User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
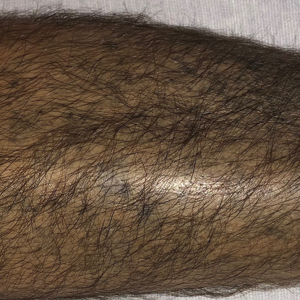
Chronic Hyperpigmented Patches on the Legs
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
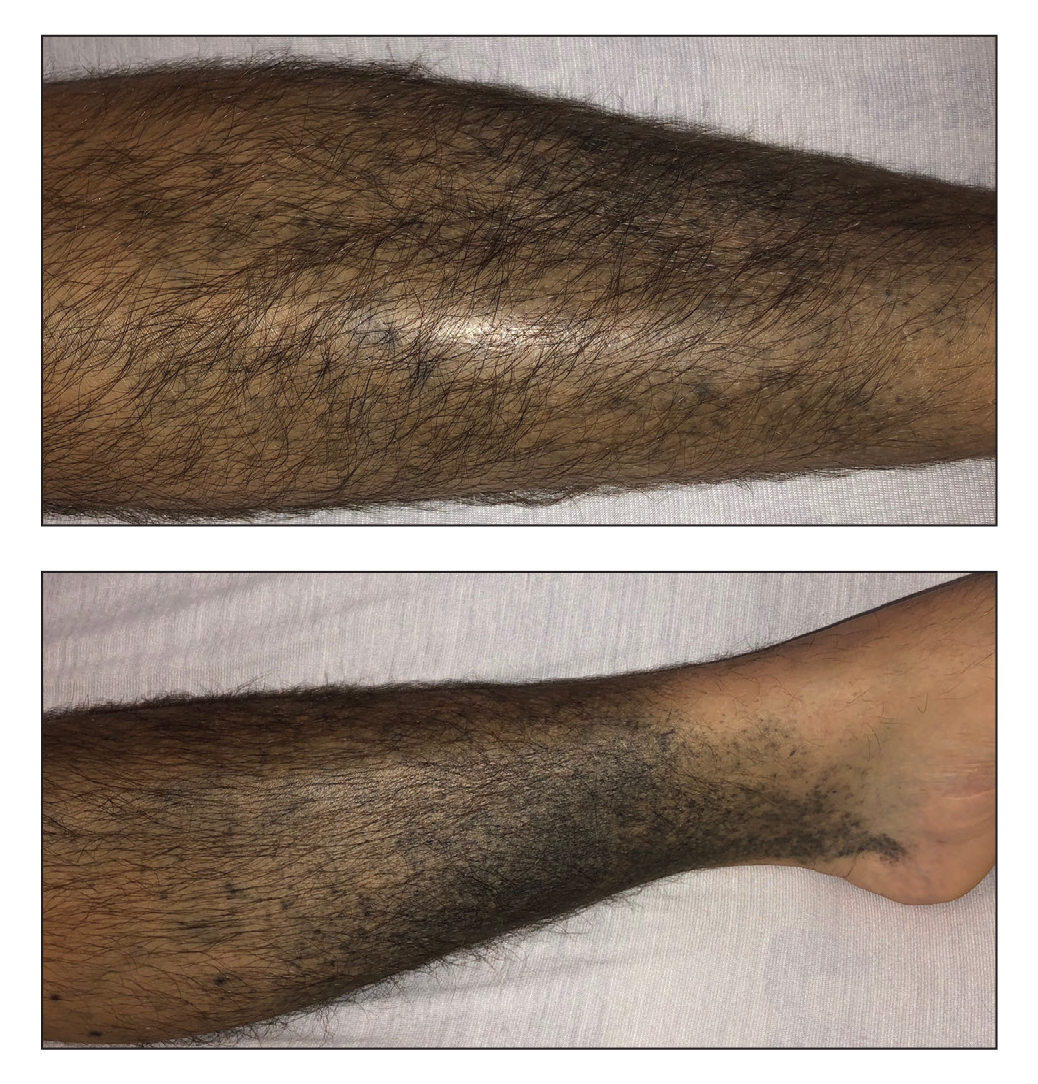
A 37-year-old man with a history of cerebral palsy, bipolar disorder, and impulse control disorder presented to the emergency department with breathing difficulty and worsening malaise. The patient subsequently was intubated due to hypoxic respiratory failure and was found to be positive for SARS-CoV-2. He was admitted to the intensive care unit, and dermatology was consulted due to concern that the cutaneous findings were demonstrative of a vasculitic process. Physical examination revealed diffuse, symmetric, dark brown to blue-gray macules coalescing into patches on the anterior tibia (top) and covering the entire lower leg (bottom). The patches were mottled and did not blanch with pressure. According to the patient’s caretaker, the leg hyperpigmentation had been present for 2 years.
Johnson & Johnson requests FDA approval for vaccine booster doses
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.
JAK inhibitor provides impressive hair growth for patients with alopecia areata
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Ruxolitinib cream meets primary endpoints in phase 3 vitiligo trial
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
Psoriasis and Psoriatic Arthritis: A Supplement to Dermatology News

Psoriasis and Psoriatic Arthritis: A Supplement to Dermatology News 2021
- Psoriasis severity redefined by expert group
- Mitigating PSA risk with biologic therapy
- Lack of diversity seen in psoriasis trials
- PSA Comorbidities effect on treatment responses
With Commentaries by Joel M. Gelfand, MD, MSCE and Alan Menter, MD
And more…
Psoriasis and Psoriatic Arthritis: A Supplement to Dermatology News 2021
- Psoriasis severity redefined by expert group
- Mitigating PSA risk with biologic therapy
- Lack of diversity seen in psoriasis trials
- PSA Comorbidities effect on treatment responses
With Commentaries by Joel M. Gelfand, MD, MSCE and Alan Menter, MD
And more…
Psoriasis and Psoriatic Arthritis: A Supplement to Dermatology News 2021
- Psoriasis severity redefined by expert group
- Mitigating PSA risk with biologic therapy
- Lack of diversity seen in psoriasis trials
- PSA Comorbidities effect on treatment responses
With Commentaries by Joel M. Gelfand, MD, MSCE and Alan Menter, MD
And more…
COVID-19: Two more cases of mucosal skin ulcers reported in male teens
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Pfizer COVID vaccine antibodies may disappear in 7 months, study says
, according to a new study published on the bioRxiv preprint server.
In the study, which hasn’t yet been peer-reviewed or formally published in a medical journal, researchers analyzed blood samples from 46 healthy young or middle-aged adults after receiving two doses, and then 6 months after the second dose.
“Our study shows vaccination with the Pfizer-BioNTech vaccine induces high levels of neutralizing antibodies against the original vaccine strain, but these levels drop by nearly 10-fold by 7 months,” the researchers told Reuters.
In about half of the adults, neutralizing antibodies were undetectable at 6 months after the second dose, particularly against coronavirus variants such as Delta, Beta, and Mu.
Neutralizing antibodies only make up part of the body’s immune defense against the virus, Reuters noted, but they are still “critically important” in protecting against coronavirus infections.
“These findings suggest that administering a booster dose at around 6 to 7 months following the initial immunization will likely enhance protection,” the study authors wrote.
BioNTech said a new vaccine formula will likely be needed by mid-2022 to protect against future mutations of the virus, according to the Financial Times.
“This year, [a different vaccine] is completely unneeded, but by mid-next year, it could be a different situation,” Ugur Sahin, MD, cofounder and CEO of BioNTech, told the news outlet.
Current variants, namely the Delta variant, are more contagious than the original coronavirus strain but not different enough to evade current vaccines, he said. But new strains may be able to evade boosters.
“This virus will stay, and the virus will further adapt,” Dr. Sahin said. “This is a continuous evolution, and that evolution has just started.”
A version of this article first appeared on WebMD.com.
, according to a new study published on the bioRxiv preprint server.
In the study, which hasn’t yet been peer-reviewed or formally published in a medical journal, researchers analyzed blood samples from 46 healthy young or middle-aged adults after receiving two doses, and then 6 months after the second dose.
“Our study shows vaccination with the Pfizer-BioNTech vaccine induces high levels of neutralizing antibodies against the original vaccine strain, but these levels drop by nearly 10-fold by 7 months,” the researchers told Reuters.
In about half of the adults, neutralizing antibodies were undetectable at 6 months after the second dose, particularly against coronavirus variants such as Delta, Beta, and Mu.
Neutralizing antibodies only make up part of the body’s immune defense against the virus, Reuters noted, but they are still “critically important” in protecting against coronavirus infections.
“These findings suggest that administering a booster dose at around 6 to 7 months following the initial immunization will likely enhance protection,” the study authors wrote.
BioNTech said a new vaccine formula will likely be needed by mid-2022 to protect against future mutations of the virus, according to the Financial Times.
“This year, [a different vaccine] is completely unneeded, but by mid-next year, it could be a different situation,” Ugur Sahin, MD, cofounder and CEO of BioNTech, told the news outlet.
Current variants, namely the Delta variant, are more contagious than the original coronavirus strain but not different enough to evade current vaccines, he said. But new strains may be able to evade boosters.
“This virus will stay, and the virus will further adapt,” Dr. Sahin said. “This is a continuous evolution, and that evolution has just started.”
A version of this article first appeared on WebMD.com.
, according to a new study published on the bioRxiv preprint server.
In the study, which hasn’t yet been peer-reviewed or formally published in a medical journal, researchers analyzed blood samples from 46 healthy young or middle-aged adults after receiving two doses, and then 6 months after the second dose.
“Our study shows vaccination with the Pfizer-BioNTech vaccine induces high levels of neutralizing antibodies against the original vaccine strain, but these levels drop by nearly 10-fold by 7 months,” the researchers told Reuters.
In about half of the adults, neutralizing antibodies were undetectable at 6 months after the second dose, particularly against coronavirus variants such as Delta, Beta, and Mu.
Neutralizing antibodies only make up part of the body’s immune defense against the virus, Reuters noted, but they are still “critically important” in protecting against coronavirus infections.
“These findings suggest that administering a booster dose at around 6 to 7 months following the initial immunization will likely enhance protection,” the study authors wrote.
BioNTech said a new vaccine formula will likely be needed by mid-2022 to protect against future mutations of the virus, according to the Financial Times.
“This year, [a different vaccine] is completely unneeded, but by mid-next year, it could be a different situation,” Ugur Sahin, MD, cofounder and CEO of BioNTech, told the news outlet.
Current variants, namely the Delta variant, are more contagious than the original coronavirus strain but not different enough to evade current vaccines, he said. But new strains may be able to evade boosters.
“This virus will stay, and the virus will further adapt,” Dr. Sahin said. “This is a continuous evolution, and that evolution has just started.”
A version of this article first appeared on WebMD.com.
Extension study finds dupilumab effective for up to 1 year in teens with AD
in a phase 3, open-label extension trial, researchers reported.

At 1 year, 86% of 50 remaining patients with weights under 60 kg (132 lb) had achieved 75% improvement on the Eczema Area and Severity Index (EASI-75, and 77% of 51 remaining patients with weights over 60 kg reached that level of clearance. Only 5 (1.7%) of 294 patients had serious treatment-emergent adverse events (TEAEs).
The findings back up a perception that patients can stay on dupilumab for some time instead of having to switch from one biologic to another after a few years, study coauthor Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, said in an interview. He added that the drug’s long-term safety profile is “very reassuring.”
The industry-funded findings of the study were released in a poster at the 2021 meeting of the World Congress of Pediatric Dermatology.
The FDA approved dupilumab (Dupixent), an interleukin-4 receptor alpha antagonist, for treating AD in adults in 2017; it is now approved for treating patients ages 6 years and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topicals.
The new study tracked patients who received at least 300 mg dupilumab subcutaneously every 4 weeks. The dose could be increased if needed to improve clinical response to once every 2 weeks (200 mg if baseline weight was <60 kg; 300 mg if ≥60 kg).
At 52 weeks, 37% of 52 patients with weights under 60 kg reached an Investigator Global Assessment (IGA) of 0/1, a level that had been fairly steady since week 16 (n = 146). Among 51 heavier patients, 49% reached an IGA of 0/1 at 52 weeks; this percentage grew steadily since baseline.
The mean percentage change in EASI was –87% in the lower-weight group (n = 50) at 52 weeks and –80.1% in the larger-weight group (n = 51). The majority of the reduction in EASI occurred in the first 4 weeks of treatment.
At 52 weeks, the mean Children’s Dermatology Life Quality Index level, which judges the effect of AD on life, was judged as “small” (low) in 71 patients. At baseline, the mean level among 189 patients was “moderate.” The levels dipped below “moderate” at week 4 and never rose above “small” after that.
“Treatment-emergent adverse events reported in ≥5% of patients were nasopharyngitis (21.1%), AD (19.4%), upper respiratory tract infection (12.4%), headache (9.4%), and oropharyngeal pain (5.7%),” the investigators wrote in the poster. They add that 6.7% of patients experienced injection-site reactions, and 8.7% of patients experienced treatment-emergent “narrow conjunctivitis,” which includes conjunctivitis, allergic conjunctivitis, bacterial conjunctivitis, viral conjunctivitis, and atopic keratoconjunctivitis.
Dr. Simpson noted that cases of conjunctivitis fell over time. It’s not clear why this adverse effect appears, he said.
He said that the findings reflect his own experience in clinic. Many of his adolescent patients took part in early dupilumab trials, he said, and dozens have been taking the drug for more than 5 years. “They just seem to get better and better,” he said.

University of Minnesota, Minneapolis, dermatologist Sheilagh Maguiness, MD, who wasn’t involved with the study, said in an interview that dupilumab remains “the safest, most effective and evidence-based therapy we had for children with moderate to severe atopic dermatitis.”
The new study’s findings are “very reassuring,” she said, and similar to those in a 2021 report that tracked long-term use of the drug in children aged 6-11.
Like Dr. Simpson, Dr. Maguiness said many pediatric patients at her clinic have stayed on the drug for more than 5 years. They still have “sustained improvement in skin disease and in their quality of life as well”
There are, however, still questions about dupilumab treatment. “For children who have responded well, when could we consider dose reduction or discontinuation? I have done this successfully just a handful of times, but I would love to see data about what percentage of pediatric patients experience rebound disease after coming off the drug and after what duration of treatment,” she said. “Another mystery that will be very interesting to unravel is the question as to whether or not early treatment with dupilumab may attenuate other atopic diseases.”
Dr. Maguiness added that “another issue specific to pediatric use of dupilumab is the recommendation surrounding vaccinations. This is an issue that should be studied in terms of antibody response and safety surrounding vaccinations, particularly as we are eagerly awaiting a pediatric FDA approval for the COVID-19 vaccine in children.”
She also urged colleagues to push back against insurers who resist paying for dupilumab. “Whether prescribing this medication on or off label, insurance companies are often requiring patients to try and fail other traditional immunosuppressive medications such as methotrexate, cyclosporine, or to pursue phototherapy,” she said. “Oftentimes, these are not practical or even safe options for children for a multitude of reasons. Don’t be shy about advocating for your patients by second- or even third-level appeals to try and gain approval for children who are in need of treatment.”
The study was funded by Sanofi Genzyme and Regeneron Pharmaceuticals. The study authors reported various disclosures. Dr. Simpson reported investigator and consultant fee relationships from various pharmaceutical companies. Dr. Maguiness was an investigator for one of the initial pediatric dupilumab trials.
A version of this article first appeared on Medscape.com.
in a phase 3, open-label extension trial, researchers reported.
At 1 year, 86% of 50 remaining patients with weights under 60 kg (132 lb) had achieved 75% improvement on the Eczema Area and Severity Index (EASI-75, and 77% of 51 remaining patients with weights over 60 kg reached that level of clearance. Only 5 (1.7%) of 294 patients had serious treatment-emergent adverse events (TEAEs).
The findings back up a perception that patients can stay on dupilumab for some time instead of having to switch from one biologic to another after a few years, study coauthor Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, said in an interview. He added that the drug’s long-term safety profile is “very reassuring.”
The industry-funded findings of the study were released in a poster at the 2021 meeting of the World Congress of Pediatric Dermatology.
The FDA approved dupilumab (Dupixent), an interleukin-4 receptor alpha antagonist, for treating AD in adults in 2017; it is now approved for treating patients ages 6 years and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topicals.
The new study tracked patients who received at least 300 mg dupilumab subcutaneously every 4 weeks. The dose could be increased if needed to improve clinical response to once every 2 weeks (200 mg if baseline weight was <60 kg; 300 mg if ≥60 kg).
At 52 weeks, 37% of 52 patients with weights under 60 kg reached an Investigator Global Assessment (IGA) of 0/1, a level that had been fairly steady since week 16 (n = 146). Among 51 heavier patients, 49% reached an IGA of 0/1 at 52 weeks; this percentage grew steadily since baseline.
The mean percentage change in EASI was –87% in the lower-weight group (n = 50) at 52 weeks and –80.1% in the larger-weight group (n = 51). The majority of the reduction in EASI occurred in the first 4 weeks of treatment.
At 52 weeks, the mean Children’s Dermatology Life Quality Index level, which judges the effect of AD on life, was judged as “small” (low) in 71 patients. At baseline, the mean level among 189 patients was “moderate.” The levels dipped below “moderate” at week 4 and never rose above “small” after that.
“Treatment-emergent adverse events reported in ≥5% of patients were nasopharyngitis (21.1%), AD (19.4%), upper respiratory tract infection (12.4%), headache (9.4%), and oropharyngeal pain (5.7%),” the investigators wrote in the poster. They add that 6.7% of patients experienced injection-site reactions, and 8.7% of patients experienced treatment-emergent “narrow conjunctivitis,” which includes conjunctivitis, allergic conjunctivitis, bacterial conjunctivitis, viral conjunctivitis, and atopic keratoconjunctivitis.
Dr. Simpson noted that cases of conjunctivitis fell over time. It’s not clear why this adverse effect appears, he said.
He said that the findings reflect his own experience in clinic. Many of his adolescent patients took part in early dupilumab trials, he said, and dozens have been taking the drug for more than 5 years. “They just seem to get better and better,” he said.
University of Minnesota, Minneapolis, dermatologist Sheilagh Maguiness, MD, who wasn’t involved with the study, said in an interview that dupilumab remains “the safest, most effective and evidence-based therapy we had for children with moderate to severe atopic dermatitis.”
The new study’s findings are “very reassuring,” she said, and similar to those in a 2021 report that tracked long-term use of the drug in children aged 6-11.
Like Dr. Simpson, Dr. Maguiness said many pediatric patients at her clinic have stayed on the drug for more than 5 years. They still have “sustained improvement in skin disease and in their quality of life as well”
There are, however, still questions about dupilumab treatment. “For children who have responded well, when could we consider dose reduction or discontinuation? I have done this successfully just a handful of times, but I would love to see data about what percentage of pediatric patients experience rebound disease after coming off the drug and after what duration of treatment,” she said. “Another mystery that will be very interesting to unravel is the question as to whether or not early treatment with dupilumab may attenuate other atopic diseases.”
Dr. Maguiness added that “another issue specific to pediatric use of dupilumab is the recommendation surrounding vaccinations. This is an issue that should be studied in terms of antibody response and safety surrounding vaccinations, particularly as we are eagerly awaiting a pediatric FDA approval for the COVID-19 vaccine in children.”
She also urged colleagues to push back against insurers who resist paying for dupilumab. “Whether prescribing this medication on or off label, insurance companies are often requiring patients to try and fail other traditional immunosuppressive medications such as methotrexate, cyclosporine, or to pursue phototherapy,” she said. “Oftentimes, these are not practical or even safe options for children for a multitude of reasons. Don’t be shy about advocating for your patients by second- or even third-level appeals to try and gain approval for children who are in need of treatment.”
The study was funded by Sanofi Genzyme and Regeneron Pharmaceuticals. The study authors reported various disclosures. Dr. Simpson reported investigator and consultant fee relationships from various pharmaceutical companies. Dr. Maguiness was an investigator for one of the initial pediatric dupilumab trials.
A version of this article first appeared on Medscape.com.
in a phase 3, open-label extension trial, researchers reported.
At 1 year, 86% of 50 remaining patients with weights under 60 kg (132 lb) had achieved 75% improvement on the Eczema Area and Severity Index (EASI-75, and 77% of 51 remaining patients with weights over 60 kg reached that level of clearance. Only 5 (1.7%) of 294 patients had serious treatment-emergent adverse events (TEAEs).
The findings back up a perception that patients can stay on dupilumab for some time instead of having to switch from one biologic to another after a few years, study coauthor Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, said in an interview. He added that the drug’s long-term safety profile is “very reassuring.”
The industry-funded findings of the study were released in a poster at the 2021 meeting of the World Congress of Pediatric Dermatology.
The FDA approved dupilumab (Dupixent), an interleukin-4 receptor alpha antagonist, for treating AD in adults in 2017; it is now approved for treating patients ages 6 years and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topicals.
The new study tracked patients who received at least 300 mg dupilumab subcutaneously every 4 weeks. The dose could be increased if needed to improve clinical response to once every 2 weeks (200 mg if baseline weight was <60 kg; 300 mg if ≥60 kg).
At 52 weeks, 37% of 52 patients with weights under 60 kg reached an Investigator Global Assessment (IGA) of 0/1, a level that had been fairly steady since week 16 (n = 146). Among 51 heavier patients, 49% reached an IGA of 0/1 at 52 weeks; this percentage grew steadily since baseline.
The mean percentage change in EASI was –87% in the lower-weight group (n = 50) at 52 weeks and –80.1% in the larger-weight group (n = 51). The majority of the reduction in EASI occurred in the first 4 weeks of treatment.
At 52 weeks, the mean Children’s Dermatology Life Quality Index level, which judges the effect of AD on life, was judged as “small” (low) in 71 patients. At baseline, the mean level among 189 patients was “moderate.” The levels dipped below “moderate” at week 4 and never rose above “small” after that.
“Treatment-emergent adverse events reported in ≥5% of patients were nasopharyngitis (21.1%), AD (19.4%), upper respiratory tract infection (12.4%), headache (9.4%), and oropharyngeal pain (5.7%),” the investigators wrote in the poster. They add that 6.7% of patients experienced injection-site reactions, and 8.7% of patients experienced treatment-emergent “narrow conjunctivitis,” which includes conjunctivitis, allergic conjunctivitis, bacterial conjunctivitis, viral conjunctivitis, and atopic keratoconjunctivitis.
Dr. Simpson noted that cases of conjunctivitis fell over time. It’s not clear why this adverse effect appears, he said.
He said that the findings reflect his own experience in clinic. Many of his adolescent patients took part in early dupilumab trials, he said, and dozens have been taking the drug for more than 5 years. “They just seem to get better and better,” he said.
University of Minnesota, Minneapolis, dermatologist Sheilagh Maguiness, MD, who wasn’t involved with the study, said in an interview that dupilumab remains “the safest, most effective and evidence-based therapy we had for children with moderate to severe atopic dermatitis.”
The new study’s findings are “very reassuring,” she said, and similar to those in a 2021 report that tracked long-term use of the drug in children aged 6-11.
Like Dr. Simpson, Dr. Maguiness said many pediatric patients at her clinic have stayed on the drug for more than 5 years. They still have “sustained improvement in skin disease and in their quality of life as well”
There are, however, still questions about dupilumab treatment. “For children who have responded well, when could we consider dose reduction or discontinuation? I have done this successfully just a handful of times, but I would love to see data about what percentage of pediatric patients experience rebound disease after coming off the drug and after what duration of treatment,” she said. “Another mystery that will be very interesting to unravel is the question as to whether or not early treatment with dupilumab may attenuate other atopic diseases.”
Dr. Maguiness added that “another issue specific to pediatric use of dupilumab is the recommendation surrounding vaccinations. This is an issue that should be studied in terms of antibody response and safety surrounding vaccinations, particularly as we are eagerly awaiting a pediatric FDA approval for the COVID-19 vaccine in children.”
She also urged colleagues to push back against insurers who resist paying for dupilumab. “Whether prescribing this medication on or off label, insurance companies are often requiring patients to try and fail other traditional immunosuppressive medications such as methotrexate, cyclosporine, or to pursue phototherapy,” she said. “Oftentimes, these are not practical or even safe options for children for a multitude of reasons. Don’t be shy about advocating for your patients by second- or even third-level appeals to try and gain approval for children who are in need of treatment.”
The study was funded by Sanofi Genzyme and Regeneron Pharmaceuticals. The study authors reported various disclosures. Dr. Simpson reported investigator and consultant fee relationships from various pharmaceutical companies. Dr. Maguiness was an investigator for one of the initial pediatric dupilumab trials.
A version of this article first appeared on Medscape.com.
Rapid response needed for rare filler injection complications
if not promptly addressed, according to an expert explaining the signs of an impending disaster at the Skin of Color Update 2021.

The most serious of the adverse events stem from vascular compromise, which is often signaled immediately by sharp pain and blanching of the skin, according to Hassan Galadari, MD, assistant professor of dermatology at the United Arab Emirates University, Dubai.
“Swift and aggressive treatment is required to avoid irreversible changes,” said Dr. Galadari, warning that blindness and vision impairment can be permanent, and that other events associated with vascular compromise include stroke and other types of embolism, as well as tissue necrosis.
To be swift, Dr. Galadari advised an immediate halt of injections and then a series of steps to abort the vascular insult. The goal is to encourage blood flow to prevent clotting and dissipate the filler.
“Massage the area like crazy. Keep on massaging. The more you massage the better. You are recruiting blood into that area so it remains viable,” Dr. Galadari said.
Hyaluronidase injections helpful
Warm compresses should also be applied for periods ranging from 5 minutes up to an hour, he added. In patients treated with hyaluronic acid, he also commonly introduces hyaluronidase injections of 200-500 IU diluted in lidocaine or saline. The injections are placed 2-3 cm apart and repeated every hour until signs and symptoms improve.
“Flush all of the filler out,” he said, emphasizing the urgency for reversing risk of vascular adverse events.
To sustain blood flow and avoid clots, he also recommends initiating aspirin with maintenance doses sustained over several days. Sildenafil to further improve conditions of blood perfusion can be “considered.”
The risks of vascular compromise, like other complications from filler injections, are low, but they are not zero, and the opportunity to prevent irreversible changes depends on acting quickly, according to Dr. Galadari.
“To prevent embolism, recognize the danger zones,” he advised, identifying the glabella region as the site of highest risk. The risk of vascular compromise from injections into the nasal region is lower but higher than injections of the nasolabial fold and forehead, which are associated with a relatively low risk.
Slow injections reduce risks
Some basic strategies he recommended for preventing vascular compromise included slow injections while keeping pressure low and using small volumes of filler per shot. Fractionated treatment and microdroplet techniques can be appropriate depending on the site of injection.
“Delivery of the filler by cannulas rather than by needles is preferable,” according to Dr. Galadari, who noted that a task force from the American Society for Dermatologic Surgery recently endorsed this approach as part of other recommendations to avoid complications of injectable fillers.
The Food and Drug Administration’s Manufacturer and User Facility Device Experience (MAUDE) database suggests that adverse events of any substantial severity from filler injections, not just those involving vascular compromise, occur at a rate of 1 per 3,600 cases. However, MAUDE is a passive surveillance system dependent on reports provided by clinicians and others, so this event rate might be an underrepresentation.
In the MAUDE database, complication rates are listed for each of the available filler products and show a variation in rates not just overall but also for each of the major types of complications, which include skin-specific complications such as nodules, discoloration, and inflammation, as well as neurologic adverse events, infection, and vascular compromise, Dr. Galadari reported.
Filler products are not interchangeable
Again, because of passive data collection, it is not clear whether the differences between products is a true representation of relative risk. Nevertheless, Dr. Galadari cautioned that these products are not necessarily interchangeable, advising clinicians to avoid products without an established safety track record.
There are a wide variety of fillers, including biostimulatory products, such as poly-L-lactic acid and calcium hydroxyapatite, and permanent fillers, such as silicone, in addition to collagen and hyaluronic acid, which function as temporary fillers, according to Dr. Galadari. He emphasized that the specific risks of each filler vary, but clinicians should always respond quickly whenever there is an adverse reaction or evidence of vascular compromise.

In flushing out filler, Cheryl M. Burgess, MD, of the Center for Dermatology and Dermatologic Surgery, Washington, who spoke at the meeting, also emphasized a prompt response. She too employs hyaluronidase injections to break down excess hyaluronic acid in the event of complications related to this filler.
Importantly, Dr. Burgess pointed out that hyaluronic acid can be considered safe for darker skin types, including Fitzpatrick skin types IV, V, and VI, but she added that speed of injection might be a particularly important variable for cosmetic procedures in skin of color.
“There is less postinflammatory hyperpigmentation with slower injection times and more with serial or multiple puncture injection technique,” she cautioned.
She further concurred with the value of cannulas over needles in most instances for facial contouring applications with filler, but she encouraged clinicians not to be overly ambitious and to move gradually toward goals.
“The desired outcome may require multiple sessions with conservative measures,” she said, indicating that conservative measures also represent a strategy to avoid adverse events.
if not promptly addressed, according to an expert explaining the signs of an impending disaster at the Skin of Color Update 2021.
The most serious of the adverse events stem from vascular compromise, which is often signaled immediately by sharp pain and blanching of the skin, according to Hassan Galadari, MD, assistant professor of dermatology at the United Arab Emirates University, Dubai.
“Swift and aggressive treatment is required to avoid irreversible changes,” said Dr. Galadari, warning that blindness and vision impairment can be permanent, and that other events associated with vascular compromise include stroke and other types of embolism, as well as tissue necrosis.
To be swift, Dr. Galadari advised an immediate halt of injections and then a series of steps to abort the vascular insult. The goal is to encourage blood flow to prevent clotting and dissipate the filler.
“Massage the area like crazy. Keep on massaging. The more you massage the better. You are recruiting blood into that area so it remains viable,” Dr. Galadari said.
Hyaluronidase injections helpful
Warm compresses should also be applied for periods ranging from 5 minutes up to an hour, he added. In patients treated with hyaluronic acid, he also commonly introduces hyaluronidase injections of 200-500 IU diluted in lidocaine or saline. The injections are placed 2-3 cm apart and repeated every hour until signs and symptoms improve.
“Flush all of the filler out,” he said, emphasizing the urgency for reversing risk of vascular adverse events.
To sustain blood flow and avoid clots, he also recommends initiating aspirin with maintenance doses sustained over several days. Sildenafil to further improve conditions of blood perfusion can be “considered.”
The risks of vascular compromise, like other complications from filler injections, are low, but they are not zero, and the opportunity to prevent irreversible changes depends on acting quickly, according to Dr. Galadari.
“To prevent embolism, recognize the danger zones,” he advised, identifying the glabella region as the site of highest risk. The risk of vascular compromise from injections into the nasal region is lower but higher than injections of the nasolabial fold and forehead, which are associated with a relatively low risk.
Slow injections reduce risks
Some basic strategies he recommended for preventing vascular compromise included slow injections while keeping pressure low and using small volumes of filler per shot. Fractionated treatment and microdroplet techniques can be appropriate depending on the site of injection.
“Delivery of the filler by cannulas rather than by needles is preferable,” according to Dr. Galadari, who noted that a task force from the American Society for Dermatologic Surgery recently endorsed this approach as part of other recommendations to avoid complications of injectable fillers.
The Food and Drug Administration’s Manufacturer and User Facility Device Experience (MAUDE) database suggests that adverse events of any substantial severity from filler injections, not just those involving vascular compromise, occur at a rate of 1 per 3,600 cases. However, MAUDE is a passive surveillance system dependent on reports provided by clinicians and others, so this event rate might be an underrepresentation.
In the MAUDE database, complication rates are listed for each of the available filler products and show a variation in rates not just overall but also for each of the major types of complications, which include skin-specific complications such as nodules, discoloration, and inflammation, as well as neurologic adverse events, infection, and vascular compromise, Dr. Galadari reported.
Filler products are not interchangeable
Again, because of passive data collection, it is not clear whether the differences between products is a true representation of relative risk. Nevertheless, Dr. Galadari cautioned that these products are not necessarily interchangeable, advising clinicians to avoid products without an established safety track record.
There are a wide variety of fillers, including biostimulatory products, such as poly-L-lactic acid and calcium hydroxyapatite, and permanent fillers, such as silicone, in addition to collagen and hyaluronic acid, which function as temporary fillers, according to Dr. Galadari. He emphasized that the specific risks of each filler vary, but clinicians should always respond quickly whenever there is an adverse reaction or evidence of vascular compromise.
In flushing out filler, Cheryl M. Burgess, MD, of the Center for Dermatology and Dermatologic Surgery, Washington, who spoke at the meeting, also emphasized a prompt response. She too employs hyaluronidase injections to break down excess hyaluronic acid in the event of complications related to this filler.
Importantly, Dr. Burgess pointed out that hyaluronic acid can be considered safe for darker skin types, including Fitzpatrick skin types IV, V, and VI, but she added that speed of injection might be a particularly important variable for cosmetic procedures in skin of color.
“There is less postinflammatory hyperpigmentation with slower injection times and more with serial or multiple puncture injection technique,” she cautioned.
She further concurred with the value of cannulas over needles in most instances for facial contouring applications with filler, but she encouraged clinicians not to be overly ambitious and to move gradually toward goals.
“The desired outcome may require multiple sessions with conservative measures,” she said, indicating that conservative measures also represent a strategy to avoid adverse events.
if not promptly addressed, according to an expert explaining the signs of an impending disaster at the Skin of Color Update 2021.
The most serious of the adverse events stem from vascular compromise, which is often signaled immediately by sharp pain and blanching of the skin, according to Hassan Galadari, MD, assistant professor of dermatology at the United Arab Emirates University, Dubai.
“Swift and aggressive treatment is required to avoid irreversible changes,” said Dr. Galadari, warning that blindness and vision impairment can be permanent, and that other events associated with vascular compromise include stroke and other types of embolism, as well as tissue necrosis.
To be swift, Dr. Galadari advised an immediate halt of injections and then a series of steps to abort the vascular insult. The goal is to encourage blood flow to prevent clotting and dissipate the filler.
“Massage the area like crazy. Keep on massaging. The more you massage the better. You are recruiting blood into that area so it remains viable,” Dr. Galadari said.
Hyaluronidase injections helpful
Warm compresses should also be applied for periods ranging from 5 minutes up to an hour, he added. In patients treated with hyaluronic acid, he also commonly introduces hyaluronidase injections of 200-500 IU diluted in lidocaine or saline. The injections are placed 2-3 cm apart and repeated every hour until signs and symptoms improve.
“Flush all of the filler out,” he said, emphasizing the urgency for reversing risk of vascular adverse events.
To sustain blood flow and avoid clots, he also recommends initiating aspirin with maintenance doses sustained over several days. Sildenafil to further improve conditions of blood perfusion can be “considered.”
The risks of vascular compromise, like other complications from filler injections, are low, but they are not zero, and the opportunity to prevent irreversible changes depends on acting quickly, according to Dr. Galadari.
“To prevent embolism, recognize the danger zones,” he advised, identifying the glabella region as the site of highest risk. The risk of vascular compromise from injections into the nasal region is lower but higher than injections of the nasolabial fold and forehead, which are associated with a relatively low risk.
Slow injections reduce risks
Some basic strategies he recommended for preventing vascular compromise included slow injections while keeping pressure low and using small volumes of filler per shot. Fractionated treatment and microdroplet techniques can be appropriate depending on the site of injection.
“Delivery of the filler by cannulas rather than by needles is preferable,” according to Dr. Galadari, who noted that a task force from the American Society for Dermatologic Surgery recently endorsed this approach as part of other recommendations to avoid complications of injectable fillers.
The Food and Drug Administration’s Manufacturer and User Facility Device Experience (MAUDE) database suggests that adverse events of any substantial severity from filler injections, not just those involving vascular compromise, occur at a rate of 1 per 3,600 cases. However, MAUDE is a passive surveillance system dependent on reports provided by clinicians and others, so this event rate might be an underrepresentation.
In the MAUDE database, complication rates are listed for each of the available filler products and show a variation in rates not just overall but also for each of the major types of complications, which include skin-specific complications such as nodules, discoloration, and inflammation, as well as neurologic adverse events, infection, and vascular compromise, Dr. Galadari reported.
Filler products are not interchangeable
Again, because of passive data collection, it is not clear whether the differences between products is a true representation of relative risk. Nevertheless, Dr. Galadari cautioned that these products are not necessarily interchangeable, advising clinicians to avoid products without an established safety track record.
There are a wide variety of fillers, including biostimulatory products, such as poly-L-lactic acid and calcium hydroxyapatite, and permanent fillers, such as silicone, in addition to collagen and hyaluronic acid, which function as temporary fillers, according to Dr. Galadari. He emphasized that the specific risks of each filler vary, but clinicians should always respond quickly whenever there is an adverse reaction or evidence of vascular compromise.
In flushing out filler, Cheryl M. Burgess, MD, of the Center for Dermatology and Dermatologic Surgery, Washington, who spoke at the meeting, also emphasized a prompt response. She too employs hyaluronidase injections to break down excess hyaluronic acid in the event of complications related to this filler.
Importantly, Dr. Burgess pointed out that hyaluronic acid can be considered safe for darker skin types, including Fitzpatrick skin types IV, V, and VI, but she added that speed of injection might be a particularly important variable for cosmetic procedures in skin of color.
“There is less postinflammatory hyperpigmentation with slower injection times and more with serial or multiple puncture injection technique,” she cautioned.
She further concurred with the value of cannulas over needles in most instances for facial contouring applications with filler, but she encouraged clinicians not to be overly ambitious and to move gradually toward goals.
“The desired outcome may require multiple sessions with conservative measures,” she said, indicating that conservative measures also represent a strategy to avoid adverse events.
FROM SOC 2021
Nonsteroidal topical found effective for psoriasis in 52-week study
Treatment with presented as a late-breaker at the virtual annual congress of the European Academy of Dermatology and Venereology.
The drug has several unique features with meaningful clinical differences from other topical psoriasis therapies, according to Linda Stein Gold, MD, director of dermatology clinical research, Henry Ford Health System, Detroit.
“The currently available nonsteroidal topical therapies are typically associated with significant irritation. We did not see that with tapinarof,” said Dr. Stein Gold. This is one of several reasons she believes this drug will be a valuable addition if it receives regulatory approval.
Tapinarof is a small-molecule aryl hydrocarbon receptor (AhR) modulating agent. AhR is widely expressed in immune cells, including macrophages, mast cells, and antigen-presenting cells. It is believed that modulation of AhR signaling by tapinarof reverses immune dysregulation that is involved in the formation of psoriatic lesions.
The newly presented PSOARING 3 data with tapinarof 1% build on the data from the 12-week PSOARING 1 and PSOARING 2 trials, which were released in August 2020 but have yet to be published.
The primary endpoint in both of the 12-week trials, each of which enrolled about 500 patients with plaque psoriasis, was a Physician Global Assessment (PGA) score of 0 (clear) or 1 (almost clear). Relative to a placebo response rate of about 6% in both trials, the proportion of patients who achieved scores of 0/1 with tapinarof 1% was 35.4% and 40.2% in the PSOARING 1 and PSOARING 2 trials, respectively (P < .0001 vs. placebo in both studies).
For the key secondary endpoint of at least 75% improvement in the Psoriasis Area and Severity Index (PASI 75), the relative advantage for tapinarof over placebo was similar. The results were highly statistically significant (P < .0001) in both of the 12-week trials.
More than 90% of the patients who participated in PSOARING 1 and PSOARING 2 and were eligible for the open-label PSOARING 3 extension trial, according to Dr. Stein Gold.
For the 79 patients with a score of 0 at the time of enrollment, tapinarof 1% was reapplied only if the PGA score reached at least 2 during the course of the study. For the 680 patients who entered with a PGA score of at least 1, once-daily applications of tapinarof 1% cream were maintained until a PGA score of 0 was achieved.
In the outcome analysis, response was defined as the proportion of patients with an initial PGA score of at least2 who achieved PGA 0. A remittive effect was defined as duration of a PGA score of 0 or 1 while off therapy after achieving a PGA score of 0. Durability of response was defined as the proportion of patients who achieved a PGA sore of 0 or 1 at least once during the study while on therapy. This last outcome provided a test of tachyphylaxis.
“Overall, 40.9% of patients achieved complete disease clearance at least once during the trial, and 58.2% who entered the study with a PGA score of 2 or higher achieved a PGA score of 0 or 1,” Dr. Stein Gold reported.
For the 79 patients who entered PSOARING 3 with a PGA score of 0 and were off treatment, the median duration of a remittive effect was 115 days. For the patients who entered the trial with a higher PGA score but who achieved a score of 0 during the study (312 patients), the mean remittive effect after discontinuing therapy was 130 days.
There was no evidence of tachyphylaxis. Rather, “there was no loss of effect despite intermittent therapy observed over the course of the trial,” Dr. Stein Gold reported.
The most common treatment-emergent adverse events in PSOARING 3, as in the previous PSOARING studies, were folliculitis, which was observed in 24.0% of patients; contact dermatitis, which occurred in 5.9% of patients; and headache, which was reported in 2%. Rates of study drug discontinuations for folliculitis and contact dermatitis were 1.2% and 1.4%, respectively. Headache did not lead to any study discontinuations.
Calling tapinarof a “first-in-class nonsteroidal,” Dr. Stein Gold suggested that this is likely to be a useful adjunctive therapy for psoriasis control. It avoids the adverse events associated with long-term topical steroid use, and its tolerability might be particularly attractive for use in sensitive areas.
“This is likely to be very useful in patients who are looking for a topical therapy for skin folds or the face, where there is a need for well-tolerated topical treatments,” Dr. Stein Gold said.
There are a lot of reasons to be positive about a new, well-tolerated topical agent for psoriasis, particularly as an alternative to topical steroids, agreed Adam Friedman, MD, director of translational research and professor and chair of the department of dermatology at George Washington University, Washington. He considers the data with tapinarof promising in general, but he also likes any new, effective topical psoriasis therapy.
“Patients and physicians are always hungry for new options, especially psoriasis patients, given many have ‘been there and done that’ with topical steroids,” Dr. Friedman said.
“Topical steroids are not irritating, but long-term use beyond recommended dosing can lead to skin thinning, lightening, tachyphylaxis, and, if really abused, HPA [hypothalamic-pituitary-adrenal] axis suppression and adrenal insufficiency,” he observed.
A topical therapy with a durable effect is particularly intriguing.
“The other issue with topical steroids is that psoriatic plaques return rather easily after stopping. The data I have seen with tapinarof show more sustainability after cessation, owing to its mechanism of action,” Dr. Friedman said. Rather than its potential for application to sensitive areas, such as the face, the durability “to me is more interesting.”
He suspects that, owing to “the incurable steroid phobia that haunts many of our patients,” an effective nonsteroidal topical option is also likely to lead to better compliance with topical treatment over time.
“A well-tolerated nonsteroidal topical drug will probably find an important place in the future management of chronic inflammatory diseases,” Marius-Anton Ionescu, MD, PhD, a dermatologist at the Hôpital Saint Louis, Paris, said in an interview. He referred to the positive effects of treatment with tapinarof in clinical trials in adults with atopic dermatitis, in addition to psoriasis.
Tapinarof 1% is also being investigated in a phase 3 study involving patients with moderate to severe atopic dermatitis. In that study, patients are as young as age 2 years. The drug is under review at the Food and Drug Administration for the plaque psoriasis indication in adults.
Dr. Stein Gold has financial relationships with Arcutis, Amgen, Bristol-Myers Squibb, Eli Lilly, Leo Pharma Ortho Dermatologic, UCB, and Dermavant Sciences, which is developing tapinarof and is provided funding for the PSOARING 3 trial. Dr. Friedman reported financial relationships with Amgen, Biogen, Encore, Galderma, GlaxoSmithKline, IntraDerm, Johnson & Johnson, Nerium, Novartis, Oculus, Onset, Pfizer, Sanova, and Valeant Pharmaceuticals. Dr. Ionescu has been a speaker or investigator (honoraria) for Celgene, Novartis, Lilly, and Uriage Cosmetics.
A version of this article first appeared on Medscape.com.
Treatment with presented as a late-breaker at the virtual annual congress of the European Academy of Dermatology and Venereology.
The drug has several unique features with meaningful clinical differences from other topical psoriasis therapies, according to Linda Stein Gold, MD, director of dermatology clinical research, Henry Ford Health System, Detroit.
“The currently available nonsteroidal topical therapies are typically associated with significant irritation. We did not see that with tapinarof,” said Dr. Stein Gold. This is one of several reasons she believes this drug will be a valuable addition if it receives regulatory approval.
Tapinarof is a small-molecule aryl hydrocarbon receptor (AhR) modulating agent. AhR is widely expressed in immune cells, including macrophages, mast cells, and antigen-presenting cells. It is believed that modulation of AhR signaling by tapinarof reverses immune dysregulation that is involved in the formation of psoriatic lesions.
The newly presented PSOARING 3 data with tapinarof 1% build on the data from the 12-week PSOARING 1 and PSOARING 2 trials, which were released in August 2020 but have yet to be published.
The primary endpoint in both of the 12-week trials, each of which enrolled about 500 patients with plaque psoriasis, was a Physician Global Assessment (PGA) score of 0 (clear) or 1 (almost clear). Relative to a placebo response rate of about 6% in both trials, the proportion of patients who achieved scores of 0/1 with tapinarof 1% was 35.4% and 40.2% in the PSOARING 1 and PSOARING 2 trials, respectively (P < .0001 vs. placebo in both studies).
For the key secondary endpoint of at least 75% improvement in the Psoriasis Area and Severity Index (PASI 75), the relative advantage for tapinarof over placebo was similar. The results were highly statistically significant (P < .0001) in both of the 12-week trials.
More than 90% of the patients who participated in PSOARING 1 and PSOARING 2 and were eligible for the open-label PSOARING 3 extension trial, according to Dr. Stein Gold.
For the 79 patients with a score of 0 at the time of enrollment, tapinarof 1% was reapplied only if the PGA score reached at least 2 during the course of the study. For the 680 patients who entered with a PGA score of at least 1, once-daily applications of tapinarof 1% cream were maintained until a PGA score of 0 was achieved.
In the outcome analysis, response was defined as the proportion of patients with an initial PGA score of at least2 who achieved PGA 0. A remittive effect was defined as duration of a PGA score of 0 or 1 while off therapy after achieving a PGA score of 0. Durability of response was defined as the proportion of patients who achieved a PGA sore of 0 or 1 at least once during the study while on therapy. This last outcome provided a test of tachyphylaxis.
“Overall, 40.9% of patients achieved complete disease clearance at least once during the trial, and 58.2% who entered the study with a PGA score of 2 or higher achieved a PGA score of 0 or 1,” Dr. Stein Gold reported.
For the 79 patients who entered PSOARING 3 with a PGA score of 0 and were off treatment, the median duration of a remittive effect was 115 days. For the patients who entered the trial with a higher PGA score but who achieved a score of 0 during the study (312 patients), the mean remittive effect after discontinuing therapy was 130 days.
There was no evidence of tachyphylaxis. Rather, “there was no loss of effect despite intermittent therapy observed over the course of the trial,” Dr. Stein Gold reported.
The most common treatment-emergent adverse events in PSOARING 3, as in the previous PSOARING studies, were folliculitis, which was observed in 24.0% of patients; contact dermatitis, which occurred in 5.9% of patients; and headache, which was reported in 2%. Rates of study drug discontinuations for folliculitis and contact dermatitis were 1.2% and 1.4%, respectively. Headache did not lead to any study discontinuations.
Calling tapinarof a “first-in-class nonsteroidal,” Dr. Stein Gold suggested that this is likely to be a useful adjunctive therapy for psoriasis control. It avoids the adverse events associated with long-term topical steroid use, and its tolerability might be particularly attractive for use in sensitive areas.
“This is likely to be very useful in patients who are looking for a topical therapy for skin folds or the face, where there is a need for well-tolerated topical treatments,” Dr. Stein Gold said.
There are a lot of reasons to be positive about a new, well-tolerated topical agent for psoriasis, particularly as an alternative to topical steroids, agreed Adam Friedman, MD, director of translational research and professor and chair of the department of dermatology at George Washington University, Washington. He considers the data with tapinarof promising in general, but he also likes any new, effective topical psoriasis therapy.
“Patients and physicians are always hungry for new options, especially psoriasis patients, given many have ‘been there and done that’ with topical steroids,” Dr. Friedman said.
“Topical steroids are not irritating, but long-term use beyond recommended dosing can lead to skin thinning, lightening, tachyphylaxis, and, if really abused, HPA [hypothalamic-pituitary-adrenal] axis suppression and adrenal insufficiency,” he observed.
A topical therapy with a durable effect is particularly intriguing.
“The other issue with topical steroids is that psoriatic plaques return rather easily after stopping. The data I have seen with tapinarof show more sustainability after cessation, owing to its mechanism of action,” Dr. Friedman said. Rather than its potential for application to sensitive areas, such as the face, the durability “to me is more interesting.”
He suspects that, owing to “the incurable steroid phobia that haunts many of our patients,” an effective nonsteroidal topical option is also likely to lead to better compliance with topical treatment over time.
“A well-tolerated nonsteroidal topical drug will probably find an important place in the future management of chronic inflammatory diseases,” Marius-Anton Ionescu, MD, PhD, a dermatologist at the Hôpital Saint Louis, Paris, said in an interview. He referred to the positive effects of treatment with tapinarof in clinical trials in adults with atopic dermatitis, in addition to psoriasis.
Tapinarof 1% is also being investigated in a phase 3 study involving patients with moderate to severe atopic dermatitis. In that study, patients are as young as age 2 years. The drug is under review at the Food and Drug Administration for the plaque psoriasis indication in adults.
Dr. Stein Gold has financial relationships with Arcutis, Amgen, Bristol-Myers Squibb, Eli Lilly, Leo Pharma Ortho Dermatologic, UCB, and Dermavant Sciences, which is developing tapinarof and is provided funding for the PSOARING 3 trial. Dr. Friedman reported financial relationships with Amgen, Biogen, Encore, Galderma, GlaxoSmithKline, IntraDerm, Johnson & Johnson, Nerium, Novartis, Oculus, Onset, Pfizer, Sanova, and Valeant Pharmaceuticals. Dr. Ionescu has been a speaker or investigator (honoraria) for Celgene, Novartis, Lilly, and Uriage Cosmetics.
A version of this article first appeared on Medscape.com.
Treatment with presented as a late-breaker at the virtual annual congress of the European Academy of Dermatology and Venereology.
The drug has several unique features with meaningful clinical differences from other topical psoriasis therapies, according to Linda Stein Gold, MD, director of dermatology clinical research, Henry Ford Health System, Detroit.
“The currently available nonsteroidal topical therapies are typically associated with significant irritation. We did not see that with tapinarof,” said Dr. Stein Gold. This is one of several reasons she believes this drug will be a valuable addition if it receives regulatory approval.
Tapinarof is a small-molecule aryl hydrocarbon receptor (AhR) modulating agent. AhR is widely expressed in immune cells, including macrophages, mast cells, and antigen-presenting cells. It is believed that modulation of AhR signaling by tapinarof reverses immune dysregulation that is involved in the formation of psoriatic lesions.
The newly presented PSOARING 3 data with tapinarof 1% build on the data from the 12-week PSOARING 1 and PSOARING 2 trials, which were released in August 2020 but have yet to be published.
The primary endpoint in both of the 12-week trials, each of which enrolled about 500 patients with plaque psoriasis, was a Physician Global Assessment (PGA) score of 0 (clear) or 1 (almost clear). Relative to a placebo response rate of about 6% in both trials, the proportion of patients who achieved scores of 0/1 with tapinarof 1% was 35.4% and 40.2% in the PSOARING 1 and PSOARING 2 trials, respectively (P < .0001 vs. placebo in both studies).
For the key secondary endpoint of at least 75% improvement in the Psoriasis Area and Severity Index (PASI 75), the relative advantage for tapinarof over placebo was similar. The results were highly statistically significant (P < .0001) in both of the 12-week trials.
More than 90% of the patients who participated in PSOARING 1 and PSOARING 2 and were eligible for the open-label PSOARING 3 extension trial, according to Dr. Stein Gold.
For the 79 patients with a score of 0 at the time of enrollment, tapinarof 1% was reapplied only if the PGA score reached at least 2 during the course of the study. For the 680 patients who entered with a PGA score of at least 1, once-daily applications of tapinarof 1% cream were maintained until a PGA score of 0 was achieved.
In the outcome analysis, response was defined as the proportion of patients with an initial PGA score of at least2 who achieved PGA 0. A remittive effect was defined as duration of a PGA score of 0 or 1 while off therapy after achieving a PGA score of 0. Durability of response was defined as the proportion of patients who achieved a PGA sore of 0 or 1 at least once during the study while on therapy. This last outcome provided a test of tachyphylaxis.
“Overall, 40.9% of patients achieved complete disease clearance at least once during the trial, and 58.2% who entered the study with a PGA score of 2 or higher achieved a PGA score of 0 or 1,” Dr. Stein Gold reported.
For the 79 patients who entered PSOARING 3 with a PGA score of 0 and were off treatment, the median duration of a remittive effect was 115 days. For the patients who entered the trial with a higher PGA score but who achieved a score of 0 during the study (312 patients), the mean remittive effect after discontinuing therapy was 130 days.
There was no evidence of tachyphylaxis. Rather, “there was no loss of effect despite intermittent therapy observed over the course of the trial,” Dr. Stein Gold reported.
The most common treatment-emergent adverse events in PSOARING 3, as in the previous PSOARING studies, were folliculitis, which was observed in 24.0% of patients; contact dermatitis, which occurred in 5.9% of patients; and headache, which was reported in 2%. Rates of study drug discontinuations for folliculitis and contact dermatitis were 1.2% and 1.4%, respectively. Headache did not lead to any study discontinuations.
Calling tapinarof a “first-in-class nonsteroidal,” Dr. Stein Gold suggested that this is likely to be a useful adjunctive therapy for psoriasis control. It avoids the adverse events associated with long-term topical steroid use, and its tolerability might be particularly attractive for use in sensitive areas.
“This is likely to be very useful in patients who are looking for a topical therapy for skin folds or the face, where there is a need for well-tolerated topical treatments,” Dr. Stein Gold said.
There are a lot of reasons to be positive about a new, well-tolerated topical agent for psoriasis, particularly as an alternative to topical steroids, agreed Adam Friedman, MD, director of translational research and professor and chair of the department of dermatology at George Washington University, Washington. He considers the data with tapinarof promising in general, but he also likes any new, effective topical psoriasis therapy.
“Patients and physicians are always hungry for new options, especially psoriasis patients, given many have ‘been there and done that’ with topical steroids,” Dr. Friedman said.
“Topical steroids are not irritating, but long-term use beyond recommended dosing can lead to skin thinning, lightening, tachyphylaxis, and, if really abused, HPA [hypothalamic-pituitary-adrenal] axis suppression and adrenal insufficiency,” he observed.
A topical therapy with a durable effect is particularly intriguing.
“The other issue with topical steroids is that psoriatic plaques return rather easily after stopping. The data I have seen with tapinarof show more sustainability after cessation, owing to its mechanism of action,” Dr. Friedman said. Rather than its potential for application to sensitive areas, such as the face, the durability “to me is more interesting.”
He suspects that, owing to “the incurable steroid phobia that haunts many of our patients,” an effective nonsteroidal topical option is also likely to lead to better compliance with topical treatment over time.
“A well-tolerated nonsteroidal topical drug will probably find an important place in the future management of chronic inflammatory diseases,” Marius-Anton Ionescu, MD, PhD, a dermatologist at the Hôpital Saint Louis, Paris, said in an interview. He referred to the positive effects of treatment with tapinarof in clinical trials in adults with atopic dermatitis, in addition to psoriasis.
Tapinarof 1% is also being investigated in a phase 3 study involving patients with moderate to severe atopic dermatitis. In that study, patients are as young as age 2 years. The drug is under review at the Food and Drug Administration for the plaque psoriasis indication in adults.
Dr. Stein Gold has financial relationships with Arcutis, Amgen, Bristol-Myers Squibb, Eli Lilly, Leo Pharma Ortho Dermatologic, UCB, and Dermavant Sciences, which is developing tapinarof and is provided funding for the PSOARING 3 trial. Dr. Friedman reported financial relationships with Amgen, Biogen, Encore, Galderma, GlaxoSmithKline, IntraDerm, Johnson & Johnson, Nerium, Novartis, Oculus, Onset, Pfizer, Sanova, and Valeant Pharmaceuticals. Dr. Ionescu has been a speaker or investigator (honoraria) for Celgene, Novartis, Lilly, and Uriage Cosmetics.
A version of this article first appeared on Medscape.com.