User login
Extensive Purpura and Necrosis of the Leg
The Diagnosis: Disseminated Mucormycosis
Histopathologic examination of a 6-mm punch biopsy of the edge of the lesion revealed numerous intravascular, broad, nonseptate hyphae in the deep vessels and perivascular dermis that stained bright red with periodic acid-Schiff (Figure). Acid-fast bacilli and Gram stains were negative. Tissue culture grew Rhizopus species. Given the patient's overall poor prognosis, her family decided to pursue hospice care following this diagnosis.
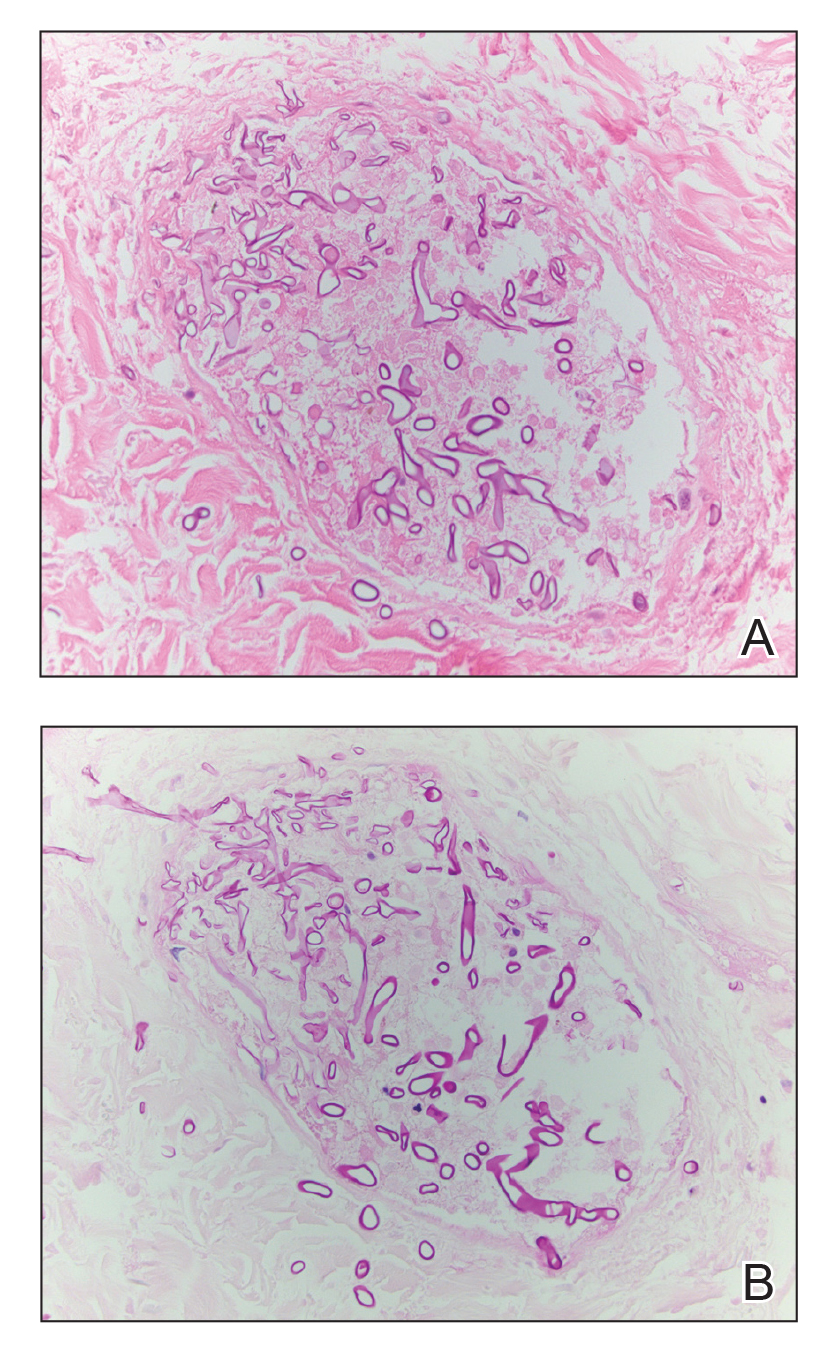
Mucormycosis (formerly zygomycosis) refers to infections from a variety of genera of fungi, most commonly Mucor and Rhizopus, that cause infections primarily in immunocompromised individuals.1 Mucormycosis infections are characterized by tissue necrosis that results from invasion of the vasculature and subsequent thrombosis. The typical presentation of cutaneous mucormycosis is a necrotic eschar accompanied by surrounding erythema and induration.2 Diagnosis is based on clinical suspicion, requiring additional testing with skin biopsy and tissue cultures for confirmation.
Cutaneous infection is the third most common presentation of mucormycosis, following rhinocerebral and pulmonary involvement.1 Although rhinocerebral and pulmonary infections normally are caused by inhalation of spores, cutaneous mucormycosis typically is caused by local inoculation, often following skin trauma.2 The skin is the most common location of iatrogenic mucormycosis, often from skin injury related to surgery, catheters, and adhesive tape.3 Most patients with cutaneous mucormycosis have underlying conditions such as hematologic malignancies, diabetes mellitus, or immunosuppression.1 However, outbreaks have occurred in immunocompetent patients following natural disasters.4 Cutaneous mucormycosis disseminates in 13% to 20% of cases in which mortality rates typically exceed 90%.1
Treatment consists of prompt surgical debridement and antifungal agents such as amphotericin B, posaconazole, and isavuconazonium sulfate.1 Our patient had multiple risk factors for infection, including hematopoietic stem cell transplantation, prolonged neutropenia, and treatment with eculizumab, a monoclonal antibody against C5 that blocks the terminal complement cascade. Eculizumab has been associated with increased risk for meningococcemia,5 but the association with mucormycosis is rare. We highlight the importance of recognizing and promptly diagnosing cutaneous mucormycosis given the difficulty of treating this disease and its poor prognosis.
Disseminated aspergillosis demonstrates septate rather than nonseptate hyphae on biopsy. Disseminated intravascular coagulation and purpura fulminans may be associated with thrombocytopenia but demonstrate thrombotic microangiopathy on biopsy. Pyoderma gangrenosum demonstrates neutrophilic infiltrate on biopsy.
- Roden MM, Zaoutis TE, Buchanan WL, et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin Infect Dis. 2005;41:634-653.
- Petrikkos G, Skiada A, Lortholary O, et al. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S23-S34.
- Rammaert B, Lanternier F, Zahar JR, et al. Healthcare-associated mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S44-S54.
- Neblett Fanfair R, Benedict K, Bos J, et al. Necrotizing cutaneous mucormycosis after a tornado in Joplin, Missouri, in 2011. N Engl J Med. 2012;367:2214-2225.
- McNamara LA, Topaz N, Wang X, et al. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2017;66:734-737.
The Diagnosis: Disseminated Mucormycosis
Histopathologic examination of a 6-mm punch biopsy of the edge of the lesion revealed numerous intravascular, broad, nonseptate hyphae in the deep vessels and perivascular dermis that stained bright red with periodic acid-Schiff (Figure). Acid-fast bacilli and Gram stains were negative. Tissue culture grew Rhizopus species. Given the patient's overall poor prognosis, her family decided to pursue hospice care following this diagnosis.
Mucormycosis (formerly zygomycosis) refers to infections from a variety of genera of fungi, most commonly Mucor and Rhizopus, that cause infections primarily in immunocompromised individuals.1 Mucormycosis infections are characterized by tissue necrosis that results from invasion of the vasculature and subsequent thrombosis. The typical presentation of cutaneous mucormycosis is a necrotic eschar accompanied by surrounding erythema and induration.2 Diagnosis is based on clinical suspicion, requiring additional testing with skin biopsy and tissue cultures for confirmation.
Cutaneous infection is the third most common presentation of mucormycosis, following rhinocerebral and pulmonary involvement.1 Although rhinocerebral and pulmonary infections normally are caused by inhalation of spores, cutaneous mucormycosis typically is caused by local inoculation, often following skin trauma.2 The skin is the most common location of iatrogenic mucormycosis, often from skin injury related to surgery, catheters, and adhesive tape.3 Most patients with cutaneous mucormycosis have underlying conditions such as hematologic malignancies, diabetes mellitus, or immunosuppression.1 However, outbreaks have occurred in immunocompetent patients following natural disasters.4 Cutaneous mucormycosis disseminates in 13% to 20% of cases in which mortality rates typically exceed 90%.1
Treatment consists of prompt surgical debridement and antifungal agents such as amphotericin B, posaconazole, and isavuconazonium sulfate.1 Our patient had multiple risk factors for infection, including hematopoietic stem cell transplantation, prolonged neutropenia, and treatment with eculizumab, a monoclonal antibody against C5 that blocks the terminal complement cascade. Eculizumab has been associated with increased risk for meningococcemia,5 but the association with mucormycosis is rare. We highlight the importance of recognizing and promptly diagnosing cutaneous mucormycosis given the difficulty of treating this disease and its poor prognosis.
Disseminated aspergillosis demonstrates septate rather than nonseptate hyphae on biopsy. Disseminated intravascular coagulation and purpura fulminans may be associated with thrombocytopenia but demonstrate thrombotic microangiopathy on biopsy. Pyoderma gangrenosum demonstrates neutrophilic infiltrate on biopsy.
The Diagnosis: Disseminated Mucormycosis
Histopathologic examination of a 6-mm punch biopsy of the edge of the lesion revealed numerous intravascular, broad, nonseptate hyphae in the deep vessels and perivascular dermis that stained bright red with periodic acid-Schiff (Figure). Acid-fast bacilli and Gram stains were negative. Tissue culture grew Rhizopus species. Given the patient's overall poor prognosis, her family decided to pursue hospice care following this diagnosis.
Mucormycosis (formerly zygomycosis) refers to infections from a variety of genera of fungi, most commonly Mucor and Rhizopus, that cause infections primarily in immunocompromised individuals.1 Mucormycosis infections are characterized by tissue necrosis that results from invasion of the vasculature and subsequent thrombosis. The typical presentation of cutaneous mucormycosis is a necrotic eschar accompanied by surrounding erythema and induration.2 Diagnosis is based on clinical suspicion, requiring additional testing with skin biopsy and tissue cultures for confirmation.
Cutaneous infection is the third most common presentation of mucormycosis, following rhinocerebral and pulmonary involvement.1 Although rhinocerebral and pulmonary infections normally are caused by inhalation of spores, cutaneous mucormycosis typically is caused by local inoculation, often following skin trauma.2 The skin is the most common location of iatrogenic mucormycosis, often from skin injury related to surgery, catheters, and adhesive tape.3 Most patients with cutaneous mucormycosis have underlying conditions such as hematologic malignancies, diabetes mellitus, or immunosuppression.1 However, outbreaks have occurred in immunocompetent patients following natural disasters.4 Cutaneous mucormycosis disseminates in 13% to 20% of cases in which mortality rates typically exceed 90%.1
Treatment consists of prompt surgical debridement and antifungal agents such as amphotericin B, posaconazole, and isavuconazonium sulfate.1 Our patient had multiple risk factors for infection, including hematopoietic stem cell transplantation, prolonged neutropenia, and treatment with eculizumab, a monoclonal antibody against C5 that blocks the terminal complement cascade. Eculizumab has been associated with increased risk for meningococcemia,5 but the association with mucormycosis is rare. We highlight the importance of recognizing and promptly diagnosing cutaneous mucormycosis given the difficulty of treating this disease and its poor prognosis.
Disseminated aspergillosis demonstrates septate rather than nonseptate hyphae on biopsy. Disseminated intravascular coagulation and purpura fulminans may be associated with thrombocytopenia but demonstrate thrombotic microangiopathy on biopsy. Pyoderma gangrenosum demonstrates neutrophilic infiltrate on biopsy.
- Roden MM, Zaoutis TE, Buchanan WL, et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin Infect Dis. 2005;41:634-653.
- Petrikkos G, Skiada A, Lortholary O, et al. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S23-S34.
- Rammaert B, Lanternier F, Zahar JR, et al. Healthcare-associated mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S44-S54.
- Neblett Fanfair R, Benedict K, Bos J, et al. Necrotizing cutaneous mucormycosis after a tornado in Joplin, Missouri, in 2011. N Engl J Med. 2012;367:2214-2225.
- McNamara LA, Topaz N, Wang X, et al. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2017;66:734-737.
- Roden MM, Zaoutis TE, Buchanan WL, et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin Infect Dis. 2005;41:634-653.
- Petrikkos G, Skiada A, Lortholary O, et al. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S23-S34.
- Rammaert B, Lanternier F, Zahar JR, et al. Healthcare-associated mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S44-S54.
- Neblett Fanfair R, Benedict K, Bos J, et al. Necrotizing cutaneous mucormycosis after a tornado in Joplin, Missouri, in 2011. N Engl J Med. 2012;367:2214-2225.
- McNamara LA, Topaz N, Wang X, et al. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2017;66:734-737.
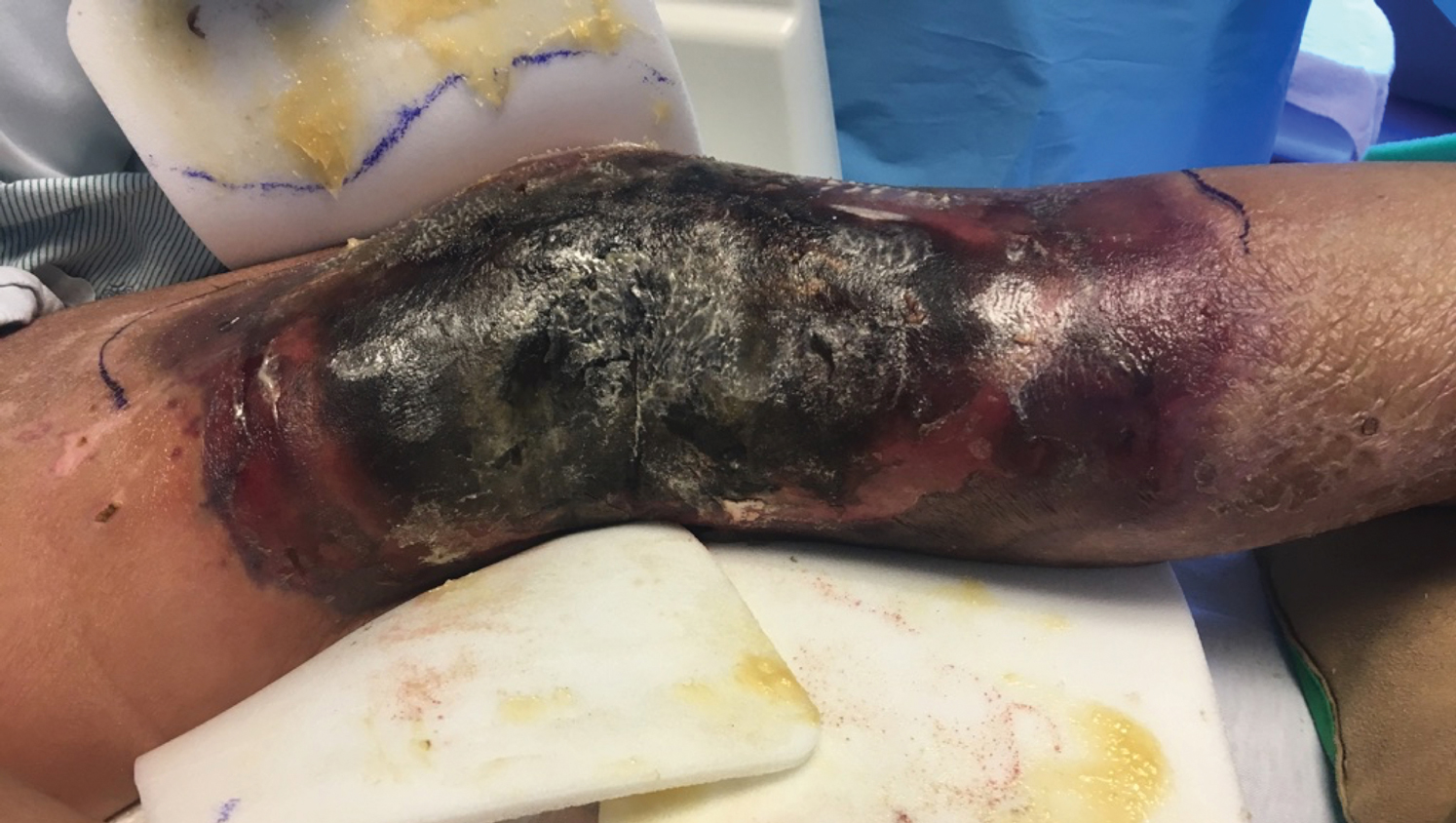
A 57-year-old woman presented with expanding purpura on the left leg of 2 weeks’ duration following a recent hematopoietic stem cell transplant for refractory diffuse large B-cell lymphoma. Prior to dermatologic consultation, the patient had been hospitalized for 2 months following the transplant due to Clostridium difficile colitis, Enterococcus faecium bacteremia, cardiac arrest, delayed engraftment with pancytopenia, and atypical hemolytic uremic syndrome with acute renal failure requiring hemodialysis and treatment with eculizumab. Her care team in the hospital initially noticed a small purpuric lesion on the posterior aspect of the left knee. The patient subsequently developed persistent fevers and expansion of the lesion, which prompted consultation of the dermatology service. Physical examination revealed a 22×10-cm, rectangular, indurated, purpuric plaque with central dusky, violaceous to black necrosis with superficial skin sloughing and peripheral dusky erythema extending from the inner thigh to the lower leg. The left distal leg felt cool, and both dorsalis pedis and posterior tibial pulses were absent. Laboratory test results revealed neutropenia and thrombocytopenia (white blood cell count, 0.2×103 /mm3 [reference range, 5–10×103 /mm3 ]; hematocrit, 23.2% [reference range, 41%–50%]; platelet count, 105×103 /µL [reference range, 150–350×103 /µL]). A punch biopsy was performed.
Antibiotic resistance: Personal responsibility in somewhat short supply
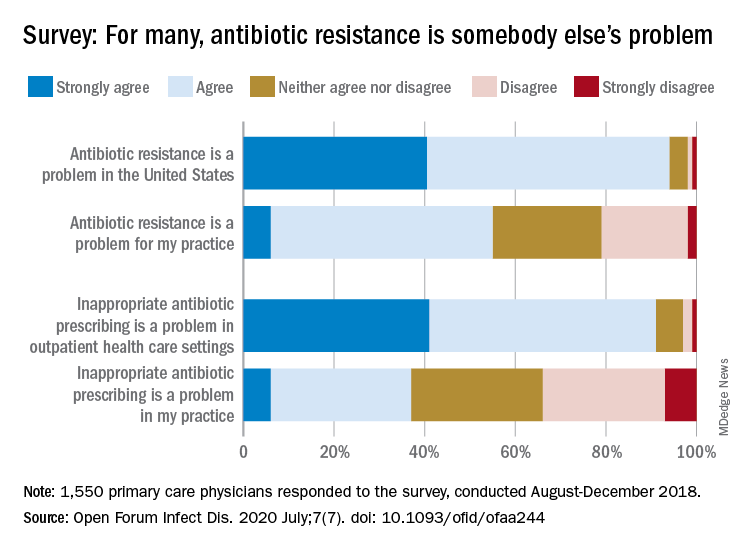
Most primary care physicians agree that antibiotic resistance and inappropriate prescribing are problems in the United States, but they are much less inclined to recognize these issues in their own practices, according to the results of a nationwide survey.
Rachel M. Zetts, MPH, of the Pew Charitable Trusts, Washington, D.C., and associates wrote in Open Forum Infectious Diseases.
Almost all (94%) of the 1,550 internists, family physicians, and pediatricians who responded to the survey said that antibiotic resistance is a national problem, and nearly that many (91%) agreed that “inappropriate antibiotic prescribing is a problem in outpatient health care settings,” the investigators acknowledged.
Narrowing the focus to their own practices, however, changed some opinions. At that level, only 55% of the respondents said that resistance was a problem for their practices, and just 37% said that there any sort of inappropriate prescribing going on, based on data from the survey, which was conducted from August to October 2018 by Pew and the American Medical Association.
Antibiotic stewardship, defined as activities meant to ensure appropriate prescribing of antibiotics, should include “staff and patient education, clinician-level antibiotic prescribing feedback, and communications training on how to discuss antibiotic prescribing with patients,” Ms. Zetts and associates explained.
The need for such stewardship in health care settings was acknowledged by 72% of respondents, but 53% of those surveyed also said that all they need to do to support such efforts “is to talk with their patients about the value of an antibiotic for their symptoms,” they noted.
The bacteria, it seems, are not the only ones with some resistance. Half of the primary care physicians believe that it would be difficult to fairly and accurately track the appropriate use of antibiotics, and 52% agreed with the statement that “practice-based reporting requirements for antibiotic use would be too onerous,” the researchers pointed out.
“Antibiotic resistance is an impending public health crisis. We are seeing today, as we respond to the COVID-19 pandemic, what our health system looks like with no or limited treatments available to tackle an outbreak. … We must all remain vigilant in combating the spread of antibiotic resistant bacteria and be prudent when prescribing antibiotics,” AMA President Susan R. Bailey, MD, said in a written statement.
SOURCE: Zetts RM et al. Open Forum Infect Dis. 2020 July;7(7). doi: 10.1093/ofid/ofaa244.
Most primary care physicians agree that antibiotic resistance and inappropriate prescribing are problems in the United States, but they are much less inclined to recognize these issues in their own practices, according to the results of a nationwide survey.
Rachel M. Zetts, MPH, of the Pew Charitable Trusts, Washington, D.C., and associates wrote in Open Forum Infectious Diseases.
Almost all (94%) of the 1,550 internists, family physicians, and pediatricians who responded to the survey said that antibiotic resistance is a national problem, and nearly that many (91%) agreed that “inappropriate antibiotic prescribing is a problem in outpatient health care settings,” the investigators acknowledged.
Narrowing the focus to their own practices, however, changed some opinions. At that level, only 55% of the respondents said that resistance was a problem for their practices, and just 37% said that there any sort of inappropriate prescribing going on, based on data from the survey, which was conducted from August to October 2018 by Pew and the American Medical Association.
Antibiotic stewardship, defined as activities meant to ensure appropriate prescribing of antibiotics, should include “staff and patient education, clinician-level antibiotic prescribing feedback, and communications training on how to discuss antibiotic prescribing with patients,” Ms. Zetts and associates explained.
The need for such stewardship in health care settings was acknowledged by 72% of respondents, but 53% of those surveyed also said that all they need to do to support such efforts “is to talk with their patients about the value of an antibiotic for their symptoms,” they noted.
The bacteria, it seems, are not the only ones with some resistance. Half of the primary care physicians believe that it would be difficult to fairly and accurately track the appropriate use of antibiotics, and 52% agreed with the statement that “practice-based reporting requirements for antibiotic use would be too onerous,” the researchers pointed out.
“Antibiotic resistance is an impending public health crisis. We are seeing today, as we respond to the COVID-19 pandemic, what our health system looks like with no or limited treatments available to tackle an outbreak. … We must all remain vigilant in combating the spread of antibiotic resistant bacteria and be prudent when prescribing antibiotics,” AMA President Susan R. Bailey, MD, said in a written statement.
SOURCE: Zetts RM et al. Open Forum Infect Dis. 2020 July;7(7). doi: 10.1093/ofid/ofaa244.
Most primary care physicians agree that antibiotic resistance and inappropriate prescribing are problems in the United States, but they are much less inclined to recognize these issues in their own practices, according to the results of a nationwide survey.
Rachel M. Zetts, MPH, of the Pew Charitable Trusts, Washington, D.C., and associates wrote in Open Forum Infectious Diseases.
Almost all (94%) of the 1,550 internists, family physicians, and pediatricians who responded to the survey said that antibiotic resistance is a national problem, and nearly that many (91%) agreed that “inappropriate antibiotic prescribing is a problem in outpatient health care settings,” the investigators acknowledged.
Narrowing the focus to their own practices, however, changed some opinions. At that level, only 55% of the respondents said that resistance was a problem for their practices, and just 37% said that there any sort of inappropriate prescribing going on, based on data from the survey, which was conducted from August to October 2018 by Pew and the American Medical Association.
Antibiotic stewardship, defined as activities meant to ensure appropriate prescribing of antibiotics, should include “staff and patient education, clinician-level antibiotic prescribing feedback, and communications training on how to discuss antibiotic prescribing with patients,” Ms. Zetts and associates explained.
The need for such stewardship in health care settings was acknowledged by 72% of respondents, but 53% of those surveyed also said that all they need to do to support such efforts “is to talk with their patients about the value of an antibiotic for their symptoms,” they noted.
The bacteria, it seems, are not the only ones with some resistance. Half of the primary care physicians believe that it would be difficult to fairly and accurately track the appropriate use of antibiotics, and 52% agreed with the statement that “practice-based reporting requirements for antibiotic use would be too onerous,” the researchers pointed out.
“Antibiotic resistance is an impending public health crisis. We are seeing today, as we respond to the COVID-19 pandemic, what our health system looks like with no or limited treatments available to tackle an outbreak. … We must all remain vigilant in combating the spread of antibiotic resistant bacteria and be prudent when prescribing antibiotics,” AMA President Susan R. Bailey, MD, said in a written statement.
SOURCE: Zetts RM et al. Open Forum Infect Dis. 2020 July;7(7). doi: 10.1093/ofid/ofaa244.
FROM OPEN FORUM INFECTIOUS DISEASES
Guidance covers glycemia in dexamethasone-treated COVID-19 patients
New guidance from the U.K. National Diabetes COVID-19 Response Group addresses glucose management in patients with COVID-19 who are receiving dexamethasone therapy.
Although there are already guidelines that address inpatient management of steroid-induced hyperglycemia, the authors of the new document wrote that this new expert opinion paper was needed “given the ‘triple insult’ of dexamethasone-induced–impaired glucose metabolism, COVID-19–induced insulin resistance, and COVID-19–impaired insulin production.”
RECOVERY trial spurs response
The document, which is the latest in a series from the Association of British Clinical Diabetologists, was published online Aug. 2 in Diabetic Medicine. The group is chaired by Gerry Rayman, MD, consultant physician at the diabetes centre and diabetes research unit, East Suffolk (England) and North East NHS Foundation Trust.
The guidance was developed in response to the recent “breakthrough” Randomised Evaluation of COVID-19 Therapy (RECOVERY) trial, which showed that dexamethasone reduced deaths in patients with COVID-19 on ventilators or receiving oxygen therapy. The advice is not intended for critical care units but can be adapted for that use.
The dose used in RECOVERY – 6 mg daily for 10 days – is 400%-500% greater than the therapeutic glucocorticoid replacement dose. High glucocorticoid doses can exacerbate hyperglycemia in people with established diabetes, unmask undiagnosed diabetes, precipitate hyperglycemia or new-onset diabetes, and can also cause hyperglycemic hyperosmolar state (HHS), the authors explained.
They recommended a target glucose of 6.0-10.0 mmol/L (108-180 mg/dL), although they say up to 12 mmol/L (216 mg/dL) is “acceptable.” They then gave advice on frequency of monitoring for people with and without known diabetes, exclusion of diabetic ketoacidosis and HHS, correction of initial hyperglycemia and maintenance of glycemic control using subcutaneous insulin, and prevention of hypoglycemia at the end of dexamethasone therapy (day 10) with insulin down-titration, discharge, and follow-up.
The detailed insulin guidance covers dose escalation for both insulin-treated and insulin-naive patients. A table suggests increasing correction doses of rapid-acting insulin based on prior total daily dose or weight.
Use of once- or twice-daily NPH insulin is recommended for patients whose glucose has risen above 12 mmol/L, in some cases with the addition of a long-acting analog. A second chart gives dose adjustments for those insulins. Additional guidance addresses patients on insulin pumps.
Guidance useful for U.S. physicians
Francisco Pasquel, MD, assistant professor of medicine in the division of endocrinology at Emory University, Atlanta, said in an interview that he believes the guidance is “acceptable” for worldwide use, and that “it’s coherent and consistent with what we typically do.”
However, Dr. Pasquel, who founded COVID-in-Diabetes, an online repository of published guidance and shared experience – to which this new document has now been added – did take issue with one piece of advice. The guidance says that patients already taking premixed insulin formulations can continue using them while increasing the dose by 20%-40%. Given the risk of hypoglycemia associated with those formulations, Dr. Pasquel said he would switch those patients to NPH during the time that they’re on dexamethasone.
He also noted that the rapid-acting insulin dose range of 2-10 units provided in the first table, for correction of initial hyperglycemia, are more conservative than those used at his hospital, where correction doses of up to 14-16 units are sometimes necessary.
But Dr. Pasquel praised the group’s overall efforts since the pandemic began, noting that “they’re very organized and constantly updating their recommendations. They have a unified system in the [National Health Service], so it’s easier to standardize. They have a unique [electronic health record] which is far superior to what we do from a public health perspective.”
Dr. Rayman reported no relevant financial relationships. Dr. Pasquel reported receiving research funding from Dexcom, Merck, and the National Institutes of Health, and consulting for AstraZeneca, Eli Lilly, Merck, and Boehringer Ingelheim.
A version of this article originally appeared on Medscape.com.
New guidance from the U.K. National Diabetes COVID-19 Response Group addresses glucose management in patients with COVID-19 who are receiving dexamethasone therapy.
Although there are already guidelines that address inpatient management of steroid-induced hyperglycemia, the authors of the new document wrote that this new expert opinion paper was needed “given the ‘triple insult’ of dexamethasone-induced–impaired glucose metabolism, COVID-19–induced insulin resistance, and COVID-19–impaired insulin production.”
RECOVERY trial spurs response
The document, which is the latest in a series from the Association of British Clinical Diabetologists, was published online Aug. 2 in Diabetic Medicine. The group is chaired by Gerry Rayman, MD, consultant physician at the diabetes centre and diabetes research unit, East Suffolk (England) and North East NHS Foundation Trust.
The guidance was developed in response to the recent “breakthrough” Randomised Evaluation of COVID-19 Therapy (RECOVERY) trial, which showed that dexamethasone reduced deaths in patients with COVID-19 on ventilators or receiving oxygen therapy. The advice is not intended for critical care units but can be adapted for that use.
The dose used in RECOVERY – 6 mg daily for 10 days – is 400%-500% greater than the therapeutic glucocorticoid replacement dose. High glucocorticoid doses can exacerbate hyperglycemia in people with established diabetes, unmask undiagnosed diabetes, precipitate hyperglycemia or new-onset diabetes, and can also cause hyperglycemic hyperosmolar state (HHS), the authors explained.
They recommended a target glucose of 6.0-10.0 mmol/L (108-180 mg/dL), although they say up to 12 mmol/L (216 mg/dL) is “acceptable.” They then gave advice on frequency of monitoring for people with and without known diabetes, exclusion of diabetic ketoacidosis and HHS, correction of initial hyperglycemia and maintenance of glycemic control using subcutaneous insulin, and prevention of hypoglycemia at the end of dexamethasone therapy (day 10) with insulin down-titration, discharge, and follow-up.
The detailed insulin guidance covers dose escalation for both insulin-treated and insulin-naive patients. A table suggests increasing correction doses of rapid-acting insulin based on prior total daily dose or weight.
Use of once- or twice-daily NPH insulin is recommended for patients whose glucose has risen above 12 mmol/L, in some cases with the addition of a long-acting analog. A second chart gives dose adjustments for those insulins. Additional guidance addresses patients on insulin pumps.
Guidance useful for U.S. physicians
Francisco Pasquel, MD, assistant professor of medicine in the division of endocrinology at Emory University, Atlanta, said in an interview that he believes the guidance is “acceptable” for worldwide use, and that “it’s coherent and consistent with what we typically do.”
However, Dr. Pasquel, who founded COVID-in-Diabetes, an online repository of published guidance and shared experience – to which this new document has now been added – did take issue with one piece of advice. The guidance says that patients already taking premixed insulin formulations can continue using them while increasing the dose by 20%-40%. Given the risk of hypoglycemia associated with those formulations, Dr. Pasquel said he would switch those patients to NPH during the time that they’re on dexamethasone.
He also noted that the rapid-acting insulin dose range of 2-10 units provided in the first table, for correction of initial hyperglycemia, are more conservative than those used at his hospital, where correction doses of up to 14-16 units are sometimes necessary.
But Dr. Pasquel praised the group’s overall efforts since the pandemic began, noting that “they’re very organized and constantly updating their recommendations. They have a unified system in the [National Health Service], so it’s easier to standardize. They have a unique [electronic health record] which is far superior to what we do from a public health perspective.”
Dr. Rayman reported no relevant financial relationships. Dr. Pasquel reported receiving research funding from Dexcom, Merck, and the National Institutes of Health, and consulting for AstraZeneca, Eli Lilly, Merck, and Boehringer Ingelheim.
A version of this article originally appeared on Medscape.com.
New guidance from the U.K. National Diabetes COVID-19 Response Group addresses glucose management in patients with COVID-19 who are receiving dexamethasone therapy.
Although there are already guidelines that address inpatient management of steroid-induced hyperglycemia, the authors of the new document wrote that this new expert opinion paper was needed “given the ‘triple insult’ of dexamethasone-induced–impaired glucose metabolism, COVID-19–induced insulin resistance, and COVID-19–impaired insulin production.”
RECOVERY trial spurs response
The document, which is the latest in a series from the Association of British Clinical Diabetologists, was published online Aug. 2 in Diabetic Medicine. The group is chaired by Gerry Rayman, MD, consultant physician at the diabetes centre and diabetes research unit, East Suffolk (England) and North East NHS Foundation Trust.
The guidance was developed in response to the recent “breakthrough” Randomised Evaluation of COVID-19 Therapy (RECOVERY) trial, which showed that dexamethasone reduced deaths in patients with COVID-19 on ventilators or receiving oxygen therapy. The advice is not intended for critical care units but can be adapted for that use.
The dose used in RECOVERY – 6 mg daily for 10 days – is 400%-500% greater than the therapeutic glucocorticoid replacement dose. High glucocorticoid doses can exacerbate hyperglycemia in people with established diabetes, unmask undiagnosed diabetes, precipitate hyperglycemia or new-onset diabetes, and can also cause hyperglycemic hyperosmolar state (HHS), the authors explained.
They recommended a target glucose of 6.0-10.0 mmol/L (108-180 mg/dL), although they say up to 12 mmol/L (216 mg/dL) is “acceptable.” They then gave advice on frequency of monitoring for people with and without known diabetes, exclusion of diabetic ketoacidosis and HHS, correction of initial hyperglycemia and maintenance of glycemic control using subcutaneous insulin, and prevention of hypoglycemia at the end of dexamethasone therapy (day 10) with insulin down-titration, discharge, and follow-up.
The detailed insulin guidance covers dose escalation for both insulin-treated and insulin-naive patients. A table suggests increasing correction doses of rapid-acting insulin based on prior total daily dose or weight.
Use of once- or twice-daily NPH insulin is recommended for patients whose glucose has risen above 12 mmol/L, in some cases with the addition of a long-acting analog. A second chart gives dose adjustments for those insulins. Additional guidance addresses patients on insulin pumps.
Guidance useful for U.S. physicians
Francisco Pasquel, MD, assistant professor of medicine in the division of endocrinology at Emory University, Atlanta, said in an interview that he believes the guidance is “acceptable” for worldwide use, and that “it’s coherent and consistent with what we typically do.”
However, Dr. Pasquel, who founded COVID-in-Diabetes, an online repository of published guidance and shared experience – to which this new document has now been added – did take issue with one piece of advice. The guidance says that patients already taking premixed insulin formulations can continue using them while increasing the dose by 20%-40%. Given the risk of hypoglycemia associated with those formulations, Dr. Pasquel said he would switch those patients to NPH during the time that they’re on dexamethasone.
He also noted that the rapid-acting insulin dose range of 2-10 units provided in the first table, for correction of initial hyperglycemia, are more conservative than those used at his hospital, where correction doses of up to 14-16 units are sometimes necessary.
But Dr. Pasquel praised the group’s overall efforts since the pandemic began, noting that “they’re very organized and constantly updating their recommendations. They have a unified system in the [National Health Service], so it’s easier to standardize. They have a unique [electronic health record] which is far superior to what we do from a public health perspective.”
Dr. Rayman reported no relevant financial relationships. Dr. Pasquel reported receiving research funding from Dexcom, Merck, and the National Institutes of Health, and consulting for AstraZeneca, Eli Lilly, Merck, and Boehringer Ingelheim.
A version of this article originally appeared on Medscape.com.
Educational intervention curbs use of antibiotics for respiratory infections
A clinician education program significantly reduced overall antibiotic prescribing during pediatric visits for acute respiratory tract infections, according to data from 57 clinicians who participated in an intervention.

In a study published in Pediatrics, Matthew P. Kronman, MD, of the University of Washington, Seattle, and associates randomized 57 clinicians at 19 pediatric practices to a stepped-wedge clinical trial. The study included visits for acute otitis media, bronchitis, pharyngitis, sinusitis, and upper respiratory infections (defined as ARTI visits) for children aged 6 months to less than 11 years, for a total of 72,723 ARTI visits by 29,762 patients. The primary outcome was overall antibiotic prescribing for ARTI visits.
For the intervention, known as the Dialogue Around Respiratory Illness Treatment (DART) quality improvement (QI) program, clinicians received three program modules containing online tutorials and webinars. These professionally-produced modules included a combination of evidence-based communication strategies and antibiotic prescribing, booster video vignettes, and individualized antibiotic prescribing feedback reports over 11 months.
Overall, the probability of antibiotic prescribing for ARTI visits decreased by 7% (adjusted relative risk 0.93) from baseline to a 2- to 8-month postintervention in an adjusted intent-to-treat analysis.
Analysis of secondary outcomes revealed that prescribing any antibiotics for viral ARTI decreased by 40% during the postintervention period compared to baseline (aRR 0.60).
In addition, second-line antibiotic prescribing decreased from baseline by 34% for streptococcal pharyngitis (aRR 0.66), and by 41% for sinusitis (aRR 0.59); however there was no significant change in prescribing for acute otitis media, the researchers said.
The study findings were limited by several factors including the potential for biased results because of the randomization of clinicians from multiple practices and the potential for clinicians to change their prescribing habits after the start of the study, Dr. Kronman and colleagues noted.
In addition, the study did not include complete data on rapid streptococcal antigen testing, which might eliminate some children from the study population, and the relatively short postintervention period “may not represent the true long-term intervention durability may not represent the true long-term intervention durability,” they said.
However, the results support the potential of the DART program. “The 7% reduction in antibiotic prescribing for all ARTIs, if extrapolated to all ambulatory ARTI visits to pediatricians nationally, would represent 1.5 million fewer antibiotic prescriptions for children with ARTI annually,” they wrote.
“Providing online communication training and evidence-based antibiotic prescribing education in combination with individualized antibiotic prescribing feedback reports may help achieve national goals of reducing unnecessary outpatient antibiotic prescribing for children,” Dr. Kronman and associates concluded.
Combining interventions are key to reducing unnecessary antibiotics use in pediatric ambulatory care, Rana F. Hamdy, MD, MPH, of Children’s National Hospital, Washington, , and Sophie E. Katz, MD, of Vanderbilt University, Nashville, Tenn., wrote in an accompanying editorial (Pediatrics. 2020 Aug 3. doi: 10.1542/peds.2020-012922).
The researchers in the current study “seem to recognize that clinicians are adult learners, and they combine interventions to implement these adult learning theory tenets to improve appropriate antibiotic prescribing,” they wrote. The DART intervention combined best practices training, communications training, and individualized antibiotic prescribing feedback reports to improve communication between providers and families “especially when faced with a situation in which a parent or guardian might expect an antibiotic prescription but the provider does not think one is necessary,” Dr. Hamdy and Dr. Katz said.
Overall, the findings suggest that the interventions work best in combination vs. being used alone, although the study did not evaluate the separate contributions of each intervention, the editorialists wrote.
“In the current study, nonengaged physicians had an increase in second-line antibiotic prescribing, whereas the engaged physicians had a decrease in second-line antibiotic prescribing,” they noted. “This suggests that the addition of communications training could mitigate the undesirable effects that may result from solely using feedback reports.”
“Each year, U.S. children are prescribed as many as 10 million unnecessary antibiotic courses for acute respiratory tract infections,” Kristina A. Bryant, MD, of the University of Louisville, Ky., said in an interview. “Some of these prescriptions result in side effects or allergic reactions, and they contribute to growing antibiotic resistance. We need effective interventions to reduce antibiotic prescribing.”
Although the DART modules are free and available online, busy clinicians might struggle to find time to view them consistently, said Dr. Bryant.
“One advantage of the study design was that information was pushed to clinicians along with communication booster videos,” she said. “We know that education and reinforcement over time works better than a one and done approach.
“Study participants also received feedback over time about their prescribing habits, which can be a powerful motivator for change, although not all clinicians may have easy access to these reports,” she noted.
To overcome some of the barriers to using the modules, clinicians who are “interested in improving their prescribing could work with their office managers to develop antibiotic prescribing reports and schedule reminders to review them,” said Dr. Bryant.
“An individual could commit to education and review of his or her own prescribing patterns, but support from one’s partners and shared accountability is likely to be even more effective,” she said. “Sharing data within a practice and exploring differences in prescribing patterns can drive improvement.
“Spaced education and regular feedback about prescribing patterns can improve antibiotic prescribing for pharyngitis and sinusitis, and reduce antibiotic prescriptions for ARTIs,” Dr. Bryant said. The take-home from the study is that it should prompt anyone who prescribes antibiotics for children to ask themselves how they can improve their own prescribing habits.
“In this study, prescribing for viral ARTIs was reduced but not eliminated. We need additional studies to further reduce unnecessary antibiotic use,” Dr. Bryant said.
In addition, areas for future research could include longer-term follow-up. “Study participants were followed for 2 to 8 months after the intervention ended in June 2018. It would be interesting to know about their prescribing practices now, and if the changes observed in the study were durable,” she concluded.
The study was supported by the National Institutes of Health, along with additional infrastructure funding from the American Academy of Pediatrics and the Health Resources and Services Administration of the Department of Health and Human Services. The researchers had no financial conflicts to disclose.
Dr. Hamdy and Dr. Katz had no financial conflicts to disclose, but Dr. Katz disclosed grant support through the Centers for Disease Control and Prevention as a recipient of the Leadership in Epidemiology, Antimicrobial Stewardship, and Public Health fellowship, sponsored by the Society for Healthcare Epidemiology of America, Infectious Diseases Society of America, and Pediatric Infectious Diseases Society.
Dr. Bryant disclosed serving as an investigator on multicenter clinical vaccine trials funded by Pfizer (but not in the last year). She also serves as the current president of the Pediatric Infectious Diseases Society, but the opinions expressed here are her own and do not necessarily reflect the views of PIDS.
SOURCE: Kronman MP et al. Pediatrics. 2020 Aug 3. doi: 10.1542/peds.2020-0038.
A clinician education program significantly reduced overall antibiotic prescribing during pediatric visits for acute respiratory tract infections, according to data from 57 clinicians who participated in an intervention.
In a study published in Pediatrics, Matthew P. Kronman, MD, of the University of Washington, Seattle, and associates randomized 57 clinicians at 19 pediatric practices to a stepped-wedge clinical trial. The study included visits for acute otitis media, bronchitis, pharyngitis, sinusitis, and upper respiratory infections (defined as ARTI visits) for children aged 6 months to less than 11 years, for a total of 72,723 ARTI visits by 29,762 patients. The primary outcome was overall antibiotic prescribing for ARTI visits.
For the intervention, known as the Dialogue Around Respiratory Illness Treatment (DART) quality improvement (QI) program, clinicians received three program modules containing online tutorials and webinars. These professionally-produced modules included a combination of evidence-based communication strategies and antibiotic prescribing, booster video vignettes, and individualized antibiotic prescribing feedback reports over 11 months.
Overall, the probability of antibiotic prescribing for ARTI visits decreased by 7% (adjusted relative risk 0.93) from baseline to a 2- to 8-month postintervention in an adjusted intent-to-treat analysis.
Analysis of secondary outcomes revealed that prescribing any antibiotics for viral ARTI decreased by 40% during the postintervention period compared to baseline (aRR 0.60).
In addition, second-line antibiotic prescribing decreased from baseline by 34% for streptococcal pharyngitis (aRR 0.66), and by 41% for sinusitis (aRR 0.59); however there was no significant change in prescribing for acute otitis media, the researchers said.
The study findings were limited by several factors including the potential for biased results because of the randomization of clinicians from multiple practices and the potential for clinicians to change their prescribing habits after the start of the study, Dr. Kronman and colleagues noted.
In addition, the study did not include complete data on rapid streptococcal antigen testing, which might eliminate some children from the study population, and the relatively short postintervention period “may not represent the true long-term intervention durability may not represent the true long-term intervention durability,” they said.
However, the results support the potential of the DART program. “The 7% reduction in antibiotic prescribing for all ARTIs, if extrapolated to all ambulatory ARTI visits to pediatricians nationally, would represent 1.5 million fewer antibiotic prescriptions for children with ARTI annually,” they wrote.
“Providing online communication training and evidence-based antibiotic prescribing education in combination with individualized antibiotic prescribing feedback reports may help achieve national goals of reducing unnecessary outpatient antibiotic prescribing for children,” Dr. Kronman and associates concluded.
Combining interventions are key to reducing unnecessary antibiotics use in pediatric ambulatory care, Rana F. Hamdy, MD, MPH, of Children’s National Hospital, Washington, , and Sophie E. Katz, MD, of Vanderbilt University, Nashville, Tenn., wrote in an accompanying editorial (Pediatrics. 2020 Aug 3. doi: 10.1542/peds.2020-012922).
The researchers in the current study “seem to recognize that clinicians are adult learners, and they combine interventions to implement these adult learning theory tenets to improve appropriate antibiotic prescribing,” they wrote. The DART intervention combined best practices training, communications training, and individualized antibiotic prescribing feedback reports to improve communication between providers and families “especially when faced with a situation in which a parent or guardian might expect an antibiotic prescription but the provider does not think one is necessary,” Dr. Hamdy and Dr. Katz said.
Overall, the findings suggest that the interventions work best in combination vs. being used alone, although the study did not evaluate the separate contributions of each intervention, the editorialists wrote.
“In the current study, nonengaged physicians had an increase in second-line antibiotic prescribing, whereas the engaged physicians had a decrease in second-line antibiotic prescribing,” they noted. “This suggests that the addition of communications training could mitigate the undesirable effects that may result from solely using feedback reports.”
“Each year, U.S. children are prescribed as many as 10 million unnecessary antibiotic courses for acute respiratory tract infections,” Kristina A. Bryant, MD, of the University of Louisville, Ky., said in an interview. “Some of these prescriptions result in side effects or allergic reactions, and they contribute to growing antibiotic resistance. We need effective interventions to reduce antibiotic prescribing.”
Although the DART modules are free and available online, busy clinicians might struggle to find time to view them consistently, said Dr. Bryant.
“One advantage of the study design was that information was pushed to clinicians along with communication booster videos,” she said. “We know that education and reinforcement over time works better than a one and done approach.
“Study participants also received feedback over time about their prescribing habits, which can be a powerful motivator for change, although not all clinicians may have easy access to these reports,” she noted.
To overcome some of the barriers to using the modules, clinicians who are “interested in improving their prescribing could work with their office managers to develop antibiotic prescribing reports and schedule reminders to review them,” said Dr. Bryant.
“An individual could commit to education and review of his or her own prescribing patterns, but support from one’s partners and shared accountability is likely to be even more effective,” she said. “Sharing data within a practice and exploring differences in prescribing patterns can drive improvement.
“Spaced education and regular feedback about prescribing patterns can improve antibiotic prescribing for pharyngitis and sinusitis, and reduce antibiotic prescriptions for ARTIs,” Dr. Bryant said. The take-home from the study is that it should prompt anyone who prescribes antibiotics for children to ask themselves how they can improve their own prescribing habits.
“In this study, prescribing for viral ARTIs was reduced but not eliminated. We need additional studies to further reduce unnecessary antibiotic use,” Dr. Bryant said.
In addition, areas for future research could include longer-term follow-up. “Study participants were followed for 2 to 8 months after the intervention ended in June 2018. It would be interesting to know about their prescribing practices now, and if the changes observed in the study were durable,” she concluded.
The study was supported by the National Institutes of Health, along with additional infrastructure funding from the American Academy of Pediatrics and the Health Resources and Services Administration of the Department of Health and Human Services. The researchers had no financial conflicts to disclose.
Dr. Hamdy and Dr. Katz had no financial conflicts to disclose, but Dr. Katz disclosed grant support through the Centers for Disease Control and Prevention as a recipient of the Leadership in Epidemiology, Antimicrobial Stewardship, and Public Health fellowship, sponsored by the Society for Healthcare Epidemiology of America, Infectious Diseases Society of America, and Pediatric Infectious Diseases Society.
Dr. Bryant disclosed serving as an investigator on multicenter clinical vaccine trials funded by Pfizer (but not in the last year). She also serves as the current president of the Pediatric Infectious Diseases Society, but the opinions expressed here are her own and do not necessarily reflect the views of PIDS.
SOURCE: Kronman MP et al. Pediatrics. 2020 Aug 3. doi: 10.1542/peds.2020-0038.
A clinician education program significantly reduced overall antibiotic prescribing during pediatric visits for acute respiratory tract infections, according to data from 57 clinicians who participated in an intervention.
In a study published in Pediatrics, Matthew P. Kronman, MD, of the University of Washington, Seattle, and associates randomized 57 clinicians at 19 pediatric practices to a stepped-wedge clinical trial. The study included visits for acute otitis media, bronchitis, pharyngitis, sinusitis, and upper respiratory infections (defined as ARTI visits) for children aged 6 months to less than 11 years, for a total of 72,723 ARTI visits by 29,762 patients. The primary outcome was overall antibiotic prescribing for ARTI visits.
For the intervention, known as the Dialogue Around Respiratory Illness Treatment (DART) quality improvement (QI) program, clinicians received three program modules containing online tutorials and webinars. These professionally-produced modules included a combination of evidence-based communication strategies and antibiotic prescribing, booster video vignettes, and individualized antibiotic prescribing feedback reports over 11 months.
Overall, the probability of antibiotic prescribing for ARTI visits decreased by 7% (adjusted relative risk 0.93) from baseline to a 2- to 8-month postintervention in an adjusted intent-to-treat analysis.
Analysis of secondary outcomes revealed that prescribing any antibiotics for viral ARTI decreased by 40% during the postintervention period compared to baseline (aRR 0.60).
In addition, second-line antibiotic prescribing decreased from baseline by 34% for streptococcal pharyngitis (aRR 0.66), and by 41% for sinusitis (aRR 0.59); however there was no significant change in prescribing for acute otitis media, the researchers said.
The study findings were limited by several factors including the potential for biased results because of the randomization of clinicians from multiple practices and the potential for clinicians to change their prescribing habits after the start of the study, Dr. Kronman and colleagues noted.
In addition, the study did not include complete data on rapid streptococcal antigen testing, which might eliminate some children from the study population, and the relatively short postintervention period “may not represent the true long-term intervention durability may not represent the true long-term intervention durability,” they said.
However, the results support the potential of the DART program. “The 7% reduction in antibiotic prescribing for all ARTIs, if extrapolated to all ambulatory ARTI visits to pediatricians nationally, would represent 1.5 million fewer antibiotic prescriptions for children with ARTI annually,” they wrote.
“Providing online communication training and evidence-based antibiotic prescribing education in combination with individualized antibiotic prescribing feedback reports may help achieve national goals of reducing unnecessary outpatient antibiotic prescribing for children,” Dr. Kronman and associates concluded.
Combining interventions are key to reducing unnecessary antibiotics use in pediatric ambulatory care, Rana F. Hamdy, MD, MPH, of Children’s National Hospital, Washington, , and Sophie E. Katz, MD, of Vanderbilt University, Nashville, Tenn., wrote in an accompanying editorial (Pediatrics. 2020 Aug 3. doi: 10.1542/peds.2020-012922).
The researchers in the current study “seem to recognize that clinicians are adult learners, and they combine interventions to implement these adult learning theory tenets to improve appropriate antibiotic prescribing,” they wrote. The DART intervention combined best practices training, communications training, and individualized antibiotic prescribing feedback reports to improve communication between providers and families “especially when faced with a situation in which a parent or guardian might expect an antibiotic prescription but the provider does not think one is necessary,” Dr. Hamdy and Dr. Katz said.
Overall, the findings suggest that the interventions work best in combination vs. being used alone, although the study did not evaluate the separate contributions of each intervention, the editorialists wrote.
“In the current study, nonengaged physicians had an increase in second-line antibiotic prescribing, whereas the engaged physicians had a decrease in second-line antibiotic prescribing,” they noted. “This suggests that the addition of communications training could mitigate the undesirable effects that may result from solely using feedback reports.”
“Each year, U.S. children are prescribed as many as 10 million unnecessary antibiotic courses for acute respiratory tract infections,” Kristina A. Bryant, MD, of the University of Louisville, Ky., said in an interview. “Some of these prescriptions result in side effects or allergic reactions, and they contribute to growing antibiotic resistance. We need effective interventions to reduce antibiotic prescribing.”
Although the DART modules are free and available online, busy clinicians might struggle to find time to view them consistently, said Dr. Bryant.
“One advantage of the study design was that information was pushed to clinicians along with communication booster videos,” she said. “We know that education and reinforcement over time works better than a one and done approach.
“Study participants also received feedback over time about their prescribing habits, which can be a powerful motivator for change, although not all clinicians may have easy access to these reports,” she noted.
To overcome some of the barriers to using the modules, clinicians who are “interested in improving their prescribing could work with their office managers to develop antibiotic prescribing reports and schedule reminders to review them,” said Dr. Bryant.
“An individual could commit to education and review of his or her own prescribing patterns, but support from one’s partners and shared accountability is likely to be even more effective,” she said. “Sharing data within a practice and exploring differences in prescribing patterns can drive improvement.
“Spaced education and regular feedback about prescribing patterns can improve antibiotic prescribing for pharyngitis and sinusitis, and reduce antibiotic prescriptions for ARTIs,” Dr. Bryant said. The take-home from the study is that it should prompt anyone who prescribes antibiotics for children to ask themselves how they can improve their own prescribing habits.
“In this study, prescribing for viral ARTIs was reduced but not eliminated. We need additional studies to further reduce unnecessary antibiotic use,” Dr. Bryant said.
In addition, areas for future research could include longer-term follow-up. “Study participants were followed for 2 to 8 months after the intervention ended in June 2018. It would be interesting to know about their prescribing practices now, and if the changes observed in the study were durable,” she concluded.
The study was supported by the National Institutes of Health, along with additional infrastructure funding from the American Academy of Pediatrics and the Health Resources and Services Administration of the Department of Health and Human Services. The researchers had no financial conflicts to disclose.
Dr. Hamdy and Dr. Katz had no financial conflicts to disclose, but Dr. Katz disclosed grant support through the Centers for Disease Control and Prevention as a recipient of the Leadership in Epidemiology, Antimicrobial Stewardship, and Public Health fellowship, sponsored by the Society for Healthcare Epidemiology of America, Infectious Diseases Society of America, and Pediatric Infectious Diseases Society.
Dr. Bryant disclosed serving as an investigator on multicenter clinical vaccine trials funded by Pfizer (but not in the last year). She also serves as the current president of the Pediatric Infectious Diseases Society, but the opinions expressed here are her own and do not necessarily reflect the views of PIDS.
SOURCE: Kronman MP et al. Pediatrics. 2020 Aug 3. doi: 10.1542/peds.2020-0038.
FROM PEDIATRICS

Diagnostic testing for COVID-19: A quick summary for PCPs
Information about COVID has evolved so quickly that it can be difficult for clinicians to feel confident that they are staying current. These summaries include links to our reference article on diagnosis of COVID-19, which is constantly updated to make sure you have the latest information.
Diagnostic testing for COVID-19 is critical. No one disputes that. But what is in dispute is whom to test, when to test, how to test, what to do while waiting for results, and how accurate those results are when you finally get them.
Here are the answers to those questions, based on the current information.
Whom to test. This is the (relatively) easy part. The ideal answer is that everyone should be tested. The Infectious Diseases Society of America issued tier-based recommendations way back in March, and they still apply. First priority continues to be patients who are ill, healthcare workers, and those with known exposure. But to truly figure out the amount of community spread in a given area, we need to test people who do not have a clear indication for testing. That is particularly true as more people return to work and the Centers for Disease Control and Prevention (CDC) has issued guidelines for workplaces to establish testing programs. Universal testing is recommended for some high-risk settings, such as nursing homes.
One key change: CDC no longer recommends testing to determine whether someone with a known infection is still infectious.
When to test. People with any symptoms suggestive of COVID should be tested, ideally as soon as feasible. But given the ongoing shortages of tests, that may not be possible, particularly for those requiring only symptomatic care. Rather, these patients should be treated as probable cases, with appropriate instructions regarding quarantine. Testing of those with known exposures ideally should be done about 5 days after exposure.
How to test. Only viral nucleic acid or antigen tests should be used to diagnose acute illness. CDC does not currently recommend using serologic assays, now broadly available, for diagnosis of acute infection, though they obviously play an important role in understanding the transmission dynamic of the virus in the general population.
Testing strategies vary from state to state and even within communities in a single state. It is recommended that clinicians check with their own local or state health department for specifics on tests available, indications for testing, and processing details. While often forgotten, it is worth emphasizing that no diagnostic tests have been approved by the US Food and Drug Administration (FDA). Rather, they are available under emergency use authorization (EUA), meaning that they have not been fully vetted by the FDA.
In late July, the FDA expanded authorization for real-time reverse transcription–polymerase chain reaction (rRT-PCR) molecular assays, utilizing nasal or nasopharyngeal swabs, to permit testing of all persons, regardless of exposure history or symptoms. The FDA maintains a list of all approved diagnostic tests and corresponding labs. Patients will have to get what is available via their health department or insurance plan.
Two point-of-care antigen tests using nasopharyngeal or nasal samples have been issued an EUA. These tests can be used only in settings with a valid CLIA certificate.
Several commercial laboratories have received approval to process diagnostic tests using patients’ self-collected saliva rather than swabs. One lab has now received authorization for in-home testing without any input from a clinician. These testing options can be a boon for patients who have symptoms or exposure and for whatever reason are unable to get to a diagnostic site. These samples are collected at home and mailed to a lab. Note that these tests are not yet widely available.
Waiting for results. If waiting for results meant a day or even a couple of days, the answer to this one would be easier. But if the wait extends to 1 and even sometimes 2 weeks, then the test is not able to meaningfully guide clinical decisions. The latest guidance from the CDC is that individuals with symptoms suggestive of COVID who do not require hospitalization should remain at home in self-quarantine for at least 10 days from symptom onset. Asymptomatic individuals with a known exposure to someone else with COVID, or participation in a high-risk event like an indoor gathering involving more than 10 persons, should self-quarantine either until they receive a negative test result or 14 days after the exposure.
Accuracy of results. A positive rRT-PCR antigen test is highly accurate, indicating presence of SARS-CoV-2 RNA. There appears to be no significant cross-reactivity with other respiratory viruses or even other coronaviruses. A small study conducted in Korea suggests that patients with persistent positive tests who are beyond 10 days from the initial positive test and are now symptom free are no longer infectious.
For patients with a high suspicion of COVID-19, a negative test should not rule out the infection. The number of false-negative results is not well known, though the resultant risk is “substantial.” A number of factors affect the likelihood of a false-negative test, including when the sample was collected relative to the timing of illness and the type of specimen collected; for example, nasopharyngeal swabs are more likely to be accurate vs nasal or throat specimens. Repeat or serial testing increases the sensitivity but may not always be available. Although rRT-PCR is the current criterion standard, more inclusive consensus-based criteria are likely to emerge because of the concern about these false-negative results.
This article first appeared on Medscape.com.
Information about COVID has evolved so quickly that it can be difficult for clinicians to feel confident that they are staying current. These summaries include links to our reference article on diagnosis of COVID-19, which is constantly updated to make sure you have the latest information.
Diagnostic testing for COVID-19 is critical. No one disputes that. But what is in dispute is whom to test, when to test, how to test, what to do while waiting for results, and how accurate those results are when you finally get them.
Here are the answers to those questions, based on the current information.
Whom to test. This is the (relatively) easy part. The ideal answer is that everyone should be tested. The Infectious Diseases Society of America issued tier-based recommendations way back in March, and they still apply. First priority continues to be patients who are ill, healthcare workers, and those with known exposure. But to truly figure out the amount of community spread in a given area, we need to test people who do not have a clear indication for testing. That is particularly true as more people return to work and the Centers for Disease Control and Prevention (CDC) has issued guidelines for workplaces to establish testing programs. Universal testing is recommended for some high-risk settings, such as nursing homes.
One key change: CDC no longer recommends testing to determine whether someone with a known infection is still infectious.
When to test. People with any symptoms suggestive of COVID should be tested, ideally as soon as feasible. But given the ongoing shortages of tests, that may not be possible, particularly for those requiring only symptomatic care. Rather, these patients should be treated as probable cases, with appropriate instructions regarding quarantine. Testing of those with known exposures ideally should be done about 5 days after exposure.
How to test. Only viral nucleic acid or antigen tests should be used to diagnose acute illness. CDC does not currently recommend using serologic assays, now broadly available, for diagnosis of acute infection, though they obviously play an important role in understanding the transmission dynamic of the virus in the general population.
Testing strategies vary from state to state and even within communities in a single state. It is recommended that clinicians check with their own local or state health department for specifics on tests available, indications for testing, and processing details. While often forgotten, it is worth emphasizing that no diagnostic tests have been approved by the US Food and Drug Administration (FDA). Rather, they are available under emergency use authorization (EUA), meaning that they have not been fully vetted by the FDA.
In late July, the FDA expanded authorization for real-time reverse transcription–polymerase chain reaction (rRT-PCR) molecular assays, utilizing nasal or nasopharyngeal swabs, to permit testing of all persons, regardless of exposure history or symptoms. The FDA maintains a list of all approved diagnostic tests and corresponding labs. Patients will have to get what is available via their health department or insurance plan.
Two point-of-care antigen tests using nasopharyngeal or nasal samples have been issued an EUA. These tests can be used only in settings with a valid CLIA certificate.
Several commercial laboratories have received approval to process diagnostic tests using patients’ self-collected saliva rather than swabs. One lab has now received authorization for in-home testing without any input from a clinician. These testing options can be a boon for patients who have symptoms or exposure and for whatever reason are unable to get to a diagnostic site. These samples are collected at home and mailed to a lab. Note that these tests are not yet widely available.
Waiting for results. If waiting for results meant a day or even a couple of days, the answer to this one would be easier. But if the wait extends to 1 and even sometimes 2 weeks, then the test is not able to meaningfully guide clinical decisions. The latest guidance from the CDC is that individuals with symptoms suggestive of COVID who do not require hospitalization should remain at home in self-quarantine for at least 10 days from symptom onset. Asymptomatic individuals with a known exposure to someone else with COVID, or participation in a high-risk event like an indoor gathering involving more than 10 persons, should self-quarantine either until they receive a negative test result or 14 days after the exposure.
Accuracy of results. A positive rRT-PCR antigen test is highly accurate, indicating presence of SARS-CoV-2 RNA. There appears to be no significant cross-reactivity with other respiratory viruses or even other coronaviruses. A small study conducted in Korea suggests that patients with persistent positive tests who are beyond 10 days from the initial positive test and are now symptom free are no longer infectious.
For patients with a high suspicion of COVID-19, a negative test should not rule out the infection. The number of false-negative results is not well known, though the resultant risk is “substantial.” A number of factors affect the likelihood of a false-negative test, including when the sample was collected relative to the timing of illness and the type of specimen collected; for example, nasopharyngeal swabs are more likely to be accurate vs nasal or throat specimens. Repeat or serial testing increases the sensitivity but may not always be available. Although rRT-PCR is the current criterion standard, more inclusive consensus-based criteria are likely to emerge because of the concern about these false-negative results.
This article first appeared on Medscape.com.
Information about COVID has evolved so quickly that it can be difficult for clinicians to feel confident that they are staying current. These summaries include links to our reference article on diagnosis of COVID-19, which is constantly updated to make sure you have the latest information.
Diagnostic testing for COVID-19 is critical. No one disputes that. But what is in dispute is whom to test, when to test, how to test, what to do while waiting for results, and how accurate those results are when you finally get them.
Here are the answers to those questions, based on the current information.
Whom to test. This is the (relatively) easy part. The ideal answer is that everyone should be tested. The Infectious Diseases Society of America issued tier-based recommendations way back in March, and they still apply. First priority continues to be patients who are ill, healthcare workers, and those with known exposure. But to truly figure out the amount of community spread in a given area, we need to test people who do not have a clear indication for testing. That is particularly true as more people return to work and the Centers for Disease Control and Prevention (CDC) has issued guidelines for workplaces to establish testing programs. Universal testing is recommended for some high-risk settings, such as nursing homes.
One key change: CDC no longer recommends testing to determine whether someone with a known infection is still infectious.
When to test. People with any symptoms suggestive of COVID should be tested, ideally as soon as feasible. But given the ongoing shortages of tests, that may not be possible, particularly for those requiring only symptomatic care. Rather, these patients should be treated as probable cases, with appropriate instructions regarding quarantine. Testing of those with known exposures ideally should be done about 5 days after exposure.
How to test. Only viral nucleic acid or antigen tests should be used to diagnose acute illness. CDC does not currently recommend using serologic assays, now broadly available, for diagnosis of acute infection, though they obviously play an important role in understanding the transmission dynamic of the virus in the general population.
Testing strategies vary from state to state and even within communities in a single state. It is recommended that clinicians check with their own local or state health department for specifics on tests available, indications for testing, and processing details. While often forgotten, it is worth emphasizing that no diagnostic tests have been approved by the US Food and Drug Administration (FDA). Rather, they are available under emergency use authorization (EUA), meaning that they have not been fully vetted by the FDA.
In late July, the FDA expanded authorization for real-time reverse transcription–polymerase chain reaction (rRT-PCR) molecular assays, utilizing nasal or nasopharyngeal swabs, to permit testing of all persons, regardless of exposure history or symptoms. The FDA maintains a list of all approved diagnostic tests and corresponding labs. Patients will have to get what is available via their health department or insurance plan.
Two point-of-care antigen tests using nasopharyngeal or nasal samples have been issued an EUA. These tests can be used only in settings with a valid CLIA certificate.
Several commercial laboratories have received approval to process diagnostic tests using patients’ self-collected saliva rather than swabs. One lab has now received authorization for in-home testing without any input from a clinician. These testing options can be a boon for patients who have symptoms or exposure and for whatever reason are unable to get to a diagnostic site. These samples are collected at home and mailed to a lab. Note that these tests are not yet widely available.
Waiting for results. If waiting for results meant a day or even a couple of days, the answer to this one would be easier. But if the wait extends to 1 and even sometimes 2 weeks, then the test is not able to meaningfully guide clinical decisions. The latest guidance from the CDC is that individuals with symptoms suggestive of COVID who do not require hospitalization should remain at home in self-quarantine for at least 10 days from symptom onset. Asymptomatic individuals with a known exposure to someone else with COVID, or participation in a high-risk event like an indoor gathering involving more than 10 persons, should self-quarantine either until they receive a negative test result or 14 days after the exposure.
Accuracy of results. A positive rRT-PCR antigen test is highly accurate, indicating presence of SARS-CoV-2 RNA. There appears to be no significant cross-reactivity with other respiratory viruses or even other coronaviruses. A small study conducted in Korea suggests that patients with persistent positive tests who are beyond 10 days from the initial positive test and are now symptom free are no longer infectious.
For patients with a high suspicion of COVID-19, a negative test should not rule out the infection. The number of false-negative results is not well known, though the resultant risk is “substantial.” A number of factors affect the likelihood of a false-negative test, including when the sample was collected relative to the timing of illness and the type of specimen collected; for example, nasopharyngeal swabs are more likely to be accurate vs nasal or throat specimens. Repeat or serial testing increases the sensitivity but may not always be available. Although rRT-PCR is the current criterion standard, more inclusive consensus-based criteria are likely to emerge because of the concern about these false-negative results.
This article first appeared on Medscape.com.
Cutaneous clues linked to COVID-19 coagulation risk
, new evidence suggests.
Researchers at Weill Cornell Medicine NewYork–Presbyterian Medical Center in New York linked livedoid and purpuric skin eruptions to a greater likelihood for occlusive vascular disease associated with SARS-CoV-2 infection in a small case series.
These skin signs could augment coagulation assays in this patient population. “Physicians should consider a hematology consult for potential anticoagulation in patients with these skin presentations and severe COVID-19,” senior author Joanna Harp, MD, said in an interview.
“Physicians should also consider D-dimer, fibrinogen, coagulation studies, and a skin biopsy given that there are other diagnoses on the differential as well.”
The research letter was published online on Aug. 5 in JAMA Dermatology.
The findings build on multiple previous reports of skin manifestations associated with COVID-19, including a study of 375 patients in Spain. Among people with suspected or confirmed SARS-CoV-2 infection, senior author of the Spanish research, Ignacio Garcia-Doval, MD, PhD, also observed livedoid and necrotic skin eruptions more commonly in severe disease.
“I think that this case series [from Harp and colleagues] confirms the findings of our previous paper – that patients with livedoid or necrotic lesions have a worse prognosis, as these are markers of vascular occlusion,” he said in an interview.
Dr. Harp and colleagues reported their observations with four patients aged 40-80 years. Each had severe COVID-19 with acute respiratory distress syndrome and required intubation. Treating clinicians requested a dermatology consult to assess acral fixed livedo racemosa and retiform purpura presentations.
D-dimer levels exceeded 3 mcg/mL in each case. All four patients had a suspected pulmonary embolism within 1-5 days of the dermatologic findings. Prophylactic anticoagulation at admission was changed to therapeutic anticoagulation because of increasing D-dimer levels and the suspected thrombotic events.
“I think that the paper is interesting because it shows the associated histopathological findings and has important clinical implications due to the association with pulmonary embolism,” said Dr. Garcia-Doval, a researcher at the Spanish Academy of Dermatology in Madrid. “These patients should probably be anticoagulated.”
Skin biopsy results
Punch biopsies revealed pauci-inflammatory thrombogenic vasculopathy involving capillaries, venules, arterioles, or small arteries.
Livedo racemosa skin findings point to partial occlusion of cutaneous blood vessels, whereas retiform purpura indicate full occlusion of cutaneous blood vessels.
An inability to confirm the exact timing of the onset of the skin rash was a limitation of the study.
“The findings suggest that clinicians caring for patients with COVID-19 should be aware of livedoid and purpuric rashes as potential manifestations of an underlying hypercoagulable state,” the authors noted. “If these skin findings are identified, a skin biopsy should be considered because the result may guide anticoagulation management.”
Observations during an outbreak
The researchers observed these cases between March 13 and April 3, during the peak of the COVID-19 outbreak in New York.
“We did see additional cases since our study period. However, it has decreased significantly with the falling number of COVID-19 cases in the city,” said Dr. Harp, a dermatologist at NewYork–Presbyterian.
Another contributing factor in the drop in cases was “implementation of earlier, more aggressive anticoagulation in many of these patients at our institution,” she added.
The investigators plan to continue the research. “We are working on a more formalized study,” lead author Caren Droesch, MD, said in an interview.
“But given very low patient numbers in our area we have not started recruiting patients,” said Dr. Droesch, a resident at Weill Cornell Medicine and NewYork–Presbyterian at the time of the study. She is now a dermatologist at Mass General Brigham in Wellesley, Mass.
Consider a dermatology consult
“This is a small case series of four patients, but mirrors what we have seen at our institution and what others have reported about individual patients around the world,” Anthony Fernandez, MD, PhD, a dermatologist at Cleveland Clinic, said in an interview. “The skin, like many other organ systems, can be affected by thrombotic events within the setting of COVID-19 disease.”
As in the current study, Dr. Fernandez observed skin manifestations in people with severe COVID-19 with elevated D-dimer levels. These patients typically require mechanical ventilation in the intensive care unit, he added.
“As these authors point out, it is important for all clinicians caring for COVID-19 patients to look for these rashes,” said Dr. Fernandez, who coauthored a report on skin manifestations in this patient population. “We also agree that clinicians should have a low threshold for consulting dermatology. A skin biopsy is minimally invasive and can be important in confirming or refuting that such rashes are truly reflective of thrombotic vasculopathy.”
Dr. Harp, Dr. Droesch and Dr. Garcia-Doval have disclosed no relevant financial relationships. Dr. Fernandez received funding from the Clinical and Translational Science Collaborative at Case Western Reserve University to study skin manifestations of COVID-19.
A version of this article originally appeared on Medscape.com.
, new evidence suggests.
Researchers at Weill Cornell Medicine NewYork–Presbyterian Medical Center in New York linked livedoid and purpuric skin eruptions to a greater likelihood for occlusive vascular disease associated with SARS-CoV-2 infection in a small case series.
These skin signs could augment coagulation assays in this patient population. “Physicians should consider a hematology consult for potential anticoagulation in patients with these skin presentations and severe COVID-19,” senior author Joanna Harp, MD, said in an interview.
“Physicians should also consider D-dimer, fibrinogen, coagulation studies, and a skin biopsy given that there are other diagnoses on the differential as well.”
The research letter was published online on Aug. 5 in JAMA Dermatology.
The findings build on multiple previous reports of skin manifestations associated with COVID-19, including a study of 375 patients in Spain. Among people with suspected or confirmed SARS-CoV-2 infection, senior author of the Spanish research, Ignacio Garcia-Doval, MD, PhD, also observed livedoid and necrotic skin eruptions more commonly in severe disease.
“I think that this case series [from Harp and colleagues] confirms the findings of our previous paper – that patients with livedoid or necrotic lesions have a worse prognosis, as these are markers of vascular occlusion,” he said in an interview.
Dr. Harp and colleagues reported their observations with four patients aged 40-80 years. Each had severe COVID-19 with acute respiratory distress syndrome and required intubation. Treating clinicians requested a dermatology consult to assess acral fixed livedo racemosa and retiform purpura presentations.
D-dimer levels exceeded 3 mcg/mL in each case. All four patients had a suspected pulmonary embolism within 1-5 days of the dermatologic findings. Prophylactic anticoagulation at admission was changed to therapeutic anticoagulation because of increasing D-dimer levels and the suspected thrombotic events.
“I think that the paper is interesting because it shows the associated histopathological findings and has important clinical implications due to the association with pulmonary embolism,” said Dr. Garcia-Doval, a researcher at the Spanish Academy of Dermatology in Madrid. “These patients should probably be anticoagulated.”
Skin biopsy results
Punch biopsies revealed pauci-inflammatory thrombogenic vasculopathy involving capillaries, venules, arterioles, or small arteries.
Livedo racemosa skin findings point to partial occlusion of cutaneous blood vessels, whereas retiform purpura indicate full occlusion of cutaneous blood vessels.
An inability to confirm the exact timing of the onset of the skin rash was a limitation of the study.
“The findings suggest that clinicians caring for patients with COVID-19 should be aware of livedoid and purpuric rashes as potential manifestations of an underlying hypercoagulable state,” the authors noted. “If these skin findings are identified, a skin biopsy should be considered because the result may guide anticoagulation management.”
Observations during an outbreak
The researchers observed these cases between March 13 and April 3, during the peak of the COVID-19 outbreak in New York.
“We did see additional cases since our study period. However, it has decreased significantly with the falling number of COVID-19 cases in the city,” said Dr. Harp, a dermatologist at NewYork–Presbyterian.
Another contributing factor in the drop in cases was “implementation of earlier, more aggressive anticoagulation in many of these patients at our institution,” she added.
The investigators plan to continue the research. “We are working on a more formalized study,” lead author Caren Droesch, MD, said in an interview.
“But given very low patient numbers in our area we have not started recruiting patients,” said Dr. Droesch, a resident at Weill Cornell Medicine and NewYork–Presbyterian at the time of the study. She is now a dermatologist at Mass General Brigham in Wellesley, Mass.
Consider a dermatology consult
“This is a small case series of four patients, but mirrors what we have seen at our institution and what others have reported about individual patients around the world,” Anthony Fernandez, MD, PhD, a dermatologist at Cleveland Clinic, said in an interview. “The skin, like many other organ systems, can be affected by thrombotic events within the setting of COVID-19 disease.”
As in the current study, Dr. Fernandez observed skin manifestations in people with severe COVID-19 with elevated D-dimer levels. These patients typically require mechanical ventilation in the intensive care unit, he added.
“As these authors point out, it is important for all clinicians caring for COVID-19 patients to look for these rashes,” said Dr. Fernandez, who coauthored a report on skin manifestations in this patient population. “We also agree that clinicians should have a low threshold for consulting dermatology. A skin biopsy is minimally invasive and can be important in confirming or refuting that such rashes are truly reflective of thrombotic vasculopathy.”
Dr. Harp, Dr. Droesch and Dr. Garcia-Doval have disclosed no relevant financial relationships. Dr. Fernandez received funding from the Clinical and Translational Science Collaborative at Case Western Reserve University to study skin manifestations of COVID-19.
A version of this article originally appeared on Medscape.com.
, new evidence suggests.
Researchers at Weill Cornell Medicine NewYork–Presbyterian Medical Center in New York linked livedoid and purpuric skin eruptions to a greater likelihood for occlusive vascular disease associated with SARS-CoV-2 infection in a small case series.
These skin signs could augment coagulation assays in this patient population. “Physicians should consider a hematology consult for potential anticoagulation in patients with these skin presentations and severe COVID-19,” senior author Joanna Harp, MD, said in an interview.
“Physicians should also consider D-dimer, fibrinogen, coagulation studies, and a skin biopsy given that there are other diagnoses on the differential as well.”
The research letter was published online on Aug. 5 in JAMA Dermatology.
The findings build on multiple previous reports of skin manifestations associated with COVID-19, including a study of 375 patients in Spain. Among people with suspected or confirmed SARS-CoV-2 infection, senior author of the Spanish research, Ignacio Garcia-Doval, MD, PhD, also observed livedoid and necrotic skin eruptions more commonly in severe disease.
“I think that this case series [from Harp and colleagues] confirms the findings of our previous paper – that patients with livedoid or necrotic lesions have a worse prognosis, as these are markers of vascular occlusion,” he said in an interview.
Dr. Harp and colleagues reported their observations with four patients aged 40-80 years. Each had severe COVID-19 with acute respiratory distress syndrome and required intubation. Treating clinicians requested a dermatology consult to assess acral fixed livedo racemosa and retiform purpura presentations.
D-dimer levels exceeded 3 mcg/mL in each case. All four patients had a suspected pulmonary embolism within 1-5 days of the dermatologic findings. Prophylactic anticoagulation at admission was changed to therapeutic anticoagulation because of increasing D-dimer levels and the suspected thrombotic events.
“I think that the paper is interesting because it shows the associated histopathological findings and has important clinical implications due to the association with pulmonary embolism,” said Dr. Garcia-Doval, a researcher at the Spanish Academy of Dermatology in Madrid. “These patients should probably be anticoagulated.”
Skin biopsy results
Punch biopsies revealed pauci-inflammatory thrombogenic vasculopathy involving capillaries, venules, arterioles, or small arteries.
Livedo racemosa skin findings point to partial occlusion of cutaneous blood vessels, whereas retiform purpura indicate full occlusion of cutaneous blood vessels.
An inability to confirm the exact timing of the onset of the skin rash was a limitation of the study.
“The findings suggest that clinicians caring for patients with COVID-19 should be aware of livedoid and purpuric rashes as potential manifestations of an underlying hypercoagulable state,” the authors noted. “If these skin findings are identified, a skin biopsy should be considered because the result may guide anticoagulation management.”
Observations during an outbreak
The researchers observed these cases between March 13 and April 3, during the peak of the COVID-19 outbreak in New York.
“We did see additional cases since our study period. However, it has decreased significantly with the falling number of COVID-19 cases in the city,” said Dr. Harp, a dermatologist at NewYork–Presbyterian.
Another contributing factor in the drop in cases was “implementation of earlier, more aggressive anticoagulation in many of these patients at our institution,” she added.
The investigators plan to continue the research. “We are working on a more formalized study,” lead author Caren Droesch, MD, said in an interview.
“But given very low patient numbers in our area we have not started recruiting patients,” said Dr. Droesch, a resident at Weill Cornell Medicine and NewYork–Presbyterian at the time of the study. She is now a dermatologist at Mass General Brigham in Wellesley, Mass.
Consider a dermatology consult
“This is a small case series of four patients, but mirrors what we have seen at our institution and what others have reported about individual patients around the world,” Anthony Fernandez, MD, PhD, a dermatologist at Cleveland Clinic, said in an interview. “The skin, like many other organ systems, can be affected by thrombotic events within the setting of COVID-19 disease.”
As in the current study, Dr. Fernandez observed skin manifestations in people with severe COVID-19 with elevated D-dimer levels. These patients typically require mechanical ventilation in the intensive care unit, he added.
“As these authors point out, it is important for all clinicians caring for COVID-19 patients to look for these rashes,” said Dr. Fernandez, who coauthored a report on skin manifestations in this patient population. “We also agree that clinicians should have a low threshold for consulting dermatology. A skin biopsy is minimally invasive and can be important in confirming or refuting that such rashes are truly reflective of thrombotic vasculopathy.”
Dr. Harp, Dr. Droesch and Dr. Garcia-Doval have disclosed no relevant financial relationships. Dr. Fernandez received funding from the Clinical and Translational Science Collaborative at Case Western Reserve University to study skin manifestations of COVID-19.
A version of this article originally appeared on Medscape.com.
FROM JAMA DERMATOLOGY
Septicemia first among hospital inpatient costs
according to a recent analysis from the Agency for Healthcare Research and Quality.
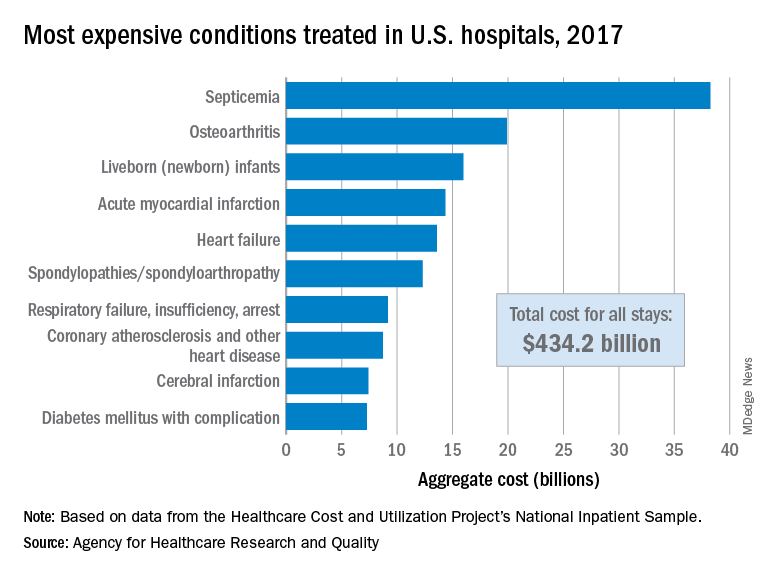
The single most expensive inpatient condition that year, representing about 8.8% of all hospital costs, was septicemia at $38.2 billion, nearly double the $19.9 billion spent on the next most expensive condition, osteoarthritis, Lan Liang, PhD, of the AHRQ, and associates said in a statistical brief.
These figures “represent the hospital’s costs to produce the services – not the amount paid for services by payers – and they do not include separately billed physician fees associated with the hospitalization,” they noted.
Third in overall cost for 2017 but first in total number of stays were live-born infants, with 3.7 million admissions costing just under $16 billion. Hospital costs for acute myocardial infarction ($14.3 billion) made it the fourth most expensive condition, with heart failure fifth at $13.6 billion, based on data from the Healthcare Cost and Utilization Project’s National Inpatient Sample.
The 20 most expensive conditions, which also included coronary atherosclerosis, pneumonia, renal failure, and lower-limb fracture, accounted for close to 47% of all hospital costs and over 43% of all stays in 2017. The total amount spent by hospitals that year, $1.1 trillion, constituted nearly a third of all health care expenditures and was 4.7% higher than in 2016, Dr. Liang and associates reported.
“Although this growth represented deceleration, compared with the 5.8% increase between 2014 and 2015, the consistent year-to-year rise in hospital-related expenses remains a central concern among policymakers,” they wrote.
according to a recent analysis from the Agency for Healthcare Research and Quality.
The single most expensive inpatient condition that year, representing about 8.8% of all hospital costs, was septicemia at $38.2 billion, nearly double the $19.9 billion spent on the next most expensive condition, osteoarthritis, Lan Liang, PhD, of the AHRQ, and associates said in a statistical brief.
These figures “represent the hospital’s costs to produce the services – not the amount paid for services by payers – and they do not include separately billed physician fees associated with the hospitalization,” they noted.
Third in overall cost for 2017 but first in total number of stays were live-born infants, with 3.7 million admissions costing just under $16 billion. Hospital costs for acute myocardial infarction ($14.3 billion) made it the fourth most expensive condition, with heart failure fifth at $13.6 billion, based on data from the Healthcare Cost and Utilization Project’s National Inpatient Sample.
The 20 most expensive conditions, which also included coronary atherosclerosis, pneumonia, renal failure, and lower-limb fracture, accounted for close to 47% of all hospital costs and over 43% of all stays in 2017. The total amount spent by hospitals that year, $1.1 trillion, constituted nearly a third of all health care expenditures and was 4.7% higher than in 2016, Dr. Liang and associates reported.
“Although this growth represented deceleration, compared with the 5.8% increase between 2014 and 2015, the consistent year-to-year rise in hospital-related expenses remains a central concern among policymakers,” they wrote.
according to a recent analysis from the Agency for Healthcare Research and Quality.
The single most expensive inpatient condition that year, representing about 8.8% of all hospital costs, was septicemia at $38.2 billion, nearly double the $19.9 billion spent on the next most expensive condition, osteoarthritis, Lan Liang, PhD, of the AHRQ, and associates said in a statistical brief.
These figures “represent the hospital’s costs to produce the services – not the amount paid for services by payers – and they do not include separately billed physician fees associated with the hospitalization,” they noted.
Third in overall cost for 2017 but first in total number of stays were live-born infants, with 3.7 million admissions costing just under $16 billion. Hospital costs for acute myocardial infarction ($14.3 billion) made it the fourth most expensive condition, with heart failure fifth at $13.6 billion, based on data from the Healthcare Cost and Utilization Project’s National Inpatient Sample.
The 20 most expensive conditions, which also included coronary atherosclerosis, pneumonia, renal failure, and lower-limb fracture, accounted for close to 47% of all hospital costs and over 43% of all stays in 2017. The total amount spent by hospitals that year, $1.1 trillion, constituted nearly a third of all health care expenditures and was 4.7% higher than in 2016, Dr. Liang and associates reported.
“Although this growth represented deceleration, compared with the 5.8% increase between 2014 and 2015, the consistent year-to-year rise in hospital-related expenses remains a central concern among policymakers,” they wrote.
Weight gain persists as HIV-treatment issue
People living with HIV who put on extra pounds and develop metabolic syndrome or related disorders linked in part to certain antiretroviral agents remain a concern today, even as the drugs used to suppress HIV infection have evolved over the decades.
Linkage of HIV treatment with lipodystrophy and insulin resistance or diabetes began in the 1990s with protease inhibitors (Clin Infect Dis. 2000 Jun;30[suppl 2]:s135-42). Several reports over the years also tied any form of effective antiretroviral therapy to weight gain in HIV patients (Antivir Ther. 2012;17[7]:1281-9). More recently, reports have rattled the HIV-treatment community by associating alarmingly high levels of weight gain with a useful and relatively new drug, tenofovir alafenamide fumarate (TAF) – a nucleoside reverse transcriptase inhibitor (NRTI) approved for use in the United States in late 2016, as well as certain agents from an entirely different antiretroviral therapy (ART) class, the integrase strand transfer inhibitors (INSTIs). Both TAF and the INSTIs have come to play major roles in the HIV-treatment landscape, despite relevant and concerning recent weight gain observations with these drugs, such as in a 2019 meta-analysis of eight trials with 5,680 treatment-naive patients who started ART during 2003-2015 (Clin Infect Dis. 2019 Oct 14;doi: 10.1093/cid/ciz999).
“Weight gain is clearly seen in studies of dolutegravir [DTG] or bictegravir [BTG] with TAF,” wrote W.D. Francois Venter, PhD and Andrew Hill, PhD in a recent published commentary on the topic (Lancet HIV. 2020 Jun 1;7[6]:e389-400). Both DTG and BTG are INSTI class members.
“Excessive weight gain, defined as more than 10% over baseline, has recently been observed among people with HIV initiating or switching to regimens incorporating TAF, an INSTI, or both, particularly DTG,” wrote Jordan E. Lake, MD, an HIV specialist at the University of Texas Health Science Center at Houston, in a recent commentary posted online. Women and Black patients “are at even greater risk for excessive weight gain,” Dr. Lake added.
“In recent times, it has emerged that weight gain is more pronounced with the integrase inhibitor class of agents, especially dolutegravir and bictegravir, the so-called second-generation” INSTIs, said Anna Maria Geretti, MD, a professor of clinical infection, microbiology, and immunology at the University of Liverpool, England. ”The effect is more pronounced in women and people of non-White ethnicity, and is of concern because of the associated potential risk of metabolic syndrome, cardiovascular disease, etc.,” Dr. Geretti said in an interview.
The unprecedented susceptibility to weight gain seen recently in non-White women may in part have resulted from the tendency of many earlier treatment trials to have cohorts comprised predominantly of White men, Dr. Venter noted in an interview.
Alarming weight gains reported
Perhaps the most eye-popping example of the potential for weight gain with the combination of TAF with an INSTI came in a recent report from the ADVANCE trial, a randomized, head-to-head comparison of three regimens in 1,053 HIV patients in South Africa. After 144 weeks on a regimen of TAF (Vemlidy), DTG (Tivicay), and FTC (emtricitabine, Emtriva), another NRTI, women gained an averaged of more than 12 kg, compared with their baseline weight, significantly more than in two comparator groups, Simiso Sokhela, MB, reported at the virtual meeting of the International AIDS conference. The women in ADVANCE on the TAF-DTG-FTC regimen also had an 11% rate of incident metabolic syndrome during their first 96 weeks on treatment, compared with rates of 8% among patients on a different form of tenofovir, tenofovir disoproxil fumarate (TDF), along with DTG-FTC, and 5% among those on TDF–EFV (efavirenz, Sustiva)–FTC said Dr. Sokhela, an HIV researcher at Ezintsha, a division of the University of the Witwatersrand in Johannesburg, South Africa.
“We believe that these results support the World Health Organization guidelines that reserve TAF for only patients with osteoporosis or impaired renal function,” Dr. Sokhela said during a press briefing at the conference. The WHO guidelines list the first-line regimen as TDF-DTG-3TC (lamivudine; Epivir) or FTC. “The risk for becoming obese continued to increase after 96 weeks” of chronic use of these drugs, she added.
“All regimens are now brilliant at viral control. Finding the ones that don’t make patients obese or have other long-term side effects is now the priority,” noted Dr. Venter, a professor and HIV researcher at University of the Witwatersrand, head of Ezintsha, and lead investigator of ADVANCE. Clinicians and researchers have recently thought that combining TAF and an INSTI plus FTC or a similar NRTI “would be the ultimate regimen to replace the nonnucleoside reverse transcriptase inhibitors (NNRTIs)” such as EFV, “but now we have a major headache” with unexpectedly high weight gains in some patients, Dr. Venter said.
Weight gains “over 10 kg are unlikely to be acceptable in any circumstances, especially when starting body mass index is already borderline overweight,” wrote Dr. Venter along with Dr. Hill in their commentary. Until recently, many clinicians chalked up weight gain on newly begun ART as a manifestation of the patient’s “return-to-health,” but this interpretation “gives a positive spin to a potentially serious and common side effect,” they added.
More from ADVANCE
The primary efficacy endpoint of ADVANCE was suppression of viral load to less than 50 RNA copies/mL after 48 weeks on treatment, and the result showed that the TAF-DTG-FTC regimen and the TDF-DTG-FTC regimen were each noninferior to the control regimen of TDF-EFV-FTC (New Engl J Med. 2019 Aug 29;381[9]:803-15). Virtually all of the enrolled patients were Black, and 59% were women. Planned follow-up of all patients ran for 96 weeks. After 48 weeks, weight gain among the women averaged 6.4 kg, 3.2 kg, and 1.7 kg in the TAF-DTG, TDF-DTG, and TDF-EFV arms respectively. After 96 weeks, the average weight gains among women were 8.2 kg, 4.6 kg, and 3.2 kg, respectively, in new results reported by Dr. Sokhela at the IAC. Follow-up to 144 weeks was partial and included about a quarter of the enrolled women, with gains averaging 12.3 kg, 7.4 kg, and 5.5 kg respectively. The pattern of weight gain among men tracked the pattern in women, but the magnitude of gain was less. Among men followed for 144 weeks, average gain among those on TAF-DTG-FTC was 7.2 kg, the largest gain seen among men on any regimen and at any follow-up time in the study.
Dr. Sokhela also reported data on body composition analyses, which showed that the weight gains were largely in fat rather than lean tissue, fat accumulation was significantly greater in women than men, and that in both sexes fat accumulated roughly equally in the trunk and on limbs.
An additional analysis looked at the incidence of new-onset obesity among the women who had a normal body mass index at baseline. After 96 weeks, incident obesity occurred in 14% of women on the TAG-DTG-FTC regimen, 8% on TDF-DTG-FTC, and in 2% of women maintained on TDF-EFV-FTC, said Dr. Hill in a separate report at the conference.
Weight starts to weigh in
“I am very mindful of weight gain potential, and I talk to patients about it. It doesn’t determine what regimen I choose for a patient” right now, “but it’s only a matter of time before it starts influencing what we do, particularly if we can achieve efficacy with fewer drugs,” commented Babafemi O. Taiwo, MD, professor of medicine and chief of infectious diseases at Northwestern University in Chicago. “I’ve had some patients show up with a weight gain of 20 kg, and that shouldn’t happen,” he said during a recent online educational session. Dr. Taiwo said his recent practice has been to warn patients about possible weight gain and to urge them to get back in touch with him quickly if it happens.
“Virologic suppression is the most important goal with ART, and the U.S. Department of Health and Human Services currently recommends INSTI-based ART for most PWH [people with HIV],” wrote Dr. Lake in April 2020. “I counsel all PWH initiating ART about the potential for weight gain, and I discuss their current diet and healthy lifestyle habits. I explain to patients that we will monitor their weight, and if weight gain seems more than either of us are comfortable with then we will reassess. Only a small percentage of patients experience excessive weight gain after starting ART.” Dr. Lake also stressed that she had not yet begun to change the regimen a patient is on solely because of weight gain. “We do not know whether this weight gain is reversible,” she noted.
“I do not anticipate that a risk of weight gain at present will dictate a change in guidelines,” said Dr. Geretti. “Drugs such as dolutegravir and bictegravir are very effective, and they are unlikely to cause drug resistance. Further data on the mechanism of weight gain and the reversibility after a change of treatment will help refine drug selection in the near future,” she predicted.
“I consider weight gain when prescribing because my patients hear about this. It’s a side effect that my patients really care about, and I don’t blame them,” said Lisa Hightow-Weidman, MD, a professor and HIV specialist at the University of North Carolina at Chapel Hill, during an on-line educational session. “If you don’t discuss it with a patient and then weight gain happens and the patient finds out [the known risk from their treatment] they may have an issue,” she noted. But weight gain is not a reason to avoid these drugs. “They are great medications in many ways, with once-daily regimens and few side effects.”
Weight gain during pregnancy a special concern
An additional analysis of data from ADVANCE presented at the conference highlighted what the observed weight gain on ART could mean for women who become pregnant while on treatment. Based on a systematic literature review, the ADVANCE investigators calculated the relative risk for six obesity-related pregnancy complications, compared with nonobese women: preterm delivery, gestational diabetes, gestational hypertension, preeclampsia, postpartum hemorrhage, and caesarean delivery. Based on the obesity changes among women on their assigned ART in ADVANCE, the researchers calculated the predicted incidence of these six complications. The analysis showed that for every 1,000 women, those on TAG-DTG-FTC would have an excess of 53 obesity-related pregnancy complications, those on TDF-DTG-FTC would develop 28 excess pregnancy complications, and those on TDG-EFV-FTC would have four excess complications, reported Dr. Hill at the International AIDS conference.
The researchers also ran a similar simulation for the incidence of neonatal complications that could result when mothers are obese because of their ART. The six neonatal complications included in this analysis were small for gestational age, large for gestational age, macrosomia, neonatal death, stillbirth, and neural tube defects. Based on the excess rate of incident obesity, they calculated that for every 1,000 pregnancies women on TAD-DTG-FTC would have 24 additional infants born with one of these complications, women on TDF-DTG-FTC would have an excess of 13 of these events, and women on TDG-EFV-FTC would have an excess of three such obesity-related neonatal complications, Dr. Hill said.
Sorting out the drugs
Results from several additional studies reported at the conference have started trying to discern exactly which ART drugs and regimens pose the greatest weight gain risk and which have the least risk while retaining high efficacy and resistance barriers.
Further evidence implicating any type of ART as a driver of increased weight came from a review of 8,256 adults infected with HIV and members of the Kaiser Permanente health system in three U.S. regions during 2000-2016. Researchers matched these cases using several demographic factors with just under 130,000 members without HIV. Those infected by HIV had half the prevalence of obesity as the matched controls at baseline. During 12 years of follow-up, those infected with HIV had a threefold higher rate of weight gain than those who were uninfected. Annual weight gain averaged 0.06 kg/year among the uninfected people and 0.22 kg/year among those infected with HIV, a statistically significant difference that was consistent regardless of whether people started the study at a normal body mass index, overweight, or obese, reported Michael J. Silverberg, PhD, an epidemiologist with Kaiser Permanente in Oakland, Calif.
Another study tried to focus on the weight gain impact when patients on three-drug ART regimens changed from taking TDF to TAF. This analysis used data collected in the OPERA (Observational Pharmaco-Epidemiology Research & Analysis) longitudinal cohort of about 115,000 U.S. PWH. The observational cohort included nearly 7,000 patients who made a TDF-to-TAF switch, including 3,288 patients who maintained treatment during this switch with an INSTI, 1,454 who maintained a background regimen based on a NNRTI, 1,430 patients who also switched from an INSTI to a different drug, and 747 patients maintained on a boosted dose of a protease inhibitor. All patients were well controlled on their baseline regimen, with at least two consecutive measures showing undetectable viral load.
Patients who maintained their background regimens while changing from TDF to TAF had a 2.0-2.6 kg increase in weight during the 9 months immediately following their switch to TAF, reported Patrick Mallon, MB, a professor of microbial diseases at University College Dublin. Among the patients who both switched to TAF and also switched to treatment with an INSTI, weight gain during the 9 months after the switch averaged 2.6-4.5 kg, depending on which INSTI was started. Patients who switched to treatment with elvitegravir/cobicistat (an INSTI plus a boosting agent) averaged a gain of 2.6 kg during 9 months, those who switched to DTG averaged a 3.1-kg gain, and those who switched to BTG averaged a 4.6-kg increase, Dr. Mallon reported at the conference.
These findings “give us a good sense that the weight gain is real. This is not just overeating or not exercising, but weight changes coincidental with a change in HIV treatment,” commented David Wohl, MD, professor of medicine and site leader of the HIV Prevention and Treatment Clinical Trials Unit at the University of North Carolina at Chapel Hill, during an online educational session.
Contrary to this evidence suggesting a consistent uptick in weight when patients start TAF treatment was a recent report on 629 HIV patients randomized to treatment with TAF-BTG-FTC or abacavir (an NRTI, Ziagen)–DTG-3TC, which found similar weight gains between these two regimens after 144 weeks on treatment (Lancet HIV. 2020 Jun;7[6]:e389-400). This finding had the effect of “strengthening the argument that TAF is simply an innocent bystander” and does not play a central role in weight gain, and supporting the notion that the alternative tenofovir formulation, TDF, differs from TAF by promoting weight loss, Dr. Venter and Dr. Hill suggested in their commentary that accompanied this report.
The new findings from Dr. Mallon raise “serious questions about the way we have moved to TAF as a replacement for TDF, especially because the benefits [from TAF] are for a small subgroup – patients with renal disease or osteoporosis,” Dr. Venter said in an interview. “The question is, will we see weight gain like this” if TAF was combined with a non-INSTI drug? he wondered.
While some study results have suggested a mitigating effect from TDF on weight gain, that wasn’t the case in the AFRICOS (African Cohort Study) study of 1,954 PWH who started treatment with TDF-DTG-FTC (742 patients) or a different three-drug regimen. After a median of 225 days on treatment, those who started on TDF-DTG-FTC had an adjusted, 85% higher rate of developing a high body mass index, compared with patients on a different ART regimen, Julie Ake, MD, reported in a talk at the conference. Her conclusion focused on the possible involvement of DTG: “Consistent with previous reports, dolutegravir was significantly associated with an increased risk of developing high body mass index,” said Dr. Ake, director of the U.S. Military HIV Research Program in Bethesda, Md. and leader of AFRICOS.
A potential workaround to some drugs that cause excessive the weight gain is to just not use them. That was part of the rationale for the TANGO study, which took 741 HIV-infected patients with successful viral suppression on a regimen of TAF-FTC plus one or two additional agents and switched half of them to a TAF-less, two-drug regimen of DTG-FTC. This open-label study’s primary endpoint was noninferiority for viral suppression of the DTG-FTC regimen, compared with patients who stayed on their starting regimen, and the results proved that DTG-FTC was just as effective over 48 weeks for this outcome (Clin Infect Dis. 2020 Jan 6. doi: 10.1093/cid/ciz1243).
At the conference, TANGO’s lead investigator, Jean van Wyk, MD, reported the weight and metabolic effects of the switch. The results showed a similar and small weight gain (on average less than 1 kg) during 48 week follow-up regardless of whether patients remained on their baseline, TAF-containing regimen or switched to DTG-FTC, said Dr. van Wyk, global medical lead for HIV treatment at Viiv Healthcare, the company that markets DTG. About three-quarters of patients in both arms received “boosted” dosages of their drugs, and in this subgroup, patients on DTG-FTC showed statistically significant benefits in several lipid levels, fasting glucose level, and in their degree of insulin resistance. Dr. van Wyk said. These between-group differences were not statistically significant among the “unboosted” patients, and the results failed to show a significant between-group difference in the incidence of metabolic syndrome.
Dr. Venter called these results “exciting,” and noted that he already uses the DTG-FTC two-drug combination “a lot” to treat PWH and renal disease.
A second alternative regimen showcased in a talk at the conference used the three-drug regimen of TDF-FTC plus the NNRTI, DOR (doravirine, Pifeltro). The DRIVE-SHIFT trial enrolled 670 HIV patients with successfully suppressed viral load on conventional regimens who were either switched to TDF-DOR-FTC or maintained on their baseline treatment. After 48 weeks, results confirmed the primary efficacy endpoint of noninferiority for maintenance of suppression with the investigational regimen (J Acquir Immune Defic Syndr. 2019 Aug;81[4]:463-72).
A post-hoc analysis looked at weight changes among these patients after as much as 144 weeks of follow-up. The results showed that patients switched to TDF-DOR-FTC had an average weight increase of 1.2-1.4 kg after more than 2 years on the new regimen, with fewer than 10% of patients having a 10% or greater weight gain with DOR, a “next-generation” NNRTI, reported Princy N. Kumar, MD, professor at Georgetown University and chief of infectious diseases at MedStar Georgetown University Hospital in Washington. “Weight gain was minimal, even over the long term,” she noted.
The tested DOR-based regimen also looks “very exciting,” but the populations it’s been tested have also been largely limited to White men, and limited data exist about the regimen’s performance in pregnant women, commented Dr. Venter. The DRIVE-SHIRT patient cohort was about 85% men, and about three-quarters White.
More weight data needed
HIV-treatment researchers and clinicians seem agreed that weight gain and other metabolic effects from HIV treatment need more assessment and evidence because current data, while suggestive, is also inconclusive.
“Clinical trials are desperately needed to understand the mechanisms of and potential therapeutic options for excessive weight gain on ART,” wrote Dr. Lake in her commentary in April. “While more research is needed,” the new data reported at the virtual International AIDS conference “get us closer to understanding the effects of integrase inhibitors and TAF on weight and the potential metabolic consequences,” she commented as chair of the conference session where these reports occurred.
“Further data on the mechanism of weight gain and its reversibility after a change of treatment will help refine drug selection in the near future,” predicted Dr. Geretti.
“It’s hard to understand physiologically how drugs from such different classes all seem to have weight effects; it’s maddening,” said Dr. Venter. “We need decent studies in all patient populations. That will now be the priority,” he declared. “Patients shouldn’t have to choose” between drugs that most effectively control their HIV infection and drugs that don’t pose a risk for weight gain or metabolic derangements. PWH “should not have to face obesity as their new epidemic,” he wrote with Dr. Hill.
ADVANCE was funded in part by Viiv, the company that markets dolutegravir (Tivicay), and received drugs supplied by Gilead and Viiv. TANGO was sponsored by Viiv. DRIVE-SHIFT was funded by Merck, the company that markets doravirine (Pifeltro). Dr. Lake, Dr. Sokhela, Dr. Ake, and Dr. Kumar had no disclosures, Dr. Venter has received personal fees from Adcock Ingraham, Aspen Healthcare, Johnson and Johnson, Merck, Mylan, Roche, and Viiv. Dr. Hill has received payments from Merck. Dr. Geretti has received honoraria and research funding from Gilead, Jansse, Roche, and Viiv. Dr. Taiwo has had financial relationships with Gilead, Janssen, and Viiv. Dr. Hightow-Weidman has received honoraria from Gilead and Jansse. Dr. Wohl has been a consultant to Gilead, Johnson and Johnson, and Merck. Dr. Silverberg received research funding from Gilead. Dr. Mallon has been an advisor to and speaker on behalf of Bristol-Myers Squibb, Cilag, Gilead, Jansse, Merck Sharp & Dohme, and Viiv. Dr. van Wyk is a Viiv employee.
People living with HIV who put on extra pounds and develop metabolic syndrome or related disorders linked in part to certain antiretroviral agents remain a concern today, even as the drugs used to suppress HIV infection have evolved over the decades.
Linkage of HIV treatment with lipodystrophy and insulin resistance or diabetes began in the 1990s with protease inhibitors (Clin Infect Dis. 2000 Jun;30[suppl 2]:s135-42). Several reports over the years also tied any form of effective antiretroviral therapy to weight gain in HIV patients (Antivir Ther. 2012;17[7]:1281-9). More recently, reports have rattled the HIV-treatment community by associating alarmingly high levels of weight gain with a useful and relatively new drug, tenofovir alafenamide fumarate (TAF) – a nucleoside reverse transcriptase inhibitor (NRTI) approved for use in the United States in late 2016, as well as certain agents from an entirely different antiretroviral therapy (ART) class, the integrase strand transfer inhibitors (INSTIs). Both TAF and the INSTIs have come to play major roles in the HIV-treatment landscape, despite relevant and concerning recent weight gain observations with these drugs, such as in a 2019 meta-analysis of eight trials with 5,680 treatment-naive patients who started ART during 2003-2015 (Clin Infect Dis. 2019 Oct 14;doi: 10.1093/cid/ciz999).
“Weight gain is clearly seen in studies of dolutegravir [DTG] or bictegravir [BTG] with TAF,” wrote W.D. Francois Venter, PhD and Andrew Hill, PhD in a recent published commentary on the topic (Lancet HIV. 2020 Jun 1;7[6]:e389-400). Both DTG and BTG are INSTI class members.
“Excessive weight gain, defined as more than 10% over baseline, has recently been observed among people with HIV initiating or switching to regimens incorporating TAF, an INSTI, or both, particularly DTG,” wrote Jordan E. Lake, MD, an HIV specialist at the University of Texas Health Science Center at Houston, in a recent commentary posted online. Women and Black patients “are at even greater risk for excessive weight gain,” Dr. Lake added.
“In recent times, it has emerged that weight gain is more pronounced with the integrase inhibitor class of agents, especially dolutegravir and bictegravir, the so-called second-generation” INSTIs, said Anna Maria Geretti, MD, a professor of clinical infection, microbiology, and immunology at the University of Liverpool, England. ”The effect is more pronounced in women and people of non-White ethnicity, and is of concern because of the associated potential risk of metabolic syndrome, cardiovascular disease, etc.,” Dr. Geretti said in an interview.
The unprecedented susceptibility to weight gain seen recently in non-White women may in part have resulted from the tendency of many earlier treatment trials to have cohorts comprised predominantly of White men, Dr. Venter noted in an interview.
Alarming weight gains reported
Perhaps the most eye-popping example of the potential for weight gain with the combination of TAF with an INSTI came in a recent report from the ADVANCE trial, a randomized, head-to-head comparison of three regimens in 1,053 HIV patients in South Africa. After 144 weeks on a regimen of TAF (Vemlidy), DTG (Tivicay), and FTC (emtricitabine, Emtriva), another NRTI, women gained an averaged of more than 12 kg, compared with their baseline weight, significantly more than in two comparator groups, Simiso Sokhela, MB, reported at the virtual meeting of the International AIDS conference. The women in ADVANCE on the TAF-DTG-FTC regimen also had an 11% rate of incident metabolic syndrome during their first 96 weeks on treatment, compared with rates of 8% among patients on a different form of tenofovir, tenofovir disoproxil fumarate (TDF), along with DTG-FTC, and 5% among those on TDF–EFV (efavirenz, Sustiva)–FTC said Dr. Sokhela, an HIV researcher at Ezintsha, a division of the University of the Witwatersrand in Johannesburg, South Africa.
“We believe that these results support the World Health Organization guidelines that reserve TAF for only patients with osteoporosis or impaired renal function,” Dr. Sokhela said during a press briefing at the conference. The WHO guidelines list the first-line regimen as TDF-DTG-3TC (lamivudine; Epivir) or FTC. “The risk for becoming obese continued to increase after 96 weeks” of chronic use of these drugs, she added.
“All regimens are now brilliant at viral control. Finding the ones that don’t make patients obese or have other long-term side effects is now the priority,” noted Dr. Venter, a professor and HIV researcher at University of the Witwatersrand, head of Ezintsha, and lead investigator of ADVANCE. Clinicians and researchers have recently thought that combining TAF and an INSTI plus FTC or a similar NRTI “would be the ultimate regimen to replace the nonnucleoside reverse transcriptase inhibitors (NNRTIs)” such as EFV, “but now we have a major headache” with unexpectedly high weight gains in some patients, Dr. Venter said.
Weight gains “over 10 kg are unlikely to be acceptable in any circumstances, especially when starting body mass index is already borderline overweight,” wrote Dr. Venter along with Dr. Hill in their commentary. Until recently, many clinicians chalked up weight gain on newly begun ART as a manifestation of the patient’s “return-to-health,” but this interpretation “gives a positive spin to a potentially serious and common side effect,” they added.
More from ADVANCE
The primary efficacy endpoint of ADVANCE was suppression of viral load to less than 50 RNA copies/mL after 48 weeks on treatment, and the result showed that the TAF-DTG-FTC regimen and the TDF-DTG-FTC regimen were each noninferior to the control regimen of TDF-EFV-FTC (New Engl J Med. 2019 Aug 29;381[9]:803-15). Virtually all of the enrolled patients were Black, and 59% were women. Planned follow-up of all patients ran for 96 weeks. After 48 weeks, weight gain among the women averaged 6.4 kg, 3.2 kg, and 1.7 kg in the TAF-DTG, TDF-DTG, and TDF-EFV arms respectively. After 96 weeks, the average weight gains among women were 8.2 kg, 4.6 kg, and 3.2 kg, respectively, in new results reported by Dr. Sokhela at the IAC. Follow-up to 144 weeks was partial and included about a quarter of the enrolled women, with gains averaging 12.3 kg, 7.4 kg, and 5.5 kg respectively. The pattern of weight gain among men tracked the pattern in women, but the magnitude of gain was less. Among men followed for 144 weeks, average gain among those on TAF-DTG-FTC was 7.2 kg, the largest gain seen among men on any regimen and at any follow-up time in the study.
Dr. Sokhela also reported data on body composition analyses, which showed that the weight gains were largely in fat rather than lean tissue, fat accumulation was significantly greater in women than men, and that in both sexes fat accumulated roughly equally in the trunk and on limbs.
An additional analysis looked at the incidence of new-onset obesity among the women who had a normal body mass index at baseline. After 96 weeks, incident obesity occurred in 14% of women on the TAG-DTG-FTC regimen, 8% on TDF-DTG-FTC, and in 2% of women maintained on TDF-EFV-FTC, said Dr. Hill in a separate report at the conference.
Weight starts to weigh in
“I am very mindful of weight gain potential, and I talk to patients about it. It doesn’t determine what regimen I choose for a patient” right now, “but it’s only a matter of time before it starts influencing what we do, particularly if we can achieve efficacy with fewer drugs,” commented Babafemi O. Taiwo, MD, professor of medicine and chief of infectious diseases at Northwestern University in Chicago. “I’ve had some patients show up with a weight gain of 20 kg, and that shouldn’t happen,” he said during a recent online educational session. Dr. Taiwo said his recent practice has been to warn patients about possible weight gain and to urge them to get back in touch with him quickly if it happens.
“Virologic suppression is the most important goal with ART, and the U.S. Department of Health and Human Services currently recommends INSTI-based ART for most PWH [people with HIV],” wrote Dr. Lake in April 2020. “I counsel all PWH initiating ART about the potential for weight gain, and I discuss their current diet and healthy lifestyle habits. I explain to patients that we will monitor their weight, and if weight gain seems more than either of us are comfortable with then we will reassess. Only a small percentage of patients experience excessive weight gain after starting ART.” Dr. Lake also stressed that she had not yet begun to change the regimen a patient is on solely because of weight gain. “We do not know whether this weight gain is reversible,” she noted.
“I do not anticipate that a risk of weight gain at present will dictate a change in guidelines,” said Dr. Geretti. “Drugs such as dolutegravir and bictegravir are very effective, and they are unlikely to cause drug resistance. Further data on the mechanism of weight gain and the reversibility after a change of treatment will help refine drug selection in the near future,” she predicted.
“I consider weight gain when prescribing because my patients hear about this. It’s a side effect that my patients really care about, and I don’t blame them,” said Lisa Hightow-Weidman, MD, a professor and HIV specialist at the University of North Carolina at Chapel Hill, during an on-line educational session. “If you don’t discuss it with a patient and then weight gain happens and the patient finds out [the known risk from their treatment] they may have an issue,” she noted. But weight gain is not a reason to avoid these drugs. “They are great medications in many ways, with once-daily regimens and few side effects.”
Weight gain during pregnancy a special concern
An additional analysis of data from ADVANCE presented at the conference highlighted what the observed weight gain on ART could mean for women who become pregnant while on treatment. Based on a systematic literature review, the ADVANCE investigators calculated the relative risk for six obesity-related pregnancy complications, compared with nonobese women: preterm delivery, gestational diabetes, gestational hypertension, preeclampsia, postpartum hemorrhage, and caesarean delivery. Based on the obesity changes among women on their assigned ART in ADVANCE, the researchers calculated the predicted incidence of these six complications. The analysis showed that for every 1,000 women, those on TAG-DTG-FTC would have an excess of 53 obesity-related pregnancy complications, those on TDF-DTG-FTC would develop 28 excess pregnancy complications, and those on TDG-EFV-FTC would have four excess complications, reported Dr. Hill at the International AIDS conference.
The researchers also ran a similar simulation for the incidence of neonatal complications that could result when mothers are obese because of their ART. The six neonatal complications included in this analysis were small for gestational age, large for gestational age, macrosomia, neonatal death, stillbirth, and neural tube defects. Based on the excess rate of incident obesity, they calculated that for every 1,000 pregnancies women on TAD-DTG-FTC would have 24 additional infants born with one of these complications, women on TDF-DTG-FTC would have an excess of 13 of these events, and women on TDG-EFV-FTC would have an excess of three such obesity-related neonatal complications, Dr. Hill said.
Sorting out the drugs
Results from several additional studies reported at the conference have started trying to discern exactly which ART drugs and regimens pose the greatest weight gain risk and which have the least risk while retaining high efficacy and resistance barriers.
Further evidence implicating any type of ART as a driver of increased weight came from a review of 8,256 adults infected with HIV and members of the Kaiser Permanente health system in three U.S. regions during 2000-2016. Researchers matched these cases using several demographic factors with just under 130,000 members without HIV. Those infected by HIV had half the prevalence of obesity as the matched controls at baseline. During 12 years of follow-up, those infected with HIV had a threefold higher rate of weight gain than those who were uninfected. Annual weight gain averaged 0.06 kg/year among the uninfected people and 0.22 kg/year among those infected with HIV, a statistically significant difference that was consistent regardless of whether people started the study at a normal body mass index, overweight, or obese, reported Michael J. Silverberg, PhD, an epidemiologist with Kaiser Permanente in Oakland, Calif.
Another study tried to focus on the weight gain impact when patients on three-drug ART regimens changed from taking TDF to TAF. This analysis used data collected in the OPERA (Observational Pharmaco-Epidemiology Research & Analysis) longitudinal cohort of about 115,000 U.S. PWH. The observational cohort included nearly 7,000 patients who made a TDF-to-TAF switch, including 3,288 patients who maintained treatment during this switch with an INSTI, 1,454 who maintained a background regimen based on a NNRTI, 1,430 patients who also switched from an INSTI to a different drug, and 747 patients maintained on a boosted dose of a protease inhibitor. All patients were well controlled on their baseline regimen, with at least two consecutive measures showing undetectable viral load.
Patients who maintained their background regimens while changing from TDF to TAF had a 2.0-2.6 kg increase in weight during the 9 months immediately following their switch to TAF, reported Patrick Mallon, MB, a professor of microbial diseases at University College Dublin. Among the patients who both switched to TAF and also switched to treatment with an INSTI, weight gain during the 9 months after the switch averaged 2.6-4.5 kg, depending on which INSTI was started. Patients who switched to treatment with elvitegravir/cobicistat (an INSTI plus a boosting agent) averaged a gain of 2.6 kg during 9 months, those who switched to DTG averaged a 3.1-kg gain, and those who switched to BTG averaged a 4.6-kg increase, Dr. Mallon reported at the conference.
These findings “give us a good sense that the weight gain is real. This is not just overeating or not exercising, but weight changes coincidental with a change in HIV treatment,” commented David Wohl, MD, professor of medicine and site leader of the HIV Prevention and Treatment Clinical Trials Unit at the University of North Carolina at Chapel Hill, during an online educational session.
Contrary to this evidence suggesting a consistent uptick in weight when patients start TAF treatment was a recent report on 629 HIV patients randomized to treatment with TAF-BTG-FTC or abacavir (an NRTI, Ziagen)–DTG-3TC, which found similar weight gains between these two regimens after 144 weeks on treatment (Lancet HIV. 2020 Jun;7[6]:e389-400). This finding had the effect of “strengthening the argument that TAF is simply an innocent bystander” and does not play a central role in weight gain, and supporting the notion that the alternative tenofovir formulation, TDF, differs from TAF by promoting weight loss, Dr. Venter and Dr. Hill suggested in their commentary that accompanied this report.
The new findings from Dr. Mallon raise “serious questions about the way we have moved to TAF as a replacement for TDF, especially because the benefits [from TAF] are for a small subgroup – patients with renal disease or osteoporosis,” Dr. Venter said in an interview. “The question is, will we see weight gain like this” if TAF was combined with a non-INSTI drug? he wondered.
While some study results have suggested a mitigating effect from TDF on weight gain, that wasn’t the case in the AFRICOS (African Cohort Study) study of 1,954 PWH who started treatment with TDF-DTG-FTC (742 patients) or a different three-drug regimen. After a median of 225 days on treatment, those who started on TDF-DTG-FTC had an adjusted, 85% higher rate of developing a high body mass index, compared with patients on a different ART regimen, Julie Ake, MD, reported in a talk at the conference. Her conclusion focused on the possible involvement of DTG: “Consistent with previous reports, dolutegravir was significantly associated with an increased risk of developing high body mass index,” said Dr. Ake, director of the U.S. Military HIV Research Program in Bethesda, Md. and leader of AFRICOS.
A potential workaround to some drugs that cause excessive the weight gain is to just not use them. That was part of the rationale for the TANGO study, which took 741 HIV-infected patients with successful viral suppression on a regimen of TAF-FTC plus one or two additional agents and switched half of them to a TAF-less, two-drug regimen of DTG-FTC. This open-label study’s primary endpoint was noninferiority for viral suppression of the DTG-FTC regimen, compared with patients who stayed on their starting regimen, and the results proved that DTG-FTC was just as effective over 48 weeks for this outcome (Clin Infect Dis. 2020 Jan 6. doi: 10.1093/cid/ciz1243).
At the conference, TANGO’s lead investigator, Jean van Wyk, MD, reported the weight and metabolic effects of the switch. The results showed a similar and small weight gain (on average less than 1 kg) during 48 week follow-up regardless of whether patients remained on their baseline, TAF-containing regimen or switched to DTG-FTC, said Dr. van Wyk, global medical lead for HIV treatment at Viiv Healthcare, the company that markets DTG. About three-quarters of patients in both arms received “boosted” dosages of their drugs, and in this subgroup, patients on DTG-FTC showed statistically significant benefits in several lipid levels, fasting glucose level, and in their degree of insulin resistance. Dr. van Wyk said. These between-group differences were not statistically significant among the “unboosted” patients, and the results failed to show a significant between-group difference in the incidence of metabolic syndrome.
Dr. Venter called these results “exciting,” and noted that he already uses the DTG-FTC two-drug combination “a lot” to treat PWH and renal disease.
A second alternative regimen showcased in a talk at the conference used the three-drug regimen of TDF-FTC plus the NNRTI, DOR (doravirine, Pifeltro). The DRIVE-SHIFT trial enrolled 670 HIV patients with successfully suppressed viral load on conventional regimens who were either switched to TDF-DOR-FTC or maintained on their baseline treatment. After 48 weeks, results confirmed the primary efficacy endpoint of noninferiority for maintenance of suppression with the investigational regimen (J Acquir Immune Defic Syndr. 2019 Aug;81[4]:463-72).
A post-hoc analysis looked at weight changes among these patients after as much as 144 weeks of follow-up. The results showed that patients switched to TDF-DOR-FTC had an average weight increase of 1.2-1.4 kg after more than 2 years on the new regimen, with fewer than 10% of patients having a 10% or greater weight gain with DOR, a “next-generation” NNRTI, reported Princy N. Kumar, MD, professor at Georgetown University and chief of infectious diseases at MedStar Georgetown University Hospital in Washington. “Weight gain was minimal, even over the long term,” she noted.
The tested DOR-based regimen also looks “very exciting,” but the populations it’s been tested have also been largely limited to White men, and limited data exist about the regimen’s performance in pregnant women, commented Dr. Venter. The DRIVE-SHIRT patient cohort was about 85% men, and about three-quarters White.
More weight data needed
HIV-treatment researchers and clinicians seem agreed that weight gain and other metabolic effects from HIV treatment need more assessment and evidence because current data, while suggestive, is also inconclusive.
“Clinical trials are desperately needed to understand the mechanisms of and potential therapeutic options for excessive weight gain on ART,” wrote Dr. Lake in her commentary in April. “While more research is needed,” the new data reported at the virtual International AIDS conference “get us closer to understanding the effects of integrase inhibitors and TAF on weight and the potential metabolic consequences,” she commented as chair of the conference session where these reports occurred.
“Further data on the mechanism of weight gain and its reversibility after a change of treatment will help refine drug selection in the near future,” predicted Dr. Geretti.
“It’s hard to understand physiologically how drugs from such different classes all seem to have weight effects; it’s maddening,” said Dr. Venter. “We need decent studies in all patient populations. That will now be the priority,” he declared. “Patients shouldn’t have to choose” between drugs that most effectively control their HIV infection and drugs that don’t pose a risk for weight gain or metabolic derangements. PWH “should not have to face obesity as their new epidemic,” he wrote with Dr. Hill.
ADVANCE was funded in part by Viiv, the company that markets dolutegravir (Tivicay), and received drugs supplied by Gilead and Viiv. TANGO was sponsored by Viiv. DRIVE-SHIFT was funded by Merck, the company that markets doravirine (Pifeltro). Dr. Lake, Dr. Sokhela, Dr. Ake, and Dr. Kumar had no disclosures, Dr. Venter has received personal fees from Adcock Ingraham, Aspen Healthcare, Johnson and Johnson, Merck, Mylan, Roche, and Viiv. Dr. Hill has received payments from Merck. Dr. Geretti has received honoraria and research funding from Gilead, Jansse, Roche, and Viiv. Dr. Taiwo has had financial relationships with Gilead, Janssen, and Viiv. Dr. Hightow-Weidman has received honoraria from Gilead and Jansse. Dr. Wohl has been a consultant to Gilead, Johnson and Johnson, and Merck. Dr. Silverberg received research funding from Gilead. Dr. Mallon has been an advisor to and speaker on behalf of Bristol-Myers Squibb, Cilag, Gilead, Jansse, Merck Sharp & Dohme, and Viiv. Dr. van Wyk is a Viiv employee.
People living with HIV who put on extra pounds and develop metabolic syndrome or related disorders linked in part to certain antiretroviral agents remain a concern today, even as the drugs used to suppress HIV infection have evolved over the decades.
Linkage of HIV treatment with lipodystrophy and insulin resistance or diabetes began in the 1990s with protease inhibitors (Clin Infect Dis. 2000 Jun;30[suppl 2]:s135-42). Several reports over the years also tied any form of effective antiretroviral therapy to weight gain in HIV patients (Antivir Ther. 2012;17[7]:1281-9). More recently, reports have rattled the HIV-treatment community by associating alarmingly high levels of weight gain with a useful and relatively new drug, tenofovir alafenamide fumarate (TAF) – a nucleoside reverse transcriptase inhibitor (NRTI) approved for use in the United States in late 2016, as well as certain agents from an entirely different antiretroviral therapy (ART) class, the integrase strand transfer inhibitors (INSTIs). Both TAF and the INSTIs have come to play major roles in the HIV-treatment landscape, despite relevant and concerning recent weight gain observations with these drugs, such as in a 2019 meta-analysis of eight trials with 5,680 treatment-naive patients who started ART during 2003-2015 (Clin Infect Dis. 2019 Oct 14;doi: 10.1093/cid/ciz999).
“Weight gain is clearly seen in studies of dolutegravir [DTG] or bictegravir [BTG] with TAF,” wrote W.D. Francois Venter, PhD and Andrew Hill, PhD in a recent published commentary on the topic (Lancet HIV. 2020 Jun 1;7[6]:e389-400). Both DTG and BTG are INSTI class members.
“Excessive weight gain, defined as more than 10% over baseline, has recently been observed among people with HIV initiating or switching to regimens incorporating TAF, an INSTI, or both, particularly DTG,” wrote Jordan E. Lake, MD, an HIV specialist at the University of Texas Health Science Center at Houston, in a recent commentary posted online. Women and Black patients “are at even greater risk for excessive weight gain,” Dr. Lake added.
“In recent times, it has emerged that weight gain is more pronounced with the integrase inhibitor class of agents, especially dolutegravir and bictegravir, the so-called second-generation” INSTIs, said Anna Maria Geretti, MD, a professor of clinical infection, microbiology, and immunology at the University of Liverpool, England. ”The effect is more pronounced in women and people of non-White ethnicity, and is of concern because of the associated potential risk of metabolic syndrome, cardiovascular disease, etc.,” Dr. Geretti said in an interview.
The unprecedented susceptibility to weight gain seen recently in non-White women may in part have resulted from the tendency of many earlier treatment trials to have cohorts comprised predominantly of White men, Dr. Venter noted in an interview.
Alarming weight gains reported
Perhaps the most eye-popping example of the potential for weight gain with the combination of TAF with an INSTI came in a recent report from the ADVANCE trial, a randomized, head-to-head comparison of three regimens in 1,053 HIV patients in South Africa. After 144 weeks on a regimen of TAF (Vemlidy), DTG (Tivicay), and FTC (emtricitabine, Emtriva), another NRTI, women gained an averaged of more than 12 kg, compared with their baseline weight, significantly more than in two comparator groups, Simiso Sokhela, MB, reported at the virtual meeting of the International AIDS conference. The women in ADVANCE on the TAF-DTG-FTC regimen also had an 11% rate of incident metabolic syndrome during their first 96 weeks on treatment, compared with rates of 8% among patients on a different form of tenofovir, tenofovir disoproxil fumarate (TDF), along with DTG-FTC, and 5% among those on TDF–EFV (efavirenz, Sustiva)–FTC said Dr. Sokhela, an HIV researcher at Ezintsha, a division of the University of the Witwatersrand in Johannesburg, South Africa.
“We believe that these results support the World Health Organization guidelines that reserve TAF for only patients with osteoporosis or impaired renal function,” Dr. Sokhela said during a press briefing at the conference. The WHO guidelines list the first-line regimen as TDF-DTG-3TC (lamivudine; Epivir) or FTC. “The risk for becoming obese continued to increase after 96 weeks” of chronic use of these drugs, she added.
“All regimens are now brilliant at viral control. Finding the ones that don’t make patients obese or have other long-term side effects is now the priority,” noted Dr. Venter, a professor and HIV researcher at University of the Witwatersrand, head of Ezintsha, and lead investigator of ADVANCE. Clinicians and researchers have recently thought that combining TAF and an INSTI plus FTC or a similar NRTI “would be the ultimate regimen to replace the nonnucleoside reverse transcriptase inhibitors (NNRTIs)” such as EFV, “but now we have a major headache” with unexpectedly high weight gains in some patients, Dr. Venter said.
Weight gains “over 10 kg are unlikely to be acceptable in any circumstances, especially when starting body mass index is already borderline overweight,” wrote Dr. Venter along with Dr. Hill in their commentary. Until recently, many clinicians chalked up weight gain on newly begun ART as a manifestation of the patient’s “return-to-health,” but this interpretation “gives a positive spin to a potentially serious and common side effect,” they added.
More from ADVANCE
The primary efficacy endpoint of ADVANCE was suppression of viral load to less than 50 RNA copies/mL after 48 weeks on treatment, and the result showed that the TAF-DTG-FTC regimen and the TDF-DTG-FTC regimen were each noninferior to the control regimen of TDF-EFV-FTC (New Engl J Med. 2019 Aug 29;381[9]:803-15). Virtually all of the enrolled patients were Black, and 59% were women. Planned follow-up of all patients ran for 96 weeks. After 48 weeks, weight gain among the women averaged 6.4 kg, 3.2 kg, and 1.7 kg in the TAF-DTG, TDF-DTG, and TDF-EFV arms respectively. After 96 weeks, the average weight gains among women were 8.2 kg, 4.6 kg, and 3.2 kg, respectively, in new results reported by Dr. Sokhela at the IAC. Follow-up to 144 weeks was partial and included about a quarter of the enrolled women, with gains averaging 12.3 kg, 7.4 kg, and 5.5 kg respectively. The pattern of weight gain among men tracked the pattern in women, but the magnitude of gain was less. Among men followed for 144 weeks, average gain among those on TAF-DTG-FTC was 7.2 kg, the largest gain seen among men on any regimen and at any follow-up time in the study.
Dr. Sokhela also reported data on body composition analyses, which showed that the weight gains were largely in fat rather than lean tissue, fat accumulation was significantly greater in women than men, and that in both sexes fat accumulated roughly equally in the trunk and on limbs.
An additional analysis looked at the incidence of new-onset obesity among the women who had a normal body mass index at baseline. After 96 weeks, incident obesity occurred in 14% of women on the TAG-DTG-FTC regimen, 8% on TDF-DTG-FTC, and in 2% of women maintained on TDF-EFV-FTC, said Dr. Hill in a separate report at the conference.
Weight starts to weigh in
“I am very mindful of weight gain potential, and I talk to patients about it. It doesn’t determine what regimen I choose for a patient” right now, “but it’s only a matter of time before it starts influencing what we do, particularly if we can achieve efficacy with fewer drugs,” commented Babafemi O. Taiwo, MD, professor of medicine and chief of infectious diseases at Northwestern University in Chicago. “I’ve had some patients show up with a weight gain of 20 kg, and that shouldn’t happen,” he said during a recent online educational session. Dr. Taiwo said his recent practice has been to warn patients about possible weight gain and to urge them to get back in touch with him quickly if it happens.
“Virologic suppression is the most important goal with ART, and the U.S. Department of Health and Human Services currently recommends INSTI-based ART for most PWH [people with HIV],” wrote Dr. Lake in April 2020. “I counsel all PWH initiating ART about the potential for weight gain, and I discuss their current diet and healthy lifestyle habits. I explain to patients that we will monitor their weight, and if weight gain seems more than either of us are comfortable with then we will reassess. Only a small percentage of patients experience excessive weight gain after starting ART.” Dr. Lake also stressed that she had not yet begun to change the regimen a patient is on solely because of weight gain. “We do not know whether this weight gain is reversible,” she noted.
“I do not anticipate that a risk of weight gain at present will dictate a change in guidelines,” said Dr. Geretti. “Drugs such as dolutegravir and bictegravir are very effective, and they are unlikely to cause drug resistance. Further data on the mechanism of weight gain and the reversibility after a change of treatment will help refine drug selection in the near future,” she predicted.
“I consider weight gain when prescribing because my patients hear about this. It’s a side effect that my patients really care about, and I don’t blame them,” said Lisa Hightow-Weidman, MD, a professor and HIV specialist at the University of North Carolina at Chapel Hill, during an on-line educational session. “If you don’t discuss it with a patient and then weight gain happens and the patient finds out [the known risk from their treatment] they may have an issue,” she noted. But weight gain is not a reason to avoid these drugs. “They are great medications in many ways, with once-daily regimens and few side effects.”
Weight gain during pregnancy a special concern
An additional analysis of data from ADVANCE presented at the conference highlighted what the observed weight gain on ART could mean for women who become pregnant while on treatment. Based on a systematic literature review, the ADVANCE investigators calculated the relative risk for six obesity-related pregnancy complications, compared with nonobese women: preterm delivery, gestational diabetes, gestational hypertension, preeclampsia, postpartum hemorrhage, and caesarean delivery. Based on the obesity changes among women on their assigned ART in ADVANCE, the researchers calculated the predicted incidence of these six complications. The analysis showed that for every 1,000 women, those on TAG-DTG-FTC would have an excess of 53 obesity-related pregnancy complications, those on TDF-DTG-FTC would develop 28 excess pregnancy complications, and those on TDG-EFV-FTC would have four excess complications, reported Dr. Hill at the International AIDS conference.
The researchers also ran a similar simulation for the incidence of neonatal complications that could result when mothers are obese because of their ART. The six neonatal complications included in this analysis were small for gestational age, large for gestational age, macrosomia, neonatal death, stillbirth, and neural tube defects. Based on the excess rate of incident obesity, they calculated that for every 1,000 pregnancies women on TAD-DTG-FTC would have 24 additional infants born with one of these complications, women on TDF-DTG-FTC would have an excess of 13 of these events, and women on TDG-EFV-FTC would have an excess of three such obesity-related neonatal complications, Dr. Hill said.
Sorting out the drugs
Results from several additional studies reported at the conference have started trying to discern exactly which ART drugs and regimens pose the greatest weight gain risk and which have the least risk while retaining high efficacy and resistance barriers.
Further evidence implicating any type of ART as a driver of increased weight came from a review of 8,256 adults infected with HIV and members of the Kaiser Permanente health system in three U.S. regions during 2000-2016. Researchers matched these cases using several demographic factors with just under 130,000 members without HIV. Those infected by HIV had half the prevalence of obesity as the matched controls at baseline. During 12 years of follow-up, those infected with HIV had a threefold higher rate of weight gain than those who were uninfected. Annual weight gain averaged 0.06 kg/year among the uninfected people and 0.22 kg/year among those infected with HIV, a statistically significant difference that was consistent regardless of whether people started the study at a normal body mass index, overweight, or obese, reported Michael J. Silverberg, PhD, an epidemiologist with Kaiser Permanente in Oakland, Calif.
Another study tried to focus on the weight gain impact when patients on three-drug ART regimens changed from taking TDF to TAF. This analysis used data collected in the OPERA (Observational Pharmaco-Epidemiology Research & Analysis) longitudinal cohort of about 115,000 U.S. PWH. The observational cohort included nearly 7,000 patients who made a TDF-to-TAF switch, including 3,288 patients who maintained treatment during this switch with an INSTI, 1,454 who maintained a background regimen based on a NNRTI, 1,430 patients who also switched from an INSTI to a different drug, and 747 patients maintained on a boosted dose of a protease inhibitor. All patients were well controlled on their baseline regimen, with at least two consecutive measures showing undetectable viral load.
Patients who maintained their background regimens while changing from TDF to TAF had a 2.0-2.6 kg increase in weight during the 9 months immediately following their switch to TAF, reported Patrick Mallon, MB, a professor of microbial diseases at University College Dublin. Among the patients who both switched to TAF and also switched to treatment with an INSTI, weight gain during the 9 months after the switch averaged 2.6-4.5 kg, depending on which INSTI was started. Patients who switched to treatment with elvitegravir/cobicistat (an INSTI plus a boosting agent) averaged a gain of 2.6 kg during 9 months, those who switched to DTG averaged a 3.1-kg gain, and those who switched to BTG averaged a 4.6-kg increase, Dr. Mallon reported at the conference.
These findings “give us a good sense that the weight gain is real. This is not just overeating or not exercising, but weight changes coincidental with a change in HIV treatment,” commented David Wohl, MD, professor of medicine and site leader of the HIV Prevention and Treatment Clinical Trials Unit at the University of North Carolina at Chapel Hill, during an online educational session.
Contrary to this evidence suggesting a consistent uptick in weight when patients start TAF treatment was a recent report on 629 HIV patients randomized to treatment with TAF-BTG-FTC or abacavir (an NRTI, Ziagen)–DTG-3TC, which found similar weight gains between these two regimens after 144 weeks on treatment (Lancet HIV. 2020 Jun;7[6]:e389-400). This finding had the effect of “strengthening the argument that TAF is simply an innocent bystander” and does not play a central role in weight gain, and supporting the notion that the alternative tenofovir formulation, TDF, differs from TAF by promoting weight loss, Dr. Venter and Dr. Hill suggested in their commentary that accompanied this report.
The new findings from Dr. Mallon raise “serious questions about the way we have moved to TAF as a replacement for TDF, especially because the benefits [from TAF] are for a small subgroup – patients with renal disease or osteoporosis,” Dr. Venter said in an interview. “The question is, will we see weight gain like this” if TAF was combined with a non-INSTI drug? he wondered.
While some study results have suggested a mitigating effect from TDF on weight gain, that wasn’t the case in the AFRICOS (African Cohort Study) study of 1,954 PWH who started treatment with TDF-DTG-FTC (742 patients) or a different three-drug regimen. After a median of 225 days on treatment, those who started on TDF-DTG-FTC had an adjusted, 85% higher rate of developing a high body mass index, compared with patients on a different ART regimen, Julie Ake, MD, reported in a talk at the conference. Her conclusion focused on the possible involvement of DTG: “Consistent with previous reports, dolutegravir was significantly associated with an increased risk of developing high body mass index,” said Dr. Ake, director of the U.S. Military HIV Research Program in Bethesda, Md. and leader of AFRICOS.
A potential workaround to some drugs that cause excessive the weight gain is to just not use them. That was part of the rationale for the TANGO study, which took 741 HIV-infected patients with successful viral suppression on a regimen of TAF-FTC plus one or two additional agents and switched half of them to a TAF-less, two-drug regimen of DTG-FTC. This open-label study’s primary endpoint was noninferiority for viral suppression of the DTG-FTC regimen, compared with patients who stayed on their starting regimen, and the results proved that DTG-FTC was just as effective over 48 weeks for this outcome (Clin Infect Dis. 2020 Jan 6. doi: 10.1093/cid/ciz1243).
At the conference, TANGO’s lead investigator, Jean van Wyk, MD, reported the weight and metabolic effects of the switch. The results showed a similar and small weight gain (on average less than 1 kg) during 48 week follow-up regardless of whether patients remained on their baseline, TAF-containing regimen or switched to DTG-FTC, said Dr. van Wyk, global medical lead for HIV treatment at Viiv Healthcare, the company that markets DTG. About three-quarters of patients in both arms received “boosted” dosages of their drugs, and in this subgroup, patients on DTG-FTC showed statistically significant benefits in several lipid levels, fasting glucose level, and in their degree of insulin resistance. Dr. van Wyk said. These between-group differences were not statistically significant among the “unboosted” patients, and the results failed to show a significant between-group difference in the incidence of metabolic syndrome.
Dr. Venter called these results “exciting,” and noted that he already uses the DTG-FTC two-drug combination “a lot” to treat PWH and renal disease.
A second alternative regimen showcased in a talk at the conference used the three-drug regimen of TDF-FTC plus the NNRTI, DOR (doravirine, Pifeltro). The DRIVE-SHIFT trial enrolled 670 HIV patients with successfully suppressed viral load on conventional regimens who were either switched to TDF-DOR-FTC or maintained on their baseline treatment. After 48 weeks, results confirmed the primary efficacy endpoint of noninferiority for maintenance of suppression with the investigational regimen (J Acquir Immune Defic Syndr. 2019 Aug;81[4]:463-72).
A post-hoc analysis looked at weight changes among these patients after as much as 144 weeks of follow-up. The results showed that patients switched to TDF-DOR-FTC had an average weight increase of 1.2-1.4 kg after more than 2 years on the new regimen, with fewer than 10% of patients having a 10% or greater weight gain with DOR, a “next-generation” NNRTI, reported Princy N. Kumar, MD, professor at Georgetown University and chief of infectious diseases at MedStar Georgetown University Hospital in Washington. “Weight gain was minimal, even over the long term,” she noted.
The tested DOR-based regimen also looks “very exciting,” but the populations it’s been tested have also been largely limited to White men, and limited data exist about the regimen’s performance in pregnant women, commented Dr. Venter. The DRIVE-SHIRT patient cohort was about 85% men, and about three-quarters White.
More weight data needed
HIV-treatment researchers and clinicians seem agreed that weight gain and other metabolic effects from HIV treatment need more assessment and evidence because current data, while suggestive, is also inconclusive.
“Clinical trials are desperately needed to understand the mechanisms of and potential therapeutic options for excessive weight gain on ART,” wrote Dr. Lake in her commentary in April. “While more research is needed,” the new data reported at the virtual International AIDS conference “get us closer to understanding the effects of integrase inhibitors and TAF on weight and the potential metabolic consequences,” she commented as chair of the conference session where these reports occurred.
“Further data on the mechanism of weight gain and its reversibility after a change of treatment will help refine drug selection in the near future,” predicted Dr. Geretti.
“It’s hard to understand physiologically how drugs from such different classes all seem to have weight effects; it’s maddening,” said Dr. Venter. “We need decent studies in all patient populations. That will now be the priority,” he declared. “Patients shouldn’t have to choose” between drugs that most effectively control their HIV infection and drugs that don’t pose a risk for weight gain or metabolic derangements. PWH “should not have to face obesity as their new epidemic,” he wrote with Dr. Hill.
ADVANCE was funded in part by Viiv, the company that markets dolutegravir (Tivicay), and received drugs supplied by Gilead and Viiv. TANGO was sponsored by Viiv. DRIVE-SHIFT was funded by Merck, the company that markets doravirine (Pifeltro). Dr. Lake, Dr. Sokhela, Dr. Ake, and Dr. Kumar had no disclosures, Dr. Venter has received personal fees from Adcock Ingraham, Aspen Healthcare, Johnson and Johnson, Merck, Mylan, Roche, and Viiv. Dr. Hill has received payments from Merck. Dr. Geretti has received honoraria and research funding from Gilead, Jansse, Roche, and Viiv. Dr. Taiwo has had financial relationships with Gilead, Janssen, and Viiv. Dr. Hightow-Weidman has received honoraria from Gilead and Jansse. Dr. Wohl has been a consultant to Gilead, Johnson and Johnson, and Merck. Dr. Silverberg received research funding from Gilead. Dr. Mallon has been an advisor to and speaker on behalf of Bristol-Myers Squibb, Cilag, Gilead, Jansse, Merck Sharp & Dohme, and Viiv. Dr. van Wyk is a Viiv employee.
FROM AIDS 2020
Infection ups mortality risk in patients with dementia
Infection increases mortality risk among patients with dementia, new research suggests. A large, registry-based cohort study showed that
“This is the first study to our knowledge to show that increased mortality is observed across all infection types in people with dementia and that increased mortality is seen both short and long term,” said coinvestigator Janet Janbek, a PhD student at the Danish Dementia Research Center, Rigshospitalet, University of Copenhagen.
The findings were presented at the virtual annual meeting of the Alzheimer’s Association International Conference.
Large Danish cohort
The investigators analyzed data from Danish national health registries for nearly 1.5 million individuals aged 65 years and older who had visited the hospital with an infection. There were 575,260 deaths during more than 12.7 million person-years of follow-up.
Patients with dementia who also had a hospital visit for infection died at a 6.5 times higher rate than participants without dementia or an infection. Those with either dementia alone or infection-related contacts alone had a threefold increased rate of death.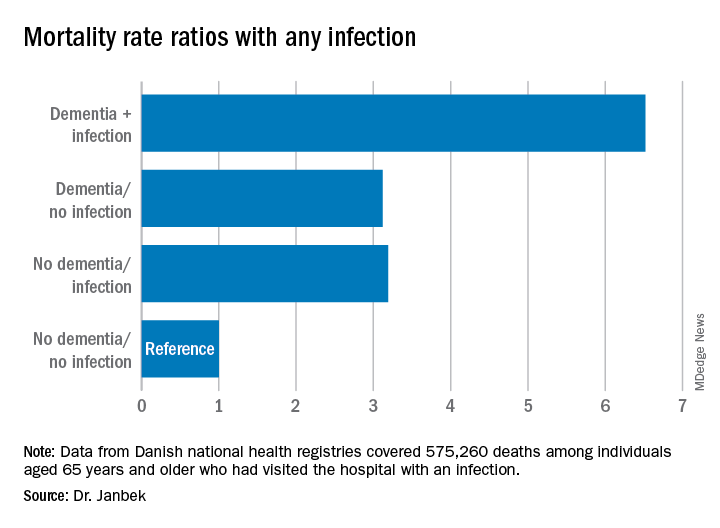
The mortality rate was highest within the first 30 days following the hospital visit for infection. However, the rate remained elevated for 10 years after the initial infection-related hospital visit.
Mortality rates from all infections, including major infections, such as sepsis, down to minor ear infections were elevated in patients with dementia, compared with people who did not have dementia or an infection-related hospital visit.
Ms. Janbek said there are several possible explanations for the association of infection and increased mortality risk in those with dementia. “After a hospital contact with a severe infection, people with dementia may become more reliant on external care, become more frail, and have declined functional levels, which might explain the observed association.”
It might also be that patients with dementia have more severe infections than those without dementia at the time of hospital contact, possibly because of delayed diagnosis, which could explain the higher mortality rates, said Ms. Janbek.
“It is also plausible that infections play a role in worsening dementia and subsequently lead to increased mortality,” she noted.
“Clinicians and health care personnel need to pay closer attention to infections of all types in people with dementia, and steps toward better clinical management and improved posthospital care need to be explored and undertaken. We need to identify possible preventive measures and targeted interventions in people with dementia and infections,” Ms. Janbek said.
‘Interesting observation’
Commenting on the study, Rebecca M. Edelmayer, PhD, director of scientific engagement for the Alzheimer’s Association, said it presents “an interesting observation.” However, “we can’t make any direct assumptions from this research per se about infections and dementia and whether they are causative in any way,” noted Dr. Edelmayer, who was not involved with the study.
Instead, the study highlighted the importance of “taking care of our overall health and making sure that individuals that might be vulnerable to infection, like those who are already living with dementia, are getting the best care possible,” she said.
Ms. Janbek and Dr. Edelmayer have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Infection increases mortality risk among patients with dementia, new research suggests. A large, registry-based cohort study showed that
“This is the first study to our knowledge to show that increased mortality is observed across all infection types in people with dementia and that increased mortality is seen both short and long term,” said coinvestigator Janet Janbek, a PhD student at the Danish Dementia Research Center, Rigshospitalet, University of Copenhagen.
The findings were presented at the virtual annual meeting of the Alzheimer’s Association International Conference.
Large Danish cohort
The investigators analyzed data from Danish national health registries for nearly 1.5 million individuals aged 65 years and older who had visited the hospital with an infection. There were 575,260 deaths during more than 12.7 million person-years of follow-up.
Patients with dementia who also had a hospital visit for infection died at a 6.5 times higher rate than participants without dementia or an infection. Those with either dementia alone or infection-related contacts alone had a threefold increased rate of death.
The mortality rate was highest within the first 30 days following the hospital visit for infection. However, the rate remained elevated for 10 years after the initial infection-related hospital visit.
Mortality rates from all infections, including major infections, such as sepsis, down to minor ear infections were elevated in patients with dementia, compared with people who did not have dementia or an infection-related hospital visit.
Ms. Janbek said there are several possible explanations for the association of infection and increased mortality risk in those with dementia. “After a hospital contact with a severe infection, people with dementia may become more reliant on external care, become more frail, and have declined functional levels, which might explain the observed association.”
It might also be that patients with dementia have more severe infections than those without dementia at the time of hospital contact, possibly because of delayed diagnosis, which could explain the higher mortality rates, said Ms. Janbek.
“It is also plausible that infections play a role in worsening dementia and subsequently lead to increased mortality,” she noted.
“Clinicians and health care personnel need to pay closer attention to infections of all types in people with dementia, and steps toward better clinical management and improved posthospital care need to be explored and undertaken. We need to identify possible preventive measures and targeted interventions in people with dementia and infections,” Ms. Janbek said.
‘Interesting observation’
Commenting on the study, Rebecca M. Edelmayer, PhD, director of scientific engagement for the Alzheimer’s Association, said it presents “an interesting observation.” However, “we can’t make any direct assumptions from this research per se about infections and dementia and whether they are causative in any way,” noted Dr. Edelmayer, who was not involved with the study.
Instead, the study highlighted the importance of “taking care of our overall health and making sure that individuals that might be vulnerable to infection, like those who are already living with dementia, are getting the best care possible,” she said.
Ms. Janbek and Dr. Edelmayer have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Infection increases mortality risk among patients with dementia, new research suggests. A large, registry-based cohort study showed that
“This is the first study to our knowledge to show that increased mortality is observed across all infection types in people with dementia and that increased mortality is seen both short and long term,” said coinvestigator Janet Janbek, a PhD student at the Danish Dementia Research Center, Rigshospitalet, University of Copenhagen.
The findings were presented at the virtual annual meeting of the Alzheimer’s Association International Conference.
Large Danish cohort
The investigators analyzed data from Danish national health registries for nearly 1.5 million individuals aged 65 years and older who had visited the hospital with an infection. There were 575,260 deaths during more than 12.7 million person-years of follow-up.
Patients with dementia who also had a hospital visit for infection died at a 6.5 times higher rate than participants without dementia or an infection. Those with either dementia alone or infection-related contacts alone had a threefold increased rate of death.
The mortality rate was highest within the first 30 days following the hospital visit for infection. However, the rate remained elevated for 10 years after the initial infection-related hospital visit.
Mortality rates from all infections, including major infections, such as sepsis, down to minor ear infections were elevated in patients with dementia, compared with people who did not have dementia or an infection-related hospital visit.
Ms. Janbek said there are several possible explanations for the association of infection and increased mortality risk in those with dementia. “After a hospital contact with a severe infection, people with dementia may become more reliant on external care, become more frail, and have declined functional levels, which might explain the observed association.”
It might also be that patients with dementia have more severe infections than those without dementia at the time of hospital contact, possibly because of delayed diagnosis, which could explain the higher mortality rates, said Ms. Janbek.
“It is also plausible that infections play a role in worsening dementia and subsequently lead to increased mortality,” she noted.
“Clinicians and health care personnel need to pay closer attention to infections of all types in people with dementia, and steps toward better clinical management and improved posthospital care need to be explored and undertaken. We need to identify possible preventive measures and targeted interventions in people with dementia and infections,” Ms. Janbek said.
‘Interesting observation’
Commenting on the study, Rebecca M. Edelmayer, PhD, director of scientific engagement for the Alzheimer’s Association, said it presents “an interesting observation.” However, “we can’t make any direct assumptions from this research per se about infections and dementia and whether they are causative in any way,” noted Dr. Edelmayer, who was not involved with the study.
Instead, the study highlighted the importance of “taking care of our overall health and making sure that individuals that might be vulnerable to infection, like those who are already living with dementia, are getting the best care possible,” she said.
Ms. Janbek and Dr. Edelmayer have reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM AAIC 2020
ACS disagrees with CDC on HPV vaccination in adults
The ACS has endorsed two recommendations made by the CDC’s Advisory Committee on Immunization Practices, but the ACS does not agree with a third recommendation for older adults.
The ACIP recommends shared clinical decision-making regarding human papillomavirus (HPV) vaccination in some adults aged 27-45 years who are not adequately vaccinated. The ACS does not endorse this recommendation “because of the low effectiveness and low cancer prevention potential of vaccination in this age group, the burden of decision-making on patients and clinicians, and the lack of sufficient guidance on the selection of individuals who might benefit,” wrote Debbie Saslow, PhD, of the ACS’s section on human papillomavirus and gynecologic cancers, and colleagues.
Dr. Saslow and colleagues detailed the ACS recommendations in CA: A Cancer Journal for Clinicians.
The HPV vaccine protects against the virus that can cause cervical, oropharyngeal, anal, vaginal, vulvar, and penile cancers. For younger people, the ACIP recommends routine HPV vaccination of boys and girls aged 9-12 years and catch-up vaccination in everyone up to age 26 who has not been fully immunized against HPV.
The ACS endorses both of these recommendations. It also advises clinicians to tell patients aged 22-26 years who haven’t received the HPV vaccine or completed the series that the vaccine is less effective at reducing the risk of cancer at older ages.
After the Food and Drug Administration approved the HPV vaccine for adults aged 27-45 years, the ACIP updated its recommendations to state that routine catch-up vaccination is not recommended for anyone aged over 26 years. However, the ACIP recommended that these older adults talk with their providers about the risks and benefits of the vaccine to determine whether to get it.
The ACS subsequently conducted a methodological review of the ACIP’s recommendations and published its own adapted guidance, stating that the ACS does not endorse the shared decision-making. Administering the HPV vaccine to adults aged over 26 years would only prevent an estimated 0.5% of additional cancer cases, 0.4% additional cases of cervical precancer, and 0.3% additional cases of genital warts over the next 100 years, compared with vaccination under age 26.
“In addition to the low effectiveness and low cancer prevention potential of vaccination in this age group, other considerations included the burden of decision-making on patients and clinicians and the lack of sufficient guidance on the selection of individuals who might benefit,” according to the guidance. The ACS also expressed concern that these provider-patient discussions could interfere with the public health goal of increasing HPV vaccination in younger people.
HPV vaccination rates have lagged substantially behind other routinely recommended childhood vaccinations. Just over half (51%) of U.S. teens aged 13-17 years were up to date with HPV vaccination, and 68% had received one dose of the vaccine in 2018, according to the National Immunization Survey.
It’s very uncommon for a professional medical organization to not endorse recommendations from the CDC, particularly with vaccines, according to Robert A. Bednarczyk, PhD, an assistant professor of public health at Emory University, Atlanta, who specializes in HPV vaccination research but was not involved with the ACS statement or the ACIP recommendations.
“Often, for vaccination recommendations, there is a harmonization between health care provider organizations, such as the American Academy of Pediatrics, American Academy of Family Physicians, etc., when new vaccination schedules are released,” Dr. Bednarczyk said.
He acknowledged the ACS’s reasons for not endorsing the ACIP’s HPV recommendations in older adults: the burden of shared decision-making given the communication issues, the vaccine’s lower effectiveness in this population, and the ongoing HPV vaccine shortage.
But Dr. Bednarczyk also pointed out that the ACIP’s recommendation opens the door to these discussions when they may actually be needed, such as in adults at greater risk for HPV. He cited data suggesting that, in 2015, divorces occurred in 24 out of 1,000 married people aged 25-39 years and 21 out of 1,000 people aged 40-49.
“When you consider these marriages that end, in addition to marriages that end when one spouse dies, there is a potential for individuals who previously had a low risk of HPV acquisition now entering into new potential sexual relationships,” Dr. Bednarczyk said. “Additionally, it has been estimated that approximately 4% of the U.S. population are in open or consensually nonmonogamous relationships, where exposure to more sexual partners may increase their risk for HPV. These are just some examples of where conversations with health care providers, and shared clinical decision-making, can help with a targeted reduction of HPV risk.”
The ACIP recommendation regarding adults aged 27-45 years also provides people in this age group with insurance coverage for the HPV vaccine if they choose to get it, Dr. Bednarczyk pointed out. Insurance companies may not be required to cover HPV vaccination in people aged over 26 years without the CDC’s recommendation, even if it’s not for routine immunization.
Dr. Bednarczyk agreed, however, with how the ACS adapted the CDC’s recommendation for routine vaccination in youth. The CDC’s routine recommendation is at ages 11-12 but can begin at 9 years, according to the ACIP. The ACS guidance qualifies this statement to place more emphasis on encouraging the vaccine earlier.
“Routine HPV vaccination between ages 9-12 is expected to achieve higher on-time vaccination rates, resulting in increased numbers of cancers prevented,” according to the ACS. “Health care providers are encouraged to start offering the HPV vaccine at age 9 or 10.”
Dr. Bednarczyk pointed to some of his past research finding low proportions of teens fully vaccinated against HPV by age 13 years (J Infect Dis. 2019 Jul 31;220[5]:730-4). Therefore, “any efforts to encourage vaccination, including starting the series at ages 9-10 years may help,” he said.
He also agreed that there may be diminished effectiveness with vaccinating adults aged 22-26, “but this should also be considered relative to an individual’s risk of acquiring HPV.”
While an HPV vaccine shortage is a major concern and HPV vaccination efforts should remain most focused on young teens, adults should not necessarily be neglected, Dr. Bednarczyk noted.
“Given how common HPV infection is in the population, open discussion between patients and health care providers can help identify those adults for whom HPV vaccination can be effective,” he said.
The development of the ACS guideline was supported by ACS operational funds. The ACS has received an independent educational grant from Merck Sharp & Dohme for a project intended to increase HPV vaccination rates. Dr. Saslow is the principal investigator for a cooperative agreement between the ACS and the CDC to support the National HPV Vaccination Roundtable and is coprincipal investigator of a cooperative agreement between the ACS and CDC to support initiatives to increase HPV vaccination. The remaining authors and Dr. Bednarczyk reported no relevant disclosures.
SOURCE: Saslow D et al. CA Cancer J Clin. 2020 Jul 8. doi: 10.3322/caac.21616.
The ACS has endorsed two recommendations made by the CDC’s Advisory Committee on Immunization Practices, but the ACS does not agree with a third recommendation for older adults.
The ACIP recommends shared clinical decision-making regarding human papillomavirus (HPV) vaccination in some adults aged 27-45 years who are not adequately vaccinated. The ACS does not endorse this recommendation “because of the low effectiveness and low cancer prevention potential of vaccination in this age group, the burden of decision-making on patients and clinicians, and the lack of sufficient guidance on the selection of individuals who might benefit,” wrote Debbie Saslow, PhD, of the ACS’s section on human papillomavirus and gynecologic cancers, and colleagues.
Dr. Saslow and colleagues detailed the ACS recommendations in CA: A Cancer Journal for Clinicians.
The HPV vaccine protects against the virus that can cause cervical, oropharyngeal, anal, vaginal, vulvar, and penile cancers. For younger people, the ACIP recommends routine HPV vaccination of boys and girls aged 9-12 years and catch-up vaccination in everyone up to age 26 who has not been fully immunized against HPV.
The ACS endorses both of these recommendations. It also advises clinicians to tell patients aged 22-26 years who haven’t received the HPV vaccine or completed the series that the vaccine is less effective at reducing the risk of cancer at older ages.
After the Food and Drug Administration approved the HPV vaccine for adults aged 27-45 years, the ACIP updated its recommendations to state that routine catch-up vaccination is not recommended for anyone aged over 26 years. However, the ACIP recommended that these older adults talk with their providers about the risks and benefits of the vaccine to determine whether to get it.
The ACS subsequently conducted a methodological review of the ACIP’s recommendations and published its own adapted guidance, stating that the ACS does not endorse the shared decision-making. Administering the HPV vaccine to adults aged over 26 years would only prevent an estimated 0.5% of additional cancer cases, 0.4% additional cases of cervical precancer, and 0.3% additional cases of genital warts over the next 100 years, compared with vaccination under age 26.
“In addition to the low effectiveness and low cancer prevention potential of vaccination in this age group, other considerations included the burden of decision-making on patients and clinicians and the lack of sufficient guidance on the selection of individuals who might benefit,” according to the guidance. The ACS also expressed concern that these provider-patient discussions could interfere with the public health goal of increasing HPV vaccination in younger people.
HPV vaccination rates have lagged substantially behind other routinely recommended childhood vaccinations. Just over half (51%) of U.S. teens aged 13-17 years were up to date with HPV vaccination, and 68% had received one dose of the vaccine in 2018, according to the National Immunization Survey.
It’s very uncommon for a professional medical organization to not endorse recommendations from the CDC, particularly with vaccines, according to Robert A. Bednarczyk, PhD, an assistant professor of public health at Emory University, Atlanta, who specializes in HPV vaccination research but was not involved with the ACS statement or the ACIP recommendations.
“Often, for vaccination recommendations, there is a harmonization between health care provider organizations, such as the American Academy of Pediatrics, American Academy of Family Physicians, etc., when new vaccination schedules are released,” Dr. Bednarczyk said.
He acknowledged the ACS’s reasons for not endorsing the ACIP’s HPV recommendations in older adults: the burden of shared decision-making given the communication issues, the vaccine’s lower effectiveness in this population, and the ongoing HPV vaccine shortage.
But Dr. Bednarczyk also pointed out that the ACIP’s recommendation opens the door to these discussions when they may actually be needed, such as in adults at greater risk for HPV. He cited data suggesting that, in 2015, divorces occurred in 24 out of 1,000 married people aged 25-39 years and 21 out of 1,000 people aged 40-49.
“When you consider these marriages that end, in addition to marriages that end when one spouse dies, there is a potential for individuals who previously had a low risk of HPV acquisition now entering into new potential sexual relationships,” Dr. Bednarczyk said. “Additionally, it has been estimated that approximately 4% of the U.S. population are in open or consensually nonmonogamous relationships, where exposure to more sexual partners may increase their risk for HPV. These are just some examples of where conversations with health care providers, and shared clinical decision-making, can help with a targeted reduction of HPV risk.”
The ACIP recommendation regarding adults aged 27-45 years also provides people in this age group with insurance coverage for the HPV vaccine if they choose to get it, Dr. Bednarczyk pointed out. Insurance companies may not be required to cover HPV vaccination in people aged over 26 years without the CDC’s recommendation, even if it’s not for routine immunization.
Dr. Bednarczyk agreed, however, with how the ACS adapted the CDC’s recommendation for routine vaccination in youth. The CDC’s routine recommendation is at ages 11-12 but can begin at 9 years, according to the ACIP. The ACS guidance qualifies this statement to place more emphasis on encouraging the vaccine earlier.
“Routine HPV vaccination between ages 9-12 is expected to achieve higher on-time vaccination rates, resulting in increased numbers of cancers prevented,” according to the ACS. “Health care providers are encouraged to start offering the HPV vaccine at age 9 or 10.”
Dr. Bednarczyk pointed to some of his past research finding low proportions of teens fully vaccinated against HPV by age 13 years (J Infect Dis. 2019 Jul 31;220[5]:730-4). Therefore, “any efforts to encourage vaccination, including starting the series at ages 9-10 years may help,” he said.
He also agreed that there may be diminished effectiveness with vaccinating adults aged 22-26, “but this should also be considered relative to an individual’s risk of acquiring HPV.”
While an HPV vaccine shortage is a major concern and HPV vaccination efforts should remain most focused on young teens, adults should not necessarily be neglected, Dr. Bednarczyk noted.
“Given how common HPV infection is in the population, open discussion between patients and health care providers can help identify those adults for whom HPV vaccination can be effective,” he said.
The development of the ACS guideline was supported by ACS operational funds. The ACS has received an independent educational grant from Merck Sharp & Dohme for a project intended to increase HPV vaccination rates. Dr. Saslow is the principal investigator for a cooperative agreement between the ACS and the CDC to support the National HPV Vaccination Roundtable and is coprincipal investigator of a cooperative agreement between the ACS and CDC to support initiatives to increase HPV vaccination. The remaining authors and Dr. Bednarczyk reported no relevant disclosures.
SOURCE: Saslow D et al. CA Cancer J Clin. 2020 Jul 8. doi: 10.3322/caac.21616.
The ACS has endorsed two recommendations made by the CDC’s Advisory Committee on Immunization Practices, but the ACS does not agree with a third recommendation for older adults.
The ACIP recommends shared clinical decision-making regarding human papillomavirus (HPV) vaccination in some adults aged 27-45 years who are not adequately vaccinated. The ACS does not endorse this recommendation “because of the low effectiveness and low cancer prevention potential of vaccination in this age group, the burden of decision-making on patients and clinicians, and the lack of sufficient guidance on the selection of individuals who might benefit,” wrote Debbie Saslow, PhD, of the ACS’s section on human papillomavirus and gynecologic cancers, and colleagues.
Dr. Saslow and colleagues detailed the ACS recommendations in CA: A Cancer Journal for Clinicians.
The HPV vaccine protects against the virus that can cause cervical, oropharyngeal, anal, vaginal, vulvar, and penile cancers. For younger people, the ACIP recommends routine HPV vaccination of boys and girls aged 9-12 years and catch-up vaccination in everyone up to age 26 who has not been fully immunized against HPV.
The ACS endorses both of these recommendations. It also advises clinicians to tell patients aged 22-26 years who haven’t received the HPV vaccine or completed the series that the vaccine is less effective at reducing the risk of cancer at older ages.
After the Food and Drug Administration approved the HPV vaccine for adults aged 27-45 years, the ACIP updated its recommendations to state that routine catch-up vaccination is not recommended for anyone aged over 26 years. However, the ACIP recommended that these older adults talk with their providers about the risks and benefits of the vaccine to determine whether to get it.
The ACS subsequently conducted a methodological review of the ACIP’s recommendations and published its own adapted guidance, stating that the ACS does not endorse the shared decision-making. Administering the HPV vaccine to adults aged over 26 years would only prevent an estimated 0.5% of additional cancer cases, 0.4% additional cases of cervical precancer, and 0.3% additional cases of genital warts over the next 100 years, compared with vaccination under age 26.
“In addition to the low effectiveness and low cancer prevention potential of vaccination in this age group, other considerations included the burden of decision-making on patients and clinicians and the lack of sufficient guidance on the selection of individuals who might benefit,” according to the guidance. The ACS also expressed concern that these provider-patient discussions could interfere with the public health goal of increasing HPV vaccination in younger people.
HPV vaccination rates have lagged substantially behind other routinely recommended childhood vaccinations. Just over half (51%) of U.S. teens aged 13-17 years were up to date with HPV vaccination, and 68% had received one dose of the vaccine in 2018, according to the National Immunization Survey.
It’s very uncommon for a professional medical organization to not endorse recommendations from the CDC, particularly with vaccines, according to Robert A. Bednarczyk, PhD, an assistant professor of public health at Emory University, Atlanta, who specializes in HPV vaccination research but was not involved with the ACS statement or the ACIP recommendations.
“Often, for vaccination recommendations, there is a harmonization between health care provider organizations, such as the American Academy of Pediatrics, American Academy of Family Physicians, etc., when new vaccination schedules are released,” Dr. Bednarczyk said.
He acknowledged the ACS’s reasons for not endorsing the ACIP’s HPV recommendations in older adults: the burden of shared decision-making given the communication issues, the vaccine’s lower effectiveness in this population, and the ongoing HPV vaccine shortage.
But Dr. Bednarczyk also pointed out that the ACIP’s recommendation opens the door to these discussions when they may actually be needed, such as in adults at greater risk for HPV. He cited data suggesting that, in 2015, divorces occurred in 24 out of 1,000 married people aged 25-39 years and 21 out of 1,000 people aged 40-49.
“When you consider these marriages that end, in addition to marriages that end when one spouse dies, there is a potential for individuals who previously had a low risk of HPV acquisition now entering into new potential sexual relationships,” Dr. Bednarczyk said. “Additionally, it has been estimated that approximately 4% of the U.S. population are in open or consensually nonmonogamous relationships, where exposure to more sexual partners may increase their risk for HPV. These are just some examples of where conversations with health care providers, and shared clinical decision-making, can help with a targeted reduction of HPV risk.”
The ACIP recommendation regarding adults aged 27-45 years also provides people in this age group with insurance coverage for the HPV vaccine if they choose to get it, Dr. Bednarczyk pointed out. Insurance companies may not be required to cover HPV vaccination in people aged over 26 years without the CDC’s recommendation, even if it’s not for routine immunization.
Dr. Bednarczyk agreed, however, with how the ACS adapted the CDC’s recommendation for routine vaccination in youth. The CDC’s routine recommendation is at ages 11-12 but can begin at 9 years, according to the ACIP. The ACS guidance qualifies this statement to place more emphasis on encouraging the vaccine earlier.
“Routine HPV vaccination between ages 9-12 is expected to achieve higher on-time vaccination rates, resulting in increased numbers of cancers prevented,” according to the ACS. “Health care providers are encouraged to start offering the HPV vaccine at age 9 or 10.”
Dr. Bednarczyk pointed to some of his past research finding low proportions of teens fully vaccinated against HPV by age 13 years (J Infect Dis. 2019 Jul 31;220[5]:730-4). Therefore, “any efforts to encourage vaccination, including starting the series at ages 9-10 years may help,” he said.
He also agreed that there may be diminished effectiveness with vaccinating adults aged 22-26, “but this should also be considered relative to an individual’s risk of acquiring HPV.”
While an HPV vaccine shortage is a major concern and HPV vaccination efforts should remain most focused on young teens, adults should not necessarily be neglected, Dr. Bednarczyk noted.
“Given how common HPV infection is in the population, open discussion between patients and health care providers can help identify those adults for whom HPV vaccination can be effective,” he said.
The development of the ACS guideline was supported by ACS operational funds. The ACS has received an independent educational grant from Merck Sharp & Dohme for a project intended to increase HPV vaccination rates. Dr. Saslow is the principal investigator for a cooperative agreement between the ACS and the CDC to support the National HPV Vaccination Roundtable and is coprincipal investigator of a cooperative agreement between the ACS and CDC to support initiatives to increase HPV vaccination. The remaining authors and Dr. Bednarczyk reported no relevant disclosures.
SOURCE: Saslow D et al. CA Cancer J Clin. 2020 Jul 8. doi: 10.3322/caac.21616.
FROM CA: A CANCER JOURNAL FOR CLINICIANS