User login
Chronic itch on the upper back, with pain
A 47-year-old man had had a chronic itch on his back for 2 years. He had no history of trauma to the site, nor did he recall applying topical products to that area.
He was otherwise healthy. He worked as an electrician and said he occasionally experienced cervical and back pain while working.
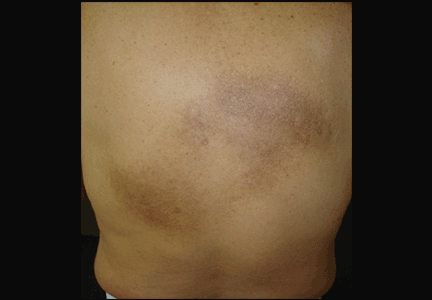
An examination revealed two grayish-brown ovoid patches on the upper back, each 5 cm to 7 cm in diameter (Figure 1).
DIAGNOSIS: NOTALGIA PARESTHETICA
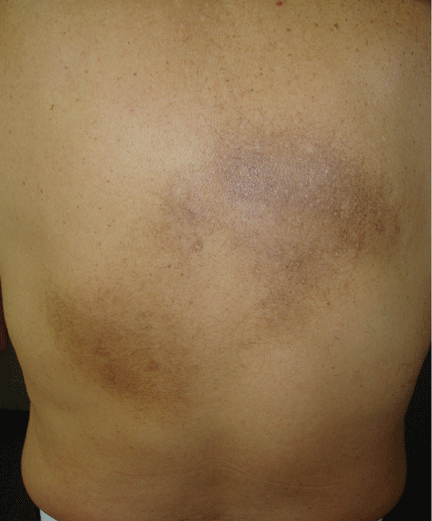
Chronic, brown-gray, itching patches on the back in an adult patient are characteristic of notalgia paresthetica.
Conditions that may be included in the differential diagnosis but that do not match the presentation in this patient include the following:
- Cutaneous sarcoidosis, which may exhibit several morphologies, but itching would be unusual
- Chronic discoid lupus erythematosus, characterized by scarring and atrophic plaques, but mainly on the face and scalp
- Contact dermatitis, an itchy eczematous condition, characterized by scaly erythematous plaques
- Lichen amyloidosis, a variant of cutaneous amyloidosis characterized by the deposition of amyloid or amyloid-like proteins in the dermis, resulting in red-brown hyperkeratotic lichenoid papules, usually on the pretibial surfaces.
CAUSES AND MANAGEMENT
Notalgia paresthetica is a neuropathic syndrome of the skin of the middle of the back characterized by localized pruritus.1–3 Although common, it often goes undiagnosed.1,3,4 It tends to be chronic, with periodic remissions and exacerbations.
Notalgia paresthetica is thought to be a sensory neuropathy and may result from compression of the posterior rami of spinal nerve segments T2 to T6. Slight degenerative changes are often but not always observed, and their clinical significance is uncertain.1,2,4 The condition affects people of all races and both sexes, usually adults ages 40 to 80.
Clinically, it presents as localized pruritus on the back, usually within the dermatomes T2 to T6.5 Examination reveals a hyperpigmented patch, sometimes with excoriations.5
Diagnosis is based on clinical findings. Laboratory tests are not useful. Imaging is not needed, but magnetic resonance imaging and evaluation by an orthopedic surgeon are appropriate when there is chronic focal pain. Skin biopsy is usually not necessary, although it may be useful in some patients to exclude other conditions. When biopsy is done, macular amyloidosis or postinflammatory hyperpigmentation is seen.
Treatment is difficult. Topical steroids and oral antihistamines are usually ineffective,5 but topical capsaicin may provide temporary relief.3 The most recommended treatment in patients with notalgia paresthetica and underlying spinal disease is evaluation and conservative management of the spinal disease, including progressive exercise and rehabilitation.2 Other therapies include oxcarbazepine, gabapentin, transcutaneous electrical nerve stimulation, phototherapy,6 and botulinum toxin injection.
TREATMENT OF OUR PATIENT
In our patient, an orthopedic evaluation revealed cervicothoracic scoliosis. He underwent 6 months of conservative treatment under the care of his family physician and a dermatologist. Treatment consisted of exercise and rehabilitation for his scoliosis, and daily application of topical mometasone. The pain and itch gradual improved.
- Pérez-Pérez LC. General features and treatment of notalgia paresthetica. Skinmed 2011; 9:353–358.
- Fleischer AB, Meade TJ, Fleischer AB. Notalgia paresthetica: successful treatment with exercises. Acta Derm Venereol 2011; 91:356–357.
- Wallengren J, Klinker M. Successful treatment of notalgia paresthetica with topical capsaicin: vehicle-controlled, double-blind, crossover study. J Am Acad Dermatol 1995; 32:287–289.
- Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol 2005; 52:1085–1087.
- Raison-Peyron N, Meunier L, Acevedo M, Meynadier J. Notalgia paresthetica: clinical, physiopathological and therapeutic aspects. A study of 12 cases. J Eur Acad Dermatol Venereol 1999; 12:215–221.
- Pérez-Pérez L, Allegue F, Fabeiro JM, Caeiro JL, Zulaica A. Notalgia paresthesica successfully treated with narrow-band UVB: report of five cases. J Eur Acad Dermatol Venereol 2010; 24:730–732.
A 47-year-old man had had a chronic itch on his back for 2 years. He had no history of trauma to the site, nor did he recall applying topical products to that area.
He was otherwise healthy. He worked as an electrician and said he occasionally experienced cervical and back pain while working.
An examination revealed two grayish-brown ovoid patches on the upper back, each 5 cm to 7 cm in diameter (Figure 1).
DIAGNOSIS: NOTALGIA PARESTHETICA
Chronic, brown-gray, itching patches on the back in an adult patient are characteristic of notalgia paresthetica.
Conditions that may be included in the differential diagnosis but that do not match the presentation in this patient include the following:
- Cutaneous sarcoidosis, which may exhibit several morphologies, but itching would be unusual
- Chronic discoid lupus erythematosus, characterized by scarring and atrophic plaques, but mainly on the face and scalp
- Contact dermatitis, an itchy eczematous condition, characterized by scaly erythematous plaques
- Lichen amyloidosis, a variant of cutaneous amyloidosis characterized by the deposition of amyloid or amyloid-like proteins in the dermis, resulting in red-brown hyperkeratotic lichenoid papules, usually on the pretibial surfaces.
CAUSES AND MANAGEMENT
Notalgia paresthetica is a neuropathic syndrome of the skin of the middle of the back characterized by localized pruritus.1–3 Although common, it often goes undiagnosed.1,3,4 It tends to be chronic, with periodic remissions and exacerbations.
Notalgia paresthetica is thought to be a sensory neuropathy and may result from compression of the posterior rami of spinal nerve segments T2 to T6. Slight degenerative changes are often but not always observed, and their clinical significance is uncertain.1,2,4 The condition affects people of all races and both sexes, usually adults ages 40 to 80.
Clinically, it presents as localized pruritus on the back, usually within the dermatomes T2 to T6.5 Examination reveals a hyperpigmented patch, sometimes with excoriations.5
Diagnosis is based on clinical findings. Laboratory tests are not useful. Imaging is not needed, but magnetic resonance imaging and evaluation by an orthopedic surgeon are appropriate when there is chronic focal pain. Skin biopsy is usually not necessary, although it may be useful in some patients to exclude other conditions. When biopsy is done, macular amyloidosis or postinflammatory hyperpigmentation is seen.
Treatment is difficult. Topical steroids and oral antihistamines are usually ineffective,5 but topical capsaicin may provide temporary relief.3 The most recommended treatment in patients with notalgia paresthetica and underlying spinal disease is evaluation and conservative management of the spinal disease, including progressive exercise and rehabilitation.2 Other therapies include oxcarbazepine, gabapentin, transcutaneous electrical nerve stimulation, phototherapy,6 and botulinum toxin injection.
TREATMENT OF OUR PATIENT
In our patient, an orthopedic evaluation revealed cervicothoracic scoliosis. He underwent 6 months of conservative treatment under the care of his family physician and a dermatologist. Treatment consisted of exercise and rehabilitation for his scoliosis, and daily application of topical mometasone. The pain and itch gradual improved.
A 47-year-old man had had a chronic itch on his back for 2 years. He had no history of trauma to the site, nor did he recall applying topical products to that area.
He was otherwise healthy. He worked as an electrician and said he occasionally experienced cervical and back pain while working.
An examination revealed two grayish-brown ovoid patches on the upper back, each 5 cm to 7 cm in diameter (Figure 1).
DIAGNOSIS: NOTALGIA PARESTHETICA
Chronic, brown-gray, itching patches on the back in an adult patient are characteristic of notalgia paresthetica.
Conditions that may be included in the differential diagnosis but that do not match the presentation in this patient include the following:
- Cutaneous sarcoidosis, which may exhibit several morphologies, but itching would be unusual
- Chronic discoid lupus erythematosus, characterized by scarring and atrophic plaques, but mainly on the face and scalp
- Contact dermatitis, an itchy eczematous condition, characterized by scaly erythematous plaques
- Lichen amyloidosis, a variant of cutaneous amyloidosis characterized by the deposition of amyloid or amyloid-like proteins in the dermis, resulting in red-brown hyperkeratotic lichenoid papules, usually on the pretibial surfaces.
CAUSES AND MANAGEMENT
Notalgia paresthetica is a neuropathic syndrome of the skin of the middle of the back characterized by localized pruritus.1–3 Although common, it often goes undiagnosed.1,3,4 It tends to be chronic, with periodic remissions and exacerbations.
Notalgia paresthetica is thought to be a sensory neuropathy and may result from compression of the posterior rami of spinal nerve segments T2 to T6. Slight degenerative changes are often but not always observed, and their clinical significance is uncertain.1,2,4 The condition affects people of all races and both sexes, usually adults ages 40 to 80.
Clinically, it presents as localized pruritus on the back, usually within the dermatomes T2 to T6.5 Examination reveals a hyperpigmented patch, sometimes with excoriations.5
Diagnosis is based on clinical findings. Laboratory tests are not useful. Imaging is not needed, but magnetic resonance imaging and evaluation by an orthopedic surgeon are appropriate when there is chronic focal pain. Skin biopsy is usually not necessary, although it may be useful in some patients to exclude other conditions. When biopsy is done, macular amyloidosis or postinflammatory hyperpigmentation is seen.
Treatment is difficult. Topical steroids and oral antihistamines are usually ineffective,5 but topical capsaicin may provide temporary relief.3 The most recommended treatment in patients with notalgia paresthetica and underlying spinal disease is evaluation and conservative management of the spinal disease, including progressive exercise and rehabilitation.2 Other therapies include oxcarbazepine, gabapentin, transcutaneous electrical nerve stimulation, phototherapy,6 and botulinum toxin injection.
TREATMENT OF OUR PATIENT
In our patient, an orthopedic evaluation revealed cervicothoracic scoliosis. He underwent 6 months of conservative treatment under the care of his family physician and a dermatologist. Treatment consisted of exercise and rehabilitation for his scoliosis, and daily application of topical mometasone. The pain and itch gradual improved.
- Pérez-Pérez LC. General features and treatment of notalgia paresthetica. Skinmed 2011; 9:353–358.
- Fleischer AB, Meade TJ, Fleischer AB. Notalgia paresthetica: successful treatment with exercises. Acta Derm Venereol 2011; 91:356–357.
- Wallengren J, Klinker M. Successful treatment of notalgia paresthetica with topical capsaicin: vehicle-controlled, double-blind, crossover study. J Am Acad Dermatol 1995; 32:287–289.
- Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol 2005; 52:1085–1087.
- Raison-Peyron N, Meunier L, Acevedo M, Meynadier J. Notalgia paresthetica: clinical, physiopathological and therapeutic aspects. A study of 12 cases. J Eur Acad Dermatol Venereol 1999; 12:215–221.
- Pérez-Pérez L, Allegue F, Fabeiro JM, Caeiro JL, Zulaica A. Notalgia paresthesica successfully treated with narrow-band UVB: report of five cases. J Eur Acad Dermatol Venereol 2010; 24:730–732.
- Pérez-Pérez LC. General features and treatment of notalgia paresthetica. Skinmed 2011; 9:353–358.
- Fleischer AB, Meade TJ, Fleischer AB. Notalgia paresthetica: successful treatment with exercises. Acta Derm Venereol 2011; 91:356–357.
- Wallengren J, Klinker M. Successful treatment of notalgia paresthetica with topical capsaicin: vehicle-controlled, double-blind, crossover study. J Am Acad Dermatol 1995; 32:287–289.
- Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol 2005; 52:1085–1087.
- Raison-Peyron N, Meunier L, Acevedo M, Meynadier J. Notalgia paresthetica: clinical, physiopathological and therapeutic aspects. A study of 12 cases. J Eur Acad Dermatol Venereol 1999; 12:215–221.
- Pérez-Pérez L, Allegue F, Fabeiro JM, Caeiro JL, Zulaica A. Notalgia paresthesica successfully treated with narrow-band UVB: report of five cases. J Eur Acad Dermatol Venereol 2010; 24:730–732.
Lung air-fluid level in a smoker
A 49-year-old man was referred for evaluation of an abnormal chest radiograph. A 25-pack-year smoker, he had a history of chronic shortness of breath on exertion with occasional coughing and whitish sputum production. He also had a history of hypertension. He had not had hemoptysis, fever, chills, weight loss, or other symptoms, and he had not traveled recently.
On examination, he appeared comfortable. His breath sounds were decreased bilaterally; the rest of his physical examination was normal. His medical history, social history, and review of systems were otherwise unremarkable.
His white blood cell count was 9.4 × 109/L (reference range 4.5–11.0), with a normal differential. His hemoglobin concentration was 166 g/L (140–175).
Pulmonary function testing demonstrated moderate obstruction, with the following values:
- Forced expiratory volume in the first second of expiration/ forced vital capacity 0.65
- Forced expiratory volume in the first second of expiration 2.40 L (72% of predicted)
- Total lung capacity 7.11 L (92% of predicted)
- Diffusing capacity of lung for carbon monoxide 58% of predicted.
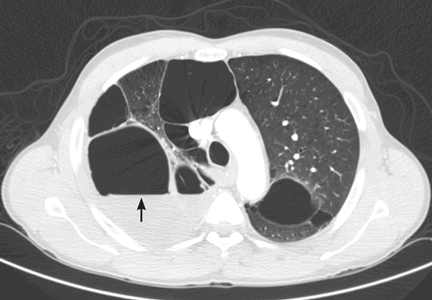
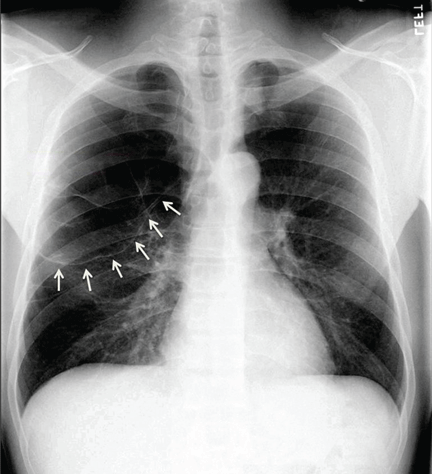
He underwent radiography (Figure 1) and computed tomography of the chest (Figure 2).
DIAGNOSIS: INFECTED EMPHYSEMATOUS BULLAE
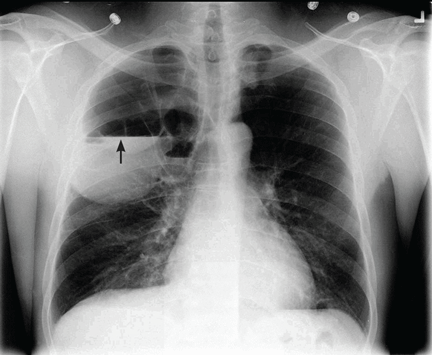
The patient had infected emphysematous bullae.
The diagnosis can typically be made by the new development of an air-fluid level in a patient known to have preexisting emphysematous bullae.1 If previous images are not available, the presence of other bullae in a patient with established chronic obstructive pulmonary disease, a thin-walled cavity, and a disproportionate presentation with impressive radiographic findings along with a subtle clinical picture can support the diagnosis.2 In most reported cases, patients are not significantly symptomatic or ill.3 The differential diagnosis includes loculated parapneumonic pleural effusion,4 lung abscess,5 tuberculosis,6 and infected pneumatocele.
Since percutaneous aspiration of the bullae has been discouraged,2 the causative organism is often not identified. Also, the role of bronchoscopy in the diagnostic evaluation and treatment of infected emphysematous bullae appears to be limited.7
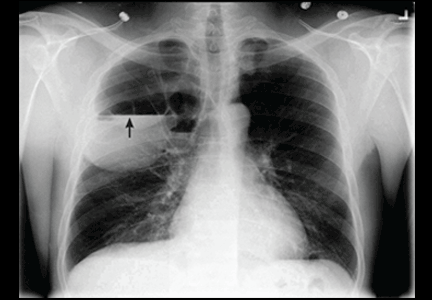
Our patient had minimal symptoms and did not appear ill; he had a relatively unremarkable physical examination, no leukocytosis, and negative blood and sputum cultures, suggesting a benign presentation. In addition, chest radiography a few months before this presentation showed multiple large emphysematous bullae (Figure 3). The current chest radiograph suggested multiple thin-walled cavitary lesions with an air-fluid level, which was confirmed on computed tomography.
TREATMENT OF INFECTED EMPHYSEMATOUS BULLAE
Currently, there is no established therapy for infected emphysematous bullae. Because the presentation is usually relatively benign in most case series, conservative treatment with a prolonged course of antibiotics alone seems to be the most appropriate initial course of action. A follow-up evaluation with chest imaging is recommended. On the other hand, in patients with worse symptoms, percutaneous aspiration of the bullae should be considered, as it may guide antibiotic therapy.8
We started our patient on clindamycin and scheduled him for follow-up chest imaging in 6 weeks.
- Burgener FA. Pulmonary cavitary and cystic lesions. In:Burgener FA, Kormano M, Pudas T, editors. Differential Diagnosis in Conventional Radiology. 3rd ed. New York, Thieme; 2008: chap.24.
- Chandra D, Soubra SH, Musher DM. A 57-year-old man with a fluid-containing lung cavity: infection of an emphysematous bulla with methicillin-resistant Staphylococcus aureus. Chest 2006; 130:1942–1946.
- Leatherman JW, McDonald FM, Niewohner DE. Fluid-containing bullae in the lung. South Med J 1985; 78:708–710.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Hammond JM, Potgieter PD, Hanslo D, Scott H, Roditi D. The etiology and antimicrobial susceptibility patterns of microorganisms in acute community-acquired lung abscess. Chest 1995; 108:937–941.
- Woodring JH, Vandiviere HM, Fried AM, Dillon ML, Williams TD, Melvin IG. Update: the radiographic features of pulmonary tuberculosis. AJR Am J Roentgenol 1986; 146:497–506.
- Chandra D, Rose SR, Carter RB, Musher DM, Hamill RJ. Fluid-containing emphysematous bullae: a spectrum of illness. Eur Respir J 2008; 32:303–306.
- Henao-Martinez AF, Fernandez JF, Adams SG, Restrepo C. Lung bullae with air-fluid levels: what is the appropriate therapeutic approach? Respir Care 2012; 57:642–645.
A 49-year-old man was referred for evaluation of an abnormal chest radiograph. A 25-pack-year smoker, he had a history of chronic shortness of breath on exertion with occasional coughing and whitish sputum production. He also had a history of hypertension. He had not had hemoptysis, fever, chills, weight loss, or other symptoms, and he had not traveled recently.
On examination, he appeared comfortable. His breath sounds were decreased bilaterally; the rest of his physical examination was normal. His medical history, social history, and review of systems were otherwise unremarkable.
His white blood cell count was 9.4 × 109/L (reference range 4.5–11.0), with a normal differential. His hemoglobin concentration was 166 g/L (140–175).
Pulmonary function testing demonstrated moderate obstruction, with the following values:
- Forced expiratory volume in the first second of expiration/ forced vital capacity 0.65
- Forced expiratory volume in the first second of expiration 2.40 L (72% of predicted)
- Total lung capacity 7.11 L (92% of predicted)
- Diffusing capacity of lung for carbon monoxide 58% of predicted.
He underwent radiography (Figure 1) and computed tomography of the chest (Figure 2).
DIAGNOSIS: INFECTED EMPHYSEMATOUS BULLAE
The patient had infected emphysematous bullae.
The diagnosis can typically be made by the new development of an air-fluid level in a patient known to have preexisting emphysematous bullae.1 If previous images are not available, the presence of other bullae in a patient with established chronic obstructive pulmonary disease, a thin-walled cavity, and a disproportionate presentation with impressive radiographic findings along with a subtle clinical picture can support the diagnosis.2 In most reported cases, patients are not significantly symptomatic or ill.3 The differential diagnosis includes loculated parapneumonic pleural effusion,4 lung abscess,5 tuberculosis,6 and infected pneumatocele.
Since percutaneous aspiration of the bullae has been discouraged,2 the causative organism is often not identified. Also, the role of bronchoscopy in the diagnostic evaluation and treatment of infected emphysematous bullae appears to be limited.7
Our patient had minimal symptoms and did not appear ill; he had a relatively unremarkable physical examination, no leukocytosis, and negative blood and sputum cultures, suggesting a benign presentation. In addition, chest radiography a few months before this presentation showed multiple large emphysematous bullae (Figure 3). The current chest radiograph suggested multiple thin-walled cavitary lesions with an air-fluid level, which was confirmed on computed tomography.
TREATMENT OF INFECTED EMPHYSEMATOUS BULLAE
Currently, there is no established therapy for infected emphysematous bullae. Because the presentation is usually relatively benign in most case series, conservative treatment with a prolonged course of antibiotics alone seems to be the most appropriate initial course of action. A follow-up evaluation with chest imaging is recommended. On the other hand, in patients with worse symptoms, percutaneous aspiration of the bullae should be considered, as it may guide antibiotic therapy.8
We started our patient on clindamycin and scheduled him for follow-up chest imaging in 6 weeks.
A 49-year-old man was referred for evaluation of an abnormal chest radiograph. A 25-pack-year smoker, he had a history of chronic shortness of breath on exertion with occasional coughing and whitish sputum production. He also had a history of hypertension. He had not had hemoptysis, fever, chills, weight loss, or other symptoms, and he had not traveled recently.
On examination, he appeared comfortable. His breath sounds were decreased bilaterally; the rest of his physical examination was normal. His medical history, social history, and review of systems were otherwise unremarkable.
His white blood cell count was 9.4 × 109/L (reference range 4.5–11.0), with a normal differential. His hemoglobin concentration was 166 g/L (140–175).
Pulmonary function testing demonstrated moderate obstruction, with the following values:
- Forced expiratory volume in the first second of expiration/ forced vital capacity 0.65
- Forced expiratory volume in the first second of expiration 2.40 L (72% of predicted)
- Total lung capacity 7.11 L (92% of predicted)
- Diffusing capacity of lung for carbon monoxide 58% of predicted.
He underwent radiography (Figure 1) and computed tomography of the chest (Figure 2).
DIAGNOSIS: INFECTED EMPHYSEMATOUS BULLAE
The patient had infected emphysematous bullae.
The diagnosis can typically be made by the new development of an air-fluid level in a patient known to have preexisting emphysematous bullae.1 If previous images are not available, the presence of other bullae in a patient with established chronic obstructive pulmonary disease, a thin-walled cavity, and a disproportionate presentation with impressive radiographic findings along with a subtle clinical picture can support the diagnosis.2 In most reported cases, patients are not significantly symptomatic or ill.3 The differential diagnosis includes loculated parapneumonic pleural effusion,4 lung abscess,5 tuberculosis,6 and infected pneumatocele.
Since percutaneous aspiration of the bullae has been discouraged,2 the causative organism is often not identified. Also, the role of bronchoscopy in the diagnostic evaluation and treatment of infected emphysematous bullae appears to be limited.7
Our patient had minimal symptoms and did not appear ill; he had a relatively unremarkable physical examination, no leukocytosis, and negative blood and sputum cultures, suggesting a benign presentation. In addition, chest radiography a few months before this presentation showed multiple large emphysematous bullae (Figure 3). The current chest radiograph suggested multiple thin-walled cavitary lesions with an air-fluid level, which was confirmed on computed tomography.
TREATMENT OF INFECTED EMPHYSEMATOUS BULLAE
Currently, there is no established therapy for infected emphysematous bullae. Because the presentation is usually relatively benign in most case series, conservative treatment with a prolonged course of antibiotics alone seems to be the most appropriate initial course of action. A follow-up evaluation with chest imaging is recommended. On the other hand, in patients with worse symptoms, percutaneous aspiration of the bullae should be considered, as it may guide antibiotic therapy.8
We started our patient on clindamycin and scheduled him for follow-up chest imaging in 6 weeks.
- Burgener FA. Pulmonary cavitary and cystic lesions. In:Burgener FA, Kormano M, Pudas T, editors. Differential Diagnosis in Conventional Radiology. 3rd ed. New York, Thieme; 2008: chap.24.
- Chandra D, Soubra SH, Musher DM. A 57-year-old man with a fluid-containing lung cavity: infection of an emphysematous bulla with methicillin-resistant Staphylococcus aureus. Chest 2006; 130:1942–1946.
- Leatherman JW, McDonald FM, Niewohner DE. Fluid-containing bullae in the lung. South Med J 1985; 78:708–710.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Hammond JM, Potgieter PD, Hanslo D, Scott H, Roditi D. The etiology and antimicrobial susceptibility patterns of microorganisms in acute community-acquired lung abscess. Chest 1995; 108:937–941.
- Woodring JH, Vandiviere HM, Fried AM, Dillon ML, Williams TD, Melvin IG. Update: the radiographic features of pulmonary tuberculosis. AJR Am J Roentgenol 1986; 146:497–506.
- Chandra D, Rose SR, Carter RB, Musher DM, Hamill RJ. Fluid-containing emphysematous bullae: a spectrum of illness. Eur Respir J 2008; 32:303–306.
- Henao-Martinez AF, Fernandez JF, Adams SG, Restrepo C. Lung bullae with air-fluid levels: what is the appropriate therapeutic approach? Respir Care 2012; 57:642–645.
- Burgener FA. Pulmonary cavitary and cystic lesions. In:Burgener FA, Kormano M, Pudas T, editors. Differential Diagnosis in Conventional Radiology. 3rd ed. New York, Thieme; 2008: chap.24.
- Chandra D, Soubra SH, Musher DM. A 57-year-old man with a fluid-containing lung cavity: infection of an emphysematous bulla with methicillin-resistant Staphylococcus aureus. Chest 2006; 130:1942–1946.
- Leatherman JW, McDonald FM, Niewohner DE. Fluid-containing bullae in the lung. South Med J 1985; 78:708–710.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Hammond JM, Potgieter PD, Hanslo D, Scott H, Roditi D. The etiology and antimicrobial susceptibility patterns of microorganisms in acute community-acquired lung abscess. Chest 1995; 108:937–941.
- Woodring JH, Vandiviere HM, Fried AM, Dillon ML, Williams TD, Melvin IG. Update: the radiographic features of pulmonary tuberculosis. AJR Am J Roentgenol 1986; 146:497–506.
- Chandra D, Rose SR, Carter RB, Musher DM, Hamill RJ. Fluid-containing emphysematous bullae: a spectrum of illness. Eur Respir J 2008; 32:303–306.
- Henao-Martinez AF, Fernandez JF, Adams SG, Restrepo C. Lung bullae with air-fluid levels: what is the appropriate therapeutic approach? Respir Care 2012; 57:642–645.
Multiple intracardiac thrombi
A 60-year-old woman presented with sudden swelling and pain in her right arm. She reported progressive lower-extremity edema and abdominal girth over the past month, associated with shortness of breath and orthopnea. She had a remote history of two spontaneous abortions.
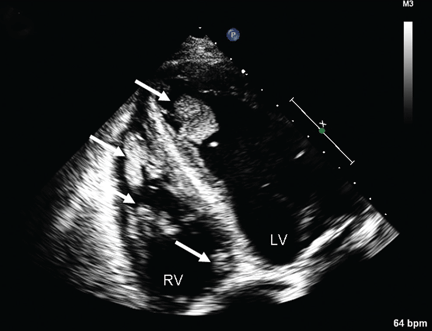
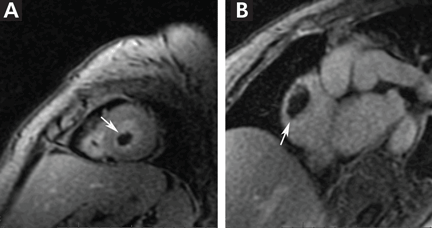
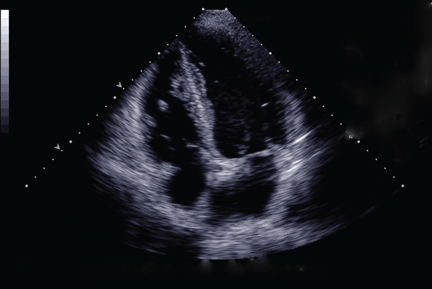
Duplex ultrasonography revealed massive venous thrombosis extending from the antecubital fossa to the right atrium. Transthoracic echocardiography revealed severe left ventricular (LV) dysfunction and multiple echo-dense masses in the LV apex, the right ventricle, and the left atrium, as well as at the base of the tricuspid valve (Figure 1). There was no evidence of a structural heart defect, eg, patent foramen ovale, atrial septal defect, or ventricular septal defect. Cardiovascular magnetic resonance imaging (MRI) confirmed the densities as thrombi (Figure 2). Her ejection fraction was 35%.
Blood testing on admission showed a prolonged partial thromboplastin time of 55.0 sec (reference range 22.7–35.6) and a prothrombin time of 13.4 sec (reference range 11.3–14.5). Tissue thromboplastin inhibition at a dilution of 1:50 was elevated at 1.5 sec (reference range 0.7–1.3), as was the tissue thromboplastin inhibition at a dilution of 1:500—ie, 1.6 sec (0.7–1.3). Dilute Russell viper venom testing and anticardiolipin antibody immunoglobulin G and M testing were negative. The lupus antiphospholipid antibody test and the hexagonal lipid neutralization test were positive.
The patient’s clinical presentation of extensive unprovoked venous thrombosis and her laboratory profile together suggested the antiphospholipid antibody syndrome.
SURGICAL TREATMENT NOT AN OPTION
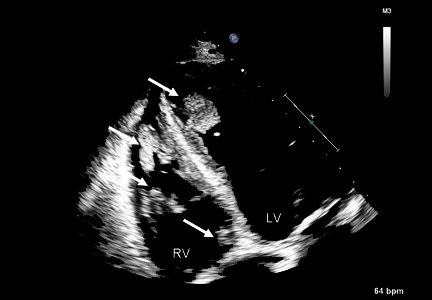
Given her extensive clot burden, surgical thrombectomy was not an option. Instead, warfarin therapy was started and resulted in a progressive diminution of the thrombi. At 4-month follow-up, the thrombi had nearly resolved (Figure 3), and her LV ejection fraction had increased to 45% to 50%. Eighteen months later, she was diagnosed with cholangiocarcinoma. In retrospect, we believe the cancer predisposed the patient to the hypercoagulable state and, subsequently, to thrombosis.
DIAGNOSING AND TREATING LEFT VENTRICULAR THROMBOSIS
Ventricular thrombosis is a serious problem, most commonly associated with extensive myocardial infarction. It is a relatively common complication of myocardial infarction and of ischemic and nonischemic cardiomy-opathies.1 In this population, the incidence of LV thrombosis is reported to be in the range of 10% to 25%, and it increases with increasing LV end-diastolic diameter, lower ejection fraction, and anterior-wall-motion akinesia, and with the presence of apical aneurysms.2 It is an important cause of morbidity and death, whether the thrombus is sessile or mobile.
How diagnostic imaging tests compare
The diagnosis of LV thrombosis requires a certain level of suspicion and has traditionally relied on echocardiography. However, several studies have raised doubt about the sensitivity of echocardiography for the detection of left or right ventricular thrombi.3 In a 2006 report, the sensitivity of transthoracic echocardiography in detecting LV thrombi was 23% and the sensitivity of transesophageal echocardiography was 40%.4 In contrast, delayed-enhancement cardiovascular MRI had a sensitivity near 90%. Similarly, in another study,5 contrast-enhanced echocardiography had a low but higher sensitivity of nearly 60%.5 Therefore, cardiovascular MRI is emerging as the new gold standard test for the detection of this important complication of ventricular dysfunction and myocardial infarction.
Treatment and screening
The optimal management of intraventricular thrombi is poorly defined. It has been suggested from case series that large, mobile, or protruding LV thrombi have more potential for embolization and, therefore, that patients with these findings may benefit from surgical thrombectomy.6 Oral anticoagulation has been reported to dissolve intraventricular thrombi, with success rates from 13% to 59%.7 A prospective study of enoxaparin in 26 patients with LV thrombi reported resolution rates close to 73% at 3-week follow-up.8
There are no guidelines at present on which to base recommendations for screening patients for intracavitary thrombi or for starting empiric anticoagulation in those at risk.
- Weinsaft JW, Kim HW, Shah DJ, et al. Detection of left ventricular thrombus by delayed-enhancement cardiovascular magnetic resonance prevalence and markers in patients with systolic dysfunction. J Am Coll Cardiol 2008; 52:148–157.
- Mollet NR, Dymarkowski S, Volders W, et al. Visualization of ventricular thrombi with contrast-enhanced magnetic resonance imaging in patients with ischemic heart disease. Circulation 2002; 106:2873–2876.
- Tsang BK, Platts DG, Javorsky G, Brown MR. Right ventricular thrombus detection and multimodality imaging using contrast echocardiography and cardiac magnetic resonance imaging. Heart Lung Circ 2012; 21:185–188.
- Srichai MB, Junor C, Rodriguez LL, et al. Clinical, imaging, and pathological characteristics of left ventricular thrombus: a comparison of contrast-enhanced magnetic resonance imaging, transthoracic echocardiography, and transesophageal echocardiography with surgical or pathological validation. Am Heart J 2006; 152:75–84.
- Weinsaft JW, Kim RJ, Ross M, et al. Contrast-enhanced anatomic imaging as compared to contrast-enhanced tissue characterization for detection of left ventricular thrombus. JACC Cardiovasc Imaging 2009; 2:969–979.
- Nili M, Deviri E, Jortner R, Strasberg B, Levy MJ. Surgical removal of a mobile, pedunculated left ventricular thrombus: report of 4 cases. Ann Thorac Surg 1988; 46:396–400.
- Heik SC, Kupper W, Hamm C, et al. Efficacy of high dose intravenous heparin for treatment of left ventricular thrombi with high embolic risk. J Am Coll Cardiol 1994; 24:1305–1309.
- Meurin P, Tabet JY, Renaud N, et al. Treatment of left ventricular thrombi with a low molecular weight heparin. Int J Cardiol 2005; 98:319–323.
A 60-year-old woman presented with sudden swelling and pain in her right arm. She reported progressive lower-extremity edema and abdominal girth over the past month, associated with shortness of breath and orthopnea. She had a remote history of two spontaneous abortions.
Duplex ultrasonography revealed massive venous thrombosis extending from the antecubital fossa to the right atrium. Transthoracic echocardiography revealed severe left ventricular (LV) dysfunction and multiple echo-dense masses in the LV apex, the right ventricle, and the left atrium, as well as at the base of the tricuspid valve (Figure 1). There was no evidence of a structural heart defect, eg, patent foramen ovale, atrial septal defect, or ventricular septal defect. Cardiovascular magnetic resonance imaging (MRI) confirmed the densities as thrombi (Figure 2). Her ejection fraction was 35%.
Blood testing on admission showed a prolonged partial thromboplastin time of 55.0 sec (reference range 22.7–35.6) and a prothrombin time of 13.4 sec (reference range 11.3–14.5). Tissue thromboplastin inhibition at a dilution of 1:50 was elevated at 1.5 sec (reference range 0.7–1.3), as was the tissue thromboplastin inhibition at a dilution of 1:500—ie, 1.6 sec (0.7–1.3). Dilute Russell viper venom testing and anticardiolipin antibody immunoglobulin G and M testing were negative. The lupus antiphospholipid antibody test and the hexagonal lipid neutralization test were positive.
The patient’s clinical presentation of extensive unprovoked venous thrombosis and her laboratory profile together suggested the antiphospholipid antibody syndrome.
SURGICAL TREATMENT NOT AN OPTION
Given her extensive clot burden, surgical thrombectomy was not an option. Instead, warfarin therapy was started and resulted in a progressive diminution of the thrombi. At 4-month follow-up, the thrombi had nearly resolved (Figure 3), and her LV ejection fraction had increased to 45% to 50%. Eighteen months later, she was diagnosed with cholangiocarcinoma. In retrospect, we believe the cancer predisposed the patient to the hypercoagulable state and, subsequently, to thrombosis.
DIAGNOSING AND TREATING LEFT VENTRICULAR THROMBOSIS
Ventricular thrombosis is a serious problem, most commonly associated with extensive myocardial infarction. It is a relatively common complication of myocardial infarction and of ischemic and nonischemic cardiomy-opathies.1 In this population, the incidence of LV thrombosis is reported to be in the range of 10% to 25%, and it increases with increasing LV end-diastolic diameter, lower ejection fraction, and anterior-wall-motion akinesia, and with the presence of apical aneurysms.2 It is an important cause of morbidity and death, whether the thrombus is sessile or mobile.
How diagnostic imaging tests compare
The diagnosis of LV thrombosis requires a certain level of suspicion and has traditionally relied on echocardiography. However, several studies have raised doubt about the sensitivity of echocardiography for the detection of left or right ventricular thrombi.3 In a 2006 report, the sensitivity of transthoracic echocardiography in detecting LV thrombi was 23% and the sensitivity of transesophageal echocardiography was 40%.4 In contrast, delayed-enhancement cardiovascular MRI had a sensitivity near 90%. Similarly, in another study,5 contrast-enhanced echocardiography had a low but higher sensitivity of nearly 60%.5 Therefore, cardiovascular MRI is emerging as the new gold standard test for the detection of this important complication of ventricular dysfunction and myocardial infarction.
Treatment and screening
The optimal management of intraventricular thrombi is poorly defined. It has been suggested from case series that large, mobile, or protruding LV thrombi have more potential for embolization and, therefore, that patients with these findings may benefit from surgical thrombectomy.6 Oral anticoagulation has been reported to dissolve intraventricular thrombi, with success rates from 13% to 59%.7 A prospective study of enoxaparin in 26 patients with LV thrombi reported resolution rates close to 73% at 3-week follow-up.8
There are no guidelines at present on which to base recommendations for screening patients for intracavitary thrombi or for starting empiric anticoagulation in those at risk.
A 60-year-old woman presented with sudden swelling and pain in her right arm. She reported progressive lower-extremity edema and abdominal girth over the past month, associated with shortness of breath and orthopnea. She had a remote history of two spontaneous abortions.
Duplex ultrasonography revealed massive venous thrombosis extending from the antecubital fossa to the right atrium. Transthoracic echocardiography revealed severe left ventricular (LV) dysfunction and multiple echo-dense masses in the LV apex, the right ventricle, and the left atrium, as well as at the base of the tricuspid valve (Figure 1). There was no evidence of a structural heart defect, eg, patent foramen ovale, atrial septal defect, or ventricular septal defect. Cardiovascular magnetic resonance imaging (MRI) confirmed the densities as thrombi (Figure 2). Her ejection fraction was 35%.
Blood testing on admission showed a prolonged partial thromboplastin time of 55.0 sec (reference range 22.7–35.6) and a prothrombin time of 13.4 sec (reference range 11.3–14.5). Tissue thromboplastin inhibition at a dilution of 1:50 was elevated at 1.5 sec (reference range 0.7–1.3), as was the tissue thromboplastin inhibition at a dilution of 1:500—ie, 1.6 sec (0.7–1.3). Dilute Russell viper venom testing and anticardiolipin antibody immunoglobulin G and M testing were negative. The lupus antiphospholipid antibody test and the hexagonal lipid neutralization test were positive.
The patient’s clinical presentation of extensive unprovoked venous thrombosis and her laboratory profile together suggested the antiphospholipid antibody syndrome.
SURGICAL TREATMENT NOT AN OPTION
Given her extensive clot burden, surgical thrombectomy was not an option. Instead, warfarin therapy was started and resulted in a progressive diminution of the thrombi. At 4-month follow-up, the thrombi had nearly resolved (Figure 3), and her LV ejection fraction had increased to 45% to 50%. Eighteen months later, she was diagnosed with cholangiocarcinoma. In retrospect, we believe the cancer predisposed the patient to the hypercoagulable state and, subsequently, to thrombosis.
DIAGNOSING AND TREATING LEFT VENTRICULAR THROMBOSIS
Ventricular thrombosis is a serious problem, most commonly associated with extensive myocardial infarction. It is a relatively common complication of myocardial infarction and of ischemic and nonischemic cardiomy-opathies.1 In this population, the incidence of LV thrombosis is reported to be in the range of 10% to 25%, and it increases with increasing LV end-diastolic diameter, lower ejection fraction, and anterior-wall-motion akinesia, and with the presence of apical aneurysms.2 It is an important cause of morbidity and death, whether the thrombus is sessile or mobile.
How diagnostic imaging tests compare
The diagnosis of LV thrombosis requires a certain level of suspicion and has traditionally relied on echocardiography. However, several studies have raised doubt about the sensitivity of echocardiography for the detection of left or right ventricular thrombi.3 In a 2006 report, the sensitivity of transthoracic echocardiography in detecting LV thrombi was 23% and the sensitivity of transesophageal echocardiography was 40%.4 In contrast, delayed-enhancement cardiovascular MRI had a sensitivity near 90%. Similarly, in another study,5 contrast-enhanced echocardiography had a low but higher sensitivity of nearly 60%.5 Therefore, cardiovascular MRI is emerging as the new gold standard test for the detection of this important complication of ventricular dysfunction and myocardial infarction.
Treatment and screening
The optimal management of intraventricular thrombi is poorly defined. It has been suggested from case series that large, mobile, or protruding LV thrombi have more potential for embolization and, therefore, that patients with these findings may benefit from surgical thrombectomy.6 Oral anticoagulation has been reported to dissolve intraventricular thrombi, with success rates from 13% to 59%.7 A prospective study of enoxaparin in 26 patients with LV thrombi reported resolution rates close to 73% at 3-week follow-up.8
There are no guidelines at present on which to base recommendations for screening patients for intracavitary thrombi or for starting empiric anticoagulation in those at risk.
- Weinsaft JW, Kim HW, Shah DJ, et al. Detection of left ventricular thrombus by delayed-enhancement cardiovascular magnetic resonance prevalence and markers in patients with systolic dysfunction. J Am Coll Cardiol 2008; 52:148–157.
- Mollet NR, Dymarkowski S, Volders W, et al. Visualization of ventricular thrombi with contrast-enhanced magnetic resonance imaging in patients with ischemic heart disease. Circulation 2002; 106:2873–2876.
- Tsang BK, Platts DG, Javorsky G, Brown MR. Right ventricular thrombus detection and multimodality imaging using contrast echocardiography and cardiac magnetic resonance imaging. Heart Lung Circ 2012; 21:185–188.
- Srichai MB, Junor C, Rodriguez LL, et al. Clinical, imaging, and pathological characteristics of left ventricular thrombus: a comparison of contrast-enhanced magnetic resonance imaging, transthoracic echocardiography, and transesophageal echocardiography with surgical or pathological validation. Am Heart J 2006; 152:75–84.
- Weinsaft JW, Kim RJ, Ross M, et al. Contrast-enhanced anatomic imaging as compared to contrast-enhanced tissue characterization for detection of left ventricular thrombus. JACC Cardiovasc Imaging 2009; 2:969–979.
- Nili M, Deviri E, Jortner R, Strasberg B, Levy MJ. Surgical removal of a mobile, pedunculated left ventricular thrombus: report of 4 cases. Ann Thorac Surg 1988; 46:396–400.
- Heik SC, Kupper W, Hamm C, et al. Efficacy of high dose intravenous heparin for treatment of left ventricular thrombi with high embolic risk. J Am Coll Cardiol 1994; 24:1305–1309.
- Meurin P, Tabet JY, Renaud N, et al. Treatment of left ventricular thrombi with a low molecular weight heparin. Int J Cardiol 2005; 98:319–323.
- Weinsaft JW, Kim HW, Shah DJ, et al. Detection of left ventricular thrombus by delayed-enhancement cardiovascular magnetic resonance prevalence and markers in patients with systolic dysfunction. J Am Coll Cardiol 2008; 52:148–157.
- Mollet NR, Dymarkowski S, Volders W, et al. Visualization of ventricular thrombi with contrast-enhanced magnetic resonance imaging in patients with ischemic heart disease. Circulation 2002; 106:2873–2876.
- Tsang BK, Platts DG, Javorsky G, Brown MR. Right ventricular thrombus detection and multimodality imaging using contrast echocardiography and cardiac magnetic resonance imaging. Heart Lung Circ 2012; 21:185–188.
- Srichai MB, Junor C, Rodriguez LL, et al. Clinical, imaging, and pathological characteristics of left ventricular thrombus: a comparison of contrast-enhanced magnetic resonance imaging, transthoracic echocardiography, and transesophageal echocardiography with surgical or pathological validation. Am Heart J 2006; 152:75–84.
- Weinsaft JW, Kim RJ, Ross M, et al. Contrast-enhanced anatomic imaging as compared to contrast-enhanced tissue characterization for detection of left ventricular thrombus. JACC Cardiovasc Imaging 2009; 2:969–979.
- Nili M, Deviri E, Jortner R, Strasberg B, Levy MJ. Surgical removal of a mobile, pedunculated left ventricular thrombus: report of 4 cases. Ann Thorac Surg 1988; 46:396–400.
- Heik SC, Kupper W, Hamm C, et al. Efficacy of high dose intravenous heparin for treatment of left ventricular thrombi with high embolic risk. J Am Coll Cardiol 1994; 24:1305–1309.
- Meurin P, Tabet JY, Renaud N, et al. Treatment of left ventricular thrombi with a low molecular weight heparin. Int J Cardiol 2005; 98:319–323.
Stiff, numb hands
A 45-year-old woman with no chronic medical problems presented to the emergency room with a 1-day history of cramps and paresthesias in both hands and feet, mainly involving the fingers and toes. She said that after an argument with her daughter she began feeling anxious, and this was accompanied by shortness of breath and palpitations as well as generalized weakness, fatigue, and body aches. She also reported nausea and repeated vomiting but no abdominal pain, distention or change in bowel movements. She had had no loss of consciousness, confusion, incontinence, headache, dizziness, diplopia, or facial paresthesia.
She is a cigarette smoker, is alcohol-dependent, but does not use illicit drugs and is not on any medications.
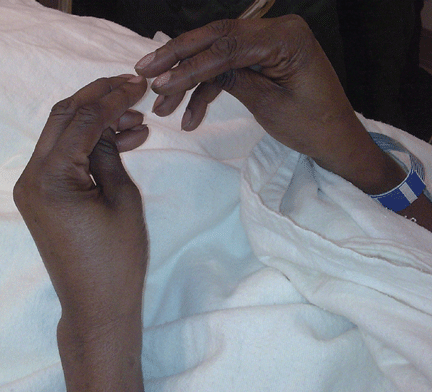
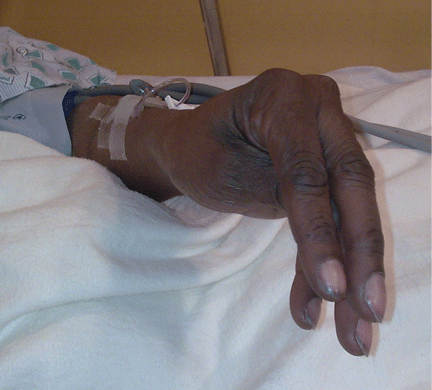
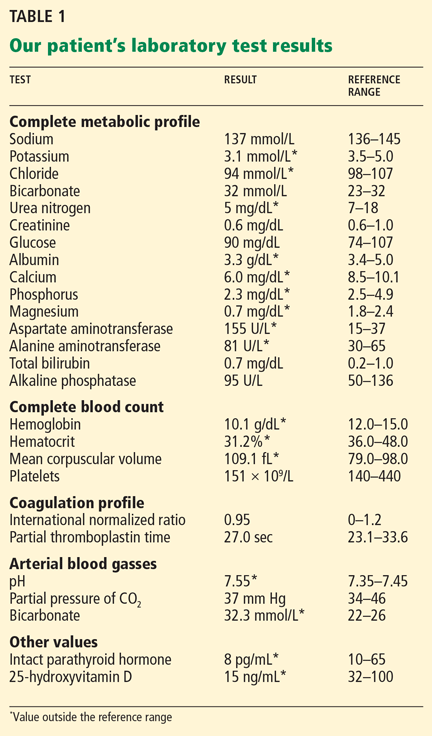
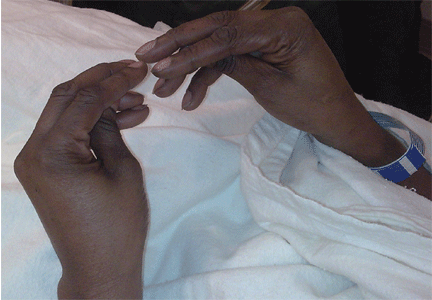
Examination revealed a temperature of 37.1°C (98.8°F), blood pressure 150/75 mm Hg, heart rate 105 bpm, respiratory rate 24 breaths per minute, and oxygen saturation 97% on room air. She appeared very fatigued, thin, and in mild distress due to her cramps. Her mucous membranes were dry, but she had no orthostatic changes. She had noticeable carpopedal spasms (Figure 1), reproducible by inflating a blood-pressure cuff placed on her arm (Trousseau sign) (Figure 2). Also noted was the Chvostek sign—contraction of the ipsilateral facial muscles when the facial nerve is tapped just in front of the ear. The rest of the systemic evaluation was normal. Laboratory investigations were as listed in Table 1. Electrocardiography showed a prolonged QTc interval (0.5 sec). The chest radiograph was normal.
HYPERVENTILATION AND TETANY
The presumptive diagnosis was latent tetany caused by an electrolyte derangement, in this case a combination of hypocalcemia, hypomagnesemia, and hypokalemia as the result of alcohol abuse, repeated vomiting, and hyperventilation brought on by a severe attack of anxiety.
Tetany results from increased excitability of nerves and muscles, leading to painful muscle cramps.1,2 Typical symptoms include circumoral and distal paresthesias, stiffness, clumsiness, myalgia, carpopedal spasm, laryngospasm, bronchospasm, and generalized seizure. The Chvostek and Trousseau signs help to confirm the diagnosis of tetany.3,4
The differential diagnosis of carpopedal spasm includes other conditions of involuntary muscle contraction, such as myotonic disorders; myokymia from Isaac syndrome (writhing movements of the muscles under the skin visualized by continuous “rippling” movements of the muscle); stiff-man syndrome (an autoimmune-antiglutamic acid decarboxylase antibody-associated muscle rigidity that waxes and wanes with concurrent spasms); and snake envenomation.
In addition, our patient’s symptoms were probably brought on by hyperventilation. In general, patients with hyperventilation syndrome are young females who display various manifestations of underlying anxiety and can develop tetany even after a brief episode of hyperventilation. At the time of presentation, our patient was found to have mixed respiratory and metabolic alkalosis. The anxiety-induced hyperventilation likely contributed to the respiratory alkalosis. She had no other symptoms or signs to suggest an acute organic respiratory illness such as pulmonary embolism, pneumothorax, or infection. Vomiting likely caused the metabolic alkalosis and hypokalemia.
Tetany is usually triggered by acute hypocalcemia and is uncommon unless the serum ionized calcium concentration falls below 4.3 mg/dL (1.1 mmol/L), which usually corresponds to a serum total calcium concentration of 7.0 to 7.5 mg/dL (1.8 to 1.9 mmol/L). Patients with a gradual onset of hypocalcemia tend to have fewer symptoms.3,4
Although alkalosis alone can cause tetany, it also enhances tetany by reducing the level of ionized calcium in the serum. Alkalemia causes hypocalcemia by an intravascular chelative mechanism in which the decrease in concentration of hydrogen ions leaves the negatively charged binding sites on albumin available to bind ionized calcium.3
The same happens to the magnesium, a cation with the same size and valence. Significant hypomagnesemia is common in tetanic patients with hyperventilation attacks and may, by itself or in combination with hypocalcemia, cause tetany.2,5,6 Hypokalemia can develop in patients with hypomagnesemia or metabolic alkalosis and may lead to tetany.6,7 Furthermore, our patient was dependent on alcohol, and this is known to cause hypomagnesemia from the excessive urinary excretion of magnesium. This defect of alcohol-induced tubular dysfunction is reversible within 4 weeks of abstinence. Magnesium depletion can cause hypocalcemia by producing resistance to parathyroid hormone or by decreasing its secretion, and this occurs in severe hypomagnesemia, ie, when the serum magnesium concentration falls below 1.0 mg/dL (0.4 mmol/L).
IDENTIFY AND TREAT THE UNDERLYING CAUSE
The management of tetany consists of identifying and treating the underlying cause. Infusion of calcium or magnesium is effective as acute therapy for tetany, and, if appropriate, vitamin D supplementation should also be provided.3,4,7 However, if associated hyperventilation syndrome is present, patients benefit from reassurance and treatment for underlying psychological stress. The traditional treatment of rebreathing into a paper bag is no longer recommended because of the potential risk of hypoxia. Sedatives and antidepressants should be reserved for patients who have not responded to conservative treatment.
Our patient was given an explanation of the condition together with breathing exercises. She received lorazepam and was immediately treated with intravenous hydration, along with intravenous infusion of magnesium, calcium, and potassium. These interventions led to an immediate resolution of her symptoms.
Her low level of intact parathyroid hormone may also have been caused by hypomagnesemia. She was given oral magnesium, potassium, calcium, and vitamin D to continue at home. In addition, she was advised to avoid excessive alcohol consumption and to see us or her primary care doctor should the symptoms recur. As expected, all the laboratory values normalized within 1 month of abstinence from alcohol, and she has been well since.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care 2008; 35:215–237.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Smets YF, Bokani N, de Meijer PH, Meinders AE. Tetany due to excessive use of alcohol: a possible magnesium deficiency [in Dutch]. Ned Tijdschr Geneeskd 2004; 148:641–644.
- Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 2007; 18:2649–2652.
A 45-year-old woman with no chronic medical problems presented to the emergency room with a 1-day history of cramps and paresthesias in both hands and feet, mainly involving the fingers and toes. She said that after an argument with her daughter she began feeling anxious, and this was accompanied by shortness of breath and palpitations as well as generalized weakness, fatigue, and body aches. She also reported nausea and repeated vomiting but no abdominal pain, distention or change in bowel movements. She had had no loss of consciousness, confusion, incontinence, headache, dizziness, diplopia, or facial paresthesia.
She is a cigarette smoker, is alcohol-dependent, but does not use illicit drugs and is not on any medications.
Examination revealed a temperature of 37.1°C (98.8°F), blood pressure 150/75 mm Hg, heart rate 105 bpm, respiratory rate 24 breaths per minute, and oxygen saturation 97% on room air. She appeared very fatigued, thin, and in mild distress due to her cramps. Her mucous membranes were dry, but she had no orthostatic changes. She had noticeable carpopedal spasms (Figure 1), reproducible by inflating a blood-pressure cuff placed on her arm (Trousseau sign) (Figure 2). Also noted was the Chvostek sign—contraction of the ipsilateral facial muscles when the facial nerve is tapped just in front of the ear. The rest of the systemic evaluation was normal. Laboratory investigations were as listed in Table 1. Electrocardiography showed a prolonged QTc interval (0.5 sec). The chest radiograph was normal.
HYPERVENTILATION AND TETANY
The presumptive diagnosis was latent tetany caused by an electrolyte derangement, in this case a combination of hypocalcemia, hypomagnesemia, and hypokalemia as the result of alcohol abuse, repeated vomiting, and hyperventilation brought on by a severe attack of anxiety.
Tetany results from increased excitability of nerves and muscles, leading to painful muscle cramps.1,2 Typical symptoms include circumoral and distal paresthesias, stiffness, clumsiness, myalgia, carpopedal spasm, laryngospasm, bronchospasm, and generalized seizure. The Chvostek and Trousseau signs help to confirm the diagnosis of tetany.3,4
The differential diagnosis of carpopedal spasm includes other conditions of involuntary muscle contraction, such as myotonic disorders; myokymia from Isaac syndrome (writhing movements of the muscles under the skin visualized by continuous “rippling” movements of the muscle); stiff-man syndrome (an autoimmune-antiglutamic acid decarboxylase antibody-associated muscle rigidity that waxes and wanes with concurrent spasms); and snake envenomation.
In addition, our patient’s symptoms were probably brought on by hyperventilation. In general, patients with hyperventilation syndrome are young females who display various manifestations of underlying anxiety and can develop tetany even after a brief episode of hyperventilation. At the time of presentation, our patient was found to have mixed respiratory and metabolic alkalosis. The anxiety-induced hyperventilation likely contributed to the respiratory alkalosis. She had no other symptoms or signs to suggest an acute organic respiratory illness such as pulmonary embolism, pneumothorax, or infection. Vomiting likely caused the metabolic alkalosis and hypokalemia.
Tetany is usually triggered by acute hypocalcemia and is uncommon unless the serum ionized calcium concentration falls below 4.3 mg/dL (1.1 mmol/L), which usually corresponds to a serum total calcium concentration of 7.0 to 7.5 mg/dL (1.8 to 1.9 mmol/L). Patients with a gradual onset of hypocalcemia tend to have fewer symptoms.3,4
Although alkalosis alone can cause tetany, it also enhances tetany by reducing the level of ionized calcium in the serum. Alkalemia causes hypocalcemia by an intravascular chelative mechanism in which the decrease in concentration of hydrogen ions leaves the negatively charged binding sites on albumin available to bind ionized calcium.3
The same happens to the magnesium, a cation with the same size and valence. Significant hypomagnesemia is common in tetanic patients with hyperventilation attacks and may, by itself or in combination with hypocalcemia, cause tetany.2,5,6 Hypokalemia can develop in patients with hypomagnesemia or metabolic alkalosis and may lead to tetany.6,7 Furthermore, our patient was dependent on alcohol, and this is known to cause hypomagnesemia from the excessive urinary excretion of magnesium. This defect of alcohol-induced tubular dysfunction is reversible within 4 weeks of abstinence. Magnesium depletion can cause hypocalcemia by producing resistance to parathyroid hormone or by decreasing its secretion, and this occurs in severe hypomagnesemia, ie, when the serum magnesium concentration falls below 1.0 mg/dL (0.4 mmol/L).
IDENTIFY AND TREAT THE UNDERLYING CAUSE
The management of tetany consists of identifying and treating the underlying cause. Infusion of calcium or magnesium is effective as acute therapy for tetany, and, if appropriate, vitamin D supplementation should also be provided.3,4,7 However, if associated hyperventilation syndrome is present, patients benefit from reassurance and treatment for underlying psychological stress. The traditional treatment of rebreathing into a paper bag is no longer recommended because of the potential risk of hypoxia. Sedatives and antidepressants should be reserved for patients who have not responded to conservative treatment.
Our patient was given an explanation of the condition together with breathing exercises. She received lorazepam and was immediately treated with intravenous hydration, along with intravenous infusion of magnesium, calcium, and potassium. These interventions led to an immediate resolution of her symptoms.
Her low level of intact parathyroid hormone may also have been caused by hypomagnesemia. She was given oral magnesium, potassium, calcium, and vitamin D to continue at home. In addition, she was advised to avoid excessive alcohol consumption and to see us or her primary care doctor should the symptoms recur. As expected, all the laboratory values normalized within 1 month of abstinence from alcohol, and she has been well since.
A 45-year-old woman with no chronic medical problems presented to the emergency room with a 1-day history of cramps and paresthesias in both hands and feet, mainly involving the fingers and toes. She said that after an argument with her daughter she began feeling anxious, and this was accompanied by shortness of breath and palpitations as well as generalized weakness, fatigue, and body aches. She also reported nausea and repeated vomiting but no abdominal pain, distention or change in bowel movements. She had had no loss of consciousness, confusion, incontinence, headache, dizziness, diplopia, or facial paresthesia.
She is a cigarette smoker, is alcohol-dependent, but does not use illicit drugs and is not on any medications.
Examination revealed a temperature of 37.1°C (98.8°F), blood pressure 150/75 mm Hg, heart rate 105 bpm, respiratory rate 24 breaths per minute, and oxygen saturation 97% on room air. She appeared very fatigued, thin, and in mild distress due to her cramps. Her mucous membranes were dry, but she had no orthostatic changes. She had noticeable carpopedal spasms (Figure 1), reproducible by inflating a blood-pressure cuff placed on her arm (Trousseau sign) (Figure 2). Also noted was the Chvostek sign—contraction of the ipsilateral facial muscles when the facial nerve is tapped just in front of the ear. The rest of the systemic evaluation was normal. Laboratory investigations were as listed in Table 1. Electrocardiography showed a prolonged QTc interval (0.5 sec). The chest radiograph was normal.
HYPERVENTILATION AND TETANY
The presumptive diagnosis was latent tetany caused by an electrolyte derangement, in this case a combination of hypocalcemia, hypomagnesemia, and hypokalemia as the result of alcohol abuse, repeated vomiting, and hyperventilation brought on by a severe attack of anxiety.
Tetany results from increased excitability of nerves and muscles, leading to painful muscle cramps.1,2 Typical symptoms include circumoral and distal paresthesias, stiffness, clumsiness, myalgia, carpopedal spasm, laryngospasm, bronchospasm, and generalized seizure. The Chvostek and Trousseau signs help to confirm the diagnosis of tetany.3,4
The differential diagnosis of carpopedal spasm includes other conditions of involuntary muscle contraction, such as myotonic disorders; myokymia from Isaac syndrome (writhing movements of the muscles under the skin visualized by continuous “rippling” movements of the muscle); stiff-man syndrome (an autoimmune-antiglutamic acid decarboxylase antibody-associated muscle rigidity that waxes and wanes with concurrent spasms); and snake envenomation.
In addition, our patient’s symptoms were probably brought on by hyperventilation. In general, patients with hyperventilation syndrome are young females who display various manifestations of underlying anxiety and can develop tetany even after a brief episode of hyperventilation. At the time of presentation, our patient was found to have mixed respiratory and metabolic alkalosis. The anxiety-induced hyperventilation likely contributed to the respiratory alkalosis. She had no other symptoms or signs to suggest an acute organic respiratory illness such as pulmonary embolism, pneumothorax, or infection. Vomiting likely caused the metabolic alkalosis and hypokalemia.
Tetany is usually triggered by acute hypocalcemia and is uncommon unless the serum ionized calcium concentration falls below 4.3 mg/dL (1.1 mmol/L), which usually corresponds to a serum total calcium concentration of 7.0 to 7.5 mg/dL (1.8 to 1.9 mmol/L). Patients with a gradual onset of hypocalcemia tend to have fewer symptoms.3,4
Although alkalosis alone can cause tetany, it also enhances tetany by reducing the level of ionized calcium in the serum. Alkalemia causes hypocalcemia by an intravascular chelative mechanism in which the decrease in concentration of hydrogen ions leaves the negatively charged binding sites on albumin available to bind ionized calcium.3
The same happens to the magnesium, a cation with the same size and valence. Significant hypomagnesemia is common in tetanic patients with hyperventilation attacks and may, by itself or in combination with hypocalcemia, cause tetany.2,5,6 Hypokalemia can develop in patients with hypomagnesemia or metabolic alkalosis and may lead to tetany.6,7 Furthermore, our patient was dependent on alcohol, and this is known to cause hypomagnesemia from the excessive urinary excretion of magnesium. This defect of alcohol-induced tubular dysfunction is reversible within 4 weeks of abstinence. Magnesium depletion can cause hypocalcemia by producing resistance to parathyroid hormone or by decreasing its secretion, and this occurs in severe hypomagnesemia, ie, when the serum magnesium concentration falls below 1.0 mg/dL (0.4 mmol/L).
IDENTIFY AND TREAT THE UNDERLYING CAUSE
The management of tetany consists of identifying and treating the underlying cause. Infusion of calcium or magnesium is effective as acute therapy for tetany, and, if appropriate, vitamin D supplementation should also be provided.3,4,7 However, if associated hyperventilation syndrome is present, patients benefit from reassurance and treatment for underlying psychological stress. The traditional treatment of rebreathing into a paper bag is no longer recommended because of the potential risk of hypoxia. Sedatives and antidepressants should be reserved for patients who have not responded to conservative treatment.
Our patient was given an explanation of the condition together with breathing exercises. She received lorazepam and was immediately treated with intravenous hydration, along with intravenous infusion of magnesium, calcium, and potassium. These interventions led to an immediate resolution of her symptoms.
Her low level of intact parathyroid hormone may also have been caused by hypomagnesemia. She was given oral magnesium, potassium, calcium, and vitamin D to continue at home. In addition, she was advised to avoid excessive alcohol consumption and to see us or her primary care doctor should the symptoms recur. As expected, all the laboratory values normalized within 1 month of abstinence from alcohol, and she has been well since.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care 2008; 35:215–237.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Smets YF, Bokani N, de Meijer PH, Meinders AE. Tetany due to excessive use of alcohol: a possible magnesium deficiency [in Dutch]. Ned Tijdschr Geneeskd 2004; 148:641–644.
- Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 2007; 18:2649–2652.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care 2008; 35:215–237.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Smets YF, Bokani N, de Meijer PH, Meinders AE. Tetany due to excessive use of alcohol: a possible magnesium deficiency [in Dutch]. Ned Tijdschr Geneeskd 2004; 148:641–644.
- Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 2007; 18:2649–2652.
Hyperpigmented patches on the neck, shoulder, and back
During a routine examination, a 17-year-old boy was noted to have unilateral hyperpigmented patches on his left neck, shoulder, and back. The lesions had been present since birth, had not changed in size or color, and were asymptomatic. His mother had noted an increase in the size of his left jaw starting at age 1.

The hyperpigmented patches had irregular borders (Figure 1). The skin was otherwise clear. Laboratory testing from 5 years earlier showed normal levels of dehydroepiandrosterone, prolactin, parathyroid hormone, thyroxine, and thyroid-stimulating hormone. Computed tomography showed a “ground-glass” appearance of the bones at the base of the skull, consistent with polyostotic fibrous dysplasia (Figure 2).
Q: Which is the most likely diagnosis?
- Neurofibromatosis
- Congenital melanocytic nevus
- McCune-Albright syndrome
- Tuberous sclerosis

A: This patient had typical features of McCune-Albright syndrome (or Albright syndrome), the classic triad of fibrous dysplasia of bone, large unilateral café-au-lait macules or patches, and precocious puberty or other endocrinopathy.1 The syndrome is rare, with an estimated prevalence of 1/100,000 to 1/1,000,000.2 It results from somatic mutation of the GNAS gene (chromosome 20q13) during embryonic development, which causes constitutive activation of intracellular cyclic adenosine monophosphate (cAMP) signaling and cellular dysplasia.3
THE DIAGNOSIS IS CLINICAL
McCune-Albright syndrome is a clinical diagnosis based on the presence of at least two features of the classic triad.1,4
Other conditions, such as neurofibromatosis, also cause café-au-lait macules in children; but the lesions of McCune-Albright syndrome are fewer in number, larger, and darker and may follow Blaschko lines, with a linear or segmental configuration.5 McCune-Albright lesions tend to have jagged, “coast-of-Maine” borders, as opposed to the smoother “coast-of-California” borders of the lesions of neurofibromatosis.1
Nevertheless, because café-au-lait macules of McCune-Albright syndrome are sometimes indistinguishable from those of neurofibromatosis, the endocrine and skeletal manifestations are essential to making the diagnosis.5
SIGNS OF GENETIC MOSAICISM
The somatic (postzygotic) nature of the GNAS mutation means that patients have normal and abnormal cell lines, ie, mosaicism. Therefore, the extent of disease depends on the precise stage in development during which the mutation occurred. This determines which tissues contain mutated cells and the proportion and distribution of affected cells at these loci.1,4
In addition, differential sensitivity to cAMP signaling between cell types and tissue-specific imprinting of GNAS may contribute to the phenotypic variation seen in McCune-Albright syndrome.4 This means that the clinical features often vary, and the classic clinical triad is not always present.6
The most common clinical features are fibrous dysplasia, which occurs in 46% to 98% of patients, and café-au-lait macules, which occur in 53.1% to 92.5% of patients.1,2 Fibrous dysplasia is typically polyostotic, ie, it involves multiple skeletal sites, with the proximal femur and skull base being the most common.6 It presents as bone pain, asymmetry, or pathologic fracture (or a combination of these) and shows a characteristic “ground-glass” appearance on computed tomography (Figure 2).1 Café-au-lait lesions present at birth or shortly thereafter are often unappreciated as a potential presenting sign.1 These hyperpigmented lesions are typically large and unilateral, often favoring the forehead, nuchal area, sacrum, and buttocks.5.6
Precocious puberty is the most common endocrinopathy in McCune-Albright syndrome, seen in 64% to 79% of cases, and is more common in girls than in boys.2 Other associated endocrinopathies include hyperthyroidism (20% to 30%), excess growth hormone, renal phosphate wasting, and Cushing syndrome.1,6
SCREEN FOR OTHER MANIFESTATIONS
McCune-Albright syndrome can involve a broad spectrum of tissues. Therefore, once the diagnosis is made, the patient should be thoroughly evaluated for other manifestations.1 The evaluation may include imaging studies and biochemical testing and may necessitate referral to an endocrinologist, a radiologist, and an orthopedic surgeon.
Young girls with premature vaginal bleeding or recurrent follicular cysts should always be evaluated for McCune-Albright syndrome, since ovarian enlargement can be mistaken for an ovarian tumor.7 Likewise, adults with isolated fibrous dysplasia or large unilateral café-au-lait macules should also be evaluated, since patients with McCune-Albright syndrome have a normal life span and so may present later in life.4,6
TREATMENT
Drug treatment of this syndrome aims to block the effects of prolonged exposure of end-organs to sex steroids. Since precocious puberty of McCune-Alright syndrome is typically peripheral in origin, it is unresponsive to gonadotropin-releasing hormone agonist drugs; instead, aromatase inhibitors (testolactone) with antiestrogens (tamoxifen) may be used in girls, or antiandrogens (spironolactone) in boys.8
Unfortunately, despite our advanced mechanistic understanding of this disease, medical management remains challenging, with poor long-term efficacy and few studies on long-term outcomes, such as skeletal growth.
GENETIC TESTING HAS LIMITED VALUE
Although genetic testing for GNAS mutations is available, the mosaic nature of McCune-Albright syndrome makes the detection of mutant alleles in affected tissues and circulating cells exceedingly difficult.1,4 These constraints, coupled with high costs, have limited the clinical utility of genetic testing at present. In addition, the lack of a known genotype-phenotype correlation in this syndrome limits the value of genetic testing.1 In the future, improvements in molecular techniques may make genetic testing more useful in the diagnosis and management of McCune-Albright syndrome, especially if clinically relevant genotype-phenotype correlates are identified.4 At this time, although genetic testing is not a standard of care, genetic counseling should be offered to all patients with this syndrome.
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; 3:12.
- Aycan Z, Önder A, Çetinkaya S. Eight-year follow-up of a girl with McCune-Albright syndrome. J Clin Res Pediatr Endocrinol 2011; 3:40–42.
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991; 325:1688–1695.
- Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev 2007; 4(suppl 4):380–385.
- Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2nd ed. St. Louis, MO: Mosby/Elsevier; 2008.
- Spitz JL, editor. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Nabhan ZM, West KW, Eugster EA. Oophorectomy in McCune-Albright syndrome: a case of mistaken identity. J Pediatr Surg 2007; 42:1578–1583.
- Haddad N, Eugster E. An update on the treatment of precocious puberty in McCune-Albright syndrome and testotoxicosis. J Pediatr Endocrinol Metab 2007; 20:653–661.
During a routine examination, a 17-year-old boy was noted to have unilateral hyperpigmented patches on his left neck, shoulder, and back. The lesions had been present since birth, had not changed in size or color, and were asymptomatic. His mother had noted an increase in the size of his left jaw starting at age 1.
The hyperpigmented patches had irregular borders (Figure 1). The skin was otherwise clear. Laboratory testing from 5 years earlier showed normal levels of dehydroepiandrosterone, prolactin, parathyroid hormone, thyroxine, and thyroid-stimulating hormone. Computed tomography showed a “ground-glass” appearance of the bones at the base of the skull, consistent with polyostotic fibrous dysplasia (Figure 2).
Q: Which is the most likely diagnosis?
- Neurofibromatosis
- Congenital melanocytic nevus
- McCune-Albright syndrome
- Tuberous sclerosis
A: This patient had typical features of McCune-Albright syndrome (or Albright syndrome), the classic triad of fibrous dysplasia of bone, large unilateral café-au-lait macules or patches, and precocious puberty or other endocrinopathy.1 The syndrome is rare, with an estimated prevalence of 1/100,000 to 1/1,000,000.2 It results from somatic mutation of the GNAS gene (chromosome 20q13) during embryonic development, which causes constitutive activation of intracellular cyclic adenosine monophosphate (cAMP) signaling and cellular dysplasia.3
THE DIAGNOSIS IS CLINICAL
McCune-Albright syndrome is a clinical diagnosis based on the presence of at least two features of the classic triad.1,4
Other conditions, such as neurofibromatosis, also cause café-au-lait macules in children; but the lesions of McCune-Albright syndrome are fewer in number, larger, and darker and may follow Blaschko lines, with a linear or segmental configuration.5 McCune-Albright lesions tend to have jagged, “coast-of-Maine” borders, as opposed to the smoother “coast-of-California” borders of the lesions of neurofibromatosis.1
Nevertheless, because café-au-lait macules of McCune-Albright syndrome are sometimes indistinguishable from those of neurofibromatosis, the endocrine and skeletal manifestations are essential to making the diagnosis.5
SIGNS OF GENETIC MOSAICISM
The somatic (postzygotic) nature of the GNAS mutation means that patients have normal and abnormal cell lines, ie, mosaicism. Therefore, the extent of disease depends on the precise stage in development during which the mutation occurred. This determines which tissues contain mutated cells and the proportion and distribution of affected cells at these loci.1,4
In addition, differential sensitivity to cAMP signaling between cell types and tissue-specific imprinting of GNAS may contribute to the phenotypic variation seen in McCune-Albright syndrome.4 This means that the clinical features often vary, and the classic clinical triad is not always present.6
The most common clinical features are fibrous dysplasia, which occurs in 46% to 98% of patients, and café-au-lait macules, which occur in 53.1% to 92.5% of patients.1,2 Fibrous dysplasia is typically polyostotic, ie, it involves multiple skeletal sites, with the proximal femur and skull base being the most common.6 It presents as bone pain, asymmetry, or pathologic fracture (or a combination of these) and shows a characteristic “ground-glass” appearance on computed tomography (Figure 2).1 Café-au-lait lesions present at birth or shortly thereafter are often unappreciated as a potential presenting sign.1 These hyperpigmented lesions are typically large and unilateral, often favoring the forehead, nuchal area, sacrum, and buttocks.5.6
Precocious puberty is the most common endocrinopathy in McCune-Albright syndrome, seen in 64% to 79% of cases, and is more common in girls than in boys.2 Other associated endocrinopathies include hyperthyroidism (20% to 30%), excess growth hormone, renal phosphate wasting, and Cushing syndrome.1,6
SCREEN FOR OTHER MANIFESTATIONS
McCune-Albright syndrome can involve a broad spectrum of tissues. Therefore, once the diagnosis is made, the patient should be thoroughly evaluated for other manifestations.1 The evaluation may include imaging studies and biochemical testing and may necessitate referral to an endocrinologist, a radiologist, and an orthopedic surgeon.
Young girls with premature vaginal bleeding or recurrent follicular cysts should always be evaluated for McCune-Albright syndrome, since ovarian enlargement can be mistaken for an ovarian tumor.7 Likewise, adults with isolated fibrous dysplasia or large unilateral café-au-lait macules should also be evaluated, since patients with McCune-Albright syndrome have a normal life span and so may present later in life.4,6
TREATMENT
Drug treatment of this syndrome aims to block the effects of prolonged exposure of end-organs to sex steroids. Since precocious puberty of McCune-Alright syndrome is typically peripheral in origin, it is unresponsive to gonadotropin-releasing hormone agonist drugs; instead, aromatase inhibitors (testolactone) with antiestrogens (tamoxifen) may be used in girls, or antiandrogens (spironolactone) in boys.8
Unfortunately, despite our advanced mechanistic understanding of this disease, medical management remains challenging, with poor long-term efficacy and few studies on long-term outcomes, such as skeletal growth.
GENETIC TESTING HAS LIMITED VALUE
Although genetic testing for GNAS mutations is available, the mosaic nature of McCune-Albright syndrome makes the detection of mutant alleles in affected tissues and circulating cells exceedingly difficult.1,4 These constraints, coupled with high costs, have limited the clinical utility of genetic testing at present. In addition, the lack of a known genotype-phenotype correlation in this syndrome limits the value of genetic testing.1 In the future, improvements in molecular techniques may make genetic testing more useful in the diagnosis and management of McCune-Albright syndrome, especially if clinically relevant genotype-phenotype correlates are identified.4 At this time, although genetic testing is not a standard of care, genetic counseling should be offered to all patients with this syndrome.
During a routine examination, a 17-year-old boy was noted to have unilateral hyperpigmented patches on his left neck, shoulder, and back. The lesions had been present since birth, had not changed in size or color, and were asymptomatic. His mother had noted an increase in the size of his left jaw starting at age 1.
The hyperpigmented patches had irregular borders (Figure 1). The skin was otherwise clear. Laboratory testing from 5 years earlier showed normal levels of dehydroepiandrosterone, prolactin, parathyroid hormone, thyroxine, and thyroid-stimulating hormone. Computed tomography showed a “ground-glass” appearance of the bones at the base of the skull, consistent with polyostotic fibrous dysplasia (Figure 2).
Q: Which is the most likely diagnosis?
- Neurofibromatosis
- Congenital melanocytic nevus
- McCune-Albright syndrome
- Tuberous sclerosis
A: This patient had typical features of McCune-Albright syndrome (or Albright syndrome), the classic triad of fibrous dysplasia of bone, large unilateral café-au-lait macules or patches, and precocious puberty or other endocrinopathy.1 The syndrome is rare, with an estimated prevalence of 1/100,000 to 1/1,000,000.2 It results from somatic mutation of the GNAS gene (chromosome 20q13) during embryonic development, which causes constitutive activation of intracellular cyclic adenosine monophosphate (cAMP) signaling and cellular dysplasia.3
THE DIAGNOSIS IS CLINICAL
McCune-Albright syndrome is a clinical diagnosis based on the presence of at least two features of the classic triad.1,4
Other conditions, such as neurofibromatosis, also cause café-au-lait macules in children; but the lesions of McCune-Albright syndrome are fewer in number, larger, and darker and may follow Blaschko lines, with a linear or segmental configuration.5 McCune-Albright lesions tend to have jagged, “coast-of-Maine” borders, as opposed to the smoother “coast-of-California” borders of the lesions of neurofibromatosis.1
Nevertheless, because café-au-lait macules of McCune-Albright syndrome are sometimes indistinguishable from those of neurofibromatosis, the endocrine and skeletal manifestations are essential to making the diagnosis.5
SIGNS OF GENETIC MOSAICISM
The somatic (postzygotic) nature of the GNAS mutation means that patients have normal and abnormal cell lines, ie, mosaicism. Therefore, the extent of disease depends on the precise stage in development during which the mutation occurred. This determines which tissues contain mutated cells and the proportion and distribution of affected cells at these loci.1,4
In addition, differential sensitivity to cAMP signaling between cell types and tissue-specific imprinting of GNAS may contribute to the phenotypic variation seen in McCune-Albright syndrome.4 This means that the clinical features often vary, and the classic clinical triad is not always present.6
The most common clinical features are fibrous dysplasia, which occurs in 46% to 98% of patients, and café-au-lait macules, which occur in 53.1% to 92.5% of patients.1,2 Fibrous dysplasia is typically polyostotic, ie, it involves multiple skeletal sites, with the proximal femur and skull base being the most common.6 It presents as bone pain, asymmetry, or pathologic fracture (or a combination of these) and shows a characteristic “ground-glass” appearance on computed tomography (Figure 2).1 Café-au-lait lesions present at birth or shortly thereafter are often unappreciated as a potential presenting sign.1 These hyperpigmented lesions are typically large and unilateral, often favoring the forehead, nuchal area, sacrum, and buttocks.5.6
Precocious puberty is the most common endocrinopathy in McCune-Albright syndrome, seen in 64% to 79% of cases, and is more common in girls than in boys.2 Other associated endocrinopathies include hyperthyroidism (20% to 30%), excess growth hormone, renal phosphate wasting, and Cushing syndrome.1,6
SCREEN FOR OTHER MANIFESTATIONS
McCune-Albright syndrome can involve a broad spectrum of tissues. Therefore, once the diagnosis is made, the patient should be thoroughly evaluated for other manifestations.1 The evaluation may include imaging studies and biochemical testing and may necessitate referral to an endocrinologist, a radiologist, and an orthopedic surgeon.
Young girls with premature vaginal bleeding or recurrent follicular cysts should always be evaluated for McCune-Albright syndrome, since ovarian enlargement can be mistaken for an ovarian tumor.7 Likewise, adults with isolated fibrous dysplasia or large unilateral café-au-lait macules should also be evaluated, since patients with McCune-Albright syndrome have a normal life span and so may present later in life.4,6
TREATMENT
Drug treatment of this syndrome aims to block the effects of prolonged exposure of end-organs to sex steroids. Since precocious puberty of McCune-Alright syndrome is typically peripheral in origin, it is unresponsive to gonadotropin-releasing hormone agonist drugs; instead, aromatase inhibitors (testolactone) with antiestrogens (tamoxifen) may be used in girls, or antiandrogens (spironolactone) in boys.8
Unfortunately, despite our advanced mechanistic understanding of this disease, medical management remains challenging, with poor long-term efficacy and few studies on long-term outcomes, such as skeletal growth.
GENETIC TESTING HAS LIMITED VALUE
Although genetic testing for GNAS mutations is available, the mosaic nature of McCune-Albright syndrome makes the detection of mutant alleles in affected tissues and circulating cells exceedingly difficult.1,4 These constraints, coupled with high costs, have limited the clinical utility of genetic testing at present. In addition, the lack of a known genotype-phenotype correlation in this syndrome limits the value of genetic testing.1 In the future, improvements in molecular techniques may make genetic testing more useful in the diagnosis and management of McCune-Albright syndrome, especially if clinically relevant genotype-phenotype correlates are identified.4 At this time, although genetic testing is not a standard of care, genetic counseling should be offered to all patients with this syndrome.
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; 3:12.
- Aycan Z, Önder A, Çetinkaya S. Eight-year follow-up of a girl with McCune-Albright syndrome. J Clin Res Pediatr Endocrinol 2011; 3:40–42.
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991; 325:1688–1695.
- Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev 2007; 4(suppl 4):380–385.
- Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2nd ed. St. Louis, MO: Mosby/Elsevier; 2008.
- Spitz JL, editor. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Nabhan ZM, West KW, Eugster EA. Oophorectomy in McCune-Albright syndrome: a case of mistaken identity. J Pediatr Surg 2007; 42:1578–1583.
- Haddad N, Eugster E. An update on the treatment of precocious puberty in McCune-Albright syndrome and testotoxicosis. J Pediatr Endocrinol Metab 2007; 20:653–661.
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; 3:12.
- Aycan Z, Önder A, Çetinkaya S. Eight-year follow-up of a girl with McCune-Albright syndrome. J Clin Res Pediatr Endocrinol 2011; 3:40–42.
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991; 325:1688–1695.
- Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev 2007; 4(suppl 4):380–385.
- Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2nd ed. St. Louis, MO: Mosby/Elsevier; 2008.
- Spitz JL, editor. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Nabhan ZM, West KW, Eugster EA. Oophorectomy in McCune-Albright syndrome: a case of mistaken identity. J Pediatr Surg 2007; 42:1578–1583.
- Haddad N, Eugster E. An update on the treatment of precocious puberty in McCune-Albright syndrome and testotoxicosis. J Pediatr Endocrinol Metab 2007; 20:653–661.

Not all joint pain is arthritis
A 47-year-old man who had been diagnosed with rheumatoid arthritis 5 years previously was referred to us for management of bilateral pleural effusions.
At the time of his diagnosis, his symptoms included pain and swelling of both wrists and the metacarpal joints of both hands. His serum C-reactive protein level had been elevated at that time, but he had no detectable rheumatoid factor. Findings on magnetic resonance imaging of the hand were very suggestive of rheumatoid arthritis (Figure 1).
He had been started on the anti-tumor necrosis factor agent etanercept but his symptoms improved only slightly, and therefore a glucocorticoid had been added.
Two years later, he developed abdominal pain, for which he underwent cholecystectomy. However, he continued to have chronic, generalized abdominal pain, and over the next 4 years he lost 25 lb. Upper endoscopy showed no mucosal changes, and multiple random biopsy samples were obtained for histologic evaluation (FIGURE 2) as part of his workup for chronic abdominal pain.
Q: What is the diagnosis?
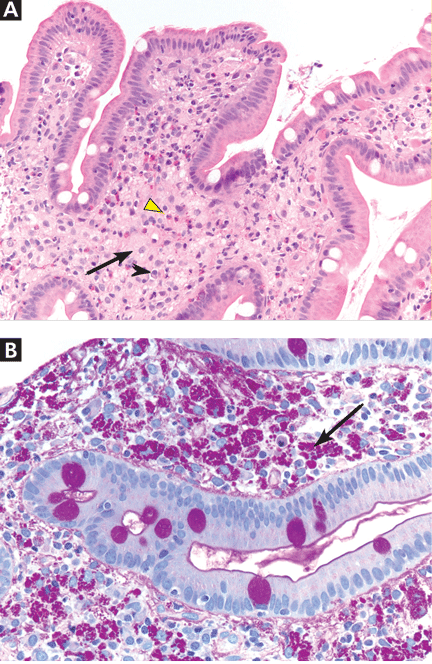
A: As shown in Figure 2, staining of duodenal specimens showed intact villous architecture, with focal expansion of the lamina propria by “foamy” macrophages, rare plasma cells, and eosinophils, a key feature of Whipple disease. Periodic acid-Schiff staining showed numerous bacilli within the macrophages, thus confirming the diagnosis of Whipple disease. The diagnosis was also confirmed by polymerase chain reaction testing. Staining for acid-fast bacilli was negative.
WHEN TO CONSIDER WHIPPLE DISEASE
Whipple disease is a rare systemic disease with a very low incidence rate worldwide. Thus, its prevalence is difficult to estimate accurately. It is caused by a gram-positive bacterium, Tropheryma whippelii.1,2 The typical clinical manifestations are diarrhea, abdominal pain, weight loss, and fever. In most patients, these are often preceded by articular symptoms,3 as in our patient, who had articular symptoms for 5 years before he was diagnosed with Whipple disease.
Interestingly, our patient also had pleural effusion, which is uncommon in Whipple disease.4
The pathogenesis of Whipple disease is thought to be related to bacterial replication within macrophages, which leads to a systemic immune response and tissue infiltration by the organism.5 Histologic evaluation is the most common way to confirm the diagnosis.
As our patient’s disease course illustrates, Whipple disease should be part of the differential diagnosis of arthritis, as antibiotic therapy alone leads to a dramatic clinical response.
Our patient was started on a 2-week course of intravenous ceftriaxone followed by oral sulfamethoxazole and trimethoprim, and his abdominal and articular symptoms completely resolved within 4 weeks.
- Dutly F, Altwegg M. Whipple’s disease and ‘Tropheryma whippelii.’ Clin Microbiol Rev 2001; 14:561–583.
- Raoult D, Birg ML, La Scola B, et al. Cultivation of the bacillus of Whipple’s disease. N Engl J Med 2000; 342:620–625.
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med 1992; 327:293–301.
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore) 1997; 76:170–184.
- Dobbins WO, Ruffin JM. A light- and electron-microscopic study of bacterial invasion in Whipple’s disease. Am J Pathol 1967; 51:225–242.
A 47-year-old man who had been diagnosed with rheumatoid arthritis 5 years previously was referred to us for management of bilateral pleural effusions.
At the time of his diagnosis, his symptoms included pain and swelling of both wrists and the metacarpal joints of both hands. His serum C-reactive protein level had been elevated at that time, but he had no detectable rheumatoid factor. Findings on magnetic resonance imaging of the hand were very suggestive of rheumatoid arthritis (Figure 1).
He had been started on the anti-tumor necrosis factor agent etanercept but his symptoms improved only slightly, and therefore a glucocorticoid had been added.
Two years later, he developed abdominal pain, for which he underwent cholecystectomy. However, he continued to have chronic, generalized abdominal pain, and over the next 4 years he lost 25 lb. Upper endoscopy showed no mucosal changes, and multiple random biopsy samples were obtained for histologic evaluation (FIGURE 2) as part of his workup for chronic abdominal pain.
Q: What is the diagnosis?
A: As shown in Figure 2, staining of duodenal specimens showed intact villous architecture, with focal expansion of the lamina propria by “foamy” macrophages, rare plasma cells, and eosinophils, a key feature of Whipple disease. Periodic acid-Schiff staining showed numerous bacilli within the macrophages, thus confirming the diagnosis of Whipple disease. The diagnosis was also confirmed by polymerase chain reaction testing. Staining for acid-fast bacilli was negative.
WHEN TO CONSIDER WHIPPLE DISEASE
Whipple disease is a rare systemic disease with a very low incidence rate worldwide. Thus, its prevalence is difficult to estimate accurately. It is caused by a gram-positive bacterium, Tropheryma whippelii.1,2 The typical clinical manifestations are diarrhea, abdominal pain, weight loss, and fever. In most patients, these are often preceded by articular symptoms,3 as in our patient, who had articular symptoms for 5 years before he was diagnosed with Whipple disease.
Interestingly, our patient also had pleural effusion, which is uncommon in Whipple disease.4
The pathogenesis of Whipple disease is thought to be related to bacterial replication within macrophages, which leads to a systemic immune response and tissue infiltration by the organism.5 Histologic evaluation is the most common way to confirm the diagnosis.
As our patient’s disease course illustrates, Whipple disease should be part of the differential diagnosis of arthritis, as antibiotic therapy alone leads to a dramatic clinical response.
Our patient was started on a 2-week course of intravenous ceftriaxone followed by oral sulfamethoxazole and trimethoprim, and his abdominal and articular symptoms completely resolved within 4 weeks.
A 47-year-old man who had been diagnosed with rheumatoid arthritis 5 years previously was referred to us for management of bilateral pleural effusions.
At the time of his diagnosis, his symptoms included pain and swelling of both wrists and the metacarpal joints of both hands. His serum C-reactive protein level had been elevated at that time, but he had no detectable rheumatoid factor. Findings on magnetic resonance imaging of the hand were very suggestive of rheumatoid arthritis (Figure 1).
He had been started on the anti-tumor necrosis factor agent etanercept but his symptoms improved only slightly, and therefore a glucocorticoid had been added.
Two years later, he developed abdominal pain, for which he underwent cholecystectomy. However, he continued to have chronic, generalized abdominal pain, and over the next 4 years he lost 25 lb. Upper endoscopy showed no mucosal changes, and multiple random biopsy samples were obtained for histologic evaluation (FIGURE 2) as part of his workup for chronic abdominal pain.
Q: What is the diagnosis?
A: As shown in Figure 2, staining of duodenal specimens showed intact villous architecture, with focal expansion of the lamina propria by “foamy” macrophages, rare plasma cells, and eosinophils, a key feature of Whipple disease. Periodic acid-Schiff staining showed numerous bacilli within the macrophages, thus confirming the diagnosis of Whipple disease. The diagnosis was also confirmed by polymerase chain reaction testing. Staining for acid-fast bacilli was negative.
WHEN TO CONSIDER WHIPPLE DISEASE
Whipple disease is a rare systemic disease with a very low incidence rate worldwide. Thus, its prevalence is difficult to estimate accurately. It is caused by a gram-positive bacterium, Tropheryma whippelii.1,2 The typical clinical manifestations are diarrhea, abdominal pain, weight loss, and fever. In most patients, these are often preceded by articular symptoms,3 as in our patient, who had articular symptoms for 5 years before he was diagnosed with Whipple disease.
Interestingly, our patient also had pleural effusion, which is uncommon in Whipple disease.4
The pathogenesis of Whipple disease is thought to be related to bacterial replication within macrophages, which leads to a systemic immune response and tissue infiltration by the organism.5 Histologic evaluation is the most common way to confirm the diagnosis.
As our patient’s disease course illustrates, Whipple disease should be part of the differential diagnosis of arthritis, as antibiotic therapy alone leads to a dramatic clinical response.
Our patient was started on a 2-week course of intravenous ceftriaxone followed by oral sulfamethoxazole and trimethoprim, and his abdominal and articular symptoms completely resolved within 4 weeks.
- Dutly F, Altwegg M. Whipple’s disease and ‘Tropheryma whippelii.’ Clin Microbiol Rev 2001; 14:561–583.
- Raoult D, Birg ML, La Scola B, et al. Cultivation of the bacillus of Whipple’s disease. N Engl J Med 2000; 342:620–625.
