User login
Study Suggests Improvement Needed in AED Treatment
About 50% of patients with epilepsy remained untreated for 6 months after their initial diagnosis according to a retrospective analysis of more than 58,000 cases.
- At 6 months after diagnosis, 46.8% were receiving treatment with antiepilepsy medication; at 12 months, that statistic had only climbed to 52.2%.
- Among the 29,226 patients who were receiving medication, nearly three quarters received monotherapy and 1.6% polytherapy as first treatment for 90 days or longer.
- The likelihood of patients remaining on antiepilepsy medication after a year was 61% for those on a single agent and 36.5% for those on more than one drug.
Faught E, Helmers S, Thurman D, et al. Patient characteristics and treatment patterns in patients with newly diagnosed epilepsy: A US database analysis. Epilepsy Behav. 2018;85:37-44.
About 50% of patients with epilepsy remained untreated for 6 months after their initial diagnosis according to a retrospective analysis of more than 58,000 cases.
- At 6 months after diagnosis, 46.8% were receiving treatment with antiepilepsy medication; at 12 months, that statistic had only climbed to 52.2%.
- Among the 29,226 patients who were receiving medication, nearly three quarters received monotherapy and 1.6% polytherapy as first treatment for 90 days or longer.
- The likelihood of patients remaining on antiepilepsy medication after a year was 61% for those on a single agent and 36.5% for those on more than one drug.
Faught E, Helmers S, Thurman D, et al. Patient characteristics and treatment patterns in patients with newly diagnosed epilepsy: A US database analysis. Epilepsy Behav. 2018;85:37-44.
About 50% of patients with epilepsy remained untreated for 6 months after their initial diagnosis according to a retrospective analysis of more than 58,000 cases.
- At 6 months after diagnosis, 46.8% were receiving treatment with antiepilepsy medication; at 12 months, that statistic had only climbed to 52.2%.
- Among the 29,226 patients who were receiving medication, nearly three quarters received monotherapy and 1.6% polytherapy as first treatment for 90 days or longer.
- The likelihood of patients remaining on antiepilepsy medication after a year was 61% for those on a single agent and 36.5% for those on more than one drug.
Faught E, Helmers S, Thurman D, et al. Patient characteristics and treatment patterns in patients with newly diagnosed epilepsy: A US database analysis. Epilepsy Behav. 2018;85:37-44.
Anxiety Plagues Many Patients with Epilepsy
Nearly half of patients with epilepsy have symptoms of high anxiety according to a study of adults treated in tertiary care centers.
- The study, which included 540 patients, evaluated the presence of anxiety with the Symptoms Checklist 90-R anxiety subscale. It also evaluated patients for depression with separate scales.
- 250 patients (46.1%) reported high anxiety.
- Focal epilepsy and epilepsy of unknown type, as well as depression scores, were independently linked to high anxiety.
- In patients with focal epilepsy, mesial temporal sclerosis was independently associated with high anxiety.
- Other factors linked to high anxiety included lower education level, being non-white, having Spanish as a native language, prior head trauma, and polydrug therapy for epilepsy.
- The researchers suggest that screening for anxiety in an epilepsy clinic can help spot patients in need of treatment.
Munger Clary HM, Snively BM, Hamberger MJ. Anxiety is common and independently associated with clinical features of epilepsy. Epilepsy Behav. 2018;85:64-71.
Nearly half of patients with epilepsy have symptoms of high anxiety according to a study of adults treated in tertiary care centers.
- The study, which included 540 patients, evaluated the presence of anxiety with the Symptoms Checklist 90-R anxiety subscale. It also evaluated patients for depression with separate scales.
- 250 patients (46.1%) reported high anxiety.
- Focal epilepsy and epilepsy of unknown type, as well as depression scores, were independently linked to high anxiety.
- In patients with focal epilepsy, mesial temporal sclerosis was independently associated with high anxiety.
- Other factors linked to high anxiety included lower education level, being non-white, having Spanish as a native language, prior head trauma, and polydrug therapy for epilepsy.
- The researchers suggest that screening for anxiety in an epilepsy clinic can help spot patients in need of treatment.
Munger Clary HM, Snively BM, Hamberger MJ. Anxiety is common and independently associated with clinical features of epilepsy. Epilepsy Behav. 2018;85:64-71.
Nearly half of patients with epilepsy have symptoms of high anxiety according to a study of adults treated in tertiary care centers.
- The study, which included 540 patients, evaluated the presence of anxiety with the Symptoms Checklist 90-R anxiety subscale. It also evaluated patients for depression with separate scales.
- 250 patients (46.1%) reported high anxiety.
- Focal epilepsy and epilepsy of unknown type, as well as depression scores, were independently linked to high anxiety.
- In patients with focal epilepsy, mesial temporal sclerosis was independently associated with high anxiety.
- Other factors linked to high anxiety included lower education level, being non-white, having Spanish as a native language, prior head trauma, and polydrug therapy for epilepsy.
- The researchers suggest that screening for anxiety in an epilepsy clinic can help spot patients in need of treatment.
Munger Clary HM, Snively BM, Hamberger MJ. Anxiety is common and independently associated with clinical features of epilepsy. Epilepsy Behav. 2018;85:64-71.
Algorithm shows promise in calculating CV risk in sleep apnea patients
BALTIMORE – Researchers have developed an algorithm to calculate circulation time during sleep that may provide another tool to identify the risk of underlying cardiac vascular disease in patients with sleep apnea, one of the study’s lead investigators reported at the annual meeting of the Associated Professional Sleep Societies.
“There’s always been a question that there could be some global or untapped physiological indices that might give us some glimpse into future cardiovascular events or instantaneous cardiovascular vulnerability during sleep apnea events,” said Younghoon Kwon, MD, assistant professor of cardiovascular medicine at the University of Virginia, Charlottesville. “Circulation time that can be derived from a sleep study may be one of these novel indices. Although it has been examined in patients with heart failure with Cheyne-Stokes respiration, it has rarely been studied in patients with obstructive sleep apnea without heart failure.”

He noted that in this study, which utilized a cohort of 686 patients from the Multi-Ethnic Study of Atherosclerosis (MESA), all with an apnea-hypopnea index greater than 15, the automated algorithm the researchers developed to calculate lung-to-finger circulation was correlated highly with visual measurement.
The algorithm used randomly selected polysomnograms from the MESA cohort. It employed the airflow/nasal signal and the oxygen saturation signal, using the visually scored start and endpoint of apnea/hypopnea as a starting point. For each event, the calculation identified two key points: the endpoint of apnea/hypopnea and the endpoint of desaturation to arrive at a calculation of lung-to-finger circulation, Dr. Kwon explained.
The significance of the findings was the correlation between the visual and automated methods of calculating lung-to-finder circulation time. In a matched subgroup of 25 subjects, the correlation was around 95% (P less than .0001); in all cases, the correlation was around 69% (P less than .001). In matched cases, the average lung-to-finger circulation times were identical with visual and automated techniques: 19.5 seconds (P = .92), whereas in all cases the averages differed: 19.6 seconds for visual versus 18.6 seconds for automated (P = .42). “The results showed that the visual against the automated circulatory time measurement was very good,” Dr. Kwon said.
With this algorithm, multiple circulation time measures were automatically derived for each sleep study. Subsequently, average circulation time was derived for each study participant. The average circulation time was 19.4 seconds in the entire cohort, versus 21.0 seconds in those with apnea and 17.6 seconds in patients with hypopnea.
“Older age, male gender, and higher obstructive sleep apnea severity appeared to be independently associated with higher than average lung-to-finger circulation times,” Dr. Kwon said. “However, there was no apparent association between the obstructive event length or the severity of oxygen desaturation and the respective circulation time within subjects. Similarly, sleep positions and sleep stages do not seem to bear any association.”
One of the limitations of the study, he noted, was its assumption of the automated algorithm as the threshold and somewhat limited candidate variables. Future studies should involve more diverse cohorts with prevalent cardiovascular disease to determine the utility of the algorithm in predicting cardiovascular events, he said.
Dr. Kwon reported having no financial relationships, and the American Academy of Sleep Medicine Foundation provided study funding.
SOURCE: Kwon Y et al. SLEEP 2018, Abstract #0450.
BALTIMORE – Researchers have developed an algorithm to calculate circulation time during sleep that may provide another tool to identify the risk of underlying cardiac vascular disease in patients with sleep apnea, one of the study’s lead investigators reported at the annual meeting of the Associated Professional Sleep Societies.
“There’s always been a question that there could be some global or untapped physiological indices that might give us some glimpse into future cardiovascular events or instantaneous cardiovascular vulnerability during sleep apnea events,” said Younghoon Kwon, MD, assistant professor of cardiovascular medicine at the University of Virginia, Charlottesville. “Circulation time that can be derived from a sleep study may be one of these novel indices. Although it has been examined in patients with heart failure with Cheyne-Stokes respiration, it has rarely been studied in patients with obstructive sleep apnea without heart failure.”
He noted that in this study, which utilized a cohort of 686 patients from the Multi-Ethnic Study of Atherosclerosis (MESA), all with an apnea-hypopnea index greater than 15, the automated algorithm the researchers developed to calculate lung-to-finger circulation was correlated highly with visual measurement.
The algorithm used randomly selected polysomnograms from the MESA cohort. It employed the airflow/nasal signal and the oxygen saturation signal, using the visually scored start and endpoint of apnea/hypopnea as a starting point. For each event, the calculation identified two key points: the endpoint of apnea/hypopnea and the endpoint of desaturation to arrive at a calculation of lung-to-finger circulation, Dr. Kwon explained.
The significance of the findings was the correlation between the visual and automated methods of calculating lung-to-finder circulation time. In a matched subgroup of 25 subjects, the correlation was around 95% (P less than .0001); in all cases, the correlation was around 69% (P less than .001). In matched cases, the average lung-to-finger circulation times were identical with visual and automated techniques: 19.5 seconds (P = .92), whereas in all cases the averages differed: 19.6 seconds for visual versus 18.6 seconds for automated (P = .42). “The results showed that the visual against the automated circulatory time measurement was very good,” Dr. Kwon said.
With this algorithm, multiple circulation time measures were automatically derived for each sleep study. Subsequently, average circulation time was derived for each study participant. The average circulation time was 19.4 seconds in the entire cohort, versus 21.0 seconds in those with apnea and 17.6 seconds in patients with hypopnea.
“Older age, male gender, and higher obstructive sleep apnea severity appeared to be independently associated with higher than average lung-to-finger circulation times,” Dr. Kwon said. “However, there was no apparent association between the obstructive event length or the severity of oxygen desaturation and the respective circulation time within subjects. Similarly, sleep positions and sleep stages do not seem to bear any association.”
One of the limitations of the study, he noted, was its assumption of the automated algorithm as the threshold and somewhat limited candidate variables. Future studies should involve more diverse cohorts with prevalent cardiovascular disease to determine the utility of the algorithm in predicting cardiovascular events, he said.
Dr. Kwon reported having no financial relationships, and the American Academy of Sleep Medicine Foundation provided study funding.
SOURCE: Kwon Y et al. SLEEP 2018, Abstract #0450.
BALTIMORE – Researchers have developed an algorithm to calculate circulation time during sleep that may provide another tool to identify the risk of underlying cardiac vascular disease in patients with sleep apnea, one of the study’s lead investigators reported at the annual meeting of the Associated Professional Sleep Societies.
“There’s always been a question that there could be some global or untapped physiological indices that might give us some glimpse into future cardiovascular events or instantaneous cardiovascular vulnerability during sleep apnea events,” said Younghoon Kwon, MD, assistant professor of cardiovascular medicine at the University of Virginia, Charlottesville. “Circulation time that can be derived from a sleep study may be one of these novel indices. Although it has been examined in patients with heart failure with Cheyne-Stokes respiration, it has rarely been studied in patients with obstructive sleep apnea without heart failure.”
He noted that in this study, which utilized a cohort of 686 patients from the Multi-Ethnic Study of Atherosclerosis (MESA), all with an apnea-hypopnea index greater than 15, the automated algorithm the researchers developed to calculate lung-to-finger circulation was correlated highly with visual measurement.
The algorithm used randomly selected polysomnograms from the MESA cohort. It employed the airflow/nasal signal and the oxygen saturation signal, using the visually scored start and endpoint of apnea/hypopnea as a starting point. For each event, the calculation identified two key points: the endpoint of apnea/hypopnea and the endpoint of desaturation to arrive at a calculation of lung-to-finger circulation, Dr. Kwon explained.
The significance of the findings was the correlation between the visual and automated methods of calculating lung-to-finder circulation time. In a matched subgroup of 25 subjects, the correlation was around 95% (P less than .0001); in all cases, the correlation was around 69% (P less than .001). In matched cases, the average lung-to-finger circulation times were identical with visual and automated techniques: 19.5 seconds (P = .92), whereas in all cases the averages differed: 19.6 seconds for visual versus 18.6 seconds for automated (P = .42). “The results showed that the visual against the automated circulatory time measurement was very good,” Dr. Kwon said.
With this algorithm, multiple circulation time measures were automatically derived for each sleep study. Subsequently, average circulation time was derived for each study participant. The average circulation time was 19.4 seconds in the entire cohort, versus 21.0 seconds in those with apnea and 17.6 seconds in patients with hypopnea.
