User login
Tofersen linked to slow, positive effects in ALS
caused by superoxide dismutase 1 (SOD1) gene mutations.
The 1-year results, presented at the European Network for the Cure of Amyotrophic Lateral Sclerosis (ENCALS) 2022 meeting, show a deceleration in functional decline that is similar, but “more pronounced” than the previously reported 6-month results, which were not statistically significant, said lead investigator Timothy Miller, MD, PhD, professor of neurology and director of the ALS Center, Washington University, St. Louis.
“What I thought we saw in the first data cut is confirmed by what we saw in the longer data,” he said in an interview. “There were trends [showing] those treated with tofersen did a bit better, but it was hard to be sure. It was hard to be confident in what we were seeing at that early time point.”
Now, with 6 more months of data, Dr. Miller says he is confident that tofersen is slowing down the neurodegenerative disease process. “I see results that I’m encouraged by,” he said. “As a clinician who treats people with ALS with this mutation I would want this drug to be available to people that I see in my clinic.”
One-year VALOR study results
The primary efficacy objective of the VALOR study was to show the 28-week impact of 100 mg tofersen (three doses given about 2 weeks apart, then five doses given every 4 weeks), versus placebo, on function, measured on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R). The open-label extension switched placebo-treated patients to tofersen (delayed-start group) and continued to compare them with the early-start group up to 1 year. This open-label extension phase included 49 patients who had been on early-start tofersen and 18 patients in the delayed-start group.
For the primary endpoint, change from baseline in 48-point ALSFRS-R score, there was a statistically significant benefit for the early-start patients with these patients scoring 3.5 points higher than the delayed-start group (P = .0272, 95% confidence interval [CI], 0.4-6.7). This means that both groups declined in function, which is expected in ALS, but the early-start group declined more slowly.
There was also a benefit associated with early-start tofersen for a number of secondary endpoints, including change from baseline in total SOD1 cerebrospinal fluid concentration (CSF SOD1), plasma neurofilament light chain (NfL) levels, and respiratory function.
“This drug targets the MRNA of SOD1, so it lowers the MRNA and then the SOD1 protein falls,” explained Dr. Miller, adding that these levels dropped 21% in the delayed start group, and 33% in the early-start group. “I think the data pretty clearly show that [tofersen] does what it is supposed to do, and that is the first step.”
Neurofilament light chain, a marker of neurodegeneration, also dropped by 41% in the delayed-start group, and 51% in the early-start group.
Respiratory function, as measured by percent predicted slow vital capacity (SVC), also declined 9.2% more slowly in the early- versus delayed-start group (P = .0159).
Finally, muscle strength, as measured by handheld dynamometry (HHD) score, declined more slowly in the early-start group compared with the late-start group, with an adjusted mean difference in score of 2.8 (P = 0.0186).
Dr. Miller said that the data show that it takes time for tofersen to impact clinical function, but there are signs of benefit before that. “I think what you see is that just starting on the drug, the first thing that happens is SOD1 goes down, the next thing is that neurofilament decreases, but clinical function is not yet changing. It takes time. What I see in these data is that it takes time for us to see that effect on clinical function.”
The bigger picture
While acknowledging that tofersen acts on a genetic mutation found in only about 2% of ALS, Dr. Miller said the study findings carry significance for the wider ALS patient population.
“Assuming we agree there is a clinical effect here, assuming we agree that there is real stabilization of clinical function, I think if we agree on that point then we know that ALS is now a treatable disorder. And that’s a really important point. I’m not sure that we knew that before,” he said. “Yes, there are FDA-approved medications that slow down ALS a bit, but they don’t stabilize it, and if we get the target correct – and we have the correct genetic target here – there can be a substantial influence on slowing down the disease, so that’s one thing to learn for the whole ALS community.”
What lies ahead?
Asked to comment on the study, Richard Bedlack, MD, PhD, who was not involved in the research, said the findings are important and show “clinically meaningful” results. “Based on the new benefit-to-burden ratio, I believe most of my patients with SOD1 mutations will want to try this drug. I would like to be able to offer it to them. But I am curious to see what the FDA will do with these data,” said Dr. Bedlack, professor of neurology at Duke University in Durham, N.C., and director of the Duke ALS Clinic.
“Sometimes that open-label extension gives us time to see differences between patients who initially got drug and those who initially got placebo. That seems to be the case with tofersen here, and it was also the case with AMX0035 [Amylyx Pharmaceuticals Inc.], which did not show a survival benefit in the first 6 months but did in the open-label extension.” A recent FDA advisory board panel concluded there was insufficient evidence of benefit for AMX0035, he noted. “I wonder if the same concern will be raised here, necessitating confirmation in another trial. I hope not, but only time will tell.”
Dr. Miller added that these results “highlight how difficult ALS drug development still is. Among the many uncertainties in setting up a trial (targets, doses, inclusion criteria, outcomes), we still do not know how long we need to treat patients in order to see statistically significant changes in the clinical measures we use (ALSFRS-R, respiratory function, strength, survival, etc.). Most American studies are 6 months long and most European studies are 12 months long. Longer studies may be more likely to show benefits on certain measures (e.g., survival), but they cost more, they are challenged by dropouts as the disease progresses, and the idea of randomizing someone to a placebo for a whole year is psychologically difficult for patients, families, and many clinicians (myself included). So, we are seeing more studies like this one where the first 6 months are randomized, blinded, and placebo controlled, and then there is an open-label extension that lasts many months more.”
The study was sponsored by Biogen. Writing and editorial support was provided by Excel Scientific Solutions. Tofersen was discovered by Ionis Pharmaceuticals Inc. Dr. Miller disclosed ties with Biogen, Ionis Pharmaceuticals Inc., Cytokinetics, C2N, Disarm Therapeutics, and UCB Pharma. Dr. Bedlack disclosed ties with Biogen.
caused by superoxide dismutase 1 (SOD1) gene mutations.
The 1-year results, presented at the European Network for the Cure of Amyotrophic Lateral Sclerosis (ENCALS) 2022 meeting, show a deceleration in functional decline that is similar, but “more pronounced” than the previously reported 6-month results, which were not statistically significant, said lead investigator Timothy Miller, MD, PhD, professor of neurology and director of the ALS Center, Washington University, St. Louis.
“What I thought we saw in the first data cut is confirmed by what we saw in the longer data,” he said in an interview. “There were trends [showing] those treated with tofersen did a bit better, but it was hard to be sure. It was hard to be confident in what we were seeing at that early time point.”
Now, with 6 more months of data, Dr. Miller says he is confident that tofersen is slowing down the neurodegenerative disease process. “I see results that I’m encouraged by,” he said. “As a clinician who treats people with ALS with this mutation I would want this drug to be available to people that I see in my clinic.”
One-year VALOR study results
The primary efficacy objective of the VALOR study was to show the 28-week impact of 100 mg tofersen (three doses given about 2 weeks apart, then five doses given every 4 weeks), versus placebo, on function, measured on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R). The open-label extension switched placebo-treated patients to tofersen (delayed-start group) and continued to compare them with the early-start group up to 1 year. This open-label extension phase included 49 patients who had been on early-start tofersen and 18 patients in the delayed-start group.
For the primary endpoint, change from baseline in 48-point ALSFRS-R score, there was a statistically significant benefit for the early-start patients with these patients scoring 3.5 points higher than the delayed-start group (P = .0272, 95% confidence interval [CI], 0.4-6.7). This means that both groups declined in function, which is expected in ALS, but the early-start group declined more slowly.
There was also a benefit associated with early-start tofersen for a number of secondary endpoints, including change from baseline in total SOD1 cerebrospinal fluid concentration (CSF SOD1), plasma neurofilament light chain (NfL) levels, and respiratory function.
“This drug targets the MRNA of SOD1, so it lowers the MRNA and then the SOD1 protein falls,” explained Dr. Miller, adding that these levels dropped 21% in the delayed start group, and 33% in the early-start group. “I think the data pretty clearly show that [tofersen] does what it is supposed to do, and that is the first step.”
Neurofilament light chain, a marker of neurodegeneration, also dropped by 41% in the delayed-start group, and 51% in the early-start group.
Respiratory function, as measured by percent predicted slow vital capacity (SVC), also declined 9.2% more slowly in the early- versus delayed-start group (P = .0159).
Finally, muscle strength, as measured by handheld dynamometry (HHD) score, declined more slowly in the early-start group compared with the late-start group, with an adjusted mean difference in score of 2.8 (P = 0.0186).
Dr. Miller said that the data show that it takes time for tofersen to impact clinical function, but there are signs of benefit before that. “I think what you see is that just starting on the drug, the first thing that happens is SOD1 goes down, the next thing is that neurofilament decreases, but clinical function is not yet changing. It takes time. What I see in these data is that it takes time for us to see that effect on clinical function.”
The bigger picture
While acknowledging that tofersen acts on a genetic mutation found in only about 2% of ALS, Dr. Miller said the study findings carry significance for the wider ALS patient population.
“Assuming we agree there is a clinical effect here, assuming we agree that there is real stabilization of clinical function, I think if we agree on that point then we know that ALS is now a treatable disorder. And that’s a really important point. I’m not sure that we knew that before,” he said. “Yes, there are FDA-approved medications that slow down ALS a bit, but they don’t stabilize it, and if we get the target correct – and we have the correct genetic target here – there can be a substantial influence on slowing down the disease, so that’s one thing to learn for the whole ALS community.”
What lies ahead?
Asked to comment on the study, Richard Bedlack, MD, PhD, who was not involved in the research, said the findings are important and show “clinically meaningful” results. “Based on the new benefit-to-burden ratio, I believe most of my patients with SOD1 mutations will want to try this drug. I would like to be able to offer it to them. But I am curious to see what the FDA will do with these data,” said Dr. Bedlack, professor of neurology at Duke University in Durham, N.C., and director of the Duke ALS Clinic.
“Sometimes that open-label extension gives us time to see differences between patients who initially got drug and those who initially got placebo. That seems to be the case with tofersen here, and it was also the case with AMX0035 [Amylyx Pharmaceuticals Inc.], which did not show a survival benefit in the first 6 months but did in the open-label extension.” A recent FDA advisory board panel concluded there was insufficient evidence of benefit for AMX0035, he noted. “I wonder if the same concern will be raised here, necessitating confirmation in another trial. I hope not, but only time will tell.”
Dr. Miller added that these results “highlight how difficult ALS drug development still is. Among the many uncertainties in setting up a trial (targets, doses, inclusion criteria, outcomes), we still do not know how long we need to treat patients in order to see statistically significant changes in the clinical measures we use (ALSFRS-R, respiratory function, strength, survival, etc.). Most American studies are 6 months long and most European studies are 12 months long. Longer studies may be more likely to show benefits on certain measures (e.g., survival), but they cost more, they are challenged by dropouts as the disease progresses, and the idea of randomizing someone to a placebo for a whole year is psychologically difficult for patients, families, and many clinicians (myself included). So, we are seeing more studies like this one where the first 6 months are randomized, blinded, and placebo controlled, and then there is an open-label extension that lasts many months more.”
The study was sponsored by Biogen. Writing and editorial support was provided by Excel Scientific Solutions. Tofersen was discovered by Ionis Pharmaceuticals Inc. Dr. Miller disclosed ties with Biogen, Ionis Pharmaceuticals Inc., Cytokinetics, C2N, Disarm Therapeutics, and UCB Pharma. Dr. Bedlack disclosed ties with Biogen.
caused by superoxide dismutase 1 (SOD1) gene mutations.
The 1-year results, presented at the European Network for the Cure of Amyotrophic Lateral Sclerosis (ENCALS) 2022 meeting, show a deceleration in functional decline that is similar, but “more pronounced” than the previously reported 6-month results, which were not statistically significant, said lead investigator Timothy Miller, MD, PhD, professor of neurology and director of the ALS Center, Washington University, St. Louis.
“What I thought we saw in the first data cut is confirmed by what we saw in the longer data,” he said in an interview. “There were trends [showing] those treated with tofersen did a bit better, but it was hard to be sure. It was hard to be confident in what we were seeing at that early time point.”
Now, with 6 more months of data, Dr. Miller says he is confident that tofersen is slowing down the neurodegenerative disease process. “I see results that I’m encouraged by,” he said. “As a clinician who treats people with ALS with this mutation I would want this drug to be available to people that I see in my clinic.”
One-year VALOR study results
The primary efficacy objective of the VALOR study was to show the 28-week impact of 100 mg tofersen (three doses given about 2 weeks apart, then five doses given every 4 weeks), versus placebo, on function, measured on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R). The open-label extension switched placebo-treated patients to tofersen (delayed-start group) and continued to compare them with the early-start group up to 1 year. This open-label extension phase included 49 patients who had been on early-start tofersen and 18 patients in the delayed-start group.
For the primary endpoint, change from baseline in 48-point ALSFRS-R score, there was a statistically significant benefit for the early-start patients with these patients scoring 3.5 points higher than the delayed-start group (P = .0272, 95% confidence interval [CI], 0.4-6.7). This means that both groups declined in function, which is expected in ALS, but the early-start group declined more slowly.
There was also a benefit associated with early-start tofersen for a number of secondary endpoints, including change from baseline in total SOD1 cerebrospinal fluid concentration (CSF SOD1), plasma neurofilament light chain (NfL) levels, and respiratory function.
“This drug targets the MRNA of SOD1, so it lowers the MRNA and then the SOD1 protein falls,” explained Dr. Miller, adding that these levels dropped 21% in the delayed start group, and 33% in the early-start group. “I think the data pretty clearly show that [tofersen] does what it is supposed to do, and that is the first step.”
Neurofilament light chain, a marker of neurodegeneration, also dropped by 41% in the delayed-start group, and 51% in the early-start group.
Respiratory function, as measured by percent predicted slow vital capacity (SVC), also declined 9.2% more slowly in the early- versus delayed-start group (P = .0159).
Finally, muscle strength, as measured by handheld dynamometry (HHD) score, declined more slowly in the early-start group compared with the late-start group, with an adjusted mean difference in score of 2.8 (P = 0.0186).
Dr. Miller said that the data show that it takes time for tofersen to impact clinical function, but there are signs of benefit before that. “I think what you see is that just starting on the drug, the first thing that happens is SOD1 goes down, the next thing is that neurofilament decreases, but clinical function is not yet changing. It takes time. What I see in these data is that it takes time for us to see that effect on clinical function.”
The bigger picture
While acknowledging that tofersen acts on a genetic mutation found in only about 2% of ALS, Dr. Miller said the study findings carry significance for the wider ALS patient population.
“Assuming we agree there is a clinical effect here, assuming we agree that there is real stabilization of clinical function, I think if we agree on that point then we know that ALS is now a treatable disorder. And that’s a really important point. I’m not sure that we knew that before,” he said. “Yes, there are FDA-approved medications that slow down ALS a bit, but they don’t stabilize it, and if we get the target correct – and we have the correct genetic target here – there can be a substantial influence on slowing down the disease, so that’s one thing to learn for the whole ALS community.”
What lies ahead?
Asked to comment on the study, Richard Bedlack, MD, PhD, who was not involved in the research, said the findings are important and show “clinically meaningful” results. “Based on the new benefit-to-burden ratio, I believe most of my patients with SOD1 mutations will want to try this drug. I would like to be able to offer it to them. But I am curious to see what the FDA will do with these data,” said Dr. Bedlack, professor of neurology at Duke University in Durham, N.C., and director of the Duke ALS Clinic.
“Sometimes that open-label extension gives us time to see differences between patients who initially got drug and those who initially got placebo. That seems to be the case with tofersen here, and it was also the case with AMX0035 [Amylyx Pharmaceuticals Inc.], which did not show a survival benefit in the first 6 months but did in the open-label extension.” A recent FDA advisory board panel concluded there was insufficient evidence of benefit for AMX0035, he noted. “I wonder if the same concern will be raised here, necessitating confirmation in another trial. I hope not, but only time will tell.”
Dr. Miller added that these results “highlight how difficult ALS drug development still is. Among the many uncertainties in setting up a trial (targets, doses, inclusion criteria, outcomes), we still do not know how long we need to treat patients in order to see statistically significant changes in the clinical measures we use (ALSFRS-R, respiratory function, strength, survival, etc.). Most American studies are 6 months long and most European studies are 12 months long. Longer studies may be more likely to show benefits on certain measures (e.g., survival), but they cost more, they are challenged by dropouts as the disease progresses, and the idea of randomizing someone to a placebo for a whole year is psychologically difficult for patients, families, and many clinicians (myself included). So, we are seeing more studies like this one where the first 6 months are randomized, blinded, and placebo controlled, and then there is an open-label extension that lasts many months more.”
The study was sponsored by Biogen. Writing and editorial support was provided by Excel Scientific Solutions. Tofersen was discovered by Ionis Pharmaceuticals Inc. Dr. Miller disclosed ties with Biogen, Ionis Pharmaceuticals Inc., Cytokinetics, C2N, Disarm Therapeutics, and UCB Pharma. Dr. Bedlack disclosed ties with Biogen.
FROM THE ENCALS MEETING 2022
Pandemic public health measures may have mitigated Kawasaki disease
The social behavior associated with the COVID-19 pandemic may have reduced the incidence of Kawasaki disease, according to results of a cohort study of nearly 4,000 children.
The incidence of Kawasaki disease in the United States declined by 28.2% between 2018 and 2020, possibly as a result of factors including school closures, mask mandates, and reduced ambient pollution that might reduce exposure to Kawasaki disease (KD) in the environment, but a potential association has not been explored, wrote Jennifer A. Burney, PhD, of the University of California, San Diego, and colleagues.
KD received greater attention in the public and medical communities because of the emergence of multisystem inflammatory syndrome in children (MIS-C), which is similar to, but distinct from, KD, and because of the noticeable drop in KD cases during the pandemic, the researchers said.
