User login
Syncope during a pharmacologic nuclear stress test
A 60-year-old woman was referred for pharmacologic nuclear stress testing before treatment for breast cancer. She had hypertension, diabetes mellitus, coronary artery disease, and a remote history of stroke, and she was taking clonidine (Catapres), labetalol (Normodyne, Trandate), furosemide (Lasix), hydralazine, valsartan (Diovan), insulin, and the aspirin-dipyridamole combination Aggrenox. Her vital signs and electrocardiogram before the stress test were normal.
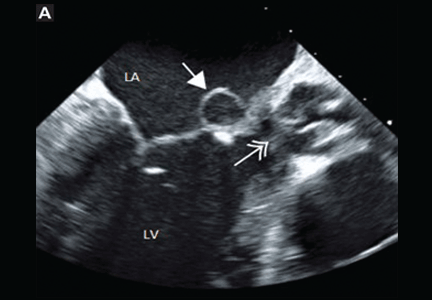
The stress test was started with a standard protocol of adenosine (Adenoscan) infused intravenously over 4 minutes. For the first 2 minutes, she was stable and had no symptoms, but then sinus pauses and second-degree atrioventricular block type 2 developed, after which her heart stopped beating (Figure 1). The infusion was immediately stopped, but she became unresponsive and remained pulseless.
Cardiopulmonary resuscitation was started, aminophylline 100 mg was given intravenously, and she regained a pulse and blood pressure within a few minutes. She was then transferred to the emergency room, where she returned to her baseline clinical and neurologic status without symptoms.
AN UNRECOGNIZED DRUG INTERACTION
Asystole occurred in this patient because of the interaction of intravenous adenosine with the dipyridamole in the medication Aggrenox. Although adenosine, given during pharmacologic stress testing, is known to interact with various medications, the potential for this interaction may be overlooked if the culprit is present in a combination drug. Aggrenox is commonly given for secondary stroke prevention and should be discontinued before pharmacologic nuclear stress testing.
Pharmacologic stress testing involves two commonly used stress agents, adenosine and regadenoson (Lexiscan), which cause coronary vasodilation through their action on A2A receptors in the heart. Coronary vasodilation results in flow heterogeneity in the region of a stenotic artery, which can be detected with nuclear perfusion agents. In addition, adenosine has a short-lived effect on the A1 receptors that block atrioventricular conduction.1
Dipyridamole (Persantine) is contraindicated when either adenosine or regadenoson is used. Dipyridamole enhances the effect of exogenous and endogenous adenosine by inhibiting its uptake by cardiac cells, thus enhancing the action of these coronary vasodilators.2 Atrioventricular block is common during adenosine stress testing but is transient because adenosine has a short half-life (< 10 seconds), and complete heart block or asystole, as seen in this patient, is rare. Giving intravenous adenosine or regadenoson to patients on dipyridamole may have a marked effect on adenosine receptors, so that profound bradycardia and even asystole leading to cardiac collapse may occur. No data are available on the specific interaction of dipyridamole and regadenoson.
Even though the pharmacodynamics of the interaction between dipyridamole and adenosine are known,3 few reports are available detailing serious adverse events. The contraindication to pharmacologic stress testing in patients taking dipyridamole is noted in the American Society of Nuclear Cardiology Guidelines for stress protocols,4 which advise discontinuing dipyridamole-containing drugs at least 48 hours before the use of adenosine or regadenoson. Similarly, the American Heart Association guidelines5 for the management of supraventricular tachycardia recommend an initial dose of 3 mg of adenosine rather than 6 mg in patients who have been taking dipyridamole.
The dose of aminophylline for reversing the adverse effects of adenosine or regadenoson is 50 to 250 mg intravenously over 30 to 60 seconds. But since these adverse effects are short-lived once the infusion is stopped, aminophylline is usually given only if the adverse effects are severe, as in this patient.
Pharmacologic nuclear stress testing with adenosine receptor agonists (eg, adenosine or regadenoson) is contraindicated in patients taking dipyridamole or the combination pill Aggrenox because of the potential for profound bradyarrhythmias or asystole.
- Zoghbi GJ, Iskandrian AE. Selective adenosine agonists and myocardial perfusion imaging. J Nucl Cardiol 2012; 19:126–141.
- Lerman BB, Wesley RC, Belardinelli L. Electrophysiologic effects of dipyridamole on atrioventricular nodal conduction and supraventricular tachycardia. Role of endogenous adenosine. Circulation 1989; 80:1536–1543.
- Biaggioni I, Onrot J, Hollister AS, Robertson D. Cardiovascular effects of adenosine infusion in man and their modulation by dipyridamole. Life Sci 1986; 39:2229–2236.
- Henzlova MJ, Cerqueira MD, Mahmarian JJ, Yao SS; Quality Assurance Committee of the American Society of Nuclear Cardiology. Stress protocols and tracers. J Nucl Cardiol 2006; 13:e80–e90.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Part 7.3: management of symptomatic bradycardia and tachycardia. Circulation 2005; 112(suppl 24):IV67–IV77.
A 60-year-old woman was referred for pharmacologic nuclear stress testing before treatment for breast cancer. She had hypertension, diabetes mellitus, coronary artery disease, and a remote history of stroke, and she was taking clonidine (Catapres), labetalol (Normodyne, Trandate), furosemide (Lasix), hydralazine, valsartan (Diovan), insulin, and the aspirin-dipyridamole combination Aggrenox. Her vital signs and electrocardiogram before the stress test were normal.
The stress test was started with a standard protocol of adenosine (Adenoscan) infused intravenously over 4 minutes. For the first 2 minutes, she was stable and had no symptoms, but then sinus pauses and second-degree atrioventricular block type 2 developed, after which her heart stopped beating (Figure 1). The infusion was immediately stopped, but she became unresponsive and remained pulseless.
Cardiopulmonary resuscitation was started, aminophylline 100 mg was given intravenously, and she regained a pulse and blood pressure within a few minutes. She was then transferred to the emergency room, where she returned to her baseline clinical and neurologic status without symptoms.
AN UNRECOGNIZED DRUG INTERACTION
Asystole occurred in this patient because of the interaction of intravenous adenosine with the dipyridamole in the medication Aggrenox. Although adenosine, given during pharmacologic stress testing, is known to interact with various medications, the potential for this interaction may be overlooked if the culprit is present in a combination drug. Aggrenox is commonly given for secondary stroke prevention and should be discontinued before pharmacologic nuclear stress testing.
Pharmacologic stress testing involves two commonly used stress agents, adenosine and regadenoson (Lexiscan), which cause coronary vasodilation through their action on A2A receptors in the heart. Coronary vasodilation results in flow heterogeneity in the region of a stenotic artery, which can be detected with nuclear perfusion agents. In addition, adenosine has a short-lived effect on the A1 receptors that block atrioventricular conduction.1
Dipyridamole (Persantine) is contraindicated when either adenosine or regadenoson is used. Dipyridamole enhances the effect of exogenous and endogenous adenosine by inhibiting its uptake by cardiac cells, thus enhancing the action of these coronary vasodilators.2 Atrioventricular block is common during adenosine stress testing but is transient because adenosine has a short half-life (< 10 seconds), and complete heart block or asystole, as seen in this patient, is rare. Giving intravenous adenosine or regadenoson to patients on dipyridamole may have a marked effect on adenosine receptors, so that profound bradycardia and even asystole leading to cardiac collapse may occur. No data are available on the specific interaction of dipyridamole and regadenoson.
Even though the pharmacodynamics of the interaction between dipyridamole and adenosine are known,3 few reports are available detailing serious adverse events. The contraindication to pharmacologic stress testing in patients taking dipyridamole is noted in the American Society of Nuclear Cardiology Guidelines for stress protocols,4 which advise discontinuing dipyridamole-containing drugs at least 48 hours before the use of adenosine or regadenoson. Similarly, the American Heart Association guidelines5 for the management of supraventricular tachycardia recommend an initial dose of 3 mg of adenosine rather than 6 mg in patients who have been taking dipyridamole.
The dose of aminophylline for reversing the adverse effects of adenosine or regadenoson is 50 to 250 mg intravenously over 30 to 60 seconds. But since these adverse effects are short-lived once the infusion is stopped, aminophylline is usually given only if the adverse effects are severe, as in this patient.
Pharmacologic nuclear stress testing with adenosine receptor agonists (eg, adenosine or regadenoson) is contraindicated in patients taking dipyridamole or the combination pill Aggrenox because of the potential for profound bradyarrhythmias or asystole.
A 60-year-old woman was referred for pharmacologic nuclear stress testing before treatment for breast cancer. She had hypertension, diabetes mellitus, coronary artery disease, and a remote history of stroke, and she was taking clonidine (Catapres), labetalol (Normodyne, Trandate), furosemide (Lasix), hydralazine, valsartan (Diovan), insulin, and the aspirin-dipyridamole combination Aggrenox. Her vital signs and electrocardiogram before the stress test were normal.
The stress test was started with a standard protocol of adenosine (Adenoscan) infused intravenously over 4 minutes. For the first 2 minutes, she was stable and had no symptoms, but then sinus pauses and second-degree atrioventricular block type 2 developed, after which her heart stopped beating (Figure 1). The infusion was immediately stopped, but she became unresponsive and remained pulseless.
Cardiopulmonary resuscitation was started, aminophylline 100 mg was given intravenously, and she regained a pulse and blood pressure within a few minutes. She was then transferred to the emergency room, where she returned to her baseline clinical and neurologic status without symptoms.
AN UNRECOGNIZED DRUG INTERACTION
Asystole occurred in this patient because of the interaction of intravenous adenosine with the dipyridamole in the medication Aggrenox. Although adenosine, given during pharmacologic stress testing, is known to interact with various medications, the potential for this interaction may be overlooked if the culprit is present in a combination drug. Aggrenox is commonly given for secondary stroke prevention and should be discontinued before pharmacologic nuclear stress testing.
Pharmacologic stress testing involves two commonly used stress agents, adenosine and regadenoson (Lexiscan), which cause coronary vasodilation through their action on A2A receptors in the heart. Coronary vasodilation results in flow heterogeneity in the region of a stenotic artery, which can be detected with nuclear perfusion agents. In addition, adenosine has a short-lived effect on the A1 receptors that block atrioventricular conduction.1
Dipyridamole (Persantine) is contraindicated when either adenosine or regadenoson is used. Dipyridamole enhances the effect of exogenous and endogenous adenosine by inhibiting its uptake by cardiac cells, thus enhancing the action of these coronary vasodilators.2 Atrioventricular block is common during adenosine stress testing but is transient because adenosine has a short half-life (< 10 seconds), and complete heart block or asystole, as seen in this patient, is rare. Giving intravenous adenosine or regadenoson to patients on dipyridamole may have a marked effect on adenosine receptors, so that profound bradycardia and even asystole leading to cardiac collapse may occur. No data are available on the specific interaction of dipyridamole and regadenoson.
Even though the pharmacodynamics of the interaction between dipyridamole and adenosine are known,3 few reports are available detailing serious adverse events. The contraindication to pharmacologic stress testing in patients taking dipyridamole is noted in the American Society of Nuclear Cardiology Guidelines for stress protocols,4 which advise discontinuing dipyridamole-containing drugs at least 48 hours before the use of adenosine or regadenoson. Similarly, the American Heart Association guidelines5 for the management of supraventricular tachycardia recommend an initial dose of 3 mg of adenosine rather than 6 mg in patients who have been taking dipyridamole.
The dose of aminophylline for reversing the adverse effects of adenosine or regadenoson is 50 to 250 mg intravenously over 30 to 60 seconds. But since these adverse effects are short-lived once the infusion is stopped, aminophylline is usually given only if the adverse effects are severe, as in this patient.
Pharmacologic nuclear stress testing with adenosine receptor agonists (eg, adenosine or regadenoson) is contraindicated in patients taking dipyridamole or the combination pill Aggrenox because of the potential for profound bradyarrhythmias or asystole.
- Zoghbi GJ, Iskandrian AE. Selective adenosine agonists and myocardial perfusion imaging. J Nucl Cardiol 2012; 19:126–141.
- Lerman BB, Wesley RC, Belardinelli L. Electrophysiologic effects of dipyridamole on atrioventricular nodal conduction and supraventricular tachycardia. Role of endogenous adenosine. Circulation 1989; 80:1536–1543.
- Biaggioni I, Onrot J, Hollister AS, Robertson D. Cardiovascular effects of adenosine infusion in man and their modulation by dipyridamole. Life Sci 1986; 39:2229–2236.
- Henzlova MJ, Cerqueira MD, Mahmarian JJ, Yao SS; Quality Assurance Committee of the American Society of Nuclear Cardiology. Stress protocols and tracers. J Nucl Cardiol 2006; 13:e80–e90.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Part 7.3: management of symptomatic bradycardia and tachycardia. Circulation 2005; 112(suppl 24):IV67–IV77.
- Zoghbi GJ, Iskandrian AE. Selective adenosine agonists and myocardial perfusion imaging. J Nucl Cardiol 2012; 19:126–141.
- Lerman BB, Wesley RC, Belardinelli L. Electrophysiologic effects of dipyridamole on atrioventricular nodal conduction and supraventricular tachycardia. Role of endogenous adenosine. Circulation 1989; 80:1536–1543.
- Biaggioni I, Onrot J, Hollister AS, Robertson D. Cardiovascular effects of adenosine infusion in man and their modulation by dipyridamole. Life Sci 1986; 39:2229–2236.
- Henzlova MJ, Cerqueira MD, Mahmarian JJ, Yao SS; Quality Assurance Committee of the American Society of Nuclear Cardiology. Stress protocols and tracers. J Nucl Cardiol 2006; 13:e80–e90.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Part 7.3: management of symptomatic bradycardia and tachycardia. Circulation 2005; 112(suppl 24):IV67–IV77.
An intravenous drug user with persistent dyspnea and lung infiltrates
A 58-year-old-man with a history of intravenous drug abuse, chronic hepatitis C, and anxiety presented to our emergency department twice in 4 weeks with progressive dyspnea and night sweats. He was a nonsmoker and had been an electrician for 15 years.
The first time he came in, chest radiography revealed bilateral reticulonodular infiltrates in the lung bases. He was treated with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax) for presumed community-acquired pneumonia and was then sent home on a 10-day course of oral amoxicillin-clavulanate (Augmentin). The antibiotics did not improve his symptoms, and 3 weeks later he presented again to the emergency department.
On his second presentation, he was in respiratory distress (oxygen saturation 78% on room air) and was afebrile and tachypneic. Physical examination revealed numerous injection marks or “tracks” on the skin of both arms, and auscultation revealed diminished intensity of breath sounds in both lung bases.
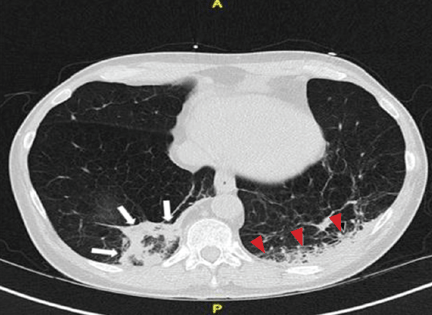
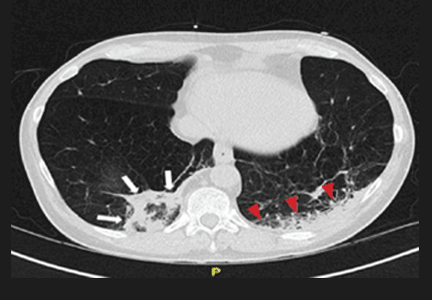
Repeat chest radiography demonstrated that the infiltrates were still there. Computed tomography was ordered and showed mild centrilobular emphysematous changes in both lungs, bibasilar opacifications, and a mass-like lesion (3.3 × 1.9 cm) in the right lower lobe (Figure 1).
