User login
Remote-Onset Alopecia Areata Attributed to Ipilimumab
Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) is a key co-stimulatory receptor expressed on activated T cells that negatively regulates T-cell activation.1-3 It exerts its effects in part by the prevention of IL-2 transcription and inhibition of cell-cycle progression.4 Cytotoxic T-lymphocyte–associated antigen 4 also is expressed by a subset of CD25+CD4+ regulatory T cells (Tregs), where it plays a role in immune tolerance.5 Blockade has demonstrated antitumor activity as well as immune activation, and CTLA-4 dysregulation has been implicated in autoimmune diseases such as alopecia areata (AA).6
Ipilimumab is a fully humanized monoclonal antibody against CTLA-4 and one of a growing class of immune checkpoint inhibitor therapies for metastatic melanoma. Phase 2 and 3 clinical trials have shown an improved survival effect of ipilimumab in patients with advanced melanoma,7-10 with 3-year survival rates ranging from 20.8% to 46.5%.10,11 The US Food and Drug Administration approved ipilimumab in 2011 for treatment of unresectable or metastatic melanoma.12 The most common toxicities of ipilimumab are immune-related adverse effects (irAEs), which represent loss of tolerance to self-antigens.13 Immune-related adverse effects occur in 64.2% of patients,14 with severe or life-threatening irAEs in 17.8% of patients.14 Rates of irAEs appear dose dependent but consistent across increased doses.15 Cutaneous irAEs occur in more than 47% of patients16 and commonly manifest as pruritus with or without a diffuse morbilliform rash,10,17 though less common skin reactions, including vitiligo, vasculitis, and Stevens-Johnson syndrome/toxic epidermal necrolysis, have been documented.9,18
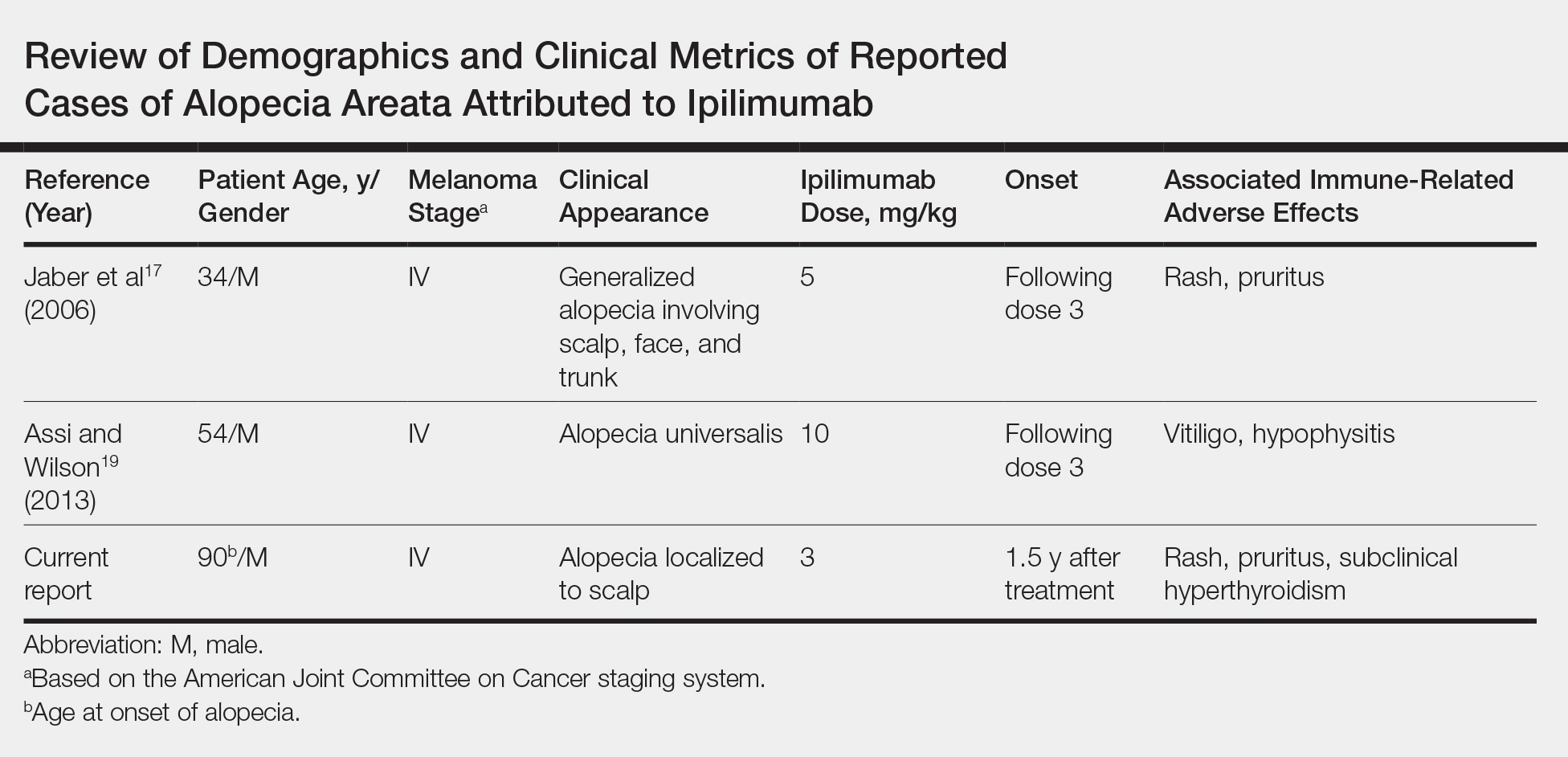
Generalized AA and its more widespread variant, alopecia universalis, have been reported as adverse effects of ipilimumab monotherapy in 2 prior cases in the English-language literature (Table).17,19 Alopecia areata also has been attributed to combination immune checkpoint inhibitor therapy.20,21 We report a case of AA attributable to ipilimumab monotherapy that was localized exclusively to the scalp and remote in onset following treatment.
Case Report
An 88-year-old man with pT3bpN3 nodular melanoma of the back demonstrated multiple lung metastases by positron emission tomography–computed tomography. Lactate dehydrogenase was within reference range, and his Eastern Cooperative Oncology Group performance status was 0 (fully active). One month later, he was started on ipilimumab 3 mg/kg intravenous infusion every 3 weeks for a total of 4 doses. At approximately week 6, his course was complicated by mild fatigue, a faintly erythematous morbilliform rash, and mild pruritus, with laboratory evidence of subclinical hyperthyroidism. Follow-up positron emission tomography–computed tomography at the conclusion of treatment demonstrated complete regression of previously noted hypermetabolic foci. His symptoms and subclinical hyperthyroidism resolved several months later.
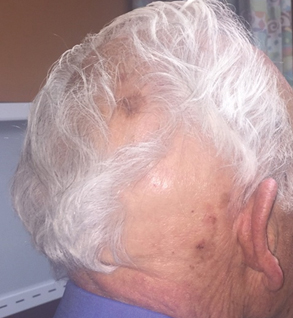
Seventeen months after completion of ipilimumab therapy (at age 90 years), the patient’s barber noted new-onset hair loss on the right occipital scalp. Physical examination demonstrated a well-circumscribed patch of nonscarring alopecia (approximately 6 cm) that was clinically consistent with AA (Figure). There were no associated symptoms or other involved areas of hair loss. He denied any personal or family history of AA. The patient’s melanoma has remained in remission to date.
Comment
This case is unique in that AA was localized to a single circumscribed patch on the scalp and occurred nearly 1.5 years after treatment with ipilimumab, which may indicate a robust blockade of CTLA-4 given the remote development of autoimmunity in the setting of persistent remission of melanoma. Although the appearance of AA may be coincidental, onset at 90 years of age would be unusual. The mean age of onset of AA has been reported between 25.2 and 36.3 years,22,23 and its incidence in men older than 60 years is only 6.4 per 100,000 person-years.24
Although AA is a rare irAE of CTLA-4 blockade, the disease has been increasingly linked to CTLA-4 dysregulation in both animal models and humans.6,25,26 A genome-wide association study of 1054 patients with AA and 3278 controls implicated several genes controlling activation and proliferation of Tregs, including CTLA-4.27 More specifically, single-nucleotide polymorphisms of the CTLA-4 gene were found to be associated with AA in a study of 1196 unrelated patients and 1280 controls,28 and Megiorni et al
Given the role of CTLA-4 dysregulation in the pathogenesis of AA, the very low rates of AA in ipilimumab are somewhat surprising, which may represent a reporting bias. Alternatively, there may be sufficient Treg activity to prevent high rates of AA at a lower ipilimumab dose of 3 mg/kg but insufficient activity to prevent development of other irAEs. With US Food and Drug Administration approval of ipilimumab at a higher dose of 10 mg/kg for use as adjuvant therapy for stage III melanomas,12 less common cutaneous irAEs such as AA may be seen with increased frequency. Clinicians planning ipilimumab therapy should discuss this side effect and other potential irAEs with their patients before initiation of treatment.
- Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature. 1987;328:267-270.
- Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143-155.
- Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125:3377-3383.
- Brunner MC, Chambers CA, Chan FK, et al. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813-5820.
- Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303-310.
- Carroll JM, McElwee KJ, E King L, et al. Gene array profiling and immunomodulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans. J Invest Dermatol. 2002;119:392-402.
- Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591-5598.
- O’Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21:1712-1717.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522-530.
- Yervoy (ipilimumab)[package insert]. Princeton, NJ: Bristol-Myers Squibb; 2019.
- Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864-872.
- Ibrahim RA, Berman DM, DePril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma [abstract]. J Clin Oncol. 2011;29(suppl):8583.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155-164.
- Kähler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9:277-286.
- Jaber SH, Cowen EW, Haworth LR, et al. Skin reactions in a subset of patients with stage IV melanoma treated with anti-cytotoxic T-lymphocyte antigen 4 monoclonal antibody as a single agent. Arch Dermatol. 2006;142:166-172.
- Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:E537545.
- Assi H, Wilson KS. Immune toxicities and long remission duration after ipilimumab therapy for metastatic melanoma: two illustrative cases. Curr Oncol. 2013;20:E165-E169.
- Zarbo A, Belum VR, Sibaud V, et al. Immune-related alopecia (areata and universalis) in cancer patients receiving immune checkpoint inhibitors. Br J Dermatol. 2017;176:1649-1652.
- Lakhmiri M, Cavelier-Balloy B, Lacoste C, et al. Nivolumab-induced alopecia areata: a reversible factor of good prognosis? JAAD Case Rep. 2018;4:761-765.
- Tan E, Tay YK, Goh CL, et al. The pattern and profile of alopecia areata in Singapore–a study of 219 Asians. Int J Dermatol. 2002;41:748-753.
- Goh C, Finkel M, Christos PJ, et al. Profile of 513 patients with alopecia areata: associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol. 2006;20:1055-1060.
- Mirzoyev SA, Schrum AG, Davis MD, et al. Lifetime incidence risk of alopecia areata estimated at 2.1% by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Zöller M, McElwee KJ, Engel P, et al. Transient CD44 variant isoform expression and reduction in CD4(+)/CD25(+) regulatory T cells in C3H/HeJ mice with alopecia areata. J Invest Dermatol. 2002;118:983-992.
- Zöller M, McElwee KJ, Vitacolonna M, et al. The progressive state, in contrast to the stable or regressive state of alopecia areata, is reflected in peripheral blood mononuclear cells. Exp Dermatol. 2004;13:435-444.
- Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113-117.
- John KK, Brockschmidt FF, Redler S, et al. Genetic variants in CTLA4 are strongly associated with alopecia areata. J Invest Dermatol. 2011;131:1169-1172.
- Megiorni F, Mora B, Maxia C, et al. Cytotoxic T-lymphocyte antigen 4 (CTLA4) +49AG and CT60 gene polymorphisms in alopecia areata: a case-control association study in the Italian population. Arch Dermatol Res. 2013;305:665-670
Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) is a key co-stimulatory receptor expressed on activated T cells that negatively regulates T-cell activation.1-3 It exerts its effects in part by the prevention of IL-2 transcription and inhibition of cell-cycle progression.4 Cytotoxic T-lymphocyte–associated antigen 4 also is expressed by a subset of CD25+CD4+ regulatory T cells (Tregs), where it plays a role in immune tolerance.5 Blockade has demonstrated antitumor activity as well as immune activation, and CTLA-4 dysregulation has been implicated in autoimmune diseases such as alopecia areata (AA).6
Ipilimumab is a fully humanized monoclonal antibody against CTLA-4 and one of a growing class of immune checkpoint inhibitor therapies for metastatic melanoma. Phase 2 and 3 clinical trials have shown an improved survival effect of ipilimumab in patients with advanced melanoma,7-10 with 3-year survival rates ranging from 20.8% to 46.5%.10,11 The US Food and Drug Administration approved ipilimumab in 2011 for treatment of unresectable or metastatic melanoma.12 The most common toxicities of ipilimumab are immune-related adverse effects (irAEs), which represent loss of tolerance to self-antigens.13 Immune-related adverse effects occur in 64.2% of patients,14 with severe or life-threatening irAEs in 17.8% of patients.14 Rates of irAEs appear dose dependent but consistent across increased doses.15 Cutaneous irAEs occur in more than 47% of patients16 and commonly manifest as pruritus with or without a diffuse morbilliform rash,10,17 though less common skin reactions, including vitiligo, vasculitis, and Stevens-Johnson syndrome/toxic epidermal necrolysis, have been documented.9,18
Generalized AA and its more widespread variant, alopecia universalis, have been reported as adverse effects of ipilimumab monotherapy in 2 prior cases in the English-language literature (Table).17,19 Alopecia areata also has been attributed to combination immune checkpoint inhibitor therapy.20,21 We report a case of AA attributable to ipilimumab monotherapy that was localized exclusively to the scalp and remote in onset following treatment.
Case Report
An 88-year-old man with pT3bpN3 nodular melanoma of the back demonstrated multiple lung metastases by positron emission tomography–computed tomography. Lactate dehydrogenase was within reference range, and his Eastern Cooperative Oncology Group performance status was 0 (fully active). One month later, he was started on ipilimumab 3 mg/kg intravenous infusion every 3 weeks for a total of 4 doses. At approximately week 6, his course was complicated by mild fatigue, a faintly erythematous morbilliform rash, and mild pruritus, with laboratory evidence of subclinical hyperthyroidism. Follow-up positron emission tomography–computed tomography at the conclusion of treatment demonstrated complete regression of previously noted hypermetabolic foci. His symptoms and subclinical hyperthyroidism resolved several months later.
Seventeen months after completion of ipilimumab therapy (at age 90 years), the patient’s barber noted new-onset hair loss on the right occipital scalp. Physical examination demonstrated a well-circumscribed patch of nonscarring alopecia (approximately 6 cm) that was clinically consistent with AA (Figure). There were no associated symptoms or other involved areas of hair loss. He denied any personal or family history of AA. The patient’s melanoma has remained in remission to date.
Comment
This case is unique in that AA was localized to a single circumscribed patch on the scalp and occurred nearly 1.5 years after treatment with ipilimumab, which may indicate a robust blockade of CTLA-4 given the remote development of autoimmunity in the setting of persistent remission of melanoma. Although the appearance of AA may be coincidental, onset at 90 years of age would be unusual. The mean age of onset of AA has been reported between 25.2 and 36.3 years,22,23 and its incidence in men older than 60 years is only 6.4 per 100,000 person-years.24
Although AA is a rare irAE of CTLA-4 blockade, the disease has been increasingly linked to CTLA-4 dysregulation in both animal models and humans.6,25,26 A genome-wide association study of 1054 patients with AA and 3278 controls implicated several genes controlling activation and proliferation of Tregs, including CTLA-4.27 More specifically, single-nucleotide polymorphisms of the CTLA-4 gene were found to be associated with AA in a study of 1196 unrelated patients and 1280 controls,28 and Megiorni et al
Given the role of CTLA-4 dysregulation in the pathogenesis of AA, the very low rates of AA in ipilimumab are somewhat surprising, which may represent a reporting bias. Alternatively, there may be sufficient Treg activity to prevent high rates of AA at a lower ipilimumab dose of 3 mg/kg but insufficient activity to prevent development of other irAEs. With US Food and Drug Administration approval of ipilimumab at a higher dose of 10 mg/kg for use as adjuvant therapy for stage III melanomas,12 less common cutaneous irAEs such as AA may be seen with increased frequency. Clinicians planning ipilimumab therapy should discuss this side effect and other potential irAEs with their patients before initiation of treatment.
Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) is a key co-stimulatory receptor expressed on activated T cells that negatively regulates T-cell activation.1-3 It exerts its effects in part by the prevention of IL-2 transcription and inhibition of cell-cycle progression.4 Cytotoxic T-lymphocyte–associated antigen 4 also is expressed by a subset of CD25+CD4+ regulatory T cells (Tregs), where it plays a role in immune tolerance.5 Blockade has demonstrated antitumor activity as well as immune activation, and CTLA-4 dysregulation has been implicated in autoimmune diseases such as alopecia areata (AA).6
Ipilimumab is a fully humanized monoclonal antibody against CTLA-4 and one of a growing class of immune checkpoint inhibitor therapies for metastatic melanoma. Phase 2 and 3 clinical trials have shown an improved survival effect of ipilimumab in patients with advanced melanoma,7-10 with 3-year survival rates ranging from 20.8% to 46.5%.10,11 The US Food and Drug Administration approved ipilimumab in 2011 for treatment of unresectable or metastatic melanoma.12 The most common toxicities of ipilimumab are immune-related adverse effects (irAEs), which represent loss of tolerance to self-antigens.13 Immune-related adverse effects occur in 64.2% of patients,14 with severe or life-threatening irAEs in 17.8% of patients.14 Rates of irAEs appear dose dependent but consistent across increased doses.15 Cutaneous irAEs occur in more than 47% of patients16 and commonly manifest as pruritus with or without a diffuse morbilliform rash,10,17 though less common skin reactions, including vitiligo, vasculitis, and Stevens-Johnson syndrome/toxic epidermal necrolysis, have been documented.9,18
Generalized AA and its more widespread variant, alopecia universalis, have been reported as adverse effects of ipilimumab monotherapy in 2 prior cases in the English-language literature (Table).17,19 Alopecia areata also has been attributed to combination immune checkpoint inhibitor therapy.20,21 We report a case of AA attributable to ipilimumab monotherapy that was localized exclusively to the scalp and remote in onset following treatment.
Case Report
An 88-year-old man with pT3bpN3 nodular melanoma of the back demonstrated multiple lung metastases by positron emission tomography–computed tomography. Lactate dehydrogenase was within reference range, and his Eastern Cooperative Oncology Group performance status was 0 (fully active). One month later, he was started on ipilimumab 3 mg/kg intravenous infusion every 3 weeks for a total of 4 doses. At approximately week 6, his course was complicated by mild fatigue, a faintly erythematous morbilliform rash, and mild pruritus, with laboratory evidence of subclinical hyperthyroidism. Follow-up positron emission tomography–computed tomography at the conclusion of treatment demonstrated complete regression of previously noted hypermetabolic foci. His symptoms and subclinical hyperthyroidism resolved several months later.
Seventeen months after completion of ipilimumab therapy (at age 90 years), the patient’s barber noted new-onset hair loss on the right occipital scalp. Physical examination demonstrated a well-circumscribed patch of nonscarring alopecia (approximately 6 cm) that was clinically consistent with AA (Figure). There were no associated symptoms or other involved areas of hair loss. He denied any personal or family history of AA. The patient’s melanoma has remained in remission to date.
Comment
This case is unique in that AA was localized to a single circumscribed patch on the scalp and occurred nearly 1.5 years after treatment with ipilimumab, which may indicate a robust blockade of CTLA-4 given the remote development of autoimmunity in the setting of persistent remission of melanoma. Although the appearance of AA may be coincidental, onset at 90 years of age would be unusual. The mean age of onset of AA has been reported between 25.2 and 36.3 years,22,23 and its incidence in men older than 60 years is only 6.4 per 100,000 person-years.24
Although AA is a rare irAE of CTLA-4 blockade, the disease has been increasingly linked to CTLA-4 dysregulation in both animal models and humans.6,25,26 A genome-wide association study of 1054 patients with AA and 3278 controls implicated several genes controlling activation and proliferation of Tregs, including CTLA-4.27 More specifically, single-nucleotide polymorphisms of the CTLA-4 gene were found to be associated with AA in a study of 1196 unrelated patients and 1280 controls,28 and Megiorni et al
Given the role of CTLA-4 dysregulation in the pathogenesis of AA, the very low rates of AA in ipilimumab are somewhat surprising, which may represent a reporting bias. Alternatively, there may be sufficient Treg activity to prevent high rates of AA at a lower ipilimumab dose of 3 mg/kg but insufficient activity to prevent development of other irAEs. With US Food and Drug Administration approval of ipilimumab at a higher dose of 10 mg/kg for use as adjuvant therapy for stage III melanomas,12 less common cutaneous irAEs such as AA may be seen with increased frequency. Clinicians planning ipilimumab therapy should discuss this side effect and other potential irAEs with their patients before initiation of treatment.
- Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature. 1987;328:267-270.
- Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143-155.
- Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125:3377-3383.
- Brunner MC, Chambers CA, Chan FK, et al. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813-5820.
- Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303-310.
- Carroll JM, McElwee KJ, E King L, et al. Gene array profiling and immunomodulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans. J Invest Dermatol. 2002;119:392-402.
- Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591-5598.
- O’Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21:1712-1717.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522-530.
- Yervoy (ipilimumab)[package insert]. Princeton, NJ: Bristol-Myers Squibb; 2019.
- Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864-872.
- Ibrahim RA, Berman DM, DePril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma [abstract]. J Clin Oncol. 2011;29(suppl):8583.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155-164.
- Kähler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9:277-286.
- Jaber SH, Cowen EW, Haworth LR, et al. Skin reactions in a subset of patients with stage IV melanoma treated with anti-cytotoxic T-lymphocyte antigen 4 monoclonal antibody as a single agent. Arch Dermatol. 2006;142:166-172.
- Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:E537545.
- Assi H, Wilson KS. Immune toxicities and long remission duration after ipilimumab therapy for metastatic melanoma: two illustrative cases. Curr Oncol. 2013;20:E165-E169.
- Zarbo A, Belum VR, Sibaud V, et al. Immune-related alopecia (areata and universalis) in cancer patients receiving immune checkpoint inhibitors. Br J Dermatol. 2017;176:1649-1652.
- Lakhmiri M, Cavelier-Balloy B, Lacoste C, et al. Nivolumab-induced alopecia areata: a reversible factor of good prognosis? JAAD Case Rep. 2018;4:761-765.
- Tan E, Tay YK, Goh CL, et al. The pattern and profile of alopecia areata in Singapore–a study of 219 Asians. Int J Dermatol. 2002;41:748-753.
- Goh C, Finkel M, Christos PJ, et al. Profile of 513 patients with alopecia areata: associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol. 2006;20:1055-1060.
- Mirzoyev SA, Schrum AG, Davis MD, et al. Lifetime incidence risk of alopecia areata estimated at 2.1% by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Zöller M, McElwee KJ, Engel P, et al. Transient CD44 variant isoform expression and reduction in CD4(+)/CD25(+) regulatory T cells in C3H/HeJ mice with alopecia areata. J Invest Dermatol. 2002;118:983-992.
- Zöller M, McElwee KJ, Vitacolonna M, et al. The progressive state, in contrast to the stable or regressive state of alopecia areata, is reflected in peripheral blood mononuclear cells. Exp Dermatol. 2004;13:435-444.
- Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113-117.
- John KK, Brockschmidt FF, Redler S, et al. Genetic variants in CTLA4 are strongly associated with alopecia areata. J Invest Dermatol. 2011;131:1169-1172.
- Megiorni F, Mora B, Maxia C, et al. Cytotoxic T-lymphocyte antigen 4 (CTLA4) +49AG and CT60 gene polymorphisms in alopecia areata: a case-control association study in the Italian population. Arch Dermatol Res. 2013;305:665-670
- Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature. 1987;328:267-270.
- Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143-155.
- Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125:3377-3383.
- Brunner MC, Chambers CA, Chan FK, et al. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813-5820.
- Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303-310.
- Carroll JM, McElwee KJ, E King L, et al. Gene array profiling and immunomodulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans. J Invest Dermatol. 2002;119:392-402.
- Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591-5598.
- O’Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21:1712-1717.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522-530.
- Yervoy (ipilimumab)[package insert]. Princeton, NJ: Bristol-Myers Squibb; 2019.
- Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864-872.
- Ibrahim RA, Berman DM, DePril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma [abstract]. J Clin Oncol. 2011;29(suppl):8583.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155-164.
- Kähler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9:277-286.
- Jaber SH, Cowen EW, Haworth LR, et al. Skin reactions in a subset of patients with stage IV melanoma treated with anti-cytotoxic T-lymphocyte antigen 4 monoclonal antibody as a single agent. Arch Dermatol. 2006;142:166-172.
- Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:E537545.
- Assi H, Wilson KS. Immune toxicities and long remission duration after ipilimumab therapy for metastatic melanoma: two illustrative cases. Curr Oncol. 2013;20:E165-E169.
- Zarbo A, Belum VR, Sibaud V, et al. Immune-related alopecia (areata and universalis) in cancer patients receiving immune checkpoint inhibitors. Br J Dermatol. 2017;176:1649-1652.
- Lakhmiri M, Cavelier-Balloy B, Lacoste C, et al. Nivolumab-induced alopecia areata: a reversible factor of good prognosis? JAAD Case Rep. 2018;4:761-765.
- Tan E, Tay YK, Goh CL, et al. The pattern and profile of alopecia areata in Singapore–a study of 219 Asians. Int J Dermatol. 2002;41:748-753.
- Goh C, Finkel M, Christos PJ, et al. Profile of 513 patients with alopecia areata: associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol. 2006;20:1055-1060.
- Mirzoyev SA, Schrum AG, Davis MD, et al. Lifetime incidence risk of alopecia areata estimated at 2.1% by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Zöller M, McElwee KJ, Engel P, et al. Transient CD44 variant isoform expression and reduction in CD4(+)/CD25(+) regulatory T cells in C3H/HeJ mice with alopecia areata. J Invest Dermatol. 2002;118:983-992.
- Zöller M, McElwee KJ, Vitacolonna M, et al. The progressive state, in contrast to the stable or regressive state of alopecia areata, is reflected in peripheral blood mononuclear cells. Exp Dermatol. 2004;13:435-444.
- Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113-117.
- John KK, Brockschmidt FF, Redler S, et al. Genetic variants in CTLA4 are strongly associated with alopecia areata. J Invest Dermatol. 2011;131:1169-1172.
- Megiorni F, Mora B, Maxia C, et al. Cytotoxic T-lymphocyte antigen 4 (CTLA4) +49AG and CT60 gene polymorphisms in alopecia areata: a case-control association study in the Italian population. Arch Dermatol Res. 2013;305:665-670
Practice Points
- Cutaneous immune-related adverse effects (irAEs) are among the most common adverse effects of ipilimumab, a fully humanized monoclonal antibody directed against cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) used to treat advanced-stage melanoma.
- Alopecia areata is a rarely reported irAE, but its connection to CTLA-4 dysregulation may mean that clinicians see an increased incidence at higher ipilimumab doses.
Updates in Our Understanding of Central Centrifugal Cicatricial Alopecia
It has been more than 50 years since central centrifugal cicatricial alopecia (CCCA) was first defined by LoPresti and colleagues1 as hot comb alopecia. Fifty years later, we are only just starting to understand the pathogenesis of CCCA and its systemic implications.
Then and Now
The use of hot combs, a metal device used to straighten naturally curly hair, was ubiquitous in the households of black women in the 1960s. It is no surprise then that this styling process was labeled as the culprit of this disease that affects black women almost exclusively. As the use of hot combs waned but the prevalence of CCCA persisted, its name evolved to chemically induced alopecia—an ode to the popular styling product of the 1990s, the chemical relaxer—and eventually CCCA, a name that reflects its clinical progression and histologic findings.2
Since then, research has explored the association with systemic diseases, some noting increased rates of type 2 diabetes mellitus and thyroid disease, and more recently, an increased rate of fibroids in affected patients.3,4
Clues to Pathogenesis
Compared to other primary cicatricial alopecias, CCCA is unique in that active progression is difficult to detect. Symptoms, such as pruritus, often are minimal or absent, rendering clinical assessment quite difficult.5 Unlike other forms of scarring hair loss, fibrosis, not inflammation, is the predominant clinical feature. The clinical presentation is not unlike a group of disorders termed fibroproliferative disorders, which includes systemic sclerosis, uterine fibroids, atherosclerosis, and keloids, among others. It has been postulated that diseases of aberrant scarring are more common in black individuals due to the protective effect profibrotic alleles have against endemic helminthic infections of sub-Saharan infections, including oncocerciasis.6
A recent study showed an increased expression of fibroproliferative genes, particularly those implicated in other fibroproliferative disorders, in affected scalp of patients with CCCA.7 Most notably, an expression in gene overlap was noted between fibroids and CCCA in this study, though the relationship between these two diseases needs to be further explored.
Gene Variants Identified in CCCA
More recently, a new study has identified a gene variant of peptidyl arginine deiminase 3, PADI3, that is present in approximately one-quarter of studied patients with CCCA.8PADI3 plays a role in hair shaft formation and has been implicated in another hair disorder, uncombable hair syndrome, though the latter presents in children, improves with age, and is not associated with a scarring phenotype.9 However, this study has provided greater insight into our understanding of CCCA by establishing a possible genetic predisposition in patients affected with this disease.8
What’s Next for CCCA?
For years, many patients with CCCA have been turned away with few answers and left thinking that it is their own styling habits that have led to their hair loss,
The future is bright for CCCA. Although our understanding of CCCA is still in its infancy, it is my hope that with greater understanding of this disease will come greater empathy for our patients.
- LoPresti P, Papa CM, Kligman AM. Hot comb alopecia. Arch Dermatol. 1968;98:234-238.
- Gathers RC, Lim HW. Central centrifugal cicatricial alopecia: past, present, and future. J Am Acad Dermatol. 2009;60:660-668.
- Kyei A, Bergfeld WF, Piliang M, et al. Medical and environmental risk factors for the development of central centrifugal cicatricial alopecia: a population study. Arch Dermatol. 2011;147:909-914.
- Dina Y, Okoye GA, Aguh C. Association of uterine leiomyomas with central centrifugal cicatricial alopecia. JAMA Dermatol. 2018;154:213-214.
- Whiting DA, Olsen EA. Central centrifugal cicatricial alopecia. Dermatol Ther. 2008;21:268-278.
- Hellwege JN, Torstenson ES, Russell SB, et al. Evidence of selection as a cause for racial disparities in fibroproliferative disease. PLoS One. 2017;12:e0182791. doi:10.1371/journal.pone.0182791.
- Aguh C, Dina Y, Talbot CC Jr, et al. Fibroproliferative genes are preferentially expressed in central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2018;79:904.e1-912.e1.
- Malki L, Sarig O, Romano MT, et al. Variant PADI3 in central centrifugal cicatricial alopecia. N Engl J Med. 2019;380:833-841.
- Matis WL, Baden H, Green R, et al. Uncombable-hair syndrome. Pediatr Dermatol. 1987;4:215-219.
It has been more than 50 years since central centrifugal cicatricial alopecia (CCCA) was first defined by LoPresti and colleagues1 as hot comb alopecia. Fifty years later, we are only just starting to understand the pathogenesis of CCCA and its systemic implications.
Then and Now
The use of hot combs, a metal device used to straighten naturally curly hair, was ubiquitous in the households of black women in the 1960s. It is no surprise then that this styling process was labeled as the culprit of this disease that affects black women almost exclusively. As the use of hot combs waned but the prevalence of CCCA persisted, its name evolved to chemically induced alopecia—an ode to the popular styling product of the 1990s, the chemical relaxer—and eventually CCCA, a name that reflects its clinical progression and histologic findings.2
Since then, research has explored the association with systemic diseases, some noting increased rates of type 2 diabetes mellitus and thyroid disease, and more recently, an increased rate of fibroids in affected patients.3,4
Clues to Pathogenesis
Compared to other primary cicatricial alopecias, CCCA is unique in that active progression is difficult to detect. Symptoms, such as pruritus, often are minimal or absent, rendering clinical assessment quite difficult.5 Unlike other forms of scarring hair loss, fibrosis, not inflammation, is the predominant clinical feature. The clinical presentation is not unlike a group of disorders termed fibroproliferative disorders, which includes systemic sclerosis, uterine fibroids, atherosclerosis, and keloids, among others. It has been postulated that diseases of aberrant scarring are more common in black individuals due to the protective effect profibrotic alleles have against endemic helminthic infections of sub-Saharan infections, including oncocerciasis.6
A recent study showed an increased expression of fibroproliferative genes, particularly those implicated in other fibroproliferative disorders, in affected scalp of patients with CCCA.7 Most notably, an expression in gene overlap was noted between fibroids and CCCA in this study, though the relationship between these two diseases needs to be further explored.
Gene Variants Identified in CCCA
More recently, a new study has identified a gene variant of peptidyl arginine deiminase 3, PADI3, that is present in approximately one-quarter of studied patients with CCCA.8PADI3 plays a role in hair shaft formation and has been implicated in another hair disorder, uncombable hair syndrome, though the latter presents in children, improves with age, and is not associated with a scarring phenotype.9 However, this study has provided greater insight into our understanding of CCCA by establishing a possible genetic predisposition in patients affected with this disease.8
What’s Next for CCCA?
For years, many patients with CCCA have been turned away with few answers and left thinking that it is their own styling habits that have led to their hair loss,
The future is bright for CCCA. Although our understanding of CCCA is still in its infancy, it is my hope that with greater understanding of this disease will come greater empathy for our patients.
It has been more than 50 years since central centrifugal cicatricial alopecia (CCCA) was first defined by LoPresti and colleagues1 as hot comb alopecia. Fifty years later, we are only just starting to understand the pathogenesis of CCCA and its systemic implications.
Then and Now
The use of hot combs, a metal device used to straighten naturally curly hair, was ubiquitous in the households of black women in the 1960s. It is no surprise then that this styling process was labeled as the culprit of this disease that affects black women almost exclusively. As the use of hot combs waned but the prevalence of CCCA persisted, its name evolved to chemically induced alopecia—an ode to the popular styling product of the 1990s, the chemical relaxer—and eventually CCCA, a name that reflects its clinical progression and histologic findings.2
Since then, research has explored the association with systemic diseases, some noting increased rates of type 2 diabetes mellitus and thyroid disease, and more recently, an increased rate of fibroids in affected patients.3,4
Clues to Pathogenesis
Compared to other primary cicatricial alopecias, CCCA is unique in that active progression is difficult to detect. Symptoms, such as pruritus, often are minimal or absent, rendering clinical assessment quite difficult.5 Unlike other forms of scarring hair loss, fibrosis, not inflammation, is the predominant clinical feature. The clinical presentation is not unlike a group of disorders termed fibroproliferative disorders, which includes systemic sclerosis, uterine fibroids, atherosclerosis, and keloids, among others. It has been postulated that diseases of aberrant scarring are more common in black individuals due to the protective effect profibrotic alleles have against endemic helminthic infections of sub-Saharan infections, including oncocerciasis.6
A recent study showed an increased expression of fibroproliferative genes, particularly those implicated in other fibroproliferative disorders, in affected scalp of patients with CCCA.7 Most notably, an expression in gene overlap was noted between fibroids and CCCA in this study, though the relationship between these two diseases needs to be further explored.
Gene Variants Identified in CCCA
More recently, a new study has identified a gene variant of peptidyl arginine deiminase 3, PADI3, that is present in approximately one-quarter of studied patients with CCCA.8PADI3 plays a role in hair shaft formation and has been implicated in another hair disorder, uncombable hair syndrome, though the latter presents in children, improves with age, and is not associated with a scarring phenotype.9 However, this study has provided greater insight into our understanding of CCCA by establishing a possible genetic predisposition in patients affected with this disease.8
What’s Next for CCCA?
For years, many patients with CCCA have been turned away with few answers and left thinking that it is their own styling habits that have led to their hair loss,
The future is bright for CCCA. Although our understanding of CCCA is still in its infancy, it is my hope that with greater understanding of this disease will come greater empathy for our patients.
- LoPresti P, Papa CM, Kligman AM. Hot comb alopecia. Arch Dermatol. 1968;98:234-238.
- Gathers RC, Lim HW. Central centrifugal cicatricial alopecia: past, present, and future. J Am Acad Dermatol. 2009;60:660-668.
- Kyei A, Bergfeld WF, Piliang M, et al. Medical and environmental risk factors for the development of central centrifugal cicatricial alopecia: a population study. Arch Dermatol. 2011;147:909-914.
- Dina Y, Okoye GA, Aguh C. Association of uterine leiomyomas with central centrifugal cicatricial alopecia. JAMA Dermatol. 2018;154:213-214.
- Whiting DA, Olsen EA. Central centrifugal cicatricial alopecia. Dermatol Ther. 2008;21:268-278.
- Hellwege JN, Torstenson ES, Russell SB, et al. Evidence of selection as a cause for racial disparities in fibroproliferative disease. PLoS One. 2017;12:e0182791. doi:10.1371/journal.pone.0182791.
- Aguh C, Dina Y, Talbot CC Jr, et al. Fibroproliferative genes are preferentially expressed in central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2018;79:904.e1-912.e1.
- Malki L, Sarig O, Romano MT, et al. Variant PADI3 in central centrifugal cicatricial alopecia. N Engl J Med. 2019;380:833-841.
- Matis WL, Baden H, Green R, et al. Uncombable-hair syndrome. Pediatr Dermatol. 1987;4:215-219.
- LoPresti P, Papa CM, Kligman AM. Hot comb alopecia. Arch Dermatol. 1968;98:234-238.
- Gathers RC, Lim HW. Central centrifugal cicatricial alopecia: past, present, and future. J Am Acad Dermatol. 2009;60:660-668.
- Kyei A, Bergfeld WF, Piliang M, et al. Medical and environmental risk factors for the development of central centrifugal cicatricial alopecia: a population study. Arch Dermatol. 2011;147:909-914.
- Dina Y, Okoye GA, Aguh C. Association of uterine leiomyomas with central centrifugal cicatricial alopecia. JAMA Dermatol. 2018;154:213-214.
- Whiting DA, Olsen EA. Central centrifugal cicatricial alopecia. Dermatol Ther. 2008;21:268-278.
- Hellwege JN, Torstenson ES, Russell SB, et al. Evidence of selection as a cause for racial disparities in fibroproliferative disease. PLoS One. 2017;12:e0182791. doi:10.1371/journal.pone.0182791.
- Aguh C, Dina Y, Talbot CC Jr, et al. Fibroproliferative genes are preferentially expressed in central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2018;79:904.e1-912.e1.
- Malki L, Sarig O, Romano MT, et al. Variant PADI3 in central centrifugal cicatricial alopecia. N Engl J Med. 2019;380:833-841.
- Matis WL, Baden H, Green R, et al. Uncombable-hair syndrome. Pediatr Dermatol. 1987;4:215-219.

Pediatric dermatology update: New research offers insight into psoriasis, alopecia
LAS VEGAS – Recent research is offering new insights into psoriasis and alopecia in the pediatric population, a dermatologist told colleagues, and it’s time to be on the lookout for psoriasis linked to treatment with tumor necrosis factor (TNF) inhibitors.

Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics, at the University of California, San Diego, offered these tips and comments in a presentation at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar:
Psoriasis
It’s a brand new day for adult psoriasis sufferers, but it seems to be only a brand new morning for their pediatric counterparts. “Kids and teenagers were left behind in the biologic revolution,” Dr. Eichenfield said. “Only two systemic biologics have been approved for psoriasis in children.” They are ustekinumab (Stelara), approved by the Food and Drug Administration for treating psoriasis in children aged 12 years and older, and etanercept, approved for aged 4 years and older.
The good news, he said, is that “our new biologic agents are now being studied in children.”
Research is also providing new insight into pediatric psoriasis, said Dr. Eichenfield, who is also chief of pediatric and adolescent dermatology at Rady Children’s Hospital in San Diego. It’s now clear that “there’s a lot more facial involvement, and a high involvement of scalp and nail,” he noted.
It’s also clear, he said, that inflammation begins early in pediatric psoriasis. That raises the question of whether it’s a good idea to launch aggressive treatment to stop the “psoriatic march” toward cardiovascular and other medical problems down the line, he commented.
“Keep an open mind to getting aggressive in therapy,” he advised, although he acknowledged that “it’s hard to get beyond the two biologics, and only one is approved for children under 12.”
Dr. Eichenfield advised colleagues to keep an eye out for TNF inhibitor–induced psoriasis. “We’re seeing it pretty regularly,” he said, commonly in children who are treated with TNF inhibitors for rheumatoid arthritis or Crohn’s disease.
The lesions “look like dermatitis but are very psoriasiform,” he said, and research suggests this can appear after a single dose or after as many as 63 months of treatment. Topical and light therapy can be helpful. But if those treatments do not help, he said, it’s time to consider changing the biologic that the patient is taking. “Is the biologic adequately controlling their underlying disease? If not, you can help find one that would be great for their underlying disease and clear up their psoriasis.”
Alopecia
Pediatric alopecia “is a problem I see pretty regularly in practice,” Dr. Eichenfield said. When he sees patients with alopecia, he says that, “‘if your child doesn’t have 50% hair loss, you’re in the good group. It will generally heal up and never come back again.’ ”
He referred to a recent study, where investigators at the Children’s Hospital of Philadelphia retrospectively studied 125 children under age 4 years who were diagnosed with alopecia areata and followed for 2 years. Over time, those children with over 50% of hair loss initially were more likely to have worsening Severity of Alopecia Tool (SALT) scores over the follow-up period. But a high proportion of those with mild alopecia initially continued to have mild alopecia at follow-up (Pediatr Dermatol. 2019 Aug 29. doi: 10.1111/pde.13990).
