User login
Severe headache • neck pain • intermittent cough • Dx?
THE CASE
A 32-year-old Chinese woman sought care from our family medicine clinic because she had a headache, neck pain, and an intermittent cough that had produced white sputum for 7 days. She described the headache as severe and pressure-like, and said that it had progressively worsened over the previous 3 weeks, coinciding with her first trip outside of China to the United States. The patient indicated that she also had occasional vomiting, dizziness, a low-grade fever, chills, night sweats, and increasing fatigue.
Prior to this visit, the patient had gone to the emergency department (ED) twice in one week, but was told that she had a migraine headache and a viral syndrome and was sent home. She was also told to make a follow-up appointment at our family medicine outpatient clinic.
Besides the symptoms that brought her to our clinic, the only other notable element of the patient’s history was a “neck mass” resection in China 8 years earlier. (The diagnosis of the neck mass was unknown.)
Concerned about her presenting signs and symptoms, we sent the patient to the ED, where she was admitted for further evaluation and treatment of possible meningitis. In the ED, she had a temperature of 101.5° F; her other vital signs were normal. A physical exam revealed mild neck stiffness.
THE DIAGNOSIS
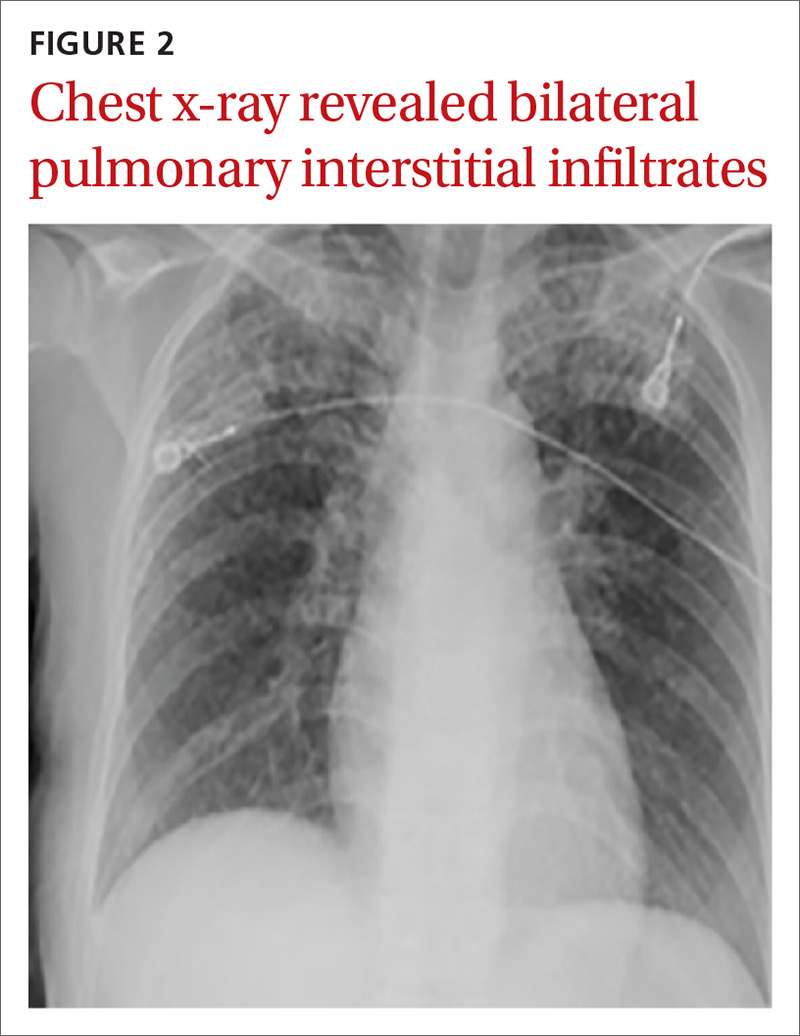
A chest computed tomography (CT) scan demonstrated extensive confluent nodular infiltrates in the lung apices bilaterally with the largest confluent nodule measuring 6 cm (FIGURE 1). A chest x-ray demonstrated extensive bilateral pulmonary interstitial infiltrates that were most pronounced in the upper lung fields (FIGURE 2).

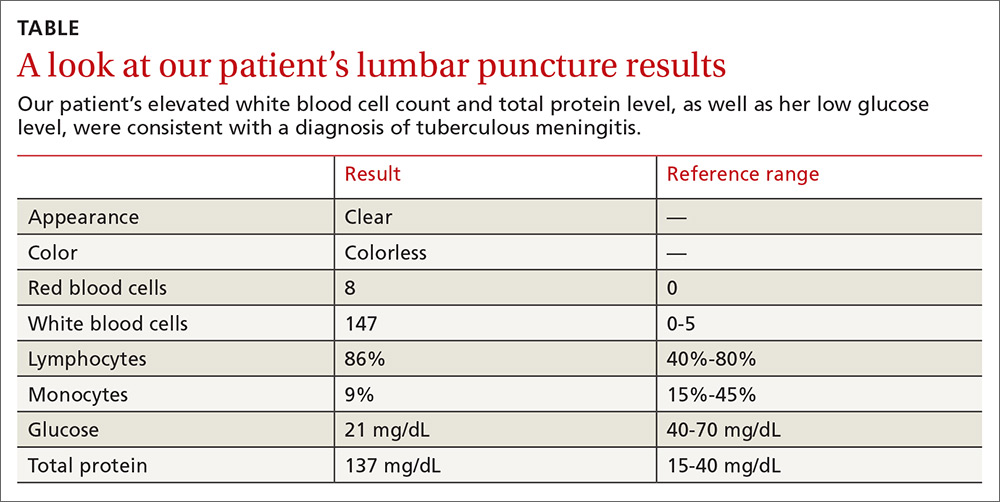
Lumbar puncture results revealed lymphocytic pleocytosis with elevated protein and low glucose levels (TABLE). Based on these results, the family medicine team suspected that our patient had tuberculous meningitis (TBM).
The team consulted with Infectious Diseases for management of TBM, and they placed our patient in a negative pressure room on airborne isolation. In addition, she was started on rifampin 450 mg/d, pyrazinamide 1000 mg/d, ethambutol 800 mg/d, and isoniazid (INH) 800 mg/d, as well as pyridoxine and intravenous dexamethasone.
DISCUSSION
TBM accounts for approximately 1% of all cases of TB and 5% of extrapulmonary diseases in immunocompetent individuals.1 In 2015, there were approximately 10.4 million cases of TB worldwide, and 6 countries accounted for 60% of the global total: India, Indonesia, China, Nigeria, Pakistan, and South Africa.2 TBM is typically a subacute disease with symptoms that can persist for weeks before diagnosis.3 An early diagnosis is critical, as the mortality rate remains relatively high (as high as nearly 70% in underdeveloped and developed countries) despite effective treatment regimens.3 (For updated recommendations on TB screening, see this month’s Practice Alert.)
Most health care facilities use AFB smears to determine when patients with suspected TB should be isolated. However, AFB smears are positive in only 60% of TB cases.4 One study indicated that nucleic acid amplification by PCR can improve sensitivity from 60% to 87% and specificity from 98% to 100%.5
The presentation of TBM varies by phase of disease:
- The prodromal phase typically lasts for 2 to 3 weeks. It is characterized by an insidious onset of malaise, headache, low-grade fever, irritability, and personality changes.
- The meningitis phase is characterized by pronounced neurologic features such as meningismus, protracted headache, confusion, myelopathy, and sensory deficits, as well as vomiting, lethargy, and urinary retention.
- During the paralytic phase, patients experience profound confusion, followed by stupor, coma, seizures, progressive paraplegia, and often, hemiparesis.1,3,6
Treatment should be given for a total of 9 to 12 months
Initiate treatment for TB based on a strong clinical suspicion for the disease. Treatment of TBM consists of an intensive phase with 4 anti-TB drugs for 2 months (typically INH 800 mg/d, rifampin 450 mg/d, pyrazinamide 1000 mg/d, and ethambutol 800 mg/d) and a continuation phase with 2 drugs (INH and rifampin) for 7 to 10 additional months, resulting in a total treatment duration of 9 to 12 months.
Our patient was discharged from the hospital after 2 weeks on an anti-TB medication regimen of INH, rifampin, and pyrazinamide, along with pyridoxine and a tapering dose of dexamethasone. After the initial 2 months of intensive phase therapy, she was switched to INH 300 mg/d and rifampin 450 mg/d for the continuation phase. The patient followed up at our family medicine outpatient clinic with slow improvement of her muscle weakness before returning to China once she was placed on the continuation phase drugs.
THE TAKEAWAY
Suspect TB in high-risk patients traveling from endemic areas. Our patient, a Chinese woman visiting Brooklyn, New York, should’ve been considered high risk for TB even without her travel history from China because Brooklyn has a high rate of TB, as well. (In 2015, Sunset Park, Brooklyn had 18.2 cases of TB per 100,000 people, which was more than double the citywide rate.7)
TBM is a subacute disease with an often subtle presentation. Once you suspect TBM, isolate the patient, obtain appropriate cultures and smears, and start anti-TB drugs and adjunctive corticosteroids immediately, while the results of studies for AFB are still pending. Prompt diagnosis and treatment can save a patient’s life.
1. Garcia-Monco JC. Central nervous system tuberculosis. Neurol Clin. 1999;17:737-759.
2. World Health Organization. Global tuberculosis report, 2016. Available at: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1. Accessed March 29, 2017.
3. Marx GE, Chan ED. Tuberculous meningitis: diagnosis and treatment overview. Tuberc Res Treat. 2011;2011:798764.
4. Siddiqui AH, Perl TM, Conlon M, et al. Preventing nosocomial transmission of pulmonary tuberculosis: when may isolation be discontinued for patients with suspected tuberculosis? Infect Control Hosp Epidemiol. 2002;23:141-144.
5. Tang YW, Meng S, Li H, et al. PCR enhances acid-fast bacillus stain-based rapid detection of Mycobacterium tuberculosis. J Clin Microbiol. 2004;42:1849-1850.
6. Long R, Gardam M. Tumour necrosis factor-alpha inhibitors and the reactivation of latent tuberculosis infection. CMAJ. 2003;168:1153-1156.
7. New York City Department of Health and Mental Hygiene. Tuberculosis in New York City, 2015. New York City Bureau of Tuberculosis Control Annual Summary. Available at: http://www1.nyc.gov/assets/doh/downloads/pdf/tb/tb2015.pdf. Accessed April 7, 2017.
THE CASE
A 32-year-old Chinese woman sought care from our family medicine clinic because she had a headache, neck pain, and an intermittent cough that had produced white sputum for 7 days. She described the headache as severe and pressure-like, and said that it had progressively worsened over the previous 3 weeks, coinciding with her first trip outside of China to the United States. The patient indicated that she also had occasional vomiting, dizziness, a low-grade fever, chills, night sweats, and increasing fatigue.
Prior to this visit, the patient had gone to the emergency department (ED) twice in one week, but was told that she had a migraine headache and a viral syndrome and was sent home. She was also told to make a follow-up appointment at our family medicine outpatient clinic.
Besides the symptoms that brought her to our clinic, the only other notable element of the patient’s history was a “neck mass” resection in China 8 years earlier. (The diagnosis of the neck mass was unknown.)
Concerned about her presenting signs and symptoms, we sent the patient to the ED, where she was admitted for further evaluation and treatment of possible meningitis. In the ED, she had a temperature of 101.5° F; her other vital signs were normal. A physical exam revealed mild neck stiffness.
THE DIAGNOSIS
A chest computed tomography (CT) scan demonstrated extensive confluent nodular infiltrates in the lung apices bilaterally with the largest confluent nodule measuring 6 cm (FIGURE 1). A chest x-ray demonstrated extensive bilateral pulmonary interstitial infiltrates that were most pronounced in the upper lung fields (FIGURE 2).
Lumbar puncture results revealed lymphocytic pleocytosis with elevated protein and low glucose levels (TABLE). Based on these results, the family medicine team suspected that our patient had tuberculous meningitis (TBM).
The team consulted with Infectious Diseases for management of TBM, and they placed our patient in a negative pressure room on airborne isolation. In addition, she was started on rifampin 450 mg/d, pyrazinamide 1000 mg/d, ethambutol 800 mg/d, and isoniazid (INH) 800 mg/d, as well as pyridoxine and intravenous dexamethasone.
DISCUSSION
TBM accounts for approximately 1% of all cases of TB and 5% of extrapulmonary diseases in immunocompetent individuals.1 In 2015, there were approximately 10.4 million cases of TB worldwide, and 6 countries accounted for 60% of the global total: India, Indonesia, China, Nigeria, Pakistan, and South Africa.2 TBM is typically a subacute disease with symptoms that can persist for weeks before diagnosis.3 An early diagnosis is critical, as the mortality rate remains relatively high (as high as nearly 70% in underdeveloped and developed countries) despite effective treatment regimens.3 (For updated recommendations on TB screening, see this month’s Practice Alert.)
Most health care facilities use AFB smears to determine when patients with suspected TB should be isolated. However, AFB smears are positive in only 60% of TB cases.4 One study indicated that nucleic acid amplification by PCR can improve sensitivity from 60% to 87% and specificity from 98% to 100%.5
The presentation of TBM varies by phase of disease:
- The prodromal phase typically lasts for 2 to 3 weeks. It is characterized by an insidious onset of malaise, headache, low-grade fever, irritability, and personality changes.
- The meningitis phase is characterized by pronounced neurologic features such as meningismus, protracted headache, confusion, myelopathy, and sensory deficits, as well as vomiting, lethargy, and urinary retention.
- During the paralytic phase, patients experience profound confusion, followed by stupor, coma, seizures, progressive paraplegia, and often, hemiparesis.1,3,6
Treatment should be given for a total of 9 to 12 months
Initiate treatment for TB based on a strong clinical suspicion for the disease. Treatment of TBM consists of an intensive phase with 4 anti-TB drugs for 2 months (typically INH 800 mg/d, rifampin 450 mg/d, pyrazinamide 1000 mg/d, and ethambutol 800 mg/d) and a continuation phase with 2 drugs (INH and rifampin) for 7 to 10 additional months, resulting in a total treatment duration of 9 to 12 months.
Our patient was discharged from the hospital after 2 weeks on an anti-TB medication regimen of INH, rifampin, and pyrazinamide, along with pyridoxine and a tapering dose of dexamethasone. After the initial 2 months of intensive phase therapy, she was switched to INH 300 mg/d and rifampin 450 mg/d for the continuation phase. The patient followed up at our family medicine outpatient clinic with slow improvement of her muscle weakness before returning to China once she was placed on the continuation phase drugs.
THE TAKEAWAY
Suspect TB in high-risk patients traveling from endemic areas. Our patient, a Chinese woman visiting Brooklyn, New York, should’ve been considered high risk for TB even without her travel history from China because Brooklyn has a high rate of TB, as well. (In 2015, Sunset Park, Brooklyn had 18.2 cases of TB per 100,000 people, which was more than double the citywide rate.7)
TBM is a subacute disease with an often subtle presentation. Once you suspect TBM, isolate the patient, obtain appropriate cultures and smears, and start anti-TB drugs and adjunctive corticosteroids immediately, while the results of studies for AFB are still pending. Prompt diagnosis and treatment can save a patient’s life.
THE CASE
A 32-year-old Chinese woman sought care from our family medicine clinic because she had a headache, neck pain, and an intermittent cough that had produced white sputum for 7 days. She described the headache as severe and pressure-like, and said that it had progressively worsened over the previous 3 weeks, coinciding with her first trip outside of China to the United States. The patient indicated that she also had occasional vomiting, dizziness, a low-grade fever, chills, night sweats, and increasing fatigue.
Prior to this visit, the patient had gone to the emergency department (ED) twice in one week, but was told that she had a migraine headache and a viral syndrome and was sent home. She was also told to make a follow-up appointment at our family medicine outpatient clinic.
Besides the symptoms that brought her to our clinic, the only other notable element of the patient’s history was a “neck mass” resection in China 8 years earlier. (The diagnosis of the neck mass was unknown.)
Concerned about her presenting signs and symptoms, we sent the patient to the ED, where she was admitted for further evaluation and treatment of possible meningitis. In the ED, she had a temperature of 101.5° F; her other vital signs were normal. A physical exam revealed mild neck stiffness.
THE DIAGNOSIS
A chest computed tomography (CT) scan demonstrated extensive confluent nodular infiltrates in the lung apices bilaterally with the largest confluent nodule measuring 6 cm (FIGURE 1). A chest x-ray demonstrated extensive bilateral pulmonary interstitial infiltrates that were most pronounced in the upper lung fields (FIGURE 2).
Lumbar puncture results revealed lymphocytic pleocytosis with elevated protein and low glucose levels (TABLE). Based on these results, the family medicine team suspected that our patient had tuberculous meningitis (TBM).
The team consulted with Infectious Diseases for management of TBM, and they placed our patient in a negative pressure room on airborne isolation. In addition, she was started on rifampin 450 mg/d, pyrazinamide 1000 mg/d, ethambutol 800 mg/d, and isoniazid (INH) 800 mg/d, as well as pyridoxine and intravenous dexamethasone.
DISCUSSION
TBM accounts for approximately 1% of all cases of TB and 5% of extrapulmonary diseases in immunocompetent individuals.1 In 2015, there were approximately 10.4 million cases of TB worldwide, and 6 countries accounted for 60% of the global total: India, Indonesia, China, Nigeria, Pakistan, and South Africa.2 TBM is typically a subacute disease with symptoms that can persist for weeks before diagnosis.3 An early diagnosis is critical, as the mortality rate remains relatively high (as high as nearly 70% in underdeveloped and developed countries) despite effective treatment regimens.3 (For updated recommendations on TB screening, see this month’s Practice Alert.)
Most health care facilities use AFB smears to determine when patients with suspected TB should be isolated. However, AFB smears are positive in only 60% of TB cases.4 One study indicated that nucleic acid amplification by PCR can improve sensitivity from 60% to 87% and specificity from 98% to 100%.5
The presentation of TBM varies by phase of disease:
- The prodromal phase typically lasts for 2 to 3 weeks. It is characterized by an insidious onset of malaise, headache, low-grade fever, irritability, and personality changes.
- The meningitis phase is characterized by pronounced neurologic features such as meningismus, protracted headache, confusion, myelopathy, and sensory deficits, as well as vomiting, lethargy, and urinary retention.
- During the paralytic phase, patients experience profound confusion, followed by stupor, coma, seizures, progressive paraplegia, and often, hemiparesis.1,3,6
Treatment should be given for a total of 9 to 12 months
Initiate treatment for TB based on a strong clinical suspicion for the disease. Treatment of TBM consists of an intensive phase with 4 anti-TB drugs for 2 months (typically INH 800 mg/d, rifampin 450 mg/d, pyrazinamide 1000 mg/d, and ethambutol 800 mg/d) and a continuation phase with 2 drugs (INH and rifampin) for 7 to 10 additional months, resulting in a total treatment duration of 9 to 12 months.
Our patient was discharged from the hospital after 2 weeks on an anti-TB medication regimen of INH, rifampin, and pyrazinamide, along with pyridoxine and a tapering dose of dexamethasone. After the initial 2 months of intensive phase therapy, she was switched to INH 300 mg/d and rifampin 450 mg/d for the continuation phase. The patient followed up at our family medicine outpatient clinic with slow improvement of her muscle weakness before returning to China once she was placed on the continuation phase drugs.
THE TAKEAWAY
Suspect TB in high-risk patients traveling from endemic areas. Our patient, a Chinese woman visiting Brooklyn, New York, should’ve been considered high risk for TB even without her travel history from China because Brooklyn has a high rate of TB, as well. (In 2015, Sunset Park, Brooklyn had 18.2 cases of TB per 100,000 people, which was more than double the citywide rate.7)
TBM is a subacute disease with an often subtle presentation. Once you suspect TBM, isolate the patient, obtain appropriate cultures and smears, and start anti-TB drugs and adjunctive corticosteroids immediately, while the results of studies for AFB are still pending. Prompt diagnosis and treatment can save a patient’s life.
1. Garcia-Monco JC. Central nervous system tuberculosis. Neurol Clin. 1999;17:737-759.
2. World Health Organization. Global tuberculosis report, 2016. Available at: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1. Accessed March 29, 2017.
3. Marx GE, Chan ED. Tuberculous meningitis: diagnosis and treatment overview. Tuberc Res Treat. 2011;2011:798764.
4. Siddiqui AH, Perl TM, Conlon M, et al. Preventing nosocomial transmission of pulmonary tuberculosis: when may isolation be discontinued for patients with suspected tuberculosis? Infect Control Hosp Epidemiol. 2002;23:141-144.
5. Tang YW, Meng S, Li H, et al. PCR enhances acid-fast bacillus stain-based rapid detection of Mycobacterium tuberculosis. J Clin Microbiol. 2004;42:1849-1850.
6. Long R, Gardam M. Tumour necrosis factor-alpha inhibitors and the reactivation of latent tuberculosis infection. CMAJ. 2003;168:1153-1156.
7. New York City Department of Health and Mental Hygiene. Tuberculosis in New York City, 2015. New York City Bureau of Tuberculosis Control Annual Summary. Available at: http://www1.nyc.gov/assets/doh/downloads/pdf/tb/tb2015.pdf. Accessed April 7, 2017.
1. Garcia-Monco JC. Central nervous system tuberculosis. Neurol Clin. 1999;17:737-759.
2. World Health Organization. Global tuberculosis report, 2016. Available at: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1. Accessed March 29, 2017.
3. Marx GE, Chan ED. Tuberculous meningitis: diagnosis and treatment overview. Tuberc Res Treat. 2011;2011:798764.
4. Siddiqui AH, Perl TM, Conlon M, et al. Preventing nosocomial transmission of pulmonary tuberculosis: when may isolation be discontinued for patients with suspected tuberculosis? Infect Control Hosp Epidemiol. 2002;23:141-144.
5. Tang YW, Meng S, Li H, et al. PCR enhances acid-fast bacillus stain-based rapid detection of Mycobacterium tuberculosis. J Clin Microbiol. 2004;42:1849-1850.
6. Long R, Gardam M. Tumour necrosis factor-alpha inhibitors and the reactivation of latent tuberculosis infection. CMAJ. 2003;168:1153-1156.
7. New York City Department of Health and Mental Hygiene. Tuberculosis in New York City, 2015. New York City Bureau of Tuberculosis Control Annual Summary. Available at: http://www1.nyc.gov/assets/doh/downloads/pdf/tb/tb2015.pdf. Accessed April 7, 2017.

Epistaxis and Death by the Trigeminocardiac Reflex: A Cautionary Report
Epistaxis is a relatively common event that is estimated to occur at least once in 60% of the U.S. population. Epistaxis is also reported to cause 1.7 emergency department (ED) visits per 1,000 population annually.1 Although epistaxis can occur at any age, it typically occurs with a bimodal age distribution and most commonly affects individuals aged < 18 years and adults aged > 50 years.2 The episodes of epistaxis involving the younger age group are more often minor and self-limited. Most bleeds occur along the anterior nasal septum from Kiesselbach’s plexus.
Posterior bleeds occur more often in older patients.2 In addition, epistaxis in the older population tends to be more severe.3 Medical intervention is required in 6% of those experiencing epistaxis. Because the median age for male veterans was 64 years in 2011 compared with a median age of 37.2 years for the average U.S. population in 2010, veterans are among those at greatest risk to develop epistaxis that requires intervention.4,5
Most episodes of epistaxis are not life-threatening, particularly when modern methods of diagnosis and treatment are used. Nevertheless, comorbid diseases, complications of treatment, and normal physiologic responses can sometimes combine to create an adverse outcome.6 This report reviews the case of a veteran patient who experienced a fatal cardiopulmonary arrest after therapeutic interventions for epistaxis. It is believed that his death was due to the well-described but little known trigeminocardiac reflex (TCR).
Case Report
A 65-year-old man visited the ED and reported that his nose had been bleeding intermittently for 1 day. He estimated that he had lost 1 cup of blood over a 24-hour period. He reported no rhinosinusitis, nasal congestion, or recent allergy or upper respiratory infections. He also reported no nasal trauma. In the ED, blood was oozing from his right nares and into his throat, causing him to cough. External compression failed to control the oozing. Topical vasoconstrictors were not applied.
His past medical history included a pulmonary embolism, well-controlled chronic obstructive pulmonary disease (COPD), and sleep apnea. The thrombotic site of origin for his pulmonary embolism had not been identified, despite a thorough examination. He had been on warfarin therapy for 3 months, and his international normalized ratio (INR) had been monitored in an anticoagulation clinic and was well regulated. He was also on inhaled medications for COPD (formoterol and budesonide as a combination preparation twice daily and albuterol every 6 hours as needed as a rescue medication). He adhered to his noninvasive positive airway pressure (PAP) device treatment for sleep apnea. He did not take aspirin or other antiplatelet medications. He reported no use of topical nasal preparations. He also reported no use of illicit drugs or over-the-counter medications, including nonsteroidal anti-inflammatory medications and herbal remedies. He reported no bleeding from other sites or easy bruising.
The patient was alert, oriented, and in no distress. His vital signs were normal. Examination of his nasal passages failed to identify an active site of bleeding. Fresh blood was present in the right nasal passage and the posterior pharynx. Examination of his chest was normal. His hemoglobin was 13.1 g/dL (13.6-17.3 g/dL) with 216 x 103/μL platelets (166-383 x 103/μL). His INR was therapeutic at 2.38. Laboratory assessments of his electrolytes, liver function, and renal function were normal. A chest radiograph demonstrated no acute process. A computed tomography failed to demonstrate sinusitis or an anatomical abnormality that could account for his epistaxis.
Due to the amount of blood loss by epistaxis complicated by anticoagulation for his recent pulmonary embolism, the patient was admitted to the hospital for observation. Reversal of the anticoagulation was considered by the admitting service, but because the patient was only oozing blood, this intervention was not undertaken. Instead, he was continued on warfarin, was treated with an oral antibiotic, and was continued on his inhaled medications for his COPD. He also used his noninvasive PAP device to sleep.
The next day, the patient began to bleed freely from his right nares. The bleeding was initially controlled with compression and positioning and resolved without additional intervention. An otolaryngologist performed silver nitrate cauterization of Kiesselbach’s plexus. The patient experienced no further bleeding, and his hemoglobin remained stable.
The next day, his nose began to bleed briskly. He passed large clots from his nose and mouth. The patient was alert and oriented. He remained hemodynamically stable. His INR was 2.1. Nasal packing was proposed, and the procedure, including the risks and benefits, were explained to the patient.
After obtaining consent from thepatient, the nasal mucosa was prepared with topical 2% lidocaine and 1% phenylephrine. Anterior and posterior nasal packing was successfully achieved with paraffin gauze. This procedure was completed in a monitored environment by an experienced otolaryngologist. However, the patient became agitated 15 to 20 minutes after the nasal packing had been accomplished. He rapidly became apneic, bradycardic, and hypotensive. His oxygen saturation on room air as measured by pulse oximetry decreased precipitously to 50%. These developments were quickly followed by asystole.
Advanced cardiac life support measures were initiated. His airway was secured by oral endotracheal intubation, and oxygen was delivered at 100% fraction of inspired oxygen by bag ventilation. At intubation, only a few small clots were present in the posterior pharynx. No blood was suctioned from the endotracheal tube; therefore, active bleeding was not suspected. The nasal packing remained in place and was not removed. The patient failed to regain spontaneous circulation and died. An arterial blood gas analysis obtained during cardiopulmonary resuscitation demonstrated no methemoglobin on co-oximetry.
Discussion
Because of the high prevalence of epistaxis in the general population, many health care providers (HCPs) are confronted with this problem. Epistaxis in most patients remits without consequence. However, HCPs may be required to intervene. Treatment modalities include simple compression and positioning maneuvers, the application of topical medications, anterior and posterior nasal packing, chemical cauterization, endoscopic electric cauterization, embolization therapy, and surgical arterial ligation.7 The choice of therapy depends on several factors, including the site of the bleeding, the severity of the bleeding, the availability of resources, and the expertise of the HCP. A localized cause of epistaxis is discovered in only 15% of patients, making a conservative therapeutic approach an attractive initial intervention.8
Nasal packing is a successful intervention in 70% of patients with posterior epistaxis. In addition, nasal packing is the preferred method for hemostasis in anterior epistaxis when cauterization fails.3,9 This patient failed simple compression and positioning maneuvers as well as chemical cauterization. For this reason, nasal packing was proposed as a therapeutic intervention. He was hemodynamically stable when the nasal packing procedure was initiated.
Although epistaxis may often have the appearance of significant blood loss and can be frightening for both the patient and HCP, most episodes are not life threatening. Death, when it occurs in association with epistaxis, is very rarely due to exsanguination.3 More commonly, death from epistaxis is related to complications of the treatment intervention or to an exacerbation of an underlying comorbid disease.10 The external overt blood loss in this patient was not significant enough to explain his cardiopulmonary collapse. Although he had experienced a recent pulmonary embolism, he had been on continuous anticoagulation for 3 months and remained adequately anticoagulated during his hospitalization. It therefore seems unlikely that he had experienced a recurrent pulmonary
embolism.
Complications
The treatment of epistaxis can be associated with serious infectious complications, including toxic shock syndrome due to nasal packing and infective endocarditis.11,12 Because patients with malignancies, autoimmune disorders, or organ failure may have epistaxis from decreased platelet production or increased platelet destruction, an infection can be devastating. Many HCPs anticipate this occurrence and provide the patient with epistaxis prophylactic antibiotics.13 Life-threatening infectious complications are usually delayed events and are generally easily recognized. An infectious process was not suspected in this patient. Nevertheless, he was treated with an oral antibiotic.
Dislodgement of the nasal packing with resultant aspiration and asphyxiation has been described as a fatal complication associated with the treatment of epistaxis.14 This complication was not observed in this patient. The otolaryngologist responsible for the placement of the nasal packing was in attendance during the cardiopulmonary resuscitation and insured oral pharyngeal airway patency. Moreover, endotracheal intubation also failed to identify an upper airway obstruction. Aspiration of the packing material was not the cause of this patient’s hemodynamic collapse.
Epidemiology
Florian Kratschmer (1843-1922) was the first researcher to provide a comprehensive analysis of changes in breathing, blood pressure, and heart rate that can occur when mucosa of the nasal airways are stimulated mechanically or chemically.15 His report is considered the first description of trigeminal-mediated bradycardia and asystole, a phenomenon that is sometimes referred to as Kratschmer’s reflex. In current terminology, it is referred to as the nasopulmonary reflex or TCR.
The trigeminal nerve is the largest of the cranial nerves. It provides sensory innervation to the face, scalp, and mucosa of the nose and mouth. The TCR may occur with manipulation of the branches of the trigeminal nerve anywhere along its intracranial or extracranial course. The TCR is described as a sudden onset of parasympathetic arrhythmia, sympathetic hypotension, or apnea elicited by central or peripheral stimulation of any of the sensory branches of the trigeminal nerve.16 The TCR may result in an immediate decrease of the mean arterial blood pressure and heart rate of > 20% when compared with the baseline levels with surgical, mechanical, electrical, or chemical stimulation of the central part of the sensory branches of the trigeminal nerve.17 The TCR represents one of the most powerful autonomous reflexes.18,19
Stimulation of trigeminal receptors that innervate the nose and nasal passage in animals can be an important stimulus for respiratory dysfunction and cardiac arrhythmias.15,16 However, the inability to accurately document the neuroanatomy of this reflex coupled with its variable and rare expression in humans has hindered the appreciation of the importance of the TCR. These observations have led some researchers to dismiss the reflex as inconsequential in humans. However, other physicians, particularly surgeons who manipulate craniofacial structures, have witnessed the effects of the TCR firsthand.20-25
In studies of neurosurgical procedures utilizing the nasal passages and transsphenoid approaches, the TCR has occurred in 10% to 18% of patients.16,24 The TCR has been consistently, although infrequently, noted by otolaryngologists in the management of epistaxis.10,26 Even when performed properly, posterior nasal packing has been reported to cause apnea, hypoxemia, and dysrhythmia.10 Although there has been debate about the importance of the TCR in humans, this response explains the sequence of events in and the death of this patient.27
The mechanism of the TCR is not well understood. The available data suggest that the response of the TCR when triggered by peripheral stimulation is different from the response when the TCR is triggered by central stimulation.18 There is additional anatomic evidence that different areas can be distinguished within the nasal mucosa with regard to stimulation site and stimulus properties.25 Specifically, it has been demonstrated in animals that mechanoreceptors are not equally sensitive throughout the nasal mucosa. The most sensitive areas for mechanical stimuli are located in the posterior parts of the nasal passages. In many animals, including humans, pronounced respiratory and cardiovascular responses can be elicited by appropriate stimulation of the nasal mucosa. These responses have been studied by many researchers in various animals and may be evoked by mechanical, electrical, and chemical stimuli.18,25
Risk Factors
Several risk factors for heightening the TCR have been described.25 Risk factors known to enhance the expression of TCR include hypercapnia, hypoxemia, light general anesthesia, the nature of provoking stimulus, the strength and duration of the stimulus, and medications. The specific pharmaceutical agents known to increase the manifestation of the TCR are narcotics, such as sufentanil and alfentanil, beta blockers, and calcium channel blockers.16,24 This patient was not on any of these medications. In addition, he had not been hypoxemic. He had no known risk for elevation of the TCR.
Evidence suggests that the intensity of the TCR corresponds with the intensity of the mechanical stimulation of the trigeminal pathway.24 Abrupt and sustained traction is more likely to evoke the TCR than is smooth and gentle manipulation. Immediate cessation of the stimulus, such as removal of the nasal packing, may be helpful in the prevention of fatal complications.16 Unfortunately, this was not accomplished in this patient. Other interventions, including the administration of atropine, local anesthetic infiltrations, or blockage of the nerve, may be helpful in preventing fatal complications.
The TCR may be elicited without prior hemodynamic changes. Nevertheless, it is important to anticipate hypoxemia and bradycardia as the first indication of a cardiopulmonary response.26 Administration of the anticholinergic atropine may be required in some cases where bradycardia is severe or persists despite cessation of the stimulus.
However, premedication with intramuscular administration of an anticholinergic medication has not been effective in preventing this reflex. Moreover, the TCR may at times be refractory to the conventional methods of treatment, and use of vasopressors and immediate cardiac life support may be required. Thus, if mechanical stimulation to the trigeminal nerve is anticipated, continuous monitoring of hemodynamic parameters may allow the clinician to more readily identify the TCR and immediately interrupt the inciting stimulus.24
This patient was being monitored, but his cardiopulmonary collapse occurred suddenly and rapidly. He received immediate resuscitation following advanced cardiac life support protocols. Unfortunately, there was no attempt to remove the material that had been employed as packing to control his epistaxis. It remains conjecture whether removal of this material could have altered his outcome. However, the gauze probably should have been removed to maximize his chance of survival.
Conclusion
This case demonstrates the clinical importance of the TCR to providers in the VA health care system, particularly to those who treat epistaxis. Because they are typically older, veterans are a high-risk group. Age is important due to the higher incidence of epistaxis in the older populace, and interventions are more often necessary in older patients with epistaxis. In addition, posterior bleeds occur more frequently in older patients. The resulting stimulation of the trigeminal nerve from interventions to control a posterior bleed may be a more potent provocation for the TCR. Finally, older patients often have comorbid illnesses requiring medications that may augment the TCR. Therefore, the veteran’s age and comorbid illnesses and medications may lead to greater susceptibility of a poor outcome, should the TCR occur as a result of interventions undertaken to control epistaxis.
VA practitioners should, therefore, be aware of the possible occurrence of the TCR in all patients with epistaxis, particularly when invasive manipulations of areas innervated by the trigeminal nerve are required. Evidence suggests that complications of the TCR range from mild bradycardia that responds to simple maneuvers to severe bradycardia and asystole requiring intervention with vagolytics. In rare cases, cardiac dysfunction may lead to death if the TCR is not suspected and early appropriate measures, such as removal of packing materials, are not undertaken.
Although the estimated complication rate of epistaxis and its treatment remains low (about 3%), the authors hope that this report will alert HCPs and that they will remain aware of the TCR as a potentially serious occurrence, even with mild to moderate manipulation of areas innervated by the trigeminal nerve.6
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Pallin DJ, Chng YM, McKay MP, Emond JA, Pelletier AJ, Camargo CA Jr. Epidemiology of epistaxis in US emergency departments, 1992 to 2001. Ann Emerg Med. 2005;46(1):77-81.
2. Viducich RA, Blanda MP, Gerson LW. Posterior epistaxis: clinical features and acute complications. Ann Emerg Med. 1995;25(5):592-596.
3. Manes RP. Evaluating and managing the patient with nosebleeds. Med Clin North Am. 2010;94(5):903-912.
4. National Center for Veterans Analysis and Statistics. Profile of veterans: 2011. Data from the American Community Survey. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2011.pdf. Published March 2013. Accessed May 4, 2015.
5. U.S. Census Bureau. Age and sex composition: 2010. http://www.census.gov/prod/cen2010/briefs/c2010br-03.pdf. Issued May 2011. Accessed May 4, 2015.
6. Pollice PA, Yoder MG. Epistaxis: a retrospective review of hospitalized patients. Otolaryngol Head Neck Surg. 1997;117(1):49-53.
7. Kucik CJ, Clenney T. Management of epistaxis. Am Fam Physician. 2005;71(2):305-311.
8. Kotecha B, Fowler S, Harkness P, Walmsley J, Brown P, Topham J. Management of epistaxis: a national survey. Ann R Coll Surg Engl. 1996;78(5):444-446.
