User login
Tuesday’s satellite symposia schedule, information
Valuable Strategies to Reduce the Risk for and Clinical Impact of Hyperkalemia
7:30 – 9:30 p.m. – Canary Room 1-2
Dinner provided at 7:30 p.m.
Presenters: Biff F. Palmer, MD, professor of internal medicine, University of Texas Southwestern Medical Center, Dallas; Robert Toto, MD, associate dean, clinical and translational research, director, Center for Translational Medicine, UT Southwestern Medical Center, Dallas.
Overview: The goal of the case-based symposium is to highlight patients at increased risk of recurrent or sustained hyperkalemia and how these patients may be managed with evidence-based treatment so that they are able to continue renin-angiotensin-aldosterone system inhibitor therapy, if appropriate. The presenters will share their experiences in managing these patients, including suggestions for the hospitalist’s role in continued care.
Accreditation: This activity has been approved for AMA PRA Category 1 Credit(s)™. See final CE activity announcement for specific details.
This activity is supported by an educational grant from Relypsa Inc.
Hepatology News Tonight: Managing Complication of Cirrhosis
7:30 – 9:30 p.m. – Canary Room 3-4
Registration will be at 7:00 p.m.
Dinner provided at 7:30 p.m.
Presenters: Naoky Tsai, MD, clinical professor of medicine, John A. Burns School of Medicine, University of Hawaii, Manoa, Honolulu; Ashwani K. Singal MD, MS, associate professor of medicine and director of UAB Porphyria Center, division of gastroenterology and hepatology, University of Alabama at Birmingham.
Overview: This educational update will highlight the most clinically relevant advances in the management of patients with cirrhosis and hepatic encephalopathy (HE). The symposium will deliver emerging science and incorporate expert opinions on best practices for the diagnosis and management of patients with cirrhosis and HE.
Target audience: This activity has been designed to meet the educational needs of physicians, advanced practice providers, and allied health professionals who provide care for hospitalized patients with liver disease.
Learning objectives:
- Understand the complications and the consequences of chronic liver disease.
- Describe the economic, patient, and caregiver burdens associated with cirrhosis and HE.
- Demonstrate the ability to properly treat HE patients and prevent recurrence of disease.
Accredited by: Rehoboth McKindley Christian Health Care Services
Provided by: Chronic Liver Disease Foundation (CLDF)
Register: http://www.chronicliverdisease.org/
Supported by an educational grant from Salix Pharmaceuticals.
Valuable Strategies to Reduce the Risk for and Clinical Impact of Hyperkalemia
7:30 – 9:30 p.m. – Canary Room 1-2
Dinner provided at 7:30 p.m.
Presenters: Biff F. Palmer, MD, professor of internal medicine, University of Texas Southwestern Medical Center, Dallas; Robert Toto, MD, associate dean, clinical and translational research, director, Center for Translational Medicine, UT Southwestern Medical Center, Dallas.
Overview: The goal of the case-based symposium is to highlight patients at increased risk of recurrent or sustained hyperkalemia and how these patients may be managed with evidence-based treatment so that they are able to continue renin-angiotensin-aldosterone system inhibitor therapy, if appropriate. The presenters will share their experiences in managing these patients, including suggestions for the hospitalist’s role in continued care.
Accreditation: This activity has been approved for AMA PRA Category 1 Credit(s)™. See final CE activity announcement for specific details.
This activity is supported by an educational grant from Relypsa Inc.
Hepatology News Tonight: Managing Complication of Cirrhosis
7:30 – 9:30 p.m. – Canary Room 3-4
Registration will be at 7:00 p.m.
Dinner provided at 7:30 p.m.
Presenters: Naoky Tsai, MD, clinical professor of medicine, John A. Burns School of Medicine, University of Hawaii, Manoa, Honolulu; Ashwani K. Singal MD, MS, associate professor of medicine and director of UAB Porphyria Center, division of gastroenterology and hepatology, University of Alabama at Birmingham.
Overview: This educational update will highlight the most clinically relevant advances in the management of patients with cirrhosis and hepatic encephalopathy (HE). The symposium will deliver emerging science and incorporate expert opinions on best practices for the diagnosis and management of patients with cirrhosis and HE.
Target audience: This activity has been designed to meet the educational needs of physicians, advanced practice providers, and allied health professionals who provide care for hospitalized patients with liver disease.
Learning objectives:
- Understand the complications and the consequences of chronic liver disease.
- Describe the economic, patient, and caregiver burdens associated with cirrhosis and HE.
- Demonstrate the ability to properly treat HE patients and prevent recurrence of disease.
Accredited by: Rehoboth McKindley Christian Health Care Services
Provided by: Chronic Liver Disease Foundation (CLDF)
Register: http://www.chronicliverdisease.org/
Supported by an educational grant from Salix Pharmaceuticals.
Valuable Strategies to Reduce the Risk for and Clinical Impact of Hyperkalemia
7:30 – 9:30 p.m. – Canary Room 1-2
Dinner provided at 7:30 p.m.
Presenters: Biff F. Palmer, MD, professor of internal medicine, University of Texas Southwestern Medical Center, Dallas; Robert Toto, MD, associate dean, clinical and translational research, director, Center for Translational Medicine, UT Southwestern Medical Center, Dallas.
Overview: The goal of the case-based symposium is to highlight patients at increased risk of recurrent or sustained hyperkalemia and how these patients may be managed with evidence-based treatment so that they are able to continue renin-angiotensin-aldosterone system inhibitor therapy, if appropriate. The presenters will share their experiences in managing these patients, including suggestions for the hospitalist’s role in continued care.
