User login
High mortality rates trail tracheostomy patients
findings of a large retrospective study suggest.
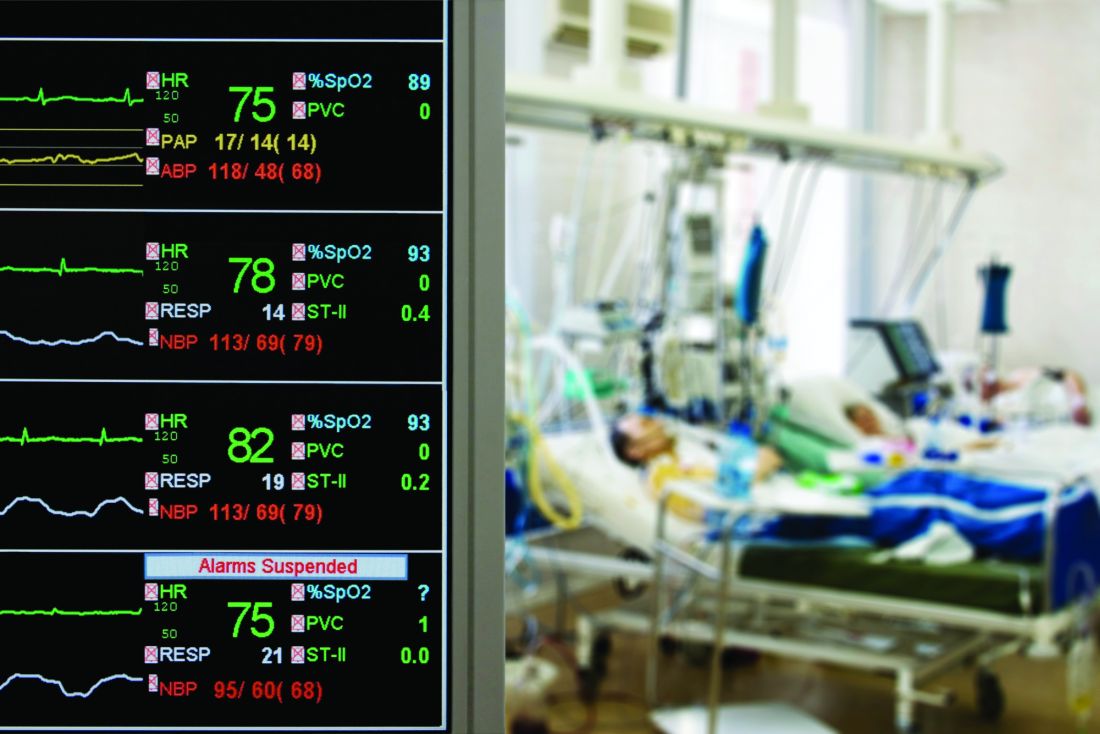
Current outcome prediction tools to support decision making regarding tracheostomies are limited, wrote Anuj B. Mehta, MD, of National Jewish Health in Denver, and colleagues. “This study provides novel and in-depth insight into mortality and health care utilization following tracheostomy not previously described at the population-level.”
In a study published in Critical Care Medicine, the researchers reviewed data from 8,343 nonsurgical patients seen in California hospitals from 2012 to 2013 who received a tracheostomy for acute respiratory failure.
Overall, the 1-year mortality rate for patients who had tracheostomies (the primary outcome) was 46.5%, with in-hospital mortality of 18.9% and 30-day mortality of 22.1%. Pneumonia was the most common diagnosis for patients with respiratory failure (79%) and some had an additional diagnosis, such as severe sepsis (56%).
Patients aged 65 years and older had significantly higher mortality than those under 65 (54.7% vs. 36.5%). The average age of the patients was 65 years; approximately 46% were women and 48% were white. The median survival for adults aged 65 years and older was 175 days, compared with median survival of more than a year for younger patients.
Secondary outcomes included discharge destination, hospital readmission, and health care utilization. A majority (86%) of patients were discharged to a long-term care facility, while 11% were sent home and approximately 3% were discharged to other destinations.
Nearly two-thirds (60%) of patients were readmitted to the hospital within a year of tracheostomy, and readmission was more common among older adults, compared with younger (66% vs. 55%).
In addition, just over one-third of all patients (36%) spent more than 50% of their days alive in the hospital in short-term acute care, and this rate was significantly higher for patients aged 65 years and older, compared with those under 65 (43% vs. 29%). On average, the total hospital cost for patients who survived the first year after tracheostomy was $215,369, with no significant difference in average cost among age groups.
The study findings were limited by several factors including the use of data from a single state, possible misclassification of billing codes, and inability to measure quality of life, the researchers noted.
However, “our findings of high mortality, low median survival for older patients, high readmission rates, potentially burdensome cost, and informative outcome trajectories provide significant insight into long-term outcomes following tracheostomy,” they concluded.
Dr. Mehta and several colleagues reported receiving funding from the National Institutes of Health. The researchers had no financial conflicts to disclose.
SOURCE: Mehta AB et al. Crit Care Med. 2019 Aug 8. doi: 10.1097/CCM.0000000000003959.
findings of a large retrospective study suggest.
Current outcome prediction tools to support decision making regarding tracheostomies are limited, wrote Anuj B. Mehta, MD, of National Jewish Health in Denver, and colleagues. “This study provides novel and in-depth insight into mortality and health care utilization following tracheostomy not previously described at the population-level.”
In a study published in Critical Care Medicine, the researchers reviewed data from 8,343 nonsurgical patients seen in California hospitals from 2012 to 2013 who received a tracheostomy for acute respiratory failure.
Overall, the 1-year mortality rate for patients who had tracheostomies (the primary outcome) was 46.5%, with in-hospital mortality of 18.9% and 30-day mortality of 22.1%. Pneumonia was the most common diagnosis for patients with respiratory failure (79%) and some had an additional diagnosis, such as severe sepsis (56%).
Patients aged 65 years and older had significantly higher mortality than those under 65 (54.7% vs. 36.5%). The average age of the patients was 65 years; approximately 46% were women and 48% were white. The median survival for adults aged 65 years and older was 175 days, compared with median survival of more than a year for younger patients.
Secondary outcomes included discharge destination, hospital readmission, and health care utilization. A majority (86%) of patients were discharged to a long-term care facility, while 11% were sent home and approximately 3% were discharged to other destinations.
Nearly two-thirds (60%) of patients were readmitted to the hospital within a year of tracheostomy, and readmission was more common among older adults, compared with younger (66% vs. 55%).
In addition, just over one-third of all patients (36%) spent more than 50% of their days alive in the hospital in short-term acute care, and this rate was significantly higher for patients aged 65 years and older, compared with those under 65 (43% vs. 29%). On average, the total hospital cost for patients who survived the first year after tracheostomy was $215,369, with no significant difference in average cost among age groups.
The study findings were limited by several factors including the use of data from a single state, possible misclassification of billing codes, and inability to measure quality of life, the researchers noted.
However, “our findings of high mortality, low median survival for older patients, high readmission rates, potentially burdensome cost, and informative outcome trajectories provide significant insight into long-term outcomes following tracheostomy,” they concluded.
Dr. Mehta and several colleagues reported receiving funding from the National Institutes of Health. The researchers had no financial conflicts to disclose.
SOURCE: Mehta AB et al. Crit Care Med. 2019 Aug 8. doi: 10.1097/CCM.0000000000003959.
findings of a large retrospective study suggest.
Current outcome prediction tools to support decision making regarding tracheostomies are limited, wrote Anuj B. Mehta, MD, of National Jewish Health in Denver, and colleagues. “This study provides novel and in-depth insight into mortality and health care utilization following tracheostomy not previously described at the population-level.”
In a study published in Critical Care Medicine, the researchers reviewed data from 8,343 nonsurgical patients seen in California hospitals from 2012 to 2013 who received a tracheostomy for acute respiratory failure.
Overall, the 1-year mortality rate for patients who had tracheostomies (the primary outcome) was 46.5%, with in-hospital mortality of 18.9% and 30-day mortality of 22.1%. Pneumonia was the most common diagnosis for patients with respiratory failure (79%) and some had an additional diagnosis, such as severe sepsis (56%).
Patients aged 65 years and older had significantly higher mortality than those under 65 (54.7% vs. 36.5%). The average age of the patients was 65 years; approximately 46% were women and 48% were white. The median survival for adults aged 65 years and older was 175 days, compared with median survival of more than a year for younger patients.
Secondary outcomes included discharge destination, hospital readmission, and health care utilization. A majority (86%) of patients were discharged to a long-term care facility, while 11% were sent home and approximately 3% were discharged to other destinations.
Nearly two-thirds (60%) of patients were readmitted to the hospital within a year of tracheostomy, and readmission was more common among older adults, compared with younger (66% vs. 55%).
In addition, just over one-third of all patients (36%) spent more than 50% of their days alive in the hospital in short-term acute care, and this rate was significantly higher for patients aged 65 years and older, compared with those under 65 (43% vs. 29%). On average, the total hospital cost for patients who survived the first year after tracheostomy was $215,369, with no significant difference in average cost among age groups.
The study findings were limited by several factors including the use of data from a single state, possible misclassification of billing codes, and inability to measure quality of life, the researchers noted.
However, “our findings of high mortality, low median survival for older patients, high readmission rates, potentially burdensome cost, and informative outcome trajectories provide significant insight into long-term outcomes following tracheostomy,” they concluded.
Dr. Mehta and several colleagues reported receiving funding from the National Institutes of Health. The researchers had no financial conflicts to disclose.
SOURCE: Mehta AB et al. Crit Care Med. 2019 Aug 8. doi: 10.1097/CCM.0000000000003959.
FROM CRITICAL CARE MEDICINE
Taking vaccines to the next level via mucosal immunity
Vaccines are marvelous, and there are many well documented success stories, including rotavirus (RV) vaccines, where a live vaccine is administered to the gastrointestinal mucosa via oral drops. Antigens presented at the mucosal/epithelial surface not only induce systemic serum IgG – as do injectable vaccines – but also induce secretory IgA (sIgA), which is most helpful in diseases that directly affect the mucosa.

Mucosal vs. systemic immunity
Antibody being present on mucosal surfaces (point of initial pathogen contact) has a chance to neutralize the pathogen before it gains a foothold. Pathogen-specific mucosal lymphoid elements (e.g. in Peyer’s patches in the gut) also appear critical for optimal protection.1 The presence of both mucosal immune elements means that infection is severely limited or at times entirely prevented. So virus entering the GI tract causes minimal to no gut lining injury. Hence, there is no or mostly reduced vomiting/diarrhea. A downside of mucosally-administered live vaccines is that preexisting antibody to the vaccine antigens can reduce or block vaccine virus replication in the vaccinee, blunting or preventing protection. Note: Preexisting antibody also affects injectable live vaccines, such as the measles vaccine, similarly.
Classic injectable live or nonlive vaccines provide their most potent protection via systemic cellular responses antibody and/or antibodies in serum and extracellular fluid (ECF) where IgG and IgM are in highest concentrations. So even successful injectable vaccines still allow mucosal infection to start but then intercept further spread and prevent most of the downstream damage (think pertussis) or neutralize an infection-generated toxin (pertussis or tetanus). It usually is only after infection-induced damage occurs that systemic IgG and IgM gain better access to respiratory epithelial surfaces, but still only at a fraction of circulating concentrations. Indeed, pertussis vaccine–induced systemic immunity allows the pathogen to attack and replicate in/on host surface cells, causing toxin release and variable amounts of local mucosal injury/inflammation before vaccine-induced systemic immunity gains adequate access to the pathogen and/or to its toxin which may enter systemic circulation.
Live attenuated influenza vaccine (LAIV) induces mucosal immunity
Another “standard” vaccine that induces mucosal immunity – LAIV – was developed to improve on protection afforded by injectable influenza vaccines (IIVs), but LAIV has had hiccups in the United States. One example is several years of negligible protection against H1N1 disease. As long as LAIV’s vaccine strain had reasonably matched the circulating strains, LAIV worked at least as well as injectable influenza vaccine, and even offered some cross-protection against mildly mismatched strains. But after a number of years of LAIV use, vaccine effectiveness in the United States vs. H1N1 strains appeared to fade due to previously undetected but significant changes in the circulating H1N1 strain. The lesson is that mucosal immunity’s advantages are lost if too much change occurs in the pathogen target for sIgA and mucosally-associated lymphoid tissue cells (MALT)).
Other vaccines likely need to induce mucosal immunity
Protection at the mucosal level will likely be needed for success against norovirus, parainfluenza, respiratory syncytial virus (RSV), Neisseria gonorrhea, and chlamydia. Another helpful aspect of mucosal immunity is that immune cells and sIgA not only reside on the mucosa where the antigen was originally presented, but there is also a reasonable chance that these components will traffic to other mucosal surfaces.2 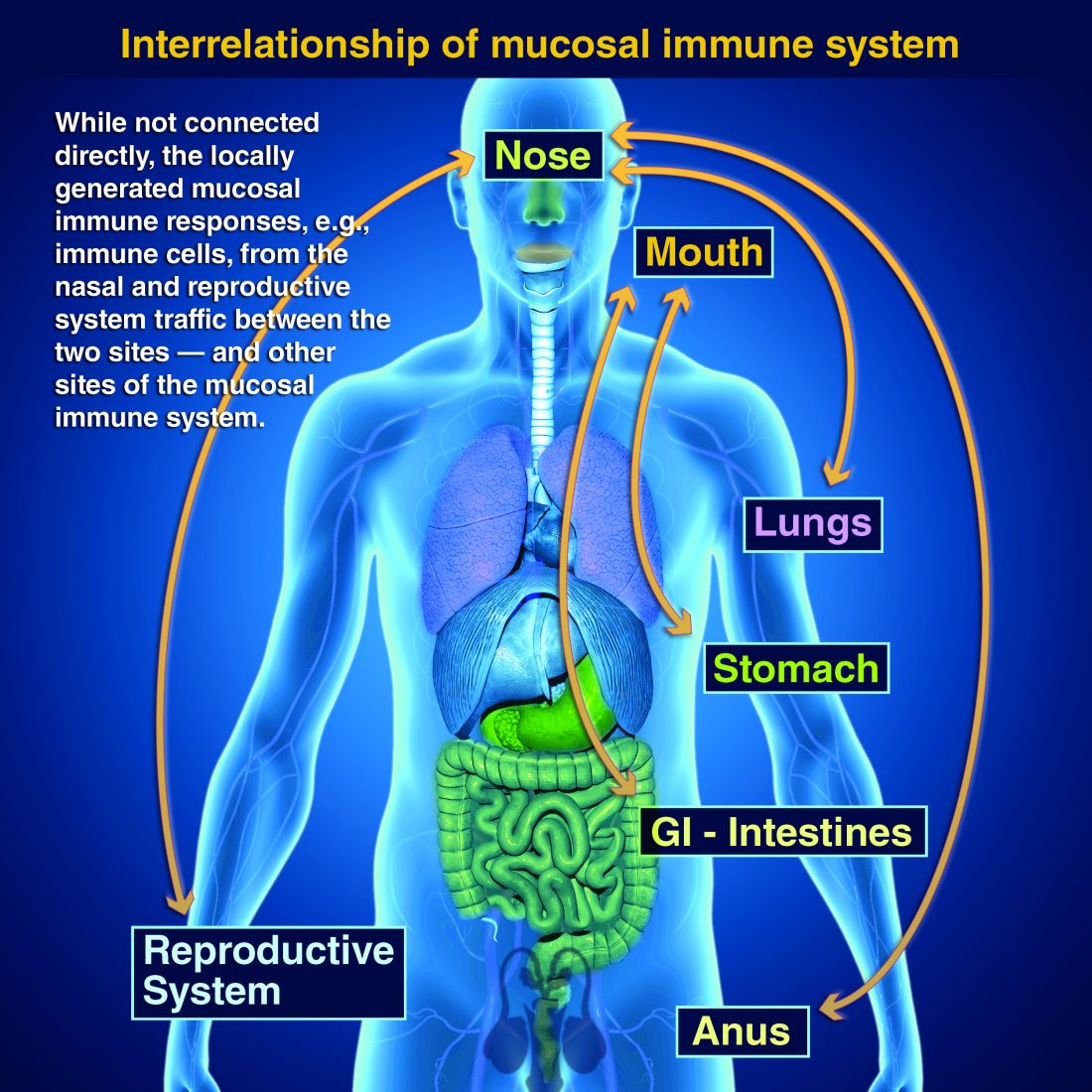
So intranasal vaccine could be expected to protect distant mucosal surfaces (urogenital, GI, and respiratory), leading to vaccine-induced systemic antibody plus mucosal immunity (sIGA and MALT responses) at each site.
