User login
Expected spike in acute flaccid myelitis did not occur in 2020
suggested researchers at the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
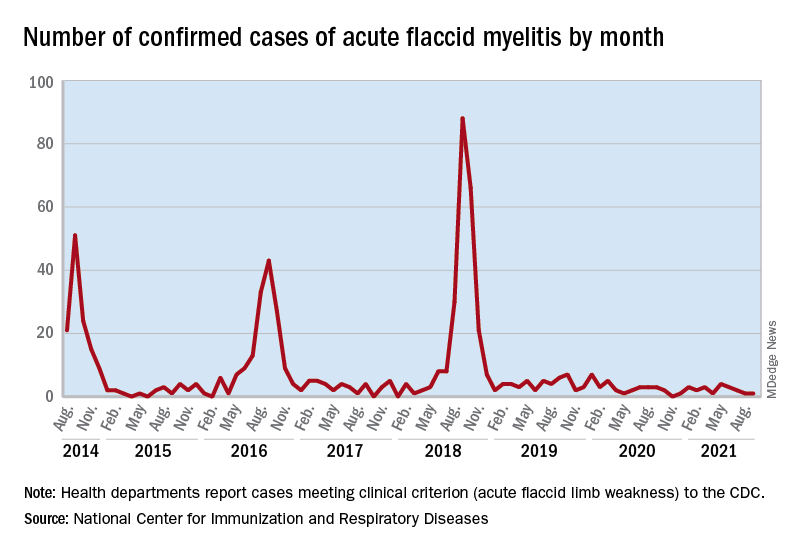
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
suggested researchers at the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
suggested researchers at the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
FROM MMWR
Which agent is best for neuromyelitis optica?
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.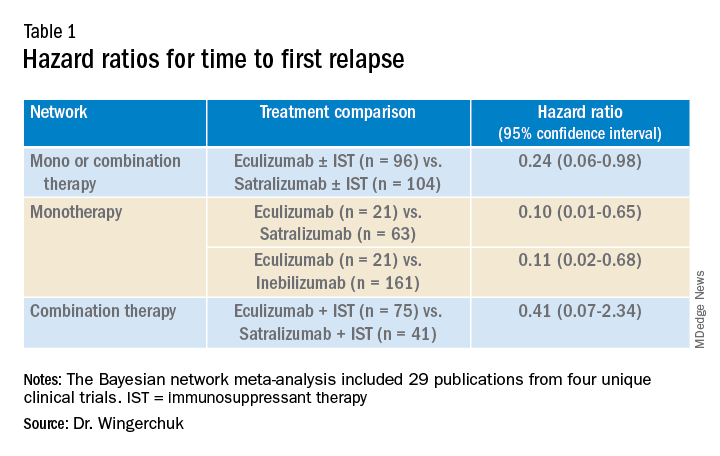
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).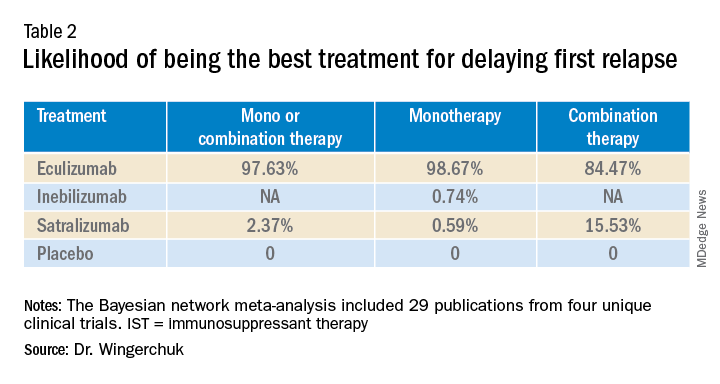
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ECTRIMS 2021
Evaluations of novel approaches to treating NF-1 tumors are underway
In the clinical experience of R. Rox Anderson, MD, currently available treatment options for benign tumors caused by neurofibromatosis type 1 (NF-1) are not acceptable.
“Simply removing the tumors with surgery is not the answer,” Dr. Anderson, a dermatologist who is the director of the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, said during a virtual course on laser and aesthetic skin therapy. “We need a way to inhibit the cutaneous neurofibromatosis early in life and prevent disfigurement that occurs when kids become adults.
“Kids with NF-1 are born looking normal,” he said. “They have café au lait macules and Lisch nodules in their eye, but they’re normal-looking kids. By early adulthood, many will grow hundreds of tumors that are disfiguring.”
In patients with NF-1, surgical excision works for cutaneous tumors but is expensive and not widely available, and is usually not covered by health insurance. “Plus, you have these adults who have already been through a lot of trauma, with the disfigurement in their lives, who have to be put under general anesthesia to remove a large number of tumors,” Dr. Anderson said at the meeting, which was named What’s the Truth and was sponsored by Harvard Medical School, Massachusetts General Hospital, and the Wellman Center for Photomedicine. Cryotherapy is a minimally invasive way to treat cutaneous neurofibroma tumors, “but this destroys the overlying skin, so you get unwanted destruction,” he said. “I like the idea of selecting heating, but we don’t know yet by what method.”
Dr. Anderson and his colleagues just launched a comparative clinical They plan to perform one or more treatment methods per patient in a single treatment session, then follow up at least 6 months later. Baseline and untreated cutaneous NF lesions will serve as controls. The researchers plan to conduct three-dimensional imaging, clinical assessments, and evaluate pain and other subjective measures.
Use of deoxycholate in a pilot trial was well tolerated and induced tumor regression in adults with cutaneous NF, he said.
Dr. Anderson noted that other researchers are studying the potential role of topical or local mitogen-activated protein kinase (MEK) inhibitors for these tumors. “Systemic MEK inhibitors are effective for plexiform neuromas, but cause significant cutaneous side effects,” he said. A “soft” MEK inhibitor, NFX-179 is rapidly metabolized such that high drug levels are achieved in skin without systemic drug levels. However, Dr. Anderson said that it remains unclear if this approach will prevent cutaneous NF tumors from forming, arrest their growth, or induce their regression.
Dr. Anderson reported having received research funding and/or consulting fees from numerous device and pharmaceutical companies.
Commentary by Lawrence F. Eichenfield, MD
Neurofibromatosis type 1 (NF1) is a common genodermatosis, associated with the development of neurofibromas derived from nerves, soft tissue, and skin. Cutaneous NFs often develop in later childhood onward and may be deforming, associated with pruritus, pain, and significant effect on quality of life. Dr. Anderson is a world leader in laser treatment, having developed the theories behind laser development for medical usage, as well as the laser technology used for vascular birthmarks and hair removal, laser and cooling techniques targeting fat, and “fractionating” laser energy, which has revolutionized scar management. We look forward to his group’s insights into better management of NF1 lesions!
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children's Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. He disclosed that he has served as an investigator and/or consultant to AbbVie, Lilly, Pfizer, Regeneron, Sanofi-Genzyme, and Verrica.
A version of this article first appeared on Medscape.com.
This article was updated 6/18/22.
In the clinical experience of R. Rox Anderson, MD, currently available treatment options for benign tumors caused by neurofibromatosis type 1 (NF-1) are not acceptable.
“Simply removing the tumors with surgery is not the answer,” Dr. Anderson, a dermatologist who is the director of the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, said during a virtual course on laser and aesthetic skin therapy. “We need a way to inhibit the cutaneous neurofibromatosis early in life and prevent disfigurement that occurs when kids become adults.
“Kids with NF-1 are born looking normal,” he said. “They have café au lait macules and Lisch nodules in their eye, but they’re normal-looking kids. By early adulthood, many will grow hundreds of tumors that are disfiguring.”
