User login
American Geriatrics Society 2023 updated Beers Criteria highlights
Every 4 years, an interprofessional panel of experts from the American Geriatrics Society provides updated guidelines on safe prescribing of medications in older adults, known as the Beers Criteria. A 2023 update was released in May 2023 after panel review of more 1,500 clinical trials and research studies published since the last update.
Anticoagulants
Notable changes to the 2023 guidelines include updated recommendations for anticoagulation. Warfarin should be avoided as initial therapy for venous thromboembolism or nonvalvular atrial fibrillation unless there are contraindications to direct oral anticoagulants (DOACs) or other substantial barriers to use.

Rivaroxaban should also be avoided, and dabigatran used with caution in favor of apixaban, which is felt to have a better safety profile in older adults. Rivaroxaban may be considered if once daily dosing is deemed to be more clinically appropriate. Financial barriers regarding drug coverage and formulary options were acknowledged as a significant barrier to equitable access to preferred direct oral anticoagulants in older adults.
Diabetes medication
Regarding diabetes management, short-acting sulfonylureas should be avoided in addition to long-acting sulfonylureas, because of the increased risk of hypoglycemia, and cardiovascular and all-cause mortality in older adults. Sodium-glucose cotransporter 2 inhibitors as an entire class are recommended to be used with caution, as older adults are at higher risk of euglycemic ketoacidosis and urogenital infections, particularly in women in the first month of initiating treatment.
Like DOACs, the panel acknowledged that financial considerations may lead to limited options for oral diabetic treatment. In circumstances where a sulfonylurea is used, short-acting forms are preferred over long acting to reduce the risk of prolonged hypoglycemia.
Aspirin for primary prevention
Alongside the U.S. Preventive Services Task Force guideline update in 2022 regarding aspirin for primary prevention of cardiovascular disease and stroke, the Beer’s Criteria recommend against initiation of aspirin for primary prevention in older adults. Ticagrelor and prasugrel should be used with caution because of the increased risk of major bleeding in older adults over the age of 75, compared with clopidogrel. If prasugrel is used, a lower dose of 5 mg is recommended, in line with guidelines by the American College of Cardiology and American Heart Association.
Pain medication
For pain management, the Beer’s Criteria updated recommendations to avoid NSAIDs, particularly when used in combination with steroids or anticoagulants. The panel highlights that even short-term use of NSAIDs is high risk when used in combination with steroids or anticoagulants. If no other alternatives are possible, patients should be placed on a proton pump inhibitor or misoprostol while taking NSAIDs.
Baclofen should be avoided in older adults with renal insufficiency (estimated glomerular filtration rate < 60 mL/min per 1.73 m2) because of the increased risk of encephalopathy, and when used, should be given at the lowest effective dose with close monitoring for mental status changes.
Androgen and estrogen replacement therapy
For androgen replacement therapy, the panel notes that testosterone supplementation should be avoided because of cardiovascular risks unless there is confirmed hypogonadism. The panel revised their recommendation on the basis of emerging data that a history of prostate cancer is not an absolute contraindication for exogenous testosterone. A risk versus benefit discussion about exogenous testosterone should be had with a medical oncologist or urologist in those with a history of prostate cancer.
Regarding estrogen, systemic formulations should not be initiated in women over the age of 60 because of increased risk of cardiovascular events, venous thromboembolism, and dementia. In women with a history of breast cancer, vaginal estrogens are generally felt to be safe to use at low doses, such as less than 25 mcg twice weekly.
Dr. Wang is a geriatrician and general internist at Harborview Medical Center, Seattle.
Every 4 years, an interprofessional panel of experts from the American Geriatrics Society provides updated guidelines on safe prescribing of medications in older adults, known as the Beers Criteria. A 2023 update was released in May 2023 after panel review of more 1,500 clinical trials and research studies published since the last update.
Anticoagulants
Notable changes to the 2023 guidelines include updated recommendations for anticoagulation. Warfarin should be avoided as initial therapy for venous thromboembolism or nonvalvular atrial fibrillation unless there are contraindications to direct oral anticoagulants (DOACs) or other substantial barriers to use.
Rivaroxaban should also be avoided, and dabigatran used with caution in favor of apixaban, which is felt to have a better safety profile in older adults. Rivaroxaban may be considered if once daily dosing is deemed to be more clinically appropriate. Financial barriers regarding drug coverage and formulary options were acknowledged as a significant barrier to equitable access to preferred direct oral anticoagulants in older adults.
Diabetes medication
Regarding diabetes management, short-acting sulfonylureas should be avoided in addition to long-acting sulfonylureas, because of the increased risk of hypoglycemia, and cardiovascular and all-cause mortality in older adults. Sodium-glucose cotransporter 2 inhibitors as an entire class are recommended to be used with caution, as older adults are at higher risk of euglycemic ketoacidosis and urogenital infections, particularly in women in the first month of initiating treatment.
Like DOACs, the panel acknowledged that financial considerations may lead to limited options for oral diabetic treatment. In circumstances where a sulfonylurea is used, short-acting forms are preferred over long acting to reduce the risk of prolonged hypoglycemia.
Aspirin for primary prevention
Alongside the U.S. Preventive Services Task Force guideline update in 2022 regarding aspirin for primary prevention of cardiovascular disease and stroke, the Beer’s Criteria recommend against initiation of aspirin for primary prevention in older adults. Ticagrelor and prasugrel should be used with caution because of the increased risk of major bleeding in older adults over the age of 75, compared with clopidogrel. If prasugrel is used, a lower dose of 5 mg is recommended, in line with guidelines by the American College of Cardiology and American Heart Association.
Pain medication
For pain management, the Beer’s Criteria updated recommendations to avoid NSAIDs, particularly when used in combination with steroids or anticoagulants. The panel highlights that even short-term use of NSAIDs is high risk when used in combination with steroids or anticoagulants. If no other alternatives are possible, patients should be placed on a proton pump inhibitor or misoprostol while taking NSAIDs.
Baclofen should be avoided in older adults with renal insufficiency (estimated glomerular filtration rate < 60 mL/min per 1.73 m2) because of the increased risk of encephalopathy, and when used, should be given at the lowest effective dose with close monitoring for mental status changes.
Androgen and estrogen replacement therapy
For androgen replacement therapy, the panel notes that testosterone supplementation should be avoided because of cardiovascular risks unless there is confirmed hypogonadism. The panel revised their recommendation on the basis of emerging data that a history of prostate cancer is not an absolute contraindication for exogenous testosterone. A risk versus benefit discussion about exogenous testosterone should be had with a medical oncologist or urologist in those with a history of prostate cancer.
Regarding estrogen, systemic formulations should not be initiated in women over the age of 60 because of increased risk of cardiovascular events, venous thromboembolism, and dementia. In women with a history of breast cancer, vaginal estrogens are generally felt to be safe to use at low doses, such as less than 25 mcg twice weekly.
Dr. Wang is a geriatrician and general internist at Harborview Medical Center, Seattle.
Every 4 years, an interprofessional panel of experts from the American Geriatrics Society provides updated guidelines on safe prescribing of medications in older adults, known as the Beers Criteria. A 2023 update was released in May 2023 after panel review of more 1,500 clinical trials and research studies published since the last update.
Anticoagulants
Notable changes to the 2023 guidelines include updated recommendations for anticoagulation. Warfarin should be avoided as initial therapy for venous thromboembolism or nonvalvular atrial fibrillation unless there are contraindications to direct oral anticoagulants (DOACs) or other substantial barriers to use.
Rivaroxaban should also be avoided, and dabigatran used with caution in favor of apixaban, which is felt to have a better safety profile in older adults. Rivaroxaban may be considered if once daily dosing is deemed to be more clinically appropriate. Financial barriers regarding drug coverage and formulary options were acknowledged as a significant barrier to equitable access to preferred direct oral anticoagulants in older adults.
Diabetes medication
Regarding diabetes management, short-acting sulfonylureas should be avoided in addition to long-acting sulfonylureas, because of the increased risk of hypoglycemia, and cardiovascular and all-cause mortality in older adults. Sodium-glucose cotransporter 2 inhibitors as an entire class are recommended to be used with caution, as older adults are at higher risk of euglycemic ketoacidosis and urogenital infections, particularly in women in the first month of initiating treatment.
Like DOACs, the panel acknowledged that financial considerations may lead to limited options for oral diabetic treatment. In circumstances where a sulfonylurea is used, short-acting forms are preferred over long acting to reduce the risk of prolonged hypoglycemia.
Aspirin for primary prevention
Alongside the U.S. Preventive Services Task Force guideline update in 2022 regarding aspirin for primary prevention of cardiovascular disease and stroke, the Beer’s Criteria recommend against initiation of aspirin for primary prevention in older adults. Ticagrelor and prasugrel should be used with caution because of the increased risk of major bleeding in older adults over the age of 75, compared with clopidogrel. If prasugrel is used, a lower dose of 5 mg is recommended, in line with guidelines by the American College of Cardiology and American Heart Association.
Pain medication
For pain management, the Beer’s Criteria updated recommendations to avoid NSAIDs, particularly when used in combination with steroids or anticoagulants. The panel highlights that even short-term use of NSAIDs is high risk when used in combination with steroids or anticoagulants. If no other alternatives are possible, patients should be placed on a proton pump inhibitor or misoprostol while taking NSAIDs.
Baclofen should be avoided in older adults with renal insufficiency (estimated glomerular filtration rate < 60 mL/min per 1.73 m2) because of the increased risk of encephalopathy, and when used, should be given at the lowest effective dose with close monitoring for mental status changes.
Androgen and estrogen replacement therapy
For androgen replacement therapy, the panel notes that testosterone supplementation should be avoided because of cardiovascular risks unless there is confirmed hypogonadism. The panel revised their recommendation on the basis of emerging data that a history of prostate cancer is not an absolute contraindication for exogenous testosterone. A risk versus benefit discussion about exogenous testosterone should be had with a medical oncologist or urologist in those with a history of prostate cancer.
Regarding estrogen, systemic formulations should not be initiated in women over the age of 60 because of increased risk of cardiovascular events, venous thromboembolism, and dementia. In women with a history of breast cancer, vaginal estrogens are generally felt to be safe to use at low doses, such as less than 25 mcg twice weekly.
Dr. Wang is a geriatrician and general internist at Harborview Medical Center, Seattle.
Morning vs. afternoon exercise debate: A false dichotomy
Should we be exercising in the morning or afternoon? Before a meal or after a meal?
Popular media outlets, researchers, and clinicians seem to love these debates. I hate them. For me, it’s a false dichotomy. A false dichotomy is when people argue two sides as if only one option exists. A winner must be crowned, and a loser exists. But
Some but not all research suggests that morning fasted exercise may be the best time of day and condition to work out for weight control and training adaptations. Morning exercise may be a bit better for logistical reasons if you like to get up early. Some of us are indeed early chronotypes who rise early, get as much done as we can, including all our fitness and work-related activities, and then head to bed early (for me that is about 10 PM). Getting an early morning workout seems to fit with our schedules as morning larks.
But if you are a late-day chronotype, early exercise may not be in sync with your low morning energy levels or your preference for leisure-time activities later in the day. And lots of people with diabetes prefer to eat and then exercise. Late chronotypes are less physically active in general, compared with early chronotypes, and those who train in the morning tend to have better training adherence and expend more energy overall throughout the day. According to Dr. Normand Boulé from the University of Alberta, Edmonton, who presented on the topic of exercise time of day at the recent scientific sessions of the American Diabetes Association in San Diego, morning exercise in the fasted state tends to be associated with higher rates of fat oxidation, better weight control, and better skeletal muscle adaptations over time, compared with exercise performed later in the day. Dr Boulé also proposed that fasted exercise might be superior for training adaptations and long-term glycemia if you have type 2 diabetes.
But the argument for morning-only exercise falls short when we look specifically at postmeal glycemia, according to Dr. Jenna Gillen from the University of Toronto, who faced off against Dr. Boulé at a debate at the meeting and also publishes on the topic. She pointed out that mild to moderate intensity exercising done soon after meals typically results in fewer glucose spikes after meals in people with diabetes, and her argument is supported by at least one recent meta-analysis where postmeal walking was best for improving glycemia in those with prediabetes and type 2 diabetes.
The notion that postmeal or afternoon exercise is best for people with type 2 diabetes is also supported by a recent reexamination of the original Look AHEAD Trial of over 2,400 adults with type 2 diabetes, wherein the role of lifestyle intervention on cardiovascular outcomes was the original goal. In this recent secondary analysis of the Look AHEAD Trial, those most active in the afternoon (between 1:43 p.m. and 5:00 p.m.) had the greatest improvements in their overall glucose control after 1 year of the intensive lifestyle intervention, compared with exercise at other times of day. Afternoon exercisers were also more likely to have complete “remission” of their diabetes, as defined by no longer needing any glucose-lowering agents to control their glucose levels. But this was not a study that was designed for determining whether exercise time of day matters for glycemia because the participants were not randomly assigned to a set time of day for their activity, and glycemic control was not the primary endpoint (cardiovascular events were).
But hold on a minute. I said this was a false-dichotomy argument. It is. Just because it may or may not be “better” for your glucose to exercise in the morning vs. afternoon, if you have diabetes, it doesn’t mean you have to choose one or the other. You could choose neither (okay, that’s bad), both, or you could alternate between the two. For me this argument is like saying; “There only one time of day to save money”; “to tell a joke”; “to eat a meal” (okay, that’s another useless debate); or “do my laundry” (my mother once told me it’s technically cheaper after 6 p.m.!).
I live with diabetes, and I take insulin. I like how morning exercise in the form of a run with my dog wakes me up, sets me up for the day with positive thoughts, helps generate lots of creative ideas, and perhaps more importantly for me, it tends not to result in hypoglycemia because my insulin on board is lowest then.
Exercise later in the day is tricky when taking insulin because it tends to result in a higher insulin “potency effect” with prandial insulins. However, I still like midday activity and late-day exercise. For example, taking an activity break after lunch blunts the rise in my glucose and breaks up my prolonged sitting time in the office. After-dinner exercise allows me to spend a little more time with my wife, dog, or friends outdoors as the hot summer day begins to cool off. On Monday nights, I play basketball because that’s the only time we can book the gymnasium and that may not end until 9:45 p.m. (15 minutes before I want to go to bed; if you remember, I am a lark). That can result in two frustrating things related to my diabetes: It can cause an immediate rise in my glucose because of a competitive stress response and then a drop in my glucose overnight when I’m sleeping. But I still do it. I know that the training I’m doing at any point of the day will benefit me in lots of little ways, and I think we all need to take as many opportunities to be physically active as we possibly can. My kids and I coin this our daily “fitness opportunities,” and it does not matter to me if its morning, noon, or night!
It’s time to make the headlines and arguments stop. There is no wrong time of day to exercise. At least not in my opinion.
Dr. Riddle is a full professor in the school of kinesiology and health science at York University and senior scientist at LMC Diabetes & Endocrinology, both in Toronto. He has disclosed financial relationships with Dexcom, Eli Lilly, Indigo Diabetes, Insulet, Novo Nordisk, Sanofi, Supersapiens, and Zucara Therapeutics.
A version of this article first appeared on Medscape.com.
Should we be exercising in the morning or afternoon? Before a meal or after a meal?
Popular media outlets, researchers, and clinicians seem to love these debates. I hate them. For me, it’s a false dichotomy. A false dichotomy is when people argue two sides as if only one option exists. A winner must be crowned, and a loser exists. But
Some but not all research suggests that morning fasted exercise may be the best time of day and condition to work out for weight control and training adaptations. Morning exercise may be a bit better for logistical reasons if you like to get up early. Some of us are indeed early chronotypes who rise early, get as much done as we can, including all our fitness and work-related activities, and then head to bed early (for me that is about 10 PM). Getting an early morning workout seems to fit with our schedules as morning larks.
But if you are a late-day chronotype, early exercise may not be in sync with your low morning energy levels or your preference for leisure-time activities later in the day. And lots of people with diabetes prefer to eat and then exercise. Late chronotypes are less physically active in general, compared with early chronotypes, and those who train in the morning tend to have better training adherence and expend more energy overall throughout the day. According to Dr. Normand Boulé from the University of Alberta, Edmonton, who presented on the topic of exercise time of day at the recent scientific sessions of the American Diabetes Association in San Diego, morning exercise in the fasted state tends to be associated with higher rates of fat oxidation, better weight control, and better skeletal muscle adaptations over time, compared with exercise performed later in the day. Dr Boulé also proposed that fasted exercise might be superior for training adaptations and long-term glycemia if you have type 2 diabetes.
But the argument for morning-only exercise falls short when we look specifically at postmeal glycemia, according to Dr. Jenna Gillen from the University of Toronto, who faced off against Dr. Boulé at a debate at the meeting and also publishes on the topic. She pointed out that mild to moderate intensity exercising done soon after meals typically results in fewer glucose spikes after meals in people with diabetes, and her argument is supported by at least one recent meta-analysis where postmeal walking was best for improving glycemia in those with prediabetes and type 2 diabetes.
The notion that postmeal or afternoon exercise is best for people with type 2 diabetes is also supported by a recent reexamination of the original Look AHEAD Trial of over 2,400 adults with type 2 diabetes, wherein the role of lifestyle intervention on cardiovascular outcomes was the original goal. In this recent secondary analysis of the Look AHEAD Trial, those most active in the afternoon (between 1:43 p.m. and 5:00 p.m.) had the greatest improvements in their overall glucose control after 1 year of the intensive lifestyle intervention, compared with exercise at other times of day. Afternoon exercisers were also more likely to have complete “remission” of their diabetes, as defined by no longer needing any glucose-lowering agents to control their glucose levels. But this was not a study that was designed for determining whether exercise time of day matters for glycemia because the participants were not randomly assigned to a set time of day for their activity, and glycemic control was not the primary endpoint (cardiovascular events were).
But hold on a minute. I said this was a false-dichotomy argument. It is. Just because it may or may not be “better” for your glucose to exercise in the morning vs. afternoon, if you have diabetes, it doesn’t mean you have to choose one or the other. You could choose neither (okay, that’s bad), both, or you could alternate between the two. For me this argument is like saying; “There only one time of day to save money”; “to tell a joke”; “to eat a meal” (okay, that’s another useless debate); or “do my laundry” (my mother once told me it’s technically cheaper after 6 p.m.!).
I live with diabetes, and I take insulin. I like how morning exercise in the form of a run with my dog wakes me up, sets me up for the day with positive thoughts, helps generate lots of creative ideas, and perhaps more importantly for me, it tends not to result in hypoglycemia because my insulin on board is lowest then.
Exercise later in the day is tricky when taking insulin because it tends to result in a higher insulin “potency effect” with prandial insulins. However, I still like midday activity and late-day exercise. For example, taking an activity break after lunch blunts the rise in my glucose and breaks up my prolonged sitting time in the office. After-dinner exercise allows me to spend a little more time with my wife, dog, or friends outdoors as the hot summer day begins to cool off. On Monday nights, I play basketball because that’s the only time we can book the gymnasium and that may not end until 9:45 p.m. (15 minutes before I want to go to bed; if you remember, I am a lark). That can result in two frustrating things related to my diabetes: It can cause an immediate rise in my glucose because of a competitive stress response and then a drop in my glucose overnight when I’m sleeping. But I still do it. I know that the training I’m doing at any point of the day will benefit me in lots of little ways, and I think we all need to take as many opportunities to be physically active as we possibly can. My kids and I coin this our daily “fitness opportunities,” and it does not matter to me if its morning, noon, or night!
It’s time to make the headlines and arguments stop. There is no wrong time of day to exercise. At least not in my opinion.
Dr. Riddle is a full professor in the school of kinesiology and health science at York University and senior scientist at LMC Diabetes & Endocrinology, both in Toronto. He has disclosed financial relationships with Dexcom, Eli Lilly, Indigo Diabetes, Insulet, Novo Nordisk, Sanofi, Supersapiens, and Zucara Therapeutics.
A version of this article first appeared on Medscape.com.
Should we be exercising in the morning or afternoon? Before a meal or after a meal?
Popular media outlets, researchers, and clinicians seem to love these debates. I hate them. For me, it’s a false dichotomy. A false dichotomy is when people argue two sides as if only one option exists. A winner must be crowned, and a loser exists. But
Some but not all research suggests that morning fasted exercise may be the best time of day and condition to work out for weight control and training adaptations. Morning exercise may be a bit better for logistical reasons if you like to get up early. Some of us are indeed early chronotypes who rise early, get as much done as we can, including all our fitness and work-related activities, and then head to bed early (for me that is about 10 PM). Getting an early morning workout seems to fit with our schedules as morning larks.
But if you are a late-day chronotype, early exercise may not be in sync with your low morning energy levels or your preference for leisure-time activities later in the day. And lots of people with diabetes prefer to eat and then exercise. Late chronotypes are less physically active in general, compared with early chronotypes, and those who train in the morning tend to have better training adherence and expend more energy overall throughout the day. According to Dr. Normand Boulé from the University of Alberta, Edmonton, who presented on the topic of exercise time of day at the recent scientific sessions of the American Diabetes Association in San Diego, morning exercise in the fasted state tends to be associated with higher rates of fat oxidation, better weight control, and better skeletal muscle adaptations over time, compared with exercise performed later in the day. Dr Boulé also proposed that fasted exercise might be superior for training adaptations and long-term glycemia if you have type 2 diabetes.
But the argument for morning-only exercise falls short when we look specifically at postmeal glycemia, according to Dr. Jenna Gillen from the University of Toronto, who faced off against Dr. Boulé at a debate at the meeting and also publishes on the topic. She pointed out that mild to moderate intensity exercising done soon after meals typically results in fewer glucose spikes after meals in people with diabetes, and her argument is supported by at least one recent meta-analysis where postmeal walking was best for improving glycemia in those with prediabetes and type 2 diabetes.
The notion that postmeal or afternoon exercise is best for people with type 2 diabetes is also supported by a recent reexamination of the original Look AHEAD Trial of over 2,400 adults with type 2 diabetes, wherein the role of lifestyle intervention on cardiovascular outcomes was the original goal. In this recent secondary analysis of the Look AHEAD Trial, those most active in the afternoon (between 1:43 p.m. and 5:00 p.m.) had the greatest improvements in their overall glucose control after 1 year of the intensive lifestyle intervention, compared with exercise at other times of day. Afternoon exercisers were also more likely to have complete “remission” of their diabetes, as defined by no longer needing any glucose-lowering agents to control their glucose levels. But this was not a study that was designed for determining whether exercise time of day matters for glycemia because the participants were not randomly assigned to a set time of day for their activity, and glycemic control was not the primary endpoint (cardiovascular events were).
But hold on a minute. I said this was a false-dichotomy argument. It is. Just because it may or may not be “better” for your glucose to exercise in the morning vs. afternoon, if you have diabetes, it doesn’t mean you have to choose one or the other. You could choose neither (okay, that’s bad), both, or you could alternate between the two. For me this argument is like saying; “There only one time of day to save money”; “to tell a joke”; “to eat a meal” (okay, that’s another useless debate); or “do my laundry” (my mother once told me it’s technically cheaper after 6 p.m.!).
I live with diabetes, and I take insulin. I like how morning exercise in the form of a run with my dog wakes me up, sets me up for the day with positive thoughts, helps generate lots of creative ideas, and perhaps more importantly for me, it tends not to result in hypoglycemia because my insulin on board is lowest then.
Exercise later in the day is tricky when taking insulin because it tends to result in a higher insulin “potency effect” with prandial insulins. However, I still like midday activity and late-day exercise. For example, taking an activity break after lunch blunts the rise in my glucose and breaks up my prolonged sitting time in the office. After-dinner exercise allows me to spend a little more time with my wife, dog, or friends outdoors as the hot summer day begins to cool off. On Monday nights, I play basketball because that’s the only time we can book the gymnasium and that may not end until 9:45 p.m. (15 minutes before I want to go to bed; if you remember, I am a lark). That can result in two frustrating things related to my diabetes: It can cause an immediate rise in my glucose because of a competitive stress response and then a drop in my glucose overnight when I’m sleeping. But I still do it. I know that the training I’m doing at any point of the day will benefit me in lots of little ways, and I think we all need to take as many opportunities to be physically active as we possibly can. My kids and I coin this our daily “fitness opportunities,” and it does not matter to me if its morning, noon, or night!
It’s time to make the headlines and arguments stop. There is no wrong time of day to exercise. At least not in my opinion.
Dr. Riddle is a full professor in the school of kinesiology and health science at York University and senior scientist at LMC Diabetes & Endocrinology, both in Toronto. He has disclosed financial relationships with Dexcom, Eli Lilly, Indigo Diabetes, Insulet, Novo Nordisk, Sanofi, Supersapiens, and Zucara Therapeutics.
A version of this article first appeared on Medscape.com.
Can we be too efficient?
“We were all of us cogs in a great machine which sometimes rolled forward, nobody knew where, sometimes backwards, nobody knew why.” – Ernst Toller
A nice feature of the Apple watch is the stopwatch. With it, I can discreetly click the timer and watch seconds tick away. Tap. There’s one lap. Tap. Two. Tap. That was a quick visit, 6 minutes and 42 seconds. Tap. Under 2 minutes to close the chart. Let’s see if I can beat it. Tap. Tap. What if I moved my Mayo stand over to this side of the room? How about a sign, “All patients must have clothes off if you want a skin exam.” You think ob.gyns. are quick from skin to baby in a stat C-section? You should see how fast I can go from alcohol wipe to Drysol on a biopsy. Seconds. Tick, tick, tap.
Every day I look for ways to go faster. This is not so I can be out the door by 3. Rather, it’s simply to make it through the day without having to log on after we put the kids to bed at night.
Speaking of bedtimes, another nice feature of the Apple watch is the timer. With it, I can set a timer and a lovely chimey alarm will go off. This comes in handy with 3-year-olds. “Sloan, in two minutes we are going to brush your teeth.” Ding. “Sloan, you have one minute to get your pajamas on.” Ding. “Sloanie, I’ll give you 3 more minutes to put the kitties away, then get into bed.” Ding, ding, ding ...
As you can see, using the stopwatch to time a bedtime routine would be demoralizing. If you’ve tried to put a toddler to bed in summer you know. They explore every option to avoid sleeping: one more book (that would make 3), “accidentally” putting their pajamas on backwards, offering to brush their teeth a second time. And once the light is off, “Papa, I have to potty.” No, bedtime routines cannot be standardized. They resist being made efficient.
In contrast, , Frederick Taylor. Taylor, a mechanical engineer, observed inefficiencies on the factory floor. His work was seminal in the development of the second industrial revolution. Before then no one had applied scientific rigor to productivity. His book, “The Principles of Scientific Management,” written in 1909, is considered the most influential management book of the 20th century. He was the first to use stopwatches to perform time studies, noting how long each task took with the belief that there was one best way. The worker was an extension of the machine, tuned by management such that he was as efficient as possible.

Others built on this idea including Frank and Lillian Gilbreth who added video recording, creating time and motion studies to further drive efficiency. This technique is still used in manufacturing and service industries today, including health care. In the 1980s, W. Edwards Deming modernized this effort, empowering workers with techniques taken from Japanese manufacturing. This, too, has been widely adopted in health care and evolved into the Lean and Lean Six Sigma quality movements about a decade ago. The common theme is to reduce waste to make health care as efficient as possible. Lately, this idea seems to have failed us.
The difficulty lies in the belief that efficient is always better. I’m unsure. Efficiency helps to reduce costs. It can also improve access. Yet, it comes at a cost. Eliminating slack concomitantly eliminates resilience. As such, when unexpected and significant changes impact a system, the gears of productivity jam. It’s in part why we are seeing rising wait times and patient dissatisfaction post pandemic. There was no slack and our system was too brittle.

A more insidious downside on the drive to efficiency lies in the nature of what we do. We aren’t factory workers punching out widgets, we’re physicians caring for people and people cannot be standardized. In this way, seeing patients is more like putting a toddler to bed than like assembling an iPhone. There will always be by-the-ways, basal cells hiding behind the ear, traffic jams, and bags of products that they want to review. Not sure how to use your fluorouracil? Let’s go over it again. Need to talk more about why you have granuloma annulare? Let me explain. Despite Taylor’s vision, some work simply cannot be optimized. And shouldn’t.
“Where’s my 11:30 patient who checked in half an hour ago?!” I asked my medical assistant. “Oh, she had to go to the bathroom.” Tap.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected].
“We were all of us cogs in a great machine which sometimes rolled forward, nobody knew where, sometimes backwards, nobody knew why.” – Ernst Toller
A nice feature of the Apple watch is the stopwatch. With it, I can discreetly click the timer and watch seconds tick away. Tap. There’s one lap. Tap. Two. Tap. That was a quick visit, 6 minutes and 42 seconds. Tap. Under 2 minutes to close the chart. Let’s see if I can beat it. Tap. Tap. What if I moved my Mayo stand over to this side of the room? How about a sign, “All patients must have clothes off if you want a skin exam.” You think ob.gyns. are quick from skin to baby in a stat C-section? You should see how fast I can go from alcohol wipe to Drysol on a biopsy. Seconds. Tick, tick, tap.
Every day I look for ways to go faster. This is not so I can be out the door by 3. Rather, it’s simply to make it through the day without having to log on after we put the kids to bed at night.
Speaking of bedtimes, another nice feature of the Apple watch is the timer. With it, I can set a timer and a lovely chimey alarm will go off. This comes in handy with 3-year-olds. “Sloan, in two minutes we are going to brush your teeth.” Ding. “Sloan, you have one minute to get your pajamas on.” Ding. “Sloanie, I’ll give you 3 more minutes to put the kitties away, then get into bed.” Ding, ding, ding ...
As you can see, using the stopwatch to time a bedtime routine would be demoralizing. If you’ve tried to put a toddler to bed in summer you know. They explore every option to avoid sleeping: one more book (that would make 3), “accidentally” putting their pajamas on backwards, offering to brush their teeth a second time. And once the light is off, “Papa, I have to potty.” No, bedtime routines cannot be standardized. They resist being made efficient.
In contrast, , Frederick Taylor. Taylor, a mechanical engineer, observed inefficiencies on the factory floor. His work was seminal in the development of the second industrial revolution. Before then no one had applied scientific rigor to productivity. His book, “The Principles of Scientific Management,” written in 1909, is considered the most influential management book of the 20th century. He was the first to use stopwatches to perform time studies, noting how long each task took with the belief that there was one best way. The worker was an extension of the machine, tuned by management such that he was as efficient as possible.
Others built on this idea including Frank and Lillian Gilbreth who added video recording, creating time and motion studies to further drive efficiency. This technique is still used in manufacturing and service industries today, including health care. In the 1980s, W. Edwards Deming modernized this effort, empowering workers with techniques taken from Japanese manufacturing. This, too, has been widely adopted in health care and evolved into the Lean and Lean Six Sigma quality movements about a decade ago. The common theme is to reduce waste to make health care as efficient as possible. Lately, this idea seems to have failed us.
The difficulty lies in the belief that efficient is always better. I’m unsure. Efficiency helps to reduce costs. It can also improve access. Yet, it comes at a cost. Eliminating slack concomitantly eliminates resilience. As such, when unexpected and significant changes impact a system, the gears of productivity jam. It’s in part why we are seeing rising wait times and patient dissatisfaction post pandemic. There was no slack and our system was too brittle.
A more insidious downside on the drive to efficiency lies in the nature of what we do. We aren’t factory workers punching out widgets, we’re physicians caring for people and people cannot be standardized. In this way, seeing patients is more like putting a toddler to bed than like assembling an iPhone. There will always be by-the-ways, basal cells hiding behind the ear, traffic jams, and bags of products that they want to review. Not sure how to use your fluorouracil? Let’s go over it again. Need to talk more about why you have granuloma annulare? Let me explain. Despite Taylor’s vision, some work simply cannot be optimized. And shouldn’t.
“Where’s my 11:30 patient who checked in half an hour ago?!” I asked my medical assistant. “Oh, she had to go to the bathroom.” Tap.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected].
“We were all of us cogs in a great machine which sometimes rolled forward, nobody knew where, sometimes backwards, nobody knew why.” – Ernst Toller
A nice feature of the Apple watch is the stopwatch. With it, I can discreetly click the timer and watch seconds tick away. Tap. There’s one lap. Tap. Two. Tap. That was a quick visit, 6 minutes and 42 seconds. Tap. Under 2 minutes to close the chart. Let’s see if I can beat it. Tap. Tap. What if I moved my Mayo stand over to this side of the room? How about a sign, “All patients must have clothes off if you want a skin exam.” You think ob.gyns. are quick from skin to baby in a stat C-section? You should see how fast I can go from alcohol wipe to Drysol on a biopsy. Seconds. Tick, tick, tap.
Every day I look for ways to go faster. This is not so I can be out the door by 3. Rather, it’s simply to make it through the day without having to log on after we put the kids to bed at night.
Speaking of bedtimes, another nice feature of the Apple watch is the timer. With it, I can set a timer and a lovely chimey alarm will go off. This comes in handy with 3-year-olds. “Sloan, in two minutes we are going to brush your teeth.” Ding. “Sloan, you have one minute to get your pajamas on.” Ding. “Sloanie, I’ll give you 3 more minutes to put the kitties away, then get into bed.” Ding, ding, ding ...
As you can see, using the stopwatch to time a bedtime routine would be demoralizing. If you’ve tried to put a toddler to bed in summer you know. They explore every option to avoid sleeping: one more book (that would make 3), “accidentally” putting their pajamas on backwards, offering to brush their teeth a second time. And once the light is off, “Papa, I have to potty.” No, bedtime routines cannot be standardized. They resist being made efficient.
In contrast, , Frederick Taylor. Taylor, a mechanical engineer, observed inefficiencies on the factory floor. His work was seminal in the development of the second industrial revolution. Before then no one had applied scientific rigor to productivity. His book, “The Principles of Scientific Management,” written in 1909, is considered the most influential management book of the 20th century. He was the first to use stopwatches to perform time studies, noting how long each task took with the belief that there was one best way. The worker was an extension of the machine, tuned by management such that he was as efficient as possible.
Others built on this idea including Frank and Lillian Gilbreth who added video recording, creating time and motion studies to further drive efficiency. This technique is still used in manufacturing and service industries today, including health care. In the 1980s, W. Edwards Deming modernized this effort, empowering workers with techniques taken from Japanese manufacturing. This, too, has been widely adopted in health care and evolved into the Lean and Lean Six Sigma quality movements about a decade ago. The common theme is to reduce waste to make health care as efficient as possible. Lately, this idea seems to have failed us.
The difficulty lies in the belief that efficient is always better. I’m unsure. Efficiency helps to reduce costs. It can also improve access. Yet, it comes at a cost. Eliminating slack concomitantly eliminates resilience. As such, when unexpected and significant changes impact a system, the gears of productivity jam. It’s in part why we are seeing rising wait times and patient dissatisfaction post pandemic. There was no slack and our system was too brittle.
A more insidious downside on the drive to efficiency lies in the nature of what we do. We aren’t factory workers punching out widgets, we’re physicians caring for people and people cannot be standardized. In this way, seeing patients is more like putting a toddler to bed than like assembling an iPhone. There will always be by-the-ways, basal cells hiding behind the ear, traffic jams, and bags of products that they want to review. Not sure how to use your fluorouracil? Let’s go over it again. Need to talk more about why you have granuloma annulare? Let me explain. Despite Taylor’s vision, some work simply cannot be optimized. And shouldn’t.
“Where’s my 11:30 patient who checked in half an hour ago?!” I asked my medical assistant. “Oh, she had to go to the bathroom.” Tap.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected].
What can you do during a mass shooting? This MD found out
Sunday night. Las Vegas. Jason Aldean had just started playing.
My wife and I were at the 2017 Route 91 Harvest Festival with three other couples; two of them were our close friends. We were sitting in the VIP section, a tented area right next to the stage. We started hearing what I was convinced were fireworks.
I’ve been in the Army for 20 some years. I’ve been deployed and shot at multiple times. But these shots were far away. And you don’t expect people to be shooting at you at a concert.
I was on the edge of the VIP area, so I could see around the corner of the tent. I looked up at the Mandalay Bay and saw the muzzle flash in the hotel window. That’s when I knew.
I screamed: “Somebody’s shooting at us! Everybody get down!”
It took a while for people to realize what was going on. When the first couple volleys sprayed into the crowd, nobody understood. But once enough people had been hit and dropped, everyone knew, and it was just mass exodus.
People screamed and ran everywhere. Some of them tried to jump over the front barrier so they could get underneath the stage. Others were trying to pick up loved ones who’d been shot.
The next 15 minutes are a little foggy. I was helping my wife and the people around us to get down. Funny things come back to you afterward. One of my friends was carrying a 16-ounce beer in his hand. Somebody’s shooting at him and he’s walking around with his beer like he’s afraid to put it down. It was so surreal.
We got everybody underneath the tent, and then we just sat there. There would be shooting and then a pause. You’d think it was over. And then there would be more shooting and another pause. It felt like it never was going to stop.
After a short period of time, somebody came in with an official badge, maybe FBI, who knows. They said: “Okay, everybody up. We’ve got to get you out of here.” So, we all got up and headed across the stage. The gate they were taking us to was in full view of the shooter, so it wasn’t very safe.
As I got up, I looked out at the field. Bodies were scattered everywhere. I’m a trauma surgeon by trade. I couldn’t just leave.
I told my two best friends to take my wife with them. My wife lost her mind at that point. She didn’t want me to run out on the field. But I had to. I saw the injured and they needed help. Another buddy and I jumped over the fence and started taking care of people.
The feeling of being out on the field was one of complete frustration. I was in sandals, shorts, and a t-shirt. We had no stretchers, no medical supplies, no nothing. I didn’t have a belt to use as a tourniquet. I didn’t even have a bandage.
Worse: We were seeing high-velocity gunshot wounds that I’ve seen for 20 years in the Army. I know how to take care of them. I know how to fix them. But there wasn’t a single thing I could do.
We had to get people off the field, so we started gathering up as many as we could. We didn’t know if we were going to get shot at again, so we were trying to hide behind things as we ran. Our main objective was just to get people to a place of safety.
A lot of it is a blur. But a few patients stick out in my mind.
A father and son. The father had been shot through the abdomen, exited out through his back. He was in severe pain and couldn’t walk.
A young girl shot in the arm. Her parents carrying her.
A group of people doing CPR on a young lady. She had a gunshot wound to the head or neck. She was obviously dead. But they were still doing chest compressions in the middle of the field. I had to say to them: “She’s dead. You can’t save her. You need to get off the field.” But they wouldn’t stop. We picked her up and took her out while they continued to do CPR.
Later, I realized I knew that woman. She was part of a group of friends that we would see at the festival. I hadn’t recognized her. I also didn’t know that my friend Marco was there. A month or 2 later, we figured out that he was one of the people doing CPR. And I was the guy who came up and said his friend was dead.
Some people were so badly injured we couldn’t lift them. We started tearing apart the fencing used to separate the crowd and slid sections of the barricades under the wounded to carry them. We also carried off a bunch of people who were dead.
We were moving patients to a covered bar area where we thought they would be safer. What we didn’t know was there was an ambulance rally point at the very far end of the field. Unfortunately, we had no idea it was there.
I saw a lot of other first responders out there, people from the fire department, corpsmen from the Navy, medics. I ran into an anesthesia provider and a series of nurses.
When we got everybody off the field, we started moving them into vehicles. People were bringing their trucks up. One guy even stole a truck so he could drive people to the ED. There wasn’t a lot of triage. We were just stacking whoever we could into the backs of these pickups.
I tried to help a nurse taking care of a lady who had been shot in the neck. She was sitting sort of half upright with the patient lying in her arms. When I reached to help her, she said: “You can’t move her.”
“We need to get her to the hospital,” I replied.
“This is the only position that this lady has an airway,” she said. “You’re going to have to move both of us together. If I move at all, she loses her airway.”
So, a group of us managed to slide something underneath and lift them into the back of a truck.
Loading the wounded went on for a while. And then, just like that, everybody was gone.
I walked back out onto this field which not too long ago held 30,000 people. It was as if aliens had just suddenly beamed everyone out.
There was stuff on the ground everywhere – blankets, clothing, single boots, wallets, purses. I walked past a food stand with food still cooking on the grill. There was a beer tap still running. It was the weirdest feeling I’d ever had in my life.
After that, things got a little crazy again. There had been a report of a second shooter, and no one knew if it was real or not. The police started herding a group of us across the street to the Tropicana. We were still trying to take cover as we walked there. We went past a big lion statue in front of one of the casinos. I have a picture from two years earlier of me sitting on the back of that lion. I remember thinking: Now I’m hunkered down behind the same lion hiding from a shooter. Times change.
They brought about 50 of us into a food court, which was closed. They wouldn’t tell us what was going on. And they wouldn’t let us leave. This went on for hours. Meanwhile, I had dropped my cell phone on the field, so my wife couldn’t get hold of me, and later she told me she assumed I’d been shot. I was just hoping that she was safe.
People were huddled together, crying, holding each other. Most were wearing Western concert–going stuff, which for a lot of them wasn’t very much clothing. The hotel eventually brought some blankets.
I was covered in blood. My shirt, shorts, and sandals were soaked. It was running down my legs. I couldn’t find anything to eat or drink. At one point, I sat down at a slot machine, put a hundred dollars in, and started playing slots. I didn’t know what else to do. It didn’t take me very long to lose it all.
Finally, I started looking for a way to get out. I checked all the exits, but there were security and police there. Then I ran into a guy who said he had found a fire exit. When we opened the fire door, there was a big security guard there, and he said: “You can’t leave.”
We said: “Try to stop us. We’re out of here.”
Another thing I’ll always remember – after I broke out of the Tropicana, I was low crawling through the bushes along the Strip toward my hotel. I got a block away and stood up to cross the street. I pushed the crosswalk button and waited. There were no cars, no people. I’ve just broken all the rules, violated police orders, and now I’m standing there waiting for a blinking light to allow me to cross the street!
I made it back to my hotel room around 3:30 or 4:00 in the morning. My wife was hysterical because I hadn’t been answering my cell phone. I came in, and she gave me a big hug, and I got in the shower. Our plane was leaving in a few hours, so we laid down, but didn’t sleep.
As we were getting ready to leave, my wife’s phone rang, and it was my number. A guy at the same hotel had found my phone on the field and called the “in case of emergency” number. So, I got my phone back.
It wasn’t easy to deal with the aftermath. It really affected everybody’s life. To this day, I’m particular about where we sit at concerts. My wife isn’t comfortable if she can’t see an exit. I now have a med bag in my car with tourniquets, pressure dressings, airway masks for CPR.
I’ll never forget that feeling of absolute frustration. That lady without an airway – I could’ve put a trach in her very quickly and made a difference. Were they able to keep her airway? Did she live?
The father and son – did the father make it? I have no idea what happened to any of them. Later, I went through and looked at the pictures of all the people who had died, but I couldn’t recognize anybody.
The hardest part was being there with my wife. I’ve been in places where people are shooting at you, in vehicles that are getting bombed. I’ve always believed that when it’s your time, it’s your time. If I get shot, well, okay, that happens. But if she got shot or my friends ... that would be really tough.
A year later, I gave a talk about it at a conference. I thought I had worked through everything. But all of those feelings, all of that helplessness, that anger, everything came roaring back to the surface again. They asked me how I deal with it, and I said: “Well ... poorly.” I’m the guy who sticks it in a box in the back of his brain, tucks it in and buries it with a bunch of other boxes, and hopes it never comes out again. But every once in a while, it does.
There were all kinds of people out on that field, some with medical training, some without, all determined to help, trying to get those injured people where they needed to be. In retrospect, it does make you feel good. Somebody was shooting at us, but people were still willing to stand up and risk their lives to help others.
We still talk with our friends about what happened that night. Over the years, it’s become less and less. But there’s still a text sent out every year on that day: “Today is the anniversary. Glad we’re all alive. Thanks for being our friends.”
Dr. Sebesta is a bariatric surgeon with MultiCare Health System in Tacoma, Wash.
A version of this article first appeared on Medscape.com.
Sunday night. Las Vegas. Jason Aldean had just started playing.
My wife and I were at the 2017 Route 91 Harvest Festival with three other couples; two of them were our close friends. We were sitting in the VIP section, a tented area right next to the stage. We started hearing what I was convinced were fireworks.
I’ve been in the Army for 20 some years. I’ve been deployed and shot at multiple times. But these shots were far away. And you don’t expect people to be shooting at you at a concert.
I was on the edge of the VIP area, so I could see around the corner of the tent. I looked up at the Mandalay Bay and saw the muzzle flash in the hotel window. That’s when I knew.
I screamed: “Somebody’s shooting at us! Everybody get down!”
It took a while for people to realize what was going on. When the first couple volleys sprayed into the crowd, nobody understood. But once enough people had been hit and dropped, everyone knew, and it was just mass exodus.
People screamed and ran everywhere. Some of them tried to jump over the front barrier so they could get underneath the stage. Others were trying to pick up loved ones who’d been shot.
The next 15 minutes are a little foggy. I was helping my wife and the people around us to get down. Funny things come back to you afterward. One of my friends was carrying a 16-ounce beer in his hand. Somebody’s shooting at him and he’s walking around with his beer like he’s afraid to put it down. It was so surreal.
We got everybody underneath the tent, and then we just sat there. There would be shooting and then a pause. You’d think it was over. And then there would be more shooting and another pause. It felt like it never was going to stop.
After a short period of time, somebody came in with an official badge, maybe FBI, who knows. They said: “Okay, everybody up. We’ve got to get you out of here.” So, we all got up and headed across the stage. The gate they were taking us to was in full view of the shooter, so it wasn’t very safe.
As I got up, I looked out at the field. Bodies were scattered everywhere. I’m a trauma surgeon by trade. I couldn’t just leave.
I told my two best friends to take my wife with them. My wife lost her mind at that point. She didn’t want me to run out on the field. But I had to. I saw the injured and they needed help. Another buddy and I jumped over the fence and started taking care of people.
The feeling of being out on the field was one of complete frustration. I was in sandals, shorts, and a t-shirt. We had no stretchers, no medical supplies, no nothing. I didn’t have a belt to use as a tourniquet. I didn’t even have a bandage.
Worse: We were seeing high-velocity gunshot wounds that I’ve seen for 20 years in the Army. I know how to take care of them. I know how to fix them. But there wasn’t a single thing I could do.
We had to get people off the field, so we started gathering up as many as we could. We didn’t know if we were going to get shot at again, so we were trying to hide behind things as we ran. Our main objective was just to get people to a place of safety.
A lot of it is a blur. But a few patients stick out in my mind.
A father and son. The father had been shot through the abdomen, exited out through his back. He was in severe pain and couldn’t walk.
A young girl shot in the arm. Her parents carrying her.
A group of people doing CPR on a young lady. She had a gunshot wound to the head or neck. She was obviously dead. But they were still doing chest compressions in the middle of the field. I had to say to them: “She’s dead. You can’t save her. You need to get off the field.” But they wouldn’t stop. We picked her up and took her out while they continued to do CPR.
Later, I realized I knew that woman. She was part of a group of friends that we would see at the festival. I hadn’t recognized her. I also didn’t know that my friend Marco was there. A month or 2 later, we figured out that he was one of the people doing CPR. And I was the guy who came up and said his friend was dead.
Some people were so badly injured we couldn’t lift them. We started tearing apart the fencing used to separate the crowd and slid sections of the barricades under the wounded to carry them. We also carried off a bunch of people who were dead.
We were moving patients to a covered bar area where we thought they would be safer. What we didn’t know was there was an ambulance rally point at the very far end of the field. Unfortunately, we had no idea it was there.
I saw a lot of other first responders out there, people from the fire department, corpsmen from the Navy, medics. I ran into an anesthesia provider and a series of nurses.
When we got everybody off the field, we started moving them into vehicles. People were bringing their trucks up. One guy even stole a truck so he could drive people to the ED. There wasn’t a lot of triage. We were just stacking whoever we could into the backs of these pickups.
I tried to help a nurse taking care of a lady who had been shot in the neck. She was sitting sort of half upright with the patient lying in her arms. When I reached to help her, she said: “You can’t move her.”
“We need to get her to the hospital,” I replied.
“This is the only position that this lady has an airway,” she said. “You’re going to have to move both of us together. If I move at all, she loses her airway.”
So, a group of us managed to slide something underneath and lift them into the back of a truck.
Loading the wounded went on for a while. And then, just like that, everybody was gone.
I walked back out onto this field which not too long ago held 30,000 people. It was as if aliens had just suddenly beamed everyone out.
There was stuff on the ground everywhere – blankets, clothing, single boots, wallets, purses. I walked past a food stand with food still cooking on the grill. There was a beer tap still running. It was the weirdest feeling I’d ever had in my life.
After that, things got a little crazy again. There had been a report of a second shooter, and no one knew if it was real or not. The police started herding a group of us across the street to the Tropicana. We were still trying to take cover as we walked there. We went past a big lion statue in front of one of the casinos. I have a picture from two years earlier of me sitting on the back of that lion. I remember thinking: Now I’m hunkered down behind the same lion hiding from a shooter. Times change.
They brought about 50 of us into a food court, which was closed. They wouldn’t tell us what was going on. And they wouldn’t let us leave. This went on for hours. Meanwhile, I had dropped my cell phone on the field, so my wife couldn’t get hold of me, and later she told me she assumed I’d been shot. I was just hoping that she was safe.
People were huddled together, crying, holding each other. Most were wearing Western concert–going stuff, which for a lot of them wasn’t very much clothing. The hotel eventually brought some blankets.
I was covered in blood. My shirt, shorts, and sandals were soaked. It was running down my legs. I couldn’t find anything to eat or drink. At one point, I sat down at a slot machine, put a hundred dollars in, and started playing slots. I didn’t know what else to do. It didn’t take me very long to lose it all.
Finally, I started looking for a way to get out. I checked all the exits, but there were security and police there. Then I ran into a guy who said he had found a fire exit. When we opened the fire door, there was a big security guard there, and he said: “You can’t leave.”
We said: “Try to stop us. We’re out of here.”
Another thing I’ll always remember – after I broke out of the Tropicana, I was low crawling through the bushes along the Strip toward my hotel. I got a block away and stood up to cross the street. I pushed the crosswalk button and waited. There were no cars, no people. I’ve just broken all the rules, violated police orders, and now I’m standing there waiting for a blinking light to allow me to cross the street!
I made it back to my hotel room around 3:30 or 4:00 in the morning. My wife was hysterical because I hadn’t been answering my cell phone. I came in, and she gave me a big hug, and I got in the shower. Our plane was leaving in a few hours, so we laid down, but didn’t sleep.
As we were getting ready to leave, my wife’s phone rang, and it was my number. A guy at the same hotel had found my phone on the field and called the “in case of emergency” number. So, I got my phone back.
It wasn’t easy to deal with the aftermath. It really affected everybody’s life. To this day, I’m particular about where we sit at concerts. My wife isn’t comfortable if she can’t see an exit. I now have a med bag in my car with tourniquets, pressure dressings, airway masks for CPR.
I’ll never forget that feeling of absolute frustration. That lady without an airway – I could’ve put a trach in her very quickly and made a difference. Were they able to keep her airway? Did she live?
The father and son – did the father make it? I have no idea what happened to any of them. Later, I went through and looked at the pictures of all the people who had died, but I couldn’t recognize anybody.
The hardest part was being there with my wife. I’ve been in places where people are shooting at you, in vehicles that are getting bombed. I’ve always believed that when it’s your time, it’s your time. If I get shot, well, okay, that happens. But if she got shot or my friends ... that would be really tough.
A year later, I gave a talk about it at a conference. I thought I had worked through everything. But all of those feelings, all of that helplessness, that anger, everything came roaring back to the surface again. They asked me how I deal with it, and I said: “Well ... poorly.” I’m the guy who sticks it in a box in the back of his brain, tucks it in and buries it with a bunch of other boxes, and hopes it never comes out again. But every once in a while, it does.
There were all kinds of people out on that field, some with medical training, some without, all determined to help, trying to get those injured people where they needed to be. In retrospect, it does make you feel good. Somebody was shooting at us, but people were still willing to stand up and risk their lives to help others.
We still talk with our friends about what happened that night. Over the years, it’s become less and less. But there’s still a text sent out every year on that day: “Today is the anniversary. Glad we’re all alive. Thanks for being our friends.”
Dr. Sebesta is a bariatric surgeon with MultiCare Health System in Tacoma, Wash.
A version of this article first appeared on Medscape.com.
Sunday night. Las Vegas. Jason Aldean had just started playing.
My wife and I were at the 2017 Route 91 Harvest Festival with three other couples; two of them were our close friends. We were sitting in the VIP section, a tented area right next to the stage. We started hearing what I was convinced were fireworks.
I’ve been in the Army for 20 some years. I’ve been deployed and shot at multiple times. But these shots were far away. And you don’t expect people to be shooting at you at a concert.
I was on the edge of the VIP area, so I could see around the corner of the tent. I looked up at the Mandalay Bay and saw the muzzle flash in the hotel window. That’s when I knew.
I screamed: “Somebody’s shooting at us! Everybody get down!”
It took a while for people to realize what was going on. When the first couple volleys sprayed into the crowd, nobody understood. But once enough people had been hit and dropped, everyone knew, and it was just mass exodus.
People screamed and ran everywhere. Some of them tried to jump over the front barrier so they could get underneath the stage. Others were trying to pick up loved ones who’d been shot.
The next 15 minutes are a little foggy. I was helping my wife and the people around us to get down. Funny things come back to you afterward. One of my friends was carrying a 16-ounce beer in his hand. Somebody’s shooting at him and he’s walking around with his beer like he’s afraid to put it down. It was so surreal.
We got everybody underneath the tent, and then we just sat there. There would be shooting and then a pause. You’d think it was over. And then there would be more shooting and another pause. It felt like it never was going to stop.
After a short period of time, somebody came in with an official badge, maybe FBI, who knows. They said: “Okay, everybody up. We’ve got to get you out of here.” So, we all got up and headed across the stage. The gate they were taking us to was in full view of the shooter, so it wasn’t very safe.
As I got up, I looked out at the field. Bodies were scattered everywhere. I’m a trauma surgeon by trade. I couldn’t just leave.
I told my two best friends to take my wife with them. My wife lost her mind at that point. She didn’t want me to run out on the field. But I had to. I saw the injured and they needed help. Another buddy and I jumped over the fence and started taking care of people.
The feeling of being out on the field was one of complete frustration. I was in sandals, shorts, and a t-shirt. We had no stretchers, no medical supplies, no nothing. I didn’t have a belt to use as a tourniquet. I didn’t even have a bandage.
Worse: We were seeing high-velocity gunshot wounds that I’ve seen for 20 years in the Army. I know how to take care of them. I know how to fix them. But there wasn’t a single thing I could do.
We had to get people off the field, so we started gathering up as many as we could. We didn’t know if we were going to get shot at again, so we were trying to hide behind things as we ran. Our main objective was just to get people to a place of safety.
A lot of it is a blur. But a few patients stick out in my mind.
A father and son. The father had been shot through the abdomen, exited out through his back. He was in severe pain and couldn’t walk.
A young girl shot in the arm. Her parents carrying her.
A group of people doing CPR on a young lady. She had a gunshot wound to the head or neck. She was obviously dead. But they were still doing chest compressions in the middle of the field. I had to say to them: “She’s dead. You can’t save her. You need to get off the field.” But they wouldn’t stop. We picked her up and took her out while they continued to do CPR.
Later, I realized I knew that woman. She was part of a group of friends that we would see at the festival. I hadn’t recognized her. I also didn’t know that my friend Marco was there. A month or 2 later, we figured out that he was one of the people doing CPR. And I was the guy who came up and said his friend was dead.
Some people were so badly injured we couldn’t lift them. We started tearing apart the fencing used to separate the crowd and slid sections of the barricades under the wounded to carry them. We also carried off a bunch of people who were dead.
We were moving patients to a covered bar area where we thought they would be safer. What we didn’t know was there was an ambulance rally point at the very far end of the field. Unfortunately, we had no idea it was there.
I saw a lot of other first responders out there, people from the fire department, corpsmen from the Navy, medics. I ran into an anesthesia provider and a series of nurses.
When we got everybody off the field, we started moving them into vehicles. People were bringing their trucks up. One guy even stole a truck so he could drive people to the ED. There wasn’t a lot of triage. We were just stacking whoever we could into the backs of these pickups.
I tried to help a nurse taking care of a lady who had been shot in the neck. She was sitting sort of half upright with the patient lying in her arms. When I reached to help her, she said: “You can’t move her.”
“We need to get her to the hospital,” I replied.
“This is the only position that this lady has an airway,” she said. “You’re going to have to move both of us together. If I move at all, she loses her airway.”
So, a group of us managed to slide something underneath and lift them into the back of a truck.
Loading the wounded went on for a while. And then, just like that, everybody was gone.
I walked back out onto this field which not too long ago held 30,000 people. It was as if aliens had just suddenly beamed everyone out.
There was stuff on the ground everywhere – blankets, clothing, single boots, wallets, purses. I walked past a food stand with food still cooking on the grill. There was a beer tap still running. It was the weirdest feeling I’d ever had in my life.
After that, things got a little crazy again. There had been a report of a second shooter, and no one knew if it was real or not. The police started herding a group of us across the street to the Tropicana. We were still trying to take cover as we walked there. We went past a big lion statue in front of one of the casinos. I have a picture from two years earlier of me sitting on the back of that lion. I remember thinking: Now I’m hunkered down behind the same lion hiding from a shooter. Times change.
They brought about 50 of us into a food court, which was closed. They wouldn’t tell us what was going on. And they wouldn’t let us leave. This went on for hours. Meanwhile, I had dropped my cell phone on the field, so my wife couldn’t get hold of me, and later she told me she assumed I’d been shot. I was just hoping that she was safe.
People were huddled together, crying, holding each other. Most were wearing Western concert–going stuff, which for a lot of them wasn’t very much clothing. The hotel eventually brought some blankets.
I was covered in blood. My shirt, shorts, and sandals were soaked. It was running down my legs. I couldn’t find anything to eat or drink. At one point, I sat down at a slot machine, put a hundred dollars in, and started playing slots. I didn’t know what else to do. It didn’t take me very long to lose it all.
Finally, I started looking for a way to get out. I checked all the exits, but there were security and police there. Then I ran into a guy who said he had found a fire exit. When we opened the fire door, there was a big security guard there, and he said: “You can’t leave.”
We said: “Try to stop us. We’re out of here.”
Another thing I’ll always remember – after I broke out of the Tropicana, I was low crawling through the bushes along the Strip toward my hotel. I got a block away and stood up to cross the street. I pushed the crosswalk button and waited. There were no cars, no people. I’ve just broken all the rules, violated police orders, and now I’m standing there waiting for a blinking light to allow me to cross the street!
I made it back to my hotel room around 3:30 or 4:00 in the morning. My wife was hysterical because I hadn’t been answering my cell phone. I came in, and she gave me a big hug, and I got in the shower. Our plane was leaving in a few hours, so we laid down, but didn’t sleep.
As we were getting ready to leave, my wife’s phone rang, and it was my number. A guy at the same hotel had found my phone on the field and called the “in case of emergency” number. So, I got my phone back.
It wasn’t easy to deal with the aftermath. It really affected everybody’s life. To this day, I’m particular about where we sit at concerts. My wife isn’t comfortable if she can’t see an exit. I now have a med bag in my car with tourniquets, pressure dressings, airway masks for CPR.
I’ll never forget that feeling of absolute frustration. That lady without an airway – I could’ve put a trach in her very quickly and made a difference. Were they able to keep her airway? Did she live?
The father and son – did the father make it? I have no idea what happened to any of them. Later, I went through and looked at the pictures of all the people who had died, but I couldn’t recognize anybody.
The hardest part was being there with my wife. I’ve been in places where people are shooting at you, in vehicles that are getting bombed. I’ve always believed that when it’s your time, it’s your time. If I get shot, well, okay, that happens. But if she got shot or my friends ... that would be really tough.
A year later, I gave a talk about it at a conference. I thought I had worked through everything. But all of those feelings, all of that helplessness, that anger, everything came roaring back to the surface again. They asked me how I deal with it, and I said: “Well ... poorly.” I’m the guy who sticks it in a box in the back of his brain, tucks it in and buries it with a bunch of other boxes, and hopes it never comes out again. But every once in a while, it does.
There were all kinds of people out on that field, some with medical training, some without, all determined to help, trying to get those injured people where they needed to be. In retrospect, it does make you feel good. Somebody was shooting at us, but people were still willing to stand up and risk their lives to help others.
We still talk with our friends about what happened that night. Over the years, it’s become less and less. But there’s still a text sent out every year on that day: “Today is the anniversary. Glad we’re all alive. Thanks for being our friends.”
Dr. Sebesta is a bariatric surgeon with MultiCare Health System in Tacoma, Wash.
A version of this article first appeared on Medscape.com.
A 75-year-old White woman presented with diffuse erythema, scale, and pruritus on her scalp
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
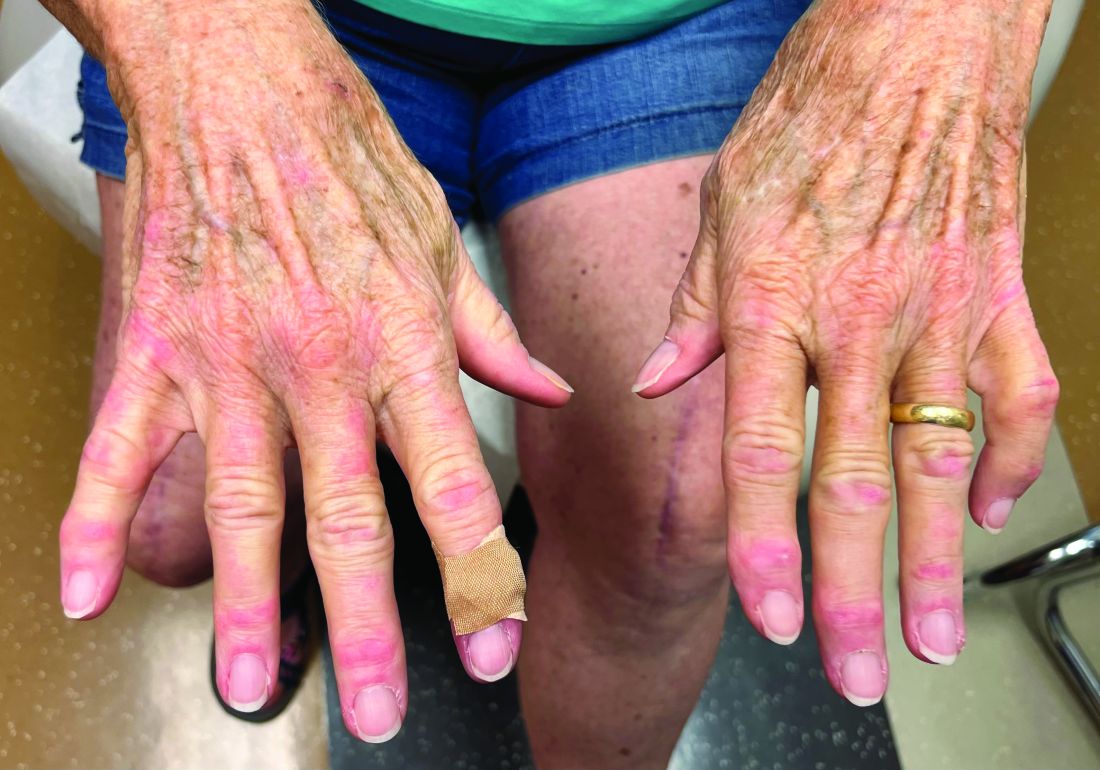
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.

Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
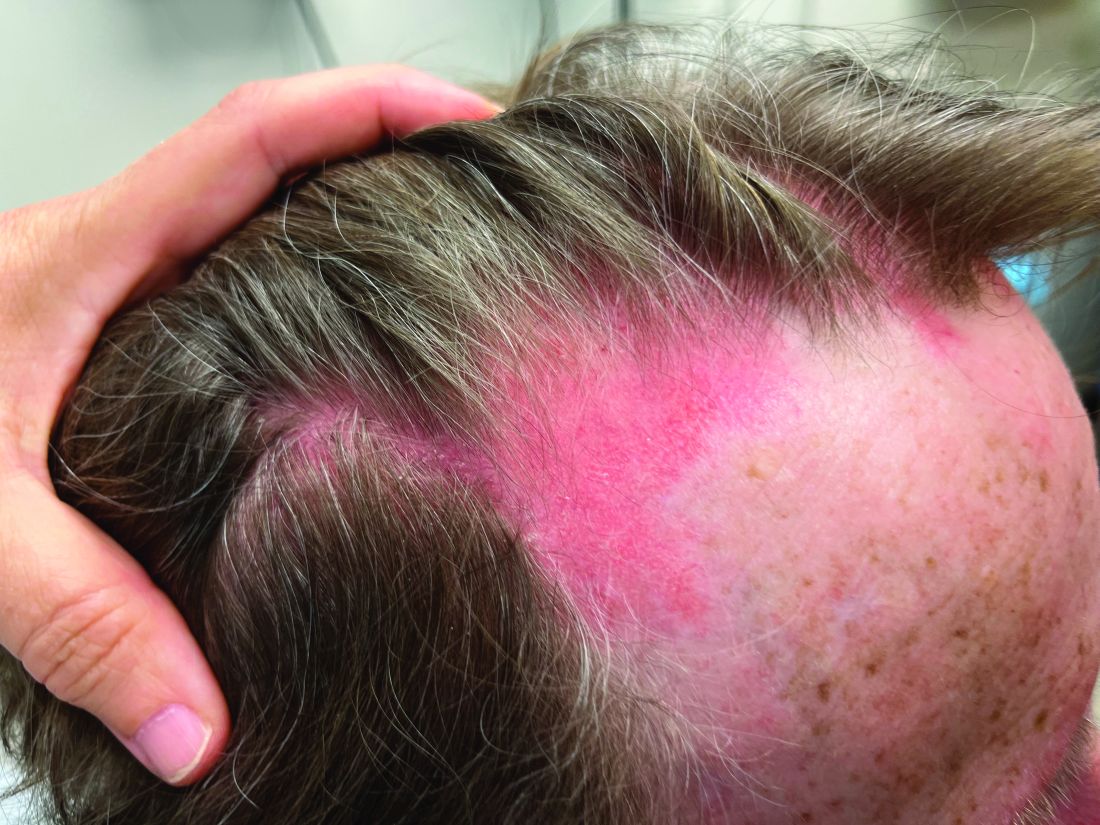
First-line therapy in T2D: Has metformin been ‘dethroned’?
Initially approved by the U.S. Food and Drug Administration (FDA) in 1994, metformin has been the preferred first-line glucose-lowering agent for patients with type 2 diabetes (T2D) owing to its effectiveness, low hypoglycemia risk, weight neutrality, long clinical track record of safety, and affordability. However, the advent of newer glucose-lowering agents with evidence-based cardiovascular (CV) and renal benefits calls into question whether metformin should continue to be the initial pharmacotherapy for all patients with T2D.
Cardiovascular outcome trials transform standard of care
In 2008, the FDA issued guidance to industry to ensure that CV risk is more thoroughly addressed during development of T2D therapies. This guidance document required dedicated trials to establish CV safety of new glucose-lowering therapies. Findings from subsequent cardiovascular outcome trials (CVOTs) and subsequent large renal and heart failure (HF) outcome trials have since prompted frequent and substantial updates to major guidelines. On the basis of recent evidence from CVOT and renal trials, contemporary clinical practice guidelines have transitioned from a traditional glucocentric treatment approach to a holistic management approach that emphasizes organ protection through heart-kidney-metabolic risk reduction.
Per the 2008 FDA guidance, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagonlike peptide-1 (GLP-1) receptor agonists, and sodium-glucose cotransporter-2 (SGLT2) inhibitors were evaluated in large dedicated CVOTs. Findings from several CVOTs established GLP-1 receptor agonist and SGLT2 inhibitor CV safety, and unexpectedly demonstrated reduced rates of major adverse cardiovascular events (MACE) relative to placebo. The LEADER and EMPA-REG OUTCOME trials were the first CVOTs to report cardioprotective benefits of the GLP-1 receptor agonist liraglutide and the SGLT2 inhibitor empagliflozin, respectively. The LEADER trial reported a 13% significant relative risk reduction for its primary composite MACE outcome, and the EMPA-REG OUTCOME trial similarly reported a 14% relative risk reduction for MACE. After CVOTs on other GLP-1 receptor agonists and SGLT2 inhibitors reported CV benefit, clinical practice guidelines began to recommend use of these agents in at-risk patients to mitigate CV risk.
During the period when most CVOTs were designed and conducted, a majority of trial participants were receiving metformin at baseline. Inclusion of a small subset of metformin-naive participants in these trials allowed for several post hoc and meta-analyses investigating the impact of background metformin use on the overall CV benefits reported. Depending on the trial, baseline metformin use in large GLP-1 receptor agonist CVOTs ranged from 66% to 81%. For instance, 76% of participants in the LEADER trial were receiving metformin at baseline, but a post hoc analysis found no heterogeneity for the observed CV benefit based on background metformin use. Similarly, a subgroup analysis of pooled data from the SUSTAIN-6 and PIONEER 6 trials of injectable and oral formulations of semaglutide, respectively, reported similar CV outcomes for participants, regardless of concomitant metformin use. When looking at the GLP-1 receptor agonist class overall, a meta-analysis of seven CVOTs, which included participants with established atherosclerotic cardiovascular disease (ASCVD) and those with multiple ASCVD risk factors, concluded that GLP-1 receptor agonist therapy reduced the overall incidence of MACE in participants not receiving concomitant metformin at baseline.
Similar analyses have examined the impact of background metformin use on CV outcomes with SGLT2 inhibitors. An analysis of EMPA-REG OUTCOME found that empagliflozin improved CV outcomes and reduced mortality irrespective of background metformin, sulfonylurea, or insulin use. Of note, this analysis suggested a greater risk reduction for incident or worsening nephropathy in patients not on concomitant metformin (hazard ratio, 0.47; 95% confidence interval, 0.37-0.59; P = .01), when compared with those taking metformin at baseline (HR, 0.68; 95% CI, 0.58-0.79; P = .01). In addition, a meta-analysis of six large outcome trials found consistent benefits of SGLT2 inhibition on CV, kidney, and mortality outcomes regardless of background metformin treatment. Therefore, although CVOTs on GLP-1 receptor agonists and SGLT2 inhibitors were not designed to assess the impact of background metformin use on CV outcomes, available evidence supports the CV benefits of these agents independent of metformin use.
Individualizing care to attain cardiorenal-metabolic goals
Three dedicated SGLT2 inhibitor renal outcome trials have been published to date: CREDENCE, DAPA-CKD, and EMPA-KIDNEY. All three studies confirmed the positive secondary renal outcomes observed in SGLT2 inhibitor CVOTs: reduced progression of kidney disease, HF-associated hospital admissions, and CV-related death. The observed renal and CV benefits from the CREDENCE trial were consistent across different levels of kidney function. Similarly, a meta-analysis of five SGLT2 inhibitor trials of patients with HF demonstrated a decreased risk for CV-related death and admission for HF, irrespective of baseline heart function. The ongoing FLOW is the first dedicated kidney-outcome trial to evaluate the effectiveness of a GLP-1 receptor agonist (semaglutide) in slowing the progression and worsening of chronic kidney disease (CKD) in patients with T2D.
As previously noted, findings from the LEADER and EMPA-REG OUTCOME trials demonstrated the beneficial effects of GLP-1 receptor agonists and SGLT2 inhibitors not only on MACE but also on secondary HF and kidney disease outcomes. These findings have supported a series of dedicated HF and kidney outcome trials further informing the standard of care for patients with these key comorbidities. Indeed, the American Diabetes Association’s 2023 Standards of Care in Diabetes updated its recommendations and algorithm for the use of glucose-lowering medications in the management of T2D. The current ADA recommendations stress cardiorenal risk reduction while concurrently achieving and maintaining glycemic and weight management goals. On the basis of evolving outcome trial data, GLP-1 receptor agonists and SGLT2 inhibitors with evidence of benefit are recommended for patients with established or at high risk for ASCVD. Further, the Standards preferentially recommend SGLT2 inhibitors for patients with HF and/or CKD. Because evidence suggests no heterogeneity of benefit based on hemoglobin A1c for MACE outcomes with GLP-1 receptor agonists and no heterogeneity of benefit for HF or CKD benefits with SGLT2 inhibitors, these agents are recommended for cardiorenal risk reduction regardless of the need to lower glucose.
The 2023 update to the American Association of Clinical Endocrinology Consensus Statement: Type 2 Diabetes Management Algorithm similarly recommends the use of GLP-1 receptor agonists and SGLT2 inhibitors to improve cardiorenal outcomes. To further emphasize the importance of prescribing agents with proven organ-protective benefits, the AACE consensus statement provides a complications-centric algorithm to guide therapeutic decisions for risk reduction in patients with key comorbidities (for instance, ASCVD, HF, CKD) and a separate glucocentric algorithm to guide selection and intensification of glucose-lowering agents in patients without key comorbidities to meet individualized glycemic targets. Within the complications-centric algorithm, AACE recommends GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment for cardiorenal risk reduction regardless of background metformin use or A1c level.
In addition to the emphasis on the use of GLP-1 receptor agonists and SGLT2 inhibitors for organ protection, guidelines now recommend SGLT2 inhibitors as the standard-of-care therapy in patients with T2D and CKD with an estimated glomerular filtration rate ≥ 20 mL/min per 1.73 m2, and irrespective of ejection fraction or a diagnosis of diabetes in the setting of HF. Overall, a common thread within current guidelines is the importance of individualized therapy based on patient- and medication-specific factors.
Optimizing guideline-directed medical therapy
Results from the DISCOVER trial found that GLP-1 receptor agonist and SGLT2 inhibitor use was less likely in the key patient subgroups most likely to benefit from therapy, including patients with peripheral artery disease and CKD. Factors contributing to underutilization of newer cardiorenal protective glucose-lowering therapies range from cost and access barriers to clinician-level barriers (for example, lack of knowledge on CKD, lack of familiarity with CKD practice guidelines). Addressing these issues and helping patients work through financial and other access barriers is essential to optimize the utilization of these therapies and improve cardiorenal and metabolic outcomes.
So, has metformin been “dethroned” as a first-line therapy for T2D? As is often the case in medicine, the answer depends on the individual patient and clinical situation. Metformin remains an important first-line treatment in combination with lifestyle interventions to help patients with T2D without key cardiorenal comorbidities achieve individualized glycemic targets. However, based on evidence demonstrating cardiorenal protective benefits and improved glycemia and weight loss, GLP-1 agonists and SGLT2 inhibitors may be considered as first-line treatment for patients with T2D with or at high risk for ASCVD, HF, or CKD, regardless of the need for additional glucose-lowering agents and independent of background metformin. Ultimately, the choice of first-line therapy for patients with T2D should be informed by individualized treatment goals, preferences, and cost-related access. Continued efforts to increase patient access to GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment when indicated are essential to ensure optimal treatment and outcomes.
Dr. Neumiller is professor, department of pharmacotherapy, Washington State University, Spokane. He disclosed ties with Bayer, Boehringer Ingelheim, and Eli Lilly. Dr. Alicic is clinical professor, department of medicine, University of Washington; and associate director of research, Inland Northwest Washington, Providence St. Joseph Health, Spokane. She disclosed ties with Providence St. Joseph Health, Boehringer Ingelheim/Lilly, and Bayer.
A version of this article appeared on Medscape.com.
Initially approved by the U.S. Food and Drug Administration (FDA) in 1994, metformin has been the preferred first-line glucose-lowering agent for patients with type 2 diabetes (T2D) owing to its effectiveness, low hypoglycemia risk, weight neutrality, long clinical track record of safety, and affordability. However, the advent of newer glucose-lowering agents with evidence-based cardiovascular (CV) and renal benefits calls into question whether metformin should continue to be the initial pharmacotherapy for all patients with T2D.
Cardiovascular outcome trials transform standard of care
In 2008, the FDA issued guidance to industry to ensure that CV risk is more thoroughly addressed during development of T2D therapies. This guidance document required dedicated trials to establish CV safety of new glucose-lowering therapies. Findings from subsequent cardiovascular outcome trials (CVOTs) and subsequent large renal and heart failure (HF) outcome trials have since prompted frequent and substantial updates to major guidelines. On the basis of recent evidence from CVOT and renal trials, contemporary clinical practice guidelines have transitioned from a traditional glucocentric treatment approach to a holistic management approach that emphasizes organ protection through heart-kidney-metabolic risk reduction.
Per the 2008 FDA guidance, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagonlike peptide-1 (GLP-1) receptor agonists, and sodium-glucose cotransporter-2 (SGLT2) inhibitors were evaluated in large dedicated CVOTs. Findings from several CVOTs established GLP-1 receptor agonist and SGLT2 inhibitor CV safety, and unexpectedly demonstrated reduced rates of major adverse cardiovascular events (MACE) relative to placebo. The LEADER and EMPA-REG OUTCOME trials were the first CVOTs to report cardioprotective benefits of the GLP-1 receptor agonist liraglutide and the SGLT2 inhibitor empagliflozin, respectively. The LEADER trial reported a 13% significant relative risk reduction for its primary composite MACE outcome, and the EMPA-REG OUTCOME trial similarly reported a 14% relative risk reduction for MACE. After CVOTs on other GLP-1 receptor agonists and SGLT2 inhibitors reported CV benefit, clinical practice guidelines began to recommend use of these agents in at-risk patients to mitigate CV risk.
During the period when most CVOTs were designed and conducted, a majority of trial participants were receiving metformin at baseline. Inclusion of a small subset of metformin-naive participants in these trials allowed for several post hoc and meta-analyses investigating the impact of background metformin use on the overall CV benefits reported. Depending on the trial, baseline metformin use in large GLP-1 receptor agonist CVOTs ranged from 66% to 81%. For instance, 76% of participants in the LEADER trial were receiving metformin at baseline, but a post hoc analysis found no heterogeneity for the observed CV benefit based on background metformin use. Similarly, a subgroup analysis of pooled data from the SUSTAIN-6 and PIONEER 6 trials of injectable and oral formulations of semaglutide, respectively, reported similar CV outcomes for participants, regardless of concomitant metformin use. When looking at the GLP-1 receptor agonist class overall, a meta-analysis of seven CVOTs, which included participants with established atherosclerotic cardiovascular disease (ASCVD) and those with multiple ASCVD risk factors, concluded that GLP-1 receptor agonist therapy reduced the overall incidence of MACE in participants not receiving concomitant metformin at baseline.
Similar analyses have examined the impact of background metformin use on CV outcomes with SGLT2 inhibitors. An analysis of EMPA-REG OUTCOME found that empagliflozin improved CV outcomes and reduced mortality irrespective of background metformin, sulfonylurea, or insulin use. Of note, this analysis suggested a greater risk reduction for incident or worsening nephropathy in patients not on concomitant metformin (hazard ratio, 0.47; 95% confidence interval, 0.37-0.59; P = .01), when compared with those taking metformin at baseline (HR, 0.68; 95% CI, 0.58-0.79; P = .01). In addition, a meta-analysis of six large outcome trials found consistent benefits of SGLT2 inhibition on CV, kidney, and mortality outcomes regardless of background metformin treatment. Therefore, although CVOTs on GLP-1 receptor agonists and SGLT2 inhibitors were not designed to assess the impact of background metformin use on CV outcomes, available evidence supports the CV benefits of these agents independent of metformin use.
Individualizing care to attain cardiorenal-metabolic goals
Three dedicated SGLT2 inhibitor renal outcome trials have been published to date: CREDENCE, DAPA-CKD, and EMPA-KIDNEY. All three studies confirmed the positive secondary renal outcomes observed in SGLT2 inhibitor CVOTs: reduced progression of kidney disease, HF-associated hospital admissions, and CV-related death. The observed renal and CV benefits from the CREDENCE trial were consistent across different levels of kidney function. Similarly, a meta-analysis of five SGLT2 inhibitor trials of patients with HF demonstrated a decreased risk for CV-related death and admission for HF, irrespective of baseline heart function. The ongoing FLOW is the first dedicated kidney-outcome trial to evaluate the effectiveness of a GLP-1 receptor agonist (semaglutide) in slowing the progression and worsening of chronic kidney disease (CKD) in patients with T2D.
As previously noted, findings from the LEADER and EMPA-REG OUTCOME trials demonstrated the beneficial effects of GLP-1 receptor agonists and SGLT2 inhibitors not only on MACE but also on secondary HF and kidney disease outcomes. These findings have supported a series of dedicated HF and kidney outcome trials further informing the standard of care for patients with these key comorbidities. Indeed, the American Diabetes Association’s 2023 Standards of Care in Diabetes updated its recommendations and algorithm for the use of glucose-lowering medications in the management of T2D. The current ADA recommendations stress cardiorenal risk reduction while concurrently achieving and maintaining glycemic and weight management goals. On the basis of evolving outcome trial data, GLP-1 receptor agonists and SGLT2 inhibitors with evidence of benefit are recommended for patients with established or at high risk for ASCVD. Further, the Standards preferentially recommend SGLT2 inhibitors for patients with HF and/or CKD. Because evidence suggests no heterogeneity of benefit based on hemoglobin A1c for MACE outcomes with GLP-1 receptor agonists and no heterogeneity of benefit for HF or CKD benefits with SGLT2 inhibitors, these agents are recommended for cardiorenal risk reduction regardless of the need to lower glucose.
The 2023 update to the American Association of Clinical Endocrinology Consensus Statement: Type 2 Diabetes Management Algorithm similarly recommends the use of GLP-1 receptor agonists and SGLT2 inhibitors to improve cardiorenal outcomes. To further emphasize the importance of prescribing agents with proven organ-protective benefits, the AACE consensus statement provides a complications-centric algorithm to guide therapeutic decisions for risk reduction in patients with key comorbidities (for instance, ASCVD, HF, CKD) and a separate glucocentric algorithm to guide selection and intensification of glucose-lowering agents in patients without key comorbidities to meet individualized glycemic targets. Within the complications-centric algorithm, AACE recommends GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment for cardiorenal risk reduction regardless of background metformin use or A1c level.
In addition to the emphasis on the use of GLP-1 receptor agonists and SGLT2 inhibitors for organ protection, guidelines now recommend SGLT2 inhibitors as the standard-of-care therapy in patients with T2D and CKD with an estimated glomerular filtration rate ≥ 20 mL/min per 1.73 m2, and irrespective of ejection fraction or a diagnosis of diabetes in the setting of HF. Overall, a common thread within current guidelines is the importance of individualized therapy based on patient- and medication-specific factors.
Optimizing guideline-directed medical therapy
Results from the DISCOVER trial found that GLP-1 receptor agonist and SGLT2 inhibitor use was less likely in the key patient subgroups most likely to benefit from therapy, including patients with peripheral artery disease and CKD. Factors contributing to underutilization of newer cardiorenal protective glucose-lowering therapies range from cost and access barriers to clinician-level barriers (for example, lack of knowledge on CKD, lack of familiarity with CKD practice guidelines). Addressing these issues and helping patients work through financial and other access barriers is essential to optimize the utilization of these therapies and improve cardiorenal and metabolic outcomes.
So, has metformin been “dethroned” as a first-line therapy for T2D? As is often the case in medicine, the answer depends on the individual patient and clinical situation. Metformin remains an important first-line treatment in combination with lifestyle interventions to help patients with T2D without key cardiorenal comorbidities achieve individualized glycemic targets. However, based on evidence demonstrating cardiorenal protective benefits and improved glycemia and weight loss, GLP-1 agonists and SGLT2 inhibitors may be considered as first-line treatment for patients with T2D with or at high risk for ASCVD, HF, or CKD, regardless of the need for additional glucose-lowering agents and independent of background metformin. Ultimately, the choice of first-line therapy for patients with T2D should be informed by individualized treatment goals, preferences, and cost-related access. Continued efforts to increase patient access to GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment when indicated are essential to ensure optimal treatment and outcomes.
Dr. Neumiller is professor, department of pharmacotherapy, Washington State University, Spokane. He disclosed ties with Bayer, Boehringer Ingelheim, and Eli Lilly. Dr. Alicic is clinical professor, department of medicine, University of Washington; and associate director of research, Inland Northwest Washington, Providence St. Joseph Health, Spokane. She disclosed ties with Providence St. Joseph Health, Boehringer Ingelheim/Lilly, and Bayer.
A version of this article appeared on Medscape.com.
Initially approved by the U.S. Food and Drug Administration (FDA) in 1994, metformin has been the preferred first-line glucose-lowering agent for patients with type 2 diabetes (T2D) owing to its effectiveness, low hypoglycemia risk, weight neutrality, long clinical track record of safety, and affordability. However, the advent of newer glucose-lowering agents with evidence-based cardiovascular (CV) and renal benefits calls into question whether metformin should continue to be the initial pharmacotherapy for all patients with T2D.
Cardiovascular outcome trials transform standard of care
In 2008, the FDA issued guidance to industry to ensure that CV risk is more thoroughly addressed during development of T2D therapies. This guidance document required dedicated trials to establish CV safety of new glucose-lowering therapies. Findings from subsequent cardiovascular outcome trials (CVOTs) and subsequent large renal and heart failure (HF) outcome trials have since prompted frequent and substantial updates to major guidelines. On the basis of recent evidence from CVOT and renal trials, contemporary clinical practice guidelines have transitioned from a traditional glucocentric treatment approach to a holistic management approach that emphasizes organ protection through heart-kidney-metabolic risk reduction.
Per the 2008 FDA guidance, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagonlike peptide-1 (GLP-1) receptor agonists, and sodium-glucose cotransporter-2 (SGLT2) inhibitors were evaluated in large dedicated CVOTs. Findings from several CVOTs established GLP-1 receptor agonist and SGLT2 inhibitor CV safety, and unexpectedly demonstrated reduced rates of major adverse cardiovascular events (MACE) relative to placebo. The LEADER and EMPA-REG OUTCOME trials were the first CVOTs to report cardioprotective benefits of the GLP-1 receptor agonist liraglutide and the SGLT2 inhibitor empagliflozin, respectively. The LEADER trial reported a 13% significant relative risk reduction for its primary composite MACE outcome, and the EMPA-REG OUTCOME trial similarly reported a 14% relative risk reduction for MACE. After CVOTs on other GLP-1 receptor agonists and SGLT2 inhibitors reported CV benefit, clinical practice guidelines began to recommend use of these agents in at-risk patients to mitigate CV risk.
During the period when most CVOTs were designed and conducted, a majority of trial participants were receiving metformin at baseline. Inclusion of a small subset of metformin-naive participants in these trials allowed for several post hoc and meta-analyses investigating the impact of background metformin use on the overall CV benefits reported. Depending on the trial, baseline metformin use in large GLP-1 receptor agonist CVOTs ranged from 66% to 81%. For instance, 76% of participants in the LEADER trial were receiving metformin at baseline, but a post hoc analysis found no heterogeneity for the observed CV benefit based on background metformin use. Similarly, a subgroup analysis of pooled data from the SUSTAIN-6 and PIONEER 6 trials of injectable and oral formulations of semaglutide, respectively, reported similar CV outcomes for participants, regardless of concomitant metformin use. When looking at the GLP-1 receptor agonist class overall, a meta-analysis of seven CVOTs, which included participants with established atherosclerotic cardiovascular disease (ASCVD) and those with multiple ASCVD risk factors, concluded that GLP-1 receptor agonist therapy reduced the overall incidence of MACE in participants not receiving concomitant metformin at baseline.
Similar analyses have examined the impact of background metformin use on CV outcomes with SGLT2 inhibitors. An analysis of EMPA-REG OUTCOME found that empagliflozin improved CV outcomes and reduced mortality irrespective of background metformin, sulfonylurea, or insulin use. Of note, this analysis suggested a greater risk reduction for incident or worsening nephropathy in patients not on concomitant metformin (hazard ratio, 0.47; 95% confidence interval, 0.37-0.59; P = .01), when compared with those taking metformin at baseline (HR, 0.68; 95% CI, 0.58-0.79; P = .01). In addition, a meta-analysis of six large outcome trials found consistent benefits of SGLT2 inhibition on CV, kidney, and mortality outcomes regardless of background metformin treatment. Therefore, although CVOTs on GLP-1 receptor agonists and SGLT2 inhibitors were not designed to assess the impact of background metformin use on CV outcomes, available evidence supports the CV benefits of these agents independent of metformin use.
Individualizing care to attain cardiorenal-metabolic goals
Three dedicated SGLT2 inhibitor renal outcome trials have been published to date: CREDENCE, DAPA-CKD, and EMPA-KIDNEY. All three studies confirmed the positive secondary renal outcomes observed in SGLT2 inhibitor CVOTs: reduced progression of kidney disease, HF-associated hospital admissions, and CV-related death. The observed renal and CV benefits from the CREDENCE trial were consistent across different levels of kidney function. Similarly, a meta-analysis of five SGLT2 inhibitor trials of patients with HF demonstrated a decreased risk for CV-related death and admission for HF, irrespective of baseline heart function. The ongoing FLOW is the first dedicated kidney-outcome trial to evaluate the effectiveness of a GLP-1 receptor agonist (semaglutide) in slowing the progression and worsening of chronic kidney disease (CKD) in patients with T2D.
As previously noted, findings from the LEADER and EMPA-REG OUTCOME trials demonstrated the beneficial effects of GLP-1 receptor agonists and SGLT2 inhibitors not only on MACE but also on secondary HF and kidney disease outcomes. These findings have supported a series of dedicated HF and kidney outcome trials further informing the standard of care for patients with these key comorbidities. Indeed, the American Diabetes Association’s 2023 Standards of Care in Diabetes updated its recommendations and algorithm for the use of glucose-lowering medications in the management of T2D. The current ADA recommendations stress cardiorenal risk reduction while concurrently achieving and maintaining glycemic and weight management goals. On the basis of evolving outcome trial data, GLP-1 receptor agonists and SGLT2 inhibitors with evidence of benefit are recommended for patients with established or at high risk for ASCVD. Further, the Standards preferentially recommend SGLT2 inhibitors for patients with HF and/or CKD. Because evidence suggests no heterogeneity of benefit based on hemoglobin A1c for MACE outcomes with GLP-1 receptor agonists and no heterogeneity of benefit for HF or CKD benefits with SGLT2 inhibitors, these agents are recommended for cardiorenal risk reduction regardless of the need to lower glucose.
The 2023 update to the American Association of Clinical Endocrinology Consensus Statement: Type 2 Diabetes Management Algorithm similarly recommends the use of GLP-1 receptor agonists and SGLT2 inhibitors to improve cardiorenal outcomes. To further emphasize the importance of prescribing agents with proven organ-protective benefits, the AACE consensus statement provides a complications-centric algorithm to guide therapeutic decisions for risk reduction in patients with key comorbidities (for instance, ASCVD, HF, CKD) and a separate glucocentric algorithm to guide selection and intensification of glucose-lowering agents in patients without key comorbidities to meet individualized glycemic targets. Within the complications-centric algorithm, AACE recommends GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment for cardiorenal risk reduction regardless of background metformin use or A1c level.
In addition to the emphasis on the use of GLP-1 receptor agonists and SGLT2 inhibitors for organ protection, guidelines now recommend SGLT2 inhibitors as the standard-of-care therapy in patients with T2D and CKD with an estimated glomerular filtration rate ≥ 20 mL/min per 1.73 m2, and irrespective of ejection fraction or a diagnosis of diabetes in the setting of HF. Overall, a common thread within current guidelines is the importance of individualized therapy based on patient- and medication-specific factors.
Optimizing guideline-directed medical therapy
Results from the DISCOVER trial found that GLP-1 receptor agonist and SGLT2 inhibitor use was less likely in the key patient subgroups most likely to benefit from therapy, including patients with peripheral artery disease and CKD. Factors contributing to underutilization of newer cardiorenal protective glucose-lowering therapies range from cost and access barriers to clinician-level barriers (for example, lack of knowledge on CKD, lack of familiarity with CKD practice guidelines). Addressing these issues and helping patients work through financial and other access barriers is essential to optimize the utilization of these therapies and improve cardiorenal and metabolic outcomes.
So, has metformin been “dethroned” as a first-line therapy for T2D? As is often the case in medicine, the answer depends on the individual patient and clinical situation. Metformin remains an important first-line treatment in combination with lifestyle interventions to help patients with T2D without key cardiorenal comorbidities achieve individualized glycemic targets. However, based on evidence demonstrating cardiorenal protective benefits and improved glycemia and weight loss, GLP-1 agonists and SGLT2 inhibitors may be considered as first-line treatment for patients with T2D with or at high risk for ASCVD, HF, or CKD, regardless of the need for additional glucose-lowering agents and independent of background metformin. Ultimately, the choice of first-line therapy for patients with T2D should be informed by individualized treatment goals, preferences, and cost-related access. Continued efforts to increase patient access to GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment when indicated are essential to ensure optimal treatment and outcomes.
Dr. Neumiller is professor, department of pharmacotherapy, Washington State University, Spokane. He disclosed ties with Bayer, Boehringer Ingelheim, and Eli Lilly. Dr. Alicic is clinical professor, department of medicine, University of Washington; and associate director of research, Inland Northwest Washington, Providence St. Joseph Health, Spokane. She disclosed ties with Providence St. Joseph Health, Boehringer Ingelheim/Lilly, and Bayer.
A version of this article appeared on Medscape.com.
More cuts to physician payment ahead
In July, the Centers for Medicare and Medicaid Services released the 2024 Physician Fee Schedule (PFS) proposed rule on proposed policy changes for Medicare payments. The proposed rule contains 2,883 pages of proposals for physician, hospital outpatient department, and ambulatory surgery center (ASC) payments for calendar year 2024. For gastroenterologists, there was good news and bad news.
Medicare physician payments have been cut each year for the better part of a decade, with additional cuts proposed for 2024.
According to the American Medical Assocition, Medicare physician payment has already declined 26% in the last 22 years when adjusting for inflation, and that’s before factoring in the proposed cuts for 2024. Physicians are one of the only health care providers without an automatic inflationary increase, the AMA reports.
AGA opposes additional cuts to physician payments and will continue to advocate to stop them. AGA and many other specialty societies support H.R. 2474, the Strengthening Medicare for Patients and Providers Act. This bill would provide a permanent, annual update equal to the increase in the Medicare Economic Index, which is how the government measures inflation in medical practice. We will continue to advocate for permanent positive annual inflation updates, which would allow physicians to invest in their practices and implement new strategies to provide high-value care.
But in some positive news from the 2024 Medicare PFS, the Hospital Outpatient Prospective Payment System (OPPS) and the ASC proposed rules include increased hospital outpatient departments and ASC payments, continued telemedicine reimbursement and coverage through 2024, and a second one-year delay in changes to rules governing split/shared visits. Specifically:
OPPS Conversion Factor: The proposed CY 2024 Medicare conversion factor for outpatient hospital departments is $87.488, an increase of 2.8%, for hospitals that meet applicable quality reporting requirements.
ASC Conversion Factor: The proposed CY 2024 Ambulatory Surgical Center conversion factor is $53.397, an increase of 2.8%, for ASCs that meet applicable quality reporting requirements. The AGA and our sister societies continue to urge CMS to reduce this gap in the ASC facility fees, when compared to the outpatient hospital facility rates, which are estimated to be a roughly 48% differential in CY 2024.
Telehealth: CMS proposes to continue reimbursing telehealth services at current levels through 2024. Payment for audio-only evaluation and management (E/M) codes will continue at parity with follow-up in-person visits as it has throughout the pandemic. Additionally, CMS is implementing telehealth flexibilities that were included in the Consolidated Appropriations Act 2023 by allowing telehealth visits to originate at any site in the United States. This will allow patients throughout the country to maintain access to needed telehealth services without facing the logistical and safety challenges that can surround in-person visits. CMS is proposing to pay telehealth services at the nonfacility payment rate, which is the same rate as in-person office visits, lift the frequency limits on telehealth visits for subsequent hospital and skilled nursing facility visits, and allow direct supervision to be provided virtually.
Split (or shared) visits: CMS has proposed a second one-year delay to its proposed split/shared visits policy. The original proposal required that the billing provider in split/shared visits be whoever spent more than half of the total time with the patient (making time the only way to define substantive portion). CMS plans to delay that through at least Dec. 31, 2024. In the interim, practices can continue to use one of the three key components (history, exam, or medical decision-making) or more than half of the total time spent to determine who can bill for the visit. The GI societies will continue to advocate for appropriate reimbursement to align with new team-based models of care delivery.
Notably, the split (or shared) visits policy was also delayed in 2023 because of widespread concerns and feedback that the policy would disrupt team-based care and care delivery in the hospital setting. The American Medical Association CPT editorial panel, the body responsible for creating and maintaining CPT codes, has approved revisions to E/M guidelines that may help address some of CMS’s concerns.
For more information on issues affecting gastroenterologists in the 2024 Medicare PFS and OPPS/ASC proposed rules, visit the AGA news website.
Dr. Garcia serves as an advisor to the AGA AMA Relative-value Update Committee. She is clinical associate professor of medicine at Stanford (Calif.) University, where she is director of the neurogastroenterology and motility laboratory in the division of gastroenterology and hepatology, and associate chief medical information officer in ambulatory care at Stanford Health Care.
In July, the Centers for Medicare and Medicaid Services released the 2024 Physician Fee Schedule (PFS) proposed rule on proposed policy changes for Medicare payments. The proposed rule contains 2,883 pages of proposals for physician, hospital outpatient department, and ambulatory surgery center (ASC) payments for calendar year 2024. For gastroenterologists, there was good news and bad news.
Medicare physician payments have been cut each year for the better part of a decade, with additional cuts proposed for 2024.
According to the American Medical Assocition, Medicare physician payment has already declined 26% in the last 22 years when adjusting for inflation, and that’s before factoring in the proposed cuts for 2024. Physicians are one of the only health care providers without an automatic inflationary increase, the AMA reports.
AGA opposes additional cuts to physician payments and will continue to advocate to stop them. AGA and many other specialty societies support H.R. 2474, the Strengthening Medicare for Patients and Providers Act. This bill would provide a permanent, annual update equal to the increase in the Medicare Economic Index, which is how the government measures inflation in medical practice. We will continue to advocate for permanent positive annual inflation updates, which would allow physicians to invest in their practices and implement new strategies to provide high-value care.
But in some positive news from the 2024 Medicare PFS, the Hospital Outpatient Prospective Payment System (OPPS) and the ASC proposed rules include increased hospital outpatient departments and ASC payments, continued telemedicine reimbursement and coverage through 2024, and a second one-year delay in changes to rules governing split/shared visits. Specifically:
OPPS Conversion Factor: The proposed CY 2024 Medicare conversion factor for outpatient hospital departments is $87.488, an increase of 2.8%, for hospitals that meet applicable quality reporting requirements.
ASC Conversion Factor: The proposed CY 2024 Ambulatory Surgical Center conversion factor is $53.397, an increase of 2.8%, for ASCs that meet applicable quality reporting requirements. The AGA and our sister societies continue to urge CMS to reduce this gap in the ASC facility fees, when compared to the outpatient hospital facility rates, which are estimated to be a roughly 48% differential in CY 2024.
Telehealth: CMS proposes to continue reimbursing telehealth services at current levels through 2024. Payment for audio-only evaluation and management (E/M) codes will continue at parity with follow-up in-person visits as it has throughout the pandemic. Additionally, CMS is implementing telehealth flexibilities that were included in the Consolidated Appropriations Act 2023 by allowing telehealth visits to originate at any site in the United States. This will allow patients throughout the country to maintain access to needed telehealth services without facing the logistical and safety challenges that can surround in-person visits. CMS is proposing to pay telehealth services at the nonfacility payment rate, which is the same rate as in-person office visits, lift the frequency limits on telehealth visits for subsequent hospital and skilled nursing facility visits, and allow direct supervision to be provided virtually.
Split (or shared) visits: CMS has proposed a second one-year delay to its proposed split/shared visits policy. The original proposal required that the billing provider in split/shared visits be whoever spent more than half of the total time with the patient (making time the only way to define substantive portion). CMS plans to delay that through at least Dec. 31, 2024. In the interim, practices can continue to use one of the three key components (history, exam, or medical decision-making) or more than half of the total time spent to determine who can bill for the visit. The GI societies will continue to advocate for appropriate reimbursement to align with new team-based models of care delivery.
Notably, the split (or shared) visits policy was also delayed in 2023 because of widespread concerns and feedback that the policy would disrupt team-based care and care delivery in the hospital setting. The American Medical Association CPT editorial panel, the body responsible for creating and maintaining CPT codes, has approved revisions to E/M guidelines that may help address some of CMS’s concerns.
For more information on issues affecting gastroenterologists in the 2024 Medicare PFS and OPPS/ASC proposed rules, visit the AGA news website.
Dr. Garcia serves as an advisor to the AGA AMA Relative-value Update Committee. She is clinical associate professor of medicine at Stanford (Calif.) University, where she is director of the neurogastroenterology and motility laboratory in the division of gastroenterology and hepatology, and associate chief medical information officer in ambulatory care at Stanford Health Care.
In July, the Centers for Medicare and Medicaid Services released the 2024 Physician Fee Schedule (PFS) proposed rule on proposed policy changes for Medicare payments. The proposed rule contains 2,883 pages of proposals for physician, hospital outpatient department, and ambulatory surgery center (ASC) payments for calendar year 2024. For gastroenterologists, there was good news and bad news.
Medicare physician payments have been cut each year for the better part of a decade, with additional cuts proposed for 2024.
According to the American Medical Assocition, Medicare physician payment has already declined 26% in the last 22 years when adjusting for inflation, and that’s before factoring in the proposed cuts for 2024. Physicians are one of the only health care providers without an automatic inflationary increase, the AMA reports.
AGA opposes additional cuts to physician payments and will continue to advocate to stop them. AGA and many other specialty societies support H.R. 2474, the Strengthening Medicare for Patients and Providers Act. This bill would provide a permanent, annual update equal to the increase in the Medicare Economic Index, which is how the government measures inflation in medical practice. We will continue to advocate for permanent positive annual inflation updates, which would allow physicians to invest in their practices and implement new strategies to provide high-value care.
But in some positive news from the 2024 Medicare PFS, the Hospital Outpatient Prospective Payment System (OPPS) and the ASC proposed rules include increased hospital outpatient departments and ASC payments, continued telemedicine reimbursement and coverage through 2024, and a second one-year delay in changes to rules governing split/shared visits. Specifically:
OPPS Conversion Factor: The proposed CY 2024 Medicare conversion factor for outpatient hospital departments is $87.488, an increase of 2.8%, for hospitals that meet applicable quality reporting requirements.
ASC Conversion Factor: The proposed CY 2024 Ambulatory Surgical Center conversion factor is $53.397, an increase of 2.8%, for ASCs that meet applicable quality reporting requirements. The AGA and our sister societies continue to urge CMS to reduce this gap in the ASC facility fees, when compared to the outpatient hospital facility rates, which are estimated to be a roughly 48% differential in CY 2024.
Telehealth: CMS proposes to continue reimbursing telehealth services at current levels through 2024. Payment for audio-only evaluation and management (E/M) codes will continue at parity with follow-up in-person visits as it has throughout the pandemic. Additionally, CMS is implementing telehealth flexibilities that were included in the Consolidated Appropriations Act 2023 by allowing telehealth visits to originate at any site in the United States. This will allow patients throughout the country to maintain access to needed telehealth services without facing the logistical and safety challenges that can surround in-person visits. CMS is proposing to pay telehealth services at the nonfacility payment rate, which is the same rate as in-person office visits, lift the frequency limits on telehealth visits for subsequent hospital and skilled nursing facility visits, and allow direct supervision to be provided virtually.
Split (or shared) visits: CMS has proposed a second one-year delay to its proposed split/shared visits policy. The original proposal required that the billing provider in split/shared visits be whoever spent more than half of the total time with the patient (making time the only way to define substantive portion). CMS plans to delay that through at least Dec. 31, 2024. In the interim, practices can continue to use one of the three key components (history, exam, or medical decision-making) or more than half of the total time spent to determine who can bill for the visit. The GI societies will continue to advocate for appropriate reimbursement to align with new team-based models of care delivery.
Notably, the split (or shared) visits policy was also delayed in 2023 because of widespread concerns and feedback that the policy would disrupt team-based care and care delivery in the hospital setting. The American Medical Association CPT editorial panel, the body responsible for creating and maintaining CPT codes, has approved revisions to E/M guidelines that may help address some of CMS’s concerns.
For more information on issues affecting gastroenterologists in the 2024 Medicare PFS and OPPS/ASC proposed rules, visit the AGA news website.
Dr. Garcia serves as an advisor to the AGA AMA Relative-value Update Committee. She is clinical associate professor of medicine at Stanford (Calif.) University, where she is director of the neurogastroenterology and motility laboratory in the division of gastroenterology and hepatology, and associate chief medical information officer in ambulatory care at Stanford Health Care.
The multitasking myth
, and that we are accomplishing more in less time. In fact, there is no credible evidence that this is true, and a mountain of evidence showing exactly the opposite.
According to this study and others, multitasking results in an average of 2 hours per day of lost productivity. It decreases the quality of work performed and increases cortisol levels, which impedes cognitive functioning, leading to a further decrease in productivity in a vicious cycle, making you increasingly ineffective and destroying your motivation and mood.

On the surface, the reasons for this are not intuitively obvious. After all, simple and routine tasks are easy to perform simultaneously; we can all walk and chew gum at the same time or eat a snack while watching TV. The problems arise when we try to multitask more complex tasks that require thought and decision-making.
It turns out that the pressures of our modern world have evolved faster than our brains. We are still hard-wired for monotasking. When we think we are completing two tasks simultaneously, we are actually performing individual actions in rapid succession. Each time you switch tasks, your brain must turn off the cognitive rules of the previous task and turn on new rules for the next one. When you switch back, the process repeats in reverse. Each of those mental gear shifts takes time and costs us productivity. According to one psychologist, even brief mental blocks created by shifting between tasks can cost as much as 40% of someone’s productive time. We are also far more likely to make mistakes while we are doing it.
Furthermore, you are stifling your creativity and innovation because you don’t focus on one task long enough to come up with original insights. Multitasking also slows down your general cognitive functions, in the same way that keeping many windows are open on your computer slows down the entire system. A study from my alma mater, the University of California, San Francisco, concluded that multitasking negativity affects memory in both younger and older adults (although the effects were greater in older adults) .
So, what to do? The fact remains that, all too often, there really are too many tasks and not enough hours in the day. How can you get through them without falling into the multitasking trap?
The first rule is to prioritize. In his book “The Seven Habits of Highly Effective People,” Stephen Covey makes an important distinction between tasks that are important and those that are merely urgent. Tasks that are important and urgent tend to make time for themselves, because they must be taken care of immediately.
Jobs that are important but not urgent are the ones we tend to try to multitask. Because there is no immediate deadline, we think we can do two or more of them simultaneously, or we fall into the other major productivity trap: procrastination. Neither of those strategies tends to end well. Identify those important but not urgent tasks and force yourself to go through them one by one.
Urgent but unimportant tasks are the productivity thieves. They demand your attention but are not worthy of it. Most tasks in this category can be delegated. I have written about physicians’ workaholic and perfectionist tendencies that drive our conviction that no one else can do anything as well as we can. Does that unimportant task, even if urgent, really demand your time, skills, education, and medical license? Is there someone in your office, or possibly an outside contractor, who could do it just as well, and maybe faster?
In fact, that is the question you should ask every time a project triggers your urge to multitask: “Who could be doing this job – or at least a major part of it – instead of me?”
If your multitasking urges are deeply ingrained – particularly those that involve phones, laptops, and the cloud – you might consider employing electronic aids. SelfControl, for example, is a free, open-sourced app that lets you block your own access to distracting websites, your email servers, social media, or anything else on the Internet. You list the sites you wish to block and set a period of time to block them. Until the set time expires, you will be unable to access those sites, even if you restart your computer or delete the application.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].
, and that we are accomplishing more in less time. In fact, there is no credible evidence that this is true, and a mountain of evidence showing exactly the opposite.
According to this study and others, multitasking results in an average of 2 hours per day of lost productivity. It decreases the quality of work performed and increases cortisol levels, which impedes cognitive functioning, leading to a further decrease in productivity in a vicious cycle, making you increasingly ineffective and destroying your motivation and mood.
On the surface, the reasons for this are not intuitively obvious. After all, simple and routine tasks are easy to perform simultaneously; we can all walk and chew gum at the same time or eat a snack while watching TV. The problems arise when we try to multitask more complex tasks that require thought and decision-making.
It turns out that the pressures of our modern world have evolved faster than our brains. We are still hard-wired for monotasking. When we think we are completing two tasks simultaneously, we are actually performing individual actions in rapid succession. Each time you switch tasks, your brain must turn off the cognitive rules of the previous task and turn on new rules for the next one. When you switch back, the process repeats in reverse. Each of those mental gear shifts takes time and costs us productivity. According to one psychologist, even brief mental blocks created by shifting between tasks can cost as much as 40% of someone’s productive time. We are also far more likely to make mistakes while we are doing it.
Furthermore, you are stifling your creativity and innovation because you don’t focus on one task long enough to come up with original insights. Multitasking also slows down your general cognitive functions, in the same way that keeping many windows are open on your computer slows down the entire system. A study from my alma mater, the University of California, San Francisco, concluded that multitasking negativity affects memory in both younger and older adults (although the effects were greater in older adults) .
So, what to do? The fact remains that, all too often, there really are too many tasks and not enough hours in the day. How can you get through them without falling into the multitasking trap?
The first rule is to prioritize. In his book “The Seven Habits of Highly Effective People,” Stephen Covey makes an important distinction between tasks that are important and those that are merely urgent. Tasks that are important and urgent tend to make time for themselves, because they must be taken care of immediately.
Jobs that are important but not urgent are the ones we tend to try to multitask. Because there is no immediate deadline, we think we can do two or more of them simultaneously, or we fall into the other major productivity trap: procrastination. Neither of those strategies tends to end well. Identify those important but not urgent tasks and force yourself to go through them one by one.
Urgent but unimportant tasks are the productivity thieves. They demand your attention but are not worthy of it. Most tasks in this category can be delegated. I have written about physicians’ workaholic and perfectionist tendencies that drive our conviction that no one else can do anything as well as we can. Does that unimportant task, even if urgent, really demand your time, skills, education, and medical license? Is there someone in your office, or possibly an outside contractor, who could do it just as well, and maybe faster?
In fact, that is the question you should ask every time a project triggers your urge to multitask: “Who could be doing this job – or at least a major part of it – instead of me?”
If your multitasking urges are deeply ingrained – particularly those that involve phones, laptops, and the cloud – you might consider employing electronic aids. SelfControl, for example, is a free, open-sourced app that lets you block your own access to distracting websites, your email servers, social media, or anything else on the Internet. You list the sites you wish to block and set a period of time to block them. Until the set time expires, you will be unable to access those sites, even if you restart your computer or delete the application.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].
, and that we are accomplishing more in less time. In fact, there is no credible evidence that this is true, and a mountain of evidence showing exactly the opposite.
According to this study and others, multitasking results in an average of 2 hours per day of lost productivity. It decreases the quality of work performed and increases cortisol levels, which impedes cognitive functioning, leading to a further decrease in productivity in a vicious cycle, making you increasingly ineffective and destroying your motivation and mood.
On the surface, the reasons for this are not intuitively obvious. After all, simple and routine tasks are easy to perform simultaneously; we can all walk and chew gum at the same time or eat a snack while watching TV. The problems arise when we try to multitask more complex tasks that require thought and decision-making.
It turns out that the pressures of our modern world have evolved faster than our brains. We are still hard-wired for monotasking. When we think we are completing two tasks simultaneously, we are actually performing individual actions in rapid succession. Each time you switch tasks, your brain must turn off the cognitive rules of the previous task and turn on new rules for the next one. When you switch back, the process repeats in reverse. Each of those mental gear shifts takes time and costs us productivity. According to one psychologist, even brief mental blocks created by shifting between tasks can cost as much as 40% of someone’s productive time. We are also far more likely to make mistakes while we are doing it.
Furthermore, you are stifling your creativity and innovation because you don’t focus on one task long enough to come up with original insights. Multitasking also slows down your general cognitive functions, in the same way that keeping many windows are open on your computer slows down the entire system. A study from my alma mater, the University of California, San Francisco, concluded that multitasking negativity affects memory in both younger and older adults (although the effects were greater in older adults) .
So, what to do? The fact remains that, all too often, there really are too many tasks and not enough hours in the day. How can you get through them without falling into the multitasking trap?
The first rule is to prioritize. In his book “The Seven Habits of Highly Effective People,” Stephen Covey makes an important distinction between tasks that are important and those that are merely urgent. Tasks that are important and urgent tend to make time for themselves, because they must be taken care of immediately.
Jobs that are important but not urgent are the ones we tend to try to multitask. Because there is no immediate deadline, we think we can do two or more of them simultaneously, or we fall into the other major productivity trap: procrastination. Neither of those strategies tends to end well. Identify those important but not urgent tasks and force yourself to go through them one by one.
Urgent but unimportant tasks are the productivity thieves. They demand your attention but are not worthy of it. Most tasks in this category can be delegated. I have written about physicians’ workaholic and perfectionist tendencies that drive our conviction that no one else can do anything as well as we can. Does that unimportant task, even if urgent, really demand your time, skills, education, and medical license? Is there someone in your office, or possibly an outside contractor, who could do it just as well, and maybe faster?
In fact, that is the question you should ask every time a project triggers your urge to multitask: “Who could be doing this job – or at least a major part of it – instead of me?”
If your multitasking urges are deeply ingrained – particularly those that involve phones, laptops, and the cloud – you might consider employing electronic aids. SelfControl, for example, is a free, open-sourced app that lets you block your own access to distracting websites, your email servers, social media, or anything else on the Internet. You list the sites you wish to block and set a period of time to block them. Until the set time expires, you will be unable to access those sites, even if you restart your computer or delete the application.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].

Nutritional psychiatry: Does it exist?
Matt was diagnosed with ADHD combined type when he was 6 years old. Given his age, the family was reluctant to try medications, but after a couple years of parenting classes and reward charts, the parents requested a stimulant. He had significant improvement in focus and impulsivity but also reduced appetite. Now at age 13, irritability and depressive symptoms have been increasing for 9 months. Skeptical of adding another medication, his parents ask whether nutrition might be an alternative tool to treat his symptoms?
What a healthy diet even consists of seems to be a moving target over decades and years. Trendy research, supplements, and dietary approaches proliferate alongside appealing theories of action. In the end, weighing which intervention is effective for which disorder and at what cost becomes murky.
Yet several fundamental principles seem clear and consistent over time and across studies.

Starting early
There is reliable evidence that in the perinatal environment, nutrition sets the stage for many aspects of healthy development. These effects are likely mediated variously through the hypothalamic-pituitary-adrenal axis, the trillions of gut bacteria that make up the microbiome, gene-environment interactions, and more. Maternal malnutrition and stress prenatally puts infants at risk for not only poor birth outcomes but also psychiatric challenges throughout childhood, such as ADHD, anxiety, depression, and autism.1
Intervening in the perinatal period has long-term benefits. A first step includes assessing food security, beginning with consistent access to nutritious food. It is important to inquire about the role of food and nutrition in the family’s history and culture, as well as identifying resources to support access to affordable nutrition. This can be paired with parenting interventions, such as family meals without screens. This may require scaffolding positive conversations in high-conflict family settings (see The Family Dinner Project).
Healthy diets promote mental health
If food security is achieved, what is next? Clinicians can inquire about the who, what, where, when, and why of nutrition to learn about a family’s eating habits.2 While randomized controlled data is very limited, both cross-sectional and longitudinal studies show that healthy diets in youth correlate with mental health – more healthy foods reducing internalizing and externalizing disorders, and more typical Western diets increasing the risk. On average, dietary interventions include higher levels of fruits and vegetables, fish, and nuts, and lower levels of processed foods.2 There is not evidence that restrictive diets or fasting is appropriate or safe for youth. Additionally, involving children in getting, growing, or preparing food with gradually increasing autonomy fosters self-confidence and skill development.
In those struggling with restrictive eating disorders, food is medicine – helping those with restrictive diets to develop more balanced and adequate intake for metabolic needs. Outside of diagnosable eating disorders, weight or body mass index is less of a goal or marker when it comes to mental health. Instead, look for participation in enjoyable activities, opportunities to move and rest, and a body image that supports self-care and self-confidence (see the National Institutes of Health’s We Can! Program). Creating dissonance with cultural ideals of appearance centered on thinness can prevent future eating disorders.3
Nutraceutical options
Outside of eating disorders, specific foods and plants with health or medicinal properties – variously called nutraceuticals, phytoceuticals, or micronutrients – have emerging evidence in mental health. A 2022 expert academic consensus panel reviewed the literature to create clinical guidelines in this area.4 For major depression, adding omega-3 fatty acids to standard antidepressant treatment or standalone St. John’s wort have adequate evidence to recommend, while adjunctive probiotics, zinc, saffron, and curcumin have sufficient though less robust evidence. S-adenosyl methionine, vitamin D, and methyfolate showed only weak evidence for depression, while vitamin C, magnesium, creatine, N-acetylcysteine, folate, and monotherapy omega-3s do not have sufficient evidence to be recommended. For ADHD there was weak support for vitamin D, but no clear evidence for omega-3s, zinc, gingko, or acetyl L-carnitine. For anxiety, there is moderate evidence for ashwagandha and lavender in adults. A child psychiatry review suggests also trying chamomile for generalized anxiety based on the evidence in young adults, and underscores some data for N-acetylcysteine for OCD in particular.5
Many of these nutraceuticals exhibit small or moderate effects in a limited number of trials, with generally much less data for youth, compared with adults. While the same could be said for many on- and off-label uses of psychiatric medications for kids, clinicians would be wise to consider these highly specific nutritional interventions as items on the menu of treatment options rather than stand-alone treatments.
Revisitng the case study
Reflecting on Matt’s care, his pediatrician first assessed his dietary patterns, noting late-night eating and caffeine use with minimal hydration or fiber across the day. Recommendations for keeping fruit and vegetable snacks easily accessible as well as carrying a water flask are well received. They also discuss adding omega-3 fatty acids and probiotics with his morning stimulant while he awaits a referral for cognitive-behavioral therapy in order to address his depressive symptoms and minimize medication needs.
Beyond addressing food security and balanced family meals, specific interventions may be appropriate as initial treatment adjuncts for mild and some moderate mental illness. For more intense moderate to severe illness, nutritional psychiatry may be considered in combination with treatments with stronger evidence. At a community level, clinicians can help advocate for universal school meal programs to address food security, and so-called salad bar interventions to increase fruit/vegetable uptake among school-age children.
Dr. Rosenfeld is associate professor of psychiatry and pediatrics at University of Vermont and the Vermont Center for Children, Youth, and Families, both in Burlington. He has no disclosures.
References
1 Vohr BR et al. Pediatrics. 2017;139:S38-49.
2. Hosker DK et al. Child Adol Psychiatr Clin N Am. 2019;28(2):171-93.
3. Stice E et al. Int J Eat Disord. 2013;46(5):478-85.
4. Sarris J et al. World J Biol Psychiatry. 2022;23(6):424-55.
5. Simkin DR et al. Child Adolesc Psychiatric Clin N Am. 2023;32:193-216.
Matt was diagnosed with ADHD combined type when he was 6 years old. Given his age, the family was reluctant to try medications, but after a couple years of parenting classes and reward charts, the parents requested a stimulant. He had significant improvement in focus and impulsivity but also reduced appetite. Now at age 13, irritability and depressive symptoms have been increasing for 9 months. Skeptical of adding another medication, his parents ask whether nutrition might be an alternative tool to treat his symptoms?
What a healthy diet even consists of seems to be a moving target over decades and years. Trendy research, supplements, and dietary approaches proliferate alongside appealing theories of action. In the end, weighing which intervention is effective for which disorder and at what cost becomes murky.
Yet several fundamental principles seem clear and consistent over time and across studies.
Starting early
There is reliable evidence that in the perinatal environment, nutrition sets the stage for many aspects of healthy development. These effects are likely mediated variously through the hypothalamic-pituitary-adrenal axis, the trillions of gut bacteria that make up the microbiome, gene-environment interactions, and more. Maternal malnutrition and stress prenatally puts infants at risk for not only poor birth outcomes but also psychiatric challenges throughout childhood, such as ADHD, anxiety, depression, and autism.1
Intervening in the perinatal period has long-term benefits. A first step includes assessing food security, beginning with consistent access to nutritious food. It is important to inquire about the role of food and nutrition in the family’s history and culture, as well as identifying resources to support access to affordable nutrition. This can be paired with parenting interventions, such as family meals without screens. This may require scaffolding positive conversations in high-conflict family settings (see The Family Dinner Project).
Healthy diets promote mental health
If food security is achieved, what is next? Clinicians can inquire about the who, what, where, when, and why of nutrition to learn about a family’s eating habits.2 While randomized controlled data is very limited, both cross-sectional and longitudinal studies show that healthy diets in youth correlate with mental health – more healthy foods reducing internalizing and externalizing disorders, and more typical Western diets increasing the risk. On average, dietary interventions include higher levels of fruits and vegetables, fish, and nuts, and lower levels of processed foods.2 There is not evidence that restrictive diets or fasting is appropriate or safe for youth. Additionally, involving children in getting, growing, or preparing food with gradually increasing autonomy fosters self-confidence and skill development.
In those struggling with restrictive eating disorders, food is medicine – helping those with restrictive diets to develop more balanced and adequate intake for metabolic needs. Outside of diagnosable eating disorders, weight or body mass index is less of a goal or marker when it comes to mental health. Instead, look for participation in enjoyable activities, opportunities to move and rest, and a body image that supports self-care and self-confidence (see the National Institutes of Health’s We Can! Program). Creating dissonance with cultural ideals of appearance centered on thinness can prevent future eating disorders.3
Nutraceutical options
Outside of eating disorders, specific foods and plants with health or medicinal properties – variously called nutraceuticals, phytoceuticals, or micronutrients – have emerging evidence in mental health. A 2022 expert academic consensus panel reviewed the literature to create clinical guidelines in this area.4 For major depression, adding omega-3 fatty acids to standard antidepressant treatment or standalone St. John’s wort have adequate evidence to recommend, while adjunctive probiotics, zinc, saffron, and curcumin have sufficient though less robust evidence. S-adenosyl methionine, vitamin D, and methyfolate showed only weak evidence for depression, while vitamin C, magnesium, creatine, N-acetylcysteine, folate, and monotherapy omega-3s do not have sufficient evidence to be recommended. For ADHD there was weak support for vitamin D, but no clear evidence for omega-3s, zinc, gingko, or acetyl L-carnitine. For anxiety, there is moderate evidence for ashwagandha and lavender in adults. A child psychiatry review suggests also trying chamomile for generalized anxiety based on the evidence in young adults, and underscores some data for N-acetylcysteine for OCD in particular.5
Many of these nutraceuticals exhibit small or moderate effects in a limited number of trials, with generally much less data for youth, compared with adults. While the same could be said for many on- and off-label uses of psychiatric medications for kids, clinicians would be wise to consider these highly specific nutritional interventions as items on the menu of treatment options rather than stand-alone treatments.
Revisitng the case study
Reflecting on Matt’s care, his pediatrician first assessed his dietary patterns, noting late-night eating and caffeine use with minimal hydration or fiber across the day. Recommendations for keeping fruit and vegetable snacks easily accessible as well as carrying a water flask are well received. They also discuss adding omega-3 fatty acids and probiotics with his morning stimulant while he awaits a referral for cognitive-behavioral therapy in order to address his depressive symptoms and minimize medication needs.
Beyond addressing food security and balanced family meals, specific interventions may be appropriate as initial treatment adjuncts for mild and some moderate mental illness. For more intense moderate to severe illness, nutritional psychiatry may be considered in combination with treatments with stronger evidence. At a community level, clinicians can help advocate for universal school meal programs to address food security, and so-called salad bar interventions to increase fruit/vegetable uptake among school-age children.
Dr. Rosenfeld is associate professor of psychiatry and pediatrics at University of Vermont and the Vermont Center for Children, Youth, and Families, both in Burlington. He has no disclosures.
References
1 Vohr BR et al. Pediatrics. 2017;139:S38-49.
2. Hosker DK et al. Child Adol Psychiatr Clin N Am. 2019;28(2):171-93.
3. Stice E et al. Int J Eat Disord. 2013;46(5):478-85.
4. Sarris J et al. World J Biol Psychiatry. 2022;23(6):424-55.
5. Simkin DR et al. Child Adolesc Psychiatric Clin N Am. 2023;32:193-216.
Matt was diagnosed with ADHD combined type when he was 6 years old. Given his age, the family was reluctant to try medications, but after a couple years of parenting classes and reward charts, the parents requested a stimulant. He had significant improvement in focus and impulsivity but also reduced appetite. Now at age 13, irritability and depressive symptoms have been increasing for 9 months. Skeptical of adding another medication, his parents ask whether nutrition might be an alternative tool to treat his symptoms?
What a healthy diet even consists of seems to be a moving target over decades and years. Trendy research, supplements, and dietary approaches proliferate alongside appealing theories of action. In the end, weighing which intervention is effective for which disorder and at what cost becomes murky.
Yet several fundamental principles seem clear and consistent over time and across studies.
Starting early
There is reliable evidence that in the perinatal environment, nutrition sets the stage for many aspects of healthy development. These effects are likely mediated variously through the hypothalamic-pituitary-adrenal axis, the trillions of gut bacteria that make up the microbiome, gene-environment interactions, and more. Maternal malnutrition and stress prenatally puts infants at risk for not only poor birth outcomes but also psychiatric challenges throughout childhood, such as ADHD, anxiety, depression, and autism.1
Intervening in the perinatal period has long-term benefits. A first step includes assessing food security, beginning with consistent access to nutritious food. It is important to inquire about the role of food and nutrition in the family’s history and culture, as well as identifying resources to support access to affordable nutrition. This can be paired with parenting interventions, such as family meals without screens. This may require scaffolding positive conversations in high-conflict family settings (see The Family Dinner Project).
Healthy diets promote mental health
If food security is achieved, what is next? Clinicians can inquire about the who, what, where, when, and why of nutrition to learn about a family’s eating habits.2 While randomized controlled data is very limited, both cross-sectional and longitudinal studies show that healthy diets in youth correlate with mental health – more healthy foods reducing internalizing and externalizing disorders, and more typical Western diets increasing the risk. On average, dietary interventions include higher levels of fruits and vegetables, fish, and nuts, and lower levels of processed foods.2 There is not evidence that restrictive diets or fasting is appropriate or safe for youth. Additionally, involving children in getting, growing, or preparing food with gradually increasing autonomy fosters self-confidence and skill development.
In those struggling with restrictive eating disorders, food is medicine – helping those with restrictive diets to develop more balanced and adequate intake for metabolic needs. Outside of diagnosable eating disorders, weight or body mass index is less of a goal or marker when it comes to mental health. Instead, look for participation in enjoyable activities, opportunities to move and rest, and a body image that supports self-care and self-confidence (see the National Institutes of Health’s We Can! Program). Creating dissonance with cultural ideals of appearance centered on thinness can prevent future eating disorders.3
Nutraceutical options
Outside of eating disorders, specific foods and plants with health or medicinal properties – variously called nutraceuticals, phytoceuticals, or micronutrients – have emerging evidence in mental health. A 2022 expert academic consensus panel reviewed the literature to create clinical guidelines in this area.4 For major depression, adding omega-3 fatty acids to standard antidepressant treatment or standalone St. John’s wort have adequate evidence to recommend, while adjunctive probiotics, zinc, saffron, and curcumin have sufficient though less robust evidence. S-adenosyl methionine, vitamin D, and methyfolate showed only weak evidence for depression, while vitamin C, magnesium, creatine, N-acetylcysteine, folate, and monotherapy omega-3s do not have sufficient evidence to be recommended. For ADHD there was weak support for vitamin D, but no clear evidence for omega-3s, zinc, gingko, or acetyl L-carnitine. For anxiety, there is moderate evidence for ashwagandha and lavender in adults. A child psychiatry review suggests also trying chamomile for generalized anxiety based on the evidence in young adults, and underscores some data for N-acetylcysteine for OCD in particular.5
Many of these nutraceuticals exhibit small or moderate effects in a limited number of trials, with generally much less data for youth, compared with adults. While the same could be said for many on- and off-label uses of psychiatric medications for kids, clinicians would be wise to consider these highly specific nutritional interventions as items on the menu of treatment options rather than stand-alone treatments.
Revisitng the case study
Reflecting on Matt’s care, his pediatrician first assessed his dietary patterns, noting late-night eating and caffeine use with minimal hydration or fiber across the day. Recommendations for keeping fruit and vegetable snacks easily accessible as well as carrying a water flask are well received. They also discuss adding omega-3 fatty acids and probiotics with his morning stimulant while he awaits a referral for cognitive-behavioral therapy in order to address his depressive symptoms and minimize medication needs.
Beyond addressing food security and balanced family meals, specific interventions may be appropriate as initial treatment adjuncts for mild and some moderate mental illness. For more intense moderate to severe illness, nutritional psychiatry may be considered in combination with treatments with stronger evidence. At a community level, clinicians can help advocate for universal school meal programs to address food security, and so-called salad bar interventions to increase fruit/vegetable uptake among school-age children.
Dr. Rosenfeld is associate professor of psychiatry and pediatrics at University of Vermont and the Vermont Center for Children, Youth, and Families, both in Burlington. He has no disclosures.
References
1 Vohr BR et al. Pediatrics. 2017;139:S38-49.
2. Hosker DK et al. Child Adol Psychiatr Clin N Am. 2019;28(2):171-93.
3. Stice E et al. Int J Eat Disord. 2013;46(5):478-85.
4. Sarris J et al. World J Biol Psychiatry. 2022;23(6):424-55.
5. Simkin DR et al. Child Adolesc Psychiatric Clin N Am. 2023;32:193-216.
Zuranolone: A novel postpartum depression treatment, with lingering questions
Postpartum depression (PPD) remains the most common complication in modern obstetrics, and a leading cause of postpartum mortality in the first year of life. The last 15 years have brought considerable progress with respect to adoption of systematic screening for PPD across America. Screening for PPD, most often using the Edinburgh Postnatal Depression Scale (EPDS), has become part of routine obstetrical care, and is also widely used in pediatric settings.
That is the good news. But the flip side of the identification of those women whose scores on the EPDS suggest significant depressive symptoms is that the number of these patients who, following identification, receive referrals for adequate treatment that gets them well is unfortunately low. This “perinatal treatment cascade” refers to the majority of women who, on the other side of identification of PPD, fail to receive adequate treatment and continue to have persistent depression (Cox E. et al. J Clin Psychiatry. 2016 Sep;77[9]:1189-1200). This is perhaps the greatest challenge to the field and to clinicians – how do we, on the other side of screening, see that these women get access to care and get well with the available treatments at hand?

Recently, a widely read and circulated article was published in The Wall Street Journal about the challenges associated with navigating care resources for women suffering from PPD. In that article, it was made clear, based on clinical vignette after clinical vignette from postpartum women across America, that neither obstetricians, mental health professionals, nor pediatricians are the “clinical home” for women suffering from postpartum mood and anxiety disorders. The article painfully highlights the system-wide failure to coordinate mental health care for women suffering from postpartum psychiatric illness.
Within a day of the publication of The Wall Street Journal article, the Food and Drug Administration approved zuranolone (Zurzuvae; Sage Therapeutics; Cambridge, Mass.) for the treatment of PPD following the review of two studies demonstrating the superiority of the new medicine over placebo. Women who were enrolled met criteria for major depressive disorder based on Diagnostic and Statistical Manual of Mental Disorders criteria beginning in no earlier than the third trimester of pregnancy or later than 4 weeks of delivery. The two studies included a combined sample size of approximately 350 patients suffering from severe PPD. In the studies, women received either 50 mg or 40 mg of zuranolone, or placebo for 14 days. Treatment was associated with a significant change in the Hamilton Depression Rating Scale at day 15, and treatment response was maintained at day 42, which was 4 weeks after the last dose of study medication.
Zuranolone is a neuroactive steroid, which is taken orally, unlike brexanolone (Zulresso; Sage Therapeutics; Cambridge, Mass.), which requires intravenous administration. Zuranolone will be commercially available based on estimates around the fourth quarter of 2023. The most common side effects are drowsiness, dizziness, and sedation, and the FDA label will have a boxed warning about zuranolone’s potential to impact a person’s driving ability, and performance of potentially hazardous activities.
It is noteworthy that while this new medication received FDA approval for the PPD indication, it did not receive FDA approval for the treatment of major depressive disorder (MDD), and the agency issued a Complete Response Letter to the manufacturers noting their application did not provide substantial evidence of effectiveness in MDD. The FDA said in the Complete Response Letter that an additional study or studies will be needed; the manufacturers are currently evaluating next steps.
Where zuranolone fits into the treatment algorithm for severe PPD
Many clinicians who support women with PPD will wonder, upon hearing this news, where zuranolone fits into the treatment algorithm for severe postpartum major depression. Some relevant issues that may determine the answer are the following:
Cost. The cost of brexanolone was substantial, at $34,000 per year, and was viewed by some as a limiting factor in terms of its very limited uptake. As of this column’s publication, zuranolone’s manufacturer has not stated how much the medication will cost.
Breastfeeding. Unlike selective serotonin reuptake inhibitors, which have been demonstrated to be effective for the treatment of PPD and safe during pregnancy and lactation, we have sparse data on the safety of zuranolone for women who wish to breastfeed. It is also unclear whether women eligible for zuranolone would, based on the limited data on safety in lactation, choose deferral of breastfeeding for 14 days in exchange for treatment.
Duration of treatment. While zuranolone was studied in the context of 14 days of acute treatment, then out to day 42, we have no published data on what happens on the other side of this brief interval. As a simple example, in a patient with a history of recurrent major depression previously treated with antidepressants, but where antidepressants were perhaps deferred during pregnancy, is PPD to be treated with zuranolone for 14 days? Or, hypothetically, should it be followed by empiric antidepressant treatment at day 14? Alternatively, are patient and clinician supposed to wait until recurrence occurs before pursuing adjunctive antidepressant therapy whether it is pharmacologic, nonpharmacologic, or both?
Treatment in patients with bipolar disorder. It is also unclear whether treatment with zuranolone applies to other populations of postpartum women. Certainly, for women with bipolar depression, which is common in postpartum women given the vulnerability of bipolar women to new onset of depression or postpartum depressive relapse of underlying disorder, we simply have no data regarding where zuranolone might fit in with respect to this group of patients.
The answers to these questions may help to determine whether zuranolone, a new antidepressant with efficacy, quick time to onset, and a novel mechanism of action is a “game changer.” The article in The Wall Street Journal provided me with some optimism, as it gave PPD and the issues surrounding PPD the attention it deserves in a major periodical. As a new treatment, it may help alleviate suffering at a critical time for patients and their families. We are inching closer to mitigation of stigma associated with this common illness.
Thinking back across the last 3 decades of my treating women suffering from PPD, I have reflected on what has gotten these patients well. I concluded that , along with family and community-based support groups, as well as a culture that reduces stigma and by so doing lessens the toll of this important and too frequently incompletely-treated illness.
Dr. Cohen is the director of the Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital (MGH) in Boston, which provides information resources and conducts clinical care and research in reproductive mental health. He has been a consultant to manufacturers of psychiatric medications. The Center for Women’s Mental Health at MGH was a non-enrolling site for the pivotal phase 3 SKYLARK trial evaluating zuranolone. Full disclosure information for Dr. Cohen is available at womensmentalhealth.org. Email Dr. Cohen at [email protected].
Postpartum depression (PPD) remains the most common complication in modern obstetrics, and a leading cause of postpartum mortality in the first year of life. The last 15 years have brought considerable progress with respect to adoption of systematic screening for PPD across America. Screening for PPD, most often using the Edinburgh Postnatal Depression Scale (EPDS), has become part of routine obstetrical care, and is also widely used in pediatric settings.
That is the good news. But the flip side of the identification of those women whose scores on the EPDS suggest significant depressive symptoms is that the number of these patients who, following identification, receive referrals for adequate treatment that gets them well is unfortunately low. This “perinatal treatment cascade” refers to the majority of women who, on the other side of identification of PPD, fail to receive adequate treatment and continue to have persistent depression (Cox E. et al. J Clin Psychiatry. 2016 Sep;77[9]:1189-1200). This is perhaps the greatest challenge to the field and to clinicians – how do we, on the other side of screening, see that these women get access to care and get well with the available treatments at hand?
Recently, a widely read and circulated article was published in The Wall Street Journal about the challenges associated with navigating care resources for women suffering from PPD. In that article, it was made clear, based on clinical vignette after clinical vignette from postpartum women across America, that neither obstetricians, mental health professionals, nor pediatricians are the “clinical home” for women suffering from postpartum mood and anxiety disorders. The article painfully highlights the system-wide failure to coordinate mental health care for women suffering from postpartum psychiatric illness.
Within a day of the publication of The Wall Street Journal article, the Food and Drug Administration approved zuranolone (Zurzuvae; Sage Therapeutics; Cambridge, Mass.) for the treatment of PPD following the review of two studies demonstrating the superiority of the new medicine over placebo. Women who were enrolled met criteria for major depressive disorder based on Diagnostic and Statistical Manual of Mental Disorders criteria beginning in no earlier than the third trimester of pregnancy or later than 4 weeks of delivery. The two studies included a combined sample size of approximately 350 patients suffering from severe PPD. In the studies, women received either 50 mg or 40 mg of zuranolone, or placebo for 14 days. Treatment was associated with a significant change in the Hamilton Depression Rating Scale at day 15, and treatment response was maintained at day 42, which was 4 weeks after the last dose of study medication.
Zuranolone is a neuroactive steroid, which is taken orally, unlike brexanolone (Zulresso; Sage Therapeutics; Cambridge, Mass.), which requires intravenous administration. Zuranolone will be commercially available based on estimates around the fourth quarter of 2023. The most common side effects are drowsiness, dizziness, and sedation, and the FDA label will have a boxed warning about zuranolone’s potential to impact a person’s driving ability, and performance of potentially hazardous activities.
It is noteworthy that while this new medication received FDA approval for the PPD indication, it did not receive FDA approval for the treatment of major depressive disorder (MDD), and the agency issued a Complete Response Letter to the manufacturers noting their application did not provide substantial evidence of effectiveness in MDD. The FDA said in the Complete Response Letter that an additional study or studies will be needed; the manufacturers are currently evaluating next steps.
Where zuranolone fits into the treatment algorithm for severe PPD
Many clinicians who support women with PPD will wonder, upon hearing this news, where zuranolone fits into the treatment algorithm for severe postpartum major depression. Some relevant issues that may determine the answer are the following:
Cost. The cost of brexanolone was substantial, at $34,000 per year, and was viewed by some as a limiting factor in terms of its very limited uptake. As of this column’s publication, zuranolone’s manufacturer has not stated how much the medication will cost.
Breastfeeding. Unlike selective serotonin reuptake inhibitors, which have been demonstrated to be effective for the treatment of PPD and safe during pregnancy and lactation, we have sparse data on the safety of zuranolone for women who wish to breastfeed. It is also unclear whether women eligible for zuranolone would, based on the limited data on safety in lactation, choose deferral of breastfeeding for 14 days in exchange for treatment.
Duration of treatment. While zuranolone was studied in the context of 14 days of acute treatment, then out to day 42, we have no published data on what happens on the other side of this brief interval. As a simple example, in a patient with a history of recurrent major depression previously treated with antidepressants, but where antidepressants were perhaps deferred during pregnancy, is PPD to be treated with zuranolone for 14 days? Or, hypothetically, should it be followed by empiric antidepressant treatment at day 14? Alternatively, are patient and clinician supposed to wait until recurrence occurs before pursuing adjunctive antidepressant therapy whether it is pharmacologic, nonpharmacologic, or both?
Treatment in patients with bipolar disorder. It is also unclear whether treatment with zuranolone applies to other populations of postpartum women. Certainly, for women with bipolar depression, which is common in postpartum women given the vulnerability of bipolar women to new onset of depression or postpartum depressive relapse of underlying disorder, we simply have no data regarding where zuranolone might fit in with respect to this group of patients.
The answers to these questions may help to determine whether zuranolone, a new antidepressant with efficacy, quick time to onset, and a novel mechanism of action is a “game changer.” The article in The Wall Street Journal provided me with some optimism, as it gave PPD and the issues surrounding PPD the attention it deserves in a major periodical. As a new treatment, it may help alleviate suffering at a critical time for patients and their families. We are inching closer to mitigation of stigma associated with this common illness.
Thinking back across the last 3 decades of my treating women suffering from PPD, I have reflected on what has gotten these patients well. I concluded that , along with family and community-based support groups, as well as a culture that reduces stigma and by so doing lessens the toll of this important and too frequently incompletely-treated illness.
Dr. Cohen is the director of the Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital (MGH) in Boston, which provides information resources and conducts clinical care and research in reproductive mental health. He has been a consultant to manufacturers of psychiatric medications. The Center for Women’s Mental Health at MGH was a non-enrolling site for the pivotal phase 3 SKYLARK trial evaluating zuranolone. Full disclosure information for Dr. Cohen is available at womensmentalhealth.org. Email Dr. Cohen at [email protected].
Postpartum depression (PPD) remains the most common complication in modern obstetrics, and a leading cause of postpartum mortality in the first year of life. The last 15 years have brought considerable progress with respect to adoption of systematic screening for PPD across America. Screening for PPD, most often using the Edinburgh Postnatal Depression Scale (EPDS), has become part of routine obstetrical care, and is also widely used in pediatric settings.
That is the good news. But the flip side of the identification of those women whose scores on the EPDS suggest significant depressive symptoms is that the number of these patients who, following identification, receive referrals for adequate treatment that gets them well is unfortunately low. This “perinatal treatment cascade” refers to the majority of women who, on the other side of identification of PPD, fail to receive adequate treatment and continue to have persistent depression (Cox E. et al. J Clin Psychiatry. 2016 Sep;77[9]:1189-1200). This is perhaps the greatest challenge to the field and to clinicians – how do we, on the other side of screening, see that these women get access to care and get well with the available treatments at hand?
Recently, a widely read and circulated article was published in The Wall Street Journal about the challenges associated with navigating care resources for women suffering from PPD. In that article, it was made clear, based on clinical vignette after clinical vignette from postpartum women across America, that neither obstetricians, mental health professionals, nor pediatricians are the “clinical home” for women suffering from postpartum mood and anxiety disorders. The article painfully highlights the system-wide failure to coordinate mental health care for women suffering from postpartum psychiatric illness.
Within a day of the publication of The Wall Street Journal article, the Food and Drug Administration approved zuranolone (Zurzuvae; Sage Therapeutics; Cambridge, Mass.) for the treatment of PPD following the review of two studies demonstrating the superiority of the new medicine over placebo. Women who were enrolled met criteria for major depressive disorder based on Diagnostic and Statistical Manual of Mental Disorders criteria beginning in no earlier than the third trimester of pregnancy or later than 4 weeks of delivery. The two studies included a combined sample size of approximately 350 patients suffering from severe PPD. In the studies, women received either 50 mg or 40 mg of zuranolone, or placebo for 14 days. Treatment was associated with a significant change in the Hamilton Depression Rating Scale at day 15, and treatment response was maintained at day 42, which was 4 weeks after the last dose of study medication.
Zuranolone is a neuroactive steroid, which is taken orally, unlike brexanolone (Zulresso; Sage Therapeutics; Cambridge, Mass.), which requires intravenous administration. Zuranolone will be commercially available based on estimates around the fourth quarter of 2023. The most common side effects are drowsiness, dizziness, and sedation, and the FDA label will have a boxed warning about zuranolone’s potential to impact a person’s driving ability, and performance of potentially hazardous activities.
It is noteworthy that while this new medication received FDA approval for the PPD indication, it did not receive FDA approval for the treatment of major depressive disorder (MDD), and the agency issued a Complete Response Letter to the manufacturers noting their application did not provide substantial evidence of effectiveness in MDD. The FDA said in the Complete Response Letter that an additional study or studies will be needed; the manufacturers are currently evaluating next steps.
Where zuranolone fits into the treatment algorithm for severe PPD
Many clinicians who support women with PPD will wonder, upon hearing this news, where zuranolone fits into the treatment algorithm for severe postpartum major depression. Some relevant issues that may determine the answer are the following:
Cost. The cost of brexanolone was substantial, at $34,000 per year, and was viewed by some as a limiting factor in terms of its very limited uptake. As of this column’s publication, zuranolone’s manufacturer has not stated how much the medication will cost.
Breastfeeding. Unlike selective serotonin reuptake inhibitors, which have been demonstrated to be effective for the treatment of PPD and safe during pregnancy and lactation, we have sparse data on the safety of zuranolone for women who wish to breastfeed. It is also unclear whether women eligible for zuranolone would, based on the limited data on safety in lactation, choose deferral of breastfeeding for 14 days in exchange for treatment.
Duration of treatment. While zuranolone was studied in the context of 14 days of acute treatment, then out to day 42, we have no published data on what happens on the other side of this brief interval. As a simple example, in a patient with a history of recurrent major depression previously treated with antidepressants, but where antidepressants were perhaps deferred during pregnancy, is PPD to be treated with zuranolone for 14 days? Or, hypothetically, should it be followed by empiric antidepressant treatment at day 14? Alternatively, are patient and clinician supposed to wait until recurrence occurs before pursuing adjunctive antidepressant therapy whether it is pharmacologic, nonpharmacologic, or both?
Treatment in patients with bipolar disorder. It is also unclear whether treatment with zuranolone applies to other populations of postpartum women. Certainly, for women with bipolar depression, which is common in postpartum women given the vulnerability of bipolar women to new onset of depression or postpartum depressive relapse of underlying disorder, we simply have no data regarding where zuranolone might fit in with respect to this group of patients.
The answers to these questions may help to determine whether zuranolone, a new antidepressant with efficacy, quick time to onset, and a novel mechanism of action is a “game changer.” The article in The Wall Street Journal provided me with some optimism, as it gave PPD and the issues surrounding PPD the attention it deserves in a major periodical. As a new treatment, it may help alleviate suffering at a critical time for patients and their families. We are inching closer to mitigation of stigma associated with this common illness.
Thinking back across the last 3 decades of my treating women suffering from PPD, I have reflected on what has gotten these patients well. I concluded that , along with family and community-based support groups, as well as a culture that reduces stigma and by so doing lessens the toll of this important and too frequently incompletely-treated illness.
Dr. Cohen is the director of the Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital (MGH) in Boston, which provides information resources and conducts clinical care and research in reproductive mental health. He has been a consultant to manufacturers of psychiatric medications. The Center for Women’s Mental Health at MGH was a non-enrolling site for the pivotal phase 3 SKYLARK trial evaluating zuranolone. Full disclosure information for Dr. Cohen is available at womensmentalhealth.org. Email Dr. Cohen at [email protected].