User login
Can a repurposed Parkinson’s drug slow ALS progression?
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.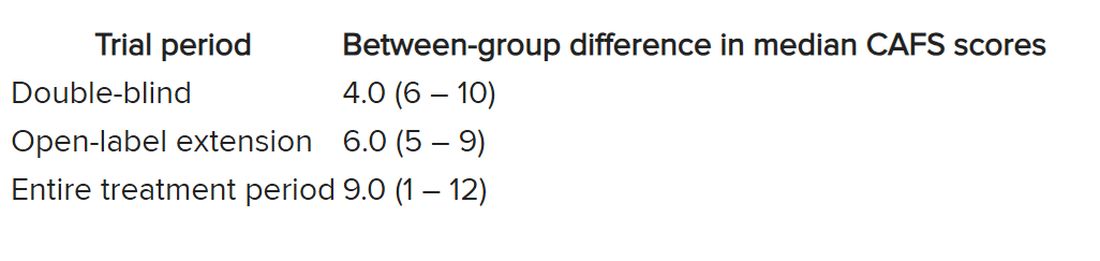
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM CELL STEM CELL
Starting indicated heart failure meds in-hospital: Progress, opportunities
Most patients aren’t receiving all the medications they should based on guidelines, nor are they getting them at the most effective time in their disease course, suggests a registry study of patients in the United States hospitalized with heart failure with reduced ejection fraction (HFrEF).
On average, one such medication was initiated per patient for every 6 days in the hospital.
Shortfalls in predischarge GDMT initiation disproportionately landed on women, patients at rural centers, and those with renal failure or other comorbidities. But they didn’t seem related to patient race or ethnicity in the study reported in JACC: Heart Failure.
The analysis covers the 3 years preceding the May 2020 first-time approval of a sodium-glucose cotransporter 2 (SGLT2) inhibitor for nondiabetic patients with HFrEF, and therefore doesn’t cover such drugs for that indication. The SGLT2 inhibitors would later join beta-blockers, renin-angiotensin system (RAS) inhibitors, and mineralocorticoid receptor antagonists (MRAs) in the quartet of core GDMT medications broadly indicated for HFrEF.
In-hospital initiation of GDMT for HFrEF is considered a predictor of being on those medications after discharge and is itself guideline recommended. There’s clear evidence that treatment with the four core medications boosts survival and cuts rehospitalization risk, and that “getting those on board as soon as possible will eventually benefit many patients,” Paul L. Hess, MD, MHS, said in an interview.
Dr. Hess, University of Colorado Anschutz Medical Campus, Aurora, is senior author on the report from the Get With The Guidelines–Heart Failure (GWTG-HF) quality improvement program of the American Heart Association.
Broad uptake of new medical therapies into practice may sometimes take 15 or more years from first publication, Dr. Hess said, so, “I find it encouraging in the study that over a shorter time period, 2017-2020, there was improvement.”
Indeed, the odds of in-hospital initiation of an indicated med during that period on average climbed a significant 8% every 3 months, the report states.
The finding suggests that “heart failure hospitalization is, in and of itself, an important intervention for getting folks on the appropriate medications,” Dr. Hess said. It also means “we’re getting better at it,” at least at the study’s 160 GWTG-HF participating hospitals nationwide.
Those centers, the report acknowledges, varied in size, geography, and teaching status but were not necessarily representative of all U.S. hospitals. In another potential limitation, the study couldn’t account for patients who weren’t prescribed all indicated medications for clinically valid reasons. It excluded patients with “clear contraindications,” Dr. Hess said. But there could have been “legitimate reasons” some indicated medications weren’t always prescribed, including patient frailty, hemodynamic intolerance, renal dysfunction, or polypharmacy concerns.
“Positive takeaways” from the analysis, notes an accompanying editorial, include improved prescription rates for key GDMT categories across more than 3 years of data, and evidence that in-hospital initiation “was feasible and, at least for some medications, reliably undertaken.”
Of note, new GDMT prescriptions from admission to discharge went from 70% to almost 98% for beta-blockers, from 59% to about 91% for RAS inhibitors, from about 26% to 56% for MRAs, and from 15.5% to 27.4% for hydralazine/nitrates, wrote Karen E. Joynt Maddox, MD, MPH, and Daniel K. Fox, MD, PhD, both of Washington University in St. Louis.
“Key areas for improvement,” they noted, include prescriptions for women, who were 12% less likely than men to have appropriate GDMT initiated during hospitalization (P < .001); and practice at rural hospitals, which were 40% less likely than urban centers to have patients on full GDMT by discharge (P = .017).
Although only 2.6% of the GWTG-HF centers were in rural locations, “rural hospitals make up approximately one-third of general acute care hospitals in this country,” the editorial states. They therefore “represent a key source of health disparity” in the United States in need of further study.
The analysis of 50,170 patients hospitalized with HFrEF compared the number of GDMT medications for which they were eligible, on at-hospital admission, and by discharge.
The drug categories included “evidence based beta blockers,” that is, bisoprolol, carvedilol, or sustained-release metoprolol; RAS inhibitors, specifically ACE inhibitors, angiotensin receptor blockers, or sacubitril/valsartan (Entresto); MRAs; SGLT2 inhibitors in patients with diabetes; diuretics for congestion; oral anticoagulants for atrial fibrillation; and hydralazine/nitrates in African Americans.
About 15% of the patients at hospital admission were on all indicated HFrEF medications for which they were eligible. The proportion more than doubled to 32.8% by discharge.
Factors significantly associated with reduced odds for in-hospital GDMT initiation include older age (odds ratio, 0.94 per 5-year increment), being female versus male (OR, 0.88), rural location (OR, 0.60), Medicaid versus Medicare or private insurance (OR, 0.93), stroke history (OR, 0.91), peripheral artery disease (OR, 0.93), chronic obstructive pulmonary disease or asthma (OR, 0.86), and renal insufficiency (OR, 0.77).
The findings suggest that there has been at least some progress in getting hospitalized patients “on the right meds” by discharge, Dr. Hess observed. To help address shortfalls in some patient groups, “there is interest in engaging pharmacists in helping us encourage providers on the front lines to initiate and titrate medications.”
The GWTG-HF program is sponsored, in part, by Novartis, Boehringer Ingelheim, Novo Nordisk, AstraZeneca, Bayer, Tylenol, and Alnylam Pharmaceuticals. Dr. Hess disclosed no relevant financial relationships. Dr. Maddox disclosed serving on the Health Policy Advisory Council for Centene. Dr. Fox reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Most patients aren’t receiving all the medications they should based on guidelines, nor are they getting them at the most effective time in their disease course, suggests a registry study of patients in the United States hospitalized with heart failure with reduced ejection fraction (HFrEF).
On average, one such medication was initiated per patient for every 6 days in the hospital.
Shortfalls in predischarge GDMT initiation disproportionately landed on women, patients at rural centers, and those with renal failure or other comorbidities. But they didn’t seem related to patient race or ethnicity in the study reported in JACC: Heart Failure.
The analysis covers the 3 years preceding the May 2020 first-time approval of a sodium-glucose cotransporter 2 (SGLT2) inhibitor for nondiabetic patients with HFrEF, and therefore doesn’t cover such drugs for that indication. The SGLT2 inhibitors would later join beta-blockers, renin-angiotensin system (RAS) inhibitors, and mineralocorticoid receptor antagonists (MRAs) in the quartet of core GDMT medications broadly indicated for HFrEF.
In-hospital initiation of GDMT for HFrEF is considered a predictor of being on those medications after discharge and is itself guideline recommended. There’s clear evidence that treatment with the four core medications boosts survival and cuts rehospitalization risk, and that “getting those on board as soon as possible will eventually benefit many patients,” Paul L. Hess, MD, MHS, said in an interview.
Dr. Hess, University of Colorado Anschutz Medical Campus, Aurora, is senior author on the report from the Get With The Guidelines–Heart Failure (GWTG-HF) quality improvement program of the American Heart Association.
Broad uptake of new medical therapies into practice may sometimes take 15 or more years from first publication, Dr. Hess said, so, “I find it encouraging in the study that over a shorter time period, 2017-2020, there was improvement.”
Indeed, the odds of in-hospital initiation of an indicated med during that period on average climbed a significant 8% every 3 months, the report states.
The finding suggests that “heart failure hospitalization is, in and of itself, an important intervention for getting folks on the appropriate medications,” Dr. Hess said. It also means “we’re getting better at it,” at least at the study’s 160 GWTG-HF participating hospitals nationwide.
Those centers, the report acknowledges, varied in size, geography, and teaching status but were not necessarily representative of all U.S. hospitals. In another potential limitation, the study couldn’t account for patients who weren’t prescribed all indicated medications for clinically valid reasons. It excluded patients with “clear contraindications,” Dr. Hess said. But there could have been “legitimate reasons” some indicated medications weren’t always prescribed, including patient frailty, hemodynamic intolerance, renal dysfunction, or polypharmacy concerns.
“Positive takeaways” from the analysis, notes an accompanying editorial, include improved prescription rates for key GDMT categories across more than 3 years of data, and evidence that in-hospital initiation “was feasible and, at least for some medications, reliably undertaken.”
Of note, new GDMT prescriptions from admission to discharge went from 70% to almost 98% for beta-blockers, from 59% to about 91% for RAS inhibitors, from about 26% to 56% for MRAs, and from 15.5% to 27.4% for hydralazine/nitrates, wrote Karen E. Joynt Maddox, MD, MPH, and Daniel K. Fox, MD, PhD, both of Washington University in St. Louis.
“Key areas for improvement,” they noted, include prescriptions for women, who were 12% less likely than men to have appropriate GDMT initiated during hospitalization (P < .001); and practice at rural hospitals, which were 40% less likely than urban centers to have patients on full GDMT by discharge (P = .017).
Although only 2.6% of the GWTG-HF centers were in rural locations, “rural hospitals make up approximately one-third of general acute care hospitals in this country,” the editorial states. They therefore “represent a key source of health disparity” in the United States in need of further study.
The analysis of 50,170 patients hospitalized with HFrEF compared the number of GDMT medications for which they were eligible, on at-hospital admission, and by discharge.
The drug categories included “evidence based beta blockers,” that is, bisoprolol, carvedilol, or sustained-release metoprolol; RAS inhibitors, specifically ACE inhibitors, angiotensin receptor blockers, or sacubitril/valsartan (Entresto); MRAs; SGLT2 inhibitors in patients with diabetes; diuretics for congestion; oral anticoagulants for atrial fibrillation; and hydralazine/nitrates in African Americans.
About 15% of the patients at hospital admission were on all indicated HFrEF medications for which they were eligible. The proportion more than doubled to 32.8% by discharge.
Factors significantly associated with reduced odds for in-hospital GDMT initiation include older age (odds ratio, 0.94 per 5-year increment), being female versus male (OR, 0.88), rural location (OR, 0.60), Medicaid versus Medicare or private insurance (OR, 0.93), stroke history (OR, 0.91), peripheral artery disease (OR, 0.93), chronic obstructive pulmonary disease or asthma (OR, 0.86), and renal insufficiency (OR, 0.77).
The findings suggest that there has been at least some progress in getting hospitalized patients “on the right meds” by discharge, Dr. Hess observed. To help address shortfalls in some patient groups, “there is interest in engaging pharmacists in helping us encourage providers on the front lines to initiate and titrate medications.”
The GWTG-HF program is sponsored, in part, by Novartis, Boehringer Ingelheim, Novo Nordisk, AstraZeneca, Bayer, Tylenol, and Alnylam Pharmaceuticals. Dr. Hess disclosed no relevant financial relationships. Dr. Maddox disclosed serving on the Health Policy Advisory Council for Centene. Dr. Fox reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Most patients aren’t receiving all the medications they should based on guidelines, nor are they getting them at the most effective time in their disease course, suggests a registry study of patients in the United States hospitalized with heart failure with reduced ejection fraction (HFrEF).
On average, one such medication was initiated per patient for every 6 days in the hospital.
Shortfalls in predischarge GDMT initiation disproportionately landed on women, patients at rural centers, and those with renal failure or other comorbidities. But they didn’t seem related to patient race or ethnicity in the study reported in JACC: Heart Failure.
The analysis covers the 3 years preceding the May 2020 first-time approval of a sodium-glucose cotransporter 2 (SGLT2) inhibitor for nondiabetic patients with HFrEF, and therefore doesn’t cover such drugs for that indication. The SGLT2 inhibitors would later join beta-blockers, renin-angiotensin system (RAS) inhibitors, and mineralocorticoid receptor antagonists (MRAs) in the quartet of core GDMT medications broadly indicated for HFrEF.
In-hospital initiation of GDMT for HFrEF is considered a predictor of being on those medications after discharge and is itself guideline recommended. There’s clear evidence that treatment with the four core medications boosts survival and cuts rehospitalization risk, and that “getting those on board as soon as possible will eventually benefit many patients,” Paul L. Hess, MD, MHS, said in an interview.
Dr. Hess, University of Colorado Anschutz Medical Campus, Aurora, is senior author on the report from the Get With The Guidelines–Heart Failure (GWTG-HF) quality improvement program of the American Heart Association.
Broad uptake of new medical therapies into practice may sometimes take 15 or more years from first publication, Dr. Hess said, so, “I find it encouraging in the study that over a shorter time period, 2017-2020, there was improvement.”
Indeed, the odds of in-hospital initiation of an indicated med during that period on average climbed a significant 8% every 3 months, the report states.
The finding suggests that “heart failure hospitalization is, in and of itself, an important intervention for getting folks on the appropriate medications,” Dr. Hess said. It also means “we’re getting better at it,” at least at the study’s 160 GWTG-HF participating hospitals nationwide.
Those centers, the report acknowledges, varied in size, geography, and teaching status but were not necessarily representative of all U.S. hospitals. In another potential limitation, the study couldn’t account for patients who weren’t prescribed all indicated medications for clinically valid reasons. It excluded patients with “clear contraindications,” Dr. Hess said. But there could have been “legitimate reasons” some indicated medications weren’t always prescribed, including patient frailty, hemodynamic intolerance, renal dysfunction, or polypharmacy concerns.
“Positive takeaways” from the analysis, notes an accompanying editorial, include improved prescription rates for key GDMT categories across more than 3 years of data, and evidence that in-hospital initiation “was feasible and, at least for some medications, reliably undertaken.”
Of note, new GDMT prescriptions from admission to discharge went from 70% to almost 98% for beta-blockers, from 59% to about 91% for RAS inhibitors, from about 26% to 56% for MRAs, and from 15.5% to 27.4% for hydralazine/nitrates, wrote Karen E. Joynt Maddox, MD, MPH, and Daniel K. Fox, MD, PhD, both of Washington University in St. Louis.
“Key areas for improvement,” they noted, include prescriptions for women, who were 12% less likely than men to have appropriate GDMT initiated during hospitalization (P < .001); and practice at rural hospitals, which were 40% less likely than urban centers to have patients on full GDMT by discharge (P = .017).
Although only 2.6% of the GWTG-HF centers were in rural locations, “rural hospitals make up approximately one-third of general acute care hospitals in this country,” the editorial states. They therefore “represent a key source of health disparity” in the United States in need of further study.
The analysis of 50,170 patients hospitalized with HFrEF compared the number of GDMT medications for which they were eligible, on at-hospital admission, and by discharge.
The drug categories included “evidence based beta blockers,” that is, bisoprolol, carvedilol, or sustained-release metoprolol; RAS inhibitors, specifically ACE inhibitors, angiotensin receptor blockers, or sacubitril/valsartan (Entresto); MRAs; SGLT2 inhibitors in patients with diabetes; diuretics for congestion; oral anticoagulants for atrial fibrillation; and hydralazine/nitrates in African Americans.
About 15% of the patients at hospital admission were on all indicated HFrEF medications for which they were eligible. The proportion more than doubled to 32.8% by discharge.
Factors significantly associated with reduced odds for in-hospital GDMT initiation include older age (odds ratio, 0.94 per 5-year increment), being female versus male (OR, 0.88), rural location (OR, 0.60), Medicaid versus Medicare or private insurance (OR, 0.93), stroke history (OR, 0.91), peripheral artery disease (OR, 0.93), chronic obstructive pulmonary disease or asthma (OR, 0.86), and renal insufficiency (OR, 0.77).