Let’s look at a novel “two-site” chlamydia vaccine
Recently a phase 1 chlamydia vaccine that used a novel two-pronged administration site/schedule was successful at inducing both mucosal and systemic immunity in a proof-of-concept study – achieving the best of both worlds.3 This may be a template for vaccines in years to come. British investigators studied 50 healthy women aged 19-45 years in a double-blind, parallel, randomized, placebo-controlled trial that used a recombinant chlamydia protein subunit antigen (CTH522). The vaccine schedule involved three injectable priming doses followed soon thereafter by two intranasal boosting doses. There were three groups:
1. CTH522 adjuvanted with CAF01 liposomes (CTH522:CAF01).
2. CTH522 adjuvanted with aluminum hydroxide (CTH522:AH).
3. Placebo (saline).
The intramuscular (IM) priming schedule was 0, 1, and 4 months. The intranasal vaccine booster doses or placebo were given at 4.5 and 5 months. No related serious adverse reactions occurred. For injectable dosing, the most frequent adverse event was mild local injection-site reactions in all subjects in both vaccine groups vs. in 60% of placebo recipients (P = .053). The adjuvants were the likely cause for local reactions. Intranasal doses had local reactions in 47% of both vaccine groups and 60% of placebo recipients; P = 1.000).
Both vaccines produced systemic IgG seroconversion (including neutralizing antibody) plus small amounts of IgG in the nasal cavity and genital tract in all vaccine recipients; no placebo recipient seroconverted. Interestingly, liposomally-adjuvanted vaccine produced a more rapid systemic IgG response and higher serum titers than the alum-adjuvanted vaccine. Likewise, the IM liposomal vaccine also induced higher but still small mucosal IgG antibody responses (P = .0091). Intranasal IM-induced IgG titers were not boosted by later intranasal vaccine dosing.
Subjects getting liposomal vaccine (but not alum vaccine or placebo) boosters had detectable sIgA titers in both nasal and genital tract secretions. Liposomal vaccine recipients also had fivefold to sixfold higher median titers than alum vaccine recipients after the priming dose, and these higher titers persisted to the end of the study. All liposomal vaccine recipients developed antichlamydial cell-mediated responses vs. 57% alum-adjuvanted vaccine recipients. (P = .01). So both use of two-site dosing and the liposomal adjuvant appeared critical to better responses.

In summary
While this candidate vaccine has hurdles to overcome before coming into routine use, the proof-of-principle that a combination injectable-intranasal vaccine schedule can induce robust systemic and mucosal immunity when given with an appropriate adjuvant is very promising. Adding more vaccines to the schedule then becomes an issue, but that is one of those “good” problems we can deal with later.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines, receives funding from GlaxoSmithKline for studies on pneumococcal and rotavirus vaccines, and from Pfizer for a study on pneumococcal vaccine on which Dr. Harrison is a sub-investigator. The hospital also receives Centers for Disease Control and Prevention funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus, and also for rotavirus. Email Dr. Harrison at [email protected].
References
1. PLOS Biology. 2012 Sep 1. doi: 10.1371/journal.pbio.1001397.
2. Mucosal Immunity in the Human Female Reproductive Tract in “Mucosal Immunology,” 4th ed., Volume 2 (Cambridge, MA: Academic Press, 2015, pp. 2097-124).
3. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30279-8.
Vaccines are marvelous, and there are many well documented success stories, including rotavirus (RV) vaccines, where a live vaccine is administered to the gastrointestinal mucosa via oral drops. Antigens presented at the mucosal/epithelial surface not only induce systemic serum IgG – as do injectable vaccines – but also induce secretory IgA (sIgA), which is most helpful in diseases that directly affect the mucosa.
Mucosal vs. systemic immunity
Antibody being present on mucosal surfaces (point of initial pathogen contact) has a chance to neutralize the pathogen before it gains a foothold. Pathogen-specific mucosal lymphoid elements (e.g. in Peyer’s patches in the gut) also appear critical for optimal protection.1 The presence of both mucosal immune elements means that infection is severely limited or at times entirely prevented. So virus entering the GI tract causes minimal to no gut lining injury. Hence, there is no or mostly reduced vomiting/diarrhea. A downside of mucosally-administered live vaccines is that preexisting antibody to the vaccine antigens can reduce or block vaccine virus replication in the vaccinee, blunting or preventing protection. Note: Preexisting antibody also affects injectable live vaccines, such as the measles vaccine, similarly.
Classic injectable live or nonlive vaccines provide their most potent protection via systemic cellular responses antibody and/or antibodies in serum and extracellular fluid (ECF) where IgG and IgM are in highest concentrations. So even successful injectable vaccines still allow mucosal infection to start but then intercept further spread and prevent most of the downstream damage (think pertussis) or neutralize an infection-generated toxin (pertussis or tetanus). It usually is only after infection-induced damage occurs that systemic IgG and IgM gain better access to respiratory epithelial surfaces, but still only at a fraction of circulating concentrations. Indeed, pertussis vaccine–induced systemic immunity allows the pathogen to attack and replicate in/on host surface cells, causing toxin release and variable amounts of local mucosal injury/inflammation before vaccine-induced systemic immunity gains adequate access to the pathogen and/or to its toxin which may enter systemic circulation.
Live attenuated influenza vaccine (LAIV) induces mucosal immunity
Another “standard” vaccine that induces mucosal immunity – LAIV – was developed to improve on protection afforded by injectable influenza vaccines (IIVs), but LAIV has had hiccups in the United States. One example is several years of negligible protection against H1N1 disease. As long as LAIV’s vaccine strain had reasonably matched the circulating strains, LAIV worked at least as well as injectable influenza vaccine, and even offered some cross-protection against mildly mismatched strains. But after a number of years of LAIV use, vaccine effectiveness in the United States vs. H1N1 strains appeared to fade due to previously undetected but significant changes in the circulating H1N1 strain. The lesson is that mucosal immunity’s advantages are lost if too much change occurs in the pathogen target for sIgA and mucosally-associated lymphoid tissue cells (MALT)).
Other vaccines likely need to induce mucosal immunity
Protection at the mucosal level will likely be needed for success against norovirus, parainfluenza, respiratory syncytial virus (RSV), Neisseria gonorrhea, and chlamydia. Another helpful aspect of mucosal immunity is that immune cells and sIgA not only reside on the mucosa where the antigen was originally presented, but there is also a reasonable chance that these components will traffic to other mucosal surfaces.2
So intranasal vaccine could be expected to protect distant mucosal surfaces (urogenital, GI, and respiratory), leading to vaccine-induced systemic antibody plus mucosal immunity (sIGA and MALT responses) at each site.
Let’s look at a novel “two-site” chlamydia vaccine
Recently a phase 1 chlamydia vaccine that used a novel two-pronged administration site/schedule was successful at inducing both mucosal and systemic immunity in a proof-of-concept study – achieving the best of both worlds.3 This may be a template for vaccines in years to come. British investigators studied 50 healthy women aged 19-45 years in a double-blind, parallel, randomized, placebo-controlled trial that used a recombinant chlamydia protein subunit antigen (CTH522). The vaccine schedule involved three injectable priming doses followed soon thereafter by two intranasal boosting doses. There were three groups:
1. CTH522 adjuvanted with CAF01 liposomes (CTH522:CAF01).
2. CTH522 adjuvanted with aluminum hydroxide (CTH522:AH).
3. Placebo (saline).
The intramuscular (IM) priming schedule was 0, 1, and 4 months. The intranasal vaccine booster doses or placebo were given at 4.5 and 5 months. No related serious adverse reactions occurred. For injectable dosing, the most frequent adverse event was mild local injection-site reactions in all subjects in both vaccine groups vs. in 60% of placebo recipients (P = .053). The adjuvants were the likely cause for local reactions. Intranasal doses had local reactions in 47% of both vaccine groups and 60% of placebo recipients; P = 1.000).
Both vaccines produced systemic IgG seroconversion (including neutralizing antibody) plus small amounts of IgG in the nasal cavity and genital tract in all vaccine recipients; no placebo recipient seroconverted. Interestingly, liposomally-adjuvanted vaccine produced a more rapid systemic IgG response and higher serum titers than the alum-adjuvanted vaccine. Likewise, the IM liposomal vaccine also induced higher but still small mucosal IgG antibody responses (P = .0091). Intranasal IM-induced IgG titers were not boosted by later intranasal vaccine dosing.
Subjects getting liposomal vaccine (but not alum vaccine or placebo) boosters had detectable sIgA titers in both nasal and genital tract secretions. Liposomal vaccine recipients also had fivefold to sixfold higher median titers than alum vaccine recipients after the priming dose, and these higher titers persisted to the end of the study. All liposomal vaccine recipients developed antichlamydial cell-mediated responses vs. 57% alum-adjuvanted vaccine recipients. (P = .01). So both use of two-site dosing and the liposomal adjuvant appeared critical to better responses.
In summary
While this candidate vaccine has hurdles to overcome before coming into routine use, the proof-of-principle that a combination injectable-intranasal vaccine schedule can induce robust systemic and mucosal immunity when given with an appropriate adjuvant is very promising. Adding more vaccines to the schedule then becomes an issue, but that is one of those “good” problems we can deal with later.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines, receives funding from GlaxoSmithKline for studies on pneumococcal and rotavirus vaccines, and from Pfizer for a study on pneumococcal vaccine on which Dr. Harrison is a sub-investigator. The hospital also receives Centers for Disease Control and Prevention funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus, and also for rotavirus. Email Dr. Harrison at [email protected].
References
1. PLOS Biology. 2012 Sep 1. doi: 10.1371/journal.pbio.1001397.
2. Mucosal Immunity in the Human Female Reproductive Tract in “Mucosal Immunology,” 4th ed., Volume 2 (Cambridge, MA: Academic Press, 2015, pp. 2097-124).
3. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30279-8.
Vaccines are marvelous, and there are many well documented success stories, including rotavirus (RV) vaccines, where a live vaccine is administered to the gastrointestinal mucosa via oral drops. Antigens presented at the mucosal/epithelial surface not only induce systemic serum IgG – as do injectable vaccines – but also induce secretory IgA (sIgA), which is most helpful in diseases that directly affect the mucosa.
Mucosal vs. systemic immunity
Antibody being present on mucosal surfaces (point of initial pathogen contact) has a chance to neutralize the pathogen before it gains a foothold. Pathogen-specific mucosal lymphoid elements (e.g. in Peyer’s patches in the gut) also appear critical for optimal protection.1 The presence of both mucosal immune elements means that infection is severely limited or at times entirely prevented. So virus entering the GI tract causes minimal to no gut lining injury. Hence, there is no or mostly reduced vomiting/diarrhea. A downside of mucosally-administered live vaccines is that preexisting antibody to the vaccine antigens can reduce or block vaccine virus replication in the vaccinee, blunting or preventing protection. Note: Preexisting antibody also affects injectable live vaccines, such as the measles vaccine, similarly.
Classic injectable live or nonlive vaccines provide their most potent protection via systemic cellular responses antibody and/or antibodies in serum and extracellular fluid (ECF) where IgG and IgM are in highest concentrations. So even successful injectable vaccines still allow mucosal infection to start but then intercept further spread and prevent most of the downstream damage (think pertussis) or neutralize an infection-generated toxin (pertussis or tetanus). It usually is only after infection-induced damage occurs that systemic IgG and IgM gain better access to respiratory epithelial surfaces, but still only at a fraction of circulating concentrations. Indeed, pertussis vaccine–induced systemic immunity allows the pathogen to attack and replicate in/on host surface cells, causing toxin release and variable amounts of local mucosal injury/inflammation before vaccine-induced systemic immunity gains adequate access to the pathogen and/or to its toxin which may enter systemic circulation.
Live attenuated influenza vaccine (LAIV) induces mucosal immunity
Another “standard” vaccine that induces mucosal immunity – LAIV – was developed to improve on protection afforded by injectable influenza vaccines (IIVs), but LAIV has had hiccups in the United States. One example is several years of negligible protection against H1N1 disease. As long as LAIV’s vaccine strain had reasonably matched the circulating strains, LAIV worked at least as well as injectable influenza vaccine, and even offered some cross-protection against mildly mismatched strains. But after a number of years of LAIV use, vaccine effectiveness in the United States vs. H1N1 strains appeared to fade due to previously undetected but significant changes in the circulating H1N1 strain. The lesson is that mucosal immunity’s advantages are lost if too much change occurs in the pathogen target for sIgA and mucosally-associated lymphoid tissue cells (MALT)).
Other vaccines likely need to induce mucosal immunity
Protection at the mucosal level will likely be needed for success against norovirus, parainfluenza, respiratory syncytial virus (RSV), Neisseria gonorrhea, and chlamydia. Another helpful aspect of mucosal immunity is that immune cells and sIgA not only reside on the mucosa where the antigen was originally presented, but there is also a reasonable chance that these components will traffic to other mucosal surfaces.2
So intranasal vaccine could be expected to protect distant mucosal surfaces (urogenital, GI, and respiratory), leading to vaccine-induced systemic antibody plus mucosal immunity (sIGA and MALT responses) at each site.
