we do not know if this is the case. The risks of bridge therapy must be weighed heavily each time we consider using it.
In my practice I reserve bridge therapy, which we know is associated with increased harm for patients in whom we think there is the very highest risk of thromboembolism, ie, those with very recent arterial or venous thrombosis, and by recently, I mean within the past 1 to 3 months; patients who have very severe thrombophilia like antitphospholipid antibody syndrome, some cancer patients, and those with mechanical valves in the mitral position or those with high-risk mechanical aortic valves.
I don’t bridge most AF patients. In fact, I can’t remember the last time I bridged an AF patient. I do know that this is somewhat discordant with the ACC recommendations, but absent the data to support bridge therapy, I’m really concerned that the risk outweighs the benefit. This is particularly true in our VA population where the bleeding risk is high because of CKD or a history of bleeding or thrombocytopenia or concomitant aspirin or something else.
Dr. Barnes. We know from some studies that have been published that the biggest driver of a clinician deciding whether or not a patient should bridge is actually whether or not they’ve had a stroke. The truth is that the BRIDGE trial enrolled a sizeable population; it was about 15% of patients with a prior TIA or stroke. So it gave us some insight. Despite that, our event rates for thromboembolism, arterial thromboembolism, including stroke, were quite low.
I tend to look far more at the collection of risk factors and not just at a history of stroke. Now as you mentioned, Dr. Minichiello, a stroke within the past 1 to 3 months, I ask, “does this procedure even need to happen?” But outside of that, it’s not a history of stroke that’s going to make the decision for me: It’s all the risk factors together.
There’s an ongoing study, the PERIOP 2 study, that is enrolling patients at higher risk for stroke and patients with mechanical valves. This study may give us more insight into exactly what kind of risks these patients are at and whether they get benefit from bridging. But in the meantime, I’m really reserving bridging for my highest risk patients, those with multiple risk factors, CHADS2 scores of 5 and 6 or CHA2DS2-VASc of 7, 8, 9, and those with a recent VT or mechanical mitral valves.
Click here to read the digital edition.
Case 1
Tracy Minichiello, MD. The first case we’ll discuss is a 75-year-old man with mild chronic kidney disease (CKD). His calculated creatinine clearance (CrCl) is about 52 mL/min, and he has a remote history of a gastrointestinal (GI) bleeding 3 years previously from a peptic ulcer. He presents with new onset nonvalvular atrial fibrillation (AF), and he’s already on aspirin for his stable coronary artery disease (CAD).
How do we think about anticoagulant selection in this patient? We have a number of new oral anticoagulants and we have warfarin. How do we decide between warfarin vs one of the direct-acting oral anticoagulants (DOACs)? If we choose a DOAC, which one would we select?
David Parra, PharmD. The first step for anticoagulation is to assess a patient’s thromboembolic risk utilizing the CHA2DS2-VASc and bleeding risk using a HAS-BLED score, or something similar. The next question is which oral anticoagulant to use. We have widespread experience with warfarin and can measure the anticoagulant effect easily. Warfarin has a long duration of action, so perhaps it’s more forgiving if you miss a dose. It also has an antidote. Lastly, organ dysfunction doesn’t preclude use of warfarin as you can still monitor the anticoagulant effect. So there still may be patients that may benefit significantly from warfarin vs a DOAC.
On the flip side, DOACs are easier to use and perform quite acceptably in comparison with warfarin in nonvalvular AF. There are some scenarios where a specific DOAC may be preferred over another, such as recent GI bleeding.
Dr. Minichiello. Do you consider renal function, bleeding history, or concomitant antiplatelet therapy?
Geoffrey Barnes, MD, MSc. A couple of factors are relevant. I think we should consider renal function for this gentleman. However, I look at some of the other features as Dr. Parra suggested. What’s the likelihood that this patient is going to take the medicine as prescribed? Is a twice-a-day regimen going to be something that’s particularly challenging? I also look at the real-world vs randomized trial experience.
This patient has a remote GI bleeding history. Some of the real-world data suggest there might be some more GI bleeding with rivaroxaban, but across the board, apixaban (in both the randomized trials and much of the real-world data) seem to have a favorable bleeding risk profile. For a patient who is open and reliable for taking medicine twice a day, apixaban might be a good option as long as we make sure that the dose is appropriate.
Arthur L. Allen, PharmD, CACP. In pivotal trial experience, dabigatran and rivaroxaban demonstrated an increased incidence of GI bleeding compared with warfarin. In some of the real-world studies, rivaroxaban mirrors warfarin with regard to bleeding, whereas dabigatran and apixaban have a lower incidence. In the pivotal trials, apixaban did not have a trigger of increased GI bleeding, but I would let the details of this patient’s GI bleeding history help me determine how important an issue this is at this point.
The other thing that is important to understand when considering choice of agents: As Dr. Parra mentioned, we do have quite a bit of experience with warfarin. But comparing the quality of evidence, the DOACs have been investigated in a far more rigorous fashion and in far more patients than warfarin ever was in its more than 60 years on the market. For example, the RE-LY trial alone enrolled more than 18,000 patients. Each of the DOACs have been studied in tens of thousands of patients for their approved indications. Further, we shouldn’t forget that the risk of intracranial hemorrhage is reduced by roughly 50% by choosing a DOAC over warfarin, which should be a consideration in this elderly gentleman.
Dr. Minichiello. In the veteran population, there is a sense of comfort with warfarin, and some concerns have been raised over a lack of reversibility for the newer agents. We have patients who have trepidation about starting one of the new anticoagulants. However, there is a marked reduction in the risk of the most devastating bleeding complication, namely intracranial hemorrhage, making the use of these agents most compelling. And when they did have bleeding complications, at least in the trials, their outcomes were no worse than they were with warfarin, where there is a reversal agent. In most cases the outcomes were actually better.
Dr. Barnes. You often have to remind patients that there was no reversal agent in these huge trials where the DOACs showed similar or safer bleeding risk profiles, especially for the most serious bleeding, such as intracranial hemorrhage. I find patients often are reassured by knowing that.
Dr. Allen. I agree that there is concern about the lack of reversibility, but I think it has been completely overplayed. In the pivotal trials, patients who bled on DOAC therapy actually had better outcomes than those that bled on warfarin. This includes intracranial hemorrhage. There was a paper published in Stroke in 2012 that evaluated the subgroup of patients in the RE-LY trial that suffered intracranial hemorrhage. Patients on dabigatran actually fared better despite a lack of a specific reversal agent. When evaluating the available data about reversal of the DOACs, I’m not 100% convinced that we’re significantly impacting outcomes by reversing these agents. We’re certainly running up the bill, but are we treating the patient or treating the providers? As long as the renal function remains intact, the DOACs clear quickly, perhaps more quickly than warfarin can typically be reversed with standard reversal agents.
Dr. Minichiello. Remember that this patient has a history of a GI bleed. We are going to start him on full-dose anticoagulation for stroke prevention for his nonvalvular AF. He’s also on aspirin, and he has stable coronary disease. He does not have any stents in place but he did have a remote non-ST elevation myocardial infarction (MI) a number of years ago. Do we feel that the risk of dual therapy—anticoagulation combined with antiplatelet therapy—outweighs the risks? And how do we approach that risk?
Dr. Barnes. This is an important point to discuss. There has been a lot of discussion in the literature recently. When I start this type of patient on an oral anticoagulant, I try to discontinue the antiplatelet agent because I know how much bleeding risk that brings. The European guidelines (for example, Eur Heart J. 2014;35[45]:3155-3179) have been forward thinking with this for the last couple of years and have highlighted that if there’s an indication for anticoagulation for patients with stable coronary disease, meaning no MI and no stent within the past year, then we should stop the antiplatelet agents after a year in order to reduce the risk of MI. This is based on a lot of older literature where warfarin was compared with aspirin and shown to be protective in coronary patients, but at the risk of bleeding.
It’s important because there have been recent studies that have raised questions, including a recent Swedish article in (Circulation. 2017;136:1183-1192) that suggested discontinuing aspirin led to increased mortality. But it’s important to look at the details. While that was true for most patients, it was not true for the group of patients who were on an oral anticoagulant. Many colleagues ask me questions about that particular paper and its media coverage. I tell them that for our patients on chronic oral anticoagulants, the paper supports the notion that there is not increased mortality when aspirin use is stopped. We know that aspirin plus an anticoagulant leads to increased bleeding, so I try to stop it for patients who have stable CAD but are on long-term anticoagulation.
Dr. Allen. This isn’t a new thought. Back in early 2012, the 9th edition of the American College of Chest Physicians (CHEST) Antithrombotic Guidelines probably gave us the best guidance that we had ever seen to help us address this issue. Since that time the cardiology guidelines have caught up to recommend that we do not need additional antiplatelet therapy for stable CAD, and, in fact, it should be limited even in the setting of acute coronary syndromes and percutaneous coronary intervention (PCI).
Dr. Minichiello. That’s a good point because people are not necessarily clear about when there would be an indication to continue dual therapy and when it is safe to go to monotherapy. Scenarios where benefit of dual therapy may outweigh risk suggested in the CHEST 2012 guidelines include acute coronary syndrome or a recent stent, high-risk mechanical valves, and history of coronary artery bypass surgery.
I think the important thing is to consider each case individually and not to reflexively continue aspirin therapy. Often what we see is once on aspirin—always on aspirin. Being thoughtful about it, we should acknowledge that it likely results in a 2-fold increased risk of bleeding and make sure that we believe that the benefit outweighs the risk.
Dr. Allen. I agree. We probably have better evidence in the CAD population, but what do we do for patients with significant peripheral vascular disease, or those patients with symptomatic carotid stenosis or history of strokes? Some of the European guidance suggests taking a similar approach to CAD, but these are the patients for whom stopping aspirin makes me more nervous.
Dr. Parra. This is a perfect example of where less is more. All too often the reflex is to continue aspirin treatment indefinitely because the patient has a history of acute coronary syndrome or even peripheral arterial disease, when the best thing to do would be to drop the aspirin. It involves an individualized risk assessment and underscores the need to periodically do a risk/benefit assessment in all patients on anticoagulants, whether it’s warfarin or a DOAC.