He subsequently underwent bronchoscopy, which showed no endobronchial abnormalities. Transbronchial lung biopsy was performed, and histopathologic analysis of the specimen (Figure 2) revealed rodlike, birefringent crystals under polarized light, with an extensive foreign-body giant-cell reaction outside pulmonary capillaries, suggestive of intravascular pulmonary talcosis. Blood and sputum cultures were negative for pathologic organisms. Bronchoalveolar lavage samples were negative for pathologic organisms and malignant cells.
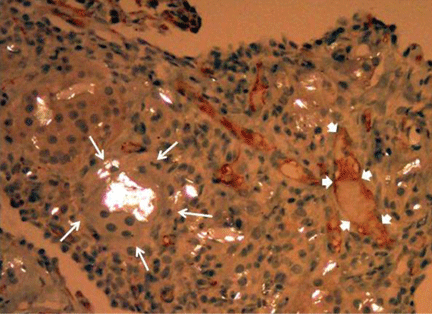
On further questioning, the patient revealed that he intravenously injected various drugs intended for oral use, such as crushed meperidine (Demerol), methylphenidate (Ritalin), and methadone tablets.
Pulmonary function tests indicated a severe obstructive pattern. The predicted forced expiratory volume in the first second of expiration (FEV1) was 25%, and the ratio of FEV1 to forced vital capacity was 27%.
Transthoracic echocardiography revealed mild pulmonary hypertension with a right ventricular systolic pressure of 28 mm Hg at rest.
Based on the results of the histologic examination, a diagnosis of intravascular pulmonary talcosis was made. Antibiotics were discontinued, and treatment with albuterol and ipratropium bromide (Combivent) inhalers was started. The patient remained oxygen-dependent at the time of hospital discharge.
INTRAVASCULAR PULMONARY TALCOSIS
Intravascular pulmonary talcosis is seen predominantly in those who chronically inject intravenous drugs intended for oral use.1,2
Many oral medications contain talc as a filler and lubricant to prevent the tablet from sticking to equipment during the manufacturing process. When oral medications containing talc are crushed, dissolved in water, and injected intravenously, the talc crystals and other particles lodge in the pulmonary vascular bed, resulting in microscopic pulmonary embolizations.
Over time, these particles migrate to the pulmonary interstitium and incite a foreign-body granulomatous reaction, which may be associated with progressive pulmonary fibrosis. The severity of this immune reaction and fibrosis may vary; hence, some patients remain asymptomatic, whereas some present with dyspnea from extensive fibrosis and pulmonary hypertension.
Persistent dyspnea along with persistent infiltrates on chest imaging in an intravenous drug abuser should prompt suspicion for intravascular pulmonary talcosis as well as consideration of other diagnoses, such as pneumonia, malignancy, and septic pulmonary emboli.
There is no established treatment for intravascular pulmonary talcosis; treatment is often supportive. A few studies and case reports have indicated varied success with systemic and inhaled corticosteroids.3–5 In extreme cases, lung transplantation may be necessary; however, this would require a comprehensive psychiatric assessment to minimize the risk of addiction relapse after transplantation.
- Arnett EN, Battle WE, Russo JV, Roberts WC. Intravenous injection of talc-containing drugs intended for oral use. A cause of pulmonary granulomatosis and pulmonary hypertension. Am J Med 1976; 60:711–718.
- Griffith CC, Raval JS, Nichols L. Intravascular talcosis due to intravenous drug use is an underrecognized cause of pulmonary hypertension. Pulm Med 2012; 2012:617531.
- Chau CH, Yew WW, Lee J. Inhaled budesonide in the treatment of talc-induced pulmonary granulomatosis. Respiration 2003; 70:439.
- Gysbrechts C, Michiels E, Verbeken E, et al. Interstitial lung disease more than 40 years after a 5 year occupational exposure to talc. Eur Respir J 1998; 11:1412–1415.
- Marchiori E, Lourenço S, Gasparetto TD, Zanetti G, Mano CM, Nobre LF. Pulmonary talcosis: imaging findings. Lung 2010; 188:165–171.
A 58-year-old-man with a history of intravenous drug abuse, chronic hepatitis C, and anxiety presented to our emergency department twice in 4 weeks with progressive dyspnea and night sweats. He was a nonsmoker and had been an electrician for 15 years.
The first time he came in, chest radiography revealed bilateral reticulonodular infiltrates in the lung bases. He was treated with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax) for presumed community-acquired pneumonia and was then sent home on a 10-day course of oral amoxicillin-clavulanate (Augmentin). The antibiotics did not improve his symptoms, and 3 weeks later he presented again to the emergency department.
On his second presentation, he was in respiratory distress (oxygen saturation 78% on room air) and was afebrile and tachypneic. Physical examination revealed numerous injection marks or “tracks” on the skin of both arms, and auscultation revealed diminished intensity of breath sounds in both lung bases.
Repeat chest radiography demonstrated that the infiltrates were still there. Computed tomography was ordered and showed mild centrilobular emphysematous changes in both lungs, bibasilar opacifications, and a mass-like lesion (3.3 × 1.9 cm) in the right lower lobe (Figure 1).
He subsequently underwent bronchoscopy, which showed no endobronchial abnormalities. Transbronchial lung biopsy was performed, and histopathologic analysis of the specimen (Figure 2) revealed rodlike, birefringent crystals under polarized light, with an extensive foreign-body giant-cell reaction outside pulmonary capillaries, suggestive of intravascular pulmonary talcosis. Blood and sputum cultures were negative for pathologic organisms. Bronchoalveolar lavage samples were negative for pathologic organisms and malignant cells.
On further questioning, the patient revealed that he intravenously injected various drugs intended for oral use, such as crushed meperidine (Demerol), methylphenidate (Ritalin), and methadone tablets.
Pulmonary function tests indicated a severe obstructive pattern. The predicted forced expiratory volume in the first second of expiration (FEV1) was 25%, and the ratio of FEV1 to forced vital capacity was 27%.
Transthoracic echocardiography revealed mild pulmonary hypertension with a right ventricular systolic pressure of 28 mm Hg at rest.
Based on the results of the histologic examination, a diagnosis of intravascular pulmonary talcosis was made. Antibiotics were discontinued, and treatment with albuterol and ipratropium bromide (Combivent) inhalers was started. The patient remained oxygen-dependent at the time of hospital discharge.
INTRAVASCULAR PULMONARY TALCOSIS
Intravascular pulmonary talcosis is seen predominantly in those who chronically inject intravenous drugs intended for oral use.1,2
Many oral medications contain talc as a filler and lubricant to prevent the tablet from sticking to equipment during the manufacturing process. When oral medications containing talc are crushed, dissolved in water, and injected intravenously, the talc crystals and other particles lodge in the pulmonary vascular bed, resulting in microscopic pulmonary embolizations.
Over time, these particles migrate to the pulmonary interstitium and incite a foreign-body granulomatous reaction, which may be associated with progressive pulmonary fibrosis. The severity of this immune reaction and fibrosis may vary; hence, some patients remain asymptomatic, whereas some present with dyspnea from extensive fibrosis and pulmonary hypertension.
Persistent dyspnea along with persistent infiltrates on chest imaging in an intravenous drug abuser should prompt suspicion for intravascular pulmonary talcosis as well as consideration of other diagnoses, such as pneumonia, malignancy, and septic pulmonary emboli.
There is no established treatment for intravascular pulmonary talcosis; treatment is often supportive. A few studies and case reports have indicated varied success with systemic and inhaled corticosteroids.3–5 In extreme cases, lung transplantation may be necessary; however, this would require a comprehensive psychiatric assessment to minimize the risk of addiction relapse after transplantation.
A 58-year-old-man with a history of intravenous drug abuse, chronic hepatitis C, and anxiety presented to our emergency department twice in 4 weeks with progressive dyspnea and night sweats. He was a nonsmoker and had been an electrician for 15 years.
The first time he came in, chest radiography revealed bilateral reticulonodular infiltrates in the lung bases. He was treated with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax) for presumed community-acquired pneumonia and was then sent home on a 10-day course of oral amoxicillin-clavulanate (Augmentin). The antibiotics did not improve his symptoms, and 3 weeks later he presented again to the emergency department.
On his second presentation, he was in respiratory distress (oxygen saturation 78% on room air) and was afebrile and tachypneic. Physical examination revealed numerous injection marks or “tracks” on the skin of both arms, and auscultation revealed diminished intensity of breath sounds in both lung bases.
Repeat chest radiography demonstrated that the infiltrates were still there. Computed tomography was ordered and showed mild centrilobular emphysematous changes in both lungs, bibasilar opacifications, and a mass-like lesion (3.3 × 1.9 cm) in the right lower lobe (Figure 1).
He subsequently underwent bronchoscopy, which showed no endobronchial abnormalities. Transbronchial lung biopsy was performed, and histopathologic analysis of the specimen (Figure 2) revealed rodlike, birefringent crystals under polarized light, with an extensive foreign-body giant-cell reaction outside pulmonary capillaries, suggestive of intravascular pulmonary talcosis. Blood and sputum cultures were negative for pathologic organisms. Bronchoalveolar lavage samples were negative for pathologic organisms and malignant cells.
On further questioning, the patient revealed that he intravenously injected various drugs intended for oral use, such as crushed meperidine (Demerol), methylphenidate (Ritalin), and methadone tablets.
Pulmonary function tests indicated a severe obstructive pattern. The predicted forced expiratory volume in the first second of expiration (FEV1) was 25%, and the ratio of FEV1 to forced vital capacity was 27%.
Transthoracic echocardiography revealed mild pulmonary hypertension with a right ventricular systolic pressure of 28 mm Hg at rest.
Based on the results of the histologic examination, a diagnosis of intravascular pulmonary talcosis was made. Antibiotics were discontinued, and treatment with albuterol and ipratropium bromide (Combivent) inhalers was started. The patient remained oxygen-dependent at the time of hospital discharge.
INTRAVASCULAR PULMONARY TALCOSIS
Intravascular pulmonary talcosis is seen predominantly in those who chronically inject intravenous drugs intended for oral use.1,2
Many oral medications contain talc as a filler and lubricant to prevent the tablet from sticking to equipment during the manufacturing process. When oral medications containing talc are crushed, dissolved in water, and injected intravenously, the talc crystals and other particles lodge in the pulmonary vascular bed, resulting in microscopic pulmonary embolizations.
Over time, these particles migrate to the pulmonary interstitium and incite a foreign-body granulomatous reaction, which may be associated with progressive pulmonary fibrosis. The severity of this immune reaction and fibrosis may vary; hence, some patients remain asymptomatic, whereas some present with dyspnea from extensive fibrosis and pulmonary hypertension.
Persistent dyspnea along with persistent infiltrates on chest imaging in an intravenous drug abuser should prompt suspicion for intravascular pulmonary talcosis as well as consideration of other diagnoses, such as pneumonia, malignancy, and septic pulmonary emboli.
There is no established treatment for intravascular pulmonary talcosis; treatment is often supportive. A few studies and case reports have indicated varied success with systemic and inhaled corticosteroids.3–5 In extreme cases, lung transplantation may be necessary; however, this would require a comprehensive psychiatric assessment to minimize the risk of addiction relapse after transplantation.
- Arnett EN, Battle WE, Russo JV, Roberts WC. Intravenous injection of talc-containing drugs intended for oral use. A cause of pulmonary granulomatosis and pulmonary hypertension. Am J Med 1976; 60:711–718.
- Griffith CC, Raval JS, Nichols L. Intravascular talcosis due to intravenous drug use is an underrecognized cause of pulmonary hypertension. Pulm Med 2012; 2012:617531.
- Chau CH, Yew WW, Lee J. Inhaled budesonide in the treatment of talc-induced pulmonary granulomatosis. Respiration 2003; 70:439.
- Gysbrechts C, Michiels E, Verbeken E, et al. Interstitial lung disease more than 40 years after a 5 year occupational exposure to talc. Eur Respir J 1998; 11:1412–1415.
- Marchiori E, Lourenço S, Gasparetto TD, Zanetti G, Mano CM, Nobre LF. Pulmonary talcosis: imaging findings. Lung 2010; 188:165–171.
- Arnett EN, Battle WE, Russo JV, Roberts WC. Intravenous injection of talc-containing drugs intended for oral use. A cause of pulmonary granulomatosis and pulmonary hypertension. Am J Med 1976; 60:711–718.
- Griffith CC, Raval JS, Nichols L. Intravascular talcosis due to intravenous drug use is an underrecognized cause of pulmonary hypertension. Pulm Med 2012; 2012:617531.
- Chau CH, Yew WW, Lee J. Inhaled budesonide in the treatment of talc-induced pulmonary granulomatosis. Respiration 2003; 70:439.
- Gysbrechts C, Michiels E, Verbeken E, et al. Interstitial lung disease more than 40 years after a 5 year occupational exposure to talc. Eur Respir J 1998; 11:1412–1415.
- Marchiori E, Lourenço S, Gasparetto TD, Zanetti G, Mano CM, Nobre LF. Pulmonary talcosis: imaging findings. Lung 2010; 188:165–171.