9. Gifford TO, Orlandi RR. Epistaxis. Otolaryngol Clin North Am. 2008;41(3):525-536.
10. Fairbanks DN. Complications of nasal packing. Otolaryngol Head Neck Surg. 1986;94(3):412-415.
11. Aeumjaturapat S, Supanakorn S, Cutchavaree A. Toxic shock syndrome after anterior-posterior nasal packing. J Med Assoc Thai. 2001;84(3):453-458.
12. Jayawardena S, Eisdorfer J, Indulkar S, Zarkaria M. Infective endocarditis of native valve after anterior nasal packing. Am J Ther. 2006;13(5):460-462.
13. Derkay CS, Hirsch BE, Johnson JT, Wagner RL. Posterior nasal packing. Are intravenous antibiotics really necessary? Arch Otolaryngol Head Neck Surg.1989;115(4):439-441.
14. Koudounarakis E, Chatzakis N, Papadakis I, Panagiotaki I, Velegrakis G. Nasal packing aspiration in a patient with Alzheimer’s disease: a rare complication. Int J Gen Med. 2012;5:643-645.
15. Kratschmer F. On reflexes from the nasal mucous membrane on respiration and circulation. Respir Physiol. 2001;127(2-3):93-104.
16. Spiriev T, Sandu N, Arasho B, Kondoff S, Tzekov C, Schaller B. A new predisposing factor for trigeminocardiac reflex during subdural empyema drainage: a case report. J Med Case Reports. 2010;4:391.
17. Schaller B. Trigemino-cardiac reflex during microvascular trigeminal decompression in cases of trigeminal neuralgia. J Neurosurg Anesthesiol. 2005;17(1):45-48.
18. Schaller B, Cornelius JF, Prabhakar H, et al; Trigemino-Cardiac Reflex Examination Group (TCREG). The trigemino-cardiac reflex: an update of the current knowledge. J Neurosurg Anesthesiol. 2009;21(3):187-195.
19. Sandu N, Spiriev T, Lemaitre F, Filis A, Schaller B; Trigemino-Cardiac Reflex Examination Group (TCREG). New molecular knowledge towards the trigemino-cardiac reflex as a cerebral oxygenconserving reflex. Sci World J. 2010;10:811-817.
20. Nirmala J, Dilip KK, Padmaja D, Gopinath R. “Kratschmer” reflex during rhinoplasty. Anesth Analg. 2006;103(5):1337-1338.
21. Jacobs JR, Levine LA, Davis H, Lefrak SS, Druck NS, Ogura JH. Posterior packs and the nasopulmonary reflex. Laryngoscope. 1981;91(2):279-284.
22. Larsen K, Juul A. Arterial blood gases and pneumatic nasal packing in epistaxis. Laryngoscope.1982;92(5):586-588.
23. Loftus BC, Blitzer A, Cozine K. Epistaxis, medical history, and the nasopulmonary reflex: what is clinically relevant? Otolaryngol Head Neck Surg. 1994;110(4):363-369.
24. Arasho B, Sandu N, Spiriev T, Prabhakar H, Schaller B. Management of the trigeminocardiac reflex: facts and own experience. Neurol India. 2009;57(4):375-380.
25. Schaller BJ, Filis A, Buchfelder M. Trigeminocardiac reflex in humans initiated by peripheral
stimulation during neurosurgical skull-base operations. Its first description. Acta Neurochir (Wien). 2008;150(7):715-717; discussion 718.
26. Stemm RA. Complications of nasal packing. Ear Nose Throat J. 1981;60(10):461-462.
27. Widdicombe J. Reflexes from the lungs and airways: historical perspective. J Appl Physiol (1985). 2006;101(2):628-634.
Epistaxis is a relatively common event that is estimated to occur at least once in 60% of the U.S. population. Epistaxis is also reported to cause 1.7 emergency department (ED) visits per 1,000 population annually.1 Although epistaxis can occur at any age, it typically occurs with a bimodal age distribution and most commonly affects individuals aged < 18 years and adults aged > 50 years.2 The episodes of epistaxis involving the younger age group are more often minor and self-limited. Most bleeds occur along the anterior nasal septum from Kiesselbach’s plexus.
Posterior bleeds occur more often in older patients.2 In addition, epistaxis in the older population tends to be more severe.3 Medical intervention is required in 6% of those experiencing epistaxis. Because the median age for male veterans was 64 years in 2011 compared with a median age of 37.2 years for the average U.S. population in 2010, veterans are among those at greatest risk to develop epistaxis that requires intervention.4,5
Most episodes of epistaxis are not life-threatening, particularly when modern methods of diagnosis and treatment are used. Nevertheless, comorbid diseases, complications of treatment, and normal physiologic responses can sometimes combine to create an adverse outcome.6 This report reviews the case of a veteran patient who experienced a fatal cardiopulmonary arrest after therapeutic interventions for epistaxis. It is believed that his death was due to the well-described but little known trigeminocardiac reflex (TCR).
Case Report
A 65-year-old man visited the ED and reported that his nose had been bleeding intermittently for 1 day. He estimated that he had lost 1 cup of blood over a 24-hour period. He reported no rhinosinusitis, nasal congestion, or recent allergy or upper respiratory infections. He also reported no nasal trauma. In the ED, blood was oozing from his right nares and into his throat, causing him to cough. External compression failed to control the oozing. Topical vasoconstrictors were not applied.
His past medical history included a pulmonary embolism, well-controlled chronic obstructive pulmonary disease (COPD), and sleep apnea. The thrombotic site of origin for his pulmonary embolism had not been identified, despite a thorough examination. He had been on warfarin therapy for 3 months, and his international normalized ratio (INR) had been monitored in an anticoagulation clinic and was well regulated. He was also on inhaled medications for COPD (formoterol and budesonide as a combination preparation twice daily and albuterol every 6 hours as needed as a rescue medication). He adhered to his noninvasive positive airway pressure (PAP) device treatment for sleep apnea. He did not take aspirin or other antiplatelet medications. He reported no use of topical nasal preparations. He also reported no use of illicit drugs or over-the-counter medications, including nonsteroidal anti-inflammatory medications and herbal remedies. He reported no bleeding from other sites or easy bruising.
The patient was alert, oriented, and in no distress. His vital signs were normal. Examination of his nasal passages failed to identify an active site of bleeding. Fresh blood was present in the right nasal passage and the posterior pharynx. Examination of his chest was normal. His hemoglobin was 13.1 g/dL (13.6-17.3 g/dL) with 216 x 103/μL platelets (166-383 x 103/μL). His INR was therapeutic at 2.38. Laboratory assessments of his electrolytes, liver function, and renal function were normal. A chest radiograph demonstrated no acute process. A computed tomography failed to demonstrate sinusitis or an anatomical abnormality that could account for his epistaxis.
Due to the amount of blood loss by epistaxis complicated by anticoagulation for his recent pulmonary embolism, the patient was admitted to the hospital for observation. Reversal of the anticoagulation was considered by the admitting service, but because the patient was only oozing blood, this intervention was not undertaken. Instead, he was continued on warfarin, was treated with an oral antibiotic, and was continued on his inhaled medications for his COPD. He also used his noninvasive PAP device to sleep.
The next day, the patient began to bleed freely from his right nares. The bleeding was initially controlled with compression and positioning and resolved without additional intervention. An otolaryngologist performed silver nitrate cauterization of Kiesselbach’s plexus. The patient experienced no further bleeding, and his hemoglobin remained stable.
The next day, his nose began to bleed briskly. He passed large clots from his nose and mouth. The patient was alert and oriented. He remained hemodynamically stable. His INR was 2.1. Nasal packing was proposed, and the procedure, including the risks and benefits, were explained to the patient.
After obtaining consent from thepatient, the nasal mucosa was prepared with topical 2% lidocaine and 1% phenylephrine. Anterior and posterior nasal packing was successfully achieved with paraffin gauze. This procedure was completed in a monitored environment by an experienced otolaryngologist. However, the patient became agitated 15 to 20 minutes after the nasal packing had been accomplished. He rapidly became apneic, bradycardic, and hypotensive. His oxygen saturation on room air as measured by pulse oximetry decreased precipitously to 50%. These developments were quickly followed by asystole.
Advanced cardiac life support measures were initiated. His airway was secured by oral endotracheal intubation, and oxygen was delivered at 100% fraction of inspired oxygen by bag ventilation. At intubation, only a few small clots were present in the posterior pharynx. No blood was suctioned from the endotracheal tube; therefore, active bleeding was not suspected. The nasal packing remained in place and was not removed. The patient failed to regain spontaneous circulation and died. An arterial blood gas analysis obtained during cardiopulmonary resuscitation demonstrated no methemoglobin on co-oximetry.
Discussion
Because of the high prevalence of epistaxis in the general population, many health care providers (HCPs) are confronted with this problem. Epistaxis in most patients remits without consequence. However, HCPs may be required to intervene. Treatment modalities include simple compression and positioning maneuvers, the application of topical medications, anterior and posterior nasal packing, chemical cauterization, endoscopic electric cauterization, embolization therapy, and surgical arterial ligation.7 The choice of therapy depends on several factors, including the site of the bleeding, the severity of the bleeding, the availability of resources, and the expertise of the HCP. A localized cause of epistaxis is discovered in only 15% of patients, making a conservative therapeutic approach an attractive initial intervention.8
Nasal packing is a successful intervention in 70% of patients with posterior epistaxis. In addition, nasal packing is the preferred method for hemostasis in anterior epistaxis when cauterization fails.3,9 This patient failed simple compression and positioning maneuvers as well as chemical cauterization. For this reason, nasal packing was proposed as a therapeutic intervention. He was hemodynamically stable when the nasal packing procedure was initiated.
Although epistaxis may often have the appearance of significant blood loss and can be frightening for both the patient and HCP, most episodes are not life threatening. Death, when it occurs in association with epistaxis, is very rarely due to exsanguination.3 More commonly, death from epistaxis is related to complications of the treatment intervention or to an exacerbation of an underlying comorbid disease.10 The external overt blood loss in this patient was not significant enough to explain his cardiopulmonary collapse. Although he had experienced a recent pulmonary embolism, he had been on continuous anticoagulation for 3 months and remained adequately anticoagulated during his hospitalization. It therefore seems unlikely that he had experienced a recurrent pulmonary
embolism.
Complications
The treatment of epistaxis can be associated with serious infectious complications, including toxic shock syndrome due to nasal packing and infective endocarditis.11,12 Because patients with malignancies, autoimmune disorders, or organ failure may have epistaxis from decreased platelet production or increased platelet destruction, an infection can be devastating. Many HCPs anticipate this occurrence and provide the patient with epistaxis prophylactic antibiotics.13 Life-threatening infectious complications are usually delayed events and are generally easily recognized. An infectious process was not suspected in this patient. Nevertheless, he was treated with an oral antibiotic.
Dislodgement of the nasal packing with resultant aspiration and asphyxiation has been described as a fatal complication associated with the treatment of epistaxis.14 This complication was not observed in this patient. The otolaryngologist responsible for the placement of the nasal packing was in attendance during the cardiopulmonary resuscitation and insured oral pharyngeal airway patency. Moreover, endotracheal intubation also failed to identify an upper airway obstruction. Aspiration of the packing material was not the cause of this patient’s hemodynamic collapse.
Epidemiology
Florian Kratschmer (1843-1922) was the first researcher to provide a comprehensive analysis of changes in breathing, blood pressure, and heart rate that can occur when mucosa of the nasal airways are stimulated mechanically or chemically.15 His report is considered the first description of trigeminal-mediated bradycardia and asystole, a phenomenon that is sometimes referred to as Kratschmer’s reflex. In current terminology, it is referred to as the nasopulmonary reflex or TCR.
The trigeminal nerve is the largest of the cranial nerves. It provides sensory innervation to the face, scalp, and mucosa of the nose and mouth. The TCR may occur with manipulation of the branches of the trigeminal nerve anywhere along its intracranial or extracranial course. The TCR is described as a sudden onset of parasympathetic arrhythmia, sympathetic hypotension, or apnea elicited by central or peripheral stimulation of any of the sensory branches of the trigeminal nerve.16 The TCR may result in an immediate decrease of the mean arterial blood pressure and heart rate of > 20% when compared with the baseline levels with surgical, mechanical, electrical, or chemical stimulation of the central part of the sensory branches of the trigeminal nerve.17 The TCR represents one of the most powerful autonomous reflexes.18,19
Stimulation of trigeminal receptors that innervate the nose and nasal passage in animals can be an important stimulus for respiratory dysfunction and cardiac arrhythmias.15,16 However, the inability to accurately document the neuroanatomy of this reflex coupled with its variable and rare expression in humans has hindered the appreciation of the importance of the TCR. These observations have led some researchers to dismiss the reflex as inconsequential in humans. However, other physicians, particularly surgeons who manipulate craniofacial structures, have witnessed the effects of the TCR firsthand.20-25
In studies of neurosurgical procedures utilizing the nasal passages and transsphenoid approaches, the TCR has occurred in 10% to 18% of patients.16,24 The TCR has been consistently, although infrequently, noted by otolaryngologists in the management of epistaxis.10,26 Even when performed properly, posterior nasal packing has been reported to cause apnea, hypoxemia, and dysrhythmia.10 Although there has been debate about the importance of the TCR in humans, this response explains the sequence of events in and the death of this patient.27
The mechanism of the TCR is not well understood. The available data suggest that the response of the TCR when triggered by peripheral stimulation is different from the response when the TCR is triggered by central stimulation.18 There is additional anatomic evidence that different areas can be distinguished within the nasal mucosa with regard to stimulation site and stimulus properties.25 Specifically, it has been demonstrated in animals that mechanoreceptors are not equally sensitive throughout the nasal mucosa. The most sensitive areas for mechanical stimuli are located in the posterior parts of the nasal passages. In many animals, including humans, pronounced respiratory and cardiovascular responses can be elicited by appropriate stimulation of the nasal mucosa. These responses have been studied by many researchers in various animals and may be evoked by mechanical, electrical, and chemical stimuli.18,25
Risk Factors
Several risk factors for heightening the TCR have been described.25 Risk factors known to enhance the expression of TCR include hypercapnia, hypoxemia, light general anesthesia, the nature of provoking stimulus, the strength and duration of the stimulus, and medications. The specific pharmaceutical agents known to increase the manifestation of the TCR are narcotics, such as sufentanil and alfentanil, beta blockers, and calcium channel blockers.16,24 This patient was not on any of these medications. In addition, he had not been hypoxemic. He had no known risk for elevation of the TCR.
Evidence suggests that the intensity of the TCR corresponds with the intensity of the mechanical stimulation of the trigeminal pathway.24 Abrupt and sustained traction is more likely to evoke the TCR than is smooth and gentle manipulation. Immediate cessation of the stimulus, such as removal of the nasal packing, may be helpful in the prevention of fatal complications.16 Unfortunately, this was not accomplished in this patient. Other interventions, including the administration of atropine, local anesthetic infiltrations, or blockage of the nerve, may be helpful in preventing fatal complications.
The TCR may be elicited without prior hemodynamic changes. Nevertheless, it is important to anticipate hypoxemia and bradycardia as the first indication of a cardiopulmonary response.26 Administration of the anticholinergic atropine may be required in some cases where bradycardia is severe or persists despite cessation of the stimulus.
However, premedication with intramuscular administration of an anticholinergic medication has not been effective in preventing this reflex. Moreover, the TCR may at times be refractory to the conventional methods of treatment, and use of vasopressors and immediate cardiac life support may be required. Thus, if mechanical stimulation to the trigeminal nerve is anticipated, continuous monitoring of hemodynamic parameters may allow the clinician to more readily identify the TCR and immediately interrupt the inciting stimulus.24
This patient was being monitored, but his cardiopulmonary collapse occurred suddenly and rapidly. He received immediate resuscitation following advanced cardiac life support protocols. Unfortunately, there was no attempt to remove the material that had been employed as packing to control his epistaxis. It remains conjecture whether removal of this material could have altered his outcome. However, the gauze probably should have been removed to maximize his chance of survival.
Conclusion
This case demonstrates the clinical importance of the TCR to providers in the VA health care system, particularly to those who treat epistaxis. Because they are typically older, veterans are a high-risk group. Age is important due to the higher incidence of epistaxis in the older populace, and interventions are more often necessary in older patients with epistaxis. In addition, posterior bleeds occur more frequently in older patients. The resulting stimulation of the trigeminal nerve from interventions to control a posterior bleed may be a more potent provocation for the TCR. Finally, older patients often have comorbid illnesses requiring medications that may augment the TCR. Therefore, the veteran’s age and comorbid illnesses and medications may lead to greater susceptibility of a poor outcome, should the TCR occur as a result of interventions undertaken to control epistaxis.
VA practitioners should, therefore, be aware of the possible occurrence of the TCR in all patients with epistaxis, particularly when invasive manipulations of areas innervated by the trigeminal nerve are required. Evidence suggests that complications of the TCR range from mild bradycardia that responds to simple maneuvers to severe bradycardia and asystole requiring intervention with vagolytics. In rare cases, cardiac dysfunction may lead to death if the TCR is not suspected and early appropriate measures, such as removal of packing materials, are not undertaken.
Although the estimated complication rate of epistaxis and its treatment remains low (about 3%), the authors hope that this report will alert HCPs and that they will remain aware of the TCR as a potentially serious occurrence, even with mild to moderate manipulation of areas innervated by the trigeminal nerve.6
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Epistaxis is a relatively common event that is estimated to occur at least once in 60% of the U.S. population. Epistaxis is also reported to cause 1.7 emergency department (ED) visits per 1,000 population annually.1 Although epistaxis can occur at any age, it typically occurs with a bimodal age distribution and most commonly affects individuals aged < 18 years and adults aged > 50 years.2 The episodes of epistaxis involving the younger age group are more often minor and self-limited. Most bleeds occur along the anterior nasal septum from Kiesselbach’s plexus.
Posterior bleeds occur more often in older patients.2 In addition, epistaxis in the older population tends to be more severe.3 Medical intervention is required in 6% of those experiencing epistaxis. Because the median age for male veterans was 64 years in 2011 compared with a median age of 37.2 years for the average U.S. population in 2010, veterans are among those at greatest risk to develop epistaxis that requires intervention.4,5
Most episodes of epistaxis are not life-threatening, particularly when modern methods of diagnosis and treatment are used. Nevertheless, comorbid diseases, complications of treatment, and normal physiologic responses can sometimes combine to create an adverse outcome.6 This report reviews the case of a veteran patient who experienced a fatal cardiopulmonary arrest after therapeutic interventions for epistaxis. It is believed that his death was due to the well-described but little known trigeminocardiac reflex (TCR).
Case Report
A 65-year-old man visited the ED and reported that his nose had been bleeding intermittently for 1 day. He estimated that he had lost 1 cup of blood over a 24-hour period. He reported no rhinosinusitis, nasal congestion, or recent allergy or upper respiratory infections. He also reported no nasal trauma. In the ED, blood was oozing from his right nares and into his throat, causing him to cough. External compression failed to control the oozing. Topical vasoconstrictors were not applied.
His past medical history included a pulmonary embolism, well-controlled chronic obstructive pulmonary disease (COPD), and sleep apnea. The thrombotic site of origin for his pulmonary embolism had not been identified, despite a thorough examination. He had been on warfarin therapy for 3 months, and his international normalized ratio (INR) had been monitored in an anticoagulation clinic and was well regulated. He was also on inhaled medications for COPD (formoterol and budesonide as a combination preparation twice daily and albuterol every 6 hours as needed as a rescue medication). He adhered to his noninvasive positive airway pressure (PAP) device treatment for sleep apnea. He did not take aspirin or other antiplatelet medications. He reported no use of topical nasal preparations. He also reported no use of illicit drugs or over-the-counter medications, including nonsteroidal anti-inflammatory medications and herbal remedies. He reported no bleeding from other sites or easy bruising.
The patient was alert, oriented, and in no distress. His vital signs were normal. Examination of his nasal passages failed to identify an active site of bleeding. Fresh blood was present in the right nasal passage and the posterior pharynx. Examination of his chest was normal. His hemoglobin was 13.1 g/dL (13.6-17.3 g/dL) with 216 x 103/μL platelets (166-383 x 103/μL). His INR was therapeutic at 2.38. Laboratory assessments of his electrolytes, liver function, and renal function were normal. A chest radiograph demonstrated no acute process. A computed tomography failed to demonstrate sinusitis or an anatomical abnormality that could account for his epistaxis.
Due to the amount of blood loss by epistaxis complicated by anticoagulation for his recent pulmonary embolism, the patient was admitted to the hospital for observation. Reversal of the anticoagulation was considered by the admitting service, but because the patient was only oozing blood, this intervention was not undertaken. Instead, he was continued on warfarin, was treated with an oral antibiotic, and was continued on his inhaled medications for his COPD. He also used his noninvasive PAP device to sleep.
The next day, the patient began to bleed freely from his right nares. The bleeding was initially controlled with compression and positioning and resolved without additional intervention. An otolaryngologist performed silver nitrate cauterization of Kiesselbach’s plexus. The patient experienced no further bleeding, and his hemoglobin remained stable.
The next day, his nose began to bleed briskly. He passed large clots from his nose and mouth. The patient was alert and oriented. He remained hemodynamically stable. His INR was 2.1. Nasal packing was proposed, and the procedure, including the risks and benefits, were explained to the patient.
After obtaining consent from thepatient, the nasal mucosa was prepared with topical 2% lidocaine and 1% phenylephrine. Anterior and posterior nasal packing was successfully achieved with paraffin gauze. This procedure was completed in a monitored environment by an experienced otolaryngologist. However, the patient became agitated 15 to 20 minutes after the nasal packing had been accomplished. He rapidly became apneic, bradycardic, and hypotensive. His oxygen saturation on room air as measured by pulse oximetry decreased precipitously to 50%. These developments were quickly followed by asystole.
Advanced cardiac life support measures were initiated. His airway was secured by oral endotracheal intubation, and oxygen was delivered at 100% fraction of inspired oxygen by bag ventilation. At intubation, only a few small clots were present in the posterior pharynx. No blood was suctioned from the endotracheal tube; therefore, active bleeding was not suspected. The nasal packing remained in place and was not removed. The patient failed to regain spontaneous circulation and died. An arterial blood gas analysis obtained during cardiopulmonary resuscitation demonstrated no methemoglobin on co-oximetry.
Discussion
Because of the high prevalence of epistaxis in the general population, many health care providers (HCPs) are confronted with this problem. Epistaxis in most patients remits without consequence. However, HCPs may be required to intervene. Treatment modalities include simple compression and positioning maneuvers, the application of topical medications, anterior and posterior nasal packing, chemical cauterization, endoscopic electric cauterization, embolization therapy, and surgical arterial ligation.7 The choice of therapy depends on several factors, including the site of the bleeding, the severity of the bleeding, the availability of resources, and the expertise of the HCP. A localized cause of epistaxis is discovered in only 15% of patients, making a conservative therapeutic approach an attractive initial intervention.8
Nasal packing is a successful intervention in 70% of patients with posterior epistaxis. In addition, nasal packing is the preferred method for hemostasis in anterior epistaxis when cauterization fails.3,9 This patient failed simple compression and positioning maneuvers as well as chemical cauterization. For this reason, nasal packing was proposed as a therapeutic intervention. He was hemodynamically stable when the nasal packing procedure was initiated.
Although epistaxis may often have the appearance of significant blood loss and can be frightening for both the patient and HCP, most episodes are not life threatening. Death, when it occurs in association with epistaxis, is very rarely due to exsanguination.3 More commonly, death from epistaxis is related to complications of the treatment intervention or to an exacerbation of an underlying comorbid disease.10 The external overt blood loss in this patient was not significant enough to explain his cardiopulmonary collapse. Although he had experienced a recent pulmonary embolism, he had been on continuous anticoagulation for 3 months and remained adequately anticoagulated during his hospitalization. It therefore seems unlikely that he had experienced a recurrent pulmonary
embolism.
Complications
The treatment of epistaxis can be associated with serious infectious complications, including toxic shock syndrome due to nasal packing and infective endocarditis.11,12 Because patients with malignancies, autoimmune disorders, or organ failure may have epistaxis from decreased platelet production or increased platelet destruction, an infection can be devastating. Many HCPs anticipate this occurrence and provide the patient with epistaxis prophylactic antibiotics.13 Life-threatening infectious complications are usually delayed events and are generally easily recognized. An infectious process was not suspected in this patient. Nevertheless, he was treated with an oral antibiotic.
Dislodgement of the nasal packing with resultant aspiration and asphyxiation has been described as a fatal complication associated with the treatment of epistaxis.14 This complication was not observed in this patient. The otolaryngologist responsible for the placement of the nasal packing was in attendance during the cardiopulmonary resuscitation and insured oral pharyngeal airway patency. Moreover, endotracheal intubation also failed to identify an upper airway obstruction. Aspiration of the packing material was not the cause of this patient’s hemodynamic collapse.
Epidemiology
Florian Kratschmer (1843-1922) was the first researcher to provide a comprehensive analysis of changes in breathing, blood pressure, and heart rate that can occur when mucosa of the nasal airways are stimulated mechanically or chemically.15 His report is considered the first description of trigeminal-mediated bradycardia and asystole, a phenomenon that is sometimes referred to as Kratschmer’s reflex. In current terminology, it is referred to as the nasopulmonary reflex or TCR.
The trigeminal nerve is the largest of the cranial nerves. It provides sensory innervation to the face, scalp, and mucosa of the nose and mouth. The TCR may occur with manipulation of the branches of the trigeminal nerve anywhere along its intracranial or extracranial course. The TCR is described as a sudden onset of parasympathetic arrhythmia, sympathetic hypotension, or apnea elicited by central or peripheral stimulation of any of the sensory branches of the trigeminal nerve.16 The TCR may result in an immediate decrease of the mean arterial blood pressure and heart rate of > 20% when compared with the baseline levels with surgical, mechanical, electrical, or chemical stimulation of the central part of the sensory branches of the trigeminal nerve.17 The TCR represents one of the most powerful autonomous reflexes.18,19
Stimulation of trigeminal receptors that innervate the nose and nasal passage in animals can be an important stimulus for respiratory dysfunction and cardiac arrhythmias.15,16 However, the inability to accurately document the neuroanatomy of this reflex coupled with its variable and rare expression in humans has hindered the appreciation of the importance of the TCR. These observations have led some researchers to dismiss the reflex as inconsequential in humans. However, other physicians, particularly surgeons who manipulate craniofacial structures, have witnessed the effects of the TCR firsthand.20-25
In studies of neurosurgical procedures utilizing the nasal passages and transsphenoid approaches, the TCR has occurred in 10% to 18% of patients.16,24 The TCR has been consistently, although infrequently, noted by otolaryngologists in the management of epistaxis.10,26 Even when performed properly, posterior nasal packing has been reported to cause apnea, hypoxemia, and dysrhythmia.10 Although there has been debate about the importance of the TCR in humans, this response explains the sequence of events in and the death of this patient.27
The mechanism of the TCR is not well understood. The available data suggest that the response of the TCR when triggered by peripheral stimulation is different from the response when the TCR is triggered by central stimulation.18 There is additional anatomic evidence that different areas can be distinguished within the nasal mucosa with regard to stimulation site and stimulus properties.25 Specifically, it has been demonstrated in animals that mechanoreceptors are not equally sensitive throughout the nasal mucosa. The most sensitive areas for mechanical stimuli are located in the posterior parts of the nasal passages. In many animals, including humans, pronounced respiratory and cardiovascular responses can be elicited by appropriate stimulation of the nasal mucosa. These responses have been studied by many researchers in various animals and may be evoked by mechanical, electrical, and chemical stimuli.18,25
Risk Factors
Several risk factors for heightening the TCR have been described.25 Risk factors known to enhance the expression of TCR include hypercapnia, hypoxemia, light general anesthesia, the nature of provoking stimulus, the strength and duration of the stimulus, and medications. The specific pharmaceutical agents known to increase the manifestation of the TCR are narcotics, such as sufentanil and alfentanil, beta blockers, and calcium channel blockers.16,24 This patient was not on any of these medications. In addition, he had not been hypoxemic. He had no known risk for elevation of the TCR.
Evidence suggests that the intensity of the TCR corresponds with the intensity of the mechanical stimulation of the trigeminal pathway.24 Abrupt and sustained traction is more likely to evoke the TCR than is smooth and gentle manipulation. Immediate cessation of the stimulus, such as removal of the nasal packing, may be helpful in the prevention of fatal complications.16 Unfortunately, this was not accomplished in this patient. Other interventions, including the administration of atropine, local anesthetic infiltrations, or blockage of the nerve, may be helpful in preventing fatal complications.
The TCR may be elicited without prior hemodynamic changes. Nevertheless, it is important to anticipate hypoxemia and bradycardia as the first indication of a cardiopulmonary response.26 Administration of the anticholinergic atropine may be required in some cases where bradycardia is severe or persists despite cessation of the stimulus.
However, premedication with intramuscular administration of an anticholinergic medication has not been effective in preventing this reflex. Moreover, the TCR may at times be refractory to the conventional methods of treatment, and use of vasopressors and immediate cardiac life support may be required. Thus, if mechanical stimulation to the trigeminal nerve is anticipated, continuous monitoring of hemodynamic parameters may allow the clinician to more readily identify the TCR and immediately interrupt the inciting stimulus.24
This patient was being monitored, but his cardiopulmonary collapse occurred suddenly and rapidly. He received immediate resuscitation following advanced cardiac life support protocols. Unfortunately, there was no attempt to remove the material that had been employed as packing to control his epistaxis. It remains conjecture whether removal of this material could have altered his outcome. However, the gauze probably should have been removed to maximize his chance of survival.
Conclusion
This case demonstrates the clinical importance of the TCR to providers in the VA health care system, particularly to those who treat epistaxis. Because they are typically older, veterans are a high-risk group. Age is important due to the higher incidence of epistaxis in the older populace, and interventions are more often necessary in older patients with epistaxis. In addition, posterior bleeds occur more frequently in older patients. The resulting stimulation of the trigeminal nerve from interventions to control a posterior bleed may be a more potent provocation for the TCR. Finally, older patients often have comorbid illnesses requiring medications that may augment the TCR. Therefore, the veteran’s age and comorbid illnesses and medications may lead to greater susceptibility of a poor outcome, should the TCR occur as a result of interventions undertaken to control epistaxis.
VA practitioners should, therefore, be aware of the possible occurrence of the TCR in all patients with epistaxis, particularly when invasive manipulations of areas innervated by the trigeminal nerve are required. Evidence suggests that complications of the TCR range from mild bradycardia that responds to simple maneuvers to severe bradycardia and asystole requiring intervention with vagolytics. In rare cases, cardiac dysfunction may lead to death if the TCR is not suspected and early appropriate measures, such as removal of packing materials, are not undertaken.
Although the estimated complication rate of epistaxis and its treatment remains low (about 3%), the authors hope that this report will alert HCPs and that they will remain aware of the TCR as a potentially serious occurrence, even with mild to moderate manipulation of areas innervated by the trigeminal nerve.6
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Pallin DJ, Chng YM, McKay MP, Emond JA, Pelletier AJ, Camargo CA Jr. Epidemiology of epistaxis in US emergency departments, 1992 to 2001. Ann Emerg Med. 2005;46(1):77-81.
2. Viducich RA, Blanda MP, Gerson LW. Posterior epistaxis: clinical features and acute complications. Ann Emerg Med. 1995;25(5):592-596.
3. Manes RP. Evaluating and managing the patient with nosebleeds. Med Clin North Am. 2010;94(5):903-912.
4. National Center for Veterans Analysis and Statistics. Profile of veterans: 2011. Data from the American Community Survey. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2011.pdf. Published March 2013. Accessed May 4, 2015.
5. U.S. Census Bureau. Age and sex composition: 2010. http://www.census.gov/prod/cen2010/briefs/c2010br-03.pdf. Issued May 2011. Accessed May 4, 2015.
6. Pollice PA, Yoder MG. Epistaxis: a retrospective review of hospitalized patients. Otolaryngol Head Neck Surg. 1997;117(1):49-53.
7. Kucik CJ, Clenney T. Management of epistaxis. Am Fam Physician. 2005;71(2):305-311.
8. Kotecha B, Fowler S, Harkness P, Walmsley J, Brown P, Topham J. Management of epistaxis: a national survey. Ann R Coll Surg Engl. 1996;78(5):444-446.
9. Gifford TO, Orlandi RR. Epistaxis. Otolaryngol Clin North Am. 2008;41(3):525-536.
10. Fairbanks DN. Complications of nasal packing. Otolaryngol Head Neck Surg. 1986;94(3):412-415.
11. Aeumjaturapat S, Supanakorn S, Cutchavaree A. Toxic shock syndrome after anterior-posterior nasal packing. J Med Assoc Thai. 2001;84(3):453-458.
12. Jayawardena S, Eisdorfer J, Indulkar S, Zarkaria M. Infective endocarditis of native valve after anterior nasal packing. Am J Ther. 2006;13(5):460-462.
13. Derkay CS, Hirsch BE, Johnson JT, Wagner RL. Posterior nasal packing. Are intravenous antibiotics really necessary? Arch Otolaryngol Head Neck Surg.1989;115(4):439-441.
14. Koudounarakis E, Chatzakis N, Papadakis I, Panagiotaki I, Velegrakis G. Nasal packing aspiration in a patient with Alzheimer’s disease: a rare complication. Int J Gen Med. 2012;5:643-645.
15. Kratschmer F. On reflexes from the nasal mucous membrane on respiration and circulation. Respir Physiol. 2001;127(2-3):93-104.
16. Spiriev T, Sandu N, Arasho B, Kondoff S, Tzekov C, Schaller B. A new predisposing factor for trigeminocardiac reflex during subdural empyema drainage: a case report. J Med Case Reports. 2010;4:391.
17. Schaller B. Trigemino-cardiac reflex during microvascular trigeminal decompression in cases of trigeminal neuralgia. J Neurosurg Anesthesiol. 2005;17(1):45-48.
18. Schaller B, Cornelius JF, Prabhakar H, et al; Trigemino-Cardiac Reflex Examination Group (TCREG). The trigemino-cardiac reflex: an update of the current knowledge. J Neurosurg Anesthesiol. 2009;21(3):187-195.
19. Sandu N, Spiriev T, Lemaitre F, Filis A, Schaller B; Trigemino-Cardiac Reflex Examination Group (TCREG). New molecular knowledge towards the trigemino-cardiac reflex as a cerebral oxygenconserving reflex. Sci World J. 2010;10:811-817.
20. Nirmala J, Dilip KK, Padmaja D, Gopinath R. “Kratschmer” reflex during rhinoplasty. Anesth Analg. 2006;103(5):1337-1338.
21. Jacobs JR, Levine LA, Davis H, Lefrak SS, Druck NS, Ogura JH. Posterior packs and the nasopulmonary reflex. Laryngoscope. 1981;91(2):279-284.
22. Larsen K, Juul A. Arterial blood gases and pneumatic nasal packing in epistaxis. Laryngoscope.1982;92(5):586-588.
23. Loftus BC, Blitzer A, Cozine K. Epistaxis, medical history, and the nasopulmonary reflex: what is clinically relevant? Otolaryngol Head Neck Surg. 1994;110(4):363-369.
24. Arasho B, Sandu N, Spiriev T, Prabhakar H, Schaller B. Management of the trigeminocardiac reflex: facts and own experience. Neurol India. 2009;57(4):375-380.
25. Schaller BJ, Filis A, Buchfelder M. Trigeminocardiac reflex in humans initiated by peripheral
stimulation during neurosurgical skull-base operations. Its first description. Acta Neurochir (Wien). 2008;150(7):715-717; discussion 718.
26. Stemm RA. Complications of nasal packing. Ear Nose Throat J. 1981;60(10):461-462.
27. Widdicombe J. Reflexes from the lungs and airways: historical perspective. J Appl Physiol (1985). 2006;101(2):628-634.
1. Pallin DJ, Chng YM, McKay MP, Emond JA, Pelletier AJ, Camargo CA Jr. Epidemiology of epistaxis in US emergency departments, 1992 to 2001. Ann Emerg Med. 2005;46(1):77-81.
2. Viducich RA, Blanda MP, Gerson LW. Posterior epistaxis: clinical features and acute complications. Ann Emerg Med. 1995;25(5):592-596.
3. Manes RP. Evaluating and managing the patient with nosebleeds. Med Clin North Am. 2010;94(5):903-912.
4. National Center for Veterans Analysis and Statistics. Profile of veterans: 2011. Data from the American Community Survey. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2011.pdf. Published March 2013. Accessed May 4, 2015.
5. U.S. Census Bureau. Age and sex composition: 2010. http://www.census.gov/prod/cen2010/briefs/c2010br-03.pdf. Issued May 2011. Accessed May 4, 2015.
6. Pollice PA, Yoder MG. Epistaxis: a retrospective review of hospitalized patients. Otolaryngol Head Neck Surg. 1997;117(1):49-53.
7. Kucik CJ, Clenney T. Management of epistaxis. Am Fam Physician. 2005;71(2):305-311.
8. Kotecha B, Fowler S, Harkness P, Walmsley J, Brown P, Topham J. Management of epistaxis: a national survey. Ann R Coll Surg Engl. 1996;78(5):444-446.
9. Gifford TO, Orlandi RR. Epistaxis. Otolaryngol Clin North Am. 2008;41(3):525-536.
10. Fairbanks DN. Complications of nasal packing. Otolaryngol Head Neck Surg. 1986;94(3):412-415.
11. Aeumjaturapat S, Supanakorn S, Cutchavaree A. Toxic shock syndrome after anterior-posterior nasal packing. J Med Assoc Thai. 2001;84(3):453-458.
12. Jayawardena S, Eisdorfer J, Indulkar S, Zarkaria M. Infective endocarditis of native valve after anterior nasal packing. Am J Ther. 2006;13(5):460-462.
13. Derkay CS, Hirsch BE, Johnson JT, Wagner RL. Posterior nasal packing. Are intravenous antibiotics really necessary? Arch Otolaryngol Head Neck Surg.1989;115(4):439-441.
14. Koudounarakis E, Chatzakis N, Papadakis I, Panagiotaki I, Velegrakis G. Nasal packing aspiration in a patient with Alzheimer’s disease: a rare complication. Int J Gen Med. 2012;5:643-645.
15. Kratschmer F. On reflexes from the nasal mucous membrane on respiration and circulation. Respir Physiol. 2001;127(2-3):93-104.
16. Spiriev T, Sandu N, Arasho B, Kondoff S, Tzekov C, Schaller B. A new predisposing factor for trigeminocardiac reflex during subdural empyema drainage: a case report. J Med Case Reports. 2010;4:391.
17. Schaller B. Trigemino-cardiac reflex during microvascular trigeminal decompression in cases of trigeminal neuralgia. J Neurosurg Anesthesiol. 2005;17(1):45-48.
18. Schaller B, Cornelius JF, Prabhakar H, et al; Trigemino-Cardiac Reflex Examination Group (TCREG). The trigemino-cardiac reflex: an update of the current knowledge. J Neurosurg Anesthesiol. 2009;21(3):187-195.
19. Sandu N, Spiriev T, Lemaitre F, Filis A, Schaller B; Trigemino-Cardiac Reflex Examination Group (TCREG). New molecular knowledge towards the trigemino-cardiac reflex as a cerebral oxygenconserving reflex. Sci World J. 2010;10:811-817.
20. Nirmala J, Dilip KK, Padmaja D, Gopinath R. “Kratschmer” reflex during rhinoplasty. Anesth Analg. 2006;103(5):1337-1338.
21. Jacobs JR, Levine LA, Davis H, Lefrak SS, Druck NS, Ogura JH. Posterior packs and the nasopulmonary reflex. Laryngoscope. 1981;91(2):279-284.
22. Larsen K, Juul A. Arterial blood gases and pneumatic nasal packing in epistaxis. Laryngoscope.1982;92(5):586-588.
23. Loftus BC, Blitzer A, Cozine K. Epistaxis, medical history, and the nasopulmonary reflex: what is clinically relevant? Otolaryngol Head Neck Surg. 1994;110(4):363-369.
24. Arasho B, Sandu N, Spiriev T, Prabhakar H, Schaller B. Management of the trigeminocardiac reflex: facts and own experience. Neurol India. 2009;57(4):375-380.
25. Schaller BJ, Filis A, Buchfelder M. Trigeminocardiac reflex in humans initiated by peripheral
stimulation during neurosurgical skull-base operations. Its first description. Acta Neurochir (Wien). 2008;150(7):715-717; discussion 718.
26. Stemm RA. Complications of nasal packing. Ear Nose Throat J. 1981;60(10):461-462.
27. Widdicombe J. Reflexes from the lungs and airways: historical perspective. J Appl Physiol (1985). 2006;101(2):628-634.
Suspected Clozapine-Induced Cardiomyopathy and Heart Failure With Reduced Ejection Fraction
Clozapine is an atypical antipsychotic that is usually reserved for use in patients with treatment-resistant schizophrenia or schizoaffective disorder with suicidalit
Clozapine-induced cardiomyopathy is a diagnosis of exclusion that requires the absence of other etiologies of cardiac dysfunction (ie, coronary artery disease, hypertension, valvular disease, congenital heart disease, etc). Diagnosing a clozapine-related cardiomyopathy may be a long and laborious task. Patients with cardiomyopathy may present with many nonspecific signs and symptoms, such as fatigue, dyspnea, edema, and/or nausea and vomiting, which are present in other diseases; therefore, multiple encounters and lab tests may be needed until a cardiac source is implicated. The exact mechanism is unknown; however, Chow and colleagues believe that clozapine is a direct toxin of the myocardium.5-7
Case Presentation
A 30-year-old woman with a history of asthma, hypothyroidism (euthyroid with supplementation), posttraumatic stress disorder, and schizoaffective disorder was started on clozapine due to major depression and increased suicidal ideation despite previous treatment with several other antipsychotic agents. Clozapine was gradually titrated to a dose of 150 mg twice a day during an inpatient psychiatric admission. Prior to starting clozapine, this patient had been admitted to the psychiatry unit 11 times within the prior 2 years. After initiating and titrating clozapine over 4 months, her psychiatric symptoms markedly improved.
More than 4 years after the initiation of clozapine and after various treatments for multiple symptoms (Sidebar), the patient was diagnosed with heart failure (HF) with a reduced ejection fraction (EF) of 10% to 15%. She was referred to the cardiology HF clinic. Her dose of clozapine 150 mg at bedtime was discontinued after a discussion with psychiatry. She had a negative workup for other HF etiologies and was started on HF medications that included carvedilol, losartan, and spironolactone. After discontinuation of clozapine, her psychiatric symptoms worsened, and she was admitted to the psychiatry unit twice within a year. Two months after clozapine was discontinued, a repeat echocardiogram (ECHO) was obtained and was essentially unchanged. A chest X-ray (CXR) obtained 4 months after clozapine discontinuation demonstrated a normalized cardio-mediastinal silhouette. A third ECHO was ordered during her second psychiatric admission, which was 11 months after clozapine discontinuation; this revealed an improved left ventricular EF (LVEF) of 30% to 35% and resolution of left ventricular (LV) dilation. 
![]()
This patient’s clinical course led to an extensive chart review that investigated whether there may have been earlier signs and symptoms of HF or cardiomyopathy. It was discovered that the initial HF signs and symptoms were likely present for about 1 year before the diagnosis was made and after having been on clozapine for about 40 months (Patient’s ECHO before and after clozapine discontinuation, click here for additional ECHO perspectives ).
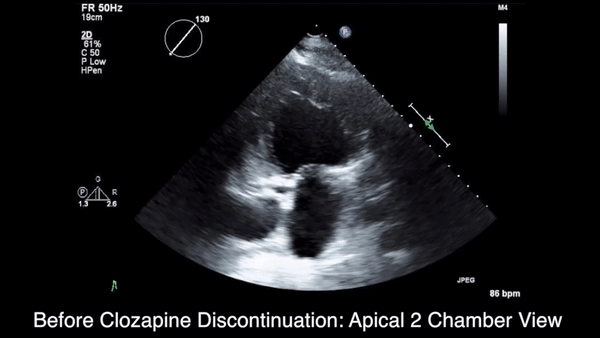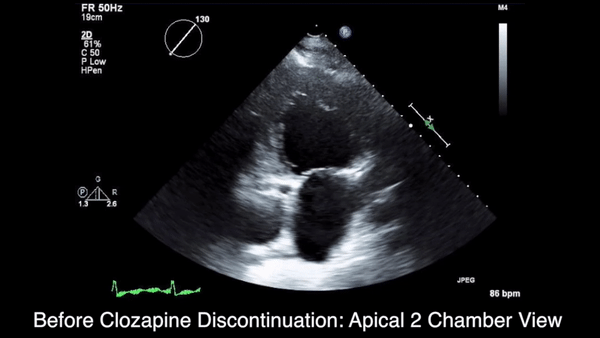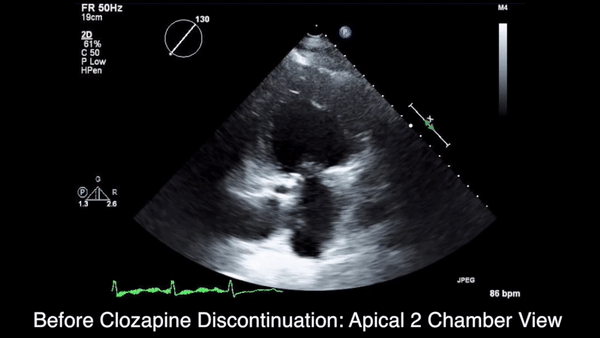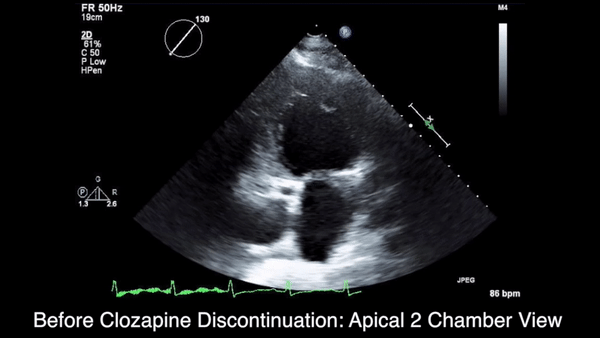
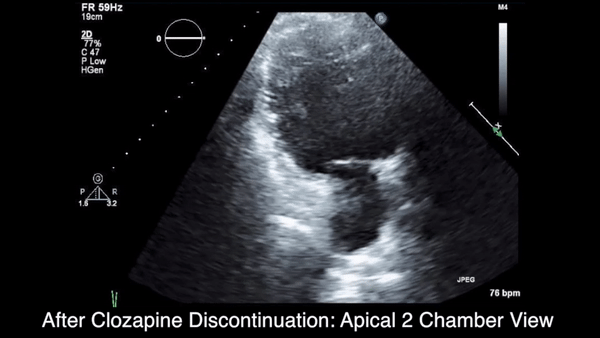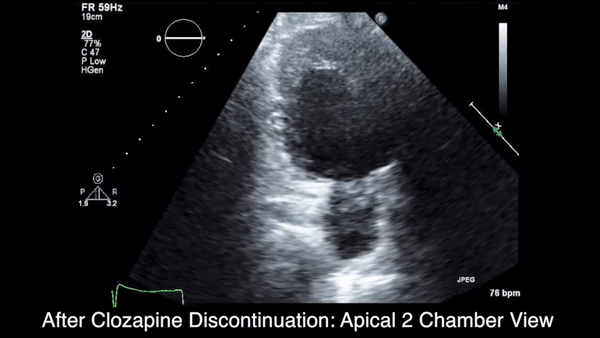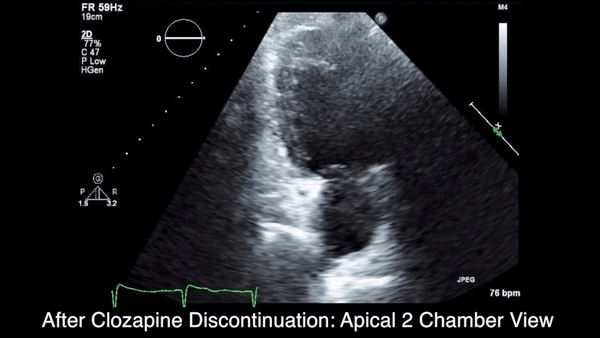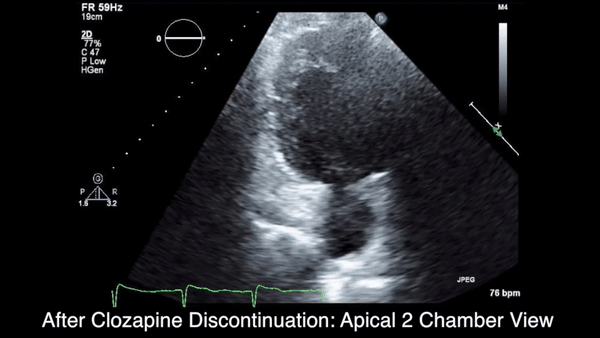
Discussion
In retrospect, this patient likely had HF for many months prior to the official diagnosis; however, given this patient’s young age, prior history of asthma, respiratory disorders, underlying severe psychiatric disease, and confounding symptoms, it is easy to understand why the diagnosis was initially overlooked and delayed.
This patient did not have significant lower extremity edema, but she reported nausea, vomiting, and weight loss. Typical patients with HF exhibit edema and weight gain unless they experience cardiac cachexia. It is not clear whether this patient had a coexisting gastrointestinal (GI) disorder or whether the GI symptoms were secondary to cardiac cachexia. Additionally, weight gain and metabolic syndrome have been documented with clozapine therapy.
It is interesting that a repeat ECHO within 2 months of clozapine discontinuation did not show an improvement, whereas a CXR at 4 months showed a normal cardiac silhouette, and an ECHO at 11 months showed an improvement in EF and normalization of LV size while on appropriate HF medications. It would have been interesting if an ECHO had been completed at 4 months to correspond with the time when the CXR normalized.
There does not seem to be a high level of awareness regarding this potentially fatal diagnosis of cardiomyopathy related to the use of clozapine. A recent review of clozapine-induced cardiomyopathy by Alawami and colleagues revealed characteristics, including median age of diagnosis of 33.5 years, a mean daily dose of 360 mg (range 125-700 mg/d), average time of therapy until the development of symptoms 14.4 months (range between 3 weeks and 4 years), and the presence of ventricular dilation in 39%.8 The most common symptoms on clinical presentation were shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).8
It is interesting that edema was not present in the patients studied in their review; this difference from the usual presentation of severe HF may lower clinical suspicion and makes diagnosing this type of cardiomyopathy more difficult. Alawami also noted that patients with an LVEF < 25% at the time of diagnosis tended to have a poorer prognosis with the highest risk of mortality and limited recovery. Fortunately, in this case, the patient’s LVEF improved significantly, and it will be interesting to continue to monitor the patient for further improvement.
As a result of this case, the authors have performed a chart review on all patients prescribed clozapine at VA Loma Linda Healthcare System. Additionally, this case supports the implementation of better cardiomyopathy monitoring of all clozapine patients. It may be recommended to obtain a baseline CXR in all patients starting clozapine, conduct monthly cardiomyopathy symptom screening that coincides with ANC monitoring, and perform an ECHO immediately on any clinical suspicion of cardiomyopathy.
Conclusion
Better awareness and regular screening for signs and symptoms of HF may help prevent a delay in diagnosing a rare but serious and potentially fatal condition associated with clozapine. Chest X-rays demonstrating cardiomegaly can be helpful when the early diagnosis of HF is suspected and may be the first diagnostic imaging test to normalize after clozapine discontinuation.
Since clozapine is a REMS medication and all patients are scheduled for regular ANC follow-up, it would seem prudent that patients also should be screened for signs and symptoms of HF, including the new onset or worsening of baseline shortness of breath, palpitations, cough, fatigue, chest pain, edema, gastroparesis, and perhaps extreme weight loss. Once a physician suspects HF, an ECHO should be obtained immediately.
In addition to the clozapine boxed warning for cardiomyopathy, it would be helpful if the clozapine patient counseling information section had a specific warning that advises patients to contact their clinician if they develop the signs and symptoms of HF.
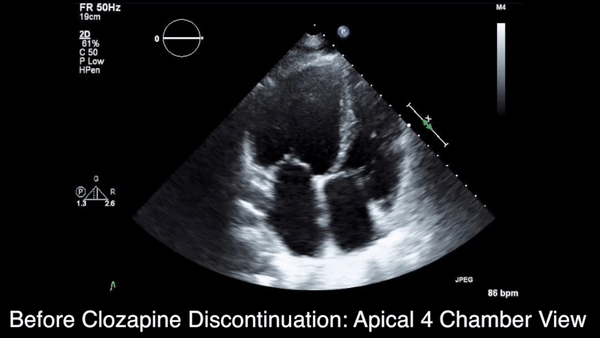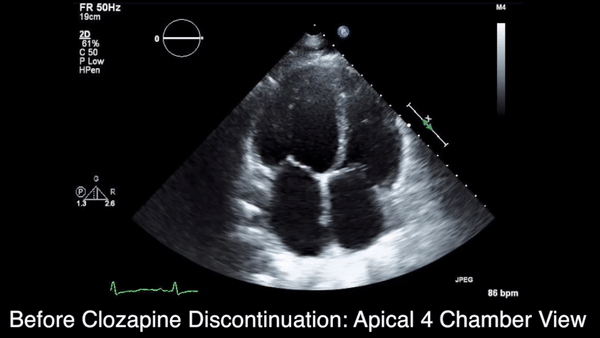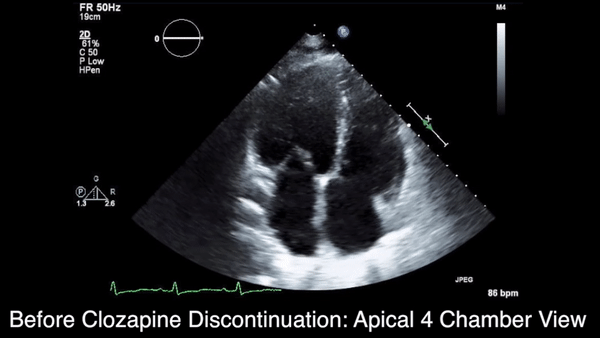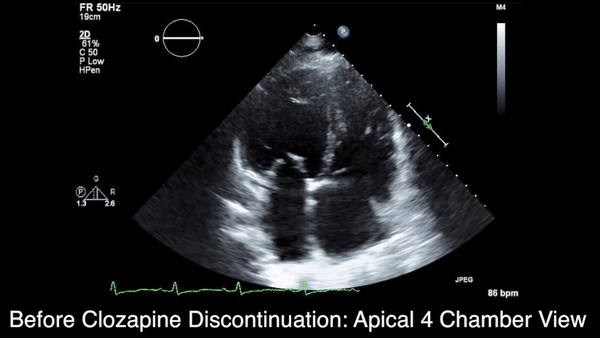
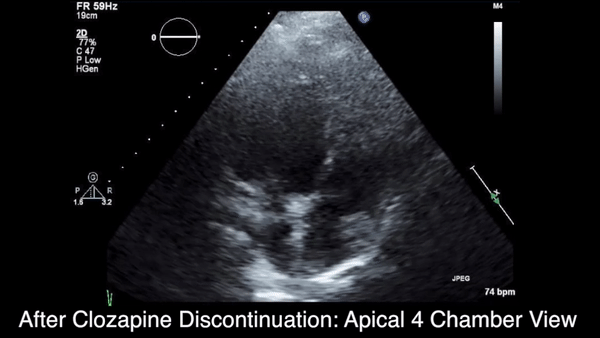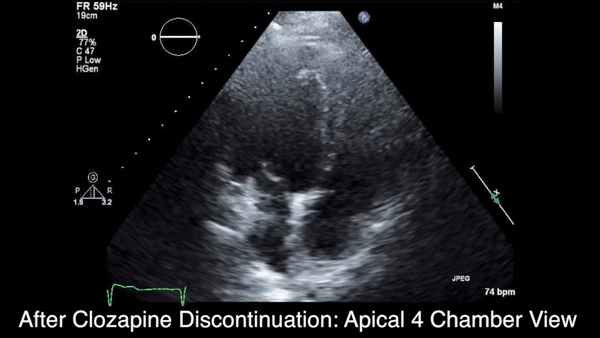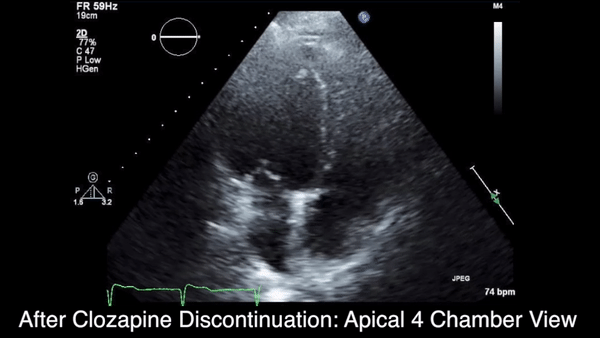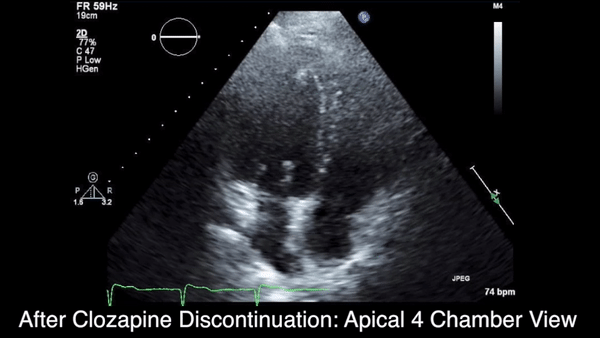
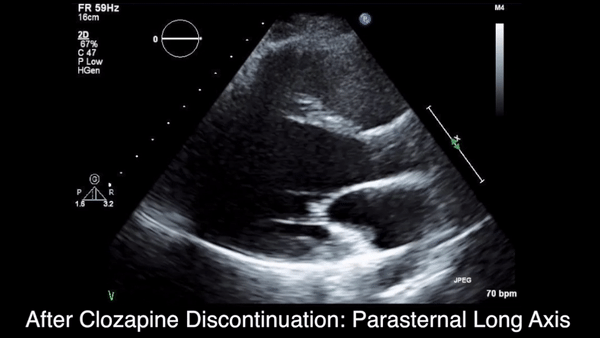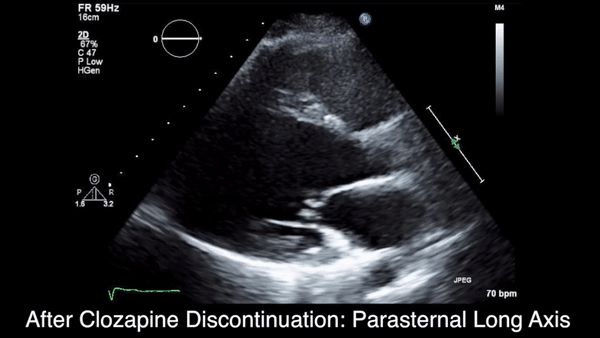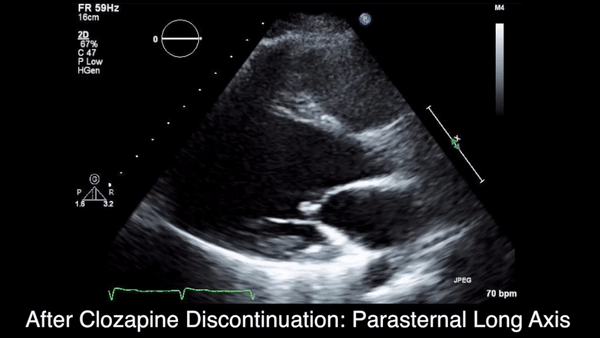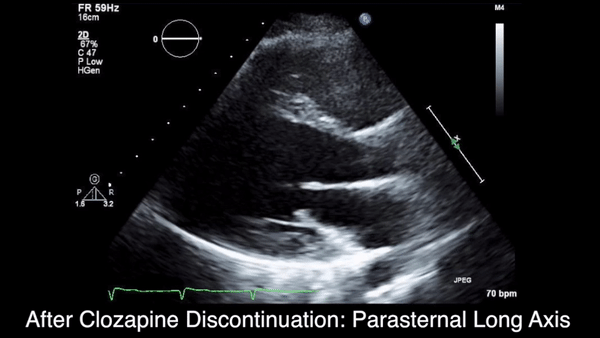
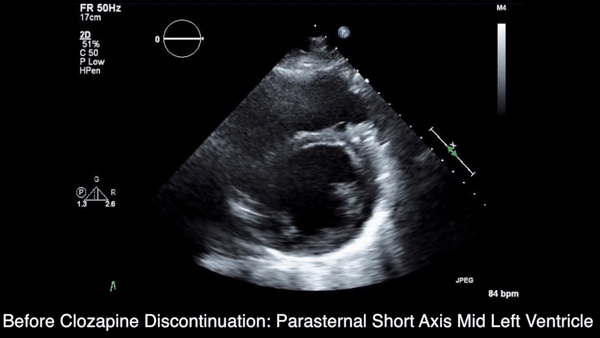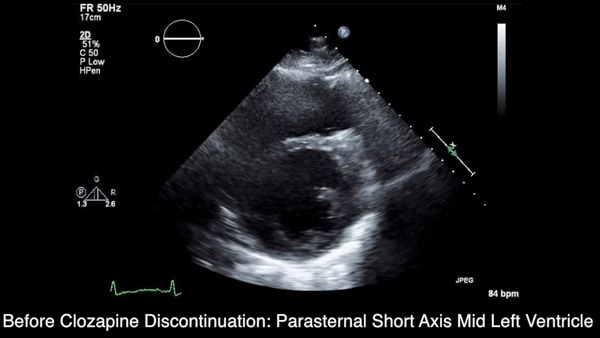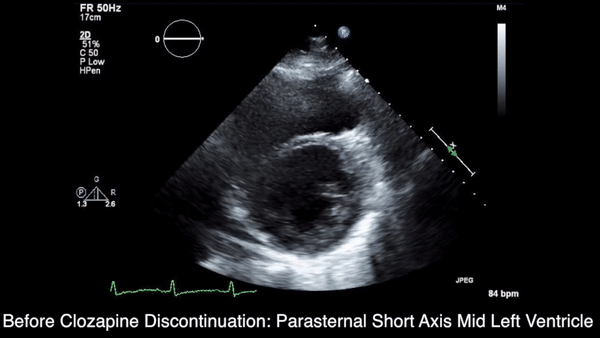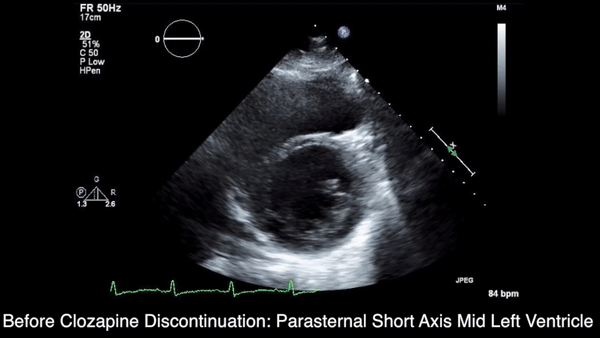
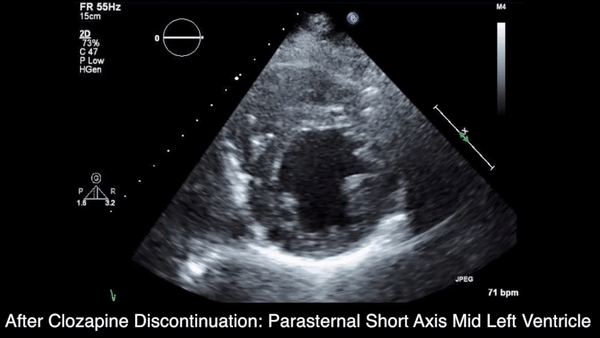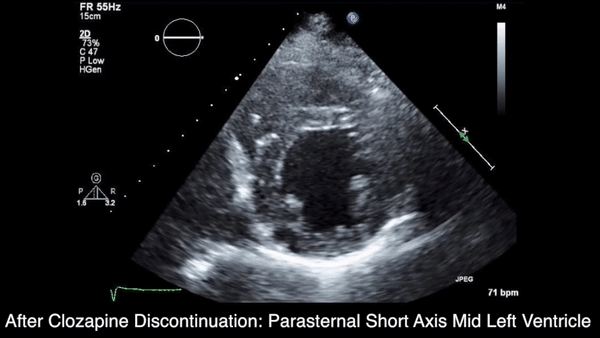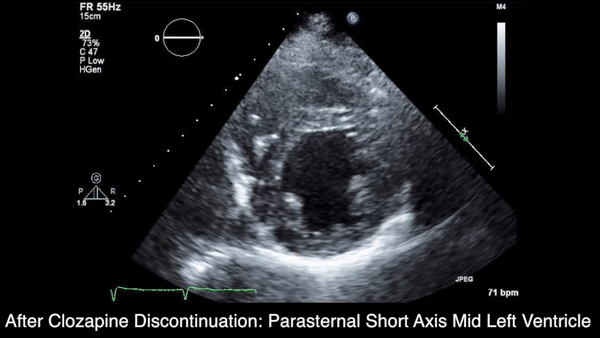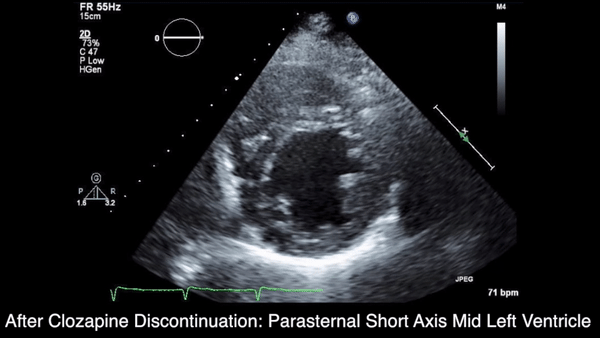
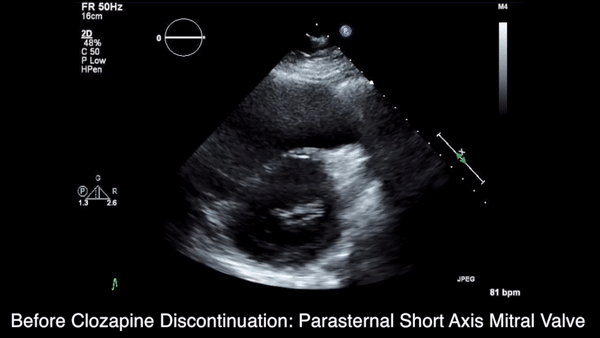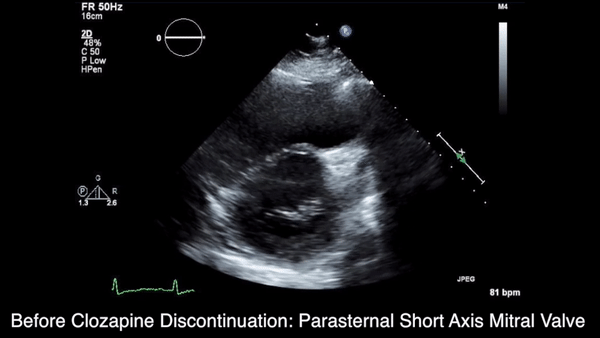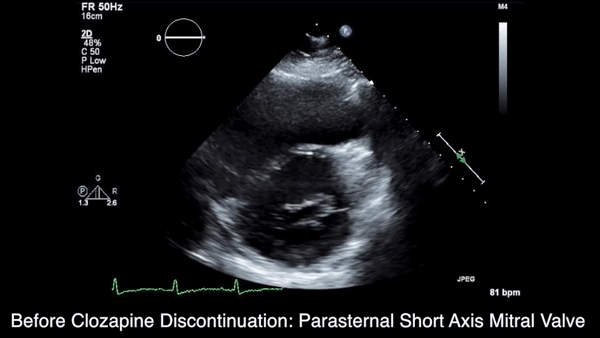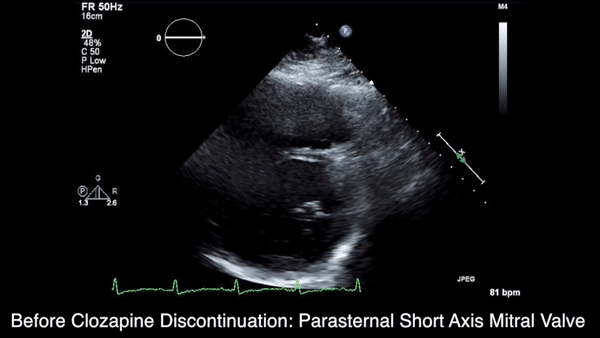
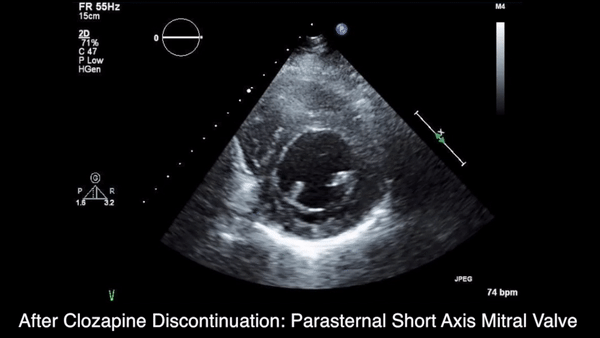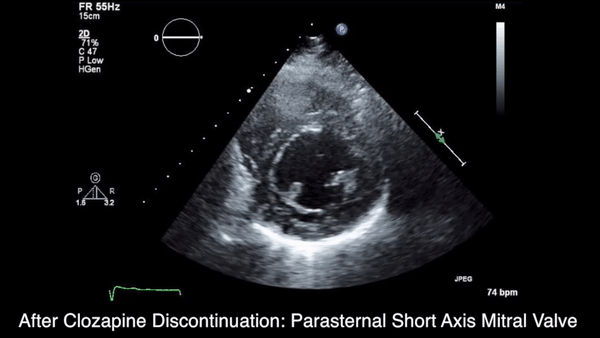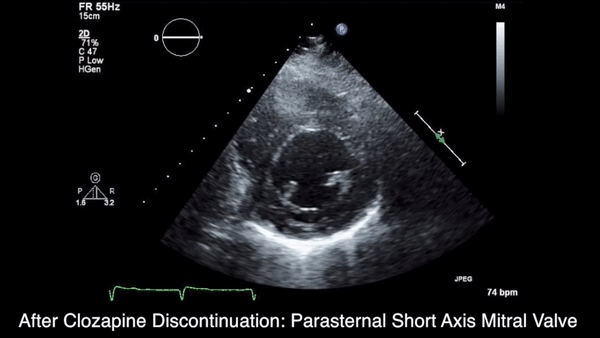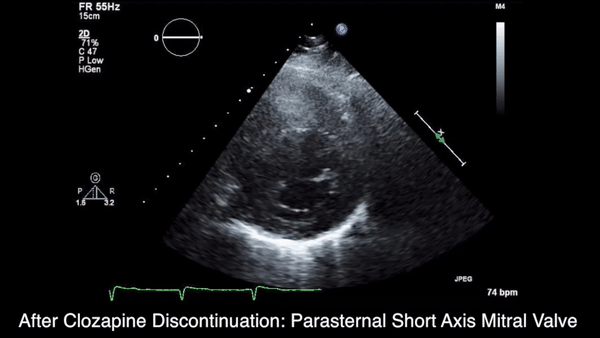
1. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc; 2015.
2. Youssef DL, Narayanan P, Gill N. Incidence and risk factors for clozapine-induced myocarditis and cardiomyopathy at a regional mental health service in Australia. Austalas Psychiatry. 2016;24(2):176-180.
3. La Grenade L, Graham D, Trontell A. Myocarditis and cardiomyopathy associated with clozapine use in the United States. N Eng J Med. 2001;345(3):224-225.
4. Kilian JG, Kerr K, Lawrence C, Celermajer DS. Myocarditis and cardiomyopathy associated with clozapine. Lancet. 1999;354(9193):1841-1845.
5. Chow V, Yeoh T, Ng AC, et al. Asymptomatic left ventricular dysfunction with long-term clozapine treatment for schizophrenia: a multicenter cross-sectional cohort study. Open Heart 2014;1(1):e000030.
6. Scelsa SN, Simpson DM, McQuistion HL, Fineman A, Ault K, Reichler B. Clozapine-induced myotoxicity in patients with chronic psychotic disorders. Neurology. 1996;47(6):1518-1523.
7. Reznik I, Volchek L, Mester R, et al. Myotoxicity and neurotoxicity during clozapine treatment. Clin Neuropharmacol. 2000;23(5):276-280.
8. Alawami A, Wasywich C, Cicovic A, Kenedi C. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
Clozapine is an atypical antipsychotic that is usually reserved for use in patients with treatment-resistant schizophrenia or schizoaffective disorder with suicidalit
Clozapine-induced cardiomyopathy is a diagnosis of exclusion that requires the absence of other etiologies of cardiac dysfunction (ie, coronary artery disease, hypertension, valvular disease, congenital heart disease, etc). Diagnosing a clozapine-related cardiomyopathy may be a long and laborious task. Patients with cardiomyopathy may present with many nonspecific signs and symptoms, such as fatigue, dyspnea, edema, and/or nausea and vomiting, which are present in other diseases; therefore, multiple encounters and lab tests may be needed until a cardiac source is implicated. The exact mechanism is unknown; however, Chow and colleagues believe that clozapine is a direct toxin of the myocardium.5-7
Case Presentation
A 30-year-old woman with a history of asthma, hypothyroidism (euthyroid with supplementation), posttraumatic stress disorder, and schizoaffective disorder was started on clozapine due to major depression and increased suicidal ideation despite previous treatment with several other antipsychotic agents. Clozapine was gradually titrated to a dose of 150 mg twice a day during an inpatient psychiatric admission. Prior to starting clozapine, this patient had been admitted to the psychiatry unit 11 times within the prior 2 years. After initiating and titrating clozapine over 4 months, her psychiatric symptoms markedly improved.
More than 4 years after the initiation of clozapine and after various treatments for multiple symptoms (Sidebar), the patient was diagnosed with heart failure (HF) with a reduced ejection fraction (EF) of 10% to 15%. She was referred to the cardiology HF clinic. Her dose of clozapine 150 mg at bedtime was discontinued after a discussion with psychiatry. She had a negative workup for other HF etiologies and was started on HF medications that included carvedilol, losartan, and spironolactone. After discontinuation of clozapine, her psychiatric symptoms worsened, and she was admitted to the psychiatry unit twice within a year. Two months after clozapine was discontinued, a repeat echocardiogram (ECHO) was obtained and was essentially unchanged. A chest X-ray (CXR) obtained 4 months after clozapine discontinuation demonstrated a normalized cardio-mediastinal silhouette. A third ECHO was ordered during her second psychiatric admission, which was 11 months after clozapine discontinuation; this revealed an improved left ventricular EF (LVEF) of 30% to 35% and resolution of left ventricular (LV) dilation.