Let’s look at a novel “two-site” chlamydia vaccine
Recently a phase 1 chlamydia vaccine that used a novel two-pronged administration site/schedule was successful at inducing both mucosal and systemic immunity in a proof-of-concept study – achieving the best of both worlds.3 This may be a template for vaccines in years to come. British investigators studied 50 healthy women aged 19-45 years in a double-blind, parallel, randomized, placebo-controlled trial that used a recombinant chlamydia protein subunit antigen (CTH522). The vaccine schedule involved three injectable priming doses followed soon thereafter by two intranasal boosting doses. There were three groups:
1. CTH522 adjuvanted with CAF01 liposomes (CTH522:CAF01).
2. CTH522 adjuvanted with aluminum hydroxide (CTH522:AH).
3. Placebo (saline).
The intramuscular (IM) priming schedule was 0, 1, and 4 months. The intranasal vaccine booster doses or placebo were given at 4.5 and 5 months. No related serious adverse reactions occurred. For injectable dosing, the most frequent adverse event was mild local injection-site reactions in all subjects in both vaccine groups vs. in 60% of placebo recipients (P = .053). The adjuvants were the likely cause for local reactions. Intranasal doses had local reactions in 47% of both vaccine groups and 60% of placebo recipients; P = 1.000).
Both vaccines produced systemic IgG seroconversion (including neutralizing antibody) plus small amounts of IgG in the nasal cavity and genital tract in all vaccine recipients; no placebo recipient seroconverted. Interestingly, liposomally-adjuvanted vaccine produced a more rapid systemic IgG response and higher serum titers than the alum-adjuvanted vaccine. Likewise, the IM liposomal vaccine also induced higher but still small mucosal IgG antibody responses (P = .0091). Intranasal IM-induced IgG titers were not boosted by later intranasal vaccine dosing.
Subjects getting liposomal vaccine (but not alum vaccine or placebo) boosters had detectable sIgA titers in both nasal and genital tract secretions. Liposomal vaccine recipients also had fivefold to sixfold higher median titers than alum vaccine recipients after the priming dose, and these higher titers persisted to the end of the study. All liposomal vaccine recipients developed antichlamydial cell-mediated responses vs. 57% alum-adjuvanted vaccine recipients. (P = .01). So both use of two-site dosing and the liposomal adjuvant appeared critical to better responses.
In summary
While this candidate vaccine has hurdles to overcome before coming into routine use, the proof-of-principle that a combination injectable-intranasal vaccine schedule can induce robust systemic and mucosal immunity when given with an appropriate adjuvant is very promising. Adding more vaccines to the schedule then becomes an issue, but that is one of those “good” problems we can deal with later.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines, receives funding from GlaxoSmithKline for studies on pneumococcal and rotavirus vaccines, and from Pfizer for a study on pneumococcal vaccine on which Dr. Harrison is a sub-investigator. The hospital also receives Centers for Disease Control and Prevention funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus, and also for rotavirus. Email Dr. Harrison at [email protected].
References
1. PLOS Biology. 2012 Sep 1. doi: 10.1371/journal.pbio.1001397.
2. Mucosal Immunity in the Human Female Reproductive Tract in “Mucosal Immunology,” 4th ed., Volume 2 (Cambridge, MA: Academic Press, 2015, pp. 2097-124).
3. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30279-8.
Growing vaping habit may lead to nicotine addiction in adolescents
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
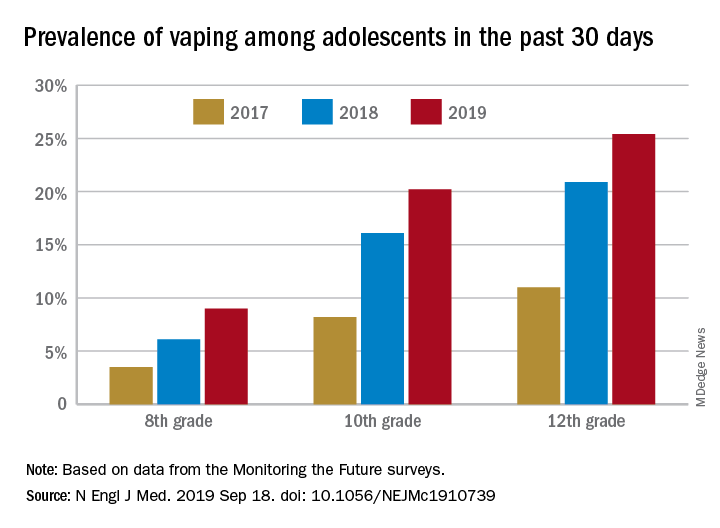
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Adolescents who use e-cigarettes every day may be developing nicotine addiction.
Major finding: In 2019, almost 12% of high school seniors were vaping every day.
Study details: Monitoring the Future surveys nationally representative samples of 8th-, 10th-, and 12th-grade students each year.
Disclosures: The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
Source: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
Wildfire smoke has acute cardiorespiratory impact, but long-term effects still under study
The 2019 wildfire season is underway in many locales across the United States, exposing millions of individuals to smoky conditions that will have health consequences ranging from stinging eyes to scratchy throats to a trip to the ED for asthma or chronic obstructive pulmonary disease (COPD) exacerbation. Questions about long-term health impacts are on the minds of many, including physicians and their patients who live with cardiorespiratory conditions.

John R. Balmes, MD, a pulmonologist at the University of California, San Francisco, and an expert on the respiratory and cardiovascular effects of air pollutants, suggested that the best available published literature points to “pretty strong evidence for acute effects of wildfire smoke on respiratory health, meaning people with preexisting asthma and COPD are at risk for exacerbations, and probably for respiratory tract infections as well.” He said, “It’s a little less clear, but there’s good biological plausibility for increased risk of respiratory tract infections because when your alveolar macrophages are overloaded with carbon particles that are toxic to those cells, they don’t function as well as a first line of defense against bacterial infection, for example.”
The new normal of wildfires

Warmer, drier summers in recent years in the western United States and many other regions, attributed by climate experts to global climate change, have produced catastrophic wildfires (PNAS;2016 Oct 18;113[42]11770-5; Science 2006 Aug 18;313:940-3). The Camp Fire in Northern California broke out in November 2018, took the lives of at least 85 people, and cost more than $16 billion in damage. Smoke from that blaze reached hazardous levels in San Francisco, Sacramento, Fresno, and many other smaller towns. Other forest fires in that year caused heavy smoke conditions in Portland, Seattle, Vancouver, and Anchorage. Such events are expected to be repeated often in the coming years (Int J Environ Res Public Health. 2019 Jul 6;16[13]).

Wildfire smoke can contain a wide range of substances, chemicals, and gases with known and unknown cardiorespiratory implications. “Smoke is composed primarily of carbon dioxide, water vapor, carbon monoxide, particulate matter, hydrocarbons and other organic chemicals, nitrogen oxides, trace minerals and several thousand other compounds,” according to the U.S. Environmental Protection Agency (Wildfire smoke: A guide for public health officials 2019. Washington, D.C.: EPA, 2019). The EPA report noted, “Particles with diameters less than 10 mcm (particulate matter, or PM10) can be inhaled into the lungs and affect the lungs, heart, and blood vessels. The smallest particles, those less than 2.5 mcm in diameter (PM2.5), are the greatest risk to public health because they can reach deep into the lungs and may even make it into the bloodstream.”
Research on health impact
In early June of 2008, Wayne Cascio, MD, awoke in his Greenville, N.C., home to the stench of smoke emanating from a large peat fire burning some 65 miles away. By the time he reached the parking lot at East Carolina University in Greenville to begin his workday as chief of cardiology, the haze of smoke had thickened to the point where he could only see a few feet in front of him.

Over the next several weeks, the fire scorched 41,000 acres and produced haze and air pollution that far exceeded National Ambient Air Quality Standards for particulate matter and blanketed rural communities in the state’s eastern region. The price tag for management of the blaze reached $20 million. Because of his interest in the health effects of wildfire smoke and because of his relationship with investigators at the EPA, Dr. Cascio initiated an epidemiology study to investigate the effects of exposure on cardiorespiratory outcomes in the population affected by the fire (Environ Health Perspect. 2011 Oct;119[10]:1415-20).
By combining satellite data with syndromic surveillance drawn from hospital records in 41 counties contained in the North Carolina Disease Event Tracking and Epidemiologic Collection Tool, he and his colleagues found that exposure to the peat wildfire smoke led to increases in the cumulative risk ratio for asthma (relative risk, 1.65), chronic obstructive pulmonary disease (RR, 1.73), and pneumonia and acute bronchitis (RR, 1.59). ED visits related to cardiopulmonary symptoms and heart failure also were significantly increased (RR, 1.23 and 1.37, respectively). “That was really the first study to strongly identify a cardiac endpoint related to wildfire smoke exposure,” said Dr. Cascio, who now directs the EPA’s National Health and Environmental Effects Research Laboratory. “It really pointed out how little we knew about the health effects of wildfire up until that time.”
Those early findings have been replicated in subsequent research about the acute health effects of exposure to wildfire smoke, which contains PM2.5 and other toxic substances from structures, electronic devices, and automobiles destroyed in the path of flames, including heavy metals and asbestos. Most of the work has focused on smoke-related cardiovascular and respiratory ED visits and hospitalizations.
A study of the 2008 California wildfire impact on ED visits accounted for ozone levels in addition to PM2.5 in the smoke. During the active fire periods, PM2.5 was significantly associated with exacerbations of asthma and COPD and these effects remained after controlling for ozone levels. PM2.5 inhalation during the wildfires was associated with increased risk of an ED visit for asthma (RR, 1.112; 95% confidence interval, 1.087-1.138) for a 10 mcg/m3 increase in PM2.5 and COPD (RR, 1.05; 95% CI, 1.019-1.0825), as well as for combined respiratory visits (RR, 1.035; 95% CI, 1.023-1.046) (Environ Int. 2109 Aug;129:291-8).
Researchers who evaluated the health impacts of wildfires in California during the 2015 fire season found an increase in all-cause cardiovascular and respiratory ED visits, especially among those aged 65 years and older during smoke days. The population-based study included 1,196,233 ED visits during May 1–Sept. 30 that year. PM2.5 concentrations were categorized as light, medium, or dense. Relative risk rose with the amount of smoke in the air. Rates of all-cause cardiovascular ED visits were elevated across levels of smoke density, with the greatest increase on dense smoke days and among those aged 65 years or older (RR,1.15; 95% CI, 1.09-1.22). All-cause cerebrovascular visits were associated with dense smoke days, especially among those aged 65 years and older (RR, 1.22; 95% CI, 1.00-1.49). Respiratory conditions also were increased on dense smoke days (RR, 1.18; 95% CI, 1.08-1.28) (J Am Heart Assoc. 2018 Apr 11;7:e007492. doi: 10.1161/JAHA.117.007492).
Long-term effects unknown
When it comes to the long-term effects of wildfire smoke on human health outcomes, much less is known. In a recent literature review, Colleen E. Reid, PhD, and Melissa May Maestas, PhD, found only one study that investigated long-term respiratory health impacts of wildfire smoke, and only a few studies that have estimated future health impacts of wildfires under likely climate change scenarios (Curr Opin Pulm Med. 2019 Mar;25:179-87).

“We know that there are immediate respiratory health effects from wildfire smoke,” said Dr. Reid of the department of geography at the University of Colorado Boulder. “What’s less known is everything else. That’s challenging, because people want to know about the long-term health effects.”
Evidence from the scientific literature suggests that exposure to air pollution adversely affects cardiovascular health, but whether exposure to wildfire smoke confers a similar risk is less clear. “Until just a few years ago we haven’t been able to study wildfire exposure measures on a large scale,” said EPA scientist Ana G. Rappold, PhD, a statistician there in the environmental public health division of the National Health and Environmental Effects Research Laboratory. “It’s also hard to predict wildfires, so it’s hard to plan for an epidemiologic study if you don’t know where they’re going to occur.”

Dr. Rappold and colleagues examined cardiopulmonary hospitalizations among adults aged 65 years and older in 692 U.S. counties within 200 km of 123 large wildfires during 2008-2010 (Environ Health Perspect. 2019;127[3]:37006. doi: 10.1289/EHP3860). They observed that an increased risk of PM2.5-related cardiopulmonary hospitalizations was similar on smoke and nonsmoke days across multiple lags and exposure metrics, while risk for asthma-related hospitalizations was higher during smoke days. “One hypothesis is that this was an older study population, so naturally if you’re inhaling smoke, the first organ that’s impacted in an older population is the lungs,” Dr. Rappold said. “If you go to the hospital for asthma, wheezing, or bronchitis, you are taken out of the risk pool for cardiovascular and other diseases. That could explain why in other studies we don’t see a clear cardiovascular signal as we have for air pollution studies in general. Another aspect to this study is, the exposure metric was PM2.5, but smoke contains many other components, particularly gases, which are respiratory irritants. It could be that this triggers a higher risk for respiratory [effects] than regular episodes of high PM2.5 exposure, just because of the additional gases that people are exposed to.”
Another complicating factor is the paucity of data about solutions to long-term exposure to wildfire smoke. “If you’re impacted by high-exposure levels for 60 days, that is not something we have experienced before,” Dr. Rappold noted. “What are the solutions for that community? What works? Can we show that by implementing community-level resilience plans with HEPA [high-efficiency particulate air] filters or other interventions, do the overall outcomes improve? Doctors are the first ones to talk with their patients about their symptoms and about how to take care of their conditions. They can clearly make a difference in emphasizing reducing exposures in a way that fits their patients individually, either reducing the amount of time spent outside, the duration of exposure, and the level of exposure. Maybe change activities based on the intensity of exposure. Don’t go for a run outside when it’s smoky, because your ventilation rate is higher and you will breathe in more smoke. Become aware of those things.”
Advising vulnerable patients
While research in this field advances, the unforgiving wildfire season looms, assuring more destruction of property and threats to cardiorespiratory health. “There are a lot of questions that research will have an opportunity to address as we go forward, including the utility and the benefit of N95 masks, the utility of HEPA filters used in the house, and even with HVAC [heating, ventilation, and air conditioning] systems,” Dr. Cascio said. “Can we really clean up the indoor air well enough to protect us from wildfire smoke?”
The way he sees it, the time is ripe for clinicians and officials in public and private practice settings to refine how they distribute information to people living in areas affected by wildfire smoke. “We can’t force people do anything, but at least if they’re informed, then they understand they can make an informed decision about how they might want to affect what they do that would limit their exposure,” he said. “As a patient, my health care system sends text and email messages to me. So, why couldn’t the hospital send out a text message or an email to all of the patients with COPD, coronary disease, and heart failure when an area is impacted by smoke, saying, ‘Check your air quality and take action if air quality is poor?’ Physicians don’t have time to do this kind of education in the office for all of their patients. I know that from experience. But if one were to only focus on those at highest risk, and encourage them to follow our guidelines, which might include doing HEPA filter treatment in the home, we probably would reduce the number of clinical events in a cost-effective way.”
The 2019 wildfire season is underway in many locales across the United States, exposing millions of individuals to smoky conditions that will have health consequences ranging from stinging eyes to scratchy throats to a trip to the ED for asthma or chronic obstructive pulmonary disease (COPD) exacerbation. Questions about long-term health impacts are on the minds of many, including physicians and their patients who live with cardiorespiratory conditions.
John R. Balmes, MD, a pulmonologist at the University of California, San Francisco, and an expert on the respiratory and cardiovascular effects of air pollutants, suggested that the best available published literature points to “pretty strong evidence for acute effects of wildfire smoke on respiratory health, meaning people with preexisting asthma and COPD are at risk for exacerbations, and probably for respiratory tract infections as well.” He said, “It’s a little less clear, but there’s good biological plausibility for increased risk of respiratory tract infections because when your alveolar macrophages are overloaded with carbon particles that are toxic to those cells, they don’t function as well as a first line of defense against bacterial infection, for example.”
The new normal of wildfires
Warmer, drier summers in recent years in the western United States and many other regions, attributed by climate experts to global climate change, have produced catastrophic wildfires (PNAS;2016 Oct 18;113[42]11770-5; Science 2006 Aug 18;313:940-3). The Camp Fire in Northern California broke out in November 2018, took the lives of at least 85 people, and cost more than $16 billion in damage. Smoke from that blaze reached hazardous levels in San Francisco, Sacramento, Fresno, and many other smaller towns. Other forest fires in that year caused heavy smoke conditions in Portland, Seattle, Vancouver, and Anchorage. Such events are expected to be repeated often in the coming years (Int J Environ Res Public Health. 2019 Jul 6;16[13]).
Wildfire smoke can contain a wide range of substances, chemicals, and gases with known and unknown cardiorespiratory implications. “Smoke is composed primarily of carbon dioxide, water vapor, carbon monoxide, particulate matter, hydrocarbons and other organic chemicals, nitrogen oxides, trace minerals and several thousand other compounds,” according to the U.S. Environmental Protection Agency (Wildfire smoke: A guide for public health officials 2019. Washington, D.C.: EPA, 2019). The EPA report noted, “Particles with diameters less than 10 mcm (particulate matter, or PM10) can be inhaled into the lungs and affect the lungs, heart, and blood vessels. The smallest particles, those less than 2.5 mcm in diameter (PM2.5), are the greatest risk to public health because they can reach deep into the lungs and may even make it into the bloodstream.”
Research on health impact
In early June of 2008, Wayne Cascio, MD, awoke in his Greenville, N.C., home to the stench of smoke emanating from a large peat fire burning some 65 miles away. By the time he reached the parking lot at East Carolina University in Greenville to begin his workday as chief of cardiology, the haze of smoke had thickened to the point where he could only see a few feet in front of him.
Over the next several weeks, the fire scorched 41,000 acres and produced haze and air pollution that far exceeded National Ambient Air Quality Standards for particulate matter and blanketed rural communities in the state’s eastern region. The price tag for management of the blaze reached $20 million. Because of his interest in the health effects of wildfire smoke and because of his relationship with investigators at the EPA, Dr. Cascio initiated an epidemiology study to investigate the effects of exposure on cardiorespiratory outcomes in the population affected by the fire (Environ Health Perspect. 2011 Oct;119[10]:1415-20).
By combining satellite data with syndromic surveillance drawn from hospital records in 41 counties contained in the North Carolina Disease Event Tracking and Epidemiologic Collection Tool, he and his colleagues found that exposure to the peat wildfire smoke led to increases in the cumulative risk ratio for asthma (relative risk, 1.65), chronic obstructive pulmonary disease (RR, 1.73), and pneumonia and acute bronchitis (RR, 1.59). ED visits related to cardiopulmonary symptoms and heart failure also were significantly increased (RR, 1.23 and 1.37, respectively). “That was really the first study to strongly identify a cardiac endpoint related to wildfire smoke exposure,” said Dr. Cascio, who now directs the EPA’s National Health and Environmental Effects Research Laboratory. “It really pointed out how little we knew about the health effects of wildfire up until that time.”
Those early findings have been replicated in subsequent research about the acute health effects of exposure to wildfire smoke, which contains PM2.5 and other toxic substances from structures, electronic devices, and automobiles destroyed in the path of flames, including heavy metals and asbestos. Most of the work has focused on smoke-related cardiovascular and respiratory ED visits and hospitalizations.
A study of the 2008 California wildfire impact on ED visits accounted for ozone levels in addition to PM2.5 in the smoke. During the active fire periods, PM2.5 was significantly associated with exacerbations of asthma and COPD and these effects remained after controlling for ozone levels. PM2.5 inhalation during the wildfires was associated with increased risk of an ED visit for asthma (RR, 1.112; 95% confidence interval, 1.087-1.138) for a 10 mcg/m3 increase in PM2.5 and COPD (RR, 1.05; 95% CI, 1.019-1.0825), as well as for combined respiratory visits (RR, 1.035; 95% CI, 1.023-1.046) (Environ Int. 2109 Aug;129:291-8).
Researchers who evaluated the health impacts of wildfires in California during the 2015 fire season found an increase in all-cause cardiovascular and respiratory ED visits, especially among those aged 65 years and older during smoke days. The population-based study included 1,196,233 ED visits during May 1–Sept. 30 that year. PM2.5 concentrations were categorized as light, medium, or dense. Relative risk rose with the amount of smoke in the air. Rates of all-cause cardiovascular ED visits were elevated across levels of smoke density, with the greatest increase on dense smoke days and among those aged 65 years or older (RR,1.15; 95% CI, 1.09-1.22). All-cause cerebrovascular visits were associated with dense smoke days, especially among those aged 65 years and older (RR, 1.22; 95% CI, 1.00-1.49). Respiratory conditions also were increased on dense smoke days (RR, 1.18; 95% CI, 1.08-1.28) (J Am Heart Assoc. 2018 Apr 11;7:e007492. doi: 10.1161/JAHA.117.007492).
Long-term effects unknown
When it comes to the long-term effects of wildfire smoke on human health outcomes, much less is known. In a recent literature review, Colleen E. Reid, PhD, and Melissa May Maestas, PhD, found only one study that investigated long-term respiratory health impacts of wildfire smoke, and only a few studies that have estimated future health impacts of wildfires under likely climate change scenarios (Curr Opin Pulm Med. 2019 Mar;25:179-87).
“We know that there are immediate respiratory health effects from wildfire smoke,” said Dr. Reid of the department of geography at the University of Colorado Boulder. “What’s less known is everything else. That’s challenging, because people want to know about the long-term health effects.”
Evidence from the scientific literature suggests that exposure to air pollution adversely affects cardiovascular health, but whether exposure to wildfire smoke confers a similar risk is less clear. “Until just a few years ago we haven’t been able to study wildfire exposure measures on a large scale,” said EPA scientist Ana G. Rappold, PhD, a statistician there in the environmental public health division of the National Health and Environmental Effects Research Laboratory. “It’s also hard to predict wildfires, so it’s hard to plan for an epidemiologic study if you don’t know where they’re going to occur.”
Dr. Rappold and colleagues examined cardiopulmonary hospitalizations among adults aged 65 years and older in 692 U.S. counties within 200 km of 123 large wildfires during 2008-2010 (Environ Health Perspect. 2019;127[3]:37006. doi: 10.1289/EHP3860). They observed that an increased risk of PM2.5-related cardiopulmonary hospitalizations was similar on smoke and nonsmoke days across multiple lags and exposure metrics, while risk for asthma-related hospitalizations was higher during smoke days. “One hypothesis is that this was an older study population, so naturally if you’re inhaling smoke, the first organ that’s impacted in an older population is the lungs,” Dr. Rappold said. “If you go to the hospital for asthma, wheezing, or bronchitis, you are taken out of the risk pool for cardiovascular and other diseases. That could explain why in other studies we don’t see a clear cardiovascular signal as we have for air pollution studies in general. Another aspect to this study is, the exposure metric was PM2.5, but smoke contains many other components, particularly gases, which are respiratory irritants. It could be that this triggers a higher risk for respiratory [effects] than regular episodes of high PM2.5 exposure, just because of the additional gases that people are exposed to.”
Another complicating factor is the paucity of data about solutions to long-term exposure to wildfire smoke. “If you’re impacted by high-exposure levels for 60 days, that is not something we have experienced before,” Dr. Rappold noted. “What are the solutions for that community? What works? Can we show that by implementing community-level resilience plans with HEPA [high-efficiency particulate air] filters or other interventions, do the overall outcomes improve? Doctors are the first ones to talk with their patients about their symptoms and about how to take care of their conditions. They can clearly make a difference in emphasizing reducing exposures in a way that fits their patients individually, either reducing the amount of time spent outside, the duration of exposure, and the level of exposure. Maybe change activities based on the intensity of exposure. Don’t go for a run outside when it’s smoky, because your ventilation rate is higher and you will breathe in more smoke. Become aware of those things.”
Advising vulnerable patients
While research in this field advances, the unforgiving wildfire season looms, assuring more destruction of property and threats to cardiorespiratory health. “There are a lot of questions that research will have an opportunity to address as we go forward, including the utility and the benefit of N95 masks, the utility of HEPA filters used in the house, and even with HVAC [heating, ventilation, and air conditioning] systems,” Dr. Cascio said. “Can we really clean up the indoor air well enough to protect us from wildfire smoke?”
The way he sees it, the time is ripe for clinicians and officials in public and private practice settings to refine how they distribute information to people living in areas affected by wildfire smoke. “We can’t force people do anything, but at least if they’re informed, then they understand they can make an informed decision about how they might want to affect what they do that would limit their exposure,” he said. “As a patient, my health care system sends text and email messages to me. So, why couldn’t the hospital send out a text message or an email to all of the patients with COPD, coronary disease, and heart failure when an area is impacted by smoke, saying, ‘Check your air quality and take action if air quality is poor?’ Physicians don’t have time to do this kind of education in the office for all of their patients. I know that from experience. But if one were to only focus on those at highest risk, and encourage them to follow our guidelines, which might include doing HEPA filter treatment in the home, we probably would reduce the number of clinical events in a cost-effective way.”
The 2019 wildfire season is underway in many locales across the United States, exposing millions of individuals to smoky conditions that will have health consequences ranging from stinging eyes to scratchy throats to a trip to the ED for asthma or chronic obstructive pulmonary disease (COPD) exacerbation. Questions about long-term health impacts are on the minds of many, including physicians and their patients who live with cardiorespiratory conditions.
John R. Balmes, MD, a pulmonologist at the University of California, San Francisco, and an expert on the respiratory and cardiovascular effects of air pollutants, suggested that the best available published literature points to “pretty strong evidence for acute effects of wildfire smoke on respiratory health, meaning people with preexisting asthma and COPD are at risk for exacerbations, and probably for respiratory tract infections as well.” He said, “It’s a little less clear, but there’s good biological plausibility for increased risk of respiratory tract infections because when your alveolar macrophages are overloaded with carbon particles that are toxic to those cells, they don’t function as well as a first line of defense against bacterial infection, for example.”
The new normal of wildfires
Warmer, drier summers in recent years in the western United States and many other regions, attributed by climate experts to global climate change, have produced catastrophic wildfires (PNAS;2016 Oct 18;113[42]11770-5; Science 2006 Aug 18;313:940-3). The Camp Fire in Northern California broke out in November 2018, took the lives of at least 85 people, and cost more than $16 billion in damage. Smoke from that blaze reached hazardous levels in San Francisco, Sacramento, Fresno, and many other smaller towns. Other forest fires in that year caused heavy smoke conditions in Portland, Seattle, Vancouver, and Anchorage. Such events are expected to be repeated often in the coming years (Int J Environ Res Public Health. 2019 Jul 6;16[13]).
Wildfire smoke can contain a wide range of substances, chemicals, and gases with known and unknown cardiorespiratory implications. “Smoke is composed primarily of carbon dioxide, water vapor, carbon monoxide, particulate matter, hydrocarbons and other organic chemicals, nitrogen oxides, trace minerals and several thousand other compounds,” according to the U.S. Environmental Protection Agency (Wildfire smoke: A guide for public health officials 2019. Washington, D.C.: EPA, 2019). The EPA report noted, “Particles with diameters less than 10 mcm (particulate matter, or PM10) can be inhaled into the lungs and affect the lungs, heart, and blood vessels. The smallest particles, those less than 2.5 mcm in diameter (PM2.5), are the greatest risk to public health because they can reach deep into the lungs and may even make it into the bloodstream.”
Research on health impact
In early June of 2008, Wayne Cascio, MD, awoke in his Greenville, N.C., home to the stench of smoke emanating from a large peat fire burning some 65 miles away. By the time he reached the parking lot at East Carolina University in Greenville to begin his workday as chief of cardiology, the haze of smoke had thickened to the point where he could only see a few feet in front of him.
Over the next several weeks, the fire scorched 41,000 acres and produced haze and air pollution that far exceeded National Ambient Air Quality Standards for particulate matter and blanketed rural communities in the state’s eastern region. The price tag for management of the blaze reached $20 million. Because of his interest in the health effects of wildfire smoke and because of his relationship with investigators at the EPA, Dr. Cascio initiated an epidemiology study to investigate the effects of exposure on cardiorespiratory outcomes in the population affected by the fire (Environ Health Perspect. 2011 Oct;119[10]:1415-20).
By combining satellite data with syndromic surveillance drawn from hospital records in 41 counties contained in the North Carolina Disease Event Tracking and Epidemiologic Collection Tool, he and his colleagues found that exposure to the peat wildfire smoke led to increases in the cumulative risk ratio for asthma (relative risk, 1.65), chronic obstructive pulmonary disease (RR, 1.73), and pneumonia and acute bronchitis (RR, 1.59). ED visits related to cardiopulmonary symptoms and heart failure also were significantly increased (RR, 1.23 and 1.37, respectively). “That was really the first study to strongly identify a cardiac endpoint related to wildfire smoke exposure,” said Dr. Cascio, who now directs the EPA’s National Health and Environmental Effects Research Laboratory. “It really pointed out how little we knew about the health effects of wildfire up until that time.”
Those early findings have been replicated in subsequent research about the acute health effects of exposure to wildfire smoke, which contains PM2.5 and other toxic substances from structures, electronic devices, and automobiles destroyed in the path of flames, including heavy metals and asbestos. Most of the work has focused on smoke-related cardiovascular and respiratory ED visits and hospitalizations.
A study of the 2008 California wildfire impact on ED visits accounted for ozone levels in addition to PM2.5 in the smoke. During the active fire periods, PM2.5 was significantly associated with exacerbations of asthma and COPD and these effects remained after controlling for ozone levels. PM2.5 inhalation during the wildfires was associated with increased risk of an ED visit for asthma (RR, 1.112; 95% confidence interval, 1.087-1.138) for a 10 mcg/m3 increase in PM2.5 and COPD (RR, 1.05; 95% CI, 1.019-1.0825), as well as for combined respiratory visits (RR, 1.035; 95% CI, 1.023-1.046) (Environ Int. 2109 Aug;129:291-8).
Researchers who evaluated the health impacts of wildfires in California during the 2015 fire season found an increase in all-cause cardiovascular and respiratory ED visits, especially among those aged 65 years and older during smoke days. The population-based study included 1,196,233 ED visits during May 1–Sept. 30 that year. PM2.5 concentrations were categorized as light, medium, or dense. Relative risk rose with the amount of smoke in the air. Rates of all-cause cardiovascular ED visits were elevated across levels of smoke density, with the greatest increase on dense smoke days and among those aged 65 years or older (RR,1.15; 95% CI, 1.09-1.22). All-cause cerebrovascular visits were associated with dense smoke days, especially among those aged 65 years and older (RR, 1.22; 95% CI, 1.00-1.49). Respiratory conditions also were increased on dense smoke days (RR, 1.18; 95% CI, 1.08-1.28) (J Am Heart Assoc. 2018 Apr 11;7:e007492. doi: 10.1161/JAHA.117.007492).
Long-term effects unknown
When it comes to the long-term effects of wildfire smoke on human health outcomes, much less is known. In a recent literature review, Colleen E. Reid, PhD, and Melissa May Maestas, PhD, found only one study that investigated long-term respiratory health impacts of wildfire smoke, and only a few studies that have estimated future health impacts of wildfires under likely climate change scenarios (Curr Opin Pulm Med. 2019 Mar;25:179-87).
“We know that there are immediate respiratory health effects from wildfire smoke,” said Dr. Reid of the department of geography at the University of Colorado Boulder. “What’s less known is everything else. That’s challenging, because people want to know about the long-term health effects.”
Evidence from the scientific literature suggests that exposure to air pollution adversely affects cardiovascular health, but whether exposure to wildfire smoke confers a similar risk is less clear. “Until just a few years ago we haven’t been able to study wildfire exposure measures on a large scale,” said EPA scientist Ana G. Rappold, PhD, a statistician there in the environmental public health division of the National Health and Environmental Effects Research Laboratory. “It’s also hard to predict wildfires, so it’s hard to plan for an epidemiologic study if you don’t know where they’re going to occur.”
Dr. Rappold and colleagues examined cardiopulmonary hospitalizations among adults aged 65 years and older in 692 U.S. counties within 200 km of 123 large wildfires during 2008-2010 (Environ Health Perspect. 2019;127[3]:37006. doi: 10.1289/EHP3860). They observed that an increased risk of PM2.5-related cardiopulmonary hospitalizations was similar on smoke and nonsmoke days across multiple lags and exposure metrics, while risk for asthma-related hospitalizations was higher during smoke days. “One hypothesis is that this was an older study population, so naturally if you’re inhaling smoke, the first organ that’s impacted in an older population is the lungs,” Dr. Rappold said. “If you go to the hospital for asthma, wheezing, or bronchitis, you are taken out of the risk pool for cardiovascular and other diseases. That could explain why in other studies we don’t see a clear cardiovascular signal as we have for air pollution studies in general. Another aspect to this study is, the exposure metric was PM2.5, but smoke contains many other components, particularly gases, which are respiratory irritants. It could be that this triggers a higher risk for respiratory [effects] than regular episodes of high PM2.5 exposure, just because of the additional gases that people are exposed to.”
Another complicating factor is the paucity of data about solutions to long-term exposure to wildfire smoke. “If you’re impacted by high-exposure levels for 60 days, that is not something we have experienced before,” Dr. Rappold noted. “What are the solutions for that community? What works? Can we show that by implementing community-level resilience plans with HEPA [high-efficiency particulate air] filters or other interventions, do the overall outcomes improve? Doctors are the first ones to talk with their patients about their symptoms and about how to take care of their conditions. They can clearly make a difference in emphasizing reducing exposures in a way that fits their patients individually, either reducing the amount of time spent outside, the duration of exposure, and the level of exposure. Maybe change activities based on the intensity of exposure. Don’t go for a run outside when it’s smoky, because your ventilation rate is higher and you will breathe in more smoke. Become aware of those things.”
Advising vulnerable patients
While research in this field advances, the unforgiving wildfire season looms, assuring more destruction of property and threats to cardiorespiratory health. “There are a lot of questions that research will have an opportunity to address as we go forward, including the utility and the benefit of N95 masks, the utility of HEPA filters used in the house, and even with HVAC [heating, ventilation, and air conditioning] systems,” Dr. Cascio said. “Can we really clean up the indoor air well enough to protect us from wildfire smoke?”
The way he sees it, the time is ripe for clinicians and officials in public and private practice settings to refine how they distribute information to people living in areas affected by wildfire smoke. “We can’t force people do anything, but at least if they’re informed, then they understand they can make an informed decision about how they might want to affect what they do that would limit their exposure,” he said. “As a patient, my health care system sends text and email messages to me. So, why couldn’t the hospital send out a text message or an email to all of the patients with COPD, coronary disease, and heart failure when an area is impacted by smoke, saying, ‘Check your air quality and take action if air quality is poor?’ Physicians don’t have time to do this kind of education in the office for all of their patients. I know that from experience. But if one were to only focus on those at highest risk, and encourage them to follow our guidelines, which might include doing HEPA filter treatment in the home, we probably would reduce the number of clinical events in a cost-effective way.”
Benefits of peanut desensitization may not last
based on data from a phase 2 randomized trial of individuals with confirmed peanut allergies.

Previous studies have shown that desensitization to peanuts can be successful, but sustained response to oral immunotherapy after treatment reduction or discontinuation has not been well studied, wrote R. Sharon Chinthrajah, MD, of Stanford (Calif.)University, and colleagues.
“We found that OIT with peanut was able to desensitise people with peanut allergy to 4,000 mg of peanut protein, but that discontinuation of peanut, or even a reduction to 300 mg daily, increased the likelihood of regaining clinical reactivity to peanut,” they wrote. “With peanut allergy therapies in varying stages of clinical development, and some nearing [Food and Drug Administration] approval, vital questions remain regarding the durability of treatment effects and the appropriate maintenance doses.”
In the Peanut Oral Immunotherapy Study: Safety Efficacy and Discovery (POISED), published in The Lancet, the researchers randomized 120 participants to three groups:
• 60 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by total discontinuation (peanut-0).
• 35 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by a 300-mg maintenance dose of peanut protein in the form of peanut flour (peanut-300).
• 25 patients to an oat flour placebo.
All participants were trained on how and when to use epinephrine autoinjector devices to treat allergic symptoms such as respiratory problems (cough, shortness of breath, or change in voice), widespread hives or erythema, repetitive vomiting, persistent abdominal pain, angioedema of the face, or feeling faint.
The primary outcome was passing a double-blind, placebo-controlled, food challenge (DBPCFC) to 4,000 mg of peanut protein, which was measured at baseline and at weeks 104, 117, 130, 143, and 156.
Overall, 35% of the peanut-0 group passed the challenge at 104 and 117 weeks, compared with 4% of the placebo group. At week 156 after discontinuing OIT, 13% of the peanut-0 group met the DBPCFC challenge, compared with 4% of the placebo group. However, 37% of participants randomized to a reduced peanut protein dose of 300 mg passed the challenge at 156 weeks, suggesting that more data are needed on optimal maintenance dosing strategies.
Baseline demographics were similar across all groups. The median age at study enrollment was 11 years and the median allergy duration was 9 years. The most common adverse events were mild gastrointestinal and respiratory problems. Adverse events decreased over time in all three groups.
“Higher levels of peanut-specific IgE to total IgE ratio, peanut sIgE, Ara h 1, Ara h 2, and Ara h 1 IgE to peanut-specific IgE ratio at baseline in participants were associated with increased frequencies of adverse events during active peanut OIT,” the researchers noted.
The study findings were limited by several factors including the ability of participants to tolerate 4,000 mg of peanut protein after achieving a maintenance dose but conducting serial testing only for those who passed the challenge. In addition, the results may be limited to peanut and not generalizable to other food allergies, the researchers said.
However, the results suggest that OIT remains a promising treatment for peanut allergies, and the association of biomarkers with clinical outcomes “might help the practitioner in identifying good candidates for OIT and those individuals who warrant increased vigilance against allergic reactions during OIT,” they said.
The National Institutes of Health supported the study. The researchers had no financial conflicts to disclose.
SOURCE: Chinthrajah RS et al. Lancet. 2019 Sep 12. doi: 10.1016/S0140-6736(19)31793-3.
based on data from a phase 2 randomized trial of individuals with confirmed peanut allergies.
Previous studies have shown that desensitization to peanuts can be successful, but sustained response to oral immunotherapy after treatment reduction or discontinuation has not been well studied, wrote R. Sharon Chinthrajah, MD, of Stanford (Calif.)University, and colleagues.
“We found that OIT with peanut was able to desensitise people with peanut allergy to 4,000 mg of peanut protein, but that discontinuation of peanut, or even a reduction to 300 mg daily, increased the likelihood of regaining clinical reactivity to peanut,” they wrote. “With peanut allergy therapies in varying stages of clinical development, and some nearing [Food and Drug Administration] approval, vital questions remain regarding the durability of treatment effects and the appropriate maintenance doses.”
In the Peanut Oral Immunotherapy Study: Safety Efficacy and Discovery (POISED), published in The Lancet, the researchers randomized 120 participants to three groups:
• 60 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by total discontinuation (peanut-0).
• 35 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by a 300-mg maintenance dose of peanut protein in the form of peanut flour (peanut-300).
• 25 patients to an oat flour placebo.
All participants were trained on how and when to use epinephrine autoinjector devices to treat allergic symptoms such as respiratory problems (cough, shortness of breath, or change in voice), widespread hives or erythema, repetitive vomiting, persistent abdominal pain, angioedema of the face, or feeling faint.
The primary outcome was passing a double-blind, placebo-controlled, food challenge (DBPCFC) to 4,000 mg of peanut protein, which was measured at baseline and at weeks 104, 117, 130, 143, and 156.
Overall, 35% of the peanut-0 group passed the challenge at 104 and 117 weeks, compared with 4% of the placebo group. At week 156 after discontinuing OIT, 13% of the peanut-0 group met the DBPCFC challenge, compared with 4% of the placebo group. However, 37% of participants randomized to a reduced peanut protein dose of 300 mg passed the challenge at 156 weeks, suggesting that more data are needed on optimal maintenance dosing strategies.
Baseline demographics were similar across all groups. The median age at study enrollment was 11 years and the median allergy duration was 9 years. The most common adverse events were mild gastrointestinal and respiratory problems. Adverse events decreased over time in all three groups.
“Higher levels of peanut-specific IgE to total IgE ratio, peanut sIgE, Ara h 1, Ara h 2, and Ara h 1 IgE to peanut-specific IgE ratio at baseline in participants were associated with increased frequencies of adverse events during active peanut OIT,” the researchers noted.
The study findings were limited by several factors including the ability of participants to tolerate 4,000 mg of peanut protein after achieving a maintenance dose but conducting serial testing only for those who passed the challenge. In addition, the results may be limited to peanut and not generalizable to other food allergies, the researchers said.
However, the results suggest that OIT remains a promising treatment for peanut allergies, and the association of biomarkers with clinical outcomes “might help the practitioner in identifying good candidates for OIT and those individuals who warrant increased vigilance against allergic reactions during OIT,” they said.
The National Institutes of Health supported the study. The researchers had no financial conflicts to disclose.
SOURCE: Chinthrajah RS et al. Lancet. 2019 Sep 12. doi: 10.1016/S0140-6736(19)31793-3.
based on data from a phase 2 randomized trial of individuals with confirmed peanut allergies.
Previous studies have shown that desensitization to peanuts can be successful, but sustained response to oral immunotherapy after treatment reduction or discontinuation has not been well studied, wrote R. Sharon Chinthrajah, MD, of Stanford (Calif.)University, and colleagues.
“We found that OIT with peanut was able to desensitise people with peanut allergy to 4,000 mg of peanut protein, but that discontinuation of peanut, or even a reduction to 300 mg daily, increased the likelihood of regaining clinical reactivity to peanut,” they wrote. “With peanut allergy therapies in varying stages of clinical development, and some nearing [Food and Drug Administration] approval, vital questions remain regarding the durability of treatment effects and the appropriate maintenance doses.”
In the Peanut Oral Immunotherapy Study: Safety Efficacy and Discovery (POISED), published in The Lancet, the researchers randomized 120 participants to three groups:
• 60 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by total discontinuation (peanut-0).
• 35 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by a 300-mg maintenance dose of peanut protein in the form of peanut flour (peanut-300).
• 25 patients to an oat flour placebo.
All participants were trained on how and when to use epinephrine autoinjector devices to treat allergic symptoms such as respiratory problems (cough, shortness of breath, or change in voice), widespread hives or erythema, repetitive vomiting, persistent abdominal pain, angioedema of the face, or feeling faint.
The primary outcome was passing a double-blind, placebo-controlled, food challenge (DBPCFC) to 4,000 mg of peanut protein, which was measured at baseline and at weeks 104, 117, 130, 143, and 156.
Overall, 35% of the peanut-0 group passed the challenge at 104 and 117 weeks, compared with 4% of the placebo group. At week 156 after discontinuing OIT, 13% of the peanut-0 group met the DBPCFC challenge, compared with 4% of the placebo group. However, 37% of participants randomized to a reduced peanut protein dose of 300 mg passed the challenge at 156 weeks, suggesting that more data are needed on optimal maintenance dosing strategies.
Baseline demographics were similar across all groups. The median age at study enrollment was 11 years and the median allergy duration was 9 years. The most common adverse events were mild gastrointestinal and respiratory problems. Adverse events decreased over time in all three groups.
“Higher levels of peanut-specific IgE to total IgE ratio, peanut sIgE, Ara h 1, Ara h 2, and Ara h 1 IgE to peanut-specific IgE ratio at baseline in participants were associated with increased frequencies of adverse events during active peanut OIT,” the researchers noted.
The study findings were limited by several factors including the ability of participants to tolerate 4,000 mg of peanut protein after achieving a maintenance dose but conducting serial testing only for those who passed the challenge. In addition, the results may be limited to peanut and not generalizable to other food allergies, the researchers said.
However, the results suggest that OIT remains a promising treatment for peanut allergies, and the association of biomarkers with clinical outcomes “might help the practitioner in identifying good candidates for OIT and those individuals who warrant increased vigilance against allergic reactions during OIT,” they said.
The National Institutes of Health supported the study. The researchers had no financial conflicts to disclose.
SOURCE: Chinthrajah RS et al. Lancet. 2019 Sep 12. doi: 10.1016/S0140-6736(19)31793-3.
FROM THE LANCET
CDC activates Emergency Operations Center to investigate vaping-associated lung injury
![]()
This move allows the CDC “to provide increased operational support” to CDC staff to meet the evolving challenges of the outbreak of vaping-related injuries and deaths, says a statement from the CDC.
“CDC has made it a priority to find out what is causing this outbreak,” noted CDC Director Robert Redfield, MD, in the statement.
The agency “continues to work closely with the U.S. Food and Drug Administration to collect information about recent e-cigarette product use, or vaping, among patients and to test the substances or chemicals within e-cigarette products used by case patients,” according to the statement.
The CDC provided email addresses and site addresses for gathering information and communicating about e-cigarettes.
Information about the collection of e-cigarettes for possible testing by FDA can be obtained through contacting [email protected].
To communicate with CDC about this public health response, clinicians and health officials can contact [email protected].
More information on the current outbreak related to e-cigarettes is available at https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html.
General information on electronic cigarette products, can be found at www.cdc.gov/e-cigarettes.
Individuals concerned about health risks of vaping should consider refraining from e-cigarette use while the cases of lung injury are being investigated, the CDC said.
![]()
This move allows the CDC “to provide increased operational support” to CDC staff to meet the evolving challenges of the outbreak of vaping-related injuries and deaths, says a statement from the CDC.
“CDC has made it a priority to find out what is causing this outbreak,” noted CDC Director Robert Redfield, MD, in the statement.
The agency “continues to work closely with the U.S. Food and Drug Administration to collect information about recent e-cigarette product use, or vaping, among patients and to test the substances or chemicals within e-cigarette products used by case patients,” according to the statement.
The CDC provided email addresses and site addresses for gathering information and communicating about e-cigarettes.
Information about the collection of e-cigarettes for possible testing by FDA can be obtained through contacting [email protected].
To communicate with CDC about this public health response, clinicians and health officials can contact [email protected].
More information on the current outbreak related to e-cigarettes is available at https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html.
General information on electronic cigarette products, can be found at www.cdc.gov/e-cigarettes.
Individuals concerned about health risks of vaping should consider refraining from e-cigarette use while the cases of lung injury are being investigated, the CDC said.
![]()
This move allows the CDC “to provide increased operational support” to CDC staff to meet the evolving challenges of the outbreak of vaping-related injuries and deaths, says a statement from the CDC.
“CDC has made it a priority to find out what is causing this outbreak,” noted CDC Director Robert Redfield, MD, in the statement.
The agency “continues to work closely with the U.S. Food and Drug Administration to collect information about recent e-cigarette product use, or vaping, among patients and to test the substances or chemicals within e-cigarette products used by case patients,” according to the statement.
The CDC provided email addresses and site addresses for gathering information and communicating about e-cigarettes.
Information about the collection of e-cigarettes for possible testing by FDA can be obtained through contacting [email protected].
To communicate with CDC about this public health response, clinicians and health officials can contact [email protected].
More information on the current outbreak related to e-cigarettes is available at https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html.
General information on electronic cigarette products, can be found at www.cdc.gov/e-cigarettes.
Individuals concerned about health risks of vaping should consider refraining from e-cigarette use while the cases of lung injury are being investigated, the CDC said.
Pulegone levels in e-liquids, smokeless tobacco products exceed FDA limits
A group of mint- and menthol-flavored e-liquids and smokeless tobacco products contained significantly more pulegone – a known carcinogen that causes hepatic carcinomas, pulmonary metaplasia, and other neoplasms – than the Food and Drug Administration considers acceptable, according to new findings.
Pulegone, an oil extract from mint plants such as peppermint, spearmint, and pennyroyal, was banned as a food additive by the agency in 2018, and the tobacco industry has taken steps to minimize pulegone levels in cigarettes because of the toxicity concerns.
Studies from the Centers for Disease Control and Prevention, however, have indicated that mint- and menthol-flavored e-cigarette liquids and smokeless tobacco products marketed in the United States contain substantial amounts of the substance, Sairam V. Jabba, DVM, PhD, and Sven-Eric Jordt, PhD, said in a research letter published in JAMA Internal Medicine.
Dr. Jabba and Dr. Jordt, both with the department of anesthesiology at Duke University, Durham, N.C., calculated the margin of exposure in five e-liquids (V2 Menthol, V2 Peppermint, Premium Menthol, South Beach Smoke Menthol, and South Beach Smoke Peppermint) and one smokeless tobacco product (Skoal Xtra Mint snuff) by dividing the no–observed adverse event level (13.39 mg/kg of bodyweight per day) by the mean human exposure to e-liquids or smokeless tobacco. The FDA considers margin-of-exposure values of 10,000 or less to require mitigation strategies.
The six products included in the analysis had pulegone concentration levels ranging from 25.7 to 119.0 mcg/g (a menthol cigarette has a pulegone concentration of 0.037-0.290 mcg/g). Based on those levels, light daily use (5 mL e-liquid, 10 g smokeless tobacco, half a pack of cigarettes) exposed e-cigarette users to 44-198 times more pulegone, compared with menthol cigarettes, and exposed smokeless tobacco users to 168-1,319 times as much pulegone. The margin of exposure ranged from 1,298 to 6,012, all below 10,000 threshhold the FDA deems acceptable.
For heavy daily use (20 mL e-liquid, 30 g smokeless tobacco, two packs of cigarettes), e-cigarette users were exposed to 282-1,608 times more pulegone, compared with menthol cigarettes; smokeless tobacco users were exposed to 126-990 times more pulegone. The margin of exposure ranged from 325 to 1,503.
The study findings “appear to establish health risks associated with pulegone intake and concerns that the FDA should address before suggesting mint- and menthol-flavored e-cigarettes and smokeless tobacco products as alternatives for people who use combustible tobacco products,” Dr. Jabba and Dr. Jordt concluded.
The study was funded by a grant from the National Institute of Environmental Health Sciences. Dr. Jordt reported receiving grants from the NIEHS and the National Institute on Drug Abuse, personal fees from Hydra Biosciences and Sanofi, and nonfinancial support from GlaxoSmithKline. Dr. Jabba reported no disclosures.
SOURCE: Jabba SV, Jordt S-E. JAMA Intern Med. 2019 Sep 16. doi: 10.1001/jamainternmed.2019.3649.
A group of mint- and menthol-flavored e-liquids and smokeless tobacco products contained significantly more pulegone – a known carcinogen that causes hepatic carcinomas, pulmonary metaplasia, and other neoplasms – than the Food and Drug Administration considers acceptable, according to new findings.
Pulegone, an oil extract from mint plants such as peppermint, spearmint, and pennyroyal, was banned as a food additive by the agency in 2018, and the tobacco industry has taken steps to minimize pulegone levels in cigarettes because of the toxicity concerns.
Studies from the Centers for Disease Control and Prevention, however, have indicated that mint- and menthol-flavored e-cigarette liquids and smokeless tobacco products marketed in the United States contain substantial amounts of the substance, Sairam V. Jabba, DVM, PhD, and Sven-Eric Jordt, PhD, said in a research letter published in JAMA Internal Medicine.
Dr. Jabba and Dr. Jordt, both with the department of anesthesiology at Duke University, Durham, N.C., calculated the margin of exposure in five e-liquids (V2 Menthol, V2 Peppermint, Premium Menthol, South Beach Smoke Menthol, and South Beach Smoke Peppermint) and one smokeless tobacco product (Skoal Xtra Mint snuff) by dividing the no–observed adverse event level (13.39 mg/kg of bodyweight per day) by the mean human exposure to e-liquids or smokeless tobacco. The FDA considers margin-of-exposure values of 10,000 or less to require mitigation strategies.
The six products included in the analysis had pulegone concentration levels ranging from 25.7 to 119.0 mcg/g (a menthol cigarette has a pulegone concentration of 0.037-0.290 mcg/g). Based on those levels, light daily use (5 mL e-liquid, 10 g smokeless tobacco, half a pack of cigarettes) exposed e-cigarette users to 44-198 times more pulegone, compared with menthol cigarettes, and exposed smokeless tobacco users to 168-1,319 times as much pulegone. The margin of exposure ranged from 1,298 to 6,012, all below 10,000 threshhold the FDA deems acceptable.
For heavy daily use (20 mL e-liquid, 30 g smokeless tobacco, two packs of cigarettes), e-cigarette users were exposed to 282-1,608 times more pulegone, compared with menthol cigarettes; smokeless tobacco users were exposed to 126-990 times more pulegone. The margin of exposure ranged from 325 to 1,503.
The study findings “appear to establish health risks associated with pulegone intake and concerns that the FDA should address before suggesting mint- and menthol-flavored e-cigarettes and smokeless tobacco products as alternatives for people who use combustible tobacco products,” Dr. Jabba and Dr. Jordt concluded.
The study was funded by a grant from the National Institute of Environmental Health Sciences. Dr. Jordt reported receiving grants from the NIEHS and the National Institute on Drug Abuse, personal fees from Hydra Biosciences and Sanofi, and nonfinancial support from GlaxoSmithKline. Dr. Jabba reported no disclosures.
SOURCE: Jabba SV, Jordt S-E. JAMA Intern Med. 2019 Sep 16. doi: 10.1001/jamainternmed.2019.3649.
A group of mint- and menthol-flavored e-liquids and smokeless tobacco products contained significantly more pulegone – a known carcinogen that causes hepatic carcinomas, pulmonary metaplasia, and other neoplasms – than the Food and Drug Administration considers acceptable, according to new findings.
Pulegone, an oil extract from mint plants such as peppermint, spearmint, and pennyroyal, was banned as a food additive by the agency in 2018, and the tobacco industry has taken steps to minimize pulegone levels in cigarettes because of the toxicity concerns.
Studies from the Centers for Disease Control and Prevention, however, have indicated that mint- and menthol-flavored e-cigarette liquids and smokeless tobacco products marketed in the United States contain substantial amounts of the substance, Sairam V. Jabba, DVM, PhD, and Sven-Eric Jordt, PhD, said in a research letter published in JAMA Internal Medicine.
Dr. Jabba and Dr. Jordt, both with the department of anesthesiology at Duke University, Durham, N.C., calculated the margin of exposure in five e-liquids (V2 Menthol, V2 Peppermint, Premium Menthol, South Beach Smoke Menthol, and South Beach Smoke Peppermint) and one smokeless tobacco product (Skoal Xtra Mint snuff) by dividing the no–observed adverse event level (13.39 mg/kg of bodyweight per day) by the mean human exposure to e-liquids or smokeless tobacco. The FDA considers margin-of-exposure values of 10,000 or less to require mitigation strategies.
The six products included in the analysis had pulegone concentration levels ranging from 25.7 to 119.0 mcg/g (a menthol cigarette has a pulegone concentration of 0.037-0.290 mcg/g). Based on those levels, light daily use (5 mL e-liquid, 10 g smokeless tobacco, half a pack of cigarettes) exposed e-cigarette users to 44-198 times more pulegone, compared with menthol cigarettes, and exposed smokeless tobacco users to 168-1,319 times as much pulegone. The margin of exposure ranged from 1,298 to 6,012, all below 10,000 threshhold the FDA deems acceptable.
For heavy daily use (20 mL e-liquid, 30 g smokeless tobacco, two packs of cigarettes), e-cigarette users were exposed to 282-1,608 times more pulegone, compared with menthol cigarettes; smokeless tobacco users were exposed to 126-990 times more pulegone. The margin of exposure ranged from 325 to 1,503.
The study findings “appear to establish health risks associated with pulegone intake and concerns that the FDA should address before suggesting mint- and menthol-flavored e-cigarettes and smokeless tobacco products as alternatives for people who use combustible tobacco products,” Dr. Jabba and Dr. Jordt concluded.
The study was funded by a grant from the National Institute of Environmental Health Sciences. Dr. Jordt reported receiving grants from the NIEHS and the National Institute on Drug Abuse, personal fees from Hydra Biosciences and Sanofi, and nonfinancial support from GlaxoSmithKline. Dr. Jabba reported no disclosures.
SOURCE: Jabba SV, Jordt S-E. JAMA Intern Med. 2019 Sep 16. doi: 10.1001/jamainternmed.2019.3649.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Mint- and menthol-flavored e-liquids and smokeless tobacco contain levels of pulegone that are much higher than those deemed acceptable by the Food and Drug Administration.
Major finding:
Study details: An assessment of pulegone in five e-liquids and one brand of smokeless tobacco.
Disclosures: The study was funded by a grant from the National Institute of Environmental Health Sciences. Dr. Jordt reported receiving grants from the NIEHS and the National Institute on Drug Abuse, personal fees from Hydra Biosciences and Sanofi, and nonfinancial support from GlaxoSmithKline. Dr. Jabba reported no disclosures.
Source: Jabba SV, Jordt S-E. JAMA Intern Med. 2019 Sep 16. doi: 10.1001/jamainternmed.2019.3649.
Peanut allergy pill gets thumbs-up from FDA advisory panel
A pill designed to desensitize peanut-allergic children and teenagers may be on the way.
aged 4-17 years old with a confirmed peanut allergy. Conditions for approval include stipulations that a black-box warning and medication use guide are included in the packaging, the panel said. The FDA usually follows the recommendations of its advisory panels.

The committee members voted 7-2 that the drug was effective and 8-1 that it was safe.
John Kelso, MD, the sole dissenter on safety, voiced concerns about the dearth of long-term follow-up in Aimmune Therapeutic’s body of research and the finding that children who received the treatment during the dose-escalation and maintenance periods had twice the number of allergic reactions requiring epinephrine, compared with those who received placebo. There are no long-term safety data to rely on yet, he added.
“Efficacy has not been demonstrated, except on the day the peanut challenge is administered,” said Dr. Kelso, an allergist at the Scripps Clinic, San Diego, adding that only long-term follow-up data would fully convince him that the drug’s benefits outweigh the risks.
In the discussion, however, other committee members pointed out that new drugs are often approved without long-term efficacy and safety data. Those data are extrapolated from clinical trials, and only real-world experience will confirm the data, they noted.
Company representatives did not explicitly address the potential cost of the therapy, but a recent review by the Institute for Clinical and Economic Review estimated the cost to be $4,200 a year. Palforzia would have to be taken every day, for an unknown amount of time, to maintain peanut tolerance.
“Using prices from analysts for AR101 ($4,200 a year), we estimated that only 41% of eligible patients could be treated in a given year without exceeding ICER’s budget impact threshold,” the institute concluded in a publicly released analysis.
Palforzia comes in individual packs of capsules filled with peanut protein, not flour. The capsules come in doses of 0.5, 1, 10, 20, and 100, and 300 mg. A single-dose sachet contains 300 mg. Treatment begins with 0.5-6 mg over 1 day and escalates every 2 weeks until 300 mg is reached or there is a reaction requiring epinephrine. Passing at least a 300-mg dose was the requirement for exiting the escalation phase and moving on to the daily, year-long maintenance phase.
The four efficacy studies presented showed that 96% of patients tolerated 300 mg, 84% tolerated 600 mg, and 63% 1,000 mg – about 10 times the reactive dose observed in the placebo controls.
“Only 125 mg of peanut protein – the amount in about half a peanut kernel – can be enough to provoke a reaction,” said Daniel Adelman, MD, chief medical officer of Aimmune. If patients can tolerate 600 mg of protein – the equivalent of two kernels, accidental ingestion will result in a “predictable, manageable” reaction.
“This is truly a clinically significant result for patients and families who report lives dictated by the allergy,” Dr. Adelman said.
Consistent manufacturing processes and positive safety data should reassure clinicians and patients that they are receiving a safe, effective, and well-regulated treatment, he added.
The capsule, however, is not a panacea. The company advises that families continue with the peanut avoidance diet. “It’s important to remember that reactive episodes can occur with dosing, and accidental exposures can occur at unpredictable times, away from home, and despite the best efforts at avoidance,” Dr. Adelman said. “This is not a drug for everyone, but it is an effective desensitization tool and would clearly be the first therapy to treat a food allergy, providing statistically significant and clinically important improvement. Outcomes align with patients’ goals.”
Safety was assessed in 709 treated patients who received the medication and 292 who received placebo. Treatment-related adverse events were most common in initial dosing: 89% of the treatment group and 58% of the placebo group experienced at least one adverse event during that time. Adverse events were mostly mild to moderate and decreased in severity over the study period. They included abdominal pain (45% active vs. 18% placebo), throat irritation (40% vs. 17%), pruritus (33% vs. 20%), vomiting (37% vs. 16%), cough (32% vs. 24%), nausea (32% vs. 14%), urticaria (28% vs. 19%), and upper abdominal pain (30% vs. 14%).
Discontinuations caused by these adverse events were infrequent, with only 1.8% of participants who were taking the active capsule and 1% of those taking placebo discontinuing the study product during initial dose escalation. Only the severe systemic allergic reactions were considered to be anaphylaxis, and they had to affect at least two body systems.
Respiratory events were more common in those in the active group, especially in children with asthma. These events included cough, wheezing, dyspnea, dysphonia, throat irritation and tightness, and exercise-induced asthma. There was, however, no “concerning change” in asthma control.
Systemic allergic reactions and anaphylaxis were more common in the active-dose group. Systemic reactions during dose escalation occurred in 9.4% of active patients and 3.8% those taking placebo. During the maintenance phase, they occurred in 8.7% and 1.7% of patients, respectively. Three patients in the active group had a serious systemic reaction – two during up-dosing and one during maintenance.
During initial dose escalation and up-dosing combined, 6.1% of patients in the active group and 3.1% in the placebo group had a systemic reaction requiring epinephrine. This was most often administered outside of the clinic.
There were 12 cases of eosinophilic esophagitis, all of which resolved after withdrawal from the study medication.
Aimmune submitted a risk-management proposal that includes the following:
- The first dose of each progressive dose must be administered in a facility that is equipped to treat systemic allergic reactions.
- Families must have a valid prescription for injectable epinephrine before treatment starts and must demonstrate that they know how to use it.
- There must be distribution controls in every pharmacy that dispenses the product.
- Packaging will be dose specific to ensure proper at-home administration.
- Pharmacologic questionnaires will be used as data collection instruments.
- Professional and patient-focused labeling will include a medication guide and educational material.
Palforzia is not the only peanut desensitization product in the works. DBC Technology has resubmitted its biologics license application to the FDA in the hope of getting approval for its peanut allergy treatment, the Viaskin Peanut patch.
The patch is designed to desensitize allergic children aged 4-11 years through a skin-patch method known as epicutaneous immunotherapy. Results from two controlled clinical trials were included in the submission. The company is also investigating the potential of this form of skin-patch therapy for milk and egg allergies.
Correction, 9/15/19: An earlier version of this article did not clearly state that the panel recommended approval rather than granted approval for the drug.
A pill designed to desensitize peanut-allergic children and teenagers may be on the way.
aged 4-17 years old with a confirmed peanut allergy. Conditions for approval include stipulations that a black-box warning and medication use guide are included in the packaging, the panel said. The FDA usually follows the recommendations of its advisory panels.
The committee members voted 7-2 that the drug was effective and 8-1 that it was safe.
John Kelso, MD, the sole dissenter on safety, voiced concerns about the dearth of long-term follow-up in Aimmune Therapeutic’s body of research and the finding that children who received the treatment during the dose-escalation and maintenance periods had twice the number of allergic reactions requiring epinephrine, compared with those who received placebo. There are no long-term safety data to rely on yet, he added.
“Efficacy has not been demonstrated, except on the day the peanut challenge is administered,” said Dr. Kelso, an allergist at the Scripps Clinic, San Diego, adding that only long-term follow-up data would fully convince him that the drug’s benefits outweigh the risks.
In the discussion, however, other committee members pointed out that new drugs are often approved without long-term efficacy and safety data. Those data are extrapolated from clinical trials, and only real-world experience will confirm the data, they noted.
Company representatives did not explicitly address the potential cost of the therapy, but a recent review by the Institute for Clinical and Economic Review estimated the cost to be $4,200 a year. Palforzia would have to be taken every day, for an unknown amount of time, to maintain peanut tolerance.
“Using prices from analysts for AR101 ($4,200 a year), we estimated that only 41% of eligible patients could be treated in a given year without exceeding ICER’s budget impact threshold,” the institute concluded in a publicly released analysis.
Palforzia comes in individual packs of capsules filled with peanut protein, not flour. The capsules come in doses of 0.5, 1, 10, 20, and 100, and 300 mg. A single-dose sachet contains 300 mg. Treatment begins with 0.5-6 mg over 1 day and escalates every 2 weeks until 300 mg is reached or there is a reaction requiring epinephrine. Passing at least a 300-mg dose was the requirement for exiting the escalation phase and moving on to the daily, year-long maintenance phase.
The four efficacy studies presented showed that 96% of patients tolerated 300 mg, 84% tolerated 600 mg, and 63% 1,000 mg – about 10 times the reactive dose observed in the placebo controls.
“Only 125 mg of peanut protein – the amount in about half a peanut kernel – can be enough to provoke a reaction,” said Daniel Adelman, MD, chief medical officer of Aimmune. If patients can tolerate 600 mg of protein – the equivalent of two kernels, accidental ingestion will result in a “predictable, manageable” reaction.
“This is truly a clinically significant result for patients and families who report lives dictated by the allergy,” Dr. Adelman said.
Consistent manufacturing processes and positive safety data should reassure clinicians and patients that they are receiving a safe, effective, and well-regulated treatment, he added.
The capsule, however, is not a panacea. The company advises that families continue with the peanut avoidance diet. “It’s important to remember that reactive episodes can occur with dosing, and accidental exposures can occur at unpredictable times, away from home, and despite the best efforts at avoidance,” Dr. Adelman said. “This is not a drug for everyone, but it is an effective desensitization tool and would clearly be the first therapy to treat a food allergy, providing statistically significant and clinically important improvement. Outcomes align with patients’ goals.”
Safety was assessed in 709 treated patients who received the medication and 292 who received placebo. Treatment-related adverse events were most common in initial dosing: 89% of the treatment group and 58% of the placebo group experienced at least one adverse event during that time. Adverse events were mostly mild to moderate and decreased in severity over the study period. They included abdominal pain (45% active vs. 18% placebo), throat irritation (40% vs. 17%), pruritus (33% vs. 20%), vomiting (37% vs. 16%), cough (32% vs. 24%), nausea (32% vs. 14%), urticaria (28% vs. 19%), and upper abdominal pain (30% vs. 14%).
Discontinuations caused by these adverse events were infrequent, with only 1.8% of participants who were taking the active capsule and 1% of those taking placebo discontinuing the study product during initial dose escalation. Only the severe systemic allergic reactions were considered to be anaphylaxis, and they had to affect at least two body systems.
Respiratory events were more common in those in the active group, especially in children with asthma. These events included cough, wheezing, dyspnea, dysphonia, throat irritation and tightness, and exercise-induced asthma. There was, however, no “concerning change” in asthma control.
Systemic allergic reactions and anaphylaxis were more common in the active-dose group. Systemic reactions during dose escalation occurred in 9.4% of active patients and 3.8% those taking placebo. During the maintenance phase, they occurred in 8.7% and 1.7% of patients, respectively. Three patients in the active group had a serious systemic reaction – two during up-dosing and one during maintenance.
During initial dose escalation and up-dosing combined, 6.1% of patients in the active group and 3.1% in the placebo group had a systemic reaction requiring epinephrine. This was most often administered outside of the clinic.
There were 12 cases of eosinophilic esophagitis, all of which resolved after withdrawal from the study medication.
Aimmune submitted a risk-management proposal that includes the following:
- The first dose of each progressive dose must be administered in a facility that is equipped to treat systemic allergic reactions.
- Families must have a valid prescription for injectable epinephrine before treatment starts and must demonstrate that they know how to use it.
- There must be distribution controls in every pharmacy that dispenses the product.
- Packaging will be dose specific to ensure proper at-home administration.
- Pharmacologic questionnaires will be used as data collection instruments.
- Professional and patient-focused labeling will include a medication guide and educational material.
Palforzia is not the only peanut desensitization product in the works. DBC Technology has resubmitted its biologics license application to the FDA in the hope of getting approval for its peanut allergy treatment, the Viaskin Peanut patch.
The patch is designed to desensitize allergic children aged 4-11 years through a skin-patch method known as epicutaneous immunotherapy. Results from two controlled clinical trials were included in the submission. The company is also investigating the potential of this form of skin-patch therapy for milk and egg allergies.
Correction, 9/15/19: An earlier version of this article did not clearly state that the panel recommended approval rather than granted approval for the drug.
A pill designed to desensitize peanut-allergic children and teenagers may be on the way.
aged 4-17 years old with a confirmed peanut allergy. Conditions for approval include stipulations that a black-box warning and medication use guide are included in the packaging, the panel said. The FDA usually follows the recommendations of its advisory panels.
The committee members voted 7-2 that the drug was effective and 8-1 that it was safe.
John Kelso, MD, the sole dissenter on safety, voiced concerns about the dearth of long-term follow-up in Aimmune Therapeutic’s body of research and the finding that children who received the treatment during the dose-escalation and maintenance periods had twice the number of allergic reactions requiring epinephrine, compared with those who received placebo. There are no long-term safety data to rely on yet, he added.
“Efficacy has not been demonstrated, except on the day the peanut challenge is administered,” said Dr. Kelso, an allergist at the Scripps Clinic, San Diego, adding that only long-term follow-up data would fully convince him that the drug’s benefits outweigh the risks.
In the discussion, however, other committee members pointed out that new drugs are often approved without long-term efficacy and safety data. Those data are extrapolated from clinical trials, and only real-world experience will confirm the data, they noted.
Company representatives did not explicitly address the potential cost of the therapy, but a recent review by the Institute for Clinical and Economic Review estimated the cost to be $4,200 a year. Palforzia would have to be taken every day, for an unknown amount of time, to maintain peanut tolerance.
“Using prices from analysts for AR101 ($4,200 a year), we estimated that only 41% of eligible patients could be treated in a given year without exceeding ICER’s budget impact threshold,” the institute concluded in a publicly released analysis.
Palforzia comes in individual packs of capsules filled with peanut protein, not flour. The capsules come in doses of 0.5, 1, 10, 20, and 100, and 300 mg. A single-dose sachet contains 300 mg. Treatment begins with 0.5-6 mg over 1 day and escalates every 2 weeks until 300 mg is reached or there is a reaction requiring epinephrine. Passing at least a 300-mg dose was the requirement for exiting the escalation phase and moving on to the daily, year-long maintenance phase.
The four efficacy studies presented showed that 96% of patients tolerated 300 mg, 84% tolerated 600 mg, and 63% 1,000 mg – about 10 times the reactive dose observed in the placebo controls.
“Only 125 mg of peanut protein – the amount in about half a peanut kernel – can be enough to provoke a reaction,” said Daniel Adelman, MD, chief medical officer of Aimmune. If patients can tolerate 600 mg of protein – the equivalent of two kernels, accidental ingestion will result in a “predictable, manageable” reaction.
“This is truly a clinically significant result for patients and families who report lives dictated by the allergy,” Dr. Adelman said.
Consistent manufacturing processes and positive safety data should reassure clinicians and patients that they are receiving a safe, effective, and well-regulated treatment, he added.
The capsule, however, is not a panacea. The company advises that families continue with the peanut avoidance diet. “It’s important to remember that reactive episodes can occur with dosing, and accidental exposures can occur at unpredictable times, away from home, and despite the best efforts at avoidance,” Dr. Adelman said. “This is not a drug for everyone, but it is an effective desensitization tool and would clearly be the first therapy to treat a food allergy, providing statistically significant and clinically important improvement. Outcomes align with patients’ goals.”
Safety was assessed in 709 treated patients who received the medication and 292 who received placebo. Treatment-related adverse events were most common in initial dosing: 89% of the treatment group and 58% of the placebo group experienced at least one adverse event during that time. Adverse events were mostly mild to moderate and decreased in severity over the study period. They included abdominal pain (45% active vs. 18% placebo), throat irritation (40% vs. 17%), pruritus (33% vs. 20%), vomiting (37% vs. 16%), cough (32% vs. 24%), nausea (32% vs. 14%), urticaria (28% vs. 19%), and upper abdominal pain (30% vs. 14%).
Discontinuations caused by these adverse events were infrequent, with only 1.8% of participants who were taking the active capsule and 1% of those taking placebo discontinuing the study product during initial dose escalation. Only the severe systemic allergic reactions were considered to be anaphylaxis, and they had to affect at least two body systems.
Respiratory events were more common in those in the active group, especially in children with asthma. These events included cough, wheezing, dyspnea, dysphonia, throat irritation and tightness, and exercise-induced asthma. There was, however, no “concerning change” in asthma control.
Systemic allergic reactions and anaphylaxis were more common in the active-dose group. Systemic reactions during dose escalation occurred in 9.4% of active patients and 3.8% those taking placebo. During the maintenance phase, they occurred in 8.7% and 1.7% of patients, respectively. Three patients in the active group had a serious systemic reaction – two during up-dosing and one during maintenance.
During initial dose escalation and up-dosing combined, 6.1% of patients in the active group and 3.1% in the placebo group had a systemic reaction requiring epinephrine. This was most often administered outside of the clinic.
There were 12 cases of eosinophilic esophagitis, all of which resolved after withdrawal from the study medication.
Aimmune submitted a risk-management proposal that includes the following:
- The first dose of each progressive dose must be administered in a facility that is equipped to treat systemic allergic reactions.
- Families must have a valid prescription for injectable epinephrine before treatment starts and must demonstrate that they know how to use it.
- There must be distribution controls in every pharmacy that dispenses the product.
- Packaging will be dose specific to ensure proper at-home administration.
- Pharmacologic questionnaires will be used as data collection instruments.
- Professional and patient-focused labeling will include a medication guide and educational material.
Palforzia is not the only peanut desensitization product in the works. DBC Technology has resubmitted its biologics license application to the FDA in the hope of getting approval for its peanut allergy treatment, the Viaskin Peanut patch.
The patch is designed to desensitize allergic children aged 4-11 years through a skin-patch method known as epicutaneous immunotherapy. Results from two controlled clinical trials were included in the submission. The company is also investigating the potential of this form of skin-patch therapy for milk and egg allergies.
Correction, 9/15/19: An earlier version of this article did not clearly state that the panel recommended approval rather than granted approval for the drug.
FROM THE FDA ALLERGENIC PRODUCTS ADVISORY COMMITTEE MEETING
FDA issues warning for CDK 4/6 inhibitors
The Food and Drug Administration is warning that the entire class of the cyclin-dependent kinase 4/6 (CDK 4/6) inhibitors used to treat advanced breast cancer may cause rare but severe inflammation of the lungs.

“We reviewed CDK 4/6 inhibitors cases from completed and ongoing clinical trials undertaken by manufacturers and their postmarket safety databases that described specific types of inflammation of the lungs, called interstitial lung disease and pneumonitis. Across the entire drug class, there were reports of serious cases, including fatalities,” the FDA said in a press statement.
The overall benefit of CDK 4/6 inhibitors, however, is still greater than the risks when used as prescribed, the agency said.
CDK 4/6 inhibitors are used in combination with hormone therapies to treat adults with hormone receptor–positive, human epidermal growth factor 2–negative advanced or metastatic breast cancer that has spread to other parts of the body. The FDA approved the CDK 4/6 inhibitors palbociclib (Ibrance) in 2015 and ribociclib (Kisqali) and abemaciclib (Verzenio) in 2017, based on improvements in progression-free survival.
Health care professionals should monitor patients regularly for pulmonary symptoms indicative of interstitial lung disease and/or pneumonitis. Signs and symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams in patients in whom infectious, neoplastic, and other causes have been excluded. Interrupt CDK 4/6 inhibitor treatment in patients who have new or worsening respiratory symptoms, and permanently discontinue treatment in patients with severe interstitial lung disease and/or pneumonitis, the FDA said.
The Food and Drug Administration is warning that the entire class of the cyclin-dependent kinase 4/6 (CDK 4/6) inhibitors used to treat advanced breast cancer may cause rare but severe inflammation of the lungs.
“We reviewed CDK 4/6 inhibitors cases from completed and ongoing clinical trials undertaken by manufacturers and their postmarket safety databases that described specific types of inflammation of the lungs, called interstitial lung disease and pneumonitis. Across the entire drug class, there were reports of serious cases, including fatalities,” the FDA said in a press statement.
The overall benefit of CDK 4/6 inhibitors, however, is still greater than the risks when used as prescribed, the agency said.
CDK 4/6 inhibitors are used in combination with hormone therapies to treat adults with hormone receptor–positive, human epidermal growth factor 2–negative advanced or metastatic breast cancer that has spread to other parts of the body. The FDA approved the CDK 4/6 inhibitors palbociclib (Ibrance) in 2015 and ribociclib (Kisqali) and abemaciclib (Verzenio) in 2017, based on improvements in progression-free survival.
Health care professionals should monitor patients regularly for pulmonary symptoms indicative of interstitial lung disease and/or pneumonitis. Signs and symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams in patients in whom infectious, neoplastic, and other causes have been excluded. Interrupt CDK 4/6 inhibitor treatment in patients who have new or worsening respiratory symptoms, and permanently discontinue treatment in patients with severe interstitial lung disease and/or pneumonitis, the FDA said.
The Food and Drug Administration is warning that the entire class of the cyclin-dependent kinase 4/6 (CDK 4/6) inhibitors used to treat advanced breast cancer may cause rare but severe inflammation of the lungs.
“We reviewed CDK 4/6 inhibitors cases from completed and ongoing clinical trials undertaken by manufacturers and their postmarket safety databases that described specific types of inflammation of the lungs, called interstitial lung disease and pneumonitis. Across the entire drug class, there were reports of serious cases, including fatalities,” the FDA said in a press statement.
The overall benefit of CDK 4/6 inhibitors, however, is still greater than the risks when used as prescribed, the agency said.
CDK 4/6 inhibitors are used in combination with hormone therapies to treat adults with hormone receptor–positive, human epidermal growth factor 2–negative advanced or metastatic breast cancer that has spread to other parts of the body. The FDA approved the CDK 4/6 inhibitors palbociclib (Ibrance) in 2015 and ribociclib (Kisqali) and abemaciclib (Verzenio) in 2017, based on improvements in progression-free survival.
Health care professionals should monitor patients regularly for pulmonary symptoms indicative of interstitial lung disease and/or pneumonitis. Signs and symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams in patients in whom infectious, neoplastic, and other causes have been excluded. Interrupt CDK 4/6 inhibitor treatment in patients who have new or worsening respiratory symptoms, and permanently discontinue treatment in patients with severe interstitial lung disease and/or pneumonitis, the FDA said.
Benralizumab trials cast doubt on eosinophil depletion’s role in COPD treatment
and eosinophilic inflammation, according to results from two phase 3 trials. The data were published in the New England Journal of Medicine.

Benralizumab, an interleukin-5 receptor alpha–directed cytolytic monoclonal antibody, is approved for the treatment of patients with severe eosinophilic asthma. To assess whether the treatment may prevent COPD exacerbations, Gerard J. Criner, MD, chair and professor of thoracic medicine and surgery at Temple University in Philadelphia and colleagues conducted two randomized, double-blind, parallel-group studies: GALATHEA and TERRANOVA. Researchers enrolled patients with frequent moderate or severe COPD exacerbations and blood eosinophil counts of at least 220 per mm3.
A 56-week treatment period
“An eosinophil threshold of 220 per mm3 was selected on the basis of the phase 2 trial of benralizumab in patients with COPD, in which modeling of annual exacerbations according to baseline blood eosinophil count indicated that patients with eosinophil counts above a similar threshold were more likely to have a response to benralizumab,” the authors wrote. “The doses selected were 30 mg, the approved dose for asthma treatment; 100 mg, to inform the safety margin; and 10 mg (in TERRANOVA), to evaluate the dose-efficacy relationship.”
Patients received placebo or benralizumab via subcutaneous injection every 4 weeks for the first three doses, then every 8 weeks for the rest of the 56-week treatment period. The primary end point was the annualized COPD exacerbation rate ratio (benralizumab vs. placebo) at week 56.
The primary analysis populations included 1,120 patients in GALATHEA and 1,545 patients in TERRANOVA. Most patients were white men, and the average age was 65 years. The percentages of patients with current asthma (5.4% in GALATHEA and 3.3% in TERRANOVA) or past asthma (8.3% in GALATHEA and 6.1% in TERRANOVA) were low.
In GALATHEA, the estimated annualized exacerbation rates were 1.19 per year in the 30-mg benralizumab group, 1.03 per year in the 100-mg benralizumab group, and 1.24 per year in the placebo group. Compared with placebo, the rate ratio was 0.96 for 30 mg of benralizumab and 0.83 for 100 mg of benralizumab.
In TERRANOVA, the estimated annualized exacerbation rates for 10 mg, 30 mg, and 100 mg of benralizumab and for placebo were 0.99 per year, 1.21 per year, 1.09 per year, and 1.17 per year, respectively. The corresponding rate ratios were 0.85, 1.04, and 0.93. “At 56 weeks, none of the annualized COPD exacerbation rate ratios for any dose of benralizumab as compared with placebo reached significance in either trial,” the researchers said. “Types and frequencies of adverse events were similar with benralizumab and placebo.”
Depletion of eosinophils in blood and sputum
By week 4, benralizumab substantially depleted blood eosinophils. In addition, treatment substantially depleted sputum eosinophils by week 24. “However, in contrast to the results in benralizumab-treated patients with severe eosinophilic asthma, this eosinophil depletion did not correspond to a significant difference in the rate of exacerbations. This finding, together with the effect on eosinophils – with minimal effect on the COPD exacerbation rate – that was observed in the mepolizumab trials, suggests that eosinophil depletion is unlikely to ameliorate exacerbation outcomes for the majority of patients with COPD,” Dr. Criner and his coauthors concluded. “Future investigation is required to identify additional clinical factors or biomarkers that may characterize the patients with COPD who are most likely to benefit from anti–interleukin-5 receptor antibody therapy.”
The trials were sponsored by AstraZeneca, which manufactures benralizumab (Fasenra), and by Kyowa Hakko Kirin. One author is supported by the National Institute for Health Research Manchester Biomedical Research Centre. The authors’ disclosures included grants and personal fees from AstraZeneca and other pharmaceutical companies.
SOURCE: Criner GJ et al. N Engl J Med. 2019;382(11):1023-34. doi: 10.1056/NEJMoa1905248.
and eosinophilic inflammation, according to results from two phase 3 trials. The data were published in the New England Journal of Medicine.
Benralizumab, an interleukin-5 receptor alpha–directed cytolytic monoclonal antibody, is approved for the treatment of patients with severe eosinophilic asthma. To assess whether the treatment may prevent COPD exacerbations, Gerard J. Criner, MD, chair and professor of thoracic medicine and surgery at Temple University in Philadelphia and colleagues conducted two randomized, double-blind, parallel-group studies: GALATHEA and TERRANOVA. Researchers enrolled patients with frequent moderate or severe COPD exacerbations and blood eosinophil counts of at least 220 per mm3.
A 56-week treatment period
“An eosinophil threshold of 220 per mm3 was selected on the basis of the phase 2 trial of benralizumab in patients with COPD, in which modeling of annual exacerbations according to baseline blood eosinophil count indicated that patients with eosinophil counts above a similar threshold were more likely to have a response to benralizumab,” the authors wrote. “The doses selected were 30 mg, the approved dose for asthma treatment; 100 mg, to inform the safety margin; and 10 mg (in TERRANOVA), to evaluate the dose-efficacy relationship.”
Patients received placebo or benralizumab via subcutaneous injection every 4 weeks for the first three doses, then every 8 weeks for the rest of the 56-week treatment period. The primary end point was the annualized COPD exacerbation rate ratio (benralizumab vs. placebo) at week 56.
The primary analysis populations included 1,120 patients in GALATHEA and 1,545 patients in TERRANOVA. Most patients were white men, and the average age was 65 years. The percentages of patients with current asthma (5.4% in GALATHEA and 3.3% in TERRANOVA) or past asthma (8.3% in GALATHEA and 6.1% in TERRANOVA) were low.
In GALATHEA, the estimated annualized exacerbation rates were 1.19 per year in the 30-mg benralizumab group, 1.03 per year in the 100-mg benralizumab group, and 1.24 per year in the placebo group. Compared with placebo, the rate ratio was 0.96 for 30 mg of benralizumab and 0.83 for 100 mg of benralizumab.
In TERRANOVA, the estimated annualized exacerbation rates for 10 mg, 30 mg, and 100 mg of benralizumab and for placebo were 0.99 per year, 1.21 per year, 1.09 per year, and 1.17 per year, respectively. The corresponding rate ratios were 0.85, 1.04, and 0.93. “At 56 weeks, none of the annualized COPD exacerbation rate ratios for any dose of benralizumab as compared with placebo reached significance in either trial,” the researchers said. “Types and frequencies of adverse events were similar with benralizumab and placebo.”
Depletion of eosinophils in blood and sputum
By week 4, benralizumab substantially depleted blood eosinophils. In addition, treatment substantially depleted sputum eosinophils by week 24. “However, in contrast to the results in benralizumab-treated patients with severe eosinophilic asthma, this eosinophil depletion did not correspond to a significant difference in the rate of exacerbations. This finding, together with the effect on eosinophils – with minimal effect on the COPD exacerbation rate – that was observed in the mepolizumab trials, suggests that eosinophil depletion is unlikely to ameliorate exacerbation outcomes for the majority of patients with COPD,” Dr. Criner and his coauthors concluded. “Future investigation is required to identify additional clinical factors or biomarkers that may characterize the patients with COPD who are most likely to benefit from anti–interleukin-5 receptor antibody therapy.”
The trials were sponsored by AstraZeneca, which manufactures benralizumab (Fasenra), and by Kyowa Hakko Kirin. One author is supported by the National Institute for Health Research Manchester Biomedical Research Centre. The authors’ disclosures included grants and personal fees from AstraZeneca and other pharmaceutical companies.
SOURCE: Criner GJ et al. N Engl J Med. 2019;382(11):1023-34. doi: 10.1056/NEJMoa1905248.
and eosinophilic inflammation, according to results from two phase 3 trials. The data were published in the New England Journal of Medicine.
Benralizumab, an interleukin-5 receptor alpha–directed cytolytic monoclonal antibody, is approved for the treatment of patients with severe eosinophilic asthma. To assess whether the treatment may prevent COPD exacerbations, Gerard J. Criner, MD, chair and professor of thoracic medicine and surgery at Temple University in Philadelphia and colleagues conducted two randomized, double-blind, parallel-group studies: GALATHEA and TERRANOVA. Researchers enrolled patients with frequent moderate or severe COPD exacerbations and blood eosinophil counts of at least 220 per mm3.
A 56-week treatment period
“An eosinophil threshold of 220 per mm3 was selected on the basis of the phase 2 trial of benralizumab in patients with COPD, in which modeling of annual exacerbations according to baseline blood eosinophil count indicated that patients with eosinophil counts above a similar threshold were more likely to have a response to benralizumab,” the authors wrote. “The doses selected were 30 mg, the approved dose for asthma treatment; 100 mg, to inform the safety margin; and 10 mg (in TERRANOVA), to evaluate the dose-efficacy relationship.”
Patients received placebo or benralizumab via subcutaneous injection every 4 weeks for the first three doses, then every 8 weeks for the rest of the 56-week treatment period. The primary end point was the annualized COPD exacerbation rate ratio (benralizumab vs. placebo) at week 56.
The primary analysis populations included 1,120 patients in GALATHEA and 1,545 patients in TERRANOVA. Most patients were white men, and the average age was 65 years. The percentages of patients with current asthma (5.4% in GALATHEA and 3.3% in TERRANOVA) or past asthma (8.3% in GALATHEA and 6.1% in TERRANOVA) were low.
In GALATHEA, the estimated annualized exacerbation rates were 1.19 per year in the 30-mg benralizumab group, 1.03 per year in the 100-mg benralizumab group, and 1.24 per year in the placebo group. Compared with placebo, the rate ratio was 0.96 for 30 mg of benralizumab and 0.83 for 100 mg of benralizumab.
In TERRANOVA, the estimated annualized exacerbation rates for 10 mg, 30 mg, and 100 mg of benralizumab and for placebo were 0.99 per year, 1.21 per year, 1.09 per year, and 1.17 per year, respectively. The corresponding rate ratios were 0.85, 1.04, and 0.93. “At 56 weeks, none of the annualized COPD exacerbation rate ratios for any dose of benralizumab as compared with placebo reached significance in either trial,” the researchers said. “Types and frequencies of adverse events were similar with benralizumab and placebo.”
Depletion of eosinophils in blood and sputum
By week 4, benralizumab substantially depleted blood eosinophils. In addition, treatment substantially depleted sputum eosinophils by week 24. “However, in contrast to the results in benralizumab-treated patients with severe eosinophilic asthma, this eosinophil depletion did not correspond to a significant difference in the rate of exacerbations. This finding, together with the effect on eosinophils – with minimal effect on the COPD exacerbation rate – that was observed in the mepolizumab trials, suggests that eosinophil depletion is unlikely to ameliorate exacerbation outcomes for the majority of patients with COPD,” Dr. Criner and his coauthors concluded. “Future investigation is required to identify additional clinical factors or biomarkers that may characterize the patients with COPD who are most likely to benefit from anti–interleukin-5 receptor antibody therapy.”
The trials were sponsored by AstraZeneca, which manufactures benralizumab (Fasenra), and by Kyowa Hakko Kirin. One author is supported by the National Institute for Health Research Manchester Biomedical Research Centre. The authors’ disclosures included grants and personal fees from AstraZeneca and other pharmaceutical companies.
SOURCE: Criner GJ et al. N Engl J Med. 2019;382(11):1023-34. doi: 10.1056/NEJMoa1905248.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE