User login
Minidose edoxaban may safely cut AFib stroke risk in the frail, very elderly
suggests a randomized trial conducted in Japan.
Many of the study’s 984 mostly octogenarian patients were objectively frail with poor renal function, low body weight, a history of serious bleeding, or other conditions that made them poor candidates for regular-dose oral anticoagulation. Yet those who took the factor Xa inhibitor edoxaban (Savaysa) at the off-label dosage of 15 mg once daily showed a two-thirds drop in risk for stroke or systemic embolism (P < .001), compared with patients who received placebo. There were no fatal bleeds and virtually no intracranial hemorrhages.
For such high-risk patients with nonvalvular AFib who otherwise would not be given an OAC, edoxaban 15 mg “can be an acceptable treatment option in decreasing the risk of devastating stroke”; however, “it may increase the risk of gastrointestinal bleeding, so care should be given in every patient,” said Ken Okumura, MD, PhD. Indeed, the rate of gastrointestinal bleeding tripled among the patients who received edoxaban, compared with those given placebo, at about 2.3% per year versus 0.8% per year.
Although their 87% increased risk for major bleeding did not reach significance, it hit close, with a P value of .09 in the trial, called Edoxaban Low-Dose for Elder Care Atrial Fibrillation Patients (ELDERCARE-AF).
Dr. Okumura, of Saiseikai Kumamoto (Japan) Hospital, presented the study August 30 during the virtual annual congress of the European Society of Cardiology. He is lead author of an article describing the study, which was simultaneously published in the New England Journal of Medicine.
Many patients with AFib suffer strokes if they are not given oral anticoagulation because of “fear of major bleeding caused by standard OAC therapy,” Dr. Okumura noted. Others are inappropriately administered antiplatelets or anticoagulants at conventional dosages. “There is no standard of practice in Japan for patients like those in the present trial,” Dr. Okumura said. “However, I believe the present study opens a new possible path of thromboprophylaxis in such high-risk patients.”
Even with its relatively few bleeding events, ELDERCARE-AF “does suggest that the risk of the worst types of bleeds is not that high,” said Daniel E. Singer, MD, of Massachusetts General Hospital, Boston. “Gastrointestinal bleeding is annoying, and it will probably stop people from taking their edoxaban, but for the most part it doesn’t kill people.”
Moreover, he added, the trial suggests that low-dose edoxaban, in exchange for a steep reduction in thromboembolic risk, “doesn’t add to your risk of intracranial hemorrhage!”
ELDERCARE-AF may give practitioners “yet another reason to rethink” whether a low-dose DOAC such as edoxaban 15 mg/day may well be a good approach for such patients with AFib who are not receiving standard-dose OAC because of a perceived high risk for serious bleeding, said Dr. Singer, who was not involved in the study.
The trial randomly and evenly assigned 984 patients with AF in Japan to take either edoxaban 15 mg/day or placebo. The patients, who were at least 80 years old and had a CHADS2 score of 2 or higher, were judged inappropriate candidates for OAC at dosages approved for stroke prevention.
The mean age of the patients was 86.6, more than a decade older than patients “in the previous landmark clinical trials of direct oral anticoagulants,” and were 5-10 years older than the general AFib population, reported Dr. Okumura and colleagues.
Their mean weight was 52 kg, and mean creatinine clearance was 36.3 mL/min; 41% were classified as frail according to validated assessment tools.
Of the 303 patients who did not complete the trial, 158 voluntarily withdrew for various reasons. The withdrawal rate was similar in the two treatment arms. Outcomes were analyzed by intention to treat, the report noted.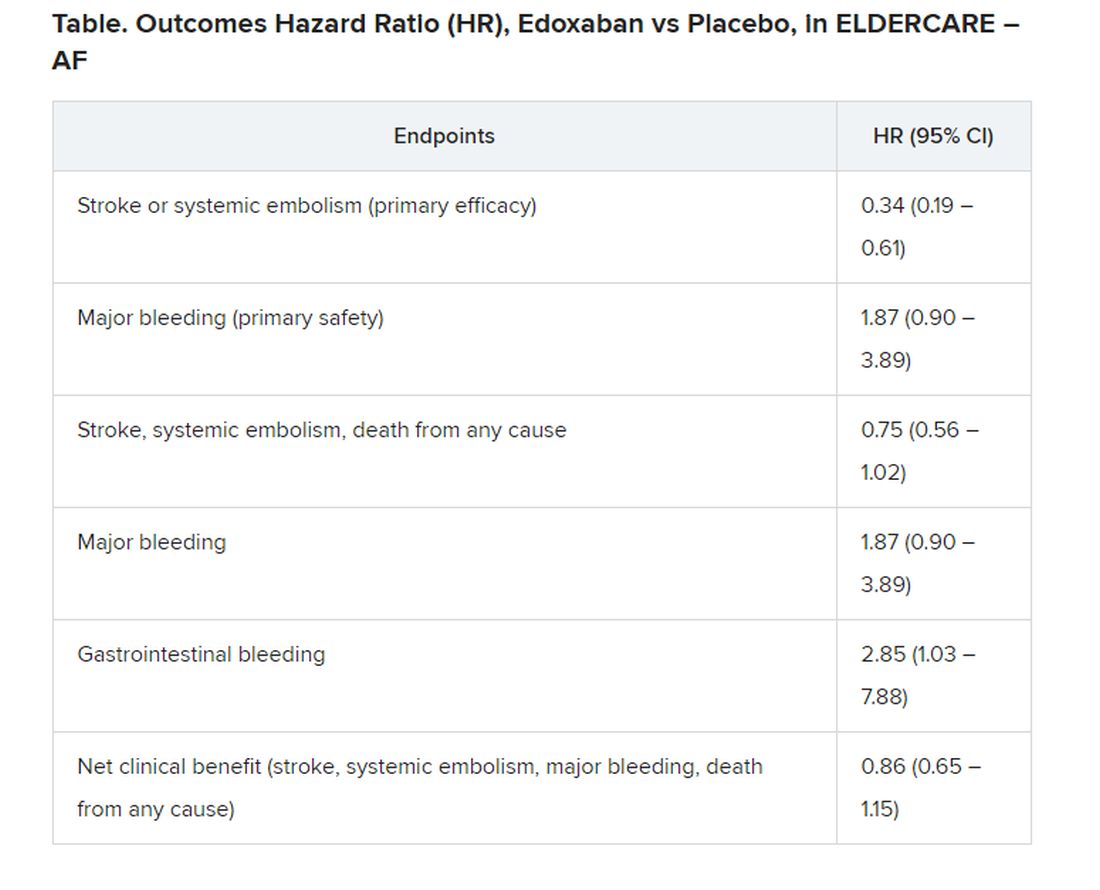
The annualized rate of stroke or systemic embolism, the primary efficacy endpoint, was 2.3% for those who received edoxaban and 6.7% for the control group. Corresponding rates for the primary safety endpoint, major bleeding as determined by International Society on Thrombosis and Hemostasis criteria, were 3.3% and 1.8%, respectively.
“The question is, can the Food and Drug Administration act on this information? I doubt it can. What will be needed is to reproduce the study in a U.S. population to see if it holds,” Dr. Singer proposed.
“Edoxaban isn’t used much in the U.S. This could heighten interest. And who knows, there may be a gold rush,” he said, if the strategy were to pan out for the other DOACs, rivaroxaban (Xarelto), apixaban (Eliquis), and dabigatran (Pradaxa).
ELDERCARE-AF was funded by Daiichi Sankyo, from which Dr. Okumura reported receiving grants and personal fees; he also disclosed personal fees from Daiichi Sankyo, Boehringer Ingelheim, Bristol-Myers Squibb, Medtronic, Johnson & Johnson, and Bayer.
A version of this article originally appeared on Medscape.com.
suggests a randomized trial conducted in Japan.
Many of the study’s 984 mostly octogenarian patients were objectively frail with poor renal function, low body weight, a history of serious bleeding, or other conditions that made them poor candidates for regular-dose oral anticoagulation. Yet those who took the factor Xa inhibitor edoxaban (Savaysa) at the off-label dosage of 15 mg once daily showed a two-thirds drop in risk for stroke or systemic embolism (P < .001), compared with patients who received placebo. There were no fatal bleeds and virtually no intracranial hemorrhages.
For such high-risk patients with nonvalvular AFib who otherwise would not be given an OAC, edoxaban 15 mg “can be an acceptable treatment option in decreasing the risk of devastating stroke”; however, “it may increase the risk of gastrointestinal bleeding, so care should be given in every patient,” said Ken Okumura, MD, PhD. Indeed, the rate of gastrointestinal bleeding tripled among the patients who received edoxaban, compared with those given placebo, at about 2.3% per year versus 0.8% per year.
Although their 87% increased risk for major bleeding did not reach significance, it hit close, with a P value of .09 in the trial, called Edoxaban Low-Dose for Elder Care Atrial Fibrillation Patients (ELDERCARE-AF).
Dr. Okumura, of Saiseikai Kumamoto (Japan) Hospital, presented the study August 30 during the virtual annual congress of the European Society of Cardiology. He is lead author of an article describing the study, which was simultaneously published in the New England Journal of Medicine.
Many patients with AFib suffer strokes if they are not given oral anticoagulation because of “fear of major bleeding caused by standard OAC therapy,” Dr. Okumura noted. Others are inappropriately administered antiplatelets or anticoagulants at conventional dosages. “There is no standard of practice in Japan for patients like those in the present trial,” Dr. Okumura said. “However, I believe the present study opens a new possible path of thromboprophylaxis in such high-risk patients.”
Even with its relatively few bleeding events, ELDERCARE-AF “does suggest that the risk of the worst types of bleeds is not that high,” said Daniel E. Singer, MD, of Massachusetts General Hospital, Boston. “Gastrointestinal bleeding is annoying, and it will probably stop people from taking their edoxaban, but for the most part it doesn’t kill people.”
Moreover, he added, the trial suggests that low-dose edoxaban, in exchange for a steep reduction in thromboembolic risk, “doesn’t add to your risk of intracranial hemorrhage!”
ELDERCARE-AF may give practitioners “yet another reason to rethink” whether a low-dose DOAC such as edoxaban 15 mg/day may well be a good approach for such patients with AFib who are not receiving standard-dose OAC because of a perceived high risk for serious bleeding, said Dr. Singer, who was not involved in the study.
The trial randomly and evenly assigned 984 patients with AF in Japan to take either edoxaban 15 mg/day or placebo. The patients, who were at least 80 years old and had a CHADS2 score of 2 or higher, were judged inappropriate candidates for OAC at dosages approved for stroke prevention.
The mean age of the patients was 86.6, more than a decade older than patients “in the previous landmark clinical trials of direct oral anticoagulants,” and were 5-10 years older than the general AFib population, reported Dr. Okumura and colleagues.
Their mean weight was 52 kg, and mean creatinine clearance was 36.3 mL/min; 41% were classified as frail according to validated assessment tools.
Of the 303 patients who did not complete the trial, 158 voluntarily withdrew for various reasons. The withdrawal rate was similar in the two treatment arms. Outcomes were analyzed by intention to treat, the report noted.
The annualized rate of stroke or systemic embolism, the primary efficacy endpoint, was 2.3% for those who received edoxaban and 6.7% for the control group. Corresponding rates for the primary safety endpoint, major bleeding as determined by International Society on Thrombosis and Hemostasis criteria, were 3.3% and 1.8%, respectively.
“The question is, can the Food and Drug Administration act on this information? I doubt it can. What will be needed is to reproduce the study in a U.S. population to see if it holds,” Dr. Singer proposed.
“Edoxaban isn’t used much in the U.S. This could heighten interest. And who knows, there may be a gold rush,” he said, if the strategy were to pan out for the other DOACs, rivaroxaban (Xarelto), apixaban (Eliquis), and dabigatran (Pradaxa).
ELDERCARE-AF was funded by Daiichi Sankyo, from which Dr. Okumura reported receiving grants and personal fees; he also disclosed personal fees from Daiichi Sankyo, Boehringer Ingelheim, Bristol-Myers Squibb, Medtronic, Johnson & Johnson, and Bayer.
A version of this article originally appeared on Medscape.com.
suggests a randomized trial conducted in Japan.
Many of the study’s 984 mostly octogenarian patients were objectively frail with poor renal function, low body weight, a history of serious bleeding, or other conditions that made them poor candidates for regular-dose oral anticoagulation. Yet those who took the factor Xa inhibitor edoxaban (Savaysa) at the off-label dosage of 15 mg once daily showed a two-thirds drop in risk for stroke or systemic embolism (P < .001), compared with patients who received placebo. There were no fatal bleeds and virtually no intracranial hemorrhages.
For such high-risk patients with nonvalvular AFib who otherwise would not be given an OAC, edoxaban 15 mg “can be an acceptable treatment option in decreasing the risk of devastating stroke”; however, “it may increase the risk of gastrointestinal bleeding, so care should be given in every patient,” said Ken Okumura, MD, PhD. Indeed, the rate of gastrointestinal bleeding tripled among the patients who received edoxaban, compared with those given placebo, at about 2.3% per year versus 0.8% per year.
Although their 87% increased risk for major bleeding did not reach significance, it hit close, with a P value of .09 in the trial, called Edoxaban Low-Dose for Elder Care Atrial Fibrillation Patients (ELDERCARE-AF).
Dr. Okumura, of Saiseikai Kumamoto (Japan) Hospital, presented the study August 30 during the virtual annual congress of the European Society of Cardiology. He is lead author of an article describing the study, which was simultaneously published in the New England Journal of Medicine.
Many patients with AFib suffer strokes if they are not given oral anticoagulation because of “fear of major bleeding caused by standard OAC therapy,” Dr. Okumura noted. Others are inappropriately administered antiplatelets or anticoagulants at conventional dosages. “There is no standard of practice in Japan for patients like those in the present trial,” Dr. Okumura said. “However, I believe the present study opens a new possible path of thromboprophylaxis in such high-risk patients.”
Even with its relatively few bleeding events, ELDERCARE-AF “does suggest that the risk of the worst types of bleeds is not that high,” said Daniel E. Singer, MD, of Massachusetts General Hospital, Boston. “Gastrointestinal bleeding is annoying, and it will probably stop people from taking their edoxaban, but for the most part it doesn’t kill people.”
Moreover, he added, the trial suggests that low-dose edoxaban, in exchange for a steep reduction in thromboembolic risk, “doesn’t add to your risk of intracranial hemorrhage!”
ELDERCARE-AF may give practitioners “yet another reason to rethink” whether a low-dose DOAC such as edoxaban 15 mg/day may well be a good approach for such patients with AFib who are not receiving standard-dose OAC because of a perceived high risk for serious bleeding, said Dr. Singer, who was not involved in the study.
The trial randomly and evenly assigned 984 patients with AF in Japan to take either edoxaban 15 mg/day or placebo. The patients, who were at least 80 years old and had a CHADS2 score of 2 or higher, were judged inappropriate candidates for OAC at dosages approved for stroke prevention.
The mean age of the patients was 86.6, more than a decade older than patients “in the previous landmark clinical trials of direct oral anticoagulants,” and were 5-10 years older than the general AFib population, reported Dr. Okumura and colleagues.
Their mean weight was 52 kg, and mean creatinine clearance was 36.3 mL/min; 41% were classified as frail according to validated assessment tools.
Of the 303 patients who did not complete the trial, 158 voluntarily withdrew for various reasons. The withdrawal rate was similar in the two treatment arms. Outcomes were analyzed by intention to treat, the report noted.
The annualized rate of stroke or systemic embolism, the primary efficacy endpoint, was 2.3% for those who received edoxaban and 6.7% for the control group. Corresponding rates for the primary safety endpoint, major bleeding as determined by International Society on Thrombosis and Hemostasis criteria, were 3.3% and 1.8%, respectively.
“The question is, can the Food and Drug Administration act on this information? I doubt it can. What will be needed is to reproduce the study in a U.S. population to see if it holds,” Dr. Singer proposed.
“Edoxaban isn’t used much in the U.S. This could heighten interest. And who knows, there may be a gold rush,” he said, if the strategy were to pan out for the other DOACs, rivaroxaban (Xarelto), apixaban (Eliquis), and dabigatran (Pradaxa).
ELDERCARE-AF was funded by Daiichi Sankyo, from which Dr. Okumura reported receiving grants and personal fees; he also disclosed personal fees from Daiichi Sankyo, Boehringer Ingelheim, Bristol-Myers Squibb, Medtronic, Johnson & Johnson, and Bayer.
A version of this article originally appeared on Medscape.com.
FROM ESC CONGRESS 2020
Nightmares: An independent risk factor for heart disease?
, new research shows. In what researchers describe as “surprising” findings, results from a large study of relatively young military veterans showed those who had nightmares two or more times per week had significantly increased risks for hypertension, myocardial infarction, or other heart problems.
“A diagnosis of PTSD incorporates sleep disturbance as a symptom. Thus, we were surprised to find that nightmares continued to be associated with CVD after controlling not only for PTSD and demographic factors, but also smoking and depression diagnosis,” said Christi Ulmer, PhD, of the department of psychiatry and behavioral sciences, Duke University Medical Center, Durham, N.C.
The findings were presented at the virtual annual meeting of the Associated Professional Sleep Societies.
Unclear mechanism
The study included 3,468 veterans (77% male) with a mean age of 38 years who had served one or two tours of duty since Sept. 11, 2001. Nearly one-third (31%) met criteria for PTSD, and 33% self-reported having at least one cardiovascular condition, such as heart problems, hypertension, stroke, and MI.
Nightmare frequency and severity was assessed using the Davidson Trauma Scale. Nightmares were considered frequent if they occurred two or more times per week and moderate to severe if they were at least moderately distressing. About 31% of veterans reported having frequent nightmares, and 35% reported moderately distressing nightmares over the past week.
After adjusting for age, race, and sex, frequent nightmares were associated with hypertension (odds ratio, 1.51; 95% confidence interval, 1.28-1.78), heart problems (OR, 1.50; 95% CI, 1.11-2.02), and MI (OR, 2.32; 95% CI, 1.18-4.54).
Associations between frequent nightmares and hypertension (OR, 1.43; 95% CI, 1.17-1.73) and heart problems (OR, 1.43; 95% CI, 1.00-2.05) remained significant after further adjusting for smoking, depression, and PTSD.
“Our cross-sectional findings set the stage for future research examining the possibility that nightmares may confer cardiovascular disease risks beyond those conferred by PTSD diagnosis alone,” Dr. Ulmer said in a news release.
Dr. Ulmer also said that, because the study was based on self-reported data, the findings are “very preliminary.” Before doctors adjust clinical practices, it’s important that our findings be replicated using longitudinal studies, clinically diagnosed medical conditions, and objectively assessed sleep,” she said.
She added that more research is needed to uncover mechanisms explaining these associations and determine if reducing the frequency and severity of nightmares can lead to improved cardiovascular health.
Timely research
Reached for comment, Rajkumar (Raj) Dasgupta, MD, of the University of Southern California, Los Angeles, noted “the correlation between nightmares and heart disease is a timely topic right now with COVID-19 as more people may be having nightmares.”
“If a patient mentions nightmares, I do think it’s important not to just glaze over it, but to talk more about it and document it in the patient record, especially in patients with cardiovascular disease, atrial fibrillation, diabetes, and hypertension,” said Dr. Dasgupta, who wasn’t involved in the study.
The research was supported by the Veterans Integrated Service Network 6 Mental Illness Research, Education and Clinical Center and the Department of Veterans Affairs HSR&D ADAPT Center at the Durham VA Health Care System. Dr. Ulmer and Dr. Dasgupta have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
, new research shows. In what researchers describe as “surprising” findings, results from a large study of relatively young military veterans showed those who had nightmares two or more times per week had significantly increased risks for hypertension, myocardial infarction, or other heart problems.
“A diagnosis of PTSD incorporates sleep disturbance as a symptom. Thus, we were surprised to find that nightmares continued to be associated with CVD after controlling not only for PTSD and demographic factors, but also smoking and depression diagnosis,” said Christi Ulmer, PhD, of the department of psychiatry and behavioral sciences, Duke University Medical Center, Durham, N.C.
The findings were presented at the virtual annual meeting of the Associated Professional Sleep Societies.
Unclear mechanism
The study included 3,468 veterans (77% male) with a mean age of 38 years who had served one or two tours of duty since Sept. 11, 2001. Nearly one-third (31%) met criteria for PTSD, and 33% self-reported having at least one cardiovascular condition, such as heart problems, hypertension, stroke, and MI.
