User login
Translucent Periorbital Papules
The Diagnosis: Apocrine Hidrocystoma
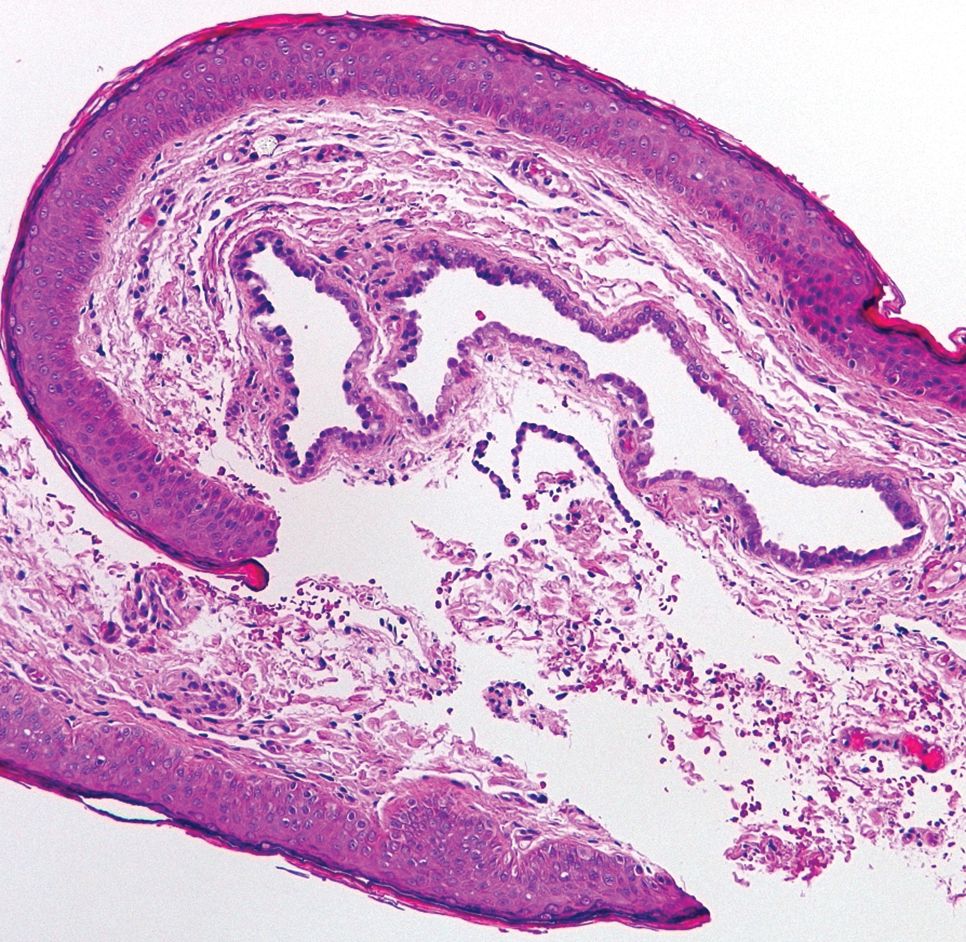
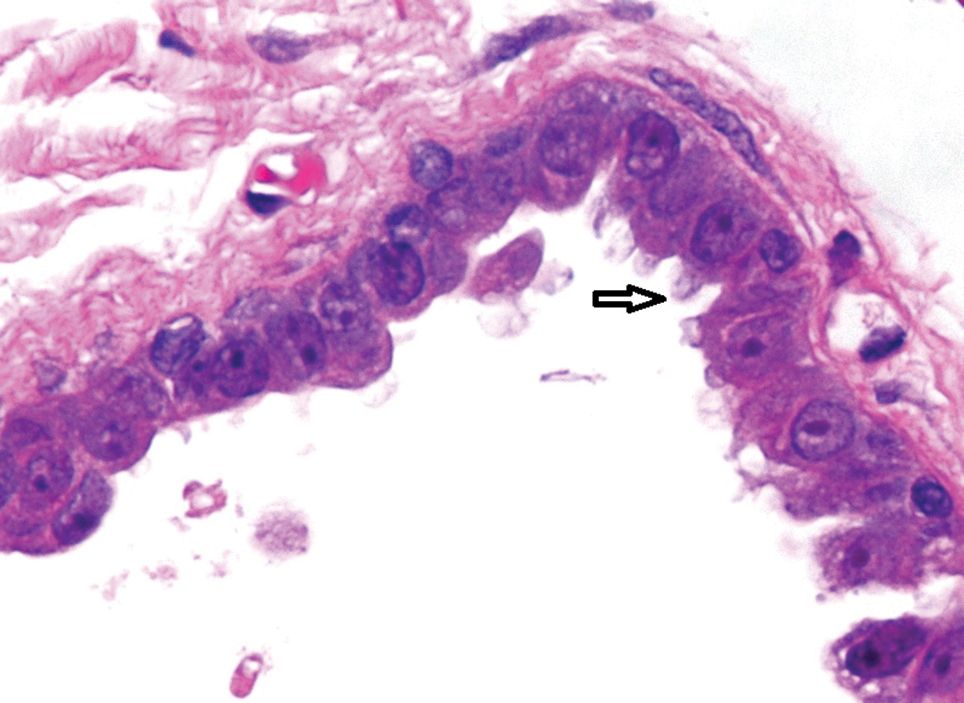
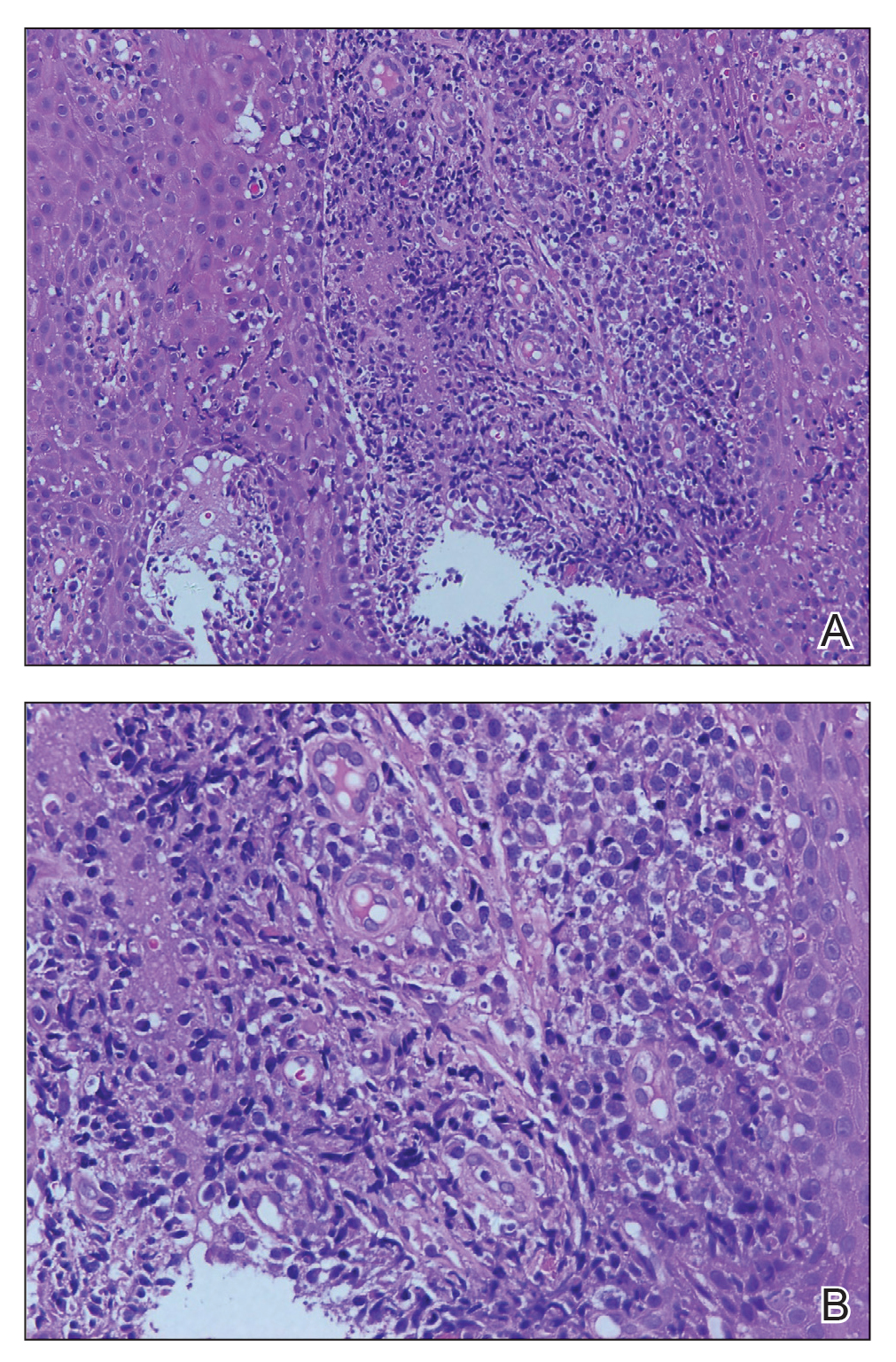
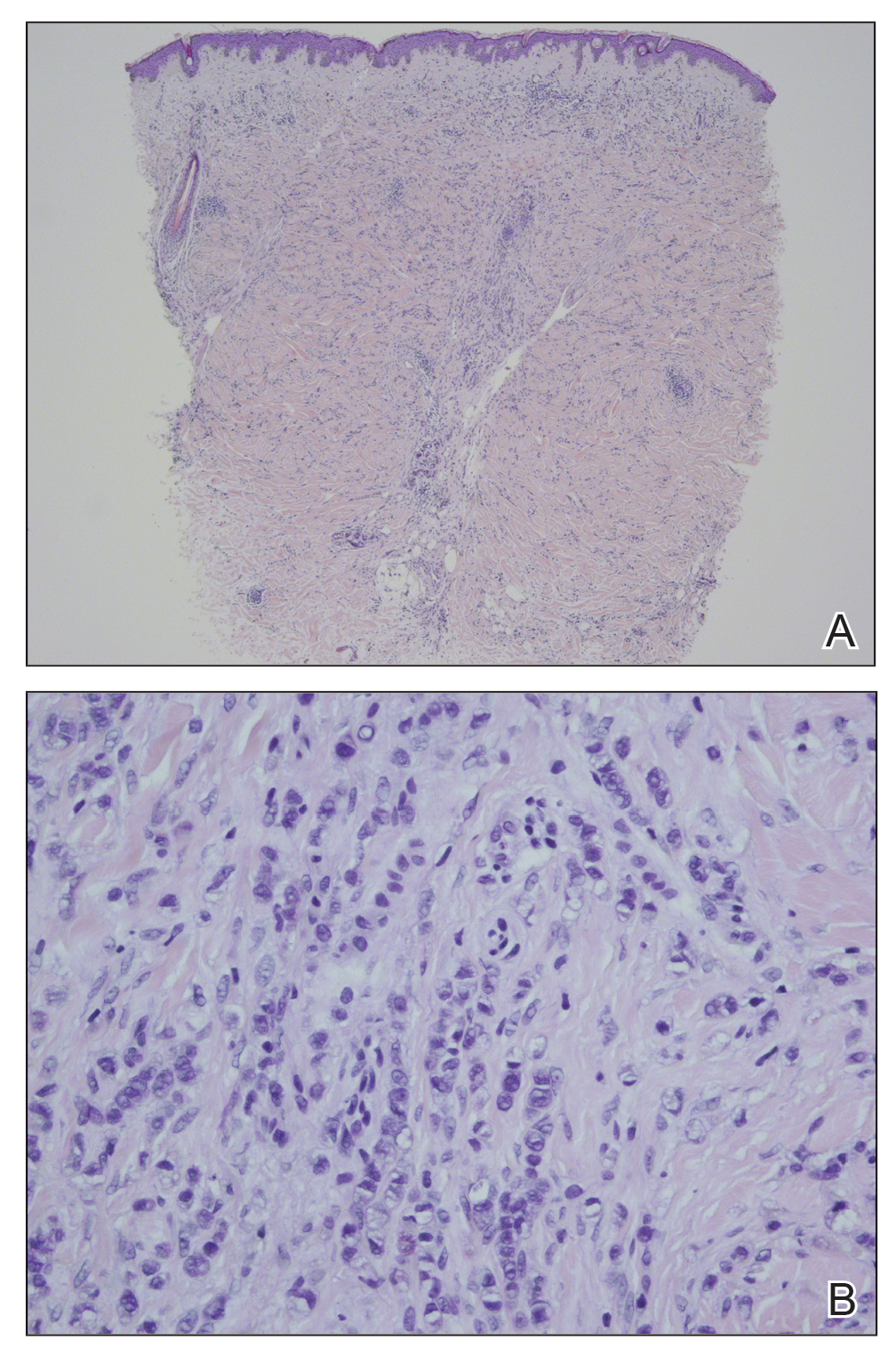
Histopathologic examination of one of the papules revealed cystic cavities located within the dermis (Figure 1) lined by a cuboidal epithelium demonstrating decapitation secretion (Figure 2), confirming the diagnosis of apocrine hidrocystomas. The presence of multiple lesions prompted further examination for an underlying genetic disorder; however, the patient's hair, nails, and teeth were normal. There also was no evidence of palmoplantar keratoderma or blaschkoid dermatosis.
Hidrocystomas are benign cysts of the sudoriferous apparatus that can be subdivided based on histogenesis (apocrine vs eccrine) or lesion count (single vs multiple).1 Multiple lesions may be associated with disorders of ectodermal dysplasia, including Goltz syndrome and Schopf-Schulz-Passarge syndrome. Apocrine hidrocystomas tend to present as solitary, translucent, flesh-colored to bluish facial papules, and the occurrence of multiple lesions is rare in contrast to its eccrine counterpart.2 Various extrafacial sites have been described including the trunk, axillae, umbilicus, genitalia, and digits.3 Apocrine hidrocystomas do not demonstrate aggravation with exposure to heat, unlike their eccrine counterparts.2
A review of 107 patients with 215 histologically proven hidrocystomas demonstrated a preponderance for women in their mid 50s; 74.8% of patients had unilateral disease, and 69.8% of all lesions affected either the lower eyelid or lateral canthus. Recurrence following conventional surgical excision was observed in 2.3% of lesions.1
A review from Japan recounted an incidence of 5 cases per year from 1999 to 2003.4 Patients ranged in age from 30 to 70 years, but there was no gender predilection. Individual apocrine hidrocystomas were mostly less than 2 cm and varied from flesh colored to light red, brown, blue, or purple; 61% of lesions arose periorbitally. Within their cohort, patients with multiple lesions were uncommon, with only 2 cases presenting with 2 lesions simultaneously.4
Apocrine hidrocystomas are thought to result from a cystic proliferation of the secretory component of apocrine sweat glands, though the exact pathogenesis still is unclear.3 Histologic features include a unilocular or multilocular cystic cavity within the dermis lined by columnar cells demonstrating decapitation secretion, followed by a peripheral rim of flattened myoepithelial cells.
Treatment of apocrine hidrocystomas includes topical anticholinergics, surgical excision, electrodesiccation, 1450-nm diode or CO2 lasers, and trichloroacetic acid.2 The novel use of cryotherapy,5 botulinum toxin,2 and intralesional injections of 50% glucose (as a sclerosant)6 also have been reported. Caution should be exercised when managing digital lesions, as digital papillary carcinoma has been described as a clinical and histopathologic mimicker.7
Lipoid proteinosis is a rare autosomal-recessive disorder. Cutaneous lesions manifest in 2 overlapping stages, typically within the first 2 years of life. The first stage consists of vesicles and hemorrhagic crusts on the face and extremities and intraorally, which may heal with scarring. In the second stage, the skin becomes diffusely thickened and waxy, with the appearance of papules, nodules, or plaques along the eyelid margins (moniliform blepharosis), face, axillae, or scrotum. Verrucous lesions also may develop on the knee or elbow extensors.8
Lymphangioma circumscriptum represents microcystic lymphatic malformations that can arise anywhere on the skin or oral mucosa. They present as clusters of clear or hemorrhagic vesicles of variable size and number favoring the proximal extremities and chest. Histologically, dilated lymphatic channels are seen in the upper dermis.8
Syringomas are common benign tumors of the sweat ducts characterized histologically by superficial dermal proliferations of small comma-shaped ducts set in a fibrotic stroma. Clinically, syringomas appear as small, firm, flesh-colored papules with a predilection for the periorbital area. An eruptive onset may be observed, most commonly affecting the trunk. Syringomas may be associated with Down syndrome, while the clear cell variant may be associated with diabetes mellitus.8
Primary systemic amyloidosis may present with a variety of systemic manifestations. Skin involvement can present as waxy, translucent, or purpuric papulonodules or plaques characteristically affecting the periorbital region. Other mucocutaneous signs include macroglossia with or without translucent to hemorrhagic papulovesicles; bruising, especially on the eyelids, neck, axillae, or anogenital area; vesiculobullous skin lesions; or diffuse cutaneous infiltration imparting a sclerodermoid appearance.8
- Maeng M, Petrakos P, Zhou M, et al. Bi-institutional retrospective study on the demographics and basic clinical presentation of hidrocystomas. Orbit. 2017;36:433-435.
- Bordelon JR, Tang N, Elston D, et al. Multiple apocrine hidrocystomas successfully treated with botulinum toxin A. Br J Dermatol. 2017;176:488-490.
- Hafsi W, Badri T. Apocrine hidrocystoma. StatPearls. Treasure Island, FL: StatPearls Publishing; 2017.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703.
- Panagiotopoulos A, Vasalou V, Sgontzou T, et al. Multiple apocrine hidrocystomas successfully treated with cryotherapy. Dermatol Surg. 2017;43:993-995.
- Osaki TH, Osaki MH, Osaki T, et al. A minimally invasive approach for apocrine hidrocystomas of the eyelid. Dermatol Surg. 2016;42:134-136.
- Molina-Ruiz AM, Llamas-Velasco M, Rütten A, et al. 'Apocrine hidrocystoma and cystadenoma'-like tumor of the digits or toes: a potential diagnostic pitfall of digital papillary adenocarcinoma. Am J Surg Pathol. 2016;40:410-418.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
The Diagnosis: Apocrine Hidrocystoma
Histopathologic examination of one of the papules revealed cystic cavities located within the dermis (Figure 1) lined by a cuboidal epithelium demonstrating decapitation secretion (Figure 2), confirming the diagnosis of apocrine hidrocystomas. The presence of multiple lesions prompted further examination for an underlying genetic disorder; however, the patient's hair, nails, and teeth were normal. There also was no evidence of palmoplantar keratoderma or blaschkoid dermatosis.
Hidrocystomas are benign cysts of the sudoriferous apparatus that can be subdivided based on histogenesis (apocrine vs eccrine) or lesion count (single vs multiple).1 Multiple lesions may be associated with disorders of ectodermal dysplasia, including Goltz syndrome and Schopf-Schulz-Passarge syndrome. Apocrine hidrocystomas tend to present as solitary, translucent, flesh-colored to bluish facial papules, and the occurrence of multiple lesions is rare in contrast to its eccrine counterpart.2 Various extrafacial sites have been described including the trunk, axillae, umbilicus, genitalia, and digits.3 Apocrine hidrocystomas do not demonstrate aggravation with exposure to heat, unlike their eccrine counterparts.2
A review of 107 patients with 215 histologically proven hidrocystomas demonstrated a preponderance for women in their mid 50s; 74.8% of patients had unilateral disease, and 69.8% of all lesions affected either the lower eyelid or lateral canthus. Recurrence following conventional surgical excision was observed in 2.3% of lesions.1
A review from Japan recounted an incidence of 5 cases per year from 1999 to 2003.4 Patients ranged in age from 30 to 70 years, but there was no gender predilection. Individual apocrine hidrocystomas were mostly less than 2 cm and varied from flesh colored to light red, brown, blue, or purple; 61% of lesions arose periorbitally. Within their cohort, patients with multiple lesions were uncommon, with only 2 cases presenting with 2 lesions simultaneously.4
Apocrine hidrocystomas are thought to result from a cystic proliferation of the secretory component of apocrine sweat glands, though the exact pathogenesis still is unclear.3 Histologic features include a unilocular or multilocular cystic cavity within the dermis lined by columnar cells demonstrating decapitation secretion, followed by a peripheral rim of flattened myoepithelial cells.
Treatment of apocrine hidrocystomas includes topical anticholinergics, surgical excision, electrodesiccation, 1450-nm diode or CO2 lasers, and trichloroacetic acid.2 The novel use of cryotherapy,5 botulinum toxin,2 and intralesional injections of 50% glucose (as a sclerosant)6 also have been reported. Caution should be exercised when managing digital lesions, as digital papillary carcinoma has been described as a clinical and histopathologic mimicker.7
Lipoid proteinosis is a rare autosomal-recessive disorder. Cutaneous lesions manifest in 2 overlapping stages, typically within the first 2 years of life. The first stage consists of vesicles and hemorrhagic crusts on the face and extremities and intraorally, which may heal with scarring. In the second stage, the skin becomes diffusely thickened and waxy, with the appearance of papules, nodules, or plaques along the eyelid margins (moniliform blepharosis), face, axillae, or scrotum. Verrucous lesions also may develop on the knee or elbow extensors.8
Lymphangioma circumscriptum represents microcystic lymphatic malformations that can arise anywhere on the skin or oral mucosa. They present as clusters of clear or hemorrhagic vesicles of variable size and number favoring the proximal extremities and chest. Histologically, dilated lymphatic channels are seen in the upper dermis.8
Syringomas are common benign tumors of the sweat ducts characterized histologically by superficial dermal proliferations of small comma-shaped ducts set in a fibrotic stroma. Clinically, syringomas appear as small, firm, flesh-colored papules with a predilection for the periorbital area. An eruptive onset may be observed, most commonly affecting the trunk. Syringomas may be associated with Down syndrome, while the clear cell variant may be associated with diabetes mellitus.8
Primary systemic amyloidosis may present with a variety of systemic manifestations. Skin involvement can present as waxy, translucent, or purpuric papulonodules or plaques characteristically affecting the periorbital region. Other mucocutaneous signs include macroglossia with or without translucent to hemorrhagic papulovesicles; bruising, especially on the eyelids, neck, axillae, or anogenital area; vesiculobullous skin lesions; or diffuse cutaneous infiltration imparting a sclerodermoid appearance.8
The Diagnosis: Apocrine Hidrocystoma
Histopathologic examination of one of the papules revealed cystic cavities located within the dermis (Figure 1) lined by a cuboidal epithelium demonstrating decapitation secretion (Figure 2), confirming the diagnosis of apocrine hidrocystomas. The presence of multiple lesions prompted further examination for an underlying genetic disorder; however, the patient's hair, nails, and teeth were normal. There also was no evidence of palmoplantar keratoderma or blaschkoid dermatosis.
Hidrocystomas are benign cysts of the sudoriferous apparatus that can be subdivided based on histogenesis (apocrine vs eccrine) or lesion count (single vs multiple).1 Multiple lesions may be associated with disorders of ectodermal dysplasia, including Goltz syndrome and Schopf-Schulz-Passarge syndrome. Apocrine hidrocystomas tend to present as solitary, translucent, flesh-colored to bluish facial papules, and the occurrence of multiple lesions is rare in contrast to its eccrine counterpart.2 Various extrafacial sites have been described including the trunk, axillae, umbilicus, genitalia, and digits.3 Apocrine hidrocystomas do not demonstrate aggravation with exposure to heat, unlike their eccrine counterparts.2
A review of 107 patients with 215 histologically proven hidrocystomas demonstrated a preponderance for women in their mid 50s; 74.8% of patients had unilateral disease, and 69.8% of all lesions affected either the lower eyelid or lateral canthus. Recurrence following conventional surgical excision was observed in 2.3% of lesions.1
A review from Japan recounted an incidence of 5 cases per year from 1999 to 2003.4 Patients ranged in age from 30 to 70 years, but there was no gender predilection. Individual apocrine hidrocystomas were mostly less than 2 cm and varied from flesh colored to light red, brown, blue, or purple; 61% of lesions arose periorbitally. Within their cohort, patients with multiple lesions were uncommon, with only 2 cases presenting with 2 lesions simultaneously.4
Apocrine hidrocystomas are thought to result from a cystic proliferation of the secretory component of apocrine sweat glands, though the exact pathogenesis still is unclear.3 Histologic features include a unilocular or multilocular cystic cavity within the dermis lined by columnar cells demonstrating decapitation secretion, followed by a peripheral rim of flattened myoepithelial cells.
Treatment of apocrine hidrocystomas includes topical anticholinergics, surgical excision, electrodesiccation, 1450-nm diode or CO2 lasers, and trichloroacetic acid.2 The novel use of cryotherapy,5 botulinum toxin,2 and intralesional injections of 50% glucose (as a sclerosant)6 also have been reported. Caution should be exercised when managing digital lesions, as digital papillary carcinoma has been described as a clinical and histopathologic mimicker.7
Lipoid proteinosis is a rare autosomal-recessive disorder. Cutaneous lesions manifest in 2 overlapping stages, typically within the first 2 years of life. The first stage consists of vesicles and hemorrhagic crusts on the face and extremities and intraorally, which may heal with scarring. In the second stage, the skin becomes diffusely thickened and waxy, with the appearance of papules, nodules, or plaques along the eyelid margins (moniliform blepharosis), face, axillae, or scrotum. Verrucous lesions also may develop on the knee or elbow extensors.8
Lymphangioma circumscriptum represents microcystic lymphatic malformations that can arise anywhere on the skin or oral mucosa. They present as clusters of clear or hemorrhagic vesicles of variable size and number favoring the proximal extremities and chest. Histologically, dilated lymphatic channels are seen in the upper dermis.8
Syringomas are common benign tumors of the sweat ducts characterized histologically by superficial dermal proliferations of small comma-shaped ducts set in a fibrotic stroma. Clinically, syringomas appear as small, firm, flesh-colored papules with a predilection for the periorbital area. An eruptive onset may be observed, most commonly affecting the trunk. Syringomas may be associated with Down syndrome, while the clear cell variant may be associated with diabetes mellitus.8
Primary systemic amyloidosis may present with a variety of systemic manifestations. Skin involvement can present as waxy, translucent, or purpuric papulonodules or plaques characteristically affecting the periorbital region. Other mucocutaneous signs include macroglossia with or without translucent to hemorrhagic papulovesicles; bruising, especially on the eyelids, neck, axillae, or anogenital area; vesiculobullous skin lesions; or diffuse cutaneous infiltration imparting a sclerodermoid appearance.8
- Maeng M, Petrakos P, Zhou M, et al. Bi-institutional retrospective study on the demographics and basic clinical presentation of hidrocystomas. Orbit. 2017;36:433-435.
- Bordelon JR, Tang N, Elston D, et al. Multiple apocrine hidrocystomas successfully treated with botulinum toxin A. Br J Dermatol. 2017;176:488-490.
- Hafsi W, Badri T. Apocrine hidrocystoma. StatPearls. Treasure Island, FL: StatPearls Publishing; 2017.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703.
- Panagiotopoulos A, Vasalou V, Sgontzou T, et al. Multiple apocrine hidrocystomas successfully treated with cryotherapy. Dermatol Surg. 2017;43:993-995.
- Osaki TH, Osaki MH, Osaki T, et al. A minimally invasive approach for apocrine hidrocystomas of the eyelid. Dermatol Surg. 2016;42:134-136.
- Molina-Ruiz AM, Llamas-Velasco M, Rütten A, et al. 'Apocrine hidrocystoma and cystadenoma'-like tumor of the digits or toes: a potential diagnostic pitfall of digital papillary adenocarcinoma. Am J Surg Pathol. 2016;40:410-418.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
- Maeng M, Petrakos P, Zhou M, et al. Bi-institutional retrospective study on the demographics and basic clinical presentation of hidrocystomas. Orbit. 2017;36:433-435.
- Bordelon JR, Tang N, Elston D, et al. Multiple apocrine hidrocystomas successfully treated with botulinum toxin A. Br J Dermatol. 2017;176:488-490.
- Hafsi W, Badri T. Apocrine hidrocystoma. StatPearls. Treasure Island, FL: StatPearls Publishing; 2017.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703.
- Panagiotopoulos A, Vasalou V, Sgontzou T, et al. Multiple apocrine hidrocystomas successfully treated with cryotherapy. Dermatol Surg. 2017;43:993-995.
- Osaki TH, Osaki MH, Osaki T, et al. A minimally invasive approach for apocrine hidrocystomas of the eyelid. Dermatol Surg. 2016;42:134-136.
- Molina-Ruiz AM, Llamas-Velasco M, Rütten A, et al. 'Apocrine hidrocystoma and cystadenoma'-like tumor of the digits or toes: a potential diagnostic pitfall of digital papillary adenocarcinoma. Am J Surg Pathol. 2016;40:410-418.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
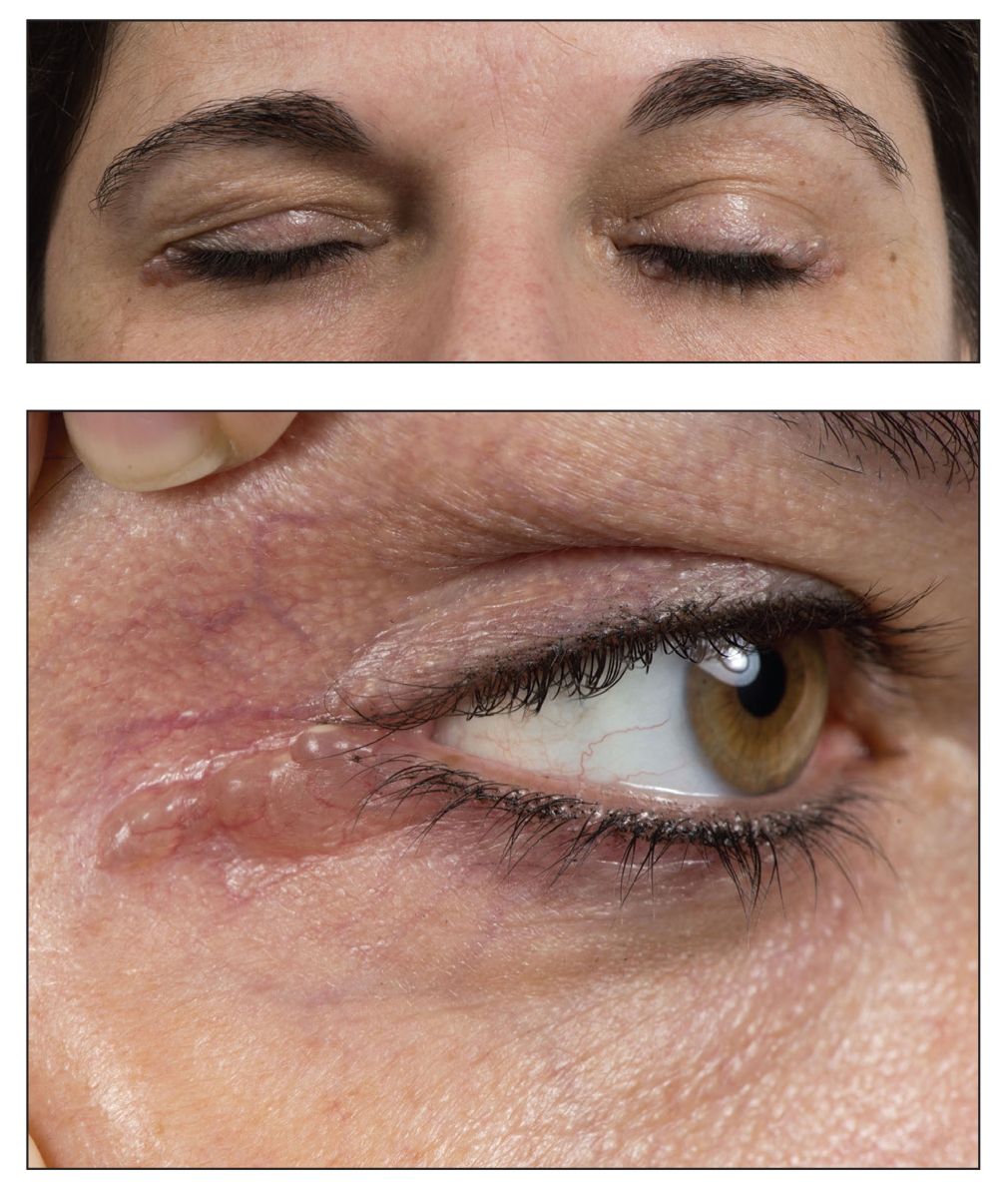
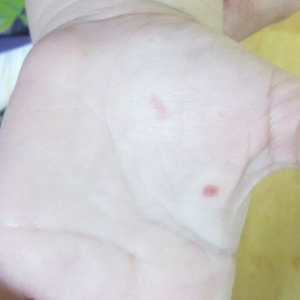
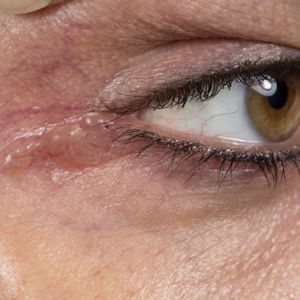
A 40-year-old woman was referred to dermatology for evaluation of occasionally pruritic periorbital papules that had gradually increased in size and number over the last 7 to 8 months (top). She had a similar solitary lesion on the left lower eyelid that was removed twice: 10 years and 10 months prior. She was taking an oral contraceptive (desogestrel) but otherwise had no notable medical history or drug allergies. Physical examination revealed individual and clustered translucent papules along the eyelid margins, left medial canthus, and both lateral canthi (bottom).
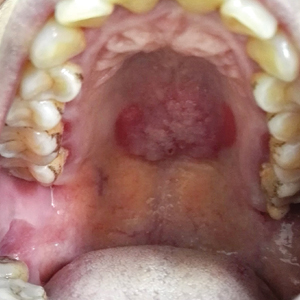
Solitary Warty Mucosal Lesion on the Hard Palate
The Diagnosis: Solitary Oral Condyloma Lata of Secondary Syphilis
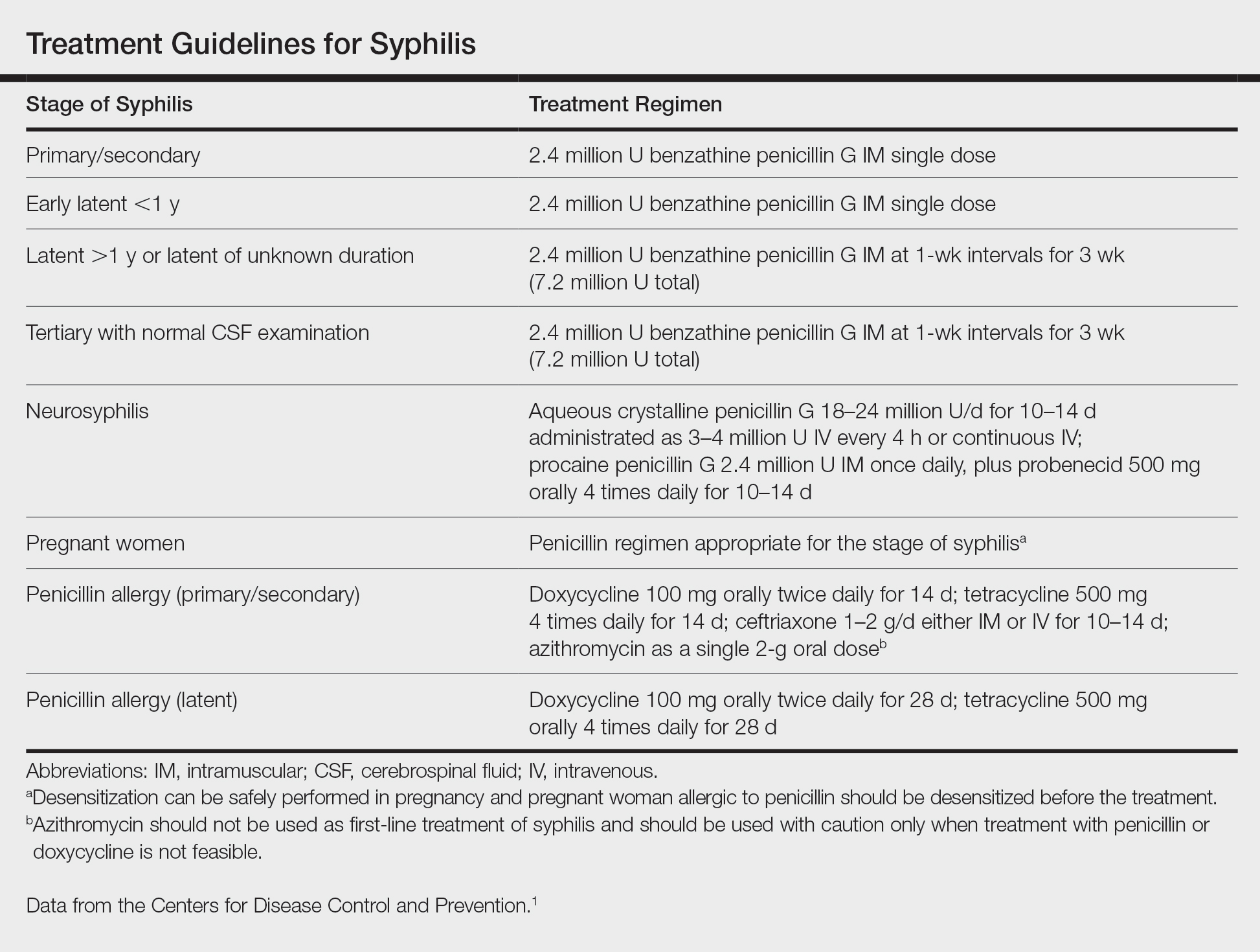
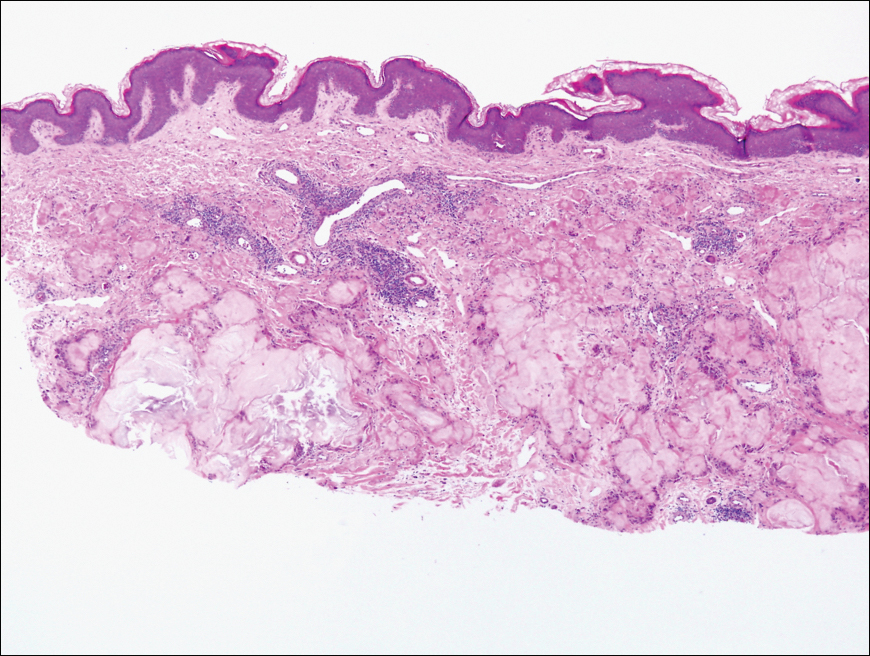
A punch biopsy of the lesion revealed acanthosis with elongation of rete ridges; interface dermatitis; and a moderately dense, predominantly lymphoid dermal infiltration (Figure). Based on a serologic toluidine red unheated serum test (TRUST) titer of 1:64 and positive Treponema antibodies, a diagnosis of secondary syphilitic infection was made. A test for human immunodeficiency virus infection was negative, and the patient was not immunocompromised. Due to allergy to benzathine penicillin G, she was prescribed oral minocycline 100 mg twice daily for 15 days. (See the Table for current recommended regimens from the Centers for Disease Control and Prevention for the treatment of syphilis.1) The hard palate plaque began to fade after 2 days of treatment and completely regressed 2 weeks later. The TRUST titer decreased to 1:4 after 6 months.
