User login
Meaningful endometriosis treatment requires a holistic approach and an understanding of chronic pain
Although it has been more than 100 years since endometriosis was first described in the literature, deciphering the mechanisms that cause pain in women with this enigmatic disease is an ongoing pursuit.
Pain is the most debilitating symptom of endometriosis.1,2 In many cases, it has a profoundly negative impact on a patient’s quality of life, and contributes significantly to disease burden, as well as to personal and societal costs from lost productivity.3,4 Women with endometriosis often experience chronic pelvic pain, deep dyspareunia, dysmenorrhea, and subfertility.5 The majority of women with the disease also have one or more comorbidities, including adenomyosis, adhesive disease, and other pelvic pain conditions such as interstitial cystitis, irritable bowel disease, inflammatory bowel disease, and pelvic floor myalgia.6-8
Recent studies have yielded new insights into the development of endometriosis-associated pelvic pain. The role of peritoneal inflammation, de novo innervation of endometriosis implants, and changes in the central nervous system are becoming increasingly clear.5,9,10 These discoveries have important treatment implications.
In this article, Andrea J. Rapkin, MD, Professor of Obstetrics and Gynecology at the University of California, Los Angeles, and Founder and Director of the UCLA Pelvic Pain Center, offers her expert opinion on the findings of key studies and their clinical implications, including the importance of a multidisciplinary treatment approach that focuses on the whole patient.
Q What mechanisms underlie the chronic pain that many women with endometriosis feel?
Although pain is the primary symptom experienced by women with endometriosis, the disease burden and symptom severity do not often correlate.11,12 “This was the first conundrum presented to clinicians,” noted Dr. Rapkin. “In fact, we do not know the true prevalence of endometriosis because women with endometriosis only come to diagnosis either based on pain or infertility. When infertility is the problem, very often we are surprised by how much disease is present in an individual with either no pain or minimal pain. Conversely, in other individuals with very severe pain, upon laparoscopic surgery, have minimal or mild endometriosis.”
Efforts to solve this clinical puzzle began decades ago. “Dr. Michael Vernon discovered that the small, red, endometriosis implants that looked like petechial hemorrhages produced more prostaglandin E2 (PGE2) in vitro than the older black-brown lesions. PGE2 is a pain-producing (algesic) chemical produced after cytokines stimulation,” said Dr. Rapkin. “This was the first evidence that, yes, there is a reason for pain in many individuals with lower-stage disease.”
“Prostaglandins are known to be a major cause of dysmenorrhea. Prostaglandins induce uterine cramping, sensitize nerve endings, and promote other inflammatory factors responsible for attracting monocytes that become macrophages, further contributing to inflammation,” Dr. Rapkin continued. “PGE2 also stimulates the enzyme aromatase, which allows androgens to be converted to estrogen, which promotes growth of endometriotic lesions. This is a self-feeding aspect of endometriosis.”
Continue to: These discoveries were followed by the realization that deeply infiltrating endometriosis...
These discoveries were followed by the realization that deeply infiltrating endometriosis (defined by disease infiltration of more than 5 mm, often in the uterosacral ligaments) was more likely to be painful than superficial disease, said Dr. Rapkin. “In some women with endometriosis, the disease we see laparoscopically is really the tip of the iceberg.”
In 2005, landmark studies performed by Karen J. Berkley, PhD, were summarized in a paper coauthored by Dr. Berkley, Dr. Rapkin, and Raymond E. Papka, PhD.13 “In a rodent model where endometriosis was developed by suturing pieces of endometrium in the mesentery, the endometriosis implants developed a vascular supply and a nerve supply. These nerves were not just functioning to govern the dilation and contraction of the blood vessels (in other words the sympathetic type nerves), but these nerves stained for neurotransmitters associated with pain (algesic agents, such as substance P and CGRP),” said Dr. Rapkin. “At UCLA, we acquired tissue from women with endometriosis and analyzed in Dr. Papka’s lab. Those tissues also showed nerves staining for pain-producing chemicals.” Other studies performed worldwide also demonstrated nerve endings with neurotrophic and algesic chemicals in endometriotic tissues. In addition to prostaglandins and cytokines, increased expression of various neuropeptides, neurotrophins, and alterations in ion channels contribute to hypersensitivity and pain.
Q What other chronic pain conditions might women with endometriosis experience?
Overlapping chronic pain conditions are common in women with endometriosis. “There is a very high co-occurrence of interstitial cystitis/painful bladder syndrome,” said Dr. Rapkin. “Irritable bowel syndrome is more common in women with endometriosis, as is vulvodynia. Fibromyalgia, migraine headache, temporo-mandibular joint pain (TMJ), anxiety, and depression also commonly co-occur in women with endometriosis.”
“Two concepts may be relevant to why these overlapping pain conditions develop,” Dr. Rapkin continued. “First, visceral sensitization: If one organ or tissue is inflamed and becomes hyperalgesic then other organs in the adjacent region with shared thoracolumbar and sacral innervation can become sensitized through shared cell bodies in the spinal cord, cross-sensitization in the cord, or at higher regions of the CNS. In addition, visceral somatic conversion occurs, whereby somatic tissues such as muscles and subcutaneous tissues with the same nerve supply as the affected organs become sensitized. This process may explain why abdominal wall and pelvic floor muscles become painful. The involvement of surrounding musculature is an important contributor to the pain in many women with endometriosis.”
“Finally, genetic studies of alterations in genes that encode for chemicals affecting the sensitivity and perception of pain are shedding light on the development of chronic pain. Ultimately these studies will advance our understanding of pain related to endometriosis.”
Continue to: Q How have new understandings about the pain mechanisms...
Q How have new understandings about the pain mechanisms involved with endometriosis-caused pelvic pain improved treatment?
According to Dr. Rapkin, the increased understanding of the mechanisms involved in endometriosis-associated pain gained from these key studies led to a paradigm shift, with endometriosis being viewed not just as a condition with mechanical hypersensitivity due to altered anatomy and inflammation but also as a neurologic condition, or a nerve pain condition with peripheral and central sensitization. “This means there is upregulation or hyperactivity both in the periphery (in the pelvis) and centrally (in the spinal cord and brain),” said Dr. Rapkin.
“In the periphery, the endometriotic lesions develop an afferent sensory innervation and communicate with the brain. Stimulation of these nerves by the inflammatory milieu contributes to pain.” Dr. Rapkin noted research by Maria Adele Giamberardino, which demonstrated that women with endometriosis and pain have a lower threshold for feeling pain in the tissues overlying the pelvis (the abdominal wall and back).14 This also has been shown by Dr. Berkley in rodents given endometriosis.
“The muscles develop trigger points and tender hyperalgesic points as part of the sensitization process. In addition, distant sensitization develops—women with pelvic pain and endometriosis have a lower threshold for sensing experimental pain in areas outside the pelvis, for example the back, leg, or shoulder. These discoveries clearly reflect up regulation for pain processing in the central nervous system.”
Dr. Rapkin also pointed to research published in 2016 by Sawson As-Sanie, MD, MPH, that showed an association between endometriosis-associated pelvic pain and altered brain chemistry and function.16 “Dr. As-Sanie demonstrated a decrease in gray matter volume in key neural pain processing areas in the brain in women with pain with endometriosis. This was not found in women with endometriosis who did not have pain,” she said. “Altered connectivity in brain areas related to perception and inhibition of pain is important in maintaining pain. Dr. As-Sanie’s studies also found that these changes are correlated with anxiety, depression, and pain intensity in patients with endometriosis and chronic pain.”
Continue to: Q What are some newer treatment approaches to chronic pain with endometriosis?
Q What are some newer treatment approaches to chronic pain with endometriosis?
“Multidisciplinary approaches to endometriosis-related pain are important,” said Dr. Rapkin. “Although it is important to excise or cauterize endometriosis lesions, or debulk as much as can safely be removed during laparoscopic surgery, it is now standard of care that medical therapy, not surgery, is the first approach to treatment. Endometriosis is a chronic condition. Inflammatory factors will continue to proliferate in patients who menstruate and produce high levels of estrogen with ovulation. The goal of medical therapy is to decrease the levels of estrogen that contribute to maintenance and proliferation of the implants. We want to suppress estrogen in a way that is compatible with long-term quality of life for our patients. Wiping out estrogen and placing patients into a chemical or surgical menopause for most of their reproductive years is not desirable.”
Approaches to hormonally modulate endometriosis include combined hormonal contraceptives and progestin-only medications, such as the levonogestrol-containing IUD, progestin-containing contraceptive implants, injections, or tablets. Second-line medical therapy consists of gonadotropin-releasing hormone agonists and antagonists that can be used for 6 months to 2 years and allows for further lowering of estrogen levels. These may not provide sufficient pain relief for some patients. “There is some evidence from Dr. Giamberadino’s studies that after women with dysmenorrhea were treated with oral contraceptives, the abdominal wall hyperalgesia decreased,” said Dr. Rapkin. “The question is, why don’t we see this in all patients? We come to the realization that endometriosis has to be treated as a neurologically mediated disorder. We have to treat the peripheral and central sensitization in a multidisciplinary way.”
A holistic approach to endometriosis is a new and exciting area for the field, said Dr. Rapkin. “We have to treat ‘bottom-up’, and ‘top-down.’ Bottom-up means we are addressing the peripheral factors that contribute to pain: endometriotic lesions, other pelvic organ pain, myofascial pain, trigger points, the tender points, and the muscle dysfunction in the abdominal wall, the back, and the pelvic floor. Pelvic floor physical therapists help women with pain and endometriosis. Often, women with endometriosis have myofascial pain and pain related to the other comorbid pain conditions they may have developed. Peripheral nerve blocks and medications used for neuropathic pain that alter nerve firing can be helpful in many situations. Pain can be augmented by cognitions and beliefs about pain, and by anxiety and depression. So the top-down approach addresses the cognitions, depression, and anxiety. We do not consider endometriosis a psychosomatic condition, but we know that if you do not address the central upregulation, including anxiety and depression, we may not get anywhere.”
“Interestingly, neurotransmitters and brain regions governing mood contribute to nerve pain. Medications such as tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors, anticonvulsants, and calcium channel blocking agents may prove fruitful. Cognitive behavioral therapy is another approach—to stimulate the prefrontal cortex, the area that is involved in pain inhibition, and other areas of the brain that may produce endogenous opioids to help with inhibiting pain. Bringing in complementary approaches is very important—for example, mindfulness-based meditation or yoga. There is growing evidence for acupuncture as well. Physical therapists, pain psychologists, anesthesiologists, or gynecologists who are facile with nerve blocks, to help tone down hyperalgesic tissues, in addition to medical and surgical therapy, have the possibility of really improving the lives of women with endometriosis.”
Q What key pearls would you like to share with readers?
“It is important to evaluate the entire individual,” she said. “Do not just viscerally focus on the uterus, the ovaries, fallopian tubes, and the peritoneum; investigate the adjacent organs and somatic tissues. Think about the abdominal wall, think about the pelvic floor. Learn how to evaluate these structures. There are simple evaluation techniques that gynecologists can learn and should include with every patient with pelvic pain, whether or not they are suspected of having endometriosis. You also want to get a complete history to determine if there are other co-occurring pain conditions. If there are, it is already a sign that there may be central sensitization.”
“Very often, it is necessary to bring in a pain psychologist—not because the disease is psychosomatic but because therapy can help the patient to learn how to use their brain to erase pain memory, and of course to address the concomitant anxiety, depression, and social isolation that happens with pain.”
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Olive DL, Lindheim SR, Pritts EA. New medical treatments for endometriosis. Best Pract Res Clin Obstet Gynaecol. 2004;18(2):319-328.
- Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364(9447): 1789-1799.
- Nnoaham KE, Hummelshoj L, Webster P, et al; World Endometriosis Research Foundation Global Study of Women’s Health consortium. Impact of endometriosis on quality of life and work productivity: a multicenter study across ten countries. Fertil Steril. 2011;96(2):366-373.e8.
- Simoens S, Dunselman G, Dirksen C, et al. The burden of endometriosis: costs and quality of life of women with endometriosis and treated in referral centres. Hum Reprod. 2012;27(5):1292–1299.
- Bruner-Tran KL, Mokshagundam S, Herington JL. Rodent models of experimental endometriosis: identifying mechanisms of disease and therapeutic targets. Curr Womens Health Rev. 2018;14(2):173-188.
- Sinaii N, Cleary SD, Ballweg ML, Nieman LK, Stratton P. High rates of autoimmune and endocrine disorders, fibromyalgia, chronic fatigue syndrome and atopic diseases among women with endometriosis: a survey analysis. Hum Reprod. 2002;17(10):2715-2724.
- Struble J, Reid S, Bedaiwy MA. Adenomyosis: a clinical review of a challenging gynecologic condition. J Minim Invasive Gynecol. 2016;23(2):164-185.
- Tirlapur SA, Kuhrt K, Chaliha C. The ‘evil twin syndrome’ in chronic pelvic pain: a systematic review of prevalence studies of bladder pain syndrome and endometriosis. Int J Surg. 2013;11(3):233-237.
- Coxon L, Horne AW, Vincent K. Pathophysiology of endometriosis-associated pain: a review of pelvic and central nervous system mechanisms. Best Pract Res Clin Obstet Gynaecol. 2018 Feb 15. pii: S1521-6934(18)30032-4. doi: 10.1016/j.bpobgyn.2018.01.014. [Epub ahead of print]
- Yan D, Liu X, Guo SW. Nerve fibers and endometriotic lesions: partners in crime in inflicting pains in women with endometriosis. Eur J Obstet Gynecol Reprod Biol. 2017;209:14-24.
- Vercellini P, Fedele L, Aimi G, Pietropaolo G, Consonni D, Crosignani PG. Association between endometriosis stage, lesion type, patient characteristics and severity of pelvic pain symptoms: a multivariate analysis of over 1000 patients. Hum Reprod. 2007;22(1):266-271.
- Fedele L, Parazzini F, Bianchi S. Stage and localization of pelvic endometriosis and pain. Fertil Steril. 1990;53(1):155-158.
- Berkley KJ, Rapkin AJ, Papka RE. The pains of endometriosis. Science. 2005;308(5728):1587-1589.
- Giamberardino MA, Tana C, Costantini R. Pain thresholds in women with chronic pelvic pain. Curr Opin Obstet Gynecol. 2014;26(4):253-259.
- Giamberardino MA, Berkley KJ, Affaitati G. Influence of endometriosis on pain behaviors and muscle hyperalgesia induced by a ureteral calculosis in female rats. Pain. 2002;95(3):247-257.
- As-Sanie S, Kim J, Schmidt-Wilcke T. Functional connectivity is associated with altered brain chemistry in women with endometriosis-associated chronic pelvic pain. J Pain. 2016;17(1):1-13.
Although it has been more than 100 years since endometriosis was first described in the literature, deciphering the mechanisms that cause pain in women with this enigmatic disease is an ongoing pursuit.
Pain is the most debilitating symptom of endometriosis.1,2 In many cases, it has a profoundly negative impact on a patient’s quality of life, and contributes significantly to disease burden, as well as to personal and societal costs from lost productivity.3,4 Women with endometriosis often experience chronic pelvic pain, deep dyspareunia, dysmenorrhea, and subfertility.5 The majority of women with the disease also have one or more comorbidities, including adenomyosis, adhesive disease, and other pelvic pain conditions such as interstitial cystitis, irritable bowel disease, inflammatory bowel disease, and pelvic floor myalgia.6-8
Recent studies have yielded new insights into the development of endometriosis-associated pelvic pain. The role of peritoneal inflammation, de novo innervation of endometriosis implants, and changes in the central nervous system are becoming increasingly clear.5,9,10 These discoveries have important treatment implications.
In this article, Andrea J. Rapkin, MD, Professor of Obstetrics and Gynecology at the University of California, Los Angeles, and Founder and Director of the UCLA Pelvic Pain Center, offers her expert opinion on the findings of key studies and their clinical implications, including the importance of a multidisciplinary treatment approach that focuses on the whole patient.
Q What mechanisms underlie the chronic pain that many women with endometriosis feel?
Although pain is the primary symptom experienced by women with endometriosis, the disease burden and symptom severity do not often correlate.11,12 “This was the first conundrum presented to clinicians,” noted Dr. Rapkin. “In fact, we do not know the true prevalence of endometriosis because women with endometriosis only come to diagnosis either based on pain or infertility. When infertility is the problem, very often we are surprised by how much disease is present in an individual with either no pain or minimal pain. Conversely, in other individuals with very severe pain, upon laparoscopic surgery, have minimal or mild endometriosis.”
Efforts to solve this clinical puzzle began decades ago. “Dr. Michael Vernon discovered that the small, red, endometriosis implants that looked like petechial hemorrhages produced more prostaglandin E2 (PGE2) in vitro than the older black-brown lesions. PGE2 is a pain-producing (algesic) chemical produced after cytokines stimulation,” said Dr. Rapkin. “This was the first evidence that, yes, there is a reason for pain in many individuals with lower-stage disease.”
“Prostaglandins are known to be a major cause of dysmenorrhea. Prostaglandins induce uterine cramping, sensitize nerve endings, and promote other inflammatory factors responsible for attracting monocytes that become macrophages, further contributing to inflammation,” Dr. Rapkin continued. “PGE2 also stimulates the enzyme aromatase, which allows androgens to be converted to estrogen, which promotes growth of endometriotic lesions. This is a self-feeding aspect of endometriosis.”
Continue to: These discoveries were followed by the realization that deeply infiltrating endometriosis...
These discoveries were followed by the realization that deeply infiltrating endometriosis (defined by disease infiltration of more than 5 mm, often in the uterosacral ligaments) was more likely to be painful than superficial disease, said Dr. Rapkin. “In some women with endometriosis, the disease we see laparoscopically is really the tip of the iceberg.”
In 2005, landmark studies performed by Karen J. Berkley, PhD, were summarized in a paper coauthored by Dr. Berkley, Dr. Rapkin, and Raymond E. Papka, PhD.13 “In a rodent model where endometriosis was developed by suturing pieces of endometrium in the mesentery, the endometriosis implants developed a vascular supply and a nerve supply. These nerves were not just functioning to govern the dilation and contraction of the blood vessels (in other words the sympathetic type nerves), but these nerves stained for neurotransmitters associated with pain (algesic agents, such as substance P and CGRP),” said Dr. Rapkin. “At UCLA, we acquired tissue from women with endometriosis and analyzed in Dr. Papka’s lab. Those tissues also showed nerves staining for pain-producing chemicals.” Other studies performed worldwide also demonstrated nerve endings with neurotrophic and algesic chemicals in endometriotic tissues. In addition to prostaglandins and cytokines, increased expression of various neuropeptides, neurotrophins, and alterations in ion channels contribute to hypersensitivity and pain.
Q What other chronic pain conditions might women with endometriosis experience?
Overlapping chronic pain conditions are common in women with endometriosis. “There is a very high co-occurrence of interstitial cystitis/painful bladder syndrome,” said Dr. Rapkin. “Irritable bowel syndrome is more common in women with endometriosis, as is vulvodynia. Fibromyalgia, migraine headache, temporo-mandibular joint pain (TMJ), anxiety, and depression also commonly co-occur in women with endometriosis.”
“Two concepts may be relevant to why these overlapping pain conditions develop,” Dr. Rapkin continued. “First, visceral sensitization: If one organ or tissue is inflamed and becomes hyperalgesic then other organs in the adjacent region with shared thoracolumbar and sacral innervation can become sensitized through shared cell bodies in the spinal cord, cross-sensitization in the cord, or at higher regions of the CNS. In addition, visceral somatic conversion occurs, whereby somatic tissues such as muscles and subcutaneous tissues with the same nerve supply as the affected organs become sensitized. This process may explain why abdominal wall and pelvic floor muscles become painful. The involvement of surrounding musculature is an important contributor to the pain in many women with endometriosis.”
“Finally, genetic studies of alterations in genes that encode for chemicals affecting the sensitivity and perception of pain are shedding light on the development of chronic pain. Ultimately these studies will advance our understanding of pain related to endometriosis.”
Continue to: Q How have new understandings about the pain mechanisms...
Q How have new understandings about the pain mechanisms involved with endometriosis-caused pelvic pain improved treatment?
According to Dr. Rapkin, the increased understanding of the mechanisms involved in endometriosis-associated pain gained from these key studies led to a paradigm shift, with endometriosis being viewed not just as a condition with mechanical hypersensitivity due to altered anatomy and inflammation but also as a neurologic condition, or a nerve pain condition with peripheral and central sensitization. “This means there is upregulation or hyperactivity both in the periphery (in the pelvis) and centrally (in the spinal cord and brain),” said Dr. Rapkin.
“In the periphery, the endometriotic lesions develop an afferent sensory innervation and communicate with the brain. Stimulation of these nerves by the inflammatory milieu contributes to pain.” Dr. Rapkin noted research by Maria Adele Giamberardino, which demonstrated that women with endometriosis and pain have a lower threshold for feeling pain in the tissues overlying the pelvis (the abdominal wall and back).14 This also has been shown by Dr. Berkley in rodents given endometriosis.
“The muscles develop trigger points and tender hyperalgesic points as part of the sensitization process. In addition, distant sensitization develops—women with pelvic pain and endometriosis have a lower threshold for sensing experimental pain in areas outside the pelvis, for example the back, leg, or shoulder. These discoveries clearly reflect up regulation for pain processing in the central nervous system.”
Dr. Rapkin also pointed to research published in 2016 by Sawson As-Sanie, MD, MPH, that showed an association between endometriosis-associated pelvic pain and altered brain chemistry and function.16 “Dr. As-Sanie demonstrated a decrease in gray matter volume in key neural pain processing areas in the brain in women with pain with endometriosis. This was not found in women with endometriosis who did not have pain,” she said. “Altered connectivity in brain areas related to perception and inhibition of pain is important in maintaining pain. Dr. As-Sanie’s studies also found that these changes are correlated with anxiety, depression, and pain intensity in patients with endometriosis and chronic pain.”
Continue to: Q What are some newer treatment approaches to chronic pain with endometriosis?
Q What are some newer treatment approaches to chronic pain with endometriosis?
“Multidisciplinary approaches to endometriosis-related pain are important,” said Dr. Rapkin. “Although it is important to excise or cauterize endometriosis lesions, or debulk as much as can safely be removed during laparoscopic surgery, it is now standard of care that medical therapy, not surgery, is the first approach to treatment. Endometriosis is a chronic condition. Inflammatory factors will continue to proliferate in patients who menstruate and produce high levels of estrogen with ovulation. The goal of medical therapy is to decrease the levels of estrogen that contribute to maintenance and proliferation of the implants. We want to suppress estrogen in a way that is compatible with long-term quality of life for our patients. Wiping out estrogen and placing patients into a chemical or surgical menopause for most of their reproductive years is not desirable.”
Approaches to hormonally modulate endometriosis include combined hormonal contraceptives and progestin-only medications, such as the levonogestrol-containing IUD, progestin-containing contraceptive implants, injections, or tablets. Second-line medical therapy consists of gonadotropin-releasing hormone agonists and antagonists that can be used for 6 months to 2 years and allows for further lowering of estrogen levels. These may not provide sufficient pain relief for some patients. “There is some evidence from Dr. Giamberadino’s studies that after women with dysmenorrhea were treated with oral contraceptives, the abdominal wall hyperalgesia decreased,” said Dr. Rapkin. “The question is, why don’t we see this in all patients? We come to the realization that endometriosis has to be treated as a neurologically mediated disorder. We have to treat the peripheral and central sensitization in a multidisciplinary way.”
A holistic approach to endometriosis is a new and exciting area for the field, said Dr. Rapkin. “We have to treat ‘bottom-up’, and ‘top-down.’ Bottom-up means we are addressing the peripheral factors that contribute to pain: endometriotic lesions, other pelvic organ pain, myofascial pain, trigger points, the tender points, and the muscle dysfunction in the abdominal wall, the back, and the pelvic floor. Pelvic floor physical therapists help women with pain and endometriosis. Often, women with endometriosis have myofascial pain and pain related to the other comorbid pain conditions they may have developed. Peripheral nerve blocks and medications used for neuropathic pain that alter nerve firing can be helpful in many situations. Pain can be augmented by cognitions and beliefs about pain, and by anxiety and depression. So the top-down approach addresses the cognitions, depression, and anxiety. We do not consider endometriosis a psychosomatic condition, but we know that if you do not address the central upregulation, including anxiety and depression, we may not get anywhere.”
“Interestingly, neurotransmitters and brain regions governing mood contribute to nerve pain. Medications such as tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors, anticonvulsants, and calcium channel blocking agents may prove fruitful. Cognitive behavioral therapy is another approach—to stimulate the prefrontal cortex, the area that is involved in pain inhibition, and other areas of the brain that may produce endogenous opioids to help with inhibiting pain. Bringing in complementary approaches is very important—for example, mindfulness-based meditation or yoga. There is growing evidence for acupuncture as well. Physical therapists, pain psychologists, anesthesiologists, or gynecologists who are facile with nerve blocks, to help tone down hyperalgesic tissues, in addition to medical and surgical therapy, have the possibility of really improving the lives of women with endometriosis.”
Q What key pearls would you like to share with readers?
“It is important to evaluate the entire individual,” she said. “Do not just viscerally focus on the uterus, the ovaries, fallopian tubes, and the peritoneum; investigate the adjacent organs and somatic tissues. Think about the abdominal wall, think about the pelvic floor. Learn how to evaluate these structures. There are simple evaluation techniques that gynecologists can learn and should include with every patient with pelvic pain, whether or not they are suspected of having endometriosis. You also want to get a complete history to determine if there are other co-occurring pain conditions. If there are, it is already a sign that there may be central sensitization.”
“Very often, it is necessary to bring in a pain psychologist—not because the disease is psychosomatic but because therapy can help the patient to learn how to use their brain to erase pain memory, and of course to address the concomitant anxiety, depression, and social isolation that happens with pain.”
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Although it has been more than 100 years since endometriosis was first described in the literature, deciphering the mechanisms that cause pain in women with this enigmatic disease is an ongoing pursuit.
Pain is the most debilitating symptom of endometriosis.1,2 In many cases, it has a profoundly negative impact on a patient’s quality of life, and contributes significantly to disease burden, as well as to personal and societal costs from lost productivity.3,4 Women with endometriosis often experience chronic pelvic pain, deep dyspareunia, dysmenorrhea, and subfertility.5 The majority of women with the disease also have one or more comorbidities, including adenomyosis, adhesive disease, and other pelvic pain conditions such as interstitial cystitis, irritable bowel disease, inflammatory bowel disease, and pelvic floor myalgia.6-8
Recent studies have yielded new insights into the development of endometriosis-associated pelvic pain. The role of peritoneal inflammation, de novo innervation of endometriosis implants, and changes in the central nervous system are becoming increasingly clear.5,9,10 These discoveries have important treatment implications.
In this article, Andrea J. Rapkin, MD, Professor of Obstetrics and Gynecology at the University of California, Los Angeles, and Founder and Director of the UCLA Pelvic Pain Center, offers her expert opinion on the findings of key studies and their clinical implications, including the importance of a multidisciplinary treatment approach that focuses on the whole patient.
Q What mechanisms underlie the chronic pain that many women with endometriosis feel?
Although pain is the primary symptom experienced by women with endometriosis, the disease burden and symptom severity do not often correlate.11,12 “This was the first conundrum presented to clinicians,” noted Dr. Rapkin. “In fact, we do not know the true prevalence of endometriosis because women with endometriosis only come to diagnosis either based on pain or infertility. When infertility is the problem, very often we are surprised by how much disease is present in an individual with either no pain or minimal pain. Conversely, in other individuals with very severe pain, upon laparoscopic surgery, have minimal or mild endometriosis.”
Efforts to solve this clinical puzzle began decades ago. “Dr. Michael Vernon discovered that the small, red, endometriosis implants that looked like petechial hemorrhages produced more prostaglandin E2 (PGE2) in vitro than the older black-brown lesions. PGE2 is a pain-producing (algesic) chemical produced after cytokines stimulation,” said Dr. Rapkin. “This was the first evidence that, yes, there is a reason for pain in many individuals with lower-stage disease.”
“Prostaglandins are known to be a major cause of dysmenorrhea. Prostaglandins induce uterine cramping, sensitize nerve endings, and promote other inflammatory factors responsible for attracting monocytes that become macrophages, further contributing to inflammation,” Dr. Rapkin continued. “PGE2 also stimulates the enzyme aromatase, which allows androgens to be converted to estrogen, which promotes growth of endometriotic lesions. This is a self-feeding aspect of endometriosis.”
Continue to: These discoveries were followed by the realization that deeply infiltrating endometriosis...
These discoveries were followed by the realization that deeply infiltrating endometriosis (defined by disease infiltration of more than 5 mm, often in the uterosacral ligaments) was more likely to be painful than superficial disease, said Dr. Rapkin. “In some women with endometriosis, the disease we see laparoscopically is really the tip of the iceberg.”
In 2005, landmark studies performed by Karen J. Berkley, PhD, were summarized in a paper coauthored by Dr. Berkley, Dr. Rapkin, and Raymond E. Papka, PhD.13 “In a rodent model where endometriosis was developed by suturing pieces of endometrium in the mesentery, the endometriosis implants developed a vascular supply and a nerve supply. These nerves were not just functioning to govern the dilation and contraction of the blood vessels (in other words the sympathetic type nerves), but these nerves stained for neurotransmitters associated with pain (algesic agents, such as substance P and CGRP),” said Dr. Rapkin. “At UCLA, we acquired tissue from women with endometriosis and analyzed in Dr. Papka’s lab. Those tissues also showed nerves staining for pain-producing chemicals.” Other studies performed worldwide also demonstrated nerve endings with neurotrophic and algesic chemicals in endometriotic tissues. In addition to prostaglandins and cytokines, increased expression of various neuropeptides, neurotrophins, and alterations in ion channels contribute to hypersensitivity and pain.
Q What other chronic pain conditions might women with endometriosis experience?
Overlapping chronic pain conditions are common in women with endometriosis. “There is a very high co-occurrence of interstitial cystitis/painful bladder syndrome,” said Dr. Rapkin. “Irritable bowel syndrome is more common in women with endometriosis, as is vulvodynia. Fibromyalgia, migraine headache, temporo-mandibular joint pain (TMJ), anxiety, and depression also commonly co-occur in women with endometriosis.”
“Two concepts may be relevant to why these overlapping pain conditions develop,” Dr. Rapkin continued. “First, visceral sensitization: If one organ or tissue is inflamed and becomes hyperalgesic then other organs in the adjacent region with shared thoracolumbar and sacral innervation can become sensitized through shared cell bodies in the spinal cord, cross-sensitization in the cord, or at higher regions of the CNS. In addition, visceral somatic conversion occurs, whereby somatic tissues such as muscles and subcutaneous tissues with the same nerve supply as the affected organs become sensitized. This process may explain why abdominal wall and pelvic floor muscles become painful. The involvement of surrounding musculature is an important contributor to the pain in many women with endometriosis.”
“Finally, genetic studies of alterations in genes that encode for chemicals affecting the sensitivity and perception of pain are shedding light on the development of chronic pain. Ultimately these studies will advance our understanding of pain related to endometriosis.”
Continue to: Q How have new understandings about the pain mechanisms...
Q How have new understandings about the pain mechanisms involved with endometriosis-caused pelvic pain improved treatment?
According to Dr. Rapkin, the increased understanding of the mechanisms involved in endometriosis-associated pain gained from these key studies led to a paradigm shift, with endometriosis being viewed not just as a condition with mechanical hypersensitivity due to altered anatomy and inflammation but also as a neurologic condition, or a nerve pain condition with peripheral and central sensitization. “This means there is upregulation or hyperactivity both in the periphery (in the pelvis) and centrally (in the spinal cord and brain),” said Dr. Rapkin.
“In the periphery, the endometriotic lesions develop an afferent sensory innervation and communicate with the brain. Stimulation of these nerves by the inflammatory milieu contributes to pain.” Dr. Rapkin noted research by Maria Adele Giamberardino, which demonstrated that women with endometriosis and pain have a lower threshold for feeling pain in the tissues overlying the pelvis (the abdominal wall and back).14 This also has been shown by Dr. Berkley in rodents given endometriosis.
“The muscles develop trigger points and tender hyperalgesic points as part of the sensitization process. In addition, distant sensitization develops—women with pelvic pain and endometriosis have a lower threshold for sensing experimental pain in areas outside the pelvis, for example the back, leg, or shoulder. These discoveries clearly reflect up regulation for pain processing in the central nervous system.”
Dr. Rapkin also pointed to research published in 2016 by Sawson As-Sanie, MD, MPH, that showed an association between endometriosis-associated pelvic pain and altered brain chemistry and function.16 “Dr. As-Sanie demonstrated a decrease in gray matter volume in key neural pain processing areas in the brain in women with pain with endometriosis. This was not found in women with endometriosis who did not have pain,” she said. “Altered connectivity in brain areas related to perception and inhibition of pain is important in maintaining pain. Dr. As-Sanie’s studies also found that these changes are correlated with anxiety, depression, and pain intensity in patients with endometriosis and chronic pain.”
Continue to: Q What are some newer treatment approaches to chronic pain with endometriosis?
Q What are some newer treatment approaches to chronic pain with endometriosis?
“Multidisciplinary approaches to endometriosis-related pain are important,” said Dr. Rapkin. “Although it is important to excise or cauterize endometriosis lesions, or debulk as much as can safely be removed during laparoscopic surgery, it is now standard of care that medical therapy, not surgery, is the first approach to treatment. Endometriosis is a chronic condition. Inflammatory factors will continue to proliferate in patients who menstruate and produce high levels of estrogen with ovulation. The goal of medical therapy is to decrease the levels of estrogen that contribute to maintenance and proliferation of the implants. We want to suppress estrogen in a way that is compatible with long-term quality of life for our patients. Wiping out estrogen and placing patients into a chemical or surgical menopause for most of their reproductive years is not desirable.”
Approaches to hormonally modulate endometriosis include combined hormonal contraceptives and progestin-only medications, such as the levonogestrol-containing IUD, progestin-containing contraceptive implants, injections, or tablets. Second-line medical therapy consists of gonadotropin-releasing hormone agonists and antagonists that can be used for 6 months to 2 years and allows for further lowering of estrogen levels. These may not provide sufficient pain relief for some patients. “There is some evidence from Dr. Giamberadino’s studies that after women with dysmenorrhea were treated with oral contraceptives, the abdominal wall hyperalgesia decreased,” said Dr. Rapkin. “The question is, why don’t we see this in all patients? We come to the realization that endometriosis has to be treated as a neurologically mediated disorder. We have to treat the peripheral and central sensitization in a multidisciplinary way.”
A holistic approach to endometriosis is a new and exciting area for the field, said Dr. Rapkin. “We have to treat ‘bottom-up’, and ‘top-down.’ Bottom-up means we are addressing the peripheral factors that contribute to pain: endometriotic lesions, other pelvic organ pain, myofascial pain, trigger points, the tender points, and the muscle dysfunction in the abdominal wall, the back, and the pelvic floor. Pelvic floor physical therapists help women with pain and endometriosis. Often, women with endometriosis have myofascial pain and pain related to the other comorbid pain conditions they may have developed. Peripheral nerve blocks and medications used for neuropathic pain that alter nerve firing can be helpful in many situations. Pain can be augmented by cognitions and beliefs about pain, and by anxiety and depression. So the top-down approach addresses the cognitions, depression, and anxiety. We do not consider endometriosis a psychosomatic condition, but we know that if you do not address the central upregulation, including anxiety and depression, we may not get anywhere.”
“Interestingly, neurotransmitters and brain regions governing mood contribute to nerve pain. Medications such as tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors, anticonvulsants, and calcium channel blocking agents may prove fruitful. Cognitive behavioral therapy is another approach—to stimulate the prefrontal cortex, the area that is involved in pain inhibition, and other areas of the brain that may produce endogenous opioids to help with inhibiting pain. Bringing in complementary approaches is very important—for example, mindfulness-based meditation or yoga. There is growing evidence for acupuncture as well. Physical therapists, pain psychologists, anesthesiologists, or gynecologists who are facile with nerve blocks, to help tone down hyperalgesic tissues, in addition to medical and surgical therapy, have the possibility of really improving the lives of women with endometriosis.”
Q What key pearls would you like to share with readers?
“It is important to evaluate the entire individual,” she said. “Do not just viscerally focus on the uterus, the ovaries, fallopian tubes, and the peritoneum; investigate the adjacent organs and somatic tissues. Think about the abdominal wall, think about the pelvic floor. Learn how to evaluate these structures. There are simple evaluation techniques that gynecologists can learn and should include with every patient with pelvic pain, whether or not they are suspected of having endometriosis. You also want to get a complete history to determine if there are other co-occurring pain conditions. If there are, it is already a sign that there may be central sensitization.”
“Very often, it is necessary to bring in a pain psychologist—not because the disease is psychosomatic but because therapy can help the patient to learn how to use their brain to erase pain memory, and of course to address the concomitant anxiety, depression, and social isolation that happens with pain.”
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Olive DL, Lindheim SR, Pritts EA. New medical treatments for endometriosis. Best Pract Res Clin Obstet Gynaecol. 2004;18(2):319-328.
- Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364(9447): 1789-1799.
- Nnoaham KE, Hummelshoj L, Webster P, et al; World Endometriosis Research Foundation Global Study of Women’s Health consortium. Impact of endometriosis on quality of life and work productivity: a multicenter study across ten countries. Fertil Steril. 2011;96(2):366-373.e8.