In a multicenter cohort study published in JAMA Network Open , the researchers reviewed data from 2,461 consecutive patients with KD who were diagnosed between Jan. 1, 2018, and Dec. 31, 2020. They conducted a detailed analysis of analysis of 1,461 children with KD who were diagnosed between Jan. 1, 2002, and Nov. 15, 2021, at Rady Children’s Hospital San Diego (RCHSD), using data from before, during, and after the height of the pandemic. The median age of the children in the RCHSD analysis was 2.8 years, 62% were male, and 35% were Hispanic.
Overall, the prevalence of KD declined from 894 in 2018 to 646 in 2020, across the United States, but the decline was uneven, the researchers noted.
In the RCHSD cohort in San Diego, KD cases in children aged 1-5 years decreased significantly from 2020 to 2021 compared to the mean number of cases in previous years (22 vs. 44.9, P = .02). KD cases also decreased significantly among males and Asian children.
Notably, the occurrence of the KD clinical features of strawberry tongue, enlarged cervical lymph node, and subacute periungual desquamation decreased during 2020 compared with the baseline period, although only strawberry tongue reached statistical significance (39% vs. 63%, P = .04). The prevalence of patients with an enlarged lymph node was 21% in 2020 vs. 32% prior to the pandemic (P = .09); the prevalence of periungual desquamation during these periods was 47% vs. 58%, P = .16).
The researchers also used data from Census Block Groups (CBGs) to assess the impact of mobility metrics and environmental exposures on KD during the pandemic for the San Diego patient cohort. They found that KD cases during the pandemic were more likely to occur in neighborhoods of higher socioeconomic status, and that neighborhoods with lower levels of nitrous oxides had fewer KD cases.
Overall, “The reduction in KD case numbers coincided with masking, school closures, reduced circulation of respiratory viruses, and reduced air pollution,” the researchers wrote in their discussion of the findings. “A rebound in KD case numbers to prepandemic levels coincided with the lifting of mask mandates and, subsequently, the return to in-person schooling,” they wrote.
The study findings were limited by several factors including the small sample sizes, which also limit the interpretation of mobility and pollution data, the researchers noted. Other limitations include the high interannual variability of KD and the inclusion of 2021 rebound data from the San Diego region only.
“Although our original hypothesis was that shelter-in-place measures would track with reduced KD cases, this was not borne out by the San Diego region data. Instead, the San Diego case occurrence data suggest that exposures that triggered KD were more likely to occur in the home, with a shift toward households with higher SES during the pandemic,” the researchers noted. However, “The results presented here are consistent with a respiratory portal of entry for the trigger(s) of KD,” they said.
Study fails to validate its conclusions
“This study attempts to test the hypothesis that various social restrictions were associated with a decrease in rate of diagnosed Kawasaki disease cases during portions of the SARS-CoV-2 pandemic,” Mark Gorelik, MD, assistant professor of pediatrics at Columbia University, New York, said in an interview.

“However, it appears that it fails to achieve this conclusion and I disagree with the findings,” said Dr. Gorelik, who was not involved in the study but served as first author on an updated Kawasaki disease treatment guideline published earlier this spring in Arthritis & Rheumatology.
“The study does not find statistically significant associations either with shelter in place orders or with cell phone mobility data, as stated in the conclusion, directly contradicting its own claim,” Dr. Gorelik said. “Secondly, the study makes an assumption that various methods, especially the wearing of masks by children and school closures, had a significant effect on the spread of respiratory viruses. There are no prospective, population based, controlled real world studies that validate this claim, and two prospective controlled real-world studies that dispute this,” he emphasized. “Cloth masks and surgical masks, which were the types of masks worn by school students, are also known to have a nonsignificant and paltry – in the latter, certainly less than 50%, and perhaps as little as 10% – effect on the reduction of respiratory viral spread,” he added.
“Mechanistic studies on mask wearing may suggest some mask efficacy, but these studies are as valid as mechanistic studies showing the effect of various antifungal pharmaceuticals on the replication of SARS-CoV-2 virus in culture, meaning only valid as hypothesis generating, and ultimately the latter hypothesis failed to bear out,” Dr. Gorelik explained. “We do not know the reason why other respiratory viruses and non-SARS-CoV-2 coronaviruses declined during the pandemic, but we do know that despite this, the SARS-CoV-2 coronavirus itself did not appear to suffer the same fate. Thus, it is very possible that another factor was at work, and we know that during other viral pandemics, typically circulating viruses decline, potentially due to induction of interferon responses in hosts, in a general effect known as ‘viral interference,’ ” he said.
“Overall, we must have robust evidence to support benefits of hypotheses that have demonstrated clear damage to children during this pandemic (such as school closures), and this study fails to live up to that requirement,” Dr. Gorelik said.
The study was supported by the Gordon and Marilyn Macklin Foundation and the Patient-Centered Outcomes Research Institute. Dr. Burney and Dr. Gorelik had no financial conflicts to disclose.
The social behavior associated with the COVID-19 pandemic may have reduced the incidence of Kawasaki disease, according to results of a cohort study of nearly 4,000 children.
The incidence of Kawasaki disease in the United States declined by 28.2% between 2018 and 2020, possibly as a result of factors including school closures, mask mandates, and reduced ambient pollution that might reduce exposure to Kawasaki disease (KD) in the environment, but a potential association has not been explored, wrote Jennifer A. Burney, PhD, of the University of California, San Diego, and colleagues.
KD received greater attention in the public and medical communities because of the emergence of multisystem inflammatory syndrome in children (MIS-C), which is similar to, but distinct from, KD, and because of the noticeable drop in KD cases during the pandemic, the researchers said.
In a multicenter cohort study published in JAMA Network Open , the researchers reviewed data from 2,461 consecutive patients with KD who were diagnosed between Jan. 1, 2018, and Dec. 31, 2020. They conducted a detailed analysis of analysis of 1,461 children with KD who were diagnosed between Jan. 1, 2002, and Nov. 15, 2021, at Rady Children’s Hospital San Diego (RCHSD), using data from before, during, and after the height of the pandemic. The median age of the children in the RCHSD analysis was 2.8 years, 62% were male, and 35% were Hispanic.
Overall, the prevalence of KD declined from 894 in 2018 to 646 in 2020, across the United States, but the decline was uneven, the researchers noted.
In the RCHSD cohort in San Diego, KD cases in children aged 1-5 years decreased significantly from 2020 to 2021 compared to the mean number of cases in previous years (22 vs. 44.9, P = .02). KD cases also decreased significantly among males and Asian children.
Notably, the occurrence of the KD clinical features of strawberry tongue, enlarged cervical lymph node, and subacute periungual desquamation decreased during 2020 compared with the baseline period, although only strawberry tongue reached statistical significance (39% vs. 63%, P = .04). The prevalence of patients with an enlarged lymph node was 21% in 2020 vs. 32% prior to the pandemic (P = .09); the prevalence of periungual desquamation during these periods was 47% vs. 58%, P = .16).
The researchers also used data from Census Block Groups (CBGs) to assess the impact of mobility metrics and environmental exposures on KD during the pandemic for the San Diego patient cohort. They found that KD cases during the pandemic were more likely to occur in neighborhoods of higher socioeconomic status, and that neighborhoods with lower levels of nitrous oxides had fewer KD cases.
Overall, “The reduction in KD case numbers coincided with masking, school closures, reduced circulation of respiratory viruses, and reduced air pollution,” the researchers wrote in their discussion of the findings. “A rebound in KD case numbers to prepandemic levels coincided with the lifting of mask mandates and, subsequently, the return to in-person schooling,” they wrote.
The study findings were limited by several factors including the small sample sizes, which also limit the interpretation of mobility and pollution data, the researchers noted. Other limitations include the high interannual variability of KD and the inclusion of 2021 rebound data from the San Diego region only.
“Although our original hypothesis was that shelter-in-place measures would track with reduced KD cases, this was not borne out by the San Diego region data. Instead, the San Diego case occurrence data suggest that exposures that triggered KD were more likely to occur in the home, with a shift toward households with higher SES during the pandemic,” the researchers noted. However, “The results presented here are consistent with a respiratory portal of entry for the trigger(s) of KD,” they said.
Study fails to validate its conclusions
“This study attempts to test the hypothesis that various social restrictions were associated with a decrease in rate of diagnosed Kawasaki disease cases during portions of the SARS-CoV-2 pandemic,” Mark Gorelik, MD, assistant professor of pediatrics at Columbia University, New York, said in an interview.
“However, it appears that it fails to achieve this conclusion and I disagree with the findings,” said Dr. Gorelik, who was not involved in the study but served as first author on an updated Kawasaki disease treatment guideline published earlier this spring in Arthritis & Rheumatology.
“The study does not find statistically significant associations either with shelter in place orders or with cell phone mobility data, as stated in the conclusion, directly contradicting its own claim,” Dr. Gorelik said. “Secondly, the study makes an assumption that various methods, especially the wearing of masks by children and school closures, had a significant effect on the spread of respiratory viruses. There are no prospective, population based, controlled real world studies that validate this claim, and two prospective controlled real-world studies that dispute this,” he emphasized. “Cloth masks and surgical masks, which were the types of masks worn by school students, are also known to have a nonsignificant and paltry – in the latter, certainly less than 50%, and perhaps as little as 10% – effect on the reduction of respiratory viral spread,” he added.
“Mechanistic studies on mask wearing may suggest some mask efficacy, but these studies are as valid as mechanistic studies showing the effect of various antifungal pharmaceuticals on the replication of SARS-CoV-2 virus in culture, meaning only valid as hypothesis generating, and ultimately the latter hypothesis failed to bear out,” Dr. Gorelik explained. “We do not know the reason why other respiratory viruses and non-SARS-CoV-2 coronaviruses declined during the pandemic, but we do know that despite this, the SARS-CoV-2 coronavirus itself did not appear to suffer the same fate. Thus, it is very possible that another factor was at work, and we know that during other viral pandemics, typically circulating viruses decline, potentially due to induction of interferon responses in hosts, in a general effect known as ‘viral interference,’ ” he said.
“Overall, we must have robust evidence to support benefits of hypotheses that have demonstrated clear damage to children during this pandemic (such as school closures), and this study fails to live up to that requirement,” Dr. Gorelik said.
The study was supported by the Gordon and Marilyn Macklin Foundation and the Patient-Centered Outcomes Research Institute. Dr. Burney and Dr. Gorelik had no financial conflicts to disclose.
The social behavior associated with the COVID-19 pandemic may have reduced the incidence of Kawasaki disease, according to results of a cohort study of nearly 4,000 children.
The incidence of Kawasaki disease in the United States declined by 28.2% between 2018 and 2020, possibly as a result of factors including school closures, mask mandates, and reduced ambient pollution that might reduce exposure to Kawasaki disease (KD) in the environment, but a potential association has not been explored, wrote Jennifer A. Burney, PhD, of the University of California, San Diego, and colleagues.
KD received greater attention in the public and medical communities because of the emergence of multisystem inflammatory syndrome in children (MIS-C), which is similar to, but distinct from, KD, and because of the noticeable drop in KD cases during the pandemic, the researchers said.
In a multicenter cohort study published in JAMA Network Open , the researchers reviewed data from 2,461 consecutive patients with KD who were diagnosed between Jan. 1, 2018, and Dec. 31, 2020. They conducted a detailed analysis of analysis of 1,461 children with KD who were diagnosed between Jan. 1, 2002, and Nov. 15, 2021, at Rady Children’s Hospital San Diego (RCHSD), using data from before, during, and after the height of the pandemic. The median age of the children in the RCHSD analysis was 2.8 years, 62% were male, and 35% were Hispanic.
Overall, the prevalence of KD declined from 894 in 2018 to 646 in 2020, across the United States, but the decline was uneven, the researchers noted.
In the RCHSD cohort in San Diego, KD cases in children aged 1-5 years decreased significantly from 2020 to 2021 compared to the mean number of cases in previous years (22 vs. 44.9, P = .02). KD cases also decreased significantly among males and Asian children.
Notably, the occurrence of the KD clinical features of strawberry tongue, enlarged cervical lymph node, and subacute periungual desquamation decreased during 2020 compared with the baseline period, although only strawberry tongue reached statistical significance (39% vs. 63%, P = .04). The prevalence of patients with an enlarged lymph node was 21% in 2020 vs. 32% prior to the pandemic (P = .09); the prevalence of periungual desquamation during these periods was 47% vs. 58%, P = .16).
The researchers also used data from Census Block Groups (CBGs) to assess the impact of mobility metrics and environmental exposures on KD during the pandemic for the San Diego patient cohort. They found that KD cases during the pandemic were more likely to occur in neighborhoods of higher socioeconomic status, and that neighborhoods with lower levels of nitrous oxides had fewer KD cases.
Overall, “The reduction in KD case numbers coincided with masking, school closures, reduced circulation of respiratory viruses, and reduced air pollution,” the researchers wrote in their discussion of the findings. “A rebound in KD case numbers to prepandemic levels coincided with the lifting of mask mandates and, subsequently, the return to in-person schooling,” they wrote.
The study findings were limited by several factors including the small sample sizes, which also limit the interpretation of mobility and pollution data, the researchers noted. Other limitations include the high interannual variability of KD and the inclusion of 2021 rebound data from the San Diego region only.
“Although our original hypothesis was that shelter-in-place measures would track with reduced KD cases, this was not borne out by the San Diego region data. Instead, the San Diego case occurrence data suggest that exposures that triggered KD were more likely to occur in the home, with a shift toward households with higher SES during the pandemic,” the researchers noted. However, “The results presented here are consistent with a respiratory portal of entry for the trigger(s) of KD,” they said.
Study fails to validate its conclusions
“This study attempts to test the hypothesis that various social restrictions were associated with a decrease in rate of diagnosed Kawasaki disease cases during portions of the SARS-CoV-2 pandemic,” Mark Gorelik, MD, assistant professor of pediatrics at Columbia University, New York, said in an interview.
“However, it appears that it fails to achieve this conclusion and I disagree with the findings,” said Dr. Gorelik, who was not involved in the study but served as first author on an updated Kawasaki disease treatment guideline published earlier this spring in Arthritis & Rheumatology.
“The study does not find statistically significant associations either with shelter in place orders or with cell phone mobility data, as stated in the conclusion, directly contradicting its own claim,” Dr. Gorelik said. “Secondly, the study makes an assumption that various methods, especially the wearing of masks by children and school closures, had a significant effect on the spread of respiratory viruses. There are no prospective, population based, controlled real world studies that validate this claim, and two prospective controlled real-world studies that dispute this,” he emphasized. “Cloth masks and surgical masks, which were the types of masks worn by school students, are also known to have a nonsignificant and paltry – in the latter, certainly less than 50%, and perhaps as little as 10% – effect on the reduction of respiratory viral spread,” he added.
“Mechanistic studies on mask wearing may suggest some mask efficacy, but these studies are as valid as mechanistic studies showing the effect of various antifungal pharmaceuticals on the replication of SARS-CoV-2 virus in culture, meaning only valid as hypothesis generating, and ultimately the latter hypothesis failed to bear out,” Dr. Gorelik explained. “We do not know the reason why other respiratory viruses and non-SARS-CoV-2 coronaviruses declined during the pandemic, but we do know that despite this, the SARS-CoV-2 coronavirus itself did not appear to suffer the same fate. Thus, it is very possible that another factor was at work, and we know that during other viral pandemics, typically circulating viruses decline, potentially due to induction of interferon responses in hosts, in a general effect known as ‘viral interference,’ ” he said.
“Overall, we must have robust evidence to support benefits of hypotheses that have demonstrated clear damage to children during this pandemic (such as school closures), and this study fails to live up to that requirement,” Dr. Gorelik said.
The study was supported by the Gordon and Marilyn Macklin Foundation and the Patient-Centered Outcomes Research Institute. Dr. Burney and Dr. Gorelik had no financial conflicts to disclose.
FROM JAMA NETWORK OPEN
FDA approves setmelanotide for obesity in Bardet-Biedl syndrome
The Food and Drug Administration has approved a supplemental indication for setmelanotide (Imcivree, Rhythm Pharmaceuticals) injection for chronic weight management in adults and pediatric patients age 6 and older with obesity due to Bardet-Biedl Syndrome (BBS).

Setmelanotide, a melanocortin-4 receptor (MC4R) agonist, is the first FDA-approved therapy for BBS, a rare genetic disorder that impairs a hunger signal along the melanocortin-4 receptor (MC4R) pathway.
BBS affects an estimated 1,500-2,500 people in the United States.
Individuals with BBS typically have obesity that starts at age 1 along with insatiable hunger (hyperphagia). Available weight management options are generally unsuccessful.
Other symptoms may include retinal degeneration, reduced kidney function, or extra digits of the hands or feet.
Setmelanotide received priority review, orphan drug designation, and breakthrough designation for this new indication.
As previously reported, in November 2020, the FDA approved setmelanotide for weight management in adults and children as young as 6 years with obesity due to proopiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency confirmed by genetic testing – who also have impaired hunger signaling from the brain.
These individuals have a normal weight at birth but develop persistent, severe obesity within months due to hyperphagia.
The FDA approval of Imcivree for BBS “represents a significant milestone for Rhythm [Pharmaceuticals], validating our strategy of developing Imcivree for people with hyperphagia and severe obesity caused by rare MC4R-pathway diseases and allowing us to provide our precision therapy to an established community of patients living with BBS and their families who are eagerly awaiting a new treatment option,” said David Meeker, MD, chair, president and CEO of Rhythm, in a press release.
Safety, effectiveness in 66-week trial in 44 patients
The safety and effectiveness of setmelanotidewas evaluated in a 66-week phase 3 clinical trial that enrolled 44 patients age 6 and older who had a diagnosis of BBS and obesity – defined as a body mass index greater than or equal to 30 kg/m2 or greater than or equal to 97th percentile for pediatric patients.