![]()
This patient’s clinical course led to an extensive chart review that investigated whether there may have been earlier signs and symptoms of HF or cardiomyopathy. It was discovered that the initial HF signs and symptoms were likely present for about 1 year before the diagnosis was made and after having been on clozapine for about 40 months (Patient’s ECHO before and after clozapine discontinuation, click here for additional ECHO perspectives ).
Discussion
In retrospect, this patient likely had HF for many months prior to the official diagnosis; however, given this patient’s young age, prior history of asthma, respiratory disorders, underlying severe psychiatric disease, and confounding symptoms, it is easy to understand why the diagnosis was initially overlooked and delayed.
This patient did not have significant lower extremity edema, but she reported nausea, vomiting, and weight loss. Typical patients with HF exhibit edema and weight gain unless they experience cardiac cachexia. It is not clear whether this patient had a coexisting gastrointestinal (GI) disorder or whether the GI symptoms were secondary to cardiac cachexia. Additionally, weight gain and metabolic syndrome have been documented with clozapine therapy.
It is interesting that a repeat ECHO within 2 months of clozapine discontinuation did not show an improvement, whereas a CXR at 4 months showed a normal cardiac silhouette, and an ECHO at 11 months showed an improvement in EF and normalization of LV size while on appropriate HF medications. It would have been interesting if an ECHO had been completed at 4 months to correspond with the time when the CXR normalized.
There does not seem to be a high level of awareness regarding this potentially fatal diagnosis of cardiomyopathy related to the use of clozapine. A recent review of clozapine-induced cardiomyopathy by Alawami and colleagues revealed characteristics, including median age of diagnosis of 33.5 years, a mean daily dose of 360 mg (range 125-700 mg/d), average time of therapy until the development of symptoms 14.4 months (range between 3 weeks and 4 years), and the presence of ventricular dilation in 39%.8 The most common symptoms on clinical presentation were shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).8
It is interesting that edema was not present in the patients studied in their review; this difference from the usual presentation of severe HF may lower clinical suspicion and makes diagnosing this type of cardiomyopathy more difficult. Alawami also noted that patients with an LVEF < 25% at the time of diagnosis tended to have a poorer prognosis with the highest risk of mortality and limited recovery. Fortunately, in this case, the patient’s LVEF improved significantly, and it will be interesting to continue to monitor the patient for further improvement.
As a result of this case, the authors have performed a chart review on all patients prescribed clozapine at VA Loma Linda Healthcare System. Additionally, this case supports the implementation of better cardiomyopathy monitoring of all clozapine patients. It may be recommended to obtain a baseline CXR in all patients starting clozapine, conduct monthly cardiomyopathy symptom screening that coincides with ANC monitoring, and perform an ECHO immediately on any clinical suspicion of cardiomyopathy.
Conclusion
Better awareness and regular screening for signs and symptoms of HF may help prevent a delay in diagnosing a rare but serious and potentially fatal condition associated with clozapine. Chest X-rays demonstrating cardiomegaly can be helpful when the early diagnosis of HF is suspected and may be the first diagnostic imaging test to normalize after clozapine discontinuation.
Since clozapine is a REMS medication and all patients are scheduled for regular ANC follow-up, it would seem prudent that patients also should be screened for signs and symptoms of HF, including the new onset or worsening of baseline shortness of breath, palpitations, cough, fatigue, chest pain, edema, gastroparesis, and perhaps extreme weight loss. Once a physician suspects HF, an ECHO should be obtained immediately.
In addition to the clozapine boxed warning for cardiomyopathy, it would be helpful if the clozapine patient counseling information section had a specific warning that advises patients to contact their clinician if they develop the signs and symptoms of HF.
Clozapine is an atypical antipsychotic that is usually reserved for use in patients with treatment-resistant schizophrenia or schizoaffective disorder with suicidalit
Clozapine-induced cardiomyopathy is a diagnosis of exclusion that requires the absence of other etiologies of cardiac dysfunction (ie, coronary artery disease, hypertension, valvular disease, congenital heart disease, etc). Diagnosing a clozapine-related cardiomyopathy may be a long and laborious task. Patients with cardiomyopathy may present with many nonspecific signs and symptoms, such as fatigue, dyspnea, edema, and/or nausea and vomiting, which are present in other diseases; therefore, multiple encounters and lab tests may be needed until a cardiac source is implicated. The exact mechanism is unknown; however, Chow and colleagues believe that clozapine is a direct toxin of the myocardium.5-7
Case Presentation
A 30-year-old woman with a history of asthma, hypothyroidism (euthyroid with supplementation), posttraumatic stress disorder, and schizoaffective disorder was started on clozapine due to major depression and increased suicidal ideation despite previous treatment with several other antipsychotic agents. Clozapine was gradually titrated to a dose of 150 mg twice a day during an inpatient psychiatric admission. Prior to starting clozapine, this patient had been admitted to the psychiatry unit 11 times within the prior 2 years. After initiating and titrating clozapine over 4 months, her psychiatric symptoms markedly improved.
More than 4 years after the initiation of clozapine and after various treatments for multiple symptoms (Sidebar), the patient was diagnosed with heart failure (HF) with a reduced ejection fraction (EF) of 10% to 15%. She was referred to the cardiology HF clinic. Her dose of clozapine 150 mg at bedtime was discontinued after a discussion with psychiatry. She had a negative workup for other HF etiologies and was started on HF medications that included carvedilol, losartan, and spironolactone. After discontinuation of clozapine, her psychiatric symptoms worsened, and she was admitted to the psychiatry unit twice within a year. Two months after clozapine was discontinued, a repeat echocardiogram (ECHO) was obtained and was essentially unchanged. A chest X-ray (CXR) obtained 4 months after clozapine discontinuation demonstrated a normalized cardio-mediastinal silhouette. A third ECHO was ordered during her second psychiatric admission, which was 11 months after clozapine discontinuation; this revealed an improved left ventricular EF (LVEF) of 30% to 35% and resolution of left ventricular (LV) dilation.
![]()
This patient’s clinical course led to an extensive chart review that investigated whether there may have been earlier signs and symptoms of HF or cardiomyopathy. It was discovered that the initial HF signs and symptoms were likely present for about 1 year before the diagnosis was made and after having been on clozapine for about 40 months (Patient’s ECHO before and after clozapine discontinuation, click here for additional ECHO perspectives ).
Discussion
In retrospect, this patient likely had HF for many months prior to the official diagnosis; however, given this patient’s young age, prior history of asthma, respiratory disorders, underlying severe psychiatric disease, and confounding symptoms, it is easy to understand why the diagnosis was initially overlooked and delayed.
This patient did not have significant lower extremity edema, but she reported nausea, vomiting, and weight loss. Typical patients with HF exhibit edema and weight gain unless they experience cardiac cachexia. It is not clear whether this patient had a coexisting gastrointestinal (GI) disorder or whether the GI symptoms were secondary to cardiac cachexia. Additionally, weight gain and metabolic syndrome have been documented with clozapine therapy.
It is interesting that a repeat ECHO within 2 months of clozapine discontinuation did not show an improvement, whereas a CXR at 4 months showed a normal cardiac silhouette, and an ECHO at 11 months showed an improvement in EF and normalization of LV size while on appropriate HF medications. It would have been interesting if an ECHO had been completed at 4 months to correspond with the time when the CXR normalized.
There does not seem to be a high level of awareness regarding this potentially fatal diagnosis of cardiomyopathy related to the use of clozapine. A recent review of clozapine-induced cardiomyopathy by Alawami and colleagues revealed characteristics, including median age of diagnosis of 33.5 years, a mean daily dose of 360 mg (range 125-700 mg/d), average time of therapy until the development of symptoms 14.4 months (range between 3 weeks and 4 years), and the presence of ventricular dilation in 39%.8 The most common symptoms on clinical presentation were shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).8
It is interesting that edema was not present in the patients studied in their review; this difference from the usual presentation of severe HF may lower clinical suspicion and makes diagnosing this type of cardiomyopathy more difficult. Alawami also noted that patients with an LVEF < 25% at the time of diagnosis tended to have a poorer prognosis with the highest risk of mortality and limited recovery. Fortunately, in this case, the patient’s LVEF improved significantly, and it will be interesting to continue to monitor the patient for further improvement.
As a result of this case, the authors have performed a chart review on all patients prescribed clozapine at VA Loma Linda Healthcare System. Additionally, this case supports the implementation of better cardiomyopathy monitoring of all clozapine patients. It may be recommended to obtain a baseline CXR in all patients starting clozapine, conduct monthly cardiomyopathy symptom screening that coincides with ANC monitoring, and perform an ECHO immediately on any clinical suspicion of cardiomyopathy.
Conclusion
Better awareness and regular screening for signs and symptoms of HF may help prevent a delay in diagnosing a rare but serious and potentially fatal condition associated with clozapine. Chest X-rays demonstrating cardiomegaly can be helpful when the early diagnosis of HF is suspected and may be the first diagnostic imaging test to normalize after clozapine discontinuation.
Since clozapine is a REMS medication and all patients are scheduled for regular ANC follow-up, it would seem prudent that patients also should be screened for signs and symptoms of HF, including the new onset or worsening of baseline shortness of breath, palpitations, cough, fatigue, chest pain, edema, gastroparesis, and perhaps extreme weight loss. Once a physician suspects HF, an ECHO should be obtained immediately.
In addition to the clozapine boxed warning for cardiomyopathy, it would be helpful if the clozapine patient counseling information section had a specific warning that advises patients to contact their clinician if they develop the signs and symptoms of HF.
1. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc; 2015.
2. Youssef DL, Narayanan P, Gill N. Incidence and risk factors for clozapine-induced myocarditis and cardiomyopathy at a regional mental health service in Australia. Austalas Psychiatry. 2016;24(2):176-180.
3. La Grenade L, Graham D, Trontell A. Myocarditis and cardiomyopathy associated with clozapine use in the United States. N Eng J Med. 2001;345(3):224-225.
4. Kilian JG, Kerr K, Lawrence C, Celermajer DS. Myocarditis and cardiomyopathy associated with clozapine. Lancet. 1999;354(9193):1841-1845.
5. Chow V, Yeoh T, Ng AC, et al. Asymptomatic left ventricular dysfunction with long-term clozapine treatment for schizophrenia: a multicenter cross-sectional cohort study. Open Heart 2014;1(1):e000030.
6. Scelsa SN, Simpson DM, McQuistion HL, Fineman A, Ault K, Reichler B. Clozapine-induced myotoxicity in patients with chronic psychotic disorders. Neurology. 1996;47(6):1518-1523.
7. Reznik I, Volchek L, Mester R, et al. Myotoxicity and neurotoxicity during clozapine treatment. Clin Neuropharmacol. 2000;23(5):276-280.
8. Alawami A, Wasywich C, Cicovic A, Kenedi C. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
1. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc; 2015.
2. Youssef DL, Narayanan P, Gill N. Incidence and risk factors for clozapine-induced myocarditis and cardiomyopathy at a regional mental health service in Australia. Austalas Psychiatry. 2016;24(2):176-180.
3. La Grenade L, Graham D, Trontell A. Myocarditis and cardiomyopathy associated with clozapine use in the United States. N Eng J Med. 2001;345(3):224-225.
4. Kilian JG, Kerr K, Lawrence C, Celermajer DS. Myocarditis and cardiomyopathy associated with clozapine. Lancet. 1999;354(9193):1841-1845.
5. Chow V, Yeoh T, Ng AC, et al. Asymptomatic left ventricular dysfunction with long-term clozapine treatment for schizophrenia: a multicenter cross-sectional cohort study. Open Heart 2014;1(1):e000030.
6. Scelsa SN, Simpson DM, McQuistion HL, Fineman A, Ault K, Reichler B. Clozapine-induced myotoxicity in patients with chronic psychotic disorders. Neurology. 1996;47(6):1518-1523.
7. Reznik I, Volchek L, Mester R, et al. Myotoxicity and neurotoxicity during clozapine treatment. Clin Neuropharmacol. 2000;23(5):276-280.
8. Alawami A, Wasywich C, Cicovic A, Kenedi C. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
Complex Malignancies: A Diagnostic and Therapeutic Trilemma
Mr. F. is a 67-year-old man with a medical history significant for hypertension, hyperlipidemia, gastroesophageal reflux disease, abdominal aortic aneurysm repair, and lung nodules (8-mm nodules in his right lower lobe and left upper lobe) since 2009 for which he had been followed in the pulmonary clinic. His last radiologic imaging had been performed in November 2010. The patient had failed to show for a serial scan and follow-up pulmonary appointments. He had seen his primary care practitioner annually. He reporteded a 60 pack-year smoking history and drank up to 10 to 15 beers a day, 3 days a week. He also reported exposure to asbestos while in the U.S. Navy as well as having worked in a sheet metal yard. He reported no family history of lung cancer.
In January 2014, Mr. F. presented to his primary care doctor with a nonpainful neck lump noted by the patient for a few months. On physical examination, the patient appeared healthy and without change in weight, loss of appetite, dysphagia, hemoptysis, or any other remarkable symptoms except for the pain in the left-sided neck mass. On oral examination, the patient did not have any oral lesions but was noted to have poor dentition. He had decreased breath sounds on the right side but no adventitious breath sounds. His cardiac status was within normal limits with a regular heart rate, warm skin, and well-perfused extremities. His abdomen was soft and nontender with good bowel sounds.
A neck computed tomography (CT) done on January 17, 2014, showed 2 left neck masses in the tail of the parotid gland or in level 2 adjacent to the parotid gland. The ear, nose, and throat (ENT) clinic noted that he had a 3-cm level 2 mass, which was mobile and nontender. A fine needle aspiration biopsy showed poorly differentiated rare malignant cells, but the small size of the biopsy precluded identification of the cells. For that reason, the patient was scheduled for an operative laryngoscopy.
A combined positron emission tomography and CT (PET/CT) scan on February 5, 2014, showed multiple areas of hypermetabolic activity. A highly metabolic 2.6 x 1.9-cm mass was noted in the left neck inseparable from the inferior border of the left parotid gland; whether this represented a parotid neoplasm or neoplastic lymph node could not be determined. Positive level 5 neck lymph nodes were suggestive of metastatic disease. There were also 2 foci of mild nonspecific metabolic activity in the right parotid gland. A spiculated mass in the right lower lung, measuring 2.3 x 3.5 cm, demonstrated intense fludeoxyglucose F 18 (FDG) uptake with maximum standardized uptake value (SUV) of 12.7, which was consistent with neoplasm. Thickening of the middistal esophageal wall with intense FDG uptake (SUV of 7.9) was suggestive of esophageal carcinoma.
The patient underwent an endoscopic ultrasound (EUS) esophageal biopsy, which demonstrated poorly differentiated adenocarcinoma (Figure 1). The tumor seemed to have originated at this site, as the biopsy fragments also contained foci of Barrett’s metaplasia and in situ carcinoma. He was presented in the hospital tumor board about whether he should undergo a high-risk lung biopsy due to the location of the mass or have his left neck mass rebiopsied, assuming that he had metastatic esophageal cancer in both the lung and neck. The safer procedure of redoing the neck biopsy in the Ear, Nose, and Throat clinic was pursued, and the patient underwent a biopsy and surgical excision of his left neck mass with positive pathology of highgrade sarcoma (Figure 2). 
The tumor consisted of spindle and epithelioid cell proliferation within a lymph node. The neoplastic cells exhibited prominent nuclear atypia, multinucleation, frequent mitoses, and focal necrosis. By immunohistochemistry, the tumor cells were diffusely positive for vimentin, CD68, and CD34 and were negative for pankeratin, CK7, CAM 5.2, CD30, CD15, tyrosine, melan-a, HMB-45, epithelial membrane antigen, keratin 903, desmin protein, CD56, CD99, actin protein, and CD1a. A differential diagnosis included malignant fibrous histiocytoma and histiocytic sarcoma. 
Knowing the patient had 2 tumors, a percutaneous lung biopsy was scheduled. It was assumed that the lung mass would be metastatic from esophagus vs sarcoma, but the biopsy showed small cell cancer (Figure 3). The tumor was composed of small cells with finely granular chromatin and a sparse amount of cytoplasm. By immunohistochemistry, the tumor cells were positive for TTF-1, CK7, CAM 5.2, CD56, and synaptophysin and were negative for CK5, CK6, and p63. The patient was represented at the tumor board with diagnostic recommendations to undergo a magnetic resonance imaging of the brain (negative for metastasis) and a PET/CT scan (stable disease). 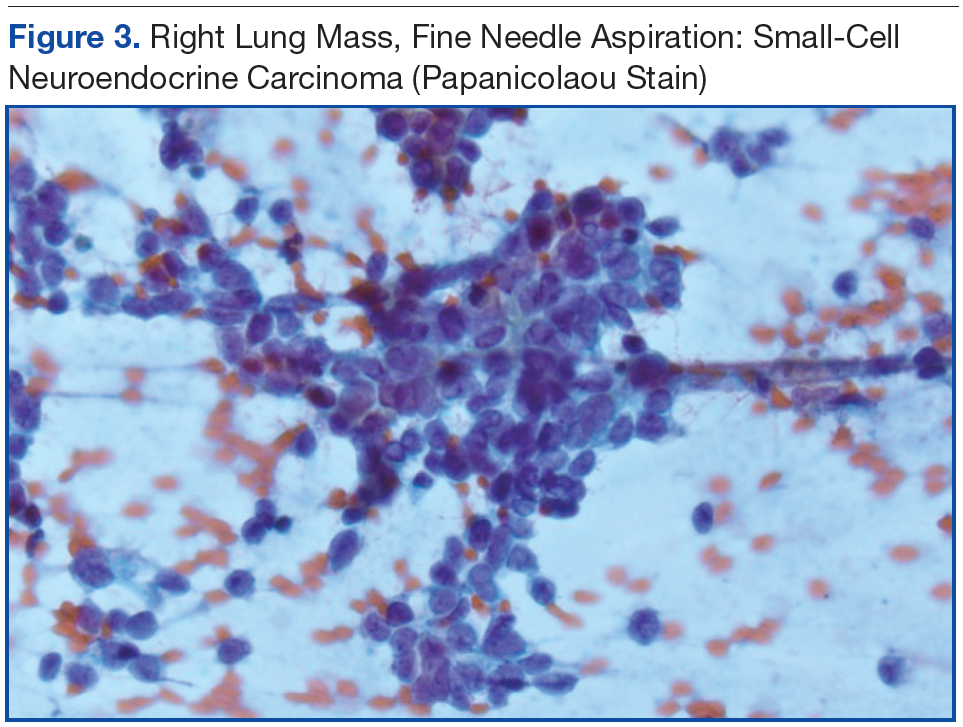
A lengthy discussion ensued regarding treatment for the 3 cancers. The AJCC Cancer Staging Manual, 7th edition, was used as the framework for all staging.1 The patient had a stage IIA (T1bN0MX), grade 3 sarcoma, which could be removed by neck dissection. He had a stage IIIB esophageal carcinoma (T3N2MX), which would require concurrent chemoradiation. His limited stage small cell lung cancer might have been amenable to a lobectomy if a mediastinoscopy was negative. The standard of care for limited stage small cell lung cancer is that if the cancer is precisely staged and the patient is a good surgical candidate, then surgery can be considered. The rationale for considering resection in this case was that if his other cancers could be cured, then surgery would be an excellent option.2 These options were presented to the patient, who opted for chemotherapy. The oncologist treating the patient elected to target the small cell cancer initially, because it was likely the fastest growing cancer. The patient started treatment with etoposide/carboplatin shortly thereafter. Mr. F. was also referred to palliative care.
Discussion
When a patient presents with several tumor masses detected either clinically or radiologically, it is reasonable to assume, at least initially, that all the lesions can be attributed to the same disease (ie, a primary neoplasm and distant metastases). Based on this assumption, it is rational to establish a tissue diagnosis by sampling the most accessible lesion. In the correct clinical context, this may be sufficient to initiate an appropriate therapy. However, health care providers should be aware that unrelated neoplasms can develop simultaneously, and about 8% of patients have > 1 malignancy at presentation.3
This patient presented with 3 different malignant neoplasms: esophageal adenocarcinoma, pulmonary small-cell carcinoma, and high-grade sarcoma. These types of tumors have completely different histogenesis. Esophageal adenocarcinoma develops from glandular cells of Barrett’s esophagus, small-cell carcinoma develops from neuroendocrine Kulchitsky cells, and sarcoma arises from mesenchymal cells. This constellation of neoplasms does not represent any known multicancer syndrome, such as Li-Fraumeni syndrome or Lynch syndrome. Whether these concomitant tumors represent coincidental neoplasms or have common underlying molecular alterations remains unknown.
The treatment for each of this patient’s cancers was very different and required a multidisciplinary, staged approach for management. This could have been arranged for him had he wanted to pursue multiple resections of the neck and lung plus have chemotherapy and radiation for the esophageal cancer. The patient opted for chemotherapy for the most aggressive cancer, but other options would have been possible had he wanted to pursue them.
Conclusion
This complex case demonstrates the role for careful review of all data and the importance of not making assumptions when analyzing images and history despite all the procedures and time to diagnosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer-Verlag; 2010.
2. Koletsis EN, Prokakis C, Karanikolas M, Apostolakis E, Dougenis D. Current role of surgery in small cell lung carcinoma. J Cardiothorac Surg. 2009;4:30.
3. Brock MV, Alberg AJ, Hooker CM, et al. Risk of subsequent primary neoplasms developing in lung cancer patients with prior malignancies. J Thorac Cardiovasc Surg. 2004;127(4):1119-1125.
Mr. F. is a 67-year-old man with a medical history significant for hypertension, hyperlipidemia, gastroesophageal reflux disease, abdominal aortic aneurysm repair, and lung nodules (8-mm nodules in his right lower lobe and left upper lobe) since 2009 for which he had been followed in the pulmonary clinic. His last radiologic imaging had been performed in November 2010. The patient had failed to show for a serial scan and follow-up pulmonary appointments. He had seen his primary care practitioner annually. He reporteded a 60 pack-year smoking history and drank up to 10 to 15 beers a day, 3 days a week. He also reported exposure to asbestos while in the U.S. Navy as well as having worked in a sheet metal yard. He reported no family history of lung cancer.
In January 2014, Mr. F. presented to his primary care doctor with a nonpainful neck lump noted by the patient for a few months. On physical examination, the patient appeared healthy and without change in weight, loss of appetite, dysphagia, hemoptysis, or any other remarkable symptoms except for the pain in the left-sided neck mass. On oral examination, the patient did not have any oral lesions but was noted to have poor dentition. He had decreased breath sounds on the right side but no adventitious breath sounds. His cardiac status was within normal limits with a regular heart rate, warm skin, and well-perfused extremities. His abdomen was soft and nontender with good bowel sounds.
A neck computed tomography (CT) done on January 17, 2014, showed 2 left neck masses in the tail of the parotid gland or in level 2 adjacent to the parotid gland. The ear, nose, and throat (ENT) clinic noted that he had a 3-cm level 2 mass, which was mobile and nontender. A fine needle aspiration biopsy showed poorly differentiated rare malignant cells, but the small size of the biopsy precluded identification of the cells. For that reason, the patient was scheduled for an operative laryngoscopy.
A combined positron emission tomography and CT (PET/CT) scan on February 5, 2014, showed multiple areas of hypermetabolic activity. A highly metabolic 2.6 x 1.9-cm mass was noted in the left neck inseparable from the inferior border of the left parotid gland; whether this represented a parotid neoplasm or neoplastic lymph node could not be determined. Positive level 5 neck lymph nodes were suggestive of metastatic disease. There were also 2 foci of mild nonspecific metabolic activity in the right parotid gland. A spiculated mass in the right lower lung, measuring 2.3 x 3.5 cm, demonstrated intense fludeoxyglucose F 18 (FDG) uptake with maximum standardized uptake value (SUV) of 12.7, which was consistent with neoplasm. Thickening of the middistal esophageal wall with intense FDG uptake (SUV of 7.9) was suggestive of esophageal carcinoma.
The patient underwent an endoscopic ultrasound (EUS) esophageal biopsy, which demonstrated poorly differentiated adenocarcinoma (Figure 1). The tumor seemed to have originated at this site, as the biopsy fragments also contained foci of Barrett’s metaplasia and in situ carcinoma. He was presented in the hospital tumor board about whether he should undergo a high-risk lung biopsy due to the location of the mass or have his left neck mass rebiopsied, assuming that he had metastatic esophageal cancer in both the lung and neck. The safer procedure of redoing the neck biopsy in the Ear, Nose, and Throat clinic was pursued, and the patient underwent a biopsy and surgical excision of his left neck mass with positive pathology of highgrade sarcoma (Figure 2).
The tumor consisted of spindle and epithelioid cell proliferation within a lymph node. The neoplastic cells exhibited prominent nuclear atypia, multinucleation, frequent mitoses, and focal necrosis. By immunohistochemistry, the tumor cells were diffusely positive for vimentin, CD68, and CD34 and were negative for pankeratin, CK7, CAM 5.2, CD30, CD15, tyrosine, melan-a, HMB-45, epithelial membrane antigen, keratin 903, desmin protein, CD56, CD99, actin protein, and CD1a. A differential diagnosis included malignant fibrous histiocytoma and histiocytic sarcoma.
Knowing the patient had 2 tumors, a percutaneous lung biopsy was scheduled. It was assumed that the lung mass would be metastatic from esophagus vs sarcoma, but the biopsy showed small cell cancer (Figure 3). The tumor was composed of small cells with finely granular chromatin and a sparse amount of cytoplasm. By immunohistochemistry, the tumor cells were positive for TTF-1, CK7, CAM 5.2, CD56, and synaptophysin and were negative for CK5, CK6, and p63. The patient was represented at the tumor board with diagnostic recommendations to undergo a magnetic resonance imaging of the brain (negative for metastasis) and a PET/CT scan (stable disease).
A lengthy discussion ensued regarding treatment for the 3 cancers. The AJCC Cancer Staging Manual, 7th edition, was used as the framework for all staging.1 The patient had a stage IIA (T1bN0MX), grade 3 sarcoma, which could be removed by neck dissection. He had a stage IIIB esophageal carcinoma (T3N2MX), which would require concurrent chemoradiation. His limited stage small cell lung cancer might have been amenable to a lobectomy if a mediastinoscopy was negative. The standard of care for limited stage small cell lung cancer is that if the cancer is precisely staged and the patient is a good surgical candidate, then surgery can be considered. The rationale for considering resection in this case was that if his other cancers could be cured, then surgery would be an excellent option.2 These options were presented to the patient, who opted for chemotherapy. The oncologist treating the patient elected to target the small cell cancer initially, because it was likely the fastest growing cancer. The patient started treatment with etoposide/carboplatin shortly thereafter. Mr. F. was also referred to palliative care.
Discussion
When a patient presents with several tumor masses detected either clinically or radiologically, it is reasonable to assume, at least initially, that all the lesions can be attributed to the same disease (ie, a primary neoplasm and distant metastases). Based on this assumption, it is rational to establish a tissue diagnosis by sampling the most accessible lesion. In the correct clinical context, this may be sufficient to initiate an appropriate therapy. However, health care providers should be aware that unrelated neoplasms can develop simultaneously, and about 8% of patients have > 1 malignancy at presentation.3
This patient presented with 3 different malignant neoplasms: esophageal adenocarcinoma, pulmonary small-cell carcinoma, and high-grade sarcoma. These types of tumors have completely different histogenesis. Esophageal adenocarcinoma develops from glandular cells of Barrett’s esophagus, small-cell carcinoma develops from neuroendocrine Kulchitsky cells, and sarcoma arises from mesenchymal cells. This constellation of neoplasms does not represent any known multicancer syndrome, such as Li-Fraumeni syndrome or Lynch syndrome. Whether these concomitant tumors represent coincidental neoplasms or have common underlying molecular alterations remains unknown.
The treatment for each of this patient’s cancers was very different and required a multidisciplinary, staged approach for management. This could have been arranged for him had he wanted to pursue multiple resections of the neck and lung plus have chemotherapy and radiation for the esophageal cancer. The patient opted for chemotherapy for the most aggressive cancer, but other options would have been possible had he wanted to pursue them.
Conclusion
This complex case demonstrates the role for careful review of all data and the importance of not making assumptions when analyzing images and history despite all the procedures and time to diagnosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Mr. F. is a 67-year-old man with a medical history significant for hypertension, hyperlipidemia, gastroesophageal reflux disease, abdominal aortic aneurysm repair, and lung nodules (8-mm nodules in his right lower lobe and left upper lobe) since 2009 for which he had been followed in the pulmonary clinic. His last radiologic imaging had been performed in November 2010. The patient had failed to show for a serial scan and follow-up pulmonary appointments. He had seen his primary care practitioner annually. He reporteded a 60 pack-year smoking history and drank up to 10 to 15 beers a day, 3 days a week. He also reported exposure to asbestos while in the U.S. Navy as well as having worked in a sheet metal yard. He reported no family history of lung cancer.
In January 2014, Mr. F. presented to his primary care doctor with a nonpainful neck lump noted by the patient for a few months. On physical examination, the patient appeared healthy and without change in weight, loss of appetite, dysphagia, hemoptysis, or any other remarkable symptoms except for the pain in the left-sided neck mass. On oral examination, the patient did not have any oral lesions but was noted to have poor dentition. He had decreased breath sounds on the right side but no adventitious breath sounds. His cardiac status was within normal limits with a regular heart rate, warm skin, and well-perfused extremities. His abdomen was soft and nontender with good bowel sounds.
A neck computed tomography (CT) done on January 17, 2014, showed 2 left neck masses in the tail of the parotid gland or in level 2 adjacent to the parotid gland. The ear, nose, and throat (ENT) clinic noted that he had a 3-cm level 2 mass, which was mobile and nontender. A fine needle aspiration biopsy showed poorly differentiated rare malignant cells, but the small size of the biopsy precluded identification of the cells. For that reason, the patient was scheduled for an operative laryngoscopy.
A combined positron emission tomography and CT (PET/CT) scan on February 5, 2014, showed multiple areas of hypermetabolic activity. A highly metabolic 2.6 x 1.9-cm mass was noted in the left neck inseparable from the inferior border of the left parotid gland; whether this represented a parotid neoplasm or neoplastic lymph node could not be determined. Positive level 5 neck lymph nodes were suggestive of metastatic disease. There were also 2 foci of mild nonspecific metabolic activity in the right parotid gland. A spiculated mass in the right lower lung, measuring 2.3 x 3.5 cm, demonstrated intense fludeoxyglucose F 18 (FDG) uptake with maximum standardized uptake value (SUV) of 12.7, which was consistent with neoplasm. Thickening of the middistal esophageal wall with intense FDG uptake (SUV of 7.9) was suggestive of esophageal carcinoma.
The patient underwent an endoscopic ultrasound (EUS) esophageal biopsy, which demonstrated poorly differentiated adenocarcinoma (Figure 1). The tumor seemed to have originated at this site, as the biopsy fragments also contained foci of Barrett’s metaplasia and in situ carcinoma. He was presented in the hospital tumor board about whether he should undergo a high-risk lung biopsy due to the location of the mass or have his left neck mass rebiopsied, assuming that he had metastatic esophageal cancer in both the lung and neck. The safer procedure of redoing the neck biopsy in the Ear, Nose, and Throat clinic was pursued, and the patient underwent a biopsy and surgical excision of his left neck mass with positive pathology of highgrade sarcoma (Figure 2).
The tumor consisted of spindle and epithelioid cell proliferation within a lymph node. The neoplastic cells exhibited prominent nuclear atypia, multinucleation, frequent mitoses, and focal necrosis. By immunohistochemistry, the tumor cells were diffusely positive for vimentin, CD68, and CD34 and were negative for pankeratin, CK7, CAM 5.2, CD30, CD15, tyrosine, melan-a, HMB-45, epithelial membrane antigen, keratin 903, desmin protein, CD56, CD99, actin protein, and CD1a. A differential diagnosis included malignant fibrous histiocytoma and histiocytic sarcoma.
Knowing the patient had 2 tumors, a percutaneous lung biopsy was scheduled. It was assumed that the lung mass would be metastatic from esophagus vs sarcoma, but the biopsy showed small cell cancer (Figure 3). The tumor was composed of small cells with finely granular chromatin and a sparse amount of cytoplasm. By immunohistochemistry, the tumor cells were positive for TTF-1, CK7, CAM 5.2, CD56, and synaptophysin and were negative for CK5, CK6, and p63. The patient was represented at the tumor board with diagnostic recommendations to undergo a magnetic resonance imaging of the brain (negative for metastasis) and a PET/CT scan (stable disease).
A lengthy discussion ensued regarding treatment for the 3 cancers. The AJCC Cancer Staging Manual, 7th edition, was used as the framework for all staging.1 The patient had a stage IIA (T1bN0MX), grade 3 sarcoma, which could be removed by neck dissection. He had a stage IIIB esophageal carcinoma (T3N2MX), which would require concurrent chemoradiation. His limited stage small cell lung cancer might have been amenable to a lobectomy if a mediastinoscopy was negative. The standard of care for limited stage small cell lung cancer is that if the cancer is precisely staged and the patient is a good surgical candidate, then surgery can be considered. The rationale for considering resection in this case was that if his other cancers could be cured, then surgery would be an excellent option.2 These options were presented to the patient, who opted for chemotherapy. The oncologist treating the patient elected to target the small cell cancer initially, because it was likely the fastest growing cancer. The patient started treatment with etoposide/carboplatin shortly thereafter. Mr. F. was also referred to palliative care.
Discussion
When a patient presents with several tumor masses detected either clinically or radiologically, it is reasonable to assume, at least initially, that all the lesions can be attributed to the same disease (ie, a primary neoplasm and distant metastases). Based on this assumption, it is rational to establish a tissue diagnosis by sampling the most accessible lesion. In the correct clinical context, this may be sufficient to initiate an appropriate therapy. However, health care providers should be aware that unrelated neoplasms can develop simultaneously, and about 8% of patients have > 1 malignancy at presentation.3
This patient presented with 3 different malignant neoplasms: esophageal adenocarcinoma, pulmonary small-cell carcinoma, and high-grade sarcoma. These types of tumors have completely different histogenesis. Esophageal adenocarcinoma develops from glandular cells of Barrett’s esophagus, small-cell carcinoma develops from neuroendocrine Kulchitsky cells, and sarcoma arises from mesenchymal cells. This constellation of neoplasms does not represent any known multicancer syndrome, such as Li-Fraumeni syndrome or Lynch syndrome. Whether these concomitant tumors represent coincidental neoplasms or have common underlying molecular alterations remains unknown.
The treatment for each of this patient’s cancers was very different and required a multidisciplinary, staged approach for management. This could have been arranged for him had he wanted to pursue multiple resections of the neck and lung plus have chemotherapy and radiation for the esophageal cancer. The patient opted for chemotherapy for the most aggressive cancer, but other options would have been possible had he wanted to pursue them.
Conclusion
This complex case demonstrates the role for careful review of all data and the importance of not making assumptions when analyzing images and history despite all the procedures and time to diagnosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer-Verlag; 2010.
2. Koletsis EN, Prokakis C, Karanikolas M, Apostolakis E, Dougenis D. Current role of surgery in small cell lung carcinoma. J Cardiothorac Surg. 2009;4:30.
3. Brock MV, Alberg AJ, Hooker CM, et al. Risk of subsequent primary neoplasms developing in lung cancer patients with prior malignancies. J Thorac Cardiovasc Surg. 2004;127(4):1119-1125.
1. Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer-Verlag; 2010.
2. Koletsis EN, Prokakis C, Karanikolas M, Apostolakis E, Dougenis D. Current role of surgery in small cell lung carcinoma. J Cardiothorac Surg. 2009;4:30.
3. Brock MV, Alberg AJ, Hooker CM, et al. Risk of subsequent primary neoplasms developing in lung cancer patients with prior malignancies. J Thorac Cardiovasc Surg. 2004;127(4):1119-1125.
Single-, low-dose cyclophosphamide-associated severe hyponatremia with seizures in a patient with breast cancer
Cyclophosphamide, an agent used to treat various malignant and autoimmune disorders, can cause severe hyponatremia with seizures in rare cases. The exact mechanism of cyclophosphamide-induced hyponatremia is poorly understood, but is thought to occur from a drug- associated antidiuretic hormone (ADH) release leading to free water retention.1 This unusual phenomenon of cyclophosphamide-associated syndrome of inappropriate antidiuretic hormone secretion (SIADH) has been described only in case reports, most of which reported the development of severe hyponatremia within a week after administration of cyclophosphamide.2-5 We report a unique case of a patient who developed severe, symptomatic hyponatremia with seizures, with her serum sodium decreasing from 137 mEq to 112 mEq within 30 hours after her first dose of low-dose cyclophosphamide (600 mg/m2).
Case presentation and summary
A 68-year-old white woman with a history of bilateral invasive ductal carcinoma of the breast (status-post bilateral mastectomy) presented to the emergency department (ED) at our facility with new onset seizure. The patient had been diagnosed 8 months earlier with stage I (T1c, N0, M0) poorly differentiated infiltrating ductal carcinoma (triple negative) of the left breast for which she underwent left segmental mastectomy about 1 month after diagnosis. She was subsequently found to have progressive disease with stage IIIC (T2, N3, and M0) infiltrating ductal carcinoma with lobular features (ER/PR+, Her2) of the right breast. She underwent a right modified radical mastectomy 5 months after her stage IIIC breast cancer diagnosis. She received her first cycle of adjuvant chemotherapy with intravenous doxorubicin (60 mg/m2) and cyclophosphamide (600 mg/m2), which included pre-hydration, a day before presenting to our facility.
According to the patient’s family who provided the initial history, the patient reported tightness in her left arm while sitting at the dinner table. She was confused and subsequently had jerking movement of her right upper extremity with left facial twitching which lasted about 40 seconds. There was no loss of consciousness, or bowel or bladder control. She became unresponsive after the episode. Review of systems was negative except for a report of nausea a few hours before the onset of seizures, which resolved with ondansetron. Her past medical history was significant for breast cancer as already mentioned, seasonal allergic rhinitis, and hypertension. Home medications included hydrochlorothiazide 12.5 mg oral daily, aspirin 81 mg oral daily, and fexofenadine and loratadine oral daily as needed for allergies. There were no other significant surgical history other than already stated. The patient lived at home with her family and was independent with her instrumental activities of daily living. She is a former smoker of tobacco and quit smoking 30 years ago.
On arrival at our facility, the patient had normal vital signs. Significant findings on physical examination were an elderly female who seemed somnolent; not able to follow commands with a documented Glasgow Coma Scale of 10 with eyes opening spontaneously, incomprehensible sounds, and flexion withdrawal from pain as her best responses. She had an increased tone in her left upper extremity and had a brisk, deep tendon reflexes without clonus or 3+ (range, 0-5+, with 2+ being normal). The remainder of her physical exam was unremarkable. Laboratory testing revealed a glucose level of 120 mg/dL (normal, 65-110 mg/dL), sodium of 112 mEq/L (normal, 135-145 mmol/L), and chloride of 78 mEq/L (normal, 95-105 mmol/L). Serum osmolality and urine osmolality were 242 mOsm/kg (normal, 282-295 mOsm/kg) and 449 mOsm/kg (normal, 500-800 mOsm/kg) respectively, indicative of suboptimally dilute urine despite relatively low serum osmolality or SIADH. Urine electrolytes were not obtained.
Imaging studies including computed-tomography scans of the head and chest x-ray performed in the ED were unremarkable. After a phenytoin load, an electroencephalogram was obtained which showed diffuse encephalopathy without active seizure foci. A non-contrast magnetic-resonance imaging (MRI) of the brain was performed but it failed to show acute infarct, mass, mass effect, or brain herniation. There was nonspecific white matter abnormality with compromise of the bilateral cerebral hemispheres, calloseptal junction, left posterior pillar, and bilateral anterior pillars of the fornix, possibly representative of chronic white matter microvascular ischemic changes or less likely vasculitis or demyelination. Correction of her hyponatremia with normal saline was started in the ED with a change in serum sodium from 112 mEq/L to 115 mEq/L within 2 hours. She was admitted to the intensive care unit (ICU) where her sodium correction with normal saline and free water restriction was continued with a goal correction rate of 8-12 mEq/L in 24 hours. The patient’s serum sodium as well as level of consciousness improved gradually over the course of her ICU stay. After 64 hours in the hospital, her sodium had corrected to 137 mEq/L (normal, 135-145 mmol/L; Figure). She was then alert and oriented to person, place, and time. All motor findings noted on presentation had resolved. Her saline infusion was discontinued and serum sodium remained within normal range. She was discharged to a rehabilitation facility. Her hydrochlorothiazide was also discontinued.

Discussion
Hyponatremia is a common finding in cancer patients caused usually by paraneoplastic syndrome, chemotherapy, immunotherapy, or other associated treatment.6 SIADH is a frequent cause of hyponatremia in cancer patients and should be suspected in patients with hyponatremia, hypo-osmolality, and a urine osmolality above 100 mOsmol/kg.7
Our patient’s presentation and laboratory findings suggested SIADH as the likely cause of hyponatremia with a low sodium, a serum osmolality 242 mOsm/kg and urine osmolality of 449 mOsm/kg.8-10 She had no known underlying contributory comorbid condition relating to her serum lipids, thyroid, adrenal, kidney, or heart to date. Her use of a thiazide diuretic was the only confounding factor. The most plausible cause of hyponatremia/SIADH in our patient was likely cyclophosphamide based on her history, timeline of symptoms, and the absence of other possible causes. Though the mechanism for many of the previously mentioned etiologies are known, the mechanism of cyclophosphamide-induced SIADH is difficult to elucidate since the imminent complication of hemorrhagic cystitis means patients receiving this drug are often aggressively hydrated to prevent this complication.11,12 The result is that there is marked retention of water leading to potentially fatal hyponatremia in selected cases.11 This phenomenon has been fairly well described in patients receiving doses of 6 g/m2 as given in the STAMP protocol for stem cell mobilization or at doses of 30-50 mg/kg used to treat malignancy.12 Our patient clearly falls in this category given that she received a dose of 600 mg/m2. We found no evidence in her history to suggest post-operative, genetic or other cause for her hyponatremia. Our case mirrors a report by Koo and colleagues who described severe hyponatremia occurring within 24 hours following a single dose of intravenous cyclophosphamide 700 mg followed by saline infusion.13 In the case reported by Jayachandra and colleagues in which suspected cyclophosphamide-induced hyponatremia led to seizures, the patient received 500 mg IV of cyclophosphamide and had serum sodium as low as 106 mEq/L within a 24-hour period,2 similar to our patient.
There is a paucity of data on cyclophosphamide-induced SIADH. The mechanism by which cyclophosphamide causes SIADH is currently unknown. In addition, there are currently no set criteria that help identify at-risk patients who may develop such an event, including the dosage of cyclophosphamide that may trigger the SIADH, because lower doses of the drug have been associated with this complication.14
In a retrospective analysis by Lee and colleagues, cyclophosphamide-induced hyponatremia was found to be associated with male sex on a univariate analysis, but no risk factors were found in a multivariate analysis.15 It is likely that the concomitant use of diuretics, hydration, and high-dose cyclophosphamide contributed to hyponatremia/SIADH in our patient, though it is not clear through what mechanism. Harlow and colleagues proposed a mechanism for this phenomenon in 1979 based on the autopsy of a patient who had received high-dose cyclophosphamide involving degranulation of hypothalamic neurosecretory organelles and loss of Herring’s bodies. They inferred that metabolites of cyclophosphamide indirectly triggered inappropriate secretion of antidiuretic hormone as seen with a use of the structurally related analogue ifosfamide,16 but to our knowledge, this has yet to be replicated. Cyclophosphamide metabolite may have a direct tubular effect on the collecting duct epithelium leading to water retention15 as established by Campbell and colleagues. In one case, an established diabetes insipidus patient developed cyclophosphamide-induced antidiuresis without vasopressin secretion.17 It is imperative that the scientific community conduct research into the risk factors, underlying mechanisms, and methods of prevention to reduce and/or eliminate SIADH associated with use of cyclophosphamide.
1. Gilbar PJ, Richmond J, Wood J, Sullivan A. Syndrome of inappropriate antidiuretic hormone secretion induced by a single dose of oral cyclophosphamide. Ann Pharmacother. 2012.46(9):e23.
2. Jayachandran NV, Chandrasekhara PK, Thomas J, Agrawal S, Narsimulu G. Cyclophosphamide-associated complications: we need to be aware of SIADH and central pontine myelinolysis. Rheumatology (Oxford). 2009;48(1):89-90.
3. Baker M, Markman M, Niu J. Cyclophosphamide-induced severe acute hyponatremic encephalopathy in patients with breast cancer: report of two cases. Case Rep Oncol. 2014;7(2):550-554.
4. Lazarevic V, Hägg E, Wahlin A. Hiccups and severe hyponatremia associated with high-dose cyclophosphamide in conditioning regimen for allogeneic stem cell transplantation. Am J Hematol. 2007;82(1):88.
5. Geng C, Tang P, Zhang Y, Gao W. Hyponatremia induced by low-dose cyclophosphamide in two patients with breast cancer. Breast J. 2014; 20(4):442-443.
6. Kamoi K, Ebe T, Hasegawa A, et al. Hyponatremia in small cell lung cancer. Mechanisms not involving inappropriate ADH secretion. Cancer. 1987;60(5):1089-1093.
7. Matwiejczuk S, Püsküllüoğlu M, Zygulska AL. Oncological emergencies: syndrome of inappropriate antidiuretic hormone secretion (SIADH). Przegl Lek. 2014;71(10):541-543.
8. Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. 2006;119(7 Suppl 1):S36-42.
9. Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int. 1976;10(1):25-37.
10. Decaux G, Musch W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin J Am Soc Nephrol. 2008;3(4):1175-1184.
11. Bressler RB, Huston DP. Water intoxication following moderate-dose intravenous cyclophosphamide. Arch Intern Med. 1985;145(3):548-549.
12. Salido M, Macarron P, Hernández-García C, D’Cruz DP, Khamashta MA, Hughes GR. Water intoxication induced by low-dose cyclophosphamide in two patients with systemic lupus erythematosus. Lupus. 2003;12(8):636-639.
13. Koo TY, Bae SC, Park JS, et al. Water intoxication following low-dose intravenous cyclophosphamide. Electrolyte Blood Press. 2007;5(1):50-54.
14. [No authors listed]. Nausea and vasopressin. Lancet. 1991;337(8750):1133-1134.
15 Lee YC1, Park JS, Lee CH, et al. Hyponatraemia induced by low-dose intravenous pulse cyclophosphamide. Nephrol Dial Transplant. 2010;25(5):1520-1524.
16. Harlow PJ, DeClerck YA, Shore NA, Ortega JA, Carranza A, Heuser E. A fatal case of inappropriate ADH secretion induced by cyclophosphamide therapy. Cancer. 1979;44(3):896-898.
17. Campbell DM, Atkinson A, Gillis D, Sochett EB. Cyclophosphamide and water retention: mechanism revisited. J Pediatr Endocrinol Metab. 2000;13(6):673-675.
Cyclophosphamide, an agent used to treat various malignant and autoimmune disorders, can cause severe hyponatremia with seizures in rare cases. The exact mechanism of cyclophosphamide-induced hyponatremia is poorly understood, but is thought to occur from a drug- associated antidiuretic hormone (ADH) release leading to free water retention.1 This unusual phenomenon of cyclophosphamide-associated syndrome of inappropriate antidiuretic hormone secretion (SIADH) has been described only in case reports, most of which reported the development of severe hyponatremia within a week after administration of cyclophosphamide.2-5 We report a unique case of a patient who developed severe, symptomatic hyponatremia with seizures, with her serum sodium decreasing from 137 mEq to 112 mEq within 30 hours after her first dose of low-dose cyclophosphamide (600 mg/m2).
Case presentation and summary
A 68-year-old white woman with a history of bilateral invasive ductal carcinoma of the breast (status-post bilateral mastectomy) presented to the emergency department (ED) at our facility with new onset seizure. The patient had been diagnosed 8 months earlier with stage I (T1c, N0, M0) poorly differentiated infiltrating ductal carcinoma (triple negative) of the left breast for which she underwent left segmental mastectomy about 1 month after diagnosis. She was subsequently found to have progressive disease with stage IIIC (T2, N3, and M0) infiltrating ductal carcinoma with lobular features (ER/PR+, Her2) of the right breast. She underwent a right modified radical mastectomy 5 months after her stage IIIC breast cancer diagnosis. She received her first cycle of adjuvant chemotherapy with intravenous doxorubicin (60 mg/m2) and cyclophosphamide (600 mg/m2), which included pre-hydration, a day before presenting to our facility.
According to the patient’s family who provided the initial history, the patient reported tightness in her left arm while sitting at the dinner table. She was confused and subsequently had jerking movement of her right upper extremity with left facial twitching which lasted about 40 seconds. There was no loss of consciousness, or bowel or bladder control. She became unresponsive after the episode. Review of systems was negative except for a report of nausea a few hours before the onset of seizures, which resolved with ondansetron. Her past medical history was significant for breast cancer as already mentioned, seasonal allergic rhinitis, and hypertension. Home medications included hydrochlorothiazide 12.5 mg oral daily, aspirin 81 mg oral daily, and fexofenadine and loratadine oral daily as needed for allergies. There were no other significant surgical history other than already stated. The patient lived at home with her family and was independent with her instrumental activities of daily living. She is a former smoker of tobacco and quit smoking 30 years ago.
On arrival at our facility, the patient had normal vital signs. Significant findings on physical examination were an elderly female who seemed somnolent; not able to follow commands with a documented Glasgow Coma Scale of 10 with eyes opening spontaneously, incomprehensible sounds, and flexion withdrawal from pain as her best responses. She had an increased tone in her left upper extremity and had a brisk, deep tendon reflexes without clonus or 3+ (range, 0-5+, with 2+ being normal). The remainder of her physical exam was unremarkable. Laboratory testing revealed a glucose level of 120 mg/dL (normal, 65-110 mg/dL), sodium of 112 mEq/L (normal, 135-145 mmol/L), and chloride of 78 mEq/L (normal, 95-105 mmol/L). Serum osmolality and urine osmolality were 242 mOsm/kg (normal, 282-295 mOsm/kg) and 449 mOsm/kg (normal, 500-800 mOsm/kg) respectively, indicative of suboptimally dilute urine despite relatively low serum osmolality or SIADH. Urine electrolytes were not obtained.
Imaging studies including computed-tomography scans of the head and chest x-ray performed in the ED were unremarkable. After a phenytoin load, an electroencephalogram was obtained which showed diffuse encephalopathy without active seizure foci. A non-contrast magnetic-resonance imaging (MRI) of the brain was performed but it failed to show acute infarct, mass, mass effect, or brain herniation. There was nonspecific white matter abnormality with compromise of the bilateral cerebral hemispheres, calloseptal junction, left posterior pillar, and bilateral anterior pillars of the fornix, possibly representative of chronic white matter microvascular ischemic changes or less likely vasculitis or demyelination. Correction of her hyponatremia with normal saline was started in the ED with a change in serum sodium from 112 mEq/L to 115 mEq/L within 2 hours. She was admitted to the intensive care unit (ICU) where her sodium correction with normal saline and free water restriction was continued with a goal correction rate of 8-12 mEq/L in 24 hours. The patient’s serum sodium as well as level of consciousness improved gradually over the course of her ICU stay. After 64 hours in the hospital, her sodium had corrected to 137 mEq/L (normal, 135-145 mmol/L; Figure). She was then alert and oriented to person, place, and time. All motor findings noted on presentation had resolved. Her saline infusion was discontinued and serum sodium remained within normal range. She was discharged to a rehabilitation facility. Her hydrochlorothiazide was also discontinued.
Discussion
Hyponatremia is a common finding in cancer patients caused usually by paraneoplastic syndrome, chemotherapy, immunotherapy, or other associated treatment.6 SIADH is a frequent cause of hyponatremia in cancer patients and should be suspected in patients with hyponatremia, hypo-osmolality, and a urine osmolality above 100 mOsmol/kg.7
Our patient’s presentation and laboratory findings suggested SIADH as the likely cause of hyponatremia with a low sodium, a serum osmolality 242 mOsm/kg and urine osmolality of 449 mOsm/kg.8-10 She had no known underlying contributory comorbid condition relating to her serum lipids, thyroid, adrenal, kidney, or heart to date. Her use of a thiazide diuretic was the only confounding factor. The most plausible cause of hyponatremia/SIADH in our patient was likely cyclophosphamide based on her history, timeline of symptoms, and the absence of other possible causes. Though the mechanism for many of the previously mentioned etiologies are known, the mechanism of cyclophosphamide-induced SIADH is difficult to elucidate since the imminent complication of hemorrhagic cystitis means patients receiving this drug are often aggressively hydrated to prevent this complication.11,12 The result is that there is marked retention of water leading to potentially fatal hyponatremia in selected cases.11 This phenomenon has been fairly well described in patients receiving doses of 6 g/m2 as given in the STAMP protocol for stem cell mobilization or at doses of 30-50 mg/kg used to treat malignancy.12 Our patient clearly falls in this category given that she received a dose of 600 mg/m2. We found no evidence in her history to suggest post-operative, genetic or other cause for her hyponatremia. Our case mirrors a report by Koo and colleagues who described severe hyponatremia occurring within 24 hours following a single dose of intravenous cyclophosphamide 700 mg followed by saline infusion.13 In the case reported by Jayachandra and colleagues in which suspected cyclophosphamide-induced hyponatremia led to seizures, the patient received 500 mg IV of cyclophosphamide and had serum sodium as low as 106 mEq/L within a 24-hour period,2 similar to our patient.
There is a paucity of data on cyclophosphamide-induced SIADH. The mechanism by which cyclophosphamide causes SIADH is currently unknown. In addition, there are currently no set criteria that help identify at-risk patients who may develop such an event, including the dosage of cyclophosphamide that may trigger the SIADH, because lower doses of the drug have been associated with this complication.14
In a retrospective analysis by Lee and colleagues, cyclophosphamide-induced hyponatremia was found to be associated with male sex on a univariate analysis, but no risk factors were found in a multivariate analysis.15 It is likely that the concomitant use of diuretics, hydration, and high-dose cyclophosphamide contributed to hyponatremia/SIADH in our patient, though it is not clear through what mechanism. Harlow and colleagues proposed a mechanism for this phenomenon in 1979 based on the autopsy of a patient who had received high-dose cyclophosphamide involving degranulation of hypothalamic neurosecretory organelles and loss of Herring’s bodies. They inferred that metabolites of cyclophosphamide indirectly triggered inappropriate secretion of antidiuretic hormone as seen with a use of the structurally related analogue ifosfamide,16 but to our knowledge, this has yet to be replicated. Cyclophosphamide metabolite may have a direct tubular effect on the collecting duct epithelium leading to water retention15 as established by Campbell and colleagues. In one case, an established diabetes insipidus patient developed cyclophosphamide-induced antidiuresis without vasopressin secretion.17 It is imperative that the scientific community conduct research into the risk factors, underlying mechanisms, and methods of prevention to reduce and/or eliminate SIADH associated with use of cyclophosphamide.
Cyclophosphamide, an agent used to treat various malignant and autoimmune disorders, can cause severe hyponatremia with seizures in rare cases. The exact mechanism of cyclophosphamide-induced hyponatremia is poorly understood, but is thought to occur from a drug- associated antidiuretic hormone (ADH) release leading to free water retention.1 This unusual phenomenon of cyclophosphamide-associated syndrome of inappropriate antidiuretic hormone secretion (SIADH) has been described only in case reports, most of which reported the development of severe hyponatremia within a week after administration of cyclophosphamide.2-5 We report a unique case of a patient who developed severe, symptomatic hyponatremia with seizures, with her serum sodium decreasing from 137 mEq to 112 mEq within 30 hours after her first dose of low-dose cyclophosphamide (600 mg/m2).
Case presentation and summary
A 68-year-old white woman with a history of bilateral invasive ductal carcinoma of the breast (status-post bilateral mastectomy) presented to the emergency department (ED) at our facility with new onset seizure. The patient had been diagnosed 8 months earlier with stage I (T1c, N0, M0) poorly differentiated infiltrating ductal carcinoma (triple negative) of the left breast for which she underwent left segmental mastectomy about 1 month after diagnosis. She was subsequently found to have progressive disease with stage IIIC (T2, N3, and M0) infiltrating ductal carcinoma with lobular features (ER/PR+, Her2) of the right breast. She underwent a right modified radical mastectomy 5 months after her stage IIIC breast cancer diagnosis. She received her first cycle of adjuvant chemotherapy with intravenous doxorubicin (60 mg/m2) and cyclophosphamide (600 mg/m2), which included pre-hydration, a day before presenting to our facility.
According to the patient’s family who provided the initial history, the patient reported tightness in her left arm while sitting at the dinner table. She was confused and subsequently had jerking movement of her right upper extremity with left facial twitching which lasted about 40 seconds. There was no loss of consciousness, or bowel or bladder control. She became unresponsive after the episode. Review of systems was negative except for a report of nausea a few hours before the onset of seizures, which resolved with ondansetron. Her past medical history was significant for breast cancer as already mentioned, seasonal allergic rhinitis, and hypertension. Home medications included hydrochlorothiazide 12.5 mg oral daily, aspirin 81 mg oral daily, and fexofenadine and loratadine oral daily as needed for allergies. There were no other significant surgical history other than already stated. The patient lived at home with her family and was independent with her instrumental activities of daily living. She is a former smoker of tobacco and quit smoking 30 years ago.
On arrival at our facility, the patient had normal vital signs. Significant findings on physical examination were an elderly female who seemed somnolent; not able to follow commands with a documented Glasgow Coma Scale of 10 with eyes opening spontaneously, incomprehensible sounds, and flexion withdrawal from pain as her best responses. She had an increased tone in her left upper extremity and had a brisk, deep tendon reflexes without clonus or 3+ (range, 0-5+, with 2+ being normal). The remainder of her physical exam was unremarkable. Laboratory testing revealed a glucose level of 120 mg/dL (normal, 65-110 mg/dL), sodium of 112 mEq/L (normal, 135-145 mmol/L), and chloride of 78 mEq/L (normal, 95-105 mmol/L). Serum osmolality and urine osmolality were 242 mOsm/kg (normal, 282-295 mOsm/kg) and 449 mOsm/kg (normal, 500-800 mOsm/kg) respectively, indicative of suboptimally dilute urine despite relatively low serum osmolality or SIADH. Urine electrolytes were not obtained.
Imaging studies including computed-tomography scans of the head and chest x-ray performed in the ED were unremarkable. After a phenytoin load, an electroencephalogram was obtained which showed diffuse encephalopathy without active seizure foci. A non-contrast magnetic-resonance imaging (MRI) of the brain was performed but it failed to show acute infarct, mass, mass effect, or brain herniation. There was nonspecific white matter abnormality with compromise of the bilateral cerebral hemispheres, calloseptal junction, left posterior pillar, and bilateral anterior pillars of the fornix, possibly representative of chronic white matter microvascular ischemic changes or less likely vasculitis or demyelination. Correction of her hyponatremia with normal saline was started in the ED with a change in serum sodium from 112 mEq/L to 115 mEq/L within 2 hours. She was admitted to the intensive care unit (ICU) where her sodium correction with normal saline and free water restriction was continued with a goal correction rate of 8-12 mEq/L in 24 hours. The patient’s serum sodium as well as level of consciousness improved gradually over the course of her ICU stay. After 64 hours in the hospital, her sodium had corrected to 137 mEq/L (normal, 135-145 mmol/L; Figure). She was then alert and oriented to person, place, and time. All motor findings noted on presentation had resolved. Her saline infusion was discontinued and serum sodium remained within normal range. She was discharged to a rehabilitation facility. Her hydrochlorothiazide was also discontinued.
Discussion
Hyponatremia is a common finding in cancer patients caused usually by paraneoplastic syndrome, chemotherapy, immunotherapy, or other associated treatment.6 SIADH is a frequent cause of hyponatremia in cancer patients and should be suspected in patients with hyponatremia, hypo-osmolality, and a urine osmolality above 100 mOsmol/kg.7
Our patient’s presentation and laboratory findings suggested SIADH as the likely cause of hyponatremia with a low sodium, a serum osmolality 242 mOsm/kg and urine osmolality of 449 mOsm/kg.8-10 She had no known underlying contributory comorbid condition relating to her serum lipids, thyroid, adrenal, kidney, or heart to date. Her use of a thiazide diuretic was the only confounding factor. The most plausible cause of hyponatremia/SIADH in our patient was likely cyclophosphamide based on her history, timeline of symptoms, and the absence of other possible causes. Though the mechanism for many of the previously mentioned etiologies are known, the mechanism of cyclophosphamide-induced SIADH is difficult to elucidate since the imminent complication of hemorrhagic cystitis means patients receiving this drug are often aggressively hydrated to prevent this complication.11,12 The result is that there is marked retention of water leading to potentially fatal hyponatremia in selected cases.11 This phenomenon has been fairly well described in patients receiving doses of 6 g/m2 as given in the STAMP protocol for stem cell mobilization or at doses of 30-50 mg/kg used to treat malignancy.12 Our patient clearly falls in this category given that she received a dose of 600 mg/m2. We found no evidence in her history to suggest post-operative, genetic or other cause for her hyponatremia. Our case mirrors a report by Koo and colleagues who described severe hyponatremia occurring within 24 hours following a single dose of intravenous cyclophosphamide 700 mg followed by saline infusion.13 In the case reported by Jayachandra and colleagues in which suspected cyclophosphamide-induced hyponatremia led to seizures, the patient received 500 mg IV of cyclophosphamide and had serum sodium as low as 106 mEq/L within a 24-hour period,2 similar to our patient.
There is a paucity of data on cyclophosphamide-induced SIADH. The mechanism by which cyclophosphamide causes SIADH is currently unknown. In addition, there are currently no set criteria that help identify at-risk patients who may develop such an event, including the dosage of cyclophosphamide that may trigger the SIADH, because lower doses of the drug have been associated with this complication.14
In a retrospective analysis by Lee and colleagues, cyclophosphamide-induced hyponatremia was found to be associated with male sex on a univariate analysis, but no risk factors were found in a multivariate analysis.15 It is likely that the concomitant use of diuretics, hydration, and high-dose cyclophosphamide contributed to hyponatremia/SIADH in our patient, though it is not clear through what mechanism. Harlow and colleagues proposed a mechanism for this phenomenon in 1979 based on the autopsy of a patient who had received high-dose cyclophosphamide involving degranulation of hypothalamic neurosecretory organelles and loss of Herring’s bodies. They inferred that metabolites of cyclophosphamide indirectly triggered inappropriate secretion of antidiuretic hormone as seen with a use of the structurally related analogue ifosfamide,16 but to our knowledge, this has yet to be replicated. Cyclophosphamide metabolite may have a direct tubular effect on the collecting duct epithelium leading to water retention15 as established by Campbell and colleagues. In one case, an established diabetes insipidus patient developed cyclophosphamide-induced antidiuresis without vasopressin secretion.17 It is imperative that the scientific community conduct research into the risk factors, underlying mechanisms, and methods of prevention to reduce and/or eliminate SIADH associated with use of cyclophosphamide.
1. Gilbar PJ, Richmond J, Wood J, Sullivan A. Syndrome of inappropriate antidiuretic hormone secretion induced by a single dose of oral cyclophosphamide. Ann Pharmacother. 2012.46(9):e23.
2. Jayachandran NV, Chandrasekhara PK, Thomas J, Agrawal S, Narsimulu G. Cyclophosphamide-associated complications: we need to be aware of SIADH and central pontine myelinolysis. Rheumatology (Oxford). 2009;48(1):89-90.
3. Baker M, Markman M, Niu J. Cyclophosphamide-induced severe acute hyponatremic encephalopathy in patients with breast cancer: report of two cases. Case Rep Oncol. 2014;7(2):550-554.
4. Lazarevic V, Hägg E, Wahlin A. Hiccups and severe hyponatremia associated with high-dose cyclophosphamide in conditioning regimen for allogeneic stem cell transplantation. Am J Hematol. 2007;82(1):88.
5. Geng C, Tang P, Zhang Y, Gao W. Hyponatremia induced by low-dose cyclophosphamide in two patients with breast cancer. Breast J. 2014; 20(4):442-443.
6. Kamoi K, Ebe T, Hasegawa A, et al. Hyponatremia in small cell lung cancer. Mechanisms not involving inappropriate ADH secretion. Cancer. 1987;60(5):1089-1093.
7. Matwiejczuk S, Püsküllüoğlu M, Zygulska AL. Oncological emergencies: syndrome of inappropriate antidiuretic hormone secretion (SIADH). Przegl Lek. 2014;71(10):541-543.
8. Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. 2006;119(7 Suppl 1):S36-42.
9. Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int. 1976;10(1):25-37.
10. Decaux G, Musch W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin J Am Soc Nephrol. 2008;3(4):1175-1184.
11. Bressler RB, Huston DP. Water intoxication following moderate-dose intravenous cyclophosphamide. Arch Intern Med. 1985;145(3):548-549.
12. Salido M, Macarron P, Hernández-García C, D’Cruz DP, Khamashta MA, Hughes GR. Water intoxication induced by low-dose cyclophosphamide in two patients with systemic lupus erythematosus. Lupus. 2003;12(8):636-639.
13. Koo TY, Bae SC, Park JS, et al. Water intoxication following low-dose intravenous cyclophosphamide. Electrolyte Blood Press. 2007;5(1):50-54.
14. [No authors listed]. Nausea and vasopressin. Lancet. 1991;337(8750):1133-1134.
15 Lee YC1, Park JS, Lee CH, et al. Hyponatraemia induced by low-dose intravenous pulse cyclophosphamide. Nephrol Dial Transplant. 2010;25(5):1520-1524.
16. Harlow PJ, DeClerck YA, Shore NA, Ortega JA, Carranza A, Heuser E. A fatal case of inappropriate ADH secretion induced by cyclophosphamide therapy. Cancer. 1979;44(3):896-898.
17. Campbell DM, Atkinson A, Gillis D, Sochett EB. Cyclophosphamide and water retention: mechanism revisited. J Pediatr Endocrinol Metab. 2000;13(6):673-675.
1. Gilbar PJ, Richmond J, Wood J, Sullivan A. Syndrome of inappropriate antidiuretic hormone secretion induced by a single dose of oral cyclophosphamide. Ann Pharmacother. 2012.46(9):e23.
2. Jayachandran NV, Chandrasekhara PK, Thomas J, Agrawal S, Narsimulu G. Cyclophosphamide-associated complications: we need to be aware of SIADH and central pontine myelinolysis. Rheumatology (Oxford). 2009;48(1):89-90.
3. Baker M, Markman M, Niu J. Cyclophosphamide-induced severe acute hyponatremic encephalopathy in patients with breast cancer: report of two cases. Case Rep Oncol. 2014;7(2):550-554.
4. Lazarevic V, Hägg E, Wahlin A. Hiccups and severe hyponatremia associated with high-dose cyclophosphamide in conditioning regimen for allogeneic stem cell transplantation. Am J Hematol. 2007;82(1):88.
5. Geng C, Tang P, Zhang Y, Gao W. Hyponatremia induced by low-dose cyclophosphamide in two patients with breast cancer. Breast J. 2014; 20(4):442-443.
6. Kamoi K, Ebe T, Hasegawa A, et al. Hyponatremia in small cell lung cancer. Mechanisms not involving inappropriate ADH secretion. Cancer. 1987;60(5):1089-1093.
7. Matwiejczuk S, Püsküllüoğlu M, Zygulska AL. Oncological emergencies: syndrome of inappropriate antidiuretic hormone secretion (SIADH). Przegl Lek. 2014;71(10):541-543.
8. Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. 2006;119(7 Suppl 1):S36-42.
9. Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int. 1976;10(1):25-37.
10. Decaux G, Musch W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin J Am Soc Nephrol. 2008;3(4):1175-1184.
11. Bressler RB, Huston DP. Water intoxication following moderate-dose intravenous cyclophosphamide. Arch Intern Med. 1985;145(3):548-549.
12. Salido M, Macarron P, Hernández-García C, D’Cruz DP, Khamashta MA, Hughes GR. Water intoxication induced by low-dose cyclophosphamide in two patients with systemic lupus erythematosus. Lupus. 2003;12(8):636-639.
13. Koo TY, Bae SC, Park JS, et al. Water intoxication following low-dose intravenous cyclophosphamide. Electrolyte Blood Press. 2007;5(1):50-54.
14. [No authors listed]. Nausea and vasopressin. Lancet. 1991;337(8750):1133-1134.
15 Lee YC1, Park JS, Lee CH, et al. Hyponatraemia induced by low-dose intravenous pulse cyclophosphamide. Nephrol Dial Transplant. 2010;25(5):1520-1524.
16. Harlow PJ, DeClerck YA, Shore NA, Ortega JA, Carranza A, Heuser E. A fatal case of inappropriate ADH secretion induced by cyclophosphamide therapy. Cancer. 1979;44(3):896-898.
17. Campbell DM, Atkinson A, Gillis D, Sochett EB. Cyclophosphamide and water retention: mechanism revisited. J Pediatr Endocrinol Metab. 2000;13(6):673-675.
Paraneoplastic leukemoid reaction – poor prognostic marker in urothelial bladder carcinoma
Certain cancers have been observed to cause symptoms called paraneoplastic syndromes that are not directly attributed to tumor invasion or compression. This phenomenon is believed to be secondary to a tumor’s secretion of functional hormones, peptides, cytokines, or its immune cross-reactivity. One such variant is a paraneoplastic leukemoid reaction (PLR), defined as a leukocytosis level of >50 x 103 cells/mm³, where the white blood cell (WBC) count differential exhibits a neutrophilia or left-shift, in which a predominance of early neutrophil precursors is observed. A PLR is believed to be incited by the tumor cell’s production of its own growth factors such as granulocyte-colony stimulating factor (G-CSF) and a number of different cytokines. These reactions may at first be mistaken for infectious processes, and it is only after an infection has been ruled out or when a leukocytosis is disproportionately high in the setting of a treated infection, that a paraneoplastic leukemoid reaction (PLR) is considered and an oncologic work-up pursued.
PLR has been previously described in a variety of malignancies including lung, esophageal, nasopharyngeal and laryngeal, gastric, cholangiocarcinoma, melanoma, multiple myeloma, renal, prostate, and hepatocellular carcinoma, but has rarely been described in urothelial carcinoma.1 Leukemoid reactions and autocrine growth induced by paraneoplastic production of G-CSF have rarely been associated with urothelial carcinoma of the bladder,2 as in the case we present here of a 67-year-old white man with invasive high-grade urothelial carcinoma of the bladder. The case highlights PLR as a negative prognostic marker, secondary to urothelial bladder cancer cells’ presumed production of G-CSF, rarely reported as in the literature previously.
Case presentation and summary
A 67-year-old white man was diagnosed with clinical stage III (T3N0M0), invasive high-grade urothelial carcinoma of the bladder (Figure 1). He received neoadjuvant chemotherapy with the standard gemcitabine-cisplatin combination (1,000 mg/m2 of gemcitabine on days 1, 8, and 15 with 70 mg/m2 of cisplatin on day 1 of a 28-day cycle for 3 cycles), and had less than partial response at the end of a 3-month course. A computed tomography scan of his pelvis obtained at treatment completion revealed persistent disease with noted enhancement of the right distal ureter and a right posterior bladder mass at the ureterovesical junction measuring 1.7 x 2.6 x 2.6 cm (0.6 x 1 x 1 in), for which, a cystoprostatectomy was recommended to remove remaining disease. The patient was seen in routine follow-up 3 weeks after his last chemotherapy treatment, when his WBC count was noted to be within normal limits at a value of 8.4 x 103 cells/mm³ (normal range, 4.5-11 x 103 cells/mm³).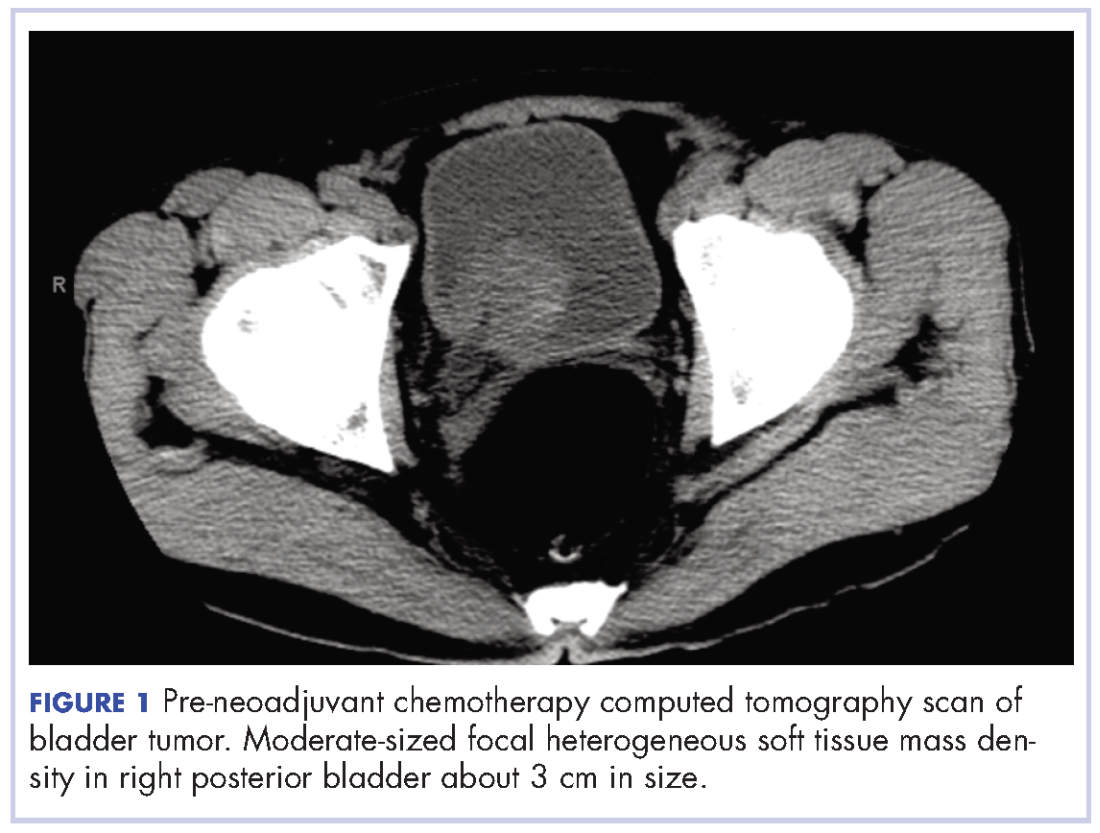
One week later (a month after treatment completion), the patient presented to the emergency department with complaints of dysuria, urinary frequency, and suprapubic pain. He was found to have leukocytosis, with a WBC count of 47 x 103 cells/mm³, (normal range, 4.5-11 x 103 cells/mm³), with an elevated neutrophil count of 82.7% (normal range 40%-60%), without clinical signs of systemic infection (fevers, chills, or rigors). A urinalysis revealed pyuria with 25-50 WBC/high power field, negative nitrite, positive leukocyte esterase, and moderate bacteria, consistent with what was presumed to be a urinary tract infection. The patient was discharged home with a 1-week course of the antibiotic levofloxacin and the alpha-blocker tamsulosin to make urination easier. Of note, the final results of the urine culture, which returned 48 hours after discharge, showed no growth.
One week prior to surgery, the patient underwent a cystoscopy for ureteral stent exchange, which revealed a necrotic tumor at the right bladder base surrounding the ureteral orifice and stent. Renal pelvis urine, sampled during stent exchange, revealed >100,000 CFU/ml (colony forming units; normal value, <10³) Candida albicans, for which the patient was started on intravenous fluconazole for fungal infection. We consulted with our colleagues in the infectious disease department and continued to follow the patient throughout his hospital course, which included several antibiotic regimens, a comprehensive hematological work-up and eventual urologic surgery. Work-ups for myeloproliferative disorders, leukemia, JAK-2, and BCR-ABL were all negative. A peripheral blood smear analysis showed mostly neutrophils, no immature cells, and occasional hypersegmented neutrophils, but was overall inconsistent with myeloproliferative disease. Despite the patient’s persistent leukocytosis, he remained completely asymptomatic and his neutrophilia was attributed to his malignancy.
The patient subsequently underwent a cystoprostatectomy with ileal conduit. The surgical pathologic analysis showed a high-grade, invasive urothelial (transitional cell) carcinoma measuring 5.2 x 5.0 x 4.5 cm (2 x 1.9 x 1.8 in) with squamous differentiation, extensive tumor necrosis, lymphovascular invasion, invasion into the adjacent seminal vesicle and prostatic stroma, and negative margins (pT4a pN0 pMX; Figure 2). On the day of surgical resection, the patient’s leukocytosis was 70 x 103 cells/mm³.
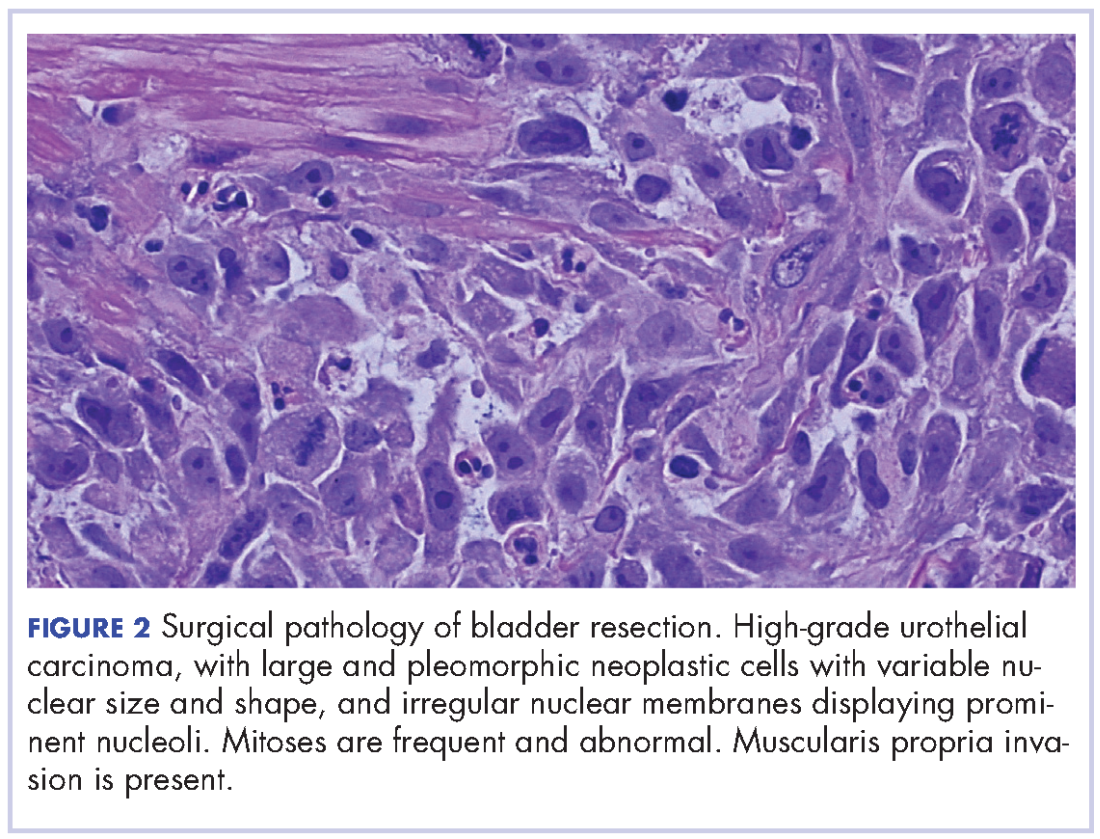
Despite a transient improvement in his leukocytosis to 37.7 x 103 cells/mm³ on postoperative day 1, the patient remained in the medical intensive care unit for uncontrolled pain and management of his leukocytosis. It is worth noting that the patient remained afebrile throughout his entire 45-day hospitalization, with negative culture data, and despite receiving an extensive broad spectrum antibiotic regimen (levofloxacin, piperacillin-tazobactam, cefazolin, metronidazole, ceftriaxone and fluconazole), his leukocytosis continued to progress, peaking at 161.5 x 103 cells/mm³ less than a month after his surgery (Figure 3).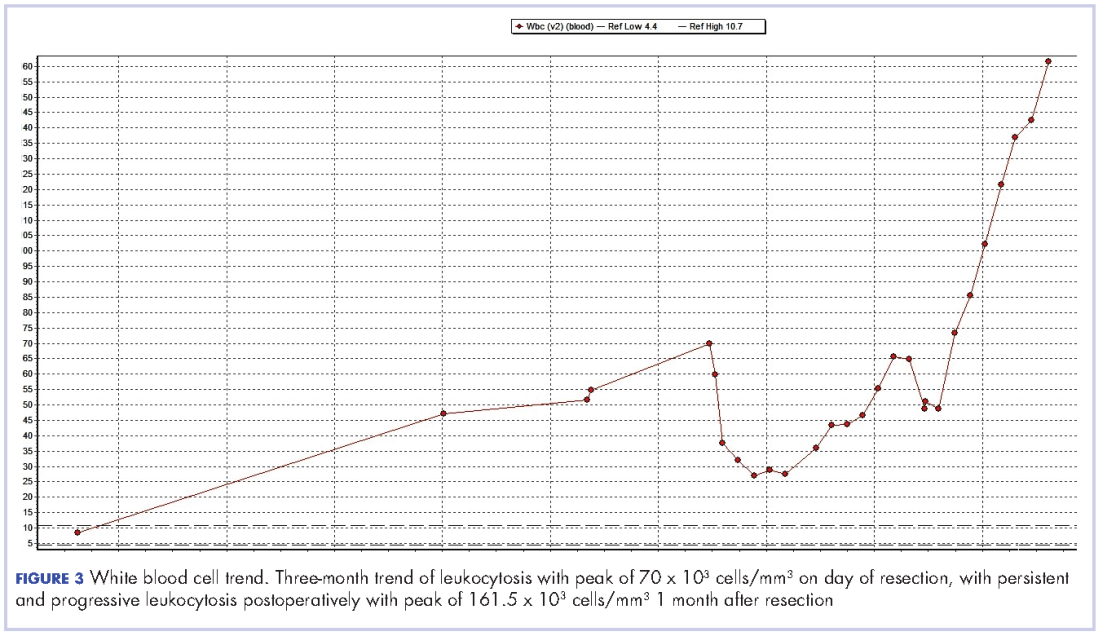
The patient continued to deteriorate rapidly, with a progressive leukocytosis, and developing metastases to the lung, perineum, and penis. He died a month after surgery (2 months after completion of neoadjuvant chemotherapy). The leukocytosis exhibited in this patient and the aggressive tumor cell growth are believed to have been secondary to a paraneoplastic leukemoid reaction incited by the urothelial bladder cancer cells’ presumed production of G-CSF, which has been rarely reported in the literature previously.
Discussion
We report here on the rare occurrence of PLR in a urothelial bladder cancer. Several mechanisms have been proposed to explain the pathophysiology of PLR. The levels of IL-1[alpha and beta], IL-3, G-CSF, GM-CSF, IL-6, and TNF-[alpha] have all been reported to be elevated in various solid tumors and suggested to contribute to an elevated leukocyte count.3 With previous reports that receptors for G-CSF have been found on cell surfaces of several nonhematopoietic cell types, Tachibana and Murai have proposed the mechanism of a cancer cell’s simultaneous acquisition of the ligand promotion and its receptor expression conferring an autocrine growth advantage.4 They have also reported on the capability of bladder cancer cells to induce a leukemoid reaction in their host through the stimulation of leukocyte production, which has been associated with aggressive tumor cell growth and a poor clinical outcome. In addition, He and colleagues have also described the correlation between PLR and high degree of malignancy, high probability of metastasis, recurrence, and poor prognosis.5
We observed the leukocytosis of 70 x 103 cells/mm³ on the day of resection with a slight drop postoperatively, peaking at 161.5 x 103 cells/mm³ less than a month after resection of the tumor. There is no clear understanding of the cause of the persistent and rapid progression of leukocytosis seen in this patient postoperatively. There is also a dearth of literature describing similar occurrences, with even fewer attempting to explain the pathophysiology of this occurrence.
When faced with similar occurrences in patients, clinicians usually treat for occult infection. Once infections and myeloproliferative diseases have been ruled out, clinicians may consider obtaining a patient’s serum G-CSF level or performing an immunohistochemistry analysis of urothelial cells for overexpression of G-CSF, when available.5 However, despite any efforts to diagnose earlier, there is little clinicians currently have to offer these patients as treatment.
As presented in this report, PLR portends a worse prognosis for patients because of its ability to not only masquerade as an infection leading to a delay in the proper treatment, but also because of its association with a more aggressive tumor cell behavior and growth, making it critically important for clinicians to be able to identify these patients early on. With further investigation into immune regulation and G-CSF receptor signaling, there may be future discoveries of novel methods to diagnose this condition and also advancements in the treatment options made available to these patients.
1. Chakraborty S, Keenportz B, Woodward S, Anderson J, Colan D. Paraneoplastic leukemoid reaction in solid tumors. Am J Clin Oncol. 2015;38(3):326-330.
2. Kumar AK, Satyan MT, Holzbeierlein J, Mirza M, Van Veldhuizen P. Leukemoid reaction and autocrine growth of bladder cancer induced by paraneoplastic production of granulocyte colony-stimulating factor-a potential neoplastic marker: a case report and review of the literature. J Med Case Rep. 2014;8(1):147.
3. Azuma T, Sakai I, Matsumoto T, et al. Leukemoid reaction in association with bone marrow necrosis due to metastatic prostate cancer. Intern Med. 2005;44(10):1093-1096.
4. Tachibana M, Murai M. G-CSF production in human bladder cancer and its ability to promote autocrine growth: a review. Cytokines Cell Mol Ther. 1998;4(2):113-120.
5. He H, Zhang Z, Ge J, Zhou W. Leukemoid reaction associated with transitional cell carcinoma: a case report and literature review. Niger J Clin Pract. 2014;17(3):391-394.
Certain cancers have been observed to cause symptoms called paraneoplastic syndromes that are not directly attributed to tumor invasion or compression. This phenomenon is believed to be secondary to a tumor’s secretion of functional hormones, peptides, cytokines, or its immune cross-reactivity. One such variant is a paraneoplastic leukemoid reaction (PLR), defined as a leukocytosis level of >50 x 103 cells/mm³, where the white blood cell (WBC) count differential exhibits a neutrophilia or left-shift, in which a predominance of early neutrophil precursors is observed. A PLR is believed to be incited by the tumor cell’s production of its own growth factors such as granulocyte-colony stimulating factor (G-CSF) and a number of different cytokines. These reactions may at first be mistaken for infectious processes, and it is only after an infection has been ruled out or when a leukocytosis is disproportionately high in the setting of a treated infection, that a paraneoplastic leukemoid reaction (PLR) is considered and an oncologic work-up pursued.
PLR has been previously described in a variety of malignancies including lung, esophageal, nasopharyngeal and laryngeal, gastric, cholangiocarcinoma, melanoma, multiple myeloma, renal, prostate, and hepatocellular carcinoma, but has rarely been described in urothelial carcinoma.1 Leukemoid reactions and autocrine growth induced by paraneoplastic production of G-CSF have rarely been associated with urothelial carcinoma of the bladder,2 as in the case we present here of a 67-year-old white man with invasive high-grade urothelial carcinoma of the bladder. The case highlights PLR as a negative prognostic marker, secondary to urothelial bladder cancer cells’ presumed production of G-CSF, rarely reported as in the literature previously.
Case presentation and summary
A 67-year-old white man was diagnosed with clinical stage III (T3N0M0), invasive high-grade urothelial carcinoma of the bladder (Figure 1). He received neoadjuvant chemotherapy with the standard gemcitabine-cisplatin combination (1,000 mg/m2 of gemcitabine on days 1, 8, and 15 with 70 mg/m2 of cisplatin on day 1 of a 28-day cycle for 3 cycles), and had less than partial response at the end of a 3-month course. A computed tomography scan of his pelvis obtained at treatment completion revealed persistent disease with noted enhancement of the right distal ureter and a right posterior bladder mass at the ureterovesical junction measuring 1.7 x 2.6 x 2.6 cm (0.6 x 1 x 1 in), for which, a cystoprostatectomy was recommended to remove remaining disease. The patient was seen in routine follow-up 3 weeks after his last chemotherapy treatment, when his WBC count was noted to be within normal limits at a value of 8.4 x 103 cells/mm³ (normal range, 4.5-11 x 103 cells/mm³).
One week later (a month after treatment completion), the patient presented to the emergency department with complaints of dysuria, urinary frequency, and suprapubic pain. He was found to have leukocytosis, with a WBC count of 47 x 103 cells/mm³, (normal range, 4.5-11 x 103 cells/mm³), with an elevated neutrophil count of 82.7% (normal range 40%-60%), without clinical signs of systemic infection (fevers, chills, or rigors). A urinalysis revealed pyuria with 25-50 WBC/high power field, negative nitrite, positive leukocyte esterase, and moderate bacteria, consistent with what was presumed to be a urinary tract infection. The patient was discharged home with a 1-week course of the antibiotic levofloxacin and the alpha-blocker tamsulosin to make urination easier. Of note, the final results of the urine culture, which returned 48 hours after discharge, showed no growth.
One week prior to surgery, the patient underwent a cystoscopy for ureteral stent exchange, which revealed a necrotic tumor at the right bladder base surrounding the ureteral orifice and stent. Renal pelvis urine, sampled during stent exchange, revealed >100,000 CFU/ml (colony forming units; normal value, <10³) Candida albicans, for which the patient was started on intravenous fluconazole for fungal infection. We consulted with our colleagues in the infectious disease department and continued to follow the patient throughout his hospital course, which included several antibiotic regimens, a comprehensive hematological work-up and eventual urologic surgery. Work-ups for myeloproliferative disorders, leukemia, JAK-2, and BCR-ABL were all negative. A peripheral blood smear analysis showed mostly neutrophils, no immature cells, and occasional hypersegmented neutrophils, but was overall inconsistent with myeloproliferative disease. Despite the patient’s persistent leukocytosis, he remained completely asymptomatic and his neutrophilia was attributed to his malignancy.
The patient subsequently underwent a cystoprostatectomy with ileal conduit. The surgical pathologic analysis showed a high-grade, invasive urothelial (transitional cell) carcinoma measuring 5.2 x 5.0 x 4.5 cm (2 x 1.9 x 1.8 in) with squamous differentiation, extensive tumor necrosis, lymphovascular invasion, invasion into the adjacent seminal vesicle and prostatic stroma, and negative margins (pT4a pN0 pMX; Figure 2). On the day of surgical resection, the patient’s leukocytosis was 70 x 103 cells/mm³.
Despite a transient improvement in his leukocytosis to 37.7 x 103 cells/mm³ on postoperative day 1, the patient remained in the medical intensive care unit for uncontrolled pain and management of his leukocytosis. It is worth noting that the patient remained afebrile throughout his entire 45-day hospitalization, with negative culture data, and despite receiving an extensive broad spectrum antibiotic regimen (levofloxacin, piperacillin-tazobactam, cefazolin, metronidazole, ceftriaxone and fluconazole), his leukocytosis continued to progress, peaking at 161.5 x 103 cells/mm³ less than a month after his surgery (Figure 3).
The patient continued to deteriorate rapidly, with a progressive leukocytosis, and developing metastases to the lung, perineum, and penis. He died a month after surgery (2 months after completion of neoadjuvant chemotherapy). The leukocytosis exhibited in this patient and the aggressive tumor cell growth are believed to have been secondary to a paraneoplastic leukemoid reaction incited by the urothelial bladder cancer cells’ presumed production of G-CSF, which has been rarely reported in the literature previously.
Discussion
We report here on the rare occurrence of PLR in a urothelial bladder cancer. Several mechanisms have been proposed to explain the pathophysiology of PLR. The levels of IL-1[alpha and beta], IL-3, G-CSF, GM-CSF, IL-6, and TNF-[alpha] have all been reported to be elevated in various solid tumors and suggested to contribute to an elevated leukocyte count.3 With previous reports that receptors for G-CSF have been found on cell surfaces of several nonhematopoietic cell types, Tachibana and Murai have proposed the mechanism of a cancer cell’s simultaneous acquisition of the ligand promotion and its receptor expression conferring an autocrine growth advantage.4 They have also reported on the capability of bladder cancer cells to induce a leukemoid reaction in their host through the stimulation of leukocyte production, which has been associated with aggressive tumor cell growth and a poor clinical outcome. In addition, He and colleagues have also described the correlation between PLR and high degree of malignancy, high probability of metastasis, recurrence, and poor prognosis.5
We observed the leukocytosis of 70 x 103 cells/mm³ on the day of resection with a slight drop postoperatively, peaking at 161.5 x 103 cells/mm³ less than a month after resection of the tumor. There is no clear understanding of the cause of the persistent and rapid progression of leukocytosis seen in this patient postoperatively. There is also a dearth of literature describing similar occurrences, with even fewer attempting to explain the pathophysiology of this occurrence.
When faced with similar occurrences in patients, clinicians usually treat for occult infection. Once infections and myeloproliferative diseases have been ruled out, clinicians may consider obtaining a patient’s serum G-CSF level or performing an immunohistochemistry analysis of urothelial cells for overexpression of G-CSF, when available.5 However, despite any efforts to diagnose earlier, there is little clinicians currently have to offer these patients as treatment.
As presented in this report, PLR portends a worse prognosis for patients because of its ability to not only masquerade as an infection leading to a delay in the proper treatment, but also because of its association with a more aggressive tumor cell behavior and growth, making it critically important for clinicians to be able to identify these patients early on. With further investigation into immune regulation and G-CSF receptor signaling, there may be future discoveries of novel methods to diagnose this condition and also advancements in the treatment options made available to these patients.
Certain cancers have been observed to cause symptoms called paraneoplastic syndromes that are not directly attributed to tumor invasion or compression. This phenomenon is believed to be secondary to a tumor’s secretion of functional hormones, peptides, cytokines, or its immune cross-reactivity. One such variant is a paraneoplastic leukemoid reaction (PLR), defined as a leukocytosis level of >50 x 103 cells/mm³, where the white blood cell (WBC) count differential exhibits a neutrophilia or left-shift, in which a predominance of early neutrophil precursors is observed. A PLR is believed to be incited by the tumor cell’s production of its own growth factors such as granulocyte-colony stimulating factor (G-CSF) and a number of different cytokines. These reactions may at first be mistaken for infectious processes, and it is only after an infection has been ruled out or when a leukocytosis is disproportionately high in the setting of a treated infection, that a paraneoplastic leukemoid reaction (PLR) is considered and an oncologic work-up pursued.
PLR has been previously described in a variety of malignancies including lung, esophageal, nasopharyngeal and laryngeal, gastric, cholangiocarcinoma, melanoma, multiple myeloma, renal, prostate, and hepatocellular carcinoma, but has rarely been described in urothelial carcinoma.1 Leukemoid reactions and autocrine growth induced by paraneoplastic production of G-CSF have rarely been associated with urothelial carcinoma of the bladder,2 as in the case we present here of a 67-year-old white man with invasive high-grade urothelial carcinoma of the bladder. The case highlights PLR as a negative prognostic marker, secondary to urothelial bladder cancer cells’ presumed production of G-CSF, rarely reported as in the literature previously.
Case presentation and summary
A 67-year-old white man was diagnosed with clinical stage III (T3N0M0), invasive high-grade urothelial carcinoma of the bladder (Figure 1). He received neoadjuvant chemotherapy with the standard gemcitabine-cisplatin combination (1,000 mg/m2 of gemcitabine on days 1, 8, and 15 with 70 mg/m2 of cisplatin on day 1 of a 28-day cycle for 3 cycles), and had less than partial response at the end of a 3-month course. A computed tomography scan of his pelvis obtained at treatment completion revealed persistent disease with noted enhancement of the right distal ureter and a right posterior bladder mass at the ureterovesical junction measuring 1.7 x 2.6 x 2.6 cm (0.6 x 1 x 1 in), for which, a cystoprostatectomy was recommended to remove remaining disease. The patient was seen in routine follow-up 3 weeks after his last chemotherapy treatment, when his WBC count was noted to be within normal limits at a value of 8.4 x 103 cells/mm³ (normal range, 4.5-11 x 103 cells/mm³).
One week later (a month after treatment completion), the patient presented to the emergency department with complaints of dysuria, urinary frequency, and suprapubic pain. He was found to have leukocytosis, with a WBC count of 47 x 103 cells/mm³, (normal range, 4.5-11 x 103 cells/mm³), with an elevated neutrophil count of 82.7% (normal range 40%-60%), without clinical signs of systemic infection (fevers, chills, or rigors). A urinalysis revealed pyuria with 25-50 WBC/high power field, negative nitrite, positive leukocyte esterase, and moderate bacteria, consistent with what was presumed to be a urinary tract infection. The patient was discharged home with a 1-week course of the antibiotic levofloxacin and the alpha-blocker tamsulosin to make urination easier. Of note, the final results of the urine culture, which returned 48 hours after discharge, showed no growth.
One week prior to surgery, the patient underwent a cystoscopy for ureteral stent exchange, which revealed a necrotic tumor at the right bladder base surrounding the ureteral orifice and stent. Renal pelvis urine, sampled during stent exchange, revealed >100,000 CFU/ml (colony forming units; normal value, <10³) Candida albicans, for which the patient was started on intravenous fluconazole for fungal infection. We consulted with our colleagues in the infectious disease department and continued to follow the patient throughout his hospital course, which included several antibiotic regimens, a comprehensive hematological work-up and eventual urologic surgery. Work-ups for myeloproliferative disorders, leukemia, JAK-2, and BCR-ABL were all negative. A peripheral blood smear analysis showed mostly neutrophils, no immature cells, and occasional hypersegmented neutrophils, but was overall inconsistent with myeloproliferative disease. Despite the patient’s persistent leukocytosis, he remained completely asymptomatic and his neutrophilia was attributed to his malignancy.
The patient subsequently underwent a cystoprostatectomy with ileal conduit. The surgical pathologic analysis showed a high-grade, invasive urothelial (transitional cell) carcinoma measuring 5.2 x 5.0 x 4.5 cm (2 x 1.9 x 1.8 in) with squamous differentiation, extensive tumor necrosis, lymphovascular invasion, invasion into the adjacent seminal vesicle and prostatic stroma, and negative margins (pT4a pN0 pMX; Figure 2). On the day of surgical resection, the patient’s leukocytosis was 70 x 103 cells/mm³.
Despite a transient improvement in his leukocytosis to 37.7 x 103 cells/mm³ on postoperative day 1, the patient remained in the medical intensive care unit for uncontrolled pain and management of his leukocytosis. It is worth noting that the patient remained afebrile throughout his entire 45-day hospitalization, with negative culture data, and despite receiving an extensive broad spectrum antibiotic regimen (levofloxacin, piperacillin-tazobactam, cefazolin, metronidazole, ceftriaxone and fluconazole), his leukocytosis continued to progress, peaking at 161.5 x 103 cells/mm³ less than a month after his surgery (Figure 3).
The patient continued to deteriorate rapidly, with a progressive leukocytosis, and developing metastases to the lung, perineum, and penis. He died a month after surgery (2 months after completion of neoadjuvant chemotherapy). The leukocytosis exhibited in this patient and the aggressive tumor cell growth are believed to have been secondary to a paraneoplastic leukemoid reaction incited by the urothelial bladder cancer cells’ presumed production of G-CSF, which has been rarely reported in the literature previously.
Discussion
We report here on the rare occurrence of PLR in a urothelial bladder cancer. Several mechanisms have been proposed to explain the pathophysiology of PLR. The levels of IL-1[alpha and beta], IL-3, G-CSF, GM-CSF, IL-6, and TNF-[alpha] have all been reported to be elevated in various solid tumors and suggested to contribute to an elevated leukocyte count.3 With previous reports that receptors for G-CSF have been found on cell surfaces of several nonhematopoietic cell types, Tachibana and Murai have proposed the mechanism of a cancer cell’s simultaneous acquisition of the ligand promotion and its receptor expression conferring an autocrine growth advantage.4 They have also reported on the capability of bladder cancer cells to induce a leukemoid reaction in their host through the stimulation of leukocyte production, which has been associated with aggressive tumor cell growth and a poor clinical outcome. In addition, He and colleagues have also described the correlation between PLR and high degree of malignancy, high probability of metastasis, recurrence, and poor prognosis.5
We observed the leukocytosis of 70 x 103 cells/mm³ on the day of resection with a slight drop postoperatively, peaking at 161.5 x 103 cells/mm³ less than a month after resection of the tumor. There is no clear understanding of the cause of the persistent and rapid progression of leukocytosis seen in this patient postoperatively. There is also a dearth of literature describing similar occurrences, with even fewer attempting to explain the pathophysiology of this occurrence.
When faced with similar occurrences in patients, clinicians usually treat for occult infection. Once infections and myeloproliferative diseases have been ruled out, clinicians may consider obtaining a patient’s serum G-CSF level or performing an immunohistochemistry analysis of urothelial cells for overexpression of G-CSF, when available.5 However, despite any efforts to diagnose earlier, there is little clinicians currently have to offer these patients as treatment.
As presented in this report, PLR portends a worse prognosis for patients because of its ability to not only masquerade as an infection leading to a delay in the proper treatment, but also because of its association with a more aggressive tumor cell behavior and growth, making it critically important for clinicians to be able to identify these patients early on. With further investigation into immune regulation and G-CSF receptor signaling, there may be future discoveries of novel methods to diagnose this condition and also advancements in the treatment options made available to these patients.
1. Chakraborty S, Keenportz B, Woodward S, Anderson J, Colan D. Paraneoplastic leukemoid reaction in solid tumors. Am J Clin Oncol. 2015;38(3):326-330.
2. Kumar AK, Satyan MT, Holzbeierlein J, Mirza M, Van Veldhuizen P. Leukemoid reaction and autocrine growth of bladder cancer induced by paraneoplastic production of granulocyte colony-stimulating factor-a potential neoplastic marker: a case report and review of the literature. J Med Case Rep. 2014;8(1):147.
3. Azuma T, Sakai I, Matsumoto T, et al. Leukemoid reaction in association with bone marrow necrosis due to metastatic prostate cancer. Intern Med. 2005;44(10):1093-1096.
4. Tachibana M, Murai M. G-CSF production in human bladder cancer and its ability to promote autocrine growth: a review. Cytokines Cell Mol Ther. 1998;4(2):113-120.
5. He H, Zhang Z, Ge J, Zhou W. Leukemoid reaction associated with transitional cell carcinoma: a case report and literature review. Niger J Clin Pract. 2014;17(3):391-394.
1. Chakraborty S, Keenportz B, Woodward S, Anderson J, Colan D. Paraneoplastic leukemoid reaction in solid tumors. Am J Clin Oncol. 2015;38(3):326-330.
2. Kumar AK, Satyan MT, Holzbeierlein J, Mirza M, Van Veldhuizen P. Leukemoid reaction and autocrine growth of bladder cancer induced by paraneoplastic production of granulocyte colony-stimulating factor-a potential neoplastic marker: a case report and review of the literature. J Med Case Rep. 2014;8(1):147.
3. Azuma T, Sakai I, Matsumoto T, et al. Leukemoid reaction in association with bone marrow necrosis due to metastatic prostate cancer. Intern Med. 2005;44(10):1093-1096.
4. Tachibana M, Murai M. G-CSF production in human bladder cancer and its ability to promote autocrine growth: a review. Cytokines Cell Mol Ther. 1998;4(2):113-120.
5. He H, Zhang Z, Ge J, Zhou W. Leukemoid reaction associated with transitional cell carcinoma: a case report and literature review. Niger J Clin Pract. 2014;17(3):391-394.
Disseminated Superficial Actinic Porokeratosis Treated With Ingenol Mebutate Gel 0.05%
Disseminated superficial actinic porokeratosis (DSAP) is a chronic condition characterized by numerous atrophic papules and patches with a distinctive peripheral keratotic ridge, typically found on sun-exposed areas.1,2 Treatment of DSAP is warranted not only for cosmetic and symptomatic benefits but also to prevent malignant transformation.3,4 Successful treatment of DSAP often is difficult and frequently requires the use of multiple modalities. Ingenol mebutate gel 0.05% is a topical medication primarily used for the treatment of actinic keratosis (AK) by inducing cell death.5 We report a case of DSAP treated effectively with ingenol mebutate gel 0.05%.
Case Report
A 37-year-old woman was referred to the dermatology department for counseling for pseudoxanthoma elasticum (PXE), which had been proven on biopsy by an outside dermatologist 2 years prior. Physical examination revealed yellow papules on the neck that were characteristic of PXE, but no lesions were noted on the arms or legs. The only other cutaneous finding was a soft nodule on the right hip consistent with a lipoma. The patient returned to our institution 6 years later with lesions on both lower legs. She reported that these lesions had been present for 3 years and were exacerbated by sun exposure. On physical examination, multiple scattered, erythematous, annular, scaling papules and plaques were noted on the bilateral legs. A biopsy showed the histopathologic findings of DSAP (Figure 1). The patient had no family history of DSAP or PXE.
To determine the best treatment modality, we treated 4 test areas on both upper and lower legs: one with trichloroacetic acid (TCA), one with cryotherapy, one with imiquimod cream 5%, and one with tretinoin cream 0.1%. The patient returned 4 weeks later and showed modest response to TCA, cryotherapy, and tretinoin cream. Because cryotherapy was determined to be most effective, 20 more lesions were frozen at that visit. Over the next 2 years, the patient was treated with TCA, imiquimod cream 5%, and tretinoin cream 0.1%, but all ultimately proved ineffective for DSAP.
The patient returned 2 years after treatment failure (age 47 years) and was prescribed ingenol mebutate gel 0.05% for 2 days over an area of 25 cm2 on the right lower leg (Figure 2A). She returned for follow-up at days 3, 15, 30, and 60. At day 3, the patient developed an inflammatory response to the medication with moderate erythema and scaling of individual lesions. No vesiculation, pustulation, edema, or ulceration was exhibited (Figure 2B). At day 30, there was a marked reduction in scaling with some postinflammatory erythema (Figure 2C). At day 60, much of the erythema had faded and the scale remained notably reduced (Figure 2D).
Comment
Disseminated superficial actinic porokeratosis is the most common subtype of porokeratosis, a keratinization disorder. There are 6 subtypes of porokeratosis identified in the literature: DSAP, disseminated superficial porokeratosis, classic porokeratosis of Mibelli, porokeratosis plantaris palmaris et disseminata, linear porokeratosis, and punctate porokeratosis.6 Disseminated superficial actinic porokeratosis has a female predominance (1.8:1 ratio)7 and generally appears in the third or fourth decades of life. Clonal proliferations of atypical keratinocytes have been implicated in the etiology of DSAP; however, the exact pathogenesis is unclear. Risk factors for DSAP include genetic susceptibility (eg, autosomal-dominant inheritance pattern), exposure to UV radiation, and drug-related immunosuppression or immunodeficiency.7 Other proposed etiologic risk factors include trauma and infection.8 Clinical diagnosis of DSAP is confirmed by the histological presence of a cornoid lamella (a thin column ofparakeratotic cells), a thinning epidermis, an absent or thinned granular cell layer, and a prominent dermal lymphocytic infiltrate.9,10
Disseminated superficial actinic porokeratosis clinically presents as small atrophic scaly papules and/or patches with raised peripheral ridges symmetrically dispersed on sun-exposed areas of the arms, legs, back, and shoulders. Although these lesions are extensive, they typically spare the mucous membranes, palms, and soles11; only a small percentage of cases report facial lesions,12 which often are asymptomatic but cosmetically bothersome. Additionally, approximately half of patients report symptoms of pruritus and/or stinging,13 thus treatment of DSAP is mainly indicated for symptomatic relief and cosmetic purposes. Malignant degeneration14,15 occurs in approximately 7.5% to 11% of porokeratosis cases,10,16 warranting treatment for preventative measures.
Management of DSAP is dependent on the extent of the disease and the level of concern for malignant transformation. Localized disease can be treated with cryotherapy, CO2 laser, and/or ablative techniques (eg, excision, curettage, dermabrasion) with variable degrees of success but high risk for scarring.1 More extensive disease requires treatment with topical retinoids, topical 5-fluorouracil, imiquimod cream 5%, diclofenac gel 3%, topical vitamin D3 analogues, and photodynamic therapy.1 Several other therapies have been reported in the literature with partial and/or complete success, including systemic retinoids (eg, acitretin), Q-switched ruby laser, Nd:YAG laser, fractional photothermolysis, Grenz rays, pulsed dye laser, fractional photothermolysis, topical corticosteroids, and fluor-hydroxy pulse peel.6 Although there is an extensive array of therapies for DSAP, treatment results are variable with mostly limited success. Successful treatment of DSAP is difficult and often requires the use of multiple modalities.
Ingenol mebutate is the active compound found in the sap of Euphorbia peplus used for the topical treatment of various skin conditions, including AKs.17 Ingenol mebutate gel 0.05% once daily for 2 days has been approved by the US Food and Drug Administration for the topical treatment of AKs. The mechanism of action of ingenol mebutate in AK therapy is not yet fully understood. In vivo and in vitro models have demonstrated both an induction of local lesion cell death and promotion of lesion-specific inflammatory response.18 When used in the treatment of AKs, ingenol mebutate gel 0.05% may cause a mild to moderate localized inflammatory response (eg, erythema, flaking/scaling, crusting, vesiculation/pustulation, erosion/ulceration, edema).
Our case is a rare report of successful treatment of DSAP with ingenol mebutate gel 0.05%. We found that treatment with ingenol mebutate gel 0.05% resulted in clinical improvement of DSAP lesions with minimal discomfort and good cosmetic response. This 2-day regimen is easy to use and patient friendly, improving medication compliance in such a cumbersome disease. We hope this case suggests that ingenol mebutate gel 0.05% could be a useful treatment alternative for DSAP, but future clinical studies should be conducted.
- Martin-Clavijo A, Kanelleas A, Vlachou C, et al. Porokeratoses. In: Lebwohl M, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease Comprehensive Therapeutic Strategies. 3rd ed. China: Elsevier Limited; 2010:584-586.
- Rouhani P, Fischer M, Meehan S, et al. Disseminated superficial actinic porokeratosis. Dermatology Online J. 2012;18:24.
- Sasson M, Krain AD. Porokeratosis and cutaneous malignancy. a review. Dermatol Surg. 1996;22:339-342.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536-538.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- O’Regan GM, Irvine AD. Porokeratosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:442-446.
- Sertznig P, von Felbert V, Megahed M. Porokeratosis: present concepts. J Eur Acad Dermatol Venereol. 2012;26:404-412.
- Brauer JA, Mandal R, Walters R, et al. Disseminated superficial porokeratosis. Dermatology Online J. 2010;16:20.
- Tallon B. Porokeratosis pathology. DermNet New Zealand website. http://www.dermnet.org.nz/pathology/porokeratosis-path.html. Updated December 2016. Accessed January 12, 2017.
- Skupsky H, Skupsky J, Goldenberg G. Disseminated superficial actinic porokeratosis: a treatment review [published online October 22, 2010]. J Dermatolog Treat. 2012;23:52-56.
- Spencer LV. Porokeratosis. UpToDate web site. https://eresources.library.mssm.edu:3285/contents/porokeratosis?source=search_result&search=porokeratosis&selectedTitle=1~22. Updated September 1, 2016. Accessed April 3, 2017.
- Sawyer R, Picou KA. Facial presentation of disseminated superficial actinic porokeratosis. Ear Nose Throat J. 1989;68:57-59.
- Schwarz T, Seiser A, Gschnait F. Disseminated superficial “actinic” porokeratosis. J Am Acad Dermatol. 1984;11(4, pt 2):724-730.
- Maubec E, Duvillard P, Margulis A, et al. Common skin cancers in porokeratosis. Br J Dermatol. 2005;152:1389-1391.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis [published online November 3, 2011]. Ann Dermatol. 2011;23:536-538.
- Kumari S, Mathur M. Disseminated superficial actinic porokeratosis. Nepal J Dermatol Venereol Leprol. 2010;9:22-24.
- Lebwohl M, Shumack S, Stein Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratosis. JAMA Dermatol. 2013;149:666-670.
- Stahlhut M, Bertelsen M, Hoyer-Hansen M, et al. Ingenol mebutate: induced cell death patterns in normal and cancer epithelial cells. J Drugs Dermatol. 2012;11:1181-1192.
