User login
Topical treatment for EB recommended for approval in the EU
A topical (EMA’s) Committee for Medicinal Products for Human Use.
“The benefit of Filsuvez is its ability to promote healing of EB partial thickness wounds,” the EMA said in an announcement on April 22. “It is thought to work by modulating inflammatory mediators and stimulating keratinocyte differentiation and migration, thereby promoting wound health and closure,” the statement adds.
The recommended indication for the product – developed by Amryt Pharmaceuticals DAC and currently designated as an orphan drug – is for the treatment of partial-thickness wounds associated with dystrophic and junctional EB in patients aged 6 months and older. The recommendation for approval came after the EMA sought and received external advice from independent physicians treating EB and from patients with the rare disease.
The most common side effects, according to the EMA announcement, are wound complications, application site reactions, wound infections, pruritus, and hypersensitivity reactions.
In February 2022, the Food and Drug Administration declined to approve the company’s new drug application as it was presented and asked the company to submit additional evidence of effectiveness for Oleogel-S10 in EB, the company announced at that time. The statement noted that the company was committed to working with the FDA to identify "the most expeditious pathway towards a potential approval.”
The company’s pivotal phase 3 trial enrolled 223 patients with EB, including 156 pediatric patients. The patients variously had three types of EB. The trial has two components: A 3-month, double-blind, randomized controlled phase, which has been completed, and an ongoing 24-month open-label, single-arm phase. The trial is being performed at 58 sites in 28 countries.
Results from the randomized controlled phase, reported in 2020, include a statistically significant increase in the proportion of patients achieving complete closure of an EB target wound within 45 days: 41.3% in the Oleogel-S10 group and 28.9% in the control group (P = .013). (Target wounds measured 10 cm² to 50 cm² and were present for at least 21 days but less than 9 months.) The safety profile of the treatment gel was acceptable and was well tolerated, compared with the control gel, according to Amryt’s press release. The results were presented at the European Academy of Dermatology and Venereology Congress in October 2020.
Data from a 12-month interim analysis of the follow-up phase were presented at the annual meeting of the American Academy of Dermatology in March 2022. Results showed further reductions in total body surface area percentage wounding to 5.4% among (from 7.4% at the end of the double-blind period and 12.1% at the beginning of the study) among the patients who continued treatment and who underwent assessment, according to a company press release. Treatment was well tolerated, and no new safety signals were identified, the release said.
A decision by the European Commission is expected within the next 2 months.
A version of this article first appeared on Medscape.com.
A topical (EMA’s) Committee for Medicinal Products for Human Use.
“The benefit of Filsuvez is its ability to promote healing of EB partial thickness wounds,” the EMA said in an announcement on April 22. “It is thought to work by modulating inflammatory mediators and stimulating keratinocyte differentiation and migration, thereby promoting wound health and closure,” the statement adds.
The recommended indication for the product – developed by Amryt Pharmaceuticals DAC and currently designated as an orphan drug – is for the treatment of partial-thickness wounds associated with dystrophic and junctional EB in patients aged 6 months and older. The recommendation for approval came after the EMA sought and received external advice from independent physicians treating EB and from patients with the rare disease.
The most common side effects, according to the EMA announcement, are wound complications, application site reactions, wound infections, pruritus, and hypersensitivity reactions.
In February 2022, the Food and Drug Administration declined to approve the company’s new drug application as it was presented and asked the company to submit additional evidence of effectiveness for Oleogel-S10 in EB, the company announced at that time. The statement noted that the company was committed to working with the FDA to identify "the most expeditious pathway towards a potential approval.”
The company’s pivotal phase 3 trial enrolled 223 patients with EB, including 156 pediatric patients. The patients variously had three types of EB. The trial has two components: A 3-month, double-blind, randomized controlled phase, which has been completed, and an ongoing 24-month open-label, single-arm phase. The trial is being performed at 58 sites in 28 countries.
Results from the randomized controlled phase, reported in 2020, include a statistically significant increase in the proportion of patients achieving complete closure of an EB target wound within 45 days: 41.3% in the Oleogel-S10 group and 28.9% in the control group (P = .013). (Target wounds measured 10 cm² to 50 cm² and were present for at least 21 days but less than 9 months.) The safety profile of the treatment gel was acceptable and was well tolerated, compared with the control gel, according to Amryt’s press release. The results were presented at the European Academy of Dermatology and Venereology Congress in October 2020.
Data from a 12-month interim analysis of the follow-up phase were presented at the annual meeting of the American Academy of Dermatology in March 2022. Results showed further reductions in total body surface area percentage wounding to 5.4% among (from 7.4% at the end of the double-blind period and 12.1% at the beginning of the study) among the patients who continued treatment and who underwent assessment, according to a company press release. Treatment was well tolerated, and no new safety signals were identified, the release said.
A decision by the European Commission is expected within the next 2 months.
A version of this article first appeared on Medscape.com.
A topical (EMA’s) Committee for Medicinal Products for Human Use.
“The benefit of Filsuvez is its ability to promote healing of EB partial thickness wounds,” the EMA said in an announcement on April 22. “It is thought to work by modulating inflammatory mediators and stimulating keratinocyte differentiation and migration, thereby promoting wound health and closure,” the statement adds.
The recommended indication for the product – developed by Amryt Pharmaceuticals DAC and currently designated as an orphan drug – is for the treatment of partial-thickness wounds associated with dystrophic and junctional EB in patients aged 6 months and older. The recommendation for approval came after the EMA sought and received external advice from independent physicians treating EB and from patients with the rare disease.
The most common side effects, according to the EMA announcement, are wound complications, application site reactions, wound infections, pruritus, and hypersensitivity reactions.
In February 2022, the Food and Drug Administration declined to approve the company’s new drug application as it was presented and asked the company to submit additional evidence of effectiveness for Oleogel-S10 in EB, the company announced at that time. The statement noted that the company was committed to working with the FDA to identify "the most expeditious pathway towards a potential approval.”
The company’s pivotal phase 3 trial enrolled 223 patients with EB, including 156 pediatric patients. The patients variously had three types of EB. The trial has two components: A 3-month, double-blind, randomized controlled phase, which has been completed, and an ongoing 24-month open-label, single-arm phase. The trial is being performed at 58 sites in 28 countries.
Results from the randomized controlled phase, reported in 2020, include a statistically significant increase in the proportion of patients achieving complete closure of an EB target wound within 45 days: 41.3% in the Oleogel-S10 group and 28.9% in the control group (P = .013). (Target wounds measured 10 cm² to 50 cm² and were present for at least 21 days but less than 9 months.) The safety profile of the treatment gel was acceptable and was well tolerated, compared with the control gel, according to Amryt’s press release. The results were presented at the European Academy of Dermatology and Venereology Congress in October 2020.
Data from a 12-month interim analysis of the follow-up phase were presented at the annual meeting of the American Academy of Dermatology in March 2022. Results showed further reductions in total body surface area percentage wounding to 5.4% among (from 7.4% at the end of the double-blind period and 12.1% at the beginning of the study) among the patients who continued treatment and who underwent assessment, according to a company press release. Treatment was well tolerated, and no new safety signals were identified, the release said.
A decision by the European Commission is expected within the next 2 months.
A version of this article first appeared on Medscape.com.
Which solid organ transplant recipients face the highest risk of skin cancer?
BOSTON – .
White patients who meet these criteria should be screening within 2 years after transplant, while Black patients should be screened within 5 years after transplant, Ally-Khan Somani, MD, PhD, said at the annual meeting of the American Academy of Dermatology.

Dr. Somani, director of dermatologic surgery and the division of cutaneous oncology at Indiana University, Indianapolis, based his remarks on consensus screening guidelines assembled from three rounds of Delphi method surveys with 47 dermatologists and 37 transplant physicians, with the goal of establishing skin cancer screening recommendations for SOTRs. Among the dermatologists surveyed, 45% were Mohs surgeons and 55% were general dermatologists.
The panel recommended that the transplant team should perform risk assessment for SOTRs to risk stratify patients for skin cancer screening (high risk vs. low risk). They also proposed that dermatologists perform skin cancer screening by full-body skin examinations, and that SOTRs with a history of skin cancer should continue with routine skin cancer surveillance as recommended by their dermatologists.
Those at low risk for skin cancer include abdominal organ recipients, SOTR age of younger than 50 at time of transplant, and female gender. The guidelines recommend that White, Asian, and Hispanic patients who meet those criteria should be screened within 5 years after transplant, while no consensus was reached for Black patients who meet those criteria.
Based on posttransplant skin cancer incidence rates, risk is increased among males, Whites, thoracic organ recipients, and being age 50 or older, Dr. Somani said. “At our institution, we make sure there’s a good connection between our transplant teams and dermatologists. We recommend rapid referral for suspicious lesions and we educate patients and screen them within 1 year of transplant, or sooner for high-risk patients. Surveillance is increased to every 3 or 4 months for patients with a history of multiple or high-risk cancers or sooner, followed by routine surveillance as recommended by the patient’s dermatologist.”
To risk stratify patients on the development of their first skin cancer post transplantation, researchers developed the Skin and Ultraviolet Neoplasia Transplant Risk Assessment Calculator (SUNTRAC), a prediction tool with a freely available app. Data for the tool were drawn from the Transplant Skin Cancer Network study, a 5-year analysis of 6,340 adult recipients of a first solid organ transplant at 26 transplant centers in the United States. It generates a risk score for SOTRs (low, medium, high, or very high), which informs transplant care providers of a patient’s risk of skin cancer.
Dr. Somani disclosed that he has received grants and funding from Castle Biosciences. He is an adviser to Cook Biotech and a consultant to Sanara MedTech.
BOSTON – .
White patients who meet these criteria should be screening within 2 years after transplant, while Black patients should be screened within 5 years after transplant, Ally-Khan Somani, MD, PhD, said at the annual meeting of the American Academy of Dermatology.
Dr. Somani, director of dermatologic surgery and the division of cutaneous oncology at Indiana University, Indianapolis, based his remarks on consensus screening guidelines assembled from three rounds of Delphi method surveys with 47 dermatologists and 37 transplant physicians, with the goal of establishing skin cancer screening recommendations for SOTRs. Among the dermatologists surveyed, 45% were Mohs surgeons and 55% were general dermatologists.
The panel recommended that the transplant team should perform risk assessment for SOTRs to risk stratify patients for skin cancer screening (high risk vs. low risk). They also proposed that dermatologists perform skin cancer screening by full-body skin examinations, and that SOTRs with a history of skin cancer should continue with routine skin cancer surveillance as recommended by their dermatologists.
Those at low risk for skin cancer include abdominal organ recipients, SOTR age of younger than 50 at time of transplant, and female gender. The guidelines recommend that White, Asian, and Hispanic patients who meet those criteria should be screened within 5 years after transplant, while no consensus was reached for Black patients who meet those criteria.
Based on posttransplant skin cancer incidence rates, risk is increased among males, Whites, thoracic organ recipients, and being age 50 or older, Dr. Somani said. “At our institution, we make sure there’s a good connection between our transplant teams and dermatologists. We recommend rapid referral for suspicious lesions and we educate patients and screen them within 1 year of transplant, or sooner for high-risk patients. Surveillance is increased to every 3 or 4 months for patients with a history of multiple or high-risk cancers or sooner, followed by routine surveillance as recommended by the patient’s dermatologist.”
To risk stratify patients on the development of their first skin cancer post transplantation, researchers developed the Skin and Ultraviolet Neoplasia Transplant Risk Assessment Calculator (SUNTRAC), a prediction tool with a freely available app. Data for the tool were drawn from the Transplant Skin Cancer Network study, a 5-year analysis of 6,340 adult recipients of a first solid organ transplant at 26 transplant centers in the United States. It generates a risk score for SOTRs (low, medium, high, or very high), which informs transplant care providers of a patient’s risk of skin cancer.
Dr. Somani disclosed that he has received grants and funding from Castle Biosciences. He is an adviser to Cook Biotech and a consultant to Sanara MedTech.
BOSTON – .
White patients who meet these criteria should be screening within 2 years after transplant, while Black patients should be screened within 5 years after transplant, Ally-Khan Somani, MD, PhD, said at the annual meeting of the American Academy of Dermatology.
Dr. Somani, director of dermatologic surgery and the division of cutaneous oncology at Indiana University, Indianapolis, based his remarks on consensus screening guidelines assembled from three rounds of Delphi method surveys with 47 dermatologists and 37 transplant physicians, with the goal of establishing skin cancer screening recommendations for SOTRs. Among the dermatologists surveyed, 45% were Mohs surgeons and 55% were general dermatologists.
The panel recommended that the transplant team should perform risk assessment for SOTRs to risk stratify patients for skin cancer screening (high risk vs. low risk). They also proposed that dermatologists perform skin cancer screening by full-body skin examinations, and that SOTRs with a history of skin cancer should continue with routine skin cancer surveillance as recommended by their dermatologists.
Those at low risk for skin cancer include abdominal organ recipients, SOTR age of younger than 50 at time of transplant, and female gender. The guidelines recommend that White, Asian, and Hispanic patients who meet those criteria should be screened within 5 years after transplant, while no consensus was reached for Black patients who meet those criteria.
Based on posttransplant skin cancer incidence rates, risk is increased among males, Whites, thoracic organ recipients, and being age 50 or older, Dr. Somani said. “At our institution, we make sure there’s a good connection between our transplant teams and dermatologists. We recommend rapid referral for suspicious lesions and we educate patients and screen them within 1 year of transplant, or sooner for high-risk patients. Surveillance is increased to every 3 or 4 months for patients with a history of multiple or high-risk cancers or sooner, followed by routine surveillance as recommended by the patient’s dermatologist.”
To risk stratify patients on the development of their first skin cancer post transplantation, researchers developed the Skin and Ultraviolet Neoplasia Transplant Risk Assessment Calculator (SUNTRAC), a prediction tool with a freely available app. Data for the tool were drawn from the Transplant Skin Cancer Network study, a 5-year analysis of 6,340 adult recipients of a first solid organ transplant at 26 transplant centers in the United States. It generates a risk score for SOTRs (low, medium, high, or very high), which informs transplant care providers of a patient’s risk of skin cancer.
Dr. Somani disclosed that he has received grants and funding from Castle Biosciences. He is an adviser to Cook Biotech and a consultant to Sanara MedTech.
AT AAD 22
Hair loss: Consider a patient’s supplement use
BOSTON – .
This is an important question because patients consider supplements as “natural and healthy,” not as drugs or chemicals, Wilma F. Bergfeld, MD, said at the annual meeting of the American Academy of Dermatology.

Some of these products contain botanicals, which are not always safe, added Dr. Bergfeld, professor of dermatology and pathology at the Cleveland Clinic. “They have many activities, and they are being touted as having some activity in helping the hair or enhancing hair growth,” including having 5-alpha-reductase inhibitors as an ingredient. “Saw palmetto is probably the most common one, but there are a host of natural ingredients that are being put into these supplements, including those that promote androgen induction, as well as antioxidants and anti-inflammatories.”
In the opinion of Dr. Bergfeld, a nutrition-focused physical assessment should include an examination of the scalp and all hairy areas. “It’s also important to see the symmetry and shape of hair growth or hair loss areas, the distribution, hair color, the thickness and texture of the hair fibers,” she added.
Besides asking about what supplements patients are taking, other questions to ask during the visit include: Are you noticing more hair on your brush, pillow, and shoulders, or in the shower? Do you think your hair is thinning? What are your medical problems? Have you experienced rapid weight loss? Have you started any new medications? What medication(s) are you on? What foods do you eat? Do you have a family history of hair loss?
Possible causes of hair loss or changes include environmental factors, stress, hormonal changes, medications, and nutrition.
Common ingredients contained in healthy hair supplements include biotin, folic acid, L-cysteine, L-methionine, MSM (methylsulfonylmethane), vitamin B complex, and vitamins A, C, D, and E. “Vitamin D and A are associated on the hair follicle receptor sites, and they balance each other, so if one is down the other is usually down,” said Dr. Bergfeld, who directs Cleveland Clinic’s hair disorders clinic and its dermatopathology program. Other important ingredients include iron, zinc, manganese, amino acids including L-Lysine, and fatty acids.
Iron deficiency is a known cause of hair loss. “The absorption of iron relies on vitamin C and sometimes lysine,” she said. Red meat has a high iron content and since many patients are restricting red meat intake, “they do need to think about that.” Zinc deficiency is less common in Western countries, she continued, “but when you find it, it’s revolutionary because if they’re shedding hair and their hair character is changing, often some supplementation will do the trick. But remember: Zinc is not only an anti-inflammatory, it’s also an antiandrogen. It has 5-alpha-reductase inhibitor capabilities.”.
Dr. Bergfeld noted that biotin, also known as vitamin B7 and found in many foods, is used in many vitamin supplements marketed for hair loss. The recommended daily allowance (RDA) is 30 mcg/day in adults but the amount in hair supplements can be up to 650% of RDA. “Biotin at high levels is believed to be safe, but can interfere with troponin and other lab testing,” she cautioned. “This can lead to dangerous false laboratory results.”
To date, insufficient data exist to recommend supplementation with zinc, riboflavin, folic acid, or vitamin B12 for hair loss, “but they may help in cases of deficiency,” said Dr. Bergfeld, a past president of the American Hair Research Society. The use of vitamin E and biotin supplementation is not supported in the literature for treating androgenetic alopecia or telogen effluvium. Excessive vitamin A (not beta carotene) and selenium can contribute to hair loss and studies have shown a relationship between androgenetic alopecia and low vitamin D levels. “Vitamin D should be supplemented if serum levels are low, but more studies are needed to determine the effect of iron and zinc supplementation” in patients with androgenetic alopecia, she said.
While there are not enough data to support a recommendation for supplementation of folic or B12 for alopecia, she said, “vitamin B12 deficiency may occur in androgenetic alopecia patients, associated with pernicious anemia.”
She added that the use biotin supplementation for the treatment of androgenetic alopecia is not supported by available data, and “it is also unclear if selenium plays a role in this disease.”
Dr. Bergfeld reported having no disclosures related to her presentation.
BOSTON – .
This is an important question because patients consider supplements as “natural and healthy,” not as drugs or chemicals, Wilma F. Bergfeld, MD, said at the annual meeting of the American Academy of Dermatology.
Some of these products contain botanicals, which are not always safe, added Dr. Bergfeld, professor of dermatology and pathology at the Cleveland Clinic. “They have many activities, and they are being touted as having some activity in helping the hair or enhancing hair growth,” including having 5-alpha-reductase inhibitors as an ingredient. “Saw palmetto is probably the most common one, but there are a host of natural ingredients that are being put into these supplements, including those that promote androgen induction, as well as antioxidants and anti-inflammatories.”
In the opinion of Dr. Bergfeld, a nutrition-focused physical assessment should include an examination of the scalp and all hairy areas. “It’s also important to see the symmetry and shape of hair growth or hair loss areas, the distribution, hair color, the thickness and texture of the hair fibers,” she added.
Besides asking about what supplements patients are taking, other questions to ask during the visit include: Are you noticing more hair on your brush, pillow, and shoulders, or in the shower? Do you think your hair is thinning? What are your medical problems? Have you experienced rapid weight loss? Have you started any new medications? What medication(s) are you on? What foods do you eat? Do you have a family history of hair loss?
Possible causes of hair loss or changes include environmental factors, stress, hormonal changes, medications, and nutrition.
Common ingredients contained in healthy hair supplements include biotin, folic acid, L-cysteine, L-methionine, MSM (methylsulfonylmethane), vitamin B complex, and vitamins A, C, D, and E. “Vitamin D and A are associated on the hair follicle receptor sites, and they balance each other, so if one is down the other is usually down,” said Dr. Bergfeld, who directs Cleveland Clinic’s hair disorders clinic and its dermatopathology program. Other important ingredients include iron, zinc, manganese, amino acids including L-Lysine, and fatty acids.
Iron deficiency is a known cause of hair loss. “The absorption of iron relies on vitamin C and sometimes lysine,” she said. Red meat has a high iron content and since many patients are restricting red meat intake, “they do need to think about that.” Zinc deficiency is less common in Western countries, she continued, “but when you find it, it’s revolutionary because if they’re shedding hair and their hair character is changing, often some supplementation will do the trick. But remember: Zinc is not only an anti-inflammatory, it’s also an antiandrogen. It has 5-alpha-reductase inhibitor capabilities.”.
Dr. Bergfeld noted that biotin, also known as vitamin B7 and found in many foods, is used in many vitamin supplements marketed for hair loss. The recommended daily allowance (RDA) is 30 mcg/day in adults but the amount in hair supplements can be up to 650% of RDA. “Biotin at high levels is believed to be safe, but can interfere with troponin and other lab testing,” she cautioned. “This can lead to dangerous false laboratory results.”
To date, insufficient data exist to recommend supplementation with zinc, riboflavin, folic acid, or vitamin B12 for hair loss, “but they may help in cases of deficiency,” said Dr. Bergfeld, a past president of the American Hair Research Society. The use of vitamin E and biotin supplementation is not supported in the literature for treating androgenetic alopecia or telogen effluvium. Excessive vitamin A (not beta carotene) and selenium can contribute to hair loss and studies have shown a relationship between androgenetic alopecia and low vitamin D levels. “Vitamin D should be supplemented if serum levels are low, but more studies are needed to determine the effect of iron and zinc supplementation” in patients with androgenetic alopecia, she said.
While there are not enough data to support a recommendation for supplementation of folic or B12 for alopecia, she said, “vitamin B12 deficiency may occur in androgenetic alopecia patients, associated with pernicious anemia.”
She added that the use biotin supplementation for the treatment of androgenetic alopecia is not supported by available data, and “it is also unclear if selenium plays a role in this disease.”
Dr. Bergfeld reported having no disclosures related to her presentation.
BOSTON – .
This is an important question because patients consider supplements as “natural and healthy,” not as drugs or chemicals, Wilma F. Bergfeld, MD, said at the annual meeting of the American Academy of Dermatology.
Some of these products contain botanicals, which are not always safe, added Dr. Bergfeld, professor of dermatology and pathology at the Cleveland Clinic. “They have many activities, and they are being touted as having some activity in helping the hair or enhancing hair growth,” including having 5-alpha-reductase inhibitors as an ingredient. “Saw palmetto is probably the most common one, but there are a host of natural ingredients that are being put into these supplements, including those that promote androgen induction, as well as antioxidants and anti-inflammatories.”
In the opinion of Dr. Bergfeld, a nutrition-focused physical assessment should include an examination of the scalp and all hairy areas. “It’s also important to see the symmetry and shape of hair growth or hair loss areas, the distribution, hair color, the thickness and texture of the hair fibers,” she added.
Besides asking about what supplements patients are taking, other questions to ask during the visit include: Are you noticing more hair on your brush, pillow, and shoulders, or in the shower? Do you think your hair is thinning? What are your medical problems? Have you experienced rapid weight loss? Have you started any new medications? What medication(s) are you on? What foods do you eat? Do you have a family history of hair loss?
Possible causes of hair loss or changes include environmental factors, stress, hormonal changes, medications, and nutrition.
Common ingredients contained in healthy hair supplements include biotin, folic acid, L-cysteine, L-methionine, MSM (methylsulfonylmethane), vitamin B complex, and vitamins A, C, D, and E. “Vitamin D and A are associated on the hair follicle receptor sites, and they balance each other, so if one is down the other is usually down,” said Dr. Bergfeld, who directs Cleveland Clinic’s hair disorders clinic and its dermatopathology program. Other important ingredients include iron, zinc, manganese, amino acids including L-Lysine, and fatty acids.
Iron deficiency is a known cause of hair loss. “The absorption of iron relies on vitamin C and sometimes lysine,” she said. Red meat has a high iron content and since many patients are restricting red meat intake, “they do need to think about that.” Zinc deficiency is less common in Western countries, she continued, “but when you find it, it’s revolutionary because if they’re shedding hair and their hair character is changing, often some supplementation will do the trick. But remember: Zinc is not only an anti-inflammatory, it’s also an antiandrogen. It has 5-alpha-reductase inhibitor capabilities.”.
Dr. Bergfeld noted that biotin, also known as vitamin B7 and found in many foods, is used in many vitamin supplements marketed for hair loss. The recommended daily allowance (RDA) is 30 mcg/day in adults but the amount in hair supplements can be up to 650% of RDA. “Biotin at high levels is believed to be safe, but can interfere with troponin and other lab testing,” she cautioned. “This can lead to dangerous false laboratory results.”
To date, insufficient data exist to recommend supplementation with zinc, riboflavin, folic acid, or vitamin B12 for hair loss, “but they may help in cases of deficiency,” said Dr. Bergfeld, a past president of the American Hair Research Society. The use of vitamin E and biotin supplementation is not supported in the literature for treating androgenetic alopecia or telogen effluvium. Excessive vitamin A (not beta carotene) and selenium can contribute to hair loss and studies have shown a relationship between androgenetic alopecia and low vitamin D levels. “Vitamin D should be supplemented if serum levels are low, but more studies are needed to determine the effect of iron and zinc supplementation” in patients with androgenetic alopecia, she said.
While there are not enough data to support a recommendation for supplementation of folic or B12 for alopecia, she said, “vitamin B12 deficiency may occur in androgenetic alopecia patients, associated with pernicious anemia.”
She added that the use biotin supplementation for the treatment of androgenetic alopecia is not supported by available data, and “it is also unclear if selenium plays a role in this disease.”
Dr. Bergfeld reported having no disclosures related to her presentation.
AT AAD 22
Long-term efficacy, safety data for ixekizumab in pediatric psoriasis reported
with the interleukin (IL)-17 inhibitor, investigators reported.
In addition, findings of a substudy, which evaluated randomized withdrawal of treatment after 60 weeks, suggest patients were able to regain benefit after not being treated for a period.
Ixekizumab (Taltz) was approved by the U.S. Food and Drug Administration for treating pediatric psoriasis in March 2020 for patients aged 6 years and older with moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
The trial (IXORA-PEDS) involved 171 patients aged 6-17 years (mean age, 13.5 years; 99 females and 72 males), who were randomly assigned to receive ixekizumab via subcutaneous administration every 4 weeks (115) or placebo for 12 weeks (56). Thereafter, 166 patients continued in an open-label maintenance period in which they were treated every 4 weeks for 12-60 weeks. This was followed by an extension period of up to 108 weeks, which was completed by 139 patients (83.7%). At baseline, the patients’ Psoriasis Area and Severity Index (PASI) score was 12 or higher, the static Physician’s Global Assessment (sPGA) score was 3 or higher, and 10% or more of body surface area was affected.
In the study, at 12 weeks, treatment with ixekizumab was superior to placebo, with sustained responses through 48 weeks. In the follow-up phase, primary and secondary endpoints were sustained through week 108, with patients achieving or maintaining PASI 75 (91.7%), PASI 90 (79%), PASI 100 (55.1%), sPGA 0 or 1 (78.3%), and sPGA 0 (52.4%). Significant improvements in itch were seen at 12 weeks and were sustained with “meaningful improvements in itch for 78.5% of these patients at week 108,” the investigators report.
Among the patients who received ixekizumab, clearance rates in areas that are difficult to treat increased from week 12 to week 108 among those affected. During this time, clearance of nail psoriasis increased from 22.8% to 68.1%, clearance of palmoplantar psoriasis increased from 46.2% to 90%, clearance of scalp psoriasis increased from 70.7% to 76.2%, and clearance of genital psoriasis increased from 83.3% to 87.5%.
No new safety findings during weeks 48-108 of the trial were reported, including no new cases of inflammatory bowel disease (IBD) or Candida infections. The results were reported in JAMA Dermatology.
“Safety is really what we think of most when we are talking about pediatric patients, especially since they may be on these for decades and ... since they most commonly start these therapies in adolescence,” said Amy Paller, MD, the study’s lead author, in an interview. “To be able to take this out 108 weeks, 2 years, is starting to get to a point where we are getting more comfortable with safety. Clearly, no new signals arose.” Dr. Paller is chair of the department of dermatology and professor of dermatology and pediatrics, Northwestern University, Chicago.

One of the biggest concerns with using IL-17 inhibitors such as ixekizumab to manage psoriasis is the development of IBD, said Dr. Paller. She noted that four cases of IBD were reported before the extension phase of the trial but that no new IBD cases were reported after week 48.
“We would not start this as a treatment of choice in someone with Crohn’s disease, or perhaps we would think twice about using it in someone with a strong family history [of Crohn’s disease],” said Dr. Paller, who is also the director of the Skin Biology and Diseases Resource-Based Center at Northwestern. “Otherwise, it does not make me concerned about its use.”
Commenting on the study, Kelly M. Cordoro, MD, professor of dermatology and pediatrics at the University of California, San Francisco, said that the trial’s results provide additional evidence regarding the optimal management of pediatric psoriasis.

“The landscape has shifted toward involving more pediatric patients in clinical trials, thereby providing dermatologists with data to select safe and effective therapies to manage children with psoriasis,” Dr. Cordoro said in an interview. “We have data showing that children with psoriasis have been undertreated, likely because of concerns about safety. The more evidence available from trials such as this, the more likely children are to receive necessary treatment.”
The efficacy data from the study on difficult-to-treat areas of psoriasis, in addition to improvements in BSA and PASI measures, are significant for clinicians deciding on a therapy for patients with psoriasis concentrated in specific body sites. “It was very valuable that the efficacy data was provided by site, such as scalp, palmoplantar, nails, and genital psoriasis, as these are low-BSA but high-impact areas for patients,” said Dr. Cordoro.
The trial data on Crohn’s disease buttress her decision to continue to refrain from initiating ixekizumab in a child with IBD or who is at high risk for IBD. “I was happy to see that there was not a signal for Candida infection,” she added.
Interestingly, in the substudy in the European population, in which there was a double-blind, randomized withdrawal period, fewer patients who were reassigned to receive ixekizumab experienced relapse, compared with those who were reassigned to receive placebo. A total of 90.9% of patients who received placebo experienced relapse, compared with 17.6% of patients treated with ixekizumab. The median time to relapse in the placebo group was 149 days.
“There are data in the adult population that suggest intermittent treatment does allow for recapture of clinical response,” said Dr. Cordoro. “While it is not a large enough dataset to know definitively, this substudy of patients suggests the possibility of intermittent treatment and the ability to regain control [of psoriasis] after a period off drug.”
The study was funded by Eli Lilly. Dr. Paller is an investigator and consultant for Eli Lilly. Several other authors have received grants, personal fees, and/or were a consultant to Eli Lilly, and two authors are Eli Lilly employees. Dr. Cordoro reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
with the interleukin (IL)-17 inhibitor, investigators reported.
In addition, findings of a substudy, which evaluated randomized withdrawal of treatment after 60 weeks, suggest patients were able to regain benefit after not being treated for a period.
Ixekizumab (Taltz) was approved by the U.S. Food and Drug Administration for treating pediatric psoriasis in March 2020 for patients aged 6 years and older with moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
The trial (IXORA-PEDS) involved 171 patients aged 6-17 years (mean age, 13.5 years; 99 females and 72 males), who were randomly assigned to receive ixekizumab via subcutaneous administration every 4 weeks (115) or placebo for 12 weeks (56). Thereafter, 166 patients continued in an open-label maintenance period in which they were treated every 4 weeks for 12-60 weeks. This was followed by an extension period of up to 108 weeks, which was completed by 139 patients (83.7%). At baseline, the patients’ Psoriasis Area and Severity Index (PASI) score was 12 or higher, the static Physician’s Global Assessment (sPGA) score was 3 or higher, and 10% or more of body surface area was affected.
In the study, at 12 weeks, treatment with ixekizumab was superior to placebo, with sustained responses through 48 weeks. In the follow-up phase, primary and secondary endpoints were sustained through week 108, with patients achieving or maintaining PASI 75 (91.7%), PASI 90 (79%), PASI 100 (55.1%), sPGA 0 or 1 (78.3%), and sPGA 0 (52.4%). Significant improvements in itch were seen at 12 weeks and were sustained with “meaningful improvements in itch for 78.5% of these patients at week 108,” the investigators report.
Among the patients who received ixekizumab, clearance rates in areas that are difficult to treat increased from week 12 to week 108 among those affected. During this time, clearance of nail psoriasis increased from 22.8% to 68.1%, clearance of palmoplantar psoriasis increased from 46.2% to 90%, clearance of scalp psoriasis increased from 70.7% to 76.2%, and clearance of genital psoriasis increased from 83.3% to 87.5%.
No new safety findings during weeks 48-108 of the trial were reported, including no new cases of inflammatory bowel disease (IBD) or Candida infections. The results were reported in JAMA Dermatology.
“Safety is really what we think of most when we are talking about pediatric patients, especially since they may be on these for decades and ... since they most commonly start these therapies in adolescence,” said Amy Paller, MD, the study’s lead author, in an interview. “To be able to take this out 108 weeks, 2 years, is starting to get to a point where we are getting more comfortable with safety. Clearly, no new signals arose.” Dr. Paller is chair of the department of dermatology and professor of dermatology and pediatrics, Northwestern University, Chicago.
One of the biggest concerns with using IL-17 inhibitors such as ixekizumab to manage psoriasis is the development of IBD, said Dr. Paller. She noted that four cases of IBD were reported before the extension phase of the trial but that no new IBD cases were reported after week 48.
“We would not start this as a treatment of choice in someone with Crohn’s disease, or perhaps we would think twice about using it in someone with a strong family history [of Crohn’s disease],” said Dr. Paller, who is also the director of the Skin Biology and Diseases Resource-Based Center at Northwestern. “Otherwise, it does not make me concerned about its use.”
Commenting on the study, Kelly M. Cordoro, MD, professor of dermatology and pediatrics at the University of California, San Francisco, said that the trial’s results provide additional evidence regarding the optimal management of pediatric psoriasis.
“The landscape has shifted toward involving more pediatric patients in clinical trials, thereby providing dermatologists with data to select safe and effective therapies to manage children with psoriasis,” Dr. Cordoro said in an interview. “We have data showing that children with psoriasis have been undertreated, likely because of concerns about safety. The more evidence available from trials such as this, the more likely children are to receive necessary treatment.”
The efficacy data from the study on difficult-to-treat areas of psoriasis, in addition to improvements in BSA and PASI measures, are significant for clinicians deciding on a therapy for patients with psoriasis concentrated in specific body sites. “It was very valuable that the efficacy data was provided by site, such as scalp, palmoplantar, nails, and genital psoriasis, as these are low-BSA but high-impact areas for patients,” said Dr. Cordoro.
The trial data on Crohn’s disease buttress her decision to continue to refrain from initiating ixekizumab in a child with IBD or who is at high risk for IBD. “I was happy to see that there was not a signal for Candida infection,” she added.
Interestingly, in the substudy in the European population, in which there was a double-blind, randomized withdrawal period, fewer patients who were reassigned to receive ixekizumab experienced relapse, compared with those who were reassigned to receive placebo. A total of 90.9% of patients who received placebo experienced relapse, compared with 17.6% of patients treated with ixekizumab. The median time to relapse in the placebo group was 149 days.
“There are data in the adult population that suggest intermittent treatment does allow for recapture of clinical response,” said Dr. Cordoro. “While it is not a large enough dataset to know definitively, this substudy of patients suggests the possibility of intermittent treatment and the ability to regain control [of psoriasis] after a period off drug.”
The study was funded by Eli Lilly. Dr. Paller is an investigator and consultant for Eli Lilly. Several other authors have received grants, personal fees, and/or were a consultant to Eli Lilly, and two authors are Eli Lilly employees. Dr. Cordoro reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
with the interleukin (IL)-17 inhibitor, investigators reported.
In addition, findings of a substudy, which evaluated randomized withdrawal of treatment after 60 weeks, suggest patients were able to regain benefit after not being treated for a period.
Ixekizumab (Taltz) was approved by the U.S. Food and Drug Administration for treating pediatric psoriasis in March 2020 for patients aged 6 years and older with moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
The trial (IXORA-PEDS) involved 171 patients aged 6-17 years (mean age, 13.5 years; 99 females and 72 males), who were randomly assigned to receive ixekizumab via subcutaneous administration every 4 weeks (115) or placebo for 12 weeks (56). Thereafter, 166 patients continued in an open-label maintenance period in which they were treated every 4 weeks for 12-60 weeks. This was followed by an extension period of up to 108 weeks, which was completed by 139 patients (83.7%). At baseline, the patients’ Psoriasis Area and Severity Index (PASI) score was 12 or higher, the static Physician’s Global Assessment (sPGA) score was 3 or higher, and 10% or more of body surface area was affected.
In the study, at 12 weeks, treatment with ixekizumab was superior to placebo, with sustained responses through 48 weeks. In the follow-up phase, primary and secondary endpoints were sustained through week 108, with patients achieving or maintaining PASI 75 (91.7%), PASI 90 (79%), PASI 100 (55.1%), sPGA 0 or 1 (78.3%), and sPGA 0 (52.4%). Significant improvements in itch were seen at 12 weeks and were sustained with “meaningful improvements in itch for 78.5% of these patients at week 108,” the investigators report.
Among the patients who received ixekizumab, clearance rates in areas that are difficult to treat increased from week 12 to week 108 among those affected. During this time, clearance of nail psoriasis increased from 22.8% to 68.1%, clearance of palmoplantar psoriasis increased from 46.2% to 90%, clearance of scalp psoriasis increased from 70.7% to 76.2%, and clearance of genital psoriasis increased from 83.3% to 87.5%.
No new safety findings during weeks 48-108 of the trial were reported, including no new cases of inflammatory bowel disease (IBD) or Candida infections. The results were reported in JAMA Dermatology.
“Safety is really what we think of most when we are talking about pediatric patients, especially since they may be on these for decades and ... since they most commonly start these therapies in adolescence,” said Amy Paller, MD, the study’s lead author, in an interview. “To be able to take this out 108 weeks, 2 years, is starting to get to a point where we are getting more comfortable with safety. Clearly, no new signals arose.” Dr. Paller is chair of the department of dermatology and professor of dermatology and pediatrics, Northwestern University, Chicago.
One of the biggest concerns with using IL-17 inhibitors such as ixekizumab to manage psoriasis is the development of IBD, said Dr. Paller. She noted that four cases of IBD were reported before the extension phase of the trial but that no new IBD cases were reported after week 48.
“We would not start this as a treatment of choice in someone with Crohn’s disease, or perhaps we would think twice about using it in someone with a strong family history [of Crohn’s disease],” said Dr. Paller, who is also the director of the Skin Biology and Diseases Resource-Based Center at Northwestern. “Otherwise, it does not make me concerned about its use.”
Commenting on the study, Kelly M. Cordoro, MD, professor of dermatology and pediatrics at the University of California, San Francisco, said that the trial’s results provide additional evidence regarding the optimal management of pediatric psoriasis.
“The landscape has shifted toward involving more pediatric patients in clinical trials, thereby providing dermatologists with data to select safe and effective therapies to manage children with psoriasis,” Dr. Cordoro said in an interview. “We have data showing that children with psoriasis have been undertreated, likely because of concerns about safety. The more evidence available from trials such as this, the more likely children are to receive necessary treatment.”
The efficacy data from the study on difficult-to-treat areas of psoriasis, in addition to improvements in BSA and PASI measures, are significant for clinicians deciding on a therapy for patients with psoriasis concentrated in specific body sites. “It was very valuable that the efficacy data was provided by site, such as scalp, palmoplantar, nails, and genital psoriasis, as these are low-BSA but high-impact areas for patients,” said Dr. Cordoro.
The trial data on Crohn’s disease buttress her decision to continue to refrain from initiating ixekizumab in a child with IBD or who is at high risk for IBD. “I was happy to see that there was not a signal for Candida infection,” she added.
Interestingly, in the substudy in the European population, in which there was a double-blind, randomized withdrawal period, fewer patients who were reassigned to receive ixekizumab experienced relapse, compared with those who were reassigned to receive placebo. A total of 90.9% of patients who received placebo experienced relapse, compared with 17.6% of patients treated with ixekizumab. The median time to relapse in the placebo group was 149 days.
“There are data in the adult population that suggest intermittent treatment does allow for recapture of clinical response,” said Dr. Cordoro. “While it is not a large enough dataset to know definitively, this substudy of patients suggests the possibility of intermittent treatment and the ability to regain control [of psoriasis] after a period off drug.”
The study was funded by Eli Lilly. Dr. Paller is an investigator and consultant for Eli Lilly. Several other authors have received grants, personal fees, and/or were a consultant to Eli Lilly, and two authors are Eli Lilly employees. Dr. Cordoro reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Secukinumab’s antipsoriatic effects confirmed in U.S. patient population
and those who up-titrated to 300 mg from the lower approved dose of 150 mg also saw benefits obtained at that level.
Researchers conducted a postmarketing trial of secukinumab in patients at U.S. centers, called CHOICE, after it was approved for psoriasis and PsA in 2015 and 2016 based on trials mainly conducted outside of the United States. The American patients in those studies “had a baseline clinical profile indicating harder-to-treat disease than the total study population, including higher body mass index (BMI), higher tender and swollen joint counts, increased prevalence of enthesitis and dactylitis, and more tumor necrosis factor inhibitor (TNFi) experience,” Tien Q. Nguyen, MD, a dermatologist in private practice in Irvine, Calif., and colleagues wrote in the Journal of Rheumatology.
In order to get a better sense of how secukinumab performs in U.S. patients who have not been treated with biologics, the researchers conducted the multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 4 CHOICE trial. It recruited patients for about 26 months at 67 U.S. centers during 2016-2018. The 258 patients randomized in the study to 300 mg (n = 103), 150 mg secukinumab (n = 103), or placebo (n = 52) had a mean time since PsA diagnosis of 3.0-3.9 years and all had a mean BMI of greater than 30 kg/m2, with dactylitis present in 48% and enthesitis in 73%. About one-third were taking methotrexate at baseline.
At week 16, patients taking secukinumab 300 mg were about 3.5 times more likely to have 20% improvement in American College of Rheumatology response criteria than with placebo (51.5% vs. 23.1%), whereas the response rate with 150 mg was not significantly different from placebo (36.9%). Rates of achieving ACR50 were significantly greater for both 300- and 150-mg doses versus placebo (28.2% and 24.3% vs. 5.8%), but only 300 mg led to a statistically significant difference in the rate of ACR70 responses, compared with placebo (17.5% vs. 1.9%).
In general, efficacy based on ACR20/50/70 responses and either remission or low disease activity on the Disease Activity in Psoriatic Arthritis index was lower among patients with less than 10 tender joints and less than 10 swollen joints at baseline. Methotrexate use at baseline did not affect ACR20 rates at week 16 in patients taking secukinumab, but the effect of methotrexate on ACR20 rates was noticeable among placebo-treated patients (38.9% vs. 14.7%). Enthesitis appeared to resolve significantly more often among patients on secukinumab, and more patients on secukinumab also had their dactylitis resolve, but the difference was not statistically significant.
Patients with psoriasis affecting more than 3% of their body surface area experienced higher response rates on the Psoriasis Area Severity Index (PASI) for 75%, 90%, and 100% skin lesion clearance than did patients taking placebo.
Patients who switched from 150 mg to 300 mg secukinumab after week 16 in the second treatment period of the trial more often achieved ACR20/50/70 responses by week 52, going from 2.4% to 65.9% of the up-titration subset for ACR20 and from 0% to 34.1% for ACR50 and to 12.2% for ACR70. Patients on placebo who switched also experienced increases in these response rates out to week 52. However, BMI above 30 kg/m2 led to numerically lower ACR50, ACR70, and PASI response rates at week 52.
The researchers noted that the response rates observed in CHOICE were lower than for the pivotal trials used for Food and Drug Administration approval for PsA, which “may have been due to patients in CHOICE having higher disease activity scores at baseline, compared with TNFi-naive patients” in the pivotal trials.
The safety profile of secukinumab appeared to be no different from what has been reported previously. The researchers said that, throughout the 52-week study, the most common adverse events in patients receiving secukinumab were upper respiratory tract infection in about 13% and diarrhea in about 7%. Most adverse events were mild or moderate, with serious adverse events occurring in 9.6% of patients taking secukinumab 300 mg and in 7.8% of patients taking secukinumab 150 mg over the 52 weeks.
“Overall, the findings from CHOICE were consistent with previous studies and demonstrated that secukinumab provides significant and sustained improvements in signs and symptoms of psoriatic arthritis. Our findings suggest that secukinumab 300 mg is safe and efficacious as a first-line biologic treatment for patients with PsA. Further studies will also help determine the optimal dose of secukinumab for treating overweight patients or those with high disease activity at treatment initiation,” the authors wrote.
The study was funded by Novartis, which manufactures secukinumab. Dr. Nguyen and some coauthors reported serving as a consultant, investigator, and/or speaker for numerous pharmaceutical companies, including Novartis.
and those who up-titrated to 300 mg from the lower approved dose of 150 mg also saw benefits obtained at that level.
Researchers conducted a postmarketing trial of secukinumab in patients at U.S. centers, called CHOICE, after it was approved for psoriasis and PsA in 2015 and 2016 based on trials mainly conducted outside of the United States. The American patients in those studies “had a baseline clinical profile indicating harder-to-treat disease than the total study population, including higher body mass index (BMI), higher tender and swollen joint counts, increased prevalence of enthesitis and dactylitis, and more tumor necrosis factor inhibitor (TNFi) experience,” Tien Q. Nguyen, MD, a dermatologist in private practice in Irvine, Calif., and colleagues wrote in the Journal of Rheumatology.
In order to get a better sense of how secukinumab performs in U.S. patients who have not been treated with biologics, the researchers conducted the multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 4 CHOICE trial. It recruited patients for about 26 months at 67 U.S. centers during 2016-2018. The 258 patients randomized in the study to 300 mg (n = 103), 150 mg secukinumab (n = 103), or placebo (n = 52) had a mean time since PsA diagnosis of 3.0-3.9 years and all had a mean BMI of greater than 30 kg/m2, with dactylitis present in 48% and enthesitis in 73%. About one-third were taking methotrexate at baseline.
At week 16, patients taking secukinumab 300 mg were about 3.5 times more likely to have 20% improvement in American College of Rheumatology response criteria than with placebo (51.5% vs. 23.1%), whereas the response rate with 150 mg was not significantly different from placebo (36.9%). Rates of achieving ACR50 were significantly greater for both 300- and 150-mg doses versus placebo (28.2% and 24.3% vs. 5.8%), but only 300 mg led to a statistically significant difference in the rate of ACR70 responses, compared with placebo (17.5% vs. 1.9%).
In general, efficacy based on ACR20/50/70 responses and either remission or low disease activity on the Disease Activity in Psoriatic Arthritis index was lower among patients with less than 10 tender joints and less than 10 swollen joints at baseline. Methotrexate use at baseline did not affect ACR20 rates at week 16 in patients taking secukinumab, but the effect of methotrexate on ACR20 rates was noticeable among placebo-treated patients (38.9% vs. 14.7%). Enthesitis appeared to resolve significantly more often among patients on secukinumab, and more patients on secukinumab also had their dactylitis resolve, but the difference was not statistically significant.
Patients with psoriasis affecting more than 3% of their body surface area experienced higher response rates on the Psoriasis Area Severity Index (PASI) for 75%, 90%, and 100% skin lesion clearance than did patients taking placebo.
Patients who switched from 150 mg to 300 mg secukinumab after week 16 in the second treatment period of the trial more often achieved ACR20/50/70 responses by week 52, going from 2.4% to 65.9% of the up-titration subset for ACR20 and from 0% to 34.1% for ACR50 and to 12.2% for ACR70. Patients on placebo who switched also experienced increases in these response rates out to week 52. However, BMI above 30 kg/m2 led to numerically lower ACR50, ACR70, and PASI response rates at week 52.
The researchers noted that the response rates observed in CHOICE were lower than for the pivotal trials used for Food and Drug Administration approval for PsA, which “may have been due to patients in CHOICE having higher disease activity scores at baseline, compared with TNFi-naive patients” in the pivotal trials.
The safety profile of secukinumab appeared to be no different from what has been reported previously. The researchers said that, throughout the 52-week study, the most common adverse events in patients receiving secukinumab were upper respiratory tract infection in about 13% and diarrhea in about 7%. Most adverse events were mild or moderate, with serious adverse events occurring in 9.6% of patients taking secukinumab 300 mg and in 7.8% of patients taking secukinumab 150 mg over the 52 weeks.
“Overall, the findings from CHOICE were consistent with previous studies and demonstrated that secukinumab provides significant and sustained improvements in signs and symptoms of psoriatic arthritis. Our findings suggest that secukinumab 300 mg is safe and efficacious as a first-line biologic treatment for patients with PsA. Further studies will also help determine the optimal dose of secukinumab for treating overweight patients or those with high disease activity at treatment initiation,” the authors wrote.
The study was funded by Novartis, which manufactures secukinumab. Dr. Nguyen and some coauthors reported serving as a consultant, investigator, and/or speaker for numerous pharmaceutical companies, including Novartis.
and those who up-titrated to 300 mg from the lower approved dose of 150 mg also saw benefits obtained at that level.
Researchers conducted a postmarketing trial of secukinumab in patients at U.S. centers, called CHOICE, after it was approved for psoriasis and PsA in 2015 and 2016 based on trials mainly conducted outside of the United States. The American patients in those studies “had a baseline clinical profile indicating harder-to-treat disease than the total study population, including higher body mass index (BMI), higher tender and swollen joint counts, increased prevalence of enthesitis and dactylitis, and more tumor necrosis factor inhibitor (TNFi) experience,” Tien Q. Nguyen, MD, a dermatologist in private practice in Irvine, Calif., and colleagues wrote in the Journal of Rheumatology.
In order to get a better sense of how secukinumab performs in U.S. patients who have not been treated with biologics, the researchers conducted the multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 4 CHOICE trial. It recruited patients for about 26 months at 67 U.S. centers during 2016-2018. The 258 patients randomized in the study to 300 mg (n = 103), 150 mg secukinumab (n = 103), or placebo (n = 52) had a mean time since PsA diagnosis of 3.0-3.9 years and all had a mean BMI of greater than 30 kg/m2, with dactylitis present in 48% and enthesitis in 73%. About one-third were taking methotrexate at baseline.
At week 16, patients taking secukinumab 300 mg were about 3.5 times more likely to have 20% improvement in American College of Rheumatology response criteria than with placebo (51.5% vs. 23.1%), whereas the response rate with 150 mg was not significantly different from placebo (36.9%). Rates of achieving ACR50 were significantly greater for both 300- and 150-mg doses versus placebo (28.2% and 24.3% vs. 5.8%), but only 300 mg led to a statistically significant difference in the rate of ACR70 responses, compared with placebo (17.5% vs. 1.9%).
In general, efficacy based on ACR20/50/70 responses and either remission or low disease activity on the Disease Activity in Psoriatic Arthritis index was lower among patients with less than 10 tender joints and less than 10 swollen joints at baseline. Methotrexate use at baseline did not affect ACR20 rates at week 16 in patients taking secukinumab, but the effect of methotrexate on ACR20 rates was noticeable among placebo-treated patients (38.9% vs. 14.7%). Enthesitis appeared to resolve significantly more often among patients on secukinumab, and more patients on secukinumab also had their dactylitis resolve, but the difference was not statistically significant.
Patients with psoriasis affecting more than 3% of their body surface area experienced higher response rates on the Psoriasis Area Severity Index (PASI) for 75%, 90%, and 100% skin lesion clearance than did patients taking placebo.
Patients who switched from 150 mg to 300 mg secukinumab after week 16 in the second treatment period of the trial more often achieved ACR20/50/70 responses by week 52, going from 2.4% to 65.9% of the up-titration subset for ACR20 and from 0% to 34.1% for ACR50 and to 12.2% for ACR70. Patients on placebo who switched also experienced increases in these response rates out to week 52. However, BMI above 30 kg/m2 led to numerically lower ACR50, ACR70, and PASI response rates at week 52.
The researchers noted that the response rates observed in CHOICE were lower than for the pivotal trials used for Food and Drug Administration approval for PsA, which “may have been due to patients in CHOICE having higher disease activity scores at baseline, compared with TNFi-naive patients” in the pivotal trials.
The safety profile of secukinumab appeared to be no different from what has been reported previously. The researchers said that, throughout the 52-week study, the most common adverse events in patients receiving secukinumab were upper respiratory tract infection in about 13% and diarrhea in about 7%. Most adverse events were mild or moderate, with serious adverse events occurring in 9.6% of patients taking secukinumab 300 mg and in 7.8% of patients taking secukinumab 150 mg over the 52 weeks.
“Overall, the findings from CHOICE were consistent with previous studies and demonstrated that secukinumab provides significant and sustained improvements in signs and symptoms of psoriatic arthritis. Our findings suggest that secukinumab 300 mg is safe and efficacious as a first-line biologic treatment for patients with PsA. Further studies will also help determine the optimal dose of secukinumab for treating overweight patients or those with high disease activity at treatment initiation,” the authors wrote.
The study was funded by Novartis, which manufactures secukinumab. Dr. Nguyen and some coauthors reported serving as a consultant, investigator, and/or speaker for numerous pharmaceutical companies, including Novartis.
FROM THE JOURNAL OF RHEUMATOLOGY
Scaly rash

Scaly plaques on sun-exposed skin with hyperpigmentation and dyspigmentation are classic signs of cutaneous lupus erythematosus (CLE). (The dyspigmentation seen in this case signaled that she likely had chronic cutaneous lupus erythematosus [CCLE]—a subtype of CLE.) At the patient’s follow-up primary care visit, her antinuclear antibodies titer was 1:1280 (≥ 1:160 is considered a positive test) and her 24-hour urine protein was 1188 mg (normal levels in adults, < 150 mg/d). In light of the patient’s joint pain, lab findings, and skin manifestations, she was also given a diagnosis of systemic lupus erythematosus (SLE).
Lupus erythematosus has an increased prevalence in women and typically occurs between the ages of 20 to 50 years.1 The incidence and prevalence of this condition is also greater in Black patients. CLE can either occur with SLE or independently. Patients with CLE should be monitored for the development of SLE. A diagnosis of CLE is based mainly on clinical features; biopsy is only indicated if there is a high degree of uncertainty.
Patients with CLE may suffer from a lower quality of life compared to patients with other dermatologic conditions due to the often disfiguring and disabling nature of the condition.1,2 Additionally, Black patients have an even higher chance of developing depressive symptoms associated with CCLE.2
Therapeutic management for CLE involves photoprotection by wearing sun-protective clothing, sunscreen, and limiting sun exposure.1 Initial treatment includes topical or intralesional corticosteroids, or topical calcineurin inhibitors. Systemic therapy is similar to that used for SLE. Oral glucocorticoids, and antimalarial agents are considered first-line systemic therapy.1 Second-line treatment includes methotrexate, mycophenolate mofetil, systemic retinoids, and azathioprine. Other immunosuppressive agents that are less commonly used include clofazimine, cyclophosphamide, and rituximab.1
The patient was treated sequentially with trials of oral azathioprine 50 mg bid, then prednisone 10 mg once daily, and then hydroxychloroquine 400 mg daily, without significant change in her condition. Additionally, topical steroids did not improve the patient’s symptoms. She was subsequently started on rituximab 1000 mg intravenously with a second dose repeated 2 weeks later, and another treatment 6 months after that. One year after her visit to the ED, the patient was experiencing marked improvement in her lesions.
Photo courtesy of Christy Nwankwo BA. Text courtesy of Christy Nwankwo, BA, University of Missouri-Kansas City School of Medicine and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque
1. Hejazi EZ, Werth VP. Cutaneous lupus erythematosus: an update on pathogenesis, diagnosis and treatment. Am J Clin Dermatol. 2016;17:135-146. doi:10.1007/s40257-016-0173-9
2. Hong J, Aspey L, Bao G, et al. Chronic cutaneous lupus erythematosus: depression burden and associated factors. Am J Clin Dermatol. 2019;20:465-475. doi:10.1007/s40257-019-00429-7
Scaly plaques on sun-exposed skin with hyperpigmentation and dyspigmentation are classic signs of cutaneous lupus erythematosus (CLE). (The dyspigmentation seen in this case signaled that she likely had chronic cutaneous lupus erythematosus [CCLE]—a subtype of CLE.) At the patient’s follow-up primary care visit, her antinuclear antibodies titer was 1:1280 (≥ 1:160 is considered a positive test) and her 24-hour urine protein was 1188 mg (normal levels in adults, < 150 mg/d). In light of the patient’s joint pain, lab findings, and skin manifestations, she was also given a diagnosis of systemic lupus erythematosus (SLE).
Lupus erythematosus has an increased prevalence in women and typically occurs between the ages of 20 to 50 years.1 The incidence and prevalence of this condition is also greater in Black patients. CLE can either occur with SLE or independently. Patients with CLE should be monitored for the development of SLE. A diagnosis of CLE is based mainly on clinical features; biopsy is only indicated if there is a high degree of uncertainty.
Patients with CLE may suffer from a lower quality of life compared to patients with other dermatologic conditions due to the often disfiguring and disabling nature of the condition.1,2 Additionally, Black patients have an even higher chance of developing depressive symptoms associated with CCLE.2
Therapeutic management for CLE involves photoprotection by wearing sun-protective clothing, sunscreen, and limiting sun exposure.1 Initial treatment includes topical or intralesional corticosteroids, or topical calcineurin inhibitors. Systemic therapy is similar to that used for SLE. Oral glucocorticoids, and antimalarial agents are considered first-line systemic therapy.1 Second-line treatment includes methotrexate, mycophenolate mofetil, systemic retinoids, and azathioprine. Other immunosuppressive agents that are less commonly used include clofazimine, cyclophosphamide, and rituximab.1
The patient was treated sequentially with trials of oral azathioprine 50 mg bid, then prednisone 10 mg once daily, and then hydroxychloroquine 400 mg daily, without significant change in her condition. Additionally, topical steroids did not improve the patient’s symptoms. She was subsequently started on rituximab 1000 mg intravenously with a second dose repeated 2 weeks later, and another treatment 6 months after that. One year after her visit to the ED, the patient was experiencing marked improvement in her lesions.
Photo courtesy of Christy Nwankwo BA. Text courtesy of Christy Nwankwo, BA, University of Missouri-Kansas City School of Medicine and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque
Scaly plaques on sun-exposed skin with hyperpigmentation and dyspigmentation are classic signs of cutaneous lupus erythematosus (CLE). (The dyspigmentation seen in this case signaled that she likely had chronic cutaneous lupus erythematosus [CCLE]—a subtype of CLE.) At the patient’s follow-up primary care visit, her antinuclear antibodies titer was 1:1280 (≥ 1:160 is considered a positive test) and her 24-hour urine protein was 1188 mg (normal levels in adults, < 150 mg/d). In light of the patient’s joint pain, lab findings, and skin manifestations, she was also given a diagnosis of systemic lupus erythematosus (SLE).
Lupus erythematosus has an increased prevalence in women and typically occurs between the ages of 20 to 50 years.1 The incidence and prevalence of this condition is also greater in Black patients. CLE can either occur with SLE or independently. Patients with CLE should be monitored for the development of SLE. A diagnosis of CLE is based mainly on clinical features; biopsy is only indicated if there is a high degree of uncertainty.
Patients with CLE may suffer from a lower quality of life compared to patients with other dermatologic conditions due to the often disfiguring and disabling nature of the condition.1,2 Additionally, Black patients have an even higher chance of developing depressive symptoms associated with CCLE.2
Therapeutic management for CLE involves photoprotection by wearing sun-protective clothing, sunscreen, and limiting sun exposure.1 Initial treatment includes topical or intralesional corticosteroids, or topical calcineurin inhibitors. Systemic therapy is similar to that used for SLE. Oral glucocorticoids, and antimalarial agents are considered first-line systemic therapy.1 Second-line treatment includes methotrexate, mycophenolate mofetil, systemic retinoids, and azathioprine. Other immunosuppressive agents that are less commonly used include clofazimine, cyclophosphamide, and rituximab.1
The patient was treated sequentially with trials of oral azathioprine 50 mg bid, then prednisone 10 mg once daily, and then hydroxychloroquine 400 mg daily, without significant change in her condition. Additionally, topical steroids did not improve the patient’s symptoms. She was subsequently started on rituximab 1000 mg intravenously with a second dose repeated 2 weeks later, and another treatment 6 months after that. One year after her visit to the ED, the patient was experiencing marked improvement in her lesions.
Photo courtesy of Christy Nwankwo BA. Text courtesy of Christy Nwankwo, BA, University of Missouri-Kansas City School of Medicine and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque
1. Hejazi EZ, Werth VP. Cutaneous lupus erythematosus: an update on pathogenesis, diagnosis and treatment. Am J Clin Dermatol. 2016;17:135-146. doi:10.1007/s40257-016-0173-9
2. Hong J, Aspey L, Bao G, et al. Chronic cutaneous lupus erythematosus: depression burden and associated factors. Am J Clin Dermatol. 2019;20:465-475. doi:10.1007/s40257-019-00429-7
1. Hejazi EZ, Werth VP. Cutaneous lupus erythematosus: an update on pathogenesis, diagnosis and treatment. Am J Clin Dermatol. 2016;17:135-146. doi:10.1007/s40257-016-0173-9
2. Hong J, Aspey L, Bao G, et al. Chronic cutaneous lupus erythematosus: depression burden and associated factors. Am J Clin Dermatol. 2019;20:465-475. doi:10.1007/s40257-019-00429-7

FDA warns companies selling OTC skin lighteners
The as the active ingredient, and don’t meet the requirements to be sold legally over the counter. The letters were dated April 13.
The 12 products with hydroquinone are “unapproved drugs and are not generally recognized as safe and effective” (abbreviated as GRASE), the FDA said.
Among the side effects associated with hydroquinone products reported to the FDA are skin rashes, facial swelling, and skin discoloration or ochronosis. The discoloration can be permanent, the FDA said. The lighteners are marketed for use on age or dark spots on the skin associated with melasma.
Tri-Luma, a prescription product for the treatment of moderate to severe melasma of the face, is the only FDA-approved drug containing hydroquinone, according to the FDA. It contains 4% hydroquinone and two other ingredients. It is meant to be used under the supervision of a health care professional. Tri-Luma is indicated for up to 8 weeks of treatment for moderate to severe melasma of the face. The OTC products contain up to 2%. (Generic versions of 4% hydroquinone are available by prescription, dermatologists said.)
“Hydroquinone is a very effective medication, and that’s exactly what it is, a medication,” said Lily Talakoub, MD, a dermatologist in McLean, Va., who supports the FDA action. “It’s very effective and very safe to use in the right hands, but when it is overused or used in the wrong situation, it can cause problems.” Those problems often occur, she said, when there is no health care professional overseeing the use of the OTC products, and when people use them over the long term.
The FDA action to ban the OTC products is “very appropriate,” said dermatologist Pooja Sodha, MD, assistant professor and director of the Center for Laser and Cosmetic Dermatology at George Washington University, Washington. “We know patients pick this up [an OTC product] and use it without physician oversight.” When patients use the products longer than is appropriate, which is also common, it can worsen the initial skin issue, she said.
The action follows reforms finalized under the CARES Act (Coronavirus Aid, Relief and Economic Security Act), which included not only COVID-19 response efforts but also updated the method in which certain OTC drugs are regulated. Manufacturers of the skin lightening products that don’t have FDA approval had been told to remove the products from the market by September 2020.
The recent letters were sent to a dozen companies still marketing their products without an FDA new drug approval. The agency asked the companies to take prompt action and respond with 15 days, stating what they have done to correct the violations.
The 12 companies are AMBI Enterprises, Clinical Formula, Elements Brands Inc., Genomma Lab USA, Intilight/Dr Thomas Balshi, M&M Beauty and Wellness, Neoteric Cosmetics/Scott’s Liquid Gold, Skin Authority, Skin Pro, Skin PS Brands, True Earth Health Products, and Ultimark Products.
Health care professionals and consumers can report adverse reactions associated with these products to the FDA’s MedWatch Adverse Event Reporting program.
A version of this article first appeared on Medscape.com.
The as the active ingredient, and don’t meet the requirements to be sold legally over the counter. The letters were dated April 13.
The 12 products with hydroquinone are “unapproved drugs and are not generally recognized as safe and effective” (abbreviated as GRASE), the FDA said.
Among the side effects associated with hydroquinone products reported to the FDA are skin rashes, facial swelling, and skin discoloration or ochronosis. The discoloration can be permanent, the FDA said. The lighteners are marketed for use on age or dark spots on the skin associated with melasma.
Tri-Luma, a prescription product for the treatment of moderate to severe melasma of the face, is the only FDA-approved drug containing hydroquinone, according to the FDA. It contains 4% hydroquinone and two other ingredients. It is meant to be used under the supervision of a health care professional. Tri-Luma is indicated for up to 8 weeks of treatment for moderate to severe melasma of the face. The OTC products contain up to 2%. (Generic versions of 4% hydroquinone are available by prescription, dermatologists said.)
“Hydroquinone is a very effective medication, and that’s exactly what it is, a medication,” said Lily Talakoub, MD, a dermatologist in McLean, Va., who supports the FDA action. “It’s very effective and very safe to use in the right hands, but when it is overused or used in the wrong situation, it can cause problems.” Those problems often occur, she said, when there is no health care professional overseeing the use of the OTC products, and when people use them over the long term.
The FDA action to ban the OTC products is “very appropriate,” said dermatologist Pooja Sodha, MD, assistant professor and director of the Center for Laser and Cosmetic Dermatology at George Washington University, Washington. “We know patients pick this up [an OTC product] and use it without physician oversight.” When patients use the products longer than is appropriate, which is also common, it can worsen the initial skin issue, she said.
The action follows reforms finalized under the CARES Act (Coronavirus Aid, Relief and Economic Security Act), which included not only COVID-19 response efforts but also updated the method in which certain OTC drugs are regulated. Manufacturers of the skin lightening products that don’t have FDA approval had been told to remove the products from the market by September 2020.
The recent letters were sent to a dozen companies still marketing their products without an FDA new drug approval. The agency asked the companies to take prompt action and respond with 15 days, stating what they have done to correct the violations.
The 12 companies are AMBI Enterprises, Clinical Formula, Elements Brands Inc., Genomma Lab USA, Intilight/Dr Thomas Balshi, M&M Beauty and Wellness, Neoteric Cosmetics/Scott’s Liquid Gold, Skin Authority, Skin Pro, Skin PS Brands, True Earth Health Products, and Ultimark Products.
Health care professionals and consumers can report adverse reactions associated with these products to the FDA’s MedWatch Adverse Event Reporting program.
A version of this article first appeared on Medscape.com.
The as the active ingredient, and don’t meet the requirements to be sold legally over the counter. The letters were dated April 13.
The 12 products with hydroquinone are “unapproved drugs and are not generally recognized as safe and effective” (abbreviated as GRASE), the FDA said.
Among the side effects associated with hydroquinone products reported to the FDA are skin rashes, facial swelling, and skin discoloration or ochronosis. The discoloration can be permanent, the FDA said. The lighteners are marketed for use on age or dark spots on the skin associated with melasma.
Tri-Luma, a prescription product for the treatment of moderate to severe melasma of the face, is the only FDA-approved drug containing hydroquinone, according to the FDA. It contains 4% hydroquinone and two other ingredients. It is meant to be used under the supervision of a health care professional. Tri-Luma is indicated for up to 8 weeks of treatment for moderate to severe melasma of the face. The OTC products contain up to 2%. (Generic versions of 4% hydroquinone are available by prescription, dermatologists said.)
“Hydroquinone is a very effective medication, and that’s exactly what it is, a medication,” said Lily Talakoub, MD, a dermatologist in McLean, Va., who supports the FDA action. “It’s very effective and very safe to use in the right hands, but when it is overused or used in the wrong situation, it can cause problems.” Those problems often occur, she said, when there is no health care professional overseeing the use of the OTC products, and when people use them over the long term.
The FDA action to ban the OTC products is “very appropriate,” said dermatologist Pooja Sodha, MD, assistant professor and director of the Center for Laser and Cosmetic Dermatology at George Washington University, Washington. “We know patients pick this up [an OTC product] and use it without physician oversight.” When patients use the products longer than is appropriate, which is also common, it can worsen the initial skin issue, she said.
The action follows reforms finalized under the CARES Act (Coronavirus Aid, Relief and Economic Security Act), which included not only COVID-19 response efforts but also updated the method in which certain OTC drugs are regulated. Manufacturers of the skin lightening products that don’t have FDA approval had been told to remove the products from the market by September 2020.
The recent letters were sent to a dozen companies still marketing their products without an FDA new drug approval. The agency asked the companies to take prompt action and respond with 15 days, stating what they have done to correct the violations.
The 12 companies are AMBI Enterprises, Clinical Formula, Elements Brands Inc., Genomma Lab USA, Intilight/Dr Thomas Balshi, M&M Beauty and Wellness, Neoteric Cosmetics/Scott’s Liquid Gold, Skin Authority, Skin Pro, Skin PS Brands, True Earth Health Products, and Ultimark Products.
Health care professionals and consumers can report adverse reactions associated with these products to the FDA’s MedWatch Adverse Event Reporting program.
A version of this article first appeared on Medscape.com.
24-year-old female presents with a 3-month history of nonpruritic rash
, typically characterized by symmetrical, nonblanching, purpuric, telangiectatic, and atrophic patches with a predilection for the lower extremities and buttocks.
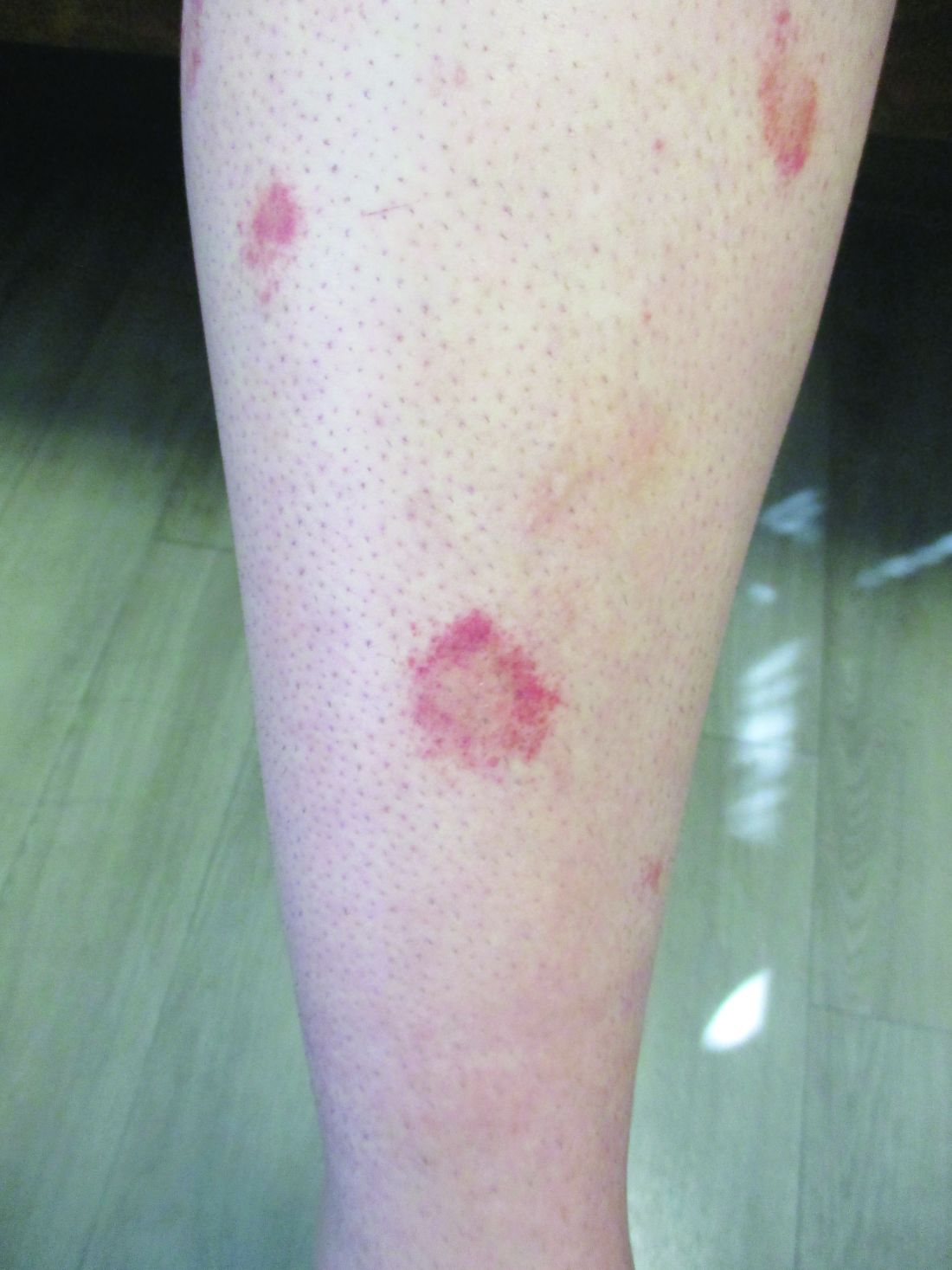
Plaques are usually 1-3 cm in diameter and annular with punctate telangiectasias and cayenne pepper petechiae in the border. The annular patches may form concentric rings. It is most commonly seen in children and young females.
The etiology of Majocchi’s disease is largely unknown and idiopathic.
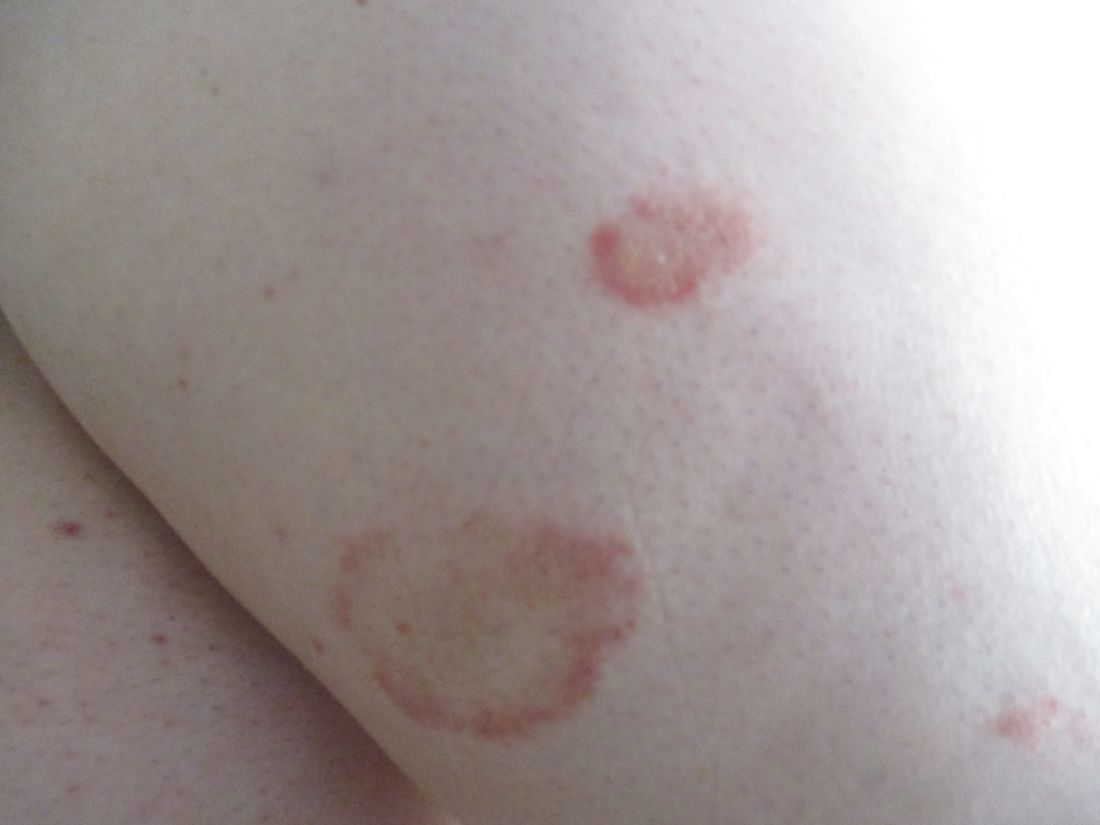
Triggers are not always detected but may be associated with viral infections, chronic comorbidities, and medications. Levofloxacin and isotretinoin have been described in as reports as causing PATM. Other medications reported to cause PPD include sedatives, stimulants, antibiotics, NSAIDS, and cardiovascular drugs.
Diagnosis of PATM is clinical and histopathologic. Direct immunofluorescence (DIF) may show fibrinogen, IgM, and/or C3 deposition in superficial dermal vessels. Histopathologic findings show lymphocytic infiltrate involving the superficial small vessels, extravasated red blood cells, and hemosiderin-laden macrophages.

There is no consensus regarding treatment with variable responses to proposed treatment based on reports and case studies. The first line of treatment is topical corticosteroids and compression hose. Additional treatments, including narrowband UVB phototherapy (NBUVB), griseofulvin, pentoxifylline, cyclosporine, colchicine, rutoside with ascorbic acid, and methotrexate, have been used with varying success.
In this patient, a punch biopsy was performed, which revealed lymphocytes and extravasated erythrocytes and siderophages in the dermis. She was treated with topical steroids with improvement. She started NBUVB, a short course of griseofulvin, and vitamin C supplements.

This case and the photos were photo submitted by Ms. Xu, of the University of California, San Diego, and Dr. Sateesh, of San Diego Family Dermatology. Dr. Donna Bilu Martin edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Garcez A et al. An Bras Dermatol. Sep-Oct 2020;95(5):664-6. doi: 10.1016/j.abd.2020.02.007.
2. Asadbeigi S, Momtahen S. Pigmented purpuric dermatosis. PathologyOutlines.com website.
3. Martínez P et al. Actas Dermosifiliogr (Engl Ed). 2020 Apr;111(3):196-204. doi: 10.1016/j.ad.2019.02.013.
4. Hoesly FJ et al. Int J Dermatol. 2009 Oct;48(10):1129-33. doi: 10.1111/j.1365-4632.2009.04160.x.
, typically characterized by symmetrical, nonblanching, purpuric, telangiectatic, and atrophic patches with a predilection for the lower extremities and buttocks.
Plaques are usually 1-3 cm in diameter and annular with punctate telangiectasias and cayenne pepper petechiae in the border. The annular patches may form concentric rings. It is most commonly seen in children and young females.
The etiology of Majocchi’s disease is largely unknown and idiopathic.
Triggers are not always detected but may be associated with viral infections, chronic comorbidities, and medications. Levofloxacin and isotretinoin have been described in as reports as causing PATM. Other medications reported to cause PPD include sedatives, stimulants, antibiotics, NSAIDS, and cardiovascular drugs.
Diagnosis of PATM is clinical and histopathologic. Direct immunofluorescence (DIF) may show fibrinogen, IgM, and/or C3 deposition in superficial dermal vessels. Histopathologic findings show lymphocytic infiltrate involving the superficial small vessels, extravasated red blood cells, and hemosiderin-laden macrophages.
There is no consensus regarding treatment with variable responses to proposed treatment based on reports and case studies. The first line of treatment is topical corticosteroids and compression hose. Additional treatments, including narrowband UVB phototherapy (NBUVB), griseofulvin, pentoxifylline, cyclosporine, colchicine, rutoside with ascorbic acid, and methotrexate, have been used with varying success.
In this patient, a punch biopsy was performed, which revealed lymphocytes and extravasated erythrocytes and siderophages in the dermis. She was treated with topical steroids with improvement. She started NBUVB, a short course of griseofulvin, and vitamin C supplements.
This case and the photos were photo submitted by Ms. Xu, of the University of California, San Diego, and Dr. Sateesh, of San Diego Family Dermatology. Dr. Donna Bilu Martin edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Garcez A et al. An Bras Dermatol. Sep-Oct 2020;95(5):664-6. doi: 10.1016/j.abd.2020.02.007.
2. Asadbeigi S, Momtahen S. Pigmented purpuric dermatosis. PathologyOutlines.com website.
3. Martínez P et al. Actas Dermosifiliogr (Engl Ed). 2020 Apr;111(3):196-204. doi: 10.1016/j.ad.2019.02.013.
4. Hoesly FJ et al. Int J Dermatol. 2009 Oct;48(10):1129-33. doi: 10.1111/j.1365-4632.2009.04160.x.
, typically characterized by symmetrical, nonblanching, purpuric, telangiectatic, and atrophic patches with a predilection for the lower extremities and buttocks.
Plaques are usually 1-3 cm in diameter and annular with punctate telangiectasias and cayenne pepper petechiae in the border. The annular patches may form concentric rings. It is most commonly seen in children and young females.
The etiology of Majocchi’s disease is largely unknown and idiopathic.
Triggers are not always detected but may be associated with viral infections, chronic comorbidities, and medications. Levofloxacin and isotretinoin have been described in as reports as causing PATM. Other medications reported to cause PPD include sedatives, stimulants, antibiotics, NSAIDS, and cardiovascular drugs.
Diagnosis of PATM is clinical and histopathologic. Direct immunofluorescence (DIF) may show fibrinogen, IgM, and/or C3 deposition in superficial dermal vessels. Histopathologic findings show lymphocytic infiltrate involving the superficial small vessels, extravasated red blood cells, and hemosiderin-laden macrophages.
There is no consensus regarding treatment with variable responses to proposed treatment based on reports and case studies. The first line of treatment is topical corticosteroids and compression hose. Additional treatments, including narrowband UVB phototherapy (NBUVB), griseofulvin, pentoxifylline, cyclosporine, colchicine, rutoside with ascorbic acid, and methotrexate, have been used with varying success.
In this patient, a punch biopsy was performed, which revealed lymphocytes and extravasated erythrocytes and siderophages in the dermis. She was treated with topical steroids with improvement. She started NBUVB, a short course of griseofulvin, and vitamin C supplements.
This case and the photos were photo submitted by Ms. Xu, of the University of California, San Diego, and Dr. Sateesh, of San Diego Family Dermatology. Dr. Donna Bilu Martin edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Garcez A et al. An Bras Dermatol. Sep-Oct 2020;95(5):664-6. doi: 10.1016/j.abd.2020.02.007.
2. Asadbeigi S, Momtahen S. Pigmented purpuric dermatosis. PathologyOutlines.com website.
3. Martínez P et al. Actas Dermosifiliogr (Engl Ed). 2020 Apr;111(3):196-204. doi: 10.1016/j.ad.2019.02.013.
4. Hoesly FJ et al. Int J Dermatol. 2009 Oct;48(10):1129-33. doi: 10.1111/j.1365-4632.2009.04160.x.
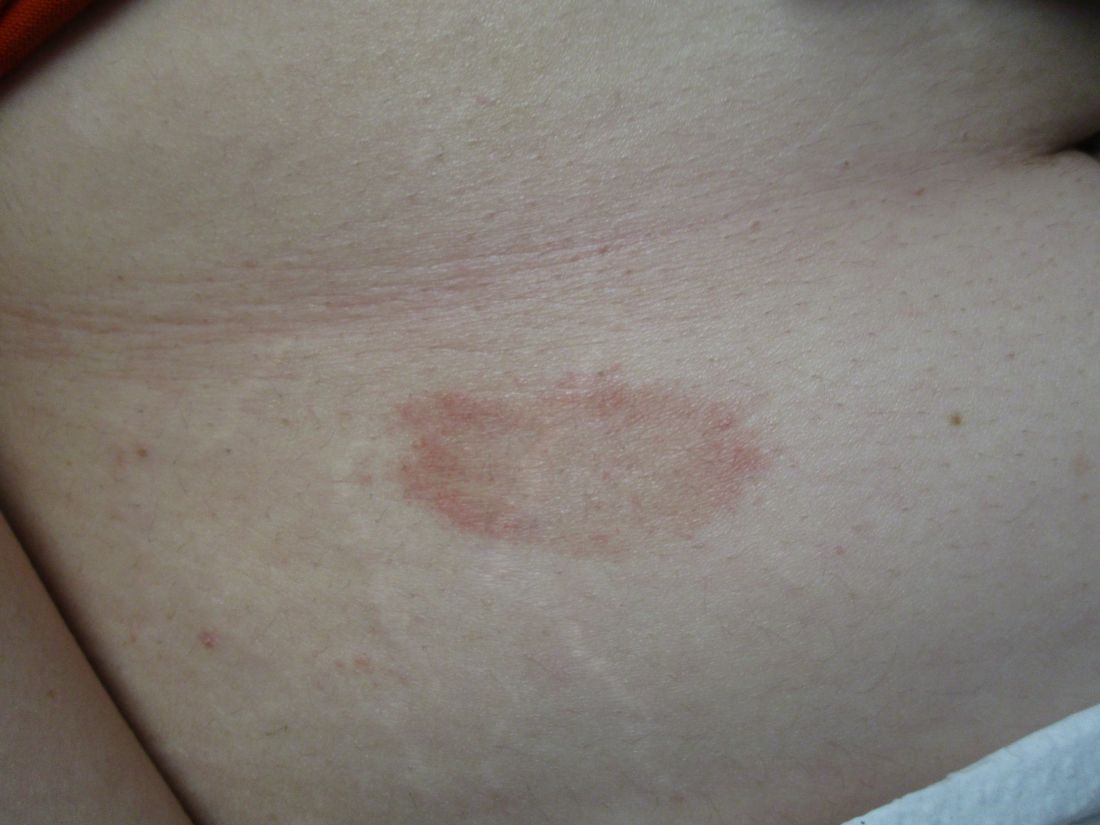
Probiotic LGG doesn’t lessen eczema, asthma, or rhinitis risk by age 7
Giving the probiotic supplement Lactobacillus rhamnosus GG (LGG) to high-risk infants in the first 6 months of life is not effective in lessening incidence of eczema, asthma, or rhinitis in later childhood, researchers have found.
The researchers, led by Michael D. Cabana, MD, MPH, with the Children’s Hospital of Montefiore, New York, said they cannot support its use in this population of children at high risk for allergic disease. Findings were published in Pediatrics.
Jonathan Spergel, MD, PhD, chief of the allergy program at Children’s Hospital of Philadelphia, who was not part of the study, said the “small, but very interesting study adds to the literature indicating that allergy prevention needs to be a multifactorial approach and simply adding LGG in a select population makes no difference.”
He noted that the study of probiotics for allergic conditions is complex as it depends on many factors, such as the child’s environment, including exposure to pets and pollution, and whether the child was delivered vaginally or by cesarean section.
Study builds on previous work
The new study builds on the same researchers’ randomized, double-masked, parallel-arm, controlled Trial of Infant Probiotic Supplementation (TIPS). That study investigated whether daily administration of LGG in the first 6 months to children at high risk for allergic disease because of asthma in a parent, could decrease their cumulative incidence of eczema. Investigators found LGG had no effect.
These additional results included participants at least 7 years old and also included physician-diagnosed asthma and physician-diagnosed rhinitis as secondary outcomes.
Retention rate over the 7-year follow-up was 56%; 49 (53%) of 92 in the intervention group and 54 (59%) of 92 in the control group.
The researchers performed modified intention-to-treat analyses with all children who received treatment in the study arm to which they had been randomized.
Eczema was diagnosed in 78 participants, asthma in 32, and rhinitis in 15. Incidence of eczema was high in infancy, but low thereafter. Incidence rates for asthma and rhinitis were constant throughout childhood.
The researchers used modeling to compare the incidence of each outcome between the intervention and control groups, adjusting for mode of delivery and how long a child was breastfed.
Cesarean delivery was linked to a greater incidence of rhinitis, with a hazard ratio of 3.33 (95% confidence interval, 1.21-9.21).
Finding the right strain
Heather Cassell, MD, a pediatric allergist and immunologist at University of Arizona, Tucson, who was not part of the study, said in an interview that many researchers, including those at her institution, are trying to find which strain of probiotic might be beneficial in lowering risk for allergic disease.
Though it appears LGG doesn’t have an effect, she said, another strain might be successful and this helps zero in on the right one.
The TIPS trial showed that there were no significant side effects from giving LGG early, which is good information to have as the search resumes for the right strain, she said.
“We know that there’s probably some immune dysregulation in kids with asthma, eczema, other allergies, but we don’t fully know the extent of it,” she said, adding that it may be that skin flora or respiratory flora and microbiomes in other parts of the body play a role.
“We don’t have bacteria just in our guts,” she noted. “It may be a combination of strains or a combination of bacteria.”
The authors, Dr. Spergel, and Dr. Cassell reported no relevant financial relationships.
Giving the probiotic supplement Lactobacillus rhamnosus GG (LGG) to high-risk infants in the first 6 months of life is not effective in lessening incidence of eczema, asthma, or rhinitis in later childhood, researchers have found.
The researchers, led by Michael D. Cabana, MD, MPH, with the Children’s Hospital of Montefiore, New York, said they cannot support its use in this population of children at high risk for allergic disease. Findings were published in Pediatrics.
Jonathan Spergel, MD, PhD, chief of the allergy program at Children’s Hospital of Philadelphia, who was not part of the study, said the “small, but very interesting study adds to the literature indicating that allergy prevention needs to be a multifactorial approach and simply adding LGG in a select population makes no difference.”
He noted that the study of probiotics for allergic conditions is complex as it depends on many factors, such as the child’s environment, including exposure to pets and pollution, and whether the child was delivered vaginally or by cesarean section.
Study builds on previous work
The new study builds on the same researchers’ randomized, double-masked, parallel-arm, controlled Trial of Infant Probiotic Supplementation (TIPS). That study investigated whether daily administration of LGG in the first 6 months to children at high risk for allergic disease because of asthma in a parent, could decrease their cumulative incidence of eczema. Investigators found LGG had no effect.
These additional results included participants at least 7 years old and also included physician-diagnosed asthma and physician-diagnosed rhinitis as secondary outcomes.
Retention rate over the 7-year follow-up was 56%; 49 (53%) of 92 in the intervention group and 54 (59%) of 92 in the control group.
The researchers performed modified intention-to-treat analyses with all children who received treatment in the study arm to which they had been randomized.
Eczema was diagnosed in 78 participants, asthma in 32, and rhinitis in 15. Incidence of eczema was high in infancy, but low thereafter. Incidence rates for asthma and rhinitis were constant throughout childhood.
The researchers used modeling to compare the incidence of each outcome between the intervention and control groups, adjusting for mode of delivery and how long a child was breastfed.
Cesarean delivery was linked to a greater incidence of rhinitis, with a hazard ratio of 3.33 (95% confidence interval, 1.21-9.21).
Finding the right strain
Heather Cassell, MD, a pediatric allergist and immunologist at University of Arizona, Tucson, who was not part of the study, said in an interview that many researchers, including those at her institution, are trying to find which strain of probiotic might be beneficial in lowering risk for allergic disease.
Though it appears LGG doesn’t have an effect, she said, another strain might be successful and this helps zero in on the right one.
The TIPS trial showed that there were no significant side effects from giving LGG early, which is good information to have as the search resumes for the right strain, she said.
“We know that there’s probably some immune dysregulation in kids with asthma, eczema, other allergies, but we don’t fully know the extent of it,” she said, adding that it may be that skin flora or respiratory flora and microbiomes in other parts of the body play a role.
“We don’t have bacteria just in our guts,” she noted. “It may be a combination of strains or a combination of bacteria.”
The authors, Dr. Spergel, and Dr. Cassell reported no relevant financial relationships.
Giving the probiotic supplement Lactobacillus rhamnosus GG (LGG) to high-risk infants in the first 6 months of life is not effective in lessening incidence of eczema, asthma, or rhinitis in later childhood, researchers have found.
The researchers, led by Michael D. Cabana, MD, MPH, with the Children’s Hospital of Montefiore, New York, said they cannot support its use in this population of children at high risk for allergic disease. Findings were published in Pediatrics.
Jonathan Spergel, MD, PhD, chief of the allergy program at Children’s Hospital of Philadelphia, who was not part of the study, said the “small, but very interesting study adds to the literature indicating that allergy prevention needs to be a multifactorial approach and simply adding LGG in a select population makes no difference.”
He noted that the study of probiotics for allergic conditions is complex as it depends on many factors, such as the child’s environment, including exposure to pets and pollution, and whether the child was delivered vaginally or by cesarean section.
Study builds on previous work
The new study builds on the same researchers’ randomized, double-masked, parallel-arm, controlled Trial of Infant Probiotic Supplementation (TIPS). That study investigated whether daily administration of LGG in the first 6 months to children at high risk for allergic disease because of asthma in a parent, could decrease their cumulative incidence of eczema. Investigators found LGG had no effect.
These additional results included participants at least 7 years old and also included physician-diagnosed asthma and physician-diagnosed rhinitis as secondary outcomes.
Retention rate over the 7-year follow-up was 56%; 49 (53%) of 92 in the intervention group and 54 (59%) of 92 in the control group.
The researchers performed modified intention-to-treat analyses with all children who received treatment in the study arm to which they had been randomized.
Eczema was diagnosed in 78 participants, asthma in 32, and rhinitis in 15. Incidence of eczema was high in infancy, but low thereafter. Incidence rates for asthma and rhinitis were constant throughout childhood.
The researchers used modeling to compare the incidence of each outcome between the intervention and control groups, adjusting for mode of delivery and how long a child was breastfed.
Cesarean delivery was linked to a greater incidence of rhinitis, with a hazard ratio of 3.33 (95% confidence interval, 1.21-9.21).
Finding the right strain
Heather Cassell, MD, a pediatric allergist and immunologist at University of Arizona, Tucson, who was not part of the study, said in an interview that many researchers, including those at her institution, are trying to find which strain of probiotic might be beneficial in lowering risk for allergic disease.
Though it appears LGG doesn’t have an effect, she said, another strain might be successful and this helps zero in on the right one.
The TIPS trial showed that there were no significant side effects from giving LGG early, which is good information to have as the search resumes for the right strain, she said.
“We know that there’s probably some immune dysregulation in kids with asthma, eczema, other allergies, but we don’t fully know the extent of it,” she said, adding that it may be that skin flora or respiratory flora and microbiomes in other parts of the body play a role.
“We don’t have bacteria just in our guts,” she noted. “It may be a combination of strains or a combination of bacteria.”
The authors, Dr. Spergel, and Dr. Cassell reported no relevant financial relationships.
FROM PEDIATRICS
A 14-year-old male presents to clinic with a new-onset rash of the hands
Photosensitivity due to doxycycline
As the patient’s rash presented in sun-exposed areas with both skin and nail changes, our patient was diagnosed with a phototoxic reaction to doxycycline, the oral antibiotic used to treat his acne.
Photosensitive cutaneous drug eruptions are reactions that occur after exposure to a medication and subsequent exposure to UV radiation or visible light. Reactions can be classified into two ways based on their mechanism of action: phototoxic or photoallergic.1 Phototoxic reactions are more common and are a result of direct keratinocyte damage and cellular necrosis. Many classes of medications may cause this adverse effect, but the tetracycline class of antibiotics is a common culprit.2 Photoallergic reactions are less common and are a result of a type IV immune reaction to the offending agent.1
Phototoxic reactions generally present shortly after sun or UV exposure with a photo-distributed eruption pattern.3 Commonly involved areas include the face, the neck, and the extensor surfaces of extremities, with sparing of relatively protected skin such as the upper eyelids and the skin folds.2 Erythema may initially develop in the exposed skin areas, followed by appearance of edema, vesicles, or bullae.1-3 The eruption may be painful and itchy, with some patients reporting severe pain.3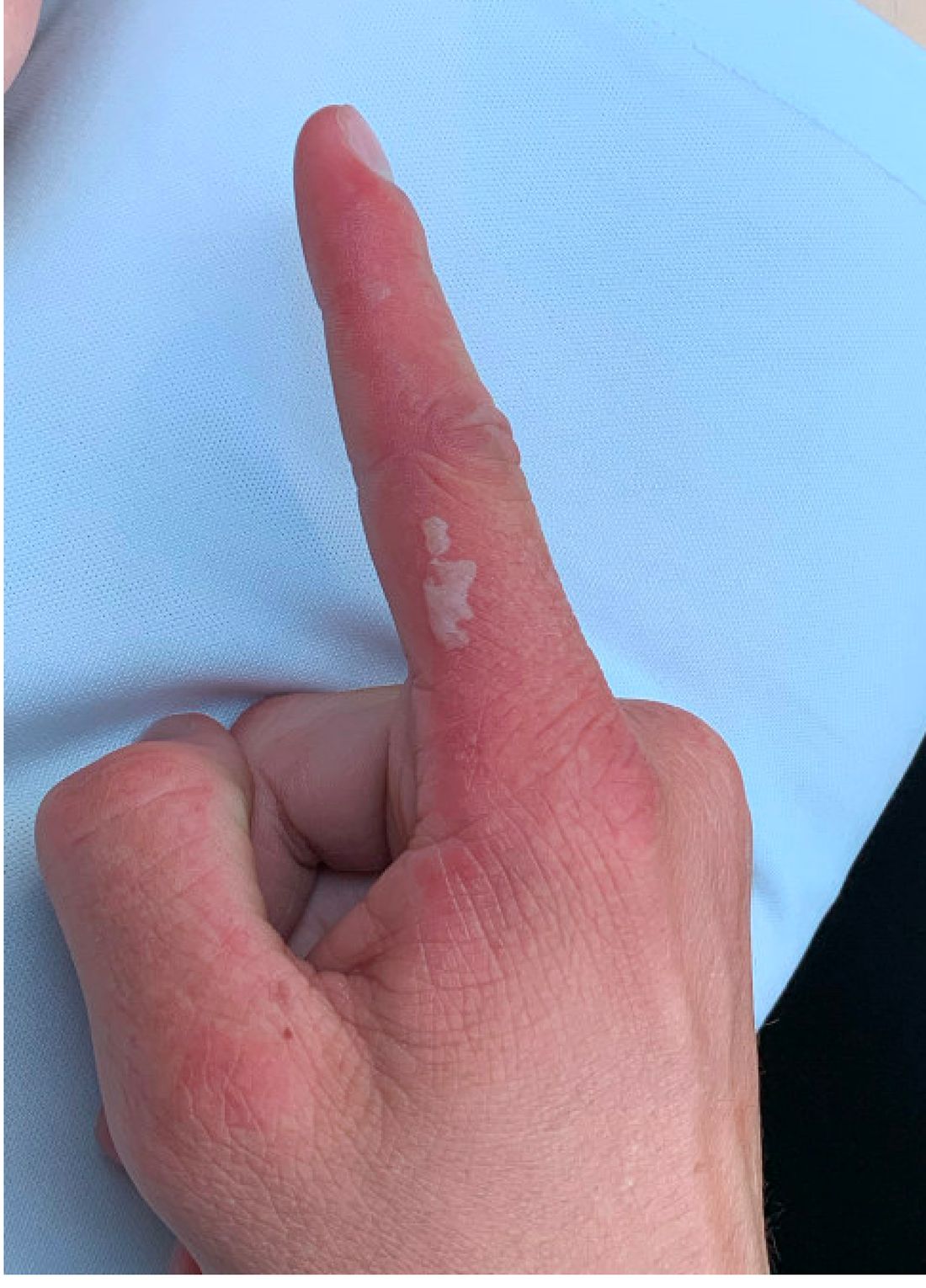
Doxycycline phototoxicity may also cause onycholysis of the nails.2 The reaction is dose dependent, with higher doses of medication leading to a higher likelihood of symptoms.1,2 It is also more prevalent in patients with Fitzpatrick skin type I and II. The usual UVA wavelength required to induce this reaction appears to be in the 320-400 nm range of the UV spectrum.4 By contrast, photoallergic reactions are dose independent, and require a sensitization period prior to the eruption.1 An eczematous eruption is most commonly seen with photoallergic reactions.3
Treatment of drug-induced photosensitivity reactions requires proper identification of the diagnosis and the offending agent, followed by cessation of the medication. If cessation is not possible, then lowering the dose can help to minimize worsening of the condition. However, for photoallergic reactions, the reaction is dose independent so switching to another tolerated agent is likely required. For persistent symptoms following medication withdrawal, topical or systemic steroids and oral antihistamine can help with symptom management.1 For patients with photo-onycholysis, treatment involves stopping the medication and waiting for the intact nail plate to grow.
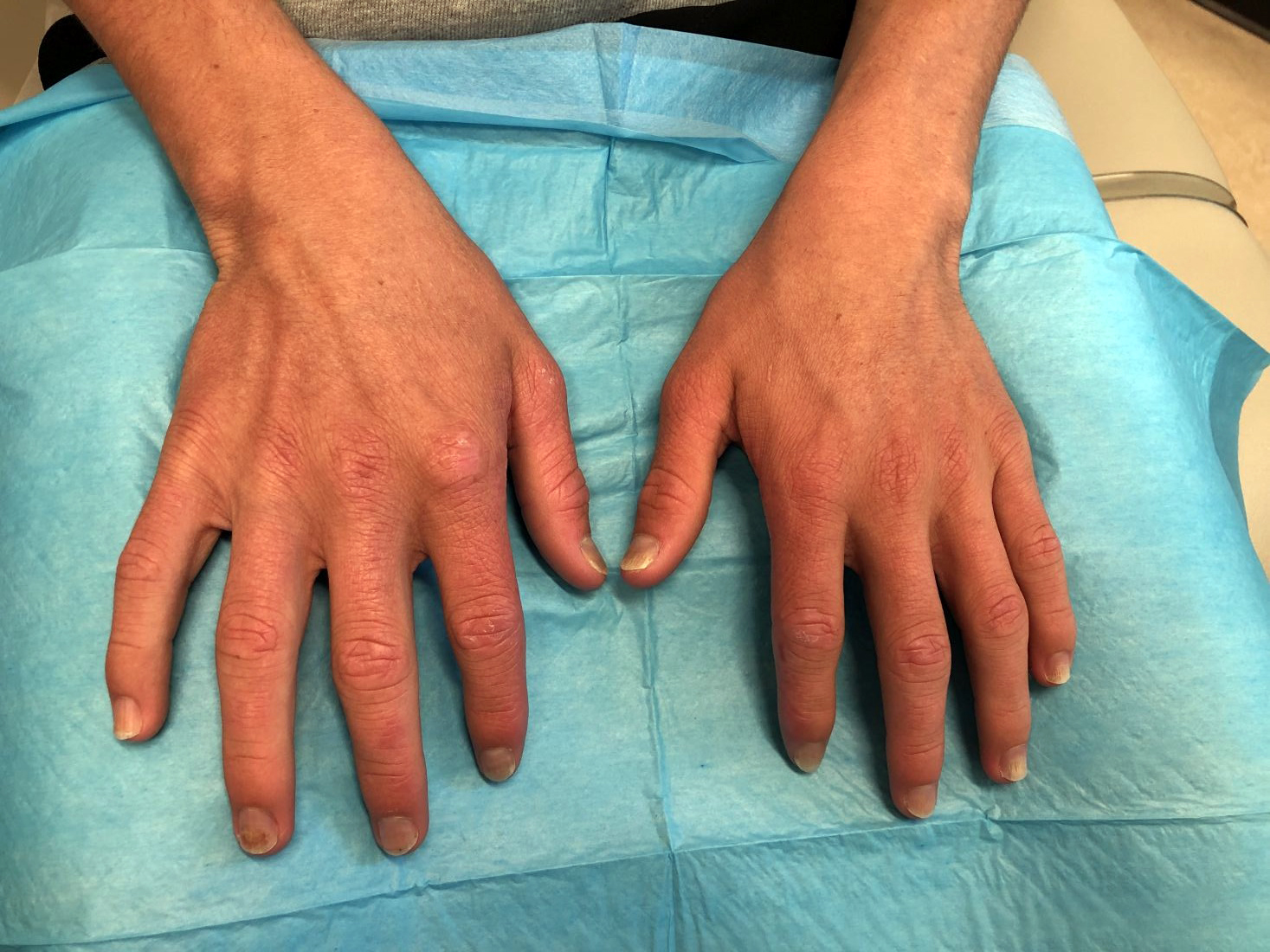
Prevention is key in the management of photosensitivity reactions. Patients should be counseled about the increased risk of photosensitivity while on tetracycline medications and encouraged to engage in enhanced sun protection measures such as wearing sun protective hats and clothing, increasing use of sunscreen that provides mainly UVA but also UVB protection, and avoiding the sun during the midday when the UV index is highest.1-3
Dermatomyositis
Dermatomyositis is an autoimmune condition that presents with skin lesions as well as systemic findings such as myositis. The cutaneous findings are variable, but pathognomonic findings include Gottron papules of the hands, Gottron’s sign on the elbows, knees, and ankles, and the heliotrope rash of the face. Eighty percent of patients have myopathy presenting as muscle weakness, and commonly have elevated creatine kinase, aspartate transaminase, and alanine transaminase values.5 Diagnosis may be confirmed through skin or muscle biopsy, though antibody studies can also play a helpful role in diagnosis. Treatment is generally with oral corticosteroids or other immunosuppressants as well as sun protection.6 The rash seen in our patient could have been seen in patients with dermatomyositis, though it was not in the typical location on the knuckles (Gottron papules) as it also affected the lateral sides of the fingers.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune condition characterized by systemic and cutaneous manifestations. Systemic symptoms may include weight loss, fever, fatigue, arthralgia, or arthritis; patients are at risk of renal, cardiovascular, pulmonary, and neurologic complications of SLE.7 The most common cutaneous finding is malar rash, though there are myriad dermatologic manifestations that can occur associated with photosensitivity. Diagnosis is made based on history, physical, and laboratory testing. Treatment options include NSAIDs, oral glucocorticoids, antimalarial drugs, and immunosuppressants.7 Though our patient exhibited photosensitivity, he had none of the systemic findings associated with SLE, making this diagnosis unlikely.

Allergic contact dermatitis
Allergic contact dermatitis (ACD) is a type IV hypersensitivity reaction, and may present as acute, subacute, or chronic dermatitis. The clinical findings vary based on chronicity. Acute ACD presents as pruritic erythematous papules and vesicles or bullae, similar to how it occurred in our patient, though our patient’s lesions were more tender than pruritic. Chronic ACD presents with erythematous lesions with pruritis, lichenification, scaling, and/or fissuring. Observing shapes or sharp demarcation of lesions may help with diagnosis. Patch testing is also useful in the diagnosis of ACD.

Treatment generally involves avoiding the offending agent with topical corticosteroids for symptom management.8
Polymorphous light eruption
Polymorphous light eruption (PLE) is a delayed, type IV hypersensitivity reaction to UV-induced antigens, though these antigens are unknown. PLE presents hours to days following solar or UV exposure and presents only in sun-exposed areas. Itching and burning are always present, but lesion morphology varies from erythema and papules to vesico-papules and blisters. Notably, PLE must be distinguished from drug photosensitivity through history. Treatment generally involves symptom management with topical steroids and sun protective measures for prevention.9 While PLE may present similarly to drug photosensitivity reactions, our patient’s use of a known phototoxic agent makes PLE a less likely diagnosis.
Ms. Appiah is a pediatric dermatology research associate and medical student at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Neither Dr. Matiz nor Ms. Appiah has any relevant financial disclosures.
References
1. Montgomery S et al. Clin Dermatol. 2022;40(1):57-63.
2. Blakely KM et al. Drug Saf. 2019;42(7):827-47.
3. Goetze S et al. Skin Pharmacol Physiol. 2017;30(2):76-80.
4. Odorici G et al. Dermatol Ther. 2021;34(4):e14978.
5. DeWane ME et al. J Am Acad Dermatol. 2020;82(2):267-81.
6. Waldman R et al. J Am Acad Dermatol. 2020;82(2):283-96.
7. Kiriakidou M et al. Ann Intern Med. 2020;172(11):ITC81-ITC96.
8. Nassau S et al. Med Clin North Am. 2020;104(1):61-76.
9. Guarrera M. Adv Exp Med Biol. 2017;996:61-70.
Photosensitivity due to doxycycline
As the patient’s rash presented in sun-exposed areas with both skin and nail changes, our patient was diagnosed with a phototoxic reaction to doxycycline, the oral antibiotic used to treat his acne.
Photosensitive cutaneous drug eruptions are reactions that occur after exposure to a medication and subsequent exposure to UV radiation or visible light. Reactions can be classified into two ways based on their mechanism of action: phototoxic or photoallergic.1 Phototoxic reactions are more common and are a result of direct keratinocyte damage and cellular necrosis. Many classes of medications may cause this adverse effect, but the tetracycline class of antibiotics is a common culprit.2 Photoallergic reactions are less common and are a result of a type IV immune reaction to the offending agent.1
Phototoxic reactions generally present shortly after sun or UV exposure with a photo-distributed eruption pattern.3 Commonly involved areas include the face, the neck, and the extensor surfaces of extremities, with sparing of relatively protected skin such as the upper eyelids and the skin folds.2 Erythema may initially develop in the exposed skin areas, followed by appearance of edema, vesicles, or bullae.1-3 The eruption may be painful and itchy, with some patients reporting severe pain.3
Doxycycline phototoxicity may also cause onycholysis of the nails.2 The reaction is dose dependent, with higher doses of medication leading to a higher likelihood of symptoms.1,2 It is also more prevalent in patients with Fitzpatrick skin type I and II. The usual UVA wavelength required to induce this reaction appears to be in the 320-400 nm range of the UV spectrum.4 By contrast, photoallergic reactions are dose independent, and require a sensitization period prior to the eruption.1 An eczematous eruption is most commonly seen with photoallergic reactions.3
Treatment of drug-induced photosensitivity reactions requires proper identification of the diagnosis and the offending agent, followed by cessation of the medication. If cessation is not possible, then lowering the dose can help to minimize worsening of the condition. However, for photoallergic reactions, the reaction is dose independent so switching to another tolerated agent is likely required. For persistent symptoms following medication withdrawal, topical or systemic steroids and oral antihistamine can help with symptom management.1 For patients with photo-onycholysis, treatment involves stopping the medication and waiting for the intact nail plate to grow.
Prevention is key in the management of photosensitivity reactions. Patients should be counseled about the increased risk of photosensitivity while on tetracycline medications and encouraged to engage in enhanced sun protection measures such as wearing sun protective hats and clothing, increasing use of sunscreen that provides mainly UVA but also UVB protection, and avoiding the sun during the midday when the UV index is highest.1-3
Dermatomyositis
Dermatomyositis is an autoimmune condition that presents with skin lesions as well as systemic findings such as myositis. The cutaneous findings are variable, but pathognomonic findings include Gottron papules of the hands, Gottron’s sign on the elbows, knees, and ankles, and the heliotrope rash of the face. Eighty percent of patients have myopathy presenting as muscle weakness, and commonly have elevated creatine kinase, aspartate transaminase, and alanine transaminase values.5 Diagnosis may be confirmed through skin or muscle biopsy, though antibody studies can also play a helpful role in diagnosis. Treatment is generally with oral corticosteroids or other immunosuppressants as well as sun protection.6 The rash seen in our patient could have been seen in patients with dermatomyositis, though it was not in the typical location on the knuckles (Gottron papules) as it also affected the lateral sides of the fingers.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune condition characterized by systemic and cutaneous manifestations. Systemic symptoms may include weight loss, fever, fatigue, arthralgia, or arthritis; patients are at risk of renal, cardiovascular, pulmonary, and neurologic complications of SLE.7 The most common cutaneous finding is malar rash, though there are myriad dermatologic manifestations that can occur associated with photosensitivity. Diagnosis is made based on history, physical, and laboratory testing. Treatment options include NSAIDs, oral glucocorticoids, antimalarial drugs, and immunosuppressants.7 Though our patient exhibited photosensitivity, he had none of the systemic findings associated with SLE, making this diagnosis unlikely.
Allergic contact dermatitis
Allergic contact dermatitis (ACD) is a type IV hypersensitivity reaction, and may present as acute, subacute, or chronic dermatitis. The clinical findings vary based on chronicity. Acute ACD presents as pruritic erythematous papules and vesicles or bullae, similar to how it occurred in our patient, though our patient’s lesions were more tender than pruritic. Chronic ACD presents with erythematous lesions with pruritis, lichenification, scaling, and/or fissuring. Observing shapes or sharp demarcation of lesions may help with diagnosis. Patch testing is also useful in the diagnosis of ACD.
Treatment generally involves avoiding the offending agent with topical corticosteroids for symptom management.8
Polymorphous light eruption
Polymorphous light eruption (PLE) is a delayed, type IV hypersensitivity reaction to UV-induced antigens, though these antigens are unknown. PLE presents hours to days following solar or UV exposure and presents only in sun-exposed areas. Itching and burning are always present, but lesion morphology varies from erythema and papules to vesico-papules and blisters. Notably, PLE must be distinguished from drug photosensitivity through history. Treatment generally involves symptom management with topical steroids and sun protective measures for prevention.9 While PLE may present similarly to drug photosensitivity reactions, our patient’s use of a known phototoxic agent makes PLE a less likely diagnosis.
Ms. Appiah is a pediatric dermatology research associate and medical student at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Neither Dr. Matiz nor Ms. Appiah has any relevant financial disclosures.
References
1. Montgomery S et al. Clin Dermatol. 2022;40(1):57-63.
2. Blakely KM et al. Drug Saf. 2019;42(7):827-47.
3. Goetze S et al. Skin Pharmacol Physiol. 2017;30(2):76-80.
4. Odorici G et al. Dermatol Ther. 2021;34(4):e14978.
5. DeWane ME et al. J Am Acad Dermatol. 2020;82(2):267-81.
6. Waldman R et al. J Am Acad Dermatol. 2020;82(2):283-96.
7. Kiriakidou M et al. Ann Intern Med. 2020;172(11):ITC81-ITC96.
8. Nassau S et al. Med Clin North Am. 2020;104(1):61-76.
9. Guarrera M. Adv Exp Med Biol. 2017;996:61-70.
Photosensitivity due to doxycycline
As the patient’s rash presented in sun-exposed areas with both skin and nail changes, our patient was diagnosed with a phototoxic reaction to doxycycline, the oral antibiotic used to treat his acne.
Photosensitive cutaneous drug eruptions are reactions that occur after exposure to a medication and subsequent exposure to UV radiation or visible light. Reactions can be classified into two ways based on their mechanism of action: phototoxic or photoallergic.1 Phototoxic reactions are more common and are a result of direct keratinocyte damage and cellular necrosis. Many classes of medications may cause this adverse effect, but the tetracycline class of antibiotics is a common culprit.2 Photoallergic reactions are less common and are a result of a type IV immune reaction to the offending agent.1
Phototoxic reactions generally present shortly after sun or UV exposure with a photo-distributed eruption pattern.3 Commonly involved areas include the face, the neck, and the extensor surfaces of extremities, with sparing of relatively protected skin such as the upper eyelids and the skin folds.2 Erythema may initially develop in the exposed skin areas, followed by appearance of edema, vesicles, or bullae.1-3 The eruption may be painful and itchy, with some patients reporting severe pain.3
Doxycycline phototoxicity may also cause onycholysis of the nails.2 The reaction is dose dependent, with higher doses of medication leading to a higher likelihood of symptoms.1,2 It is also more prevalent in patients with Fitzpatrick skin type I and II. The usual UVA wavelength required to induce this reaction appears to be in the 320-400 nm range of the UV spectrum.4 By contrast, photoallergic reactions are dose independent, and require a sensitization period prior to the eruption.1 An eczematous eruption is most commonly seen with photoallergic reactions.3
Treatment of drug-induced photosensitivity reactions requires proper identification of the diagnosis and the offending agent, followed by cessation of the medication. If cessation is not possible, then lowering the dose can help to minimize worsening of the condition. However, for photoallergic reactions, the reaction is dose independent so switching to another tolerated agent is likely required. For persistent symptoms following medication withdrawal, topical or systemic steroids and oral antihistamine can help with symptom management.1 For patients with photo-onycholysis, treatment involves stopping the medication and waiting for the intact nail plate to grow.
Prevention is key in the management of photosensitivity reactions. Patients should be counseled about the increased risk of photosensitivity while on tetracycline medications and encouraged to engage in enhanced sun protection measures such as wearing sun protective hats and clothing, increasing use of sunscreen that provides mainly UVA but also UVB protection, and avoiding the sun during the midday when the UV index is highest.1-3
Dermatomyositis
Dermatomyositis is an autoimmune condition that presents with skin lesions as well as systemic findings such as myositis. The cutaneous findings are variable, but pathognomonic findings include Gottron papules of the hands, Gottron’s sign on the elbows, knees, and ankles, and the heliotrope rash of the face. Eighty percent of patients have myopathy presenting as muscle weakness, and commonly have elevated creatine kinase, aspartate transaminase, and alanine transaminase values.5 Diagnosis may be confirmed through skin or muscle biopsy, though antibody studies can also play a helpful role in diagnosis. Treatment is generally with oral corticosteroids or other immunosuppressants as well as sun protection.6 The rash seen in our patient could have been seen in patients with dermatomyositis, though it was not in the typical location on the knuckles (Gottron papules) as it also affected the lateral sides of the fingers.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune condition characterized by systemic and cutaneous manifestations. Systemic symptoms may include weight loss, fever, fatigue, arthralgia, or arthritis; patients are at risk of renal, cardiovascular, pulmonary, and neurologic complications of SLE.7 The most common cutaneous finding is malar rash, though there are myriad dermatologic manifestations that can occur associated with photosensitivity. Diagnosis is made based on history, physical, and laboratory testing. Treatment options include NSAIDs, oral glucocorticoids, antimalarial drugs, and immunosuppressants.7 Though our patient exhibited photosensitivity, he had none of the systemic findings associated with SLE, making this diagnosis unlikely.
Allergic contact dermatitis
Allergic contact dermatitis (ACD) is a type IV hypersensitivity reaction, and may present as acute, subacute, or chronic dermatitis. The clinical findings vary based on chronicity. Acute ACD presents as pruritic erythematous papules and vesicles or bullae, similar to how it occurred in our patient, though our patient’s lesions were more tender than pruritic. Chronic ACD presents with erythematous lesions with pruritis, lichenification, scaling, and/or fissuring. Observing shapes or sharp demarcation of lesions may help with diagnosis. Patch testing is also useful in the diagnosis of ACD.
Treatment generally involves avoiding the offending agent with topical corticosteroids for symptom management.8
Polymorphous light eruption
Polymorphous light eruption (PLE) is a delayed, type IV hypersensitivity reaction to UV-induced antigens, though these antigens are unknown. PLE presents hours to days following solar or UV exposure and presents only in sun-exposed areas. Itching and burning are always present, but lesion morphology varies from erythema and papules to vesico-papules and blisters. Notably, PLE must be distinguished from drug photosensitivity through history. Treatment generally involves symptom management with topical steroids and sun protective measures for prevention.9 While PLE may present similarly to drug photosensitivity reactions, our patient’s use of a known phototoxic agent makes PLE a less likely diagnosis.
Ms. Appiah is a pediatric dermatology research associate and medical student at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Neither Dr. Matiz nor Ms. Appiah has any relevant financial disclosures.
References
1. Montgomery S et al. Clin Dermatol. 2022;40(1):57-63.
2. Blakely KM et al. Drug Saf. 2019;42(7):827-47.
3. Goetze S et al. Skin Pharmacol Physiol. 2017;30(2):76-80.
4. Odorici G et al. Dermatol Ther. 2021;34(4):e14978.
5. DeWane ME et al. J Am Acad Dermatol. 2020;82(2):267-81.
6. Waldman R et al. J Am Acad Dermatol. 2020;82(2):283-96.
7. Kiriakidou M et al. Ann Intern Med. 2020;172(11):ITC81-ITC96.
8. Nassau S et al. Med Clin North Am. 2020;104(1):61-76.
9. Guarrera M. Adv Exp Med Biol. 2017;996:61-70.
He reported no hiking or gardening, no new topical products such as new sunscreens or lotions, and no new medications. The patient had a history of acne, for which he used over-the-counter benzoyl peroxide wash, adapalene gel, and an oral antibiotic for 3 months. His review of systems was negative for fevers, chills, muscle weakness, mouth sores, or joint pain and no prior rashes following sun exposure.
On physical exam he presented with pink plaques with thin vesicles on the dorsum of the hands that were more noticeable on the lateral aspect of both the first and second fingers (Figures 1 and 2). His nails also had a yellow discoloration.