User login
VIDEO: Don’t miss reservoirs when treating recurrent onychomycosis
WAILEA, HAWAII – Patients attribute every nail problem to nail fungus, but part of the problem with nails is concomitant tinea pedis, tinea corporis, and other reservoirs of infection, according to Neal Bhatia, MD, director of clinical dermatology at Therapeutics Clinical Research in San Diego.
Failure to locate and treat these other reservoirs can lead to recurrence of the nail infection, Dr. Bhatia said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“If we aren’t treating the skin as well as the nails, it is creating a reservoir effect that reaccumulates in the nail itself,” he said. Similarly, the nails can serve as a reservoir of infection for the skin.
“That makes it all the more important” to treat both skin and nails and “treat through the disease state,” he commented in a video interview. Cost can influence treatment decisions, and there is a role for both topical and systemic therapy, he said. However, “if there’s not adequate testing or if there’s not a good proof of the diagnosis, we are just losing out no matter what we use,” he cautioned.
Dr. Bhatia disclosed relationships with companies including Actavis, Aqua, Allergan, Anacor, Bayer, Biofrontera, Biopharmx, Dermira, Dusa, Exceltis, Ferndale, Foamix, Galderma, Intraderm, ISDIN, LaRoche-Posay, Leo, Novartis, Sanofi, and Valeant.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, HAWAII – Patients attribute every nail problem to nail fungus, but part of the problem with nails is concomitant tinea pedis, tinea corporis, and other reservoirs of infection, according to Neal Bhatia, MD, director of clinical dermatology at Therapeutics Clinical Research in San Diego.
Failure to locate and treat these other reservoirs can lead to recurrence of the nail infection, Dr. Bhatia said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“If we aren’t treating the skin as well as the nails, it is creating a reservoir effect that reaccumulates in the nail itself,” he said. Similarly, the nails can serve as a reservoir of infection for the skin.
“That makes it all the more important” to treat both skin and nails and “treat through the disease state,” he commented in a video interview. Cost can influence treatment decisions, and there is a role for both topical and systemic therapy, he said. However, “if there’s not adequate testing or if there’s not a good proof of the diagnosis, we are just losing out no matter what we use,” he cautioned.
Dr. Bhatia disclosed relationships with companies including Actavis, Aqua, Allergan, Anacor, Bayer, Biofrontera, Biopharmx, Dermira, Dusa, Exceltis, Ferndale, Foamix, Galderma, Intraderm, ISDIN, LaRoche-Posay, Leo, Novartis, Sanofi, and Valeant.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, HAWAII – Patients attribute every nail problem to nail fungus, but part of the problem with nails is concomitant tinea pedis, tinea corporis, and other reservoirs of infection, according to Neal Bhatia, MD, director of clinical dermatology at Therapeutics Clinical Research in San Diego.
Failure to locate and treat these other reservoirs can lead to recurrence of the nail infection, Dr. Bhatia said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“If we aren’t treating the skin as well as the nails, it is creating a reservoir effect that reaccumulates in the nail itself,” he said. Similarly, the nails can serve as a reservoir of infection for the skin.
“That makes it all the more important” to treat both skin and nails and “treat through the disease state,” he commented in a video interview. Cost can influence treatment decisions, and there is a role for both topical and systemic therapy, he said. However, “if there’s not adequate testing or if there’s not a good proof of the diagnosis, we are just losing out no matter what we use,” he cautioned.
Dr. Bhatia disclosed relationships with companies including Actavis, Aqua, Allergan, Anacor, Bayer, Biofrontera, Biopharmx, Dermira, Dusa, Exceltis, Ferndale, Foamix, Galderma, Intraderm, ISDIN, LaRoche-Posay, Leo, Novartis, Sanofi, and Valeant.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT SDEF HAWAII DERMATOLOGY SEMINAR

Bilateral Symmetric Onycholysis of Distal Fingernails
The Diagnosis: Allergic Contact Dermatitis
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
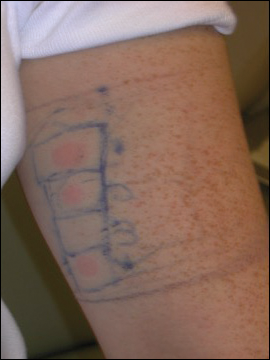
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
The Diagnosis: Allergic Contact Dermatitis
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
The Diagnosis: Allergic Contact Dermatitis
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
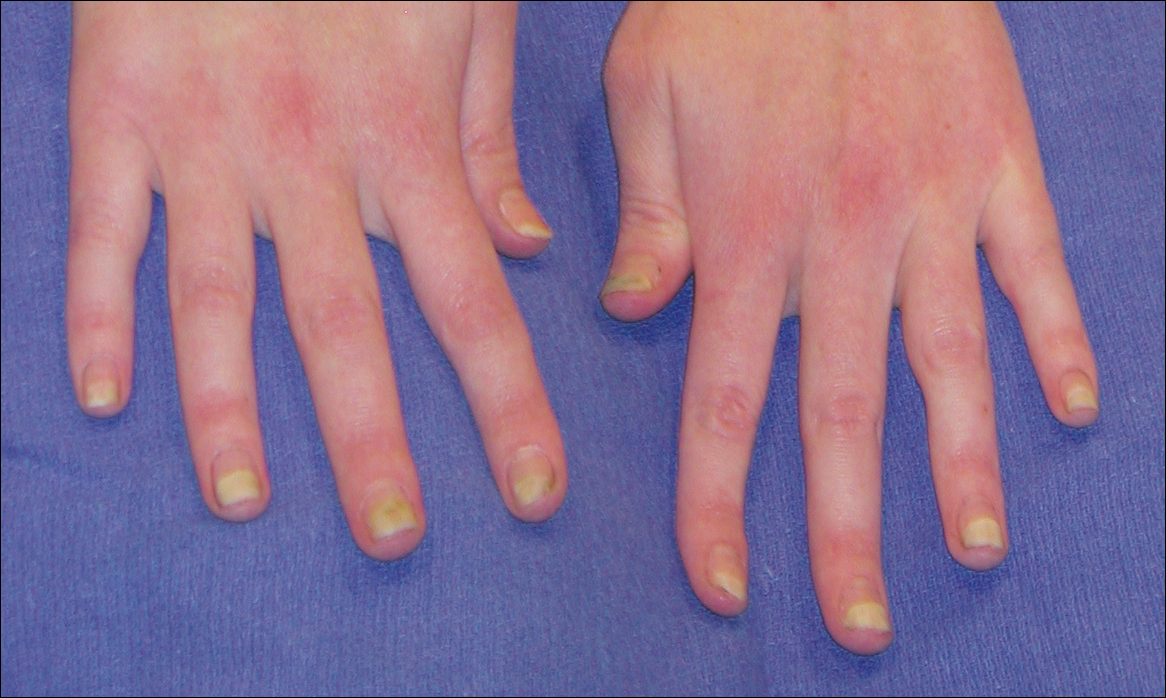
A 28-year-old woman presented with distal onycholysis of all 10 fingernails. The patient started to notice brittleness in the first, second, and third fingernails of the right hand 2 months prior. She had a 10-year history of wearing acrylic nails and reported a history of periungual eczema. On physical examination, all 10 fingernails had distal onycholysis and there was a green discoloration of the first fingernail on the left hand. On blood analysis, thyroid-stimulating hormone and free thyroxine were within reference range. A nail clipping showed onychodystrophy and a negative periodic acid-Schiff stain.
‘Anxiety sensitivity’ tied to psychodermatologic disorders
Adult patients who experience stress in the form of “anxiety sensitivity” are more likely to develop psychodermatological conditions than those that are not psychodermatological, a cross-sectional study of 115 participants shows.
“The results suggest that [anxiety sensitivity] interventions combined with dermatology treatments may be beneficial for psychodermatological patients,” wrote Laura J. Dixon, PhD, of the University of Mississippi Medical Center, Jackson, and her associates. “There is strong evidence that cognitive-behavioral therapy significantly reduces [anxiety sensitivity] through strategies such as psychoeducation, interoceptive exposure, and cognitive therapy.”
Dr. Dixon and her associates recruited 123 dermatologic patients aged 18-83 years over 30 weeks through three outpatient university dermatology clinics in Central Mississippi. Sixty-five percent of the participants were white, 33% were black, 1% were Asian, and 1% were Native American; 65% were female. Most of the patients were married and living with their spouses. The final sample of participants comprised 63 psychodermatological patients and 52 nonpsychodermatological patients (Psychosomatics. 2016;57:498-504).
The investigators assessed general anxiety symptoms using the 7-item depression, anxiety, and stress subscale (DASS-A) from the 21-item version of the questionnaire (DASS-21). Anxiety sensitivity – which refers to the “extent of beliefs that anxiety symptoms or arousal can have harmful consequences” (Turk Psikiyatri Derg. 2011 Fall;22[3]:187-93) – was measured using the Anxiety Sensitivity Index–3 (ASI-3, an 18-item self-report instrument that assesses physical manifestations of anxiety, such as blushing and fast heart beating.
Psychodermatological conditions were classified as disorders that might be rooted in or made worse by psychological, behavioral, or stress-related factors. Conditions in this category include acne, alopecia, atopic dermatitis, eczema, hidradenitis, prurigo, psoriasis, and rosacea. Dermatologic conditions not tied to psychological factors and classified as biologically based include brittle fingernails, cysts, keloids, rashes, skin cancer, skin lesions, spider veins, and warts, reported Dr. Dixon.
No significant differences were observed on the DASS-A scores between the two groups.
The mean scores of psychodermatological patients on the ASI-3 were significantly higher than the scores of patients with nonpsychodermatological conditions (21.1 vs. 13.7; P = .013). In fact, Dr. Dixon and her associates found that “each 1-unit increment in the ASI-3 social subscale score was associated with a 12.7% increased odds of patients having a psychodermatological condition.”
“Taken together, these results are supported by existing theoretical models of psychodermatological disorders that highlight the importance of stress among patients with certain dermatological conditions,” the researchers wrote.
One of the authors, dermatologist Robert T. Brodell, disclosed receiving honoraria from Allergan, Galderma Laboratories, and PharmaDerm; he also disclosed receiving consultant fees and performing clinical trials for other pharmaceutical companies. Neither Dr. Dixon nor any of the other authors declared relevant financial disclosures.
[email protected]
On Twitter @ginalhenderson
Adult patients who experience stress in the form of “anxiety sensitivity” are more likely to develop psychodermatological conditions than those that are not psychodermatological, a cross-sectional study of 115 participants shows.
“The results suggest that [anxiety sensitivity] interventions combined with dermatology treatments may be beneficial for psychodermatological patients,” wrote Laura J. Dixon, PhD, of the University of Mississippi Medical Center, Jackson, and her associates. “There is strong evidence that cognitive-behavioral therapy significantly reduces [anxiety sensitivity] through strategies such as psychoeducation, interoceptive exposure, and cognitive therapy.”
Dr. Dixon and her associates recruited 123 dermatologic patients aged 18-83 years over 30 weeks through three outpatient university dermatology clinics in Central Mississippi. Sixty-five percent of the participants were white, 33% were black, 1% were Asian, and 1% were Native American; 65% were female. Most of the patients were married and living with their spouses. The final sample of participants comprised 63 psychodermatological patients and 52 nonpsychodermatological patients (Psychosomatics. 2016;57:498-504).
The investigators assessed general anxiety symptoms using the 7-item depression, anxiety, and stress subscale (DASS-A) from the 21-item version of the questionnaire (DASS-21). Anxiety sensitivity – which refers to the “extent of beliefs that anxiety symptoms or arousal can have harmful consequences” (Turk Psikiyatri Derg. 2011 Fall;22[3]:187-93) – was measured using the Anxiety Sensitivity Index–3 (ASI-3, an 18-item self-report instrument that assesses physical manifestations of anxiety, such as blushing and fast heart beating.
Psychodermatological conditions were classified as disorders that might be rooted in or made worse by psychological, behavioral, or stress-related factors. Conditions in this category include acne, alopecia, atopic dermatitis, eczema, hidradenitis, prurigo, psoriasis, and rosacea. Dermatologic conditions not tied to psychological factors and classified as biologically based include brittle fingernails, cysts, keloids, rashes, skin cancer, skin lesions, spider veins, and warts, reported Dr. Dixon.
No significant differences were observed on the DASS-A scores between the two groups.
The mean scores of psychodermatological patients on the ASI-3 were significantly higher than the scores of patients with nonpsychodermatological conditions (21.1 vs. 13.7; P = .013). In fact, Dr. Dixon and her associates found that “each 1-unit increment in the ASI-3 social subscale score was associated with a 12.7% increased odds of patients having a psychodermatological condition.”
“Taken together, these results are supported by existing theoretical models of psychodermatological disorders that highlight the importance of stress among patients with certain dermatological conditions,” the researchers wrote.
One of the authors, dermatologist Robert T. Brodell, disclosed receiving honoraria from Allergan, Galderma Laboratories, and PharmaDerm; he also disclosed receiving consultant fees and performing clinical trials for other pharmaceutical companies. Neither Dr. Dixon nor any of the other authors declared relevant financial disclosures.
[email protected]
On Twitter @ginalhenderson
Adult patients who experience stress in the form of “anxiety sensitivity” are more likely to develop psychodermatological conditions than those that are not psychodermatological, a cross-sectional study of 115 participants shows.
“The results suggest that [anxiety sensitivity] interventions combined with dermatology treatments may be beneficial for psychodermatological patients,” wrote Laura J. Dixon, PhD, of the University of Mississippi Medical Center, Jackson, and her associates. “There is strong evidence that cognitive-behavioral therapy significantly reduces [anxiety sensitivity] through strategies such as psychoeducation, interoceptive exposure, and cognitive therapy.”
Dr. Dixon and her associates recruited 123 dermatologic patients aged 18-83 years over 30 weeks through three outpatient university dermatology clinics in Central Mississippi. Sixty-five percent of the participants were white, 33% were black, 1% were Asian, and 1% were Native American; 65% were female. Most of the patients were married and living with their spouses. The final sample of participants comprised 63 psychodermatological patients and 52 nonpsychodermatological patients (Psychosomatics. 2016;57:498-504).
The investigators assessed general anxiety symptoms using the 7-item depression, anxiety, and stress subscale (DASS-A) from the 21-item version of the questionnaire (DASS-21). Anxiety sensitivity – which refers to the “extent of beliefs that anxiety symptoms or arousal can have harmful consequences” (Turk Psikiyatri Derg. 2011 Fall;22[3]:187-93) – was measured using the Anxiety Sensitivity Index–3 (ASI-3, an 18-item self-report instrument that assesses physical manifestations of anxiety, such as blushing and fast heart beating.
Psychodermatological conditions were classified as disorders that might be rooted in or made worse by psychological, behavioral, or stress-related factors. Conditions in this category include acne, alopecia, atopic dermatitis, eczema, hidradenitis, prurigo, psoriasis, and rosacea. Dermatologic conditions not tied to psychological factors and classified as biologically based include brittle fingernails, cysts, keloids, rashes, skin cancer, skin lesions, spider veins, and warts, reported Dr. Dixon.
No significant differences were observed on the DASS-A scores between the two groups.
The mean scores of psychodermatological patients on the ASI-3 were significantly higher than the scores of patients with nonpsychodermatological conditions (21.1 vs. 13.7; P = .013). In fact, Dr. Dixon and her associates found that “each 1-unit increment in the ASI-3 social subscale score was associated with a 12.7% increased odds of patients having a psychodermatological condition.”
“Taken together, these results are supported by existing theoretical models of psychodermatological disorders that highlight the importance of stress among patients with certain dermatological conditions,” the researchers wrote.
One of the authors, dermatologist Robert T. Brodell, disclosed receiving honoraria from Allergan, Galderma Laboratories, and PharmaDerm; he also disclosed receiving consultant fees and performing clinical trials for other pharmaceutical companies. Neither Dr. Dixon nor any of the other authors declared relevant financial disclosures.
[email protected]
On Twitter @ginalhenderson
Shedding Light on Onychomadesis
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23

Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
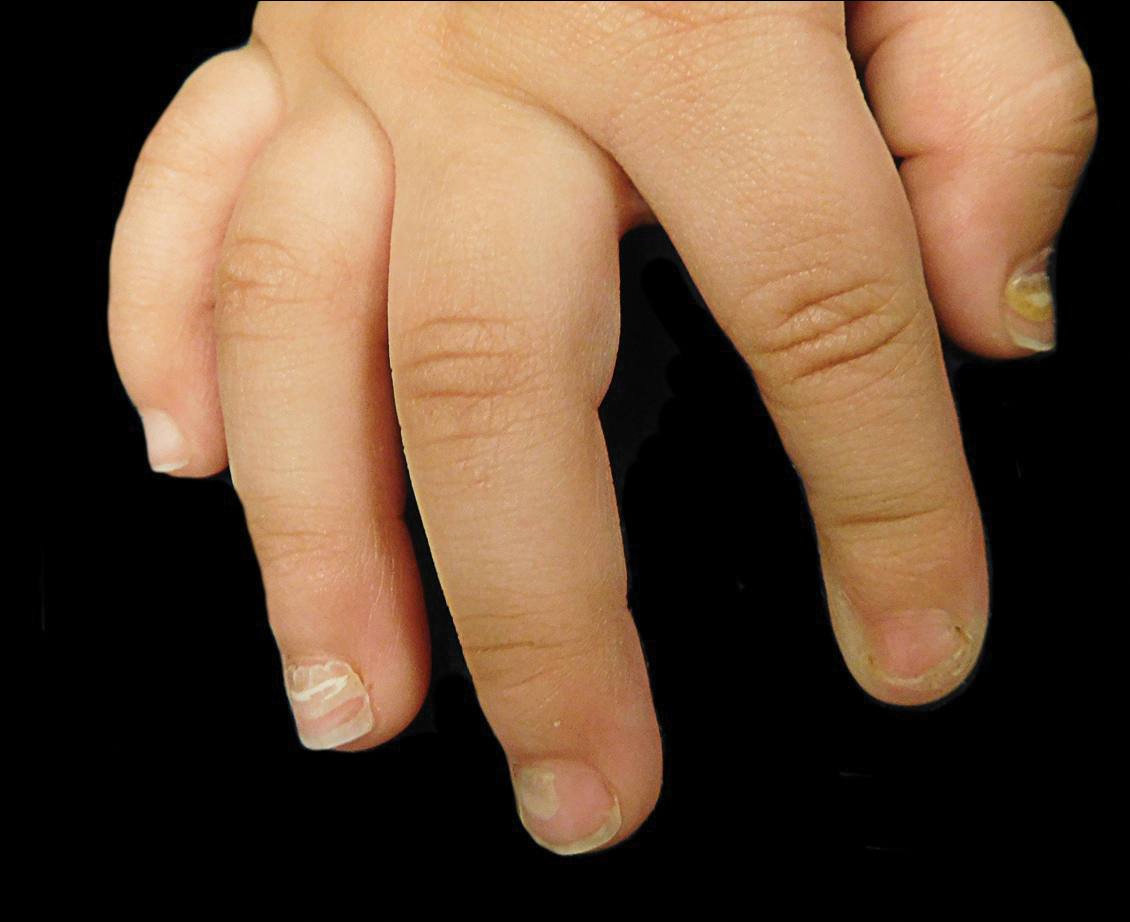
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23
Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23
Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
Practice Points
- Onychomadesis in a child may be a cutaneous sign of systemic disease.
- In childhood, onychomadesis is sometimes linked with hand-foot-and-mouth disease.
- Spontaneous nail regrowth usually occurs within 12 weeks but may occur faster in children.
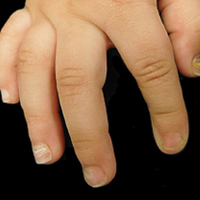
Common Hair Disorders
Review the PDF of the fact sheet on common hair disorders with board-relevant, easy-to-review material. This fact sheet reviews information about the most common hair disorders, including clinical and histopathological features, trichoscopy, and management of these diseases.
Practice Questions
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Answers to practice questions provided on next page
Practice Question Answers
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Review the PDF of the fact sheet on common hair disorders with board-relevant, easy-to-review material. This fact sheet reviews information about the most common hair disorders, including clinical and histopathological features, trichoscopy, and management of these diseases.
Practice Questions
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Answers to practice questions provided on next page
Practice Question Answers
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Review the PDF of the fact sheet on common hair disorders with board-relevant, easy-to-review material. This fact sheet reviews information about the most common hair disorders, including clinical and histopathological features, trichoscopy, and management of these diseases.
Practice Questions
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Answers to practice questions provided on next page
Practice Question Answers
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania

Over-the-counter and Natural Remedies for Onychomycosis: Do They Really Work?
Onychomycosis is a fungal infection of the nail unit by dermatophytes, yeasts, and nondermatophyte molds. It is characterized by a white or yellow discoloration of the nail plate; hyperkeratosis of the nail bed; distal detachment of the nail plate from its bed (onycholysis); and nail plate dystrophy, including thickening, crumbling, and ridging. Onychomycosis is an important problem, representing 30% of all superficial fungal infections and an estimated 50% of all nail diseases.1 Reported prevalence rates of onychomycosis in the United States and worldwide are varied, but the mean prevalence based on population-based studies in Europe and North America is estimated to be 4.3%.2 It is more common in older individuals, with an incidence rate of 20% in those older than 60 years and 50% in those older than 70 years.3 Onychomycosis is more common in patients with diabetes and 1.9 to 2.8 times higher than the general population.4 Dermatophytes are responsible for the majority of cases of onychomycosis, particularly Trichophyton rubrum and Trichophyton mentagrophytes.5
Onychomycosis is divided into different subtypes based on clinical presentation, which in turn are characterized by varying infecting organisms and prognoses. The subtypes of onychomycosis are distal and lateral subungual (DLSO), proximal subungual, superficial, endonyx, mixed pattern, total dystrophic, and secondary. Distal and lateral subungual onychomycosis are by far the most common presentation and begins when the infecting organism invades the hyponychium and distal or lateral nail bed. Trichophyton rubrum is the most common organism and T mentagrophytes is second, but Candida parapsilosis and Candida albicans also are possibilities. Proximal subungual onychomycosis is far less frequent than DLSO and is usually caused by T rubrum. The fungus invades the proximal nail folds and penetrates the newly growing nail plate.6 This pattern is more common in immunosuppressed patients and should prompt testing for human immunodeficiency virus.7 Total dystrophic onychomycosis is the end stage of fungal nail plate invasion, may follow DLSO or proximal subungual onychomycosis, and is difficult to treat.6
Onychomycosis causes pain, paresthesia, and difficulty with ambulation.8 In patients with peripheral neuropathy and vascular problems, including diabetes, onychomycosis can increase the risk for foot ulcers, with amputation in severe cases.9 Patients also may present with aesthetic concerns that may impact their quality of life.10
Given the effect on quality of life along with medical risks associated with onychomycosis, a safe and successful treatment modality with a low risk of recurrence is desirable. Unfortunately, treatment of nail fungus is quite challenging for a number of reasons. First, the thickness of the nail and/or the fungal mass may be a barrier to the delivery of topical and systemic drugs at the source of the infection. In addition, the nail plate does not have intrinsic immunity. Also, recurrence after treatment is common due to residual hyphae or spores that were not previously eliminated.11 Finally, many topical medications require long treatment courses, which may limit patient compliance, especially in patients who want to use nail polish for cosmesis or camouflage.
Currently Approved Therapies for Onychomycosis
Several definitions are needed to better interpret the results of onychomycosis clinical trials. Complete cure is defined as a negative potassium hydroxide preparation and negative fungal culture with a completely normal appearance of the nail. Mycological cure is defined as potassium hydroxide microscopy and fungal culture negative. Clinical cure is stated as 0% nail plate involvement but at times is reported as less than 5% and less than 10% involvement.
Terbinafine and itraconazole are the only US Food and Drug Administration (FDA)–approved systemic therapies, and ciclopirox, efinaconazole, and tavaborole are the only FDA-approved topicals. Advantages of systemic agents generally are higher cure rates and shorter treatment courses, thus better compliance. Disadvantages include greater incidence of systemic side effects and drug-drug interactions as well as the need for laboratory monitoring. Pros of topical therapies are low potential for adverse effects, no drug-drug interactions, and no monitoring of blood work. Cons include lower efficacy, long treatment courses, and poor patient compliance.