“Older age, male gender, and higher obstructive sleep apnea severity appeared to be independently associated with higher than average lung-to-finger circulation times,” Dr. Kwon said. “However, there was no apparent association between the obstructive event length or the severity of oxygen desaturation and the respective circulation time within subjects. Similarly, sleep positions and sleep stages do not seem to bear any association.”
One of the limitations of the study, he noted, was its assumption of the automated algorithm as the threshold and somewhat limited candidate variables. Future studies should involve more diverse cohorts with prevalent cardiovascular disease to determine the utility of the algorithm in predicting cardiovascular events, he said.
Dr. Kwon reported having no financial relationships, and the American Academy of Sleep Medicine Foundation provided study funding.
SOURCE: Kwon Y et al. SLEEP 2018, Abstract #0450.
REPORTING FROM SLEEP 2018
Key clinical point: An automated algorithm to calculate sleep-study circulation times correlated with visual review.
Major finding: The correlation between visual and automated methods for calculating lung-to-finger circulation times was around 95% (P less than .0001).
Study details: A cohort of 686 participants in the Multi-Ethnic Study of Atherosclerosis.
Disclosures: Dr. Kwon reported no financial disclosures, and the American Academy of Sleep Medicine Foundation provided study funding.
Source: Kwon Y et al. SLEEP 2018, Abstract #0450.
Sunscreen use in grade schoolers: Wide racial, ethnic disparities seen
and the figures were much lower for non-Hispanic black children.
Just 23% of fifth graders almost always used sunscreen, according to data drawn from the Healthy Passages study, which surveyed the parents or caregivers of 5,119 fifth graders. That figure was similar in the 1,802 Hispanic respondents, but fell to just 6% of the 1,748 non-Hispanic black respondents.
Some other factors that were associated with less chance of adherence to sunscreen use included being male and having lower socioeconomic status, wrote Christina M. Correnti, MD, and her study coauthors. The report was published in in Pediatric Dermatology. Perhaps surprisingly, they said, “School-based sun-safety education and involvement in team sports were not significant factors.”
Healthy Passages is a prospective multisite cohort study of child and adolescent health. Dr. Correnti, a dermatology resident at the University of Maryland, Baltimore, and her colleagues used baseline Healthy Passages data collected from the period of 2004-2006. Children enrolled in fifth grade at public schools in Birmingham, Ala., Houston, and Los Angeles, together with their caregivers, participated in the survey. Deidentified demographic data were collected, and participants were asked about four preventive health behaviors in addition to sunscreen use and flossing teeth: brushing teeth, helmet use, seatbelt use, and well-child examinations.
Dr. Correnti and her colleagues used multivariable analysis to calculate odds ratios for the association between the various demographic factors and other preventive behaviors and sunscreen use. They found that sunscreen adherence was correlated with all other preventive behaviors (P less than .001), but that the interrelationship with helmet use was confounded by racial and ethnic variables. Seatbelt use was not significantly correlated with sunscreen use for non-Hispanic black or Hispanic respondents.
“Children from more-educated and affluent households were more likely to use sun protection. Perhaps they had greater parental awareness and practice of sun safe habits,” wrote Dr. Correnti and her colleagues, noting that other work has shown that even low-income parents generally don’t see the cost of sunscreen as a barrier to use.
Although overall use of sunscreen among non-Hispanic black children was low, both non-Hispanic black and Hispanic children were more likely to use sunscreen if they had three or more sunburns within the prior 12 months. “Although darker skin tones may afford some sun protection, melanoma incidence is growing in Hispanic populations,” the researchers wrote.
To address these overall low rates of sunscreen use, the investigators discussed the utility of a variety of education options. The well-child visit affords an opportunity to reinforce the importance of preventive behaviors, but physicians may run into a time crunch and forgo thorough sun safety education, they said. Written materials can be a useful adjunct for clinicians in this setting.
“Health care practitioners may use absence of other preventive behaviors as potential markers for inadequate sunscreen use, prompting a point-of-care sun-safety intervention,” they suggested.
A school-based public health approach offers another route for education. “School sun-safety programs may alleviate the primary care burden,” wrote Dr. Correnti and her coinvestigators. The opportunity to deliver repeated, age-tailored messages as children progress through school may be effective in promoting healthy sun behaviors. Messaging that focuses on the negative effects of sun exposure on appearance such as age spots and wrinkles have been more effective than those warning of the risk of skin cancer for teens; investigating appearance-based content for this age group might be a good idea, the authors said.
The fact that the survey sites were in southern cities may mean that national rates of consistent sunscreen use for elementary schoolers may be even lower, said Dr. Correnti and her coauthors. Many other real-world factors, such as frequency and amount of sunscreen applied and the use of sun-protective clothing, couldn’t be captured by the survey, they acknowledged.
“Even in the most adherent group, non-Hispanic whites, only 44.8% always used sunscreen,” the researchers wrote. The study’s findings leave plenty of room for implementation of broad-based programs, especially in low-resource communities.
The National Institutes of Health funded the research. Dr. Correnti was supported by NIH awards.
SOURCE: Correnti CM et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13550.
and the figures were much lower for non-Hispanic black children.
Just 23% of fifth graders almost always used sunscreen, according to data drawn from the Healthy Passages study, which surveyed the parents or caregivers of 5,119 fifth graders. That figure was similar in the 1,802 Hispanic respondents, but fell to just 6% of the 1,748 non-Hispanic black respondents.
Some other factors that were associated with less chance of adherence to sunscreen use included being male and having lower socioeconomic status, wrote Christina M. Correnti, MD, and her study coauthors. The report was published in in Pediatric Dermatology. Perhaps surprisingly, they said, “School-based sun-safety education and involvement in team sports were not significant factors.”
Healthy Passages is a prospective multisite cohort study of child and adolescent health. Dr. Correnti, a dermatology resident at the University of Maryland, Baltimore, and her colleagues used baseline Healthy Passages data collected from the period of 2004-2006. Children enrolled in fifth grade at public schools in Birmingham, Ala., Houston, and Los Angeles, together with their caregivers, participated in the survey. Deidentified demographic data were collected, and participants were asked about four preventive health behaviors in addition to sunscreen use and flossing teeth: brushing teeth, helmet use, seatbelt use, and well-child examinations.
Dr. Correnti and her colleagues used multivariable analysis to calculate odds ratios for the association between the various demographic factors and other preventive behaviors and sunscreen use. They found that sunscreen adherence was correlated with all other preventive behaviors (P less than .001), but that the interrelationship with helmet use was confounded by racial and ethnic variables. Seatbelt use was not significantly correlated with sunscreen use for non-Hispanic black or Hispanic respondents.
“Children from more-educated and affluent households were more likely to use sun protection. Perhaps they had greater parental awareness and practice of sun safe habits,” wrote Dr. Correnti and her colleagues, noting that other work has shown that even low-income parents generally don’t see the cost of sunscreen as a barrier to use.
Although overall use of sunscreen among non-Hispanic black children was low, both non-Hispanic black and Hispanic children were more likely to use sunscreen if they had three or more sunburns within the prior 12 months. “Although darker skin tones may afford some sun protection, melanoma incidence is growing in Hispanic populations,” the researchers wrote.
To address these overall low rates of sunscreen use, the investigators discussed the utility of a variety of education options. The well-child visit affords an opportunity to reinforce the importance of preventive behaviors, but physicians may run into a time crunch and forgo thorough sun safety education, they said. Written materials can be a useful adjunct for clinicians in this setting.
“Health care practitioners may use absence of other preventive behaviors as potential markers for inadequate sunscreen use, prompting a point-of-care sun-safety intervention,” they suggested.
A school-based public health approach offers another route for education. “School sun-safety programs may alleviate the primary care burden,” wrote Dr. Correnti and her coinvestigators. The opportunity to deliver repeated, age-tailored messages as children progress through school may be effective in promoting healthy sun behaviors. Messaging that focuses on the negative effects of sun exposure on appearance such as age spots and wrinkles have been more effective than those warning of the risk of skin cancer for teens; investigating appearance-based content for this age group might be a good idea, the authors said.
The fact that the survey sites were in southern cities may mean that national rates of consistent sunscreen use for elementary schoolers may be even lower, said Dr. Correnti and her coauthors. Many other real-world factors, such as frequency and amount of sunscreen applied and the use of sun-protective clothing, couldn’t be captured by the survey, they acknowledged.
“Even in the most adherent group, non-Hispanic whites, only 44.8% always used sunscreen,” the researchers wrote. The study’s findings leave plenty of room for implementation of broad-based programs, especially in low-resource communities.
The National Institutes of Health funded the research. Dr. Correnti was supported by NIH awards.
SOURCE: Correnti CM et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13550.
and the figures were much lower for non-Hispanic black children.
Just 23% of fifth graders almost always used sunscreen, according to data drawn from the Healthy Passages study, which surveyed the parents or caregivers of 5,119 fifth graders. That figure was similar in the 1,802 Hispanic respondents, but fell to just 6% of the 1,748 non-Hispanic black respondents.
Some other factors that were associated with less chance of adherence to sunscreen use included being male and having lower socioeconomic status, wrote Christina M. Correnti, MD, and her study coauthors. The report was published in in Pediatric Dermatology. Perhaps surprisingly, they said, “School-based sun-safety education and involvement in team sports were not significant factors.”
Healthy Passages is a prospective multisite cohort study of child and adolescent health. Dr. Correnti, a dermatology resident at the University of Maryland, Baltimore, and her colleagues used baseline Healthy Passages data collected from the period of 2004-2006. Children enrolled in fifth grade at public schools in Birmingham, Ala., Houston, and Los Angeles, together with their caregivers, participated in the survey. Deidentified demographic data were collected, and participants were asked about four preventive health behaviors in addition to sunscreen use and flossing teeth: brushing teeth, helmet use, seatbelt use, and well-child examinations.
Dr. Correnti and her colleagues used multivariable analysis to calculate odds ratios for the association between the various demographic factors and other preventive behaviors and sunscreen use. They found that sunscreen adherence was correlated with all other preventive behaviors (P less than .001), but that the interrelationship with helmet use was confounded by racial and ethnic variables. Seatbelt use was not significantly correlated with sunscreen use for non-Hispanic black or Hispanic respondents.
“Children from more-educated and affluent households were more likely to use sun protection. Perhaps they had greater parental awareness and practice of sun safe habits,” wrote Dr. Correnti and her colleagues, noting that other work has shown that even low-income parents generally don’t see the cost of sunscreen as a barrier to use.
Although overall use of sunscreen among non-Hispanic black children was low, both non-Hispanic black and Hispanic children were more likely to use sunscreen if they had three or more sunburns within the prior 12 months. “Although darker skin tones may afford some sun protection, melanoma incidence is growing in Hispanic populations,” the researchers wrote.
To address these overall low rates of sunscreen use, the investigators discussed the utility of a variety of education options. The well-child visit affords an opportunity to reinforce the importance of preventive behaviors, but physicians may run into a time crunch and forgo thorough sun safety education, they said. Written materials can be a useful adjunct for clinicians in this setting.
“Health care practitioners may use absence of other preventive behaviors as potential markers for inadequate sunscreen use, prompting a point-of-care sun-safety intervention,” they suggested.
A school-based public health approach offers another route for education. “School sun-safety programs may alleviate the primary care burden,” wrote Dr. Correnti and her coinvestigators. The opportunity to deliver repeated, age-tailored messages as children progress through school may be effective in promoting healthy sun behaviors. Messaging that focuses on the negative effects of sun exposure on appearance such as age spots and wrinkles have been more effective than those warning of the risk of skin cancer for teens; investigating appearance-based content for this age group might be a good idea, the authors said.
The fact that the survey sites were in southern cities may mean that national rates of consistent sunscreen use for elementary schoolers may be even lower, said Dr. Correnti and her coauthors. Many other real-world factors, such as frequency and amount of sunscreen applied and the use of sun-protective clothing, couldn’t be captured by the survey, they acknowledged.
“Even in the most adherent group, non-Hispanic whites, only 44.8% always used sunscreen,” the researchers wrote. The study’s findings leave plenty of room for implementation of broad-based programs, especially in low-resource communities.
The National Institutes of Health funded the research. Dr. Correnti was supported by NIH awards.
SOURCE: Correnti CM et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13550.
FROM PEDIATRIC DERMATOLOGY
Key clinical point: Most parents surveyed said their children didn’t use sunscreen consistently.
Major finding: Of non-Hispanic black children, 6% almost always used sunscreen.
Study details: Data drawn from Healthy Passages, a prospective cohort study of 5,119 fifth-graders and their parents or caregivers.
Disclosures: The National Institutes of Health funded the research. Dr. Correnti was supported by NIH awards.
Source: Correnti C et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13550.
Glucocorticoids linked with surgical infections in RA patients
AMSTERDAM – Patients with rheumatoid arthritis who underwent elective knee or hip arthroplasty had a doubled rate of hospitalization for infection when they averaged more than 10 mg/day oral prednisone during the 3 months before surgery, based on a review of about 11,000 U.S. insurance claims.

“Limiting glucocorticoid exposure before surgery should be a focus of perioperative management,” Michael D. George, MD, said at the European Congress of Rheumatology. “Glucocorticoid use, especially greater than 10 mg/day, is associated with a greater risk of infection and hospital readmission,” said Dr. George, a rheumatologist at the University of Pennsylvania in Philadelphia.
The analysis also showed that treatment with any biologic drug – including abatacept (Orencia), rituximab (Rituxan), tocilizumab (Actemra), and any of several tumor necrosis factor (TNF) inhibitors – had a similar impact on both postsurgical infections requiring hospitalization and 30-day hospital readmissions.
The findings suggest “it’s more important to reduce glucocorticoids than biological drugs,” commented John D. Isaacs, MD, professor of clinical rheumatology at Newcastle University in Newcastle upon Tyne, England. “This is a really important question that has been very difficult to answer.”

Dr. George and his associates used data from patients with rheumatoid arthritis during 2006-2015 who underwent knee or hip arthroplasty and were in databases from Medicare, or MarketScan, which includes commercial insurers. This identified 11,021 RA patients on any of several biologic drugs before their surgery: 16% on abatacept, 4% on rituximab, 4% on tocilizumab, and the remaining 76% on a TNF inhibitor, either adalimumab (Humira), etanercept (Enbrel), or infliximab (Remicade). About 43% of all patients were on a glucocorticoid during the 3 months before surgery. Biologic use was defined as a minimum of one dose within 8 weeks of surgery, and at least three total dosages during the prior year, except for rituximab, which was at least one dose given 16 weeks before surgery and at least two doses during the prior year.
The rate of hospitalized infections ranged from 6.6% to 8.5% depending on the biologic drug used, and 30-day readmissions ranged from 4.8% to 6.8%. A third outcome the analysis assessed was prosthetic joint infection during 1-year follow-up, which was again similar across most of the biologics, except for patients on tocilizumab, who had prosthetic joint infections roughly threefold more often than the other patients. Although this was a statistically significant difference, Dr. George discounted the finding given the very small number of tocilizumab-treated patients who had these infections and said that any conclusion about tocilizumab’s effect on this outcome had to await data from more patients.
The glucocorticoid analysis divided patients into four subgroups: those not on a glucocorticoid, those on an average daily dosage of 5 mg/day prednisone or equivalent or less, patients on 6-10 mg/day prednisone, and those on more than 10 mg/day. In a propensity-weighted analysis, these three escalating levels of glucocorticoid use showed a dose-response relationship to the rates of both hospitalized infections and 30-day readmissions. At the highest level of glucocorticoid use, hospitalized infections occurred twice as often as in patients not on a glucocorticoid, and 30-day readmissions were more than 50% higher than in those not on an oral steroid, both statistically significant differences. For the outcome of 1-year prosthetic joint infections, the analysis again showed a dose-related link among glucocorticoid users, topping out with a greater than 50% increased rate among those on the highest glucocorticoid dosages when compared with nonusers, but this difference was not statistically significant.
The study was partially funded by Bristol-Myers Squibb, the company that markets abatacept. Dr. George has received research funding from Bristol-Myers Squibb, and some of his coauthors on the study are employees of the company.
SOURCE: George MD et al. EULAR 2018. Abstract OP0228.
AMSTERDAM – Patients with rheumatoid arthritis who underwent elective knee or hip arthroplasty had a doubled rate of hospitalization for infection when they averaged more than 10 mg/day oral prednisone during the 3 months before surgery, based on a review of about 11,000 U.S. insurance claims.
“Limiting glucocorticoid exposure before surgery should be a focus of perioperative management,” Michael D. George, MD, said at the European Congress of Rheumatology. “Glucocorticoid use, especially greater than 10 mg/day, is associated with a greater risk of infection and hospital readmission,” said Dr. George, a rheumatologist at the University of Pennsylvania in Philadelphia.
The analysis also showed that treatment with any biologic drug – including abatacept (Orencia), rituximab (Rituxan), tocilizumab (Actemra), and any of several tumor necrosis factor (TNF) inhibitors – had a similar impact on both postsurgical infections requiring hospitalization and 30-day hospital readmissions.
The findings suggest “it’s more important to reduce glucocorticoids than biological drugs,” commented John D. Isaacs, MD, professor of clinical rheumatology at Newcastle University in Newcastle upon Tyne, England. “This is a really important question that has been very difficult to answer.”
Dr. George and his associates used data from patients with rheumatoid arthritis during 2006-2015 who underwent knee or hip arthroplasty and were in databases from Medicare, or MarketScan, which includes commercial insurers. This identified 11,021 RA patients on any of several biologic drugs before their surgery: 16% on abatacept, 4% on rituximab, 4% on tocilizumab, and the remaining 76% on a TNF inhibitor, either adalimumab (Humira), etanercept (Enbrel), or infliximab (Remicade). About 43% of all patients were on a glucocorticoid during the 3 months before surgery. Biologic use was defined as a minimum of one dose within 8 weeks of surgery, and at least three total dosages during the prior year, except for rituximab, which was at least one dose given 16 weeks before surgery and at least two doses during the prior year.
The rate of hospitalized infections ranged from 6.6% to 8.5% depending on the biologic drug used, and 30-day readmissions ranged from 4.8% to 6.8%. A third outcome the analysis assessed was prosthetic joint infection during 1-year follow-up, which was again similar across most of the biologics, except for patients on tocilizumab, who had prosthetic joint infections roughly threefold more often than the other patients. Although this was a statistically significant difference, Dr. George discounted the finding given the very small number of tocilizumab-treated patients who had these infections and said that any conclusion about tocilizumab’s effect on this outcome had to await data from more patients.
The glucocorticoid analysis divided patients into four subgroups: those not on a glucocorticoid, those on an average daily dosage of 5 mg/day prednisone or equivalent or less, patients on 6-10 mg/day prednisone, and those on more than 10 mg/day. In a propensity-weighted analysis, these three escalating levels of glucocorticoid use showed a dose-response relationship to the rates of both hospitalized infections and 30-day readmissions. At the highest level of glucocorticoid use, hospitalized infections occurred twice as often as in patients not on a glucocorticoid, and 30-day readmissions were more than 50% higher than in those not on an oral steroid, both statistically significant differences. For the outcome of 1-year prosthetic joint infections, the analysis again showed a dose-related link among glucocorticoid users, topping out with a greater than 50% increased rate among those on the highest glucocorticoid dosages when compared with nonusers, but this difference was not statistically significant.
The study was partially funded by Bristol-Myers Squibb, the company that markets abatacept. Dr. George has received research funding from Bristol-Myers Squibb, and some of his coauthors on the study are employees of the company.
SOURCE: George MD et al. EULAR 2018. Abstract OP0228.
AMSTERDAM – Patients with rheumatoid arthritis who underwent elective knee or hip arthroplasty had a doubled rate of hospitalization for infection when they averaged more than 10 mg/day oral prednisone during the 3 months before surgery, based on a review of about 11,000 U.S. insurance claims.
“Limiting glucocorticoid exposure before surgery should be a focus of perioperative management,” Michael D. George, MD, said at the European Congress of Rheumatology. “Glucocorticoid use, especially greater than 10 mg/day, is associated with a greater risk of infection and hospital readmission,” said Dr. George, a rheumatologist at the University of Pennsylvania in Philadelphia.
The analysis also showed that treatment with any biologic drug – including abatacept (Orencia), rituximab (Rituxan), tocilizumab (Actemra), and any of several tumor necrosis factor (TNF) inhibitors – had a similar impact on both postsurgical infections requiring hospitalization and 30-day hospital readmissions.
The findings suggest “it’s more important to reduce glucocorticoids than biological drugs,” commented John D. Isaacs, MD, professor of clinical rheumatology at Newcastle University in Newcastle upon Tyne, England. “This is a really important question that has been very difficult to answer.”
Dr. George and his associates used data from patients with rheumatoid arthritis during 2006-2015 who underwent knee or hip arthroplasty and were in databases from Medicare, or MarketScan, which includes commercial insurers. This identified 11,021 RA patients on any of several biologic drugs before their surgery: 16% on abatacept, 4% on rituximab, 4% on tocilizumab, and the remaining 76% on a TNF inhibitor, either adalimumab (Humira), etanercept (Enbrel), or infliximab (Remicade). About 43% of all patients were on a glucocorticoid during the 3 months before surgery. Biologic use was defined as a minimum of one dose within 8 weeks of surgery, and at least three total dosages during the prior year, except for rituximab, which was at least one dose given 16 weeks before surgery and at least two doses during the prior year.
The rate of hospitalized infections ranged from 6.6% to 8.5% depending on the biologic drug used, and 30-day readmissions ranged from 4.8% to 6.8%. A third outcome the analysis assessed was prosthetic joint infection during 1-year follow-up, which was again similar across most of the biologics, except for patients on tocilizumab, who had prosthetic joint infections roughly threefold more often than the other patients. Although this was a statistically significant difference, Dr. George discounted the finding given the very small number of tocilizumab-treated patients who had these infections and said that any conclusion about tocilizumab’s effect on this outcome had to await data from more patients.
The glucocorticoid analysis divided patients into four subgroups: those not on a glucocorticoid, those on an average daily dosage of 5 mg/day prednisone or equivalent or less, patients on 6-10 mg/day prednisone, and those on more than 10 mg/day. In a propensity-weighted analysis, these three escalating levels of glucocorticoid use showed a dose-response relationship to the rates of both hospitalized infections and 30-day readmissions. At the highest level of glucocorticoid use, hospitalized infections occurred twice as often as in patients not on a glucocorticoid, and 30-day readmissions were more than 50% higher than in those not on an oral steroid, both statistically significant differences. For the outcome of 1-year prosthetic joint infections, the analysis again showed a dose-related link among glucocorticoid users, topping out with a greater than 50% increased rate among those on the highest glucocorticoid dosages when compared with nonusers, but this difference was not statistically significant.
The study was partially funded by Bristol-Myers Squibb, the company that markets abatacept. Dr. George has received research funding from Bristol-Myers Squibb, and some of his coauthors on the study are employees of the company.
SOURCE: George MD et al. EULAR 2018. Abstract OP0228.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point:
Major finding: RA patients on more than 10 mg/day prednisone had a more than twofold higher rate of postsurgical hospitalized infections.
Study details: Review of Medicare and MarketScan administrative claims data for 11,021 patients with rheumatoid arthritis who underwent joint surgery.
Disclosures: The study was partially funded by Bristol-Myers Squibb, the company that markets abatacept (Orencia). Dr. George has received research funding from Bristol-Myers Squibb, and some of his coauthors on the study are employees of the company.
Source: George MD et al. EULAR 2018. Abstract OP0228.
Older black patients die sooner after in-hospital cardiac arrest
Older black adults who experienced in-hospital cardiac arrest had a 28% lower relative risk of living to 1 year and a 33% lower relative risk of living to 5 years after discharge, compared with older white adults, suggesting potential racial differences in postdischarge care, according to an article published in Circulation.
The investigators examined data for 8,764 patients in the Get With The Guidelines–Resuscitation registry who were at least 65 years of age and experienced in-hospital cardiac arrest (IHCA) during 2000-2011 and then survived to be discharged. They linked patients to Medicare claims data and tracked survival outcomes for 1 year, 3 years, and 5 years, and evaluated the “proportion of racial differences explained by patient, hospital, and unmeasured factors.”
After discharge and adjustment for hospital site, investigators found older black patients had a 28% lower survival rate, compared with older white patients, at 1 year (43.6% vs. 60.2%), a 29% lower rate at 3 years (31.6% vs. 45.3%), and a 33% lower rate at 5 years (23.5% vs. 35.4%), all statistically significant at P less than .001. While patient factors accounted for 29% and hospital treatment factors accounted for 17% of racial differences at 1 year after discharge, about one-half of the differences remained unexplained. Investigators said these racial differences were potentially “result of differences in care after discharge or unmeasured confounding,” and “generally similar” results were seen in patients at 3 years and 5 years post discharge.
“This finding suggests a need to examine whether racial differences in postdischarge care explain a substantial proportion of racial differences in long-term survival after in-hospital cardiac arrest,” Lena M. Chen, MD, of the University of Michigan, Ann Arbor, and her colleagues wrote in Circulation.
The investigators noted they were unable to determine patient socioeconomic status, income, and social support with the available data, and did not know whether patients used or had an outpatient medical center near them after discharge. Furthermore, the study comprised older adults with fee-for-service Medicare and isn’t necessarily indicative of care for younger adults.
“Further investigation is warranted to better understand whether modifiable aspects of postdischarge care account for the remaining differences and whether interventions can be developed to eliminate racial disparities in care and survival for cardiac arrest survivors,” Dr. Chen and colleagues wrote.
This project was supported by a grant from the American Heart Association Young Investigator Research Seed. Dr. Chen received funding support from the Agency for Healthcare Research and Quality and the National Institute on Aging. Dr. Chen and another investigator received funding support from the National Heart, Lung, and Blood Institute.
SOURCE: Chen LM. Circulation. 2018 Jul 9. doi: 10.1161/circulationaha.117.033211.
Older black adults who experienced in-hospital cardiac arrest had a 28% lower relative risk of living to 1 year and a 33% lower relative risk of living to 5 years after discharge, compared with older white adults, suggesting potential racial differences in postdischarge care, according to an article published in Circulation.
The investigators examined data for 8,764 patients in the Get With The Guidelines–Resuscitation registry who were at least 65 years of age and experienced in-hospital cardiac arrest (IHCA) during 2000-2011 and then survived to be discharged. They linked patients to Medicare claims data and tracked survival outcomes for 1 year, 3 years, and 5 years, and evaluated the “proportion of racial differences explained by patient, hospital, and unmeasured factors.”
After discharge and adjustment for hospital site, investigators found older black patients had a 28% lower survival rate, compared with older white patients, at 1 year (43.6% vs. 60.2%), a 29% lower rate at 3 years (31.6% vs. 45.3%), and a 33% lower rate at 5 years (23.5% vs. 35.4%), all statistically significant at P less than .001. While patient factors accounted for 29% and hospital treatment factors accounted for 17% of racial differences at 1 year after discharge, about one-half of the differences remained unexplained. Investigators said these racial differences were potentially “result of differences in care after discharge or unmeasured confounding,” and “generally similar” results were seen in patients at 3 years and 5 years post discharge.
“This finding suggests a need to examine whether racial differences in postdischarge care explain a substantial proportion of racial differences in long-term survival after in-hospital cardiac arrest,” Lena M. Chen, MD, of the University of Michigan, Ann Arbor, and her colleagues wrote in Circulation.
The investigators noted they were unable to determine patient socioeconomic status, income, and social support with the available data, and did not know whether patients used or had an outpatient medical center near them after discharge. Furthermore, the study comprised older adults with fee-for-service Medicare and isn’t necessarily indicative of care for younger adults.
“Further investigation is warranted to better understand whether modifiable aspects of postdischarge care account for the remaining differences and whether interventions can be developed to eliminate racial disparities in care and survival for cardiac arrest survivors,” Dr. Chen and colleagues wrote.
This project was supported by a grant from the American Heart Association Young Investigator Research Seed. Dr. Chen received funding support from the Agency for Healthcare Research and Quality and the National Institute on Aging. Dr. Chen and another investigator received funding support from the National Heart, Lung, and Blood Institute.
SOURCE: Chen LM. Circulation. 2018 Jul 9. doi: 10.1161/circulationaha.117.033211.
Older black adults who experienced in-hospital cardiac arrest had a 28% lower relative risk of living to 1 year and a 33% lower relative risk of living to 5 years after discharge, compared with older white adults, suggesting potential racial differences in postdischarge care, according to an article published in Circulation.
The investigators examined data for 8,764 patients in the Get With The Guidelines–Resuscitation registry who were at least 65 years of age and experienced in-hospital cardiac arrest (IHCA) during 2000-2011 and then survived to be discharged. They linked patients to Medicare claims data and tracked survival outcomes for 1 year, 3 years, and 5 years, and evaluated the “proportion of racial differences explained by patient, hospital, and unmeasured factors.”
After discharge and adjustment for hospital site, investigators found older black patients had a 28% lower survival rate, compared with older white patients, at 1 year (43.6% vs. 60.2%), a 29% lower rate at 3 years (31.6% vs. 45.3%), and a 33% lower rate at 5 years (23.5% vs. 35.4%), all statistically significant at P less than .001. While patient factors accounted for 29% and hospital treatment factors accounted for 17% of racial differences at 1 year after discharge, about one-half of the differences remained unexplained. Investigators said these racial differences were potentially “result of differences in care after discharge or unmeasured confounding,” and “generally similar” results were seen in patients at 3 years and 5 years post discharge.
“This finding suggests a need to examine whether racial differences in postdischarge care explain a substantial proportion of racial differences in long-term survival after in-hospital cardiac arrest,” Lena M. Chen, MD, of the University of Michigan, Ann Arbor, and her colleagues wrote in Circulation.
The investigators noted they were unable to determine patient socioeconomic status, income, and social support with the available data, and did not know whether patients used or had an outpatient medical center near them after discharge. Furthermore, the study comprised older adults with fee-for-service Medicare and isn’t necessarily indicative of care for younger adults.
“Further investigation is warranted to better understand whether modifiable aspects of postdischarge care account for the remaining differences and whether interventions can be developed to eliminate racial disparities in care and survival for cardiac arrest survivors,” Dr. Chen and colleagues wrote.
This project was supported by a grant from the American Heart Association Young Investigator Research Seed. Dr. Chen received funding support from the Agency for Healthcare Research and Quality and the National Institute on Aging. Dr. Chen and another investigator received funding support from the National Heart, Lung, and Blood Institute.
SOURCE: Chen LM. Circulation. 2018 Jul 9. doi: 10.1161/circulationaha.117.033211.
FROM CIRCULATION
Key clinical point: Older black adults who had an in-hospital cardiac event had a lower chance of 1-year, 3-year, and 5-year survival post discharge, compared with white counterparts.
Major finding: in the study.
Data source: A longitudinal study of 8,764 patients at least 65 years of age in the Get With The Guidelines–Resuscitation registry who were discharged after experiencing in-hospital cardiac arrest.
Disclosures: This project was supported by a grant from the American Heart Association Young Investigator Research Seed. Dr. Chen received funding support from the Agency for Healthcare Research and Quality and the National Institute on Aging. Dr. Chen and another investigator received funding support from the National Heart, Lung, and Blood Institute.
Source: Chen LM. Circulation. 2018 Jul 9. doi: 10.1161/circulationaha.117.033211.
Original Study: Should Nitrofurantoin Be Used to Treat Alkaline Urinary Tract Infection?
Background
Urinary tract infections (UTIs), one of the most common human bacterial infections, affect approximately 150 million people annually worldwide.1,2 In the United States, UTIs account for approximately 1% of all outpatient clinic visits and about 2 to 3 million ED visits annually.1,3-5
Although the urine pH level is frequently assessed in urinalysis, it is rarely considered in the management of a patient with a UTI.
Reports correlating urine pH with urine culture data from ED patients with UTIs are lacking. While poorly studied, there are multiple factors that could potentially alter the urine pH of patients with a UTI, including blood pH, diabetes, dehydration, ketosis, drugs, and renal function, as well as factors related to the infecting microorganism. For instance, Proteus mirabilis produces urease, an enzyme that hydrolyzes urea to ammonia and carbon dioxide.6-8
Objective
The objective of this study is to assess the relationship between the urine pH and the infecting microbe in ED patients diagnosed with UTIs, and to determine if P mirabilis is associated with alkaline urine.
Methods
We obtained approval from our Institutional Review Board to retrospectively obtain electronic medical record data from patients aged 18 years and older who presented to our institution’s ED and who were diagnosed with either cystitis or a UTI between January 1, 2012 and March 31, 2015. Both urine pH level and a urine culture were obtained for all patients.
The results of all of the patients’ urinary cultures in our study were positive for one bacterial species or genera (≥100,000 CFU/mL). The International Classification of Disease, Ninth Revision/Tenth Revision codes used to identify patients with cystitis and UTI were as follows: 595.0, 595.1, 595.9, 599.0, N30.91, N30.90, N30.80, N30.81, N30.00, N30.01, N30.20, N30.20, and N39.0.
To ensure that the focus of our study was limited to cystitis and UTIs, we excluded patients who were diagnosed with pyelonephritis, sexually transmitted infection, pelvic inflammatory disease, or vaginal discharge. The urine pH values reported from the clinical laboratory were 5.0, 5.5, 6.0, 6.5, 7.0. 7.5, 8.0, 8.5, and 9.0. Our dataset contained 1,331 clinical encounters. We used descriptive statistics and unpaired t-tests to evaluate the associations between urine pH values and the different microbes.
Results
Data were categorized into 16 different bacterial genera or species. Acinetobacter (n = 1), Kluyvera ascorbata (n = 2), and Stenotrophomonas maltophilia (n = 3) were underrepresented in the dataset and therefore were not included in the data analysis. The data are summarized in the Table.
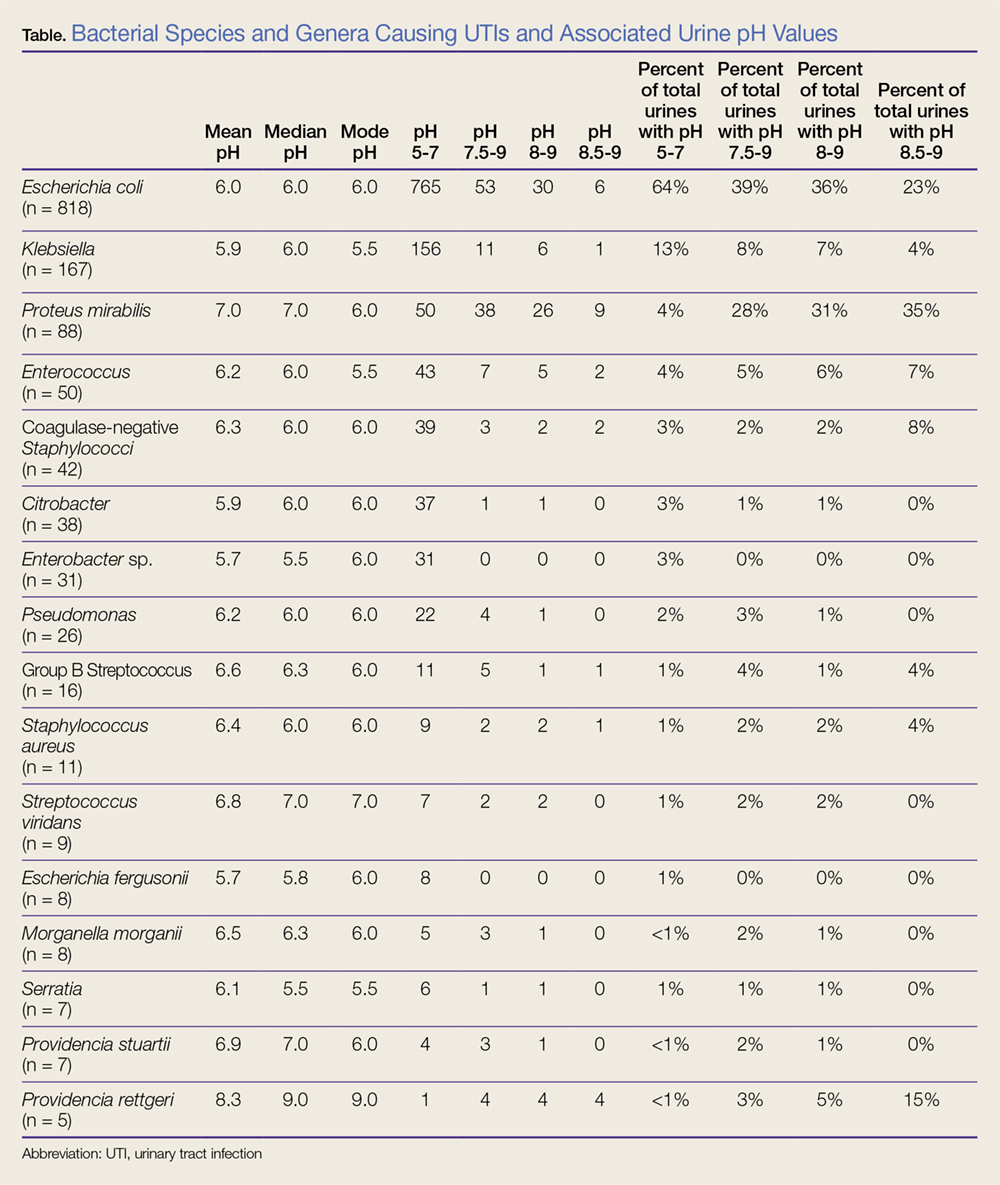
In our dataset, the most common bacteria associated with UTI, irrespective of urine pH, were Escherichia coli (n = 818/1,331; 62%), Klebsiella (n = 167/1,331; 13%), and P mirabilis (n = 88/1,331; 7%). The mean urine pH in our cohort was 6.1 (range, 5.0-9.0; SD, 0.88; median, 6; and mode, 6), and 1,194/1,331 (90%) of all urine samples had a urine pH of 5.0 to 7.0. Among patients who had a urine pH of 7.5 to 9.0, E coli was the cause of UTI in 39% (53/137) and P mirabilis was the cause of UTI in 28% (38/137). Likewise, among patients who had a urine pH of 8.0 to 9.0, E coli was the cause of UTI in 36% (30/83), and P mirabilis was the cause of UTI in 31% (26/83). Lastly, in patients who had a urine pH of 8.5 to 9.0, P mirabilis was the most common cause of UTI, present in 35% (9/26) patients.
The mean urine pH in our dataset for P mirabilis (n = 88) was 7.0, with a standard deviation (SD) of 1.03 and the standard error of the mean (SEM) of 0.11. The majority, 50/88 (57%) of P mirabilis UTIs were associated with a urine pH of 5.0 to 7.0. However, the urine pH for P mirabilis was significantly more alkaline than the combined urine pH from all of the other bacterial genera and species in our cohort (P < .0001). The mean urine pH in our cohort, excluding the P mirabilis data, was 6.01 with an SD of 0.828 and a SEM of 0.023.
Limitations
Our data were obtained retrospectively from a single ED, and did not include the following information: patient age and gender, and mode in which urine samples were obtained (eg, Foley catheter, clean catch). In addition, no reports were available regarding the sensitivity of the urine cultures with respect to urine pH.
Discussion
While alkaline urine was present in only 10% of patients, a high percentage of alkaline UTIs were associated with P mirabilis, an organism with intrinsic resistance to nitrofurantoin. Therefore, health care providers could consider obtaining a urine culture and/or prescribing an antibiotic other than nitrofurantoin for treating uncomplicated UTIs with alkaline urine. In addition, nitrofurantoin has been shown to be less effective against otherwise susceptible organisms in an alkaline urine.9
Conclusion
Our data demonstrates that urine pH of UTIs diagnosed in ED patients varied with the associated bacterial pathogen, and thus urine pH potentially could affect ED provider choice of antibiotics for the treatment of UTIs. Additional research is needed to confirm our results from a larger, more diverse dataset before changes in practice are recommended.
1. Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol. 2015;13(5):269-284. doi:10.1038/nrmicro3432.
2. Stamm WE, Norrby SR. Urinary tract infections: disease panorama and challenges. J Infect Dis. 2001;183 Suppl 1:S1-S4. doi:10.1086/318850.
3. Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. Vital Health Stat 13. 2011;13(169):1-38.
4. Foxman B. Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am. 2014;28(1):1-13. doi:10.1016/j.idc.2013.09.003.
5. Foxman B. The epidemiology of urinary tract infection. Nat Rev Urol. 2010;7(12):653-660. doi:10.1038/nrurol.2010.190.
6. Coker C, Poore CA, Li X, Mobley HL. Pathogenesis of Proteus mirabilis urinary tract infection. Microbes Infect. 2000;2(12):1497-1505.
7. Armbruster CE, Mobley HL. Merging mythology and morphology: the multifaceted lifestyle of Proteus mirabilis. Nat Rev Microbiol. 2012;10(11):743-754. doi:10.1038/nrmicro2890.
8. Schaffer JN, Pearson MM. Proteus mirabilis and urinary tract infections. Microbiol Spectr. 2015;3(5). doi:10.1128/microbiolspec.UTI-0017-2013.
9. Yang L, Wang K, Li H, Denstedt JD, Cadieux PA. The influence of urinary pH on antibiotic efficacy against bacterial uropathogens. Urology. 2014;84(3):731.e1-e7. doi:10.1016/j.urology.2014.04.048.
Background
Urinary tract infections (UTIs), one of the most common human bacterial infections, affect approximately 150 million people annually worldwide.1,2 In the United States, UTIs account for approximately 1% of all outpatient clinic visits and about 2 to 3 million ED visits annually.1,3-5
Although the urine pH level is frequently assessed in urinalysis, it is rarely considered in the management of a patient with a UTI.
Reports correlating urine pH with urine culture data from ED patients with UTIs are lacking. While poorly studied, there are multiple factors that could potentially alter the urine pH of patients with a UTI, including blood pH, diabetes, dehydration, ketosis, drugs, and renal function, as well as factors related to the infecting microorganism. For instance, Proteus mirabilis produces urease, an enzyme that hydrolyzes urea to ammonia and carbon dioxide.6-8
Objective
The objective of this study is to assess the relationship between the urine pH and the infecting microbe in ED patients diagnosed with UTIs, and to determine if P mirabilis is associated with alkaline urine.
Methods
We obtained approval from our Institutional Review Board to retrospectively obtain electronic medical record data from patients aged 18 years and older who presented to our institution’s ED and who were diagnosed with either cystitis or a UTI between January 1, 2012 and March 31, 2015. Both urine pH level and a urine culture were obtained for all patients.
The results of all of the patients’ urinary cultures in our study were positive for one bacterial species or genera (≥100,000 CFU/mL). The International Classification of Disease, Ninth Revision/Tenth Revision codes used to identify patients with cystitis and UTI were as follows: 595.0, 595.1, 595.9, 599.0, N30.91, N30.90, N30.80, N30.81, N30.00, N30.01, N30.20, N30.20, and N39.0.
To ensure that the focus of our study was limited to cystitis and UTIs, we excluded patients who were diagnosed with pyelonephritis, sexually transmitted infection, pelvic inflammatory disease, or vaginal discharge. The urine pH values reported from the clinical laboratory were 5.0, 5.5, 6.0, 6.5, 7.0. 7.5, 8.0, 8.5, and 9.0. Our dataset contained 1,331 clinical encounters. We used descriptive statistics and unpaired t-tests to evaluate the associations between urine pH values and the different microbes.
Results
Data were categorized into 16 different bacterial genera or species. Acinetobacter (n = 1), Kluyvera ascorbata (n = 2), and Stenotrophomonas maltophilia (n = 3) were underrepresented in the dataset and therefore were not included in the data analysis. The data are summarized in the Table.
In our dataset, the most common bacteria associated with UTI, irrespective of urine pH, were Escherichia coli (n = 818/1,331; 62%), Klebsiella (n = 167/1,331; 13%), and P mirabilis (n = 88/1,331; 7%). The mean urine pH in our cohort was 6.1 (range, 5.0-9.0; SD, 0.88; median, 6; and mode, 6), and 1,194/1,331 (90%) of all urine samples had a urine pH of 5.0 to 7.0. Among patients who had a urine pH of 7.5 to 9.0, E coli was the cause of UTI in 39% (53/137) and P mirabilis was the cause of UTI in 28% (38/137). Likewise, among patients who had a urine pH of 8.0 to 9.0, E coli was the cause of UTI in 36% (30/83), and P mirabilis was the cause of UTI in 31% (26/83). Lastly, in patients who had a urine pH of 8.5 to 9.0, P mirabilis was the most common cause of UTI, present in 35% (9/26) patients.
The mean urine pH in our dataset for P mirabilis (n = 88) was 7.0, with a standard deviation (SD) of 1.03 and the standard error of the mean (SEM) of 0.11. The majority, 50/88 (57%) of P mirabilis UTIs were associated with a urine pH of 5.0 to 7.0. However, the urine pH for P mirabilis was significantly more alkaline than the combined urine pH from all of the other bacterial genera and species in our cohort (P < .0001). The mean urine pH in our cohort, excluding the P mirabilis data, was 6.01 with an SD of 0.828 and a SEM of 0.023.
Limitations
Our data were obtained retrospectively from a single ED, and did not include the following information: patient age and gender, and mode in which urine samples were obtained (eg, Foley catheter, clean catch). In addition, no reports were available regarding the sensitivity of the urine cultures with respect to urine pH.
Discussion
While alkaline urine was present in only 10% of patients, a high percentage of alkaline UTIs were associated with P mirabilis, an organism with intrinsic resistance to nitrofurantoin. Therefore, health care providers could consider obtaining a urine culture and/or prescribing an antibiotic other than nitrofurantoin for treating uncomplicated UTIs with alkaline urine. In addition, nitrofurantoin has been shown to be less effective against otherwise susceptible organisms in an alkaline urine.9
Conclusion
Our data demonstrates that urine pH of UTIs diagnosed in ED patients varied with the associated bacterial pathogen, and thus urine pH potentially could affect ED provider choice of antibiotics for the treatment of UTIs. Additional research is needed to confirm our results from a larger, more diverse dataset before changes in practice are recommended.
Background
Urinary tract infections (UTIs), one of the most common human bacterial infections, affect approximately 150 million people annually worldwide.1,2 In the United States, UTIs account for approximately 1% of all outpatient clinic visits and about 2 to 3 million ED visits annually.1,3-5
Although the urine pH level is frequently assessed in urinalysis, it is rarely considered in the management of a patient with a UTI.
Reports correlating urine pH with urine culture data from ED patients with UTIs are lacking. While poorly studied, there are multiple factors that could potentially alter the urine pH of patients with a UTI, including blood pH, diabetes, dehydration, ketosis, drugs, and renal function, as well as factors related to the infecting microorganism. For instance, Proteus mirabilis produces urease, an enzyme that hydrolyzes urea to ammonia and carbon dioxide.6-8
Objective
The objective of this study is to assess the relationship between the urine pH and the infecting microbe in ED patients diagnosed with UTIs, and to determine if P mirabilis is associated with alkaline urine.
Methods
We obtained approval from our Institutional Review Board to retrospectively obtain electronic medical record data from patients aged 18 years and older who presented to our institution’s ED and who were diagnosed with either cystitis or a UTI between January 1, 2012 and March 31, 2015. Both urine pH level and a urine culture were obtained for all patients.
The results of all of the patients’ urinary cultures in our study were positive for one bacterial species or genera (≥100,000 CFU/mL). The International Classification of Disease, Ninth Revision/Tenth Revision codes used to identify patients with cystitis and UTI were as follows: 595.0, 595.1, 595.9, 599.0, N30.91, N30.90, N30.80, N30.81, N30.00, N30.01, N30.20, N30.20, and N39.0.
To ensure that the focus of our study was limited to cystitis and UTIs, we excluded patients who were diagnosed with pyelonephritis, sexually transmitted infection, pelvic inflammatory disease, or vaginal discharge. The urine pH values reported from the clinical laboratory were 5.0, 5.5, 6.0, 6.5, 7.0. 7.5, 8.0, 8.5, and 9.0. Our dataset contained 1,331 clinical encounters. We used descriptive statistics and unpaired t-tests to evaluate the associations between urine pH values and the different microbes.
Results
Data were categorized into 16 different bacterial genera or species. Acinetobacter (n = 1), Kluyvera ascorbata (n = 2), and Stenotrophomonas maltophilia (n = 3) were underrepresented in the dataset and therefore were not included in the data analysis. The data are summarized in the Table.
In our dataset, the most common bacteria associated with UTI, irrespective of urine pH, were Escherichia coli (n = 818/1,331; 62%), Klebsiella (n = 167/1,331; 13%), and P mirabilis (n = 88/1,331; 7%). The mean urine pH in our cohort was 6.1 (range, 5.0-9.0; SD, 0.88; median, 6; and mode, 6), and 1,194/1,331 (90%) of all urine samples had a urine pH of 5.0 to 7.0. Among patients who had a urine pH of 7.5 to 9.0, E coli was the cause of UTI in 39% (53/137) and P mirabilis was the cause of UTI in 28% (38/137). Likewise, among patients who had a urine pH of 8.0 to 9.0, E coli was the cause of UTI in 36% (30/83), and P mirabilis was the cause of UTI in 31% (26/83). Lastly, in patients who had a urine pH of 8.5 to 9.0, P mirabilis was the most common cause of UTI, present in 35% (9/26) patients.
The mean urine pH in our dataset for P mirabilis (n = 88) was 7.0, with a standard deviation (SD) of 1.03 and the standard error of the mean (SEM) of 0.11. The majority, 50/88 (57%) of P mirabilis UTIs were associated with a urine pH of 5.0 to 7.0. However, the urine pH for P mirabilis was significantly more alkaline than the combined urine pH from all of the other bacterial genera and species in our cohort (P < .0001). The mean urine pH in our cohort, excluding the P mirabilis data, was 6.01 with an SD of 0.828 and a SEM of 0.023.
Limitations
Our data were obtained retrospectively from a single ED, and did not include the following information: patient age and gender, and mode in which urine samples were obtained (eg, Foley catheter, clean catch). In addition, no reports were available regarding the sensitivity of the urine cultures with respect to urine pH.
Discussion
While alkaline urine was present in only 10% of patients, a high percentage of alkaline UTIs were associated with P mirabilis, an organism with intrinsic resistance to nitrofurantoin. Therefore, health care providers could consider obtaining a urine culture and/or prescribing an antibiotic other than nitrofurantoin for treating uncomplicated UTIs with alkaline urine. In addition, nitrofurantoin has been shown to be less effective against otherwise susceptible organisms in an alkaline urine.9
Conclusion
Our data demonstrates that urine pH of UTIs diagnosed in ED patients varied with the associated bacterial pathogen, and thus urine pH potentially could affect ED provider choice of antibiotics for the treatment of UTIs. Additional research is needed to confirm our results from a larger, more diverse dataset before changes in practice are recommended.
1. Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol. 2015;13(5):269-284. doi:10.1038/nrmicro3432.
2. Stamm WE, Norrby SR. Urinary tract infections: disease panorama and challenges. J Infect Dis. 2001;183 Suppl 1:S1-S4. doi:10.1086/318850.
3. Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. Vital Health Stat 13. 2011;13(169):1-38.
4. Foxman B. Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am. 2014;28(1):1-13. doi:10.1016/j.idc.2013.09.003.
5. Foxman B. The epidemiology of urinary tract infection. Nat Rev Urol. 2010;7(12):653-660. doi:10.1038/nrurol.2010.190.
6. Coker C, Poore CA, Li X, Mobley HL. Pathogenesis of Proteus mirabilis urinary tract infection. Microbes Infect. 2000;2(12):1497-1505.
7. Armbruster CE, Mobley HL. Merging mythology and morphology: the multifaceted lifestyle of Proteus mirabilis. Nat Rev Microbiol. 2012;10(11):743-754. doi:10.1038/nrmicro2890.
8. Schaffer JN, Pearson MM. Proteus mirabilis and urinary tract infections. Microbiol Spectr. 2015;3(5). doi:10.1128/microbiolspec.UTI-0017-2013.
9. Yang L, Wang K, Li H, Denstedt JD, Cadieux PA. The influence of urinary pH on antibiotic efficacy against bacterial uropathogens. Urology. 2014;84(3):731.e1-e7. doi:10.1016/j.urology.2014.04.048.
1. Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol. 2015;13(5):269-284. doi:10.1038/nrmicro3432.
2. Stamm WE, Norrby SR. Urinary tract infections: disease panorama and challenges. J Infect Dis. 2001;183 Suppl 1:S1-S4. doi:10.1086/318850.
3. Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. Vital Health Stat 13. 2011;13(169):1-38.
