User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Florida-based doctor arrested in Haiti president’s assassination
About two dozen people have been arrested as suspects, the newspaper reported, though police believe Christian Emmanuel Sanon, 63, was plotting to become president.
“He arrived by private plane in June with political objectives and contacted a private security firm to recruit the people who committed this act,” Léon Charles, Haiti’s national police chief, said during a news conference on July 11.
The firm, called CTU Security, is a Venezuelan company based in Miami, Mr. Charles said. During a raid at Mr. Sanon’s home in Port-au-Prince, police found six rifles, 20 boxes of bullets, 24 unused shooting targets, pistol holsters, and a hat with a U.S. Drug Enforcement Agency logo.
“This initial mission that was given to these assailants was to protect the individual named Emmanuel Sanon, but afterwards, the mission changed,” Mr. Charles said.
The new “mission” was to arrest President Moïse and install Mr. Sanon as president, The New York Times reported, though Mr. Charles didn’t explain when the mission changed to assassination or how Mr. Sanon could have taken control of the government.
President Moïse was shot to death on July 7 at his home in Port-au-Prince by a “team of commandos,” according to The Washington Post. On July 9, Haiti asked the U.S. to send troops to the country to protect its airport and key infrastructure.
The announcement of Mr. Sanon’s arrest came hours after FBI and Department of Homeland Security officials arrived in Haiti on July 11 to discuss how the U.S. can offer assistance, the newspaper reported.
Mr. Sanon has a YouTube channel with three political campaign videos from 2011, which include discussions about Haitian politics, according to Forbes. In one of the videos, titled “Dr. Christian Sanon – Leadership for Haiti,” Mr. Sanon talks about corruption in the country and presents himself as a potential leader.
Mr. Sanon lived in Florida for more than 20 years, ranging from the Tampa Bay area to South Florida, according to the Miami Herald. Public records show that he had more than a dozen businesses registered in the state, including medical services and real estate, though most are inactive.
Mr. Sanon is the third person with links to the U.S. who has been arrested in connection with the assassination, the Miami Herald reported. Two Haitian-Americans from southern Florida – James Solages, 35, and Joseph G. Vincent, 55 – were arrested by local police. They claimed they were working as translators for the assassins.
The first lady, Martine Moïse, was wounded in the attack and is now receiving treatment at a hospital in Miami, the newspaper reported.
A version of this article first appeared on WebMD.com.
About two dozen people have been arrested as suspects, the newspaper reported, though police believe Christian Emmanuel Sanon, 63, was plotting to become president.
“He arrived by private plane in June with political objectives and contacted a private security firm to recruit the people who committed this act,” Léon Charles, Haiti’s national police chief, said during a news conference on July 11.
The firm, called CTU Security, is a Venezuelan company based in Miami, Mr. Charles said. During a raid at Mr. Sanon’s home in Port-au-Prince, police found six rifles, 20 boxes of bullets, 24 unused shooting targets, pistol holsters, and a hat with a U.S. Drug Enforcement Agency logo.
“This initial mission that was given to these assailants was to protect the individual named Emmanuel Sanon, but afterwards, the mission changed,” Mr. Charles said.
The new “mission” was to arrest President Moïse and install Mr. Sanon as president, The New York Times reported, though Mr. Charles didn’t explain when the mission changed to assassination or how Mr. Sanon could have taken control of the government.
President Moïse was shot to death on July 7 at his home in Port-au-Prince by a “team of commandos,” according to The Washington Post. On July 9, Haiti asked the U.S. to send troops to the country to protect its airport and key infrastructure.
The announcement of Mr. Sanon’s arrest came hours after FBI and Department of Homeland Security officials arrived in Haiti on July 11 to discuss how the U.S. can offer assistance, the newspaper reported.
Mr. Sanon has a YouTube channel with three political campaign videos from 2011, which include discussions about Haitian politics, according to Forbes. In one of the videos, titled “Dr. Christian Sanon – Leadership for Haiti,” Mr. Sanon talks about corruption in the country and presents himself as a potential leader.
Mr. Sanon lived in Florida for more than 20 years, ranging from the Tampa Bay area to South Florida, according to the Miami Herald. Public records show that he had more than a dozen businesses registered in the state, including medical services and real estate, though most are inactive.
Mr. Sanon is the third person with links to the U.S. who has been arrested in connection with the assassination, the Miami Herald reported. Two Haitian-Americans from southern Florida – James Solages, 35, and Joseph G. Vincent, 55 – were arrested by local police. They claimed they were working as translators for the assassins.
The first lady, Martine Moïse, was wounded in the attack and is now receiving treatment at a hospital in Miami, the newspaper reported.
A version of this article first appeared on WebMD.com.
About two dozen people have been arrested as suspects, the newspaper reported, though police believe Christian Emmanuel Sanon, 63, was plotting to become president.
“He arrived by private plane in June with political objectives and contacted a private security firm to recruit the people who committed this act,” Léon Charles, Haiti’s national police chief, said during a news conference on July 11.
The firm, called CTU Security, is a Venezuelan company based in Miami, Mr. Charles said. During a raid at Mr. Sanon’s home in Port-au-Prince, police found six rifles, 20 boxes of bullets, 24 unused shooting targets, pistol holsters, and a hat with a U.S. Drug Enforcement Agency logo.
“This initial mission that was given to these assailants was to protect the individual named Emmanuel Sanon, but afterwards, the mission changed,” Mr. Charles said.
The new “mission” was to arrest President Moïse and install Mr. Sanon as president, The New York Times reported, though Mr. Charles didn’t explain when the mission changed to assassination or how Mr. Sanon could have taken control of the government.
President Moïse was shot to death on July 7 at his home in Port-au-Prince by a “team of commandos,” according to The Washington Post. On July 9, Haiti asked the U.S. to send troops to the country to protect its airport and key infrastructure.
The announcement of Mr. Sanon’s arrest came hours after FBI and Department of Homeland Security officials arrived in Haiti on July 11 to discuss how the U.S. can offer assistance, the newspaper reported.
Mr. Sanon has a YouTube channel with three political campaign videos from 2011, which include discussions about Haitian politics, according to Forbes. In one of the videos, titled “Dr. Christian Sanon – Leadership for Haiti,” Mr. Sanon talks about corruption in the country and presents himself as a potential leader.
Mr. Sanon lived in Florida for more than 20 years, ranging from the Tampa Bay area to South Florida, according to the Miami Herald. Public records show that he had more than a dozen businesses registered in the state, including medical services and real estate, though most are inactive.
Mr. Sanon is the third person with links to the U.S. who has been arrested in connection with the assassination, the Miami Herald reported. Two Haitian-Americans from southern Florida – James Solages, 35, and Joseph G. Vincent, 55 – were arrested by local police. They claimed they were working as translators for the assassins.
The first lady, Martine Moïse, was wounded in the attack and is now receiving treatment at a hospital in Miami, the newspaper reported.
A version of this article first appeared on WebMD.com.
UV light linked to prevention of allergic disease in infants
Higher direct ultraviolet light exposure in the first 3 months of life was linked to lower incidence of proinflammatory immune markers and lower incidence of eczema in an early-stage double-blind, randomized controlled trial.
Kristina Rueter, MD, with the University of Western Australia, Perth, who presented her team’s findings on Sunday at the European Academy of Allergy and Clinical Immunology (EAACI) Hybrid Congress 2021, said their study is the first to demonstrate the association.
“There has been a significant rise in allergic diseases, particularly within the last 20-30 years,” Dr. Rueter noted.
“Changes to the genetic pool take thousands of years to have an impact,” she said, “so the question is why do we have the significant, very recent rise of allergic diseases?”
Suboptimal vitamin D levels during infancy, lifestyle changes, nutritional changes, and living at higher latitudes have emerged as explanations.
In this study, 195 high-risk newborns were randomized to receive oral vitamin D supplements (400 IU/day) or placebo until 6 months of age.
Researchers found that UV light exposure appears more beneficial than vitamin D supplements as an allergy prevention strategy in the critical early years of immune system development.
The researchers used a novel approach of attaching a personal UV dosimeter to the infants’ clothing to measure direct UV light exposure (290-380 nm). Vitamin D levels were measured at 3, 6, 12, and 30 months of age. Immune function was assessed at 6 months of age, and food allergy, eczema, and wheeze were assessed at 6, 12, and 30 months of age.
At 3 (P < .01) and 6 (P = .02) months of age, vitamin D levels were greater in the children who received vitamin D supplements than those who received placebo, but there was no difference in eczema incidence between groups. The finding matched those of previous studies that compared the supplements with placebo, Dr. Rueter said.
However, infants with eczema were found to have had less UV light exposure compared to those without eczema (median interquartile range [IQR], 555 J/m2 vs. 998 J/m2; P = .023).
“We also found an inverse correlation between total UV light exposure and toll-like receptor cytokine production,” Dr. Rueter said.
“The more direct UV light exposure a child got, the less the chance to develop eczema,” she said.
Researchers then extended their analysis to see whether the effect of direct UV light exposure on reduced eczema would be maintained in the first 2.5 years of life, “and we could see again a significant difference, that the children who received higher UV light exposure had less eczema,” Dr. Rueter said.
Barbara Rogala, MD, PhD, professor at the Medical University of Silesia, Katowice, Poland, told this news organization that, just as in studies on vitamin D in adult populations, there must be a balance in infant studies between potential benefit of a therapeutic strategy of vitamin D and sunlight and risk of side effects. (Dr. Rogala was not involved in Dr. Rueter’s study.)
Although vitamin D supplements are a standard part of infant care, exposure to sunlight can come with cancer risk, she noted.
Dr. Rueter agreed caution is necessary.
“You have to follow the cancer guidelines,” she said. “Sunlight may play a role in causing skin cancer, and lots of research needs to be done to find the right balance between what is a good amount which may influence the immune system in a positive way and what, on the other hand, might be too much.”
As for vitamin D supplements, Dr. Rueter said, toxic levels require “extremely high doses,” so with 400 IU/day used in the study, children are likely not being overtreated by combining sunlight and vitamin D supplements.
The study was supported by grants from Telethon–New Children’s Hospital Research Fund, Australia; Asthma Foundation of Western Australia; and the Princess Margaret Hospital Foundation, Australia. Dr. Rueter and Dr. Rogala have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Higher direct ultraviolet light exposure in the first 3 months of life was linked to lower incidence of proinflammatory immune markers and lower incidence of eczema in an early-stage double-blind, randomized controlled trial.
Kristina Rueter, MD, with the University of Western Australia, Perth, who presented her team’s findings on Sunday at the European Academy of Allergy and Clinical Immunology (EAACI) Hybrid Congress 2021, said their study is the first to demonstrate the association.
“There has been a significant rise in allergic diseases, particularly within the last 20-30 years,” Dr. Rueter noted.
“Changes to the genetic pool take thousands of years to have an impact,” she said, “so the question is why do we have the significant, very recent rise of allergic diseases?”
Suboptimal vitamin D levels during infancy, lifestyle changes, nutritional changes, and living at higher latitudes have emerged as explanations.
In this study, 195 high-risk newborns were randomized to receive oral vitamin D supplements (400 IU/day) or placebo until 6 months of age.
Researchers found that UV light exposure appears more beneficial than vitamin D supplements as an allergy prevention strategy in the critical early years of immune system development.
The researchers used a novel approach of attaching a personal UV dosimeter to the infants’ clothing to measure direct UV light exposure (290-380 nm). Vitamin D levels were measured at 3, 6, 12, and 30 months of age. Immune function was assessed at 6 months of age, and food allergy, eczema, and wheeze were assessed at 6, 12, and 30 months of age.
At 3 (P < .01) and 6 (P = .02) months of age, vitamin D levels were greater in the children who received vitamin D supplements than those who received placebo, but there was no difference in eczema incidence between groups. The finding matched those of previous studies that compared the supplements with placebo, Dr. Rueter said.
However, infants with eczema were found to have had less UV light exposure compared to those without eczema (median interquartile range [IQR], 555 J/m2 vs. 998 J/m2; P = .023).
“We also found an inverse correlation between total UV light exposure and toll-like receptor cytokine production,” Dr. Rueter said.
“The more direct UV light exposure a child got, the less the chance to develop eczema,” she said.
Researchers then extended their analysis to see whether the effect of direct UV light exposure on reduced eczema would be maintained in the first 2.5 years of life, “and we could see again a significant difference, that the children who received higher UV light exposure had less eczema,” Dr. Rueter said.
Barbara Rogala, MD, PhD, professor at the Medical University of Silesia, Katowice, Poland, told this news organization that, just as in studies on vitamin D in adult populations, there must be a balance in infant studies between potential benefit of a therapeutic strategy of vitamin D and sunlight and risk of side effects. (Dr. Rogala was not involved in Dr. Rueter’s study.)
Although vitamin D supplements are a standard part of infant care, exposure to sunlight can come with cancer risk, she noted.
Dr. Rueter agreed caution is necessary.
“You have to follow the cancer guidelines,” she said. “Sunlight may play a role in causing skin cancer, and lots of research needs to be done to find the right balance between what is a good amount which may influence the immune system in a positive way and what, on the other hand, might be too much.”
As for vitamin D supplements, Dr. Rueter said, toxic levels require “extremely high doses,” so with 400 IU/day used in the study, children are likely not being overtreated by combining sunlight and vitamin D supplements.
The study was supported by grants from Telethon–New Children’s Hospital Research Fund, Australia; Asthma Foundation of Western Australia; and the Princess Margaret Hospital Foundation, Australia. Dr. Rueter and Dr. Rogala have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Higher direct ultraviolet light exposure in the first 3 months of life was linked to lower incidence of proinflammatory immune markers and lower incidence of eczema in an early-stage double-blind, randomized controlled trial.
Kristina Rueter, MD, with the University of Western Australia, Perth, who presented her team’s findings on Sunday at the European Academy of Allergy and Clinical Immunology (EAACI) Hybrid Congress 2021, said their study is the first to demonstrate the association.
“There has been a significant rise in allergic diseases, particularly within the last 20-30 years,” Dr. Rueter noted.
“Changes to the genetic pool take thousands of years to have an impact,” she said, “so the question is why do we have the significant, very recent rise of allergic diseases?”
Suboptimal vitamin D levels during infancy, lifestyle changes, nutritional changes, and living at higher latitudes have emerged as explanations.
In this study, 195 high-risk newborns were randomized to receive oral vitamin D supplements (400 IU/day) or placebo until 6 months of age.
Researchers found that UV light exposure appears more beneficial than vitamin D supplements as an allergy prevention strategy in the critical early years of immune system development.
The researchers used a novel approach of attaching a personal UV dosimeter to the infants’ clothing to measure direct UV light exposure (290-380 nm). Vitamin D levels were measured at 3, 6, 12, and 30 months of age. Immune function was assessed at 6 months of age, and food allergy, eczema, and wheeze were assessed at 6, 12, and 30 months of age.
At 3 (P < .01) and 6 (P = .02) months of age, vitamin D levels were greater in the children who received vitamin D supplements than those who received placebo, but there was no difference in eczema incidence between groups. The finding matched those of previous studies that compared the supplements with placebo, Dr. Rueter said.
However, infants with eczema were found to have had less UV light exposure compared to those without eczema (median interquartile range [IQR], 555 J/m2 vs. 998 J/m2; P = .023).
“We also found an inverse correlation between total UV light exposure and toll-like receptor cytokine production,” Dr. Rueter said.
“The more direct UV light exposure a child got, the less the chance to develop eczema,” she said.
Researchers then extended their analysis to see whether the effect of direct UV light exposure on reduced eczema would be maintained in the first 2.5 years of life, “and we could see again a significant difference, that the children who received higher UV light exposure had less eczema,” Dr. Rueter said.
Barbara Rogala, MD, PhD, professor at the Medical University of Silesia, Katowice, Poland, told this news organization that, just as in studies on vitamin D in adult populations, there must be a balance in infant studies between potential benefit of a therapeutic strategy of vitamin D and sunlight and risk of side effects. (Dr. Rogala was not involved in Dr. Rueter’s study.)
Although vitamin D supplements are a standard part of infant care, exposure to sunlight can come with cancer risk, she noted.
Dr. Rueter agreed caution is necessary.
“You have to follow the cancer guidelines,” she said. “Sunlight may play a role in causing skin cancer, and lots of research needs to be done to find the right balance between what is a good amount which may influence the immune system in a positive way and what, on the other hand, might be too much.”
As for vitamin D supplements, Dr. Rueter said, toxic levels require “extremely high doses,” so with 400 IU/day used in the study, children are likely not being overtreated by combining sunlight and vitamin D supplements.
The study was supported by grants from Telethon–New Children’s Hospital Research Fund, Australia; Asthma Foundation of Western Australia; and the Princess Margaret Hospital Foundation, Australia. Dr. Rueter and Dr. Rogala have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Subcutaneous Nodule on the Chest
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.

Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6

Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6

Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12

In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.
Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6
Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6
Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12
In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.
Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6
Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6
Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12
In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.


A healthy 45-year-old man presented to the dermatology clinic with a slow-growing subcutaneous nodule on the left chest that had been present for years.
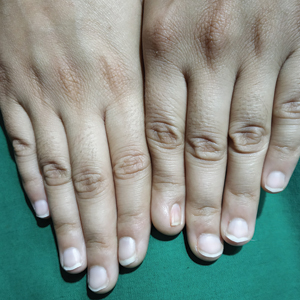
Agminated Nodules on the Scalp
The Diagnosis: Cutaneous Angiosarcoma
Biopsy revealed a cellular neoplasm consisting of atypical polygonal cells with a hobnailed appearance, vasoformative characteristics, and rare extravasated erythrocytes. The tumor had an infiltrative growth pattern as demonstrated by dissecting dermal collagen and a poorly defined border with adjacent normal tissue (Figure 1). Immunohistochemistry revealed that the lesion was positive for CD31 and D2-40 (Figure 2) but negative for cytokeratin, CD10, CD68, human herpesvirus 8, CD34, and Melan A, thus confirming the endothelial origin of the tumor cells and the diagnosis of cutaneous angiosarcoma (CAS). The patient was treated with extended surgical excision and radiation therapy. No recurrence or metastasis was found throughout 2 years of follow-up.
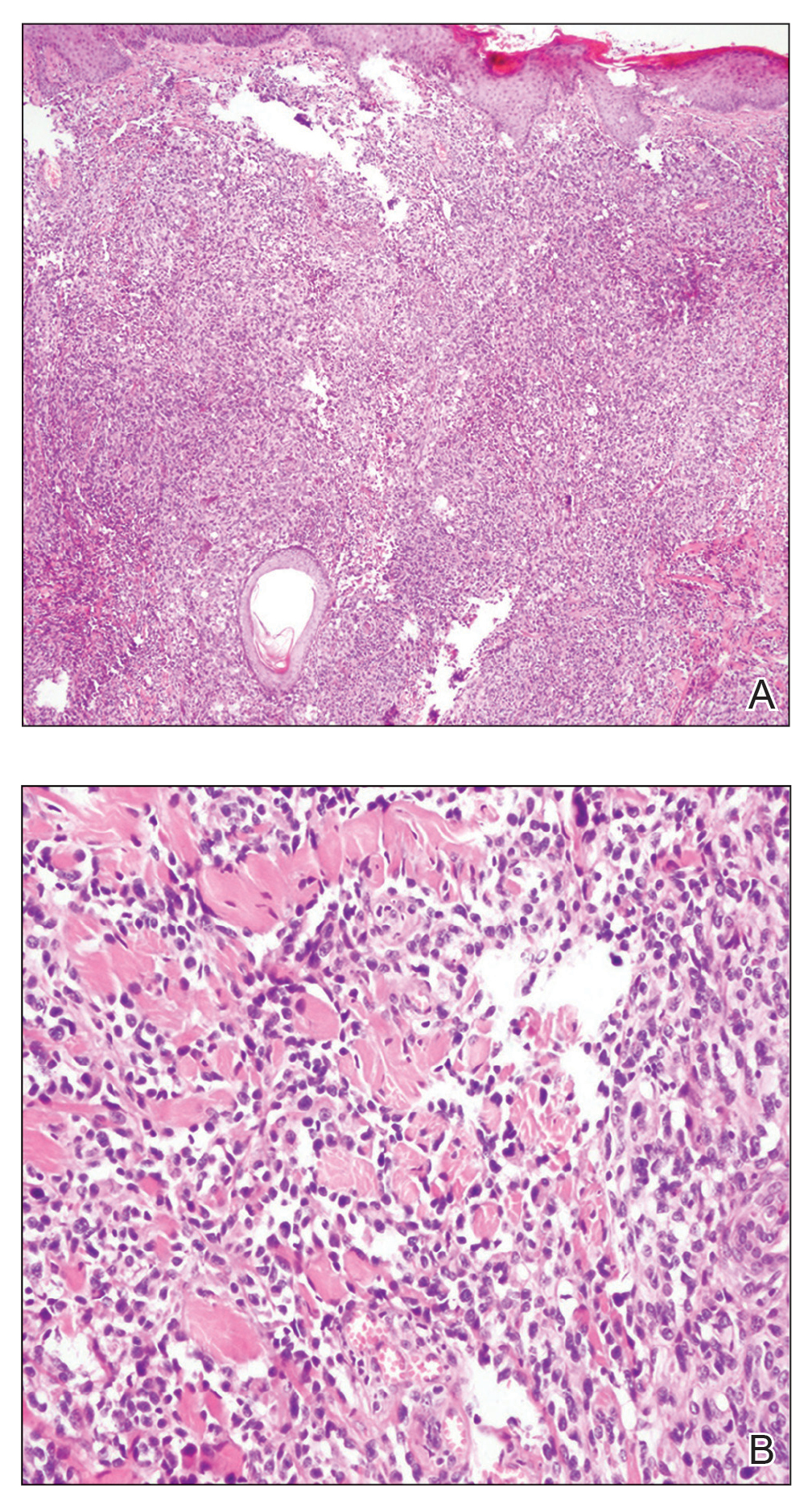
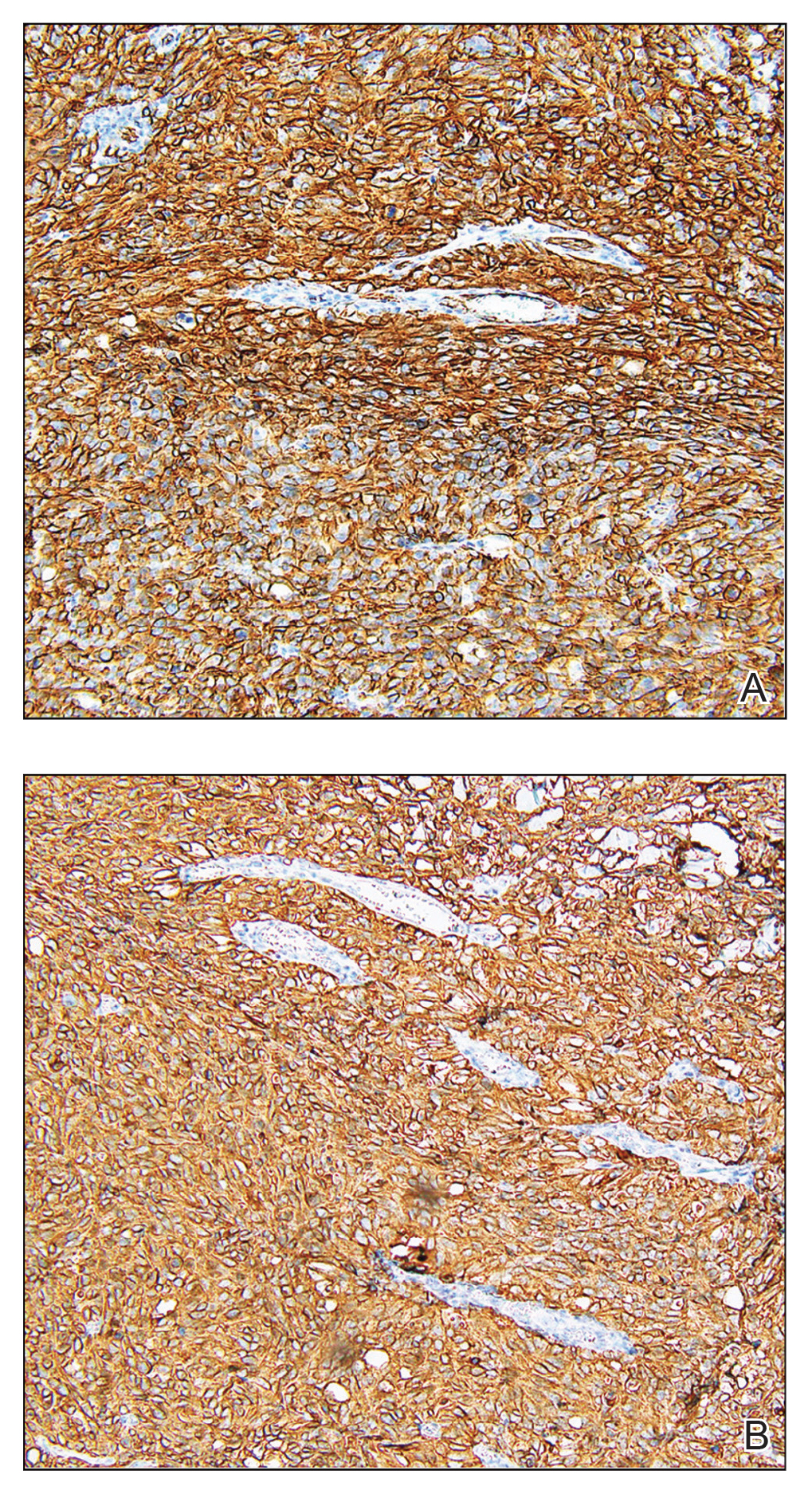
Angiosarcoma is a highly aggressive malignant neoplasm derived from vascular endothelial cells, most commonly involving the skin and superficial soft tissue. Angiosarcoma can be subdivided into CAS and visceral angiosarcoma according to the primary site of the tumor.1 Accurate and timely diagnosis of CAS is paramount due to its poor prognostic outcomes despite aggressive treatments. Clinically, CAS most frequently presents asymptomatically as an enlarging purple-red or bruiselike lesion with poorly defined margins. Cutaneous angiosarcoma often is misdiagnosed as an ecchymosis or hematoma due to its initial subtle presentation. It also may resemble eczema, hemangioma, and cellulitis; advanced lesions can mimic epithelial or mesenchymal neoplasms, including squamous cell carcinoma, keratoacanthoma, basal cell carcinoma, atypical fibroxanthoma (AFX), and malignant melanoma.2 Our patient lacked the classic clinical presentation of a hematomalike lesion and characteristic histologic features of anastomosing vascular structures with abundant extravasated erythrocytes at low magnification. However, the presence of erythrocytes in vascular channels along with CD31 and D2-40 immunoreactivity confirmed its vascular origin.
The prognosis of CAS is poor even with localized lesions. Age is a substantial prognostic factor, as a near 50% reduction of overall survival rate has been observed in patients older than 50 years.3 Other reported poor predictors for prognosis include male sex, the presence of cardiovascular diseases, location on the scalp, history of smoking, tumor size larger than 5 cm, and the presence of satellite lesions. Distant metastases are common, primarily affecting the lungs but also the bones and liver.4
Radical resection with a negative margin is considered the first-line treatment of choice. Although there is a paucity of studies assessing the specific width of surgical margins, application of no less than a 3-cm peripheral margin as well as a clear deep margin is recommended.5 Adjuvant radiation therapy also is essential to prevent local recurrence. Patients receiving combination therapy have a superior overall survival rate when compared to those undergoing surgery or radiation therapy alone.4
Cutaneous follicle center lymphoma also may present as 1 or more localized erythematous papules, plaques, and/or nodules, commonly arising on the scalp/forehead or trunk of middle-aged men. Despite being a low-grade lymphoma with a favorable prognosis, it may have a relatively fast growth and locally aggressive course if left untreated. The distinguishing histologic feature is a dense proliferation of neoplastic infiltrates in the dermis, which is separated from the epidermis by the grenz zone.6
The clinical presentation of cutaneous metastatic carcinomas varies greatly, with 1 or multiple localized or widespread lesions commonly involving the abdominal wall, scalp, and face. The lesions also may mimic benign dermatologic conditions, thus potentially resulting in erroneous clinical diagnosis and delayed therapy of the primary malignancy. Obtaining clinical history is crucial; however, a precise diagnosis may require histologic examination.7
Atypical fibroxanthoma is a rare superficial cutaneous sarcoma that typically occurs on the head and neck in sun-damaged elderly individuals. Clinically, AFX presents as well-circumscribed red or pink nodules or plaques with or without ulceration, crust, or scale.8 Atypical fibroxanthoma lesions usually are small, with a median diameter of 1 cm, while those greater than 2 cm reportedly account for less than 5% of cases.9 Atypical fibroxanthoma typically grows rapidly with no pain or discomfort. Histologically, AFX is characterized by a well-circumscribed dermal nodule consisting of pleomorphic spindle cells and multinucleated giant cells that can stain positively for CD10 and procollagen 1.10
Cutaneous pseudolymphoma is a benign inflammatory response process that stimulates polyclonal T- or B-cell lymphoproliferation. The clinical presentation may appear as localized or disseminated flesh-colored or red papules, infiltrated plaques, and nodules.11 Histopathology will show mixtures of B and T cells along with dendritic cells and macrophages, but irregular vascular structure and dissecting dermal collagen are not involved.
We present an unusual case of CAS with multiple pink nodules on the scalp. Early biopsy of these lesions is important to reach a correct diagnosis and to initiate appropriate treatment.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Albores-Saavedra J, Schwartz AM, Henson DE, et al. Cutaneous angiosarcoma. analysis of 434 cases from the surveillance, epidemiology, and end results program, 1973-2007. Ann Diagn Pathol. 2011;15:93-97.
- Guadagnolo BA, Zagars GK, Araujo D, et al. Outcomes after definitive treatment for cutaneous angiosarcoma of the face and scalp. Head Neck. 2011;33:661-667.
- Lindford A, Böhling T, Vaalavirta L, et al. Surgical management of radiation-associated cutaneous breast angiosarcoma. J Plast Reconstr Aesthet Surg. 2011;64:1036-1042.
- Costa EPW, Lu.0cena BD, Amin GA, et al. Primary cutaneous follicle center lymphoma. An Bras Dermatol. 2017;92:701-703.
- Menon AR, Thomas AS, Suresh N, et al. Cutaneous metastasis: an unusual presenting feature of urologic malignancies. Urol Ann. 2016;8:377-380.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls. StatPearls Publishing; 2020.
- Sarac E, Yuksel M, Turkmen IC, et al. Case for diagnosis. atypical fibroxanthoma. An Bras Dermatol. 2019;94:239-241.
- Miguel D, Peckruhn M, Elsner P. Treatment of cutaneous pseudolymphoma: a systematic review. Acta Derm Venereol. 2018;98:310-317.
The Diagnosis: Cutaneous Angiosarcoma
Biopsy revealed a cellular neoplasm consisting of atypical polygonal cells with a hobnailed appearance, vasoformative characteristics, and rare extravasated erythrocytes. The tumor had an infiltrative growth pattern as demonstrated by dissecting dermal collagen and a poorly defined border with adjacent normal tissue (Figure 1). Immunohistochemistry revealed that the lesion was positive for CD31 and D2-40 (Figure 2) but negative for cytokeratin, CD10, CD68, human herpesvirus 8, CD34, and Melan A, thus confirming the endothelial origin of the tumor cells and the diagnosis of cutaneous angiosarcoma (CAS). The patient was treated with extended surgical excision and radiation therapy. No recurrence or metastasis was found throughout 2 years of follow-up.
Angiosarcoma is a highly aggressive malignant neoplasm derived from vascular endothelial cells, most commonly involving the skin and superficial soft tissue. Angiosarcoma can be subdivided into CAS and visceral angiosarcoma according to the primary site of the tumor.1 Accurate and timely diagnosis of CAS is paramount due to its poor prognostic outcomes despite aggressive treatments. Clinically, CAS most frequently presents asymptomatically as an enlarging purple-red or bruiselike lesion with poorly defined margins. Cutaneous angiosarcoma often is misdiagnosed as an ecchymosis or hematoma due to its initial subtle presentation. It also may resemble eczema, hemangioma, and cellulitis; advanced lesions can mimic epithelial or mesenchymal neoplasms, including squamous cell carcinoma, keratoacanthoma, basal cell carcinoma, atypical fibroxanthoma (AFX), and malignant melanoma.2 Our patient lacked the classic clinical presentation of a hematomalike lesion and characteristic histologic features of anastomosing vascular structures with abundant extravasated erythrocytes at low magnification. However, the presence of erythrocytes in vascular channels along with CD31 and D2-40 immunoreactivity confirmed its vascular origin.
The prognosis of CAS is poor even with localized lesions. Age is a substantial prognostic factor, as a near 50% reduction of overall survival rate has been observed in patients older than 50 years.3 Other reported poor predictors for prognosis include male sex, the presence of cardiovascular diseases, location on the scalp, history of smoking, tumor size larger than 5 cm, and the presence of satellite lesions. Distant metastases are common, primarily affecting the lungs but also the bones and liver.4
Radical resection with a negative margin is considered the first-line treatment of choice. Although there is a paucity of studies assessing the specific width of surgical margins, application of no less than a 3-cm peripheral margin as well as a clear deep margin is recommended.5 Adjuvant radiation therapy also is essential to prevent local recurrence. Patients receiving combination therapy have a superior overall survival rate when compared to those undergoing surgery or radiation therapy alone.4
Cutaneous follicle center lymphoma also may present as 1 or more localized erythematous papules, plaques, and/or nodules, commonly arising on the scalp/forehead or trunk of middle-aged men. Despite being a low-grade lymphoma with a favorable prognosis, it may have a relatively fast growth and locally aggressive course if left untreated. The distinguishing histologic feature is a dense proliferation of neoplastic infiltrates in the dermis, which is separated from the epidermis by the grenz zone.6
The clinical presentation of cutaneous metastatic carcinomas varies greatly, with 1 or multiple localized or widespread lesions commonly involving the abdominal wall, scalp, and face. The lesions also may mimic benign dermatologic conditions, thus potentially resulting in erroneous clinical diagnosis and delayed therapy of the primary malignancy. Obtaining clinical history is crucial; however, a precise diagnosis may require histologic examination.7
Atypical fibroxanthoma is a rare superficial cutaneous sarcoma that typically occurs on the head and neck in sun-damaged elderly individuals. Clinically, AFX presents as well-circumscribed red or pink nodules or plaques with or without ulceration, crust, or scale.8 Atypical fibroxanthoma lesions usually are small, with a median diameter of 1 cm, while those greater than 2 cm reportedly account for less than 5% of cases.9 Atypical fibroxanthoma typically grows rapidly with no pain or discomfort. Histologically, AFX is characterized by a well-circumscribed dermal nodule consisting of pleomorphic spindle cells and multinucleated giant cells that can stain positively for CD10 and procollagen 1.10
Cutaneous pseudolymphoma is a benign inflammatory response process that stimulates polyclonal T- or B-cell lymphoproliferation. The clinical presentation may appear as localized or disseminated flesh-colored or red papules, infiltrated plaques, and nodules.11 Histopathology will show mixtures of B and T cells along with dendritic cells and macrophages, but irregular vascular structure and dissecting dermal collagen are not involved.
We present an unusual case of CAS with multiple pink nodules on the scalp. Early biopsy of these lesions is important to reach a correct diagnosis and to initiate appropriate treatment.
The Diagnosis: Cutaneous Angiosarcoma
Biopsy revealed a cellular neoplasm consisting of atypical polygonal cells with a hobnailed appearance, vasoformative characteristics, and rare extravasated erythrocytes. The tumor had an infiltrative growth pattern as demonstrated by dissecting dermal collagen and a poorly defined border with adjacent normal tissue (Figure 1). Immunohistochemistry revealed that the lesion was positive for CD31 and D2-40 (Figure 2) but negative for cytokeratin, CD10, CD68, human herpesvirus 8, CD34, and Melan A, thus confirming the endothelial origin of the tumor cells and the diagnosis of cutaneous angiosarcoma (CAS). The patient was treated with extended surgical excision and radiation therapy. No recurrence or metastasis was found throughout 2 years of follow-up.
Angiosarcoma is a highly aggressive malignant neoplasm derived from vascular endothelial cells, most commonly involving the skin and superficial soft tissue. Angiosarcoma can be subdivided into CAS and visceral angiosarcoma according to the primary site of the tumor.1 Accurate and timely diagnosis of CAS is paramount due to its poor prognostic outcomes despite aggressive treatments. Clinically, CAS most frequently presents asymptomatically as an enlarging purple-red or bruiselike lesion with poorly defined margins. Cutaneous angiosarcoma often is misdiagnosed as an ecchymosis or hematoma due to its initial subtle presentation. It also may resemble eczema, hemangioma, and cellulitis; advanced lesions can mimic epithelial or mesenchymal neoplasms, including squamous cell carcinoma, keratoacanthoma, basal cell carcinoma, atypical fibroxanthoma (AFX), and malignant melanoma.2 Our patient lacked the classic clinical presentation of a hematomalike lesion and characteristic histologic features of anastomosing vascular structures with abundant extravasated erythrocytes at low magnification. However, the presence of erythrocytes in vascular channels along with CD31 and D2-40 immunoreactivity confirmed its vascular origin.
The prognosis of CAS is poor even with localized lesions. Age is a substantial prognostic factor, as a near 50% reduction of overall survival rate has been observed in patients older than 50 years.3 Other reported poor predictors for prognosis include male sex, the presence of cardiovascular diseases, location on the scalp, history of smoking, tumor size larger than 5 cm, and the presence of satellite lesions. Distant metastases are common, primarily affecting the lungs but also the bones and liver.4
Radical resection with a negative margin is considered the first-line treatment of choice. Although there is a paucity of studies assessing the specific width of surgical margins, application of no less than a 3-cm peripheral margin as well as a clear deep margin is recommended.5 Adjuvant radiation therapy also is essential to prevent local recurrence. Patients receiving combination therapy have a superior overall survival rate when compared to those undergoing surgery or radiation therapy alone.4
Cutaneous follicle center lymphoma also may present as 1 or more localized erythematous papules, plaques, and/or nodules, commonly arising on the scalp/forehead or trunk of middle-aged men. Despite being a low-grade lymphoma with a favorable prognosis, it may have a relatively fast growth and locally aggressive course if left untreated. The distinguishing histologic feature is a dense proliferation of neoplastic infiltrates in the dermis, which is separated from the epidermis by the grenz zone.6
The clinical presentation of cutaneous metastatic carcinomas varies greatly, with 1 or multiple localized or widespread lesions commonly involving the abdominal wall, scalp, and face. The lesions also may mimic benign dermatologic conditions, thus potentially resulting in erroneous clinical diagnosis and delayed therapy of the primary malignancy. Obtaining clinical history is crucial; however, a precise diagnosis may require histologic examination.7
Atypical fibroxanthoma is a rare superficial cutaneous sarcoma that typically occurs on the head and neck in sun-damaged elderly individuals. Clinically, AFX presents as well-circumscribed red or pink nodules or plaques with or without ulceration, crust, or scale.8 Atypical fibroxanthoma lesions usually are small, with a median diameter of 1 cm, while those greater than 2 cm reportedly account for less than 5% of cases.9 Atypical fibroxanthoma typically grows rapidly with no pain or discomfort. Histologically, AFX is characterized by a well-circumscribed dermal nodule consisting of pleomorphic spindle cells and multinucleated giant cells that can stain positively for CD10 and procollagen 1.10
Cutaneous pseudolymphoma is a benign inflammatory response process that stimulates polyclonal T- or B-cell lymphoproliferation. The clinical presentation may appear as localized or disseminated flesh-colored or red papules, infiltrated plaques, and nodules.11 Histopathology will show mixtures of B and T cells along with dendritic cells and macrophages, but irregular vascular structure and dissecting dermal collagen are not involved.
We present an unusual case of CAS with multiple pink nodules on the scalp. Early biopsy of these lesions is important to reach a correct diagnosis and to initiate appropriate treatment.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Albores-Saavedra J, Schwartz AM, Henson DE, et al. Cutaneous angiosarcoma. analysis of 434 cases from the surveillance, epidemiology, and end results program, 1973-2007. Ann Diagn Pathol. 2011;15:93-97.
- Guadagnolo BA, Zagars GK, Araujo D, et al. Outcomes after definitive treatment for cutaneous angiosarcoma of the face and scalp. Head Neck. 2011;33:661-667.
- Lindford A, Böhling T, Vaalavirta L, et al. Surgical management of radiation-associated cutaneous breast angiosarcoma. J Plast Reconstr Aesthet Surg. 2011;64:1036-1042.
- Costa EPW, Lu.0cena BD, Amin GA, et al. Primary cutaneous follicle center lymphoma. An Bras Dermatol. 2017;92:701-703.
- Menon AR, Thomas AS, Suresh N, et al. Cutaneous metastasis: an unusual presenting feature of urologic malignancies. Urol Ann. 2016;8:377-380.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls. StatPearls Publishing; 2020.
- Sarac E, Yuksel M, Turkmen IC, et al. Case for diagnosis. atypical fibroxanthoma. An Bras Dermatol. 2019;94:239-241.
- Miguel D, Peckruhn M, Elsner P. Treatment of cutaneous pseudolymphoma: a systematic review. Acta Derm Venereol. 2018;98:310-317.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Albores-Saavedra J, Schwartz AM, Henson DE, et al. Cutaneous angiosarcoma. analysis of 434 cases from the surveillance, epidemiology, and end results program, 1973-2007. Ann Diagn Pathol. 2011;15:93-97.
- Guadagnolo BA, Zagars GK, Araujo D, et al. Outcomes after definitive treatment for cutaneous angiosarcoma of the face and scalp. Head Neck. 2011;33:661-667.
- Lindford A, Böhling T, Vaalavirta L, et al. Surgical management of radiation-associated cutaneous breast angiosarcoma. J Plast Reconstr Aesthet Surg. 2011;64:1036-1042.
- Costa EPW, Lu.0cena BD, Amin GA, et al. Primary cutaneous follicle center lymphoma. An Bras Dermatol. 2017;92:701-703.
- Menon AR, Thomas AS, Suresh N, et al. Cutaneous metastasis: an unusual presenting feature of urologic malignancies. Urol Ann. 2016;8:377-380.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls. StatPearls Publishing; 2020.
- Sarac E, Yuksel M, Turkmen IC, et al. Case for diagnosis. atypical fibroxanthoma. An Bras Dermatol. 2019;94:239-241.
- Miguel D, Peckruhn M, Elsner P. Treatment of cutaneous pseudolymphoma: a systematic review. Acta Derm Venereol. 2018;98:310-317.

A 67-year-old man presented with pink nodules on the scalp that were enlarging and increasing over the course of 2 months. The patient was otherwise healthy, had no constitutional symptoms such as fever or weight loss, and did not note pruritus or pain. His medications included telmisartan and Salvia miltiorrhiza for hypertension and coronary heart disease, respectively. He had been a heavy smoker for 44 years. Physical examination revealed several dome-shaped, pink nodules with smooth surfaces distributed in an agminated appearance on the scalp. The lesions were indurated and ranged from 1 to 5 cm in diameter.
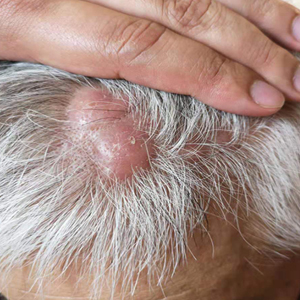
Comment on “Distribution of Skin-Type Diversity in Photographs in AAD Online Educational Modules”
To the Editor:
We read with great interest the article by Chu et al1 (Cutis. 2021;107:157-159) and commend them for noting the underrepresentation of skin of color (SOC) in the American Academy of Dermatology (AAD) Basic Dermatology Curriculum. The AAD Basic Dermatology Curriculum represents one introductory resource that is ubiquitously utilized by medical students. Herein, we add an analysis of the representation of SOC in the following resources that also comprise the first exposure medical students have to dermatology: Dermatology Clinics Clinical Advisor articles (https://www.clinicaladvisor.com/home/dermatology/dermatology-clinics/), Learn Derm Module (LDM) by VisualDx (https://www.visualdx.com/learnderm/), Lookingbill and Marks’ Principles of Dermatology (6th ed)(LB&M),2 and DermNet NZ (https://dermnetnz.org/). We performed a focused search of the DermNet NZ database for images of the following common dermatologic conditions: acne, rosacea, alopecia, urticaria, arthropod bites, blistering diseases (bullous pemphigoid and pemphigus vulgaris), connective tissue diseases (dermatomyositis and lupus), inflammatory conditions (atopic dermatitis, contact dermatitis, and psoriasis), keloids, benign and malignant neoplasms (nevi, seborrheic keratosis, actinic keratosis, basal and squamous cell carcinomas, and melanoma including acral melanoma), bacterial skin infections (impetigo, erysipelas, cellulitis, staphylococcal scalded skin syndrome, and syphilis), fungal infections (dermatophyte infections), and viral skin infections (herpes, molluscum contagiosum, varicella-zoster virus, and warts). We classified images as light (Fitzpatrick phototypes I–IV) or dark (Fitzpatrick phototypes V or VI). We excluded images without visible background skin (eg, images of oral mucosa, genitalia, nails, palms and soles, dermoscopic images, histopathologic images).
We found the representation of SOC in the resources we selected to be as follows: Dermatology Clinics Clinical Advisor articles (70/367 or 19%); LDM (26/150 or 17%); LB&M (52/374 or 14%); DermNet NZ (11/310 or 4%). Representation of SOC in common dermatologic conditions such as actinic keratosis, alopecia, rosacea, urticaria, and warts was entirely absent across all resources. Other common skin diseases were represented in only one of the resources we analyzed: acne (represented only in LB&M, where only 3/11 images of acne were depicted in SOC); contact dermatitis (represented only in LB&M, where only 1/6 images of contact dermatitis were depicted in SOC); psoriasis (represented only on DermNet NZ, where only 2/25 images of psoriasis were depicted in SOC); seborrheic keratosis (represented only in LB&M, where 1/2 images of seborrheic keratosis were depicted in SOC). Furthermore, none of the resources we analyzed depicted malignancy (basal cell carcinoma, squamous cell carcinoma, and melanoma) in SOC. Although the poor representation of SOC in malignancies can be explained by the predilection of skin cancer for light skin, other dermatologic conditions that are more common in SOC also were poorly represented in these resources in SOC: acral melanoma, not represented in any of the resources we analyzed; subacute cutaneous lupus erythematosus and systemic lupus erythematosus, also not represented in any of the resources we analyzed; keloids, represented only in LB&M.
Although no study has investigated the true prevalence of Fitzpatrick phototypes in the United States, He et al3 demonstrated the prevalence of Fitzpatrick phototypes V and VI to be 25.0% and 18.8%, respectively, in an ethnically diverse study of 3386 participants. Indeed, the representation of SOC in the resources we analyzed falls short of this plausible estimate of SOC in an increasingly diverse US population.
Our work adds to the growing body of literature exposing the deficiencies in SOC representation in dermatology. As Lester et al4 noted, such poor representation of SOC is deleterious not just to patients, who may be misdiagnosed, but also more generally to the integrity of the field of dermatology. Moreover, our study, which analyzes introductory resources referenced by the junior medical student, highlights a potential danger of poor SOC representation for trainees—limited exposure to SOC may leave medical students unprepared to recognize lesions in SOC during clerkships and residency. Furthermore, we note an additional concern with minimal SOC representation in online modules such as the AAD and LDM module as well as online databases such as DermNet NZ; images from these resources may be used as training sets for machine learning (ML) software (indeed, DermNet NZ has been used as a training set for ML programs5). However, if data sets with poor representation of SOC are used to train ML algorithms, then ML software may be unable to recognize lesions in SOC.6 Thus, inadequate representation of SOC in online modules and databases may exacerbate existing inequities in dermatology.
To address the paucity of SOC representation, students can be directed to resources devoted to depicting SOC; however, as discussed eloquently by Chu et al,1 an attempt to update existing resources also must be made. The senior author in our study (S.J.K.) embraced such an approach, updating the dermatology lectures given to medical students to include more images of SOC. Such a top-down approach may represent a major step in dismantling the systemic biases that pervade dermatology.
A limitation of our analysis was use of the Fitzpatrick scale, which was conceived as a phenotypic scale to assess cutaneous responses to UV irradiation.7 Although it is the most commonly used scale to describe race/ethnicity and/or constitute skin color, it is not possible to include all non-White skin types and classify strictly under this umbrella term.
References
1. Chu B, Fathy R, Onyekaba G, et al. Distribution of skin-type diversity in photographs in AAD online educational modules. Cutis. 2021;107:157-159. doi:10.12788/cutis.0196
2. Marks JG Jr, Miller JJ. Lookingbill and Marks’ Principles of Dermatology. 6th ed. Saunders Elsevier; 2018.
3. He SY, McCulloch CE, Boscardin WJ, et al. Self-reported pigmentary phenotypes and race are significant but incomplete predictors of Fitzpatrick skin phototype in an ethnically diverse population. J Am Acad Dermatol. 2014;71:731-737. doi:10.1016/j.jaad.2014.05.023
4. Lester JC, Taylor SC, Chren M-M. Under‐representation of skin of colour in dermatology images: not just an educational issue. Br J Dermatol. 2019;180:1521-1522. doi:10.1111/bjd.17608
5. Aggarwal P. Data augmentation in dermatology image recognition using machine learning. Skin Res Technol. 2019;25:815-820. doi:10.1111/srt.12726
6. Adamson AS, Smith A. Machine learning and health care disparities in dermatology. JAMA Dermatol. 2018;154:1247-1248. doi:10.1001/jamadermatol.2018.2348
7. Ware OR, Dawson JE, Shinohara MM, et al. Racial limitations of Fitzpatrick skin type. Cutis. 2020;105:77-80.
Authors’ Response
We thank Mr. Joshi and Dr. Kim for their reply to our article and their added contribution to the literature on inadequate representation of skin of color (SOC) in dermatology educational materials. In recent years, multiple analyses have reviewed textbooks and popular online resources for SOC representation.1 These resources encompass all levels of education—from the laypatient to the medical student, and to residency and beyond—demonstrating the significant challenges to overcome.
In addition, as Mr. Joshi and Dr. Kim state, the potential for these inadequately representative resources to serve as training data for prediction and classification tools adds further urgency to the broader task at hand, as we do not wish to perpetuate disparities. Several tools already exist, including Derm Assist, a recent Google-produced tool that suggests a list of diagnoses from patient-provided images.2 Although Derm Assist has been marked as a CE Class I (low risk) medical device in the European Union, the original research it is built on relied on training data with low representation of darker skin types (2.7% Fitzpatrick V and 0% Fitzpatrick VI),3 drawing concern for its generalizability.
These concerns about SOC representation are not new; dermatology advocates, scholars, and organizations such as the Skin of Color Society have been working to address these deficiencies for many years, contributing to education (including writing of resources and textbooks) and academic research. This work continues today. For instance, Lester et al4 described best practices for clinical photography in SOC; this guidance was not yet published at the time of our original submission. Not only should dermatology strive for increased quantity of representation but also quality. This metric is particularly important if the images are intended not just for education but also for use as training data for prediction and classification tools.
Examples of more recent actions at the organizational level include the American Academy of Dermatology (AAD) announcing a 3-year plan to promote diversity, equity, and inclusion5 and VisualDx establishing #ProjectIMPACT, a collaboration to reduce health care biases in SOC.6 In the AAD 3-year plan, one goal is to “[i]ncrease use of images reflecting full spectrum of skin types and highlight topics on skin of color, health disparities, and cultural competency across all AAD education.”5 Although not specifically mentioned, we hope that the AAD has included updating the Basic Dermatology Curriculum, given its inadequate SOC representation, as part of its short-term goals. The greater recognition of these issues through more prevalent analyses published in leading dermatology journals is encouraging, and we hope both that improvements can be successfully implemented and that future studies will reveal improvements in representation.
Brian Chu, BS; Ramie Fathy, AB; Ginikanwa Onyekaba, BS; Jules B. Lipoff, MD
From the Perelman School of Medicine, University of Pennsylvania, Philadelphia. Dr. Lipoff is from the Department of Dermatology and the Leonard Davis Institute of Health Economics.
The authors report no conflict of interest.
Correspondence: Jules B. Lipoff, MD, Department of Dermatology, University of Pennsylvania, Penn Medicine University City, 3737 Market St, Ste 1100, Philadelphia, PA 19104 ([email protected]).
References
1. Perlman KL, Williams NM, Egbeto IA, et al. Skin of color lacks representation in medical student resources: a cross-sectional study. Int J Womens Dermatol. 2021;7:195-196. doi:10.1016/j.ijwd.2020.12.018
2. Bui P, Liu Y. Using AI to help find answers to common skin conditions. Published May 18, 2021. Accessed June 12, 2021. https://blog.google/technology/health/ai-dermatology-preview-io-2021
3. Liu Y, Jain A, Eng C, et al. A deep learning system for differential diagnosis of skin diseases. Nature Medicine. 2020;26:900-908. doi:10.1038/s41591-020-0842-3
4. Lester JC, Clark L, Linos E, et al. Clinical photography in skin of colour: tips and best practices. Br J Dermatol. 2021;184:1177-1179. doi:10.1111/bjd.19811
5. American Academy of Dermatology Association. Diversity in dermatology: diversity committee approved plan 2021-2023. Published January 26, 2021. Accessed June 24, 2021. https://assets.ctfassets.net/1ny4yoiyrqia/xQgnCE6ji5skUlcZQHS2b/65f0a9072811e11afcc33d043e02cd4d/DEI_Plan.pdf
6. VisualDx. #ProjectIMPACT. Accessed June 24, 2021. https://www.visualdx.com/projectimpact/
To the Editor:
We read with great interest the article by Chu et al1 (Cutis. 2021;107:157-159) and commend them for noting the underrepresentation of skin of color (SOC) in the American Academy of Dermatology (AAD) Basic Dermatology Curriculum. The AAD Basic Dermatology Curriculum represents one introductory resource that is ubiquitously utilized by medical students. Herein, we add an analysis of the representation of SOC in the following resources that also comprise the first exposure medical students have to dermatology: Dermatology Clinics Clinical Advisor articles (https://www.clinicaladvisor.com/home/dermatology/dermatology-clinics/), Learn Derm Module (LDM) by VisualDx (https://www.visualdx.com/learnderm/), Lookingbill and Marks’ Principles of Dermatology (6th ed)(LB&M),2 and DermNet NZ (https://dermnetnz.org/). We performed a focused search of the DermNet NZ database for images of the following common dermatologic conditions: acne, rosacea, alopecia, urticaria, arthropod bites, blistering diseases (bullous pemphigoid and pemphigus vulgaris), connective tissue diseases (dermatomyositis and lupus), inflammatory conditions (atopic dermatitis, contact dermatitis, and psoriasis), keloids, benign and malignant neoplasms (nevi, seborrheic keratosis, actinic keratosis, basal and squamous cell carcinomas, and melanoma including acral melanoma), bacterial skin infections (impetigo, erysipelas, cellulitis, staphylococcal scalded skin syndrome, and syphilis), fungal infections (dermatophyte infections), and viral skin infections (herpes, molluscum contagiosum, varicella-zoster virus, and warts). We classified images as light (Fitzpatrick phototypes I–IV) or dark (Fitzpatrick phototypes V or VI). We excluded images without visible background skin (eg, images of oral mucosa, genitalia, nails, palms and soles, dermoscopic images, histopathologic images).
We found the representation of SOC in the resources we selected to be as follows: Dermatology Clinics Clinical Advisor articles (70/367 or 19%); LDM (26/150 or 17%); LB&M (52/374 or 14%); DermNet NZ (11/310 or 4%). Representation of SOC in common dermatologic conditions such as actinic keratosis, alopecia, rosacea, urticaria, and warts was entirely absent across all resources. Other common skin diseases were represented in only one of the resources we analyzed: acne (represented only in LB&M, where only 3/11 images of acne were depicted in SOC); contact dermatitis (represented only in LB&M, where only 1/6 images of contact dermatitis were depicted in SOC); psoriasis (represented only on DermNet NZ, where only 2/25 images of psoriasis were depicted in SOC); seborrheic keratosis (represented only in LB&M, where 1/2 images of seborrheic keratosis were depicted in SOC). Furthermore, none of the resources we analyzed depicted malignancy (basal cell carcinoma, squamous cell carcinoma, and melanoma) in SOC. Although the poor representation of SOC in malignancies can be explained by the predilection of skin cancer for light skin, other dermatologic conditions that are more common in SOC also were poorly represented in these resources in SOC: acral melanoma, not represented in any of the resources we analyzed; subacute cutaneous lupus erythematosus and systemic lupus erythematosus, also not represented in any of the resources we analyzed; keloids, represented only in LB&M.
Although no study has investigated the true prevalence of Fitzpatrick phototypes in the United States, He et al3 demonstrated the prevalence of Fitzpatrick phototypes V and VI to be 25.0% and 18.8%, respectively, in an ethnically diverse study of 3386 participants. Indeed, the representation of SOC in the resources we analyzed falls short of this plausible estimate of SOC in an increasingly diverse US population.
Our work adds to the growing body of literature exposing the deficiencies in SOC representation in dermatology. As Lester et al4 noted, such poor representation of SOC is deleterious not just to patients, who may be misdiagnosed, but also more generally to the integrity of the field of dermatology. Moreover, our study, which analyzes introductory resources referenced by the junior medical student, highlights a potential danger of poor SOC representation for trainees—limited exposure to SOC may leave medical students unprepared to recognize lesions in SOC during clerkships and residency. Furthermore, we note an additional concern with minimal SOC representation in online modules such as the AAD and LDM module as well as online databases such as DermNet NZ; images from these resources may be used as training sets for machine learning (ML) software (indeed, DermNet NZ has been used as a training set for ML programs5). However, if data sets with poor representation of SOC are used to train ML algorithms, then ML software may be unable to recognize lesions in SOC.6 Thus, inadequate representation of SOC in online modules and databases may exacerbate existing inequities in dermatology.
To address the paucity of SOC representation, students can be directed to resources devoted to depicting SOC; however, as discussed eloquently by Chu et al,1 an attempt to update existing resources also must be made. The senior author in our study (S.J.K.) embraced such an approach, updating the dermatology lectures given to medical students to include more images of SOC. Such a top-down approach may represent a major step in dismantling the systemic biases that pervade dermatology.
A limitation of our analysis was use of the Fitzpatrick scale, which was conceived as a phenotypic scale to assess cutaneous responses to UV irradiation.7 Although it is the most commonly used scale to describe race/ethnicity and/or constitute skin color, it is not possible to include all non-White skin types and classify strictly under this umbrella term.
References
1. Chu B, Fathy R, Onyekaba G, et al. Distribution of skin-type diversity in photographs in AAD online educational modules. Cutis. 2021;107:157-159. doi:10.12788/cutis.0196
2. Marks JG Jr, Miller JJ. Lookingbill and Marks’ Principles of Dermatology. 6th ed. Saunders Elsevier; 2018.
3. He SY, McCulloch CE, Boscardin WJ, et al. Self-reported pigmentary phenotypes and race are significant but incomplete predictors of Fitzpatrick skin phototype in an ethnically diverse population. J Am Acad Dermatol. 2014;71:731-737. doi:10.1016/j.jaad.2014.05.023
4. Lester JC, Taylor SC, Chren M-M. Under‐representation of skin of colour in dermatology images: not just an educational issue. Br J Dermatol. 2019;180:1521-1522. doi:10.1111/bjd.17608
5. Aggarwal P. Data augmentation in dermatology image recognition using machine learning. Skin Res Technol. 2019;25:815-820. doi:10.1111/srt.12726
6. Adamson AS, Smith A. Machine learning and health care disparities in dermatology. JAMA Dermatol. 2018;154:1247-1248. doi:10.1001/jamadermatol.2018.2348
7. Ware OR, Dawson JE, Shinohara MM, et al. Racial limitations of Fitzpatrick skin type. Cutis. 2020;105:77-80.
Authors’ Response
We thank Mr. Joshi and Dr. Kim for their reply to our article and their added contribution to the literature on inadequate representation of skin of color (SOC) in dermatology educational materials. In recent years, multiple analyses have reviewed textbooks and popular online resources for SOC representation.1 These resources encompass all levels of education—from the laypatient to the medical student, and to residency and beyond—demonstrating the significant challenges to overcome.
In addition, as Mr. Joshi and Dr. Kim state, the potential for these inadequately representative resources to serve as training data for prediction and classification tools adds further urgency to the broader task at hand, as we do not wish to perpetuate disparities. Several tools already exist, including Derm Assist, a recent Google-produced tool that suggests a list of diagnoses from patient-provided images.2 Although Derm Assist has been marked as a CE Class I (low risk) medical device in the European Union, the original research it is built on relied on training data with low representation of darker skin types (2.7% Fitzpatrick V and 0% Fitzpatrick VI),3 drawing concern for its generalizability.
These concerns about SOC representation are not new; dermatology advocates, scholars, and organizations such as the Skin of Color Society have been working to address these deficiencies for many years, contributing to education (including writing of resources and textbooks) and academic research. This work continues today. For instance, Lester et al4 described best practices for clinical photography in SOC; this guidance was not yet published at the time of our original submission. Not only should dermatology strive for increased quantity of representation but also quality. This metric is particularly important if the images are intended not just for education but also for use as training data for prediction and classification tools.
Examples of more recent actions at the organizational level include the American Academy of Dermatology (AAD) announcing a 3-year plan to promote diversity, equity, and inclusion5 and VisualDx establishing #ProjectIMPACT, a collaboration to reduce health care biases in SOC.6 In the AAD 3-year plan, one goal is to “[i]ncrease use of images reflecting full spectrum of skin types and highlight topics on skin of color, health disparities, and cultural competency across all AAD education.”5 Although not specifically mentioned, we hope that the AAD has included updating the Basic Dermatology Curriculum, given its inadequate SOC representation, as part of its short-term goals. The greater recognition of these issues through more prevalent analyses published in leading dermatology journals is encouraging, and we hope both that improvements can be successfully implemented and that future studies will reveal improvements in representation.
Brian Chu, BS; Ramie Fathy, AB; Ginikanwa Onyekaba, BS; Jules B. Lipoff, MD
From the Perelman School of Medicine, University of Pennsylvania, Philadelphia. Dr. Lipoff is from the Department of Dermatology and the Leonard Davis Institute of Health Economics.
The authors report no conflict of interest.
Correspondence: Jules B. Lipoff, MD, Department of Dermatology, University of Pennsylvania, Penn Medicine University City, 3737 Market St, Ste 1100, Philadelphia, PA 19104 ([email protected]).
References
1. Perlman KL, Williams NM, Egbeto IA, et al. Skin of color lacks representation in medical student resources: a cross-sectional study. Int J Womens Dermatol. 2021;7:195-196. doi:10.1016/j.ijwd.2020.12.018
2. Bui P, Liu Y. Using AI to help find answers to common skin conditions. Published May 18, 2021. Accessed June 12, 2021. https://blog.google/technology/health/ai-dermatology-preview-io-2021
3. Liu Y, Jain A, Eng C, et al. A deep learning system for differential diagnosis of skin diseases. Nature Medicine. 2020;26:900-908. doi:10.1038/s41591-020-0842-3
4. Lester JC, Clark L, Linos E, et al. Clinical photography in skin of colour: tips and best practices. Br J Dermatol. 2021;184:1177-1179. doi:10.1111/bjd.19811
5. American Academy of Dermatology Association. Diversity in dermatology: diversity committee approved plan 2021-2023. Published January 26, 2021. Accessed June 24, 2021. https://assets.ctfassets.net/1ny4yoiyrqia/xQgnCE6ji5skUlcZQHS2b/65f0a9072811e11afcc33d043e02cd4d/DEI_Plan.pdf
6. VisualDx. #ProjectIMPACT. Accessed June 24, 2021. https://www.visualdx.com/projectimpact/
To the Editor:
We read with great interest the article by Chu et al1 (Cutis. 2021;107:157-159) and commend them for noting the underrepresentation of skin of color (SOC) in the American Academy of Dermatology (AAD) Basic Dermatology Curriculum. The AAD Basic Dermatology Curriculum represents one introductory resource that is ubiquitously utilized by medical students. Herein, we add an analysis of the representation of SOC in the following resources that also comprise the first exposure medical students have to dermatology: Dermatology Clinics Clinical Advisor articles (https://www.clinicaladvisor.com/home/dermatology/dermatology-clinics/), Learn Derm Module (LDM) by VisualDx (https://www.visualdx.com/learnderm/), Lookingbill and Marks’ Principles of Dermatology (6th ed)(LB&M),2 and DermNet NZ (https://dermnetnz.org/). We performed a focused search of the DermNet NZ database for images of the following common dermatologic conditions: acne, rosacea, alopecia, urticaria, arthropod bites, blistering diseases (bullous pemphigoid and pemphigus vulgaris), connective tissue diseases (dermatomyositis and lupus), inflammatory conditions (atopic dermatitis, contact dermatitis, and psoriasis), keloids, benign and malignant neoplasms (nevi, seborrheic keratosis, actinic keratosis, basal and squamous cell carcinomas, and melanoma including acral melanoma), bacterial skin infections (impetigo, erysipelas, cellulitis, staphylococcal scalded skin syndrome, and syphilis), fungal infections (dermatophyte infections), and viral skin infections (herpes, molluscum contagiosum, varicella-zoster virus, and warts). We classified images as light (Fitzpatrick phototypes I–IV) or dark (Fitzpatrick phototypes V or VI). We excluded images without visible background skin (eg, images of oral mucosa, genitalia, nails, palms and soles, dermoscopic images, histopathologic images).
We found the representation of SOC in the resources we selected to be as follows: Dermatology Clinics Clinical Advisor articles (70/367 or 19%); LDM (26/150 or 17%); LB&M (52/374 or 14%); DermNet NZ (11/310 or 4%). Representation of SOC in common dermatologic conditions such as actinic keratosis, alopecia, rosacea, urticaria, and warts was entirely absent across all resources. Other common skin diseases were represented in only one of the resources we analyzed: acne (represented only in LB&M, where only 3/11 images of acne were depicted in SOC); contact dermatitis (represented only in LB&M, where only 1/6 images of contact dermatitis were depicted in SOC); psoriasis (represented only on DermNet NZ, where only 2/25 images of psoriasis were depicted in SOC); seborrheic keratosis (represented only in LB&M, where 1/2 images of seborrheic keratosis were depicted in SOC). Furthermore, none of the resources we analyzed depicted malignancy (basal cell carcinoma, squamous cell carcinoma, and melanoma) in SOC. Although the poor representation of SOC in malignancies can be explained by the predilection of skin cancer for light skin, other dermatologic conditions that are more common in SOC also were poorly represented in these resources in SOC: acral melanoma, not represented in any of the resources we analyzed; subacute cutaneous lupus erythematosus and systemic lupus erythematosus, also not represented in any of the resources we analyzed; keloids, represented only in LB&M.
Although no study has investigated the true prevalence of Fitzpatrick phototypes in the United States, He et al3 demonstrated the prevalence of Fitzpatrick phototypes V and VI to be 25.0% and 18.8%, respectively, in an ethnically diverse study of 3386 participants. Indeed, the representation of SOC in the resources we analyzed falls short of this plausible estimate of SOC in an increasingly diverse US population.
Our work adds to the growing body of literature exposing the deficiencies in SOC representation in dermatology. As Lester et al4 noted, such poor representation of SOC is deleterious not just to patients, who may be misdiagnosed, but also more generally to the integrity of the field of dermatology. Moreover, our study, which analyzes introductory resources referenced by the junior medical student, highlights a potential danger of poor SOC representation for trainees—limited exposure to SOC may leave medical students unprepared to recognize lesions in SOC during clerkships and residency. Furthermore, we note an additional concern with minimal SOC representation in online modules such as the AAD and LDM module as well as online databases such as DermNet NZ; images from these resources may be used as training sets for machine learning (ML) software (indeed, DermNet NZ has been used as a training set for ML programs5). However, if data sets with poor representation of SOC are used to train ML algorithms, then ML software may be unable to recognize lesions in SOC.6 Thus, inadequate representation of SOC in online modules and databases may exacerbate existing inequities in dermatology.
To address the paucity of SOC representation, students can be directed to resources devoted to depicting SOC; however, as discussed eloquently by Chu et al,1 an attempt to update existing resources also must be made. The senior author in our study (S.J.K.) embraced such an approach, updating the dermatology lectures given to medical students to include more images of SOC. Such a top-down approach may represent a major step in dismantling the systemic biases that pervade dermatology.
A limitation of our analysis was use of the Fitzpatrick scale, which was conceived as a phenotypic scale to assess cutaneous responses to UV irradiation.7 Although it is the most commonly used scale to describe race/ethnicity and/or constitute skin color, it is not possible to include all non-White skin types and classify strictly under this umbrella term.
References
1. Chu B, Fathy R, Onyekaba G, et al. Distribution of skin-type diversity in photographs in AAD online educational modules. Cutis. 2021;107:157-159. doi:10.12788/cutis.0196
2. Marks JG Jr, Miller JJ. Lookingbill and Marks’ Principles of Dermatology. 6th ed. Saunders Elsevier; 2018.
3. He SY, McCulloch CE, Boscardin WJ, et al. Self-reported pigmentary phenotypes and race are significant but incomplete predictors of Fitzpatrick skin phototype in an ethnically diverse population. J Am Acad Dermatol. 2014;71:731-737. doi:10.1016/j.jaad.2014.05.023
4. Lester JC, Taylor SC, Chren M-M. Under‐representation of skin of colour in dermatology images: not just an educational issue. Br J Dermatol. 2019;180:1521-1522. doi:10.1111/bjd.17608
5. Aggarwal P. Data augmentation in dermatology image recognition using machine learning. Skin Res Technol. 2019;25:815-820. doi:10.1111/srt.12726
6. Adamson AS, Smith A. Machine learning and health care disparities in dermatology. JAMA Dermatol. 2018;154:1247-1248. doi:10.1001/jamadermatol.2018.2348
7. Ware OR, Dawson JE, Shinohara MM, et al. Racial limitations of Fitzpatrick skin type. Cutis. 2020;105:77-80.
Authors’ Response
We thank Mr. Joshi and Dr. Kim for their reply to our article and their added contribution to the literature on inadequate representation of skin of color (SOC) in dermatology educational materials. In recent years, multiple analyses have reviewed textbooks and popular online resources for SOC representation.1 These resources encompass all levels of education—from the laypatient to the medical student, and to residency and beyond—demonstrating the significant challenges to overcome.
In addition, as Mr. Joshi and Dr. Kim state, the potential for these inadequately representative resources to serve as training data for prediction and classification tools adds further urgency to the broader task at hand, as we do not wish to perpetuate disparities. Several tools already exist, including Derm Assist, a recent Google-produced tool that suggests a list of diagnoses from patient-provided images.2 Although Derm Assist has been marked as a CE Class I (low risk) medical device in the European Union, the original research it is built on relied on training data with low representation of darker skin types (2.7% Fitzpatrick V and 0% Fitzpatrick VI),3 drawing concern for its generalizability.
These concerns about SOC representation are not new; dermatology advocates, scholars, and organizations such as the Skin of Color Society have been working to address these deficiencies for many years, contributing to education (including writing of resources and textbooks) and academic research. This work continues today. For instance, Lester et al4 described best practices for clinical photography in SOC; this guidance was not yet published at the time of our original submission. Not only should dermatology strive for increased quantity of representation but also quality. This metric is particularly important if the images are intended not just for education but also for use as training data for prediction and classification tools.
Examples of more recent actions at the organizational level include the American Academy of Dermatology (AAD) announcing a 3-year plan to promote diversity, equity, and inclusion5 and VisualDx establishing #ProjectIMPACT, a collaboration to reduce health care biases in SOC.6 In the AAD 3-year plan, one goal is to “[i]ncrease use of images reflecting full spectrum of skin types and highlight topics on skin of color, health disparities, and cultural competency across all AAD education.”5 Although not specifically mentioned, we hope that the AAD has included updating the Basic Dermatology Curriculum, given its inadequate SOC representation, as part of its short-term goals. The greater recognition of these issues through more prevalent analyses published in leading dermatology journals is encouraging, and we hope both that improvements can be successfully implemented and that future studies will reveal improvements in representation.
Brian Chu, BS; Ramie Fathy, AB; Ginikanwa Onyekaba, BS; Jules B. Lipoff, MD
From the Perelman School of Medicine, University of Pennsylvania, Philadelphia. Dr. Lipoff is from the Department of Dermatology and the Leonard Davis Institute of Health Economics.
The authors report no conflict of interest.
Correspondence: Jules B. Lipoff, MD, Department of Dermatology, University of Pennsylvania, Penn Medicine University City, 3737 Market St, Ste 1100, Philadelphia, PA 19104 ([email protected]).
References
1. Perlman KL, Williams NM, Egbeto IA, et al. Skin of color lacks representation in medical student resources: a cross-sectional study. Int J Womens Dermatol. 2021;7:195-196. doi:10.1016/j.ijwd.2020.12.018
2. Bui P, Liu Y. Using AI to help find answers to common skin conditions. Published May 18, 2021. Accessed June 12, 2021. https://blog.google/technology/health/ai-dermatology-preview-io-2021
3. Liu Y, Jain A, Eng C, et al. A deep learning system for differential diagnosis of skin diseases. Nature Medicine. 2020;26:900-908. doi:10.1038/s41591-020-0842-3
4. Lester JC, Clark L, Linos E, et al. Clinical photography in skin of colour: tips and best practices. Br J Dermatol. 2021;184:1177-1179. doi:10.1111/bjd.19811
5. American Academy of Dermatology Association. Diversity in dermatology: diversity committee approved plan 2021-2023. Published January 26, 2021. Accessed June 24, 2021. https://assets.ctfassets.net/1ny4yoiyrqia/xQgnCE6ji5skUlcZQHS2b/65f0a9072811e11afcc33d043e02cd4d/DEI_Plan.pdf
6. VisualDx. #ProjectIMPACT. Accessed June 24, 2021. https://www.visualdx.com/projectimpact/
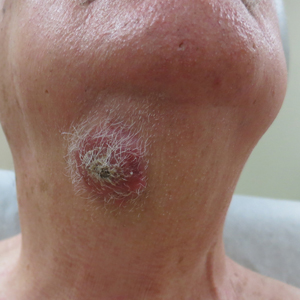
Unexpected Complications: A Case of Rosacea Fulminans in Pregnancy
Rosacea fulminans (RF) is a rare facial dermatosis characterized by its fulminating course. 1 It presents with superficial and deep-seated papules, pustules, and nodules combined with an intense reddish or cyanotic erythema localized to the face. Furthermore, there is an absence of comedones and involvement of the chest or back. 2 Rosacea fulminans primarily affects women and often is, but not always, proceeded by seborrhea, chronic acne vulgaris, or rosacea. Although the etiology of RF remains unknown, immunologic, hormonal, and vascular factors have been implicated. 3 We report a case of RF in a pregnant patient with a history of mild acne as a teenager that was long ago resolved.
Case Report
A 32-year-old pregnant woman (10 weeks’ gestation) presented with a rapidly progressing inflammatory disorder of the face of 1 month’s duration. The lesions developed 3 weeks after beginning progesterone therapy (200 mg vaginal suppository) for infertility due to polycystic ovary syndrome. Despite discontinuing progesterone for the last month, the patient’s lesions had dramatically worsened (Figure 1). Empiric cephalosporin treatment prescribed by her primary care physician yielded no improvement. Physical examination at the current presentation revealed erythematous nodules and pustules all over the face, coalescing into large thick plaques on the patient’s right cheek and chin. Submental nodes were palpable and tender. Based on the initial clinical findings, acne conglobata secondary to progesterone therapy was considered. The patient was given intralesional triamcinolone (2.5 mg/cc) injections to all larger nodules and several blue light treatments.
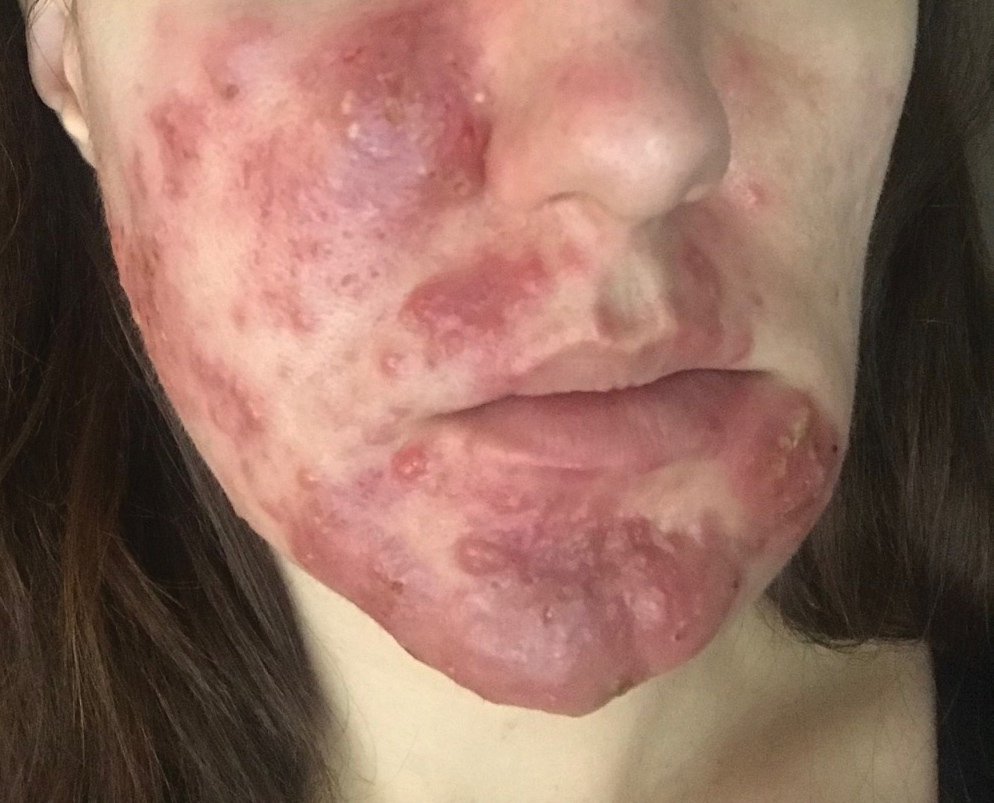
The injected areas had improved 5 days after the initial visit; however, the chin and right paranasal cheek developed even more nodules and papules coalescing into large plaques. After consulting the patient’s obstetrician, prednisone (20 mg once daily) was initiated. Three weeks later, the patient’s nodular lesions had improved, but there was a showering of more than 100 pustules and increased general erythema of the entire face (Figure 2). Crotamiton cream 10% (every day before noon), ivermectin cream 1% (every night at bedtime), and sodium sulfacetamide cleanser 10% once daily were added to the treatment plan.

At 16 weeks’ gestation, there was slight improvement; however, there was still erythema on the entire face with scattered pustules and multiple papules and nodules. Many small ice-pick scars were seen on the cheeks and forehead. No comedones were observed. A punch biopsy of an intact papule showed a prominent inflammatory infiltrate with granulomatous reaction and numerous neutrophils predominantly affecting hair follicles. Based on the clinical presentation and histopathology, a diagnosis of RF was made. Azithromycin (250 mg once daily) and metronidazole cream 0.75% twice daily were added. Two weeks later there were fewer nodules but many papules, edema, and intense erythema. The prednisone dosage was increased to 40 mg once daily. Two weeks later, the patient showed improvement with fewer lesions, less edema, and less erythema. The patient was instructed to finish the azithromycin course and discontinue use. At 28 weeks’ gestation, a prednisone taper was started with the intention to reduce the daily dose by delivery.
The patient delivered a healthy girl (birth weight, 1.985 kg) prematurely at 34 weeks’ gestation. At 2 months postpartum, the patient’s existing lesions continued to spontaneously improve; however, she still had numerous nodules and papules and continued to develop new lesions and form additional scars. Isotretinoin was instituted at 3 months postpartum upon cessation of nursing. Three months later (40 mg/d isotretinoin), the patient was nearly clear. At 8 months postpartum, isotretinoin was discontinued after a course of 150 mg/kg.
Comment
Rosacea fulminans initially was called pyoderma faciale but was later regarded as a severe form of rosacea and was renamed rosacea fulminans.2 According to a PubMed search of articles indexed for MEDLINE using the terms pregnancy and rosacea fulminans or pyoderma faciale, we identified 12 publications reporting 20 cases of RF associated with pregnancy (Table). Although there is no substantial evidence regarding the exact mechanism, these cases indicate that pregnancy can be an exacerbating or causative factor in the pathogenesis of RF.
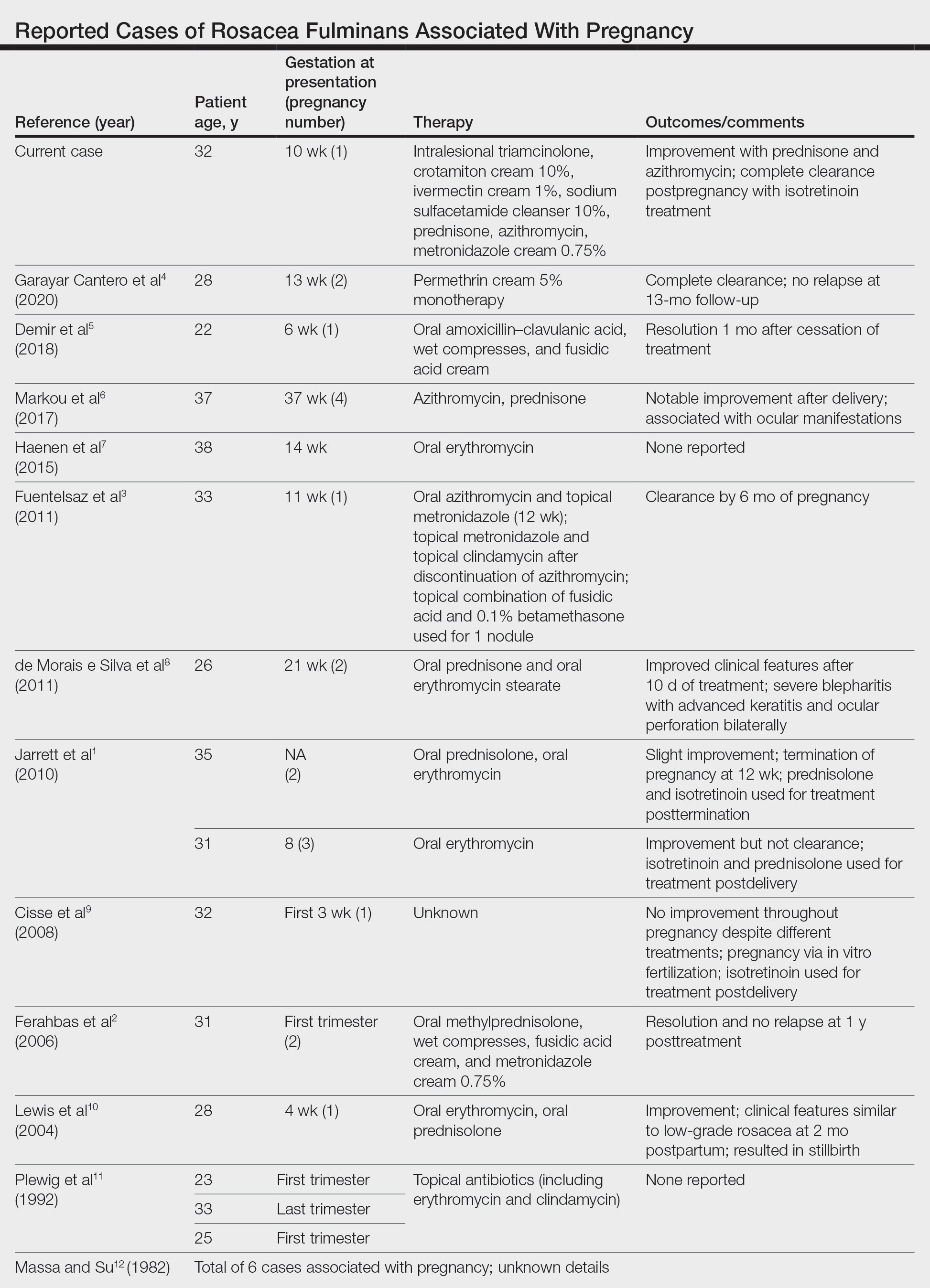
In addition to pregnancy, RF has been associated with inflammatory bowel disease, thyroid and liver disease, erythema nodosum, and severe emotional trauma. However, no organism has been consistently isolated, and no evidence of family history has been reported.1 Histopathologic findings are dependent on the stage of disease. Massive infiltrates of neutrophils may be observed in early stages. In older lesions, infiltrates take the form of epithelioid cell granulomas.2
Treatment of RF during pregnancy is challenging. Early and aggressive treatment with retinoids, tetracycline antibiotics, antiandrogenic contraceptives, and dapsone is recommended in patients who are not pregnant; these therapies are all contraindicated in pregnancy. Topical steroids can be safely used; however, systemic steroids usually are required to control RF. The use of systemic steroids can only be justified if the risks for intrauterine growth retardation, maternal diabetes mellitus, and hypertension outweigh the benefits of treating this severe disfiguring skin condition.10 A study by Bakar et al13 indicated that azithromycin is an effective and safe alternative in the treatment of RF. It has a superior pharmacokinetic profile compared to other macrolides and does not pose increased risks for congenital malformation or miscarriage. Because of the concomitant use of both azithromycin and prednisone, it is not possible to determine which had the larger role in the patient’s improvement.
Isotretinoin therapy in our patient led to substantial improvement of RF. Time will tell if the response will be durable. Also unknown is the risk for recurrence with subsequent pregnancies, which has not been reported in the literature. Although it is difficult to confidently say that pregnancy was the inciting factor in this patient’s RF, this case certainly provides more evidence for a link between pregnancy and RF.
- Jarrett R, Gonsalves R, Anstey AV. Differing obstetric outcomes of rosacea fulminans in pregnancy: report of three cases with review of pathogenesis and management. Clin Exp Dermatol. 2010;35:888-891. doi:10.1111/j.1365-2230.2010.03846.x
- Ferahbas A, Utas S, Mistik S, et al. Rosacea fulminans in pregnancy: case report and review of the literature. Am J Clin Dermatol. 2006;7:141-144. doi:10.2165/00128071-200607020-00007
- Fuentelsaz V, Ara M, Corredera C, et al. Rosacea fulminans in pregnancy: successful treatment with azithromycin. Clin Exp Dermatol. 2011;36:674-676. doi:10.1111/j.1365-2230.2011.04042.x
- Garayar Cantero M, Garabito Solovera E, Aguado García Á, et al. Use of permethrin in the treatment of rosacea fulminans during pregnancy: one case report. Dermatol Ther. 2020;33:E13436. doi:10.1111/dth.13436
- Demir O, Tas IS, Gunay B, et al. A rare dermatologic disease in pregnancy: rosacea fulminans—case report and review of the literature. Open Access Maced J Med Sci. 2018;6:1438-1441. doi:10.3889/oamjms.2018.267
- Markou AG, Alessandrini V, Muray JM, et al. Rosacea fulminans during pregnancy. Clin Exp Obstet Gynecol. 2017;44:157-159.
- Haenen CCP, Kouwenhoven STP, van Doorn R. Rosacea fulminans in pregnancy [in Dutch]. Ned Tijdschr Geneeskd. 2015;159:A8334.
- de Morais e Silva FA, Bonassi M, Steiner D, et al. Rosacea fulminans in pregnancy with ocular perforation. J Dtsch Dermatol Ges. 2011;9:542-543. doi:10.1111/j.1610-0387.2011.07616.x
- Cisse M, Maruani A, Bré C. Rosacea fulminans in the early course of a pregnancy by in vitro fertilization with embryo transfer [in French]. Ann Dermatol Venereol. 2008;135:675-678. doi:10.1016/j.annder.2008.04.015
- Lewis VJ, Holme SA, Wright A, et al. Rosacea fulminans in pregnancy. Br J Dermatol. 2004;151:917-919. doi:10.1111/j.1365-2133.2004.06190.x
- Plewig G, Jansen T, Kligman AM. Pyoderma faciale. a review and report of 20 additional cases: is it rosacea? Arch Dermatol. 1992;128:1611-1617. doi:10.1001/archderm.128.12.1611
- Massa MC, Su WP. Pyoderma faciale: a clinical study of twenty-nine patients. J Am Acad Dermatol. 1982;6:84-91. doi:10.1016/s0190-9622(82)70008-8
- Bakar O, Demirçay Z, Gürbüz O. Therapeutic potential of azithromycin in rosacea. Int J Dermatol. 2004;43:151-154. doi:10.1111/j.1365-4632.2004.01958.x
Rosacea fulminans (RF) is a rare facial dermatosis characterized by its fulminating course. 1 It presents with superficial and deep-seated papules, pustules, and nodules combined with an intense reddish or cyanotic erythema localized to the face. Furthermore, there is an absence of comedones and involvement of the chest or back. 2 Rosacea fulminans primarily affects women and often is, but not always, proceeded by seborrhea, chronic acne vulgaris, or rosacea. Although the etiology of RF remains unknown, immunologic, hormonal, and vascular factors have been implicated. 3 We report a case of RF in a pregnant patient with a history of mild acne as a teenager that was long ago resolved.
Case Report
A 32-year-old pregnant woman (10 weeks’ gestation) presented with a rapidly progressing inflammatory disorder of the face of 1 month’s duration. The lesions developed 3 weeks after beginning progesterone therapy (200 mg vaginal suppository) for infertility due to polycystic ovary syndrome. Despite discontinuing progesterone for the last month, the patient’s lesions had dramatically worsened (Figure 1). Empiric cephalosporin treatment prescribed by her primary care physician yielded no improvement. Physical examination at the current presentation revealed erythematous nodules and pustules all over the face, coalescing into large thick plaques on the patient’s right cheek and chin. Submental nodes were palpable and tender. Based on the initial clinical findings, acne conglobata secondary to progesterone therapy was considered. The patient was given intralesional triamcinolone (2.5 mg/cc) injections to all larger nodules and several blue light treatments.
The injected areas had improved 5 days after the initial visit; however, the chin and right paranasal cheek developed even more nodules and papules coalescing into large plaques. After consulting the patient’s obstetrician, prednisone (20 mg once daily) was initiated. Three weeks later, the patient’s nodular lesions had improved, but there was a showering of more than 100 pustules and increased general erythema of the entire face (Figure 2). Crotamiton cream 10% (every day before noon), ivermectin cream 1% (every night at bedtime), and sodium sulfacetamide cleanser 10% once daily were added to the treatment plan.
At 16 weeks’ gestation, there was slight improvement; however, there was still erythema on the entire face with scattered pustules and multiple papules and nodules. Many small ice-pick scars were seen on the cheeks and forehead. No comedones were observed. A punch biopsy of an intact papule showed a prominent inflammatory infiltrate with granulomatous reaction and numerous neutrophils predominantly affecting hair follicles. Based on the clinical presentation and histopathology, a diagnosis of RF was made. Azithromycin (250 mg once daily) and metronidazole cream 0.75% twice daily were added. Two weeks later there were fewer nodules but many papules, edema, and intense erythema. The prednisone dosage was increased to 40 mg once daily. Two weeks later, the patient showed improvement with fewer lesions, less edema, and less erythema. The patient was instructed to finish the azithromycin course and discontinue use. At 28 weeks’ gestation, a prednisone taper was started with the intention to reduce the daily dose by delivery.
The patient delivered a healthy girl (birth weight, 1.985 kg) prematurely at 34 weeks’ gestation. At 2 months postpartum, the patient’s existing lesions continued to spontaneously improve; however, she still had numerous nodules and papules and continued to develop new lesions and form additional scars. Isotretinoin was instituted at 3 months postpartum upon cessation of nursing. Three months later (40 mg/d isotretinoin), the patient was nearly clear. At 8 months postpartum, isotretinoin was discontinued after a course of 150 mg/kg.
Comment
Rosacea fulminans initially was called pyoderma faciale but was later regarded as a severe form of rosacea and was renamed rosacea fulminans.2 According to a PubMed search of articles indexed for MEDLINE using the terms pregnancy and rosacea fulminans or pyoderma faciale, we identified 12 publications reporting 20 cases of RF associated with pregnancy (Table). Although there is no substantial evidence regarding the exact mechanism, these cases indicate that pregnancy can be an exacerbating or causative factor in the pathogenesis of RF.
In addition to pregnancy, RF has been associated with inflammatory bowel disease, thyroid and liver disease, erythema nodosum, and severe emotional trauma. However, no organism has been consistently isolated, and no evidence of family history has been reported.1 Histopathologic findings are dependent on the stage of disease. Massive infiltrates of neutrophils may be observed in early stages. In older lesions, infiltrates take the form of epithelioid cell granulomas.2
Treatment of RF during pregnancy is challenging. Early and aggressive treatment with retinoids, tetracycline antibiotics, antiandrogenic contraceptives, and dapsone is recommended in patients who are not pregnant; these therapies are all contraindicated in pregnancy. Topical steroids can be safely used; however, systemic steroids usually are required to control RF. The use of systemic steroids can only be justified if the risks for intrauterine growth retardation, maternal diabetes mellitus, and hypertension outweigh the benefits of treating this severe disfiguring skin condition.10 A study by Bakar et al13 indicated that azithromycin is an effective and safe alternative in the treatment of RF. It has a superior pharmacokinetic profile compared to other macrolides and does not pose increased risks for congenital malformation or miscarriage. Because of the concomitant use of both azithromycin and prednisone, it is not possible to determine which had the larger role in the patient’s improvement.
Isotretinoin therapy in our patient led to substantial improvement of RF. Time will tell if the response will be durable. Also unknown is the risk for recurrence with subsequent pregnancies, which has not been reported in the literature. Although it is difficult to confidently say that pregnancy was the inciting factor in this patient’s RF, this case certainly provides more evidence for a link between pregnancy and RF.
Rosacea fulminans (RF) is a rare facial dermatosis characterized by its fulminating course. 1 It presents with superficial and deep-seated papules, pustules, and nodules combined with an intense reddish or cyanotic erythema localized to the face. Furthermore, there is an absence of comedones and involvement of the chest or back. 2 Rosacea fulminans primarily affects women and often is, but not always, proceeded by seborrhea, chronic acne vulgaris, or rosacea. Although the etiology of RF remains unknown, immunologic, hormonal, and vascular factors have been implicated. 3 We report a case of RF in a pregnant patient with a history of mild acne as a teenager that was long ago resolved.
Case Report
A 32-year-old pregnant woman (10 weeks’ gestation) presented with a rapidly progressing inflammatory disorder of the face of 1 month’s duration. The lesions developed 3 weeks after beginning progesterone therapy (200 mg vaginal suppository) for infertility due to polycystic ovary syndrome. Despite discontinuing progesterone for the last month, the patient’s lesions had dramatically worsened (Figure 1). Empiric cephalosporin treatment prescribed by her primary care physician yielded no improvement. Physical examination at the current presentation revealed erythematous nodules and pustules all over the face, coalescing into large thick plaques on the patient’s right cheek and chin. Submental nodes were palpable and tender. Based on the initial clinical findings, acne conglobata secondary to progesterone therapy was considered. The patient was given intralesional triamcinolone (2.5 mg/cc) injections to all larger nodules and several blue light treatments.
The injected areas had improved 5 days after the initial visit; however, the chin and right paranasal cheek developed even more nodules and papules coalescing into large plaques. After consulting the patient’s obstetrician, prednisone (20 mg once daily) was initiated. Three weeks later, the patient’s nodular lesions had improved, but there was a showering of more than 100 pustules and increased general erythema of the entire face (Figure 2). Crotamiton cream 10% (every day before noon), ivermectin cream 1% (every night at bedtime), and sodium sulfacetamide cleanser 10% once daily were added to the treatment plan.
At 16 weeks’ gestation, there was slight improvement; however, there was still erythema on the entire face with scattered pustules and multiple papules and nodules. Many small ice-pick scars were seen on the cheeks and forehead. No comedones were observed. A punch biopsy of an intact papule showed a prominent inflammatory infiltrate with granulomatous reaction and numerous neutrophils predominantly affecting hair follicles. Based on the clinical presentation and histopathology, a diagnosis of RF was made. Azithromycin (250 mg once daily) and metronidazole cream 0.75% twice daily were added. Two weeks later there were fewer nodules but many papules, edema, and intense erythema. The prednisone dosage was increased to 40 mg once daily. Two weeks later, the patient showed improvement with fewer lesions, less edema, and less erythema. The patient was instructed to finish the azithromycin course and discontinue use. At 28 weeks’ gestation, a prednisone taper was started with the intention to reduce the daily dose by delivery.
The patient delivered a healthy girl (birth weight, 1.985 kg) prematurely at 34 weeks’ gestation. At 2 months postpartum, the patient’s existing lesions continued to spontaneously improve; however, she still had numerous nodules and papules and continued to develop new lesions and form additional scars. Isotretinoin was instituted at 3 months postpartum upon cessation of nursing. Three months later (40 mg/d isotretinoin), the patient was nearly clear. At 8 months postpartum, isotretinoin was discontinued after a course of 150 mg/kg.
Comment
Rosacea fulminans initially was called pyoderma faciale but was later regarded as a severe form of rosacea and was renamed rosacea fulminans.2 According to a PubMed search of articles indexed for MEDLINE using the terms pregnancy and rosacea fulminans or pyoderma faciale, we identified 12 publications reporting 20 cases of RF associated with pregnancy (Table). Although there is no substantial evidence regarding the exact mechanism, these cases indicate that pregnancy can be an exacerbating or causative factor in the pathogenesis of RF.
In addition to pregnancy, RF has been associated with inflammatory bowel disease, thyroid and liver disease, erythema nodosum, and severe emotional trauma. However, no organism has been consistently isolated, and no evidence of family history has been reported.1 Histopathologic findings are dependent on the stage of disease. Massive infiltrates of neutrophils may be observed in early stages. In older lesions, infiltrates take the form of epithelioid cell granulomas.2
Treatment of RF during pregnancy is challenging. Early and aggressive treatment with retinoids, tetracycline antibiotics, antiandrogenic contraceptives, and dapsone is recommended in patients who are not pregnant; these therapies are all contraindicated in pregnancy. Topical steroids can be safely used; however, systemic steroids usually are required to control RF. The use of systemic steroids can only be justified if the risks for intrauterine growth retardation, maternal diabetes mellitus, and hypertension outweigh the benefits of treating this severe disfiguring skin condition.10 A study by Bakar et al13 indicated that azithromycin is an effective and safe alternative in the treatment of RF. It has a superior pharmacokinetic profile compared to other macrolides and does not pose increased risks for congenital malformation or miscarriage. Because of the concomitant use of both azithromycin and prednisone, it is not possible to determine which had the larger role in the patient’s improvement.
Isotretinoin therapy in our patient led to substantial improvement of RF. Time will tell if the response will be durable. Also unknown is the risk for recurrence with subsequent pregnancies, which has not been reported in the literature. Although it is difficult to confidently say that pregnancy was the inciting factor in this patient’s RF, this case certainly provides more evidence for a link between pregnancy and RF.
- Jarrett R, Gonsalves R, Anstey AV. Differing obstetric outcomes of rosacea fulminans in pregnancy: report of three cases with review of pathogenesis and management. Clin Exp Dermatol. 2010;35:888-891. doi:10.1111/j.1365-2230.2010.03846.x
- Ferahbas A, Utas S, Mistik S, et al. Rosacea fulminans in pregnancy: case report and review of the literature. Am J Clin Dermatol. 2006;7:141-144. doi:10.2165/00128071-200607020-00007
- Fuentelsaz V, Ara M, Corredera C, et al. Rosacea fulminans in pregnancy: successful treatment with azithromycin. Clin Exp Dermatol. 2011;36:674-676. doi:10.1111/j.1365-2230.2011.04042.x
- Garayar Cantero M, Garabito Solovera E, Aguado García Á, et al. Use of permethrin in the treatment of rosacea fulminans during pregnancy: one case report. Dermatol Ther. 2020;33:E13436. doi:10.1111/dth.13436
- Demir O, Tas IS, Gunay B, et al. A rare dermatologic disease in pregnancy: rosacea fulminans—case report and review of the literature. Open Access Maced J Med Sci. 2018;6:1438-1441. doi:10.3889/oamjms.2018.267
- Markou AG, Alessandrini V, Muray JM, et al. Rosacea fulminans during pregnancy. Clin Exp Obstet Gynecol. 2017;44:157-159.
- Haenen CCP, Kouwenhoven STP, van Doorn R. Rosacea fulminans in pregnancy [in Dutch]. Ned Tijdschr Geneeskd. 2015;159:A8334.
- de Morais e Silva FA, Bonassi M, Steiner D, et al. Rosacea fulminans in pregnancy with ocular perforation. J Dtsch Dermatol Ges. 2011;9:542-543. doi:10.1111/j.1610-0387.2011.07616.x
- Cisse M, Maruani A, Bré C. Rosacea fulminans in the early course of a pregnancy by in vitro fertilization with embryo transfer [in French]. Ann Dermatol Venereol. 2008;135:675-678. doi:10.1016/j.annder.2008.04.015
- Lewis VJ, Holme SA, Wright A, et al. Rosacea fulminans in pregnancy. Br J Dermatol. 2004;151:917-919. doi:10.1111/j.1365-2133.2004.06190.x
- Plewig G, Jansen T, Kligman AM. Pyoderma faciale. a review and report of 20 additional cases: is it rosacea? Arch Dermatol. 1992;128:1611-1617. doi:10.1001/archderm.128.12.1611
- Massa MC, Su WP. Pyoderma faciale: a clinical study of twenty-nine patients. J Am Acad Dermatol. 1982;6:84-91. doi:10.1016/s0190-9622(82)70008-8
- Bakar O, Demirçay Z, Gürbüz O. Therapeutic potential of azithromycin in rosacea. Int J Dermatol. 2004;43:151-154. doi:10.1111/j.1365-4632.2004.01958.x
- Jarrett R, Gonsalves R, Anstey AV. Differing obstetric outcomes of rosacea fulminans in pregnancy: report of three cases with review of pathogenesis and management. Clin Exp Dermatol. 2010;35:888-891. doi:10.1111/j.1365-2230.2010.03846.x
- Ferahbas A, Utas S, Mistik S, et al. Rosacea fulminans in pregnancy: case report and review of the literature. Am J Clin Dermatol. 2006;7:141-144. doi:10.2165/00128071-200607020-00007
- Fuentelsaz V, Ara M, Corredera C, et al. Rosacea fulminans in pregnancy: successful treatment with azithromycin. Clin Exp Dermatol. 2011;36:674-676. doi:10.1111/j.1365-2230.2011.04042.x
- Garayar Cantero M, Garabito Solovera E, Aguado García Á, et al. Use of permethrin in the treatment of rosacea fulminans during pregnancy: one case report. Dermatol Ther. 2020;33:E13436. doi:10.1111/dth.13436
- Demir O, Tas IS, Gunay B, et al. A rare dermatologic disease in pregnancy: rosacea fulminans—case report and review of the literature. Open Access Maced J Med Sci. 2018;6:1438-1441. doi:10.3889/oamjms.2018.267
- Markou AG, Alessandrini V, Muray JM, et al. Rosacea fulminans during pregnancy. Clin Exp Obstet Gynecol. 2017;44:157-159.
- Haenen CCP, Kouwenhoven STP, van Doorn R. Rosacea fulminans in pregnancy [in Dutch]. Ned Tijdschr Geneeskd. 2015;159:A8334.
- de Morais e Silva FA, Bonassi M, Steiner D, et al. Rosacea fulminans in pregnancy with ocular perforation. J Dtsch Dermatol Ges. 2011;9:542-543. doi:10.1111/j.1610-0387.2011.07616.x
- Cisse M, Maruani A, Bré C. Rosacea fulminans in the early course of a pregnancy by in vitro fertilization with embryo transfer [in French]. Ann Dermatol Venereol. 2008;135:675-678. doi:10.1016/j.annder.2008.04.015
- Lewis VJ, Holme SA, Wright A, et al. Rosacea fulminans in pregnancy. Br J Dermatol. 2004;151:917-919. doi:10.1111/j.1365-2133.2004.06190.x
- Plewig G, Jansen T, Kligman AM. Pyoderma faciale. a review and report of 20 additional cases: is it rosacea? Arch Dermatol. 1992;128:1611-1617. doi:10.1001/archderm.128.12.1611
- Massa MC, Su WP. Pyoderma faciale: a clinical study of twenty-nine patients. J Am Acad Dermatol. 1982;6:84-91. doi:10.1016/s0190-9622(82)70008-8
- Bakar O, Demirçay Z, Gürbüz O. Therapeutic potential of azithromycin in rosacea. Int J Dermatol. 2004;43:151-154. doi:10.1111/j.1365-4632.2004.01958.x
Practice Points
- Rosacea fulminans (RF) is a rare facial dermatosis that can present in pregnant patients.
- Treatment of RF in a pregnant patient requires special considerations because typical therapies are contraindicated in pregnancy.

Utilizing a Sleep Mask to Reduce Patient Anxiety During Nail Surgery
Practice Gap
Perioperative anxiety is common in patients undergoing nail surgery. Patients might worry about seeing blood; about the procedure itself, including nail avulsion; and about associated pain and disfigurement. Nail surgery causes a high level of anxiety that correlates positively with postoperative pain1 and overall patient dissatisfaction. Furthermore, surgery-related anxiety is a predictor of increased postoperative analgesic use2 and delayed recovery.3
Therefore, implementing strategies that reduce perioperative anxiety may help minimize postoperative pain. Squeezing a stress ball, hand-holding, virtual reality, and music are tools that have been studied to reduce anxiety in the context of Mohs micrographic surgery; these strategies have not been studied for nail surgery.
The Technique
Using a sleep mask is a practical solution to reduce patient anxiety during nail surgery. A minority of patients will choose to watch their surgical procedure; most become unnerved observing their nail surgery. Using a sleep mask diverts visual attention from the surgical field without physically interfering with the nail surgeon. Utilizing a sleep mask is cost-effective, with disposable sleep masks available online for less than $0.30 each. Patients can bring their own mask, or a mask can be offered prior to surgery.
If desired, patients are instructed to wear the sleep mask during the entirety of the procedure, starting from anesthetic infiltration until wound closure and dressing application. Any adjustments can be made with the patient’s free hand. The sleep mask can be offered to patients of all ages undergoing nail surgery under local anesthesia, except babies and young children, who require general anesthesia.
Practical Implications
Distraction is an important strategy to reduce anxiety and pain in patients undergoing surgical procedures. In an observational study of 3087 surgical patients, 36% reported that self-distraction was the most helpful strategy for coping with preoperative anxiety.4 In a randomized, open-label clinical trial of 72 patients undergoing peripheral venous catheterization, asking the patients simple questions during the procedure was more effective than local anesthesia in reducing the perception of pain.5
It is crucial to implement strategies to reduce anxiety in patients undergoing nail surgery. Using a sleep mask impedes direct visualization of the surgical field, thus distracting the patient’s sight and attention from the procedure. Furthermore, this technique is safe and cost-effective.
Controlled clinical trials are necessary to assess the efficacy of this method in reducing nail surgery–related anxiety in comparison to other techniques.
- Navarro-Gastón D, Munuera-Martínez PV. Prevalence of preoperative anxiety and its relationship with postoperative pain in foot nail surgery: a cross-sectional study. Int J Environ Res Public Health. 2020;17:4481. doi:10.3390/ijerph17124481
- Ip HYV, Abrishami A, Peng PWH, et al. Predictors of postoperative pain and analgesic consumption: a qualitative systematic review. Anesthesiology. 2009;111:657-677. doi:10.1097/ALN.0b013e3181aae87a
- Mavros MN, Athanasiou S, Gkegkes ID, et al. Do psychological variables affect early surgical recovery? PLoS One. 2011;6:E20306. doi:10.1371/journal.pone.0020306
- Aust H, Rüsch D, Schuster M, et al. Coping strategies in anxious surgical patients. BMC Health Serv Res. 2016;16:250. doi:10.1186/s12913-016-1492-5
- Balanyuk I, Ledonne G, Provenzano M, et al. Distraction technique for pain reduction in peripheral venous catheterization: randomized, controlled trial. Acta Biomed. 2018;89(suppl 4):55-63. doi:10.23750/abmv89i4-S.7115
Practice Gap
Perioperative anxiety is common in patients undergoing nail surgery. Patients might worry about seeing blood; about the procedure itself, including nail avulsion; and about associated pain and disfigurement. Nail surgery causes a high level of anxiety that correlates positively with postoperative pain1 and overall patient dissatisfaction. Furthermore, surgery-related anxiety is a predictor of increased postoperative analgesic use2 and delayed recovery.3
Therefore, implementing strategies that reduce perioperative anxiety may help minimize postoperative pain. Squeezing a stress ball, hand-holding, virtual reality, and music are tools that have been studied to reduce anxiety in the context of Mohs micrographic surgery; these strategies have not been studied for nail surgery.
The Technique
Using a sleep mask is a practical solution to reduce patient anxiety during nail surgery. A minority of patients will choose to watch their surgical procedure; most become unnerved observing their nail surgery. Using a sleep mask diverts visual attention from the surgical field without physically interfering with the nail surgeon. Utilizing a sleep mask is cost-effective, with disposable sleep masks available online for less than $0.30 each. Patients can bring their own mask, or a mask can be offered prior to surgery.
If desired, patients are instructed to wear the sleep mask during the entirety of the procedure, starting from anesthetic infiltration until wound closure and dressing application. Any adjustments can be made with the patient’s free hand. The sleep mask can be offered to patients of all ages undergoing nail surgery under local anesthesia, except babies and young children, who require general anesthesia.
Practical Implications
Distraction is an important strategy to reduce anxiety and pain in patients undergoing surgical procedures. In an observational study of 3087 surgical patients, 36% reported that self-distraction was the most helpful strategy for coping with preoperative anxiety.4 In a randomized, open-label clinical trial of 72 patients undergoing peripheral venous catheterization, asking the patients simple questions during the procedure was more effective than local anesthesia in reducing the perception of pain.5
It is crucial to implement strategies to reduce anxiety in patients undergoing nail surgery. Using a sleep mask impedes direct visualization of the surgical field, thus distracting the patient’s sight and attention from the procedure. Furthermore, this technique is safe and cost-effective.
Controlled clinical trials are necessary to assess the efficacy of this method in reducing nail surgery–related anxiety in comparison to other techniques.
Practice Gap
Perioperative anxiety is common in patients undergoing nail surgery. Patients might worry about seeing blood; about the procedure itself, including nail avulsion; and about associated pain and disfigurement. Nail surgery causes a high level of anxiety that correlates positively with postoperative pain1 and overall patient dissatisfaction. Furthermore, surgery-related anxiety is a predictor of increased postoperative analgesic use2 and delayed recovery.3
Therefore, implementing strategies that reduce perioperative anxiety may help minimize postoperative pain. Squeezing a stress ball, hand-holding, virtual reality, and music are tools that have been studied to reduce anxiety in the context of Mohs micrographic surgery; these strategies have not been studied for nail surgery.
The Technique
Using a sleep mask is a practical solution to reduce patient anxiety during nail surgery. A minority of patients will choose to watch their surgical procedure; most become unnerved observing their nail surgery. Using a sleep mask diverts visual attention from the surgical field without physically interfering with the nail surgeon. Utilizing a sleep mask is cost-effective, with disposable sleep masks available online for less than $0.30 each. Patients can bring their own mask, or a mask can be offered prior to surgery.
If desired, patients are instructed to wear the sleep mask during the entirety of the procedure, starting from anesthetic infiltration until wound closure and dressing application. Any adjustments can be made with the patient’s free hand. The sleep mask can be offered to patients of all ages undergoing nail surgery under local anesthesia, except babies and young children, who require general anesthesia.
Practical Implications
Distraction is an important strategy to reduce anxiety and pain in patients undergoing surgical procedures. In an observational study of 3087 surgical patients, 36% reported that self-distraction was the most helpful strategy for coping with preoperative anxiety.4 In a randomized, open-label clinical trial of 72 patients undergoing peripheral venous catheterization, asking the patients simple questions during the procedure was more effective than local anesthesia in reducing the perception of pain.5
It is crucial to implement strategies to reduce anxiety in patients undergoing nail surgery. Using a sleep mask impedes direct visualization of the surgical field, thus distracting the patient’s sight and attention from the procedure. Furthermore, this technique is safe and cost-effective.
Controlled clinical trials are necessary to assess the efficacy of this method in reducing nail surgery–related anxiety in comparison to other techniques.
- Navarro-Gastón D, Munuera-Martínez PV. Prevalence of preoperative anxiety and its relationship with postoperative pain in foot nail surgery: a cross-sectional study. Int J Environ Res Public Health. 2020;17:4481. doi:10.3390/ijerph17124481
- Ip HYV, Abrishami A, Peng PWH, et al. Predictors of postoperative pain and analgesic consumption: a qualitative systematic review. Anesthesiology. 2009;111:657-677. doi:10.1097/ALN.0b013e3181aae87a
- Mavros MN, Athanasiou S, Gkegkes ID, et al. Do psychological variables affect early surgical recovery? PLoS One. 2011;6:E20306. doi:10.1371/journal.pone.0020306
- Aust H, Rüsch D, Schuster M, et al. Coping strategies in anxious surgical patients. BMC Health Serv Res. 2016;16:250. doi:10.1186/s12913-016-1492-5
- Balanyuk I, Ledonne G, Provenzano M, et al. Distraction technique for pain reduction in peripheral venous catheterization: randomized, controlled trial. Acta Biomed. 2018;89(suppl 4):55-63. doi:10.23750/abmv89i4-S.7115
- Navarro-Gastón D, Munuera-Martínez PV. Prevalence of preoperative anxiety and its relationship with postoperative pain in foot nail surgery: a cross-sectional study. Int J Environ Res Public Health. 2020;17:4481. doi:10.3390/ijerph17124481
- Ip HYV, Abrishami A, Peng PWH, et al. Predictors of postoperative pain and analgesic consumption: a qualitative systematic review. Anesthesiology. 2009;111:657-677. doi:10.1097/ALN.0b013e3181aae87a
- Mavros MN, Athanasiou S, Gkegkes ID, et al. Do psychological variables affect early surgical recovery? PLoS One. 2011;6:E20306. doi:10.1371/journal.pone.0020306
- Aust H, Rüsch D, Schuster M, et al. Coping strategies in anxious surgical patients. BMC Health Serv Res. 2016;16:250. doi:10.1186/s12913-016-1492-5
- Balanyuk I, Ledonne G, Provenzano M, et al. Distraction technique for pain reduction in peripheral venous catheterization: randomized, controlled trial. Acta Biomed. 2018;89(suppl 4):55-63. doi:10.23750/abmv89i4-S.7115
Micronychia of the Index Finger
Congenital onychodysplasia of the index finger (COIF), or Iso-Kikuchi syndrome, is a rare disorder characterized by malformation of one or both nails of the index fingers. The various anomalies described are anonychia, micronychia, polyonychia, malalignment, or hemi-onychogryphosis. It may be associated with abnormalities of the underlying phalangeal bone, the most masked being bifurcation of the terminal phalange.1 Initially thought to be nonhereditary and nonfamilial,2 it is now known that COIF can be inherited in an autosomal-dominant fashion.3 Millman and Strier3 described a family of 9 patients with COIF. It rarely is described outside of Japan. Padmavathy et al4 described a case in an Indian patient with COIF that was associated with the absence of a ring finger in addition to anomalies of the metacarpal bones.
Congenital onychodysplasia of the index finger has a broad spectrum regarding its etiology and clinical features.5 The pathogenesis of COIF still is poorly understood. Deficient circulation in digital arteries is thought to be a putative mechanism for developing a deformed nail. The nail is affected on the radial side of the index finger, likely because of the smaller caliber of the artery on that side.5 Hereditary as well as nonhereditary sporadic cases have been reported. In addition to the various fingernail anomalies, skeletal abnormalities also have been reported. Baran and Stroud6 have reported deformed lunulae as a manifestation of COIF.
The Diagnosis: Congenital Onychodysplasia of the Index Finger
The differential diagnosis of COIF includes hidrotic ectodermal dysplasia, nail-patella syndrome, Poland syndrome, and DOOR syndrome. Hidrotic ectodermal dysplasia exhibits onychodystrophy, generalized hypotrichosis, palmoplantar keratoderma, and dental anomalies.7 Nail-patella syndrome presents with hypoplasia of the fingernails and toenails, triangular nail lunulae, absent or hypoplastic patellae, and elbow and iliac horn dysplasia. Poland syndrome is distinguished from COIF by the congenital absence of the pectoralis major muscle on the ipsilateral side of the involved digits. The DOOR syndrome tetrad is comprised of deafness, onychodystrophy, osteodystrophy, and mental retardation.8 Unlike these conditions, COIF does not involve systems other than the nails and phalanges.
Treatment of this condition is mainly conservative, as patients typically do not have symptoms.9 Surgical interventions can be considered for cosmetic concerns. Knowledge of this congenital entity and its clinical findings is essential to prevent unnecessary procedures and workup.
- De Berker AR, Baran R. Science of the nail apparatus. Diseases of the Nails and Their Management. In: Baran R, De Berker AR, Holzberg M, et al, eds. 4th ed. Willey-Blackwell; 2012:1-50.
- Kikuchi I, Horikawa S, Amano F. Congenital onychodysplasia of the index fingers. Arch Dermatol. 1974;110:743-746.
- Millman AJ, Strier RP. Congenital onychodysplasia of the index fingers: report of a family. J Am Acad Dermatol. 1982;7:57-65.
- Padmavathy L, Rao L, Ethirajan N, et al. Iso-Kikuchi syndrome with absence of ring fingers and metacarpal bone abnormality. Indian J Dermatol Venereol Leprol. 2008;74:513.
- Hadj-Rabia S, Juhlin L, Baran R. Hereditary and congenital nail disorders. In: Baran R, De Berker AR, Holzberg M, et al, eds. Diseases of the Nails and Their Management. 4th ed. Wiley-Blackwell; 2012:485-490.
- Baran R, Stroud JD. Congenital onychodysplasia of the index fingers: Iso and Kikuchi syndrome. Arch Dermatol. 1984;120:243-244.
- Valerio E, Favot F, Mattei I, et al. Congenital isolated Iso-Kikuchi syndrome in a newborn. Clin Case Rep. 2015;3:866.
- Danarti R, Rahmayani S, Wirohadidjojo YW, et al. Deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures (DOORS) syndrome: a new case report from Indonesia and review of the literature. Eur J Dermatol. 2020;30:404-407.
- Milani-Nejad N, Mosser-Goldfarb J. Congenital onychodysplasia of index fingers: Iso-Kikuchi syndrome. J Pediatr. 2020;218:254.
Congenital onychodysplasia of the index finger (COIF), or Iso-Kikuchi syndrome, is a rare disorder characterized by malformation of one or both nails of the index fingers. The various anomalies described are anonychia, micronychia, polyonychia, malalignment, or hemi-onychogryphosis. It may be associated with abnormalities of the underlying phalangeal bone, the most masked being bifurcation of the terminal phalange.1 Initially thought to be nonhereditary and nonfamilial,2 it is now known that COIF can be inherited in an autosomal-dominant fashion.3 Millman and Strier3 described a family of 9 patients with COIF. It rarely is described outside of Japan. Padmavathy et al4 described a case in an Indian patient with COIF that was associated with the absence of a ring finger in addition to anomalies of the metacarpal bones.
Congenital onychodysplasia of the index finger has a broad spectrum regarding its etiology and clinical features.5 The pathogenesis of COIF still is poorly understood. Deficient circulation in digital arteries is thought to be a putative mechanism for developing a deformed nail. The nail is affected on the radial side of the index finger, likely because of the smaller caliber of the artery on that side.5 Hereditary as well as nonhereditary sporadic cases have been reported. In addition to the various fingernail anomalies, skeletal abnormalities also have been reported. Baran and Stroud6 have reported deformed lunulae as a manifestation of COIF.
The Diagnosis: Congenital Onychodysplasia of the Index Finger
The differential diagnosis of COIF includes hidrotic ectodermal dysplasia, nail-patella syndrome, Poland syndrome, and DOOR syndrome. Hidrotic ectodermal dysplasia exhibits onychodystrophy, generalized hypotrichosis, palmoplantar keratoderma, and dental anomalies.7 Nail-patella syndrome presents with hypoplasia of the fingernails and toenails, triangular nail lunulae, absent or hypoplastic patellae, and elbow and iliac horn dysplasia. Poland syndrome is distinguished from COIF by the congenital absence of the pectoralis major muscle on the ipsilateral side of the involved digits. The DOOR syndrome tetrad is comprised of deafness, onychodystrophy, osteodystrophy, and mental retardation.8 Unlike these conditions, COIF does not involve systems other than the nails and phalanges.
Treatment of this condition is mainly conservative, as patients typically do not have symptoms.9 Surgical interventions can be considered for cosmetic concerns. Knowledge of this congenital entity and its clinical findings is essential to prevent unnecessary procedures and workup.
Congenital onychodysplasia of the index finger (COIF), or Iso-Kikuchi syndrome, is a rare disorder characterized by malformation of one or both nails of the index fingers. The various anomalies described are anonychia, micronychia, polyonychia, malalignment, or hemi-onychogryphosis. It may be associated with abnormalities of the underlying phalangeal bone, the most masked being bifurcation of the terminal phalange.1 Initially thought to be nonhereditary and nonfamilial,2 it is now known that COIF can be inherited in an autosomal-dominant fashion.3 Millman and Strier3 described a family of 9 patients with COIF. It rarely is described outside of Japan. Padmavathy et al4 described a case in an Indian patient with COIF that was associated with the absence of a ring finger in addition to anomalies of the metacarpal bones.
Congenital onychodysplasia of the index finger has a broad spectrum regarding its etiology and clinical features.5 The pathogenesis of COIF still is poorly understood. Deficient circulation in digital arteries is thought to be a putative mechanism for developing a deformed nail. The nail is affected on the radial side of the index finger, likely because of the smaller caliber of the artery on that side.5 Hereditary as well as nonhereditary sporadic cases have been reported. In addition to the various fingernail anomalies, skeletal abnormalities also have been reported. Baran and Stroud6 have reported deformed lunulae as a manifestation of COIF.
The Diagnosis: Congenital Onychodysplasia of the Index Finger
The differential diagnosis of COIF includes hidrotic ectodermal dysplasia, nail-patella syndrome, Poland syndrome, and DOOR syndrome. Hidrotic ectodermal dysplasia exhibits onychodystrophy, generalized hypotrichosis, palmoplantar keratoderma, and dental anomalies.7 Nail-patella syndrome presents with hypoplasia of the fingernails and toenails, triangular nail lunulae, absent or hypoplastic patellae, and elbow and iliac horn dysplasia. Poland syndrome is distinguished from COIF by the congenital absence of the pectoralis major muscle on the ipsilateral side of the involved digits. The DOOR syndrome tetrad is comprised of deafness, onychodystrophy, osteodystrophy, and mental retardation.8 Unlike these conditions, COIF does not involve systems other than the nails and phalanges.
Treatment of this condition is mainly conservative, as patients typically do not have symptoms.9 Surgical interventions can be considered for cosmetic concerns. Knowledge of this congenital entity and its clinical findings is essential to prevent unnecessary procedures and workup.
- De Berker AR, Baran R. Science of the nail apparatus. Diseases of the Nails and Their Management. In: Baran R, De Berker AR, Holzberg M, et al, eds. 4th ed. Willey-Blackwell; 2012:1-50.
- Kikuchi I, Horikawa S, Amano F. Congenital onychodysplasia of the index fingers. Arch Dermatol. 1974;110:743-746.
- Millman AJ, Strier RP. Congenital onychodysplasia of the index fingers: report of a family. J Am Acad Dermatol. 1982;7:57-65.
- Padmavathy L, Rao L, Ethirajan N, et al. Iso-Kikuchi syndrome with absence of ring fingers and metacarpal bone abnormality. Indian J Dermatol Venereol Leprol. 2008;74:513.
- Hadj-Rabia S, Juhlin L, Baran R. Hereditary and congenital nail disorders. In: Baran R, De Berker AR, Holzberg M, et al, eds. Diseases of the Nails and Their Management. 4th ed. Wiley-Blackwell; 2012:485-490.
- Baran R, Stroud JD. Congenital onychodysplasia of the index fingers: Iso and Kikuchi syndrome. Arch Dermatol. 1984;120:243-244.
- Valerio E, Favot F, Mattei I, et al. Congenital isolated Iso-Kikuchi syndrome in a newborn. Clin Case Rep. 2015;3:866.
- Danarti R, Rahmayani S, Wirohadidjojo YW, et al. Deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures (DOORS) syndrome: a new case report from Indonesia and review of the literature. Eur J Dermatol. 2020;30:404-407.
- Milani-Nejad N, Mosser-Goldfarb J. Congenital onychodysplasia of index fingers: Iso-Kikuchi syndrome. J Pediatr. 2020;218:254.
- De Berker AR, Baran R. Science of the nail apparatus. Diseases of the Nails and Their Management. In: Baran R, De Berker AR, Holzberg M, et al, eds. 4th ed. Willey-Blackwell; 2012:1-50.
- Kikuchi I, Horikawa S, Amano F. Congenital onychodysplasia of the index fingers. Arch Dermatol. 1974;110:743-746.
- Millman AJ, Strier RP. Congenital onychodysplasia of the index fingers: report of a family. J Am Acad Dermatol. 1982;7:57-65.
- Padmavathy L, Rao L, Ethirajan N, et al. Iso-Kikuchi syndrome with absence of ring fingers and metacarpal bone abnormality. Indian J Dermatol Venereol Leprol. 2008;74:513.
- Hadj-Rabia S, Juhlin L, Baran R. Hereditary and congenital nail disorders. In: Baran R, De Berker AR, Holzberg M, et al, eds. Diseases of the Nails and Their Management. 4th ed. Wiley-Blackwell; 2012:485-490.
- Baran R, Stroud JD. Congenital onychodysplasia of the index fingers: Iso and Kikuchi syndrome. Arch Dermatol. 1984;120:243-244.
- Valerio E, Favot F, Mattei I, et al. Congenital isolated Iso-Kikuchi syndrome in a newborn. Clin Case Rep. 2015;3:866.
- Danarti R, Rahmayani S, Wirohadidjojo YW, et al. Deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures (DOORS) syndrome: a new case report from Indonesia and review of the literature. Eur J Dermatol. 2020;30:404-407.
- Milani-Nejad N, Mosser-Goldfarb J. Congenital onychodysplasia of index fingers: Iso-Kikuchi syndrome. J Pediatr. 2020;218:254.
A 21-year-old Indian woman who was initially seeking dermatology consultation for acne also was noted to have micronychia of the nail of the left index finger. The affected nail was narrow and half as broad as the unaffected normal nail on the right index finger. The patient confirmed that this finding had been present since birth; she faced no cosmetic disability and had not sought medical care for diagnosis or treatment. There was no history of trauma, complications during pregnancy, family history of micronychia or similar eruptions, or any other inciting event. The teeth, hair, and skin as well as the patient’s height, weight, and physical and mental development were normal. Systemic examination revealed no abnormalities. Radiography of the hands did not reveal any apparent bony abnormalities.
Medicare proposes direct payments to PAs, telehealth expansion
It also intends to change the approach to payments for office visits and for coaching programs for diabetes prevention.
The Centers for Medicare & Medicaid Services recently posted its proposed 2022 physician fee schedule. Running to more than 1,700 pages, the draft rule contains myriad other changes in how the giant federal health program pays for medical care, including revisions to its approach to evaluation and management (E/M) services, which represent many office visits. In addition, Medicare is seeking to increase participation in a program intended to prevent people from developing diabetes.
Physician groups posted quick complaints about a proposed 3.75% reduction to the conversion factor because of budget neutrality requirements. The cut reinstates a reduction Congress prevented in late 2020.
In a statement, Anders Gilberg, senior vice president of government affairs for the Medical Group Management Association, called the draft rule a “mixed bag for physician practices.” Mr. Gilberg said the MGMA will seek congressional intervention to avert the cut for services in 2022.
In keeping with a provision Congress included in a massive spending bill enacted in December, Medicare will let PAs directly bill, as nurse practitioners already can. In a press release, CMS on July 13 described this as a move likely to expand access to care and reduce administrative burden. In 2020, the American Academy of PAs praised the inclusion in the spending bill of the provision allowing its members to directly bill Medicare.
In the draft rule, CMS also intends to remove certain geographic restrictions regarding use of telehealth services for diagnosis, evaluation, and treatment of mental health disorders. CMS also is proposing to allow payment to eligible clinicians for certain mental health and behavioral health services to patients via audio-only telephone calls. These services would include counseling and therapy services provided through opioid treatment programs.
“These changes would be particularly helpful for those in areas with poor broadband infrastructure and among people with Medicare who are not capable of, or do not consent to the use of, devices that permit a two-way, audio/video interaction for their health care visits,” CMS said in a statement.
Slimmer Medicare enrollees, bigger payments for coaches?
CMS is seeking to draw more participants to the Medicare Diabetes Prevention Program (MDPP). This program includes organizations that provide structured, coach-led sessions in community and health care settings to help people lose weight and exercise more. During the COVID-19 public health emergency, CMS waived an enrollment fee for new suppliers of services in MDPP. CMS now is proposing to waive this fee for all organizations that submit an application to enroll in Medicare as an MDPP supplier on or after Jan. 1, 2022.
Another proposed change in MDPP services is a restructuring of payments so that organizations involved in coaching would receive larger payments when their participants reach milestones for attendance and for becoming slimmer.
“We propose to increase performance payments for MDPP beneficiary achievement of the 5% weight-loss goal, as well as continued attendance during each core maintenance interval,” CMS said in a statement.
Medicare remains engaged in a review of its payments for E/M services. In the draft rule, CMS is proposing a number of refinements to current policies for split, or shared, E/M visits, critical care services, and services furnished by teaching physicians involving residents. The intention of these changes is to “better reflect the current practice of medicine, the evolving role of nonphysician practitioners as members of the medical team, and to clarify conditions of payment that must be met to bill Medicare for these services,” CMS said.
A version of this article first appeared on Medscape.com.
It also intends to change the approach to payments for office visits and for coaching programs for diabetes prevention.
The Centers for Medicare & Medicaid Services recently posted its proposed 2022 physician fee schedule. Running to more than 1,700 pages, the draft rule contains myriad other changes in how the giant federal health program pays for medical care, including revisions to its approach to evaluation and management (E/M) services, which represent many office visits. In addition, Medicare is seeking to increase participation in a program intended to prevent people from developing diabetes.
Physician groups posted quick complaints about a proposed 3.75% reduction to the conversion factor because of budget neutrality requirements. The cut reinstates a reduction Congress prevented in late 2020.
In a statement, Anders Gilberg, senior vice president of government affairs for the Medical Group Management Association, called the draft rule a “mixed bag for physician practices.” Mr. Gilberg said the MGMA will seek congressional intervention to avert the cut for services in 2022.
In keeping with a provision Congress included in a massive spending bill enacted in December, Medicare will let PAs directly bill, as nurse practitioners already can. In a press release, CMS on July 13 described this as a move likely to expand access to care and reduce administrative burden. In 2020, the American Academy of PAs praised the inclusion in the spending bill of the provision allowing its members to directly bill Medicare.
In the draft rule, CMS also intends to remove certain geographic restrictions regarding use of telehealth services for diagnosis, evaluation, and treatment of mental health disorders. CMS also is proposing to allow payment to eligible clinicians for certain mental health and behavioral health services to patients via audio-only telephone calls. These services would include counseling and therapy services provided through opioid treatment programs.
“These changes would be particularly helpful for those in areas with poor broadband infrastructure and among people with Medicare who are not capable of, or do not consent to the use of, devices that permit a two-way, audio/video interaction for their health care visits,” CMS said in a statement.
Slimmer Medicare enrollees, bigger payments for coaches?
CMS is seeking to draw more participants to the Medicare Diabetes Prevention Program (MDPP). This program includes organizations that provide structured, coach-led sessions in community and health care settings to help people lose weight and exercise more. During the COVID-19 public health emergency, CMS waived an enrollment fee for new suppliers of services in MDPP. CMS now is proposing to waive this fee for all organizations that submit an application to enroll in Medicare as an MDPP supplier on or after Jan. 1, 2022.
Another proposed change in MDPP services is a restructuring of payments so that organizations involved in coaching would receive larger payments when their participants reach milestones for attendance and for becoming slimmer.
“We propose to increase performance payments for MDPP beneficiary achievement of the 5% weight-loss goal, as well as continued attendance during each core maintenance interval,” CMS said in a statement.
Medicare remains engaged in a review of its payments for E/M services. In the draft rule, CMS is proposing a number of refinements to current policies for split, or shared, E/M visits, critical care services, and services furnished by teaching physicians involving residents. The intention of these changes is to “better reflect the current practice of medicine, the evolving role of nonphysician practitioners as members of the medical team, and to clarify conditions of payment that must be met to bill Medicare for these services,” CMS said.
A version of this article first appeared on Medscape.com.
It also intends to change the approach to payments for office visits and for coaching programs for diabetes prevention.
The Centers for Medicare & Medicaid Services recently posted its proposed 2022 physician fee schedule. Running to more than 1,700 pages, the draft rule contains myriad other changes in how the giant federal health program pays for medical care, including revisions to its approach to evaluation and management (E/M) services, which represent many office visits. In addition, Medicare is seeking to increase participation in a program intended to prevent people from developing diabetes.
Physician groups posted quick complaints about a proposed 3.75% reduction to the conversion factor because of budget neutrality requirements. The cut reinstates a reduction Congress prevented in late 2020.
In a statement, Anders Gilberg, senior vice president of government affairs for the Medical Group Management Association, called the draft rule a “mixed bag for physician practices.” Mr. Gilberg said the MGMA will seek congressional intervention to avert the cut for services in 2022.
In keeping with a provision Congress included in a massive spending bill enacted in December, Medicare will let PAs directly bill, as nurse practitioners already can. In a press release, CMS on July 13 described this as a move likely to expand access to care and reduce administrative burden. In 2020, the American Academy of PAs praised the inclusion in the spending bill of the provision allowing its members to directly bill Medicare.
In the draft rule, CMS also intends to remove certain geographic restrictions regarding use of telehealth services for diagnosis, evaluation, and treatment of mental health disorders. CMS also is proposing to allow payment to eligible clinicians for certain mental health and behavioral health services to patients via audio-only telephone calls. These services would include counseling and therapy services provided through opioid treatment programs.
“These changes would be particularly helpful for those in areas with poor broadband infrastructure and among people with Medicare who are not capable of, or do not consent to the use of, devices that permit a two-way, audio/video interaction for their health care visits,” CMS said in a statement.
Slimmer Medicare enrollees, bigger payments for coaches?
CMS is seeking to draw more participants to the Medicare Diabetes Prevention Program (MDPP). This program includes organizations that provide structured, coach-led sessions in community and health care settings to help people lose weight and exercise more. During the COVID-19 public health emergency, CMS waived an enrollment fee for new suppliers of services in MDPP. CMS now is proposing to waive this fee for all organizations that submit an application to enroll in Medicare as an MDPP supplier on or after Jan. 1, 2022.
Another proposed change in MDPP services is a restructuring of payments so that organizations involved in coaching would receive larger payments when their participants reach milestones for attendance and for becoming slimmer.
“We propose to increase performance payments for MDPP beneficiary achievement of the 5% weight-loss goal, as well as continued attendance during each core maintenance interval,” CMS said in a statement.
Medicare remains engaged in a review of its payments for E/M services. In the draft rule, CMS is proposing a number of refinements to current policies for split, or shared, E/M visits, critical care services, and services furnished by teaching physicians involving residents. The intention of these changes is to “better reflect the current practice of medicine, the evolving role of nonphysician practitioners as members of the medical team, and to clarify conditions of payment that must be met to bill Medicare for these services,” CMS said.
A version of this article first appeared on Medscape.com.
Proposed classification framework for atopic dermatitis unveiled
The heterogeneous clinical course of atopic dermatitis (AD) and its differing signs, symptoms, burden, and response to treatment can pose a quandary for physicians.

This is behind facilitate tailoring of therapy to individual patient characteristics, and better identify therapeutically relevant disease subsets.
Dr. Silverberg, director of clinical research in the department of dermatology at George Washington University, Washington, debuted DESCRIBE-AD during the Revolutionizing Atopic Dermatitis symposium. The “D” in the mnemonic stands for dermatitis morphology and phenotype, the “E” for evolution of disease, the “S” for symptom severity, the “C” for comorbid health disorders, the “R” for response to therapy, the “I” for intensity of lesions, the “B” for burden of disease, and the “E” for extent of lesions.
At the meeting, he discussed the concepts behind each letter of the mnemonic.
Dermatitis morphology and phenotype
In the dermatitis morphology and phenotype component of DESCRIBE-AD, “there’s a lot to consider,” he said. “There are chronic signs like lichenification and prurigo nodules, which have treatment ramifications,” such as the length of time patients may need to be treated, and possibly “the use of more potent, targeted options to go after some of these lesions.”
Recent studies suggest that nummular lesions have a different underlying pathogenesis suggesting an overlap between Th2 and Th17 cell–mediated lesions. “How does that impact response to targeted therapies?” he asked. “We have no idea. We need to learn that.” He noted that psoriasiform lesions are not limited to Asian patients, but also appear in elderly patients with AD. “They look different [in elderly patients] and they may respond differently; they have more psoriasiform lesions and it’s not exactly clear why.”
Other morphologic variants of AD to be aware of include follicular eczema, xerosis, and the itch-dominant form, which Dr. Silverberg and colleagues addressed in a recently published study. “There are some patients who have milder-looking lesions, but their itch is just out of control,” he said. “This is a pattern that we need to recognize.”
Evolution of disease, symptom severity
Factors to consider for the evolution of disease component of the proposed classification include age of AD onset or disease recurrence, frequency and duration of flares, disease activity between flares, periods of disease clearance, and the overall disease trajectory. “We do get patients who say that every year their disease seems to get worse over time, for reasons that are not always clear,” Dr. Silverberg said.
Assessment tools he recommends for the symptom severity component of DESCRIBE-AD include the patient-reported global AD severity, numerical rating scale (NRS) worst or average itch in the past 7 days, the Skin Pain NRS, and the Sleep Quality NRS, which each take fewer than 30 seconds to complete. “You can have your nurses do this or can you have the patients fill out the form in the waiting area before they see you,” Dr. Silverberg said.
He also advises asking patients about the number of nights they experience sleep disturbance and if they have difficulty falling asleep or have nighttime awakenings because of their AD. Symptoms of anxiety and depression can be assessed with the Hospital Anxiety and Depression Scale and the Patient-Health Questionnaire–9, which each take 2-3 minutes to complete.
Recommended assessment tools for other symptoms – such as bleeding, oozing, and xerosis – include the Patient-Oriented Eczema Measure, which takes 2-3 minutes to complete, and the Atopic Dermatitis Control Tool or the Recap of Atopic Eczema, which each take 2-3 minutes to complete.
Comorbid health disorders
Comorbid health disorders linked to AD are varied and include atopic comorbidities such as asthma or wheeze, hay fever or oculonasal symptoms, food allergy, recurrent infections such as herpes simplex virus, mental health disorders, alopecia areata, Th1-mediated comorbidities, and adverse events to medication such as venous thromboembolism, hypertension, and impaired renal or liver function. “All of these are important because if the patients have these at baseline, they may not be good candidates for some therapies that cause these types of side effects,” Dr. Silverberg said.
Response to therapy, intensity of lesions
As for response to therapy, clinicians can ask patients, “How do you feel you’re improving?” But it’s also important to assess the signs, symptoms, frequency of flares, and comorbidities as part of that response to therapy, “and of course the adverse events and treatment burden,” he said.
For the intensity of lesions component of DESCRIBE-AD, Dr. Silverberg said that the Investigator’s Global Assessment–AD is an effective tool for clinical use. “You can also use tools like the Eczema Area and Severity Index or the Scoring AD, but recognize these are challenging,” and can be difficult to use if not well trained to use them, he said. “At the very least, do an Investigator’s Global Assessment and do a body surface area measurement.”
In his opinion, four key signs that should be assessed in clinical trials are erythema, edema/papulation, excoriation, and lichenification/prurigo nodules.
Burden of disease
In terms of assessing AD disease burden, guidelines from the American Academy of Dermatology don’t give a specific tool to use, but recommend asking open-ended questions, Dr. Silverberg said. “I would not recommend that, because when you ask an open-ended question, the flood gates open up because most patients are suffering miserably with this disease when it’s uncontrolled.
“That’s why it’s valuable to use structured, validated tools like the Dermatology Life Quality Index and the Patient Reported Outcome Measurement Information System. They don’t take a lot of time to complete, and you can look at the score and determine how burdensome their disease is, even in a busy clinical practice. They’re not going to slow you down; they’re going to speed you up and make you better at your therapeutic decision-making. I can guarantee you that most patients will love you for it. Sometimes patients say to me, ‘you’re the first doctor to ask these questions.’ ”
Extent of disease
Finally, for the extent of disease component of DESCRIBE-AD, he emphasized the importance of doing a full-body exam to appreciate the affected body surface area, flexural versus extensor distribution, and involvement and severity of disease on special sites such as the face, hands, feet, genitals, and scalp.
Dr. Silverberg reported that he is a consultant to and/or an advisory board member for several pharmaceutical companies. He is also a speaker for Regeneron and Sanofi and has received a grant from Galderma.
The heterogeneous clinical course of atopic dermatitis (AD) and its differing signs, symptoms, burden, and response to treatment can pose a quandary for physicians.
This is behind facilitate tailoring of therapy to individual patient characteristics, and better identify therapeutically relevant disease subsets.
Dr. Silverberg, director of clinical research in the department of dermatology at George Washington University, Washington, debuted DESCRIBE-AD during the Revolutionizing Atopic Dermatitis symposium. The “D” in the mnemonic stands for dermatitis morphology and phenotype, the “E” for evolution of disease, the “S” for symptom severity, the “C” for comorbid health disorders, the “R” for response to therapy, the “I” for intensity of lesions, the “B” for burden of disease, and the “E” for extent of lesions.
At the meeting, he discussed the concepts behind each letter of the mnemonic.
Dermatitis morphology and phenotype
In the dermatitis morphology and phenotype component of DESCRIBE-AD, “there’s a lot to consider,” he said. “There are chronic signs like lichenification and prurigo nodules, which have treatment ramifications,” such as the length of time patients may need to be treated, and possibly “the use of more potent, targeted options to go after some of these lesions.”
Recent studies suggest that nummular lesions have a different underlying pathogenesis suggesting an overlap between Th2 and Th17 cell–mediated lesions. “How does that impact response to targeted therapies?” he asked. “We have no idea. We need to learn that.” He noted that psoriasiform lesions are not limited to Asian patients, but also appear in elderly patients with AD. “They look different [in elderly patients] and they may respond differently; they have more psoriasiform lesions and it’s not exactly clear why.”
Other morphologic variants of AD to be aware of include follicular eczema, xerosis, and the itch-dominant form, which Dr. Silverberg and colleagues addressed in a recently published study. “There are some patients who have milder-looking lesions, but their itch is just out of control,” he said. “This is a pattern that we need to recognize.”
Evolution of disease, symptom severity
Factors to consider for the evolution of disease component of the proposed classification include age of AD onset or disease recurrence, frequency and duration of flares, disease activity between flares, periods of disease clearance, and the overall disease trajectory. “We do get patients who say that every year their disease seems to get worse over time, for reasons that are not always clear,” Dr. Silverberg said.
Assessment tools he recommends for the symptom severity component of DESCRIBE-AD include the patient-reported global AD severity, numerical rating scale (NRS) worst or average itch in the past 7 days, the Skin Pain NRS, and the Sleep Quality NRS, which each take fewer than 30 seconds to complete. “You can have your nurses do this or can you have the patients fill out the form in the waiting area before they see you,” Dr. Silverberg said.
He also advises asking patients about the number of nights they experience sleep disturbance and if they have difficulty falling asleep or have nighttime awakenings because of their AD. Symptoms of anxiety and depression can be assessed with the Hospital Anxiety and Depression Scale and the Patient-Health Questionnaire–9, which each take 2-3 minutes to complete.
Recommended assessment tools for other symptoms – such as bleeding, oozing, and xerosis – include the Patient-Oriented Eczema Measure, which takes 2-3 minutes to complete, and the Atopic Dermatitis Control Tool or the Recap of Atopic Eczema, which each take 2-3 minutes to complete.
Comorbid health disorders
Comorbid health disorders linked to AD are varied and include atopic comorbidities such as asthma or wheeze, hay fever or oculonasal symptoms, food allergy, recurrent infections such as herpes simplex virus, mental health disorders, alopecia areata, Th1-mediated comorbidities, and adverse events to medication such as venous thromboembolism, hypertension, and impaired renal or liver function. “All of these are important because if the patients have these at baseline, they may not be good candidates for some therapies that cause these types of side effects,” Dr. Silverberg said.
Response to therapy, intensity of lesions
As for response to therapy, clinicians can ask patients, “How do you feel you’re improving?” But it’s also important to assess the signs, symptoms, frequency of flares, and comorbidities as part of that response to therapy, “and of course the adverse events and treatment burden,” he said.
For the intensity of lesions component of DESCRIBE-AD, Dr. Silverberg said that the Investigator’s Global Assessment–AD is an effective tool for clinical use. “You can also use tools like the Eczema Area and Severity Index or the Scoring AD, but recognize these are challenging,” and can be difficult to use if not well trained to use them, he said. “At the very least, do an Investigator’s Global Assessment and do a body surface area measurement.”
In his opinion, four key signs that should be assessed in clinical trials are erythema, edema/papulation, excoriation, and lichenification/prurigo nodules.
Burden of disease
In terms of assessing AD disease burden, guidelines from the American Academy of Dermatology don’t give a specific tool to use, but recommend asking open-ended questions, Dr. Silverberg said. “I would not recommend that, because when you ask an open-ended question, the flood gates open up because most patients are suffering miserably with this disease when it’s uncontrolled.
“That’s why it’s valuable to use structured, validated tools like the Dermatology Life Quality Index and the Patient Reported Outcome Measurement Information System. They don’t take a lot of time to complete, and you can look at the score and determine how burdensome their disease is, even in a busy clinical practice. They’re not going to slow you down; they’re going to speed you up and make you better at your therapeutic decision-making. I can guarantee you that most patients will love you for it. Sometimes patients say to me, ‘you’re the first doctor to ask these questions.’ ”
Extent of disease
Finally, for the extent of disease component of DESCRIBE-AD, he emphasized the importance of doing a full-body exam to appreciate the affected body surface area, flexural versus extensor distribution, and involvement and severity of disease on special sites such as the face, hands, feet, genitals, and scalp.
Dr. Silverberg reported that he is a consultant to and/or an advisory board member for several pharmaceutical companies. He is also a speaker for Regeneron and Sanofi and has received a grant from Galderma.
The heterogeneous clinical course of atopic dermatitis (AD) and its differing signs, symptoms, burden, and response to treatment can pose a quandary for physicians.
This is behind facilitate tailoring of therapy to individual patient characteristics, and better identify therapeutically relevant disease subsets.
Dr. Silverberg, director of clinical research in the department of dermatology at George Washington University, Washington, debuted DESCRIBE-AD during the Revolutionizing Atopic Dermatitis symposium. The “D” in the mnemonic stands for dermatitis morphology and phenotype, the “E” for evolution of disease, the “S” for symptom severity, the “C” for comorbid health disorders, the “R” for response to therapy, the “I” for intensity of lesions, the “B” for burden of disease, and the “E” for extent of lesions.
At the meeting, he discussed the concepts behind each letter of the mnemonic.
Dermatitis morphology and phenotype
In the dermatitis morphology and phenotype component of DESCRIBE-AD, “there’s a lot to consider,” he said. “There are chronic signs like lichenification and prurigo nodules, which have treatment ramifications,” such as the length of time patients may need to be treated, and possibly “the use of more potent, targeted options to go after some of these lesions.”
Recent studies suggest that nummular lesions have a different underlying pathogenesis suggesting an overlap between Th2 and Th17 cell–mediated lesions. “How does that impact response to targeted therapies?” he asked. “We have no idea. We need to learn that.” He noted that psoriasiform lesions are not limited to Asian patients, but also appear in elderly patients with AD. “They look different [in elderly patients] and they may respond differently; they have more psoriasiform lesions and it’s not exactly clear why.”
Other morphologic variants of AD to be aware of include follicular eczema, xerosis, and the itch-dominant form, which Dr. Silverberg and colleagues addressed in a recently published study. “There are some patients who have milder-looking lesions, but their itch is just out of control,” he said. “This is a pattern that we need to recognize.”
Evolution of disease, symptom severity
Factors to consider for the evolution of disease component of the proposed classification include age of AD onset or disease recurrence, frequency and duration of flares, disease activity between flares, periods of disease clearance, and the overall disease trajectory. “We do get patients who say that every year their disease seems to get worse over time, for reasons that are not always clear,” Dr. Silverberg said.
Assessment tools he recommends for the symptom severity component of DESCRIBE-AD include the patient-reported global AD severity, numerical rating scale (NRS) worst or average itch in the past 7 days, the Skin Pain NRS, and the Sleep Quality NRS, which each take fewer than 30 seconds to complete. “You can have your nurses do this or can you have the patients fill out the form in the waiting area before they see you,” Dr. Silverberg said.
He also advises asking patients about the number of nights they experience sleep disturbance and if they have difficulty falling asleep or have nighttime awakenings because of their AD. Symptoms of anxiety and depression can be assessed with the Hospital Anxiety and Depression Scale and the Patient-Health Questionnaire–9, which each take 2-3 minutes to complete.
Recommended assessment tools for other symptoms – such as bleeding, oozing, and xerosis – include the Patient-Oriented Eczema Measure, which takes 2-3 minutes to complete, and the Atopic Dermatitis Control Tool or the Recap of Atopic Eczema, which each take 2-3 minutes to complete.
Comorbid health disorders
Comorbid health disorders linked to AD are varied and include atopic comorbidities such as asthma or wheeze, hay fever or oculonasal symptoms, food allergy, recurrent infections such as herpes simplex virus, mental health disorders, alopecia areata, Th1-mediated comorbidities, and adverse events to medication such as venous thromboembolism, hypertension, and impaired renal or liver function. “All of these are important because if the patients have these at baseline, they may not be good candidates for some therapies that cause these types of side effects,” Dr. Silverberg said.
Response to therapy, intensity of lesions
As for response to therapy, clinicians can ask patients, “How do you feel you’re improving?” But it’s also important to assess the signs, symptoms, frequency of flares, and comorbidities as part of that response to therapy, “and of course the adverse events and treatment burden,” he said.
For the intensity of lesions component of DESCRIBE-AD, Dr. Silverberg said that the Investigator’s Global Assessment–AD is an effective tool for clinical use. “You can also use tools like the Eczema Area and Severity Index or the Scoring AD, but recognize these are challenging,” and can be difficult to use if not well trained to use them, he said. “At the very least, do an Investigator’s Global Assessment and do a body surface area measurement.”
In his opinion, four key signs that should be assessed in clinical trials are erythema, edema/papulation, excoriation, and lichenification/prurigo nodules.
Burden of disease
In terms of assessing AD disease burden, guidelines from the American Academy of Dermatology don’t give a specific tool to use, but recommend asking open-ended questions, Dr. Silverberg said. “I would not recommend that, because when you ask an open-ended question, the flood gates open up because most patients are suffering miserably with this disease when it’s uncontrolled.
“That’s why it’s valuable to use structured, validated tools like the Dermatology Life Quality Index and the Patient Reported Outcome Measurement Information System. They don’t take a lot of time to complete, and you can look at the score and determine how burdensome their disease is, even in a busy clinical practice. They’re not going to slow you down; they’re going to speed you up and make you better at your therapeutic decision-making. I can guarantee you that most patients will love you for it. Sometimes patients say to me, ‘you’re the first doctor to ask these questions.’ ”
Extent of disease
Finally, for the extent of disease component of DESCRIBE-AD, he emphasized the importance of doing a full-body exam to appreciate the affected body surface area, flexural versus extensor distribution, and involvement and severity of disease on special sites such as the face, hands, feet, genitals, and scalp.
Dr. Silverberg reported that he is a consultant to and/or an advisory board member for several pharmaceutical companies. He is also a speaker for Regeneron and Sanofi and has received a grant from Galderma.
FROM REVOLUTIONIZING AD 2021