- Simoens S, Dunselman G, Dirksen C, et al. The burden of endometriosis: costs and quality of life of women with endometriosis and treated in referral centres. Hum Reprod. 2012;27(5):1292–1299.
- Bruner-Tran KL, Mokshagundam S, Herington JL. Rodent models of experimental endometriosis: identifying mechanisms of disease and therapeutic targets. Curr Womens Health Rev. 2018;14(2):173-188.
- Sinaii N, Cleary SD, Ballweg ML, Nieman LK, Stratton P. High rates of autoimmune and endocrine disorders, fibromyalgia, chronic fatigue syndrome and atopic diseases among women with endometriosis: a survey analysis. Hum Reprod. 2002;17(10):2715-2724.
- Struble J, Reid S, Bedaiwy MA. Adenomyosis: a clinical review of a challenging gynecologic condition. J Minim Invasive Gynecol. 2016;23(2):164-185.
- Tirlapur SA, Kuhrt K, Chaliha C. The ‘evil twin syndrome’ in chronic pelvic pain: a systematic review of prevalence studies of bladder pain syndrome and endometriosis. Int J Surg. 2013;11(3):233-237.
- Coxon L, Horne AW, Vincent K. Pathophysiology of endometriosis-associated pain: a review of pelvic and central nervous system mechanisms. Best Pract Res Clin Obstet Gynaecol. 2018 Feb 15. pii: S1521-6934(18)30032-4. doi: 10.1016/j.bpobgyn.2018.01.014. [Epub ahead of print]
- Yan D, Liu X, Guo SW. Nerve fibers and endometriotic lesions: partners in crime in inflicting pains in women with endometriosis. Eur J Obstet Gynecol Reprod Biol. 2017;209:14-24.
- Vercellini P, Fedele L, Aimi G, Pietropaolo G, Consonni D, Crosignani PG. Association between endometriosis stage, lesion type, patient characteristics and severity of pelvic pain symptoms: a multivariate analysis of over 1000 patients. Hum Reprod. 2007;22(1):266-271.
- Fedele L, Parazzini F, Bianchi S. Stage and localization of pelvic endometriosis and pain. Fertil Steril. 1990;53(1):155-158.
- Berkley KJ, Rapkin AJ, Papka RE. The pains of endometriosis. Science. 2005;308(5728):1587-1589.
- Giamberardino MA, Tana C, Costantini R. Pain thresholds in women with chronic pelvic pain. Curr Opin Obstet Gynecol. 2014;26(4):253-259.
- Giamberardino MA, Berkley KJ, Affaitati G. Influence of endometriosis on pain behaviors and muscle hyperalgesia induced by a ureteral calculosis in female rats. Pain. 2002;95(3):247-257.
- As-Sanie S, Kim J, Schmidt-Wilcke T. Functional connectivity is associated with altered brain chemistry in women with endometriosis-associated chronic pelvic pain. J Pain. 2016;17(1):1-13.
- Olive DL, Lindheim SR, Pritts EA. New medical treatments for endometriosis. Best Pract Res Clin Obstet Gynaecol. 2004;18(2):319-328.
- Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364(9447): 1789-1799.
- Nnoaham KE, Hummelshoj L, Webster P, et al; World Endometriosis Research Foundation Global Study of Women’s Health consortium. Impact of endometriosis on quality of life and work productivity: a multicenter study across ten countries. Fertil Steril. 2011;96(2):366-373.e8.
- Simoens S, Dunselman G, Dirksen C, et al. The burden of endometriosis: costs and quality of life of women with endometriosis and treated in referral centres. Hum Reprod. 2012;27(5):1292–1299.
- Bruner-Tran KL, Mokshagundam S, Herington JL. Rodent models of experimental endometriosis: identifying mechanisms of disease and therapeutic targets. Curr Womens Health Rev. 2018;14(2):173-188.
- Sinaii N, Cleary SD, Ballweg ML, Nieman LK, Stratton P. High rates of autoimmune and endocrine disorders, fibromyalgia, chronic fatigue syndrome and atopic diseases among women with endometriosis: a survey analysis. Hum Reprod. 2002;17(10):2715-2724.
- Struble J, Reid S, Bedaiwy MA. Adenomyosis: a clinical review of a challenging gynecologic condition. J Minim Invasive Gynecol. 2016;23(2):164-185.
- Tirlapur SA, Kuhrt K, Chaliha C. The ‘evil twin syndrome’ in chronic pelvic pain: a systematic review of prevalence studies of bladder pain syndrome and endometriosis. Int J Surg. 2013;11(3):233-237.
- Coxon L, Horne AW, Vincent K. Pathophysiology of endometriosis-associated pain: a review of pelvic and central nervous system mechanisms. Best Pract Res Clin Obstet Gynaecol. 2018 Feb 15. pii: S1521-6934(18)30032-4. doi: 10.1016/j.bpobgyn.2018.01.014. [Epub ahead of print]
- Yan D, Liu X, Guo SW. Nerve fibers and endometriotic lesions: partners in crime in inflicting pains in women with endometriosis. Eur J Obstet Gynecol Reprod Biol. 2017;209:14-24.
- Vercellini P, Fedele L, Aimi G, Pietropaolo G, Consonni D, Crosignani PG. Association between endometriosis stage, lesion type, patient characteristics and severity of pelvic pain symptoms: a multivariate analysis of over 1000 patients. Hum Reprod. 2007;22(1):266-271.
- Fedele L, Parazzini F, Bianchi S. Stage and localization of pelvic endometriosis and pain. Fertil Steril. 1990;53(1):155-158.
- Berkley KJ, Rapkin AJ, Papka RE. The pains of endometriosis. Science. 2005;308(5728):1587-1589.
- Giamberardino MA, Tana C, Costantini R. Pain thresholds in women with chronic pelvic pain. Curr Opin Obstet Gynecol. 2014;26(4):253-259.
- Giamberardino MA, Berkley KJ, Affaitati G. Influence of endometriosis on pain behaviors and muscle hyperalgesia induced by a ureteral calculosis in female rats. Pain. 2002;95(3):247-257.
- As-Sanie S, Kim J, Schmidt-Wilcke T. Functional connectivity is associated with altered brain chemistry in women with endometriosis-associated chronic pelvic pain. J Pain. 2016;17(1):1-13.

Coding and reimbursement 101: How to maximize your payments
While reimbursement for ObGyn services seemingly should be a simple matter of putting codes on a claim form, the reality is that it is complex, and it requires a team approach to accomplish timely filing to receive fair and accurate reimbursement.
Reimbursement occurs over the length of the revenue cycle for a patient encounter and involves many steps. It starts when the patient makes an appointment for services and ends when the practice receives payment. Along the way, there must be good clinician documentation and sound knowledge about the billing process (including the Current Procedural Terminology [CPT] or Healthcare Common Procedure Coding System [HCPCS] codes for services), the International Classification of Diseases, Tenth Revision, Clinical Modification [ICD-10-CM] codes that establish medical necessity, the modifiers that alter the meaning of the codes, and, of course, the bundling issues that now accompany many coding situations.
In addition, ObGyn practices must contend with a multitude of payers—from federal to commercial—and must understand and adhere to each payer’s rules and policies to maximize and retain reimbursement.
In this article, I detail stumbling blocks to maximizing reimbursement and how to avoid them.
Coding considerations for office services
Good documentation before, during, and after a patient’s office visit is essential, along with accurate codes, modifiers, and order of services on the claims you submit.
Prep paperwork before the patient encounter
Once a patient makes an appointment, the front-end staff can handle some of the tasks in the cycle. This includes ensuring that the patient’s insurance coverage information is current, informing the patient of any additional information to bring at the time of the visit (such as a patient history form for a new patient visit or a list of current prescriptions), or, if an established patient will be having a procedure, making sure that prior authorization is complete. This streamlines the process, assists the clinician with documentation housekeeping, and ensures that incorrect or missing information does not cause a claim to be denied or not be filed in a timely manner (many payers require submission of an initial claim 30 days from the date of service).
Continue to: Document details of the clinician-patient interaction
Document details of the clinician-patient interaction
At the time of the encounter, you are responsible for documenting your contact with the patient in enough detail to support billing a CPT evaluation and management (E/M) code at the level selected and/or any procedures or other services performed. The TABLE provides an overview of the requirements for each level of office service.
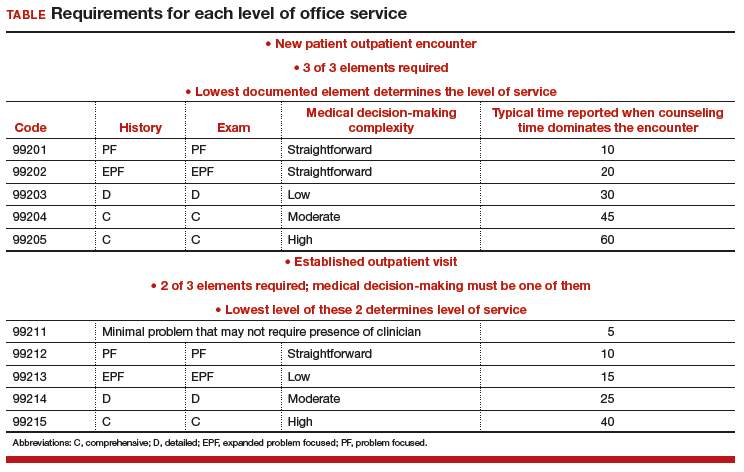
If both an E/M and a procedure are performed on the same date of service, the E/M must be documented to show it was separate from the procedure and that the work was significantly more than would be required to accomplish the procedure. Documentation of the procedure should include the indication, steps performed, findings, the patient’s condition afterward, and instructions for aftercare or follow-up.
If you use an electronic health record for reporting, you may be the one responsible for selecting both the CPT code for services performed and an ICD-10-CM code(s) to establish the medical need for them. Select the most accurate CPT codes, and clearly link them to a supporting diagnosis for each service that will be billed. If more than one diagnosis is applicable, the first one linked to any given service should represent the most important justification, as not all payers will accept more than one diagnosis code on the claim per service billed.
If the billing staff is assigned the task of selecting the CPT and/or ICD-10-CM diagnostic codes based on your documentation, they should be well versed in the services, procedures, and diagnoses reported for their ObGyn practice.
The actual code selection may end up being a joint venture between the clinician and the staff to ensure that accurate information will be entered on the claim. Good and frequent clinician-staff communication on billing of services can transform average reimbursement into maximized reimbursement.
Be aware of bundles
Sometimes more than one service or procedure is listed on a claim on the same date of service. However, it is important to identify all potential bundles before billing to ensure correct payment. For instance, payers like to bundle an E/M service and a procedure, or you may be in the global period (defined below) of a surgery but need to report an unrelated service.
You and your staff must work together to ensure the claim is submitted with the correct modifiers; on the other hand, you may decide that a better method of coding is in order. Some payers, for example, will not reimburse both an insertion and a removal of an intrauterine device (IUD) on the same date of service. If that does happen, a modifier on the removal code might save the day, rather than billing 2 codes.
Continue to: Manage the modifiers
Manage the modifiers
Sometimes the code billed requires a modifier to ensure payment. Typical modifiers used in an ObGyn office setting include the following:
- 22, Increased procedural services (the clinician must assign a fee that is higher than the usual fee for the procedure and be able to document CPT equivalents to the work involved)
- 24, Unrelated E/M during the postoperative period (note that this modifier does not apply during the antepartum period for pregnancy)
- 25, Significant and separate E/M on the same date as another service or minor procedure
- 52, Reduced services (generally, the payer will expect an explanation of the reduced service and will determine payment accordingly)
- 57, Decision to perform major surgery the day of or the day before the surgery
- 59, Distinct procedural service (used when 2 procedures are bundled and a modifier is allowed). Note that payment reductions for multiple procedures will still apply.
- 79, Unrelated procedure during the postoperative period (usually paid at the full allowable).
Organize the order of services on the claim
For an outpatient claim that includes both an E/M service and procedures, the order of the services—not the order in which they were performed—may be important to obtaining maximum reimbursement. In general, payers will pay in full for a supported E/M service no matter where it appears on the claim, but they apply reductions only for multiple procedures.
For instance, if you insert levonorgestrel implants on the same date as you remove a large polyp from the cervix, you would want to report the code with the highest relative value unit (RVU) first. In this case, it would be 11981 (4.05 RVUs), 57500 (3.61 RVUs).
In the IUD case mentioned earlier (removal and insertion of IUDs on the same date), the order of the codes, assuming the payer reimburses for both, will be even more important since removal usually has a higher payment: 58301 (2.70 RUVs), 58300 (1.54 RVUs).
Coding considerations for surgical services
Surgical services performed in a hospital or ambulatory surgical center present another set of must-dos to ensure timely and fair reimbursement.
Grasp the ‘global package’ concept
Understanding this concept can be crucial to getting paid for additional services during this time period and correct billing for any E/M services performed prior to surgery. In general, the routine history and physical examination performed prior to a major surgery is considered included in the work and should not be billed separately. Surgical clearance for a patient’s condition, such as hypertension, a heart condition, or lung issues, can be billed separately, but these generally are performed by someone other than the operating surgeon.
Procedures performed in the hospital setting generally will have a 10- or 90-day global period. During this time, any related E/M service should not be billed separately, and the use of modifiers becomes even more important than with office services.
Applicable modifiers for use with hospital surgery can include all those for outpatient services plus:
- 50, Bilateral procedure (for which you may be paid up to 150% of the allowable)
- 58, Staged or related procedure during the postoperative period (this may be paid at the full allowable)
- 62, Co-surgeons (both surgeons bill the same CPT code and both document their involvement in the surgery). Medicare will reimburse each surgeon 62.5% of the allowable.
- 78, Return to the operating room for an unplanned related procedure (the full allowable may be reduced by some payers owing to their belief that this is soon after the original procedure so intraoperative time only is considered).
Be savvy about surgical bundles
Here, it is important to understand all published bundling edits for multiple procedures performed by the same surgeon at the same surgical session. If a code combination is never allowed but the surgery is more intense due to additional work required, a modifier -22 may be your only option. Again, clear, concise documentation of the additional work is imperative to receive the additional payment.
When a modifier is allowed, it generally will be one that denotes a procedure done on bilateral organs (such as the ovaries) when there is no extensive code to cover all of the work or when the additional procedure is “distinct” and meets the criteria for using a modifier 59.
Medicare has expanded the modifier -59 into additional modifiers to further explain the situation. These additional modifiers are:
- XE, A service that is distinct because it occurred during a separate encounter on the same date of service
- XS, A service that is distinct because it was performed on a separate organ/structure
- XP, A service that is distinct because it was performed by a different practitioner
- XU, The use of a service that is distinct because it does not overlap usual components of the main service.
Standards of care: Some steps are inherent to the surgery
Expect to receive claim denials if you bill separately for adhesiolysis during a surgical procedure. Every payer considers this procedure related to access to the surgical site and will deny separate coding. If the lysis was truly significant in terms of work, try reporting the modifier 22 and provide adequate documentation.
Other procedures at the time of surgery that generally are not paid for include 1) examination under anesthesia, 2) any procedure done to check the surgeon’s work (for example, cystoscopy, especially when done after urinary or pelvic reconstruction procedures, or chromotubation following extensive ovariolysis), 3) placement of catheters, and 4) placement of devices to alleviate postsurgical pain.
Bottom line
Maximizing reimbursement involves good documentation, correct CPT codes linked to specific and accurate medical indications, the use of appropriate modifiers, and listing codes in order of their relative values from highest to lowest.
Should a denial or unfair reduction in payment come your way, analyze the rejection to determine the cause and make billing and reporting changes as needed to improve your future reimbursements.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.

The author reports no financial relationships relevant to this article.
The author reports no financial relationships relevant to this article.
The author reports no financial relationships relevant to this article.
While reimbursement for ObGyn services seemingly should be a simple matter of putting codes on a claim form, the reality is that it is complex, and it requires a team approach to accomplish timely filing to receive fair and accurate reimbursement.
Reimbursement occurs over the length of the revenue cycle for a patient encounter and involves many steps. It starts when the patient makes an appointment for services and ends when the practice receives payment. Along the way, there must be good clinician documentation and sound knowledge about the billing process (including the Current Procedural Terminology [CPT] or Healthcare Common Procedure Coding System [HCPCS] codes for services), the International Classification of Diseases, Tenth Revision, Clinical Modification [ICD-10-CM] codes that establish medical necessity, the modifiers that alter the meaning of the codes, and, of course, the bundling issues that now accompany many coding situations.
In addition, ObGyn practices must contend with a multitude of payers—from federal to commercial—and must understand and adhere to each payer’s rules and policies to maximize and retain reimbursement.
In this article, I detail stumbling blocks to maximizing reimbursement and how to avoid them.
Coding considerations for office services
Good documentation before, during, and after a patient’s office visit is essential, along with accurate codes, modifiers, and order of services on the claims you submit.
Prep paperwork before the patient encounter
Once a patient makes an appointment, the front-end staff can handle some of the tasks in the cycle. This includes ensuring that the patient’s insurance coverage information is current, informing the patient of any additional information to bring at the time of the visit (such as a patient history form for a new patient visit or a list of current prescriptions), or, if an established patient will be having a procedure, making sure that prior authorization is complete. This streamlines the process, assists the clinician with documentation housekeeping, and ensures that incorrect or missing information does not cause a claim to be denied or not be filed in a timely manner (many payers require submission of an initial claim 30 days from the date of service).
Continue to: Document details of the clinician-patient interaction
Document details of the clinician-patient interaction
At the time of the encounter, you are responsible for documenting your contact with the patient in enough detail to support billing a CPT evaluation and management (E/M) code at the level selected and/or any procedures or other services performed. The TABLE provides an overview of the requirements for each level of office service.
If both an E/M and a procedure are performed on the same date of service, the E/M must be documented to show it was separate from the procedure and that the work was significantly more than would be required to accomplish the procedure. Documentation of the procedure should include the indication, steps performed, findings, the patient’s condition afterward, and instructions for aftercare or follow-up.
If you use an electronic health record for reporting, you may be the one responsible for selecting both the CPT code for services performed and an ICD-10-CM code(s) to establish the medical need for them. Select the most accurate CPT codes, and clearly link them to a supporting diagnosis for each service that will be billed. If more than one diagnosis is applicable, the first one linked to any given service should represent the most important justification, as not all payers will accept more than one diagnosis code on the claim per service billed.
If the billing staff is assigned the task of selecting the CPT and/or ICD-10-CM diagnostic codes based on your documentation, they should be well versed in the services, procedures, and diagnoses reported for their ObGyn practice.
The actual code selection may end up being a joint venture between the clinician and the staff to ensure that accurate information will be entered on the claim. Good and frequent clinician-staff communication on billing of services can transform average reimbursement into maximized reimbursement.
Be aware of bundles
Sometimes more than one service or procedure is listed on a claim on the same date of service. However, it is important to identify all potential bundles before billing to ensure correct payment. For instance, payers like to bundle an E/M service and a procedure, or you may be in the global period (defined below) of a surgery but need to report an unrelated service.