4. Foxman B. Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am. 2014;28(1):1-13. doi:10.1016/j.idc.2013.09.003.
5. Foxman B. The epidemiology of urinary tract infection. Nat Rev Urol. 2010;7(12):653-660. doi:10.1038/nrurol.2010.190.
6. Coker C, Poore CA, Li X, Mobley HL. Pathogenesis of Proteus mirabilis urinary tract infection. Microbes Infect. 2000;2(12):1497-1505.
7. Armbruster CE, Mobley HL. Merging mythology and morphology: the multifaceted lifestyle of Proteus mirabilis. Nat Rev Microbiol. 2012;10(11):743-754. doi:10.1038/nrmicro2890.
8. Schaffer JN, Pearson MM. Proteus mirabilis and urinary tract infections. Microbiol Spectr. 2015;3(5). doi:10.1128/microbiolspec.UTI-0017-2013.
9. Yang L, Wang K, Li H, Denstedt JD, Cadieux PA. The influence of urinary pH on antibiotic efficacy against bacterial uropathogens. Urology. 2014;84(3):731.e1-e7. doi:10.1016/j.urology.2014.04.048.
How to Prevent Mosquito and Tick-Borne Disease

Not Another Missed Spinal Epidural Abscess
A 55-year-old man presented for evaluation of a 2-day history of worsening left lower back pain.
Delay in the diagnosis and treatment of spinal epidural abscess (SEA) increases the likelihood of permanent disability (eg, residual motor weakness) or even death.1-5 Studies suggest that the incidence of SEA may be on the rise,6,7 which is especially troubling in an era of emerging antibiotic resistance.8 The pervasive theme among the medical literature stresses the challenges with early recognition; however, missed SEA is a theoretical mishap in a manner akin to Schrödinger’s cat or Heisenberg’s uncertainty principle.9,10
One will recall Erwin Schrödinger’s thought experiment of 1935 when he challenged the theory of quantum mechanics by asking whether or not the cat in a box is still alive if there is a 50/50 chance poisonous gas has been released. He suggested that before one looks in the box, the cat is both alive and dead—a state of superposition.
Unfortunately, medical diagnoses do not exist in dual states. Tests are either positive or negative; disease is either present or absent; and in medicine, the cat is either alive or dead. Moreover, when SEA is diagnosed after the initial presentation and workup, (ie, the “bounce-back”), the clinician cannot categorically assume the condition was present, but missed, at the initial evaluation. We present the following case, not as a miraculous catch, or a “zebra-hunting guide” but rather as a rare glimpse into the evolution of a disease process.
Case
A 55-year-old man with history of type 2 diabetes mellitus (DM), hypertension, and hyperlipidemia presented to the ED with a 2-day history of progressively worsening left lower back pain. Although the patient denied a recent history of trauma, he did state that he helped one of his friends move furniture 1 day prior to presentation and had attributed the worsening pain to this event. The patient described his pain as mild and dull when he was at rest, rating it as a 2 on a pain scale of 1 to 10; and sharp-feeling and at its worst upon movement, rating it as a 9 on a pain scale of 1 to 10. The patient noted experiencing only mild relief when he shifted to certain positions.
The patient’s pain was nonradiating and associated with dull pain in the left anterior proximal thigh. The patient denied any numbness or weakness in any of his extremities. He also denied any perineal numbness or urinary or bowel incontinence; however, he did note experiencing a sense of incomplete evacuation of stools over the past 5 mornings.
The patient denied any recent history of fever, chills, numbness, weakness, difficulty with balance, direct trauma, instrumentation or chiropractic manipulation, or unexplained or unintentional weight loss. He had no history of malignancy and vehemently denied intravenous (IV) drug use.
On physical examination, the patient’s vital signs were: blood pressure, 143/93 mm Hg; heart rate, 108 beats/min; respiratory rate, 16 breaths/min; and temperature, 97.6°F. Oxygen saturation was 95% on room air. Upon examination, the patient was in no acute distress and was resting comfortably and quietly. Pertinent findings included a supple neck examination, without lymphadenopathy or meningismus. There was no midline tenderness to palpation of the cervical spine and no step-off deformities. The lungs were clear to auscultation bilaterally and without wheezing, rhonchi, or rales. Examination of the heart revealed a regular rhythm with borderline tachycardia, but without murmurs, rubs, or gallops. The patient had 2+ pulses in all four extremities, and capillary refill was less than 2 seconds. The abdomen was soft and nontender, without rebound, guarding, or rigidity. There were no pulsatile abdominal masses or bruits, and bowel sounds were present. The patient had no costovertebral angle tenderness on percussion, and had full range of motion of all four extremities, with no tenderness to palpation and no bony deformities.
Examination of the back revealed a positive straight leg raise on the right, but there was no midline tenderness to palpation or step-off deformity of the thoracic or lumbar spine. The patient exhibited mild left-sided upper lumbar paraspinal tenderness to palpation, but had no associated muscle spasm or overlying skin changes. On neurological assessment, the patient was alert and oriented with cranial nerves II-XII intact. He had 5/5 motor strength in all four extremities, with careful attention to hip flexion and extension, knee flexion and extension, and dorsiflexion and plantar flexion at the ankle. The sensory examination was normal, as were patella and ankle reflexes. The patient was able to ambulate with a steady gait.
Laboratory evaluation included a complete blood count, basic metabolic profile (BMP), urinalysis, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level. The urinalysis was negative for blood or signs of infection. The BMP demonstrated hyperglycemia without acidosis, but no additional electrolyte abnormalities or renal insufficiency. The patient did have leukocytosis (white blood cell [WBC], 19.8×109/L) with a left shift. The ESR was within normal limits (14 mm/h), but the CRP was mildly elevated (37.33 nmol/L).
Blood cultures were ordered, and the patient was given 2,500 mg vancomycin and 2,000 mg ceftriaxone IV. Given the patient’s abnormal back examination and the presence of a leukocytosis, a magnetic resonance imaging (MRI) study of the thoracic and lumbar spine was ordered. Radiology services reported the following findings from the MRI:
- Unremarkable thoracic spine MRI. No evidence of thoracic spine infection or significant degenerative changes.
- No evidence of infection involving the lumbar spine.
- L3-4 and L4-5 disc bulges and posterior element degenerative changes with moderate canal stenoses.
- Edema in the posterior paraspinous musculature on the left at the L3 and L4 levels.
The patient had DM with multiple systemic inflammatory response syndrome criteria and was admitted to the hospital for undifferentiated sepsis, clinical uncertainty, and pain control. Within 24 hours, blood cultures were positive for gram-positive cocci, later identified as methicillin-sensitive Staphylococcus aureus. However, despite treatment with antibiotics and analgesics, the patient’s back pain persisted. A repeat MRI of the lumbar spine obtained on hospital day 4 revealed the following:
At L3-4, since the comparison study, there has been development of two epidural abscesses with abnormal peripheral enhancement, one located dorsally measuring 6.8 x 8.1 x 14 mm and another located in the left lateral recess measuring 8.1 x 9.7 x 10.9 mm. The combination of the broad-based disk bulge, epidural abscesses, and hypertrophic facets resulted in severe spinal canal stenosis.
After receiving this report, the hospitalist contacted neurosurgery services. Shortly thereafter the patient underwent unilateral laminotomy with bilateral canal decompression on hospital day 5. He was discharged home on hospital day 10 without any neurological deficits, and continued IV antibiotics as an outpatient for an additional 5 weeks.
Discussion
Only a minority of patients with SEA present with the classic triad of back pain, fever, and progressive neurological findings associated with this condition.1Careful history-taking therefore is essential to identify high-risk patients. Risk factors for SEA include diabetes, IV drug abuse, immunosuppression, chronic renal failure, liver disease, alcoholism, indwelling catheter, recent invasive spinal procedure, recent vertebral fracture, cancer, and distant site of infection.1-3,5,7,11
Leukocytosis (WBC >10×109/L) is only found in two-thirds or less of patients with SEA at the time of admission.1,3,12 Inflammatory markers such as CRP and ESR are more sensitive but not specific to SEAs.1,2,5,7,11-13
An MRI study with gadolinium is the diagnostic modality of choice over computed tomography myelography to assess for SEAs due to its noninvasive nature and ability to better delineate the extent of disease.5,7,14 An MRI of the entire spine is recommended to delineate longitudinal and paraspinal extension as SEA can traverse multiple vertebral levels.15 While awaiting the results of blood cultures, patients should be treated with broad spectrum antibiotics that include coverage of the most common etiology of SEA, S aureus.1,3,4,7,11,13 While some cases of SEA may be managed medically, the emergency physician should always treat SEA as a neurosurgical emergency and obtain consultation with the appropriate services (eg, neurosurgery, infectious disease, neurology radiology).1,5
Our patient represents an unusual case of SEAs in that he presented with S aureus bacteremia while afebrile, along with back pain and tachycardia. He subsequently developed SEA, which was recognized only through serial MRI studies. The patient’s tachycardia alone could have been easily attributed to pain and anxiety associated with the ED environment. As such, he could have easily been discharged home with a prescription [for] nonsteroidal anti-inflammatory drugs and/or muscle relaxers for pain management—though it is likely that he would have returned to the ED 3 days later with persistent and even worsening symptoms, during which he would have undergone additional testing, possibly MRI, which would have revealed the missed SEA.
Our case clearly demonstrates that no SEA was present at the time of the
Studies show that half of all patients with SEAs are not diagnosed until after two or more visits to the ED.1,11 The literature posits that most cases are misdiagnosed at the time of initial evaluation. It has even been postulated that “misdiagnosis of spinal epidural abscess is the rule rather than the exception.”1 Although our patient was eventually diagnosed with a SEA, it was not present on the first MRI taken during the initial evaluation.
Summary
Unlike the rules of quantum mechanics and the paradox of Schrödinger’s cat, SEA follows a progression of disease.3,7,16 There is no superposition—the MRI is either positive or negative. However, excellent care requires the practitioner to know the risk factors of SEA, apply the appropriate screening tests, obtain MRI when necessary, and if diagnostic uncertainty remains, discuss with the patient or family signs and symptoms to monitor as well as reasons to return for re-evaluation.
1. Davis DP, Wold RM, Patel RJ, et al. The clinical presentation and impact of diagnostic delays on emergency department patients with spinal epidural abscess. J Emerg Med. 2004;26(3):285-291.
2. Bhise V, Meyer A, Singh H, et al. errors in diagnosis of spinal epidural abscesses in the era of electronic health records. Am J Med. 2017;130(8):975-981. doi:10.1016/j.amjmed.2017.03.009.
3. Darouiche RO, Hamill RJ, Greenberg SB, Weathers SW, Musher DM. Bacterial spinal epidural abscess. Review of 43 cases and literature survey. Medicine (Baltimore). 1992;71(6):369-385.
4. Baker AS, Ojemann RG, Swartz MN, Richardson EP Jr. Spinal epidural abscess. N Engl J Med. 1975;293(10):463-468. doi:10.1056/NEJM197509042931001.
5. Nussbaum ES, Rigamonti D, Standiford H, Numaguchi Y, Wolf AL, Robinson WL. Spinal epidural abscess: a report of 40 cases and review. Surg Neurol. 1992;38(3):225-231.
6. Vakili M, Crum-Cianflone NF. Spinal epidural abscess: a series of 101 cases. Am J Med. 2017;130(12):1458-1463. doi:10.1016/j.amjmed.2017.07.017.
7. Rigamonti D, Liem L, Sampath P, et al. Spinal epidural abscess: contemporary trends in etiology, evaluation, and management. Surg Neurol. 1999;52(2):189-196; discussion 197.
8. The World Health Organization. Antimicrobial resistance: global report on surveillance. http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf?ua=1. Accessed June 12, 2018.
9. Schrödinger E. Die gegenwärtige situation in der quantenmechanik. Maturwissenschaften. 1935;23(48):807-812; 823-828; 844-849. doi:10.1007/BF01491891.
10. Heisenberg H. Über quantentheoretische umdeutung kinematischer und mechanischer beziehungen. Zeitschrift für Physik. 1925;33(1):879-893. doi:10.1007/BF01328377.
11. Tang HJ, Lin HJ, Liu YC, Li CM. Spinal epidural abscess—experience with 46 patients and evaluation of prognostic factors. J Infect. 2002;45(2):76-81.
12. Soehle M, Wallenfang T. Spinal epidural abscesses: clinical manifestations, prognostic factors, and outcomes. Neurosurgery. 2002;51(1):79-85; discussion 86-87.
13. Del Curling O Jr, Gower DJ, McWhorter JM. Changing concepts in spinal epidural abscess: a report of 29 cases. Neurosurgery. 1990;27(2):185-192.
14. Hlavin ML, Kaminski HJ, Ross JS, Ganz E. Spinal epidural abscess: a ten-year perspective. Neurosurgery. 1990;27(2):177-184.
15. Parkinson JF, Sekhon LH. Spinal epidural abscess: appearance on magnetic resonance imaging as a guide to surgical management. Report of five cases. Neurosurg Focus. 2004;17(6):E12.
16. Heusner AP. Nontuberculous spinal epidural infections. N Engl J Med. 1948;239(23):845-854. doi:10.1056/NEJM194812022392301.
A 55-year-old man presented for evaluation of a 2-day history of worsening left lower back pain.
A 55-year-old man presented for evaluation of a 2-day history of worsening left lower back pain.
Delay in the diagnosis and treatment of spinal epidural abscess (SEA) increases the likelihood of permanent disability (eg, residual motor weakness) or even death.1-5 Studies suggest that the incidence of SEA may be on the rise,6,7 which is especially troubling in an era of emerging antibiotic resistance.8 The pervasive theme among the medical literature stresses the challenges with early recognition; however, missed SEA is a theoretical mishap in a manner akin to Schrödinger’s cat or Heisenberg’s uncertainty principle.9,10
One will recall Erwin Schrödinger’s thought experiment of 1935 when he challenged the theory of quantum mechanics by asking whether or not the cat in a box is still alive if there is a 50/50 chance poisonous gas has been released. He suggested that before one looks in the box, the cat is both alive and dead—a state of superposition.
Unfortunately, medical diagnoses do not exist in dual states. Tests are either positive or negative; disease is either present or absent; and in medicine, the cat is either alive or dead. Moreover, when SEA is diagnosed after the initial presentation and workup, (ie, the “bounce-back”), the clinician cannot categorically assume the condition was present, but missed, at the initial evaluation. We present the following case, not as a miraculous catch, or a “zebra-hunting guide” but rather as a rare glimpse into the evolution of a disease process.
Case
A 55-year-old man with history of type 2 diabetes mellitus (DM), hypertension, and hyperlipidemia presented to the ED with a 2-day history of progressively worsening left lower back pain. Although the patient denied a recent history of trauma, he did state that he helped one of his friends move furniture 1 day prior to presentation and had attributed the worsening pain to this event. The patient described his pain as mild and dull when he was at rest, rating it as a 2 on a pain scale of 1 to 10; and sharp-feeling and at its worst upon movement, rating it as a 9 on a pain scale of 1 to 10. The patient noted experiencing only mild relief when he shifted to certain positions.
The patient’s pain was nonradiating and associated with dull pain in the left anterior proximal thigh. The patient denied any numbness or weakness in any of his extremities. He also denied any perineal numbness or urinary or bowel incontinence; however, he did note experiencing a sense of incomplete evacuation of stools over the past 5 mornings.
The patient denied any recent history of fever, chills, numbness, weakness, difficulty with balance, direct trauma, instrumentation or chiropractic manipulation, or unexplained or unintentional weight loss. He had no history of malignancy and vehemently denied intravenous (IV) drug use.
On physical examination, the patient’s vital signs were: blood pressure, 143/93 mm Hg; heart rate, 108 beats/min; respiratory rate, 16 breaths/min; and temperature, 97.6°F. Oxygen saturation was 95% on room air. Upon examination, the patient was in no acute distress and was resting comfortably and quietly. Pertinent findings included a supple neck examination, without lymphadenopathy or meningismus. There was no midline tenderness to palpation of the cervical spine and no step-off deformities. The lungs were clear to auscultation bilaterally and without wheezing, rhonchi, or rales. Examination of the heart revealed a regular rhythm with borderline tachycardia, but without murmurs, rubs, or gallops. The patient had 2+ pulses in all four extremities, and capillary refill was less than 2 seconds. The abdomen was soft and nontender, without rebound, guarding, or rigidity. There were no pulsatile abdominal masses or bruits, and bowel sounds were present. The patient had no costovertebral angle tenderness on percussion, and had full range of motion of all four extremities, with no tenderness to palpation and no bony deformities.
Examination of the back revealed a positive straight leg raise on the right, but there was no midline tenderness to palpation or step-off deformity of the thoracic or lumbar spine. The patient exhibited mild left-sided upper lumbar paraspinal tenderness to palpation, but had no associated muscle spasm or overlying skin changes. On neurological assessment, the patient was alert and oriented with cranial nerves II-XII intact. He had 5/5 motor strength in all four extremities, with careful attention to hip flexion and extension, knee flexion and extension, and dorsiflexion and plantar flexion at the ankle. The sensory examination was normal, as were patella and ankle reflexes. The patient was able to ambulate with a steady gait.
Laboratory evaluation included a complete blood count, basic metabolic profile (BMP), urinalysis, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level. The urinalysis was negative for blood or signs of infection. The BMP demonstrated hyperglycemia without acidosis, but no additional electrolyte abnormalities or renal insufficiency. The patient did have leukocytosis (white blood cell [WBC], 19.8×109/L) with a left shift. The ESR was within normal limits (14 mm/h), but the CRP was mildly elevated (37.33 nmol/L).
Blood cultures were ordered, and the patient was given 2,500 mg vancomycin and 2,000 mg ceftriaxone IV. Given the patient’s abnormal back examination and the presence of a leukocytosis, a magnetic resonance imaging (MRI) study of the thoracic and lumbar spine was ordered. Radiology services reported the following findings from the MRI:
- Unremarkable thoracic spine MRI. No evidence of thoracic spine infection or significant degenerative changes.
- No evidence of infection involving the lumbar spine.
- L3-4 and L4-5 disc bulges and posterior element degenerative changes with moderate canal stenoses.
- Edema in the posterior paraspinous musculature on the left at the L3 and L4 levels.
The patient had DM with multiple systemic inflammatory response syndrome criteria and was admitted to the hospital for undifferentiated sepsis, clinical uncertainty, and pain control. Within 24 hours, blood cultures were positive for gram-positive cocci, later identified as methicillin-sensitive Staphylococcus aureus. However, despite treatment with antibiotics and analgesics, the patient’s back pain persisted. A repeat MRI of the lumbar spine obtained on hospital day 4 revealed the following:
At L3-4, since the comparison study, there has been development of two epidural abscesses with abnormal peripheral enhancement, one located dorsally measuring 6.8 x 8.1 x 14 mm and another located in the left lateral recess measuring 8.1 x 9.7 x 10.9 mm. The combination of the broad-based disk bulge, epidural abscesses, and hypertrophic facets resulted in severe spinal canal stenosis.
After receiving this report, the hospitalist contacted neurosurgery services. Shortly thereafter the patient underwent unilateral laminotomy with bilateral canal decompression on hospital day 5. He was discharged home on hospital day 10 without any neurological deficits, and continued IV antibiotics as an outpatient for an additional 5 weeks.
Discussion
Only a minority of patients with SEA present with the classic triad of back pain, fever, and progressive neurological findings associated with this condition.1Careful history-taking therefore is essential to identify high-risk patients. Risk factors for SEA include diabetes, IV drug abuse, immunosuppression, chronic renal failure, liver disease, alcoholism, indwelling catheter, recent invasive spinal procedure, recent vertebral fracture, cancer, and distant site of infection.1-3,5,7,11
Leukocytosis (WBC >10×109/L) is only found in two-thirds or less of patients with SEA at the time of admission.1,3,12 Inflammatory markers such as CRP and ESR are more sensitive but not specific to SEAs.1,2,5,7,11-13
An MRI study with gadolinium is the diagnostic modality of choice over computed tomography myelography to assess for SEAs due to its noninvasive nature and ability to better delineate the extent of disease.5,7,14 An MRI of the entire spine is recommended to delineate longitudinal and paraspinal extension as SEA can traverse multiple vertebral levels.15 While awaiting the results of blood cultures, patients should be treated with broad spectrum antibiotics that include coverage of the most common etiology of SEA, S aureus.1,3,4,7,11,13 While some cases of SEA may be managed medically, the emergency physician should always treat SEA as a neurosurgical emergency and obtain consultation with the appropriate services (eg, neurosurgery, infectious disease, neurology radiology).1,5
Our patient represents an unusual case of SEAs in that he presented with S aureus bacteremia while afebrile, along with back pain and tachycardia. He subsequently developed SEA, which was recognized only through serial MRI studies. The patient’s tachycardia alone could have been easily attributed to pain and anxiety associated with the ED environment. As such, he could have easily been discharged home with a prescription [for] nonsteroidal anti-inflammatory drugs and/or muscle relaxers for pain management—though it is likely that he would have returned to the ED 3 days later with persistent and even worsening symptoms, during which he would have undergone additional testing, possibly MRI, which would have revealed the missed SEA.
Our case clearly demonstrates that no SEA was present at the time of the
Studies show that half of all patients with SEAs are not diagnosed until after two or more visits to the ED.1,11 The literature posits that most cases are misdiagnosed at the time of initial evaluation. It has even been postulated that “misdiagnosis of spinal epidural abscess is the rule rather than the exception.”1 Although our patient was eventually diagnosed with a SEA, it was not present on the first MRI taken during the initial evaluation.
Summary
Unlike the rules of quantum mechanics and the paradox of Schrödinger’s cat, SEA follows a progression of disease.3,7,16 There is no superposition—the MRI is either positive or negative. However, excellent care requires the practitioner to know the risk factors of SEA, apply the appropriate screening tests, obtain MRI when necessary, and if diagnostic uncertainty remains, discuss with the patient or family signs and symptoms to monitor as well as reasons to return for re-evaluation.
Delay in the diagnosis and treatment of spinal epidural abscess (SEA) increases the likelihood of permanent disability (eg, residual motor weakness) or even death.1-5 Studies suggest that the incidence of SEA may be on the rise,6,7 which is especially troubling in an era of emerging antibiotic resistance.8 The pervasive theme among the medical literature stresses the challenges with early recognition; however, missed SEA is a theoretical mishap in a manner akin to Schrödinger’s cat or Heisenberg’s uncertainty principle.9,10
One will recall Erwin Schrödinger’s thought experiment of 1935 when he challenged the theory of quantum mechanics by asking whether or not the cat in a box is still alive if there is a 50/50 chance poisonous gas has been released. He suggested that before one looks in the box, the cat is both alive and dead—a state of superposition.
Unfortunately, medical diagnoses do not exist in dual states. Tests are either positive or negative; disease is either present or absent; and in medicine, the cat is either alive or dead. Moreover, when SEA is diagnosed after the initial presentation and workup, (ie, the “bounce-back”), the clinician cannot categorically assume the condition was present, but missed, at the initial evaluation. We present the following case, not as a miraculous catch, or a “zebra-hunting guide” but rather as a rare glimpse into the evolution of a disease process.
Case
A 55-year-old man with history of type 2 diabetes mellitus (DM), hypertension, and hyperlipidemia presented to the ED with a 2-day history of progressively worsening left lower back pain. Although the patient denied a recent history of trauma, he did state that he helped one of his friends move furniture 1 day prior to presentation and had attributed the worsening pain to this event. The patient described his pain as mild and dull when he was at rest, rating it as a 2 on a pain scale of 1 to 10; and sharp-feeling and at its worst upon movement, rating it as a 9 on a pain scale of 1 to 10. The patient noted experiencing only mild relief when he shifted to certain positions.
The patient’s pain was nonradiating and associated with dull pain in the left anterior proximal thigh. The patient denied any numbness or weakness in any of his extremities. He also denied any perineal numbness or urinary or bowel incontinence; however, he did note experiencing a sense of incomplete evacuation of stools over the past 5 mornings.
The patient denied any recent history of fever, chills, numbness, weakness, difficulty with balance, direct trauma, instrumentation or chiropractic manipulation, or unexplained or unintentional weight loss. He had no history of malignancy and vehemently denied intravenous (IV) drug use.
On physical examination, the patient’s vital signs were: blood pressure, 143/93 mm Hg; heart rate, 108 beats/min; respiratory rate, 16 breaths/min; and temperature, 97.6°F. Oxygen saturation was 95% on room air. Upon examination, the patient was in no acute distress and was resting comfortably and quietly. Pertinent findings included a supple neck examination, without lymphadenopathy or meningismus. There was no midline tenderness to palpation of the cervical spine and no step-off deformities. The lungs were clear to auscultation bilaterally and without wheezing, rhonchi, or rales. Examination of the heart revealed a regular rhythm with borderline tachycardia, but without murmurs, rubs, or gallops. The patient had 2+ pulses in all four extremities, and capillary refill was less than 2 seconds. The abdomen was soft and nontender, without rebound, guarding, or rigidity. There were no pulsatile abdominal masses or bruits, and bowel sounds were present. The patient had no costovertebral angle tenderness on percussion, and had full range of motion of all four extremities, with no tenderness to palpation and no bony deformities.
Examination of the back revealed a positive straight leg raise on the right, but there was no midline tenderness to palpation or step-off deformity of the thoracic or lumbar spine. The patient exhibited mild left-sided upper lumbar paraspinal tenderness to palpation, but had no associated muscle spasm or overlying skin changes. On neurological assessment, the patient was alert and oriented with cranial nerves II-XII intact. He had 5/5 motor strength in all four extremities, with careful attention to hip flexion and extension, knee flexion and extension, and dorsiflexion and plantar flexion at the ankle. The sensory examination was normal, as were patella and ankle reflexes. The patient was able to ambulate with a steady gait.
Laboratory evaluation included a complete blood count, basic metabolic profile (BMP), urinalysis, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level. The urinalysis was negative for blood or signs of infection. The BMP demonstrated hyperglycemia without acidosis, but no additional electrolyte abnormalities or renal insufficiency. The patient did have leukocytosis (white blood cell [WBC], 19.8×109/L) with a left shift. The ESR was within normal limits (14 mm/h), but the CRP was mildly elevated (37.33 nmol/L).
Blood cultures were ordered, and the patient was given 2,500 mg vancomycin and 2,000 mg ceftriaxone IV. Given the patient’s abnormal back examination and the presence of a leukocytosis, a magnetic resonance imaging (MRI) study of the thoracic and lumbar spine was ordered. Radiology services reported the following findings from the MRI:
- Unremarkable thoracic spine MRI. No evidence of thoracic spine infection or significant degenerative changes.