Wide QRS complex rhythm with pulseless electrical activity
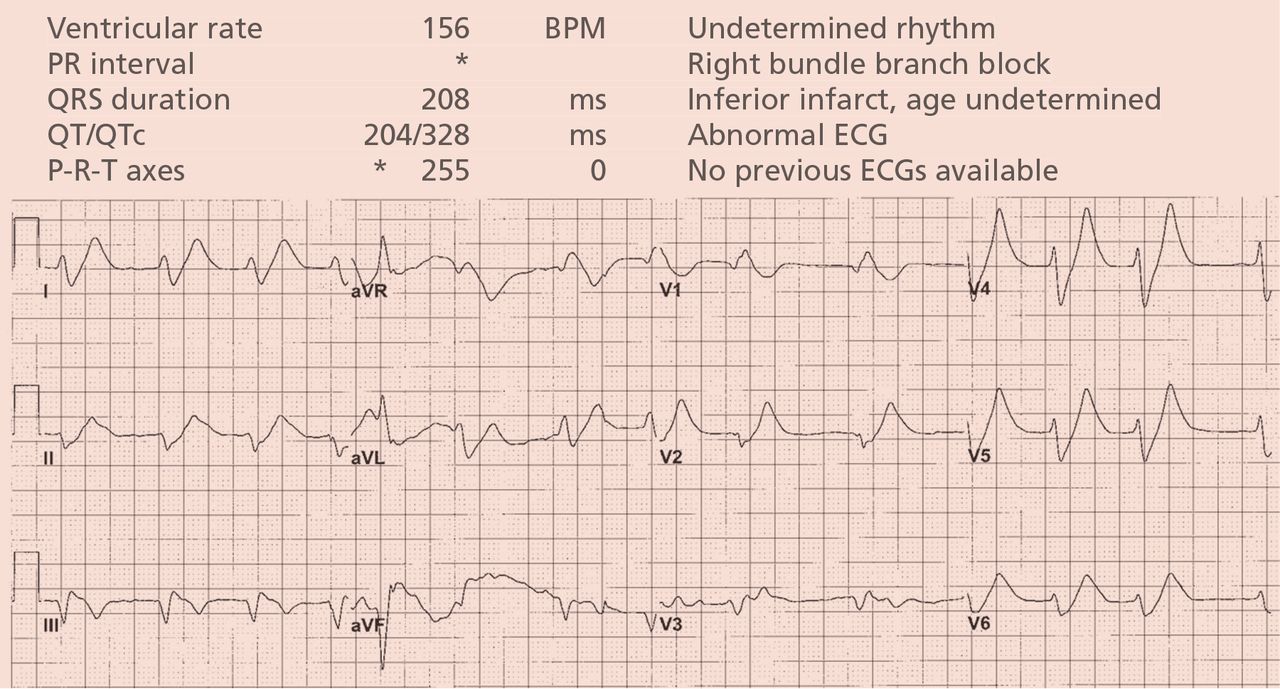
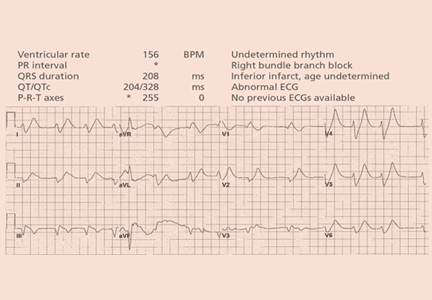
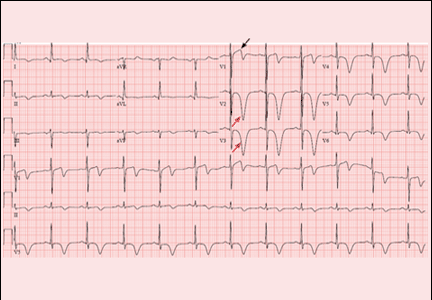
A 64-year-old man with chronic kidney disease and recent upper gastrointestinal hemorrhage suffered pulseless electrical activity and cardiac arrest. Cardiopulmonary resuscitation was started, with three attempted but failed electrical cardioversions. Return of spontaneous circulation required prolonged resuscitation efforts, including multiple rounds of epinephrine, calcium, and sodium bicarbonate. The standard 12-lead electrocardiogram (Figure 1) showed an irregular wide-QRS-complex rhythm, with right bundle branch block and right-superior-axis deviation.
What was the cause of the pulseless electrical activity and the features on the electrocardiogram?
The presentation of cardiac arrest with pulseless electrical activity usually has a grave prognosis, and in the acute setting, the cause may be difficult to establish. However, several conditions that cause this presentation have treatments that, applied immediately, can lead to quick and sustained recovery.1
Electrocardiography can be a powerful tool in the urgent evaluation of pulseless electrical activity.2,3 Narrow-QRS-complex pulseless electrical activity is often caused by mechanical factors such as cardiac tamponade, tension pneumothorax, pulmonary embolism, and major hemorrhage.3 Pulseless electrical activity associated with a wide QRS complex and marked axis deviation, as in this patient, is usually the result of a metabolic abnormality, most often hyperkalemia3; additional indicators of severe hyperkalemia include ST-segment elevation in the anterior chest leads (including the Brugada pattern4) and, as in this patient, “double counting” of the heart rate by the interpretation software (Figure 1).5,6
Based on the suspicion of a metabolic cause, the serum potassium was tested and was 8.9 mmol/L (reference range 3.5–5.0). The patient was given intravenous calcium, sodium bicarbonate, glucose, and insulin, and 2 hours later the serum potassium had decreased to 7.1 mmol/L. At that time, the electrocardiogram (Figure 2) showed a regular rhythm with ectopic P waves, probably an ectopic atrial tachycardia. There were now narrow QRS complexes with J-point depression, upsloping ST segments, and tall, hyperacute T waves in the chest leads—a pattern recently described in proximal left anterior descending coronary artery occlusion.7 The electrocardiographic similarities in hyperkalemia and acute myocardial infarction are probably the result of potassium accumulation in the ischemic myocardium associated with acute coronary occlusion.7
The patient had a full recovery, both clinically and on electrocardiography.

- Saarinen S, Nurmi J, Toivio T, Fredman D, Virkkunen I, Castrén M. Does appropriate treatment of the primary underlying cause of PEA during resuscitation improve patients’ survival? Resuscitation 2012; 83:819–822.
- Mehta C, Brady W. Pulseless electrical activity in cardiac arrest: electrocardiographic presentations and management considerations based on the electrocardiogram. Am J Emerg Med 2012; 30:236–239.
- Littmann L, Bustin DJ, Haley MW. A simplified and structured teaching tool for the evaluation and management of pulseless electrical activity. Med Princ Pract 2014; 23:1–6.
- Littmann L, Monroe MH, Taylor L, Brearley WD. The hyperkalemic Brugada sign. J Electrocardiol 2007; 40:53–59.
- Littmann L, Brearley WD, Taylor L, Monroe MH. Double counting of heart rate by interpretation software: a new electrocardiographic sign of severe hyperkalemia. Am J Emerg Med 2007; 25:584–586.
- Tomcsányi J, Wágner V, Bózsik B. Littmann sign in hyperkalemia: double counting of heart rate. Am J Emerg Med 2007; 25:1077–1078.
- de Winter RJ, Verouden NJ, Wellens HJ, Wilde AA; Interventional Cardiology Group of the Academic Medical Center. A new ECG sign of proximal LAD occlusion. N Engl J Med 2008; 359:2071–2073.
A 64-year-old man with chronic kidney disease and recent upper gastrointestinal hemorrhage suffered pulseless electrical activity and cardiac arrest. Cardiopulmonary resuscitation was started, with three attempted but failed electrical cardioversions. Return of spontaneous circulation required prolonged resuscitation efforts, including multiple rounds of epinephrine, calcium, and sodium bicarbonate. The standard 12-lead electrocardiogram (Figure 1) showed an irregular wide-QRS-complex rhythm, with right bundle branch block and right-superior-axis deviation.
What was the cause of the pulseless electrical activity and the features on the electrocardiogram?
The presentation of cardiac arrest with pulseless electrical activity usually has a grave prognosis, and in the acute setting, the cause may be difficult to establish. However, several conditions that cause this presentation have treatments that, applied immediately, can lead to quick and sustained recovery.1
Electrocardiography can be a powerful tool in the urgent evaluation of pulseless electrical activity.2,3 Narrow-QRS-complex pulseless electrical activity is often caused by mechanical factors such as cardiac tamponade, tension pneumothorax, pulmonary embolism, and major hemorrhage.3 Pulseless electrical activity associated with a wide QRS complex and marked axis deviation, as in this patient, is usually the result of a metabolic abnormality, most often hyperkalemia3; additional indicators of severe hyperkalemia include ST-segment elevation in the anterior chest leads (including the Brugada pattern4) and, as in this patient, “double counting” of the heart rate by the interpretation software (Figure 1).5,6
Based on the suspicion of a metabolic cause, the serum potassium was tested and was 8.9 mmol/L (reference range 3.5–5.0). The patient was given intravenous calcium, sodium bicarbonate, glucose, and insulin, and 2 hours later the serum potassium had decreased to 7.1 mmol/L. At that time, the electrocardiogram (Figure 2) showed a regular rhythm with ectopic P waves, probably an ectopic atrial tachycardia. There were now narrow QRS complexes with J-point depression, upsloping ST segments, and tall, hyperacute T waves in the chest leads—a pattern recently described in proximal left anterior descending coronary artery occlusion.7 The electrocardiographic similarities in hyperkalemia and acute myocardial infarction are probably the result of potassium accumulation in the ischemic myocardium associated with acute coronary occlusion.7
The patient had a full recovery, both clinically and on electrocardiography.
A 64-year-old man with chronic kidney disease and recent upper gastrointestinal hemorrhage suffered pulseless electrical activity and cardiac arrest. Cardiopulmonary resuscitation was started, with three attempted but failed electrical cardioversions. Return of spontaneous circulation required prolonged resuscitation efforts, including multiple rounds of epinephrine, calcium, and sodium bicarbonate. The standard 12-lead electrocardiogram (Figure 1) showed an irregular wide-QRS-complex rhythm, with right bundle branch block and right-superior-axis deviation.
What was the cause of the pulseless electrical activity and the features on the electrocardiogram?
The presentation of cardiac arrest with pulseless electrical activity usually has a grave prognosis, and in the acute setting, the cause may be difficult to establish. However, several conditions that cause this presentation have treatments that, applied immediately, can lead to quick and sustained recovery.1
Electrocardiography can be a powerful tool in the urgent evaluation of pulseless electrical activity.2,3 Narrow-QRS-complex pulseless electrical activity is often caused by mechanical factors such as cardiac tamponade, tension pneumothorax, pulmonary embolism, and major hemorrhage.3 Pulseless electrical activity associated with a wide QRS complex and marked axis deviation, as in this patient, is usually the result of a metabolic abnormality, most often hyperkalemia3; additional indicators of severe hyperkalemia include ST-segment elevation in the anterior chest leads (including the Brugada pattern4) and, as in this patient, “double counting” of the heart rate by the interpretation software (Figure 1).5,6
Based on the suspicion of a metabolic cause, the serum potassium was tested and was 8.9 mmol/L (reference range 3.5–5.0). The patient was given intravenous calcium, sodium bicarbonate, glucose, and insulin, and 2 hours later the serum potassium had decreased to 7.1 mmol/L. At that time, the electrocardiogram (Figure 2) showed a regular rhythm with ectopic P waves, probably an ectopic atrial tachycardia. There were now narrow QRS complexes with J-point depression, upsloping ST segments, and tall, hyperacute T waves in the chest leads—a pattern recently described in proximal left anterior descending coronary artery occlusion.7 The electrocardiographic similarities in hyperkalemia and acute myocardial infarction are probably the result of potassium accumulation in the ischemic myocardium associated with acute coronary occlusion.7
The patient had a full recovery, both clinically and on electrocardiography.
- Saarinen S, Nurmi J, Toivio T, Fredman D, Virkkunen I, Castrén M. Does appropriate treatment of the primary underlying cause of PEA during resuscitation improve patients’ survival? Resuscitation 2012; 83:819–822.
- Mehta C, Brady W. Pulseless electrical activity in cardiac arrest: electrocardiographic presentations and management considerations based on the electrocardiogram. Am J Emerg Med 2012; 30:236–239.
- Littmann L, Bustin DJ, Haley MW. A simplified and structured teaching tool for the evaluation and management of pulseless electrical activity. Med Princ Pract 2014; 23:1–6.
- Littmann L, Monroe MH, Taylor L, Brearley WD. The hyperkalemic Brugada sign. J Electrocardiol 2007; 40:53–59.
- Littmann L, Brearley WD, Taylor L, Monroe MH. Double counting of heart rate by interpretation software: a new electrocardiographic sign of severe hyperkalemia. Am J Emerg Med 2007; 25:584–586.
- Tomcsányi J, Wágner V, Bózsik B. Littmann sign in hyperkalemia: double counting of heart rate. Am J Emerg Med 2007; 25:1077–1078.
- de Winter RJ, Verouden NJ, Wellens HJ, Wilde AA; Interventional Cardiology Group of the Academic Medical Center. A new ECG sign of proximal LAD occlusion. N Engl J Med 2008; 359:2071–2073.
- Saarinen S, Nurmi J, Toivio T, Fredman D, Virkkunen I, Castrén M. Does appropriate treatment of the primary underlying cause of PEA during resuscitation improve patients’ survival? Resuscitation 2012; 83:819–822.
- Mehta C, Brady W. Pulseless electrical activity in cardiac arrest: electrocardiographic presentations and management considerations based on the electrocardiogram. Am J Emerg Med 2012; 30:236–239.
- Littmann L, Bustin DJ, Haley MW. A simplified and structured teaching tool for the evaluation and management of pulseless electrical activity. Med Princ Pract 2014; 23:1–6.
- Littmann L, Monroe MH, Taylor L, Brearley WD. The hyperkalemic Brugada sign. J Electrocardiol 2007; 40:53–59.
- Littmann L, Brearley WD, Taylor L, Monroe MH. Double counting of heart rate by interpretation software: a new electrocardiographic sign of severe hyperkalemia. Am J Emerg Med 2007; 25:584–586.
- Tomcsányi J, Wágner V, Bózsik B. Littmann sign in hyperkalemia: double counting of heart rate. Am J Emerg Med 2007; 25:1077–1078.
- de Winter RJ, Verouden NJ, Wellens HJ, Wilde AA; Interventional Cardiology Group of the Academic Medical Center. A new ECG sign of proximal LAD occlusion. N Engl J Med 2008; 359:2071–2073.
Slow-growing angiomatous lesions on the limbs
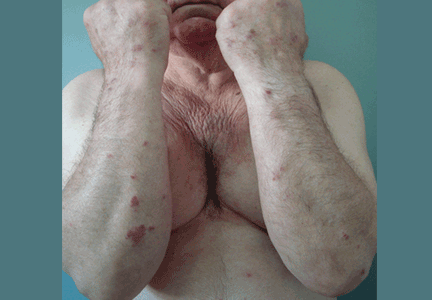
A 70-year-old man presented with multiple erythematous plaques on the arms and legs (Figure 1). The plaques had infiltrated the skin and were poorly demarcated.
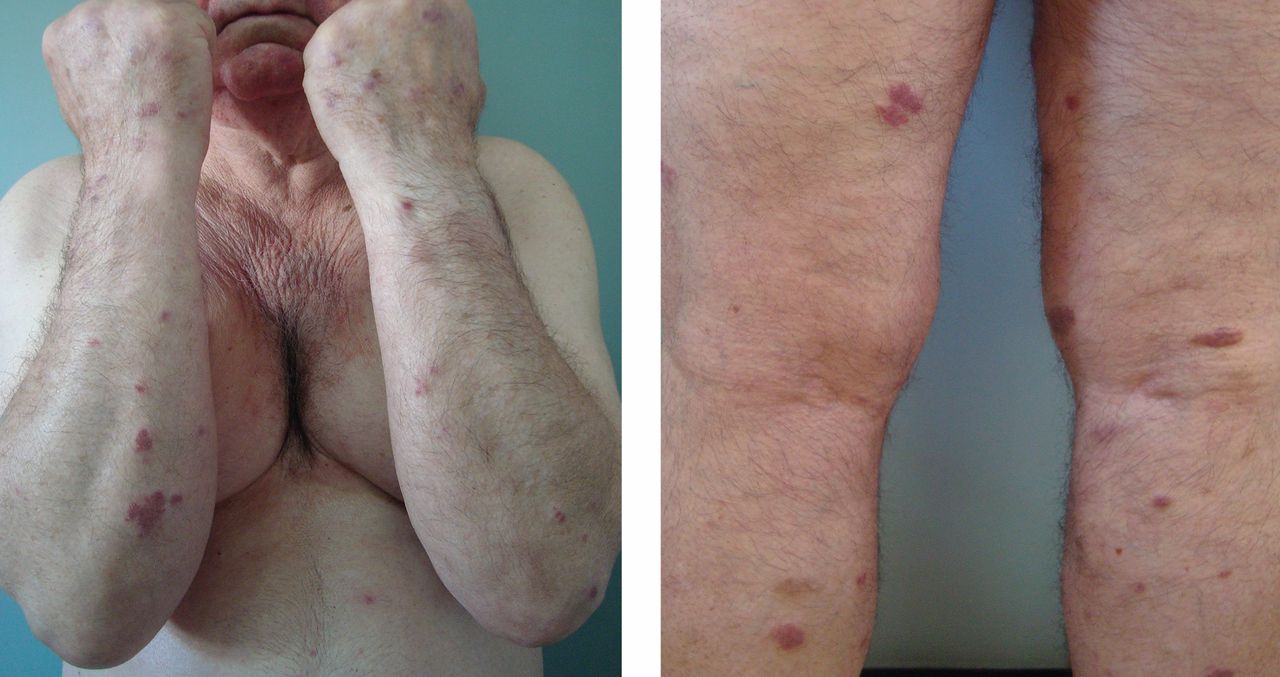
He had hypertension but no history of other relevant medical conditions, and he was not taking any medication. He was not neutropenic or immunocompromised.