Dr. Eichenfield noted that the study found that 41% of the patients also had atopic dermatitis.
He also highlighted two other recent studies on pediatric alopecia: One found that while vitamin D levels were low in a majority of children with alopecia in the study, the proportion who had a deficiency was similar to the proportion in a larger pediatric population, at about 22% in both groups (J Am Acad Dermatol. 2018 Sep;79(3):e43-e44). Supplementation doesn’t seem to help. “It’s not important to test levels,” he said.
Another study examined whether it’s a good idea to test patients for celiac disease in children with alopecia (Pediatr Dermatol. 2018 Jul;35[4]:535-8). Some parents may ask this question, but the answer, he said, is generally no.
What’s next? “We were hoping oral and topical JAK inhibitors would work well” in this population, Dr. Eichenfield said, but study findings haven’t been promising.
Still, oral tofacitinib (Xeljanz) showed some “pretty impressive” success in a recent study in four children, he noted. Based on the results, the authors wrote that “we suggest that, after proper counseling regarding the risks, including severe infection and malignancy, the use of tofacitinib may be considered for preadolescent children with AA [alopecia areata] who are experiencing psychosocial impairment” (J Am Acad Dermatol. 2019 Feb;80[2]:568-70).
In general, Dr. Eichenfield said, research on pediatric alopecia “will be secondary, especially with JAK inhibitors because of the risk of side effects. But [children will] probably tolerate them better than adults do because they have fewer medical problems.”
Meanwhile, he added, controversy continues to swirl around how to treat children over age 10 years who have lost 50% or more of their hair. “I’ve seen hundreds of kids with alopecia areata,” he said, “and I can’t predict what the course may be.”
Dr. Eichenfield reports multiple relationships (consultant or investigator) with various pharmaceutical companies, including Abbvie, Allergan, Lilly, Novartis, and others. SDEF and this news organization are owned by the same parent company.
LAS VEGAS – Recent research is offering new insights into psoriasis and alopecia in the pediatric population, a dermatologist told colleagues, and it’s time to be on the lookout for psoriasis linked to treatment with tumor necrosis factor (TNF) inhibitors.
Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics, at the University of California, San Diego, offered these tips and comments in a presentation at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar:
Psoriasis
It’s a brand new day for adult psoriasis sufferers, but it seems to be only a brand new morning for their pediatric counterparts. “Kids and teenagers were left behind in the biologic revolution,” Dr. Eichenfield said. “Only two systemic biologics have been approved for psoriasis in children.” They are ustekinumab (Stelara), approved by the Food and Drug Administration for treating psoriasis in children aged 12 years and older, and etanercept, approved for aged 4 years and older.
The good news, he said, is that “our new biologic agents are now being studied in children.”
Research is also providing new insight into pediatric psoriasis, said Dr. Eichenfield, who is also chief of pediatric and adolescent dermatology at Rady Children’s Hospital in San Diego. It’s now clear that “there’s a lot more facial involvement, and a high involvement of scalp and nail,” he noted.
It’s also clear, he said, that inflammation begins early in pediatric psoriasis. That raises the question of whether it’s a good idea to launch aggressive treatment to stop the “psoriatic march” toward cardiovascular and other medical problems down the line, he commented.
“Keep an open mind to getting aggressive in therapy,” he advised, although he acknowledged that “it’s hard to get beyond the two biologics, and only one is approved for children under 12.”
Dr. Eichenfield advised colleagues to keep an eye out for TNF inhibitor–induced psoriasis. “We’re seeing it pretty regularly,” he said, commonly in children who are treated with TNF inhibitors for rheumatoid arthritis or Crohn’s disease.
The lesions “look like dermatitis but are very psoriasiform,” he said, and research suggests this can appear after a single dose or after as many as 63 months of treatment. Topical and light therapy can be helpful. But if those treatments do not help, he said, it’s time to consider changing the biologic that the patient is taking. “Is the biologic adequately controlling their underlying disease? If not, you can help find one that would be great for their underlying disease and clear up their psoriasis.”
Alopecia
Pediatric alopecia “is a problem I see pretty regularly in practice,” Dr. Eichenfield said. When he sees patients with alopecia, he says that, “‘if your child doesn’t have 50% hair loss, you’re in the good group. It will generally heal up and never come back again.’ ”
He referred to a recent study, where investigators at the Children’s Hospital of Philadelphia retrospectively studied 125 children under age 4 years who were diagnosed with alopecia areata and followed for 2 years. Over time, those children with over 50% of hair loss initially were more likely to have worsening Severity of Alopecia Tool (SALT) scores over the follow-up period. But a high proportion of those with mild alopecia initially continued to have mild alopecia at follow-up (Pediatr Dermatol. 2019 Aug 29. doi: 10.1111/pde.13990).
Dr. Eichenfield noted that the study found that 41% of the patients also had atopic dermatitis.
He also highlighted two other recent studies on pediatric alopecia: One found that while vitamin D levels were low in a majority of children with alopecia in the study, the proportion who had a deficiency was similar to the proportion in a larger pediatric population, at about 22% in both groups (J Am Acad Dermatol. 2018 Sep;79(3):e43-e44). Supplementation doesn’t seem to help. “It’s not important to test levels,” he said.
Another study examined whether it’s a good idea to test patients for celiac disease in children with alopecia (Pediatr Dermatol. 2018 Jul;35[4]:535-8). Some parents may ask this question, but the answer, he said, is generally no.
What’s next? “We were hoping oral and topical JAK inhibitors would work well” in this population, Dr. Eichenfield said, but study findings haven’t been promising.
Still, oral tofacitinib (Xeljanz) showed some “pretty impressive” success in a recent study in four children, he noted. Based on the results, the authors wrote that “we suggest that, after proper counseling regarding the risks, including severe infection and malignancy, the use of tofacitinib may be considered for preadolescent children with AA [alopecia areata] who are experiencing psychosocial impairment” (J Am Acad Dermatol. 2019 Feb;80[2]:568-70).
In general, Dr. Eichenfield said, research on pediatric alopecia “will be secondary, especially with JAK inhibitors because of the risk of side effects. But [children will] probably tolerate them better than adults do because they have fewer medical problems.”
Meanwhile, he added, controversy continues to swirl around how to treat children over age 10 years who have lost 50% or more of their hair. “I’ve seen hundreds of kids with alopecia areata,” he said, “and I can’t predict what the course may be.”
Dr. Eichenfield reports multiple relationships (consultant or investigator) with various pharmaceutical companies, including Abbvie, Allergan, Lilly, Novartis, and others. SDEF and this news organization are owned by the same parent company.
LAS VEGAS – Recent research is offering new insights into psoriasis and alopecia in the pediatric population, a dermatologist told colleagues, and it’s time to be on the lookout for psoriasis linked to treatment with tumor necrosis factor (TNF) inhibitors.
Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics, at the University of California, San Diego, offered these tips and comments in a presentation at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar:
Psoriasis
It’s a brand new day for adult psoriasis sufferers, but it seems to be only a brand new morning for their pediatric counterparts. “Kids and teenagers were left behind in the biologic revolution,” Dr. Eichenfield said. “Only two systemic biologics have been approved for psoriasis in children.” They are ustekinumab (Stelara), approved by the Food and Drug Administration for treating psoriasis in children aged 12 years and older, and etanercept, approved for aged 4 years and older.
The good news, he said, is that “our new biologic agents are now being studied in children.”
Research is also providing new insight into pediatric psoriasis, said Dr. Eichenfield, who is also chief of pediatric and adolescent dermatology at Rady Children’s Hospital in San Diego. It’s now clear that “there’s a lot more facial involvement, and a high involvement of scalp and nail,” he noted.
It’s also clear, he said, that inflammation begins early in pediatric psoriasis. That raises the question of whether it’s a good idea to launch aggressive treatment to stop the “psoriatic march” toward cardiovascular and other medical problems down the line, he commented.
“Keep an open mind to getting aggressive in therapy,” he advised, although he acknowledged that “it’s hard to get beyond the two biologics, and only one is approved for children under 12.”
Dr. Eichenfield advised colleagues to keep an eye out for TNF inhibitor–induced psoriasis. “We’re seeing it pretty regularly,” he said, commonly in children who are treated with TNF inhibitors for rheumatoid arthritis or Crohn’s disease.
The lesions “look like dermatitis but are very psoriasiform,” he said, and research suggests this can appear after a single dose or after as many as 63 months of treatment. Topical and light therapy can be helpful. But if those treatments do not help, he said, it’s time to consider changing the biologic that the patient is taking. “Is the biologic adequately controlling their underlying disease? If not, you can help find one that would be great for their underlying disease and clear up their psoriasis.”
Alopecia
Pediatric alopecia “is a problem I see pretty regularly in practice,” Dr. Eichenfield said. When he sees patients with alopecia, he says that, “‘if your child doesn’t have 50% hair loss, you’re in the good group. It will generally heal up and never come back again.’ ”
He referred to a recent study, where investigators at the Children’s Hospital of Philadelphia retrospectively studied 125 children under age 4 years who were diagnosed with alopecia areata and followed for 2 years. Over time, those children with over 50% of hair loss initially were more likely to have worsening Severity of Alopecia Tool (SALT) scores over the follow-up period. But a high proportion of those with mild alopecia initially continued to have mild alopecia at follow-up (Pediatr Dermatol. 2019 Aug 29. doi: 10.1111/pde.13990).
Dr. Eichenfield noted that the study found that 41% of the patients also had atopic dermatitis.
He also highlighted two other recent studies on pediatric alopecia: One found that while vitamin D levels were low in a majority of children with alopecia in the study, the proportion who had a deficiency was similar to the proportion in a larger pediatric population, at about 22% in both groups (J Am Acad Dermatol. 2018 Sep;79(3):e43-e44). Supplementation doesn’t seem to help. “It’s not important to test levels,” he said.
Another study examined whether it’s a good idea to test patients for celiac disease in children with alopecia (Pediatr Dermatol. 2018 Jul;35[4]:535-8). Some parents may ask this question, but the answer, he said, is generally no.
What’s next? “We were hoping oral and topical JAK inhibitors would work well” in this population, Dr. Eichenfield said, but study findings haven’t been promising.
Still, oral tofacitinib (Xeljanz) showed some “pretty impressive” success in a recent study in four children, he noted. Based on the results, the authors wrote that “we suggest that, after proper counseling regarding the risks, including severe infection and malignancy, the use of tofacitinib may be considered for preadolescent children with AA [alopecia areata] who are experiencing psychosocial impairment” (J Am Acad Dermatol. 2019 Feb;80[2]:568-70).
In general, Dr. Eichenfield said, research on pediatric alopecia “will be secondary, especially with JAK inhibitors because of the risk of side effects. But [children will] probably tolerate them better than adults do because they have fewer medical problems.”
Meanwhile, he added, controversy continues to swirl around how to treat children over age 10 years who have lost 50% or more of their hair. “I’ve seen hundreds of kids with alopecia areata,” he said, “and I can’t predict what the course may be.”
Dr. Eichenfield reports multiple relationships (consultant or investigator) with various pharmaceutical companies, including Abbvie, Allergan, Lilly, Novartis, and others. SDEF and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
FDA still concerned about biotin affecting troponin tests
The

However, not all troponin tests are affected, according to the update. “Since the FDA’s safety communication on this topic in 2017, some lab test developers have been successful at mitigating the biotin interference of their assays, but others have not yet addressed it,” according to the new communication, issued in early November.
Also known as vitamin B7 and appearing in many dietary supplements, including prenatal multivitamins and supplements for hair, skin, and nail growth, biotin can lead to falsely low results on some troponin tests, especially at high levels. The worry is that biotin interference could therefore lead to missed diagnoses. The FDA has provided a list of those tests that have not taken biotin’s effects into account, titled “Biotin Interference with Troponin Lab Tests – Assays Subject to Biotin Interference.”
The daily recommended allowance for biotin, according to the communication, is about 0.3 mg, but it isn’t always clear how much is actually included in supplements – some can contain 20 mg or even as much as 100 mg per pill of biotin. The communication includes recommendations for patients, health care professionals, laboratory personnel, and lab test manufacturers and developers.
The full safety communication can be found on the FDA website, and problems with tests can be reported via the FDA’s MedWatch Voluntary Reporting Form.
The
However, not all troponin tests are affected, according to the update. “Since the FDA’s safety communication on this topic in 2017, some lab test developers have been successful at mitigating the biotin interference of their assays, but others have not yet addressed it,” according to the new communication, issued in early November.
Also known as vitamin B7 and appearing in many dietary supplements, including prenatal multivitamins and supplements for hair, skin, and nail growth, biotin can lead to falsely low results on some troponin tests, especially at high levels. The worry is that biotin interference could therefore lead to missed diagnoses. The FDA has provided a list of those tests that have not taken biotin’s effects into account, titled “Biotin Interference with Troponin Lab Tests – Assays Subject to Biotin Interference.”
The daily recommended allowance for biotin, according to the communication, is about 0.3 mg, but it isn’t always clear how much is actually included in supplements – some can contain 20 mg or even as much as 100 mg per pill of biotin. The communication includes recommendations for patients, health care professionals, laboratory personnel, and lab test manufacturers and developers.
The full safety communication can be found on the FDA website, and problems with tests can be reported via the FDA’s MedWatch Voluntary Reporting Form.
The
However, not all troponin tests are affected, according to the update. “Since the FDA’s safety communication on this topic in 2017, some lab test developers have been successful at mitigating the biotin interference of their assays, but others have not yet addressed it,” according to the new communication, issued in early November.
Also known as vitamin B7 and appearing in many dietary supplements, including prenatal multivitamins and supplements for hair, skin, and nail growth, biotin can lead to falsely low results on some troponin tests, especially at high levels. The worry is that biotin interference could therefore lead to missed diagnoses. The FDA has provided a list of those tests that have not taken biotin’s effects into account, titled “Biotin Interference with Troponin Lab Tests – Assays Subject to Biotin Interference.”
The daily recommended allowance for biotin, according to the communication, is about 0.3 mg, but it isn’t always clear how much is actually included in supplements – some can contain 20 mg or even as much as 100 mg per pill of biotin. The communication includes recommendations for patients, health care professionals, laboratory personnel, and lab test manufacturers and developers.
The full safety communication can be found on the FDA website, and problems with tests can be reported via the FDA’s MedWatch Voluntary Reporting Form.
Expert reviews strategies for diagnosing, treating onychomycosis
LAS VEGAS – The way Neal Bhatia, MD, sees it, there is no such thing as a classical presentation of onychomycosis.

“This is where proving your diagnosis is half the battle, even though we are sometimes using empiric therapy in suspected cases,” he said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. “Prove the diagnosis and get the extra tests necessary.”
According to Dr. Bhatia, director of clinical dermatology research at San Diego-based Therapeutics Clinical Research, . With onychomycosis, the ultimate treatment goal from the standpoint of clinicians is no more fungus, he noted, while the desired endpoint from the standpoint of some patients is normal-looking nails.
“Endpoint failures in a research trial are not the same as what we tell patients in the clinic,” Dr. Bhatia said. “If you have a patient using something topical for 52 weeks and they see two-thirds of their nail improve, you’re not going to say, ‘Stop; you’re a failure now.’ You tell them, ‘Keep going and we’ll keep watching.’ But in the research world, it’s very different when you look at all of the different endpoints that have to be met at the finish line. It’s very important to measure success based on what that patient’s experiencing.”
According to Dr. Bhatia, diagnosing onychomycosis by visual assessment has a sensitivity of 77% and a specificity of 47%, while KOH has a sensitivity between 67% and 93% and specificity between 38% and 78%. PCR, meanwhile, “is quick, but it’s expensive and it has a high false-positive rate. So, in those difficult patients who aren’t responding [to treatment], maybe we can’t just trust our eyes alone [to make a diagnosis].”
Tests such as a KOH stain have low sensitivity, with high costs of more sensitive tests such as the periodic acid–Schiff (PAS) stain. In a recent study, researchers conducted a retrospective cohort analysis of 600 patients with toenail clippings sent for PAS stain during January 2000–December 2013 (J Am Acad Dermatol. 2015 May; 72(5):AB116). They reviewed records to determine which PAS stains were performed to confirm probable clinical diagnosis of onychomycosis.
The researchers found that 30% of toenail clippings were sent for confirmatory PAS staining by dermatologists, compared with 37% by podiatrists and 34% by other clinicians. Of these tests, 75% ordered by dermatologists were positive for fungus, compared with 81% ordered by podiatrists, and 66% ordered by other physicians. “The positive predictive value of clinical suspicion for true onychomycosis was high, and the findings question whether or not a confirmatory test is really necessary,” Dr. Bhatia said.
Preventative strategies to control recurrence of onychomycosis include using maintenance regimens of the recommended antifungal agent, discarding old shoes, alternating wearing different pairs of shoes, periodically disinfecting shoes, washing feet regularly, and alerting the physician to the first sign of infection.
In an effort to investigate strategies to minimize recurrence of onychomycosis, Dr. Bhatia and colleagues evaluated 73 patients over the course of 7 years who were taking either terbinafine or itraconazole (J Drugs Dermatol. 2016;15[3]:279-82). Thirty-six months later, the overall mean recurrence rate among patients was 14%. Prognostic factors influencing recurrence included patient’s family history; lifestyle; underlying physiology (presence of a very thick nail, extensive involvement of the entire nail unit, lateral nail disease and yellow spikes); physical trauma, especially in the elderly; concomitant disease, such as peripheral artery disease and/or diabetes; immunocompromised or immunosuppressed patients, and the presence of tinea pedis.
Based on their analysis, they recommended the following strategies to prevent or limit recurrence: prophylactic use of a topical antifungal twice-weekly for 2-3 years; periodic application of a topical antifungal to plantar surface and/or interdigital spaces; treatment of any coexisting tinea pedis; treating immediate family members; footwear and sock decontamination with antifungal powder; ultraviolet light or ozone; avoidance of activities known to risk spread of disease, such as communal swimming pools.
Dr. Bhatia concluded his presentation by noting that the ideal treatment for onychomycosis would not pose a systemic risk to the liver, heart, or other organs; would not require monitoring of labs; would not require debridement; and would not interact with other drugs. It would also penetrate the nail plate – especially the diseased nail – and would be quick drying.
SDEF and this news organization are owned by the same parent company.
Dr. Bhatia disclosed having affiliations with Actavis, Allergan, Anacor, Aqua, Bayer, Biofrontera, BiopharmX, Cipher, Dermira, Dusa, Exeltis, Ferndale, Foamix, Galderma, Intraderm, ISDIN, LaRoche-Posay, Leo, Novan, Novartis, PharmaDerm, Promius, Regeneron, Sanofi, Sun Pharma, and Valeant.
LAS VEGAS – The way Neal Bhatia, MD, sees it, there is no such thing as a classical presentation of onychomycosis.
“This is where proving your diagnosis is half the battle, even though we are sometimes using empiric therapy in suspected cases,” he said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. “Prove the diagnosis and get the extra tests necessary.”
According to Dr. Bhatia, director of clinical dermatology research at San Diego-based Therapeutics Clinical Research, . With onychomycosis, the ultimate treatment goal from the standpoint of clinicians is no more fungus, he noted, while the desired endpoint from the standpoint of some patients is normal-looking nails.
“Endpoint failures in a research trial are not the same as what we tell patients in the clinic,” Dr. Bhatia said. “If you have a patient using something topical for 52 weeks and they see two-thirds of their nail improve, you’re not going to say, ‘Stop; you’re a failure now.’ You tell them, ‘Keep going and we’ll keep watching.’ But in the research world, it’s very different when you look at all of the different endpoints that have to be met at the finish line. It’s very important to measure success based on what that patient’s experiencing.”
According to Dr. Bhatia, diagnosing onychomycosis by visual assessment has a sensitivity of 77% and a specificity of 47%, while KOH has a sensitivity between 67% and 93% and specificity between 38% and 78%. PCR, meanwhile, “is quick, but it’s expensive and it has a high false-positive rate. So, in those difficult patients who aren’t responding [to treatment], maybe we can’t just trust our eyes alone [to make a diagnosis].”
Tests such as a KOH stain have low sensitivity, with high costs of more sensitive tests such as the periodic acid–Schiff (PAS) stain. In a recent study, researchers conducted a retrospective cohort analysis of 600 patients with toenail clippings sent for PAS stain during January 2000–December 2013 (J Am Acad Dermatol. 2015 May; 72(5):AB116). They reviewed records to determine which PAS stains were performed to confirm probable clinical diagnosis of onychomycosis.