The patient's husband was examined following confirmation of his wife's infection; his TRUST titer was 1:64 and Treponema antibodies were positive. No skin lesions were detected. A test for human immunodeficiency virus infection also was negative. Further inquiry revealed that he had had sexual intercourse with a prostitute about 3 months prior. He was diagnosed with latent syphilis and prescribed the same medication regimen as his wife. However, after 6 months, his TRUST titer was still 1:64, possibly due to irregular medication use.
Secondary syphilis often is preceded by flulike symptoms of fever, sore throat, headache, malaise, generalized painless lymphadenopathy, and myalgia 4 to 10 weeks after onset of infection.2-5 Condyloma lata can be one of the characteristic mucosal signs of secondary syphilis; however, it is typically located in the anogenital area or less commonly in atypical areas such as the umbilicus, axillae, inframammary folds, and toe web spaces.6 Condyloma lata in the oral cavity is rare. In fact, this unusual manifestation prompted the patient to suspect cancer and she initially presented to a local tumor hospital. However, oral computed tomography did not detect any tumor cells, and subsequent testing yielded the diagnosis of secondary syphilis.
The differential diagnosis for a warty oral mass includes squamous cell carcinoma, condyloma acuminatum, oral submucous fibrosis, and Wegener granulomatosis.
Similar to other nontreponemal tests, TRUST is a flocculation-based quantitative test that can be used to follow treatment response, as its antibody titers may correlate with disease activity.7 Clinically, a 4-fold change in titer (equivalent to a change of 2 dilutions) is considered necessary to demonstrate a notable difference between 2 nontreponemal test results obtained using the same serologic test. The TRUST titers for the case patient decreased from 1:64 to 1:4, indicating a good response to minocycline. In contrast, the TRUST of her husband remained as high at 6-month follow-up as it had been at initial examination. This serofast state was most likely related to his irregular medication use; however, other possibilities should be considered, including confounding nontreponemal inflammatory conditions in the host, the variability of host response to infection, or even persistent low-level infection with Treponema pallidum.8 Because treponemal antibodies typically remain positive for life and most patients who have a reactive treponemal test will have a reactive report for the remainder of their lives, regardless of treatment or disease activity, treponemal antibody titers should not be used to monitor treatment response.9
China has experienced a resurgence in the incidence and prevalence of syphilis in recent decades. According to the national reporting database, the annual rate of syphilis in China has increased 14.3% since 2009 (6.5 cases per 100,000 population in 1999 vs 24.66 cases per 100,000 population in 2009).10 This re-emergence is truly remarkable, given this infection was virtually eradicated in the country 60 years ago. Recognizing this syphilis epidemic as a public health threat, the Ministry of Health of the People's Republic of China in 2010 announced a 10-year plan for syphilis control and prevention to curb the spread of syphilis and other sexually transmitted diseases. Currently, the syphilis burden is still great, with 25.54 cases per 100,000 population in 2016,11 but the situation has been stabilized and the annual increase is less than 1% since the plan's introduction.
Globally, there has been a marked resurgence of syphilis in the last decade, largely attributed to changing social and behavioral factors, especially among the population of men who have sex with men. Despite the availability of effective treatments and previously reliable prevention strategies, there are an estimated 6 million new cases of syphilis in those aged 15 to 49 years, and congenital syphilis causes more than 300,000 fetal and neonatal deaths each year.12 Continued vigilance and investment is needed to combat syphilis worldwide, and recognition of syphilis, with its versatile presentations, is of vital importance today.13
The presentation of secondary syphilis can be highly variable and requires a high level of awareness.4-6 Solitary oral involvement in secondary syphilis is rare and can lead to misdiagnosis; therefore, a high level of suspicion for syphilis should be maintained when evaluating oral lesions.
- Centers for Disease Control and Prevention. 2015 SexuallyTransmitted Diseases Treatment Guidelines: Syphilis. https://www.cdc.gov/std/tg2015/syphilis.htm. Accessed March 25, 2020.
- Lombardo J, Alhashim M. Secondary syphilis: an atypical presentation complicated by a false negative rapid plasma reagin test. Cutis. 2018;101:E11-E13.
- Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68:283-290.
- Dourmishev LA, Assen L. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Martin DH, Mroczkowski TF. Dermatological manifestations of sexually transmitted diseases other than HIV. Infect Dis Clin North Am. 1994;8:533-583.
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277.
- Morshed MG, Singh AE. Recent trends in the serologic diagnosis of syphilis. Clin Vaccine Immunol. 2015;22:137-147.
- Seña AC, Wolff M, Behets F, et al. Response to therapy following retreatment of serofast early syphilis patients with benzathine penicillin. Clin Infect Dis. 2013;56:420-422.
- Rhoads DD, Genzen JR, Bashleben CP, et al. Prevalence of traditional and reverse-algorithm syphilis screening in laboratory practice: a survey of participants in the College of American Pathologists syphilis serology proficiency testing program. Arch Pathol Lab Med. 2017;141:93-97.
- Tucker JD, Cohen MS. China's syphilis epidemic: epidemiology, proximate determinants of spread, and control responses. Curr Opin Infect Dis. 2011;24:50-55.
- Yang S, Wu J, Ding C, et al. Epidemiological features of and changes in incidence of infectious diseases in China in the first decade after the SARS outbreak: an observational trend study. Lancet Infect Dis. 2016;17:716-725.
- Noah K, Jeffrey DK. An update on the global epidemiology of syphilis. Curr Epidemiol Rep. 2018;5:24-38.
- Ghanem KG, Ram S, Rice PA. The modern epidemic of syphilis. N Engl J Med. 2020;382:845-854.
The Diagnosis: Solitary Oral Condyloma Lata of Secondary Syphilis
A punch biopsy of the lesion revealed acanthosis with elongation of rete ridges; interface dermatitis; and a moderately dense, predominantly lymphoid dermal infiltration (Figure). Based on a serologic toluidine red unheated serum test (TRUST) titer of 1:64 and positive Treponema antibodies, a diagnosis of secondary syphilitic infection was made. A test for human immunodeficiency virus infection was negative, and the patient was not immunocompromised. Due to allergy to benzathine penicillin G, she was prescribed oral minocycline 100 mg twice daily for 15 days. (See the Table for current recommended regimens from the Centers for Disease Control and Prevention for the treatment of syphilis.1) The hard palate plaque began to fade after 2 days of treatment and completely regressed 2 weeks later. The TRUST titer decreased to 1:4 after 6 months.
The patient's husband was examined following confirmation of his wife's infection; his TRUST titer was 1:64 and Treponema antibodies were positive. No skin lesions were detected. A test for human immunodeficiency virus infection also was negative. Further inquiry revealed that he had had sexual intercourse with a prostitute about 3 months prior. He was diagnosed with latent syphilis and prescribed the same medication regimen as his wife. However, after 6 months, his TRUST titer was still 1:64, possibly due to irregular medication use.
Secondary syphilis often is preceded by flulike symptoms of fever, sore throat, headache, malaise, generalized painless lymphadenopathy, and myalgia 4 to 10 weeks after onset of infection.2-5 Condyloma lata can be one of the characteristic mucosal signs of secondary syphilis; however, it is typically located in the anogenital area or less commonly in atypical areas such as the umbilicus, axillae, inframammary folds, and toe web spaces.6 Condyloma lata in the oral cavity is rare. In fact, this unusual manifestation prompted the patient to suspect cancer and she initially presented to a local tumor hospital. However, oral computed tomography did not detect any tumor cells, and subsequent testing yielded the diagnosis of secondary syphilis.
The differential diagnosis for a warty oral mass includes squamous cell carcinoma, condyloma acuminatum, oral submucous fibrosis, and Wegener granulomatosis.
Similar to other nontreponemal tests, TRUST is a flocculation-based quantitative test that can be used to follow treatment response, as its antibody titers may correlate with disease activity.7 Clinically, a 4-fold change in titer (equivalent to a change of 2 dilutions) is considered necessary to demonstrate a notable difference between 2 nontreponemal test results obtained using the same serologic test. The TRUST titers for the case patient decreased from 1:64 to 1:4, indicating a good response to minocycline. In contrast, the TRUST of her husband remained as high at 6-month follow-up as it had been at initial examination. This serofast state was most likely related to his irregular medication use; however, other possibilities should be considered, including confounding nontreponemal inflammatory conditions in the host, the variability of host response to infection, or even persistent low-level infection with Treponema pallidum.8 Because treponemal antibodies typically remain positive for life and most patients who have a reactive treponemal test will have a reactive report for the remainder of their lives, regardless of treatment or disease activity, treponemal antibody titers should not be used to monitor treatment response.9
China has experienced a resurgence in the incidence and prevalence of syphilis in recent decades. According to the national reporting database, the annual rate of syphilis in China has increased 14.3% since 2009 (6.5 cases per 100,000 population in 1999 vs 24.66 cases per 100,000 population in 2009).10 This re-emergence is truly remarkable, given this infection was virtually eradicated in the country 60 years ago. Recognizing this syphilis epidemic as a public health threat, the Ministry of Health of the People's Republic of China in 2010 announced a 10-year plan for syphilis control and prevention to curb the spread of syphilis and other sexually transmitted diseases. Currently, the syphilis burden is still great, with 25.54 cases per 100,000 population in 2016,11 but the situation has been stabilized and the annual increase is less than 1% since the plan's introduction.
Globally, there has been a marked resurgence of syphilis in the last decade, largely attributed to changing social and behavioral factors, especially among the population of men who have sex with men. Despite the availability of effective treatments and previously reliable prevention strategies, there are an estimated 6 million new cases of syphilis in those aged 15 to 49 years, and congenital syphilis causes more than 300,000 fetal and neonatal deaths each year.12 Continued vigilance and investment is needed to combat syphilis worldwide, and recognition of syphilis, with its versatile presentations, is of vital importance today.13
The presentation of secondary syphilis can be highly variable and requires a high level of awareness.4-6 Solitary oral involvement in secondary syphilis is rare and can lead to misdiagnosis; therefore, a high level of suspicion for syphilis should be maintained when evaluating oral lesions.
The Diagnosis: Solitary Oral Condyloma Lata of Secondary Syphilis
A punch biopsy of the lesion revealed acanthosis with elongation of rete ridges; interface dermatitis; and a moderately dense, predominantly lymphoid dermal infiltration (Figure). Based on a serologic toluidine red unheated serum test (TRUST) titer of 1:64 and positive Treponema antibodies, a diagnosis of secondary syphilitic infection was made. A test for human immunodeficiency virus infection was negative, and the patient was not immunocompromised. Due to allergy to benzathine penicillin G, she was prescribed oral minocycline 100 mg twice daily for 15 days. (See the Table for current recommended regimens from the Centers for Disease Control and Prevention for the treatment of syphilis.1) The hard palate plaque began to fade after 2 days of treatment and completely regressed 2 weeks later. The TRUST titer decreased to 1:4 after 6 months.
The patient's husband was examined following confirmation of his wife's infection; his TRUST titer was 1:64 and Treponema antibodies were positive. No skin lesions were detected. A test for human immunodeficiency virus infection also was negative. Further inquiry revealed that he had had sexual intercourse with a prostitute about 3 months prior. He was diagnosed with latent syphilis and prescribed the same medication regimen as his wife. However, after 6 months, his TRUST titer was still 1:64, possibly due to irregular medication use.
Secondary syphilis often is preceded by flulike symptoms of fever, sore throat, headache, malaise, generalized painless lymphadenopathy, and myalgia 4 to 10 weeks after onset of infection.2-5 Condyloma lata can be one of the characteristic mucosal signs of secondary syphilis; however, it is typically located in the anogenital area or less commonly in atypical areas such as the umbilicus, axillae, inframammary folds, and toe web spaces.6 Condyloma lata in the oral cavity is rare. In fact, this unusual manifestation prompted the patient to suspect cancer and she initially presented to a local tumor hospital. However, oral computed tomography did not detect any tumor cells, and subsequent testing yielded the diagnosis of secondary syphilis.
The differential diagnosis for a warty oral mass includes squamous cell carcinoma, condyloma acuminatum, oral submucous fibrosis, and Wegener granulomatosis.
Similar to other nontreponemal tests, TRUST is a flocculation-based quantitative test that can be used to follow treatment response, as its antibody titers may correlate with disease activity.7 Clinically, a 4-fold change in titer (equivalent to a change of 2 dilutions) is considered necessary to demonstrate a notable difference between 2 nontreponemal test results obtained using the same serologic test. The TRUST titers for the case patient decreased from 1:64 to 1:4, indicating a good response to minocycline. In contrast, the TRUST of her husband remained as high at 6-month follow-up as it had been at initial examination. This serofast state was most likely related to his irregular medication use; however, other possibilities should be considered, including confounding nontreponemal inflammatory conditions in the host, the variability of host response to infection, or even persistent low-level infection with Treponema pallidum.8 Because treponemal antibodies typically remain positive for life and most patients who have a reactive treponemal test will have a reactive report for the remainder of their lives, regardless of treatment or disease activity, treponemal antibody titers should not be used to monitor treatment response.9
China has experienced a resurgence in the incidence and prevalence of syphilis in recent decades. According to the national reporting database, the annual rate of syphilis in China has increased 14.3% since 2009 (6.5 cases per 100,000 population in 1999 vs 24.66 cases per 100,000 population in 2009).10 This re-emergence is truly remarkable, given this infection was virtually eradicated in the country 60 years ago. Recognizing this syphilis epidemic as a public health threat, the Ministry of Health of the People's Republic of China in 2010 announced a 10-year plan for syphilis control and prevention to curb the spread of syphilis and other sexually transmitted diseases. Currently, the syphilis burden is still great, with 25.54 cases per 100,000 population in 2016,11 but the situation has been stabilized and the annual increase is less than 1% since the plan's introduction.
Globally, there has been a marked resurgence of syphilis in the last decade, largely attributed to changing social and behavioral factors, especially among the population of men who have sex with men. Despite the availability of effective treatments and previously reliable prevention strategies, there are an estimated 6 million new cases of syphilis in those aged 15 to 49 years, and congenital syphilis causes more than 300,000 fetal and neonatal deaths each year.12 Continued vigilance and investment is needed to combat syphilis worldwide, and recognition of syphilis, with its versatile presentations, is of vital importance today.13
The presentation of secondary syphilis can be highly variable and requires a high level of awareness.4-6 Solitary oral involvement in secondary syphilis is rare and can lead to misdiagnosis; therefore, a high level of suspicion for syphilis should be maintained when evaluating oral lesions.
- Centers for Disease Control and Prevention. 2015 SexuallyTransmitted Diseases Treatment Guidelines: Syphilis. https://www.cdc.gov/std/tg2015/syphilis.htm. Accessed March 25, 2020.
- Lombardo J, Alhashim M. Secondary syphilis: an atypical presentation complicated by a false negative rapid plasma reagin test. Cutis. 2018;101:E11-E13.
- Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68:283-290.
- Dourmishev LA, Assen L. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Martin DH, Mroczkowski TF. Dermatological manifestations of sexually transmitted diseases other than HIV. Infect Dis Clin North Am. 1994;8:533-583.
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277.
- Morshed MG, Singh AE. Recent trends in the serologic diagnosis of syphilis. Clin Vaccine Immunol. 2015;22:137-147.
- Seña AC, Wolff M, Behets F, et al. Response to therapy following retreatment of serofast early syphilis patients with benzathine penicillin. Clin Infect Dis. 2013;56:420-422.
- Rhoads DD, Genzen JR, Bashleben CP, et al. Prevalence of traditional and reverse-algorithm syphilis screening in laboratory practice: a survey of participants in the College of American Pathologists syphilis serology proficiency testing program. Arch Pathol Lab Med. 2017;141:93-97.
- Tucker JD, Cohen MS. China's syphilis epidemic: epidemiology, proximate determinants of spread, and control responses. Curr Opin Infect Dis. 2011;24:50-55.
- Yang S, Wu J, Ding C, et al. Epidemiological features of and changes in incidence of infectious diseases in China in the first decade after the SARS outbreak: an observational trend study. Lancet Infect Dis. 2016;17:716-725.
- Noah K, Jeffrey DK. An update on the global epidemiology of syphilis. Curr Epidemiol Rep. 2018;5:24-38.
- Ghanem KG, Ram S, Rice PA. The modern epidemic of syphilis. N Engl J Med. 2020;382:845-854.
- Centers for Disease Control and Prevention. 2015 SexuallyTransmitted Diseases Treatment Guidelines: Syphilis. https://www.cdc.gov/std/tg2015/syphilis.htm. Accessed March 25, 2020.
- Lombardo J, Alhashim M. Secondary syphilis: an atypical presentation complicated by a false negative rapid plasma reagin test. Cutis. 2018;101:E11-E13.
- Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68:283-290.
- Dourmishev LA, Assen L. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Martin DH, Mroczkowski TF. Dermatological manifestations of sexually transmitted diseases other than HIV. Infect Dis Clin North Am. 1994;8:533-583.
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277.
- Morshed MG, Singh AE. Recent trends in the serologic diagnosis of syphilis. Clin Vaccine Immunol. 2015;22:137-147.
- Seña AC, Wolff M, Behets F, et al. Response to therapy following retreatment of serofast early syphilis patients with benzathine penicillin. Clin Infect Dis. 2013;56:420-422.
- Rhoads DD, Genzen JR, Bashleben CP, et al. Prevalence of traditional and reverse-algorithm syphilis screening in laboratory practice: a survey of participants in the College of American Pathologists syphilis serology proficiency testing program. Arch Pathol Lab Med. 2017;141:93-97.
- Tucker JD, Cohen MS. China's syphilis epidemic: epidemiology, proximate determinants of spread, and control responses. Curr Opin Infect Dis. 2011;24:50-55.
- Yang S, Wu J, Ding C, et al. Epidemiological features of and changes in incidence of infectious diseases in China in the first decade after the SARS outbreak: an observational trend study. Lancet Infect Dis. 2016;17:716-725.
- Noah K, Jeffrey DK. An update on the global epidemiology of syphilis. Curr Epidemiol Rep. 2018;5:24-38.
- Ghanem KG, Ram S, Rice PA. The modern epidemic of syphilis. N Engl J Med. 2020;382:845-854.
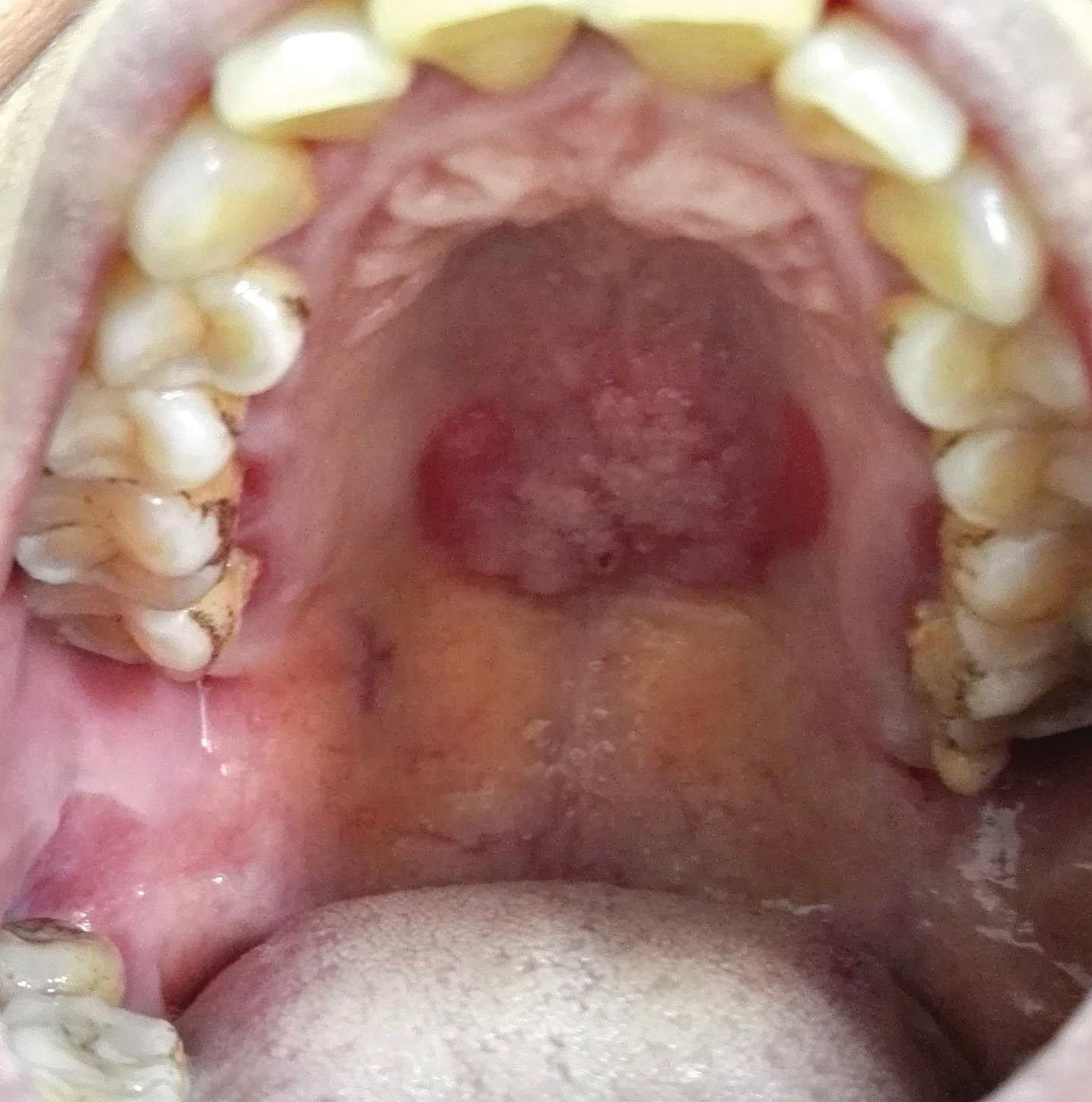
A 50-year-old Chinese woman presented with a painless, well-demarcated, nontender, elevated, flat-topped verrucous plaque on the hard palate of 1 month's duration. The lesion measured 2 cm in diameter. The patient reported no other dermatologic or systemic concerns, and no other skin or genital lesions were observed.
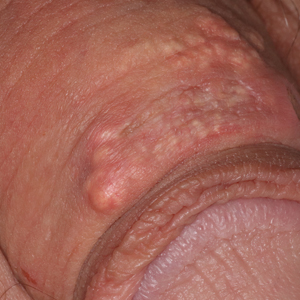
Multinodular Plaque on the Penis
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
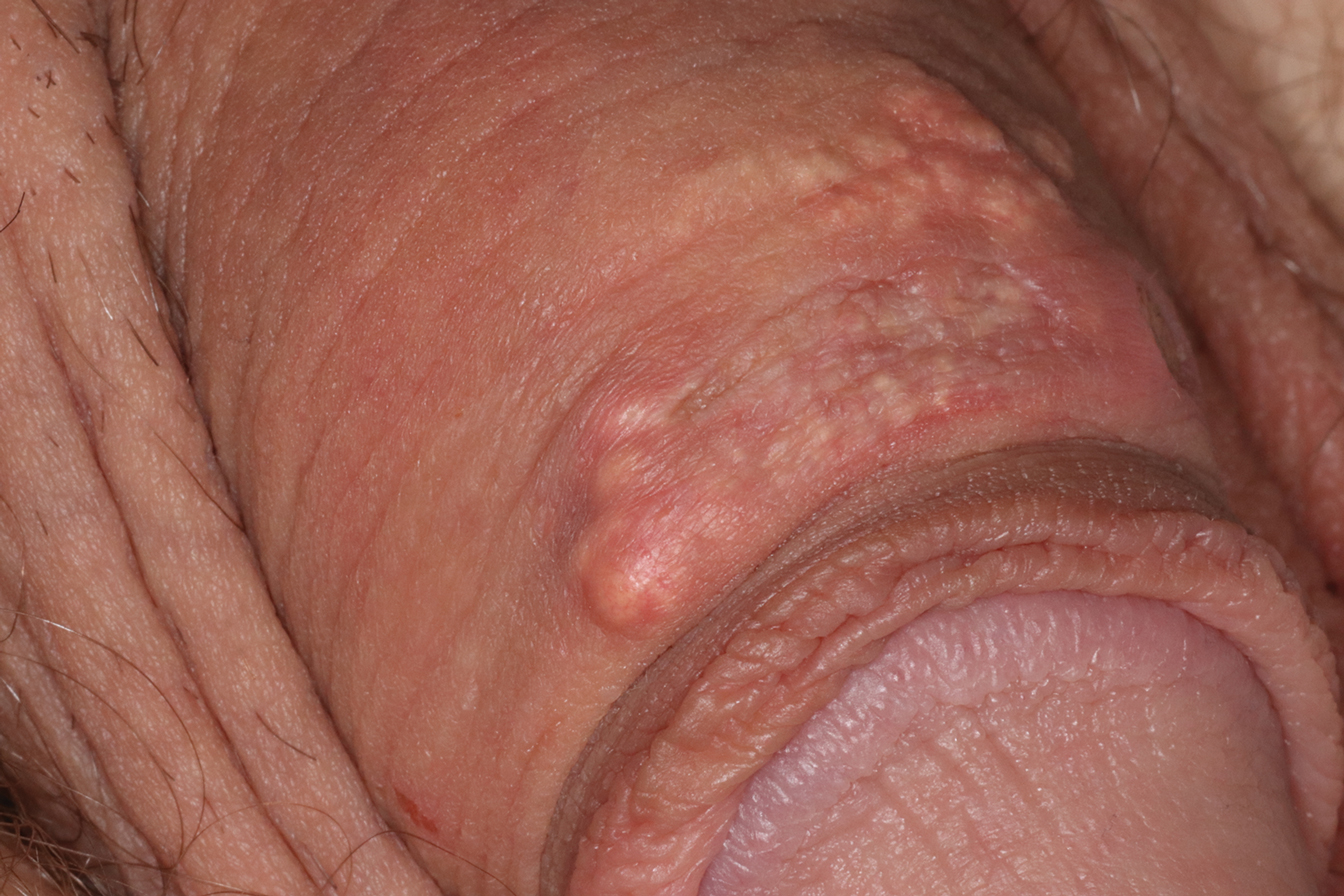
A 34-year-old man presented for evaluation of a slowly growing group of firm white bumps on the penis. The lesions were nontender and asymptomatic. Medical and family history was notable for gout, though he was not being treated. Physical examination revealed a 3-cm, firm, multinodular, chalky white plaque on the dorsal aspect of the penile shaft. A tangential biopsy was performed and sent for hematoxylin and eosin staining.
Tense Bullae on the Hands
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
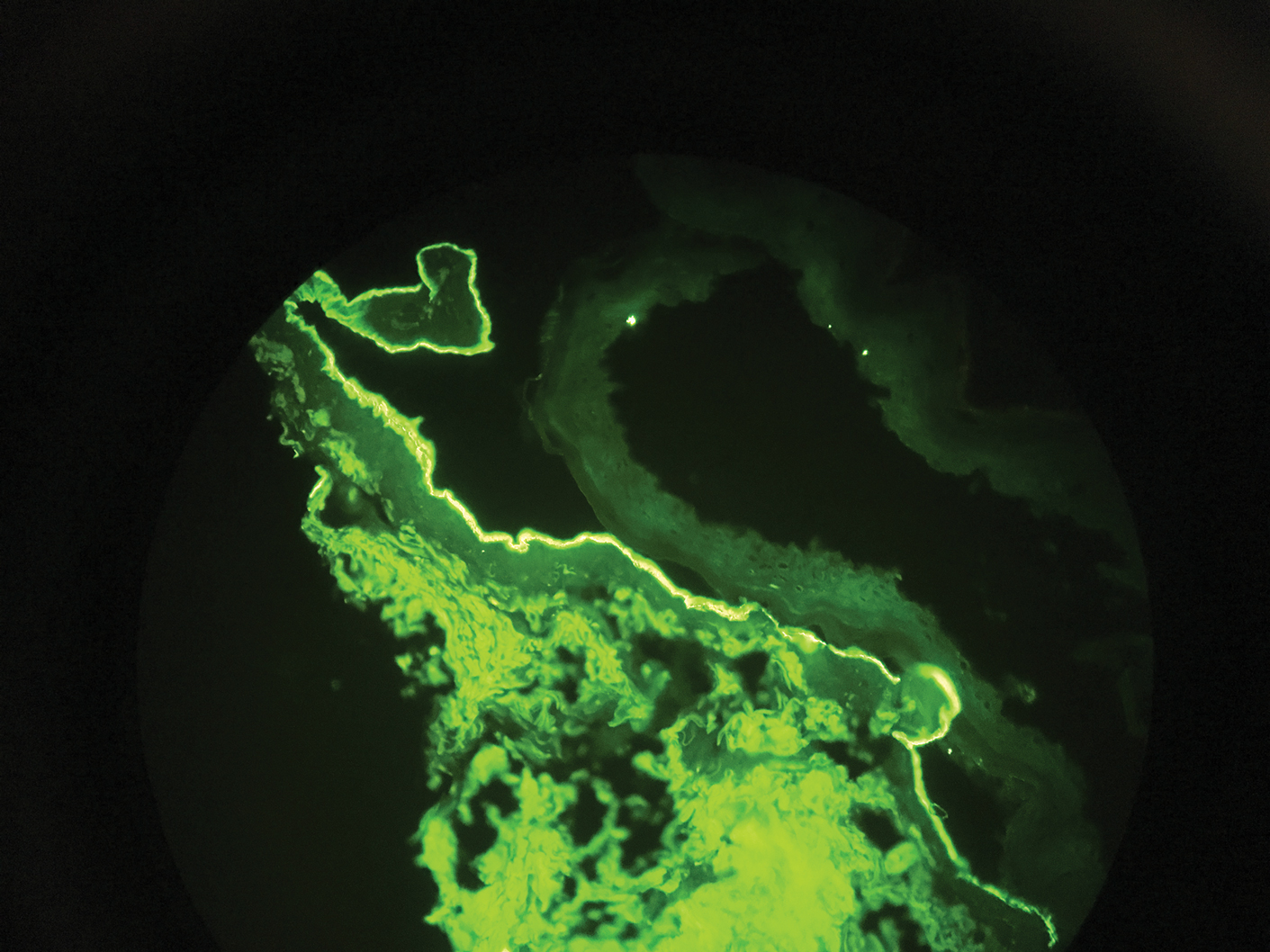
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
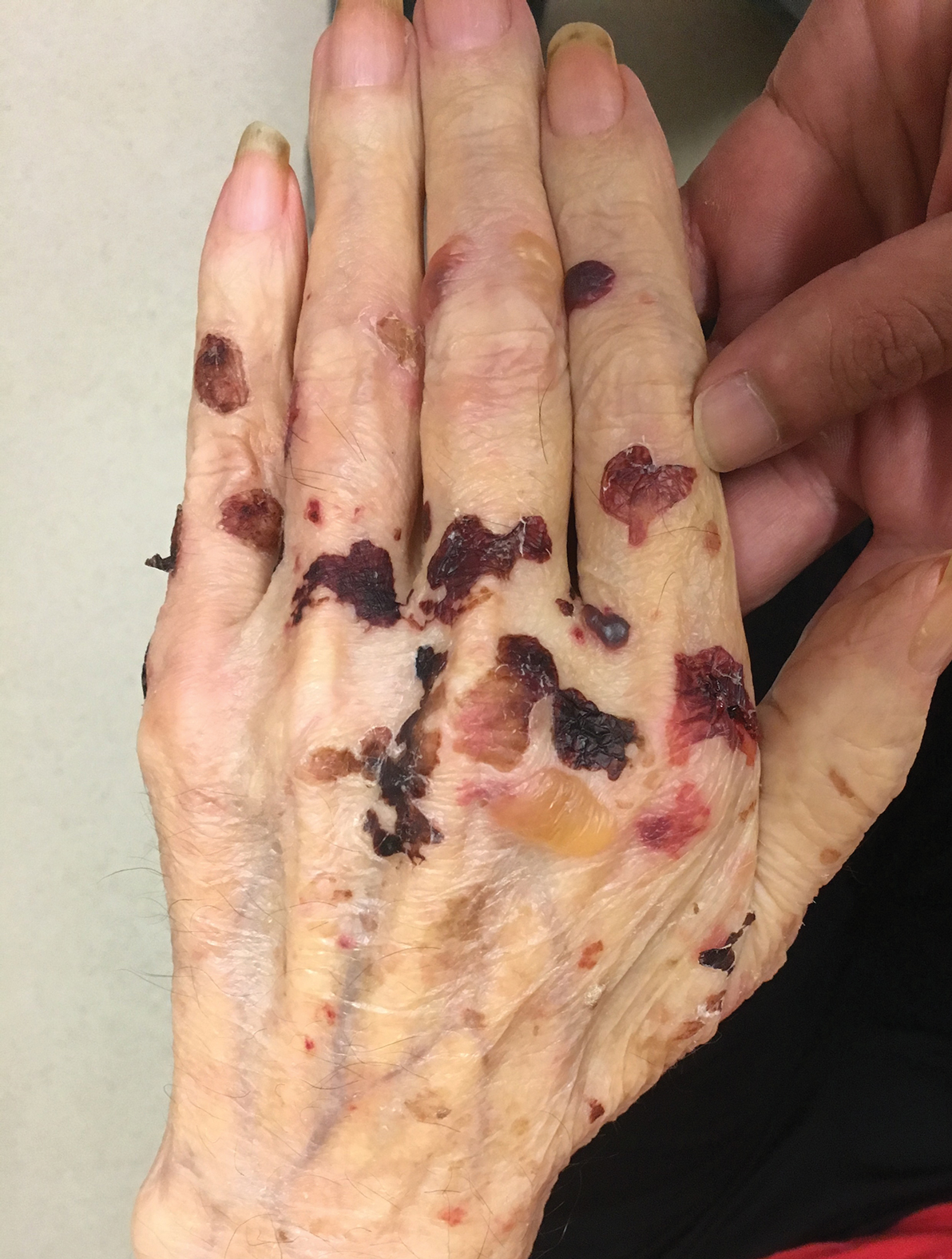
A 75-year-old man presented to our clinic with nonpainful, nonpruritic, tense bullae and erosions on the dorsal aspects of the hands and extensor surfaces of the elbows of 1 month's duration. The patient also had erythematous erosions and crusted papules on the left cheek and surrounding the left eye. He denied any new medications, history of liver or kidney disease, or history of hepatitis or human immunodeficiency virus. There were no obvious exacerbating factors, including exposure to sunlight. Direct immunofluorescence using a salt-split preparation was performed on a biopsy of the perilesional skin.