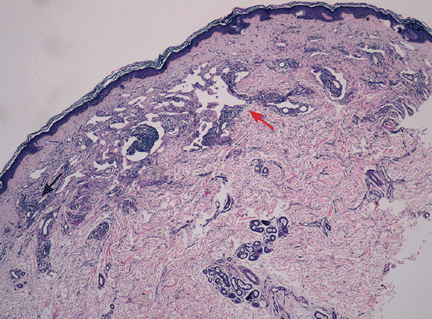
In a patient after the sixth decade of life, erythematous plate-shaped lesions on the legs that become apparent on palpation should raise suspicion of classic Kaposi sarcoma. A biopsy confirmed this diagnosis (Figure 2). Immunohistochemical staining was positive for human herpesvirus 8 latent nuclear antigen. Clinical examination, computed tomography, and blood tests showed no extracutaneous involvement or other associated pathology. He was treated with paclitaxel, which resulted in improvement of his symptoms.
DIFFERENTIAL DIAGNOSIS
Bacillary angiomatosis, acroangiodermatitis (“pseudo-Kaposi sarcoma”) and atypical mycobacterial infections such as Mycobacterium marinum infection may present as papules or nodules on the legs.
Bacillary angiomatosis
Bacillary angiomatosis is more common in patients with acquired immunodeficiency syndrome and other forms of immunosuppression. Bacilli are produced by Bartonella henselae and B quintana and are a manifestation of cat-scratch disease in an immunocompromised host.1 The disease manifests as pyogenic granuloma-like lesions or subcutaneous nodules and may be associated with liver damage and systemic impairment.
Acroangiodermatitis
Acroangiodermatitis, or pseudo-Kaposi sarcoma, is caused by hyperplasia of the venous vasculature or chronic venous stasis. It is an entity observed in amputees, in hemodialysis patients with an arteriovenous fistula who are inflected with hepatitis C virus, and in patients with vascular malformations.2–3 It presents as plaques or violaceous papules on the legs.
M marinum infection
This syndrome presents clinically with erythematous papular and nodular lesions on the skin surface. They can appear on the knees and feet of people infected while swimming in pools, or on the hands of aquarium owners.4–5 A high index of suspicion and a complete medical history are key to properly diagnosing this disease.
CLASSIC KAPOSI SARCOMA
Kaposi sarcoma is a neoplasm of lymphatic endothelial cells. Four types have been described: classic, endemic, iatrogenic, and associated with human immunodeficiency virus infection.
The disease affects men and women around the sixth decade of life. It is more common in Ashkenazi Jews and people of Mediterranean origin. It presents as slow-growing plaques or nodules on the lower extremities; a disseminated form or internal organ involvement is rare.6
Histologic study reveals vascular proliferation with superficial perivascular lymphoplasmacytic infiltration, displaying the classic formation of new vessels from pre-existing vessels. It has a benign course that may last 10 years. Single lesions can be removed surgically or can be treated with chemotherapy. Advanced disease requires systemic chemotherapy with pegylated liposomal doxorubicin, interferon alfa-2a, paclitaxel, or bleomycin and vinblastine.7,8
- Zarraga M, Rosen L, Herschthal D. Bacillary angiomatosis in an immunocompetent child: a case report and review of the literature. Am J Dermatopathol 2011; 33:513–515.
- Brenner S, Martinez de Morentin E. What’s new in pseudo-Kaposi’s sarcoma. J Eur Acad Dermatol Venereol 2001; 15:382–384.
- Mehta AA, Pereira RR, Nayak CS, Dhurat RS. Acroangiodermatitis of mali: a rare vascular phenomenon. IIndian J Dermatol Venereol Leprol 2010; 76:553–556.
- Petrini B. Mycobacterium marinum: ubiquitous agent of waterborne granulomatous skin infections. Eur J Clin Microbiol Infect Dis 2006; 25:609–613.
- Aboutalebi A, Shen A, Katta R, Allen SE. Primary cutaneous infection by Mycobacterium avium: a case report and literature review. Cutis 2012; 89:175–179.
- Kandemir NO, Yurdakan G, Bektas S, Tekin NS. Classic Kaposi sarcoma with sarcoid-like granulomas: a case report and literature review. Exp Mol Pathol 2009; 87:89–93.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol 2013; 68:313–331.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer 2008; 112:1147–1152.
A 70-year-old man presented with multiple erythematous plaques on the arms and legs (Figure 1). The plaques had infiltrated the skin and were poorly demarcated.
He had hypertension but no history of other relevant medical conditions, and he was not taking any medication. He was not neutropenic or immunocompromised.
In a patient after the sixth decade of life, erythematous plate-shaped lesions on the legs that become apparent on palpation should raise suspicion of classic Kaposi sarcoma. A biopsy confirmed this diagnosis (Figure 2). Immunohistochemical staining was positive for human herpesvirus 8 latent nuclear antigen. Clinical examination, computed tomography, and blood tests showed no extracutaneous involvement or other associated pathology. He was treated with paclitaxel, which resulted in improvement of his symptoms.
DIFFERENTIAL DIAGNOSIS
Bacillary angiomatosis, acroangiodermatitis (“pseudo-Kaposi sarcoma”) and atypical mycobacterial infections such as Mycobacterium marinum infection may present as papules or nodules on the legs.
Bacillary angiomatosis
Bacillary angiomatosis is more common in patients with acquired immunodeficiency syndrome and other forms of immunosuppression. Bacilli are produced by Bartonella henselae and B quintana and are a manifestation of cat-scratch disease in an immunocompromised host.1 The disease manifests as pyogenic granuloma-like lesions or subcutaneous nodules and may be associated with liver damage and systemic impairment.
Acroangiodermatitis
Acroangiodermatitis, or pseudo-Kaposi sarcoma, is caused by hyperplasia of the venous vasculature or chronic venous stasis. It is an entity observed in amputees, in hemodialysis patients with an arteriovenous fistula who are inflected with hepatitis C virus, and in patients with vascular malformations.2–3 It presents as plaques or violaceous papules on the legs.
M marinum infection
This syndrome presents clinically with erythematous papular and nodular lesions on the skin surface. They can appear on the knees and feet of people infected while swimming in pools, or on the hands of aquarium owners.4–5 A high index of suspicion and a complete medical history are key to properly diagnosing this disease.
CLASSIC KAPOSI SARCOMA
Kaposi sarcoma is a neoplasm of lymphatic endothelial cells. Four types have been described: classic, endemic, iatrogenic, and associated with human immunodeficiency virus infection.
The disease affects men and women around the sixth decade of life. It is more common in Ashkenazi Jews and people of Mediterranean origin. It presents as slow-growing plaques or nodules on the lower extremities; a disseminated form or internal organ involvement is rare.6
Histologic study reveals vascular proliferation with superficial perivascular lymphoplasmacytic infiltration, displaying the classic formation of new vessels from pre-existing vessels. It has a benign course that may last 10 years. Single lesions can be removed surgically or can be treated with chemotherapy. Advanced disease requires systemic chemotherapy with pegylated liposomal doxorubicin, interferon alfa-2a, paclitaxel, or bleomycin and vinblastine.7,8
A 70-year-old man presented with multiple erythematous plaques on the arms and legs (Figure 1). The plaques had infiltrated the skin and were poorly demarcated.
He had hypertension but no history of other relevant medical conditions, and he was not taking any medication. He was not neutropenic or immunocompromised.
In a patient after the sixth decade of life, erythematous plate-shaped lesions on the legs that become apparent on palpation should raise suspicion of classic Kaposi sarcoma. A biopsy confirmed this diagnosis (Figure 2). Immunohistochemical staining was positive for human herpesvirus 8 latent nuclear antigen. Clinical examination, computed tomography, and blood tests showed no extracutaneous involvement or other associated pathology. He was treated with paclitaxel, which resulted in improvement of his symptoms.
DIFFERENTIAL DIAGNOSIS
Bacillary angiomatosis, acroangiodermatitis (“pseudo-Kaposi sarcoma”) and atypical mycobacterial infections such as Mycobacterium marinum infection may present as papules or nodules on the legs.
Bacillary angiomatosis
Bacillary angiomatosis is more common in patients with acquired immunodeficiency syndrome and other forms of immunosuppression. Bacilli are produced by Bartonella henselae and B quintana and are a manifestation of cat-scratch disease in an immunocompromised host.1 The disease manifests as pyogenic granuloma-like lesions or subcutaneous nodules and may be associated with liver damage and systemic impairment.
Acroangiodermatitis
Acroangiodermatitis, or pseudo-Kaposi sarcoma, is caused by hyperplasia of the venous vasculature or chronic venous stasis. It is an entity observed in amputees, in hemodialysis patients with an arteriovenous fistula who are inflected with hepatitis C virus, and in patients with vascular malformations.2–3 It presents as plaques or violaceous papules on the legs.
M marinum infection
This syndrome presents clinically with erythematous papular and nodular lesions on the skin surface. They can appear on the knees and feet of people infected while swimming in pools, or on the hands of aquarium owners.4–5 A high index of suspicion and a complete medical history are key to properly diagnosing this disease.
CLASSIC KAPOSI SARCOMA
Kaposi sarcoma is a neoplasm of lymphatic endothelial cells. Four types have been described: classic, endemic, iatrogenic, and associated with human immunodeficiency virus infection.
The disease affects men and women around the sixth decade of life. It is more common in Ashkenazi Jews and people of Mediterranean origin. It presents as slow-growing plaques or nodules on the lower extremities; a disseminated form or internal organ involvement is rare.6
Histologic study reveals vascular proliferation with superficial perivascular lymphoplasmacytic infiltration, displaying the classic formation of new vessels from pre-existing vessels. It has a benign course that may last 10 years. Single lesions can be removed surgically or can be treated with chemotherapy. Advanced disease requires systemic chemotherapy with pegylated liposomal doxorubicin, interferon alfa-2a, paclitaxel, or bleomycin and vinblastine.7,8
- Zarraga M, Rosen L, Herschthal D. Bacillary angiomatosis in an immunocompetent child: a case report and review of the literature. Am J Dermatopathol 2011; 33:513–515.
- Brenner S, Martinez de Morentin E. What’s new in pseudo-Kaposi’s sarcoma. J Eur Acad Dermatol Venereol 2001; 15:382–384.
- Mehta AA, Pereira RR, Nayak CS, Dhurat RS. Acroangiodermatitis of mali: a rare vascular phenomenon. IIndian J Dermatol Venereol Leprol 2010; 76:553–556.
- Petrini B. Mycobacterium marinum: ubiquitous agent of waterborne granulomatous skin infections. Eur J Clin Microbiol Infect Dis 2006; 25:609–613.
- Aboutalebi A, Shen A, Katta R, Allen SE. Primary cutaneous infection by Mycobacterium avium: a case report and literature review. Cutis 2012; 89:175–179.
- Kandemir NO, Yurdakan G, Bektas S, Tekin NS. Classic Kaposi sarcoma with sarcoid-like granulomas: a case report and literature review. Exp Mol Pathol 2009; 87:89–93.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol 2013; 68:313–331.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer 2008; 112:1147–1152.
- Zarraga M, Rosen L, Herschthal D. Bacillary angiomatosis in an immunocompetent child: a case report and review of the literature. Am J Dermatopathol 2011; 33:513–515.
- Brenner S, Martinez de Morentin E. What’s new in pseudo-Kaposi’s sarcoma. J Eur Acad Dermatol Venereol 2001; 15:382–384.
- Mehta AA, Pereira RR, Nayak CS, Dhurat RS. Acroangiodermatitis of mali: a rare vascular phenomenon. IIndian J Dermatol Venereol Leprol 2010; 76:553–556.
- Petrini B. Mycobacterium marinum: ubiquitous agent of waterborne granulomatous skin infections. Eur J Clin Microbiol Infect Dis 2006; 25:609–613.
- Aboutalebi A, Shen A, Katta R, Allen SE. Primary cutaneous infection by Mycobacterium avium: a case report and literature review. Cutis 2012; 89:175–179.
- Kandemir NO, Yurdakan G, Bektas S, Tekin NS. Classic Kaposi sarcoma with sarcoid-like granulomas: a case report and literature review. Exp Mol Pathol 2009; 87:89–93.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol 2013; 68:313–331.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer 2008; 112:1147–1152.
Deep T waves and chest pain
A 67-year-old man with a history of hypertension and hyperlipidemia presented to the emergency department after 3 hours of what he described as a burning sensation in his chest that woke him from sleep. He attributed it at first to a late-night meal and treated himself with some milk and yogurt, which seemed to relieve the symptoms. However, the pain recurred and was associated with difficulty breathing. At that point, he drove himself to the emergency department.
On arrival, his temperature was 36.5°C (97.7°F), blood pressure 134/67 mm Hg, heart rate 89 bpm, respirations 18/min, and oxygen saturation 98% on room air. His cardiovascular, lung, and neurologic examinations were normal. His cardiac enzyme levels (creatine kinase, creatine kinase MB fraction, and troponin T) were within normal limits.
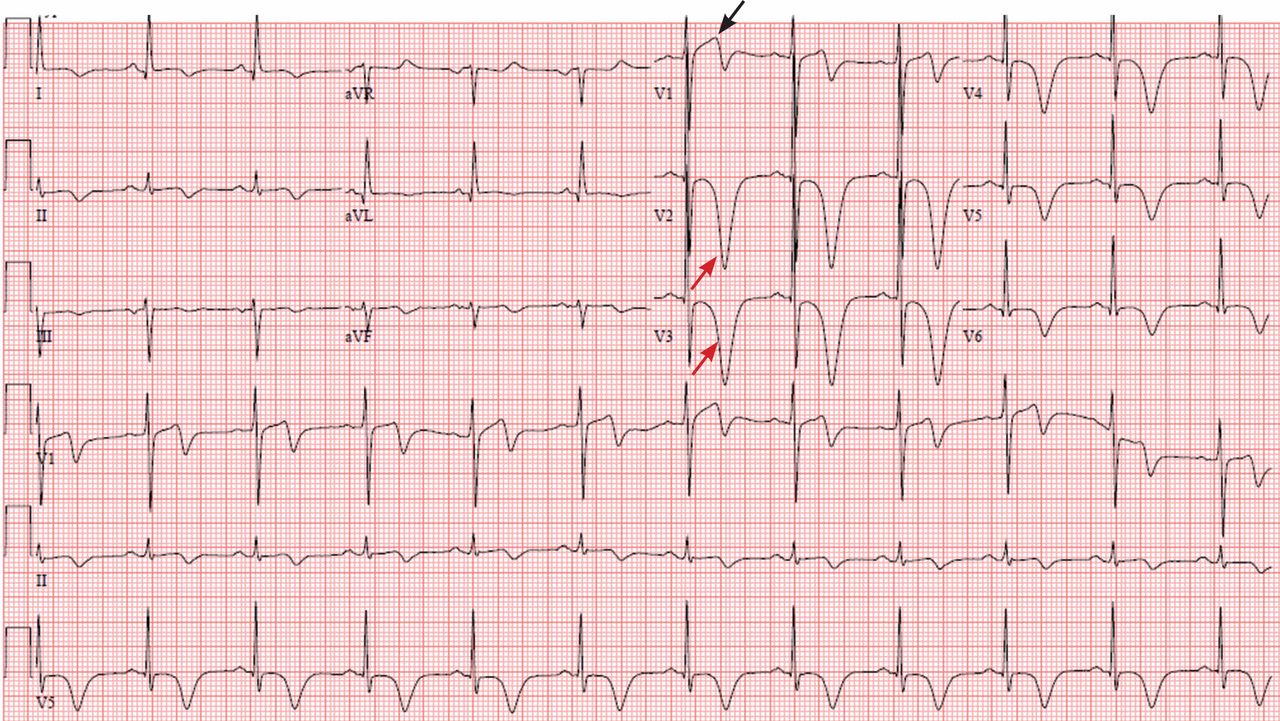
Figure 1 depicts his initial electrocardiogram. It showed deep, symmetric T-wave inversions in the precordial leads especially in V2 and V3, changes known as Wellens syndrome. The ST-T changes in lead V1 suggested a very proximal lesion in the left anterior descending artery (LAD), before the first septal perforator. Also, lateral and high lateral (V5 and V6) findings indicated stenoses of the branching diagonals and left circumflex myocardial territory. Furthermore, the inferior ST-T changes indicated that his LAD may have wrapped around the cardiac apex. All of these findings were prognostically significant.
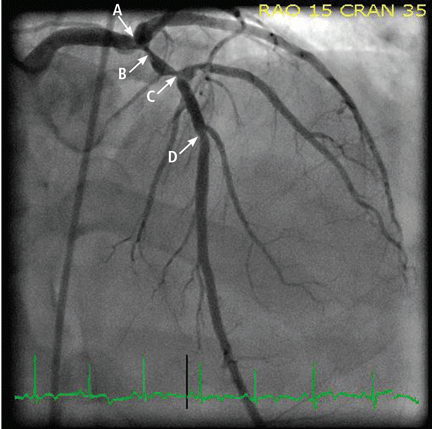
The patient was given aspirin and was started on intravenous unfractionated heparin and nitroglycerin. He was sent for urgent left-heart catheterization, which showed a 50% to 60% stenosis in the left main coronary artery, with involvement of the left circumflex artery proximally, in addition to a “tight” first-diagonal stenosis, a 90% stenosis in a large (> 3.0-mm) proximal segment of the LAD, an 80% stenosis in a large (> 3.0-mm) mid-LAD segment, and a 40% stenosis in a large (> 3.0-mm) second diagonal artery (Figure 2).
He was referred for cardiac surgery and underwent triple coronary artery bypass grafting: the left internal thoracic artery was grafted to the LAD, a reverse saphenous vein graft was performed to the diagonal artery, and a reverse saphenous vein graft was performed to the obtuse marginal artery.
A PRECURSOR TO INFARCTION
Wellens et al described specific precordial T-wave changes in patients with unstable angina who subsequently developed anterior wall myocardial infarction.1
The importance of Wellens syndrome is that it occurs in the pain-free interval when no other evidence of ischemia or angina may be present.1 Cardiac enzyme levels are typically normal or only minimally elevated; only 12% of patients with this syndrome have elevated cardiac biomarker levels.2
Given the extent of myocardial injury, urgent echocardiography can show a wall-motion abnormality even if cardiac enzyme levels are normal. This gives important insight into electrocardiographic changes and should prompt consideration of revascularization.
Even with extensive medical management, Wellens syndrome progresses to acute anterior wall ischemia. About 75% of patients with Wellens syndrome who receive medical management but do not undergo revascularization (eg, coronary artery bypass grafting, percutaneous coronary intervention) develop extensive anterior wall infarction within days.1,3 Despite negative cardiac biomarkers, Wellens syndrome is considered an acute coronary syndrome requiring urgent cardiac intervention.
- Movahed MR. Wellens’ syndrome or inverted U-waves? Clin Cardiol 2008; 31:133–134.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
A 67-year-old man with a history of hypertension and hyperlipidemia presented to the emergency department after 3 hours of what he described as a burning sensation in his chest that woke him from sleep. He attributed it at first to a late-night meal and treated himself with some milk and yogurt, which seemed to relieve the symptoms. However, the pain recurred and was associated with difficulty breathing. At that point, he drove himself to the emergency department.
On arrival, his temperature was 36.5°C (97.7°F), blood pressure 134/67 mm Hg, heart rate 89 bpm, respirations 18/min, and oxygen saturation 98% on room air. His cardiovascular, lung, and neurologic examinations were normal. His cardiac enzyme levels (creatine kinase, creatine kinase MB fraction, and troponin T) were within normal limits.
Figure 1 depicts his initial electrocardiogram. It showed deep, symmetric T-wave inversions in the precordial leads especially in V2 and V3, changes known as Wellens syndrome. The ST-T changes in lead V1 suggested a very proximal lesion in the left anterior descending artery (LAD), before the first septal perforator. Also, lateral and high lateral (V5 and V6) findings indicated stenoses of the branching diagonals and left circumflex myocardial territory. Furthermore, the inferior ST-T changes indicated that his LAD may have wrapped around the cardiac apex. All of these findings were prognostically significant.
The patient was given aspirin and was started on intravenous unfractionated heparin and nitroglycerin. He was sent for urgent left-heart catheterization, which showed a 50% to 60% stenosis in the left main coronary artery, with involvement of the left circumflex artery proximally, in addition to a “tight” first-diagonal stenosis, a 90% stenosis in a large (> 3.0-mm) proximal segment of the LAD, an 80% stenosis in a large (> 3.0-mm) mid-LAD segment, and a 40% stenosis in a large (> 3.0-mm) second diagonal artery (Figure 2).
He was referred for cardiac surgery and underwent triple coronary artery bypass grafting: the left internal thoracic artery was grafted to the LAD, a reverse saphenous vein graft was performed to the diagonal artery, and a reverse saphenous vein graft was performed to the obtuse marginal artery.
A PRECURSOR TO INFARCTION
Wellens et al described specific precordial T-wave changes in patients with unstable angina who subsequently developed anterior wall myocardial infarction.1
The importance of Wellens syndrome is that it occurs in the pain-free interval when no other evidence of ischemia or angina may be present.1 Cardiac enzyme levels are typically normal or only minimally elevated; only 12% of patients with this syndrome have elevated cardiac biomarker levels.2
Given the extent of myocardial injury, urgent echocardiography can show a wall-motion abnormality even if cardiac enzyme levels are normal. This gives important insight into electrocardiographic changes and should prompt consideration of revascularization.
Even with extensive medical management, Wellens syndrome progresses to acute anterior wall ischemia. About 75% of patients with Wellens syndrome who receive medical management but do not undergo revascularization (eg, coronary artery bypass grafting, percutaneous coronary intervention) develop extensive anterior wall infarction within days.1,3 Despite negative cardiac biomarkers, Wellens syndrome is considered an acute coronary syndrome requiring urgent cardiac intervention.
A 67-year-old man with a history of hypertension and hyperlipidemia presented to the emergency department after 3 hours of what he described as a burning sensation in his chest that woke him from sleep. He attributed it at first to a late-night meal and treated himself with some milk and yogurt, which seemed to relieve the symptoms. However, the pain recurred and was associated with difficulty breathing. At that point, he drove himself to the emergency department.
On arrival, his temperature was 36.5°C (97.7°F), blood pressure 134/67 mm Hg, heart rate 89 bpm, respirations 18/min, and oxygen saturation 98% on room air. His cardiovascular, lung, and neurologic examinations were normal. His cardiac enzyme levels (creatine kinase, creatine kinase MB fraction, and troponin T) were within normal limits.
Figure 1 depicts his initial electrocardiogram. It showed deep, symmetric T-wave inversions in the precordial leads especially in V2 and V3, changes known as Wellens syndrome. The ST-T changes in lead V1 suggested a very proximal lesion in the left anterior descending artery (LAD), before the first septal perforator. Also, lateral and high lateral (V5 and V6) findings indicated stenoses of the branching diagonals and left circumflex myocardial territory. Furthermore, the inferior ST-T changes indicated that his LAD may have wrapped around the cardiac apex. All of these findings were prognostically significant.
The patient was given aspirin and was started on intravenous unfractionated heparin and nitroglycerin. He was sent for urgent left-heart catheterization, which showed a 50% to 60% stenosis in the left main coronary artery, with involvement of the left circumflex artery proximally, in addition to a “tight” first-diagonal stenosis, a 90% stenosis in a large (> 3.0-mm) proximal segment of the LAD, an 80% stenosis in a large (> 3.0-mm) mid-LAD segment, and a 40% stenosis in a large (> 3.0-mm) second diagonal artery (Figure 2).
He was referred for cardiac surgery and underwent triple coronary artery bypass grafting: the left internal thoracic artery was grafted to the LAD, a reverse saphenous vein graft was performed to the diagonal artery, and a reverse saphenous vein graft was performed to the obtuse marginal artery.
A PRECURSOR TO INFARCTION
Wellens et al described specific precordial T-wave changes in patients with unstable angina who subsequently developed anterior wall myocardial infarction.1
The importance of Wellens syndrome is that it occurs in the pain-free interval when no other evidence of ischemia or angina may be present.1 Cardiac enzyme levels are typically normal or only minimally elevated; only 12% of patients with this syndrome have elevated cardiac biomarker levels.2
Given the extent of myocardial injury, urgent echocardiography can show a wall-motion abnormality even if cardiac enzyme levels are normal. This gives important insight into electrocardiographic changes and should prompt consideration of revascularization.
Even with extensive medical management, Wellens syndrome progresses to acute anterior wall ischemia. About 75% of patients with Wellens syndrome who receive medical management but do not undergo revascularization (eg, coronary artery bypass grafting, percutaneous coronary intervention) develop extensive anterior wall infarction within days.1,3 Despite negative cardiac biomarkers, Wellens syndrome is considered an acute coronary syndrome requiring urgent cardiac intervention.
- Movahed MR. Wellens’ syndrome or inverted U-waves? Clin Cardiol 2008; 31:133–134.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
- Movahed MR. Wellens’ syndrome or inverted U-waves? Clin Cardiol 2008; 31:133–134.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
A 66-year-old woman with an enlarged tongue
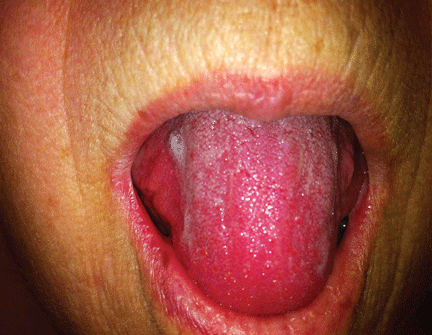
A 66-year-old woman was prompted by her dentist to seek medical attention for an unusually enlarged, smooth-appearing tongue (Figure 1). She also complained of fatigue, dyspnea on exertion, and tingling of her hands.
Basic laboratory tests showed normocytic anemia and renal insufficiency. Her thyrotropin level was within normal limits. Serum protein electrophoresis showed a monoclonal M-spike, which prompted a bone marrow biopsy that was diagnostic of multiple myeloma.
Transthoracic echocardiography revealed diffuse hypokinesis with a restrictive filling pattern, myocardial thickening, and moderate mitral and tricuspid regurgitation, highly suggestive of an infiltrative cardiomyopathy. Biopsy of the right ventricle confirmed cardiac amyloidosis of the amyloid immunoglobulin light chain (AL) subtype.
The patient underwent chemotherapy, followed by autologous stem-cell transplantation. She achieved successful remission, and her cardiomyopathy was compensated.
AMYLOIDOSIS IS HETEROGENEOUS
Amyloidosis is a heterogeneous syndrome characterized by abnormal folding of proteins that deposit as insoluble fibrils in different tissues, impairing both structure and function. Virchow was the first to describe amyloid (from amylon, Greek for starch) as an abnormal material seen in postmortem examination of the liver. On Congo red staining, the extracellular proteins appear as salmon-red conglomerates, which also show apple-green birefringence under polarized light.
Amyloidosis can be localized but more often represents a systemic process, often associated with a plasma cell dyscrasia such as multiple myeloma.
Modern classification is based on the precursor protein,1 eg:
- Light chains (AL)
- Acute-phase protein (AA)
- Beta-2-microglobulin (Aß2M)
- Transthyretin (ATTR; occurring in senile systemic amyloidosis)
- Other proteins (occurring in various forms of hereditary systemic amyloidosis).
AMYLOIDOSIS AND THE TONGUE
Macroglossia is defined as protrusion of the tongue beyond the alveolar ridge of the teeth at rest. When caused by amyloidosis, it is most often associated with the systemic AL variant and is present in 10% to 23% of patients with this subtype.2
On physical examination, tongue enlargement can present with lateral indentations, with a smooth contour or with nodular deposits. Less often, bullous lesions, vesicles, and ulcers can also be seen, particularly on the lips. Infiltration of salivary glands can result in xerostomia. Functional symptoms, such as hypogeusia, dysarthria, dysphagia, dysphonia, and, in advanced cases, upper-airway dysfunction can result from restricted mobility of the tongue and tethering to deeper structures.
Surgical management may be necessary if severe obstructive symptoms are present, but infiltrative lesions tend to recur.
AMYLOIDOSIS AND THE HEART
Cardiac involvement in amyloidosis is currently the primary determinant of prognosis.3 It is more often seen in the AL, senile, and hereditary forms. It usually manifests as diastolic heart failure, but angina, orthostatic hypotension, dysrhythmias, and syncope can also occur. Systolic dysfunction is typically a late finding in the course of the disease.
Although an electrocardiographic pattern of low voltage in the precordial and limb leads has been classically associated with cardiac amyloidosis, only 30% of patients with the senile and hereditary forms show this feature.4 Left ventricular hypertrophy on electrocardiography is thought to be uncommon, but it has been reported in 16% of patients with AL amyloidosis and biopsy-proven cardiac involvement.5 Cardiac troponin levels can be elevated, as seen in other infiltrative cardiomyopathies.3 The diagnosis can be established by endomyocardial biopsy or indirectly by cardiac imaging and a positive extracardiac biopsy.
Drug therapy is supportive and mainly involves diuretics, since angiotensin-converting enzyme inhibitors, beta-blockers, and calcium channel blockers may cause hypotension and exacerbate myocardial dysfunction. The specific treatment varies depending on the underlying cause of amyloidosis.
- Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid 2007; 14:179–183.
- Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995; 32:45–59.
- Kapoor P, Thenappan T, Singh E, Kumar S, Greipp PR. Cardiac amyloidosis: a practical approach to diagnosis and management. Am J Med 2011; 124:1006–1015.
- Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med 2005; 165:1425–1429.
- Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, Grogan M. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 2005; 95:535–537.
A 66-year-old woman was prompted by her dentist to seek medical attention for an unusually enlarged, smooth-appearing tongue (Figure 1). She also complained of fatigue, dyspnea on exertion, and tingling of her hands.
Basic laboratory tests showed normocytic anemia and renal insufficiency. Her thyrotropin level was within normal limits. Serum protein electrophoresis showed a monoclonal M-spike, which prompted a bone marrow biopsy that was diagnostic of multiple myeloma.
Transthoracic echocardiography revealed diffuse hypokinesis with a restrictive filling pattern, myocardial thickening, and moderate mitral and tricuspid regurgitation, highly suggestive of an infiltrative cardiomyopathy. Biopsy of the right ventricle confirmed cardiac amyloidosis of the amyloid immunoglobulin light chain (AL) subtype.
The patient underwent chemotherapy, followed by autologous stem-cell transplantation. She achieved successful remission, and her cardiomyopathy was compensated.
AMYLOIDOSIS IS HETEROGENEOUS
Amyloidosis is a heterogeneous syndrome characterized by abnormal folding of proteins that deposit as insoluble fibrils in different tissues, impairing both structure and function. Virchow was the first to describe amyloid (from amylon, Greek for starch) as an abnormal material seen in postmortem examination of the liver. On Congo red staining, the extracellular proteins appear as salmon-red conglomerates, which also show apple-green birefringence under polarized light.
Amyloidosis can be localized but more often represents a systemic process, often associated with a plasma cell dyscrasia such as multiple myeloma.
Modern classification is based on the precursor protein,1 eg:
- Light chains (AL)
- Acute-phase protein (AA)
- Beta-2-microglobulin (Aß2M)
- Transthyretin (ATTR; occurring in senile systemic amyloidosis)
- Other proteins (occurring in various forms of hereditary systemic amyloidosis).
AMYLOIDOSIS AND THE TONGUE
Macroglossia is defined as protrusion of the tongue beyond the alveolar ridge of the teeth at rest. When caused by amyloidosis, it is most often associated with the systemic AL variant and is present in 10% to 23% of patients with this subtype.2
On physical examination, tongue enlargement can present with lateral indentations, with a smooth contour or with nodular deposits. Less often, bullous lesions, vesicles, and ulcers can also be seen, particularly on the lips. Infiltration of salivary glands can result in xerostomia. Functional symptoms, such as hypogeusia, dysarthria, dysphagia, dysphonia, and, in advanced cases, upper-airway dysfunction can result from restricted mobility of the tongue and tethering to deeper structures.
Surgical management may be necessary if severe obstructive symptoms are present, but infiltrative lesions tend to recur.
AMYLOIDOSIS AND THE HEART
Cardiac involvement in amyloidosis is currently the primary determinant of prognosis.3 It is more often seen in the AL, senile, and hereditary forms. It usually manifests as diastolic heart failure, but angina, orthostatic hypotension, dysrhythmias, and syncope can also occur. Systolic dysfunction is typically a late finding in the course of the disease.
Although an electrocardiographic pattern of low voltage in the precordial and limb leads has been classically associated with cardiac amyloidosis, only 30% of patients with the senile and hereditary forms show this feature.4 Left ventricular hypertrophy on electrocardiography is thought to be uncommon, but it has been reported in 16% of patients with AL amyloidosis and biopsy-proven cardiac involvement.5 Cardiac troponin levels can be elevated, as seen in other infiltrative cardiomyopathies.3 The diagnosis can be established by endomyocardial biopsy or indirectly by cardiac imaging and a positive extracardiac biopsy.
Drug therapy is supportive and mainly involves diuretics, since angiotensin-converting enzyme inhibitors, beta-blockers, and calcium channel blockers may cause hypotension and exacerbate myocardial dysfunction. The specific treatment varies depending on the underlying cause of amyloidosis.
A 66-year-old woman was prompted by her dentist to seek medical attention for an unusually enlarged, smooth-appearing tongue (Figure 1). She also complained of fatigue, dyspnea on exertion, and tingling of her hands.
Basic laboratory tests showed normocytic anemia and renal insufficiency. Her thyrotropin level was within normal limits. Serum protein electrophoresis showed a monoclonal M-spike, which prompted a bone marrow biopsy that was diagnostic of multiple myeloma.
Transthoracic echocardiography revealed diffuse hypokinesis with a restrictive filling pattern, myocardial thickening, and moderate mitral and tricuspid regurgitation, highly suggestive of an infiltrative cardiomyopathy. Biopsy of the right ventricle confirmed cardiac amyloidosis of the amyloid immunoglobulin light chain (AL) subtype.
The patient underwent chemotherapy, followed by autologous stem-cell transplantation. She achieved successful remission, and her cardiomyopathy was compensated.
AMYLOIDOSIS IS HETEROGENEOUS
Amyloidosis is a heterogeneous syndrome characterized by abnormal folding of proteins that deposit as insoluble fibrils in different tissues, impairing both structure and function. Virchow was the first to describe amyloid (from amylon, Greek for starch) as an abnormal material seen in postmortem examination of the liver. On Congo red staining, the extracellular proteins appear as salmon-red conglomerates, which also show apple-green birefringence under polarized light.
Amyloidosis can be localized but more often represents a systemic process, often associated with a plasma cell dyscrasia such as multiple myeloma.
Modern classification is based on the precursor protein,1 eg:
- Light chains (AL)
- Acute-phase protein (AA)
- Beta-2-microglobulin (Aß2M)
- Transthyretin (ATTR; occurring in senile systemic amyloidosis)
- Other proteins (occurring in various forms of hereditary systemic amyloidosis).
AMYLOIDOSIS AND THE TONGUE
Macroglossia is defined as protrusion of the tongue beyond the alveolar ridge of the teeth at rest. When caused by amyloidosis, it is most often associated with the systemic AL variant and is present in 10% to 23% of patients with this subtype.2
On physical examination, tongue enlargement can present with lateral indentations, with a smooth contour or with nodular deposits. Less often, bullous lesions, vesicles, and ulcers can also be seen, particularly on the lips. Infiltration of salivary glands can result in xerostomia. Functional symptoms, such as hypogeusia, dysarthria, dysphagia, dysphonia, and, in advanced cases, upper-airway dysfunction can result from restricted mobility of the tongue and tethering to deeper structures.
Surgical management may be necessary if severe obstructive symptoms are present, but infiltrative lesions tend to recur.
AMYLOIDOSIS AND THE HEART
Cardiac involvement in amyloidosis is currently the primary determinant of prognosis.3 It is more often seen in the AL, senile, and hereditary forms. It usually manifests as diastolic heart failure, but angina, orthostatic hypotension, dysrhythmias, and syncope can also occur. Systolic dysfunction is typically a late finding in the course of the disease.
Although an electrocardiographic pattern of low voltage in the precordial and limb leads has been classically associated with cardiac amyloidosis, only 30% of patients with the senile and hereditary forms show this feature.4 Left ventricular hypertrophy on electrocardiography is thought to be uncommon, but it has been reported in 16% of patients with AL amyloidosis and biopsy-proven cardiac involvement.5 Cardiac troponin levels can be elevated, as seen in other infiltrative cardiomyopathies.3 The diagnosis can be established by endomyocardial biopsy or indirectly by cardiac imaging and a positive extracardiac biopsy.
Drug therapy is supportive and mainly involves diuretics, since angiotensin-converting enzyme inhibitors, beta-blockers, and calcium channel blockers may cause hypotension and exacerbate myocardial dysfunction. The specific treatment varies depending on the underlying cause of amyloidosis.
- Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid 2007; 14:179–183.
- Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995; 32:45–59.
- Kapoor P, Thenappan T, Singh E, Kumar S, Greipp PR. Cardiac amyloidosis: a practical approach to diagnosis and management. Am J Med 2011; 124:1006–1015.
- Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med 2005; 165:1425–1429.
- Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, Grogan M. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 2005; 95:535–537.
- Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid 2007; 14:179–183.
- Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995; 32:45–59.
- Kapoor P, Thenappan T, Singh E, Kumar S, Greipp PR. Cardiac amyloidosis: a practical approach to diagnosis and management. Am J Med 2011; 124:1006–1015.
- Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med 2005; 165:1425–1429.
- Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, Grogan M. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 2005; 95:535–537.
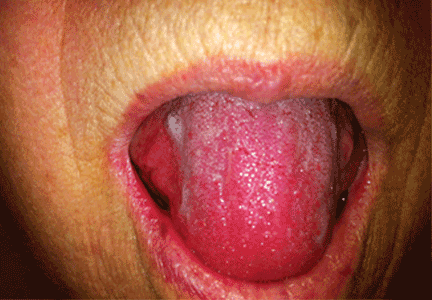
Dyspnea after treatment of recurrent urinary tract infection
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
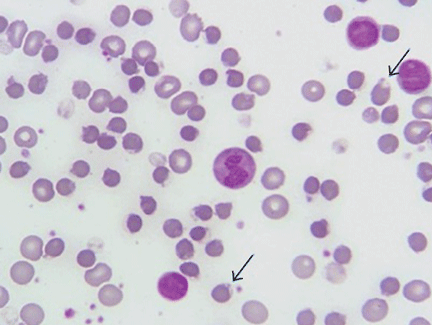
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.

More than skin-deep
A 68-year-old man presented for evaluation of a diffuse rash with mucosal involvement. During the past 9 months, he had had oral ulcers, a truncal rash, and blistering lesions on his hands with fingernail erosion, and all of these had been getting worse (Figure 1). He also reported cycles of fever and a weight loss of 50 lb.
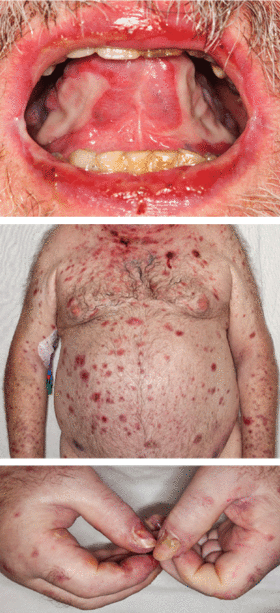
Computed tomography revealed diffuse lymphadenopathy, including a bulky 18-cm mediastinal mass. Lymph node biopsy confirmed follicular non-Hodgkin lymphoma.
On serum enzyme-linked immunosorbent assay (ELISA), the desmoglein 1 antibody titer was 62.4 U (< 14 is negative, > 20 is positive) and the desmoglein 3 antibody titer was 36.0 U (< 9 is negative, > 20 is positive). Indirect immunofluorescence testing on monkey esophagus substrate was reactive.
ELISA detected no immunoglobulin G (IgG) reactivity against the bullous pemphigoid autoantigens BP180 and BP230. Direct immunofluorescence testing revealed strong IgG and C3 deposition on epithelial cell surfaces, and indirect immunofluorescence on rat bladder substrate was positive, suggesting the diagnosis of paraneoplastic pemphigus.
The patient was treated with dexamethasone and bendamustine-rituximab. Five months later, desmoglein 1 and 3 antibody titers were measured again and were within normal limits. His lesions had improved significantly (Figure 2), but he had endured multiple medical setbacks, including Pseudomonas peritonitis, fistulizing cytomegalovirus perianal ulceration, septic shock secondary to fulminant Clostridium difficile colitis, and toxic megacolon necessitating total colectomy with end-ileostomy.
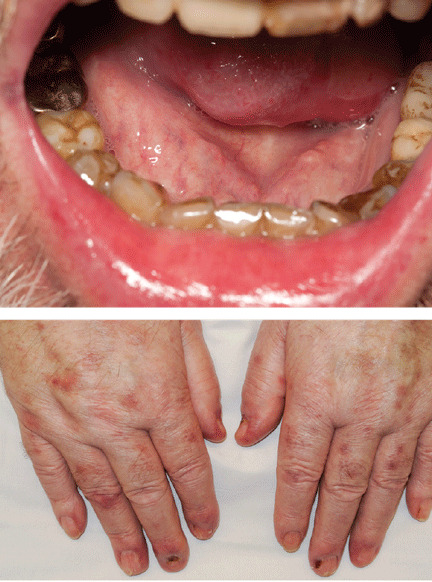
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus usually occurs in the presence of underlying lymphoproliferative neoplasm, most often non-Hodgkin lymphoma, chronic lymphocytic leukemia, or Castleman disease. It may also occur with epithelial carcinoma, mesenchymal sarcoma, and, rarely, malignant melanoma.1 The association between malignancy and autoimmune mucocutaneous disease was first described in 1990.2 Autoantibodies against periplakin, envoplakin, and desmoglein 3 are often present and may play a role in pathogenesis.
Paraneoplastic pemphigus associated with malignancy portends a poor prognosis. Although historically reported to have an overall death rate ranging from 75% to 90%, with a mean survival of less than 1 year, a recent retrospective cohort has shown slightly improved outcomes, with 49% of patients remaining alive at 1 year and 38% alive at 5 years.3 The most common causes of death are sepsis, respiratory failure, and the underlying malignancy. Bronchiolitis obliterans may occur late in the disease course and is an ominous prognostic factor, with a death rate of 41% after a median interval of 13 months.4
Immunosuppressive drugs are often used to control the disease. First-line treatment is with high-dose steroids. Immunosuppressives such as azathioprine and cyclosporin may be used in conjunction to reduce the steroid dose that is required and to limit the adverse effects of steroid therapy. Treating the underlying malignancy may decrease autoantibody production and lead to clinical improvement.
- Kaplan I, Hodak E, Ackerman L, Mimouni D, Anhalt GJ, Calderon S. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol 2004; 40:553–562.
- Anhalt GJ, Kim SC, Stanley JR, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med 1990; 323:1729–1735.
- Leger S, Picard D, Ingen-Housz-Oro S, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol 2012; 148:1165–1172.
- Maldonado F, Pittelkow MR, Ryu JH. Constrictive bronchiolitis associated with pareaneoplastic autoimmune multi-organ syndrome. Respirology 2009; 14:129–133.
A 68-year-old man presented for evaluation of a diffuse rash with mucosal involvement. During the past 9 months, he had had oral ulcers, a truncal rash, and blistering lesions on his hands with fingernail erosion, and all of these had been getting worse (Figure 1). He also reported cycles of fever and a weight loss of 50 lb.
Computed tomography revealed diffuse lymphadenopathy, including a bulky 18-cm mediastinal mass. Lymph node biopsy confirmed follicular non-Hodgkin lymphoma.
On serum enzyme-linked immunosorbent assay (ELISA), the desmoglein 1 antibody titer was 62.4 U (< 14 is negative, > 20 is positive) and the desmoglein 3 antibody titer was 36.0 U (< 9 is negative, > 20 is positive). Indirect immunofluorescence testing on monkey esophagus substrate was reactive.
ELISA detected no immunoglobulin G (IgG) reactivity against the bullous pemphigoid autoantigens BP180 and BP230. Direct immunofluorescence testing revealed strong IgG and C3 deposition on epithelial cell surfaces, and indirect immunofluorescence on rat bladder substrate was positive, suggesting the diagnosis of paraneoplastic pemphigus.
The patient was treated with dexamethasone and bendamustine-rituximab. Five months later, desmoglein 1 and 3 antibody titers were measured again and were within normal limits. His lesions had improved significantly (Figure 2), but he had endured multiple medical setbacks, including Pseudomonas peritonitis, fistulizing cytomegalovirus perianal ulceration, septic shock secondary to fulminant Clostridium difficile colitis, and toxic megacolon necessitating total colectomy with end-ileostomy.