The researchers found that 30% of toenail clippings were sent for confirmatory PAS staining by dermatologists, compared with 37% by podiatrists and 34% by other clinicians. Of these tests, 75% ordered by dermatologists were positive for fungus, compared with 81% ordered by podiatrists, and 66% ordered by other physicians. “The positive predictive value of clinical suspicion for true onychomycosis was high, and the findings question whether or not a confirmatory test is really necessary,” Dr. Bhatia said.
Preventative strategies to control recurrence of onychomycosis include using maintenance regimens of the recommended antifungal agent, discarding old shoes, alternating wearing different pairs of shoes, periodically disinfecting shoes, washing feet regularly, and alerting the physician to the first sign of infection.
In an effort to investigate strategies to minimize recurrence of onychomycosis, Dr. Bhatia and colleagues evaluated 73 patients over the course of 7 years who were taking either terbinafine or itraconazole (J Drugs Dermatol. 2016;15[3]:279-82). Thirty-six months later, the overall mean recurrence rate among patients was 14%. Prognostic factors influencing recurrence included patient’s family history; lifestyle; underlying physiology (presence of a very thick nail, extensive involvement of the entire nail unit, lateral nail disease and yellow spikes); physical trauma, especially in the elderly; concomitant disease, such as peripheral artery disease and/or diabetes; immunocompromised or immunosuppressed patients, and the presence of tinea pedis.
Based on their analysis, they recommended the following strategies to prevent or limit recurrence: prophylactic use of a topical antifungal twice-weekly for 2-3 years; periodic application of a topical antifungal to plantar surface and/or interdigital spaces; treatment of any coexisting tinea pedis; treating immediate family members; footwear and sock decontamination with antifungal powder; ultraviolet light or ozone; avoidance of activities known to risk spread of disease, such as communal swimming pools.
Dr. Bhatia concluded his presentation by noting that the ideal treatment for onychomycosis would not pose a systemic risk to the liver, heart, or other organs; would not require monitoring of labs; would not require debridement; and would not interact with other drugs. It would also penetrate the nail plate – especially the diseased nail – and would be quick drying.
SDEF and this news organization are owned by the same parent company.
Dr. Bhatia disclosed having affiliations with Actavis, Allergan, Anacor, Aqua, Bayer, Biofrontera, BiopharmX, Cipher, Dermira, Dusa, Exeltis, Ferndale, Foamix, Galderma, Intraderm, ISDIN, LaRoche-Posay, Leo, Novan, Novartis, PharmaDerm, Promius, Regeneron, Sanofi, Sun Pharma, and Valeant.
LAS VEGAS – The way Neal Bhatia, MD, sees it, there is no such thing as a classical presentation of onychomycosis.
“This is where proving your diagnosis is half the battle, even though we are sometimes using empiric therapy in suspected cases,” he said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. “Prove the diagnosis and get the extra tests necessary.”
According to Dr. Bhatia, director of clinical dermatology research at San Diego-based Therapeutics Clinical Research, . With onychomycosis, the ultimate treatment goal from the standpoint of clinicians is no more fungus, he noted, while the desired endpoint from the standpoint of some patients is normal-looking nails.
“Endpoint failures in a research trial are not the same as what we tell patients in the clinic,” Dr. Bhatia said. “If you have a patient using something topical for 52 weeks and they see two-thirds of their nail improve, you’re not going to say, ‘Stop; you’re a failure now.’ You tell them, ‘Keep going and we’ll keep watching.’ But in the research world, it’s very different when you look at all of the different endpoints that have to be met at the finish line. It’s very important to measure success based on what that patient’s experiencing.”
According to Dr. Bhatia, diagnosing onychomycosis by visual assessment has a sensitivity of 77% and a specificity of 47%, while KOH has a sensitivity between 67% and 93% and specificity between 38% and 78%. PCR, meanwhile, “is quick, but it’s expensive and it has a high false-positive rate. So, in those difficult patients who aren’t responding [to treatment], maybe we can’t just trust our eyes alone [to make a diagnosis].”
Tests such as a KOH stain have low sensitivity, with high costs of more sensitive tests such as the periodic acid–Schiff (PAS) stain. In a recent study, researchers conducted a retrospective cohort analysis of 600 patients with toenail clippings sent for PAS stain during January 2000–December 2013 (J Am Acad Dermatol. 2015 May; 72(5):AB116). They reviewed records to determine which PAS stains were performed to confirm probable clinical diagnosis of onychomycosis.
The researchers found that 30% of toenail clippings were sent for confirmatory PAS staining by dermatologists, compared with 37% by podiatrists and 34% by other clinicians. Of these tests, 75% ordered by dermatologists were positive for fungus, compared with 81% ordered by podiatrists, and 66% ordered by other physicians. “The positive predictive value of clinical suspicion for true onychomycosis was high, and the findings question whether or not a confirmatory test is really necessary,” Dr. Bhatia said.
Preventative strategies to control recurrence of onychomycosis include using maintenance regimens of the recommended antifungal agent, discarding old shoes, alternating wearing different pairs of shoes, periodically disinfecting shoes, washing feet regularly, and alerting the physician to the first sign of infection.
In an effort to investigate strategies to minimize recurrence of onychomycosis, Dr. Bhatia and colleagues evaluated 73 patients over the course of 7 years who were taking either terbinafine or itraconazole (J Drugs Dermatol. 2016;15[3]:279-82). Thirty-six months later, the overall mean recurrence rate among patients was 14%. Prognostic factors influencing recurrence included patient’s family history; lifestyle; underlying physiology (presence of a very thick nail, extensive involvement of the entire nail unit, lateral nail disease and yellow spikes); physical trauma, especially in the elderly; concomitant disease, such as peripheral artery disease and/or diabetes; immunocompromised or immunosuppressed patients, and the presence of tinea pedis.
Based on their analysis, they recommended the following strategies to prevent or limit recurrence: prophylactic use of a topical antifungal twice-weekly for 2-3 years; periodic application of a topical antifungal to plantar surface and/or interdigital spaces; treatment of any coexisting tinea pedis; treating immediate family members; footwear and sock decontamination with antifungal powder; ultraviolet light or ozone; avoidance of activities known to risk spread of disease, such as communal swimming pools.
Dr. Bhatia concluded his presentation by noting that the ideal treatment for onychomycosis would not pose a systemic risk to the liver, heart, or other organs; would not require monitoring of labs; would not require debridement; and would not interact with other drugs. It would also penetrate the nail plate – especially the diseased nail – and would be quick drying.
SDEF and this news organization are owned by the same parent company.
Dr. Bhatia disclosed having affiliations with Actavis, Allergan, Anacor, Aqua, Bayer, Biofrontera, BiopharmX, Cipher, Dermira, Dusa, Exeltis, Ferndale, Foamix, Galderma, Intraderm, ISDIN, LaRoche-Posay, Leo, Novan, Novartis, PharmaDerm, Promius, Regeneron, Sanofi, Sun Pharma, and Valeant.
AT THE SDEF LAS VEGAS DERMATOLOGY SEMINAR
Oral JAK1/2 inhibitor promising in alopecia areata
MADRID – In a phase 2, dose-ranging study, 78% of patients with long-standing moderate or severe alopecia areata rated their condition as “much improved” or “very much improved” after 24 weeks on the top dose of an investigational oral selective Janus kinase 1 and 2 (JAK1/2) inhibitor, compared with 21% of placebo-treated controls, James V. Cassella, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.

The primary study endpoint – at least a 50% reduction in the Severity of Alopecia Tool (SALT) score between baseline and 24 weeks – was achieved in 58% of patients on the JAK1/2 inhibitor (known for now as CTP-543) at 12 mg twice a day, 47% of those on CTP-543 at 8 mg twice a day, 21% on 4 mg twice a day, and in 9% of controls on placebo in the double-blind randomized trial. The 4-mg twice-daily dosing will not move on to phase 3 studies because of its demonstrated lack of efficacy, according to Dr. Cassella, chief development officer at Concert Pharmaceuticals, the study sponsor.
The study included 149 adults whose current episode of alopecia areata was of 3-6 years’ duration, with an average lifetime 15-year history of active disease. Their average baseline score on the 0-100 SALT scale was in the upper 80s, indicative of 80% or greater hair loss.
A SALT 75 response, meaning at least a 75% reduction in SALT score from baseline, was achieved in a dose-dependent fashion: In 42% of patients at the top dose of CTP-543, 29% of those on 8 mg twice a day, 14% with 4 mg twice a day, and in 7% of controls. An even more rigorous SALT 90 response was attained in 36%, 16%, 2%, and no controls, respectively.
The 12-mg twice-daily dosing produced faster onset and greater magnitude of response than did the 8-mg twice-daily dosing, but this dose-ranging study is not the final word on that score, according to Dr. Cassella.
“Week 24 is not a magic number,” he said. “The slope of the efficacy line for 8 mg [twice a day] looked like it was still going up at week 24, and the 12-mg BID curve hadn’t completely plateaued. Those are things we will consider for the future in terms of long-term trials.”
Changes in the eyebrows and lashes weren’t formally assessed in this study, although they will be in future trials. Anecdotally, however, patients with alopecia areata at those sites typically experienced complete or nearly complete regrowth in response to the higher doses of CTP-543, he said.
Safety-wise, there was no trend for increased adverse events with increasing doses of CTP-543. The observed treatment side effects were those typical of JAK inhibitors as a class effect, mainly headache, nasopharyngitis, upper respiratory infections, and new-onset acne. In terms of hematologic findings of special interest, there was one case of reversible neutropenia in the control group and another in the 8-mg twice-daily group, which resolved upon temporary suspension of treatment.
“Nothing surprising to us, and nothing very serious,” Dr. Cassella said.
Most patients in the 12-mg twice daily group have enrolled in an ongoing long-term extension study. In addition, two phase 2 studies are ongoing, with a focus on once-daily versus twice-daily dosing at 8 mg or 12 mg. Phase 3 studies are in the planning stage, he added.
The phase 2 dose-ranging study was sponsored by Concert Pharmaceuticals.
CTP-543 is one of an array of oral JAK inhibitors now in the developmental pipeline for alopecia areata, a severe, psychosocially devastating disease for which at present there is no Food and Drug Administration–approved therapy.
MADRID – In a phase 2, dose-ranging study, 78% of patients with long-standing moderate or severe alopecia areata rated their condition as “much improved” or “very much improved” after 24 weeks on the top dose of an investigational oral selective Janus kinase 1 and 2 (JAK1/2) inhibitor, compared with 21% of placebo-treated controls, James V. Cassella, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The primary study endpoint – at least a 50% reduction in the Severity of Alopecia Tool (SALT) score between baseline and 24 weeks – was achieved in 58% of patients on the JAK1/2 inhibitor (known for now as CTP-543) at 12 mg twice a day, 47% of those on CTP-543 at 8 mg twice a day, 21% on 4 mg twice a day, and in 9% of controls on placebo in the double-blind randomized trial. The 4-mg twice-daily dosing will not move on to phase 3 studies because of its demonstrated lack of efficacy, according to Dr. Cassella, chief development officer at Concert Pharmaceuticals, the study sponsor.
The study included 149 adults whose current episode of alopecia areata was of 3-6 years’ duration, with an average lifetime 15-year history of active disease. Their average baseline score on the 0-100 SALT scale was in the upper 80s, indicative of 80% or greater hair loss.
A SALT 75 response, meaning at least a 75% reduction in SALT score from baseline, was achieved in a dose-dependent fashion: In 42% of patients at the top dose of CTP-543, 29% of those on 8 mg twice a day, 14% with 4 mg twice a day, and in 7% of controls. An even more rigorous SALT 90 response was attained in 36%, 16%, 2%, and no controls, respectively.
The 12-mg twice-daily dosing produced faster onset and greater magnitude of response than did the 8-mg twice-daily dosing, but this dose-ranging study is not the final word on that score, according to Dr. Cassella.
“Week 24 is not a magic number,” he said. “The slope of the efficacy line for 8 mg [twice a day] looked like it was still going up at week 24, and the 12-mg BID curve hadn’t completely plateaued. Those are things we will consider for the future in terms of long-term trials.”
Changes in the eyebrows and lashes weren’t formally assessed in this study, although they will be in future trials. Anecdotally, however, patients with alopecia areata at those sites typically experienced complete or nearly complete regrowth in response to the higher doses of CTP-543, he said.
Safety-wise, there was no trend for increased adverse events with increasing doses of CTP-543. The observed treatment side effects were those typical of JAK inhibitors as a class effect, mainly headache, nasopharyngitis, upper respiratory infections, and new-onset acne. In terms of hematologic findings of special interest, there was one case of reversible neutropenia in the control group and another in the 8-mg twice-daily group, which resolved upon temporary suspension of treatment.
“Nothing surprising to us, and nothing very serious,” Dr. Cassella said.
Most patients in the 12-mg twice daily group have enrolled in an ongoing long-term extension study. In addition, two phase 2 studies are ongoing, with a focus on once-daily versus twice-daily dosing at 8 mg or 12 mg. Phase 3 studies are in the planning stage, he added.
The phase 2 dose-ranging study was sponsored by Concert Pharmaceuticals.
CTP-543 is one of an array of oral JAK inhibitors now in the developmental pipeline for alopecia areata, a severe, psychosocially devastating disease for which at present there is no Food and Drug Administration–approved therapy.
MADRID – In a phase 2, dose-ranging study, 78% of patients with long-standing moderate or severe alopecia areata rated their condition as “much improved” or “very much improved” after 24 weeks on the top dose of an investigational oral selective Janus kinase 1 and 2 (JAK1/2) inhibitor, compared with 21% of placebo-treated controls, James V. Cassella, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The primary study endpoint – at least a 50% reduction in the Severity of Alopecia Tool (SALT) score between baseline and 24 weeks – was achieved in 58% of patients on the JAK1/2 inhibitor (known for now as CTP-543) at 12 mg twice a day, 47% of those on CTP-543 at 8 mg twice a day, 21% on 4 mg twice a day, and in 9% of controls on placebo in the double-blind randomized trial. The 4-mg twice-daily dosing will not move on to phase 3 studies because of its demonstrated lack of efficacy, according to Dr. Cassella, chief development officer at Concert Pharmaceuticals, the study sponsor.
The study included 149 adults whose current episode of alopecia areata was of 3-6 years’ duration, with an average lifetime 15-year history of active disease. Their average baseline score on the 0-100 SALT scale was in the upper 80s, indicative of 80% or greater hair loss.
A SALT 75 response, meaning at least a 75% reduction in SALT score from baseline, was achieved in a dose-dependent fashion: In 42% of patients at the top dose of CTP-543, 29% of those on 8 mg twice a day, 14% with 4 mg twice a day, and in 7% of controls. An even more rigorous SALT 90 response was attained in 36%, 16%, 2%, and no controls, respectively.
The 12-mg twice-daily dosing produced faster onset and greater magnitude of response than did the 8-mg twice-daily dosing, but this dose-ranging study is not the final word on that score, according to Dr. Cassella.
“Week 24 is not a magic number,” he said. “The slope of the efficacy line for 8 mg [twice a day] looked like it was still going up at week 24, and the 12-mg BID curve hadn’t completely plateaued. Those are things we will consider for the future in terms of long-term trials.”
Changes in the eyebrows and lashes weren’t formally assessed in this study, although they will be in future trials. Anecdotally, however, patients with alopecia areata at those sites typically experienced complete or nearly complete regrowth in response to the higher doses of CTP-543, he said.
Safety-wise, there was no trend for increased adverse events with increasing doses of CTP-543. The observed treatment side effects were those typical of JAK inhibitors as a class effect, mainly headache, nasopharyngitis, upper respiratory infections, and new-onset acne. In terms of hematologic findings of special interest, there was one case of reversible neutropenia in the control group and another in the 8-mg twice-daily group, which resolved upon temporary suspension of treatment.
“Nothing surprising to us, and nothing very serious,” Dr. Cassella said.
Most patients in the 12-mg twice daily group have enrolled in an ongoing long-term extension study. In addition, two phase 2 studies are ongoing, with a focus on once-daily versus twice-daily dosing at 8 mg or 12 mg. Phase 3 studies are in the planning stage, he added.
The phase 2 dose-ranging study was sponsored by Concert Pharmaceuticals.
CTP-543 is one of an array of oral JAK inhibitors now in the developmental pipeline for alopecia areata, a severe, psychosocially devastating disease for which at present there is no Food and Drug Administration–approved therapy.
REPORTING FROM THE EADV CONGRESS
Central centrifugal cicatricial alopecia called epidemic in skin of color
NEW YORK – For unclear reasons, the prevalence of , creating an urgent need for early diagnosis and treatment, according to an expert who described this phenomenon at the Skin of Color Update 2019.

“CCCA has reached epidemic proportions,” contended Susan Taylor, MD, director of diversity, department of dermatology, Penn Medicine, Philadelphia.
Published data place prevalence rates of CCCA somewhere in the range of 3% to 6% among black women, but Dr. Taylor reported that she believes the condition is underdiagnosed. “I am seeing far more patients now than I was 30 years ago,” she maintained.
Others participating in the Skin of Color Update 2019 agreed. Heather Woolery-Lloyd, MD, director of ethnic skin care, department of dermatology and cutaneous surgery, University of Miami, also called the rising incidence of CCCA “an epidemic.” She, like Dr. Taylor, emphasized the critical importance of early diagnosis and treatment.
“I tell patients that if we can prevent hair loss over the next 10 years, this is a treatment success,” Dr. Woolery-Lloyd said. Although hair regrowth can be achieved in a minority of patients with treatments such as minoxidil, “the first goal is to prevent hair loss.”
Upon diagnosis, Dr. Woolery-Lloyd recommends treatment with intralesional triamcinolone and topical steroids immediately, adding other agents, such as oral antibiotics, if needed. Even in cases where CCCA has been identified before hair loss is visible, Dr. Woolery-Lloyd advised immediate therapy. Given that CCCA is a disease of reversible hair loss, she said, “do not take a wait-and-see approach.”
One potential obstacle for early diagnosis of CCCA, shared by other types of alopecia that are common in skin of color, is the failure of many clinicians to employ a standardized diagnosis in this patient population.
“If you do not have tightly coiled hair, it does not mean you cannot understand tightly coiled hair, but you have to learn, and you have to let patients know that you understand and have experience,” said Dr. Taylor, emphasizing the role of reassuring patients so they can be confident in the clinical recommendations.
Part of this reassurance will be derived from “interacting with patients in a culturally competent manner,” Dr. Taylor said. Clinicians must develop comfort and confidence in physically examining the hair and scalp, in asking patients to remove weaves or braids for a thorough inspection, and in avoiding comments that might be misinterpreted. Among these, she advised tactful questions about shampooing to avoid any implication that the clinician is implying inadequate hygiene.
When CCCA is suspected, a “biopsy is important” even in circumstances when the diagnosis seems straightforward. For one reason, a substantial proportion of patients may have a concomitant diagnosis. Dr. Taylor cited data from one study in which nearly 20% of CCCA patients also had traction alopecia and more than 10% had androgenic alopecia. Other coexisting problems identified on biopsy, including infection or seborrheic dermatitis, can help clinicians tailor a more effective intervention.
The initial signs of CCCA are typically hair breakage in the vertex of the scalp, which then expands in a central centrifugal pattern, according to Dr. Taylor. Although not all patients have signs of inflammation, such as itching and pustules, inhibition of inflammation represents the first line of therapy.
Relative to hair in the white population, the hair of black individuals grows more slowly and is more fragile, with greater amounts of breakage, said Dr. Taylor, citing published studies that support these differences. To improve early diagnosis of CCCA, understanding the hair in the black population is the first step for spotting problems in routine physical examinations. It may be this lack of familiarity that is contributing to underdiagnosis of CCCA.
“Almost 70% of African-American patients feel that physicians do not understand their hair,” Dr. Taylor said. “Let’s begin to change that.”
Cautioning that it is too often assumed that hairstyles and hair care, such as relaxants or hot combs, are the source of hair loss in black women, Dr. Taylor advised not to jump to conclusions. As an example, she described a case where weaves, braids, and other strategies were employed to mask alopecia after CCCA developed, not before.
“CCCA is the most important cause of scarring alopecia in African-American women,” Dr. Taylor said. Reiterating that hair loss can be prevented or modified with treatment, she added that this places the emphasis on first obtaining an accurate diagnosis.
Dr. Taylor has a financial relationship with Biersdorf; Dr. Woolery-Lloyd has financial relationships with Allergan, Galderma, Ortho Diagnostics, Pfizer, and Somabella Laboratories.
NEW YORK – For unclear reasons, the prevalence of , creating an urgent need for early diagnosis and treatment, according to an expert who described this phenomenon at the Skin of Color Update 2019.
“CCCA has reached epidemic proportions,” contended Susan Taylor, MD, director of diversity, department of dermatology, Penn Medicine, Philadelphia.
Published data place prevalence rates of CCCA somewhere in the range of 3% to 6% among black women, but Dr. Taylor reported that she believes the condition is underdiagnosed. “I am seeing far more patients now than I was 30 years ago,” she maintained.
Others participating in the Skin of Color Update 2019 agreed. Heather Woolery-Lloyd, MD, director of ethnic skin care, department of dermatology and cutaneous surgery, University of Miami, also called the rising incidence of CCCA “an epidemic.” She, like Dr. Taylor, emphasized the critical importance of early diagnosis and treatment.
“I tell patients that if we can prevent hair loss over the next 10 years, this is a treatment success,” Dr. Woolery-Lloyd said. Although hair regrowth can be achieved in a minority of patients with treatments such as minoxidil, “the first goal is to prevent hair loss.”
Upon diagnosis, Dr. Woolery-Lloyd recommends treatment with intralesional triamcinolone and topical steroids immediately, adding other agents, such as oral antibiotics, if needed. Even in cases where CCCA has been identified before hair loss is visible, Dr. Woolery-Lloyd advised immediate therapy. Given that CCCA is a disease of reversible hair loss, she said, “do not take a wait-and-see approach.”
One potential obstacle for early diagnosis of CCCA, shared by other types of alopecia that are common in skin of color, is the failure of many clinicians to employ a standardized diagnosis in this patient population.
“If you do not have tightly coiled hair, it does not mean you cannot understand tightly coiled hair, but you have to learn, and you have to let patients know that you understand and have experience,” said Dr. Taylor, emphasizing the role of reassuring patients so they can be confident in the clinical recommendations.
Part of this reassurance will be derived from “interacting with patients in a culturally competent manner,” Dr. Taylor said. Clinicians must develop comfort and confidence in physically examining the hair and scalp, in asking patients to remove weaves or braids for a thorough inspection, and in avoiding comments that might be misinterpreted. Among these, she advised tactful questions about shampooing to avoid any implication that the clinician is implying inadequate hygiene.
When CCCA is suspected, a “biopsy is important” even in circumstances when the diagnosis seems straightforward. For one reason, a substantial proportion of patients may have a concomitant diagnosis. Dr. Taylor cited data from one study in which nearly 20% of CCCA patients also had traction alopecia and more than 10% had androgenic alopecia. Other coexisting problems identified on biopsy, including infection or seborrheic dermatitis, can help clinicians tailor a more effective intervention.
The initial signs of CCCA are typically hair breakage in the vertex of the scalp, which then expands in a central centrifugal pattern, according to Dr. Taylor. Although not all patients have signs of inflammation, such as itching and pustules, inhibition of inflammation represents the first line of therapy.
Relative to hair in the white population, the hair of black individuals grows more slowly and is more fragile, with greater amounts of breakage, said Dr. Taylor, citing published studies that support these differences. To improve early diagnosis of CCCA, understanding the hair in the black population is the first step for spotting problems in routine physical examinations. It may be this lack of familiarity that is contributing to underdiagnosis of CCCA.
“Almost 70% of African-American patients feel that physicians do not understand their hair,” Dr. Taylor said. “Let’s begin to change that.”
Cautioning that it is too often assumed that hairstyles and hair care, such as relaxants or hot combs, are the source of hair loss in black women, Dr. Taylor advised not to jump to conclusions. As an example, she described a case where weaves, braids, and other strategies were employed to mask alopecia after CCCA developed, not before.
“CCCA is the most important cause of scarring alopecia in African-American women,” Dr. Taylor said. Reiterating that hair loss can be prevented or modified with treatment, she added that this places the emphasis on first obtaining an accurate diagnosis.
Dr. Taylor has a financial relationship with Biersdorf; Dr. Woolery-Lloyd has financial relationships with Allergan, Galderma, Ortho Diagnostics, Pfizer, and Somabella Laboratories.
NEW YORK – For unclear reasons, the prevalence of , creating an urgent need for early diagnosis and treatment, according to an expert who described this phenomenon at the Skin of Color Update 2019.
“CCCA has reached epidemic proportions,” contended Susan Taylor, MD, director of diversity, department of dermatology, Penn Medicine, Philadelphia.
Published data place prevalence rates of CCCA somewhere in the range of 3% to 6% among black women, but Dr. Taylor reported that she believes the condition is underdiagnosed. “I am seeing far more patients now than I was 30 years ago,” she maintained.
Others participating in the Skin of Color Update 2019 agreed. Heather Woolery-Lloyd, MD, director of ethnic skin care, department of dermatology and cutaneous surgery, University of Miami, also called the rising incidence of CCCA “an epidemic.” She, like Dr. Taylor, emphasized the critical importance of early diagnosis and treatment.
“I tell patients that if we can prevent hair loss over the next 10 years, this is a treatment success,” Dr. Woolery-Lloyd said. Although hair regrowth can be achieved in a minority of patients with treatments such as minoxidil, “the first goal is to prevent hair loss.”
Upon diagnosis, Dr. Woolery-Lloyd recommends treatment with intralesional triamcinolone and topical steroids immediately, adding other agents, such as oral antibiotics, if needed. Even in cases where CCCA has been identified before hair loss is visible, Dr. Woolery-Lloyd advised immediate therapy. Given that CCCA is a disease of reversible hair loss, she said, “do not take a wait-and-see approach.”
One potential obstacle for early diagnosis of CCCA, shared by other types of alopecia that are common in skin of color, is the failure of many clinicians to employ a standardized diagnosis in this patient population.
“If you do not have tightly coiled hair, it does not mean you cannot understand tightly coiled hair, but you have to learn, and you have to let patients know that you understand and have experience,” said Dr. Taylor, emphasizing the role of reassuring patients so they can be confident in the clinical recommendations.
Part of this reassurance will be derived from “interacting with patients in a culturally competent manner,” Dr. Taylor said. Clinicians must develop comfort and confidence in physically examining the hair and scalp, in asking patients to remove weaves or braids for a thorough inspection, and in avoiding comments that might be misinterpreted. Among these, she advised tactful questions about shampooing to avoid any implication that the clinician is implying inadequate hygiene.
When CCCA is suspected, a “biopsy is important” even in circumstances when the diagnosis seems straightforward. For one reason, a substantial proportion of patients may have a concomitant diagnosis. Dr. Taylor cited data from one study in which nearly 20% of CCCA patients also had traction alopecia and more than 10% had androgenic alopecia. Other coexisting problems identified on biopsy, including infection or seborrheic dermatitis, can help clinicians tailor a more effective intervention.
The initial signs of CCCA are typically hair breakage in the vertex of the scalp, which then expands in a central centrifugal pattern, according to Dr. Taylor. Although not all patients have signs of inflammation, such as itching and pustules, inhibition of inflammation represents the first line of therapy.
Relative to hair in the white population, the hair of black individuals grows more slowly and is more fragile, with greater amounts of breakage, said Dr. Taylor, citing published studies that support these differences. To improve early diagnosis of CCCA, understanding the hair in the black population is the first step for spotting problems in routine physical examinations. It may be this lack of familiarity that is contributing to underdiagnosis of CCCA.
“Almost 70% of African-American patients feel that physicians do not understand their hair,” Dr. Taylor said. “Let’s begin to change that.”
Cautioning that it is too often assumed that hairstyles and hair care, such as relaxants or hot combs, are the source of hair loss in black women, Dr. Taylor advised not to jump to conclusions. As an example, she described a case where weaves, braids, and other strategies were employed to mask alopecia after CCCA developed, not before.
“CCCA is the most important cause of scarring alopecia in African-American women,” Dr. Taylor said. Reiterating that hair loss can be prevented or modified with treatment, she added that this places the emphasis on first obtaining an accurate diagnosis.
Dr. Taylor has a financial relationship with Biersdorf; Dr. Woolery-Lloyd has financial relationships with Allergan, Galderma, Ortho Diagnostics, Pfizer, and Somabella Laboratories.
REPORTING FROM SOC 2019
The Role of Vitamins and Supplements on Skin Appearance
As the largest and most exposed organ in the body, the skin experiences trauma from both extrinsic and intrinsic aging factors, resulting in loss of elasticity, increased laxity, wrinkling, and rough-textured appearance.1 Chronologically aged skin appears dry, thin, and finely wrinkled; photoaged skin appears leathery with coarse wrinkles and uneven pigmentation.2 In recent years, numerous systemic nutrients have been proposed to improve skin appearance. This article reviews the efficacy of these vitamins and supplements.
Carotenoids
Carotenoids are a group of lipophilic molecules derived from vitamin A.3,4 Ingestion of carotenoids may play a role in photoprotection against UV radiation (UVR) by acting as acceptors of reactive oxygen species.4-6 Stahl et al7 investigated lycopene’s usefulness in protection against UVR-induced erythema. Over 10 weeks, 9 volunteers received 40 g of tomato paste containing 16 mg daily of lycopene while 10 controls received placebo. A solar simulator was used to induce erythema of the skin at weeks 0, 4, and 10. At week 10, erythema formation was 40% lower in the lycopene group compared to controls (P=.02).7
In another study assessing the photoprotective effects of a novel nutritional and phytonutrient blend of carotenoids, 36 women with Fitzpatrick skin types I and II were treated for 8 weeks.8 Presupplementation, UVR-induced erythema, and skin carotenoid concentrations were determined along with facial skin attributes and characteristics. Results showed protection against UVR-induced skin damage, with reductions in erythema at 3 minimal erythema doses (MEDs)(P=.01). Additionally, significant improvements were noted in facial skin elasticity, radiance, and overall appearance (all P<.05).8
In 2013, Meinke et al9 conducted an 8-week, double-blind, placebo-controlled study on 24 volunteers whose diets were supplemented with moderate amounts of carotenoids, including lutein, beta-carotene, and lycopene. Utilizing novel techniques to measure the skin’s ability to scavenge free radicals, they discovered that dietary carotenoids provided notable protection against stress-induced radical formation and increased baseline radical scavenging activity of the skin by 34%. The authors concluded that dietary supplementation could avoid premature skin aging.9
Vitamins C and E
Vitamin C is an essential vitamin that must be obtained through dietary sources.10 It functions as a free radical scavenger and is a necessary cofactor for the synthesis and stabilization of collagen.
A study evaluated the effect of UVR-induced oxidative stress and the association with vitamin C supplementation among 20 white patients with Fitzpatrick skin types II or III.11 The volunteers were treated with UVR on two 1-cm sites on the buttock. Six punch biopsies of these sites and 2 control biopsies from nonexposed skin were taken. Volunteers took vitamin C supplements (500 mg) for 8 weeks, and the exposure and biopsy were repeated. Researchers concluded that supplementation with vitamin C had no effect on the MED, with identical concentrations at baseline and after 8 weeks of supplementation. Additionally, there was no evidence that vitamin C affects UVR-induced oxidative stress.11
In 2007, Cosgrove et al12 conducted a study to assess the associations between nutrient intake and skin aging in more than 4000 women aged 40 to 74 years. Higher dietary vitamin C intakes were associated with a significantly lower likelihood of senile xerosis and wrinkled appearance (P<.009).12
Vitamin E is a lipid-soluble, membrane-bound vitamin, and its most active form is α-tocopherol.11,13 Vitamin E functions as an antioxidant and protects cellular membranes from lipid peroxidation by free radicals.13-15 Once oxidized, vitamin E can be regenerated to its reduced form by vitamin C.11 Their synergistic effects on skin protection have been studied extensively. A double-blind, placebo-controlled study of 10 patients compared 2 g of vitamin C combined with 1000 IU of vitamin E vs placebo.16 The patients’ skin reaction before and after 8 days of treatment were assessed by determination of MED and the cutaneous blood flow of skin irradiated with UV light. Results showed that the median MED of those taking vitamins increased from 80 to 96.5 mJ/cm2 (P<.01) and decreased for the placebo group. Investigators concluded that the combination of vitamins C and E reduces the sunburn reaction and leads to a reduction in the sequelae of UV-induced skin damage.16 A prospective, randomized, placebo-controlled study by Fuchs and Kern17 replicated these findings, also concluding that combinations of vitamins C and E provide improved photoprotective effects than either vitamin alone.
Vitamin D
Vitamin D is a fat-soluble vitamin obtained through dietary intake and exposure to UV light.3,18,19 Precursors of vitamin D require interaction with UV light for conversion into active forms. The highest concentrations of 7-dehydrocholesterol are found in keratinocytes in the basal cell and spinous cell layers of the skin where they are protected from UV light by melanin. As such, individuals with higher melanin content in their skin require more exposure to UV light to produce the same levels of vitamin D as those with less melanin,20 leading to a high rate of vitamin D deficiency in dark-skinned individuals. Because of their prodifferentiating and antiproliferative effects, vitamin D analogs have been very effective in the treatment of psoriasis.20,21 Vitamin D deficiency also has been implicated in the pathogenesis of vitiligo. A systematic review and meta-analysis conducted in 2016 found that a significant relationship existed between low 25-hydroxyvitamin D levels and vitiligo (P<.01), but no causal relationship could be established.22
A 2017 double-blind, placebo-controlled study performed by Scott et al23 aimed to elucidate the relationship between vitamin D concentrations and sunburn. Twenty adults received either placebo or high-dose vitamin D3 (200,000 IU) 1 hour after experimental sunburn induced by an erythemogenic dose of UVR. Investigators measured participants’ concentrations of the proinflammatory mediators tumor necrosis factor α and nitric oxide synthase via skin biopsy 48 hours later. Patients in the experimental group were found to have significantly reduced expression of both tumor necrosis factor α (P=.04) and nitric oxide synthase (P=.02). Additionally, participants with significantly higher vitamin D3 levels following supplementation (P=.007) demonstrated increased skin expression of the anti-inflammatory marker arginase-1 (P=.005) as well as a persistent reduction in skin redness (P=.02). Investigators concluded that vitamin D plays a large role in skin homeostasis and implicated vitamin D’s upregulation of arginase-1 as a potent mechanism of its anti-inflammatory effects.23
Collagen
As humans age, the density of collagen in the dermis decreases, leading to sagging and wrinkling of skin.24 Oral supplementation of collagen has been examined for its dermatologic benefits, primarily increasing the thickness and density of collagen in the dermal layer. In 2014, Proksch et al25 performed a double-blind, placebo-controlled trial in which 69 women were randomized to receive 2.5 or 5 g of collagen peptides or placebo for 8 weeks. Both treatment groups demonstrated improvements in skin elasticity as well as improved skin moisture and decreased skin evaporation; however, changes in the latter 2 qualities failed to reach statistical significance.25
The results of this study were replicated by Asserin et al.26 One hundred six female patients were randomly assigned to receive 10 g of collagen peptides or placebo daily for 8 weeks. The collagen group demonstrated significantly improved skin hydration (P=.003) and increased density of collagen in the dermis (P=.007) relative to placebo.26
In another randomized, double-blind, placebo-controlled study, 71 women consumed a 20-mL beverage containing either 3000 mg of collagen peptides or placebo for 12 weeks.27 Participants in the treatment group demonstrated significant decreases in periorbital wrinkles (P<.05) and enhanced facial skin moisture (P<.001) and elasticity (P<.001) after 12 weeks. Researchers concluded that oral supplementation with collagen peptides holds promise as a natural supplement to provide cutaneous antiaging properties.27
Ceramides
Ceramides are lipids composed of a sphingoid base conjugated to a fatty acid and serve as the main component of the stratum corneum of the skin. Ceramides are crucial for the maintenance of skin barrier integrity and for preventing transepidermal water loss.28 In a 3-month study of 51 women with dry skin, Guillou et al29 showed that a ceramide wheat extract capsule significantly increased corneometry measurements of skin hydration on the arms (P<.001) and the legs (P=.012) compared to placebo.
Mixed Supplements
The discovery that nutritional contents can affect skin appearance has energized the development of combination supplements containing multiple vitamins and micronutrients. Imedeen is a biomarine complex and antioxidant supplement with several different formulations, including Prime Renewal, Time Perfection, and Derma One (Pfizer Inc). The ingredients include a combination of a biomarine complex (blend of fish proteins and polysaccharides), lycopene, grape seed extract, vitamin C, vitamin E, and zinc. Several trials have been conducted to assess the efficacy of the supplements on improving the appearance of photodamaged and aged skin (Table).
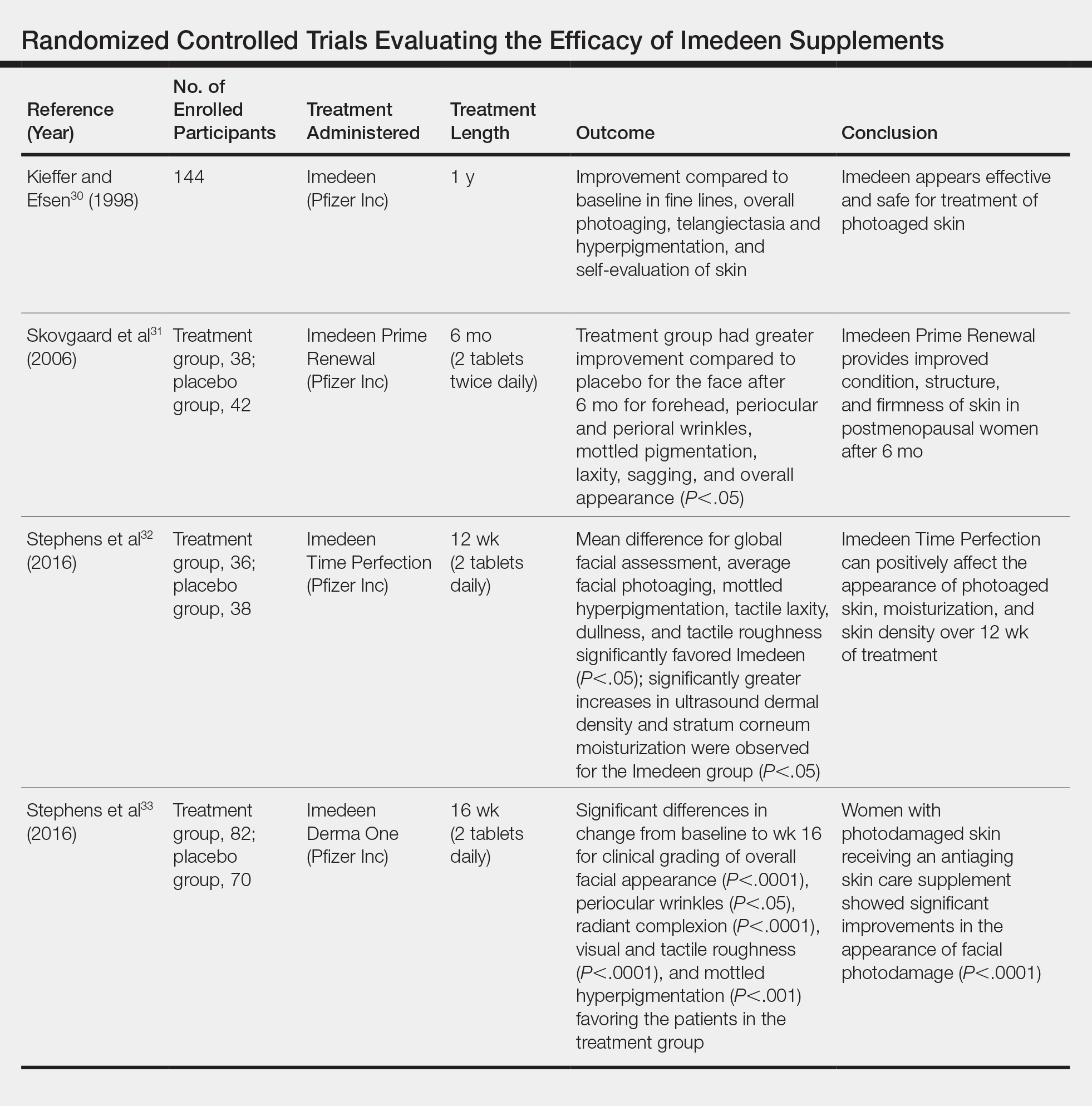
A placebo-controlled, randomized study of 144 participants conducted by Kieffer and Efsen30 assessed the efficacy of Imedeen supplements over 12 months. The trial included a 3-month placebo-controlled study and 9-month uncontrolled continuation. Imedeen’s efficacy was measured using clinical evaluation, transepidermal water loss, self-evaluation, and photograph evaluation. After 1 year of treatment, improvement occurred in photograph evaluation of fine lines, overall photoaging, telangiectasia and hyperpigmentation, and self-evaluation of skin condition.30 Additional double-blind, placebo-controlled, randomized studies assessing the efficacy of Imedeen have shown increased dermal and epidermal thickness, improvement of stratum corneum moisturization, and improved overall facial complexion.31-33
Several combined supplements containing collagen peptide as the main ingredient have been created for use in skin care. Collagen is found in the extracellular matrix of the dermis and is responsible for the resiliency and strength of skin.34,35 Damage to the dermis can occur with prolonged UV light exposure and is seen histologically as disorganized collagen fibrils and grossly as wrinkles and photoaged skin.35,36
A study assessed the effect of BioCell Collagen (BioCell Technology, LLC), a supplement containing type II collagen, on skin aging.37 Twenty-six women underwent baseline visual assessments of their skin before taking 2 tablets of the supplement daily. Twelve weeks of supplementation led to significant reduction in global lines and wrinkles (13.2%; P=.028) as well as skin dryness and scaling (76%; P=.002). Assessment of collagen content at 6 weeks revealed a significant increase from baseline (6.3%; P=.002), though the difference after 12 weeks was not significant (3.5%; P=.134). The authors concluded that although preliminary data suggested that BioCell Collagen may reduce visible signs of aging, a controlled study was necessary to verify this finding.37
A single-blind, case-controlled study assessed a similar supplement, Celergen, that contained marine collagen peptides.38 Forty-one adults took 2 capsules each day for 60 days. Assessment of their skin physiology was conducted at the enrollment visit, 2 months later, and after the treatment period ended. Skin elasticity, transepidermal water loss, epidermal and dermal thickness, and density were measured. Investigators found that Celergen administration significantly enhanced skin elasticity and sebum production (P<.0001) but did not influence cutaneous moisture. The dermal thickness and homogenous distribution of collagen fibers were enhanced in 11 patients while properties of the epidermis remained unchanged. The study determined that supplementation remarkably improved skin elasticity, sebum production, and dermal ultrasonic markers.38
A double-blind, randomized, placebo-controlled study assessed a collagen- and antioxidant-containing supplement, Gold Collagen Forte, on skin properties.39 The treatment and placebo groups each consisted of 60 patients who consumed 1 bottle (50 mL) of the product each day for 90 days. Patients completed a self-assessment of their skin regarding photoaging, focusing on the crow’s-feet area and nasolabial folds, while skin elasticity was assessed with the SkinLab USB elasticity module. Results showed a significant increase in skin elasticity (+7.5%; P≤.001). Self-assessment results showed improvements in both the treatment and placebo groups, and investigators concluded that Gold Collagen Forte may have photoprotective effects and help improve skin health.39
Safety
Although trials have demonstrated vitamin supplementation to be safe and effective for skin enhancement, it is important to consider potential vitamin toxicities. High doses of vitamin C supplementation have been shown to cause damage via lipid peroxidation.40 In a study assessing if high levels of beta-carotene and vitamin E were associated with a lower risk for lung cancer, data showed that these supplements may actually have harmful effects.40,41 Additionally, consumption of high-dose dietary supplements has been associated with an increased risk for severe medical events, including disability and death among adolescents and young adults.42
Conclusion
Numerous trials have indicated that the use of systemic vitamins can have beneficial effects on the protection and appearance of skin. Photodamage from UV light–induced erythema can be decreased by carotenoids and vitamins C and E. Similarly, supplements that combine multiple nutrients with collagen have been shown to improve the appearance of aging skin by decreasing the prominence of wrinkles. Given the growing number of products and advertisements that exist in the supplement marketplace, it is crucial for clinicians to ground their recommendations to patients in the scientific data of robust studies.
- Zhang S, Duan E. Fighting against skin aging: the way from bench to bedside. Cell Transplant. 2018;27:729-738.
- Rittié L, Fisher GJ. Natural and sun-induced aging of human skin. Cold Spring Harb Perspect Med. 2015;5:a015370.
- Draelos ZD. Nutrition and enhancing youthful-appearing skin. Clin Dermatol. 2010;28:400-408.
- Anunciato TP, da Rocha Filho PA. Carotenoids and polyphenols in nutricosmetics, nutraceuticals, and cosmeceuticals. J Cosmet Dermatol. 2012;11:51-54.
- Stahl W, Heinrich U, Jungmann H, et al. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am J Clin Nutr. 2000;71:795-798.
- Anstey AV. Systemic photoprotection with alpha-tocopherol (vitamin E) and beta-carotene. Clin Exp Dermatol. 2002;27:170-176.
- Stahl W, Heinrich U, Wiseman S, et al. Dietary tomato paste protects against ultraviolet light-induced erythema in humans. J Nutr. 2001;131:1449-1451.
- Wood SM, Mastaloudis AF, Hester SN, et al. Protective effects of a novel nutritional and phytonutrient blend on ultraviolet radiation-induced skin damage and inflammatory response through aging defense mechanisms. J Cosmet Dermatol. 2017;16:491-499.
- Meinke MC, Friedrich A, Tscherch K, et al. Influence of dietary carotenoids on radical scavenging capacity of the skin and skin lipids. Eur J Pharm Biopharm. 2013;84:365-373.
- Manela-Azulay M, Bagatin E. Cosmeceuticals vitamins. Clin Dermatol. 2009;27:469-474.
- McArdle F, Rhodes LE, Parslew R, et al. UVR-induced oxidative stress in human skin in vivo: effects of oral vitamin C supplementation. Free Radic Biol Med. 2002;33:1355-1362.
- Cosgrove MC, Franco OH, Granger SP, et al. Dietary nutrient intakes and skin-aging appearance among middle-aged American women. Am J Clin Nutr. 2007;86:1225-1231.
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667.
- Schagen SK, Zampeli VA, Makrantonaki E, et al. Discovering the link between nutrition and skin aging. Dermatoendocrinol. 2012;4:298-307.
- Chan AC. Partners in defense, vitamin E and vitamin C. Can J Physiol Pharmacol. 1993;71:725-731.
- Eberlein-Konig B, Placzek M, Przybilla B. Protective effect against sunburn of combined systemic ascorbic acid (vitamin C) and d-alpha-tocopherol (vitamin E). J Am Acad Dermatol. 1998;38:45-48.
- Fuchs J, Kern H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: a clinical study using solar simulated radiation. Free Radic Biol Med. 1998;25:1006-1012.
- Shahriari M, Kerr PE, Slade K, et al. Vitamin D and the skin. Clin Dermatol. 2010;28:663-668.
- Soleymani T, Hung T, Soung J. The role of vitamin D in psoriasis: a review. Int J Dermatol. 2015;54:383-392.
- Lehmann B, Querings K, Reichrath J. Vitamin D and skin: new aspects for dermatology. Exp Dermatol. 2004;13(suppl 4):11-15.
- Kannan S, Lim HW. Photoprotection and vitamin D: a review. Photodermatol Photoimmunol Photomed. 2014;30:137-145.
- Upala S, Sanguankeo A. Low 25-hydroxyvitamin D levels are associated with vitiligo: a systematic review and meta-analysis. Photodermatol Photoimmunol Photomed. 2016;32:181-190.
- Scott JF, Das LM, Ahsanuddin S, et al. Oral vitamin D rapidly attenuates inflammation from sunburn: an interventional study. J Invest Dermatol. 2017;137:2078-2086.
- Varani J, Dame MK, Rittie L, et al. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861-1868.
- Proksch E, Segger D, Degwert J, et al. Oral supplementation of specific collagen peptides has beneficial effects on human skin physiology: a double-blind, placebo-controlled study. Skin Pharmacol Physiol. 2014;27:47-55.
- Asserin J, Lati E, Shioya T, et al. The effect of oral collagen peptide supplementation on skin moisture and the dermal collagen network: evidence from an ex vivo model and randomized, placebo-controlled clinical trials. J Cosmet Dermatol. 2015;14:291-301.
- Koizumi S, Inoue N, Shimizu M, et al. Effects of dietary supplementation with fish scales-derived collagen peptides on skin parameters and condition: a randomized, placebo-controlled, double-blind study. Int J Peptide Res Ther. 2018;24:397-402.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19. doi:10.3390/ijms19103059.
- Guillou S, Ghabri S, Jannot C, et al. The moisturizing effect of a wheat extract food supplement on women’s skin: a randomized, double-blind placebo-controlled trial. Int J Cosmet Sci. 2011;33:138-143.
- Kieffer ME, Efsen J. Imedeen in the treatment of photoaged skin: an efficacy and safety trial over 12 months. J Eur Acad Dermatol Venereol. 1998;11:129-136.
- Skovgaard GR, Jensen AS, Sigler ML. Effect of a novel dietary supplement on skin aging in post-menopausal women. Eur J Clin Nutr. 2006;60:1201-1206.
- Stephens TJ, Sigler ML, Herndon JH Jr, et al. A placebo-controlled, double-blind clinical trial to evaluate the efficacy of Imedeen(®) Time Perfection(®) for improving the appearance of photodamaged skin. Clin Cosmet Investig Dermatol. 2016;9:63-70.
- Stephens TJ, Sigler ML, Hino PD, et al. A randomized, double-blind, placebo-controlled clinical trial evaluating an oral anti-aging skin care supplement for treating photodamaged skin. J Clin Aesthet Dermatol. 2016;9:25-32.
- El-Domyati M, Attia S, Saleh F, et al. Intrinsic aging vs. photoaging: a comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp Dermatol. 2002;11:398-405.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Kang MC, Yumnam S, Kim SY. Oral intake of collagen peptide attenuates ultraviolet B irradiation-induced skin dehydration in vivo by regulating hyaluronic acid synthesis. Int J Mol Sci. 2018;19. doi:10.3390/ijms19113551.
- Schwartz SR, Park J. Ingestion of BioCell Collagen(®), a novel hydrolyzed chicken sternal cartilage extract; enhanced blood microcirculation and reduced facial aging signs. Clin Interv Aging. 2012;7:267-273.
- De Luca C, Mikhal’chik EV, Suprun MV, et al. Skin antiageing and systemic redox effects of supplementation with marine collagen peptides and plant-derived antioxidants: a single-blind case-control clinical study. Oxid Med Cell Longev. 2016;2016:4389410.
- Genovese L, Corbo A, Sibilla S. An insight into the changes in skin texture and properties following dietary intervention with a nutricosmeceutical containing a blend of collagen bioactive peptides and antioxidants. Skin Pharmacol Physiol. 2017;30:146-158.
- Hamishehkar H, Ranjdoost F, Asgharian P, et al. Vitamins, are they safe? Adv Pharm Bull. 2016;6:467-477.
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029-1035.
- Or F, Yongjoo K, Simms J, et al. Taking stock of dietary supplements’ harmful effects on children, adolescents, and young adults [published online June 3, 2019]. J Adolesc Health. S1054-139X(19)30163-6. doi:10.1016/j.jadohealth.2019.03.005.
As the largest and most exposed organ in the body, the skin experiences trauma from both extrinsic and intrinsic aging factors, resulting in loss of elasticity, increased laxity, wrinkling, and rough-textured appearance.1 Chronologically aged skin appears dry, thin, and finely wrinkled; photoaged skin appears leathery with coarse wrinkles and uneven pigmentation.2 In recent years, numerous systemic nutrients have been proposed to improve skin appearance. This article reviews the efficacy of these vitamins and supplements.
Carotenoids
Carotenoids are a group of lipophilic molecules derived from vitamin A.3,4 Ingestion of carotenoids may play a role in photoprotection against UV radiation (UVR) by acting as acceptors of reactive oxygen species.4-6 Stahl et al7 investigated lycopene’s usefulness in protection against UVR-induced erythema. Over 10 weeks, 9 volunteers received 40 g of tomato paste containing 16 mg daily of lycopene while 10 controls received placebo. A solar simulator was used to induce erythema of the skin at weeks 0, 4, and 10. At week 10, erythema formation was 40% lower in the lycopene group compared to controls (P=.02).7
In another study assessing the photoprotective effects of a novel nutritional and phytonutrient blend of carotenoids, 36 women with Fitzpatrick skin types I and II were treated for 8 weeks.8 Presupplementation, UVR-induced erythema, and skin carotenoid concentrations were determined along with facial skin attributes and characteristics. Results showed protection against UVR-induced skin damage, with reductions in erythema at 3 minimal erythema doses (MEDs)(P=.01). Additionally, significant improvements were noted in facial skin elasticity, radiance, and overall appearance (all P<.05).8
In 2013, Meinke et al9 conducted an 8-week, double-blind, placebo-controlled study on 24 volunteers whose diets were supplemented with moderate amounts of carotenoids, including lutein, beta-carotene, and lycopene. Utilizing novel techniques to measure the skin’s ability to scavenge free radicals, they discovered that dietary carotenoids provided notable protection against stress-induced radical formation and increased baseline radical scavenging activity of the skin by 34%. The authors concluded that dietary supplementation could avoid premature skin aging.9
Vitamins C and E
Vitamin C is an essential vitamin that must be obtained through dietary sources.10 It functions as a free radical scavenger and is a necessary cofactor for the synthesis and stabilization of collagen.
A study evaluated the effect of UVR-induced oxidative stress and the association with vitamin C supplementation among 20 white patients with Fitzpatrick skin types II or III.11 The volunteers were treated with UVR on two 1-cm sites on the buttock. Six punch biopsies of these sites and 2 control biopsies from nonexposed skin were taken. Volunteers took vitamin C supplements (500 mg) for 8 weeks, and the exposure and biopsy were repeated. Researchers concluded that supplementation with vitamin C had no effect on the MED, with identical concentrations at baseline and after 8 weeks of supplementation. Additionally, there was no evidence that vitamin C affects UVR-induced oxidative stress.11
In 2007, Cosgrove et al12 conducted a study to assess the associations between nutrient intake and skin aging in more than 4000 women aged 40 to 74 years. Higher dietary vitamin C intakes were associated with a significantly lower likelihood of senile xerosis and wrinkled appearance (P<.009).12
Vitamin E is a lipid-soluble, membrane-bound vitamin, and its most active form is α-tocopherol.11,13 Vitamin E functions as an antioxidant and protects cellular membranes from lipid peroxidation by free radicals.13-15 Once oxidized, vitamin E can be regenerated to its reduced form by vitamin C.11 Their synergistic effects on skin protection have been studied extensively. A double-blind, placebo-controlled study of 10 patients compared 2 g of vitamin C combined with 1000 IU of vitamin E vs placebo.16 The patients’ skin reaction before and after 8 days of treatment were assessed by determination of MED and the cutaneous blood flow of skin irradiated with UV light. Results showed that the median MED of those taking vitamins increased from 80 to 96.5 mJ/cm2 (P<.01) and decreased for the placebo group. Investigators concluded that the combination of vitamins C and E reduces the sunburn reaction and leads to a reduction in the sequelae of UV-induced skin damage.16 A prospective, randomized, placebo-controlled study by Fuchs and Kern17 replicated these findings, also concluding that combinations of vitamins C and E provide improved photoprotective effects than either vitamin alone.
Vitamin D
Vitamin D is a fat-soluble vitamin obtained through dietary intake and exposure to UV light.3,18,19 Precursors of vitamin D require interaction with UV light for conversion into active forms. The highest concentrations of 7-dehydrocholesterol are found in keratinocytes in the basal cell and spinous cell layers of the skin where they are protected from UV light by melanin. As such, individuals with higher melanin content in their skin require more exposure to UV light to produce the same levels of vitamin D as those with less melanin,20 leading to a high rate of vitamin D deficiency in dark-skinned individuals. Because of their prodifferentiating and antiproliferative effects, vitamin D analogs have been very effective in the treatment of psoriasis.20,21 Vitamin D deficiency also has been implicated in the pathogenesis of vitiligo. A systematic review and meta-analysis conducted in 2016 found that a significant relationship existed between low 25-hydroxyvitamin D levels and vitiligo (P<.01), but no causal relationship could be established.22
A 2017 double-blind, placebo-controlled study performed by Scott et al23 aimed to elucidate the relationship between vitamin D concentrations and sunburn. Twenty adults received either placebo or high-dose vitamin D3 (200,000 IU) 1 hour after experimental sunburn induced by an erythemogenic dose of UVR. Investigators measured participants’ concentrations of the proinflammatory mediators tumor necrosis factor α and nitric oxide synthase via skin biopsy 48 hours later. Patients in the experimental group were found to have significantly reduced expression of both tumor necrosis factor α (P=.04) and nitric oxide synthase (P=.02). Additionally, participants with significantly higher vitamin D3 levels following supplementation (P=.007) demonstrated increased skin expression of the anti-inflammatory marker arginase-1 (P=.005) as well as a persistent reduction in skin redness (P=.02). Investigators concluded that vitamin D plays a large role in skin homeostasis and implicated vitamin D’s upregulation of arginase-1 as a potent mechanism of its anti-inflammatory effects.23
Collagen
As humans age, the density of collagen in the dermis decreases, leading to sagging and wrinkling of skin.24 Oral supplementation of collagen has been examined for its dermatologic benefits, primarily increasing the thickness and density of collagen in the dermal layer. In 2014, Proksch et al25 performed a double-blind, placebo-controlled trial in which 69 women were randomized to receive 2.5 or 5 g of collagen peptides or placebo for 8 weeks. Both treatment groups demonstrated improvements in skin elasticity as well as improved skin moisture and decreased skin evaporation; however, changes in the latter 2 qualities failed to reach statistical significance.25
The results of this study were replicated by Asserin et al.26 One hundred six female patients were randomly assigned to receive 10 g of collagen peptides or placebo daily for 8 weeks. The collagen group demonstrated significantly improved skin hydration (P=.003) and increased density of collagen in the dermis (P=.007) relative to placebo.26
In another randomized, double-blind, placebo-controlled study, 71 women consumed a 20-mL beverage containing either 3000 mg of collagen peptides or placebo for 12 weeks.27 Participants in the treatment group demonstrated significant decreases in periorbital wrinkles (P<.05) and enhanced facial skin moisture (P<.001) and elasticity (P<.001) after 12 weeks. Researchers concluded that oral supplementation with collagen peptides holds promise as a natural supplement to provide cutaneous antiaging properties.27
Ceramides
Ceramides are lipids composed of a sphingoid base conjugated to a fatty acid and serve as the main component of the stratum corneum of the skin. Ceramides are crucial for the maintenance of skin barrier integrity and for preventing transepidermal water loss.28 In a 3-month study of 51 women with dry skin, Guillou et al29 showed that a ceramide wheat extract capsule significantly increased corneometry measurements of skin hydration on the arms (P<.001) and the legs (P=.012) compared to placebo.
Mixed Supplements
The discovery that nutritional contents can affect skin appearance has energized the development of combination supplements containing multiple vitamins and micronutrients. Imedeen is a biomarine complex and antioxidant supplement with several different formulations, including Prime Renewal, Time Perfection, and Derma One (Pfizer Inc). The ingredients include a combination of a biomarine complex (blend of fish proteins and polysaccharides), lycopene, grape seed extract, vitamin C, vitamin E, and zinc. Several trials have been conducted to assess the efficacy of the supplements on improving the appearance of photodamaged and aged skin (Table).
A placebo-controlled, randomized study of 144 participants conducted by Kieffer and Efsen30 assessed the efficacy of Imedeen supplements over 12 months. The trial included a 3-month placebo-controlled study and 9-month uncontrolled continuation. Imedeen’s efficacy was measured using clinical evaluation, transepidermal water loss, self-evaluation, and photograph evaluation. After 1 year of treatment, improvement occurred in photograph evaluation of fine lines, overall photoaging, telangiectasia and hyperpigmentation, and self-evaluation of skin condition.30 Additional double-blind, placebo-controlled, randomized studies assessing the efficacy of Imedeen have shown increased dermal and epidermal thickness, improvement of stratum corneum moisturization, and improved overall facial complexion.31-33
Several combined supplements containing collagen peptide as the main ingredient have been created for use in skin care. Collagen is found in the extracellular matrix of the dermis and is responsible for the resiliency and strength of skin.34,35 Damage to the dermis can occur with prolonged UV light exposure and is seen histologically as disorganized collagen fibrils and grossly as wrinkles and photoaged skin.35,36
A study assessed the effect of BioCell Collagen (BioCell Technology, LLC), a supplement containing type II collagen, on skin aging.37 Twenty-six women underwent baseline visual assessments of their skin before taking 2 tablets of the supplement daily. Twelve weeks of supplementation led to significant reduction in global lines and wrinkles (13.2%; P=.028) as well as skin dryness and scaling (76%; P=.002). Assessment of collagen content at 6 weeks revealed a significant increase from baseline (6.3%; P=.002), though the difference after 12 weeks was not significant (3.5%; P=.134). The authors concluded that although preliminary data suggested that BioCell Collagen may reduce visible signs of aging, a controlled study was necessary to verify this finding.37
A single-blind, case-controlled study assessed a similar supplement, Celergen, that contained marine collagen peptides.38 Forty-one adults took 2 capsules each day for 60 days. Assessment of their skin physiology was conducted at the enrollment visit, 2 months later, and after the treatment period ended. Skin elasticity, transepidermal water loss, epidermal and dermal thickness, and density were measured. Investigators found that Celergen administration significantly enhanced skin elasticity and sebum production (P<.0001) but did not influence cutaneous moisture. The dermal thickness and homogenous distribution of collagen fibers were enhanced in 11 patients while properties of the epidermis remained unchanged. The study determined that supplementation remarkably improved skin elasticity, sebum production, and dermal ultrasonic markers.38
A double-blind, randomized, placebo-controlled study assessed a collagen- and antioxidant-containing supplement, Gold Collagen Forte, on skin properties.39 The treatment and placebo groups each consisted of 60 patients who consumed 1 bottle (50 mL) of the product each day for 90 days. Patients completed a self-assessment of their skin regarding photoaging, focusing on the crow’s-feet area and nasolabial folds, while skin elasticity was assessed with the SkinLab USB elasticity module. Results showed a significant increase in skin elasticity (+7.5%; P≤.001). Self-assessment results showed improvements in both the treatment and placebo groups, and investigators concluded that Gold Collagen Forte may have photoprotective effects and help improve skin health.39
Safety
Although trials have demonstrated vitamin supplementation to be safe and effective for skin enhancement, it is important to consider potential vitamin toxicities. High doses of vitamin C supplementation have been shown to cause damage via lipid peroxidation.40 In a study assessing if high levels of beta-carotene and vitamin E were associated with a lower risk for lung cancer, data showed that these supplements may actually have harmful effects.40,41 Additionally, consumption of high-dose dietary supplements has been associated with an increased risk for severe medical events, including disability and death among adolescents and young adults.42
Conclusion
Numerous trials have indicated that the use of systemic vitamins can have beneficial effects on the protection and appearance of skin. Photodamage from UV light–induced erythema can be decreased by carotenoids and vitamins C and E. Similarly, supplements that combine multiple nutrients with collagen have been shown to improve the appearance of aging skin by decreasing the prominence of wrinkles. Given the growing number of products and advertisements that exist in the supplement marketplace, it is crucial for clinicians to ground their recommendations to patients in the scientific data of robust studies.
As the largest and most exposed organ in the body, the skin experiences trauma from both extrinsic and intrinsic aging factors, resulting in loss of elasticity, increased laxity, wrinkling, and rough-textured appearance.1 Chronologically aged skin appears dry, thin, and finely wrinkled; photoaged skin appears leathery with coarse wrinkles and uneven pigmentation.2 In recent years, numerous systemic nutrients have been proposed to improve skin appearance. This article reviews the efficacy of these vitamins and supplements.
Carotenoids
Carotenoids are a group of lipophilic molecules derived from vitamin A.3,4 Ingestion of carotenoids may play a role in photoprotection against UV radiation (UVR) by acting as acceptors of reactive oxygen species.4-6 Stahl et al7 investigated lycopene’s usefulness in protection against UVR-induced erythema. Over 10 weeks, 9 volunteers received 40 g of tomato paste containing 16 mg daily of lycopene while 10 controls received placebo. A solar simulator was used to induce erythema of the skin at weeks 0, 4, and 10. At week 10, erythema formation was 40% lower in the lycopene group compared to controls (P=.02).7
In another study assessing the photoprotective effects of a novel nutritional and phytonutrient blend of carotenoids, 36 women with Fitzpatrick skin types I and II were treated for 8 weeks.8 Presupplementation, UVR-induced erythema, and skin carotenoid concentrations were determined along with facial skin attributes and characteristics. Results showed protection against UVR-induced skin damage, with reductions in erythema at 3 minimal erythema doses (MEDs)(P=.01). Additionally, significant improvements were noted in facial skin elasticity, radiance, and overall appearance (all P<.05).8
In 2013, Meinke et al9 conducted an 8-week, double-blind, placebo-controlled study on 24 volunteers whose diets were supplemented with moderate amounts of carotenoids, including lutein, beta-carotene, and lycopene. Utilizing novel techniques to measure the skin’s ability to scavenge free radicals, they discovered that dietary carotenoids provided notable protection against stress-induced radical formation and increased baseline radical scavenging activity of the skin by 34%. The authors concluded that dietary supplementation could avoid premature skin aging.9
Vitamins C and E
Vitamin C is an essential vitamin that must be obtained through dietary sources.10 It functions as a free radical scavenger and is a necessary cofactor for the synthesis and stabilization of collagen.
A study evaluated the effect of UVR-induced oxidative stress and the association with vitamin C supplementation among 20 white patients with Fitzpatrick skin types II or III.11 The volunteers were treated with UVR on two 1-cm sites on the buttock. Six punch biopsies of these sites and 2 control biopsies from nonexposed skin were taken. Volunteers took vitamin C supplements (500 mg) for 8 weeks, and the exposure and biopsy were repeated. Researchers concluded that supplementation with vitamin C had no effect on the MED, with identical concentrations at baseline and after 8 weeks of supplementation. Additionally, there was no evidence that vitamin C affects UVR-induced oxidative stress.11
In 2007, Cosgrove et al12 conducted a study to assess the associations between nutrient intake and skin aging in more than 4000 women aged 40 to 74 years. Higher dietary vitamin C intakes were associated with a significantly lower likelihood of senile xerosis and wrinkled appearance (P<.009).12
Vitamin E is a lipid-soluble, membrane-bound vitamin, and its most active form is α-tocopherol.11,13 Vitamin E functions as an antioxidant and protects cellular membranes from lipid peroxidation by free radicals.13-15 Once oxidized, vitamin E can be regenerated to its reduced form by vitamin C.11 Their synergistic effects on skin protection have been studied extensively. A double-blind, placebo-controlled study of 10 patients compared 2 g of vitamin C combined with 1000 IU of vitamin E vs placebo.16 The patients’ skin reaction before and after 8 days of treatment were assessed by determination of MED and the cutaneous blood flow of skin irradiated with UV light. Results showed that the median MED of those taking vitamins increased from 80 to 96.5 mJ/cm2 (P<.01) and decreased for the placebo group. Investigators concluded that the combination of vitamins C and E reduces the sunburn reaction and leads to a reduction in the sequelae of UV-induced skin damage.16 A prospective, randomized, placebo-controlled study by Fuchs and Kern17 replicated these findings, also concluding that combinations of vitamins C and E provide improved photoprotective effects than either vitamin alone.
Vitamin D
Vitamin D is a fat-soluble vitamin obtained through dietary intake and exposure to UV light.3,18,19 Precursors of vitamin D require interaction with UV light for conversion into active forms. The highest concentrations of 7-dehydrocholesterol are found in keratinocytes in the basal cell and spinous cell layers of the skin where they are protected from UV light by melanin. As such, individuals with higher melanin content in their skin require more exposure to UV light to produce the same levels of vitamin D as those with less melanin,20 leading to a high rate of vitamin D deficiency in dark-skinned individuals. Because of their prodifferentiating and antiproliferative effects, vitamin D analogs have been very effective in the treatment of psoriasis.20,21 Vitamin D deficiency also has been implicated in the pathogenesis of vitiligo. A systematic review and meta-analysis conducted in 2016 found that a significant relationship existed between low 25-hydroxyvitamin D levels and vitiligo (P<.01), but no causal relationship could be established.22
A 2017 double-blind, placebo-controlled study performed by Scott et al23 aimed to elucidate the relationship between vitamin D concentrations and sunburn. Twenty adults received either placebo or high-dose vitamin D3 (200,000 IU) 1 hour after experimental sunburn induced by an erythemogenic dose of UVR. Investigators measured participants’ concentrations of the proinflammatory mediators tumor necrosis factor α and nitric oxide synthase via skin biopsy 48 hours later. Patients in the experimental group were found to have significantly reduced expression of both tumor necrosis factor α (P=.04) and nitric oxide synthase (P=.02). Additionally, participants with significantly higher vitamin D3 levels following supplementation (P=.007) demonstrated increased skin expression of the anti-inflammatory marker arginase-1 (P=.005) as well as a persistent reduction in skin redness (P=.02). Investigators concluded that vitamin D plays a large role in skin homeostasis and implicated vitamin D’s upregulation of arginase-1 as a potent mechanism of its anti-inflammatory effects.23
Collagen
As humans age, the density of collagen in the dermis decreases, leading to sagging and wrinkling of skin.24 Oral supplementation of collagen has been examined for its dermatologic benefits, primarily increasing the thickness and density of collagen in the dermal layer. In 2014, Proksch et al25 performed a double-blind, placebo-controlled trial in which 69 women were randomized to receive 2.5 or 5 g of collagen peptides or placebo for 8 weeks. Both treatment groups demonstrated improvements in skin elasticity as well as improved skin moisture and decreased skin evaporation; however, changes in the latter 2 qualities failed to reach statistical significance.25
The results of this study were replicated by Asserin et al.26 One hundred six female patients were randomly assigned to receive 10 g of collagen peptides or placebo daily for 8 weeks. The collagen group demonstrated significantly improved skin hydration (P=.003) and increased density of collagen in the dermis (P=.007) relative to placebo.26
In another randomized, double-blind, placebo-controlled study, 71 women consumed a 20-mL beverage containing either 3000 mg of collagen peptides or placebo for 12 weeks.27 Participants in the treatment group demonstrated significant decreases in periorbital wrinkles (P<.05) and enhanced facial skin moisture (P<.001) and elasticity (P<.001) after 12 weeks. Researchers concluded that oral supplementation with collagen peptides holds promise as a natural supplement to provide cutaneous antiaging properties.27
Ceramides
Ceramides are lipids composed of a sphingoid base conjugated to a fatty acid and serve as the main component of the stratum corneum of the skin. Ceramides are crucial for the maintenance of skin barrier integrity and for preventing transepidermal water loss.28 In a 3-month study of 51 women with dry skin, Guillou et al29 showed that a ceramide wheat extract capsule significantly increased corneometry measurements of skin hydration on the arms (P<.001) and the legs (P=.012) compared to placebo.
Mixed Supplements
The discovery that nutritional contents can affect skin appearance has energized the development of combination supplements containing multiple vitamins and micronutrients. Imedeen is a biomarine complex and antioxidant supplement with several different formulations, including Prime Renewal, Time Perfection, and Derma One (Pfizer Inc). The ingredients include a combination of a biomarine complex (blend of fish proteins and polysaccharides), lycopene, grape seed extract, vitamin C, vitamin E, and zinc. Several trials have been conducted to assess the efficacy of the supplements on improving the appearance of photodamaged and aged skin (Table).
A placebo-controlled, randomized study of 144 participants conducted by Kieffer and Efsen30 assessed the efficacy of Imedeen supplements over 12 months. The trial included a 3-month placebo-controlled study and 9-month uncontrolled continuation. Imedeen’s efficacy was measured using clinical evaluation, transepidermal water loss, self-evaluation, and photograph evaluation. After 1 year of treatment, improvement occurred in photograph evaluation of fine lines, overall photoaging, telangiectasia and hyperpigmentation, and self-evaluation of skin condition.30 Additional double-blind, placebo-controlled, randomized studies assessing the efficacy of Imedeen have shown increased dermal and epidermal thickness, improvement of stratum corneum moisturization, and improved overall facial complexion.31-33
Several combined supplements containing collagen peptide as the main ingredient have been created for use in skin care. Collagen is found in the extracellular matrix of the dermis and is responsible for the resiliency and strength of skin.34,35 Damage to the dermis can occur with prolonged UV light exposure and is seen histologically as disorganized collagen fibrils and grossly as wrinkles and photoaged skin.35,36
A study assessed the effect of BioCell Collagen (BioCell Technology, LLC), a supplement containing type II collagen, on skin aging.37 Twenty-six women underwent baseline visual assessments of their skin before taking 2 tablets of the supplement daily. Twelve weeks of supplementation led to significant reduction in global lines and wrinkles (13.2%; P=.028) as well as skin dryness and scaling (76%; P=.002). Assessment of collagen content at 6 weeks revealed a significant increase from baseline (6.3%; P=.002), though the difference after 12 weeks was not significant (3.5%; P=.134). The authors concluded that although preliminary data suggested that BioCell Collagen may reduce visible signs of aging, a controlled study was necessary to verify this finding.37
A single-blind, case-controlled study assessed a similar supplement, Celergen, that contained marine collagen peptides.38 Forty-one adults took 2 capsules each day for 60 days. Assessment of their skin physiology was conducted at the enrollment visit, 2 months later, and after the treatment period ended. Skin elasticity, transepidermal water loss, epidermal and dermal thickness, and density were measured. Investigators found that Celergen administration significantly enhanced skin elasticity and sebum production (P<.0001) but did not influence cutaneous moisture. The dermal thickness and homogenous distribution of collagen fibers were enhanced in 11 patients while properties of the epidermis remained unchanged. The study determined that supplementation remarkably improved skin elasticity, sebum production, and dermal ultrasonic markers.38
A double-blind, randomized, placebo-controlled study assessed a collagen- and antioxidant-containing supplement, Gold Collagen Forte, on skin properties.39 The treatment and placebo groups each consisted of 60 patients who consumed 1 bottle (50 mL) of the product each day for 90 days. Patients completed a self-assessment of their skin regarding photoaging, focusing on the crow’s-feet area and nasolabial folds, while skin elasticity was assessed with the SkinLab USB elasticity module. Results showed a significant increase in skin elasticity (+7.5%; P≤.001). Self-assessment results showed improvements in both the treatment and placebo groups, and investigators concluded that Gold Collagen Forte may have photoprotective effects and help improve skin health.39
Safety
Although trials have demonstrated vitamin supplementation to be safe and effective for skin enhancement, it is important to consider potential vitamin toxicities. High doses of vitamin C supplementation have been shown to cause damage via lipid peroxidation.40 In a study assessing if high levels of beta-carotene and vitamin E were associated with a lower risk for lung cancer, data showed that these supplements may actually have harmful effects.40,41 Additionally, consumption of high-dose dietary supplements has been associated with an increased risk for severe medical events, including disability and death among adolescents and young adults.42
Conclusion
Numerous trials have indicated that the use of systemic vitamins can have beneficial effects on the protection and appearance of skin. Photodamage from UV light–induced erythema can be decreased by carotenoids and vitamins C and E. Similarly, supplements that combine multiple nutrients with collagen have been shown to improve the appearance of aging skin by decreasing the prominence of wrinkles. Given the growing number of products and advertisements that exist in the supplement marketplace, it is crucial for clinicians to ground their recommendations to patients in the scientific data of robust studies.
- Zhang S, Duan E. Fighting against skin aging: the way from bench to bedside. Cell Transplant. 2018;27:729-738.
- Rittié L, Fisher GJ. Natural and sun-induced aging of human skin. Cold Spring Harb Perspect Med. 2015;5:a015370.
- Draelos ZD. Nutrition and enhancing youthful-appearing skin. Clin Dermatol. 2010;28:400-408.
- Anunciato TP, da Rocha Filho PA. Carotenoids and polyphenols in nutricosmetics, nutraceuticals, and cosmeceuticals. J Cosmet Dermatol. 2012;11:51-54.
- Stahl W, Heinrich U, Jungmann H, et al. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am J Clin Nutr. 2000;71:795-798.
- Anstey AV. Systemic photoprotection with alpha-tocopherol (vitamin E) and beta-carotene. Clin Exp Dermatol. 2002;27:170-176.
- Stahl W, Heinrich U, Wiseman S, et al. Dietary tomato paste protects against ultraviolet light-induced erythema in humans. J Nutr. 2001;131:1449-1451.
- Wood SM, Mastaloudis AF, Hester SN, et al. Protective effects of a novel nutritional and phytonutrient blend on ultraviolet radiation-induced skin damage and inflammatory response through aging defense mechanisms. J Cosmet Dermatol. 2017;16:491-499.
- Meinke MC, Friedrich A, Tscherch K, et al. Influence of dietary carotenoids on radical scavenging capacity of the skin and skin lipids. Eur J Pharm Biopharm. 2013;84:365-373.
- Manela-Azulay M, Bagatin E. Cosmeceuticals vitamins. Clin Dermatol. 2009;27:469-474.
- McArdle F, Rhodes LE, Parslew R, et al. UVR-induced oxidative stress in human skin in vivo: effects of oral vitamin C supplementation. Free Radic Biol Med. 2002;33:1355-1362.
- Cosgrove MC, Franco OH, Granger SP, et al. Dietary nutrient intakes and skin-aging appearance among middle-aged American women. Am J Clin Nutr. 2007;86:1225-1231.
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667.
- Schagen SK, Zampeli VA, Makrantonaki E, et al. Discovering the link between nutrition and skin aging. Dermatoendocrinol. 2012;4:298-307.
- Chan AC. Partners in defense, vitamin E and vitamin C. Can J Physiol Pharmacol. 1993;71:725-731.
- Eberlein-Konig B, Placzek M, Przybilla B. Protective effect against sunburn of combined systemic ascorbic acid (vitamin C) and d-alpha-tocopherol (vitamin E). J Am Acad Dermatol. 1998;38:45-48.
- Fuchs J, Kern H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: a clinical study using solar simulated radiation. Free Radic Biol Med. 1998;25:1006-1012.
- Shahriari M, Kerr PE, Slade K, et al. Vitamin D and the skin. Clin Dermatol. 2010;28:663-668.
- Soleymani T, Hung T, Soung J. The role of vitamin D in psoriasis: a review. Int J Dermatol. 2015;54:383-392.
- Lehmann B, Querings K, Reichrath J. Vitamin D and skin: new aspects for dermatology. Exp Dermatol. 2004;13(suppl 4):11-15.
- Kannan S, Lim HW. Photoprotection and vitamin D: a review. Photodermatol Photoimmunol Photomed. 2014;30:137-145.
- Upala S, Sanguankeo A. Low 25-hydroxyvitamin D levels are associated with vitiligo: a systematic review and meta-analysis. Photodermatol Photoimmunol Photomed. 2016;32:181-190.
- Scott JF, Das LM, Ahsanuddin S, et al. Oral vitamin D rapidly attenuates inflammation from sunburn: an interventional study. J Invest Dermatol. 2017;137:2078-2086.
- Varani J, Dame MK, Rittie L, et al. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861-1868.
- Proksch E, Segger D, Degwert J, et al. Oral supplementation of specific collagen peptides has beneficial effects on human skin physiology: a double-blind, placebo-controlled study. Skin Pharmacol Physiol. 2014;27:47-55.
- Asserin J, Lati E, Shioya T, et al. The effect of oral collagen peptide supplementation on skin moisture and the dermal collagen network: evidence from an ex vivo model and randomized, placebo-controlled clinical trials. J Cosmet Dermatol. 2015;14:291-301.
- Koizumi S, Inoue N, Shimizu M, et al. Effects of dietary supplementation with fish scales-derived collagen peptides on skin parameters and condition: a randomized, placebo-controlled, double-blind study. Int J Peptide Res Ther. 2018;24:397-402.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19. doi:10.3390/ijms19103059.
- Guillou S, Ghabri S, Jannot C, et al. The moisturizing effect of a wheat extract food supplement on women’s skin: a randomized, double-blind placebo-controlled trial. Int J Cosmet Sci. 2011;33:138-143.
- Kieffer ME, Efsen J. Imedeen in the treatment of photoaged skin: an efficacy and safety trial over 12 months. J Eur Acad Dermatol Venereol. 1998;11:129-136.
- Skovgaard GR, Jensen AS, Sigler ML. Effect of a novel dietary supplement on skin aging in post-menopausal women. Eur J Clin Nutr. 2006;60:1201-1206.
- Stephens TJ, Sigler ML, Herndon JH Jr, et al. A placebo-controlled, double-blind clinical trial to evaluate the efficacy of Imedeen(®) Time Perfection(®) for improving the appearance of photodamaged skin. Clin Cosmet Investig Dermatol. 2016;9:63-70.
- Stephens TJ, Sigler ML, Hino PD, et al. A randomized, double-blind, placebo-controlled clinical trial evaluating an oral anti-aging skin care supplement for treating photodamaged skin. J Clin Aesthet Dermatol. 2016;9:25-32.
- El-Domyati M, Attia S, Saleh F, et al. Intrinsic aging vs. photoaging: a comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp Dermatol. 2002;11:398-405.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Kang MC, Yumnam S, Kim SY. Oral intake of collagen peptide attenuates ultraviolet B irradiation-induced skin dehydration in vivo by regulating hyaluronic acid synthesis. Int J Mol Sci. 2018;19. doi:10.3390/ijms19113551.
- Schwartz SR, Park J. Ingestion of BioCell Collagen(®), a novel hydrolyzed chicken sternal cartilage extract; enhanced blood microcirculation and reduced facial aging signs. Clin Interv Aging. 2012;7:267-273.
- De Luca C, Mikhal’chik EV, Suprun MV, et al. Skin antiageing and systemic redox effects of supplementation with marine collagen peptides and plant-derived antioxidants: a single-blind case-control clinical study. Oxid Med Cell Longev. 2016;2016:4389410.
- Genovese L, Corbo A, Sibilla S. An insight into the changes in skin texture and properties following dietary intervention with a nutricosmeceutical containing a blend of collagen bioactive peptides and antioxidants. Skin Pharmacol Physiol. 2017;30:146-158.
- Hamishehkar H, Ranjdoost F, Asgharian P, et al. Vitamins, are they safe? Adv Pharm Bull. 2016;6:467-477.
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029-1035.
- Or F, Yongjoo K, Simms J, et al. Taking stock of dietary supplements’ harmful effects on children, adolescents, and young adults [published online June 3, 2019]. J Adolesc Health. S1054-139X(19)30163-6. doi:10.1016/j.jadohealth.2019.03.005.
- Zhang S, Duan E. Fighting against skin aging: the way from bench to bedside. Cell Transplant. 2018;27:729-738.
- Rittié L, Fisher GJ. Natural and sun-induced aging of human skin. Cold Spring Harb Perspect Med. 2015;5:a015370.
- Draelos ZD. Nutrition and enhancing youthful-appearing skin. Clin Dermatol. 2010;28:400-408.
- Anunciato TP, da Rocha Filho PA. Carotenoids and polyphenols in nutricosmetics, nutraceuticals, and cosmeceuticals. J Cosmet Dermatol. 2012;11:51-54.
- Stahl W, Heinrich U, Jungmann H, et al. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am J Clin Nutr. 2000;71:795-798.
- Anstey AV. Systemic photoprotection with alpha-tocopherol (vitamin E) and beta-carotene. Clin Exp Dermatol. 2002;27:170-176.
- Stahl W, Heinrich U, Wiseman S, et al. Dietary tomato paste protects against ultraviolet light-induced erythema in humans. J Nutr. 2001;131:1449-1451.
- Wood SM, Mastaloudis AF, Hester SN, et al. Protective effects of a novel nutritional and phytonutrient blend on ultraviolet radiation-induced skin damage and inflammatory response through aging defense mechanisms. J Cosmet Dermatol. 2017;16:491-499.
- Meinke MC, Friedrich A, Tscherch K, et al. Influence of dietary carotenoids on radical scavenging capacity of the skin and skin lipids. Eur J Pharm Biopharm. 2013;84:365-373.
- Manela-Azulay M, Bagatin E. Cosmeceuticals vitamins. Clin Dermatol. 2009;27:469-474.
- McArdle F, Rhodes LE, Parslew R, et al. UVR-induced oxidative stress in human skin in vivo: effects of oral vitamin C supplementation. Free Radic Biol Med. 2002;33:1355-1362.
- Cosgrove MC, Franco OH, Granger SP, et al. Dietary nutrient intakes and skin-aging appearance among middle-aged American women. Am J Clin Nutr. 2007;86:1225-1231.
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667.
- Schagen SK, Zampeli VA, Makrantonaki E, et al. Discovering the link between nutrition and skin aging. Dermatoendocrinol. 2012;4:298-307.
- Chan AC. Partners in defense, vitamin E and vitamin C. Can J Physiol Pharmacol. 1993;71:725-731.
- Eberlein-Konig B, Placzek M, Przybilla B. Protective effect against sunburn of combined systemic ascorbic acid (vitamin C) and d-alpha-tocopherol (vitamin E). J Am Acad Dermatol. 1998;38:45-48.
- Fuchs J, Kern H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: a clinical study using solar simulated radiation. Free Radic Biol Med. 1998;25:1006-1012.
- Shahriari M, Kerr PE, Slade K, et al. Vitamin D and the skin. Clin Dermatol. 2010;28:663-668.
- Soleymani T, Hung T, Soung J. The role of vitamin D in psoriasis: a review. Int J Dermatol. 2015;54:383-392.
- Lehmann B, Querings K, Reichrath J. Vitamin D and skin: new aspects for dermatology. Exp Dermatol. 2004;13(suppl 4):11-15.
- Kannan S, Lim HW. Photoprotection and vitamin D: a review. Photodermatol Photoimmunol Photomed. 2014;30:137-145.
- Upala S, Sanguankeo A. Low 25-hydroxyvitamin D levels are associated with vitiligo: a systematic review and meta-analysis. Photodermatol Photoimmunol Photomed. 2016;32:181-190.
- Scott JF, Das LM, Ahsanuddin S, et al. Oral vitamin D rapidly attenuates inflammation from sunburn: an interventional study. J Invest Dermatol. 2017;137:2078-2086.
- Varani J, Dame MK, Rittie L, et al. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861-1868.
- Proksch E, Segger D, Degwert J, et al. Oral supplementation of specific collagen peptides has beneficial effects on human skin physiology: a double-blind, placebo-controlled study. Skin Pharmacol Physiol. 2014;27:47-55.
- Asserin J, Lati E, Shioya T, et al. The effect of oral collagen peptide supplementation on skin moisture and the dermal collagen network: evidence from an ex vivo model and randomized, placebo-controlled clinical trials. J Cosmet Dermatol. 2015;14:291-301.
- Koizumi S, Inoue N, Shimizu M, et al. Effects of dietary supplementation with fish scales-derived collagen peptides on skin parameters and condition: a randomized, placebo-controlled, double-blind study. Int J Peptide Res Ther. 2018;24:397-402.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19. doi:10.3390/ijms19103059.
- Guillou S, Ghabri S, Jannot C, et al. The moisturizing effect of a wheat extract food supplement on women’s skin: a randomized, double-blind placebo-controlled trial. Int J Cosmet Sci. 2011;33:138-143.
- Kieffer ME, Efsen J. Imedeen in the treatment of photoaged skin: an efficacy and safety trial over 12 months. J Eur Acad Dermatol Venereol. 1998;11:129-136.
- Skovgaard GR, Jensen AS, Sigler ML. Effect of a novel dietary supplement on skin aging in post-menopausal women. Eur J Clin Nutr. 2006;60:1201-1206.
- Stephens TJ, Sigler ML, Herndon JH Jr, et al. A placebo-controlled, double-blind clinical trial to evaluate the efficacy of Imedeen(®) Time Perfection(®) for improving the appearance of photodamaged skin. Clin Cosmet Investig Dermatol. 2016;9:63-70.
- Stephens TJ, Sigler ML, Hino PD, et al. A randomized, double-blind, placebo-controlled clinical trial evaluating an oral anti-aging skin care supplement for treating photodamaged skin. J Clin Aesthet Dermatol. 2016;9:25-32.
- El-Domyati M, Attia S, Saleh F, et al. Intrinsic aging vs. photoaging: a comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp Dermatol. 2002;11:398-405.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Kang MC, Yumnam S, Kim SY. Oral intake of collagen peptide attenuates ultraviolet B irradiation-induced skin dehydration in vivo by regulating hyaluronic acid synthesis. Int J Mol Sci. 2018;19. doi:10.3390/ijms19113551.
- Schwartz SR, Park J. Ingestion of BioCell Collagen(®), a novel hydrolyzed chicken sternal cartilage extract; enhanced blood microcirculation and reduced facial aging signs. Clin Interv Aging. 2012;7:267-273.
- De Luca C, Mikhal’chik EV, Suprun MV, et al. Skin antiageing and systemic redox effects of supplementation with marine collagen peptides and plant-derived antioxidants: a single-blind case-control clinical study. Oxid Med Cell Longev. 2016;2016:4389410.
- Genovese L, Corbo A, Sibilla S. An insight into the changes in skin texture and properties following dietary intervention with a nutricosmeceutical containing a blend of collagen bioactive peptides and antioxidants. Skin Pharmacol Physiol. 2017;30:146-158.
- Hamishehkar H, Ranjdoost F, Asgharian P, et al. Vitamins, are they safe? Adv Pharm Bull. 2016;6:467-477.
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029-1035.
- Or F, Yongjoo K, Simms J, et al. Taking stock of dietary supplements’ harmful effects on children, adolescents, and young adults [published online June 3, 2019]. J Adolesc Health. S1054-139X(19)30163-6. doi:10.1016/j.jadohealth.2019.03.005.
Practice Points
- Multiple vitamins and supplements have demonstrated evidence in improving skin appearance.
- Carotenoids, along with vitamins C and E, have been shown to protect skin from UV-induced photodamage, while supplements containing collagen decrease the appearance of wrinkles.

Barber’s Sinus Between the Toes of a Female Hairdresser
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
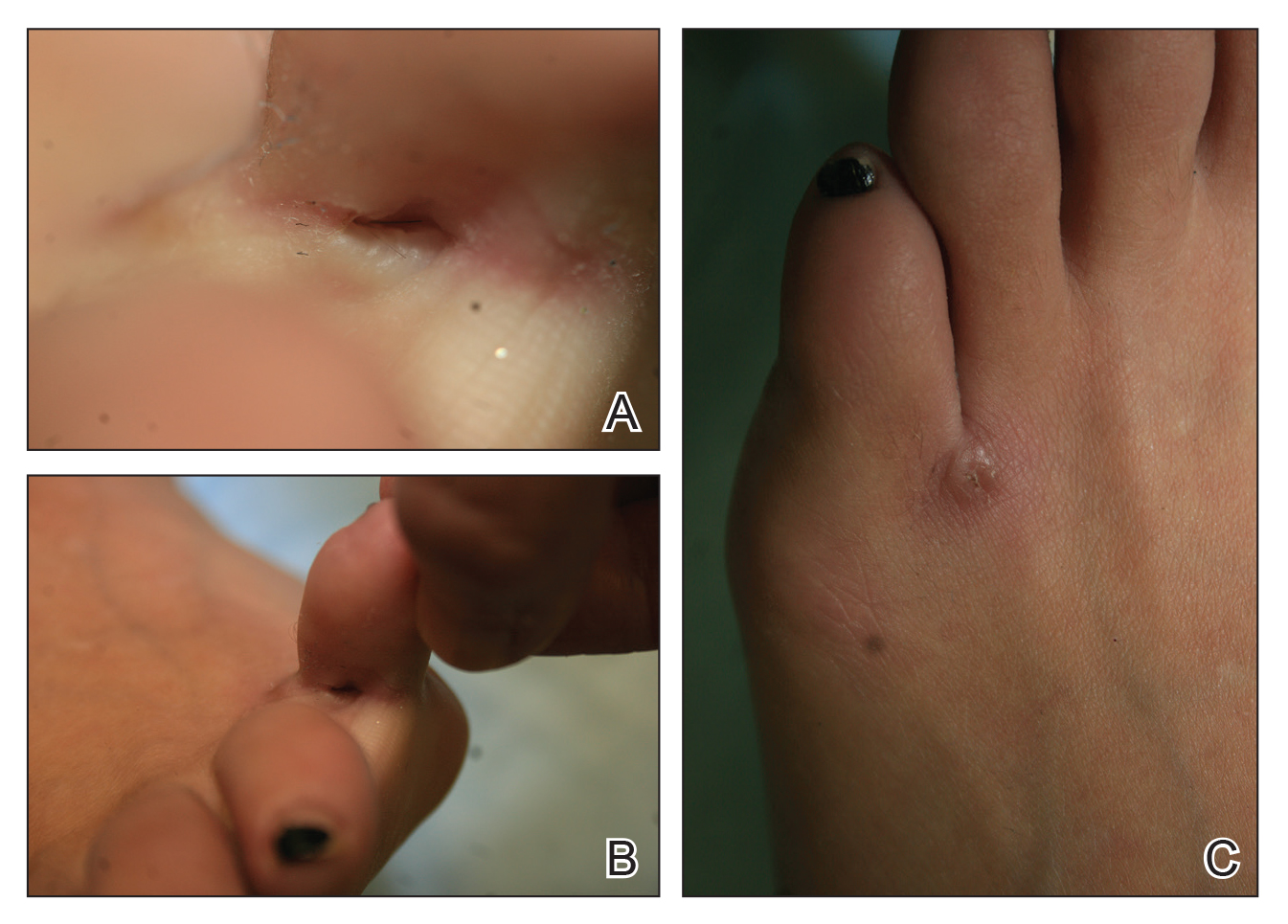
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
Practice Points
- This case illustrates a disease in which a medical history and simple clinical examination can lead to the diagnosis.
- Patients may value a diagnosis without treatment. A patient with barber’s sinus may be satisfied with watchful waiting.
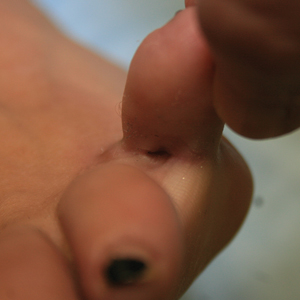
Review looks at natural course of alopecia areata in young children
, according to a retrospective chart review of 125 children.
Almost 90% of the children presented between aged 2 and 4 years, compared with 11.9% between ages 1 and 2 years, and 1.6% aged under 1 year, “in keeping with the existing literature,” the study authors reported in Pediatric Dermatology. “A high percentage of patients continued to have mild, patchy alopecia at their follow‐up visits,” they added.
Epidemiologic studies of children with alopecia areata are few and have not focused on the youngest patients, said Sneha Rangu, of the section of dermatology at Children’s Hospital of Philadelphia, and coauthors. They performed a retrospective chart review of 125 patients, who initially presented at the hospital with alopecia areata between Jan. 1, 2016, and June 1, 2018, when they were younger than 4 years. Patients who received systemic therapy or topical JAK inhibitors for alopecia were excluded. Severity was measured with the Severity of Alopecia Tool (SALT) score, to monitor progression of hair loss, analyzing scores at the initial presentation, at 3-6 months, at 1 year, and at 2 or more years.
Almost 70% were female, which the authors said was similar to other studies that have found alopecia areata is more prevalent in females; and 86.6% were between ages 2 and 4 years when they first presented. The initial diagnosis was alopecia areata in 72.0%, alopecia totalis in 8.8%, and alopecia universalis in 19.2%. Of the 41 boys, 39% had alopecia totalis or alopecia universalis, as did 22% of the girls, which suggested that boys presenting under aged 4 years were more likely to have more severe disease, or that “guardians of boys are more likely to present for therapy when disease is more severe,” the authors wrote.
About 40% of the children presented with a history of atopic dermatitis, and 4% had an autoimmune disease (vitiligo, celiac disease, or type 1 diabetes). Twenty-eight percent of patients had a family history of alopecia areata, 27.2% had a family history of other autoimmune diseases, and 32% had a family history of hypothyroidism.
At the first visit, 57.6% had patch‐stage alopecia and SALT scores in the mild range (0%‐24% hair loss), which was present in a high proportion of these patients at follow-up: 49.4% at 3-6 months, 39.5% at 1 year, and 42.9% at two or more years.
At the first visit, 28% had high SALT scores (50%-100% hair loss), increasing to 36% at 3-6 months, 41.8% at 1 year, and 46.4% at 2 or more years. They calculated that for those with more than 50% hair loss at the initial presentation, the likelihood of being in a high category of hair loss, as measured by increasing SALT scores, was significantly higher at 1 year (odds ratio, 1.85, P =.033) and at 2 or more years (OR, 2.29, P = .038).
“While there is a likelihood of increasing disease severity, those with higher severity at initial presentation are likely to stay severe after one or 2 years,” the authors noted.
They concluded that their results add to the understanding of the epidemiology of alopecia areata in children “and perhaps can provide clinicians and families with a better sense of prognosis for progression in the youngest patients presenting with alopecia areata.”
They said the retrospective design and small sample size were among the study’s limitations. They had no conflicts of interest to disclose.
SOURCE: Rangu S et al. Pediatr Dermatol. 2019 Aug 29. doi: 10.1111/pde.13990.
, according to a retrospective chart review of 125 children.
Almost 90% of the children presented between aged 2 and 4 years, compared with 11.9% between ages 1 and 2 years, and 1.6% aged under 1 year, “in keeping with the existing literature,” the study authors reported in Pediatric Dermatology. “A high percentage of patients continued to have mild, patchy alopecia at their follow‐up visits,” they added.
Epidemiologic studies of children with alopecia areata are few and have not focused on the youngest patients, said Sneha Rangu, of the section of dermatology at Children’s Hospital of Philadelphia, and coauthors. They performed a retrospective chart review of 125 patients, who initially presented at the hospital with alopecia areata between Jan. 1, 2016, and June 1, 2018, when they were younger than 4 years. Patients who received systemic therapy or topical JAK inhibitors for alopecia were excluded. Severity was measured with the Severity of Alopecia Tool (SALT) score, to monitor progression of hair loss, analyzing scores at the initial presentation, at 3-6 months, at 1 year, and at 2 or more years.
Almost 70% were female, which the authors said was similar to other studies that have found alopecia areata is more prevalent in females; and 86.6% were between ages 2 and 4 years when they first presented. The initial diagnosis was alopecia areata in 72.0%, alopecia totalis in 8.8%, and alopecia universalis in 19.2%. Of the 41 boys, 39% had alopecia totalis or alopecia universalis, as did 22% of the girls, which suggested that boys presenting under aged 4 years were more likely to have more severe disease, or that “guardians of boys are more likely to present for therapy when disease is more severe,” the authors wrote.
About 40% of the children presented with a history of atopic dermatitis, and 4% had an autoimmune disease (vitiligo, celiac disease, or type 1 diabetes). Twenty-eight percent of patients had a family history of alopecia areata, 27.2% had a family history of other autoimmune diseases, and 32% had a family history of hypothyroidism.
At the first visit, 57.6% had patch‐stage alopecia and SALT scores in the mild range (0%‐24% hair loss), which was present in a high proportion of these patients at follow-up: 49.4% at 3-6 months, 39.5% at 1 year, and 42.9% at two or more years.
At the first visit, 28% had high SALT scores (50%-100% hair loss), increasing to 36% at 3-6 months, 41.8% at 1 year, and 46.4% at 2 or more years. They calculated that for those with more than 50% hair loss at the initial presentation, the likelihood of being in a high category of hair loss, as measured by increasing SALT scores, was significantly higher at 1 year (odds ratio, 1.85, P =.033) and at 2 or more years (OR, 2.29, P = .038).
“While there is a likelihood of increasing disease severity, those with higher severity at initial presentation are likely to stay severe after one or 2 years,” the authors noted.
They concluded that their results add to the understanding of the epidemiology of alopecia areata in children “and perhaps can provide clinicians and families with a better sense of prognosis for progression in the youngest patients presenting with alopecia areata.”
They said the retrospective design and small sample size were among the study’s limitations. They had no conflicts of interest to disclose.
SOURCE: Rangu S et al. Pediatr Dermatol. 2019 Aug 29. doi: 10.1111/pde.13990.
, according to a retrospective chart review of 125 children.
Almost 90% of the children presented between aged 2 and 4 years, compared with 11.9% between ages 1 and 2 years, and 1.6% aged under 1 year, “in keeping with the existing literature,” the study authors reported in Pediatric Dermatology. “A high percentage of patients continued to have mild, patchy alopecia at their follow‐up visits,” they added.
Epidemiologic studies of children with alopecia areata are few and have not focused on the youngest patients, said Sneha Rangu, of the section of dermatology at Children’s Hospital of Philadelphia, and coauthors. They performed a retrospective chart review of 125 patients, who initially presented at the hospital with alopecia areata between Jan. 1, 2016, and June 1, 2018, when they were younger than 4 years. Patients who received systemic therapy or topical JAK inhibitors for alopecia were excluded. Severity was measured with the Severity of Alopecia Tool (SALT) score, to monitor progression of hair loss, analyzing scores at the initial presentation, at 3-6 months, at 1 year, and at 2 or more years.
Almost 70% were female, which the authors said was similar to other studies that have found alopecia areata is more prevalent in females; and 86.6% were between ages 2 and 4 years when they first presented. The initial diagnosis was alopecia areata in 72.0%, alopecia totalis in 8.8%, and alopecia universalis in 19.2%. Of the 41 boys, 39% had alopecia totalis or alopecia universalis, as did 22% of the girls, which suggested that boys presenting under aged 4 years were more likely to have more severe disease, or that “guardians of boys are more likely to present for therapy when disease is more severe,” the authors wrote.
About 40% of the children presented with a history of atopic dermatitis, and 4% had an autoimmune disease (vitiligo, celiac disease, or type 1 diabetes). Twenty-eight percent of patients had a family history of alopecia areata, 27.2% had a family history of other autoimmune diseases, and 32% had a family history of hypothyroidism.
At the first visit, 57.6% had patch‐stage alopecia and SALT scores in the mild range (0%‐24% hair loss), which was present in a high proportion of these patients at follow-up: 49.4% at 3-6 months, 39.5% at 1 year, and 42.9% at two or more years.
At the first visit, 28% had high SALT scores (50%-100% hair loss), increasing to 36% at 3-6 months, 41.8% at 1 year, and 46.4% at 2 or more years. They calculated that for those with more than 50% hair loss at the initial presentation, the likelihood of being in a high category of hair loss, as measured by increasing SALT scores, was significantly higher at 1 year (odds ratio, 1.85, P =.033) and at 2 or more years (OR, 2.29, P = .038).
“While there is a likelihood of increasing disease severity, those with higher severity at initial presentation are likely to stay severe after one or 2 years,” the authors noted.
They concluded that their results add to the understanding of the epidemiology of alopecia areata in children “and perhaps can provide clinicians and families with a better sense of prognosis for progression in the youngest patients presenting with alopecia areata.”
They said the retrospective design and small sample size were among the study’s limitations. They had no conflicts of interest to disclose.
SOURCE: Rangu S et al. Pediatr Dermatol. 2019 Aug 29. doi: 10.1111/pde.13990.
FROM PEDIATRIC DERMATOLOGY