In patients with NF-1, surgical excision works for cutaneous tumors but is expensive and not widely available, and is usually not covered by health insurance. “Plus, you have these adults who have already been through a lot of trauma, with the disfigurement in their lives, who have to be put under general anesthesia to remove a large number of tumors,” Dr. Anderson said at the meeting, which was named What’s the Truth and was sponsored by Harvard Medical School, Massachusetts General Hospital, and the Wellman Center for Photomedicine. Cryotherapy is a minimally invasive way to treat cutaneous neurofibroma tumors, “but this destroys the overlying skin, so you get unwanted destruction,” he said. “I like the idea of selecting heating, but we don’t know yet by what method.”
Dr. Anderson and his colleagues just launched a comparative clinical They plan to perform one or more treatment methods per patient in a single treatment session, then follow up at least 6 months later. Baseline and untreated cutaneous NF lesions will serve as controls. The researchers plan to conduct three-dimensional imaging, clinical assessments, and evaluate pain and other subjective measures.
Use of deoxycholate in a pilot trial was well tolerated and induced tumor regression in adults with cutaneous NF, he said.
Dr. Anderson noted that other researchers are studying the potential role of topical or local mitogen-activated protein kinase (MEK) inhibitors for these tumors. “Systemic MEK inhibitors are effective for plexiform neuromas, but cause significant cutaneous side effects,” he said. A “soft” MEK inhibitor, NFX-179 is rapidly metabolized such that high drug levels are achieved in skin without systemic drug levels. However, Dr. Anderson said that it remains unclear if this approach will prevent cutaneous NF tumors from forming, arrest their growth, or induce their regression.
Dr. Anderson reported having received research funding and/or consulting fees from numerous device and pharmaceutical companies.
Commentary by Lawrence F. Eichenfield, MD
Neurofibromatosis type 1 (NF1) is a common genodermatosis, associated with the development of neurofibromas derived from nerves, soft tissue, and skin. Cutaneous NFs often develop in later childhood onward and may be deforming, associated with pruritus, pain, and significant effect on quality of life. Dr. Anderson is a world leader in laser treatment, having developed the theories behind laser development for medical usage, as well as the laser technology used for vascular birthmarks and hair removal, laser and cooling techniques targeting fat, and “fractionating” laser energy, which has revolutionized scar management. We look forward to his group’s insights into better management of NF1 lesions!
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children's Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. He disclosed that he has served as an investigator and/or consultant to AbbVie, Lilly, Pfizer, Regeneron, Sanofi-Genzyme, and Verrica.
A version of this article first appeared on Medscape.com.
This article was updated 6/18/22.
In the clinical experience of R. Rox Anderson, MD, currently available treatment options for benign tumors caused by neurofibromatosis type 1 (NF-1) are not acceptable.
“Simply removing the tumors with surgery is not the answer,” Dr. Anderson, a dermatologist who is the director of the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, said during a virtual course on laser and aesthetic skin therapy. “We need a way to inhibit the cutaneous neurofibromatosis early in life and prevent disfigurement that occurs when kids become adults.
“Kids with NF-1 are born looking normal,” he said. “They have café au lait macules and Lisch nodules in their eye, but they’re normal-looking kids. By early adulthood, many will grow hundreds of tumors that are disfiguring.”
In patients with NF-1, surgical excision works for cutaneous tumors but is expensive and not widely available, and is usually not covered by health insurance. “Plus, you have these adults who have already been through a lot of trauma, with the disfigurement in their lives, who have to be put under general anesthesia to remove a large number of tumors,” Dr. Anderson said at the meeting, which was named What’s the Truth and was sponsored by Harvard Medical School, Massachusetts General Hospital, and the Wellman Center for Photomedicine. Cryotherapy is a minimally invasive way to treat cutaneous neurofibroma tumors, “but this destroys the overlying skin, so you get unwanted destruction,” he said. “I like the idea of selecting heating, but we don’t know yet by what method.”
Dr. Anderson and his colleagues just launched a comparative clinical They plan to perform one or more treatment methods per patient in a single treatment session, then follow up at least 6 months later. Baseline and untreated cutaneous NF lesions will serve as controls. The researchers plan to conduct three-dimensional imaging, clinical assessments, and evaluate pain and other subjective measures.
Use of deoxycholate in a pilot trial was well tolerated and induced tumor regression in adults with cutaneous NF, he said.
Dr. Anderson noted that other researchers are studying the potential role of topical or local mitogen-activated protein kinase (MEK) inhibitors for these tumors. “Systemic MEK inhibitors are effective for plexiform neuromas, but cause significant cutaneous side effects,” he said. A “soft” MEK inhibitor, NFX-179 is rapidly metabolized such that high drug levels are achieved in skin without systemic drug levels. However, Dr. Anderson said that it remains unclear if this approach will prevent cutaneous NF tumors from forming, arrest their growth, or induce their regression.
Dr. Anderson reported having received research funding and/or consulting fees from numerous device and pharmaceutical companies.
Commentary by Lawrence F. Eichenfield, MD
Neurofibromatosis type 1 (NF1) is a common genodermatosis, associated with the development of neurofibromas derived from nerves, soft tissue, and skin. Cutaneous NFs often develop in later childhood onward and may be deforming, associated with pruritus, pain, and significant effect on quality of life. Dr. Anderson is a world leader in laser treatment, having developed the theories behind laser development for medical usage, as well as the laser technology used for vascular birthmarks and hair removal, laser and cooling techniques targeting fat, and “fractionating” laser energy, which has revolutionized scar management. We look forward to his group’s insights into better management of NF1 lesions!
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children's Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. He disclosed that he has served as an investigator and/or consultant to AbbVie, Lilly, Pfizer, Regeneron, Sanofi-Genzyme, and Verrica.
A version of this article first appeared on Medscape.com.
This article was updated 6/18/22.
FROM A LASER & AESTHETIC SKIN THERAPY COURSE
WHO unveils global roadmap to defeat meningitis by 2030
The World Health Organization and its partners recently released an ambitious plan, Defeating meningitis by 2030: A global road map. The goal is to reduce deaths and disabilities from bacterial meningitis, which kills about 250,000 people annually of the 1.2 million estimated to be infected.
This type of infection around the brain and spinal cord also causes long-term disabilities – deafness, learning problems, seizures, loss of limbs – in about one-quarter of survivors.
The leading causes of bacterial meningitis are Neisseria meningitidis (meningococcus), Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, and group B streptococcus. As with malaria, about half of the cases are in children under age 5 years. The most severely affected area for both infections is sub-Saharan Africa.
The main goal of the roadmap is to reduce vaccine-preventable cases of bacterial meningitis by 50% and deaths by 70% by 2030. WHO’s partners included the Centers for Disease Control and Prevention, the London School of Hygiene and Tropical Medicine, Médecins Sans Frontières (Doctors Without Borders), the Meningitis Research Foundation, PATH, UNICEF, and numerous global consultants.
For primary prevention and epidemic control, a major goal is to achieve higher vaccine coverage. Another goal is developing and deploying rapid diagnostic tests to guide treatment and prevention activities and measure the impact of vaccination. The lack of laboratory capacity to confirm the bacteria is a significant challenge. Also, patients often receive antibiotics before appropriate tests are conducted, and lumbar punctures are frequently not done.
The commitment to this project emerged in 2017. It was followed by a baseline analysis in 2018 and a draft roadmap the following year. Consultations with experts and with more than 600 patient groups in more than 90 countries followed.
Prevention through greater vaccine uptake was the top priority. Vaccination is considered the first line of defense against antibiotic resistance among the targeted bacteria.
Another goal is to quantify the decrease in antibiotic use for invasive infections or prophylaxis and the subsequent reduction in antimicrobial resistance in relation to increased vaccination.
Surveillance is weak in many regions, limiting the ability to detect epidemics and to respond appropriately. Similarly, there are limited data on the burden of sequelae, such as deafness, on meningitis survivors.