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus usually occurs in the presence of underlying lymphoproliferative neoplasm, most often non-Hodgkin lymphoma, chronic lymphocytic leukemia, or Castleman disease. It may also occur with epithelial carcinoma, mesenchymal sarcoma, and, rarely, malignant melanoma.1 The association between malignancy and autoimmune mucocutaneous disease was first described in 1990.2 Autoantibodies against periplakin, envoplakin, and desmoglein 3 are often present and may play a role in pathogenesis.
Paraneoplastic pemphigus associated with malignancy portends a poor prognosis. Although historically reported to have an overall death rate ranging from 75% to 90%, with a mean survival of less than 1 year, a recent retrospective cohort has shown slightly improved outcomes, with 49% of patients remaining alive at 1 year and 38% alive at 5 years.3 The most common causes of death are sepsis, respiratory failure, and the underlying malignancy. Bronchiolitis obliterans may occur late in the disease course and is an ominous prognostic factor, with a death rate of 41% after a median interval of 13 months.4
Immunosuppressive drugs are often used to control the disease. First-line treatment is with high-dose steroids. Immunosuppressives such as azathioprine and cyclosporin may be used in conjunction to reduce the steroid dose that is required and to limit the adverse effects of steroid therapy. Treating the underlying malignancy may decrease autoantibody production and lead to clinical improvement.
A 68-year-old man presented for evaluation of a diffuse rash with mucosal involvement. During the past 9 months, he had had oral ulcers, a truncal rash, and blistering lesions on his hands with fingernail erosion, and all of these had been getting worse (Figure 1). He also reported cycles of fever and a weight loss of 50 lb.
Computed tomography revealed diffuse lymphadenopathy, including a bulky 18-cm mediastinal mass. Lymph node biopsy confirmed follicular non-Hodgkin lymphoma.
On serum enzyme-linked immunosorbent assay (ELISA), the desmoglein 1 antibody titer was 62.4 U (< 14 is negative, > 20 is positive) and the desmoglein 3 antibody titer was 36.0 U (< 9 is negative, > 20 is positive). Indirect immunofluorescence testing on monkey esophagus substrate was reactive.
ELISA detected no immunoglobulin G (IgG) reactivity against the bullous pemphigoid autoantigens BP180 and BP230. Direct immunofluorescence testing revealed strong IgG and C3 deposition on epithelial cell surfaces, and indirect immunofluorescence on rat bladder substrate was positive, suggesting the diagnosis of paraneoplastic pemphigus.
The patient was treated with dexamethasone and bendamustine-rituximab. Five months later, desmoglein 1 and 3 antibody titers were measured again and were within normal limits. His lesions had improved significantly (Figure 2), but he had endured multiple medical setbacks, including Pseudomonas peritonitis, fistulizing cytomegalovirus perianal ulceration, septic shock secondary to fulminant Clostridium difficile colitis, and toxic megacolon necessitating total colectomy with end-ileostomy.
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus usually occurs in the presence of underlying lymphoproliferative neoplasm, most often non-Hodgkin lymphoma, chronic lymphocytic leukemia, or Castleman disease. It may also occur with epithelial carcinoma, mesenchymal sarcoma, and, rarely, malignant melanoma.1 The association between malignancy and autoimmune mucocutaneous disease was first described in 1990.2 Autoantibodies against periplakin, envoplakin, and desmoglein 3 are often present and may play a role in pathogenesis.
Paraneoplastic pemphigus associated with malignancy portends a poor prognosis. Although historically reported to have an overall death rate ranging from 75% to 90%, with a mean survival of less than 1 year, a recent retrospective cohort has shown slightly improved outcomes, with 49% of patients remaining alive at 1 year and 38% alive at 5 years.3 The most common causes of death are sepsis, respiratory failure, and the underlying malignancy. Bronchiolitis obliterans may occur late in the disease course and is an ominous prognostic factor, with a death rate of 41% after a median interval of 13 months.4
Immunosuppressive drugs are often used to control the disease. First-line treatment is with high-dose steroids. Immunosuppressives such as azathioprine and cyclosporin may be used in conjunction to reduce the steroid dose that is required and to limit the adverse effects of steroid therapy. Treating the underlying malignancy may decrease autoantibody production and lead to clinical improvement.
- Kaplan I, Hodak E, Ackerman L, Mimouni D, Anhalt GJ, Calderon S. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol 2004; 40:553–562.
- Anhalt GJ, Kim SC, Stanley JR, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med 1990; 323:1729–1735.
- Leger S, Picard D, Ingen-Housz-Oro S, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol 2012; 148:1165–1172.
- Maldonado F, Pittelkow MR, Ryu JH. Constrictive bronchiolitis associated with pareaneoplastic autoimmune multi-organ syndrome. Respirology 2009; 14:129–133.
- Kaplan I, Hodak E, Ackerman L, Mimouni D, Anhalt GJ, Calderon S. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol 2004; 40:553–562.
- Anhalt GJ, Kim SC, Stanley JR, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med 1990; 323:1729–1735.
- Leger S, Picard D, Ingen-Housz-Oro S, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol 2012; 148:1165–1172.
- Maldonado F, Pittelkow MR, Ryu JH. Constrictive bronchiolitis associated with pareaneoplastic autoimmune multi-organ syndrome. Respirology 2009; 14:129–133.
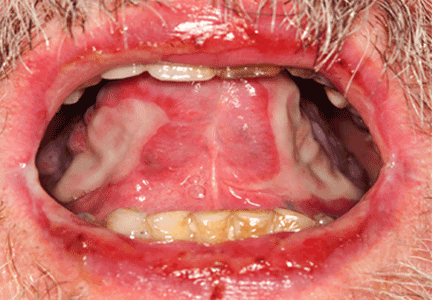
Peripheral opacity on plain chest radiography
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
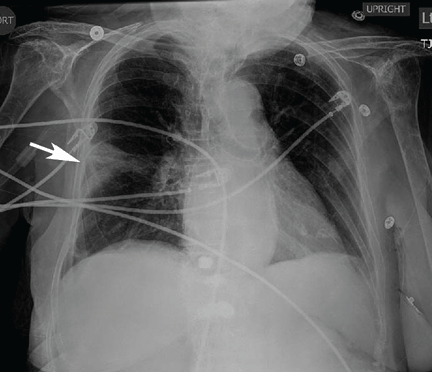
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
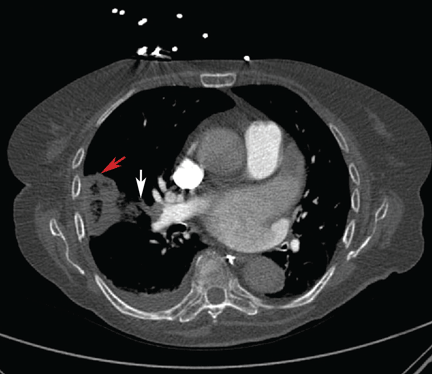
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
Fever, dyspnea, and a new heart murmur
A 35-year-old man presented to the emergency department because of night sweats, fever, chills, and shortness of breath. He also had an acute onset of blue discoloration of his right fourth finger. His symptoms (except for the finger discoloration) had begun about 6 months previously and had rapidly progressed despite several courses of different antibiotics of different types, given both intravenously in the hospital and orally at home. He had lost 20 lb during this time. Previously, he had been healthy.
About 1 month after his symptoms began, he had consulted his primary care physician, who detected a new grade 4/6 systolic and diastolic murmur. Transthoracic echocardiography about 2 months after that demonstrated mild aortic and mitral insufficiency but no echocardiographic features supporting infective endocarditis. Of note, the patient had no risk factors for endocarditis such as illicit drug use or poor dental health.
In the emergency department, his temperature was 99.4°F (37.4°C), pulse 109 beats per minute, and blood pressure 126/60 mm Hg. He had a grade 3/6 harsh holosystolic murmur best heard at the right upper sternal border, a grade 3/4 holodiastolic murmur audible across the precordium, and a grade 3/4 holosystolic blowing murmur best heard at the cardiac apex. Other findings included signs of aortic insufficiency—the Duroziez sign (a diastolic murmur heard over the femoral artery when compressed), Watson’s water-hammer pulse (indicating a wide pulse pressure), and the Müller sign (pulsation of the uvula)—and small Janeway lesions on the inner aspect of his right arm and palm.
Electrocardiography showed normal sinus rhythm, PR interval 128 ms, QRS complex 100 ms, QT interval 360 ms, and corrected QT interval 473 ms.
Blood cultures grew Streptococcus sanguinis. Both transthoracic and transesophageal echocardiography were done promptly and revealed multiple mobile echodensities attached to a trileaflet aortic valve, consistent with vegetations and valve leaflet destruction; severe (4+) aortic regurgitation with flow reversal in the abdominal aorta; mild mitral regurgitation; and a mitral valve aneurysm with mild mitral regurgitation (Figure 1).
INFECTIVE ENDOCARDITIS: WORTH CONSIDERING
S sanguinis is a member of the group of viridans streptococci. As a normal inhabitant of the healthy human mouth, it is found in dental plaque. It may enter the bloodstream during dental cleaning and may colonize the heart valves, particularly the mitral and aortic valves, where it is the most common cause of subacute bacterial endocarditis.
Infective endocarditis is often diagnosed clinically with the Duke criteria (www.med-calc.com/endocarditis.html).1 However, the variability of the clinical presentation and the nonspecific nature of the initial workup often create a diagnostic challenge for the evaluating physician.1,2
In cases of recurrent persistent fever and a new heart murmur, infective endocarditis must always be considered. Blood cultures should be ordered early and repeatedly. If blood cultures are positive, transesophageal echocardiography should be done without delay if transthoracic echocardiography was unremarkable. Prompt diagnosis and surgical intervention prevent complications.
MITRAL VALVE ANEURYSM IN AORTIC VALVE ENDOCARDITIS
Aortic valve endocarditis often also involves the mitral valve; mitral valve endocarditis is seen in 17% of patients undergoing surgery for aortic valve endocarditis.3 Proposed mechanisms for this association include jet lesions from aortic regurgitation, vegetation prolapse with direct contact between the aortic valve and anterior mitral leaflet (“kissing lesions”), and direct local spread of infection.4–7
One of every five patients with mitral valve involvement in aortic valve endocarditis has a mitral valve aneurysm.3 This is a serious finding, as it can lead to septic embolization. Also, the weakened lining of the mitral valve aneurysm can rupture, resulting in severe mitral regurgitation, acute pulmonary edema, and precipitous cardiopulmonary decompensation.5
Transesophageal echocardiography is more sensitive than transthoracic echocardiography for detecting mitral valve aneurysm.8 On two-dimensional echocardiography, the lesion appears as a narrow-necked, saccular echolucency with systolic protrusion into the left atrium. Color Doppler imaging often shows turbulent, high-velocity flow.
Differential diagnosis of mitral valve aneurysm
Differential diagnostic considerations include a valvular blood cyst, a congenital cardiac diverticulum, and mitral valve prolapse.
Valvular blood cysts are extremely rare in adults.9 These benign, congenital tumors are most often found on the atrioventricular valves in infants, in whom the reported incidence is between 25% and 100%. In almost all cases, these cysts are believed to regress spontaneously with time.
In almost all reported cases, the cyst involved the valvular apparatus or papillary muscle of the tricuspid, pulmonary, or mitral valve.10 Cysts consist of a benign diverticulum lined with flattened, cobblestone-shaped endothelium and are filled with blood. They can cause heart murmurs in otherwise asymptomatic patients.
On echocardiography, a blood cyst appears as an oval mass (often at the interatrial septum), often with normal cardiac function. In the rare case in which a blood cyst is found incidentally during echocardiography, the hemodynamic impact, if any, should be determined by Doppler techniques.
When benign, a valvular blood cyst can be safely monitored with echocardiographic follow-up.11 Treatment involves surgical resection of the mass in symptomatic patients in whom cardiac function is impaired by the presence of the cyst.
Congenital cardiac diverticuli are extremely rare, most often seen in children, and associated with a midline thoracoabdominal defect. Echocardiography can differentiate a ventricular diverticulum from an aneurysm or a pseudoaneurysm.
A ventricular diverticulum has a fibrous, narrow neck connecting with the ventricle, and a small circular echo-free space that communicates with the ventricle via this narrow neck.2 Doppler imaging shows systolic flow from the diverticulum to the ventricle, and systolic contractility may also be seen during cardiac catheterization. Congenital diverticulum is typically confused with ventricular aneurysm and, to a lesser degree, with mitral valve aneurysm.
Mitral valve prolapse is characterized by interchordal ballooning or hooding of the mitral valve leaflets that occurs when one or both floppy, enlarged leaflets prolapse into the left atrium during systole.
BACK TO OUR PATIENT
The patient underwent open heart surgery, with successful repair of the aortic root, replacement of the aortic valve, and repair of the mitral valve. An abscess was found within the aneurysmal cavity.
- Durack DT, Lukes AS, Bright DK. New criteria for diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Duke Endocarditis Service. Am J Med 1994; 96:200–209.
- Prendergast BD. Diagnostic criteria and problems in infective endocarditis. Heart 2004; 90:611–613.
- Gonzalez-Lavin L, Lise M, Ross D. The importance of the ‘jet lesion’ in bacterial endocarditis involving the left heart. Surgical considerations. J Thorac Cardiovasc Surg 1970; 59:185–192.
- Silbiger JJ. Review: mitral valve aneurysms in infective endocarditis: mechanisms, clinical recognition, and treatment. J Heart Valve Dis 2009; 18:476–480.
- Reid CL, Chandraratna AN, Harrison E, et al. Mitral valve aneurysm: clinical features, echocardiographic-pathologic correlations. J Am Coll Cardiol 1983; 2:460–464.
- Rodbard S. Blood velocity and endocarditis. Circulation 1963; 27:18–28.
- Piper C, Hetzer R, Körfer R, Bergemann R, Horstkotte D. The importance of secondary mitral valve involvement in primary aortic valve endocarditis; the mitral kissing vegetation. Eur Heart J 2002; 23:79–86.
- Cziner DG, Rosenzweig BP, Katz ES, Keller AM, Daniel WG, Kronzon I. Transesophageal versus transthoracic echocardiography for diagnosing mitral valve perforation. Am J Cardiol 1992; 69:1495–1497.
- Roberts PF, Serra AJ, McNicholas KW, Shapira N, Lemole GM. Atrial blood cyst: a rare finding. Ann Thorac Surg 1996; 62:880–882.
- Grimaldi A, Capritti E, Pappalardo F, et al. Images in cardiovascular medicine: blood cyst of the mitral valve. J Cardiovasc Med 2012; 3:46.
- Boyd WC, Rosengart TK, Hartman GS. Isolated left ventricular diverticulum in an adult. J Cardiothorac Vasc Anesth 1999; 13:468–470.
A 35-year-old man presented to the emergency department because of night sweats, fever, chills, and shortness of breath. He also had an acute onset of blue discoloration of his right fourth finger. His symptoms (except for the finger discoloration) had begun about 6 months previously and had rapidly progressed despite several courses of different antibiotics of different types, given both intravenously in the hospital and orally at home. He had lost 20 lb during this time. Previously, he had been healthy.