Nightmare frequency and severity was assessed using the Davidson Trauma Scale. Nightmares were considered frequent if they occurred two or more times per week and moderate to severe if they were at least moderately distressing. About 31% of veterans reported having frequent nightmares, and 35% reported moderately distressing nightmares over the past week.
After adjusting for age, race, and sex, frequent nightmares were associated with hypertension (odds ratio, 1.51; 95% confidence interval, 1.28-1.78), heart problems (OR, 1.50; 95% CI, 1.11-2.02), and MI (OR, 2.32; 95% CI, 1.18-4.54).
Associations between frequent nightmares and hypertension (OR, 1.43; 95% CI, 1.17-1.73) and heart problems (OR, 1.43; 95% CI, 1.00-2.05) remained significant after further adjusting for smoking, depression, and PTSD.
“Our cross-sectional findings set the stage for future research examining the possibility that nightmares may confer cardiovascular disease risks beyond those conferred by PTSD diagnosis alone,” Dr. Ulmer said in a news release.
Dr. Ulmer also said that, because the study was based on self-reported data, the findings are “very preliminary.” Before doctors adjust clinical practices, it’s important that our findings be replicated using longitudinal studies, clinically diagnosed medical conditions, and objectively assessed sleep,” she said.
She added that more research is needed to uncover mechanisms explaining these associations and determine if reducing the frequency and severity of nightmares can lead to improved cardiovascular health.
Timely research
Reached for comment, Rajkumar (Raj) Dasgupta, MD, of the University of Southern California, Los Angeles, noted “the correlation between nightmares and heart disease is a timely topic right now with COVID-19 as more people may be having nightmares.”
“If a patient mentions nightmares, I do think it’s important not to just glaze over it, but to talk more about it and document it in the patient record, especially in patients with cardiovascular disease, atrial fibrillation, diabetes, and hypertension,” said Dr. Dasgupta, who wasn’t involved in the study.
The research was supported by the Veterans Integrated Service Network 6 Mental Illness Research, Education and Clinical Center and the Department of Veterans Affairs HSR&D ADAPT Center at the Durham VA Health Care System. Dr. Ulmer and Dr. Dasgupta have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
, new research shows. In what researchers describe as “surprising” findings, results from a large study of relatively young military veterans showed those who had nightmares two or more times per week had significantly increased risks for hypertension, myocardial infarction, or other heart problems.
“A diagnosis of PTSD incorporates sleep disturbance as a symptom. Thus, we were surprised to find that nightmares continued to be associated with CVD after controlling not only for PTSD and demographic factors, but also smoking and depression diagnosis,” said Christi Ulmer, PhD, of the department of psychiatry and behavioral sciences, Duke University Medical Center, Durham, N.C.
The findings were presented at the virtual annual meeting of the Associated Professional Sleep Societies.
Unclear mechanism
The study included 3,468 veterans (77% male) with a mean age of 38 years who had served one or two tours of duty since Sept. 11, 2001. Nearly one-third (31%) met criteria for PTSD, and 33% self-reported having at least one cardiovascular condition, such as heart problems, hypertension, stroke, and MI.
Nightmare frequency and severity was assessed using the Davidson Trauma Scale. Nightmares were considered frequent if they occurred two or more times per week and moderate to severe if they were at least moderately distressing. About 31% of veterans reported having frequent nightmares, and 35% reported moderately distressing nightmares over the past week.
After adjusting for age, race, and sex, frequent nightmares were associated with hypertension (odds ratio, 1.51; 95% confidence interval, 1.28-1.78), heart problems (OR, 1.50; 95% CI, 1.11-2.02), and MI (OR, 2.32; 95% CI, 1.18-4.54).
Associations between frequent nightmares and hypertension (OR, 1.43; 95% CI, 1.17-1.73) and heart problems (OR, 1.43; 95% CI, 1.00-2.05) remained significant after further adjusting for smoking, depression, and PTSD.
“Our cross-sectional findings set the stage for future research examining the possibility that nightmares may confer cardiovascular disease risks beyond those conferred by PTSD diagnosis alone,” Dr. Ulmer said in a news release.
Dr. Ulmer also said that, because the study was based on self-reported data, the findings are “very preliminary.” Before doctors adjust clinical practices, it’s important that our findings be replicated using longitudinal studies, clinically diagnosed medical conditions, and objectively assessed sleep,” she said.
She added that more research is needed to uncover mechanisms explaining these associations and determine if reducing the frequency and severity of nightmares can lead to improved cardiovascular health.
Timely research
Reached for comment, Rajkumar (Raj) Dasgupta, MD, of the University of Southern California, Los Angeles, noted “the correlation between nightmares and heart disease is a timely topic right now with COVID-19 as more people may be having nightmares.”
“If a patient mentions nightmares, I do think it’s important not to just glaze over it, but to talk more about it and document it in the patient record, especially in patients with cardiovascular disease, atrial fibrillation, diabetes, and hypertension,” said Dr. Dasgupta, who wasn’t involved in the study.
The research was supported by the Veterans Integrated Service Network 6 Mental Illness Research, Education and Clinical Center and the Department of Veterans Affairs HSR&D ADAPT Center at the Durham VA Health Care System. Dr. Ulmer and Dr. Dasgupta have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM SLEEP 2020
COVID-19 at home: What does optimal care look like?
Marilyn Stebbins, PharmD, fell ill at the end of February 2020. Initially diagnosed with multifocal pneumonia and treated with antibiotics, she later developed severe gastrointestinal symptoms, fatigue, and shortness of breath. She was hospitalized in early March and was diagnosed with COVID-19.
It was still early in the pandemic, and testing was not available for her husband. After she was discharged, her husband isolated himself as much as possible. But that limited the amount of care he could offer.
“When I came home after 8 days in the ICU, I felt completely alone and terrified of not being able to care for myself and not knowing how much care my husband could provide,” said Dr. Stebbins, professor of clinical pharmacy at the University of California, San Francisco.
“I can’t even imagine what it would have been like if I had been home alone without my husband in the house,” she said. “I think about the people who died at home and understand how that might happen.”
Dr. Stebbins is one of tens of thousands of people who, whether hospitalized and discharged or never admitted for inpatient care, needed to find ways to convalesce at home. Data from the Centers for Medicare & Medicaid Services show that, of 326,674 beneficiaries who tested positive for COVID-19 between May 16 and June 11, 2020, 109,607 were hospitalized, suggesting that two-thirds were outpatients.
Most attention has focused on the sickest patients, leaving less severe cases to fall through the cracks. Despite fever, cough, difficulty breathing, and a surfeit of other symptoms, there are few available resources and all too little support to help patients navigate the physical and emotional struggles of contending with COVID-19 at home.
No ‘cookie-cutter’ approach
The speed with which the pandemic progressed caught public health systems off guard, but now, “it is essential to put into place the infrastructure to care for the physical and mental health needs of patients at home because most are in the community and many, if not most, still aren’t receiving sufficient support at home,” said Dr. Stebbins.
said Gary LeRoy, MD, a family physician in Dayton, Ohio. He emphasized that there is “no cookie-cutter formula” for home care, because every patient’s situation is different.
“I begin by having a detailed conversation with each patient to ascertain whether their home environment is safe and to paint a picture of their circumstances,” Dr. LeRoy, who is the president of the American Academy of Family Physicians, said in an interview.
Dr. LeRoy suggested questions that constitute “not just a ‘medical’ checklist but a ‘whole life’ checklist.”
- Do you have access to food, water, medications, sanitation/cleaning supplies, a thermometer, and other necessities? If not, who might assist in providing those?
- Do you need help with activities of daily living and self-care?
- Who else lives in your household? Do they have signs and symptoms of the virus? Have they been tested?
- Do you have enough physical space between you and other household members?
- Do you have children? How are they being cared for?
- What type of work do you do? What are the implications for your employment if you are unable to work for an extended period?
- Do you have an emotional, social, and spiritual support system (e.g., family, friends, community, church)?
- Do you have concerns I haven’t mentioned?
Patients’ responses will inform the management plan and determine what medical and social resources are needed, he said.
Daily check-in
Dr. Stebbins said the nurse case manager from her insurance company called her daily after she came home from the hospital. She was told that a public health nurse would also call, but no one from the health department called for days – a situation she hopes has improved.
One way or another, she said, “health care providers [or their staff] should check in with patients daily, either telephonically or via video.” She noted that video is superior, because “someone who isn’t a family member needs to put eyes on a patient and might be able to detect warning signs that a family member without healthcare training might not notice.”
Dr. LeRoy, who is also an associate professor of medicine at Wright State University, Dayton, Ohio, said that, given his time constraints, a nurse or medical assistant in his practice conducts the daily check-ins and notifies him if the patient has fever or other symptoms.
“Under ordinary circumstances, when a patient comes to see me for some type of medical condition, I get to meet the patient, consider what might be going on, then order a test, wait for the results, and suggest a treatment plan. But these are anything but ordinary circumstances,” said Matthew Exline, MD, a pulmonary and critical care specialist at the Ohio State University Wexner Medical Center, Columbus.
“That traditional structure broke down with COVID-19, when we may have test results without even seeing the patient. And without this interaction, it is harder to know as a physician what course of action to take,” he said in an interview.
Once a diagnosis has been made, the physician has at least some data to help guide next steps, even if there has been no prior meeting with the patient.
For example, a positive test raises a host of issues, not the least of which is the risk of spreading the infection to other household members and questions about whether to go the hospital. Moreover, for patients, positive tests can have serious ramifications.
“Severe shortness of breath at rest is not typical of the flu, nor is loss of taste or smell,” said Dr. Exline. Practitioners must educate patients and families about specific symptoms of COVID-19, including shortness of breath, loss of taste or smell, and gastrointestinal or neurologic symptoms, and when to seek emergency care.
Dr. LeRoy suggests buying a pulse oximeter to gauge blood oxygen levels and pulse rate. Together with a thermometer, a portable blood pressure monitor, and, if indicated, a blood glucose monitor, these devices provide a comprehensive and accurate assessment of vital signs.
Dr. LeRoy also educates patients and their families about when to seek medical attention.
Dr. Stebbins takes a similar approach. “Family members are part of, not apart from, the care of patients with COVID-19, and it’s our responsibility as healthcare providers to consider them in the patient’s care plan.”
Keeping family safe
Beyond care, family members need a plan to keep themselves healthy, too.
“A patient with COVID-19 at home should self-quarantine as much as possible to keep other family members safe, if they continue to live in the same house,” Dr. Exline said.
Ideally, uninfected family members should stay with relatives or friends. When that’s not possible, everyone in the household should wear a mask, be vigilant about hand washing, and wipe down all surfaces – including doorknobs, light switches, faucet handles, cellphones, and utensils – regularly with bleach or an alcohol solution.
Caregivers should also minimize the amount of time they are exposed to the patient.
“Set food, water, and medication on the night table and leave the room rather than spending hours at the bedside, since limiting exposure to viral load reduces the chances of contagion,” said Dr. Exline.
The Centers for Disease Control and Prevention offers guidance for household members caring for COVID-19 patients at home. It provides tips on how to help patients follow the doctor’s instructions and ways to ensure adequate hydration and rest, among others.
Patients with COVID-19 who live alone face more formidable challenges.
Dr. LeRoy says physicians can help patients by educating themselves about available social services in their community so they can provide appropriate referrals and connections. Such initiatives can include meal programs, friendly visit and financial assistance programs, as well as childcare and home health agencies.
He noted that Aunt Bertha, a social care network, provides a guide to social services throughout the United States. Additional resources are available on USA.gov.
Comfort and support
Patients with COVID-19 need to be as comfortable and as supported as possible, both physically and emotionally.
“While I was sick, my dogs curled up next to me and didn’t leave my side, and they were my saving grace. There’s not enough to be said about emotional support,” Dr. Stebbins said.
Although important, emotional support is not enough. For patients with respiratory disorders, such as chronic obstructive pulmonary disease, asthma, heart failure, or pneumonia, their subjective symptoms of shortness of breath, air hunger, or cough may improve with supplemental oxygen at home. Other measures include repositioning of the patient to lessen the body weight over the lungs or the use of lung percussion, Leroy said.
He added that improvement may also come from drainage of sputum from the airway passages, the use of agents to liquefy thick sputum (mucolytics), or aerosolized bronchodilator medications.
However, Dr. LeRoy cautioned, “one remedy does not work for everyone – an individual can improve gradually by using these home support interventions, or their respiratory status can deteriorate rapidly despite all these interventions.”
For this reason, he says patients should consult their personal physician to determine which, if any, of these home treatments would be best for their particular situation.
Patients who need emotional support, psychotherapy, or psychotropic medications may find teletherapy helpful. Guidance for psychiatrists, psychologists, and social workers regarding the treatment of COVID-19 patients via teletherapy can be found on the American Psychiatric Association, the American Psychological Association, and the National Association of Social Workers websites.
Pharmacists can also help ensure patient safety, Dr. Stebbins said.
If a patient has not picked up their usual medications, Dr. Stebbins said, “they may need a check-in call. Some may be ill and alone and may need encouragement to seek medical attention, and some may have no means of getting to the pharmacy and may need medications delivered.”
A home healthcare agency may also be helpful for homebound patients. David Bersson, director of operations at Synergy Home Care of Bergen County, N.J., has arranged in-home caregivers for patients with COVID-19.
The amount of care that professional caregivers provide can range from several hours per week to full-time, depending on the patient’s needs and budget, and can include companionship, Mr. Bersson said in an interview.
Because patient and caregiver safety are paramount, caregivers are thoroughly trained in protection and decontamination procedures and are regularly tested for COVID-19 prior to being sent into a client’s home.
Health insurance companies do not cover this service, Mr. Bersson noted, but the VetAssist program covers home care for veterans and their spouses who meet income requirements.
Caregiving and companionship are both vital pieces of the at-home care puzzle. “It was the virtual emotional support I got from friends, family, coworkers, and healthcare professionals that meant so much to me, and I know they played an important part in my recovery,” Dr. Stebbins said.
Dr. LeRoy agreed, noting that he calls patients, even if they only have mild symptoms and his nurse has already spoken to them. “The call doesn’t take much time – maybe just a 5-minute conversation – but it makes patients aware that I care.”
Dr. Stebbins, Dr. Exline, and Dr. LeRoy report no relevant financial relationships. Mr. Bersson is the director of operations at Synergy Home Care of Bergen County, New Jersey.
This story first appeared on Medscape.com.
Marilyn Stebbins, PharmD, fell ill at the end of February 2020. Initially diagnosed with multifocal pneumonia and treated with antibiotics, she later developed severe gastrointestinal symptoms, fatigue, and shortness of breath. She was hospitalized in early March and was diagnosed with COVID-19.
It was still early in the pandemic, and testing was not available for her husband. After she was discharged, her husband isolated himself as much as possible. But that limited the amount of care he could offer.
“When I came home after 8 days in the ICU, I felt completely alone and terrified of not being able to care for myself and not knowing how much care my husband could provide,” said Dr. Stebbins, professor of clinical pharmacy at the University of California, San Francisco.
“I can’t even imagine what it would have been like if I had been home alone without my husband in the house,” she said. “I think about the people who died at home and understand how that might happen.”
Dr. Stebbins is one of tens of thousands of people who, whether hospitalized and discharged or never admitted for inpatient care, needed to find ways to convalesce at home. Data from the Centers for Medicare & Medicaid Services show that, of 326,674 beneficiaries who tested positive for COVID-19 between May 16 and June 11, 2020, 109,607 were hospitalized, suggesting that two-thirds were outpatients.
Most attention has focused on the sickest patients, leaving less severe cases to fall through the cracks. Despite fever, cough, difficulty breathing, and a surfeit of other symptoms, there are few available resources and all too little support to help patients navigate the physical and emotional struggles of contending with COVID-19 at home.
No ‘cookie-cutter’ approach
The speed with which the pandemic progressed caught public health systems off guard, but now, “it is essential to put into place the infrastructure to care for the physical and mental health needs of patients at home because most are in the community and many, if not most, still aren’t receiving sufficient support at home,” said Dr. Stebbins.
said Gary LeRoy, MD, a family physician in Dayton, Ohio. He emphasized that there is “no cookie-cutter formula” for home care, because every patient’s situation is different.
“I begin by having a detailed conversation with each patient to ascertain whether their home environment is safe and to paint a picture of their circumstances,” Dr. LeRoy, who is the president of the American Academy of Family Physicians, said in an interview.
Dr. LeRoy suggested questions that constitute “not just a ‘medical’ checklist but a ‘whole life’ checklist.”
- Do you have access to food, water, medications, sanitation/cleaning supplies, a thermometer, and other necessities? If not, who might assist in providing those?
- Do you need help with activities of daily living and self-care?
- Who else lives in your household? Do they have signs and symptoms of the virus? Have they been tested?
- Do you have enough physical space between you and other household members?
- Do you have children? How are they being cared for?
- What type of work do you do? What are the implications for your employment if you are unable to work for an extended period?
- Do you have an emotional, social, and spiritual support system (e.g., family, friends, community, church)?
- Do you have concerns I haven’t mentioned?
Patients’ responses will inform the management plan and determine what medical and social resources are needed, he said.
Daily check-in
Dr. Stebbins said the nurse case manager from her insurance company called her daily after she came home from the hospital. She was told that a public health nurse would also call, but no one from the health department called for days – a situation she hopes has improved.
One way or another, she said, “health care providers [or their staff] should check in with patients daily, either telephonically or via video.” She noted that video is superior, because “someone who isn’t a family member needs to put eyes on a patient and might be able to detect warning signs that a family member without healthcare training might not notice.”
Dr. LeRoy, who is also an associate professor of medicine at Wright State University, Dayton, Ohio, said that, given his time constraints, a nurse or medical assistant in his practice conducts the daily check-ins and notifies him if the patient has fever or other symptoms.
“Under ordinary circumstances, when a patient comes to see me for some type of medical condition, I get to meet the patient, consider what might be going on, then order a test, wait for the results, and suggest a treatment plan. But these are anything but ordinary circumstances,” said Matthew Exline, MD, a pulmonary and critical care specialist at the Ohio State University Wexner Medical Center, Columbus.
“That traditional structure broke down with COVID-19, when we may have test results without even seeing the patient. And without this interaction, it is harder to know as a physician what course of action to take,” he said in an interview.
Once a diagnosis has been made, the physician has at least some data to help guide next steps, even if there has been no prior meeting with the patient.
For example, a positive test raises a host of issues, not the least of which is the risk of spreading the infection to other household members and questions about whether to go the hospital. Moreover, for patients, positive tests can have serious ramifications.
“Severe shortness of breath at rest is not typical of the flu, nor is loss of taste or smell,” said Dr. Exline. Practitioners must educate patients and families about specific symptoms of COVID-19, including shortness of breath, loss of taste or smell, and gastrointestinal or neurologic symptoms, and when to seek emergency care.
Dr. LeRoy suggests buying a pulse oximeter to gauge blood oxygen levels and pulse rate. Together with a thermometer, a portable blood pressure monitor, and, if indicated, a blood glucose monitor, these devices provide a comprehensive and accurate assessment of vital signs.
Dr. LeRoy also educates patients and their families about when to seek medical attention.
Dr. Stebbins takes a similar approach. “Family members are part of, not apart from, the care of patients with COVID-19, and it’s our responsibility as healthcare providers to consider them in the patient’s care plan.”
Keeping family safe
Beyond care, family members need a plan to keep themselves healthy, too.
“A patient with COVID-19 at home should self-quarantine as much as possible to keep other family members safe, if they continue to live in the same house,” Dr. Exline said.
Ideally, uninfected family members should stay with relatives or friends. When that’s not possible, everyone in the household should wear a mask, be vigilant about hand washing, and wipe down all surfaces – including doorknobs, light switches, faucet handles, cellphones, and utensils – regularly with bleach or an alcohol solution.
Caregivers should also minimize the amount of time they are exposed to the patient.
“Set food, water, and medication on the night table and leave the room rather than spending hours at the bedside, since limiting exposure to viral load reduces the chances of contagion,” said Dr. Exline.
The Centers for Disease Control and Prevention offers guidance for household members caring for COVID-19 patients at home. It provides tips on how to help patients follow the doctor’s instructions and ways to ensure adequate hydration and rest, among others.
Patients with COVID-19 who live alone face more formidable challenges.
Dr. LeRoy says physicians can help patients by educating themselves about available social services in their community so they can provide appropriate referrals and connections. Such initiatives can include meal programs, friendly visit and financial assistance programs, as well as childcare and home health agencies.
He noted that Aunt Bertha, a social care network, provides a guide to social services throughout the United States. Additional resources are available on USA.gov.
Comfort and support
Patients with COVID-19 need to be as comfortable and as supported as possible, both physically and emotionally.
“While I was sick, my dogs curled up next to me and didn’t leave my side, and they were my saving grace. There’s not enough to be said about emotional support,” Dr. Stebbins said.
Although important, emotional support is not enough. For patients with respiratory disorders, such as chronic obstructive pulmonary disease, asthma, heart failure, or pneumonia, their subjective symptoms of shortness of breath, air hunger, or cough may improve with supplemental oxygen at home. Other measures include repositioning of the patient to lessen the body weight over the lungs or the use of lung percussion, Leroy said.
He added that improvement may also come from drainage of sputum from the airway passages, the use of agents to liquefy thick sputum (mucolytics), or aerosolized bronchodilator medications.
However, Dr. LeRoy cautioned, “one remedy does not work for everyone – an individual can improve gradually by using these home support interventions, or their respiratory status can deteriorate rapidly despite all these interventions.”
For this reason, he says patients should consult their personal physician to determine which, if any, of these home treatments would be best for their particular situation.
Patients who need emotional support, psychotherapy, or psychotropic medications may find teletherapy helpful. Guidance for psychiatrists, psychologists, and social workers regarding the treatment of COVID-19 patients via teletherapy can be found on the American Psychiatric Association, the American Psychological Association, and the National Association of Social Workers websites.
Pharmacists can also help ensure patient safety, Dr. Stebbins said.
If a patient has not picked up their usual medications, Dr. Stebbins said, “they may need a check-in call. Some may be ill and alone and may need encouragement to seek medical attention, and some may have no means of getting to the pharmacy and may need medications delivered.”
A home healthcare agency may also be helpful for homebound patients. David Bersson, director of operations at Synergy Home Care of Bergen County, N.J., has arranged in-home caregivers for patients with COVID-19.
The amount of care that professional caregivers provide can range from several hours per week to full-time, depending on the patient’s needs and budget, and can include companionship, Mr. Bersson said in an interview.
Because patient and caregiver safety are paramount, caregivers are thoroughly trained in protection and decontamination procedures and are regularly tested for COVID-19 prior to being sent into a client’s home.
Health insurance companies do not cover this service, Mr. Bersson noted, but the VetAssist program covers home care for veterans and their spouses who meet income requirements.
Caregiving and companionship are both vital pieces of the at-home care puzzle. “It was the virtual emotional support I got from friends, family, coworkers, and healthcare professionals that meant so much to me, and I know they played an important part in my recovery,” Dr. Stebbins said.
Dr. LeRoy agreed, noting that he calls patients, even if they only have mild symptoms and his nurse has already spoken to them. “The call doesn’t take much time – maybe just a 5-minute conversation – but it makes patients aware that I care.”
Dr. Stebbins, Dr. Exline, and Dr. LeRoy report no relevant financial relationships. Mr. Bersson is the director of operations at Synergy Home Care of Bergen County, New Jersey.
This story first appeared on Medscape.com.
Marilyn Stebbins, PharmD, fell ill at the end of February 2020. Initially diagnosed with multifocal pneumonia and treated with antibiotics, she later developed severe gastrointestinal symptoms, fatigue, and shortness of breath. She was hospitalized in early March and was diagnosed with COVID-19.
It was still early in the pandemic, and testing was not available for her husband. After she was discharged, her husband isolated himself as much as possible. But that limited the amount of care he could offer.
“When I came home after 8 days in the ICU, I felt completely alone and terrified of not being able to care for myself and not knowing how much care my husband could provide,” said Dr. Stebbins, professor of clinical pharmacy at the University of California, San Francisco.
“I can’t even imagine what it would have been like if I had been home alone without my husband in the house,” she said. “I think about the people who died at home and understand how that might happen.”
Dr. Stebbins is one of tens of thousands of people who, whether hospitalized and discharged or never admitted for inpatient care, needed to find ways to convalesce at home. Data from the Centers for Medicare & Medicaid Services show that, of 326,674 beneficiaries who tested positive for COVID-19 between May 16 and June 11, 2020, 109,607 were hospitalized, suggesting that two-thirds were outpatients.
Most attention has focused on the sickest patients, leaving less severe cases to fall through the cracks. Despite fever, cough, difficulty breathing, and a surfeit of other symptoms, there are few available resources and all too little support to help patients navigate the physical and emotional struggles of contending with COVID-19 at home.
No ‘cookie-cutter’ approach
The speed with which the pandemic progressed caught public health systems off guard, but now, “it is essential to put into place the infrastructure to care for the physical and mental health needs of patients at home because most are in the community and many, if not most, still aren’t receiving sufficient support at home,” said Dr. Stebbins.
said Gary LeRoy, MD, a family physician in Dayton, Ohio. He emphasized that there is “no cookie-cutter formula” for home care, because every patient’s situation is different.
“I begin by having a detailed conversation with each patient to ascertain whether their home environment is safe and to paint a picture of their circumstances,” Dr. LeRoy, who is the president of the American Academy of Family Physicians, said in an interview.
Dr. LeRoy suggested questions that constitute “not just a ‘medical’ checklist but a ‘whole life’ checklist.”
- Do you have access to food, water, medications, sanitation/cleaning supplies, a thermometer, and other necessities? If not, who might assist in providing those?
- Do you need help with activities of daily living and self-care?
- Who else lives in your household? Do they have signs and symptoms of the virus? Have they been tested?
- Do you have enough physical space between you and other household members?
- Do you have children? How are they being cared for?
- What type of work do you do? What are the implications for your employment if you are unable to work for an extended period?
- Do you have an emotional, social, and spiritual support system (e.g., family, friends, community, church)?
- Do you have concerns I haven’t mentioned?
Patients’ responses will inform the management plan and determine what medical and social resources are needed, he said.
Daily check-in
Dr. Stebbins said the nurse case manager from her insurance company called her daily after she came home from the hospital. She was told that a public health nurse would also call, but no one from the health department called for days – a situation she hopes has improved.
One way or another, she said, “health care providers [or their staff] should check in with patients daily, either telephonically or via video.” She noted that video is superior, because “someone who isn’t a family member needs to put eyes on a patient and might be able to detect warning signs that a family member without healthcare training might not notice.”
Dr. LeRoy, who is also an associate professor of medicine at Wright State University, Dayton, Ohio, said that, given his time constraints, a nurse or medical assistant in his practice conducts the daily check-ins and notifies him if the patient has fever or other symptoms.
“Under ordinary circumstances, when a patient comes to see me for some type of medical condition, I get to meet the patient, consider what might be going on, then order a test, wait for the results, and suggest a treatment plan. But these are anything but ordinary circumstances,” said Matthew Exline, MD, a pulmonary and critical care specialist at the Ohio State University Wexner Medical Center, Columbus.
“That traditional structure broke down with COVID-19, when we may have test results without even seeing the patient. And without this interaction, it is harder to know as a physician what course of action to take,” he said in an interview.
Once a diagnosis has been made, the physician has at least some data to help guide next steps, even if there has been no prior meeting with the patient.
For example, a positive test raises a host of issues, not the least of which is the risk of spreading the infection to other household members and questions about whether to go the hospital. Moreover, for patients, positive tests can have serious ramifications.
“Severe shortness of breath at rest is not typical of the flu, nor is loss of taste or smell,” said Dr. Exline. Practitioners must educate patients and families about specific symptoms of COVID-19, including shortness of breath, loss of taste or smell, and gastrointestinal or neurologic symptoms, and when to seek emergency care.
Dr. LeRoy suggests buying a pulse oximeter to gauge blood oxygen levels and pulse rate. Together with a thermometer, a portable blood pressure monitor, and, if indicated, a blood glucose monitor, these devices provide a comprehensive and accurate assessment of vital signs.
Dr. LeRoy also educates patients and their families about when to seek medical attention.
Dr. Stebbins takes a similar approach. “Family members are part of, not apart from, the care of patients with COVID-19, and it’s our responsibility as healthcare providers to consider them in the patient’s care plan.”
Keeping family safe
Beyond care, family members need a plan to keep themselves healthy, too.
“A patient with COVID-19 at home should self-quarantine as much as possible to keep other family members safe, if they continue to live in the same house,” Dr. Exline said.
Ideally, uninfected family members should stay with relatives or friends. When that’s not possible, everyone in the household should wear a mask, be vigilant about hand washing, and wipe down all surfaces – including doorknobs, light switches, faucet handles, cellphones, and utensils – regularly with bleach or an alcohol solution.
Caregivers should also minimize the amount of time they are exposed to the patient.
“Set food, water, and medication on the night table and leave the room rather than spending hours at the bedside, since limiting exposure to viral load reduces the chances of contagion,” said Dr. Exline.
The Centers for Disease Control and Prevention offers guidance for household members caring for COVID-19 patients at home. It provides tips on how to help patients follow the doctor’s instructions and ways to ensure adequate hydration and rest, among others.
Patients with COVID-19 who live alone face more formidable challenges.
Dr. LeRoy says physicians can help patients by educating themselves about available social services in their community so they can provide appropriate referrals and connections. Such initiatives can include meal programs, friendly visit and financial assistance programs, as well as childcare and home health agencies.
He noted that Aunt Bertha, a social care network, provides a guide to social services throughout the United States. Additional resources are available on USA.gov.
Comfort and support
Patients with COVID-19 need to be as comfortable and as supported as possible, both physically and emotionally.
“While I was sick, my dogs curled up next to me and didn’t leave my side, and they were my saving grace. There’s not enough to be said about emotional support,” Dr. Stebbins said.
Although important, emotional support is not enough. For patients with respiratory disorders, such as chronic obstructive pulmonary disease, asthma, heart failure, or pneumonia, their subjective symptoms of shortness of breath, air hunger, or cough may improve with supplemental oxygen at home. Other measures include repositioning of the patient to lessen the body weight over the lungs or the use of lung percussion, Leroy said.
He added that improvement may also come from drainage of sputum from the airway passages, the use of agents to liquefy thick sputum (mucolytics), or aerosolized bronchodilator medications.
However, Dr. LeRoy cautioned, “one remedy does not work for everyone – an individual can improve gradually by using these home support interventions, or their respiratory status can deteriorate rapidly despite all these interventions.”
For this reason, he says patients should consult their personal physician to determine which, if any, of these home treatments would be best for their particular situation.
Patients who need emotional support, psychotherapy, or psychotropic medications may find teletherapy helpful. Guidance for psychiatrists, psychologists, and social workers regarding the treatment of COVID-19 patients via teletherapy can be found on the American Psychiatric Association, the American Psychological Association, and the National Association of Social Workers websites.
Pharmacists can also help ensure patient safety, Dr. Stebbins said.
If a patient has not picked up their usual medications, Dr. Stebbins said, “they may need a check-in call. Some may be ill and alone and may need encouragement to seek medical attention, and some may have no means of getting to the pharmacy and may need medications delivered.”
A home healthcare agency may also be helpful for homebound patients. David Bersson, director of operations at Synergy Home Care of Bergen County, N.J., has arranged in-home caregivers for patients with COVID-19.
The amount of care that professional caregivers provide can range from several hours per week to full-time, depending on the patient’s needs and budget, and can include companionship, Mr. Bersson said in an interview.
Because patient and caregiver safety are paramount, caregivers are thoroughly trained in protection and decontamination procedures and are regularly tested for COVID-19 prior to being sent into a client’s home.
Health insurance companies do not cover this service, Mr. Bersson noted, but the VetAssist program covers home care for veterans and their spouses who meet income requirements.
Caregiving and companionship are both vital pieces of the at-home care puzzle. “It was the virtual emotional support I got from friends, family, coworkers, and healthcare professionals that meant so much to me, and I know they played an important part in my recovery,” Dr. Stebbins said.
Dr. LeRoy agreed, noting that he calls patients, even if they only have mild symptoms and his nurse has already spoken to them. “The call doesn’t take much time – maybe just a 5-minute conversation – but it makes patients aware that I care.”
Dr. Stebbins, Dr. Exline, and Dr. LeRoy report no relevant financial relationships. Mr. Bersson is the director of operations at Synergy Home Care of Bergen County, New Jersey.
This story first appeared on Medscape.com.
Mortality burden of dementia may be greater than estimated
This burden may be greatest among non-Hispanic black older adults, compared with Hispanic and non-Hispanic whites. This burden also is significantly greater among people with less than a high school education, compared with those with a college education.

The study results underscore the importance of broadening access to population-based interventions that focus on dementia prevention and care, the investigators wrote. “Future research could examine the extent to which deaths attributable to dementia and underestimation of dementia as an underlying cause of death on death certificates might have changed over time,” wrote Andrew C. Stokes, PhD, assistant professor of global health at the Boston University School of Public Health, and colleagues.
The study was published online Aug. 24 in JAMA Neurology.
In 2019, approximately 5.6 million adults in the United States who were aged 65 years or older had Alzheimer’s disease, vascular dementia, or mixed-cause dementia. A further 18.8% of Americans in this age group had cognitive impairment without dementia (CIND). About one third of patients with CIND may develop Alzheimer’s disease or related dementias (ADRD) within 5 years.
Research suggests that medical examiners significantly underreport ADRD on death certificates. One community-based study, for example, found that only 25% of deaths in patients with dementia had Alzheimer’s disease listed on the death certificates. Other research found that deaths in patients with dementia were often coded using more proximate causes, such as cardiovascular disease, sepsis, and pneumonia.
Health and retirement study
Dr. Stokes and colleagues examined data from the Health and Retirement Study (HRS) to evaluate the association of dementia and CIND with all-cause mortality. The HRS is a longitudinal cohort study of adults older than 50 years who live in the community. Its sample is nationally representative. The HRS investigators also initiated the Aging, Demographics, and Memory study to develop a procedure for assessing cognitive status in the HRS sample.
In their study, Dr. Stokes and colleagues included adults who had been sampled in the 2000 wave of HRS. They focused on participants between ages 70 and 99 years at baseline, and their final sample included 7,342 older adults. To identify dementia status, the researchers used the Langa–Weir score cutoff, which is based on tests of immediate and delayed recall of 10 words, a serial 7-second task, and a backward counting task. They also classified dementia status using the Herzog–Wallace, Wu, Hurd, and modified Hurd algorithms.
At baseline, the researchers measured age, sex, race or ethnicity, educational attainment, smoking status, self-reported disease diagnoses, and U.S. Census division as covariates. The National Center for Health Statistics linked HRS data with National Death Index records. These linked records include underlying cause of death and any mention of a condition or cause of death on the death certificate. The researchers compared the percentage of deaths attributable to ADRD according to a population attributable fraction estimate with the proportion of dementia-related deaths according to underlying causes and with any mention of dementia on death certificates.
The sample of 7,342 older adults included 4,348 (60.3%) women. Data for 1,030 (13.4%) people were reported by proxy. At baseline, most participants (64.0%) were between ages 70 and 79 years, 31% were between ages 80 and 89, and 5% were between ages 90 and 99 years. The prevalence of dementia in the complete sample was 14.3%, and the prevalence of CIND was 24.7%. The prevalence of dementia (22.4%) and CIND (29.3%) was higher among decedents than among the full population.
The hazard ratio (HR) for mortality was 2.53 among participants with dementia and 1.53 among patients with CIND. Although 13.6% of deaths were attributable to dementia, the proportion of deaths assigned to dementia as an underlying cause on death certificates was 5.0%. This discrepancy suggests that dementia is underreported by more than a factor of 2.7.
The mortality burden of dementia was 24.7% in non-Hispanic black older adults, 20.7% in Hispanic white participants, and 12.2% in non-Hispanic white participants. In addition, the mortality burden of dementia was significantly greater among participants with less than a high school education (16.2%) than among participants with a college education (9.8%).
The degree to which the underlying cause of death underestimated the mortality burden of dementia varied by sociodemographic characteristics, health status, and geography. The burden was underestimated by a factor of 7.1 among non-Hispanic black participants, a factor of 4.1 among Hispanic participants, and a factor of 2.3 among non-Hispanic white participants. The burden was underestimated by a factor of 3.5 in men and a factor of 2.4 in women. In addition, the burden was underestimated by a factor of 3.0 among participants with less than a high school education, by a factor of 2.3 among participants with a high school education, by a factor of 1.9 in participants with some college, and by a factor of 2.5 among participants with a college or higher education.
One of the study’s strengths was its population attributable fraction analysis, which reduced the risk of overestimating the mortality burden of dementia, Dr. Stokes and colleagues wrote. Examining CIND is valuable because of its high prevalence and consequent influence on outcomes in the population, even though CIND is associated with a lower mortality risk, they added. Nevertheless, the investigators were unable to assess mortality for dementia subtypes, and the classifications of dementia status and CIND may be subject to measurement error.
Underestimation is systematic
“This study is eye-opening in that it highlights the systematic underestimation of deaths attributable to dementia,” said Costantino Iadecola, MD, Anne Parrish Titzell professor of neurology and director and chair of the Feil Family Brain and Mind Research Institute at Weill Cornell Medicine in New York. The study’s main strength is that it is nationally representative, but the data must be confirmed in a larger population, he added.
The results will clarify the effect of dementia on mortality for neurologists, and geriatricians should be made aware of them, said Dr. Iadecola. “These data should be valuable to rationalize public health efforts and related funding decisions concerning research and community support.”
Further research could determine the mortality of dementia subgroups, “especially dementias linked to vascular factors in which prevention may be effective,” said Dr. Iadecola. “In the older population, vascular factors may play a more preeminent role, and it may help focus preventive approaches.”
The study was supported by a grant from the National Institute on Aging. Dr. Stokes received grants from Ethicon that were unrelated to this study. Dr. Iadecola serves on the scientific advisory board of Broadview Venture.
SOURCE: Stokes AC et al. JAMA Neurol. 2020 Aug 24. doi: 10.1001/jamaneurol.2020.2831.
This burden may be greatest among non-Hispanic black older adults, compared with Hispanic and non-Hispanic whites. This burden also is significantly greater among people with less than a high school education, compared with those with a college education.
The study results underscore the importance of broadening access to population-based interventions that focus on dementia prevention and care, the investigators wrote. “Future research could examine the extent to which deaths attributable to dementia and underestimation of dementia as an underlying cause of death on death certificates might have changed over time,” wrote Andrew C. Stokes, PhD, assistant professor of global health at the Boston University School of Public Health, and colleagues.
The study was published online Aug. 24 in JAMA Neurology.
In 2019, approximately 5.6 million adults in the United States who were aged 65 years or older had Alzheimer’s disease, vascular dementia, or mixed-cause dementia. A further 18.8% of Americans in this age group had cognitive impairment without dementia (CIND). About one third of patients with CIND may develop Alzheimer’s disease or related dementias (ADRD) within 5 years.
Research suggests that medical examiners significantly underreport ADRD on death certificates. One community-based study, for example, found that only 25% of deaths in patients with dementia had Alzheimer’s disease listed on the death certificates. Other research found that deaths in patients with dementia were often coded using more proximate causes, such as cardiovascular disease, sepsis, and pneumonia.
Health and retirement study
Dr. Stokes and colleagues examined data from the Health and Retirement Study (HRS) to evaluate the association of dementia and CIND with all-cause mortality. The HRS is a longitudinal cohort study of adults older than 50 years who live in the community. Its sample is nationally representative. The HRS investigators also initiated the Aging, Demographics, and Memory study to develop a procedure for assessing cognitive status in the HRS sample.
In their study, Dr. Stokes and colleagues included adults who had been sampled in the 2000 wave of HRS. They focused on participants between ages 70 and 99 years at baseline, and their final sample included 7,342 older adults. To identify dementia status, the researchers used the Langa–Weir score cutoff, which is based on tests of immediate and delayed recall of 10 words, a serial 7-second task, and a backward counting task. They also classified dementia status using the Herzog–Wallace, Wu, Hurd, and modified Hurd algorithms.
At baseline, the researchers measured age, sex, race or ethnicity, educational attainment, smoking status, self-reported disease diagnoses, and U.S. Census division as covariates. The National Center for Health Statistics linked HRS data with National Death Index records. These linked records include underlying cause of death and any mention of a condition or cause of death on the death certificate. The researchers compared the percentage of deaths attributable to ADRD according to a population attributable fraction estimate with the proportion of dementia-related deaths according to underlying causes and with any mention of dementia on death certificates.
The sample of 7,342 older adults included 4,348 (60.3%) women. Data for 1,030 (13.4%) people were reported by proxy. At baseline, most participants (64.0%) were between ages 70 and 79 years, 31% were between ages 80 and 89, and 5% were between ages 90 and 99 years. The prevalence of dementia in the complete sample was 14.3%, and the prevalence of CIND was 24.7%. The prevalence of dementia (22.4%) and CIND (29.3%) was higher among decedents than among the full population.
The hazard ratio (HR) for mortality was 2.53 among participants with dementia and 1.53 among patients with CIND. Although 13.6% of deaths were attributable to dementia, the proportion of deaths assigned to dementia as an underlying cause on death certificates was 5.0%. This discrepancy suggests that dementia is underreported by more than a factor of 2.7.
The mortality burden of dementia was 24.7% in non-Hispanic black older adults, 20.7% in Hispanic white participants, and 12.2% in non-Hispanic white participants. In addition, the mortality burden of dementia was significantly greater among participants with less than a high school education (16.2%) than among participants with a college education (9.8%).
The degree to which the underlying cause of death underestimated the mortality burden of dementia varied by sociodemographic characteristics, health status, and geography. The burden was underestimated by a factor of 7.1 among non-Hispanic black participants, a factor of 4.1 among Hispanic participants, and a factor of 2.3 among non-Hispanic white participants. The burden was underestimated by a factor of 3.5 in men and a factor of 2.4 in women. In addition, the burden was underestimated by a factor of 3.0 among participants with less than a high school education, by a factor of 2.3 among participants with a high school education, by a factor of 1.9 in participants with some college, and by a factor of 2.5 among participants with a college or higher education.
One of the study’s strengths was its population attributable fraction analysis, which reduced the risk of overestimating the mortality burden of dementia, Dr. Stokes and colleagues wrote. Examining CIND is valuable because of its high prevalence and consequent influence on outcomes in the population, even though CIND is associated with a lower mortality risk, they added. Nevertheless, the investigators were unable to assess mortality for dementia subtypes, and the classifications of dementia status and CIND may be subject to measurement error.
Underestimation is systematic
“This study is eye-opening in that it highlights the systematic underestimation of deaths attributable to dementia,” said Costantino Iadecola, MD, Anne Parrish Titzell professor of neurology and director and chair of the Feil Family Brain and Mind Research Institute at Weill Cornell Medicine in New York. The study’s main strength is that it is nationally representative, but the data must be confirmed in a larger population, he added.
The results will clarify the effect of dementia on mortality for neurologists, and geriatricians should be made aware of them, said Dr. Iadecola. “These data should be valuable to rationalize public health efforts and related funding decisions concerning research and community support.”
Further research could determine the mortality of dementia subgroups, “especially dementias linked to vascular factors in which prevention may be effective,” said Dr. Iadecola. “In the older population, vascular factors may play a more preeminent role, and it may help focus preventive approaches.”
The study was supported by a grant from the National Institute on Aging. Dr. Stokes received grants from Ethicon that were unrelated to this study. Dr. Iadecola serves on the scientific advisory board of Broadview Venture.
SOURCE: Stokes AC et al. JAMA Neurol. 2020 Aug 24. doi: 10.1001/jamaneurol.2020.2831.
This burden may be greatest among non-Hispanic black older adults, compared with Hispanic and non-Hispanic whites. This burden also is significantly greater among people with less than a high school education, compared with those with a college education.
The study results underscore the importance of broadening access to population-based interventions that focus on dementia prevention and care, the investigators wrote. “Future research could examine the extent to which deaths attributable to dementia and underestimation of dementia as an underlying cause of death on death certificates might have changed over time,” wrote Andrew C. Stokes, PhD, assistant professor of global health at the Boston University School of Public Health, and colleagues.
The study was published online Aug. 24 in JAMA Neurology.
In 2019, approximately 5.6 million adults in the United States who were aged 65 years or older had Alzheimer’s disease, vascular dementia, or mixed-cause dementia. A further 18.8% of Americans in this age group had cognitive impairment without dementia (CIND). About one third of patients with CIND may develop Alzheimer’s disease or related dementias (ADRD) within 5 years.
Research suggests that medical examiners significantly underreport ADRD on death certificates. One community-based study, for example, found that only 25% of deaths in patients with dementia had Alzheimer’s disease listed on the death certificates. Other research found that deaths in patients with dementia were often coded using more proximate causes, such as cardiovascular disease, sepsis, and pneumonia.
Health and retirement study
Dr. Stokes and colleagues examined data from the Health and Retirement Study (HRS) to evaluate the association of dementia and CIND with all-cause mortality. The HRS is a longitudinal cohort study of adults older than 50 years who live in the community. Its sample is nationally representative. The HRS investigators also initiated the Aging, Demographics, and Memory study to develop a procedure for assessing cognitive status in the HRS sample.
In their study, Dr. Stokes and colleagues included adults who had been sampled in the 2000 wave of HRS. They focused on participants between ages 70 and 99 years at baseline, and their final sample included 7,342 older adults. To identify dementia status, the researchers used the Langa–Weir score cutoff, which is based on tests of immediate and delayed recall of 10 words, a serial 7-second task, and a backward counting task. They also classified dementia status using the Herzog–Wallace, Wu, Hurd, and modified Hurd algorithms.
At baseline, the researchers measured age, sex, race or ethnicity, educational attainment, smoking status, self-reported disease diagnoses, and U.S. Census division as covariates. The National Center for Health Statistics linked HRS data with National Death Index records. These linked records include underlying cause of death and any mention of a condition or cause of death on the death certificate. The researchers compared the percentage of deaths attributable to ADRD according to a population attributable fraction estimate with the proportion of dementia-related deaths according to underlying causes and with any mention of dementia on death certificates.
The sample of 7,342 older adults included 4,348 (60.3%) women. Data for 1,030 (13.4%) people were reported by proxy. At baseline, most participants (64.0%) were between ages 70 and 79 years, 31% were between ages 80 and 89, and 5% were between ages 90 and 99 years. The prevalence of dementia in the complete sample was 14.3%, and the prevalence of CIND was 24.7%. The prevalence of dementia (22.4%) and CIND (29.3%) was higher among decedents than among the full population.
The hazard ratio (HR) for mortality was 2.53 among participants with dementia and 1.53 among patients with CIND. Although 13.6% of deaths were attributable to dementia, the proportion of deaths assigned to dementia as an underlying cause on death certificates was 5.0%. This discrepancy suggests that dementia is underreported by more than a factor of 2.7.
The mortality burden of dementia was 24.7% in non-Hispanic black older adults, 20.7% in Hispanic white participants, and 12.2% in non-Hispanic white participants. In addition, the mortality burden of dementia was significantly greater among participants with less than a high school education (16.2%) than among participants with a college education (9.8%).
The degree to which the underlying cause of death underestimated the mortality burden of dementia varied by sociodemographic characteristics, health status, and geography. The burden was underestimated by a factor of 7.1 among non-Hispanic black participants, a factor of 4.1 among Hispanic participants, and a factor of 2.3 among non-Hispanic white participants. The burden was underestimated by a factor of 3.5 in men and a factor of 2.4 in women. In addition, the burden was underestimated by a factor of 3.0 among participants with less than a high school education, by a factor of 2.3 among participants with a high school education, by a factor of 1.9 in participants with some college, and by a factor of 2.5 among participants with a college or higher education.
One of the study’s strengths was its population attributable fraction analysis, which reduced the risk of overestimating the mortality burden of dementia, Dr. Stokes and colleagues wrote. Examining CIND is valuable because of its high prevalence and consequent influence on outcomes in the population, even though CIND is associated with a lower mortality risk, they added. Nevertheless, the investigators were unable to assess mortality for dementia subtypes, and the classifications of dementia status and CIND may be subject to measurement error.
Underestimation is systematic
“This study is eye-opening in that it highlights the systematic underestimation of deaths attributable to dementia,” said Costantino Iadecola, MD, Anne Parrish Titzell professor of neurology and director and chair of the Feil Family Brain and Mind Research Institute at Weill Cornell Medicine in New York. The study’s main strength is that it is nationally representative, but the data must be confirmed in a larger population, he added.
The results will clarify the effect of dementia on mortality for neurologists, and geriatricians should be made aware of them, said Dr. Iadecola. “These data should be valuable to rationalize public health efforts and related funding decisions concerning research and community support.”
Further research could determine the mortality of dementia subgroups, “especially dementias linked to vascular factors in which prevention may be effective,” said Dr. Iadecola. “In the older population, vascular factors may play a more preeminent role, and it may help focus preventive approaches.”
The study was supported by a grant from the National Institute on Aging. Dr. Stokes received grants from Ethicon that were unrelated to this study. Dr. Iadecola serves on the scientific advisory board of Broadview Venture.
SOURCE: Stokes AC et al. JAMA Neurol. 2020 Aug 24. doi: 10.1001/jamaneurol.2020.2831.
FROM JAMA NEUROLOGY
TNF inhibitors linked to inflammatory CNS events
, new research suggests
The nested case-control study included more than 200 participants with diseases such as rheumatoid arthritis, psoriasis, and Crohn’s disease. Results showed that exposure to TNF inhibitors was significantly associated with increased risk for demyelinating CNS events, such as multiple sclerosis, and nondemyelinating events, such as meningitis and encephalitis.
Interestingly, disease-specific secondary analyses showed that the strongest association for inflammatory events was in patients with rheumatoid arthritis.
Lead author Amy Kunchok, MD, of Mayo Clinic, Rochester, Minn., noted that “these are highly effective therapies for patients” and that these CNS events are likely uncommon.
“Our study has observed an association, but this does not imply causality. Therefore, we are not cautioning against using these therapies in appropriate patients,” Dr. Kunchok said in an interview.
“Rather, we recommend that clinicians assessing patients with both inflammatory demyelinating and nondemyelinating CNS events consider a detailed evaluation of the medication history, particularly in patients with coexistent autoimmune diseases who may have a current or past history of biological therapies,” she said.
The findings were published in JAMA Neurology.
Poorly understood
TNF inhibitors “are common therapies for certain autoimmune diseases,” the investigators noted.
Previously, a link between exposure to these inhibitors and inflammatory CNS events “has been postulated but is poorly understood,” they wrote.
In the current study, they examined records for 106 patients who were treated at Mayo clinics in Minnesota, Arizona, or Florida from January 2003 through February 2019. All participants had been diagnosed with an autoimmune disease that the Food and Drug Administration has listed as an indication for TNF inhibitor use. This included rheumatoid arthritis (n = 48), ankylosing spondylitis (n = 4), psoriasis and psoriatic arthritis (n = 21), Crohn’s disease (n = 27), and ulcerative colitis (n = 6). Their records also showed diagnostic codes for the inflammatory demyelinating CNS events of relapsing-remitting or primary progressive MS, clinically isolated syndrome, radiologically isolated syndrome, neuromyelitis optica spectrum disorder, and transverse myelitis or for the inflammatory nondemyelinating CNS events of meningitis, meningoencephalitis, encephalitis, neurosarcoidosis, and CNS vasculitis. The investigators also included 106 age-, sex-, and autoimmune disease–matched participants 1:1 to act as the control group.
In the total study population, 64% were women and the median age at disease onset was 52 years. In addition, 60% of the patient group and 40% of the control group were exposed to TNF inhibitors.
Novel finding?
Results showed that TNF inhibitor exposure was significantly linked to increased risk for developing any inflammatory CNS event (adjusted odds ratio, 3.01; 95% CI, 1.55-5.82; P = .001). When the outcomes were stratified by class of inflammatory event, these results were similar. The aOR was 3.09 (95% CI, 1.19-8.04; P = .02) for inflammatory demyelinating CNS events and was 2.97 (95% CI, 1.15-7.65; P = .02) for inflammatory nondemyelinating events.
Dr. Kunchok noted that the association between the inhibitors and nondemyelinating events was “a novel finding from this study.”
In secondary analyses, patients with rheumatoid arthritis and exposure to TNF inhibitors had the strongest association with any inflammatory CNS event (aOR, 4.82; 95% CI, 1.62-14.36; P = .005).
A pooled cohort comprising only the participants with the other autoimmune diseases did not show a significant association between exposure to TNF inhibitors and development of CNS events (P = .09).
“Because of the lack of power, further stratification by individual autoimmune diseases was not analyzed,” the investigators reported.
Although the overall findings showed that exposure to TNF inhibitors was linked to increased risk for inflammatory events, whether this association “represents de novo or exacerbated inflammatory pathways requires further research,” the authors wrote.
Dr. Kunchok added that more research, especially population-based studies, is also needed to examine the incidence of these inflammatory CNS events in patients exposed to TNF-alpha inhibitors.
Adds to the literature
In an accompanying editorial, Jeffrey M. Gelfand, MD, department of neurology at the University of California, San Francisco, and Jinoos Yazdany, MD, Zuckerberg San Francisco General Hospital at UCSF, noted that although the study adds to the literature, the magnitude of the risk found “remains unclear.”
“Randomized clinical trials are not suited to the study of rare adverse events,” Dr. Gelfand and Dr. Yazdany wrote. They agree with Dr. Kunchok that “next steps should include population-based observational studies that control for disease severity.”
Still, the current study provides additional evidence of rare adverse events in patients receiving TNF inhibitors, they noted. So how should prescribers proceed?
“As with all treatments, the risk-benefit ratio for the individual patient’s situation must be weighed and appropriate counseling must be given to facilitate shared decision-making discussions,” wrote the editorialists.
“Given what is known about the risk of harm, avoiding TNF inhibitors is advisable in patients with known MS,” they wrote.
In addition, neurologic consultation can be helpful for clarifying diagnoses and providing advice on monitoring strategies for TNF inhibitor treatment in those with possible MS or other demyelinating conditions, noted the editorialists.
“In patients who develop new concerning neurological symptoms while receiving TNF inhibitor treatment, timely evaluation is indicated, including consideration of neuroinflammatory, infectious, and neurological diagnoses that may be unrelated to treatment,” they added.
“Broader awareness of risks that studies such as this one by Kunchok et al provide can ... encourage timelier recognition of potential TNF inhibitor–associated neuroinflammatory events and may improve outcomes for patients,” Dr. Gelfand and Dr. Yazdany concluded.
The study was funded by a grant from the National Center for Advancing Translational Sciences. Dr. Kunchok reports having received research funding from Biogen outside this study. A full list of disclosures for the other study authors is in the original article. Dr. Gelfand reports having received g rants for a clinical trial from Genentech and consulting fees from Biogen, Alexion, Theranica, Impel Neuropharma, Advanced Clinical, Biohaven, and Satsuma. Dr. Yazdany reports having received grants from Pfizer and consulting fees from AstraZeneca and Eli Lilly outside the submitted work.
A version of this article originally appeared on Medscape.com.
, new research suggests
The nested case-control study included more than 200 participants with diseases such as rheumatoid arthritis, psoriasis, and Crohn’s disease. Results showed that exposure to TNF inhibitors was significantly associated with increased risk for demyelinating CNS events, such as multiple sclerosis, and nondemyelinating events, such as meningitis and encephalitis.
Interestingly, disease-specific secondary analyses showed that the strongest association for inflammatory events was in patients with rheumatoid arthritis.
Lead author Amy Kunchok, MD, of Mayo Clinic, Rochester, Minn., noted that “these are highly effective therapies for patients” and that these CNS events are likely uncommon.
“Our study has observed an association, but this does not imply causality. Therefore, we are not cautioning against using these therapies in appropriate patients,” Dr. Kunchok said in an interview.
“Rather, we recommend that clinicians assessing patients with both inflammatory demyelinating and nondemyelinating CNS events consider a detailed evaluation of the medication history, particularly in patients with coexistent autoimmune diseases who may have a current or past history of biological therapies,” she said.
The findings were published in JAMA Neurology.
Poorly understood
TNF inhibitors “are common therapies for certain autoimmune diseases,” the investigators noted.
Previously, a link between exposure to these inhibitors and inflammatory CNS events “has been postulated but is poorly understood,” they wrote.
In the current study, they examined records for 106 patients who were treated at Mayo clinics in Minnesota, Arizona, or Florida from January 2003 through February 2019. All participants had been diagnosed with an autoimmune disease that the Food and Drug Administration has listed as an indication for TNF inhibitor use. This included rheumatoid arthritis (n = 48), ankylosing spondylitis (n = 4), psoriasis and psoriatic arthritis (n = 21), Crohn’s disease (n = 27), and ulcerative colitis (n = 6). Their records also showed diagnostic codes for the inflammatory demyelinating CNS events of relapsing-remitting or primary progressive MS, clinically isolated syndrome, radiologically isolated syndrome, neuromyelitis optica spectrum disorder, and transverse myelitis or for the inflammatory nondemyelinating CNS events of meningitis, meningoencephalitis, encephalitis, neurosarcoidosis, and CNS vasculitis. The investigators also included 106 age-, sex-, and autoimmune disease–matched participants 1:1 to act as the control group.
In the total study population, 64% were women and the median age at disease onset was 52 years. In addition, 60% of the patient group and 40% of the control group were exposed to TNF inhibitors.
Novel finding?
Results showed that TNF inhibitor exposure was significantly linked to increased risk for developing any inflammatory CNS event (adjusted odds ratio, 3.01; 95% CI, 1.55-5.82; P = .001). When the outcomes were stratified by class of inflammatory event, these results were similar. The aOR was 3.09 (95% CI, 1.19-8.04; P = .02) for inflammatory demyelinating CNS events and was 2.97 (95% CI, 1.15-7.65; P = .02) for inflammatory nondemyelinating events.
Dr. Kunchok noted that the association between the inhibitors and nondemyelinating events was “a novel finding from this study.”
In secondary analyses, patients with rheumatoid arthritis and exposure to TNF inhibitors had the strongest association with any inflammatory CNS event (aOR, 4.82; 95% CI, 1.62-14.36; P = .005).
A pooled cohort comprising only the participants with the other autoimmune diseases did not show a significant association between exposure to TNF inhibitors and development of CNS events (P = .09).
“Because of the lack of power, further stratification by individual autoimmune diseases was not analyzed,” the investigators reported.
Although the overall findings showed that exposure to TNF inhibitors was linked to increased risk for inflammatory events, whether this association “represents de novo or exacerbated inflammatory pathways requires further research,” the authors wrote.
Dr. Kunchok added that more research, especially population-based studies, is also needed to examine the incidence of these inflammatory CNS events in patients exposed to TNF-alpha inhibitors.
Adds to the literature
In an accompanying editorial, Jeffrey M. Gelfand, MD, department of neurology at the University of California, San Francisco, and Jinoos Yazdany, MD, Zuckerberg San Francisco General Hospital at UCSF, noted that although the study adds to the literature, the magnitude of the risk found “remains unclear.”
“Randomized clinical trials are not suited to the study of rare adverse events,” Dr. Gelfand and Dr. Yazdany wrote. They agree with Dr. Kunchok that “next steps should include population-based observational studies that control for disease severity.”
Still, the current study provides additional evidence of rare adverse events in patients receiving TNF inhibitors, they noted. So how should prescribers proceed?
“As with all treatments, the risk-benefit ratio for the individual patient’s situation must be weighed and appropriate counseling must be given to facilitate shared decision-making discussions,” wrote the editorialists.
“Given what is known about the risk of harm, avoiding TNF inhibitors is advisable in patients with known MS,” they wrote.
In addition, neurologic consultation can be helpful for clarifying diagnoses and providing advice on monitoring strategies for TNF inhibitor treatment in those with possible MS or other demyelinating conditions, noted the editorialists.
“In patients who develop new concerning neurological symptoms while receiving TNF inhibitor treatment, timely evaluation is indicated, including consideration of neuroinflammatory, infectious, and neurological diagnoses that may be unrelated to treatment,” they added.
“Broader awareness of risks that studies such as this one by Kunchok et al provide can ... encourage timelier recognition of potential TNF inhibitor–associated neuroinflammatory events and may improve outcomes for patients,” Dr. Gelfand and Dr. Yazdany concluded.
The study was funded by a grant from the National Center for Advancing Translational Sciences. Dr. Kunchok reports having received research funding from Biogen outside this study. A full list of disclosures for the other study authors is in the original article. Dr. Gelfand reports having received g rants for a clinical trial from Genentech and consulting fees from Biogen, Alexion, Theranica, Impel Neuropharma, Advanced Clinical, Biohaven, and Satsuma. Dr. Yazdany reports having received grants from Pfizer and consulting fees from AstraZeneca and Eli Lilly outside the submitted work.
A version of this article originally appeared on Medscape.com.
, new research suggests
The nested case-control study included more than 200 participants with diseases such as rheumatoid arthritis, psoriasis, and Crohn’s disease. Results showed that exposure to TNF inhibitors was significantly associated with increased risk for demyelinating CNS events, such as multiple sclerosis, and nondemyelinating events, such as meningitis and encephalitis.
Interestingly, disease-specific secondary analyses showed that the strongest association for inflammatory events was in patients with rheumatoid arthritis.
Lead author Amy Kunchok, MD, of Mayo Clinic, Rochester, Minn., noted that “these are highly effective therapies for patients” and that these CNS events are likely uncommon.
“Our study has observed an association, but this does not imply causality. Therefore, we are not cautioning against using these therapies in appropriate patients,” Dr. Kunchok said in an interview.
“Rather, we recommend that clinicians assessing patients with both inflammatory demyelinating and nondemyelinating CNS events consider a detailed evaluation of the medication history, particularly in patients with coexistent autoimmune diseases who may have a current or past history of biological therapies,” she said.
The findings were published in JAMA Neurology.
Poorly understood
TNF inhibitors “are common therapies for certain autoimmune diseases,” the investigators noted.
Previously, a link between exposure to these inhibitors and inflammatory CNS events “has been postulated but is poorly understood,” they wrote.
In the current study, they examined records for 106 patients who were treated at Mayo clinics in Minnesota, Arizona, or Florida from January 2003 through February 2019. All participants had been diagnosed with an autoimmune disease that the Food and Drug Administration has listed as an indication for TNF inhibitor use. This included rheumatoid arthritis (n = 48), ankylosing spondylitis (n = 4), psoriasis and psoriatic arthritis (n = 21), Crohn’s disease (n = 27), and ulcerative colitis (n = 6). Their records also showed diagnostic codes for the inflammatory demyelinating CNS events of relapsing-remitting or primary progressive MS, clinically isolated syndrome, radiologically isolated syndrome, neuromyelitis optica spectrum disorder, and transverse myelitis or for the inflammatory nondemyelinating CNS events of meningitis, meningoencephalitis, encephalitis, neurosarcoidosis, and CNS vasculitis. The investigators also included 106 age-, sex-, and autoimmune disease–matched participants 1:1 to act as the control group.
In the total study population, 64% were women and the median age at disease onset was 52 years. In addition, 60% of the patient group and 40% of the control group were exposed to TNF inhibitors.
Novel finding?
Results showed that TNF inhibitor exposure was significantly linked to increased risk for developing any inflammatory CNS event (adjusted odds ratio, 3.01; 95% CI, 1.55-5.82; P = .001). When the outcomes were stratified by class of inflammatory event, these results were similar. The aOR was 3.09 (95% CI, 1.19-8.04; P = .02) for inflammatory demyelinating CNS events and was 2.97 (95% CI, 1.15-7.65; P = .02) for inflammatory nondemyelinating events.
Dr. Kunchok noted that the association between the inhibitors and nondemyelinating events was “a novel finding from this study.”
In secondary analyses, patients with rheumatoid arthritis and exposure to TNF inhibitors had the strongest association with any inflammatory CNS event (aOR, 4.82; 95% CI, 1.62-14.36; P = .005).
A pooled cohort comprising only the participants with the other autoimmune diseases did not show a significant association between exposure to TNF inhibitors and development of CNS events (P = .09).
“Because of the lack of power, further stratification by individual autoimmune diseases was not analyzed,” the investigators reported.
Although the overall findings showed that exposure to TNF inhibitors was linked to increased risk for inflammatory events, whether this association “represents de novo or exacerbated inflammatory pathways requires further research,” the authors wrote.
Dr. Kunchok added that more research, especially population-based studies, is also needed to examine the incidence of these inflammatory CNS events in patients exposed to TNF-alpha inhibitors.
Adds to the literature
In an accompanying editorial, Jeffrey M. Gelfand, MD, department of neurology at the University of California, San Francisco, and Jinoos Yazdany, MD, Zuckerberg San Francisco General Hospital at UCSF, noted that although the study adds to the literature, the magnitude of the risk found “remains unclear.”
“Randomized clinical trials are not suited to the study of rare adverse events,” Dr. Gelfand and Dr. Yazdany wrote. They agree with Dr. Kunchok that “next steps should include population-based observational studies that control for disease severity.”
Still, the current study provides additional evidence of rare adverse events in patients receiving TNF inhibitors, they noted. So how should prescribers proceed?
“As with all treatments, the risk-benefit ratio for the individual patient’s situation must be weighed and appropriate counseling must be given to facilitate shared decision-making discussions,” wrote the editorialists.
“Given what is known about the risk of harm, avoiding TNF inhibitors is advisable in patients with known MS,” they wrote.
In addition, neurologic consultation can be helpful for clarifying diagnoses and providing advice on monitoring strategies for TNF inhibitor treatment in those with possible MS or other demyelinating conditions, noted the editorialists.
“In patients who develop new concerning neurological symptoms while receiving TNF inhibitor treatment, timely evaluation is indicated, including consideration of neuroinflammatory, infectious, and neurological diagnoses that may be unrelated to treatment,” they added.
“Broader awareness of risks that studies such as this one by Kunchok et al provide can ... encourage timelier recognition of potential TNF inhibitor–associated neuroinflammatory events and may improve outcomes for patients,” Dr. Gelfand and Dr. Yazdany concluded.
The study was funded by a grant from the National Center for Advancing Translational Sciences. Dr. Kunchok reports having received research funding from Biogen outside this study. A full list of disclosures for the other study authors is in the original article. Dr. Gelfand reports having received g rants for a clinical trial from Genentech and consulting fees from Biogen, Alexion, Theranica, Impel Neuropharma, Advanced Clinical, Biohaven, and Satsuma. Dr. Yazdany reports having received grants from Pfizer and consulting fees from AstraZeneca and Eli Lilly outside the submitted work.
A version of this article originally appeared on Medscape.com.
FDA clears first brain stimulation device to help smokers quit
The Food and Drug Administration has granted marketing approval for the BrainsWay deep transcranial magnetic stimulation (TMS) system to help adult smokers kick tobacco.

in a press release.
As previously reported, the system has already been approved by the FDA as a treatment for patients suffering from obsessive-compulsive disorder and major depressive disorder.
The BrainsWay deep TMS system with H4-coil is designed to target addiction-related brain circuits.
It was evaluated as an aid to short-term smoking cessation in a prospective, double-blind, randomized, sham-controlled, multicenter study that involved 262 adults who had a history of smoking an average of more than 26 years and had attempted to quit multiple times but failed.
Active and sham treatments were performed daily 5 days a week for 3 weeks, followed by an additional three sessions once weekly for 3 weeks, for a total of 18 sessions over 6 weeks.
In the full intention-to-treat population (all 262 participants), the 4-week continuous quit rate (CQR, the primary endpoint) was higher in the active deep TMS group than in the sham TMS group (17.1% vs. 7.9%; P = .0238).
Among participants who completed the study, that is, those who underwent treatment for 4 weeks, who kept daily records, and for whom confirmatory urine samples were available, the CQR was 28.4% in the active deep TMS group, compared with 11.7% in the sham treatment group (P = .0063).
The average number of cigarettes smoked per day, as determined on the basis of daily records (secondary endpoint), was statistically significantly lower in the active deep TMS group, compared with the sham treatment group (P = .0311).
No patient suffered a seizure. The most common adverse event was headache, for which there was no statistical difference between the active and sham treatment groups. Other side effects included application site discomfort, back pain, muscle twitching, and discomfort.
“This FDA clearance represents a significant milestone for BrainsWay and our deep TMS platform technology,” Christopher von Jako, PhD, president and CEO of the company, said in the release.
“While other therapies are currently available, a substantial medical need continues to exist for treatments that can increase the continuous quit rate among smokers,” Dr. von Jako noted.
“Based on the compelling data from our large, randomized pivotal study of 262 subjects, we are confident that our deep TMS technology can play an important role in treating cigarette smokers who seek to quit,” he added.
The company plans a “controlled” U.S. market release of the system for this indication early next year.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has granted marketing approval for the BrainsWay deep transcranial magnetic stimulation (TMS) system to help adult smokers kick tobacco.
in a press release.
As previously reported, the system has already been approved by the FDA as a treatment for patients suffering from obsessive-compulsive disorder and major depressive disorder.
The BrainsWay deep TMS system with H4-coil is designed to target addiction-related brain circuits.
It was evaluated as an aid to short-term smoking cessation in a prospective, double-blind, randomized, sham-controlled, multicenter study that involved 262 adults who had a history of smoking an average of more than 26 years and had attempted to quit multiple times but failed.
Active and sham treatments were performed daily 5 days a week for 3 weeks, followed by an additional three sessions once weekly for 3 weeks, for a total of 18 sessions over 6 weeks.
In the full intention-to-treat population (all 262 participants), the 4-week continuous quit rate (CQR, the primary endpoint) was higher in the active deep TMS group than in the sham TMS group (17.1% vs. 7.9%; P = .0238).
Among participants who completed the study, that is, those who underwent treatment for 4 weeks, who kept daily records, and for whom confirmatory urine samples were available, the CQR was 28.4% in the active deep TMS group, compared with 11.7% in the sham treatment group (P = .0063).
The average number of cigarettes smoked per day, as determined on the basis of daily records (secondary endpoint), was statistically significantly lower in the active deep TMS group, compared with the sham treatment group (P = .0311).
No patient suffered a seizure. The most common adverse event was headache, for which there was no statistical difference between the active and sham treatment groups. Other side effects included application site discomfort, back pain, muscle twitching, and discomfort.
“This FDA clearance represents a significant milestone for BrainsWay and our deep TMS platform technology,” Christopher von Jako, PhD, president and CEO of the company, said in the release.
“While other therapies are currently available, a substantial medical need continues to exist for treatments that can increase the continuous quit rate among smokers,” Dr. von Jako noted.
“Based on the compelling data from our large, randomized pivotal study of 262 subjects, we are confident that our deep TMS technology can play an important role in treating cigarette smokers who seek to quit,” he added.
The company plans a “controlled” U.S. market release of the system for this indication early next year.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has granted marketing approval for the BrainsWay deep transcranial magnetic stimulation (TMS) system to help adult smokers kick tobacco.
in a press release.
As previously reported, the system has already been approved by the FDA as a treatment for patients suffering from obsessive-compulsive disorder and major depressive disorder.
The BrainsWay deep TMS system with H4-coil is designed to target addiction-related brain circuits.
It was evaluated as an aid to short-term smoking cessation in a prospective, double-blind, randomized, sham-controlled, multicenter study that involved 262 adults who had a history of smoking an average of more than 26 years and had attempted to quit multiple times but failed.
Active and sham treatments were performed daily 5 days a week for 3 weeks, followed by an additional three sessions once weekly for 3 weeks, for a total of 18 sessions over 6 weeks.
In the full intention-to-treat population (all 262 participants), the 4-week continuous quit rate (CQR, the primary endpoint) was higher in the active deep TMS group than in the sham TMS group (17.1% vs. 7.9%; P = .0238).
Among participants who completed the study, that is, those who underwent treatment for 4 weeks, who kept daily records, and for whom confirmatory urine samples were available, the CQR was 28.4% in the active deep TMS group, compared with 11.7% in the sham treatment group (P = .0063).
The average number of cigarettes smoked per day, as determined on the basis of daily records (secondary endpoint), was statistically significantly lower in the active deep TMS group, compared with the sham treatment group (P = .0311).
No patient suffered a seizure. The most common adverse event was headache, for which there was no statistical difference between the active and sham treatment groups. Other side effects included application site discomfort, back pain, muscle twitching, and discomfort.
“This FDA clearance represents a significant milestone for BrainsWay and our deep TMS platform technology,” Christopher von Jako, PhD, president and CEO of the company, said in the release.
“While other therapies are currently available, a substantial medical need continues to exist for treatments that can increase the continuous quit rate among smokers,” Dr. von Jako noted.
“Based on the compelling data from our large, randomized pivotal study of 262 subjects, we are confident that our deep TMS technology can play an important role in treating cigarette smokers who seek to quit,” he added.
The company plans a “controlled” U.S. market release of the system for this indication early next year.
A version of this article originally appeared on Medscape.com.
Clinical pearls for administering cognitive exams during the pandemic
Patients have often been labeled as “poor historians” if they are not able to recollect their own medical history, whether through illness or difficulties in communication. But Fred Ovsiew, MD, speaking at Focus on Neuropsychiatry presented by Current Psychiatry and the American Academy of Clinical Psychiatrists, sees that label as an excuse on the part of the clinician.

“I strongly advise you to drop that phrase from your vocabulary if you do use it, because the patient is not the historian. The doctor, the clinician is the historian,” Dr. Ovsiew said at the meeting, presented by Global Academy for Medical Education. “It is the clinician’s job to put the story together using the account by the patient as one source, but [also] interviewing a collateral informant and/or reviewing records, which is necessary in almost every case of a neuropsychiatric illness.”
Rather, clinicians taking history at the bedside should focus on why the patients cannot give a narrative account of their illness. Patients can have narrative incapacity on a psychogenic basis, such as in patients with conversion or somatoform disorder, he explained. “I think this is a result of the narrative incapacity that develops in people who have had trauma or adverse experiences in childhood and insecure attachment. This is shown on the adult attachment interview as a disorganized account of their childhoods.”
Other patients might not be able to recount their medical history because they are amnestic, which leaves their account vague because of a lack of access to information. “It may be frozen in time in the sense that, up to a certain point in their life, they can recount the history,” Dr. Ovsiew said. “But in recent years, their account becomes vague.”
Patients with right hemisphere lesions might not know that their account has incongruity and is implausible, while patients with dorsolateral prefrontal lesions might be aspontaneous, use few words to describe their situation, and have poor insight. Those with ventromedial prefrontal lesions can be impulsive and have poor insight, not considering alternative possibilities, Dr. Ovsiew noted.
Asking open-ended questions of the patient is the first step to identifying any potential narrative incapacity, followed by a detailed medical history by the clinician. When taking a medical history, try avoiding what Dr. Ovsiew calls the “anything like that?” problem, where a clinician asks a question about a cluster of symptoms that would make sense to a doctor, but not a patient. For example, a doctor might ask whether a patient is experiencing “chest pain or leg swelling – anything like that?” because he or she knows what those symptoms have in common, but the patient might not know the relationship between those symptoms. “You can’t count on the patient to tell you all the relevant information,” he said. “You have to know what to ask about.”
“Patients with brain disease have subtle personality changes, sometimes more obvious personality changes. These need to be inquired about,” Dr. Ovsiew said. “The patient with apathy has reduced negative as well as positive emotions. The patient with depression has reduced positive emotions, but often tells you very clearly about the negative emotions of sadness, guilt. The patient with depression has diurnal variation in mood, a very telling symptom, especially when it’s disclosed spontaneously,” Dr. Ovsiew explained. “The point is, you need to know to ask about it.”
When taking a sleep history, clinicians should be aware of sleep disturbances apart from insomnia and early waking. REM sleep behavior disorder is a condition that should be inquired about. Obstructive sleep apnea is a condition that might not be immediately apparent to the patient, but a bed partner can identify whether a patient has problems breathing throughout the night.
“This is an important condition to uncover for the neuropsychiatrist because it contributes to treatment resistance and depression, and it contributes to cognitive impairment,” Dr. Ovsiew said. “These patients commonly have mild difficulties with attention and concentration.”
Always ask about head injury in every history, which can be relevant to later onset depression, PTSD, and cognitive impairment. Every head injury follows a trajectory of retrograde amnesia and altered state of consciousness (including coma), followed by a period of posttraumatic amnesia. Duration of these states can be used to assess the severity of brain injury, but the 15-point Glasgow Coma Scale is another way to assess injury severity, Dr. Ovsiew explained.
However, the two do not always overlap, he noted. “Someone may have a Glasgow Coma Scale score that is 9-12, predicting moderate brain injury, but they may have a short duration of amnesia. These don’t always follow the same path. There are many different ways of classifying how severe the brain injury is.”
Keep probes brief, straightforward
Cognitive exams of patients with suspected psychiatric disorders should be simple, easy to administer and focused on a single domain of cognition. “Probes should be brief. They should not require specialized equipment. The Purdue Pegboard Test might be a great neuropsychological instrument, but very few of us carry a pegboard around in our medical bags,” Dr. Ovsiew said.
The probe administered should also be accessible to the patient. The serial sevens clinical test, where a patient is asked to repeatedly subtract 7 from 100, is only effective at testing concentration if the patient is capable of completing the test. “There are going to be patients who can’t do the task, but it’s not because of concentration failure, it’s because of subtraction failure,” he said.
When assessing attention, effective tasks include having the patient perform the digit span test forward and backward, count backward from 20 to 1, listing the months of the year in reverse, and performing the Mental Alternation Test. However, Dr. Ovsiew explained there may be some barriers for patients in completing these tasks. “The person may be aphasic and not know the alphabet. The person may have English as a second language and not be skilled at giving the alphabet in English. In some cases, you may want to check and not assume that the patient can count and does know the alphabet.”
In assessing language, listen for aphasic abnormalities. “The patient, of course, is speaking throughout the interview, but you need to take a moment to listen for prosody, to listen to rate of speech, to listen for paraphasic errors or word-finding problems,” Dr. Ovsiew said. Any abnormalities should be probed further through confrontation naming tasks, which can be done in person and with some success through video, but not by phone. Naming to definition (“What do you call the part of a shirt that covers the arm?”) is one way of administering the test over the phone.
Visuospatial function can be assessed by clock drawing but also carries problems. Patients who do not plan their clock before beginning to draw, for example, may have an executive function problem instead of a visuospatial problem, Dr. Ovsiew noted. Patients in whom a clinician suspects hemineglect should be given a visual search task or line by section task. “I like doing clock drawing. It’s a nice screening test. It’s becoming, I think, less useful as people count on digital clocks and have trouble even imagining what an analog clock looks like.”
An approach that is better suited to in-person assessment, but also works by video, is the Poppelreuter figure visual perceptual function test, which is a prompt for the patient that involves common household items overlaying one another “in atypical positions and atypical configurations” where the patient is instructed to describe the items they see on the card. Another approach that works over video is the interlocking finger test, where the patient is asked to copy the hand positions made by the clinician.
Dr. Ovsiew admitted that visuospatial function is nearly impossible to assess over the phone. Asking topographical questions (“If you’re driving from Chicago to Los Angeles, is the Pacific Ocean in front of you, behind you, to your left, or to your right?”) may help judge visuospatial function, but this relies on the patient having the topographic knowledge to answer the questions. Some patients who are topographically disoriented can’t do them at all,” Dr. Ovsiew said.
Bedside neuropsychiatry assesses encoding of a memory, its retention and its retrieval as well as verbal and visual cues. Each one of these aspects of memory can be impaired on its own and should be explored separately, Dr. Ovsiew explained. “Neuropsychiatric clinicians have a rough-and-ready, seat-of-the-pants way of approaching this that wouldn’t pass muster if you’re a psychologist, but is the best we can do at the bedside.”
To test retrieval and retention, the Three Words–Three Shapes test works well in person, with some difficulty by video, and is not possible to administer over the phone. In lieu of that test, giving the patient a simple word list and asking them to repeat the list in order. Using the word list, “these different stages of memory function can be parsed out pretty well at the bedside or chairside, and even by the phone. Figuring out where the memory failure is diagnostically important,” Dr. Ovsiew said.
Executive function, which involves activation, planning, sequencing, maintaining, self-monitoring, and flexible employment of action and attention, is “complicated to evaluate because there are multiple aspects of executive function, multiple deficits that can be seen with executive dysfunction, and they don’t all correlate with each other.”
Within executive function evaluation, the Mental Alternation Test can assess working memory, motor sequencing can be assessed through the ring/fist, fist/edge/palm, alternating fist, and rampart tests. The Go/No-Go test can be used to assess response inhibition. For effortful retrieval evaluation, spontaneous word-list generation – such as thinking of all the items one can buy at a supermarket– can test category fluency, while a task to name all the words starting with a certain letter can assess letter stimulus.
Executive function “is of crucial importance in the neuropsychiatric evaluation because it’s strongly correlated with how well the person functions outside the office,” Dr. Ovsiew said.
Global Academy and this news organization are owned by the same parent company. Dr. Ovsiew reported relationships with Wolters Kluwer Health in the form of consulting, receiving royalty payments, and related activities.
Patients have often been labeled as “poor historians” if they are not able to recollect their own medical history, whether through illness or difficulties in communication. But Fred Ovsiew, MD, speaking at Focus on Neuropsychiatry presented by Current Psychiatry and the American Academy of Clinical Psychiatrists, sees that label as an excuse on the part of the clinician.
“I strongly advise you to drop that phrase from your vocabulary if you do use it, because the patient is not the historian. The doctor, the clinician is the historian,” Dr. Ovsiew said at the meeting, presented by Global Academy for Medical Education. “It is the clinician’s job to put the story together using the account by the patient as one source, but [also] interviewing a collateral informant and/or reviewing records, which is necessary in almost every case of a neuropsychiatric illness.”
Rather, clinicians taking history at the bedside should focus on why the patients cannot give a narrative account of their illness. Patients can have narrative incapacity on a psychogenic basis, such as in patients with conversion or somatoform disorder, he explained. “I think this is a result of the narrative incapacity that develops in people who have had trauma or adverse experiences in childhood and insecure attachment. This is shown on the adult attachment interview as a disorganized account of their childhoods.”
Other patients might not be able to recount their medical history because they are amnestic, which leaves their account vague because of a lack of access to information. “It may be frozen in time in the sense that, up to a certain point in their life, they can recount the history,” Dr. Ovsiew said. “But in recent years, their account becomes vague.”
Patients with right hemisphere lesions might not know that their account has incongruity and is implausible, while patients with dorsolateral prefrontal lesions might be aspontaneous, use few words to describe their situation, and have poor insight. Those with ventromedial prefrontal lesions can be impulsive and have poor insight, not considering alternative possibilities, Dr. Ovsiew noted.
Asking open-ended questions of the patient is the first step to identifying any potential narrative incapacity, followed by a detailed medical history by the clinician. When taking a medical history, try avoiding what Dr. Ovsiew calls the “anything like that?” problem, where a clinician asks a question about a cluster of symptoms that would make sense to a doctor, but not a patient. For example, a doctor might ask whether a patient is experiencing “chest pain or leg swelling – anything like that?” because he or she knows what those symptoms have in common, but the patient might not know the relationship between those symptoms. “You can’t count on the patient to tell you all the relevant information,” he said. “You have to know what to ask about.”
“Patients with brain disease have subtle personality changes, sometimes more obvious personality changes. These need to be inquired about,” Dr. Ovsiew said. “The patient with apathy has reduced negative as well as positive emotions. The patient with depression has reduced positive emotions, but often tells you very clearly about the negative emotions of sadness, guilt. The patient with depression has diurnal variation in mood, a very telling symptom, especially when it’s disclosed spontaneously,” Dr. Ovsiew explained. “The point is, you need to know to ask about it.”
When taking a sleep history, clinicians should be aware of sleep disturbances apart from insomnia and early waking. REM sleep behavior disorder is a condition that should be inquired about. Obstructive sleep apnea is a condition that might not be immediately apparent to the patient, but a bed partner can identify whether a patient has problems breathing throughout the night.
“This is an important condition to uncover for the neuropsychiatrist because it contributes to treatment resistance and depression, and it contributes to cognitive impairment,” Dr. Ovsiew said. “These patients commonly have mild difficulties with attention and concentration.”
Always ask about head injury in every history, which can be relevant to later onset depression, PTSD, and cognitive impairment. Every head injury follows a trajectory of retrograde amnesia and altered state of consciousness (including coma), followed by a period of posttraumatic amnesia. Duration of these states can be used to assess the severity of brain injury, but the 15-point Glasgow Coma Scale is another way to assess injury severity, Dr. Ovsiew explained.
However, the two do not always overlap, he noted. “Someone may have a Glasgow Coma Scale score that is 9-12, predicting moderate brain injury, but they may have a short duration of amnesia. These don’t always follow the same path. There are many different ways of classifying how severe the brain injury is.”
Keep probes brief, straightforward
Cognitive exams of patients with suspected psychiatric disorders should be simple, easy to administer and focused on a single domain of cognition. “Probes should be brief. They should not require specialized equipment. The Purdue Pegboard Test might be a great neuropsychological instrument, but very few of us carry a pegboard around in our medical bags,” Dr. Ovsiew said.
The probe administered should also be accessible to the patient. The serial sevens clinical test, where a patient is asked to repeatedly subtract 7 from 100, is only effective at testing concentration if the patient is capable of completing the test. “There are going to be patients who can’t do the task, but it’s not because of concentration failure, it’s because of subtraction failure,” he said.
When assessing attention, effective tasks include having the patient perform the digit span test forward and backward, count backward from 20 to 1, listing the months of the year in reverse, and performing the Mental Alternation Test. However, Dr. Ovsiew explained there may be some barriers for patients in completing these tasks. “The person may be aphasic and not know the alphabet. The person may have English as a second language and not be skilled at giving the alphabet in English. In some cases, you may want to check and not assume that the patient can count and does know the alphabet.”
In assessing language, listen for aphasic abnormalities. “The patient, of course, is speaking throughout the interview, but you need to take a moment to listen for prosody, to listen to rate of speech, to listen for paraphasic errors or word-finding problems,” Dr. Ovsiew said. Any abnormalities should be probed further through confrontation naming tasks, which can be done in person and with some success through video, but not by phone. Naming to definition (“What do you call the part of a shirt that covers the arm?”) is one way of administering the test over the phone.
Visuospatial function can be assessed by clock drawing but also carries problems. Patients who do not plan their clock before beginning to draw, for example, may have an executive function problem instead of a visuospatial problem, Dr. Ovsiew noted. Patients in whom a clinician suspects hemineglect should be given a visual search task or line by section task. “I like doing clock drawing. It’s a nice screening test. It’s becoming, I think, less useful as people count on digital clocks and have trouble even imagining what an analog clock looks like.”
An approach that is better suited to in-person assessment, but also works by video, is the Poppelreuter figure visual perceptual function test, which is a prompt for the patient that involves common household items overlaying one another “in atypical positions and atypical configurations” where the patient is instructed to describe the items they see on the card. Another approach that works over video is the interlocking finger test, where the patient is asked to copy the hand positions made by the clinician.
Dr. Ovsiew admitted that visuospatial function is nearly impossible to assess over the phone. Asking topographical questions (“If you’re driving from Chicago to Los Angeles, is the Pacific Ocean in front of you, behind you, to your left, or to your right?”) may help judge visuospatial function, but this relies on the patient having the topographic knowledge to answer the questions. Some patients who are topographically disoriented can’t do them at all,” Dr. Ovsiew said.
Bedside neuropsychiatry assesses encoding of a memory, its retention and its retrieval as well as verbal and visual cues. Each one of these aspects of memory can be impaired on its own and should be explored separately, Dr. Ovsiew explained. “Neuropsychiatric clinicians have a rough-and-ready, seat-of-the-pants way of approaching this that wouldn’t pass muster if you’re a psychologist, but is the best we can do at the bedside.”
To test retrieval and retention, the Three Words–Three Shapes test works well in person, with some difficulty by video, and is not possible to administer over the phone. In lieu of that test, giving the patient a simple word list and asking them to repeat the list in order. Using the word list, “these different stages of memory function can be parsed out pretty well at the bedside or chairside, and even by the phone. Figuring out where the memory failure is diagnostically important,” Dr. Ovsiew said.
Executive function, which involves activation, planning, sequencing, maintaining, self-monitoring, and flexible employment of action and attention, is “complicated to evaluate because there are multiple aspects of executive function, multiple deficits that can be seen with executive dysfunction, and they don’t all correlate with each other.”
Within executive function evaluation, the Mental Alternation Test can assess working memory, motor sequencing can be assessed through the ring/fist, fist/edge/palm, alternating fist, and rampart tests. The Go/No-Go test can be used to assess response inhibition. For effortful retrieval evaluation, spontaneous word-list generation – such as thinking of all the items one can buy at a supermarket– can test category fluency, while a task to name all the words starting with a certain letter can assess letter stimulus.
Executive function “is of crucial importance in the neuropsychiatric evaluation because it’s strongly correlated with how well the person functions outside the office,” Dr. Ovsiew said.
Global Academy and this news organization are owned by the same parent company. Dr. Ovsiew reported relationships with Wolters Kluwer Health in the form of consulting, receiving royalty payments, and related activities.
Patients have often been labeled as “poor historians” if they are not able to recollect their own medical history, whether through illness or difficulties in communication. But Fred Ovsiew, MD, speaking at Focus on Neuropsychiatry presented by Current Psychiatry and the American Academy of Clinical Psychiatrists, sees that label as an excuse on the part of the clinician.
“I strongly advise you to drop that phrase from your vocabulary if you do use it, because the patient is not the historian. The doctor, the clinician is the historian,” Dr. Ovsiew said at the meeting, presented by Global Academy for Medical Education. “It is the clinician’s job to put the story together using the account by the patient as one source, but [also] interviewing a collateral informant and/or reviewing records, which is necessary in almost every case of a neuropsychiatric illness.”
Rather, clinicians taking history at the bedside should focus on why the patients cannot give a narrative account of their illness. Patients can have narrative incapacity on a psychogenic basis, such as in patients with conversion or somatoform disorder, he explained. “I think this is a result of the narrative incapacity that develops in people who have had trauma or adverse experiences in childhood and insecure attachment. This is shown on the adult attachment interview as a disorganized account of their childhoods.”
Other patients might not be able to recount their medical history because they are amnestic, which leaves their account vague because of a lack of access to information. “It may be frozen in time in the sense that, up to a certain point in their life, they can recount the history,” Dr. Ovsiew said. “But in recent years, their account becomes vague.”
Patients with right hemisphere lesions might not know that their account has incongruity and is implausible, while patients with dorsolateral prefrontal lesions might be aspontaneous, use few words to describe their situation, and have poor insight. Those with ventromedial prefrontal lesions can be impulsive and have poor insight, not considering alternative possibilities, Dr. Ovsiew noted.
Asking open-ended questions of the patient is the first step to identifying any potential narrative incapacity, followed by a detailed medical history by the clinician. When taking a medical history, try avoiding what Dr. Ovsiew calls the “anything like that?” problem, where a clinician asks a question about a cluster of symptoms that would make sense to a doctor, but not a patient. For example, a doctor might ask whether a patient is experiencing “chest pain or leg swelling – anything like that?” because he or she knows what those symptoms have in common, but the patient might not know the relationship between those symptoms. “You can’t count on the patient to tell you all the relevant information,” he said. “You have to know what to ask about.”
“Patients with brain disease have subtle personality changes, sometimes more obvious personality changes. These need to be inquired about,” Dr. Ovsiew said. “The patient with apathy has reduced negative as well as positive emotions. The patient with depression has reduced positive emotions, but often tells you very clearly about the negative emotions of sadness, guilt. The patient with depression has diurnal variation in mood, a very telling symptom, especially when it’s disclosed spontaneously,” Dr. Ovsiew explained. “The point is, you need to know to ask about it.”
When taking a sleep history, clinicians should be aware of sleep disturbances apart from insomnia and early waking. REM sleep behavior disorder is a condition that should be inquired about. Obstructive sleep apnea is a condition that might not be immediately apparent to the patient, but a bed partner can identify whether a patient has problems breathing throughout the night.
“This is an important condition to uncover for the neuropsychiatrist because it contributes to treatment resistance and depression, and it contributes to cognitive impairment,” Dr. Ovsiew said. “These patients commonly have mild difficulties with attention and concentration.”
Always ask about head injury in every history, which can be relevant to later onset depression, PTSD, and cognitive impairment. Every head injury follows a trajectory of retrograde amnesia and altered state of consciousness (including coma), followed by a period of posttraumatic amnesia. Duration of these states can be used to assess the severity of brain injury, but the 15-point Glasgow Coma Scale is another way to assess injury severity, Dr. Ovsiew explained.
However, the two do not always overlap, he noted. “Someone may have a Glasgow Coma Scale score that is 9-12, predicting moderate brain injury, but they may have a short duration of amnesia. These don’t always follow the same path. There are many different ways of classifying how severe the brain injury is.”
Keep probes brief, straightforward
Cognitive exams of patients with suspected psychiatric disorders should be simple, easy to administer and focused on a single domain of cognition. “Probes should be brief. They should not require specialized equipment. The Purdue Pegboard Test might be a great neuropsychological instrument, but very few of us carry a pegboard around in our medical bags,” Dr. Ovsiew said.
The probe administered should also be accessible to the patient. The serial sevens clinical test, where a patient is asked to repeatedly subtract 7 from 100, is only effective at testing concentration if the patient is capable of completing the test. “There are going to be patients who can’t do the task, but it’s not because of concentration failure, it’s because of subtraction failure,” he said.
When assessing attention, effective tasks include having the patient perform the digit span test forward and backward, count backward from 20 to 1, listing the months of the year in reverse, and performing the Mental Alternation Test. However, Dr. Ovsiew explained there may be some barriers for patients in completing these tasks. “The person may be aphasic and not know the alphabet. The person may have English as a second language and not be skilled at giving the alphabet in English. In some cases, you may want to check and not assume that the patient can count and does know the alphabet.”
In assessing language, listen for aphasic abnormalities. “The patient, of course, is speaking throughout the interview, but you need to take a moment to listen for prosody, to listen to rate of speech, to listen for paraphasic errors or word-finding problems,” Dr. Ovsiew said. Any abnormalities should be probed further through confrontation naming tasks, which can be done in person and with some success through video, but not by phone. Naming to definition (“What do you call the part of a shirt that covers the arm?”) is one way of administering the test over the phone.
Visuospatial function can be assessed by clock drawing but also carries problems. Patients who do not plan their clock before beginning to draw, for example, may have an executive function problem instead of a visuospatial problem, Dr. Ovsiew noted. Patients in whom a clinician suspects hemineglect should be given a visual search task or line by section task. “I like doing clock drawing. It’s a nice screening test. It’s becoming, I think, less useful as people count on digital clocks and have trouble even imagining what an analog clock looks like.”
An approach that is better suited to in-person assessment, but also works by video, is the Poppelreuter figure visual perceptual function test, which is a prompt for the patient that involves common household items overlaying one another “in atypical positions and atypical configurations” where the patient is instructed to describe the items they see on the card. Another approach that works over video is the interlocking finger test, where the patient is asked to copy the hand positions made by the clinician.
Dr. Ovsiew admitted that visuospatial function is nearly impossible to assess over the phone. Asking topographical questions (“If you’re driving from Chicago to Los Angeles, is the Pacific Ocean in front of you, behind you, to your left, or to your right?”) may help judge visuospatial function, but this relies on the patient having the topographic knowledge to answer the questions. Some patients who are topographically disoriented can’t do them at all,” Dr. Ovsiew said.
Bedside neuropsychiatry assesses encoding of a memory, its retention and its retrieval as well as verbal and visual cues. Each one of these aspects of memory can be impaired on its own and should be explored separately, Dr. Ovsiew explained. “Neuropsychiatric clinicians have a rough-and-ready, seat-of-the-pants way of approaching this that wouldn’t pass muster if you’re a psychologist, but is the best we can do at the bedside.”
To test retrieval and retention, the Three Words–Three Shapes test works well in person, with some difficulty by video, and is not possible to administer over the phone. In lieu of that test, giving the patient a simple word list and asking them to repeat the list in order. Using the word list, “these different stages of memory function can be parsed out pretty well at the bedside or chairside, and even by the phone. Figuring out where the memory failure is diagnostically important,” Dr. Ovsiew said.
Executive function, which involves activation, planning, sequencing, maintaining, self-monitoring, and flexible employment of action and attention, is “complicated to evaluate because there are multiple aspects of executive function, multiple deficits that can be seen with executive dysfunction, and they don’t all correlate with each other.”
Within executive function evaluation, the Mental Alternation Test can assess working memory, motor sequencing can be assessed through the ring/fist, fist/edge/palm, alternating fist, and rampart tests. The Go/No-Go test can be used to assess response inhibition. For effortful retrieval evaluation, spontaneous word-list generation – such as thinking of all the items one can buy at a supermarket– can test category fluency, while a task to name all the words starting with a certain letter can assess letter stimulus.
Executive function “is of crucial importance in the neuropsychiatric evaluation because it’s strongly correlated with how well the person functions outside the office,” Dr. Ovsiew said.
Global Academy and this news organization are owned by the same parent company. Dr. Ovsiew reported relationships with Wolters Kluwer Health in the form of consulting, receiving royalty payments, and related activities.
FROM FOCUS ON NEUROPSYCHIATRY 2020
COVID-19 linked to development of myasthenia gravis
Myasthenia gravis should be added to the growing list of potential neurological sequelae associated with COVID-19, new research suggests. Clinicians from Italy have described what they believe are
“I think it is possible that there could be many more cases,” said lead author Domenico Restivo, MD, of the Garibaldi Hospital, Catania, Italy. “In fact, myasthenia gravis could be underestimated especially in the course of COVID-19 infection in which a specific muscular weakness is frequently present. For this reason, this association is easy to miss if not top of mind,” Dr. Restivo said.
None of the three patients had previous neurologic or autoimmune disorders. In all three cases, symptoms of myasthenia gravis appeared within 5-7 days after onset of fever caused by SARS-CoV-2 infection. The time from presumed SARS-CoV-2 infection to myasthenia gravis symptoms “is consistent with the time from infection to symptoms in other neurologic disorders triggered by infections,” the investigators reported.
The findings were published online August 10 in Annals of Internal Medicine.
First patients
The first patient described in the report was a 64-year-old man who had a fever as high as 39° C (102.2° F) for 4 days. Five days after fever onset, he developed diplopia and muscle fatigue. The patient’s neurologic examination was “unremarkable.” Computed tomography (CT) of the thorax excluded thymoma, and findings on chest radiograph were normal. He tested positive for SARS-CoV-2 on nasopharyngeal swab and real-time reverse transcriptase polymerase chain reaction (RT-PCR).
The patient’s symptoms led the investigators to suspect myasthenia gravis. Repetitive stimulation of the patient’s facial nerve showed a 57% decrement, confirming involvement of the postsynaptic neuromuscular junction. The concentration of AChR antibodies in serum was also elevated (22.8 pmol/L; reference range, <0.4 pmol/L). The patient was treated with pyridostigmine bromide and prednisone and had a response “typical for someone with myasthenia gravis,” the researchers wrote.
The second patient was a 68-year-old man who had a fever as high as 38.8° C (101.8° F) for 7 days. On day 7, he developed muscle fatigue, diplopia, and dysphagia. Findings of a chest CT and neurologic exam were normal. Nasopharyngeal swab and RT-PCR testing for COVID-19 were positive. As with the first patient, myasthenia gravis was suspected because of the patient’s symptoms. Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the facial (52%) and ulnar (21%) nerves. His serum AChR antibody level was elevated (27.6 pmol/L). The patient improved after one cycle of intravenous immunoglobulin treatment.
Possible mechanisms
The third patient was a 71-year-old woman with cough and a fever up to 38.6° C (101.5° F) for 6 days. She initially tested negative for SARS-CoV-2 on nasopharyngeal swab and RT-PCR. Five days after her symptoms began, she developed bilateral ocular ptosis, diplopia, and hypophonia. CT of the thorax excluded thymoma but showed bilateral interstitial pneumonia. On day 6, she developed dysphagia and respiratory failure, and was transferred to the ICU where she received mechanical ventilation.
Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the ulnar nerve (56%), and her serum AChR antibody level was elevated (35.6 pmol/L). Five days later, a second nasopharyngeal swab test for SARS-CoV-2 was positive. The patient improved following plasmapheresis treatment and was successfully extubated.
The investigators noted that this patient received hydroxychloroquine the day after the onset of neurologic symptoms, but the drug was withdrawn a day later, so they do not believe that it caused the symptoms of myasthenia gravis.
The observations in these three patients are “consistent with reports of other infections that induce autoimmune disorders, as well as with the growing evidence of other neurologic disorders with presumed autoimmune mechanisms after COVID-19 onset,” the researchers wrote.
They offered several possible explanations for the link between COVID-19 and myasthenia gravis. “Antibodies that are directed against SARS-CoV-2 proteins may cross-react with AChR subunits, because the virus has epitopes that are similar to components of the neuromuscular junction; this is known to occur in other neurologic autoimmune disorders after infection. Alternatively, COVID-19 infection may break immunologic self-tolerance,” the investigators wrote.
“The main message for clinicians is that myasthenia gravis, as well as other neurological disorders associated with autoimmunity, could occur in the course of SARS-CoV-2 infection,” Dr. Restivo said. Prompt recognition of the disease “could lead to a drug treatment that limits its evolution as quickly as possible,” he added.
An “unmasking”
Commenting on the findings, Anthony Geraci, MD, director of neuromuscular medicine, Northwell Health, Great Neck, N.Y., said these case reports of myasthenia gravis after SARS-CoV-2 infection are “not unique or novel as there has been a long understanding that seropositive [AChR antibody-positive] myasthenia gravis can and is frequently ‘unmasked’ in the setting” of several viral and bacterial infections.
“Antibodies in myasthenia gravis are of a type that take several weeks to develop to measurable levels as in the reported cases by Restivo et al., giving strong support to the notion that subclinical myasthenia gravis can be immunologically upregulated in the setting of viral infection and this is a far more likely explanation of the observed association reported,” added Dr. Geraci, who was not involved with the research.
He noted that, at his institution, “we have also observed ocular myasthenia gravis emerge in patients with SARS-CoV-2 infection, with similar double vision and lid droop, as we have seen similarly in patients with Zika, West Nile, and other viral infections, as well as a multiplicity of bacterial infections.”
“Most of our observed patients have responded to treatment much the same as reported by the three cases from Restivo and colleagues,” Dr.Geraci reported.
The authors of the study disclosed no conflicts of interest.
A version of this article originally appeared on Medscape.com.
Myasthenia gravis should be added to the growing list of potential neurological sequelae associated with COVID-19, new research suggests. Clinicians from Italy have described what they believe are
“I think it is possible that there could be many more cases,” said lead author Domenico Restivo, MD, of the Garibaldi Hospital, Catania, Italy. “In fact, myasthenia gravis could be underestimated especially in the course of COVID-19 infection in which a specific muscular weakness is frequently present. For this reason, this association is easy to miss if not top of mind,” Dr. Restivo said.
None of the three patients had previous neurologic or autoimmune disorders. In all three cases, symptoms of myasthenia gravis appeared within 5-7 days after onset of fever caused by SARS-CoV-2 infection. The time from presumed SARS-CoV-2 infection to myasthenia gravis symptoms “is consistent with the time from infection to symptoms in other neurologic disorders triggered by infections,” the investigators reported.
The findings were published online August 10 in Annals of Internal Medicine.
First patients
The first patient described in the report was a 64-year-old man who had a fever as high as 39° C (102.2° F) for 4 days. Five days after fever onset, he developed diplopia and muscle fatigue. The patient’s neurologic examination was “unremarkable.” Computed tomography (CT) of the thorax excluded thymoma, and findings on chest radiograph were normal. He tested positive for SARS-CoV-2 on nasopharyngeal swab and real-time reverse transcriptase polymerase chain reaction (RT-PCR).
The patient’s symptoms led the investigators to suspect myasthenia gravis. Repetitive stimulation of the patient’s facial nerve showed a 57% decrement, confirming involvement of the postsynaptic neuromuscular junction. The concentration of AChR antibodies in serum was also elevated (22.8 pmol/L; reference range, <0.4 pmol/L). The patient was treated with pyridostigmine bromide and prednisone and had a response “typical for someone with myasthenia gravis,” the researchers wrote.
The second patient was a 68-year-old man who had a fever as high as 38.8° C (101.8° F) for 7 days. On day 7, he developed muscle fatigue, diplopia, and dysphagia. Findings of a chest CT and neurologic exam were normal. Nasopharyngeal swab and RT-PCR testing for COVID-19 were positive. As with the first patient, myasthenia gravis was suspected because of the patient’s symptoms. Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the facial (52%) and ulnar (21%) nerves. His serum AChR antibody level was elevated (27.6 pmol/L). The patient improved after one cycle of intravenous immunoglobulin treatment.
Possible mechanisms
The third patient was a 71-year-old woman with cough and a fever up to 38.6° C (101.5° F) for 6 days. She initially tested negative for SARS-CoV-2 on nasopharyngeal swab and RT-PCR. Five days after her symptoms began, she developed bilateral ocular ptosis, diplopia, and hypophonia. CT of the thorax excluded thymoma but showed bilateral interstitial pneumonia. On day 6, she developed dysphagia and respiratory failure, and was transferred to the ICU where she received mechanical ventilation.
Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the ulnar nerve (56%), and her serum AChR antibody level was elevated (35.6 pmol/L). Five days later, a second nasopharyngeal swab test for SARS-CoV-2 was positive. The patient improved following plasmapheresis treatment and was successfully extubated.
The investigators noted that this patient received hydroxychloroquine the day after the onset of neurologic symptoms, but the drug was withdrawn a day later, so they do not believe that it caused the symptoms of myasthenia gravis.
The observations in these three patients are “consistent with reports of other infections that induce autoimmune disorders, as well as with the growing evidence of other neurologic disorders with presumed autoimmune mechanisms after COVID-19 onset,” the researchers wrote.
They offered several possible explanations for the link between COVID-19 and myasthenia gravis. “Antibodies that are directed against SARS-CoV-2 proteins may cross-react with AChR subunits, because the virus has epitopes that are similar to components of the neuromuscular junction; this is known to occur in other neurologic autoimmune disorders after infection. Alternatively, COVID-19 infection may break immunologic self-tolerance,” the investigators wrote.
“The main message for clinicians is that myasthenia gravis, as well as other neurological disorders associated with autoimmunity, could occur in the course of SARS-CoV-2 infection,” Dr. Restivo said. Prompt recognition of the disease “could lead to a drug treatment that limits its evolution as quickly as possible,” he added.
An “unmasking”
Commenting on the findings, Anthony Geraci, MD, director of neuromuscular medicine, Northwell Health, Great Neck, N.Y., said these case reports of myasthenia gravis after SARS-CoV-2 infection are “not unique or novel as there has been a long understanding that seropositive [AChR antibody-positive] myasthenia gravis can and is frequently ‘unmasked’ in the setting” of several viral and bacterial infections.
“Antibodies in myasthenia gravis are of a type that take several weeks to develop to measurable levels as in the reported cases by Restivo et al., giving strong support to the notion that subclinical myasthenia gravis can be immunologically upregulated in the setting of viral infection and this is a far more likely explanation of the observed association reported,” added Dr. Geraci, who was not involved with the research.
He noted that, at his institution, “we have also observed ocular myasthenia gravis emerge in patients with SARS-CoV-2 infection, with similar double vision and lid droop, as we have seen similarly in patients with Zika, West Nile, and other viral infections, as well as a multiplicity of bacterial infections.”
“Most of our observed patients have responded to treatment much the same as reported by the three cases from Restivo and colleagues,” Dr.Geraci reported.
The authors of the study disclosed no conflicts of interest.
A version of this article originally appeared on Medscape.com.
Myasthenia gravis should be added to the growing list of potential neurological sequelae associated with COVID-19, new research suggests. Clinicians from Italy have described what they believe are
“I think it is possible that there could be many more cases,” said lead author Domenico Restivo, MD, of the Garibaldi Hospital, Catania, Italy. “In fact, myasthenia gravis could be underestimated especially in the course of COVID-19 infection in which a specific muscular weakness is frequently present. For this reason, this association is easy to miss if not top of mind,” Dr. Restivo said.
None of the three patients had previous neurologic or autoimmune disorders. In all three cases, symptoms of myasthenia gravis appeared within 5-7 days after onset of fever caused by SARS-CoV-2 infection. The time from presumed SARS-CoV-2 infection to myasthenia gravis symptoms “is consistent with the time from infection to symptoms in other neurologic disorders triggered by infections,” the investigators reported.
The findings were published online August 10 in Annals of Internal Medicine.
First patients
The first patient described in the report was a 64-year-old man who had a fever as high as 39° C (102.2° F) for 4 days. Five days after fever onset, he developed diplopia and muscle fatigue. The patient’s neurologic examination was “unremarkable.” Computed tomography (CT) of the thorax excluded thymoma, and findings on chest radiograph were normal. He tested positive for SARS-CoV-2 on nasopharyngeal swab and real-time reverse transcriptase polymerase chain reaction (RT-PCR).
The patient’s symptoms led the investigators to suspect myasthenia gravis. Repetitive stimulation of the patient’s facial nerve showed a 57% decrement, confirming involvement of the postsynaptic neuromuscular junction. The concentration of AChR antibodies in serum was also elevated (22.8 pmol/L; reference range, <0.4 pmol/L). The patient was treated with pyridostigmine bromide and prednisone and had a response “typical for someone with myasthenia gravis,” the researchers wrote.
The second patient was a 68-year-old man who had a fever as high as 38.8° C (101.8° F) for 7 days. On day 7, he developed muscle fatigue, diplopia, and dysphagia. Findings of a chest CT and neurologic exam were normal. Nasopharyngeal swab and RT-PCR testing for COVID-19 were positive. As with the first patient, myasthenia gravis was suspected because of the patient’s symptoms. Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the facial (52%) and ulnar (21%) nerves. His serum AChR antibody level was elevated (27.6 pmol/L). The patient improved after one cycle of intravenous immunoglobulin treatment.
Possible mechanisms
The third patient was a 71-year-old woman with cough and a fever up to 38.6° C (101.5° F) for 6 days. She initially tested negative for SARS-CoV-2 on nasopharyngeal swab and RT-PCR. Five days after her symptoms began, she developed bilateral ocular ptosis, diplopia, and hypophonia. CT of the thorax excluded thymoma but showed bilateral interstitial pneumonia. On day 6, she developed dysphagia and respiratory failure, and was transferred to the ICU where she received mechanical ventilation.
Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the ulnar nerve (56%), and her serum AChR antibody level was elevated (35.6 pmol/L). Five days later, a second nasopharyngeal swab test for SARS-CoV-2 was positive. The patient improved following plasmapheresis treatment and was successfully extubated.
The investigators noted that this patient received hydroxychloroquine the day after the onset of neurologic symptoms, but the drug was withdrawn a day later, so they do not believe that it caused the symptoms of myasthenia gravis.
The observations in these three patients are “consistent with reports of other infections that induce autoimmune disorders, as well as with the growing evidence of other neurologic disorders with presumed autoimmune mechanisms after COVID-19 onset,” the researchers wrote.
They offered several possible explanations for the link between COVID-19 and myasthenia gravis. “Antibodies that are directed against SARS-CoV-2 proteins may cross-react with AChR subunits, because the virus has epitopes that are similar to components of the neuromuscular junction; this is known to occur in other neurologic autoimmune disorders after infection. Alternatively, COVID-19 infection may break immunologic self-tolerance,” the investigators wrote.
“The main message for clinicians is that myasthenia gravis, as well as other neurological disorders associated with autoimmunity, could occur in the course of SARS-CoV-2 infection,” Dr. Restivo said. Prompt recognition of the disease “could lead to a drug treatment that limits its evolution as quickly as possible,” he added.
An “unmasking”
Commenting on the findings, Anthony Geraci, MD, director of neuromuscular medicine, Northwell Health, Great Neck, N.Y., said these case reports of myasthenia gravis after SARS-CoV-2 infection are “not unique or novel as there has been a long understanding that seropositive [AChR antibody-positive] myasthenia gravis can and is frequently ‘unmasked’ in the setting” of several viral and bacterial infections.
“Antibodies in myasthenia gravis are of a type that take several weeks to develop to measurable levels as in the reported cases by Restivo et al., giving strong support to the notion that subclinical myasthenia gravis can be immunologically upregulated in the setting of viral infection and this is a far more likely explanation of the observed association reported,” added Dr. Geraci, who was not involved with the research.
He noted that, at his institution, “we have also observed ocular myasthenia gravis emerge in patients with SARS-CoV-2 infection, with similar double vision and lid droop, as we have seen similarly in patients with Zika, West Nile, and other viral infections, as well as a multiplicity of bacterial infections.”
“Most of our observed patients have responded to treatment much the same as reported by the three cases from Restivo and colleagues,” Dr.Geraci reported.
The authors of the study disclosed no conflicts of interest.
A version of this article originally appeared on Medscape.com.
ADHD and dyslexia may affect evaluation of concussion
, a new study shows.
“Our results suggest kids with certain learning disorders may respond differently to concussion tests, and this needs to be taken into account when advising on recovery times and when they can return to sport,” said lead author Mathew Stokes, MD. Dr. Stokes is assistant professor of pediatrics and neurology/neurotherapeutics at the University of Texas–Southwestern Medical Center, Dallas.
The study was presented at the American Academy of Neurology Sports Concussion Virtual Conference, held online July 31 to Aug. 1.
Learning disorders affected scores
The researchers analyzed data from participants aged 10-18 years who were enrolled in the North Texas Concussion Registry (ConTex). Participants had been diagnosed with a concussion that was sustained within 30 days of enrollment. The researchers investigated whether there were differences between patients who had no history of learning disorders and those with a history of dyslexia and/or ADD/ADHD with regard to results of clinical testing following concussion.
Of the 1,298 individuals in the study, 58 had been diagnosed with dyslexia, 158 had been diagnosed with ADD/ADHD, and 35 had been diagnosed with both conditions. There was no difference in age, time since injury, or history of concussion between those with learning disorders and those without, but there were more male patients in the ADD/ADHD group.
Results showed that in the dyslexia group, mean time was slower (P = .011), and there was an increase in error scores on the King-Devick (KD) test (P = .028). That test assesses eye movements and involves the rapid naming of numbers that are spaced differently. In addition, those with ADD/ADHD had significantly higher impulse control scores (P = .007) on the ImPACT series of tests, which are commonly used in the evaluation of concussion. Participants with both dyslexia and ADHD demonstrated slower KD times (P = .009) and had higher depression scores and anxiety scores.
Dr. Stokes noted that a limiting factor of the study was that baseline scores were not available. “It is possible that kids with ADD have less impulse control even at baseline, and this would need to be taken into account,” he said. “You may perhaps also expect someone with dyslexia to have a worse score on the KD tests, so we need more data on how these scores are affected from baseline in these individuals. But our results show that when evaluating kids pre- or post concussion, it is important to know about learning disorders, as this will affect how we interpret the data.”
At 3-month follow-up, there were no longer significant differences in anxiety and depression scores for those with and those without learning disorders. “This suggests anxiety and depression may well be worse temporarily after concussion for those with ADD/ADHD but gets better with time,” Dr. Stokes said.
Follow-up data were not available for the other cognitive tests.
Are recovery times longer?
Asked whether young people with these learning disorders needed a longer time to recover after concussion, Dr. Stokes said: “That is a million-dollar question. Studies so far on this have shown conflicting results. Our results add to a growing body of literature on this.” He stressed that it is important to include anxiety and depression scores on both baseline and postconcussion tests. “People don’t tend to think of these symptoms as being associated with concussion, but they are actually very prominent in this situation,” he noted. “Our results suggest that individuals with ADHD may be more prone to anxiety and depression, and a blow to the head may tip them more into these symptoms.”
Discussing the study at a virtual press conference as part of the AAN Sports Concussion meeting, the codirector of the meeting, David Dodick, MD, Mayo Clinic, Scottsdale, Ariz., said: “This is a very interesting and important study which suggests there are differences between adolescents with a history of dyslexia/ADHD and those without these conditions in performance in concussion tests. Understanding the differences in these groups will help health care providers in evaluating these athletes and assisting in counseling them and their families with regard to their risk of injury.
“It is important to recognize that athletes with ADHD, whether or not they are on medication, may take longer to recover from a concussion,” Dr. Dodick added. They also exhibit greater reductions in cognitive skills and visual motor speed regarding hand-eye coordination, he said. There is an increase in the severity of symptoms. “Symptoms that exist in both groups tend to more severe in those individuals with ADHD,” he noted.
“Ascertaining the presence or absence of ADHD or dyslexia in those who are participating in sport is important, especially when trying to interpret the results of baseline testing, the results of postinjury testing, decisions on when to return to play, and assessing for individuals and their families the risk of long-term repeat concussions and adverse outcomes,” he concluded.
The other codirector of the AAN meeting, Brian Hainline, MD, chief medical officer of the National Collegiate Athletic Association, added: “It appears that athletes with ADHD may suffer more with concussion and have a longer recovery time. This can inform our decision making and help these individuals to understand that they are at higher risk.”
Dr. Hainline said this raises another important point: “Concussion is not a homogeneous entity. It is a brain injury that can manifest in multiple parts of the brain, and the way the brain is from a premorbid or comorbid point of view can influence the manifestation of concussion as well,” he said. “All these things need to be taken into account.”
Attentional deficit may itself make an individual more susceptible to sustaining an injury in the first place, he said. “All of this is an evolving body of research which is helping clinicians to make better-informed decisions for athletes who may manifest differently.”
A version of this article originally appeared on Medscape.com.
, a new study shows.
“Our results suggest kids with certain learning disorders may respond differently to concussion tests, and this needs to be taken into account when advising on recovery times and when they can return to sport,” said lead author Mathew Stokes, MD. Dr. Stokes is assistant professor of pediatrics and neurology/neurotherapeutics at the University of Texas–Southwestern Medical Center, Dallas.
The study was presented at the American Academy of Neurology Sports Concussion Virtual Conference, held online July 31 to Aug. 1.
Learning disorders affected scores
The researchers analyzed data from participants aged 10-18 years who were enrolled in the North Texas Concussion Registry (ConTex). Participants had been diagnosed with a concussion that was sustained within 30 days of enrollment. The researchers investigated whether there were differences between patients who had no history of learning disorders and those with a history of dyslexia and/or ADD/ADHD with regard to results of clinical testing following concussion.
Of the 1,298 individuals in the study, 58 had been diagnosed with dyslexia, 158 had been diagnosed with ADD/ADHD, and 35 had been diagnosed with both conditions. There was no difference in age, time since injury, or history of concussion between those with learning disorders and those without, but there were more male patients in the ADD/ADHD group.
Results showed that in the dyslexia group, mean time was slower (P = .011), and there was an increase in error scores on the King-Devick (KD) test (P = .028). That test assesses eye movements and involves the rapid naming of numbers that are spaced differently. In addition, those with ADD/ADHD had significantly higher impulse control scores (P = .007) on the ImPACT series of tests, which are commonly used in the evaluation of concussion. Participants with both dyslexia and ADHD demonstrated slower KD times (P = .009) and had higher depression scores and anxiety scores.
Dr. Stokes noted that a limiting factor of the study was that baseline scores were not available. “It is possible that kids with ADD have less impulse control even at baseline, and this would need to be taken into account,” he said. “You may perhaps also expect someone with dyslexia to have a worse score on the KD tests, so we need more data on how these scores are affected from baseline in these individuals. But our results show that when evaluating kids pre- or post concussion, it is important to know about learning disorders, as this will affect how we interpret the data.”
At 3-month follow-up, there were no longer significant differences in anxiety and depression scores for those with and those without learning disorders. “This suggests anxiety and depression may well be worse temporarily after concussion for those with ADD/ADHD but gets better with time,” Dr. Stokes said.
Follow-up data were not available for the other cognitive tests.
Are recovery times longer?
Asked whether young people with these learning disorders needed a longer time to recover after concussion, Dr. Stokes said: “That is a million-dollar question. Studies so far on this have shown conflicting results. Our results add to a growing body of literature on this.” He stressed that it is important to include anxiety and depression scores on both baseline and postconcussion tests. “People don’t tend to think of these symptoms as being associated with concussion, but they are actually very prominent in this situation,” he noted. “Our results suggest that individuals with ADHD may be more prone to anxiety and depression, and a blow to the head may tip them more into these symptoms.”
Discussing the study at a virtual press conference as part of the AAN Sports Concussion meeting, the codirector of the meeting, David Dodick, MD, Mayo Clinic, Scottsdale, Ariz., said: “This is a very interesting and important study which suggests there are differences between adolescents with a history of dyslexia/ADHD and those without these conditions in performance in concussion tests. Understanding the differences in these groups will help health care providers in evaluating these athletes and assisting in counseling them and their families with regard to their risk of injury.
“It is important to recognize that athletes with ADHD, whether or not they are on medication, may take longer to recover from a concussion,” Dr. Dodick added. They also exhibit greater reductions in cognitive skills and visual motor speed regarding hand-eye coordination, he said. There is an increase in the severity of symptoms. “Symptoms that exist in both groups tend to more severe in those individuals with ADHD,” he noted.
“Ascertaining the presence or absence of ADHD or dyslexia in those who are participating in sport is important, especially when trying to interpret the results of baseline testing, the results of postinjury testing, decisions on when to return to play, and assessing for individuals and their families the risk of long-term repeat concussions and adverse outcomes,” he concluded.
The other codirector of the AAN meeting, Brian Hainline, MD, chief medical officer of the National Collegiate Athletic Association, added: “It appears that athletes with ADHD may suffer more with concussion and have a longer recovery time. This can inform our decision making and help these individuals to understand that they are at higher risk.”
Dr. Hainline said this raises another important point: “Concussion is not a homogeneous entity. It is a brain injury that can manifest in multiple parts of the brain, and the way the brain is from a premorbid or comorbid point of view can influence the manifestation of concussion as well,” he said. “All these things need to be taken into account.”
Attentional deficit may itself make an individual more susceptible to sustaining an injury in the first place, he said. “All of this is an evolving body of research which is helping clinicians to make better-informed decisions for athletes who may manifest differently.”
A version of this article originally appeared on Medscape.com.
, a new study shows.
“Our results suggest kids with certain learning disorders may respond differently to concussion tests, and this needs to be taken into account when advising on recovery times and when they can return to sport,” said lead author Mathew Stokes, MD. Dr. Stokes is assistant professor of pediatrics and neurology/neurotherapeutics at the University of Texas–Southwestern Medical Center, Dallas.
The study was presented at the American Academy of Neurology Sports Concussion Virtual Conference, held online July 31 to Aug. 1.
Learning disorders affected scores
The researchers analyzed data from participants aged 10-18 years who were enrolled in the North Texas Concussion Registry (ConTex). Participants had been diagnosed with a concussion that was sustained within 30 days of enrollment. The researchers investigated whether there were differences between patients who had no history of learning disorders and those with a history of dyslexia and/or ADD/ADHD with regard to results of clinical testing following concussion.
Of the 1,298 individuals in the study, 58 had been diagnosed with dyslexia, 158 had been diagnosed with ADD/ADHD, and 35 had been diagnosed with both conditions. There was no difference in age, time since injury, or history of concussion between those with learning disorders and those without, but there were more male patients in the ADD/ADHD group.
Results showed that in the dyslexia group, mean time was slower (P = .011), and there was an increase in error scores on the King-Devick (KD) test (P = .028). That test assesses eye movements and involves the rapid naming of numbers that are spaced differently. In addition, those with ADD/ADHD had significantly higher impulse control scores (P = .007) on the ImPACT series of tests, which are commonly used in the evaluation of concussion. Participants with both dyslexia and ADHD demonstrated slower KD times (P = .009) and had higher depression scores and anxiety scores.
Dr. Stokes noted that a limiting factor of the study was that baseline scores were not available. “It is possible that kids with ADD have less impulse control even at baseline, and this would need to be taken into account,” he said. “You may perhaps also expect someone with dyslexia to have a worse score on the KD tests, so we need more data on how these scores are affected from baseline in these individuals. But our results show that when evaluating kids pre- or post concussion, it is important to know about learning disorders, as this will affect how we interpret the data.”
At 3-month follow-up, there were no longer significant differences in anxiety and depression scores for those with and those without learning disorders. “This suggests anxiety and depression may well be worse temporarily after concussion for those with ADD/ADHD but gets better with time,” Dr. Stokes said.
Follow-up data were not available for the other cognitive tests.
Are recovery times longer?
Asked whether young people with these learning disorders needed a longer time to recover after concussion, Dr. Stokes said: “That is a million-dollar question. Studies so far on this have shown conflicting results. Our results add to a growing body of literature on this.” He stressed that it is important to include anxiety and depression scores on both baseline and postconcussion tests. “People don’t tend to think of these symptoms as being associated with concussion, but they are actually very prominent in this situation,” he noted. “Our results suggest that individuals with ADHD may be more prone to anxiety and depression, and a blow to the head may tip them more into these symptoms.”
Discussing the study at a virtual press conference as part of the AAN Sports Concussion meeting, the codirector of the meeting, David Dodick, MD, Mayo Clinic, Scottsdale, Ariz., said: “This is a very interesting and important study which suggests there are differences between adolescents with a history of dyslexia/ADHD and those without these conditions in performance in concussion tests. Understanding the differences in these groups will help health care providers in evaluating these athletes and assisting in counseling them and their families with regard to their risk of injury.
“It is important to recognize that athletes with ADHD, whether or not they are on medication, may take longer to recover from a concussion,” Dr. Dodick added. They also exhibit greater reductions in cognitive skills and visual motor speed regarding hand-eye coordination, he said. There is an increase in the severity of symptoms. “Symptoms that exist in both groups tend to more severe in those individuals with ADHD,” he noted.
“Ascertaining the presence or absence of ADHD or dyslexia in those who are participating in sport is important, especially when trying to interpret the results of baseline testing, the results of postinjury testing, decisions on when to return to play, and assessing for individuals and their families the risk of long-term repeat concussions and adverse outcomes,” he concluded.
The other codirector of the AAN meeting, Brian Hainline, MD, chief medical officer of the National Collegiate Athletic Association, added: “It appears that athletes with ADHD may suffer more with concussion and have a longer recovery time. This can inform our decision making and help these individuals to understand that they are at higher risk.”
Dr. Hainline said this raises another important point: “Concussion is not a homogeneous entity. It is a brain injury that can manifest in multiple parts of the brain, and the way the brain is from a premorbid or comorbid point of view can influence the manifestation of concussion as well,” he said. “All these things need to be taken into account.”
Attentional deficit may itself make an individual more susceptible to sustaining an injury in the first place, he said. “All of this is an evolving body of research which is helping clinicians to make better-informed decisions for athletes who may manifest differently.”
A version of this article originally appeared on Medscape.com.
From AAN Sports Concussion Conference
More evidence links gum disease and dementia risk
especially in those with severe gum inflammation and edentulism, new research suggests.
Over a 20-year period, investigators prospectively followed more than 8,000 individuals aged around 63 years who did not have cognitive impairment or dementia at baseline, grouping them based on the extent and severity of their periodontal disease and number of lost teeth.
Results showed that 14% of participants with healthy gums and all their teeth at baseline developed dementia, compared with 18% of those with mild periodontal disease and 22% who had severe periodontal disease. The highest percentage (23%) of participants who developed dementia was found in those who were edentulous.
After accounting for comorbidities that might affect dementia risk, edentulous participants had a 20% higher risk for developing MCI or dementia, compared with the healthy group.
Because the study was observational, “we don’t have knowledge of causality so we cannot state that if you treat periodontal disease you can prevent or treat dementia,” said lead author Ryan T. Demmer, PhD, MPH, associate professor, division of epidemiology and community health, University of Minnesota, Minneapolis. However, “the take-home message from this paper is that it further supports the possibility that oral infections could be a risk factor for dementia.”
The study was published online July 29 in Neurology.
The ARIC trial
Prior studies have “described the interrelation of tooth loss or periodontal disease and cognitive outcomes, although many reports were cross-sectional or case-control … and often lacked robust confounder adjustment,” the investigators noted. Additionally, lack of longitudinal data impedes the “potential for baseline periodontal status to predict incident MCI.”
To explore the associations between periodontal status and incident MCI and dementia, the researchers studied participants in the ARIC study, a community-based longitudinal cohort consisting of 15,792 predominantly Black and White participants aged 45-64 years. The current analysis included 8,275 individuals (55% women; 21% black; mean age, 63 years) who at baseline did not meet criteria for dementia or MCI.
A full-mouth periodontal examination was conducted at baseline and participants were categorized according to the severity and extent of gingival inflammation and tooth attachment loss based on the Periodontal Profile Class (PPC) seven-category model. Potential confounding variables included age, race, education level, physical activity, smoking status, oral hygiene and access to care, plasma lipid levels, APOE genotype, body mass index, blood pressure, type 2 diabetes, and heart failure.
Based on PPC categorization, 22% of the patients had healthy gums, 12% had mild periodontal disease, 8% had a high gingival inflammation index, and 12% had posterior disease (with 6% having severe disease). In addition, 9% had tooth loss, 11% had severe tooth loss, and 20% were edentulous.
Infection hypothesis
Results showed that participants with worse periodontal status were more likely to have risk factors for vascular disease and dementia, such as smoking, hypertension, diabetes, and coronary heart disease. During median follow-up of 18.4 years, 19% of participants overall (n = 1,569) developed dementia, translating into 11.8 cases per 1,000 person-years. There were notable differences between the PPC categories in rates of incident dementia, with edentulous participants at twice the risk for developing dementia, compared with those who had healthy gums.
For participants with severe PPC, including severe tooth loss and severe disease, the multivariable-adjusted hazard ratio for incident dementia was 1.22 (95% confidence interval, 1.01-1.47) versus those who were periodontally healthy. For participants with edentulism, the HR was 1.21 (95% CI, 0.99-1.48). The adjusted risk ratios for the combined dementia/MCI outcome among participants with mild to intermediate PPC, severe PPC, or edentulism versus the periodontal healthy group were 1.22 (95% CI, 1.00-1.48), 1.15 (95% CI, 0.88-1.51), and 1.90 (95% CI, 1.40-2.58), respectively.
These findings were most pronounced among younger (median age at dental exam, younger than 62) versus older (62 years and older) participants (P = .02). Severe disease or total tooth loss were associated with an approximately 20% greater dementia incidence during the follow-up period, compared with healthy gums.
The investigators noted that the findings were “generally consistent” when considering the combined outcome of MCI and dementia. However, they noted that the association between edentulism and MCI was “markedly stronger,” with an approximate 100% increase in MCI or MCI plus dementia.
The association between periodontal disease and MCI or dementia “is rooted in the infection hypothesis, meaning adverse microbial exposures in the mucosal surfaces of the mouth, especially the subgingival space,” Dr. Demmer said. “One notion is that there could somehow be a direct infection of the brain with oral organisms, which posits that the oral organism could travel to the brain, colonize there, and cause damage that impairs cognition.”
Another possible mechanism is that chronic systemic inflammation in response to oral infections can eventually lead to vascular disease which, in turn, is a known risk factor for future dementia, he noted.
“Brush and floss”
Commenting on the research findings, James M. Noble, MD, associate professor of neurology, Taub Institute for Research on Alzheimer’s and the Aging Brain, Columbia University, New York, called the study “well characterized both by whole-mouth assessments and cognitive assessments performed in a standardized manner.” Moreover, “the study was sufficiently sized to allow for exploration of age and suggests that oral health may be a more important factor earlier in the course of aging, in late adulthood,” said Dr. Noble, who was not involved with the research.
The study also “makes an important contribution to this field through a rigorously followed cohort and robust design for both periodontal predictor and cognitive outcome assessments,” he said, noting that, “as always, the take-home message is ‘brush and floss.’
“Although we don’t know if treating periodontal disease can help treat dementia, this study suggests that we have to pay attention to good oral hygiene and make referrals to dentists when appropriate,” Dr. Demmer added.
The ARIC trial is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute. Dr. Demmer, the study coauthors, and Dr. Noble have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
especially in those with severe gum inflammation and edentulism, new research suggests.
Over a 20-year period, investigators prospectively followed more than 8,000 individuals aged around 63 years who did not have cognitive impairment or dementia at baseline, grouping them based on the extent and severity of their periodontal disease and number of lost teeth.
Results showed that 14% of participants with healthy gums and all their teeth at baseline developed dementia, compared with 18% of those with mild periodontal disease and 22% who had severe periodontal disease. The highest percentage (23%) of participants who developed dementia was found in those who were edentulous.
After accounting for comorbidities that might affect dementia risk, edentulous participants had a 20% higher risk for developing MCI or dementia, compared with the healthy group.
Because the study was observational, “we don’t have knowledge of causality so we cannot state that if you treat periodontal disease you can prevent or treat dementia,” said lead author Ryan T. Demmer, PhD, MPH, associate professor, division of epidemiology and community health, University of Minnesota, Minneapolis. However, “the take-home message from this paper is that it further supports the possibility that oral infections could be a risk factor for dementia.”
The study was published online July 29 in Neurology.
The ARIC trial
Prior studies have “described the interrelation of tooth loss or periodontal disease and cognitive outcomes, although many reports were cross-sectional or case-control … and often lacked robust confounder adjustment,” the investigators noted. Additionally, lack of longitudinal data impedes the “potential for baseline periodontal status to predict incident MCI.”
To explore the associations between periodontal status and incident MCI and dementia, the researchers studied participants in the ARIC study, a community-based longitudinal cohort consisting of 15,792 predominantly Black and White participants aged 45-64 years. The current analysis included 8,275 individuals (55% women; 21% black; mean age, 63 years) who at baseline did not meet criteria for dementia or MCI.
A full-mouth periodontal examination was conducted at baseline and participants were categorized according to the severity and extent of gingival inflammation and tooth attachment loss based on the Periodontal Profile Class (PPC) seven-category model. Potential confounding variables included age, race, education level, physical activity, smoking status, oral hygiene and access to care, plasma lipid levels, APOE genotype, body mass index, blood pressure, type 2 diabetes, and heart failure.
Based on PPC categorization, 22% of the patients had healthy gums, 12% had mild periodontal disease, 8% had a high gingival inflammation index, and 12% had posterior disease (with 6% having severe disease). In addition, 9% had tooth loss, 11% had severe tooth loss, and 20% were edentulous.
Infection hypothesis
Results showed that participants with worse periodontal status were more likely to have risk factors for vascular disease and dementia, such as smoking, hypertension, diabetes, and coronary heart disease. During median follow-up of 18.4 years, 19% of participants overall (n = 1,569) developed dementia, translating into 11.8 cases per 1,000 person-years. There were notable differences between the PPC categories in rates of incident dementia, with edentulous participants at twice the risk for developing dementia, compared with those who had healthy gums.
For participants with severe PPC, including severe tooth loss and severe disease, the multivariable-adjusted hazard ratio for incident dementia was 1.22 (95% confidence interval, 1.01-1.47) versus those who were periodontally healthy. For participants with edentulism, the HR was 1.21 (95% CI, 0.99-1.48). The adjusted risk ratios for the combined dementia/MCI outcome among participants with mild to intermediate PPC, severe PPC, or edentulism versus the periodontal healthy group were 1.22 (95% CI, 1.00-1.48), 1.15 (95% CI, 0.88-1.51), and 1.90 (95% CI, 1.40-2.58), respectively.
These findings were most pronounced among younger (median age at dental exam, younger than 62) versus older (62 years and older) participants (P = .02). Severe disease or total tooth loss were associated with an approximately 20% greater dementia incidence during the follow-up period, compared with healthy gums.
The investigators noted that the findings were “generally consistent” when considering the combined outcome of MCI and dementia. However, they noted that the association between edentulism and MCI was “markedly stronger,” with an approximate 100% increase in MCI or MCI plus dementia.
The association between periodontal disease and MCI or dementia “is rooted in the infection hypothesis, meaning adverse microbial exposures in the mucosal surfaces of the mouth, especially the subgingival space,” Dr. Demmer said. “One notion is that there could somehow be a direct infection of the brain with oral organisms, which posits that the oral organism could travel to the brain, colonize there, and cause damage that impairs cognition.”
Another possible mechanism is that chronic systemic inflammation in response to oral infections can eventually lead to vascular disease which, in turn, is a known risk factor for future dementia, he noted.
“Brush and floss”
Commenting on the research findings, James M. Noble, MD, associate professor of neurology, Taub Institute for Research on Alzheimer’s and the Aging Brain, Columbia University, New York, called the study “well characterized both by whole-mouth assessments and cognitive assessments performed in a standardized manner.” Moreover, “the study was sufficiently sized to allow for exploration of age and suggests that oral health may be a more important factor earlier in the course of aging, in late adulthood,” said Dr. Noble, who was not involved with the research.
The study also “makes an important contribution to this field through a rigorously followed cohort and robust design for both periodontal predictor and cognitive outcome assessments,” he said, noting that, “as always, the take-home message is ‘brush and floss.’
“Although we don’t know if treating periodontal disease can help treat dementia, this study suggests that we have to pay attention to good oral hygiene and make referrals to dentists when appropriate,” Dr. Demmer added.
The ARIC trial is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute. Dr. Demmer, the study coauthors, and Dr. Noble have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
especially in those with severe gum inflammation and edentulism, new research suggests.
Over a 20-year period, investigators prospectively followed more than 8,000 individuals aged around 63 years who did not have cognitive impairment or dementia at baseline, grouping them based on the extent and severity of their periodontal disease and number of lost teeth.
Results showed that 14% of participants with healthy gums and all their teeth at baseline developed dementia, compared with 18% of those with mild periodontal disease and 22% who had severe periodontal disease. The highest percentage (23%) of participants who developed dementia was found in those who were edentulous.
After accounting for comorbidities that might affect dementia risk, edentulous participants had a 20% higher risk for developing MCI or dementia, compared with the healthy group.
Because the study was observational, “we don’t have knowledge of causality so we cannot state that if you treat periodontal disease you can prevent or treat dementia,” said lead author Ryan T. Demmer, PhD, MPH, associate professor, division of epidemiology and community health, University of Minnesota, Minneapolis. However, “the take-home message from this paper is that it further supports the possibility that oral infections could be a risk factor for dementia.”
The study was published online July 29 in Neurology.
The ARIC trial
Prior studies have “described the interrelation of tooth loss or periodontal disease and cognitive outcomes, although many reports were cross-sectional or case-control … and often lacked robust confounder adjustment,” the investigators noted. Additionally, lack of longitudinal data impedes the “potential for baseline periodontal status to predict incident MCI.”
To explore the associations between periodontal status and incident MCI and dementia, the researchers studied participants in the ARIC study, a community-based longitudinal cohort consisting of 15,792 predominantly Black and White participants aged 45-64 years. The current analysis included 8,275 individuals (55% women; 21% black; mean age, 63 years) who at baseline did not meet criteria for dementia or MCI.
A full-mouth periodontal examination was conducted at baseline and participants were categorized according to the severity and extent of gingival inflammation and tooth attachment loss based on the Periodontal Profile Class (PPC) seven-category model. Potential confounding variables included age, race, education level, physical activity, smoking status, oral hygiene and access to care, plasma lipid levels, APOE genotype, body mass index, blood pressure, type 2 diabetes, and heart failure.
Based on PPC categorization, 22% of the patients had healthy gums, 12% had mild periodontal disease, 8% had a high gingival inflammation index, and 12% had posterior disease (with 6% having severe disease). In addition, 9% had tooth loss, 11% had severe tooth loss, and 20% were edentulous.
Infection hypothesis
Results showed that participants with worse periodontal status were more likely to have risk factors for vascular disease and dementia, such as smoking, hypertension, diabetes, and coronary heart disease. During median follow-up of 18.4 years, 19% of participants overall (n = 1,569) developed dementia, translating into 11.8 cases per 1,000 person-years. There were notable differences between the PPC categories in rates of incident dementia, with edentulous participants at twice the risk for developing dementia, compared with those who had healthy gums.
For participants with severe PPC, including severe tooth loss and severe disease, the multivariable-adjusted hazard ratio for incident dementia was 1.22 (95% confidence interval, 1.01-1.47) versus those who were periodontally healthy. For participants with edentulism, the HR was 1.21 (95% CI, 0.99-1.48). The adjusted risk ratios for the combined dementia/MCI outcome among participants with mild to intermediate PPC, severe PPC, or edentulism versus the periodontal healthy group were 1.22 (95% CI, 1.00-1.48), 1.15 (95% CI, 0.88-1.51), and 1.90 (95% CI, 1.40-2.58), respectively.
These findings were most pronounced among younger (median age at dental exam, younger than 62) versus older (62 years and older) participants (P = .02). Severe disease or total tooth loss were associated with an approximately 20% greater dementia incidence during the follow-up period, compared with healthy gums.
The investigators noted that the findings were “generally consistent” when considering the combined outcome of MCI and dementia. However, they noted that the association between edentulism and MCI was “markedly stronger,” with an approximate 100% increase in MCI or MCI plus dementia.
The association between periodontal disease and MCI or dementia “is rooted in the infection hypothesis, meaning adverse microbial exposures in the mucosal surfaces of the mouth, especially the subgingival space,” Dr. Demmer said. “One notion is that there could somehow be a direct infection of the brain with oral organisms, which posits that the oral organism could travel to the brain, colonize there, and cause damage that impairs cognition.”
Another possible mechanism is that chronic systemic inflammation in response to oral infections can eventually lead to vascular disease which, in turn, is a known risk factor for future dementia, he noted.
“Brush and floss”
Commenting on the research findings, James M. Noble, MD, associate professor of neurology, Taub Institute for Research on Alzheimer’s and the Aging Brain, Columbia University, New York, called the study “well characterized both by whole-mouth assessments and cognitive assessments performed in a standardized manner.” Moreover, “the study was sufficiently sized to allow for exploration of age and suggests that oral health may be a more important factor earlier in the course of aging, in late adulthood,” said Dr. Noble, who was not involved with the research.
The study also “makes an important contribution to this field through a rigorously followed cohort and robust design for both periodontal predictor and cognitive outcome assessments,” he said, noting that, “as always, the take-home message is ‘brush and floss.’
“Although we don’t know if treating periodontal disease can help treat dementia, this study suggests that we have to pay attention to good oral hygiene and make referrals to dentists when appropriate,” Dr. Demmer added.
The ARIC trial is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute. Dr. Demmer, the study coauthors, and Dr. Noble have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.