The findings suggest that there has been at least some progress in getting hospitalized patients “on the right meds” by discharge, Dr. Hess observed. To help address shortfalls in some patient groups, “there is interest in engaging pharmacists in helping us encourage providers on the front lines to initiate and titrate medications.”
The GWTG-HF program is sponsored, in part, by Novartis, Boehringer Ingelheim, Novo Nordisk, AstraZeneca, Bayer, Tylenol, and Alnylam Pharmaceuticals. Dr. Hess disclosed no relevant financial relationships. Dr. Maddox disclosed serving on the Health Policy Advisory Council for Centene. Dr. Fox reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JACC: HEART FAILURE
SSRI improves cognition, major depression in early dementia
TOPLINE:
METHODOLOGY:
- The multicenter MEMORY study included 82 subjects with MDD and early-stage dementia, mean age 70.3 years, mostly female (66%) and White (95%).
- Vortioxetine, a modulator of 5-hydroxytryptamine receptor activity and an inhibitor of the 5-HT transporter, initiated at 5 mg/day (recommended starting dose in older adults) with the dose up-titrated to 10 mg/day after a week and flexible dosing thereafter.
- Depression was assessed using the Montgomery-Åsberg Depression Rating Scale (MADRS), and cognition with the Digit Symbol Substitution Test (DSST) and Rey Auditory Verbal Learning Test.
TAKEAWAY:
- There was significant and clinically meaningful improvement in the severity of depressive symptoms, as measured by MADRS total score (the primary outcome), at all assessment time points (P < .0001).
- Improvements in depressive symptoms were irrespective of dementia type.
- There were also significant improvements in DSST total score (P < .0001) and in daily functioning and health-related quality of life (HRQoL).
- Vortioxetine was well tolerated; side effects, including nausea and abdominal pain, were mostly mild to moderate.
IN PRACTICE:
“Vortioxetine demonstrated effectiveness in clinically significantly improving depressive symptoms, cognitive performance, daily and global functioning, and HRQoL in patients with MDD and comorbid early-stage dementia treated for 12 weeks” the researchers noted.
STUDY DETAILS:
The study was conducted by Michael Cronquist Christensen from pharmaceutical company H. Lundbeck, Valby, Denmark, and colleagues. It was published online in the Journal of Affective Disorders.
LIMITATIONS:
The study is open label and lacked a control group. Learning effects were possible, which could contribute to improved cognitive performance, although significant improvement on the RAVLT was not observed until week 4, suggesting earning effects were minimal.
DISCLOSURES:
The study was funded by H. Lundbeck. Mr. Christensen is an employee of H. Lundbeck.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The multicenter MEMORY study included 82 subjects with MDD and early-stage dementia, mean age 70.3 years, mostly female (66%) and White (95%).
- Vortioxetine, a modulator of 5-hydroxytryptamine receptor activity and an inhibitor of the 5-HT transporter, initiated at 5 mg/day (recommended starting dose in older adults) with the dose up-titrated to 10 mg/day after a week and flexible dosing thereafter.
- Depression was assessed using the Montgomery-Åsberg Depression Rating Scale (MADRS), and cognition with the Digit Symbol Substitution Test (DSST) and Rey Auditory Verbal Learning Test.
TAKEAWAY:
- There was significant and clinically meaningful improvement in the severity of depressive symptoms, as measured by MADRS total score (the primary outcome), at all assessment time points (P < .0001).
- Improvements in depressive symptoms were irrespective of dementia type.
- There were also significant improvements in DSST total score (P < .0001) and in daily functioning and health-related quality of life (HRQoL).
- Vortioxetine was well tolerated; side effects, including nausea and abdominal pain, were mostly mild to moderate.
IN PRACTICE:
“Vortioxetine demonstrated effectiveness in clinically significantly improving depressive symptoms, cognitive performance, daily and global functioning, and HRQoL in patients with MDD and comorbid early-stage dementia treated for 12 weeks” the researchers noted.
STUDY DETAILS:
The study was conducted by Michael Cronquist Christensen from pharmaceutical company H. Lundbeck, Valby, Denmark, and colleagues. It was published online in the Journal of Affective Disorders.
LIMITATIONS:
The study is open label and lacked a control group. Learning effects were possible, which could contribute to improved cognitive performance, although significant improvement on the RAVLT was not observed until week 4, suggesting earning effects were minimal.
DISCLOSURES:
The study was funded by H. Lundbeck. Mr. Christensen is an employee of H. Lundbeck.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The multicenter MEMORY study included 82 subjects with MDD and early-stage dementia, mean age 70.3 years, mostly female (66%) and White (95%).
- Vortioxetine, a modulator of 5-hydroxytryptamine receptor activity and an inhibitor of the 5-HT transporter, initiated at 5 mg/day (recommended starting dose in older adults) with the dose up-titrated to 10 mg/day after a week and flexible dosing thereafter.
- Depression was assessed using the Montgomery-Åsberg Depression Rating Scale (MADRS), and cognition with the Digit Symbol Substitution Test (DSST) and Rey Auditory Verbal Learning Test.
TAKEAWAY:
- There was significant and clinically meaningful improvement in the severity of depressive symptoms, as measured by MADRS total score (the primary outcome), at all assessment time points (P < .0001).
- Improvements in depressive symptoms were irrespective of dementia type.
- There were also significant improvements in DSST total score (P < .0001) and in daily functioning and health-related quality of life (HRQoL).
- Vortioxetine was well tolerated; side effects, including nausea and abdominal pain, were mostly mild to moderate.
IN PRACTICE:
“Vortioxetine demonstrated effectiveness in clinically significantly improving depressive symptoms, cognitive performance, daily and global functioning, and HRQoL in patients with MDD and comorbid early-stage dementia treated for 12 weeks” the researchers noted.
STUDY DETAILS:
The study was conducted by Michael Cronquist Christensen from pharmaceutical company H. Lundbeck, Valby, Denmark, and colleagues. It was published online in the Journal of Affective Disorders.
LIMITATIONS:
The study is open label and lacked a control group. Learning effects were possible, which could contribute to improved cognitive performance, although significant improvement on the RAVLT was not observed until week 4, suggesting earning effects were minimal.
DISCLOSURES:
The study was funded by H. Lundbeck. Mr. Christensen is an employee of H. Lundbeck.
A version of this article first appeared on Medscape.com.
FDA OKs empagliflozin for children with type 2 diabetes
aged 10 years and older.
This approval represents only the second oral treatment option for children and adolescents with type 2 diabetes after metformin; the latter appears to be less effective for pediatric patients than for adults.
Injectable glucagonlike peptide–1 (GLP-1) agonists are also available for youth with type 2 diabetes. These include daily liraglutide (Victoza) and once-weekly extended-release exenatide (Bydureon/Bydureon BCise).
Jardiance has been approved for adults with type 2 diabetes since 2014, and Synjardy has been approved since 2015.
“Compared to adults, children with type 2 diabetes have limited treatment options, even though the disease and symptom onset generally progress more rapidly in children,” said Michelle Carey, MD, MPH.
“Today’s approvals provide much-needed additional treatment options for children with type 2 diabetes,” added Dr. Carey, associate director for therapeutic review for the division of diabetes, lipid disorders, and obesity in the FDA’s Center for Drug Evaluation and Research.
Type 2 diabetes rising exponentially in children, mainly non-Whites
Type 2 diabetes is rising exponentially in children and adolescents in the United States.
Data from the SEARCH for Diabetes in Youth study show that the incidence of type 2 diabetes among youth rose by about 5% per year between 2002 and 2015, and it continues to rise.
A more recent study found that a doubling of cases occurred during the pandemic, with youth often presenting with more severe disease. The majority of cases are among non-White racial groups.
Safety and efficacy data for empagliflozin for children came from the Diabetes Study of Linagliptin and Empagliflozin in Children and Adolescents (DINAMO) trial. That trial included 157 patients aged 10-17 years with A1c of 7% or above. Patients were randomly assigned to receive empagliflozin 10 mg or 25 mg daily, linagliptin (a DPP-4 inhibitor) 5 mg, or placebo for 26 weeks. Over 90% were also taking metformin, 40% in combination with insulin. All patients were given diet and exercise advice.
At week 26, the children treated with empagliflozin showed an average 0.2 percentage point decrease in A1c, compared with a 0.7-point increase among those taking placebo. Use of empagliflozin was also associated with lower fasting plasma glucose levels compared with placebo.
Side effects were similar to those seen in adults except for a higher risk of hypoglycemia, regardless of other glucose-lowering therapies that were being taken.
Reduction in A1c for participants treated with linagliptin was not statistically significant in comparison with placebo. There was a numerical reduction of 0.34% (P = .2935).
“Across the lifespan, we know that people living with type 2 diabetes have a high risk for many diabetes complications, so it’s important to recognize and treat diabetes early in its course,” Lori Laffel, MD, lead investigator of the DINAMO study, said in a press release from BI.
“These findings are particularly important given the need for more therapeutic options, especially oral agents, to manage type 2 diabetes in young people as, to date, metformin [has been] the only globally available oral treatment for youth,” added Dr. Laffel, chief of the pediatric, adolescent, and young adult section at the Joslin Diabetes Center and professor of pediatrics at Harvard Medical School, Boston.
A version of this article first appeared on Medscape.com.
aged 10 years and older.
This approval represents only the second oral treatment option for children and adolescents with type 2 diabetes after metformin; the latter appears to be less effective for pediatric patients than for adults.
Injectable glucagonlike peptide–1 (GLP-1) agonists are also available for youth with type 2 diabetes. These include daily liraglutide (Victoza) and once-weekly extended-release exenatide (Bydureon/Bydureon BCise).
Jardiance has been approved for adults with type 2 diabetes since 2014, and Synjardy has been approved since 2015.
“Compared to adults, children with type 2 diabetes have limited treatment options, even though the disease and symptom onset generally progress more rapidly in children,” said Michelle Carey, MD, MPH.
“Today’s approvals provide much-needed additional treatment options for children with type 2 diabetes,” added Dr. Carey, associate director for therapeutic review for the division of diabetes, lipid disorders, and obesity in the FDA’s Center for Drug Evaluation and Research.
Type 2 diabetes rising exponentially in children, mainly non-Whites
Type 2 diabetes is rising exponentially in children and adolescents in the United States.
Data from the SEARCH for Diabetes in Youth study show that the incidence of type 2 diabetes among youth rose by about 5% per year between 2002 and 2015, and it continues to rise.
A more recent study found that a doubling of cases occurred during the pandemic, with youth often presenting with more severe disease. The majority of cases are among non-White racial groups.
Safety and efficacy data for empagliflozin for children came from the Diabetes Study of Linagliptin and Empagliflozin in Children and Adolescents (DINAMO) trial. That trial included 157 patients aged 10-17 years with A1c of 7% or above. Patients were randomly assigned to receive empagliflozin 10 mg or 25 mg daily, linagliptin (a DPP-4 inhibitor) 5 mg, or placebo for 26 weeks. Over 90% were also taking metformin, 40% in combination with insulin. All patients were given diet and exercise advice.
At week 26, the children treated with empagliflozin showed an average 0.2 percentage point decrease in A1c, compared with a 0.7-point increase among those taking placebo. Use of empagliflozin was also associated with lower fasting plasma glucose levels compared with placebo.
Side effects were similar to those seen in adults except for a higher risk of hypoglycemia, regardless of other glucose-lowering therapies that were being taken.
Reduction in A1c for participants treated with linagliptin was not statistically significant in comparison with placebo. There was a numerical reduction of 0.34% (P = .2935).
“Across the lifespan, we know that people living with type 2 diabetes have a high risk for many diabetes complications, so it’s important to recognize and treat diabetes early in its course,” Lori Laffel, MD, lead investigator of the DINAMO study, said in a press release from BI.
“These findings are particularly important given the need for more therapeutic options, especially oral agents, to manage type 2 diabetes in young people as, to date, metformin [has been] the only globally available oral treatment for youth,” added Dr. Laffel, chief of the pediatric, adolescent, and young adult section at the Joslin Diabetes Center and professor of pediatrics at Harvard Medical School, Boston.
A version of this article first appeared on Medscape.com.
aged 10 years and older.
This approval represents only the second oral treatment option for children and adolescents with type 2 diabetes after metformin; the latter appears to be less effective for pediatric patients than for adults.
Injectable glucagonlike peptide–1 (GLP-1) agonists are also available for youth with type 2 diabetes. These include daily liraglutide (Victoza) and once-weekly extended-release exenatide (Bydureon/Bydureon BCise).
Jardiance has been approved for adults with type 2 diabetes since 2014, and Synjardy has been approved since 2015.
“Compared to adults, children with type 2 diabetes have limited treatment options, even though the disease and symptom onset generally progress more rapidly in children,” said Michelle Carey, MD, MPH.
“Today’s approvals provide much-needed additional treatment options for children with type 2 diabetes,” added Dr. Carey, associate director for therapeutic review for the division of diabetes, lipid disorders, and obesity in the FDA’s Center for Drug Evaluation and Research.
Type 2 diabetes rising exponentially in children, mainly non-Whites
Type 2 diabetes is rising exponentially in children and adolescents in the United States.
Data from the SEARCH for Diabetes in Youth study show that the incidence of type 2 diabetes among youth rose by about 5% per year between 2002 and 2015, and it continues to rise.
A more recent study found that a doubling of cases occurred during the pandemic, with youth often presenting with more severe disease. The majority of cases are among non-White racial groups.
Safety and efficacy data for empagliflozin for children came from the Diabetes Study of Linagliptin and Empagliflozin in Children and Adolescents (DINAMO) trial. That trial included 157 patients aged 10-17 years with A1c of 7% or above. Patients were randomly assigned to receive empagliflozin 10 mg or 25 mg daily, linagliptin (a DPP-4 inhibitor) 5 mg, or placebo for 26 weeks. Over 90% were also taking metformin, 40% in combination with insulin. All patients were given diet and exercise advice.
At week 26, the children treated with empagliflozin showed an average 0.2 percentage point decrease in A1c, compared with a 0.7-point increase among those taking placebo. Use of empagliflozin was also associated with lower fasting plasma glucose levels compared with placebo.
Side effects were similar to those seen in adults except for a higher risk of hypoglycemia, regardless of other glucose-lowering therapies that were being taken.
Reduction in A1c for participants treated with linagliptin was not statistically significant in comparison with placebo. There was a numerical reduction of 0.34% (P = .2935).
“Across the lifespan, we know that people living with type 2 diabetes have a high risk for many diabetes complications, so it’s important to recognize and treat diabetes early in its course,” Lori Laffel, MD, lead investigator of the DINAMO study, said in a press release from BI.
“These findings are particularly important given the need for more therapeutic options, especially oral agents, to manage type 2 diabetes in young people as, to date, metformin [has been] the only globally available oral treatment for youth,” added Dr. Laffel, chief of the pediatric, adolescent, and young adult section at the Joslin Diabetes Center and professor of pediatrics at Harvard Medical School, Boston.
A version of this article first appeared on Medscape.com.
Gilteritinib maintenance reduces relapse in MRD+ AML
The research was presented at the European Hematology Association Hybrid Congress 2023.
For the study, AML patients with the most common form of mutation in the proto-oncogene fms-like tyrosine kinase 3 (FLT3), known as the internal tandem duplication (ITD), were randomized to 24 months of maintenance therapy with either the FLT3 inhibitor gilteritinib or placebo.
The trial did not meet its primary endpoint, as there was no significant difference in relapse-free survival (RFS) between those assigned to the active drug and those given placebo, and there was no difference in overall survival rates.
However, subgroup analysis revealed that FLT3/ITD AML patients who were MRD+ after transplant, which represented approximately half of the participants, experienced a significant 48% improvement in RFS with gilteritinib versus placebo, while no benefit was seen in MRD– patients.
While acknowledging that the trial did not meet its primary endpoint, presenter Mark J. Levis, MD, PhD, program leader, hematologic malignancies and bone marrow transplant program, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, said it was nevertheless “a successful study.”