Disseminated superficial actinic porokeratosis (DSAP) is a chronic condition characterized by numerous atrophic papules and patches with a distinctive peripheral keratotic ridge, typically found on sun-exposed areas.1,2 Treatment of DSAP is warranted not only for cosmetic and symptomatic benefits but also to prevent malignant transformation.3,4 Successful treatment of DSAP often is difficult and frequently requires the use of multiple modalities. Ingenol mebutate gel 0.05% is a topical medication primarily used for the treatment of actinic keratosis (AK) by inducing cell death.5 We report a case of DSAP treated effectively with ingenol mebutate gel 0.05%.
Case Report
A 37-year-old woman was referred to the dermatology department for counseling for pseudoxanthoma elasticum (PXE), which had been proven on biopsy by an outside dermatologist 2 years prior. Physical examination revealed yellow papules on the neck that were characteristic of PXE, but no lesions were noted on the arms or legs. The only other cutaneous finding was a soft nodule on the right hip consistent with a lipoma. The patient returned to our institution 6 years later with lesions on both lower legs. She reported that these lesions had been present for 3 years and were exacerbated by sun exposure. On physical examination, multiple scattered, erythematous, annular, scaling papules and plaques were noted on the bilateral legs. A biopsy showed the histopathologic findings of DSAP (Figure 1). The patient had no family history of DSAP or PXE.
To determine the best treatment modality, we treated 4 test areas on both upper and lower legs: one with trichloroacetic acid (TCA), one with cryotherapy, one with imiquimod cream 5%, and one with tretinoin cream 0.1%. The patient returned 4 weeks later and showed modest response to TCA, cryotherapy, and tretinoin cream. Because cryotherapy was determined to be most effective, 20 more lesions were frozen at that visit. Over the next 2 years, the patient was treated with TCA, imiquimod cream 5%, and tretinoin cream 0.1%, but all ultimately proved ineffective for DSAP.
The patient returned 2 years after treatment failure (age 47 years) and was prescribed ingenol mebutate gel 0.05% for 2 days over an area of 25 cm2 on the right lower leg (Figure 2A). She returned for follow-up at days 3, 15, 30, and 60. At day 3, the patient developed an inflammatory response to the medication with moderate erythema and scaling of individual lesions. No vesiculation, pustulation, edema, or ulceration was exhibited (Figure 2B). At day 30, there was a marked reduction in scaling with some postinflammatory erythema (Figure 2C). At day 60, much of the erythema had faded and the scale remained notably reduced (Figure 2D).
Comment
Disseminated superficial actinic porokeratosis is the most common subtype of porokeratosis, a keratinization disorder. There are 6 subtypes of porokeratosis identified in the literature: DSAP, disseminated superficial porokeratosis, classic porokeratosis of Mibelli, porokeratosis plantaris palmaris et disseminata, linear porokeratosis, and punctate porokeratosis.6 Disseminated superficial actinic porokeratosis has a female predominance (1.8:1 ratio)7 and generally appears in the third or fourth decades of life. Clonal proliferations of atypical keratinocytes have been implicated in the etiology of DSAP; however, the exact pathogenesis is unclear. Risk factors for DSAP include genetic susceptibility (eg, autosomal-dominant inheritance pattern), exposure to UV radiation, and drug-related immunosuppression or immunodeficiency.7 Other proposed etiologic risk factors include trauma and infection.8 Clinical diagnosis of DSAP is confirmed by the histological presence of a cornoid lamella (a thin column ofparakeratotic cells), a thinning epidermis, an absent or thinned granular cell layer, and a prominent dermal lymphocytic infiltrate.9,10
Disseminated superficial actinic porokeratosis clinically presents as small atrophic scaly papules and/or patches with raised peripheral ridges symmetrically dispersed on sun-exposed areas of the arms, legs, back, and shoulders. Although these lesions are extensive, they typically spare the mucous membranes, palms, and soles11; only a small percentage of cases report facial lesions,12 which often are asymptomatic but cosmetically bothersome. Additionally, approximately half of patients report symptoms of pruritus and/or stinging,13 thus treatment of DSAP is mainly indicated for symptomatic relief and cosmetic purposes. Malignant degeneration14,15 occurs in approximately 7.5% to 11% of porokeratosis cases,10,16 warranting treatment for preventative measures.
Management of DSAP is dependent on the extent of the disease and the level of concern for malignant transformation. Localized disease can be treated with cryotherapy, CO2 laser, and/or ablative techniques (eg, excision, curettage, dermabrasion) with variable degrees of success but high risk for scarring.1 More extensive disease requires treatment with topical retinoids, topical 5-fluorouracil, imiquimod cream 5%, diclofenac gel 3%, topical vitamin D3 analogues, and photodynamic therapy.1 Several other therapies have been reported in the literature with partial and/or complete success, including systemic retinoids (eg, acitretin), Q-switched ruby laser, Nd:YAG laser, fractional photothermolysis, Grenz rays, pulsed dye laser, fractional photothermolysis, topical corticosteroids, and fluor-hydroxy pulse peel.6 Although there is an extensive array of therapies for DSAP, treatment results are variable with mostly limited success. Successful treatment of DSAP is difficult and often requires the use of multiple modalities.
Ingenol mebutate is the active compound found in the sap of Euphorbia peplus used for the topical treatment of various skin conditions, including AKs.17 Ingenol mebutate gel 0.05% once daily for 2 days has been approved by the US Food and Drug Administration for the topical treatment of AKs. The mechanism of action of ingenol mebutate in AK therapy is not yet fully understood. In vivo and in vitro models have demonstrated both an induction of local lesion cell death and promotion of lesion-specific inflammatory response.18 When used in the treatment of AKs, ingenol mebutate gel 0.05% may cause a mild to moderate localized inflammatory response (eg, erythema, flaking/scaling, crusting, vesiculation/pustulation, erosion/ulceration, edema).
Our case is a rare report of successful treatment of DSAP with ingenol mebutate gel 0.05%. We found that treatment with ingenol mebutate gel 0.05% resulted in clinical improvement of DSAP lesions with minimal discomfort and good cosmetic response. This 2-day regimen is easy to use and patient friendly, improving medication compliance in such a cumbersome disease. We hope this case suggests that ingenol mebutate gel 0.05% could be a useful treatment alternative for DSAP, but future clinical studies should be conducted.
Disseminated superficial actinic porokeratosis (DSAP) is a chronic condition characterized by numerous atrophic papules and patches with a distinctive peripheral keratotic ridge, typically found on sun-exposed areas.1,2 Treatment of DSAP is warranted not only for cosmetic and symptomatic benefits but also to prevent malignant transformation.3,4 Successful treatment of DSAP often is difficult and frequently requires the use of multiple modalities. Ingenol mebutate gel 0.05% is a topical medication primarily used for the treatment of actinic keratosis (AK) by inducing cell death.5 We report a case of DSAP treated effectively with ingenol mebutate gel 0.05%.
Case Report
A 37-year-old woman was referred to the dermatology department for counseling for pseudoxanthoma elasticum (PXE), which had been proven on biopsy by an outside dermatologist 2 years prior. Physical examination revealed yellow papules on the neck that were characteristic of PXE, but no lesions were noted on the arms or legs. The only other cutaneous finding was a soft nodule on the right hip consistent with a lipoma. The patient returned to our institution 6 years later with lesions on both lower legs. She reported that these lesions had been present for 3 years and were exacerbated by sun exposure. On physical examination, multiple scattered, erythematous, annular, scaling papules and plaques were noted on the bilateral legs. A biopsy showed the histopathologic findings of DSAP (Figure 1). The patient had no family history of DSAP or PXE.
To determine the best treatment modality, we treated 4 test areas on both upper and lower legs: one with trichloroacetic acid (TCA), one with cryotherapy, one with imiquimod cream 5%, and one with tretinoin cream 0.1%. The patient returned 4 weeks later and showed modest response to TCA, cryotherapy, and tretinoin cream. Because cryotherapy was determined to be most effective, 20 more lesions were frozen at that visit. Over the next 2 years, the patient was treated with TCA, imiquimod cream 5%, and tretinoin cream 0.1%, but all ultimately proved ineffective for DSAP.
The patient returned 2 years after treatment failure (age 47 years) and was prescribed ingenol mebutate gel 0.05% for 2 days over an area of 25 cm2 on the right lower leg (Figure 2A). She returned for follow-up at days 3, 15, 30, and 60. At day 3, the patient developed an inflammatory response to the medication with moderate erythema and scaling of individual lesions. No vesiculation, pustulation, edema, or ulceration was exhibited (Figure 2B). At day 30, there was a marked reduction in scaling with some postinflammatory erythema (Figure 2C). At day 60, much of the erythema had faded and the scale remained notably reduced (Figure 2D).
Comment
Disseminated superficial actinic porokeratosis is the most common subtype of porokeratosis, a keratinization disorder. There are 6 subtypes of porokeratosis identified in the literature: DSAP, disseminated superficial porokeratosis, classic porokeratosis of Mibelli, porokeratosis plantaris palmaris et disseminata, linear porokeratosis, and punctate porokeratosis.6 Disseminated superficial actinic porokeratosis has a female predominance (1.8:1 ratio)7 and generally appears in the third or fourth decades of life. Clonal proliferations of atypical keratinocytes have been implicated in the etiology of DSAP; however, the exact pathogenesis is unclear. Risk factors for DSAP include genetic susceptibility (eg, autosomal-dominant inheritance pattern), exposure to UV radiation, and drug-related immunosuppression or immunodeficiency.7 Other proposed etiologic risk factors include trauma and infection.8 Clinical diagnosis of DSAP is confirmed by the histological presence of a cornoid lamella (a thin column ofparakeratotic cells), a thinning epidermis, an absent or thinned granular cell layer, and a prominent dermal lymphocytic infiltrate.9,10
Disseminated superficial actinic porokeratosis clinically presents as small atrophic scaly papules and/or patches with raised peripheral ridges symmetrically dispersed on sun-exposed areas of the arms, legs, back, and shoulders. Although these lesions are extensive, they typically spare the mucous membranes, palms, and soles11; only a small percentage of cases report facial lesions,12 which often are asymptomatic but cosmetically bothersome. Additionally, approximately half of patients report symptoms of pruritus and/or stinging,13 thus treatment of DSAP is mainly indicated for symptomatic relief and cosmetic purposes. Malignant degeneration14,15 occurs in approximately 7.5% to 11% of porokeratosis cases,10,16 warranting treatment for preventative measures.
Management of DSAP is dependent on the extent of the disease and the level of concern for malignant transformation. Localized disease can be treated with cryotherapy, CO2 laser, and/or ablative techniques (eg, excision, curettage, dermabrasion) with variable degrees of success but high risk for scarring.1 More extensive disease requires treatment with topical retinoids, topical 5-fluorouracil, imiquimod cream 5%, diclofenac gel 3%, topical vitamin D3 analogues, and photodynamic therapy.1 Several other therapies have been reported in the literature with partial and/or complete success, including systemic retinoids (eg, acitretin), Q-switched ruby laser, Nd:YAG laser, fractional photothermolysis, Grenz rays, pulsed dye laser, fractional photothermolysis, topical corticosteroids, and fluor-hydroxy pulse peel.6 Although there is an extensive array of therapies for DSAP, treatment results are variable with mostly limited success. Successful treatment of DSAP is difficult and often requires the use of multiple modalities.
Ingenol mebutate is the active compound found in the sap of Euphorbia peplus used for the topical treatment of various skin conditions, including AKs.17 Ingenol mebutate gel 0.05% once daily for 2 days has been approved by the US Food and Drug Administration for the topical treatment of AKs. The mechanism of action of ingenol mebutate in AK therapy is not yet fully understood. In vivo and in vitro models have demonstrated both an induction of local lesion cell death and promotion of lesion-specific inflammatory response.18 When used in the treatment of AKs, ingenol mebutate gel 0.05% may cause a mild to moderate localized inflammatory response (eg, erythema, flaking/scaling, crusting, vesiculation/pustulation, erosion/ulceration, edema).
Our case is a rare report of successful treatment of DSAP with ingenol mebutate gel 0.05%. We found that treatment with ingenol mebutate gel 0.05% resulted in clinical improvement of DSAP lesions with minimal discomfort and good cosmetic response. This 2-day regimen is easy to use and patient friendly, improving medication compliance in such a cumbersome disease. We hope this case suggests that ingenol mebutate gel 0.05% could be a useful treatment alternative for DSAP, but future clinical studies should be conducted.
- Martin-Clavijo A, Kanelleas A, Vlachou C, et al. Porokeratoses. In: Lebwohl M, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease Comprehensive Therapeutic Strategies. 3rd ed. China: Elsevier Limited; 2010:584-586.
- Rouhani P, Fischer M, Meehan S, et al. Disseminated superficial actinic porokeratosis. Dermatology Online J. 2012;18:24.
- Sasson M, Krain AD. Porokeratosis and cutaneous malignancy. a review. Dermatol Surg. 1996;22:339-342.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536-538.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- O’Regan GM, Irvine AD. Porokeratosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:442-446.
- Sertznig P, von Felbert V, Megahed M. Porokeratosis: present concepts. J Eur Acad Dermatol Venereol. 2012;26:404-412.
- Brauer JA, Mandal R, Walters R, et al. Disseminated superficial porokeratosis. Dermatology Online J. 2010;16:20.
- Tallon B. Porokeratosis pathology. DermNet New Zealand website. http://www.dermnet.org.nz/pathology/porokeratosis-path.html. Updated December 2016. Accessed January 12, 2017.
- Skupsky H, Skupsky J, Goldenberg G. Disseminated superficial actinic porokeratosis: a treatment review [published online October 22, 2010]. J Dermatolog Treat. 2012;23:52-56.
- Spencer LV. Porokeratosis. UpToDate web site. https://eresources.library.mssm.edu:3285/contents/porokeratosis?source=search_result&search=porokeratosis&selectedTitle=1~22. Updated September 1, 2016. Accessed April 3, 2017.
- Sawyer R, Picou KA. Facial presentation of disseminated superficial actinic porokeratosis. Ear Nose Throat J. 1989;68:57-59.
- Schwarz T, Seiser A, Gschnait F. Disseminated superficial “actinic” porokeratosis. J Am Acad Dermatol. 1984;11(4, pt 2):724-730.
- Maubec E, Duvillard P, Margulis A, et al. Common skin cancers in porokeratosis. Br J Dermatol. 2005;152:1389-1391.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis [published online November 3, 2011]. Ann Dermatol. 2011;23:536-538.
- Kumari S, Mathur M. Disseminated superficial actinic porokeratosis. Nepal J Dermatol Venereol Leprol. 2010;9:22-24.
- Lebwohl M, Shumack S, Stein Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratosis. JAMA Dermatol. 2013;149:666-670.
- Stahlhut M, Bertelsen M, Hoyer-Hansen M, et al. Ingenol mebutate: induced cell death patterns in normal and cancer epithelial cells. J Drugs Dermatol. 2012;11:1181-1192.
- Martin-Clavijo A, Kanelleas A, Vlachou C, et al. Porokeratoses. In: Lebwohl M, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease Comprehensive Therapeutic Strategies. 3rd ed. China: Elsevier Limited; 2010:584-586.
- Rouhani P, Fischer M, Meehan S, et al. Disseminated superficial actinic porokeratosis. Dermatology Online J. 2012;18:24.
- Sasson M, Krain AD. Porokeratosis and cutaneous malignancy. a review. Dermatol Surg. 1996;22:339-342.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536-538.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- O’Regan GM, Irvine AD. Porokeratosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:442-446.
- Sertznig P, von Felbert V, Megahed M. Porokeratosis: present concepts. J Eur Acad Dermatol Venereol. 2012;26:404-412.
- Brauer JA, Mandal R, Walters R, et al. Disseminated superficial porokeratosis. Dermatology Online J. 2010;16:20.
- Tallon B. Porokeratosis pathology. DermNet New Zealand website. http://www.dermnet.org.nz/pathology/porokeratosis-path.html. Updated December 2016. Accessed January 12, 2017.
- Skupsky H, Skupsky J, Goldenberg G. Disseminated superficial actinic porokeratosis: a treatment review [published online October 22, 2010]. J Dermatolog Treat. 2012;23:52-56.
- Spencer LV. Porokeratosis. UpToDate web site. https://eresources.library.mssm.edu:3285/contents/porokeratosis?source=search_result&search=porokeratosis&selectedTitle=1~22. Updated September 1, 2016. Accessed April 3, 2017.
- Sawyer R, Picou KA. Facial presentation of disseminated superficial actinic porokeratosis. Ear Nose Throat J. 1989;68:57-59.
- Schwarz T, Seiser A, Gschnait F. Disseminated superficial “actinic” porokeratosis. J Am Acad Dermatol. 1984;11(4, pt 2):724-730.
- Maubec E, Duvillard P, Margulis A, et al. Common skin cancers in porokeratosis. Br J Dermatol. 2005;152:1389-1391.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis [published online November 3, 2011]. Ann Dermatol. 2011;23:536-538.
- Kumari S, Mathur M. Disseminated superficial actinic porokeratosis. Nepal J Dermatol Venereol Leprol. 2010;9:22-24.
- Lebwohl M, Shumack S, Stein Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratosis. JAMA Dermatol. 2013;149:666-670.
- Stahlhut M, Bertelsen M, Hoyer-Hansen M, et al. Ingenol mebutate: induced cell death patterns in normal and cancer epithelial cells. J Drugs Dermatol. 2012;11:1181-1192.
Practice Points
- Disseminated superficial actinic porokeratosis (DSAP) is an uncommon skin condition consisting of multiple annular hyperkeratotic lesions on sun-exposed areas.
- Treatment of DSAP is necessary due to its potential for progression to malignancy.
- Consider ingenol mebutate gel 0.05% for the treatment of DSAP on the arms and legs.
Hypoperfusion Retinopathy
Cardiovascular diseases are some of the most common conditions found in the geriatric population. Ocular manifestations of systemic cardiovascular conditions often are the initial presentation of the systemic disease. Identifying these findings help reveal the underlying disease and prevent more serious visual and systemic complications or even death.
Hypoperfusion retinopathy can occur as an early manifestation of carotid occlusive disease. It results from poor arterial perfusion pressure secondary to significant or complete carotid artery blockage resulting in retinal cha
Case Report
A 71-year-old white male was referred by his primary care physician (PCP) to the eye clinic for a routine comprehensive eye exam. The patient reported that his current progressive lenses, prescribed 2 years prior, were not strong enough at both distance and near, and that his eyes often felt dry. The symptoms were gradual in onset since his prior exam with no reported flashes, floaters, loss of vision, headaches, or ocular irritations.
The patient’s medical history was significant for morbid obesity, hypertension, borderline diabetes mellitus, and obstructive sleep apnea. His ocular history included recurrent conjunctivitis. At the time of the visit, the patient’s medications included 81 mg aspirin, 10 mg benazepril, 1,000 mg fish oil, 80 mg simvastatin, and use of a continuous positive airway pressure machine.
Best-corrected Snellen visual acuity was stable to his last eye exam at 20/25+2 right eye and 20/25-1 left eye with a manifest refraction of +2.25-0.75 × 077, and +2.75-1.25 × 096 in the right and left eye, respectively. Pupils were equally round and reactive to light with no afferent pupillary defect. Extraocular motility and finger counting fields were unremarkable. Anterior segment evaluation revealed lax bilateral upper lid apposition and mild cataracts in both eyes but were otherwise unremarkable (Figure 1). Dilated fundus examination revealed extensive hemorrhaging in the midperipheral retina of the right eye only (Figure 2). The left eye retina showed no abnormalities.

At this point the patient declined any additional symptoms, including eye pain, headache, transient vision loss, jaw claudication, and stroke signs. A complete blood count and hemoglobin A1c (HbA1c) was ordered, and all findings were unremarkable with no evidence of blood dyscrasia and with a HbA1c of 6.0. A carotid ultrasound (CUS) was also performed and revealed severe narrowing of the proximal section of the right internal carotid artery (ICA) with a trickle flow (Figure 3). The peak systolic velocity (PSV) at this level was 508 cm/s. There also was severe narrowing and turbulent flow in both the mid and distal portions of the right ICA. The patient was sent for a vascular evaluation 2 days following the CUS.
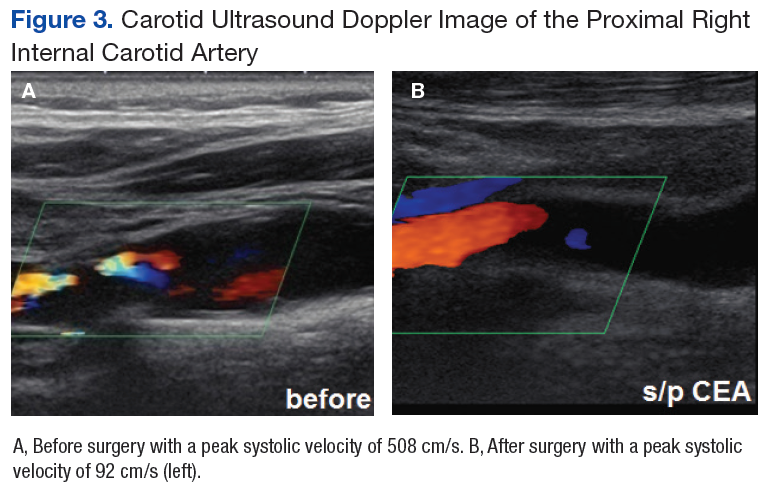
Based on the ocular findings and CUS results, the diagnosis of hypoperfusion retinopathy secondary to carotid occlusive disease was made. Because the patient was asymptomatic with no additional ocular sequelae, he was scheduled for an eye clinic follow-up in 2 months. The electrocardiogram, chest X-ray, and exercise stress test results were negative for acute cardiopulmonary disease, ischemia, or arrhythmias. A computed tomography angiography was performed and confirmed a high-grade lesion of the right ICA of > 95%. The vascular surgeon reported an 11% risk of stroke within 5 years and a 1% risk of stroke with surgery. Based on these results the patient underwent a right carotid endarterectomy (CEA) 2 weeks later. A follow-up CUS was performed 1 month post-CEA and revealed no abnormal fluid or significant plaque with a PSV of 92 cm/s (prior to surgery PSV was 508 cm/s) (Figure 3).
The patient returned to the eye clinic 1 month after the CEA. Gonioscopy revealed no neovascularization of the iris or angle and the dilated eye exam showed resolution of the midperipheral blot hemorrhages in his right eye with no evidence of retinal neovascularization.
Discussion
Hypoperfusion retinopathy is characterized by posterior retinal changes secondary to chronic ocular ischemia from decreased arterial perfusion related to significant or complete carotid artery stenosis.1-5 Early literature referred to this condition as venous stasis retinopathy; however, this term is misleading as the condition results from a reduction in arterial perfusion pressure and the term describes venous outflow obstruction.6 The terms carotid ischemic retinopathy, ischemic oculopathy, and hypotensive retinopathy also have been used interchangeably when describing hypoperfusion retinopathy.6
Incidence of hypoperfusion retinopathy is twice as high in males as it is in females due to a higher prevalence of cardiovascular disease.7 Hypoperfusion retinopathy rarely presents before the age of 50 years, with the average age of onset around 65 years.7 The exact rate of occurrence is unknown as this condition often is underdiagnosed because it mimics other vascular conditions, such as venous occlusive disease and diabetic retinopathy.1,7 Patients can present asymptomatically where findings are incidental on a dilated eye exam, or they may present with vision loss that can be gradual, sudden, or transient in nature.5,6,8
Gradual vision loss can follow a period of weeks to months and can occur secondary to posterior ischemia, macular edema, or choroidal hypoperfusion.1,3,8,9 Sudden vision loss can occur from severe hypoperfusion, creating an acute inner layer retinal ischemia. This type of vision loss often is accompanied by a cherry red spot in the macula and can be caused by an embolic plaque.1,8 Transient vision loss (TVL) also can be secondary to a plaque emboli or light induced. Patients with light-induced TVL report poor to blurry vision or prolonged after image when exposed to bright lights. In theory when the retina is exposed to light, there is an increase in metabolic demand that is unmet in those with choroidal vascular insufficiency from significant carotid stenosis.3,8,10
The clinical presentation most often is unilateral. Early stages of the disease generally affect the midperipheral retina but can be found in the posterior pole with chronicity. Early findings include microaneurysms, nerve fiber layer and inner retinal layer hemorrhages, and dilated, but generally not tortuous, veins.5 Chronic stage findings include arteriolar narrowing, extreme venous dilation, occasionally macular edema, and neovascularization of the disc and or retina.5 Disc edema or collaterals usually are not present.5
The mechanism behind hypoperfusion retinopathy results from an overall ischemic cascade and starts with comorbid cardiovascular conditions, such as hypertension, hypercholesterolemia, diabetes, heart disease, and history of smoking.1,2,5 These conditions play a role in creating atherosclerotic buildup in the arterial lumen leading to chronic narrowing and a decrease in arterial perfusion pressure. Over time, a low-grade hypoxic situation is formed, generating vascular endothelial cell damage and pericytes cell loss, thus causing leakage of fluid.1,2,5 With these chronic hypoxic states, angiogenic factor release eventually leads to posterior neovascularization.1,2,5 Further chronicity of carotid occlusive disease can create a panocular ischemia that also involves anterior structures, including iris, conjunctiva, episclera, or cornea. At this point, hypoperfusion retinopathy progresses to a more severe condition called ocular ischemic syndrome (OIS).2,5
Ocular ischemic syndrome can be associated with a 40% mortality rate within 5 years of onset as it is generally found in those with overall poor health.5 Along with posterior neovascularization, anterior structures also are involved. Sixty-seven percent of cases have iris or angle neovascularization of which 35% go on to develop neovascular glaucoma and its complications.1,8 With OIS, 90% of cases have some type of vision loss, and 40% report ipsilateral ocular pain.1,8 Visual loss can be gradual, sudden, or transient. The pain can occur from ocular ischemia, ruptured corneal epithelial microcysts secondary to acute glaucoma, elevated intraocular pressure (IOP) with neovascular glaucoma, or from ipsilateral dural ischemia.1,5,6,8 Fluorescein angiography is commonly used to diagnose and manage OIS, because it allows for the visualization of retinal and choroidal circulation and the detection of neovascular proliferation and ischemic areas.
Diagnostic Imaging
Several diagnostic testing strategies are available to evaluate for carotid occlusive disease. Carotid ultrasonography is a noninvasive, safe, and inexpensive screening tool to evaluate for high-grade stenosis. However, it can sometimes overestimate the degree of stenosis and is not reliable with severe calcifications.8 Computed tomography angiography and magnetic resonance angiography are minimally invasive tools that can be used to screen or confirm the degree of stenosis.8 These can be used in addition or instead of ultrasonography, especially in instances where patients have a short neck or high carotid bifurcation that may affect reliability. Both are contraindicated in those with renal failure as both modalities require the use of a contrast dye. Magnetic resonance angiography is far more expensive, time consuming, and not readily available.8 Carotid angiography is considered the gold standard for imaging the entire carotid artery system because it allows for the evaluation of plaque morphology, atherosclerotic disease, and collateral circulations.8 The disadvantages to this invasive and high-cost procedure include a risk of mortality that can occur secondary to an embolic stroke, myocardial infarction (MI), carotid artery dissection, or arterial thrombosis.8
Treatment
Treatment and management for carotid artery stenosis is focused on combined effort with the patient’s PCP and other specialists, including cardiologist, neurologist, and vascular surgeons.11 Treatment of comorbid conditions, education on healthy lifestyle, and smoking cessation are all imperative to the patient’s well-being. Managing ocular sequelae is based on specific findings and can include intravitreal antivascular edothelial growth factor or steroidal injections, pan retinal photocoagulation, or hypotensive drops.6,7
Restoration of arterial perfusion pressure is the main goal of treatment, and this can be done through CEA or carotid artery stents. Surgical intervention by CEA is determined based on each patient and his or her overall health. A full cardiac workup is required due to surgical risks. The North American Symptomatic Carotid Endarterectomy Trial evaluated symptomatic stenosis and the effectiveness of surgical intervention on stroke prevention. The trial reported that CEA was beneficial in symptomatic patients with 55% to 99% stenosis and especially in those with higher grade stenosis (> 70% up to 95%).5,7,8,12 With regard to asymptomatic patients with high-grade stenosis, CEA has been found to reduce the risk of stroke if there is at least 60% stenosis.5,7,8
Carotid artery stents can be used as an alternative when CEA is not effective or contraindicated due to a history of previous CEA, neck radiation, unstable angina, congestive heart failure, or recent MI.5,7,8 Neither CEA nor stenting is considered effective in complete occlusions due to the high risk of thromboembolism formation.5,7,8
Conclusion
Hypoperfusion retinopathy describes posterior retinal findings that occur secondary to poor arterial perfusion caused by carotid occlusive disease. Early intervention and restoration of this pressure can prevent the risk of developing a more serious condition characterized by a panocular ischemia called OIS. Unlike hypoperfusion retinopathy, OIS also includes anterior segment findings such as iris neovascularization, which may lead to neovascular glaucoma, whereas hypoperfusion retinopathy is localized to the posterior pole. Patients that develop OIS are at a 40% risk of mortality within 5 years due to poor overall health. Understanding the patient’s signs and symptoms can aid in the diagnosis of both conditions. Collaborative management with the patient’s PCP and specialists in treating comorbid conditions is vital to the patients’ well-being.
1. Brown GC, Magargal LE. The ocular ischemic syndrome. Int Ophthalmol. 1988;11(4):239-251.
2. Dahlman AH, McCormack D, Harrison RJ. Bilateral hypoperfuion retinopathy. J R Soc Med. 2001; 94(6):298-299.
3. Dugan JD Jr, Green WR. Ophthalmologic manifestations of carotid occlusive disease. Eye (Lond). 1991;5(pt 2):226-238.
4. Klijn CJ, Kappelle LJ, Tulleken CAF, van Gijn J. Symptomatic carotid artery occlusion. A reappraisal of hemodynamic factors. Stroke. 1997;28(10):2084-2093.
5. McCrary JA III. Venous stasis retinopathy of stenotic or occlusive caroid origin. J Clin Neuroophthalmol. 1989;9(3):195-199.
6. Sanborn GE, Magargal LE. Arterial obstructive disease of the eye. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 14.
7. Terelak-Borys B, Skonieczna K, Grabska-Liberek I. Ocular ischemic syndrome–a systematic review. Med Sci Monit. 2012;18(8):RA138-RA144.
8. Atebara NH, Brown GC. The ocular ischemic syndrome. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 12.
9. Ho AC, Lieb WE, Flaharty PM, et al. Color Doppler imaging of the ocular ischaemic syndrome. Ophthalmology. 1992;99(9):1453-1462.
10. Kahn M, Green WR, Knox DL, Miller NR. Ocular features of carotid occlusive disease. Retina. 1986;6(4):239-252.
11. Mizener JB, Podhajsky P, Hayreh SS. Ocular ischemic syndrome. Ophthalmology. 1997;104(5):859-864.
12. Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke. 1999;30(9):1751-1758.
Cardiovascular diseases are some of the most common conditions found in the geriatric population. Ocular manifestations of systemic cardiovascular conditions often are the initial presentation of the systemic disease. Identifying these findings help reveal the underlying disease and prevent more serious visual and systemic complications or even death.
Hypoperfusion retinopathy can occur as an early manifestation of carotid occlusive disease. It results from poor arterial perfusion pressure secondary to significant or complete carotid artery blockage resulting in retinal cha
Case Report
A 71-year-old white male was referred by his primary care physician (PCP) to the eye clinic for a routine comprehensive eye exam. The patient reported that his current progressive lenses, prescribed 2 years prior, were not strong enough at both distance and near, and that his eyes often felt dry. The symptoms were gradual in onset since his prior exam with no reported flashes, floaters, loss of vision, headaches, or ocular irritations.
The patient’s medical history was significant for morbid obesity, hypertension, borderline diabetes mellitus, and obstructive sleep apnea. His ocular history included recurrent conjunctivitis. At the time of the visit, the patient’s medications included 81 mg aspirin, 10 mg benazepril, 1,000 mg fish oil, 80 mg simvastatin, and use of a continuous positive airway pressure machine.
Best-corrected Snellen visual acuity was stable to his last eye exam at 20/25+2 right eye and 20/25-1 left eye with a manifest refraction of +2.25-0.75 × 077, and +2.75-1.25 × 096 in the right and left eye, respectively. Pupils were equally round and reactive to light with no afferent pupillary defect. Extraocular motility and finger counting fields were unremarkable. Anterior segment evaluation revealed lax bilateral upper lid apposition and mild cataracts in both eyes but were otherwise unremarkable (Figure 1). Dilated fundus examination revealed extensive hemorrhaging in the midperipheral retina of the right eye only (Figure 2). The left eye retina showed no abnormalities.
At this point the patient declined any additional symptoms, including eye pain, headache, transient vision loss, jaw claudication, and stroke signs. A complete blood count and hemoglobin A1c (HbA1c) was ordered, and all findings were unremarkable with no evidence of blood dyscrasia and with a HbA1c of 6.0. A carotid ultrasound (CUS) was also performed and revealed severe narrowing of the proximal section of the right internal carotid artery (ICA) with a trickle flow (Figure 3). The peak systolic velocity (PSV) at this level was 508 cm/s. There also was severe narrowing and turbulent flow in both the mid and distal portions of the right ICA. The patient was sent for a vascular evaluation 2 days following the CUS.
Based on the ocular findings and CUS results, the diagnosis of hypoperfusion retinopathy secondary to carotid occlusive disease was made. Because the patient was asymptomatic with no additional ocular sequelae, he was scheduled for an eye clinic follow-up in 2 months. The electrocardiogram, chest X-ray, and exercise stress test results were negative for acute cardiopulmonary disease, ischemia, or arrhythmias. A computed tomography angiography was performed and confirmed a high-grade lesion of the right ICA of > 95%. The vascular surgeon reported an 11% risk of stroke within 5 years and a 1% risk of stroke with surgery. Based on these results the patient underwent a right carotid endarterectomy (CEA) 2 weeks later. A follow-up CUS was performed 1 month post-CEA and revealed no abnormal fluid or significant plaque with a PSV of 92 cm/s (prior to surgery PSV was 508 cm/s) (Figure 3).
The patient returned to the eye clinic 1 month after the CEA. Gonioscopy revealed no neovascularization of the iris or angle and the dilated eye exam showed resolution of the midperipheral blot hemorrhages in his right eye with no evidence of retinal neovascularization.
Discussion
Hypoperfusion retinopathy is characterized by posterior retinal changes secondary to chronic ocular ischemia from decreased arterial perfusion related to significant or complete carotid artery stenosis.1-5 Early literature referred to this condition as venous stasis retinopathy; however, this term is misleading as the condition results from a reduction in arterial perfusion pressure and the term describes venous outflow obstruction.6 The terms carotid ischemic retinopathy, ischemic oculopathy, and hypotensive retinopathy also have been used interchangeably when describing hypoperfusion retinopathy.6
Incidence of hypoperfusion retinopathy is twice as high in males as it is in females due to a higher prevalence of cardiovascular disease.7 Hypoperfusion retinopathy rarely presents before the age of 50 years, with the average age of onset around 65 years.7 The exact rate of occurrence is unknown as this condition often is underdiagnosed because it mimics other vascular conditions, such as venous occlusive disease and diabetic retinopathy.1,7 Patients can present asymptomatically where findings are incidental on a dilated eye exam, or they may present with vision loss that can be gradual, sudden, or transient in nature.5,6,8
Gradual vision loss can follow a period of weeks to months and can occur secondary to posterior ischemia, macular edema, or choroidal hypoperfusion.1,3,8,9 Sudden vision loss can occur from severe hypoperfusion, creating an acute inner layer retinal ischemia. This type of vision loss often is accompanied by a cherry red spot in the macula and can be caused by an embolic plaque.1,8 Transient vision loss (TVL) also can be secondary to a plaque emboli or light induced. Patients with light-induced TVL report poor to blurry vision or prolonged after image when exposed to bright lights. In theory when the retina is exposed to light, there is an increase in metabolic demand that is unmet in those with choroidal vascular insufficiency from significant carotid stenosis.3,8,10
The clinical presentation most often is unilateral. Early stages of the disease generally affect the midperipheral retina but can be found in the posterior pole with chronicity. Early findings include microaneurysms, nerve fiber layer and inner retinal layer hemorrhages, and dilated, but generally not tortuous, veins.5 Chronic stage findings include arteriolar narrowing, extreme venous dilation, occasionally macular edema, and neovascularization of the disc and or retina.5 Disc edema or collaterals usually are not present.5
The mechanism behind hypoperfusion retinopathy results from an overall ischemic cascade and starts with comorbid cardiovascular conditions, such as hypertension, hypercholesterolemia, diabetes, heart disease, and history of smoking.1,2,5 These conditions play a role in creating atherosclerotic buildup in the arterial lumen leading to chronic narrowing and a decrease in arterial perfusion pressure. Over time, a low-grade hypoxic situation is formed, generating vascular endothelial cell damage and pericytes cell loss, thus causing leakage of fluid.1,2,5 With these chronic hypoxic states, angiogenic factor release eventually leads to posterior neovascularization.1,2,5 Further chronicity of carotid occlusive disease can create a panocular ischemia that also involves anterior structures, including iris, conjunctiva, episclera, or cornea. At this point, hypoperfusion retinopathy progresses to a more severe condition called ocular ischemic syndrome (OIS).2,5
Ocular ischemic syndrome can be associated with a 40% mortality rate within 5 years of onset as it is generally found in those with overall poor health.5 Along with posterior neovascularization, anterior structures also are involved. Sixty-seven percent of cases have iris or angle neovascularization of which 35% go on to develop neovascular glaucoma and its complications.1,8 With OIS, 90% of cases have some type of vision loss, and 40% report ipsilateral ocular pain.1,8 Visual loss can be gradual, sudden, or transient. The pain can occur from ocular ischemia, ruptured corneal epithelial microcysts secondary to acute glaucoma, elevated intraocular pressure (IOP) with neovascular glaucoma, or from ipsilateral dural ischemia.1,5,6,8 Fluorescein angiography is commonly used to diagnose and manage OIS, because it allows for the visualization of retinal and choroidal circulation and the detection of neovascular proliferation and ischemic areas.
Diagnostic Imaging
Several diagnostic testing strategies are available to evaluate for carotid occlusive disease. Carotid ultrasonography is a noninvasive, safe, and inexpensive screening tool to evaluate for high-grade stenosis. However, it can sometimes overestimate the degree of stenosis and is not reliable with severe calcifications.8 Computed tomography angiography and magnetic resonance angiography are minimally invasive tools that can be used to screen or confirm the degree of stenosis.8 These can be used in addition or instead of ultrasonography, especially in instances where patients have a short neck or high carotid bifurcation that may affect reliability. Both are contraindicated in those with renal failure as both modalities require the use of a contrast dye. Magnetic resonance angiography is far more expensive, time consuming, and not readily available.8 Carotid angiography is considered the gold standard for imaging the entire carotid artery system because it allows for the evaluation of plaque morphology, atherosclerotic disease, and collateral circulations.8 The disadvantages to this invasive and high-cost procedure include a risk of mortality that can occur secondary to an embolic stroke, myocardial infarction (MI), carotid artery dissection, or arterial thrombosis.8
Treatment
Treatment and management for carotid artery stenosis is focused on combined effort with the patient’s PCP and other specialists, including cardiologist, neurologist, and vascular surgeons.11 Treatment of comorbid conditions, education on healthy lifestyle, and smoking cessation are all imperative to the patient’s well-being. Managing ocular sequelae is based on specific findings and can include intravitreal antivascular edothelial growth factor or steroidal injections, pan retinal photocoagulation, or hypotensive drops.6,7
Restoration of arterial perfusion pressure is the main goal of treatment, and this can be done through CEA or carotid artery stents. Surgical intervention by CEA is determined based on each patient and his or her overall health. A full cardiac workup is required due to surgical risks. The North American Symptomatic Carotid Endarterectomy Trial evaluated symptomatic stenosis and the effectiveness of surgical intervention on stroke prevention. The trial reported that CEA was beneficial in symptomatic patients with 55% to 99% stenosis and especially in those with higher grade stenosis (> 70% up to 95%).5,7,8,12 With regard to asymptomatic patients with high-grade stenosis, CEA has been found to reduce the risk of stroke if there is at least 60% stenosis.5,7,8
Carotid artery stents can be used as an alternative when CEA is not effective or contraindicated due to a history of previous CEA, neck radiation, unstable angina, congestive heart failure, or recent MI.5,7,8 Neither CEA nor stenting is considered effective in complete occlusions due to the high risk of thromboembolism formation.5,7,8
Conclusion
Hypoperfusion retinopathy describes posterior retinal findings that occur secondary to poor arterial perfusion caused by carotid occlusive disease. Early intervention and restoration of this pressure can prevent the risk of developing a more serious condition characterized by a panocular ischemia called OIS. Unlike hypoperfusion retinopathy, OIS also includes anterior segment findings such as iris neovascularization, which may lead to neovascular glaucoma, whereas hypoperfusion retinopathy is localized to the posterior pole. Patients that develop OIS are at a 40% risk of mortality within 5 years due to poor overall health. Understanding the patient’s signs and symptoms can aid in the diagnosis of both conditions. Collaborative management with the patient’s PCP and specialists in treating comorbid conditions is vital to the patients’ well-being.
Cardiovascular diseases are some of the most common conditions found in the geriatric population. Ocular manifestations of systemic cardiovascular conditions often are the initial presentation of the systemic disease. Identifying these findings help reveal the underlying disease and prevent more serious visual and systemic complications or even death.
Hypoperfusion retinopathy can occur as an early manifestation of carotid occlusive disease. It results from poor arterial perfusion pressure secondary to significant or complete carotid artery blockage resulting in retinal cha
Case Report
A 71-year-old white male was referred by his primary care physician (PCP) to the eye clinic for a routine comprehensive eye exam. The patient reported that his current progressive lenses, prescribed 2 years prior, were not strong enough at both distance and near, and that his eyes often felt dry. The symptoms were gradual in onset since his prior exam with no reported flashes, floaters, loss of vision, headaches, or ocular irritations.
The patient’s medical history was significant for morbid obesity, hypertension, borderline diabetes mellitus, and obstructive sleep apnea. His ocular history included recurrent conjunctivitis. At the time of the visit, the patient’s medications included 81 mg aspirin, 10 mg benazepril, 1,000 mg fish oil, 80 mg simvastatin, and use of a continuous positive airway pressure machine.
Best-corrected Snellen visual acuity was stable to his last eye exam at 20/25+2 right eye and 20/25-1 left eye with a manifest refraction of +2.25-0.75 × 077, and +2.75-1.25 × 096 in the right and left eye, respectively. Pupils were equally round and reactive to light with no afferent pupillary defect. Extraocular motility and finger counting fields were unremarkable. Anterior segment evaluation revealed lax bilateral upper lid apposition and mild cataracts in both eyes but were otherwise unremarkable (Figure 1). Dilated fundus examination revealed extensive hemorrhaging in the midperipheral retina of the right eye only (Figure 2). The left eye retina showed no abnormalities.
At this point the patient declined any additional symptoms, including eye pain, headache, transient vision loss, jaw claudication, and stroke signs. A complete blood count and hemoglobin A1c (HbA1c) was ordered, and all findings were unremarkable with no evidence of blood dyscrasia and with a HbA1c of 6.0. A carotid ultrasound (CUS) was also performed and revealed severe narrowing of the proximal section of the right internal carotid artery (ICA) with a trickle flow (Figure 3). The peak systolic velocity (PSV) at this level was 508 cm/s. There also was severe narrowing and turbulent flow in both the mid and distal portions of the right ICA. The patient was sent for a vascular evaluation 2 days following the CUS.
Based on the ocular findings and CUS results, the diagnosis of hypoperfusion retinopathy secondary to carotid occlusive disease was made. Because the patient was asymptomatic with no additional ocular sequelae, he was scheduled for an eye clinic follow-up in 2 months. The electrocardiogram, chest X-ray, and exercise stress test results were negative for acute cardiopulmonary disease, ischemia, or arrhythmias. A computed tomography angiography was performed and confirmed a high-grade lesion of the right ICA of > 95%. The vascular surgeon reported an 11% risk of stroke within 5 years and a 1% risk of stroke with surgery. Based on these results the patient underwent a right carotid endarterectomy (CEA) 2 weeks later. A follow-up CUS was performed 1 month post-CEA and revealed no abnormal fluid or significant plaque with a PSV of 92 cm/s (prior to surgery PSV was 508 cm/s) (Figure 3).
The patient returned to the eye clinic 1 month after the CEA. Gonioscopy revealed no neovascularization of the iris or angle and the dilated eye exam showed resolution of the midperipheral blot hemorrhages in his right eye with no evidence of retinal neovascularization.
Discussion
Hypoperfusion retinopathy is characterized by posterior retinal changes secondary to chronic ocular ischemia from decreased arterial perfusion related to significant or complete carotid artery stenosis.1-5 Early literature referred to this condition as venous stasis retinopathy; however, this term is misleading as the condition results from a reduction in arterial perfusion pressure and the term describes venous outflow obstruction.6 The terms carotid ischemic retinopathy, ischemic oculopathy, and hypotensive retinopathy also have been used interchangeably when describing hypoperfusion retinopathy.6
Incidence of hypoperfusion retinopathy is twice as high in males as it is in females due to a higher prevalence of cardiovascular disease.7 Hypoperfusion retinopathy rarely presents before the age of 50 years, with the average age of onset around 65 years.7 The exact rate of occurrence is unknown as this condition often is underdiagnosed because it mimics other vascular conditions, such as venous occlusive disease and diabetic retinopathy.1,7 Patients can present asymptomatically where findings are incidental on a dilated eye exam, or they may present with vision loss that can be gradual, sudden, or transient in nature.5,6,8
Gradual vision loss can follow a period of weeks to months and can occur secondary to posterior ischemia, macular edema, or choroidal hypoperfusion.1,3,8,9 Sudden vision loss can occur from severe hypoperfusion, creating an acute inner layer retinal ischemia. This type of vision loss often is accompanied by a cherry red spot in the macula and can be caused by an embolic plaque.1,8 Transient vision loss (TVL) also can be secondary to a plaque emboli or light induced. Patients with light-induced TVL report poor to blurry vision or prolonged after image when exposed to bright lights. In theory when the retina is exposed to light, there is an increase in metabolic demand that is unmet in those with choroidal vascular insufficiency from significant carotid stenosis.3,8,10
The clinical presentation most often is unilateral. Early stages of the disease generally affect the midperipheral retina but can be found in the posterior pole with chronicity. Early findings include microaneurysms, nerve fiber layer and inner retinal layer hemorrhages, and dilated, but generally not tortuous, veins.5 Chronic stage findings include arteriolar narrowing, extreme venous dilation, occasionally macular edema, and neovascularization of the disc and or retina.5 Disc edema or collaterals usually are not present.5
The mechanism behind hypoperfusion retinopathy results from an overall ischemic cascade and starts with comorbid cardiovascular conditions, such as hypertension, hypercholesterolemia, diabetes, heart disease, and history of smoking.1,2,5 These conditions play a role in creating atherosclerotic buildup in the arterial lumen leading to chronic narrowing and a decrease in arterial perfusion pressure. Over time, a low-grade hypoxic situation is formed, generating vascular endothelial cell damage and pericytes cell loss, thus causing leakage of fluid.1,2,5 With these chronic hypoxic states, angiogenic factor release eventually leads to posterior neovascularization.1,2,5 Further chronicity of carotid occlusive disease can create a panocular ischemia that also involves anterior structures, including iris, conjunctiva, episclera, or cornea. At this point, hypoperfusion retinopathy progresses to a more severe condition called ocular ischemic syndrome (OIS).2,5
Ocular ischemic syndrome can be associated with a 40% mortality rate within 5 years of onset as it is generally found in those with overall poor health.5 Along with posterior neovascularization, anterior structures also are involved. Sixty-seven percent of cases have iris or angle neovascularization of which 35% go on to develop neovascular glaucoma and its complications.1,8 With OIS, 90% of cases have some type of vision loss, and 40% report ipsilateral ocular pain.1,8 Visual loss can be gradual, sudden, or transient. The pain can occur from ocular ischemia, ruptured corneal epithelial microcysts secondary to acute glaucoma, elevated intraocular pressure (IOP) with neovascular glaucoma, or from ipsilateral dural ischemia.1,5,6,8 Fluorescein angiography is commonly used to diagnose and manage OIS, because it allows for the visualization of retinal and choroidal circulation and the detection of neovascular proliferation and ischemic areas.
Diagnostic Imaging
Several diagnostic testing strategies are available to evaluate for carotid occlusive disease. Carotid ultrasonography is a noninvasive, safe, and inexpensive screening tool to evaluate for high-grade stenosis. However, it can sometimes overestimate the degree of stenosis and is not reliable with severe calcifications.8 Computed tomography angiography and magnetic resonance angiography are minimally invasive tools that can be used to screen or confirm the degree of stenosis.8 These can be used in addition or instead of ultrasonography, especially in instances where patients have a short neck or high carotid bifurcation that may affect reliability. Both are contraindicated in those with renal failure as both modalities require the use of a contrast dye. Magnetic resonance angiography is far more expensive, time consuming, and not readily available.8 Carotid angiography is considered the gold standard for imaging the entire carotid artery system because it allows for the evaluation of plaque morphology, atherosclerotic disease, and collateral circulations.8 The disadvantages to this invasive and high-cost procedure include a risk of mortality that can occur secondary to an embolic stroke, myocardial infarction (MI), carotid artery dissection, or arterial thrombosis.8
Treatment
Treatment and management for carotid artery stenosis is focused on combined effort with the patient’s PCP and other specialists, including cardiologist, neurologist, and vascular surgeons.11 Treatment of comorbid conditions, education on healthy lifestyle, and smoking cessation are all imperative to the patient’s well-being. Managing ocular sequelae is based on specific findings and can include intravitreal antivascular edothelial growth factor or steroidal injections, pan retinal photocoagulation, or hypotensive drops.6,7
Restoration of arterial perfusion pressure is the main goal of treatment, and this can be done through CEA or carotid artery stents. Surgical intervention by CEA is determined based on each patient and his or her overall health. A full cardiac workup is required due to surgical risks. The North American Symptomatic Carotid Endarterectomy Trial evaluated symptomatic stenosis and the effectiveness of surgical intervention on stroke prevention. The trial reported that CEA was beneficial in symptomatic patients with 55% to 99% stenosis and especially in those with higher grade stenosis (> 70% up to 95%).5,7,8,12 With regard to asymptomatic patients with high-grade stenosis, CEA has been found to reduce the risk of stroke if there is at least 60% stenosis.5,7,8
Carotid artery stents can be used as an alternative when CEA is not effective or contraindicated due to a history of previous CEA, neck radiation, unstable angina, congestive heart failure, or recent MI.5,7,8 Neither CEA nor stenting is considered effective in complete occlusions due to the high risk of thromboembolism formation.5,7,8
Conclusion
Hypoperfusion retinopathy describes posterior retinal findings that occur secondary to poor arterial perfusion caused by carotid occlusive disease. Early intervention and restoration of this pressure can prevent the risk of developing a more serious condition characterized by a panocular ischemia called OIS. Unlike hypoperfusion retinopathy, OIS also includes anterior segment findings such as iris neovascularization, which may lead to neovascular glaucoma, whereas hypoperfusion retinopathy is localized to the posterior pole. Patients that develop OIS are at a 40% risk of mortality within 5 years due to poor overall health. Understanding the patient’s signs and symptoms can aid in the diagnosis of both conditions. Collaborative management with the patient’s PCP and specialists in treating comorbid conditions is vital to the patients’ well-being.
1. Brown GC, Magargal LE. The ocular ischemic syndrome. Int Ophthalmol. 1988;11(4):239-251.
2. Dahlman AH, McCormack D, Harrison RJ. Bilateral hypoperfuion retinopathy. J R Soc Med. 2001; 94(6):298-299.
3. Dugan JD Jr, Green WR. Ophthalmologic manifestations of carotid occlusive disease. Eye (Lond). 1991;5(pt 2):226-238.
4. Klijn CJ, Kappelle LJ, Tulleken CAF, van Gijn J. Symptomatic carotid artery occlusion. A reappraisal of hemodynamic factors. Stroke. 1997;28(10):2084-2093.
5. McCrary JA III. Venous stasis retinopathy of stenotic or occlusive caroid origin. J Clin Neuroophthalmol. 1989;9(3):195-199.
6. Sanborn GE, Magargal LE. Arterial obstructive disease of the eye. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 14.
7. Terelak-Borys B, Skonieczna K, Grabska-Liberek I. Ocular ischemic syndrome–a systematic review. Med Sci Monit. 2012;18(8):RA138-RA144.
8. Atebara NH, Brown GC. The ocular ischemic syndrome. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 12.
9. Ho AC, Lieb WE, Flaharty PM, et al. Color Doppler imaging of the ocular ischaemic syndrome. Ophthalmology. 1992;99(9):1453-1462.
10. Kahn M, Green WR, Knox DL, Miller NR. Ocular features of carotid occlusive disease. Retina. 1986;6(4):239-252.
11. Mizener JB, Podhajsky P, Hayreh SS. Ocular ischemic syndrome. Ophthalmology. 1997;104(5):859-864.
12. Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke. 1999;30(9):1751-1758.
1. Brown GC, Magargal LE. The ocular ischemic syndrome. Int Ophthalmol. 1988;11(4):239-251.
2. Dahlman AH, McCormack D, Harrison RJ. Bilateral hypoperfuion retinopathy. J R Soc Med. 2001; 94(6):298-299.
3. Dugan JD Jr, Green WR. Ophthalmologic manifestations of carotid occlusive disease. Eye (Lond). 1991;5(pt 2):226-238.
4. Klijn CJ, Kappelle LJ, Tulleken CAF, van Gijn J. Symptomatic carotid artery occlusion. A reappraisal of hemodynamic factors. Stroke. 1997;28(10):2084-2093.
5. McCrary JA III. Venous stasis retinopathy of stenotic or occlusive caroid origin. J Clin Neuroophthalmol. 1989;9(3):195-199.
6. Sanborn GE, Magargal LE. Arterial obstructive disease of the eye. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 14.
7. Terelak-Borys B, Skonieczna K, Grabska-Liberek I. Ocular ischemic syndrome–a systematic review. Med Sci Monit. 2012;18(8):RA138-RA144.
8. Atebara NH, Brown GC. The ocular ischemic syndrome. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 12.
9. Ho AC, Lieb WE, Flaharty PM, et al. Color Doppler imaging of the ocular ischaemic syndrome. Ophthalmology. 1992;99(9):1453-1462.
10. Kahn M, Green WR, Knox DL, Miller NR. Ocular features of carotid occlusive disease. Retina. 1986;6(4):239-252.
11. Mizener JB, Podhajsky P, Hayreh SS. Ocular ischemic syndrome. Ophthalmology. 1997;104(5):859-864.
12. Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke. 1999;30(9):1751-1758.
Systemic Hypothermia as Treatment for an Acute Cervical Spinal Cord Injury in a Professional Football Player: 9-Year Follow-Up
Take-Home Points
- Importance of on-field management.
- Preseason drilling of spinal injury management.
- Early and rapid intervention.
- Possible benefit of moderate systemic hypothermia as treatment for acute cervical injury.
In 2010, we reported the case of a professional American football player who sustained a complete cervical spinal cord injury (SCI) while tackling an opposing player.1 He received prompt medical and surgical care based on then-current recommendations, but was also treated with systemic hypothermia soon after his injury. Although systemic hypothermia had been used in the management of other neurologic injuries at that time, it had not been used in humans with acute SCI, except as described in 2 case reports.2,3 However, Dietrich4 described early emerging animal data on the efficacy of systemic hypothermia for acute SCI. We now provide a clinical update on our patient, who provided written informed consent for print and electronic publication of this case report.
Case Report
During a National Football League game, the player sustained a C3–C4 fracture-dislocation after a helmet-to-helmet hit on an opposing player. He fell face down on the ground and did not move. The team’s physician and trainer rushed to the player’s side, immediately assessed him, and initiated the emergency spinal resuscitation protocol.
As per protocol, the assigned team leader took charge of managing the player’s head to maintain in-line traction with the helmet in place until the head was secured in place on a backboard designed to accommodate the helmet. 
Complete motor paralysis and sensory loss (American Spinal Injury Association [ASIA] level A) were noted below the clavicles during physical examination by the head athletic trainer and 2 independent physicians, and by self-report. 

On arrival at the hospital, the patient had a core temperature of 98°F, which is substantially lower than the average core temperature (≤101.7°F) of an active football player.6He had a normal level of consciousness and normal cranial nerve function but remained without any voluntary motor function in the extremities and still had no sensation below the clavicles, except crude pressure sensation in one hand while in the emergency department. After the helmet and shoulder pads were removed, per National Athletic Trainers’ Association (NATA) protocol7 (Figure 2), he was stabilized, and a hard cervical collar was placed. A lateral radiograph (Figure 4) showed a C3–C4 facet dislocation with about 46% anterior translation of C3 on C4 and obvious disruption of the facets. 
About 3 hours after injury, the patient was taken to the operating room. Although closed reduction improved alignment dramatically, it failed to completely reduce the dislocated left C3–C4 facet. An hour later, anterior C3–C4 discectomy was performed from the front with instrumented anterior interbody fusion. This was immediately followed by posterior decompressive laminectomy, bilateral facet reduction, and fusion with instrumentation. Surgery was completed within about 4 hours, almost exactly 7 hours after injury. Anesthesia records indicated a core temperature range of 94.1°F to 95.3°F with passive cooling during surgery. CT and MRI performed within 4 hours after surgery showed excellent cord decompression.
The next morning, about 14.5 hours after injury, the patient demonstrated a flicker of the adductor muscles of the lower extremities. An examination an hour later revealed 1/5 quadriceps, 2/5 adductors, and 1/5 gastrocnemius/soleus. A nurse’s hourly examinations and the surgeon’s repeat examinations revealed no other motor function. Sensory function was more difficult to evaluate because of sedation, but rudimentary sensation was noted throughout the lower extremities, and proprioception and vibratory sensation were noted as well. With passive cooling, it was difficult to consistently maintain moderate hypothermia; the patient’s core temperature ranged from 94.8°F to 98.8°F by 6:00 a.m. Therefore, the decision was made to place a Cordis sheath in the left femoral vein and introduce an intra-vena cava cooling catheter through it. This catheter was highly effective in maintaining the patient’s temperature at about 92.5°F.
Over the next 36 hours, the patient demonstrated increased motor activity in the upper and lower extremities: 1/5 biceps, 2-3/5 triceps, 3/5 quadriceps. He was slowly rewarmed and, on postoperative day 3, extubated. 
At 2 years, the patient underwent another anterior-only cervical procedure: The inferior adjacent segment (C4–C5) was fused because of neck pain and deformity. 
With respect to the original injury and the evolution in cord appearance, the patient had solid arthrodesis from C3–C5 with instrumentation in good position. There was evidence of loss of lordosis at C5–C6 with disk dessication and broad-based bulging. The spinal cord had evidence of myelomalacia; this was noted when the patient was in rehabilitation, 1 month after injury. The 2-cm × 11-mm area of myelomalacia was directly posterior to the fused C3–C4 interval (original MRI, Figure 5; 2-week MRI, Figure 6).
Conclusion
At the time this player was injured, use of systemic hypothermia with standard therapy for acute SCI was unique and controversial. Since then, smaller randomized human studies have described the tolerable safety profile, efficacy, and potential benefits of this intervention in acute SCI in humans.8-10 Now, modest systemic hypothermia can be one of many tools considered in the treatment of acute SCI. Before it can become the standard of care, however, additional larger prospective randomized studies need to be completed.
Am J Orthop. 2017;46(2):E79-E82. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Cappuccino A, Bisson LJ, Carpenter B, Marzo J, Dietrich WD 3rd, Cappuccino H. The use of systemic hypothermia for the treatment of an acute cervical spinal cord injury in a professional football player. Spine. 2010;35(2):E57-E62.
2. Goldstein J. Lowering body temp shows promise for trauma treatment. Spinal Cord Injury Information Pages news blog. http://www.sci-info-pages.com/2006/05/lowering-body-temp-shows-promise-for.html. Published May 3, 2006. Accessed March 19, 2009.
3. Hartemink KJ, Wisselink W, Rauwerda JA, Girbes AR, Polderman KH. Novel applications of therapeutic hypothermia: report of three cases. Crit Care. 2004;8(5):R343-R346.
4. Dietrich WD. Presidential address presented at: 34th Annual Meeting of the Cervical Spine Research Society; November 30, 2006; Palm Beach, FL.
5. Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405-1411.
6. Horodyski MB, LuCante K, Escobar E, et al. Intermittent Cool, Dry Air Underneath Football Shoulder Pads Assists in Temperature Homeostasis. In: The American Orthopaedic Society for Sports Medicine Proceedings 2008; 87-88.
7. Kleiner DM, Almquist JL, Bailes J, et al; Inter-Association Task Force for Appropriate Care of the Spine-Injured Athlete. Prehospital Care of the Spine-Injured Athlete. Dallas, TX: National Athletic Trainers’ Association; 2001. http://www.msata.org/Resources/Documents/PreHospitalCare4SpineInjuredAthlete.pdf. Published March 2001. Accessed January 10, 2017.
8. Dididze M, Green BA, Dietrich WD, Vanni S, Wang MY, Levi AD. Systemic hypothermia in acute cervical spinal cord injury: a case-controlled study. Spinal Cord. 2013;51(5):395-400.
9. Levi AD, Casella G, Green BA, et al. Clinical outcomes using modest intravascular hypothermia after acute cervical spinal cord injury. Neurosurgery. 2010;66(4):670-677.
10. Levi AD, Green BA, Wang MY, et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26(3):407-415.
Take-Home Points
- Importance of on-field management.
- Preseason drilling of spinal injury management.
- Early and rapid intervention.
- Possible benefit of moderate systemic hypothermia as treatment for acute cervical injury.
In 2010, we reported the case of a professional American football player who sustained a complete cervical spinal cord injury (SCI) while tackling an opposing player.1 He received prompt medical and surgical care based on then-current recommendations, but was also treated with systemic hypothermia soon after his injury. Although systemic hypothermia had been used in the management of other neurologic injuries at that time, it had not been used in humans with acute SCI, except as described in 2 case reports.2,3 However, Dietrich4 described early emerging animal data on the efficacy of systemic hypothermia for acute SCI. We now provide a clinical update on our patient, who provided written informed consent for print and electronic publication of this case report.
Case Report
During a National Football League game, the player sustained a C3–C4 fracture-dislocation after a helmet-to-helmet hit on an opposing player. He fell face down on the ground and did not move. The team’s physician and trainer rushed to the player’s side, immediately assessed him, and initiated the emergency spinal resuscitation protocol.
As per protocol, the assigned team leader took charge of managing the player’s head to maintain in-line traction with the helmet in place until the head was secured in place on a backboard designed to accommodate the helmet.
Complete motor paralysis and sensory loss (American Spinal Injury Association [ASIA] level A) were noted below the clavicles during physical examination by the head athletic trainer and 2 independent physicians, and by self-report.
On arrival at the hospital, the patient had a core temperature of 98°F, which is substantially lower than the average core temperature (≤101.7°F) of an active football player.6He had a normal level of consciousness and normal cranial nerve function but remained without any voluntary motor function in the extremities and still had no sensation below the clavicles, except crude pressure sensation in one hand while in the emergency department. After the helmet and shoulder pads were removed, per National Athletic Trainers’ Association (NATA) protocol7 (Figure 2), he was stabilized, and a hard cervical collar was placed. A lateral radiograph (Figure 4) showed a C3–C4 facet dislocation with about 46% anterior translation of C3 on C4 and obvious disruption of the facets.
About 3 hours after injury, the patient was taken to the operating room. Although closed reduction improved alignment dramatically, it failed to completely reduce the dislocated left C3–C4 facet. An hour later, anterior C3–C4 discectomy was performed from the front with instrumented anterior interbody fusion. This was immediately followed by posterior decompressive laminectomy, bilateral facet reduction, and fusion with instrumentation. Surgery was completed within about 4 hours, almost exactly 7 hours after injury. Anesthesia records indicated a core temperature range of 94.1°F to 95.3°F with passive cooling during surgery. CT and MRI performed within 4 hours after surgery showed excellent cord decompression.
The next morning, about 14.5 hours after injury, the patient demonstrated a flicker of the adductor muscles of the lower extremities. An examination an hour later revealed 1/5 quadriceps, 2/5 adductors, and 1/5 gastrocnemius/soleus. A nurse’s hourly examinations and the surgeon’s repeat examinations revealed no other motor function. Sensory function was more difficult to evaluate because of sedation, but rudimentary sensation was noted throughout the lower extremities, and proprioception and vibratory sensation were noted as well. With passive cooling, it was difficult to consistently maintain moderate hypothermia; the patient’s core temperature ranged from 94.8°F to 98.8°F by 6:00 a.m. Therefore, the decision was made to place a Cordis sheath in the left femoral vein and introduce an intra-vena cava cooling catheter through it. This catheter was highly effective in maintaining the patient’s temperature at about 92.5°F.
Over the next 36 hours, the patient demonstrated increased motor activity in the upper and lower extremities: 1/5 biceps, 2-3/5 triceps, 3/5 quadriceps. He was slowly rewarmed and, on postoperative day 3, extubated.
At 2 years, the patient underwent another anterior-only cervical procedure: The inferior adjacent segment (C4–C5) was fused because of neck pain and deformity.
With respect to the original injury and the evolution in cord appearance, the patient had solid arthrodesis from C3–C5 with instrumentation in good position. There was evidence of loss of lordosis at C5–C6 with disk dessication and broad-based bulging. The spinal cord had evidence of myelomalacia; this was noted when the patient was in rehabilitation, 1 month after injury. The 2-cm × 11-mm area of myelomalacia was directly posterior to the fused C3–C4 interval (original MRI, Figure 5; 2-week MRI, Figure 6).
Conclusion
At the time this player was injured, use of systemic hypothermia with standard therapy for acute SCI was unique and controversial. Since then, smaller randomized human studies have described the tolerable safety profile, efficacy, and potential benefits of this intervention in acute SCI in humans.8-10 Now, modest systemic hypothermia can be one of many tools considered in the treatment of acute SCI. Before it can become the standard of care, however, additional larger prospective randomized studies need to be completed.
Am J Orthop. 2017;46(2):E79-E82. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Importance of on-field management.
- Preseason drilling of spinal injury management.
- Early and rapid intervention.
- Possible benefit of moderate systemic hypothermia as treatment for acute cervical injury.
In 2010, we reported the case of a professional American football player who sustained a complete cervical spinal cord injury (SCI) while tackling an opposing player.1 He received prompt medical and surgical care based on then-current recommendations, but was also treated with systemic hypothermia soon after his injury. Although systemic hypothermia had been used in the management of other neurologic injuries at that time, it had not been used in humans with acute SCI, except as described in 2 case reports.2,3 However, Dietrich4 described early emerging animal data on the efficacy of systemic hypothermia for acute SCI. We now provide a clinical update on our patient, who provided written informed consent for print and electronic publication of this case report.
Case Report
During a National Football League game, the player sustained a C3–C4 fracture-dislocation after a helmet-to-helmet hit on an opposing player. He fell face down on the ground and did not move. The team’s physician and trainer rushed to the player’s side, immediately assessed him, and initiated the emergency spinal resuscitation protocol.
As per protocol, the assigned team leader took charge of managing the player’s head to maintain in-line traction with the helmet in place until the head was secured in place on a backboard designed to accommodate the helmet.
Complete motor paralysis and sensory loss (American Spinal Injury Association [ASIA] level A) were noted below the clavicles during physical examination by the head athletic trainer and 2 independent physicians, and by self-report.
On arrival at the hospital, the patient had a core temperature of 98°F, which is substantially lower than the average core temperature (≤101.7°F) of an active football player.6He had a normal level of consciousness and normal cranial nerve function but remained without any voluntary motor function in the extremities and still had no sensation below the clavicles, except crude pressure sensation in one hand while in the emergency department. After the helmet and shoulder pads were removed, per National Athletic Trainers’ Association (NATA) protocol7 (Figure 2), he was stabilized, and a hard cervical collar was placed. A lateral radiograph (Figure 4) showed a C3–C4 facet dislocation with about 46% anterior translation of C3 on C4 and obvious disruption of the facets.
About 3 hours after injury, the patient was taken to the operating room. Although closed reduction improved alignment dramatically, it failed to completely reduce the dislocated left C3–C4 facet. An hour later, anterior C3–C4 discectomy was performed from the front with instrumented anterior interbody fusion. This was immediately followed by posterior decompressive laminectomy, bilateral facet reduction, and fusion with instrumentation. Surgery was completed within about 4 hours, almost exactly 7 hours after injury. Anesthesia records indicated a core temperature range of 94.1°F to 95.3°F with passive cooling during surgery. CT and MRI performed within 4 hours after surgery showed excellent cord decompression.
The next morning, about 14.5 hours after injury, the patient demonstrated a flicker of the adductor muscles of the lower extremities. An examination an hour later revealed 1/5 quadriceps, 2/5 adductors, and 1/5 gastrocnemius/soleus. A nurse’s hourly examinations and the surgeon’s repeat examinations revealed no other motor function. Sensory function was more difficult to evaluate because of sedation, but rudimentary sensation was noted throughout the lower extremities, and proprioception and vibratory sensation were noted as well. With passive cooling, it was difficult to consistently maintain moderate hypothermia; the patient’s core temperature ranged from 94.8°F to 98.8°F by 6:00 a.m. Therefore, the decision was made to place a Cordis sheath in the left femoral vein and introduce an intra-vena cava cooling catheter through it. This catheter was highly effective in maintaining the patient’s temperature at about 92.5°F.
Over the next 36 hours, the patient demonstrated increased motor activity in the upper and lower extremities: 1/5 biceps, 2-3/5 triceps, 3/5 quadriceps. He was slowly rewarmed and, on postoperative day 3, extubated.
At 2 years, the patient underwent another anterior-only cervical procedure: The inferior adjacent segment (C4–C5) was fused because of neck pain and deformity.
With respect to the original injury and the evolution in cord appearance, the patient had solid arthrodesis from C3–C5 with instrumentation in good position. There was evidence of loss of lordosis at C5–C6 with disk dessication and broad-based bulging. The spinal cord had evidence of myelomalacia; this was noted when the patient was in rehabilitation, 1 month after injury. The 2-cm × 11-mm area of myelomalacia was directly posterior to the fused C3–C4 interval (original MRI, Figure 5; 2-week MRI, Figure 6).
Conclusion
At the time this player was injured, use of systemic hypothermia with standard therapy for acute SCI was unique and controversial. Since then, smaller randomized human studies have described the tolerable safety profile, efficacy, and potential benefits of this intervention in acute SCI in humans.8-10 Now, modest systemic hypothermia can be one of many tools considered in the treatment of acute SCI. Before it can become the standard of care, however, additional larger prospective randomized studies need to be completed.
Am J Orthop. 2017;46(2):E79-E82. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Cappuccino A, Bisson LJ, Carpenter B, Marzo J, Dietrich WD 3rd, Cappuccino H. The use of systemic hypothermia for the treatment of an acute cervical spinal cord injury in a professional football player. Spine. 2010;35(2):E57-E62.
2. Goldstein J. Lowering body temp shows promise for trauma treatment. Spinal Cord Injury Information Pages news blog. http://www.sci-info-pages.com/2006/05/lowering-body-temp-shows-promise-for.html. Published May 3, 2006. Accessed March 19, 2009.
3. Hartemink KJ, Wisselink W, Rauwerda JA, Girbes AR, Polderman KH. Novel applications of therapeutic hypothermia: report of three cases. Crit Care. 2004;8(5):R343-R346.
4. Dietrich WD. Presidential address presented at: 34th Annual Meeting of the Cervical Spine Research Society; November 30, 2006; Palm Beach, FL.
5. Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405-1411.
6. Horodyski MB, LuCante K, Escobar E, et al. Intermittent Cool, Dry Air Underneath Football Shoulder Pads Assists in Temperature Homeostasis. In: The American Orthopaedic Society for Sports Medicine Proceedings 2008; 87-88.
7. Kleiner DM, Almquist JL, Bailes J, et al; Inter-Association Task Force for Appropriate Care of the Spine-Injured Athlete. Prehospital Care of the Spine-Injured Athlete. Dallas, TX: National Athletic Trainers’ Association; 2001. http://www.msata.org/Resources/Documents/PreHospitalCare4SpineInjuredAthlete.pdf. Published March 2001. Accessed January 10, 2017.
8. Dididze M, Green BA, Dietrich WD, Vanni S, Wang MY, Levi AD. Systemic hypothermia in acute cervical spinal cord injury: a case-controlled study. Spinal Cord. 2013;51(5):395-400.
9. Levi AD, Casella G, Green BA, et al. Clinical outcomes using modest intravascular hypothermia after acute cervical spinal cord injury. Neurosurgery. 2010;66(4):670-677.
10. Levi AD, Green BA, Wang MY, et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26(3):407-415.
1. Cappuccino A, Bisson LJ, Carpenter B, Marzo J, Dietrich WD 3rd, Cappuccino H. The use of systemic hypothermia for the treatment of an acute cervical spinal cord injury in a professional football player. Spine. 2010;35(2):E57-E62.
2. Goldstein J. Lowering body temp shows promise for trauma treatment. Spinal Cord Injury Information Pages news blog. http://www.sci-info-pages.com/2006/05/lowering-body-temp-shows-promise-for.html. Published May 3, 2006. Accessed March 19, 2009.
3. Hartemink KJ, Wisselink W, Rauwerda JA, Girbes AR, Polderman KH. Novel applications of therapeutic hypothermia: report of three cases. Crit Care. 2004;8(5):R343-R346.
4. Dietrich WD. Presidential address presented at: 34th Annual Meeting of the Cervical Spine Research Society; November 30, 2006; Palm Beach, FL.
5. Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405-1411.
6. Horodyski MB, LuCante K, Escobar E, et al. Intermittent Cool, Dry Air Underneath Football Shoulder Pads Assists in Temperature Homeostasis. In: The American Orthopaedic Society for Sports Medicine Proceedings 2008; 87-88.
7. Kleiner DM, Almquist JL, Bailes J, et al; Inter-Association Task Force for Appropriate Care of the Spine-Injured Athlete. Prehospital Care of the Spine-Injured Athlete. Dallas, TX: National Athletic Trainers’ Association; 2001. http://www.msata.org/Resources/Documents/PreHospitalCare4SpineInjuredAthlete.pdf. Published March 2001. Accessed January 10, 2017.
8. Dididze M, Green BA, Dietrich WD, Vanni S, Wang MY, Levi AD. Systemic hypothermia in acute cervical spinal cord injury: a case-controlled study. Spinal Cord. 2013;51(5):395-400.
9. Levi AD, Casella G, Green BA, et al. Clinical outcomes using modest intravascular hypothermia after acute cervical spinal cord injury. Neurosurgery. 2010;66(4):670-677.
10. Levi AD, Green BA, Wang MY, et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26(3):407-415.