After an initial 14-week, randomized, double-blind, placebo-controlled treatment period, patients entered a 52-week, open-label period.
The trial met its primary endpoint and all key secondary endpoints, with statistically significant reductions in weight and hunger at 52 weeks on therapy.
- After 52 weeks of treatment, patients taking setmelanotide lost, on average, 7.9% of their initial BMI.
- 61% of patients lost 5% or more of their initial BMI, and 39% lost 10% or more of their initial BMI.
- In the 14-week, placebo-controlled treatment, on average, BMI dropped by 4.6% in the 22 patients treated with the study drug and dropped 0.1% in the 22 patients treated with placebo.
- At 52 weeks, the 14 patients aged 12 and older who were able to self-report their hunger had a significant –2.1 mean change in hunger score.
Setmelanotide is associated with the following warnings and precautions:
- Spontaneous penile erections in males and sexual adverse reactions in females. Instruct males with erection lasting longer than 4 hours to seek emergency medical attention.
- Depression and suicidal ideation. Monitor patients for new onset or worsening depression or suicidal thoughts or behaviors. Consider discontinuing the drug if patients have suicidal thoughts or behaviors or clinically significant or persistent depression symptoms.
- Skin pigmentation and darkening of preexisting nevi (moles). Examine skin before and during treatment.
- Setmelanotide is not approved for use in neonates or infants. Serious and fatal adverse reactions including “gasping syndrome” can occur in neonates and low-birth-weight infants treated with benzyl alcohol-preserved drugs.
The most common adverse reactions (with an incidence greater than or equal to 20%) included skin hyperpigmentation, injection site reactions, nausea, headache, diarrhea, abdominal pain, vomiting, depression, and spontaneous penile erection.
The FDA did not approve the company’s supplemental new drug application for setmelanotide in Alström syndrome.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved a supplemental indication for setmelanotide (Imcivree, Rhythm Pharmaceuticals) injection for chronic weight management in adults and pediatric patients age 6 and older with obesity due to Bardet-Biedl Syndrome (BBS).
Setmelanotide, a melanocortin-4 receptor (MC4R) agonist, is the first FDA-approved therapy for BBS, a rare genetic disorder that impairs a hunger signal along the melanocortin-4 receptor (MC4R) pathway.
BBS affects an estimated 1,500-2,500 people in the United States.
Individuals with BBS typically have obesity that starts at age 1 along with insatiable hunger (hyperphagia). Available weight management options are generally unsuccessful.
Other symptoms may include retinal degeneration, reduced kidney function, or extra digits of the hands or feet.
Setmelanotide received priority review, orphan drug designation, and breakthrough designation for this new indication.
As previously reported, in November 2020, the FDA approved setmelanotide for weight management in adults and children as young as 6 years with obesity due to proopiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency confirmed by genetic testing – who also have impaired hunger signaling from the brain.
These individuals have a normal weight at birth but develop persistent, severe obesity within months due to hyperphagia.
The FDA approval of Imcivree for BBS “represents a significant milestone for Rhythm [Pharmaceuticals], validating our strategy of developing Imcivree for people with hyperphagia and severe obesity caused by rare MC4R-pathway diseases and allowing us to provide our precision therapy to an established community of patients living with BBS and their families who are eagerly awaiting a new treatment option,” said David Meeker, MD, chair, president and CEO of Rhythm, in a press release.
Safety, effectiveness in 66-week trial in 44 patients
The safety and effectiveness of setmelanotidewas evaluated in a 66-week phase 3 clinical trial that enrolled 44 patients age 6 and older who had a diagnosis of BBS and obesity – defined as a body mass index greater than or equal to 30 kg/m2 or greater than or equal to 97th percentile for pediatric patients.
After an initial 14-week, randomized, double-blind, placebo-controlled treatment period, patients entered a 52-week, open-label period.
The trial met its primary endpoint and all key secondary endpoints, with statistically significant reductions in weight and hunger at 52 weeks on therapy.
- After 52 weeks of treatment, patients taking setmelanotide lost, on average, 7.9% of their initial BMI.
- 61% of patients lost 5% or more of their initial BMI, and 39% lost 10% or more of their initial BMI.
- In the 14-week, placebo-controlled treatment, on average, BMI dropped by 4.6% in the 22 patients treated with the study drug and dropped 0.1% in the 22 patients treated with placebo.
- At 52 weeks, the 14 patients aged 12 and older who were able to self-report their hunger had a significant –2.1 mean change in hunger score.
Setmelanotide is associated with the following warnings and precautions:
- Spontaneous penile erections in males and sexual adverse reactions in females. Instruct males with erection lasting longer than 4 hours to seek emergency medical attention.
- Depression and suicidal ideation. Monitor patients for new onset or worsening depression or suicidal thoughts or behaviors. Consider discontinuing the drug if patients have suicidal thoughts or behaviors or clinically significant or persistent depression symptoms.
- Skin pigmentation and darkening of preexisting nevi (moles). Examine skin before and during treatment.
- Setmelanotide is not approved for use in neonates or infants. Serious and fatal adverse reactions including “gasping syndrome” can occur in neonates and low-birth-weight infants treated with benzyl alcohol-preserved drugs.
The most common adverse reactions (with an incidence greater than or equal to 20%) included skin hyperpigmentation, injection site reactions, nausea, headache, diarrhea, abdominal pain, vomiting, depression, and spontaneous penile erection.
The FDA did not approve the company’s supplemental new drug application for setmelanotide in Alström syndrome.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved a supplemental indication for setmelanotide (Imcivree, Rhythm Pharmaceuticals) injection for chronic weight management in adults and pediatric patients age 6 and older with obesity due to Bardet-Biedl Syndrome (BBS).
Setmelanotide, a melanocortin-4 receptor (MC4R) agonist, is the first FDA-approved therapy for BBS, a rare genetic disorder that impairs a hunger signal along the melanocortin-4 receptor (MC4R) pathway.
BBS affects an estimated 1,500-2,500 people in the United States.
Individuals with BBS typically have obesity that starts at age 1 along with insatiable hunger (hyperphagia). Available weight management options are generally unsuccessful.
Other symptoms may include retinal degeneration, reduced kidney function, or extra digits of the hands or feet.
Setmelanotide received priority review, orphan drug designation, and breakthrough designation for this new indication.
As previously reported, in November 2020, the FDA approved setmelanotide for weight management in adults and children as young as 6 years with obesity due to proopiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency confirmed by genetic testing – who also have impaired hunger signaling from the brain.
These individuals have a normal weight at birth but develop persistent, severe obesity within months due to hyperphagia.
The FDA approval of Imcivree for BBS “represents a significant milestone for Rhythm [Pharmaceuticals], validating our strategy of developing Imcivree for people with hyperphagia and severe obesity caused by rare MC4R-pathway diseases and allowing us to provide our precision therapy to an established community of patients living with BBS and their families who are eagerly awaiting a new treatment option,” said David Meeker, MD, chair, president and CEO of Rhythm, in a press release.
Safety, effectiveness in 66-week trial in 44 patients
The safety and effectiveness of setmelanotidewas evaluated in a 66-week phase 3 clinical trial that enrolled 44 patients age 6 and older who had a diagnosis of BBS and obesity – defined as a body mass index greater than or equal to 30 kg/m2 or greater than or equal to 97th percentile for pediatric patients.
After an initial 14-week, randomized, double-blind, placebo-controlled treatment period, patients entered a 52-week, open-label period.
The trial met its primary endpoint and all key secondary endpoints, with statistically significant reductions in weight and hunger at 52 weeks on therapy.
- After 52 weeks of treatment, patients taking setmelanotide lost, on average, 7.9% of their initial BMI.
- 61% of patients lost 5% or more of their initial BMI, and 39% lost 10% or more of their initial BMI.
- In the 14-week, placebo-controlled treatment, on average, BMI dropped by 4.6% in the 22 patients treated with the study drug and dropped 0.1% in the 22 patients treated with placebo.
- At 52 weeks, the 14 patients aged 12 and older who were able to self-report their hunger had a significant –2.1 mean change in hunger score.
Setmelanotide is associated with the following warnings and precautions:
- Spontaneous penile erections in males and sexual adverse reactions in females. Instruct males with erection lasting longer than 4 hours to seek emergency medical attention.
- Depression and suicidal ideation. Monitor patients for new onset or worsening depression or suicidal thoughts or behaviors. Consider discontinuing the drug if patients have suicidal thoughts or behaviors or clinically significant or persistent depression symptoms.
- Skin pigmentation and darkening of preexisting nevi (moles). Examine skin before and during treatment.
- Setmelanotide is not approved for use in neonates or infants. Serious and fatal adverse reactions including “gasping syndrome” can occur in neonates and low-birth-weight infants treated with benzyl alcohol-preserved drugs.
The most common adverse reactions (with an incidence greater than or equal to 20%) included skin hyperpigmentation, injection site reactions, nausea, headache, diarrhea, abdominal pain, vomiting, depression, and spontaneous penile erection.
The FDA did not approve the company’s supplemental new drug application for setmelanotide in Alström syndrome.
A version of this article first appeared on Medscape.com.
Biomarkers may help to predict persistent oligoarticular JIA
Ongoing research in patients with oligoarticular juvenile idiopathic arthritis (JIA) so far suggests that a set of biomarkers in synovial fluid may help to predict which patients may be more likely to stay with persistent oligoarticular disease rather than progress to polyarticular disease, according to new research presented at the annual scientific meeting of the Childhood Arthritis and Rheumatology Research Alliance, held virtually this year. Identifying biomarkers in synovial fluid or possibly serum could aid families and physicians in being more proactive in treatment protocols, said AnneMarie C. Brescia, MD, of Nemours Children’s Hospital in Wilmington, Del.
“JIA carries the risk of permanent joint damage and disability, which can result when joint involvement evolves from oligoarticular into a polyarticular course, termed extended oligoarticular disease,” Dr. Brescia told attendees. “Since disease progression increases the risk for disability, early prediction of this course is essential.”
This group – those whose oligoarticular disease will begin recruiting joints and ultimately become extended oligoarticular JIA – is “very important because they have been shown to have worse health-related quality of life and greater risk of needing a joint replacement than even polyarticular [JIA],” Dr. Brescia said. “So, our lab has really focused on trying to predict who will fall in this group.”
Melissa Oliver, MD, assistant professor of clinical pediatrics in the division of pediatric rheumatology at Indiana University in Indianapolis, was not involved in the study but agreed that having highly sensitive and specific biomarkers could be particularly helpful in clinical care.
“Biomarkers can help guide treatment decisions and help physicians and their patients share the decision-making about next choices and when to change,” Dr. Oliver told this news organization. “If a provider and parent know that their child has these markers in their serum or synovial fluid that may predict extension of their disease, then they may be more aggressive upfront with therapy.”
The study aimed to determine whether differential levels of synovial fluid proteins could be used to predict whether JIA would evolve into an extended course before it became clinically evident. Although early aggressive treatment is common with rheumatoid arthritis and can lead to remission, JIA treatment paradigms tend to be more reactive, Dr. Brescia said.
“It would be better to switch to proactive, that if we’re able to predict that this patient may have a more difficult course with extension to polyarticular, we could be prepared, we could inform the parents, and it would just help us have a more proactive approach,” she said.
The researchers used antibody arrays to detect the following inflammatory mediators in blinded samples: CD14, interleukin (IL)-1-alpha, IL-3, IL-5, IL-6, vascular endothelial growth factor (VEGF), and angiogenin. They analyzed 37 samples with persistent disease and 32 samples from disease that had not yet extended but would become extended in that patient. The samples came from patients who were taking no medicines or only NSAIDs. The researchers assessed the sensitivity and specificity of each biomarker. Sensitivity referred the biomarker’s ability to correctly indicate that the sample would extend, and specificity referred to the biomarker’s accuracy in determining that the disease in the sample would remain persistent.
Combining samples from cohorts at Nemours Children’s Health (14 persistent and 7 extended-to-be) and Cincinnati Children’s Hospital (23 persistent and 25 extended-to-be) yielded the following results: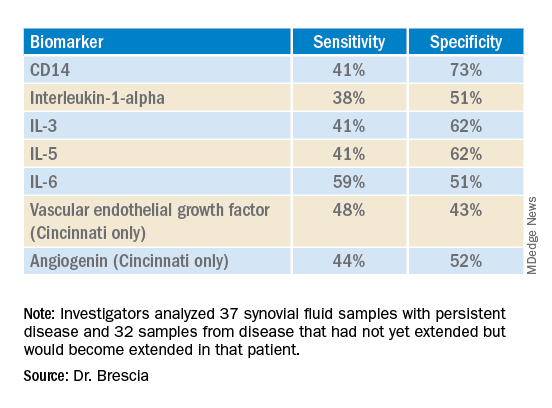
The findings revealed that the selected biomarkers were more accurate at predicting whose disease would remain persistent than predicting those that would extend, Dr. Brescia said. CD14 was the most specific biomarker, and IL-6 was the most sensitive biomarker in both groups.
When the researchers translated the findings from ELISA to the Luminex platform, positive results in synovial fluid for all these biomarkers were also positive in serum samples. Although the differences between persistent and extended-to-be samples did not reach statistical significance using Luminex, the pattern was the same for each biomarker.
“Luminex is more sensitive than ELISA. We believe that conducting an LDA [linear discriminant analysis] using these Luminex measurements will allow us to determine new cutoffs or new protein levels that are appropriate for Luminex to predict who will extend,” Dr. Brescia said. “It’s also our goal to develop a serum panel because ... being able to detect these markers in serum would expand the applicability of these markers to more patients.”
Dr. Brescia then described the group’s work in defining clinically relevant subpopulations of patients based on fibroblast-like synoviocytes (FLS) cells in the synovial intimal lining that produce inflammatory cytokines.
“Our compelling, single-cell, RNA sequencing preliminary data revealing multiple subpopulations within the total FLS population supports our hypothesis that distinct FLS subpopulations correlate with clinical outcome,” said Dr. Brescia. They looked at the percentage of chondrocyte-like, fibroblast-like, and smooth muscle-like subpopulations in samples from patients with oligoarticular JIA, extended-to-be JIA, and polyarticular JIA. Chondrocytes occurred in the largest proportion, and polyarticular JIA FLS had the largest percentage of chondrocytes, compared with the other two subpopulation groups.
“This is a work in progress,” Dr. Brescia said, “so hopefully you’ll hear about it next year.” In response to an attendee’s question, she said she believes identifying reliable biomarkers will eventually lead to refining treatment paradigms.
“I think it will at least change the guidance we can provide parents about making next choices and how quickly to accelerate to those next choices,” Dr. Brescia said. For example, if a child’s serum or synovial fluid has markers that show a very high likelihood of extension, the parent may decide to proceed to the next level medication sooner. “I do think it will push both parents and doctors to be a little more proactive instead of reactive when the poor patient comes back with 13 joints involved when they had just been an oligo for years.”
Dr. Oliver noted the promise of CD14 and IL-6 in potentially predicting which patients’ disease will stay persistent but cautioned that it’s still early in evaluating these biomarkers, especially with the limited patient samples in this study.
“I think these results are promising, and it’s great that there are groups out there working on this,” Dr. Oliver said. “Once we have a reliable, highly sensitive and specific biomarker, that will definitely help providers, parents, and patients be more informed.”
The research was supported by the Open Net Foundation, the Arthritis Foundation, Delaware Community Foundation, the Delaware Clinical and Translational Research (DE-CTR) ACCEL Program, the Nancy Taylor Foundation for Chronic Diseases, and CARRA. Dr. Brescia and Dr. Oliver have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Ongoing research in patients with oligoarticular juvenile idiopathic arthritis (JIA) so far suggests that a set of biomarkers in synovial fluid may help to predict which patients may be more likely to stay with persistent oligoarticular disease rather than progress to polyarticular disease, according to new research presented at the annual scientific meeting of the Childhood Arthritis and Rheumatology Research Alliance, held virtually this year. Identifying biomarkers in synovial fluid or possibly serum could aid families and physicians in being more proactive in treatment protocols, said AnneMarie C. Brescia, MD, of Nemours Children’s Hospital in Wilmington, Del.
“JIA carries the risk of permanent joint damage and disability, which can result when joint involvement evolves from oligoarticular into a polyarticular course, termed extended oligoarticular disease,” Dr. Brescia told attendees. “Since disease progression increases the risk for disability, early prediction of this course is essential.”
This group – those whose oligoarticular disease will begin recruiting joints and ultimately become extended oligoarticular JIA – is “very important because they have been shown to have worse health-related quality of life and greater risk of needing a joint replacement than even polyarticular [JIA],” Dr. Brescia said. “So, our lab has really focused on trying to predict who will fall in this group.”
Melissa Oliver, MD, assistant professor of clinical pediatrics in the division of pediatric rheumatology at Indiana University in Indianapolis, was not involved in the study but agreed that having highly sensitive and specific biomarkers could be particularly helpful in clinical care.
“Biomarkers can help guide treatment decisions and help physicians and their patients share the decision-making about next choices and when to change,” Dr. Oliver told this news organization. “If a provider and parent know that their child has these markers in their serum or synovial fluid that may predict extension of their disease, then they may be more aggressive upfront with therapy.”
The study aimed to determine whether differential levels of synovial fluid proteins could be used to predict whether JIA would evolve into an extended course before it became clinically evident. Although early aggressive treatment is common with rheumatoid arthritis and can lead to remission, JIA treatment paradigms tend to be more reactive, Dr. Brescia said.
“It would be better to switch to proactive, that if we’re able to predict that this patient may have a more difficult course with extension to polyarticular, we could be prepared, we could inform the parents, and it would just help us have a more proactive approach,” she said.