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med 1992; 327:293–301.
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore) 1997; 76:170–184.
- Dobbins WO, Ruffin JM. A light- and electron-microscopic study of bacterial invasion in Whipple’s disease. Am J Pathol 1967; 51:225–242.
- Dutly F, Altwegg M. Whipple’s disease and ‘Tropheryma whippelii.’ Clin Microbiol Rev 2001; 14:561–583.
- Raoult D, Birg ML, La Scola B, et al. Cultivation of the bacillus of Whipple’s disease. N Engl J Med 2000; 342:620–625.
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med 1992; 327:293–301.
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore) 1997; 76:170–184.
- Dobbins WO, Ruffin JM. A light- and electron-microscopic study of bacterial invasion in Whipple’s disease. Am J Pathol 1967; 51:225–242.
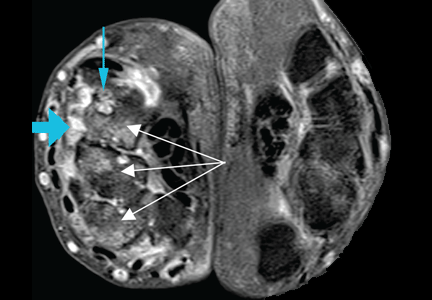
A rapidly growing crusted nodule on the lip
A 76-year-old man presented with a rapidly growing, indurated, crusted nodule on his lower lip (Figure 1). This combination—a rapidly growing nodule on a sun-exposed surface in an older patient—pointed to a diagnosis of squamous cell carcinoma, and biopsy study of the affected area confirmed this diagnosis (Figure 2). Clinical examination, computed tomography, and ultrasonography of the neck revealed no lymph node involvement; the tumor was staged as T2N0M0. Mohs microscopically controlled surgery was performed to remove the tumor with clear margins, and the patient has done well.
DIFFERENTIAL DIAGNOSIS
Malignant melanoma, Merkel cell carcinoma, and deep fungal infection can also cause a rapidly growing crusted nodule on the lower lip, but this typically is not an initial presentation for these conditions.
Malignant melanoma
Malignant melanoma should be considered for any rapidly growing cutaneous tumor, especially on sun-damaged skin. Most melanoma lesions have variations in pigment and may contain shades of blue, brown, red, pink, and white. Amelanotic melanomas may mimic squamous cell carcinomas, but the histologic features of atypical keratinocytes in this patient ruled out that diagnosis. Biopsy of amelanotic melanoma reveals nests of melanocytes, usually associated with an in situ component in the overlying epithelium.
Merkel cell carcinoma
Merkel cell carcinoma can mimic squamous cell carcinoma or can even arise in association with squamous cell carcinoma. Histologic examination allows for differentiation. In difficult cases, cytokeratin staining with cytokeratin 20 and CAM 5.2 can clarify the diagnosis, because Merkel cell carcinomas exhibit a characteristic paranuclear dot-like pattern not evident in squamous cell carcinoma.
Deep fungal infection
Colonization by Candida organisms was noted within the overlying crust in this patient’s lesion, but fungal organisms were not noted in the epidermis or dermis. Pseudoepitheliomatous hyperplasia can be prominent in deep fungal infection, but the marked cytologic atypia in this case excluded deep fungal infection.
Mucosal neuroma
Mucosal neuromas are typically smooth-surfaced, soft lesions on the lip. They may be associated with multiple endocrine neoplasia syndromes. Biopsy study reveals that they are composed of delicate spindle cells that lack atypia.
SQUAMOUS CELL CARCINOMA
Squamous cell carcinoma is one of the most common types of nonmelanoma skin cancer and is associated with increased sun exposure and light skin pigmentation.1 Unlike basal cell carcinoma, squamous cell carcinoma has a considerable potential to metastasize.2–4 The current cancer staging manual of the American Joint Commission on Cancer refects recent evidence-based information on staging and prognosis.5–7 Squamous cell carcinoma of the lip carries an increased risk of metastasis,8 and an increase in the number or the size of involved lymph nodes carries a worse prognosis.9 The lower lip is most commonly involved because of increased sun exposure. The most common route for the spread of squamous cell carcinoma of the head and neck is via lymphatics, so careful evaluation of the head and neck is indicated.10
STAGING OUR PATIENT’S LESION
Current staging of cutaneous squamous cell carcinoma takes into account a variety of different features. Our patient’s tumor was not associated with invasion of the maxilla, mandible, orbit, or temporal bone. This alone would have qualified the tumor as a T1 lesion, but its size of 3 cm indicated a higher risk and qualified it as a T2 lesion.
The histologic features of our patient’s lesion also indicate higher risk according to the current staging system.5 The Breslow thickness on histologic examination (measured from the granular cell layer of the epidermis to the deepest portion of the tumor) was 4 mm and the Clark’s level was IV (extension to the reticular dermis). The high-risk features and the location on the lip warrant a classification as T2 and carry a worse prognosis.
Wedge excision of the lower lip and Mohs surgery are accepted treatment options. The patient chose Mohs surgery and has done well, with excellent cosmetic and functional outcome. Radiation therapy can be useful as an adjuvant when lymph nodes are involved,11 but this was not necessary in our patient. Careful long-term follow up is warranted, as these patients are at higher risk of developing other, separate tumors.
- Schwartz RA. Squamous cell carcinoma. In:Schwartz RA, editor. Skin Cancer: Recognition and Management, 2nd ed. Malden, MA: Blackwell Publishing; 2008:47–65.
- D’Souza J, Clark J. Management of the neck in metastatic cutaneous squamous cell carcinoma of the head and neck. Curr Opin Otolaryngol Head Neck Surg 2011; 19:99–105.
- Zbar RI, Canady JW. MOC-PSSM CME article: Nonmelanoma facial skin malignancy. Plast Reconstr Surg 2008; 121(suppl 1):1–9.
- Morselli P, Masciotra L, Pinto V, Zollino I, Brunelli G, Carinci F. Clinical parameters in T1N0M0 lower lip squamous cell carcinoma. J Craniofac Surg 2007; 18:1079–1082.
- American Joint Commission on Cancer. Cancer Staging Manual. 7th ed. http://www.cancerstaging.org. Accessed February 3, 2013.
- Lardaro T, Shea SM, Sharfman W, Liégeois N, Sober AJ. Improvements in the staging of cutaneous squamous-cell carcinoma in the 7th edition of the AJCC Cancer Staging Manual. Ann Surg Oncol 2010; 17:1979–1980.
- Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In:Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, editors. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301–314.
- Frierson HF, Cooper PH. Prognostic factors in squamous cell carcinoma of the lower lip. Hum Pathol 1986; 17:346–354.
- Civantos FJ, Moffat FL, goodwin WJ. Lymphatic mapping and sentinel lymphadenectomy for 106 head and neck lesions: contrasts between oral cavity and cutaneous malignancy. Laryngoscope 2006; 112(3 Pt 2 suppl 109):1–15.
- Rowe De, Carroll RJ, Day CL. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol 1992; 26:976–990.
- Veness MJ, Palme CE, Smith M, Cakir B, Morgan GJ, Kalnins I. Cutaneous head and neck squamous cell carcinoma metastatic to cervical lymph nodes (nonparotid): a better outcome with surgery and adjuvant radiotherapy. Laryngoscope 2003; 113:1827–1833.
A 76-year-old man presented with a rapidly growing, indurated, crusted nodule on his lower lip (Figure 1). This combination—a rapidly growing nodule on a sun-exposed surface in an older patient—pointed to a diagnosis of squamous cell carcinoma, and biopsy study of the affected area confirmed this diagnosis (Figure 2). Clinical examination, computed tomography, and ultrasonography of the neck revealed no lymph node involvement; the tumor was staged as T2N0M0. Mohs microscopically controlled surgery was performed to remove the tumor with clear margins, and the patient has done well.
DIFFERENTIAL DIAGNOSIS
Malignant melanoma, Merkel cell carcinoma, and deep fungal infection can also cause a rapidly growing crusted nodule on the lower lip, but this typically is not an initial presentation for these conditions.
Malignant melanoma
Malignant melanoma should be considered for any rapidly growing cutaneous tumor, especially on sun-damaged skin. Most melanoma lesions have variations in pigment and may contain shades of blue, brown, red, pink, and white. Amelanotic melanomas may mimic squamous cell carcinomas, but the histologic features of atypical keratinocytes in this patient ruled out that diagnosis. Biopsy of amelanotic melanoma reveals nests of melanocytes, usually associated with an in situ component in the overlying epithelium.
Merkel cell carcinoma
Merkel cell carcinoma can mimic squamous cell carcinoma or can even arise in association with squamous cell carcinoma. Histologic examination allows for differentiation. In difficult cases, cytokeratin staining with cytokeratin 20 and CAM 5.2 can clarify the diagnosis, because Merkel cell carcinomas exhibit a characteristic paranuclear dot-like pattern not evident in squamous cell carcinoma.
Deep fungal infection
Colonization by Candida organisms was noted within the overlying crust in this patient’s lesion, but fungal organisms were not noted in the epidermis or dermis. Pseudoepitheliomatous hyperplasia can be prominent in deep fungal infection, but the marked cytologic atypia in this case excluded deep fungal infection.
Mucosal neuroma
Mucosal neuromas are typically smooth-surfaced, soft lesions on the lip. They may be associated with multiple endocrine neoplasia syndromes. Biopsy study reveals that they are composed of delicate spindle cells that lack atypia.
SQUAMOUS CELL CARCINOMA
Squamous cell carcinoma is one of the most common types of nonmelanoma skin cancer and is associated with increased sun exposure and light skin pigmentation.1 Unlike basal cell carcinoma, squamous cell carcinoma has a considerable potential to metastasize.2–4 The current cancer staging manual of the American Joint Commission on Cancer refects recent evidence-based information on staging and prognosis.5–7 Squamous cell carcinoma of the lip carries an increased risk of metastasis,8 and an increase in the number or the size of involved lymph nodes carries a worse prognosis.9 The lower lip is most commonly involved because of increased sun exposure. The most common route for the spread of squamous cell carcinoma of the head and neck is via lymphatics, so careful evaluation of the head and neck is indicated.10
STAGING OUR PATIENT’S LESION
Current staging of cutaneous squamous cell carcinoma takes into account a variety of different features. Our patient’s tumor was not associated with invasion of the maxilla, mandible, orbit, or temporal bone. This alone would have qualified the tumor as a T1 lesion, but its size of 3 cm indicated a higher risk and qualified it as a T2 lesion.
The histologic features of our patient’s lesion also indicate higher risk according to the current staging system.5 The Breslow thickness on histologic examination (measured from the granular cell layer of the epidermis to the deepest portion of the tumor) was 4 mm and the Clark’s level was IV (extension to the reticular dermis). The high-risk features and the location on the lip warrant a classification as T2 and carry a worse prognosis.
Wedge excision of the lower lip and Mohs surgery are accepted treatment options. The patient chose Mohs surgery and has done well, with excellent cosmetic and functional outcome. Radiation therapy can be useful as an adjuvant when lymph nodes are involved,11 but this was not necessary in our patient. Careful long-term follow up is warranted, as these patients are at higher risk of developing other, separate tumors.
A 76-year-old man presented with a rapidly growing, indurated, crusted nodule on his lower lip (Figure 1). This combination—a rapidly growing nodule on a sun-exposed surface in an older patient—pointed to a diagnosis of squamous cell carcinoma, and biopsy study of the affected area confirmed this diagnosis (Figure 2). Clinical examination, computed tomography, and ultrasonography of the neck revealed no lymph node involvement; the tumor was staged as T2N0M0. Mohs microscopically controlled surgery was performed to remove the tumor with clear margins, and the patient has done well.
DIFFERENTIAL DIAGNOSIS
Malignant melanoma, Merkel cell carcinoma, and deep fungal infection can also cause a rapidly growing crusted nodule on the lower lip, but this typically is not an initial presentation for these conditions.
Malignant melanoma
Malignant melanoma should be considered for any rapidly growing cutaneous tumor, especially on sun-damaged skin. Most melanoma lesions have variations in pigment and may contain shades of blue, brown, red, pink, and white. Amelanotic melanomas may mimic squamous cell carcinomas, but the histologic features of atypical keratinocytes in this patient ruled out that diagnosis. Biopsy of amelanotic melanoma reveals nests of melanocytes, usually associated with an in situ component in the overlying epithelium.
Merkel cell carcinoma
Merkel cell carcinoma can mimic squamous cell carcinoma or can even arise in association with squamous cell carcinoma. Histologic examination allows for differentiation. In difficult cases, cytokeratin staining with cytokeratin 20 and CAM 5.2 can clarify the diagnosis, because Merkel cell carcinomas exhibit a characteristic paranuclear dot-like pattern not evident in squamous cell carcinoma.
Deep fungal infection
Colonization by Candida organisms was noted within the overlying crust in this patient’s lesion, but fungal organisms were not noted in the epidermis or dermis. Pseudoepitheliomatous hyperplasia can be prominent in deep fungal infection, but the marked cytologic atypia in this case excluded deep fungal infection.
Mucosal neuroma
Mucosal neuromas are typically smooth-surfaced, soft lesions on the lip. They may be associated with multiple endocrine neoplasia syndromes. Biopsy study reveals that they are composed of delicate spindle cells that lack atypia.
SQUAMOUS CELL CARCINOMA
Squamous cell carcinoma is one of the most common types of nonmelanoma skin cancer and is associated with increased sun exposure and light skin pigmentation.1 Unlike basal cell carcinoma, squamous cell carcinoma has a considerable potential to metastasize.2–4 The current cancer staging manual of the American Joint Commission on Cancer refects recent evidence-based information on staging and prognosis.5–7 Squamous cell carcinoma of the lip carries an increased risk of metastasis,8 and an increase in the number or the size of involved lymph nodes carries a worse prognosis.9 The lower lip is most commonly involved because of increased sun exposure. The most common route for the spread of squamous cell carcinoma of the head and neck is via lymphatics, so careful evaluation of the head and neck is indicated.10
STAGING OUR PATIENT’S LESION
Current staging of cutaneous squamous cell carcinoma takes into account a variety of different features. Our patient’s tumor was not associated with invasion of the maxilla, mandible, orbit, or temporal bone. This alone would have qualified the tumor as a T1 lesion, but its size of 3 cm indicated a higher risk and qualified it as a T2 lesion.
The histologic features of our patient’s lesion also indicate higher risk according to the current staging system.5 The Breslow thickness on histologic examination (measured from the granular cell layer of the epidermis to the deepest portion of the tumor) was 4 mm and the Clark’s level was IV (extension to the reticular dermis). The high-risk features and the location on the lip warrant a classification as T2 and carry a worse prognosis.
Wedge excision of the lower lip and Mohs surgery are accepted treatment options. The patient chose Mohs surgery and has done well, with excellent cosmetic and functional outcome. Radiation therapy can be useful as an adjuvant when lymph nodes are involved,11 but this was not necessary in our patient. Careful long-term follow up is warranted, as these patients are at higher risk of developing other, separate tumors.
- Schwartz RA. Squamous cell carcinoma. In:Schwartz RA, editor. Skin Cancer: Recognition and Management, 2nd ed. Malden, MA: Blackwell Publishing; 2008:47–65.
- D’Souza J, Clark J. Management of the neck in metastatic cutaneous squamous cell carcinoma of the head and neck. Curr Opin Otolaryngol Head Neck Surg 2011; 19:99–105.
- Zbar RI, Canady JW. MOC-PSSM CME article: Nonmelanoma facial skin malignancy. Plast Reconstr Surg 2008; 121(suppl 1):1–9.
- Morselli P, Masciotra L, Pinto V, Zollino I, Brunelli G, Carinci F. Clinical parameters in T1N0M0 lower lip squamous cell carcinoma. J Craniofac Surg 2007; 18:1079–1082.
- American Joint Commission on Cancer. Cancer Staging Manual. 7th ed. http://www.cancerstaging.org. Accessed February 3, 2013.
- Lardaro T, Shea SM, Sharfman W, Liégeois N, Sober AJ. Improvements in the staging of cutaneous squamous-cell carcinoma in the 7th edition of the AJCC Cancer Staging Manual. Ann Surg Oncol 2010; 17:1979–1980.
- Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In:Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, editors. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301–314.
- Frierson HF, Cooper PH. Prognostic factors in squamous cell carcinoma of the lower lip. Hum Pathol 1986; 17:346–354.
- Civantos FJ, Moffat FL, goodwin WJ. Lymphatic mapping and sentinel lymphadenectomy for 106 head and neck lesions: contrasts between oral cavity and cutaneous malignancy. Laryngoscope 2006; 112(3 Pt 2 suppl 109):1–15.
- Rowe De, Carroll RJ, Day CL. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol 1992; 26:976–990.
- Veness MJ, Palme CE, Smith M, Cakir B, Morgan GJ, Kalnins I. Cutaneous head and neck squamous cell carcinoma metastatic to cervical lymph nodes (nonparotid): a better outcome with surgery and adjuvant radiotherapy. Laryngoscope 2003; 113:1827–1833.
- Schwartz RA. Squamous cell carcinoma. In:Schwartz RA, editor. Skin Cancer: Recognition and Management, 2nd ed. Malden, MA: Blackwell Publishing; 2008:47–65.
- D’Souza J, Clark J. Management of the neck in metastatic cutaneous squamous cell carcinoma of the head and neck. Curr Opin Otolaryngol Head Neck Surg 2011; 19:99–105.
- Zbar RI, Canady JW. MOC-PSSM CME article: Nonmelanoma facial skin malignancy. Plast Reconstr Surg 2008; 121(suppl 1):1–9.
- Morselli P, Masciotra L, Pinto V, Zollino I, Brunelli G, Carinci F. Clinical parameters in T1N0M0 lower lip squamous cell carcinoma. J Craniofac Surg 2007; 18:1079–1082.
- American Joint Commission on Cancer. Cancer Staging Manual. 7th ed. http://www.cancerstaging.org. Accessed February 3, 2013.
- Lardaro T, Shea SM, Sharfman W, Liégeois N, Sober AJ. Improvements in the staging of cutaneous squamous-cell carcinoma in the 7th edition of the AJCC Cancer Staging Manual. Ann Surg Oncol 2010; 17:1979–1980.
- Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In:Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, editors. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301–314.
- Frierson HF, Cooper PH. Prognostic factors in squamous cell carcinoma of the lower lip. Hum Pathol 1986; 17:346–354.
- Civantos FJ, Moffat FL, goodwin WJ. Lymphatic mapping and sentinel lymphadenectomy for 106 head and neck lesions: contrasts between oral cavity and cutaneous malignancy. Laryngoscope 2006; 112(3 Pt 2 suppl 109):1–15.
- Rowe De, Carroll RJ, Day CL. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol 1992; 26:976–990.
- Veness MJ, Palme CE, Smith M, Cakir B, Morgan GJ, Kalnins I. Cutaneous head and neck squamous cell carcinoma metastatic to cervical lymph nodes (nonparotid): a better outcome with surgery and adjuvant radiotherapy. Laryngoscope 2003; 113:1827–1833.
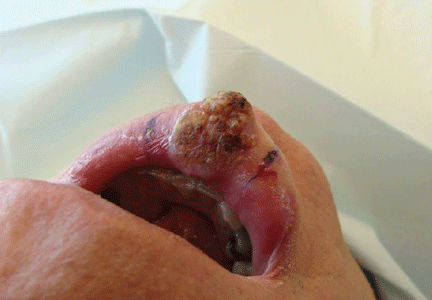
Implications of a prominent R wave in V1
A 19-year-old woman with no significant cardiac or pulmonary history presented with exertional dyspnea, which had begun a few months earlier. Auscultation revealed a loud pulmonary component of the second heart sound and a diastolic murmur heard along the upper left sternal border. Her 12-lead electrocardiogram is shown in Figure 1.
Q: Which of the following can cause prominent R waves in lead V1?
- Normal variant in young adults
- Wolff-Parkinson-White syndrome
- Posterior wall myocardial infarction
- Right ventricular hypertrophy
- All of the above
A: The correct answer is all of the above.
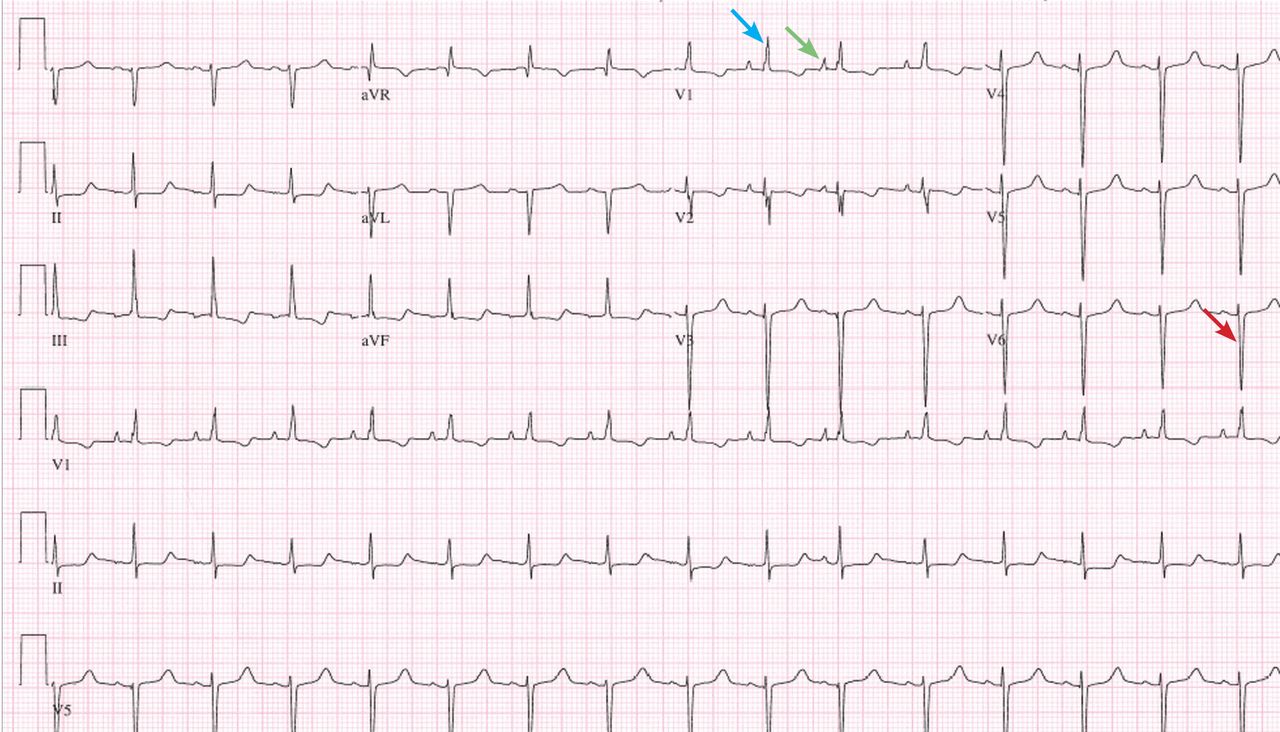
The patient’s electrocardiogram shows a right atrial abnormality and right ventricular hypertrophy. Right atrial enlargement is evidenced by a prominent initial P wave in V1 with an amplitude of at least 1.5 mm (0.15 mV). A P wave taller than 2.5 mm (0.25 mV) in lead II may also suggest a right atrial abnormality.1
Multiple criteria exist for the diagnosis of right ventricular hypertrophy. Tall R waves in V1 with an R/S ratio greater than 1 (ie, the R wave amplitude is more than the S wave depth) is commonly used.2 Deep S waves with an R/S ratio less than 1 in V6 is another criterion. Tall R waves of amplitude greater than 7 mm in V1 by themselves may represent right ventricular hypertrophy. Most of the electrocardiographic criteria are specific but not sensitive for this diagnosis.3

Other causes of tall R waves in V1 are given in Table 1.
Q: Which of the following diseases can present with an electrocardiographic pattern of right ventricular hypertrophy in young patients?
- Pulmonary hypertension
- Atrial septal defect
- Tetralogy of Fallot
- Pulmonary stenosis
- All of the above
A: The correct answer is all of the above.4
Our patient underwent multiple investigations. On echocardiography, her estimated right ventricular pressure was 80 mm Hg, and on cardiac catheterization her mean pulmonary arterial pressure was 55 mm Hg and her pulmonary capillary wedge pressure was 6 mm Hg. She was diagnosed with pulmonary arterial hypertension, which was the cause of her right ventricular hypertrophy. She eventually underwent bilateral lung transplantation.
- Hancock EW, Deal BJ, Mirvis DM, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part V: electrocardiogram changes associated with cardiac chamber hypertrophy: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation 2009; 119:e251–e261.
- Milnor WR. Electrocardiogram and vectorcardiogram in right ventricular hypertrophy and right bundle-branch block. Circulation 1957; 16:348–367.
- Lehtonen J, Sutinen S, Ikäheimo M, Pääkkö P. Electrocardiographic criteria for the diagnosis of right ventricular hypertrophy verified at autopsy. Chest 1988; 93:839–842.
- Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006; 114:1645–1653.
A 19-year-old woman with no significant cardiac or pulmonary history presented with exertional dyspnea, which had begun a few months earlier. Auscultation revealed a loud pulmonary component of the second heart sound and a diastolic murmur heard along the upper left sternal border. Her 12-lead electrocardiogram is shown in Figure 1.
Q: Which of the following can cause prominent R waves in lead V1?
- Normal variant in young adults
- Wolff-Parkinson-White syndrome
- Posterior wall myocardial infarction
- Right ventricular hypertrophy
- All of the above
A: The correct answer is all of the above.
The patient’s electrocardiogram shows a right atrial abnormality and right ventricular hypertrophy. Right atrial enlargement is evidenced by a prominent initial P wave in V1 with an amplitude of at least 1.5 mm (0.15 mV). A P wave taller than 2.5 mm (0.25 mV) in lead II may also suggest a right atrial abnormality.1
Multiple criteria exist for the diagnosis of right ventricular hypertrophy. Tall R waves in V1 with an R/S ratio greater than 1 (ie, the R wave amplitude is more than the S wave depth) is commonly used.2 Deep S waves with an R/S ratio less than 1 in V6 is another criterion. Tall R waves of amplitude greater than 7 mm in V1 by themselves may represent right ventricular hypertrophy. Most of the electrocardiographic criteria are specific but not sensitive for this diagnosis.3
Other causes of tall R waves in V1 are given in Table 1.
Q: Which of the following diseases can present with an electrocardiographic pattern of right ventricular hypertrophy in young patients?
- Pulmonary hypertension
- Atrial septal defect
- Tetralogy of Fallot
- Pulmonary stenosis
- All of the above
A: The correct answer is all of the above.4
Our patient underwent multiple investigations. On echocardiography, her estimated right ventricular pressure was 80 mm Hg, and on cardiac catheterization her mean pulmonary arterial pressure was 55 mm Hg and her pulmonary capillary wedge pressure was 6 mm Hg. She was diagnosed with pulmonary arterial hypertension, which was the cause of her right ventricular hypertrophy. She eventually underwent bilateral lung transplantation.
A 19-year-old woman with no significant cardiac or pulmonary history presented with exertional dyspnea, which had begun a few months earlier. Auscultation revealed a loud pulmonary component of the second heart sound and a diastolic murmur heard along the upper left sternal border. Her 12-lead electrocardiogram is shown in Figure 1.
Q: Which of the following can cause prominent R waves in lead V1?
- Normal variant in young adults
- Wolff-Parkinson-White syndrome
- Posterior wall myocardial infarction
- Right ventricular hypertrophy
- All of the above
A: The correct answer is all of the above.
The patient’s electrocardiogram shows a right atrial abnormality and right ventricular hypertrophy. Right atrial enlargement is evidenced by a prominent initial P wave in V1 with an amplitude of at least 1.5 mm (0.15 mV). A P wave taller than 2.5 mm (0.25 mV) in lead II may also suggest a right atrial abnormality.1
Multiple criteria exist for the diagnosis of right ventricular hypertrophy. Tall R waves in V1 with an R/S ratio greater than 1 (ie, the R wave amplitude is more than the S wave depth) is commonly used.2 Deep S waves with an R/S ratio less than 1 in V6 is another criterion. Tall R waves of amplitude greater than 7 mm in V1 by themselves may represent right ventricular hypertrophy. Most of the electrocardiographic criteria are specific but not sensitive for this diagnosis.3
Other causes of tall R waves in V1 are given in Table 1.
Q: Which of the following diseases can present with an electrocardiographic pattern of right ventricular hypertrophy in young patients?
- Pulmonary hypertension
- Atrial septal defect
- Tetralogy of Fallot
- Pulmonary stenosis
- All of the above
A: The correct answer is all of the above.4
Our patient underwent multiple investigations. On echocardiography, her estimated right ventricular pressure was 80 mm Hg, and on cardiac catheterization her mean pulmonary arterial pressure was 55 mm Hg and her pulmonary capillary wedge pressure was 6 mm Hg. She was diagnosed with pulmonary arterial hypertension, which was the cause of her right ventricular hypertrophy. She eventually underwent bilateral lung transplantation.
- Hancock EW, Deal BJ, Mirvis DM, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part V: electrocardiogram changes associated with cardiac chamber hypertrophy: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation 2009; 119:e251–e261.
- Milnor WR. Electrocardiogram and vectorcardiogram in right ventricular hypertrophy and right bundle-branch block. Circulation 1957; 16:348–367.
- Lehtonen J, Sutinen S, Ikäheimo M, Pääkkö P. Electrocardiographic criteria for the diagnosis of right ventricular hypertrophy verified at autopsy. Chest 1988; 93:839–842.
- Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006; 114:1645–1653.
- Hancock EW, Deal BJ, Mirvis DM, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part V: electrocardiogram changes associated with cardiac chamber hypertrophy: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation 2009; 119:e251–e261.
- Milnor WR. Electrocardiogram and vectorcardiogram in right ventricular hypertrophy and right bundle-branch block. Circulation 1957; 16:348–367.
- Lehtonen J, Sutinen S, Ikäheimo M, Pääkkö P. Electrocardiographic criteria for the diagnosis of right ventricular hypertrophy verified at autopsy. Chest 1988; 93:839–842.
- Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006; 114:1645–1653.
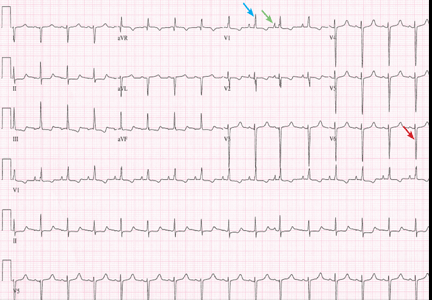
Sore throat, odynophagia, hoarseness, and a muffled, high-pitched voice
A 55-year-old man presents to the emergency department with a sore throat, odynophagia, hoarseness, and a high-pitched muffled voice for 1 day. He is otherwise healthy, with no fever, chills, or exposure to sick contacts.
Q: Which is the most likely diagnosis?
- Retropharyngeal abscess
- Croup
- Streptococcal pharyngitis (“strep throat”)
- Acute epiglottitis
- Viral upper-respiratory infection
A: The correct answer is acute epiglottitis.
Acute epiglottitis is a cellulitis of the epiglottis and adjacent tissues that, without treatment, can progress to life-threatening airway obstruction.
In children, this entity most often occurs between ages 2 and 4. Previously, it was mainly associated with Haemophilus influenzae type B infection, but since the start of vaccination for this organism, epiglottitis has become less common.
In recent years, Streptococcus pneumoniae and beta-hemolytic streptococci have become the main culprits in epiglottitis in children. In adults, common organisms include H influenzae, S pneumoniae, Staphylococcus aureus, and betahemolytic streptococci. Epiglottitis may also be caused by herpes simplex, varicella-zoster, and influenza viruses. Noninfectious causes include ingestion of a foreign body and thermal injury.
Common signs and symptoms of epiglottitis include sore throat, a high-pitched and muffled voice, labored breathing, and respiratory distress.1 Since airway obstruction can be remarkably precipitous, it is important to obtain early consultation with an otolaryngologist for laryngoscopy and visualization of the erythematous, swollen epiglottis. Signs of severe upper-airway obstruction may be absent until late in the disease process, making early diagnosis crucial.
On physical examination, the oropharynx will often appear benign. Currently, the preferred diagnostic test for epiglottitis is visualization by laryngoscopy, which has close to 100% sensitivity and specificity.2 By comparison, lateral neck radiography has poor sensitivity (38% to 88%) and specificity (78%).3,4 Bedside ultrasonography has been reported as capable of revealing the “alphabet P sign” in the longitudinal view through the thyrohyoid membrane.5
At triage, if clinical suspicion of total airway obstruction is high, the patient may be taken directly to the operating room for visualization of the epiglottis and, if necessary, for endotracheal intubation. When the risk is considered low or moderate, lateral neck radiography may be helpful,6 as it will sometimes reveal a swollen epiglottis (ie, “thumb sign”), thickened arytenoepiglottic folds, and obliteration of the epiglottic vallecula. If the radiograph is normal and clinical suspicion is still present, laryngoscopy can be done.
For low-risk, acute epiglottitis, closely monitoring the patient in an intensive care unit is recommended. For severe, high-risk cases, securing the airway is of the utmost importance. In addition to securing the airway, empiric antimicrobial therapy—such as with cefotaxime or ceftriaxone plus clindamycin or vancomycin—is often warranted in both low-risk and high-risk cases. The use of corticosteroids in epiglottitis has not been studied in a randomized, controlled trial, and, though common, it remains controversial.7 While undergoing treatment, patients should be closely monitored for respiratory compromise.
HOW OUR PATIENT WAS MANAGED
Our patient had a high-pitched, muffled voice and prominent, tender anterior cervical lymph nodes. No drooling or stridor was noted. The oropharynx appeared normal. Lateral soft-tissue radiography of the neck (Figure 1) revealed several key findings:
- A swollen epiglottis (ie, the “thumb sign”) consistent with acute epiglottitis and a narrowed airway
- Swollen arytenoids
- A shallow V-shaped epiglottic vallecula.
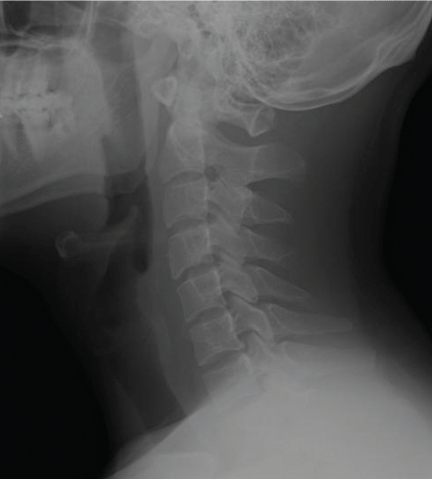
For comparison, Figure 2 shows a normal lateral soft-tissue radiograph of the neck from a different patient.
Our patient was referred to an otolaryngologist, who noted a severely inflamed epiglottis on laryngoscopy.
The patient was given intravenous vancomycin, ceftriaxone, and high-dose corticosteroids and was admitted to the intensive care unit. After his symptoms improved, repeat laryngoscopy revealed markedly diminished inflammation. He was discharged home with an additional course of oral antibiotics.
- Alcaide ML, Bisno AL. Pharyngitis and epiglottitis. Infect Dis Clin North Am 2007; 21:449–469.
- Cheung CS, Man SY, Graham CA, et al. Adult epiglottitis: 6 years experience in a university teaching hospital in Hong Kong. Eur J Emerg Med 2009; 16:221–226.
- Stankiewicz JA, Bowes AK. Croup and epiglottitis: a radiologic study. Laryngoscope 1985; 95:1159–1160.
- Solomon P, Weisbrod M, Irish JC, Gullane PJ. Adult epiglottitis: the Toronto Hospital experience. J Otolaryngol 1998; 27:332–336.
- Hung TY, Li S, Chen PS, et al. Bedside ultrasonography as a safe and effective tool to diagnose acute epiglottitis. Am J Emerg Med 2011; 29:359.e1–359.e3.
- Ragosta KG, Orr R, Detweiler MJ. Revisiting epiglottitis: a protocol—the value of lateral neck radiographs. J Am Osteopath Assoc 1997; 97:227–229.
- Glynn F, Fenton JE. Diagnosis and management of supraglottitis (epiglottitis). Curr Infect Dis Rep 2008; 10:200–204.
A 55-year-old man presents to the emergency department with a sore throat, odynophagia, hoarseness, and a high-pitched muffled voice for 1 day. He is otherwise healthy, with no fever, chills, or exposure to sick contacts.
Q: Which is the most likely diagnosis?
- Retropharyngeal abscess
- Croup
- Streptococcal pharyngitis (“strep throat”)
- Acute epiglottitis
- Viral upper-respiratory infection
A: The correct answer is acute epiglottitis.
Acute epiglottitis is a cellulitis of the epiglottis and adjacent tissues that, without treatment, can progress to life-threatening airway obstruction.
In children, this entity most often occurs between ages 2 and 4. Previously, it was mainly associated with Haemophilus influenzae type B infection, but since the start of vaccination for this organism, epiglottitis has become less common.
In recent years, Streptococcus pneumoniae and beta-hemolytic streptococci have become the main culprits in epiglottitis in children. In adults, common organisms include H influenzae, S pneumoniae, Staphylococcus aureus, and betahemolytic streptococci. Epiglottitis may also be caused by herpes simplex, varicella-zoster, and influenza viruses. Noninfectious causes include ingestion of a foreign body and thermal injury.
Common signs and symptoms of epiglottitis include sore throat, a high-pitched and muffled voice, labored breathing, and respiratory distress.1 Since airway obstruction can be remarkably precipitous, it is important to obtain early consultation with an otolaryngologist for laryngoscopy and visualization of the erythematous, swollen epiglottis. Signs of severe upper-airway obstruction may be absent until late in the disease process, making early diagnosis crucial.
On physical examination, the oropharynx will often appear benign. Currently, the preferred diagnostic test for epiglottitis is visualization by laryngoscopy, which has close to 100% sensitivity and specificity.2 By comparison, lateral neck radiography has poor sensitivity (38% to 88%) and specificity (78%).3,4 Bedside ultrasonography has been reported as capable of revealing the “alphabet P sign” in the longitudinal view through the thyrohyoid membrane.5
At triage, if clinical suspicion of total airway obstruction is high, the patient may be taken directly to the operating room for visualization of the epiglottis and, if necessary, for endotracheal intubation. When the risk is considered low or moderate, lateral neck radiography may be helpful,6 as it will sometimes reveal a swollen epiglottis (ie, “thumb sign”), thickened arytenoepiglottic folds, and obliteration of the epiglottic vallecula. If the radiograph is normal and clinical suspicion is still present, laryngoscopy can be done.
For low-risk, acute epiglottitis, closely monitoring the patient in an intensive care unit is recommended. For severe, high-risk cases, securing the airway is of the utmost importance. In addition to securing the airway, empiric antimicrobial therapy—such as with cefotaxime or ceftriaxone plus clindamycin or vancomycin—is often warranted in both low-risk and high-risk cases. The use of corticosteroids in epiglottitis has not been studied in a randomized, controlled trial, and, though common, it remains controversial.7 While undergoing treatment, patients should be closely monitored for respiratory compromise.
HOW OUR PATIENT WAS MANAGED
Our patient had a high-pitched, muffled voice and prominent, tender anterior cervical lymph nodes. No drooling or stridor was noted. The oropharynx appeared normal. Lateral soft-tissue radiography of the neck (Figure 1) revealed several key findings:
- A swollen epiglottis (ie, the “thumb sign”) consistent with acute epiglottitis and a narrowed airway
- Swollen arytenoids
- A shallow V-shaped epiglottic vallecula.
For comparison, Figure 2 shows a normal lateral soft-tissue radiograph of the neck from a different patient.
Our patient was referred to an otolaryngologist, who noted a severely inflamed epiglottis on laryngoscopy.
The patient was given intravenous vancomycin, ceftriaxone, and high-dose corticosteroids and was admitted to the intensive care unit. After his symptoms improved, repeat laryngoscopy revealed markedly diminished inflammation. He was discharged home with an additional course of oral antibiotics.
A 55-year-old man presents to the emergency department with a sore throat, odynophagia, hoarseness, and a high-pitched muffled voice for 1 day. He is otherwise healthy, with no fever, chills, or exposure to sick contacts.
Q: Which is the most likely diagnosis?
- Retropharyngeal abscess
- Croup
- Streptococcal pharyngitis (“strep throat”)
- Acute epiglottitis
- Viral upper-respiratory infection
A: The correct answer is acute epiglottitis.
Acute epiglottitis is a cellulitis of the epiglottis and adjacent tissues that, without treatment, can progress to life-threatening airway obstruction.
In children, this entity most often occurs between ages 2 and 4. Previously, it was mainly associated with Haemophilus influenzae type B infection, but since the start of vaccination for this organism, epiglottitis has become less common.
In recent years, Streptococcus pneumoniae and beta-hemolytic streptococci have become the main culprits in epiglottitis in children. In adults, common organisms include H influenzae, S pneumoniae, Staphylococcus aureus, and betahemolytic streptococci. Epiglottitis may also be caused by herpes simplex, varicella-zoster, and influenza viruses. Noninfectious causes include ingestion of a foreign body and thermal injury.
Common signs and symptoms of epiglottitis include sore throat, a high-pitched and muffled voice, labored breathing, and respiratory distress.1 Since airway obstruction can be remarkably precipitous, it is important to obtain early consultation with an otolaryngologist for laryngoscopy and visualization of the erythematous, swollen epiglottis. Signs of severe upper-airway obstruction may be absent until late in the disease process, making early diagnosis crucial.
On physical examination, the oropharynx will often appear benign. Currently, the preferred diagnostic test for epiglottitis is visualization by laryngoscopy, which has close to 100% sensitivity and specificity.2 By comparison, lateral neck radiography has poor sensitivity (38% to 88%) and specificity (78%).3,4 Bedside ultrasonography has been reported as capable of revealing the “alphabet P sign” in the longitudinal view through the thyrohyoid membrane.5
At triage, if clinical suspicion of total airway obstruction is high, the patient may be taken directly to the operating room for visualization of the epiglottis and, if necessary, for endotracheal intubation. When the risk is considered low or moderate, lateral neck radiography may be helpful,6 as it will sometimes reveal a swollen epiglottis (ie, “thumb sign”), thickened arytenoepiglottic folds, and obliteration of the epiglottic vallecula. If the radiograph is normal and clinical suspicion is still present, laryngoscopy can be done.
For low-risk, acute epiglottitis, closely monitoring the patient in an intensive care unit is recommended. For severe, high-risk cases, securing the airway is of the utmost importance. In addition to securing the airway, empiric antimicrobial therapy—such as with cefotaxime or ceftriaxone plus clindamycin or vancomycin—is often warranted in both low-risk and high-risk cases. The use of corticosteroids in epiglottitis has not been studied in a randomized, controlled trial, and, though common, it remains controversial.7 While undergoing treatment, patients should be closely monitored for respiratory compromise.
HOW OUR PATIENT WAS MANAGED
Our patient had a high-pitched, muffled voice and prominent, tender anterior cervical lymph nodes. No drooling or stridor was noted. The oropharynx appeared normal. Lateral soft-tissue radiography of the neck (Figure 1) revealed several key findings:
- A swollen epiglottis (ie, the “thumb sign”) consistent with acute epiglottitis and a narrowed airway
- Swollen arytenoids
- A shallow V-shaped epiglottic vallecula.
For comparison, Figure 2 shows a normal lateral soft-tissue radiograph of the neck from a different patient.
Our patient was referred to an otolaryngologist, who noted a severely inflamed epiglottis on laryngoscopy.
The patient was given intravenous vancomycin, ceftriaxone, and high-dose corticosteroids and was admitted to the intensive care unit. After his symptoms improved, repeat laryngoscopy revealed markedly diminished inflammation. He was discharged home with an additional course of oral antibiotics.
- Alcaide ML, Bisno AL. Pharyngitis and epiglottitis. Infect Dis Clin North Am 2007; 21:449–469.
- Cheung CS, Man SY, Graham CA, et al. Adult epiglottitis: 6 years experience in a university teaching hospital in Hong Kong. Eur J Emerg Med 2009; 16:221–226.
- Stankiewicz JA, Bowes AK. Croup and epiglottitis: a radiologic study. Laryngoscope 1985; 95:1159–1160.
- Solomon P, Weisbrod M, Irish JC, Gullane PJ. Adult epiglottitis: the Toronto Hospital experience. J Otolaryngol 1998; 27:332–336.
- Hung TY, Li S, Chen PS, et al. Bedside ultrasonography as a safe and effective tool to diagnose acute epiglottitis. Am J Emerg Med 2011; 29:359.e1–359.e3.
- Ragosta KG, Orr R, Detweiler MJ. Revisiting epiglottitis: a protocol—the value of lateral neck radiographs. J Am Osteopath Assoc 1997; 97:227–229.
- Glynn F, Fenton JE. Diagnosis and management of supraglottitis (epiglottitis). Curr Infect Dis Rep 2008; 10:200–204.
- Alcaide ML, Bisno AL. Pharyngitis and epiglottitis. Infect Dis Clin North Am 2007; 21:449–469.
- Cheung CS, Man SY, Graham CA, et al. Adult epiglottitis: 6 years experience in a university teaching hospital in Hong Kong. Eur J Emerg Med 2009; 16:221–226.
- Stankiewicz JA, Bowes AK. Croup and epiglottitis: a radiologic study. Laryngoscope 1985; 95:1159–1160.
- Solomon P, Weisbrod M, Irish JC, Gullane PJ. Adult epiglottitis: the Toronto Hospital experience. J Otolaryngol 1998; 27:332–336.
- Hung TY, Li S, Chen PS, et al. Bedside ultrasonography as a safe and effective tool to diagnose acute epiglottitis. Am J Emerg Med 2011; 29:359.e1–359.e3.
- Ragosta KG, Orr R, Detweiler MJ. Revisiting epiglottitis: a protocol—the value of lateral neck radiographs. J Am Osteopath Assoc 1997; 97:227–229.
- Glynn F, Fenton JE. Diagnosis and management of supraglottitis (epiglottitis). Curr Infect Dis Rep 2008; 10:200–204.
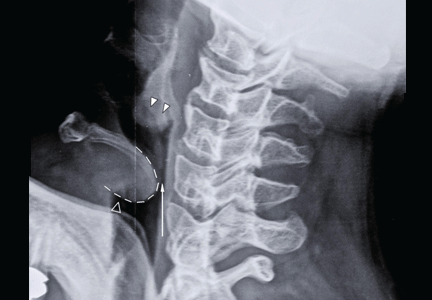
Giant inverted T waves
A 48-year-old man with hypertension was being evaluated for a noncardiac issue (progressive multifocal leukoencephalopathy). He had been an active runner and did not have any cardiovascular symptoms at the time. The electrocardiogram (ECG) shown in Figure 1 was a routine study done as a part of that evaluation. His cardiovascular examination was unremarkable, without murmur, S3, or S4. His pulse was regular at 72 beats per minute, and his blood pressure was 112/76 mm Hg.
Q: Which of the following electrocardiographic findings suggest left ventricular hypertrophy?
- Sum of the S wave in V1 and the R wave in V6 ≥ 35 mm
- Sum of the S wave in V3 and the R wave in aVL > 28 mm (men)
- Sum of the S wave in V3 and the R wave in aVL > 20 mm (women)
- All of the above
A: The correct answer is all of the above.1,2
Our patient’s ECG shows sinus bradycardia and left ventricular hypertrophy, suggested by prominent voltage (sum of S in V1 and R in V6 ≥ 35 mm) and supported by ST-segment and T-wave changes in the lateral and midprecordial leads. Classic changes of left ventricular hypertrophy often include increased voltage and downsloping ST-segment depression with negative T waves in V5 and V6 (secondary repolarization changes or “strain” pattern).
Notable on this tracing are the large, asymmetric negative T waves in leads V3 through V6. Giant T waves are defined as negative T waves with voltage greater than 10 mm.3 Although there is no specific pattern of ventricular hypertrophy on an ECG that establishes the diagnosis of hypertrophic cardiomyopathy, left ventricular hypertrophy with T waves of this quality suggest the possibility of hypertrophic cardiomyopathy with apical hypertrophy.
Q: What are the other causes of giant negative T waves?
- Subarachnoid hemorrhage
- Complete heart block
- Non-Q-wave myocardial infarction
- All of the above

A: The correct answer is all of the above. Additional causes of dramatic T-wave inversion are listed in Table 1. Clinically, non-Q-wave myocardial infarction with T-wave changes and acute central nervous system injury are probably the most commonly seen.4
Echocardiography in this patient revealed severe apical hypertrophy of the ventricle with distal cavity obliteration. The left ventricular outflow-tract gradient was normal. The mitral valve appeared normal, and there was no resting systolic anterior motion.
Cardiac magnetic resonance imaging showed the apical variant of hypertrophic cardiomyopathy but no evidence of left ventricular noncompaction, which is a differential diagnosis of apical hypertrophic obstructive cardiomyopathy. This disease was first described in Japan by Yamaguchi et al5 and Sakamoto et al6 and is regarded as a subgroup of nonobstructive hypertrophic cardiomyopathy. The prognosis of apical hypertrophic cardiomyopathy with regard to sudden cardiac death is believed to be better than that of other forms of hypertrophic cardiomyopathy.3
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. 1949. Ann Noninvasive Electrocardiol 2001; 6:343–368.
- Casale PN, Devereux RB, Alonso DR, Campo E, Kligfield P. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation 1987; 75:565–572.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Jacobson D, Schrire V. Giant T wave inversion. Br Heart J 1966; 28:768–775.
- Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol 1979; 44:401–412.
- Sakamoto T, Tei C, Murayama M, Ichiyasu H, Hada Y. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasonocardiotomographic study. Jpn Heart J 1976; 17:611–629.
A 48-year-old man with hypertension was being evaluated for a noncardiac issue (progressive multifocal leukoencephalopathy). He had been an active runner and did not have any cardiovascular symptoms at the time. The electrocardiogram (ECG) shown in Figure 1 was a routine study done as a part of that evaluation. His cardiovascular examination was unremarkable, without murmur, S3, or S4. His pulse was regular at 72 beats per minute, and his blood pressure was 112/76 mm Hg.
Q: Which of the following electrocardiographic findings suggest left ventricular hypertrophy?
- Sum of the S wave in V1 and the R wave in V6 ≥ 35 mm
- Sum of the S wave in V3 and the R wave in aVL > 28 mm (men)
- Sum of the S wave in V3 and the R wave in aVL > 20 mm (women)
- All of the above
A: The correct answer is all of the above.1,2
Our patient’s ECG shows sinus bradycardia and left ventricular hypertrophy, suggested by prominent voltage (sum of S in V1 and R in V6 ≥ 35 mm) and supported by ST-segment and T-wave changes in the lateral and midprecordial leads. Classic changes of left ventricular hypertrophy often include increased voltage and downsloping ST-segment depression with negative T waves in V5 and V6 (secondary repolarization changes or “strain” pattern).
Notable on this tracing are the large, asymmetric negative T waves in leads V3 through V6. Giant T waves are defined as negative T waves with voltage greater than 10 mm.3 Although there is no specific pattern of ventricular hypertrophy on an ECG that establishes the diagnosis of hypertrophic cardiomyopathy, left ventricular hypertrophy with T waves of this quality suggest the possibility of hypertrophic cardiomyopathy with apical hypertrophy.
Q: What are the other causes of giant negative T waves?
- Subarachnoid hemorrhage
- Complete heart block
- Non-Q-wave myocardial infarction
- All of the above
A: The correct answer is all of the above. Additional causes of dramatic T-wave inversion are listed in Table 1. Clinically, non-Q-wave myocardial infarction with T-wave changes and acute central nervous system injury are probably the most commonly seen.4
Echocardiography in this patient revealed severe apical hypertrophy of the ventricle with distal cavity obliteration. The left ventricular outflow-tract gradient was normal. The mitral valve appeared normal, and there was no resting systolic anterior motion.
Cardiac magnetic resonance imaging showed the apical variant of hypertrophic cardiomyopathy but no evidence of left ventricular noncompaction, which is a differential diagnosis of apical hypertrophic obstructive cardiomyopathy. This disease was first described in Japan by Yamaguchi et al5 and Sakamoto et al6 and is regarded as a subgroup of nonobstructive hypertrophic cardiomyopathy. The prognosis of apical hypertrophic cardiomyopathy with regard to sudden cardiac death is believed to be better than that of other forms of hypertrophic cardiomyopathy.3
A 48-year-old man with hypertension was being evaluated for a noncardiac issue (progressive multifocal leukoencephalopathy). He had been an active runner and did not have any cardiovascular symptoms at the time. The electrocardiogram (ECG) shown in Figure 1 was a routine study done as a part of that evaluation. His cardiovascular examination was unremarkable, without murmur, S3, or S4. His pulse was regular at 72 beats per minute, and his blood pressure was 112/76 mm Hg.
Q: Which of the following electrocardiographic findings suggest left ventricular hypertrophy?
- Sum of the S wave in V1 and the R wave in V6 ≥ 35 mm
- Sum of the S wave in V3 and the R wave in aVL > 28 mm (men)
- Sum of the S wave in V3 and the R wave in aVL > 20 mm (women)
- All of the above
A: The correct answer is all of the above.1,2
Our patient’s ECG shows sinus bradycardia and left ventricular hypertrophy, suggested by prominent voltage (sum of S in V1 and R in V6 ≥ 35 mm) and supported by ST-segment and T-wave changes in the lateral and midprecordial leads. Classic changes of left ventricular hypertrophy often include increased voltage and downsloping ST-segment depression with negative T waves in V5 and V6 (secondary repolarization changes or “strain” pattern).
Notable on this tracing are the large, asymmetric negative T waves in leads V3 through V6. Giant T waves are defined as negative T waves with voltage greater than 10 mm.3 Although there is no specific pattern of ventricular hypertrophy on an ECG that establishes the diagnosis of hypertrophic cardiomyopathy, left ventricular hypertrophy with T waves of this quality suggest the possibility of hypertrophic cardiomyopathy with apical hypertrophy.
Q: What are the other causes of giant negative T waves?
- Subarachnoid hemorrhage
- Complete heart block
- Non-Q-wave myocardial infarction
- All of the above
A: The correct answer is all of the above. Additional causes of dramatic T-wave inversion are listed in Table 1. Clinically, non-Q-wave myocardial infarction with T-wave changes and acute central nervous system injury are probably the most commonly seen.4
Echocardiography in this patient revealed severe apical hypertrophy of the ventricle with distal cavity obliteration. The left ventricular outflow-tract gradient was normal. The mitral valve appeared normal, and there was no resting systolic anterior motion.
Cardiac magnetic resonance imaging showed the apical variant of hypertrophic cardiomyopathy but no evidence of left ventricular noncompaction, which is a differential diagnosis of apical hypertrophic obstructive cardiomyopathy. This disease was first described in Japan by Yamaguchi et al5 and Sakamoto et al6 and is regarded as a subgroup of nonobstructive hypertrophic cardiomyopathy. The prognosis of apical hypertrophic cardiomyopathy with regard to sudden cardiac death is believed to be better than that of other forms of hypertrophic cardiomyopathy.3
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. 1949. Ann Noninvasive Electrocardiol 2001; 6:343–368.
- Casale PN, Devereux RB, Alonso DR, Campo E, Kligfield P. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation 1987; 75:565–572.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Jacobson D, Schrire V. Giant T wave inversion. Br Heart J 1966; 28:768–775.
- Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol 1979; 44:401–412.
- Sakamoto T, Tei C, Murayama M, Ichiyasu H, Hada Y. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasonocardiotomographic study. Jpn Heart J 1976; 17:611–629.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. 1949. Ann Noninvasive Electrocardiol 2001; 6:343–368.
- Casale PN, Devereux RB, Alonso DR, Campo E, Kligfield P. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation 1987; 75:565–572.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Jacobson D, Schrire V. Giant T wave inversion. Br Heart J 1966; 28:768–775.
- Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol 1979; 44:401–412.
- Sakamoto T, Tei C, Murayama M, Ichiyasu H, Hada Y. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasonocardiotomographic study. Jpn Heart J 1976; 17:611–629.