You and your staff must work together to ensure the claim is submitted with the correct modifiers; on the other hand, you may decide that a better method of coding is in order. Some payers, for example, will not reimburse both an insertion and a removal of an intrauterine device (IUD) on the same date of service. If that does happen, a modifier on the removal code might save the day, rather than billing 2 codes.
Continue to: Manage the modifiers
Manage the modifiers
Sometimes the code billed requires a modifier to ensure payment. Typical modifiers used in an ObGyn office setting include the following:
- 22, Increased procedural services (the clinician must assign a fee that is higher than the usual fee for the procedure and be able to document CPT equivalents to the work involved)
- 24, Unrelated E/M during the postoperative period (note that this modifier does not apply during the antepartum period for pregnancy)
- 25, Significant and separate E/M on the same date as another service or minor procedure
- 52, Reduced services (generally, the payer will expect an explanation of the reduced service and will determine payment accordingly)
- 57, Decision to perform major surgery the day of or the day before the surgery
- 59, Distinct procedural service (used when 2 procedures are bundled and a modifier is allowed). Note that payment reductions for multiple procedures will still apply.
- 79, Unrelated procedure during the postoperative period (usually paid at the full allowable).
Organize the order of services on the claim
For an outpatient claim that includes both an E/M service and procedures, the order of the services—not the order in which they were performed—may be important to obtaining maximum reimbursement. In general, payers will pay in full for a supported E/M service no matter where it appears on the claim, but they apply reductions only for multiple procedures.
For instance, if you insert levonorgestrel implants on the same date as you remove a large polyp from the cervix, you would want to report the code with the highest relative value unit (RVU) first. In this case, it would be 11981 (4.05 RVUs), 57500 (3.61 RVUs).
In the IUD case mentioned earlier (removal and insertion of IUDs on the same date), the order of the codes, assuming the payer reimburses for both, will be even more important since removal usually has a higher payment: 58301 (2.70 RUVs), 58300 (1.54 RVUs).
Coding considerations for surgical services
Surgical services performed in a hospital or ambulatory surgical center present another set of must-dos to ensure timely and fair reimbursement.
Grasp the ‘global package’ concept
Understanding this concept can be crucial to getting paid for additional services during this time period and correct billing for any E/M services performed prior to surgery. In general, the routine history and physical examination performed prior to a major surgery is considered included in the work and should not be billed separately. Surgical clearance for a patient’s condition, such as hypertension, a heart condition, or lung issues, can be billed separately, but these generally are performed by someone other than the operating surgeon.
Procedures performed in the hospital setting generally will have a 10- or 90-day global period. During this time, any related E/M service should not be billed separately, and the use of modifiers becomes even more important than with office services.
Applicable modifiers for use with hospital surgery can include all those for outpatient services plus:
- 50, Bilateral procedure (for which you may be paid up to 150% of the allowable)
- 58, Staged or related procedure during the postoperative period (this may be paid at the full allowable)
- 62, Co-surgeons (both surgeons bill the same CPT code and both document their involvement in the surgery). Medicare will reimburse each surgeon 62.5% of the allowable.
- 78, Return to the operating room for an unplanned related procedure (the full allowable may be reduced by some payers owing to their belief that this is soon after the original procedure so intraoperative time only is considered).
Be savvy about surgical bundles
Here, it is important to understand all published bundling edits for multiple procedures performed by the same surgeon at the same surgical session. If a code combination is never allowed but the surgery is more intense due to additional work required, a modifier -22 may be your only option. Again, clear, concise documentation of the additional work is imperative to receive the additional payment.
When a modifier is allowed, it generally will be one that denotes a procedure done on bilateral organs (such as the ovaries) when there is no extensive code to cover all of the work or when the additional procedure is “distinct” and meets the criteria for using a modifier 59.
Medicare has expanded the modifier -59 into additional modifiers to further explain the situation. These additional modifiers are:
- XE, A service that is distinct because it occurred during a separate encounter on the same date of service
- XS, A service that is distinct because it was performed on a separate organ/structure
- XP, A service that is distinct because it was performed by a different practitioner
- XU, The use of a service that is distinct because it does not overlap usual components of the main service.
Standards of care: Some steps are inherent to the surgery
Expect to receive claim denials if you bill separately for adhesiolysis during a surgical procedure. Every payer considers this procedure related to access to the surgical site and will deny separate coding. If the lysis was truly significant in terms of work, try reporting the modifier 22 and provide adequate documentation.
Other procedures at the time of surgery that generally are not paid for include 1) examination under anesthesia, 2) any procedure done to check the surgeon’s work (for example, cystoscopy, especially when done after urinary or pelvic reconstruction procedures, or chromotubation following extensive ovariolysis), 3) placement of catheters, and 4) placement of devices to alleviate postsurgical pain.
Bottom line
Maximizing reimbursement involves good documentation, correct CPT codes linked to specific and accurate medical indications, the use of appropriate modifiers, and listing codes in order of their relative values from highest to lowest.
Should a denial or unfair reduction in payment come your way, analyze the rejection to determine the cause and make billing and reporting changes as needed to improve your future reimbursements.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
While reimbursement for ObGyn services seemingly should be a simple matter of putting codes on a claim form, the reality is that it is complex, and it requires a team approach to accomplish timely filing to receive fair and accurate reimbursement.
Reimbursement occurs over the length of the revenue cycle for a patient encounter and involves many steps. It starts when the patient makes an appointment for services and ends when the practice receives payment. Along the way, there must be good clinician documentation and sound knowledge about the billing process (including the Current Procedural Terminology [CPT] or Healthcare Common Procedure Coding System [HCPCS] codes for services), the International Classification of Diseases, Tenth Revision, Clinical Modification [ICD-10-CM] codes that establish medical necessity, the modifiers that alter the meaning of the codes, and, of course, the bundling issues that now accompany many coding situations.
In addition, ObGyn practices must contend with a multitude of payers—from federal to commercial—and must understand and adhere to each payer’s rules and policies to maximize and retain reimbursement.
In this article, I detail stumbling blocks to maximizing reimbursement and how to avoid them.
Coding considerations for office services
Good documentation before, during, and after a patient’s office visit is essential, along with accurate codes, modifiers, and order of services on the claims you submit.
Prep paperwork before the patient encounter
Once a patient makes an appointment, the front-end staff can handle some of the tasks in the cycle. This includes ensuring that the patient’s insurance coverage information is current, informing the patient of any additional information to bring at the time of the visit (such as a patient history form for a new patient visit or a list of current prescriptions), or, if an established patient will be having a procedure, making sure that prior authorization is complete. This streamlines the process, assists the clinician with documentation housekeeping, and ensures that incorrect or missing information does not cause a claim to be denied or not be filed in a timely manner (many payers require submission of an initial claim 30 days from the date of service).
Continue to: Document details of the clinician-patient interaction
Document details of the clinician-patient interaction
At the time of the encounter, you are responsible for documenting your contact with the patient in enough detail to support billing a CPT evaluation and management (E/M) code at the level selected and/or any procedures or other services performed. The TABLE provides an overview of the requirements for each level of office service.
If both an E/M and a procedure are performed on the same date of service, the E/M must be documented to show it was separate from the procedure and that the work was significantly more than would be required to accomplish the procedure. Documentation of the procedure should include the indication, steps performed, findings, the patient’s condition afterward, and instructions for aftercare or follow-up.
If you use an electronic health record for reporting, you may be the one responsible for selecting both the CPT code for services performed and an ICD-10-CM code(s) to establish the medical need for them. Select the most accurate CPT codes, and clearly link them to a supporting diagnosis for each service that will be billed. If more than one diagnosis is applicable, the first one linked to any given service should represent the most important justification, as not all payers will accept more than one diagnosis code on the claim per service billed.
If the billing staff is assigned the task of selecting the CPT and/or ICD-10-CM diagnostic codes based on your documentation, they should be well versed in the services, procedures, and diagnoses reported for their ObGyn practice.
The actual code selection may end up being a joint venture between the clinician and the staff to ensure that accurate information will be entered on the claim. Good and frequent clinician-staff communication on billing of services can transform average reimbursement into maximized reimbursement.
Be aware of bundles
Sometimes more than one service or procedure is listed on a claim on the same date of service. However, it is important to identify all potential bundles before billing to ensure correct payment. For instance, payers like to bundle an E/M service and a procedure, or you may be in the global period (defined below) of a surgery but need to report an unrelated service.
You and your staff must work together to ensure the claim is submitted with the correct modifiers; on the other hand, you may decide that a better method of coding is in order. Some payers, for example, will not reimburse both an insertion and a removal of an intrauterine device (IUD) on the same date of service. If that does happen, a modifier on the removal code might save the day, rather than billing 2 codes.
Continue to: Manage the modifiers
Manage the modifiers
Sometimes the code billed requires a modifier to ensure payment. Typical modifiers used in an ObGyn office setting include the following:
- 22, Increased procedural services (the clinician must assign a fee that is higher than the usual fee for the procedure and be able to document CPT equivalents to the work involved)
- 24, Unrelated E/M during the postoperative period (note that this modifier does not apply during the antepartum period for pregnancy)
- 25, Significant and separate E/M on the same date as another service or minor procedure
- 52, Reduced services (generally, the payer will expect an explanation of the reduced service and will determine payment accordingly)
- 57, Decision to perform major surgery the day of or the day before the surgery
- 59, Distinct procedural service (used when 2 procedures are bundled and a modifier is allowed). Note that payment reductions for multiple procedures will still apply.
- 79, Unrelated procedure during the postoperative period (usually paid at the full allowable).
Organize the order of services on the claim
For an outpatient claim that includes both an E/M service and procedures, the order of the services—not the order in which they were performed—may be important to obtaining maximum reimbursement. In general, payers will pay in full for a supported E/M service no matter where it appears on the claim, but they apply reductions only for multiple procedures.
For instance, if you insert levonorgestrel implants on the same date as you remove a large polyp from the cervix, you would want to report the code with the highest relative value unit (RVU) first. In this case, it would be 11981 (4.05 RVUs), 57500 (3.61 RVUs).
In the IUD case mentioned earlier (removal and insertion of IUDs on the same date), the order of the codes, assuming the payer reimburses for both, will be even more important since removal usually has a higher payment: 58301 (2.70 RUVs), 58300 (1.54 RVUs).
Coding considerations for surgical services
Surgical services performed in a hospital or ambulatory surgical center present another set of must-dos to ensure timely and fair reimbursement.
Grasp the ‘global package’ concept
Understanding this concept can be crucial to getting paid for additional services during this time period and correct billing for any E/M services performed prior to surgery. In general, the routine history and physical examination performed prior to a major surgery is considered included in the work and should not be billed separately. Surgical clearance for a patient’s condition, such as hypertension, a heart condition, or lung issues, can be billed separately, but these generally are performed by someone other than the operating surgeon.
Procedures performed in the hospital setting generally will have a 10- or 90-day global period. During this time, any related E/M service should not be billed separately, and the use of modifiers becomes even more important than with office services.
Applicable modifiers for use with hospital surgery can include all those for outpatient services plus:
- 50, Bilateral procedure (for which you may be paid up to 150% of the allowable)
- 58, Staged or related procedure during the postoperative period (this may be paid at the full allowable)
- 62, Co-surgeons (both surgeons bill the same CPT code and both document their involvement in the surgery). Medicare will reimburse each surgeon 62.5% of the allowable.
- 78, Return to the operating room for an unplanned related procedure (the full allowable may be reduced by some payers owing to their belief that this is soon after the original procedure so intraoperative time only is considered).
Be savvy about surgical bundles
Here, it is important to understand all published bundling edits for multiple procedures performed by the same surgeon at the same surgical session. If a code combination is never allowed but the surgery is more intense due to additional work required, a modifier -22 may be your only option. Again, clear, concise documentation of the additional work is imperative to receive the additional payment.
When a modifier is allowed, it generally will be one that denotes a procedure done on bilateral organs (such as the ovaries) when there is no extensive code to cover all of the work or when the additional procedure is “distinct” and meets the criteria for using a modifier 59.
Medicare has expanded the modifier -59 into additional modifiers to further explain the situation. These additional modifiers are:
- XE, A service that is distinct because it occurred during a separate encounter on the same date of service
- XS, A service that is distinct because it was performed on a separate organ/structure
- XP, A service that is distinct because it was performed by a different practitioner
- XU, The use of a service that is distinct because it does not overlap usual components of the main service.
Standards of care: Some steps are inherent to the surgery
Expect to receive claim denials if you bill separately for adhesiolysis during a surgical procedure. Every payer considers this procedure related to access to the surgical site and will deny separate coding. If the lysis was truly significant in terms of work, try reporting the modifier 22 and provide adequate documentation.
Other procedures at the time of surgery that generally are not paid for include 1) examination under anesthesia, 2) any procedure done to check the surgeon’s work (for example, cystoscopy, especially when done after urinary or pelvic reconstruction procedures, or chromotubation following extensive ovariolysis), 3) placement of catheters, and 4) placement of devices to alleviate postsurgical pain.
Bottom line
Maximizing reimbursement involves good documentation, correct CPT codes linked to specific and accurate medical indications, the use of appropriate modifiers, and listing codes in order of their relative values from highest to lowest.
Should a denial or unfair reduction in payment come your way, analyze the rejection to determine the cause and make billing and reporting changes as needed to improve your future reimbursements.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.

Facing a lawsuit? Take the right steps early
It’s happened. A patient is suing you. Now what? Legal experts warn that a doctor’s first steps after a lawsuit can dramatically impact the outcome of the case – for better or worse.

Below, medical malpractice defense attorneys share the most important do’s and don’ts for physicians after they receive a lawsuit notice. Spoiler: Whatever you do, don’t ignore the summons.
• Do contact your insurer and/or risk manager. Once you receive notice of a lawsuit, the first step is calling your medical malpractice insurer and/or risk manager, said Steven Fitzer, a medical liability defense attorney based in Tacoma, Wash. The insurer and risk manager will take the matter from there and advise your next moves. Resist the urge to disregard the notice and hope that the challenge goes away when the patient is no longer angry, he said. Failing to notify the insurer in a timely manner could be a policy violation and affect current or future coverage.
• Don’t contact the plaintiff/patient or patient’s family. Instinctively, many physicians feel compelled to call the patient and attempt to settle the conflict verbally, particularly if they have had a longstanding relationship, Mr. Fitzer said in an interview. Don’t do it.
“In 42 years, I’ve never come across a physician who successfully talked somebody out of a lawsuit, once it was started,” he said. “It’s a pipe dream.”
Keep in mind that conversations with patients after a lawsuit filing can be used against doctors in court and certain words can easily be misconstrued as admissions of guilt.
• Do secure all medical records pertaining to the case. Obtain and print copies of all information relevant to the patient’s suit, such as history, billing records, letters, and medical chart. Store the data in a secure location in preparation for transferring to the insurer and/or attorney, said Michael Moroney, a medical liability defense attorney based in Teaneck, N.J.
• Don’t access or change the record. It may seem tempting to review the plaintiff’s medical record and fix any errors found. However, accessing the patient’s electronic data can appear as an attempt to manipulate or delete relevant data, said Joshua R. Cohen, a medical liability defense attorney based in New York.

“Avoid accessing [the] EMR or PAC system [and] leaving a digital fingerprint,” he said in an interview. “For example, if a radiologist is sued for an alleged failure to diagnose breast cancer, they should not open that study on their computer as an audit trail will show that. Worse is when they start making measurements after the lawsuit which are now discoverable as part of the lawsuit.”
Leave the record alone and let the attorneys handle the data from here on out, he advised.
• Do discuss the patient case openly with your attorney and risk manager. Honesty about all aspects of a medical case from the start sets the right tone for a positive relationship between doctor and attorney, experts say. Help your attorney understand the medicine so that they can speak intelligently about the details to the court and any retained experts, Mr. Fitzer recommended. If disagreements continually arise among physicians and attorneys, and the match fails, consider speaking to the insurer about a change in attorneys.
• Don’t discuss the case. As Mr. Fitzer puts it, “loose lips sink ships.” Physicians lose confidentiality protections when they talk about lawsuit details with third parties, and those conversations could come back to haunt them. This includes colleagues and staff members in the patient’s care loop, said Catherine Flynn, a medical liability defense attorney based in Teaneck, N.J. The third parties could later be questioned by the plaintiff’s attorney about the case, which could harm your defense.

"It’s like that kid game of telephone where you may something to the nurses and then a year later, they’re deposed, and their recollection is very different,” Ms. Flynn said in an interview. “It turns into something that you did not say.”
Your spouse is the exception. Most states protect conversations among spouses and bar husbands and wives from having to testify against their spouse.
• Do alert staff to the lawsuit and track any document requests. Following a lawsuit notice, inform staff that a claim has been filed by a patient – without going into detail. Be alert to document requests by nonpatients and make sure your attorney is aware of such requests. For example, some plaintiffs hire a private investigator to contact the medical practice and attempt to obtain records, Mr. Moroney said. In other cases, the plaintiff’s attorney or their paralegal tries to get copies of the medical chart or billing records.
• Don’t release any patient data to third parties. Ensure that staff members do not provide any patient information to the plaintiff’s attorney or other third parties, Mr. Moroney said. All relevant records should go through your attorney. No questions about the patient or the circumstances of the complaint should be divulged by the doctor or staff members to any third party, he said.
• Do seek emotional support from family and friends. Facing a lawsuit can be draining, both physically and mentally. Make time for self-care and lean on loved ones when needed, Mr. Fitzer said. Sharing your feelings – without going into detail about the case – can help relieve stress and reduce the emotional strain of being sued.
• Don’t isolate yourself. “This can be an isolating experience,” Mr. Fitzer said. “You need support. You need reinforcements. Take care of yourself and your family – they are your biggest source of support.”
It’s happened. A patient is suing you. Now what? Legal experts warn that a doctor’s first steps after a lawsuit can dramatically impact the outcome of the case – for better or worse.
Below, medical malpractice defense attorneys share the most important do’s and don’ts for physicians after they receive a lawsuit notice. Spoiler: Whatever you do, don’t ignore the summons.
• Do contact your insurer and/or risk manager. Once you receive notice of a lawsuit, the first step is calling your medical malpractice insurer and/or risk manager, said Steven Fitzer, a medical liability defense attorney based in Tacoma, Wash. The insurer and risk manager will take the matter from there and advise your next moves. Resist the urge to disregard the notice and hope that the challenge goes away when the patient is no longer angry, he said. Failing to notify the insurer in a timely manner could be a policy violation and affect current or future coverage.
• Don’t contact the plaintiff/patient or patient’s family. Instinctively, many physicians feel compelled to call the patient and attempt to settle the conflict verbally, particularly if they have had a longstanding relationship, Mr. Fitzer said in an interview. Don’t do it.
“In 42 years, I’ve never come across a physician who successfully talked somebody out of a lawsuit, once it was started,” he said. “It’s a pipe dream.”
Keep in mind that conversations with patients after a lawsuit filing can be used against doctors in court and certain words can easily be misconstrued as admissions of guilt.
• Do secure all medical records pertaining to the case. Obtain and print copies of all information relevant to the patient’s suit, such as history, billing records, letters, and medical chart. Store the data in a secure location in preparation for transferring to the insurer and/or attorney, said Michael Moroney, a medical liability defense attorney based in Teaneck, N.J.
• Don’t access or change the record. It may seem tempting to review the plaintiff’s medical record and fix any errors found. However, accessing the patient’s electronic data can appear as an attempt to manipulate or delete relevant data, said Joshua R. Cohen, a medical liability defense attorney based in New York.
“Avoid accessing [the] EMR or PAC system [and] leaving a digital fingerprint,” he said in an interview. “For example, if a radiologist is sued for an alleged failure to diagnose breast cancer, they should not open that study on their computer as an audit trail will show that. Worse is when they start making measurements after the lawsuit which are now discoverable as part of the lawsuit.”
Leave the record alone and let the attorneys handle the data from here on out, he advised.
• Do discuss the patient case openly with your attorney and risk manager. Honesty about all aspects of a medical case from the start sets the right tone for a positive relationship between doctor and attorney, experts say. Help your attorney understand the medicine so that they can speak intelligently about the details to the court and any retained experts, Mr. Fitzer recommended. If disagreements continually arise among physicians and attorneys, and the match fails, consider speaking to the insurer about a change in attorneys.
• Don’t discuss the case. As Mr. Fitzer puts it, “loose lips sink ships.” Physicians lose confidentiality protections when they talk about lawsuit details with third parties, and those conversations could come back to haunt them. This includes colleagues and staff members in the patient’s care loop, said Catherine Flynn, a medical liability defense attorney based in Teaneck, N.J. The third parties could later be questioned by the plaintiff’s attorney about the case, which could harm your defense.
"It’s like that kid game of telephone where you may something to the nurses and then a year later, they’re deposed, and their recollection is very different,” Ms. Flynn said in an interview. “It turns into something that you did not say.”
Your spouse is the exception. Most states protect conversations among spouses and bar husbands and wives from having to testify against their spouse.
• Do alert staff to the lawsuit and track any document requests. Following a lawsuit notice, inform staff that a claim has been filed by a patient – without going into detail. Be alert to document requests by nonpatients and make sure your attorney is aware of such requests. For example, some plaintiffs hire a private investigator to contact the medical practice and attempt to obtain records, Mr. Moroney said. In other cases, the plaintiff’s attorney or their paralegal tries to get copies of the medical chart or billing records.
• Don’t release any patient data to third parties. Ensure that staff members do not provide any patient information to the plaintiff’s attorney or other third parties, Mr. Moroney said. All relevant records should go through your attorney. No questions about the patient or the circumstances of the complaint should be divulged by the doctor or staff members to any third party, he said.
• Do seek emotional support from family and friends. Facing a lawsuit can be draining, both physically and mentally. Make time for self-care and lean on loved ones when needed, Mr. Fitzer said. Sharing your feelings – without going into detail about the case – can help relieve stress and reduce the emotional strain of being sued.
• Don’t isolate yourself. “This can be an isolating experience,” Mr. Fitzer said. “You need support. You need reinforcements. Take care of yourself and your family – they are your biggest source of support.”
It’s happened. A patient is suing you. Now what? Legal experts warn that a doctor’s first steps after a lawsuit can dramatically impact the outcome of the case – for better or worse.
Below, medical malpractice defense attorneys share the most important do’s and don’ts for physicians after they receive a lawsuit notice. Spoiler: Whatever you do, don’t ignore the summons.
• Do contact your insurer and/or risk manager. Once you receive notice of a lawsuit, the first step is calling your medical malpractice insurer and/or risk manager, said Steven Fitzer, a medical liability defense attorney based in Tacoma, Wash. The insurer and risk manager will take the matter from there and advise your next moves. Resist the urge to disregard the notice and hope that the challenge goes away when the patient is no longer angry, he said. Failing to notify the insurer in a timely manner could be a policy violation and affect current or future coverage.
• Don’t contact the plaintiff/patient or patient’s family. Instinctively, many physicians feel compelled to call the patient and attempt to settle the conflict verbally, particularly if they have had a longstanding relationship, Mr. Fitzer said in an interview. Don’t do it.
“In 42 years, I’ve never come across a physician who successfully talked somebody out of a lawsuit, once it was started,” he said. “It’s a pipe dream.”
Keep in mind that conversations with patients after a lawsuit filing can be used against doctors in court and certain words can easily be misconstrued as admissions of guilt.
• Do secure all medical records pertaining to the case. Obtain and print copies of all information relevant to the patient’s suit, such as history, billing records, letters, and medical chart. Store the data in a secure location in preparation for transferring to the insurer and/or attorney, said Michael Moroney, a medical liability defense attorney based in Teaneck, N.J.
• Don’t access or change the record. It may seem tempting to review the plaintiff’s medical record and fix any errors found. However, accessing the patient’s electronic data can appear as an attempt to manipulate or delete relevant data, said Joshua R. Cohen, a medical liability defense attorney based in New York.
“Avoid accessing [the] EMR or PAC system [and] leaving a digital fingerprint,” he said in an interview. “For example, if a radiologist is sued for an alleged failure to diagnose breast cancer, they should not open that study on their computer as an audit trail will show that. Worse is when they start making measurements after the lawsuit which are now discoverable as part of the lawsuit.”
Leave the record alone and let the attorneys handle the data from here on out, he advised.
• Do discuss the patient case openly with your attorney and risk manager. Honesty about all aspects of a medical case from the start sets the right tone for a positive relationship between doctor and attorney, experts say. Help your attorney understand the medicine so that they can speak intelligently about the details to the court and any retained experts, Mr. Fitzer recommended. If disagreements continually arise among physicians and attorneys, and the match fails, consider speaking to the insurer about a change in attorneys.
• Don’t discuss the case. As Mr. Fitzer puts it, “loose lips sink ships.” Physicians lose confidentiality protections when they talk about lawsuit details with third parties, and those conversations could come back to haunt them. This includes colleagues and staff members in the patient’s care loop, said Catherine Flynn, a medical liability defense attorney based in Teaneck, N.J. The third parties could later be questioned by the plaintiff’s attorney about the case, which could harm your defense.
"It’s like that kid game of telephone where you may something to the nurses and then a year later, they’re deposed, and their recollection is very different,” Ms. Flynn said in an interview. “It turns into something that you did not say.”
Your spouse is the exception. Most states protect conversations among spouses and bar husbands and wives from having to testify against their spouse.
• Do alert staff to the lawsuit and track any document requests. Following a lawsuit notice, inform staff that a claim has been filed by a patient – without going into detail. Be alert to document requests by nonpatients and make sure your attorney is aware of such requests. For example, some plaintiffs hire a private investigator to contact the medical practice and attempt to obtain records, Mr. Moroney said. In other cases, the plaintiff’s attorney or their paralegal tries to get copies of the medical chart or billing records.
• Don’t release any patient data to third parties. Ensure that staff members do not provide any patient information to the plaintiff’s attorney or other third parties, Mr. Moroney said. All relevant records should go through your attorney. No questions about the patient or the circumstances of the complaint should be divulged by the doctor or staff members to any third party, he said.
• Do seek emotional support from family and friends. Facing a lawsuit can be draining, both physically and mentally. Make time for self-care and lean on loved ones when needed, Mr. Fitzer said. Sharing your feelings – without going into detail about the case – can help relieve stress and reduce the emotional strain of being sued.
• Don’t isolate yourself. “This can be an isolating experience,” Mr. Fitzer said. “You need support. You need reinforcements. Take care of yourself and your family – they are your biggest source of support.”
Does low-dose aspirin decrease a woman’s risk of ovarian cancer?
EXPERT COMMENTARY
Epidemiologic studies conducted in ovarian cancer suggest an association between chronic inflammation and incidence of disease.1 Nonsteroidal anti-inflammatory drugs (NSAIDs) work to decrease inflammation through the inhibition of cyclo-oxygenase (COX). Therefore, anti-inflammatory agents such as NSAIDs have been proposed to play a role in the pathophysiology of ovarian cancer.
Previous studies of this association show conflicting data. The majority of these studies are retrospective, and those that are prospective do not include detailed data regarding dosing and frequency of ASA use.2-6
_
Details of the study
This study by Barnard and colleagues is a prospective cohort study evaluating a total of 205,498 women from 1980–2015 from 2 separate cohorts (the Nurses’ Health Study and the Nurses’ Health Study II). The primary outcome was “to evaluate whether regular aspirin or nonaspirin NSAID use and patterns of use are associated with lower ovarian cancer risk.” Analgesic use and data regarding covariates were obtained via self-reported questionnaires. Ovarian cancer diagnosis was confirmed via medical records.
Results demonstrated that current low-dose aspirin use was associated with a decreased risk of ovarian cancer (hazard ratio [HR], 0.77; 95% confidence interval [CI], 0.61–0.96). This significance was not maintained upon further controlling for inflammatory factors (hypertension, autoimmune disease, inflammatory diet scores, smoking, etc) (HR, 0.94; 95% CI, 0.69–1.26). Other significant findings included an increased risk of developing ovarian cancer with standard-dose ASA use of ≥5 years or standard-dose use at 6 to 9 tablets per week (HR, 1.77; 95% CI, 1.13–2.77 and HR, 2.00; 95% CI, 1.27–3.15, respectively). An increased risk of developing ovarian cancer also was found for >10-year use or use of >10 tablets per week of nonaspirin NSAIDs (HR, 2.00; 95% CI, 1.27–3.15 and HR, 1.35; 95% CI, 1.02–1.79, respectively).
The authors concluded that there was a slight inverse association for low-dose aspirin and ovarian cancer risk and that standard aspirin or NSAID use actually may be associated with an increased risk of ovarian cancer.
Study strengths and weaknesses
This study has many strengths. It was a large prospective cohort investigation with adequate power to detect clinically significant differences. The authors collected detailed exposure data, which was novel. They also considered a latency period prior to the diagnosis of ovarian cancer during which a patient may increase their analgesic use in order to treat pain caused by the impending cancer.
However, the conclusions of the authors seem to be overstated in the setting of the data. Specifically, the deduction regarding a decreased risk of ovarian cancer with low-dose aspirin use given the loss of the statistical significance when controlling for pertinent cofounders. Further, the study authors did not evaluate adverse effects associated with low-dose aspirin use, which would be clinically applicable when determining whether the results from this study should become formal recommendations. Lastly, other important clinical factors, such as the presence of genetic mutations or endometriosis, were not considered, and these considerations would greatly affect results.
In the setting of previous large prospective studies that suggest no association between ASA use and ovarian cancer risk,4-6 data from this study are not compelling enough to recommend regular low-dose aspirin use to all women.
Based on these current data, there is insufficient evidence to suggest the use of low-dose aspirin for chemoprophylaxis of ovarian cancer. In order to suggest the use of a drug for prophylaxis the benefits must outweigh the risks, and in the case of NSAIDs, this has yet to be confirmed.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Poole EM, Lee IM, Ridker PM, et al. A prospective study of circulating C-reactive protein, interleukin-6, and tumor necrosis factor alpha receptor 2 levels and risk of ovarian cancer. Am J Epidemiol. 2013;178:1256-1264.
- Trabert B, Ness RB, Lo-Ciganic WH, et al. Aspirin, nonaspirin nonsteroidal anti-inflammatory drug, and acetaminophen use and risk of invasive epithelial ovarian cancer: a pooled analysis in the Ovarian Cancer Association Consortium. J Natl Cancer Inst. 2014;106:djt431.
- Peres LC, Camacho F, Abbott SE, et al. Analgesic medication use and risk of epithelial ovarian cancer in African American women. Br J Cancer. 2016;114(7):819-825.
- Murphy MA, Trabert B, Yang HP, et al. Non-steroidal antiinflammatory drug use and ovarian cancer risk: findings from the NIH-AARP Diet and Health Study and systematic review. Cancer Causes Control. 2012;23:1839-1852.
- Brasky TM, Liu J, White E, et al. Non-steroidal antiinflammatory drugs and cancer risk in women: results from the Women’s Health Initiative. Int J Cancer. 2014;135:1869-1883.
- Lacey JV Jr, Sherman ME, Hartge P, et al. Medication use and risk of ovarian carcinoma: a prospective study. Int J Cancer. 2004;108:281-286.
EXPERT COMMENTARY
Epidemiologic studies conducted in ovarian cancer suggest an association between chronic inflammation and incidence of disease.1 Nonsteroidal anti-inflammatory drugs (NSAIDs) work to decrease inflammation through the inhibition of cyclo-oxygenase (COX). Therefore, anti-inflammatory agents such as NSAIDs have been proposed to play a role in the pathophysiology of ovarian cancer.
Previous studies of this association show conflicting data. The majority of these studies are retrospective, and those that are prospective do not include detailed data regarding dosing and frequency of ASA use.2-6
_
Details of the study
This study by Barnard and colleagues is a prospective cohort study evaluating a total of 205,498 women from 1980–2015 from 2 separate cohorts (the Nurses’ Health Study and the Nurses’ Health Study II). The primary outcome was “to evaluate whether regular aspirin or nonaspirin NSAID use and patterns of use are associated with lower ovarian cancer risk.” Analgesic use and data regarding covariates were obtained via self-reported questionnaires. Ovarian cancer diagnosis was confirmed via medical records.
Results demonstrated that current low-dose aspirin use was associated with a decreased risk of ovarian cancer (hazard ratio [HR], 0.77; 95% confidence interval [CI], 0.61–0.96). This significance was not maintained upon further controlling for inflammatory factors (hypertension, autoimmune disease, inflammatory diet scores, smoking, etc) (HR, 0.94; 95% CI, 0.69–1.26). Other significant findings included an increased risk of developing ovarian cancer with standard-dose ASA use of ≥5 years or standard-dose use at 6 to 9 tablets per week (HR, 1.77; 95% CI, 1.13–2.77 and HR, 2.00; 95% CI, 1.27–3.15, respectively). An increased risk of developing ovarian cancer also was found for >10-year use or use of >10 tablets per week of nonaspirin NSAIDs (HR, 2.00; 95% CI, 1.27–3.15 and HR, 1.35; 95% CI, 1.02–1.79, respectively).
The authors concluded that there was a slight inverse association for low-dose aspirin and ovarian cancer risk and that standard aspirin or NSAID use actually may be associated with an increased risk of ovarian cancer.
Study strengths and weaknesses
This study has many strengths. It was a large prospective cohort investigation with adequate power to detect clinically significant differences. The authors collected detailed exposure data, which was novel. They also considered a latency period prior to the diagnosis of ovarian cancer during which a patient may increase their analgesic use in order to treat pain caused by the impending cancer.
However, the conclusions of the authors seem to be overstated in the setting of the data. Specifically, the deduction regarding a decreased risk of ovarian cancer with low-dose aspirin use given the loss of the statistical significance when controlling for pertinent cofounders. Further, the study authors did not evaluate adverse effects associated with low-dose aspirin use, which would be clinically applicable when determining whether the results from this study should become formal recommendations. Lastly, other important clinical factors, such as the presence of genetic mutations or endometriosis, were not considered, and these considerations would greatly affect results.
In the setting of previous large prospective studies that suggest no association between ASA use and ovarian cancer risk,4-6 data from this study are not compelling enough to recommend regular low-dose aspirin use to all women.
Based on these current data, there is insufficient evidence to suggest the use of low-dose aspirin for chemoprophylaxis of ovarian cancer. In order to suggest the use of a drug for prophylaxis the benefits must outweigh the risks, and in the case of NSAIDs, this has yet to be confirmed.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
EXPERT COMMENTARY
Epidemiologic studies conducted in ovarian cancer suggest an association between chronic inflammation and incidence of disease.1 Nonsteroidal anti-inflammatory drugs (NSAIDs) work to decrease inflammation through the inhibition of cyclo-oxygenase (COX). Therefore, anti-inflammatory agents such as NSAIDs have been proposed to play a role in the pathophysiology of ovarian cancer.
Previous studies of this association show conflicting data. The majority of these studies are retrospective, and those that are prospective do not include detailed data regarding dosing and frequency of ASA use.2-6
_
Details of the study
This study by Barnard and colleagues is a prospective cohort study evaluating a total of 205,498 women from 1980–2015 from 2 separate cohorts (the Nurses’ Health Study and the Nurses’ Health Study II). The primary outcome was “to evaluate whether regular aspirin or nonaspirin NSAID use and patterns of use are associated with lower ovarian cancer risk.” Analgesic use and data regarding covariates were obtained via self-reported questionnaires. Ovarian cancer diagnosis was confirmed via medical records.
Results demonstrated that current low-dose aspirin use was associated with a decreased risk of ovarian cancer (hazard ratio [HR], 0.77; 95% confidence interval [CI], 0.61–0.96). This significance was not maintained upon further controlling for inflammatory factors (hypertension, autoimmune disease, inflammatory diet scores, smoking, etc) (HR, 0.94; 95% CI, 0.69–1.26). Other significant findings included an increased risk of developing ovarian cancer with standard-dose ASA use of ≥5 years or standard-dose use at 6 to 9 tablets per week (HR, 1.77; 95% CI, 1.13–2.77 and HR, 2.00; 95% CI, 1.27–3.15, respectively). An increased risk of developing ovarian cancer also was found for >10-year use or use of >10 tablets per week of nonaspirin NSAIDs (HR, 2.00; 95% CI, 1.27–3.15 and HR, 1.35; 95% CI, 1.02–1.79, respectively).
The authors concluded that there was a slight inverse association for low-dose aspirin and ovarian cancer risk and that standard aspirin or NSAID use actually may be associated with an increased risk of ovarian cancer.
Study strengths and weaknesses
This study has many strengths. It was a large prospective cohort investigation with adequate power to detect clinically significant differences. The authors collected detailed exposure data, which was novel. They also considered a latency period prior to the diagnosis of ovarian cancer during which a patient may increase their analgesic use in order to treat pain caused by the impending cancer.
However, the conclusions of the authors seem to be overstated in the setting of the data. Specifically, the deduction regarding a decreased risk of ovarian cancer with low-dose aspirin use given the loss of the statistical significance when controlling for pertinent cofounders. Further, the study authors did not evaluate adverse effects associated with low-dose aspirin use, which would be clinically applicable when determining whether the results from this study should become formal recommendations. Lastly, other important clinical factors, such as the presence of genetic mutations or endometriosis, were not considered, and these considerations would greatly affect results.
In the setting of previous large prospective studies that suggest no association between ASA use and ovarian cancer risk,4-6 data from this study are not compelling enough to recommend regular low-dose aspirin use to all women.