There is an inadequate supply of affordable vaccines to respond to epidemics. Currently, routine vaccination against Neisseria meningitidis is occurring in 18 of 26 countries in the meningitis belt. Epidemics of meningococcus occur every few years in the driest time of the year and abate with the rains. Epidemics of pneumococcal meningitis are much rarer but follow a similar pattern; they have also been associated with crowding and alcohol use.
Care for those affected by meningitis is another focus, as is affirming the right to prevention and care. There’s a need for earlier recognition of the complications of meningitis and an increase in efforts to treat those complications.
WHO’s final goal in its roadmap is to boost awareness of meningitis and make it a priority for policymakers. Similarly, there is a need to educate communities about the disease, including how to access vaccines. If someone becomes ill, they need to be aware of the symptoms, the need for early treatment, and what aftercare is available.
Marie-Pierre Préziosi, MD, the core secretariat of WHO’s Technical Taskforce, told this news organization that while the roadmap looks aspirational, “it is feasible … you have strategic goals – each has milestones with time limits and who will do it.”
Regarding vaccinations, Dr. Préziosi said that “the strategy was a victim of its success. The mass campaign knocked down transmission completely.” Some countries are now waiting for multivalent vaccines. She said that vaccine hesitancy is not a significant problem in Africa “because the disease is so feared.”
Major obstacles to implementing the roadmap include the complacency of public health leaders and the COVID-19 lockdowns, which decreased vaccination coverage rates. “The second thing is also sufficient funding to do the research and innovation so that we get the affordable tools that we need globally,” Dr. Préziosi said.
Marilyn Felkner, DrPH, School of Human Ecology, University of Texas at Austin, said in an interview, “It’s very cliché, but we have often said that communicable diseases do not respect political boundaries. So to expect a country to be able to control that by themselves is a false hope.”
Regarding the roadmap, Dr. Felkner said, “I think that organizing ideas and having them in writing is always a good first step. And it can help people move forward if they’re feeling overwhelmed ... Having a written plan can certainly provide that fundamental basis. So, the important thing is not to say, ‘Oh, we have this great plan done; hope somebody picks up the plan.’ There’s got to be some momentum behind it, and hopefully some funding.”
Dr. Préziosi and Dr. Felkner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The World Health Organization and its partners recently released an ambitious plan, Defeating meningitis by 2030: A global road map. The goal is to reduce deaths and disabilities from bacterial meningitis, which kills about 250,000 people annually of the 1.2 million estimated to be infected.
This type of infection around the brain and spinal cord also causes long-term disabilities – deafness, learning problems, seizures, loss of limbs – in about one-quarter of survivors.
The leading causes of bacterial meningitis are Neisseria meningitidis (meningococcus), Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, and group B streptococcus. As with malaria, about half of the cases are in children under age 5 years. The most severely affected area for both infections is sub-Saharan Africa.
The main goal of the roadmap is to reduce vaccine-preventable cases of bacterial meningitis by 50% and deaths by 70% by 2030. WHO’s partners included the Centers for Disease Control and Prevention, the London School of Hygiene and Tropical Medicine, Médecins Sans Frontières (Doctors Without Borders), the Meningitis Research Foundation, PATH, UNICEF, and numerous global consultants.
For primary prevention and epidemic control, a major goal is to achieve higher vaccine coverage. Another goal is developing and deploying rapid diagnostic tests to guide treatment and prevention activities and measure the impact of vaccination. The lack of laboratory capacity to confirm the bacteria is a significant challenge. Also, patients often receive antibiotics before appropriate tests are conducted, and lumbar punctures are frequently not done.
The commitment to this project emerged in 2017. It was followed by a baseline analysis in 2018 and a draft roadmap the following year. Consultations with experts and with more than 600 patient groups in more than 90 countries followed.
Prevention through greater vaccine uptake was the top priority. Vaccination is considered the first line of defense against antibiotic resistance among the targeted bacteria.
Another goal is to quantify the decrease in antibiotic use for invasive infections or prophylaxis and the subsequent reduction in antimicrobial resistance in relation to increased vaccination.
Surveillance is weak in many regions, limiting the ability to detect epidemics and to respond appropriately. Similarly, there are limited data on the burden of sequelae, such as deafness, on meningitis survivors.
There is an inadequate supply of affordable vaccines to respond to epidemics. Currently, routine vaccination against Neisseria meningitidis is occurring in 18 of 26 countries in the meningitis belt. Epidemics of meningococcus occur every few years in the driest time of the year and abate with the rains. Epidemics of pneumococcal meningitis are much rarer but follow a similar pattern; they have also been associated with crowding and alcohol use.
Care for those affected by meningitis is another focus, as is affirming the right to prevention and care. There’s a need for earlier recognition of the complications of meningitis and an increase in efforts to treat those complications.
WHO’s final goal in its roadmap is to boost awareness of meningitis and make it a priority for policymakers. Similarly, there is a need to educate communities about the disease, including how to access vaccines. If someone becomes ill, they need to be aware of the symptoms, the need for early treatment, and what aftercare is available.
Marie-Pierre Préziosi, MD, the core secretariat of WHO’s Technical Taskforce, told this news organization that while the roadmap looks aspirational, “it is feasible … you have strategic goals – each has milestones with time limits and who will do it.”
Regarding vaccinations, Dr. Préziosi said that “the strategy was a victim of its success. The mass campaign knocked down transmission completely.” Some countries are now waiting for multivalent vaccines. She said that vaccine hesitancy is not a significant problem in Africa “because the disease is so feared.”
Major obstacles to implementing the roadmap include the complacency of public health leaders and the COVID-19 lockdowns, which decreased vaccination coverage rates. “The second thing is also sufficient funding to do the research and innovation so that we get the affordable tools that we need globally,” Dr. Préziosi said.
Marilyn Felkner, DrPH, School of Human Ecology, University of Texas at Austin, said in an interview, “It’s very cliché, but we have often said that communicable diseases do not respect political boundaries. So to expect a country to be able to control that by themselves is a false hope.”
Regarding the roadmap, Dr. Felkner said, “I think that organizing ideas and having them in writing is always a good first step. And it can help people move forward if they’re feeling overwhelmed ... Having a written plan can certainly provide that fundamental basis. So, the important thing is not to say, ‘Oh, we have this great plan done; hope somebody picks up the plan.’ There’s got to be some momentum behind it, and hopefully some funding.”
Dr. Préziosi and Dr. Felkner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The World Health Organization and its partners recently released an ambitious plan, Defeating meningitis by 2030: A global road map. The goal is to reduce deaths and disabilities from bacterial meningitis, which kills about 250,000 people annually of the 1.2 million estimated to be infected.
This type of infection around the brain and spinal cord also causes long-term disabilities – deafness, learning problems, seizures, loss of limbs – in about one-quarter of survivors.
The leading causes of bacterial meningitis are Neisseria meningitidis (meningococcus), Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, and group B streptococcus. As with malaria, about half of the cases are in children under age 5 years. The most severely affected area for both infections is sub-Saharan Africa.
The main goal of the roadmap is to reduce vaccine-preventable cases of bacterial meningitis by 50% and deaths by 70% by 2030. WHO’s partners included the Centers for Disease Control and Prevention, the London School of Hygiene and Tropical Medicine, Médecins Sans Frontières (Doctors Without Borders), the Meningitis Research Foundation, PATH, UNICEF, and numerous global consultants.
For primary prevention and epidemic control, a major goal is to achieve higher vaccine coverage. Another goal is developing and deploying rapid diagnostic tests to guide treatment and prevention activities and measure the impact of vaccination. The lack of laboratory capacity to confirm the bacteria is a significant challenge. Also, patients often receive antibiotics before appropriate tests are conducted, and lumbar punctures are frequently not done.
The commitment to this project emerged in 2017. It was followed by a baseline analysis in 2018 and a draft roadmap the following year. Consultations with experts and with more than 600 patient groups in more than 90 countries followed.
Prevention through greater vaccine uptake was the top priority. Vaccination is considered the first line of defense against antibiotic resistance among the targeted bacteria.
Another goal is to quantify the decrease in antibiotic use for invasive infections or prophylaxis and the subsequent reduction in antimicrobial resistance in relation to increased vaccination.
Surveillance is weak in many regions, limiting the ability to detect epidemics and to respond appropriately. Similarly, there are limited data on the burden of sequelae, such as deafness, on meningitis survivors.
There is an inadequate supply of affordable vaccines to respond to epidemics. Currently, routine vaccination against Neisseria meningitidis is occurring in 18 of 26 countries in the meningitis belt. Epidemics of meningococcus occur every few years in the driest time of the year and abate with the rains. Epidemics of pneumococcal meningitis are much rarer but follow a similar pattern; they have also been associated with crowding and alcohol use.
Care for those affected by meningitis is another focus, as is affirming the right to prevention and care. There’s a need for earlier recognition of the complications of meningitis and an increase in efforts to treat those complications.
WHO’s final goal in its roadmap is to boost awareness of meningitis and make it a priority for policymakers. Similarly, there is a need to educate communities about the disease, including how to access vaccines. If someone becomes ill, they need to be aware of the symptoms, the need for early treatment, and what aftercare is available.
Marie-Pierre Préziosi, MD, the core secretariat of WHO’s Technical Taskforce, told this news organization that while the roadmap looks aspirational, “it is feasible … you have strategic goals – each has milestones with time limits and who will do it.”
Regarding vaccinations, Dr. Préziosi said that “the strategy was a victim of its success. The mass campaign knocked down transmission completely.” Some countries are now waiting for multivalent vaccines. She said that vaccine hesitancy is not a significant problem in Africa “because the disease is so feared.”
Major obstacles to implementing the roadmap include the complacency of public health leaders and the COVID-19 lockdowns, which decreased vaccination coverage rates. “The second thing is also sufficient funding to do the research and innovation so that we get the affordable tools that we need globally,” Dr. Préziosi said.
Marilyn Felkner, DrPH, School of Human Ecology, University of Texas at Austin, said in an interview, “It’s very cliché, but we have often said that communicable diseases do not respect political boundaries. So to expect a country to be able to control that by themselves is a false hope.”
Regarding the roadmap, Dr. Felkner said, “I think that organizing ideas and having them in writing is always a good first step. And it can help people move forward if they’re feeling overwhelmed ... Having a written plan can certainly provide that fundamental basis. So, the important thing is not to say, ‘Oh, we have this great plan done; hope somebody picks up the plan.’ There’s got to be some momentum behind it, and hopefully some funding.”
Dr. Préziosi and Dr. Felkner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FDA approves avacopan for rare ANCA autoimmune disease
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.

ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.
ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.
ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
Genes related to osteosarcoma survival identified
When they combined them into a risk score and added one additional factor – metastases at diagnosis – the model was an “excellent” predictor of 1-year survival, the team said (area under the curve, 0.947; 95% confidence interval, 0.832-0.972).
“The survival-associated” immune-related genes (IRGs) “examined in this study have potential for identifying prognosis in osteosarcoma and may be clinically useful as relevant clinical biomarkers and candidate targets for anticancer therapy,” said investigators led by Wangmi Liu, MD, of Zhejiang University in Hangzhou, China. The study was published in JAMA Network Open.
They explained that it’s often difficult to distinguish high- and low-risk patients at osteosarcoma diagnosis. To address the issue, they analyzed the genomic signatures of 84 patients in the Cancer Genome Atlas and their associated clinical information.
The team split their subjects evenly into high-risk and low-risk groups based on a score developed from their genetic signatures. A total of 26 patients in the high-risk group (61.9%) died over a median follow up of 4.1 years versus only 1 (2.4%) in the low-risk group.
The risk score also correlated positively with B-cell tumor infiltration, and negatively with infiltration of CD8 T cells and macrophages.
Overall, 16 genes were significantly up-regulated, and 187 genes were significantly down-regulated in the high-risk group, including three survival-associated IRGs: CCL2, CD79A, and FPR1.
The differentially expressed genes were most significantly associated with transmembrane signaling receptor activity and inflammatory response. The team noted that “it has been reported that inflammatory response plays a critical role in tumor initiation, promotion, malignant conversion, invasion, and metastases.”
Of the 14 survival-associated IRGs, 5 have been reported before in osteosarcoma. The other nine were deduced from computational analysis and may be potential treatment targets, including bone morphogenetic protein 8b (BMP8b). Another member of the BMP family, BMP9, has been shown to promote the proliferation of osteosarcoma cells, “which is similar to this study’s finding that BMP8b was a risk factor in osteosarcoma. Therefore, the role of BMP8b in osteosarcoma needs further research,” the team said.
“Because the database provides limited clinical information, other important factors, such as staging and grading, were not included in our analysis. Therefore, extrapolation based on the findings must be done very carefully,” they cautioned.
The work was supported by the National Natural Science Foundation of China and others. The investigators didn’t have any disclosures.
When they combined them into a risk score and added one additional factor – metastases at diagnosis – the model was an “excellent” predictor of 1-year survival, the team said (area under the curve, 0.947; 95% confidence interval, 0.832-0.972).
“The survival-associated” immune-related genes (IRGs) “examined in this study have potential for identifying prognosis in osteosarcoma and may be clinically useful as relevant clinical biomarkers and candidate targets for anticancer therapy,” said investigators led by Wangmi Liu, MD, of Zhejiang University in Hangzhou, China. The study was published in JAMA Network Open.
They explained that it’s often difficult to distinguish high- and low-risk patients at osteosarcoma diagnosis. To address the issue, they analyzed the genomic signatures of 84 patients in the Cancer Genome Atlas and their associated clinical information.
The team split their subjects evenly into high-risk and low-risk groups based on a score developed from their genetic signatures. A total of 26 patients in the high-risk group (61.9%) died over a median follow up of 4.1 years versus only 1 (2.4%) in the low-risk group.
The risk score also correlated positively with B-cell tumor infiltration, and negatively with infiltration of CD8 T cells and macrophages.
Overall, 16 genes were significantly up-regulated, and 187 genes were significantly down-regulated in the high-risk group, including three survival-associated IRGs: CCL2, CD79A, and FPR1.
The differentially expressed genes were most significantly associated with transmembrane signaling receptor activity and inflammatory response. The team noted that “it has been reported that inflammatory response plays a critical role in tumor initiation, promotion, malignant conversion, invasion, and metastases.”
Of the 14 survival-associated IRGs, 5 have been reported before in osteosarcoma. The other nine were deduced from computational analysis and may be potential treatment targets, including bone morphogenetic protein 8b (BMP8b). Another member of the BMP family, BMP9, has been shown to promote the proliferation of osteosarcoma cells, “which is similar to this study’s finding that BMP8b was a risk factor in osteosarcoma. Therefore, the role of BMP8b in osteosarcoma needs further research,” the team said.
“Because the database provides limited clinical information, other important factors, such as staging and grading, were not included in our analysis. Therefore, extrapolation based on the findings must be done very carefully,” they cautioned.
The work was supported by the National Natural Science Foundation of China and others. The investigators didn’t have any disclosures.
When they combined them into a risk score and added one additional factor – metastases at diagnosis – the model was an “excellent” predictor of 1-year survival, the team said (area under the curve, 0.947; 95% confidence interval, 0.832-0.972).
“The survival-associated” immune-related genes (IRGs) “examined in this study have potential for identifying prognosis in osteosarcoma and may be clinically useful as relevant clinical biomarkers and candidate targets for anticancer therapy,” said investigators led by Wangmi Liu, MD, of Zhejiang University in Hangzhou, China. The study was published in JAMA Network Open.
They explained that it’s often difficult to distinguish high- and low-risk patients at osteosarcoma diagnosis. To address the issue, they analyzed the genomic signatures of 84 patients in the Cancer Genome Atlas and their associated clinical information.
The team split their subjects evenly into high-risk and low-risk groups based on a score developed from their genetic signatures. A total of 26 patients in the high-risk group (61.9%) died over a median follow up of 4.1 years versus only 1 (2.4%) in the low-risk group.
The risk score also correlated positively with B-cell tumor infiltration, and negatively with infiltration of CD8 T cells and macrophages.
Overall, 16 genes were significantly up-regulated, and 187 genes were significantly down-regulated in the high-risk group, including three survival-associated IRGs: CCL2, CD79A, and FPR1.
The differentially expressed genes were most significantly associated with transmembrane signaling receptor activity and inflammatory response. The team noted that “it has been reported that inflammatory response plays a critical role in tumor initiation, promotion, malignant conversion, invasion, and metastases.”
Of the 14 survival-associated IRGs, 5 have been reported before in osteosarcoma. The other nine were deduced from computational analysis and may be potential treatment targets, including bone morphogenetic protein 8b (BMP8b). Another member of the BMP family, BMP9, has been shown to promote the proliferation of osteosarcoma cells, “which is similar to this study’s finding that BMP8b was a risk factor in osteosarcoma. Therefore, the role of BMP8b in osteosarcoma needs further research,” the team said.
“Because the database provides limited clinical information, other important factors, such as staging and grading, were not included in our analysis. Therefore, extrapolation based on the findings must be done very carefully,” they cautioned.
The work was supported by the National Natural Science Foundation of China and others. The investigators didn’t have any disclosures.
FROM JAMA NETWORK OPEN
Rare hematologic malignancy may first present to a dermatologist
in about 80% of cases.
“You won’t see blastic plasmacytoid dendritic cell neoplasm listed on our primary cutaneous lymphoma classifications because it’s not technically a primary cutaneous disease,” Brittney K. DeClerck, MD, said during the annual meeting of the Pacific Dermatologic Association. “It’s a systemic disease that has secondary cutaneous manifestations. That’s a very important distinction to make, in terms of not missing the underlying disease associated with what might be commonly first seen on the skin.”

BPDCN is a malignancy of plasmacytoid dendritic cells, which capture, process, and present antigen, and allow the remainder of the immune system to be activated. “They are mainly derived from the myeloid cell lineage, and possibly from the lymphoid line in a subset of cases,” said Dr. DeClerck, associate professor of clinical pathology and dermatology at the University of Southern California, Los Angeles. “They secrete high levels of type I interferons, which is important for antiviral immunity, but they can also be implicated in severe systemic inflammatory diseases, such as systemic lupus erythematosus and systemic sclerosis.”
BPDCN involves the skin in about 80% of cases, she added, “but invariably at some point it involves the bone marrow and has an acute leukemic presentation, whether or not it happens concurrently with what we see on the skin as dermatologists. We also see variable involvement of the peripheral blood, lymph nodes, and the central nervous system.”
The classification of BPDCN has changed over time based on evolving immunohistochemical markers and technologies. For example, in 1995 it was called agranular CD4+ NK cell leukemia, in 2001 it was called blastic NK-cell lymphoma, in 2005 it was called CD4+/CD56+ hematodermic neoplasm, and in 2008 it was called BPDCN (AML subset). In 2016 it became classified as its own entity: BPDCN.
Because of changing nomenclature, the true incidence of the disease is unknown, but according to the best available literature, 75% of cases occur in men and the median age is between 60 and 70 years, “but all ages can be affected,” Dr. DeClerck said. “Cases seem to come in clusters. Our most recent cluster has been in our pediatric population. At Children’s Hospital Los Angeles, we’ve had three cases in the last couple of years. To me, that was a bit unusual.”
She added that 10%-20% of patients will have either a history of, or will develop another, hematologic malignancy, such as myelodysplastic syndrome (MDS), chronic myelogenous leukemia (CML), or acute myelogenous leukemia (AML).
The general prognosis of BPDCN is poor, and the mean time from onset of lesions to an actual diagnosis is about 6.2 months, which underscores the importance of early diagnosis, Dr. DeClerck said. “There can be some nondescript solitary lesions that patients can present with, so don’t hesitate to biopsy.” The median overall survival is less than 20 months, but patients under 60 years of age have a slightly better prognosis.
Clinical presentation
Clinically, the malignancy presents with variable involvement of the skin, bone marrow, lymph nodes, peripheral blood, and central nervous system. “Patients may have one or all of these,” she said. Because 80% of patients have skin lesions, “dermatologists should be aware of this entity in order to communicate with our pathologists to understand that maybe one biopsy isn’t enough. Several biopsies may be required.”
The most common dermatologic presentation of BPDCN is erythematous to deeply violaceous nodules. Other patients may present with infiltrated ecchymotic plaques or petechial to hyperpigmented macules, patches, and plaques. Biopsy reveals a diffusely infiltrated dermis of markedly atypical large cells, but occasionally can be more subtle. “Early lesions may only be perivascular in nature, so going on high power on anything that looks atypical on low power is important in these cases,” Dr. DeClerck said.
The recommended histochemical stains for suspected BPDCN include CD123, CD4, and CD56. “We need to have other stains to rule out other things, such as negative stains that are going to exclude other T cell and B cell processes, and Merkel cell carcinoma, which can express CD56. We also want to have another confirmatory stain because other things can express CD123, CD4, and CD56. Commonly we use TCL1 or TCF4.”
The differential diagnosis of cutaneous findings includes leukemia cutis, mycosis fungoides, NK/T-cell lymphoma, and cutaneous gamma-delta T-cell lymphoma, while the differential diagnosis of biopsy findings includes AML, acute lymphoblastic leukemia, and NK/T-cell lymphoma.
Treatment of BPDCN
Historically, BPDCN was treated with multiagent high-dose chemotherapy. “Patients would frequently respond early but would relapse quickly, progress, and have a poor outcome,” Dr. DeClerck said. Now, first-line therapy is tagraxofusp-erzs (Elzonris) or multiagent chemotherapy based on where the patient is in the course of disease. Tagraxofusp-erzs is an IL-3 conjugated diphtheria toxic fusion protein which binds to CD123, which was approved by the Food and Drug Administration in 2018 for treating BPDCN. After that initial therapy, it is determined whether the patient has a complete response or failed response, she said. “If they have a complete response, they frequently go on to bone marrow transplantation, which is the only curative therapy at this point for these patients.”
According to Dr. DeClerck, an anti-BCL-2 therapy, venetoclax, can be used for patients with BPDCN as well. National Comprehensive Cancer Network (NCCN) guidelines for the treatment of BPDCN can be found on the NCCN website.
Dr. DeClerck emphasized the importance of reviewing biopsy results with a hematopathologist, “because there are complex leukemias that are beyond what dermatopathologists have been trained in.” Once a patient is diagnosed with BPDCN, she recommends rapid referral to a large center for treatment and possible bone marrow transplantation.
Dr. DeClerck disclosed that she is an adviser for tagraxofusp-erzs manufacturer Stemline Therapeutics.
in about 80% of cases.
“You won’t see blastic plasmacytoid dendritic cell neoplasm listed on our primary cutaneous lymphoma classifications because it’s not technically a primary cutaneous disease,” Brittney K. DeClerck, MD, said during the annual meeting of the Pacific Dermatologic Association. “It’s a systemic disease that has secondary cutaneous manifestations. That’s a very important distinction to make, in terms of not missing the underlying disease associated with what might be commonly first seen on the skin.”
BPDCN is a malignancy of plasmacytoid dendritic cells, which capture, process, and present antigen, and allow the remainder of the immune system to be activated. “They are mainly derived from the myeloid cell lineage, and possibly from the lymphoid line in a subset of cases,” said Dr. DeClerck, associate professor of clinical pathology and dermatology at the University of Southern California, Los Angeles. “They secrete high levels of type I interferons, which is important for antiviral immunity, but they can also be implicated in severe systemic inflammatory diseases, such as systemic lupus erythematosus and systemic sclerosis.”
BPDCN involves the skin in about 80% of cases, she added, “but invariably at some point it involves the bone marrow and has an acute leukemic presentation, whether or not it happens concurrently with what we see on the skin as dermatologists. We also see variable involvement of the peripheral blood, lymph nodes, and the central nervous system.”
The classification of BPDCN has changed over time based on evolving immunohistochemical markers and technologies. For example, in 1995 it was called agranular CD4+ NK cell leukemia, in 2001 it was called blastic NK-cell lymphoma, in 2005 it was called CD4+/CD56+ hematodermic neoplasm, and in 2008 it was called BPDCN (AML subset). In 2016 it became classified as its own entity: BPDCN.
Because of changing nomenclature, the true incidence of the disease is unknown, but according to the best available literature, 75% of cases occur in men and the median age is between 60 and 70 years, “but all ages can be affected,” Dr. DeClerck said. “Cases seem to come in clusters. Our most recent cluster has been in our pediatric population. At Children’s Hospital Los Angeles, we’ve had three cases in the last couple of years. To me, that was a bit unusual.”
She added that 10%-20% of patients will have either a history of, or will develop another, hematologic malignancy, such as myelodysplastic syndrome (MDS), chronic myelogenous leukemia (CML), or acute myelogenous leukemia (AML).
The general prognosis of BPDCN is poor, and the mean time from onset of lesions to an actual diagnosis is about 6.2 months, which underscores the importance of early diagnosis, Dr. DeClerck said. “There can be some nondescript solitary lesions that patients can present with, so don’t hesitate to biopsy.” The median overall survival is less than 20 months, but patients under 60 years of age have a slightly better prognosis.
Clinical presentation
Clinically, the malignancy presents with variable involvement of the skin, bone marrow, lymph nodes, peripheral blood, and central nervous system. “Patients may have one or all of these,” she said. Because 80% of patients have skin lesions, “dermatologists should be aware of this entity in order to communicate with our pathologists to understand that maybe one biopsy isn’t enough. Several biopsies may be required.”
The most common dermatologic presentation of BPDCN is erythematous to deeply violaceous nodules. Other patients may present with infiltrated ecchymotic plaques or petechial to hyperpigmented macules, patches, and plaques. Biopsy reveals a diffusely infiltrated dermis of markedly atypical large cells, but occasionally can be more subtle. “Early lesions may only be perivascular in nature, so going on high power on anything that looks atypical on low power is important in these cases,” Dr. DeClerck said.
The recommended histochemical stains for suspected BPDCN include CD123, CD4, and CD56. “We need to have other stains to rule out other things, such as negative stains that are going to exclude other T cell and B cell processes, and Merkel cell carcinoma, which can express CD56. We also want to have another confirmatory stain because other things can express CD123, CD4, and CD56. Commonly we use TCL1 or TCF4.”
The differential diagnosis of cutaneous findings includes leukemia cutis, mycosis fungoides, NK/T-cell lymphoma, and cutaneous gamma-delta T-cell lymphoma, while the differential diagnosis of biopsy findings includes AML, acute lymphoblastic leukemia, and NK/T-cell lymphoma.
Treatment of BPDCN
Historically, BPDCN was treated with multiagent high-dose chemotherapy. “Patients would frequently respond early but would relapse quickly, progress, and have a poor outcome,” Dr. DeClerck said. Now, first-line therapy is tagraxofusp-erzs (Elzonris) or multiagent chemotherapy based on where the patient is in the course of disease. Tagraxofusp-erzs is an IL-3 conjugated diphtheria toxic fusion protein which binds to CD123, which was approved by the Food and Drug Administration in 2018 for treating BPDCN. After that initial therapy, it is determined whether the patient has a complete response or failed response, she said. “If they have a complete response, they frequently go on to bone marrow transplantation, which is the only curative therapy at this point for these patients.”
According to Dr. DeClerck, an anti-BCL-2 therapy, venetoclax, can be used for patients with BPDCN as well. National Comprehensive Cancer Network (NCCN) guidelines for the treatment of BPDCN can be found on the NCCN website.
Dr. DeClerck emphasized the importance of reviewing biopsy results with a hematopathologist, “because there are complex leukemias that are beyond what dermatopathologists have been trained in.” Once a patient is diagnosed with BPDCN, she recommends rapid referral to a large center for treatment and possible bone marrow transplantation.
Dr. DeClerck disclosed that she is an adviser for tagraxofusp-erzs manufacturer Stemline Therapeutics.
in about 80% of cases.
“You won’t see blastic plasmacytoid dendritic cell neoplasm listed on our primary cutaneous lymphoma classifications because it’s not technically a primary cutaneous disease,” Brittney K. DeClerck, MD, said during the annual meeting of the Pacific Dermatologic Association. “It’s a systemic disease that has secondary cutaneous manifestations. That’s a very important distinction to make, in terms of not missing the underlying disease associated with what might be commonly first seen on the skin.”
BPDCN is a malignancy of plasmacytoid dendritic cells, which capture, process, and present antigen, and allow the remainder of the immune system to be activated. “They are mainly derived from the myeloid cell lineage, and possibly from the lymphoid line in a subset of cases,” said Dr. DeClerck, associate professor of clinical pathology and dermatology at the University of Southern California, Los Angeles. “They secrete high levels of type I interferons, which is important for antiviral immunity, but they can also be implicated in severe systemic inflammatory diseases, such as systemic lupus erythematosus and systemic sclerosis.”
BPDCN involves the skin in about 80% of cases, she added, “but invariably at some point it involves the bone marrow and has an acute leukemic presentation, whether or not it happens concurrently with what we see on the skin as dermatologists. We also see variable involvement of the peripheral blood, lymph nodes, and the central nervous system.”
The classification of BPDCN has changed over time based on evolving immunohistochemical markers and technologies. For example, in 1995 it was called agranular CD4+ NK cell leukemia, in 2001 it was called blastic NK-cell lymphoma, in 2005 it was called CD4+/CD56+ hematodermic neoplasm, and in 2008 it was called BPDCN (AML subset). In 2016 it became classified as its own entity: BPDCN.
Because of changing nomenclature, the true incidence of the disease is unknown, but according to the best available literature, 75% of cases occur in men and the median age is between 60 and 70 years, “but all ages can be affected,” Dr. DeClerck said. “Cases seem to come in clusters. Our most recent cluster has been in our pediatric population. At Children’s Hospital Los Angeles, we’ve had three cases in the last couple of years. To me, that was a bit unusual.”
She added that 10%-20% of patients will have either a history of, or will develop another, hematologic malignancy, such as myelodysplastic syndrome (MDS), chronic myelogenous leukemia (CML), or acute myelogenous leukemia (AML).
The general prognosis of BPDCN is poor, and the mean time from onset of lesions to an actual diagnosis is about 6.2 months, which underscores the importance of early diagnosis, Dr. DeClerck said. “There can be some nondescript solitary lesions that patients can present with, so don’t hesitate to biopsy.” The median overall survival is less than 20 months, but patients under 60 years of age have a slightly better prognosis.
Clinical presentation
Clinically, the malignancy presents with variable involvement of the skin, bone marrow, lymph nodes, peripheral blood, and central nervous system. “Patients may have one or all of these,” she said. Because 80% of patients have skin lesions, “dermatologists should be aware of this entity in order to communicate with our pathologists to understand that maybe one biopsy isn’t enough. Several biopsies may be required.”
The most common dermatologic presentation of BPDCN is erythematous to deeply violaceous nodules. Other patients may present with infiltrated ecchymotic plaques or petechial to hyperpigmented macules, patches, and plaques. Biopsy reveals a diffusely infiltrated dermis of markedly atypical large cells, but occasionally can be more subtle. “Early lesions may only be perivascular in nature, so going on high power on anything that looks atypical on low power is important in these cases,” Dr. DeClerck said.
The recommended histochemical stains for suspected BPDCN include CD123, CD4, and CD56. “We need to have other stains to rule out other things, such as negative stains that are going to exclude other T cell and B cell processes, and Merkel cell carcinoma, which can express CD56. We also want to have another confirmatory stain because other things can express CD123, CD4, and CD56. Commonly we use TCL1 or TCF4.”
The differential diagnosis of cutaneous findings includes leukemia cutis, mycosis fungoides, NK/T-cell lymphoma, and cutaneous gamma-delta T-cell lymphoma, while the differential diagnosis of biopsy findings includes AML, acute lymphoblastic leukemia, and NK/T-cell lymphoma.
Treatment of BPDCN
Historically, BPDCN was treated with multiagent high-dose chemotherapy. “Patients would frequently respond early but would relapse quickly, progress, and have a poor outcome,” Dr. DeClerck said. Now, first-line therapy is tagraxofusp-erzs (Elzonris) or multiagent chemotherapy based on where the patient is in the course of disease. Tagraxofusp-erzs is an IL-3 conjugated diphtheria toxic fusion protein which binds to CD123, which was approved by the Food and Drug Administration in 2018 for treating BPDCN. After that initial therapy, it is determined whether the patient has a complete response or failed response, she said. “If they have a complete response, they frequently go on to bone marrow transplantation, which is the only curative therapy at this point for these patients.”
According to Dr. DeClerck, an anti-BCL-2 therapy, venetoclax, can be used for patients with BPDCN as well. National Comprehensive Cancer Network (NCCN) guidelines for the treatment of BPDCN can be found on the NCCN website.
Dr. DeClerck emphasized the importance of reviewing biopsy results with a hematopathologist, “because there are complex leukemias that are beyond what dermatopathologists have been trained in.” Once a patient is diagnosed with BPDCN, she recommends rapid referral to a large center for treatment and possible bone marrow transplantation.
Dr. DeClerck disclosed that she is an adviser for tagraxofusp-erzs manufacturer Stemline Therapeutics.
FROM PDA 2021
Synthetic triglyceride shows potential in Huntington’s disease
, according to data presented at the International Congress of Parkinson’s Disease and Movement Disorders.

Reporting results of TRIHEP3 and an extension study, Fanny Mochel, MD, PhD, of Sorbonne University in Paris and the Paris Brain Institute, said in an interview that her group is the only one investigating triheptanoin to target caudate atrophy in Huntington’s disease. The Food and Drug Administration last year approved triheptanoin for the treatment of long-chain fatty acid oxidation disorders.
“The main findings are two observations: that patients were clinically stable based on their gradation of total motor score (TMS) on UHDRS (Unified Huntington’s Disease Rating Scale) after 1 year,” Dr. Mochel said in an interview. “The other is that we observed a reduction of the caudate atrophy progression that we usually see over 1 year by about 50%.”
TRIHEP3 randomized 100 patients with early-stage Huntington’s disease to triheptanoin 1g/kg daily and placebo. It followed on previous research in which the group used 31-phosphorus brain MR spectroscopy to demonstrate triheptanoin restored a normal brain energetic profile in patients with Huntington’s disease. TRIHEP3 was a 6-month randomized controlled trial at two centers, followed by a 6-month open-label phase. After that, 42 patients opted to participate in the 1-year extension study.
TRIHEP3 found no difference in caudate boundary shift integral (cBSI) at 6 months – the primary endpoint. But in the extension study, TMS tended to stabilize in patients treated for 1 year (0.6 ± 5.1), compared with those treated for 6 months (2.5 ± 4.5, P = .072).
Using a placebo control group from an external study of patients with Huntington’s disease with what Dr. Mochel described as “identical clinical characteristics,” she said the research confirmed TMS clinical stability in treated patients at 1 year (2.6 ± 4.6 vs. 0.6 ± 5.1, P = .057) and found significantly lower caudate atrophy (–3% vs. –6.7%, compared with baseline, P < .001).
Dr. Mochel also noted that Diffusion Tensor Imaging and Fixed-based analyses (FBA) showed fewer alterations in fiber metrics at 24 months in patients treated from baseline. FBA also showed improved fiber trophicity at 24 months in both groups.
‘The first good news’
Dr. Mochel noted that the Huntington’s disease community had been shaken in the spring by the failure of three trials of gene-targeting therapies for Huntington’s disease. Roche halted a phase 3 study of its antisense oligonucleotide (ASO) tominersen, and Wave Life Sciences scuttled two ASO programs in phase 1/2 trials.
“Triheptanoin is not going to cure Huntington’s disease; it’s a disease with many components, but it does work on the energy aspects and that seems to stabilize patients over the time of observation,” Dr. Mochel said. “That’s the first good news.”
She also noted that side effects were mainly gastrointestinal in nature, and they typically resolved with dietary management.

As a target in Huntington disease, the caudate nucleus is highly desirable, and caudate atrophy has been shown to occur even before the onset of motor symptoms, said N. Ahmad Aziz, MD, PhD, a neurologist and epidemiologist at the German Center for Neurodegenerative Diseases at the University of Bonn (Germany). “In this light, the findings of the trial conducted by Dr. Mochel and colleagues, which suggest that triheptanoin intake may slow down the rate of caudate atrophy in patients with early-stage Huntington’s disease, are highly promising,” Dr. Aziz said in an interview.
However, he noted that the improvement in caudate atrophy was only a secondary endpoint in the extension study. “Nevertheless, given triheptanoin’s biologically plausible mechanism of action – i.e., provision of substrates to the Krebs cycle and at least partial restoration of the well-documented defective mitochondrial function in Huntington’s disease – combined with its apparently relatively mild side-effect profile and good tolerability, I think that the preliminary findings of this trial are very promising and justify a larger phase 3 trial,” Dr. Aziz said.
Dr. Mochel said that the findings are prompting the investigators to consider just that.
Dr. Mochel has received consulting fees from and conducted investigator‐sponsored studies supported by Ultragenyx Pharmaceuticals. Dr. Aziz has no relevant financial relationships to disclose.
, according to data presented at the International Congress of Parkinson’s Disease and Movement Disorders.
Reporting results of TRIHEP3 and an extension study, Fanny Mochel, MD, PhD, of Sorbonne University in Paris and the Paris Brain Institute, said in an interview that her group is the only one investigating triheptanoin to target caudate atrophy in Huntington’s disease. The Food and Drug Administration last year approved triheptanoin for the treatment of long-chain fatty acid oxidation disorders.
“The main findings are two observations: that patients were clinically stable based on their gradation of total motor score (TMS) on UHDRS (Unified Huntington’s Disease Rating Scale) after 1 year,” Dr. Mochel said in an interview. “The other is that we observed a reduction of the caudate atrophy progression that we usually see over 1 year by about 50%.”
TRIHEP3 randomized 100 patients with early-stage Huntington’s disease to triheptanoin 1g/kg daily and placebo. It followed on previous research in which the group used 31-phosphorus brain MR spectroscopy to demonstrate triheptanoin restored a normal brain energetic profile in patients with Huntington’s disease. TRIHEP3 was a 6-month randomized controlled trial at two centers, followed by a 6-month open-label phase. After that, 42 patients opted to participate in the 1-year extension study.
TRIHEP3 found no difference in caudate boundary shift integral (cBSI) at 6 months – the primary endpoint. But in the extension study, TMS tended to stabilize in patients treated for 1 year (0.6 ± 5.1), compared with those treated for 6 months (2.5 ± 4.5, P = .072).
Using a placebo control group from an external study of patients with Huntington’s disease with what Dr. Mochel described as “identical clinical characteristics,” she said the research confirmed TMS clinical stability in treated patients at 1 year (2.6 ± 4.6 vs. 0.6 ± 5.1, P = .057) and found significantly lower caudate atrophy (–3% vs. –6.7%, compared with baseline, P < .001).
Dr. Mochel also noted that Diffusion Tensor Imaging and Fixed-based analyses (FBA) showed fewer alterations in fiber metrics at 24 months in patients treated from baseline. FBA also showed improved fiber trophicity at 24 months in both groups.
‘The first good news’
Dr. Mochel noted that the Huntington’s disease community had been shaken in the spring by the failure of three trials of gene-targeting therapies for Huntington’s disease. Roche halted a phase 3 study of its antisense oligonucleotide (ASO) tominersen, and Wave Life Sciences scuttled two ASO programs in phase 1/2 trials.
“Triheptanoin is not going to cure Huntington’s disease; it’s a disease with many components, but it does work on the energy aspects and that seems to stabilize patients over the time of observation,” Dr. Mochel said. “That’s the first good news.”
She also noted that side effects were mainly gastrointestinal in nature, and they typically resolved with dietary management.
As a target in Huntington disease, the caudate nucleus is highly desirable, and caudate atrophy has been shown to occur even before the onset of motor symptoms, said N. Ahmad Aziz, MD, PhD, a neurologist and epidemiologist at the German Center for Neurodegenerative Diseases at the University of Bonn (Germany). “In this light, the findings of the trial conducted by Dr. Mochel and colleagues, which suggest that triheptanoin intake may slow down the rate of caudate atrophy in patients with early-stage Huntington’s disease, are highly promising,” Dr. Aziz said in an interview.
However, he noted that the improvement in caudate atrophy was only a secondary endpoint in the extension study. “Nevertheless, given triheptanoin’s biologically plausible mechanism of action – i.e., provision of substrates to the Krebs cycle and at least partial restoration of the well-documented defective mitochondrial function in Huntington’s disease – combined with its apparently relatively mild side-effect profile and good tolerability, I think that the preliminary findings of this trial are very promising and justify a larger phase 3 trial,” Dr. Aziz said.
Dr. Mochel said that the findings are prompting the investigators to consider just that.
Dr. Mochel has received consulting fees from and conducted investigator‐sponsored studies supported by Ultragenyx Pharmaceuticals. Dr. Aziz has no relevant financial relationships to disclose.
, according to data presented at the International Congress of Parkinson’s Disease and Movement Disorders.
Reporting results of TRIHEP3 and an extension study, Fanny Mochel, MD, PhD, of Sorbonne University in Paris and the Paris Brain Institute, said in an interview that her group is the only one investigating triheptanoin to target caudate atrophy in Huntington’s disease. The Food and Drug Administration last year approved triheptanoin for the treatment of long-chain fatty acid oxidation disorders.
“The main findings are two observations: that patients were clinically stable based on their gradation of total motor score (TMS) on UHDRS (Unified Huntington’s Disease Rating Scale) after 1 year,” Dr. Mochel said in an interview. “The other is that we observed a reduction of the caudate atrophy progression that we usually see over 1 year by about 50%.”
TRIHEP3 randomized 100 patients with early-stage Huntington’s disease to triheptanoin 1g/kg daily and placebo. It followed on previous research in which the group used 31-phosphorus brain MR spectroscopy to demonstrate triheptanoin restored a normal brain energetic profile in patients with Huntington’s disease. TRIHEP3 was a 6-month randomized controlled trial at two centers, followed by a 6-month open-label phase. After that, 42 patients opted to participate in the 1-year extension study.
TRIHEP3 found no difference in caudate boundary shift integral (cBSI) at 6 months – the primary endpoint. But in the extension study, TMS tended to stabilize in patients treated for 1 year (0.6 ± 5.1), compared with those treated for 6 months (2.5 ± 4.5, P = .072).
Using a placebo control group from an external study of patients with Huntington’s disease with what Dr. Mochel described as “identical clinical characteristics,” she said the research confirmed TMS clinical stability in treated patients at 1 year (2.6 ± 4.6 vs. 0.6 ± 5.1, P = .057) and found significantly lower caudate atrophy (–3% vs. –6.7%, compared with baseline, P < .001).
Dr. Mochel also noted that Diffusion Tensor Imaging and Fixed-based analyses (FBA) showed fewer alterations in fiber metrics at 24 months in patients treated from baseline. FBA also showed improved fiber trophicity at 24 months in both groups.
‘The first good news’
Dr. Mochel noted that the Huntington’s disease community had been shaken in the spring by the failure of three trials of gene-targeting therapies for Huntington’s disease. Roche halted a phase 3 study of its antisense oligonucleotide (ASO) tominersen, and Wave Life Sciences scuttled two ASO programs in phase 1/2 trials.
“Triheptanoin is not going to cure Huntington’s disease; it’s a disease with many components, but it does work on the energy aspects and that seems to stabilize patients over the time of observation,” Dr. Mochel said. “That’s the first good news.”
She also noted that side effects were mainly gastrointestinal in nature, and they typically resolved with dietary management.
As a target in Huntington disease, the caudate nucleus is highly desirable, and caudate atrophy has been shown to occur even before the onset of motor symptoms, said N. Ahmad Aziz, MD, PhD, a neurologist and epidemiologist at the German Center for Neurodegenerative Diseases at the University of Bonn (Germany). “In this light, the findings of the trial conducted by Dr. Mochel and colleagues, which suggest that triheptanoin intake may slow down the rate of caudate atrophy in patients with early-stage Huntington’s disease, are highly promising,” Dr. Aziz said in an interview.
However, he noted that the improvement in caudate atrophy was only a secondary endpoint in the extension study. “Nevertheless, given triheptanoin’s biologically plausible mechanism of action – i.e., provision of substrates to the Krebs cycle and at least partial restoration of the well-documented defective mitochondrial function in Huntington’s disease – combined with its apparently relatively mild side-effect profile and good tolerability, I think that the preliminary findings of this trial are very promising and justify a larger phase 3 trial,” Dr. Aziz said.
Dr. Mochel said that the findings are prompting the investigators to consider just that.
Dr. Mochel has received consulting fees from and conducted investigator‐sponsored studies supported by Ultragenyx Pharmaceuticals. Dr. Aziz has no relevant financial relationships to disclose.
FROM MDS VIRTUAL CONGRESS 2021
Overlapping Phenotypic Features of PTEN Hamartoma Tumor Syndrome and Birt-Hogg-Dubé Syndrome
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.

Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).

Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.

The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10

Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.

Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.
Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).
Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.
The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10
Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.
Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.
Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).
Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.
The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10
Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.
Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
PRACTICE POINTS
- PTEN hamartoma tumor syndrome (PHTS) represents a spectrum of disorders caused by autosomal-dominant germline mutations in PTEN.
- Our patient presented with phenotypic features of PHTS and Birt-Hogg-Dubé syndrome. Given that both syndromes cause alterations in mammalian target of rapamycin signaling, overlapping phenotypic features may be seen.
- Recognizing overlapping phenotypic features of these syndromes will allow for timely diagnosis and surveillance for malignancy.
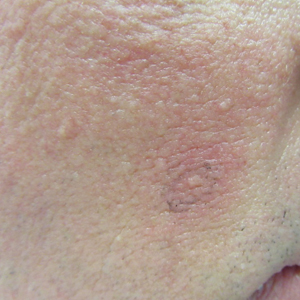
Friedreich’s ataxia treatment shows extended benefit
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.

The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.
The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.
The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
FROM MDS VIRTUAL CONGRESS 2021