User login
Miss the ear, and you may miss the diagnosis
A 52-year-old woman presented with pain in both ears associated with redness and swelling. The symptoms appeared 3 weeks earlier. The pain had started on one side, then spread to the other over a period of 2 weeks. She denied fever, chills, rigor, rash, or upper respiratory symptoms. She had experienced similar but unilateral ear pain months before. Her medical history included bilateral knee pain and swelling (treated as osteoarthritis), hypertension, hyperlipidemia, and hypothyroidism. She also reported progressive bilateral hearing loss, for which she now uses hearing aids. She had no history of conjunctivitis or uveitis.
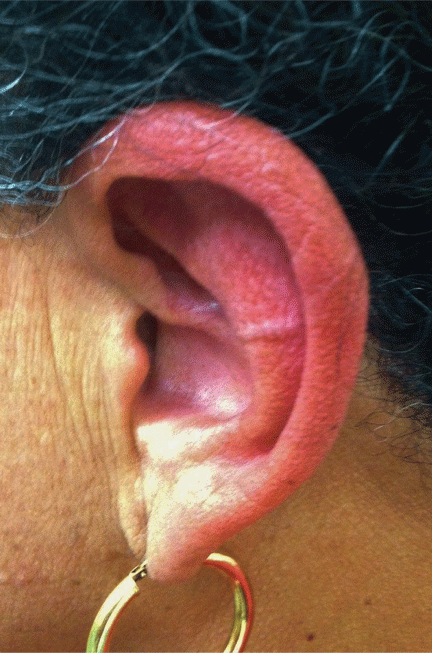
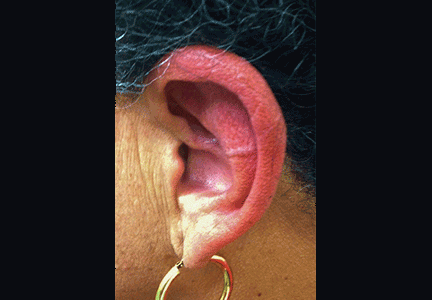
Physical examination showed swelling and erythema of both ears, sparing the earlobes (Figure 1), as well as bilateral knee-joint tenderness and restricted joint movement. The erythrocyte sedimentation rate was elevated at 52 mm/h (reference range 0–20); the complete blood cell count, creatinine, and liver enzyme levels were normal. An autoimmune panel was negative for antinuclear antibody, antineutrophil cytoplasmic antibody, and rheumatoid factor.
A clinical diagnosis of relapsing polychondritis was made based on the McAdam criteria.1 The patient was initially started on steroids and then was maintained on methotrexate. Her symptoms improved dramatically by 3 weeks.
RELAPSING POLYCHONDRITIS
Relapsing polychondritis is a rare, chronic, and potentially multisystem disorder characterized by recurrent episodes of cartilaginous inflammation that often lead to progressive destruction of the cartilage.2,3
Auricular chondritis is the initial presentation in 43% of cases and eventually develops in 89% of patients.2,4 The earlobes are spared, as they are devoid of cartilage, and this feature helps to differentiate the condition from an infection.
If the condition is not treated, recurrent attacks can result in irreversible cartilage damage and drooping of the pinna (ie, “cauliflower ear”). Biopsy is usually avoided, as it may further damage the ear. The diagnostic criteria for relapsing polychondritis formulated by McAdam et al1 accommodate the different presentations in order to limit the need for biopsy. Systemic involvement may include external eye structures, vasculitis affecting the eighth cranial (vestibulocochlear) nerve, noninflammatory large-joint arthritis, and the trachea. There is also an association with myelodysplasia.
- McAdam LP, O’Hanlan MA, Bluestone R, Pearson CM. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore) 1976; 55:193–215.
- Mathew SD, Battafarano DF, Morris MJ. Relapsing polychondritis in the Department of Defense population and review of the literature. Semin Arthritis Rheum 2012; 42:70–83.
- Letko E, Zafirakis P, Baltatzis S, Voudouri A, Livir-Rallatos C, Foster CS. Relapsing polychondritis: a clinical review. Semin Arthritis Rheum 2002; 31:384–395.
- Kent PD, Michet CJ, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol 2004; 16:56–61.
A 52-year-old woman presented with pain in both ears associated with redness and swelling. The symptoms appeared 3 weeks earlier. The pain had started on one side, then spread to the other over a period of 2 weeks. She denied fever, chills, rigor, rash, or upper respiratory symptoms. She had experienced similar but unilateral ear pain months before. Her medical history included bilateral knee pain and swelling (treated as osteoarthritis), hypertension, hyperlipidemia, and hypothyroidism. She also reported progressive bilateral hearing loss, for which she now uses hearing aids. She had no history of conjunctivitis or uveitis.
Physical examination showed swelling and erythema of both ears, sparing the earlobes (Figure 1), as well as bilateral knee-joint tenderness and restricted joint movement. The erythrocyte sedimentation rate was elevated at 52 mm/h (reference range 0–20); the complete blood cell count, creatinine, and liver enzyme levels were normal. An autoimmune panel was negative for antinuclear antibody, antineutrophil cytoplasmic antibody, and rheumatoid factor.
A clinical diagnosis of relapsing polychondritis was made based on the McAdam criteria.1 The patient was initially started on steroids and then was maintained on methotrexate. Her symptoms improved dramatically by 3 weeks.
RELAPSING POLYCHONDRITIS
Relapsing polychondritis is a rare, chronic, and potentially multisystem disorder characterized by recurrent episodes of cartilaginous inflammation that often lead to progressive destruction of the cartilage.2,3
Auricular chondritis is the initial presentation in 43% of cases and eventually develops in 89% of patients.2,4 The earlobes are spared, as they are devoid of cartilage, and this feature helps to differentiate the condition from an infection.
If the condition is not treated, recurrent attacks can result in irreversible cartilage damage and drooping of the pinna (ie, “cauliflower ear”). Biopsy is usually avoided, as it may further damage the ear. The diagnostic criteria for relapsing polychondritis formulated by McAdam et al1 accommodate the different presentations in order to limit the need for biopsy. Systemic involvement may include external eye structures, vasculitis affecting the eighth cranial (vestibulocochlear) nerve, noninflammatory large-joint arthritis, and the trachea. There is also an association with myelodysplasia.
A 52-year-old woman presented with pain in both ears associated with redness and swelling. The symptoms appeared 3 weeks earlier. The pain had started on one side, then spread to the other over a period of 2 weeks. She denied fever, chills, rigor, rash, or upper respiratory symptoms. She had experienced similar but unilateral ear pain months before. Her medical history included bilateral knee pain and swelling (treated as osteoarthritis), hypertension, hyperlipidemia, and hypothyroidism. She also reported progressive bilateral hearing loss, for which she now uses hearing aids. She had no history of conjunctivitis or uveitis.
Physical examination showed swelling and erythema of both ears, sparing the earlobes (Figure 1), as well as bilateral knee-joint tenderness and restricted joint movement. The erythrocyte sedimentation rate was elevated at 52 mm/h (reference range 0–20); the complete blood cell count, creatinine, and liver enzyme levels were normal. An autoimmune panel was negative for antinuclear antibody, antineutrophil cytoplasmic antibody, and rheumatoid factor.
A clinical diagnosis of relapsing polychondritis was made based on the McAdam criteria.1 The patient was initially started on steroids and then was maintained on methotrexate. Her symptoms improved dramatically by 3 weeks.
RELAPSING POLYCHONDRITIS
Relapsing polychondritis is a rare, chronic, and potentially multisystem disorder characterized by recurrent episodes of cartilaginous inflammation that often lead to progressive destruction of the cartilage.2,3
Auricular chondritis is the initial presentation in 43% of cases and eventually develops in 89% of patients.2,4 The earlobes are spared, as they are devoid of cartilage, and this feature helps to differentiate the condition from an infection.
If the condition is not treated, recurrent attacks can result in irreversible cartilage damage and drooping of the pinna (ie, “cauliflower ear”). Biopsy is usually avoided, as it may further damage the ear. The diagnostic criteria for relapsing polychondritis formulated by McAdam et al1 accommodate the different presentations in order to limit the need for biopsy. Systemic involvement may include external eye structures, vasculitis affecting the eighth cranial (vestibulocochlear) nerve, noninflammatory large-joint arthritis, and the trachea. There is also an association with myelodysplasia.
- McAdam LP, O’Hanlan MA, Bluestone R, Pearson CM. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore) 1976; 55:193–215.
- Mathew SD, Battafarano DF, Morris MJ. Relapsing polychondritis in the Department of Defense population and review of the literature. Semin Arthritis Rheum 2012; 42:70–83.
- Letko E, Zafirakis P, Baltatzis S, Voudouri A, Livir-Rallatos C, Foster CS. Relapsing polychondritis: a clinical review. Semin Arthritis Rheum 2002; 31:384–395.
- Kent PD, Michet CJ, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol 2004; 16:56–61.
- McAdam LP, O’Hanlan MA, Bluestone R, Pearson CM. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore) 1976; 55:193–215.
- Mathew SD, Battafarano DF, Morris MJ. Relapsing polychondritis in the Department of Defense population and review of the literature. Semin Arthritis Rheum 2012; 42:70–83.
- Letko E, Zafirakis P, Baltatzis S, Voudouri A, Livir-Rallatos C, Foster CS. Relapsing polychondritis: a clinical review. Semin Arthritis Rheum 2002; 31:384–395.
- Kent PD, Michet CJ, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol 2004; 16:56–61.
Terry nails in a patient with chronic alcoholic liver disease
A 45-year-old man with chronic alcoholism for the past 20 years and chronic liver disease for the past 2 years was admitted to the hospital with abdominal distention, yellowish discoloration of the eyes, itching all over the body, decreased appetite, and fresh rectal bleeding.
He had the classic signs of chronic liver disease, including icterus, pallor, parotid swelling, gynecomastia, spider angiomata, sparse axillary and pubic hair, transverse stretched umbilicus, divarication of the rectus abdominis muscles, caput medusae, small testes, and bilateral pedal edema.

Examination of the fingernails revealed a distal thin pink-to-brown transverse band 0.5 to 2.0 mm in width, a white nail bed, and no lunula (Figure 1)—the characteristic findings of Terry nails.
Systemic examination revealed moderate ascites (shifting dullness present), splenomegaly, and external hemorrhoids suggestive of portal hypertension.
TERRY NAILS
In 1954, Dr. Richard Terry first reported the finding of a white nail bed with ground-glass opacity in patients who had hepatic cirrhosis.1 The condition is bilaterally symmetrical, with a tendency to be more marked in the thumb and forefinger.1
In 1984, Holzberg and Walker2 consecutively studied 512 hospitalized patients and observed Terry nails in 25.2% of them. Based on their findings, they redefined the criteria for Terry nail as follows:
- Distal thin pink-to-brown transverse band, 0.5 to 3.0 mm in width
- Decreased venous return not obscuring the distal band
- White or light pink proximal nail
- Lunula possibly absent
- At least 4 of 10 nails with the above criteria.
Patients who do not have the findings on all fingernails commonly have involvement of the thumb and forefinger. Holzberg and Walker confirmed a statistically significant association of Terry nails with cirrhosis, chronic heart failure, and adult-onset diabetes, especially in younger patients.2 Terry nails have also been observed in thyrotoxicosis, pulmonary eosinophilia, malnutrition, actinic keratosis, and advanced age.1–4
Using the updated diagnostic criteria, Park et al5 studied fingernails in 444 medical inpatients with chronic systemic disease, and only 30.6% had Terry nails. There were statistically significant associations with cirrhosis (57%), congestive heart failure (51.5%), and diabetes mellitus (49%); the associations with chronic renal failure (19%) and cancer (18%) were not statistically significant.5 They were more common in older patients. The average number of nails affected per patient tended to be higher in frequency close to the thumb; 28.7% patients had all nails affected.5
Terry nails should alert the clinician to the possibility of an underlying systemic disease, especially advanced liver disease. Possible explanations for the clinical changes observed in Terry nails include abnormal steroid metabolism, abnormal estrogen-androgen ratio, alteration of nail bed-to-nail plate attachment, hypoalbuminemia, increased digital blood flow, and overgrowth of the connective tissue between nail bed and the growth plate. The pathologic study of longitudinal nail biopsy specimens shows telangiectasia in the upper dermis of the distal nail band.3
Important differential diagnoses are Lindsay (half-and-half) nails, associated with chronic renal failure, and Neapolitan nails, associated with aging.3
- Terry R. White nails in hepatic cirrhosis. Lancet 1954; 266:757–759.
- Holzberg M, Walker HK. Terry’s nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Holzberg M. The nail in systemic disease. In:Baran R, de Berker DAR, Holzberg M, Thomas L, editors. Baran & Dawber’s Diseases of the Nails and Their Management, 4th ed. Oxford, UK: Blackwell Publishing Ltd.; 2012.
- Nia AM, Ederer S, Dahlem KM, Gassanov N, Er F. Terry’s nails: a window to systemic diseases. Am J Med 2011; 124:602–604.
- Park KY, Kim SW, Cho JS. Research on the frequency of Terry’s nail in the medical inpatients with chronic illnesses. Korean J Dermatol 1992; 30:864–870.
A 45-year-old man with chronic alcoholism for the past 20 years and chronic liver disease for the past 2 years was admitted to the hospital with abdominal distention, yellowish discoloration of the eyes, itching all over the body, decreased appetite, and fresh rectal bleeding.
He had the classic signs of chronic liver disease, including icterus, pallor, parotid swelling, gynecomastia, spider angiomata, sparse axillary and pubic hair, transverse stretched umbilicus, divarication of the rectus abdominis muscles, caput medusae, small testes, and bilateral pedal edema.
Examination of the fingernails revealed a distal thin pink-to-brown transverse band 0.5 to 2.0 mm in width, a white nail bed, and no lunula (Figure 1)—the characteristic findings of Terry nails.
Systemic examination revealed moderate ascites (shifting dullness present), splenomegaly, and external hemorrhoids suggestive of portal hypertension.
TERRY NAILS
In 1954, Dr. Richard Terry first reported the finding of a white nail bed with ground-glass opacity in patients who had hepatic cirrhosis.1 The condition is bilaterally symmetrical, with a tendency to be more marked in the thumb and forefinger.1
In 1984, Holzberg and Walker2 consecutively studied 512 hospitalized patients and observed Terry nails in 25.2% of them. Based on their findings, they redefined the criteria for Terry nail as follows:
- Distal thin pink-to-brown transverse band, 0.5 to 3.0 mm in width
- Decreased venous return not obscuring the distal band
- White or light pink proximal nail
- Lunula possibly absent
- At least 4 of 10 nails with the above criteria.
Patients who do not have the findings on all fingernails commonly have involvement of the thumb and forefinger. Holzberg and Walker confirmed a statistically significant association of Terry nails with cirrhosis, chronic heart failure, and adult-onset diabetes, especially in younger patients.2 Terry nails have also been observed in thyrotoxicosis, pulmonary eosinophilia, malnutrition, actinic keratosis, and advanced age.1–4
Using the updated diagnostic criteria, Park et al5 studied fingernails in 444 medical inpatients with chronic systemic disease, and only 30.6% had Terry nails. There were statistically significant associations with cirrhosis (57%), congestive heart failure (51.5%), and diabetes mellitus (49%); the associations with chronic renal failure (19%) and cancer (18%) were not statistically significant.5 They were more common in older patients. The average number of nails affected per patient tended to be higher in frequency close to the thumb; 28.7% patients had all nails affected.5
Terry nails should alert the clinician to the possibility of an underlying systemic disease, especially advanced liver disease. Possible explanations for the clinical changes observed in Terry nails include abnormal steroid metabolism, abnormal estrogen-androgen ratio, alteration of nail bed-to-nail plate attachment, hypoalbuminemia, increased digital blood flow, and overgrowth of the connective tissue between nail bed and the growth plate. The pathologic study of longitudinal nail biopsy specimens shows telangiectasia in the upper dermis of the distal nail band.3
Important differential diagnoses are Lindsay (half-and-half) nails, associated with chronic renal failure, and Neapolitan nails, associated with aging.3
A 45-year-old man with chronic alcoholism for the past 20 years and chronic liver disease for the past 2 years was admitted to the hospital with abdominal distention, yellowish discoloration of the eyes, itching all over the body, decreased appetite, and fresh rectal bleeding.
He had the classic signs of chronic liver disease, including icterus, pallor, parotid swelling, gynecomastia, spider angiomata, sparse axillary and pubic hair, transverse stretched umbilicus, divarication of the rectus abdominis muscles, caput medusae, small testes, and bilateral pedal edema.
Examination of the fingernails revealed a distal thin pink-to-brown transverse band 0.5 to 2.0 mm in width, a white nail bed, and no lunula (Figure 1)—the characteristic findings of Terry nails.
Systemic examination revealed moderate ascites (shifting dullness present), splenomegaly, and external hemorrhoids suggestive of portal hypertension.
TERRY NAILS
In 1954, Dr. Richard Terry first reported the finding of a white nail bed with ground-glass opacity in patients who had hepatic cirrhosis.1 The condition is bilaterally symmetrical, with a tendency to be more marked in the thumb and forefinger.1
In 1984, Holzberg and Walker2 consecutively studied 512 hospitalized patients and observed Terry nails in 25.2% of them. Based on their findings, they redefined the criteria for Terry nail as follows:
- Distal thin pink-to-brown transverse band, 0.5 to 3.0 mm in width
- Decreased venous return not obscuring the distal band
- White or light pink proximal nail
- Lunula possibly absent
- At least 4 of 10 nails with the above criteria.
Patients who do not have the findings on all fingernails commonly have involvement of the thumb and forefinger. Holzberg and Walker confirmed a statistically significant association of Terry nails with cirrhosis, chronic heart failure, and adult-onset diabetes, especially in younger patients.2 Terry nails have also been observed in thyrotoxicosis, pulmonary eosinophilia, malnutrition, actinic keratosis, and advanced age.1–4
Using the updated diagnostic criteria, Park et al5 studied fingernails in 444 medical inpatients with chronic systemic disease, and only 30.6% had Terry nails. There were statistically significant associations with cirrhosis (57%), congestive heart failure (51.5%), and diabetes mellitus (49%); the associations with chronic renal failure (19%) and cancer (18%) were not statistically significant.5 They were more common in older patients. The average number of nails affected per patient tended to be higher in frequency close to the thumb; 28.7% patients had all nails affected.5
Terry nails should alert the clinician to the possibility of an underlying systemic disease, especially advanced liver disease. Possible explanations for the clinical changes observed in Terry nails include abnormal steroid metabolism, abnormal estrogen-androgen ratio, alteration of nail bed-to-nail plate attachment, hypoalbuminemia, increased digital blood flow, and overgrowth of the connective tissue between nail bed and the growth plate. The pathologic study of longitudinal nail biopsy specimens shows telangiectasia in the upper dermis of the distal nail band.3
Important differential diagnoses are Lindsay (half-and-half) nails, associated with chronic renal failure, and Neapolitan nails, associated with aging.3
- Terry R. White nails in hepatic cirrhosis. Lancet 1954; 266:757–759.
- Holzberg M, Walker HK. Terry’s nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Holzberg M. The nail in systemic disease. In:Baran R, de Berker DAR, Holzberg M, Thomas L, editors. Baran & Dawber’s Diseases of the Nails and Their Management, 4th ed. Oxford, UK: Blackwell Publishing Ltd.; 2012.
- Nia AM, Ederer S, Dahlem KM, Gassanov N, Er F. Terry’s nails: a window to systemic diseases. Am J Med 2011; 124:602–604.
- Park KY, Kim SW, Cho JS. Research on the frequency of Terry’s nail in the medical inpatients with chronic illnesses. Korean J Dermatol 1992; 30:864–870.
- Terry R. White nails in hepatic cirrhosis. Lancet 1954; 266:757–759.
- Holzberg M, Walker HK. Terry’s nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Holzberg M. The nail in systemic disease. In:Baran R, de Berker DAR, Holzberg M, Thomas L, editors. Baran & Dawber’s Diseases of the Nails and Their Management, 4th ed. Oxford, UK: Blackwell Publishing Ltd.; 2012.
- Nia AM, Ederer S, Dahlem KM, Gassanov N, Er F. Terry’s nails: a window to systemic diseases. Am J Med 2011; 124:602–604.
- Park KY, Kim SW, Cho JS. Research on the frequency of Terry’s nail in the medical inpatients with chronic illnesses. Korean J Dermatol 1992; 30:864–870.

Erythema and atrophy on the tongue
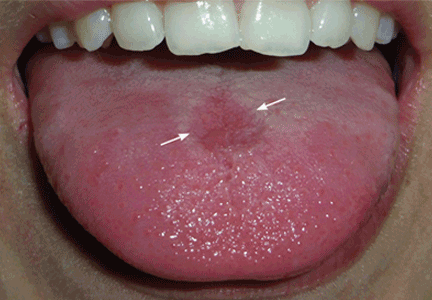
A 26-year-old woman was referred to the dermatology department with a 6-month history of a painful burning sensation on the tongue. Examination revealed a reddish, atrophic area on the dorsum of the tongue (Figure 1).
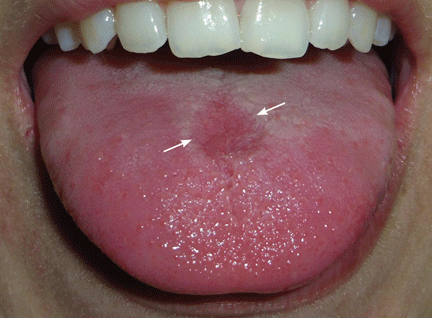
She had been treated unsuccessfully with topical antifungal drugs (clotrimazole and nystatin) for a presumed diagnosis of oral candidiasis. Otherwise, her medical history was notable only for occasional episodes of epigastric pain. She did not smoke or drink alcohol.
Fungal culture and oral exfoliative cytology studies were negative.
Laboratory results:
- Red blood cell count 3.9 × 1012/L (reference range 4.2–5.4)
- Hemoglobin 11.3 g/dL (12–16)
- Mean corpuscular volume 92 fL (80–99)
- Mean corpuscular hemoglobin 29 pg (27–34)
- Iron 14 μg/dL (37–145),
- Vitamin B12 119 pg/dL (250–900)
- Zinc 33 μg/dL (66–110)
- Serum gastric parietal cell antibody positive
- Serum creatinine and liver enzyme tests were normal.
Biopsy of the gastric mucosa revealed severe atrophic gastritis, so the possibility of atrophy related to gastroesophageal reflux was considered. But the laboratory results and the patient’s presentation pointed to iron deficiency and pernicious anemia (due to deficiency of vitamin B12). Zinc deficiency is associated with oral burning but not atrophic glossitis.
Based on the patient’s symptoms and the testing results, she was given the diagnosis of atrophic glossitis. She was treated with oral iron supplementation, intramuscular injections of vitamin B12, and oral zinc supplementation. The glossitis resolved, and the gastric symptoms improved within 2 months, thus supporting our diagnosis of atrophic glossitis.
ATROPHIC GLOSSITIS
The diagnosis of abnormalities of the tongue requires a thorough history, including onset and duration, antecedent symptoms, and tobacco and alcohol use. Examination of tongue morphology is also important.1 Tongue abnormalities related to tobacco use and to alcohol use include leukoplakia, erythroplakia, oral submucosal fibrosis, lichen planus, and oral squamous cell carcinoma.
Atrophic glossitis is often linked to an underlying nutritional deficiency of iron, folic acid, vitamin B12, riboflavin, or niacin, although other nutritional deficiencies can be implicated. As noted, zinc deficiency can cause oral burning but not atrophic glossitis, and it resolves with correction of the underlying deficiency.2 Cobalamin deficiency is the main cause of atrophic glossitis.
As our patient’s presentation illustrated, oral symptoms can be multifactorial. Oral conditions may be an early clinical manifestation of a nutritional deficiency, but they can also reflect an alteration of the gastric mucosa3; a bacterial, viral, or fungal infection; neoplastic disease; autoimmune disease; endocrine disorder; local mechanical trauma; exposure to an irritant; or an allergic reaction.2
- Reamy BV, Derby R, Bunt CW. Common tongue conditions in primary care. Am Fam Physician 2010; 81:627–634.
- Chi AC, Neville BW, Krayer JW, Gonsalves WC. Oral manifestations of systemic disease. Am Fam Physician 2010; 82:1381–1388.
- Sun A, Lin HP, Wang YP, Chiang CP. Significant association of deficiency of hemoglobin, iron and vitamin B12, high homocysteine level, and gastric parietal cell antibody positivity with atrophic glossitis. J Oral Pathol Med 2012; 41:500–504.
A 26-year-old woman was referred to the dermatology department with a 6-month history of a painful burning sensation on the tongue. Examination revealed a reddish, atrophic area on the dorsum of the tongue (Figure 1).
She had been treated unsuccessfully with topical antifungal drugs (clotrimazole and nystatin) for a presumed diagnosis of oral candidiasis. Otherwise, her medical history was notable only for occasional episodes of epigastric pain. She did not smoke or drink alcohol.
Fungal culture and oral exfoliative cytology studies were negative.
Laboratory results:
- Red blood cell count 3.9 × 1012/L (reference range 4.2–5.4)
- Hemoglobin 11.3 g/dL (12–16)
- Mean corpuscular volume 92 fL (80–99)
- Mean corpuscular hemoglobin 29 pg (27–34)
- Iron 14 μg/dL (37–145),
- Vitamin B12 119 pg/dL (250–900)
- Zinc 33 μg/dL (66–110)
- Serum gastric parietal cell antibody positive
- Serum creatinine and liver enzyme tests were normal.
Biopsy of the gastric mucosa revealed severe atrophic gastritis, so the possibility of atrophy related to gastroesophageal reflux was considered. But the laboratory results and the patient’s presentation pointed to iron deficiency and pernicious anemia (due to deficiency of vitamin B12). Zinc deficiency is associated with oral burning but not atrophic glossitis.
Based on the patient’s symptoms and the testing results, she was given the diagnosis of atrophic glossitis. She was treated with oral iron supplementation, intramuscular injections of vitamin B12, and oral zinc supplementation. The glossitis resolved, and the gastric symptoms improved within 2 months, thus supporting our diagnosis of atrophic glossitis.
ATROPHIC GLOSSITIS
The diagnosis of abnormalities of the tongue requires a thorough history, including onset and duration, antecedent symptoms, and tobacco and alcohol use. Examination of tongue morphology is also important.1 Tongue abnormalities related to tobacco use and to alcohol use include leukoplakia, erythroplakia, oral submucosal fibrosis, lichen planus, and oral squamous cell carcinoma.
Atrophic glossitis is often linked to an underlying nutritional deficiency of iron, folic acid, vitamin B12, riboflavin, or niacin, although other nutritional deficiencies can be implicated. As noted, zinc deficiency can cause oral burning but not atrophic glossitis, and it resolves with correction of the underlying deficiency.2 Cobalamin deficiency is the main cause of atrophic glossitis.
As our patient’s presentation illustrated, oral symptoms can be multifactorial. Oral conditions may be an early clinical manifestation of a nutritional deficiency, but they can also reflect an alteration of the gastric mucosa3; a bacterial, viral, or fungal infection; neoplastic disease; autoimmune disease; endocrine disorder; local mechanical trauma; exposure to an irritant; or an allergic reaction.2
A 26-year-old woman was referred to the dermatology department with a 6-month history of a painful burning sensation on the tongue. Examination revealed a reddish, atrophic area on the dorsum of the tongue (Figure 1).
She had been treated unsuccessfully with topical antifungal drugs (clotrimazole and nystatin) for a presumed diagnosis of oral candidiasis. Otherwise, her medical history was notable only for occasional episodes of epigastric pain. She did not smoke or drink alcohol.
Fungal culture and oral exfoliative cytology studies were negative.
Laboratory results:
- Red blood cell count 3.9 × 1012/L (reference range 4.2–5.4)
- Hemoglobin 11.3 g/dL (12–16)
- Mean corpuscular volume 92 fL (80–99)
- Mean corpuscular hemoglobin 29 pg (27–34)
- Iron 14 μg/dL (37–145),
- Vitamin B12 119 pg/dL (250–900)
- Zinc 33 μg/dL (66–110)
- Serum gastric parietal cell antibody positive
- Serum creatinine and liver enzyme tests were normal.
Biopsy of the gastric mucosa revealed severe atrophic gastritis, so the possibility of atrophy related to gastroesophageal reflux was considered. But the laboratory results and the patient’s presentation pointed to iron deficiency and pernicious anemia (due to deficiency of vitamin B12). Zinc deficiency is associated with oral burning but not atrophic glossitis.
Based on the patient’s symptoms and the testing results, she was given the diagnosis of atrophic glossitis. She was treated with oral iron supplementation, intramuscular injections of vitamin B12, and oral zinc supplementation. The glossitis resolved, and the gastric symptoms improved within 2 months, thus supporting our diagnosis of atrophic glossitis.
ATROPHIC GLOSSITIS
The diagnosis of abnormalities of the tongue requires a thorough history, including onset and duration, antecedent symptoms, and tobacco and alcohol use. Examination of tongue morphology is also important.1 Tongue abnormalities related to tobacco use and to alcohol use include leukoplakia, erythroplakia, oral submucosal fibrosis, lichen planus, and oral squamous cell carcinoma.
Atrophic glossitis is often linked to an underlying nutritional deficiency of iron, folic acid, vitamin B12, riboflavin, or niacin, although other nutritional deficiencies can be implicated. As noted, zinc deficiency can cause oral burning but not atrophic glossitis, and it resolves with correction of the underlying deficiency.2 Cobalamin deficiency is the main cause of atrophic glossitis.
As our patient’s presentation illustrated, oral symptoms can be multifactorial. Oral conditions may be an early clinical manifestation of a nutritional deficiency, but they can also reflect an alteration of the gastric mucosa3; a bacterial, viral, or fungal infection; neoplastic disease; autoimmune disease; endocrine disorder; local mechanical trauma; exposure to an irritant; or an allergic reaction.2
- Reamy BV, Derby R, Bunt CW. Common tongue conditions in primary care. Am Fam Physician 2010; 81:627–634.
- Chi AC, Neville BW, Krayer JW, Gonsalves WC. Oral manifestations of systemic disease. Am Fam Physician 2010; 82:1381–1388.
- Sun A, Lin HP, Wang YP, Chiang CP. Significant association of deficiency of hemoglobin, iron and vitamin B12, high homocysteine level, and gastric parietal cell antibody positivity with atrophic glossitis. J Oral Pathol Med 2012; 41:500–504.
- Reamy BV, Derby R, Bunt CW. Common tongue conditions in primary care. Am Fam Physician 2010; 81:627–634.
- Chi AC, Neville BW, Krayer JW, Gonsalves WC. Oral manifestations of systemic disease. Am Fam Physician 2010; 82:1381–1388.
- Sun A, Lin HP, Wang YP, Chiang CP. Significant association of deficiency of hemoglobin, iron and vitamin B12, high homocysteine level, and gastric parietal cell antibody positivity with atrophic glossitis. J Oral Pathol Med 2012; 41:500–504.
'Allergic to the sun'
A 54-year-old white man presents to the emergency department with burning pain in his left upper arm for the past 2 to 3 days. His medical history includes seizure disorder, for which he takes levetiracetam (Keppra); hypertension, for which he takes metoprolol succinate (Toprol); and in the remote past, a gunshot wound to the head that left his right arm with residual contracture and weakness.
He says he is homeless, has been “allergic to the sun for a while,” and has had dark-colored urine and intermittent abdominal pain. He states that he does not use illicit substances but that he drinks 6 to 12 beers per night and smokes 1 pack of cigarettes per day.
Initial vital signs:
- Temperature 37.7°C (99.9°F)
- Blood pressure 217/114 mm Hg
- Heart rate 82 bpm
- Respiratory rate 18 per minute
- Capillary oxygen saturation 98% while breathing room air.
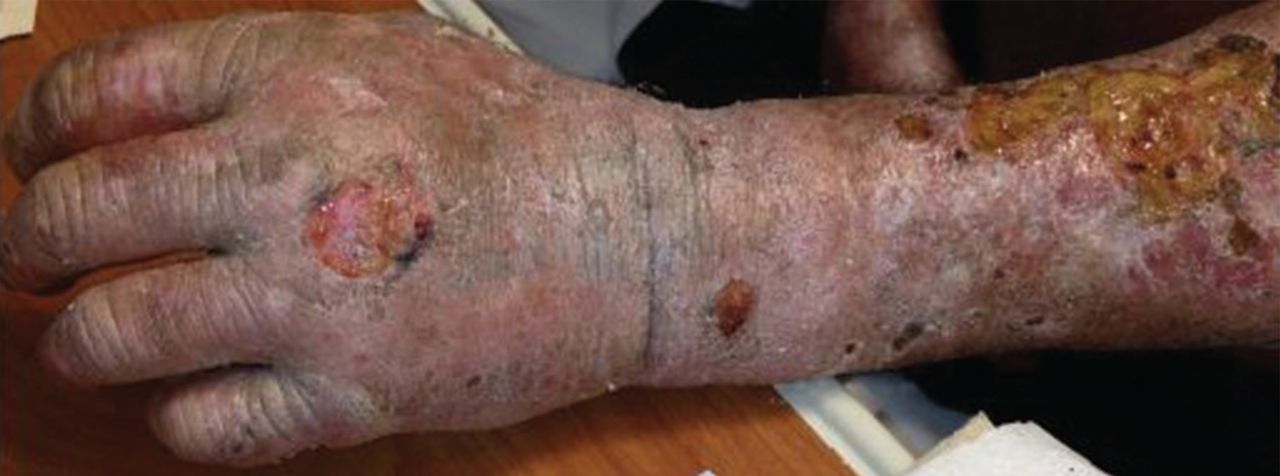
On examination, his right arm is significantly weak and contracted. His left arm has decreased sensation to pinprick and light touch from elbow to fingers. His face and both arms show hyperpigmentation alternating with atrophic scarring, which also affects his lips. There is no overt mucosal involvement. His hands and forearms have a sclerotic texture and patchy hair loss. Several small bullae are present on the dorsum of the left forearm and hand. There is a 6-inch, irregular, open lesion on the left forearm and a 1-inch lesion on the left hand (Figure 1).
Initial laboratory studies show:
- Chemistries and complete blood cell count within normal limits
- Platelet count 305 × 109/L (reference range 150–350)
- Orange-colored urine
- Hepatitis C virus (HCV) antibody positive (new finding)
- Human immunodeficiency virus antibody, hepatitis B surface antigen, and antinuclear antibody negative
- Phenytoin and urine drug screen negative
- Aspartate aminotransferase 70 U/L (reference range 5–34)
- Alanine aminotransferase 73 U/L (reference range 0–55)
- Prothrombin time 10.8 seconds (reference range 8.3–13.0), international normalized ratio 0.98 (reference range 0.8–1.2)
- Iron studies within normal limits.
The patient is admitted to the hospital and is started on cefazolin and clindamycin. Urine is collected for a porphyrin screen, and punch-biopsy samples from the forearms are sent for study. Ultrasonography shows splenomegaly, as well as increased echogenicity of the liver without structural abnormalities. Blood and urine cultures, drawn upon admission, are negative by discharge.
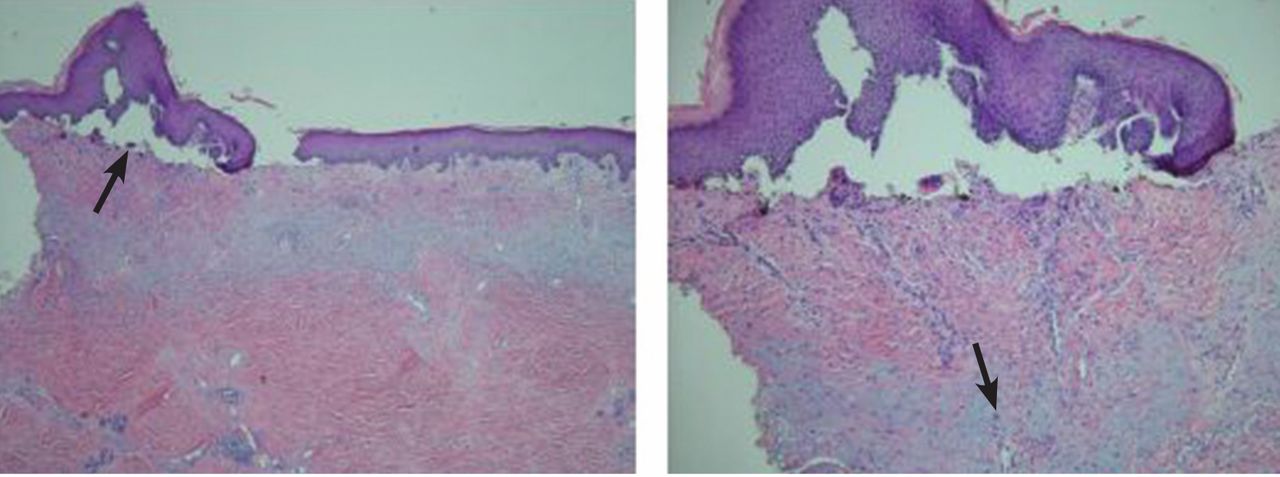
Pathologic study of the punch-biopsy specimens (Figure 2) shows the formation of subepidermal vesicles with extensive reticular and dermal fibrosis.
DIAGNOSIS: PORPHYRIA CUTANEA TARDA
Because of the patient’s history, examination, and pathology results, he was preliminarily diagnosed with porphyria cutanea tarda (PCT).1,2 The diagnosis was confirmed after he was discharged when his urine uroporphyrin level was found to be 157.5 μmol/mol of creatinine (reference range < 4) and his urine heptacarboxylporphyrin level was 118.0 μmol/mol of creatinine (reference range < 2).
This patient’s clinical presentation is classic for sporadic (ie, type 1) PCT. Sporadic PCT is an acquired deficiency of uroporphyrinogen decarboxylase, an enzyme that catalyzes the fifth step in heme metabolism.3 The deficiency of this enzyme is exclusively hepatic and is strongly associated with chronic hepatitis C infection. Mutations of the hemochromatosis gene (HFE), human immunodeficiency virus infection, alcohol use, and smoking are also risk factors.4 The prevalence in the United States is about 1:25,000; nearly 80% of cases are sporadic (type 1), and 20% are familial (type 2).5
Manifestations of PCT include photosensitive dermatitis, facial hypertrichosis, and orange urine.3 The photosensitivity dermatitis heals slowly and leads to sclerosis and hyperpigmentation.
Repeated phlebotomy is the first-line treatment, and hydroxychloroquine (Plaquenil) is the second-line treatment.6 Patients with PCT and hepatitis C should be considered for antiviral therapy according to standard guidelines. Treatment of hepatitis C may reduce the symptoms of PCT, even without a sustained viral response. However, not enough evidence exists to make treatment recommendations for this group.7
Because we were uncertain that the patient would return for follow-up, we did not start phlebotomy or treatment for hepatitis C. However, we did prescribe hydroxychloroquine 100 mg three times a week and instructed him to cover his skin when outside and to use effective sunblock. An outpatient visit was scheduled prior to discharge. Unfortunately, the patient was lost to follow-up.
Acknowledgment: The authors would like to personally thank Dr. Karen DeSouza from the University of Tennessee, Graduate School of Medicine, Department of Pathology, for her clinical expertise and kind advice.
- The University of Iowa, Department of Pathology, Laboratory Services Handbook. Porphyrins & Porphobilinogen, Urine (24 hr or random). www.healthcare.uiowa.edu/path_handbook/handbook/test2893.html. Accessed August 8, 2014.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol 1992; 19:40–47.
- Thunell S, Harper P. Porphyrins, porphyrin metabolism, porphyrias. III. Diagnosis, care and monitoring in porphyria cutanea tarda—suggestions for a handling programme. Scand J Clin Lab Invest 2000; 60:561–579.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- Kushner JP, Barbuto AJ, Lee GR. An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity. J Clin Invest 1976; 58:1089–1097.
- Singal AK, Kormos-Hallberg C, Lee C, et al. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin Gastroenterol Hepatol 2012; 10:1402–1409.
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 2012; 32:880–893.
A 54-year-old white man presents to the emergency department with burning pain in his left upper arm for the past 2 to 3 days. His medical history includes seizure disorder, for which he takes levetiracetam (Keppra); hypertension, for which he takes metoprolol succinate (Toprol); and in the remote past, a gunshot wound to the head that left his right arm with residual contracture and weakness.
He says he is homeless, has been “allergic to the sun for a while,” and has had dark-colored urine and intermittent abdominal pain. He states that he does not use illicit substances but that he drinks 6 to 12 beers per night and smokes 1 pack of cigarettes per day.
Initial vital signs:
- Temperature 37.7°C (99.9°F)
- Blood pressure 217/114 mm Hg
- Heart rate 82 bpm
- Respiratory rate 18 per minute
- Capillary oxygen saturation 98% while breathing room air.
On examination, his right arm is significantly weak and contracted. His left arm has decreased sensation to pinprick and light touch from elbow to fingers. His face and both arms show hyperpigmentation alternating with atrophic scarring, which also affects his lips. There is no overt mucosal involvement. His hands and forearms have a sclerotic texture and patchy hair loss. Several small bullae are present on the dorsum of the left forearm and hand. There is a 6-inch, irregular, open lesion on the left forearm and a 1-inch lesion on the left hand (Figure 1).
Initial laboratory studies show:
- Chemistries and complete blood cell count within normal limits
- Platelet count 305 × 109/L (reference range 150–350)
- Orange-colored urine
- Hepatitis C virus (HCV) antibody positive (new finding)
- Human immunodeficiency virus antibody, hepatitis B surface antigen, and antinuclear antibody negative
- Phenytoin and urine drug screen negative
- Aspartate aminotransferase 70 U/L (reference range 5–34)
- Alanine aminotransferase 73 U/L (reference range 0–55)
- Prothrombin time 10.8 seconds (reference range 8.3–13.0), international normalized ratio 0.98 (reference range 0.8–1.2)
- Iron studies within normal limits.
The patient is admitted to the hospital and is started on cefazolin and clindamycin. Urine is collected for a porphyrin screen, and punch-biopsy samples from the forearms are sent for study. Ultrasonography shows splenomegaly, as well as increased echogenicity of the liver without structural abnormalities. Blood and urine cultures, drawn upon admission, are negative by discharge.
Pathologic study of the punch-biopsy specimens (Figure 2) shows the formation of subepidermal vesicles with extensive reticular and dermal fibrosis.
DIAGNOSIS: PORPHYRIA CUTANEA TARDA
Because of the patient’s history, examination, and pathology results, he was preliminarily diagnosed with porphyria cutanea tarda (PCT).1,2 The diagnosis was confirmed after he was discharged when his urine uroporphyrin level was found to be 157.5 μmol/mol of creatinine (reference range < 4) and his urine heptacarboxylporphyrin level was 118.0 μmol/mol of creatinine (reference range < 2).
This patient’s clinical presentation is classic for sporadic (ie, type 1) PCT. Sporadic PCT is an acquired deficiency of uroporphyrinogen decarboxylase, an enzyme that catalyzes the fifth step in heme metabolism.3 The deficiency of this enzyme is exclusively hepatic and is strongly associated with chronic hepatitis C infection. Mutations of the hemochromatosis gene (HFE), human immunodeficiency virus infection, alcohol use, and smoking are also risk factors.4 The prevalence in the United States is about 1:25,000; nearly 80% of cases are sporadic (type 1), and 20% are familial (type 2).5
Manifestations of PCT include photosensitive dermatitis, facial hypertrichosis, and orange urine.3 The photosensitivity dermatitis heals slowly and leads to sclerosis and hyperpigmentation.
Repeated phlebotomy is the first-line treatment, and hydroxychloroquine (Plaquenil) is the second-line treatment.6 Patients with PCT and hepatitis C should be considered for antiviral therapy according to standard guidelines. Treatment of hepatitis C may reduce the symptoms of PCT, even without a sustained viral response. However, not enough evidence exists to make treatment recommendations for this group.7
Because we were uncertain that the patient would return for follow-up, we did not start phlebotomy or treatment for hepatitis C. However, we did prescribe hydroxychloroquine 100 mg three times a week and instructed him to cover his skin when outside and to use effective sunblock. An outpatient visit was scheduled prior to discharge. Unfortunately, the patient was lost to follow-up.
Acknowledgment: The authors would like to personally thank Dr. Karen DeSouza from the University of Tennessee, Graduate School of Medicine, Department of Pathology, for her clinical expertise and kind advice.
A 54-year-old white man presents to the emergency department with burning pain in his left upper arm for the past 2 to 3 days. His medical history includes seizure disorder, for which he takes levetiracetam (Keppra); hypertension, for which he takes metoprolol succinate (Toprol); and in the remote past, a gunshot wound to the head that left his right arm with residual contracture and weakness.
He says he is homeless, has been “allergic to the sun for a while,” and has had dark-colored urine and intermittent abdominal pain. He states that he does not use illicit substances but that he drinks 6 to 12 beers per night and smokes 1 pack of cigarettes per day.
Initial vital signs:
- Temperature 37.7°C (99.9°F)
- Blood pressure 217/114 mm Hg
- Heart rate 82 bpm
- Respiratory rate 18 per minute
- Capillary oxygen saturation 98% while breathing room air.
On examination, his right arm is significantly weak and contracted. His left arm has decreased sensation to pinprick and light touch from elbow to fingers. His face and both arms show hyperpigmentation alternating with atrophic scarring, which also affects his lips. There is no overt mucosal involvement. His hands and forearms have a sclerotic texture and patchy hair loss. Several small bullae are present on the dorsum of the left forearm and hand. There is a 6-inch, irregular, open lesion on the left forearm and a 1-inch lesion on the left hand (Figure 1).
Initial laboratory studies show:
- Chemistries and complete blood cell count within normal limits
- Platelet count 305 × 109/L (reference range 150–350)
- Orange-colored urine
- Hepatitis C virus (HCV) antibody positive (new finding)
- Human immunodeficiency virus antibody, hepatitis B surface antigen, and antinuclear antibody negative
- Phenytoin and urine drug screen negative
- Aspartate aminotransferase 70 U/L (reference range 5–34)
- Alanine aminotransferase 73 U/L (reference range 0–55)
- Prothrombin time 10.8 seconds (reference range 8.3–13.0), international normalized ratio 0.98 (reference range 0.8–1.2)
- Iron studies within normal limits.
The patient is admitted to the hospital and is started on cefazolin and clindamycin. Urine is collected for a porphyrin screen, and punch-biopsy samples from the forearms are sent for study. Ultrasonography shows splenomegaly, as well as increased echogenicity of the liver without structural abnormalities. Blood and urine cultures, drawn upon admission, are negative by discharge.
Pathologic study of the punch-biopsy specimens (Figure 2) shows the formation of subepidermal vesicles with extensive reticular and dermal fibrosis.
DIAGNOSIS: PORPHYRIA CUTANEA TARDA
Because of the patient’s history, examination, and pathology results, he was preliminarily diagnosed with porphyria cutanea tarda (PCT).1,2 The diagnosis was confirmed after he was discharged when his urine uroporphyrin level was found to be 157.5 μmol/mol of creatinine (reference range < 4) and his urine heptacarboxylporphyrin level was 118.0 μmol/mol of creatinine (reference range < 2).
This patient’s clinical presentation is classic for sporadic (ie, type 1) PCT. Sporadic PCT is an acquired deficiency of uroporphyrinogen decarboxylase, an enzyme that catalyzes the fifth step in heme metabolism.3 The deficiency of this enzyme is exclusively hepatic and is strongly associated with chronic hepatitis C infection. Mutations of the hemochromatosis gene (HFE), human immunodeficiency virus infection, alcohol use, and smoking are also risk factors.4 The prevalence in the United States is about 1:25,000; nearly 80% of cases are sporadic (type 1), and 20% are familial (type 2).5
Manifestations of PCT include photosensitive dermatitis, facial hypertrichosis, and orange urine.3 The photosensitivity dermatitis heals slowly and leads to sclerosis and hyperpigmentation.
Repeated phlebotomy is the first-line treatment, and hydroxychloroquine (Plaquenil) is the second-line treatment.6 Patients with PCT and hepatitis C should be considered for antiviral therapy according to standard guidelines. Treatment of hepatitis C may reduce the symptoms of PCT, even without a sustained viral response. However, not enough evidence exists to make treatment recommendations for this group.7
Because we were uncertain that the patient would return for follow-up, we did not start phlebotomy or treatment for hepatitis C. However, we did prescribe hydroxychloroquine 100 mg three times a week and instructed him to cover his skin when outside and to use effective sunblock. An outpatient visit was scheduled prior to discharge. Unfortunately, the patient was lost to follow-up.
Acknowledgment: The authors would like to personally thank Dr. Karen DeSouza from the University of Tennessee, Graduate School of Medicine, Department of Pathology, for her clinical expertise and kind advice.
- The University of Iowa, Department of Pathology, Laboratory Services Handbook. Porphyrins & Porphobilinogen, Urine (24 hr or random). www.healthcare.uiowa.edu/path_handbook/handbook/test2893.html. Accessed August 8, 2014.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol 1992; 19:40–47.
- Thunell S, Harper P. Porphyrins, porphyrin metabolism, porphyrias. III. Diagnosis, care and monitoring in porphyria cutanea tarda—suggestions for a handling programme. Scand J Clin Lab Invest 2000; 60:561–579.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- Kushner JP, Barbuto AJ, Lee GR. An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity. J Clin Invest 1976; 58:1089–1097.
- Singal AK, Kormos-Hallberg C, Lee C, et al. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin Gastroenterol Hepatol 2012; 10:1402–1409.
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 2012; 32:880–893.
- The University of Iowa, Department of Pathology, Laboratory Services Handbook. Porphyrins & Porphobilinogen, Urine (24 hr or random). www.healthcare.uiowa.edu/path_handbook/handbook/test2893.html. Accessed August 8, 2014.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol 1992; 19:40–47.
- Thunell S, Harper P. Porphyrins, porphyrin metabolism, porphyrias. III. Diagnosis, care and monitoring in porphyria cutanea tarda—suggestions for a handling programme. Scand J Clin Lab Invest 2000; 60:561–579.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- Kushner JP, Barbuto AJ, Lee GR. An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity. J Clin Invest 1976; 58:1089–1097.
- Singal AK, Kormos-Hallberg C, Lee C, et al. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin Gastroenterol Hepatol 2012; 10:1402–1409.
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 2012; 32:880–893.
Hypertrophic cardiomyopathy apical variant
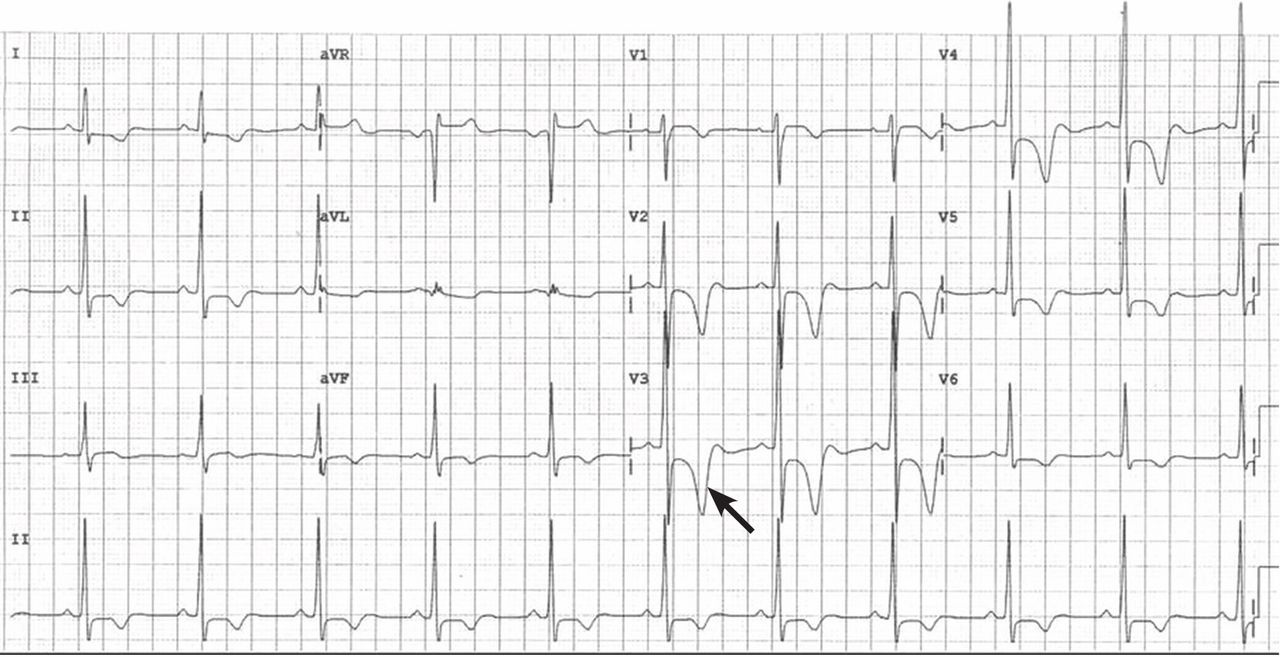
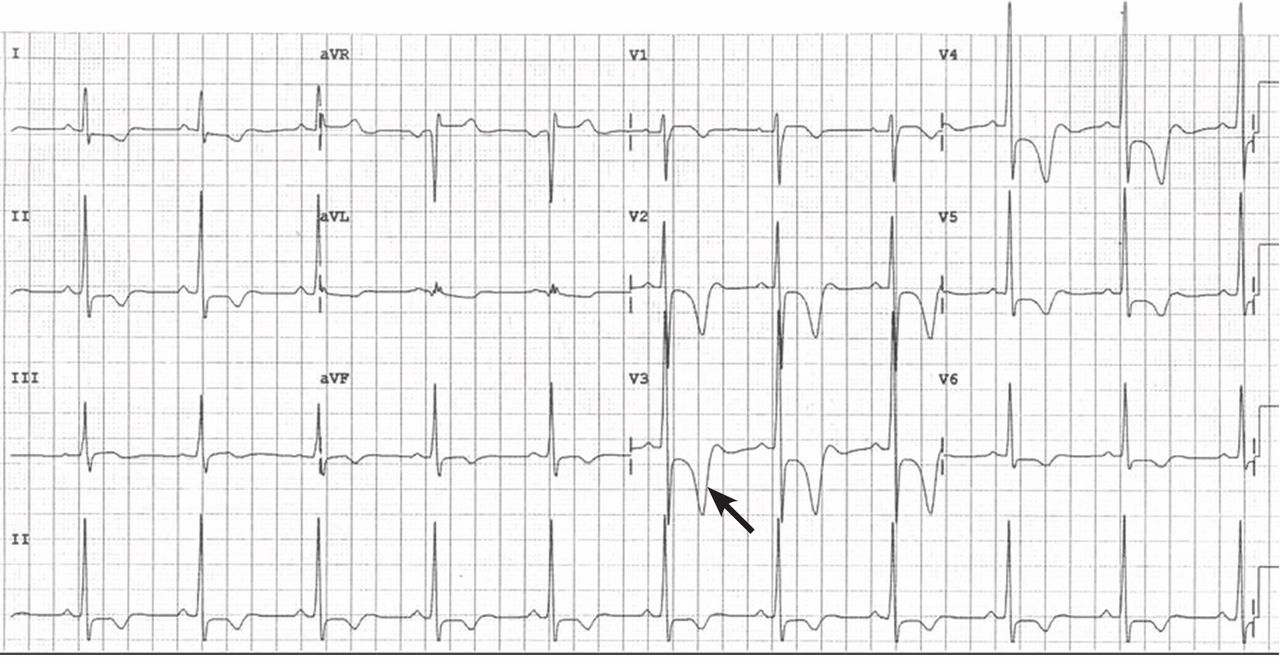
He had had an isolated syncopal episode while intensely training a year ago, but his medical history was otherwise unremarkable.
On examination, he appeared fit. His vital signs were normal. The apical pulse was sustained on palpation and was not displaced. Auscultation revealed an S4 heart sound.
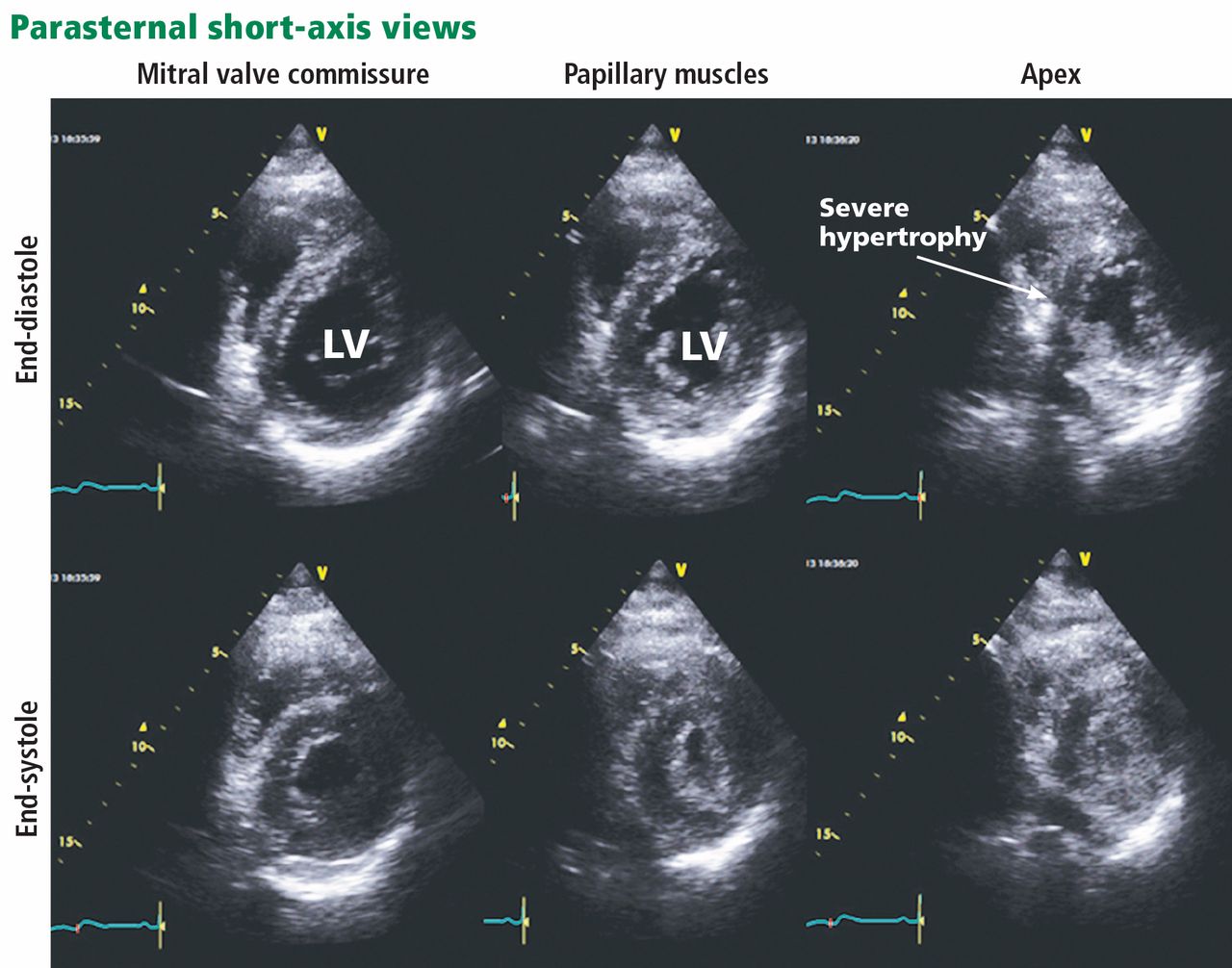
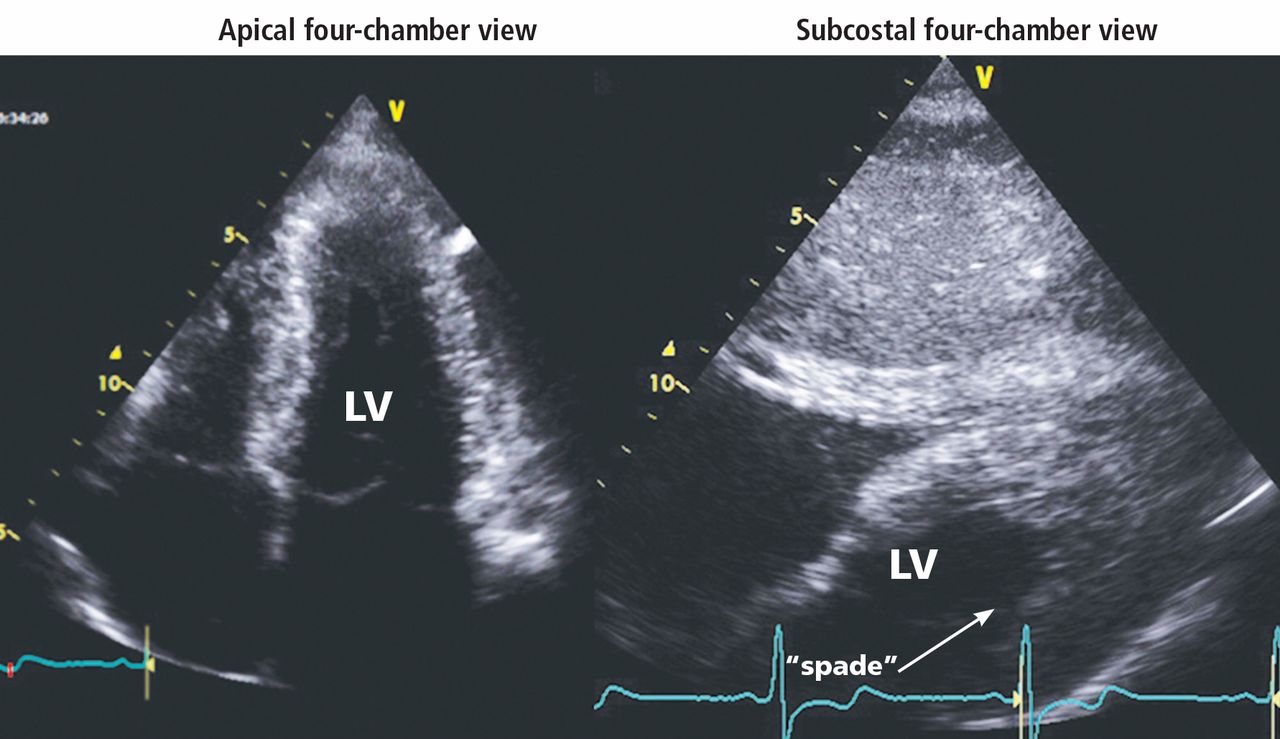
HYPERTROPHIC CARDIOMYOPATHY: THE APICAL VARIANT
In typical hypertrophic cardiomyopathy, the left ventricle, especially the interventricular septum, is thickened, but the left ventricular chamber size is normal or small. In severe cases, the left ventricular outflow tract can become very narrowed, resulting in accelerated blood flow, which may further increase in the presence of hypovolemia, peripheral vasodilation, and increased cardiac contractility. The Venturi effect thus created may entrain a typically malformed anterior mitral valve leaflet toward the aortic valve (systolic anterior motion), causing mitral insufficiency and exacerbating obstruction of the left ventricular outflow tract. Systolic anterior motion may play an important role in exercise-induced syncope and sudden death in young people with hypertrophic cardiomyopathy.4
In the apical variant, hypertrophy is confined to the left ventricular apex.1–3 There is no dynamic outflow tract obstruction. Still, unexplained syncope has been reported, and recent data challenge the conventional wisdom that the apical variant of hypertrophic cardiomyopathy has a benign prognosis.2 In patients without a history of recurrent syncope, chest pain, or heart failure, perioperative risk is probably not significantly increased. The differential diagnosis includes myocardial ischemia or infarction, electrolyte disturbances, effects of drugs (eg, digoxin), and subarachnoid hemorrhage.1–3,5 Plain or contrast-enhanced echocardiography or cardiac magnetic resonance imaging, or both, can help confirm the diagnosis. Long-term management should be guided by the patient’s symptoms.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Kasirye Y, Manne JR, Epperla N, Bapani S, Garcia-Montilla R. Apical hypertrophic cardiomyopathy presenting as recurrent unexplained syncope. Clin Med Res 2012; 10:26–31.
- Lee CH, Liu PY, Lin LJ, Chen JH, Tsai LM. Clinical features and outcome of patients with apical hypertrophic cardiomyopathy in Taiwan. Cardiology 2006; 106:29–35.
- Nishimura RA, Holmes DR Clinical practice. Hypertrophic obstructive cardiomyopathy. N Engl J Med 2004; 350:1320–1327.
- Lin CS, Chen CH, Ding PY. Apical hypertrophic cardiomyopathy mimicking acute myocardial infarction. Int J Cardiol 1998; 64:305–307.
He had had an isolated syncopal episode while intensely training a year ago, but his medical history was otherwise unremarkable.
On examination, he appeared fit. His vital signs were normal. The apical pulse was sustained on palpation and was not displaced. Auscultation revealed an S4 heart sound.
HYPERTROPHIC CARDIOMYOPATHY: THE APICAL VARIANT
In typical hypertrophic cardiomyopathy, the left ventricle, especially the interventricular septum, is thickened, but the left ventricular chamber size is normal or small. In severe cases, the left ventricular outflow tract can become very narrowed, resulting in accelerated blood flow, which may further increase in the presence of hypovolemia, peripheral vasodilation, and increased cardiac contractility. The Venturi effect thus created may entrain a typically malformed anterior mitral valve leaflet toward the aortic valve (systolic anterior motion), causing mitral insufficiency and exacerbating obstruction of the left ventricular outflow tract. Systolic anterior motion may play an important role in exercise-induced syncope and sudden death in young people with hypertrophic cardiomyopathy.4
In the apical variant, hypertrophy is confined to the left ventricular apex.1–3 There is no dynamic outflow tract obstruction. Still, unexplained syncope has been reported, and recent data challenge the conventional wisdom that the apical variant of hypertrophic cardiomyopathy has a benign prognosis.2 In patients without a history of recurrent syncope, chest pain, or heart failure, perioperative risk is probably not significantly increased. The differential diagnosis includes myocardial ischemia or infarction, electrolyte disturbances, effects of drugs (eg, digoxin), and subarachnoid hemorrhage.1–3,5 Plain or contrast-enhanced echocardiography or cardiac magnetic resonance imaging, or both, can help confirm the diagnosis. Long-term management should be guided by the patient’s symptoms.
He had had an isolated syncopal episode while intensely training a year ago, but his medical history was otherwise unremarkable.
On examination, he appeared fit. His vital signs were normal. The apical pulse was sustained on palpation and was not displaced. Auscultation revealed an S4 heart sound.
HYPERTROPHIC CARDIOMYOPATHY: THE APICAL VARIANT
In typical hypertrophic cardiomyopathy, the left ventricle, especially the interventricular septum, is thickened, but the left ventricular chamber size is normal or small. In severe cases, the left ventricular outflow tract can become very narrowed, resulting in accelerated blood flow, which may further increase in the presence of hypovolemia, peripheral vasodilation, and increased cardiac contractility. The Venturi effect thus created may entrain a typically malformed anterior mitral valve leaflet toward the aortic valve (systolic anterior motion), causing mitral insufficiency and exacerbating obstruction of the left ventricular outflow tract. Systolic anterior motion may play an important role in exercise-induced syncope and sudden death in young people with hypertrophic cardiomyopathy.4
In the apical variant, hypertrophy is confined to the left ventricular apex.1–3 There is no dynamic outflow tract obstruction. Still, unexplained syncope has been reported, and recent data challenge the conventional wisdom that the apical variant of hypertrophic cardiomyopathy has a benign prognosis.2 In patients without a history of recurrent syncope, chest pain, or heart failure, perioperative risk is probably not significantly increased. The differential diagnosis includes myocardial ischemia or infarction, electrolyte disturbances, effects of drugs (eg, digoxin), and subarachnoid hemorrhage.1–3,5 Plain or contrast-enhanced echocardiography or cardiac magnetic resonance imaging, or both, can help confirm the diagnosis. Long-term management should be guided by the patient’s symptoms.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Kasirye Y, Manne JR, Epperla N, Bapani S, Garcia-Montilla R. Apical hypertrophic cardiomyopathy presenting as recurrent unexplained syncope. Clin Med Res 2012; 10:26–31.
- Lee CH, Liu PY, Lin LJ, Chen JH, Tsai LM. Clinical features and outcome of patients with apical hypertrophic cardiomyopathy in Taiwan. Cardiology 2006; 106:29–35.
- Nishimura RA, Holmes DR Clinical practice. Hypertrophic obstructive cardiomyopathy. N Engl J Med 2004; 350:1320–1327.
- Lin CS, Chen CH, Ding PY. Apical hypertrophic cardiomyopathy mimicking acute myocardial infarction. Int J Cardiol 1998; 64:305–307.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Kasirye Y, Manne JR, Epperla N, Bapani S, Garcia-Montilla R. Apical hypertrophic cardiomyopathy presenting as recurrent unexplained syncope. Clin Med Res 2012; 10:26–31.
- Lee CH, Liu PY, Lin LJ, Chen JH, Tsai LM. Clinical features and outcome of patients with apical hypertrophic cardiomyopathy in Taiwan. Cardiology 2006; 106:29–35.
- Nishimura RA, Holmes DR Clinical practice. Hypertrophic obstructive cardiomyopathy. N Engl J Med 2004; 350:1320–1327.
- Lin CS, Chen CH, Ding PY. Apical hypertrophic cardiomyopathy mimicking acute myocardial infarction. Int J Cardiol 1998; 64:305–307.
A 78-year-old smoker with an incidental pulmonary mass
When a 78-year-old man underwent magnetic resonance imaging of the lumbar spine because of back pain, the scan revealed a mass in the right lung. He had no respiratory symptoms but had a 40-pack-year smoking history. Physical examination and routine blood tests were unremarkable.
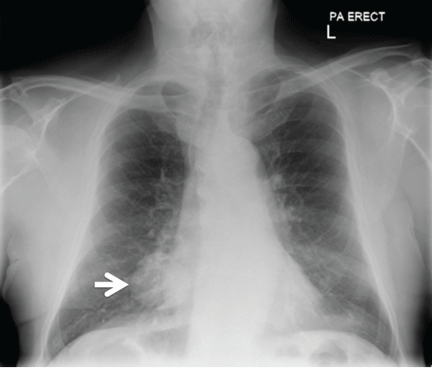
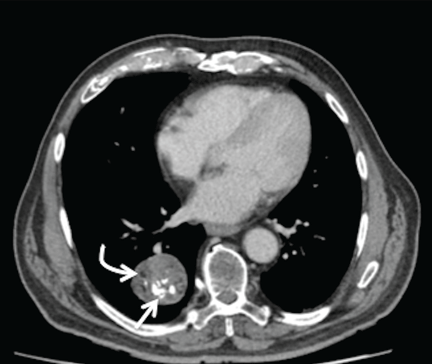
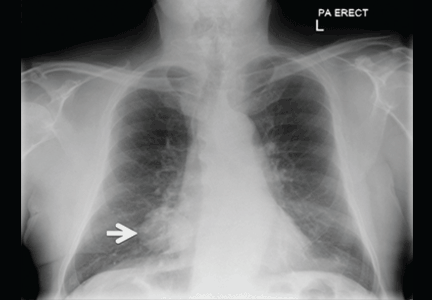
Radiography (Figure 1) showed a large rounded opacity in the right lower lobe. The patient’s age, smoking history, and imaging findings raised concern for lung cancer, so computed tomography (CT) was performed (Figure 2).
DIAGNOSIS: PULMONARY HAMARTOMA
The findings of a well-circumscribed solitary pulmonary nodule or mass containing areas of fat, either as focal islands or more generally distributed, and chondroid “popcorn” calcification are virtually pathognomonic for pulmonary hamartoma.1,2 Unfortunately, although this pattern of calcification is strongly diagnostic, it is present in only a minority of cases of hamartoma.
Pulmonary hamartoma is the most common benign tumor of the lung, accounting for approximately 75% of benign neoplasms and 6% to 8% of all focal lung parenchymal masses.3
Like hamartoma elsewhere in the body, pulmonary hamartoma consists of disorganized overgrowth and aberrant arrangement of normal tissues, including cartilage (which may calcify), smooth muscle, epithelium, and fibrostroma. Pulmonary hamartoma is twice as common in men as in women, and it has a peak incidence in the seventh decade of life.4
Although size ranged from 0.2 to 6 cm in a large case series,4 hamartomas are usually less than 2.5 cm in diameter. As noted in Figure 1, our patient’s lesion was 5 cm.
Pulmonary hamartomas grow slowly and are often asymptomatic, although up to 39% of patients may have symptoms such as cough, dyspnea, and chest tightness.5 The nonspecific nature of these symptoms makes it difficult to be certain that they are caused by the hamartoma; in many cases, they are likely to be coincidental. Lesions tend to occur in the periphery of the lobe and do not favor a particular lobe. Endobronchial lesions can occur but are uncommon.
The internal heterogeneous elements are difficult to see on radiography; CT is usually required to further characterize the lesion and to exclude more sinister differential diagnoses. In some cases the characteristic features of fat and calcification are absent, making a certain diagnosis difficult or impossible radiologically; in such cases, biopsy or resection may be required.
Hamartomas usually do not take up fluorodeoxyglucose avidly on positron-emission tomography CT. However, nuclear medicine studies such as this are superfluous if the classic features are present on CT.
FOLLOW-UP AND TREATMENT
Given the benign nature, slow growth, and usually incidental detection of pulmonary hamartoma in patients without symptoms, no follow-up imaging or treatment is usually required. In the few cases in which symptoms are attributable to the lesion, the lesion can be resected.5 Resection is also an option when the patient is very anxious about the mass, or when imaging studies do not provide a clear diagnosis and tissue needs to be obtained for study.
Because patients often present to different institutions during their lifetime, it is important to counsel them about the natural history of pulmonary hamartomas. Giving them a copy of their imaging may help avoid unnecessary repetition.
- Erasmus JJ, Connolly JE, McAdams HP, Roggli VL. Solitary pulmonary nodules: Part I. Morphologic evaluation for differentiation of benign and malignant lesions. Radiographics 2000; 20:43–58.
- Khan AN, Al-Jahdali HH, Allen CM, Irion KL, Al Ghanem S, Koteyar SS. The calcified lung nodule: what does it mean? Ann Thorac Med 2010; 5:67–79.
- Siegelman SS, Khouri NF, Scott WW, et al. Pulmonary hamartoma: CT findings. Radiology 1986; 160:313–317.
- Gjevre JA, Myers JL, Prakash UB. Pulmonary hamartomas. Mayo Clin Proc 1996; 71:14–20.
- Hansen CP, Holtveg H, Francis D, Rasch L, Bertelsen S. Pulmonary hamartoma. J Thorac Cardiovasc Surg 1992; 104:674–678.
When a 78-year-old man underwent magnetic resonance imaging of the lumbar spine because of back pain, the scan revealed a mass in the right lung. He had no respiratory symptoms but had a 40-pack-year smoking history. Physical examination and routine blood tests were unremarkable.
Radiography (Figure 1) showed a large rounded opacity in the right lower lobe. The patient’s age, smoking history, and imaging findings raised concern for lung cancer, so computed tomography (CT) was performed (Figure 2).
DIAGNOSIS: PULMONARY HAMARTOMA
The findings of a well-circumscribed solitary pulmonary nodule or mass containing areas of fat, either as focal islands or more generally distributed, and chondroid “popcorn” calcification are virtually pathognomonic for pulmonary hamartoma.1,2 Unfortunately, although this pattern of calcification is strongly diagnostic, it is present in only a minority of cases of hamartoma.
Pulmonary hamartoma is the most common benign tumor of the lung, accounting for approximately 75% of benign neoplasms and 6% to 8% of all focal lung parenchymal masses.3
Like hamartoma elsewhere in the body, pulmonary hamartoma consists of disorganized overgrowth and aberrant arrangement of normal tissues, including cartilage (which may calcify), smooth muscle, epithelium, and fibrostroma. Pulmonary hamartoma is twice as common in men as in women, and it has a peak incidence in the seventh decade of life.4
Although size ranged from 0.2 to 6 cm in a large case series,4 hamartomas are usually less than 2.5 cm in diameter. As noted in Figure 1, our patient’s lesion was 5 cm.
Pulmonary hamartomas grow slowly and are often asymptomatic, although up to 39% of patients may have symptoms such as cough, dyspnea, and chest tightness.5 The nonspecific nature of these symptoms makes it difficult to be certain that they are caused by the hamartoma; in many cases, they are likely to be coincidental. Lesions tend to occur in the periphery of the lobe and do not favor a particular lobe. Endobronchial lesions can occur but are uncommon.
The internal heterogeneous elements are difficult to see on radiography; CT is usually required to further characterize the lesion and to exclude more sinister differential diagnoses. In some cases the characteristic features of fat and calcification are absent, making a certain diagnosis difficult or impossible radiologically; in such cases, biopsy or resection may be required.
Hamartomas usually do not take up fluorodeoxyglucose avidly on positron-emission tomography CT. However, nuclear medicine studies such as this are superfluous if the classic features are present on CT.
FOLLOW-UP AND TREATMENT
Given the benign nature, slow growth, and usually incidental detection of pulmonary hamartoma in patients without symptoms, no follow-up imaging or treatment is usually required. In the few cases in which symptoms are attributable to the lesion, the lesion can be resected.5 Resection is also an option when the patient is very anxious about the mass, or when imaging studies do not provide a clear diagnosis and tissue needs to be obtained for study.
Because patients often present to different institutions during their lifetime, it is important to counsel them about the natural history of pulmonary hamartomas. Giving them a copy of their imaging may help avoid unnecessary repetition.
When a 78-year-old man underwent magnetic resonance imaging of the lumbar spine because of back pain, the scan revealed a mass in the right lung. He had no respiratory symptoms but had a 40-pack-year smoking history. Physical examination and routine blood tests were unremarkable.
Radiography (Figure 1) showed a large rounded opacity in the right lower lobe. The patient’s age, smoking history, and imaging findings raised concern for lung cancer, so computed tomography (CT) was performed (Figure 2).
DIAGNOSIS: PULMONARY HAMARTOMA
The findings of a well-circumscribed solitary pulmonary nodule or mass containing areas of fat, either as focal islands or more generally distributed, and chondroid “popcorn” calcification are virtually pathognomonic for pulmonary hamartoma.1,2 Unfortunately, although this pattern of calcification is strongly diagnostic, it is present in only a minority of cases of hamartoma.
Pulmonary hamartoma is the most common benign tumor of the lung, accounting for approximately 75% of benign neoplasms and 6% to 8% of all focal lung parenchymal masses.3
Like hamartoma elsewhere in the body, pulmonary hamartoma consists of disorganized overgrowth and aberrant arrangement of normal tissues, including cartilage (which may calcify), smooth muscle, epithelium, and fibrostroma. Pulmonary hamartoma is twice as common in men as in women, and it has a peak incidence in the seventh decade of life.4
Although size ranged from 0.2 to 6 cm in a large case series,4 hamartomas are usually less than 2.5 cm in diameter. As noted in Figure 1, our patient’s lesion was 5 cm.
Pulmonary hamartomas grow slowly and are often asymptomatic, although up to 39% of patients may have symptoms such as cough, dyspnea, and chest tightness.5 The nonspecific nature of these symptoms makes it difficult to be certain that they are caused by the hamartoma; in many cases, they are likely to be coincidental. Lesions tend to occur in the periphery of the lobe and do not favor a particular lobe. Endobronchial lesions can occur but are uncommon.
The internal heterogeneous elements are difficult to see on radiography; CT is usually required to further characterize the lesion and to exclude more sinister differential diagnoses. In some cases the characteristic features of fat and calcification are absent, making a certain diagnosis difficult or impossible radiologically; in such cases, biopsy or resection may be required.
Hamartomas usually do not take up fluorodeoxyglucose avidly on positron-emission tomography CT. However, nuclear medicine studies such as this are superfluous if the classic features are present on CT.
FOLLOW-UP AND TREATMENT
Given the benign nature, slow growth, and usually incidental detection of pulmonary hamartoma in patients without symptoms, no follow-up imaging or treatment is usually required. In the few cases in which symptoms are attributable to the lesion, the lesion can be resected.5 Resection is also an option when the patient is very anxious about the mass, or when imaging studies do not provide a clear diagnosis and tissue needs to be obtained for study.
Because patients often present to different institutions during their lifetime, it is important to counsel them about the natural history of pulmonary hamartomas. Giving them a copy of their imaging may help avoid unnecessary repetition.
- Erasmus JJ, Connolly JE, McAdams HP, Roggli VL. Solitary pulmonary nodules: Part I. Morphologic evaluation for differentiation of benign and malignant lesions. Radiographics 2000; 20:43–58.
- Khan AN, Al-Jahdali HH, Allen CM, Irion KL, Al Ghanem S, Koteyar SS. The calcified lung nodule: what does it mean? Ann Thorac Med 2010; 5:67–79.
- Siegelman SS, Khouri NF, Scott WW, et al. Pulmonary hamartoma: CT findings. Radiology 1986; 160:313–317.
- Gjevre JA, Myers JL, Prakash UB. Pulmonary hamartomas. Mayo Clin Proc 1996; 71:14–20.
- Hansen CP, Holtveg H, Francis D, Rasch L, Bertelsen S. Pulmonary hamartoma. J Thorac Cardiovasc Surg 1992; 104:674–678.
- Erasmus JJ, Connolly JE, McAdams HP, Roggli VL. Solitary pulmonary nodules: Part I. Morphologic evaluation for differentiation of benign and malignant lesions. Radiographics 2000; 20:43–58.
- Khan AN, Al-Jahdali HH, Allen CM, Irion KL, Al Ghanem S, Koteyar SS. The calcified lung nodule: what does it mean? Ann Thorac Med 2010; 5:67–79.
- Siegelman SS, Khouri NF, Scott WW, et al. Pulmonary hamartoma: CT findings. Radiology 1986; 160:313–317.
- Gjevre JA, Myers JL, Prakash UB. Pulmonary hamartomas. Mayo Clin Proc 1996; 71:14–20.
- Hansen CP, Holtveg H, Francis D, Rasch L, Bertelsen S. Pulmonary hamartoma. J Thorac Cardiovasc Surg 1992; 104:674–678.
Alveolar proteinosis: A slow drowning in mud
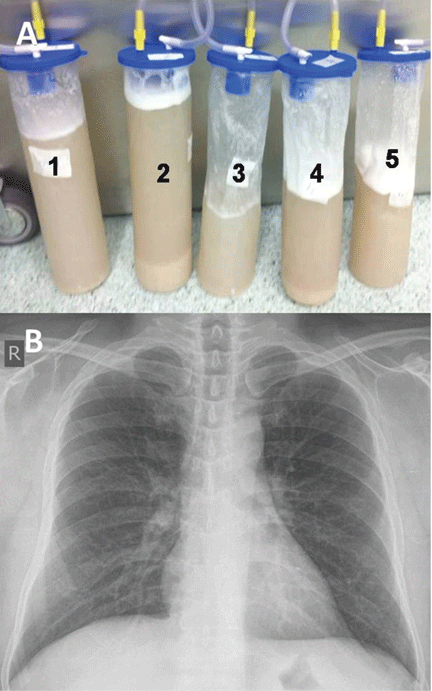
A 30-year-old man presented with progressive dyspnea and dry cough, which had developed over the last 6 months. His oxygen saturation was 88% on room air, and he had diffuse bilateral crackles on auscultation. Imaging showed a mixture of diffuse airspace and interstitial abnormalities (Figure 1).
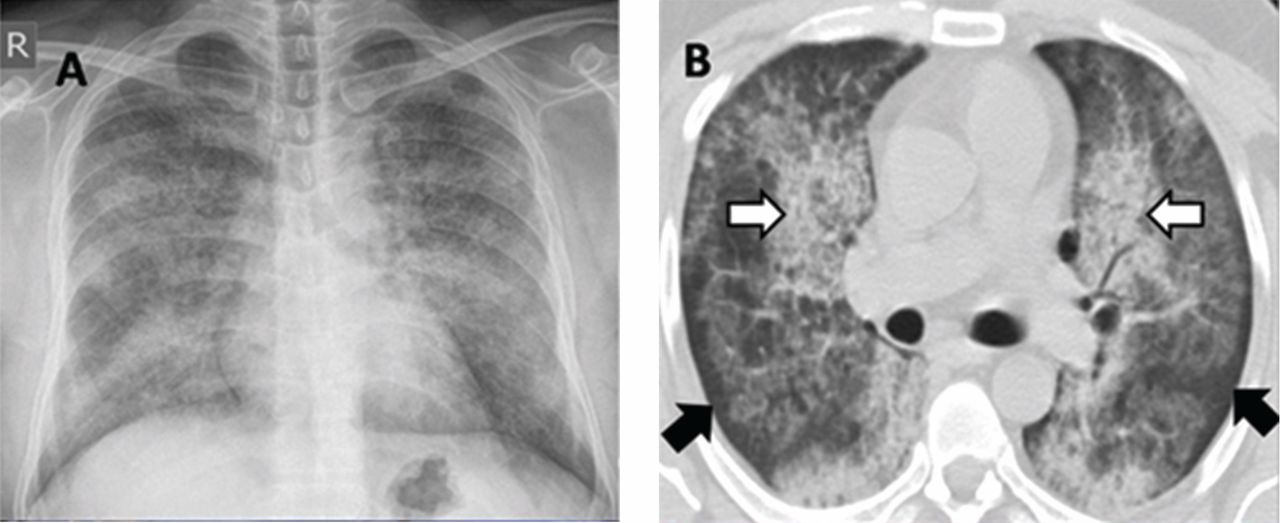
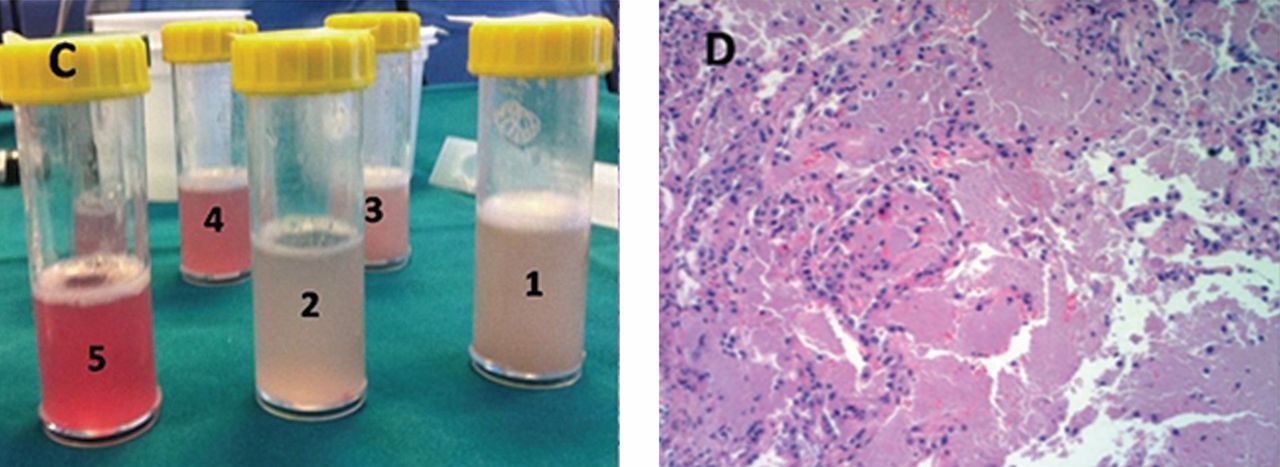
He underwent bronchoscopy. The bronchoalveolar lavage fluid had a turbid appearance that gradually cleared with successive aliquots. Transbronchial biopsy studies confirmed the diagnosis of pulmonary alveolar proteinosis (Figure 2). Sequential whole-lung lavage recovered significant amounts of thick, proteinaceous effluent that slowly cleared. After the procedure, the patient’s symptoms, oxygen saturation, and chest radiographic appearance (Figure 3) improved markedly, with no recurrence at 1 year of follow-up.
ALVEOLAR PROTEINOSIS
Pulmonary alveolar proteinosis is a rare disease characterized by the accumulation of lipoproteinaceous material in the alveolar space secondary to alveolar macrophage dysfunction. The condition can be congenital, secondary, or acquired. Patients typically present with progressive exertional dyspnea, nonproductive cough, variable restrictive ventilatory defects, and diffusion limitation on pulmonary function testing.
Plain chest radiographs usually resemble those seen in pulmonary edema but without features of heart failure, ie, cardiomegaly, Kerley B lines, and effusion.
A “crazy-paving” pattern on computed tomography—a combination of geographic ground-glass appearance and interseptal thickening—suggests alveolar proteinosis, but is not specific for it. Other differential diagnoses for the crazy-paving pattern include Pneumocystis jirovecii infection, invasive mucinous adenocarcinoma, cardiogenic pulmonary edema, alveolar hemorrhage, sarcoidosis, cryptogenic organizing pneumonia, exogenous lipoid pneumonia, drug-induced lung disease, acute radiation pneumonitis, and nonspecific interstitial pneumonia.1
Laboratory testing is not very helpful in the diagnosis, although the serum lactate dehydrogenase level may be mildly elevated. Circulating antibodies to granulocyte macrophage colony-stimulating factor may support the diagnosis, but they are only present in the acquired form. Communication with a research laboratory is usually needed to test for these antibodies.
The bronchoalveolar lavage fluid typically has an opaque, milky, or muddy appearance. The diagnosis is confirmed by demonstration of alveolar filling with material that is periodic acid-Schiff-positive and that is amorphous, eosinophilic, and granular.
Whole-lung lavage2 is the physical removal of surfactant by repeated flooding of the lungs with warmed saline, done under general anesthesia with single-lung ventilation. It remains the standard of care and is indicated in patients with the confirmed diagnosis and one of the following: severe dyspnea, resting hypoxemia (Pao2 < 60 mm Hg at sea level), alveolar-arterial gradient > 40 mm Hg, or a shunt fraction of more than 10%. Successful bronchoscopic lavage has also been reported.3
Other treatments include granulocyte-macrophage colony-stimulating factor, rituximab (Rituxan, an anti-CD20 monoclonal antibody), plasmapheresis, and lung transplantation. Systemic corticosteroids are usually ineffective unless indicated for secondary types of alveolar proteinosis.
Inhalation rather than subcutaneous administration of granulocyte-macrophage colony-stimulating factor seems preferred as it ensures a high concentration in the target organ, avoids systemic complications (injection-site edema, erythema, neutropenia, malaise, and shortness of breath) and achieves lower levels of autoantibodies in bronchoalveolar lavage fluid, which correlates with disease activity.
Data are sparse as to the recurrence of autoimmune pulmonary alveolar proteinosis after whole-lung lavage, yet about 40% of patients require a repeat procedure within 18 months. Recurrence has also been reported after double-lung transplantation.4
Adjuvant therapy with rituximab or, to a lesser extent, with inhaled granulocyte-macrophage colony-stimulating factor has recently been shown to diminish the need for repeated lavage.5 These treatments can also be used when whole-lung lavage cannot be performed or proves to be ineffective.5
Acknowledgment: I would like to thank Dr. Kamelia Velikova for providing the pathology image.
- Rossi SE, Erasmus JJ, Volpacchio M, Franquet T, Castiglioni T, McAdams HP. ‘Crazy-paving’ pattern at thin-section CT of the lungs: radiologic-pathologic overview. Radiographics 2003; 23:1509–1519.
- Michaud G, Reddy C, Ernst A. Whole-lung lavage for pulmonary alveolar proteinosis. Chest 2009; 136:1678–1681.
- Cheng SL, Chang HT, Lau HP, Lee LN, Yang PC. Pulmonary alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest 2002; 122:1480–1485.
- Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest 1997; 111:1457–1458.
- Leth S, Bendstrup E, Vestergaard H, Hilberg O. Autoimmune pulmonary alveolar proteinosis: treatment options in year 2013. Respirology 2013; 18:82–91.
SUGGESTED READING
Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis.Eur Respir Rev 2011; 20:98–107.
Carey B, Trapnell BC. The molecular basis of pulmonary alveolarproteinosis. Clin Immunol 2010; 135:223–235.
Ioachimescu OC, Kavuru MS. Pulmonary alveolar proteinosis.Chron Respir Dis 2006; 3:149–159.
Luisetti M, Kadija Z, Mariani F, Rodi G, Campo I, Trapnell BC.Therapy options in pulmonary alveolar proteinosis. TherAdv Respir Dis 2010; 4:239–248.
A 30-year-old man presented with progressive dyspnea and dry cough, which had developed over the last 6 months. His oxygen saturation was 88% on room air, and he had diffuse bilateral crackles on auscultation. Imaging showed a mixture of diffuse airspace and interstitial abnormalities (Figure 1).
He underwent bronchoscopy. The bronchoalveolar lavage fluid had a turbid appearance that gradually cleared with successive aliquots. Transbronchial biopsy studies confirmed the diagnosis of pulmonary alveolar proteinosis (Figure 2). Sequential whole-lung lavage recovered significant amounts of thick, proteinaceous effluent that slowly cleared. After the procedure, the patient’s symptoms, oxygen saturation, and chest radiographic appearance (Figure 3) improved markedly, with no recurrence at 1 year of follow-up.
ALVEOLAR PROTEINOSIS
Pulmonary alveolar proteinosis is a rare disease characterized by the accumulation of lipoproteinaceous material in the alveolar space secondary to alveolar macrophage dysfunction. The condition can be congenital, secondary, or acquired. Patients typically present with progressive exertional dyspnea, nonproductive cough, variable restrictive ventilatory defects, and diffusion limitation on pulmonary function testing.
Plain chest radiographs usually resemble those seen in pulmonary edema but without features of heart failure, ie, cardiomegaly, Kerley B lines, and effusion.
A “crazy-paving” pattern on computed tomography—a combination of geographic ground-glass appearance and interseptal thickening—suggests alveolar proteinosis, but is not specific for it. Other differential diagnoses for the crazy-paving pattern include Pneumocystis jirovecii infection, invasive mucinous adenocarcinoma, cardiogenic pulmonary edema, alveolar hemorrhage, sarcoidosis, cryptogenic organizing pneumonia, exogenous lipoid pneumonia, drug-induced lung disease, acute radiation pneumonitis, and nonspecific interstitial pneumonia.1
Laboratory testing is not very helpful in the diagnosis, although the serum lactate dehydrogenase level may be mildly elevated. Circulating antibodies to granulocyte macrophage colony-stimulating factor may support the diagnosis, but they are only present in the acquired form. Communication with a research laboratory is usually needed to test for these antibodies.
The bronchoalveolar lavage fluid typically has an opaque, milky, or muddy appearance. The diagnosis is confirmed by demonstration of alveolar filling with material that is periodic acid-Schiff-positive and that is amorphous, eosinophilic, and granular.
Whole-lung lavage2 is the physical removal of surfactant by repeated flooding of the lungs with warmed saline, done under general anesthesia with single-lung ventilation. It remains the standard of care and is indicated in patients with the confirmed diagnosis and one of the following: severe dyspnea, resting hypoxemia (Pao2 < 60 mm Hg at sea level), alveolar-arterial gradient > 40 mm Hg, or a shunt fraction of more than 10%. Successful bronchoscopic lavage has also been reported.3
Other treatments include granulocyte-macrophage colony-stimulating factor, rituximab (Rituxan, an anti-CD20 monoclonal antibody), plasmapheresis, and lung transplantation. Systemic corticosteroids are usually ineffective unless indicated for secondary types of alveolar proteinosis.
Inhalation rather than subcutaneous administration of granulocyte-macrophage colony-stimulating factor seems preferred as it ensures a high concentration in the target organ, avoids systemic complications (injection-site edema, erythema, neutropenia, malaise, and shortness of breath) and achieves lower levels of autoantibodies in bronchoalveolar lavage fluid, which correlates with disease activity.
Data are sparse as to the recurrence of autoimmune pulmonary alveolar proteinosis after whole-lung lavage, yet about 40% of patients require a repeat procedure within 18 months. Recurrence has also been reported after double-lung transplantation.4
Adjuvant therapy with rituximab or, to a lesser extent, with inhaled granulocyte-macrophage colony-stimulating factor has recently been shown to diminish the need for repeated lavage.5 These treatments can also be used when whole-lung lavage cannot be performed or proves to be ineffective.5
Acknowledgment: I would like to thank Dr. Kamelia Velikova for providing the pathology image.
A 30-year-old man presented with progressive dyspnea and dry cough, which had developed over the last 6 months. His oxygen saturation was 88% on room air, and he had diffuse bilateral crackles on auscultation. Imaging showed a mixture of diffuse airspace and interstitial abnormalities (Figure 1).
He underwent bronchoscopy. The bronchoalveolar lavage fluid had a turbid appearance that gradually cleared with successive aliquots. Transbronchial biopsy studies confirmed the diagnosis of pulmonary alveolar proteinosis (Figure 2). Sequential whole-lung lavage recovered significant amounts of thick, proteinaceous effluent that slowly cleared. After the procedure, the patient’s symptoms, oxygen saturation, and chest radiographic appearance (Figure 3) improved markedly, with no recurrence at 1 year of follow-up.
ALVEOLAR PROTEINOSIS
Pulmonary alveolar proteinosis is a rare disease characterized by the accumulation of lipoproteinaceous material in the alveolar space secondary to alveolar macrophage dysfunction. The condition can be congenital, secondary, or acquired. Patients typically present with progressive exertional dyspnea, nonproductive cough, variable restrictive ventilatory defects, and diffusion limitation on pulmonary function testing.
Plain chest radiographs usually resemble those seen in pulmonary edema but without features of heart failure, ie, cardiomegaly, Kerley B lines, and effusion.
A “crazy-paving” pattern on computed tomography—a combination of geographic ground-glass appearance and interseptal thickening—suggests alveolar proteinosis, but is not specific for it. Other differential diagnoses for the crazy-paving pattern include Pneumocystis jirovecii infection, invasive mucinous adenocarcinoma, cardiogenic pulmonary edema, alveolar hemorrhage, sarcoidosis, cryptogenic organizing pneumonia, exogenous lipoid pneumonia, drug-induced lung disease, acute radiation pneumonitis, and nonspecific interstitial pneumonia.1
Laboratory testing is not very helpful in the diagnosis, although the serum lactate dehydrogenase level may be mildly elevated. Circulating antibodies to granulocyte macrophage colony-stimulating factor may support the diagnosis, but they are only present in the acquired form. Communication with a research laboratory is usually needed to test for these antibodies.
The bronchoalveolar lavage fluid typically has an opaque, milky, or muddy appearance. The diagnosis is confirmed by demonstration of alveolar filling with material that is periodic acid-Schiff-positive and that is amorphous, eosinophilic, and granular.
Whole-lung lavage2 is the physical removal of surfactant by repeated flooding of the lungs with warmed saline, done under general anesthesia with single-lung ventilation. It remains the standard of care and is indicated in patients with the confirmed diagnosis and one of the following: severe dyspnea, resting hypoxemia (Pao2 < 60 mm Hg at sea level), alveolar-arterial gradient > 40 mm Hg, or a shunt fraction of more than 10%. Successful bronchoscopic lavage has also been reported.3
Other treatments include granulocyte-macrophage colony-stimulating factor, rituximab (Rituxan, an anti-CD20 monoclonal antibody), plasmapheresis, and lung transplantation. Systemic corticosteroids are usually ineffective unless indicated for secondary types of alveolar proteinosis.
Inhalation rather than subcutaneous administration of granulocyte-macrophage colony-stimulating factor seems preferred as it ensures a high concentration in the target organ, avoids systemic complications (injection-site edema, erythema, neutropenia, malaise, and shortness of breath) and achieves lower levels of autoantibodies in bronchoalveolar lavage fluid, which correlates with disease activity.
Data are sparse as to the recurrence of autoimmune pulmonary alveolar proteinosis after whole-lung lavage, yet about 40% of patients require a repeat procedure within 18 months. Recurrence has also been reported after double-lung transplantation.4
Adjuvant therapy with rituximab or, to a lesser extent, with inhaled granulocyte-macrophage colony-stimulating factor has recently been shown to diminish the need for repeated lavage.5 These treatments can also be used when whole-lung lavage cannot be performed or proves to be ineffective.5
Acknowledgment: I would like to thank Dr. Kamelia Velikova for providing the pathology image.
- Rossi SE, Erasmus JJ, Volpacchio M, Franquet T, Castiglioni T, McAdams HP. ‘Crazy-paving’ pattern at thin-section CT of the lungs: radiologic-pathologic overview. Radiographics 2003; 23:1509–1519.
- Michaud G, Reddy C, Ernst A. Whole-lung lavage for pulmonary alveolar proteinosis. Chest 2009; 136:1678–1681.
- Cheng SL, Chang HT, Lau HP, Lee LN, Yang PC. Pulmonary alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest 2002; 122:1480–1485.
- Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest 1997; 111:1457–1458.
- Leth S, Bendstrup E, Vestergaard H, Hilberg O. Autoimmune pulmonary alveolar proteinosis: treatment options in year 2013. Respirology 2013; 18:82–91.
SUGGESTED READING
Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis.Eur Respir Rev 2011; 20:98–107.
Carey B, Trapnell BC. The molecular basis of pulmonary alveolarproteinosis. Clin Immunol 2010; 135:223–235.
Ioachimescu OC, Kavuru MS. Pulmonary alveolar proteinosis.Chron Respir Dis 2006; 3:149–159.
Luisetti M, Kadija Z, Mariani F, Rodi G, Campo I, Trapnell BC.Therapy options in pulmonary alveolar proteinosis. TherAdv Respir Dis 2010; 4:239–248.
- Rossi SE, Erasmus JJ, Volpacchio M, Franquet T, Castiglioni T, McAdams HP. ‘Crazy-paving’ pattern at thin-section CT of the lungs: radiologic-pathologic overview. Radiographics 2003; 23:1509–1519.
- Michaud G, Reddy C, Ernst A. Whole-lung lavage for pulmonary alveolar proteinosis. Chest 2009; 136:1678–1681.
- Cheng SL, Chang HT, Lau HP, Lee LN, Yang PC. Pulmonary alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest 2002; 122:1480–1485.
- Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest 1997; 111:1457–1458.
- Leth S, Bendstrup E, Vestergaard H, Hilberg O. Autoimmune pulmonary alveolar proteinosis: treatment options in year 2013. Respirology 2013; 18:82–91.
SUGGESTED READING
Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis.Eur Respir Rev 2011; 20:98–107.
Carey B, Trapnell BC. The molecular basis of pulmonary alveolarproteinosis. Clin Immunol 2010; 135:223–235.
Ioachimescu OC, Kavuru MS. Pulmonary alveolar proteinosis.Chron Respir Dis 2006; 3:149–159.
Luisetti M, Kadija Z, Mariani F, Rodi G, Campo I, Trapnell BC.Therapy options in pulmonary alveolar proteinosis. TherAdv Respir Dis 2010; 4:239–248.

Double trouble: Simultaneous complications of therapeutic thoracentesis
A 51-year-old man with end-stage liver disease from alcohol abuse presented with worsening dyspnea on exertion. He had a history of ascites requiring diuretic therapy and intermittent paracentesis, as well as symptomatic hepatic hydrothorax requiring thoracentesis. Chest radiography showed a large right hydrothorax (Figure 1).
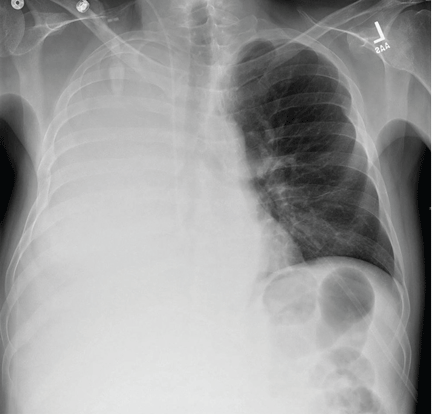
The patient underwent high-volume thoracentesis, and 3.2 L of clear fluid was removed. Chest radiography after the procedure revealed a right-sided pneumothorax (Figure 2, arrow). The patient was mildly short of breath and was treated with high-flow oxygen. Later the same day, his shortness of breath worsened, and repeat chest radiography showed an unchanged pneumothorax that was now complicated by reexpansion pulmonary edema after thoracentesis (Figure 3, star). The reexpansion pulmonary edema resolved by the following day, and the pneumothorax resolved after placement of a pig-tail catheter into the pleural space (Figure 4).
Iatrogenic pneumothorax after thoracentesis occurs in 6% of cases.1 Iatrogenic reexpansion pulmonary edema after thoracentesis occurs in fewer than 1% of cases.2,3 Simultaneous pneumothorax and reexpansion pulmonary edema arising from the same procedure appears to be extremely rare.
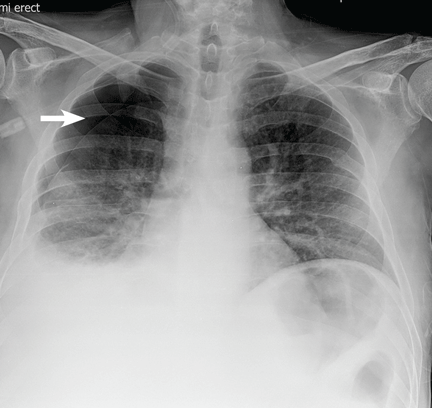
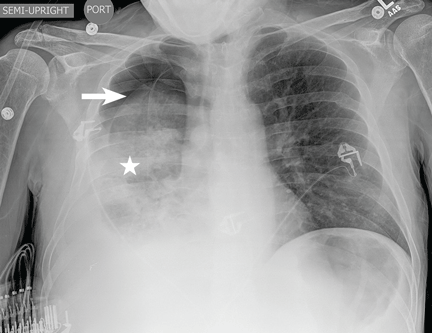
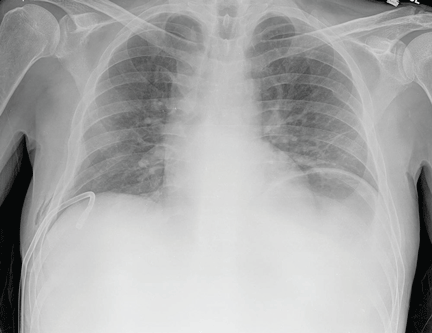
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Ragozzino MW, Greene R. Bilateral reexpansion pulmonary edema following unilateral pleurocentesis. Chest 1991; 99:506–508.
- Dias OM, Teixeira LR, Vargas FS. Reexpansion pulmonary edema after therapeutic thoracentesis. Clinics (Sao Paulo) 2010; 65:1387–1389.
A 51-year-old man with end-stage liver disease from alcohol abuse presented with worsening dyspnea on exertion. He had a history of ascites requiring diuretic therapy and intermittent paracentesis, as well as symptomatic hepatic hydrothorax requiring thoracentesis. Chest radiography showed a large right hydrothorax (Figure 1).
The patient underwent high-volume thoracentesis, and 3.2 L of clear fluid was removed. Chest radiography after the procedure revealed a right-sided pneumothorax (Figure 2, arrow). The patient was mildly short of breath and was treated with high-flow oxygen. Later the same day, his shortness of breath worsened, and repeat chest radiography showed an unchanged pneumothorax that was now complicated by reexpansion pulmonary edema after thoracentesis (Figure 3, star). The reexpansion pulmonary edema resolved by the following day, and the pneumothorax resolved after placement of a pig-tail catheter into the pleural space (Figure 4).
Iatrogenic pneumothorax after thoracentesis occurs in 6% of cases.1 Iatrogenic reexpansion pulmonary edema after thoracentesis occurs in fewer than 1% of cases.2,3 Simultaneous pneumothorax and reexpansion pulmonary edema arising from the same procedure appears to be extremely rare.
A 51-year-old man with end-stage liver disease from alcohol abuse presented with worsening dyspnea on exertion. He had a history of ascites requiring diuretic therapy and intermittent paracentesis, as well as symptomatic hepatic hydrothorax requiring thoracentesis. Chest radiography showed a large right hydrothorax (Figure 1).
The patient underwent high-volume thoracentesis, and 3.2 L of clear fluid was removed. Chest radiography after the procedure revealed a right-sided pneumothorax (Figure 2, arrow). The patient was mildly short of breath and was treated with high-flow oxygen. Later the same day, his shortness of breath worsened, and repeat chest radiography showed an unchanged pneumothorax that was now complicated by reexpansion pulmonary edema after thoracentesis (Figure 3, star). The reexpansion pulmonary edema resolved by the following day, and the pneumothorax resolved after placement of a pig-tail catheter into the pleural space (Figure 4).
Iatrogenic pneumothorax after thoracentesis occurs in 6% of cases.1 Iatrogenic reexpansion pulmonary edema after thoracentesis occurs in fewer than 1% of cases.2,3 Simultaneous pneumothorax and reexpansion pulmonary edema arising from the same procedure appears to be extremely rare.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Ragozzino MW, Greene R. Bilateral reexpansion pulmonary edema following unilateral pleurocentesis. Chest 1991; 99:506–508.
- Dias OM, Teixeira LR, Vargas FS. Reexpansion pulmonary edema after therapeutic thoracentesis. Clinics (Sao Paulo) 2010; 65:1387–1389.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Ragozzino MW, Greene R. Bilateral reexpansion pulmonary edema following unilateral pleurocentesis. Chest 1991; 99:506–508.
- Dias OM, Teixeira LR, Vargas FS. Reexpansion pulmonary edema after therapeutic thoracentesis. Clinics (Sao Paulo) 2010; 65:1387–1389.
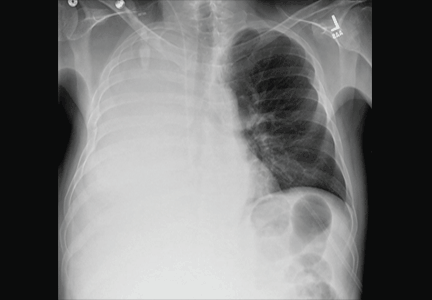
Encephalopathy despite thiamine repletion during alcohol withdrawal
Managing a patient with chronic alcohol abuse who is beginning to withdraw is a situation in which to expect the unexpected.
A 61-year-old man with a 40-year history of alcohol abuse was admitted to the hospital after presenting with a 6-month history of weakness and increasing frequency of falls, as well as a 2-day history of myalgias and fatigue. He had anorexia and chronic diarrhea, and he reported an unintentional 100-lb weight loss over the past year. He consumed at least three to five alcoholic drinks daily, including a daily “eye-opener,” and his diet was essentially devoid of meat, bread, fruits, and vegetables. His last drink had been on the morning of admission.
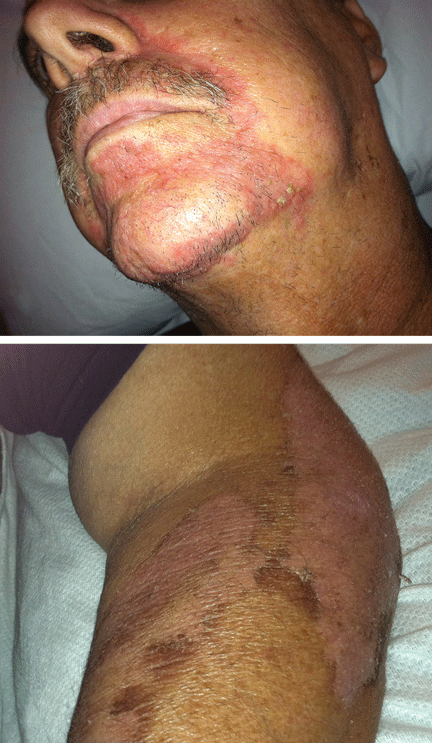
On physical examination, his vital signs were notable for marked orthostatic hypotension. He had angular cheilitis, an erythematous, scaly facial rash, and poorly healing severe sunburn on the dorsal surface of the left forearm, acquired 1 month earlier after minimal sun exposure while driving his car (Figure 1).
The neurologic examination noted decreased sensation in a stocking distribution, reduced proprioception in both great toes, and an intention tremor. His upper extremities were slightly hyperreflexic, he had down-going toes, he did not have clonus, and his gait was wide-based.
Laboratory results were notable for mild anemia, with an elevated mean corpuscular volume of 109.2 fL and a normal thyrotropin level. Urinalysis and urine culture were normal. Stool testing for Clostridium difficile toxin was negative. Computed tomography of the head was unremarkable.
He was placed on the Clinical Institute Withdrawal Assessment for Alcohol revised protocol,1 and he was started on careful fluid, electrolyte, and nutritional management, including intravenous supplementation with thiamine 100 mg daily, folic acid 1 mg twice daily, and an oral multivitamin. Despite these interventions, he became delirious and developed horizontal nystagmus, visual hallucinations, dizziness, and increasing weakness. Benzodiazepine toxicity, which may cause similar findings, was thought unlikely, as he had received only a maximum of 4 mg of lorazepam in any single 24-hour period. His ongoing confusion, orthostatic hypotension, ataxia, and nystagmus raised concern for Wernicke encephalopathy. Thiamine was increased to 500 mg intravenously three times daily for 2 days, followed by 500 mg daily for 2 days, as recommended for acute Wernicke encephalopathy by the Royal College of Physicians.2
Although his ocular findings resolved and his blood pressure improved with high-dose thiamine, he remained confused. Other causes of delirium were considered, including other micronutrient deficiencies. He was evaluated for magnesium deficiency, as magnesium is a necessary cofactor in thiamine metabolism,3 but his level was within normal limits. The constellation of symptoms, including persistent delirium, diarrhea, and skin findings, appeared to be consistent with pellagra, and niacin deficiency became a concern.
PELLAGRA AND ALCOHOL ABUSE
Niacin is a water-soluble vitamin converted to two important coenzymes involved in carbohydrate metabolism and fatty-acid synthesis.4 Body stores are minimal, although the liver can convert tryptophan to niacin.4
In the United States, pellagra has become rare since niacin enrichment of bread and flour began in the 1940s. However, the risk is increased in those with chronic alcohol dependency, metabolic disease such as carcinoid syndrome, and malabsorptive states such as Crohn disease.5 Alcohol abuse is commonly associated with micronutrient deficiencies. Even in alcoholics with sufficient food intake, their ability to absorb niacin may be impaired both directly and indirectly because of the toxic effects of alcohol on the gastrointestinal tract.5
The three D’s of niacin deficiency
The clinical picture of pellagra includes the three “D’s” of niacin deficiency—diarrhea, dementia, and dermatitis—representing the organ systems most commonly affected (ie, the gastrointestinal tract, nervous system, and skin). Gastrointestinal signs and symptoms may include anorexia, diarrhea, stomatitis, and abdominal discomfort.4 Neurologic signs and symptoms are the most frequent clinical manifestations of pellagra but are often overlooked in alcoholics with altered mental status.5,6 Early neurologic findings may include apathy, depression, paresthesias, dizziness, and falling.4,5 Later findings may include hallucinations, seizures, and psychosis.4,5 If severe enough, this may progress to coma and to a fourth “D”—death—if left untreated.
Skin findings, if present, are typically the most characteristic of pellagra.5,6 They include a photosensitive eruption that most commonly appears on the face, arms, and the “V” of the neck (“Casal’s necklace”).3,6 Erythema develops on areas of sun-exposed skin and may be painful or pruritic.6 It may take up to four times as long to resolve as a normal sunburn.6 On repeated exposure, the skin becomes thickened, hyperkeratotic, and hyperpigmented.6
DIAGNOSIS AND TREATMENT OF PELLAGRA
Suspicion of pellagra should be high in any patient with chronic alcohol abuse with changes in mental status.5 Pellagra is typically diagnosed clinically. Although testing of serum levels of niacin or urine levels of niacin metabolites can be performed,4,5 these tests are not commonly used, as they are neither widely available nor reliable.3,4 Because there is little downside to giving niacin, treatment should be started if there is clinical suspicion of pellagra.3,4 The diagnosis is confirmed by clinical improvement after niacin replacement begins.4
Pellagra is easy and inexpensive to treat once the diagnosis has been established. Supplementation can be given orally at 50 mg to 100 mg three times daily, with a maximum of 500 mg daily for 3 to 4 weeks.3,5 The multivitamin our patient initially received provided only 30 mg of niacin, which was insufficient to treat his deficiency.
Other micronutrient deficiencies are also common in pellagra, such as the suspected thiamine deficiency in this patient. Although rare in adults, pyridoxine deficiency should be considered because pyridoxine plays an important role in niacin metabolism, and in patients such as this, the signs and symptoms of pellagra will not resolve with niacin administration alone.3,4
Neurologic symptoms can begin to resolve as soon as 24 to 48 hours after replacement therapy is started, although skin symptoms take longer to respond.6
OUR PATIENT’S MANAGEMENT AND LESSONS LEARNED
Our patient was treated with extended-release niacin 500 mg daily. He was significantly less confused within 24 hours, and 2 days later he was no longer delirious. His diarrhea resolved, but his peripheral neuropathy improved only modestly, and we believe that this was the result of his long history of alcohol abuse.
Although there are guidelines to help with the management of patients undergoing alcohol withdrawal, our experience with this patient illustrates that important clinical conditions, including micronutrient deficiencies, can easily be overlooked or misattributed, with potentially dangerous consequences. It is crucial to be mindful of these considerations.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Thomson AD, Cook CC, Touquet R, Henry JA; Royal College of Physicians, London. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and Emergency Department. Alcohol Alcohol 2002; 37:513–521.
- Weathers AL, Lewis SL. Rare and unusual … or are they? Less commonly diagnosed encephalopathies associated with systemic disease. Semin Neurol 2009; 29:136–153.
- World Health Organization, United Nations High Commissions for Refugees. Pellagra and its prevention and control in major emergencies. 2000. http://www.who.int/nutrition/publications/emergencies/WHO_NHD_00.10/en/index.html. Accessed April 26, 2014.
- Oldham MA, Ivkovic A. Pellagrous encephalopathy presenting as alcohol withdrawal delirium: a case series and literature review. Addict Sci Clin Pract 2012; 7:12.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol 2002; 41:476–481.
Managing a patient with chronic alcohol abuse who is beginning to withdraw is a situation in which to expect the unexpected.
A 61-year-old man with a 40-year history of alcohol abuse was admitted to the hospital after presenting with a 6-month history of weakness and increasing frequency of falls, as well as a 2-day history of myalgias and fatigue. He had anorexia and chronic diarrhea, and he reported an unintentional 100-lb weight loss over the past year. He consumed at least three to five alcoholic drinks daily, including a daily “eye-opener,” and his diet was essentially devoid of meat, bread, fruits, and vegetables. His last drink had been on the morning of admission.
On physical examination, his vital signs were notable for marked orthostatic hypotension. He had angular cheilitis, an erythematous, scaly facial rash, and poorly healing severe sunburn on the dorsal surface of the left forearm, acquired 1 month earlier after minimal sun exposure while driving his car (Figure 1).
The neurologic examination noted decreased sensation in a stocking distribution, reduced proprioception in both great toes, and an intention tremor. His upper extremities were slightly hyperreflexic, he had down-going toes, he did not have clonus, and his gait was wide-based.
Laboratory results were notable for mild anemia, with an elevated mean corpuscular volume of 109.2 fL and a normal thyrotropin level. Urinalysis and urine culture were normal. Stool testing for Clostridium difficile toxin was negative. Computed tomography of the head was unremarkable.
He was placed on the Clinical Institute Withdrawal Assessment for Alcohol revised protocol,1 and he was started on careful fluid, electrolyte, and nutritional management, including intravenous supplementation with thiamine 100 mg daily, folic acid 1 mg twice daily, and an oral multivitamin. Despite these interventions, he became delirious and developed horizontal nystagmus, visual hallucinations, dizziness, and increasing weakness. Benzodiazepine toxicity, which may cause similar findings, was thought unlikely, as he had received only a maximum of 4 mg of lorazepam in any single 24-hour period. His ongoing confusion, orthostatic hypotension, ataxia, and nystagmus raised concern for Wernicke encephalopathy. Thiamine was increased to 500 mg intravenously three times daily for 2 days, followed by 500 mg daily for 2 days, as recommended for acute Wernicke encephalopathy by the Royal College of Physicians.2
Although his ocular findings resolved and his blood pressure improved with high-dose thiamine, he remained confused. Other causes of delirium were considered, including other micronutrient deficiencies. He was evaluated for magnesium deficiency, as magnesium is a necessary cofactor in thiamine metabolism,3 but his level was within normal limits. The constellation of symptoms, including persistent delirium, diarrhea, and skin findings, appeared to be consistent with pellagra, and niacin deficiency became a concern.
PELLAGRA AND ALCOHOL ABUSE
Niacin is a water-soluble vitamin converted to two important coenzymes involved in carbohydrate metabolism and fatty-acid synthesis.4 Body stores are minimal, although the liver can convert tryptophan to niacin.4
In the United States, pellagra has become rare since niacin enrichment of bread and flour began in the 1940s. However, the risk is increased in those with chronic alcohol dependency, metabolic disease such as carcinoid syndrome, and malabsorptive states such as Crohn disease.5 Alcohol abuse is commonly associated with micronutrient deficiencies. Even in alcoholics with sufficient food intake, their ability to absorb niacin may be impaired both directly and indirectly because of the toxic effects of alcohol on the gastrointestinal tract.5
The three D’s of niacin deficiency
The clinical picture of pellagra includes the three “D’s” of niacin deficiency—diarrhea, dementia, and dermatitis—representing the organ systems most commonly affected (ie, the gastrointestinal tract, nervous system, and skin). Gastrointestinal signs and symptoms may include anorexia, diarrhea, stomatitis, and abdominal discomfort.4 Neurologic signs and symptoms are the most frequent clinical manifestations of pellagra but are often overlooked in alcoholics with altered mental status.5,6 Early neurologic findings may include apathy, depression, paresthesias, dizziness, and falling.4,5 Later findings may include hallucinations, seizures, and psychosis.4,5 If severe enough, this may progress to coma and to a fourth “D”—death—if left untreated.
Skin findings, if present, are typically the most characteristic of pellagra.5,6 They include a photosensitive eruption that most commonly appears on the face, arms, and the “V” of the neck (“Casal’s necklace”).3,6 Erythema develops on areas of sun-exposed skin and may be painful or pruritic.6 It may take up to four times as long to resolve as a normal sunburn.6 On repeated exposure, the skin becomes thickened, hyperkeratotic, and hyperpigmented.6
DIAGNOSIS AND TREATMENT OF PELLAGRA
Suspicion of pellagra should be high in any patient with chronic alcohol abuse with changes in mental status.5 Pellagra is typically diagnosed clinically. Although testing of serum levels of niacin or urine levels of niacin metabolites can be performed,4,5 these tests are not commonly used, as they are neither widely available nor reliable.3,4 Because there is little downside to giving niacin, treatment should be started if there is clinical suspicion of pellagra.3,4 The diagnosis is confirmed by clinical improvement after niacin replacement begins.4
Pellagra is easy and inexpensive to treat once the diagnosis has been established. Supplementation can be given orally at 50 mg to 100 mg three times daily, with a maximum of 500 mg daily for 3 to 4 weeks.3,5 The multivitamin our patient initially received provided only 30 mg of niacin, which was insufficient to treat his deficiency.
Other micronutrient deficiencies are also common in pellagra, such as the suspected thiamine deficiency in this patient. Although rare in adults, pyridoxine deficiency should be considered because pyridoxine plays an important role in niacin metabolism, and in patients such as this, the signs and symptoms of pellagra will not resolve with niacin administration alone.3,4
Neurologic symptoms can begin to resolve as soon as 24 to 48 hours after replacement therapy is started, although skin symptoms take longer to respond.6
OUR PATIENT’S MANAGEMENT AND LESSONS LEARNED
Our patient was treated with extended-release niacin 500 mg daily. He was significantly less confused within 24 hours, and 2 days later he was no longer delirious. His diarrhea resolved, but his peripheral neuropathy improved only modestly, and we believe that this was the result of his long history of alcohol abuse.
Although there are guidelines to help with the management of patients undergoing alcohol withdrawal, our experience with this patient illustrates that important clinical conditions, including micronutrient deficiencies, can easily be overlooked or misattributed, with potentially dangerous consequences. It is crucial to be mindful of these considerations.
Managing a patient with chronic alcohol abuse who is beginning to withdraw is a situation in which to expect the unexpected.
A 61-year-old man with a 40-year history of alcohol abuse was admitted to the hospital after presenting with a 6-month history of weakness and increasing frequency of falls, as well as a 2-day history of myalgias and fatigue. He had anorexia and chronic diarrhea, and he reported an unintentional 100-lb weight loss over the past year. He consumed at least three to five alcoholic drinks daily, including a daily “eye-opener,” and his diet was essentially devoid of meat, bread, fruits, and vegetables. His last drink had been on the morning of admission.
On physical examination, his vital signs were notable for marked orthostatic hypotension. He had angular cheilitis, an erythematous, scaly facial rash, and poorly healing severe sunburn on the dorsal surface of the left forearm, acquired 1 month earlier after minimal sun exposure while driving his car (Figure 1).
The neurologic examination noted decreased sensation in a stocking distribution, reduced proprioception in both great toes, and an intention tremor. His upper extremities were slightly hyperreflexic, he had down-going toes, he did not have clonus, and his gait was wide-based.
Laboratory results were notable for mild anemia, with an elevated mean corpuscular volume of 109.2 fL and a normal thyrotropin level. Urinalysis and urine culture were normal. Stool testing for Clostridium difficile toxin was negative. Computed tomography of the head was unremarkable.
He was placed on the Clinical Institute Withdrawal Assessment for Alcohol revised protocol,1 and he was started on careful fluid, electrolyte, and nutritional management, including intravenous supplementation with thiamine 100 mg daily, folic acid 1 mg twice daily, and an oral multivitamin. Despite these interventions, he became delirious and developed horizontal nystagmus, visual hallucinations, dizziness, and increasing weakness. Benzodiazepine toxicity, which may cause similar findings, was thought unlikely, as he had received only a maximum of 4 mg of lorazepam in any single 24-hour period. His ongoing confusion, orthostatic hypotension, ataxia, and nystagmus raised concern for Wernicke encephalopathy. Thiamine was increased to 500 mg intravenously three times daily for 2 days, followed by 500 mg daily for 2 days, as recommended for acute Wernicke encephalopathy by the Royal College of Physicians.2
Although his ocular findings resolved and his blood pressure improved with high-dose thiamine, he remained confused. Other causes of delirium were considered, including other micronutrient deficiencies. He was evaluated for magnesium deficiency, as magnesium is a necessary cofactor in thiamine metabolism,3 but his level was within normal limits. The constellation of symptoms, including persistent delirium, diarrhea, and skin findings, appeared to be consistent with pellagra, and niacin deficiency became a concern.
PELLAGRA AND ALCOHOL ABUSE
Niacin is a water-soluble vitamin converted to two important coenzymes involved in carbohydrate metabolism and fatty-acid synthesis.4 Body stores are minimal, although the liver can convert tryptophan to niacin.4
In the United States, pellagra has become rare since niacin enrichment of bread and flour began in the 1940s. However, the risk is increased in those with chronic alcohol dependency, metabolic disease such as carcinoid syndrome, and malabsorptive states such as Crohn disease.5 Alcohol abuse is commonly associated with micronutrient deficiencies. Even in alcoholics with sufficient food intake, their ability to absorb niacin may be impaired both directly and indirectly because of the toxic effects of alcohol on the gastrointestinal tract.5
The three D’s of niacin deficiency
The clinical picture of pellagra includes the three “D’s” of niacin deficiency—diarrhea, dementia, and dermatitis—representing the organ systems most commonly affected (ie, the gastrointestinal tract, nervous system, and skin). Gastrointestinal signs and symptoms may include anorexia, diarrhea, stomatitis, and abdominal discomfort.4 Neurologic signs and symptoms are the most frequent clinical manifestations of pellagra but are often overlooked in alcoholics with altered mental status.5,6 Early neurologic findings may include apathy, depression, paresthesias, dizziness, and falling.4,5 Later findings may include hallucinations, seizures, and psychosis.4,5 If severe enough, this may progress to coma and to a fourth “D”—death—if left untreated.
Skin findings, if present, are typically the most characteristic of pellagra.5,6 They include a photosensitive eruption that most commonly appears on the face, arms, and the “V” of the neck (“Casal’s necklace”).3,6 Erythema develops on areas of sun-exposed skin and may be painful or pruritic.6 It may take up to four times as long to resolve as a normal sunburn.6 On repeated exposure, the skin becomes thickened, hyperkeratotic, and hyperpigmented.6
DIAGNOSIS AND TREATMENT OF PELLAGRA
Suspicion of pellagra should be high in any patient with chronic alcohol abuse with changes in mental status.5 Pellagra is typically diagnosed clinically. Although testing of serum levels of niacin or urine levels of niacin metabolites can be performed,4,5 these tests are not commonly used, as they are neither widely available nor reliable.3,4 Because there is little downside to giving niacin, treatment should be started if there is clinical suspicion of pellagra.3,4 The diagnosis is confirmed by clinical improvement after niacin replacement begins.4
Pellagra is easy and inexpensive to treat once the diagnosis has been established. Supplementation can be given orally at 50 mg to 100 mg three times daily, with a maximum of 500 mg daily for 3 to 4 weeks.3,5 The multivitamin our patient initially received provided only 30 mg of niacin, which was insufficient to treat his deficiency.
Other micronutrient deficiencies are also common in pellagra, such as the suspected thiamine deficiency in this patient. Although rare in adults, pyridoxine deficiency should be considered because pyridoxine plays an important role in niacin metabolism, and in patients such as this, the signs and symptoms of pellagra will not resolve with niacin administration alone.3,4
Neurologic symptoms can begin to resolve as soon as 24 to 48 hours after replacement therapy is started, although skin symptoms take longer to respond.6
OUR PATIENT’S MANAGEMENT AND LESSONS LEARNED
Our patient was treated with extended-release niacin 500 mg daily. He was significantly less confused within 24 hours, and 2 days later he was no longer delirious. His diarrhea resolved, but his peripheral neuropathy improved only modestly, and we believe that this was the result of his long history of alcohol abuse.
Although there are guidelines to help with the management of patients undergoing alcohol withdrawal, our experience with this patient illustrates that important clinical conditions, including micronutrient deficiencies, can easily be overlooked or misattributed, with potentially dangerous consequences. It is crucial to be mindful of these considerations.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Thomson AD, Cook CC, Touquet R, Henry JA; Royal College of Physicians, London. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and Emergency Department. Alcohol Alcohol 2002; 37:513–521.
- Weathers AL, Lewis SL. Rare and unusual … or are they? Less commonly diagnosed encephalopathies associated with systemic disease. Semin Neurol 2009; 29:136–153.
- World Health Organization, United Nations High Commissions for Refugees. Pellagra and its prevention and control in major emergencies. 2000. http://www.who.int/nutrition/publications/emergencies/WHO_NHD_00.10/en/index.html. Accessed April 26, 2014.
- Oldham MA, Ivkovic A. Pellagrous encephalopathy presenting as alcohol withdrawal delirium: a case series and literature review. Addict Sci Clin Pract 2012; 7:12.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol 2002; 41:476–481.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Thomson AD, Cook CC, Touquet R, Henry JA; Royal College of Physicians, London. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and Emergency Department. Alcohol Alcohol 2002; 37:513–521.
- Weathers AL, Lewis SL. Rare and unusual … or are they? Less commonly diagnosed encephalopathies associated with systemic disease. Semin Neurol 2009; 29:136–153.
- World Health Organization, United Nations High Commissions for Refugees. Pellagra and its prevention and control in major emergencies. 2000. http://www.who.int/nutrition/publications/emergencies/WHO_NHD_00.10/en/index.html. Accessed April 26, 2014.
- Oldham MA, Ivkovic A. Pellagrous encephalopathy presenting as alcohol withdrawal delirium: a case series and literature review. Addict Sci Clin Pract 2012; 7:12.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol 2002; 41:476–481.
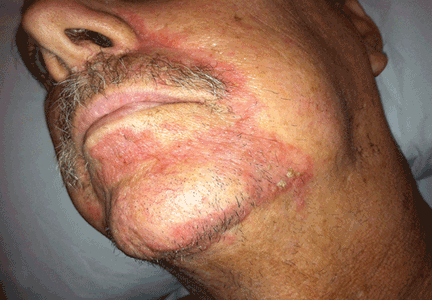
An 18-year-old woman with hepatic cysts
An 18-year-old woman presents with 3 days of epigastric abdominal pain, with no fever or constitutional symptoms. She was born in the United States and reports yearly trips since age 3 to her family’s farm in a rural area of Mexico, where she is exposed to dogs and horses.
Ultrasonography reveals two large hepatic cysts measuring 5.8 × 6.8 × 5.4 cm and 5.3 × 4.9 × 7 cm, with thickened walls and internal debris (Figure 1). The debris moves to dependent areas when the patient is asked to move onto her side.
Laboratory values at the time of presentation are as follows:
- White blood cell count 11.9 × 109/L (reference range 4.5–11.0), with 20% eosinophils
- Alkaline phosphatase 116 U/L (30–100)
- Total protein 7.3 g/dL (6.0–8.0)
- Albumin 4.3 g/dL (3.5–5.0)
- Aspartate aminotransferase (AST) 19 U/L (10–40)
- Alanine aminotransferase (ALT) 18 U/L (5–40)
- Total bilirubin 0.2 mg/dL (0.3–1.2)
- Direct bilirubin 0.1 mg/dL (0.1–0.3)
- Echinococcus antibody (IgG) testing is positive.
