User login
MIS-C follow-up proves challenging across pediatric hospitals
The discovery of any novel disease or condition means a steep learning curve as physicians must develop protocols for diagnosis, management, and follow-up on the fly in the midst of admitting and treating patients. Medical society task forces and committees often release interim guidance during the learning process, but each institution ultimately has to determine what works for them based on their resources, clinical experience, and patient population.

But when the novel condition demands the involvement of multiple different specialties, the challenge of management grows even more complex – as does follow-up after patients are discharged. Such has been the story with multisystem inflammatory syndrome in children (MIS-C), a complication of COVID-19 that shares some features with Kawasaki disease.
The similarities to Kawasaki provided physicians a place to start in developing appropriate treatment regimens and involved a similar interdisciplinary team from, at the least, cardiology and rheumatology, plus infectious disease since MIS-C results from COVID-19.
“It literally has it in the name – multisystem essentially hints that there are multiple specialties involved, multiple hands in the pot trying to manage the kids, and so each specialty has their own kind of unique role in the patient’s care even on the outpatient side,” said Samina S. Bhumbra, MD, an infectious disease pediatrician at Riley Hospital for Children and assistant professor of clinical pediatrics at Indiana University in Indianapolis. “This isn’t a disease that falls under one specialty.”

By July, the American College of Rheumatology had issued interim clinical guidance for management that most children’s hospitals have followed or slightly adapted. But ACR guidelines could not address how each institution should handle outpatient follow-up visits, especially since those visits required, again, at least cardiology and rheumatology if not infectious disease or other specialties as well.
“When their kids are admitted to the hospital, to be told at discharge you have to be followed up by all these specialists is a lot to handle,” Dr. Bhumbra said. But just as it’s difficult for parents to deal with the need to see several different doctors after discharge, it can be difficult at some institutions for physicians to design a follow-up schedule that can accommodate families, especially families who live far from the hospital in the first place.
“Some of our follow-up is disjointed because all of our clinics had never been on the same day just because of staff availability,” Dr. Bhumbra said. “But it can be a 2- to 3-hour drive for some of our patients, depending on how far they’re coming.”
Many of them can’t make that drive more than once in the same month, much less the same week.
“If you have multiple visits, it makes it more likely that they’re not showing up,” said Ryan M. Serrano, MD, a pediatric cardiologist at Riley and assistant professor of pediatrics at Indiana University. Riley used telehealth when possible, especially if families could get labs done near home. But pediatric echocardiograms require technicians who have experience with children, so families need to come to the hospital.

Children’s hospitals have therefore had to adapt scheduling strategies or develop pediatric specialty clinics to coordinate across the multiple departments and accommodate a complex follow-up regimen that is still evolving as physicians learn more about MIS-C.
Determining a follow-up regimen
Even before determining how to coordinate appointments, hospitals had to decide what follow-up itself should be.
“How long do we follow these patients and how often do we follow them?” said Melissa S. Oliver, MD, a rheumatologist at Riley and assistant professor of clinical pediatrics at Indiana University.

“We’re seeing that a lot of our patients rapidly respond when they get appropriate therapy, but we don’t know about long-term outcomes yet. We’re all still learning.”
At Children’s Hospital of Philadelphia, infectious disease follows up 4-6 weeks post discharge. The cardiology division came up with a follow-up plan that has evolved over time, said Matthew Elias, MD, an attending cardiologist at CHOP’s Cardiac Center and clinical assistant professor of pediatrics at the University of Pennsylvania, Philadelphia.

Patients get an EKG and echocardiogram at 2 weeks and, if their condition is stable, 6 weeks after discharge. After that, it depends on the patient’s clinical situation. Patients with moderately diminished left ventricular systolic function are recommended to get an MRI scan 3 months after discharge and, if old enough, exercise stress tests. Otherwise, they are seen at 6 months, but that appointment is optional for those whose prior echos have consistently been normal.
Other institutions, including Riley, are following a similar schedule of 2-week, 6-week, and 6-month postdischarge follow-ups, and most plan to do a 1-year follow-up as well, although that 1-year mark hasn’t arrived yet for most. Most do rheumatology labs at the 2-week appointment and use that to determine steroids management and whether labs are needed at the 6-week appointment. If labs have normalized, they aren’t done at 6 months. Small variations in follow-up management exist across institutions, but all are remaining open to changes. Riley, for example, is considering MRI screening for ongoing cardiac inflammation at 6 months to a year for all patients, Dr. Serrano said.
The dedicated clinic model
The two challenges Riley needed to address were the lack of a clear consensus on what MIS-C follow-up should look like and the need for continuity of care, Dr. Serrano said.
Regular discussion in departmental meetings at Riley “progressed from how do we take care of them and what treatments do we give them to how do we follow them and manage them in outpatient,” Dr. Oliver said. In the inpatient setting, they had an interdisciplinary team, but how could they maintain that for outpatients without overwhelming the families?
“I think the main challenge is for the families to identify who is leading the care for them,” said Martha M. Rodriguez, MD, a rheumatologist at Riley and assistant professor of clinical pediatrics at Indiana University. That sometimes led to families picking which follow-up appointments they would attend and which they would skip if they could not make them all – and sometimes they skipped the more important ones. “They would go to the appointment with me and then miss the cardiology appointments and the echocardiogram, which was more important to follow any abnormalities in the heart,” Dr. Rodriguez said.
After trying to coordinate separate follow-up appointments for months, Riley ultimately decided to form a dedicated clinic for MIS-C follow-up – a “one-stop shop” single appointment at each follow-up, Dr. Bhumbra said, that covers labs, EKG, echocardiogram, and any other necessary tests.
“Our goal with the clinic is to make life easier for the families and to be able to coordinate the appointments,” Dr. Rodriguez said. “They will be able to see the three of us, and it would be easier for us to communicate with each other about their plan.”
The clinic began Feb. 11 and occurs twice a month. Though it’s just begun, Dr. Oliver said the first clinic went well, and it’s helping them figure out the role each specialty needs to play in follow-up care.
“For us with rheumatology, after lab values have returned to normal and they’re off steroids, sometimes we think there isn’t much more we can contribute to,” she said. And then there are the patients who didn’t see any rheumatologists while inpatients.
“That’s what we’re trying to figure out as well,” Dr. Oliver said. “Should we be seeing every single kid regardless of whether we were involved in their inpatient [stay] or only seeing the ones we’ve seen?” She expects the coming months will help them work that out.
Texas Children’s Hospital in Houston also uses a dedicated clinic, but they set it up before the first MIS-C patient came through the doors, said Sara Kristen Sexson Tejtel, MD, a pediatric cardiologist at Texas Children’s. The hospital already has other types of multidisciplinary clinics, and they anticipated the challenge of getting families to come to too many appointments in a short period of time.

“Getting someone to come back once is hard enough,” Dr. Sexson Tejtel said. “Getting them to come back twice is impossible.”
Infectious disease is less involved at Texas Children’s, so it’s primarily Dr. Sexson Tejtel and her rheumatologist colleague who see the patients. They hold the clinic once a week, twice if needed.
“It does make the appointment a little longer, but I think the patients appreciate that everything can be addressed with that one visit,” Dr. Sexson Tejtel said. “Being in the hospital as long as some of these kids are is so hard, so making any of that easy as possible is so helpful.” A single appointment also allows the doctors to work together on what labs are needed so that children don’t need multiple labs drawn.
At the appointment, she and the rheumatologist enter the patient’s room and take the patient’s history together.
“It’s nice because it makes the family not to have to repeat things and tell the same story over and over,” she said. “Sometimes I ask questions that then the rheumatologist jumps off of, and then sometimes he’ll ask questions, and I’ll think, ‘Ooh, I’ll ask more questions about that.’ ”
In fact, this team approach at all clinics has made her a more thoughtful, well-rounded physician, she said.
“I have learned so much going to all of my multidisciplinary clinics, and I think I’m able to better care for my patients because I’m not just thinking about it from a cardiac perspective,” she said. “It takes some work, but it’s not hard and I think it is beneficial both for the patient and for the physician. This team approach is definitely where we’re trying to live right now.”
Separate but coordinated appointments
A dedicated clinic isn’t the answer for all institutions, however. At Children’s Hospital of Philadelphia, the size of the networks and all its satellites made a one-stop shop impractical.
“We talked about a consolidated clinic early on, when MIS-C was first emerging and all our groups were collaborating and coming up with our inpatient and outpatient care pathways,” said Sanjeev K. Swami, MD, an infectious disease pediatrician at CHOP and associate professor of clinical pediatrics at the University of Pennsylvania. But timing varies on when each specialist wants to see the families return, and existing clinic schedules and locations varied too much.

So CHOP coordinates appointments individually for each patient, depending on where the patient lives and sometimes stacking them on the same day when possible. Sometimes infectious disease or rheumatology use telehealth, and CHOP, like the other hospitals, prioritizes cardiology, especially for the patients who had cardiac abnormalities in the hospital, Dr. Swami said.
“All three of our groups try to be as flexible as possible. We’ve had a really good collaboration between our groups,” he said, and spreading out follow-up allows specialists to ask about concerns raised at previous appointments, ensuring stronger continuity of care.
“We can make sure things are getting followed up on,” Dr. Swami said. “I think that has been beneficial to make sure things aren’t falling through the cracks.”
CHOP cardiologist Dr. Elias said that ongoing communication, among providers and with families, has been absolutely crucial.
“Everyone’s been talking so frequently about our MIS-C patients while inpatient that by the time they’re an outpatient, it seems to work smoothly, where families are hearing similar items but with a different flair, one from infectious, one from rheumatology, and one from cardiology,” he said.
Children’s Mercy in Kansas City, Mo., also has multiple satellite clinics and follows a model similar to that of CHOP. They discussed having a dedicated multidisciplinary team for each MIS-C patient, but even the logistics of that were difficult, said Emily J. Fox, MD, a rheumatologist and assistant professor of pediatrics at the University of Missouri-Kansas City.

Instead, Children’s Mercy tries to coordinate follow-up appointments to be on the same day and often use telehealth for the rheumatology appointments. Families that live closer to the hospital’s location in Joplin, Mo., go in for their cardiology appointment there, and then Dr. Fox conducts a telehealth appointment with the help of nurses in Joplin.
“We really do try hard, especially since these kids are in the hospital for a long time, to make the coordination as easy as possible,” Dr. Fox said. “This was all was very new, especially in the beginning, but I think at least our group is getting a little bit more comfortable in managing these patients.”
Looking ahead
The biggest question that still looms is what happens to these children, if anything, down the line.
“What was unique about this was this was a new disease we were all learning about together with no baseline,” Dr. Swami said. “None of us had ever seen this condition before.”
So far, the prognosis for the vast majority of children is good. “Most of these kids survive, most of them are doing well, and they almost all recover,” Dr. Serrano said. Labs tend to normalize by 6 weeks post discharge, if not much earlier, and not much cardiac involvement is showing up at later follow-ups. But not even a year has passed, so there’s plenty to learn. “We don’t know if there’s long-term risk. I would not be surprised if 20 years down the road we’re finding out things about this that we had no idea” about, Dr. Serrano said. “Everybody wants answers, and nobody has any, and the answers we have may end up being wrong. That’s how it goes when you’re dealing with something you’ve never seen.”
Research underway will ideally begin providing those answers soon. CHOP is a participating site in an NIH-NHLBI–sponsored study, called COVID MUSIC, that is tracking long-term outcomes for MIS-C at 30 centers across the United States and Canada for 5 years.
“That will really definitely be helpful in answering some of the questions about long-term outcomes,” Dr. Elias said. “We hope this is going to be a transient issue and that patients won’t have any long-term manifestations, but we don’t know that yet.”
Meanwhile, one benefit that has come out of the pandemic is strong collaboration, Dr. Bhumbra said.
“The biggest thing we’re all eagerly waiting and hoping for is standard guidelines on how best to follow-up on these kids, but I know that’s a ways away,” Dr. Bhumbra said. So for now, each institution is doing what it can to develop protocols that they feel best serve the patients’ needs, such as Riley’s new dedicated MIS-C clinic. “It takes a village to take care of these kids, and MIS-C has proven that having a clinic with all three specialties at one clinic is going to be great for the families.”
Dr. Fox serves on a committee for Pfizer unrelated to MIS-C. No other doctors interviewed for this story had relevant conflicts of interest to disclose.
The discovery of any novel disease or condition means a steep learning curve as physicians must develop protocols for diagnosis, management, and follow-up on the fly in the midst of admitting and treating patients. Medical society task forces and committees often release interim guidance during the learning process, but each institution ultimately has to determine what works for them based on their resources, clinical experience, and patient population.
But when the novel condition demands the involvement of multiple different specialties, the challenge of management grows even more complex – as does follow-up after patients are discharged. Such has been the story with multisystem inflammatory syndrome in children (MIS-C), a complication of COVID-19 that shares some features with Kawasaki disease.
The similarities to Kawasaki provided physicians a place to start in developing appropriate treatment regimens and involved a similar interdisciplinary team from, at the least, cardiology and rheumatology, plus infectious disease since MIS-C results from COVID-19.
“It literally has it in the name – multisystem essentially hints that there are multiple specialties involved, multiple hands in the pot trying to manage the kids, and so each specialty has their own kind of unique role in the patient’s care even on the outpatient side,” said Samina S. Bhumbra, MD, an infectious disease pediatrician at Riley Hospital for Children and assistant professor of clinical pediatrics at Indiana University in Indianapolis. “This isn’t a disease that falls under one specialty.”
By July, the American College of Rheumatology had issued interim clinical guidance for management that most children’s hospitals have followed or slightly adapted. But ACR guidelines could not address how each institution should handle outpatient follow-up visits, especially since those visits required, again, at least cardiology and rheumatology if not infectious disease or other specialties as well.
“When their kids are admitted to the hospital, to be told at discharge you have to be followed up by all these specialists is a lot to handle,” Dr. Bhumbra said. But just as it’s difficult for parents to deal with the need to see several different doctors after discharge, it can be difficult at some institutions for physicians to design a follow-up schedule that can accommodate families, especially families who live far from the hospital in the first place.
“Some of our follow-up is disjointed because all of our clinics had never been on the same day just because of staff availability,” Dr. Bhumbra said. “But it can be a 2- to 3-hour drive for some of our patients, depending on how far they’re coming.”
Many of them can’t make that drive more than once in the same month, much less the same week.
“If you have multiple visits, it makes it more likely that they’re not showing up,” said Ryan M. Serrano, MD, a pediatric cardiologist at Riley and assistant professor of pediatrics at Indiana University. Riley used telehealth when possible, especially if families could get labs done near home. But pediatric echocardiograms require technicians who have experience with children, so families need to come to the hospital.
Children’s hospitals have therefore had to adapt scheduling strategies or develop pediatric specialty clinics to coordinate across the multiple departments and accommodate a complex follow-up regimen that is still evolving as physicians learn more about MIS-C.
Determining a follow-up regimen
Even before determining how to coordinate appointments, hospitals had to decide what follow-up itself should be.
“How long do we follow these patients and how often do we follow them?” said Melissa S. Oliver, MD, a rheumatologist at Riley and assistant professor of clinical pediatrics at Indiana University.
“We’re seeing that a lot of our patients rapidly respond when they get appropriate therapy, but we don’t know about long-term outcomes yet. We’re all still learning.”
At Children’s Hospital of Philadelphia, infectious disease follows up 4-6 weeks post discharge. The cardiology division came up with a follow-up plan that has evolved over time, said Matthew Elias, MD, an attending cardiologist at CHOP’s Cardiac Center and clinical assistant professor of pediatrics at the University of Pennsylvania, Philadelphia.
Patients get an EKG and echocardiogram at 2 weeks and, if their condition is stable, 6 weeks after discharge. After that, it depends on the patient’s clinical situation. Patients with moderately diminished left ventricular systolic function are recommended to get an MRI scan 3 months after discharge and, if old enough, exercise stress tests. Otherwise, they are seen at 6 months, but that appointment is optional for those whose prior echos have consistently been normal.
Other institutions, including Riley, are following a similar schedule of 2-week, 6-week, and 6-month postdischarge follow-ups, and most plan to do a 1-year follow-up as well, although that 1-year mark hasn’t arrived yet for most. Most do rheumatology labs at the 2-week appointment and use that to determine steroids management and whether labs are needed at the 6-week appointment. If labs have normalized, they aren’t done at 6 months. Small variations in follow-up management exist across institutions, but all are remaining open to changes. Riley, for example, is considering MRI screening for ongoing cardiac inflammation at 6 months to a year for all patients, Dr. Serrano said.
The dedicated clinic model
The two challenges Riley needed to address were the lack of a clear consensus on what MIS-C follow-up should look like and the need for continuity of care, Dr. Serrano said.
Regular discussion in departmental meetings at Riley “progressed from how do we take care of them and what treatments do we give them to how do we follow them and manage them in outpatient,” Dr. Oliver said. In the inpatient setting, they had an interdisciplinary team, but how could they maintain that for outpatients without overwhelming the families?
“I think the main challenge is for the families to identify who is leading the care for them,” said Martha M. Rodriguez, MD, a rheumatologist at Riley and assistant professor of clinical pediatrics at Indiana University. That sometimes led to families picking which follow-up appointments they would attend and which they would skip if they could not make them all – and sometimes they skipped the more important ones. “They would go to the appointment with me and then miss the cardiology appointments and the echocardiogram, which was more important to follow any abnormalities in the heart,” Dr. Rodriguez said.
After trying to coordinate separate follow-up appointments for months, Riley ultimately decided to form a dedicated clinic for MIS-C follow-up – a “one-stop shop” single appointment at each follow-up, Dr. Bhumbra said, that covers labs, EKG, echocardiogram, and any other necessary tests.
“Our goal with the clinic is to make life easier for the families and to be able to coordinate the appointments,” Dr. Rodriguez said. “They will be able to see the three of us, and it would be easier for us to communicate with each other about their plan.”
The clinic began Feb. 11 and occurs twice a month. Though it’s just begun, Dr. Oliver said the first clinic went well, and it’s helping them figure out the role each specialty needs to play in follow-up care.
“For us with rheumatology, after lab values have returned to normal and they’re off steroids, sometimes we think there isn’t much more we can contribute to,” she said. And then there are the patients who didn’t see any rheumatologists while inpatients.
“That’s what we’re trying to figure out as well,” Dr. Oliver said. “Should we be seeing every single kid regardless of whether we were involved in their inpatient [stay] or only seeing the ones we’ve seen?” She expects the coming months will help them work that out.
Texas Children’s Hospital in Houston also uses a dedicated clinic, but they set it up before the first MIS-C patient came through the doors, said Sara Kristen Sexson Tejtel, MD, a pediatric cardiologist at Texas Children’s. The hospital already has other types of multidisciplinary clinics, and they anticipated the challenge of getting families to come to too many appointments in a short period of time.
“Getting someone to come back once is hard enough,” Dr. Sexson Tejtel said. “Getting them to come back twice is impossible.”
Infectious disease is less involved at Texas Children’s, so it’s primarily Dr. Sexson Tejtel and her rheumatologist colleague who see the patients. They hold the clinic once a week, twice if needed.
“It does make the appointment a little longer, but I think the patients appreciate that everything can be addressed with that one visit,” Dr. Sexson Tejtel said. “Being in the hospital as long as some of these kids are is so hard, so making any of that easy as possible is so helpful.” A single appointment also allows the doctors to work together on what labs are needed so that children don’t need multiple labs drawn.
At the appointment, she and the rheumatologist enter the patient’s room and take the patient’s history together.
“It’s nice because it makes the family not to have to repeat things and tell the same story over and over,” she said. “Sometimes I ask questions that then the rheumatologist jumps off of, and then sometimes he’ll ask questions, and I’ll think, ‘Ooh, I’ll ask more questions about that.’ ”
In fact, this team approach at all clinics has made her a more thoughtful, well-rounded physician, she said.
“I have learned so much going to all of my multidisciplinary clinics, and I think I’m able to better care for my patients because I’m not just thinking about it from a cardiac perspective,” she said. “It takes some work, but it’s not hard and I think it is beneficial both for the patient and for the physician. This team approach is definitely where we’re trying to live right now.”
Separate but coordinated appointments
A dedicated clinic isn’t the answer for all institutions, however. At Children’s Hospital of Philadelphia, the size of the networks and all its satellites made a one-stop shop impractical.
“We talked about a consolidated clinic early on, when MIS-C was first emerging and all our groups were collaborating and coming up with our inpatient and outpatient care pathways,” said Sanjeev K. Swami, MD, an infectious disease pediatrician at CHOP and associate professor of clinical pediatrics at the University of Pennsylvania. But timing varies on when each specialist wants to see the families return, and existing clinic schedules and locations varied too much.
So CHOP coordinates appointments individually for each patient, depending on where the patient lives and sometimes stacking them on the same day when possible. Sometimes infectious disease or rheumatology use telehealth, and CHOP, like the other hospitals, prioritizes cardiology, especially for the patients who had cardiac abnormalities in the hospital, Dr. Swami said.
“All three of our groups try to be as flexible as possible. We’ve had a really good collaboration between our groups,” he said, and spreading out follow-up allows specialists to ask about concerns raised at previous appointments, ensuring stronger continuity of care.
“We can make sure things are getting followed up on,” Dr. Swami said. “I think that has been beneficial to make sure things aren’t falling through the cracks.”
CHOP cardiologist Dr. Elias said that ongoing communication, among providers and with families, has been absolutely crucial.
“Everyone’s been talking so frequently about our MIS-C patients while inpatient that by the time they’re an outpatient, it seems to work smoothly, where families are hearing similar items but with a different flair, one from infectious, one from rheumatology, and one from cardiology,” he said.
Children’s Mercy in Kansas City, Mo., also has multiple satellite clinics and follows a model similar to that of CHOP. They discussed having a dedicated multidisciplinary team for each MIS-C patient, but even the logistics of that were difficult, said Emily J. Fox, MD, a rheumatologist and assistant professor of pediatrics at the University of Missouri-Kansas City.
Instead, Children’s Mercy tries to coordinate follow-up appointments to be on the same day and often use telehealth for the rheumatology appointments. Families that live closer to the hospital’s location in Joplin, Mo., go in for their cardiology appointment there, and then Dr. Fox conducts a telehealth appointment with the help of nurses in Joplin.
“We really do try hard, especially since these kids are in the hospital for a long time, to make the coordination as easy as possible,” Dr. Fox said. “This was all was very new, especially in the beginning, but I think at least our group is getting a little bit more comfortable in managing these patients.”
Looking ahead
The biggest question that still looms is what happens to these children, if anything, down the line.
“What was unique about this was this was a new disease we were all learning about together with no baseline,” Dr. Swami said. “None of us had ever seen this condition before.”
So far, the prognosis for the vast majority of children is good. “Most of these kids survive, most of them are doing well, and they almost all recover,” Dr. Serrano said. Labs tend to normalize by 6 weeks post discharge, if not much earlier, and not much cardiac involvement is showing up at later follow-ups. But not even a year has passed, so there’s plenty to learn. “We don’t know if there’s long-term risk. I would not be surprised if 20 years down the road we’re finding out things about this that we had no idea” about, Dr. Serrano said. “Everybody wants answers, and nobody has any, and the answers we have may end up being wrong. That’s how it goes when you’re dealing with something you’ve never seen.”
Research underway will ideally begin providing those answers soon. CHOP is a participating site in an NIH-NHLBI–sponsored study, called COVID MUSIC, that is tracking long-term outcomes for MIS-C at 30 centers across the United States and Canada for 5 years.
“That will really definitely be helpful in answering some of the questions about long-term outcomes,” Dr. Elias said. “We hope this is going to be a transient issue and that patients won’t have any long-term manifestations, but we don’t know that yet.”
Meanwhile, one benefit that has come out of the pandemic is strong collaboration, Dr. Bhumbra said.
“The biggest thing we’re all eagerly waiting and hoping for is standard guidelines on how best to follow-up on these kids, but I know that’s a ways away,” Dr. Bhumbra said. So for now, each institution is doing what it can to develop protocols that they feel best serve the patients’ needs, such as Riley’s new dedicated MIS-C clinic. “It takes a village to take care of these kids, and MIS-C has proven that having a clinic with all three specialties at one clinic is going to be great for the families.”
Dr. Fox serves on a committee for Pfizer unrelated to MIS-C. No other doctors interviewed for this story had relevant conflicts of interest to disclose.
The discovery of any novel disease or condition means a steep learning curve as physicians must develop protocols for diagnosis, management, and follow-up on the fly in the midst of admitting and treating patients. Medical society task forces and committees often release interim guidance during the learning process, but each institution ultimately has to determine what works for them based on their resources, clinical experience, and patient population.
But when the novel condition demands the involvement of multiple different specialties, the challenge of management grows even more complex – as does follow-up after patients are discharged. Such has been the story with multisystem inflammatory syndrome in children (MIS-C), a complication of COVID-19 that shares some features with Kawasaki disease.
The similarities to Kawasaki provided physicians a place to start in developing appropriate treatment regimens and involved a similar interdisciplinary team from, at the least, cardiology and rheumatology, plus infectious disease since MIS-C results from COVID-19.
“It literally has it in the name – multisystem essentially hints that there are multiple specialties involved, multiple hands in the pot trying to manage the kids, and so each specialty has their own kind of unique role in the patient’s care even on the outpatient side,” said Samina S. Bhumbra, MD, an infectious disease pediatrician at Riley Hospital for Children and assistant professor of clinical pediatrics at Indiana University in Indianapolis. “This isn’t a disease that falls under one specialty.”
By July, the American College of Rheumatology had issued interim clinical guidance for management that most children’s hospitals have followed or slightly adapted. But ACR guidelines could not address how each institution should handle outpatient follow-up visits, especially since those visits required, again, at least cardiology and rheumatology if not infectious disease or other specialties as well.
“When their kids are admitted to the hospital, to be told at discharge you have to be followed up by all these specialists is a lot to handle,” Dr. Bhumbra said. But just as it’s difficult for parents to deal with the need to see several different doctors after discharge, it can be difficult at some institutions for physicians to design a follow-up schedule that can accommodate families, especially families who live far from the hospital in the first place.
“Some of our follow-up is disjointed because all of our clinics had never been on the same day just because of staff availability,” Dr. Bhumbra said. “But it can be a 2- to 3-hour drive for some of our patients, depending on how far they’re coming.”
Many of them can’t make that drive more than once in the same month, much less the same week.
“If you have multiple visits, it makes it more likely that they’re not showing up,” said Ryan M. Serrano, MD, a pediatric cardiologist at Riley and assistant professor of pediatrics at Indiana University. Riley used telehealth when possible, especially if families could get labs done near home. But pediatric echocardiograms require technicians who have experience with children, so families need to come to the hospital.
Children’s hospitals have therefore had to adapt scheduling strategies or develop pediatric specialty clinics to coordinate across the multiple departments and accommodate a complex follow-up regimen that is still evolving as physicians learn more about MIS-C.
Determining a follow-up regimen
Even before determining how to coordinate appointments, hospitals had to decide what follow-up itself should be.
“How long do we follow these patients and how often do we follow them?” said Melissa S. Oliver, MD, a rheumatologist at Riley and assistant professor of clinical pediatrics at Indiana University.
“We’re seeing that a lot of our patients rapidly respond when they get appropriate therapy, but we don’t know about long-term outcomes yet. We’re all still learning.”
At Children’s Hospital of Philadelphia, infectious disease follows up 4-6 weeks post discharge. The cardiology division came up with a follow-up plan that has evolved over time, said Matthew Elias, MD, an attending cardiologist at CHOP’s Cardiac Center and clinical assistant professor of pediatrics at the University of Pennsylvania, Philadelphia.
Patients get an EKG and echocardiogram at 2 weeks and, if their condition is stable, 6 weeks after discharge. After that, it depends on the patient’s clinical situation. Patients with moderately diminished left ventricular systolic function are recommended to get an MRI scan 3 months after discharge and, if old enough, exercise stress tests. Otherwise, they are seen at 6 months, but that appointment is optional for those whose prior echos have consistently been normal.
Other institutions, including Riley, are following a similar schedule of 2-week, 6-week, and 6-month postdischarge follow-ups, and most plan to do a 1-year follow-up as well, although that 1-year mark hasn’t arrived yet for most. Most do rheumatology labs at the 2-week appointment and use that to determine steroids management and whether labs are needed at the 6-week appointment. If labs have normalized, they aren’t done at 6 months. Small variations in follow-up management exist across institutions, but all are remaining open to changes. Riley, for example, is considering MRI screening for ongoing cardiac inflammation at 6 months to a year for all patients, Dr. Serrano said.
The dedicated clinic model
The two challenges Riley needed to address were the lack of a clear consensus on what MIS-C follow-up should look like and the need for continuity of care, Dr. Serrano said.
Regular discussion in departmental meetings at Riley “progressed from how do we take care of them and what treatments do we give them to how do we follow them and manage them in outpatient,” Dr. Oliver said. In the inpatient setting, they had an interdisciplinary team, but how could they maintain that for outpatients without overwhelming the families?
“I think the main challenge is for the families to identify who is leading the care for them,” said Martha M. Rodriguez, MD, a rheumatologist at Riley and assistant professor of clinical pediatrics at Indiana University. That sometimes led to families picking which follow-up appointments they would attend and which they would skip if they could not make them all – and sometimes they skipped the more important ones. “They would go to the appointment with me and then miss the cardiology appointments and the echocardiogram, which was more important to follow any abnormalities in the heart,” Dr. Rodriguez said.
After trying to coordinate separate follow-up appointments for months, Riley ultimately decided to form a dedicated clinic for MIS-C follow-up – a “one-stop shop” single appointment at each follow-up, Dr. Bhumbra said, that covers labs, EKG, echocardiogram, and any other necessary tests.
“Our goal with the clinic is to make life easier for the families and to be able to coordinate the appointments,” Dr. Rodriguez said. “They will be able to see the three of us, and it would be easier for us to communicate with each other about their plan.”
The clinic began Feb. 11 and occurs twice a month. Though it’s just begun, Dr. Oliver said the first clinic went well, and it’s helping them figure out the role each specialty needs to play in follow-up care.
“For us with rheumatology, after lab values have returned to normal and they’re off steroids, sometimes we think there isn’t much more we can contribute to,” she said. And then there are the patients who didn’t see any rheumatologists while inpatients.
“That’s what we’re trying to figure out as well,” Dr. Oliver said. “Should we be seeing every single kid regardless of whether we were involved in their inpatient [stay] or only seeing the ones we’ve seen?” She expects the coming months will help them work that out.
Texas Children’s Hospital in Houston also uses a dedicated clinic, but they set it up before the first MIS-C patient came through the doors, said Sara Kristen Sexson Tejtel, MD, a pediatric cardiologist at Texas Children’s. The hospital already has other types of multidisciplinary clinics, and they anticipated the challenge of getting families to come to too many appointments in a short period of time.
“Getting someone to come back once is hard enough,” Dr. Sexson Tejtel said. “Getting them to come back twice is impossible.”
Infectious disease is less involved at Texas Children’s, so it’s primarily Dr. Sexson Tejtel and her rheumatologist colleague who see the patients. They hold the clinic once a week, twice if needed.
“It does make the appointment a little longer, but I think the patients appreciate that everything can be addressed with that one visit,” Dr. Sexson Tejtel said. “Being in the hospital as long as some of these kids are is so hard, so making any of that easy as possible is so helpful.” A single appointment also allows the doctors to work together on what labs are needed so that children don’t need multiple labs drawn.
At the appointment, she and the rheumatologist enter the patient’s room and take the patient’s history together.
“It’s nice because it makes the family not to have to repeat things and tell the same story over and over,” she said. “Sometimes I ask questions that then the rheumatologist jumps off of, and then sometimes he’ll ask questions, and I’ll think, ‘Ooh, I’ll ask more questions about that.’ ”
In fact, this team approach at all clinics has made her a more thoughtful, well-rounded physician, she said.
“I have learned so much going to all of my multidisciplinary clinics, and I think I’m able to better care for my patients because I’m not just thinking about it from a cardiac perspective,” she said. “It takes some work, but it’s not hard and I think it is beneficial both for the patient and for the physician. This team approach is definitely where we’re trying to live right now.”
Separate but coordinated appointments
A dedicated clinic isn’t the answer for all institutions, however. At Children’s Hospital of Philadelphia, the size of the networks and all its satellites made a one-stop shop impractical.
“We talked about a consolidated clinic early on, when MIS-C was first emerging and all our groups were collaborating and coming up with our inpatient and outpatient care pathways,” said Sanjeev K. Swami, MD, an infectious disease pediatrician at CHOP and associate professor of clinical pediatrics at the University of Pennsylvania. But timing varies on when each specialist wants to see the families return, and existing clinic schedules and locations varied too much.
So CHOP coordinates appointments individually for each patient, depending on where the patient lives and sometimes stacking them on the same day when possible. Sometimes infectious disease or rheumatology use telehealth, and CHOP, like the other hospitals, prioritizes cardiology, especially for the patients who had cardiac abnormalities in the hospital, Dr. Swami said.
“All three of our groups try to be as flexible as possible. We’ve had a really good collaboration between our groups,” he said, and spreading out follow-up allows specialists to ask about concerns raised at previous appointments, ensuring stronger continuity of care.
“We can make sure things are getting followed up on,” Dr. Swami said. “I think that has been beneficial to make sure things aren’t falling through the cracks.”
CHOP cardiologist Dr. Elias said that ongoing communication, among providers and with families, has been absolutely crucial.
“Everyone’s been talking so frequently about our MIS-C patients while inpatient that by the time they’re an outpatient, it seems to work smoothly, where families are hearing similar items but with a different flair, one from infectious, one from rheumatology, and one from cardiology,” he said.
Children’s Mercy in Kansas City, Mo., also has multiple satellite clinics and follows a model similar to that of CHOP. They discussed having a dedicated multidisciplinary team for each MIS-C patient, but even the logistics of that were difficult, said Emily J. Fox, MD, a rheumatologist and assistant professor of pediatrics at the University of Missouri-Kansas City.
Instead, Children’s Mercy tries to coordinate follow-up appointments to be on the same day and often use telehealth for the rheumatology appointments. Families that live closer to the hospital’s location in Joplin, Mo., go in for their cardiology appointment there, and then Dr. Fox conducts a telehealth appointment with the help of nurses in Joplin.
“We really do try hard, especially since these kids are in the hospital for a long time, to make the coordination as easy as possible,” Dr. Fox said. “This was all was very new, especially in the beginning, but I think at least our group is getting a little bit more comfortable in managing these patients.”
Looking ahead
The biggest question that still looms is what happens to these children, if anything, down the line.
“What was unique about this was this was a new disease we were all learning about together with no baseline,” Dr. Swami said. “None of us had ever seen this condition before.”
So far, the prognosis for the vast majority of children is good. “Most of these kids survive, most of them are doing well, and they almost all recover,” Dr. Serrano said. Labs tend to normalize by 6 weeks post discharge, if not much earlier, and not much cardiac involvement is showing up at later follow-ups. But not even a year has passed, so there’s plenty to learn. “We don’t know if there’s long-term risk. I would not be surprised if 20 years down the road we’re finding out things about this that we had no idea” about, Dr. Serrano said. “Everybody wants answers, and nobody has any, and the answers we have may end up being wrong. That’s how it goes when you’re dealing with something you’ve never seen.”
Research underway will ideally begin providing those answers soon. CHOP is a participating site in an NIH-NHLBI–sponsored study, called COVID MUSIC, that is tracking long-term outcomes for MIS-C at 30 centers across the United States and Canada for 5 years.
“That will really definitely be helpful in answering some of the questions about long-term outcomes,” Dr. Elias said. “We hope this is going to be a transient issue and that patients won’t have any long-term manifestations, but we don’t know that yet.”
Meanwhile, one benefit that has come out of the pandemic is strong collaboration, Dr. Bhumbra said.
“The biggest thing we’re all eagerly waiting and hoping for is standard guidelines on how best to follow-up on these kids, but I know that’s a ways away,” Dr. Bhumbra said. So for now, each institution is doing what it can to develop protocols that they feel best serve the patients’ needs, such as Riley’s new dedicated MIS-C clinic. “It takes a village to take care of these kids, and MIS-C has proven that having a clinic with all three specialties at one clinic is going to be great for the families.”
Dr. Fox serves on a committee for Pfizer unrelated to MIS-C. No other doctors interviewed for this story had relevant conflicts of interest to disclose.
Tocilizumab (Actemra) scores FDA approval for systemic sclerosis–associated interstitial lung disease
The Food and Drug Administration has approved subcutaneously-injected tocilizumab (Actemra) to reduce the rate of pulmonary function decline in systemic sclerosis–associated interstitial lung disease (SSc-ILD) patients, according to a press release from manufacturer Genentech.

Tocilizumab is the first biologic to be approved by the agency for adults with SSc-ILD, a rare and potentially life-threatening condition that may affect up to 80% of SSc patients and lead to lung inflammation and scarring.
The approval was based primarily on data from a phase 3 randomized, double-blind, placebo-controlled clinical trial (the focuSSced trial) that included 212 adults with SSc. Although that study failed to meet its primary endpoint of change from baseline to 48 weeks in the modified Rodnan Skin Score, the researchers observed a significantly reduced lung function decline as measured by forced vital capacity (FVC) and percent predicted forced vital capacity (ppFVC) among tocilizumab-treated patients, compared with those who received placebo. A total of 68 patients (65%) in the tocilizumab group and 68 patients (64%) in the placebo group had SSc-ILD at baseline.
In a subgroup analysis, patients taking tocilizumab had a smaller decline in mean ppFVC, compared with placebo patients (0.07% vs. –6.4%; mean difference, 6.47%), and a smaller decline in FVC (mean change –14 mL vs. –255 mL with placebo; mean difference, 241 mL).
The mean change from baseline to week 48 in modified Rodnan Skin Score was –5.88 for patients on tocilizumab and –3.77 with placebo.
Safety data were similar between tocilizumab and placebo groups through 48 weeks, and similar for patients with and without SSc-ILD. In general, tocilizumab side effects include increased susceptibility to infections, and serious side effects may include stomach tears, hepatotoxicity, and increased risk of cancer and hepatitis B, according to the prescribing information. However, the most common side effects are upper respiratory tract infections, headache, hypertension, and injection-site reactions.
Tocilizumab, an interleukin-6 receptor antagonist, is already approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis, as well as for adult patients with giant cell arteritis; patients aged 2 years and older with active polyarticular juvenile idiopathic arthritis or active systemic juvenile idiopathic arthritis; and adults and pediatric patients 2 years of age and older with chimeric antigen receptor T-cell–induced severe or life-threatening cytokine release syndrome.
Prescribing information is available here.
The Food and Drug Administration has approved subcutaneously-injected tocilizumab (Actemra) to reduce the rate of pulmonary function decline in systemic sclerosis–associated interstitial lung disease (SSc-ILD) patients, according to a press release from manufacturer Genentech.
Tocilizumab is the first biologic to be approved by the agency for adults with SSc-ILD, a rare and potentially life-threatening condition that may affect up to 80% of SSc patients and lead to lung inflammation and scarring.
The approval was based primarily on data from a phase 3 randomized, double-blind, placebo-controlled clinical trial (the focuSSced trial) that included 212 adults with SSc. Although that study failed to meet its primary endpoint of change from baseline to 48 weeks in the modified Rodnan Skin Score, the researchers observed a significantly reduced lung function decline as measured by forced vital capacity (FVC) and percent predicted forced vital capacity (ppFVC) among tocilizumab-treated patients, compared with those who received placebo. A total of 68 patients (65%) in the tocilizumab group and 68 patients (64%) in the placebo group had SSc-ILD at baseline.
In a subgroup analysis, patients taking tocilizumab had a smaller decline in mean ppFVC, compared with placebo patients (0.07% vs. –6.4%; mean difference, 6.47%), and a smaller decline in FVC (mean change –14 mL vs. –255 mL with placebo; mean difference, 241 mL).
The mean change from baseline to week 48 in modified Rodnan Skin Score was –5.88 for patients on tocilizumab and –3.77 with placebo.
Safety data were similar between tocilizumab and placebo groups through 48 weeks, and similar for patients with and without SSc-ILD. In general, tocilizumab side effects include increased susceptibility to infections, and serious side effects may include stomach tears, hepatotoxicity, and increased risk of cancer and hepatitis B, according to the prescribing information. However, the most common side effects are upper respiratory tract infections, headache, hypertension, and injection-site reactions.
Tocilizumab, an interleukin-6 receptor antagonist, is already approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis, as well as for adult patients with giant cell arteritis; patients aged 2 years and older with active polyarticular juvenile idiopathic arthritis or active systemic juvenile idiopathic arthritis; and adults and pediatric patients 2 years of age and older with chimeric antigen receptor T-cell–induced severe or life-threatening cytokine release syndrome.
Prescribing information is available here.
The Food and Drug Administration has approved subcutaneously-injected tocilizumab (Actemra) to reduce the rate of pulmonary function decline in systemic sclerosis–associated interstitial lung disease (SSc-ILD) patients, according to a press release from manufacturer Genentech.
Tocilizumab is the first biologic to be approved by the agency for adults with SSc-ILD, a rare and potentially life-threatening condition that may affect up to 80% of SSc patients and lead to lung inflammation and scarring.
The approval was based primarily on data from a phase 3 randomized, double-blind, placebo-controlled clinical trial (the focuSSced trial) that included 212 adults with SSc. Although that study failed to meet its primary endpoint of change from baseline to 48 weeks in the modified Rodnan Skin Score, the researchers observed a significantly reduced lung function decline as measured by forced vital capacity (FVC) and percent predicted forced vital capacity (ppFVC) among tocilizumab-treated patients, compared with those who received placebo. A total of 68 patients (65%) in the tocilizumab group and 68 patients (64%) in the placebo group had SSc-ILD at baseline.
In a subgroup analysis, patients taking tocilizumab had a smaller decline in mean ppFVC, compared with placebo patients (0.07% vs. –6.4%; mean difference, 6.47%), and a smaller decline in FVC (mean change –14 mL vs. –255 mL with placebo; mean difference, 241 mL).
The mean change from baseline to week 48 in modified Rodnan Skin Score was –5.88 for patients on tocilizumab and –3.77 with placebo.
Safety data were similar between tocilizumab and placebo groups through 48 weeks, and similar for patients with and without SSc-ILD. In general, tocilizumab side effects include increased susceptibility to infections, and serious side effects may include stomach tears, hepatotoxicity, and increased risk of cancer and hepatitis B, according to the prescribing information. However, the most common side effects are upper respiratory tract infections, headache, hypertension, and injection-site reactions.
Tocilizumab, an interleukin-6 receptor antagonist, is already approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis, as well as for adult patients with giant cell arteritis; patients aged 2 years and older with active polyarticular juvenile idiopathic arthritis or active systemic juvenile idiopathic arthritis; and adults and pediatric patients 2 years of age and older with chimeric antigen receptor T-cell–induced severe or life-threatening cytokine release syndrome.
Prescribing information is available here.
FDA approves first targeted treatment for rare DMD mutation
, the agency has announced.
This particular mutation of the DMD gene “is amenable to exon 45 skipping,” the FDA noted in a press release. The agency added that this is its first approval of a targeted treatment for patients with the mutation.
“Developing drugs designed for patients with specific mutations is a critical part of personalized medicine,” Eric Bastings, MD, deputy director of the Office of Neuroscience at the FDA’s Center for Drug Evaluation and Research, said in a statement.
The approval was based on results from a 43-person randomized controlled trial. Patients who received casimersen had a greater increase in production of the muscle-fiber protein dystrophin compared with their counterparts who received placebo.
Approved – with cautions
The FDA noted that DMD prevalence worldwide is about 1 in 3,600 boys – although it can also affect girls in rare cases. Symptoms of the disorder are commonly first observed around age 3 years but worsen steadily over time. DMD gene mutations lead to a decrease in dystrophin.
As reported by Medscape Medical News in August, the FDA approved viltolarsen (Viltepso, NS Pharma) for the treatment of DMD in patients with a confirmed mutation amenable to exon 53 skipping, following approval of golodirsen injection (Vyondys 53, Sarepta Therapeutics) for the same indication in December 2019.
The DMD gene mutation that is amenable to exon 45 skipping is present in about 8% of patients with DMD.
The trial that carried weight with the FDA included 43 male participants with DMD aged 7-20 years. All were confirmed to have the exon 45-skipping gene mutation and all were randomly assigned 2:1 to received IV casimersen 30 mg/kg or matching placebo.
Results showed that, between baseline and 48 weeks post treatment, the casimersen group showed a significantly higher increase in levels of dystrophin protein than in the placebo group.
Upper respiratory tract infections, fever, joint and throat pain, headache, and cough were the most common adverse events experienced by the active-treatment group.
Although the clinical studies assessing casimersen did not show any reports of kidney toxicity, the adverse event was observed in some nonclinical studies. Therefore, clinicians should monitor kidney function in any patient receiving this treatment, the FDA recommended.
Overall, “the FDA has concluded that the data submitted by the applicant demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit” in this patient population, the agency said in its press release.
However, it noted that definitive clinical benefits such as improved motor function were not “established.”
“In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of [other] available therapy,” the agency said.
It added that the manufacturer is currently conducting a multicenter study focused on the safety and efficacy of the drug in ambulatory patients with DMD.
The FDA approved casimersen using its Accelerated Approval pathway, granted Fast Track and Priority Review designations to its applications, and gave the treatment Orphan Drug designation.
A version of this article first appeared on Medscape.com.
, the agency has announced.
This particular mutation of the DMD gene “is amenable to exon 45 skipping,” the FDA noted in a press release. The agency added that this is its first approval of a targeted treatment for patients with the mutation.
“Developing drugs designed for patients with specific mutations is a critical part of personalized medicine,” Eric Bastings, MD, deputy director of the Office of Neuroscience at the FDA’s Center for Drug Evaluation and Research, said in a statement.
The approval was based on results from a 43-person randomized controlled trial. Patients who received casimersen had a greater increase in production of the muscle-fiber protein dystrophin compared with their counterparts who received placebo.
Approved – with cautions
The FDA noted that DMD prevalence worldwide is about 1 in 3,600 boys – although it can also affect girls in rare cases. Symptoms of the disorder are commonly first observed around age 3 years but worsen steadily over time. DMD gene mutations lead to a decrease in dystrophin.
As reported by Medscape Medical News in August, the FDA approved viltolarsen (Viltepso, NS Pharma) for the treatment of DMD in patients with a confirmed mutation amenable to exon 53 skipping, following approval of golodirsen injection (Vyondys 53, Sarepta Therapeutics) for the same indication in December 2019.
The DMD gene mutation that is amenable to exon 45 skipping is present in about 8% of patients with DMD.
The trial that carried weight with the FDA included 43 male participants with DMD aged 7-20 years. All were confirmed to have the exon 45-skipping gene mutation and all were randomly assigned 2:1 to received IV casimersen 30 mg/kg or matching placebo.
Results showed that, between baseline and 48 weeks post treatment, the casimersen group showed a significantly higher increase in levels of dystrophin protein than in the placebo group.
Upper respiratory tract infections, fever, joint and throat pain, headache, and cough were the most common adverse events experienced by the active-treatment group.
Although the clinical studies assessing casimersen did not show any reports of kidney toxicity, the adverse event was observed in some nonclinical studies. Therefore, clinicians should monitor kidney function in any patient receiving this treatment, the FDA recommended.
Overall, “the FDA has concluded that the data submitted by the applicant demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit” in this patient population, the agency said in its press release.
However, it noted that definitive clinical benefits such as improved motor function were not “established.”
“In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of [other] available therapy,” the agency said.
It added that the manufacturer is currently conducting a multicenter study focused on the safety and efficacy of the drug in ambulatory patients with DMD.
The FDA approved casimersen using its Accelerated Approval pathway, granted Fast Track and Priority Review designations to its applications, and gave the treatment Orphan Drug designation.
A version of this article first appeared on Medscape.com.
, the agency has announced.
This particular mutation of the DMD gene “is amenable to exon 45 skipping,” the FDA noted in a press release. The agency added that this is its first approval of a targeted treatment for patients with the mutation.
“Developing drugs designed for patients with specific mutations is a critical part of personalized medicine,” Eric Bastings, MD, deputy director of the Office of Neuroscience at the FDA’s Center for Drug Evaluation and Research, said in a statement.
The approval was based on results from a 43-person randomized controlled trial. Patients who received casimersen had a greater increase in production of the muscle-fiber protein dystrophin compared with their counterparts who received placebo.
Approved – with cautions
The FDA noted that DMD prevalence worldwide is about 1 in 3,600 boys – although it can also affect girls in rare cases. Symptoms of the disorder are commonly first observed around age 3 years but worsen steadily over time. DMD gene mutations lead to a decrease in dystrophin.
As reported by Medscape Medical News in August, the FDA approved viltolarsen (Viltepso, NS Pharma) for the treatment of DMD in patients with a confirmed mutation amenable to exon 53 skipping, following approval of golodirsen injection (Vyondys 53, Sarepta Therapeutics) for the same indication in December 2019.
The DMD gene mutation that is amenable to exon 45 skipping is present in about 8% of patients with DMD.
The trial that carried weight with the FDA included 43 male participants with DMD aged 7-20 years. All were confirmed to have the exon 45-skipping gene mutation and all were randomly assigned 2:1 to received IV casimersen 30 mg/kg or matching placebo.
Results showed that, between baseline and 48 weeks post treatment, the casimersen group showed a significantly higher increase in levels of dystrophin protein than in the placebo group.
Upper respiratory tract infections, fever, joint and throat pain, headache, and cough were the most common adverse events experienced by the active-treatment group.
Although the clinical studies assessing casimersen did not show any reports of kidney toxicity, the adverse event was observed in some nonclinical studies. Therefore, clinicians should monitor kidney function in any patient receiving this treatment, the FDA recommended.
Overall, “the FDA has concluded that the data submitted by the applicant demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit” in this patient population, the agency said in its press release.
However, it noted that definitive clinical benefits such as improved motor function were not “established.”
“In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of [other] available therapy,” the agency said.
It added that the manufacturer is currently conducting a multicenter study focused on the safety and efficacy of the drug in ambulatory patients with DMD.
The FDA approved casimersen using its Accelerated Approval pathway, granted Fast Track and Priority Review designations to its applications, and gave the treatment Orphan Drug designation.
A version of this article first appeared on Medscape.com.
New steroid dosing regimen for myasthenia gravis
. The trial showed that the conventional slow tapering regimen enabled discontinuation of prednisone earlier than previously reported but the new rapid-tapering regimen enabled an even faster discontinuation.
Noting that although both regimens led to a comparable myasthenia gravis status and prednisone dose at 15 months, the authors stated: “We think that the reduction of the cumulative dose over a year (equivalent to 5 mg/day) is a clinically relevant reduction, since the risk of complications is proportional to the daily or cumulative doses of prednisone.
“Our results warrant testing of a more rapid-tapering regimen in a future trial. In the meantime, our trial provides useful information on how prednisone tapering could be managed in patients with generalized myasthenia gravis treated with azathioprine,” they concluded.
The trial was published online Feb. 8 in JAMA Neurology.
Myasthenia gravis is a disorder of neuromuscular transmission, resulting from autoantibodies to components of the neuromuscular junction, most commonly the acetylcholine receptor. The incidence ranges from 0.3 to 2.8 per 100,000, and it is estimated to affect more than 700,000 people worldwide.
The authors of the current paper, led by Tarek Sharshar, MD, PhD, Groupe Hospitalier Universitaire, Paris, explained that many patients whose symptoms are not controlled by cholinesterase inhibitors are treated with corticosteroids and an immunosuppressant, usually azathioprine. No specific dosing protocol for prednisone has been validated, but it is commonly gradually increased to 0.75 mg/kg on alternate days and reduced progressively when minimal manifestation status (MMS; no symptoms or functional limitations) is reached.
They noted that this regimen leads to high and prolonged corticosteroid treatment – often for several years – with the mean daily prednisone dose exceeding 30 mg/day at 15 months and 20 mg/day at 36 months. As long-term use of corticosteroids is often associated with significant complications, reducing or even discontinuing prednisone treatment without destabilizing myasthenia gravis is therefore a therapeutic goal.
Evaluating dosage regimens
To investigate whether different dosage regimens could help wean patients with generalized myasthenia gravis from corticosteroid therapy without compromising efficacy, the researchers conducted this study in which the current recommended regimen was compared with an approach using higher initial corticosteroid doses followed by rapid tapering.
In the conventional slow-tapering group (control group), prednisone was given on alternate days, starting at a dose of 10 mg then increased by increments of 10 mg every 2 days up to 1.5 mg/kg on alternate days without exceeding 100 mg. This dose was maintained until MMS was reached and then reduced by 10 mg every 2 weeks until a dosage of 40 mg was reached, with subsequent slowing of the taper to 5 mg monthly. If MMS was not maintained, the alternate-day prednisone dose was increased by 10 mg every 2 weeks until MMS was restored, and the tapering resumed 4 weeks later.
In the new rapid-tapering group, oral prednisone was immediately started at 0.75 mg/kg per day, and this was followed by an earlier and rapid decrease once improved myasthenia gravis status was attained. Three different tapering schedules were applied dependent on the improvement status of the patient.
First, If the patient reached MMS at 1 month, the dose of prednisone was reduced by 0.1 mg/kg every 10 days up to 0.45 mg/kg per day, then 0.05 mg/kg every 10 days up to 0.25 mg/kg per day, then in decrements of 1 mg by adjusting the duration of the decrements according to the participant’s weight with the aim of achieving complete cessation of corticosteroid therapy within 18-20 weeks for this third stage of tapering.
Second, if the state of MMS was not reached at 1 month but the participant had improved, a slower tapering was conducted, with the dosage reduced in a similar way to the first instance but with each reduction introduced every 20 days. If the participant reached MMS during this tapering process, the tapering of prednisone was similar to the sequence described in the first group.
Third, if MMS was not reached and the participant had not improved, the initial dose was maintained for the first 3 months; beyond that time, a decrease in the prednisone dose was undertaken as in the second group to a minimum dose of 0.25 mg/kg per day, after which the prednisone dose was not reduced further. If the patient improved, the tapering of prednisone followed the sequence described in the second category.
Reductions in prednisone dose could be accelerated in the case of severe prednisone adverse effects, according to the prescriber’s decision.
In the event of a myasthenia gravis exacerbation, the patient was hospitalized and the dose of prednisone was routinely doubled, or for a more moderate aggravation, the dose was increased to the previous dose recommended in the tapering regimen.
Azathioprine, up to a maximum dose of 3 mg/kg per day, was prescribed for all participants. In all, 117 patients were randomly assigned, and 113 completed the study.
The primary outcome was the proportion of participants having reached MMS without prednisone at 12 months and having not relapsed or taken prednisone between months 12 and 15. This was achieved by significantly more patients in the rapid-tapering group (39% vs. 9%; risk ratio, 3.61; P < .001).
Rapid tapering allowed sparing of a mean of 1,898 mg of prednisone over 1 year (5.3 mg/day) per patient.
The rate of myasthenia gravis exacerbation or worsening did not differ significantly between the two groups, nor did the use of plasmapheresis or IVIG or the doses of azathioprine.
The overall number of serious adverse events did not differ significantly between the two groups (slow tapering, 22% vs. rapid-tapering, 36%; P = .15).
The researchers said it is possible that prednisone tapering would differ with another immunosuppressive agent but as azathioprine is the first-line immunosuppressant usually recommended, these results are relevant for a large proportion of patients.
They said the better outcome of the intervention group could have been related to one or more of four differences in prednisone administration: An immediate high dose versus a slow increase of the prednisone dose; daily versus alternate-day dosing; earlier tapering initiation; and faster tapering. However, the structure of the study did not allow identification of which of these factors was responsible.
“Researching the best prednisone-tapering scheme is not only a major issue for patients with myasthenia gravis but also for other autoimmune or inflammatory diseases, because validated prednisone-tapering regimens are scarce,” the authors said.
The rapid tapering of prednisone therapy appears to be feasible, beneficial, and safe in patients with generalized myasthenia gravis and “warrants testing in other autoimmune diseases,” they added.
Particularly relevant to late-onset disease
Commenting on the study, Raffi Topakian, MD, Klinikum Wels-Grieskirchen, Wels, Austria, said the results showed that in patients with moderate to severe generalized myasthenia gravis requiring high-dose prednisone, azathioprine, a widely used immunosuppressant, may have a quicker steroid-sparing effect than previously thought, and that rapid steroid tapering can be achieved safely, resulting in a reduction of the cumulative steroid dose over a year despite higher initial doses.
Dr. Topakian, who was not involved with the research, pointed out that the median age was advanced (around 56 years), and the benefit of a regimen that leads to a reduction of the cumulative steroid dose over a year may be disproportionately larger for older, sicker patients with many comorbidities who are at considerably higher risk for a prednisone-induced increase in cardiovascular complications, osteoporotic fractures, and gastrointestinal bleeding.
“The study findings are particularly relevant for the management of late-onset myasthenia gravis (when first symptoms start after age 45-50 years), which is being encountered more frequently over the past years,” he said.
“But the holy grail of myasthenia gravis treatment has not been found yet,” Dr. Topakian noted. “Disappointingly, rapid tapering of steroids (compared to slow tapering) resulted in a reduction of the cumulative steroid dose only, but was not associated with better myasthenia gravis functional status or lower doses of steroids at 15 months. To my view, this finding points to the limited immunosuppressive efficacy of azathioprine.”
He added that the study findings should not be extrapolated to patients with mild presentations or to those with muscle-specific kinase myasthenia gravis.
Dr. Sharshar disclosed no relevant financial relationships. Disclosures for the study coauthors appear in the original article.
A version of this article first appeared on Medscape.com.
. The trial showed that the conventional slow tapering regimen enabled discontinuation of prednisone earlier than previously reported but the new rapid-tapering regimen enabled an even faster discontinuation.
Noting that although both regimens led to a comparable myasthenia gravis status and prednisone dose at 15 months, the authors stated: “We think that the reduction of the cumulative dose over a year (equivalent to 5 mg/day) is a clinically relevant reduction, since the risk of complications is proportional to the daily or cumulative doses of prednisone.
“Our results warrant testing of a more rapid-tapering regimen in a future trial. In the meantime, our trial provides useful information on how prednisone tapering could be managed in patients with generalized myasthenia gravis treated with azathioprine,” they concluded.
The trial was published online Feb. 8 in JAMA Neurology.
Myasthenia gravis is a disorder of neuromuscular transmission, resulting from autoantibodies to components of the neuromuscular junction, most commonly the acetylcholine receptor. The incidence ranges from 0.3 to 2.8 per 100,000, and it is estimated to affect more than 700,000 people worldwide.
The authors of the current paper, led by Tarek Sharshar, MD, PhD, Groupe Hospitalier Universitaire, Paris, explained that many patients whose symptoms are not controlled by cholinesterase inhibitors are treated with corticosteroids and an immunosuppressant, usually azathioprine. No specific dosing protocol for prednisone has been validated, but it is commonly gradually increased to 0.75 mg/kg on alternate days and reduced progressively when minimal manifestation status (MMS; no symptoms or functional limitations) is reached.
They noted that this regimen leads to high and prolonged corticosteroid treatment – often for several years – with the mean daily prednisone dose exceeding 30 mg/day at 15 months and 20 mg/day at 36 months. As long-term use of corticosteroids is often associated with significant complications, reducing or even discontinuing prednisone treatment without destabilizing myasthenia gravis is therefore a therapeutic goal.
Evaluating dosage regimens
To investigate whether different dosage regimens could help wean patients with generalized myasthenia gravis from corticosteroid therapy without compromising efficacy, the researchers conducted this study in which the current recommended regimen was compared with an approach using higher initial corticosteroid doses followed by rapid tapering.
In the conventional slow-tapering group (control group), prednisone was given on alternate days, starting at a dose of 10 mg then increased by increments of 10 mg every 2 days up to 1.5 mg/kg on alternate days without exceeding 100 mg. This dose was maintained until MMS was reached and then reduced by 10 mg every 2 weeks until a dosage of 40 mg was reached, with subsequent slowing of the taper to 5 mg monthly. If MMS was not maintained, the alternate-day prednisone dose was increased by 10 mg every 2 weeks until MMS was restored, and the tapering resumed 4 weeks later.
In the new rapid-tapering group, oral prednisone was immediately started at 0.75 mg/kg per day, and this was followed by an earlier and rapid decrease once improved myasthenia gravis status was attained. Three different tapering schedules were applied dependent on the improvement status of the patient.
First, If the patient reached MMS at 1 month, the dose of prednisone was reduced by 0.1 mg/kg every 10 days up to 0.45 mg/kg per day, then 0.05 mg/kg every 10 days up to 0.25 mg/kg per day, then in decrements of 1 mg by adjusting the duration of the decrements according to the participant’s weight with the aim of achieving complete cessation of corticosteroid therapy within 18-20 weeks for this third stage of tapering.
Second, if the state of MMS was not reached at 1 month but the participant had improved, a slower tapering was conducted, with the dosage reduced in a similar way to the first instance but with each reduction introduced every 20 days. If the participant reached MMS during this tapering process, the tapering of prednisone was similar to the sequence described in the first group.
Third, if MMS was not reached and the participant had not improved, the initial dose was maintained for the first 3 months; beyond that time, a decrease in the prednisone dose was undertaken as in the second group to a minimum dose of 0.25 mg/kg per day, after which the prednisone dose was not reduced further. If the patient improved, the tapering of prednisone followed the sequence described in the second category.
Reductions in prednisone dose could be accelerated in the case of severe prednisone adverse effects, according to the prescriber’s decision.
In the event of a myasthenia gravis exacerbation, the patient was hospitalized and the dose of prednisone was routinely doubled, or for a more moderate aggravation, the dose was increased to the previous dose recommended in the tapering regimen.
Azathioprine, up to a maximum dose of 3 mg/kg per day, was prescribed for all participants. In all, 117 patients were randomly assigned, and 113 completed the study.
The primary outcome was the proportion of participants having reached MMS without prednisone at 12 months and having not relapsed or taken prednisone between months 12 and 15. This was achieved by significantly more patients in the rapid-tapering group (39% vs. 9%; risk ratio, 3.61; P < .001).
Rapid tapering allowed sparing of a mean of 1,898 mg of prednisone over 1 year (5.3 mg/day) per patient.
The rate of myasthenia gravis exacerbation or worsening did not differ significantly between the two groups, nor did the use of plasmapheresis or IVIG or the doses of azathioprine.
The overall number of serious adverse events did not differ significantly between the two groups (slow tapering, 22% vs. rapid-tapering, 36%; P = .15).
The researchers said it is possible that prednisone tapering would differ with another immunosuppressive agent but as azathioprine is the first-line immunosuppressant usually recommended, these results are relevant for a large proportion of patients.
They said the better outcome of the intervention group could have been related to one or more of four differences in prednisone administration: An immediate high dose versus a slow increase of the prednisone dose; daily versus alternate-day dosing; earlier tapering initiation; and faster tapering. However, the structure of the study did not allow identification of which of these factors was responsible.
“Researching the best prednisone-tapering scheme is not only a major issue for patients with myasthenia gravis but also for other autoimmune or inflammatory diseases, because validated prednisone-tapering regimens are scarce,” the authors said.
The rapid tapering of prednisone therapy appears to be feasible, beneficial, and safe in patients with generalized myasthenia gravis and “warrants testing in other autoimmune diseases,” they added.
Particularly relevant to late-onset disease
Commenting on the study, Raffi Topakian, MD, Klinikum Wels-Grieskirchen, Wels, Austria, said the results showed that in patients with moderate to severe generalized myasthenia gravis requiring high-dose prednisone, azathioprine, a widely used immunosuppressant, may have a quicker steroid-sparing effect than previously thought, and that rapid steroid tapering can be achieved safely, resulting in a reduction of the cumulative steroid dose over a year despite higher initial doses.
Dr. Topakian, who was not involved with the research, pointed out that the median age was advanced (around 56 years), and the benefit of a regimen that leads to a reduction of the cumulative steroid dose over a year may be disproportionately larger for older, sicker patients with many comorbidities who are at considerably higher risk for a prednisone-induced increase in cardiovascular complications, osteoporotic fractures, and gastrointestinal bleeding.
“The study findings are particularly relevant for the management of late-onset myasthenia gravis (when first symptoms start after age 45-50 years), which is being encountered more frequently over the past years,” he said.
“But the holy grail of myasthenia gravis treatment has not been found yet,” Dr. Topakian noted. “Disappointingly, rapid tapering of steroids (compared to slow tapering) resulted in a reduction of the cumulative steroid dose only, but was not associated with better myasthenia gravis functional status or lower doses of steroids at 15 months. To my view, this finding points to the limited immunosuppressive efficacy of azathioprine.”
He added that the study findings should not be extrapolated to patients with mild presentations or to those with muscle-specific kinase myasthenia gravis.
Dr. Sharshar disclosed no relevant financial relationships. Disclosures for the study coauthors appear in the original article.
A version of this article first appeared on Medscape.com.
. The trial showed that the conventional slow tapering regimen enabled discontinuation of prednisone earlier than previously reported but the new rapid-tapering regimen enabled an even faster discontinuation.
Noting that although both regimens led to a comparable myasthenia gravis status and prednisone dose at 15 months, the authors stated: “We think that the reduction of the cumulative dose over a year (equivalent to 5 mg/day) is a clinically relevant reduction, since the risk of complications is proportional to the daily or cumulative doses of prednisone.
“Our results warrant testing of a more rapid-tapering regimen in a future trial. In the meantime, our trial provides useful information on how prednisone tapering could be managed in patients with generalized myasthenia gravis treated with azathioprine,” they concluded.
The trial was published online Feb. 8 in JAMA Neurology.
Myasthenia gravis is a disorder of neuromuscular transmission, resulting from autoantibodies to components of the neuromuscular junction, most commonly the acetylcholine receptor. The incidence ranges from 0.3 to 2.8 per 100,000, and it is estimated to affect more than 700,000 people worldwide.
The authors of the current paper, led by Tarek Sharshar, MD, PhD, Groupe Hospitalier Universitaire, Paris, explained that many patients whose symptoms are not controlled by cholinesterase inhibitors are treated with corticosteroids and an immunosuppressant, usually azathioprine. No specific dosing protocol for prednisone has been validated, but it is commonly gradually increased to 0.75 mg/kg on alternate days and reduced progressively when minimal manifestation status (MMS; no symptoms or functional limitations) is reached.
They noted that this regimen leads to high and prolonged corticosteroid treatment – often for several years – with the mean daily prednisone dose exceeding 30 mg/day at 15 months and 20 mg/day at 36 months. As long-term use of corticosteroids is often associated with significant complications, reducing or even discontinuing prednisone treatment without destabilizing myasthenia gravis is therefore a therapeutic goal.
Evaluating dosage regimens
To investigate whether different dosage regimens could help wean patients with generalized myasthenia gravis from corticosteroid therapy without compromising efficacy, the researchers conducted this study in which the current recommended regimen was compared with an approach using higher initial corticosteroid doses followed by rapid tapering.
In the conventional slow-tapering group (control group), prednisone was given on alternate days, starting at a dose of 10 mg then increased by increments of 10 mg every 2 days up to 1.5 mg/kg on alternate days without exceeding 100 mg. This dose was maintained until MMS was reached and then reduced by 10 mg every 2 weeks until a dosage of 40 mg was reached, with subsequent slowing of the taper to 5 mg monthly. If MMS was not maintained, the alternate-day prednisone dose was increased by 10 mg every 2 weeks until MMS was restored, and the tapering resumed 4 weeks later.
In the new rapid-tapering group, oral prednisone was immediately started at 0.75 mg/kg per day, and this was followed by an earlier and rapid decrease once improved myasthenia gravis status was attained. Three different tapering schedules were applied dependent on the improvement status of the patient.
First, If the patient reached MMS at 1 month, the dose of prednisone was reduced by 0.1 mg/kg every 10 days up to 0.45 mg/kg per day, then 0.05 mg/kg every 10 days up to 0.25 mg/kg per day, then in decrements of 1 mg by adjusting the duration of the decrements according to the participant’s weight with the aim of achieving complete cessation of corticosteroid therapy within 18-20 weeks for this third stage of tapering.
Second, if the state of MMS was not reached at 1 month but the participant had improved, a slower tapering was conducted, with the dosage reduced in a similar way to the first instance but with each reduction introduced every 20 days. If the participant reached MMS during this tapering process, the tapering of prednisone was similar to the sequence described in the first group.
Third, if MMS was not reached and the participant had not improved, the initial dose was maintained for the first 3 months; beyond that time, a decrease in the prednisone dose was undertaken as in the second group to a minimum dose of 0.25 mg/kg per day, after which the prednisone dose was not reduced further. If the patient improved, the tapering of prednisone followed the sequence described in the second category.
Reductions in prednisone dose could be accelerated in the case of severe prednisone adverse effects, according to the prescriber’s decision.
In the event of a myasthenia gravis exacerbation, the patient was hospitalized and the dose of prednisone was routinely doubled, or for a more moderate aggravation, the dose was increased to the previous dose recommended in the tapering regimen.
Azathioprine, up to a maximum dose of 3 mg/kg per day, was prescribed for all participants. In all, 117 patients were randomly assigned, and 113 completed the study.
The primary outcome was the proportion of participants having reached MMS without prednisone at 12 months and having not relapsed or taken prednisone between months 12 and 15. This was achieved by significantly more patients in the rapid-tapering group (39% vs. 9%; risk ratio, 3.61; P < .001).
Rapid tapering allowed sparing of a mean of 1,898 mg of prednisone over 1 year (5.3 mg/day) per patient.
The rate of myasthenia gravis exacerbation or worsening did not differ significantly between the two groups, nor did the use of plasmapheresis or IVIG or the doses of azathioprine.
The overall number of serious adverse events did not differ significantly between the two groups (slow tapering, 22% vs. rapid-tapering, 36%; P = .15).
The researchers said it is possible that prednisone tapering would differ with another immunosuppressive agent but as azathioprine is the first-line immunosuppressant usually recommended, these results are relevant for a large proportion of patients.
They said the better outcome of the intervention group could have been related to one or more of four differences in prednisone administration: An immediate high dose versus a slow increase of the prednisone dose; daily versus alternate-day dosing; earlier tapering initiation; and faster tapering. However, the structure of the study did not allow identification of which of these factors was responsible.
“Researching the best prednisone-tapering scheme is not only a major issue for patients with myasthenia gravis but also for other autoimmune or inflammatory diseases, because validated prednisone-tapering regimens are scarce,” the authors said.
The rapid tapering of prednisone therapy appears to be feasible, beneficial, and safe in patients with generalized myasthenia gravis and “warrants testing in other autoimmune diseases,” they added.
Particularly relevant to late-onset disease
Commenting on the study, Raffi Topakian, MD, Klinikum Wels-Grieskirchen, Wels, Austria, said the results showed that in patients with moderate to severe generalized myasthenia gravis requiring high-dose prednisone, azathioprine, a widely used immunosuppressant, may have a quicker steroid-sparing effect than previously thought, and that rapid steroid tapering can be achieved safely, resulting in a reduction of the cumulative steroid dose over a year despite higher initial doses.
Dr. Topakian, who was not involved with the research, pointed out that the median age was advanced (around 56 years), and the benefit of a regimen that leads to a reduction of the cumulative steroid dose over a year may be disproportionately larger for older, sicker patients with many comorbidities who are at considerably higher risk for a prednisone-induced increase in cardiovascular complications, osteoporotic fractures, and gastrointestinal bleeding.
“The study findings are particularly relevant for the management of late-onset myasthenia gravis (when first symptoms start after age 45-50 years), which is being encountered more frequently over the past years,” he said.
“But the holy grail of myasthenia gravis treatment has not been found yet,” Dr. Topakian noted. “Disappointingly, rapid tapering of steroids (compared to slow tapering) resulted in a reduction of the cumulative steroid dose only, but was not associated with better myasthenia gravis functional status or lower doses of steroids at 15 months. To my view, this finding points to the limited immunosuppressive efficacy of azathioprine.”
He added that the study findings should not be extrapolated to patients with mild presentations or to those with muscle-specific kinase myasthenia gravis.
Dr. Sharshar disclosed no relevant financial relationships. Disclosures for the study coauthors appear in the original article.
A version of this article first appeared on Medscape.com.
FROM JAMA NEUROLOGY
Outcomes have improved for PAH in connective tissue disease
Survival rates for patients with pulmonary arterial hypertension associated with connective tissue diseases have improved significantly in recent years, and there is growing evidence that treatments for idiopathic pulmonary arterial hypertension can also benefit this group.

In an article published online Feb. 3, 2021, in Arthritis & Rheumatology, researchers report the outcomes of a meta-analysis to explore the effect of more modern pulmonary arterial hypertension treatments on patients with conditions such as systemic sclerosis.
First author Dinesh Khanna, MBBS, MSc, of the division of rheumatology at the University of Michigan, Ann Arbor, said in an interview that connective tissue disease–associated pulmonary arterial hypertension (CTD-PAH) was a leading cause of death, but earlier clinical trials had found poor outcomes in patients with CTD, compared with those with idiopathic PAH.
“Recent clinical trial data show that aggressive, up-front PAH treatments have better outcomes in those with CTD-PAH, and we wanted to explore these observations carefully in a systematic review and meta-analysis,” Dr. Khanna said.
The analysis included 11 randomized, controlled trials, involving 4,329 patients with PAH (1,267 with CTD), and 19 registries with a total of 9,739 patients with PAH, including 4,008 with CTD. Trials were required to report long-term clinical outcomes with a median enrollment time of greater than 6 months, and outcomes measured between 3-6 months after the patients started treatment.
Patients with CTDs had an older mean age and a lower 6-minute walk distance than did those with idiopathic PAH.
Five randomized, controlled trials – involving 3,172 patients, 941 of whom had a CTD – found that additional PAH treatment was associated with a 36% reduction in the risk of morbidity or mortality events, compared with controls both in the overall PAH group and in those with CTD.
Additional therapy was also associated with a 34.6-meter increase in 6-minute walk distance in the general PAH population, and a 20.4-meter increase in those with CTD.
The authors commented that the smaller improvement in 6-minute walk distance among patients with CTD may be influenced by comorbidities such as musculoskeletal involvement that would be independent of their cardiopulmonary function.
Differential patient survival among PAH etiologies
“Our meta-analysis of RCTs demonstrated that patients with CTD-PAH derive a clinically significant benefit from currently available PAH therapies which, in many patients, comprised the addition of a drug targeting a second or third pathway involved in the pathophysiology of PAH,” the authors wrote.
When researchers analyzed data from nine registries that included a wide range of PAH etiologies, they found the overall survival rates were lower among patients with CTD, compared with the overall population. The analysis also suggested that patients with systemic sclerosis and PAH had lower survival rates than did those with systemic lupus erythematosus.
Dr. Khanna said this may relate to different pathophysiology of PAH in patients with CTDs, but could also be a reflection of other differences, such as older age and the involvement of other comorbidities, including lung fibrosis and heart involvement.
Data across all 19 registries also showed that survival rates among those with CTD were higher in registries where more than 50% of the registry study period was during or after 2010, compared with registries where 50% or more of the study period was before 2010.
The authors suggested the differences in survival rates may relate to increased screening for PAH, particularly among people with CTDs. They noted that increased screening leads to earlier diagnosis, which could introduce a lead-time bias such that later registries would have younger participants with less severe disease. However, their analysis found that the later registries had older patients but also with less severe disease, and they suggested that it wasn’t possible to determine if lead-time bias was playing a role in their results.
Improvements in treatment options could also account for differences in survival over time, although the authors commented that only six registries in the study included patients from 2015 or later, when currently available treatments came into use and early combination therapy was used more.
“These data also support the 2018 World Symposium on Pulmonary Hypertension recommendations to initiate up-front combination pulmonary arterial hypertension therapy in majority of cases with CTD-PAH,” Dr. Khanna said.
‘Still have to be aggressive at identifying the high-risk patients’
Commenting on the findings, Virginia Steen, MD, of the division of rheumatology at Georgetown University, Washington, said clinicians were finally seeing some significant changes over time in scleroderma-associated PAH.

“Although some of it may be just early diagnosis, I think that the combination of early diagnosis and more aggressive treatment with combination medication is definitely making a difference,” Dr. Steen said in an interview. “The bottom line is that we as rheumatologists still have to be aggressive at identifying the high-risk patients, making an early diagnosis, and working with our pulmonary hypertension colleagues and aggressively treating these patients so we can make a long-term difference.”
The authors of an accompanying editorial said the meta-analysis’ findings showed the positive impact of early combination therapy and early diagnosis through proactive screening.
“It is notable because the present analysis again confirms that outcomes are worse in CTD-PAH than in idiopathic or familial forms of PAH, the impact of treatments should no longer be regarded as insignificant,” the editorial’s authors wrote. “This is a practice changing observation, especially now that many of the drugs are available in generic formulations and so the cost of modern PAH treatment has fallen at the same time as its true value is convincingly demonstrated.”
They also argued there was strong evidence for the value of combination therapies, both for PAH-targeted drugs used in combination and concurrent use of immunosuppression and drugs specifically for PAH in some patients with CTD-PAH.
However, they pointed out that not all treatments for idiopathic PAH were suitable for patients with CTDs, highlighting the example of anticoagulation that can improve survival in the first but worsen it in the second.
The study was funded by Actelion. Six authors declared funding and grants from the pharmaceutical sector, including the study sponsor, and three authors were employees of Actelion.
Survival rates for patients with pulmonary arterial hypertension associated with connective tissue diseases have improved significantly in recent years, and there is growing evidence that treatments for idiopathic pulmonary arterial hypertension can also benefit this group.
In an article published online Feb. 3, 2021, in Arthritis & Rheumatology, researchers report the outcomes of a meta-analysis to explore the effect of more modern pulmonary arterial hypertension treatments on patients with conditions such as systemic sclerosis.
First author Dinesh Khanna, MBBS, MSc, of the division of rheumatology at the University of Michigan, Ann Arbor, said in an interview that connective tissue disease–associated pulmonary arterial hypertension (CTD-PAH) was a leading cause of death, but earlier clinical trials had found poor outcomes in patients with CTD, compared with those with idiopathic PAH.
“Recent clinical trial data show that aggressive, up-front PAH treatments have better outcomes in those with CTD-PAH, and we wanted to explore these observations carefully in a systematic review and meta-analysis,” Dr. Khanna said.
The analysis included 11 randomized, controlled trials, involving 4,329 patients with PAH (1,267 with CTD), and 19 registries with a total of 9,739 patients with PAH, including 4,008 with CTD. Trials were required to report long-term clinical outcomes with a median enrollment time of greater than 6 months, and outcomes measured between 3-6 months after the patients started treatment.
Patients with CTDs had an older mean age and a lower 6-minute walk distance than did those with idiopathic PAH.
Five randomized, controlled trials – involving 3,172 patients, 941 of whom had a CTD – found that additional PAH treatment was associated with a 36% reduction in the risk of morbidity or mortality events, compared with controls both in the overall PAH group and in those with CTD.
Additional therapy was also associated with a 34.6-meter increase in 6-minute walk distance in the general PAH population, and a 20.4-meter increase in those with CTD.
The authors commented that the smaller improvement in 6-minute walk distance among patients with CTD may be influenced by comorbidities such as musculoskeletal involvement that would be independent of their cardiopulmonary function.
Differential patient survival among PAH etiologies
“Our meta-analysis of RCTs demonstrated that patients with CTD-PAH derive a clinically significant benefit from currently available PAH therapies which, in many patients, comprised the addition of a drug targeting a second or third pathway involved in the pathophysiology of PAH,” the authors wrote.
When researchers analyzed data from nine registries that included a wide range of PAH etiologies, they found the overall survival rates were lower among patients with CTD, compared with the overall population. The analysis also suggested that patients with systemic sclerosis and PAH had lower survival rates than did those with systemic lupus erythematosus.
Dr. Khanna said this may relate to different pathophysiology of PAH in patients with CTDs, but could also be a reflection of other differences, such as older age and the involvement of other comorbidities, including lung fibrosis and heart involvement.
Data across all 19 registries also showed that survival rates among those with CTD were higher in registries where more than 50% of the registry study period was during or after 2010, compared with registries where 50% or more of the study period was before 2010.
The authors suggested the differences in survival rates may relate to increased screening for PAH, particularly among people with CTDs. They noted that increased screening leads to earlier diagnosis, which could introduce a lead-time bias such that later registries would have younger participants with less severe disease. However, their analysis found that the later registries had older patients but also with less severe disease, and they suggested that it wasn’t possible to determine if lead-time bias was playing a role in their results.
Improvements in treatment options could also account for differences in survival over time, although the authors commented that only six registries in the study included patients from 2015 or later, when currently available treatments came into use and early combination therapy was used more.
“These data also support the 2018 World Symposium on Pulmonary Hypertension recommendations to initiate up-front combination pulmonary arterial hypertension therapy in majority of cases with CTD-PAH,” Dr. Khanna said.
‘Still have to be aggressive at identifying the high-risk patients’
Commenting on the findings, Virginia Steen, MD, of the division of rheumatology at Georgetown University, Washington, said clinicians were finally seeing some significant changes over time in scleroderma-associated PAH.
“Although some of it may be just early diagnosis, I think that the combination of early diagnosis and more aggressive treatment with combination medication is definitely making a difference,” Dr. Steen said in an interview. “The bottom line is that we as rheumatologists still have to be aggressive at identifying the high-risk patients, making an early diagnosis, and working with our pulmonary hypertension colleagues and aggressively treating these patients so we can make a long-term difference.”
The authors of an accompanying editorial said the meta-analysis’ findings showed the positive impact of early combination therapy and early diagnosis through proactive screening.
“It is notable because the present analysis again confirms that outcomes are worse in CTD-PAH than in idiopathic or familial forms of PAH, the impact of treatments should no longer be regarded as insignificant,” the editorial’s authors wrote. “This is a practice changing observation, especially now that many of the drugs are available in generic formulations and so the cost of modern PAH treatment has fallen at the same time as its true value is convincingly demonstrated.”
They also argued there was strong evidence for the value of combination therapies, both for PAH-targeted drugs used in combination and concurrent use of immunosuppression and drugs specifically for PAH in some patients with CTD-PAH.
However, they pointed out that not all treatments for idiopathic PAH were suitable for patients with CTDs, highlighting the example of anticoagulation that can improve survival in the first but worsen it in the second.
The study was funded by Actelion. Six authors declared funding and grants from the pharmaceutical sector, including the study sponsor, and three authors were employees of Actelion.
Survival rates for patients with pulmonary arterial hypertension associated with connective tissue diseases have improved significantly in recent years, and there is growing evidence that treatments for idiopathic pulmonary arterial hypertension can also benefit this group.
In an article published online Feb. 3, 2021, in Arthritis & Rheumatology, researchers report the outcomes of a meta-analysis to explore the effect of more modern pulmonary arterial hypertension treatments on patients with conditions such as systemic sclerosis.
First author Dinesh Khanna, MBBS, MSc, of the division of rheumatology at the University of Michigan, Ann Arbor, said in an interview that connective tissue disease–associated pulmonary arterial hypertension (CTD-PAH) was a leading cause of death, but earlier clinical trials had found poor outcomes in patients with CTD, compared with those with idiopathic PAH.
“Recent clinical trial data show that aggressive, up-front PAH treatments have better outcomes in those with CTD-PAH, and we wanted to explore these observations carefully in a systematic review and meta-analysis,” Dr. Khanna said.
The analysis included 11 randomized, controlled trials, involving 4,329 patients with PAH (1,267 with CTD), and 19 registries with a total of 9,739 patients with PAH, including 4,008 with CTD. Trials were required to report long-term clinical outcomes with a median enrollment time of greater than 6 months, and outcomes measured between 3-6 months after the patients started treatment.
Patients with CTDs had an older mean age and a lower 6-minute walk distance than did those with idiopathic PAH.
Five randomized, controlled trials – involving 3,172 patients, 941 of whom had a CTD – found that additional PAH treatment was associated with a 36% reduction in the risk of morbidity or mortality events, compared with controls both in the overall PAH group and in those with CTD.
Additional therapy was also associated with a 34.6-meter increase in 6-minute walk distance in the general PAH population, and a 20.4-meter increase in those with CTD.
The authors commented that the smaller improvement in 6-minute walk distance among patients with CTD may be influenced by comorbidities such as musculoskeletal involvement that would be independent of their cardiopulmonary function.
Differential patient survival among PAH etiologies
“Our meta-analysis of RCTs demonstrated that patients with CTD-PAH derive a clinically significant benefit from currently available PAH therapies which, in many patients, comprised the addition of a drug targeting a second or third pathway involved in the pathophysiology of PAH,” the authors wrote.
When researchers analyzed data from nine registries that included a wide range of PAH etiologies, they found the overall survival rates were lower among patients with CTD, compared with the overall population. The analysis also suggested that patients with systemic sclerosis and PAH had lower survival rates than did those with systemic lupus erythematosus.
Dr. Khanna said this may relate to different pathophysiology of PAH in patients with CTDs, but could also be a reflection of other differences, such as older age and the involvement of other comorbidities, including lung fibrosis and heart involvement.
Data across all 19 registries also showed that survival rates among those with CTD were higher in registries where more than 50% of the registry study period was during or after 2010, compared with registries where 50% or more of the study period was before 2010.
The authors suggested the differences in survival rates may relate to increased screening for PAH, particularly among people with CTDs. They noted that increased screening leads to earlier diagnosis, which could introduce a lead-time bias such that later registries would have younger participants with less severe disease. However, their analysis found that the later registries had older patients but also with less severe disease, and they suggested that it wasn’t possible to determine if lead-time bias was playing a role in their results.
Improvements in treatment options could also account for differences in survival over time, although the authors commented that only six registries in the study included patients from 2015 or later, when currently available treatments came into use and early combination therapy was used more.
“These data also support the 2018 World Symposium on Pulmonary Hypertension recommendations to initiate up-front combination pulmonary arterial hypertension therapy in majority of cases with CTD-PAH,” Dr. Khanna said.
‘Still have to be aggressive at identifying the high-risk patients’
Commenting on the findings, Virginia Steen, MD, of the division of rheumatology at Georgetown University, Washington, said clinicians were finally seeing some significant changes over time in scleroderma-associated PAH.
“Although some of it may be just early diagnosis, I think that the combination of early diagnosis and more aggressive treatment with combination medication is definitely making a difference,” Dr. Steen said in an interview. “The bottom line is that we as rheumatologists still have to be aggressive at identifying the high-risk patients, making an early diagnosis, and working with our pulmonary hypertension colleagues and aggressively treating these patients so we can make a long-term difference.”
The authors of an accompanying editorial said the meta-analysis’ findings showed the positive impact of early combination therapy and early diagnosis through proactive screening.
“It is notable because the present analysis again confirms that outcomes are worse in CTD-PAH than in idiopathic or familial forms of PAH, the impact of treatments should no longer be regarded as insignificant,” the editorial’s authors wrote. “This is a practice changing observation, especially now that many of the drugs are available in generic formulations and so the cost of modern PAH treatment has fallen at the same time as its true value is convincingly demonstrated.”
They also argued there was strong evidence for the value of combination therapies, both for PAH-targeted drugs used in combination and concurrent use of immunosuppression and drugs specifically for PAH in some patients with CTD-PAH.
However, they pointed out that not all treatments for idiopathic PAH were suitable for patients with CTDs, highlighting the example of anticoagulation that can improve survival in the first but worsen it in the second.
The study was funded by Actelion. Six authors declared funding and grants from the pharmaceutical sector, including the study sponsor, and three authors were employees of Actelion.
FROM ARTHRITIS & RHEUMATOLOGY
Tocilizumab may improve lung function in early systemic sclerosis
Treatment with tocilizumab (Actemra) could stabilize or improve lung function in people with early interstitial lung disease associated with systemic sclerosis (SSc-ILD), a new study has found.
A paper published online Feb. 3 in Arthritis & Rheumatology presents the results of a post hoc analysis of data from a phase 3, placebo-controlled, double-blind trial of subcutaneous tocilizumab in patients with SSc and progressive skin disease, which included high-resolution chest CT to assess lung involvement and fibrosis.
Tocilizumab is a monoclonal antibody that targets interleukin-6 and is currently approved for the treatment of immune-mediated diseases such as rheumatoid arthritis, giant cell arteritis, cytokine release syndrome, and systemic and polyarticular course juvenile idiopathic arthritis.
Two previous studies of tocilizumab in patients with early, diffuse cutaneous SSc had also found that the treatment was associated with preservation of lung function but did not characterize that effect using radiography.
Of the 210 participants in the trial, called focuSSced, 136 were found to have interstitial lung disease at baseline and were randomized to 162 mg tocilizumab weekly or placebo for 48 weeks.
At baseline, around three-quarters of those with interstitial lung disease had moderate to severe lung involvement, defined as ground glass opacities, honeycombing, and fibrotic reticulation across at least 20% of the whole lung.
Those in the tocilizumab group showed a 0.1% mean decline in forced vital capacity (FVC) over the 48-week study, while those in the placebo group had a mean decline of 6.3%.
When stratified by severity of lung involvement, those with mild lung disease group treated with tocilizumab had a 4.1% decline in FVC, compared with a 10% decline in the placebo group; those with moderate disease in the treatment group had an 0.7% mean increase in FVC, compared with a 5.7% decrease in the placebo group, and those with severe lung involvement in the treatment arm had a 2.1% increase in FVC, compared with a 6.7% decrease in the placebo arm.
Those treated with tocilizumab also showed a statistically significant 1.8% improvement in the amount of lung involvement, which was largely seen in those with more extensive lung involvement at baseline. Those with more than 20% of the lung affected had a significant 4.9% reduction in lung area affected, while those in the placebo arm showed a significant increase in fibrosis.
First author David Roofeh, MD, of the University of Michigan Scleroderma Program, and colleagues wrote that most patients with SSc will develop interstitial lung disease – particularly those with early, diffuse cutaneous SSc and elevated markers such as C-reactive protein.
“Patients with these high-risk features, especially those with disease in the initial phase of development, represent an important target for early intervention as ILD is largely irreversible in SSc,” the authors wrote.
Findings from a specific patient population may not be generalizable
Commenting on the findings, Lorinda Chung, MD, of Stanford (Calif.) University, said in an interview that the study demonstrated that tocilizumab could prevent radiographic progression of ILD in early diffuse SSc patients with mild to severe lung disease and evidence of active skin disease, as well as elevated inflammatory markers.
“This was a very specific patient population who was studied in the focuSSced clinical trial, and this paper only evaluated a subset of these patients,” Dr. Chung said. “The results may not be generalizable to all SSc-ILD patients and further studies are needed.”
The authors suggested that the patients with progressive skin disease and elevated acute phase reactants may represent a group in the immunoinflammatory phase of the disease rather than the advanced fibrotic stage, and that this might be a “window of therapeutic opportunity to preserve lung function.”
Dr. Chung noted that the radiographic improvement induced by tocilizumab treatment was greatest in those with the most radiographic disease at baseline.
“This may reflect tocilizumab’s impact on decreasing inflammation, but we are not provided the data on the effects of tocilizumab on the individual components of the QILD [quantitative ILD: summation of ground glass opacities, honeycombing, and fibrotic reticulation],” she said.
The study’s authors also made a point about the utility of screening patients with high-resolution chest CT to detect early signs of ILD.
“Our data demonstrate the value of obtaining HRCT at the time of diagnosis: PFTs [pulmonary function tests] are not sensitive enough to accurately assess the presence of ILD and delays in treatment initiation may lead to irreversible disease,” they wrote.
Describing the results as ‘hypothesis-generating’ owing to the post hoc nature of the analysis, the authors said that FVC was an indirect measure of the flow-resistive properties of the lung, and that other aspects of SSc – such as hide-bound chest thickness – could cause thoracic restriction.
Two authors were funded by the National Institutes of Health. Six authors declared grants, funding, and other support from the pharmaceutical sector, including Roche, which sponsored the original focuSSced trial.
Treatment with tocilizumab (Actemra) could stabilize or improve lung function in people with early interstitial lung disease associated with systemic sclerosis (SSc-ILD), a new study has found.
A paper published online Feb. 3 in Arthritis & Rheumatology presents the results of a post hoc analysis of data from a phase 3, placebo-controlled, double-blind trial of subcutaneous tocilizumab in patients with SSc and progressive skin disease, which included high-resolution chest CT to assess lung involvement and fibrosis.
Tocilizumab is a monoclonal antibody that targets interleukin-6 and is currently approved for the treatment of immune-mediated diseases such as rheumatoid arthritis, giant cell arteritis, cytokine release syndrome, and systemic and polyarticular course juvenile idiopathic arthritis.
Two previous studies of tocilizumab in patients with early, diffuse cutaneous SSc had also found that the treatment was associated with preservation of lung function but did not characterize that effect using radiography.
Of the 210 participants in the trial, called focuSSced, 136 were found to have interstitial lung disease at baseline and were randomized to 162 mg tocilizumab weekly or placebo for 48 weeks.
At baseline, around three-quarters of those with interstitial lung disease had moderate to severe lung involvement, defined as ground glass opacities, honeycombing, and fibrotic reticulation across at least 20% of the whole lung.
Those in the tocilizumab group showed a 0.1% mean decline in forced vital capacity (FVC) over the 48-week study, while those in the placebo group had a mean decline of 6.3%.
When stratified by severity of lung involvement, those with mild lung disease group treated with tocilizumab had a 4.1% decline in FVC, compared with a 10% decline in the placebo group; those with moderate disease in the treatment group had an 0.7% mean increase in FVC, compared with a 5.7% decrease in the placebo group, and those with severe lung involvement in the treatment arm had a 2.1% increase in FVC, compared with a 6.7% decrease in the placebo arm.
Those treated with tocilizumab also showed a statistically significant 1.8% improvement in the amount of lung involvement, which was largely seen in those with more extensive lung involvement at baseline. Those with more than 20% of the lung affected had a significant 4.9% reduction in lung area affected, while those in the placebo arm showed a significant increase in fibrosis.
First author David Roofeh, MD, of the University of Michigan Scleroderma Program, and colleagues wrote that most patients with SSc will develop interstitial lung disease – particularly those with early, diffuse cutaneous SSc and elevated markers such as C-reactive protein.
“Patients with these high-risk features, especially those with disease in the initial phase of development, represent an important target for early intervention as ILD is largely irreversible in SSc,” the authors wrote.
Findings from a specific patient population may not be generalizable
Commenting on the findings, Lorinda Chung, MD, of Stanford (Calif.) University, said in an interview that the study demonstrated that tocilizumab could prevent radiographic progression of ILD in early diffuse SSc patients with mild to severe lung disease and evidence of active skin disease, as well as elevated inflammatory markers.
“This was a very specific patient population who was studied in the focuSSced clinical trial, and this paper only evaluated a subset of these patients,” Dr. Chung said. “The results may not be generalizable to all SSc-ILD patients and further studies are needed.”
The authors suggested that the patients with progressive skin disease and elevated acute phase reactants may represent a group in the immunoinflammatory phase of the disease rather than the advanced fibrotic stage, and that this might be a “window of therapeutic opportunity to preserve lung function.”
Dr. Chung noted that the radiographic improvement induced by tocilizumab treatment was greatest in those with the most radiographic disease at baseline.
“This may reflect tocilizumab’s impact on decreasing inflammation, but we are not provided the data on the effects of tocilizumab on the individual components of the QILD [quantitative ILD: summation of ground glass opacities, honeycombing, and fibrotic reticulation],” she said.
The study’s authors also made a point about the utility of screening patients with high-resolution chest CT to detect early signs of ILD.
“Our data demonstrate the value of obtaining HRCT at the time of diagnosis: PFTs [pulmonary function tests] are not sensitive enough to accurately assess the presence of ILD and delays in treatment initiation may lead to irreversible disease,” they wrote.
Describing the results as ‘hypothesis-generating’ owing to the post hoc nature of the analysis, the authors said that FVC was an indirect measure of the flow-resistive properties of the lung, and that other aspects of SSc – such as hide-bound chest thickness – could cause thoracic restriction.
Two authors were funded by the National Institutes of Health. Six authors declared grants, funding, and other support from the pharmaceutical sector, including Roche, which sponsored the original focuSSced trial.
Treatment with tocilizumab (Actemra) could stabilize or improve lung function in people with early interstitial lung disease associated with systemic sclerosis (SSc-ILD), a new study has found.
A paper published online Feb. 3 in Arthritis & Rheumatology presents the results of a post hoc analysis of data from a phase 3, placebo-controlled, double-blind trial of subcutaneous tocilizumab in patients with SSc and progressive skin disease, which included high-resolution chest CT to assess lung involvement and fibrosis.
Tocilizumab is a monoclonal antibody that targets interleukin-6 and is currently approved for the treatment of immune-mediated diseases such as rheumatoid arthritis, giant cell arteritis, cytokine release syndrome, and systemic and polyarticular course juvenile idiopathic arthritis.
Two previous studies of tocilizumab in patients with early, diffuse cutaneous SSc had also found that the treatment was associated with preservation of lung function but did not characterize that effect using radiography.
Of the 210 participants in the trial, called focuSSced, 136 were found to have interstitial lung disease at baseline and were randomized to 162 mg tocilizumab weekly or placebo for 48 weeks.
At baseline, around three-quarters of those with interstitial lung disease had moderate to severe lung involvement, defined as ground glass opacities, honeycombing, and fibrotic reticulation across at least 20% of the whole lung.
Those in the tocilizumab group showed a 0.1% mean decline in forced vital capacity (FVC) over the 48-week study, while those in the placebo group had a mean decline of 6.3%.
When stratified by severity of lung involvement, those with mild lung disease group treated with tocilizumab had a 4.1% decline in FVC, compared with a 10% decline in the placebo group; those with moderate disease in the treatment group had an 0.7% mean increase in FVC, compared with a 5.7% decrease in the placebo group, and those with severe lung involvement in the treatment arm had a 2.1% increase in FVC, compared with a 6.7% decrease in the placebo arm.
Those treated with tocilizumab also showed a statistically significant 1.8% improvement in the amount of lung involvement, which was largely seen in those with more extensive lung involvement at baseline. Those with more than 20% of the lung affected had a significant 4.9% reduction in lung area affected, while those in the placebo arm showed a significant increase in fibrosis.
First author David Roofeh, MD, of the University of Michigan Scleroderma Program, and colleagues wrote that most patients with SSc will develop interstitial lung disease – particularly those with early, diffuse cutaneous SSc and elevated markers such as C-reactive protein.
“Patients with these high-risk features, especially those with disease in the initial phase of development, represent an important target for early intervention as ILD is largely irreversible in SSc,” the authors wrote.
Findings from a specific patient population may not be generalizable
Commenting on the findings, Lorinda Chung, MD, of Stanford (Calif.) University, said in an interview that the study demonstrated that tocilizumab could prevent radiographic progression of ILD in early diffuse SSc patients with mild to severe lung disease and evidence of active skin disease, as well as elevated inflammatory markers.
“This was a very specific patient population who was studied in the focuSSced clinical trial, and this paper only evaluated a subset of these patients,” Dr. Chung said. “The results may not be generalizable to all SSc-ILD patients and further studies are needed.”
The authors suggested that the patients with progressive skin disease and elevated acute phase reactants may represent a group in the immunoinflammatory phase of the disease rather than the advanced fibrotic stage, and that this might be a “window of therapeutic opportunity to preserve lung function.”
Dr. Chung noted that the radiographic improvement induced by tocilizumab treatment was greatest in those with the most radiographic disease at baseline.
“This may reflect tocilizumab’s impact on decreasing inflammation, but we are not provided the data on the effects of tocilizumab on the individual components of the QILD [quantitative ILD: summation of ground glass opacities, honeycombing, and fibrotic reticulation],” she said.
The study’s authors also made a point about the utility of screening patients with high-resolution chest CT to detect early signs of ILD.
“Our data demonstrate the value of obtaining HRCT at the time of diagnosis: PFTs [pulmonary function tests] are not sensitive enough to accurately assess the presence of ILD and delays in treatment initiation may lead to irreversible disease,” they wrote.
Describing the results as ‘hypothesis-generating’ owing to the post hoc nature of the analysis, the authors said that FVC was an indirect measure of the flow-resistive properties of the lung, and that other aspects of SSc – such as hide-bound chest thickness – could cause thoracic restriction.
Two authors were funded by the National Institutes of Health. Six authors declared grants, funding, and other support from the pharmaceutical sector, including Roche, which sponsored the original focuSSced trial.
FROM ARTHRITIS & RHEUMATOLOGY
A 35-year-old male who takes antiseizure medications, has asymptomatic lesions on his nose and cheeks, present since birth
although up to 75% of cases may be caused by a spontaneous mutation. It is caused by mutations in the TSC1 gene on chromosome 9q34–encoding hamartin or the TSC2 gene on chromosome I6pl3–encoding tuberin. Patients present at birth and males and females are affected equally.
There are multiple skin findings in TS that may herald the diagnosis. The earliest findings are hypopigmented macules, found in 85% of patients. They may be in an ash-leaf shape or confetti pattern. Adenoma sebaceum, or angiofibromas, are present on the forehead, nose, and cheeks, and often present in childhood. Periungual angiofibromas called Koenen tumors tend to occur at puberty. Connective-tissue nevi called Shagreen plaques, or collagenomas, may be present, which is what our patient exhibits on his back. The lumbosacral region is the most common area for these to appear in the first decade of life.
TS can affect other organ systems in the body. Seizures, neuropsychiatric diseases, and mental deficiency are common. Cortical tumors, gliomas, and astrocytomas may develop in the brain. Congenital retinal hamartomas (phakomas) occur. Renal cysts and angiomyolipomas may occur in the kidneys. In the lungs, patients may develop lymphangiomyomatosis. Rhabdomyomas can occur in the heart in infancy and may regress spontaneously over time. Bony changes such as cysts and sclerosis may occur.
Treatment and monitoring of TS requires a multidisciplinary approach with neurology, pulmonology, cardiology, ophthalmology, orthopedics, and dermatology. Cosmetic treatment for angiofibromas includes CO2 laser, shaving, and dermabrasion. Topical rapamycin use has been described in the literature to improve the appearance of angiofibromas. Our patient has been using rapamycin 1% cream for more than 5 years and has had a substantial reduction in the size and number of angiofibromas.

This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References:
Spitz J. Genodermatoses. A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins, 2005.
James W et al. Andrews’ Diseases of the Skin. Philadelphia: Saunders, 2006.
Bolognia JL et al. Dermatology. London: Mosby Elsevier, 2008.
although up to 75% of cases may be caused by a spontaneous mutation. It is caused by mutations in the TSC1 gene on chromosome 9q34–encoding hamartin or the TSC2 gene on chromosome I6pl3–encoding tuberin. Patients present at birth and males and females are affected equally.
There are multiple skin findings in TS that may herald the diagnosis. The earliest findings are hypopigmented macules, found in 85% of patients. They may be in an ash-leaf shape or confetti pattern. Adenoma sebaceum, or angiofibromas, are present on the forehead, nose, and cheeks, and often present in childhood. Periungual angiofibromas called Koenen tumors tend to occur at puberty. Connective-tissue nevi called Shagreen plaques, or collagenomas, may be present, which is what our patient exhibits on his back. The lumbosacral region is the most common area for these to appear in the first decade of life.
TS can affect other organ systems in the body. Seizures, neuropsychiatric diseases, and mental deficiency are common. Cortical tumors, gliomas, and astrocytomas may develop in the brain. Congenital retinal hamartomas (phakomas) occur. Renal cysts and angiomyolipomas may occur in the kidneys. In the lungs, patients may develop lymphangiomyomatosis. Rhabdomyomas can occur in the heart in infancy and may regress spontaneously over time. Bony changes such as cysts and sclerosis may occur.
Treatment and monitoring of TS requires a multidisciplinary approach with neurology, pulmonology, cardiology, ophthalmology, orthopedics, and dermatology. Cosmetic treatment for angiofibromas includes CO2 laser, shaving, and dermabrasion. Topical rapamycin use has been described in the literature to improve the appearance of angiofibromas. Our patient has been using rapamycin 1% cream for more than 5 years and has had a substantial reduction in the size and number of angiofibromas.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References:
Spitz J. Genodermatoses. A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins, 2005.
James W et al. Andrews’ Diseases of the Skin. Philadelphia: Saunders, 2006.
Bolognia JL et al. Dermatology. London: Mosby Elsevier, 2008.
although up to 75% of cases may be caused by a spontaneous mutation. It is caused by mutations in the TSC1 gene on chromosome 9q34–encoding hamartin or the TSC2 gene on chromosome I6pl3–encoding tuberin. Patients present at birth and males and females are affected equally.
There are multiple skin findings in TS that may herald the diagnosis. The earliest findings are hypopigmented macules, found in 85% of patients. They may be in an ash-leaf shape or confetti pattern. Adenoma sebaceum, or angiofibromas, are present on the forehead, nose, and cheeks, and often present in childhood. Periungual angiofibromas called Koenen tumors tend to occur at puberty. Connective-tissue nevi called Shagreen plaques, or collagenomas, may be present, which is what our patient exhibits on his back. The lumbosacral region is the most common area for these to appear in the first decade of life.
TS can affect other organ systems in the body. Seizures, neuropsychiatric diseases, and mental deficiency are common. Cortical tumors, gliomas, and astrocytomas may develop in the brain. Congenital retinal hamartomas (phakomas) occur. Renal cysts and angiomyolipomas may occur in the kidneys. In the lungs, patients may develop lymphangiomyomatosis. Rhabdomyomas can occur in the heart in infancy and may regress spontaneously over time. Bony changes such as cysts and sclerosis may occur.
Treatment and monitoring of TS requires a multidisciplinary approach with neurology, pulmonology, cardiology, ophthalmology, orthopedics, and dermatology. Cosmetic treatment for angiofibromas includes CO2 laser, shaving, and dermabrasion. Topical rapamycin use has been described in the literature to improve the appearance of angiofibromas. Our patient has been using rapamycin 1% cream for more than 5 years and has had a substantial reduction in the size and number of angiofibromas.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References:
Spitz J. Genodermatoses. A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins, 2005.
James W et al. Andrews’ Diseases of the Skin. Philadelphia: Saunders, 2006.
Bolognia JL et al. Dermatology. London: Mosby Elsevier, 2008.
Dan Kastner wins Crafoord Prize in Polyarthritis
“for establishing the concept of autoinflammatory diseases.” The prize, named after the donor Holger Crafoord because of his bout with severe rheumatoid arthritis toward the end of his life, is for 6 million Swedish kronor (approximately USD $700,000).
Dr. Kastner, scientific director at the U.S. National Human Genome Research Institute’s division of intramural research, received the award for identifying the mechanisms responsible for familial Mediterranean fever, tumor necrosis factor receptor–associated periodic syndrome, and other diagnoses within the group of autoinflammatory diseases.
“Dan Kastner is often called the father of autoinflammatory diseases, a title that he thoroughly deserves. His discoveries have taught us a great deal about the immune system and its functions, contributing to effective treatments that reduce the symptoms of diseases from which patients previously suffered enormously, sometimes leading to premature death,” Olle Kämpe, chair of the prize committee, said in a press announcement.
While the Crafoord Prize normally is awarded on a 3-year rotating basis for achievements in mathematics and astronomy, geosciences, and biosciences, the prize in polyarthritis is “only awarded when there has been scientific progress that motivates a prize,” according to the press release.
“for establishing the concept of autoinflammatory diseases.” The prize, named after the donor Holger Crafoord because of his bout with severe rheumatoid arthritis toward the end of his life, is for 6 million Swedish kronor (approximately USD $700,000).
Dr. Kastner, scientific director at the U.S. National Human Genome Research Institute’s division of intramural research, received the award for identifying the mechanisms responsible for familial Mediterranean fever, tumor necrosis factor receptor–associated periodic syndrome, and other diagnoses within the group of autoinflammatory diseases.
“Dan Kastner is often called the father of autoinflammatory diseases, a title that he thoroughly deserves. His discoveries have taught us a great deal about the immune system and its functions, contributing to effective treatments that reduce the symptoms of diseases from which patients previously suffered enormously, sometimes leading to premature death,” Olle Kämpe, chair of the prize committee, said in a press announcement.
While the Crafoord Prize normally is awarded on a 3-year rotating basis for achievements in mathematics and astronomy, geosciences, and biosciences, the prize in polyarthritis is “only awarded when there has been scientific progress that motivates a prize,” according to the press release.
“for establishing the concept of autoinflammatory diseases.” The prize, named after the donor Holger Crafoord because of his bout with severe rheumatoid arthritis toward the end of his life, is for 6 million Swedish kronor (approximately USD $700,000).
Dr. Kastner, scientific director at the U.S. National Human Genome Research Institute’s division of intramural research, received the award for identifying the mechanisms responsible for familial Mediterranean fever, tumor necrosis factor receptor–associated periodic syndrome, and other diagnoses within the group of autoinflammatory diseases.
“Dan Kastner is often called the father of autoinflammatory diseases, a title that he thoroughly deserves. His discoveries have taught us a great deal about the immune system and its functions, contributing to effective treatments that reduce the symptoms of diseases from which patients previously suffered enormously, sometimes leading to premature death,” Olle Kämpe, chair of the prize committee, said in a press announcement.
While the Crafoord Prize normally is awarded on a 3-year rotating basis for achievements in mathematics and astronomy, geosciences, and biosciences, the prize in polyarthritis is “only awarded when there has been scientific progress that motivates a prize,” according to the press release.
Consensus statement issued on retinoids for ichthyosis, disorders of cornification
Clinicians using advised the authors of a new consensus statement.

In the statement, published in Pediatric Dermatology, they also addressed the effects of topical and systemic retinoid use on bone, eye, cardiovascular, and mental health, and the risks some retinoids pose to reproductive health.
Many patients with these chronic conditions, driven by multiple genetic mutations, respond to topical and/or systemic retinoids. However, to date, no specific guidance has addressed the safety, efficacy, or overall precautions for their use in the pediatric population, one of the statement authors, Moise L. Levy, MD, professor of pediatrics and medicine at the University of Texas at Austin, said in an interview.
Dr. Levy was one of the physicians on the multidisciplinary panel, The Pediatric Dermatology Research Alliance Use of Retinoids in Ichthyosis Work Group, formed to devise best practice recommendations on the use of retinoids in the management of ichthyoses and other cornification disorders in children and adolescents. The panel conducted an extensive evidence-based literature review and met in person to arrive at their conclusions. Representation from the Foundation for Ichthyosis and Related Skin Types (FIRST) was also key to this work. “Additionally, the teratogenic effects of retinoids prompted examination of gynecologic considerations and the role of the iPLEDGE program in the United States on patient access to isotretinoin,” the authors wrote.
Retinoid effects, dosing
“Both topical and systemic retinoids can improve scaling in patients with select forms of ichthyosis,” and some subtypes of disease respond better to treatment than others, they noted. Oral or topical retinoids are known to improve cases of congenital ichthyosiform erythroderma (select genotypes), Sjögren-Larsson syndrome, ichthyosis follicularis–alopecia-photophobia syndromes and keratitis-ichthyosis-deafness syndrome, erythrokeratodermia variabilis, harlequin ichthyosis, ichthyosis with confetti, and other subtypes.
Comparatively, they added, there are no data on the use of retinoids, or data showing no improvement with retinoids for several ichthyosis subtypes, including congenital hemidysplasia with ichthyosiform erythroderma and limb defects, CHIME syndrome, Conradi-Hünermann-Happle syndrome, ichthyosis-hypotrichosis syndrome, ichthyosis-hypotrichosis-sclerosing cholangitis, ichthyosis prematurity syndrome, MEDNIK syndrome, peeling skin disease, Refsum syndrome, and trichothiodystrophy though the response to such cases may vary.
Retinoids may worsen conditions that lead to peeling or skin fragility, atopic diathesis, or excessive desquamation, “and should be used with caution,” the authors advised.
Pediatric and adult patients with moderate to severe disease and significant functional or psychological impairment “should be offered the opportunity to make a benefit/risk assessment of treatment” with a systemic retinoid, they added, noting that topical retinoids have a lower risk profile and may be a better choice for milder disease.
Clinicians should aim for the lowest dose possible “that will achieve and maintain the desired therapeutic effect with acceptable mucocutaneous and systemic toxicities,” the panel recommended. Lower doses work especially well in patients with epidermolytic ichthyoses and erythrokeratodermia variabilis.
“Given the cutaneous and extracutaneous toxicities of oral retinoids, lower doses were found to achieve the most acceptable risk-benefit result. Few individuals now receive more than 1 mg/kg per day of isotretinoin or 0.5 mg/kg per day of acitretin,” according to the panel.
Dosing decisions call for a group conversation between physicians, patients and caregivers, addressing skin care, comfort and appearance issues, risk of adverse effects, and tolerance of the therapy.
Retinoid effects on organs
The impact of retinoids on the body varies by organ system, type of therapy and dosage. Dose and duration of therapy, for example, help determine the toxic effects of retinoids on bone. “Long-term use of systemic retinoids in ichthyosis/DOC is associated with skeletal concerns,” noted the authors, adding that clinicians should still consider this therapeutic approach if there is a strong clinical case for using it in a patient.
Children on long-term systemic therapy should undergo a series of tests and evaluations for bone monitoring, including an annual growth assessment. The group also recommended a baseline skeletal radiographic survey when children are on long-term systemic retinoid therapy, repeated after 3-5 years or when symptoms are present. Clinicians should also inquire about diet and discuss with patients factors that impact susceptibility to retinoid bone toxicity, such as genetic risk, diet and physical activity.
They also recommended monitoring patients taking systemic retinoids for psychiatric symptoms.
Adolescents of childbearing potential using systemic retinoids, who are sexually active, should receive counseling about contraceptive options, and should use two forms of contraception, including one highly effective method, the statement advises.
In the United States, all patients and prescribers of isotretinoin must comply with iPLEDGE guidelines; the statement addresses the issue that iPLEDGE was not designed for long-term use of isotretinoin in patients with ichthyosis, and “imposes a significant burden” in this group.
Other practice gaps and unmet needs in this area of study were discussed, calling for a closer examination of optimal timing of therapy initiation, and the adverse effects of long-term retinoid treatment. “The work, as a whole, is a starting point for these important management issues,” said Dr. Levy.
Unrestricted educational grants from Sun Pharmaceuticals and FIRST funded this effort. Dr. Levy’s disclosed serving on the advisory board and as a consultant for Cassiopea, Regeneron, and UCB, and an investigator for Fibrocell, Galderma, Janssen, and Pfizer. The other authors disclosed serving as investigators, advisers, consultants, and/or other relationships with various pharmaceutical companies.
Clinicians using advised the authors of a new consensus statement.
In the statement, published in Pediatric Dermatology, they also addressed the effects of topical and systemic retinoid use on bone, eye, cardiovascular, and mental health, and the risks some retinoids pose to reproductive health.
Many patients with these chronic conditions, driven by multiple genetic mutations, respond to topical and/or systemic retinoids. However, to date, no specific guidance has addressed the safety, efficacy, or overall precautions for their use in the pediatric population, one of the statement authors, Moise L. Levy, MD, professor of pediatrics and medicine at the University of Texas at Austin, said in an interview.
Dr. Levy was one of the physicians on the multidisciplinary panel, The Pediatric Dermatology Research Alliance Use of Retinoids in Ichthyosis Work Group, formed to devise best practice recommendations on the use of retinoids in the management of ichthyoses and other cornification disorders in children and adolescents. The panel conducted an extensive evidence-based literature review and met in person to arrive at their conclusions. Representation from the Foundation for Ichthyosis and Related Skin Types (FIRST) was also key to this work. “Additionally, the teratogenic effects of retinoids prompted examination of gynecologic considerations and the role of the iPLEDGE program in the United States on patient access to isotretinoin,” the authors wrote.
Retinoid effects, dosing
“Both topical and systemic retinoids can improve scaling in patients with select forms of ichthyosis,” and some subtypes of disease respond better to treatment than others, they noted. Oral or topical retinoids are known to improve cases of congenital ichthyosiform erythroderma (select genotypes), Sjögren-Larsson syndrome, ichthyosis follicularis–alopecia-photophobia syndromes and keratitis-ichthyosis-deafness syndrome, erythrokeratodermia variabilis, harlequin ichthyosis, ichthyosis with confetti, and other subtypes.
Comparatively, they added, there are no data on the use of retinoids, or data showing no improvement with retinoids for several ichthyosis subtypes, including congenital hemidysplasia with ichthyosiform erythroderma and limb defects, CHIME syndrome, Conradi-Hünermann-Happle syndrome, ichthyosis-hypotrichosis syndrome, ichthyosis-hypotrichosis-sclerosing cholangitis, ichthyosis prematurity syndrome, MEDNIK syndrome, peeling skin disease, Refsum syndrome, and trichothiodystrophy though the response to such cases may vary.
Retinoids may worsen conditions that lead to peeling or skin fragility, atopic diathesis, or excessive desquamation, “and should be used with caution,” the authors advised.
Pediatric and adult patients with moderate to severe disease and significant functional or psychological impairment “should be offered the opportunity to make a benefit/risk assessment of treatment” with a systemic retinoid, they added, noting that topical retinoids have a lower risk profile and may be a better choice for milder disease.
Clinicians should aim for the lowest dose possible “that will achieve and maintain the desired therapeutic effect with acceptable mucocutaneous and systemic toxicities,” the panel recommended. Lower doses work especially well in patients with epidermolytic ichthyoses and erythrokeratodermia variabilis.
“Given the cutaneous and extracutaneous toxicities of oral retinoids, lower doses were found to achieve the most acceptable risk-benefit result. Few individuals now receive more than 1 mg/kg per day of isotretinoin or 0.5 mg/kg per day of acitretin,” according to the panel.
Dosing decisions call for a group conversation between physicians, patients and caregivers, addressing skin care, comfort and appearance issues, risk of adverse effects, and tolerance of the therapy.
Retinoid effects on organs
The impact of retinoids on the body varies by organ system, type of therapy and dosage. Dose and duration of therapy, for example, help determine the toxic effects of retinoids on bone. “Long-term use of systemic retinoids in ichthyosis/DOC is associated with skeletal concerns,” noted the authors, adding that clinicians should still consider this therapeutic approach if there is a strong clinical case for using it in a patient.
Children on long-term systemic therapy should undergo a series of tests and evaluations for bone monitoring, including an annual growth assessment. The group also recommended a baseline skeletal radiographic survey when children are on long-term systemic retinoid therapy, repeated after 3-5 years or when symptoms are present. Clinicians should also inquire about diet and discuss with patients factors that impact susceptibility to retinoid bone toxicity, such as genetic risk, diet and physical activity.
They also recommended monitoring patients taking systemic retinoids for psychiatric symptoms.
Adolescents of childbearing potential using systemic retinoids, who are sexually active, should receive counseling about contraceptive options, and should use two forms of contraception, including one highly effective method, the statement advises.
In the United States, all patients and prescribers of isotretinoin must comply with iPLEDGE guidelines; the statement addresses the issue that iPLEDGE was not designed for long-term use of isotretinoin in patients with ichthyosis, and “imposes a significant burden” in this group.
Other practice gaps and unmet needs in this area of study were discussed, calling for a closer examination of optimal timing of therapy initiation, and the adverse effects of long-term retinoid treatment. “The work, as a whole, is a starting point for these important management issues,” said Dr. Levy.
Unrestricted educational grants from Sun Pharmaceuticals and FIRST funded this effort. Dr. Levy’s disclosed serving on the advisory board and as a consultant for Cassiopea, Regeneron, and UCB, and an investigator for Fibrocell, Galderma, Janssen, and Pfizer. The other authors disclosed serving as investigators, advisers, consultants, and/or other relationships with various pharmaceutical companies.
Clinicians using advised the authors of a new consensus statement.
In the statement, published in Pediatric Dermatology, they also addressed the effects of topical and systemic retinoid use on bone, eye, cardiovascular, and mental health, and the risks some retinoids pose to reproductive health.
Many patients with these chronic conditions, driven by multiple genetic mutations, respond to topical and/or systemic retinoids. However, to date, no specific guidance has addressed the safety, efficacy, or overall precautions for their use in the pediatric population, one of the statement authors, Moise L. Levy, MD, professor of pediatrics and medicine at the University of Texas at Austin, said in an interview.
Dr. Levy was one of the physicians on the multidisciplinary panel, The Pediatric Dermatology Research Alliance Use of Retinoids in Ichthyosis Work Group, formed to devise best practice recommendations on the use of retinoids in the management of ichthyoses and other cornification disorders in children and adolescents. The panel conducted an extensive evidence-based literature review and met in person to arrive at their conclusions. Representation from the Foundation for Ichthyosis and Related Skin Types (FIRST) was also key to this work. “Additionally, the teratogenic effects of retinoids prompted examination of gynecologic considerations and the role of the iPLEDGE program in the United States on patient access to isotretinoin,” the authors wrote.
Retinoid effects, dosing
“Both topical and systemic retinoids can improve scaling in patients with select forms of ichthyosis,” and some subtypes of disease respond better to treatment than others, they noted. Oral or topical retinoids are known to improve cases of congenital ichthyosiform erythroderma (select genotypes), Sjögren-Larsson syndrome, ichthyosis follicularis–alopecia-photophobia syndromes and keratitis-ichthyosis-deafness syndrome, erythrokeratodermia variabilis, harlequin ichthyosis, ichthyosis with confetti, and other subtypes.
Comparatively, they added, there are no data on the use of retinoids, or data showing no improvement with retinoids for several ichthyosis subtypes, including congenital hemidysplasia with ichthyosiform erythroderma and limb defects, CHIME syndrome, Conradi-Hünermann-Happle syndrome, ichthyosis-hypotrichosis syndrome, ichthyosis-hypotrichosis-sclerosing cholangitis, ichthyosis prematurity syndrome, MEDNIK syndrome, peeling skin disease, Refsum syndrome, and trichothiodystrophy though the response to such cases may vary.
Retinoids may worsen conditions that lead to peeling or skin fragility, atopic diathesis, or excessive desquamation, “and should be used with caution,” the authors advised.
Pediatric and adult patients with moderate to severe disease and significant functional or psychological impairment “should be offered the opportunity to make a benefit/risk assessment of treatment” with a systemic retinoid, they added, noting that topical retinoids have a lower risk profile and may be a better choice for milder disease.
Clinicians should aim for the lowest dose possible “that will achieve and maintain the desired therapeutic effect with acceptable mucocutaneous and systemic toxicities,” the panel recommended. Lower doses work especially well in patients with epidermolytic ichthyoses and erythrokeratodermia variabilis.
“Given the cutaneous and extracutaneous toxicities of oral retinoids, lower doses were found to achieve the most acceptable risk-benefit result. Few individuals now receive more than 1 mg/kg per day of isotretinoin or 0.5 mg/kg per day of acitretin,” according to the panel.
Dosing decisions call for a group conversation between physicians, patients and caregivers, addressing skin care, comfort and appearance issues, risk of adverse effects, and tolerance of the therapy.
Retinoid effects on organs
The impact of retinoids on the body varies by organ system, type of therapy and dosage. Dose and duration of therapy, for example, help determine the toxic effects of retinoids on bone. “Long-term use of systemic retinoids in ichthyosis/DOC is associated with skeletal concerns,” noted the authors, adding that clinicians should still consider this therapeutic approach if there is a strong clinical case for using it in a patient.
Children on long-term systemic therapy should undergo a series of tests and evaluations for bone monitoring, including an annual growth assessment. The group also recommended a baseline skeletal radiographic survey when children are on long-term systemic retinoid therapy, repeated after 3-5 years or when symptoms are present. Clinicians should also inquire about diet and discuss with patients factors that impact susceptibility to retinoid bone toxicity, such as genetic risk, diet and physical activity.
They also recommended monitoring patients taking systemic retinoids for psychiatric symptoms.
Adolescents of childbearing potential using systemic retinoids, who are sexually active, should receive counseling about contraceptive options, and should use two forms of contraception, including one highly effective method, the statement advises.
In the United States, all patients and prescribers of isotretinoin must comply with iPLEDGE guidelines; the statement addresses the issue that iPLEDGE was not designed for long-term use of isotretinoin in patients with ichthyosis, and “imposes a significant burden” in this group.
Other practice gaps and unmet needs in this area of study were discussed, calling for a closer examination of optimal timing of therapy initiation, and the adverse effects of long-term retinoid treatment. “The work, as a whole, is a starting point for these important management issues,” said Dr. Levy.
Unrestricted educational grants from Sun Pharmaceuticals and FIRST funded this effort. Dr. Levy’s disclosed serving on the advisory board and as a consultant for Cassiopea, Regeneron, and UCB, and an investigator for Fibrocell, Galderma, Janssen, and Pfizer. The other authors disclosed serving as investigators, advisers, consultants, and/or other relationships with various pharmaceutical companies.
FROM PEDIATRIC DERMATOLOGY
Cutaneous Insulin-Derived Amyloidosis Presenting as Hyperkeratotic Nodules
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
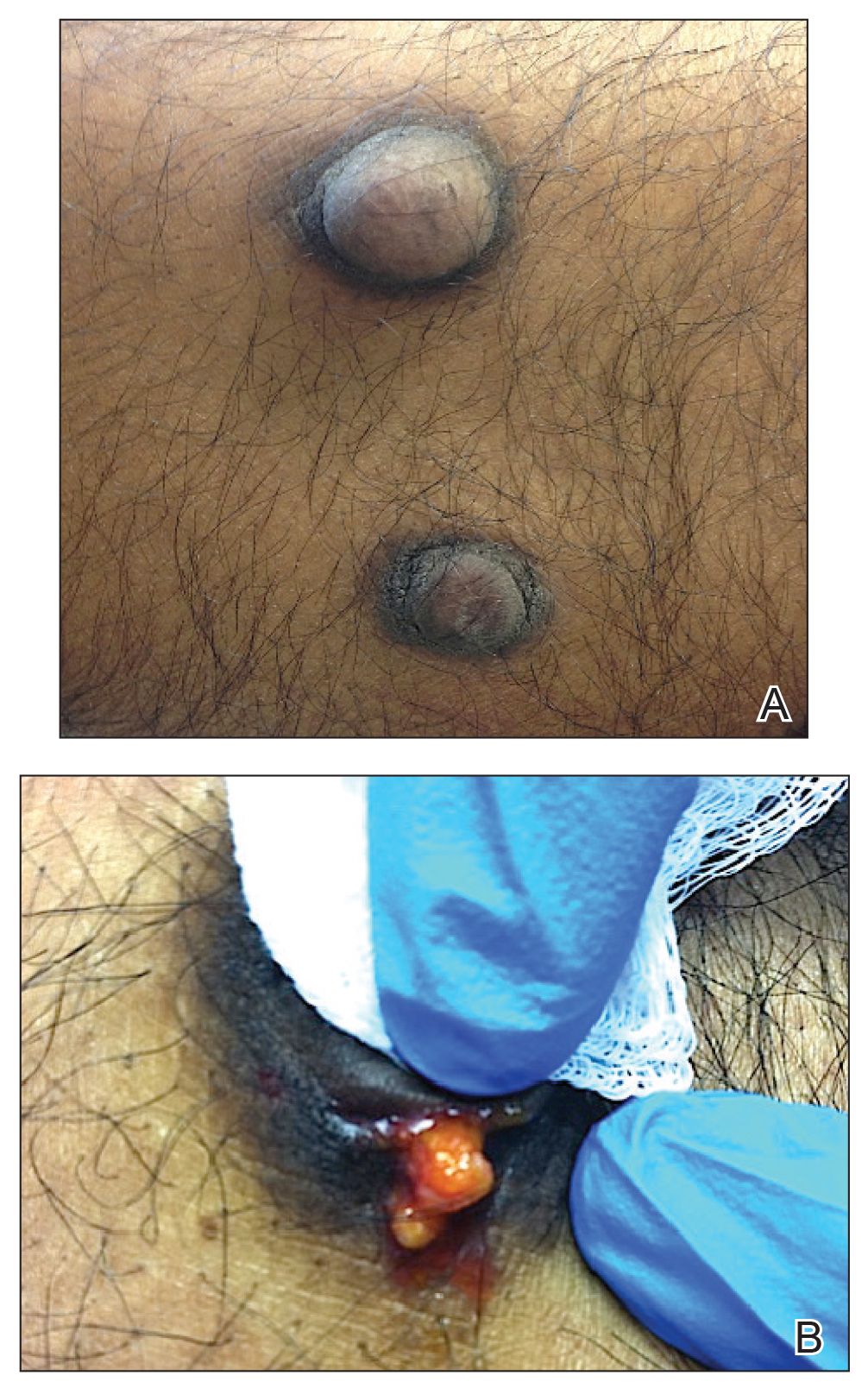
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
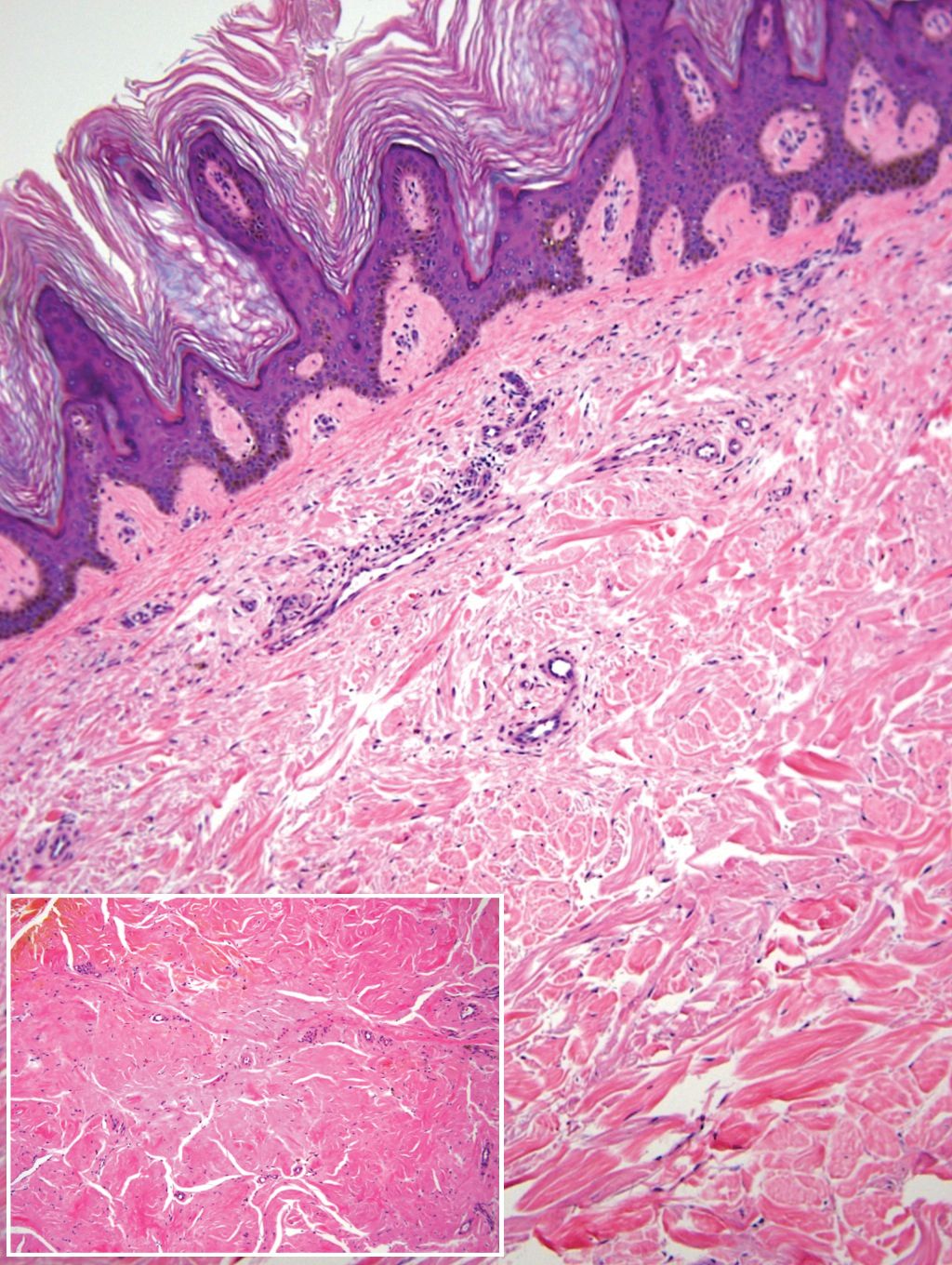
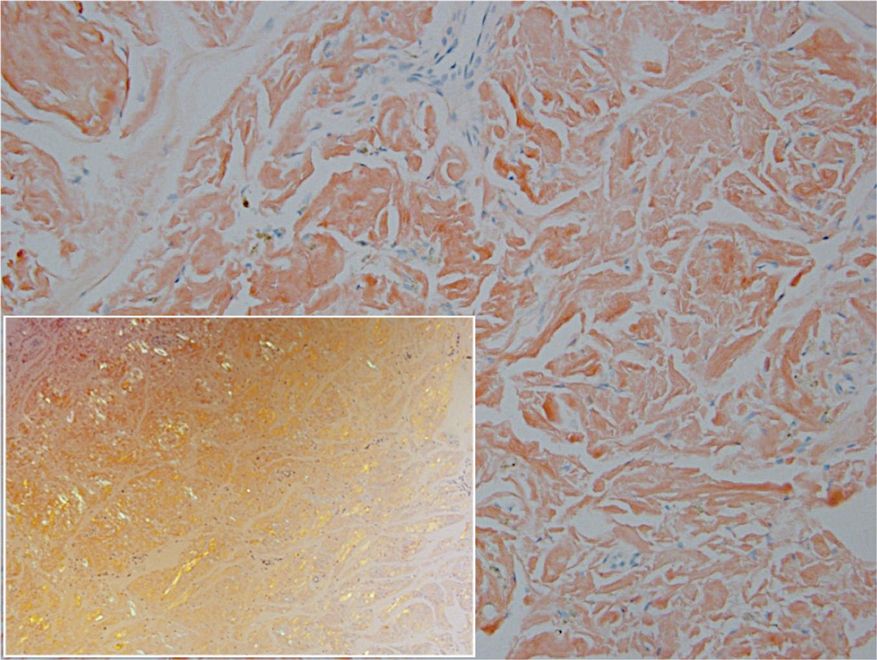
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
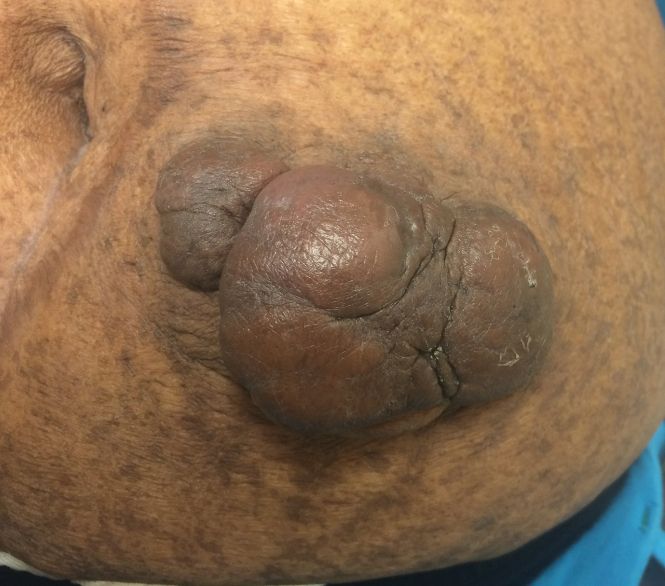
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Practice Points
- Deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.
- Patients with insulin-derived (AIns) amyloidosis may initially present after noticing nodular deposits.
- Insulin injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis.