User login
Biologics better solo than with methotrexate in psoriatic arthritis
Ustekinumab or a tumor necrosis factor inhibitor (TNFi) are better used alone than with methotrexate in the treatment of psoriatic arthritis suggest the results of PsABio (A Study on Assessment of STELARA and Tumor Necrosis Factor Alpha Inhibitor Therapies in Participants With Psoriatic Arthritis), a large, ongoing, prospective observational study.

The percentages of patients achieving multiple psoriatic arthritis disease activity outcome measures at 6 months were higher if biologic monotherapy was used rather than a biologic in combination with methotrexate.
For example, minimal disease activity (MDA) was achieved by 27.5% of patients taking ustekinumab as monotherapy and by 32.1% of those taking a TNFi alone. When methotrexate was used in combination, the respective percentages of patients achieving MDA were 23.7% and 27.8%.
A similar pattern was seen for very-low disease activity (VLDA), with 9.8% of patients in the ustekinumab monotherapy arm and 12% of those in the TNFi monotherapy arm achieving this target, compared with 5.7% and 5.4% when these drugs were combined with methotrexate.
MDA is defined as meeting five or more cutoffs for seven domains of disease activity, and VLDA for all seven: 0-1 tender joints, 0-1 swollen joints, Psoriasis Area Severity Index 1 or less or body surface area involved 3% or less, 0-1 tender entheseal points, Health Assessment Questionnaire score of 0.5 or less, patient global disease activity visual analog scale score of 20 or lower, and patient pain visual analog scale score of 15 or lower.
Other outcome measures used that showed no advantage of adding methotrexate to these biologics were the Clinical Disease Activity in Psoriatic Arthritis low disease activity and remission scores, the patient acceptable symptoms rate of the 12-item Psoriatic Arthritis Impact of Disease Questionnaire, and improvement in skin involvement.
“Patients were no more likely to achieve lower disease activity or a remission target having received methotrexate than they did just on the biologic drug on its own,” Stefan Siebert, MBBCh, PhD, one of the PsABio investigators, said in an interview.
Dr. Siebert, who is clinical senior lecturer in inflammation and rheumatology at the University of Glasgow (Scotland), was scheduled to present the findings at the British Society for Rheumatology annual conference. The meeting was canceled because of the ongoing COVID-19 crisis. Abstracts and ePosters from the meeting have since been released in a supplement to Rheumatology and via the BSR’s conference app.
First data for ustekinumab
“There certainly doesn’t appear to be any added benefit from using methotrexate on a group level in patients getting ustekinumab and TNF inhibitors,” Dr. Siebert said. “We’ve looked at everything,” he emphasized, and “none of the single domains or composite measures were improved by the addition of methotrexate. I think we knew that for TNF inhibitors, but the key thing is we’ve never known that for ustekinumab, and this is the first study to show that.”
Indeed, the findings match up with those from the SEAM-PsA (Etanercept and Methotrexate in Subjects with Psoriatic Arthritis) study in which patients who were treated with the TNFi etanercept as monotherapy did much better than those given the TNFi in combination with methotrexate or methotrexate alone. While not a randomized trial, PsABio now shows that the same is true for ustekinumab.
Obviously, there are some clear differences between a clinical trial and an observational study such as PsABio. For one thing, there was no randomization and patients taking methotrexate were presumably doing so for good reason, Dr. Siebert said. Secondly, there was no methotrexate-only arm.
PsABio recruited patients who were starting treatment with either ustekinumab or a new TNFi as first-, second-, or third-line biologic disease-modifying antirheumatic therapy (DMARD). “These are all people starting on a biologic, so they’ve already got severe disease and have failed methotrexate on some level. So everything we’ve done is biologic without methotrexate or biologic with methotrexate,” Dr. Siebert explained. Patients may not have been taking methotrexate for a variety of reasons, such as inefficacy or side effects, so PsABio “doesn’t tell us anything about methotrexate on its own.”
Time to rethink ingrained methotrexate use
The rationale for using methotrexate in combination with biologics in psoriatic arthritis comes from its long-standing use in rheumatoid arthritis. Much of what is advocated in guidelines comes from experience in RA, Dr. Siebert said.
“In rheumatoid arthritis, we know that the TNF inhibitors work much better if you use methotrexate, that’s a given,” he noted. “We’ve been trained that you have to have methotrexate to have a biologic. However, PsABio, together with other studies, show that you don’t have to, and you should have a good reason to add methotrexate.”
Individual patients may still benefit from methotrexate use, but the decision to treat all patients the same is not supported by the current evidence. “It’s good that it shows that, actually, once you get someone on a decent biologic, it’s working: It’s doing what it ‘says on the tin’ for a lot of patients. I really think that is the key message, here, that you don’t have to; this reassures clinicians and actually makes them think ‘should this patient be on methotrexate?’ ” Dr. Siebert said.
The PsABio study was funded by Janssen. Dr. Siebert has acted as a consultant to and received research funding from Janssen, UCB, Pfizer, Boehringer Ingelheim, Novartis, and Celgene. He has also acted as a consultant for AbbVie and received research support from Bristol-Myers Squibb.
SOURCE: Siebert S et al. Rheumatology. 2020;59(Suppl 2). doi: 10.1093/rheumatology/keaa110.023, Abstract O24.
Ustekinumab or a tumor necrosis factor inhibitor (TNFi) are better used alone than with methotrexate in the treatment of psoriatic arthritis suggest the results of PsABio (A Study on Assessment of STELARA and Tumor Necrosis Factor Alpha Inhibitor Therapies in Participants With Psoriatic Arthritis), a large, ongoing, prospective observational study.
The percentages of patients achieving multiple psoriatic arthritis disease activity outcome measures at 6 months were higher if biologic monotherapy was used rather than a biologic in combination with methotrexate.
For example, minimal disease activity (MDA) was achieved by 27.5% of patients taking ustekinumab as monotherapy and by 32.1% of those taking a TNFi alone. When methotrexate was used in combination, the respective percentages of patients achieving MDA were 23.7% and 27.8%.
A similar pattern was seen for very-low disease activity (VLDA), with 9.8% of patients in the ustekinumab monotherapy arm and 12% of those in the TNFi monotherapy arm achieving this target, compared with 5.7% and 5.4% when these drugs were combined with methotrexate.
MDA is defined as meeting five or more cutoffs for seven domains of disease activity, and VLDA for all seven: 0-1 tender joints, 0-1 swollen joints, Psoriasis Area Severity Index 1 or less or body surface area involved 3% or less, 0-1 tender entheseal points, Health Assessment Questionnaire score of 0.5 or less, patient global disease activity visual analog scale score of 20 or lower, and patient pain visual analog scale score of 15 or lower.
Other outcome measures used that showed no advantage of adding methotrexate to these biologics were the Clinical Disease Activity in Psoriatic Arthritis low disease activity and remission scores, the patient acceptable symptoms rate of the 12-item Psoriatic Arthritis Impact of Disease Questionnaire, and improvement in skin involvement.
“Patients were no more likely to achieve lower disease activity or a remission target having received methotrexate than they did just on the biologic drug on its own,” Stefan Siebert, MBBCh, PhD, one of the PsABio investigators, said in an interview.
Dr. Siebert, who is clinical senior lecturer in inflammation and rheumatology at the University of Glasgow (Scotland), was scheduled to present the findings at the British Society for Rheumatology annual conference. The meeting was canceled because of the ongoing COVID-19 crisis. Abstracts and ePosters from the meeting have since been released in a supplement to Rheumatology and via the BSR’s conference app.
First data for ustekinumab
“There certainly doesn’t appear to be any added benefit from using methotrexate on a group level in patients getting ustekinumab and TNF inhibitors,” Dr. Siebert said. “We’ve looked at everything,” he emphasized, and “none of the single domains or composite measures were improved by the addition of methotrexate. I think we knew that for TNF inhibitors, but the key thing is we’ve never known that for ustekinumab, and this is the first study to show that.”
Indeed, the findings match up with those from the SEAM-PsA (Etanercept and Methotrexate in Subjects with Psoriatic Arthritis) study in which patients who were treated with the TNFi etanercept as monotherapy did much better than those given the TNFi in combination with methotrexate or methotrexate alone. While not a randomized trial, PsABio now shows that the same is true for ustekinumab.
Obviously, there are some clear differences between a clinical trial and an observational study such as PsABio. For one thing, there was no randomization and patients taking methotrexate were presumably doing so for good reason, Dr. Siebert said. Secondly, there was no methotrexate-only arm.
PsABio recruited patients who were starting treatment with either ustekinumab or a new TNFi as first-, second-, or third-line biologic disease-modifying antirheumatic therapy (DMARD). “These are all people starting on a biologic, so they’ve already got severe disease and have failed methotrexate on some level. So everything we’ve done is biologic without methotrexate or biologic with methotrexate,” Dr. Siebert explained. Patients may not have been taking methotrexate for a variety of reasons, such as inefficacy or side effects, so PsABio “doesn’t tell us anything about methotrexate on its own.”
Time to rethink ingrained methotrexate use
The rationale for using methotrexate in combination with biologics in psoriatic arthritis comes from its long-standing use in rheumatoid arthritis. Much of what is advocated in guidelines comes from experience in RA, Dr. Siebert said.
“In rheumatoid arthritis, we know that the TNF inhibitors work much better if you use methotrexate, that’s a given,” he noted. “We’ve been trained that you have to have methotrexate to have a biologic. However, PsABio, together with other studies, show that you don’t have to, and you should have a good reason to add methotrexate.”
Individual patients may still benefit from methotrexate use, but the decision to treat all patients the same is not supported by the current evidence. “It’s good that it shows that, actually, once you get someone on a decent biologic, it’s working: It’s doing what it ‘says on the tin’ for a lot of patients. I really think that is the key message, here, that you don’t have to; this reassures clinicians and actually makes them think ‘should this patient be on methotrexate?’ ” Dr. Siebert said.
The PsABio study was funded by Janssen. Dr. Siebert has acted as a consultant to and received research funding from Janssen, UCB, Pfizer, Boehringer Ingelheim, Novartis, and Celgene. He has also acted as a consultant for AbbVie and received research support from Bristol-Myers Squibb.
SOURCE: Siebert S et al. Rheumatology. 2020;59(Suppl 2). doi: 10.1093/rheumatology/keaa110.023, Abstract O24.
Ustekinumab or a tumor necrosis factor inhibitor (TNFi) are better used alone than with methotrexate in the treatment of psoriatic arthritis suggest the results of PsABio (A Study on Assessment of STELARA and Tumor Necrosis Factor Alpha Inhibitor Therapies in Participants With Psoriatic Arthritis), a large, ongoing, prospective observational study.
The percentages of patients achieving multiple psoriatic arthritis disease activity outcome measures at 6 months were higher if biologic monotherapy was used rather than a biologic in combination with methotrexate.
For example, minimal disease activity (MDA) was achieved by 27.5% of patients taking ustekinumab as monotherapy and by 32.1% of those taking a TNFi alone. When methotrexate was used in combination, the respective percentages of patients achieving MDA were 23.7% and 27.8%.
A similar pattern was seen for very-low disease activity (VLDA), with 9.8% of patients in the ustekinumab monotherapy arm and 12% of those in the TNFi monotherapy arm achieving this target, compared with 5.7% and 5.4% when these drugs were combined with methotrexate.
MDA is defined as meeting five or more cutoffs for seven domains of disease activity, and VLDA for all seven: 0-1 tender joints, 0-1 swollen joints, Psoriasis Area Severity Index 1 or less or body surface area involved 3% or less, 0-1 tender entheseal points, Health Assessment Questionnaire score of 0.5 or less, patient global disease activity visual analog scale score of 20 or lower, and patient pain visual analog scale score of 15 or lower.
Other outcome measures used that showed no advantage of adding methotrexate to these biologics were the Clinical Disease Activity in Psoriatic Arthritis low disease activity and remission scores, the patient acceptable symptoms rate of the 12-item Psoriatic Arthritis Impact of Disease Questionnaire, and improvement in skin involvement.
“Patients were no more likely to achieve lower disease activity or a remission target having received methotrexate than they did just on the biologic drug on its own,” Stefan Siebert, MBBCh, PhD, one of the PsABio investigators, said in an interview.
Dr. Siebert, who is clinical senior lecturer in inflammation and rheumatology at the University of Glasgow (Scotland), was scheduled to present the findings at the British Society for Rheumatology annual conference. The meeting was canceled because of the ongoing COVID-19 crisis. Abstracts and ePosters from the meeting have since been released in a supplement to Rheumatology and via the BSR’s conference app.
First data for ustekinumab
“There certainly doesn’t appear to be any added benefit from using methotrexate on a group level in patients getting ustekinumab and TNF inhibitors,” Dr. Siebert said. “We’ve looked at everything,” he emphasized, and “none of the single domains or composite measures were improved by the addition of methotrexate. I think we knew that for TNF inhibitors, but the key thing is we’ve never known that for ustekinumab, and this is the first study to show that.”
Indeed, the findings match up with those from the SEAM-PsA (Etanercept and Methotrexate in Subjects with Psoriatic Arthritis) study in which patients who were treated with the TNFi etanercept as monotherapy did much better than those given the TNFi in combination with methotrexate or methotrexate alone. While not a randomized trial, PsABio now shows that the same is true for ustekinumab.
Obviously, there are some clear differences between a clinical trial and an observational study such as PsABio. For one thing, there was no randomization and patients taking methotrexate were presumably doing so for good reason, Dr. Siebert said. Secondly, there was no methotrexate-only arm.
PsABio recruited patients who were starting treatment with either ustekinumab or a new TNFi as first-, second-, or third-line biologic disease-modifying antirheumatic therapy (DMARD). “These are all people starting on a biologic, so they’ve already got severe disease and have failed methotrexate on some level. So everything we’ve done is biologic without methotrexate or biologic with methotrexate,” Dr. Siebert explained. Patients may not have been taking methotrexate for a variety of reasons, such as inefficacy or side effects, so PsABio “doesn’t tell us anything about methotrexate on its own.”
Time to rethink ingrained methotrexate use
The rationale for using methotrexate in combination with biologics in psoriatic arthritis comes from its long-standing use in rheumatoid arthritis. Much of what is advocated in guidelines comes from experience in RA, Dr. Siebert said.
“In rheumatoid arthritis, we know that the TNF inhibitors work much better if you use methotrexate, that’s a given,” he noted. “We’ve been trained that you have to have methotrexate to have a biologic. However, PsABio, together with other studies, show that you don’t have to, and you should have a good reason to add methotrexate.”
Individual patients may still benefit from methotrexate use, but the decision to treat all patients the same is not supported by the current evidence. “It’s good that it shows that, actually, once you get someone on a decent biologic, it’s working: It’s doing what it ‘says on the tin’ for a lot of patients. I really think that is the key message, here, that you don’t have to; this reassures clinicians and actually makes them think ‘should this patient be on methotrexate?’ ” Dr. Siebert said.
The PsABio study was funded by Janssen. Dr. Siebert has acted as a consultant to and received research funding from Janssen, UCB, Pfizer, Boehringer Ingelheim, Novartis, and Celgene. He has also acted as a consultant for AbbVie and received research support from Bristol-Myers Squibb.
SOURCE: Siebert S et al. Rheumatology. 2020;59(Suppl 2). doi: 10.1093/rheumatology/keaa110.023, Abstract O24.
FROM BSR 2020
Angiotensin drugs and COVID-19: More reassuring data
Initial data from one Chinese center on the use of angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) in patients hospitalized with COVID-19 appear to give some further reassurance about continued use of these drugs.
The report from one hospital in Wuhan found that among patients with hypertension hospitalized with the COVID-19 virus, there was no difference in disease severity or death rate in patients taking ACE inhibitors or ARBs and those not taking such medications.
The data were published online April 23 in JAMA Cardiology.
The study adds to another recent report in a larger number of COVID-19 patients from nine Chinese hospitals that suggested a beneficial effect of ACE inhibitors or ARBs on mortality.
Additional studies
Two other similar studies have also been recently released. Another study from China, published online March 31 in Emerging Microbes & Infections, included a small sample of 42 hospitalized patients with COVID-19 on antihypertensive therapy. Those on ACE inhibitor/ARB therapy had a lower rate of severe disease and a trend toward a lower level of IL-6 in peripheral blood. In addition, patients on ACE inhibitor/ARB therapy had increased CD3+ and CD8+ T-cell counts in peripheral blood and decreased peak viral load compared with other antihypertensive drugs.
And a preliminary study from the UK, which has not yet been peer reviewed, found that treatment with ACE inhibitors was associated with a reduced risk of rapidly deteriorating severe COVID-19 disease.
The study, available online on MedRxiv, a preprint server for health sciences, reports on 205 acute inpatients with COVID-19 at King’s College Hospital and Princess Royal University Hospital, London.
Of these, 51.2% had hypertension, 30.2% had diabetes, and 14.6% had ischemic heart disease or heart failure. Of the 37 patients on ACE inhibitors, five (14%) died or required critical care support compared with 29% (48/168) of patients not taking an ACE inhibitor.
New Wuhan study
The authors of the new article published in JAMA Cardiology, led by Juyi Li, MD, reported on a case series of 1,178 patients hospitalized with COVID-19 at the Central Hospital of Wuhan, Hubei, China, between Jan. 15 and March 15, 2020.
Patients were a median age of 55 years, and 46% were men. They had an overall in-hospital mortality rate of 11%.
Of the 1,178 patients, 362 (30.7%) had a diagnosis of hypertension. These patients were older (median age, 66 years) and had a greater prevalence of chronic diseases. Patients with hypertension also had more severe manifestations of COVID-19 compared to those without hypertension, including higher rates of acute respiratory distress syndrome and in-hospital mortality (21.3% vs. 6.5%).
Of the 362 patients with hypertension, 31.8% were taking ACE inhibitors or ARBs.
Apart from a greater prevalence of coronary artery disease, patients taking ACE inhibitors or ARBs had similar comorbidities to those not taking these medications, and also similar laboratory profile results including blood counts, inflammatory markers, renal and liver function tests, and cardiac biomarkers, although those taking ACE inhibitors/ARBs had higher levels of alkaline phosphatase.
The most commonly used antihypertensive drugs were calcium blockers. The percentage of patients with hypertension taking any drug or drug combination did not differ between those with severe and nonsevere infections and between those who survived and those who died.
Specifically regarding ACE inhibitors/ARBs, there was no difference between those with severe versus nonsevere illness in the use of ACE inhibitors (9.2% vs. 10.1%; P = .80), ARBs (24.9% vs. 21.2%; P = .40), or the composite of ACE inhibitors or ARBs (32.9% vs. 30.7%; P = .65).
Similarly, there were no differences in nonsurvivors and survivors in the use of ACE inhibitors (9.1% vs. 9.8%; P = .85); ARBs (19.5% vs. 23.9%; P = .42), or the composite of ACE inhibitors or ARBs (27.3% vs. 33.0%; P = .34).
The frequency of severe illness and death also did not differ between those treated with and without ACE inhibitors/ARBs in patients with hypertension and other various chronic conditions including coronary heart disease, cerebrovascular disease, diabetes, neurological disease, and chronic renal disease.
The authors noted that these data confirm previous reports showing that patients with hypertension have more severe illness and higher mortality rates associated with COVID-19 than those without hypertension.
But they added: “Our data provide some reassurance that ACE inhibitors/ARBs are not associated with the progression or outcome of COVID-19 hospitalizations in patients with hypertension.”
They also noted that these results support the recommendations from almost all major cardiovascular societies that patients do not discontinue ACE inhibitors or ARBs because of worries about COVID-19.
However, the authors did point out some limitations of their study, which included a small number of patients with hypertension taking ACE inhibitors or ARBs and the fact that a nonsevere disease course was still severe enough to require hospitalization. In addition, it was not clear whether ACE inhibitor/ARB treatment at baseline was maintained throughout hospitalization for all patients.
This was also an observational comparison and may be biased by differences in patients taking versus not taking ACE inhibitors or ARBs at the time of hospitalization, although the measured baseline characteristics were similar in both groups.
But the authors also highlighted the finding that, in this cohort, patients with hypertension had three times the mortality rate of all other patients hospitalized with COVID-19.
“Hypertension combined with cardiovascular and cerebrovascular disease, diabetes, and chronic kidney disease would predispose patients to an increased risk of severity and mortality of COVID-19. Therefore, patients with these underlying conditions who develop COVID-19 require particularly intensive surveillance and care,” they wrote.
Experts cautiously optimistic
Some cardiovascular experts were cautiously optimistic about these latest results.
Michael A. Weber, MD, professor of medicine at the State University of New York, Brooklyn, and editor-in-chief of the Journal of Clinical Hypertension, said: “This new report from Wuhan, China, gives modest reassurance that the use of ACE inhibitors or ARBs in hypertensive patients with COVID-19 disease does not increase the risk of clinical deterioration or death.
“Ongoing, more definitive studies should help resolve competing hypotheses regarding the effects of these agents: whether the increased ACE2 enzyme levels they produce can worsen outcomes by increasing access of the COVID virus to lung tissue; or whether there is a benefit linked to a protective effect of increased ACE2 on alveolar cell function,” Dr. Weber noted.
“Though the number of patients included in this new report is small, it is startling that hypertensive patients were three times as likely as nonhypertensives to have a fatal outcome, presumably reflecting vulnerability due to the cardiovascular and metabolic comorbidities associated with hypertension,” he added.
“In any case, for now, clinicians should continue treating hypertensive patients with whichever drugs, including ACE inhibitors and ARBs, best provide protection from adverse outcomes,” Dr. Weber concluded.
John McMurray, MD, professor of medical cardiology, University of Glasgow, Scotland, commented: “This study from Wuhan provides some reassurance about one of the two questions about ACEI/ARBs: Do these drugs increase susceptibility to infection? And if [the patient is] infected, do they increase the severity of infection? This study addresses the latter question and appears to suggest no increased severity.”
However, Dr. McMurray pointed out that the study had many limitations. There were only small patient numbers and the data were unadjusted, “although it looks like the ACE inhibitor/ARB treated patients were higher risk to start with.” It was an observational study, and patients were not randomized and were predominantly treated with ARBs, and not ACE inhibitors, so “we don’t know if the concerns apply equally to these two classes of drug.
“Other data published and unpublished supporting this (even showing better outcomes in patients treated with an ACE inhibitor/ARB), and, to date, any concerns about these drugs remain unsubstantiated and the guidance from medical societies to continue treatment with these agents in patients prescribed them seems wise,” Dr. McMurray added.
Franz H. Messerli, MD, professor of medicine at the University of Bern, Switzerland, commented: “The study from Wuhan is not a great study. They didn’t even do a multivariable analysis. They could have done a bit more with the data, but it still gives some reassurance.”
Dr. Messerli said it was “interesting” that 30% of the patients hospitalized with COVID-19 in the sample had hypertension. “That corresponds to the general population, so does not suggest that having hypertension increases susceptibility to infection – but it does seem to increase the risk of a bad outcome.”
Dr. Messerli noted that there are two more similar studies due to be published soon, both said to suggest either a beneficial or neutral effect of ACE inhibitors/ARBs on COVID-19 outcomes in hospitalized patients.
“This does help with confidence in prescribing these agents and reinforces the recommendations for patients to stay on these drugs,” he said.
“However, none of these studies address the infectivity issue – whether their use upregulates the ACE2 receptor, which the virus uses to gain entry to cells, thereby increasing susceptibility to the infection,” Dr. Messerli cautioned. “But the similar or better outcomes on these drugs are encouraging,” he added.
The Wuhan study was supported by the Health and Family Planning Commission of Wuhan City, China. The authors have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Initial data from one Chinese center on the use of angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) in patients hospitalized with COVID-19 appear to give some further reassurance about continued use of these drugs.
The report from one hospital in Wuhan found that among patients with hypertension hospitalized with the COVID-19 virus, there was no difference in disease severity or death rate in patients taking ACE inhibitors or ARBs and those not taking such medications.
The data were published online April 23 in JAMA Cardiology.
The study adds to another recent report in a larger number of COVID-19 patients from nine Chinese hospitals that suggested a beneficial effect of ACE inhibitors or ARBs on mortality.
Additional studies
Two other similar studies have also been recently released. Another study from China, published online March 31 in Emerging Microbes & Infections, included a small sample of 42 hospitalized patients with COVID-19 on antihypertensive therapy. Those on ACE inhibitor/ARB therapy had a lower rate of severe disease and a trend toward a lower level of IL-6 in peripheral blood. In addition, patients on ACE inhibitor/ARB therapy had increased CD3+ and CD8+ T-cell counts in peripheral blood and decreased peak viral load compared with other antihypertensive drugs.
And a preliminary study from the UK, which has not yet been peer reviewed, found that treatment with ACE inhibitors was associated with a reduced risk of rapidly deteriorating severe COVID-19 disease.
The study, available online on MedRxiv, a preprint server for health sciences, reports on 205 acute inpatients with COVID-19 at King’s College Hospital and Princess Royal University Hospital, London.
Of these, 51.2% had hypertension, 30.2% had diabetes, and 14.6% had ischemic heart disease or heart failure. Of the 37 patients on ACE inhibitors, five (14%) died or required critical care support compared with 29% (48/168) of patients not taking an ACE inhibitor.
New Wuhan study
The authors of the new article published in JAMA Cardiology, led by Juyi Li, MD, reported on a case series of 1,178 patients hospitalized with COVID-19 at the Central Hospital of Wuhan, Hubei, China, between Jan. 15 and March 15, 2020.
Patients were a median age of 55 years, and 46% were men. They had an overall in-hospital mortality rate of 11%.
Of the 1,178 patients, 362 (30.7%) had a diagnosis of hypertension. These patients were older (median age, 66 years) and had a greater prevalence of chronic diseases. Patients with hypertension also had more severe manifestations of COVID-19 compared to those without hypertension, including higher rates of acute respiratory distress syndrome and in-hospital mortality (21.3% vs. 6.5%).
Of the 362 patients with hypertension, 31.8% were taking ACE inhibitors or ARBs.
Apart from a greater prevalence of coronary artery disease, patients taking ACE inhibitors or ARBs had similar comorbidities to those not taking these medications, and also similar laboratory profile results including blood counts, inflammatory markers, renal and liver function tests, and cardiac biomarkers, although those taking ACE inhibitors/ARBs had higher levels of alkaline phosphatase.
The most commonly used antihypertensive drugs were calcium blockers. The percentage of patients with hypertension taking any drug or drug combination did not differ between those with severe and nonsevere infections and between those who survived and those who died.
Specifically regarding ACE inhibitors/ARBs, there was no difference between those with severe versus nonsevere illness in the use of ACE inhibitors (9.2% vs. 10.1%; P = .80), ARBs (24.9% vs. 21.2%; P = .40), or the composite of ACE inhibitors or ARBs (32.9% vs. 30.7%; P = .65).
Similarly, there were no differences in nonsurvivors and survivors in the use of ACE inhibitors (9.1% vs. 9.8%; P = .85); ARBs (19.5% vs. 23.9%; P = .42), or the composite of ACE inhibitors or ARBs (27.3% vs. 33.0%; P = .34).
The frequency of severe illness and death also did not differ between those treated with and without ACE inhibitors/ARBs in patients with hypertension and other various chronic conditions including coronary heart disease, cerebrovascular disease, diabetes, neurological disease, and chronic renal disease.
The authors noted that these data confirm previous reports showing that patients with hypertension have more severe illness and higher mortality rates associated with COVID-19 than those without hypertension.
But they added: “Our data provide some reassurance that ACE inhibitors/ARBs are not associated with the progression or outcome of COVID-19 hospitalizations in patients with hypertension.”
They also noted that these results support the recommendations from almost all major cardiovascular societies that patients do not discontinue ACE inhibitors or ARBs because of worries about COVID-19.
However, the authors did point out some limitations of their study, which included a small number of patients with hypertension taking ACE inhibitors or ARBs and the fact that a nonsevere disease course was still severe enough to require hospitalization. In addition, it was not clear whether ACE inhibitor/ARB treatment at baseline was maintained throughout hospitalization for all patients.
This was also an observational comparison and may be biased by differences in patients taking versus not taking ACE inhibitors or ARBs at the time of hospitalization, although the measured baseline characteristics were similar in both groups.
But the authors also highlighted the finding that, in this cohort, patients with hypertension had three times the mortality rate of all other patients hospitalized with COVID-19.
“Hypertension combined with cardiovascular and cerebrovascular disease, diabetes, and chronic kidney disease would predispose patients to an increased risk of severity and mortality of COVID-19. Therefore, patients with these underlying conditions who develop COVID-19 require particularly intensive surveillance and care,” they wrote.
Experts cautiously optimistic
Some cardiovascular experts were cautiously optimistic about these latest results.
Michael A. Weber, MD, professor of medicine at the State University of New York, Brooklyn, and editor-in-chief of the Journal of Clinical Hypertension, said: “This new report from Wuhan, China, gives modest reassurance that the use of ACE inhibitors or ARBs in hypertensive patients with COVID-19 disease does not increase the risk of clinical deterioration or death.
“Ongoing, more definitive studies should help resolve competing hypotheses regarding the effects of these agents: whether the increased ACE2 enzyme levels they produce can worsen outcomes by increasing access of the COVID virus to lung tissue; or whether there is a benefit linked to a protective effect of increased ACE2 on alveolar cell function,” Dr. Weber noted.
“Though the number of patients included in this new report is small, it is startling that hypertensive patients were three times as likely as nonhypertensives to have a fatal outcome, presumably reflecting vulnerability due to the cardiovascular and metabolic comorbidities associated with hypertension,” he added.
“In any case, for now, clinicians should continue treating hypertensive patients with whichever drugs, including ACE inhibitors and ARBs, best provide protection from adverse outcomes,” Dr. Weber concluded.
John McMurray, MD, professor of medical cardiology, University of Glasgow, Scotland, commented: “This study from Wuhan provides some reassurance about one of the two questions about ACEI/ARBs: Do these drugs increase susceptibility to infection? And if [the patient is] infected, do they increase the severity of infection? This study addresses the latter question and appears to suggest no increased severity.”
However, Dr. McMurray pointed out that the study had many limitations. There were only small patient numbers and the data were unadjusted, “although it looks like the ACE inhibitor/ARB treated patients were higher risk to start with.” It was an observational study, and patients were not randomized and were predominantly treated with ARBs, and not ACE inhibitors, so “we don’t know if the concerns apply equally to these two classes of drug.
“Other data published and unpublished supporting this (even showing better outcomes in patients treated with an ACE inhibitor/ARB), and, to date, any concerns about these drugs remain unsubstantiated and the guidance from medical societies to continue treatment with these agents in patients prescribed them seems wise,” Dr. McMurray added.
Franz H. Messerli, MD, professor of medicine at the University of Bern, Switzerland, commented: “The study from Wuhan is not a great study. They didn’t even do a multivariable analysis. They could have done a bit more with the data, but it still gives some reassurance.”
Dr. Messerli said it was “interesting” that 30% of the patients hospitalized with COVID-19 in the sample had hypertension. “That corresponds to the general population, so does not suggest that having hypertension increases susceptibility to infection – but it does seem to increase the risk of a bad outcome.”
Dr. Messerli noted that there are two more similar studies due to be published soon, both said to suggest either a beneficial or neutral effect of ACE inhibitors/ARBs on COVID-19 outcomes in hospitalized patients.
“This does help with confidence in prescribing these agents and reinforces the recommendations for patients to stay on these drugs,” he said.
“However, none of these studies address the infectivity issue – whether their use upregulates the ACE2 receptor, which the virus uses to gain entry to cells, thereby increasing susceptibility to the infection,” Dr. Messerli cautioned. “But the similar or better outcomes on these drugs are encouraging,” he added.
The Wuhan study was supported by the Health and Family Planning Commission of Wuhan City, China. The authors have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Initial data from one Chinese center on the use of angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) in patients hospitalized with COVID-19 appear to give some further reassurance about continued use of these drugs.
The report from one hospital in Wuhan found that among patients with hypertension hospitalized with the COVID-19 virus, there was no difference in disease severity or death rate in patients taking ACE inhibitors or ARBs and those not taking such medications.
The data were published online April 23 in JAMA Cardiology.
The study adds to another recent report in a larger number of COVID-19 patients from nine Chinese hospitals that suggested a beneficial effect of ACE inhibitors or ARBs on mortality.
Additional studies
Two other similar studies have also been recently released. Another study from China, published online March 31 in Emerging Microbes & Infections, included a small sample of 42 hospitalized patients with COVID-19 on antihypertensive therapy. Those on ACE inhibitor/ARB therapy had a lower rate of severe disease and a trend toward a lower level of IL-6 in peripheral blood. In addition, patients on ACE inhibitor/ARB therapy had increased CD3+ and CD8+ T-cell counts in peripheral blood and decreased peak viral load compared with other antihypertensive drugs.
And a preliminary study from the UK, which has not yet been peer reviewed, found that treatment with ACE inhibitors was associated with a reduced risk of rapidly deteriorating severe COVID-19 disease.
The study, available online on MedRxiv, a preprint server for health sciences, reports on 205 acute inpatients with COVID-19 at King’s College Hospital and Princess Royal University Hospital, London.
Of these, 51.2% had hypertension, 30.2% had diabetes, and 14.6% had ischemic heart disease or heart failure. Of the 37 patients on ACE inhibitors, five (14%) died or required critical care support compared with 29% (48/168) of patients not taking an ACE inhibitor.
New Wuhan study
The authors of the new article published in JAMA Cardiology, led by Juyi Li, MD, reported on a case series of 1,178 patients hospitalized with COVID-19 at the Central Hospital of Wuhan, Hubei, China, between Jan. 15 and March 15, 2020.
Patients were a median age of 55 years, and 46% were men. They had an overall in-hospital mortality rate of 11%.
Of the 1,178 patients, 362 (30.7%) had a diagnosis of hypertension. These patients were older (median age, 66 years) and had a greater prevalence of chronic diseases. Patients with hypertension also had more severe manifestations of COVID-19 compared to those without hypertension, including higher rates of acute respiratory distress syndrome and in-hospital mortality (21.3% vs. 6.5%).
Of the 362 patients with hypertension, 31.8% were taking ACE inhibitors or ARBs.
Apart from a greater prevalence of coronary artery disease, patients taking ACE inhibitors or ARBs had similar comorbidities to those not taking these medications, and also similar laboratory profile results including blood counts, inflammatory markers, renal and liver function tests, and cardiac biomarkers, although those taking ACE inhibitors/ARBs had higher levels of alkaline phosphatase.
The most commonly used antihypertensive drugs were calcium blockers. The percentage of patients with hypertension taking any drug or drug combination did not differ between those with severe and nonsevere infections and between those who survived and those who died.
Specifically regarding ACE inhibitors/ARBs, there was no difference between those with severe versus nonsevere illness in the use of ACE inhibitors (9.2% vs. 10.1%; P = .80), ARBs (24.9% vs. 21.2%; P = .40), or the composite of ACE inhibitors or ARBs (32.9% vs. 30.7%; P = .65).
Similarly, there were no differences in nonsurvivors and survivors in the use of ACE inhibitors (9.1% vs. 9.8%; P = .85); ARBs (19.5% vs. 23.9%; P = .42), or the composite of ACE inhibitors or ARBs (27.3% vs. 33.0%; P = .34).
The frequency of severe illness and death also did not differ between those treated with and without ACE inhibitors/ARBs in patients with hypertension and other various chronic conditions including coronary heart disease, cerebrovascular disease, diabetes, neurological disease, and chronic renal disease.
The authors noted that these data confirm previous reports showing that patients with hypertension have more severe illness and higher mortality rates associated with COVID-19 than those without hypertension.
But they added: “Our data provide some reassurance that ACE inhibitors/ARBs are not associated with the progression or outcome of COVID-19 hospitalizations in patients with hypertension.”
They also noted that these results support the recommendations from almost all major cardiovascular societies that patients do not discontinue ACE inhibitors or ARBs because of worries about COVID-19.
However, the authors did point out some limitations of their study, which included a small number of patients with hypertension taking ACE inhibitors or ARBs and the fact that a nonsevere disease course was still severe enough to require hospitalization. In addition, it was not clear whether ACE inhibitor/ARB treatment at baseline was maintained throughout hospitalization for all patients.
This was also an observational comparison and may be biased by differences in patients taking versus not taking ACE inhibitors or ARBs at the time of hospitalization, although the measured baseline characteristics were similar in both groups.
But the authors also highlighted the finding that, in this cohort, patients with hypertension had three times the mortality rate of all other patients hospitalized with COVID-19.
“Hypertension combined with cardiovascular and cerebrovascular disease, diabetes, and chronic kidney disease would predispose patients to an increased risk of severity and mortality of COVID-19. Therefore, patients with these underlying conditions who develop COVID-19 require particularly intensive surveillance and care,” they wrote.
Experts cautiously optimistic
Some cardiovascular experts were cautiously optimistic about these latest results.
Michael A. Weber, MD, professor of medicine at the State University of New York, Brooklyn, and editor-in-chief of the Journal of Clinical Hypertension, said: “This new report from Wuhan, China, gives modest reassurance that the use of ACE inhibitors or ARBs in hypertensive patients with COVID-19 disease does not increase the risk of clinical deterioration or death.
“Ongoing, more definitive studies should help resolve competing hypotheses regarding the effects of these agents: whether the increased ACE2 enzyme levels they produce can worsen outcomes by increasing access of the COVID virus to lung tissue; or whether there is a benefit linked to a protective effect of increased ACE2 on alveolar cell function,” Dr. Weber noted.
“Though the number of patients included in this new report is small, it is startling that hypertensive patients were three times as likely as nonhypertensives to have a fatal outcome, presumably reflecting vulnerability due to the cardiovascular and metabolic comorbidities associated with hypertension,” he added.
“In any case, for now, clinicians should continue treating hypertensive patients with whichever drugs, including ACE inhibitors and ARBs, best provide protection from adverse outcomes,” Dr. Weber concluded.
John McMurray, MD, professor of medical cardiology, University of Glasgow, Scotland, commented: “This study from Wuhan provides some reassurance about one of the two questions about ACEI/ARBs: Do these drugs increase susceptibility to infection? And if [the patient is] infected, do they increase the severity of infection? This study addresses the latter question and appears to suggest no increased severity.”
However, Dr. McMurray pointed out that the study had many limitations. There were only small patient numbers and the data were unadjusted, “although it looks like the ACE inhibitor/ARB treated patients were higher risk to start with.” It was an observational study, and patients were not randomized and were predominantly treated with ARBs, and not ACE inhibitors, so “we don’t know if the concerns apply equally to these two classes of drug.
“Other data published and unpublished supporting this (even showing better outcomes in patients treated with an ACE inhibitor/ARB), and, to date, any concerns about these drugs remain unsubstantiated and the guidance from medical societies to continue treatment with these agents in patients prescribed them seems wise,” Dr. McMurray added.
Franz H. Messerli, MD, professor of medicine at the University of Bern, Switzerland, commented: “The study from Wuhan is not a great study. They didn’t even do a multivariable analysis. They could have done a bit more with the data, but it still gives some reassurance.”
Dr. Messerli said it was “interesting” that 30% of the patients hospitalized with COVID-19 in the sample had hypertension. “That corresponds to the general population, so does not suggest that having hypertension increases susceptibility to infection – but it does seem to increase the risk of a bad outcome.”
Dr. Messerli noted that there are two more similar studies due to be published soon, both said to suggest either a beneficial or neutral effect of ACE inhibitors/ARBs on COVID-19 outcomes in hospitalized patients.
“This does help with confidence in prescribing these agents and reinforces the recommendations for patients to stay on these drugs,” he said.
“However, none of these studies address the infectivity issue – whether their use upregulates the ACE2 receptor, which the virus uses to gain entry to cells, thereby increasing susceptibility to the infection,” Dr. Messerli cautioned. “But the similar or better outcomes on these drugs are encouraging,” he added.
The Wuhan study was supported by the Health and Family Planning Commission of Wuhan City, China. The authors have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Mislabeled clopidogrel lot recalled, may contain simvastatin
International Laboratories has initiated a voluntary recall to the consumer level in the United States of a single lot of the antiplatelet clopidogrel because it is mislabeled and may contain simvastatin, a cholesterol-lowering drug, instead of clopidogrel.
The recalled product ― lot number 117099A of clopidogrel tablets (USP 75 mg) packaged in bottles of 30 tablets ― may contain clopidogrel 75 mg tablets or it could contain simvastatin tablets (USP 10 mg), according to a company announcement posted on the US Food and Drug Administration (FDA) website.
“Missed doses of clopidogrel increases the risk of heart attack and stroke which can be life threatening. Additionally, unintentional consumption of simvastatin could include the common side effects associated with its use and may cause fetal harm when administered to a pregnant woman,” the company cautions.
To date, the company has not received any reports of harm arising from the problem that prompted the recall.
The recalled product was distributed nationwide and was delivered to distribution centers in Arkansas, Georgia, Indiana, California, and Maryland and to retail stores in all US states.
International Laboratories is notifying distributors and customers by letter and is arranging for the return of all recalled products.
For questions regarding this recall, contact Inmar by phone 855-258-7280 (weekdays between 9:00 AM and 5:00 PM EST) or by email at [email protected].
Adverse reactions or quality problems experienced with the use of this product should be reported to the FDA’s MedWatch adverse event reporting program.
This article first appeared on Medscape.com.
International Laboratories has initiated a voluntary recall to the consumer level in the United States of a single lot of the antiplatelet clopidogrel because it is mislabeled and may contain simvastatin, a cholesterol-lowering drug, instead of clopidogrel.
The recalled product ― lot number 117099A of clopidogrel tablets (USP 75 mg) packaged in bottles of 30 tablets ― may contain clopidogrel 75 mg tablets or it could contain simvastatin tablets (USP 10 mg), according to a company announcement posted on the US Food and Drug Administration (FDA) website.
“Missed doses of clopidogrel increases the risk of heart attack and stroke which can be life threatening. Additionally, unintentional consumption of simvastatin could include the common side effects associated with its use and may cause fetal harm when administered to a pregnant woman,” the company cautions.
To date, the company has not received any reports of harm arising from the problem that prompted the recall.
The recalled product was distributed nationwide and was delivered to distribution centers in Arkansas, Georgia, Indiana, California, and Maryland and to retail stores in all US states.
International Laboratories is notifying distributors and customers by letter and is arranging for the return of all recalled products.
For questions regarding this recall, contact Inmar by phone 855-258-7280 (weekdays between 9:00 AM and 5:00 PM EST) or by email at [email protected].
Adverse reactions or quality problems experienced with the use of this product should be reported to the FDA’s MedWatch adverse event reporting program.
This article first appeared on Medscape.com.
International Laboratories has initiated a voluntary recall to the consumer level in the United States of a single lot of the antiplatelet clopidogrel because it is mislabeled and may contain simvastatin, a cholesterol-lowering drug, instead of clopidogrel.
The recalled product ― lot number 117099A of clopidogrel tablets (USP 75 mg) packaged in bottles of 30 tablets ― may contain clopidogrel 75 mg tablets or it could contain simvastatin tablets (USP 10 mg), according to a company announcement posted on the US Food and Drug Administration (FDA) website.
“Missed doses of clopidogrel increases the risk of heart attack and stroke which can be life threatening. Additionally, unintentional consumption of simvastatin could include the common side effects associated with its use and may cause fetal harm when administered to a pregnant woman,” the company cautions.
To date, the company has not received any reports of harm arising from the problem that prompted the recall.
The recalled product was distributed nationwide and was delivered to distribution centers in Arkansas, Georgia, Indiana, California, and Maryland and to retail stores in all US states.
International Laboratories is notifying distributors and customers by letter and is arranging for the return of all recalled products.
For questions regarding this recall, contact Inmar by phone 855-258-7280 (weekdays between 9:00 AM and 5:00 PM EST) or by email at [email protected].
Adverse reactions or quality problems experienced with the use of this product should be reported to the FDA’s MedWatch adverse event reporting program.
This article first appeared on Medscape.com.
Cardiology groups push back on hydroxychloroquine, azithromycin for COVID-19
The .

“Hydroxychloroquine and azithromycin have been touted for potential prophylaxis or treatment for COVID-19; both drugs are listed as definite causes of torsade de pointes” and increase in the risk of other arrhythmias and sudden death, the American Heart Association, the American College of Cardiology, and the Heart Rhythm Society said in a joint statement April 8 in Circulation.
The statement came amid ongoing promotion by the Trump administration of hydroxychloroquine, in particular, for COVID-19 despite lack of strong data.
In addition to underlying cardiovascular disease, “seriously ill patients often have comorbidities that can increase risk of serious arrhythmias,” including hypokalemia, hypomagnesemia, fever, and systemic inflammation, the groups said.
They recommended withholding the drugs in patients with baseline QT prolongation (e.g., QTc of at least 500 msec) or with known congenital long QT syndrome; monitoring cardiac rhythm and QT interval and withdrawing hydroxychloroquine and azithromycin if QTc exceeds 500 msec; correcting hypokalemia to levels greater than 4 mEq/L and hypomagnesemia to more than 2 mg/dL; and avoiding other QTc-prolonging agents when possible.
The groups noted that, “in patients critically ill with COVID-19 infection, frequent caregiver contact may need to be minimized, so optimal electrocardiographic interval and rhythm monitoring may not be possible.” There is also a possible compounding arrhythmic effect when hydroxychloroquine and azithromycin are used together, but that has not been studied.
There’s a known risk of torsade de pointes with chloroquine and a possible risk with the antiviral HIV combination drug lopinavir-ritonavir, two other candidates for COVID-19 treatment. Hydroxychloroquine and chloroquine, both antimalarials, might help prevent or treat infection by interfering with angiotensin-converting enzyme 2 receptors, which the COVID-19 virus uses for cell entry, the groups said.
“The urgency of COVID-19 must not diminish the scientific rigor with which we approach COVID-19 treatment. While these medications may work against COVID-19 individually or in combination, we recommend caution with these medications for patients with existing cardiovascular disease,” Robert A. Harrington, MD, AHA president and chair of the department of medicine at Stanford (Calif.) University, emphasized in a press release.
SOURCE: Roden DM et al. Circulation. 2020 Apr 8. doi:10.1161/CIRCULATIONAHA.120.047521.
The .
“Hydroxychloroquine and azithromycin have been touted for potential prophylaxis or treatment for COVID-19; both drugs are listed as definite causes of torsade de pointes” and increase in the risk of other arrhythmias and sudden death, the American Heart Association, the American College of Cardiology, and the Heart Rhythm Society said in a joint statement April 8 in Circulation.
The statement came amid ongoing promotion by the Trump administration of hydroxychloroquine, in particular, for COVID-19 despite lack of strong data.
In addition to underlying cardiovascular disease, “seriously ill patients often have comorbidities that can increase risk of serious arrhythmias,” including hypokalemia, hypomagnesemia, fever, and systemic inflammation, the groups said.
They recommended withholding the drugs in patients with baseline QT prolongation (e.g., QTc of at least 500 msec) or with known congenital long QT syndrome; monitoring cardiac rhythm and QT interval and withdrawing hydroxychloroquine and azithromycin if QTc exceeds 500 msec; correcting hypokalemia to levels greater than 4 mEq/L and hypomagnesemia to more than 2 mg/dL; and avoiding other QTc-prolonging agents when possible.
The groups noted that, “in patients critically ill with COVID-19 infection, frequent caregiver contact may need to be minimized, so optimal electrocardiographic interval and rhythm monitoring may not be possible.” There is also a possible compounding arrhythmic effect when hydroxychloroquine and azithromycin are used together, but that has not been studied.
There’s a known risk of torsade de pointes with chloroquine and a possible risk with the antiviral HIV combination drug lopinavir-ritonavir, two other candidates for COVID-19 treatment. Hydroxychloroquine and chloroquine, both antimalarials, might help prevent or treat infection by interfering with angiotensin-converting enzyme 2 receptors, which the COVID-19 virus uses for cell entry, the groups said.
“The urgency of COVID-19 must not diminish the scientific rigor with which we approach COVID-19 treatment. While these medications may work against COVID-19 individually or in combination, we recommend caution with these medications for patients with existing cardiovascular disease,” Robert A. Harrington, MD, AHA president and chair of the department of medicine at Stanford (Calif.) University, emphasized in a press release.
SOURCE: Roden DM et al. Circulation. 2020 Apr 8. doi:10.1161/CIRCULATIONAHA.120.047521.
The .
“Hydroxychloroquine and azithromycin have been touted for potential prophylaxis or treatment for COVID-19; both drugs are listed as definite causes of torsade de pointes” and increase in the risk of other arrhythmias and sudden death, the American Heart Association, the American College of Cardiology, and the Heart Rhythm Society said in a joint statement April 8 in Circulation.
The statement came amid ongoing promotion by the Trump administration of hydroxychloroquine, in particular, for COVID-19 despite lack of strong data.
In addition to underlying cardiovascular disease, “seriously ill patients often have comorbidities that can increase risk of serious arrhythmias,” including hypokalemia, hypomagnesemia, fever, and systemic inflammation, the groups said.
They recommended withholding the drugs in patients with baseline QT prolongation (e.g., QTc of at least 500 msec) or with known congenital long QT syndrome; monitoring cardiac rhythm and QT interval and withdrawing hydroxychloroquine and azithromycin if QTc exceeds 500 msec; correcting hypokalemia to levels greater than 4 mEq/L and hypomagnesemia to more than 2 mg/dL; and avoiding other QTc-prolonging agents when possible.
The groups noted that, “in patients critically ill with COVID-19 infection, frequent caregiver contact may need to be minimized, so optimal electrocardiographic interval and rhythm monitoring may not be possible.” There is also a possible compounding arrhythmic effect when hydroxychloroquine and azithromycin are used together, but that has not been studied.
There’s a known risk of torsade de pointes with chloroquine and a possible risk with the antiviral HIV combination drug lopinavir-ritonavir, two other candidates for COVID-19 treatment. Hydroxychloroquine and chloroquine, both antimalarials, might help prevent or treat infection by interfering with angiotensin-converting enzyme 2 receptors, which the COVID-19 virus uses for cell entry, the groups said.
“The urgency of COVID-19 must not diminish the scientific rigor with which we approach COVID-19 treatment. While these medications may work against COVID-19 individually or in combination, we recommend caution with these medications for patients with existing cardiovascular disease,” Robert A. Harrington, MD, AHA president and chair of the department of medicine at Stanford (Calif.) University, emphasized in a press release.
SOURCE: Roden DM et al. Circulation. 2020 Apr 8. doi:10.1161/CIRCULATIONAHA.120.047521.
Aerosolization of COVID-19 and Contamination Risks During Respiratory Treatments
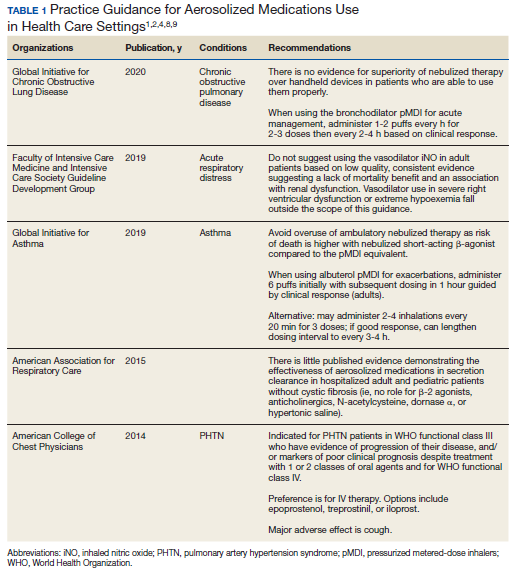
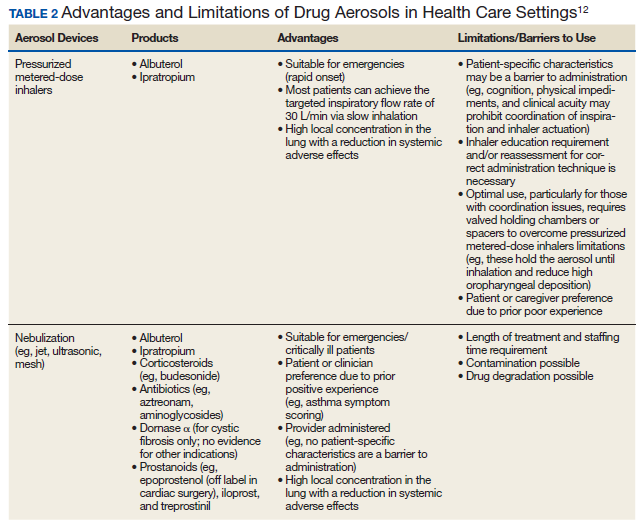
Beyond asthma and chronic obstructive pulmonary disease (COPD), inhalation therapy is a mainstay in the management of bronchiectasis, cystic fibrosis, and pulmonary artery hypertension. Several US Food and Drug Administration off-label indications for inhalational medications include hypoxia secondary to acute respiratory distress syndrome (ARDS) and intraoperative and postoperative pulmonary hypertension during and following cardiac surgery, respectively.1-11 Therapeutic delivery of aerosols to the lung may be provided via nebulization, pressurized metered-dose inhalers (pMDI), and other devices (eg, dry powder inhalers, soft-mist inhalers, and smart inhalers).12 The most common aerosolized medications given in the clinical setting are bronchodilators.12
Product selection is often guided by practice guidelines (Table 1), consideration of the formulation’s advantages and disadvantages (Table 2), and/or formulary considerations. For example, current guidelines for COPD state that there is no evidence for superiority of nebulized bronchodilator therapy over handheld devices in patients who can use them properly.2 Due to equivalence, nebulized formulations are commonly used in hospitals, emergency departments (EDs) and ambulatory clinics based on the drug’s unit cost. In contrast, a pMDI is often more cost-effective for use in ambulatory patients who are administering multiple doses from the same canister.
The World Health Organization (WHO) and the Centers for Disease Control and Prevention (CDC) recommend droplet and contact precautions for all patients suspected or diagnosed with novel coronavirus-19 (COVID-19).13,14 Airborne precautions must be applied when performing aerosol-generating medical procedures (AGMPs), including but not limited to, open suctioning of the respiratory tract, intubation, bronchoscopy, and cardiopulmonary resuscitation (CPR). Data from the severe acute respiratory syndrome (SARS-CoV) epidemic suggest that nebulization of medication is also an AGMP.15-17
Institutions must ensure that their health care workers (HCWs) are wearing appropriate personal protective equipment (PPE) including gloves, long-sleeved gowns, eye protection, and fit-tested particulate respirators (N95 mask) for airborne procedures and are carefully discarding PPE after use.13,14 Due to severe shortages in available respirators in the US supply chain, the CDC has temporarily modified WHO recommendations. Face masks are now an acceptable alternative to protect HCWs from splashes and sprays from procedures not likely to generate aerosols and for cleaning of rooms, although there is no evidence to support this decision.
Internationally, HCWs are falling ill with COVID-19. Data from Italy and Spain show that about 9% to 13% of these countries’ cases are HCWs.18,19 Within the US, the Ohio health department reports approximately 16% of cases are HCWs.20 It is possible that 20% of frontline HCWs will become infected.21 Evolving laboratory research shows that COVID-19 remains viable in aerosols for up to 3 hours postaerosolization, thus making aerosol transmission plausible.22 Nebulizers convert liquids into aerosols and during dispersal may potentially cause secondary inhalation of fugitive emissions.23 Since interim CDC infection control guidance is to allow only essential personnel to enter the room of patients with COVID-19, many facilities will rely on their frontline nursing staff to clean and disinfect high-touch surfaces following routine care activities.24
Achieving adequate fomite disinfection following viral aerosolization may pose a significant problem for any patient receiving scheduled doses of nebulized medications. Additionally, for personnel who clean rooms following intermittent drug nebulization while wearing PPE that includes a face mask, protection from aerosolized virus may be inadequate. Subsequently, fugitive emissions from nebulized medications may potentially contribute to both nosocomial COVID-19 transmission and viral infections in the medical staff until proven otherwise by studies conducted outside of the laboratory. Prevention of infection in the medical staff is imperative since federal health care systems cannot sustain a significant loss of its workforce.
Recommendations
We recommend that health care systems stop business as usual and adopt public health recommendations issued by Canadian and Hong Kong health care authorities for the management of suspected or confirmed COVID-19 disease.25-28 We have further clarified and expanded on these interventions. During viral pandemics, prescribers and health care systems should:
- Deprescribe nebulized therapies on medical wards and intensive care units as an infection control measure. Also avoid use in any outpatient health care setting (eg, community-based clinics, EDs, triage).
- Avoid initiation of nebulized unproven therapies (eg, n-acetylcysteine, hypertonic saline).1
- Use alternative bronchodilator formulations as appropriate (eg, oral β-2 agonist, recognizing its slower onset) before prescribing nebulized agents to patients who are uncooperative or unable to follow directions needed to use a pMDI with a spacer or have experienced a prior poor response to a pMDI with spacer (eg, OptiChamber Diamond, Philips).25,27
- Limit nebulized drug utilization (eg, bronchodilators, epoprostenol) to patients who are on mechanical ventilation and will receive nebulized therapies via a closed system or to patients housed in negative pressure hospital rooms.22 Use a viral filter (eg, Salter Labs system) to decrease the spread of infection for those receiving epoprostenol via face mask.25
- Adjust procurement practices (eg, pharmacy, logistics) to address the transition from nebulized drugs to alternatives.
- Add a safety net to the drug-ordering process by restricting new orders for nebulized therapies to the prior authorization process.27 Apply the exclusion criterion of suspected or definite COVID-19.
- Add a safety net to environmental service practices. Nursing staff should track patients who received ≥ 1 nebulizations via open (before diagnosis) or closed systems so that staff wear suitable PPE to include a N-95 mask while cleaning the room.
Conclusions
To implement the aggressive infection control guidance promulgated here, we recommend collaboration with infection control, pharmacy service (eg, prior authorization team, clinical pharmacy team, and procurement team), respiratory therapy, pulmonary and other critical care physicians, EDs, CPR committee, and other stakeholders. When making significant transitions in clinical care during a viral pandemic, guidelines must be timely, use imperative wording, and consist of easily identifiable education and/or instructions for the affected frontline staff in order to change attitudes.29 Additionally, when transitioning from nebulized bronchodilators to pMDI, educational in-services should be provided to frontline staff to avoid misconceptions regarding pMDI treatment efficacy and patients’ ability to use their pMDI with spacer.30
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the VA Tennessee Valley Healthcare System in Nashville.
1. Strickland SL, Rubin BK, Haas CF, Volsko TA, Drescher GS, O’Malley CA. AARC Clinical Practice Guideline: effectiveness of pharmacologic airway clearance therapies in hospitalized patients. Respir Care. 2015;60(7):1071-1077.
2. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. 2020 GOLD Report. https://goldcopd.org/gold-reports/. Accessed March 26, 2020.
3. Van Geffen WH, Douma WR, Slebos DJ, Kerstjens HAM. Bronchodilators delivered by nebulizer versus pMDI with spacer or DPI for exacerbations of COPD (Review). Cochrane Database Syst Rev. 2016;8:CD011826.
4. Global Initiative for Asthma. https://ginasthma.org/wp-content/uploads/2019/06/GINA-2019-main-report-June-2019-wms.pdf. Accessed March 26, 2020.
5. Global Initiative for Asthma. Difficult-to-treat and severe asthma in adolescent and adult patients: diagnosis and management. https://ginasthma.org/wp-content/uploads/2019/04/GINA-Severe-asthma-Pocket-Guide-v2.0-wms-1.pdf. Accessed March 26, 2020.
6. Cates CJ, Welsh EJ, Rowe BH. Holding chambers (spacers) versus nebulizers for beta-agonist treatment of acute asthma. Cochrane Database Syst Rev. 2013;9:CD000052.
7. Welsh EJ, Evans DJ, Fowler SJ, Spencer S. Interventions for bronchiectasis: an overview of Cochrane systematic reviews. Cochrane Database Syst Rev. 2015;7:CD010337.
8. Taichman DB, Ornelas J, Chung L, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST Guideline and Expert Panel Report. CHEST. 2014;146(2):449-475.
9. Griffiths MJD, McAuley DF, Perkins GD, et al. Guidelines on the management of acute respiratory distress syndrome. BMJ Open Resp Res. 2019;6(1):e000420.
10. McGinn K, Reichert M. A comparison of inhaled nitric oxide versus inhaled epoprostenol for acute pulmonary hypertension following cardiac surgery. Ann Pharmacother. 2016;50(1):22-26.
11. Dzierba AL, Abel EE, Buckley MS, Lat I. A review of inhaled nitric oxide and aerosolized epoprostenol in acute lung injury or acute respiratory distress syndrome. Pharmacotherapy. 2014;34(3):279-290.
12. Pleasants RA, Hess DR. Aerosol delivery devices for obstructive lung diseases. Respir Care. 2018;63(6):708-733.
13. World Health Organization. Clinical management of severe acute respiratory infection when novel coronavirus (nCoV) infection is suspected. https://www.who.int/publications-detail/clinical-management-of-severe-acute-respiratory-infection-when-novel-coronavirus-(ncov)-infection-is-suspected Accessed March 26, 2020.
14. Centers for Disease Control and Prevention. Interim clinical guidance for management of patients with confirmed coronavirus disease (COVID-19). https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-guidance-management-patients.html. Revised March 7, 2020. Accessed March 26, 2020.
15. Wong RSM, Hui DS. Index patient and SARS outbreak in Hong Kong. Emerg Infect Dis. 2004;10(2):339-341.
16. Wong T-W, Lee C-K, Tam W, et al; Outbreak Study Group. Emerg Infect Dis. 2004;10(2):269-276.
17. Seto WH, Tsang D, Yung RWH, et al; Advisors of Expert SARS group of Hospital Authority. Effectiveness of precautions against droplets and contact in prevention of nosocomial transmission of severe acute respiratory syndrome (SARS). Lancet. 2003;361(9368):1519-1520.
18. Livingston E, Bucher K. Coronavirus Disease 2019 (COVID-19) in Italy. https://jamanetwork.com/journals/jama/fullarticle/2763401?resultClick=1. Published March 17, 2020. Accessed March 26, 2020.
19. Jones S. Spain: doctors struggle to cope as 514 die from coronavirus in a day. The Guardian. March 24, 2020. https://www.theguardian.com/world/2020/mar/24/spain-doctors-lack-protection-coronavirus-covid-19. Accessed March 27, 2020.
20. 16% of Ohio’s diagnosed COVID-19 cases are healthcare workers. https://www.wlwt.com/article/16-of-ohio-s-diagnosed-covid-19-cases-are-healthcare-workers/31930566#. Updated March 25, 2020. Accessed March 27, 2020.
21. Remuzzi A, Remuzzi G. COVID-19 and Italy: what next? Lancet. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)30627-9/fulltext. Accessed March 27, 2020.
22. van Doremalen N, Bushmaker T, Morris DH, et al. Aerosol and surface stability of SARS-CoV-2 as Compared with SARS-CoV-1 [published online ahead of print, 2020 Mar 17]. N Engl J Med. 2020;10.1056/NEJMc2004973.
23. McGrath JA, O’Sullivan A, Bennett G, et al. Investigation of the quantity of exhaled aerosol released into the environment during nebulization. Pharmaceutics. 2019;11(2):75.
24. Centers for Disease Control and Prevention. Healthcare Infection prevention and control FAQs for COVID-19. https://www.cdc.gov/coronavirus/2019-ncov/infection-control/infection-prevention-control-faq.html. Revised March 24, 2020. Accessed March 26, 2020.
25. Practice standards of respiratory procedures: post SARS era. Use of aerosolized medications. December 2003. http://www.hkresp.com/hkts.php?page=page/hkts/detail&meid=93742. Accessed March 26, 2020.
26. Wax RS, Christian MD. Practical recommendations for critical care and anesthesiology teams caring for novel coronavirus (2019-nCoV) patients. Can J Anesth. 2020. [ePub ahead of print.]
27. Newhouse MT. RE: transmission of coronavirus by nebulizer- as serious, underappreciated risk! https://www.cmaj.ca/content/re-transmission-corona-virus-nebulizer-serious-underappreciated-risk. Accessed March 26, 2020. [ePub ahead of print.]
28. Moira C-Y. Severe acute respiratory syndrome (SARS) and healthcare workers. Int J Occup Environ Health. 2004;10(4):421-427.
29. Timen A, Hulscher MEJL, Rust L, et al. Barriers to implementing infection prevention and control guidelines during crises: experiences of health care professionals. Am J Infect Control. 2010;38(9):726-733.
30. Khoo SM, Tan LK, Said N, Lim TK. Metered-dose inhaler with spacer instead of nebulizer during the outbreak of severe acute respiratory syndrome in Singapore. Respir Care. 2009;54(7):855-860.
Beyond asthma and chronic obstructive pulmonary disease (COPD), inhalation therapy is a mainstay in the management of bronchiectasis, cystic fibrosis, and pulmonary artery hypertension. Several US Food and Drug Administration off-label indications for inhalational medications include hypoxia secondary to acute respiratory distress syndrome (ARDS) and intraoperative and postoperative pulmonary hypertension during and following cardiac surgery, respectively.1-11 Therapeutic delivery of aerosols to the lung may be provided via nebulization, pressurized metered-dose inhalers (pMDI), and other devices (eg, dry powder inhalers, soft-mist inhalers, and smart inhalers).12 The most common aerosolized medications given in the clinical setting are bronchodilators.12
Product selection is often guided by practice guidelines (Table 1), consideration of the formulation’s advantages and disadvantages (Table 2), and/or formulary considerations. For example, current guidelines for COPD state that there is no evidence for superiority of nebulized bronchodilator therapy over handheld devices in patients who can use them properly.2 Due to equivalence, nebulized formulations are commonly used in hospitals, emergency departments (EDs) and ambulatory clinics based on the drug’s unit cost. In contrast, a pMDI is often more cost-effective for use in ambulatory patients who are administering multiple doses from the same canister.
The World Health Organization (WHO) and the Centers for Disease Control and Prevention (CDC) recommend droplet and contact precautions for all patients suspected or diagnosed with novel coronavirus-19 (COVID-19).13,14 Airborne precautions must be applied when performing aerosol-generating medical procedures (AGMPs), including but not limited to, open suctioning of the respiratory tract, intubation, bronchoscopy, and cardiopulmonary resuscitation (CPR). Data from the severe acute respiratory syndrome (SARS-CoV) epidemic suggest that nebulization of medication is also an AGMP.15-17
Institutions must ensure that their health care workers (HCWs) are wearing appropriate personal protective equipment (PPE) including gloves, long-sleeved gowns, eye protection, and fit-tested particulate respirators (N95 mask) for airborne procedures and are carefully discarding PPE after use.13,14 Due to severe shortages in available respirators in the US supply chain, the CDC has temporarily modified WHO recommendations. Face masks are now an acceptable alternative to protect HCWs from splashes and sprays from procedures not likely to generate aerosols and for cleaning of rooms, although there is no evidence to support this decision.
Internationally, HCWs are falling ill with COVID-19. Data from Italy and Spain show that about 9% to 13% of these countries’ cases are HCWs.18,19 Within the US, the Ohio health department reports approximately 16% of cases are HCWs.20 It is possible that 20% of frontline HCWs will become infected.21 Evolving laboratory research shows that COVID-19 remains viable in aerosols for up to 3 hours postaerosolization, thus making aerosol transmission plausible.22 Nebulizers convert liquids into aerosols and during dispersal may potentially cause secondary inhalation of fugitive emissions.23 Since interim CDC infection control guidance is to allow only essential personnel to enter the room of patients with COVID-19, many facilities will rely on their frontline nursing staff to clean and disinfect high-touch surfaces following routine care activities.24
Achieving adequate fomite disinfection following viral aerosolization may pose a significant problem for any patient receiving scheduled doses of nebulized medications. Additionally, for personnel who clean rooms following intermittent drug nebulization while wearing PPE that includes a face mask, protection from aerosolized virus may be inadequate. Subsequently, fugitive emissions from nebulized medications may potentially contribute to both nosocomial COVID-19 transmission and viral infections in the medical staff until proven otherwise by studies conducted outside of the laboratory. Prevention of infection in the medical staff is imperative since federal health care systems cannot sustain a significant loss of its workforce.
Recommendations
We recommend that health care systems stop business as usual and adopt public health recommendations issued by Canadian and Hong Kong health care authorities for the management of suspected or confirmed COVID-19 disease.25-28 We have further clarified and expanded on these interventions. During viral pandemics, prescribers and health care systems should:
- Deprescribe nebulized therapies on medical wards and intensive care units as an infection control measure. Also avoid use in any outpatient health care setting (eg, community-based clinics, EDs, triage).
- Avoid initiation of nebulized unproven therapies (eg, n-acetylcysteine, hypertonic saline).1
- Use alternative bronchodilator formulations as appropriate (eg, oral β-2 agonist, recognizing its slower onset) before prescribing nebulized agents to patients who are uncooperative or unable to follow directions needed to use a pMDI with a spacer or have experienced a prior poor response to a pMDI with spacer (eg, OptiChamber Diamond, Philips).25,27
- Limit nebulized drug utilization (eg, bronchodilators, epoprostenol) to patients who are on mechanical ventilation and will receive nebulized therapies via a closed system or to patients housed in negative pressure hospital rooms.22 Use a viral filter (eg, Salter Labs system) to decrease the spread of infection for those receiving epoprostenol via face mask.25
- Adjust procurement practices (eg, pharmacy, logistics) to address the transition from nebulized drugs to alternatives.
- Add a safety net to the drug-ordering process by restricting new orders for nebulized therapies to the prior authorization process.27 Apply the exclusion criterion of suspected or definite COVID-19.
- Add a safety net to environmental service practices. Nursing staff should track patients who received ≥ 1 nebulizations via open (before diagnosis) or closed systems so that staff wear suitable PPE to include a N-95 mask while cleaning the room.
Conclusions
To implement the aggressive infection control guidance promulgated here, we recommend collaboration with infection control, pharmacy service (eg, prior authorization team, clinical pharmacy team, and procurement team), respiratory therapy, pulmonary and other critical care physicians, EDs, CPR committee, and other stakeholders. When making significant transitions in clinical care during a viral pandemic, guidelines must be timely, use imperative wording, and consist of easily identifiable education and/or instructions for the affected frontline staff in order to change attitudes.29 Additionally, when transitioning from nebulized bronchodilators to pMDI, educational in-services should be provided to frontline staff to avoid misconceptions regarding pMDI treatment efficacy and patients’ ability to use their pMDI with spacer.30
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the VA Tennessee Valley Healthcare System in Nashville.
Beyond asthma and chronic obstructive pulmonary disease (COPD), inhalation therapy is a mainstay in the management of bronchiectasis, cystic fibrosis, and pulmonary artery hypertension. Several US Food and Drug Administration off-label indications for inhalational medications include hypoxia secondary to acute respiratory distress syndrome (ARDS) and intraoperative and postoperative pulmonary hypertension during and following cardiac surgery, respectively.1-11 Therapeutic delivery of aerosols to the lung may be provided via nebulization, pressurized metered-dose inhalers (pMDI), and other devices (eg, dry powder inhalers, soft-mist inhalers, and smart inhalers).12 The most common aerosolized medications given in the clinical setting are bronchodilators.12
Product selection is often guided by practice guidelines (Table 1), consideration of the formulation’s advantages and disadvantages (Table 2), and/or formulary considerations. For example, current guidelines for COPD state that there is no evidence for superiority of nebulized bronchodilator therapy over handheld devices in patients who can use them properly.2 Due to equivalence, nebulized formulations are commonly used in hospitals, emergency departments (EDs) and ambulatory clinics based on the drug’s unit cost. In contrast, a pMDI is often more cost-effective for use in ambulatory patients who are administering multiple doses from the same canister.
The World Health Organization (WHO) and the Centers for Disease Control and Prevention (CDC) recommend droplet and contact precautions for all patients suspected or diagnosed with novel coronavirus-19 (COVID-19).13,14 Airborne precautions must be applied when performing aerosol-generating medical procedures (AGMPs), including but not limited to, open suctioning of the respiratory tract, intubation, bronchoscopy, and cardiopulmonary resuscitation (CPR). Data from the severe acute respiratory syndrome (SARS-CoV) epidemic suggest that nebulization of medication is also an AGMP.15-17
Institutions must ensure that their health care workers (HCWs) are wearing appropriate personal protective equipment (PPE) including gloves, long-sleeved gowns, eye protection, and fit-tested particulate respirators (N95 mask) for airborne procedures and are carefully discarding PPE after use.13,14 Due to severe shortages in available respirators in the US supply chain, the CDC has temporarily modified WHO recommendations. Face masks are now an acceptable alternative to protect HCWs from splashes and sprays from procedures not likely to generate aerosols and for cleaning of rooms, although there is no evidence to support this decision.
Internationally, HCWs are falling ill with COVID-19. Data from Italy and Spain show that about 9% to 13% of these countries’ cases are HCWs.18,19 Within the US, the Ohio health department reports approximately 16% of cases are HCWs.20 It is possible that 20% of frontline HCWs will become infected.21 Evolving laboratory research shows that COVID-19 remains viable in aerosols for up to 3 hours postaerosolization, thus making aerosol transmission plausible.22 Nebulizers convert liquids into aerosols and during dispersal may potentially cause secondary inhalation of fugitive emissions.23 Since interim CDC infection control guidance is to allow only essential personnel to enter the room of patients with COVID-19, many facilities will rely on their frontline nursing staff to clean and disinfect high-touch surfaces following routine care activities.24
Achieving adequate fomite disinfection following viral aerosolization may pose a significant problem for any patient receiving scheduled doses of nebulized medications. Additionally, for personnel who clean rooms following intermittent drug nebulization while wearing PPE that includes a face mask, protection from aerosolized virus may be inadequate. Subsequently, fugitive emissions from nebulized medications may potentially contribute to both nosocomial COVID-19 transmission and viral infections in the medical staff until proven otherwise by studies conducted outside of the laboratory. Prevention of infection in the medical staff is imperative since federal health care systems cannot sustain a significant loss of its workforce.
Recommendations
We recommend that health care systems stop business as usual and adopt public health recommendations issued by Canadian and Hong Kong health care authorities for the management of suspected or confirmed COVID-19 disease.25-28 We have further clarified and expanded on these interventions. During viral pandemics, prescribers and health care systems should:
- Deprescribe nebulized therapies on medical wards and intensive care units as an infection control measure. Also avoid use in any outpatient health care setting (eg, community-based clinics, EDs, triage).
- Avoid initiation of nebulized unproven therapies (eg, n-acetylcysteine, hypertonic saline).1
- Use alternative bronchodilator formulations as appropriate (eg, oral β-2 agonist, recognizing its slower onset) before prescribing nebulized agents to patients who are uncooperative or unable to follow directions needed to use a pMDI with a spacer or have experienced a prior poor response to a pMDI with spacer (eg, OptiChamber Diamond, Philips).25,27
- Limit nebulized drug utilization (eg, bronchodilators, epoprostenol) to patients who are on mechanical ventilation and will receive nebulized therapies via a closed system or to patients housed in negative pressure hospital rooms.22 Use a viral filter (eg, Salter Labs system) to decrease the spread of infection for those receiving epoprostenol via face mask.25
- Adjust procurement practices (eg, pharmacy, logistics) to address the transition from nebulized drugs to alternatives.
- Add a safety net to the drug-ordering process by restricting new orders for nebulized therapies to the prior authorization process.27 Apply the exclusion criterion of suspected or definite COVID-19.
- Add a safety net to environmental service practices. Nursing staff should track patients who received ≥ 1 nebulizations via open (before diagnosis) or closed systems so that staff wear suitable PPE to include a N-95 mask while cleaning the room.
Conclusions
To implement the aggressive infection control guidance promulgated here, we recommend collaboration with infection control, pharmacy service (eg, prior authorization team, clinical pharmacy team, and procurement team), respiratory therapy, pulmonary and other critical care physicians, EDs, CPR committee, and other stakeholders. When making significant transitions in clinical care during a viral pandemic, guidelines must be timely, use imperative wording, and consist of easily identifiable education and/or instructions for the affected frontline staff in order to change attitudes.29 Additionally, when transitioning from nebulized bronchodilators to pMDI, educational in-services should be provided to frontline staff to avoid misconceptions regarding pMDI treatment efficacy and patients’ ability to use their pMDI with spacer.30
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the VA Tennessee Valley Healthcare System in Nashville.
1. Strickland SL, Rubin BK, Haas CF, Volsko TA, Drescher GS, O’Malley CA. AARC Clinical Practice Guideline: effectiveness of pharmacologic airway clearance therapies in hospitalized patients. Respir Care. 2015;60(7):1071-1077.
2. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. 2020 GOLD Report. https://goldcopd.org/gold-reports/. Accessed March 26, 2020.
3. Van Geffen WH, Douma WR, Slebos DJ, Kerstjens HAM. Bronchodilators delivered by nebulizer versus pMDI with spacer or DPI for exacerbations of COPD (Review). Cochrane Database Syst Rev. 2016;8:CD011826.
4. Global Initiative for Asthma. https://ginasthma.org/wp-content/uploads/2019/06/GINA-2019-main-report-June-2019-wms.pdf. Accessed March 26, 2020.
5. Global Initiative for Asthma. Difficult-to-treat and severe asthma in adolescent and adult patients: diagnosis and management. https://ginasthma.org/wp-content/uploads/2019/04/GINA-Severe-asthma-Pocket-Guide-v2.0-wms-1.pdf. Accessed March 26, 2020.
6. Cates CJ, Welsh EJ, Rowe BH. Holding chambers (spacers) versus nebulizers for beta-agonist treatment of acute asthma. Cochrane Database Syst Rev. 2013;9:CD000052.
7. Welsh EJ, Evans DJ, Fowler SJ, Spencer S. Interventions for bronchiectasis: an overview of Cochrane systematic reviews. Cochrane Database Syst Rev. 2015;7:CD010337.
8. Taichman DB, Ornelas J, Chung L, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST Guideline and Expert Panel Report. CHEST. 2014;146(2):449-475.
9. Griffiths MJD, McAuley DF, Perkins GD, et al. Guidelines on the management of acute respiratory distress syndrome. BMJ Open Resp Res. 2019;6(1):e000420.
10. McGinn K, Reichert M. A comparison of inhaled nitric oxide versus inhaled epoprostenol for acute pulmonary hypertension following cardiac surgery. Ann Pharmacother. 2016;50(1):22-26.
11. Dzierba AL, Abel EE, Buckley MS, Lat I. A review of inhaled nitric oxide and aerosolized epoprostenol in acute lung injury or acute respiratory distress syndrome. Pharmacotherapy. 2014;34(3):279-290.
12. Pleasants RA, Hess DR. Aerosol delivery devices for obstructive lung diseases. Respir Care. 2018;63(6):708-733.
13. World Health Organization. Clinical management of severe acute respiratory infection when novel coronavirus (nCoV) infection is suspected. https://www.who.int/publications-detail/clinical-management-of-severe-acute-respiratory-infection-when-novel-coronavirus-(ncov)-infection-is-suspected Accessed March 26, 2020.
14. Centers for Disease Control and Prevention. Interim clinical guidance for management of patients with confirmed coronavirus disease (COVID-19). https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-guidance-management-patients.html. Revised March 7, 2020. Accessed March 26, 2020.
15. Wong RSM, Hui DS. Index patient and SARS outbreak in Hong Kong. Emerg Infect Dis. 2004;10(2):339-341.
16. Wong T-W, Lee C-K, Tam W, et al; Outbreak Study Group. Emerg Infect Dis. 2004;10(2):269-276.
17. Seto WH, Tsang D, Yung RWH, et al; Advisors of Expert SARS group of Hospital Authority. Effectiveness of precautions against droplets and contact in prevention of nosocomial transmission of severe acute respiratory syndrome (SARS). Lancet. 2003;361(9368):1519-1520.
18. Livingston E, Bucher K. Coronavirus Disease 2019 (COVID-19) in Italy. https://jamanetwork.com/journals/jama/fullarticle/2763401?resultClick=1. Published March 17, 2020. Accessed March 26, 2020.
19. Jones S. Spain: doctors struggle to cope as 514 die from coronavirus in a day. The Guardian. March 24, 2020. https://www.theguardian.com/world/2020/mar/24/spain-doctors-lack-protection-coronavirus-covid-19. Accessed March 27, 2020.
20. 16% of Ohio’s diagnosed COVID-19 cases are healthcare workers. https://www.wlwt.com/article/16-of-ohio-s-diagnosed-covid-19-cases-are-healthcare-workers/31930566#. Updated March 25, 2020. Accessed March 27, 2020.
21. Remuzzi A, Remuzzi G. COVID-19 and Italy: what next? Lancet. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)30627-9/fulltext. Accessed March 27, 2020.
22. van Doremalen N, Bushmaker T, Morris DH, et al. Aerosol and surface stability of SARS-CoV-2 as Compared with SARS-CoV-1 [published online ahead of print, 2020 Mar 17]. N Engl J Med. 2020;10.1056/NEJMc2004973.
23. McGrath JA, O’Sullivan A, Bennett G, et al. Investigation of the quantity of exhaled aerosol released into the environment during nebulization. Pharmaceutics. 2019;11(2):75.
24. Centers for Disease Control and Prevention. Healthcare Infection prevention and control FAQs for COVID-19. https://www.cdc.gov/coronavirus/2019-ncov/infection-control/infection-prevention-control-faq.html. Revised March 24, 2020. Accessed March 26, 2020.
25. Practice standards of respiratory procedures: post SARS era. Use of aerosolized medications. December 2003. http://www.hkresp.com/hkts.php?page=page/hkts/detail&meid=93742. Accessed March 26, 2020.
26. Wax RS, Christian MD. Practical recommendations for critical care and anesthesiology teams caring for novel coronavirus (2019-nCoV) patients. Can J Anesth. 2020. [ePub ahead of print.]
27. Newhouse MT. RE: transmission of coronavirus by nebulizer- as serious, underappreciated risk! https://www.cmaj.ca/content/re-transmission-corona-virus-nebulizer-serious-underappreciated-risk. Accessed March 26, 2020. [ePub ahead of print.]
28. Moira C-Y. Severe acute respiratory syndrome (SARS) and healthcare workers. Int J Occup Environ Health. 2004;10(4):421-427.
29. Timen A, Hulscher MEJL, Rust L, et al. Barriers to implementing infection prevention and control guidelines during crises: experiences of health care professionals. Am J Infect Control. 2010;38(9):726-733.
30. Khoo SM, Tan LK, Said N, Lim TK. Metered-dose inhaler with spacer instead of nebulizer during the outbreak of severe acute respiratory syndrome in Singapore. Respir Care. 2009;54(7):855-860.
1. Strickland SL, Rubin BK, Haas CF, Volsko TA, Drescher GS, O’Malley CA. AARC Clinical Practice Guideline: effectiveness of pharmacologic airway clearance therapies in hospitalized patients. Respir Care. 2015;60(7):1071-1077.
2. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. 2020 GOLD Report. https://goldcopd.org/gold-reports/. Accessed March 26, 2020.
3. Van Geffen WH, Douma WR, Slebos DJ, Kerstjens HAM. Bronchodilators delivered by nebulizer versus pMDI with spacer or DPI for exacerbations of COPD (Review). Cochrane Database Syst Rev. 2016;8:CD011826.
4. Global Initiative for Asthma. https://ginasthma.org/wp-content/uploads/2019/06/GINA-2019-main-report-June-2019-wms.pdf. Accessed March 26, 2020.
5. Global Initiative for Asthma. Difficult-to-treat and severe asthma in adolescent and adult patients: diagnosis and management. https://ginasthma.org/wp-content/uploads/2019/04/GINA-Severe-asthma-Pocket-Guide-v2.0-wms-1.pdf. Accessed March 26, 2020.
6. Cates CJ, Welsh EJ, Rowe BH. Holding chambers (spacers) versus nebulizers for beta-agonist treatment of acute asthma. Cochrane Database Syst Rev. 2013;9:CD000052.
7. Welsh EJ, Evans DJ, Fowler SJ, Spencer S. Interventions for bronchiectasis: an overview of Cochrane systematic reviews. Cochrane Database Syst Rev. 2015;7:CD010337.
8. Taichman DB, Ornelas J, Chung L, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST Guideline and Expert Panel Report. CHEST. 2014;146(2):449-475.
9. Griffiths MJD, McAuley DF, Perkins GD, et al. Guidelines on the management of acute respiratory distress syndrome. BMJ Open Resp Res. 2019;6(1):e000420.
10. McGinn K, Reichert M. A comparison of inhaled nitric oxide versus inhaled epoprostenol for acute pulmonary hypertension following cardiac surgery. Ann Pharmacother. 2016;50(1):22-26.
11. Dzierba AL, Abel EE, Buckley MS, Lat I. A review of inhaled nitric oxide and aerosolized epoprostenol in acute lung injury or acute respiratory distress syndrome. Pharmacotherapy. 2014;34(3):279-290.
12. Pleasants RA, Hess DR. Aerosol delivery devices for obstructive lung diseases. Respir Care. 2018;63(6):708-733.
13. World Health Organization. Clinical management of severe acute respiratory infection when novel coronavirus (nCoV) infection is suspected. https://www.who.int/publications-detail/clinical-management-of-severe-acute-respiratory-infection-when-novel-coronavirus-(ncov)-infection-is-suspected Accessed March 26, 2020.
14. Centers for Disease Control and Prevention. Interim clinical guidance for management of patients with confirmed coronavirus disease (COVID-19). https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-guidance-management-patients.html. Revised March 7, 2020. Accessed March 26, 2020.
15. Wong RSM, Hui DS. Index patient and SARS outbreak in Hong Kong. Emerg Infect Dis. 2004;10(2):339-341.
16. Wong T-W, Lee C-K, Tam W, et al; Outbreak Study Group. Emerg Infect Dis. 2004;10(2):269-276.
17. Seto WH, Tsang D, Yung RWH, et al; Advisors of Expert SARS group of Hospital Authority. Effectiveness of precautions against droplets and contact in prevention of nosocomial transmission of severe acute respiratory syndrome (SARS). Lancet. 2003;361(9368):1519-1520.
18. Livingston E, Bucher K. Coronavirus Disease 2019 (COVID-19) in Italy. https://jamanetwork.com/journals/jama/fullarticle/2763401?resultClick=1. Published March 17, 2020. Accessed March 26, 2020.
19. Jones S. Spain: doctors struggle to cope as 514 die from coronavirus in a day. The Guardian. March 24, 2020. https://www.theguardian.com/world/2020/mar/24/spain-doctors-lack-protection-coronavirus-covid-19. Accessed March 27, 2020.
20. 16% of Ohio’s diagnosed COVID-19 cases are healthcare workers. https://www.wlwt.com/article/16-of-ohio-s-diagnosed-covid-19-cases-are-healthcare-workers/31930566#. Updated March 25, 2020. Accessed March 27, 2020.
21. Remuzzi A, Remuzzi G. COVID-19 and Italy: what next? Lancet. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)30627-9/fulltext. Accessed March 27, 2020.
22. van Doremalen N, Bushmaker T, Morris DH, et al. Aerosol and surface stability of SARS-CoV-2 as Compared with SARS-CoV-1 [published online ahead of print, 2020 Mar 17]. N Engl J Med. 2020;10.1056/NEJMc2004973.
23. McGrath JA, O’Sullivan A, Bennett G, et al. Investigation of the quantity of exhaled aerosol released into the environment during nebulization. Pharmaceutics. 2019;11(2):75.
24. Centers for Disease Control and Prevention. Healthcare Infection prevention and control FAQs for COVID-19. https://www.cdc.gov/coronavirus/2019-ncov/infection-control/infection-prevention-control-faq.html. Revised March 24, 2020. Accessed March 26, 2020.
25. Practice standards of respiratory procedures: post SARS era. Use of aerosolized medications. December 2003. http://www.hkresp.com/hkts.php?page=page/hkts/detail&meid=93742. Accessed March 26, 2020.
26. Wax RS, Christian MD. Practical recommendations for critical care and anesthesiology teams caring for novel coronavirus (2019-nCoV) patients. Can J Anesth. 2020. [ePub ahead of print.]
27. Newhouse MT. RE: transmission of coronavirus by nebulizer- as serious, underappreciated risk! https://www.cmaj.ca/content/re-transmission-corona-virus-nebulizer-serious-underappreciated-risk. Accessed March 26, 2020. [ePub ahead of print.]
28. Moira C-Y. Severe acute respiratory syndrome (SARS) and healthcare workers. Int J Occup Environ Health. 2004;10(4):421-427.
29. Timen A, Hulscher MEJL, Rust L, et al. Barriers to implementing infection prevention and control guidelines during crises: experiences of health care professionals. Am J Infect Control. 2010;38(9):726-733.
30. Khoo SM, Tan LK, Said N, Lim TK. Metered-dose inhaler with spacer instead of nebulizer during the outbreak of severe acute respiratory syndrome in Singapore. Respir Care. 2009;54(7):855-860.
FDA OKs durvalumab combo for extensive-stage SCLC
The US Food and Drug Administration has approved the immunotherapy durvalumab (Imfinzi, AstraZeneca) in combination with etoposide and either carboplatin or cisplatin as first-line treatment of patients with extensive-stage small cell lung cancer (ES-SCLC).
Durvalumab plus chemotherapy “can be considered a new standard in ES-SCLC,” said Myung-Ju Ahn, MD, Sungkyunkwan University, Seoul, South Korea, last year at the European Society of Medical Oncology (ESMO) annual meeting, where he discussed results from the phase 3 trial known as CASPIAN.
The new approval is based on efficacy and safety data from that trial, conducted in patients with previously untreated ES-SCLC. In the experimental group (n = 268), durvalumab plus etoposide and a platinum agent (EP) was followed by maintenance durvalumab, and in the control group (n = 269) patients received the EP regimen alone.
Median overall survival (OS) was 13 months in the durvalumab plus chemotherapy group compared with 10.3 months in the chemotherapy alone group (hazard ratio 0.73; 95% confidence interval, 0.59-0.91; P = .0047).
Reporting these results, trial investigator Luis Paz-Ares, MD, Hospital Universitario 12 de Octubre, Madrid, put the new survival benefit in the context of standard treatments at the ESMO meeting last year.
“Initial response rates to etoposide plus a platinum are high, but responses are not durable and patients treated with EP typically relapse within 6 months of starting treatment with a median OS of approximately 10 months,” he said.
In addition to the primary endpoint of OS, additional efficacy outcome measures were investigator-assessed progression-free survival (PFS) and objective response rate (ORR).
Median PFS was not statistically significant with immunotherapy; it was 5.1 months (95% CI, 4.7-6.2) in the durvalumab plus chemotherapy group and 5.4 months (95% CI, 4.8-6.2) in the chemotherapy alone group (HR, 0.78; 95% CI, 0.65-0.94).
The investigator-assessed ORR was 68% in the durvalumab plus chemotherapy group and 58% in the chemotherapy alone group.
The most common adverse reactions (≥ 20%) in patients with ES-SCLC were nausea, fatigue/asthenia, and alopecia, according to the FDA.
At ESMO, Paz-Ares reported that rates of serious adverse events (AEs) were comparable at 30.9% and 36.1% for the durvalumab plus EP group vs. EP alone, respectively; rates of AEs leading to discontinuation were identical in both groups at 9.4%. Unsurprisingly, immune-mediated AEs were higher at 19.6% in the durvalumab combination group vs. 2.6% in the EP alone group.
In this setting, durvalumab is administered prior to chemotherapy on the same day. The recommended durvalumab dose (when administered with etoposide and carboplatin or cisplatin) is 1,500 mg every 3 weeks prior to chemotherapy and then every 4 weeks as a single-agent maintenance therapy.
Durvalumab is already approved for metastatic non–small cell lung cancer in patients whose tumors have only spread in the chest, and is also approved for use in urothelial cancer.
This article first appeared on Medscape.com.
The US Food and Drug Administration has approved the immunotherapy durvalumab (Imfinzi, AstraZeneca) in combination with etoposide and either carboplatin or cisplatin as first-line treatment of patients with extensive-stage small cell lung cancer (ES-SCLC).
Durvalumab plus chemotherapy “can be considered a new standard in ES-SCLC,” said Myung-Ju Ahn, MD, Sungkyunkwan University, Seoul, South Korea, last year at the European Society of Medical Oncology (ESMO) annual meeting, where he discussed results from the phase 3 trial known as CASPIAN.
The new approval is based on efficacy and safety data from that trial, conducted in patients with previously untreated ES-SCLC. In the experimental group (n = 268), durvalumab plus etoposide and a platinum agent (EP) was followed by maintenance durvalumab, and in the control group (n = 269) patients received the EP regimen alone.
Median overall survival (OS) was 13 months in the durvalumab plus chemotherapy group compared with 10.3 months in the chemotherapy alone group (hazard ratio 0.73; 95% confidence interval, 0.59-0.91; P = .0047).
Reporting these results, trial investigator Luis Paz-Ares, MD, Hospital Universitario 12 de Octubre, Madrid, put the new survival benefit in the context of standard treatments at the ESMO meeting last year.
“Initial response rates to etoposide plus a platinum are high, but responses are not durable and patients treated with EP typically relapse within 6 months of starting treatment with a median OS of approximately 10 months,” he said.
In addition to the primary endpoint of OS, additional efficacy outcome measures were investigator-assessed progression-free survival (PFS) and objective response rate (ORR).
Median PFS was not statistically significant with immunotherapy; it was 5.1 months (95% CI, 4.7-6.2) in the durvalumab plus chemotherapy group and 5.4 months (95% CI, 4.8-6.2) in the chemotherapy alone group (HR, 0.78; 95% CI, 0.65-0.94).
The investigator-assessed ORR was 68% in the durvalumab plus chemotherapy group and 58% in the chemotherapy alone group.
The most common adverse reactions (≥ 20%) in patients with ES-SCLC were nausea, fatigue/asthenia, and alopecia, according to the FDA.
At ESMO, Paz-Ares reported that rates of serious adverse events (AEs) were comparable at 30.9% and 36.1% for the durvalumab plus EP group vs. EP alone, respectively; rates of AEs leading to discontinuation were identical in both groups at 9.4%. Unsurprisingly, immune-mediated AEs were higher at 19.6% in the durvalumab combination group vs. 2.6% in the EP alone group.
In this setting, durvalumab is administered prior to chemotherapy on the same day. The recommended durvalumab dose (when administered with etoposide and carboplatin or cisplatin) is 1,500 mg every 3 weeks prior to chemotherapy and then every 4 weeks as a single-agent maintenance therapy.
Durvalumab is already approved for metastatic non–small cell lung cancer in patients whose tumors have only spread in the chest, and is also approved for use in urothelial cancer.
This article first appeared on Medscape.com.
The US Food and Drug Administration has approved the immunotherapy durvalumab (Imfinzi, AstraZeneca) in combination with etoposide and either carboplatin or cisplatin as first-line treatment of patients with extensive-stage small cell lung cancer (ES-SCLC).
Durvalumab plus chemotherapy “can be considered a new standard in ES-SCLC,” said Myung-Ju Ahn, MD, Sungkyunkwan University, Seoul, South Korea, last year at the European Society of Medical Oncology (ESMO) annual meeting, where he discussed results from the phase 3 trial known as CASPIAN.
The new approval is based on efficacy and safety data from that trial, conducted in patients with previously untreated ES-SCLC. In the experimental group (n = 268), durvalumab plus etoposide and a platinum agent (EP) was followed by maintenance durvalumab, and in the control group (n = 269) patients received the EP regimen alone.
Median overall survival (OS) was 13 months in the durvalumab plus chemotherapy group compared with 10.3 months in the chemotherapy alone group (hazard ratio 0.73; 95% confidence interval, 0.59-0.91; P = .0047).
Reporting these results, trial investigator Luis Paz-Ares, MD, Hospital Universitario 12 de Octubre, Madrid, put the new survival benefit in the context of standard treatments at the ESMO meeting last year.
“Initial response rates to etoposide plus a platinum are high, but responses are not durable and patients treated with EP typically relapse within 6 months of starting treatment with a median OS of approximately 10 months,” he said.
In addition to the primary endpoint of OS, additional efficacy outcome measures were investigator-assessed progression-free survival (PFS) and objective response rate (ORR).
Median PFS was not statistically significant with immunotherapy; it was 5.1 months (95% CI, 4.7-6.2) in the durvalumab plus chemotherapy group and 5.4 months (95% CI, 4.8-6.2) in the chemotherapy alone group (HR, 0.78; 95% CI, 0.65-0.94).
The investigator-assessed ORR was 68% in the durvalumab plus chemotherapy group and 58% in the chemotherapy alone group.
The most common adverse reactions (≥ 20%) in patients with ES-SCLC were nausea, fatigue/asthenia, and alopecia, according to the FDA.
At ESMO, Paz-Ares reported that rates of serious adverse events (AEs) were comparable at 30.9% and 36.1% for the durvalumab plus EP group vs. EP alone, respectively; rates of AEs leading to discontinuation were identical in both groups at 9.4%. Unsurprisingly, immune-mediated AEs were higher at 19.6% in the durvalumab combination group vs. 2.6% in the EP alone group.
In this setting, durvalumab is administered prior to chemotherapy on the same day. The recommended durvalumab dose (when administered with etoposide and carboplatin or cisplatin) is 1,500 mg every 3 weeks prior to chemotherapy and then every 4 weeks as a single-agent maintenance therapy.
Durvalumab is already approved for metastatic non–small cell lung cancer in patients whose tumors have only spread in the chest, and is also approved for use in urothelial cancer.
This article first appeared on Medscape.com.
Monthly injection therapy for HIV found noninferior to daily oral dosing
Two international phase 3 randomized trials of according to reports published in the New England Journal of Medicine.
The Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection (FLAIR) study and the Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression (ATLAS) study looked at a separate facet of the use of a monthly therapeutic injection as a replacement for daily oral HIV therapy.
The FLAIR trial (ClinicalTrials.gov number, NCT02938520) was a phase 3, randomized, open-label study in which adults with HIV-1 infection who had not previously received antiretroviral therapy were given 20 weeks of daily oral induction therapy with dolutegravir–abacavir–lamivudine. Those patients with an HIV-1 RNA level of less than 50 copies per milliliter after 16 weeks were then randomly assigned (1:1) to continue their current oral therapy or switch to oral cabotegravir plus rilpivirine for 1 month followed by monthly intramuscular injections of long-acting cabotegravir, an HIV-1 integrase strand-transfer inhibitor, and rilpivirine, a nonnucleoside reverse-transcriptase inhibitor.
At week 48, an HIV-1 RNA level of 50 copies per milliliter or higher was found in 6 of 283 patients (2.1%) who received the long-acting therapy and in 7 of 283 (2.5%) who received oral therapy, which met the criterion for noninferiority for the primary endpoint. An HIV-1 RNA level of less than 50 copies per milliliter at week 48 was found in 93.6% of patients who received long-acting therapy and in 93.3% who received oral therapy, which also met the criterion for noninferiority, according to the study published in the New England Journal of Medicine.
Injection site reactions were reported in 86% of the long-acting therapy patients, 4 of whom withdrew from the trial for injection-related reasons. Grade 3 or higher adverse events and events that met liver-related stopping criteria occurred in 11% and 2%, respectively, of those who received long-acting therapy and in 4% and 1% of those who received oral therapy.
An assessment of treatment satisfaction at 48 weeks showed that 91% of the patients who switched to long-acting therapy preferred it to their daily oral therapy.
The ATLAS trial (ClinicalTrials.gov number, NCT02951052) was a phase 3, open-label, multicenter, noninferiority trial involving patients who had plasma HIV-1 RNA levels of less than 50 copies per milliliter for at least 6 months while taking standard oral antiretroviral therapy. These patients were randomized (308 in each group) to the long-acting cabotegravir plus rilpivirine injection therapy or daily oral therapy.
At 48 weeks, HIV-1 RNA levels of 50 copies per milliliter or higher were found in five participants (1.6%) receiving long-acting therapy and in three (1.0%) receiving oral therapy, which met the criterion for noninferiority for the primary endpoint, according to a study reported in the New England Journal of Medicine.
HIV-1 RNA levels of less than 50 copies per milliliter at week 48 occurred in 92.5% of patients on long-acting therapy and in 95.5% of those receiving oral therapy, which also met the criterion for noninferiority for this endpoint. Three patients in the long-acting therapy group had virologic failure, compared with four participants who received oral therapy.
Adverse events were more common in the long-acting–therapy group and included injection-site pain, which occurred in 231 recipients (75%) of long-acting therapy. This was mild or moderate in most cases, according to the authors. However, 1% of the participants in this group withdrew because of it. Overall, serious adverse events were reported in no more than 5% of participants in each group.
Together, the ATLAS and the FLAIR trials show that long-acting intramuscular injection therapy is noninferior to oral therapy as both an early regimen for HIV treatment, as well as for later, maintenance dosing. The use of long-acting therapy may improve patient adherence to treatment, according to both sets of study authors.
The ATLAS and FLAIR trials were funded by ViiV Healthcare and Janssen. The authors of both studies reported ties to pharmaceutical associations, and some authors are employees of the two funding sources.
SOURCE: Orkin C et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1909512 and Swindells S et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1904398.
Two international phase 3 randomized trials of according to reports published in the New England Journal of Medicine.
The Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection (FLAIR) study and the Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression (ATLAS) study looked at a separate facet of the use of a monthly therapeutic injection as a replacement for daily oral HIV therapy.
The FLAIR trial (ClinicalTrials.gov number, NCT02938520) was a phase 3, randomized, open-label study in which adults with HIV-1 infection who had not previously received antiretroviral therapy were given 20 weeks of daily oral induction therapy with dolutegravir–abacavir–lamivudine. Those patients with an HIV-1 RNA level of less than 50 copies per milliliter after 16 weeks were then randomly assigned (1:1) to continue their current oral therapy or switch to oral cabotegravir plus rilpivirine for 1 month followed by monthly intramuscular injections of long-acting cabotegravir, an HIV-1 integrase strand-transfer inhibitor, and rilpivirine, a nonnucleoside reverse-transcriptase inhibitor.
At week 48, an HIV-1 RNA level of 50 copies per milliliter or higher was found in 6 of 283 patients (2.1%) who received the long-acting therapy and in 7 of 283 (2.5%) who received oral therapy, which met the criterion for noninferiority for the primary endpoint. An HIV-1 RNA level of less than 50 copies per milliliter at week 48 was found in 93.6% of patients who received long-acting therapy and in 93.3% who received oral therapy, which also met the criterion for noninferiority, according to the study published in the New England Journal of Medicine.
Injection site reactions were reported in 86% of the long-acting therapy patients, 4 of whom withdrew from the trial for injection-related reasons. Grade 3 or higher adverse events and events that met liver-related stopping criteria occurred in 11% and 2%, respectively, of those who received long-acting therapy and in 4% and 1% of those who received oral therapy.
An assessment of treatment satisfaction at 48 weeks showed that 91% of the patients who switched to long-acting therapy preferred it to their daily oral therapy.
The ATLAS trial (ClinicalTrials.gov number, NCT02951052) was a phase 3, open-label, multicenter, noninferiority trial involving patients who had plasma HIV-1 RNA levels of less than 50 copies per milliliter for at least 6 months while taking standard oral antiretroviral therapy. These patients were randomized (308 in each group) to the long-acting cabotegravir plus rilpivirine injection therapy or daily oral therapy.
At 48 weeks, HIV-1 RNA levels of 50 copies per milliliter or higher were found in five participants (1.6%) receiving long-acting therapy and in three (1.0%) receiving oral therapy, which met the criterion for noninferiority for the primary endpoint, according to a study reported in the New England Journal of Medicine.
HIV-1 RNA levels of less than 50 copies per milliliter at week 48 occurred in 92.5% of patients on long-acting therapy and in 95.5% of those receiving oral therapy, which also met the criterion for noninferiority for this endpoint. Three patients in the long-acting therapy group had virologic failure, compared with four participants who received oral therapy.
Adverse events were more common in the long-acting–therapy group and included injection-site pain, which occurred in 231 recipients (75%) of long-acting therapy. This was mild or moderate in most cases, according to the authors. However, 1% of the participants in this group withdrew because of it. Overall, serious adverse events were reported in no more than 5% of participants in each group.
Together, the ATLAS and the FLAIR trials show that long-acting intramuscular injection therapy is noninferior to oral therapy as both an early regimen for HIV treatment, as well as for later, maintenance dosing. The use of long-acting therapy may improve patient adherence to treatment, according to both sets of study authors.
The ATLAS and FLAIR trials were funded by ViiV Healthcare and Janssen. The authors of both studies reported ties to pharmaceutical associations, and some authors are employees of the two funding sources.
SOURCE: Orkin C et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1909512 and Swindells S et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1904398.
Two international phase 3 randomized trials of according to reports published in the New England Journal of Medicine.
The Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection (FLAIR) study and the Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression (ATLAS) study looked at a separate facet of the use of a monthly therapeutic injection as a replacement for daily oral HIV therapy.
The FLAIR trial (ClinicalTrials.gov number, NCT02938520) was a phase 3, randomized, open-label study in which adults with HIV-1 infection who had not previously received antiretroviral therapy were given 20 weeks of daily oral induction therapy with dolutegravir–abacavir–lamivudine. Those patients with an HIV-1 RNA level of less than 50 copies per milliliter after 16 weeks were then randomly assigned (1:1) to continue their current oral therapy or switch to oral cabotegravir plus rilpivirine for 1 month followed by monthly intramuscular injections of long-acting cabotegravir, an HIV-1 integrase strand-transfer inhibitor, and rilpivirine, a nonnucleoside reverse-transcriptase inhibitor.
At week 48, an HIV-1 RNA level of 50 copies per milliliter or higher was found in 6 of 283 patients (2.1%) who received the long-acting therapy and in 7 of 283 (2.5%) who received oral therapy, which met the criterion for noninferiority for the primary endpoint. An HIV-1 RNA level of less than 50 copies per milliliter at week 48 was found in 93.6% of patients who received long-acting therapy and in 93.3% who received oral therapy, which also met the criterion for noninferiority, according to the study published in the New England Journal of Medicine.
Injection site reactions were reported in 86% of the long-acting therapy patients, 4 of whom withdrew from the trial for injection-related reasons. Grade 3 or higher adverse events and events that met liver-related stopping criteria occurred in 11% and 2%, respectively, of those who received long-acting therapy and in 4% and 1% of those who received oral therapy.
An assessment of treatment satisfaction at 48 weeks showed that 91% of the patients who switched to long-acting therapy preferred it to their daily oral therapy.
The ATLAS trial (ClinicalTrials.gov number, NCT02951052) was a phase 3, open-label, multicenter, noninferiority trial involving patients who had plasma HIV-1 RNA levels of less than 50 copies per milliliter for at least 6 months while taking standard oral antiretroviral therapy. These patients were randomized (308 in each group) to the long-acting cabotegravir plus rilpivirine injection therapy or daily oral therapy.
At 48 weeks, HIV-1 RNA levels of 50 copies per milliliter or higher were found in five participants (1.6%) receiving long-acting therapy and in three (1.0%) receiving oral therapy, which met the criterion for noninferiority for the primary endpoint, according to a study reported in the New England Journal of Medicine.
HIV-1 RNA levels of less than 50 copies per milliliter at week 48 occurred in 92.5% of patients on long-acting therapy and in 95.5% of those receiving oral therapy, which also met the criterion for noninferiority for this endpoint. Three patients in the long-acting therapy group had virologic failure, compared with four participants who received oral therapy.
Adverse events were more common in the long-acting–therapy group and included injection-site pain, which occurred in 231 recipients (75%) of long-acting therapy. This was mild or moderate in most cases, according to the authors. However, 1% of the participants in this group withdrew because of it. Overall, serious adverse events were reported in no more than 5% of participants in each group.
Together, the ATLAS and the FLAIR trials show that long-acting intramuscular injection therapy is noninferior to oral therapy as both an early regimen for HIV treatment, as well as for later, maintenance dosing. The use of long-acting therapy may improve patient adherence to treatment, according to both sets of study authors.
The ATLAS and FLAIR trials were funded by ViiV Healthcare and Janssen. The authors of both studies reported ties to pharmaceutical associations, and some authors are employees of the two funding sources.
SOURCE: Orkin C et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1909512 and Swindells S et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1904398.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
RT plus checkpoint blockade active in head and neck cancer
The combination of a phase 2 trial suggests.

“There are convincing arguments that radiation sensitizes patients to immunotherapy and can enhance its effects,” Jared Weiss, MD, associate professor of medicine, UNC Lineberger Comprehensive Cancer, Chapel Hill, North Carolina, said in a statement.
“And the opposite direction also seems to be true – radiation therapy needs a functional immune system to work. Our hope was that pembrolizumab might be a radiation sensitizer for these patients,” he said.
The study was presented at the Multidisciplinary Head and Neck Cancer Symposium, sponsored by the American Society for Radiation Oncology.
Both modalities have had some outstanding results in the past, observed Weiss. “If you look back to the historic studies, radiation alone often cures patients with this disease, while some of the first patients treated with pembrolizumab for recurrent/metastatic cancer are still alive many years out, with no evidence of disease,” he said.
“Our concept was that, in addition to whatever synergy the immunotherapy might provide with radiation, we also conceived of it as a ‘second shot on goal’ towards a cure, because there is durable control with drug alone,” he added.
Single-arm trial
The single-arm trial included 29 patients with locally advanced HNSCC.
Only about 10% of patients were current smokers, but more than half of the study group had a history of smoking. Of those, more than 55% had a history of 10 pack-years or more.
In slightly more than one third of patients, the primary site of the cancer was the base of the tongue. The tonsils were the primary site in slightly more than one third.
Platinum ineligibility was defined by provider and standard measures.
More than two thirds of patients were ineligible to receive cisplatin because of preexisting otopathy, including hearing impairment and tinnitus.
The combination of cisplatin and definitive-dose radiotherapy is standard treatment for locally advanced head and neck cancer, but contraindications to cisplatin are common in everyday clinical practice. Weiss noted that contraindications are present in about one third of his patients.
“We replaced standard, every-3-week cisplatin with pembrolizumab every 3 weeks,” Weiss explained, “and we hypothesized that with the ongoing effects of radiation therapy after completion, that additional adjuvant cycles could further sensitize patients [to the effects of radiation] without impairing recovery, so we added three adjuvant cycles as well,” he added.
With six cycles of an every-3-week drug, patients received 18 weeks of pembrolizumab in total.
Echoing results from the previously reported KEYNOTE-48 trial, pembrolizumab given with radiotherapy instead of chemotherapy led to an overall progression-free survival (PFS) rate of 76% at 1 year and an estimated PFS of 71% at 2 years.
At 1 year, 86% of patients were still alive, and at 2 years, an estimated 75% of patients were still alive, Weiss added.
For patients with human papillomavirus 16–positive cancer, rates of PFS and overall survival were slightly better, at 88% and 94%, respectively.
With regard to toxicities, “For the most part, this [treatment regimen] looks like radiation alone with one very notable exception, which was lymphopenia,” Weiss observed. Grade 3-4 lymphopenia affected 59% of patients.
Lymphocyte count hit bottom at week 4, he added, with only partial recovery at week 20 and no further recovery at 40 weeks. Lymphocyte count alone or any change in it was not predictive of early progression.
However, in comparing patients who experienced early disease progression to patients who did not experience progression, levels of baseline naive B cells in peripheral blood were higher and levels of circulating marginal zone B cells were lower in patients with progressive disease, Weiss reported.
Patient-reported outcomes indicated that common symptoms of treatment peaked at week 10, and there was relative recovery by week 20.
As reflected by Functional Assessment of Cancer Therapy (FACT) scores, which include social, emotional, and functional well-being, as well as the head and neck cancer scale, “we again see a nadir at 10 weeks with relative recovery at 20 weeks,” Weiss noted.
“We found that concurrent pembrolizumab with radiotherapy is a safe and feasible option for locally advanced head and neck cancer patients with cisplatin ineligibility,” Weiss concluded.
More research is being conducted in this area, and multiple ongoing studies will further elucidate the value of PD-1 or PD-L1 checkpoint blockade with definitive radiation therapy, he added.
The study was funded by Merck & Co. Weiss’ institution has received research funding from Celgene, Pfizer, Merck, AZ/Medimmmune, Amgen, Carefusion, G1 Therapeutics, Immunicum, Loxo/Lilly, and the Jimmy V Foundation. Weiss has received honoraria for consulting from AstraZeneca, EMD Serono, Genentech, Inivata, Celgene, G1 Therapeutics, Jounce Therapeutics, Abbvie, Rakuten, Nanobiotix, Azitra, Loxo/Lilly, Pfizer, and Blueprint had has stock in Nektar and Vesselon.
This article first appeared on Medscape.com.
The combination of a phase 2 trial suggests.
“There are convincing arguments that radiation sensitizes patients to immunotherapy and can enhance its effects,” Jared Weiss, MD, associate professor of medicine, UNC Lineberger Comprehensive Cancer, Chapel Hill, North Carolina, said in a statement.
“And the opposite direction also seems to be true – radiation therapy needs a functional immune system to work. Our hope was that pembrolizumab might be a radiation sensitizer for these patients,” he said.
The study was presented at the Multidisciplinary Head and Neck Cancer Symposium, sponsored by the American Society for Radiation Oncology.
Both modalities have had some outstanding results in the past, observed Weiss. “If you look back to the historic studies, radiation alone often cures patients with this disease, while some of the first patients treated with pembrolizumab for recurrent/metastatic cancer are still alive many years out, with no evidence of disease,” he said.
“Our concept was that, in addition to whatever synergy the immunotherapy might provide with radiation, we also conceived of it as a ‘second shot on goal’ towards a cure, because there is durable control with drug alone,” he added.
Single-arm trial
The single-arm trial included 29 patients with locally advanced HNSCC.
Only about 10% of patients were current smokers, but more than half of the study group had a history of smoking. Of those, more than 55% had a history of 10 pack-years or more.
In slightly more than one third of patients, the primary site of the cancer was the base of the tongue. The tonsils were the primary site in slightly more than one third.
Platinum ineligibility was defined by provider and standard measures.
More than two thirds of patients were ineligible to receive cisplatin because of preexisting otopathy, including hearing impairment and tinnitus.
The combination of cisplatin and definitive-dose radiotherapy is standard treatment for locally advanced head and neck cancer, but contraindications to cisplatin are common in everyday clinical practice. Weiss noted that contraindications are present in about one third of his patients.
“We replaced standard, every-3-week cisplatin with pembrolizumab every 3 weeks,” Weiss explained, “and we hypothesized that with the ongoing effects of radiation therapy after completion, that additional adjuvant cycles could further sensitize patients [to the effects of radiation] without impairing recovery, so we added three adjuvant cycles as well,” he added.
With six cycles of an every-3-week drug, patients received 18 weeks of pembrolizumab in total.
Echoing results from the previously reported KEYNOTE-48 trial, pembrolizumab given with radiotherapy instead of chemotherapy led to an overall progression-free survival (PFS) rate of 76% at 1 year and an estimated PFS of 71% at 2 years.
At 1 year, 86% of patients were still alive, and at 2 years, an estimated 75% of patients were still alive, Weiss added.
For patients with human papillomavirus 16–positive cancer, rates of PFS and overall survival were slightly better, at 88% and 94%, respectively.
With regard to toxicities, “For the most part, this [treatment regimen] looks like radiation alone with one very notable exception, which was lymphopenia,” Weiss observed. Grade 3-4 lymphopenia affected 59% of patients.
Lymphocyte count hit bottom at week 4, he added, with only partial recovery at week 20 and no further recovery at 40 weeks. Lymphocyte count alone or any change in it was not predictive of early progression.
However, in comparing patients who experienced early disease progression to patients who did not experience progression, levels of baseline naive B cells in peripheral blood were higher and levels of circulating marginal zone B cells were lower in patients with progressive disease, Weiss reported.
Patient-reported outcomes indicated that common symptoms of treatment peaked at week 10, and there was relative recovery by week 20.
As reflected by Functional Assessment of Cancer Therapy (FACT) scores, which include social, emotional, and functional well-being, as well as the head and neck cancer scale, “we again see a nadir at 10 weeks with relative recovery at 20 weeks,” Weiss noted.
“We found that concurrent pembrolizumab with radiotherapy is a safe and feasible option for locally advanced head and neck cancer patients with cisplatin ineligibility,” Weiss concluded.
More research is being conducted in this area, and multiple ongoing studies will further elucidate the value of PD-1 or PD-L1 checkpoint blockade with definitive radiation therapy, he added.
The study was funded by Merck & Co. Weiss’ institution has received research funding from Celgene, Pfizer, Merck, AZ/Medimmmune, Amgen, Carefusion, G1 Therapeutics, Immunicum, Loxo/Lilly, and the Jimmy V Foundation. Weiss has received honoraria for consulting from AstraZeneca, EMD Serono, Genentech, Inivata, Celgene, G1 Therapeutics, Jounce Therapeutics, Abbvie, Rakuten, Nanobiotix, Azitra, Loxo/Lilly, Pfizer, and Blueprint had has stock in Nektar and Vesselon.
This article first appeared on Medscape.com.
The combination of a phase 2 trial suggests.
“There are convincing arguments that radiation sensitizes patients to immunotherapy and can enhance its effects,” Jared Weiss, MD, associate professor of medicine, UNC Lineberger Comprehensive Cancer, Chapel Hill, North Carolina, said in a statement.
“And the opposite direction also seems to be true – radiation therapy needs a functional immune system to work. Our hope was that pembrolizumab might be a radiation sensitizer for these patients,” he said.
The study was presented at the Multidisciplinary Head and Neck Cancer Symposium, sponsored by the American Society for Radiation Oncology.
Both modalities have had some outstanding results in the past, observed Weiss. “If you look back to the historic studies, radiation alone often cures patients with this disease, while some of the first patients treated with pembrolizumab for recurrent/metastatic cancer are still alive many years out, with no evidence of disease,” he said.
“Our concept was that, in addition to whatever synergy the immunotherapy might provide with radiation, we also conceived of it as a ‘second shot on goal’ towards a cure, because there is durable control with drug alone,” he added.
Single-arm trial
The single-arm trial included 29 patients with locally advanced HNSCC.
Only about 10% of patients were current smokers, but more than half of the study group had a history of smoking. Of those, more than 55% had a history of 10 pack-years or more.
In slightly more than one third of patients, the primary site of the cancer was the base of the tongue. The tonsils were the primary site in slightly more than one third.
Platinum ineligibility was defined by provider and standard measures.
More than two thirds of patients were ineligible to receive cisplatin because of preexisting otopathy, including hearing impairment and tinnitus.
The combination of cisplatin and definitive-dose radiotherapy is standard treatment for locally advanced head and neck cancer, but contraindications to cisplatin are common in everyday clinical practice. Weiss noted that contraindications are present in about one third of his patients.
“We replaced standard, every-3-week cisplatin with pembrolizumab every 3 weeks,” Weiss explained, “and we hypothesized that with the ongoing effects of radiation therapy after completion, that additional adjuvant cycles could further sensitize patients [to the effects of radiation] without impairing recovery, so we added three adjuvant cycles as well,” he added.
With six cycles of an every-3-week drug, patients received 18 weeks of pembrolizumab in total.
Echoing results from the previously reported KEYNOTE-48 trial, pembrolizumab given with radiotherapy instead of chemotherapy led to an overall progression-free survival (PFS) rate of 76% at 1 year and an estimated PFS of 71% at 2 years.
At 1 year, 86% of patients were still alive, and at 2 years, an estimated 75% of patients were still alive, Weiss added.
For patients with human papillomavirus 16–positive cancer, rates of PFS and overall survival were slightly better, at 88% and 94%, respectively.
With regard to toxicities, “For the most part, this [treatment regimen] looks like radiation alone with one very notable exception, which was lymphopenia,” Weiss observed. Grade 3-4 lymphopenia affected 59% of patients.
Lymphocyte count hit bottom at week 4, he added, with only partial recovery at week 20 and no further recovery at 40 weeks. Lymphocyte count alone or any change in it was not predictive of early progression.
However, in comparing patients who experienced early disease progression to patients who did not experience progression, levels of baseline naive B cells in peripheral blood were higher and levels of circulating marginal zone B cells were lower in patients with progressive disease, Weiss reported.
Patient-reported outcomes indicated that common symptoms of treatment peaked at week 10, and there was relative recovery by week 20.
As reflected by Functional Assessment of Cancer Therapy (FACT) scores, which include social, emotional, and functional well-being, as well as the head and neck cancer scale, “we again see a nadir at 10 weeks with relative recovery at 20 weeks,” Weiss noted.
“We found that concurrent pembrolizumab with radiotherapy is a safe and feasible option for locally advanced head and neck cancer patients with cisplatin ineligibility,” Weiss concluded.
More research is being conducted in this area, and multiple ongoing studies will further elucidate the value of PD-1 or PD-L1 checkpoint blockade with definitive radiation therapy, he added.
The study was funded by Merck & Co. Weiss’ institution has received research funding from Celgene, Pfizer, Merck, AZ/Medimmmune, Amgen, Carefusion, G1 Therapeutics, Immunicum, Loxo/Lilly, and the Jimmy V Foundation. Weiss has received honoraria for consulting from AstraZeneca, EMD Serono, Genentech, Inivata, Celgene, G1 Therapeutics, Jounce Therapeutics, Abbvie, Rakuten, Nanobiotix, Azitra, Loxo/Lilly, Pfizer, and Blueprint had has stock in Nektar and Vesselon.
This article first appeared on Medscape.com.
REPORTING FROM HEAD AND NECK CANCERS SYMPOSIUM 2020
MACE benefits with dapagliflozin improve with disease duration
Treatment with the sodium-glucose transporter 2 inhibitor dapagliflozin reduced the risk for cardiovascular disease or hospitalization for heart failure (CVD/HHF) in patients with diabetes, regardless of the duration of the disease, but had a greater protective benefit against major adverse cardiovascular events (MACE) and renal events in patients with longer disease duration, according to new findings from a post hoc analysis of the DECLARE-TIMI 58 trial.
The positive effect of dapagliflozin in patients with MACE – which includes myocardial infarction (MI), CVD, and ischemic stroke – may have been driven by lower rates of MI and ischemic stroke with the drug, compared with placebo, in patients with longer disease duration, wrote Harpreet S. Bajaj, MD, and colleagues. Their report is in Diabetes, Obesity and Metabolism (2020 Feb 23. doi: 10.1111/dom.14011).
It has been previously reported that the risk for complications in diabetes increases with increasing duration of the disease. Recent studies with SGLT-2 inhibitors have shown that the drugs improve cardiovascular and renal outcomes in diabetes, and they are recommended by the American Diabetes Association as second-line therapy in patients with atherosclerotic cardiovascular disease, chronic kidney disease, or heart failure. The European Society of Cardiology and the European Association for the Study of Diabetes recommend that patients with diabetes patients who have three or more risk factors, or those with a disease duration of more than 20 years, should be deemed very high risk and be considered for early treatment with SGLT2 inhibitors.
“The MACE benefit observed with dapagliflozin in this study in patients with diabetes duration of [more than] 20 years, clearly supports that notion,” the authors wrote.
In DECLARE-TIMI 58, 17,160 patients with type 2 diabetes received dapagliflozin or placebo and were followed for a median of 4.2 years. Of those patients, 22.4% had a disease duration of fewer than 5 years; 27.6%, a duration of 5-10 years; 23.0%, 10-15 years; 14.2%, 10-15 years; and 12.9%, more than 20 years. The median duration of disease was 11 years.
Patients in all the age groups had similar reductions in CVD/HHF, compared with placebo, with hazard ratios of 0.79 (disease duration of 5 or fewer years), 0.86, 0.92, 0.81, and 0.75 (duration of 20 years), respectively (interaction trend P = .760).
Treatment with dapagliflozin reduced the incidence of MACE, but the benefit was more apparent in patients with longer-term disease: HR, 1.08; 1.02; 0.94; 0.92; and 0.67, respectively (interaction trend P = .004). Similar trends were seen with MI (interaction trend P = .019) and ischemic stroke (interaction trend P = .015).
The researchers also reported improved benefits in renal-specific outcome with increasing disease duration, with HRs ranging from 0.79 in patients with diabetes duration of fewer than 5 years, to 0.42 in those with a duration of more than 20 years (interaction trend P = .084).
Limitations of the study include the fact that the information about diabetes duration relied on patient reports, and that the original trial was not powered for all subgroup interactions. This authors emphasized that this was a post hoc analysis and as such, should be considered hypothesis generating.
All but two of the authors reported relationships with Astra Zeneca, which funded the study, and other drug companies.
SOURCE: Bajaj HS et al. Diabetes Obes Metab. 2020 Feb 23. doi: 10.1111/dom.14011.
Treatment with the sodium-glucose transporter 2 inhibitor dapagliflozin reduced the risk for cardiovascular disease or hospitalization for heart failure (CVD/HHF) in patients with diabetes, regardless of the duration of the disease, but had a greater protective benefit against major adverse cardiovascular events (MACE) and renal events in patients with longer disease duration, according to new findings from a post hoc analysis of the DECLARE-TIMI 58 trial.
The positive effect of dapagliflozin in patients with MACE – which includes myocardial infarction (MI), CVD, and ischemic stroke – may have been driven by lower rates of MI and ischemic stroke with the drug, compared with placebo, in patients with longer disease duration, wrote Harpreet S. Bajaj, MD, and colleagues. Their report is in Diabetes, Obesity and Metabolism (2020 Feb 23. doi: 10.1111/dom.14011).
It has been previously reported that the risk for complications in diabetes increases with increasing duration of the disease. Recent studies with SGLT-2 inhibitors have shown that the drugs improve cardiovascular and renal outcomes in diabetes, and they are recommended by the American Diabetes Association as second-line therapy in patients with atherosclerotic cardiovascular disease, chronic kidney disease, or heart failure. The European Society of Cardiology and the European Association for the Study of Diabetes recommend that patients with diabetes patients who have three or more risk factors, or those with a disease duration of more than 20 years, should be deemed very high risk and be considered for early treatment with SGLT2 inhibitors.
“The MACE benefit observed with dapagliflozin in this study in patients with diabetes duration of [more than] 20 years, clearly supports that notion,” the authors wrote.
In DECLARE-TIMI 58, 17,160 patients with type 2 diabetes received dapagliflozin or placebo and were followed for a median of 4.2 years. Of those patients, 22.4% had a disease duration of fewer than 5 years; 27.6%, a duration of 5-10 years; 23.0%, 10-15 years; 14.2%, 10-15 years; and 12.9%, more than 20 years. The median duration of disease was 11 years.
Patients in all the age groups had similar reductions in CVD/HHF, compared with placebo, with hazard ratios of 0.79 (disease duration of 5 or fewer years), 0.86, 0.92, 0.81, and 0.75 (duration of 20 years), respectively (interaction trend P = .760).
Treatment with dapagliflozin reduced the incidence of MACE, but the benefit was more apparent in patients with longer-term disease: HR, 1.08; 1.02; 0.94; 0.92; and 0.67, respectively (interaction trend P = .004). Similar trends were seen with MI (interaction trend P = .019) and ischemic stroke (interaction trend P = .015).
The researchers also reported improved benefits in renal-specific outcome with increasing disease duration, with HRs ranging from 0.79 in patients with diabetes duration of fewer than 5 years, to 0.42 in those with a duration of more than 20 years (interaction trend P = .084).
Limitations of the study include the fact that the information about diabetes duration relied on patient reports, and that the original trial was not powered for all subgroup interactions. This authors emphasized that this was a post hoc analysis and as such, should be considered hypothesis generating.
All but two of the authors reported relationships with Astra Zeneca, which funded the study, and other drug companies.
SOURCE: Bajaj HS et al. Diabetes Obes Metab. 2020 Feb 23. doi: 10.1111/dom.14011.
Treatment with the sodium-glucose transporter 2 inhibitor dapagliflozin reduced the risk for cardiovascular disease or hospitalization for heart failure (CVD/HHF) in patients with diabetes, regardless of the duration of the disease, but had a greater protective benefit against major adverse cardiovascular events (MACE) and renal events in patients with longer disease duration, according to new findings from a post hoc analysis of the DECLARE-TIMI 58 trial.
The positive effect of dapagliflozin in patients with MACE – which includes myocardial infarction (MI), CVD, and ischemic stroke – may have been driven by lower rates of MI and ischemic stroke with the drug, compared with placebo, in patients with longer disease duration, wrote Harpreet S. Bajaj, MD, and colleagues. Their report is in Diabetes, Obesity and Metabolism (2020 Feb 23. doi: 10.1111/dom.14011).
It has been previously reported that the risk for complications in diabetes increases with increasing duration of the disease. Recent studies with SGLT-2 inhibitors have shown that the drugs improve cardiovascular and renal outcomes in diabetes, and they are recommended by the American Diabetes Association as second-line therapy in patients with atherosclerotic cardiovascular disease, chronic kidney disease, or heart failure. The European Society of Cardiology and the European Association for the Study of Diabetes recommend that patients with diabetes patients who have three or more risk factors, or those with a disease duration of more than 20 years, should be deemed very high risk and be considered for early treatment with SGLT2 inhibitors.
“The MACE benefit observed with dapagliflozin in this study in patients with diabetes duration of [more than] 20 years, clearly supports that notion,” the authors wrote.
In DECLARE-TIMI 58, 17,160 patients with type 2 diabetes received dapagliflozin or placebo and were followed for a median of 4.2 years. Of those patients, 22.4% had a disease duration of fewer than 5 years; 27.6%, a duration of 5-10 years; 23.0%, 10-15 years; 14.2%, 10-15 years; and 12.9%, more than 20 years. The median duration of disease was 11 years.
Patients in all the age groups had similar reductions in CVD/HHF, compared with placebo, with hazard ratios of 0.79 (disease duration of 5 or fewer years), 0.86, 0.92, 0.81, and 0.75 (duration of 20 years), respectively (interaction trend P = .760).
Treatment with dapagliflozin reduced the incidence of MACE, but the benefit was more apparent in patients with longer-term disease: HR, 1.08; 1.02; 0.94; 0.92; and 0.67, respectively (interaction trend P = .004). Similar trends were seen with MI (interaction trend P = .019) and ischemic stroke (interaction trend P = .015).
The researchers also reported improved benefits in renal-specific outcome with increasing disease duration, with HRs ranging from 0.79 in patients with diabetes duration of fewer than 5 years, to 0.42 in those with a duration of more than 20 years (interaction trend P = .084).
Limitations of the study include the fact that the information about diabetes duration relied on patient reports, and that the original trial was not powered for all subgroup interactions. This authors emphasized that this was a post hoc analysis and as such, should be considered hypothesis generating.
All but two of the authors reported relationships with Astra Zeneca, which funded the study, and other drug companies.
SOURCE: Bajaj HS et al. Diabetes Obes Metab. 2020 Feb 23. doi: 10.1111/dom.14011.
FROM DIABETES, OBESITY AND METABOLISM
Pembro ups survival in NSCLC: ‘Really extraordinary’ results
More than a third (35%) of patients with relapsed non–small cell lung cancer (NSCLC) treated with pembrolizumab (Keytruda, Merck) were still alive at 3 years, according to long-term results from a pivotal clinical trial.
The results also showed that, among the 10% of patients who completed all 35 cycles of pembrolizumab, the 3-year overall survival was approximately 99%, with progression-free survival (PFS) at around 70%.
“It is too soon to say that pembrolizumab is a potential cure...and we know that it doesn’t work for all patients, but the agent remains very, very promising,” said lead investigator Roy Herbst, MD, PhD, Department of Medical Oncology, Yale Comprehensive Cancer Center, New Haven, Connecticut.
These new results come from the KEYNOTE-010 trial, conducted in more than 1000 patients with NSCLC who had progressed on chemotherapy, randomized to receive immunotherapy with pembrolizumab or chemotherapy with docetaxel.
The results were published online on February 20 in the Journal of Clinical Oncology and were previously presented at the 2018 annual meeting of the European Society of Medical Oncology.
Overall survival at 3 years was 35% in patients with PD-L1 expression ≥ 50% in the tumor, and 23% in those with PD-L1 ≥ 1%.
This compares with 3-year overall survival of 11-13% with docetaxel.
These results are “really extraordinary,” Herbst commented to Medscape Medical News.
The 3-year overall survival rate of 35% in patients with PD-L1 ≥ 50% “is huge,” he said. “It really shows the durability of the response.”
Herbst commented that the “almost 100%” survival at 3 years among patients who completed 35 cycles of pembrolizumab shows that this treatment period (of about 2 years) is “probably about the right time to treat.”
“Currently, the agent is being used in all potential settings, before any other treatment, after other treatment, and with other treatments,” he said.
“Our hope is to find the very best way to use pembrolizumab to treat individual lung cancer patients, assessing how much PD-L1 a tumor expresses, what stage the patient is in, as well as other variables and biomarkers we are working on. This is the story of tailored therapy,” Herbst said.
Approached for comment, Solange Peters, MD, PhD, Oncology Department, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland, said that the results are “very good” and “confirm the paradigms we have been seeing in melanoma,” with good long-term control, which is “very reassuring.”
However, she told Medscape Medical News that the trial raises an important question: «How long do you need to expose your patient with lung cancer to immunotherapy in order to get this long-term control?»
She said the “good news” is that, for the 10% of patients who completed 2 years of treatment per protocol, almost all of them are still alive at 3 years, “which is not observed with chemotherapy.”
The question for Peters is “more about the definition of long-term control,” as it was seen that almost one in three patients nevertheless had some form of progression.
This suggests that you have a group of people “who are nicely controlled, you stop the drug, and 1 year later a third of them have progressed.”
Peters said: “So how long do you need to treat these patients? I would say I still don’t know.”
“If I were one of these patients probably I would still want to continue [on the drug]. Of course, some might have progressed even while remaining on the drug, but the proportion who would have progressed is probably smaller than this one.”
Responses on Re-introduction of Therapy
The study also allowed patients who had completed 35 cycles of pembrolizumab to be restarted on the drug if they experienced progression.
The team found that, among 14 patients, 43% had a partial response and 36% had stable disease.
Herbst highlighted this finding and told Medscape Medical News that this «could be very important to physicians because they might want to think about using the drug again» in patients who have progressed on it.
He believes that the progression was not because of any resistance per se but rather a slowing down of the adaptive immune response.
“It’s just that it needs a boost,” he said, while noting that tissue specimens will nevertheless be required to demonstrate the theory.
Peters agreed that these results are “very promising,” but questioned their overall significance, as it is “a very small number of patients” from a subset whose disease was controlled while on treatment and then progressed after stopping.
She also pointed out that, in another study in patients with lung cancer (CheckMate-153), some patients were rechallenged with immunotherapy after having stopped treatment at 1 year “with very poor results.”
Peters said studies in melanoma have shown “rechallenge can be useful in a significant proportion of patients, but still you have not demonstrated that stopping and rechallenging is the same as not stopping.”
Study Details
KEYNOTE-010 involved patients with NSCLC from 202 centers in 24 countries with stage IIIB/IV disease expressing PD-L1 who had experienced disease progression after at least two cycles of platinum-based chemotherapy.
They were randomized 1:1:1 to open-label pembrolizumab 2 mg/kg, pembrolizumab 10 mg/kg, or docetaxel 75 mg/m2 every 3 weeks.
Pembrolizumab was continued for 35 treatment cycles over 2 years and docetaxel was continued for the maximum duration allowed by local regulators.
Patients who stopped pembrolizumab after a complete response or completing all 35 cycles, and who subsequently experienced disease progression, could receive up to 17 additional cycles over 1 year if they had not received another anticancer therapy in the meantime.
Among the 1,034 patients originally recruited between August 2013 and February 2015, 691 were assigned to pembrolizumab at 3 mg/kg or 10 mg/kg and 343 to docetaxel.
For the intention-to-treat analysis in 1033 patients, the mean duration of follow-up was 42.6 months, with a median treatment duration of 3.5 months in the pembrolizumab group and 2.0 months in the docetaxel group.
Compared with docetaxel, pembrolizumab was associated with a significant reduction in the risk of death, at a hazard ratio of 0.53 in patients with PD-L1 ≥ 50% and 0.69 in those with PD-L1 ≥ 1% (both P < .0001).
In patients with PD-L1 ≥ 50%, median overall survival was 16.9 months in those given pembrolizumab and 8.2 months with docetaxel. Among those with PD-L1 ≥ 1%, median overall survival was 11.8 months with pembrolizumab versus 8.4 months with docetaxel.
Overall survival on Kaplan-Meier analysis was 34.5% with pembrolizumab and 12.7% with docetaxel in the PD-L1 ≥ 50% group, and 22.9% versus 11.0% in the PD-L1 ≥ 1% group.
PFS significantly improved with pembrolizumab versus docetaxel, at a hazard ratio of 0.57 (P < .00001) among patients with PD-L1 ≥ 50% and 0.83 (P < .005) in those with PD-L1 ≥ 1%.
In terms of safety, 17.7% of patients who completed 2 years of pembrolizumab had grade 3-5 treatment-related adverse events, compared with 16.6% among all pembrolizumab-treated patients and 36.6% of those given docetaxel.
The team reports that 79 patients completed 35 cycles of pembrolizumab, with a median follow-up of 43.4 months.
Compared with the overall patient group, these patients were less likely to be aged ≥ 65 years and to have received two or more prior treatment lines, although they were more likely to be current or former smokers and to have squamous tumor histology.
Patients who completed 35 cycles had an objective response rate of 94.9%, and 91.0% were still alive at the data cutoff. Overall survival rates were 98.7% at 12 months and 86.3% at 24 months.
Of 71 patients eligible for analysis, 23 experienced progression after completing pembrolizumab, at PFS rates at 12 and 24 months of 72.5% and 57.7%, respectively.
A total of 14 patients were given a second course of pembrolizumab, of whom six had a partial response and five had stable disease. At the data cutoff, five patients had completed 17 additional cycles and 11 were alive.
Pembro Approved at Fixed Dose
One notable aspect of the study is that patients in the pembrolizumab arm were given two different doses of the drug based on body weight, whereas the drug is approved in the United States at a fixed dose of 200 mg.
Herbst told Medscape Medical News he considers the 200-mg dose to be appropriate.
“I didn’t think that the 3-mg versus 10-mg dose per kg that we used in our study made much difference in an average-sized person,” he said, adding that the 200-mg dose “is something a little bit more than 3 mg/kg.”
“So I think that this is clearly the right dos, and I don’t think more would make any difference,” he said.
The study was funded by Merck, the manufacturer of pembrolizumab. Herbst has reported having a consulting or advisory role for many pharmaceutical companies. Other coauthors have also reported relationships with industry, and some of the authors are Merck employees. Peters has reported receiving education grants, providing consultation, attending advisory boards, and/or providing lectures for many pharmaceutical companies.
This article first appeared on Medscape.com.
More than a third (35%) of patients with relapsed non–small cell lung cancer (NSCLC) treated with pembrolizumab (Keytruda, Merck) were still alive at 3 years, according to long-term results from a pivotal clinical trial.
The results also showed that, among the 10% of patients who completed all 35 cycles of pembrolizumab, the 3-year overall survival was approximately 99%, with progression-free survival (PFS) at around 70%.
“It is too soon to say that pembrolizumab is a potential cure...and we know that it doesn’t work for all patients, but the agent remains very, very promising,” said lead investigator Roy Herbst, MD, PhD, Department of Medical Oncology, Yale Comprehensive Cancer Center, New Haven, Connecticut.
These new results come from the KEYNOTE-010 trial, conducted in more than 1000 patients with NSCLC who had progressed on chemotherapy, randomized to receive immunotherapy with pembrolizumab or chemotherapy with docetaxel.
The results were published online on February 20 in the Journal of Clinical Oncology and were previously presented at the 2018 annual meeting of the European Society of Medical Oncology.
Overall survival at 3 years was 35% in patients with PD-L1 expression ≥ 50% in the tumor, and 23% in those with PD-L1 ≥ 1%.
This compares with 3-year overall survival of 11-13% with docetaxel.
These results are “really extraordinary,” Herbst commented to Medscape Medical News.
The 3-year overall survival rate of 35% in patients with PD-L1 ≥ 50% “is huge,” he said. “It really shows the durability of the response.”
Herbst commented that the “almost 100%” survival at 3 years among patients who completed 35 cycles of pembrolizumab shows that this treatment period (of about 2 years) is “probably about the right time to treat.”
“Currently, the agent is being used in all potential settings, before any other treatment, after other treatment, and with other treatments,” he said.
“Our hope is to find the very best way to use pembrolizumab to treat individual lung cancer patients, assessing how much PD-L1 a tumor expresses, what stage the patient is in, as well as other variables and biomarkers we are working on. This is the story of tailored therapy,” Herbst said.
Approached for comment, Solange Peters, MD, PhD, Oncology Department, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland, said that the results are “very good” and “confirm the paradigms we have been seeing in melanoma,” with good long-term control, which is “very reassuring.”
However, she told Medscape Medical News that the trial raises an important question: «How long do you need to expose your patient with lung cancer to immunotherapy in order to get this long-term control?»
She said the “good news” is that, for the 10% of patients who completed 2 years of treatment per protocol, almost all of them are still alive at 3 years, “which is not observed with chemotherapy.”
The question for Peters is “more about the definition of long-term control,” as it was seen that almost one in three patients nevertheless had some form of progression.
This suggests that you have a group of people “who are nicely controlled, you stop the drug, and 1 year later a third of them have progressed.”
Peters said: “So how long do you need to treat these patients? I would say I still don’t know.”
“If I were one of these patients probably I would still want to continue [on the drug]. Of course, some might have progressed even while remaining on the drug, but the proportion who would have progressed is probably smaller than this one.”
Responses on Re-introduction of Therapy
The study also allowed patients who had completed 35 cycles of pembrolizumab to be restarted on the drug if they experienced progression.
The team found that, among 14 patients, 43% had a partial response and 36% had stable disease.
Herbst highlighted this finding and told Medscape Medical News that this «could be very important to physicians because they might want to think about using the drug again» in patients who have progressed on it.
He believes that the progression was not because of any resistance per se but rather a slowing down of the adaptive immune response.
“It’s just that it needs a boost,” he said, while noting that tissue specimens will nevertheless be required to demonstrate the theory.
Peters agreed that these results are “very promising,” but questioned their overall significance, as it is “a very small number of patients” from a subset whose disease was controlled while on treatment and then progressed after stopping.
She also pointed out that, in another study in patients with lung cancer (CheckMate-153), some patients were rechallenged with immunotherapy after having stopped treatment at 1 year “with very poor results.”
Peters said studies in melanoma have shown “rechallenge can be useful in a significant proportion of patients, but still you have not demonstrated that stopping and rechallenging is the same as not stopping.”
Study Details
KEYNOTE-010 involved patients with NSCLC from 202 centers in 24 countries with stage IIIB/IV disease expressing PD-L1 who had experienced disease progression after at least two cycles of platinum-based chemotherapy.
They were randomized 1:1:1 to open-label pembrolizumab 2 mg/kg, pembrolizumab 10 mg/kg, or docetaxel 75 mg/m2 every 3 weeks.
Pembrolizumab was continued for 35 treatment cycles over 2 years and docetaxel was continued for the maximum duration allowed by local regulators.
Patients who stopped pembrolizumab after a complete response or completing all 35 cycles, and who subsequently experienced disease progression, could receive up to 17 additional cycles over 1 year if they had not received another anticancer therapy in the meantime.
Among the 1,034 patients originally recruited between August 2013 and February 2015, 691 were assigned to pembrolizumab at 3 mg/kg or 10 mg/kg and 343 to docetaxel.
For the intention-to-treat analysis in 1033 patients, the mean duration of follow-up was 42.6 months, with a median treatment duration of 3.5 months in the pembrolizumab group and 2.0 months in the docetaxel group.
Compared with docetaxel, pembrolizumab was associated with a significant reduction in the risk of death, at a hazard ratio of 0.53 in patients with PD-L1 ≥ 50% and 0.69 in those with PD-L1 ≥ 1% (both P < .0001).
In patients with PD-L1 ≥ 50%, median overall survival was 16.9 months in those given pembrolizumab and 8.2 months with docetaxel. Among those with PD-L1 ≥ 1%, median overall survival was 11.8 months with pembrolizumab versus 8.4 months with docetaxel.
Overall survival on Kaplan-Meier analysis was 34.5% with pembrolizumab and 12.7% with docetaxel in the PD-L1 ≥ 50% group, and 22.9% versus 11.0% in the PD-L1 ≥ 1% group.
PFS significantly improved with pembrolizumab versus docetaxel, at a hazard ratio of 0.57 (P < .00001) among patients with PD-L1 ≥ 50% and 0.83 (P < .005) in those with PD-L1 ≥ 1%.
In terms of safety, 17.7% of patients who completed 2 years of pembrolizumab had grade 3-5 treatment-related adverse events, compared with 16.6% among all pembrolizumab-treated patients and 36.6% of those given docetaxel.
The team reports that 79 patients completed 35 cycles of pembrolizumab, with a median follow-up of 43.4 months.
Compared with the overall patient group, these patients were less likely to be aged ≥ 65 years and to have received two or more prior treatment lines, although they were more likely to be current or former smokers and to have squamous tumor histology.
Patients who completed 35 cycles had an objective response rate of 94.9%, and 91.0% were still alive at the data cutoff. Overall survival rates were 98.7% at 12 months and 86.3% at 24 months.
Of 71 patients eligible for analysis, 23 experienced progression after completing pembrolizumab, at PFS rates at 12 and 24 months of 72.5% and 57.7%, respectively.
A total of 14 patients were given a second course of pembrolizumab, of whom six had a partial response and five had stable disease. At the data cutoff, five patients had completed 17 additional cycles and 11 were alive.
Pembro Approved at Fixed Dose
One notable aspect of the study is that patients in the pembrolizumab arm were given two different doses of the drug based on body weight, whereas the drug is approved in the United States at a fixed dose of 200 mg.
Herbst told Medscape Medical News he considers the 200-mg dose to be appropriate.
“I didn’t think that the 3-mg versus 10-mg dose per kg that we used in our study made much difference in an average-sized person,” he said, adding that the 200-mg dose “is something a little bit more than 3 mg/kg.”
“So I think that this is clearly the right dos, and I don’t think more would make any difference,” he said.
The study was funded by Merck, the manufacturer of pembrolizumab. Herbst has reported having a consulting or advisory role for many pharmaceutical companies. Other coauthors have also reported relationships with industry, and some of the authors are Merck employees. Peters has reported receiving education grants, providing consultation, attending advisory boards, and/or providing lectures for many pharmaceutical companies.
This article first appeared on Medscape.com.
More than a third (35%) of patients with relapsed non–small cell lung cancer (NSCLC) treated with pembrolizumab (Keytruda, Merck) were still alive at 3 years, according to long-term results from a pivotal clinical trial.
The results also showed that, among the 10% of patients who completed all 35 cycles of pembrolizumab, the 3-year overall survival was approximately 99%, with progression-free survival (PFS) at around 70%.
“It is too soon to say that pembrolizumab is a potential cure...and we know that it doesn’t work for all patients, but the agent remains very, very promising,” said lead investigator Roy Herbst, MD, PhD, Department of Medical Oncology, Yale Comprehensive Cancer Center, New Haven, Connecticut.
These new results come from the KEYNOTE-010 trial, conducted in more than 1000 patients with NSCLC who had progressed on chemotherapy, randomized to receive immunotherapy with pembrolizumab or chemotherapy with docetaxel.
The results were published online on February 20 in the Journal of Clinical Oncology and were previously presented at the 2018 annual meeting of the European Society of Medical Oncology.
Overall survival at 3 years was 35% in patients with PD-L1 expression ≥ 50% in the tumor, and 23% in those with PD-L1 ≥ 1%.
This compares with 3-year overall survival of 11-13% with docetaxel.
These results are “really extraordinary,” Herbst commented to Medscape Medical News.
The 3-year overall survival rate of 35% in patients with PD-L1 ≥ 50% “is huge,” he said. “It really shows the durability of the response.”
Herbst commented that the “almost 100%” survival at 3 years among patients who completed 35 cycles of pembrolizumab shows that this treatment period (of about 2 years) is “probably about the right time to treat.”
“Currently, the agent is being used in all potential settings, before any other treatment, after other treatment, and with other treatments,” he said.
“Our hope is to find the very best way to use pembrolizumab to treat individual lung cancer patients, assessing how much PD-L1 a tumor expresses, what stage the patient is in, as well as other variables and biomarkers we are working on. This is the story of tailored therapy,” Herbst said.
Approached for comment, Solange Peters, MD, PhD, Oncology Department, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland, said that the results are “very good” and “confirm the paradigms we have been seeing in melanoma,” with good long-term control, which is “very reassuring.”
However, she told Medscape Medical News that the trial raises an important question: «How long do you need to expose your patient with lung cancer to immunotherapy in order to get this long-term control?»
She said the “good news” is that, for the 10% of patients who completed 2 years of treatment per protocol, almost all of them are still alive at 3 years, “which is not observed with chemotherapy.”
The question for Peters is “more about the definition of long-term control,” as it was seen that almost one in three patients nevertheless had some form of progression.
This suggests that you have a group of people “who are nicely controlled, you stop the drug, and 1 year later a third of them have progressed.”
Peters said: “So how long do you need to treat these patients? I would say I still don’t know.”
“If I were one of these patients probably I would still want to continue [on the drug]. Of course, some might have progressed even while remaining on the drug, but the proportion who would have progressed is probably smaller than this one.”
Responses on Re-introduction of Therapy
The study also allowed patients who had completed 35 cycles of pembrolizumab to be restarted on the drug if they experienced progression.
The team found that, among 14 patients, 43% had a partial response and 36% had stable disease.
Herbst highlighted this finding and told Medscape Medical News that this «could be very important to physicians because they might want to think about using the drug again» in patients who have progressed on it.
He believes that the progression was not because of any resistance per se but rather a slowing down of the adaptive immune response.
“It’s just that it needs a boost,” he said, while noting that tissue specimens will nevertheless be required to demonstrate the theory.
Peters agreed that these results are “very promising,” but questioned their overall significance, as it is “a very small number of patients” from a subset whose disease was controlled while on treatment and then progressed after stopping.
She also pointed out that, in another study in patients with lung cancer (CheckMate-153), some patients were rechallenged with immunotherapy after having stopped treatment at 1 year “with very poor results.”
Peters said studies in melanoma have shown “rechallenge can be useful in a significant proportion of patients, but still you have not demonstrated that stopping and rechallenging is the same as not stopping.”
Study Details
KEYNOTE-010 involved patients with NSCLC from 202 centers in 24 countries with stage IIIB/IV disease expressing PD-L1 who had experienced disease progression after at least two cycles of platinum-based chemotherapy.
They were randomized 1:1:1 to open-label pembrolizumab 2 mg/kg, pembrolizumab 10 mg/kg, or docetaxel 75 mg/m2 every 3 weeks.
Pembrolizumab was continued for 35 treatment cycles over 2 years and docetaxel was continued for the maximum duration allowed by local regulators.
Patients who stopped pembrolizumab after a complete response or completing all 35 cycles, and who subsequently experienced disease progression, could receive up to 17 additional cycles over 1 year if they had not received another anticancer therapy in the meantime.
Among the 1,034 patients originally recruited between August 2013 and February 2015, 691 were assigned to pembrolizumab at 3 mg/kg or 10 mg/kg and 343 to docetaxel.
For the intention-to-treat analysis in 1033 patients, the mean duration of follow-up was 42.6 months, with a median treatment duration of 3.5 months in the pembrolizumab group and 2.0 months in the docetaxel group.
Compared with docetaxel, pembrolizumab was associated with a significant reduction in the risk of death, at a hazard ratio of 0.53 in patients with PD-L1 ≥ 50% and 0.69 in those with PD-L1 ≥ 1% (both P < .0001).
In patients with PD-L1 ≥ 50%, median overall survival was 16.9 months in those given pembrolizumab and 8.2 months with docetaxel. Among those with PD-L1 ≥ 1%, median overall survival was 11.8 months with pembrolizumab versus 8.4 months with docetaxel.
Overall survival on Kaplan-Meier analysis was 34.5% with pembrolizumab and 12.7% with docetaxel in the PD-L1 ≥ 50% group, and 22.9% versus 11.0% in the PD-L1 ≥ 1% group.
PFS significantly improved with pembrolizumab versus docetaxel, at a hazard ratio of 0.57 (P < .00001) among patients with PD-L1 ≥ 50% and 0.83 (P < .005) in those with PD-L1 ≥ 1%.
In terms of safety, 17.7% of patients who completed 2 years of pembrolizumab had grade 3-5 treatment-related adverse events, compared with 16.6% among all pembrolizumab-treated patients and 36.6% of those given docetaxel.
The team reports that 79 patients completed 35 cycles of pembrolizumab, with a median follow-up of 43.4 months.
Compared with the overall patient group, these patients were less likely to be aged ≥ 65 years and to have received two or more prior treatment lines, although they were more likely to be current or former smokers and to have squamous tumor histology.
Patients who completed 35 cycles had an objective response rate of 94.9%, and 91.0% were still alive at the data cutoff. Overall survival rates were 98.7% at 12 months and 86.3% at 24 months.
Of 71 patients eligible for analysis, 23 experienced progression after completing pembrolizumab, at PFS rates at 12 and 24 months of 72.5% and 57.7%, respectively.
A total of 14 patients were given a second course of pembrolizumab, of whom six had a partial response and five had stable disease. At the data cutoff, five patients had completed 17 additional cycles and 11 were alive.
Pembro Approved at Fixed Dose
One notable aspect of the study is that patients in the pembrolizumab arm were given two different doses of the drug based on body weight, whereas the drug is approved in the United States at a fixed dose of 200 mg.
Herbst told Medscape Medical News he considers the 200-mg dose to be appropriate.
“I didn’t think that the 3-mg versus 10-mg dose per kg that we used in our study made much difference in an average-sized person,” he said, adding that the 200-mg dose “is something a little bit more than 3 mg/kg.”
“So I think that this is clearly the right dos, and I don’t think more would make any difference,” he said.
The study was funded by Merck, the manufacturer of pembrolizumab. Herbst has reported having a consulting or advisory role for many pharmaceutical companies. Other coauthors have also reported relationships with industry, and some of the authors are Merck employees. Peters has reported receiving education grants, providing consultation, attending advisory boards, and/or providing lectures for many pharmaceutical companies.
This article first appeared on Medscape.com.