User login
Novel approach brings hospice-bound MM patient into remission
In a case that researchers hope might pave the way for similar responses, a hospice-bound relapsed/refractory multiple myeloma (RRMM) patient who relapsed after chimeric antigen receptor (CAR) T-cell therapy was brought back into remission with the help of next-generation genomic sequencing, targeted molecular analysis and a novel combination of MAP kinase (MAPK)–inhibiting drugs.
“We have shown that comprehensive molecular profiling of advanced myeloma patients may provide critical information to guide treatment beyond standard of care,” senior author Alessandro Lagana, PhD, of the Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, said in an interview.

“This represents proof of concept that, while not curative, targeted molecules may serve as potential bridging therapies to clinical trial enrollment,” the authors further report in the case study, published recently in the Journal of Hematology & Oncology.
The use of B-cell maturation antigen (BCMA) CAR T-cell therapy approaches has transformed the treatment of multiple myeloma and leukemias, resulting in high response rates. However, most patients ultimately relapse, and no clear treatment options beyond CAR T therapy are established.
Such was the case for a 61-year old patient described in the study, who had relapsed 6 months after undergoing anti-BCMA CAR T-cell therapy and progressed after being salvaged for a short period with autologous stem cell transplantation. The patient had developed skin extramedullary disease, manifested as subcutaneous nodules.
“The subcutaneous skin lesions in lower extremities made him [ineligible] for another clinical trial and left him with no options,” Dr. Lagana said.
Using next-generation whole-exome sequencing, Dr. Lagana and colleagues had observed that a previously identified BRAF V600E–dominant subclone had persisted, despite the CAR T-cell treatment, in the patient’s bone marrow and cutaneous plasmacytoma.
The finding was not uncommon. More than half of RRMM patients (about 53%) show emerging clones with mutations within the MAPK signaling pathway, and in about 7% of patients, those include BRAF V600E, which can be targeted, the authors noted.
Further assessment of the patient’s CD138-positive MM cells using western blot signaling pathway analysis looking at DNA and RNA markers did indeed show an increase in MAPK signaling as a consequence of the mutation. This suggested a potential benefit of triple MAPK inhibition, compared with standard strategies.
Based on that information and on insights the researchers had gained from previous research, they implemented the novel, orally administered triple-combination treatment strategy, consisting of monomeric inhibition of BRAF dabrafenib (100 mg, twice daily), as well as dimeric inhibition with the multi–kinase inhibitor regorafenib (40 mg, once daily) and a MEK inhibitor (trametinib, 1.5 mg, for 21/28 days daily).
Of note, previous efforts using only monomeric inhibition of BRAF have not shown much success, but early data has shown some potential, with the inclusion of dimeric inhibition.
“Monomeric inhibition of BRAF has been attempted in patients with V600E, but the efficacy has been limited, likely due to feedback activation of the MAPK pathway via induction of BRAF dimer formation,” Dr. Lagana explained.
Meanwhile, “previous in vitro data from our colleagues at Mount Sinai has shown that inhibition of both monomeric and dimeric forms of BRAF in combination with MEK inhibition can overcome the negative feedback and lead to more efficacious and tolerable treatment,” he said.
With the treatment, the patient achieved a very good partial response for 110 days, with prompt reduction of the subcutaneous skin lesions and an 80% reduction in lambda free light chain (27.5 mg/L).
The triple-drug combination was well tolerated with minimal side effects, primarily involving grade 1 fatigue, and the patient was able to carry out activities of daily living and return to work.
“The triple inhibition allowed us to use less of each drug, which resulted in a well-tolerated regimen without any significant side effects,” Dr. Lagana said.
While the patient relapsed about 3 months later, there was, importantly, no recurrence of the subcutaneous nodules.
“We believe that the triple MAPK inhibition completely eradicated the disease clones driving the extramedullary disease,” Dr. Lagana said.
The therapy meanwhile enabled the patient to bridge to a new clinical trial, where he went into complete remission, and still was as of Sept. 29.
“To our knowledge, this was the first reported successful case of this treatment in an RRMM patient,” Dr. Lagana explained.
Case suggests ‘hope’ for relapsing patients
Importantly, currently many patients in the same position may wind up going to hospice, until such targeted medicine gains momentum, coauthor Samir Parekh, MD, a professor of hematology-oncology at the Hess Center for Science and Medicine, Icahn School of Medicine at Mount Sinai, said in an interview.

“As precision medicine is in its infancy in myeloma, these patients are not routinely sequenced for drug options that may be identified by next-generation sequencing,” said Dr. Parekh.
But for clinicians, the message of this case should be that “there is hope for patients relapsing after CAR T,” he added.
“Precision medicine approaches may be applicable even for this relapsed patient population,” he added. “MAP kinase mutations are common and drugs targeting them may be useful in myeloma.”
Noting that “the infrastructure to test and guide application of these therapies needs to be developed for myeloma, Dr. Parekh predicted that, “in the future, more effective MAPK inhibitors and other mutation or RNA-seq guided therapies will be applicable and hopefully provide more durable remissions.”
Approach may help address unmet need
Until then, however, treatment for patients who relapse after CAR-T and BCMA-targeted therapies has emerged as a significant unmet need. Therefore, this case highlights an important potential strategy, said Hans Lee, MD, an associate professor in the department of lymphoma/myeloma, division of cancer medicine, University of Texas MD Anderson Cancer Center, Houston, commenting on the study.

“This case report provides impetus for oncologists to strongly consider performing next-generation sequencing on myeloma tumor samples to look for potential actionable mutations, such as those in the MAPK pathway – which are common in myeloma,” he said. “With limited treatment options in the post–CAR T and post-BCMA setting, identifying such actional mutations may at least provide a bridge to other effective therapies available through clinical trials such as this patient’s case.”
Dr. Lee noted that key caveats include the fact that most physicians currently don’t have access to the type of next-generation sequencing and drug sensitivity testing used in the study.
Nevertheless, considering the limited options in the post–CAR T and post-BCMA setting, “the successful use of triple MAPK pathway inhibition through monomeric and dimeric inhibition of BRAF and MEK inhibition warrants further study in multiple myeloma in a clinical trial,” he said.
Dr. Lagana and associates are doing just that.
“We are about to launch the clinical trial, where we will match advanced RRMM patients with potential targeted treatments using different DNA and RNA markers,” Dr. Lagana said.
Dr. Lagana and Dr. Parekh had no disclosures to report. Three study coauthors reported receiving research grants or consulting fees from numerous pharmaceutical companies.
In a case that researchers hope might pave the way for similar responses, a hospice-bound relapsed/refractory multiple myeloma (RRMM) patient who relapsed after chimeric antigen receptor (CAR) T-cell therapy was brought back into remission with the help of next-generation genomic sequencing, targeted molecular analysis and a novel combination of MAP kinase (MAPK)–inhibiting drugs.
“We have shown that comprehensive molecular profiling of advanced myeloma patients may provide critical information to guide treatment beyond standard of care,” senior author Alessandro Lagana, PhD, of the Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, said in an interview.
“This represents proof of concept that, while not curative, targeted molecules may serve as potential bridging therapies to clinical trial enrollment,” the authors further report in the case study, published recently in the Journal of Hematology & Oncology.
The use of B-cell maturation antigen (BCMA) CAR T-cell therapy approaches has transformed the treatment of multiple myeloma and leukemias, resulting in high response rates. However, most patients ultimately relapse, and no clear treatment options beyond CAR T therapy are established.
Such was the case for a 61-year old patient described in the study, who had relapsed 6 months after undergoing anti-BCMA CAR T-cell therapy and progressed after being salvaged for a short period with autologous stem cell transplantation. The patient had developed skin extramedullary disease, manifested as subcutaneous nodules.
“The subcutaneous skin lesions in lower extremities made him [ineligible] for another clinical trial and left him with no options,” Dr. Lagana said.
Using next-generation whole-exome sequencing, Dr. Lagana and colleagues had observed that a previously identified BRAF V600E–dominant subclone had persisted, despite the CAR T-cell treatment, in the patient’s bone marrow and cutaneous plasmacytoma.
The finding was not uncommon. More than half of RRMM patients (about 53%) show emerging clones with mutations within the MAPK signaling pathway, and in about 7% of patients, those include BRAF V600E, which can be targeted, the authors noted.
Further assessment of the patient’s CD138-positive MM cells using western blot signaling pathway analysis looking at DNA and RNA markers did indeed show an increase in MAPK signaling as a consequence of the mutation. This suggested a potential benefit of triple MAPK inhibition, compared with standard strategies.
Based on that information and on insights the researchers had gained from previous research, they implemented the novel, orally administered triple-combination treatment strategy, consisting of monomeric inhibition of BRAF dabrafenib (100 mg, twice daily), as well as dimeric inhibition with the multi–kinase inhibitor regorafenib (40 mg, once daily) and a MEK inhibitor (trametinib, 1.5 mg, for 21/28 days daily).
Of note, previous efforts using only monomeric inhibition of BRAF have not shown much success, but early data has shown some potential, with the inclusion of dimeric inhibition.
“Monomeric inhibition of BRAF has been attempted in patients with V600E, but the efficacy has been limited, likely due to feedback activation of the MAPK pathway via induction of BRAF dimer formation,” Dr. Lagana explained.
Meanwhile, “previous in vitro data from our colleagues at Mount Sinai has shown that inhibition of both monomeric and dimeric forms of BRAF in combination with MEK inhibition can overcome the negative feedback and lead to more efficacious and tolerable treatment,” he said.
With the treatment, the patient achieved a very good partial response for 110 days, with prompt reduction of the subcutaneous skin lesions and an 80% reduction in lambda free light chain (27.5 mg/L).
The triple-drug combination was well tolerated with minimal side effects, primarily involving grade 1 fatigue, and the patient was able to carry out activities of daily living and return to work.
“The triple inhibition allowed us to use less of each drug, which resulted in a well-tolerated regimen without any significant side effects,” Dr. Lagana said.
While the patient relapsed about 3 months later, there was, importantly, no recurrence of the subcutaneous nodules.
“We believe that the triple MAPK inhibition completely eradicated the disease clones driving the extramedullary disease,” Dr. Lagana said.
The therapy meanwhile enabled the patient to bridge to a new clinical trial, where he went into complete remission, and still was as of Sept. 29.
“To our knowledge, this was the first reported successful case of this treatment in an RRMM patient,” Dr. Lagana explained.
Case suggests ‘hope’ for relapsing patients
Importantly, currently many patients in the same position may wind up going to hospice, until such targeted medicine gains momentum, coauthor Samir Parekh, MD, a professor of hematology-oncology at the Hess Center for Science and Medicine, Icahn School of Medicine at Mount Sinai, said in an interview.
“As precision medicine is in its infancy in myeloma, these patients are not routinely sequenced for drug options that may be identified by next-generation sequencing,” said Dr. Parekh.
But for clinicians, the message of this case should be that “there is hope for patients relapsing after CAR T,” he added.
“Precision medicine approaches may be applicable even for this relapsed patient population,” he added. “MAP kinase mutations are common and drugs targeting them may be useful in myeloma.”
Noting that “the infrastructure to test and guide application of these therapies needs to be developed for myeloma, Dr. Parekh predicted that, “in the future, more effective MAPK inhibitors and other mutation or RNA-seq guided therapies will be applicable and hopefully provide more durable remissions.”
Approach may help address unmet need
Until then, however, treatment for patients who relapse after CAR-T and BCMA-targeted therapies has emerged as a significant unmet need. Therefore, this case highlights an important potential strategy, said Hans Lee, MD, an associate professor in the department of lymphoma/myeloma, division of cancer medicine, University of Texas MD Anderson Cancer Center, Houston, commenting on the study.
“This case report provides impetus for oncologists to strongly consider performing next-generation sequencing on myeloma tumor samples to look for potential actionable mutations, such as those in the MAPK pathway – which are common in myeloma,” he said. “With limited treatment options in the post–CAR T and post-BCMA setting, identifying such actional mutations may at least provide a bridge to other effective therapies available through clinical trials such as this patient’s case.”
Dr. Lee noted that key caveats include the fact that most physicians currently don’t have access to the type of next-generation sequencing and drug sensitivity testing used in the study.
Nevertheless, considering the limited options in the post–CAR T and post-BCMA setting, “the successful use of triple MAPK pathway inhibition through monomeric and dimeric inhibition of BRAF and MEK inhibition warrants further study in multiple myeloma in a clinical trial,” he said.
Dr. Lagana and associates are doing just that.
“We are about to launch the clinical trial, where we will match advanced RRMM patients with potential targeted treatments using different DNA and RNA markers,” Dr. Lagana said.
Dr. Lagana and Dr. Parekh had no disclosures to report. Three study coauthors reported receiving research grants or consulting fees from numerous pharmaceutical companies.
In a case that researchers hope might pave the way for similar responses, a hospice-bound relapsed/refractory multiple myeloma (RRMM) patient who relapsed after chimeric antigen receptor (CAR) T-cell therapy was brought back into remission with the help of next-generation genomic sequencing, targeted molecular analysis and a novel combination of MAP kinase (MAPK)–inhibiting drugs.
“We have shown that comprehensive molecular profiling of advanced myeloma patients may provide critical information to guide treatment beyond standard of care,” senior author Alessandro Lagana, PhD, of the Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, said in an interview.
“This represents proof of concept that, while not curative, targeted molecules may serve as potential bridging therapies to clinical trial enrollment,” the authors further report in the case study, published recently in the Journal of Hematology & Oncology.
The use of B-cell maturation antigen (BCMA) CAR T-cell therapy approaches has transformed the treatment of multiple myeloma and leukemias, resulting in high response rates. However, most patients ultimately relapse, and no clear treatment options beyond CAR T therapy are established.
Such was the case for a 61-year old patient described in the study, who had relapsed 6 months after undergoing anti-BCMA CAR T-cell therapy and progressed after being salvaged for a short period with autologous stem cell transplantation. The patient had developed skin extramedullary disease, manifested as subcutaneous nodules.
“The subcutaneous skin lesions in lower extremities made him [ineligible] for another clinical trial and left him with no options,” Dr. Lagana said.
Using next-generation whole-exome sequencing, Dr. Lagana and colleagues had observed that a previously identified BRAF V600E–dominant subclone had persisted, despite the CAR T-cell treatment, in the patient’s bone marrow and cutaneous plasmacytoma.
The finding was not uncommon. More than half of RRMM patients (about 53%) show emerging clones with mutations within the MAPK signaling pathway, and in about 7% of patients, those include BRAF V600E, which can be targeted, the authors noted.
Further assessment of the patient’s CD138-positive MM cells using western blot signaling pathway analysis looking at DNA and RNA markers did indeed show an increase in MAPK signaling as a consequence of the mutation. This suggested a potential benefit of triple MAPK inhibition, compared with standard strategies.
Based on that information and on insights the researchers had gained from previous research, they implemented the novel, orally administered triple-combination treatment strategy, consisting of monomeric inhibition of BRAF dabrafenib (100 mg, twice daily), as well as dimeric inhibition with the multi–kinase inhibitor regorafenib (40 mg, once daily) and a MEK inhibitor (trametinib, 1.5 mg, for 21/28 days daily).
Of note, previous efforts using only monomeric inhibition of BRAF have not shown much success, but early data has shown some potential, with the inclusion of dimeric inhibition.
“Monomeric inhibition of BRAF has been attempted in patients with V600E, but the efficacy has been limited, likely due to feedback activation of the MAPK pathway via induction of BRAF dimer formation,” Dr. Lagana explained.
Meanwhile, “previous in vitro data from our colleagues at Mount Sinai has shown that inhibition of both monomeric and dimeric forms of BRAF in combination with MEK inhibition can overcome the negative feedback and lead to more efficacious and tolerable treatment,” he said.
With the treatment, the patient achieved a very good partial response for 110 days, with prompt reduction of the subcutaneous skin lesions and an 80% reduction in lambda free light chain (27.5 mg/L).
The triple-drug combination was well tolerated with minimal side effects, primarily involving grade 1 fatigue, and the patient was able to carry out activities of daily living and return to work.
“The triple inhibition allowed us to use less of each drug, which resulted in a well-tolerated regimen without any significant side effects,” Dr. Lagana said.
While the patient relapsed about 3 months later, there was, importantly, no recurrence of the subcutaneous nodules.
“We believe that the triple MAPK inhibition completely eradicated the disease clones driving the extramedullary disease,” Dr. Lagana said.
The therapy meanwhile enabled the patient to bridge to a new clinical trial, where he went into complete remission, and still was as of Sept. 29.
“To our knowledge, this was the first reported successful case of this treatment in an RRMM patient,” Dr. Lagana explained.
Case suggests ‘hope’ for relapsing patients
Importantly, currently many patients in the same position may wind up going to hospice, until such targeted medicine gains momentum, coauthor Samir Parekh, MD, a professor of hematology-oncology at the Hess Center for Science and Medicine, Icahn School of Medicine at Mount Sinai, said in an interview.
“As precision medicine is in its infancy in myeloma, these patients are not routinely sequenced for drug options that may be identified by next-generation sequencing,” said Dr. Parekh.
But for clinicians, the message of this case should be that “there is hope for patients relapsing after CAR T,” he added.
“Precision medicine approaches may be applicable even for this relapsed patient population,” he added. “MAP kinase mutations are common and drugs targeting them may be useful in myeloma.”
Noting that “the infrastructure to test and guide application of these therapies needs to be developed for myeloma, Dr. Parekh predicted that, “in the future, more effective MAPK inhibitors and other mutation or RNA-seq guided therapies will be applicable and hopefully provide more durable remissions.”
Approach may help address unmet need
Until then, however, treatment for patients who relapse after CAR-T and BCMA-targeted therapies has emerged as a significant unmet need. Therefore, this case highlights an important potential strategy, said Hans Lee, MD, an associate professor in the department of lymphoma/myeloma, division of cancer medicine, University of Texas MD Anderson Cancer Center, Houston, commenting on the study.
“This case report provides impetus for oncologists to strongly consider performing next-generation sequencing on myeloma tumor samples to look for potential actionable mutations, such as those in the MAPK pathway – which are common in myeloma,” he said. “With limited treatment options in the post–CAR T and post-BCMA setting, identifying such actional mutations may at least provide a bridge to other effective therapies available through clinical trials such as this patient’s case.”
Dr. Lee noted that key caveats include the fact that most physicians currently don’t have access to the type of next-generation sequencing and drug sensitivity testing used in the study.
Nevertheless, considering the limited options in the post–CAR T and post-BCMA setting, “the successful use of triple MAPK pathway inhibition through monomeric and dimeric inhibition of BRAF and MEK inhibition warrants further study in multiple myeloma in a clinical trial,” he said.
Dr. Lagana and associates are doing just that.
“We are about to launch the clinical trial, where we will match advanced RRMM patients with potential targeted treatments using different DNA and RNA markers,” Dr. Lagana said.
Dr. Lagana and Dr. Parekh had no disclosures to report. Three study coauthors reported receiving research grants or consulting fees from numerous pharmaceutical companies.
FROM THE JOURNAL OF HEMATOLOGY & ONCOLOGY
Pivotal trials in blood cancers don’t mirror patient populations
, a new study concludes.
“Our analysis shows that, over the past 10 years, participation in pivotal clinical trials investigating therapies for leukemias and MM is unrepresentative of the U.S. population,” say the authors, led by Jorge E. Cortes, MD, of the Georgia Cancer Center at Augusta University, Ga. “Trials should represent the population with the disease,” they comment.
The study was published in the Journal of Clinical Oncology.
“This study confirms that the U.S. cancer population for select hematologic malignancies was inadequately racially and ethnically represented in studies leading to drug approval,” comment the authors of an accompanying editorial.
“The results from this study should lead to questions about the generalizability of drug safety and efficacy in populations we serve as medical hematologists and oncologists,” say Mikkael A. Sekeres, MD, along with Namrata S. Chandhok, MD, both of the division of hematology, Sylvester Comprehensive Cancer Center, University of Miami.
They pose the question, for instance, as physicians practicing in South Florida, where most of their patients are Hispanic, “can we apply the results of these pivotal studies – and drug labels – to them, without any sense of whether they metabolize the drug the same way as those included in the study or have the same biologic targets?”
Analysis of pivotal trials
For their study, Dr. Cortes and colleagues analyzed 61 pivotal trials for leukemia and MM leading to approval of the drugs from the U.S. Food and Drug Administration between 2011 and 2021.
They found that only two-thirds (67.2%) of these trials reported data pertaining to race, while about half (48.8%) reported on ethnicity.
The trials that did report data on race involved a total of 13,731 patients. The vast majority (81.6%) were White, and Black patients represented only 3.8%. Asian/Pacific Islanders made up 9.1%, and American Indians or Alaskan Natives made up just 0.12% of participants, with 1.5% categorized as other.
Among the trials reporting on ethnicity, 4.7% of patients were Hispanic, with 11.5% being Hispanic in acute lymphoblastic leukemia (ALL) trials and 7.6% Hispanic in chronic myeloid leukemia (CML) trials.
Slightly more than half (54.8%) of all trial participants were male, and patients’ average ages ranged from 41.7 to 67.3 years across all malignancies.
Of the minority groups, Asian/Pacific Islanders and Black people had the highest representation in trials involving CML, at 12.7% and 5.3%, respectively.
Their lowest representation was in chronic lymphocytic leukemia (CLL), at 3% and 1.1%, respectively.
Among the trials reporting ethnicity, Hispanic people were the highest representation, with percentages ranging from 3.8% of MM trials to 11.5% in ALL trials.
Inconsistent with patient populations
Next, the researchers compared the proportions of race/ethnic groups that were found among the participants of these pivotal trials with the proportions that would be expected in patient populations for each of these blood cancers (according to the U.S. Surveillance, Epidemiology, and End Results [SEER] database).
For example, White people made up 80.3% of participants in clinical trials of MM, whereas they represent 68.7% of patients with MM, a difference that was statistically significant (P < .0001).
The finding was similar for CML, with White people accounting for 90.5% of participants in clinical trials versus 82.5% of the patient population (P < .0001).
For AML, the difference was smaller, with respective percentages of 79.6 versus 77.3% (P = .0389).
For Black people, Asian/Pacific Islanders and Hispanic people, across all five cancer types that were analyzed, the proportion of participants in clinical trials was significantly lower than the proportion in the patient population.
The analysis also showed that females were overrepresented in clinical trials for two blood cancers. For MM, trial participation was 44.7%, while disease incidence was 41.7% (P < .0001), and for CML the proportions were 44.7% versus 39.5% (P = .0009). However, females were underrepresented in a third blood cancer: in AML, the proportions were 44.7% versus 60.5% (P < .0001).
Geographic location of trials often inaccessible
The study also highlighted an obstacle to minorities participating in clinical trials: geography.
For this analysis, the researchers looked at mortality rates for the various blood cancers.
For AML, they found mortality rates were high across the whole of the United States, but centers conducting AML clinical trials were primarily in the Northeast, with no centers in the Midwest.
Key regions with high rates of AML mortality, low access to trials, and high minority representation were notably clustered in areas including east of the Carolinas, South Georgia, Alabama, and Mississippi, the authors noted.
“In many instances, trials were absent in areas with high mortality,” they report. “This makes access to clinical trials difficult, if not impossible, to patients who do not have the financial means for travel.”
Further action needed
Racial and ethnic disparities in clinical trials have been widely reported in numerous previous studies, the authors note.
Various initiatives have been launched in recent years to tackle the problem, including the National Institutes of Health Revitalization Act, FDA race and ethnicity guidance, and the International Conference for Harmonization guidance.
For oncology, the American Society of Clinical Oncology has also taken steps with the release of the new Equity, Diversity, and Inclusion Action Plan in 2021 to improve representation of minorities in research.
Dr. Cortes and colleagues suggest another step that is needed is standardized reporting of demographics of clinical trial participants.
“More importantly, efforts to increase representation of minorities and disadvantaged populations in clinical trials should be prioritized,” they say.
Dr. Cortes reports a consulting role and receiving research funding from many pharmaceutical companies. No other coauthors have financial disclosures. Dr. Chandhok reports honoraria from Healio, Clinical Care Options, and a consulting role with Servier. Dr. Sekeres reports a consulting role with Celgene, Millennium, Pfizer, Novartis, Syros Pharmaceuticals, Kurome Therapeutics, and institutional research funding from Takeda, Pfizer, Bristol Myers Squibb, Actuate Therapeutics, Sellas Life Sciences, and Bio-Path Holdings.
A version of this article first appeared on Medscape.com.
, a new study concludes.
“Our analysis shows that, over the past 10 years, participation in pivotal clinical trials investigating therapies for leukemias and MM is unrepresentative of the U.S. population,” say the authors, led by Jorge E. Cortes, MD, of the Georgia Cancer Center at Augusta University, Ga. “Trials should represent the population with the disease,” they comment.
The study was published in the Journal of Clinical Oncology.
“This study confirms that the U.S. cancer population for select hematologic malignancies was inadequately racially and ethnically represented in studies leading to drug approval,” comment the authors of an accompanying editorial.
“The results from this study should lead to questions about the generalizability of drug safety and efficacy in populations we serve as medical hematologists and oncologists,” say Mikkael A. Sekeres, MD, along with Namrata S. Chandhok, MD, both of the division of hematology, Sylvester Comprehensive Cancer Center, University of Miami.
They pose the question, for instance, as physicians practicing in South Florida, where most of their patients are Hispanic, “can we apply the results of these pivotal studies – and drug labels – to them, without any sense of whether they metabolize the drug the same way as those included in the study or have the same biologic targets?”
Analysis of pivotal trials
For their study, Dr. Cortes and colleagues analyzed 61 pivotal trials for leukemia and MM leading to approval of the drugs from the U.S. Food and Drug Administration between 2011 and 2021.
They found that only two-thirds (67.2%) of these trials reported data pertaining to race, while about half (48.8%) reported on ethnicity.
The trials that did report data on race involved a total of 13,731 patients. The vast majority (81.6%) were White, and Black patients represented only 3.8%. Asian/Pacific Islanders made up 9.1%, and American Indians or Alaskan Natives made up just 0.12% of participants, with 1.5% categorized as other.
Among the trials reporting on ethnicity, 4.7% of patients were Hispanic, with 11.5% being Hispanic in acute lymphoblastic leukemia (ALL) trials and 7.6% Hispanic in chronic myeloid leukemia (CML) trials.
Slightly more than half (54.8%) of all trial participants were male, and patients’ average ages ranged from 41.7 to 67.3 years across all malignancies.
Of the minority groups, Asian/Pacific Islanders and Black people had the highest representation in trials involving CML, at 12.7% and 5.3%, respectively.
Their lowest representation was in chronic lymphocytic leukemia (CLL), at 3% and 1.1%, respectively.
Among the trials reporting ethnicity, Hispanic people were the highest representation, with percentages ranging from 3.8% of MM trials to 11.5% in ALL trials.
Inconsistent with patient populations
Next, the researchers compared the proportions of race/ethnic groups that were found among the participants of these pivotal trials with the proportions that would be expected in patient populations for each of these blood cancers (according to the U.S. Surveillance, Epidemiology, and End Results [SEER] database).
For example, White people made up 80.3% of participants in clinical trials of MM, whereas they represent 68.7% of patients with MM, a difference that was statistically significant (P < .0001).
The finding was similar for CML, with White people accounting for 90.5% of participants in clinical trials versus 82.5% of the patient population (P < .0001).
For AML, the difference was smaller, with respective percentages of 79.6 versus 77.3% (P = .0389).
For Black people, Asian/Pacific Islanders and Hispanic people, across all five cancer types that were analyzed, the proportion of participants in clinical trials was significantly lower than the proportion in the patient population.
The analysis also showed that females were overrepresented in clinical trials for two blood cancers. For MM, trial participation was 44.7%, while disease incidence was 41.7% (P < .0001), and for CML the proportions were 44.7% versus 39.5% (P = .0009). However, females were underrepresented in a third blood cancer: in AML, the proportions were 44.7% versus 60.5% (P < .0001).
Geographic location of trials often inaccessible
The study also highlighted an obstacle to minorities participating in clinical trials: geography.
For this analysis, the researchers looked at mortality rates for the various blood cancers.
For AML, they found mortality rates were high across the whole of the United States, but centers conducting AML clinical trials were primarily in the Northeast, with no centers in the Midwest.
Key regions with high rates of AML mortality, low access to trials, and high minority representation were notably clustered in areas including east of the Carolinas, South Georgia, Alabama, and Mississippi, the authors noted.
“In many instances, trials were absent in areas with high mortality,” they report. “This makes access to clinical trials difficult, if not impossible, to patients who do not have the financial means for travel.”
Further action needed
Racial and ethnic disparities in clinical trials have been widely reported in numerous previous studies, the authors note.
Various initiatives have been launched in recent years to tackle the problem, including the National Institutes of Health Revitalization Act, FDA race and ethnicity guidance, and the International Conference for Harmonization guidance.
For oncology, the American Society of Clinical Oncology has also taken steps with the release of the new Equity, Diversity, and Inclusion Action Plan in 2021 to improve representation of minorities in research.
Dr. Cortes and colleagues suggest another step that is needed is standardized reporting of demographics of clinical trial participants.
“More importantly, efforts to increase representation of minorities and disadvantaged populations in clinical trials should be prioritized,” they say.
Dr. Cortes reports a consulting role and receiving research funding from many pharmaceutical companies. No other coauthors have financial disclosures. Dr. Chandhok reports honoraria from Healio, Clinical Care Options, and a consulting role with Servier. Dr. Sekeres reports a consulting role with Celgene, Millennium, Pfizer, Novartis, Syros Pharmaceuticals, Kurome Therapeutics, and institutional research funding from Takeda, Pfizer, Bristol Myers Squibb, Actuate Therapeutics, Sellas Life Sciences, and Bio-Path Holdings.
A version of this article first appeared on Medscape.com.
, a new study concludes.
“Our analysis shows that, over the past 10 years, participation in pivotal clinical trials investigating therapies for leukemias and MM is unrepresentative of the U.S. population,” say the authors, led by Jorge E. Cortes, MD, of the Georgia Cancer Center at Augusta University, Ga. “Trials should represent the population with the disease,” they comment.
The study was published in the Journal of Clinical Oncology.
“This study confirms that the U.S. cancer population for select hematologic malignancies was inadequately racially and ethnically represented in studies leading to drug approval,” comment the authors of an accompanying editorial.
“The results from this study should lead to questions about the generalizability of drug safety and efficacy in populations we serve as medical hematologists and oncologists,” say Mikkael A. Sekeres, MD, along with Namrata S. Chandhok, MD, both of the division of hematology, Sylvester Comprehensive Cancer Center, University of Miami.
They pose the question, for instance, as physicians practicing in South Florida, where most of their patients are Hispanic, “can we apply the results of these pivotal studies – and drug labels – to them, without any sense of whether they metabolize the drug the same way as those included in the study or have the same biologic targets?”
Analysis of pivotal trials
For their study, Dr. Cortes and colleagues analyzed 61 pivotal trials for leukemia and MM leading to approval of the drugs from the U.S. Food and Drug Administration between 2011 and 2021.
They found that only two-thirds (67.2%) of these trials reported data pertaining to race, while about half (48.8%) reported on ethnicity.
The trials that did report data on race involved a total of 13,731 patients. The vast majority (81.6%) were White, and Black patients represented only 3.8%. Asian/Pacific Islanders made up 9.1%, and American Indians or Alaskan Natives made up just 0.12% of participants, with 1.5% categorized as other.
Among the trials reporting on ethnicity, 4.7% of patients were Hispanic, with 11.5% being Hispanic in acute lymphoblastic leukemia (ALL) trials and 7.6% Hispanic in chronic myeloid leukemia (CML) trials.
Slightly more than half (54.8%) of all trial participants were male, and patients’ average ages ranged from 41.7 to 67.3 years across all malignancies.
Of the minority groups, Asian/Pacific Islanders and Black people had the highest representation in trials involving CML, at 12.7% and 5.3%, respectively.
Their lowest representation was in chronic lymphocytic leukemia (CLL), at 3% and 1.1%, respectively.
Among the trials reporting ethnicity, Hispanic people were the highest representation, with percentages ranging from 3.8% of MM trials to 11.5% in ALL trials.
Inconsistent with patient populations
Next, the researchers compared the proportions of race/ethnic groups that were found among the participants of these pivotal trials with the proportions that would be expected in patient populations for each of these blood cancers (according to the U.S. Surveillance, Epidemiology, and End Results [SEER] database).
For example, White people made up 80.3% of participants in clinical trials of MM, whereas they represent 68.7% of patients with MM, a difference that was statistically significant (P < .0001).
The finding was similar for CML, with White people accounting for 90.5% of participants in clinical trials versus 82.5% of the patient population (P < .0001).
For AML, the difference was smaller, with respective percentages of 79.6 versus 77.3% (P = .0389).
For Black people, Asian/Pacific Islanders and Hispanic people, across all five cancer types that were analyzed, the proportion of participants in clinical trials was significantly lower than the proportion in the patient population.
The analysis also showed that females were overrepresented in clinical trials for two blood cancers. For MM, trial participation was 44.7%, while disease incidence was 41.7% (P < .0001), and for CML the proportions were 44.7% versus 39.5% (P = .0009). However, females were underrepresented in a third blood cancer: in AML, the proportions were 44.7% versus 60.5% (P < .0001).
Geographic location of trials often inaccessible
The study also highlighted an obstacle to minorities participating in clinical trials: geography.
For this analysis, the researchers looked at mortality rates for the various blood cancers.
For AML, they found mortality rates were high across the whole of the United States, but centers conducting AML clinical trials were primarily in the Northeast, with no centers in the Midwest.
Key regions with high rates of AML mortality, low access to trials, and high minority representation were notably clustered in areas including east of the Carolinas, South Georgia, Alabama, and Mississippi, the authors noted.
“In many instances, trials were absent in areas with high mortality,” they report. “This makes access to clinical trials difficult, if not impossible, to patients who do not have the financial means for travel.”
Further action needed
Racial and ethnic disparities in clinical trials have been widely reported in numerous previous studies, the authors note.
Various initiatives have been launched in recent years to tackle the problem, including the National Institutes of Health Revitalization Act, FDA race and ethnicity guidance, and the International Conference for Harmonization guidance.
For oncology, the American Society of Clinical Oncology has also taken steps with the release of the new Equity, Diversity, and Inclusion Action Plan in 2021 to improve representation of minorities in research.
Dr. Cortes and colleagues suggest another step that is needed is standardized reporting of demographics of clinical trial participants.
“More importantly, efforts to increase representation of minorities and disadvantaged populations in clinical trials should be prioritized,” they say.
Dr. Cortes reports a consulting role and receiving research funding from many pharmaceutical companies. No other coauthors have financial disclosures. Dr. Chandhok reports honoraria from Healio, Clinical Care Options, and a consulting role with Servier. Dr. Sekeres reports a consulting role with Celgene, Millennium, Pfizer, Novartis, Syros Pharmaceuticals, Kurome Therapeutics, and institutional research funding from Takeda, Pfizer, Bristol Myers Squibb, Actuate Therapeutics, Sellas Life Sciences, and Bio-Path Holdings.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Bias and other barriers to HSCT access
For example, at the June 5 plenary session of the American Society of Clinical Oncology, Paul Richardson, MD, presented results of the DETERMINATION trial. More than 40,000 attendees heard his message that, in patients with newly diagnosed multiple myeloma (MM), up-front high-dose melphalan with autologous hematopoietic stem cell transplant (HSCT) support is associated with a significantly longer median progression-free survival of 67 months, compared with 46 months for patients randomized to delayed transplantation. The 5-year overall survival is similar for both arms.

While I and many of my colleagues in the field of transplantation used this data to strongly encourage MM patients to undergo HSCT as consolidation of their initial remission, others – including many investigators on the DETERMINATION trial – reached a starkly different conclusion. They suggested that delaying transplant was a valid option, since no survival benefit was observed.
Bias, when defined as a prejudice in favor of or against a specific treatment on the part of physicians and patients, has not been carefully studied in the realm of cellular therapies. However, physician and patient perceptions or misperceptions about the value or toxicity of a specific therapy are probably major drivers of whether a patient is referred for and accepts a particular form of treatment. In my specialization, that would mean either a stem cell transplant or other forms of cell therapy.
As with other medical procedures, in my field there are significant disparities in the use of transplantation among patients of different racial, ethnic, and age groups. Rates of both auto- and allo-HSCT are significantly higher for Whites than for African Americans. Hispanic patients have the lowest rates of utilization of auto-HSCT. Patients over the age of 60 have an eightfold risk of nonreferral to an HSCT center. Obviously, these nonreferrals reduce access to HSCT for older patients, particularly if they are seen at nonacademic centers.
One must question whether these disparities are caused by the physicians not believing in the value of transplantation, or simply not understanding its value? Or do they just lack the time to refer patients to a transplant center?
Socioeconomic factors, insurance status, age, and psychosocial characteristics all impact access to HSCT, yet some older patients with fewer economic resources and less insurance coverage still undergo the procedure. Is that because their physicians spent time educating these patients about the potential value of this treatment? Is it because the physicians went the extra mile to get these patients access to HSCT?
Physician preference also plays a significant role in whether a patient receives an allo-HSCT for acute myeloid leukemia and myelodysplastic syndrome. In a large survey of hematologists and oncologists performed by Pidala and colleagues, half of those surveyed agreed with the statement: “I feel the risk (morbidity and mortality) after HSCT is very high.” Most indicated that they “feel outcomes of unrelated donor HCT are much worse than matched sibling HCT.”
More importantly, more than one-third of those surveyed agreed that, “because of the high risks of allogeneic HSCT, I refer only after failure of conventional chemotherapy.” They voiced this opinion despite the fact that mortality rates after HSCT have been reduced significantly. With modern techniques, outcomes of unrelated donors are as good as with sibling donor transplants, and national guidelines strongly recommend that patients get referred before they become refractory to chemotherapy.
What can we do about this problem? Obviously, physician and provider education is important, but primary care physicians and general oncologists are already bombarded daily with new information. Relatively rare conditions like those we treat simply may not get their attention.
Personally, I think one of the most effective ways to overcome bias among physicians would be to target patients through a direct advertising campaign and public service announcements. Only by getting the attention of patients can they be directed to current, accurate information.
This solution could reduce the impact of physician biases or misperceptions and provide patients with greater access to lifesaving cell therapies.
Dr. Giralt is deputy division head of the division of hematologic malignancies at Memorial Sloan Kettering Cancer Center in New York.
For example, at the June 5 plenary session of the American Society of Clinical Oncology, Paul Richardson, MD, presented results of the DETERMINATION trial. More than 40,000 attendees heard his message that, in patients with newly diagnosed multiple myeloma (MM), up-front high-dose melphalan with autologous hematopoietic stem cell transplant (HSCT) support is associated with a significantly longer median progression-free survival of 67 months, compared with 46 months for patients randomized to delayed transplantation. The 5-year overall survival is similar for both arms.
While I and many of my colleagues in the field of transplantation used this data to strongly encourage MM patients to undergo HSCT as consolidation of their initial remission, others – including many investigators on the DETERMINATION trial – reached a starkly different conclusion. They suggested that delaying transplant was a valid option, since no survival benefit was observed.
Bias, when defined as a prejudice in favor of or against a specific treatment on the part of physicians and patients, has not been carefully studied in the realm of cellular therapies. However, physician and patient perceptions or misperceptions about the value or toxicity of a specific therapy are probably major drivers of whether a patient is referred for and accepts a particular form of treatment. In my specialization, that would mean either a stem cell transplant or other forms of cell therapy.
As with other medical procedures, in my field there are significant disparities in the use of transplantation among patients of different racial, ethnic, and age groups. Rates of both auto- and allo-HSCT are significantly higher for Whites than for African Americans. Hispanic patients have the lowest rates of utilization of auto-HSCT. Patients over the age of 60 have an eightfold risk of nonreferral to an HSCT center. Obviously, these nonreferrals reduce access to HSCT for older patients, particularly if they are seen at nonacademic centers.
One must question whether these disparities are caused by the physicians not believing in the value of transplantation, or simply not understanding its value? Or do they just lack the time to refer patients to a transplant center?
Socioeconomic factors, insurance status, age, and psychosocial characteristics all impact access to HSCT, yet some older patients with fewer economic resources and less insurance coverage still undergo the procedure. Is that because their physicians spent time educating these patients about the potential value of this treatment? Is it because the physicians went the extra mile to get these patients access to HSCT?
Physician preference also plays a significant role in whether a patient receives an allo-HSCT for acute myeloid leukemia and myelodysplastic syndrome. In a large survey of hematologists and oncologists performed by Pidala and colleagues, half of those surveyed agreed with the statement: “I feel the risk (morbidity and mortality) after HSCT is very high.” Most indicated that they “feel outcomes of unrelated donor HCT are much worse than matched sibling HCT.”
More importantly, more than one-third of those surveyed agreed that, “because of the high risks of allogeneic HSCT, I refer only after failure of conventional chemotherapy.” They voiced this opinion despite the fact that mortality rates after HSCT have been reduced significantly. With modern techniques, outcomes of unrelated donors are as good as with sibling donor transplants, and national guidelines strongly recommend that patients get referred before they become refractory to chemotherapy.
What can we do about this problem? Obviously, physician and provider education is important, but primary care physicians and general oncologists are already bombarded daily with new information. Relatively rare conditions like those we treat simply may not get their attention.
Personally, I think one of the most effective ways to overcome bias among physicians would be to target patients through a direct advertising campaign and public service announcements. Only by getting the attention of patients can they be directed to current, accurate information.
This solution could reduce the impact of physician biases or misperceptions and provide patients with greater access to lifesaving cell therapies.
Dr. Giralt is deputy division head of the division of hematologic malignancies at Memorial Sloan Kettering Cancer Center in New York.
For example, at the June 5 plenary session of the American Society of Clinical Oncology, Paul Richardson, MD, presented results of the DETERMINATION trial. More than 40,000 attendees heard his message that, in patients with newly diagnosed multiple myeloma (MM), up-front high-dose melphalan with autologous hematopoietic stem cell transplant (HSCT) support is associated with a significantly longer median progression-free survival of 67 months, compared with 46 months for patients randomized to delayed transplantation. The 5-year overall survival is similar for both arms.
While I and many of my colleagues in the field of transplantation used this data to strongly encourage MM patients to undergo HSCT as consolidation of their initial remission, others – including many investigators on the DETERMINATION trial – reached a starkly different conclusion. They suggested that delaying transplant was a valid option, since no survival benefit was observed.
Bias, when defined as a prejudice in favor of or against a specific treatment on the part of physicians and patients, has not been carefully studied in the realm of cellular therapies. However, physician and patient perceptions or misperceptions about the value or toxicity of a specific therapy are probably major drivers of whether a patient is referred for and accepts a particular form of treatment. In my specialization, that would mean either a stem cell transplant or other forms of cell therapy.
As with other medical procedures, in my field there are significant disparities in the use of transplantation among patients of different racial, ethnic, and age groups. Rates of both auto- and allo-HSCT are significantly higher for Whites than for African Americans. Hispanic patients have the lowest rates of utilization of auto-HSCT. Patients over the age of 60 have an eightfold risk of nonreferral to an HSCT center. Obviously, these nonreferrals reduce access to HSCT for older patients, particularly if they are seen at nonacademic centers.
One must question whether these disparities are caused by the physicians not believing in the value of transplantation, or simply not understanding its value? Or do they just lack the time to refer patients to a transplant center?
Socioeconomic factors, insurance status, age, and psychosocial characteristics all impact access to HSCT, yet some older patients with fewer economic resources and less insurance coverage still undergo the procedure. Is that because their physicians spent time educating these patients about the potential value of this treatment? Is it because the physicians went the extra mile to get these patients access to HSCT?
Physician preference also plays a significant role in whether a patient receives an allo-HSCT for acute myeloid leukemia and myelodysplastic syndrome. In a large survey of hematologists and oncologists performed by Pidala and colleagues, half of those surveyed agreed with the statement: “I feel the risk (morbidity and mortality) after HSCT is very high.” Most indicated that they “feel outcomes of unrelated donor HCT are much worse than matched sibling HCT.”
More importantly, more than one-third of those surveyed agreed that, “because of the high risks of allogeneic HSCT, I refer only after failure of conventional chemotherapy.” They voiced this opinion despite the fact that mortality rates after HSCT have been reduced significantly. With modern techniques, outcomes of unrelated donors are as good as with sibling donor transplants, and national guidelines strongly recommend that patients get referred before they become refractory to chemotherapy.
What can we do about this problem? Obviously, physician and provider education is important, but primary care physicians and general oncologists are already bombarded daily with new information. Relatively rare conditions like those we treat simply may not get their attention.
Personally, I think one of the most effective ways to overcome bias among physicians would be to target patients through a direct advertising campaign and public service announcements. Only by getting the attention of patients can they be directed to current, accurate information.
This solution could reduce the impact of physician biases or misperceptions and provide patients with greater access to lifesaving cell therapies.
Dr. Giralt is deputy division head of the division of hematologic malignancies at Memorial Sloan Kettering Cancer Center in New York.
Agent Orange Exposure, Transformation From MGUS to Multiple Myeloma, and Outcomes in Veterans
Multiple myeloma (MM) accounts for 1% to 2% of all cancers and slightly more than 17% of hematologic malignancies in the United States.1 MM is characterized by the neoplastic proliferation of immunoglobulin (Ig)-producing plasma cells with ≥ 10% clonal plasma cells in the bone marrow or biopsy-proven bony or soft tissue plasmacytoma, plus presence of related organ or tissue impairment or presence of a biomarker associated with near-inevitable progression to end-organ damage.2
Background
Up to 97% of patients with MM will have a monoclonal (M) protein produced and secreted by the malignant plasma cells, which can be detected by protein electrophoresis of the serum and an aliquot of urine from a 24-hour collection combined with immunofixation of the serum and urine. The M protein in MM usually consists of IgG 50% of the time and light chains 16% of the time. Patients who lack detectable M protein are considered to have nonsecretory myeloma. MM presents with end-organ damage, which includes hypercalcemia, renal dysfunction, anemia, or lytic bone lesions. Patients with MM frequently present with renal insufficiency due to cast nephropathy or light chain deposition disease.3
MM is thought to evolve from monoclonal gammopathy of uncertain significance (MGUS), an asymptomatic premalignant stage of clonal plasma cell proliferation with a risk of progression to active myeloma at 1% per year.4,5 Epidemiologic data suggest that people who develop MM have a genetic predisposition, but risk factors may develop or be acquired, such as age, immunosuppression, and environmental exposures. To better assess what causes transformation from MGUS to MM, it is important to identify agents that may cause this second hit.6
In November 1961, President John F. Kennedy authorized the start of Operation Ranch Hand, the US Air Force’s herbicide program during the Vietnam War. Twenty million gallons of various chemicals were sprayed in Vietnam, eastern Laos, and parts of Cambodia to defoliate rural land, depriving guerillas of their support base. Agent Orange (AO) was one of these chemicals; it is a mixed herbicide with traces of dioxin, a compound that has been associated with major health problems among exposed individuals.7 Several studies have evaluated exposure to AO and its potential harmful repercussions. Studies have assessed the link between AO and MGUS as well as AO to various leukemias, such as chronic lymphocytic leukemia.8,9 Other studies have shown the relationship between AO exposure and worse outcomes in persons with MM.10 To date, only a single abstract from a US Department of Veterans Affairs (VA) medical center has investigated the relationships between AO exposure and MGUS, MM, and the rate of transformation. The VA study of patients seen from 2005 to 2015 in Detroit, Michigan, found that AO exposure led to an increase in cumulative incidence rate of MGUS/MM, suggesting possible changes in disease biology and genetics.11
In this study, we aimed to determine the incidence of transformation of MGUS to MM in patients with and without exposure to AO. We then analyzed survival as a function of AO exposure, transformation, and clinical and sociodemographic variables. We also explored the impact of psychosocial variables and hematopoietic stem cell transplantation (HSCT), a standard of treatment for MM.
Methods
This retrospective cohort study assembled electronic health record (EHR) data from the Veterans Health Administration Corporate Data Warehouse (CDW). The VA Central Texas Veterans Healthcare System Institutional Review Board granted a waiver of consent for this record review. Eligible patients were Vietnam-era veterans who were in the military during the time that AO was used (1961-1971). Veterans were included if they were being cared for and received a diagnosis for MGUS or MM between October 1, 2009, and September 30, 2015 (all prevalent cases fiscal years 2010-2015). Cases were excluded if there was illogical death data or if age, race, ethnicity, body mass index (BMI), or prior-year diagnostic data were missing.
Measures
Patients were followed through April 2020. Presence of MGUS was defined by the International Classification of Diseases, Ninth Revision (ICD-9) diagnosis code 273.1. MM was identified by ICD-9 diagnosis codes 203.00, 203.01, and 203.02. The study index date was the earliest date of diagnosis of MGUS or MM in fiscal years 2010-2015. It was suspected that some patients with MM may have had a history of MGUS prior to this period. Therefore, for patients with MM, historical diagnosis of MGUS was extracted going back through the earliest data in the CDW (October 1999). Patients diagnosed with both MGUS and MM were considered transformation patients.
Other measures included age at index date, sex, race, ethnicity, VA priority status (a value 1 to 8 summarizing why the veteran qualified for VA care, such as military service-connected disability or very low income), and AO exposure authenticated per VA enrollment files and disability records. Service years were separated into 1961 to 1968 and 1969 to 1971 to match a change in the formulation of AO associated with decreased carcinogenic effect. Comorbidity data from the year prior to first MGUS/MM diagnosis in the observation period were extracted. Lifestyle factors associated with development of MGUS/MM were determined using the following codes: obesity per BMI calculation or diagnosis (ICD-9, 278.0), tobacco use per diagnosis (ICD-9, 305.1, V15.82), and survival from MGUS/MM diagnosis index date to date of death from any cause. Comorbidity was assessed using ICD-9 diagnosis codes to calculate the Charlson Comorbidity Index (CCI), which includes cardiovascular diseases, diabetes mellitus, liver and kidney diseases, cancers, and metastatic solid tumors. Cancers were omitted from our adapted CCI to avoid collinearity in the multivariable models. The theoretical maximum CCI score in this study was 25.12,13 Additional conditions known to be associated with variation in outcomes among veterans using the VA were indicated, including major depressive disorder, posttraumatic stress disorder (PTSD), alcohol use disorder (AUD), substance use disorder (SUD), and common chronic disease (hypertension, lipid disorders).14
Treatment with autologous HSCT was defined by Current Procedural Terminology and ICD-9 Clinical Modification procedure codes for bone marrow and autologous HSCT occurring at any time in the CDW (eAppendix). Days elapsed from MM diagnosis to HSCT were calculated.
Statistical Analysis
Sample characteristics were represented by frequencies and percentages for categorical variables and means and SDs (or medians and ranges where appropriate) for continuous variables. A χ2 test (or Fisher exact test when cell counts were low) assessed associations in bivariate comparisons. A 2-sample t test (or Wilcoxon rank sum test as appropriate) assessed differences in continuous variables between 2 groups. Kaplan-Meier curves depicted the unadjusted relationship of AO exposure to survival. Cox proportional hazards survival models examined an unadjusted model containing only the AO exposure indicator as a predictor and adjusted models were used for demographic and clinical factors for MGUS and patients with MM separately.
Predictors were age in decades, sex, Hispanic ethnicity, race, nicotine dependence, obesity, overweight, AUD, SUD, major depressive disorder, PTSD, and the adapted CCI. When modeling patients with MM, MGUS was added to the model to identify the transformation group. The interaction of AO with transformation was also analyzed for patients with MM. Results were reported as hazard ratios (HR) with their 95% CI.
Results
We identified 18,215 veterans diagnosed with either MGUS or MM during fiscal years 2010-2015 with 16,366 meeting inclusion criteria. Patients were excluded for missing data on exposure (n = 334), age (n = 12), race (n = 1058), ethnicity (n = 164), diagnosis (n = 47), treatment (n = 56), and BMI (n = 178). All were Vietnam War era veterans; 14 also served in other eras.
The cohort was 98.5% male (Table 1). Twenty-nine percent were Black veterans, 65% were White veterans, and 4% of individuals reported Hispanic ethnicity. Patients had a mean (SD) age of 66.7 (5.9) years (range, 52-96). Most patients were married (58%) or divorced/separated (27%). All were VA priority 1 to 5 (no 6, 7, or 8); 50% were priority 1 with 50% to 100% service-connected disability. Another 29% were eligible for VA care by reason of low income, 17% had 10% to 40% service-connected disability, and 4% were otherwise disabled.
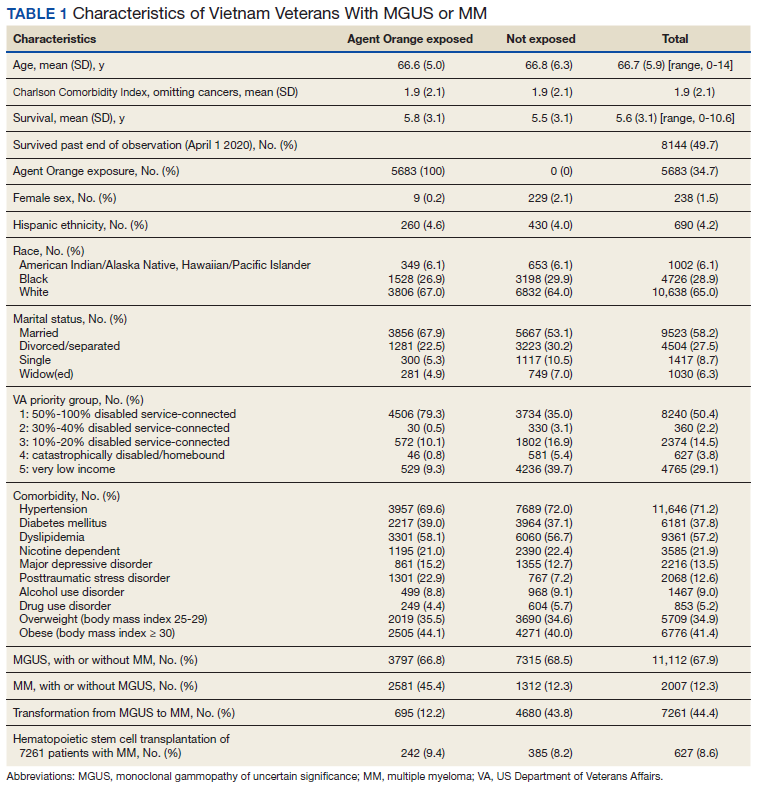
During fiscal years 2010 to 2015, 68% of our cohort had a diagnosis of MGUS (n = 11,112; 9105 had MGUS only), 44% had MM (n = 7261; 5254 had MM only), and 12% of these were transformation patients (n = 2007). AO exposure characterized 3102 MGUS-only patients (34%), 1886 MM-only patients (36%), and 695 transformation patients (35%) (χ2 = 4.92, P = .09). Among 5683 AO-exposed patients, 695 (12.2%) underwent MGUS-to-MM transformation. Among 10,683 nonexposed veterans, 1312 (12.3%) experienced transformation.
Comorbidity in the year leading up to the index MGUS/MM date determined using CCI was a mean (SD) of 1.9 (2.1) (range, 0-14). Among disorders not included in the CCI, 71% were diagnosed with hypertension, 57% with lipid disorders, 22% with nicotine dependence, 14% with major depressive disorder, 13% with PTSD, and 9% with AUD. Overweight (BMI 25 to < 30) and obesity (BMI ≥ 30) were common (35% and 41%, respectively). For 98% of patients, weight was measured within 90 days of their index MGUS/MM date. Most of the cohort (70%) were in Vietnam in 1961 to 1968.
HSCT was provided to 632 patients with MM (8.7%), including 441 patients who were treated after their index date and 219 patients treated before their index date. From fiscal years 2010 to 2015, the median (IQR) number of days from MM index date to HSCT receipt was 349 (243-650) days. Historical HSCT occurred a median (IQR) of 857 (353-1592) days before the index date, per data available back to October 1999; this median suggests long histories of MM in this cohort.
The unadjusted survival model found a very small inverse association of mortality with AO exposure in the total sample, meaning patients with documented AO exposure lived longer (HR, 0.85; 95% CI, 0.81-0.89; Table 2; Figure). Among 11,112 MGUS patients, AO was similarly associated with mortality (HR, 0.79; 95% CI, 0.74-0.84). The effect was also seen among 7269 patients with MM (HR, 0.86; 95% CI, 0.81-0.91).
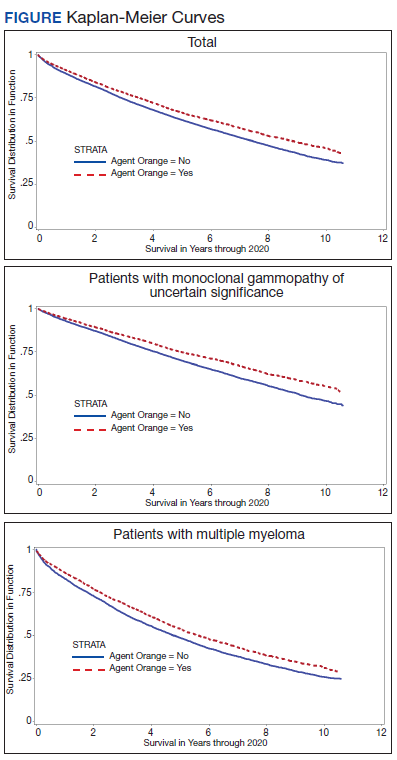
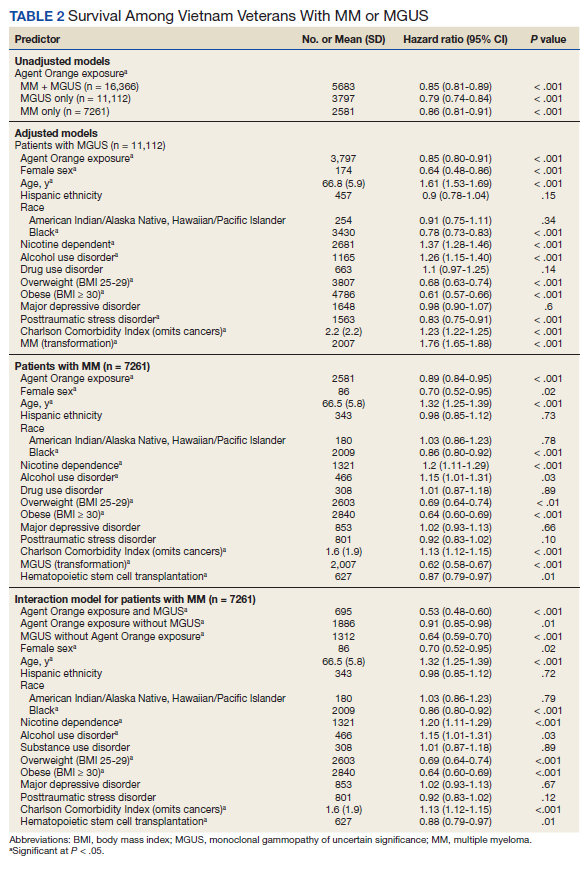
In the adjusted model of the total sample, the mortality hazard was greater for veterans who were older, with AUD and nicotine dependence, greater comorbidity per the CCI, diagnosis of MM, and transformation from MGUS to MM. Protective effects were noted for AO exposure, female sex, Black race, obesity, overweight, PTSD, and HSCT.
After adjusting for covariates, AO exposure was still associated with lower mortality among 11,112 patients with MGUS (HR, 0.85; 95% CI, 0.80-0.91). Risk factors were older age, nicotine dependence, AUD, the adapted CCI score (HR, 1.23 per point increase in the index; 95% CI, 1.22-1.25), and transformation to MM (HR, 1.76; 95% CI, 1.65-1.88). Additional protective factors were female sex, Black race, obesity, overweight, and PTSD.
After adjusting for covariates and limiting the analytic cohort to MM patients, the effect of AO exposure persisted (HR, 0.89; 95% CI, 0.84-0.95). Mortality risk factors were older age, nicotine dependence, AUD, and higher CCI score. Also protective were female sex, Black race, obesity, overweight, diagnosis of MGUS (transformation), and HSCT.
In the final model on patients with MM, the interaction term of AO exposure with transformation was significant. The combination of AO exposure with MGUS transformation had a greater protective effect than either AO exposure alone or MGUS without prior AO exposure. Additional protective factors were female sex, Black race, obesity, overweight, and HSCT. Older age, AUD, nicotine dependence, and greater comorbidity increased mortality risk.
Disscussion
Elucidating the pathophysiology and risk of transformation from MGUS to MM is an ongoing endeavor, even 35 years after the end of US involvement in the Vietnam War. Our study sought to understand a relationship between AO exposure, risk of MGUS transforming to MM, and associated mortality in US Vietnam War veterans. The rate of transformation (MGUS progressing to active MM) is well cited at 1% per year.15 Here, we found 12% of our cohort had undergone this transformation over 10 years.
Vietnam War era veterans who were exposed to AO during the Operation Ranch Hand period had 2.4 times greater risk of developing MGUS compared with veterans not exposed to AO.8 Our study was not designed to look at this association of AO exposure and MGUS/MM as this was a retrospective review to assess the difference in outcomes based on AO exposure. We found that AO exposure is associated with a decrease in mortality in contrast to a prior study showing worse survival with individuals with AO exposure.10 Another single center study found no association between AO exposure and overall survival, but it did identify an increased risk of progression from MGUS to MM.11 Our study did not show increased risk of transformation but did show positive effect on survival.
Black individuals have twice the risk of developing MM compared with White individuals and are diagnosed at a younger age (66 vs 70 years, respectively).16 Interestingly, Black race was a protective factor in our study. Given the length of time (35 years) elapsed since the Vietnam War ended, it is likely that most vulnerable Black veterans did not survive until our observation period.
HSCT, as expected, was a protective factor for veterans undergoing this treatment modality, but it is unclear why such a small number (8%) underwent HSCT as this is a standard of care in the management of MM. Obesity was also found to be a protective factor in a prior study, which was also seen in our study cohort.8
Limitations
This study was limited by its retrospective review of survivors among the Vietnam-era cohort several decades after the exposure of concern. Clinician notes and full historical data, such as date of onset for any disorder, were unavailable. These data also relied on the practitioners caring for the veterans to make the correct diagnosis with the associated code so that the data could be captured. Neither AO exposure nor diagnoses codes were verified against other sources of data; however, validation studies over the years have supported the accuracy of the diagnosis codes recorded in the VA EHR.
Conclusions
Because AO exposure is a nonmodifiable risk factor, focus should be placed on modifiable risk factors (eg, nicotine dependence, alcohol and substance use disorders, underlying comorbid conditions) as these were associated with worse outcomes. Future studies will look at the correlation of AO exposure, cytogenetics, and clinical outcomes in these veterans to learn how best to identify their disease course and optimize their care in the latter part of their life.
Acknowledgments
This research was supported by the Central Texas Veterans Health Care System and Baylor Scott and White Health, both in Temple and Veterans Affairs Central Western Massachusetts Healthcare System, Leeds.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30. doi:10.3322/caac.21442
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. doi:10.1016/S1470-2045(14)70442-5
3. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. doi:10.4065/78.1.21
4. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564- 569. doi:10.1056/NEJMoa01133202
5. International Myeloma Foundation. What Are MGUS, smoldering and active myeloma? Updated June 6, 2021. Accessed June 20, 2022. https://www.myeloma .org/what-are-mgus-smm-mm
6. Riedel DA, Pottern LM. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am. 1992;6(2):225-247. doi:10.1016/S0889-8588(18)30341-1
7. Buckingham Jr WA. Operation Ranch Hand: The Air Force and herbicides in southeast Asia, 1961-1971. Washington, DC: Office of Air Force History, United States Air Force; 1982. Accessed June 20, 2022. https://apps.dtic.mil/sti /pdfs/ADA121709.pdf
8. Landgren O, Shim YK, Michalek J, et al. Agent Orange exposure and monoclonal gammopathy of undetermined significance: an Operation Ranch Hand veteran cohort study. JAMA Oncol. 2015;1(8):1061-1068. doi:10.1001/jamaoncol.2015.2938
9. Mescher C, Gilbertson D, Randall NM, et al. The impact of Agent Orange exposure on prognosis and management in patients with chronic lymphocytic leukemia: a National Veteran Affairs Tumor Registry Study. Leuk Lymphoma. 2018;59(6):1348-1355. doi:10.1080/10428194.2017.1375109
10. Callander NS, Freytes CO, Luo S, Carson KR. Previous Agent Orange exposure is correlated with worse outcome in patients with multiple myeloma (MM) [abstract]. Blood. 2015;126(23):4194. doi:10.1182/blood.V126.23.4194.4194
11. Bumma N, Nagasaka M, Kim S, Vankayala HM, Ahmed S, Jasti P. Incidence of monoclonal gammopathy of undetermined significance (MGUS) and subsequent transformation to multiple myeloma (MM) and effect of exposure to Agent Orange (AO): a single center experience from VA Detroit [abstract]. Blood. 2017;130(suppl 1):5383. doi:10.1182/blood.V130.Suppl_1.5383.5383
12. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
13. Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45(6):613-619. doi:10.1016/0895-4356(92)90133-8
14. Copeland LA, Zeber JE, Sako EY, et al. Serious mental illnesses associated with receipt of surgery in retrospective analysis of patients in the Veterans Health Administration. BMC Surg. 2015;15:74. doi:10.1186/s12893-015-0064-7
15. Younes MA, Perez JD, Alirhayim Z, Ochoa C, Patel R, Dabak VS. MGUS Transformation into multiple myeloma in patients with solid organ transplantation [Abstract presented at American Society of Hematology Annual Meeting, November 15, 2013]. Blood. 2013;122(21):5325. doi:10.1182/blood.V122.21.5325.5325
16. Waxman AJ, Mink PJ, Devesa SS, et al. Racial disparities in incidence and outcome in multiple myeloma: a population- based study. Blood. 2010 Dec 16;116(25):5501-5506. doi:10.1182/blood-2010-07-298760
Multiple myeloma (MM) accounts for 1% to 2% of all cancers and slightly more than 17% of hematologic malignancies in the United States.1 MM is characterized by the neoplastic proliferation of immunoglobulin (Ig)-producing plasma cells with ≥ 10% clonal plasma cells in the bone marrow or biopsy-proven bony or soft tissue plasmacytoma, plus presence of related organ or tissue impairment or presence of a biomarker associated with near-inevitable progression to end-organ damage.2
Background
Up to 97% of patients with MM will have a monoclonal (M) protein produced and secreted by the malignant plasma cells, which can be detected by protein electrophoresis of the serum and an aliquot of urine from a 24-hour collection combined with immunofixation of the serum and urine. The M protein in MM usually consists of IgG 50% of the time and light chains 16% of the time. Patients who lack detectable M protein are considered to have nonsecretory myeloma. MM presents with end-organ damage, which includes hypercalcemia, renal dysfunction, anemia, or lytic bone lesions. Patients with MM frequently present with renal insufficiency due to cast nephropathy or light chain deposition disease.3
MM is thought to evolve from monoclonal gammopathy of uncertain significance (MGUS), an asymptomatic premalignant stage of clonal plasma cell proliferation with a risk of progression to active myeloma at 1% per year.4,5 Epidemiologic data suggest that people who develop MM have a genetic predisposition, but risk factors may develop or be acquired, such as age, immunosuppression, and environmental exposures. To better assess what causes transformation from MGUS to MM, it is important to identify agents that may cause this second hit.6
In November 1961, President John F. Kennedy authorized the start of Operation Ranch Hand, the US Air Force’s herbicide program during the Vietnam War. Twenty million gallons of various chemicals were sprayed in Vietnam, eastern Laos, and parts of Cambodia to defoliate rural land, depriving guerillas of their support base. Agent Orange (AO) was one of these chemicals; it is a mixed herbicide with traces of dioxin, a compound that has been associated with major health problems among exposed individuals.7 Several studies have evaluated exposure to AO and its potential harmful repercussions. Studies have assessed the link between AO and MGUS as well as AO to various leukemias, such as chronic lymphocytic leukemia.8,9 Other studies have shown the relationship between AO exposure and worse outcomes in persons with MM.10 To date, only a single abstract from a US Department of Veterans Affairs (VA) medical center has investigated the relationships between AO exposure and MGUS, MM, and the rate of transformation. The VA study of patients seen from 2005 to 2015 in Detroit, Michigan, found that AO exposure led to an increase in cumulative incidence rate of MGUS/MM, suggesting possible changes in disease biology and genetics.11
In this study, we aimed to determine the incidence of transformation of MGUS to MM in patients with and without exposure to AO. We then analyzed survival as a function of AO exposure, transformation, and clinical and sociodemographic variables. We also explored the impact of psychosocial variables and hematopoietic stem cell transplantation (HSCT), a standard of treatment for MM.
Methods
This retrospective cohort study assembled electronic health record (EHR) data from the Veterans Health Administration Corporate Data Warehouse (CDW). The VA Central Texas Veterans Healthcare System Institutional Review Board granted a waiver of consent for this record review. Eligible patients were Vietnam-era veterans who were in the military during the time that AO was used (1961-1971). Veterans were included if they were being cared for and received a diagnosis for MGUS or MM between October 1, 2009, and September 30, 2015 (all prevalent cases fiscal years 2010-2015). Cases were excluded if there was illogical death data or if age, race, ethnicity, body mass index (BMI), or prior-year diagnostic data were missing.
Measures
Patients were followed through April 2020. Presence of MGUS was defined by the International Classification of Diseases, Ninth Revision (ICD-9) diagnosis code 273.1. MM was identified by ICD-9 diagnosis codes 203.00, 203.01, and 203.02. The study index date was the earliest date of diagnosis of MGUS or MM in fiscal years 2010-2015. It was suspected that some patients with MM may have had a history of MGUS prior to this period. Therefore, for patients with MM, historical diagnosis of MGUS was extracted going back through the earliest data in the CDW (October 1999). Patients diagnosed with both MGUS and MM were considered transformation patients.
Other measures included age at index date, sex, race, ethnicity, VA priority status (a value 1 to 8 summarizing why the veteran qualified for VA care, such as military service-connected disability or very low income), and AO exposure authenticated per VA enrollment files and disability records. Service years were separated into 1961 to 1968 and 1969 to 1971 to match a change in the formulation of AO associated with decreased carcinogenic effect. Comorbidity data from the year prior to first MGUS/MM diagnosis in the observation period were extracted. Lifestyle factors associated with development of MGUS/MM were determined using the following codes: obesity per BMI calculation or diagnosis (ICD-9, 278.0), tobacco use per diagnosis (ICD-9, 305.1, V15.82), and survival from MGUS/MM diagnosis index date to date of death from any cause. Comorbidity was assessed using ICD-9 diagnosis codes to calculate the Charlson Comorbidity Index (CCI), which includes cardiovascular diseases, diabetes mellitus, liver and kidney diseases, cancers, and metastatic solid tumors. Cancers were omitted from our adapted CCI to avoid collinearity in the multivariable models. The theoretical maximum CCI score in this study was 25.12,13 Additional conditions known to be associated with variation in outcomes among veterans using the VA were indicated, including major depressive disorder, posttraumatic stress disorder (PTSD), alcohol use disorder (AUD), substance use disorder (SUD), and common chronic disease (hypertension, lipid disorders).14
Treatment with autologous HSCT was defined by Current Procedural Terminology and ICD-9 Clinical Modification procedure codes for bone marrow and autologous HSCT occurring at any time in the CDW (eAppendix). Days elapsed from MM diagnosis to HSCT were calculated.
Statistical Analysis
Sample characteristics were represented by frequencies and percentages for categorical variables and means and SDs (or medians and ranges where appropriate) for continuous variables. A χ2 test (or Fisher exact test when cell counts were low) assessed associations in bivariate comparisons. A 2-sample t test (or Wilcoxon rank sum test as appropriate) assessed differences in continuous variables between 2 groups. Kaplan-Meier curves depicted the unadjusted relationship of AO exposure to survival. Cox proportional hazards survival models examined an unadjusted model containing only the AO exposure indicator as a predictor and adjusted models were used for demographic and clinical factors for MGUS and patients with MM separately.
Predictors were age in decades, sex, Hispanic ethnicity, race, nicotine dependence, obesity, overweight, AUD, SUD, major depressive disorder, PTSD, and the adapted CCI. When modeling patients with MM, MGUS was added to the model to identify the transformation group. The interaction of AO with transformation was also analyzed for patients with MM. Results were reported as hazard ratios (HR) with their 95% CI.
Results
We identified 18,215 veterans diagnosed with either MGUS or MM during fiscal years 2010-2015 with 16,366 meeting inclusion criteria. Patients were excluded for missing data on exposure (n = 334), age (n = 12), race (n = 1058), ethnicity (n = 164), diagnosis (n = 47), treatment (n = 56), and BMI (n = 178). All were Vietnam War era veterans; 14 also served in other eras.
The cohort was 98.5% male (Table 1). Twenty-nine percent were Black veterans, 65% were White veterans, and 4% of individuals reported Hispanic ethnicity. Patients had a mean (SD) age of 66.7 (5.9) years (range, 52-96). Most patients were married (58%) or divorced/separated (27%). All were VA priority 1 to 5 (no 6, 7, or 8); 50% were priority 1 with 50% to 100% service-connected disability. Another 29% were eligible for VA care by reason of low income, 17% had 10% to 40% service-connected disability, and 4% were otherwise disabled.
During fiscal years 2010 to 2015, 68% of our cohort had a diagnosis of MGUS (n = 11,112; 9105 had MGUS only), 44% had MM (n = 7261; 5254 had MM only), and 12% of these were transformation patients (n = 2007). AO exposure characterized 3102 MGUS-only patients (34%), 1886 MM-only patients (36%), and 695 transformation patients (35%) (χ2 = 4.92, P = .09). Among 5683 AO-exposed patients, 695 (12.2%) underwent MGUS-to-MM transformation. Among 10,683 nonexposed veterans, 1312 (12.3%) experienced transformation.
Comorbidity in the year leading up to the index MGUS/MM date determined using CCI was a mean (SD) of 1.9 (2.1) (range, 0-14). Among disorders not included in the CCI, 71% were diagnosed with hypertension, 57% with lipid disorders, 22% with nicotine dependence, 14% with major depressive disorder, 13% with PTSD, and 9% with AUD. Overweight (BMI 25 to < 30) and obesity (BMI ≥ 30) were common (35% and 41%, respectively). For 98% of patients, weight was measured within 90 days of their index MGUS/MM date. Most of the cohort (70%) were in Vietnam in 1961 to 1968.
HSCT was provided to 632 patients with MM (8.7%), including 441 patients who were treated after their index date and 219 patients treated before their index date. From fiscal years 2010 to 2015, the median (IQR) number of days from MM index date to HSCT receipt was 349 (243-650) days. Historical HSCT occurred a median (IQR) of 857 (353-1592) days before the index date, per data available back to October 1999; this median suggests long histories of MM in this cohort.
The unadjusted survival model found a very small inverse association of mortality with AO exposure in the total sample, meaning patients with documented AO exposure lived longer (HR, 0.85; 95% CI, 0.81-0.89; Table 2; Figure). Among 11,112 MGUS patients, AO was similarly associated with mortality (HR, 0.79; 95% CI, 0.74-0.84). The effect was also seen among 7269 patients with MM (HR, 0.86; 95% CI, 0.81-0.91).
In the adjusted model of the total sample, the mortality hazard was greater for veterans who were older, with AUD and nicotine dependence, greater comorbidity per the CCI, diagnosis of MM, and transformation from MGUS to MM. Protective effects were noted for AO exposure, female sex, Black race, obesity, overweight, PTSD, and HSCT.
After adjusting for covariates, AO exposure was still associated with lower mortality among 11,112 patients with MGUS (HR, 0.85; 95% CI, 0.80-0.91). Risk factors were older age, nicotine dependence, AUD, the adapted CCI score (HR, 1.23 per point increase in the index; 95% CI, 1.22-1.25), and transformation to MM (HR, 1.76; 95% CI, 1.65-1.88). Additional protective factors were female sex, Black race, obesity, overweight, and PTSD.
After adjusting for covariates and limiting the analytic cohort to MM patients, the effect of AO exposure persisted (HR, 0.89; 95% CI, 0.84-0.95). Mortality risk factors were older age, nicotine dependence, AUD, and higher CCI score. Also protective were female sex, Black race, obesity, overweight, diagnosis of MGUS (transformation), and HSCT.
In the final model on patients with MM, the interaction term of AO exposure with transformation was significant. The combination of AO exposure with MGUS transformation had a greater protective effect than either AO exposure alone or MGUS without prior AO exposure. Additional protective factors were female sex, Black race, obesity, overweight, and HSCT. Older age, AUD, nicotine dependence, and greater comorbidity increased mortality risk.
Disscussion
Elucidating the pathophysiology and risk of transformation from MGUS to MM is an ongoing endeavor, even 35 years after the end of US involvement in the Vietnam War. Our study sought to understand a relationship between AO exposure, risk of MGUS transforming to MM, and associated mortality in US Vietnam War veterans. The rate of transformation (MGUS progressing to active MM) is well cited at 1% per year.15 Here, we found 12% of our cohort had undergone this transformation over 10 years.
Vietnam War era veterans who were exposed to AO during the Operation Ranch Hand period had 2.4 times greater risk of developing MGUS compared with veterans not exposed to AO.8 Our study was not designed to look at this association of AO exposure and MGUS/MM as this was a retrospective review to assess the difference in outcomes based on AO exposure. We found that AO exposure is associated with a decrease in mortality in contrast to a prior study showing worse survival with individuals with AO exposure.10 Another single center study found no association between AO exposure and overall survival, but it did identify an increased risk of progression from MGUS to MM.11 Our study did not show increased risk of transformation but did show positive effect on survival.
Black individuals have twice the risk of developing MM compared with White individuals and are diagnosed at a younger age (66 vs 70 years, respectively).16 Interestingly, Black race was a protective factor in our study. Given the length of time (35 years) elapsed since the Vietnam War ended, it is likely that most vulnerable Black veterans did not survive until our observation period.
HSCT, as expected, was a protective factor for veterans undergoing this treatment modality, but it is unclear why such a small number (8%) underwent HSCT as this is a standard of care in the management of MM. Obesity was also found to be a protective factor in a prior study, which was also seen in our study cohort.8
Limitations
This study was limited by its retrospective review of survivors among the Vietnam-era cohort several decades after the exposure of concern. Clinician notes and full historical data, such as date of onset for any disorder, were unavailable. These data also relied on the practitioners caring for the veterans to make the correct diagnosis with the associated code so that the data could be captured. Neither AO exposure nor diagnoses codes were verified against other sources of data; however, validation studies over the years have supported the accuracy of the diagnosis codes recorded in the VA EHR.
Conclusions
Because AO exposure is a nonmodifiable risk factor, focus should be placed on modifiable risk factors (eg, nicotine dependence, alcohol and substance use disorders, underlying comorbid conditions) as these were associated with worse outcomes. Future studies will look at the correlation of AO exposure, cytogenetics, and clinical outcomes in these veterans to learn how best to identify their disease course and optimize their care in the latter part of their life.
Acknowledgments
This research was supported by the Central Texas Veterans Health Care System and Baylor Scott and White Health, both in Temple and Veterans Affairs Central Western Massachusetts Healthcare System, Leeds.
Multiple myeloma (MM) accounts for 1% to 2% of all cancers and slightly more than 17% of hematologic malignancies in the United States.1 MM is characterized by the neoplastic proliferation of immunoglobulin (Ig)-producing plasma cells with ≥ 10% clonal plasma cells in the bone marrow or biopsy-proven bony or soft tissue plasmacytoma, plus presence of related organ or tissue impairment or presence of a biomarker associated with near-inevitable progression to end-organ damage.2
Background
Up to 97% of patients with MM will have a monoclonal (M) protein produced and secreted by the malignant plasma cells, which can be detected by protein electrophoresis of the serum and an aliquot of urine from a 24-hour collection combined with immunofixation of the serum and urine. The M protein in MM usually consists of IgG 50% of the time and light chains 16% of the time. Patients who lack detectable M protein are considered to have nonsecretory myeloma. MM presents with end-organ damage, which includes hypercalcemia, renal dysfunction, anemia, or lytic bone lesions. Patients with MM frequently present with renal insufficiency due to cast nephropathy or light chain deposition disease.3
MM is thought to evolve from monoclonal gammopathy of uncertain significance (MGUS), an asymptomatic premalignant stage of clonal plasma cell proliferation with a risk of progression to active myeloma at 1% per year.4,5 Epidemiologic data suggest that people who develop MM have a genetic predisposition, but risk factors may develop or be acquired, such as age, immunosuppression, and environmental exposures. To better assess what causes transformation from MGUS to MM, it is important to identify agents that may cause this second hit.6
In November 1961, President John F. Kennedy authorized the start of Operation Ranch Hand, the US Air Force’s herbicide program during the Vietnam War. Twenty million gallons of various chemicals were sprayed in Vietnam, eastern Laos, and parts of Cambodia to defoliate rural land, depriving guerillas of their support base. Agent Orange (AO) was one of these chemicals; it is a mixed herbicide with traces of dioxin, a compound that has been associated with major health problems among exposed individuals.7 Several studies have evaluated exposure to AO and its potential harmful repercussions. Studies have assessed the link between AO and MGUS as well as AO to various leukemias, such as chronic lymphocytic leukemia.8,9 Other studies have shown the relationship between AO exposure and worse outcomes in persons with MM.10 To date, only a single abstract from a US Department of Veterans Affairs (VA) medical center has investigated the relationships between AO exposure and MGUS, MM, and the rate of transformation. The VA study of patients seen from 2005 to 2015 in Detroit, Michigan, found that AO exposure led to an increase in cumulative incidence rate of MGUS/MM, suggesting possible changes in disease biology and genetics.11
In this study, we aimed to determine the incidence of transformation of MGUS to MM in patients with and without exposure to AO. We then analyzed survival as a function of AO exposure, transformation, and clinical and sociodemographic variables. We also explored the impact of psychosocial variables and hematopoietic stem cell transplantation (HSCT), a standard of treatment for MM.
Methods
This retrospective cohort study assembled electronic health record (EHR) data from the Veterans Health Administration Corporate Data Warehouse (CDW). The VA Central Texas Veterans Healthcare System Institutional Review Board granted a waiver of consent for this record review. Eligible patients were Vietnam-era veterans who were in the military during the time that AO was used (1961-1971). Veterans were included if they were being cared for and received a diagnosis for MGUS or MM between October 1, 2009, and September 30, 2015 (all prevalent cases fiscal years 2010-2015). Cases were excluded if there was illogical death data or if age, race, ethnicity, body mass index (BMI), or prior-year diagnostic data were missing.
Measures
Patients were followed through April 2020. Presence of MGUS was defined by the International Classification of Diseases, Ninth Revision (ICD-9) diagnosis code 273.1. MM was identified by ICD-9 diagnosis codes 203.00, 203.01, and 203.02. The study index date was the earliest date of diagnosis of MGUS or MM in fiscal years 2010-2015. It was suspected that some patients with MM may have had a history of MGUS prior to this period. Therefore, for patients with MM, historical diagnosis of MGUS was extracted going back through the earliest data in the CDW (October 1999). Patients diagnosed with both MGUS and MM were considered transformation patients.
Other measures included age at index date, sex, race, ethnicity, VA priority status (a value 1 to 8 summarizing why the veteran qualified for VA care, such as military service-connected disability or very low income), and AO exposure authenticated per VA enrollment files and disability records. Service years were separated into 1961 to 1968 and 1969 to 1971 to match a change in the formulation of AO associated with decreased carcinogenic effect. Comorbidity data from the year prior to first MGUS/MM diagnosis in the observation period were extracted. Lifestyle factors associated with development of MGUS/MM were determined using the following codes: obesity per BMI calculation or diagnosis (ICD-9, 278.0), tobacco use per diagnosis (ICD-9, 305.1, V15.82), and survival from MGUS/MM diagnosis index date to date of death from any cause. Comorbidity was assessed using ICD-9 diagnosis codes to calculate the Charlson Comorbidity Index (CCI), which includes cardiovascular diseases, diabetes mellitus, liver and kidney diseases, cancers, and metastatic solid tumors. Cancers were omitted from our adapted CCI to avoid collinearity in the multivariable models. The theoretical maximum CCI score in this study was 25.12,13 Additional conditions known to be associated with variation in outcomes among veterans using the VA were indicated, including major depressive disorder, posttraumatic stress disorder (PTSD), alcohol use disorder (AUD), substance use disorder (SUD), and common chronic disease (hypertension, lipid disorders).14
Treatment with autologous HSCT was defined by Current Procedural Terminology and ICD-9 Clinical Modification procedure codes for bone marrow and autologous HSCT occurring at any time in the CDW (eAppendix). Days elapsed from MM diagnosis to HSCT were calculated.
Statistical Analysis
Sample characteristics were represented by frequencies and percentages for categorical variables and means and SDs (or medians and ranges where appropriate) for continuous variables. A χ2 test (or Fisher exact test when cell counts were low) assessed associations in bivariate comparisons. A 2-sample t test (or Wilcoxon rank sum test as appropriate) assessed differences in continuous variables between 2 groups. Kaplan-Meier curves depicted the unadjusted relationship of AO exposure to survival. Cox proportional hazards survival models examined an unadjusted model containing only the AO exposure indicator as a predictor and adjusted models were used for demographic and clinical factors for MGUS and patients with MM separately.
Predictors were age in decades, sex, Hispanic ethnicity, race, nicotine dependence, obesity, overweight, AUD, SUD, major depressive disorder, PTSD, and the adapted CCI. When modeling patients with MM, MGUS was added to the model to identify the transformation group. The interaction of AO with transformation was also analyzed for patients with MM. Results were reported as hazard ratios (HR) with their 95% CI.
Results
We identified 18,215 veterans diagnosed with either MGUS or MM during fiscal years 2010-2015 with 16,366 meeting inclusion criteria. Patients were excluded for missing data on exposure (n = 334), age (n = 12), race (n = 1058), ethnicity (n = 164), diagnosis (n = 47), treatment (n = 56), and BMI (n = 178). All were Vietnam War era veterans; 14 also served in other eras.
The cohort was 98.5% male (Table 1). Twenty-nine percent were Black veterans, 65% were White veterans, and 4% of individuals reported Hispanic ethnicity. Patients had a mean (SD) age of 66.7 (5.9) years (range, 52-96). Most patients were married (58%) or divorced/separated (27%). All were VA priority 1 to 5 (no 6, 7, or 8); 50% were priority 1 with 50% to 100% service-connected disability. Another 29% were eligible for VA care by reason of low income, 17% had 10% to 40% service-connected disability, and 4% were otherwise disabled.
During fiscal years 2010 to 2015, 68% of our cohort had a diagnosis of MGUS (n = 11,112; 9105 had MGUS only), 44% had MM (n = 7261; 5254 had MM only), and 12% of these were transformation patients (n = 2007). AO exposure characterized 3102 MGUS-only patients (34%), 1886 MM-only patients (36%), and 695 transformation patients (35%) (χ2 = 4.92, P = .09). Among 5683 AO-exposed patients, 695 (12.2%) underwent MGUS-to-MM transformation. Among 10,683 nonexposed veterans, 1312 (12.3%) experienced transformation.
Comorbidity in the year leading up to the index MGUS/MM date determined using CCI was a mean (SD) of 1.9 (2.1) (range, 0-14). Among disorders not included in the CCI, 71% were diagnosed with hypertension, 57% with lipid disorders, 22% with nicotine dependence, 14% with major depressive disorder, 13% with PTSD, and 9% with AUD. Overweight (BMI 25 to < 30) and obesity (BMI ≥ 30) were common (35% and 41%, respectively). For 98% of patients, weight was measured within 90 days of their index MGUS/MM date. Most of the cohort (70%) were in Vietnam in 1961 to 1968.
HSCT was provided to 632 patients with MM (8.7%), including 441 patients who were treated after their index date and 219 patients treated before their index date. From fiscal years 2010 to 2015, the median (IQR) number of days from MM index date to HSCT receipt was 349 (243-650) days. Historical HSCT occurred a median (IQR) of 857 (353-1592) days before the index date, per data available back to October 1999; this median suggests long histories of MM in this cohort.
The unadjusted survival model found a very small inverse association of mortality with AO exposure in the total sample, meaning patients with documented AO exposure lived longer (HR, 0.85; 95% CI, 0.81-0.89; Table 2; Figure). Among 11,112 MGUS patients, AO was similarly associated with mortality (HR, 0.79; 95% CI, 0.74-0.84). The effect was also seen among 7269 patients with MM (HR, 0.86; 95% CI, 0.81-0.91).
In the adjusted model of the total sample, the mortality hazard was greater for veterans who were older, with AUD and nicotine dependence, greater comorbidity per the CCI, diagnosis of MM, and transformation from MGUS to MM. Protective effects were noted for AO exposure, female sex, Black race, obesity, overweight, PTSD, and HSCT.
After adjusting for covariates, AO exposure was still associated with lower mortality among 11,112 patients with MGUS (HR, 0.85; 95% CI, 0.80-0.91). Risk factors were older age, nicotine dependence, AUD, the adapted CCI score (HR, 1.23 per point increase in the index; 95% CI, 1.22-1.25), and transformation to MM (HR, 1.76; 95% CI, 1.65-1.88). Additional protective factors were female sex, Black race, obesity, overweight, and PTSD.
After adjusting for covariates and limiting the analytic cohort to MM patients, the effect of AO exposure persisted (HR, 0.89; 95% CI, 0.84-0.95). Mortality risk factors were older age, nicotine dependence, AUD, and higher CCI score. Also protective were female sex, Black race, obesity, overweight, diagnosis of MGUS (transformation), and HSCT.
In the final model on patients with MM, the interaction term of AO exposure with transformation was significant. The combination of AO exposure with MGUS transformation had a greater protective effect than either AO exposure alone or MGUS without prior AO exposure. Additional protective factors were female sex, Black race, obesity, overweight, and HSCT. Older age, AUD, nicotine dependence, and greater comorbidity increased mortality risk.
Disscussion
Elucidating the pathophysiology and risk of transformation from MGUS to MM is an ongoing endeavor, even 35 years after the end of US involvement in the Vietnam War. Our study sought to understand a relationship between AO exposure, risk of MGUS transforming to MM, and associated mortality in US Vietnam War veterans. The rate of transformation (MGUS progressing to active MM) is well cited at 1% per year.15 Here, we found 12% of our cohort had undergone this transformation over 10 years.
Vietnam War era veterans who were exposed to AO during the Operation Ranch Hand period had 2.4 times greater risk of developing MGUS compared with veterans not exposed to AO.8 Our study was not designed to look at this association of AO exposure and MGUS/MM as this was a retrospective review to assess the difference in outcomes based on AO exposure. We found that AO exposure is associated with a decrease in mortality in contrast to a prior study showing worse survival with individuals with AO exposure.10 Another single center study found no association between AO exposure and overall survival, but it did identify an increased risk of progression from MGUS to MM.11 Our study did not show increased risk of transformation but did show positive effect on survival.
Black individuals have twice the risk of developing MM compared with White individuals and are diagnosed at a younger age (66 vs 70 years, respectively).16 Interestingly, Black race was a protective factor in our study. Given the length of time (35 years) elapsed since the Vietnam War ended, it is likely that most vulnerable Black veterans did not survive until our observation period.
HSCT, as expected, was a protective factor for veterans undergoing this treatment modality, but it is unclear why such a small number (8%) underwent HSCT as this is a standard of care in the management of MM. Obesity was also found to be a protective factor in a prior study, which was also seen in our study cohort.8
Limitations
This study was limited by its retrospective review of survivors among the Vietnam-era cohort several decades after the exposure of concern. Clinician notes and full historical data, such as date of onset for any disorder, were unavailable. These data also relied on the practitioners caring for the veterans to make the correct diagnosis with the associated code so that the data could be captured. Neither AO exposure nor diagnoses codes were verified against other sources of data; however, validation studies over the years have supported the accuracy of the diagnosis codes recorded in the VA EHR.
Conclusions
Because AO exposure is a nonmodifiable risk factor, focus should be placed on modifiable risk factors (eg, nicotine dependence, alcohol and substance use disorders, underlying comorbid conditions) as these were associated with worse outcomes. Future studies will look at the correlation of AO exposure, cytogenetics, and clinical outcomes in these veterans to learn how best to identify their disease course and optimize their care in the latter part of their life.
Acknowledgments
This research was supported by the Central Texas Veterans Health Care System and Baylor Scott and White Health, both in Temple and Veterans Affairs Central Western Massachusetts Healthcare System, Leeds.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30. doi:10.3322/caac.21442
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. doi:10.1016/S1470-2045(14)70442-5
3. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. doi:10.4065/78.1.21
4. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564- 569. doi:10.1056/NEJMoa01133202
5. International Myeloma Foundation. What Are MGUS, smoldering and active myeloma? Updated June 6, 2021. Accessed June 20, 2022. https://www.myeloma .org/what-are-mgus-smm-mm
6. Riedel DA, Pottern LM. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am. 1992;6(2):225-247. doi:10.1016/S0889-8588(18)30341-1
7. Buckingham Jr WA. Operation Ranch Hand: The Air Force and herbicides in southeast Asia, 1961-1971. Washington, DC: Office of Air Force History, United States Air Force; 1982. Accessed June 20, 2022. https://apps.dtic.mil/sti /pdfs/ADA121709.pdf
8. Landgren O, Shim YK, Michalek J, et al. Agent Orange exposure and monoclonal gammopathy of undetermined significance: an Operation Ranch Hand veteran cohort study. JAMA Oncol. 2015;1(8):1061-1068. doi:10.1001/jamaoncol.2015.2938
9. Mescher C, Gilbertson D, Randall NM, et al. The impact of Agent Orange exposure on prognosis and management in patients with chronic lymphocytic leukemia: a National Veteran Affairs Tumor Registry Study. Leuk Lymphoma. 2018;59(6):1348-1355. doi:10.1080/10428194.2017.1375109
10. Callander NS, Freytes CO, Luo S, Carson KR. Previous Agent Orange exposure is correlated with worse outcome in patients with multiple myeloma (MM) [abstract]. Blood. 2015;126(23):4194. doi:10.1182/blood.V126.23.4194.4194
11. Bumma N, Nagasaka M, Kim S, Vankayala HM, Ahmed S, Jasti P. Incidence of monoclonal gammopathy of undetermined significance (MGUS) and subsequent transformation to multiple myeloma (MM) and effect of exposure to Agent Orange (AO): a single center experience from VA Detroit [abstract]. Blood. 2017;130(suppl 1):5383. doi:10.1182/blood.V130.Suppl_1.5383.5383
12. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
13. Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45(6):613-619. doi:10.1016/0895-4356(92)90133-8
14. Copeland LA, Zeber JE, Sako EY, et al. Serious mental illnesses associated with receipt of surgery in retrospective analysis of patients in the Veterans Health Administration. BMC Surg. 2015;15:74. doi:10.1186/s12893-015-0064-7
15. Younes MA, Perez JD, Alirhayim Z, Ochoa C, Patel R, Dabak VS. MGUS Transformation into multiple myeloma in patients with solid organ transplantation [Abstract presented at American Society of Hematology Annual Meeting, November 15, 2013]. Blood. 2013;122(21):5325. doi:10.1182/blood.V122.21.5325.5325
16. Waxman AJ, Mink PJ, Devesa SS, et al. Racial disparities in incidence and outcome in multiple myeloma: a population- based study. Blood. 2010 Dec 16;116(25):5501-5506. doi:10.1182/blood-2010-07-298760
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30. doi:10.3322/caac.21442
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. doi:10.1016/S1470-2045(14)70442-5
3. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. doi:10.4065/78.1.21
4. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564- 569. doi:10.1056/NEJMoa01133202
5. International Myeloma Foundation. What Are MGUS, smoldering and active myeloma? Updated June 6, 2021. Accessed June 20, 2022. https://www.myeloma .org/what-are-mgus-smm-mm
6. Riedel DA, Pottern LM. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am. 1992;6(2):225-247. doi:10.1016/S0889-8588(18)30341-1
7. Buckingham Jr WA. Operation Ranch Hand: The Air Force and herbicides in southeast Asia, 1961-1971. Washington, DC: Office of Air Force History, United States Air Force; 1982. Accessed June 20, 2022. https://apps.dtic.mil/sti /pdfs/ADA121709.pdf
8. Landgren O, Shim YK, Michalek J, et al. Agent Orange exposure and monoclonal gammopathy of undetermined significance: an Operation Ranch Hand veteran cohort study. JAMA Oncol. 2015;1(8):1061-1068. doi:10.1001/jamaoncol.2015.2938
9. Mescher C, Gilbertson D, Randall NM, et al. The impact of Agent Orange exposure on prognosis and management in patients with chronic lymphocytic leukemia: a National Veteran Affairs Tumor Registry Study. Leuk Lymphoma. 2018;59(6):1348-1355. doi:10.1080/10428194.2017.1375109
10. Callander NS, Freytes CO, Luo S, Carson KR. Previous Agent Orange exposure is correlated with worse outcome in patients with multiple myeloma (MM) [abstract]. Blood. 2015;126(23):4194. doi:10.1182/blood.V126.23.4194.4194
11. Bumma N, Nagasaka M, Kim S, Vankayala HM, Ahmed S, Jasti P. Incidence of monoclonal gammopathy of undetermined significance (MGUS) and subsequent transformation to multiple myeloma (MM) and effect of exposure to Agent Orange (AO): a single center experience from VA Detroit [abstract]. Blood. 2017;130(suppl 1):5383. doi:10.1182/blood.V130.Suppl_1.5383.5383
12. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
13. Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45(6):613-619. doi:10.1016/0895-4356(92)90133-8
14. Copeland LA, Zeber JE, Sako EY, et al. Serious mental illnesses associated with receipt of surgery in retrospective analysis of patients in the Veterans Health Administration. BMC Surg. 2015;15:74. doi:10.1186/s12893-015-0064-7
15. Younes MA, Perez JD, Alirhayim Z, Ochoa C, Patel R, Dabak VS. MGUS Transformation into multiple myeloma in patients with solid organ transplantation [Abstract presented at American Society of Hematology Annual Meeting, November 15, 2013]. Blood. 2013;122(21):5325. doi:10.1182/blood.V122.21.5325.5325
16. Waxman AJ, Mink PJ, Devesa SS, et al. Racial disparities in incidence and outcome in multiple myeloma: a population- based study. Blood. 2010 Dec 16;116(25):5501-5506. doi:10.1182/blood-2010-07-298760
Many die waiting for `last-chance’ therapy
Some patients with blood cancers for whom all other therapeutic options have been exhausted have one final chance of getting rid of their disease: treatment with chimeric antigen-receptor (CAR) T cells.
Described as a “living drug,” the treatment involves genetically engineering the patient’s own blood cells and reinfusing them back into their system. These CAR T cells then hunt down and destroy cancer cells; in some cases, they manage to eradicate the disease completely.
About half of patients with leukemia or lymphoma and about a third of those with multiple myeloma who receive this treatment have a complete remission and achieve a functional “cure.”
But not all patients who could benefit from this therapy are able to get it. Some are spending months on waiting lists, often deteriorating while they wait. These patients have exhausted all other therapeutic options, and many are facing hospice and death.
The scope of this problem was illustrated by a recent survey of the centers that are certified to deliver this complex therapy.
The survey was led by Yi Lin, MD, PhD, associate professor of medicine at the Mayo Clinic, Rochester, Minn., and medical director for the cellular therapy program. It was published as an abstract at the annual meeting of the American Society of Clinical Oncology recently, although it was not presented there.
“We wanted to find out just how widespread this problem is,” Dr. Lin said, adding: “There had been nothing in the literature thus far about it.”
The team contacted 20 centers across the United States and received responses from 17. Results showed that the median time on the waiting list was 6 months and that only 25% of patients eventually received CAR T-cell therapy. An additional 25% were able to enter a CAR T clinical trial. The remaining 50% of patients either were enrolled in a different type of trial, entered hospice, or died.
For patient selection, all centers reported using a committee of experienced physicians to ensure consistency. They employed different ethical principles for selection. Some centers sought to maximize the total benefit, such as selecting the patients most likely to achieve leukapheresis or a clinical response, while others based their decisions on the time patients spent on waiting list or gave priority to the patients who were the “worst off” with the most limited therapeutic options.
Shortage affecting mostly myeloma patients
The shortages in CAR T-cell therapies primarily involve the products used for patients with multiple myeloma.
The problem has not, as yet, noticeably spilled over to lymphoma and leukemia treatments, which use a slightly different type of CAR T-cell therapy (it targets CD19, whereas the cell therapies used for myeloma target BCMA).
“We have backlog of myeloma patients who don’t have access,” said Nina Shah, MD, a hematologist and professor of medicine at the University of California, San Francisco. “We have only four slots for the two myeloma products but about 50-60 eligible patients.”
Long waiting times for CAR T cells for myeloma have been an issue ever since the first of these products appeared on the market: idecabtagene vicleucel (ide-cel; Abecma), developed by Bluebird Bio and Bristol-Myers Squibb. “As soon as it became available in March 2021, we had people waiting and limits on our access to it,” Dr. Shah said.
A second CAR T-cell therapy for myeloma, ciltacabtagene autoleucel (cilta-cel, Carvykti), developed by Janssen and Legend Biotech, received approval in February 2022. While that helped provide centers with a few more slots, it wasn’t sufficient to cut waiting times, and the demand for these myeloma therapies continues to outstrip the capacity to produce CAR-T products in a timely manner.
“For myeloma, the demand is very high, as most patients are not cured from any other existing myeloma therapies, and most patients will make it to fifth-line therapy where the two CAR T-cell products are approved right now,” said Krina K. Patel, MD, medical director of the department of lymphoma/myeloma in the division of cancer medicine at the University of Texas MD Anderson Cancer Center, Houston.
“We likely have 10 eligible CAR-T myeloma patients each month at our center,” she said, “but were getting two slots per month for the past 8 months, and now are getting four slots a month.”
“Our clinic has also experienced the impact of the low number of manufacturing slots offered to each cancer center for some CAR T-cell products,” said David Maloney, MD, PhD, medical director, Cellular Immunotherapy and Bezos Family Immunotherapy Clinic, Seattle Cancer Care Alliance.
He noted that, as with other cancer centers, for multiple myeloma they are provided a specific number of manufacturing slots for each treatment. “Our providers discuss which patients are most appropriate for available slots for that month,” said Dr. Maloney.
“Additionally, juggling patient schedules may be required to address the extended manufacturing time for some products. In some cases, clinical trials may be available in a more timely fashion for appropriate patients, and in some cases, switching to an alternative product is possible,” he commented.
Complex causes behind bottleneck
The cause of the current bottleneck for myeloma patients is complex. It stems from a shortage of raw materials and supply chain restraints, among other things.
While the biggest impact of shortages has been on patients with multiple myeloma, Dr. Patel pointed out that these constraints are also affecting patients with lymphoma at her institution, but to a lesser degree.
“This is multifactorial as to why, but most of the issues arise from manufacturing,” Dr. Patel said in an interview. “Initially, the FDA limited how many slots each new product could have per month, then there was a viral vector shortage, and then the quality-control process the FDA requires takes longer than the manufacturing of the cells actually do.”
On top of that, “we have about a 5% manufacturing fail rate so far,” she added. Such failures occur when the cells taken from a patient cannot be converted into CAR T cells for therapy.
Matthew J. Frigault, MD, from the Center for Cellular Therapies, Mass General Cancer Center, Boston, explained that the growing excitement about the potential for cellular therapy and recent approvals for these products for use in earlier lines of treatment have increased demand for them.
There are also problems regarding supply. Manufacture and delivery of CAR T is complicated and takes time to scale up, Dr. Frigault pointed out. “Therefore, we are seeing limited access, more so for the BCMA-directed therapies [which are used for myeloma].”
The shortages and delays likely involve two main factors. “For the newer indications, there is a significant backlog of patients who have been waiting for these therapies and have not been able to access them in the clinical trial setting, and manufacturing is extremely complicated and not easily scaled up,” he said.
“That being said, manufacturers are trying to increase the number of available manufacturing slots and decrease the time needed to manufacture cells,” Dr. Frigault commented.
Delays in access to myeloma CAR T-cell therapy are also affecting patient care at Fox Chase Cancer Center in Philadelphia. “We have had about one slot every 2 months for Abecma,” noted Henry Fung, MD, chair of the department of bone marrow transplant and cellular therapies at Fox Chase. “For Carvykti, there are only 32 certified centers in [the] U.S., and access is very limited.”
Dr. Fung explained that they have had to offer alternative treatments to many of their patients. “There are rumors that there’s shortage in obtaining raw materials, such as the virus used for transduction, although we have not encountered any problems in other CAR T products used for lymphomas.”
Pharma companies trying to meet the demand
This news organization reached out to the manufacturers of CAR T products. All have reported that they are doing what they feasibly can to ramp up production.
“The complexity of delivering CAR T-cell therapies is unlike any other traditional biologic or small-molecule medicine, using a patient’s own cells to start a highly sophisticated and personalized manufacturing process,” commented a spokesperson for BMS, which has two CAR T-cell products currently on the market.
“In this nascent field of cell therapy, we continue to evolve every day, addressing supply and manufacturing challenges head on by applying key learnings across our three state-of-the-art cell therapy facilities and two new facilities in progress.
“We have been encouraged by a steady increase in our manufacturing capacity, and we continue efforts to ramp up further to meet the demand for our cell therapies,” the BMS spokesperson commented. “We have already seen improvements in the stabilization of vector supply and expect additional improvements in capacity in the second half of 2022.”
Novartis said much the same thing. They have a “comprehensive, integrated global CAR-T manufacturing footprint that strengthens the flexibility, resilience, and sustainability of the Novartis manufacturing and supply chain. Together with an improved manufacturing process, we are confident in our ability to meet patient demand with timely delivery,” according to a Novartis spokesperson.
The spokesperson also pointed out that the company has continuously incorporated process improvements that have significantly increased manufacturing capacity and success rates for patients in need of CAR T cells.
“Data presented at [the] American Society of Hematology annual meeting in 2021 showed the Novartis Morris Plains facility, our flagship CAR T manufacturing site, had commercial manufacturing and shipping success rates of 96% and 99%, respectively, between January and August 2021,” according to the spokesperson.
Legend and Janssen, the companies behind Carvykti, one of the two approved cell products for myeloma, which launched earlier in 2022, said that they have continued to activate certified treatment centers in a phased approach that will enable them to expand availability throughout 2022 and beyond.
“This phased approach was designed to ensure the highest level of predictability and reliability for the patient and the certified treatment centers,” the spokesperson said. “We understand the urgency for patients in need of Carvyki and are committed to doing everything we can to accelerate our ability to deliver this important cell therapy in a reliable and timely manner.”
With regard to the industry-wide supply shortage of lentivirus, Legend and Janssen say they have put in place multiple processes to address the shortage, “including enhancing our own internal manufacturing capabilities of this essential drug substance, to ensure sufficient and sustained supply.”
Incredibly exciting potential
Given the immense potential of CAR T-cell therapy, the supply shortage that myeloma patients are experiencing is all the more poignant and distressing. While not everyone benefits, some patients for whom every other therapy failed and who were facing hospice have had dramatic results.
“Incredibly exciting with unbelievable potential” was how one expert described these new therapies when the first product was about to enter the marketplace. Since then, six CAR T-cell therapies have received regulatory approval for an ever-increasing range of hematologic malignancies.
But these CAR T-cell therapies have their own set of adverse events, which can be serious and even life-threatening. In addition, not all patients become cancer free, although long-term data are impressive.
A study that included one of the longest follow-ups to date was reported at the 2020 annual meeting of the American Society of Clinical Oncology. The researchers reported that remissions lasted over 9 years for patients with relapsed/refractory B-cell lymphoma or chronic lymphocytic leukemia who underwent treatment with Kite’s axicaptagene cilleucel (Yescarta). This review included 43 patients and showed an overall remission rate of 76%. Complete remission was achieved for 54% of patients, and partial remission was achieved for 22%.
The results with CAR T-cell therapy in multiple myeloma are not quite as impressive, but even so, the clinical data that supported the approval of Abecma showed that a third of patients, who had previously received a median of six prior therapies, achieved a complete response.
At the time of the Abecma approval, the lead investigator of the study, Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, commented: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
A version of this article first appeared on Medscape.com.
Some patients with blood cancers for whom all other therapeutic options have been exhausted have one final chance of getting rid of their disease: treatment with chimeric antigen-receptor (CAR) T cells.
Described as a “living drug,” the treatment involves genetically engineering the patient’s own blood cells and reinfusing them back into their system. These CAR T cells then hunt down and destroy cancer cells; in some cases, they manage to eradicate the disease completely.
About half of patients with leukemia or lymphoma and about a third of those with multiple myeloma who receive this treatment have a complete remission and achieve a functional “cure.”
But not all patients who could benefit from this therapy are able to get it. Some are spending months on waiting lists, often deteriorating while they wait. These patients have exhausted all other therapeutic options, and many are facing hospice and death.
The scope of this problem was illustrated by a recent survey of the centers that are certified to deliver this complex therapy.
The survey was led by Yi Lin, MD, PhD, associate professor of medicine at the Mayo Clinic, Rochester, Minn., and medical director for the cellular therapy program. It was published as an abstract at the annual meeting of the American Society of Clinical Oncology recently, although it was not presented there.
“We wanted to find out just how widespread this problem is,” Dr. Lin said, adding: “There had been nothing in the literature thus far about it.”
The team contacted 20 centers across the United States and received responses from 17. Results showed that the median time on the waiting list was 6 months and that only 25% of patients eventually received CAR T-cell therapy. An additional 25% were able to enter a CAR T clinical trial. The remaining 50% of patients either were enrolled in a different type of trial, entered hospice, or died.
For patient selection, all centers reported using a committee of experienced physicians to ensure consistency. They employed different ethical principles for selection. Some centers sought to maximize the total benefit, such as selecting the patients most likely to achieve leukapheresis or a clinical response, while others based their decisions on the time patients spent on waiting list or gave priority to the patients who were the “worst off” with the most limited therapeutic options.
Shortage affecting mostly myeloma patients
The shortages in CAR T-cell therapies primarily involve the products used for patients with multiple myeloma.
The problem has not, as yet, noticeably spilled over to lymphoma and leukemia treatments, which use a slightly different type of CAR T-cell therapy (it targets CD19, whereas the cell therapies used for myeloma target BCMA).
“We have backlog of myeloma patients who don’t have access,” said Nina Shah, MD, a hematologist and professor of medicine at the University of California, San Francisco. “We have only four slots for the two myeloma products but about 50-60 eligible patients.”
Long waiting times for CAR T cells for myeloma have been an issue ever since the first of these products appeared on the market: idecabtagene vicleucel (ide-cel; Abecma), developed by Bluebird Bio and Bristol-Myers Squibb. “As soon as it became available in March 2021, we had people waiting and limits on our access to it,” Dr. Shah said.
A second CAR T-cell therapy for myeloma, ciltacabtagene autoleucel (cilta-cel, Carvykti), developed by Janssen and Legend Biotech, received approval in February 2022. While that helped provide centers with a few more slots, it wasn’t sufficient to cut waiting times, and the demand for these myeloma therapies continues to outstrip the capacity to produce CAR-T products in a timely manner.
“For myeloma, the demand is very high, as most patients are not cured from any other existing myeloma therapies, and most patients will make it to fifth-line therapy where the two CAR T-cell products are approved right now,” said Krina K. Patel, MD, medical director of the department of lymphoma/myeloma in the division of cancer medicine at the University of Texas MD Anderson Cancer Center, Houston.
“We likely have 10 eligible CAR-T myeloma patients each month at our center,” she said, “but were getting two slots per month for the past 8 months, and now are getting four slots a month.”
“Our clinic has also experienced the impact of the low number of manufacturing slots offered to each cancer center for some CAR T-cell products,” said David Maloney, MD, PhD, medical director, Cellular Immunotherapy and Bezos Family Immunotherapy Clinic, Seattle Cancer Care Alliance.
He noted that, as with other cancer centers, for multiple myeloma they are provided a specific number of manufacturing slots for each treatment. “Our providers discuss which patients are most appropriate for available slots for that month,” said Dr. Maloney.
“Additionally, juggling patient schedules may be required to address the extended manufacturing time for some products. In some cases, clinical trials may be available in a more timely fashion for appropriate patients, and in some cases, switching to an alternative product is possible,” he commented.
Complex causes behind bottleneck
The cause of the current bottleneck for myeloma patients is complex. It stems from a shortage of raw materials and supply chain restraints, among other things.
While the biggest impact of shortages has been on patients with multiple myeloma, Dr. Patel pointed out that these constraints are also affecting patients with lymphoma at her institution, but to a lesser degree.
“This is multifactorial as to why, but most of the issues arise from manufacturing,” Dr. Patel said in an interview. “Initially, the FDA limited how many slots each new product could have per month, then there was a viral vector shortage, and then the quality-control process the FDA requires takes longer than the manufacturing of the cells actually do.”
On top of that, “we have about a 5% manufacturing fail rate so far,” she added. Such failures occur when the cells taken from a patient cannot be converted into CAR T cells for therapy.
Matthew J. Frigault, MD, from the Center for Cellular Therapies, Mass General Cancer Center, Boston, explained that the growing excitement about the potential for cellular therapy and recent approvals for these products for use in earlier lines of treatment have increased demand for them.
There are also problems regarding supply. Manufacture and delivery of CAR T is complicated and takes time to scale up, Dr. Frigault pointed out. “Therefore, we are seeing limited access, more so for the BCMA-directed therapies [which are used for myeloma].”
The shortages and delays likely involve two main factors. “For the newer indications, there is a significant backlog of patients who have been waiting for these therapies and have not been able to access them in the clinical trial setting, and manufacturing is extremely complicated and not easily scaled up,” he said.
“That being said, manufacturers are trying to increase the number of available manufacturing slots and decrease the time needed to manufacture cells,” Dr. Frigault commented.
Delays in access to myeloma CAR T-cell therapy are also affecting patient care at Fox Chase Cancer Center in Philadelphia. “We have had about one slot every 2 months for Abecma,” noted Henry Fung, MD, chair of the department of bone marrow transplant and cellular therapies at Fox Chase. “For Carvykti, there are only 32 certified centers in [the] U.S., and access is very limited.”
Dr. Fung explained that they have had to offer alternative treatments to many of their patients. “There are rumors that there’s shortage in obtaining raw materials, such as the virus used for transduction, although we have not encountered any problems in other CAR T products used for lymphomas.”
Pharma companies trying to meet the demand
This news organization reached out to the manufacturers of CAR T products. All have reported that they are doing what they feasibly can to ramp up production.
“The complexity of delivering CAR T-cell therapies is unlike any other traditional biologic or small-molecule medicine, using a patient’s own cells to start a highly sophisticated and personalized manufacturing process,” commented a spokesperson for BMS, which has two CAR T-cell products currently on the market.
“In this nascent field of cell therapy, we continue to evolve every day, addressing supply and manufacturing challenges head on by applying key learnings across our three state-of-the-art cell therapy facilities and two new facilities in progress.
“We have been encouraged by a steady increase in our manufacturing capacity, and we continue efforts to ramp up further to meet the demand for our cell therapies,” the BMS spokesperson commented. “We have already seen improvements in the stabilization of vector supply and expect additional improvements in capacity in the second half of 2022.”
Novartis said much the same thing. They have a “comprehensive, integrated global CAR-T manufacturing footprint that strengthens the flexibility, resilience, and sustainability of the Novartis manufacturing and supply chain. Together with an improved manufacturing process, we are confident in our ability to meet patient demand with timely delivery,” according to a Novartis spokesperson.
The spokesperson also pointed out that the company has continuously incorporated process improvements that have significantly increased manufacturing capacity and success rates for patients in need of CAR T cells.
“Data presented at [the] American Society of Hematology annual meeting in 2021 showed the Novartis Morris Plains facility, our flagship CAR T manufacturing site, had commercial manufacturing and shipping success rates of 96% and 99%, respectively, between January and August 2021,” according to the spokesperson.
Legend and Janssen, the companies behind Carvykti, one of the two approved cell products for myeloma, which launched earlier in 2022, said that they have continued to activate certified treatment centers in a phased approach that will enable them to expand availability throughout 2022 and beyond.
“This phased approach was designed to ensure the highest level of predictability and reliability for the patient and the certified treatment centers,” the spokesperson said. “We understand the urgency for patients in need of Carvyki and are committed to doing everything we can to accelerate our ability to deliver this important cell therapy in a reliable and timely manner.”
With regard to the industry-wide supply shortage of lentivirus, Legend and Janssen say they have put in place multiple processes to address the shortage, “including enhancing our own internal manufacturing capabilities of this essential drug substance, to ensure sufficient and sustained supply.”
Incredibly exciting potential
Given the immense potential of CAR T-cell therapy, the supply shortage that myeloma patients are experiencing is all the more poignant and distressing. While not everyone benefits, some patients for whom every other therapy failed and who were facing hospice have had dramatic results.
“Incredibly exciting with unbelievable potential” was how one expert described these new therapies when the first product was about to enter the marketplace. Since then, six CAR T-cell therapies have received regulatory approval for an ever-increasing range of hematologic malignancies.
But these CAR T-cell therapies have their own set of adverse events, which can be serious and even life-threatening. In addition, not all patients become cancer free, although long-term data are impressive.
A study that included one of the longest follow-ups to date was reported at the 2020 annual meeting of the American Society of Clinical Oncology. The researchers reported that remissions lasted over 9 years for patients with relapsed/refractory B-cell lymphoma or chronic lymphocytic leukemia who underwent treatment with Kite’s axicaptagene cilleucel (Yescarta). This review included 43 patients and showed an overall remission rate of 76%. Complete remission was achieved for 54% of patients, and partial remission was achieved for 22%.
The results with CAR T-cell therapy in multiple myeloma are not quite as impressive, but even so, the clinical data that supported the approval of Abecma showed that a third of patients, who had previously received a median of six prior therapies, achieved a complete response.
At the time of the Abecma approval, the lead investigator of the study, Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, commented: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
A version of this article first appeared on Medscape.com.
Some patients with blood cancers for whom all other therapeutic options have been exhausted have one final chance of getting rid of their disease: treatment with chimeric antigen-receptor (CAR) T cells.
Described as a “living drug,” the treatment involves genetically engineering the patient’s own blood cells and reinfusing them back into their system. These CAR T cells then hunt down and destroy cancer cells; in some cases, they manage to eradicate the disease completely.
About half of patients with leukemia or lymphoma and about a third of those with multiple myeloma who receive this treatment have a complete remission and achieve a functional “cure.”
But not all patients who could benefit from this therapy are able to get it. Some are spending months on waiting lists, often deteriorating while they wait. These patients have exhausted all other therapeutic options, and many are facing hospice and death.
The scope of this problem was illustrated by a recent survey of the centers that are certified to deliver this complex therapy.
The survey was led by Yi Lin, MD, PhD, associate professor of medicine at the Mayo Clinic, Rochester, Minn., and medical director for the cellular therapy program. It was published as an abstract at the annual meeting of the American Society of Clinical Oncology recently, although it was not presented there.
“We wanted to find out just how widespread this problem is,” Dr. Lin said, adding: “There had been nothing in the literature thus far about it.”
The team contacted 20 centers across the United States and received responses from 17. Results showed that the median time on the waiting list was 6 months and that only 25% of patients eventually received CAR T-cell therapy. An additional 25% were able to enter a CAR T clinical trial. The remaining 50% of patients either were enrolled in a different type of trial, entered hospice, or died.
For patient selection, all centers reported using a committee of experienced physicians to ensure consistency. They employed different ethical principles for selection. Some centers sought to maximize the total benefit, such as selecting the patients most likely to achieve leukapheresis or a clinical response, while others based their decisions on the time patients spent on waiting list or gave priority to the patients who were the “worst off” with the most limited therapeutic options.
Shortage affecting mostly myeloma patients
The shortages in CAR T-cell therapies primarily involve the products used for patients with multiple myeloma.
The problem has not, as yet, noticeably spilled over to lymphoma and leukemia treatments, which use a slightly different type of CAR T-cell therapy (it targets CD19, whereas the cell therapies used for myeloma target BCMA).
“We have backlog of myeloma patients who don’t have access,” said Nina Shah, MD, a hematologist and professor of medicine at the University of California, San Francisco. “We have only four slots for the two myeloma products but about 50-60 eligible patients.”
Long waiting times for CAR T cells for myeloma have been an issue ever since the first of these products appeared on the market: idecabtagene vicleucel (ide-cel; Abecma), developed by Bluebird Bio and Bristol-Myers Squibb. “As soon as it became available in March 2021, we had people waiting and limits on our access to it,” Dr. Shah said.
A second CAR T-cell therapy for myeloma, ciltacabtagene autoleucel (cilta-cel, Carvykti), developed by Janssen and Legend Biotech, received approval in February 2022. While that helped provide centers with a few more slots, it wasn’t sufficient to cut waiting times, and the demand for these myeloma therapies continues to outstrip the capacity to produce CAR-T products in a timely manner.
“For myeloma, the demand is very high, as most patients are not cured from any other existing myeloma therapies, and most patients will make it to fifth-line therapy where the two CAR T-cell products are approved right now,” said Krina K. Patel, MD, medical director of the department of lymphoma/myeloma in the division of cancer medicine at the University of Texas MD Anderson Cancer Center, Houston.
“We likely have 10 eligible CAR-T myeloma patients each month at our center,” she said, “but were getting two slots per month for the past 8 months, and now are getting four slots a month.”
“Our clinic has also experienced the impact of the low number of manufacturing slots offered to each cancer center for some CAR T-cell products,” said David Maloney, MD, PhD, medical director, Cellular Immunotherapy and Bezos Family Immunotherapy Clinic, Seattle Cancer Care Alliance.
He noted that, as with other cancer centers, for multiple myeloma they are provided a specific number of manufacturing slots for each treatment. “Our providers discuss which patients are most appropriate for available slots for that month,” said Dr. Maloney.
“Additionally, juggling patient schedules may be required to address the extended manufacturing time for some products. In some cases, clinical trials may be available in a more timely fashion for appropriate patients, and in some cases, switching to an alternative product is possible,” he commented.
Complex causes behind bottleneck
The cause of the current bottleneck for myeloma patients is complex. It stems from a shortage of raw materials and supply chain restraints, among other things.
While the biggest impact of shortages has been on patients with multiple myeloma, Dr. Patel pointed out that these constraints are also affecting patients with lymphoma at her institution, but to a lesser degree.
“This is multifactorial as to why, but most of the issues arise from manufacturing,” Dr. Patel said in an interview. “Initially, the FDA limited how many slots each new product could have per month, then there was a viral vector shortage, and then the quality-control process the FDA requires takes longer than the manufacturing of the cells actually do.”
On top of that, “we have about a 5% manufacturing fail rate so far,” she added. Such failures occur when the cells taken from a patient cannot be converted into CAR T cells for therapy.
Matthew J. Frigault, MD, from the Center for Cellular Therapies, Mass General Cancer Center, Boston, explained that the growing excitement about the potential for cellular therapy and recent approvals for these products for use in earlier lines of treatment have increased demand for them.
There are also problems regarding supply. Manufacture and delivery of CAR T is complicated and takes time to scale up, Dr. Frigault pointed out. “Therefore, we are seeing limited access, more so for the BCMA-directed therapies [which are used for myeloma].”
The shortages and delays likely involve two main factors. “For the newer indications, there is a significant backlog of patients who have been waiting for these therapies and have not been able to access them in the clinical trial setting, and manufacturing is extremely complicated and not easily scaled up,” he said.
“That being said, manufacturers are trying to increase the number of available manufacturing slots and decrease the time needed to manufacture cells,” Dr. Frigault commented.
Delays in access to myeloma CAR T-cell therapy are also affecting patient care at Fox Chase Cancer Center in Philadelphia. “We have had about one slot every 2 months for Abecma,” noted Henry Fung, MD, chair of the department of bone marrow transplant and cellular therapies at Fox Chase. “For Carvykti, there are only 32 certified centers in [the] U.S., and access is very limited.”
Dr. Fung explained that they have had to offer alternative treatments to many of their patients. “There are rumors that there’s shortage in obtaining raw materials, such as the virus used for transduction, although we have not encountered any problems in other CAR T products used for lymphomas.”
Pharma companies trying to meet the demand
This news organization reached out to the manufacturers of CAR T products. All have reported that they are doing what they feasibly can to ramp up production.
“The complexity of delivering CAR T-cell therapies is unlike any other traditional biologic or small-molecule medicine, using a patient’s own cells to start a highly sophisticated and personalized manufacturing process,” commented a spokesperson for BMS, which has two CAR T-cell products currently on the market.
“In this nascent field of cell therapy, we continue to evolve every day, addressing supply and manufacturing challenges head on by applying key learnings across our three state-of-the-art cell therapy facilities and two new facilities in progress.
“We have been encouraged by a steady increase in our manufacturing capacity, and we continue efforts to ramp up further to meet the demand for our cell therapies,” the BMS spokesperson commented. “We have already seen improvements in the stabilization of vector supply and expect additional improvements in capacity in the second half of 2022.”
Novartis said much the same thing. They have a “comprehensive, integrated global CAR-T manufacturing footprint that strengthens the flexibility, resilience, and sustainability of the Novartis manufacturing and supply chain. Together with an improved manufacturing process, we are confident in our ability to meet patient demand with timely delivery,” according to a Novartis spokesperson.
The spokesperson also pointed out that the company has continuously incorporated process improvements that have significantly increased manufacturing capacity and success rates for patients in need of CAR T cells.
“Data presented at [the] American Society of Hematology annual meeting in 2021 showed the Novartis Morris Plains facility, our flagship CAR T manufacturing site, had commercial manufacturing and shipping success rates of 96% and 99%, respectively, between January and August 2021,” according to the spokesperson.
Legend and Janssen, the companies behind Carvykti, one of the two approved cell products for myeloma, which launched earlier in 2022, said that they have continued to activate certified treatment centers in a phased approach that will enable them to expand availability throughout 2022 and beyond.
“This phased approach was designed to ensure the highest level of predictability and reliability for the patient and the certified treatment centers,” the spokesperson said. “We understand the urgency for patients in need of Carvyki and are committed to doing everything we can to accelerate our ability to deliver this important cell therapy in a reliable and timely manner.”
With regard to the industry-wide supply shortage of lentivirus, Legend and Janssen say they have put in place multiple processes to address the shortage, “including enhancing our own internal manufacturing capabilities of this essential drug substance, to ensure sufficient and sustained supply.”
Incredibly exciting potential
Given the immense potential of CAR T-cell therapy, the supply shortage that myeloma patients are experiencing is all the more poignant and distressing. While not everyone benefits, some patients for whom every other therapy failed and who were facing hospice have had dramatic results.
“Incredibly exciting with unbelievable potential” was how one expert described these new therapies when the first product was about to enter the marketplace. Since then, six CAR T-cell therapies have received regulatory approval for an ever-increasing range of hematologic malignancies.
But these CAR T-cell therapies have their own set of adverse events, which can be serious and even life-threatening. In addition, not all patients become cancer free, although long-term data are impressive.
A study that included one of the longest follow-ups to date was reported at the 2020 annual meeting of the American Society of Clinical Oncology. The researchers reported that remissions lasted over 9 years for patients with relapsed/refractory B-cell lymphoma or chronic lymphocytic leukemia who underwent treatment with Kite’s axicaptagene cilleucel (Yescarta). This review included 43 patients and showed an overall remission rate of 76%. Complete remission was achieved for 54% of patients, and partial remission was achieved for 22%.
The results with CAR T-cell therapy in multiple myeloma are not quite as impressive, but even so, the clinical data that supported the approval of Abecma showed that a third of patients, who had previously received a median of six prior therapies, achieved a complete response.
At the time of the Abecma approval, the lead investigator of the study, Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, commented: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
A version of this article first appeared on Medscape.com.
Fewer transplants for MM with quadruplet therapy?
“It is not a big leap of faith to imagine that, in the near future, with the availability of quadruplets and T-cell therapies, the role of high-dose melphalan and autologous stem cell transplant will be diminished,” said Dickran Kazandjian, MD, and Ola Landgren, MD, PhD, of the myeloma division, Sylvester Comprehensive Cancer Center, University of Miami.
They commented in a editorial in JAMA Oncology, prompted by a paper describing new results with a novel quadruple combination of therapies. These treatments included the monoclonal antibody elotuzumab (Empliciti) added onto the established backbone of carfilzomib (Kyprolis), lenalidomide (Revlimid), and dexamethasone (known as KRd).
“Regardless of what the future holds for elotuzumab-based combinations, it is clear that the new treatment paradigm of newly diagnosed MM will incorporate antibody-based quadruplet regimens,” the editorialists commented.
“Novel immunotherapies are here to stay,” they added, “as they are already transforming the lives of patients with multiple MM and bringing a bright horizon to the treatment landscape.”
Study details
The trial of the novel quadruplet regimen was a multicenter, single-arm, phase 2 study that involved 46 patients with newly diagnosed multiple myeloma, explain first author Benjamin A. Derman, MD, of the University of Chicago Medical Center, and colleagues.
These patients had a median age of 62; more than two-thirds were male (72%) and White (70%). About half (48%) had high-risk cytogenetic abnormalities.
All patients were treated with 12 cycles of the quadruple therapy Elo-KRd regimen. They underwent bone marrow assessment of measurable residual disease (MRD; with 10-5 sensitivity) after cycle 8 and cycle 12.
“An MRD-adapted treatment approach is rational because it may identify which patients can be administered shorter courses of intensive therapy without compromising efficacy,” the authors explained.
Patients who had MRD negativity at both time points did not receive further Elo-KRd, while patients who converted from MRD positivity to negativity in between cycles 8 and 12 received 6 additional cycles of Elo-KRd. Those who remained MRD positive or converted to positivity after 12 cycles received an additional 12 cycles of Elo-KRd.
Following Elo-KRd treatment, all patients transitioned to triple therapy with Elo-Rd (with no carfilzomib), for indefinite maintenance therapy or until disease progression.
For the primary endpoint, the rate of stringent complete response and/or MRD-negativity after cycle 8 was 58% (26 of 45), meeting the predefined definition of efficacy.
Importantly, 26% of patients converted from MRD positivity after cycle 8 to negativity at a later time point, while 50% of patients reached 1-year sustained MRD negativity.
Overall, the estimated 3-year, progression-free survival was 72%, and the rate was 92% for patients with MRD-negativity at cycle 8. The overall survival rate was 78%.
The most common grade 3 or 4 adverse events were lung and nonpulmonary infections (13% and 11%, respectively), and one patient had a grade 5 MI. Three patients discontinued the treatment because of intolerance.
“An MRD-adapted design using elotuzumab and weekly KRd without autologous stem cell transplantation showed a high rate of stringent complete response (sCR) and/or MRD-negativity and durable responses,” the authors wrote.
“This approach provides support for further evaluation of MRD-guided de-escalation of therapy to decrease treatment exposure while sustaining deep responses.”
To better assess the difference of the therapy versus treatment including stem cell transplantation, a phase 3, randomized trial is currently underway to compare the Elo-KRd regimen against KRd with autologous stem cell transplant in newly diagnosed MM.
“If Elo-KRd proves superior, a randomized comparison of Elo versus anti-CD38 mAb-based quadruplets would help determine the optimal combination of therapies in the frontline setting,” the authors noted.
Randomized trial anticipated to clarify benefit
In their editorial, Dr. Kazandjian and Dr. Landgren agreed with the authors that the role of elotuzumab needs to be better clarified in a randomized trial setting.
Elotuzumab received FDA approval in 2015 based on results from the ELOQUENT-2 study, which showed improved progression-free survival and overall survival with the addition of elotuzumab to lenalidomide and dexamethasone in patients with multiple myeloma who have previously received one to three other therapies.
However, the editorialists pointed out that recently published results from the randomized ELOQUENT-1 trial of lenalidomide and dexamethasone with and without elotuzumab showed the addition of elotuzumab was not associated with a statistically significant difference in progression-free survival.
The editorialists also pointed out that, in the setting of newly diagnosed multiple myeloma, another recent, similarly designed study found that the backbone regimen of carfilzomib, lenalidomide, and dexamethasone – on its own – was also associated with a favorable MRD-negative rate of 62%.
In addition, several studies involving novel quadruple treatments with the monoclonal antibody daratumumab (Darzalex) instead of elotuzumab, have also shown benefit in newly diagnosed multiple myeloma, resulting in high rates of MRD negativity.
Collectively, the findings bode well for the quadruple regimens in the treatment of MM, the editorialists emphasized.
“Importantly, with the rate of deep remissions observed with antibody-based quadruplet therapies, one may question the role of using early high-dose melphalan and autologous stem cell transplant in every patient, especially in those who have achieved MRD negativity with the quadruplet alone,” they added.
The study was sponsored in part by Amgen, Bristol-Myers Squibb, and the Multiple Myeloma Research Consortium. Dr. Derman reported advisory board fees from Sanofi, Janssen, and COTA Healthcare; honoraria from PleXus Communications and MJH Life Sciences. Dr. Kazandjian declares receiving advisory board or consulting fees from Bristol-Myers Squibb, Sanofi, and Arcellx outside the submitted work. Dr. Landgren has received grant support from numerous organizations and pharmaceutical companies. Dr. Landgren has also received honoraria for scientific talks/participated in advisory boards for Adaptive Biotech, Amgen, Binding Site, Bristol-Myers Squibb, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer, and served on independent data monitoring committees for international randomized trials by Takeda, Merck, Janssen, and Theradex.
A version of this article first appeared on Medscape.com.
“It is not a big leap of faith to imagine that, in the near future, with the availability of quadruplets and T-cell therapies, the role of high-dose melphalan and autologous stem cell transplant will be diminished,” said Dickran Kazandjian, MD, and Ola Landgren, MD, PhD, of the myeloma division, Sylvester Comprehensive Cancer Center, University of Miami.
They commented in a editorial in JAMA Oncology, prompted by a paper describing new results with a novel quadruple combination of therapies. These treatments included the monoclonal antibody elotuzumab (Empliciti) added onto the established backbone of carfilzomib (Kyprolis), lenalidomide (Revlimid), and dexamethasone (known as KRd).
“Regardless of what the future holds for elotuzumab-based combinations, it is clear that the new treatment paradigm of newly diagnosed MM will incorporate antibody-based quadruplet regimens,” the editorialists commented.
“Novel immunotherapies are here to stay,” they added, “as they are already transforming the lives of patients with multiple MM and bringing a bright horizon to the treatment landscape.”
Study details
The trial of the novel quadruplet regimen was a multicenter, single-arm, phase 2 study that involved 46 patients with newly diagnosed multiple myeloma, explain first author Benjamin A. Derman, MD, of the University of Chicago Medical Center, and colleagues.
These patients had a median age of 62; more than two-thirds were male (72%) and White (70%). About half (48%) had high-risk cytogenetic abnormalities.
All patients were treated with 12 cycles of the quadruple therapy Elo-KRd regimen. They underwent bone marrow assessment of measurable residual disease (MRD; with 10-5 sensitivity) after cycle 8 and cycle 12.
“An MRD-adapted treatment approach is rational because it may identify which patients can be administered shorter courses of intensive therapy without compromising efficacy,” the authors explained.
Patients who had MRD negativity at both time points did not receive further Elo-KRd, while patients who converted from MRD positivity to negativity in between cycles 8 and 12 received 6 additional cycles of Elo-KRd. Those who remained MRD positive or converted to positivity after 12 cycles received an additional 12 cycles of Elo-KRd.
Following Elo-KRd treatment, all patients transitioned to triple therapy with Elo-Rd (with no carfilzomib), for indefinite maintenance therapy or until disease progression.
For the primary endpoint, the rate of stringent complete response and/or MRD-negativity after cycle 8 was 58% (26 of 45), meeting the predefined definition of efficacy.
Importantly, 26% of patients converted from MRD positivity after cycle 8 to negativity at a later time point, while 50% of patients reached 1-year sustained MRD negativity.
Overall, the estimated 3-year, progression-free survival was 72%, and the rate was 92% for patients with MRD-negativity at cycle 8. The overall survival rate was 78%.
The most common grade 3 or 4 adverse events were lung and nonpulmonary infections (13% and 11%, respectively), and one patient had a grade 5 MI. Three patients discontinued the treatment because of intolerance.
“An MRD-adapted design using elotuzumab and weekly KRd without autologous stem cell transplantation showed a high rate of stringent complete response (sCR) and/or MRD-negativity and durable responses,” the authors wrote.
“This approach provides support for further evaluation of MRD-guided de-escalation of therapy to decrease treatment exposure while sustaining deep responses.”
To better assess the difference of the therapy versus treatment including stem cell transplantation, a phase 3, randomized trial is currently underway to compare the Elo-KRd regimen against KRd with autologous stem cell transplant in newly diagnosed MM.
“If Elo-KRd proves superior, a randomized comparison of Elo versus anti-CD38 mAb-based quadruplets would help determine the optimal combination of therapies in the frontline setting,” the authors noted.
Randomized trial anticipated to clarify benefit
In their editorial, Dr. Kazandjian and Dr. Landgren agreed with the authors that the role of elotuzumab needs to be better clarified in a randomized trial setting.
Elotuzumab received FDA approval in 2015 based on results from the ELOQUENT-2 study, which showed improved progression-free survival and overall survival with the addition of elotuzumab to lenalidomide and dexamethasone in patients with multiple myeloma who have previously received one to three other therapies.
However, the editorialists pointed out that recently published results from the randomized ELOQUENT-1 trial of lenalidomide and dexamethasone with and without elotuzumab showed the addition of elotuzumab was not associated with a statistically significant difference in progression-free survival.
The editorialists also pointed out that, in the setting of newly diagnosed multiple myeloma, another recent, similarly designed study found that the backbone regimen of carfilzomib, lenalidomide, and dexamethasone – on its own – was also associated with a favorable MRD-negative rate of 62%.
In addition, several studies involving novel quadruple treatments with the monoclonal antibody daratumumab (Darzalex) instead of elotuzumab, have also shown benefit in newly diagnosed multiple myeloma, resulting in high rates of MRD negativity.
Collectively, the findings bode well for the quadruple regimens in the treatment of MM, the editorialists emphasized.
“Importantly, with the rate of deep remissions observed with antibody-based quadruplet therapies, one may question the role of using early high-dose melphalan and autologous stem cell transplant in every patient, especially in those who have achieved MRD negativity with the quadruplet alone,” they added.
The study was sponsored in part by Amgen, Bristol-Myers Squibb, and the Multiple Myeloma Research Consortium. Dr. Derman reported advisory board fees from Sanofi, Janssen, and COTA Healthcare; honoraria from PleXus Communications and MJH Life Sciences. Dr. Kazandjian declares receiving advisory board or consulting fees from Bristol-Myers Squibb, Sanofi, and Arcellx outside the submitted work. Dr. Landgren has received grant support from numerous organizations and pharmaceutical companies. Dr. Landgren has also received honoraria for scientific talks/participated in advisory boards for Adaptive Biotech, Amgen, Binding Site, Bristol-Myers Squibb, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer, and served on independent data monitoring committees for international randomized trials by Takeda, Merck, Janssen, and Theradex.
A version of this article first appeared on Medscape.com.
“It is not a big leap of faith to imagine that, in the near future, with the availability of quadruplets and T-cell therapies, the role of high-dose melphalan and autologous stem cell transplant will be diminished,” said Dickran Kazandjian, MD, and Ola Landgren, MD, PhD, of the myeloma division, Sylvester Comprehensive Cancer Center, University of Miami.
They commented in a editorial in JAMA Oncology, prompted by a paper describing new results with a novel quadruple combination of therapies. These treatments included the monoclonal antibody elotuzumab (Empliciti) added onto the established backbone of carfilzomib (Kyprolis), lenalidomide (Revlimid), and dexamethasone (known as KRd).
“Regardless of what the future holds for elotuzumab-based combinations, it is clear that the new treatment paradigm of newly diagnosed MM will incorporate antibody-based quadruplet regimens,” the editorialists commented.
“Novel immunotherapies are here to stay,” they added, “as they are already transforming the lives of patients with multiple MM and bringing a bright horizon to the treatment landscape.”
Study details
The trial of the novel quadruplet regimen was a multicenter, single-arm, phase 2 study that involved 46 patients with newly diagnosed multiple myeloma, explain first author Benjamin A. Derman, MD, of the University of Chicago Medical Center, and colleagues.
These patients had a median age of 62; more than two-thirds were male (72%) and White (70%). About half (48%) had high-risk cytogenetic abnormalities.
All patients were treated with 12 cycles of the quadruple therapy Elo-KRd regimen. They underwent bone marrow assessment of measurable residual disease (MRD; with 10-5 sensitivity) after cycle 8 and cycle 12.
“An MRD-adapted treatment approach is rational because it may identify which patients can be administered shorter courses of intensive therapy without compromising efficacy,” the authors explained.
Patients who had MRD negativity at both time points did not receive further Elo-KRd, while patients who converted from MRD positivity to negativity in between cycles 8 and 12 received 6 additional cycles of Elo-KRd. Those who remained MRD positive or converted to positivity after 12 cycles received an additional 12 cycles of Elo-KRd.
Following Elo-KRd treatment, all patients transitioned to triple therapy with Elo-Rd (with no carfilzomib), for indefinite maintenance therapy or until disease progression.
For the primary endpoint, the rate of stringent complete response and/or MRD-negativity after cycle 8 was 58% (26 of 45), meeting the predefined definition of efficacy.
Importantly, 26% of patients converted from MRD positivity after cycle 8 to negativity at a later time point, while 50% of patients reached 1-year sustained MRD negativity.
Overall, the estimated 3-year, progression-free survival was 72%, and the rate was 92% for patients with MRD-negativity at cycle 8. The overall survival rate was 78%.
The most common grade 3 or 4 adverse events were lung and nonpulmonary infections (13% and 11%, respectively), and one patient had a grade 5 MI. Three patients discontinued the treatment because of intolerance.
“An MRD-adapted design using elotuzumab and weekly KRd without autologous stem cell transplantation showed a high rate of stringent complete response (sCR) and/or MRD-negativity and durable responses,” the authors wrote.
“This approach provides support for further evaluation of MRD-guided de-escalation of therapy to decrease treatment exposure while sustaining deep responses.”
To better assess the difference of the therapy versus treatment including stem cell transplantation, a phase 3, randomized trial is currently underway to compare the Elo-KRd regimen against KRd with autologous stem cell transplant in newly diagnosed MM.
“If Elo-KRd proves superior, a randomized comparison of Elo versus anti-CD38 mAb-based quadruplets would help determine the optimal combination of therapies in the frontline setting,” the authors noted.
Randomized trial anticipated to clarify benefit
In their editorial, Dr. Kazandjian and Dr. Landgren agreed with the authors that the role of elotuzumab needs to be better clarified in a randomized trial setting.
Elotuzumab received FDA approval in 2015 based on results from the ELOQUENT-2 study, which showed improved progression-free survival and overall survival with the addition of elotuzumab to lenalidomide and dexamethasone in patients with multiple myeloma who have previously received one to three other therapies.
However, the editorialists pointed out that recently published results from the randomized ELOQUENT-1 trial of lenalidomide and dexamethasone with and without elotuzumab showed the addition of elotuzumab was not associated with a statistically significant difference in progression-free survival.
The editorialists also pointed out that, in the setting of newly diagnosed multiple myeloma, another recent, similarly designed study found that the backbone regimen of carfilzomib, lenalidomide, and dexamethasone – on its own – was also associated with a favorable MRD-negative rate of 62%.
In addition, several studies involving novel quadruple treatments with the monoclonal antibody daratumumab (Darzalex) instead of elotuzumab, have also shown benefit in newly diagnosed multiple myeloma, resulting in high rates of MRD negativity.
Collectively, the findings bode well for the quadruple regimens in the treatment of MM, the editorialists emphasized.
“Importantly, with the rate of deep remissions observed with antibody-based quadruplet therapies, one may question the role of using early high-dose melphalan and autologous stem cell transplant in every patient, especially in those who have achieved MRD negativity with the quadruplet alone,” they added.
The study was sponsored in part by Amgen, Bristol-Myers Squibb, and the Multiple Myeloma Research Consortium. Dr. Derman reported advisory board fees from Sanofi, Janssen, and COTA Healthcare; honoraria from PleXus Communications and MJH Life Sciences. Dr. Kazandjian declares receiving advisory board or consulting fees from Bristol-Myers Squibb, Sanofi, and Arcellx outside the submitted work. Dr. Landgren has received grant support from numerous organizations and pharmaceutical companies. Dr. Landgren has also received honoraria for scientific talks/participated in advisory boards for Adaptive Biotech, Amgen, Binding Site, Bristol-Myers Squibb, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer, and served on independent data monitoring committees for international randomized trials by Takeda, Merck, Janssen, and Theradex.
A version of this article first appeared on Medscape.com.
FROM JAMA ONCOLOGY
Simultaneous Cases of Carfilzomib-Induced Thrombotic Microangiopathy in 2 Patients With Multiple Myeloma
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
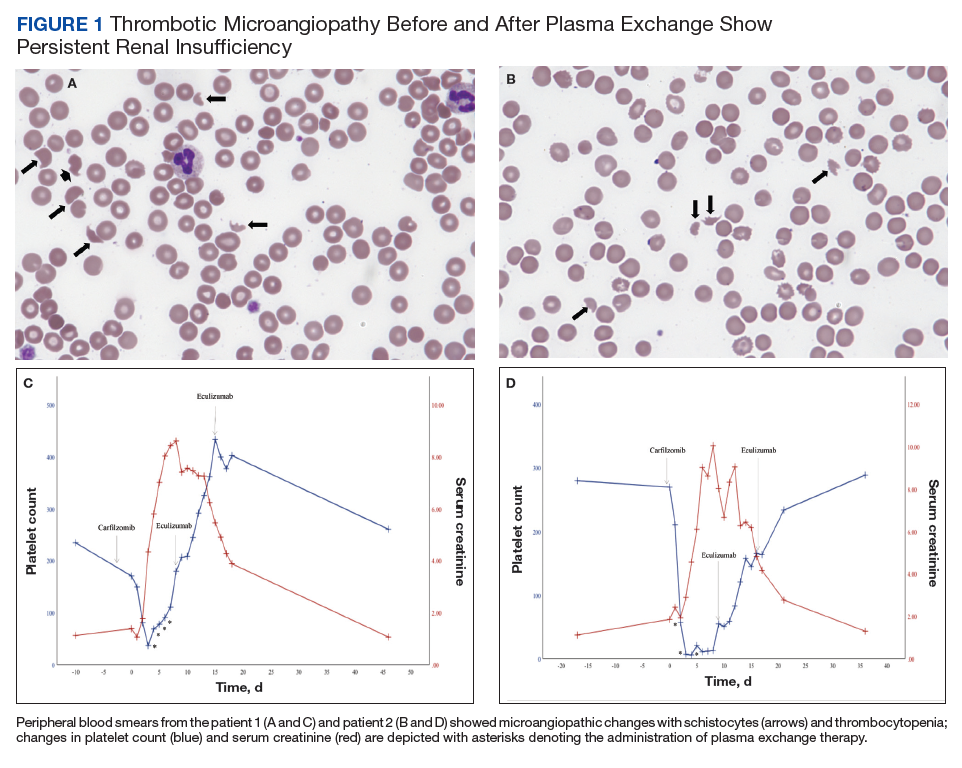
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
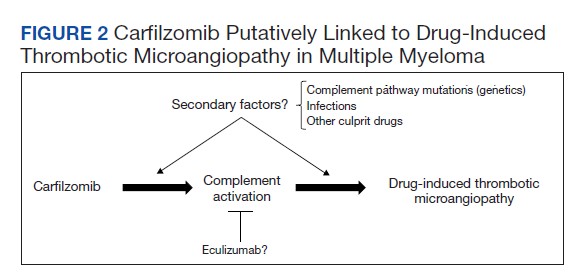
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
Evidence still lacking that vitamins prevent CVD, cancer: USPSTF
There is not enough evidence to recommend for or against taking most vitamin and mineral supplements to prevent heart disease, stroke, and cancer, a new report by the U.S. Preventive Services Task Force concludes.
However, there are two vitamins – vitamin E and beta-carotene – that the task force recommends against for the prevention of heart disease, stroke, and cancer. Evidence shows that there is no benefit to taking vitamin E and that beta-carotene can increase the risk for lung cancer in people already at risk, such as smokers and those with occupational exposure to asbestos.
These are the main findings of the USPSTF’s final recommendation statement on vitamin, mineral, and multivitamin supplementation to prevent cardiovascular disease and cancer. The statement was published in JAMA.
“This is essentially the same recommendation that the task force made in 2014,” USPSTF member John Wong, MD, professor of medicine at Tufts University, Boston, said in an interview.
“We recognize that over half of people in the U.S. take a vitamin supplement of some sort every day and 30% take a vitamin/mineral combination. We wanted to review the evidence again to see if there was any benefit in terms of reducing the risk of cardiovascular disease or cancer or increasing the chances of living longer,” Dr. Wong explained.
“We looked hard for evidence, reviewing 84 studies in total. But we did not find sufficient evidence in favor of taking or not taking vitamins, with the two exceptions of beta-carotene and vitamin E, which we recommend against taking,” he noted.
Although there is evidence of some harm with beta-carotene, the main reason behind the recommendation against taking vitamin E is the consistent evidence of no benefit, Dr. Wong explained.
“While the evidence for some other vitamins is conflicting, there is more consistent evidence of no benefit for vitamin E,” he said.
The bulk of new evidence since the last review in 2014 was predominately for vitamin D supplementation, but despite the inclusion of 32 new randomized, controlled trials and two cohort studies, pooled estimates for all-cause mortality were similar to those in the previous review, with confidence intervals only slightly crossing 1, and point estimates that suggest at most a very small benefit, the task force noted.
“Apart from beta-carotene and vitamin E, after reviewing 84 studies – including 78 randomized controlled trials – in over a million patients, we can find no clear demonstration of benefit or harm of taking vitamins in terms of developing cardiovascular disease or cancer or the effect on all-cause mortality. So, we don’t know whether people should take vitamins or not, and we need more research,” Dr. Wong added.
On the use of a multivitamin supplement, Dr. Wong noted that the complete body of evidence did not find any benefit of taking a multivitamin on cardiovascular or cancer mortality. But there was a small reduction in cancer incidence.
However, he pointed out that the three studies that suggested a reduction in cancer incidence all had issues regarding generalizability.
“The recently published COSMOS trial had an average follow-up of only 3.6 years, which isn’t really long enough when thinking about the prevention of cancer, one of the other studies only used antioxidants, and the third study was conducted only in U.S. male physicians. So those limitations regarding generalizability limited our confidence in making recommendations about multivitamins,” Dr. Wong explained.
But he noted that the task force did not find any significant harms from taking multivitamins.
“There are possible harms from taking high doses of vitamin A and vitamin D, but generally the doses contained in a multivitamin tablet are lower than these. But if the goal for taking a multivitamin is to lower your risk of cancer or cardiovascular disease, we didn’t find sufficient evidence to be able to make a recommendation,” he said.
Asked what he would say to all the people currently taking multivitamins, Dr. Wong responded that he would advise them to have a conversation with a trusted health care professional about their particular circumstances.
“Our statement has quite a narrow focus. It is directed toward community-dwelling, nonpregnant adults. This recommendation does not apply to children, persons who are pregnant or may become pregnant, or persons who are chronically ill, are hospitalized, or have a known nutritional deficiency,” he commented.
‘Any benefit likely to be small’
In an editorial accompanying the publication of the USPSTF statement, Jenny Jia, MD; Natalie Cameron, MD; and Jeffrey Linder, MD – all from Northwestern University, Chicago – noted that the current evidence base includes 52 additional studies not available when the last USPSTF recommendation on this topic was published in 2014.
The editorialists pointed out that for multivitamins, proving the absence of a benefit is challenging, but at best, current evidence suggests that any potential benefits of a multivitamin to reduce mortality are likely to be small.
They gave an example of a healthy 65-year-old woman with a 9-year estimated mortality risk of about 8%, and note that taking a multivitamin for 5-10 years might reduce her estimated mortality risk to 7.5% (based on an odds ratio of 0.94).
“In addition to showing small potential benefit, this estimate is based on imperfect evidence, is imprecise, and is highly sensitive to how the data are interpreted and analyzed,” they said.
The editorialists recommended that lifestyle counseling to prevent chronic diseases should continue to focus on evidence-based approaches, including balanced diets that are high in fruits and vegetables and physical activity.
However, they added that healthy eating can be a challenge when the American industrialized food system does not prioritize health, and healthy foods tend to be more expensive, leading to access problems and food insecurity.
The editorialists suggested that, rather than focusing money, time, and attention on supplements, it would be better to emphasize lower-risk, higher-benefit activities, such as getting exercise, maintaining a healthy weight, and avoiding smoking, in addition to following a healthful diet.
Possible benefit for older adults?
Commenting on the USPSTF statement, JoAnn Manson, MD, chief, division of preventive medicine, Brigham and Women’s Hospital, Boston, who led the recent COSMOS study, said that vitamin and mineral supplements should not be perceived as a substitute for a healthful diet.
“The emphasis needs to be on getting nutritional needs from a healthy diet that is high in plant-based and whole foods that don’t strip the vitamins and minerals through excessive processing,” she said. “Although it’s easier to pop a pill each day than to focus on healthful dietary patterns, the mixture of phytochemicals, fiber, and all the other nutrients in actual foods just can’t be packaged into a pill. Also, vitamins and minerals tend to be better absorbed from food than from supplements and healthy foods can replace calories from less healthy foods, such as red meat and processed foods.”
However, Dr. Manson noted that the evidence is mounting that taking a tablet containing moderate doses of a wide range of vitamins and minerals is safe and may actually have benefits for some people.
She pointed out that the COSMOS and COSMOS-Mind studies showed benefits of multivitamins in slowing cognitive decline in older adults, but the findings need to be replicated.
“The USPSTF did see a statistically significant 7% reduction in cancer with multivitamins in their meta-analysis of four randomized trials and a borderline 6% reduction in all-cause mortality,” she noted. “Plus, multivitamins have been shown to be quite safe in several large and long-term randomized trials. I agree the evidence is not sufficient to make a blanket recommendation for everyone to take multivitamins, but the evidence is mounting that this would be a prudent approach for many older adults,” Dr. Manson said.
“Many people view multivitamins as a form of insurance, as a way to hedge their bets,” she added. “Although this is a rational approach, especially for those who have concerns about the adequacy of their diet, it’s important that this mindset not lead to complacency about following healthy lifestyle practices, including healthy eating, regular physical activity, not smoking, making sure that blood pressure and cholesterol levels are well controlled, and many other practices that critically important for health but are more challenging than simply popping a pill each day.”
A version of this article first appeared on Medscape.com.
There is not enough evidence to recommend for or against taking most vitamin and mineral supplements to prevent heart disease, stroke, and cancer, a new report by the U.S. Preventive Services Task Force concludes.
However, there are two vitamins – vitamin E and beta-carotene – that the task force recommends against for the prevention of heart disease, stroke, and cancer. Evidence shows that there is no benefit to taking vitamin E and that beta-carotene can increase the risk for lung cancer in people already at risk, such as smokers and those with occupational exposure to asbestos.
These are the main findings of the USPSTF’s final recommendation statement on vitamin, mineral, and multivitamin supplementation to prevent cardiovascular disease and cancer. The statement was published in JAMA.
“This is essentially the same recommendation that the task force made in 2014,” USPSTF member John Wong, MD, professor of medicine at Tufts University, Boston, said in an interview.
“We recognize that over half of people in the U.S. take a vitamin supplement of some sort every day and 30% take a vitamin/mineral combination. We wanted to review the evidence again to see if there was any benefit in terms of reducing the risk of cardiovascular disease or cancer or increasing the chances of living longer,” Dr. Wong explained.
“We looked hard for evidence, reviewing 84 studies in total. But we did not find sufficient evidence in favor of taking or not taking vitamins, with the two exceptions of beta-carotene and vitamin E, which we recommend against taking,” he noted.
Although there is evidence of some harm with beta-carotene, the main reason behind the recommendation against taking vitamin E is the consistent evidence of no benefit, Dr. Wong explained.
“While the evidence for some other vitamins is conflicting, there is more consistent evidence of no benefit for vitamin E,” he said.
The bulk of new evidence since the last review in 2014 was predominately for vitamin D supplementation, but despite the inclusion of 32 new randomized, controlled trials and two cohort studies, pooled estimates for all-cause mortality were similar to those in the previous review, with confidence intervals only slightly crossing 1, and point estimates that suggest at most a very small benefit, the task force noted.
“Apart from beta-carotene and vitamin E, after reviewing 84 studies – including 78 randomized controlled trials – in over a million patients, we can find no clear demonstration of benefit or harm of taking vitamins in terms of developing cardiovascular disease or cancer or the effect on all-cause mortality. So, we don’t know whether people should take vitamins or not, and we need more research,” Dr. Wong added.
On the use of a multivitamin supplement, Dr. Wong noted that the complete body of evidence did not find any benefit of taking a multivitamin on cardiovascular or cancer mortality. But there was a small reduction in cancer incidence.
However, he pointed out that the three studies that suggested a reduction in cancer incidence all had issues regarding generalizability.
“The recently published COSMOS trial had an average follow-up of only 3.6 years, which isn’t really long enough when thinking about the prevention of cancer, one of the other studies only used antioxidants, and the third study was conducted only in U.S. male physicians. So those limitations regarding generalizability limited our confidence in making recommendations about multivitamins,” Dr. Wong explained.
But he noted that the task force did not find any significant harms from taking multivitamins.
“There are possible harms from taking high doses of vitamin A and vitamin D, but generally the doses contained in a multivitamin tablet are lower than these. But if the goal for taking a multivitamin is to lower your risk of cancer or cardiovascular disease, we didn’t find sufficient evidence to be able to make a recommendation,” he said.
Asked what he would say to all the people currently taking multivitamins, Dr. Wong responded that he would advise them to have a conversation with a trusted health care professional about their particular circumstances.
“Our statement has quite a narrow focus. It is directed toward community-dwelling, nonpregnant adults. This recommendation does not apply to children, persons who are pregnant or may become pregnant, or persons who are chronically ill, are hospitalized, or have a known nutritional deficiency,” he commented.
‘Any benefit likely to be small’
In an editorial accompanying the publication of the USPSTF statement, Jenny Jia, MD; Natalie Cameron, MD; and Jeffrey Linder, MD – all from Northwestern University, Chicago – noted that the current evidence base includes 52 additional studies not available when the last USPSTF recommendation on this topic was published in 2014.
The editorialists pointed out that for multivitamins, proving the absence of a benefit is challenging, but at best, current evidence suggests that any potential benefits of a multivitamin to reduce mortality are likely to be small.
They gave an example of a healthy 65-year-old woman with a 9-year estimated mortality risk of about 8%, and note that taking a multivitamin for 5-10 years might reduce her estimated mortality risk to 7.5% (based on an odds ratio of 0.94).
“In addition to showing small potential benefit, this estimate is based on imperfect evidence, is imprecise, and is highly sensitive to how the data are interpreted and analyzed,” they said.
The editorialists recommended that lifestyle counseling to prevent chronic diseases should continue to focus on evidence-based approaches, including balanced diets that are high in fruits and vegetables and physical activity.
However, they added that healthy eating can be a challenge when the American industrialized food system does not prioritize health, and healthy foods tend to be more expensive, leading to access problems and food insecurity.
The editorialists suggested that, rather than focusing money, time, and attention on supplements, it would be better to emphasize lower-risk, higher-benefit activities, such as getting exercise, maintaining a healthy weight, and avoiding smoking, in addition to following a healthful diet.
Possible benefit for older adults?
Commenting on the USPSTF statement, JoAnn Manson, MD, chief, division of preventive medicine, Brigham and Women’s Hospital, Boston, who led the recent COSMOS study, said that vitamin and mineral supplements should not be perceived as a substitute for a healthful diet.
“The emphasis needs to be on getting nutritional needs from a healthy diet that is high in plant-based and whole foods that don’t strip the vitamins and minerals through excessive processing,” she said. “Although it’s easier to pop a pill each day than to focus on healthful dietary patterns, the mixture of phytochemicals, fiber, and all the other nutrients in actual foods just can’t be packaged into a pill. Also, vitamins and minerals tend to be better absorbed from food than from supplements and healthy foods can replace calories from less healthy foods, such as red meat and processed foods.”
However, Dr. Manson noted that the evidence is mounting that taking a tablet containing moderate doses of a wide range of vitamins and minerals is safe and may actually have benefits for some people.
She pointed out that the COSMOS and COSMOS-Mind studies showed benefits of multivitamins in slowing cognitive decline in older adults, but the findings need to be replicated.
“The USPSTF did see a statistically significant 7% reduction in cancer with multivitamins in their meta-analysis of four randomized trials and a borderline 6% reduction in all-cause mortality,” she noted. “Plus, multivitamins have been shown to be quite safe in several large and long-term randomized trials. I agree the evidence is not sufficient to make a blanket recommendation for everyone to take multivitamins, but the evidence is mounting that this would be a prudent approach for many older adults,” Dr. Manson said.
“Many people view multivitamins as a form of insurance, as a way to hedge their bets,” she added. “Although this is a rational approach, especially for those who have concerns about the adequacy of their diet, it’s important that this mindset not lead to complacency about following healthy lifestyle practices, including healthy eating, regular physical activity, not smoking, making sure that blood pressure and cholesterol levels are well controlled, and many other practices that critically important for health but are more challenging than simply popping a pill each day.”
A version of this article first appeared on Medscape.com.
There is not enough evidence to recommend for or against taking most vitamin and mineral supplements to prevent heart disease, stroke, and cancer, a new report by the U.S. Preventive Services Task Force concludes.
However, there are two vitamins – vitamin E and beta-carotene – that the task force recommends against for the prevention of heart disease, stroke, and cancer. Evidence shows that there is no benefit to taking vitamin E and that beta-carotene can increase the risk for lung cancer in people already at risk, such as smokers and those with occupational exposure to asbestos.
These are the main findings of the USPSTF’s final recommendation statement on vitamin, mineral, and multivitamin supplementation to prevent cardiovascular disease and cancer. The statement was published in JAMA.
“This is essentially the same recommendation that the task force made in 2014,” USPSTF member John Wong, MD, professor of medicine at Tufts University, Boston, said in an interview.
“We recognize that over half of people in the U.S. take a vitamin supplement of some sort every day and 30% take a vitamin/mineral combination. We wanted to review the evidence again to see if there was any benefit in terms of reducing the risk of cardiovascular disease or cancer or increasing the chances of living longer,” Dr. Wong explained.
“We looked hard for evidence, reviewing 84 studies in total. But we did not find sufficient evidence in favor of taking or not taking vitamins, with the two exceptions of beta-carotene and vitamin E, which we recommend against taking,” he noted.
Although there is evidence of some harm with beta-carotene, the main reason behind the recommendation against taking vitamin E is the consistent evidence of no benefit, Dr. Wong explained.
“While the evidence for some other vitamins is conflicting, there is more consistent evidence of no benefit for vitamin E,” he said.
The bulk of new evidence since the last review in 2014 was predominately for vitamin D supplementation, but despite the inclusion of 32 new randomized, controlled trials and two cohort studies, pooled estimates for all-cause mortality were similar to those in the previous review, with confidence intervals only slightly crossing 1, and point estimates that suggest at most a very small benefit, the task force noted.
“Apart from beta-carotene and vitamin E, after reviewing 84 studies – including 78 randomized controlled trials – in over a million patients, we can find no clear demonstration of benefit or harm of taking vitamins in terms of developing cardiovascular disease or cancer or the effect on all-cause mortality. So, we don’t know whether people should take vitamins or not, and we need more research,” Dr. Wong added.
On the use of a multivitamin supplement, Dr. Wong noted that the complete body of evidence did not find any benefit of taking a multivitamin on cardiovascular or cancer mortality. But there was a small reduction in cancer incidence.
However, he pointed out that the three studies that suggested a reduction in cancer incidence all had issues regarding generalizability.
“The recently published COSMOS trial had an average follow-up of only 3.6 years, which isn’t really long enough when thinking about the prevention of cancer, one of the other studies only used antioxidants, and the third study was conducted only in U.S. male physicians. So those limitations regarding generalizability limited our confidence in making recommendations about multivitamins,” Dr. Wong explained.
But he noted that the task force did not find any significant harms from taking multivitamins.
“There are possible harms from taking high doses of vitamin A and vitamin D, but generally the doses contained in a multivitamin tablet are lower than these. But if the goal for taking a multivitamin is to lower your risk of cancer or cardiovascular disease, we didn’t find sufficient evidence to be able to make a recommendation,” he said.
Asked what he would say to all the people currently taking multivitamins, Dr. Wong responded that he would advise them to have a conversation with a trusted health care professional about their particular circumstances.
“Our statement has quite a narrow focus. It is directed toward community-dwelling, nonpregnant adults. This recommendation does not apply to children, persons who are pregnant or may become pregnant, or persons who are chronically ill, are hospitalized, or have a known nutritional deficiency,” he commented.
‘Any benefit likely to be small’
In an editorial accompanying the publication of the USPSTF statement, Jenny Jia, MD; Natalie Cameron, MD; and Jeffrey Linder, MD – all from Northwestern University, Chicago – noted that the current evidence base includes 52 additional studies not available when the last USPSTF recommendation on this topic was published in 2014.
The editorialists pointed out that for multivitamins, proving the absence of a benefit is challenging, but at best, current evidence suggests that any potential benefits of a multivitamin to reduce mortality are likely to be small.
They gave an example of a healthy 65-year-old woman with a 9-year estimated mortality risk of about 8%, and note that taking a multivitamin for 5-10 years might reduce her estimated mortality risk to 7.5% (based on an odds ratio of 0.94).
“In addition to showing small potential benefit, this estimate is based on imperfect evidence, is imprecise, and is highly sensitive to how the data are interpreted and analyzed,” they said.
The editorialists recommended that lifestyle counseling to prevent chronic diseases should continue to focus on evidence-based approaches, including balanced diets that are high in fruits and vegetables and physical activity.
However, they added that healthy eating can be a challenge when the American industrialized food system does not prioritize health, and healthy foods tend to be more expensive, leading to access problems and food insecurity.
The editorialists suggested that, rather than focusing money, time, and attention on supplements, it would be better to emphasize lower-risk, higher-benefit activities, such as getting exercise, maintaining a healthy weight, and avoiding smoking, in addition to following a healthful diet.
Possible benefit for older adults?
Commenting on the USPSTF statement, JoAnn Manson, MD, chief, division of preventive medicine, Brigham and Women’s Hospital, Boston, who led the recent COSMOS study, said that vitamin and mineral supplements should not be perceived as a substitute for a healthful diet.
“The emphasis needs to be on getting nutritional needs from a healthy diet that is high in plant-based and whole foods that don’t strip the vitamins and minerals through excessive processing,” she said. “Although it’s easier to pop a pill each day than to focus on healthful dietary patterns, the mixture of phytochemicals, fiber, and all the other nutrients in actual foods just can’t be packaged into a pill. Also, vitamins and minerals tend to be better absorbed from food than from supplements and healthy foods can replace calories from less healthy foods, such as red meat and processed foods.”
However, Dr. Manson noted that the evidence is mounting that taking a tablet containing moderate doses of a wide range of vitamins and minerals is safe and may actually have benefits for some people.
She pointed out that the COSMOS and COSMOS-Mind studies showed benefits of multivitamins in slowing cognitive decline in older adults, but the findings need to be replicated.
“The USPSTF did see a statistically significant 7% reduction in cancer with multivitamins in their meta-analysis of four randomized trials and a borderline 6% reduction in all-cause mortality,” she noted. “Plus, multivitamins have been shown to be quite safe in several large and long-term randomized trials. I agree the evidence is not sufficient to make a blanket recommendation for everyone to take multivitamins, but the evidence is mounting that this would be a prudent approach for many older adults,” Dr. Manson said.
“Many people view multivitamins as a form of insurance, as a way to hedge their bets,” she added. “Although this is a rational approach, especially for those who have concerns about the adequacy of their diet, it’s important that this mindset not lead to complacency about following healthy lifestyle practices, including healthy eating, regular physical activity, not smoking, making sure that blood pressure and cholesterol levels are well controlled, and many other practices that critically important for health but are more challenging than simply popping a pill each day.”
A version of this article first appeared on Medscape.com.
FROM JAMA
‘Extremely exciting’ study results guide MM treatment options
CHICAGO – New results from a trial in patients with newly diagnosed multiple myeloma (MM) offer some answers to questions about which treatment route to choose.
Patients who received the triplet of lenalidomide, bortezomib, and dexamethasone (RVD) plus ASCT had a median PFS of 67.5 months, compared with 46.2 months for those who received RVD but did not have a transplant soon after.
However, patients were just as likely to be alive more than 6 years after treatment regardless of whether or not they underwent an immediate stem cell transplant.
In addition, treatment-related adverse events of grade 3 or above were higher in the group that received the transplant immediately after the triplet therapy.
The results were presented during a plenary session at the American Society of Clinical Oncology annual meeting and simultaneously published in the New England Journal of Medicine.
“Our findings confirm the PFS benefit of transplantation as first-line treatment for patients with myeloma and confirms stem cell transplant as a standard of care with certain triplet therapy,” said lead author Paul G. Richardson, MD, professor of medicine, Harvard Medical School, and clinical program leader and director of clinical research at the Jerome Lipper Multiple Myeloma Center at Dana Farber Cancer Institute, Boston.
Another finding from the trial was that the use of maintenance lenalidomide in both groups continuously until progression conferred substantial clinical benefit.
“We can also say that the use of lenalidomide maintenance therapy is also a standard of care,” he added.
Study details
In this trial, Dr. Richardson and colleagues randomly assigned 873 patients newly diagnosed with multiple myeloma to the RVD-alone group (n = 357) or the transplantation group (n = 365). All patients had received one cycle of RVD prior to randomization and then received two additional RVD cycles plus stem-cell mobilization followed by either five additional RVD cycles (the RVD-alone group) or high-dose melphalan plus ASCT followed by two additional RVD cycles (the transplantation group). Lenalidomide was administered to all patients until disease progression, unacceptable side effects, or both.
At a median follow-up of 76.0 months, the risk of disease progression or death was 53% higher among patients who received RVD alone versus the transplantation group (hazard ratio [HR], 1.53; P < .001). The median duration of PFS among patients with a high-risk cytogenetic profile was 55.5 vs. 17.1 months, favoring the transplantation group.
The percentage of patients who were alive without progression at 5 years was 58.4% vs 41.6%, respectively (HR, 1.66) and median duration of response was 56.4 vs 38.9 months, also favoring transplantation (HR, 1.45).
The estimated 5-year overall survival was similar between groups: 80.7% for transplantation and 79.2% for RVD alone (HR for death, 1.10; P > .99). For patients with a high-risk cytogenetic profile, 5-year survival was 63.4% versus 54.3%, respectively.
“This tells us that for patients who had kept transplant in reserve, they had the same overall survival as those who had had a transplant right away, despite there being such impressive initial disease control for the patients in whom transplant was used early,” Dr. Richardson said in a press release from his institution.
Patients who did not undergo immediate transplant received treatment when their disease progressed with newer and active therapies, such as monoclonal antibodies and/or next-generation novel agents, he noted. Only 28% of patients used the reserve option of a transplant.
“It demonstrates the extent to which patients now have options and that we have new data to guide them in balancing the pluses and minuses of each approach,” he added.
When looking at safety, the authors noted that the most common treatment-related adverse events of grade 3 or higher occurred in 279 patients (78.2%) in the RVD-alone group and 344 patients (94.2%) in the transplantation group. Of those patients, 60.5% and 89.9%, respectively, reported hematologic events of grade 3 or higher (P < .001). The 5-year cumulative incidence of invasive second primary cancers was similar in both cohorts (RVD-alone group, 4.9%; transplantation group, 6.5%).
However, while the risk of secondary cancers was similar between groups, Dr. Richardson noted that there was a higher incidence of acute myeloid leukemia and myelodysplastic syndromes in the transplant cohort.
“There was also a significant drop in quality of life across transplant procedures, but the good news is that it was recoverable rapidly,” he said. “What is also really important is that we have prospective, multicenter, national comparative data on toxicity. That’s very important for providing patients with a choice as they move forward with their treatment plan.”
He noted that treatment continues to evolve. “This study was designed in 2009, begun in 2010, and now there is mature data in 2022,” Dr. Richardson said. “This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies and novel next-generation therapies. The results from these studies are extremely exciting.
“Now more than ever, treatment for multiple myeloma can be adapted for each patient,” Dr. Richardson said. “Our study provides important information about the benefits of transplant in the era of highly effective novel therapies and continuous maintenance, as well as the potential risks, to help patients and their physicians decide what approach may be best for them. This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies, such as RVD combined with daratumumab.”
Lack of difference in overall survival
These new results further support an already established role of autologous hematopoietic stem cell transplantation in the management of patients with multiple myeloma, said Samer Al-Homsi, MD, clinical professor of medicine and director of the blood and marrow transplant program at Perlmutter Cancer Center, NYU Langone, New York, who was approached for comment.
“The treatment regimen is applicable to patients who are determined by an expert in transplantation to be fit to receive autologous hematopoietic transplantation,” he added. “Although this study, like many others, establishes hematopoietic stem cell transplantation as part of the standard of care in multiple myeloma, only a fraction of patients are actually offered this important modality of treatment for a variety of reasons, including provider bias,” he noted. “In fact, although improvement in supportive care has enhanced the safety of the procedure, many patients are denied this therapy.”
Dr. Al-Homsi noted that the lack of difference in overall survival might be due to the fact that some patients (28%) in the RVD-alone group did end up undergoing transplantation at the time of progression. “Also, longer follow-up might reveal a difference in overall survival,” he said.
The toxicities are manageable, and the incidence of secondary malignancies was not significantly different between cohorts. “However,” he emphasized, “lenalidomide has been associated in other studies with increased incidence of secondary malignancies and it must be noted that this study used extended administration of lenalidomide until progression.”
Support for this study was provided by grants to the Blood and Marrow Transplant Clinical Trials Network from the National Heart, Lung, and Blood Institute, the National Cancer Institute, R. J. Corman Multiple Myeloma Foundation, Celgene/Bristol Myers Squibb, and Millennium/Takeda Pharmaceutical. Dr. Richardson has reported relationships with Celgene, Janssen, Jazz Pharmaceuticals, Karyopharm Therapeutics, Oncopeptides, Sanofi, Secura Bio, Takeda, and Bristol Myers Squibb. Dr. Al-Homsi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
CHICAGO – New results from a trial in patients with newly diagnosed multiple myeloma (MM) offer some answers to questions about which treatment route to choose.
Patients who received the triplet of lenalidomide, bortezomib, and dexamethasone (RVD) plus ASCT had a median PFS of 67.5 months, compared with 46.2 months for those who received RVD but did not have a transplant soon after.
However, patients were just as likely to be alive more than 6 years after treatment regardless of whether or not they underwent an immediate stem cell transplant.
In addition, treatment-related adverse events of grade 3 or above were higher in the group that received the transplant immediately after the triplet therapy.
The results were presented during a plenary session at the American Society of Clinical Oncology annual meeting and simultaneously published in the New England Journal of Medicine.
“Our findings confirm the PFS benefit of transplantation as first-line treatment for patients with myeloma and confirms stem cell transplant as a standard of care with certain triplet therapy,” said lead author Paul G. Richardson, MD, professor of medicine, Harvard Medical School, and clinical program leader and director of clinical research at the Jerome Lipper Multiple Myeloma Center at Dana Farber Cancer Institute, Boston.
Another finding from the trial was that the use of maintenance lenalidomide in both groups continuously until progression conferred substantial clinical benefit.
“We can also say that the use of lenalidomide maintenance therapy is also a standard of care,” he added.
Study details
In this trial, Dr. Richardson and colleagues randomly assigned 873 patients newly diagnosed with multiple myeloma to the RVD-alone group (n = 357) or the transplantation group (n = 365). All patients had received one cycle of RVD prior to randomization and then received two additional RVD cycles plus stem-cell mobilization followed by either five additional RVD cycles (the RVD-alone group) or high-dose melphalan plus ASCT followed by two additional RVD cycles (the transplantation group). Lenalidomide was administered to all patients until disease progression, unacceptable side effects, or both.
At a median follow-up of 76.0 months, the risk of disease progression or death was 53% higher among patients who received RVD alone versus the transplantation group (hazard ratio [HR], 1.53; P < .001). The median duration of PFS among patients with a high-risk cytogenetic profile was 55.5 vs. 17.1 months, favoring the transplantation group.
The percentage of patients who were alive without progression at 5 years was 58.4% vs 41.6%, respectively (HR, 1.66) and median duration of response was 56.4 vs 38.9 months, also favoring transplantation (HR, 1.45).
The estimated 5-year overall survival was similar between groups: 80.7% for transplantation and 79.2% for RVD alone (HR for death, 1.10; P > .99). For patients with a high-risk cytogenetic profile, 5-year survival was 63.4% versus 54.3%, respectively.
“This tells us that for patients who had kept transplant in reserve, they had the same overall survival as those who had had a transplant right away, despite there being such impressive initial disease control for the patients in whom transplant was used early,” Dr. Richardson said in a press release from his institution.
Patients who did not undergo immediate transplant received treatment when their disease progressed with newer and active therapies, such as monoclonal antibodies and/or next-generation novel agents, he noted. Only 28% of patients used the reserve option of a transplant.
“It demonstrates the extent to which patients now have options and that we have new data to guide them in balancing the pluses and minuses of each approach,” he added.
When looking at safety, the authors noted that the most common treatment-related adverse events of grade 3 or higher occurred in 279 patients (78.2%) in the RVD-alone group and 344 patients (94.2%) in the transplantation group. Of those patients, 60.5% and 89.9%, respectively, reported hematologic events of grade 3 or higher (P < .001). The 5-year cumulative incidence of invasive second primary cancers was similar in both cohorts (RVD-alone group, 4.9%; transplantation group, 6.5%).
However, while the risk of secondary cancers was similar between groups, Dr. Richardson noted that there was a higher incidence of acute myeloid leukemia and myelodysplastic syndromes in the transplant cohort.
“There was also a significant drop in quality of life across transplant procedures, but the good news is that it was recoverable rapidly,” he said. “What is also really important is that we have prospective, multicenter, national comparative data on toxicity. That’s very important for providing patients with a choice as they move forward with their treatment plan.”
He noted that treatment continues to evolve. “This study was designed in 2009, begun in 2010, and now there is mature data in 2022,” Dr. Richardson said. “This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies and novel next-generation therapies. The results from these studies are extremely exciting.
“Now more than ever, treatment for multiple myeloma can be adapted for each patient,” Dr. Richardson said. “Our study provides important information about the benefits of transplant in the era of highly effective novel therapies and continuous maintenance, as well as the potential risks, to help patients and their physicians decide what approach may be best for them. This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies, such as RVD combined with daratumumab.”
Lack of difference in overall survival
These new results further support an already established role of autologous hematopoietic stem cell transplantation in the management of patients with multiple myeloma, said Samer Al-Homsi, MD, clinical professor of medicine and director of the blood and marrow transplant program at Perlmutter Cancer Center, NYU Langone, New York, who was approached for comment.
“The treatment regimen is applicable to patients who are determined by an expert in transplantation to be fit to receive autologous hematopoietic transplantation,” he added. “Although this study, like many others, establishes hematopoietic stem cell transplantation as part of the standard of care in multiple myeloma, only a fraction of patients are actually offered this important modality of treatment for a variety of reasons, including provider bias,” he noted. “In fact, although improvement in supportive care has enhanced the safety of the procedure, many patients are denied this therapy.”
Dr. Al-Homsi noted that the lack of difference in overall survival might be due to the fact that some patients (28%) in the RVD-alone group did end up undergoing transplantation at the time of progression. “Also, longer follow-up might reveal a difference in overall survival,” he said.
The toxicities are manageable, and the incidence of secondary malignancies was not significantly different between cohorts. “However,” he emphasized, “lenalidomide has been associated in other studies with increased incidence of secondary malignancies and it must be noted that this study used extended administration of lenalidomide until progression.”
Support for this study was provided by grants to the Blood and Marrow Transplant Clinical Trials Network from the National Heart, Lung, and Blood Institute, the National Cancer Institute, R. J. Corman Multiple Myeloma Foundation, Celgene/Bristol Myers Squibb, and Millennium/Takeda Pharmaceutical. Dr. Richardson has reported relationships with Celgene, Janssen, Jazz Pharmaceuticals, Karyopharm Therapeutics, Oncopeptides, Sanofi, Secura Bio, Takeda, and Bristol Myers Squibb. Dr. Al-Homsi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
CHICAGO – New results from a trial in patients with newly diagnosed multiple myeloma (MM) offer some answers to questions about which treatment route to choose.
Patients who received the triplet of lenalidomide, bortezomib, and dexamethasone (RVD) plus ASCT had a median PFS of 67.5 months, compared with 46.2 months for those who received RVD but did not have a transplant soon after.
However, patients were just as likely to be alive more than 6 years after treatment regardless of whether or not they underwent an immediate stem cell transplant.
In addition, treatment-related adverse events of grade 3 or above were higher in the group that received the transplant immediately after the triplet therapy.
The results were presented during a plenary session at the American Society of Clinical Oncology annual meeting and simultaneously published in the New England Journal of Medicine.
“Our findings confirm the PFS benefit of transplantation as first-line treatment for patients with myeloma and confirms stem cell transplant as a standard of care with certain triplet therapy,” said lead author Paul G. Richardson, MD, professor of medicine, Harvard Medical School, and clinical program leader and director of clinical research at the Jerome Lipper Multiple Myeloma Center at Dana Farber Cancer Institute, Boston.
Another finding from the trial was that the use of maintenance lenalidomide in both groups continuously until progression conferred substantial clinical benefit.
“We can also say that the use of lenalidomide maintenance therapy is also a standard of care,” he added.
Study details
In this trial, Dr. Richardson and colleagues randomly assigned 873 patients newly diagnosed with multiple myeloma to the RVD-alone group (n = 357) or the transplantation group (n = 365). All patients had received one cycle of RVD prior to randomization and then received two additional RVD cycles plus stem-cell mobilization followed by either five additional RVD cycles (the RVD-alone group) or high-dose melphalan plus ASCT followed by two additional RVD cycles (the transplantation group). Lenalidomide was administered to all patients until disease progression, unacceptable side effects, or both.
At a median follow-up of 76.0 months, the risk of disease progression or death was 53% higher among patients who received RVD alone versus the transplantation group (hazard ratio [HR], 1.53; P < .001). The median duration of PFS among patients with a high-risk cytogenetic profile was 55.5 vs. 17.1 months, favoring the transplantation group.
The percentage of patients who were alive without progression at 5 years was 58.4% vs 41.6%, respectively (HR, 1.66) and median duration of response was 56.4 vs 38.9 months, also favoring transplantation (HR, 1.45).
The estimated 5-year overall survival was similar between groups: 80.7% for transplantation and 79.2% for RVD alone (HR for death, 1.10; P > .99). For patients with a high-risk cytogenetic profile, 5-year survival was 63.4% versus 54.3%, respectively.
“This tells us that for patients who had kept transplant in reserve, they had the same overall survival as those who had had a transplant right away, despite there being such impressive initial disease control for the patients in whom transplant was used early,” Dr. Richardson said in a press release from his institution.
Patients who did not undergo immediate transplant received treatment when their disease progressed with newer and active therapies, such as monoclonal antibodies and/or next-generation novel agents, he noted. Only 28% of patients used the reserve option of a transplant.
“It demonstrates the extent to which patients now have options and that we have new data to guide them in balancing the pluses and minuses of each approach,” he added.
When looking at safety, the authors noted that the most common treatment-related adverse events of grade 3 or higher occurred in 279 patients (78.2%) in the RVD-alone group and 344 patients (94.2%) in the transplantation group. Of those patients, 60.5% and 89.9%, respectively, reported hematologic events of grade 3 or higher (P < .001). The 5-year cumulative incidence of invasive second primary cancers was similar in both cohorts (RVD-alone group, 4.9%; transplantation group, 6.5%).
However, while the risk of secondary cancers was similar between groups, Dr. Richardson noted that there was a higher incidence of acute myeloid leukemia and myelodysplastic syndromes in the transplant cohort.
“There was also a significant drop in quality of life across transplant procedures, but the good news is that it was recoverable rapidly,” he said. “What is also really important is that we have prospective, multicenter, national comparative data on toxicity. That’s very important for providing patients with a choice as they move forward with their treatment plan.”
He noted that treatment continues to evolve. “This study was designed in 2009, begun in 2010, and now there is mature data in 2022,” Dr. Richardson said. “This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies and novel next-generation therapies. The results from these studies are extremely exciting.
“Now more than ever, treatment for multiple myeloma can be adapted for each patient,” Dr. Richardson said. “Our study provides important information about the benefits of transplant in the era of highly effective novel therapies and continuous maintenance, as well as the potential risks, to help patients and their physicians decide what approach may be best for them. This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies, such as RVD combined with daratumumab.”
Lack of difference in overall survival
These new results further support an already established role of autologous hematopoietic stem cell transplantation in the management of patients with multiple myeloma, said Samer Al-Homsi, MD, clinical professor of medicine and director of the blood and marrow transplant program at Perlmutter Cancer Center, NYU Langone, New York, who was approached for comment.
“The treatment regimen is applicable to patients who are determined by an expert in transplantation to be fit to receive autologous hematopoietic transplantation,” he added. “Although this study, like many others, establishes hematopoietic stem cell transplantation as part of the standard of care in multiple myeloma, only a fraction of patients are actually offered this important modality of treatment for a variety of reasons, including provider bias,” he noted. “In fact, although improvement in supportive care has enhanced the safety of the procedure, many patients are denied this therapy.”
Dr. Al-Homsi noted that the lack of difference in overall survival might be due to the fact that some patients (28%) in the RVD-alone group did end up undergoing transplantation at the time of progression. “Also, longer follow-up might reveal a difference in overall survival,” he said.
The toxicities are manageable, and the incidence of secondary malignancies was not significantly different between cohorts. “However,” he emphasized, “lenalidomide has been associated in other studies with increased incidence of secondary malignancies and it must be noted that this study used extended administration of lenalidomide until progression.”
Support for this study was provided by grants to the Blood and Marrow Transplant Clinical Trials Network from the National Heart, Lung, and Blood Institute, the National Cancer Institute, R. J. Corman Multiple Myeloma Foundation, Celgene/Bristol Myers Squibb, and Millennium/Takeda Pharmaceutical. Dr. Richardson has reported relationships with Celgene, Janssen, Jazz Pharmaceuticals, Karyopharm Therapeutics, Oncopeptides, Sanofi, Secura Bio, Takeda, and Bristol Myers Squibb. Dr. Al-Homsi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ASCO 2022
‘Very impressive’ data promise new blood cancer option
“We have limited treatment options for triple-class exposed and refractory multiple myeloma patients, especially for use in the community,” coauthor Dr. Saad Z. Usmani, of Memorial Sloan Kettering Cancer Center, New York, said in an interview. “Teclistamab is a BCMA directed bispecific antibody that is showing high response rates at the recommended subcutaneous phase 2 doses (RP2D),” and has a strong safety profile, he explained.
Teclistamab tackles two targets – both CD3 on the surface of T cells and B-cell maturation antigen (BCMA) on the surface of myeloma cells – said Dr. Ajay K. Nooka of Emory University, Atlanta, in the meeting presentation. The study was published simultaneously in the New England Journal of Medicine.
After teclistamab showed promising efficacy and an acceptable level of side effects in phase 1, researchers enrolled 165 adults aged 33-84 years with relapsed or refractory multiple myeloma (MM). The patients had experienced at least three previous lines of therapy (LOT). All patients received a weekly subcutaneous injection of 1.5 mg/kg of body weight following step-up doses of 0.06 mg/kg and 0.3 mg/kg. The primary endpoint of the study was overall response.
The median age of the patients was 64 years; 58% were male, 81.2% were White. The median prior LOT was 5; all of the patients were triple-class exposed (100%); 70% were penta-drug exposed, 78% were triple-class refractory, and 30% penta-drug refractory.
The overall response rate (ORR) was 63% over a median follow-up period of approximately 14.1 months. In addition, 39.4% of patients had a complete response or better, and 26.7% had no minimal residual disease, for a negative minimal residual disease rate of 46.2% in patients with complete response. The median durations of response and progression-free survival were 18.4 months and 11.3 months, respectively.
“The ORR was consistent across clinically relevant subgroups, including high cytogenetic risk and penta-drug refractory subgroups,” Dr. Nooka said in his presentation.
The most common adverse event was cytokine release syndrome, which occurred in 72.1% of patients; however, only 0.6% of these events were grade 3, and none were grade 4. Other adverse events included neutropenia in 70.9% (64.2% of events were grade 3 or 4), anemia (52.1%, 37.0% of events were grade 3 or 4, respectively) and thrombocytopenia (40%, 21.2% of events were grade 3 or 4). Infections occurred in 76.4% of patients overall, 44.8% of which were grade 3 or 4, and neurotoxic events occurred in 24 patients (14.5%). The five cases of immune effector cell–associated neurotoxicity syndrome (CRS) were grade 1 or 2.
A total of 2 patients (1.2%) discontinued the study because of adverse events, but no discontinuations or dose reductions occurred as a result of neurotoxic events.
A total of 5 deaths attributed to teclistamab occurred during the study: 2 caused by COVID-19, 1 pneumonia, 1 hepatic failure, and 1 progressive multifocal leukoencephalopathy (PML).
The responses were durable and persisted over time, said Dr. Nooka. At the point of data cutoff, 64.4% of patients who responded maintained that response.
Overall, the data support teclistamab as “a promising new, off-the-shelf, T-cell redirecting therapy targeting BCMA for patients with relapsed or refractory MM,” with phase 3 studies ongoing and early access programs in progress, Dr. Nooka concluded.
“The ORR and durability of response seen with teclistamab is very impressive when one sees the data for other single agents approved for relapsed/refractory MM in the past,” Dr. Usmani said in an interview. “I hope the current data will help get a regulatory approval for the triple class exposed MM population.”
However, potential barriers to widespread use of teclistamab in practice include logistics and a learning curve for practicing hematologists/oncologists, Dr. Usmani noted. “While the CRS appears to be grade 1 or 2 and very manageable, the logistics of giving bispecific antibodies in the community setting and managing CRS during the first cycle of therapy in the community will need to be worked out, and partnership with academic centers that have experience in managing these patients will be needed, he added.
As for additional research, “teclistamab is being combined with other MM therapies and being explored in earlier lines of treatment,” Dr. Usmani said.
Be ready to manage infections
Despite promising early findings, the use of teclistamab and other BCMA-targeting biospecific therapies is “not a free lunch” for refractory and relapsed MM patients, said discussant Dr. Madhav V. Dhodapkar of Emory University, Atlanta, during the discussion period after the ASCO presentation.
Although the risk of CRS and ICANS appears low, “infections are emerging as a major adverse event” that need to be recognized and managed, he said.
A distinct pattern of infections may be emerging, based on data from the current study and other studies of similar therapies, with infections such as Pneumocystis jirovecii (PJP) and cytomegalovirus (CMV) reactivation, Dr. Dhodapkar added.
He noted other considerations for studies of teclistamab and similar therapies, including the need to address both host-related and tumor-related factors, as well as seasonal and opportunistic threats such as COVID-19.
Future research questions include whether there is a role for pathogen-specific surveillance to help mitigate infection risk, including COVID-19 risk management strategies, he emphasized.
The study was funded by Janssen Research and Development.
Dr. Usmani disclosed relationships as a consultant or advisor, speakers’ bureau member, and/or recipient of research funding from serving as a consultant or advisor for Abbvie, Amgen, Bristol-Myers Squibb/Celgene, Celgene, Genentech, Gilead Sciences, GlaxoSmithKline, Janssen Oncology, Karyopharm Therapeutics, Merck, Oncopeptides, Seattle Genetics, Skyline Diagnostics, and Takeda. Lead author of the New England Journal paper Dr. Philippe Moreau disclosed relationships with companies including Abbvie, Amgen, Celgene, GlaxoSmithKline, Janssen-Cilag, Oncopeptides, and Sanofi. ASCO presenting author Dr. Nooka disclosed relationships with companies including Adaptive Biotechnologies, Amgen, BeyondSpring Pharmaceuticals, Bristol-Myers Squibb/Celgene, Genzyme, GlaxoSmithKline, Janssen Oncology, Karyopharm Therapeutics, Oncopeptides, Secura Bio, Arch Oncology, and Takeda.
“We have limited treatment options for triple-class exposed and refractory multiple myeloma patients, especially for use in the community,” coauthor Dr. Saad Z. Usmani, of Memorial Sloan Kettering Cancer Center, New York, said in an interview. “Teclistamab is a BCMA directed bispecific antibody that is showing high response rates at the recommended subcutaneous phase 2 doses (RP2D),” and has a strong safety profile, he explained.
Teclistamab tackles two targets – both CD3 on the surface of T cells and B-cell maturation antigen (BCMA) on the surface of myeloma cells – said Dr. Ajay K. Nooka of Emory University, Atlanta, in the meeting presentation. The study was published simultaneously in the New England Journal of Medicine.
After teclistamab showed promising efficacy and an acceptable level of side effects in phase 1, researchers enrolled 165 adults aged 33-84 years with relapsed or refractory multiple myeloma (MM). The patients had experienced at least three previous lines of therapy (LOT). All patients received a weekly subcutaneous injection of 1.5 mg/kg of body weight following step-up doses of 0.06 mg/kg and 0.3 mg/kg. The primary endpoint of the study was overall response.
The median age of the patients was 64 years; 58% were male, 81.2% were White. The median prior LOT was 5; all of the patients were triple-class exposed (100%); 70% were penta-drug exposed, 78% were triple-class refractory, and 30% penta-drug refractory.
The overall response rate (ORR) was 63% over a median follow-up period of approximately 14.1 months. In addition, 39.4% of patients had a complete response or better, and 26.7% had no minimal residual disease, for a negative minimal residual disease rate of 46.2% in patients with complete response. The median durations of response and progression-free survival were 18.4 months and 11.3 months, respectively.
“The ORR was consistent across clinically relevant subgroups, including high cytogenetic risk and penta-drug refractory subgroups,” Dr. Nooka said in his presentation.
The most common adverse event was cytokine release syndrome, which occurred in 72.1% of patients; however, only 0.6% of these events were grade 3, and none were grade 4. Other adverse events included neutropenia in 70.9% (64.2% of events were grade 3 or 4), anemia (52.1%, 37.0% of events were grade 3 or 4, respectively) and thrombocytopenia (40%, 21.2% of events were grade 3 or 4). Infections occurred in 76.4% of patients overall, 44.8% of which were grade 3 or 4, and neurotoxic events occurred in 24 patients (14.5%). The five cases of immune effector cell–associated neurotoxicity syndrome (CRS) were grade 1 or 2.
A total of 2 patients (1.2%) discontinued the study because of adverse events, but no discontinuations or dose reductions occurred as a result of neurotoxic events.
A total of 5 deaths attributed to teclistamab occurred during the study: 2 caused by COVID-19, 1 pneumonia, 1 hepatic failure, and 1 progressive multifocal leukoencephalopathy (PML).
The responses were durable and persisted over time, said Dr. Nooka. At the point of data cutoff, 64.4% of patients who responded maintained that response.
Overall, the data support teclistamab as “a promising new, off-the-shelf, T-cell redirecting therapy targeting BCMA for patients with relapsed or refractory MM,” with phase 3 studies ongoing and early access programs in progress, Dr. Nooka concluded.
“The ORR and durability of response seen with teclistamab is very impressive when one sees the data for other single agents approved for relapsed/refractory MM in the past,” Dr. Usmani said in an interview. “I hope the current data will help get a regulatory approval for the triple class exposed MM population.”
However, potential barriers to widespread use of teclistamab in practice include logistics and a learning curve for practicing hematologists/oncologists, Dr. Usmani noted. “While the CRS appears to be grade 1 or 2 and very manageable, the logistics of giving bispecific antibodies in the community setting and managing CRS during the first cycle of therapy in the community will need to be worked out, and partnership with academic centers that have experience in managing these patients will be needed, he added.
As for additional research, “teclistamab is being combined with other MM therapies and being explored in earlier lines of treatment,” Dr. Usmani said.
Be ready to manage infections
Despite promising early findings, the use of teclistamab and other BCMA-targeting biospecific therapies is “not a free lunch” for refractory and relapsed MM patients, said discussant Dr. Madhav V. Dhodapkar of Emory University, Atlanta, during the discussion period after the ASCO presentation.
Although the risk of CRS and ICANS appears low, “infections are emerging as a major adverse event” that need to be recognized and managed, he said.
A distinct pattern of infections may be emerging, based on data from the current study and other studies of similar therapies, with infections such as Pneumocystis jirovecii (PJP) and cytomegalovirus (CMV) reactivation, Dr. Dhodapkar added.
He noted other considerations for studies of teclistamab and similar therapies, including the need to address both host-related and tumor-related factors, as well as seasonal and opportunistic threats such as COVID-19.
Future research questions include whether there is a role for pathogen-specific surveillance to help mitigate infection risk, including COVID-19 risk management strategies, he emphasized.
The study was funded by Janssen Research and Development.
Dr. Usmani disclosed relationships as a consultant or advisor, speakers’ bureau member, and/or recipient of research funding from serving as a consultant or advisor for Abbvie, Amgen, Bristol-Myers Squibb/Celgene, Celgene, Genentech, Gilead Sciences, GlaxoSmithKline, Janssen Oncology, Karyopharm Therapeutics, Merck, Oncopeptides, Seattle Genetics, Skyline Diagnostics, and Takeda. Lead author of the New England Journal paper Dr. Philippe Moreau disclosed relationships with companies including Abbvie, Amgen, Celgene, GlaxoSmithKline, Janssen-Cilag, Oncopeptides, and Sanofi. ASCO presenting author Dr. Nooka disclosed relationships with companies including Adaptive Biotechnologies, Amgen, BeyondSpring Pharmaceuticals, Bristol-Myers Squibb/Celgene, Genzyme, GlaxoSmithKline, Janssen Oncology, Karyopharm Therapeutics, Oncopeptides, Secura Bio, Arch Oncology, and Takeda.
“We have limited treatment options for triple-class exposed and refractory multiple myeloma patients, especially for use in the community,” coauthor Dr. Saad Z. Usmani, of Memorial Sloan Kettering Cancer Center, New York, said in an interview. “Teclistamab is a BCMA directed bispecific antibody that is showing high response rates at the recommended subcutaneous phase 2 doses (RP2D),” and has a strong safety profile, he explained.
Teclistamab tackles two targets – both CD3 on the surface of T cells and B-cell maturation antigen (BCMA) on the surface of myeloma cells – said Dr. Ajay K. Nooka of Emory University, Atlanta, in the meeting presentation. The study was published simultaneously in the New England Journal of Medicine.
After teclistamab showed promising efficacy and an acceptable level of side effects in phase 1, researchers enrolled 165 adults aged 33-84 years with relapsed or refractory multiple myeloma (MM). The patients had experienced at least three previous lines of therapy (LOT). All patients received a weekly subcutaneous injection of 1.5 mg/kg of body weight following step-up doses of 0.06 mg/kg and 0.3 mg/kg. The primary endpoint of the study was overall response.
The median age of the patients was 64 years; 58% were male, 81.2% were White. The median prior LOT was 5; all of the patients were triple-class exposed (100%); 70% were penta-drug exposed, 78% were triple-class refractory, and 30% penta-drug refractory.
The overall response rate (ORR) was 63% over a median follow-up period of approximately 14.1 months. In addition, 39.4% of patients had a complete response or better, and 26.7% had no minimal residual disease, for a negative minimal residual disease rate of 46.2% in patients with complete response. The median durations of response and progression-free survival were 18.4 months and 11.3 months, respectively.
“The ORR was consistent across clinically relevant subgroups, including high cytogenetic risk and penta-drug refractory subgroups,” Dr. Nooka said in his presentation.
The most common adverse event was cytokine release syndrome, which occurred in 72.1% of patients; however, only 0.6% of these events were grade 3, and none were grade 4. Other adverse events included neutropenia in 70.9% (64.2% of events were grade 3 or 4), anemia (52.1%, 37.0% of events were grade 3 or 4, respectively) and thrombocytopenia (40%, 21.2% of events were grade 3 or 4). Infections occurred in 76.4% of patients overall, 44.8% of which were grade 3 or 4, and neurotoxic events occurred in 24 patients (14.5%). The five cases of immune effector cell–associated neurotoxicity syndrome (CRS) were grade 1 or 2.
A total of 2 patients (1.2%) discontinued the study because of adverse events, but no discontinuations or dose reductions occurred as a result of neurotoxic events.
A total of 5 deaths attributed to teclistamab occurred during the study: 2 caused by COVID-19, 1 pneumonia, 1 hepatic failure, and 1 progressive multifocal leukoencephalopathy (PML).
The responses were durable and persisted over time, said Dr. Nooka. At the point of data cutoff, 64.4% of patients who responded maintained that response.
Overall, the data support teclistamab as “a promising new, off-the-shelf, T-cell redirecting therapy targeting BCMA for patients with relapsed or refractory MM,” with phase 3 studies ongoing and early access programs in progress, Dr. Nooka concluded.
“The ORR and durability of response seen with teclistamab is very impressive when one sees the data for other single agents approved for relapsed/refractory MM in the past,” Dr. Usmani said in an interview. “I hope the current data will help get a regulatory approval for the triple class exposed MM population.”
However, potential barriers to widespread use of teclistamab in practice include logistics and a learning curve for practicing hematologists/oncologists, Dr. Usmani noted. “While the CRS appears to be grade 1 or 2 and very manageable, the logistics of giving bispecific antibodies in the community setting and managing CRS during the first cycle of therapy in the community will need to be worked out, and partnership with academic centers that have experience in managing these patients will be needed, he added.
As for additional research, “teclistamab is being combined with other MM therapies and being explored in earlier lines of treatment,” Dr. Usmani said.
Be ready to manage infections
Despite promising early findings, the use of teclistamab and other BCMA-targeting biospecific therapies is “not a free lunch” for refractory and relapsed MM patients, said discussant Dr. Madhav V. Dhodapkar of Emory University, Atlanta, during the discussion period after the ASCO presentation.
Although the risk of CRS and ICANS appears low, “infections are emerging as a major adverse event” that need to be recognized and managed, he said.
A distinct pattern of infections may be emerging, based on data from the current study and other studies of similar therapies, with infections such as Pneumocystis jirovecii (PJP) and cytomegalovirus (CMV) reactivation, Dr. Dhodapkar added.
He noted other considerations for studies of teclistamab and similar therapies, including the need to address both host-related and tumor-related factors, as well as seasonal and opportunistic threats such as COVID-19.
Future research questions include whether there is a role for pathogen-specific surveillance to help mitigate infection risk, including COVID-19 risk management strategies, he emphasized.
The study was funded by Janssen Research and Development.
Dr. Usmani disclosed relationships as a consultant or advisor, speakers’ bureau member, and/or recipient of research funding from serving as a consultant or advisor for Abbvie, Amgen, Bristol-Myers Squibb/Celgene, Celgene, Genentech, Gilead Sciences, GlaxoSmithKline, Janssen Oncology, Karyopharm Therapeutics, Merck, Oncopeptides, Seattle Genetics, Skyline Diagnostics, and Takeda. Lead author of the New England Journal paper Dr. Philippe Moreau disclosed relationships with companies including Abbvie, Amgen, Celgene, GlaxoSmithKline, Janssen-Cilag, Oncopeptides, and Sanofi. ASCO presenting author Dr. Nooka disclosed relationships with companies including Adaptive Biotechnologies, Amgen, BeyondSpring Pharmaceuticals, Bristol-Myers Squibb/Celgene, Genzyme, GlaxoSmithKline, Janssen Oncology, Karyopharm Therapeutics, Oncopeptides, Secura Bio, Arch Oncology, and Takeda.
FROM ASCO 2022