CYSTIC ECHINOCOCCOSIS
The two clinically relevant species of Echinococcus that cause human infection are E granulosus (in cystic echinococcosis) and E multilocularis (in alveolar echinococcosis). Based on clinical and radiographic findings, hepatic hydatid disease from cystic echinococcosis can usually be differentiated from the alveolar form.
E granulosus is a parasitic tapeworm that requires an intermediate host (sheep, goats, cows) and a definite host (dogs, foxes, and related species) for its life cycle. Humans become infected when they ingest food contaminated with feces that contain the eggs of the tapeworm or when they handle carnivorous animals, usually dogs, and accidentally ingest the tapeworm eggs. Once ingested, the egg releases an oncosphere that penetrates the intestinal wall, enters the circulation, and develops into a cyst, most often in the liver and the lungs.1 Human-to-human transmission does not occur.2
Hydatid cysts grow slowly, at a rate of 1 to 50 mm per year,3 so most patients remain asymptomatic for several years. Symptoms occur when a cyst ruptures or impinges on structures.3 Fever and constitutional symptoms usually occur only if there is rupture or bacterial superinfection of the cyst. Tests of liver function tend to be normal unless a cyst obstructs biliary flow. Eosinophilia occurs in 25% of patients.1 Eosinophilia along with the abrupt onset of abdominal pain suggests cyst rupture.
Making the diagnosis
Diagnosis is made by characteristic ultrasonographic findings and by serologic testing. Antibody assays for Echinococcus include indirect hemagglutination, enzyme-linked immunosorbent assay, and latex agglutination. However, these serologic antibody assays for immunoglobulin G cross-react to different echinococcal species as well as to other helminthic infections. Specific serologic studies such as an enzyme-linked immunosorbent assay for E multilocularis are available to confirm the species of Echinococcus but are only used to distinguish cystic echinococcosis from alveolar echinococcosis.
Treatment options
Treatment options include surgery, percutaneous procedures, drug therapy, and observation.
Currently, there is no clear consensus on treatment. To guide treatment decisions, the World Health Organization Informal Working Group on Echinococcosis (WHO-IWGE) recommends management of hepatic hydatid cysts based on classification, size, symptoms, location, and available resources.3
Two percutaneous treatments are aspiration, injection, and re-aspiration to destroy the germinal matrix, and percutaneous therapy to destroy the endocyst. Percutaneous aspiration, injection, and re-aspiration is increasingly used as the first-line treatment for single or easily accessible cysts and for patients who cannot undergo surgery. Surgery is considered for multiple cysts, large cysts, and cysts not easily accessible with a percutaneous technique.3 Complication rates and length of hospital stay with percutaneous aspiration are lower than with surgery.4 Observation is recommended for small, asymptomatic, inactive cysts.
Leakage of cyst contents during surgical or percutaneous intervention or spontaneous rupture can cause a recurrence,5 and anaphylaxis is a potential complication of cyst rupture.1 For this reason, giving oral albendazole (Albenza) is recommended before any intervention. Sterilization of the cyst contents with a protoscolicidal agent (20% NaCl) before evacuation of cyst contents is also standard practice.
The rate of cyst recurrence is 16.2% with open surgery and 3.5% with percutaneous intervention.6 A higher incidence of recurrence in patients who undergo surgical cystectomy likely reflects the more complicated and active nature of the cysts in patients who undergo surgery.6
Albendazole is the drug of choice for hepatic hydatid disease.3 The optimal duration of treatment is unclear but should be guided by a combination of clinical response, medication side effects, serologic titers, and imaging. The most common adverse effects of albendazole are hepatotoxicity, abdominal pain, and nausea.
OUR PATIENT’S DIAGNOSIS AND TREATMENT
In our patient, ultrasonography confirmed the diagnosis of cystic echinococcosis by the finding of active anechoic cysts with echogenic internal debris and with a well-delineated cyst wall. The WHO-IWGE classification was CE1, ie, active anechoic cysts with internal echogenic debris.
Our patient underwent surgical rather than percutaneous cystectomy because of concern about possible cyst leakage, since she had presented with the acute onset of pain and eosinophilia. We were also concerned about the subdiaphragmatic location of the larger cyst, which could have been difficult to reach percutaneously.
Open total pericystectomy involved opening the cyst cavity, sterilizing the contents with 20% NaCl, evacuating the cyst contents, and removing the cyst tissue. Two large cysts were excised and sent for histologic examination, which confirmed E granulosus. Percutaneous aspiration was necessary 4 months later because of a recurrence, and albendazole 400 mg twice daily was continued for another 5 months. Ultrasonography 3 years later showed no evidence of echinococcal cysts, and her antibody titers remain undetectable.
- McManus DP, Gray DJ, Zhang W, Yang Y. Diagnosis, treatment, and management of echinococcosis. BMJ 2012; 344:e3866.
- McManus DP, Zhang W, Li J, Bartley PB. Echinococcosis. Lancet 2003; 362:1295–1304.
- Brunetti E, Kern P, Vuitton DA; Writing Panel for the WHO-IWGE. Expert consensus for the diagnosis and treatment of cystic and alveolar echinococcosis in humans. Acta Trop 2010; 114:1–16.
- Khuroo MS, Wani NA, Javid G, et al. Percutaneous drainage compared with surgery for hepatic hydatid cysts. N Engl J Med 1997; 337:881–887.
- Kayaalp C, Sengul N, Akoglu M. Importance of cyst content in hydatid liver surgery. Arch Surg 2002; 137:159–163.
- Yagci G, Ustunsoz B, Kaymakcioglu N, et al. Results of surgical, laparoscopic, and percutaneous treatment for hydatid disease of the liver: 10 years experience with 355 patients. World J Surg 2005; 29:1670–1679.
An 18-year-old woman presents with 3 days of epigastric abdominal pain, with no fever or constitutional symptoms. She was born in the United States and reports yearly trips since age 3 to her family’s farm in a rural area of Mexico, where she is exposed to dogs and horses.
Ultrasonography reveals two large hepatic cysts measuring 5.8 × 6.8 × 5.4 cm and 5.3 × 4.9 × 7 cm, with thickened walls and internal debris (Figure 1). The debris moves to dependent areas when the patient is asked to move onto her side.
Laboratory values at the time of presentation are as follows:
- White blood cell count 11.9 × 109/L (reference range 4.5–11.0), with 20% eosinophils
- Alkaline phosphatase 116 U/L (30–100)
- Total protein 7.3 g/dL (6.0–8.0)
- Albumin 4.3 g/dL (3.5–5.0)
- Aspartate aminotransferase (AST) 19 U/L (10–40)
- Alanine aminotransferase (ALT) 18 U/L (5–40)
- Total bilirubin 0.2 mg/dL (0.3–1.2)
- Direct bilirubin 0.1 mg/dL (0.1–0.3)
- Echinococcus antibody (IgG) testing is positive.
CYSTIC ECHINOCOCCOSIS
The two clinically relevant species of Echinococcus that cause human infection are E granulosus (in cystic echinococcosis) and E multilocularis (in alveolar echinococcosis). Based on clinical and radiographic findings, hepatic hydatid disease from cystic echinococcosis can usually be differentiated from the alveolar form.
E granulosus is a parasitic tapeworm that requires an intermediate host (sheep, goats, cows) and a definite host (dogs, foxes, and related species) for its life cycle. Humans become infected when they ingest food contaminated with feces that contain the eggs of the tapeworm or when they handle carnivorous animals, usually dogs, and accidentally ingest the tapeworm eggs. Once ingested, the egg releases an oncosphere that penetrates the intestinal wall, enters the circulation, and develops into a cyst, most often in the liver and the lungs.1 Human-to-human transmission does not occur.2
Hydatid cysts grow slowly, at a rate of 1 to 50 mm per year,3 so most patients remain asymptomatic for several years. Symptoms occur when a cyst ruptures or impinges on structures.3 Fever and constitutional symptoms usually occur only if there is rupture or bacterial superinfection of the cyst. Tests of liver function tend to be normal unless a cyst obstructs biliary flow. Eosinophilia occurs in 25% of patients.1 Eosinophilia along with the abrupt onset of abdominal pain suggests cyst rupture.
Making the diagnosis
Diagnosis is made by characteristic ultrasonographic findings and by serologic testing. Antibody assays for Echinococcus include indirect hemagglutination, enzyme-linked immunosorbent assay, and latex agglutination. However, these serologic antibody assays for immunoglobulin G cross-react to different echinococcal species as well as to other helminthic infections. Specific serologic studies such as an enzyme-linked immunosorbent assay for E multilocularis are available to confirm the species of Echinococcus but are only used to distinguish cystic echinococcosis from alveolar echinococcosis.
Treatment options
Treatment options include surgery, percutaneous procedures, drug therapy, and observation.
Currently, there is no clear consensus on treatment. To guide treatment decisions, the World Health Organization Informal Working Group on Echinococcosis (WHO-IWGE) recommends management of hepatic hydatid cysts based on classification, size, symptoms, location, and available resources.3
Two percutaneous treatments are aspiration, injection, and re-aspiration to destroy the germinal matrix, and percutaneous therapy to destroy the endocyst. Percutaneous aspiration, injection, and re-aspiration is increasingly used as the first-line treatment for single or easily accessible cysts and for patients who cannot undergo surgery. Surgery is considered for multiple cysts, large cysts, and cysts not easily accessible with a percutaneous technique.3 Complication rates and length of hospital stay with percutaneous aspiration are lower than with surgery.4 Observation is recommended for small, asymptomatic, inactive cysts.
Leakage of cyst contents during surgical or percutaneous intervention or spontaneous rupture can cause a recurrence,5 and anaphylaxis is a potential complication of cyst rupture.1 For this reason, giving oral albendazole (Albenza) is recommended before any intervention. Sterilization of the cyst contents with a protoscolicidal agent (20% NaCl) before evacuation of cyst contents is also standard practice.
The rate of cyst recurrence is 16.2% with open surgery and 3.5% with percutaneous intervention.6 A higher incidence of recurrence in patients who undergo surgical cystectomy likely reflects the more complicated and active nature of the cysts in patients who undergo surgery.6
Albendazole is the drug of choice for hepatic hydatid disease.3 The optimal duration of treatment is unclear but should be guided by a combination of clinical response, medication side effects, serologic titers, and imaging. The most common adverse effects of albendazole are hepatotoxicity, abdominal pain, and nausea.
OUR PATIENT’S DIAGNOSIS AND TREATMENT
In our patient, ultrasonography confirmed the diagnosis of cystic echinococcosis by the finding of active anechoic cysts with echogenic internal debris and with a well-delineated cyst wall. The WHO-IWGE classification was CE1, ie, active anechoic cysts with internal echogenic debris.
Our patient underwent surgical rather than percutaneous cystectomy because of concern about possible cyst leakage, since she had presented with the acute onset of pain and eosinophilia. We were also concerned about the subdiaphragmatic location of the larger cyst, which could have been difficult to reach percutaneously.
Open total pericystectomy involved opening the cyst cavity, sterilizing the contents with 20% NaCl, evacuating the cyst contents, and removing the cyst tissue. Two large cysts were excised and sent for histologic examination, which confirmed E granulosus. Percutaneous aspiration was necessary 4 months later because of a recurrence, and albendazole 400 mg twice daily was continued for another 5 months. Ultrasonography 3 years later showed no evidence of echinococcal cysts, and her antibody titers remain undetectable.
An 18-year-old woman presents with 3 days of epigastric abdominal pain, with no fever or constitutional symptoms. She was born in the United States and reports yearly trips since age 3 to her family’s farm in a rural area of Mexico, where she is exposed to dogs and horses.
Ultrasonography reveals two large hepatic cysts measuring 5.8 × 6.8 × 5.4 cm and 5.3 × 4.9 × 7 cm, with thickened walls and internal debris (Figure 1). The debris moves to dependent areas when the patient is asked to move onto her side.
Laboratory values at the time of presentation are as follows:
- White blood cell count 11.9 × 109/L (reference range 4.5–11.0), with 20% eosinophils
- Alkaline phosphatase 116 U/L (30–100)
- Total protein 7.3 g/dL (6.0–8.0)
- Albumin 4.3 g/dL (3.5–5.0)
- Aspartate aminotransferase (AST) 19 U/L (10–40)
- Alanine aminotransferase (ALT) 18 U/L (5–40)
- Total bilirubin 0.2 mg/dL (0.3–1.2)
- Direct bilirubin 0.1 mg/dL (0.1–0.3)
- Echinococcus antibody (IgG) testing is positive.
CYSTIC ECHINOCOCCOSIS
The two clinically relevant species of Echinococcus that cause human infection are E granulosus (in cystic echinococcosis) and E multilocularis (in alveolar echinococcosis). Based on clinical and radiographic findings, hepatic hydatid disease from cystic echinococcosis can usually be differentiated from the alveolar form.
E granulosus is a parasitic tapeworm that requires an intermediate host (sheep, goats, cows) and a definite host (dogs, foxes, and related species) for its life cycle. Humans become infected when they ingest food contaminated with feces that contain the eggs of the tapeworm or when they handle carnivorous animals, usually dogs, and accidentally ingest the tapeworm eggs. Once ingested, the egg releases an oncosphere that penetrates the intestinal wall, enters the circulation, and develops into a cyst, most often in the liver and the lungs.1 Human-to-human transmission does not occur.2
Hydatid cysts grow slowly, at a rate of 1 to 50 mm per year,3 so most patients remain asymptomatic for several years. Symptoms occur when a cyst ruptures or impinges on structures.3 Fever and constitutional symptoms usually occur only if there is rupture or bacterial superinfection of the cyst. Tests of liver function tend to be normal unless a cyst obstructs biliary flow. Eosinophilia occurs in 25% of patients.1 Eosinophilia along with the abrupt onset of abdominal pain suggests cyst rupture.
Making the diagnosis
Diagnosis is made by characteristic ultrasonographic findings and by serologic testing. Antibody assays for Echinococcus include indirect hemagglutination, enzyme-linked immunosorbent assay, and latex agglutination. However, these serologic antibody assays for immunoglobulin G cross-react to different echinococcal species as well as to other helminthic infections. Specific serologic studies such as an enzyme-linked immunosorbent assay for E multilocularis are available to confirm the species of Echinococcus but are only used to distinguish cystic echinococcosis from alveolar echinococcosis.
Treatment options
Treatment options include surgery, percutaneous procedures, drug therapy, and observation.
Currently, there is no clear consensus on treatment. To guide treatment decisions, the World Health Organization Informal Working Group on Echinococcosis (WHO-IWGE) recommends management of hepatic hydatid cysts based on classification, size, symptoms, location, and available resources.3
Two percutaneous treatments are aspiration, injection, and re-aspiration to destroy the germinal matrix, and percutaneous therapy to destroy the endocyst. Percutaneous aspiration, injection, and re-aspiration is increasingly used as the first-line treatment for single or easily accessible cysts and for patients who cannot undergo surgery. Surgery is considered for multiple cysts, large cysts, and cysts not easily accessible with a percutaneous technique.3 Complication rates and length of hospital stay with percutaneous aspiration are lower than with surgery.4 Observation is recommended for small, asymptomatic, inactive cysts.
Leakage of cyst contents during surgical or percutaneous intervention or spontaneous rupture can cause a recurrence,5 and anaphylaxis is a potential complication of cyst rupture.1 For this reason, giving oral albendazole (Albenza) is recommended before any intervention. Sterilization of the cyst contents with a protoscolicidal agent (20% NaCl) before evacuation of cyst contents is also standard practice.
The rate of cyst recurrence is 16.2% with open surgery and 3.5% with percutaneous intervention.6 A higher incidence of recurrence in patients who undergo surgical cystectomy likely reflects the more complicated and active nature of the cysts in patients who undergo surgery.6
Albendazole is the drug of choice for hepatic hydatid disease.3 The optimal duration of treatment is unclear but should be guided by a combination of clinical response, medication side effects, serologic titers, and imaging. The most common adverse effects of albendazole are hepatotoxicity, abdominal pain, and nausea.
OUR PATIENT’S DIAGNOSIS AND TREATMENT
In our patient, ultrasonography confirmed the diagnosis of cystic echinococcosis by the finding of active anechoic cysts with echogenic internal debris and with a well-delineated cyst wall. The WHO-IWGE classification was CE1, ie, active anechoic cysts with internal echogenic debris.
Our patient underwent surgical rather than percutaneous cystectomy because of concern about possible cyst leakage, since she had presented with the acute onset of pain and eosinophilia. We were also concerned about the subdiaphragmatic location of the larger cyst, which could have been difficult to reach percutaneously.
Open total pericystectomy involved opening the cyst cavity, sterilizing the contents with 20% NaCl, evacuating the cyst contents, and removing the cyst tissue. Two large cysts were excised and sent for histologic examination, which confirmed E granulosus. Percutaneous aspiration was necessary 4 months later because of a recurrence, and albendazole 400 mg twice daily was continued for another 5 months. Ultrasonography 3 years later showed no evidence of echinococcal cysts, and her antibody titers remain undetectable.
- McManus DP, Gray DJ, Zhang W, Yang Y. Diagnosis, treatment, and management of echinococcosis. BMJ 2012; 344:e3866.
- McManus DP, Zhang W, Li J, Bartley PB. Echinococcosis. Lancet 2003; 362:1295–1304.
- Brunetti E, Kern P, Vuitton DA; Writing Panel for the WHO-IWGE. Expert consensus for the diagnosis and treatment of cystic and alveolar echinococcosis in humans. Acta Trop 2010; 114:1–16.
- Khuroo MS, Wani NA, Javid G, et al. Percutaneous drainage compared with surgery for hepatic hydatid cysts. N Engl J Med 1997; 337:881–887.
- Kayaalp C, Sengul N, Akoglu M. Importance of cyst content in hydatid liver surgery. Arch Surg 2002; 137:159–163.
- Yagci G, Ustunsoz B, Kaymakcioglu N, et al. Results of surgical, laparoscopic, and percutaneous treatment for hydatid disease of the liver: 10 years experience with 355 patients. World J Surg 2005; 29:1670–1679.
- McManus DP, Gray DJ, Zhang W, Yang Y. Diagnosis, treatment, and management of echinococcosis. BMJ 2012; 344:e3866.
- McManus DP, Zhang W, Li J, Bartley PB. Echinococcosis. Lancet 2003; 362:1295–1304.
- Brunetti E, Kern P, Vuitton DA; Writing Panel for the WHO-IWGE. Expert consensus for the diagnosis and treatment of cystic and alveolar echinococcosis in humans. Acta Trop 2010; 114:1–16.
- Khuroo MS, Wani NA, Javid G, et al. Percutaneous drainage compared with surgery for hepatic hydatid cysts. N Engl J Med 1997; 337:881–887.
- Kayaalp C, Sengul N, Akoglu M. Importance of cyst content in hydatid liver surgery. Arch Surg 2002; 137:159–163.
- Yagci G, Ustunsoz B, Kaymakcioglu N, et al. Results of surgical, laparoscopic, and percutaneous treatment for hydatid disease of the liver: 10 years experience with 355 patients. World J Surg 2005; 29:1670–1679.