Based on these current data, there is insufficient evidence to suggest the use of low-dose aspirin for chemoprophylaxis of ovarian cancer. In order to suggest the use of a drug for prophylaxis the benefits must outweigh the risks, and in the case of NSAIDs, this has yet to be confirmed.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Poole EM, Lee IM, Ridker PM, et al. A prospective study of circulating C-reactive protein, interleukin-6, and tumor necrosis factor alpha receptor 2 levels and risk of ovarian cancer. Am J Epidemiol. 2013;178:1256-1264.
- Trabert B, Ness RB, Lo-Ciganic WH, et al. Aspirin, nonaspirin nonsteroidal anti-inflammatory drug, and acetaminophen use and risk of invasive epithelial ovarian cancer: a pooled analysis in the Ovarian Cancer Association Consortium. J Natl Cancer Inst. 2014;106:djt431.
- Peres LC, Camacho F, Abbott SE, et al. Analgesic medication use and risk of epithelial ovarian cancer in African American women. Br J Cancer. 2016;114(7):819-825.
- Murphy MA, Trabert B, Yang HP, et al. Non-steroidal antiinflammatory drug use and ovarian cancer risk: findings from the NIH-AARP Diet and Health Study and systematic review. Cancer Causes Control. 2012;23:1839-1852.
- Brasky TM, Liu J, White E, et al. Non-steroidal antiinflammatory drugs and cancer risk in women: results from the Women’s Health Initiative. Int J Cancer. 2014;135:1869-1883.
- Lacey JV Jr, Sherman ME, Hartge P, et al. Medication use and risk of ovarian carcinoma: a prospective study. Int J Cancer. 2004;108:281-286.
- Poole EM, Lee IM, Ridker PM, et al. A prospective study of circulating C-reactive protein, interleukin-6, and tumor necrosis factor alpha receptor 2 levels and risk of ovarian cancer. Am J Epidemiol. 2013;178:1256-1264.
- Trabert B, Ness RB, Lo-Ciganic WH, et al. Aspirin, nonaspirin nonsteroidal anti-inflammatory drug, and acetaminophen use and risk of invasive epithelial ovarian cancer: a pooled analysis in the Ovarian Cancer Association Consortium. J Natl Cancer Inst. 2014;106:djt431.
- Peres LC, Camacho F, Abbott SE, et al. Analgesic medication use and risk of epithelial ovarian cancer in African American women. Br J Cancer. 2016;114(7):819-825.
- Murphy MA, Trabert B, Yang HP, et al. Non-steroidal antiinflammatory drug use and ovarian cancer risk: findings from the NIH-AARP Diet and Health Study and systematic review. Cancer Causes Control. 2012;23:1839-1852.
- Brasky TM, Liu J, White E, et al. Non-steroidal antiinflammatory drugs and cancer risk in women: results from the Women’s Health Initiative. Int J Cancer. 2014;135:1869-1883.
- Lacey JV Jr, Sherman ME, Hartge P, et al. Medication use and risk of ovarian carcinoma: a prospective study. Int J Cancer. 2004;108:281-286.
No boost in OS with addition of capecitabine for early TNBC
SAN ANTONIO – A phase 3 randomized controlled trial jointly conducted by GEICAM and CIBOMA is negative, showing that adding adjuvant capecitabine (Xeloda) to surgery and standard chemotherapy does not improve disease-free or overall survival in women with early-stage triple-negative breast cancer, reported lead investigator Miguel Martín, MD, PhD.
At the San Antonio Breast Cancer Symposium, he discussed the overall findings and intriguing subgroup results suggesting that there was a benefit in women with tumors having the nonbasal phenotype. Dr. Martín also detailed implications in the context of the CREATE-X trial findings and the era of personalized medicine, and outlined next avenues of research.
The trial was supported by Roche, which also provided capecitabine. Dr. Martín disclosed that he has received speakers honoraria from Pfizer and Lilly; honoraria for participation in advisory boards from AstraZeneca, Novartis, Roche-Genentech, Pfizer, GlaxoSmithKline, PharmaMar, Taiho Oncology, and Lilly; and research grants from Novartis and Roche.
SAN ANTONIO – A phase 3 randomized controlled trial jointly conducted by GEICAM and CIBOMA is negative, showing that adding adjuvant capecitabine (Xeloda) to surgery and standard chemotherapy does not improve disease-free or overall survival in women with early-stage triple-negative breast cancer, reported lead investigator Miguel Martín, MD, PhD.
At the San Antonio Breast Cancer Symposium, he discussed the overall findings and intriguing subgroup results suggesting that there was a benefit in women with tumors having the nonbasal phenotype. Dr. Martín also detailed implications in the context of the CREATE-X trial findings and the era of personalized medicine, and outlined next avenues of research.
The trial was supported by Roche, which also provided capecitabine. Dr. Martín disclosed that he has received speakers honoraria from Pfizer and Lilly; honoraria for participation in advisory boards from AstraZeneca, Novartis, Roche-Genentech, Pfizer, GlaxoSmithKline, PharmaMar, Taiho Oncology, and Lilly; and research grants from Novartis and Roche.
SAN ANTONIO – A phase 3 randomized controlled trial jointly conducted by GEICAM and CIBOMA is negative, showing that adding adjuvant capecitabine (Xeloda) to surgery and standard chemotherapy does not improve disease-free or overall survival in women with early-stage triple-negative breast cancer, reported lead investigator Miguel Martín, MD, PhD.
At the San Antonio Breast Cancer Symposium, he discussed the overall findings and intriguing subgroup results suggesting that there was a benefit in women with tumors having the nonbasal phenotype. Dr. Martín also detailed implications in the context of the CREATE-X trial findings and the era of personalized medicine, and outlined next avenues of research.
The trial was supported by Roche, which also provided capecitabine. Dr. Martín disclosed that he has received speakers honoraria from Pfizer and Lilly; honoraria for participation in advisory boards from AstraZeneca, Novartis, Roche-Genentech, Pfizer, GlaxoSmithKline, PharmaMar, Taiho Oncology, and Lilly; and research grants from Novartis and Roche.
REPORTING FROM SABCS 2018

Treatment Challenges When Headache Has Central and Peripheral Involvement
Diagnosis and treatment can be complicated when the headache history includes evidence of central and peripheral causes of pain.
ASHEVILLE, NC—Although neurologists tend to classify disorders as problems of either the CNS or the peripheral nervous system, patients with headache may have symptoms that indicate the involvement of both systems, according to an overview provided at the Eighth Annual Scientific Meeting of the Southern Headache Society. Research has revealed anatomic connections between extracranial and intracranial spaces that could contribute to the generation of headaches. Thus, the central and peripheral nervous systems “do not have to be two separate spaces and two separate pathologies,” said Pamela Blake, MD, Director of the Headache Center of Greater Heights in Houston.
Problems With Prevention and Acute Treatment
One patient presented to Dr. Blake with an eight-year history of throbbing headaches and constant pain and tightness in the neck and occiput. The pain radiated to her temples and forehead two to four times per week with accompanying photophobia, phonophobia, and nausea. The patient also had mild allodynia. The frontal pain and accompanying symptoms were consistent with episodic migraine, but the allodynia and pain in the neck and occiput were not, said Dr. Blake. A possible diagnosis was episodic migraine without aura with chronic tension-type headaches and neck pain, she added.

A 2017 study published in the Journal of Headache and Pain suggested that this headache type is problematic. Among 148 migraineurs, the researchers identified 100 patients who also had tension-type headache and chronic neck pain. Compared with healthy controls, these patients had less physical activity, less psychologic well-being, more perceived stress, and poorer self-rated health. Pain reduced these patients’ ability to perform physical activity, which could make treatment more difficult, according to the authors.
Patients with these symptoms have trigeminal and occipital pain. “These symptoms do not appear to be solely, or even primarily, central,” said Dr. Blake. The frontal pain responds to triptans, but the occipital pain, which is the more constant pain, does not. “Preventive medications do not work well in this population, and that’s why they have chronic headaches,” said Dr. Blake.
Physiologic and Pathophysiologic Mechanisms
Research by Schueler and colleagues suggested a potential physiologic explanation for combined central and peripheral involvement in headache. They applied a fluorescent tracer to proximally cut meningeal nerves in rat skulls and to distal branches of the spinosus nerve in human calvaria that was lined with dura mater. They observed that branches of the spinosus nerve travel “along the middle meningeal artery, supplying the dura, entering the cranial bone, and running through the calvarium,” said Dr. Blake. Branches of the spinosus nerve also “entered the tenderness junctions of the pericranial muscles, including in the neck.”
This work indicates a connection between intracranial and extracranial areas but does not shed light on the pathophysiology of a headache with central and peripheral symptoms, said Dr. Blake. In 2016, she and her colleagues took perivascular biopsies from healthy controls and subjects with chronic migraine and predominantly occipital headache. They found a significant increase in the expression of proinflammatory genes and a decrease in the expression of anti-inflammatory genes among migraineurs, compared with controls. “This was the first evidence of localized extracranial pathophysiology in chronic migraine,” said Dr. Blake.
This inflammation could result from compression of the occipital nerves. A 2013 study by Schmid et al found that progressive nerve compression results in chronic local and remote immune-mediated inflammation. Stress also can cause inflammation. “Many patients who present with occipital nerve compression headaches had the onset of their pain during a time of intense stress,” said Dr. Blake.
The Role of the Occipital Nerve
Occipital nerve compression headache is characterized by daily or near-daily pain in the distribution of the occipital nerve. Patients describe the pain as a tight, imploding pressure that sometimes radiates to frontal areas and becomes a throbbing pain. This headache rarely has a neuropathic component.
Allodynia is “almost a requirement of this diagnosis,” said Dr. Blake. The allodynia symptom checklist, however, does not capture it well in these patients because it focuses on pain in the trigeminal nerve distribution. Patients report that the back of the head is tender to the touch and that it hurts to rest the head on a pillow.
The cervical muscles compress the nerve and contribute to the symptoms as well. Patients often report that moving the head or neck exacerbates the pain. The headache also may have migrainous features. It takes skill and expertise to elicit an adequate history from these patients, said Dr. Blake. “Careful questioning is helpful. I often find … that a second visit is more helpful to obtain this history after reviewing the anatomy with the patient, reviewing this pathophysiology, and sending them back out to keep a careful log for two weeks.”
Nerve Blocks and Nerve Decompression
Occipital nerve blocks provide relief for these patients, but they may not be easy to administer. A large dorsal occipital nerve may be mistaken for the greater occipital nerve, for example. Physiologic abnormalities in some patients also can complicate this treatment.
Another effective treatment is nerve decompression surgery. Dr. Blake and colleagues conducted a retrospective review of patients who had undergone decompression of the greater occipital nerves at the point where they traverse the musculature of the posterior neck. The intervention provided complete relief for three to six years in one patient with new daily persistent headache and two patients with chronic posttraumatic headache. Two patients with chronic headache or migraine had partial relief. Surgery provided no relief for two patients with episodic migraine, one patient with chronic migraine, and one patient with chronic tension-type headache.
“In our experience, … 75% to 80% of patients experience a greater than 50% reduction in their headaches, measured by headache frequency and intensity,” said Dr. Blake. She and her colleagues compared outcomes between 18 chronic migraineurs with predominantly occipital pain who underwent surgical decompression of the occipital nerve and 23 patients who were referred for surgery but unable to receive it. In the surgical group, the number of predominantly occipital chronic migraine days per month decreased from 28.9 at baseline to 7.28 at a mean of 46 months later. The outcome in the control group did not change.
Correct patient selection “is the first, most important step” toward treatment success, said Dr. Blake. This process includes a preoperative psychologic evaluation that screens surgical candidates for somatic symptom disorder, mood disorders, a history of trauma, and catastrophizing. If indicated, neurologists may begin providing cognitive behavioral therapy or supportive psychotherapy before surgery. “We have a comprehensive program with postoperative management, including physical therapy and gradual taper of medications,” said Dr. Blake. Central migraine processes may contribute to headache in some of these patients. “Menstrual migraines are not going to go away with nerve decompression. Chronic migraine sometimes does not go away. This is a complex group of patients who definitely require a lot of follow-up.”
—Erik Greb
Suggested Reading
Blake P, Nir RR, Perry CJ, Burstein R. Tracking patients with chronic occipital headache after occipital nerve decompression surgery: a case series. Cephalalgia. 2018 Sep 14 [Epub ahead of print].
Krøll LS, Hammarlund CS, Westergaard ML, et al. Level of physical activity, well-being, stress and self-rated health in persons with migraine and co-existing tension-type headache and neck pain. J Headache Pain. 2017;18(1):46. Perry CJ, Blake P, Buettner C, et al. Upregulation of inflammatory gene transcripts in periosteum of chronic migraineurs: implications for extracranial origin of headache. Ann Neurol. 2016;79(6):1000-1013.
Schmid AB, Coppieters MW, Ruitenberg MJ, McLachlan EM. Local and remote immune-mediated inflammation after mild peripheral nerve compression in rats. J Neuropathol Exp Neurol. 2013;72(7):662-680.
Schueler M, Neuhuber WL, De Col R, Messlinger K. Innervation of rat and human dura mater and pericranial tissues in the parieto-temporal region by meningeal afferents. Headache. 2014;54(6):996-1009.
Diagnosis and treatment can be complicated when the headache history includes evidence of central and peripheral causes of pain.
Diagnosis and treatment can be complicated when the headache history includes evidence of central and peripheral causes of pain.
ASHEVILLE, NC—Although neurologists tend to classify disorders as problems of either the CNS or the peripheral nervous system, patients with headache may have symptoms that indicate the involvement of both systems, according to an overview provided at the Eighth Annual Scientific Meeting of the Southern Headache Society. Research has revealed anatomic connections between extracranial and intracranial spaces that could contribute to the generation of headaches. Thus, the central and peripheral nervous systems “do not have to be two separate spaces and two separate pathologies,” said Pamela Blake, MD, Director of the Headache Center of Greater Heights in Houston.
Problems With Prevention and Acute Treatment
One patient presented to Dr. Blake with an eight-year history of throbbing headaches and constant pain and tightness in the neck and occiput. The pain radiated to her temples and forehead two to four times per week with accompanying photophobia, phonophobia, and nausea. The patient also had mild allodynia. The frontal pain and accompanying symptoms were consistent with episodic migraine, but the allodynia and pain in the neck and occiput were not, said Dr. Blake. A possible diagnosis was episodic migraine without aura with chronic tension-type headaches and neck pain, she added.
A 2017 study published in the Journal of Headache and Pain suggested that this headache type is problematic. Among 148 migraineurs, the researchers identified 100 patients who also had tension-type headache and chronic neck pain. Compared with healthy controls, these patients had less physical activity, less psychologic well-being, more perceived stress, and poorer self-rated health. Pain reduced these patients’ ability to perform physical activity, which could make treatment more difficult, according to the authors.
Patients with these symptoms have trigeminal and occipital pain. “These symptoms do not appear to be solely, or even primarily, central,” said Dr. Blake. The frontal pain responds to triptans, but the occipital pain, which is the more constant pain, does not. “Preventive medications do not work well in this population, and that’s why they have chronic headaches,” said Dr. Blake.
Physiologic and Pathophysiologic Mechanisms
Research by Schueler and colleagues suggested a potential physiologic explanation for combined central and peripheral involvement in headache. They applied a fluorescent tracer to proximally cut meningeal nerves in rat skulls and to distal branches of the spinosus nerve in human calvaria that was lined with dura mater. They observed that branches of the spinosus nerve travel “along the middle meningeal artery, supplying the dura, entering the cranial bone, and running through the calvarium,” said Dr. Blake. Branches of the spinosus nerve also “entered the tenderness junctions of the pericranial muscles, including in the neck.”
This work indicates a connection between intracranial and extracranial areas but does not shed light on the pathophysiology of a headache with central and peripheral symptoms, said Dr. Blake. In 2016, she and her colleagues took perivascular biopsies from healthy controls and subjects with chronic migraine and predominantly occipital headache. They found a significant increase in the expression of proinflammatory genes and a decrease in the expression of anti-inflammatory genes among migraineurs, compared with controls. “This was the first evidence of localized extracranial pathophysiology in chronic migraine,” said Dr. Blake.
This inflammation could result from compression of the occipital nerves. A 2013 study by Schmid et al found that progressive nerve compression results in chronic local and remote immune-mediated inflammation. Stress also can cause inflammation. “Many patients who present with occipital nerve compression headaches had the onset of their pain during a time of intense stress,” said Dr. Blake.
The Role of the Occipital Nerve
Occipital nerve compression headache is characterized by daily or near-daily pain in the distribution of the occipital nerve. Patients describe the pain as a tight, imploding pressure that sometimes radiates to frontal areas and becomes a throbbing pain. This headache rarely has a neuropathic component.
Allodynia is “almost a requirement of this diagnosis,” said Dr. Blake. The allodynia symptom checklist, however, does not capture it well in these patients because it focuses on pain in the trigeminal nerve distribution. Patients report that the back of the head is tender to the touch and that it hurts to rest the head on a pillow.
The cervical muscles compress the nerve and contribute to the symptoms as well. Patients often report that moving the head or neck exacerbates the pain. The headache also may have migrainous features. It takes skill and expertise to elicit an adequate history from these patients, said Dr. Blake. “Careful questioning is helpful. I often find … that a second visit is more helpful to obtain this history after reviewing the anatomy with the patient, reviewing this pathophysiology, and sending them back out to keep a careful log for two weeks.”
Nerve Blocks and Nerve Decompression
Occipital nerve blocks provide relief for these patients, but they may not be easy to administer. A large dorsal occipital nerve may be mistaken for the greater occipital nerve, for example. Physiologic abnormalities in some patients also can complicate this treatment.
Another effective treatment is nerve decompression surgery. Dr. Blake and colleagues conducted a retrospective review of patients who had undergone decompression of the greater occipital nerves at the point where they traverse the musculature of the posterior neck. The intervention provided complete relief for three to six years in one patient with new daily persistent headache and two patients with chronic posttraumatic headache. Two patients with chronic headache or migraine had partial relief. Surgery provided no relief for two patients with episodic migraine, one patient with chronic migraine, and one patient with chronic tension-type headache.
“In our experience, … 75% to 80% of patients experience a greater than 50% reduction in their headaches, measured by headache frequency and intensity,” said Dr. Blake. She and her colleagues compared outcomes between 18 chronic migraineurs with predominantly occipital pain who underwent surgical decompression of the occipital nerve and 23 patients who were referred for surgery but unable to receive it. In the surgical group, the number of predominantly occipital chronic migraine days per month decreased from 28.9 at baseline to 7.28 at a mean of 46 months later. The outcome in the control group did not change.
Correct patient selection “is the first, most important step” toward treatment success, said Dr. Blake. This process includes a preoperative psychologic evaluation that screens surgical candidates for somatic symptom disorder, mood disorders, a history of trauma, and catastrophizing. If indicated, neurologists may begin providing cognitive behavioral therapy or supportive psychotherapy before surgery. “We have a comprehensive program with postoperative management, including physical therapy and gradual taper of medications,” said Dr. Blake. Central migraine processes may contribute to headache in some of these patients. “Menstrual migraines are not going to go away with nerve decompression. Chronic migraine sometimes does not go away. This is a complex group of patients who definitely require a lot of follow-up.”
—Erik Greb
Suggested Reading
Blake P, Nir RR, Perry CJ, Burstein R. Tracking patients with chronic occipital headache after occipital nerve decompression surgery: a case series. Cephalalgia. 2018 Sep 14 [Epub ahead of print].
Krøll LS, Hammarlund CS, Westergaard ML, et al. Level of physical activity, well-being, stress and self-rated health in persons with migraine and co-existing tension-type headache and neck pain. J Headache Pain. 2017;18(1):46. Perry CJ, Blake P, Buettner C, et al. Upregulation of inflammatory gene transcripts in periosteum of chronic migraineurs: implications for extracranial origin of headache. Ann Neurol. 2016;79(6):1000-1013.
Schmid AB, Coppieters MW, Ruitenberg MJ, McLachlan EM. Local and remote immune-mediated inflammation after mild peripheral nerve compression in rats. J Neuropathol Exp Neurol. 2013;72(7):662-680.
Schueler M, Neuhuber WL, De Col R, Messlinger K. Innervation of rat and human dura mater and pericranial tissues in the parieto-temporal region by meningeal afferents. Headache. 2014;54(6):996-1009.
ASHEVILLE, NC—Although neurologists tend to classify disorders as problems of either the CNS or the peripheral nervous system, patients with headache may have symptoms that indicate the involvement of both systems, according to an overview provided at the Eighth Annual Scientific Meeting of the Southern Headache Society. Research has revealed anatomic connections between extracranial and intracranial spaces that could contribute to the generation of headaches. Thus, the central and peripheral nervous systems “do not have to be two separate spaces and two separate pathologies,” said Pamela Blake, MD, Director of the Headache Center of Greater Heights in Houston.
Problems With Prevention and Acute Treatment
One patient presented to Dr. Blake with an eight-year history of throbbing headaches and constant pain and tightness in the neck and occiput. The pain radiated to her temples and forehead two to four times per week with accompanying photophobia, phonophobia, and nausea. The patient also had mild allodynia. The frontal pain and accompanying symptoms were consistent with episodic migraine, but the allodynia and pain in the neck and occiput were not, said Dr. Blake. A possible diagnosis was episodic migraine without aura with chronic tension-type headaches and neck pain, she added.
A 2017 study published in the Journal of Headache and Pain suggested that this headache type is problematic. Among 148 migraineurs, the researchers identified 100 patients who also had tension-type headache and chronic neck pain. Compared with healthy controls, these patients had less physical activity, less psychologic well-being, more perceived stress, and poorer self-rated health. Pain reduced these patients’ ability to perform physical activity, which could make treatment more difficult, according to the authors.
Patients with these symptoms have trigeminal and occipital pain. “These symptoms do not appear to be solely, or even primarily, central,” said Dr. Blake. The frontal pain responds to triptans, but the occipital pain, which is the more constant pain, does not. “Preventive medications do not work well in this population, and that’s why they have chronic headaches,” said Dr. Blake.
Physiologic and Pathophysiologic Mechanisms
Research by Schueler and colleagues suggested a potential physiologic explanation for combined central and peripheral involvement in headache. They applied a fluorescent tracer to proximally cut meningeal nerves in rat skulls and to distal branches of the spinosus nerve in human calvaria that was lined with dura mater. They observed that branches of the spinosus nerve travel “along the middle meningeal artery, supplying the dura, entering the cranial bone, and running through the calvarium,” said Dr. Blake. Branches of the spinosus nerve also “entered the tenderness junctions of the pericranial muscles, including in the neck.”
This work indicates a connection between intracranial and extracranial areas but does not shed light on the pathophysiology of a headache with central and peripheral symptoms, said Dr. Blake. In 2016, she and her colleagues took perivascular biopsies from healthy controls and subjects with chronic migraine and predominantly occipital headache. They found a significant increase in the expression of proinflammatory genes and a decrease in the expression of anti-inflammatory genes among migraineurs, compared with controls. “This was the first evidence of localized extracranial pathophysiology in chronic migraine,” said Dr. Blake.
This inflammation could result from compression of the occipital nerves. A 2013 study by Schmid et al found that progressive nerve compression results in chronic local and remote immune-mediated inflammation. Stress also can cause inflammation. “Many patients who present with occipital nerve compression headaches had the onset of their pain during a time of intense stress,” said Dr. Blake.
The Role of the Occipital Nerve
Occipital nerve compression headache is characterized by daily or near-daily pain in the distribution of the occipital nerve. Patients describe the pain as a tight, imploding pressure that sometimes radiates to frontal areas and becomes a throbbing pain. This headache rarely has a neuropathic component.
Allodynia is “almost a requirement of this diagnosis,” said Dr. Blake. The allodynia symptom checklist, however, does not capture it well in these patients because it focuses on pain in the trigeminal nerve distribution. Patients report that the back of the head is tender to the touch and that it hurts to rest the head on a pillow.
The cervical muscles compress the nerve and contribute to the symptoms as well. Patients often report that moving the head or neck exacerbates the pain. The headache also may have migrainous features. It takes skill and expertise to elicit an adequate history from these patients, said Dr. Blake. “Careful questioning is helpful. I often find … that a second visit is more helpful to obtain this history after reviewing the anatomy with the patient, reviewing this pathophysiology, and sending them back out to keep a careful log for two weeks.”
Nerve Blocks and Nerve Decompression
Occipital nerve blocks provide relief for these patients, but they may not be easy to administer. A large dorsal occipital nerve may be mistaken for the greater occipital nerve, for example. Physiologic abnormalities in some patients also can complicate this treatment.
Another effective treatment is nerve decompression surgery. Dr. Blake and colleagues conducted a retrospective review of patients who had undergone decompression of the greater occipital nerves at the point where they traverse the musculature of the posterior neck. The intervention provided complete relief for three to six years in one patient with new daily persistent headache and two patients with chronic posttraumatic headache. Two patients with chronic headache or migraine had partial relief. Surgery provided no relief for two patients with episodic migraine, one patient with chronic migraine, and one patient with chronic tension-type headache.
“In our experience, … 75% to 80% of patients experience a greater than 50% reduction in their headaches, measured by headache frequency and intensity,” said Dr. Blake. She and her colleagues compared outcomes between 18 chronic migraineurs with predominantly occipital pain who underwent surgical decompression of the occipital nerve and 23 patients who were referred for surgery but unable to receive it. In the surgical group, the number of predominantly occipital chronic migraine days per month decreased from 28.9 at baseline to 7.28 at a mean of 46 months later. The outcome in the control group did not change.
Correct patient selection “is the first, most important step” toward treatment success, said Dr. Blake. This process includes a preoperative psychologic evaluation that screens surgical candidates for somatic symptom disorder, mood disorders, a history of trauma, and catastrophizing. If indicated, neurologists may begin providing cognitive behavioral therapy or supportive psychotherapy before surgery. “We have a comprehensive program with postoperative management, including physical therapy and gradual taper of medications,” said Dr. Blake. Central migraine processes may contribute to headache in some of these patients. “Menstrual migraines are not going to go away with nerve decompression. Chronic migraine sometimes does not go away. This is a complex group of patients who definitely require a lot of follow-up.”
—Erik Greb
Suggested Reading
Blake P, Nir RR, Perry CJ, Burstein R. Tracking patients with chronic occipital headache after occipital nerve decompression surgery: a case series. Cephalalgia. 2018 Sep 14 [Epub ahead of print].
Krøll LS, Hammarlund CS, Westergaard ML, et al. Level of physical activity, well-being, stress and self-rated health in persons with migraine and co-existing tension-type headache and neck pain. J Headache Pain. 2017;18(1):46. Perry CJ, Blake P, Buettner C, et al. Upregulation of inflammatory gene transcripts in periosteum of chronic migraineurs: implications for extracranial origin of headache. Ann Neurol. 2016;79(6):1000-1013.
Schmid AB, Coppieters MW, Ruitenberg MJ, McLachlan EM. Local and remote immune-mediated inflammation after mild peripheral nerve compression in rats. J Neuropathol Exp Neurol. 2013;72(7):662-680.
Schueler M, Neuhuber WL, De Col R, Messlinger K. Innervation of rat and human dura mater and pericranial tissues in the parieto-temporal region by meningeal afferents. Headache. 2014;54(6):996-1009.
New devices can monitor personalized light exposure for radiation
, according to three studies of the millimeter-scale near-field communication (mm-NFC) devices.

“These studies highlight the differences between mm-NFC dosimeters and commercial devices in real-world, practical scenarios,” wrote lead author Seung Yun Heo of the department of biomedical engineering at the Center for Bio-Integrated Electronics at Northwestern University, Chicago, and her coauthors. “The former operate in continuous, uninterrupted modes, whereas the latter capture instantaneous values of intensity at preprogrammed intervals,”they noted. The study was published in Science Translational Medicine.
Separate studies to assess the performance of these “flexible” dosimeters took place in Rio de Janeiro, Brazil, and St. Petersburg, Fla. The Florida study included 13 healthy participants who wore skin-mounted mm-NFC ultraviolet A (UVA) dosimeters on the right back hand, left back hand, left inner arm, and left outer arm, plus a commercial dosimeter on the right wrist. The volunteers walked a 6.44-km path three times: a morning and subsequent afternoon stroll, plus an evening walk 4 days later. Four devices failed during the afternoon exercise, but otherwise, participants received data on their smartphones via the dosimeters at 30-minute intervals.
The Brazilian study was made up of nine healthy participants who wore mm-NFC UVA dosimeters on the thumbnail or the middle fingernail; commercial dosimeters were worn on the wrist of the ipsilateral side. These volunteers engaged in rooftop recreational activities that corresponded to solar zenith angles, along with showering and swimming with the use of soap and skin creams. All sensors remained functional over the 4 days of testing, and 14 of 20 devices remained adhered to the fingernail. Accumulated doses ranged widely, “as expected on the basis of the differences in behaviors,” the authors wrote. These observations imply highly variable UV-associated risks between participants, due not only to differences in Fitzpatrick skin types but also to individual behavior patterns,” they added.
The third study of mm-NFC blue light dosimeters comprised three newborns in an Urbana, Ill., neonatal ICU undergoing blue light phototherapy treatments. Nurses mounted dosimeters on the patients’ chests before phototherapy; an antenna underneath the incubator mattress transmitted continuous wireless measurements of blue intensity and dosage at 20-minute intervals for 20 hours.
The authors acknowledged that these devices and their designs have limitations, including a small detection area as compared to the surface area of a human body. The study’s results “represent localized measurements of exposure, whereas the sun irradiance profile across the body surface is not uniform and varies by position of the sun in the sky over the course of a day.” They recommended that future research could “create anatomic specific risk assessment of UV exposure” via multinodal sensing with UVA/UVB dosimeters on several parts of the body.
The Brazilian UV study was sponsored by La Roche Posay and L’Oreal California Research Center. Research was supported by the National Cancer Institute. Five of the authors reported commercial interests in the technology. Another author reported paid consultation for Aclaris Therapeutics.
SOURCE: Heo SY et al. Sci. Transl. Med. 2018 Dec 5. doi: 10.1126/scitranslmed.aau1643.
, according to three studies of the millimeter-scale near-field communication (mm-NFC) devices.
“These studies highlight the differences between mm-NFC dosimeters and commercial devices in real-world, practical scenarios,” wrote lead author Seung Yun Heo of the department of biomedical engineering at the Center for Bio-Integrated Electronics at Northwestern University, Chicago, and her coauthors. “The former operate in continuous, uninterrupted modes, whereas the latter capture instantaneous values of intensity at preprogrammed intervals,”they noted. The study was published in Science Translational Medicine.
Separate studies to assess the performance of these “flexible” dosimeters took place in Rio de Janeiro, Brazil, and St. Petersburg, Fla. The Florida study included 13 healthy participants who wore skin-mounted mm-NFC ultraviolet A (UVA) dosimeters on the right back hand, left back hand, left inner arm, and left outer arm, plus a commercial dosimeter on the right wrist. The volunteers walked a 6.44-km path three times: a morning and subsequent afternoon stroll, plus an evening walk 4 days later. Four devices failed during the afternoon exercise, but otherwise, participants received data on their smartphones via the dosimeters at 30-minute intervals.
The Brazilian study was made up of nine healthy participants who wore mm-NFC UVA dosimeters on the thumbnail or the middle fingernail; commercial dosimeters were worn on the wrist of the ipsilateral side. These volunteers engaged in rooftop recreational activities that corresponded to solar zenith angles, along with showering and swimming with the use of soap and skin creams. All sensors remained functional over the 4 days of testing, and 14 of 20 devices remained adhered to the fingernail. Accumulated doses ranged widely, “as expected on the basis of the differences in behaviors,” the authors wrote. These observations imply highly variable UV-associated risks between participants, due not only to differences in Fitzpatrick skin types but also to individual behavior patterns,” they added.
The third study of mm-NFC blue light dosimeters comprised three newborns in an Urbana, Ill., neonatal ICU undergoing blue light phototherapy treatments. Nurses mounted dosimeters on the patients’ chests before phototherapy; an antenna underneath the incubator mattress transmitted continuous wireless measurements of blue intensity and dosage at 20-minute intervals for 20 hours.
The authors acknowledged that these devices and their designs have limitations, including a small detection area as compared to the surface area of a human body. The study’s results “represent localized measurements of exposure, whereas the sun irradiance profile across the body surface is not uniform and varies by position of the sun in the sky over the course of a day.” They recommended that future research could “create anatomic specific risk assessment of UV exposure” via multinodal sensing with UVA/UVB dosimeters on several parts of the body.
The Brazilian UV study was sponsored by La Roche Posay and L’Oreal California Research Center. Research was supported by the National Cancer Institute. Five of the authors reported commercial interests in the technology. Another author reported paid consultation for Aclaris Therapeutics.
SOURCE: Heo SY et al. Sci. Transl. Med. 2018 Dec 5. doi: 10.1126/scitranslmed.aau1643.
, according to three studies of the millimeter-scale near-field communication (mm-NFC) devices.
“These studies highlight the differences between mm-NFC dosimeters and commercial devices in real-world, practical scenarios,” wrote lead author Seung Yun Heo of the department of biomedical engineering at the Center for Bio-Integrated Electronics at Northwestern University, Chicago, and her coauthors. “The former operate in continuous, uninterrupted modes, whereas the latter capture instantaneous values of intensity at preprogrammed intervals,”they noted. The study was published in Science Translational Medicine.
Separate studies to assess the performance of these “flexible” dosimeters took place in Rio de Janeiro, Brazil, and St. Petersburg, Fla. The Florida study included 13 healthy participants who wore skin-mounted mm-NFC ultraviolet A (UVA) dosimeters on the right back hand, left back hand, left inner arm, and left outer arm, plus a commercial dosimeter on the right wrist. The volunteers walked a 6.44-km path three times: a morning and subsequent afternoon stroll, plus an evening walk 4 days later. Four devices failed during the afternoon exercise, but otherwise, participants received data on their smartphones via the dosimeters at 30-minute intervals.
The Brazilian study was made up of nine healthy participants who wore mm-NFC UVA dosimeters on the thumbnail or the middle fingernail; commercial dosimeters were worn on the wrist of the ipsilateral side. These volunteers engaged in rooftop recreational activities that corresponded to solar zenith angles, along with showering and swimming with the use of soap and skin creams. All sensors remained functional over the 4 days of testing, and 14 of 20 devices remained adhered to the fingernail. Accumulated doses ranged widely, “as expected on the basis of the differences in behaviors,” the authors wrote. These observations imply highly variable UV-associated risks between participants, due not only to differences in Fitzpatrick skin types but also to individual behavior patterns,” they added.
The third study of mm-NFC blue light dosimeters comprised three newborns in an Urbana, Ill., neonatal ICU undergoing blue light phototherapy treatments. Nurses mounted dosimeters on the patients’ chests before phototherapy; an antenna underneath the incubator mattress transmitted continuous wireless measurements of blue intensity and dosage at 20-minute intervals for 20 hours.
The authors acknowledged that these devices and their designs have limitations, including a small detection area as compared to the surface area of a human body. The study’s results “represent localized measurements of exposure, whereas the sun irradiance profile across the body surface is not uniform and varies by position of the sun in the sky over the course of a day.” They recommended that future research could “create anatomic specific risk assessment of UV exposure” via multinodal sensing with UVA/UVB dosimeters on several parts of the body.
The Brazilian UV study was sponsored by La Roche Posay and L’Oreal California Research Center. Research was supported by the National Cancer Institute. Five of the authors reported commercial interests in the technology. Another author reported paid consultation for Aclaris Therapeutics.
SOURCE: Heo SY et al. Sci. Transl. Med. 2018 Dec 5. doi: 10.1126/scitranslmed.aau1643.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point: Newly designed flexible dosimeters can track personalized light exposure and electromagnetic radiation via wireless sensor technology.
Major finding: In one study, during four days of testing – including recreational activities, showering, and swimming—all mm-NFC UVA dosimeter sensors remained functional and 14 of 20 devices remained adhered to the fingernail.
Study details: Three studies of millimeter-scale near-field communication dosimeters, comprising healthy volunteers from Rio de Janeiro, Brazil; St. Petersburg, Florida; and neonates undergoing blue light phototherapy treatments in an Urbana, Ill., neonatal ICU.
Disclosures: The UV study in Brazil, was sponsored by La Roche Posay and the L’Oreal California Research Center. Research was supported by the National Cancer Institute. Five of the authors reported commercial interests in the technology. Another author reported paid consultation for Aclaris Therapeutics.
Source: Heo SY et al. Sci. Transl. Med. 2018 Dec 5 doi: 10.1126/scitranslmed.aau1643.
Anticoagulant choice, PPI cotherapy impact risk of upper GI bleeding
Patients receiving oral anticoagulant treatment had the lowest risk of gastrointestinal bleeding when taking apixaban, compared with rivaroxaban, dabigatran, and warfarin, according to a recent study.
Further, patients who received proton pump inhibitor (PPI) cotherapy had a lower overall risk of gastrointestinal bleeding, according to Wayne A. Ray, PhD, from the department of health policy at Vanderbilt University, Nashville, Tenn., and his colleagues.
“These findings indicate the potential benefits of a gastrointestinal bleeding risk assessment before initiating anticoagulant treatment,” Dr. Ray and his colleagues wrote in their study, which was published in JAMA.
Dr. Ray and his colleagues performed a retrospective, population-based study of 1,643,123 Medicare beneficiaries (mean age, 76.4 years) who received 1,713,183 new episodes of oral anticoagulant treatment between January 2011 and September 2015. They analyzed how patients reacted to apixaban, dabigatran, rivaroxaban, or warfarin both with and without PPI cotherapy.
Overall, the risk of gastrointestinal bleeding across 754,389 person-years without PPI therapy was 115 per 10,000 person-years (95% confidence interval, 112-118) in 7,119 patients. The researchers found the risk of gastrointestinal bleeding was highest in patients taking rivaroxaban (1,278 patients; 144 per 10,000 person-years; 95% CI, 136-152) and lowest when taking apixaban (279 patients; 120 per 10,000 person-years; incidence rate ratio, 1,97; 95% CI, 1.73-2.25), compared with dabigatran (629 patients; 120 per 10,000 person-years; IRR, 1.19; 95% CI, 1.08-1.32) and warfarin (4,933 patients; 113 per 10,000 person-years; IRR, 1.27; 95% CI, 1.19-1.35). There was a significantly lower incidence of gastrointestinal bleeding for apixaban, compared with warfarin (IRR, 0.64; 95% CI, 0.57-0.73) and dabigatran (IRR, 0.61; 95% CI, 0.52-0.70).
There was a lower overall incidence of gastrointestinal bleeding when receiving PPI cotherapy (264,447 person-years; 76 per 10,000 person-years), compared with patients who received anticoagulant treatment without PPI cotherapy (IRR, 0.66; 95% CI, 0.62-0.69). This reduced incidence of gastrointestinal bleeding was also seen in patients receiving PPI cotherapy and taking apixaban (IRR, 0.66; 95% CI, 0.52-0.85), dabigatran (IRR, 0.49; 95% CI, 0.41-0.59), rivaroxaban (IRR, 0.75; 95% CI, 0.68-0.84), and warfarin (IRR, 0.65; 95% CI, 0.62-0.69).
The researchers noted that limitations in this study included potential misclassification of anticoagulant treatment, PPI cotherapy, and NSAIDs because of a reliance on filled prescription data; confounding by unmeasured factors such as aspirin exposure or Helicobacter pylori infection; and gastrointestinal bleeding being measured using a disease risk score.
This study was supported by a grant from the National Heart, Lung, and Blood Institute. The authors reported no relevant conflicts of interest.
SOURCE: Ray WA et al. JAMA. 2018 Dec 4. doi: 10.1001/jama.2018.17242.
Patients receiving oral anticoagulant treatment had the lowest risk of gastrointestinal bleeding when taking apixaban, compared with rivaroxaban, dabigatran, and warfarin, according to a recent study.
Further, patients who received proton pump inhibitor (PPI) cotherapy had a lower overall risk of gastrointestinal bleeding, according to Wayne A. Ray, PhD, from the department of health policy at Vanderbilt University, Nashville, Tenn., and his colleagues.
“These findings indicate the potential benefits of a gastrointestinal bleeding risk assessment before initiating anticoagulant treatment,” Dr. Ray and his colleagues wrote in their study, which was published in JAMA.
Dr. Ray and his colleagues performed a retrospective, population-based study of 1,643,123 Medicare beneficiaries (mean age, 76.4 years) who received 1,713,183 new episodes of oral anticoagulant treatment between January 2011 and September 2015. They analyzed how patients reacted to apixaban, dabigatran, rivaroxaban, or warfarin both with and without PPI cotherapy.
Overall, the risk of gastrointestinal bleeding across 754,389 person-years without PPI therapy was 115 per 10,000 person-years (95% confidence interval, 112-118) in 7,119 patients. The researchers found the risk of gastrointestinal bleeding was highest in patients taking rivaroxaban (1,278 patients; 144 per 10,000 person-years; 95% CI, 136-152) and lowest when taking apixaban (279 patients; 120 per 10,000 person-years; incidence rate ratio, 1,97; 95% CI, 1.73-2.25), compared with dabigatran (629 patients; 120 per 10,000 person-years; IRR, 1.19; 95% CI, 1.08-1.32) and warfarin (4,933 patients; 113 per 10,000 person-years; IRR, 1.27; 95% CI, 1.19-1.35). There was a significantly lower incidence of gastrointestinal bleeding for apixaban, compared with warfarin (IRR, 0.64; 95% CI, 0.57-0.73) and dabigatran (IRR, 0.61; 95% CI, 0.52-0.70).
There was a lower overall incidence of gastrointestinal bleeding when receiving PPI cotherapy (264,447 person-years; 76 per 10,000 person-years), compared with patients who received anticoagulant treatment without PPI cotherapy (IRR, 0.66; 95% CI, 0.62-0.69). This reduced incidence of gastrointestinal bleeding was also seen in patients receiving PPI cotherapy and taking apixaban (IRR, 0.66; 95% CI, 0.52-0.85), dabigatran (IRR, 0.49; 95% CI, 0.41-0.59), rivaroxaban (IRR, 0.75; 95% CI, 0.68-0.84), and warfarin (IRR, 0.65; 95% CI, 0.62-0.69).
The researchers noted that limitations in this study included potential misclassification of anticoagulant treatment, PPI cotherapy, and NSAIDs because of a reliance on filled prescription data; confounding by unmeasured factors such as aspirin exposure or Helicobacter pylori infection; and gastrointestinal bleeding being measured using a disease risk score.
This study was supported by a grant from the National Heart, Lung, and Blood Institute. The authors reported no relevant conflicts of interest.
SOURCE: Ray WA et al. JAMA. 2018 Dec 4. doi: 10.1001/jama.2018.17242.
Patients receiving oral anticoagulant treatment had the lowest risk of gastrointestinal bleeding when taking apixaban, compared with rivaroxaban, dabigatran, and warfarin, according to a recent study.
Further, patients who received proton pump inhibitor (PPI) cotherapy had a lower overall risk of gastrointestinal bleeding, according to Wayne A. Ray, PhD, from the department of health policy at Vanderbilt University, Nashville, Tenn., and his colleagues.
“These findings indicate the potential benefits of a gastrointestinal bleeding risk assessment before initiating anticoagulant treatment,” Dr. Ray and his colleagues wrote in their study, which was published in JAMA.
Dr. Ray and his colleagues performed a retrospective, population-based study of 1,643,123 Medicare beneficiaries (mean age, 76.4 years) who received 1,713,183 new episodes of oral anticoagulant treatment between January 2011 and September 2015. They analyzed how patients reacted to apixaban, dabigatran, rivaroxaban, or warfarin both with and without PPI cotherapy.
Overall, the risk of gastrointestinal bleeding across 754,389 person-years without PPI therapy was 115 per 10,000 person-years (95% confidence interval, 112-118) in 7,119 patients. The researchers found the risk of gastrointestinal bleeding was highest in patients taking rivaroxaban (1,278 patients; 144 per 10,000 person-years; 95% CI, 136-152) and lowest when taking apixaban (279 patients; 120 per 10,000 person-years; incidence rate ratio, 1,97; 95% CI, 1.73-2.25), compared with dabigatran (629 patients; 120 per 10,000 person-years; IRR, 1.19; 95% CI, 1.08-1.32) and warfarin (4,933 patients; 113 per 10,000 person-years; IRR, 1.27; 95% CI, 1.19-1.35). There was a significantly lower incidence of gastrointestinal bleeding for apixaban, compared with warfarin (IRR, 0.64; 95% CI, 0.57-0.73) and dabigatran (IRR, 0.61; 95% CI, 0.52-0.70).
There was a lower overall incidence of gastrointestinal bleeding when receiving PPI cotherapy (264,447 person-years; 76 per 10,000 person-years), compared with patients who received anticoagulant treatment without PPI cotherapy (IRR, 0.66; 95% CI, 0.62-0.69). This reduced incidence of gastrointestinal bleeding was also seen in patients receiving PPI cotherapy and taking apixaban (IRR, 0.66; 95% CI, 0.52-0.85), dabigatran (IRR, 0.49; 95% CI, 0.41-0.59), rivaroxaban (IRR, 0.75; 95% CI, 0.68-0.84), and warfarin (IRR, 0.65; 95% CI, 0.62-0.69).
The researchers noted that limitations in this study included potential misclassification of anticoagulant treatment, PPI cotherapy, and NSAIDs because of a reliance on filled prescription data; confounding by unmeasured factors such as aspirin exposure or Helicobacter pylori infection; and gastrointestinal bleeding being measured using a disease risk score.
This study was supported by a grant from the National Heart, Lung, and Blood Institute. The authors reported no relevant conflicts of interest.
SOURCE: Ray WA et al. JAMA. 2018 Dec 4. doi: 10.1001/jama.2018.17242.
FROM JAMA
Key clinical point: In patients receiving oral anticoagulant treatment, risk of gastrointestinal bleeding was highest in patients taking rivaroxaban, lowest when taking apixaban, and there was a lower overall incidence of gastrointestinal bleeding when receiving proton pump inhibitor cotherapy.
Major finding: Per 10,000 person-years, the incidence rate of gastrointestinal bleeding was 144 for rivaroxaban, 73 for apixaban, 120 for dabigatran, and 113 for warfarin; there was a gastrointestinal bleeding incidence rate ratio of 0.66 for patients using protein pump inhibitor cotherapy.
Study details: A retrospective, population-based study of 1,643,123 Medicare beneficiaries who received oral anticoagulant treatment between January 2011 and September 2015.
Disclosures: This study was supported by a grant from the National Heart, Lung, and Blood Institute. The authors reported no relevant conflicts of interest.
Source: Ray WA et al. JAMA. 2018 Dec 4. doi: 10.1001/jama.2018.17242.
ONC releases draft strategy on reducing EHR burden
A new federal proposal aims to move you away from the keyboard and back face-to-face with your patients.

The draft strategy from the Office of the National Coordinator for Health IT has three aims: to reduce the time and effort to record information in EHRs; to reduce the time and effort required to meet regulatory requirements; and to improve the functionality and ease of use of EHRs.
“This draft strategy includes recommendations that will allow physicians and other clinicians to provide effective care to their patients with a renewed sense of satisfaction for them and their patients,” Andrew Gettinger, MD, chief clinical officer at ONC, and Kate Goodrich, MD, chief medical officer at the Centers for Medicare & Medicaid Services, wrote in a recent blog post. “We are taking one more step toward improving the interoperability and usability of health information by establishing a goal, strategy, and recommendations to reduce regulatory and administrative burdens relating to the use of EHRs.”
To ease documentation burdens, the proposal seeks to “mitigate the EHR-related burden associated with a variety of administrative processes,” the draft strategy notes. “We are considering how reforming certain administrative requirements or optimizing out-of-date requirements for health IT–enabled health care provider work flows can reduce the burden of clinical documentation.”
Specifically, ONC proposes to reduce the overall regulatory burden, leverage data present in the electronic record to reduce the redocumentation, waive certain documentation requirements for participants in advanced alternative payment models (APMs), and promote standardized documentation for ordering and prior authorization.
To improve health IT usability, the draft strategy aims to “address how improvements in the design and use of health IT systems” can reduce burden and calls on clinicians, software developers, and other vendors to collaborate.
To do so, ONC recommends better alignment between EHR design and clinical work flow and making improvements to clinical decision support, as well as improving the presentation of clinical data within EHRs and clinical documentation functionality.
ONC also recommends standardizing basic clinical operations across all EHRs, designing EHR interfaces that are standard to health care delivery, and better integration of the EHR with the exam room.
The draft strategy also includes recommendations to help doctors better understand the financial requirements for successful implementation and optimize the log-in procedures to help reduce burden.
EHR reporting strategies “are designed to address many of the programmatic, technical, and operational challenges raised by stakeholders to reduce EHR-related burden associated with program reporting.”
ONC wants to simplify scoring for the “promoting interoperability” performance category in the Merit-based Incentive Payment System (MIPS) track of the Quality Payment Program and improving other measures of health IT usage; applying additional data standards to make data access, extraction, and integration across multiple systems easier and less costly; and exploring alternate, less burdensome approaches to electronic quality measurement through pilot programs and reporting program incentives.
Finally, public health reporting strategies “look at a set of topics linked to federal, state, local, territorial, and tribal government policies and public health programs, with a specific focus on EPCS [electronic prescribing for controlled substances] and PDMPs [prescription drug monitoring programs]. Where EHR-related burden remains a key barrier to progress in these areas, there are several recommendations for how stakeholders can advance these burden reduction goals related to public health.”
In this area, ONC is recommending increasing adoption of e-prescribing of controlled substances with access to medication history to better inform prescribing of controlled substances, harmonizing reporting requirements across federally funded programs to streamline reporting requirements, and providing better guidance about HIPPA and federal confidentially requirements governing substance abuse disorder to better facilitate the electronic exchange of health information for patient care.
Comments on the report may be submitted electronically through Jan. 28, 2019.
A new federal proposal aims to move you away from the keyboard and back face-to-face with your patients.
The draft strategy from the Office of the National Coordinator for Health IT has three aims: to reduce the time and effort to record information in EHRs; to reduce the time and effort required to meet regulatory requirements; and to improve the functionality and ease of use of EHRs.
“This draft strategy includes recommendations that will allow physicians and other clinicians to provide effective care to their patients with a renewed sense of satisfaction for them and their patients,” Andrew Gettinger, MD, chief clinical officer at ONC, and Kate Goodrich, MD, chief medical officer at the Centers for Medicare & Medicaid Services, wrote in a recent blog post. “We are taking one more step toward improving the interoperability and usability of health information by establishing a goal, strategy, and recommendations to reduce regulatory and administrative burdens relating to the use of EHRs.”
To ease documentation burdens, the proposal seeks to “mitigate the EHR-related burden associated with a variety of administrative processes,” the draft strategy notes. “We are considering how reforming certain administrative requirements or optimizing out-of-date requirements for health IT–enabled health care provider work flows can reduce the burden of clinical documentation.”
Specifically, ONC proposes to reduce the overall regulatory burden, leverage data present in the electronic record to reduce the redocumentation, waive certain documentation requirements for participants in advanced alternative payment models (APMs), and promote standardized documentation for ordering and prior authorization.
To improve health IT usability, the draft strategy aims to “address how improvements in the design and use of health IT systems” can reduce burden and calls on clinicians, software developers, and other vendors to collaborate.
To do so, ONC recommends better alignment between EHR design and clinical work flow and making improvements to clinical decision support, as well as improving the presentation of clinical data within EHRs and clinical documentation functionality.
ONC also recommends standardizing basic clinical operations across all EHRs, designing EHR interfaces that are standard to health care delivery, and better integration of the EHR with the exam room.
The draft strategy also includes recommendations to help doctors better understand the financial requirements for successful implementation and optimize the log-in procedures to help reduce burden.
EHR reporting strategies “are designed to address many of the programmatic, technical, and operational challenges raised by stakeholders to reduce EHR-related burden associated with program reporting.”
ONC wants to simplify scoring for the “promoting interoperability” performance category in the Merit-based Incentive Payment System (MIPS) track of the Quality Payment Program and improving other measures of health IT usage; applying additional data standards to make data access, extraction, and integration across multiple systems easier and less costly; and exploring alternate, less burdensome approaches to electronic quality measurement through pilot programs and reporting program incentives.
Finally, public health reporting strategies “look at a set of topics linked to federal, state, local, territorial, and tribal government policies and public health programs, with a specific focus on EPCS [electronic prescribing for controlled substances] and PDMPs [prescription drug monitoring programs]. Where EHR-related burden remains a key barrier to progress in these areas, there are several recommendations for how stakeholders can advance these burden reduction goals related to public health.”
In this area, ONC is recommending increasing adoption of e-prescribing of controlled substances with access to medication history to better inform prescribing of controlled substances, harmonizing reporting requirements across federally funded programs to streamline reporting requirements, and providing better guidance about HIPPA and federal confidentially requirements governing substance abuse disorder to better facilitate the electronic exchange of health information for patient care.
Comments on the report may be submitted electronically through Jan. 28, 2019.
A new federal proposal aims to move you away from the keyboard and back face-to-face with your patients.
The draft strategy from the Office of the National Coordinator for Health IT has three aims: to reduce the time and effort to record information in EHRs; to reduce the time and effort required to meet regulatory requirements; and to improve the functionality and ease of use of EHRs.
“This draft strategy includes recommendations that will allow physicians and other clinicians to provide effective care to their patients with a renewed sense of satisfaction for them and their patients,” Andrew Gettinger, MD, chief clinical officer at ONC, and Kate Goodrich, MD, chief medical officer at the Centers for Medicare & Medicaid Services, wrote in a recent blog post. “We are taking one more step toward improving the interoperability and usability of health information by establishing a goal, strategy, and recommendations to reduce regulatory and administrative burdens relating to the use of EHRs.”
To ease documentation burdens, the proposal seeks to “mitigate the EHR-related burden associated with a variety of administrative processes,” the draft strategy notes. “We are considering how reforming certain administrative requirements or optimizing out-of-date requirements for health IT–enabled health care provider work flows can reduce the burden of clinical documentation.”
Specifically, ONC proposes to reduce the overall regulatory burden, leverage data present in the electronic record to reduce the redocumentation, waive certain documentation requirements for participants in advanced alternative payment models (APMs), and promote standardized documentation for ordering and prior authorization.
To improve health IT usability, the draft strategy aims to “address how improvements in the design and use of health IT systems” can reduce burden and calls on clinicians, software developers, and other vendors to collaborate.
To do so, ONC recommends better alignment between EHR design and clinical work flow and making improvements to clinical decision support, as well as improving the presentation of clinical data within EHRs and clinical documentation functionality.
ONC also recommends standardizing basic clinical operations across all EHRs, designing EHR interfaces that are standard to health care delivery, and better integration of the EHR with the exam room.
The draft strategy also includes recommendations to help doctors better understand the financial requirements for successful implementation and optimize the log-in procedures to help reduce burden.
EHR reporting strategies “are designed to address many of the programmatic, technical, and operational challenges raised by stakeholders to reduce EHR-related burden associated with program reporting.”
ONC wants to simplify scoring for the “promoting interoperability” performance category in the Merit-based Incentive Payment System (MIPS) track of the Quality Payment Program and improving other measures of health IT usage; applying additional data standards to make data access, extraction, and integration across multiple systems easier and less costly; and exploring alternate, less burdensome approaches to electronic quality measurement through pilot programs and reporting program incentives.
Finally, public health reporting strategies “look at a set of topics linked to federal, state, local, territorial, and tribal government policies and public health programs, with a specific focus on EPCS [electronic prescribing for controlled substances] and PDMPs [prescription drug monitoring programs]. Where EHR-related burden remains a key barrier to progress in these areas, there are several recommendations for how stakeholders can advance these burden reduction goals related to public health.”
In this area, ONC is recommending increasing adoption of e-prescribing of controlled substances with access to medication history to better inform prescribing of controlled substances, harmonizing reporting requirements across federally funded programs to streamline reporting requirements, and providing better guidance about HIPPA and federal confidentially requirements governing substance abuse disorder to better facilitate the electronic exchange of health information for patient care.
Comments on the report may be submitted electronically through Jan. 28, 2019.
2018: A banner year for hematology drug approvals
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
REPORTING FROM ASH 2018