About 1 month after his symptoms began, he had consulted his primary care physician, who detected a new grade 4/6 systolic and diastolic murmur. Transthoracic echocardiography about 2 months after that demonstrated mild aortic and mitral insufficiency but no echocardiographic features supporting infective endocarditis. Of note, the patient had no risk factors for endocarditis such as illicit drug use or poor dental health.
In the emergency department, his temperature was 99.4°F (37.4°C), pulse 109 beats per minute, and blood pressure 126/60 mm Hg. He had a grade 3/6 harsh holosystolic murmur best heard at the right upper sternal border, a grade 3/4 holodiastolic murmur audible across the precordium, and a grade 3/4 holosystolic blowing murmur best heard at the cardiac apex. Other findings included signs of aortic insufficiency—the Duroziez sign (a diastolic murmur heard over the femoral artery when compressed), Watson’s water-hammer pulse (indicating a wide pulse pressure), and the Müller sign (pulsation of the uvula)—and small Janeway lesions on the inner aspect of his right arm and palm.
Electrocardiography showed normal sinus rhythm, PR interval 128 ms, QRS complex 100 ms, QT interval 360 ms, and corrected QT interval 473 ms.
Blood cultures grew Streptococcus sanguinis. Both transthoracic and transesophageal echocardiography were done promptly and revealed multiple mobile echodensities attached to a trileaflet aortic valve, consistent with vegetations and valve leaflet destruction; severe (4+) aortic regurgitation with flow reversal in the abdominal aorta; mild mitral regurgitation; and a mitral valve aneurysm with mild mitral regurgitation (Figure 1).
INFECTIVE ENDOCARDITIS: WORTH CONSIDERING
S sanguinis is a member of the group of viridans streptococci. As a normal inhabitant of the healthy human mouth, it is found in dental plaque. It may enter the bloodstream during dental cleaning and may colonize the heart valves, particularly the mitral and aortic valves, where it is the most common cause of subacute bacterial endocarditis.
Infective endocarditis is often diagnosed clinically with the Duke criteria (www.med-calc.com/endocarditis.html).1 However, the variability of the clinical presentation and the nonspecific nature of the initial workup often create a diagnostic challenge for the evaluating physician.1,2
In cases of recurrent persistent fever and a new heart murmur, infective endocarditis must always be considered. Blood cultures should be ordered early and repeatedly. If blood cultures are positive, transesophageal echocardiography should be done without delay if transthoracic echocardiography was unremarkable. Prompt diagnosis and surgical intervention prevent complications.
MITRAL VALVE ANEURYSM IN AORTIC VALVE ENDOCARDITIS
Aortic valve endocarditis often also involves the mitral valve; mitral valve endocarditis is seen in 17% of patients undergoing surgery for aortic valve endocarditis.3 Proposed mechanisms for this association include jet lesions from aortic regurgitation, vegetation prolapse with direct contact between the aortic valve and anterior mitral leaflet (“kissing lesions”), and direct local spread of infection.4–7
One of every five patients with mitral valve involvement in aortic valve endocarditis has a mitral valve aneurysm.3 This is a serious finding, as it can lead to septic embolization. Also, the weakened lining of the mitral valve aneurysm can rupture, resulting in severe mitral regurgitation, acute pulmonary edema, and precipitous cardiopulmonary decompensation.5
Transesophageal echocardiography is more sensitive than transthoracic echocardiography for detecting mitral valve aneurysm.8 On two-dimensional echocardiography, the lesion appears as a narrow-necked, saccular echolucency with systolic protrusion into the left atrium. Color Doppler imaging often shows turbulent, high-velocity flow.
Differential diagnosis of mitral valve aneurysm
Differential diagnostic considerations include a valvular blood cyst, a congenital cardiac diverticulum, and mitral valve prolapse.
Valvular blood cysts are extremely rare in adults.9 These benign, congenital tumors are most often found on the atrioventricular valves in infants, in whom the reported incidence is between 25% and 100%. In almost all cases, these cysts are believed to regress spontaneously with time.
In almost all reported cases, the cyst involved the valvular apparatus or papillary muscle of the tricuspid, pulmonary, or mitral valve.10 Cysts consist of a benign diverticulum lined with flattened, cobblestone-shaped endothelium and are filled with blood. They can cause heart murmurs in otherwise asymptomatic patients.
On echocardiography, a blood cyst appears as an oval mass (often at the interatrial septum), often with normal cardiac function. In the rare case in which a blood cyst is found incidentally during echocardiography, the hemodynamic impact, if any, should be determined by Doppler techniques.
When benign, a valvular blood cyst can be safely monitored with echocardiographic follow-up.11 Treatment involves surgical resection of the mass in symptomatic patients in whom cardiac function is impaired by the presence of the cyst.
Congenital cardiac diverticuli are extremely rare, most often seen in children, and associated with a midline thoracoabdominal defect. Echocardiography can differentiate a ventricular diverticulum from an aneurysm or a pseudoaneurysm.
A ventricular diverticulum has a fibrous, narrow neck connecting with the ventricle, and a small circular echo-free space that communicates with the ventricle via this narrow neck.2 Doppler imaging shows systolic flow from the diverticulum to the ventricle, and systolic contractility may also be seen during cardiac catheterization. Congenital diverticulum is typically confused with ventricular aneurysm and, to a lesser degree, with mitral valve aneurysm.
Mitral valve prolapse is characterized by interchordal ballooning or hooding of the mitral valve leaflets that occurs when one or both floppy, enlarged leaflets prolapse into the left atrium during systole.
BACK TO OUR PATIENT
The patient underwent open heart surgery, with successful repair of the aortic root, replacement of the aortic valve, and repair of the mitral valve. An abscess was found within the aneurysmal cavity.
A 35-year-old man presented to the emergency department because of night sweats, fever, chills, and shortness of breath. He also had an acute onset of blue discoloration of his right fourth finger. His symptoms (except for the finger discoloration) had begun about 6 months previously and had rapidly progressed despite several courses of different antibiotics of different types, given both intravenously in the hospital and orally at home. He had lost 20 lb during this time. Previously, he had been healthy.
About 1 month after his symptoms began, he had consulted his primary care physician, who detected a new grade 4/6 systolic and diastolic murmur. Transthoracic echocardiography about 2 months after that demonstrated mild aortic and mitral insufficiency but no echocardiographic features supporting infective endocarditis. Of note, the patient had no risk factors for endocarditis such as illicit drug use or poor dental health.
In the emergency department, his temperature was 99.4°F (37.4°C), pulse 109 beats per minute, and blood pressure 126/60 mm Hg. He had a grade 3/6 harsh holosystolic murmur best heard at the right upper sternal border, a grade 3/4 holodiastolic murmur audible across the precordium, and a grade 3/4 holosystolic blowing murmur best heard at the cardiac apex. Other findings included signs of aortic insufficiency—the Duroziez sign (a diastolic murmur heard over the femoral artery when compressed), Watson’s water-hammer pulse (indicating a wide pulse pressure), and the Müller sign (pulsation of the uvula)—and small Janeway lesions on the inner aspect of his right arm and palm.
Electrocardiography showed normal sinus rhythm, PR interval 128 ms, QRS complex 100 ms, QT interval 360 ms, and corrected QT interval 473 ms.
Blood cultures grew Streptococcus sanguinis. Both transthoracic and transesophageal echocardiography were done promptly and revealed multiple mobile echodensities attached to a trileaflet aortic valve, consistent with vegetations and valve leaflet destruction; severe (4+) aortic regurgitation with flow reversal in the abdominal aorta; mild mitral regurgitation; and a mitral valve aneurysm with mild mitral regurgitation (Figure 1).
INFECTIVE ENDOCARDITIS: WORTH CONSIDERING
S sanguinis is a member of the group of viridans streptococci. As a normal inhabitant of the healthy human mouth, it is found in dental plaque. It may enter the bloodstream during dental cleaning and may colonize the heart valves, particularly the mitral and aortic valves, where it is the most common cause of subacute bacterial endocarditis.
Infective endocarditis is often diagnosed clinically with the Duke criteria (www.med-calc.com/endocarditis.html).1 However, the variability of the clinical presentation and the nonspecific nature of the initial workup often create a diagnostic challenge for the evaluating physician.1,2
In cases of recurrent persistent fever and a new heart murmur, infective endocarditis must always be considered. Blood cultures should be ordered early and repeatedly. If blood cultures are positive, transesophageal echocardiography should be done without delay if transthoracic echocardiography was unremarkable. Prompt diagnosis and surgical intervention prevent complications.
MITRAL VALVE ANEURYSM IN AORTIC VALVE ENDOCARDITIS
Aortic valve endocarditis often also involves the mitral valve; mitral valve endocarditis is seen in 17% of patients undergoing surgery for aortic valve endocarditis.3 Proposed mechanisms for this association include jet lesions from aortic regurgitation, vegetation prolapse with direct contact between the aortic valve and anterior mitral leaflet (“kissing lesions”), and direct local spread of infection.4–7
One of every five patients with mitral valve involvement in aortic valve endocarditis has a mitral valve aneurysm.3 This is a serious finding, as it can lead to septic embolization. Also, the weakened lining of the mitral valve aneurysm can rupture, resulting in severe mitral regurgitation, acute pulmonary edema, and precipitous cardiopulmonary decompensation.5
Transesophageal echocardiography is more sensitive than transthoracic echocardiography for detecting mitral valve aneurysm.8 On two-dimensional echocardiography, the lesion appears as a narrow-necked, saccular echolucency with systolic protrusion into the left atrium. Color Doppler imaging often shows turbulent, high-velocity flow.
Differential diagnosis of mitral valve aneurysm
Differential diagnostic considerations include a valvular blood cyst, a congenital cardiac diverticulum, and mitral valve prolapse.
Valvular blood cysts are extremely rare in adults.9 These benign, congenital tumors are most often found on the atrioventricular valves in infants, in whom the reported incidence is between 25% and 100%. In almost all cases, these cysts are believed to regress spontaneously with time.
In almost all reported cases, the cyst involved the valvular apparatus or papillary muscle of the tricuspid, pulmonary, or mitral valve.10 Cysts consist of a benign diverticulum lined with flattened, cobblestone-shaped endothelium and are filled with blood. They can cause heart murmurs in otherwise asymptomatic patients.
On echocardiography, a blood cyst appears as an oval mass (often at the interatrial septum), often with normal cardiac function. In the rare case in which a blood cyst is found incidentally during echocardiography, the hemodynamic impact, if any, should be determined by Doppler techniques.
When benign, a valvular blood cyst can be safely monitored with echocardiographic follow-up.11 Treatment involves surgical resection of the mass in symptomatic patients in whom cardiac function is impaired by the presence of the cyst.
Congenital cardiac diverticuli are extremely rare, most often seen in children, and associated with a midline thoracoabdominal defect. Echocardiography can differentiate a ventricular diverticulum from an aneurysm or a pseudoaneurysm.
A ventricular diverticulum has a fibrous, narrow neck connecting with the ventricle, and a small circular echo-free space that communicates with the ventricle via this narrow neck.2 Doppler imaging shows systolic flow from the diverticulum to the ventricle, and systolic contractility may also be seen during cardiac catheterization. Congenital diverticulum is typically confused with ventricular aneurysm and, to a lesser degree, with mitral valve aneurysm.
Mitral valve prolapse is characterized by interchordal ballooning or hooding of the mitral valve leaflets that occurs when one or both floppy, enlarged leaflets prolapse into the left atrium during systole.
BACK TO OUR PATIENT
The patient underwent open heart surgery, with successful repair of the aortic root, replacement of the aortic valve, and repair of the mitral valve. An abscess was found within the aneurysmal cavity.
- Durack DT, Lukes AS, Bright DK. New criteria for diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Duke Endocarditis Service. Am J Med 1994; 96:200–209.
- Prendergast BD. Diagnostic criteria and problems in infective endocarditis. Heart 2004; 90:611–613.
- Gonzalez-Lavin L, Lise M, Ross D. The importance of the ‘jet lesion’ in bacterial endocarditis involving the left heart. Surgical considerations. J Thorac Cardiovasc Surg 1970; 59:185–192.
- Silbiger JJ. Review: mitral valve aneurysms in infective endocarditis: mechanisms, clinical recognition, and treatment. J Heart Valve Dis 2009; 18:476–480.
- Reid CL, Chandraratna AN, Harrison E, et al. Mitral valve aneurysm: clinical features, echocardiographic-pathologic correlations. J Am Coll Cardiol 1983; 2:460–464.
- Rodbard S. Blood velocity and endocarditis. Circulation 1963; 27:18–28.
- Piper C, Hetzer R, Körfer R, Bergemann R, Horstkotte D. The importance of secondary mitral valve involvement in primary aortic valve endocarditis; the mitral kissing vegetation. Eur Heart J 2002; 23:79–86.
- Cziner DG, Rosenzweig BP, Katz ES, Keller AM, Daniel WG, Kronzon I. Transesophageal versus transthoracic echocardiography for diagnosing mitral valve perforation. Am J Cardiol 1992; 69:1495–1497.
- Roberts PF, Serra AJ, McNicholas KW, Shapira N, Lemole GM. Atrial blood cyst: a rare finding. Ann Thorac Surg 1996; 62:880–882.
- Grimaldi A, Capritti E, Pappalardo F, et al. Images in cardiovascular medicine: blood cyst of the mitral valve. J Cardiovasc Med 2012; 3:46.
- Boyd WC, Rosengart TK, Hartman GS. Isolated left ventricular diverticulum in an adult. J Cardiothorac Vasc Anesth 1999; 13:468–470.
- Durack DT, Lukes AS, Bright DK. New criteria for diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Duke Endocarditis Service. Am J Med 1994; 96:200–209.
- Prendergast BD. Diagnostic criteria and problems in infective endocarditis. Heart 2004; 90:611–613.
- Gonzalez-Lavin L, Lise M, Ross D. The importance of the ‘jet lesion’ in bacterial endocarditis involving the left heart. Surgical considerations. J Thorac Cardiovasc Surg 1970; 59:185–192.
- Silbiger JJ. Review: mitral valve aneurysms in infective endocarditis: mechanisms, clinical recognition, and treatment. J Heart Valve Dis 2009; 18:476–480.
- Reid CL, Chandraratna AN, Harrison E, et al. Mitral valve aneurysm: clinical features, echocardiographic-pathologic correlations. J Am Coll Cardiol 1983; 2:460–464.
- Rodbard S. Blood velocity and endocarditis. Circulation 1963; 27:18–28.
- Piper C, Hetzer R, Körfer R, Bergemann R, Horstkotte D. The importance of secondary mitral valve involvement in primary aortic valve endocarditis; the mitral kissing vegetation. Eur Heart J 2002; 23:79–86.
- Cziner DG, Rosenzweig BP, Katz ES, Keller AM, Daniel WG, Kronzon I. Transesophageal versus transthoracic echocardiography for diagnosing mitral valve perforation. Am J Cardiol 1992; 69:1495–1497.
- Roberts PF, Serra AJ, McNicholas KW, Shapira N, Lemole GM. Atrial blood cyst: a rare finding. Ann Thorac Surg 1996; 62:880–882.
- Grimaldi A, Capritti E, Pappalardo F, et al. Images in cardiovascular medicine: blood cyst of the mitral valve. J Cardiovasc Med 2012; 3:46.
- Boyd WC, Rosengart TK, Hartman GS. Isolated left ventricular diverticulum in an adult. J Cardiothorac Vasc Anesth 1999; 13:468–470.