The researchers used antibody arrays to detect the following inflammatory mediators in blinded samples: CD14, interleukin (IL)-1-alpha, IL-3, IL-5, IL-6, vascular endothelial growth factor (VEGF), and angiogenin. They analyzed 37 samples with persistent disease and 32 samples from disease that had not yet extended but would become extended in that patient. The samples came from patients who were taking no medicines or only NSAIDs. The researchers assessed the sensitivity and specificity of each biomarker. Sensitivity referred the biomarker’s ability to correctly indicate that the sample would extend, and specificity referred to the biomarker’s accuracy in determining that the disease in the sample would remain persistent.
Combining samples from cohorts at Nemours Children’s Health (14 persistent and 7 extended-to-be) and Cincinnati Children’s Hospital (23 persistent and 25 extended-to-be) yielded the following results:
The findings revealed that the selected biomarkers were more accurate at predicting whose disease would remain persistent than predicting those that would extend, Dr. Brescia said. CD14 was the most specific biomarker, and IL-6 was the most sensitive biomarker in both groups.
When the researchers translated the findings from ELISA to the Luminex platform, positive results in synovial fluid for all these biomarkers were also positive in serum samples. Although the differences between persistent and extended-to-be samples did not reach statistical significance using Luminex, the pattern was the same for each biomarker.
“Luminex is more sensitive than ELISA. We believe that conducting an LDA [linear discriminant analysis] using these Luminex measurements will allow us to determine new cutoffs or new protein levels that are appropriate for Luminex to predict who will extend,” Dr. Brescia said. “It’s also our goal to develop a serum panel because ... being able to detect these markers in serum would expand the applicability of these markers to more patients.”
Dr. Brescia then described the group’s work in defining clinically relevant subpopulations of patients based on fibroblast-like synoviocytes (FLS) cells in the synovial intimal lining that produce inflammatory cytokines.
“Our compelling, single-cell, RNA sequencing preliminary data revealing multiple subpopulations within the total FLS population supports our hypothesis that distinct FLS subpopulations correlate with clinical outcome,” said Dr. Brescia. They looked at the percentage of chondrocyte-like, fibroblast-like, and smooth muscle-like subpopulations in samples from patients with oligoarticular JIA, extended-to-be JIA, and polyarticular JIA. Chondrocytes occurred in the largest proportion, and polyarticular JIA FLS had the largest percentage of chondrocytes, compared with the other two subpopulation groups.
“This is a work in progress,” Dr. Brescia said, “so hopefully you’ll hear about it next year.” In response to an attendee’s question, she said she believes identifying reliable biomarkers will eventually lead to refining treatment paradigms.
“I think it will at least change the guidance we can provide parents about making next choices and how quickly to accelerate to those next choices,” Dr. Brescia said. For example, if a child’s serum or synovial fluid has markers that show a very high likelihood of extension, the parent may decide to proceed to the next level medication sooner. “I do think it will push both parents and doctors to be a little more proactive instead of reactive when the poor patient comes back with 13 joints involved when they had just been an oligo for years.”
Dr. Oliver noted the promise of CD14 and IL-6 in potentially predicting which patients’ disease will stay persistent but cautioned that it’s still early in evaluating these biomarkers, especially with the limited patient samples in this study.
“I think these results are promising, and it’s great that there are groups out there working on this,” Dr. Oliver said. “Once we have a reliable, highly sensitive and specific biomarker, that will definitely help providers, parents, and patients be more informed.”
The research was supported by the Open Net Foundation, the Arthritis Foundation, Delaware Community Foundation, the Delaware Clinical and Translational Research (DE-CTR) ACCEL Program, the Nancy Taylor Foundation for Chronic Diseases, and CARRA. Dr. Brescia and Dr. Oliver have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Ongoing research in patients with oligoarticular juvenile idiopathic arthritis (JIA) so far suggests that a set of biomarkers in synovial fluid may help to predict which patients may be more likely to stay with persistent oligoarticular disease rather than progress to polyarticular disease, according to new research presented at the annual scientific meeting of the Childhood Arthritis and Rheumatology Research Alliance, held virtually this year. Identifying biomarkers in synovial fluid or possibly serum could aid families and physicians in being more proactive in treatment protocols, said AnneMarie C. Brescia, MD, of Nemours Children’s Hospital in Wilmington, Del.
“JIA carries the risk of permanent joint damage and disability, which can result when joint involvement evolves from oligoarticular into a polyarticular course, termed extended oligoarticular disease,” Dr. Brescia told attendees. “Since disease progression increases the risk for disability, early prediction of this course is essential.”
This group – those whose oligoarticular disease will begin recruiting joints and ultimately become extended oligoarticular JIA – is “very important because they have been shown to have worse health-related quality of life and greater risk of needing a joint replacement than even polyarticular [JIA],” Dr. Brescia said. “So, our lab has really focused on trying to predict who will fall in this group.”
Melissa Oliver, MD, assistant professor of clinical pediatrics in the division of pediatric rheumatology at Indiana University in Indianapolis, was not involved in the study but agreed that having highly sensitive and specific biomarkers could be particularly helpful in clinical care.
“Biomarkers can help guide treatment decisions and help physicians and their patients share the decision-making about next choices and when to change,” Dr. Oliver told this news organization. “If a provider and parent know that their child has these markers in their serum or synovial fluid that may predict extension of their disease, then they may be more aggressive upfront with therapy.”
The study aimed to determine whether differential levels of synovial fluid proteins could be used to predict whether JIA would evolve into an extended course before it became clinically evident. Although early aggressive treatment is common with rheumatoid arthritis and can lead to remission, JIA treatment paradigms tend to be more reactive, Dr. Brescia said.
“It would be better to switch to proactive, that if we’re able to predict that this patient may have a more difficult course with extension to polyarticular, we could be prepared, we could inform the parents, and it would just help us have a more proactive approach,” she said.
The researchers used antibody arrays to detect the following inflammatory mediators in blinded samples: CD14, interleukin (IL)-1-alpha, IL-3, IL-5, IL-6, vascular endothelial growth factor (VEGF), and angiogenin. They analyzed 37 samples with persistent disease and 32 samples from disease that had not yet extended but would become extended in that patient. The samples came from patients who were taking no medicines or only NSAIDs. The researchers assessed the sensitivity and specificity of each biomarker. Sensitivity referred the biomarker’s ability to correctly indicate that the sample would extend, and specificity referred to the biomarker’s accuracy in determining that the disease in the sample would remain persistent.
Combining samples from cohorts at Nemours Children’s Health (14 persistent and 7 extended-to-be) and Cincinnati Children’s Hospital (23 persistent and 25 extended-to-be) yielded the following results:
The findings revealed that the selected biomarkers were more accurate at predicting whose disease would remain persistent than predicting those that would extend, Dr. Brescia said. CD14 was the most specific biomarker, and IL-6 was the most sensitive biomarker in both groups.
When the researchers translated the findings from ELISA to the Luminex platform, positive results in synovial fluid for all these biomarkers were also positive in serum samples. Although the differences between persistent and extended-to-be samples did not reach statistical significance using Luminex, the pattern was the same for each biomarker.
“Luminex is more sensitive than ELISA. We believe that conducting an LDA [linear discriminant analysis] using these Luminex measurements will allow us to determine new cutoffs or new protein levels that are appropriate for Luminex to predict who will extend,” Dr. Brescia said. “It’s also our goal to develop a serum panel because ... being able to detect these markers in serum would expand the applicability of these markers to more patients.”
Dr. Brescia then described the group’s work in defining clinically relevant subpopulations of patients based on fibroblast-like synoviocytes (FLS) cells in the synovial intimal lining that produce inflammatory cytokines.
“Our compelling, single-cell, RNA sequencing preliminary data revealing multiple subpopulations within the total FLS population supports our hypothesis that distinct FLS subpopulations correlate with clinical outcome,” said Dr. Brescia. They looked at the percentage of chondrocyte-like, fibroblast-like, and smooth muscle-like subpopulations in samples from patients with oligoarticular JIA, extended-to-be JIA, and polyarticular JIA. Chondrocytes occurred in the largest proportion, and polyarticular JIA FLS had the largest percentage of chondrocytes, compared with the other two subpopulation groups.
“This is a work in progress,” Dr. Brescia said, “so hopefully you’ll hear about it next year.” In response to an attendee’s question, she said she believes identifying reliable biomarkers will eventually lead to refining treatment paradigms.
“I think it will at least change the guidance we can provide parents about making next choices and how quickly to accelerate to those next choices,” Dr. Brescia said. For example, if a child’s serum or synovial fluid has markers that show a very high likelihood of extension, the parent may decide to proceed to the next level medication sooner. “I do think it will push both parents and doctors to be a little more proactive instead of reactive when the poor patient comes back with 13 joints involved when they had just been an oligo for years.”
Dr. Oliver noted the promise of CD14 and IL-6 in potentially predicting which patients’ disease will stay persistent but cautioned that it’s still early in evaluating these biomarkers, especially with the limited patient samples in this study.
“I think these results are promising, and it’s great that there are groups out there working on this,” Dr. Oliver said. “Once we have a reliable, highly sensitive and specific biomarker, that will definitely help providers, parents, and patients be more informed.”
The research was supported by the Open Net Foundation, the Arthritis Foundation, Delaware Community Foundation, the Delaware Clinical and Translational Research (DE-CTR) ACCEL Program, the Nancy Taylor Foundation for Chronic Diseases, and CARRA. Dr. Brescia and Dr. Oliver have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM CARRA 2022
Monkeypox: What’s a pediatrician to do?
Not long ago, a pediatrician working in a local urgent care clinic called me about a teenage girl with a pruritic rash. She described vesicles and pustules located primarily on the face and arms with no surrounding cellulitis or other exam findings.
“She probably has impetigo,” my colleague said. “But I took a travel and exposure history and learned that her grandma had recently returned home from visiting family in the Congo. Do you think I need to worry about monkeypox?”
While most pediatricians in the United States have never seen a case of monkeypox, the virus is not new. An orthopox, it belongs to the same genus that includes smallpox and cowpox viruses. It was discovered in 1958 when two colonies of monkeys kept for research developed pox-like rashes. The earliest human case was reported in 1970 in the Democratic Republic of Congo and now the virus is endemic in some counties in Central and West Africa.

Monkeypox virus is a zoonotic disease – it can spread from animals to people. Rodents and other small mammals – not monkeys – are thought to be the most likely reservoir. The virus typically spreads from person to person through close contact with skin or respiratory secretions or contact with contaminated fomites. Typical infection begins with fever, lymphadenopathy, and flulike symptoms that include headache and malaise. One to four days after the onset of fever, the characteristic rash begins as macular lesions that evolve into papules, then vesicles, and finally pustules. Pustular lesions are deep-seated, well circumscribed, and are usually the same size and in the same stage of development on a given body site. The rash often starts on the face or the mouth, and then moves to the extremities, including the palms and soles. Over time, the lesions umbilicate and ultimately crust over.
On May 20, the Centers for Disease Control and Prevention issued a Health Advisory describing a case of monkeypox in a patient in Massachusetts. A single case normally wouldn’t cause too much alarm. In fact, there were two cases reported in the United States in 2021, both in travelers returning to the United States from Nigeria, a country in which the virus is endemic. No transmissions from these individuals to close contacts were identified.
The Massachusetts case was remarkable for two reasons. It occurred in an individual who had recently returned from a trip to Canada, which is not a country in which the virus is endemic. Additionally, it occurred in the context of a global outbreak of monkey pox that has, to date, disproportionately affected individuals who identify as men who have sex with men. Patients have often lacked the characteristic prodrome and many have had rash localized to the perianal and genital area, with or without symptoms of proctitis (anorectal pain, tenesmus, and bleeding). Clinically, some lesions mimicked sexually transmitted infections that the occur in the anogenital area, including herpes, syphilis, and lymphogranuloma venereum.
As of May 31, 2022, 17 persons in nine states had been diagnosed with presumed monkeypox virus infection. They ranged in age from 28 to 61 years and 16/17 identified as MSM. Fourteen reported international travel in the 3 weeks before developing symptoms. As of June 12, that number had grown to 53, while worldwide the number of confirmed and suspected cases reached 1,584. Up-to-date case counts are available at https://ourworldindata.org/monkeypox.
Back on the phone, my colleague laughed a little nervously. “I guess I’m not really worried about monkeypox in my patient.” She paused and then asked, “This isn’t going to be the next pandemic, is it?”
Public health experts at the Centers for Disease Control and Prevention and the World Health Organization have been reassuring in that regard. Two vaccines are available for the prevention of monkeypox. JYNNEOS is a nonreplicating live viral vaccine licensed as a two-dose series to prevent both monkeypox and smallpox. ACAM 2000 is a live Vaccinia virus preparation licensed to prevent smallpox. These vaccines are effective when given before exposure but are thought to also beneficial when given as postexposure prophylaxis. According to the CDC, vaccination within 4 days of exposure can prevent the development of disease. Vaccination within 14 days of exposure may not prevent the development of disease but may lessen symptoms. Treatment is generally supportive but antiviral therapy could be considered for individuals with severe disease. Tecovirmat is Food and Drug Administration approved for the treatment of smallpox but is available under nonresearch Expanded Access Investigational New Drug (EA-IND) protocol for the treatment of children and adults with severe orthopox infections, including monkeypox.
So, what’s a pediatrician to do? Take a good travel history, as my colleague did, because that is good medicine. At this point in an outbreak though, a lack of travel does not exclude the diagnosis. Perform a thorough exam of skin and mucosal areas. When there are rashes in the genital or perianal area, consider the possibility of monkeypox in addition to typical sexually transmitted infections. Ask about exposure to other persons with similar rashes, as well as close or intimate contact with a persons in a social network experiencing monkeypox infections. This includes MSM who meet partners through an online website, app, or at social events. Monkeypox can also be spread through contact with an animal (dead or alive) that is an African endemic species or use of a product derived from such animals. Public health experts encourage clinicians to be alert for rash illnesses consistent with monkeypox, regardless of a patient’s gender or sexual orientation, history of international travel, or specific risk factors.
Pediatricians see many kids with rashes, and while cases of monkeypox climb daily, the disease is still very rare. Given the media coverage of the outbreak, pediatricians should be prepared for questions from patients and their parents. Clinicians who suspect a case of monkeypox should contact their local or state health department for guidance and the need for testing. Tips for recognizing monkeypox and distinguishing it from more common viral illnesses such as chicken pox are available at www.cdc.gov/poxvirus/monkeypox/clinicians/clinical-recognition.html.
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital, also in Louisville. She said she had no relevant financial disclosures. Email her at [email protected].
Not long ago, a pediatrician working in a local urgent care clinic called me about a teenage girl with a pruritic rash. She described vesicles and pustules located primarily on the face and arms with no surrounding cellulitis or other exam findings.
“She probably has impetigo,” my colleague said. “But I took a travel and exposure history and learned that her grandma had recently returned home from visiting family in the Congo. Do you think I need to worry about monkeypox?”
While most pediatricians in the United States have never seen a case of monkeypox, the virus is not new. An orthopox, it belongs to the same genus that includes smallpox and cowpox viruses. It was discovered in 1958 when two colonies of monkeys kept for research developed pox-like rashes. The earliest human case was reported in 1970 in the Democratic Republic of Congo and now the virus is endemic in some counties in Central and West Africa.
Monkeypox virus is a zoonotic disease – it can spread from animals to people. Rodents and other small mammals – not monkeys – are thought to be the most likely reservoir. The virus typically spreads from person to person through close contact with skin or respiratory secretions or contact with contaminated fomites. Typical infection begins with fever, lymphadenopathy, and flulike symptoms that include headache and malaise. One to four days after the onset of fever, the characteristic rash begins as macular lesions that evolve into papules, then vesicles, and finally pustules. Pustular lesions are deep-seated, well circumscribed, and are usually the same size and in the same stage of development on a given body site. The rash often starts on the face or the mouth, and then moves to the extremities, including the palms and soles. Over time, the lesions umbilicate and ultimately crust over.
On May 20, the Centers for Disease Control and Prevention issued a Health Advisory describing a case of monkeypox in a patient in Massachusetts. A single case normally wouldn’t cause too much alarm. In fact, there were two cases reported in the United States in 2021, both in travelers returning to the United States from Nigeria, a country in which the virus is endemic. No transmissions from these individuals to close contacts were identified.
The Massachusetts case was remarkable for two reasons. It occurred in an individual who had recently returned from a trip to Canada, which is not a country in which the virus is endemic. Additionally, it occurred in the context of a global outbreak of monkey pox that has, to date, disproportionately affected individuals who identify as men who have sex with men. Patients have often lacked the characteristic prodrome and many have had rash localized to the perianal and genital area, with or without symptoms of proctitis (anorectal pain, tenesmus, and bleeding). Clinically, some lesions mimicked sexually transmitted infections that the occur in the anogenital area, including herpes, syphilis, and lymphogranuloma venereum.
As of May 31, 2022, 17 persons in nine states had been diagnosed with presumed monkeypox virus infection. They ranged in age from 28 to 61 years and 16/17 identified as MSM. Fourteen reported international travel in the 3 weeks before developing symptoms. As of June 12, that number had grown to 53, while worldwide the number of confirmed and suspected cases reached 1,584. Up-to-date case counts are available at https://ourworldindata.org/monkeypox.
Back on the phone, my colleague laughed a little nervously. “I guess I’m not really worried about monkeypox in my patient.” She paused and then asked, “This isn’t going to be the next pandemic, is it?”
Public health experts at the Centers for Disease Control and Prevention and the World Health Organization have been reassuring in that regard. Two vaccines are available for the prevention of monkeypox. JYNNEOS is a nonreplicating live viral vaccine licensed as a two-dose series to prevent both monkeypox and smallpox. ACAM 2000 is a live Vaccinia virus preparation licensed to prevent smallpox. These vaccines are effective when given before exposure but are thought to also beneficial when given as postexposure prophylaxis. According to the CDC, vaccination within 4 days of exposure can prevent the development of disease. Vaccination within 14 days of exposure may not prevent the development of disease but may lessen symptoms. Treatment is generally supportive but antiviral therapy could be considered for individuals with severe disease. Tecovirmat is Food and Drug Administration approved for the treatment of smallpox but is available under nonresearch Expanded Access Investigational New Drug (EA-IND) protocol for the treatment of children and adults with severe orthopox infections, including monkeypox.