- No evidence of infection involving the lumbar spine.
- L3-4 and L4-5 disc bulges and posterior element degenerative changes with moderate canal stenoses.
- Edema in the posterior paraspinous musculature on the left at the L3 and L4 levels.
The patient had DM with multiple systemic inflammatory response syndrome criteria and was admitted to the hospital for undifferentiated sepsis, clinical uncertainty, and pain control. Within 24 hours, blood cultures were positive for gram-positive cocci, later identified as methicillin-sensitive Staphylococcus aureus. However, despite treatment with antibiotics and analgesics, the patient’s back pain persisted. A repeat MRI of the lumbar spine obtained on hospital day 4 revealed the following:
At L3-4, since the comparison study, there has been development of two epidural abscesses with abnormal peripheral enhancement, one located dorsally measuring 6.8 x 8.1 x 14 mm and another located in the left lateral recess measuring 8.1 x 9.7 x 10.9 mm. The combination of the broad-based disk bulge, epidural abscesses, and hypertrophic facets resulted in severe spinal canal stenosis.
After receiving this report, the hospitalist contacted neurosurgery services. Shortly thereafter the patient underwent unilateral laminotomy with bilateral canal decompression on hospital day 5. He was discharged home on hospital day 10 without any neurological deficits, and continued IV antibiotics as an outpatient for an additional 5 weeks.
Discussion
Only a minority of patients with SEA present with the classic triad of back pain, fever, and progressive neurological findings associated with this condition.1Careful history-taking therefore is essential to identify high-risk patients. Risk factors for SEA include diabetes, IV drug abuse, immunosuppression, chronic renal failure, liver disease, alcoholism, indwelling catheter, recent invasive spinal procedure, recent vertebral fracture, cancer, and distant site of infection.1-3,5,7,11
Leukocytosis (WBC >10×109/L) is only found in two-thirds or less of patients with SEA at the time of admission.1,3,12 Inflammatory markers such as CRP and ESR are more sensitive but not specific to SEAs.1,2,5,7,11-13
An MRI study with gadolinium is the diagnostic modality of choice over computed tomography myelography to assess for SEAs due to its noninvasive nature and ability to better delineate the extent of disease.5,7,14 An MRI of the entire spine is recommended to delineate longitudinal and paraspinal extension as SEA can traverse multiple vertebral levels.15 While awaiting the results of blood cultures, patients should be treated with broad spectrum antibiotics that include coverage of the most common etiology of SEA, S aureus.1,3,4,7,11,13 While some cases of SEA may be managed medically, the emergency physician should always treat SEA as a neurosurgical emergency and obtain consultation with the appropriate services (eg, neurosurgery, infectious disease, neurology radiology).1,5
Our patient represents an unusual case of SEAs in that he presented with S aureus bacteremia while afebrile, along with back pain and tachycardia. He subsequently developed SEA, which was recognized only through serial MRI studies. The patient’s tachycardia alone could have been easily attributed to pain and anxiety associated with the ED environment. As such, he could have easily been discharged home with a prescription [for] nonsteroidal anti-inflammatory drugs and/or muscle relaxers for pain management—though it is likely that he would have returned to the ED 3 days later with persistent and even worsening symptoms, during which he would have undergone additional testing, possibly MRI, which would have revealed the missed SEA.
Our case clearly demonstrates that no SEA was present at the time of the
Studies show that half of all patients with SEAs are not diagnosed until after two or more visits to the ED.1,11 The literature posits that most cases are misdiagnosed at the time of initial evaluation. It has even been postulated that “misdiagnosis of spinal epidural abscess is the rule rather than the exception.”1 Although our patient was eventually diagnosed with a SEA, it was not present on the first MRI taken during the initial evaluation.
Summary
Unlike the rules of quantum mechanics and the paradox of Schrödinger’s cat, SEA follows a progression of disease.3,7,16 There is no superposition—the MRI is either positive or negative. However, excellent care requires the practitioner to know the risk factors of SEA, apply the appropriate screening tests, obtain MRI when necessary, and if diagnostic uncertainty remains, discuss with the patient or family signs and symptoms to monitor as well as reasons to return for re-evaluation.
1. Davis DP, Wold RM, Patel RJ, et al. The clinical presentation and impact of diagnostic delays on emergency department patients with spinal epidural abscess. J Emerg Med. 2004;26(3):285-291.
2. Bhise V, Meyer A, Singh H, et al. errors in diagnosis of spinal epidural abscesses in the era of electronic health records. Am J Med. 2017;130(8):975-981. doi:10.1016/j.amjmed.2017.03.009.
3. Darouiche RO, Hamill RJ, Greenberg SB, Weathers SW, Musher DM. Bacterial spinal epidural abscess. Review of 43 cases and literature survey. Medicine (Baltimore). 1992;71(6):369-385.
4. Baker AS, Ojemann RG, Swartz MN, Richardson EP Jr. Spinal epidural abscess. N Engl J Med. 1975;293(10):463-468. doi:10.1056/NEJM197509042931001.
5. Nussbaum ES, Rigamonti D, Standiford H, Numaguchi Y, Wolf AL, Robinson WL. Spinal epidural abscess: a report of 40 cases and review. Surg Neurol. 1992;38(3):225-231.
6. Vakili M, Crum-Cianflone NF. Spinal epidural abscess: a series of 101 cases. Am J Med. 2017;130(12):1458-1463. doi:10.1016/j.amjmed.2017.07.017.
7. Rigamonti D, Liem L, Sampath P, et al. Spinal epidural abscess: contemporary trends in etiology, evaluation, and management. Surg Neurol. 1999;52(2):189-196; discussion 197.
8. The World Health Organization. Antimicrobial resistance: global report on surveillance. http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf?ua=1. Accessed June 12, 2018.
9. Schrödinger E. Die gegenwärtige situation in der quantenmechanik. Maturwissenschaften. 1935;23(48):807-812; 823-828; 844-849. doi:10.1007/BF01491891.
10. Heisenberg H. Über quantentheoretische umdeutung kinematischer und mechanischer beziehungen. Zeitschrift für Physik. 1925;33(1):879-893. doi:10.1007/BF01328377.
11. Tang HJ, Lin HJ, Liu YC, Li CM. Spinal epidural abscess—experience with 46 patients and evaluation of prognostic factors. J Infect. 2002;45(2):76-81.
12. Soehle M, Wallenfang T. Spinal epidural abscesses: clinical manifestations, prognostic factors, and outcomes. Neurosurgery. 2002;51(1):79-85; discussion 86-87.
13. Del Curling O Jr, Gower DJ, McWhorter JM. Changing concepts in spinal epidural abscess: a report of 29 cases. Neurosurgery. 1990;27(2):185-192.
14. Hlavin ML, Kaminski HJ, Ross JS, Ganz E. Spinal epidural abscess: a ten-year perspective. Neurosurgery. 1990;27(2):177-184.
15. Parkinson JF, Sekhon LH. Spinal epidural abscess: appearance on magnetic resonance imaging as a guide to surgical management. Report of five cases. Neurosurg Focus. 2004;17(6):E12.
16. Heusner AP. Nontuberculous spinal epidural infections. N Engl J Med. 1948;239(23):845-854. doi:10.1056/NEJM194812022392301.
1. Davis DP, Wold RM, Patel RJ, et al. The clinical presentation and impact of diagnostic delays on emergency department patients with spinal epidural abscess. J Emerg Med. 2004;26(3):285-291.
2. Bhise V, Meyer A, Singh H, et al. errors in diagnosis of spinal epidural abscesses in the era of electronic health records. Am J Med. 2017;130(8):975-981. doi:10.1016/j.amjmed.2017.03.009.
3. Darouiche RO, Hamill RJ, Greenberg SB, Weathers SW, Musher DM. Bacterial spinal epidural abscess. Review of 43 cases and literature survey. Medicine (Baltimore). 1992;71(6):369-385.
4. Baker AS, Ojemann RG, Swartz MN, Richardson EP Jr. Spinal epidural abscess. N Engl J Med. 1975;293(10):463-468. doi:10.1056/NEJM197509042931001.
5. Nussbaum ES, Rigamonti D, Standiford H, Numaguchi Y, Wolf AL, Robinson WL. Spinal epidural abscess: a report of 40 cases and review. Surg Neurol. 1992;38(3):225-231.
6. Vakili M, Crum-Cianflone NF. Spinal epidural abscess: a series of 101 cases. Am J Med. 2017;130(12):1458-1463. doi:10.1016/j.amjmed.2017.07.017.
7. Rigamonti D, Liem L, Sampath P, et al. Spinal epidural abscess: contemporary trends in etiology, evaluation, and management. Surg Neurol. 1999;52(2):189-196; discussion 197.
8. The World Health Organization. Antimicrobial resistance: global report on surveillance. http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf?ua=1. Accessed June 12, 2018.
9. Schrödinger E. Die gegenwärtige situation in der quantenmechanik. Maturwissenschaften. 1935;23(48):807-812; 823-828; 844-849. doi:10.1007/BF01491891.
10. Heisenberg H. Über quantentheoretische umdeutung kinematischer und mechanischer beziehungen. Zeitschrift für Physik. 1925;33(1):879-893. doi:10.1007/BF01328377.
11. Tang HJ, Lin HJ, Liu YC, Li CM. Spinal epidural abscess—experience with 46 patients and evaluation of prognostic factors. J Infect. 2002;45(2):76-81.
12. Soehle M, Wallenfang T. Spinal epidural abscesses: clinical manifestations, prognostic factors, and outcomes. Neurosurgery. 2002;51(1):79-85; discussion 86-87.
13. Del Curling O Jr, Gower DJ, McWhorter JM. Changing concepts in spinal epidural abscess: a report of 29 cases. Neurosurgery. 1990;27(2):185-192.
14. Hlavin ML, Kaminski HJ, Ross JS, Ganz E. Spinal epidural abscess: a ten-year perspective. Neurosurgery. 1990;27(2):177-184.
15. Parkinson JF, Sekhon LH. Spinal epidural abscess: appearance on magnetic resonance imaging as a guide to surgical management. Report of five cases. Neurosurg Focus. 2004;17(6):E12.
16. Heusner AP. Nontuberculous spinal epidural infections. N Engl J Med. 1948;239(23):845-854. doi:10.1056/NEJM194812022392301.
Transplanting HCV-infected kidneys in HCV-infected patients showed positive outcomes, costs
Transplanting a kidney infected with hepatitis C into individuals infected with HCV, followed by treatment, was more effective and less costly than transplanting an uninfected kidney, preceded by HCV treatment, according to Mark H. Eckman, MD, of the University of Cincinnati and his colleagues.
Largely because of the longer wait times for uninfected kidneys, a typical patient aged 58 years on hemodialysis would gain an average of 0.5 quality-adjusted life-years at a lifetime cost savings of $41,591 dollars, according to the model.
“In an era of increasing success for kidney transplants and demand that far outstrips supply, deferring antiviral therapy until after transplant of HCV-infected kidneys, when available, should be both cost saving and effective,” the researchers wrote.
The study was funded by grants from Merck Sharpe & Dohme and the National Center for Advancing Translational Science. Several of the authors reported having grants from Merck and grants and personal fees from a variety of other pharmaceutical companies.
SOURCE: Eckman MH et al. Ann Intern Med. 2018 Jul 10. doi: 10.7326/M17-3088.
Transplanting a kidney infected with hepatitis C into individuals infected with HCV, followed by treatment, was more effective and less costly than transplanting an uninfected kidney, preceded by HCV treatment, according to Mark H. Eckman, MD, of the University of Cincinnati and his colleagues.
Largely because of the longer wait times for uninfected kidneys, a typical patient aged 58 years on hemodialysis would gain an average of 0.5 quality-adjusted life-years at a lifetime cost savings of $41,591 dollars, according to the model.
“In an era of increasing success for kidney transplants and demand that far outstrips supply, deferring antiviral therapy until after transplant of HCV-infected kidneys, when available, should be both cost saving and effective,” the researchers wrote.
The study was funded by grants from Merck Sharpe & Dohme and the National Center for Advancing Translational Science. Several of the authors reported having grants from Merck and grants and personal fees from a variety of other pharmaceutical companies.
SOURCE: Eckman MH et al. Ann Intern Med. 2018 Jul 10. doi: 10.7326/M17-3088.
Transplanting a kidney infected with hepatitis C into individuals infected with HCV, followed by treatment, was more effective and less costly than transplanting an uninfected kidney, preceded by HCV treatment, according to Mark H. Eckman, MD, of the University of Cincinnati and his colleagues.
Largely because of the longer wait times for uninfected kidneys, a typical patient aged 58 years on hemodialysis would gain an average of 0.5 quality-adjusted life-years at a lifetime cost savings of $41,591 dollars, according to the model.
“In an era of increasing success for kidney transplants and demand that far outstrips supply, deferring antiviral therapy until after transplant of HCV-infected kidneys, when available, should be both cost saving and effective,” the researchers wrote.
The study was funded by grants from Merck Sharpe & Dohme and the National Center for Advancing Translational Science. Several of the authors reported having grants from Merck and grants and personal fees from a variety of other pharmaceutical companies.
SOURCE: Eckman MH et al. Ann Intern Med. 2018 Jul 10. doi: 10.7326/M17-3088.
FROM ANNALS OF INTERNAL MEDICINE