Terbinafine, an allylamine, taken orally once daily (250 mg) for 12 weeks for toenails and 6 weeks for fingernails currently is the preferred systemic treatment of onychomycosis, with complete cure rates of 38% and 59% and mycological cure rates of 70% and 79% for toenails and fingernails, respectively.12 Itraconazole, an azole, is dosed orally at 200 mg daily for 3 months for toenails, with a complete cure rate of 14% and mycological cure rate of 54%.13 For fingernail onychomycosis only, itraconazole is dosed at 200 mg twice daily for 1 week, followed by a treatment-free period of 3 weeks, and then another 1-week course at thesame dose. The complete cure rate is 47% and the mycological cure is 61% for this pulse regimen.13
Ciclopirox is a hydroxypyridone and the 8% nail lacquer formulation was approved in 1999, making it the first topical medication to gain FDA approval for the treatment of toenail onychomycosis. Based on 2 clinical trials, complete cure rates for toenails are 5.5% and 8.5% and mycological cure rates are 29% and 36% at 48 weeks with removal of residual lacquer and debridement.14 Efinaconazole is an azole and the 10% solution was FDA approved for the treatment of toenail onychomycosis in 2014.15 In 2 clinical trials, complete cure rates were 17.8% and 15.2% and mycological cure rates were 55.2% and 53.4% with once daily toenail application for 48 weeks.16 Tavaborole is a benzoxaborole and the 5% solution also was approved for the treatment of toenail onychomycosis in 2014.17 Two clinical trials reported complete cure rates of 6.5% and 9.1% and mycological cure rates of 31.1% and 35.9% with once daily toenail application for 48 weeks.18
Given the poor efficacy, systemic side effects, potential for drug-drug interactions, long-term treatment courses, and cost associated with current systemic and/or topical treatments, there has been a renewed interest in natural remedies and over-the-counter (OTC) therapies for onychomycosis. This review summarizes the in vitro and in vivo data, mechanisms of action, and clinical efficacy of various natural and OTC agents for the treatment of onychomycosis. Specifically, we summarize the data on tea tree oil (TTO), a popular topical cough suppressant (TCS), natural coniferous resin (NCR) lacquer, Ageratina pichinchensis (AP) extract, and ozonized sunflower oil.
Tea Tree Oil
Background
Tea tree oil is a volatile oil whose medicinal use dates back to the early 20th century when the Bundjabung aborigines of North and New South Wales extracted TTO from the dried leaves of the Melaleuca alternifolia plant and used it to treat superficial wounds.19 Tea tree oil has been shown to be an effective treatment of tinea pedis,20 and it is widely used in Australia as well as in Europe and North America.21 Tea tree oil also has been investigated as an antifungal agent for the treatment of onychomycosis, both in vitro22-28 and in clinical trials.29,30
In Vitro Data
Because TTO is composed of more than 100 active components,23 the antifungal activity of these individual components was investigated against 14 fungal isolates, including C albicans, T mentagrophytes, and Aspergillus species. The minimum inhibitory concentration (MIC) for α-pinene was less than 0.004% for T mentagrophytes and the components with the greatest MIC and minimum fungicidal concentration for the fungi tested were terpinen-4-ol and α-terpineol, respectively.22 The antifungal activity of TTO also was tested using disk diffusion assay experiments with 58 clinical isolates of fungi including C albicans, T rubrum, T mentagrophytes, and Aspergillus niger.24 Tea tree oil was most effective at inhibiting T rubrum followed by T mentagrophytes,24 which are the 2 most common etiologies of onychomycosis.5 In another report, the authors determined the MIC of TTO utilizing 4 different experiments with T rubrum as the infecting organism. Because TTO inhibited the growth of T rubrum at all concentrations greater than 0.1%, they found that the MIC was 0.1%.25 Given the lack of adequate nail penetration of most topical therapies, TTO in nanocapsules (TTO-NC), TTO nanoemulsions, and normal emulsions were tested in vitro for their ability to inhibit the growth of T rubrum inoculated into nail shavings. Colony growth decreased significantly within the first week of treatment, with TTO-NC showing maximum efficacy (P<.001). This study showed that TTO, particularly TTO-NC, was effective in inhibiting the growth of T rubrum in vitro and that using nanocapsule technology may increase nail penetration and bioavailability.31
Much of what we know about TTO’s antifungal mechanism of action comes from experiments involving C albicans. To date, it has not been studied in T rubrum or T mentagrophytes, the 2 most common etiologies of onychomycosis.5 In C albicans, TTO causes altered permeability of plasma membranes,32 dose-dependent alteration of respiration,33 decreased glucose-induced acidification of media surrounding fungi,32 and reversible inhibition of germ tube formation.19,34
Clinical Trials
A randomized, double-blind, multicenter trial was performed on 117 patients with culture-proven DLSO who were randomized to receive TTO 100% or clotrimazole solution 1% applied twice daily to affected toenails for 6 months.29 Primary outcome measures were mycologic cure, clinical assessment, and patient subjective assessment (Table 1). There were no statistical differences between the 2 treatment groups. Erythema and irritation were the most common adverse reactions occurring in 7.8% (5/64) of the TTO group.29
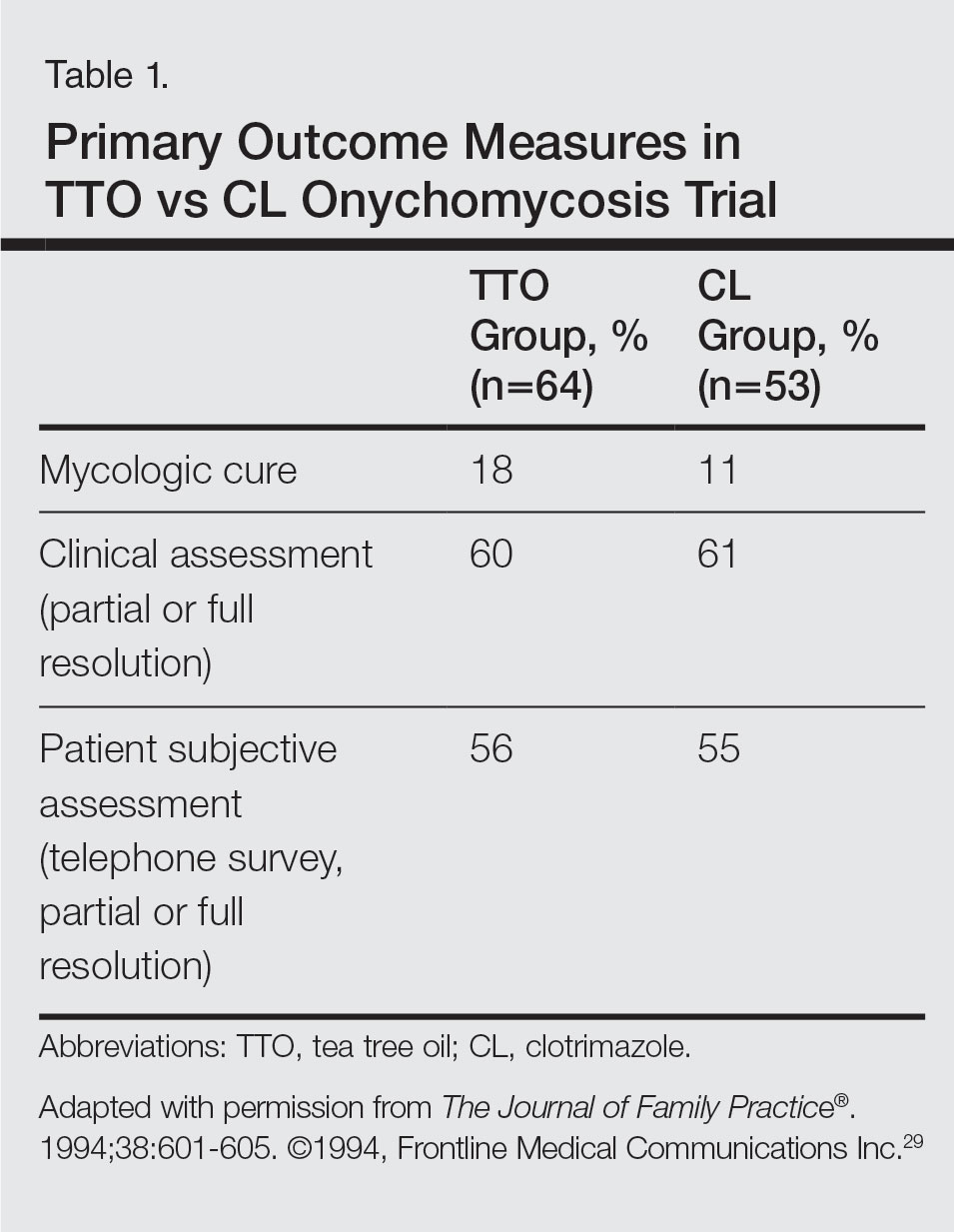
Another study was a double-blind, placebo-controlled trial involving 60 patients with clinical and mycologic evidence of DLSO who were randomized to treatment with a cream containing butenafine hydrochloride 2% and TTO 5% (n=40) or a control cream containing only TTO (n=20), with active treatment for 8 weeks and final follow-up at 36 weeks.30 Patients were instructed to apply the cream 3 times daily under occlusion for 8 weeks and the nail was debrided between weeks 4 and 6 if feasible. If the nail could not be debrided after 8 weeks, it was considered resistant to treatment. At the end of the study, the complete cure rate was 80% in the active group compared to 0% in the placebo group (P<.0001), and the mean time to complete healing with progressive nail growth was 29 weeks. There were no adverse effects in the placebo group, but 4 patients in the active group had mild skin inflammation.30
Topical Cough Suppressant
Background
Topical cough suppressants, which are made up of several natural ingredients, are OTC ointments for adults and children 2 years and older that are indicated as cough suppressants when applied to the chest and throat and as relief of mild muscle and joint pains.35 The active ingredients are camphor 4.8%, eucalyptus oil 1.2%, and menthol 2.6%, while the inactive ingredients are cedarleaf oil, nutmeg oil, petrolatum, thymol, and turpentine oil.35 Some of the active and inactive ingredients in TCSs have shown efficacy against dermatophytes in vitro,36-38 and although they are not specifically indicated for onychomycosis, they have been popularized as home remedies for fungal nail infections.36,39 A TCS has been evaluated for its efficacy for the treatment of onychomycosis in one clinical trial.40
In Vitro Data
An in vitro study was performed to evaluate the antifungal activity of the individual and combined components of TCS on 16 different dermatophytes, nondermatophytes, and molds. The zones of inhibition against these organisms were greatest for camphor, menthol, thymol, and eucalyptus oil. Interestingly, there were large zones of inhibition and a synergistic effect when a mixture of components was used against T rubrum and T mentagrophytes.36 The in vitro activity of thymol, a component of TCS, was tested against Candida species.37 The essential oil subtypes Thymus vulgaris and Thymus zygis (subspecies zygis) showed similar antifungal activity, which was superior to Thymus mastichina, and all 3 compounds had similar MIC and minimal lethal concentration values. The authors showed that the antifungal mechanism was due to cell membrane damage and inhibition of germ tube formation.37 It should be noted that Candida species are less common causes of onychomycosis, and it is not known whether this data is applicable to T rubrum. In another study, the authors investigated the antifungal activity of Thymus pulegioides and found that MIC ranged from 0.16 to 0.32 μL/mL for dermatophytes and Aspergillus strains and 0.32 to 0.64 μL/mL for Candida species. When an essential oil concentration of 0.08 μL/mL was used against T rubrum, ergosterol content decreased by 70 %, indicating that T pulegioides inhibits ergosterol biosynthesis in T rubrum.38
Clinical Observations and Clinical Trial
There is one report documenting the clinical observations on a group of patients with a clinical diagnosis of onychomycosis who were instructed to apply TCS to affected nail(s) once daily.36 Eighty-five charts were reviewed (mean age, 77 years), and although follow-up was not complete or standardized, the following data were reported: 32 (38%) cleared their fungal infection, 21 (25%) had no record of change but also no record of compliance, 19 (22%) had only 1 documented follow-up visit, 9 (11%) reported they did not use the treatment, and 4 (5%) did not return for a follow-up visit. Of the 32 patients whose nails were cured, 3 (9%) had clearance within 5 months, 8 (25%) within 7 months, 11 (34%) within 9 months, 4 (13%) within 11 months, and 6 (19%) within 16 months.36
A small pilot study was performed to evaluate the efficacy of daily application of TCS in the treatment of onychomycosis in patients 18 years and older with at least 1 great toenail affected.40 The primary end points were mycologic cure at 48 weeks and clinical cure at the end of the study graded as complete, partial, or no change. The secondary end point was patient satisfaction with the appearance of the affected nail at 48 weeks. Eighteen participants completed the study; 55% (10/18) were male, with an average age of 51 years (age range, 30–85 years). The mean initial amount of affected nail was 62% (range, 16%–100%), and cultures included dermatophytes, nondermatophytes, and molds. With TCS treatment, 27.8% (5/18) showed mycologic cure of which 4 (22.2%) had a complete clinical cure. Ten participants (55.6%) had partial clinical cure and 3 (16.7%) had no clinical improvement. Interestingly, the 4 participants who had complete clinical cure had baseline cultures positive for either T mentagrophytes or C parapsilosis. Most patients were content with the treatment, as 9 participants stated that they were very satisfied and 9 stated that they were satisfied. The average ratio of affected to total nail area declined from 63% at screening to 41% at the end of the study (P<.001). No adverse effects were reported with study drug.40
NCR Lacquer
Background
Resins are natural products derived from coniferous trees and are believed to protect trees against insects and microbial pathogens.41 Natural coniferous resin derived from the Norway spruce tree (Picea abies) mixed with boiled animal fat or butter has been used topically for centuries in Finland and Sweden to treat infections and wounds.42-44 The activity of NCR has been studied against a wide range of microbes, demonstrating broad-spectrum antimicrobial activity against both gram-positive bacteria and fungi.45-48 There are 2 published clinical trials evaluating NCR in the treatment of onychomycosis.49,50
In Vitro Data
Natural coniferous resin has shown antifungal activity against T mentagrophytes, Trichophyton tonsurans, and T rubrum in vitro, which was demonstrated using medicated disks of resin on petri dishes inoculated with these organisms.46 In another study, the authors evaluated the antifungal activity of NCR against human pathogenic fungi and yeasts using agar plate diffusion tests and showed that the resin had antifungal activity against Trichophyton species but not against Fusarium and most Candida species. Electron microscopy of T mentagrophytes exposed to NCR showed that all cells were dead inside the inhibition zone, with striking changes seen in the hyphal cell walls, while fungal cells outside the inhibition zone were morphologically normal.47 In another report, utilizing the European Pharmacopoeia challenge test, NCR was highly effective against gram-positive and gram-negative bacteria as well as C albicans.42
Clinical Trials
In one preliminary observational and prospective clinical trial, 15 participants with clinical and mycologic evidence of onychomycosis were instructed to apply NCR lacquer once daily for 9 months with a 4-week washout period, with the primary outcome measures being clinical and mycologic cure.49 Thirteen (87%) enrolled participants were male and the average age was 65 years (age range, 37–80 years). The DLSO subtype was present in 9 (60%) participants. The mycologic cure rate at the end of the study was 65% (95% CI, 42%-87%), and none achieved clinical cure, but 6 participants showed some improvement in the appearance of the nail.49
The second trial was a prospective, controlled, investigator-blinded study of 73 patients with clinical and mycologic evidence of toenail onychomycosis who were randomized to receive NCR 30%, amorolfine lacquer 5%, or 250 mg oral terbinafine.50 The primary end point was mycologic cure at 10 months, and secondary end points were clinical efficacy, cost-effectiveness, and patient compliance. Clinical efficacy was based on the proximal linear growth of healthy nail and was classified as unchanged, partial, or complete. Partial responses were described as substantial decreases in onycholysis, subungual hyperkeratosis, and streaks. A complete response was defined as a fully normal appearance of the toenail. Most patients were male in the NCR (91% [21/23]), amorolfine (80% [20/25]), and terbinafine (68% [17/25]) groups; the average ages were 64, 63, and 64 years, respectively. Trichophyton rubrum was cultured most often in all 3 groups: NCR, 87% (20/23); amorolfine, 96% (24/25); and terbinafine, 84% (21/25). The remaining cases were from T mentagrophytes. A summary of the results is shown in Table 2. Patient compliance was 100% in all except 1 patient in the amorolfine treatment group with moderate compliance. There were no adverse events, except for 2 in the terbinafine group: diarrhea and rash.50
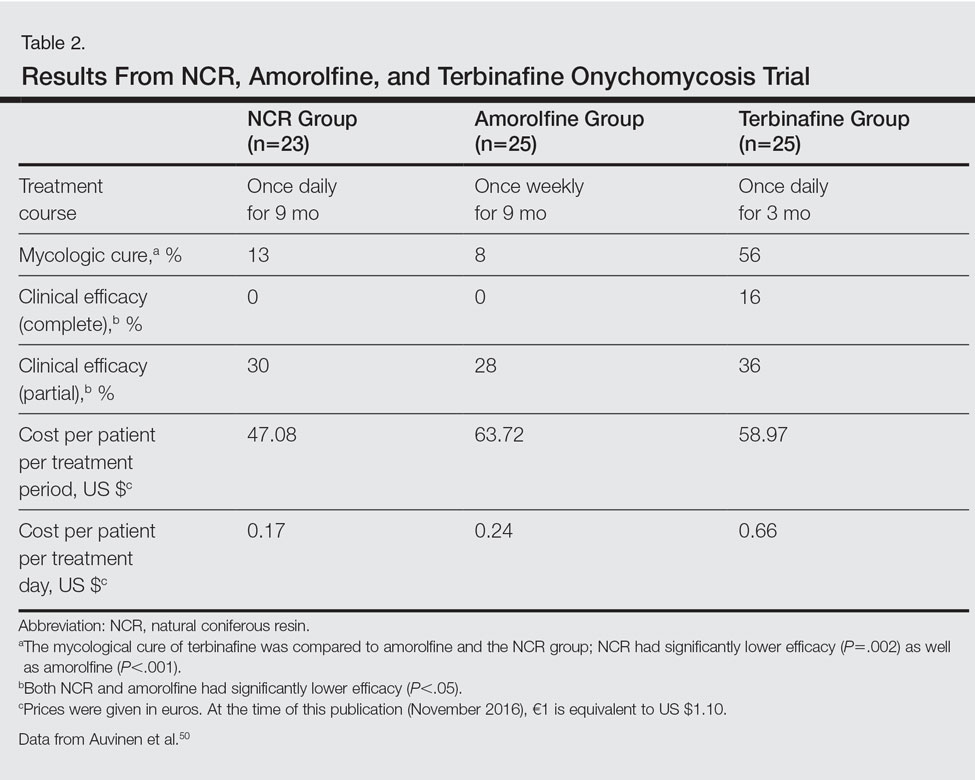
AP Extract
Background
Ageratina pichinchensis, a member of the Asteraceae family, has been used historically in Mexico for fungal infections of the skin.51,52 Fresh or dried leaves were extracted with alcohol and the product was administered topically onto damaged skin without considerable skin irritation.53 Multiple studies have demonstrated that AP extract has in vitro antifungal activity along with other members of the Asteraceae family.54-56 There also is evidence from clinical trials that AP extract is effective against superficial dermatophyte infections such as tinea pedis.57 Given the positive antifungal in vitro data, the potential use of this agent was investigated for onychomycosis treatment.53,58
In Vitro Data
The antifungal properties of the Asteraceae family have been tested in several in vitro experiments. Eupatorium aschenbornianum, described as synonymous with A pichinchensis,59 was found to be most active against the dermatophytes T rubrum and T mentagrophytes with MICs of 0.3 and 0.03 mg/mL, respectively.54 It is thought that the primary antimycotic activity is due to encecalin, an acetylchromene compound that was identified in other plants from the Asteraceae family and has activity against dermatophytes.55 In another study, Ageratum houstanianum Mill, a comparable member of the Asteraceae family, had fungitoxic activity against T rubrum and C albicans isolated from nail infections.56
Clinical Trials
A double-blind controlled trial was performed on 110 patients with clinical and mycologic evidence of mild to moderate toenail onychomycosis randomized to treatment with AP lacquer or ciclopirox lacquer 8% (control).58 Primary end points were clinical effectiveness (completely normal nails) and mycologic cure. Patients were instructed to apply the lacquer once every third day during the first month, twice a week for the second month, and once a week for 16 weeks, with removal of the lacquer weekly. Demographics were similar between the AP lacquer and control groups, with mean ages of 44.6 and 46.5 years, respectively; women made up 74.5% and 67.2%, respectively, of each treatment group, with most patients having a 2- to 5-year history of disease (41.8% and 40.1%, respectively).58 A summary of the data is shown in Table 3. No severe side effects were documented, but minimal nail fold skin pain was reported in 3 patients in the control group in the first week, resolving later in the trial.58
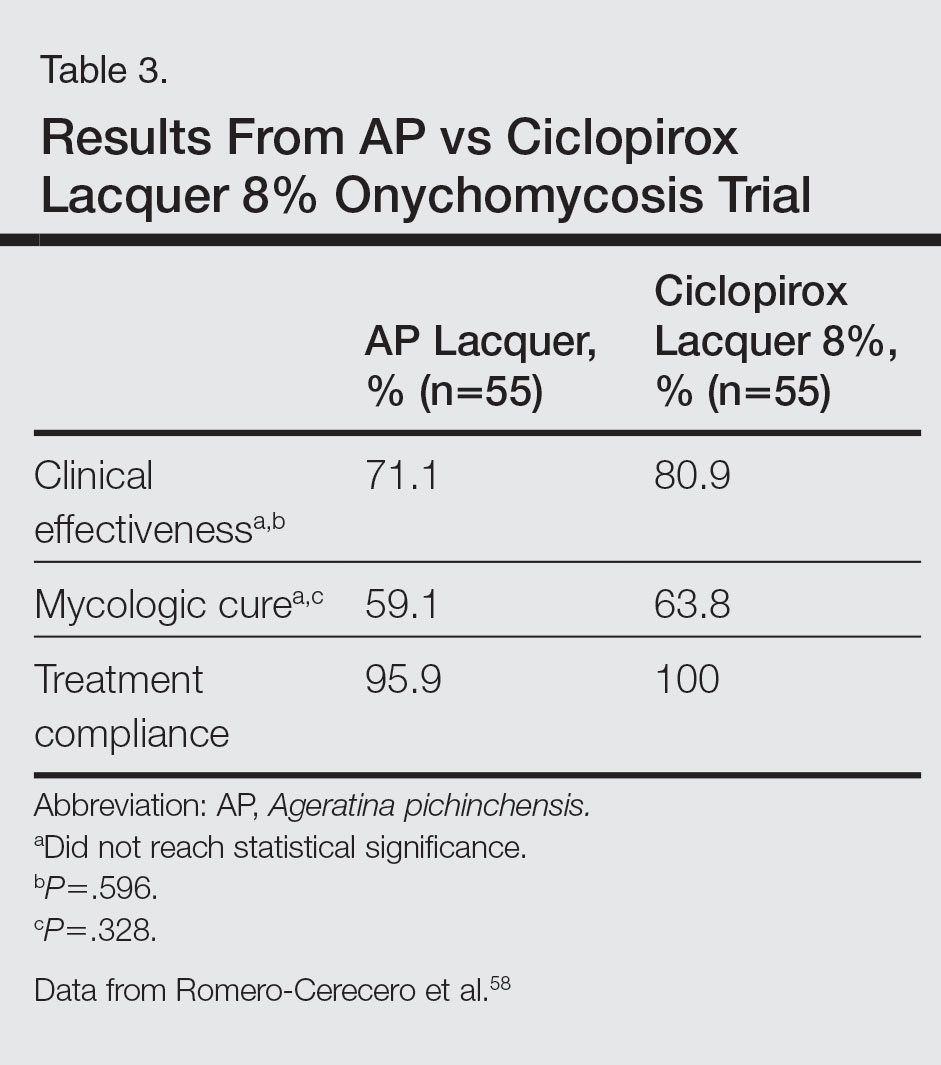
A follow-up study was performed to determine the optimal concentration of AP lacquer for the treatment of onychomycosis.53 One hundred twenty-two patients aged 19 to 65 years with clinical and mycologic evidence of mild to moderate DLSO were randomized to receive 12.6% or 16.8% AP lacquer applied once daily to the affected nails for 6 months. The nails were graded as healthy, mild, or moderately affected before and after treatment. There were no significant differences in demographics between the 2 treatment groups, and 77% of patients were women with a median age of 47 years. There were no significant side effects from either concentration of AP lacquer.53
Ozonized Sunflower Oil
Background
Ozonized sunflower oil is derived by reacting ozone (O3) with sunflower plant (Helianthus annuus) oil to form a petroleum jelly–like material.60 It was originally shown to have antibacterial properties in vitro,61 and further studies have confirmed these findings and demonstrated anti-inflammatory, wound healing, and antifungal properties.62-64 A formulation of ozonized sunflower oil used in Cuba is clinically indicated for the treatment of tinea pedis and impetigo.65 The clinical efficacy of this product has been evaluated in a clinical trial for the treatment of onychomycosis.65
In Vitro Data
A compound made up of 30% ozonized sunflower oil with 0.5% of α-lipoic acid was found to have antifungal activity against C albicans using the disk diffusion method, in addition to other bacterial organisms. The MIC values ranged from 2.0 to 3.5 mg/mL.62 Another study was designed to evaluate the in vitro antifungal activity of this formulation on samples cultured from patients with onychomycosis using the disk diffusion method. They found inhibition of growth of C albicans, C parapsilosis, and Candida tropicalis, which was inferior to amphotericin B, ketoconazole, fluconazole, and itraconazole.64
Clinical Trial
A single-blind, controlled, phase 3 study was performed on 400 patients with clinical and mycologic evidence of onychomycosis. Patients were randomized to treatment with an ozonized sunflower oil solution or ketoconazole cream 2% applied to affected nails twice daily for 3 months, with filing and massage of the affected nails upon application of treatment.65 Cured was defined as mycologic cure in addition to a healthy appearing nail, improved as an increase in healthy appearing nail in addition to a decrease in symptoms (ie, paresthesia, pain, itching) but positive mycological testing, same as no clinical change in appearance with positive mycological findings, and worse as increasing diseased nail involvement in the presence of positive mycological findings. Demographics were similar between groups with a mean age of 35 years. Men accounted for 80% of the study population, and 65% of the study population was white. The mean duration of disease was 30 months. They also reported on a 1-year follow-up, with 2.8% of patients in the ozonized sunflower oil solution group and 37.0% of patients in the ketoconazole group describing relapses. Trichophyton rubrum and C albicans were cultured from these patients.65
Comment
Due to the poor efficacy, long-term treatment courses, inability to use nail polish, and high cost associated with many FDA-approved topical treatments, along with the systemic side effects, potential for drug-drug interactions, and cost associated with many oral therapies approved for onychomycosis, there has been a renewed interest in natural remedies and OTC treatments. Overall, TTO, TCS, NCR, AP extract, and ozonized sunflower oil have shown efficacy in vitro against some dermatophytes, nondermatophytes, and molds responsible for onychomycosis. One or more clinical trials were performed with each of these agents for the treatment of onychomycosis. They were mostly small pilot studies, and due to differences in trial design, the results cannot be compared with each other or with currently FDA-approved treatments. We can conclude that because adverse events were rare with all of these therapies—most commonly skin irritation or mild skin pain—they exhibit good safety.
For TTO, there was no statistical difference between the clotrimazole and TTO treatment groups in mycologic cure, clinical assessment, or patient subjective assessment of the nails.29 Although there was an 80% complete cure in the butenafine and TTO group, it was 0% in the TTO group at week 36.30 Trial design, longer treatment periods, incorporation into nanocapsules, or combination treatment with other antifungal agents may influence our future use of TTO for onychomycosis, but based on the present data we cannot recommend this treatment in clinical practice.
With TCS, 27.8% of participants had a mycologic cure and 22.2% had complete clinical cure.40 Although it is difficult to draw firm conclusions from this small pilot study, there may be some benefit to treating toenail onychomycosis due to T mentagrophytes or C parapsilosis with TCS but no benefit in treating onychomycosis due to T rubrum, the more common cause of onychomycosis. Limitations of this study were lack of a placebo group, small sample size, wide variety of represented pathogens that may not be representative of the true population, and lack of stratification by baseline severity or involvement of nail. A larger randomized controlled clinical trial would be necessary to confirm the results of this small study and make formal recommendations.
In one clinical trial with NCR, mycologic cure was 65% at the end of the study.49 No participants achieved clinical cure, but 6 participants showed some improvement in the appearance of the nail. Because this study was small (N=15), it is difficult to draw firm conclusions.49 In another study with NCR, mycologic cure rates with NCR, amorolfine, and terbinafine were 13%, 8%, and 56%, respectively. Based on these results, NCR has similar antifungal efficacy to amorolfine but was inferior to oral terbinafine.50 A larger randomized controlled clinical trial with more homogenous and less severely affected patients and longer treatment periods would be necessary to confirm the results of these small studies and make formal recommendations.
Because there were no significant differences in clinical effectiveness of mycologic cure rates between AP lacquer 10% and ciclopirox lacquer 8% in one clinical trial,58 AP does not seem to be more effective than at least one of the current FDA-approved topical treatments; however, because AP lacquer 16.8% was shown to be more effective than AP lacquer 12.6% in one onychomycosis clinical trial, using higher concentrations of AP may yield better results in future trials.53
One trial comparing ozonized sunflower oil to ketoconazole cream 2% showed 90.5% and 13.5% cure rates, respectively.65 Although there is good in vitro antifungal activity and a clinical trial showing efficacy using ozonized sunflower oil for the treatment of onychomycosis, confirmatory studies are necessary before we can recommend this OTC treatment to our patients. Specifically, we will get the most data from large randomized controlled trials with strict inclusion/exclusion and efficacy criteria.
Conclusion
Over-the-counter and natural remedies may be an emerging area of research in the treatment of onychomycosis. This review summarizes the laboratory data and clinical trials on several of these agents and, when available, compares their clinical and mycologic efficacy with FDA-approved therapies. Shortcomings of some of these studies include a small study population, lack of adequate controls, nonstandardized mycologic testing, and abbreviated posttreatment evaluation times. It may be concluded that these products have varying degrees of efficacy and appear to be safe in the studies cited; however, at present, we cannot recommend any of them to our patients until there are larger randomized clinical trials with appropriate controls demonstrating their efficacy.
- Scher RK, Daniel CR. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005.
- Sigurgeirsson B, Baran R. The prevalence of onychomycosis in the global population: a literature study. J Eur Acad Dermatol Venereol. 2014;28:1480-1491.
- Thomas J, Jacobson GA, Narkowicz CK, et al. Toenail onychomycosis: an important global disease burden. J Clin Pharm Ther. 2010;35:497-519.
- Mayser P, Freund V, Budihardja D. Toenail onychomycosis in diabetic patients: issues and management. Am J Clin Dermatol. 2009;10:211-220.
- Ghannoum MA, Hajjeh RA, Scher R, et al. A large-scale North American study of fungal isolates from nails: the frequency of onychomycosis, fungal distribution, and antifungal susceptibility patterns. J Am Acad Dermatol. 2000;43:641-648.
- Hay RJ, Baran R. Onychomycosis: a proposed revision of the clinical classification J Am Acad Dermatol. 2011;65:1219-1227.
- Elewski B. Clinical pearl: proximal white subungual onychomycosis in AIDS. J Am Acad Dermatol. 1993;29:631-632.
- Scher RK. Onychomycosis is more than a cosmetic problem. Br J Dermatol. 1994;130(suppl 43):15.
- Boyko EJ, Ahroni JH, Cohen V, et al. Prediction of diabetic foot ulcer occurrence using commonly available clinical information: the Seattle Diabetic Foot Study. Diabetes Care. 2006;29:1202-1207.
- Szepietowski JC, Reich A, Pacan P, et al. Evaluation of quality of life in patients with toenail onychomycosis by Polish version of an international onychomycosis-specific questionnaire. J Eur Acad Dermatol Venereol. 2007;21:491-496.
- Scher RK, Baron R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
- Lamisil [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2011.
- Sporanox [package insert]. Raritan, NJ: Ortho-McNeil-Janssen Pharmaceuticals, Inc; 2001
- Penlac [package insert]. Bridgewater, NJ: Dermik Laboratories; 2006.
- Jublia [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals North America LLC; 2014.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Kerydin [package insert]. Palo Alto, CA: Anacor Pharmaceuticals, Inc; 2014
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies [published online May 5, 2015]. J Am Acad Dermatol. 2015;73:62-69.
- D’Auria FD, Laino L, Strippoli V, et al. In vitro activity of tea tree oil against Candida albicans mycelial conversion and other pathogenic fungi. J Chemother. 2001;13:377-383.
- Satchell AC, Saurajen A, Bell C, et al. Treatment of interdigital tinea pedis with 25% and 50% tea tree oil solution: a randomized, placebo-controlled, blinded study. Australas J Dermatol. 2002;43:175-178.
- Carson CF, Hammer KA, Riley TV. Melaleuca alternifolia (tea tree) oil: a review of antimicrobial and other medicinal properties. Clin Microbiol Rev. 2006;19:50-62.
- Hammer KA, Carson CF, Riley TV. Antifungal activity of the components of Melaleuca alternifolia (tea tree) oil. J Appl Microbiol. 2003;95:853-860.
- Brophy JJ, Davies NW, Southwell IA, et al. Gas chromatographic quality control for oil of Melaleuca terpinen-4-ol type (Australian tea tree). J Agric Food Chem. 1989;37:1330-1335.
- Concha JM, Moore LS, Holloway WJ. 1998 William J. Stickel Bronze Award. Antifungal activity of Melaleuca alternifolia (tea-tree) oil against various pathogenic organisms. J Am Podiatr Med Assoc. 1998;88:489-492.
- Benger S, Townsend P, Ashford RL, et al. An in vitro study to determine the minimum inhibitory concentration of Melaleuca alternifolia against the dermatophyte Trichophyton rubrum. Foot. 2004;14:86-91.
- Hammer KA, Carson CF, Riley TV. In-vitro activity of essential oils, in particular Melaleuca alternifolia (tea tree) oil and tea tree oil products, against Candida spp. J Antimicrob Chemother. 1998;42:591-595.
- Altman P. Australian tea tree oil. Aust J Pharm. 1998;69:276-278.
- Guterres SS, Alves MP, Pohlmann AR. Polymeric nanoparticles, nanospheres and nanocapsules, for cutaneous applications. Drug Target Insights. 2007;2:147-157.
- Buck DS, Nidorf DM, Addino JG. Comparison of two topical preparations for the treatment of onychomycosis: Melaleuca alternifolia (tea tree) oil and clotrimazole. J Fam Pract. 1994;38:601-605.
- Syed TA, Qureshi ZA, Ali SM, et al. Treatment of toenail onychomycosis with 2% butenafine and 5% Melaleuca alternifolia (tea tree) oil in cream. Tropical Med Int Health. 1999;4:284-287.
- Flores FC, de Lima JA, Ribeiro RF, et al. Antifungal activity of nanocapsule suspensions containing tea tree oil on the growth of Trichophyton rubrum. Mycopathologia. 2013;175:281-286.
- Hammer KA, Carson CF, Riley TV. Antifungal effects of Melaleuca alternifolia (tea tree) oil and its components on Candida albicans, Candida glabrata and Saccharomyces cerevisiae. J Antimicrob Chemother. 2004;53:1081-1085.
- Cox SD, Mann CM, Markham JL, et al. The mode of antimicrobial action of the essential oil of Melaleuca alternifolia (tea tree oil). J Appl Microbiol. 2000;88:170-175.
- Hammer KA, Carson CF, Riley TV. Melaleuca alternifolia (tea tree) oil inhibits germ tube formation by Candida albicans. Med Mycol. 2000;38:355-362.
- Vicks VapoRub [package insert]. Gross-Gerau, Germany: Proctor & Gamble; 2010.
- Ramsewak RS, Nair MG, Stommel M, et al. In vitro antagonistic activity of monoterpenes and their mixtures against ‘toe nail fungus’ pathogens. Phytother Res. 2003;17:376-379.
- Pina-Vaz C, Gonçalves Rodrigues A, Pinto E, et al. Antifungal activity of Thymus oils and their major compounds. J Eur Acad Dermatol Venereol. 2004;18:73-78.
- Pinto E, Pina-Vaz C, Salgueiro L, et al. Antifungal activity of the essential oil of Thymus pulegioides on Candida, Aspergillus and dermatophyte species. J Med Microbiol. 2006;55:1367-1373.
- Vicks VapoRub might help fight toenail fungus. Consumer Reports. 2006;71:49.
- Derby R, Rohal P, Jackson C, et al. Novel treatment of onychomycosis using over-the-counter mentholated ointment: a clinical case series. J Am Board Fam Med. 2011;24:69-74.
- Trapp S, Croteau R. Defensive resin biosynthesis in conifers. Ann Rev Plant Physiol Plant Mol Biol. 2001;52:689-724.
- Sipponen A, Laitinen K. Antimicrobial properties of natural coniferous rosin in the European Pharmacopoeia challenge test. APMIS. 2011;119:720-724.
- Sipponen A, Lohi J. Lappish gum care “new” treatment of pressure ulcers? People’s improvement at it’s best. Eng Med J. 2003;58:2775-2776.
- Benedictus O. Een Nyttigh Läkare. Malmö: Kroon; 1938.
- Rautio M, Sipponen A, Peltola R, et al. Antibacterial effects of home-made resin salve from Norway spruce (Picea abies). APMIS. 2007;115:335-340.
- Laitinen K, Sipponen A, Jokinen JJ, et al. Resin salve from Norway spruce is antifungal against dermatophytes causing nail infections. EWMA. 2009;56:289-296.
- Rautio M, Sipponen A, Lohi J, et al. In vitro fungistatic effects of natural coniferous resin from Norway spruce (Picea abies). Eur J Clin Microbiol Infect Dis. 2012;31:1783-1789.
- Sipponen A, Peltola R, Jokinen JJ, et al. Effects of Norway spruce (Picea abies) resin on cell wall and cell membrane of Staphylococcus aureus. Ultrastruct Pathol. 2009;33:128-135.
- Sipponen P, Sipponen A, Lohi J, et al. Natural coniferous resin lacquer in treatment of toenail onychomycosis: an observational study. Mycoses. 2013;56:289-296.
- Auvinen T, Tiihonen R, Soini M, et al. Efficacy of topical resin lacquer, amorolfine, and oral terbinafine for treating toenail onychomycosis: a prospective, randomized, controlled, investigator-blinded, parallel-group clinical trial. Br J Dermatol. 2015;173:940-948.
- Argueta A, Cano L, Rodarte M. Atlas de las Plantas de la Medicina Tradicional Mexicana. Vol 3. Mexico City, Mexico: Instituto Nacional Indigenista; 1994:72-680.
- Avilés M, Suárez G. Catálogo de Plantas Medicinales del Jardín Etnobotánico. Peru: Instituto Nacional de Antropología e Historia; 1994.
- Romero-Cerecero O, Roman-Ramos R, Zamilpa A, et al. Clinical trial to compare the effectiveness of two concentrations of the Ageratina pichinchensis extract in the topical treatment of onychomycosis. J Ethnopharmacol. 2009;126:74-78.
- Navarro Garcia VM, Gonzalez A, Fuentes M, et al. Antifungal activities of nine traditional Mexican medicinal plants. J Ethnopharmacol. 2003;87:85-88.
- Castañeda P, Gómez L, Mata R, et al. Phytogrowth-inhibitory and antifungal constituents of Helianthella quinquenervis. J Nat Prod. 1996;59:323-326.
- Kumar N. Inhibition of nail infecting fungi of peoples of North Eastern UP causing Tinea unguium through leaf essential oil of Ageratum houstonianum Mill. IOSR J Pharm. June 2014;4:36-42.
- Romero-Cerecero O, Rojas G, Navarro V, et al. Effectiveness and tolerability of a standardized extract from Ageratina pichinchensis on patients with tinea pedis: an explorative pilot study controlled with ketoconazole. Planta Med. 2006;72:1257-1261.
- Romero-Cerecero O, Zamilpa A, Jimenez-Ferrer JE, et al. Double-blind clinical trial for evaluating the effectiveness and tolerability of Ageratina pichinchensis extract on patients with mild to moderate onychomycosis. a comparative study with ciclopirox. Planta Med. 2008;74:1430-1435.
- Rzedowski J, De Rzedowski GC. Flora Fanerogámica del Valle de México. Mexico City, Mexico: Instituto de Ecología Escuela Nacional de Ciencias Biológicas del Instituto Politécnico Nacional; 1985.
- Bocci V. Biological and clinical effects of ozone. has ozone therapy a future in medicine? Br J Biomed Sci. 1999;56:270-279.
- Sechi LA, Lezcano I, Nunez N, et al. Antibacterial activity of ozonized sunflower oil (Oleozon). J Appl Microbiol. 2001;90:279-284.
- Rodrigues KL, Cardoso CC, Caputo LR, et al. Cicatrizing and antimicrobial properties of an ozonised oil from sunflower seeds. Inflammopharmacology. 2004;12:261-270.
- Daud FV, Ueda SMY, Navarini A, et al. The use of ozonized oil in the treatment of dermatophitosis caused by Microsporum canis in rabbits. Braz J Microbiol. 2011;42:274-281.
- Guerrer LV, Cunha KC, Nogueira MC, et al. “In vitro” antifungal activity of ozonized sunflower oil on yeasts from onychomycosis. Braz J Microbiol. 2012;43:1315-1318.
- Menéndez S, Falcón L, Maqueira Y. Therapeutic efficacy of topical OLEOZON in patients suffering from onychomycosis. Mycoses. 2011;54:E272-E277.
Onychomycosis is a fungal infection of the nail unit by dermatophytes, yeasts, and nondermatophyte molds. It is characterized by a white or yellow discoloration of the nail plate; hyperkeratosis of the nail bed; distal detachment of the nail plate from its bed (onycholysis); and nail plate dystrophy, including thickening, crumbling, and ridging. Onychomycosis is an important problem, representing 30% of all superficial fungal infections and an estimated 50% of all nail diseases.1 Reported prevalence rates of onychomycosis in the United States and worldwide are varied, but the mean prevalence based on population-based studies in Europe and North America is estimated to be 4.3%.2 It is more common in older individuals, with an incidence rate of 20% in those older than 60 years and 50% in those older than 70 years.3 Onychomycosis is more common in patients with diabetes and 1.9 to 2.8 times higher than the general population.4 Dermatophytes are responsible for the majority of cases of onychomycosis, particularly Trichophyton rubrum and Trichophyton mentagrophytes.5
Onychomycosis is divided into different subtypes based on clinical presentation, which in turn are characterized by varying infecting organisms and prognoses. The subtypes of onychomycosis are distal and lateral subungual (DLSO), proximal subungual, superficial, endonyx, mixed pattern, total dystrophic, and secondary. Distal and lateral subungual onychomycosis are by far the most common presentation and begins when the infecting organism invades the hyponychium and distal or lateral nail bed. Trichophyton rubrum is the most common organism and T mentagrophytes is second, but Candida parapsilosis and Candida albicans also are possibilities. Proximal subungual onychomycosis is far less frequent than DLSO and is usually caused by T rubrum. The fungus invades the proximal nail folds and penetrates the newly growing nail plate.6 This pattern is more common in immunosuppressed patients and should prompt testing for human immunodeficiency virus.7 Total dystrophic onychomycosis is the end stage of fungal nail plate invasion, may follow DLSO or proximal subungual onychomycosis, and is difficult to treat.6
Onychomycosis causes pain, paresthesia, and difficulty with ambulation.8 In patients with peripheral neuropathy and vascular problems, including diabetes, onychomycosis can increase the risk for foot ulcers, with amputation in severe cases.9 Patients also may present with aesthetic concerns that may impact their quality of life.10
Given the effect on quality of life along with medical risks associated with onychomycosis, a safe and successful treatment modality with a low risk of recurrence is desirable. Unfortunately, treatment of nail fungus is quite challenging for a number of reasons. First, the thickness of the nail and/or the fungal mass may be a barrier to the delivery of topical and systemic drugs at the source of the infection. In addition, the nail plate does not have intrinsic immunity. Also, recurrence after treatment is common due to residual hyphae or spores that were not previously eliminated.11 Finally, many topical medications require long treatment courses, which may limit patient compliance, especially in patients who want to use nail polish for cosmesis or camouflage.
Currently Approved Therapies for Onychomycosis
Several definitions are needed to better interpret the results of onychomycosis clinical trials. Complete cure is defined as a negative potassium hydroxide preparation and negative fungal culture with a completely normal appearance of the nail. Mycological cure is defined as potassium hydroxide microscopy and fungal culture negative. Clinical cure is stated as 0% nail plate involvement but at times is reported as less than 5% and less than 10% involvement.
Terbinafine and itraconazole are the only US Food and Drug Administration (FDA)–approved systemic therapies, and ciclopirox, efinaconazole, and tavaborole are the only FDA-approved topicals. Advantages of systemic agents generally are higher cure rates and shorter treatment courses, thus better compliance. Disadvantages include greater incidence of systemic side effects and drug-drug interactions as well as the need for laboratory monitoring. Pros of topical therapies are low potential for adverse effects, no drug-drug interactions, and no monitoring of blood work. Cons include lower efficacy, long treatment courses, and poor patient compliance.
Terbinafine, an allylamine, taken orally once daily (250 mg) for 12 weeks for toenails and 6 weeks for fingernails currently is the preferred systemic treatment of onychomycosis, with complete cure rates of 38% and 59% and mycological cure rates of 70% and 79% for toenails and fingernails, respectively.12 Itraconazole, an azole, is dosed orally at 200 mg daily for 3 months for toenails, with a complete cure rate of 14% and mycological cure rate of 54%.13 For fingernail onychomycosis only, itraconazole is dosed at 200 mg twice daily for 1 week, followed by a treatment-free period of 3 weeks, and then another 1-week course at thesame dose. The complete cure rate is 47% and the mycological cure is 61% for this pulse regimen.13
Ciclopirox is a hydroxypyridone and the 8% nail lacquer formulation was approved in 1999, making it the first topical medication to gain FDA approval for the treatment of toenail onychomycosis. Based on 2 clinical trials, complete cure rates for toenails are 5.5% and 8.5% and mycological cure rates are 29% and 36% at 48 weeks with removal of residual lacquer and debridement.14 Efinaconazole is an azole and the 10% solution was FDA approved for the treatment of toenail onychomycosis in 2014.15 In 2 clinical trials, complete cure rates were 17.8% and 15.2% and mycological cure rates were 55.2% and 53.4% with once daily toenail application for 48 weeks.16 Tavaborole is a benzoxaborole and the 5% solution also was approved for the treatment of toenail onychomycosis in 2014.17 Two clinical trials reported complete cure rates of 6.5% and 9.1% and mycological cure rates of 31.1% and 35.9% with once daily toenail application for 48 weeks.18
Given the poor efficacy, systemic side effects, potential for drug-drug interactions, long-term treatment courses, and cost associated with current systemic and/or topical treatments, there has been a renewed interest in natural remedies and over-the-counter (OTC) therapies for onychomycosis. This review summarizes the in vitro and in vivo data, mechanisms of action, and clinical efficacy of various natural and OTC agents for the treatment of onychomycosis. Specifically, we summarize the data on tea tree oil (TTO), a popular topical cough suppressant (TCS), natural coniferous resin (NCR) lacquer, Ageratina pichinchensis (AP) extract, and ozonized sunflower oil.
Tea Tree Oil
Background
Tea tree oil is a volatile oil whose medicinal use dates back to the early 20th century when the Bundjabung aborigines of North and New South Wales extracted TTO from the dried leaves of the Melaleuca alternifolia plant and used it to treat superficial wounds.19 Tea tree oil has been shown to be an effective treatment of tinea pedis,20 and it is widely used in Australia as well as in Europe and North America.21 Tea tree oil also has been investigated as an antifungal agent for the treatment of onychomycosis, both in vitro22-28 and in clinical trials.29,30
In Vitro Data
Because TTO is composed of more than 100 active components,23 the antifungal activity of these individual components was investigated against 14 fungal isolates, including C albicans, T mentagrophytes, and Aspergillus species. The minimum inhibitory concentration (MIC) for α-pinene was less than 0.004% for T mentagrophytes and the components with the greatest MIC and minimum fungicidal concentration for the fungi tested were terpinen-4-ol and α-terpineol, respectively.22 The antifungal activity of TTO also was tested using disk diffusion assay experiments with 58 clinical isolates of fungi including C albicans, T rubrum, T mentagrophytes, and Aspergillus niger.24 Tea tree oil was most effective at inhibiting T rubrum followed by T mentagrophytes,24 which are the 2 most common etiologies of onychomycosis.5 In another report, the authors determined the MIC of TTO utilizing 4 different experiments with T rubrum as the infecting organism. Because TTO inhibited the growth of T rubrum at all concentrations greater than 0.1%, they found that the MIC was 0.1%.25 Given the lack of adequate nail penetration of most topical therapies, TTO in nanocapsules (TTO-NC), TTO nanoemulsions, and normal emulsions were tested in vitro for their ability to inhibit the growth of T rubrum inoculated into nail shavings. Colony growth decreased significantly within the first week of treatment, with TTO-NC showing maximum efficacy (P<.001). This study showed that TTO, particularly TTO-NC, was effective in inhibiting the growth of T rubrum in vitro and that using nanocapsule technology may increase nail penetration and bioavailability.31
Much of what we know about TTO’s antifungal mechanism of action comes from experiments involving C albicans. To date, it has not been studied in T rubrum or T mentagrophytes, the 2 most common etiologies of onychomycosis.5 In C albicans, TTO causes altered permeability of plasma membranes,32 dose-dependent alteration of respiration,33 decreased glucose-induced acidification of media surrounding fungi,32 and reversible inhibition of germ tube formation.19,34
Clinical Trials
A randomized, double-blind, multicenter trial was performed on 117 patients with culture-proven DLSO who were randomized to receive TTO 100% or clotrimazole solution 1% applied twice daily to affected toenails for 6 months.29 Primary outcome measures were mycologic cure, clinical assessment, and patient subjective assessment (Table 1). There were no statistical differences between the 2 treatment groups. Erythema and irritation were the most common adverse reactions occurring in 7.8% (5/64) of the TTO group.29
Another study was a double-blind, placebo-controlled trial involving 60 patients with clinical and mycologic evidence of DLSO who were randomized to treatment with a cream containing butenafine hydrochloride 2% and TTO 5% (n=40) or a control cream containing only TTO (n=20), with active treatment for 8 weeks and final follow-up at 36 weeks.30 Patients were instructed to apply the cream 3 times daily under occlusion for 8 weeks and the nail was debrided between weeks 4 and 6 if feasible. If the nail could not be debrided after 8 weeks, it was considered resistant to treatment. At the end of the study, the complete cure rate was 80% in the active group compared to 0% in the placebo group (P<.0001), and the mean time to complete healing with progressive nail growth was 29 weeks. There were no adverse effects in the placebo group, but 4 patients in the active group had mild skin inflammation.30
Topical Cough Suppressant
Background
Topical cough suppressants, which are made up of several natural ingredients, are OTC ointments for adults and children 2 years and older that are indicated as cough suppressants when applied to the chest and throat and as relief of mild muscle and joint pains.35 The active ingredients are camphor 4.8%, eucalyptus oil 1.2%, and menthol 2.6%, while the inactive ingredients are cedarleaf oil, nutmeg oil, petrolatum, thymol, and turpentine oil.35 Some of the active and inactive ingredients in TCSs have shown efficacy against dermatophytes in vitro,36-38 and although they are not specifically indicated for onychomycosis, they have been popularized as home remedies for fungal nail infections.36,39 A TCS has been evaluated for its efficacy for the treatment of onychomycosis in one clinical trial.40
In Vitro Data
An in vitro study was performed to evaluate the antifungal activity of the individual and combined components of TCS on 16 different dermatophytes, nondermatophytes, and molds. The zones of inhibition against these organisms were greatest for camphor, menthol, thymol, and eucalyptus oil. Interestingly, there were large zones of inhibition and a synergistic effect when a mixture of components was used against T rubrum and T mentagrophytes.36 The in vitro activity of thymol, a component of TCS, was tested against Candida species.37 The essential oil subtypes Thymus vulgaris and Thymus zygis (subspecies zygis) showed similar antifungal activity, which was superior to Thymus mastichina, and all 3 compounds had similar MIC and minimal lethal concentration values. The authors showed that the antifungal mechanism was due to cell membrane damage and inhibition of germ tube formation.37 It should be noted that Candida species are less common causes of onychomycosis, and it is not known whether this data is applicable to T rubrum. In another study, the authors investigated the antifungal activity of Thymus pulegioides and found that MIC ranged from 0.16 to 0.32 μL/mL for dermatophytes and Aspergillus strains and 0.32 to 0.64 μL/mL for Candida species. When an essential oil concentration of 0.08 μL/mL was used against T rubrum, ergosterol content decreased by 70 %, indicating that T pulegioides inhibits ergosterol biosynthesis in T rubrum.38
Clinical Observations and Clinical Trial
There is one report documenting the clinical observations on a group of patients with a clinical diagnosis of onychomycosis who were instructed to apply TCS to affected nail(s) once daily.36 Eighty-five charts were reviewed (mean age, 77 years), and although follow-up was not complete or standardized, the following data were reported: 32 (38%) cleared their fungal infection, 21 (25%) had no record of change but also no record of compliance, 19 (22%) had only 1 documented follow-up visit, 9 (11%) reported they did not use the treatment, and 4 (5%) did not return for a follow-up visit. Of the 32 patients whose nails were cured, 3 (9%) had clearance within 5 months, 8 (25%) within 7 months, 11 (34%) within 9 months, 4 (13%) within 11 months, and 6 (19%) within 16 months.36
A small pilot study was performed to evaluate the efficacy of daily application of TCS in the treatment of onychomycosis in patients 18 years and older with at least 1 great toenail affected.40 The primary end points were mycologic cure at 48 weeks and clinical cure at the end of the study graded as complete, partial, or no change. The secondary end point was patient satisfaction with the appearance of the affected nail at 48 weeks. Eighteen participants completed the study; 55% (10/18) were male, with an average age of 51 years (age range, 30–85 years). The mean initial amount of affected nail was 62% (range, 16%–100%), and cultures included dermatophytes, nondermatophytes, and molds. With TCS treatment, 27.8% (5/18) showed mycologic cure of which 4 (22.2%) had a complete clinical cure. Ten participants (55.6%) had partial clinical cure and 3 (16.7%) had no clinical improvement. Interestingly, the 4 participants who had complete clinical cure had baseline cultures positive for either T mentagrophytes or C parapsilosis. Most patients were content with the treatment, as 9 participants stated that they were very satisfied and 9 stated that they were satisfied. The average ratio of affected to total nail area declined from 63% at screening to 41% at the end of the study (P<.001). No adverse effects were reported with study drug.40
NCR Lacquer
Background
Resins are natural products derived from coniferous trees and are believed to protect trees against insects and microbial pathogens.41 Natural coniferous resin derived from the Norway spruce tree (Picea abies) mixed with boiled animal fat or butter has been used topically for centuries in Finland and Sweden to treat infections and wounds.42-44 The activity of NCR has been studied against a wide range of microbes, demonstrating broad-spectrum antimicrobial activity against both gram-positive bacteria and fungi.45-48 There are 2 published clinical trials evaluating NCR in the treatment of onychomycosis.49,50
In Vitro Data
Natural coniferous resin has shown antifungal activity against T mentagrophytes, Trichophyton tonsurans, and T rubrum in vitro, which was demonstrated using medicated disks of resin on petri dishes inoculated with these organisms.46 In another study, the authors evaluated the antifungal activity of NCR against human pathogenic fungi and yeasts using agar plate diffusion tests and showed that the resin had antifungal activity against Trichophyton species but not against Fusarium and most Candida species. Electron microscopy of T mentagrophytes exposed to NCR showed that all cells were dead inside the inhibition zone, with striking changes seen in the hyphal cell walls, while fungal cells outside the inhibition zone were morphologically normal.47 In another report, utilizing the European Pharmacopoeia challenge test, NCR was highly effective against gram-positive and gram-negative bacteria as well as C albicans.42
Clinical Trials
In one preliminary observational and prospective clinical trial, 15 participants with clinical and mycologic evidence of onychomycosis were instructed to apply NCR lacquer once daily for 9 months with a 4-week washout period, with the primary outcome measures being clinical and mycologic cure.49 Thirteen (87%) enrolled participants were male and the average age was 65 years (age range, 37–80 years). The DLSO subtype was present in 9 (60%) participants. The mycologic cure rate at the end of the study was 65% (95% CI, 42%-87%), and none achieved clinical cure, but 6 participants showed some improvement in the appearance of the nail.49
The second trial was a prospective, controlled, investigator-blinded study of 73 patients with clinical and mycologic evidence of toenail onychomycosis who were randomized to receive NCR 30%, amorolfine lacquer 5%, or 250 mg oral terbinafine.50 The primary end point was mycologic cure at 10 months, and secondary end points were clinical efficacy, cost-effectiveness, and patient compliance. Clinical efficacy was based on the proximal linear growth of healthy nail and was classified as unchanged, partial, or complete. Partial responses were described as substantial decreases in onycholysis, subungual hyperkeratosis, and streaks. A complete response was defined as a fully normal appearance of the toenail. Most patients were male in the NCR (91% [21/23]), amorolfine (80% [20/25]), and terbinafine (68% [17/25]) groups; the average ages were 64, 63, and 64 years, respectively. Trichophyton rubrum was cultured most often in all 3 groups: NCR, 87% (20/23); amorolfine, 96% (24/25); and terbinafine, 84% (21/25). The remaining cases were from T mentagrophytes. A summary of the results is shown in Table 2. Patient compliance was 100% in all except 1 patient in the amorolfine treatment group with moderate compliance. There were no adverse events, except for 2 in the terbinafine group: diarrhea and rash.50
AP Extract
Background
Ageratina pichinchensis, a member of the Asteraceae family, has been used historically in Mexico for fungal infections of the skin.51,52 Fresh or dried leaves were extracted with alcohol and the product was administered topically onto damaged skin without considerable skin irritation.53 Multiple studies have demonstrated that AP extract has in vitro antifungal activity along with other members of the Asteraceae family.54-56 There also is evidence from clinical trials that AP extract is effective against superficial dermatophyte infections such as tinea pedis.57 Given the positive antifungal in vitro data, the potential use of this agent was investigated for onychomycosis treatment.53,58
In Vitro Data
The antifungal properties of the Asteraceae family have been tested in several in vitro experiments. Eupatorium aschenbornianum, described as synonymous with A pichinchensis,59 was found to be most active against the dermatophytes T rubrum and T mentagrophytes with MICs of 0.3 and 0.03 mg/mL, respectively.54 It is thought that the primary antimycotic activity is due to encecalin, an acetylchromene compound that was identified in other plants from the Asteraceae family and has activity against dermatophytes.55 In another study, Ageratum houstanianum Mill, a comparable member of the Asteraceae family, had fungitoxic activity against T rubrum and C albicans isolated from nail infections.56
Clinical Trials
A double-blind controlled trial was performed on 110 patients with clinical and mycologic evidence of mild to moderate toenail onychomycosis randomized to treatment with AP lacquer or ciclopirox lacquer 8% (control).58 Primary end points were clinical effectiveness (completely normal nails) and mycologic cure. Patients were instructed to apply the lacquer once every third day during the first month, twice a week for the second month, and once a week for 16 weeks, with removal of the lacquer weekly. Demographics were similar between the AP lacquer and control groups, with mean ages of 44.6 and 46.5 years, respectively; women made up 74.5% and 67.2%, respectively, of each treatment group, with most patients having a 2- to 5-year history of disease (41.8% and 40.1%, respectively).58 A summary of the data is shown in Table 3. No severe side effects were documented, but minimal nail fold skin pain was reported in 3 patients in the control group in the first week, resolving later in the trial.58
A follow-up study was performed to determine the optimal concentration of AP lacquer for the treatment of onychomycosis.53 One hundred twenty-two patients aged 19 to 65 years with clinical and mycologic evidence of mild to moderate DLSO were randomized to receive 12.6% or 16.8% AP lacquer applied once daily to the affected nails for 6 months. The nails were graded as healthy, mild, or moderately affected before and after treatment. There were no significant differences in demographics between the 2 treatment groups, and 77% of patients were women with a median age of 47 years. There were no significant side effects from either concentration of AP lacquer.53
Ozonized Sunflower Oil
Background
Ozonized sunflower oil is derived by reacting ozone (O3) with sunflower plant (Helianthus annuus) oil to form a petroleum jelly–like material.60 It was originally shown to have antibacterial properties in vitro,61 and further studies have confirmed these findings and demonstrated anti-inflammatory, wound healing, and antifungal properties.62-64 A formulation of ozonized sunflower oil used in Cuba is clinically indicated for the treatment of tinea pedis and impetigo.65 The clinical efficacy of this product has been evaluated in a clinical trial for the treatment of onychomycosis.65
In Vitro Data
A compound made up of 30% ozonized sunflower oil with 0.5% of α-lipoic acid was found to have antifungal activity against C albicans using the disk diffusion method, in addition to other bacterial organisms. The MIC values ranged from 2.0 to 3.5 mg/mL.62 Another study was designed to evaluate the in vitro antifungal activity of this formulation on samples cultured from patients with onychomycosis using the disk diffusion method. They found inhibition of growth of C albicans, C parapsilosis, and Candida tropicalis, which was inferior to amphotericin B, ketoconazole, fluconazole, and itraconazole.64
Clinical Trial
A single-blind, controlled, phase 3 study was performed on 400 patients with clinical and mycologic evidence of onychomycosis. Patients were randomized to treatment with an ozonized sunflower oil solution or ketoconazole cream 2% applied to affected nails twice daily for 3 months, with filing and massage of the affected nails upon application of treatment.65 Cured was defined as mycologic cure in addition to a healthy appearing nail, improved as an increase in healthy appearing nail in addition to a decrease in symptoms (ie, paresthesia, pain, itching) but positive mycological testing, same as no clinical change in appearance with positive mycological findings, and worse as increasing diseased nail involvement in the presence of positive mycological findings. Demographics were similar between groups with a mean age of 35 years. Men accounted for 80% of the study population, and 65% of the study population was white. The mean duration of disease was 30 months. They also reported on a 1-year follow-up, with 2.8% of patients in the ozonized sunflower oil solution group and 37.0% of patients in the ketoconazole group describing relapses. Trichophyton rubrum and C albicans were cultured from these patients.65
Comment
Due to the poor efficacy, long-term treatment courses, inability to use nail polish, and high cost associated with many FDA-approved topical treatments, along with the systemic side effects, potential for drug-drug interactions, and cost associated with many oral therapies approved for onychomycosis, there has been a renewed interest in natural remedies and OTC treatments. Overall, TTO, TCS, NCR, AP extract, and ozonized sunflower oil have shown efficacy in vitro against some dermatophytes, nondermatophytes, and molds responsible for onychomycosis. One or more clinical trials were performed with each of these agents for the treatment of onychomycosis. They were mostly small pilot studies, and due to differences in trial design, the results cannot be compared with each other or with currently FDA-approved treatments. We can conclude that because adverse events were rare with all of these therapies—most commonly skin irritation or mild skin pain—they exhibit good safety.
For TTO, there was no statistical difference between the clotrimazole and TTO treatment groups in mycologic cure, clinical assessment, or patient subjective assessment of the nails.29 Although there was an 80% complete cure in the butenafine and TTO group, it was 0% in the TTO group at week 36.30 Trial design, longer treatment periods, incorporation into nanocapsules, or combination treatment with other antifungal agents may influence our future use of TTO for onychomycosis, but based on the present data we cannot recommend this treatment in clinical practice.
With TCS, 27.8% of participants had a mycologic cure and 22.2% had complete clinical cure.40 Although it is difficult to draw firm conclusions from this small pilot study, there may be some benefit to treating toenail onychomycosis due to T mentagrophytes or C parapsilosis with TCS but no benefit in treating onychomycosis due to T rubrum, the more common cause of onychomycosis. Limitations of this study were lack of a placebo group, small sample size, wide variety of represented pathogens that may not be representative of the true population, and lack of stratification by baseline severity or involvement of nail. A larger randomized controlled clinical trial would be necessary to confirm the results of this small study and make formal recommendations.
In one clinical trial with NCR, mycologic cure was 65% at the end of the study.49 No participants achieved clinical cure, but 6 participants showed some improvement in the appearance of the nail. Because this study was small (N=15), it is difficult to draw firm conclusions.49 In another study with NCR, mycologic cure rates with NCR, amorolfine, and terbinafine were 13%, 8%, and 56%, respectively. Based on these results, NCR has similar antifungal efficacy to amorolfine but was inferior to oral terbinafine.50 A larger randomized controlled clinical trial with more homogenous and less severely affected patients and longer treatment periods would be necessary to confirm the results of these small studies and make formal recommendations.
Because there were no significant differences in clinical effectiveness of mycologic cure rates between AP lacquer 10% and ciclopirox lacquer 8% in one clinical trial,58 AP does not seem to be more effective than at least one of the current FDA-approved topical treatments; however, because AP lacquer 16.8% was shown to be more effective than AP lacquer 12.6% in one onychomycosis clinical trial, using higher concentrations of AP may yield better results in future trials.53
One trial comparing ozonized sunflower oil to ketoconazole cream 2% showed 90.5% and 13.5% cure rates, respectively.65 Although there is good in vitro antifungal activity and a clinical trial showing efficacy using ozonized sunflower oil for the treatment of onychomycosis, confirmatory studies are necessary before we can recommend this OTC treatment to our patients. Specifically, we will get the most data from large randomized controlled trials with strict inclusion/exclusion and efficacy criteria.
Conclusion
Over-the-counter and natural remedies may be an emerging area of research in the treatment of onychomycosis. This review summarizes the laboratory data and clinical trials on several of these agents and, when available, compares their clinical and mycologic efficacy with FDA-approved therapies. Shortcomings of some of these studies include a small study population, lack of adequate controls, nonstandardized mycologic testing, and abbreviated posttreatment evaluation times. It may be concluded that these products have varying degrees of efficacy and appear to be safe in the studies cited; however, at present, we cannot recommend any of them to our patients until there are larger randomized clinical trials with appropriate controls demonstrating their efficacy.
Onychomycosis is a fungal infection of the nail unit by dermatophytes, yeasts, and nondermatophyte molds. It is characterized by a white or yellow discoloration of the nail plate; hyperkeratosis of the nail bed; distal detachment of the nail plate from its bed (onycholysis); and nail plate dystrophy, including thickening, crumbling, and ridging. Onychomycosis is an important problem, representing 30% of all superficial fungal infections and an estimated 50% of all nail diseases.1 Reported prevalence rates of onychomycosis in the United States and worldwide are varied, but the mean prevalence based on population-based studies in Europe and North America is estimated to be 4.3%.2 It is more common in older individuals, with an incidence rate of 20% in those older than 60 years and 50% in those older than 70 years.3 Onychomycosis is more common in patients with diabetes and 1.9 to 2.8 times higher than the general population.4 Dermatophytes are responsible for the majority of cases of onychomycosis, particularly Trichophyton rubrum and Trichophyton mentagrophytes.5
Onychomycosis is divided into different subtypes based on clinical presentation, which in turn are characterized by varying infecting organisms and prognoses. The subtypes of onychomycosis are distal and lateral subungual (DLSO), proximal subungual, superficial, endonyx, mixed pattern, total dystrophic, and secondary. Distal and lateral subungual onychomycosis are by far the most common presentation and begins when the infecting organism invades the hyponychium and distal or lateral nail bed. Trichophyton rubrum is the most common organism and T mentagrophytes is second, but Candida parapsilosis and Candida albicans also are possibilities. Proximal subungual onychomycosis is far less frequent than DLSO and is usually caused by T rubrum. The fungus invades the proximal nail folds and penetrates the newly growing nail plate.6 This pattern is more common in immunosuppressed patients and should prompt testing for human immunodeficiency virus.7 Total dystrophic onychomycosis is the end stage of fungal nail plate invasion, may follow DLSO or proximal subungual onychomycosis, and is difficult to treat.6
Onychomycosis causes pain, paresthesia, and difficulty with ambulation.8 In patients with peripheral neuropathy and vascular problems, including diabetes, onychomycosis can increase the risk for foot ulcers, with amputation in severe cases.9 Patients also may present with aesthetic concerns that may impact their quality of life.10
Given the effect on quality of life along with medical risks associated with onychomycosis, a safe and successful treatment modality with a low risk of recurrence is desirable. Unfortunately, treatment of nail fungus is quite challenging for a number of reasons. First, the thickness of the nail and/or the fungal mass may be a barrier to the delivery of topical and systemic drugs at the source of the infection. In addition, the nail plate does not have intrinsic immunity. Also, recurrence after treatment is common due to residual hyphae or spores that were not previously eliminated.11 Finally, many topical medications require long treatment courses, which may limit patient compliance, especially in patients who want to use nail polish for cosmesis or camouflage.
Currently Approved Therapies for Onychomycosis
Several definitions are needed to better interpret the results of onychomycosis clinical trials. Complete cure is defined as a negative potassium hydroxide preparation and negative fungal culture with a completely normal appearance of the nail. Mycological cure is defined as potassium hydroxide microscopy and fungal culture negative. Clinical cure is stated as 0% nail plate involvement but at times is reported as less than 5% and less than 10% involvement.
Terbinafine and itraconazole are the only US Food and Drug Administration (FDA)–approved systemic therapies, and ciclopirox, efinaconazole, and tavaborole are the only FDA-approved topicals. Advantages of systemic agents generally are higher cure rates and shorter treatment courses, thus better compliance. Disadvantages include greater incidence of systemic side effects and drug-drug interactions as well as the need for laboratory monitoring. Pros of topical therapies are low potential for adverse effects, no drug-drug interactions, and no monitoring of blood work. Cons include lower efficacy, long treatment courses, and poor patient compliance.
Terbinafine, an allylamine, taken orally once daily (250 mg) for 12 weeks for toenails and 6 weeks for fingernails currently is the preferred systemic treatment of onychomycosis, with complete cure rates of 38% and 59% and mycological cure rates of 70% and 79% for toenails and fingernails, respectively.12 Itraconazole, an azole, is dosed orally at 200 mg daily for 3 months for toenails, with a complete cure rate of 14% and mycological cure rate of 54%.13 For fingernail onychomycosis only, itraconazole is dosed at 200 mg twice daily for 1 week, followed by a treatment-free period of 3 weeks, and then another 1-week course at thesame dose. The complete cure rate is 47% and the mycological cure is 61% for this pulse regimen.13
Ciclopirox is a hydroxypyridone and the 8% nail lacquer formulation was approved in 1999, making it the first topical medication to gain FDA approval for the treatment of toenail onychomycosis. Based on 2 clinical trials, complete cure rates for toenails are 5.5% and 8.5% and mycological cure rates are 29% and 36% at 48 weeks with removal of residual lacquer and debridement.14 Efinaconazole is an azole and the 10% solution was FDA approved for the treatment of toenail onychomycosis in 2014.15 In 2 clinical trials, complete cure rates were 17.8% and 15.2% and mycological cure rates were 55.2% and 53.4% with once daily toenail application for 48 weeks.16 Tavaborole is a benzoxaborole and the 5% solution also was approved for the treatment of toenail onychomycosis in 2014.17 Two clinical trials reported complete cure rates of 6.5% and 9.1% and mycological cure rates of 31.1% and 35.9% with once daily toenail application for 48 weeks.18
Given the poor efficacy, systemic side effects, potential for drug-drug interactions, long-term treatment courses, and cost associated with current systemic and/or topical treatments, there has been a renewed interest in natural remedies and over-the-counter (OTC) therapies for onychomycosis. This review summarizes the in vitro and in vivo data, mechanisms of action, and clinical efficacy of various natural and OTC agents for the treatment of onychomycosis. Specifically, we summarize the data on tea tree oil (TTO), a popular topical cough suppressant (TCS), natural coniferous resin (NCR) lacquer, Ageratina pichinchensis (AP) extract, and ozonized sunflower oil.
Tea Tree Oil
Background
Tea tree oil is a volatile oil whose medicinal use dates back to the early 20th century when the Bundjabung aborigines of North and New South Wales extracted TTO from the dried leaves of the Melaleuca alternifolia plant and used it to treat superficial wounds.19 Tea tree oil has been shown to be an effective treatment of tinea pedis,20 and it is widely used in Australia as well as in Europe and North America.21 Tea tree oil also has been investigated as an antifungal agent for the treatment of onychomycosis, both in vitro22-28 and in clinical trials.29,30
In Vitro Data
Because TTO is composed of more than 100 active components,23 the antifungal activity of these individual components was investigated against 14 fungal isolates, including C albicans, T mentagrophytes, and Aspergillus species. The minimum inhibitory concentration (MIC) for α-pinene was less than 0.004% for T mentagrophytes and the components with the greatest MIC and minimum fungicidal concentration for the fungi tested were terpinen-4-ol and α-terpineol, respectively.22 The antifungal activity of TTO also was tested using disk diffusion assay experiments with 58 clinical isolates of fungi including C albicans, T rubrum, T mentagrophytes, and Aspergillus niger.24 Tea tree oil was most effective at inhibiting T rubrum followed by T mentagrophytes,24 which are the 2 most common etiologies of onychomycosis.5 In another report, the authors determined the MIC of TTO utilizing 4 different experiments with T rubrum as the infecting organism. Because TTO inhibited the growth of T rubrum at all concentrations greater than 0.1%, they found that the MIC was 0.1%.25 Given the lack of adequate nail penetration of most topical therapies, TTO in nanocapsules (TTO-NC), TTO nanoemulsions, and normal emulsions were tested in vitro for their ability to inhibit the growth of T rubrum inoculated into nail shavings. Colony growth decreased significantly within the first week of treatment, with TTO-NC showing maximum efficacy (P<.001). This study showed that TTO, particularly TTO-NC, was effective in inhibiting the growth of T rubrum in vitro and that using nanocapsule technology may increase nail penetration and bioavailability.31
Much of what we know about TTO’s antifungal mechanism of action comes from experiments involving C albicans. To date, it has not been studied in T rubrum or T mentagrophytes, the 2 most common etiologies of onychomycosis.5 In C albicans, TTO causes altered permeability of plasma membranes,32 dose-dependent alteration of respiration,33 decreased glucose-induced acidification of media surrounding fungi,32 and reversible inhibition of germ tube formation.19,34
Clinical Trials
A randomized, double-blind, multicenter trial was performed on 117 patients with culture-proven DLSO who were randomized to receive TTO 100% or clotrimazole solution 1% applied twice daily to affected toenails for 6 months.29 Primary outcome measures were mycologic cure, clinical assessment, and patient subjective assessment (Table 1). There were no statistical differences between the 2 treatment groups. Erythema and irritation were the most common adverse reactions occurring in 7.8% (5/64) of the TTO group.29
Another study was a double-blind, placebo-controlled trial involving 60 patients with clinical and mycologic evidence of DLSO who were randomized to treatment with a cream containing butenafine hydrochloride 2% and TTO 5% (n=40) or a control cream containing only TTO (n=20), with active treatment for 8 weeks and final follow-up at 36 weeks.30 Patients were instructed to apply the cream 3 times daily under occlusion for 8 weeks and the nail was debrided between weeks 4 and 6 if feasible. If the nail could not be debrided after 8 weeks, it was considered resistant to treatment. At the end of the study, the complete cure rate was 80% in the active group compared to 0% in the placebo group (P<.0001), and the mean time to complete healing with progressive nail growth was 29 weeks. There were no adverse effects in the placebo group, but 4 patients in the active group had mild skin inflammation.30
Topical Cough Suppressant
Background
Topical cough suppressants, which are made up of several natural ingredients, are OTC ointments for adults and children 2 years and older that are indicated as cough suppressants when applied to the chest and throat and as relief of mild muscle and joint pains.35 The active ingredients are camphor 4.8%, eucalyptus oil 1.2%, and menthol 2.6%, while the inactive ingredients are cedarleaf oil, nutmeg oil, petrolatum, thymol, and turpentine oil.35 Some of the active and inactive ingredients in TCSs have shown efficacy against dermatophytes in vitro,36-38 and although they are not specifically indicated for onychomycosis, they have been popularized as home remedies for fungal nail infections.36,39 A TCS has been evaluated for its efficacy for the treatment of onychomycosis in one clinical trial.40
In Vitro Data
An in vitro study was performed to evaluate the antifungal activity of the individual and combined components of TCS on 16 different dermatophytes, nondermatophytes, and molds. The zones of inhibition against these organisms were greatest for camphor, menthol, thymol, and eucalyptus oil. Interestingly, there were large zones of inhibition and a synergistic effect when a mixture of components was used against T rubrum and T mentagrophytes.36 The in vitro activity of thymol, a component of TCS, was tested against Candida species.37 The essential oil subtypes Thymus vulgaris and Thymus zygis (subspecies zygis) showed similar antifungal activity, which was superior to Thymus mastichina, and all 3 compounds had similar MIC and minimal lethal concentration values. The authors showed that the antifungal mechanism was due to cell membrane damage and inhibition of germ tube formation.37 It should be noted that Candida species are less common causes of onychomycosis, and it is not known whether this data is applicable to T rubrum. In another study, the authors investigated the antifungal activity of Thymus pulegioides and found that MIC ranged from 0.16 to 0.32 μL/mL for dermatophytes and Aspergillus strains and 0.32 to 0.64 μL/mL for Candida species. When an essential oil concentration of 0.08 μL/mL was used against T rubrum, ergosterol content decreased by 70 %, indicating that T pulegioides inhibits ergosterol biosynthesis in T rubrum.38
Clinical Observations and Clinical Trial
There is one report documenting the clinical observations on a group of patients with a clinical diagnosis of onychomycosis who were instructed to apply TCS to affected nail(s) once daily.36 Eighty-five charts were reviewed (mean age, 77 years), and although follow-up was not complete or standardized, the following data were reported: 32 (38%) cleared their fungal infection, 21 (25%) had no record of change but also no record of compliance, 19 (22%) had only 1 documented follow-up visit, 9 (11%) reported they did not use the treatment, and 4 (5%) did not return for a follow-up visit. Of the 32 patients whose nails were cured, 3 (9%) had clearance within 5 months, 8 (25%) within 7 months, 11 (34%) within 9 months, 4 (13%) within 11 months, and 6 (19%) within 16 months.36
A small pilot study was performed to evaluate the efficacy of daily application of TCS in the treatment of onychomycosis in patients 18 years and older with at least 1 great toenail affected.40 The primary end points were mycologic cure at 48 weeks and clinical cure at the end of the study graded as complete, partial, or no change. The secondary end point was patient satisfaction with the appearance of the affected nail at 48 weeks. Eighteen participants completed the study; 55% (10/18) were male, with an average age of 51 years (age range, 30–85 years). The mean initial amount of affected nail was 62% (range, 16%–100%), and cultures included dermatophytes, nondermatophytes, and molds. With TCS treatment, 27.8% (5/18) showed mycologic cure of which 4 (22.2%) had a complete clinical cure. Ten participants (55.6%) had partial clinical cure and 3 (16.7%) had no clinical improvement. Interestingly, the 4 participants who had complete clinical cure had baseline cultures positive for either T mentagrophytes or C parapsilosis. Most patients were content with the treatment, as 9 participants stated that they were very satisfied and 9 stated that they were satisfied. The average ratio of affected to total nail area declined from 63% at screening to 41% at the end of the study (P<.001). No adverse effects were reported with study drug.40
NCR Lacquer
Background
Resins are natural products derived from coniferous trees and are believed to protect trees against insects and microbial pathogens.41 Natural coniferous resin derived from the Norway spruce tree (Picea abies) mixed with boiled animal fat or butter has been used topically for centuries in Finland and Sweden to treat infections and wounds.42-44 The activity of NCR has been studied against a wide range of microbes, demonstrating broad-spectrum antimicrobial activity against both gram-positive bacteria and fungi.45-48 There are 2 published clinical trials evaluating NCR in the treatment of onychomycosis.49,50
In Vitro Data
Natural coniferous resin has shown antifungal activity against T mentagrophytes, Trichophyton tonsurans, and T rubrum in vitro, which was demonstrated using medicated disks of resin on petri dishes inoculated with these organisms.46 In another study, the authors evaluated the antifungal activity of NCR against human pathogenic fungi and yeasts using agar plate diffusion tests and showed that the resin had antifungal activity against Trichophyton species but not against Fusarium and most Candida species. Electron microscopy of T mentagrophytes exposed to NCR showed that all cells were dead inside the inhibition zone, with striking changes seen in the hyphal cell walls, while fungal cells outside the inhibition zone were morphologically normal.47 In another report, utilizing the European Pharmacopoeia challenge test, NCR was highly effective against gram-positive and gram-negative bacteria as well as C albicans.42
Clinical Trials
In one preliminary observational and prospective clinical trial, 15 participants with clinical and mycologic evidence of onychomycosis were instructed to apply NCR lacquer once daily for 9 months with a 4-week washout period, with the primary outcome measures being clinical and mycologic cure.49 Thirteen (87%) enrolled participants were male and the average age was 65 years (age range, 37–80 years). The DLSO subtype was present in 9 (60%) participants. The mycologic cure rate at the end of the study was 65% (95% CI, 42%-87%), and none achieved clinical cure, but 6 participants showed some improvement in the appearance of the nail.49
The second trial was a prospective, controlled, investigator-blinded study of 73 patients with clinical and mycologic evidence of toenail onychomycosis who were randomized to receive NCR 30%, amorolfine lacquer 5%, or 250 mg oral terbinafine.50 The primary end point was mycologic cure at 10 months, and secondary end points were clinical efficacy, cost-effectiveness, and patient compliance. Clinical efficacy was based on the proximal linear growth of healthy nail and was classified as unchanged, partial, or complete. Partial responses were described as substantial decreases in onycholysis, subungual hyperkeratosis, and streaks. A complete response was defined as a fully normal appearance of the toenail. Most patients were male in the NCR (91% [21/23]), amorolfine (80% [20/25]), and terbinafine (68% [17/25]) groups; the average ages were 64, 63, and 64 years, respectively. Trichophyton rubrum was cultured most often in all 3 groups: NCR, 87% (20/23); amorolfine, 96% (24/25); and terbinafine, 84% (21/25). The remaining cases were from T mentagrophytes. A summary of the results is shown in Table 2. Patient compliance was 100% in all except 1 patient in the amorolfine treatment group with moderate compliance. There were no adverse events, except for 2 in the terbinafine group: diarrhea and rash.50
AP Extract
Background
Ageratina pichinchensis, a member of the Asteraceae family, has been used historically in Mexico for fungal infections of the skin.51,52 Fresh or dried leaves were extracted with alcohol and the product was administered topically onto damaged skin without considerable skin irritation.53 Multiple studies have demonstrated that AP extract has in vitro antifungal activity along with other members of the Asteraceae family.54-56 There also is evidence from clinical trials that AP extract is effective against superficial dermatophyte infections such as tinea pedis.57 Given the positive antifungal in vitro data, the potential use of this agent was investigated for onychomycosis treatment.53,58
In Vitro Data
The antifungal properties of the Asteraceae family have been tested in several in vitro experiments. Eupatorium aschenbornianum, described as synonymous with A pichinchensis,59 was found to be most active against the dermatophytes T rubrum and T mentagrophytes with MICs of 0.3 and 0.03 mg/mL, respectively.54 It is thought that the primary antimycotic activity is due to encecalin, an acetylchromene compound that was identified in other plants from the Asteraceae family and has activity against dermatophytes.55 In another study, Ageratum houstanianum Mill, a comparable member of the Asteraceae family, had fungitoxic activity against T rubrum and C albicans isolated from nail infections.56
Clinical Trials
A double-blind controlled trial was performed on 110 patients with clinical and mycologic evidence of mild to moderate toenail onychomycosis randomized to treatment with AP lacquer or ciclopirox lacquer 8% (control).58 Primary end points were clinical effectiveness (completely normal nails) and mycologic cure. Patients were instructed to apply the lacquer once every third day during the first month, twice a week for the second month, and once a week for 16 weeks, with removal of the lacquer weekly. Demographics were similar between the AP lacquer and control groups, with mean ages of 44.6 and 46.5 years, respectively; women made up 74.5% and 67.2%, respectively, of each treatment group, with most patients having a 2- to 5-year history of disease (41.8% and 40.1%, respectively).58 A summary of the data is shown in Table 3. No severe side effects were documented, but minimal nail fold skin pain was reported in 3 patients in the control group in the first week, resolving later in the trial.58
A follow-up study was performed to determine the optimal concentration of AP lacquer for the treatment of onychomycosis.53 One hundred twenty-two patients aged 19 to 65 years with clinical and mycologic evidence of mild to moderate DLSO were randomized to receive 12.6% or 16.8% AP lacquer applied once daily to the affected nails for 6 months. The nails were graded as healthy, mild, or moderately affected before and after treatment. There were no significant differences in demographics between the 2 treatment groups, and 77% of patients were women with a median age of 47 years. There were no significant side effects from either concentration of AP lacquer.53
Ozonized Sunflower Oil
Background
Ozonized sunflower oil is derived by reacting ozone (O3) with sunflower plant (Helianthus annuus) oil to form a petroleum jelly–like material.60 It was originally shown to have antibacterial properties in vitro,61 and further studies have confirmed these findings and demonstrated anti-inflammatory, wound healing, and antifungal properties.62-64 A formulation of ozonized sunflower oil used in Cuba is clinically indicated for the treatment of tinea pedis and impetigo.65 The clinical efficacy of this product has been evaluated in a clinical trial for the treatment of onychomycosis.65
In Vitro Data
A compound made up of 30% ozonized sunflower oil with 0.5% of α-lipoic acid was found to have antifungal activity against C albicans using the disk diffusion method, in addition to other bacterial organisms. The MIC values ranged from 2.0 to 3.5 mg/mL.62 Another study was designed to evaluate the in vitro antifungal activity of this formulation on samples cultured from patients with onychomycosis using the disk diffusion method. They found inhibition of growth of C albicans, C parapsilosis, and Candida tropicalis, which was inferior to amphotericin B, ketoconazole, fluconazole, and itraconazole.64
Clinical Trial
A single-blind, controlled, phase 3 study was performed on 400 patients with clinical and mycologic evidence of onychomycosis. Patients were randomized to treatment with an ozonized sunflower oil solution or ketoconazole cream 2% applied to affected nails twice daily for 3 months, with filing and massage of the affected nails upon application of treatment.65 Cured was defined as mycologic cure in addition to a healthy appearing nail, improved as an increase in healthy appearing nail in addition to a decrease in symptoms (ie, paresthesia, pain, itching) but positive mycological testing, same as no clinical change in appearance with positive mycological findings, and worse as increasing diseased nail involvement in the presence of positive mycological findings. Demographics were similar between groups with a mean age of 35 years. Men accounted for 80% of the study population, and 65% of the study population was white. The mean duration of disease was 30 months. They also reported on a 1-year follow-up, with 2.8% of patients in the ozonized sunflower oil solution group and 37.0% of patients in the ketoconazole group describing relapses. Trichophyton rubrum and C albicans were cultured from these patients.65
Comment
Due to the poor efficacy, long-term treatment courses, inability to use nail polish, and high cost associated with many FDA-approved topical treatments, along with the systemic side effects, potential for drug-drug interactions, and cost associated with many oral therapies approved for onychomycosis, there has been a renewed interest in natural remedies and OTC treatments. Overall, TTO, TCS, NCR, AP extract, and ozonized sunflower oil have shown efficacy in vitro against some dermatophytes, nondermatophytes, and molds responsible for onychomycosis. One or more clinical trials were performed with each of these agents for the treatment of onychomycosis. They were mostly small pilot studies, and due to differences in trial design, the results cannot be compared with each other or with currently FDA-approved treatments. We can conclude that because adverse events were rare with all of these therapies—most commonly skin irritation or mild skin pain—they exhibit good safety.
For TTO, there was no statistical difference between the clotrimazole and TTO treatment groups in mycologic cure, clinical assessment, or patient subjective assessment of the nails.29 Although there was an 80% complete cure in the butenafine and TTO group, it was 0% in the TTO group at week 36.30 Trial design, longer treatment periods, incorporation into nanocapsules, or combination treatment with other antifungal agents may influence our future use of TTO for onychomycosis, but based on the present data we cannot recommend this treatment in clinical practice.
With TCS, 27.8% of participants had a mycologic cure and 22.2% had complete clinical cure.40 Although it is difficult to draw firm conclusions from this small pilot study, there may be some benefit to treating toenail onychomycosis due to T mentagrophytes or C parapsilosis with TCS but no benefit in treating onychomycosis due to T rubrum, the more common cause of onychomycosis. Limitations of this study were lack of a placebo group, small sample size, wide variety of represented pathogens that may not be representative of the true population, and lack of stratification by baseline severity or involvement of nail. A larger randomized controlled clinical trial would be necessary to confirm the results of this small study and make formal recommendations.
In one clinical trial with NCR, mycologic cure was 65% at the end of the study.49 No participants achieved clinical cure, but 6 participants showed some improvement in the appearance of the nail. Because this study was small (N=15), it is difficult to draw firm conclusions.49 In another study with NCR, mycologic cure rates with NCR, amorolfine, and terbinafine were 13%, 8%, and 56%, respectively. Based on these results, NCR has similar antifungal efficacy to amorolfine but was inferior to oral terbinafine.50 A larger randomized controlled clinical trial with more homogenous and less severely affected patients and longer treatment periods would be necessary to confirm the results of these small studies and make formal recommendations.
Because there were no significant differences in clinical effectiveness of mycologic cure rates between AP lacquer 10% and ciclopirox lacquer 8% in one clinical trial,58 AP does not seem to be more effective than at least one of the current FDA-approved topical treatments; however, because AP lacquer 16.8% was shown to be more effective than AP lacquer 12.6% in one onychomycosis clinical trial, using higher concentrations of AP may yield better results in future trials.53
One trial comparing ozonized sunflower oil to ketoconazole cream 2% showed 90.5% and 13.5% cure rates, respectively.65 Although there is good in vitro antifungal activity and a clinical trial showing efficacy using ozonized sunflower oil for the treatment of onychomycosis, confirmatory studies are necessary before we can recommend this OTC treatment to our patients. Specifically, we will get the most data from large randomized controlled trials with strict inclusion/exclusion and efficacy criteria.
Conclusion
Over-the-counter and natural remedies may be an emerging area of research in the treatment of onychomycosis. This review summarizes the laboratory data and clinical trials on several of these agents and, when available, compares their clinical and mycologic efficacy with FDA-approved therapies. Shortcomings of some of these studies include a small study population, lack of adequate controls, nonstandardized mycologic testing, and abbreviated posttreatment evaluation times. It may be concluded that these products have varying degrees of efficacy and appear to be safe in the studies cited; however, at present, we cannot recommend any of them to our patients until there are larger randomized clinical trials with appropriate controls demonstrating their efficacy.
- Scher RK, Daniel CR. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005.
- Sigurgeirsson B, Baran R. The prevalence of onychomycosis in the global population: a literature study. J Eur Acad Dermatol Venereol. 2014;28:1480-1491.
- Thomas J, Jacobson GA, Narkowicz CK, et al. Toenail onychomycosis: an important global disease burden. J Clin Pharm Ther. 2010;35:497-519.
- Mayser P, Freund V, Budihardja D. Toenail onychomycosis in diabetic patients: issues and management. Am J Clin Dermatol. 2009;10:211-220.
- Ghannoum MA, Hajjeh RA, Scher R, et al. A large-scale North American study of fungal isolates from nails: the frequency of onychomycosis, fungal distribution, and antifungal susceptibility patterns. J Am Acad Dermatol. 2000;43:641-648.
- Hay RJ, Baran R. Onychomycosis: a proposed revision of the clinical classification J Am Acad Dermatol. 2011;65:1219-1227.
- Elewski B. Clinical pearl: proximal white subungual onychomycosis in AIDS. J Am Acad Dermatol. 1993;29:631-632.
- Scher RK. Onychomycosis is more than a cosmetic problem. Br J Dermatol. 1994;130(suppl 43):15.
- Boyko EJ, Ahroni JH, Cohen V, et al. Prediction of diabetic foot ulcer occurrence using commonly available clinical information: the Seattle Diabetic Foot Study. Diabetes Care. 2006;29:1202-1207.
- Szepietowski JC, Reich A, Pacan P, et al. Evaluation of quality of life in patients with toenail onychomycosis by Polish version of an international onychomycosis-specific questionnaire. J Eur Acad Dermatol Venereol. 2007;21:491-496.
- Scher RK, Baron R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
- Lamisil [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2011.
- Sporanox [package insert]. Raritan, NJ: Ortho-McNeil-Janssen Pharmaceuticals, Inc; 2001
- Penlac [package insert]. Bridgewater, NJ: Dermik Laboratories; 2006.
- Jublia [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals North America LLC; 2014.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Kerydin [package insert]. Palo Alto, CA: Anacor Pharmaceuticals, Inc; 2014
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies [published online May 5, 2015]. J Am Acad Dermatol. 2015;73:62-69.
- D’Auria FD, Laino L, Strippoli V, et al. In vitro activity of tea tree oil against Candida albicans mycelial conversion and other pathogenic fungi. J Chemother. 2001;13:377-383.
- Satchell AC, Saurajen A, Bell C, et al. Treatment of interdigital tinea pedis with 25% and 50% tea tree oil solution: a randomized, placebo-controlled, blinded study. Australas J Dermatol. 2002;43:175-178.
- Carson CF, Hammer KA, Riley TV. Melaleuca alternifolia (tea tree) oil: a review of antimicrobial and other medicinal properties. Clin Microbiol Rev. 2006;19:50-62.
- Hammer KA, Carson CF, Riley TV. Antifungal activity of the components of Melaleuca alternifolia (tea tree) oil. J Appl Microbiol. 2003;95:853-860.
- Brophy JJ, Davies NW, Southwell IA, et al. Gas chromatographic quality control for oil of Melaleuca terpinen-4-ol type (Australian tea tree). J Agric Food Chem. 1989;37:1330-1335.
- Concha JM, Moore LS, Holloway WJ. 1998 William J. Stickel Bronze Award. Antifungal activity of Melaleuca alternifolia (tea-tree) oil against various pathogenic organisms. J Am Podiatr Med Assoc. 1998;88:489-492.
- Benger S, Townsend P, Ashford RL, et al. An in vitro study to determine the minimum inhibitory concentration of Melaleuca alternifolia against the dermatophyte Trichophyton rubrum. Foot. 2004;14:86-91.
- Hammer KA, Carson CF, Riley TV. In-vitro activity of essential oils, in particular Melaleuca alternifolia (tea tree) oil and tea tree oil products, against Candida spp. J Antimicrob Chemother. 1998;42:591-595.
- Altman P. Australian tea tree oil. Aust J Pharm. 1998;69:276-278.
- Guterres SS, Alves MP, Pohlmann AR. Polymeric nanoparticles, nanospheres and nanocapsules, for cutaneous applications. Drug Target Insights. 2007;2:147-157.
- Buck DS, Nidorf DM, Addino JG. Comparison of two topical preparations for the treatment of onychomycosis: Melaleuca alternifolia (tea tree) oil and clotrimazole. J Fam Pract. 1994;38:601-605.
- Syed TA, Qureshi ZA, Ali SM, et al. Treatment of toenail onychomycosis with 2% butenafine and 5% Melaleuca alternifolia (tea tree) oil in cream. Tropical Med Int Health. 1999;4:284-287.
- Flores FC, de Lima JA, Ribeiro RF, et al. Antifungal activity of nanocapsule suspensions containing tea tree oil on the growth of Trichophyton rubrum. Mycopathologia. 2013;175:281-286.
- Hammer KA, Carson CF, Riley TV. Antifungal effects of Melaleuca alternifolia (tea tree) oil and its components on Candida albicans, Candida glabrata and Saccharomyces cerevisiae. J Antimicrob Chemother. 2004;53:1081-1085.
- Cox SD, Mann CM, Markham JL, et al. The mode of antimicrobial action of the essential oil of Melaleuca alternifolia (tea tree oil). J Appl Microbiol. 2000;88:170-175.
- Hammer KA, Carson CF, Riley TV. Melaleuca alternifolia (tea tree) oil inhibits germ tube formation by Candida albicans. Med Mycol. 2000;38:355-362.
- Vicks VapoRub [package insert]. Gross-Gerau, Germany: Proctor & Gamble; 2010.
- Ramsewak RS, Nair MG, Stommel M, et al. In vitro antagonistic activity of monoterpenes and their mixtures against ‘toe nail fungus’ pathogens. Phytother Res. 2003;17:376-379.
- Pina-Vaz C, Gonçalves Rodrigues A, Pinto E, et al. Antifungal activity of Thymus oils and their major compounds. J Eur Acad Dermatol Venereol. 2004;18:73-78.
- Pinto E, Pina-Vaz C, Salgueiro L, et al. Antifungal activity of the essential oil of Thymus pulegioides on Candida, Aspergillus and dermatophyte species. J Med Microbiol. 2006;55:1367-1373.
- Vicks VapoRub might help fight toenail fungus. Consumer Reports. 2006;71:49.
- Derby R, Rohal P, Jackson C, et al. Novel treatment of onychomycosis using over-the-counter mentholated ointment: a clinical case series. J Am Board Fam Med. 2011;24:69-74.
- Trapp S, Croteau R. Defensive resin biosynthesis in conifers. Ann Rev Plant Physiol Plant Mol Biol. 2001;52:689-724.
- Sipponen A, Laitinen K. Antimicrobial properties of natural coniferous rosin in the European Pharmacopoeia challenge test. APMIS. 2011;119:720-724.
- Sipponen A, Lohi J. Lappish gum care “new” treatment of pressure ulcers? People’s improvement at it’s best. Eng Med J. 2003;58:2775-2776.
- Benedictus O. Een Nyttigh Läkare. Malmö: Kroon; 1938.
- Rautio M, Sipponen A, Peltola R, et al. Antibacterial effects of home-made resin salve from Norway spruce (Picea abies). APMIS. 2007;115:335-340.
- Laitinen K, Sipponen A, Jokinen JJ, et al. Resin salve from Norway spruce is antifungal against dermatophytes causing nail infections. EWMA. 2009;56:289-296.
- Rautio M, Sipponen A, Lohi J, et al. In vitro fungistatic effects of natural coniferous resin from Norway spruce (Picea abies). Eur J Clin Microbiol Infect Dis. 2012;31:1783-1789.
- Sipponen A, Peltola R, Jokinen JJ, et al. Effects of Norway spruce (Picea abies) resin on cell wall and cell membrane of Staphylococcus aureus. Ultrastruct Pathol. 2009;33:128-135.
- Sipponen P, Sipponen A, Lohi J, et al. Natural coniferous resin lacquer in treatment of toenail onychomycosis: an observational study. Mycoses. 2013;56:289-296.
- Auvinen T, Tiihonen R, Soini M, et al. Efficacy of topical resin lacquer, amorolfine, and oral terbinafine for treating toenail onychomycosis: a prospective, randomized, controlled, investigator-blinded, parallel-group clinical trial. Br J Dermatol. 2015;173:940-948.
- Argueta A, Cano L, Rodarte M. Atlas de las Plantas de la Medicina Tradicional Mexicana. Vol 3. Mexico City, Mexico: Instituto Nacional Indigenista; 1994:72-680.
- Avilés M, Suárez G. Catálogo de Plantas Medicinales del Jardín Etnobotánico. Peru: Instituto Nacional de Antropología e Historia; 1994.
- Romero-Cerecero O, Roman-Ramos R, Zamilpa A, et al. Clinical trial to compare the effectiveness of two concentrations of the Ageratina pichinchensis extract in the topical treatment of onychomycosis. J Ethnopharmacol. 2009;126:74-78.
- Navarro Garcia VM, Gonzalez A, Fuentes M, et al. Antifungal activities of nine traditional Mexican medicinal plants. J Ethnopharmacol. 2003;87:85-88.
- Castañeda P, Gómez L, Mata R, et al. Phytogrowth-inhibitory and antifungal constituents of Helianthella quinquenervis. J Nat Prod. 1996;59:323-326.
- Kumar N. Inhibition of nail infecting fungi of peoples of North Eastern UP causing Tinea unguium through leaf essential oil of Ageratum houstonianum Mill. IOSR J Pharm. June 2014;4:36-42.
- Romero-Cerecero O, Rojas G, Navarro V, et al. Effectiveness and tolerability of a standardized extract from Ageratina pichinchensis on patients with tinea pedis: an explorative pilot study controlled with ketoconazole. Planta Med. 2006;72:1257-1261.
- Romero-Cerecero O, Zamilpa A, Jimenez-Ferrer JE, et al. Double-blind clinical trial for evaluating the effectiveness and tolerability of Ageratina pichinchensis extract on patients with mild to moderate onychomycosis. a comparative study with ciclopirox. Planta Med. 2008;74:1430-1435.
- Rzedowski J, De Rzedowski GC. Flora Fanerogámica del Valle de México. Mexico City, Mexico: Instituto de Ecología Escuela Nacional de Ciencias Biológicas del Instituto Politécnico Nacional; 1985.
- Bocci V. Biological and clinical effects of ozone. has ozone therapy a future in medicine? Br J Biomed Sci. 1999;56:270-279.
- Sechi LA, Lezcano I, Nunez N, et al. Antibacterial activity of ozonized sunflower oil (Oleozon). J Appl Microbiol. 2001;90:279-284.
- Rodrigues KL, Cardoso CC, Caputo LR, et al. Cicatrizing and antimicrobial properties of an ozonised oil from sunflower seeds. Inflammopharmacology. 2004;12:261-270.
- Daud FV, Ueda SMY, Navarini A, et al. The use of ozonized oil in the treatment of dermatophitosis caused by Microsporum canis in rabbits. Braz J Microbiol. 2011;42:274-281.
- Guerrer LV, Cunha KC, Nogueira MC, et al. “In vitro” antifungal activity of ozonized sunflower oil on yeasts from onychomycosis. Braz J Microbiol. 2012;43:1315-1318.
- Menéndez S, Falcón L, Maqueira Y. Therapeutic efficacy of topical OLEOZON in patients suffering from onychomycosis. Mycoses. 2011;54:E272-E277.
- Scher RK, Daniel CR. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005.
- Sigurgeirsson B, Baran R. The prevalence of onychomycosis in the global population: a literature study. J Eur Acad Dermatol Venereol. 2014;28:1480-1491.
- Thomas J, Jacobson GA, Narkowicz CK, et al. Toenail onychomycosis: an important global disease burden. J Clin Pharm Ther. 2010;35:497-519.
- Mayser P, Freund V, Budihardja D. Toenail onychomycosis in diabetic patients: issues and management. Am J Clin Dermatol. 2009;10:211-220.
- Ghannoum MA, Hajjeh RA, Scher R, et al. A large-scale North American study of fungal isolates from nails: the frequency of onychomycosis, fungal distribution, and antifungal susceptibility patterns. J Am Acad Dermatol. 2000;43:641-648.
- Hay RJ, Baran R. Onychomycosis: a proposed revision of the clinical classification J Am Acad Dermatol. 2011;65:1219-1227.
- Elewski B. Clinical pearl: proximal white subungual onychomycosis in AIDS. J Am Acad Dermatol. 1993;29:631-632.
- Scher RK. Onychomycosis is more than a cosmetic problem. Br J Dermatol. 1994;130(suppl 43):15.
- Boyko EJ, Ahroni JH, Cohen V, et al. Prediction of diabetic foot ulcer occurrence using commonly available clinical information: the Seattle Diabetic Foot Study. Diabetes Care. 2006;29:1202-1207.
- Szepietowski JC, Reich A, Pacan P, et al. Evaluation of quality of life in patients with toenail onychomycosis by Polish version of an international onychomycosis-specific questionnaire. J Eur Acad Dermatol Venereol. 2007;21:491-496.
- Scher RK, Baron R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
- Lamisil [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2011.
- Sporanox [package insert]. Raritan, NJ: Ortho-McNeil-Janssen Pharmaceuticals, Inc; 2001
- Penlac [package insert]. Bridgewater, NJ: Dermik Laboratories; 2006.
- Jublia [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals North America LLC; 2014.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Kerydin [package insert]. Palo Alto, CA: Anacor Pharmaceuticals, Inc; 2014
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies [published online May 5, 2015]. J Am Acad Dermatol. 2015;73:62-69.
- D’Auria FD, Laino L, Strippoli V, et al. In vitro activity of tea tree oil against Candida albicans mycelial conversion and other pathogenic fungi. J Chemother. 2001;13:377-383.
- Satchell AC, Saurajen A, Bell C, et al. Treatment of interdigital tinea pedis with 25% and 50% tea tree oil solution: a randomized, placebo-controlled, blinded study. Australas J Dermatol. 2002;43:175-178.
- Carson CF, Hammer KA, Riley TV. Melaleuca alternifolia (tea tree) oil: a review of antimicrobial and other medicinal properties. Clin Microbiol Rev. 2006;19:50-62.
- Hammer KA, Carson CF, Riley TV. Antifungal activity of the components of Melaleuca alternifolia (tea tree) oil. J Appl Microbiol. 2003;95:853-860.
- Brophy JJ, Davies NW, Southwell IA, et al. Gas chromatographic quality control for oil of Melaleuca terpinen-4-ol type (Australian tea tree). J Agric Food Chem. 1989;37:1330-1335.
- Concha JM, Moore LS, Holloway WJ. 1998 William J. Stickel Bronze Award. Antifungal activity of Melaleuca alternifolia (tea-tree) oil against various pathogenic organisms. J Am Podiatr Med Assoc. 1998;88:489-492.
- Benger S, Townsend P, Ashford RL, et al. An in vitro study to determine the minimum inhibitory concentration of Melaleuca alternifolia against the dermatophyte Trichophyton rubrum. Foot. 2004;14:86-91.
- Hammer KA, Carson CF, Riley TV. In-vitro activity of essential oils, in particular Melaleuca alternifolia (tea tree) oil and tea tree oil products, against Candida spp. J Antimicrob Chemother. 1998;42:591-595.
- Altman P. Australian tea tree oil. Aust J Pharm. 1998;69:276-278.
- Guterres SS, Alves MP, Pohlmann AR. Polymeric nanoparticles, nanospheres and nanocapsules, for cutaneous applications. Drug Target Insights. 2007;2:147-157.
- Buck DS, Nidorf DM, Addino JG. Comparison of two topical preparations for the treatment of onychomycosis: Melaleuca alternifolia (tea tree) oil and clotrimazole. J Fam Pract. 1994;38:601-605.
- Syed TA, Qureshi ZA, Ali SM, et al. Treatment of toenail onychomycosis with 2% butenafine and 5% Melaleuca alternifolia (tea tree) oil in cream. Tropical Med Int Health. 1999;4:284-287.
- Flores FC, de Lima JA, Ribeiro RF, et al. Antifungal activity of nanocapsule suspensions containing tea tree oil on the growth of Trichophyton rubrum. Mycopathologia. 2013;175:281-286.
- Hammer KA, Carson CF, Riley TV. Antifungal effects of Melaleuca alternifolia (tea tree) oil and its components on Candida albicans, Candida glabrata and Saccharomyces cerevisiae. J Antimicrob Chemother. 2004;53:1081-1085.
- Cox SD, Mann CM, Markham JL, et al. The mode of antimicrobial action of the essential oil of Melaleuca alternifolia (tea tree oil). J Appl Microbiol. 2000;88:170-175.
- Hammer KA, Carson CF, Riley TV. Melaleuca alternifolia (tea tree) oil inhibits germ tube formation by Candida albicans. Med Mycol. 2000;38:355-362.
- Vicks VapoRub [package insert]. Gross-Gerau, Germany: Proctor & Gamble; 2010.
- Ramsewak RS, Nair MG, Stommel M, et al. In vitro antagonistic activity of monoterpenes and their mixtures against ‘toe nail fungus’ pathogens. Phytother Res. 2003;17:376-379.
- Pina-Vaz C, Gonçalves Rodrigues A, Pinto E, et al. Antifungal activity of Thymus oils and their major compounds. J Eur Acad Dermatol Venereol. 2004;18:73-78.
- Pinto E, Pina-Vaz C, Salgueiro L, et al. Antifungal activity of the essential oil of Thymus pulegioides on Candida, Aspergillus and dermatophyte species. J Med Microbiol. 2006;55:1367-1373.
- Vicks VapoRub might help fight toenail fungus. Consumer Reports. 2006;71:49.
- Derby R, Rohal P, Jackson C, et al. Novel treatment of onychomycosis using over-the-counter mentholated ointment: a clinical case series. J Am Board Fam Med. 2011;24:69-74.
- Trapp S, Croteau R. Defensive resin biosynthesis in conifers. Ann Rev Plant Physiol Plant Mol Biol. 2001;52:689-724.
- Sipponen A, Laitinen K. Antimicrobial properties of natural coniferous rosin in the European Pharmacopoeia challenge test. APMIS. 2011;119:720-724.
- Sipponen A, Lohi J. Lappish gum care “new” treatment of pressure ulcers? People’s improvement at it’s best. Eng Med J. 2003;58:2775-2776.
- Benedictus O. Een Nyttigh Läkare. Malmö: Kroon; 1938.
- Rautio M, Sipponen A, Peltola R, et al. Antibacterial effects of home-made resin salve from Norway spruce (Picea abies). APMIS. 2007;115:335-340.
- Laitinen K, Sipponen A, Jokinen JJ, et al. Resin salve from Norway spruce is antifungal against dermatophytes causing nail infections. EWMA. 2009;56:289-296.
- Rautio M, Sipponen A, Lohi J, et al. In vitro fungistatic effects of natural coniferous resin from Norway spruce (Picea abies). Eur J Clin Microbiol Infect Dis. 2012;31:1783-1789.
- Sipponen A, Peltola R, Jokinen JJ, et al. Effects of Norway spruce (Picea abies) resin on cell wall and cell membrane of Staphylococcus aureus. Ultrastruct Pathol. 2009;33:128-135.
- Sipponen P, Sipponen A, Lohi J, et al. Natural coniferous resin lacquer in treatment of toenail onychomycosis: an observational study. Mycoses. 2013;56:289-296.
- Auvinen T, Tiihonen R, Soini M, et al. Efficacy of topical resin lacquer, amorolfine, and oral terbinafine for treating toenail onychomycosis: a prospective, randomized, controlled, investigator-blinded, parallel-group clinical trial. Br J Dermatol. 2015;173:940-948.
- Argueta A, Cano L, Rodarte M. Atlas de las Plantas de la Medicina Tradicional Mexicana. Vol 3. Mexico City, Mexico: Instituto Nacional Indigenista; 1994:72-680.
- Avilés M, Suárez G. Catálogo de Plantas Medicinales del Jardín Etnobotánico. Peru: Instituto Nacional de Antropología e Historia; 1994.
- Romero-Cerecero O, Roman-Ramos R, Zamilpa A, et al. Clinical trial to compare the effectiveness of two concentrations of the Ageratina pichinchensis extract in the topical treatment of onychomycosis. J Ethnopharmacol. 2009;126:74-78.
- Navarro Garcia VM, Gonzalez A, Fuentes M, et al. Antifungal activities of nine traditional Mexican medicinal plants. J Ethnopharmacol. 2003;87:85-88.
- Castañeda P, Gómez L, Mata R, et al. Phytogrowth-inhibitory and antifungal constituents of Helianthella quinquenervis. J Nat Prod. 1996;59:323-326.
- Kumar N. Inhibition of nail infecting fungi of peoples of North Eastern UP causing Tinea unguium through leaf essential oil of Ageratum houstonianum Mill. IOSR J Pharm. June 2014;4:36-42.
- Romero-Cerecero O, Rojas G, Navarro V, et al. Effectiveness and tolerability of a standardized extract from Ageratina pichinchensis on patients with tinea pedis: an explorative pilot study controlled with ketoconazole. Planta Med. 2006;72:1257-1261.
- Romero-Cerecero O, Zamilpa A, Jimenez-Ferrer JE, et al. Double-blind clinical trial for evaluating the effectiveness and tolerability of Ageratina pichinchensis extract on patients with mild to moderate onychomycosis. a comparative study with ciclopirox. Planta Med. 2008;74:1430-1435.
- Rzedowski J, De Rzedowski GC. Flora Fanerogámica del Valle de México. Mexico City, Mexico: Instituto de Ecología Escuela Nacional de Ciencias Biológicas del Instituto Politécnico Nacional; 1985.
- Bocci V. Biological and clinical effects of ozone. has ozone therapy a future in medicine? Br J Biomed Sci. 1999;56:270-279.
- Sechi LA, Lezcano I, Nunez N, et al. Antibacterial activity of ozonized sunflower oil (Oleozon). J Appl Microbiol. 2001;90:279-284.
- Rodrigues KL, Cardoso CC, Caputo LR, et al. Cicatrizing and antimicrobial properties of an ozonised oil from sunflower seeds. Inflammopharmacology. 2004;12:261-270.
- Daud FV, Ueda SMY, Navarini A, et al. The use of ozonized oil in the treatment of dermatophitosis caused by Microsporum canis in rabbits. Braz J Microbiol. 2011;42:274-281.
- Guerrer LV, Cunha KC, Nogueira MC, et al. “In vitro” antifungal activity of ozonized sunflower oil on yeasts from onychomycosis. Braz J Microbiol. 2012;43:1315-1318.
- Menéndez S, Falcón L, Maqueira Y. Therapeutic efficacy of topical OLEOZON in patients suffering from onychomycosis. Mycoses. 2011;54:E272-E277.
Practice Points
- Natural remedies, including tea tree oil, natural topical cough suppressants, natural coniferous resin lacquer, Ageratina pichinchensis extract, and ozonized sunflower oil, have shown antifungal activities in in vitro studies.
- Some of these products have efficacy and appear to be safe in clinical studies.
- Larger randomized clinical trials demonstrating efficacy are required before we can recommend these products to our patients.
Tinea Capitis Caused by Trichophyton rubrum Mimicking Favus
In 1909, Sabouraud1 published a report delineating the clinical subsets of a chronic fungal infection of the scalp known as favus. The rarest subset was termed favus papyroide and consisted of a thin, dry, gray, parchmentlike crust up to 5 cm in diameter. Hair shafts were described as piercing the crust, with the underlying skin exhibiting erythema, moisture, and erosions. Children were reported to be affected more often than adults.1 Subsequent descriptions of patients with similar presentations have not appeared in the medical literature. In this case, an elderly woman with tinea capitis (TC) due to Trichophyton rubrum exhibited features of favus papyroide.
Case Report
An 87-year-old woman with a long history of actinic keratoses and nonmelanoma skin cancers presented to our dermatology clinic with numerous growths on the head, neck, and arms. The patient resided in a nursing home and had a history of hypertension, osteoarthritis, and mild to moderate dementia. Physical examination revealed a frail elderly woman in a wheelchair. Numerous actinic keratoses were noted on the arms and face. Examination of the scalp revealed a large, white-gray, palm-sized plaque on the crown (Figure 1) with 2 yellow, quarter-sized, hyperkeratotic nodules on the left temple and left parietal scalp. The differential diagnosis for the nodules on the temple and scalp included squamous cell carcinoma and hyperkeratotic actinic keratosis, and both lesions were biopsied. Histologically, they demonstrated pronounced hyperkeratosis and parakeratosis with numerous infiltrating neutrophils. The stratum malpighii exhibited focal atypia consistent with an actinic keratosis with areas of spongiosis and pustular folliculitis but no evidence of an invasive cutaneous malignancy. Periodic acid–Schiff stains were performed on both specimens and revealed numerous fungal hyphae within the stratum corneum (Figure 2) as well as evidence of a fungal folliculitis.
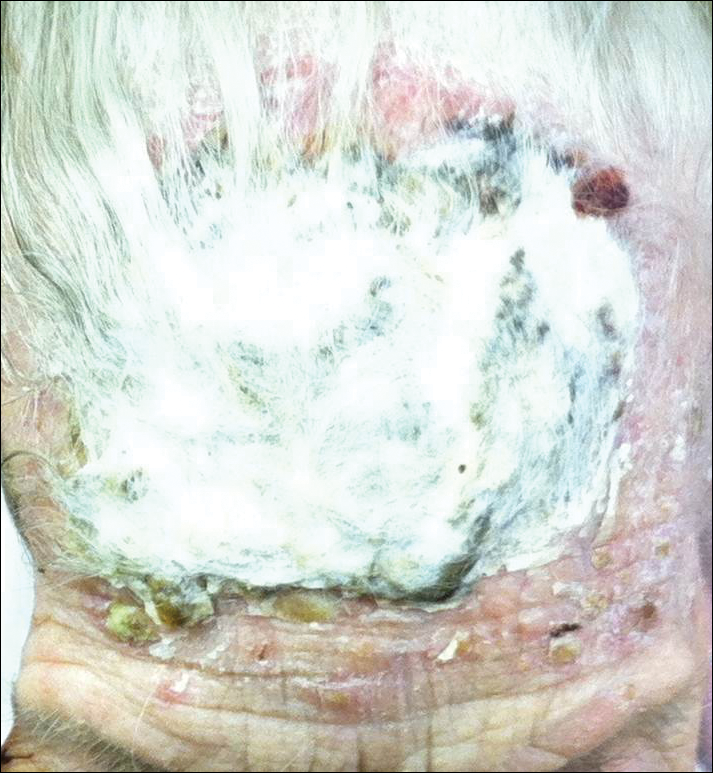
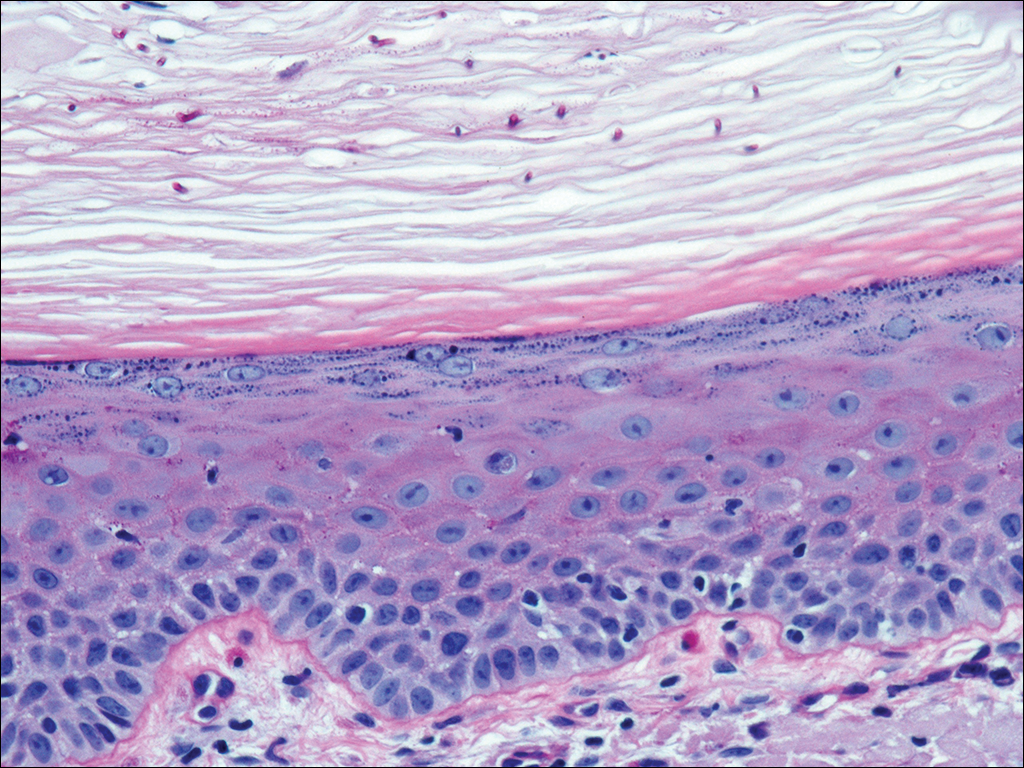
At a follow-up visit 2 weeks later, a portion of the hyperkeratotic material on the crown of the scalp was lifted free from the skin surface, removed with scissors, and submitted for histologic analysis and culture. The underlying skin exhibited substantial erythema and diffuse alopecia. The specimen consisted entirely of masses of hyperkeratotic and parakeratotic stratum corneum with numerous infiltrating neutrophils, cellular debris, and focal secondary bacterial colonization (Figure 3). Fungal hyphae and spores were readily demonstrated on Gomori methenamine-silver stain (Figure 4). A fungal culture from this material failed to demonstrate growth at 28 days. The organism was molecularly identified as T rubrum using the Sanger sequencing assay. The patient was treated with fluconazole 150 mg once daily for 3 weeks with eventual resolution of the plaque. The patient died approximately 3 months later (unrelated to her scalp infection).
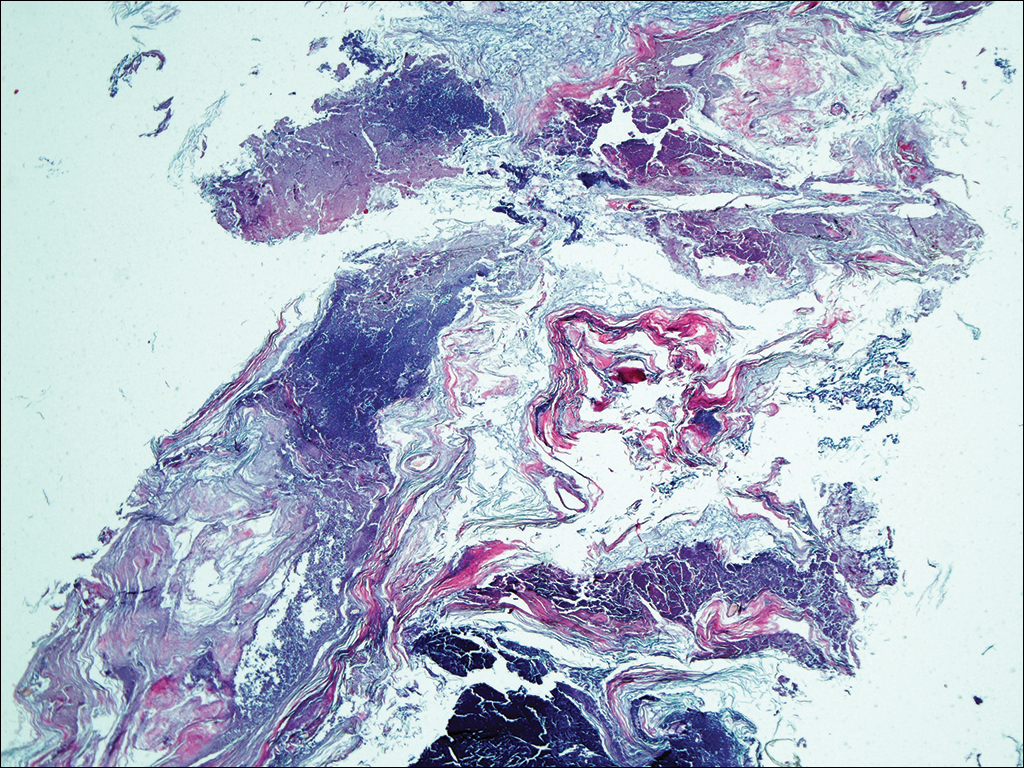

Comment
Favus, or tinea favosa, is a chronic inflammatory dermatophyte infection of the scalp, less commonly involving the skin and nails.2 The classic lesion is termed a scutulum or godet consisting of concave, cup-shaped, yellow crusts typically pierced by a single hair shaft.1 With an increase in size, the scutula may become confluent. Alopecia commonly results and infected patients may exude a “cheesy” or “mousy” odor from the lesions.3 Sabouraud1 delineated 3 clinical presentations of favus: (1) favus pityroide, the most common type consisting of a seborrheic dermatitis–like picture and scutula; (2) favus impetigoide, exhibiting honey-colored crusts reminiscent of impetigo but without appreciable scutula; and (3) favus papyroide, the rarest variant, demonstrating a dry, gray, parchmentlike crust pierced by hair shafts overlying an eroded erythematous scalp.
Favus usually is acquired in childhood or adolescence and often persists into adulthood.3 It is transmitted directly by hairs, infected keratinocytes, and fomites. Child-to-child transmission is much less common than other forms of TC.4 The responsible organism is almost always Trichophyton schoenleinii, with rare cases of Trichophyton violaceum, Trichophyton verrucosum, Trichophyton mentagrophytes var quinckeanum, Microsporum canis, and Microsporum gypseum having been reported.2,5,6 This anthropophilic dermatophyte infects only humans, is capable of surviving in the same dwelling space for generations, and is believed to require prolonged exposure for transmission. Trichophyton schoenleinii was the predominant infectious cause of TC in eastern Europe in the 19th and early 20th centuries, but its incidence has dramatically declined in the last 50 years.7 A survey conducted in 1997 and published in 2001 of TC that was culture-positive for T schoenleinii in 19 European countries found only 3 cases among 3671 isolates (0.08%).8 Between 1980 and 2005, no cases were reported in the British Isles.9 Currently, favus generally is found in impoverished geographic regions with poor hygiene, malnutrition, and limited access to health care; however, endemic foci in Kentucky, Quebec, and Montreal have been reported in North America.10 Although favus rarely resolves spontaneously, T schoenleinii was eradicated in most of the world with the introduction of griseofulvin in 1958.7 Terbinafine and itraconazole are currently the drugs of choice for therapy.10
Tinea capitis is the most common fungal infection in children, with 1 in 20 US children displaying evidence of overt infection.11 Infection in adults is rare and most affected patients typically display serious illnesses with concomitant immune compromise.12 Only 3% to 5% of cases arise in patients older than 20 years.13 Adult hair appears to be relatively resistant to dermatophyte infection, probably from the fungistatic properties of long-chain fatty acids found in sebum.13 Tinea capitis in adults usually occurs in postmenopausal women, presumably from involution of sebaceous glands associated with declining estrogen levels. Patients typically exhibit erythematous scaly patches with central clearing, alopecia, varying degrees of inflammation, and few pustules, though exudative and heavily inflammatory lesions also have been described.14
In the current case, TC was not raised in the differential diagnosis. Regardless, given that scaly red patches and papules of the scalp may represent a dermatophyte infection in this patient population, clinicians are encouraged to consider this possibility. Transmission is by direct human-to-human contact and contact with objects containing fomites including brushes, combs, bedding, clothing, toys, furniture, and telephones.15 It is frequently spread among family members and classmates.16
Prior to World War II, most cases of TC in the United States were due to M canis, with Microsporum audouinii becoming more prevalent until the 1960s and 1970s when Trichophyton tonsurans began surging in incidence.12,17 Currently, the latter organism is responsible for more than 95% of TC cases in the United States.18Microsporum canis is the main causative species in Europe but varies widely by country. In the Middle East and Africa, T violaceum is responsible for many infections.
Trichophyton rubrum–associated TC appears to be a rare occurrence. A global study in 1995 noted that less than 1% of TC cases were due to T rubrum infection, most having been described in emerging nations.12 A meta-analysis of 9 studies from developed countries found only 9 of 10,145 cases of TC with a culture positive for T rubrum.14 In adults, infected patients typically exhibit either evidence of a concomitant fungal infection of the skin and/or nails or health conditions with impaired immunity, whereas in children, interfamilial spread appears more common.11
- Sabouraud R. Les favus atypiques, clinique. Paris. 1909;4:296-299.
- Olkit M. Favus of the scalp: an overview and update. Mycopathologia. 2010;170:143-154.
- Elewski BE. Tinea capitis: a current perspective. J Am Acad Dermatol. 2000;42:1-20.
- Aly R, Hay RJ, del Palacio A, et al. Epidemiology of tinea capitis. Med Mycol. 2000;38(suppl 1):183-188.
- Joly J, Delage G, Auger P, et al. Favus: twenty indigenous cases in the province of Quebec. Arch Dermatol. 1978;114:1647-1648.
- Garcia-Sanchez MS, Pereira M, Pereira MM, et al. Favus due to Trichophyton mentagrophytes var. quinckeanum. Dermatology. 1997;194:177-179.
- Seebacher C, Bouchara JP, Mignon B. Updates on the epidemiology of dermatophyte infections. Mycopathologia. 2008;166:335-352.
- Hay RJ, Robles W, Midgley MK, et al. Tinea capitis in Europe: new perspective on an old problem. J Eur Acad Dermatol Venereol. 2001;15:229-233.
- Borman AM, Campbell CK, Fraser M, et al. Analysis of the dermatophyte species isolated in the British Isles between 1980 and 2005 and review of worldwide dermatophyte trends over the last three decades. Med Mycol. 2007;45:131-141.
- Rippon JW. Dermatophytosis and dermatomycosis. In: Rippon JW. Medical Mycology: The Pathogenic Fungi and the Pathogenic Actinomycetes. 3rd ed. Philadelphia, PA: WB Saunders; 1988:197-199.
- Abdel-Rahman SM, Penny J, Alander SW. Trichophyton rubrum tinea capitis in a young child. Ped Dermatol. 2004;21:63-65.
- Schwinn A, Ebert J, Brocker EB. Frequency of Trichophyton rubrum in tinea capitis. Mycoses. 1995;38:1-7.
- Ziemer A, Kohl K, Schroder G. Trichophyton rubrum induced inflammatory tinea capitis in a 63-year-old man. Mycoses. 2005;48:76-79.
- Anstey A, Lucke TW, Philpot C. Tinea capitis caused by Trichophyton rubrum. Br J Dermatol. 1996;135:113-115.
- Schwinn A, Ebert J, Muller I, et al. Trichophyton rubrum as the causative agent of tinea capitis in three children. Mycoses. 1995;38:9-11.
- Chang SE, Kang SK, Choi JH, et al. Tinea capitis due to Trichophyton rubrum in a neonate. Ped Dermatol. 2002;19:356-358.
- Stiller MJ, Rosenthal SA, Weinstein AS. Tinea capitis caused by Trichophyton rubrum in a 67-year-old woman with systemic lupus erythematosus. J Am Acad Dermatol. 1993;29:257-258.
- Foster KW, Ghannoum MA, Elewski BE. Epidemiologic surveillance of cutaneous fungal infection in the United States from 1999 to 2002. J Am Acad Dermatol. 2004;50:748-752.
In 1909, Sabouraud1 published a report delineating the clinical subsets of a chronic fungal infection of the scalp known as favus. The rarest subset was termed favus papyroide and consisted of a thin, dry, gray, parchmentlike crust up to 5 cm in diameter. Hair shafts were described as piercing the crust, with the underlying skin exhibiting erythema, moisture, and erosions. Children were reported to be affected more often than adults.1 Subsequent descriptions of patients with similar presentations have not appeared in the medical literature. In this case, an elderly woman with tinea capitis (TC) due to Trichophyton rubrum exhibited features of favus papyroide.
Case Report
An 87-year-old woman with a long history of actinic keratoses and nonmelanoma skin cancers presented to our dermatology clinic with numerous growths on the head, neck, and arms. The patient resided in a nursing home and had a history of hypertension, osteoarthritis, and mild to moderate dementia. Physical examination revealed a frail elderly woman in a wheelchair. Numerous actinic keratoses were noted on the arms and face. Examination of the scalp revealed a large, white-gray, palm-sized plaque on the crown (Figure 1) with 2 yellow, quarter-sized, hyperkeratotic nodules on the left temple and left parietal scalp. The differential diagnosis for the nodules on the temple and scalp included squamous cell carcinoma and hyperkeratotic actinic keratosis, and both lesions were biopsied. Histologically, they demonstrated pronounced hyperkeratosis and parakeratosis with numerous infiltrating neutrophils. The stratum malpighii exhibited focal atypia consistent with an actinic keratosis with areas of spongiosis and pustular folliculitis but no evidence of an invasive cutaneous malignancy. Periodic acid–Schiff stains were performed on both specimens and revealed numerous fungal hyphae within the stratum corneum (Figure 2) as well as evidence of a fungal folliculitis.
At a follow-up visit 2 weeks later, a portion of the hyperkeratotic material on the crown of the scalp was lifted free from the skin surface, removed with scissors, and submitted for histologic analysis and culture. The underlying skin exhibited substantial erythema and diffuse alopecia. The specimen consisted entirely of masses of hyperkeratotic and parakeratotic stratum corneum with numerous infiltrating neutrophils, cellular debris, and focal secondary bacterial colonization (Figure 3). Fungal hyphae and spores were readily demonstrated on Gomori methenamine-silver stain (Figure 4). A fungal culture from this material failed to demonstrate growth at 28 days. The organism was molecularly identified as T rubrum using the Sanger sequencing assay. The patient was treated with fluconazole 150 mg once daily for 3 weeks with eventual resolution of the plaque. The patient died approximately 3 months later (unrelated to her scalp infection).
Comment
Favus, or tinea favosa, is a chronic inflammatory dermatophyte infection of the scalp, less commonly involving the skin and nails.2 The classic lesion is termed a scutulum or godet consisting of concave, cup-shaped, yellow crusts typically pierced by a single hair shaft.1 With an increase in size, the scutula may become confluent. Alopecia commonly results and infected patients may exude a “cheesy” or “mousy” odor from the lesions.3 Sabouraud1 delineated 3 clinical presentations of favus: (1) favus pityroide, the most common type consisting of a seborrheic dermatitis–like picture and scutula; (2) favus impetigoide, exhibiting honey-colored crusts reminiscent of impetigo but without appreciable scutula; and (3) favus papyroide, the rarest variant, demonstrating a dry, gray, parchmentlike crust pierced by hair shafts overlying an eroded erythematous scalp.
Favus usually is acquired in childhood or adolescence and often persists into adulthood.3 It is transmitted directly by hairs, infected keratinocytes, and fomites. Child-to-child transmission is much less common than other forms of TC.4 The responsible organism is almost always Trichophyton schoenleinii, with rare cases of Trichophyton violaceum, Trichophyton verrucosum, Trichophyton mentagrophytes var quinckeanum, Microsporum canis, and Microsporum gypseum having been reported.2,5,6 This anthropophilic dermatophyte infects only humans, is capable of surviving in the same dwelling space for generations, and is believed to require prolonged exposure for transmission. Trichophyton schoenleinii was the predominant infectious cause of TC in eastern Europe in the 19th and early 20th centuries, but its incidence has dramatically declined in the last 50 years.7 A survey conducted in 1997 and published in 2001 of TC that was culture-positive for T schoenleinii in 19 European countries found only 3 cases among 3671 isolates (0.08%).8 Between 1980 and 2005, no cases were reported in the British Isles.9 Currently, favus generally is found in impoverished geographic regions with poor hygiene, malnutrition, and limited access to health care; however, endemic foci in Kentucky, Quebec, and Montreal have been reported in North America.10 Although favus rarely resolves spontaneously, T schoenleinii was eradicated in most of the world with the introduction of griseofulvin in 1958.7 Terbinafine and itraconazole are currently the drugs of choice for therapy.10
Tinea capitis is the most common fungal infection in children, with 1 in 20 US children displaying evidence of overt infection.11 Infection in adults is rare and most affected patients typically display serious illnesses with concomitant immune compromise.12 Only 3% to 5% of cases arise in patients older than 20 years.13 Adult hair appears to be relatively resistant to dermatophyte infection, probably from the fungistatic properties of long-chain fatty acids found in sebum.13 Tinea capitis in adults usually occurs in postmenopausal women, presumably from involution of sebaceous glands associated with declining estrogen levels. Patients typically exhibit erythematous scaly patches with central clearing, alopecia, varying degrees of inflammation, and few pustules, though exudative and heavily inflammatory lesions also have been described.14
In the current case, TC was not raised in the differential diagnosis. Regardless, given that scaly red patches and papules of the scalp may represent a dermatophyte infection in this patient population, clinicians are encouraged to consider this possibility. Transmission is by direct human-to-human contact and contact with objects containing fomites including brushes, combs, bedding, clothing, toys, furniture, and telephones.15 It is frequently spread among family members and classmates.16
Prior to World War II, most cases of TC in the United States were due to M canis, with Microsporum audouinii becoming more prevalent until the 1960s and 1970s when Trichophyton tonsurans began surging in incidence.12,17 Currently, the latter organism is responsible for more than 95% of TC cases in the United States.18Microsporum canis is the main causative species in Europe but varies widely by country. In the Middle East and Africa, T violaceum is responsible for many infections.
Trichophyton rubrum–associated TC appears to be a rare occurrence. A global study in 1995 noted that less than 1% of TC cases were due to T rubrum infection, most having been described in emerging nations.12 A meta-analysis of 9 studies from developed countries found only 9 of 10,145 cases of TC with a culture positive for T rubrum.14 In adults, infected patients typically exhibit either evidence of a concomitant fungal infection of the skin and/or nails or health conditions with impaired immunity, whereas in children, interfamilial spread appears more common.11
In 1909, Sabouraud1 published a report delineating the clinical subsets of a chronic fungal infection of the scalp known as favus. The rarest subset was termed favus papyroide and consisted of a thin, dry, gray, parchmentlike crust up to 5 cm in diameter. Hair shafts were described as piercing the crust, with the underlying skin exhibiting erythema, moisture, and erosions. Children were reported to be affected more often than adults.1 Subsequent descriptions of patients with similar presentations have not appeared in the medical literature. In this case, an elderly woman with tinea capitis (TC) due to Trichophyton rubrum exhibited features of favus papyroide.
Case Report
An 87-year-old woman with a long history of actinic keratoses and nonmelanoma skin cancers presented to our dermatology clinic with numerous growths on the head, neck, and arms. The patient resided in a nursing home and had a history of hypertension, osteoarthritis, and mild to moderate dementia. Physical examination revealed a frail elderly woman in a wheelchair. Numerous actinic keratoses were noted on the arms and face. Examination of the scalp revealed a large, white-gray, palm-sized plaque on the crown (Figure 1) with 2 yellow, quarter-sized, hyperkeratotic nodules on the left temple and left parietal scalp. The differential diagnosis for the nodules on the temple and scalp included squamous cell carcinoma and hyperkeratotic actinic keratosis, and both lesions were biopsied. Histologically, they demonstrated pronounced hyperkeratosis and parakeratosis with numerous infiltrating neutrophils. The stratum malpighii exhibited focal atypia consistent with an actinic keratosis with areas of spongiosis and pustular folliculitis but no evidence of an invasive cutaneous malignancy. Periodic acid–Schiff stains were performed on both specimens and revealed numerous fungal hyphae within the stratum corneum (Figure 2) as well as evidence of a fungal folliculitis.
At a follow-up visit 2 weeks later, a portion of the hyperkeratotic material on the crown of the scalp was lifted free from the skin surface, removed with scissors, and submitted for histologic analysis and culture. The underlying skin exhibited substantial erythema and diffuse alopecia. The specimen consisted entirely of masses of hyperkeratotic and parakeratotic stratum corneum with numerous infiltrating neutrophils, cellular debris, and focal secondary bacterial colonization (Figure 3). Fungal hyphae and spores were readily demonstrated on Gomori methenamine-silver stain (Figure 4). A fungal culture from this material failed to demonstrate growth at 28 days. The organism was molecularly identified as T rubrum using the Sanger sequencing assay. The patient was treated with fluconazole 150 mg once daily for 3 weeks with eventual resolution of the plaque. The patient died approximately 3 months later (unrelated to her scalp infection).
Comment
Favus, or tinea favosa, is a chronic inflammatory dermatophyte infection of the scalp, less commonly involving the skin and nails.2 The classic lesion is termed a scutulum or godet consisting of concave, cup-shaped, yellow crusts typically pierced by a single hair shaft.1 With an increase in size, the scutula may become confluent. Alopecia commonly results and infected patients may exude a “cheesy” or “mousy” odor from the lesions.3 Sabouraud1 delineated 3 clinical presentations of favus: (1) favus pityroide, the most common type consisting of a seborrheic dermatitis–like picture and scutula; (2) favus impetigoide, exhibiting honey-colored crusts reminiscent of impetigo but without appreciable scutula; and (3) favus papyroide, the rarest variant, demonstrating a dry, gray, parchmentlike crust pierced by hair shafts overlying an eroded erythematous scalp.
Favus usually is acquired in childhood or adolescence and often persists into adulthood.3 It is transmitted directly by hairs, infected keratinocytes, and fomites. Child-to-child transmission is much less common than other forms of TC.4 The responsible organism is almost always Trichophyton schoenleinii, with rare cases of Trichophyton violaceum, Trichophyton verrucosum, Trichophyton mentagrophytes var quinckeanum, Microsporum canis, and Microsporum gypseum having been reported.2,5,6 This anthropophilic dermatophyte infects only humans, is capable of surviving in the same dwelling space for generations, and is believed to require prolonged exposure for transmission. Trichophyton schoenleinii was the predominant infectious cause of TC in eastern Europe in the 19th and early 20th centuries, but its incidence has dramatically declined in the last 50 years.7 A survey conducted in 1997 and published in 2001 of TC that was culture-positive for T schoenleinii in 19 European countries found only 3 cases among 3671 isolates (0.08%).8 Between 1980 and 2005, no cases were reported in the British Isles.9 Currently, favus generally is found in impoverished geographic regions with poor hygiene, malnutrition, and limited access to health care; however, endemic foci in Kentucky, Quebec, and Montreal have been reported in North America.10 Although favus rarely resolves spontaneously, T schoenleinii was eradicated in most of the world with the introduction of griseofulvin in 1958.7 Terbinafine and itraconazole are currently the drugs of choice for therapy.10
Tinea capitis is the most common fungal infection in children, with 1 in 20 US children displaying evidence of overt infection.11 Infection in adults is rare and most affected patients typically display serious illnesses with concomitant immune compromise.12 Only 3% to 5% of cases arise in patients older than 20 years.13 Adult hair appears to be relatively resistant to dermatophyte infection, probably from the fungistatic properties of long-chain fatty acids found in sebum.13 Tinea capitis in adults usually occurs in postmenopausal women, presumably from involution of sebaceous glands associated with declining estrogen levels. Patients typically exhibit erythematous scaly patches with central clearing, alopecia, varying degrees of inflammation, and few pustules, though exudative and heavily inflammatory lesions also have been described.14
In the current case, TC was not raised in the differential diagnosis. Regardless, given that scaly red patches and papules of the scalp may represent a dermatophyte infection in this patient population, clinicians are encouraged to consider this possibility. Transmission is by direct human-to-human contact and contact with objects containing fomites including brushes, combs, bedding, clothing, toys, furniture, and telephones.15 It is frequently spread among family members and classmates.16
Prior to World War II, most cases of TC in the United States were due to M canis, with Microsporum audouinii becoming more prevalent until the 1960s and 1970s when Trichophyton tonsurans began surging in incidence.12,17 Currently, the latter organism is responsible for more than 95% of TC cases in the United States.18Microsporum canis is the main causative species in Europe but varies widely by country. In the Middle East and Africa, T violaceum is responsible for many infections.
Trichophyton rubrum–associated TC appears to be a rare occurrence. A global study in 1995 noted that less than 1% of TC cases were due to T rubrum infection, most having been described in emerging nations.12 A meta-analysis of 9 studies from developed countries found only 9 of 10,145 cases of TC with a culture positive for T rubrum.14 In adults, infected patients typically exhibit either evidence of a concomitant fungal infection of the skin and/or nails or health conditions with impaired immunity, whereas in children, interfamilial spread appears more common.11
- Sabouraud R. Les favus atypiques, clinique. Paris. 1909;4:296-299.
- Olkit M. Favus of the scalp: an overview and update. Mycopathologia. 2010;170:143-154.
- Elewski BE. Tinea capitis: a current perspective. J Am Acad Dermatol. 2000;42:1-20.
- Aly R, Hay RJ, del Palacio A, et al. Epidemiology of tinea capitis. Med Mycol. 2000;38(suppl 1):183-188.
- Joly J, Delage G, Auger P, et al. Favus: twenty indigenous cases in the province of Quebec. Arch Dermatol. 1978;114:1647-1648.
- Garcia-Sanchez MS, Pereira M, Pereira MM, et al. Favus due to Trichophyton mentagrophytes var. quinckeanum. Dermatology. 1997;194:177-179.
- Seebacher C, Bouchara JP, Mignon B. Updates on the epidemiology of dermatophyte infections. Mycopathologia. 2008;166:335-352.
- Hay RJ, Robles W, Midgley MK, et al. Tinea capitis in Europe: new perspective on an old problem. J Eur Acad Dermatol Venereol. 2001;15:229-233.
- Borman AM, Campbell CK, Fraser M, et al. Analysis of the dermatophyte species isolated in the British Isles between 1980 and 2005 and review of worldwide dermatophyte trends over the last three decades. Med Mycol. 2007;45:131-141.
- Rippon JW. Dermatophytosis and dermatomycosis. In: Rippon JW. Medical Mycology: The Pathogenic Fungi and the Pathogenic Actinomycetes. 3rd ed. Philadelphia, PA: WB Saunders; 1988:197-199.
- Abdel-Rahman SM, Penny J, Alander SW. Trichophyton rubrum tinea capitis in a young child. Ped Dermatol. 2004;21:63-65.
- Schwinn A, Ebert J, Brocker EB. Frequency of Trichophyton rubrum in tinea capitis. Mycoses. 1995;38:1-7.
- Ziemer A, Kohl K, Schroder G. Trichophyton rubrum induced inflammatory tinea capitis in a 63-year-old man. Mycoses. 2005;48:76-79.
- Anstey A, Lucke TW, Philpot C. Tinea capitis caused by Trichophyton rubrum. Br J Dermatol. 1996;135:113-115.
- Schwinn A, Ebert J, Muller I, et al. Trichophyton rubrum as the causative agent of tinea capitis in three children. Mycoses. 1995;38:9-11.
- Chang SE, Kang SK, Choi JH, et al. Tinea capitis due to Trichophyton rubrum in a neonate. Ped Dermatol. 2002;19:356-358.
- Stiller MJ, Rosenthal SA, Weinstein AS. Tinea capitis caused by Trichophyton rubrum in a 67-year-old woman with systemic lupus erythematosus. J Am Acad Dermatol. 1993;29:257-258.
- Foster KW, Ghannoum MA, Elewski BE. Epidemiologic surveillance of cutaneous fungal infection in the United States from 1999 to 2002. J Am Acad Dermatol. 2004;50:748-752.
- Sabouraud R. Les favus atypiques, clinique. Paris. 1909;4:296-299.
- Olkit M. Favus of the scalp: an overview and update. Mycopathologia. 2010;170:143-154.
- Elewski BE. Tinea capitis: a current perspective. J Am Acad Dermatol. 2000;42:1-20.
- Aly R, Hay RJ, del Palacio A, et al. Epidemiology of tinea capitis. Med Mycol. 2000;38(suppl 1):183-188.
- Joly J, Delage G, Auger P, et al. Favus: twenty indigenous cases in the province of Quebec. Arch Dermatol. 1978;114:1647-1648.
- Garcia-Sanchez MS, Pereira M, Pereira MM, et al. Favus due to Trichophyton mentagrophytes var. quinckeanum. Dermatology. 1997;194:177-179.
- Seebacher C, Bouchara JP, Mignon B. Updates on the epidemiology of dermatophyte infections. Mycopathologia. 2008;166:335-352.
- Hay RJ, Robles W, Midgley MK, et al. Tinea capitis in Europe: new perspective on an old problem. J Eur Acad Dermatol Venereol. 2001;15:229-233.
- Borman AM, Campbell CK, Fraser M, et al. Analysis of the dermatophyte species isolated in the British Isles between 1980 and 2005 and review of worldwide dermatophyte trends over the last three decades. Med Mycol. 2007;45:131-141.
- Rippon JW. Dermatophytosis and dermatomycosis. In: Rippon JW. Medical Mycology: The Pathogenic Fungi and the Pathogenic Actinomycetes. 3rd ed. Philadelphia, PA: WB Saunders; 1988:197-199.
- Abdel-Rahman SM, Penny J, Alander SW. Trichophyton rubrum tinea capitis in a young child. Ped Dermatol. 2004;21:63-65.
- Schwinn A, Ebert J, Brocker EB. Frequency of Trichophyton rubrum in tinea capitis. Mycoses. 1995;38:1-7.
- Ziemer A, Kohl K, Schroder G. Trichophyton rubrum induced inflammatory tinea capitis in a 63-year-old man. Mycoses. 2005;48:76-79.
- Anstey A, Lucke TW, Philpot C. Tinea capitis caused by Trichophyton rubrum. Br J Dermatol. 1996;135:113-115.
- Schwinn A, Ebert J, Muller I, et al. Trichophyton rubrum as the causative agent of tinea capitis in three children. Mycoses. 1995;38:9-11.
- Chang SE, Kang SK, Choi JH, et al. Tinea capitis due to Trichophyton rubrum in a neonate. Ped Dermatol. 2002;19:356-358.
- Stiller MJ, Rosenthal SA, Weinstein AS. Tinea capitis caused by Trichophyton rubrum in a 67-year-old woman with systemic lupus erythematosus. J Am Acad Dermatol. 1993;29:257-258.
- Foster KW, Ghannoum MA, Elewski BE. Epidemiologic surveillance of cutaneous fungal infection in the United States from 1999 to 2002. J Am Acad Dermatol. 2004;50:748-752.
Practice Points
- Although favus is uncommonly seen in developed countries, it still exists and can mimick other conditions, notably cutaneous malignancies.
- Favus may affect the skin and nails in addition to the hair.
- The lesions of favus may persist for many years.
Novel oral antifungal headed to phase III for onychomycosis
VIENNA – A once-weekly oral antifungal drug known as VT-1161 will move into phase III clinical testing in 2017 based on its impressive performance in an interim analysis of a phase IIb study, Amir Tavakkol, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“We saw robust evidence of clinical and mycologic efficacy in patients with moderate to severe onychomycosis. This was true even in patients with nails considered very difficult to treat because of dermatophytomas and spikes, which are usually poor prognostic elements,” said Dr. Tavakkol, chief development officer at Viamet Pharmaceuticals in Durham, N.C., which is developing VT-1161.

He reported on 107 patients with toenail onychomycosis who enrolled in the phase IIb, double-blind, placebo-controlled RENOVATE (Restoring Nail: An Oral VT-1161 Tablet Evaluation) study. At enrollment they averaged 47% disease involvement of the big toenail, the target nail for the trial. Participants in the five study arms had an average of 4.2-5.0 affected toenails. Both the percentage of nail involvement and the number of diseased toenails were roughly twice as great as is typical in studies of topical antifungals, underscoring that participants in the VT-1161 trial had fairly severe onychomycosis.
Patients were assigned to one of five study arms: 300 mg of VT-1161 once weekly for 12 weeks, then 12 weeks of placebo; 600 mg of VT-1161 once weekly for 12 weeks, followed by 12 weeks of placebo; either 300 or 600 mg of the antifungal agent once weekly for the full 24 weeks; or 24 weeks of once-per-week placebo. Immediately prior to the formal start of the study, however, everyone received 14 days of a once-daily loading dose of VT-1161 or placebo at the dose they would subsequently take once weekly during the trial.
The primary outcome in the ongoing study is the percentage of patients with a complete cure, both mycologic and clinical, at 48 weeks. Those data aren’t in yet, but Dr. Tavakkol presented the results of the prespecified interim analysis at 24 weeks.
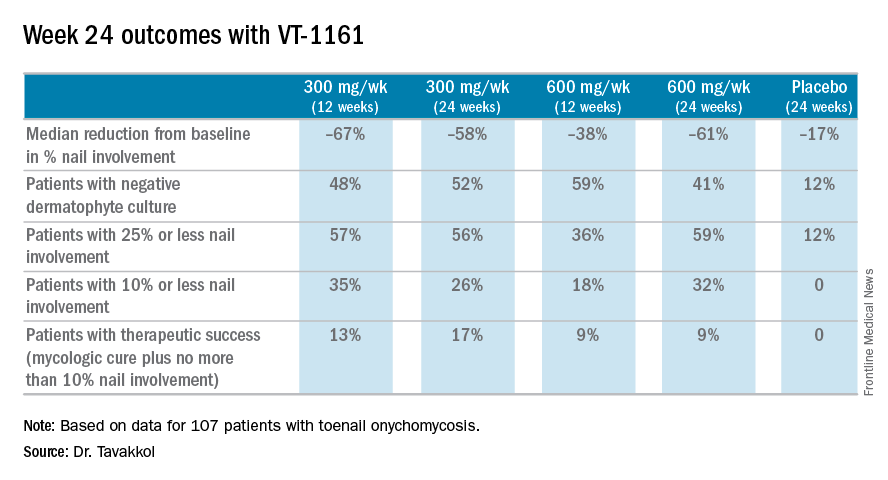
“Please keep in mind that this is only at most 24 weeks of therapy. Given the rate of nail growth at 1 mm per month when it is healthy, these are remarkable data. Ten percent or less nail involvement is basically 1-2 mm at the distal end. I believe a substantial percentage of these patients will reach clinical cure by 48 weeks,” he said.
VT-1161, a molecule with high selectivity for fungal CYP51, blocks the production of ergosterol, a key component of the fungal cell membrane, according to the company. It has no known drug interactions. At all doses studied in the trial, it was safe and well tolerated, with no abnormal liver function tests, no effect on bilirubin, and no change in QTc interval. Only 8 of the 107 patients reported adverse events deemed possibly related to the study drug by blinded investigators. No one dropped out of the study.
“VT-1161 is also being developed for recurrent vulvovaginal candidiasis. The results there are outstanding, too,” Dr. Tavakkol said.
The trial was funded by Viamet, where he is employed.
VIENNA – A once-weekly oral antifungal drug known as VT-1161 will move into phase III clinical testing in 2017 based on its impressive performance in an interim analysis of a phase IIb study, Amir Tavakkol, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“We saw robust evidence of clinical and mycologic efficacy in patients with moderate to severe onychomycosis. This was true even in patients with nails considered very difficult to treat because of dermatophytomas and spikes, which are usually poor prognostic elements,” said Dr. Tavakkol, chief development officer at Viamet Pharmaceuticals in Durham, N.C., which is developing VT-1161.
He reported on 107 patients with toenail onychomycosis who enrolled in the phase IIb, double-blind, placebo-controlled RENOVATE (Restoring Nail: An Oral VT-1161 Tablet Evaluation) study. At enrollment they averaged 47% disease involvement of the big toenail, the target nail for the trial. Participants in the five study arms had an average of 4.2-5.0 affected toenails. Both the percentage of nail involvement and the number of diseased toenails were roughly twice as great as is typical in studies of topical antifungals, underscoring that participants in the VT-1161 trial had fairly severe onychomycosis.
Patients were assigned to one of five study arms: 300 mg of VT-1161 once weekly for 12 weeks, then 12 weeks of placebo; 600 mg of VT-1161 once weekly for 12 weeks, followed by 12 weeks of placebo; either 300 or 600 mg of the antifungal agent once weekly for the full 24 weeks; or 24 weeks of once-per-week placebo. Immediately prior to the formal start of the study, however, everyone received 14 days of a once-daily loading dose of VT-1161 or placebo at the dose they would subsequently take once weekly during the trial.
The primary outcome in the ongoing study is the percentage of patients with a complete cure, both mycologic and clinical, at 48 weeks. Those data aren’t in yet, but Dr. Tavakkol presented the results of the prespecified interim analysis at 24 weeks.
“Please keep in mind that this is only at most 24 weeks of therapy. Given the rate of nail growth at 1 mm per month when it is healthy, these are remarkable data. Ten percent or less nail involvement is basically 1-2 mm at the distal end. I believe a substantial percentage of these patients will reach clinical cure by 48 weeks,” he said.
VT-1161, a molecule with high selectivity for fungal CYP51, blocks the production of ergosterol, a key component of the fungal cell membrane, according to the company. It has no known drug interactions. At all doses studied in the trial, it was safe and well tolerated, with no abnormal liver function tests, no effect on bilirubin, and no change in QTc interval. Only 8 of the 107 patients reported adverse events deemed possibly related to the study drug by blinded investigators. No one dropped out of the study.
“VT-1161 is also being developed for recurrent vulvovaginal candidiasis. The results there are outstanding, too,” Dr. Tavakkol said.
The trial was funded by Viamet, where he is employed.
VIENNA – A once-weekly oral antifungal drug known as VT-1161 will move into phase III clinical testing in 2017 based on its impressive performance in an interim analysis of a phase IIb study, Amir Tavakkol, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“We saw robust evidence of clinical and mycologic efficacy in patients with moderate to severe onychomycosis. This was true even in patients with nails considered very difficult to treat because of dermatophytomas and spikes, which are usually poor prognostic elements,” said Dr. Tavakkol, chief development officer at Viamet Pharmaceuticals in Durham, N.C., which is developing VT-1161.
He reported on 107 patients with toenail onychomycosis who enrolled in the phase IIb, double-blind, placebo-controlled RENOVATE (Restoring Nail: An Oral VT-1161 Tablet Evaluation) study. At enrollment they averaged 47% disease involvement of the big toenail, the target nail for the trial. Participants in the five study arms had an average of 4.2-5.0 affected toenails. Both the percentage of nail involvement and the number of diseased toenails were roughly twice as great as is typical in studies of topical antifungals, underscoring that participants in the VT-1161 trial had fairly severe onychomycosis.
Patients were assigned to one of five study arms: 300 mg of VT-1161 once weekly for 12 weeks, then 12 weeks of placebo; 600 mg of VT-1161 once weekly for 12 weeks, followed by 12 weeks of placebo; either 300 or 600 mg of the antifungal agent once weekly for the full 24 weeks; or 24 weeks of once-per-week placebo. Immediately prior to the formal start of the study, however, everyone received 14 days of a once-daily loading dose of VT-1161 or placebo at the dose they would subsequently take once weekly during the trial.
The primary outcome in the ongoing study is the percentage of patients with a complete cure, both mycologic and clinical, at 48 weeks. Those data aren’t in yet, but Dr. Tavakkol presented the results of the prespecified interim analysis at 24 weeks.
“Please keep in mind that this is only at most 24 weeks of therapy. Given the rate of nail growth at 1 mm per month when it is healthy, these are remarkable data. Ten percent or less nail involvement is basically 1-2 mm at the distal end. I believe a substantial percentage of these patients will reach clinical cure by 48 weeks,” he said.
VT-1161, a molecule with high selectivity for fungal CYP51, blocks the production of ergosterol, a key component of the fungal cell membrane, according to the company. It has no known drug interactions. At all doses studied in the trial, it was safe and well tolerated, with no abnormal liver function tests, no effect on bilirubin, and no change in QTc interval. Only 8 of the 107 patients reported adverse events deemed possibly related to the study drug by blinded investigators. No one dropped out of the study.
“VT-1161 is also being developed for recurrent vulvovaginal candidiasis. The results there are outstanding, too,” Dr. Tavakkol said.
The trial was funded by Viamet, where he is employed.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Among onychomycosis patients with an average 47% target toenail involvement at baseline, 35% had improved to no more than 10% nail involvement at 24 weeks after 12 weeks of once-weekly VT-1161 followed by 12 weeks of placebo.
Data source: A double-blind, randomized, phase IIb clinical trial involving 107 patients with toenail onychomycosis.
Disclosures: The trial was funded by Viamet Pharmaceuticals, where the study presenter is employed.
Survey finds high rate of misdiagnosed fungal infections
Fungal skin infections may be missed or misdiagnosed by many dermatologists, according to the results of a survey published online in the Journal of the American Academy of Dermatology.
For the interactive survey, conducted during a session on fungal infections at the 2016 Orlando Dermatology Aesthetic and Clinical Conference, board-certified dermatologists viewed 13 clinical images (which included other conditions such as secondary syphilis and pityriasis rosea) and were asked via an audience response system whether or not they thought the case was a fungal skin infection. In only 1 of the 13 cases presented did 90% of the dermatologists correctly categorize the case as either a dermatomycosis or not, reported Ramsin Joseph Yadgar of George Washington University in Washington, D.C., and colleagues.
Although most cases (8 of 13) “were appropriately categorized by more than 50% of the audience, this percentage decreased as accuracy of categorization increased,” they wrote. “For example, in only 4 of the 13 cases did audience members accurately categorize the cases with more than 75% accuracy,” they said (J Am Acad Dermatol. 2016 Nov 11. pii: S0190-9622[16]30883-0. doi: 10.1016/j.jaad.2016.09.041).

“Dermatology is full of doppelgangers,” Dr. Friedman, director of the residency program and of translational research in the department of dermatology at George Washington University, said in an interview.
“While we [dermatologists] pride ourselves on our visual prowess, there are many skin diseases which do not follow the textbook and can be quite protean in their presentations,” he said.
The variability in presentation makes diagnosing fungal infections especially challenging, he noted. “Fungal infections of the skin can have many clinical flavors and can infect skin, hair and nails. Also, inappropriate treatment can obscure the appearance of the infection, and the fact that there are multiple other conditions that can look like these [fungal] infections makes proper identification difficult.”
Although the results were limited by several factors including possible selection bias, lack of measurable response rate, and small sample size, the findings highlight how easy it can be to miss a diagnosis of fungal infection, “which can result in inappropriate therapy, worsening of symptoms, and even additional skin and soft-tissue infections,” the researchers wrote.
“Keep an open mind and cast a wider differential,” to help catch fungal infections, and use all the dermatologic tools, including slide preps, cultures, and biopsies, Dr. Friedman said. Better diagnostic tools and improved training for clinicians outside of dermatology also could reduce the misdiagnosis of fungal infections, he added. “Many of these patients are misdiagnosed in the emergency department, urgent care, or primary care settings,” and delayed treatment increases associated morbidity, he said.
Mr. Yadgar, Dr. Friedman, and another coauthor, Neal Bhatia, MD, of Therapeutics Clinical Research, San Diego, Calif., had no financial conflicts to disclose. There was no funding source.
Fungal skin infections may be missed or misdiagnosed by many dermatologists, according to the results of a survey published online in the Journal of the American Academy of Dermatology.
For the interactive survey, conducted during a session on fungal infections at the 2016 Orlando Dermatology Aesthetic and Clinical Conference, board-certified dermatologists viewed 13 clinical images (which included other conditions such as secondary syphilis and pityriasis rosea) and were asked via an audience response system whether or not they thought the case was a fungal skin infection. In only 1 of the 13 cases presented did 90% of the dermatologists correctly categorize the case as either a dermatomycosis or not, reported Ramsin Joseph Yadgar of George Washington University in Washington, D.C., and colleagues.
Although most cases (8 of 13) “were appropriately categorized by more than 50% of the audience, this percentage decreased as accuracy of categorization increased,” they wrote. “For example, in only 4 of the 13 cases did audience members accurately categorize the cases with more than 75% accuracy,” they said (J Am Acad Dermatol. 2016 Nov 11. pii: S0190-9622[16]30883-0. doi: 10.1016/j.jaad.2016.09.041).
“Dermatology is full of doppelgangers,” Dr. Friedman, director of the residency program and of translational research in the department of dermatology at George Washington University, said in an interview.
“While we [dermatologists] pride ourselves on our visual prowess, there are many skin diseases which do not follow the textbook and can be quite protean in their presentations,” he said.
The variability in presentation makes diagnosing fungal infections especially challenging, he noted. “Fungal infections of the skin can have many clinical flavors and can infect skin, hair and nails. Also, inappropriate treatment can obscure the appearance of the infection, and the fact that there are multiple other conditions that can look like these [fungal] infections makes proper identification difficult.”
Although the results were limited by several factors including possible selection bias, lack of measurable response rate, and small sample size, the findings highlight how easy it can be to miss a diagnosis of fungal infection, “which can result in inappropriate therapy, worsening of symptoms, and even additional skin and soft-tissue infections,” the researchers wrote.
“Keep an open mind and cast a wider differential,” to help catch fungal infections, and use all the dermatologic tools, including slide preps, cultures, and biopsies, Dr. Friedman said. Better diagnostic tools and improved training for clinicians outside of dermatology also could reduce the misdiagnosis of fungal infections, he added. “Many of these patients are misdiagnosed in the emergency department, urgent care, or primary care settings,” and delayed treatment increases associated morbidity, he said.
Mr. Yadgar, Dr. Friedman, and another coauthor, Neal Bhatia, MD, of Therapeutics Clinical Research, San Diego, Calif., had no financial conflicts to disclose. There was no funding source.
Fungal skin infections may be missed or misdiagnosed by many dermatologists, according to the results of a survey published online in the Journal of the American Academy of Dermatology.
For the interactive survey, conducted during a session on fungal infections at the 2016 Orlando Dermatology Aesthetic and Clinical Conference, board-certified dermatologists viewed 13 clinical images (which included other conditions such as secondary syphilis and pityriasis rosea) and were asked via an audience response system whether or not they thought the case was a fungal skin infection. In only 1 of the 13 cases presented did 90% of the dermatologists correctly categorize the case as either a dermatomycosis or not, reported Ramsin Joseph Yadgar of George Washington University in Washington, D.C., and colleagues.
Although most cases (8 of 13) “were appropriately categorized by more than 50% of the audience, this percentage decreased as accuracy of categorization increased,” they wrote. “For example, in only 4 of the 13 cases did audience members accurately categorize the cases with more than 75% accuracy,” they said (J Am Acad Dermatol. 2016 Nov 11. pii: S0190-9622[16]30883-0. doi: 10.1016/j.jaad.2016.09.041).
“Dermatology is full of doppelgangers,” Dr. Friedman, director of the residency program and of translational research in the department of dermatology at George Washington University, said in an interview.
“While we [dermatologists] pride ourselves on our visual prowess, there are many skin diseases which do not follow the textbook and can be quite protean in their presentations,” he said.
The variability in presentation makes diagnosing fungal infections especially challenging, he noted. “Fungal infections of the skin can have many clinical flavors and can infect skin, hair and nails. Also, inappropriate treatment can obscure the appearance of the infection, and the fact that there are multiple other conditions that can look like these [fungal] infections makes proper identification difficult.”
Although the results were limited by several factors including possible selection bias, lack of measurable response rate, and small sample size, the findings highlight how easy it can be to miss a diagnosis of fungal infection, “which can result in inappropriate therapy, worsening of symptoms, and even additional skin and soft-tissue infections,” the researchers wrote.
“Keep an open mind and cast a wider differential,” to help catch fungal infections, and use all the dermatologic tools, including slide preps, cultures, and biopsies, Dr. Friedman said. Better diagnostic tools and improved training for clinicians outside of dermatology also could reduce the misdiagnosis of fungal infections, he added. “Many of these patients are misdiagnosed in the emergency department, urgent care, or primary care settings,” and delayed treatment increases associated morbidity, he said.
Mr. Yadgar, Dr. Friedman, and another coauthor, Neal Bhatia, MD, of Therapeutics Clinical Research, San Diego, Calif., had no financial conflicts to disclose. There was no funding source.
FROM JAAD
Key clinical point: Fungal infections may often be missed or misdiagnosed by dermatologists.
Major finding: In 1 of 13 cases did 90% of an audience of dermatologists correctly categorize the condition.
Data source: A survey of board-certified dermatologists, asked whether or not 13 clinical images were a fungal infection or not, during a session on fungal infections at a dermatology meeting.
Disclosures: The research team had no relevant financial conflicts to disclose.
VIDEO: Topical antifungals win with patients
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM SDEF LAS VEGAS SEMINAR