So, what’s a pediatrician to do? Take a good travel history, as my colleague did, because that is good medicine. At this point in an outbreak though, a lack of travel does not exclude the diagnosis. Perform a thorough exam of skin and mucosal areas. When there are rashes in the genital or perianal area, consider the possibility of monkeypox in addition to typical sexually transmitted infections. Ask about exposure to other persons with similar rashes, as well as close or intimate contact with a persons in a social network experiencing monkeypox infections. This includes MSM who meet partners through an online website, app, or at social events. Monkeypox can also be spread through contact with an animal (dead or alive) that is an African endemic species or use of a product derived from such animals. Public health experts encourage clinicians to be alert for rash illnesses consistent with monkeypox, regardless of a patient’s gender or sexual orientation, history of international travel, or specific risk factors.
Pediatricians see many kids with rashes, and while cases of monkeypox climb daily, the disease is still very rare. Given the media coverage of the outbreak, pediatricians should be prepared for questions from patients and their parents. Clinicians who suspect a case of monkeypox should contact their local or state health department for guidance and the need for testing. Tips for recognizing monkeypox and distinguishing it from more common viral illnesses such as chicken pox are available at www.cdc.gov/poxvirus/monkeypox/clinicians/clinical-recognition.html.
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital, also in Louisville. She said she had no relevant financial disclosures. Email her at [email protected].
Not long ago, a pediatrician working in a local urgent care clinic called me about a teenage girl with a pruritic rash. She described vesicles and pustules located primarily on the face and arms with no surrounding cellulitis or other exam findings.
“She probably has impetigo,” my colleague said. “But I took a travel and exposure history and learned that her grandma had recently returned home from visiting family in the Congo. Do you think I need to worry about monkeypox?”
While most pediatricians in the United States have never seen a case of monkeypox, the virus is not new. An orthopox, it belongs to the same genus that includes smallpox and cowpox viruses. It was discovered in 1958 when two colonies of monkeys kept for research developed pox-like rashes. The earliest human case was reported in 1970 in the Democratic Republic of Congo and now the virus is endemic in some counties in Central and West Africa.
Monkeypox virus is a zoonotic disease – it can spread from animals to people. Rodents and other small mammals – not monkeys – are thought to be the most likely reservoir. The virus typically spreads from person to person through close contact with skin or respiratory secretions or contact with contaminated fomites. Typical infection begins with fever, lymphadenopathy, and flulike symptoms that include headache and malaise. One to four days after the onset of fever, the characteristic rash begins as macular lesions that evolve into papules, then vesicles, and finally pustules. Pustular lesions are deep-seated, well circumscribed, and are usually the same size and in the same stage of development on a given body site. The rash often starts on the face or the mouth, and then moves to the extremities, including the palms and soles. Over time, the lesions umbilicate and ultimately crust over.
On May 20, the Centers for Disease Control and Prevention issued a Health Advisory describing a case of monkeypox in a patient in Massachusetts. A single case normally wouldn’t cause too much alarm. In fact, there were two cases reported in the United States in 2021, both in travelers returning to the United States from Nigeria, a country in which the virus is endemic. No transmissions from these individuals to close contacts were identified.
The Massachusetts case was remarkable for two reasons. It occurred in an individual who had recently returned from a trip to Canada, which is not a country in which the virus is endemic. Additionally, it occurred in the context of a global outbreak of monkey pox that has, to date, disproportionately affected individuals who identify as men who have sex with men. Patients have often lacked the characteristic prodrome and many have had rash localized to the perianal and genital area, with or without symptoms of proctitis (anorectal pain, tenesmus, and bleeding). Clinically, some lesions mimicked sexually transmitted infections that the occur in the anogenital area, including herpes, syphilis, and lymphogranuloma venereum.
As of May 31, 2022, 17 persons in nine states had been diagnosed with presumed monkeypox virus infection. They ranged in age from 28 to 61 years and 16/17 identified as MSM. Fourteen reported international travel in the 3 weeks before developing symptoms. As of June 12, that number had grown to 53, while worldwide the number of confirmed and suspected cases reached 1,584. Up-to-date case counts are available at https://ourworldindata.org/monkeypox.
Back on the phone, my colleague laughed a little nervously. “I guess I’m not really worried about monkeypox in my patient.” She paused and then asked, “This isn’t going to be the next pandemic, is it?”
Public health experts at the Centers for Disease Control and Prevention and the World Health Organization have been reassuring in that regard. Two vaccines are available for the prevention of monkeypox. JYNNEOS is a nonreplicating live viral vaccine licensed as a two-dose series to prevent both monkeypox and smallpox. ACAM 2000 is a live Vaccinia virus preparation licensed to prevent smallpox. These vaccines are effective when given before exposure but are thought to also beneficial when given as postexposure prophylaxis. According to the CDC, vaccination within 4 days of exposure can prevent the development of disease. Vaccination within 14 days of exposure may not prevent the development of disease but may lessen symptoms. Treatment is generally supportive but antiviral therapy could be considered for individuals with severe disease. Tecovirmat is Food and Drug Administration approved for the treatment of smallpox but is available under nonresearch Expanded Access Investigational New Drug (EA-IND) protocol for the treatment of children and adults with severe orthopox infections, including monkeypox.
So, what’s a pediatrician to do? Take a good travel history, as my colleague did, because that is good medicine. At this point in an outbreak though, a lack of travel does not exclude the diagnosis. Perform a thorough exam of skin and mucosal areas. When there are rashes in the genital or perianal area, consider the possibility of monkeypox in addition to typical sexually transmitted infections. Ask about exposure to other persons with similar rashes, as well as close or intimate contact with a persons in a social network experiencing monkeypox infections. This includes MSM who meet partners through an online website, app, or at social events. Monkeypox can also be spread through contact with an animal (dead or alive) that is an African endemic species or use of a product derived from such animals. Public health experts encourage clinicians to be alert for rash illnesses consistent with monkeypox, regardless of a patient’s gender or sexual orientation, history of international travel, or specific risk factors.
Pediatricians see many kids with rashes, and while cases of monkeypox climb daily, the disease is still very rare. Given the media coverage of the outbreak, pediatricians should be prepared for questions from patients and their parents. Clinicians who suspect a case of monkeypox should contact their local or state health department for guidance and the need for testing. Tips for recognizing monkeypox and distinguishing it from more common viral illnesses such as chicken pox are available at www.cdc.gov/poxvirus/monkeypox/clinicians/clinical-recognition.html.
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital, also in Louisville. She said she had no relevant financial disclosures. Email her at [email protected].
Updated pediatric uveitis recommendations advise on expanded treatment options
Glucocorticoids should be bridging therapies in the treatment of juvenile idiopathic arthritis–associated uveitis (JIAU) and idiopathic chronic anterior uveitis (CAU), according to recently released recommendations from the Multinational Interdisciplinary Working Group for Uveitis in Childhood (MIWGUC).
The recommendations cover literature from December 2014 to June 2020 and represent an update of previously published treatment guidelines from 2018. The MIWGUC work group that formulated the new recommendations consisted of eight pediatric rheumatologists and eight ophthalmologists with expertise in pediatric uveitis.

One major shift from the previous guidelines is the lack of distinction between JIAU and CAU, said lead author Ivan Foeldvari, MD, head of the Hamburg (Germany) Center for Pediatric and Adolescent Rheumatology.
“We are considering these two conditions equivalent regarding the ophthalmological presentation,” Dr. Foeldvari said in an interview.
These guidelines have also expanded possible treatment options for these conditions in light of data from clinical trials that have pointed to new options, Dr. Foeldvari noted.
The guidelines also present new options, compared with the 2019 American College of Rheumatology/Arthritis Foundation JIA-associated uveitis guideline. The data cutoff for that guideline was 2014. “Many key papers were published since 2014,” Dr. Foeldvari said.
Another major change is in the escalation of therapy, he noted.
“We view glucocorticoids as a bridging agent, which is very important to emphasize,” Dr. Foeldvari said. “We do not want oral glucocorticoids used as a monotherapy. If you consider a child who has severe uveitis and you want to give an oral glucocorticoid treatment, then it should be considered only for bridging. We suggest to start a DMARD [disease-modifying antirheumatic drug].”
The specific recommendation is that methotrexate be the first DMARD that clinicians choose after using glucocorticoids as a bridging therapy; adalimumab is recommended as the next treatment choice for patients who do not respond to methotrexate.
The working group also calls for limited use of topical glucocorticoids in the affected eye, he said.
“We recommend no more than two or three drops long term in the eye, because there are studies that show continuous local therapy is the main reason that children may develop blindness,” Dr. Foeldvari said. “With respect to oral corticosteroids, they have a lot of systemic effects. Those effects include a high risk of infection, weight gain, and growth disturbance.”

The new recommendations can guide treatment decisions for rheumatologists and ophthalmologists alike, according to Daniel J. Lovell, MD, MPH, the Joseph E. Levinson Endowed Chair of Pediatric Rheumatology and professor of pediatrics at the University of Cincinnati and Cincinnati Children’s Hospital Medical Center. He was one of the authors of the 2019 ACR/Arthritis Foundation guideline.
“We [rheumatologists] comanage these patients with ophthalmologists,” Dr. Lovell said in an interview. “Ophthalmologists are oftentimes not as experienced in using biologics or methotrexate in terms of monitoring for safety and dosing.”
Dr. Lovell pointed out that the key message from this set of recommendations is to curb the use of topical steroids.
“Topical steroids should be used sparingly and as monotherapy for a very short period of time,” Dr. Lovell said. “Any guidelines agree that if eye inflammation is still present at 3 months, we need to move beyond topical steroid monotherapy.”
These new recommendations from MIWGUC are fairly consistent with the 2019 ACR/Arthritis Foundation guideline, he noted.
“The differences are very minor,” Dr. Lovell said. “In both instances, systemic corticosteroids should be bridging therapy. If you have a patient who needs systemic corticosteroids in addition to topical at the same time, you should be talking about adding other anti-inflammatory treatments, such as traditional and/or biologic DMARDs. Both MIWGUC and the ACR guidelines agree on that.”
The 2019 ACR/Arthritis Foundation guideline did not mention rituximab as an option, nor Janus kinase (JAK) inhibitors, Dr. Lovell said, noting there was no literature on JAK inhibitors as a possible option for JIAU when the guideline was being formulated.
Both sets of guidelines point out that there is a dearth of literature with respect to determining the safe dose of maintenance topical corticosteroids, Dr. Lovell said.
“The ACR 2019 guidelines state you should add systemic therapy if there is persistent eye inflammation despite use of up to two drops per day of topical corticosteroids, while the European [MIWGUC] guideline states you can allow up to three drops,” he said. “In both instances, they are quoting the same two sources. Both guidelines indicate that the literature is very scant as to defining a true, safe dose of topical ocular corticosteroids. They differ by one drop allowed per day. In both instances, in the presence of active uveitis, at 3 months on topical steroid monotherapy, both [guidelines] strongly recommend adding systemic therapy.”

Marinka Twilt, MD, MSCE, PhD, associate professor in the department of pediatrics at the University of Calgary (Alta.), noted in an interview that these latest recommendations from MIWGUC have included consensus views on what to do if certain medications fail to lead to remission, which is not addressed in the 2019 ACR/Arthritis Foundation guideline.
“The new manuscript provides consensus on the use of abatacept, JAK inhibitors, and rituximab if patients are refractory to adalimumab and tocilizumab, which is not discussed in the 2019 recommendations,” Dr. Twilt said.
She also pointed out that these recommendations suggest adalimumab as treatment before infliximab, whereas the 2019 guideline did not recommend using one or the other first.
In compiling the recommendations, the authors received no outside financial support. Dr. Foeldvari is a member of advisory boards for Lilly, Pfizer, Novartis, and Medac. Dr. Lovell and Dr. Twilt disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Glucocorticoids should be bridging therapies in the treatment of juvenile idiopathic arthritis–associated uveitis (JIAU) and idiopathic chronic anterior uveitis (CAU), according to recently released recommendations from the Multinational Interdisciplinary Working Group for Uveitis in Childhood (MIWGUC).
The recommendations cover literature from December 2014 to June 2020 and represent an update of previously published treatment guidelines from 2018. The MIWGUC work group that formulated the new recommendations consisted of eight pediatric rheumatologists and eight ophthalmologists with expertise in pediatric uveitis.
One major shift from the previous guidelines is the lack of distinction between JIAU and CAU, said lead author Ivan Foeldvari, MD, head of the Hamburg (Germany) Center for Pediatric and Adolescent Rheumatology.
“We are considering these two conditions equivalent regarding the ophthalmological presentation,” Dr. Foeldvari said in an interview.
These guidelines have also expanded possible treatment options for these conditions in light of data from clinical trials that have pointed to new options, Dr. Foeldvari noted.
The guidelines also present new options, compared with the 2019 American College of Rheumatology/Arthritis Foundation JIA-associated uveitis guideline. The data cutoff for that guideline was 2014. “Many key papers were published since 2014,” Dr. Foeldvari said.
Another major change is in the escalation of therapy, he noted.
“We view glucocorticoids as a bridging agent, which is very important to emphasize,” Dr. Foeldvari said. “We do not want oral glucocorticoids used as a monotherapy. If you consider a child who has severe uveitis and you want to give an oral glucocorticoid treatment, then it should be considered only for bridging. We suggest to start a DMARD [disease-modifying antirheumatic drug].”
The specific recommendation is that methotrexate be the first DMARD that clinicians choose after using glucocorticoids as a bridging therapy; adalimumab is recommended as the next treatment choice for patients who do not respond to methotrexate.
The working group also calls for limited use of topical glucocorticoids in the affected eye, he said.
“We recommend no more than two or three drops long term in the eye, because there are studies that show continuous local therapy is the main reason that children may develop blindness,” Dr. Foeldvari said. “With respect to oral corticosteroids, they have a lot of systemic effects. Those effects include a high risk of infection, weight gain, and growth disturbance.”
The new recommendations can guide treatment decisions for rheumatologists and ophthalmologists alike, according to Daniel J. Lovell, MD, MPH, the Joseph E. Levinson Endowed Chair of Pediatric Rheumatology and professor of pediatrics at the University of Cincinnati and Cincinnati Children’s Hospital Medical Center. He was one of the authors of the 2019 ACR/Arthritis Foundation guideline.
“We [rheumatologists] comanage these patients with ophthalmologists,” Dr. Lovell said in an interview. “Ophthalmologists are oftentimes not as experienced in using biologics or methotrexate in terms of monitoring for safety and dosing.”
Dr. Lovell pointed out that the key message from this set of recommendations is to curb the use of topical steroids.
“Topical steroids should be used sparingly and as monotherapy for a very short period of time,” Dr. Lovell said. “Any guidelines agree that if eye inflammation is still present at 3 months, we need to move beyond topical steroid monotherapy.”
These new recommendations from MIWGUC are fairly consistent with the 2019 ACR/Arthritis Foundation guideline, he noted.
“The differences are very minor,” Dr. Lovell said. “In both instances, systemic corticosteroids should be bridging therapy. If you have a patient who needs systemic corticosteroids in addition to topical at the same time, you should be talking about adding other anti-inflammatory treatments, such as traditional and/or biologic DMARDs. Both MIWGUC and the ACR guidelines agree on that.”
The 2019 ACR/Arthritis Foundation guideline did not mention rituximab as an option, nor Janus kinase (JAK) inhibitors, Dr. Lovell said, noting there was no literature on JAK inhibitors as a possible option for JIAU when the guideline was being formulated.
Both sets of guidelines point out that there is a dearth of literature with respect to determining the safe dose of maintenance topical corticosteroids, Dr. Lovell said.
“The ACR 2019 guidelines state you should add systemic therapy if there is persistent eye inflammation despite use of up to two drops per day of topical corticosteroids, while the European [MIWGUC] guideline states you can allow up to three drops,” he said. “In both instances, they are quoting the same two sources. Both guidelines indicate that the literature is very scant as to defining a true, safe dose of topical ocular corticosteroids. They differ by one drop allowed per day. In both instances, in the presence of active uveitis, at 3 months on topical steroid monotherapy, both [guidelines] strongly recommend adding systemic therapy.”
Marinka Twilt, MD, MSCE, PhD, associate professor in the department of pediatrics at the University of Calgary (Alta.), noted in an interview that these latest recommendations from MIWGUC have included consensus views on what to do if certain medications fail to lead to remission, which is not addressed in the 2019 ACR/Arthritis Foundation guideline.
“The new manuscript provides consensus on the use of abatacept, JAK inhibitors, and rituximab if patients are refractory to adalimumab and tocilizumab, which is not discussed in the 2019 recommendations,” Dr. Twilt said.
She also pointed out that these recommendations suggest adalimumab as treatment before infliximab, whereas the 2019 guideline did not recommend using one or the other first.
In compiling the recommendations, the authors received no outside financial support. Dr. Foeldvari is a member of advisory boards for Lilly, Pfizer, Novartis, and Medac. Dr. Lovell and Dr. Twilt disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Glucocorticoids should be bridging therapies in the treatment of juvenile idiopathic arthritis–associated uveitis (JIAU) and idiopathic chronic anterior uveitis (CAU), according to recently released recommendations from the Multinational Interdisciplinary Working Group for Uveitis in Childhood (MIWGUC).
The recommendations cover literature from December 2014 to June 2020 and represent an update of previously published treatment guidelines from 2018. The MIWGUC work group that formulated the new recommendations consisted of eight pediatric rheumatologists and eight ophthalmologists with expertise in pediatric uveitis.
One major shift from the previous guidelines is the lack of distinction between JIAU and CAU, said lead author Ivan Foeldvari, MD, head of the Hamburg (Germany) Center for Pediatric and Adolescent Rheumatology.
“We are considering these two conditions equivalent regarding the ophthalmological presentation,” Dr. Foeldvari said in an interview.
These guidelines have also expanded possible treatment options for these conditions in light of data from clinical trials that have pointed to new options, Dr. Foeldvari noted.
The guidelines also present new options, compared with the 2019 American College of Rheumatology/Arthritis Foundation JIA-associated uveitis guideline. The data cutoff for that guideline was 2014. “Many key papers were published since 2014,” Dr. Foeldvari said.
Another major change is in the escalation of therapy, he noted.
“We view glucocorticoids as a bridging agent, which is very important to emphasize,” Dr. Foeldvari said. “We do not want oral glucocorticoids used as a monotherapy. If you consider a child who has severe uveitis and you want to give an oral glucocorticoid treatment, then it should be considered only for bridging. We suggest to start a DMARD [disease-modifying antirheumatic drug].”
The specific recommendation is that methotrexate be the first DMARD that clinicians choose after using glucocorticoids as a bridging therapy; adalimumab is recommended as the next treatment choice for patients who do not respond to methotrexate.
The working group also calls for limited use of topical glucocorticoids in the affected eye, he said.
“We recommend no more than two or three drops long term in the eye, because there are studies that show continuous local therapy is the main reason that children may develop blindness,” Dr. Foeldvari said. “With respect to oral corticosteroids, they have a lot of systemic effects. Those effects include a high risk of infection, weight gain, and growth disturbance.”
The new recommendations can guide treatment decisions for rheumatologists and ophthalmologists alike, according to Daniel J. Lovell, MD, MPH, the Joseph E. Levinson Endowed Chair of Pediatric Rheumatology and professor of pediatrics at the University of Cincinnati and Cincinnati Children’s Hospital Medical Center. He was one of the authors of the 2019 ACR/Arthritis Foundation guideline.
“We [rheumatologists] comanage these patients with ophthalmologists,” Dr. Lovell said in an interview. “Ophthalmologists are oftentimes not as experienced in using biologics or methotrexate in terms of monitoring for safety and dosing.”
Dr. Lovell pointed out that the key message from this set of recommendations is to curb the use of topical steroids.
“Topical steroids should be used sparingly and as monotherapy for a very short period of time,” Dr. Lovell said. “Any guidelines agree that if eye inflammation is still present at 3 months, we need to move beyond topical steroid monotherapy.”
These new recommendations from MIWGUC are fairly consistent with the 2019 ACR/Arthritis Foundation guideline, he noted.
“The differences are very minor,” Dr. Lovell said. “In both instances, systemic corticosteroids should be bridging therapy. If you have a patient who needs systemic corticosteroids in addition to topical at the same time, you should be talking about adding other anti-inflammatory treatments, such as traditional and/or biologic DMARDs. Both MIWGUC and the ACR guidelines agree on that.”
The 2019 ACR/Arthritis Foundation guideline did not mention rituximab as an option, nor Janus kinase (JAK) inhibitors, Dr. Lovell said, noting there was no literature on JAK inhibitors as a possible option for JIAU when the guideline was being formulated.
Both sets of guidelines point out that there is a dearth of literature with respect to determining the safe dose of maintenance topical corticosteroids, Dr. Lovell said.
“The ACR 2019 guidelines state you should add systemic therapy if there is persistent eye inflammation despite use of up to two drops per day of topical corticosteroids, while the European [MIWGUC] guideline states you can allow up to three drops,” he said. “In both instances, they are quoting the same two sources. Both guidelines indicate that the literature is very scant as to defining a true, safe dose of topical ocular corticosteroids. They differ by one drop allowed per day. In both instances, in the presence of active uveitis, at 3 months on topical steroid monotherapy, both [guidelines] strongly recommend adding systemic therapy.”
Marinka Twilt, MD, MSCE, PhD, associate professor in the department of pediatrics at the University of Calgary (Alta.), noted in an interview that these latest recommendations from MIWGUC have included consensus views on what to do if certain medications fail to lead to remission, which is not addressed in the 2019 ACR/Arthritis Foundation guideline.
“The new manuscript provides consensus on the use of abatacept, JAK inhibitors, and rituximab if patients are refractory to adalimumab and tocilizumab, which is not discussed in the 2019 recommendations,” Dr. Twilt said.
She also pointed out that these recommendations suggest adalimumab as treatment before infliximab, whereas the 2019 guideline did not recommend using one or the other first.
In compiling the recommendations, the authors received no outside financial support. Dr. Foeldvari is a member of advisory boards for Lilly, Pfizer, Novartis, and Medac. Dr. Lovell and Dr. Twilt disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ARTHRITIS CARE RESEARCH
Inebilizumab beneficial across genotypes in NMOSD
NATIONAL HARBOR, MD. – – including a common genetic variation linked to reduced response to anti-CD20 therapies, new research shows.
The phase 3 N-MOmentum Study previously showed safety and efficacy for inebilizumab over placebo in more than 200 adults with NMOSD.
A new analysis focused on participants who were carriers of either the F/F allele, which is known to reduce the effectiveness of certain monoclonal antibodies, or the rs396991 V-allele, which has not been associated with a reduced response.

Results showed no significant differences between the two carrier groups in NMOSD activity, including annual rates of new/enlarging T2 lesions, during the trial and up to 6 months after treatment with inebilizumab.
“These data illustrate how mechanistic precision in treatment design can help patients gain benefit from their regimen regardless of the genetic make-up of their immune systems,” coinvestigator Bruce Cree, MD, PhD, professor of clinical neurology at the University of California, San Francisco, Weill Institute for Neurosciences, said in a press release.
“The combination of efficacy, safety, and ease of administration with twice-yearly infusions make this product an excellent choice for first-line therapy in NMOSD,” Dr. Cree said.
The findings were presented at the annual meeting of the Consortium of Multiple Sclerosis Centers.
B-cell depletion
Inebilizumab has also been approved in China, Japan, and South Korea for the treatment of NMOSD, a rare and severe autoantibody-mediated disease of the central nervous system that includes NMO and related syndromes.
The drug’s B-cell depletion capability is credited with reducing inflammation, lesion formation, and astrocyte damage. The latter can cause severe effects in an NMOSD attack, affecting the optic nerve, spinal cord, and brain.
Manifestations can range from loss of vision to paralysis, loss of sensation, bladder and bowel dysfunction, nerve pain or respiratory failure. Attacks can also result in cumulative damage and disability, the researchers noted.
Results from the original double-blind trial of 230 adults with NMOSD showed that treatment with inebilizumab demonstrated efficacy and safety over placebo. However, questions have remained regarding the treatment’s effectiveness, specifically among patients with the FCGR3A (F/F) allele, a genetic variant that encodes the low-affinity Fc gamma receptor IIIa.
This genotype is known to reduce the effectiveness of certain monoclonal antibodies and anti-CD20 therapies, notably rituximab, in disorders such as NMOSD.
With up to 40% of White and Black individuals known to carry the F/F allele, inebilizumab was designed specifically with that risk in mind, with strong binding to the allele.
Although inebilizumab joins two other Food and Drug Administration–approved treatments for NMOSD – eculizumab and satralizumab – neither of those have a mechanism involving the FCGRA3 receptor. Therefore, those drugs are not a concern for individuals with those genotypes.
To evaluate inebilizumab’s effects among patients with the F/F allele, Dr. Cree and colleagues assessed data on a subset of 142 patients from the N-MOmentum trial.
The study included a 28-week randomized controlled period in which adults with NMOSD received either 300 mg of intravenous inebilizumab or placebo on days 1 and 15, followed by an optional open-label period of at least 2 years. During the open-label phase, all patients received 300 mg of IV inebilizumab every 26 weeks.
Of the 142 patients in the genetic analysis, 104 received inebilizumab and 38 received placebo. In addition, 68 group participants were carriers of the F/F allele, while 74 carried the rs396991 V-allele.
No significant differences
Prior to the trial, annualized attack rates (AARs) and disability, as assessed by change in the Expanded Disability Status Scale (EDSS) scores, were nominally higher in the V allele group from disease onset to trial enrollment.
During the trial’s first 6 months, AARs and annual rates of new/enlarging T2 lesions were nominally lower in inebilizumab-treated V allele participants compared with the F/F allele participants, although the differences were not statistically significant.
The AAR was 0.1 for the V allele group vs. 0.3 for the F/F allele group (hazard ratio, 0.40; P = .17). The annual rate of new/enlarging T2 lesions was 1.4 vs. 1.7 (risk ratio, 0.91; P = .88), respectively.
However, at the end of the randomized controlled period, there were no significant differences between the two genotype groups. There was also little difference in clinical metrics of NMOSD activity or B-cell depletion between the two genotype groups during the open-label period involving the long-term repeated inebilizumab dosing.
“Though greater B-cell depletion was observed in inebilizumab-treated V allele participants, compared with F/F participants during the first 6 months, no significant difference in NMOSD activity was observed during this time period,” the investigators reported.
“No differences in B-cell depletion or NMOSD disease activity were observed after 6 months of inebilizumab treatment,” they added.
Dr. Cree noted the study showed that, overall, inebiluzumab’s efficacy was not adversely affected by a polymorphism in the Fc gamma receptor. “These types of genetic analyses may help inform future screening mechanisms to tailor treatment strategies that can optimize the response rate for each patient,” he said.
Dr. Cree added the higher degree of disease activity among those carrying the alleles at baseline is notable and deserves further investigation. That finding “suggests that the presence of the F/F allele is to some extent protective of the detrimental effects the auto-antibody directed against aquaporin-4 that underlies NMOSD pathogenesis,” he said.
A new era?
Commenting on the study, Marcelo Matiello, MD, assistant professor of neurology at Harvard Medical School and associate director of the Neuromyelitis Optica clinic at Massachusetts General Hospital, both in Boston, said the findings provide valuable insights into risks for key patient subgroups.
“The data is quite important because we know that with other conditions, such as rheumatoid arthritis, people with this particular genotype do have lower response and are more likely to be refractory,” said Dr. Matiello, who was not involved with the research.
He noted that rituximab is the most commonly used medication in the United States for NMOSD. “It’s not FDA approved, but because of extensive experience, and many case series and small prospective studies, most NMO patients are using rituximab,” Dr. Matiello said. However, the drug’s mechanism “can be compromised” by the F/F allele, he added.
The new findings “provide a good understanding that this medication would likely be superior to patients with this genotype,” he said.
“I think it’s a new era,” Dr. Matiello added. “Not only do we have approved medication for this very severe disease, but we can find out who can benefit most. So, I think this is exciting and is a major step in more individualized appropriate use.”
The study was funded by Horizon Therapeutics. Dr. Cree has consulted for Horizon Therapeutics. Dr. Matiello reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
NATIONAL HARBOR, MD. – – including a common genetic variation linked to reduced response to anti-CD20 therapies, new research shows.
The phase 3 N-MOmentum Study previously showed safety and efficacy for inebilizumab over placebo in more than 200 adults with NMOSD.
A new analysis focused on participants who were carriers of either the F/F allele, which is known to reduce the effectiveness of certain monoclonal antibodies, or the rs396991 V-allele, which has not been associated with a reduced response.
Results showed no significant differences between the two carrier groups in NMOSD activity, including annual rates of new/enlarging T2 lesions, during the trial and up to 6 months after treatment with inebilizumab.
“These data illustrate how mechanistic precision in treatment design can help patients gain benefit from their regimen regardless of the genetic make-up of their immune systems,” coinvestigator Bruce Cree, MD, PhD, professor of clinical neurology at the University of California, San Francisco, Weill Institute for Neurosciences, said in a press release.
“The combination of efficacy, safety, and ease of administration with twice-yearly infusions make this product an excellent choice for first-line therapy in NMOSD,” Dr. Cree said.
The findings were presented at the annual meeting of the Consortium of Multiple Sclerosis Centers.
B-cell depletion
Inebilizumab has also been approved in China, Japan, and South Korea for the treatment of NMOSD, a rare and severe autoantibody-mediated disease of the central nervous system that includes NMO and related syndromes.
The drug’s B-cell depletion capability is credited with reducing inflammation, lesion formation, and astrocyte damage. The latter can cause severe effects in an NMOSD attack, affecting the optic nerve, spinal cord, and brain.
Manifestations can range from loss of vision to paralysis, loss of sensation, bladder and bowel dysfunction, nerve pain or respiratory failure. Attacks can also result in cumulative damage and disability, the researchers noted.
Results from the original double-blind trial of 230 adults with NMOSD showed that treatment with inebilizumab demonstrated efficacy and safety over placebo. However, questions have remained regarding the treatment’s effectiveness, specifically among patients with the FCGR3A (F/F) allele, a genetic variant that encodes the low-affinity Fc gamma receptor IIIa.
This genotype is known to reduce the effectiveness of certain monoclonal antibodies and anti-CD20 therapies, notably rituximab, in disorders such as NMOSD.
With up to 40% of White and Black individuals known to carry the F/F allele, inebilizumab was designed specifically with that risk in mind, with strong binding to the allele.
Although inebilizumab joins two other Food and Drug Administration–approved treatments for NMOSD – eculizumab and satralizumab – neither of those have a mechanism involving the FCGRA3 receptor. Therefore, those drugs are not a concern for individuals with those genotypes.
To evaluate inebilizumab’s effects among patients with the F/F allele, Dr. Cree and colleagues assessed data on a subset of 142 patients from the N-MOmentum trial.
The study included a 28-week randomized controlled period in which adults with NMOSD received either 300 mg of intravenous inebilizumab or placebo on days 1 and 15, followed by an optional open-label period of at least 2 years. During the open-label phase, all patients received 300 mg of IV inebilizumab every 26 weeks.
Of the 142 patients in the genetic analysis, 104 received inebilizumab and 38 received placebo. In addition, 68 group participants were carriers of the F/F allele, while 74 carried the rs396991 V-allele.
No significant differences
Prior to the trial, annualized attack rates (AARs) and disability, as assessed by change in the Expanded Disability Status Scale (EDSS) scores, were nominally higher in the V allele group from disease onset to trial enrollment.
During the trial’s first 6 months, AARs and annual rates of new/enlarging T2 lesions were nominally lower in inebilizumab-treated V allele participants compared with the F/F allele participants, although the differences were not statistically significant.
The AAR was 0.1 for the V allele group vs. 0.3 for the F/F allele group (hazard ratio, 0.40; P = .17). The annual rate of new/enlarging T2 lesions was 1.4 vs. 1.7 (risk ratio, 0.91; P = .88), respectively.
However, at the end of the randomized controlled period, there were no significant differences between the two genotype groups. There was also little difference in clinical metrics of NMOSD activity or B-cell depletion between the two genotype groups during the open-label period involving the long-term repeated inebilizumab dosing.
“Though greater B-cell depletion was observed in inebilizumab-treated V allele participants, compared with F/F participants during the first 6 months, no significant difference in NMOSD activity was observed during this time period,” the investigators reported.
“No differences in B-cell depletion or NMOSD disease activity were observed after 6 months of inebilizumab treatment,” they added.
Dr. Cree noted the study showed that, overall, inebiluzumab’s efficacy was not adversely affected by a polymorphism in the Fc gamma receptor. “These types of genetic analyses may help inform future screening mechanisms to tailor treatment strategies that can optimize the response rate for each patient,” he said.
Dr. Cree added the higher degree of disease activity among those carrying the alleles at baseline is notable and deserves further investigation. That finding “suggests that the presence of the F/F allele is to some extent protective of the detrimental effects the auto-antibody directed against aquaporin-4 that underlies NMOSD pathogenesis,” he said.
A new era?
Commenting on the study, Marcelo Matiello, MD, assistant professor of neurology at Harvard Medical School and associate director of the Neuromyelitis Optica clinic at Massachusetts General Hospital, both in Boston, said the findings provide valuable insights into risks for key patient subgroups.
“The data is quite important because we know that with other conditions, such as rheumatoid arthritis, people with this particular genotype do have lower response and are more likely to be refractory,” said Dr. Matiello, who was not involved with the research.
He noted that rituximab is the most commonly used medication in the United States for NMOSD. “It’s not FDA approved, but because of extensive experience, and many case series and small prospective studies, most NMO patients are using rituximab,” Dr. Matiello said. However, the drug’s mechanism “can be compromised” by the F/F allele, he added.
The new findings “provide a good understanding that this medication would likely be superior to patients with this genotype,” he said.
“I think it’s a new era,” Dr. Matiello added. “Not only do we have approved medication for this very severe disease, but we can find out who can benefit most. So, I think this is exciting and is a major step in more individualized appropriate use.”
The study was funded by Horizon Therapeutics. Dr. Cree has consulted for Horizon Therapeutics. Dr. Matiello reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
NATIONAL HARBOR, MD. – – including a common genetic variation linked to reduced response to anti-CD20 therapies, new research shows.
The phase 3 N-MOmentum Study previously showed safety and efficacy for inebilizumab over placebo in more than 200 adults with NMOSD.
A new analysis focused on participants who were carriers of either the F/F allele, which is known to reduce the effectiveness of certain monoclonal antibodies, or the rs396991 V-allele, which has not been associated with a reduced response.
Results showed no significant differences between the two carrier groups in NMOSD activity, including annual rates of new/enlarging T2 lesions, during the trial and up to 6 months after treatment with inebilizumab.
“These data illustrate how mechanistic precision in treatment design can help patients gain benefit from their regimen regardless of the genetic make-up of their immune systems,” coinvestigator Bruce Cree, MD, PhD, professor of clinical neurology at the University of California, San Francisco, Weill Institute for Neurosciences, said in a press release.
“The combination of efficacy, safety, and ease of administration with twice-yearly infusions make this product an excellent choice for first-line therapy in NMOSD,” Dr. Cree said.
The findings were presented at the annual meeting of the Consortium of Multiple Sclerosis Centers.
B-cell depletion
Inebilizumab has also been approved in China, Japan, and South Korea for the treatment of NMOSD, a rare and severe autoantibody-mediated disease of the central nervous system that includes NMO and related syndromes.
The drug’s B-cell depletion capability is credited with reducing inflammation, lesion formation, and astrocyte damage. The latter can cause severe effects in an NMOSD attack, affecting the optic nerve, spinal cord, and brain.
Manifestations can range from loss of vision to paralysis, loss of sensation, bladder and bowel dysfunction, nerve pain or respiratory failure. Attacks can also result in cumulative damage and disability, the researchers noted.
Results from the original double-blind trial of 230 adults with NMOSD showed that treatment with inebilizumab demonstrated efficacy and safety over placebo. However, questions have remained regarding the treatment’s effectiveness, specifically among patients with the FCGR3A (F/F) allele, a genetic variant that encodes the low-affinity Fc gamma receptor IIIa.
This genotype is known to reduce the effectiveness of certain monoclonal antibodies and anti-CD20 therapies, notably rituximab, in disorders such as NMOSD.
With up to 40% of White and Black individuals known to carry the F/F allele, inebilizumab was designed specifically with that risk in mind, with strong binding to the allele.
Although inebilizumab joins two other Food and Drug Administration–approved treatments for NMOSD – eculizumab and satralizumab – neither of those have a mechanism involving the FCGRA3 receptor. Therefore, those drugs are not a concern for individuals with those genotypes.
To evaluate inebilizumab’s effects among patients with the F/F allele, Dr. Cree and colleagues assessed data on a subset of 142 patients from the N-MOmentum trial.
The study included a 28-week randomized controlled period in which adults with NMOSD received either 300 mg of intravenous inebilizumab or placebo on days 1 and 15, followed by an optional open-label period of at least 2 years. During the open-label phase, all patients received 300 mg of IV inebilizumab every 26 weeks.
Of the 142 patients in the genetic analysis, 104 received inebilizumab and 38 received placebo. In addition, 68 group participants were carriers of the F/F allele, while 74 carried the rs396991 V-allele.
No significant differences
Prior to the trial, annualized attack rates (AARs) and disability, as assessed by change in the Expanded Disability Status Scale (EDSS) scores, were nominally higher in the V allele group from disease onset to trial enrollment.
During the trial’s first 6 months, AARs and annual rates of new/enlarging T2 lesions were nominally lower in inebilizumab-treated V allele participants compared with the F/F allele participants, although the differences were not statistically significant.
The AAR was 0.1 for the V allele group vs. 0.3 for the F/F allele group (hazard ratio, 0.40; P = .17). The annual rate of new/enlarging T2 lesions was 1.4 vs. 1.7 (risk ratio, 0.91; P = .88), respectively.
However, at the end of the randomized controlled period, there were no significant differences between the two genotype groups. There was also little difference in clinical metrics of NMOSD activity or B-cell depletion between the two genotype groups during the open-label period involving the long-term repeated inebilizumab dosing.
“Though greater B-cell depletion was observed in inebilizumab-treated V allele participants, compared with F/F participants during the first 6 months, no significant difference in NMOSD activity was observed during this time period,” the investigators reported.
“No differences in B-cell depletion or NMOSD disease activity were observed after 6 months of inebilizumab treatment,” they added.
Dr. Cree noted the study showed that, overall, inebiluzumab’s efficacy was not adversely affected by a polymorphism in the Fc gamma receptor. “These types of genetic analyses may help inform future screening mechanisms to tailor treatment strategies that can optimize the response rate for each patient,” he said.
Dr. Cree added the higher degree of disease activity among those carrying the alleles at baseline is notable and deserves further investigation. That finding “suggests that the presence of the F/F allele is to some extent protective of the detrimental effects the auto-antibody directed against aquaporin-4 that underlies NMOSD pathogenesis,” he said.
A new era?
Commenting on the study, Marcelo Matiello, MD, assistant professor of neurology at Harvard Medical School and associate director of the Neuromyelitis Optica clinic at Massachusetts General Hospital, both in Boston, said the findings provide valuable insights into risks for key patient subgroups.
“The data is quite important because we know that with other conditions, such as rheumatoid arthritis, people with this particular genotype do have lower response and are more likely to be refractory,” said Dr. Matiello, who was not involved with the research.
He noted that rituximab is the most commonly used medication in the United States for NMOSD. “It’s not FDA approved, but because of extensive experience, and many case series and small prospective studies, most NMO patients are using rituximab,” Dr. Matiello said. However, the drug’s mechanism “can be compromised” by the F/F allele, he added.
The new findings “provide a good understanding that this medication would likely be superior to patients with this genotype,” he said.
“I think it’s a new era,” Dr. Matiello added. “Not only do we have approved medication for this very severe disease, but we can find out who can benefit most. So, I think this is exciting and is a major step in more individualized appropriate use.”
The study was funded by Horizon Therapeutics. Dr. Cree has consulted for Horizon Therapeutics. Dr. Matiello reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT CMSC 2022

FDA expands indication for spinal muscular atrophy drug
As previously reported, the FDA first approved oral risdiplam for SMA in children older than age 2 years in 2020.
The FDA expanded the indication for risdiplam to include babies younger than 2 months old because of interim safety and efficacy data from the ongoing RAINBOWFISH study. It includes 25 babies from birth to 6 weeks of age at first dose, all of whom have genetically diagnosed SMA but are not yet presenting with symptoms.
After 12 months of risdiplam treatment, the majority of presymptomatic infants with SMA reached key motor milestones, Genentech said in a news release.
Of the six babies with two or three copies of the SMN2 gene, all were able to sit after 1 year of active treatment, roughly two-thirds could stand, and half could walk independently.
All babies were alive at 12 months without permanent ventilation.
“The approval of Evrysdi for presymptomatic babies is particularly important, as early treatment of SMA, before symptoms start to arise, can help babies to achieve motor milestones,” Richard Finkel, MD, principal investigator of the trial, said in the release.
“With the inclusion of SMA in newborn screening programs, this approval provides the opportunity to start treating at home with Evrysdi soon after the diagnosis is confirmed,” added Dr. Finkel, who is director of the experimental neuroscience program, St. Jude Children’s Research Hospital, Memphis.
From newborns to older adults?
SMA is a rare and often fatal genetic disease that causes muscle weakness and progressive loss of movement.
SMA, which affects about 1 in 10,000 babies, is caused by a mutation in the survival motor neuron 1 (SMN1) gene. The gene encodes the SMN protein, which is critical for the maintenance and function of motor neurons.
Risdiplam is an orally administered, centrally and peripherally distributed small molecule that modulates survival motor neuron 2 (SMN2) premessenger RNA splicing to increase SMN protein levels.
As part of the label extension, the prescribing information for risdiplam has also been updated to include 2-year pooled data from parts 1 and 2 of the FIREFISH study, which demonstrated long-term efficacy and safety in symptomatic infants with Type 1 SMA, the company noted.
“Because of its efficacy in multiple settings, Evrysdi is now available for people with SMA, from presymptomatic newborns to older adults,” Levi Garraway, MD, PhD, chief medical officer and head of global product development at Genentech, said in the release.
“We are proud of this achievement, which has the potential to make a real difference to those living with SMA and their caregivers,” Dr. Garraway added.
A version of this article first appeared on Medscape.com.
As previously reported, the FDA first approved oral risdiplam for SMA in children older than age 2 years in 2020.
The FDA expanded the indication for risdiplam to include babies younger than 2 months old because of interim safety and efficacy data from the ongoing RAINBOWFISH study. It includes 25 babies from birth to 6 weeks of age at first dose, all of whom have genetically diagnosed SMA but are not yet presenting with symptoms.
After 12 months of risdiplam treatment, the majority of presymptomatic infants with SMA reached key motor milestones, Genentech said in a news release.
Of the six babies with two or three copies of the SMN2 gene, all were able to sit after 1 year of active treatment, roughly two-thirds could stand, and half could walk independently.
All babies were alive at 12 months without permanent ventilation.
“The approval of Evrysdi for presymptomatic babies is particularly important, as early treatment of SMA, before symptoms start to arise, can help babies to achieve motor milestones,” Richard Finkel, MD, principal investigator of the trial, said in the release.
“With the inclusion of SMA in newborn screening programs, this approval provides the opportunity to start treating at home with Evrysdi soon after the diagnosis is confirmed,” added Dr. Finkel, who is director of the experimental neuroscience program, St. Jude Children’s Research Hospital, Memphis.
From newborns to older adults?
SMA is a rare and often fatal genetic disease that causes muscle weakness and progressive loss of movement.
SMA, which affects about 1 in 10,000 babies, is caused by a mutation in the survival motor neuron 1 (SMN1) gene. The gene encodes the SMN protein, which is critical for the maintenance and function of motor neurons.
Risdiplam is an orally administered, centrally and peripherally distributed small molecule that modulates survival motor neuron 2 (SMN2) premessenger RNA splicing to increase SMN protein levels.
As part of the label extension, the prescribing information for risdiplam has also been updated to include 2-year pooled data from parts 1 and 2 of the FIREFISH study, which demonstrated long-term efficacy and safety in symptomatic infants with Type 1 SMA, the company noted.
“Because of its efficacy in multiple settings, Evrysdi is now available for people with SMA, from presymptomatic newborns to older adults,” Levi Garraway, MD, PhD, chief medical officer and head of global product development at Genentech, said in the release.
“We are proud of this achievement, which has the potential to make a real difference to those living with SMA and their caregivers,” Dr. Garraway added.
A version of this article first appeared on Medscape.com.
As previously reported, the FDA first approved oral risdiplam for SMA in children older than age 2 years in 2020.
The FDA expanded the indication for risdiplam to include babies younger than 2 months old because of interim safety and efficacy data from the ongoing RAINBOWFISH study. It includes 25 babies from birth to 6 weeks of age at first dose, all of whom have genetically diagnosed SMA but are not yet presenting with symptoms.
After 12 months of risdiplam treatment, the majority of presymptomatic infants with SMA reached key motor milestones, Genentech said in a news release.
Of the six babies with two or three copies of the SMN2 gene, all were able to sit after 1 year of active treatment, roughly two-thirds could stand, and half could walk independently.
All babies were alive at 12 months without permanent ventilation.
“The approval of Evrysdi for presymptomatic babies is particularly important, as early treatment of SMA, before symptoms start to arise, can help babies to achieve motor milestones,” Richard Finkel, MD, principal investigator of the trial, said in the release.
“With the inclusion of SMA in newborn screening programs, this approval provides the opportunity to start treating at home with Evrysdi soon after the diagnosis is confirmed,” added Dr. Finkel, who is director of the experimental neuroscience program, St. Jude Children’s Research Hospital, Memphis.
From newborns to older adults?
SMA is a rare and often fatal genetic disease that causes muscle weakness and progressive loss of movement.
SMA, which affects about 1 in 10,000 babies, is caused by a mutation in the survival motor neuron 1 (SMN1) gene. The gene encodes the SMN protein, which is critical for the maintenance and function of motor neurons.
Risdiplam is an orally administered, centrally and peripherally distributed small molecule that modulates survival motor neuron 2 (SMN2) premessenger RNA splicing to increase SMN protein levels.
As part of the label extension, the prescribing information for risdiplam has also been updated to include 2-year pooled data from parts 1 and 2 of the FIREFISH study, which demonstrated long-term efficacy and safety in symptomatic infants with Type 1 SMA, the company noted.
“Because of its efficacy in multiple settings, Evrysdi is now available for people with SMA, from presymptomatic newborns to older adults,” Levi Garraway, MD, PhD, chief medical officer and head of global product development at Genentech, said in the release.
“We are proud of this achievement, which has the potential to make a real difference to those living with SMA and their caregivers,” Dr. Garraway added.
A version of this article first appeared on Medscape.com.
Meet a miracle: Man with trisomy 13 to celebrate 20th birthday
Lloyd Tyler Rochez, born in 2002 with trisomy 13, a genetic disorder that can involve severe learning problems and health woes that affect nearly every organ.
Lloyd’s diagnosis was confirmed shortly after he was born, when his doctors noticed that his facial features weren’t measuring right for a baby of his size, he had an extra finger on his left hand, and his fingers were joined on the right. His heart was also on the right side of his chest instead of the left. When he had breathing issues, he was quickly rushed to the neonatal ICU (NICU) in the New York City hospital where he was born.
Ms. Nunez wasn’t sure exactly what was wrong with her newborn, but the next morning, a genetics expert came to her room to discuss her medical history and whether anyone in the family had Down syndrome. That same health care provider told her that the next step was to run some tests and do more bloodwork.
Four days later, when Ms. Nunez was told that Lloyd had trisomy 13 and was likely to live for only 2 weeks, she was unable to come to terms with the news.
“There was so much information being told to me at once,” recalls Ms. Nunez, now 42, who is also the mom of two daughters, ages 8 and 10. “I had just turned 22, and this was my first experience giving birth. I can’t even remember everything the doctors told me.”
But she does remember her doctor telling her something about faith.
“After he tried to explain trisomy 13 to me, the downside and the prognosis, at the end he said, ‘I don’t know if you believe in some supernatural being, but if you want to ask that someone for a miracle, I would advise you to do that. Pray for your miracle, and you may get it.’”
Prepared for the worst, Ms. Nunez, who now works from her Martinsburg, WV, home as a case manager for unaccompanied minors coming to the U.S., decided that she would commit to providing the best possible care for her new baby no matter how long he lived.
Thus began an incredible story of Lloyd defying all the odds. While he stayed in the hospital for 2 weeks, his breathing soon began to stabilize, and he could eat by mouth. With that, he was discharged and allowed to go home.
“I was this inexperienced first-time mom who had been told to watch for all sorts of things, like making sure he didn’t turn blue at night,” she says. “I spent so many sleepless nights, but I was dedicated to Lloyd.”
Then, when Lloyd was 6 months old, Ms. Nunez made another important choice.
“I decided that I wasn’t going to live each day as if he was going to die,” she says. “I decided, instead, to enjoy him every day.”
But many health complications still came about, including a serious intestine issue at 8 months, at which point Lloyd’s doctors suggested waiting until he was a year old to have surgery.
Lloyd was able to get through the procedure, but while he was in the recovery room, he stopped breathing.
“I started screaming ‘my son is dying,’” Ms. Nunez recalls. “The nurses put me in a room, and I think I was in there for 10 minutes, but it felt like an eternity of me screaming.”She soon learned that Lloyd had had a seizure. He spent the next 3 weeks in the hospital.
“That was our life,” she says. “He would have respiratory pneumonia, for example, and we would go back to the hospital. We were in and out and in and out.”
But she kept the faith, and since then, Lloyd’s health has mostly stabilized. Ms. Nunez can care for him at home on her own and with family members who help out from time to time.
And, while Lloyd is unable to speak, he smiles and laughs when he’s happy, he’s quiet when he feels ill, and, when he wants to be alone, he groans, Ms. Nunez says. He can stand up, and he crawls from place to place. He also can’t go to the bathroom on his own and is fed by a gastrostomy tube, or G-tube.
In December, when Lloyd was diagnosed with COVID-19, Ms. Nunez started worrying all over again.
“Seeing him in the ICU, all I could think of was ‘please don’t make my son suffer,’” she says. “If he goes, I want him to go in peace, and I don’t want to see him in a machine and suffering.”
But Lloyd once again defied the odds against him and came home again. He has since faced yet another health challenge: He recently had a pelvic fracture.
“When I saw the orthopedist, he told me that Lloyd has a bone deficiency and that his bones don’t have enough room to grow,” she says. “I’m afraid this will be the beginning of a new journey.”
How this mom finds strength
While Ms. Nunez doesn’t go to a support group or speak with a mental health professional about all that she’s juggling, she says she draws strength from Lloyd himself.
“I’m very private, and I come from a culture where you don’t want people feeling sorry for you,” she says. “But I want to give Lloyd everything – he goes to school, we go to church, he had a quinceañera when he was 15, we’ve been to Disney, and we’ve both gotten on a roller coaster. I haven’t limited his life.”
She also draws comfort from her daughters.
“Everyone calls him ‘Baby Lloyd,’” she says. “My girls come right home from school, wash their hands, and throw themselves on his bed and watch TV with him. They also worry about him a lot. When he goes to the hospital, they suffer more than I do.”
In the end, Ms. Nunez hopes her story inspires others to think beyond a prognosis.
“Don’t lose hope,” she says. “I want people to feel hopeful when they read about Lloyd. He’s going to be 20 years old, and no one ever believed he would be here today ... I feel blessed.”
A version of this article first appeared on WebMD.com.
Lloyd Tyler Rochez, born in 2002 with trisomy 13, a genetic disorder that can involve severe learning problems and health woes that affect nearly every organ.
Lloyd’s diagnosis was confirmed shortly after he was born, when his doctors noticed that his facial features weren’t measuring right for a baby of his size, he had an extra finger on his left hand, and his fingers were joined on the right. His heart was also on the right side of his chest instead of the left. When he had breathing issues, he was quickly rushed to the neonatal ICU (NICU) in the New York City hospital where he was born.
Ms. Nunez wasn’t sure exactly what was wrong with her newborn, but the next morning, a genetics expert came to her room to discuss her medical history and whether anyone in the family had Down syndrome. That same health care provider told her that the next step was to run some tests and do more bloodwork.
Four days later, when Ms. Nunez was told that Lloyd had trisomy 13 and was likely to live for only 2 weeks, she was unable to come to terms with the news.
“There was so much information being told to me at once,” recalls Ms. Nunez, now 42, who is also the mom of two daughters, ages 8 and 10. “I had just turned 22, and this was my first experience giving birth. I can’t even remember everything the doctors told me.”
