User login
Soft Tissue Sarcoma Chemotherapy
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Uptick in adult syphilis means congenital syphilis may be lurking
While many pediatric clinicians have not frequently managed newborns of mothers with reactive syphilis serology, increased adult syphilis may change that.1

Diagnosing/managing congenital syphilis is not always clear cut. A positive rapid plasma reagin (RPR) titer in a newborn may not indicate congenital infection but merely may reflect transplacental, passively acquired maternal IgG from the mother’s current or previous infection rather than antibodies produced by the newborn. Because currently no IgM assay for syphilis is recommended by the Centers for Disease Control and Prevention for newborn testing, we must deal with IgG test results.
Often initial management decisions are needed while the infant’s status is evolving. The questions to answer to make final decisions include the following2:
- Was the mother actively infected with Treponema pallidum during pregnancy?
- If so, was the mother appropriately treated and when?
- Does the infant have any clinical, laboratory, or radiographic evidence of syphilis?
- How do the mother’s and infant’s nontreponemal serologic titers (NTT) compare at delivery using the same test?
Note: All infants assessed for congenital syphilis need a full evaluation for HIV.
Managing the infant of a mother with positive tests3,4
All such neonates need an examination for evidence of congenital syphilis. The clinical signs of congenital syphilis in neonates include nonimmune hydrops, jaundice, hepatosplenomegaly, rhinitis, skin rash, and pseudoparalysis of extremity. Also, consider dark-field examination or polymerase chain reaction (PCR) of lesions (such as bullae) or secretions (nasal). If available, have the placenta examined histologically (silver stain) or by PCR (Clinical Laboratory Improvement Amendments–validated test). Skeletal radiographic surveys are more useful for stillborn than live born infants. (The complete algorithm can be found in Figure 3.10 of reference 4.)
Order a quantitative NTT, using the Venereal Disease Research Laboratory (VDRL) test or RPR test on neonatal serum. Umbilical cord blood is not appropriate because of potential maternal blood contamination, which could give a false-positive result, or Wharton’s jelly, which could give a false-negative result. Use of treponemal-specific tests that are used for maternal diagnosis – such as T. pallidum particle agglutination (TP-PA), T. pallidum enzyme-linked immunosorbent assay (TP-EIA), fluorescent treponemal antibody absorption (FTA-ABS) test, or T. pallidum chemiluminescence immunoassay (TP-CIA) – on neonatal serum is not recommended because of difficulties in interpretation.
Diagnostic results allow designation of an infant into one of four CDC categories: proven/highly probable syphilis; possible syphilis; syphilis less likely; and syphilis unlikely. Treatment recommendations are based on these categories.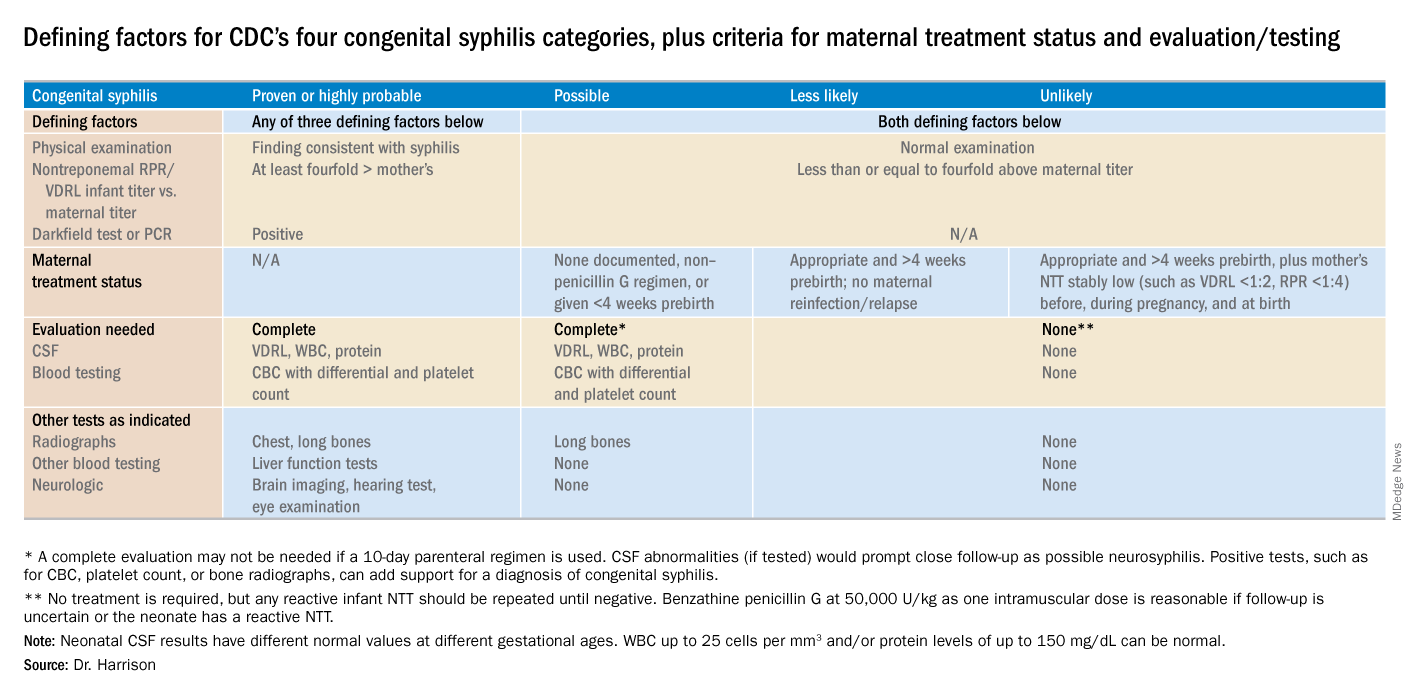
Proven or highly probable syphilis
There are two alternative recommended 10-day treatment regimens.
A. Aqueous crystalline penicillin G 100,000-150,000 U/kg per day by IV at 50,000 U/kg per dose, given every 12 hours through 7 days of age or every 8 hours if greater than 7 days old.
B. Procaine penicillin G at 50,000 U/kg per dose intramuscularly in one dose each day.
More than 1 day of missed therapy requires restarting a new 10-day course. Use of other antimicrobial agents (such as ampicillin) is not validated, so any empiric ampicillin initially given for possible sepsis does not count toward the 10-day penicillin regimen. If nonpenicillin drugs must be used, close serologic follow-up must occur to ensure adequacy of response to therapy.
Possible syphilis
There are three alternative regimens, the same two as in proven/highly probable syphilis (above) plus a single-dose option
A. Aqueous crystalline penicillin G, as described above.
B. Procaine penicillin G, as described above.
C. Benzathine penicillin G at 50,000 U/kg per dose intramuscularly in a single dose.
Note: To be eligible for regimen C, an infant must have a complete evaluation that is normal (cerebrospinal fluid [CSF] examination, long-bone radiographs, and complete blood count with platelet count) and follow-up must be assured. Exception: Neonates born to mothers with untreated early syphilis at the time of delivery are at increased risk for congenital syphilis, and the 10-day course of penicillin G may be considered even if the complete evaluation is normal and follow-up is certain.
Less likely syphilis
One antibiotic regimen is available, but no treatment also may be an option.
A. Benzathine penicillin G as described above.
B. If mother’s NTT has decreased at least fourfold after appropriate early syphilis therapy or remained stably low, which indicates latent syphilis (VDRL less than 1:2; RPR less than 1:4), no treatment is an option but requires repeat serology every 2-3 months until infant is 6 months old.
Unlikely syphilis
No treatment is recommended unless follow-up is uncertain, in which case it is appropriate to give the infant benzathine penicillin G as described above.
Infant with positive NTT at birth
All neonates with reactive NTT need careful follow-up examinations and repeat NTT every 2-3 months until nonreactive. NTT in infants who are not treated because of less likely or unlikely syphilis status should drop by 3 months and be nonreactive by 6 months; this indicates NTT was passively transferred maternal IgG. If NTT remains reactive at 6 months, the infant is likely infected and needs treatment. Persistent NTT at 6-12 months in treated neonates should trigger repeat CSF examination and infectious diseases consultation about a possible repeat of the 10-day penicillin G regimen. If the mother was seroreactive, but the newborn’s NTT was negative at birth, testing of the infant’s NTT needs repeating at 3 months to exclude the possibility that the congenital syphilis was incubating when prior testing occurred at birth. Note: Treponemal-specific tests are not useful in assessing treatment because detectable maternal IgG treponemal antibody can persist at least 15 months.
Neonates with abnormal CSF at birth
Repeat cerebrospinal fluid evaluation every 6 months until results normalize. Persistently reactive CSF VDRL or abnormal CSF indexes not caused by another known cause requires retreatment for possible neurosyphilis, as well as consultation with an expert.
Summary
NTT are the essential test for newborns and some degree of laboratory or imaging work up often are needed. Consider consulting an expert in infectious diseases and/or perinatology if the gray areas do not readily become clear. Treatment of the correct patients with the right drug for the right duration remains the goal, as usual.
Dr. Harrison is a professor of pediatrics at University of Missouri-Kansas City and Director of Research Affairs in the pediatric infectious diseases division at Children’s Mercy Hospital – Kansas City. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2015 Nov 13;64(44);1241-5.
2. “Congenital Syphilis,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
3. “Syphilis During Pregnancy,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
4. Syphilis – Section 3: Summaries of Infectious Diseases. Red Book Online. 2018.
While many pediatric clinicians have not frequently managed newborns of mothers with reactive syphilis serology, increased adult syphilis may change that.1
Diagnosing/managing congenital syphilis is not always clear cut. A positive rapid plasma reagin (RPR) titer in a newborn may not indicate congenital infection but merely may reflect transplacental, passively acquired maternal IgG from the mother’s current or previous infection rather than antibodies produced by the newborn. Because currently no IgM assay for syphilis is recommended by the Centers for Disease Control and Prevention for newborn testing, we must deal with IgG test results.
Often initial management decisions are needed while the infant’s status is evolving. The questions to answer to make final decisions include the following2:
- Was the mother actively infected with Treponema pallidum during pregnancy?
- If so, was the mother appropriately treated and when?
- Does the infant have any clinical, laboratory, or radiographic evidence of syphilis?
- How do the mother’s and infant’s nontreponemal serologic titers (NTT) compare at delivery using the same test?
Note: All infants assessed for congenital syphilis need a full evaluation for HIV.
Managing the infant of a mother with positive tests3,4
All such neonates need an examination for evidence of congenital syphilis. The clinical signs of congenital syphilis in neonates include nonimmune hydrops, jaundice, hepatosplenomegaly, rhinitis, skin rash, and pseudoparalysis of extremity. Also, consider dark-field examination or polymerase chain reaction (PCR) of lesions (such as bullae) or secretions (nasal). If available, have the placenta examined histologically (silver stain) or by PCR (Clinical Laboratory Improvement Amendments–validated test). Skeletal radiographic surveys are more useful for stillborn than live born infants. (The complete algorithm can be found in Figure 3.10 of reference 4.)
Order a quantitative NTT, using the Venereal Disease Research Laboratory (VDRL) test or RPR test on neonatal serum. Umbilical cord blood is not appropriate because of potential maternal blood contamination, which could give a false-positive result, or Wharton’s jelly, which could give a false-negative result. Use of treponemal-specific tests that are used for maternal diagnosis – such as T. pallidum particle agglutination (TP-PA), T. pallidum enzyme-linked immunosorbent assay (TP-EIA), fluorescent treponemal antibody absorption (FTA-ABS) test, or T. pallidum chemiluminescence immunoassay (TP-CIA) – on neonatal serum is not recommended because of difficulties in interpretation.
Diagnostic results allow designation of an infant into one of four CDC categories: proven/highly probable syphilis; possible syphilis; syphilis less likely; and syphilis unlikely. Treatment recommendations are based on these categories.
Proven or highly probable syphilis
There are two alternative recommended 10-day treatment regimens.
A. Aqueous crystalline penicillin G 100,000-150,000 U/kg per day by IV at 50,000 U/kg per dose, given every 12 hours through 7 days of age or every 8 hours if greater than 7 days old.
B. Procaine penicillin G at 50,000 U/kg per dose intramuscularly in one dose each day.
More than 1 day of missed therapy requires restarting a new 10-day course. Use of other antimicrobial agents (such as ampicillin) is not validated, so any empiric ampicillin initially given for possible sepsis does not count toward the 10-day penicillin regimen. If nonpenicillin drugs must be used, close serologic follow-up must occur to ensure adequacy of response to therapy.
Possible syphilis
There are three alternative regimens, the same two as in proven/highly probable syphilis (above) plus a single-dose option
A. Aqueous crystalline penicillin G, as described above.
B. Procaine penicillin G, as described above.
C. Benzathine penicillin G at 50,000 U/kg per dose intramuscularly in a single dose.
Note: To be eligible for regimen C, an infant must have a complete evaluation that is normal (cerebrospinal fluid [CSF] examination, long-bone radiographs, and complete blood count with platelet count) and follow-up must be assured. Exception: Neonates born to mothers with untreated early syphilis at the time of delivery are at increased risk for congenital syphilis, and the 10-day course of penicillin G may be considered even if the complete evaluation is normal and follow-up is certain.
Less likely syphilis
One antibiotic regimen is available, but no treatment also may be an option.
A. Benzathine penicillin G as described above.
B. If mother’s NTT has decreased at least fourfold after appropriate early syphilis therapy or remained stably low, which indicates latent syphilis (VDRL less than 1:2; RPR less than 1:4), no treatment is an option but requires repeat serology every 2-3 months until infant is 6 months old.
Unlikely syphilis
No treatment is recommended unless follow-up is uncertain, in which case it is appropriate to give the infant benzathine penicillin G as described above.
Infant with positive NTT at birth
All neonates with reactive NTT need careful follow-up examinations and repeat NTT every 2-3 months until nonreactive. NTT in infants who are not treated because of less likely or unlikely syphilis status should drop by 3 months and be nonreactive by 6 months; this indicates NTT was passively transferred maternal IgG. If NTT remains reactive at 6 months, the infant is likely infected and needs treatment. Persistent NTT at 6-12 months in treated neonates should trigger repeat CSF examination and infectious diseases consultation about a possible repeat of the 10-day penicillin G regimen. If the mother was seroreactive, but the newborn’s NTT was negative at birth, testing of the infant’s NTT needs repeating at 3 months to exclude the possibility that the congenital syphilis was incubating when prior testing occurred at birth. Note: Treponemal-specific tests are not useful in assessing treatment because detectable maternal IgG treponemal antibody can persist at least 15 months.
Neonates with abnormal CSF at birth
Repeat cerebrospinal fluid evaluation every 6 months until results normalize. Persistently reactive CSF VDRL or abnormal CSF indexes not caused by another known cause requires retreatment for possible neurosyphilis, as well as consultation with an expert.
Summary
NTT are the essential test for newborns and some degree of laboratory or imaging work up often are needed. Consider consulting an expert in infectious diseases and/or perinatology if the gray areas do not readily become clear. Treatment of the correct patients with the right drug for the right duration remains the goal, as usual.
Dr. Harrison is a professor of pediatrics at University of Missouri-Kansas City and Director of Research Affairs in the pediatric infectious diseases division at Children’s Mercy Hospital – Kansas City. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2015 Nov 13;64(44);1241-5.
2. “Congenital Syphilis,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
3. “Syphilis During Pregnancy,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
4. Syphilis – Section 3: Summaries of Infectious Diseases. Red Book Online. 2018.
While many pediatric clinicians have not frequently managed newborns of mothers with reactive syphilis serology, increased adult syphilis may change that.1
Diagnosing/managing congenital syphilis is not always clear cut. A positive rapid plasma reagin (RPR) titer in a newborn may not indicate congenital infection but merely may reflect transplacental, passively acquired maternal IgG from the mother’s current or previous infection rather than antibodies produced by the newborn. Because currently no IgM assay for syphilis is recommended by the Centers for Disease Control and Prevention for newborn testing, we must deal with IgG test results.
Often initial management decisions are needed while the infant’s status is evolving. The questions to answer to make final decisions include the following2:
- Was the mother actively infected with Treponema pallidum during pregnancy?
- If so, was the mother appropriately treated and when?
- Does the infant have any clinical, laboratory, or radiographic evidence of syphilis?
- How do the mother’s and infant’s nontreponemal serologic titers (NTT) compare at delivery using the same test?
Note: All infants assessed for congenital syphilis need a full evaluation for HIV.
Managing the infant of a mother with positive tests3,4
All such neonates need an examination for evidence of congenital syphilis. The clinical signs of congenital syphilis in neonates include nonimmune hydrops, jaundice, hepatosplenomegaly, rhinitis, skin rash, and pseudoparalysis of extremity. Also, consider dark-field examination or polymerase chain reaction (PCR) of lesions (such as bullae) or secretions (nasal). If available, have the placenta examined histologically (silver stain) or by PCR (Clinical Laboratory Improvement Amendments–validated test). Skeletal radiographic surveys are more useful for stillborn than live born infants. (The complete algorithm can be found in Figure 3.10 of reference 4.)
Order a quantitative NTT, using the Venereal Disease Research Laboratory (VDRL) test or RPR test on neonatal serum. Umbilical cord blood is not appropriate because of potential maternal blood contamination, which could give a false-positive result, or Wharton’s jelly, which could give a false-negative result. Use of treponemal-specific tests that are used for maternal diagnosis – such as T. pallidum particle agglutination (TP-PA), T. pallidum enzyme-linked immunosorbent assay (TP-EIA), fluorescent treponemal antibody absorption (FTA-ABS) test, or T. pallidum chemiluminescence immunoassay (TP-CIA) – on neonatal serum is not recommended because of difficulties in interpretation.
Diagnostic results allow designation of an infant into one of four CDC categories: proven/highly probable syphilis; possible syphilis; syphilis less likely; and syphilis unlikely. Treatment recommendations are based on these categories.
Proven or highly probable syphilis
There are two alternative recommended 10-day treatment regimens.
A. Aqueous crystalline penicillin G 100,000-150,000 U/kg per day by IV at 50,000 U/kg per dose, given every 12 hours through 7 days of age or every 8 hours if greater than 7 days old.
B. Procaine penicillin G at 50,000 U/kg per dose intramuscularly in one dose each day.
More than 1 day of missed therapy requires restarting a new 10-day course. Use of other antimicrobial agents (such as ampicillin) is not validated, so any empiric ampicillin initially given for possible sepsis does not count toward the 10-day penicillin regimen. If nonpenicillin drugs must be used, close serologic follow-up must occur to ensure adequacy of response to therapy.
Possible syphilis
There are three alternative regimens, the same two as in proven/highly probable syphilis (above) plus a single-dose option
A. Aqueous crystalline penicillin G, as described above.
B. Procaine penicillin G, as described above.
C. Benzathine penicillin G at 50,000 U/kg per dose intramuscularly in a single dose.
Note: To be eligible for regimen C, an infant must have a complete evaluation that is normal (cerebrospinal fluid [CSF] examination, long-bone radiographs, and complete blood count with platelet count) and follow-up must be assured. Exception: Neonates born to mothers with untreated early syphilis at the time of delivery are at increased risk for congenital syphilis, and the 10-day course of penicillin G may be considered even if the complete evaluation is normal and follow-up is certain.
Less likely syphilis
One antibiotic regimen is available, but no treatment also may be an option.
A. Benzathine penicillin G as described above.
B. If mother’s NTT has decreased at least fourfold after appropriate early syphilis therapy or remained stably low, which indicates latent syphilis (VDRL less than 1:2; RPR less than 1:4), no treatment is an option but requires repeat serology every 2-3 months until infant is 6 months old.
Unlikely syphilis
No treatment is recommended unless follow-up is uncertain, in which case it is appropriate to give the infant benzathine penicillin G as described above.
Infant with positive NTT at birth
All neonates with reactive NTT need careful follow-up examinations and repeat NTT every 2-3 months until nonreactive. NTT in infants who are not treated because of less likely or unlikely syphilis status should drop by 3 months and be nonreactive by 6 months; this indicates NTT was passively transferred maternal IgG. If NTT remains reactive at 6 months, the infant is likely infected and needs treatment. Persistent NTT at 6-12 months in treated neonates should trigger repeat CSF examination and infectious diseases consultation about a possible repeat of the 10-day penicillin G regimen. If the mother was seroreactive, but the newborn’s NTT was negative at birth, testing of the infant’s NTT needs repeating at 3 months to exclude the possibility that the congenital syphilis was incubating when prior testing occurred at birth. Note: Treponemal-specific tests are not useful in assessing treatment because detectable maternal IgG treponemal antibody can persist at least 15 months.
Neonates with abnormal CSF at birth
Repeat cerebrospinal fluid evaluation every 6 months until results normalize. Persistently reactive CSF VDRL or abnormal CSF indexes not caused by another known cause requires retreatment for possible neurosyphilis, as well as consultation with an expert.
Summary
NTT are the essential test for newborns and some degree of laboratory or imaging work up often are needed. Consider consulting an expert in infectious diseases and/or perinatology if the gray areas do not readily become clear. Treatment of the correct patients with the right drug for the right duration remains the goal, as usual.
Dr. Harrison is a professor of pediatrics at University of Missouri-Kansas City and Director of Research Affairs in the pediatric infectious diseases division at Children’s Mercy Hospital – Kansas City. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2015 Nov 13;64(44);1241-5.
2. “Congenital Syphilis,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
3. “Syphilis During Pregnancy,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
4. Syphilis – Section 3: Summaries of Infectious Diseases. Red Book Online. 2018.
Launching an HIV testing reminder
Trying a new tool to reduce infection rates
The world’s largest gay dating app, Grindr, changed its software earlier this year to create reminders for users to get regular HIV tests.
According to Grindr, 3.3 million users around the world visit the site daily; it sends those who opt into the service a reminder every 3-6 months to get a test. The message also directs them to the nearest testing site. Grindr also plans to give clinics, gay community centers, and other testing sites free advertising.
Among health care providers, the decision has been widely applauded. “This will ‘demedicalize’ testing and destigmatize it,” Perry N. Halkitis, PhD, dean of the Rutgers School of Public Health, in Newark, N.J., told the New York Times. “The more you make it normal, the more people are going to access it.”
Studies have shown that reminders by text or phone can triple or quadruple the chance that the recipient will get tested.
Reference
McNeil Jr. DG. Grindr App to Offer H.I.V. Test Reminders. The New York Times. March 26, 2018. Accessed April 5, 2018.
Trying a new tool to reduce infection rates
Trying a new tool to reduce infection rates
The world’s largest gay dating app, Grindr, changed its software earlier this year to create reminders for users to get regular HIV tests.
According to Grindr, 3.3 million users around the world visit the site daily; it sends those who opt into the service a reminder every 3-6 months to get a test. The message also directs them to the nearest testing site. Grindr also plans to give clinics, gay community centers, and other testing sites free advertising.
Among health care providers, the decision has been widely applauded. “This will ‘demedicalize’ testing and destigmatize it,” Perry N. Halkitis, PhD, dean of the Rutgers School of Public Health, in Newark, N.J., told the New York Times. “The more you make it normal, the more people are going to access it.”
Studies have shown that reminders by text or phone can triple or quadruple the chance that the recipient will get tested.
Reference
McNeil Jr. DG. Grindr App to Offer H.I.V. Test Reminders. The New York Times. March 26, 2018. Accessed April 5, 2018.
The world’s largest gay dating app, Grindr, changed its software earlier this year to create reminders for users to get regular HIV tests.
According to Grindr, 3.3 million users around the world visit the site daily; it sends those who opt into the service a reminder every 3-6 months to get a test. The message also directs them to the nearest testing site. Grindr also plans to give clinics, gay community centers, and other testing sites free advertising.
Among health care providers, the decision has been widely applauded. “This will ‘demedicalize’ testing and destigmatize it,” Perry N. Halkitis, PhD, dean of the Rutgers School of Public Health, in Newark, N.J., told the New York Times. “The more you make it normal, the more people are going to access it.”
Studies have shown that reminders by text or phone can triple or quadruple the chance that the recipient will get tested.
Reference
McNeil Jr. DG. Grindr App to Offer H.I.V. Test Reminders. The New York Times. March 26, 2018. Accessed April 5, 2018.
More rheumatology slots available, filled on Specialty Match Day, but numbers well below need
Although more fellowships slots in rheumatology were available and filled during the Specialty Match Day for the 2019 appointment year, the numbers are still well below what will be needed to fill the physician shortfall in this area.

For the 2019 appointment year, there were 236 fellowship positions available, with 233 of them filled, up from 221 positions available and 218 filled on the previous year’s Specialty Match Day, according to results released by the National Resident Matching Program.
Adding 10 or even 20 a year “is still not going to make a dent” in the expected rheumatologist shortfall, Beth Jonas, MD, director of the Rheumatology Fellowship Training Program at the University of North Carolina, Chapel Hill, said in an interview.
That being said, there are still positive messages to be taken away from the Specialty Match Day results.
“The rheumatology match was great this year,” Dr. Jonas said. “From the perspective of the quality of the applicant pool, it was very strong. The interest in rheumatology is very high. From the perspective of program directors, we had a lot of excellent applicants to interview and to choose from.”
She noted that in total there were 358 applicants who listed rheumatology as their preference for the 2019 appointment year, up from 313. Dr. Jonas also serves as chair of the American College of Rheumatology’s Committee on Rheumatology Training and Workforce Issues.
“The message here is that there are a lot more people who want to become rheumatologists than there are slots to train them,” she stated.
And the key reason more people aren’t being trained is money.
“There is capacity to train more people, but the challenge is funding those slots,” Dr. Jonas said, noting that the ACR is doing what it can to help fund fellowship slots, and the Arthritis Foundation has a new grant mechanism that has helped to fund new slots.
“But I think there needs to be other ways.”
She suggested that there should be more involvement from the federal government, perhaps through Medicare, and more involvement from local institutions.
Mona Singer, president and CEO of the National Resident Matching Program, highlighted rheumatology as a standout among the overall specialty matching programs for the 2019 program year.
“The specialty that has become more popular over the years is rheumatology, which I find interesting,” Ms. Singer said. “This isn’t the first year it has done well, but it has been on an upward trend.”
Overall, the programs that have been doing well, matching at a rate of 95% or better, have continued to see those high fill rates: allergy/immunology, cardiology, gastroenterology, hematology, oncology, pulmonary and critical care medicine, and endocrinology.
For the 2019 appointment year, there were a total of 5,881 applicants for 5,215 program slots, with 4,579 positions filled. The fill rate of 87.8% is unchanged from the previous year, when 5,491 applicants competed for 4,831 positions, with 4,242 slots filled.
Although more fellowships slots in rheumatology were available and filled during the Specialty Match Day for the 2019 appointment year, the numbers are still well below what will be needed to fill the physician shortfall in this area.
For the 2019 appointment year, there were 236 fellowship positions available, with 233 of them filled, up from 221 positions available and 218 filled on the previous year’s Specialty Match Day, according to results released by the National Resident Matching Program.
Adding 10 or even 20 a year “is still not going to make a dent” in the expected rheumatologist shortfall, Beth Jonas, MD, director of the Rheumatology Fellowship Training Program at the University of North Carolina, Chapel Hill, said in an interview.
That being said, there are still positive messages to be taken away from the Specialty Match Day results.
“The rheumatology match was great this year,” Dr. Jonas said. “From the perspective of the quality of the applicant pool, it was very strong. The interest in rheumatology is very high. From the perspective of program directors, we had a lot of excellent applicants to interview and to choose from.”
She noted that in total there were 358 applicants who listed rheumatology as their preference for the 2019 appointment year, up from 313. Dr. Jonas also serves as chair of the American College of Rheumatology’s Committee on Rheumatology Training and Workforce Issues.
“The message here is that there are a lot more people who want to become rheumatologists than there are slots to train them,” she stated.
And the key reason more people aren’t being trained is money.
“There is capacity to train more people, but the challenge is funding those slots,” Dr. Jonas said, noting that the ACR is doing what it can to help fund fellowship slots, and the Arthritis Foundation has a new grant mechanism that has helped to fund new slots.
“But I think there needs to be other ways.”
She suggested that there should be more involvement from the federal government, perhaps through Medicare, and more involvement from local institutions.
Mona Singer, president and CEO of the National Resident Matching Program, highlighted rheumatology as a standout among the overall specialty matching programs for the 2019 program year.
“The specialty that has become more popular over the years is rheumatology, which I find interesting,” Ms. Singer said. “This isn’t the first year it has done well, but it has been on an upward trend.”
Overall, the programs that have been doing well, matching at a rate of 95% or better, have continued to see those high fill rates: allergy/immunology, cardiology, gastroenterology, hematology, oncology, pulmonary and critical care medicine, and endocrinology.
For the 2019 appointment year, there were a total of 5,881 applicants for 5,215 program slots, with 4,579 positions filled. The fill rate of 87.8% is unchanged from the previous year, when 5,491 applicants competed for 4,831 positions, with 4,242 slots filled.
Although more fellowships slots in rheumatology were available and filled during the Specialty Match Day for the 2019 appointment year, the numbers are still well below what will be needed to fill the physician shortfall in this area.
For the 2019 appointment year, there were 236 fellowship positions available, with 233 of them filled, up from 221 positions available and 218 filled on the previous year’s Specialty Match Day, according to results released by the National Resident Matching Program.
Adding 10 or even 20 a year “is still not going to make a dent” in the expected rheumatologist shortfall, Beth Jonas, MD, director of the Rheumatology Fellowship Training Program at the University of North Carolina, Chapel Hill, said in an interview.
That being said, there are still positive messages to be taken away from the Specialty Match Day results.
“The rheumatology match was great this year,” Dr. Jonas said. “From the perspective of the quality of the applicant pool, it was very strong. The interest in rheumatology is very high. From the perspective of program directors, we had a lot of excellent applicants to interview and to choose from.”
She noted that in total there were 358 applicants who listed rheumatology as their preference for the 2019 appointment year, up from 313. Dr. Jonas also serves as chair of the American College of Rheumatology’s Committee on Rheumatology Training and Workforce Issues.
“The message here is that there are a lot more people who want to become rheumatologists than there are slots to train them,” she stated.
And the key reason more people aren’t being trained is money.
“There is capacity to train more people, but the challenge is funding those slots,” Dr. Jonas said, noting that the ACR is doing what it can to help fund fellowship slots, and the Arthritis Foundation has a new grant mechanism that has helped to fund new slots.
“But I think there needs to be other ways.”
She suggested that there should be more involvement from the federal government, perhaps through Medicare, and more involvement from local institutions.
Mona Singer, president and CEO of the National Resident Matching Program, highlighted rheumatology as a standout among the overall specialty matching programs for the 2019 program year.
“The specialty that has become more popular over the years is rheumatology, which I find interesting,” Ms. Singer said. “This isn’t the first year it has done well, but it has been on an upward trend.”
Overall, the programs that have been doing well, matching at a rate of 95% or better, have continued to see those high fill rates: allergy/immunology, cardiology, gastroenterology, hematology, oncology, pulmonary and critical care medicine, and endocrinology.
For the 2019 appointment year, there were a total of 5,881 applicants for 5,215 program slots, with 4,579 positions filled. The fill rate of 87.8% is unchanged from the previous year, when 5,491 applicants competed for 4,831 positions, with 4,242 slots filled.
Duodenoscopes contain more bacteria than expected
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.

“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
The AGA Center for GI Innovation and Technology supports innovation and the development of new technology in gastroenterology, hepatology, nutrition and obesity by guiding medical device and therapeutics innovators through the technology development and adoption process. Learn more at www.gastro.org/CGIT
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.
“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
The AGA Center for GI Innovation and Technology supports innovation and the development of new technology in gastroenterology, hepatology, nutrition and obesity by guiding medical device and therapeutics innovators through the technology development and adoption process. Learn more at www.gastro.org/CGIT
Reprocessed duodenoscopes are more contaminated than expected, with up to 3% of samples testing positive for disease-causing bacteria including Escherichia coli and Staphylococcus aureus, according to an updated safety communication issued by the Food and Drug Administration on December 10.
“Because of the higher-than-expected contamination rates and to help protect patients from bacterial infections associated with the use of duodenoscopes, we have included in today’s safety communication updated recommendations regarding steps that health care providers can take to enhance duodenoscope reprocessing,” Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in the statement.
The FDA advised clinicians to follow additional cleaning measures including microbiological culturing, sterilization, use of a liquid chemical sterilant processing system, and repeated high-level disinfection beyond what is recommended by duodenoscope manufacturers.
The interim data cited in the safety communication come from postmarket surveillance studies conducted by duodenoscope manufacturers at the FDA’s request as part of the agency’s ongoing efforts to prevent patient infections caused by contaminated duodenoscopes. In addition to the positive tests for disease-causing bacteria, up to 3% of properly collected samples contained more than 100 colony-forming units of other organisms unlikely to cause infection. However, the presence of such organisms further highlights the failure of the current reprocessing protocol to adequately clean the devices, according to the FDA.
Dr. Shuren emphasized that the risk of infection from a duodenoscope for an individual patient remains low and that infection rates have declined in recent years in response to the FDA’s enhanced safety measures and stated that the agency remains “committed to enhancing the safety margin of procedures with reprocessed medical devices.”
Read the full safety communication here: https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm628020.htm.
The AGA Center for GI Innovation and Technology supports innovation and the development of new technology in gastroenterology, hepatology, nutrition and obesity by guiding medical device and therapeutics innovators through the technology development and adoption process. Learn more at www.gastro.org/CGIT
Negative colonoscopy linked with lower risk for more than 10 years
A negative colonoscopy result is associated with a reduced risk of colorectal cancer for more than 12 years after the examination, compared with an unscreened population, new research has found.
In a retrospective cohort study published in JAMA Internal Medicine, researchers analyzed data from 1,251,318 individuals at average risk of colorectal cancer who were eligible to participate in screening over more than 9 million person-years of follow-up.
They found that screened individuals with a negative result had an adjusted 46%-95% lower risk of colorectal cancer and 29%-96% lower risk of colorectal cancer mortality than unscreened individuals across more than 12 years of follow-up.
At 10 years post colonoscopy, participants who had a negative colonoscopy result still had a significant 46% lower risk of colorectal cancer and 88% lower risk of colorectal cancer mortality. After more than 12 years, there was still a nonsignificant trend towards a lower risk of colorectal cancer incidence and mortality.
Jeffrey K. Lee, MD, of Kaiser Permanente San Francisco, and his coauthors suggested that their findings have implications for the timing of rescreening after a negative colonoscopy result.
“The current guideline-recommended 10-year rescreening interval is not based on a predetermined risk threshold, and while we observed a reduced risk of colorectal cancer and related deaths throughout the more than 12-year follow-up period, an examination of absolute risk [incidence] could provide another justification for the timing for rescreening,” they wrote. “Additional research is needed to evaluate the costs and benefits of earlier versus later rescreening, optimal rescreening tests following a negative colonoscopy result [e.g., another colonoscopy versus annual fecal immunochemical testing], and whether the benefits of rescreening vary between subgroups.”
The study showed that the rate of repeat endoscopic procedures increased at year 10, largely because of screening colonoscopies which are recommended at 10-year intervals. However in a separate analysis, the authors excluded colonoscopies for a screening indication and still found a similar reduction in the risk of colorectal cancer, compared with the unscreened group.
The data also showed a 22%-87% lower risk of proximal colorectal cancer, a 50%-99% lower risk of distal cancer, a 31%-95% lower risk of early-stage colorectal cancer, and a 56%-96% reduced risk of advanced-stage colorectal cancer among those who had a negative result, compared with those who did not undergo screening.
The authors wrote that this pattern of greater risk reductions in the distal versus proximal cancer had been seen in previous studies and could be the result of incomplete examinations, inadequate bowel cleansing, challenges in identifying right colon polyps and sessile serrated adenomas, or differences in polyp biology in the proximal versus distal colon.
The incidence rates of colorectal cancer among those who had a negative result from colonoscopy ranged from 16.6 per 100,000 person-years at 1 year after screening to 133.2 per 100,000 person-years at the 10-year mark. In comparison, the incidence rates among the unscreened population increased from 62.9 per 100,000 person-years at year 1 to 224.8 after year 12.
Mortality rates from colorectal cancer at year 1 were 6.8 per 100,000 person-years in the negative results group and 10.5 in the unscreened cohort. At year 12, that figure was 92.2 per 100,000 person-years in the negative results cohort, while after year 12 in the unscreened cohort, colorectal cancer mortality rates increased to 192 per 100,000 person years.
While the study made use of a validated cancer registry to ensure they accurately captured cancers and mortality, the authors acknowledged that they weren’t able to adjust for residual confounding factors such as red-meat intake or smoking.
The study was supported by the National Cancer Institute, the American Gastroenterological Association, and the Sylvia Allison Kaplan Foundation. No conflicts of interest were reported.
SOURCE: Lee JK et al. JAMA Intern Med. 2018 Dec 17. doi: 10.1001/jamainternmed.2018.5565.
A negative colonoscopy result is associated with a reduced risk of colorectal cancer for more than 12 years after the examination, compared with an unscreened population, new research has found.
In a retrospective cohort study published in JAMA Internal Medicine, researchers analyzed data from 1,251,318 individuals at average risk of colorectal cancer who were eligible to participate in screening over more than 9 million person-years of follow-up.
They found that screened individuals with a negative result had an adjusted 46%-95% lower risk of colorectal cancer and 29%-96% lower risk of colorectal cancer mortality than unscreened individuals across more than 12 years of follow-up.
At 10 years post colonoscopy, participants who had a negative colonoscopy result still had a significant 46% lower risk of colorectal cancer and 88% lower risk of colorectal cancer mortality. After more than 12 years, there was still a nonsignificant trend towards a lower risk of colorectal cancer incidence and mortality.
Jeffrey K. Lee, MD, of Kaiser Permanente San Francisco, and his coauthors suggested that their findings have implications for the timing of rescreening after a negative colonoscopy result.
“The current guideline-recommended 10-year rescreening interval is not based on a predetermined risk threshold, and while we observed a reduced risk of colorectal cancer and related deaths throughout the more than 12-year follow-up period, an examination of absolute risk [incidence] could provide another justification for the timing for rescreening,” they wrote. “Additional research is needed to evaluate the costs and benefits of earlier versus later rescreening, optimal rescreening tests following a negative colonoscopy result [e.g., another colonoscopy versus annual fecal immunochemical testing], and whether the benefits of rescreening vary between subgroups.”
The study showed that the rate of repeat endoscopic procedures increased at year 10, largely because of screening colonoscopies which are recommended at 10-year intervals. However in a separate analysis, the authors excluded colonoscopies for a screening indication and still found a similar reduction in the risk of colorectal cancer, compared with the unscreened group.
The data also showed a 22%-87% lower risk of proximal colorectal cancer, a 50%-99% lower risk of distal cancer, a 31%-95% lower risk of early-stage colorectal cancer, and a 56%-96% reduced risk of advanced-stage colorectal cancer among those who had a negative result, compared with those who did not undergo screening.
The authors wrote that this pattern of greater risk reductions in the distal versus proximal cancer had been seen in previous studies and could be the result of incomplete examinations, inadequate bowel cleansing, challenges in identifying right colon polyps and sessile serrated adenomas, or differences in polyp biology in the proximal versus distal colon.
The incidence rates of colorectal cancer among those who had a negative result from colonoscopy ranged from 16.6 per 100,000 person-years at 1 year after screening to 133.2 per 100,000 person-years at the 10-year mark. In comparison, the incidence rates among the unscreened population increased from 62.9 per 100,000 person-years at year 1 to 224.8 after year 12.
Mortality rates from colorectal cancer at year 1 were 6.8 per 100,000 person-years in the negative results group and 10.5 in the unscreened cohort. At year 12, that figure was 92.2 per 100,000 person-years in the negative results cohort, while after year 12 in the unscreened cohort, colorectal cancer mortality rates increased to 192 per 100,000 person years.
While the study made use of a validated cancer registry to ensure they accurately captured cancers and mortality, the authors acknowledged that they weren’t able to adjust for residual confounding factors such as red-meat intake or smoking.
The study was supported by the National Cancer Institute, the American Gastroenterological Association, and the Sylvia Allison Kaplan Foundation. No conflicts of interest were reported.
SOURCE: Lee JK et al. JAMA Intern Med. 2018 Dec 17. doi: 10.1001/jamainternmed.2018.5565.
A negative colonoscopy result is associated with a reduced risk of colorectal cancer for more than 12 years after the examination, compared with an unscreened population, new research has found.
In a retrospective cohort study published in JAMA Internal Medicine, researchers analyzed data from 1,251,318 individuals at average risk of colorectal cancer who were eligible to participate in screening over more than 9 million person-years of follow-up.
They found that screened individuals with a negative result had an adjusted 46%-95% lower risk of colorectal cancer and 29%-96% lower risk of colorectal cancer mortality than unscreened individuals across more than 12 years of follow-up.
At 10 years post colonoscopy, participants who had a negative colonoscopy result still had a significant 46% lower risk of colorectal cancer and 88% lower risk of colorectal cancer mortality. After more than 12 years, there was still a nonsignificant trend towards a lower risk of colorectal cancer incidence and mortality.
Jeffrey K. Lee, MD, of Kaiser Permanente San Francisco, and his coauthors suggested that their findings have implications for the timing of rescreening after a negative colonoscopy result.
“The current guideline-recommended 10-year rescreening interval is not based on a predetermined risk threshold, and while we observed a reduced risk of colorectal cancer and related deaths throughout the more than 12-year follow-up period, an examination of absolute risk [incidence] could provide another justification for the timing for rescreening,” they wrote. “Additional research is needed to evaluate the costs and benefits of earlier versus later rescreening, optimal rescreening tests following a negative colonoscopy result [e.g., another colonoscopy versus annual fecal immunochemical testing], and whether the benefits of rescreening vary between subgroups.”
The study showed that the rate of repeat endoscopic procedures increased at year 10, largely because of screening colonoscopies which are recommended at 10-year intervals. However in a separate analysis, the authors excluded colonoscopies for a screening indication and still found a similar reduction in the risk of colorectal cancer, compared with the unscreened group.
The data also showed a 22%-87% lower risk of proximal colorectal cancer, a 50%-99% lower risk of distal cancer, a 31%-95% lower risk of early-stage colorectal cancer, and a 56%-96% reduced risk of advanced-stage colorectal cancer among those who had a negative result, compared with those who did not undergo screening.
The authors wrote that this pattern of greater risk reductions in the distal versus proximal cancer had been seen in previous studies and could be the result of incomplete examinations, inadequate bowel cleansing, challenges in identifying right colon polyps and sessile serrated adenomas, or differences in polyp biology in the proximal versus distal colon.
The incidence rates of colorectal cancer among those who had a negative result from colonoscopy ranged from 16.6 per 100,000 person-years at 1 year after screening to 133.2 per 100,000 person-years at the 10-year mark. In comparison, the incidence rates among the unscreened population increased from 62.9 per 100,000 person-years at year 1 to 224.8 after year 12.
Mortality rates from colorectal cancer at year 1 were 6.8 per 100,000 person-years in the negative results group and 10.5 in the unscreened cohort. At year 12, that figure was 92.2 per 100,000 person-years in the negative results cohort, while after year 12 in the unscreened cohort, colorectal cancer mortality rates increased to 192 per 100,000 person years.
While the study made use of a validated cancer registry to ensure they accurately captured cancers and mortality, the authors acknowledged that they weren’t able to adjust for residual confounding factors such as red-meat intake or smoking.
The study was supported by the National Cancer Institute, the American Gastroenterological Association, and the Sylvia Allison Kaplan Foundation. No conflicts of interest were reported.
SOURCE: Lee JK et al. JAMA Intern Med. 2018 Dec 17. doi: 10.1001/jamainternmed.2018.5565.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Reduced rates of colorectal cancer after a negative colonoscopy persist beyond 10 years.
Major finding: After 10 years, a negative colonoscopy is still associated with an 88% lower risk of colorectal cancer mortality, compared with the unscreened.
Study details: A retrospective cohort study in 1,251,318 screening-eligible individuals.
Disclosures: The study was supported by the National Cancer Institute, the American Gastroenterological Association, and the Sylvia Allison Kaplan Foundation. No conflicts of interest were reported.
Source: Lee JK et al. JAMA Intern Med. 2018 Dec 17. doi: 10.1001/jamainternmed.2018.5565.
“FAST” indices help to identify fibromyalgia in routine rheumatology care
CHICAGO – Fibromyalgia assessment screening tool (FAST) indices derived from the Multidimensional Health Assessment Questionnaire (MDHAQ) provide a simple and effective method for identifying fibromyalgia in routine care, according to findings from a series of more than 500 patients.

The indices are as accurate as the existing – and more complex – diagnostic criteria for fibromyalgia, Juan Schmukler, MD, reported at the annual meeting of the American College of Rheumatology.
For example, three FAST indices developed in the course of the study had better performance versus existing diagnostic criteria than did certain individual MDHAQ scales alone (area under the curve range, 0.924-0.937 vs. 0.829-0.889, respectively), said Dr. Schmukler, who conducted the research as a medical student at Rush Medical College, Chicago, and is now a rheumatology fellow at Mount Sinai Hospital in Chicago.
The findings are notable, because the two-page MDHAQ and a summary index (the RAPID3) derived from three of the MDHAQ scales are already used routinely in clinical care for diagnosing rheumatic diseases, and the new indices could easily – and with minimal work flow disruption – also be incorporated for each patient at each visit.
“Fibromyalgia is common in the general population and it is believed to be more common in patients with other rheumatic diagnoses,” Dr. Schmukler said. “Fibromyalgia may be easily recognized in many cases, but it can also be very subtle, particularly in patients who have other rheumatic diseases.”
ACR fibromyalgia classification criteria published in 1990 were based on tender point examination and non-ACR diagnostic criteria as revised in 2011 are based entirely on patient self-report.
A one-page fibromyalgia criteria questionnaire is available and is useful in clinical trials and other research, but is “rarely, if ever” used in routine care, he said, explaining that the questionnaire contains two domains: a symptom severity score (0-12 scale) and a widespread pain index (0-19 scale).
The MDHAQ/RAPID3 is informative in RA, and has also been shown to be “useful in all rheumatic diseases in which it has been studied,” and at least three prior reports have suggested that the MDHAQ may also provide clues to the presence of fibromyalgia, Dr. Schmukler said.
“And we know from prior reports that patients with fibromyalgia reported the highest RAPID3 scores, compared to other rheumatic disease,” he added, explaining that the goal of the present study was to develop FAST cumulative indices based on the routine MDHAQ scales and using the 2011 diagnostic criteria as a reference standard.
All patients with all diagnoses seen at the Rush Medical College rheumatology clinic in Chicago complete the MDHAQ at all visits in routine care, and between April and July 2017, the fibromyalgia criteria questionnaire was also administered in 502 consecutive patients.
Of those patients, 131 met the 2011 fibromyalgia diagnostic criteria.
MDHAQ scores were analyzed for agreement with the fibromyalgia criteria questionnaire according to receiver operator characteristic curves for AUC and were compiled into three different FAST indices that included various combinations of either three or four of the MDHAQ measures that had the best agreement with the diagnostic criteria questionnaire as identified by the highest AUC values. Those were the 60-symptom checklist (AUC, 0.889), painful joint count (AUC, 0.870), fatigue visual analog scale (VAS; AUC, 0.860), and pain VAS (AUC, 0.829), Dr. Schmukler said.
Proposed cut points that reflected the optimal trade-off between specificity and sensitivity for each of the scales were scores of 6 or greater for the pain VAS and fatigue VAS, and scores of 16 or greater for the symptom checklist and painful joint count measure.
In addition to the better performance of each FAST index versus the 2011 criteria, their performance was also better than that of the RAPID3 versus the 2011 criteria (AUC, 0.848), which many clinicians use in practice without using the full MDHAQ for patient assessment, he said.
Further, the “very easily calculated” FAST indices performed as well as a “very difficult to calculate” polysymptomatic distress continuous scale derived from the fibromyalgia questionnaire to assess the degree of fibromyalgia symptoms, which had an AUC of 0.929 versus the 2011 criteria. The latter index requires complex mathematical calculations for scale conversion and thus is impractical in clinical practice, he noted.
The FAST indices also performed comparably with physician diagnoses as indicated in patient charts and with diagnoses based on tender point count as shown in prior studies.
The FAST index with the greatest sensitivity (85.5% at a cut point of 2 or greater on a scale of 1-3) was the FAST3-P, which includes pain VAS score of 6 or greater, symptom checklist score of 16 or greater, and painful joint count of 16 or greater. The FAST index with the greatest specificity (90.3% at a cut point of score of 3 or greater on a scale of 1-4) was the FAST4 index, which includes a pain VAS score of 6 or greater, fatigue VAS score of 6 or greater, symptom checklist score of 16 or greater, and painful joint count of 16 or greater.
Although the findings are limited by the lack of a gold standard for fibromyalgia diagnosis, changing diagnostic criteria, and a need for physician input for a fibromyalgia diagnosis, the study provides useful real-world data and supports findings from some prior studies with respect to the benefit of using these tools in routine practice.
With minimal extra physician time – if the MDHAQ is completed by patients at the time of registration – this approach can be used in all rheumatology patients to help identify fibromyalgia, he concluded.
Dr. Schmukler reported having no disclosures. One of his coauthors at Rush, Ted Pincus, MD, receives royalties and license fees for the copyright and trademark for the MDHAQ and RAPID3, all of which go to support clinical research.
SOURCE: Schmukler J et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 839.
CHICAGO – Fibromyalgia assessment screening tool (FAST) indices derived from the Multidimensional Health Assessment Questionnaire (MDHAQ) provide a simple and effective method for identifying fibromyalgia in routine care, according to findings from a series of more than 500 patients.
The indices are as accurate as the existing – and more complex – diagnostic criteria for fibromyalgia, Juan Schmukler, MD, reported at the annual meeting of the American College of Rheumatology.
For example, three FAST indices developed in the course of the study had better performance versus existing diagnostic criteria than did certain individual MDHAQ scales alone (area under the curve range, 0.924-0.937 vs. 0.829-0.889, respectively), said Dr. Schmukler, who conducted the research as a medical student at Rush Medical College, Chicago, and is now a rheumatology fellow at Mount Sinai Hospital in Chicago.
The findings are notable, because the two-page MDHAQ and a summary index (the RAPID3) derived from three of the MDHAQ scales are already used routinely in clinical care for diagnosing rheumatic diseases, and the new indices could easily – and with minimal work flow disruption – also be incorporated for each patient at each visit.
“Fibromyalgia is common in the general population and it is believed to be more common in patients with other rheumatic diagnoses,” Dr. Schmukler said. “Fibromyalgia may be easily recognized in many cases, but it can also be very subtle, particularly in patients who have other rheumatic diseases.”
ACR fibromyalgia classification criteria published in 1990 were based on tender point examination and non-ACR diagnostic criteria as revised in 2011 are based entirely on patient self-report.
A one-page fibromyalgia criteria questionnaire is available and is useful in clinical trials and other research, but is “rarely, if ever” used in routine care, he said, explaining that the questionnaire contains two domains: a symptom severity score (0-12 scale) and a widespread pain index (0-19 scale).
The MDHAQ/RAPID3 is informative in RA, and has also been shown to be “useful in all rheumatic diseases in which it has been studied,” and at least three prior reports have suggested that the MDHAQ may also provide clues to the presence of fibromyalgia, Dr. Schmukler said.
“And we know from prior reports that patients with fibromyalgia reported the highest RAPID3 scores, compared to other rheumatic disease,” he added, explaining that the goal of the present study was to develop FAST cumulative indices based on the routine MDHAQ scales and using the 2011 diagnostic criteria as a reference standard.
All patients with all diagnoses seen at the Rush Medical College rheumatology clinic in Chicago complete the MDHAQ at all visits in routine care, and between April and July 2017, the fibromyalgia criteria questionnaire was also administered in 502 consecutive patients.
Of those patients, 131 met the 2011 fibromyalgia diagnostic criteria.
MDHAQ scores were analyzed for agreement with the fibromyalgia criteria questionnaire according to receiver operator characteristic curves for AUC and were compiled into three different FAST indices that included various combinations of either three or four of the MDHAQ measures that had the best agreement with the diagnostic criteria questionnaire as identified by the highest AUC values. Those were the 60-symptom checklist (AUC, 0.889), painful joint count (AUC, 0.870), fatigue visual analog scale (VAS; AUC, 0.860), and pain VAS (AUC, 0.829), Dr. Schmukler said.
Proposed cut points that reflected the optimal trade-off between specificity and sensitivity for each of the scales were scores of 6 or greater for the pain VAS and fatigue VAS, and scores of 16 or greater for the symptom checklist and painful joint count measure.
In addition to the better performance of each FAST index versus the 2011 criteria, their performance was also better than that of the RAPID3 versus the 2011 criteria (AUC, 0.848), which many clinicians use in practice without using the full MDHAQ for patient assessment, he said.
Further, the “very easily calculated” FAST indices performed as well as a “very difficult to calculate” polysymptomatic distress continuous scale derived from the fibromyalgia questionnaire to assess the degree of fibromyalgia symptoms, which had an AUC of 0.929 versus the 2011 criteria. The latter index requires complex mathematical calculations for scale conversion and thus is impractical in clinical practice, he noted.
The FAST indices also performed comparably with physician diagnoses as indicated in patient charts and with diagnoses based on tender point count as shown in prior studies.
The FAST index with the greatest sensitivity (85.5% at a cut point of 2 or greater on a scale of 1-3) was the FAST3-P, which includes pain VAS score of 6 or greater, symptom checklist score of 16 or greater, and painful joint count of 16 or greater. The FAST index with the greatest specificity (90.3% at a cut point of score of 3 or greater on a scale of 1-4) was the FAST4 index, which includes a pain VAS score of 6 or greater, fatigue VAS score of 6 or greater, symptom checklist score of 16 or greater, and painful joint count of 16 or greater.
Although the findings are limited by the lack of a gold standard for fibromyalgia diagnosis, changing diagnostic criteria, and a need for physician input for a fibromyalgia diagnosis, the study provides useful real-world data and supports findings from some prior studies with respect to the benefit of using these tools in routine practice.
With minimal extra physician time – if the MDHAQ is completed by patients at the time of registration – this approach can be used in all rheumatology patients to help identify fibromyalgia, he concluded.
Dr. Schmukler reported having no disclosures. One of his coauthors at Rush, Ted Pincus, MD, receives royalties and license fees for the copyright and trademark for the MDHAQ and RAPID3, all of which go to support clinical research.
SOURCE: Schmukler J et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 839.
CHICAGO – Fibromyalgia assessment screening tool (FAST) indices derived from the Multidimensional Health Assessment Questionnaire (MDHAQ) provide a simple and effective method for identifying fibromyalgia in routine care, according to findings from a series of more than 500 patients.
The indices are as accurate as the existing – and more complex – diagnostic criteria for fibromyalgia, Juan Schmukler, MD, reported at the annual meeting of the American College of Rheumatology.
For example, three FAST indices developed in the course of the study had better performance versus existing diagnostic criteria than did certain individual MDHAQ scales alone (area under the curve range, 0.924-0.937 vs. 0.829-0.889, respectively), said Dr. Schmukler, who conducted the research as a medical student at Rush Medical College, Chicago, and is now a rheumatology fellow at Mount Sinai Hospital in Chicago.
The findings are notable, because the two-page MDHAQ and a summary index (the RAPID3) derived from three of the MDHAQ scales are already used routinely in clinical care for diagnosing rheumatic diseases, and the new indices could easily – and with minimal work flow disruption – also be incorporated for each patient at each visit.
“Fibromyalgia is common in the general population and it is believed to be more common in patients with other rheumatic diagnoses,” Dr. Schmukler said. “Fibromyalgia may be easily recognized in many cases, but it can also be very subtle, particularly in patients who have other rheumatic diseases.”
ACR fibromyalgia classification criteria published in 1990 were based on tender point examination and non-ACR diagnostic criteria as revised in 2011 are based entirely on patient self-report.
A one-page fibromyalgia criteria questionnaire is available and is useful in clinical trials and other research, but is “rarely, if ever” used in routine care, he said, explaining that the questionnaire contains two domains: a symptom severity score (0-12 scale) and a widespread pain index (0-19 scale).
The MDHAQ/RAPID3 is informative in RA, and has also been shown to be “useful in all rheumatic diseases in which it has been studied,” and at least three prior reports have suggested that the MDHAQ may also provide clues to the presence of fibromyalgia, Dr. Schmukler said.
“And we know from prior reports that patients with fibromyalgia reported the highest RAPID3 scores, compared to other rheumatic disease,” he added, explaining that the goal of the present study was to develop FAST cumulative indices based on the routine MDHAQ scales and using the 2011 diagnostic criteria as a reference standard.
All patients with all diagnoses seen at the Rush Medical College rheumatology clinic in Chicago complete the MDHAQ at all visits in routine care, and between April and July 2017, the fibromyalgia criteria questionnaire was also administered in 502 consecutive patients.
Of those patients, 131 met the 2011 fibromyalgia diagnostic criteria.
MDHAQ scores were analyzed for agreement with the fibromyalgia criteria questionnaire according to receiver operator characteristic curves for AUC and were compiled into three different FAST indices that included various combinations of either three or four of the MDHAQ measures that had the best agreement with the diagnostic criteria questionnaire as identified by the highest AUC values. Those were the 60-symptom checklist (AUC, 0.889), painful joint count (AUC, 0.870), fatigue visual analog scale (VAS; AUC, 0.860), and pain VAS (AUC, 0.829), Dr. Schmukler said.
Proposed cut points that reflected the optimal trade-off between specificity and sensitivity for each of the scales were scores of 6 or greater for the pain VAS and fatigue VAS, and scores of 16 or greater for the symptom checklist and painful joint count measure.
In addition to the better performance of each FAST index versus the 2011 criteria, their performance was also better than that of the RAPID3 versus the 2011 criteria (AUC, 0.848), which many clinicians use in practice without using the full MDHAQ for patient assessment, he said.
Further, the “very easily calculated” FAST indices performed as well as a “very difficult to calculate” polysymptomatic distress continuous scale derived from the fibromyalgia questionnaire to assess the degree of fibromyalgia symptoms, which had an AUC of 0.929 versus the 2011 criteria. The latter index requires complex mathematical calculations for scale conversion and thus is impractical in clinical practice, he noted.
The FAST indices also performed comparably with physician diagnoses as indicated in patient charts and with diagnoses based on tender point count as shown in prior studies.
The FAST index with the greatest sensitivity (85.5% at a cut point of 2 or greater on a scale of 1-3) was the FAST3-P, which includes pain VAS score of 6 or greater, symptom checklist score of 16 or greater, and painful joint count of 16 or greater. The FAST index with the greatest specificity (90.3% at a cut point of score of 3 or greater on a scale of 1-4) was the FAST4 index, which includes a pain VAS score of 6 or greater, fatigue VAS score of 6 or greater, symptom checklist score of 16 or greater, and painful joint count of 16 or greater.
Although the findings are limited by the lack of a gold standard for fibromyalgia diagnosis, changing diagnostic criteria, and a need for physician input for a fibromyalgia diagnosis, the study provides useful real-world data and supports findings from some prior studies with respect to the benefit of using these tools in routine practice.
With minimal extra physician time – if the MDHAQ is completed by patients at the time of registration – this approach can be used in all rheumatology patients to help identify fibromyalgia, he concluded.
Dr. Schmukler reported having no disclosures. One of his coauthors at Rush, Ted Pincus, MD, receives royalties and license fees for the copyright and trademark for the MDHAQ and RAPID3, all of which go to support clinical research.
SOURCE: Schmukler J et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 839.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point:
Major finding: Each fibromyalgia assessment screening tool index had better performance versus 2011 diagnostic criteria (area under the curve, 0.924-0.937) than did individual scales alone (AUCs, 0.829-0.889) and the RAPID3 summary index (AUC, 0.848).
Study details: An assessment of fibromyalgia assessment screening tool indices developed in a series of 502 patients.
Disclosures: Dr. Schmukler reported having no disclosures. One of his coauthors at Rush, Ted Pincus, MD, receives royalties and license fees for the copyright and trademark for the Multidimensional Health Assessment Questionnaire and RAPID3, all of which go to support clinical research.
Source: Schmukler J et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 839.
A Retrospective Analysis of the Modified Intervastus Approach
ABSTRACT
The subvastus (SV) approach is a well-known muscle- and tendon-sparing approach for total knee arthroplasty (TKA), which has been shown in some studies to provide better outcomes in the visual analog pain score (VAS), knee range of motion (ROM), straight-leg raise, as well as faster rehabilitation, compared with the standard medial parapatellar (MP) approach. We previously described a new knee replacement technique known as the modified intervastus (MIV) approach. The MIV approach is a muscle- and tendon-sparing approach that is extensile and simple to perform. It may be used in the majority of complex primary cases and revisions. Here we describe the surgical technique for performing the MIV approach and provide functional outcome measures. A total of 127 patients (mean age, 66.75 years) underwent TKA using the MIV approach with 1-year follow-up. Clinical outcomes were assessed by recording both a VAS and knee ROM preoperatively, and again at several postoperative time points when the length of time required to ambulate independently (without assistive devices) was also measured. The VAS decreased significantly from the preoperative period (3.69 ± 2.22) to postoperative day 1 (3.17 ± 1.97) (P < .05). Although knee ROM decreased 1 week after surgery, the ROM increased by 6 weeks after surgery compared with the preoperative ROM, and the trend continued over the 1-year follow-up. One-third (33%) of patients were able to walk independently (without assistive devices) at 2 weeks and 78% at 8 weeks. The MIV approach to the knee is a muscle- and tendon-sparing approach that offers advantages over the SV approach and may be used for complex primary and revision total knee cases.
Continue to: Total knee arthroplasty...
Total knee arthroplasty (TKA) is one of the most common orthopedic surgical procedures, with more than 600,000 TKAs performed annually in the United States, and by 2030 the number is expected to reach 3.48 million per year.1Several approaches have been described for total TKA, and the medial parapatellar (MP) approach to the knee is considered the workhorse of total knee replacements. It is an extensile approach that is easy to perform but may delay active knee extension and straight-leg raise after surgery.2 Alternative approaches such as the subvastus (SV) and midvastus approaches to the knee (Figure 1A) typically allow a more rapid straight-leg raise but may be more challenging to perform and time-consuming in morbidly obese patients, muscular patients, and patients with severe deformities.3
The intervastus approach (Figure 1A) described by Jojima4 and others utilizes the interval between the quadriceps tendon and vastus medialis. Although it is a simple approach to perform that is extensile, it is not considered tendon-sparing since the vastus medialis inserts onto the medial aspect of the quadriceps tendon (Figure 2).5 Therefore, dissecting through this interval, without elevation of the vastus medialis (Figure 3A), damages the quadriceps tendon, and strangulation of the muscle occurs with repair of the arthrotomy site (Figure 3B). This situation is even more likely in patients with a low-lying vastus medialis.6
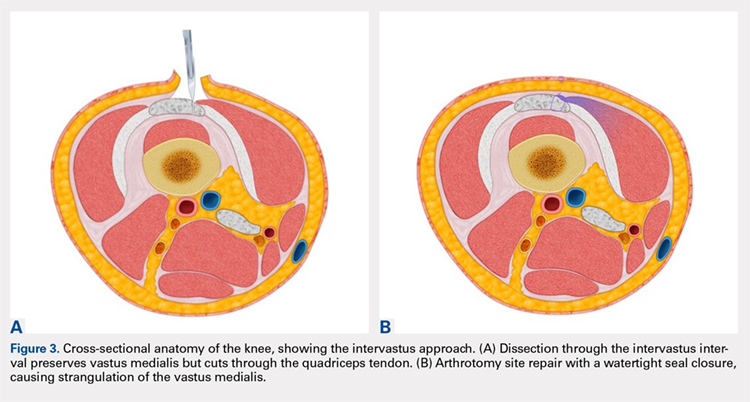
The modified intervastus (MIV) approach described previously,7 may be used in the majority of patients undergoing TKA. The advantages of this approach include its extensile nature, similarities to an MP approach, and preservation of both the extensor mechanism and the vastus medialis, leading to a more rapid return to active knee extension than is traditionally observed.2,7 The approach is simple to perform, easy to close, and is compatible with more extensile approaches such as a quadriceps snip if required in revision scenarios.7 However, functional outcomes of the MIV approach have not been quantified. It is unknown whether these outcomes will offer any advantages compared with the SV approach. Therefore, the objective of this study was to measure functional outcomes of the MIV approach and to compare the results with those previously published for the SV approach. We hypothesized that using the MIV approach for TKA surgery would lead to early straight-leg raise and increase in knee range of motion (ROM) postoperatively.
SURGICAL TECHNIQUE
The MIV approach preserves the quadriceps tendon and vastus medialis.7 After exposure of the vastus medialis muscle, the interval between the quadriceps tendon and vastus medialis is identified (Figure 4). The fascia overlying the lateral edge of the vastus medialis is incised where it meets the quadriceps tendon (Figure 5). The muscle is then bluntly elevated off the underlying capsule just enough to allow for a capsular repair later (Figure 6). An arthrotomy is then performed from cephalad to caudal (Figure 7). This interval may be extended proximally between the vastus intermedius and vastus medialis to expose the distal femur if needed. The closure is performed by repairing the capsule with absorbable suture (Figure 8A), and the vastus medialis fascia is repaired back to the medial edge of the quadriceps tendon, restoring the anatomy (Figure 8B).
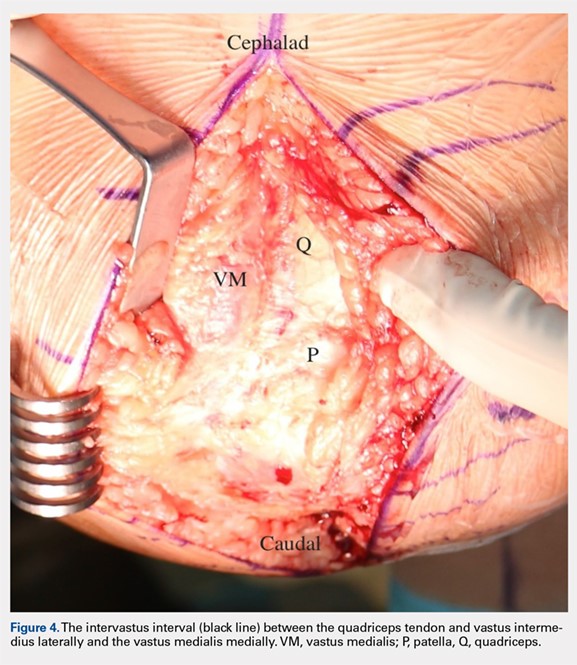
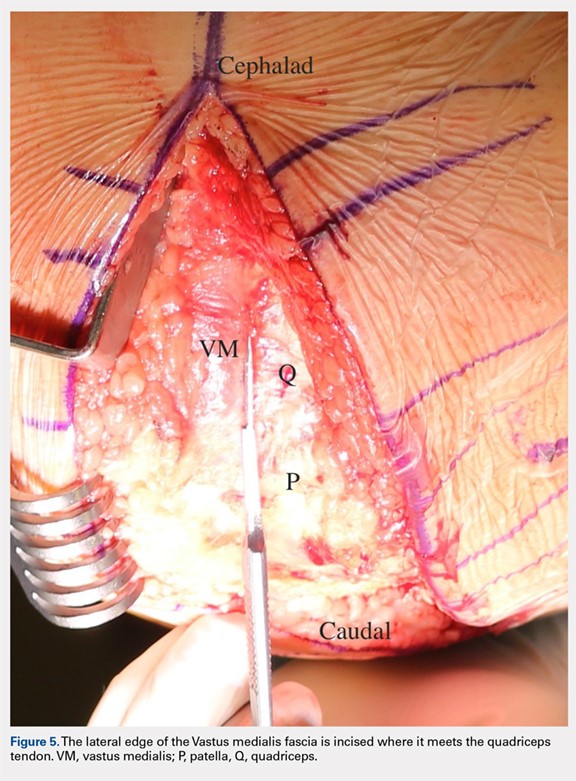
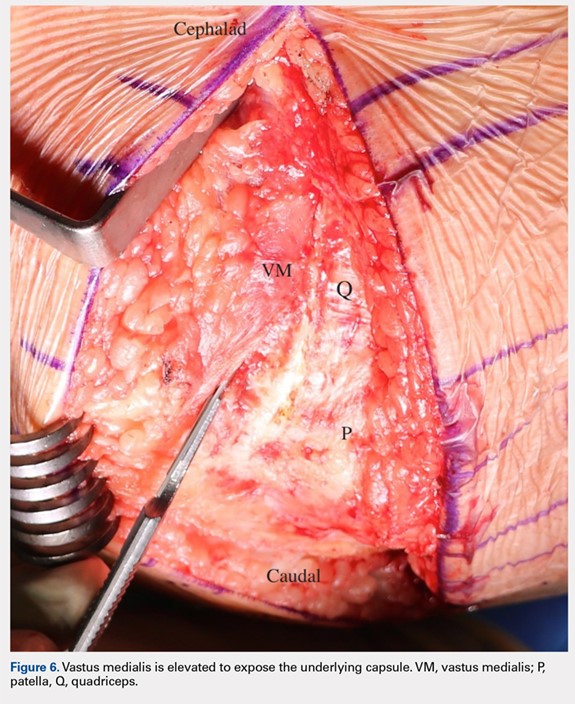
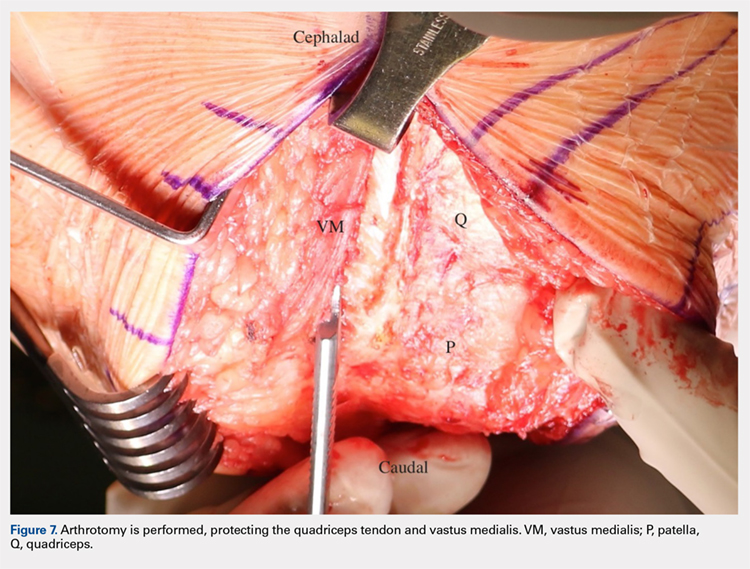
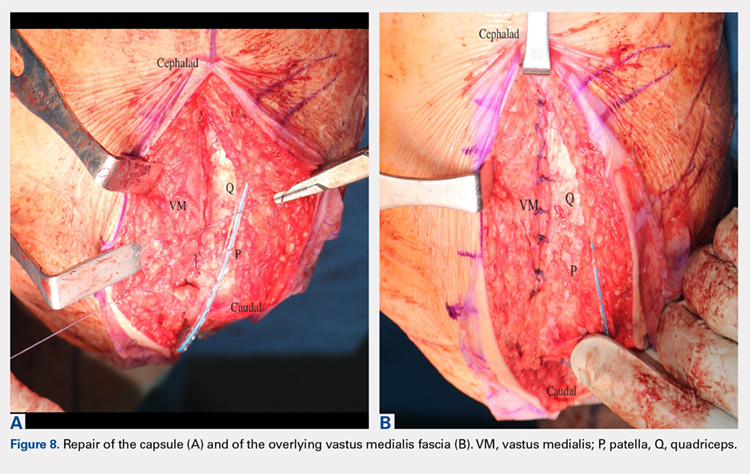
Continue to: PATIENTS AND METHODS...
PATIENTS AND METHODS
A retrospective review of functional outcomes after TKA using the MIV approach was conducted; the study was approved by the University of Illinois Institutional Review Board. A total of 127 patients of mean age 66.75 years (range, 48–86 years) with primary osteoarthritis of the knee who were indicated for a total knee replacement with 1-year follow-up were included. The patient demographics are shown in Table 1. All patients underwent TKA using the MIV approach described above by 2 experienced orthopedic surgeons at the same institution. Patients received spinal anesthesia and a periarticular pain block intraoperatively. A measured resection technique was used by 1 surgeon, and a gap-balancing technique by the other. Surgeon 1 used the Persona PS cemented knee system (Zimmer Biomet, and Surgeon 2 used the Sigma PS cemented knee system (Depuy). Patellar resurfacing was done in all cases. Patellar tracking was checked intraoperatively using the ‘no-touch’ technique, and the need for a lateral release was noted. Drains were removed on postoperative day 1. Oral opioids were given as needed. Intravenous antibiotics were continued for 24 hours. Oral anticoagulants were used for thromboembolism prophylaxis for 3 weeks. Patients were mobilized on the day of surgery with full weight-bearing under the supervision of an experienced physical therapist. Static and dynamic quadriceps exercises were started on the same day of surgery along with active knee ROM exercises. Pain score, extensor lag, ROM, walking ability, and complications were recorded in all patients.
Table 1. Patient Demographics
Total no. of patients | 127 | |
Gender | Male | 44 |
Female | 83 | |
Age (years) | Mean ± Standard deviation | 66.75 ± 9.12 |
Range | 48 – 86 | |
Weight (lb) | Mean ± Standard deviation | 218.38 ± 54.47 |
Range | 125 – 364 | |
BMI (kg/m2) | Mean ± Standard deviation | 34.10 ± 7.22 |
Range | 21.1 – 62.5 | |
The visual analog score (VAS) was obtained preoperatively and recorded on postoperative day 1. Patient walking distance with assistance was measured on the day of surgery, after surgery, and on the day of hospital discharge. Patients were assessed preoperatively and postoperatively at 1 week, 2 weeks, 6 weeks, 3 months, 6 months, and 1 year for knee ROM. A one-way ANOVA was conducted to compare the preoperative and postoperative day 1 VAS with significance set at P < .05 (OriginPro 2015, OriginLab Corporation). Differences in knee ROM between preoperative and postoperative follow-up periods (1 week, 2 weeks, 6 weeks, 3 months, 6 months, and 1 year) were identified using a 1-way ANOVA with a post hoc Tukey test. Significance was set at P < .05.
RESULTS
All patients were able to fully straight-leg raise and demonstrate functional knee ROM by postoperative day 1. The patella tracked centrally in all patients, and none required a lateral retinacular release. The majority of patients were discharged in the first 48 hours after surgery on oral narcotics. None required IV narcotics during their hospital stay or a blood transfusion. Two cases were complicated by severe knee skin blistering postoperatively due to a reaction to an adhesive dressing; one was complicated by skin necrosis leading to a flap reconstruction that became infected, requiring a 2-stage revision. A separate case had an acute postoperative infection that required irrigation and debridement with polyethylene exchange. After a 12-week course of antibiotics, the infection was eradicated. All patients reported a high satisfaction rate during their acute postoperative phase.
Postoperatively, all patients were able to walk on the day of surgery either independently or with some assistance. On the day of surgery, 10% of patients were able to walk >200 feet, and this increased to 65% of patients able to walk >200 feet on the day of discharge (compare Figure 9A and Figure 9B). Within 2 weeks of surgery, 30% of patients could walk independently (without assistive devices), and this number increased to 78% by 8 weeks after surgery (Figure 10).
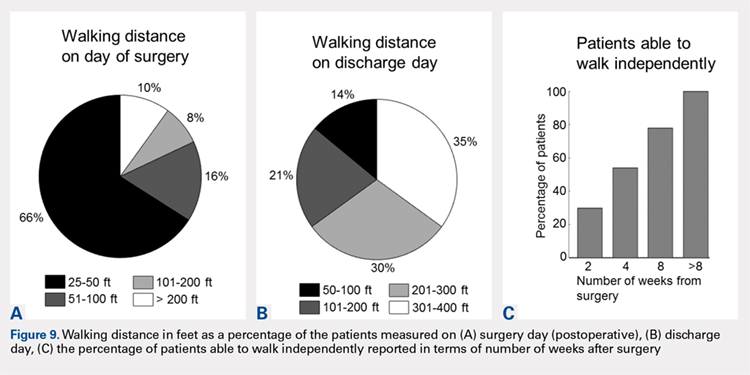
Pain assessed using the VAS was lower on postoperative day 1 (3.17 ± 1.97) than the preoperative score (3.69 ± 2.22, P< .05). Overall, knee ROM significantly increased during the follow-up after surgery. Initially, the ROM decreased 1 week after surgery (90.82 ± 10.28) compared with preoperative ROM (101.04 ± 19.48, P < .001) (Figure 10). At 2 weeks after surgery, knee ROM returned to the preoperative value (100.70 ± 13.36). By 6 weeks after surgery, knee ROM was 17° greater than the preoperative ROM (118.45 ± 11.89, P < .001). Knee ROM remained stable at 3- and 6-month assessments, and showed further improvement by 1 year (126.62 ± 9.81, P < .001) compared with the preoperative state (Figure 10). The net improvement in knee ROM was 25° of increased knee flexion by 1 year.
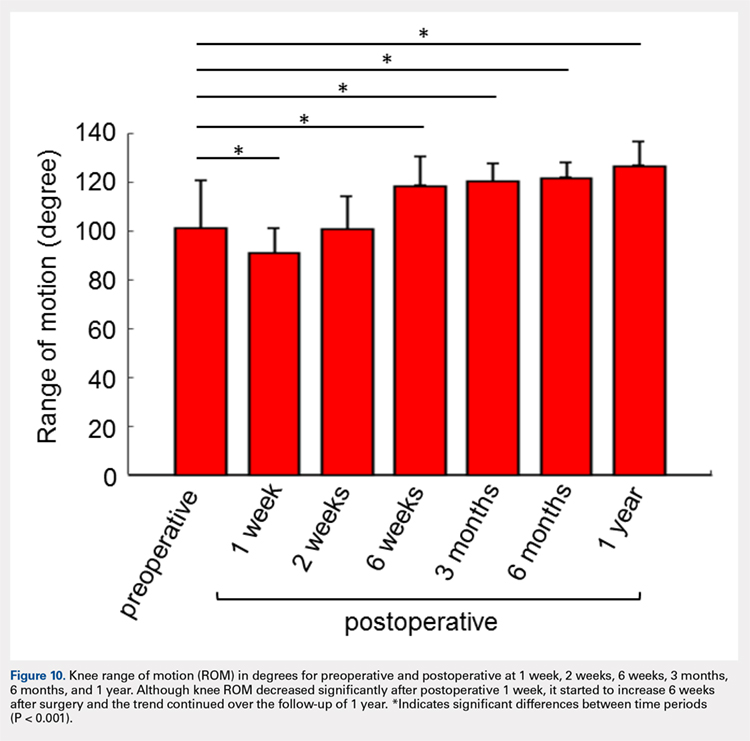
Continue to: DISCUSSION...
DISCUSSION
TKA is a successful procedure that restores knee function with pain relief in osteoarthritis patients. The SV approach for TKA has better outcomes in terms of the VAS, ROM, straight-leg raise with faster rehabilitation compared with the standard MP approach;8–12 however, it can be challenging and time-consuming when used in morbidly obese and muscular patients.3 The SV approach can also increase the risk of complications such as patellar tendon avulsion or medial collateral injury because of the difficulty in exposure specifically for knees with limited ROM.13 Here we introduce the MIV approach as an alternative to the SV approach, overcoming most of these difficulties.
With the prevalence of morbid obesity and the market demand for minimally invasive techniques, we believe the MIV approach represents a good approach for surgeons since it is easy to perform, does not require specialized instrumentation, and is a reproducible approach even on the most complex deformities. The minimal time added to ensure blunt elevation of the vastus medialis muscle and an anatomic repair of the underlying knee capsule and vastus medialis fascia to the medial edge of the quadriceps tendon allows restoration of the anatomy and a robust double-layered watertight seal closure with no strangulation of soft tissues. We believe this reproducible muscle- and tendon-sparing approach that allows gentle, soft tissue handling even in the most complex primary total knee cases may lead to less soft tissue swelling, and therefore, less postoperative pain resulting in an accelerated recovery.
The pain level in this group of patients was reduced after the MIV approach as indicated by the VAS. The VAS was significantly decreased on postoperative day 1 compared with the VAS recorded preoperatively (P < .05), indicating patients felt less pain on the day after surgery. The average VAS on postoperative day 1 from other studies for SV approach ranged from 2.1 to 5,9,12,14–19 whereas our MIV approach value was 3.17. Periarticular blocks were available for this study group, and no peripheral nerve blocks were used. Some studies of the SV approach mention the use of peripheral nerve blocks, while others did not describe the method used for treatment or control of postoperative pain. The decreased reported pain levels and the observed increased knee ROM seen in the MIV and SV approach study groups might be attributable to the treatment of postoperative pain.
Patient ambulation also increased from the day of surgery to the day of discharge for the MIV approach. Only 10% of patients were able to walk with assistance >200 feet on the day of surgery; however, this percentage increased to 65% on the discharge day showing an excellent recovery of walking ability within 2 days of surgery. Mehta and colleagues20reported that 95% of patients undergoing subvastus/midvastus approaches could walk >10 blocks at 6 months follow-up. For the MIV approach, 78% of patients were able to walk independently within 8 weeks of surgery.
Continue to: The MIV approach shows improvement...
The MIV approach shows improvement in functional outcomes in terms of knee ROM from preoperative to postoperative. Although knee ROM decreased significantly at postoperative week 1, which is most likely because of a peak in postoperative pain and swelling; the ROM started to increase significantly at postoperative week 6. After 1 year, the knee ROM increased by 25% compared with preoperative ROM for the MIV approach. We found the average knee ROM at postoperative 3 months using the MIV approach to be 120.4°. Knee ROM at postoperative 3 months using the SV approach reported by other studies ranged from 87.1° to 120°.19,21–23 Similarly, after 1 year, the average ROM by MIV approach was 126.62°, whereas average knee ROM from other studies using the SV approach was 114.1°.2,10,19,21 Achieving a knee ROM of 120° allows patients to return to their baseline function and perform activities of daily living without limitation. With the prevalence of knee osteoarthritis in the younger population, the need for increased knee ROM post-TKA becomes more important so patients can return to work and function at their baseline.
In addition to being both a muscle- and tendon-sparing approach, other advantages of the MIV approach are that it is simple to perform and gives similar exposure to the MP approach even in difficult primary cases. It is also as extensile as the medial parapatellar approach, and it can be converted to a quadriceps snip if required. There is also minimal, if any, muscle retraction preventing muscle injury that may be seen with some other muscle-sparing approaches to the knee, especially in more difficult primary total knee cases. Another advantage to this approach, as opposed to the other approaches to the knee described here, is that it allows a double-layered closure, decreasing the possibility of arthrotomy dehiscence. Because of this double-layered closure, there is no muscle strangulation with repair of the arthrotomy site as seen in some other approaches to the knee, and both vascularity and innervation to the vastus medialis are preserved.
One limitation of this study is that it is a retrospective analysis. Future studies of the consequences of using this approach need to include quantitative assessment of muscle function using electromyography or dynamometer measurements, utilization of any number of validated knee score systems to measure functional outcomes, and incorporation of a randomized controlled trial.
In summary, the new MIV approach is easy to perform and is compatible with more extensile approaches such as a quadriceps snip, if required in revision scenarios. The MIV approach preserves the extensor mechanism, vascularity, and innervation to the vastus medialis. Our study documents both safety and good clinical outcomes using the MIV approach in TKA. Therefore, the MIV approach may be used in the majority of patients undergoing TKA.
- Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785. doi:10.2106/JBJS.F.00222.
- Dutka J, Skowronek M, Sosin P, Skowronek P. Subvastus and medial parapatellar approaches in TKA: Comparison of functional results. Orthopedics. 2011;34(6):148-153. doi:10.3928/01477447-20110427-05.
- Hofmann AA, Plaster RL, Murdock LE. Subvastus (Southern) approach for primary total knee arthroplasty. Clin Orthop Relat Res. 1991;(269):70-77.
- Jojima H, Whiteside LA, Ogata K. Anatomic consideration of nerve supply to the vastus medialis in knee surgery. Clin Orthop Relat Res. 2004;423:157-160. doi:10.1097/01.blo.0000128642.61260.b3.
- Chavan SK, Wabale RN. Reviewing morphology of quadriceps femoris muscle. J Morphol Sci. 2016;33(2):112-117. doi:10.4322/jms.053513.
- Holt G, Nunn T, Allen RA, Forrester AW, Gregori A. Variation of the vastus medialis obliquus insertion and its relevance to minimally invasive total knee arthroplasty. J Arthroplasty. 2008;23(4):600-604. doi:10.1016/j.arth.2007.05.053.
- Sartawi M, Kohlman J, Valle C Della. Modified intervastus approach to the knee. J Knee Surg. 2017;31(05):422-424. doi:10.1055/s-0037-1604150.
- Hu X, Wang G, Pei F, et al. A meta-analysis of the sub-vastus approach and medial parapatellar approach in total knee arthroplasty. Knee Surg Sports Traumatol Arthrosc. 2013;21(10):2398-2404. doi:10.1007/s00167-012-2080-9.
- Jain S, Wasnik S, Mittal A, Hegde C. Outcome of subvastus approach in elderly nonobese patients undergoing bilateral simultaneous total knee arthroplasty: A randomized controlled study. Indian J Orthop. 2013;47(1):45-49. doi:10.4103/0019-5413.106900.
- Kholeif AM, Radwan YA, Mansour AM, Almoalem HA. Early functional results of the subvastus and medial parapatellar approaches in total knee arthroplasty. Med J Cairo Univ. 2017;85(3):849-854.
- Tzatzairis T, Fiska A, Ververidis A, Tilkeridis K, Kazakos K, Drosos GI. Minimally invasive versus conventional approaches in total knee replacement/arthroplasty: A review of the literature. J Orthop. 2018;15(2):459-466. doi:10.1016/j.jor.2018.03.026.
- Li Z, Cheng W, Sun L, Yao Y, Cao Q, Ye S, et al. Mini-subvastus versus medial parapatellar approach for total knee arthroplasty: a prospective randomized controlled study. Int Orthop. 2018;42(3):543-549. doi:10.1007/s00264-017-3703-z.
- Shah NA, Patil HG, Vaishnav VO, Savale A. Total knee arthroplasty using subvastus approach in stiff knee: a retrospective analysis of 110 cases. Indian J Orthop. 2016;50(2):166-171. doi:10.4103/0019-5413.177582.
- Berstock JR, Murray JR, Whitehouse MR, Blom AW, Beswick AD. Medial subvastus versus the medial parapatellar approach for total knee replacement: A systematic review and meta-analysis of randomized controlled trials. EFORT Open Rev. 2018;3(3):78-84. doi:10.1302/2058-5241.3.170030.
- Pan WM, Li XG, Tang TS, Qian ZL, Zhang Q, Zhang CM. Mini-subvastus versus a standard approach in total knee arthroplasty: A prospective, randomized, controlled study. J Int Med Res. 2010;38(3):890-900. doi:10.1177/147323001003800315.
- Bourke MG, Jull GA, Buttrum PJ, Fitzpatrick PL, Dalton PA, Russell TG. Comparing outcomes of medial parapatellar and subvastus approaches in total knee arthroplasty a randomized controlled trial. J Arthroplasty. 2011;27(3):347-353.e1. doi:10.1016/j.arth.2011.06.005.
- Tomek IM, Kantor SR, Cori LAA, et al. Early patient outcomes after primary total knee arthroplasty with quadriceps-sparing subvastus and medial parapatellar techniques: a randomized, double-blind clinical trial. J Bone Jt Surg Am. 2014;96(11):907-915. doi:10.2106/JBJS.L.01578.
- van Hemert WLW, Senden R, Grimm B, van der Linde MJA, Lataster A, Heyligers IC. Early functional outcome after subvastus or parapatellar approach in knee arthroplasty is comparable. Knee Surg Sports Traumatol Arthrosc. 2011;19(6):943-951. doi:10.1007/s00167-010-1292-0.
- Koh IJ, Kim MW, Kim MS, Jang SW, Park DC, In Y. The patient’s perception does not differ following subvastus and medial parapatellar approaches in total knee arthroplasty: a simultaneous bilateral randomized study. J Arthroplasty. 2016;31(1):112-117. doi:10.1016/j.arth.2015.08.004.
- Mehta N, Bhat MS, Goyal A, Mishra P, Joshi D, Chaudhary D. Quadriceps sparing (subvastus/midvastus) approach versus the conventional medial parapatellar approach in primary knee arthroplasty. J Arthrosc Jt Surg. 2017;4(1):15-20. doi:10.1016/j.jajs.2017.02.004.
- Bridgman SA, Walley G, MacKenzie G, Clement D, Griffiths D, Maffulli N. Sub-vastus approach is more effective than a medial parapatellar approach in primary total knee arthroplasty: A randomized controlled trial. Knee. 2009;16(3):216-222. doi:10.1016/j.knee.2008.11.012.
- Liu HW, Gu WD, Xu NW, Sun JY. Surgical approaches in total knee arthroplasty: A meta-analysis comparing the midvastus and subvastus to the medial peripatellar approach. J Arthroplasty. 2014;29(12):2298-2304. doi:10.1016/j.arth.2013.10.023.
- Varela-Egocheaga JR, Suarez-Suarez MA, Fernandez-Villan M, Gonzalez-Sastre V, Varela-Gomez JR, Rodriguez-Merchan C. Minimally invasive subvastus approach: Improving the results of total knee arthroplasty: a prospective, randomized trial. Clin Orthop Relat Res. 2010;468(5):1200-1208. doi:10.1007/s11999-009-1160-8.
ABSTRACT
The subvastus (SV) approach is a well-known muscle- and tendon-sparing approach for total knee arthroplasty (TKA), which has been shown in some studies to provide better outcomes in the visual analog pain score (VAS), knee range of motion (ROM), straight-leg raise, as well as faster rehabilitation, compared with the standard medial parapatellar (MP) approach. We previously described a new knee replacement technique known as the modified intervastus (MIV) approach. The MIV approach is a muscle- and tendon-sparing approach that is extensile and simple to perform. It may be used in the majority of complex primary cases and revisions. Here we describe the surgical technique for performing the MIV approach and provide functional outcome measures. A total of 127 patients (mean age, 66.75 years) underwent TKA using the MIV approach with 1-year follow-up. Clinical outcomes were assessed by recording both a VAS and knee ROM preoperatively, and again at several postoperative time points when the length of time required to ambulate independently (without assistive devices) was also measured. The VAS decreased significantly from the preoperative period (3.69 ± 2.22) to postoperative day 1 (3.17 ± 1.97) (P < .05). Although knee ROM decreased 1 week after surgery, the ROM increased by 6 weeks after surgery compared with the preoperative ROM, and the trend continued over the 1-year follow-up. One-third (33%) of patients were able to walk independently (without assistive devices) at 2 weeks and 78% at 8 weeks. The MIV approach to the knee is a muscle- and tendon-sparing approach that offers advantages over the SV approach and may be used for complex primary and revision total knee cases.
Continue to: Total knee arthroplasty...
Total knee arthroplasty (TKA) is one of the most common orthopedic surgical procedures, with more than 600,000 TKAs performed annually in the United States, and by 2030 the number is expected to reach 3.48 million per year.1Several approaches have been described for total TKA, and the medial parapatellar (MP) approach to the knee is considered the workhorse of total knee replacements. It is an extensile approach that is easy to perform but may delay active knee extension and straight-leg raise after surgery.2 Alternative approaches such as the subvastus (SV) and midvastus approaches to the knee (Figure 1A) typically allow a more rapid straight-leg raise but may be more challenging to perform and time-consuming in morbidly obese patients, muscular patients, and patients with severe deformities.3
The intervastus approach (Figure 1A) described by Jojima4 and others utilizes the interval between the quadriceps tendon and vastus medialis. Although it is a simple approach to perform that is extensile, it is not considered tendon-sparing since the vastus medialis inserts onto the medial aspect of the quadriceps tendon (Figure 2).5 Therefore, dissecting through this interval, without elevation of the vastus medialis (Figure 3A), damages the quadriceps tendon, and strangulation of the muscle occurs with repair of the arthrotomy site (Figure 3B). This situation is even more likely in patients with a low-lying vastus medialis.6
The modified intervastus (MIV) approach described previously,7 may be used in the majority of patients undergoing TKA. The advantages of this approach include its extensile nature, similarities to an MP approach, and preservation of both the extensor mechanism and the vastus medialis, leading to a more rapid return to active knee extension than is traditionally observed.2,7 The approach is simple to perform, easy to close, and is compatible with more extensile approaches such as a quadriceps snip if required in revision scenarios.7 However, functional outcomes of the MIV approach have not been quantified. It is unknown whether these outcomes will offer any advantages compared with the SV approach. Therefore, the objective of this study was to measure functional outcomes of the MIV approach and to compare the results with those previously published for the SV approach. We hypothesized that using the MIV approach for TKA surgery would lead to early straight-leg raise and increase in knee range of motion (ROM) postoperatively.
SURGICAL TECHNIQUE
The MIV approach preserves the quadriceps tendon and vastus medialis.7 After exposure of the vastus medialis muscle, the interval between the quadriceps tendon and vastus medialis is identified (Figure 4). The fascia overlying the lateral edge of the vastus medialis is incised where it meets the quadriceps tendon (Figure 5). The muscle is then bluntly elevated off the underlying capsule just enough to allow for a capsular repair later (Figure 6). An arthrotomy is then performed from cephalad to caudal (Figure 7). This interval may be extended proximally between the vastus intermedius and vastus medialis to expose the distal femur if needed. The closure is performed by repairing the capsule with absorbable suture (Figure 8A), and the vastus medialis fascia is repaired back to the medial edge of the quadriceps tendon, restoring the anatomy (Figure 8B).
Continue to: PATIENTS AND METHODS...
PATIENTS AND METHODS
A retrospective review of functional outcomes after TKA using the MIV approach was conducted; the study was approved by the University of Illinois Institutional Review Board. A total of 127 patients of mean age 66.75 years (range, 48–86 years) with primary osteoarthritis of the knee who were indicated for a total knee replacement with 1-year follow-up were included. The patient demographics are shown in Table 1. All patients underwent TKA using the MIV approach described above by 2 experienced orthopedic surgeons at the same institution. Patients received spinal anesthesia and a periarticular pain block intraoperatively. A measured resection technique was used by 1 surgeon, and a gap-balancing technique by the other. Surgeon 1 used the Persona PS cemented knee system (Zimmer Biomet, and Surgeon 2 used the Sigma PS cemented knee system (Depuy). Patellar resurfacing was done in all cases. Patellar tracking was checked intraoperatively using the ‘no-touch’ technique, and the need for a lateral release was noted. Drains were removed on postoperative day 1. Oral opioids were given as needed. Intravenous antibiotics were continued for 24 hours. Oral anticoagulants were used for thromboembolism prophylaxis for 3 weeks. Patients were mobilized on the day of surgery with full weight-bearing under the supervision of an experienced physical therapist. Static and dynamic quadriceps exercises were started on the same day of surgery along with active knee ROM exercises. Pain score, extensor lag, ROM, walking ability, and complications were recorded in all patients.
Table 1. Patient Demographics
Total no. of patients | 127 | |
Gender | Male | 44 |
Female | 83 | |
Age (years) | Mean ± Standard deviation | 66.75 ± 9.12 |
Range | 48 – 86 | |
Weight (lb) | Mean ± Standard deviation | 218.38 ± 54.47 |
Range | 125 – 364 | |
BMI (kg/m2) | Mean ± Standard deviation | 34.10 ± 7.22 |
Range | 21.1 – 62.5 | |
The visual analog score (VAS) was obtained preoperatively and recorded on postoperative day 1. Patient walking distance with assistance was measured on the day of surgery, after surgery, and on the day of hospital discharge. Patients were assessed preoperatively and postoperatively at 1 week, 2 weeks, 6 weeks, 3 months, 6 months, and 1 year for knee ROM. A one-way ANOVA was conducted to compare the preoperative and postoperative day 1 VAS with significance set at P < .05 (OriginPro 2015, OriginLab Corporation). Differences in knee ROM between preoperative and postoperative follow-up periods (1 week, 2 weeks, 6 weeks, 3 months, 6 months, and 1 year) were identified using a 1-way ANOVA with a post hoc Tukey test. Significance was set at P < .05.
RESULTS
All patients were able to fully straight-leg raise and demonstrate functional knee ROM by postoperative day 1. The patella tracked centrally in all patients, and none required a lateral retinacular release. The majority of patients were discharged in the first 48 hours after surgery on oral narcotics. None required IV narcotics during their hospital stay or a blood transfusion. Two cases were complicated by severe knee skin blistering postoperatively due to a reaction to an adhesive dressing; one was complicated by skin necrosis leading to a flap reconstruction that became infected, requiring a 2-stage revision. A separate case had an acute postoperative infection that required irrigation and debridement with polyethylene exchange. After a 12-week course of antibiotics, the infection was eradicated. All patients reported a high satisfaction rate during their acute postoperative phase.
Postoperatively, all patients were able to walk on the day of surgery either independently or with some assistance. On the day of surgery, 10% of patients were able to walk >200 feet, and this increased to 65% of patients able to walk >200 feet on the day of discharge (compare Figure 9A and Figure 9B). Within 2 weeks of surgery, 30% of patients could walk independently (without assistive devices), and this number increased to 78% by 8 weeks after surgery (Figure 10).
Pain assessed using the VAS was lower on postoperative day 1 (3.17 ± 1.97) than the preoperative score (3.69 ± 2.22, P< .05). Overall, knee ROM significantly increased during the follow-up after surgery. Initially, the ROM decreased 1 week after surgery (90.82 ± 10.28) compared with preoperative ROM (101.04 ± 19.48, P < .001) (Figure 10). At 2 weeks after surgery, knee ROM returned to the preoperative value (100.70 ± 13.36). By 6 weeks after surgery, knee ROM was 17° greater than the preoperative ROM (118.45 ± 11.89, P < .001). Knee ROM remained stable at 3- and 6-month assessments, and showed further improvement by 1 year (126.62 ± 9.81, P < .001) compared with the preoperative state (Figure 10). The net improvement in knee ROM was 25° of increased knee flexion by 1 year.
Continue to: DISCUSSION...
DISCUSSION
TKA is a successful procedure that restores knee function with pain relief in osteoarthritis patients. The SV approach for TKA has better outcomes in terms of the VAS, ROM, straight-leg raise with faster rehabilitation compared with the standard MP approach;8–12 however, it can be challenging and time-consuming when used in morbidly obese and muscular patients.3 The SV approach can also increase the risk of complications such as patellar tendon avulsion or medial collateral injury because of the difficulty in exposure specifically for knees with limited ROM.13 Here we introduce the MIV approach as an alternative to the SV approach, overcoming most of these difficulties.
With the prevalence of morbid obesity and the market demand for minimally invasive techniques, we believe the MIV approach represents a good approach for surgeons since it is easy to perform, does not require specialized instrumentation, and is a reproducible approach even on the most complex deformities. The minimal time added to ensure blunt elevation of the vastus medialis muscle and an anatomic repair of the underlying knee capsule and vastus medialis fascia to the medial edge of the quadriceps tendon allows restoration of the anatomy and a robust double-layered watertight seal closure with no strangulation of soft tissues. We believe this reproducible muscle- and tendon-sparing approach that allows gentle, soft tissue handling even in the most complex primary total knee cases may lead to less soft tissue swelling, and therefore, less postoperative pain resulting in an accelerated recovery.
The pain level in this group of patients was reduced after the MIV approach as indicated by the VAS. The VAS was significantly decreased on postoperative day 1 compared with the VAS recorded preoperatively (P < .05), indicating patients felt less pain on the day after surgery. The average VAS on postoperative day 1 from other studies for SV approach ranged from 2.1 to 5,9,12,14–19 whereas our MIV approach value was 3.17. Periarticular blocks were available for this study group, and no peripheral nerve blocks were used. Some studies of the SV approach mention the use of peripheral nerve blocks, while others did not describe the method used for treatment or control of postoperative pain. The decreased reported pain levels and the observed increased knee ROM seen in the MIV and SV approach study groups might be attributable to the treatment of postoperative pain.
Patient ambulation also increased from the day of surgery to the day of discharge for the MIV approach. Only 10% of patients were able to walk with assistance >200 feet on the day of surgery; however, this percentage increased to 65% on the discharge day showing an excellent recovery of walking ability within 2 days of surgery. Mehta and colleagues20reported that 95% of patients undergoing subvastus/midvastus approaches could walk >10 blocks at 6 months follow-up. For the MIV approach, 78% of patients were able to walk independently within 8 weeks of surgery.
Continue to: The MIV approach shows improvement...
The MIV approach shows improvement in functional outcomes in terms of knee ROM from preoperative to postoperative. Although knee ROM decreased significantly at postoperative week 1, which is most likely because of a peak in postoperative pain and swelling; the ROM started to increase significantly at postoperative week 6. After 1 year, the knee ROM increased by 25% compared with preoperative ROM for the MIV approach. We found the average knee ROM at postoperative 3 months using the MIV approach to be 120.4°. Knee ROM at postoperative 3 months using the SV approach reported by other studies ranged from 87.1° to 120°.19,21–23 Similarly, after 1 year, the average ROM by MIV approach was 126.62°, whereas average knee ROM from other studies using the SV approach was 114.1°.2,10,19,21 Achieving a knee ROM of 120° allows patients to return to their baseline function and perform activities of daily living without limitation. With the prevalence of knee osteoarthritis in the younger population, the need for increased knee ROM post-TKA becomes more important so patients can return to work and function at their baseline.
In addition to being both a muscle- and tendon-sparing approach, other advantages of the MIV approach are that it is simple to perform and gives similar exposure to the MP approach even in difficult primary cases. It is also as extensile as the medial parapatellar approach, and it can be converted to a quadriceps snip if required. There is also minimal, if any, muscle retraction preventing muscle injury that may be seen with some other muscle-sparing approaches to the knee, especially in more difficult primary total knee cases. Another advantage to this approach, as opposed to the other approaches to the knee described here, is that it allows a double-layered closure, decreasing the possibility of arthrotomy dehiscence. Because of this double-layered closure, there is no muscle strangulation with repair of the arthrotomy site as seen in some other approaches to the knee, and both vascularity and innervation to the vastus medialis are preserved.
One limitation of this study is that it is a retrospective analysis. Future studies of the consequences of using this approach need to include quantitative assessment of muscle function using electromyography or dynamometer measurements, utilization of any number of validated knee score systems to measure functional outcomes, and incorporation of a randomized controlled trial.
In summary, the new MIV approach is easy to perform and is compatible with more extensile approaches such as a quadriceps snip, if required in revision scenarios. The MIV approach preserves the extensor mechanism, vascularity, and innervation to the vastus medialis. Our study documents both safety and good clinical outcomes using the MIV approach in TKA. Therefore, the MIV approach may be used in the majority of patients undergoing TKA.
ABSTRACT
The subvastus (SV) approach is a well-known muscle- and tendon-sparing approach for total knee arthroplasty (TKA), which has been shown in some studies to provide better outcomes in the visual analog pain score (VAS), knee range of motion (ROM), straight-leg raise, as well as faster rehabilitation, compared with the standard medial parapatellar (MP) approach. We previously described a new knee replacement technique known as the modified intervastus (MIV) approach. The MIV approach is a muscle- and tendon-sparing approach that is extensile and simple to perform. It may be used in the majority of complex primary cases and revisions. Here we describe the surgical technique for performing the MIV approach and provide functional outcome measures. A total of 127 patients (mean age, 66.75 years) underwent TKA using the MIV approach with 1-year follow-up. Clinical outcomes were assessed by recording both a VAS and knee ROM preoperatively, and again at several postoperative time points when the length of time required to ambulate independently (without assistive devices) was also measured. The VAS decreased significantly from the preoperative period (3.69 ± 2.22) to postoperative day 1 (3.17 ± 1.97) (P < .05). Although knee ROM decreased 1 week after surgery, the ROM increased by 6 weeks after surgery compared with the preoperative ROM, and the trend continued over the 1-year follow-up. One-third (33%) of patients were able to walk independently (without assistive devices) at 2 weeks and 78% at 8 weeks. The MIV approach to the knee is a muscle- and tendon-sparing approach that offers advantages over the SV approach and may be used for complex primary and revision total knee cases.
Continue to: Total knee arthroplasty...
Total knee arthroplasty (TKA) is one of the most common orthopedic surgical procedures, with more than 600,000 TKAs performed annually in the United States, and by 2030 the number is expected to reach 3.48 million per year.1Several approaches have been described for total TKA, and the medial parapatellar (MP) approach to the knee is considered the workhorse of total knee replacements. It is an extensile approach that is easy to perform but may delay active knee extension and straight-leg raise after surgery.2 Alternative approaches such as the subvastus (SV) and midvastus approaches to the knee (Figure 1A) typically allow a more rapid straight-leg raise but may be more challenging to perform and time-consuming in morbidly obese patients, muscular patients, and patients with severe deformities.3
The intervastus approach (Figure 1A) described by Jojima4 and others utilizes the interval between the quadriceps tendon and vastus medialis. Although it is a simple approach to perform that is extensile, it is not considered tendon-sparing since the vastus medialis inserts onto the medial aspect of the quadriceps tendon (Figure 2).5 Therefore, dissecting through this interval, without elevation of the vastus medialis (Figure 3A), damages the quadriceps tendon, and strangulation of the muscle occurs with repair of the arthrotomy site (Figure 3B). This situation is even more likely in patients with a low-lying vastus medialis.6
The modified intervastus (MIV) approach described previously,7 may be used in the majority of patients undergoing TKA. The advantages of this approach include its extensile nature, similarities to an MP approach, and preservation of both the extensor mechanism and the vastus medialis, leading to a more rapid return to active knee extension than is traditionally observed.2,7 The approach is simple to perform, easy to close, and is compatible with more extensile approaches such as a quadriceps snip if required in revision scenarios.7 However, functional outcomes of the MIV approach have not been quantified. It is unknown whether these outcomes will offer any advantages compared with the SV approach. Therefore, the objective of this study was to measure functional outcomes of the MIV approach and to compare the results with those previously published for the SV approach. We hypothesized that using the MIV approach for TKA surgery would lead to early straight-leg raise and increase in knee range of motion (ROM) postoperatively.
SURGICAL TECHNIQUE
The MIV approach preserves the quadriceps tendon and vastus medialis.7 After exposure of the vastus medialis muscle, the interval between the quadriceps tendon and vastus medialis is identified (Figure 4). The fascia overlying the lateral edge of the vastus medialis is incised where it meets the quadriceps tendon (Figure 5). The muscle is then bluntly elevated off the underlying capsule just enough to allow for a capsular repair later (Figure 6). An arthrotomy is then performed from cephalad to caudal (Figure 7). This interval may be extended proximally between the vastus intermedius and vastus medialis to expose the distal femur if needed. The closure is performed by repairing the capsule with absorbable suture (Figure 8A), and the vastus medialis fascia is repaired back to the medial edge of the quadriceps tendon, restoring the anatomy (Figure 8B).
Continue to: PATIENTS AND METHODS...
PATIENTS AND METHODS
A retrospective review of functional outcomes after TKA using the MIV approach was conducted; the study was approved by the University of Illinois Institutional Review Board. A total of 127 patients of mean age 66.75 years (range, 48–86 years) with primary osteoarthritis of the knee who were indicated for a total knee replacement with 1-year follow-up were included. The patient demographics are shown in Table 1. All patients underwent TKA using the MIV approach described above by 2 experienced orthopedic surgeons at the same institution. Patients received spinal anesthesia and a periarticular pain block intraoperatively. A measured resection technique was used by 1 surgeon, and a gap-balancing technique by the other. Surgeon 1 used the Persona PS cemented knee system (Zimmer Biomet, and Surgeon 2 used the Sigma PS cemented knee system (Depuy). Patellar resurfacing was done in all cases. Patellar tracking was checked intraoperatively using the ‘no-touch’ technique, and the need for a lateral release was noted. Drains were removed on postoperative day 1. Oral opioids were given as needed. Intravenous antibiotics were continued for 24 hours. Oral anticoagulants were used for thromboembolism prophylaxis for 3 weeks. Patients were mobilized on the day of surgery with full weight-bearing under the supervision of an experienced physical therapist. Static and dynamic quadriceps exercises were started on the same day of surgery along with active knee ROM exercises. Pain score, extensor lag, ROM, walking ability, and complications were recorded in all patients.
Table 1. Patient Demographics
Total no. of patients | 127 | |
Gender | Male | 44 |
Female | 83 | |
Age (years) | Mean ± Standard deviation | 66.75 ± 9.12 |
Range | 48 – 86 | |
Weight (lb) | Mean ± Standard deviation | 218.38 ± 54.47 |
Range | 125 – 364 | |
BMI (kg/m2) | Mean ± Standard deviation | 34.10 ± 7.22 |
Range | 21.1 – 62.5 | |
The visual analog score (VAS) was obtained preoperatively and recorded on postoperative day 1. Patient walking distance with assistance was measured on the day of surgery, after surgery, and on the day of hospital discharge. Patients were assessed preoperatively and postoperatively at 1 week, 2 weeks, 6 weeks, 3 months, 6 months, and 1 year for knee ROM. A one-way ANOVA was conducted to compare the preoperative and postoperative day 1 VAS with significance set at P < .05 (OriginPro 2015, OriginLab Corporation). Differences in knee ROM between preoperative and postoperative follow-up periods (1 week, 2 weeks, 6 weeks, 3 months, 6 months, and 1 year) were identified using a 1-way ANOVA with a post hoc Tukey test. Significance was set at P < .05.
RESULTS
All patients were able to fully straight-leg raise and demonstrate functional knee ROM by postoperative day 1. The patella tracked centrally in all patients, and none required a lateral retinacular release. The majority of patients were discharged in the first 48 hours after surgery on oral narcotics. None required IV narcotics during their hospital stay or a blood transfusion. Two cases were complicated by severe knee skin blistering postoperatively due to a reaction to an adhesive dressing; one was complicated by skin necrosis leading to a flap reconstruction that became infected, requiring a 2-stage revision. A separate case had an acute postoperative infection that required irrigation and debridement with polyethylene exchange. After a 12-week course of antibiotics, the infection was eradicated. All patients reported a high satisfaction rate during their acute postoperative phase.
Postoperatively, all patients were able to walk on the day of surgery either independently or with some assistance. On the day of surgery, 10% of patients were able to walk >200 feet, and this increased to 65% of patients able to walk >200 feet on the day of discharge (compare Figure 9A and Figure 9B). Within 2 weeks of surgery, 30% of patients could walk independently (without assistive devices), and this number increased to 78% by 8 weeks after surgery (Figure 10).
Pain assessed using the VAS was lower on postoperative day 1 (3.17 ± 1.97) than the preoperative score (3.69 ± 2.22, P< .05). Overall, knee ROM significantly increased during the follow-up after surgery. Initially, the ROM decreased 1 week after surgery (90.82 ± 10.28) compared with preoperative ROM (101.04 ± 19.48, P < .001) (Figure 10). At 2 weeks after surgery, knee ROM returned to the preoperative value (100.70 ± 13.36). By 6 weeks after surgery, knee ROM was 17° greater than the preoperative ROM (118.45 ± 11.89, P < .001). Knee ROM remained stable at 3- and 6-month assessments, and showed further improvement by 1 year (126.62 ± 9.81, P < .001) compared with the preoperative state (Figure 10). The net improvement in knee ROM was 25° of increased knee flexion by 1 year.
Continue to: DISCUSSION...
DISCUSSION
TKA is a successful procedure that restores knee function with pain relief in osteoarthritis patients. The SV approach for TKA has better outcomes in terms of the VAS, ROM, straight-leg raise with faster rehabilitation compared with the standard MP approach;8–12 however, it can be challenging and time-consuming when used in morbidly obese and muscular patients.3 The SV approach can also increase the risk of complications such as patellar tendon avulsion or medial collateral injury because of the difficulty in exposure specifically for knees with limited ROM.13 Here we introduce the MIV approach as an alternative to the SV approach, overcoming most of these difficulties.
With the prevalence of morbid obesity and the market demand for minimally invasive techniques, we believe the MIV approach represents a good approach for surgeons since it is easy to perform, does not require specialized instrumentation, and is a reproducible approach even on the most complex deformities. The minimal time added to ensure blunt elevation of the vastus medialis muscle and an anatomic repair of the underlying knee capsule and vastus medialis fascia to the medial edge of the quadriceps tendon allows restoration of the anatomy and a robust double-layered watertight seal closure with no strangulation of soft tissues. We believe this reproducible muscle- and tendon-sparing approach that allows gentle, soft tissue handling even in the most complex primary total knee cases may lead to less soft tissue swelling, and therefore, less postoperative pain resulting in an accelerated recovery.
The pain level in this group of patients was reduced after the MIV approach as indicated by the VAS. The VAS was significantly decreased on postoperative day 1 compared with the VAS recorded preoperatively (P < .05), indicating patients felt less pain on the day after surgery. The average VAS on postoperative day 1 from other studies for SV approach ranged from 2.1 to 5,9,12,14–19 whereas our MIV approach value was 3.17. Periarticular blocks were available for this study group, and no peripheral nerve blocks were used. Some studies of the SV approach mention the use of peripheral nerve blocks, while others did not describe the method used for treatment or control of postoperative pain. The decreased reported pain levels and the observed increased knee ROM seen in the MIV and SV approach study groups might be attributable to the treatment of postoperative pain.
Patient ambulation also increased from the day of surgery to the day of discharge for the MIV approach. Only 10% of patients were able to walk with assistance >200 feet on the day of surgery; however, this percentage increased to 65% on the discharge day showing an excellent recovery of walking ability within 2 days of surgery. Mehta and colleagues20reported that 95% of patients undergoing subvastus/midvastus approaches could walk >10 blocks at 6 months follow-up. For the MIV approach, 78% of patients were able to walk independently within 8 weeks of surgery.
Continue to: The MIV approach shows improvement...
The MIV approach shows improvement in functional outcomes in terms of knee ROM from preoperative to postoperative. Although knee ROM decreased significantly at postoperative week 1, which is most likely because of a peak in postoperative pain and swelling; the ROM started to increase significantly at postoperative week 6. After 1 year, the knee ROM increased by 25% compared with preoperative ROM for the MIV approach. We found the average knee ROM at postoperative 3 months using the MIV approach to be 120.4°. Knee ROM at postoperative 3 months using the SV approach reported by other studies ranged from 87.1° to 120°.19,21–23 Similarly, after 1 year, the average ROM by MIV approach was 126.62°, whereas average knee ROM from other studies using the SV approach was 114.1°.2,10,19,21 Achieving a knee ROM of 120° allows patients to return to their baseline function and perform activities of daily living without limitation. With the prevalence of knee osteoarthritis in the younger population, the need for increased knee ROM post-TKA becomes more important so patients can return to work and function at their baseline.
In addition to being both a muscle- and tendon-sparing approach, other advantages of the MIV approach are that it is simple to perform and gives similar exposure to the MP approach even in difficult primary cases. It is also as extensile as the medial parapatellar approach, and it can be converted to a quadriceps snip if required. There is also minimal, if any, muscle retraction preventing muscle injury that may be seen with some other muscle-sparing approaches to the knee, especially in more difficult primary total knee cases. Another advantage to this approach, as opposed to the other approaches to the knee described here, is that it allows a double-layered closure, decreasing the possibility of arthrotomy dehiscence. Because of this double-layered closure, there is no muscle strangulation with repair of the arthrotomy site as seen in some other approaches to the knee, and both vascularity and innervation to the vastus medialis are preserved.
One limitation of this study is that it is a retrospective analysis. Future studies of the consequences of using this approach need to include quantitative assessment of muscle function using electromyography or dynamometer measurements, utilization of any number of validated knee score systems to measure functional outcomes, and incorporation of a randomized controlled trial.
In summary, the new MIV approach is easy to perform and is compatible with more extensile approaches such as a quadriceps snip, if required in revision scenarios. The MIV approach preserves the extensor mechanism, vascularity, and innervation to the vastus medialis. Our study documents both safety and good clinical outcomes using the MIV approach in TKA. Therefore, the MIV approach may be used in the majority of patients undergoing TKA.
- Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785. doi:10.2106/JBJS.F.00222.
- Dutka J, Skowronek M, Sosin P, Skowronek P. Subvastus and medial parapatellar approaches in TKA: Comparison of functional results. Orthopedics. 2011;34(6):148-153. doi:10.3928/01477447-20110427-05.
- Hofmann AA, Plaster RL, Murdock LE. Subvastus (Southern) approach for primary total knee arthroplasty. Clin Orthop Relat Res. 1991;(269):70-77.
- Jojima H, Whiteside LA, Ogata K. Anatomic consideration of nerve supply to the vastus medialis in knee surgery. Clin Orthop Relat Res. 2004;423:157-160. doi:10.1097/01.blo.0000128642.61260.b3.
- Chavan SK, Wabale RN. Reviewing morphology of quadriceps femoris muscle. J Morphol Sci. 2016;33(2):112-117. doi:10.4322/jms.053513.
- Holt G, Nunn T, Allen RA, Forrester AW, Gregori A. Variation of the vastus medialis obliquus insertion and its relevance to minimally invasive total knee arthroplasty. J Arthroplasty. 2008;23(4):600-604. doi:10.1016/j.arth.2007.05.053.
- Sartawi M, Kohlman J, Valle C Della. Modified intervastus approach to the knee. J Knee Surg. 2017;31(05):422-424. doi:10.1055/s-0037-1604150.
- Hu X, Wang G, Pei F, et al. A meta-analysis of the sub-vastus approach and medial parapatellar approach in total knee arthroplasty. Knee Surg Sports Traumatol Arthrosc. 2013;21(10):2398-2404. doi:10.1007/s00167-012-2080-9.
- Jain S, Wasnik S, Mittal A, Hegde C. Outcome of subvastus approach in elderly nonobese patients undergoing bilateral simultaneous total knee arthroplasty: A randomized controlled study. Indian J Orthop. 2013;47(1):45-49. doi:10.4103/0019-5413.106900.
- Kholeif AM, Radwan YA, Mansour AM, Almoalem HA. Early functional results of the subvastus and medial parapatellar approaches in total knee arthroplasty. Med J Cairo Univ. 2017;85(3):849-854.
- Tzatzairis T, Fiska A, Ververidis A, Tilkeridis K, Kazakos K, Drosos GI. Minimally invasive versus conventional approaches in total knee replacement/arthroplasty: A review of the literature. J Orthop. 2018;15(2):459-466. doi:10.1016/j.jor.2018.03.026.
- Li Z, Cheng W, Sun L, Yao Y, Cao Q, Ye S, et al. Mini-subvastus versus medial parapatellar approach for total knee arthroplasty: a prospective randomized controlled study. Int Orthop. 2018;42(3):543-549. doi:10.1007/s00264-017-3703-z.
- Shah NA, Patil HG, Vaishnav VO, Savale A. Total knee arthroplasty using subvastus approach in stiff knee: a retrospective analysis of 110 cases. Indian J Orthop. 2016;50(2):166-171. doi:10.4103/0019-5413.177582.
- Berstock JR, Murray JR, Whitehouse MR, Blom AW, Beswick AD. Medial subvastus versus the medial parapatellar approach for total knee replacement: A systematic review and meta-analysis of randomized controlled trials. EFORT Open Rev. 2018;3(3):78-84. doi:10.1302/2058-5241.3.170030.
- Pan WM, Li XG, Tang TS, Qian ZL, Zhang Q, Zhang CM. Mini-subvastus versus a standard approach in total knee arthroplasty: A prospective, randomized, controlled study. J Int Med Res. 2010;38(3):890-900. doi:10.1177/147323001003800315.
- Bourke MG, Jull GA, Buttrum PJ, Fitzpatrick PL, Dalton PA, Russell TG. Comparing outcomes of medial parapatellar and subvastus approaches in total knee arthroplasty a randomized controlled trial. J Arthroplasty. 2011;27(3):347-353.e1. doi:10.1016/j.arth.2011.06.005.
- Tomek IM, Kantor SR, Cori LAA, et al. Early patient outcomes after primary total knee arthroplasty with quadriceps-sparing subvastus and medial parapatellar techniques: a randomized, double-blind clinical trial. J Bone Jt Surg Am. 2014;96(11):907-915. doi:10.2106/JBJS.L.01578.
- van Hemert WLW, Senden R, Grimm B, van der Linde MJA, Lataster A, Heyligers IC. Early functional outcome after subvastus or parapatellar approach in knee arthroplasty is comparable. Knee Surg Sports Traumatol Arthrosc. 2011;19(6):943-951. doi:10.1007/s00167-010-1292-0.
- Koh IJ, Kim MW, Kim MS, Jang SW, Park DC, In Y. The patient’s perception does not differ following subvastus and medial parapatellar approaches in total knee arthroplasty: a simultaneous bilateral randomized study. J Arthroplasty. 2016;31(1):112-117. doi:10.1016/j.arth.2015.08.004.
- Mehta N, Bhat MS, Goyal A, Mishra P, Joshi D, Chaudhary D. Quadriceps sparing (subvastus/midvastus) approach versus the conventional medial parapatellar approach in primary knee arthroplasty. J Arthrosc Jt Surg. 2017;4(1):15-20. doi:10.1016/j.jajs.2017.02.004.
- Bridgman SA, Walley G, MacKenzie G, Clement D, Griffiths D, Maffulli N. Sub-vastus approach is more effective than a medial parapatellar approach in primary total knee arthroplasty: A randomized controlled trial. Knee. 2009;16(3):216-222. doi:10.1016/j.knee.2008.11.012.
- Liu HW, Gu WD, Xu NW, Sun JY. Surgical approaches in total knee arthroplasty: A meta-analysis comparing the midvastus and subvastus to the medial peripatellar approach. J Arthroplasty. 2014;29(12):2298-2304. doi:10.1016/j.arth.2013.10.023.
- Varela-Egocheaga JR, Suarez-Suarez MA, Fernandez-Villan M, Gonzalez-Sastre V, Varela-Gomez JR, Rodriguez-Merchan C. Minimally invasive subvastus approach: Improving the results of total knee arthroplasty: a prospective, randomized trial. Clin Orthop Relat Res. 2010;468(5):1200-1208. doi:10.1007/s11999-009-1160-8.
- Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785. doi:10.2106/JBJS.F.00222.
- Dutka J, Skowronek M, Sosin P, Skowronek P. Subvastus and medial parapatellar approaches in TKA: Comparison of functional results. Orthopedics. 2011;34(6):148-153. doi:10.3928/01477447-20110427-05.
- Hofmann AA, Plaster RL, Murdock LE. Subvastus (Southern) approach for primary total knee arthroplasty. Clin Orthop Relat Res. 1991;(269):70-77.
- Jojima H, Whiteside LA, Ogata K. Anatomic consideration of nerve supply to the vastus medialis in knee surgery. Clin Orthop Relat Res. 2004;423:157-160. doi:10.1097/01.blo.0000128642.61260.b3.
- Chavan SK, Wabale RN. Reviewing morphology of quadriceps femoris muscle. J Morphol Sci. 2016;33(2):112-117. doi:10.4322/jms.053513.
- Holt G, Nunn T, Allen RA, Forrester AW, Gregori A. Variation of the vastus medialis obliquus insertion and its relevance to minimally invasive total knee arthroplasty. J Arthroplasty. 2008;23(4):600-604. doi:10.1016/j.arth.2007.05.053.
- Sartawi M, Kohlman J, Valle C Della. Modified intervastus approach to the knee. J Knee Surg. 2017;31(05):422-424. doi:10.1055/s-0037-1604150.
- Hu X, Wang G, Pei F, et al. A meta-analysis of the sub-vastus approach and medial parapatellar approach in total knee arthroplasty. Knee Surg Sports Traumatol Arthrosc. 2013;21(10):2398-2404. doi:10.1007/s00167-012-2080-9.
- Jain S, Wasnik S, Mittal A, Hegde C. Outcome of subvastus approach in elderly nonobese patients undergoing bilateral simultaneous total knee arthroplasty: A randomized controlled study. Indian J Orthop. 2013;47(1):45-49. doi:10.4103/0019-5413.106900.
- Kholeif AM, Radwan YA, Mansour AM, Almoalem HA. Early functional results of the subvastus and medial parapatellar approaches in total knee arthroplasty. Med J Cairo Univ. 2017;85(3):849-854.
- Tzatzairis T, Fiska A, Ververidis A, Tilkeridis K, Kazakos K, Drosos GI. Minimally invasive versus conventional approaches in total knee replacement/arthroplasty: A review of the literature. J Orthop. 2018;15(2):459-466. doi:10.1016/j.jor.2018.03.026.
- Li Z, Cheng W, Sun L, Yao Y, Cao Q, Ye S, et al. Mini-subvastus versus medial parapatellar approach for total knee arthroplasty: a prospective randomized controlled study. Int Orthop. 2018;42(3):543-549. doi:10.1007/s00264-017-3703-z.
- Shah NA, Patil HG, Vaishnav VO, Savale A. Total knee arthroplasty using subvastus approach in stiff knee: a retrospective analysis of 110 cases. Indian J Orthop. 2016;50(2):166-171. doi:10.4103/0019-5413.177582.
- Berstock JR, Murray JR, Whitehouse MR, Blom AW, Beswick AD. Medial subvastus versus the medial parapatellar approach for total knee replacement: A systematic review and meta-analysis of randomized controlled trials. EFORT Open Rev. 2018;3(3):78-84. doi:10.1302/2058-5241.3.170030.
- Pan WM, Li XG, Tang TS, Qian ZL, Zhang Q, Zhang CM. Mini-subvastus versus a standard approach in total knee arthroplasty: A prospective, randomized, controlled study. J Int Med Res. 2010;38(3):890-900. doi:10.1177/147323001003800315.
- Bourke MG, Jull GA, Buttrum PJ, Fitzpatrick PL, Dalton PA, Russell TG. Comparing outcomes of medial parapatellar and subvastus approaches in total knee arthroplasty a randomized controlled trial. J Arthroplasty. 2011;27(3):347-353.e1. doi:10.1016/j.arth.2011.06.005.
- Tomek IM, Kantor SR, Cori LAA, et al. Early patient outcomes after primary total knee arthroplasty with quadriceps-sparing subvastus and medial parapatellar techniques: a randomized, double-blind clinical trial. J Bone Jt Surg Am. 2014;96(11):907-915. doi:10.2106/JBJS.L.01578.
- van Hemert WLW, Senden R, Grimm B, van der Linde MJA, Lataster A, Heyligers IC. Early functional outcome after subvastus or parapatellar approach in knee arthroplasty is comparable. Knee Surg Sports Traumatol Arthrosc. 2011;19(6):943-951. doi:10.1007/s00167-010-1292-0.
- Koh IJ, Kim MW, Kim MS, Jang SW, Park DC, In Y. The patient’s perception does not differ following subvastus and medial parapatellar approaches in total knee arthroplasty: a simultaneous bilateral randomized study. J Arthroplasty. 2016;31(1):112-117. doi:10.1016/j.arth.2015.08.004.
- Mehta N, Bhat MS, Goyal A, Mishra P, Joshi D, Chaudhary D. Quadriceps sparing (subvastus/midvastus) approach versus the conventional medial parapatellar approach in primary knee arthroplasty. J Arthrosc Jt Surg. 2017;4(1):15-20. doi:10.1016/j.jajs.2017.02.004.
- Bridgman SA, Walley G, MacKenzie G, Clement D, Griffiths D, Maffulli N. Sub-vastus approach is more effective than a medial parapatellar approach in primary total knee arthroplasty: A randomized controlled trial. Knee. 2009;16(3):216-222. doi:10.1016/j.knee.2008.11.012.
- Liu HW, Gu WD, Xu NW, Sun JY. Surgical approaches in total knee arthroplasty: A meta-analysis comparing the midvastus and subvastus to the medial peripatellar approach. J Arthroplasty. 2014;29(12):2298-2304. doi:10.1016/j.arth.2013.10.023.
- Varela-Egocheaga JR, Suarez-Suarez MA, Fernandez-Villan M, Gonzalez-Sastre V, Varela-Gomez JR, Rodriguez-Merchan C. Minimally invasive subvastus approach: Improving the results of total knee arthroplasty: a prospective, randomized trial. Clin Orthop Relat Res. 2010;468(5):1200-1208. doi:10.1007/s11999-009-1160-8.
TAKE-HOME POINTS
- The modified intervastus approach to the knee spares the quadriceps tendon and the vastus medialis muscle.
- The modified intervastus approach is an extensile approach.
- The modified intersvastus approach is a safe approach to the knee that can be used in total knee arthroplasty and leads to early straight-leg raise and a rapid recovery.
- The modified intervastus approach can be used on the majority of patients requiring a total knee replacement.
- The modified intervastus approach utilizes a unique closure that avoids soft tissue strangulation.
Texas judge strikes down ACA putting law in peril — again
The future of the Affordable Care Act is threatened – again – this time by a ruling Friday from a federal district court judge in Texas.
Judge Reed C. O’Connor struck down the law, siding with a group of 18 Republican state attorneys general and two GOP governors who brought the case. Judge O’Connor said the tax bill passed by Congress in December 2017 effectively rendered the entire health law unconstitutional.
That tax measure eliminated the penalty for not having insurance. An earlier Supreme Court decision upheld the ACA based on the view that the penalty was a tax and thus the law was valid because it relied on appropriate power allowed Congress under the Constitution. Judge O’Connor’s decision said that without that penalty, the law no longer met that constitutional test.
“In some ways, the question before the court involves the intent of both the 2010 and 2017 Congresses,” Judge O’Connor wrote in his 55-page decision. “The former enacted the ACA. The latter sawed off the last leg it stood on.”
The decision came just hours before the end of open enrollment for ACA plans in most states that use the federal HealthCare.gov insurance exchange. It is not expected that the ruling will impact the coverage for those people – the final decision will likely not come until the case reaches the Supreme Court again.
Seema Verma, the administrator of the Centers for Medicare & Medicaid Services, which oversees those insurance exchanges, said in a tweet: “The recent federal court decision is still moving through the courts, and the exchanges are still open for business and we will continue with open enrollment. There is no impact to current coverage or coverage in a 2019 plan.”
The 16 Democratic state attorneys general who intervened in the case to defend the health law immediately vowed to appeal.
“The ACA has already survived more than 70 unsuccessful repeal attempts and withstood scrutiny in the Supreme Court,” said a statement from California Attorney General Xavier Becerra. “Today’s misguided ruling will not deter us: our coalition will continue to fight in court for the health and wellbeing of all Americans.”
It is all but certain the case will become the third time the Supreme Court decides a constitutional question related to the ACA. In addition to upholding the law in 2012, the court rejected another challenge to the law in 2015.
It is hard to overstate what would happen to the nation’s health care system if the decision is ultimately upheld. The Affordable Care Act touched almost every aspect of health care, from Medicare and Medicaid to generic biologic drugs, the Indian Health Service, and public health changes like calorie counts on menus.
The case, Texas v. United States, was filed in February. The plaintiffs argued that because the Supreme Court upheld the ACA in 2012 as a constitutional use of its taxing power, the elimination of the tax makes the rest of the law unconstitutional.
In June, the Justice Department announced it would not fully defend the law in court. While the Trump administration said it did not agree with the plaintiffs that the tax law meant the entire ACA was unconstitutional, it said that the provisions of the law guaranteeing that people with preexisting health conditions could purchase coverage at the same price as everyone else were so inextricably linked to the tax penalty that they should be struck.
The administration urged the court to declare those provisions invalid beginning Jan. 1, 2019. That is the day the tax penalty for not having insurance disappears.
The protections for people with preexisting conditions was one of the top health issues in the midterm elections in November. While the issue mostly played to the advantage of Democrats, one of the Republican plaintiffs, Missouri Attorney General Josh Hawley, defeated Democratic incumbent Sen. Claire McCaskill. Another plaintiff, West Virginia Attorney General Patrick Morrisey, lost to Democratic incumbent Sen. Joe Manchin.
President Donald Trump was quick to take a victory lap, and pressed Senate Majority Leader Mitch McConnell (R-Ky.) and presumed incoming House Speaker Nancy Pelosi (D-Calif.) to fix the problem. He tweeted Friday night that “As I predicted all along, Obamacare has been struck down as an UNCONSTITUTIONAL disaster! Now Congress must pass a STRONG law that provides GREAT healthcare and protects pre-existing conditions. Mitch and Nancy, get it done!”
But congressional leaders were quick to point out that the suit is far from over.
“The ruling seems to be based on faulty legal reasoning and hopefully it will be overturned,” said a statement from Senate Minority Leader Chuck Schumer (D-N.Y.).
Many legal experts agreed with that. “This is insanity in print, and it will not stand up on appeal,” tweeted University of Michigan Law School professor Nicholas Bagley, an expert in health law.
Even some conservatives were left scratching their heads. “Congress acted last year to repeal the mandate, but leave everything else in place and the courts should have deferred to that,” tweeted former congressional GOP aide Chris Jacobs.
Kaiser Health News (KHN) is a national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation which is not affiliated with Kaiser Permanente.
The future of the Affordable Care Act is threatened – again – this time by a ruling Friday from a federal district court judge in Texas.
Judge Reed C. O’Connor struck down the law, siding with a group of 18 Republican state attorneys general and two GOP governors who brought the case. Judge O’Connor said the tax bill passed by Congress in December 2017 effectively rendered the entire health law unconstitutional.
That tax measure eliminated the penalty for not having insurance. An earlier Supreme Court decision upheld the ACA based on the view that the penalty was a tax and thus the law was valid because it relied on appropriate power allowed Congress under the Constitution. Judge O’Connor’s decision said that without that penalty, the law no longer met that constitutional test.
“In some ways, the question before the court involves the intent of both the 2010 and 2017 Congresses,” Judge O’Connor wrote in his 55-page decision. “The former enacted the ACA. The latter sawed off the last leg it stood on.”
The decision came just hours before the end of open enrollment for ACA plans in most states that use the federal HealthCare.gov insurance exchange. It is not expected that the ruling will impact the coverage for those people – the final decision will likely not come until the case reaches the Supreme Court again.
Seema Verma, the administrator of the Centers for Medicare & Medicaid Services, which oversees those insurance exchanges, said in a tweet: “The recent federal court decision is still moving through the courts, and the exchanges are still open for business and we will continue with open enrollment. There is no impact to current coverage or coverage in a 2019 plan.”
The 16 Democratic state attorneys general who intervened in the case to defend the health law immediately vowed to appeal.
“The ACA has already survived more than 70 unsuccessful repeal attempts and withstood scrutiny in the Supreme Court,” said a statement from California Attorney General Xavier Becerra. “Today’s misguided ruling will not deter us: our coalition will continue to fight in court for the health and wellbeing of all Americans.”
It is all but certain the case will become the third time the Supreme Court decides a constitutional question related to the ACA. In addition to upholding the law in 2012, the court rejected another challenge to the law in 2015.
It is hard to overstate what would happen to the nation’s health care system if the decision is ultimately upheld. The Affordable Care Act touched almost every aspect of health care, from Medicare and Medicaid to generic biologic drugs, the Indian Health Service, and public health changes like calorie counts on menus.
The case, Texas v. United States, was filed in February. The plaintiffs argued that because the Supreme Court upheld the ACA in 2012 as a constitutional use of its taxing power, the elimination of the tax makes the rest of the law unconstitutional.
In June, the Justice Department announced it would not fully defend the law in court. While the Trump administration said it did not agree with the plaintiffs that the tax law meant the entire ACA was unconstitutional, it said that the provisions of the law guaranteeing that people with preexisting health conditions could purchase coverage at the same price as everyone else were so inextricably linked to the tax penalty that they should be struck.
The administration urged the court to declare those provisions invalid beginning Jan. 1, 2019. That is the day the tax penalty for not having insurance disappears.
The protections for people with preexisting conditions was one of the top health issues in the midterm elections in November. While the issue mostly played to the advantage of Democrats, one of the Republican plaintiffs, Missouri Attorney General Josh Hawley, defeated Democratic incumbent Sen. Claire McCaskill. Another plaintiff, West Virginia Attorney General Patrick Morrisey, lost to Democratic incumbent Sen. Joe Manchin.
President Donald Trump was quick to take a victory lap, and pressed Senate Majority Leader Mitch McConnell (R-Ky.) and presumed incoming House Speaker Nancy Pelosi (D-Calif.) to fix the problem. He tweeted Friday night that “As I predicted all along, Obamacare has been struck down as an UNCONSTITUTIONAL disaster! Now Congress must pass a STRONG law that provides GREAT healthcare and protects pre-existing conditions. Mitch and Nancy, get it done!”
But congressional leaders were quick to point out that the suit is far from over.
“The ruling seems to be based on faulty legal reasoning and hopefully it will be overturned,” said a statement from Senate Minority Leader Chuck Schumer (D-N.Y.).
Many legal experts agreed with that. “This is insanity in print, and it will not stand up on appeal,” tweeted University of Michigan Law School professor Nicholas Bagley, an expert in health law.
Even some conservatives were left scratching their heads. “Congress acted last year to repeal the mandate, but leave everything else in place and the courts should have deferred to that,” tweeted former congressional GOP aide Chris Jacobs.
Kaiser Health News (KHN) is a national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation which is not affiliated with Kaiser Permanente.
The future of the Affordable Care Act is threatened – again – this time by a ruling Friday from a federal district court judge in Texas.
Judge Reed C. O’Connor struck down the law, siding with a group of 18 Republican state attorneys general and two GOP governors who brought the case. Judge O’Connor said the tax bill passed by Congress in December 2017 effectively rendered the entire health law unconstitutional.
That tax measure eliminated the penalty for not having insurance. An earlier Supreme Court decision upheld the ACA based on the view that the penalty was a tax and thus the law was valid because it relied on appropriate power allowed Congress under the Constitution. Judge O’Connor’s decision said that without that penalty, the law no longer met that constitutional test.
“In some ways, the question before the court involves the intent of both the 2010 and 2017 Congresses,” Judge O’Connor wrote in his 55-page decision. “The former enacted the ACA. The latter sawed off the last leg it stood on.”
The decision came just hours before the end of open enrollment for ACA plans in most states that use the federal HealthCare.gov insurance exchange. It is not expected that the ruling will impact the coverage for those people – the final decision will likely not come until the case reaches the Supreme Court again.
Seema Verma, the administrator of the Centers for Medicare & Medicaid Services, which oversees those insurance exchanges, said in a tweet: “The recent federal court decision is still moving through the courts, and the exchanges are still open for business and we will continue with open enrollment. There is no impact to current coverage or coverage in a 2019 plan.”
The 16 Democratic state attorneys general who intervened in the case to defend the health law immediately vowed to appeal.
“The ACA has already survived more than 70 unsuccessful repeal attempts and withstood scrutiny in the Supreme Court,” said a statement from California Attorney General Xavier Becerra. “Today’s misguided ruling will not deter us: our coalition will continue to fight in court for the health and wellbeing of all Americans.”
It is all but certain the case will become the third time the Supreme Court decides a constitutional question related to the ACA. In addition to upholding the law in 2012, the court rejected another challenge to the law in 2015.
It is hard to overstate what would happen to the nation’s health care system if the decision is ultimately upheld. The Affordable Care Act touched almost every aspect of health care, from Medicare and Medicaid to generic biologic drugs, the Indian Health Service, and public health changes like calorie counts on menus.
The case, Texas v. United States, was filed in February. The plaintiffs argued that because the Supreme Court upheld the ACA in 2012 as a constitutional use of its taxing power, the elimination of the tax makes the rest of the law unconstitutional.
In June, the Justice Department announced it would not fully defend the law in court. While the Trump administration said it did not agree with the plaintiffs that the tax law meant the entire ACA was unconstitutional, it said that the provisions of the law guaranteeing that people with preexisting health conditions could purchase coverage at the same price as everyone else were so inextricably linked to the tax penalty that they should be struck.
The administration urged the court to declare those provisions invalid beginning Jan. 1, 2019. That is the day the tax penalty for not having insurance disappears.
The protections for people with preexisting conditions was one of the top health issues in the midterm elections in November. While the issue mostly played to the advantage of Democrats, one of the Republican plaintiffs, Missouri Attorney General Josh Hawley, defeated Democratic incumbent Sen. Claire McCaskill. Another plaintiff, West Virginia Attorney General Patrick Morrisey, lost to Democratic incumbent Sen. Joe Manchin.
President Donald Trump was quick to take a victory lap, and pressed Senate Majority Leader Mitch McConnell (R-Ky.) and presumed incoming House Speaker Nancy Pelosi (D-Calif.) to fix the problem. He tweeted Friday night that “As I predicted all along, Obamacare has been struck down as an UNCONSTITUTIONAL disaster! Now Congress must pass a STRONG law that provides GREAT healthcare and protects pre-existing conditions. Mitch and Nancy, get it done!”
But congressional leaders were quick to point out that the suit is far from over.
“The ruling seems to be based on faulty legal reasoning and hopefully it will be overturned,” said a statement from Senate Minority Leader Chuck Schumer (D-N.Y.).
Many legal experts agreed with that. “This is insanity in print, and it will not stand up on appeal,” tweeted University of Michigan Law School professor Nicholas Bagley, an expert in health law.
Even some conservatives were left scratching their heads. “Congress acted last year to repeal the mandate, but leave everything else in place and the courts should have deferred to that,” tweeted former congressional GOP aide Chris Jacobs.
Kaiser Health News (KHN) is a national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation which is not affiliated with Kaiser Permanente.
Reports from the annual meeting of The Connective Tissue Oncology Society held in Rome, November 14-17, 2018 Sarcoma of the Year: Intimal Sarcoma
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).