“We learned how to use these drugs and in whom,” he continued, adding: “No, not everybody needs and should get a FLT3 inhibitor post-transplant, but we can use this [MRD] assay to identify who.”
Consequently, Dr. Levis believes that gilteritinib “should be a standard of care for those who are MRD positive,” although the decision to use it “should be balanced against the potential for toxicity,” compared with not adding an additional treatment after HCT.
He told a press conference that “we’re going to certainly make sure that patients who are MRD positive get [gilteritinib],” although the MRD negative patients “are going to be more questionable,” especially because the assay that they used in the study is not “perfect.”
Dr. Levis also suggested that the trial did not meet its endpoint because of regional differences in the clinical practice, such as in the number of treatment cycles prior to HCT, the time to transplant, and the previous use of a FLT3 inhibitor, all of which may have skewed the findings.
“Everybody in the world is convinced that they’re the best transplanter,” he said, and yet “they all do it differently, and the heterogeneity is astounding.”
He added: “If we’d restricted everybody [to a] pretransplant regimen, I suspect we would have had a different result than what we’re getting here, but this is releasing the drug into the world and saying: ‘Here, transplant however you want, however it’s practiced in the real world. Tell us how this works.’ ”
Approached for comment, Claudio Brunstein, MD, PhD, vice-chair of the department of hematology and oncology in the Cleveland Clinic Taussig Cancer Institute, said that while there was “some disappointment” with the results, he was “not surprised” that the trial did not meet its primary endpoint.
He said in an interview that the patient population was not of “high enough risk” to demonstrate an overall difference between gilteritinib and placebo, although he conceded that it is “hard to get to high-risk patients in a timely way” and so conduct a trial with them.
As to the notion that variations in clinical practice could have been responsible, Dr. Brunstein pointed out that it was a randomized trial, so the issue would have applied equally to both sides.
He nevertheless believes that it is “a very important study,” and “just the fact that it was done in the context of a number of drugs coming and being approved by the [U.S. Food and Drug Administration] in AML is quite remarkable.”
This is especially the case given that “many centers are already using [gilteritinib] as off-label maintenance therapy.”
Dr. Brunstein added that it is “good news” that the drug was effective in MRD+ patients, as it shows “you can overcome that with maintenance therapy rather than keeping giving more and more chemotherapy, especially as there are patients you’re worried about giving more intensive chemotherapy to make them MRD negative.”
He pointed out, however, that the assay used in the trial was “research grade” and very sensitive to MRD and “is not available everywhere, so there is an adjustment that the community will have to do to in order to apply this data.”
“But for those who are more obviously MRD positive with less sensitive assays, gilteritinib is already something that can be used,” Dr. Brunstein said.
Presenting the findings, Dr. Levis stated: “We all know that patients with FLT3/ITD AML have a high risk of relapse and are routinely referred for transplant. And we know that the detection of measurable residual disease pretransplant is highly predictive of outcome post-transplant.”
He continued that FLT3 inhibitors are “routinely given as post-transplant maintenance ... based on some prior trials, mostly with sorafenib.”
“But uncertainty exists as to the broad applicability of these trials,” Dr. Levis said. Moreover, the use of sorafenib in this context is “off label and can be difficult to tolerate,” and “we know that most patients are cured with allogeneic transplant alone.”
Gilteritinib is already known to be well tolerated as a monotherapy, and was approved by the FDA for the treatment of adult patients with FLT3 mutation–positive relapsed or refractory AML in 2018.
The investigators therefore examined whether it would be beneficial as a post-HCT maintenance therapy in FLT3-ITD AML. Patients were required to be in morphologic remission after one or two courses of induction therapy, with Dr. Levis underlining: “We did not allow patients who had been salvaged onto the study.”
They subsequently had a marrow aspirate sample taken for MRD analysis before undergoing allogeneic transplant, with any conditioning regimen, donor, or graft-versus-host disease (GVHD) prophylaxis allowed.
Between 30 and 90 days later, patients with successful engraftment who were able to take oral medication were then randomized to 24 months of maintenance therapy with either gilteritinib or placebo.
Dr. Levis showed that, among 620 patients screened at 110 centers in 16 countries, 356 were randomized between Aug. 15, 2017, and July 8, 2020. The median age was 53 years, and 49% of gilteritinib patients and 48% of those given placebo were female.
He noted that there was a “fairly even global distribution” of patients from North America, Europe, and the Asia/Pacific region, and that 60% of patients underwent a myeloablative conditioning regimen. Approximately the same proportion had received an FLT3 inhibitor prior to HCT.
MRD positivity, assessed at a cell count of ≥ 10-6, was observed pre-HCT in 47% of patients in both treatment groups, and in 50% of gilteritinib patients and 51% of placebo patients at both pre- and post-transplant assessments.
The treatment regimen was completed by 52.8% of patients assigned to gilteritinib and 53.9% in the placebo arm. Dr. Levis said that 18.5% and 20.3% of patients, respectively, experienced a grade 3/4 treatment emergent acute GVHD event, while 32.6% and 21.5%, respectively, had a grade ≥ 3 treatment emergent infection.
He noted that “adverse events were clearly more common in the gilteritinib arm and often led to either dose reduction or interruption, or withdrawal of treatment.”
The most common grade ≥ 3 treatment emergent adverse event was a decrease in neutrophil count, seen in 24.7% of gilteritinib patients and 7.9% of those given placebo, followed by reduced platelet count, in 15.2% and 5.6%, respectively, and anemia, in 6.2% and 1.7%, respectively.
Turning to the efficacy outcomes, Dr. Levis reported that the trial did not meet its primary endpoint, with no significant difference in RFS between the gilteritinib and placebo arms, at a hazard ratio of 0.679 (P = .0518). There was also no significant difference in the key secondary objective of overall survival, at a hazard ratio of 0.846 (P = .4394).
However, Dr. Levis noted that there was a “clear difference in the benefit of gilteritinib by region,” and, “at every level,” MRD predicted a benefit from gilteritinib, which he said was a “big surprise” and “really leapt out in the subgroup analysis.”
He explained that the researchers used a modified version of a two-step assay that has been used in previous studies, and was able to detect MRD at a sensitivity of approximately 1x10-6. “In our study, 98% of participants had samples pre- and post-[transplant].”
Regardless of treatment arm, MRD positivity measured at that sensitivity was associated with a significant reduction in overall survival, at a hazard ratio versus MRD– status of 0.514 (P = .0025).
When stratifying the patients by MRD status, the researchers found that, among MRD+ participants, gilteritinib was associated with a significant improvement in RFS, at a hazard ratio versus placebo of 0.515 (P = .0065), while there was no significant difference in MRD– patients.
Stratifying the patients by their conditioning regimen prior to HCT also revealed differences, with those undergoing myeloablative conditioning having significantly greater overall survival than those who underwent reduced-intensity conditioning, at a hazard ratio for death of 0.529 (P = .0027).
Dr. Levis said there is “no surprise there,” and the result could reflect the selection of fitter, younger patients to undergo the more intensive regimen.
He then showed that MRD+ patients who had undergone myeloablative conditioning had better overall survival with gilteritinib than placebo, at a hazard ratio for death of 0.418 (P = .0087). Again, the difference disappeared when looking at MRD– patients.
“So conditioning doesn’t help you in the setting of MRD,” Dr. Levis said.
Finally, he took a deeper dive into the regional differences in outcomes, noting that patients in the Asia/Pacific region, where gilteritinib showed no benefit over placebo, “were 10 years younger” than those in other regions, “tended to get myeloablative conditioning, and hardly ever used FLT3 inhibitors.”
In contrast, North American patients, who experienced a significant gilteritinib benefit in terms of RFS, underwent HCT an average of 26 days earlier than those elsewhere, and received fewer courses of chemotherapy pre-HCT. Moreover, 93.5% received an FLT3 inhibitor pretransplant.
The study was funded by Astellas Pharma Global Development. Dr. Levis declares relationships with Abbvie, Amgen, Astellas, Bristol-Myers-Squibb, Daiichi-Sankyo, GlaxoSmithKline, Jazz, Menarini, Pfizer, Sumitomo-Dainippon, Syndax, Takeda. Dr. Brunstein declares no relevant relationships.
The research was presented at the European Hematology Association Hybrid Congress 2023.
For the study, AML patients with the most common form of mutation in the proto-oncogene fms-like tyrosine kinase 3 (FLT3), known as the internal tandem duplication (ITD), were randomized to 24 months of maintenance therapy with either the FLT3 inhibitor gilteritinib or placebo.
The trial did not meet its primary endpoint, as there was no significant difference in relapse-free survival (RFS) between those assigned to the active drug and those given placebo, and there was no difference in overall survival rates.
However, subgroup analysis revealed that FLT3/ITD AML patients who were MRD+ after transplant, which represented approximately half of the participants, experienced a significant 48% improvement in RFS with gilteritinib versus placebo, while no benefit was seen in MRD– patients.
While acknowledging that the trial did not meet its primary endpoint, presenter Mark J. Levis, MD, PhD, program leader, hematologic malignancies and bone marrow transplant program, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, said it was nevertheless “a successful study.”
“We learned how to use these drugs and in whom,” he continued, adding: “No, not everybody needs and should get a FLT3 inhibitor post-transplant, but we can use this [MRD] assay to identify who.”
Consequently, Dr. Levis believes that gilteritinib “should be a standard of care for those who are MRD positive,” although the decision to use it “should be balanced against the potential for toxicity,” compared with not adding an additional treatment after HCT.
He told a press conference that “we’re going to certainly make sure that patients who are MRD positive get [gilteritinib],” although the MRD negative patients “are going to be more questionable,” especially because the assay that they used in the study is not “perfect.”
Dr. Levis also suggested that the trial did not meet its endpoint because of regional differences in the clinical practice, such as in the number of treatment cycles prior to HCT, the time to transplant, and the previous use of a FLT3 inhibitor, all of which may have skewed the findings.
“Everybody in the world is convinced that they’re the best transplanter,” he said, and yet “they all do it differently, and the heterogeneity is astounding.”
He added: “If we’d restricted everybody [to a] pretransplant regimen, I suspect we would have had a different result than what we’re getting here, but this is releasing the drug into the world and saying: ‘Here, transplant however you want, however it’s practiced in the real world. Tell us how this works.’ ”
Approached for comment, Claudio Brunstein, MD, PhD, vice-chair of the department of hematology and oncology in the Cleveland Clinic Taussig Cancer Institute, said that while there was “some disappointment” with the results, he was “not surprised” that the trial did not meet its primary endpoint.
He said in an interview that the patient population was not of “high enough risk” to demonstrate an overall difference between gilteritinib and placebo, although he conceded that it is “hard to get to high-risk patients in a timely way” and so conduct a trial with them.
As to the notion that variations in clinical practice could have been responsible, Dr. Brunstein pointed out that it was a randomized trial, so the issue would have applied equally to both sides.
He nevertheless believes that it is “a very important study,” and “just the fact that it was done in the context of a number of drugs coming and being approved by the [U.S. Food and Drug Administration] in AML is quite remarkable.”
This is especially the case given that “many centers are already using [gilteritinib] as off-label maintenance therapy.”
Dr. Brunstein added that it is “good news” that the drug was effective in MRD+ patients, as it shows “you can overcome that with maintenance therapy rather than keeping giving more and more chemotherapy, especially as there are patients you’re worried about giving more intensive chemotherapy to make them MRD negative.”
He pointed out, however, that the assay used in the trial was “research grade” and very sensitive to MRD and “is not available everywhere, so there is an adjustment that the community will have to do to in order to apply this data.”
“But for those who are more obviously MRD positive with less sensitive assays, gilteritinib is already something that can be used,” Dr. Brunstein said.
Presenting the findings, Dr. Levis stated: “We all know that patients with FLT3/ITD AML have a high risk of relapse and are routinely referred for transplant. And we know that the detection of measurable residual disease pretransplant is highly predictive of outcome post-transplant.”
He continued that FLT3 inhibitors are “routinely given as post-transplant maintenance ... based on some prior trials, mostly with sorafenib.”
“But uncertainty exists as to the broad applicability of these trials,” Dr. Levis said. Moreover, the use of sorafenib in this context is “off label and can be difficult to tolerate,” and “we know that most patients are cured with allogeneic transplant alone.”
Gilteritinib is already known to be well tolerated as a monotherapy, and was approved by the FDA for the treatment of adult patients with FLT3 mutation–positive relapsed or refractory AML in 2018.
The investigators therefore examined whether it would be beneficial as a post-HCT maintenance therapy in FLT3-ITD AML. Patients were required to be in morphologic remission after one or two courses of induction therapy, with Dr. Levis underlining: “We did not allow patients who had been salvaged onto the study.”
They subsequently had a marrow aspirate sample taken for MRD analysis before undergoing allogeneic transplant, with any conditioning regimen, donor, or graft-versus-host disease (GVHD) prophylaxis allowed.
Between 30 and 90 days later, patients with successful engraftment who were able to take oral medication were then randomized to 24 months of maintenance therapy with either gilteritinib or placebo.
Dr. Levis showed that, among 620 patients screened at 110 centers in 16 countries, 356 were randomized between Aug. 15, 2017, and July 8, 2020. The median age was 53 years, and 49% of gilteritinib patients and 48% of those given placebo were female.
He noted that there was a “fairly even global distribution” of patients from North America, Europe, and the Asia/Pacific region, and that 60% of patients underwent a myeloablative conditioning regimen. Approximately the same proportion had received an FLT3 inhibitor prior to HCT.
MRD positivity, assessed at a cell count of ≥ 10-6, was observed pre-HCT in 47% of patients in both treatment groups, and in 50% of gilteritinib patients and 51% of placebo patients at both pre- and post-transplant assessments.
The treatment regimen was completed by 52.8% of patients assigned to gilteritinib and 53.9% in the placebo arm. Dr. Levis said that 18.5% and 20.3% of patients, respectively, experienced a grade 3/4 treatment emergent acute GVHD event, while 32.6% and 21.5%, respectively, had a grade ≥ 3 treatment emergent infection.
He noted that “adverse events were clearly more common in the gilteritinib arm and often led to either dose reduction or interruption, or withdrawal of treatment.”
The most common grade ≥ 3 treatment emergent adverse event was a decrease in neutrophil count, seen in 24.7% of gilteritinib patients and 7.9% of those given placebo, followed by reduced platelet count, in 15.2% and 5.6%, respectively, and anemia, in 6.2% and 1.7%, respectively.
Turning to the efficacy outcomes, Dr. Levis reported that the trial did not meet its primary endpoint, with no significant difference in RFS between the gilteritinib and placebo arms, at a hazard ratio of 0.679 (P = .0518). There was also no significant difference in the key secondary objective of overall survival, at a hazard ratio of 0.846 (P = .4394).
However, Dr. Levis noted that there was a “clear difference in the benefit of gilteritinib by region,” and, “at every level,” MRD predicted a benefit from gilteritinib, which he said was a “big surprise” and “really leapt out in the subgroup analysis.”
He explained that the researchers used a modified version of a two-step assay that has been used in previous studies, and was able to detect MRD at a sensitivity of approximately 1x10-6. “In our study, 98% of participants had samples pre- and post-[transplant].”
Regardless of treatment arm, MRD positivity measured at that sensitivity was associated with a significant reduction in overall survival, at a hazard ratio versus MRD– status of 0.514 (P = .0025).
When stratifying the patients by MRD status, the researchers found that, among MRD+ participants, gilteritinib was associated with a significant improvement in RFS, at a hazard ratio versus placebo of 0.515 (P = .0065), while there was no significant difference in MRD– patients.
Stratifying the patients by their conditioning regimen prior to HCT also revealed differences, with those undergoing myeloablative conditioning having significantly greater overall survival than those who underwent reduced-intensity conditioning, at a hazard ratio for death of 0.529 (P = .0027).
Dr. Levis said there is “no surprise there,” and the result could reflect the selection of fitter, younger patients to undergo the more intensive regimen.
He then showed that MRD+ patients who had undergone myeloablative conditioning had better overall survival with gilteritinib than placebo, at a hazard ratio for death of 0.418 (P = .0087). Again, the difference disappeared when looking at MRD– patients.
“So conditioning doesn’t help you in the setting of MRD,” Dr. Levis said.
Finally, he took a deeper dive into the regional differences in outcomes, noting that patients in the Asia/Pacific region, where gilteritinib showed no benefit over placebo, “were 10 years younger” than those in other regions, “tended to get myeloablative conditioning, and hardly ever used FLT3 inhibitors.”
In contrast, North American patients, who experienced a significant gilteritinib benefit in terms of RFS, underwent HCT an average of 26 days earlier than those elsewhere, and received fewer courses of chemotherapy pre-HCT. Moreover, 93.5% received an FLT3 inhibitor pretransplant.
The study was funded by Astellas Pharma Global Development. Dr. Levis declares relationships with Abbvie, Amgen, Astellas, Bristol-Myers-Squibb, Daiichi-Sankyo, GlaxoSmithKline, Jazz, Menarini, Pfizer, Sumitomo-Dainippon, Syndax, Takeda. Dr. Brunstein declares no relevant relationships.
The research was presented at the European Hematology Association Hybrid Congress 2023.
For the study, AML patients with the most common form of mutation in the proto-oncogene fms-like tyrosine kinase 3 (FLT3), known as the internal tandem duplication (ITD), were randomized to 24 months of maintenance therapy with either the FLT3 inhibitor gilteritinib or placebo.
The trial did not meet its primary endpoint, as there was no significant difference in relapse-free survival (RFS) between those assigned to the active drug and those given placebo, and there was no difference in overall survival rates.
However, subgroup analysis revealed that FLT3/ITD AML patients who were MRD+ after transplant, which represented approximately half of the participants, experienced a significant 48% improvement in RFS with gilteritinib versus placebo, while no benefit was seen in MRD– patients.
While acknowledging that the trial did not meet its primary endpoint, presenter Mark J. Levis, MD, PhD, program leader, hematologic malignancies and bone marrow transplant program, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, said it was nevertheless “a successful study.”
“We learned how to use these drugs and in whom,” he continued, adding: “No, not everybody needs and should get a FLT3 inhibitor post-transplant, but we can use this [MRD] assay to identify who.”
Consequently, Dr. Levis believes that gilteritinib “should be a standard of care for those who are MRD positive,” although the decision to use it “should be balanced against the potential for toxicity,” compared with not adding an additional treatment after HCT.
He told a press conference that “we’re going to certainly make sure that patients who are MRD positive get [gilteritinib],” although the MRD negative patients “are going to be more questionable,” especially because the assay that they used in the study is not “perfect.”
Dr. Levis also suggested that the trial did not meet its endpoint because of regional differences in the clinical practice, such as in the number of treatment cycles prior to HCT, the time to transplant, and the previous use of a FLT3 inhibitor, all of which may have skewed the findings.
“Everybody in the world is convinced that they’re the best transplanter,” he said, and yet “they all do it differently, and the heterogeneity is astounding.”
He added: “If we’d restricted everybody [to a] pretransplant regimen, I suspect we would have had a different result than what we’re getting here, but this is releasing the drug into the world and saying: ‘Here, transplant however you want, however it’s practiced in the real world. Tell us how this works.’ ”
Approached for comment, Claudio Brunstein, MD, PhD, vice-chair of the department of hematology and oncology in the Cleveland Clinic Taussig Cancer Institute, said that while there was “some disappointment” with the results, he was “not surprised” that the trial did not meet its primary endpoint.
He said in an interview that the patient population was not of “high enough risk” to demonstrate an overall difference between gilteritinib and placebo, although he conceded that it is “hard to get to high-risk patients in a timely way” and so conduct a trial with them.
As to the notion that variations in clinical practice could have been responsible, Dr. Brunstein pointed out that it was a randomized trial, so the issue would have applied equally to both sides.
He nevertheless believes that it is “a very important study,” and “just the fact that it was done in the context of a number of drugs coming and being approved by the [U.S. Food and Drug Administration] in AML is quite remarkable.”
This is especially the case given that “many centers are already using [gilteritinib] as off-label maintenance therapy.”
Dr. Brunstein added that it is “good news” that the drug was effective in MRD+ patients, as it shows “you can overcome that with maintenance therapy rather than keeping giving more and more chemotherapy, especially as there are patients you’re worried about giving more intensive chemotherapy to make them MRD negative.”
He pointed out, however, that the assay used in the trial was “research grade” and very sensitive to MRD and “is not available everywhere, so there is an adjustment that the community will have to do to in order to apply this data.”
“But for those who are more obviously MRD positive with less sensitive assays, gilteritinib is already something that can be used,” Dr. Brunstein said.
Presenting the findings, Dr. Levis stated: “We all know that patients with FLT3/ITD AML have a high risk of relapse and are routinely referred for transplant. And we know that the detection of measurable residual disease pretransplant is highly predictive of outcome post-transplant.”
He continued that FLT3 inhibitors are “routinely given as post-transplant maintenance ... based on some prior trials, mostly with sorafenib.”
“But uncertainty exists as to the broad applicability of these trials,” Dr. Levis said. Moreover, the use of sorafenib in this context is “off label and can be difficult to tolerate,” and “we know that most patients are cured with allogeneic transplant alone.”
Gilteritinib is already known to be well tolerated as a monotherapy, and was approved by the FDA for the treatment of adult patients with FLT3 mutation–positive relapsed or refractory AML in 2018.
The investigators therefore examined whether it would be beneficial as a post-HCT maintenance therapy in FLT3-ITD AML. Patients were required to be in morphologic remission after one or two courses of induction therapy, with Dr. Levis underlining: “We did not allow patients who had been salvaged onto the study.”
They subsequently had a marrow aspirate sample taken for MRD analysis before undergoing allogeneic transplant, with any conditioning regimen, donor, or graft-versus-host disease (GVHD) prophylaxis allowed.
Between 30 and 90 days later, patients with successful engraftment who were able to take oral medication were then randomized to 24 months of maintenance therapy with either gilteritinib or placebo.
Dr. Levis showed that, among 620 patients screened at 110 centers in 16 countries, 356 were randomized between Aug. 15, 2017, and July 8, 2020. The median age was 53 years, and 49% of gilteritinib patients and 48% of those given placebo were female.
He noted that there was a “fairly even global distribution” of patients from North America, Europe, and the Asia/Pacific region, and that 60% of patients underwent a myeloablative conditioning regimen. Approximately the same proportion had received an FLT3 inhibitor prior to HCT.
MRD positivity, assessed at a cell count of ≥ 10-6, was observed pre-HCT in 47% of patients in both treatment groups, and in 50% of gilteritinib patients and 51% of placebo patients at both pre- and post-transplant assessments.
The treatment regimen was completed by 52.8% of patients assigned to gilteritinib and 53.9% in the placebo arm. Dr. Levis said that 18.5% and 20.3% of patients, respectively, experienced a grade 3/4 treatment emergent acute GVHD event, while 32.6% and 21.5%, respectively, had a grade ≥ 3 treatment emergent infection.
He noted that “adverse events were clearly more common in the gilteritinib arm and often led to either dose reduction or interruption, or withdrawal of treatment.”
The most common grade ≥ 3 treatment emergent adverse event was a decrease in neutrophil count, seen in 24.7% of gilteritinib patients and 7.9% of those given placebo, followed by reduced platelet count, in 15.2% and 5.6%, respectively, and anemia, in 6.2% and 1.7%, respectively.
Turning to the efficacy outcomes, Dr. Levis reported that the trial did not meet its primary endpoint, with no significant difference in RFS between the gilteritinib and placebo arms, at a hazard ratio of 0.679 (P = .0518). There was also no significant difference in the key secondary objective of overall survival, at a hazard ratio of 0.846 (P = .4394).
However, Dr. Levis noted that there was a “clear difference in the benefit of gilteritinib by region,” and, “at every level,” MRD predicted a benefit from gilteritinib, which he said was a “big surprise” and “really leapt out in the subgroup analysis.”
He explained that the researchers used a modified version of a two-step assay that has been used in previous studies, and was able to detect MRD at a sensitivity of approximately 1x10-6. “In our study, 98% of participants had samples pre- and post-[transplant].”
Regardless of treatment arm, MRD positivity measured at that sensitivity was associated with a significant reduction in overall survival, at a hazard ratio versus MRD– status of 0.514 (P = .0025).
When stratifying the patients by MRD status, the researchers found that, among MRD+ participants, gilteritinib was associated with a significant improvement in RFS, at a hazard ratio versus placebo of 0.515 (P = .0065), while there was no significant difference in MRD– patients.
Stratifying the patients by their conditioning regimen prior to HCT also revealed differences, with those undergoing myeloablative conditioning having significantly greater overall survival than those who underwent reduced-intensity conditioning, at a hazard ratio for death of 0.529 (P = .0027).
Dr. Levis said there is “no surprise there,” and the result could reflect the selection of fitter, younger patients to undergo the more intensive regimen.
He then showed that MRD+ patients who had undergone myeloablative conditioning had better overall survival with gilteritinib than placebo, at a hazard ratio for death of 0.418 (P = .0087). Again, the difference disappeared when looking at MRD– patients.
“So conditioning doesn’t help you in the setting of MRD,” Dr. Levis said.
Finally, he took a deeper dive into the regional differences in outcomes, noting that patients in the Asia/Pacific region, where gilteritinib showed no benefit over placebo, “were 10 years younger” than those in other regions, “tended to get myeloablative conditioning, and hardly ever used FLT3 inhibitors.”
In contrast, North American patients, who experienced a significant gilteritinib benefit in terms of RFS, underwent HCT an average of 26 days earlier than those elsewhere, and received fewer courses of chemotherapy pre-HCT. Moreover, 93.5% received an FLT3 inhibitor pretransplant.
The study was funded by Astellas Pharma Global Development. Dr. Levis declares relationships with Abbvie, Amgen, Astellas, Bristol-Myers-Squibb, Daiichi-Sankyo, GlaxoSmithKline, Jazz, Menarini, Pfizer, Sumitomo-Dainippon, Syndax, Takeda. Dr. Brunstein declares no relevant relationships.
AT EHA 2023
FDA OKs low-dose colchicine for broad CV indication
The Food and Drug Administration has approved the anti-inflammatory drug colchicine 0.5 mg tablets (Lodoco) as the first specific anti-inflammatory drug demonstrated to reduce the risk for myocardial infarction, stroke, coronary revascularization, and cardiovascular death in adult patients with established atherosclerotic disease or with multiple risk factors for cardiovascular disease.
The drug, which targets residual inflammation as an underlying cause of atherosclerotic cardiovascular disease, has a dosage of 0.5 mg once daily, and can be used alone or in combination with cholesterol-lowering medications. 
The drug’s manufacturer, Agepha Pharma, said it anticipates that Lodoco will be available for prescription in the second half of 2023.
Colchicine has been available for many years and used at higher doses for the acute treatment of gout and pericarditis, but the current formulation is a much lower dose for long-term use in patients with atherosclerotic heart disease.
Data supporting the approval has come from two major randomized trials, LoDoCo-2 and COLCOT.
In the LoDoCo-2 trial, the anti-inflammatory drug cut the risk of cardiovascular events by one third when added to standard prevention therapies in patients with chronic coronary disease. And in the COLCOT study, use of colchicine reduced cardiovascular events by 23% compared with placebo in patients with a recent MI.
Paul Ridker, MD, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital in Boston, who has been a pioneer in establishing inflammation as an underlying cause of atherosclerotic cardiovascular disease, welcomed the Lodoco approval.
‘A very big day for cardiology’
“This is a very big day for cardiology,” Dr. Ridker said in an interview.
“The FDA approval of colchicine for patients with atherosclerotic disease is a huge signal that physicians need to be aware of inflammation as a key player in cardiovascular disease,” he said.
Dr. Ridker was the lead author of a recent study showing that among patients receiving contemporary statins, inflammation assessed by high-sensitivity C-reactive protein (hsCRP) was a stronger predictor for risk of future cardiovascular events and death than LDL cholesterol.
He pointed out that
“That is virtually identical to the indication approved for statin therapy. That shows just how important the FDA thinks this is,” he commented.
But Dr. Ridker added that, while the label does not specify that Lodoco has to be used in addition to statin therapy, he believes that it will be used as additional therapy to statins in the vast majority of patients.
“This is not an alternative to statin therapy. In the randomized trials, the benefits were seen on top of statins,” he stressed.
Dr. Ridker believes that physicians will need time to feel comfortable with this new approach.
“Initially, I think, it will be used mainly by cardiologists who know about inflammation, but I believe over time it will be widely prescribed by internists, in much the same way as statins are used today,” he commented.
Dr. Ridker said he already uses low dose colchicine in his high-risk patients who have high levels of inflammation as seen on hsCRP testing. He believes this is where the drug will mostly be used initially, as this is where it is likely to be most effective.
The prescribing information states that Lodoco is contraindicated in patients who are taking strong CYP3A4 inhibitors or P-glycoprotein inhibitors, such as ketoconazole, fluconazole, and clarithromycin, and in patients with preexisting blood dyscrasias, renal failure, and severe hepatic impairment.
Common side effects reported in published clinical studies and literature with the use of colchicine are gastrointestinal symptoms (diarrhea, vomiting, abdominal cramping) and myalgia.
More serious adverse effects are listed as blood dyscrasias such as myelosuppression, leukopenia, granulocytopenia, thrombocytopenia, pancytopenia, and aplastic anemia; and neuromuscular toxicity in the form of myotoxicity including rhabdomyolysis, which may occur, especially in combination with other drugs known to cause this effect. If these adverse effects occur, it is recommended that the drug be stopped.
The prescribing information also notes that Lodoco may rarely and transiently impair fertility in males; and that patients with renal or hepatic impairment should be monitored closely for adverse effects of colchicine.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the anti-inflammatory drug colchicine 0.5 mg tablets (Lodoco) as the first specific anti-inflammatory drug demonstrated to reduce the risk for myocardial infarction, stroke, coronary revascularization, and cardiovascular death in adult patients with established atherosclerotic disease or with multiple risk factors for cardiovascular disease.
The drug, which targets residual inflammation as an underlying cause of atherosclerotic cardiovascular disease, has a dosage of 0.5 mg once daily, and can be used alone or in combination with cholesterol-lowering medications.
The drug’s manufacturer, Agepha Pharma, said it anticipates that Lodoco will be available for prescription in the second half of 2023.
Colchicine has been available for many years and used at higher doses for the acute treatment of gout and pericarditis, but the current formulation is a much lower dose for long-term use in patients with atherosclerotic heart disease.
Data supporting the approval has come from two major randomized trials, LoDoCo-2 and COLCOT.
In the LoDoCo-2 trial, the anti-inflammatory drug cut the risk of cardiovascular events by one third when added to standard prevention therapies in patients with chronic coronary disease. And in the COLCOT study, use of colchicine reduced cardiovascular events by 23% compared with placebo in patients with a recent MI.
Paul Ridker, MD, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital in Boston, who has been a pioneer in establishing inflammation as an underlying cause of atherosclerotic cardiovascular disease, welcomed the Lodoco approval.
‘A very big day for cardiology’
“This is a very big day for cardiology,” Dr. Ridker said in an interview.
“The FDA approval of colchicine for patients with atherosclerotic disease is a huge signal that physicians need to be aware of inflammation as a key player in cardiovascular disease,” he said.
Dr. Ridker was the lead author of a recent study showing that among patients receiving contemporary statins, inflammation assessed by high-sensitivity C-reactive protein (hsCRP) was a stronger predictor for risk of future cardiovascular events and death than LDL cholesterol.
He pointed out that
“That is virtually identical to the indication approved for statin therapy. That shows just how important the FDA thinks this is,” he commented.
But Dr. Ridker added that, while the label does not specify that Lodoco has to be used in addition to statin therapy, he believes that it will be used as additional therapy to statins in the vast majority of patients.
“This is not an alternative to statin therapy. In the randomized trials, the benefits were seen on top of statins,” he stressed.
Dr. Ridker believes that physicians will need time to feel comfortable with this new approach.
“Initially, I think, it will be used mainly by cardiologists who know about inflammation, but I believe over time it will be widely prescribed by internists, in much the same way as statins are used today,” he commented.
Dr. Ridker said he already uses low dose colchicine in his high-risk patients who have high levels of inflammation as seen on hsCRP testing. He believes this is where the drug will mostly be used initially, as this is where it is likely to be most effective.
The prescribing information states that Lodoco is contraindicated in patients who are taking strong CYP3A4 inhibitors or P-glycoprotein inhibitors, such as ketoconazole, fluconazole, and clarithromycin, and in patients with preexisting blood dyscrasias, renal failure, and severe hepatic impairment.
Common side effects reported in published clinical studies and literature with the use of colchicine are gastrointestinal symptoms (diarrhea, vomiting, abdominal cramping) and myalgia.
More serious adverse effects are listed as blood dyscrasias such as myelosuppression, leukopenia, granulocytopenia, thrombocytopenia, pancytopenia, and aplastic anemia; and neuromuscular toxicity in the form of myotoxicity including rhabdomyolysis, which may occur, especially in combination with other drugs known to cause this effect. If these adverse effects occur, it is recommended that the drug be stopped.
The prescribing information also notes that Lodoco may rarely and transiently impair fertility in males; and that patients with renal or hepatic impairment should be monitored closely for adverse effects of colchicine.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the anti-inflammatory drug colchicine 0.5 mg tablets (Lodoco) as the first specific anti-inflammatory drug demonstrated to reduce the risk for myocardial infarction, stroke, coronary revascularization, and cardiovascular death in adult patients with established atherosclerotic disease or with multiple risk factors for cardiovascular disease.
The drug, which targets residual inflammation as an underlying cause of atherosclerotic cardiovascular disease, has a dosage of 0.5 mg once daily, and can be used alone or in combination with cholesterol-lowering medications.
The drug’s manufacturer, Agepha Pharma, said it anticipates that Lodoco will be available for prescription in the second half of 2023.
Colchicine has been available for many years and used at higher doses for the acute treatment of gout and pericarditis, but the current formulation is a much lower dose for long-term use in patients with atherosclerotic heart disease.
Data supporting the approval has come from two major randomized trials, LoDoCo-2 and COLCOT.
In the LoDoCo-2 trial, the anti-inflammatory drug cut the risk of cardiovascular events by one third when added to standard prevention therapies in patients with chronic coronary disease. And in the COLCOT study, use of colchicine reduced cardiovascular events by 23% compared with placebo in patients with a recent MI.
Paul Ridker, MD, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital in Boston, who has been a pioneer in establishing inflammation as an underlying cause of atherosclerotic cardiovascular disease, welcomed the Lodoco approval.
‘A very big day for cardiology’
“This is a very big day for cardiology,” Dr. Ridker said in an interview.
“The FDA approval of colchicine for patients with atherosclerotic disease is a huge signal that physicians need to be aware of inflammation as a key player in cardiovascular disease,” he said.
Dr. Ridker was the lead author of a recent study showing that among patients receiving contemporary statins, inflammation assessed by high-sensitivity C-reactive protein (hsCRP) was a stronger predictor for risk of future cardiovascular events and death than LDL cholesterol.
He pointed out that
“That is virtually identical to the indication approved for statin therapy. That shows just how important the FDA thinks this is,” he commented.
But Dr. Ridker added that, while the label does not specify that Lodoco has to be used in addition to statin therapy, he believes that it will be used as additional therapy to statins in the vast majority of patients.
“This is not an alternative to statin therapy. In the randomized trials, the benefits were seen on top of statins,” he stressed.
Dr. Ridker believes that physicians will need time to feel comfortable with this new approach.
“Initially, I think, it will be used mainly by cardiologists who know about inflammation, but I believe over time it will be widely prescribed by internists, in much the same way as statins are used today,” he commented.
Dr. Ridker said he already uses low dose colchicine in his high-risk patients who have high levels of inflammation as seen on hsCRP testing. He believes this is where the drug will mostly be used initially, as this is where it is likely to be most effective.
The prescribing information states that Lodoco is contraindicated in patients who are taking strong CYP3A4 inhibitors or P-glycoprotein inhibitors, such as ketoconazole, fluconazole, and clarithromycin, and in patients with preexisting blood dyscrasias, renal failure, and severe hepatic impairment.
Common side effects reported in published clinical studies and literature with the use of colchicine are gastrointestinal symptoms (diarrhea, vomiting, abdominal cramping) and myalgia.
More serious adverse effects are listed as blood dyscrasias such as myelosuppression, leukopenia, granulocytopenia, thrombocytopenia, pancytopenia, and aplastic anemia; and neuromuscular toxicity in the form of myotoxicity including rhabdomyolysis, which may occur, especially in combination with other drugs known to cause this effect. If these adverse effects occur, it is recommended that the drug be stopped.
The prescribing information also notes that Lodoco may rarely and transiently impair fertility in males; and that patients with renal or hepatic impairment should be monitored closely for adverse effects of colchicine.
A version of this article first appeared on Medscape.com.
Aspirin warning: Anemia may increase with daily use
, according to results from a new randomized controlled trial.
In the study, which was published in Annals of Internal Medicine, investigators analyzed data from the Aspirin in Reducing Events in the Elderly (ASPREE) trial and examined hemoglobin concentrations among 19,114 healthy, community-dwelling older patients.
“We knew from large clinical trials, including our ASPREE trial, that daily low-dose aspirin increased the risk of clinically significant bleeding,” said Zoe McQuilten, MBBS, PhD, a hematologist at Monash University in Australia and the study’s lead author. “From our study, we found that low-dose aspirin also increased the risk of anemia during the trial, and this was most likely due to bleeding that was not clinically apparent.”
Anemia is common among elderly patients. It can cause fatigue, fast or irregular heartbeat, headache, chest pain, and pounding or whooshing sounds in the ear, according to the Cleveland Clinic. It can also worsen conditions such as heart failure, cognitive impairment, and depression in people aged 65 and older.
The U.S. Preventive Services Task Force changed its recommendation on aspirin for the primary prevention of cardiovascular disease in 2022, recommending against initiating low-dose aspirin for adults aged 60 years or older. For adults aged 40-59 who have a 10% or greater 10-year risk for cardiovascular disease, the agency recommends that patients and clinicians make the decision to initiate low-dose aspirin use on a case-by-case basis, as the net benefit is small.
Dr. McQuilten said she spent the last 5 years designing substages of anemia and conditions such as blood cancer. In many cases of anemia, doctors are unable to determine the underlying cause, she said. One study published in the Journal of American Geriatrics Society in 2021 found that in about one-third of anemia cases, the etiology was not clear.
About 50% of people older than 60 who were involved in the latest study took aspirin for prevention from 2011 to 2018. That number likely dropped after changes were made to the guidelines in 2022, according to Dr. McQuilten, but long-term use may have continued among older patients. The researchers also examined ferritin levels, which serve as a proxy for iron levels, at baseline and after 3 years.
The incidence of anemia was 51 events per 1,000 person-years in the aspirin group compared with 43 events per 1,000 person-years in the placebo group, according to the researchers. The estimated probability of experiencing anemia within 5 years was 23.5% (95% confidence interval [CI], 22.4%-24.6%) in the aspirin group and 20.3% (95% CI: 19.3% to 21.4%) in the placebo group. Aspirin therapy resulted in a 20% increase in the risk for anemia (95% CI, 1.12-1.29).
People who took aspirin were more likely to have lower serum levels of ferritin at the 3-year mark than were those who received placebo. The average decrease in ferritin among participants who took aspirin was 11.5% greater (95% CI, 9.3%-13.7%) than among those who took placebo.
Basil Eldadah, MD, PhD, supervisory medical officer at the National Institute on Aging, part of the National Institutes of Health, said the findings should encourage clinicians to pay closer attention to hemoglobin levels and have conversations with patients to discuss their need for taking aspirin.
“If somebody is already taking aspirin for any reason, keep an eye on hemoglobin,” said Dr. Eldadah, who was not involved in the study. “For somebody who’s taking aspirin and who’s older, and it’s not for an indication like cardiovascular disease, consider seriously whether that’s the best treatment option.”
The study did not examine the functional consequences of anemia on participants, which Dr. Eldadah said could be fodder for future research. The researchers said one limitation was that it was not clear whether anemia was sufficient to cause symptoms that affected participants’ quality of life or whether occult bleeding caused the anemia. The researchers also did not document whether patients saw their regular physicians and received treatment for anemia over the course of the trial.
The study was funded through grants from the National Health and Medical Research Council and the Bill and Melinda Gates Foundation. The authors reported receiving consulting fees, honoraria, and stock options, and have participated on data monitoring boards not related to the study for Vifor Pharma, ITL Biomedical, Pfizer, Boehringer Ingelheim, Bayer Healthcare, AbbVie, and Abbott Diagnostics.
A version of this article originally appeared on Medscape.com.
, according to results from a new randomized controlled trial.
In the study, which was published in Annals of Internal Medicine, investigators analyzed data from the Aspirin in Reducing Events in the Elderly (ASPREE) trial and examined hemoglobin concentrations among 19,114 healthy, community-dwelling older patients.
“We knew from large clinical trials, including our ASPREE trial, that daily low-dose aspirin increased the risk of clinically significant bleeding,” said Zoe McQuilten, MBBS, PhD, a hematologist at Monash University in Australia and the study’s lead author. “From our study, we found that low-dose aspirin also increased the risk of anemia during the trial, and this was most likely due to bleeding that was not clinically apparent.”
Anemia is common among elderly patients. It can cause fatigue, fast or irregular heartbeat, headache, chest pain, and pounding or whooshing sounds in the ear, according to the Cleveland Clinic. It can also worsen conditions such as heart failure, cognitive impairment, and depression in people aged 65 and older.
The U.S. Preventive Services Task Force changed its recommendation on aspirin for the primary prevention of cardiovascular disease in 2022, recommending against initiating low-dose aspirin for adults aged 60 years or older. For adults aged 40-59 who have a 10% or greater 10-year risk for cardiovascular disease, the agency recommends that patients and clinicians make the decision to initiate low-dose aspirin use on a case-by-case basis, as the net benefit is small.
Dr. McQuilten said she spent the last 5 years designing substages of anemia and conditions such as blood cancer. In many cases of anemia, doctors are unable to determine the underlying cause, she said. One study published in the Journal of American Geriatrics Society in 2021 found that in about one-third of anemia cases, the etiology was not clear.
About 50% of people older than 60 who were involved in the latest study took aspirin for prevention from 2011 to 2018. That number likely dropped after changes were made to the guidelines in 2022, according to Dr. McQuilten, but long-term use may have continued among older patients. The researchers also examined ferritin levels, which serve as a proxy for iron levels, at baseline and after 3 years.
The incidence of anemia was 51 events per 1,000 person-years in the aspirin group compared with 43 events per 1,000 person-years in the placebo group, according to the researchers. The estimated probability of experiencing anemia within 5 years was 23.5% (95% confidence interval [CI], 22.4%-24.6%) in the aspirin group and 20.3% (95% CI: 19.3% to 21.4%) in the placebo group. Aspirin therapy resulted in a 20% increase in the risk for anemia (95% CI, 1.12-1.29).
People who took aspirin were more likely to have lower serum levels of ferritin at the 3-year mark than were those who received placebo. The average decrease in ferritin among participants who took aspirin was 11.5% greater (95% CI, 9.3%-13.7%) than among those who took placebo.
Basil Eldadah, MD, PhD, supervisory medical officer at the National Institute on Aging, part of the National Institutes of Health, said the findings should encourage clinicians to pay closer attention to hemoglobin levels and have conversations with patients to discuss their need for taking aspirin.
“If somebody is already taking aspirin for any reason, keep an eye on hemoglobin,” said Dr. Eldadah, who was not involved in the study. “For somebody who’s taking aspirin and who’s older, and it’s not for an indication like cardiovascular disease, consider seriously whether that’s the best treatment option.”
The study did not examine the functional consequences of anemia on participants, which Dr. Eldadah said could be fodder for future research. The researchers said one limitation was that it was not clear whether anemia was sufficient to cause symptoms that affected participants’ quality of life or whether occult bleeding caused the anemia. The researchers also did not document whether patients saw their regular physicians and received treatment for anemia over the course of the trial.
The study was funded through grants from the National Health and Medical Research Council and the Bill and Melinda Gates Foundation. The authors reported receiving consulting fees, honoraria, and stock options, and have participated on data monitoring boards not related to the study for Vifor Pharma, ITL Biomedical, Pfizer, Boehringer Ingelheim, Bayer Healthcare, AbbVie, and Abbott Diagnostics.
A version of this article originally appeared on Medscape.com.
, according to results from a new randomized controlled trial.
In the study, which was published in Annals of Internal Medicine, investigators analyzed data from the Aspirin in Reducing Events in the Elderly (ASPREE) trial and examined hemoglobin concentrations among 19,114 healthy, community-dwelling older patients.
“We knew from large clinical trials, including our ASPREE trial, that daily low-dose aspirin increased the risk of clinically significant bleeding,” said Zoe McQuilten, MBBS, PhD, a hematologist at Monash University in Australia and the study’s lead author. “From our study, we found that low-dose aspirin also increased the risk of anemia during the trial, and this was most likely due to bleeding that was not clinically apparent.”
Anemia is common among elderly patients. It can cause fatigue, fast or irregular heartbeat, headache, chest pain, and pounding or whooshing sounds in the ear, according to the Cleveland Clinic. It can also worsen conditions such as heart failure, cognitive impairment, and depression in people aged 65 and older.
The U.S. Preventive Services Task Force changed its recommendation on aspirin for the primary prevention of cardiovascular disease in 2022, recommending against initiating low-dose aspirin for adults aged 60 years or older. For adults aged 40-59 who have a 10% or greater 10-year risk for cardiovascular disease, the agency recommends that patients and clinicians make the decision to initiate low-dose aspirin use on a case-by-case basis, as the net benefit is small.
Dr. McQuilten said she spent the last 5 years designing substages of anemia and conditions such as blood cancer. In many cases of anemia, doctors are unable to determine the underlying cause, she said. One study published in the Journal of American Geriatrics Society in 2021 found that in about one-third of anemia cases, the etiology was not clear.
About 50% of people older than 60 who were involved in the latest study took aspirin for prevention from 2011 to 2018. That number likely dropped after changes were made to the guidelines in 2022, according to Dr. McQuilten, but long-term use may have continued among older patients. The researchers also examined ferritin levels, which serve as a proxy for iron levels, at baseline and after 3 years.
The incidence of anemia was 51 events per 1,000 person-years in the aspirin group compared with 43 events per 1,000 person-years in the placebo group, according to the researchers. The estimated probability of experiencing anemia within 5 years was 23.5% (95% confidence interval [CI], 22.4%-24.6%) in the aspirin group and 20.3% (95% CI: 19.3% to 21.4%) in the placebo group. Aspirin therapy resulted in a 20% increase in the risk for anemia (95% CI, 1.12-1.29).
People who took aspirin were more likely to have lower serum levels of ferritin at the 3-year mark than were those who received placebo. The average decrease in ferritin among participants who took aspirin was 11.5% greater (95% CI, 9.3%-13.7%) than among those who took placebo.
Basil Eldadah, MD, PhD, supervisory medical officer at the National Institute on Aging, part of the National Institutes of Health, said the findings should encourage clinicians to pay closer attention to hemoglobin levels and have conversations with patients to discuss their need for taking aspirin.
“If somebody is already taking aspirin for any reason, keep an eye on hemoglobin,” said Dr. Eldadah, who was not involved in the study. “For somebody who’s taking aspirin and who’s older, and it’s not for an indication like cardiovascular disease, consider seriously whether that’s the best treatment option.”
The study did not examine the functional consequences of anemia on participants, which Dr. Eldadah said could be fodder for future research. The researchers said one limitation was that it was not clear whether anemia was sufficient to cause symptoms that affected participants’ quality of life or whether occult bleeding caused the anemia. The researchers also did not document whether patients saw their regular physicians and received treatment for anemia over the course of the trial.
The study was funded through grants from the National Health and Medical Research Council and the Bill and Melinda Gates Foundation. The authors reported receiving consulting fees, honoraria, and stock options, and have participated on data monitoring boards not related to the study for Vifor Pharma, ITL Biomedical, Pfizer, Boehringer Ingelheim, Bayer Healthcare, AbbVie, and Abbott Diagnostics.
A version of this article originally appeared on Medscape.com.
FROM ANNALS OF INTERNAL MEDICINE
Combo treatment eases nausea and vomiting of pregnancy
. While the benefit of either agent was clinically small for moderate to severe symptoms, the combination showed numerically larger and potentially more meaningful benefit, according to a team led by Xiao-Ke Wu, MD, PhD, of the department of obstetrics and gynecology at First Affiliated Hospital, Heilongjiang University of Chinese Medicine, and Heilongjiang Provincial Hospital in Harbin, China.

The treatments found small reductions in symptoms of less than one point to 1.6 points on an emesis scale. Nevertheless, Dr. Wu’s group wrote online June 19 in Annals of Internal Medicine that the finding “is especially significant because there is a pressing need to establish a pregnancy-safe treatment regimen and an integrative guideline for managing severe NVP.”
NVP affects as many as 85% of pregnant women, 80%-90% of whom have only mild symptoms, the authors noted. However, severe NVP and hyperemesis gravidarum, or HG, develop in about 10%. “Unfortunately, as many as 10% of wanted pregnancies with severe NVP or HG are terminated because of intolerable and untreatable symptoms and complications,” Dr. Wu told this news organization. And antiemetics may be underprescribed by general practitioners because of concerns about potential teratogenic effects, he said.
“Our findings suggest that either acupuncture or doxylamine-pyridoxine alone is a suitable for treating moderate to severe NVP, and a combination of both can be used to treat severe NVP and HG,” Dr. Wu said.
Commenting on the study but not involved in it, Catherine S. Stika, MD, a clinical professor of ob.gyn. at Northwestern University in Chicago, said the results suggest these two therapies are more suited to mild than severe symptoms. “But an RCT is important to do in order to support the use of these therapies since they’re not as widely accepted as they ought to be,” she said in an interview.

According to Dr. Stika, many pregnant women are reluctant to take drugs at all or participate in drug studies, “so the combination of nonpharmaceutical/pharmaceutical treatment might be a bit more appealing.” She noted that some women have such severe nausea they are literally starving and so weak they are bedridden or even hospitalized.
Both treatments have been recommended for some time, and the American College of Obstetricians and Gynecologists’ 2018 practice bulletin recommends acupuncture for mild nausea.
Design
The randomized, double-blind, placebo-controled 2x2 factorial trial was conducted at 13 tertiary-care hospitals in mainland China from June 2020 to February 2022. The researchers recruited 352 women in early pregnancy with moderate to severe NVP. The mean age of participants was about 29 years and the mean gestational age was about 9 weeks.
Participants were randomized into four 14-day treatment groups: active acupuncture for 30 minutes a day plus the antihistamine-vitamin B6 agent doxylamine-pyridoxine; sham acupuncture for 30 minutes daily plus doxylamine-pyridoxine; active acupuncture plus placebo; and sham acupuncture plus placebo.
The primary outcome was the reduction in Pregnancy-Unique Quantification of Emesis (PUQE) score at day 15 relative to baseline with a score of less than 6 indicating mild NVP, 6-12 indicating moderate NVP, and 13 or higher indicating severe NVP. Secondary outcomes ranged from quality of life and adverse events to maternal and perinatal complications. Acupuncture and combined treatment yielded larger though still small reductions in PUQE score, compared with control treatments. The mean differences were as follows: acupuncture, –.07; 95% confidence interval, 1.3-0.1); doxylamine-pyridoxine, –1.0: 95% CI, 1.6-0.4); combination of both, –1.6; 95% CI, 2.2-0.9). No significant interaction was detected between the interventions (P = .69).Compared with placebo treatments, pharmaceutical therapy resulted in more somnolence, while active acupuncture led to more frequent dyspnea, bruising, itching, and pain. A higher risk of babies born small for gestational age was observed in mothers who took doxylamine-pyridoxine versus placebo: odds ratio, 3.8; 95% CI, 1-14.1). Neither the placebo effects of the sham interventions nor the natural regression of symptoms experienced by many women were evaluated.
Suited to milder symptoms?
Dr. Stika called the study well-designed and well-written but cited several limitations, including the small cohort, the minor symptom improvement, and the lack of a comparator group receiving neither sham nor active treatment.
“Compared with sham combination treatments, the active combination arm was only about a point and a half better,” she said. “And would some women have got better over the 2 weeks anyway with no intervention at all? A large percentage of women with NVP do improve on their own.”
And in terms of acceptability to U.S. women, she cautioned, “The study cohort was entirely Chinese, and this is a population that already accepts acupuncture treatment.”
Countered Dr. Wu, “Medical care provided by licensed acupuncturists is approved in many countries. Certainly, it is ready to be prescribed by physicians when a pregnant patient is seeking NVP treatment.”
Dr. Stika stressed that these therapies are suited to milder NV, and would “barely take edge off severe symptoms,” for which a patient might have to “go up to a big gun like the antiemetic Zofran” (ondansetron). She is currently involved in a National Institutes of Health–funded clinical trial of the antidepressant mirtazapine (Remeron) for NVP.
Matthew Carroll, MD, a professor of obstetrics and gynecology at Baylor College of Medicine and Texas Children’s Hospital in Houston, noted that doxylamine-pyridoxine is already an effective treatment for NVP, but in his experience it is often "not enough" to help patients deal with symptoms.
"Many patients are hesitant to take additional medications," he said. "If acupuncture can be safely done in pregnancy, then it seems a reasonable option as an adjuvant treatment for NVP. I think there is a cohort of pregnant people in the US who would be excited to try a complementary and nonpharmaceutical treatment option. Unfortunately, complementary therapies are rarely evaluated at a systems level for safety and so they are hard to recommend for obstetricians in the US," he added.
Dr. Carroll, who was not involved in the study. noted that "studies like this can help us counsel patients who may be seeking these treatments even if not approved or recommended by ACOG."
This study was funded by the National Key R&D Program of China and the Project of Heilongjiang Province “TouYan” Innovation Team. Support also came from the National Clinical Research Base of Chinese Medicine, the Heilongjiang Provincial Clinical Research Centre for Ovary Diseases, and the 2023 Capability Improvement Project for Evidence-based Assessment of Traditional Chinese Medicine.
Study coauthor Ben Willem J. Mol, MD, PhD, reported consulting fees from ObsEva and Merck and travel fees from Merck.
Dr. Stika and Dr. Carroll had no competing interests to disclose.
. While the benefit of either agent was clinically small for moderate to severe symptoms, the combination showed numerically larger and potentially more meaningful benefit, according to a team led by Xiao-Ke Wu, MD, PhD, of the department of obstetrics and gynecology at First Affiliated Hospital, Heilongjiang University of Chinese Medicine, and Heilongjiang Provincial Hospital in Harbin, China.
The treatments found small reductions in symptoms of less than one point to 1.6 points on an emesis scale. Nevertheless, Dr. Wu’s group wrote online June 19 in Annals of Internal Medicine that the finding “is especially significant because there is a pressing need to establish a pregnancy-safe treatment regimen and an integrative guideline for managing severe NVP.”
NVP affects as many as 85% of pregnant women, 80%-90% of whom have only mild symptoms, the authors noted. However, severe NVP and hyperemesis gravidarum, or HG, develop in about 10%. “Unfortunately, as many as 10% of wanted pregnancies with severe NVP or HG are terminated because of intolerable and untreatable symptoms and complications,” Dr. Wu told this news organization. And antiemetics may be underprescribed by general practitioners because of concerns about potential teratogenic effects, he said.
“Our findings suggest that either acupuncture or doxylamine-pyridoxine alone is a suitable for treating moderate to severe NVP, and a combination of both can be used to treat severe NVP and HG,” Dr. Wu said.
Commenting on the study but not involved in it, Catherine S. Stika, MD, a clinical professor of ob.gyn. at Northwestern University in Chicago, said the results suggest these two therapies are more suited to mild than severe symptoms. “But an RCT is important to do in order to support the use of these therapies since they’re not as widely accepted as they ought to be,” she said in an interview.
According to Dr. Stika, many pregnant women are reluctant to take drugs at all or participate in drug studies, “so the combination of nonpharmaceutical/pharmaceutical treatment might be a bit more appealing.” She noted that some women have such severe nausea they are literally starving and so weak they are bedridden or even hospitalized.
Both treatments have been recommended for some time, and the American College of Obstetricians and Gynecologists’ 2018 practice bulletin recommends acupuncture for mild nausea.
Design
The randomized, double-blind, placebo-controled 2x2 factorial trial was conducted at 13 tertiary-care hospitals in mainland China from June 2020 to February 2022. The researchers recruited 352 women in early pregnancy with moderate to severe NVP. The mean age of participants was about 29 years and the mean gestational age was about 9 weeks.
Participants were randomized into four 14-day treatment groups: active acupuncture for 30 minutes a day plus the antihistamine-vitamin B6 agent doxylamine-pyridoxine; sham acupuncture for 30 minutes daily plus doxylamine-pyridoxine; active acupuncture plus placebo; and sham acupuncture plus placebo.
The primary outcome was the reduction in Pregnancy-Unique Quantification of Emesis (PUQE) score at day 15 relative to baseline with a score of less than 6 indicating mild NVP, 6-12 indicating moderate NVP, and 13 or higher indicating severe NVP. Secondary outcomes ranged from quality of life and adverse events to maternal and perinatal complications. Acupuncture and combined treatment yielded larger though still small reductions in PUQE score, compared with control treatments. The mean differences were as follows: acupuncture, –.07; 95% confidence interval, 1.3-0.1); doxylamine-pyridoxine, –1.0: 95% CI, 1.6-0.4); combination of both, –1.6; 95% CI, 2.2-0.9). No significant interaction was detected between the interventions (P = .69).Compared with placebo treatments, pharmaceutical therapy resulted in more somnolence, while active acupuncture led to more frequent dyspnea, bruising, itching, and pain. A higher risk of babies born small for gestational age was observed in mothers who took doxylamine-pyridoxine versus placebo: odds ratio, 3.8; 95% CI, 1-14.1). Neither the placebo effects of the sham interventions nor the natural regression of symptoms experienced by many women were evaluated.
Suited to milder symptoms?
Dr. Stika called the study well-designed and well-written but cited several limitations, including the small cohort, the minor symptom improvement, and the lack of a comparator group receiving neither sham nor active treatment.
“Compared with sham combination treatments, the active combination arm was only about a point and a half better,” she said. “And would some women have got better over the 2 weeks anyway with no intervention at all? A large percentage of women with NVP do improve on their own.”
And in terms of acceptability to U.S. women, she cautioned, “The study cohort was entirely Chinese, and this is a population that already accepts acupuncture treatment.”
Countered Dr. Wu, “Medical care provided by licensed acupuncturists is approved in many countries. Certainly, it is ready to be prescribed by physicians when a pregnant patient is seeking NVP treatment.”
Dr. Stika stressed that these therapies are suited to milder NV, and would “barely take edge off severe symptoms,” for which a patient might have to “go up to a big gun like the antiemetic Zofran” (ondansetron). She is currently involved in a National Institutes of Health–funded clinical trial of the antidepressant mirtazapine (Remeron) for NVP.
Matthew Carroll, MD, a professor of obstetrics and gynecology at Baylor College of Medicine and Texas Children’s Hospital in Houston, noted that doxylamine-pyridoxine is already an effective treatment for NVP, but in his experience it is often "not enough" to help patients deal with symptoms.
"Many patients are hesitant to take additional medications," he said. "If acupuncture can be safely done in pregnancy, then it seems a reasonable option as an adjuvant treatment for NVP. I think there is a cohort of pregnant people in the US who would be excited to try a complementary and nonpharmaceutical treatment option. Unfortunately, complementary therapies are rarely evaluated at a systems level for safety and so they are hard to recommend for obstetricians in the US," he added.
Dr. Carroll, who was not involved in the study. noted that "studies like this can help us counsel patients who may be seeking these treatments even if not approved or recommended by ACOG."
This study was funded by the National Key R&D Program of China and the Project of Heilongjiang Province “TouYan” Innovation Team. Support also came from the National Clinical Research Base of Chinese Medicine, the Heilongjiang Provincial Clinical Research Centre for Ovary Diseases, and the 2023 Capability Improvement Project for Evidence-based Assessment of Traditional Chinese Medicine.
Study coauthor Ben Willem J. Mol, MD, PhD, reported consulting fees from ObsEva and Merck and travel fees from Merck.
Dr. Stika and Dr. Carroll had no competing interests to disclose.
. While the benefit of either agent was clinically small for moderate to severe symptoms, the combination showed numerically larger and potentially more meaningful benefit, according to a team led by Xiao-Ke Wu, MD, PhD, of the department of obstetrics and gynecology at First Affiliated Hospital, Heilongjiang University of Chinese Medicine, and Heilongjiang Provincial Hospital in Harbin, China.
The treatments found small reductions in symptoms of less than one point to 1.6 points on an emesis scale. Nevertheless, Dr. Wu’s group wrote online June 19 in Annals of Internal Medicine that the finding “is especially significant because there is a pressing need to establish a pregnancy-safe treatment regimen and an integrative guideline for managing severe NVP.”
NVP affects as many as 85% of pregnant women, 80%-90% of whom have only mild symptoms, the authors noted. However, severe NVP and hyperemesis gravidarum, or HG, develop in about 10%. “Unfortunately, as many as 10% of wanted pregnancies with severe NVP or HG are terminated because of intolerable and untreatable symptoms and complications,” Dr. Wu told this news organization. And antiemetics may be underprescribed by general practitioners because of concerns about potential teratogenic effects, he said.
“Our findings suggest that either acupuncture or doxylamine-pyridoxine alone is a suitable for treating moderate to severe NVP, and a combination of both can be used to treat severe NVP and HG,” Dr. Wu said.
Commenting on the study but not involved in it, Catherine S. Stika, MD, a clinical professor of ob.gyn. at Northwestern University in Chicago, said the results suggest these two therapies are more suited to mild than severe symptoms. “But an RCT is important to do in order to support the use of these therapies since they’re not as widely accepted as they ought to be,” she said in an interview.
According to Dr. Stika, many pregnant women are reluctant to take drugs at all or participate in drug studies, “so the combination of nonpharmaceutical/pharmaceutical treatment might be a bit more appealing.” She noted that some women have such severe nausea they are literally starving and so weak they are bedridden or even hospitalized.
Both treatments have been recommended for some time, and the American College of Obstetricians and Gynecologists’ 2018 practice bulletin recommends acupuncture for mild nausea.
Design
The randomized, double-blind, placebo-controled 2x2 factorial trial was conducted at 13 tertiary-care hospitals in mainland China from June 2020 to February 2022. The researchers recruited 352 women in early pregnancy with moderate to severe NVP. The mean age of participants was about 29 years and the mean gestational age was about 9 weeks.
Participants were randomized into four 14-day treatment groups: active acupuncture for 30 minutes a day plus the antihistamine-vitamin B6 agent doxylamine-pyridoxine; sham acupuncture for 30 minutes daily plus doxylamine-pyridoxine; active acupuncture plus placebo; and sham acupuncture plus placebo.
The primary outcome was the reduction in Pregnancy-Unique Quantification of Emesis (PUQE) score at day 15 relative to baseline with a score of less than 6 indicating mild NVP, 6-12 indicating moderate NVP, and 13 or higher indicating severe NVP. Secondary outcomes ranged from quality of life and adverse events to maternal and perinatal complications. Acupuncture and combined treatment yielded larger though still small reductions in PUQE score, compared with control treatments. The mean differences were as follows: acupuncture, –.07; 95% confidence interval, 1.3-0.1); doxylamine-pyridoxine, –1.0: 95% CI, 1.6-0.4); combination of both, –1.6; 95% CI, 2.2-0.9). No significant interaction was detected between the interventions (P = .69).Compared with placebo treatments, pharmaceutical therapy resulted in more somnolence, while active acupuncture led to more frequent dyspnea, bruising, itching, and pain. A higher risk of babies born small for gestational age was observed in mothers who took doxylamine-pyridoxine versus placebo: odds ratio, 3.8; 95% CI, 1-14.1). Neither the placebo effects of the sham interventions nor the natural regression of symptoms experienced by many women were evaluated.
Suited to milder symptoms?
Dr. Stika called the study well-designed and well-written but cited several limitations, including the small cohort, the minor symptom improvement, and the lack of a comparator group receiving neither sham nor active treatment.
“Compared with sham combination treatments, the active combination arm was only about a point and a half better,” she said. “And would some women have got better over the 2 weeks anyway with no intervention at all? A large percentage of women with NVP do improve on their own.”
And in terms of acceptability to U.S. women, she cautioned, “The study cohort was entirely Chinese, and this is a population that already accepts acupuncture treatment.”
Countered Dr. Wu, “Medical care provided by licensed acupuncturists is approved in many countries. Certainly, it is ready to be prescribed by physicians when a pregnant patient is seeking NVP treatment.”
Dr. Stika stressed that these therapies are suited to milder NV, and would “barely take edge off severe symptoms,” for which a patient might have to “go up to a big gun like the antiemetic Zofran” (ondansetron). She is currently involved in a National Institutes of Health–funded clinical trial of the antidepressant mirtazapine (Remeron) for NVP.
Matthew Carroll, MD, a professor of obstetrics and gynecology at Baylor College of Medicine and Texas Children’s Hospital in Houston, noted that doxylamine-pyridoxine is already an effective treatment for NVP, but in his experience it is often "not enough" to help patients deal with symptoms.
"Many patients are hesitant to take additional medications," he said. "If acupuncture can be safely done in pregnancy, then it seems a reasonable option as an adjuvant treatment for NVP. I think there is a cohort of pregnant people in the US who would be excited to try a complementary and nonpharmaceutical treatment option. Unfortunately, complementary therapies are rarely evaluated at a systems level for safety and so they are hard to recommend for obstetricians in the US," he added.
Dr. Carroll, who was not involved in the study. noted that "studies like this can help us counsel patients who may be seeking these treatments even if not approved or recommended by ACOG."
This study was funded by the National Key R&D Program of China and the Project of Heilongjiang Province “TouYan” Innovation Team. Support also came from the National Clinical Research Base of Chinese Medicine, the Heilongjiang Provincial Clinical Research Centre for Ovary Diseases, and the 2023 Capability Improvement Project for Evidence-based Assessment of Traditional Chinese Medicine.
Study coauthor Ben Willem J. Mol, MD, PhD, reported consulting fees from ObsEva and Merck and travel fees from Merck.
Dr. Stika and Dr. Carroll had no competing interests to disclose.
FROM ANNALS OF INTERNAL MEDICINE
‘Deprescribing’: Should some older adults shed their meds?
Joanne Lynn, MD, has lost track of the number of times in her 40 years as a geriatrician she’s seen a new patient come to her office carrying a bucket full of prescription medications – many of which they don’t need.
Dr. Lynn, who is on the faculty of George Washington University,Washington, recalled one woman who unwittingly was taking two blood pressure medications with different names.
“The risks included all the side effects overdosing carries,” Dr. Lynn said, ranging from blurred vision and crankiness to organ failure and even death.
For doctors with patients who don’t know they’re taking too much of a medication, “you wonder whether the drug is causing the health problems, and it’s a symptom of the wrong medication,” rather than a symptom of an undiagnosed illness, she said.
Patients often assume their health providers check for drug interactions or assess if a medication is no longer needed, and will catch extra prescriptions. That could be a risky assumption. Some doctors may prescribe yet another prescription to manage the side effects of an unnecessary drug, instead of doing a medication review and potentially “deprescribing” or discontinuing, a treatment that’s no longer needed.
About 57% of people age 65 years or older take five or more medications regularly – a concept known as polypharmacy, a study published in 2020 in the Journal of the American Geriatrics Society shows. While doctors prescribe drugs to help patients manage various ailments, as a list of medications grows, so do potential complications.
An older adult might forget to tell their doctor what they’re taking, or maybe they don’t even know what they’re taking or why, Dr. Lynn said.
“In some cases, a doctor just added a drug to treat something, not realizing they were already taking something else for it,” she said. “Of course, the situation of whether these patients can even afford all these drugs matters a lot, too.”
Some older adults may pick and choose which medications to take based on cost, not knowing which prescriptions are necessary, Dr. Lynn said.
Finding the ‘right balance’
Indeed, if given the option, up to 80% of older adults ages 50-80 would be open to stopping one or more of their prescribed medications, according to a 2023 poll by researchers at the University of Michigan, Ann Arbor.
“A lot of drugs that people take might have been appropriate at one point, but might have outlived their usefulness for that individual,” said Michael Steinman, MD, a professor of medicine and a geriatrician at the University of California, San Francisco, and coprincipal investigator of the U.S. Deprescribing Research Network, a doctor group focused on improving medication use for older adults.
“Having fewer medications can actually be beneficial,” he said. “You can take too many medications; you can take too few. The optimal thing is finding what is the right balance for you.”
Defining how many medications is too many depends on each person, which is why caregivers and older adults can ask their doctor for a review of medications that have multiplied over time.
By reevaluating their medications, older adults can actually lower their chances of potentially harmful side effects, and avoid the spiral of being prescribed even more medications, said Sarah Vordenberg, PharmD, MPH, a clinical associate professor at the University of Michigan’s College of Pharmacy, Ann Arbor.
“It’s not really the number of medications, it’s [about] are they inappropriate or unnecessary medications for a patient,” she said.
Patients and caregivers can ask for an honest conversation with their doctor. The University of Michigan poll found that more than 90% of older adults who took prescription medications expected their health care provider to review their medicines during a regular visit.
But doctors often need prompting from patients to start a review.
“The clinical inertia, or maintaining the status quo, unfortunately is a lot of times easier than having time-intensive conversations,” Dr. Vordenberg said.
Ask questions
Sara Merwin spent many years helping manage her parents’ medical appointments and health as they transitioned from living independently in Colorado to a retirement community and finally a nursing home. Ms. Merwin, coauthor of “The Informed Patient,” said her father was taking a long list of medications, and she often asked his primary care doctor for a medication review.
“I felt that my father at his age and his frailty didn’t need as many meds as he was on,” said Ms. Merwin, who lives in Long Island, N.Y. “So we went over his meds, and I asked, ‘Does he really need to be on this?’ ‘Does he really need to be on that?’ ”
She questioned one medication in particular, a statin to lower his cholesterol and risk of a heart attack.
“I thought possibly the statin was causing some myalgia, some muscle aches in his legs, which is why I advocated for coming off it,” she said.
The primary care doctor discontinued the anticholesterol drug.
Local pharmacies can also serve as a starting point for older adults and caregivers, where a pharmacist can give them more information on whether a particular combination of the medications taken may be harmful. In states that allow for pharmacists to prescribe some medications, pharmacists may be able to consolidate some of the medications or advise that a patient stop taking one or more, Dr. Vordenberg said.
“All pharmacists have the training to do a comprehensive medication review,” she said. “All pharmacists have the ability to follow up with the patient to find out how the deprescribing is going.”
Ms. Merwin’s parents received their prescriptions from a “small mom-and-pop pharmacy, where they were on a first-name basis with the pharmacist who really looked out for them. So they had that expertise available to them,” she said.
With information in hand on potentially unnecessary medications, the work of shedding medications should be done along with health care providers, some of whom prescribed the medications in the first place.
Many older adults live in geographically isolated areas without pharmacies, or receive prescriptions from mail-order pharmacies. In this case, Medicare plans offer free medication reviews with a doctor or pharmacist – known as a medication therapy management program – and provide recommendations for taking each drug.
Ms. Merwin’s father died in early 2020. She sometimes questions whether he should have stayed on the statin for longer, or if the doctor agreed too quickly without doing more research. But overall, she doesn’t regret raising the question with his health care providers, and she advises other caregivers and older adults to pay attention to medication lists.
“It’s dangerous to be passive when it comes to one’s health care now,” Ms. Merwin said. “That’s a difficult message for older adults to hear because they have grown up with the primacy of the doctor and the authority of the doctor, as opposed to it being a collaborative relationship.”
A version of this article first appeared on WebMD.com.
Joanne Lynn, MD, has lost track of the number of times in her 40 years as a geriatrician she’s seen a new patient come to her office carrying a bucket full of prescription medications – many of which they don’t need.
Dr. Lynn, who is on the faculty of George Washington University,Washington, recalled one woman who unwittingly was taking two blood pressure medications with different names.
“The risks included all the side effects overdosing carries,” Dr. Lynn said, ranging from blurred vision and crankiness to organ failure and even death.
For doctors with patients who don’t know they’re taking too much of a medication, “you wonder whether the drug is causing the health problems, and it’s a symptom of the wrong medication,” rather than a symptom of an undiagnosed illness, she said.
Patients often assume their health providers check for drug interactions or assess if a medication is no longer needed, and will catch extra prescriptions. That could be a risky assumption. Some doctors may prescribe yet another prescription to manage the side effects of an unnecessary drug, instead of doing a medication review and potentially “deprescribing” or discontinuing, a treatment that’s no longer needed.
About 57% of people age 65 years or older take five or more medications regularly – a concept known as polypharmacy, a study published in 2020 in the Journal of the American Geriatrics Society shows. While doctors prescribe drugs to help patients manage various ailments, as a list of medications grows, so do potential complications.
An older adult might forget to tell their doctor what they’re taking, or maybe they don’t even know what they’re taking or why, Dr. Lynn said.
“In some cases, a doctor just added a drug to treat something, not realizing they were already taking something else for it,” she said. “Of course, the situation of whether these patients can even afford all these drugs matters a lot, too.”
Some older adults may pick and choose which medications to take based on cost, not knowing which prescriptions are necessary, Dr. Lynn said.
Finding the ‘right balance’
Indeed, if given the option, up to 80% of older adults ages 50-80 would be open to stopping one or more of their prescribed medications, according to a 2023 poll by researchers at the University of Michigan, Ann Arbor.
“A lot of drugs that people take might have been appropriate at one point, but might have outlived their usefulness for that individual,” said Michael Steinman, MD, a professor of medicine and a geriatrician at the University of California, San Francisco, and coprincipal investigator of the U.S. Deprescribing Research Network, a doctor group focused on improving medication use for older adults.
“Having fewer medications can actually be beneficial,” he said. “You can take too many medications; you can take too few. The optimal thing is finding what is the right balance for you.”
Defining how many medications is too many depends on each person, which is why caregivers and older adults can ask their doctor for a review of medications that have multiplied over time.
By reevaluating their medications, older adults can actually lower their chances of potentially harmful side effects, and avoid the spiral of being prescribed even more medications, said Sarah Vordenberg, PharmD, MPH, a clinical associate professor at the University of Michigan’s College of Pharmacy, Ann Arbor.
“It’s not really the number of medications, it’s [about] are they inappropriate or unnecessary medications for a patient,” she said.
Patients and caregivers can ask for an honest conversation with their doctor. The University of Michigan poll found that more than 90% of older adults who took prescription medications expected their health care provider to review their medicines during a regular visit.
But doctors often need prompting from patients to start a review.
“The clinical inertia, or maintaining the status quo, unfortunately is a lot of times easier than having time-intensive conversations,” Dr. Vordenberg said.
Ask questions
Sara Merwin spent many years helping manage her parents’ medical appointments and health as they transitioned from living independently in Colorado to a retirement community and finally a nursing home. Ms. Merwin, coauthor of “The Informed Patient,” said her father was taking a long list of medications, and she often asked his primary care doctor for a medication review.
“I felt that my father at his age and his frailty didn’t need as many meds as he was on,” said Ms. Merwin, who lives in Long Island, N.Y. “So we went over his meds, and I asked, ‘Does he really need to be on this?’ ‘Does he really need to be on that?’ ”
She questioned one medication in particular, a statin to lower his cholesterol and risk of a heart attack.
“I thought possibly the statin was causing some myalgia, some muscle aches in his legs, which is why I advocated for coming off it,” she said.
The primary care doctor discontinued the anticholesterol drug.
Local pharmacies can also serve as a starting point for older adults and caregivers, where a pharmacist can give them more information on whether a particular combination of the medications taken may be harmful. In states that allow for pharmacists to prescribe some medications, pharmacists may be able to consolidate some of the medications or advise that a patient stop taking one or more, Dr. Vordenberg said.
“All pharmacists have the training to do a comprehensive medication review,” she said. “All pharmacists have the ability to follow up with the patient to find out how the deprescribing is going.”
Ms. Merwin’s parents received their prescriptions from a “small mom-and-pop pharmacy, where they were on a first-name basis with the pharmacist who really looked out for them. So they had that expertise available to them,” she said.
With information in hand on potentially unnecessary medications, the work of shedding medications should be done along with health care providers, some of whom prescribed the medications in the first place.
Many older adults live in geographically isolated areas without pharmacies, or receive prescriptions from mail-order pharmacies. In this case, Medicare plans offer free medication reviews with a doctor or pharmacist – known as a medication therapy management program – and provide recommendations for taking each drug.
Ms. Merwin’s father died in early 2020. She sometimes questions whether he should have stayed on the statin for longer, or if the doctor agreed too quickly without doing more research. But overall, she doesn’t regret raising the question with his health care providers, and she advises other caregivers and older adults to pay attention to medication lists.
“It’s dangerous to be passive when it comes to one’s health care now,” Ms. Merwin said. “That’s a difficult message for older adults to hear because they have grown up with the primacy of the doctor and the authority of the doctor, as opposed to it being a collaborative relationship.”
A version of this article first appeared on WebMD.com.
Joanne Lynn, MD, has lost track of the number of times in her 40 years as a geriatrician she’s seen a new patient come to her office carrying a bucket full of prescription medications – many of which they don’t need.
Dr. Lynn, who is on the faculty of George Washington University,Washington, recalled one woman who unwittingly was taking two blood pressure medications with different names.
“The risks included all the side effects overdosing carries,” Dr. Lynn said, ranging from blurred vision and crankiness to organ failure and even death.
For doctors with patients who don’t know they’re taking too much of a medication, “you wonder whether the drug is causing the health problems, and it’s a symptom of the wrong medication,” rather than a symptom of an undiagnosed illness, she said.
Patients often assume their health providers check for drug interactions or assess if a medication is no longer needed, and will catch extra prescriptions. That could be a risky assumption. Some doctors may prescribe yet another prescription to manage the side effects of an unnecessary drug, instead of doing a medication review and potentially “deprescribing” or discontinuing, a treatment that’s no longer needed.
About 57% of people age 65 years or older take five or more medications regularly – a concept known as polypharmacy, a study published in 2020 in the Journal of the American Geriatrics Society shows. While doctors prescribe drugs to help patients manage various ailments, as a list of medications grows, so do potential complications.
An older adult might forget to tell their doctor what they’re taking, or maybe they don’t even know what they’re taking or why, Dr. Lynn said.
“In some cases, a doctor just added a drug to treat something, not realizing they were already taking something else for it,” she said. “Of course, the situation of whether these patients can even afford all these drugs matters a lot, too.”
Some older adults may pick and choose which medications to take based on cost, not knowing which prescriptions are necessary, Dr. Lynn said.
Finding the ‘right balance’
Indeed, if given the option, up to 80% of older adults ages 50-80 would be open to stopping one or more of their prescribed medications, according to a 2023 poll by researchers at the University of Michigan, Ann Arbor.
“A lot of drugs that people take might have been appropriate at one point, but might have outlived their usefulness for that individual,” said Michael Steinman, MD, a professor of medicine and a geriatrician at the University of California, San Francisco, and coprincipal investigator of the U.S. Deprescribing Research Network, a doctor group focused on improving medication use for older adults.
“Having fewer medications can actually be beneficial,” he said. “You can take too many medications; you can take too few. The optimal thing is finding what is the right balance for you.”
Defining how many medications is too many depends on each person, which is why caregivers and older adults can ask their doctor for a review of medications that have multiplied over time.
By reevaluating their medications, older adults can actually lower their chances of potentially harmful side effects, and avoid the spiral of being prescribed even more medications, said Sarah Vordenberg, PharmD, MPH, a clinical associate professor at the University of Michigan’s College of Pharmacy, Ann Arbor.
“It’s not really the number of medications, it’s [about] are they inappropriate or unnecessary medications for a patient,” she said.
Patients and caregivers can ask for an honest conversation with their doctor. The University of Michigan poll found that more than 90% of older adults who took prescription medications expected their health care provider to review their medicines during a regular visit.
But doctors often need prompting from patients to start a review.
“The clinical inertia, or maintaining the status quo, unfortunately is a lot of times easier than having time-intensive conversations,” Dr. Vordenberg said.
Ask questions
Sara Merwin spent many years helping manage her parents’ medical appointments and health as they transitioned from living independently in Colorado to a retirement community and finally a nursing home. Ms. Merwin, coauthor of “The Informed Patient,” said her father was taking a long list of medications, and she often asked his primary care doctor for a medication review.
“I felt that my father at his age and his frailty didn’t need as many meds as he was on,” said Ms. Merwin, who lives in Long Island, N.Y. “So we went over his meds, and I asked, ‘Does he really need to be on this?’ ‘Does he really need to be on that?’ ”
She questioned one medication in particular, a statin to lower his cholesterol and risk of a heart attack.
“I thought possibly the statin was causing some myalgia, some muscle aches in his legs, which is why I advocated for coming off it,” she said.
The primary care doctor discontinued the anticholesterol drug.
Local pharmacies can also serve as a starting point for older adults and caregivers, where a pharmacist can give them more information on whether a particular combination of the medications taken may be harmful. In states that allow for pharmacists to prescribe some medications, pharmacists may be able to consolidate some of the medications or advise that a patient stop taking one or more, Dr. Vordenberg said.
“All pharmacists have the training to do a comprehensive medication review,” she said. “All pharmacists have the ability to follow up with the patient to find out how the deprescribing is going.”
Ms. Merwin’s parents received their prescriptions from a “small mom-and-pop pharmacy, where they were on a first-name basis with the pharmacist who really looked out for them. So they had that expertise available to them,” she said.
With information in hand on potentially unnecessary medications, the work of shedding medications should be done along with health care providers, some of whom prescribed the medications in the first place.
Many older adults live in geographically isolated areas without pharmacies, or receive prescriptions from mail-order pharmacies. In this case, Medicare plans offer free medication reviews with a doctor or pharmacist – known as a medication therapy management program – and provide recommendations for taking each drug.
Ms. Merwin’s father died in early 2020. She sometimes questions whether he should have stayed on the statin for longer, or if the doctor agreed too quickly without doing more research. But overall, she doesn’t regret raising the question with his health care providers, and she advises other caregivers and older adults to pay attention to medication lists.
“It’s dangerous to be passive when it comes to one’s health care now,” Ms. Merwin said. “That’s a difficult message for older adults to hear because they have grown up with the primacy of the doctor and the authority of the doctor, as opposed to it being a collaborative relationship.”
A version of this article first appeared on WebMD.com.
New study backs up capecitabine dosing practice in metastatic BC
Both progression-free survival (PFS) and overall survival (OS) were similar between the two groups, but patients on the alternative schedule experienced fewer cases of hand-foot syndrome (HFS), diarrhea, and stomatitis, and also had fewer discontinuations and dose modifications.
The Food and Drug Administration–approved dose of capecitabine is 1,250 mg/m2, but 14 days of treatment can lead to significant toxicity, said Qamar Khan, MD, during a presentation of the study at the annual meeting of the American Society of Clinical Oncology. “Mathematical models applied to xenograft [animal model] data suggest that the maximum cytotoxic effect of capecitabine occurs after about 7 days of treatment, beyond which time only toxicity increases,” Dr. Khan said during his talk on the randomized control trial.
The researchers randomized 153 patients to receive a fixed 1,500-mg capecitabine dose twice per day on a 7-day-on, 7-day-off schedule (7/7), or the 1,250–mg/m2 dose twice per day for 14 days followed by 7 days off (14/7). The median age was 60 years, and 85.6% were White, 8.5% were African American, 3.3% were Hispanic, 0.7% were American Indian or Alaskan Native, and 2.0% were other. With respect to disease characteristics, 44% had visceral metastasis, 78% were hormone receptor positive/HER2 negative, and 11% had triple-negative breast cancer. About two-thirds (65%) had received no prior chemotherapy.
Restricted mean survival time (RMST) at 36 months for PFS was 13.9 months in the 7/7 group and 14.6 months in the 14/7 group (difference, 0.7 months; 95.5% CI, –3.14 to 4.57 months). The objective response rate was 8.9% in the 7/7 group and 19.6% in the 14/7 group (P = .11). Median OS was 19.8 months in the 7/7 group and 17.5 months in the 14/7 group (hazard ratio, 0.76; P = .17). The RMST at 47 months for OS was 24.5 months in the 7/7 group and 20.9 months in the 14/7 group (difference, –3.6 months; 95% CI, –8.89 to 1.54 months).
The researchers found no differences in subgroup analyses by visceral metastasis, breast cancer subtype, or number of lines of previous therapy.
The toxicity profile of 7/7 was better with respect to grade 2-4 diarrhea (2.5% vs. 20.5%, P = .0008), grade 2-4 HFS (3.8% vs. 15.1%; P = .0019), and grade 2-4 mucositis (0% vs. 5.5%; P =.0001).
Findings back up clinical practice
“The fixed-dose capecitabine dosing is something that’s been done a lot in practice, because a lot of practitioners recognize that giving the drug for two weeks in a row with a week break is overly toxic, so it’s something we’ve been doing in the community for quite a while,” said Michael Danso, MD, who comoderated the session.
Still, the safety and efficacy data back up that general clinical practice. “There was a randomized trial and colon cancer that didn’t show [equivalent outcomes with the alternate dosing schedule]. So to see that it’s safe and effective in breast cancer is an important [finding],” said Dr. Danso, who is the Research Director at Virginia Oncology Associates, Norfolk.
During the question-and-answer following the talk, Jeffrey Kirshner, MD, a medical oncologist at Hematology-Oncology Associates of Central New York, East Syracuse, noted that his practice has used a similar schedule for years. “I really commend you for doing that study. It really supports what many of us in the real world have been doing for many years. We figured this out empirically, both upfront and when patients can’t tolerate [the 14/7 schedule].”
Fixed dose versus body surface area
Dr. Kirshner also said his practice uses a dose of 1 g/m2 of body surface area on a 7/7 schedule rather than a fixed dose as was done in Dr. Khan’s study. “If you use the higher dose, you might have seen a higher response rate because many of our patients, as you know, have a body surface [BSA] area much greater than 1.5 g/m2.”
Dr. Khan responded that there is little data available on BSA dosing. “We selected 1,500 mg because a lot of people are practicing that, and for convenience, and that most patients who started at a higher dose eventually wound up on a dose of 1,500 mg twice daily.”
Dr. Kirshner also pointed out that the study was conducted in a population with metastatic disease. “I think we need to emphasize that we do not use the 7/7 regimen in a potentially curative setting, such as the CREATE-X regimen for triple-negative [breast cancer].”
Dr. Khan agreed. “I would use the same dose as the CREATE-X trial in the adjuvant setting,” he responded.
Dr. Danso has received honoraria from Amgen and has consulted or advised Immunomedics, Novartis, Pfizer, and Seagen. Dr. Khan and Dr. Kirshner have no relevant financial disclosures.
Both progression-free survival (PFS) and overall survival (OS) were similar between the two groups, but patients on the alternative schedule experienced fewer cases of hand-foot syndrome (HFS), diarrhea, and stomatitis, and also had fewer discontinuations and dose modifications.
The Food and Drug Administration–approved dose of capecitabine is 1,250 mg/m2, but 14 days of treatment can lead to significant toxicity, said Qamar Khan, MD, during a presentation of the study at the annual meeting of the American Society of Clinical Oncology. “Mathematical models applied to xenograft [animal model] data suggest that the maximum cytotoxic effect of capecitabine occurs after about 7 days of treatment, beyond which time only toxicity increases,” Dr. Khan said during his talk on the randomized control trial.
The researchers randomized 153 patients to receive a fixed 1,500-mg capecitabine dose twice per day on a 7-day-on, 7-day-off schedule (7/7), or the 1,250–mg/m2 dose twice per day for 14 days followed by 7 days off (14/7). The median age was 60 years, and 85.6% were White, 8.5% were African American, 3.3% were Hispanic, 0.7% were American Indian or Alaskan Native, and 2.0% were other. With respect to disease characteristics, 44% had visceral metastasis, 78% were hormone receptor positive/HER2 negative, and 11% had triple-negative breast cancer. About two-thirds (65%) had received no prior chemotherapy.
Restricted mean survival time (RMST) at 36 months for PFS was 13.9 months in the 7/7 group and 14.6 months in the 14/7 group (difference, 0.7 months; 95.5% CI, –3.14 to 4.57 months). The objective response rate was 8.9% in the 7/7 group and 19.6% in the 14/7 group (P = .11). Median OS was 19.8 months in the 7/7 group and 17.5 months in the 14/7 group (hazard ratio, 0.76; P = .17). The RMST at 47 months for OS was 24.5 months in the 7/7 group and 20.9 months in the 14/7 group (difference, –3.6 months; 95% CI, –8.89 to 1.54 months).
The researchers found no differences in subgroup analyses by visceral metastasis, breast cancer subtype, or number of lines of previous therapy.
The toxicity profile of 7/7 was better with respect to grade 2-4 diarrhea (2.5% vs. 20.5%, P = .0008), grade 2-4 HFS (3.8% vs. 15.1%; P = .0019), and grade 2-4 mucositis (0% vs. 5.5%; P =.0001).
Findings back up clinical practice
“The fixed-dose capecitabine dosing is something that’s been done a lot in practice, because a lot of practitioners recognize that giving the drug for two weeks in a row with a week break is overly toxic, so it’s something we’ve been doing in the community for quite a while,” said Michael Danso, MD, who comoderated the session.
Still, the safety and efficacy data back up that general clinical practice. “There was a randomized trial and colon cancer that didn’t show [equivalent outcomes with the alternate dosing schedule]. So to see that it’s safe and effective in breast cancer is an important [finding],” said Dr. Danso, who is the Research Director at Virginia Oncology Associates, Norfolk.
During the question-and-answer following the talk, Jeffrey Kirshner, MD, a medical oncologist at Hematology-Oncology Associates of Central New York, East Syracuse, noted that his practice has used a similar schedule for years. “I really commend you for doing that study. It really supports what many of us in the real world have been doing for many years. We figured this out empirically, both upfront and when patients can’t tolerate [the 14/7 schedule].”
Fixed dose versus body surface area
Dr. Kirshner also said his practice uses a dose of 1 g/m2 of body surface area on a 7/7 schedule rather than a fixed dose as was done in Dr. Khan’s study. “If you use the higher dose, you might have seen a higher response rate because many of our patients, as you know, have a body surface [BSA] area much greater than 1.5 g/m2.”
Dr. Khan responded that there is little data available on BSA dosing. “We selected 1,500 mg because a lot of people are practicing that, and for convenience, and that most patients who started at a higher dose eventually wound up on a dose of 1,500 mg twice daily.”
Dr. Kirshner also pointed out that the study was conducted in a population with metastatic disease. “I think we need to emphasize that we do not use the 7/7 regimen in a potentially curative setting, such as the CREATE-X regimen for triple-negative [breast cancer].”
Dr. Khan agreed. “I would use the same dose as the CREATE-X trial in the adjuvant setting,” he responded.
Dr. Danso has received honoraria from Amgen and has consulted or advised Immunomedics, Novartis, Pfizer, and Seagen. Dr. Khan and Dr. Kirshner have no relevant financial disclosures.
Both progression-free survival (PFS) and overall survival (OS) were similar between the two groups, but patients on the alternative schedule experienced fewer cases of hand-foot syndrome (HFS), diarrhea, and stomatitis, and also had fewer discontinuations and dose modifications.
The Food and Drug Administration–approved dose of capecitabine is 1,250 mg/m2, but 14 days of treatment can lead to significant toxicity, said Qamar Khan, MD, during a presentation of the study at the annual meeting of the American Society of Clinical Oncology. “Mathematical models applied to xenograft [animal model] data suggest that the maximum cytotoxic effect of capecitabine occurs after about 7 days of treatment, beyond which time only toxicity increases,” Dr. Khan said during his talk on the randomized control trial.
The researchers randomized 153 patients to receive a fixed 1,500-mg capecitabine dose twice per day on a 7-day-on, 7-day-off schedule (7/7), or the 1,250–mg/m2 dose twice per day for 14 days followed by 7 days off (14/7). The median age was 60 years, and 85.6% were White, 8.5% were African American, 3.3% were Hispanic, 0.7% were American Indian or Alaskan Native, and 2.0% were other. With respect to disease characteristics, 44% had visceral metastasis, 78% were hormone receptor positive/HER2 negative, and 11% had triple-negative breast cancer. About two-thirds (65%) had received no prior chemotherapy.
Restricted mean survival time (RMST) at 36 months for PFS was 13.9 months in the 7/7 group and 14.6 months in the 14/7 group (difference, 0.7 months; 95.5% CI, –3.14 to 4.57 months). The objective response rate was 8.9% in the 7/7 group and 19.6% in the 14/7 group (P = .11). Median OS was 19.8 months in the 7/7 group and 17.5 months in the 14/7 group (hazard ratio, 0.76; P = .17). The RMST at 47 months for OS was 24.5 months in the 7/7 group and 20.9 months in the 14/7 group (difference, –3.6 months; 95% CI, –8.89 to 1.54 months).
The researchers found no differences in subgroup analyses by visceral metastasis, breast cancer subtype, or number of lines of previous therapy.
The toxicity profile of 7/7 was better with respect to grade 2-4 diarrhea (2.5% vs. 20.5%, P = .0008), grade 2-4 HFS (3.8% vs. 15.1%; P = .0019), and grade 2-4 mucositis (0% vs. 5.5%; P =.0001).
Findings back up clinical practice
“The fixed-dose capecitabine dosing is something that’s been done a lot in practice, because a lot of practitioners recognize that giving the drug for two weeks in a row with a week break is overly toxic, so it’s something we’ve been doing in the community for quite a while,” said Michael Danso, MD, who comoderated the session.
Still, the safety and efficacy data back up that general clinical practice. “There was a randomized trial and colon cancer that didn’t show [equivalent outcomes with the alternate dosing schedule]. So to see that it’s safe and effective in breast cancer is an important [finding],” said Dr. Danso, who is the Research Director at Virginia Oncology Associates, Norfolk.
During the question-and-answer following the talk, Jeffrey Kirshner, MD, a medical oncologist at Hematology-Oncology Associates of Central New York, East Syracuse, noted that his practice has used a similar schedule for years. “I really commend you for doing that study. It really supports what many of us in the real world have been doing for many years. We figured this out empirically, both upfront and when patients can’t tolerate [the 14/7 schedule].”
Fixed dose versus body surface area
Dr. Kirshner also said his practice uses a dose of 1 g/m2 of body surface area on a 7/7 schedule rather than a fixed dose as was done in Dr. Khan’s study. “If you use the higher dose, you might have seen a higher response rate because many of our patients, as you know, have a body surface [BSA] area much greater than 1.5 g/m2.”
Dr. Khan responded that there is little data available on BSA dosing. “We selected 1,500 mg because a lot of people are practicing that, and for convenience, and that most patients who started at a higher dose eventually wound up on a dose of 1,500 mg twice daily.”
Dr. Kirshner also pointed out that the study was conducted in a population with metastatic disease. “I think we need to emphasize that we do not use the 7/7 regimen in a potentially curative setting, such as the CREATE-X regimen for triple-negative [breast cancer].”
Dr. Khan agreed. “I would use the same dose as the CREATE-X trial in the adjuvant setting,” he responded.
Dr. Danso has received honoraria from Amgen and has consulted or advised Immunomedics, Novartis, Pfizer, and Seagen. Dr. Khan and Dr. Kirshner have no relevant financial disclosures.
FROM ASCO 2023