But she does remember her doctor telling her something about faith.
“After he tried to explain trisomy 13 to me, the downside and the prognosis, at the end he said, ‘I don’t know if you believe in some supernatural being, but if you want to ask that someone for a miracle, I would advise you to do that. Pray for your miracle, and you may get it.’”
Prepared for the worst, Ms. Nunez, who now works from her Martinsburg, WV, home as a case manager for unaccompanied minors coming to the U.S., decided that she would commit to providing the best possible care for her new baby no matter how long he lived.
Thus began an incredible story of Lloyd defying all the odds. While he stayed in the hospital for 2 weeks, his breathing soon began to stabilize, and he could eat by mouth. With that, he was discharged and allowed to go home.
“I was this inexperienced first-time mom who had been told to watch for all sorts of things, like making sure he didn’t turn blue at night,” she says. “I spent so many sleepless nights, but I was dedicated to Lloyd.”
Then, when Lloyd was 6 months old, Ms. Nunez made another important choice.
“I decided that I wasn’t going to live each day as if he was going to die,” she says. “I decided, instead, to enjoy him every day.”
But many health complications still came about, including a serious intestine issue at 8 months, at which point Lloyd’s doctors suggested waiting until he was a year old to have surgery.
Lloyd was able to get through the procedure, but while he was in the recovery room, he stopped breathing.
“I started screaming ‘my son is dying,’” Ms. Nunez recalls. “The nurses put me in a room, and I think I was in there for 10 minutes, but it felt like an eternity of me screaming.”She soon learned that Lloyd had had a seizure. He spent the next 3 weeks in the hospital.
“That was our life,” she says. “He would have respiratory pneumonia, for example, and we would go back to the hospital. We were in and out and in and out.”
But she kept the faith, and since then, Lloyd’s health has mostly stabilized. Ms. Nunez can care for him at home on her own and with family members who help out from time to time.
And, while Lloyd is unable to speak, he smiles and laughs when he’s happy, he’s quiet when he feels ill, and, when he wants to be alone, he groans, Ms. Nunez says. He can stand up, and he crawls from place to place. He also can’t go to the bathroom on his own and is fed by a gastrostomy tube, or G-tube.
In December, when Lloyd was diagnosed with COVID-19, Ms. Nunez started worrying all over again.
“Seeing him in the ICU, all I could think of was ‘please don’t make my son suffer,’” she says. “If he goes, I want him to go in peace, and I don’t want to see him in a machine and suffering.”
But Lloyd once again defied the odds against him and came home again. He has since faced yet another health challenge: He recently had a pelvic fracture.
“When I saw the orthopedist, he told me that Lloyd has a bone deficiency and that his bones don’t have enough room to grow,” she says. “I’m afraid this will be the beginning of a new journey.”
How this mom finds strength
While Ms. Nunez doesn’t go to a support group or speak with a mental health professional about all that she’s juggling, she says she draws strength from Lloyd himself.
“I’m very private, and I come from a culture where you don’t want people feeling sorry for you,” she says. “But I want to give Lloyd everything – he goes to school, we go to church, he had a quinceañera when he was 15, we’ve been to Disney, and we’ve both gotten on a roller coaster. I haven’t limited his life.”
She also draws comfort from her daughters.
“Everyone calls him ‘Baby Lloyd,’” she says. “My girls come right home from school, wash their hands, and throw themselves on his bed and watch TV with him. They also worry about him a lot. When he goes to the hospital, they suffer more than I do.”
In the end, Ms. Nunez hopes her story inspires others to think beyond a prognosis.
“Don’t lose hope,” she says. “I want people to feel hopeful when they read about Lloyd. He’s going to be 20 years old, and no one ever believed he would be here today ... I feel blessed.”
A version of this article first appeared on WebMD.com.
Lloyd Tyler Rochez, born in 2002 with trisomy 13, a genetic disorder that can involve severe learning problems and health woes that affect nearly every organ.
Lloyd’s diagnosis was confirmed shortly after he was born, when his doctors noticed that his facial features weren’t measuring right for a baby of his size, he had an extra finger on his left hand, and his fingers were joined on the right. His heart was also on the right side of his chest instead of the left. When he had breathing issues, he was quickly rushed to the neonatal ICU (NICU) in the New York City hospital where he was born.
Ms. Nunez wasn’t sure exactly what was wrong with her newborn, but the next morning, a genetics expert came to her room to discuss her medical history and whether anyone in the family had Down syndrome. That same health care provider told her that the next step was to run some tests and do more bloodwork.
Four days later, when Ms. Nunez was told that Lloyd had trisomy 13 and was likely to live for only 2 weeks, she was unable to come to terms with the news.
“There was so much information being told to me at once,” recalls Ms. Nunez, now 42, who is also the mom of two daughters, ages 8 and 10. “I had just turned 22, and this was my first experience giving birth. I can’t even remember everything the doctors told me.”
But she does remember her doctor telling her something about faith.
“After he tried to explain trisomy 13 to me, the downside and the prognosis, at the end he said, ‘I don’t know if you believe in some supernatural being, but if you want to ask that someone for a miracle, I would advise you to do that. Pray for your miracle, and you may get it.’”
Prepared for the worst, Ms. Nunez, who now works from her Martinsburg, WV, home as a case manager for unaccompanied minors coming to the U.S., decided that she would commit to providing the best possible care for her new baby no matter how long he lived.
Thus began an incredible story of Lloyd defying all the odds. While he stayed in the hospital for 2 weeks, his breathing soon began to stabilize, and he could eat by mouth. With that, he was discharged and allowed to go home.
“I was this inexperienced first-time mom who had been told to watch for all sorts of things, like making sure he didn’t turn blue at night,” she says. “I spent so many sleepless nights, but I was dedicated to Lloyd.”
Then, when Lloyd was 6 months old, Ms. Nunez made another important choice.
“I decided that I wasn’t going to live each day as if he was going to die,” she says. “I decided, instead, to enjoy him every day.”
But many health complications still came about, including a serious intestine issue at 8 months, at which point Lloyd’s doctors suggested waiting until he was a year old to have surgery.
Lloyd was able to get through the procedure, but while he was in the recovery room, he stopped breathing.
“I started screaming ‘my son is dying,’” Ms. Nunez recalls. “The nurses put me in a room, and I think I was in there for 10 minutes, but it felt like an eternity of me screaming.”She soon learned that Lloyd had had a seizure. He spent the next 3 weeks in the hospital.
“That was our life,” she says. “He would have respiratory pneumonia, for example, and we would go back to the hospital. We were in and out and in and out.”
But she kept the faith, and since then, Lloyd’s health has mostly stabilized. Ms. Nunez can care for him at home on her own and with family members who help out from time to time.
And, while Lloyd is unable to speak, he smiles and laughs when he’s happy, he’s quiet when he feels ill, and, when he wants to be alone, he groans, Ms. Nunez says. He can stand up, and he crawls from place to place. He also can’t go to the bathroom on his own and is fed by a gastrostomy tube, or G-tube.
In December, when Lloyd was diagnosed with COVID-19, Ms. Nunez started worrying all over again.
“Seeing him in the ICU, all I could think of was ‘please don’t make my son suffer,’” she says. “If he goes, I want him to go in peace, and I don’t want to see him in a machine and suffering.”
But Lloyd once again defied the odds against him and came home again. He has since faced yet another health challenge: He recently had a pelvic fracture.
“When I saw the orthopedist, he told me that Lloyd has a bone deficiency and that his bones don’t have enough room to grow,” she says. “I’m afraid this will be the beginning of a new journey.”
How this mom finds strength
While Ms. Nunez doesn’t go to a support group or speak with a mental health professional about all that she’s juggling, she says she draws strength from Lloyd himself.
“I’m very private, and I come from a culture where you don’t want people feeling sorry for you,” she says. “But I want to give Lloyd everything – he goes to school, we go to church, he had a quinceañera when he was 15, we’ve been to Disney, and we’ve both gotten on a roller coaster. I haven’t limited his life.”
She also draws comfort from her daughters.
“Everyone calls him ‘Baby Lloyd,’” she says. “My girls come right home from school, wash their hands, and throw themselves on his bed and watch TV with him. They also worry about him a lot. When he goes to the hospital, they suffer more than I do.”
In the end, Ms. Nunez hopes her story inspires others to think beyond a prognosis.
“Don’t lose hope,” she says. “I want people to feel hopeful when they read about Lloyd. He’s going to be 20 years old, and no one ever believed he would be here today ... I feel blessed.”
A version of this article first appeared on WebMD.com.
CDC updates guidelines for hepatitis outbreak among children
The Centers for Disease Control and Prevention updated its recommendations for doctors and public health officials regarding the unusual outbreak of acute hepatitis among children.
As of May 5, the CDC and state health departments are investigating 109 children with hepatitis of unknown origin across 25 states and territories.
More than half have tested positive for adenovirus, the CDC said. More than 90% have been hospitalized, and 14% have had liver transplants. Five deaths are under investigation.
This week’s CDC alert provides updated recommendations for testing, given the potential association between adenovirus infection and pediatric hepatitis, or liver inflammation.
“Clinicians are recommended to consider adenovirus testing for patients with hepatitis of unknown etiology and to report such cases to their state or jurisdictional public health authorities,” the CDC said.
Doctors should also consider collecting a blood sample, respiratory sample, and stool sample. They may also collect liver tissue if a biopsy occurred or an autopsy is available.
In November 2021, clinicians at a large children’s hospital in Alabama notified the CDC about five pediatric patients with significant liver injury, including three with acute liver failure, who also tested positive for adenovirus. All children were previously healthy, and none had COVID-19, according to a CDC alert in April.
Four additional pediatric patients with hepatitis and adenovirus infection were identified. After lab testing found adenovirus infection in all nine patients in the initial cluster, public health officials began investigating a possible association between pediatric hepatitis and adenovirus. Among the five specimens that could be sequenced, they were all adenovirus type 41.
Unexplained hepatitis cases have been reported in children worldwide, reaching 450 cases and 11 deaths, according to the latest update from the European Centre for Disease Prevention and Control.
The cases have been reported in more than two dozen countries around the world, with 14 countries reporting more than five cases. The United Kingdom and the United States have reported the largest case counts so far.
In the United Kingdom, officials have identified 163 cases in children under age 16 years, including 11 that required liver transplants.
In the European Union, 14 countries have reported 106 cases collectively, with Italy reporting 35 cases and Spain reporting 22 cases. Outside of the European Union, Brazil has reported 16, Indonesia has reported 15, and Israel has reported 12.
Among the 11 deaths reported globally, the Uniyed States has reported five, Indonesia has reported five, and Palestine has reported one.
The cause of severe hepatitis remains a mystery, according to Ars Technica. Some cases have been identified retrospectively, dating back to the beginning of October 2021.
About 70% of the cases that have been tested for an adenovirus have tested positive, and subtype testing continues to show adenovirus type 41. The cases don’t appear to be linked to common causes, such as hepatitis viruses A, B, C, D, or E, which can cause liver inflammation and injury.
Adenoviruses aren’t known to cause hepatitis in healthy children, though the viruses have been linked to liver damage in children with compromised immune systems, according to Ars Technica. Adenoviruses typically cause respiratory infections in children, although type 41 tends to cause gastrointestinal illness.
“At present, the leading hypotheses remain those which involve adenovirus,” Philippa Easterbrook, a senior scientist at the WHO, said May 10 during a press briefing.
“I think [there’s] also still an important consideration about the role of COVID as well, either as a co-infection or as a past infection,” she said.
WHO officials expect data within a week from U.K. cases, Ms. Easterbrook said, which may indicate whether the adenovirus is an incidental infection or a more direct cause.
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention updated its recommendations for doctors and public health officials regarding the unusual outbreak of acute hepatitis among children.
As of May 5, the CDC and state health departments are investigating 109 children with hepatitis of unknown origin across 25 states and territories.
More than half have tested positive for adenovirus, the CDC said. More than 90% have been hospitalized, and 14% have had liver transplants. Five deaths are under investigation.
This week’s CDC alert provides updated recommendations for testing, given the potential association between adenovirus infection and pediatric hepatitis, or liver inflammation.
“Clinicians are recommended to consider adenovirus testing for patients with hepatitis of unknown etiology and to report such cases to their state or jurisdictional public health authorities,” the CDC said.
Doctors should also consider collecting a blood sample, respiratory sample, and stool sample. They may also collect liver tissue if a biopsy occurred or an autopsy is available.
In November 2021, clinicians at a large children’s hospital in Alabama notified the CDC about five pediatric patients with significant liver injury, including three with acute liver failure, who also tested positive for adenovirus. All children were previously healthy, and none had COVID-19, according to a CDC alert in April.
Four additional pediatric patients with hepatitis and adenovirus infection were identified. After lab testing found adenovirus infection in all nine patients in the initial cluster, public health officials began investigating a possible association between pediatric hepatitis and adenovirus. Among the five specimens that could be sequenced, they were all adenovirus type 41.
Unexplained hepatitis cases have been reported in children worldwide, reaching 450 cases and 11 deaths, according to the latest update from the European Centre for Disease Prevention and Control.
The cases have been reported in more than two dozen countries around the world, with 14 countries reporting more than five cases. The United Kingdom and the United States have reported the largest case counts so far.
In the United Kingdom, officials have identified 163 cases in children under age 16 years, including 11 that required liver transplants.
In the European Union, 14 countries have reported 106 cases collectively, with Italy reporting 35 cases and Spain reporting 22 cases. Outside of the European Union, Brazil has reported 16, Indonesia has reported 15, and Israel has reported 12.
Among the 11 deaths reported globally, the Uniyed States has reported five, Indonesia has reported five, and Palestine has reported one.
The cause of severe hepatitis remains a mystery, according to Ars Technica. Some cases have been identified retrospectively, dating back to the beginning of October 2021.
About 70% of the cases that have been tested for an adenovirus have tested positive, and subtype testing continues to show adenovirus type 41. The cases don’t appear to be linked to common causes, such as hepatitis viruses A, B, C, D, or E, which can cause liver inflammation and injury.
Adenoviruses aren’t known to cause hepatitis in healthy children, though the viruses have been linked to liver damage in children with compromised immune systems, according to Ars Technica. Adenoviruses typically cause respiratory infections in children, although type 41 tends to cause gastrointestinal illness.
“At present, the leading hypotheses remain those which involve adenovirus,” Philippa Easterbrook, a senior scientist at the WHO, said May 10 during a press briefing.
“I think [there’s] also still an important consideration about the role of COVID as well, either as a co-infection or as a past infection,” she said.
WHO officials expect data within a week from U.K. cases, Ms. Easterbrook said, which may indicate whether the adenovirus is an incidental infection or a more direct cause.
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention updated its recommendations for doctors and public health officials regarding the unusual outbreak of acute hepatitis among children.
As of May 5, the CDC and state health departments are investigating 109 children with hepatitis of unknown origin across 25 states and territories.
More than half have tested positive for adenovirus, the CDC said. More than 90% have been hospitalized, and 14% have had liver transplants. Five deaths are under investigation.
This week’s CDC alert provides updated recommendations for testing, given the potential association between adenovirus infection and pediatric hepatitis, or liver inflammation.
“Clinicians are recommended to consider adenovirus testing for patients with hepatitis of unknown etiology and to report such cases to their state or jurisdictional public health authorities,” the CDC said.
Doctors should also consider collecting a blood sample, respiratory sample, and stool sample. They may also collect liver tissue if a biopsy occurred or an autopsy is available.
In November 2021, clinicians at a large children’s hospital in Alabama notified the CDC about five pediatric patients with significant liver injury, including three with acute liver failure, who also tested positive for adenovirus. All children were previously healthy, and none had COVID-19, according to a CDC alert in April.
Four additional pediatric patients with hepatitis and adenovirus infection were identified. After lab testing found adenovirus infection in all nine patients in the initial cluster, public health officials began investigating a possible association between pediatric hepatitis and adenovirus. Among the five specimens that could be sequenced, they were all adenovirus type 41.
Unexplained hepatitis cases have been reported in children worldwide, reaching 450 cases and 11 deaths, according to the latest update from the European Centre for Disease Prevention and Control.
The cases have been reported in more than two dozen countries around the world, with 14 countries reporting more than five cases. The United Kingdom and the United States have reported the largest case counts so far.
In the United Kingdom, officials have identified 163 cases in children under age 16 years, including 11 that required liver transplants.
In the European Union, 14 countries have reported 106 cases collectively, with Italy reporting 35 cases and Spain reporting 22 cases. Outside of the European Union, Brazil has reported 16, Indonesia has reported 15, and Israel has reported 12.
Among the 11 deaths reported globally, the Uniyed States has reported five, Indonesia has reported five, and Palestine has reported one.
The cause of severe hepatitis remains a mystery, according to Ars Technica. Some cases have been identified retrospectively, dating back to the beginning of October 2021.
About 70% of the cases that have been tested for an adenovirus have tested positive, and subtype testing continues to show adenovirus type 41. The cases don’t appear to be linked to common causes, such as hepatitis viruses A, B, C, D, or E, which can cause liver inflammation and injury.
Adenoviruses aren’t known to cause hepatitis in healthy children, though the viruses have been linked to liver damage in children with compromised immune systems, according to Ars Technica. Adenoviruses typically cause respiratory infections in children, although type 41 tends to cause gastrointestinal illness.
“At present, the leading hypotheses remain those which involve adenovirus,” Philippa Easterbrook, a senior scientist at the WHO, said May 10 during a press briefing.
“I think [there’s] also still an important consideration about the role of COVID as well, either as a co-infection or as a past infection,” she said.
WHO officials expect data within a week from U.K. cases, Ms. Easterbrook said, which may indicate whether the adenovirus is an incidental infection or a more direct cause.
A version of this article first appeared on Medscape.com.