Firm Abdominal Papule
The Diagnosis: Cutaneous Metastatic Gastric Carcinoma
Cutaneous metastasis of primary gastric carcinoma is a rare occurrence, with the more common metastatic sites being the lymph nodes, liver, and peritoneal cavity. The incidence of visceral neoplasm metastasis to the skin ranges from 0.7% to 9% and is less than 1% for upper digestive tract carcinomas.1 Cutaneous metastases make up 2% of all tumors of the skin and commonly are located near the site of the primary tumor.2 The most common cutaneous metastasis sites for gastric carcinoma include the neck, chest, and head.3 One of the more typical sites of cutaneous metastasis from gastric cancer is the umbilicus (ie, Sister Mary Joseph nodule). Cutaneous metastases from gastric carcinoma commonly present as asymptomatic hyperpigmented nodules.1,3
In our patient, histopathologic sections showed diffuse infiltration of the dermis by atypical polygonal/round cells arranged in cords and small aggregates. Some of the neoplastic cells had signet ring morphology (Figure). Tumor cells demonstrated positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen; they were negative for S-100, MART-1 (melanoma-associated antigen recognized by T cells 1), leukocyte common antigen, gross cystic disease fluid protein 15, estrogen and progesterone receptor, and HER2/neu (human epidermal growth factor receptor 2).
Our patient's presentation was rare in that she developed an asymptomatic erythematous papule on the skin of the abdomen. However, her history of stage IIIB gastric adenocarcinoma in conjunction with the clinical picture and microscopic findings were most consistent with metastatic carcinoma of gastrointestinal origin. The histologic hallmarks of cutaneous metastatic gastric carcinoma include aggregates of neoplastic cells arranged in cords, sometimes forming glands, embedded in a fibrous stroma. Tumor cells may demonstrate signet ring morphology. These unique histologic findings, as well as positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen, rule out other potential diagnoses for an asymptomatic solitary papule.
Dermatofibrosarcoma protuberans presents as an asymptomatic, slow-growing, indurated papule or plaque that develops into a red or brownish nodule. Histologically, dermatofibrosarcoma protuberans is characterized by spindled cells, few mitotic figures, infiltration of the subcutaneous tissue in a honeycomblike pattern, and obliteration of the adnexal structures.4
Cutaneous B-cell lymphoma (CBCL) can present as single or multiple red papules or nodules located on the trunk, face, or extremities. Histologically, CBCL would show a nodular or diffuse infiltrate throughout the dermis, frequently with accentuation in the deep reticular dermis, sparing of the epidermis, and the presence of a grenz zone. The infiltrate in CBCL consists of CD20+, CD19+, and CD79a+ B cells. Identification of a monoclonal B-cell population either by immunohistochemistry or polymerase chain reaction would further support a diagnosis of CBCL.4 These specific histologic findings and the immunohistochemical staining pattern helped rule out CBCL as the diagnosis in our patient.
Amelanotic melanomas present as flesh-colored to light pink papules, making them especially challenging to diagnose clinically. Asymmetrical, poorly circumscribed nests of atypical melanocytes as well as single melanocytes within the epidermis and dermis are seen histologically; mitotic figures are common. Immunohistochemical staining for melanoma includes S-100, human melanoma black 45, MART-1/Melan-A, tyrosinase, and microphthalmia-associated transcription factor 1.4
Neurothekeomas can present as asymptomatic, solitary, flesh-colored papules located on the head, neck, and upper trunk. Histologically, neurothekeomas have a distinct appearance consisting of a well-defined mass composed of variable-sized lobules of spindled and epithelioid cells dispersed in a myxoid stroma within the reticular dermis.4 These specific histologic findings helped rule out neurothekeoma in our patient.
Following the diagnosis of cutaneous metastatic gastric carcinoma in our patient, positron emission tomography and computed tomography of the chest, abdomen, and pelvis were unremarkable for distant disease. Subsequently, the patient underwent surgical excision of the papule with clear margins, followed by a short course of radiation therapy. She currently is under close monitoring but remains in remission with no new cutaneous manifestations of the gastric carcinoma.
- Erdemir A, Atilganoglu U, Onsun N, et al. Cutaneous metastases from gastric adenocarcinoma. Indian J Dermatol. 2011;56:236-237.
- Junqueira AL, Corbett AM, Oliveira Filho Jd, et al. Cutaneous metastasis from gastrointestinal adenocarcinoma of unknown primary origin. An Bras Dermatol. 2015;90:564-566.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:E9-E13.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
The Diagnosis: Cutaneous Metastatic Gastric Carcinoma
Cutaneous metastasis of primary gastric carcinoma is a rare occurrence, with the more common metastatic sites being the lymph nodes, liver, and peritoneal cavity. The incidence of visceral neoplasm metastasis to the skin ranges from 0.7% to 9% and is less than 1% for upper digestive tract carcinomas.1 Cutaneous metastases make up 2% of all tumors of the skin and commonly are located near the site of the primary tumor.2 The most common cutaneous metastasis sites for gastric carcinoma include the neck, chest, and head.3 One of the more typical sites of cutaneous metastasis from gastric cancer is the umbilicus (ie, Sister Mary Joseph nodule). Cutaneous metastases from gastric carcinoma commonly present as asymptomatic hyperpigmented nodules.1,3
In our patient, histopathologic sections showed diffuse infiltration of the dermis by atypical polygonal/round cells arranged in cords and small aggregates. Some of the neoplastic cells had signet ring morphology (Figure). Tumor cells demonstrated positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen; they were negative for S-100, MART-1 (melanoma-associated antigen recognized by T cells 1), leukocyte common antigen, gross cystic disease fluid protein 15, estrogen and progesterone receptor, and HER2/neu (human epidermal growth factor receptor 2).
Our patient's presentation was rare in that she developed an asymptomatic erythematous papule on the skin of the abdomen. However, her history of stage IIIB gastric adenocarcinoma in conjunction with the clinical picture and microscopic findings were most consistent with metastatic carcinoma of gastrointestinal origin. The histologic hallmarks of cutaneous metastatic gastric carcinoma include aggregates of neoplastic cells arranged in cords, sometimes forming glands, embedded in a fibrous stroma. Tumor cells may demonstrate signet ring morphology. These unique histologic findings, as well as positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen, rule out other potential diagnoses for an asymptomatic solitary papule.
Dermatofibrosarcoma protuberans presents as an asymptomatic, slow-growing, indurated papule or plaque that develops into a red or brownish nodule. Histologically, dermatofibrosarcoma protuberans is characterized by spindled cells, few mitotic figures, infiltration of the subcutaneous tissue in a honeycomblike pattern, and obliteration of the adnexal structures.4
Cutaneous B-cell lymphoma (CBCL) can present as single or multiple red papules or nodules located on the trunk, face, or extremities. Histologically, CBCL would show a nodular or diffuse infiltrate throughout the dermis, frequently with accentuation in the deep reticular dermis, sparing of the epidermis, and the presence of a grenz zone. The infiltrate in CBCL consists of CD20+, CD19+, and CD79a+ B cells. Identification of a monoclonal B-cell population either by immunohistochemistry or polymerase chain reaction would further support a diagnosis of CBCL.4 These specific histologic findings and the immunohistochemical staining pattern helped rule out CBCL as the diagnosis in our patient.
Amelanotic melanomas present as flesh-colored to light pink papules, making them especially challenging to diagnose clinically. Asymmetrical, poorly circumscribed nests of atypical melanocytes as well as single melanocytes within the epidermis and dermis are seen histologically; mitotic figures are common. Immunohistochemical staining for melanoma includes S-100, human melanoma black 45, MART-1/Melan-A, tyrosinase, and microphthalmia-associated transcription factor 1.4
Neurothekeomas can present as asymptomatic, solitary, flesh-colored papules located on the head, neck, and upper trunk. Histologically, neurothekeomas have a distinct appearance consisting of a well-defined mass composed of variable-sized lobules of spindled and epithelioid cells dispersed in a myxoid stroma within the reticular dermis.4 These specific histologic findings helped rule out neurothekeoma in our patient.
Following the diagnosis of cutaneous metastatic gastric carcinoma in our patient, positron emission tomography and computed tomography of the chest, abdomen, and pelvis were unremarkable for distant disease. Subsequently, the patient underwent surgical excision of the papule with clear margins, followed by a short course of radiation therapy. She currently is under close monitoring but remains in remission with no new cutaneous manifestations of the gastric carcinoma.
The Diagnosis: Cutaneous Metastatic Gastric Carcinoma
Cutaneous metastasis of primary gastric carcinoma is a rare occurrence, with the more common metastatic sites being the lymph nodes, liver, and peritoneal cavity. The incidence of visceral neoplasm metastasis to the skin ranges from 0.7% to 9% and is less than 1% for upper digestive tract carcinomas.1 Cutaneous metastases make up 2% of all tumors of the skin and commonly are located near the site of the primary tumor.2 The most common cutaneous metastasis sites for gastric carcinoma include the neck, chest, and head.3 One of the more typical sites of cutaneous metastasis from gastric cancer is the umbilicus (ie, Sister Mary Joseph nodule). Cutaneous metastases from gastric carcinoma commonly present as asymptomatic hyperpigmented nodules.1,3
In our patient, histopathologic sections showed diffuse infiltration of the dermis by atypical polygonal/round cells arranged in cords and small aggregates. Some of the neoplastic cells had signet ring morphology (Figure). Tumor cells demonstrated positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen; they were negative for S-100, MART-1 (melanoma-associated antigen recognized by T cells 1), leukocyte common antigen, gross cystic disease fluid protein 15, estrogen and progesterone receptor, and HER2/neu (human epidermal growth factor receptor 2).
Our patient's presentation was rare in that she developed an asymptomatic erythematous papule on the skin of the abdomen. However, her history of stage IIIB gastric adenocarcinoma in conjunction with the clinical picture and microscopic findings were most consistent with metastatic carcinoma of gastrointestinal origin. The histologic hallmarks of cutaneous metastatic gastric carcinoma include aggregates of neoplastic cells arranged in cords, sometimes forming glands, embedded in a fibrous stroma. Tumor cells may demonstrate signet ring morphology. These unique histologic findings, as well as positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen, rule out other potential diagnoses for an asymptomatic solitary papule.
Dermatofibrosarcoma protuberans presents as an asymptomatic, slow-growing, indurated papule or plaque that develops into a red or brownish nodule. Histologically, dermatofibrosarcoma protuberans is characterized by spindled cells, few mitotic figures, infiltration of the subcutaneous tissue in a honeycomblike pattern, and obliteration of the adnexal structures.4
Cutaneous B-cell lymphoma (CBCL) can present as single or multiple red papules or nodules located on the trunk, face, or extremities. Histologically, CBCL would show a nodular or diffuse infiltrate throughout the dermis, frequently with accentuation in the deep reticular dermis, sparing of the epidermis, and the presence of a grenz zone. The infiltrate in CBCL consists of CD20+, CD19+, and CD79a+ B cells. Identification of a monoclonal B-cell population either by immunohistochemistry or polymerase chain reaction would further support a diagnosis of CBCL.4 These specific histologic findings and the immunohistochemical staining pattern helped rule out CBCL as the diagnosis in our patient.
Amelanotic melanomas present as flesh-colored to light pink papules, making them especially challenging to diagnose clinically. Asymmetrical, poorly circumscribed nests of atypical melanocytes as well as single melanocytes within the epidermis and dermis are seen histologically; mitotic figures are common. Immunohistochemical staining for melanoma includes S-100, human melanoma black 45, MART-1/Melan-A, tyrosinase, and microphthalmia-associated transcription factor 1.4
Neurothekeomas can present as asymptomatic, solitary, flesh-colored papules located on the head, neck, and upper trunk. Histologically, neurothekeomas have a distinct appearance consisting of a well-defined mass composed of variable-sized lobules of spindled and epithelioid cells dispersed in a myxoid stroma within the reticular dermis.4 These specific histologic findings helped rule out neurothekeoma in our patient.
Following the diagnosis of cutaneous metastatic gastric carcinoma in our patient, positron emission tomography and computed tomography of the chest, abdomen, and pelvis were unremarkable for distant disease. Subsequently, the patient underwent surgical excision of the papule with clear margins, followed by a short course of radiation therapy. She currently is under close monitoring but remains in remission with no new cutaneous manifestations of the gastric carcinoma.
- Erdemir A, Atilganoglu U, Onsun N, et al. Cutaneous metastases from gastric adenocarcinoma. Indian J Dermatol. 2011;56:236-237.
- Junqueira AL, Corbett AM, Oliveira Filho Jd, et al. Cutaneous metastasis from gastrointestinal adenocarcinoma of unknown primary origin. An Bras Dermatol. 2015;90:564-566.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:E9-E13.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
- Erdemir A, Atilganoglu U, Onsun N, et al. Cutaneous metastases from gastric adenocarcinoma. Indian J Dermatol. 2011;56:236-237.
- Junqueira AL, Corbett AM, Oliveira Filho Jd, et al. Cutaneous metastasis from gastrointestinal adenocarcinoma of unknown primary origin. An Bras Dermatol. 2015;90:564-566.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:E9-E13.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
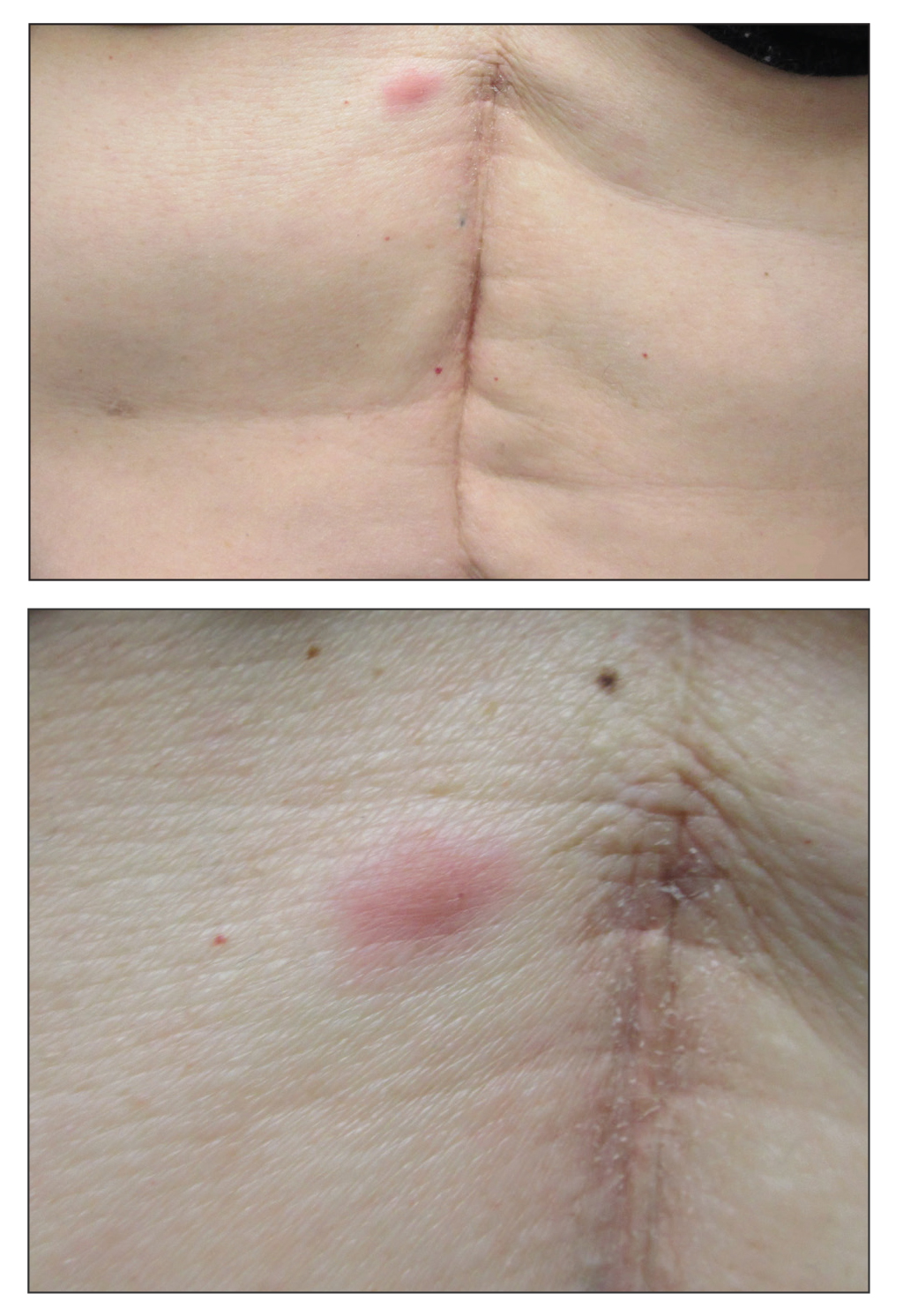
A 53-year-old woman with a history of melanoma on the right thigh, stage II Hodgkin lymphoma, and stage IIIB gastric adenocarcinoma treated with a distal gastrectomy presented with an asymptomatic but persistent skin lesion on the abdomen of 2 months' duration. The lesion arose spontaneously 6 months prior and had increased in size during that time. Physical examination revealed a 6-mm, solitary, firm, erythematous papule on the skin of the right upper quadrant of the abdomen. The patient was otherwise healthy, and a review of systems did not reveal any abnormalities. A punch biopsy was submitted for histopathologic review.
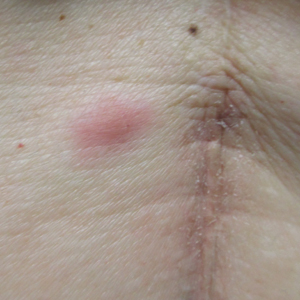
Recurrent Vesicles on the Palm
The Diagnosis: Herpes Simplex Virus Dermatitis
A swab of the lesions yielded negative varicella-zoster virus and herpes simplex virus (HSV) cultures, but polymerase chain reaction (PCR) was positive for HSV DNA. The patient was started on acyclovir, which resulted in resolution of the lesions.
Recurrent HSV dermatitis most frequently is encountered in the orolabial or genital regions. After primary infection, HSV is retrogradely taken up into the dorsal root ganglion and may be reactivated in the same dermatome upon stress induction, forming clustered vesicles that rupture to form painful erosions.1 Our patient's history of numerous recurrent episodes in the same area of the palm in the distribution of the median nerve suggests viral latency in the C5 through T1 dorsal root ganglia with reactivation rather than autoinoculation or external infection from another source. The incidence of HSV involving the hand has been estimated at 2.4 cases per 100,000 individuals per year, with finger, thumb, or palm/wrist involvement accounting for 67%, 22%, and 11% of cases, respectively.2 Of the palmar cases that have been reported, most have a positive history for genital or orolabial HSV infection.2-5
In cases of suspected HSV dermatitis with atypical presentations, diagnostic studies are of importance. Although viral culture is the diagnostic gold standard in active lesions, it has lower sensitivity in improperly handled specimens; cases of recurrent disease; and specimens from dried, crusted, or aged lesions,1 which helps to explain the negative culture result in our patient. Viral culture has been largely replaced in clinical practice by nucleic acid amplification tests using PCR, which is fast and type specific.6,7 The sensitivity of PCR approaches 100% when vesicles or wet ulcers are sampled, and PCR has better yields from dry ulcers or crusts compared to viral culture.6 However, because viral shedding is intermittent, a negative PCR result does not rule out HSV infection.8 Additional bedside diagnostic techniques include Tzanck smear, a rapid and inexpensive test in which lesions are scraped and stained with Giemsa, Wright, or Papanicolaou stains. Under light microscopy, multinucleated giant cells are seen in 60% to 75% of cases.9 This method, however, cannot distinguish HSV from varicella-zoster virus and must be followed by direct fluorescent antibody testing or immunohistochemistry for viral typing.1,9 Serologic testing also may be useful in patients who have a suspicious history for HSV infection but do not have lesions on physical examination to diagnose clinically or sample for PCR. Enzyme-linked immunosorbent assay testing can detect IgG starting 3 weeks after infection, and newer type-specific assays can distinguish between HSV types 1 and 2.6 In low-incidence populations, false positives from enzyme-linked immunosorbent assay can be seen and should be confirmed by western blot.6,7
Preferred treatment of HSV includes antiviral medications such as acyclovir, valacyclovir, and famciclovir. Regimens vary based on the site of infection, primary or recurrent nature of the infection, immune status of the patient, and whether or not viral suppression is desired to prevent recurrent outbreaks.7,10
Tinea manuum also may present with unilateral vesicles and erosions involving the palms11; however, it was less likely than HSV dermatitis in this patient presenting with a history of numerous recurrent episodes and without scaling on physical examination. Dyshidrotic eczema, contact dermatitis, and scabies are more characteristically pruritic rather than painful. Additionally, dyshidrotic eczema and scabies would be more likely to have symmetric involvement of the arms. Although vesicles are seen in both dyshidrotic eczema and HSV dermatitis, the vesicles of dyshidrotic eczema usually are noninflammatory compared to the painful vesicles on an erythematous base classically seen in HSV dermatitis.
Acknowledgment
The authors thank Elizabeth Ergen, MD (Knoxville, Tennessee), for her assistance with this case.
- Fatahzadeh M, Schwartz RA. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57:737-763; quiz 764-766.
- Gill MJ, Arlette J, Buchan K. Herpes simplex virus infection of the hand. a profile of 79 cases. Am J Med. 1988;84:89-93.
- Widenfalk B, Wallin J. Recurrent herpes simplex virus infections in the adult hand. Scand J Plast Reconstr Surg Hand Surg. 1988;22:177-180.
- Gill MJ, Arlette J, Buchan KA. Herpes simplex virus infection of the hand. J Am Acad Dermatol. 1990;22:111-116.
- Osio A, Fremont G, Petit A, et al. An unusual bipolar primary herpes simplex virus 1 infection. J Clin Virol. 2008;43:230-232.
- Gnann JW Jr, Whitley RJ. Clinical practice. genital herpes. N Engl J Med. 2016;375:666-674.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
- Downing C, Mendoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- WHO Guidelines Approved by the Guidelines Review Committee. WHO Guidelines for the Treatment of Genital Herpes Simplex Virus. Geneva, Switzerland: World Health Organization; 2016.
- Veraldi S, Schianchi R, Benzecry V, et al. Tinea manuum: a report of 18 cases observed in the metropolitan area of Milan and review of the literature. Mycoses. 2019;62:604-608.
The Diagnosis: Herpes Simplex Virus Dermatitis
A swab of the lesions yielded negative varicella-zoster virus and herpes simplex virus (HSV) cultures, but polymerase chain reaction (PCR) was positive for HSV DNA. The patient was started on acyclovir, which resulted in resolution of the lesions.
Recurrent HSV dermatitis most frequently is encountered in the orolabial or genital regions. After primary infection, HSV is retrogradely taken up into the dorsal root ganglion and may be reactivated in the same dermatome upon stress induction, forming clustered vesicles that rupture to form painful erosions.1 Our patient's history of numerous recurrent episodes in the same area of the palm in the distribution of the median nerve suggests viral latency in the C5 through T1 dorsal root ganglia with reactivation rather than autoinoculation or external infection from another source. The incidence of HSV involving the hand has been estimated at 2.4 cases per 100,000 individuals per year, with finger, thumb, or palm/wrist involvement accounting for 67%, 22%, and 11% of cases, respectively.2 Of the palmar cases that have been reported, most have a positive history for genital or orolabial HSV infection.2-5
In cases of suspected HSV dermatitis with atypical presentations, diagnostic studies are of importance. Although viral culture is the diagnostic gold standard in active lesions, it has lower sensitivity in improperly handled specimens; cases of recurrent disease; and specimens from dried, crusted, or aged lesions,1 which helps to explain the negative culture result in our patient. Viral culture has been largely replaced in clinical practice by nucleic acid amplification tests using PCR, which is fast and type specific.6,7 The sensitivity of PCR approaches 100% when vesicles or wet ulcers are sampled, and PCR has better yields from dry ulcers or crusts compared to viral culture.6 However, because viral shedding is intermittent, a negative PCR result does not rule out HSV infection.8 Additional bedside diagnostic techniques include Tzanck smear, a rapid and inexpensive test in which lesions are scraped and stained with Giemsa, Wright, or Papanicolaou stains. Under light microscopy, multinucleated giant cells are seen in 60% to 75% of cases.9 This method, however, cannot distinguish HSV from varicella-zoster virus and must be followed by direct fluorescent antibody testing or immunohistochemistry for viral typing.1,9 Serologic testing also may be useful in patients who have a suspicious history for HSV infection but do not have lesions on physical examination to diagnose clinically or sample for PCR. Enzyme-linked immunosorbent assay testing can detect IgG starting 3 weeks after infection, and newer type-specific assays can distinguish between HSV types 1 and 2.6 In low-incidence populations, false positives from enzyme-linked immunosorbent assay can be seen and should be confirmed by western blot.6,7
Preferred treatment of HSV includes antiviral medications such as acyclovir, valacyclovir, and famciclovir. Regimens vary based on the site of infection, primary or recurrent nature of the infection, immune status of the patient, and whether or not viral suppression is desired to prevent recurrent outbreaks.7,10
Tinea manuum also may present with unilateral vesicles and erosions involving the palms11; however, it was less likely than HSV dermatitis in this patient presenting with a history of numerous recurrent episodes and without scaling on physical examination. Dyshidrotic eczema, contact dermatitis, and scabies are more characteristically pruritic rather than painful. Additionally, dyshidrotic eczema and scabies would be more likely to have symmetric involvement of the arms. Although vesicles are seen in both dyshidrotic eczema and HSV dermatitis, the vesicles of dyshidrotic eczema usually are noninflammatory compared to the painful vesicles on an erythematous base classically seen in HSV dermatitis.
Acknowledgment
The authors thank Elizabeth Ergen, MD (Knoxville, Tennessee), for her assistance with this case.
The Diagnosis: Herpes Simplex Virus Dermatitis
A swab of the lesions yielded negative varicella-zoster virus and herpes simplex virus (HSV) cultures, but polymerase chain reaction (PCR) was positive for HSV DNA. The patient was started on acyclovir, which resulted in resolution of the lesions.
Recurrent HSV dermatitis most frequently is encountered in the orolabial or genital regions. After primary infection, HSV is retrogradely taken up into the dorsal root ganglion and may be reactivated in the same dermatome upon stress induction, forming clustered vesicles that rupture to form painful erosions.1 Our patient's history of numerous recurrent episodes in the same area of the palm in the distribution of the median nerve suggests viral latency in the C5 through T1 dorsal root ganglia with reactivation rather than autoinoculation or external infection from another source. The incidence of HSV involving the hand has been estimated at 2.4 cases per 100,000 individuals per year, with finger, thumb, or palm/wrist involvement accounting for 67%, 22%, and 11% of cases, respectively.2 Of the palmar cases that have been reported, most have a positive history for genital or orolabial HSV infection.2-5
In cases of suspected HSV dermatitis with atypical presentations, diagnostic studies are of importance. Although viral culture is the diagnostic gold standard in active lesions, it has lower sensitivity in improperly handled specimens; cases of recurrent disease; and specimens from dried, crusted, or aged lesions,1 which helps to explain the negative culture result in our patient. Viral culture has been largely replaced in clinical practice by nucleic acid amplification tests using PCR, which is fast and type specific.6,7 The sensitivity of PCR approaches 100% when vesicles or wet ulcers are sampled, and PCR has better yields from dry ulcers or crusts compared to viral culture.6 However, because viral shedding is intermittent, a negative PCR result does not rule out HSV infection.8 Additional bedside diagnostic techniques include Tzanck smear, a rapid and inexpensive test in which lesions are scraped and stained with Giemsa, Wright, or Papanicolaou stains. Under light microscopy, multinucleated giant cells are seen in 60% to 75% of cases.9 This method, however, cannot distinguish HSV from varicella-zoster virus and must be followed by direct fluorescent antibody testing or immunohistochemistry for viral typing.1,9 Serologic testing also may be useful in patients who have a suspicious history for HSV infection but do not have lesions on physical examination to diagnose clinically or sample for PCR. Enzyme-linked immunosorbent assay testing can detect IgG starting 3 weeks after infection, and newer type-specific assays can distinguish between HSV types 1 and 2.6 In low-incidence populations, false positives from enzyme-linked immunosorbent assay can be seen and should be confirmed by western blot.6,7
Preferred treatment of HSV includes antiviral medications such as acyclovir, valacyclovir, and famciclovir. Regimens vary based on the site of infection, primary or recurrent nature of the infection, immune status of the patient, and whether or not viral suppression is desired to prevent recurrent outbreaks.7,10
Tinea manuum also may present with unilateral vesicles and erosions involving the palms11; however, it was less likely than HSV dermatitis in this patient presenting with a history of numerous recurrent episodes and without scaling on physical examination. Dyshidrotic eczema, contact dermatitis, and scabies are more characteristically pruritic rather than painful. Additionally, dyshidrotic eczema and scabies would be more likely to have symmetric involvement of the arms. Although vesicles are seen in both dyshidrotic eczema and HSV dermatitis, the vesicles of dyshidrotic eczema usually are noninflammatory compared to the painful vesicles on an erythematous base classically seen in HSV dermatitis.
Acknowledgment
The authors thank Elizabeth Ergen, MD (Knoxville, Tennessee), for her assistance with this case.
- Fatahzadeh M, Schwartz RA. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57:737-763; quiz 764-766.
- Gill MJ, Arlette J, Buchan K. Herpes simplex virus infection of the hand. a profile of 79 cases. Am J Med. 1988;84:89-93.
- Widenfalk B, Wallin J. Recurrent herpes simplex virus infections in the adult hand. Scand J Plast Reconstr Surg Hand Surg. 1988;22:177-180.
- Gill MJ, Arlette J, Buchan KA. Herpes simplex virus infection of the hand. J Am Acad Dermatol. 1990;22:111-116.
- Osio A, Fremont G, Petit A, et al. An unusual bipolar primary herpes simplex virus 1 infection. J Clin Virol. 2008;43:230-232.
- Gnann JW Jr, Whitley RJ. Clinical practice. genital herpes. N Engl J Med. 2016;375:666-674.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
- Downing C, Mendoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- WHO Guidelines Approved by the Guidelines Review Committee. WHO Guidelines for the Treatment of Genital Herpes Simplex Virus. Geneva, Switzerland: World Health Organization; 2016.
- Veraldi S, Schianchi R, Benzecry V, et al. Tinea manuum: a report of 18 cases observed in the metropolitan area of Milan and review of the literature. Mycoses. 2019;62:604-608.
- Fatahzadeh M, Schwartz RA. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57:737-763; quiz 764-766.
- Gill MJ, Arlette J, Buchan K. Herpes simplex virus infection of the hand. a profile of 79 cases. Am J Med. 1988;84:89-93.
- Widenfalk B, Wallin J. Recurrent herpes simplex virus infections in the adult hand. Scand J Plast Reconstr Surg Hand Surg. 1988;22:177-180.
- Gill MJ, Arlette J, Buchan KA. Herpes simplex virus infection of the hand. J Am Acad Dermatol. 1990;22:111-116.
- Osio A, Fremont G, Petit A, et al. An unusual bipolar primary herpes simplex virus 1 infection. J Clin Virol. 2008;43:230-232.
- Gnann JW Jr, Whitley RJ. Clinical practice. genital herpes. N Engl J Med. 2016;375:666-674.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
- Downing C, Mendoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- WHO Guidelines Approved by the Guidelines Review Committee. WHO Guidelines for the Treatment of Genital Herpes Simplex Virus. Geneva, Switzerland: World Health Organization; 2016.
- Veraldi S, Schianchi R, Benzecry V, et al. Tinea manuum: a report of 18 cases observed in the metropolitan area of Milan and review of the literature. Mycoses. 2019;62:604-608.
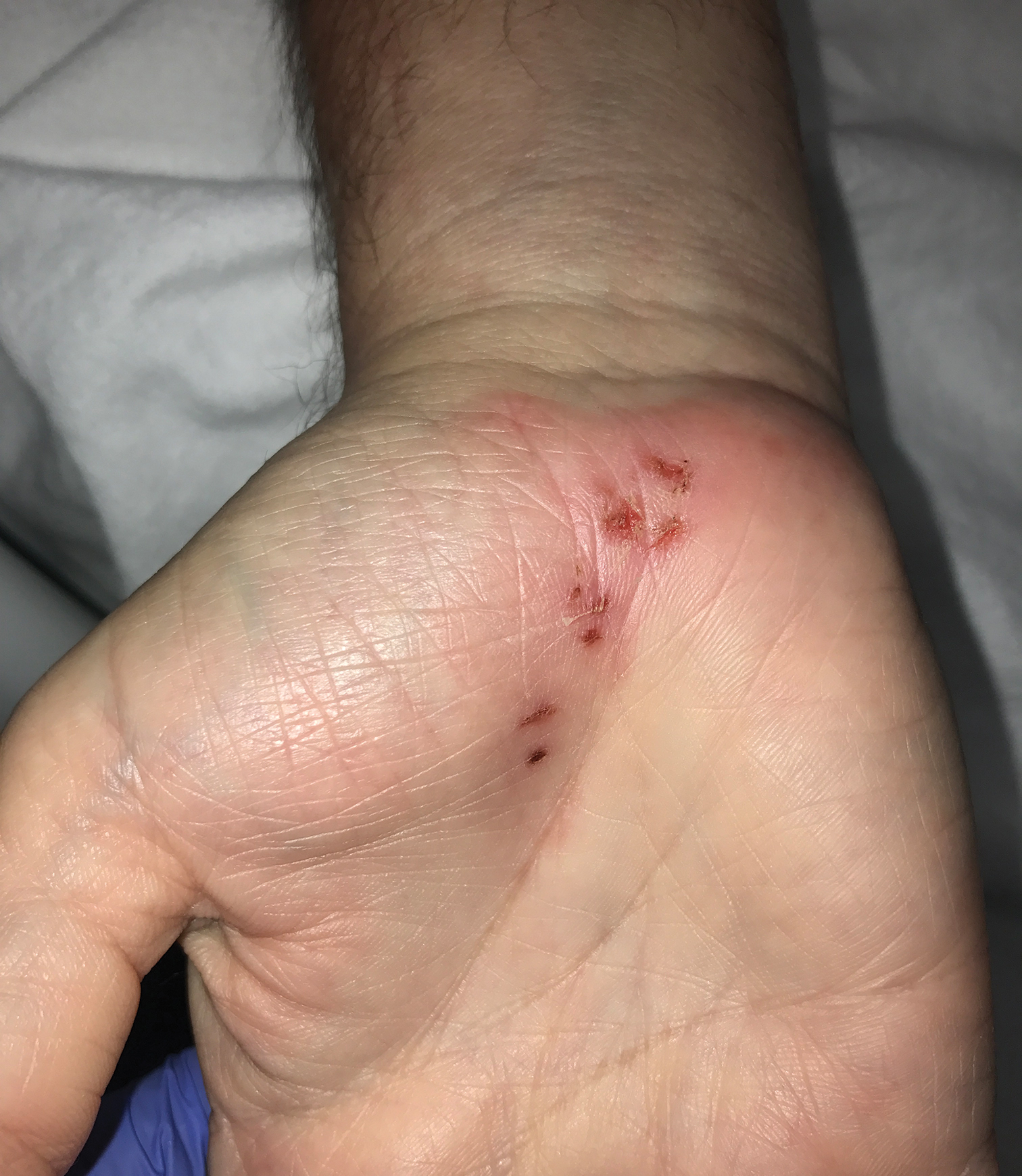
A 54-year-old man presented to the emergency department with painful lesions at the base of the right palm that had progressed to include areas of erythema and warmth migrating proximally along the right forearm and distal right arm. He stated that similar lesions had occurred episodically in the same location approximately 100 times over the last 20 years. Each time, the lesions began as painful vesicles that he subsequently popped with a sewing needle. He denied any history of orolabial or genital herpes simplex virus infection. Physical examination revealed erythematous scattered papules with dry hemorrhagic crust over the base of the right palm with expressible serous fluid upon forceful pressure. Swelling, erythema, and warmth of the distal right forearm also were observed.
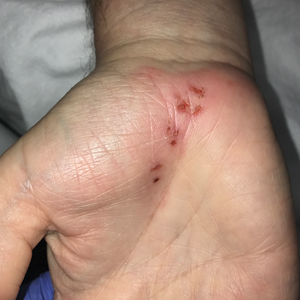
Chronic Diarrhea in an Adolescent Girl With a Genetic Skin Condition
The Diagnosis: Netherton Syndrome
Netherton syndrome (NS) is a rare autosomal-recessive disorder characterized by a clinical triad of ichthyosis linearis circumflexa; atopic diathesis; and hair shaft abnormalities, most classically trichorrhexis invaginata.1 Netherton syndrome is caused by a loss-of-function mutation in the serine peptidase inhibitor Kazal-type gene, SPINK5, which encodes LEKTI proteins and is found in all stratified epithelia as well as the thymus.2 A lack of functional LEKTI leads to the activation of a cascade of allergy and inflammation as well as uncontrolled proteolytic activity in the stratum corneum, which causes increased desquamation.1
Netherton syndrome presents with serpiginous or circinate scaling plaques with double-edged scale referred to as ichthyosis linearis circumflexa (quiz image). Skin plaques are intensely pruritic and migratory with fluctuating severity. Alternately, patients may have a generalized scaling erythroderma. Infants are at an especially high risk for recurrent infections, sepsis, hypernatremic dehydration, and failure to thrive.2
Netherton syndrome often gradually improves over time, though adults with NS usually have intensely pruritic, localized patches of redness, scaling, or ichthyosis linearis circumflexa. Lichenification and eczematous plaques of the popliteal and antecubital fossae also are common.1 Therapeutic options for NS include emollients, topical steroids, phototherapy, and intravenous immunoglobulin for severe cases.3 Because there is skin barrier dysfunction in NS, supratherapeutic serum levels of tacrolimus following topical application have been reported.4 Topical pimecrolimus has been demonstrated as an effective and safer application.5 Trichorrhexis invaginata (also known as bamboo hair) of the hair and eyebrows is a pathognomonic finding in NS, involving invagination of the distal hair shaft into the proximal shaft on light microscopy examination.1
Histopathology is variable and nonspecific with psoriasiform hyperplasia as the most frequent finding. Other histologic findings include incomplete keratinization of the epidermis, incomplete cornification with a severely reduced granular layer, and mild to moderate inflammatory dermal infiltrate.6 LEKTI immunostaining is confirmatory and shows the reduction or complete absence of LEKTI in the granular layer and inner root sheath of follicles.1 Patchy LEKTI staining would be suggestive of atopic dermatitis and psoriasis instead of NS.2
Atopic manifestations include angioedema, urticaria, and anaphylaxis, as well as chronic diarrhea or vomiting due to food allergies.1 Elevated IgE levels for staple foods (eg, milk, wheat), elevated total serum IgE, and eosinophilia frequently are seen.7 Biopsy of the esophagus and colon likely would show mucosal eosinophilia.7,8 Elimination of major food triggers through specific serum IgE testing and oral allergen desensitization can lead to the reduction of digestive symptoms.9 Cisapride and omeprazole are effective treatments for gastroesophageal reflux and poor feeding.8 Biopsy of the intestines in this patient likely would not have shown total villous atrophy, which is rare and primarily reported in infants with NS who have failure to thrive.10 There is a limited association between NS and intestinal metaplasia, intraepithelial lymphocytes, and bacterial overgrowth.
The primary morphology of dyskeratosis follicularis includes keratotic papules developing in sebaceous areas of the skin rather than scaly serpiginous plaques as seen in NS. Elastosis perforans serpiginosa is a perforating disorder seen in the context of several genetic conditions. It has a serpiginous appearance but, unlike NS, tends to be localized and features keratotic papules rather than patches with scale. Erythema marginatum is an uncommon feature of rheumatic fever and appears as pink annular macules and tends not to be pruritic. Subacute cutaneous lupus does feature scaly annular and serpiginous plaques but features trailing scale without the double-edge appearance of NS and is acquired rather than genetic.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289-300.
- Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Human Mol Genet. 2003;12:2417-2430.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Shah KN, Yan AC. Low but detectable serum levels of tacrolimus seen with the use of very dilute, extemporaneously compounded formulations of tacrolimus ointment in the treatment of patients with Netherton syndrome. Arch Dermatol. 2006;142:1362-1363.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Leclerc-Mercier S, Bodemer C, Furio L, et al. Skin biopsy in Netherton syndrome: a histological review of a large series and new findings. Am J Dermatopathol. 2016;38:83-91.
- Pauluel-Marmont C, Bellon N, Barbet P, et al. Eosinophilic esophagitis and colonic mucosal eosinophilia in Netherton syndrome. J Allergy Clin Immunol. 2017;139:2003-2005.e1.
- Hannula-Jouppi K, Laasanen SL, Heikkila H, et al. IgE allergen component-based profiling and atopic manifestations in patients with Netherton syndrome. J Allergy Clin Immunol. 2014;134:985-988.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006;4:1097-1102.
- Judge MR, Morgan G , Harper JI. A clinical and immunological study of Netherton's syndrome. Br J Dermatol. 1994;131:615-21.
The Diagnosis: Netherton Syndrome
Netherton syndrome (NS) is a rare autosomal-recessive disorder characterized by a clinical triad of ichthyosis linearis circumflexa; atopic diathesis; and hair shaft abnormalities, most classically trichorrhexis invaginata.1 Netherton syndrome is caused by a loss-of-function mutation in the serine peptidase inhibitor Kazal-type gene, SPINK5, which encodes LEKTI proteins and is found in all stratified epithelia as well as the thymus.2 A lack of functional LEKTI leads to the activation of a cascade of allergy and inflammation as well as uncontrolled proteolytic activity in the stratum corneum, which causes increased desquamation.1
Netherton syndrome presents with serpiginous or circinate scaling plaques with double-edged scale referred to as ichthyosis linearis circumflexa (quiz image). Skin plaques are intensely pruritic and migratory with fluctuating severity. Alternately, patients may have a generalized scaling erythroderma. Infants are at an especially high risk for recurrent infections, sepsis, hypernatremic dehydration, and failure to thrive.2
Netherton syndrome often gradually improves over time, though adults with NS usually have intensely pruritic, localized patches of redness, scaling, or ichthyosis linearis circumflexa. Lichenification and eczematous plaques of the popliteal and antecubital fossae also are common.1 Therapeutic options for NS include emollients, topical steroids, phototherapy, and intravenous immunoglobulin for severe cases.3 Because there is skin barrier dysfunction in NS, supratherapeutic serum levels of tacrolimus following topical application have been reported.4 Topical pimecrolimus has been demonstrated as an effective and safer application.5 Trichorrhexis invaginata (also known as bamboo hair) of the hair and eyebrows is a pathognomonic finding in NS, involving invagination of the distal hair shaft into the proximal shaft on light microscopy examination.1
Histopathology is variable and nonspecific with psoriasiform hyperplasia as the most frequent finding. Other histologic findings include incomplete keratinization of the epidermis, incomplete cornification with a severely reduced granular layer, and mild to moderate inflammatory dermal infiltrate.6 LEKTI immunostaining is confirmatory and shows the reduction or complete absence of LEKTI in the granular layer and inner root sheath of follicles.1 Patchy LEKTI staining would be suggestive of atopic dermatitis and psoriasis instead of NS.2
Atopic manifestations include angioedema, urticaria, and anaphylaxis, as well as chronic diarrhea or vomiting due to food allergies.1 Elevated IgE levels for staple foods (eg, milk, wheat), elevated total serum IgE, and eosinophilia frequently are seen.7 Biopsy of the esophagus and colon likely would show mucosal eosinophilia.7,8 Elimination of major food triggers through specific serum IgE testing and oral allergen desensitization can lead to the reduction of digestive symptoms.9 Cisapride and omeprazole are effective treatments for gastroesophageal reflux and poor feeding.8 Biopsy of the intestines in this patient likely would not have shown total villous atrophy, which is rare and primarily reported in infants with NS who have failure to thrive.10 There is a limited association between NS and intestinal metaplasia, intraepithelial lymphocytes, and bacterial overgrowth.
The primary morphology of dyskeratosis follicularis includes keratotic papules developing in sebaceous areas of the skin rather than scaly serpiginous plaques as seen in NS. Elastosis perforans serpiginosa is a perforating disorder seen in the context of several genetic conditions. It has a serpiginous appearance but, unlike NS, tends to be localized and features keratotic papules rather than patches with scale. Erythema marginatum is an uncommon feature of rheumatic fever and appears as pink annular macules and tends not to be pruritic. Subacute cutaneous lupus does feature scaly annular and serpiginous plaques but features trailing scale without the double-edge appearance of NS and is acquired rather than genetic.
The Diagnosis: Netherton Syndrome
Netherton syndrome (NS) is a rare autosomal-recessive disorder characterized by a clinical triad of ichthyosis linearis circumflexa; atopic diathesis; and hair shaft abnormalities, most classically trichorrhexis invaginata.1 Netherton syndrome is caused by a loss-of-function mutation in the serine peptidase inhibitor Kazal-type gene, SPINK5, which encodes LEKTI proteins and is found in all stratified epithelia as well as the thymus.2 A lack of functional LEKTI leads to the activation of a cascade of allergy and inflammation as well as uncontrolled proteolytic activity in the stratum corneum, which causes increased desquamation.1
Netherton syndrome presents with serpiginous or circinate scaling plaques with double-edged scale referred to as ichthyosis linearis circumflexa (quiz image). Skin plaques are intensely pruritic and migratory with fluctuating severity. Alternately, patients may have a generalized scaling erythroderma. Infants are at an especially high risk for recurrent infections, sepsis, hypernatremic dehydration, and failure to thrive.2
Netherton syndrome often gradually improves over time, though adults with NS usually have intensely pruritic, localized patches of redness, scaling, or ichthyosis linearis circumflexa. Lichenification and eczematous plaques of the popliteal and antecubital fossae also are common.1 Therapeutic options for NS include emollients, topical steroids, phototherapy, and intravenous immunoglobulin for severe cases.3 Because there is skin barrier dysfunction in NS, supratherapeutic serum levels of tacrolimus following topical application have been reported.4 Topical pimecrolimus has been demonstrated as an effective and safer application.5 Trichorrhexis invaginata (also known as bamboo hair) of the hair and eyebrows is a pathognomonic finding in NS, involving invagination of the distal hair shaft into the proximal shaft on light microscopy examination.1
Histopathology is variable and nonspecific with psoriasiform hyperplasia as the most frequent finding. Other histologic findings include incomplete keratinization of the epidermis, incomplete cornification with a severely reduced granular layer, and mild to moderate inflammatory dermal infiltrate.6 LEKTI immunostaining is confirmatory and shows the reduction or complete absence of LEKTI in the granular layer and inner root sheath of follicles.1 Patchy LEKTI staining would be suggestive of atopic dermatitis and psoriasis instead of NS.2
Atopic manifestations include angioedema, urticaria, and anaphylaxis, as well as chronic diarrhea or vomiting due to food allergies.1 Elevated IgE levels for staple foods (eg, milk, wheat), elevated total serum IgE, and eosinophilia frequently are seen.7 Biopsy of the esophagus and colon likely would show mucosal eosinophilia.7,8 Elimination of major food triggers through specific serum IgE testing and oral allergen desensitization can lead to the reduction of digestive symptoms.9 Cisapride and omeprazole are effective treatments for gastroesophageal reflux and poor feeding.8 Biopsy of the intestines in this patient likely would not have shown total villous atrophy, which is rare and primarily reported in infants with NS who have failure to thrive.10 There is a limited association between NS and intestinal metaplasia, intraepithelial lymphocytes, and bacterial overgrowth.
The primary morphology of dyskeratosis follicularis includes keratotic papules developing in sebaceous areas of the skin rather than scaly serpiginous plaques as seen in NS. Elastosis perforans serpiginosa is a perforating disorder seen in the context of several genetic conditions. It has a serpiginous appearance but, unlike NS, tends to be localized and features keratotic papules rather than patches with scale. Erythema marginatum is an uncommon feature of rheumatic fever and appears as pink annular macules and tends not to be pruritic. Subacute cutaneous lupus does feature scaly annular and serpiginous plaques but features trailing scale without the double-edge appearance of NS and is acquired rather than genetic.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289-300.
- Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Human Mol Genet. 2003;12:2417-2430.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Shah KN, Yan AC. Low but detectable serum levels of tacrolimus seen with the use of very dilute, extemporaneously compounded formulations of tacrolimus ointment in the treatment of patients with Netherton syndrome. Arch Dermatol. 2006;142:1362-1363.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Leclerc-Mercier S, Bodemer C, Furio L, et al. Skin biopsy in Netherton syndrome: a histological review of a large series and new findings. Am J Dermatopathol. 2016;38:83-91.
- Pauluel-Marmont C, Bellon N, Barbet P, et al. Eosinophilic esophagitis and colonic mucosal eosinophilia in Netherton syndrome. J Allergy Clin Immunol. 2017;139:2003-2005.e1.
- Hannula-Jouppi K, Laasanen SL, Heikkila H, et al. IgE allergen component-based profiling and atopic manifestations in patients with Netherton syndrome. J Allergy Clin Immunol. 2014;134:985-988.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006;4:1097-1102.
- Judge MR, Morgan G , Harper JI. A clinical and immunological study of Netherton's syndrome. Br J Dermatol. 1994;131:615-21.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289-300.
- Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Human Mol Genet. 2003;12:2417-2430.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Shah KN, Yan AC. Low but detectable serum levels of tacrolimus seen with the use of very dilute, extemporaneously compounded formulations of tacrolimus ointment in the treatment of patients with Netherton syndrome. Arch Dermatol. 2006;142:1362-1363.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Leclerc-Mercier S, Bodemer C, Furio L, et al. Skin biopsy in Netherton syndrome: a histological review of a large series and new findings. Am J Dermatopathol. 2016;38:83-91.
- Pauluel-Marmont C, Bellon N, Barbet P, et al. Eosinophilic esophagitis and colonic mucosal eosinophilia in Netherton syndrome. J Allergy Clin Immunol. 2017;139:2003-2005.e1.
- Hannula-Jouppi K, Laasanen SL, Heikkila H, et al. IgE allergen component-based profiling and atopic manifestations in patients with Netherton syndrome. J Allergy Clin Immunol. 2014;134:985-988.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006;4:1097-1102.
- Judge MR, Morgan G , Harper JI. A clinical and immunological study of Netherton's syndrome. Br J Dermatol. 1994;131:615-21.
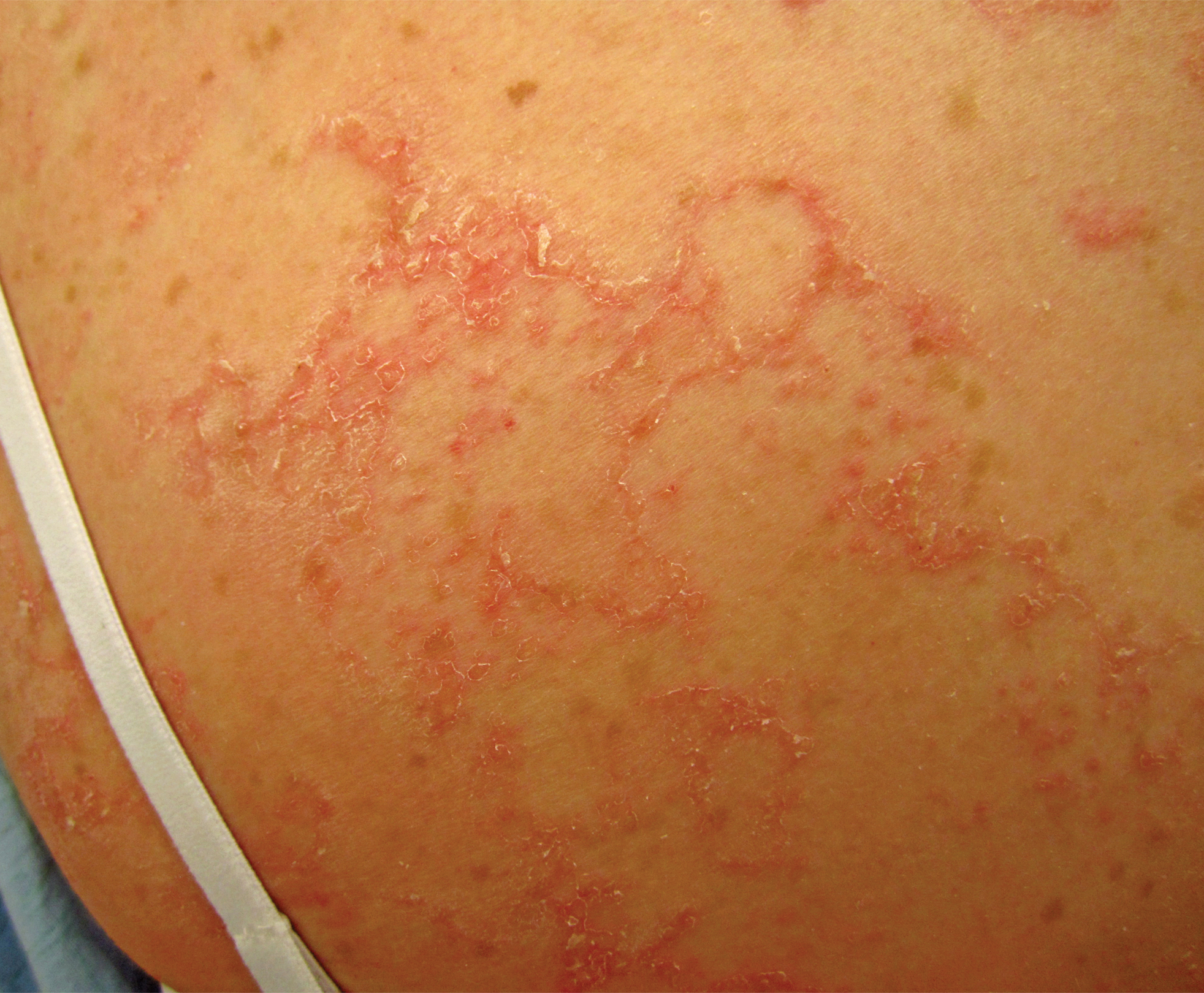
A 17-year-old adolescent girl visited our clinic to establish care for her genetic skin condition. She exhibited red scaly plaques and patches over much of the body surface area consistent with atopic dermatitis but also had areas on the trunk with serpiginous red plaques with scale on the leading and trailing edges. She also noted fragile hair with sparse eyebrows. The patient reported that she had experienced chronic diarrhea and abdominal pain since childhood. She asked if it could be related to her genetic condition.
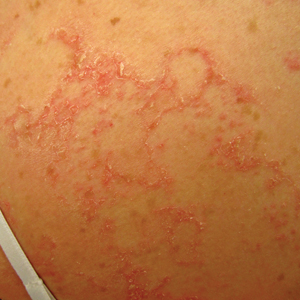
Unilateral Vesicular Eruption in a Neonate
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.

A 4-day-old female neonate presented to the dermatology clinic with a vesicular eruption on the left leg of 1 day's duration. The eruption was asymptomatic without any extracutaneous findings. This term infant was born without complication, and the mother denied any symptoms consistent with herpes simplex virus infection. Physical examination revealed yellow-red vesicles on an erythematous base in a blaschkoid distribution on the left leg. The rest of the examination was unremarkable. Herpes simplex virus polymerase chain reaction testing was negative.
Bleeding Hand Mass in an Older Man
The Diagnosis: Epithelioid Angiosarcoma
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.
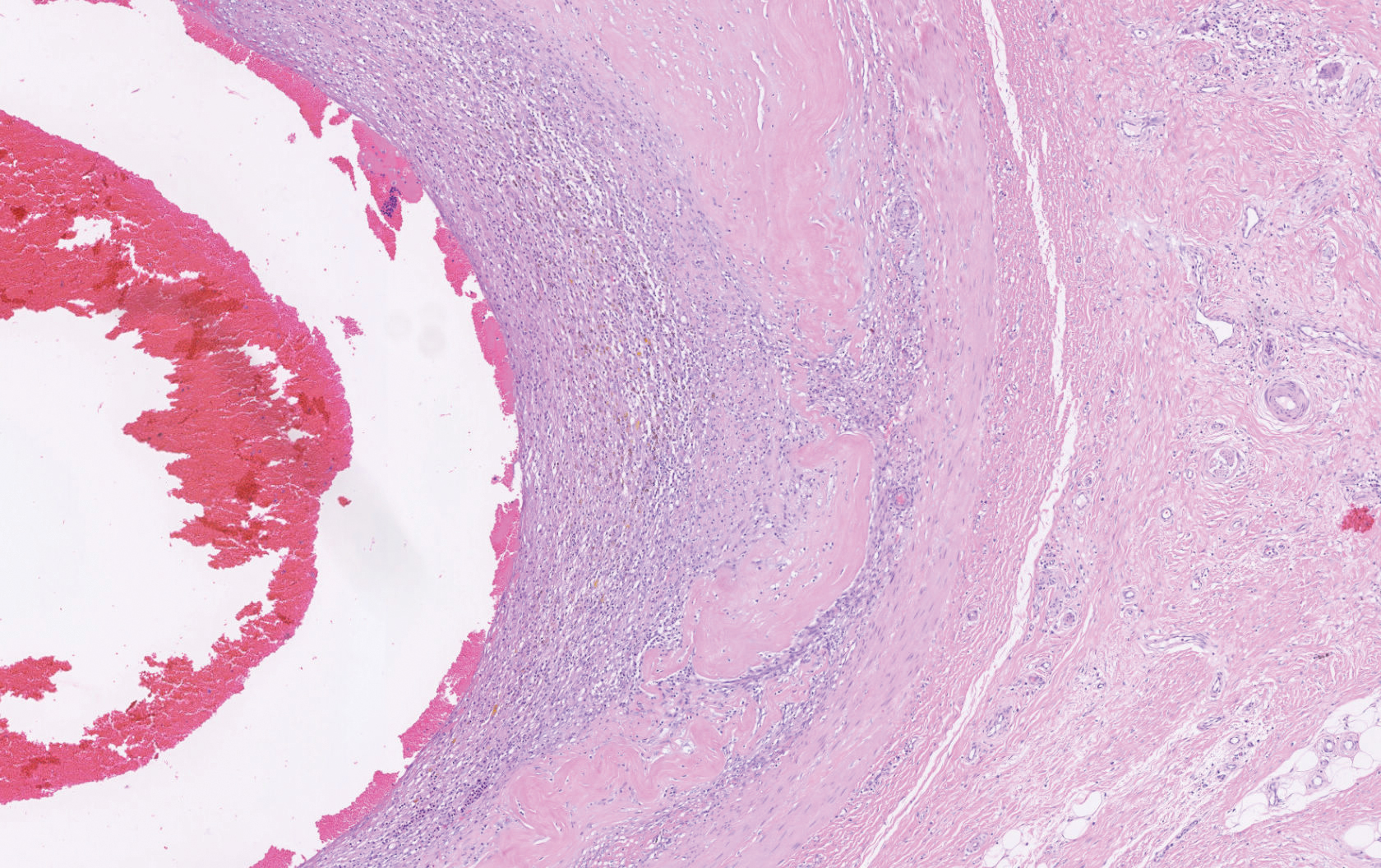
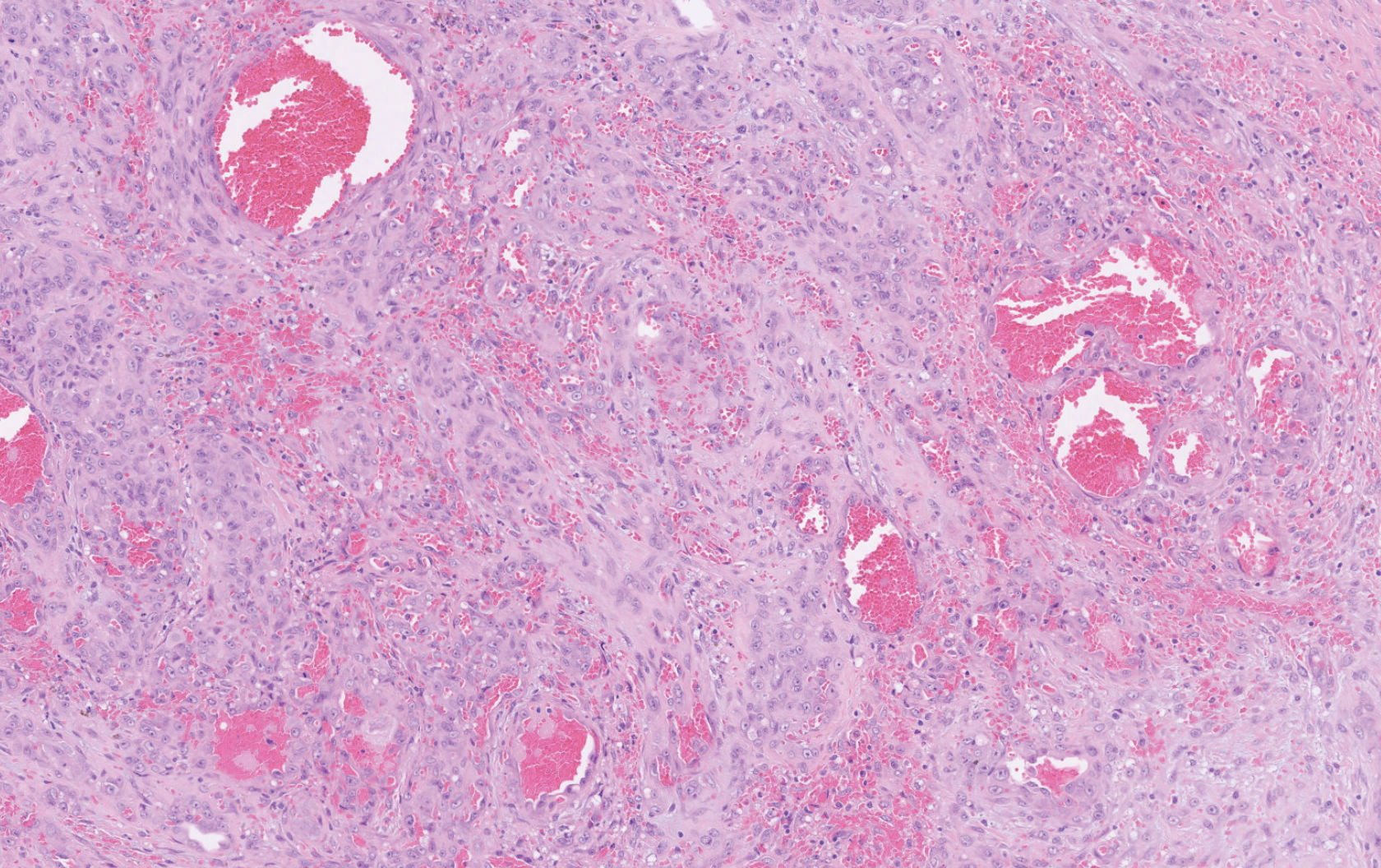
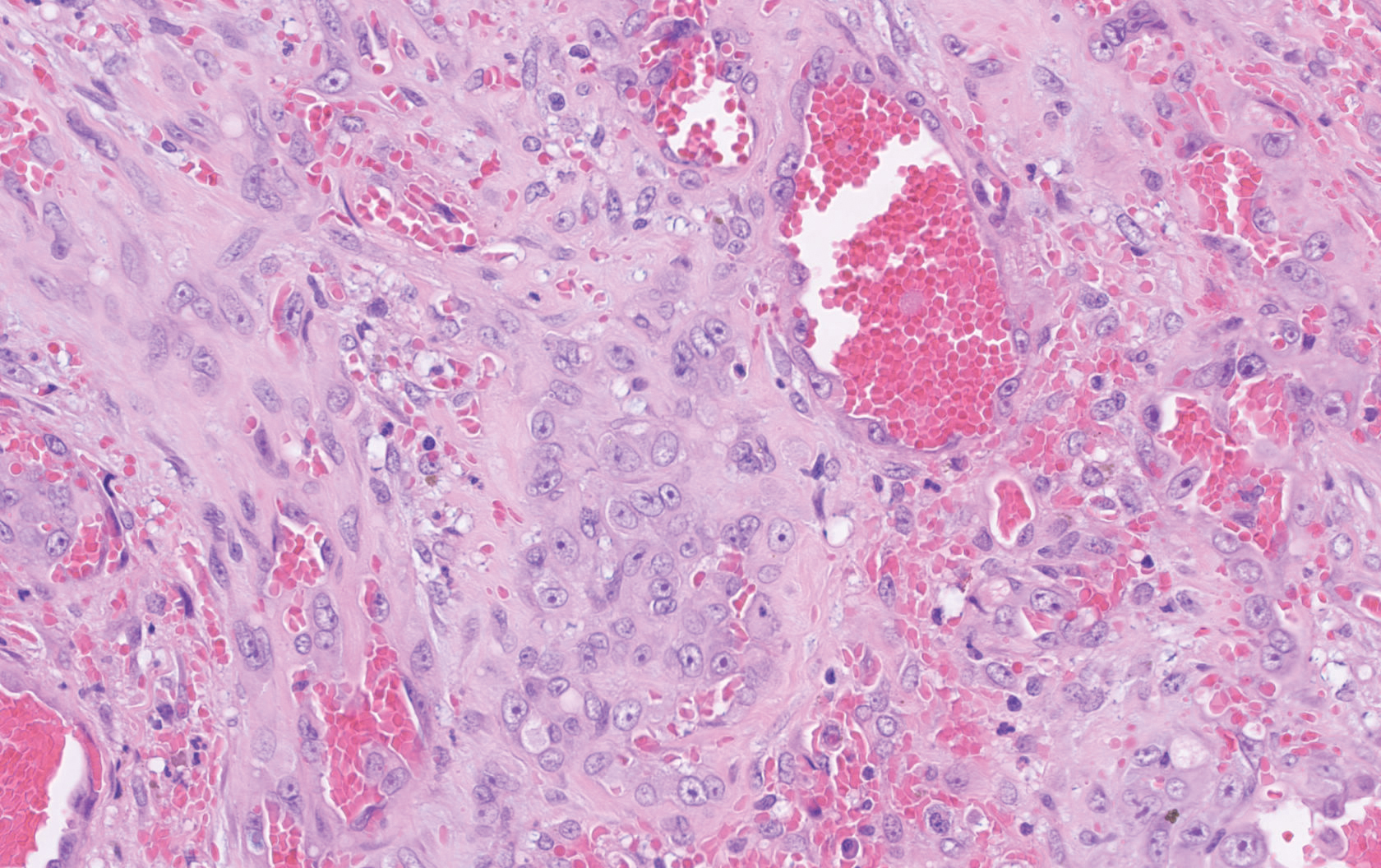

Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
The Diagnosis: Epithelioid Angiosarcoma
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.
Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
The Diagnosis: Epithelioid Angiosarcoma
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.
Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
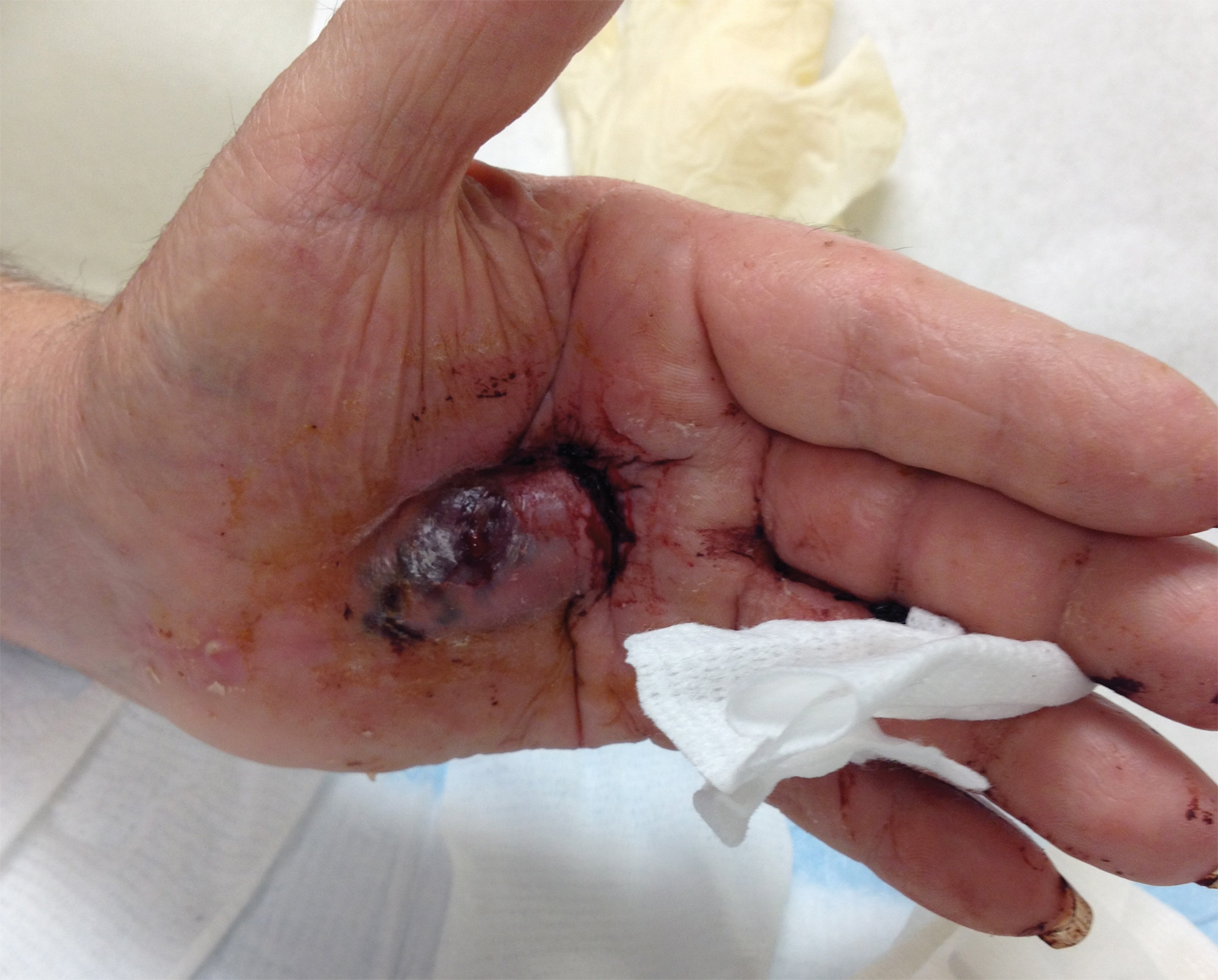
A 72-year-old man presented for evaluation of a mass on the left hand that continued to grow over the last few months and eventually bled. The patient first noticed a small firm lump on the palm approximately 1 year prior to presentation, and it was originally diagnosed as a Dupuytren contracture by his primary care physician. Months later, the lesion grew and began to bleed. Magnetic resonance imaging showed large hematomas of the hand with areas of nodular enhancement. The mass was located between the third and fourth proximal phalanges and abutted the extensor tendon. Complete excision yielded a definitive diagnosis.
Broadly Distributed Vascular Macules in a Pediatric Patient
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
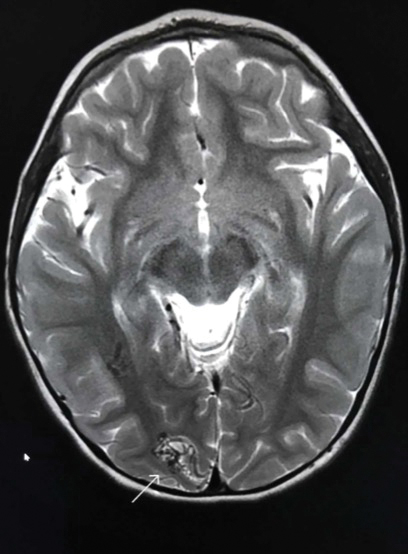
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
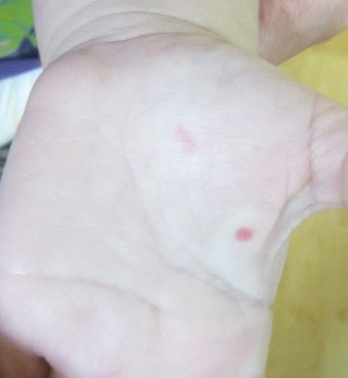
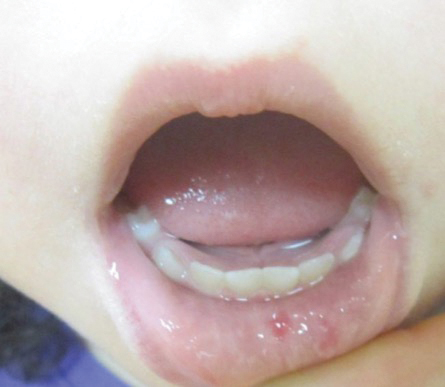
A 2-year-old girl presented with an erythematous macule on the left nasal sidewall that had been present since birth as well as other similar-appearing macules that had slowly evolved over the last 2 years. The patient was born via normal spontaneous vaginal delivery to healthy parents. She had 2 healthy siblings. Her parents reported that she was otherwise growing and developing normally. The patient had no history of epistaxis, and there was no family history of vascular anomalies. Physical examination revealed 2- to 6-mm vascular macules that blanched with pressure and filled quickly thereafter on the left nasal sidewall, upper (top) and lower extremities, and trunk. Some macules were surrounded by blanched halos. Several 1-mm red macules also were noted on the exterior and interior of the mucosal lower lip (bottom).