I’d like to take a moment and step back to the case in the context of the GI bleeding. When we look at patients with a history of GI bleeding, it is important to understand the circumstances that surround it. This individual had a GI bleed 3 years previously and peptic ulcer disease. In these situations I ask whether the patient was taking over-the-counter nonsteroidal anti-inflamitory drugs at the time, had excessive alcohol use, or was successfully treated for Helicobacter pylori. All of these may influence whether or not I think the GI bleed is significant to influence the DOAC choice.
The other thing I consider is that the overall risk of major GI bleeding in those pivotal DOAC trials was quite small, < 1.5% per year with dabigatran 150 mg twice daily and < 1% per year with apixaban. The numbers needed to harm were quite high, over 200 patients per year with dabigatran 150 mg twice daily vs warfarin and over 350 patients per year with edoxaban 60 mg daily vs warfarin. There are no head-to-head comparisons with DOACs, but this small increased risk vs warfarin may still be an important consideration in some patients. In addition, it is important to remember that intracranial hemorrhage and fatal bleeding was less in all the pivotal NVAF trials with the DOACs when compared with warfarin. So that is something we need to reinforce with patients when we discuss treatment regardless of the DOAC selected.
Case 2
Dr. Minichiello. The next case is a 63-year-old man with hypertension, diabetes mellitus, nonvalvular AF, and he is taking dabigatran for stroke prevention. He presents in the emergency department with chest pain, and he is found to have a non-ST elevation MI. He goes to the cath lab and he is found to have a lesion in his left circumflex. The patient receives a newer generation drug-eluting stent. What are we going to do with his anticoagulation? We know he’s going to get some antiplatelet therapy, but what are our thoughts on this?
Dr. Parra. This is something that we run into all too often. I think the estimates are about 20% to 30% of patients who have indications for anticoagulation also end up having ischemic heart disease that requires PCI. The second thing is that we know combining an anticoagulant with antiplatelet therapy is associated with a 4% to 16% risk of fatal and nonfatal bleeding, and we have found out in patients with ischemic heart disease that when they bleed, they also have a higher mortality rate.
We’re trying to find the optimal balance between ischemic and thrombotic risk and bleeding risk. This is where some of the risk assessment tools that we have come into play. First, we need to establish the thrombotic risk by considering the CHA2DS2-VASc score, and the factors associated with increasing bleeding risk and stent thrombosis. You have time to work this out because, initially, all patients that are at sufficiently high thrombotic risk will receive dual antiplatelet therapy and anticoagulation therapy for a given period. This gives providers time to use some of those resources and figure out a long-term plan for the patient.
Dr. Allen. The fear and loathing that this brings up comes back to some historic things that should be considered. What we did for drug-eluting stents or bare-metal stents comes from older data where different stent technology was used. The stents used today are safer with a lower risk of in-stent thrombosis. Historically, we knew what to do for ACS and PCI and we knew what to do for AF, but we didn’t know what to do when the 2 crossed paths. We would put patients on warfarin and say, “Well, for now, target the INR between 2 and 2.5 and good luck with that.” That was all we had. The good news is now we have some evidence to move away from the use of dual antiplatelet plus anticoagulant therapies.
There were 2 DOAC studies recently published: The PIONEER-AF trial used rivaroxaban and more recently the RE-DUAL PCI trial used dabigatran. Each of the studies had some issues, but they were both studied in AF populations and aimed to address this issue of triple therapy. The PIONEER-AF trial looked at a number of different scenarios on different doses of rivaroxaban with either single or dual antiplatelet therapy compared to triple therapy with warfarin. The RE-DUAL PCI trial with dabigatran was less complex. Both studies were powered to look at safety, and they did show that with single antiplatelet plus oral anticoagulation regimens, the incidence of major bleeding complications was reduced.
However, that brings up some issues about how the studies were conducted. Both studied AF populations and in some cases did not study doses approved for AF. Yet at the same time, the studies were not powered to look at stroke outcomes, which raises the question: Are we running the risk of giving up stroke efficacy for reduced bleeding? I don’t think that we’ve fully answered that, certainly not with the PIONEER AF trial.
Dr. Parra. I agree. When we look at those trials, 2 things come to mind. First, the doses of dabigatran used in the RE-DUAL PCI trial were doses that have been shown to be beneficial in the nonvalvular AF population. Second, a take-home point from those trials is the P2Y12 inhibitor that was utilized—close to 90% or more used clopidogrel in the PIONEER-AF trial. Clopidogrel remains the P2Y12 inhibitor of choice. One of the other findings is that the aspirin dosing should be low, < 100 mg daily, and that we need to consider routine use of proton pump inhibitors to protect against the bleeding that can be found with the antiplatelet agents.
Also of interest, the European Society of Cardiology (ESC) recently released a focused update with some excellent recommendations on dual antiplatelet therapy in coronary artery disease in which they incorporated the results from the PIONEER-AF-PCI trial. The RE-DUAL PCI trial had not been published when these came out. If you’re concerned about ischemic risk prevailing, ESC recommendations based upon risk stratification are triple therapy with aspirin, clopidogrel, and an oral anticoagulant for longer than 1 month and up to 6 months and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months; afterward just oral anticoagulation alone. If the concerns about bleeding prevail, then we have 2 different pathways: one limiting triple therapy to 1 month and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months. But the ESC also has a second recommendation for patients at high risk of bleeding, which is dual therapy with clopidogrel and an oral anticoagulant at the offset for up to 12 months. I found these guidelines to be particularly helpful in terms of how to put this into practice.
Dr. Allen. There’s still so much concern about in-stent thrombosis. Although a smaller trial, we knew from the WOEST trial that single antiplatelet therapy with warfarin was reasonable. We know from the PIONEER-AF and RE-DUAL PCI trials that we didn’t get significantly more in-stent thrombosis by giving up the second antiplatelet. Whether or not we answered the stroke question is another issue, but the cardiology societies are still hanging on to dual antiplatelet therapy. I question if that’s based on the older data and the older stent technologies.
Dr. Minichiello. This highlights again that often we have to consider these patients’ case-by-case analysis, and that these decisions require multidisciplinary input. It involves coming together and figuring out in this particular patient, which of those 3 options would be best. We have a lot more options than we did just a short year or year and a half ago with at least some data providing comfort that DOACs at effective doses for stroke prevention in nonvalvular AF look like they can be combined with single antiplatelet therapy for post-PCI patients.
Dr. Barnes. Speaking as a cardiologist, this is a question I encounter all the time. I think everything said here is really well taken. I’ll just summarize to say for patients who have acute coronary syndromes and AF. First, I’m okay with using warfarin and now I’m okay using the DOACs, but the anticoagulant needs to continue for stroke prevention. Second, the patient has to be on clopidogrel as a P2Y12 inhibitor because it has the lowest bleeding risk profile. Third, the patient doesn’t always need 1 year of dual antiplatelet therapy the way we used to think of it. With newer generation stents and ongoing anticoagulation, you can get away with shorter courses of your antiplatelets, albeit 3 or 6 months. Providers should have a conversation with patients and think hard about how to balance clotting vs that bleeding profile.
Case 3
Dr. Minichiello. This case involves a 66-year-old man who has nonvalvular AF and is on warfarin. He has CKD, and his CrCl is about 30. He has hypertension, diabetes mellitus, and he is going to go for a colonoscopy. The proceduralist lets you know about the date and wants to know whether he needs to be bridged. He also has a remote history of a transient ischemic attack.
Dr. Allen. Historically, we’ve had detailed guidance on how to risk assess patients with AF, venous thromboembolism (VT), and mechanical heart valves in the periprocedural period. From that risk assessment the guidance helped us determine whether or not we should offer periprocedural bridging with heparin or low-molecularweight heparin.
The issue is that the detailed guidance was always based on expert opinion not hard science and there was no great evidence that we were preventing thrombotic events or that there was a net clinical benefit to bridging. Some retrospective cohort studies started coming out around 2012 that demonstrated an increased incidence of major bleeding events associated with bridging with no reduction in thrombotic complications. Some might argue that this is because thrombotic complications are so rare that you would have to have tens of thousands of patients for adequate power. Nonetheless, these studies were adequately powered to show a significant increase in major bleeding.
The best prospective trial data we have for this population comes from the BRIDGE trial, which randomized AF patients to receive dalteparin bridge therapy vs placebo during periprocedural interruptions in warfarin therapy. It too demonstrated a significant increase in the risk of bleeding complications associated with bridging with no significant reduction in the risk of thromboembolic events. Critics point out that some of the higher risk patients were underrepresented, and the same could be said about some of the other retrospective studies in the VT and mechanical valve populations.
We have waited for quite a while for guidances to catch up with these data. The most recent guidance published would apply to this patient. It was guidance for the AF population published by the American College of Cardiology (ACC) in early 2017
To make a decision to bridge, you not only have to make an assumption that your patient is at such extraordinary thrombotic risk that you would find some reduction in thrombotic events associated with bridging, but also that the benefit is going to be so great that it would overcome this very clear increased risk of bleeding, resulting in a net clinical benefit. Only then would it make sense to bridge. Based on the available data, it is quite possible that no such patient population exists.
Dr. Parra. I agree. We found from the BRIDGE trial, with the caveat that you pointed out, that high or moderately high thrombotic risk individuals weren’t as well represented, that there is at least a 2.5-fold increase in major bleeding. We also know that higher thrombotic-risk patients tend to also have higher bleeding risk. So in high thrombotic-risk patients, while there is uncertainty about whether or not there’s going to be a thrombotic benefit from bridging, we can be confident there will be an increased risk of major bleeding perhaps even more than that seen in the BRIDGE trial.
One thing that really illustrates this is the 2017 American Heart Association/ACC-focused update on the 2014 guideline for the management of patients with valvular heart disease. The guideline has a section on bridging therapy for prosthetic valves. And that was recently downgraded from a Class I recommendation to Class IIA, recognizing that the data are limited in terms of even when to bridge prosthetic valves.
Dr. Allen. Bridging is an area where we continue to do something that we know causes harm because we hope it has some benefit. Despite what some societal guidance still says, I very rarely bridge patients. If I do, it’s because the patient and/or caregivers have heard the facts and have opted to do it.
Dr. Minichiello. Practically speaking, the data—the retrospective data, the observational data, the BRIDGE trial, etc—definitely make us step back and think about bridging and realize that it’s a HIGH RISK of intervention. We really need to be informing patients about that risk. We know that bridging increases the risk of bleeding. We do not have data showing a reduction in the risk of thromboembolic disease with bridge therapy. That’s all based on what we think would happen, what we hope would be the result of bridge therapy, but in truth we do not know if this is the case. The risks of bridge therapy must be weighed heavily each time we consider using it.
In my practice I reserve bridge therapy, which we know is associated with increased harm for patients in whom we think there is the very highest risk of thromboembolism, ie, those with very recent arterial or venous thrombosis, and by recently, I mean within the past 1 to 3 months; patients who have very severe thrombophilia like antitphospholipid antibody syndrome, some cancer patients, and those with mechanical valves in the mitral position or those with high-risk mechanical aortic valves.
I don’t bridge most AF patients. In fact, I can’t remember the last time I bridged an AF patient. I do know that this is somewhat discordant with the ACC recommendations, but absent the data to support bridge therapy, I’m really concerned that the risk outweighs the benefit. This is particularly true in our VA population where the bleeding risk is high because of CKD or a history of bleeding or thrombocytopenia or concomitant aspirin or something else.
Dr. Barnes. We know from some studies that have been published that the biggest driver of a clinician deciding whether or not a patient should bridge is actually whether or not they’ve had a stroke. The truth is that the BRIDGE trial enrolled a sizeable population; it was about 15% of patients with a prior TIA or stroke. So it gave us some insight. Despite that, our event rates for thromboembolism, arterial thromboembolism, including stroke, were quite low.
I tend to look far more at the collection of risk factors and not just at a history of stroke. Now as you mentioned, Dr. Minichiello, a stroke within the past 1 to 3 months, I ask, “does this procedure even need to happen?” But outside of that, it’s not a history of stroke that’s going to make the decision for me: It’s all the risk factors together.
There’s an ongoing study, the PERIOP 2 study, that is enrolling patients at higher risk for stroke and patients with mechanical valves. This study may give us more insight into exactly what kind of risks these patients are at and whether they get benefit from bridging. But in the meantime, I’m really reserving bridging for my highest risk patients, those with multiple risk factors, CHADS2 scores of 5 and 6 or CHA2DS2-VASc of 7, 8, 9, and those with a recent VT or mechanical mitral valves.
Click here to read the digital edition.
Eruptive Vellus Hair Cysts in Identical Triplets With Dermoscopic Findings
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
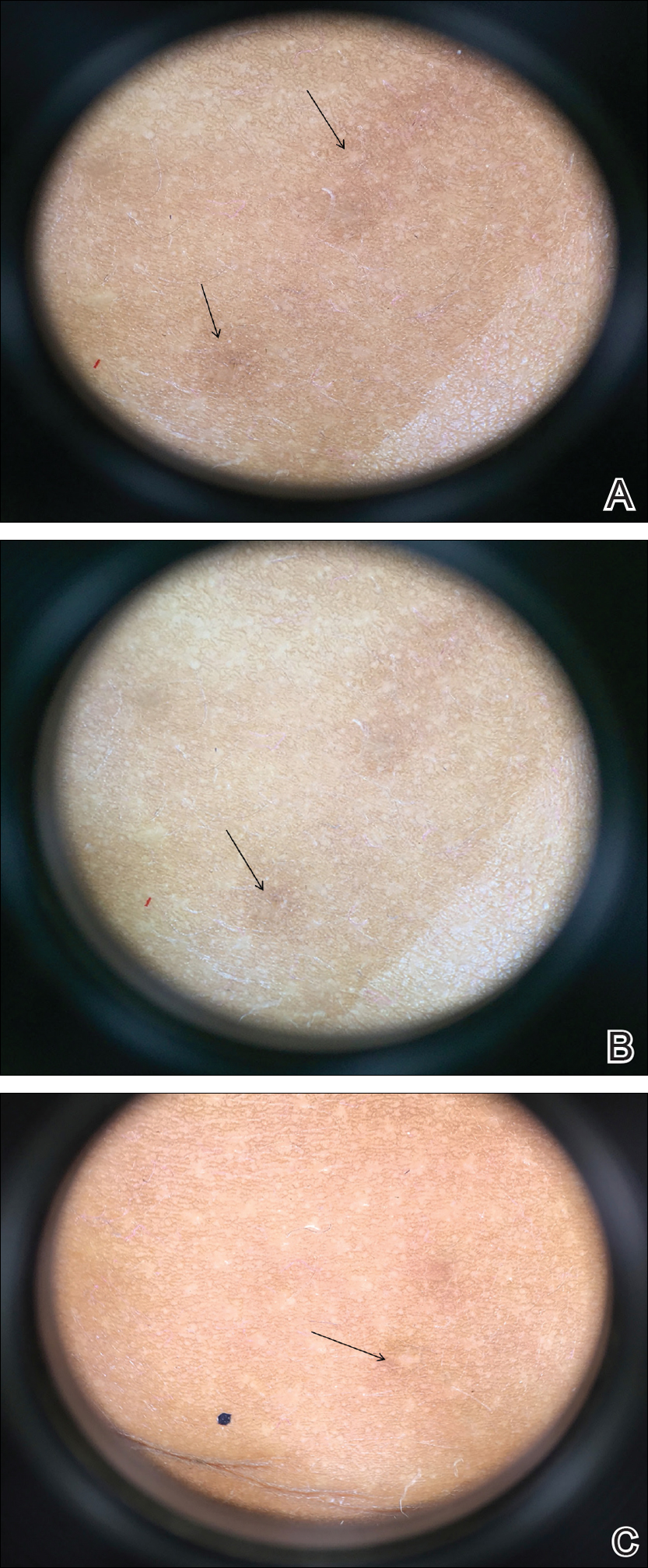
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
Practice Points
- Eruptive vellus hair cysts (EVHCs) are 1- to 3-mm round, dome-shaped, flesh-colored, asymptomatic, benign papules typically occurring on the chest and extremities.
- Pathogenesis and inheritance are unclear. Although the majority of EVHC cases are sporadic, the strong influence of genes is indicated by numerous reports of families in whom 2 or more members were affected.
- Dermoscopy is a noninvasive diagnostic procedure that should be utilized to diagnose EVHCs in the pediatric population; specifically, EVHCs exhibit light yellow, homogenous, circular structures with a maroon or erythematous halo.
- The main indication for treatment of EVHCs is cosmetic concern; however, one-quarter of cases may resolve spontaneously.
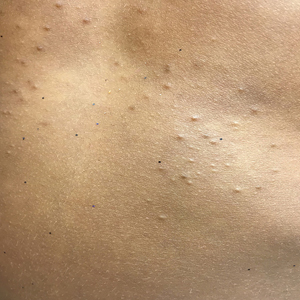
Pediatric Primary Cutaneous Blastomycosis Clinically Responsive to Itraconazole
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
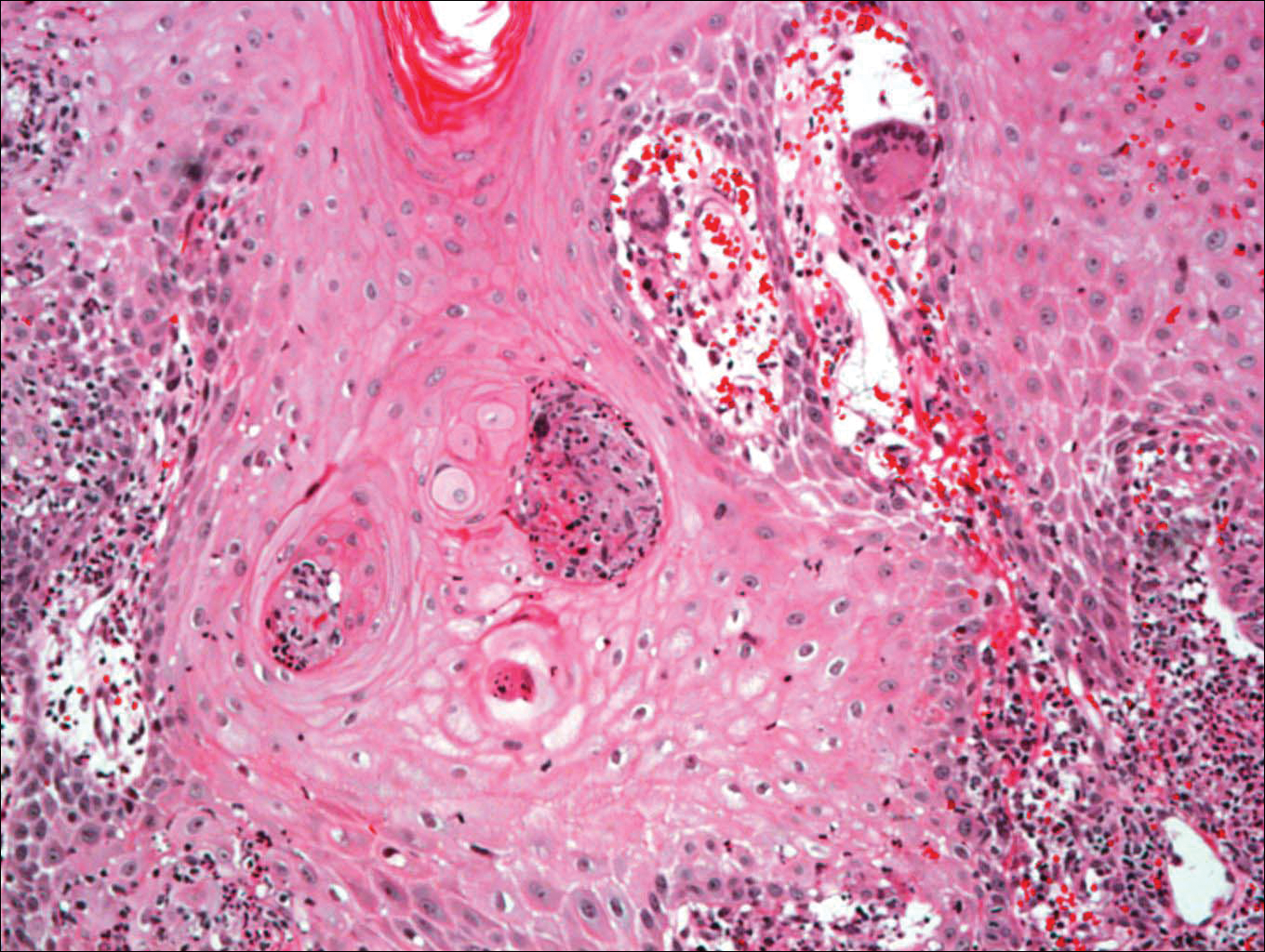
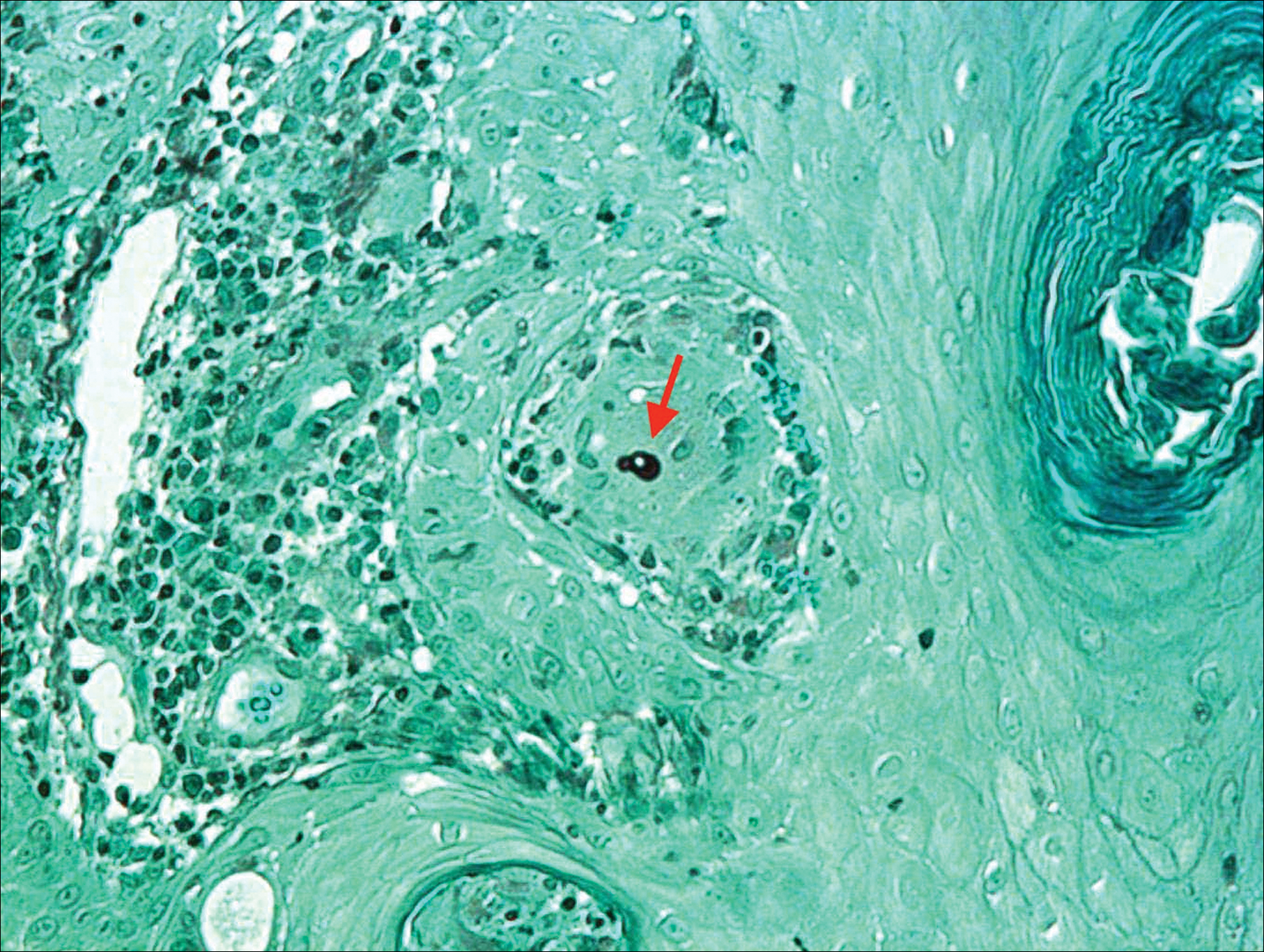
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
Practice Points
- Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin, resulting in primary cutaneous disease.
- Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.
- Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.
- Increased awareness of this rare infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
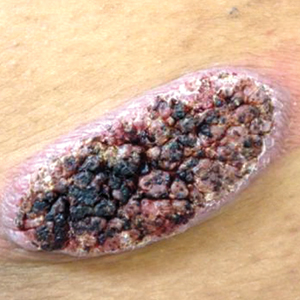
Acute Hemorrhagic Edema of Infancy: Guide to Prevent Misdiagnosis
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
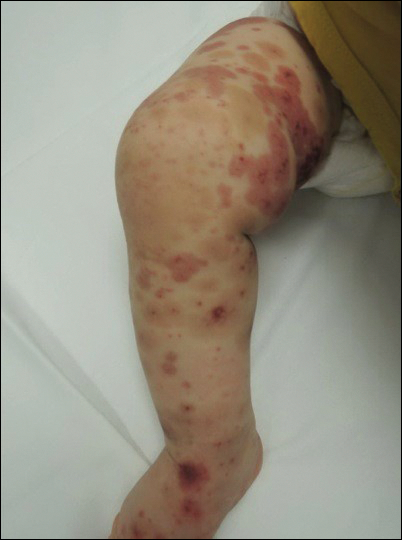
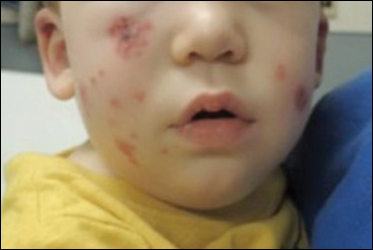
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
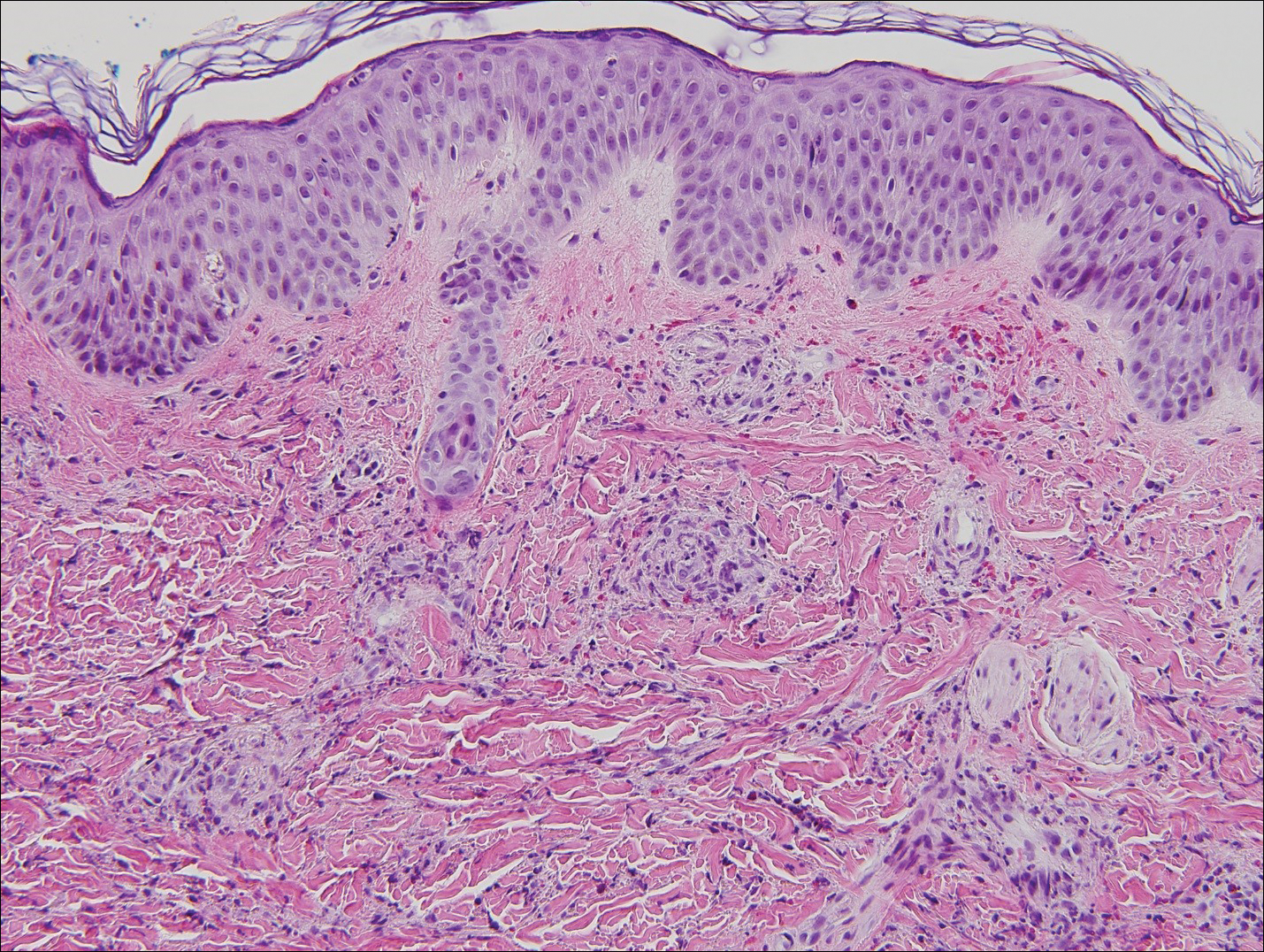
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8

Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8
Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8
Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
Practice Points
- Acute hemorrhagic edema of infancy (AHEI) is an uncommon benign leukocytoclastic vasculitis of unknown precise pathophysiology that is thought be immune complex mediated.
- Clinical history, physical examination, and histopathologic analysis combine to allow the important differentiation between AHEI and Henoch-Schönlein purpura (HSP).
- Differentiation between AHEI and HSP determines treatment decisions and indicates the need for counseling on potential associated renal and gastrointestinal risks of HSP.