Accreditation: This activity has been approved for AMA PRA Category 1 Credit(s)™. See final CE activity announcement for specific details.
This activity is supported by an educational grant from Relypsa Inc.
Hepatology News Tonight: Managing Complication of Cirrhosis
7:30 – 9:30 p.m. – Canary Room 3-4
Registration will be at 7:00 p.m.
Dinner provided at 7:30 p.m.
Presenters: Naoky Tsai, MD, clinical professor of medicine, John A. Burns School of Medicine, University of Hawaii, Manoa, Honolulu; Ashwani K. Singal MD, MS, associate professor of medicine and director of UAB Porphyria Center, division of gastroenterology and hepatology, University of Alabama at Birmingham.
Overview: This educational update will highlight the most clinically relevant advances in the management of patients with cirrhosis and hepatic encephalopathy (HE). The symposium will deliver emerging science and incorporate expert opinions on best practices for the diagnosis and management of patients with cirrhosis and HE.
Target audience: This activity has been designed to meet the educational needs of physicians, advanced practice providers, and allied health professionals who provide care for hospitalized patients with liver disease.
Learning objectives:
- Understand the complications and the consequences of chronic liver disease.
- Describe the economic, patient, and caregiver burdens associated with cirrhosis and HE.
- Demonstrate the ability to properly treat HE patients and prevent recurrence of disease.
Accredited by: Rehoboth McKindley Christian Health Care Services
Provided by: Chronic Liver Disease Foundation (CLDF)
Register: http://www.chronicliverdisease.org/
Supported by an educational grant from Salix Pharmaceuticals.
RIV plenary showcases best research
The best research in hospital medicine will be front and center today in the “Best of Research and Innovations in 2018” part of this morning’s plenary session.
New research also will figure prominently in the second “Clinical Vignettes Poster Competition” at lunchtime today.
During the plenary session, investigators will present the top-rated research among hundreds of submissions, said Ethan Cumbler, MD, FHM, chair of the Research, Innovations, Vignettes (RIV) competition and professor of medicine and medical director of the University of Colorado Acute Care Center for the Elderly, Denver, unit. Three independent, blinded reviewers chose the abstracts for oral presentation after rating them based on originality, scientific rigor, and importance to hospital medicine, he said. These oral presentations are meant not only to provide information to other hospitalists but also to inspire hospitalists to engage in research themselves.

“I think about it as a collective celebration of how far the field of hospital medicine has advanced in the last year and its potential moving forward,” Dr. Cumbler said. “My fundamental view is every hospitalist can and should be examining what they’re doing, thinking about how to do it better, and, by God, sharing it with the rest of us when they figure out that something can work better.”
Even as a veteran hospitalist, Dr. Cumbler said he still gets inspired by research presented in the RIV.
“When I see the RIV posters or come to hear the oral presentations, I get inspired, because I get to see what other people are doing in their local microenvironments, in their laboratories, in their hospitals,” he said. “And often I think ... ‘We could be doing stuff just that exciting.’ Often, it’s a chance to collaborate with the people whose work is inspiring you, or to take a great idea and run with it.”
At the Clinical Vignettes competition, research will focus on lessons learned from specific cases, Dr. Cumbler said.
“A typical clinical vignette would be a case presentation, maybe a diagnostic image or a description of the test that clinched the diagnosis, and then, most importantly, the lessons from that case, which are more widely applicable,” he said. “One of the things I love about Clinical Vignettes is it gives you a chance to highlight your best catches, but it also lets you, with humility, share your misses so that other people can learn from your experience.”
He said he hopes the sharing of research in formal oral presentations – and in the poster hall – continues to advance the hospital medicine literature.
“It’s come an incredible distance over the last 10 years, 15 years,” he said. “When I look at where we are heading next, I think it is into more multicenter research, multiple-institution quality improvement. I really see us graduating from proof-of-concept and pilot work into the kind of trials which answer questions – the big questions that face medicine.”
Best of Research and Innovations in 2018
8-9 a.m., Palms Ballroom
Clinical Vignettes #2
Poster Competition
12-1:30 p.m., Cypress Ballroom
The best research in hospital medicine will be front and center today in the “Best of Research and Innovations in 2018” part of this morning’s plenary session.
New research also will figure prominently in the second “Clinical Vignettes Poster Competition” at lunchtime today.
During the plenary session, investigators will present the top-rated research among hundreds of submissions, said Ethan Cumbler, MD, FHM, chair of the Research, Innovations, Vignettes (RIV) competition and professor of medicine and medical director of the University of Colorado Acute Care Center for the Elderly, Denver, unit. Three independent, blinded reviewers chose the abstracts for oral presentation after rating them based on originality, scientific rigor, and importance to hospital medicine, he said. These oral presentations are meant not only to provide information to other hospitalists but also to inspire hospitalists to engage in research themselves.
“I think about it as a collective celebration of how far the field of hospital medicine has advanced in the last year and its potential moving forward,” Dr. Cumbler said. “My fundamental view is every hospitalist can and should be examining what they’re doing, thinking about how to do it better, and, by God, sharing it with the rest of us when they figure out that something can work better.”
Even as a veteran hospitalist, Dr. Cumbler said he still gets inspired by research presented in the RIV.
“When I see the RIV posters or come to hear the oral presentations, I get inspired, because I get to see what other people are doing in their local microenvironments, in their laboratories, in their hospitals,” he said. “And often I think ... ‘We could be doing stuff just that exciting.’ Often, it’s a chance to collaborate with the people whose work is inspiring you, or to take a great idea and run with it.”
At the Clinical Vignettes competition, research will focus on lessons learned from specific cases, Dr. Cumbler said.
“A typical clinical vignette would be a case presentation, maybe a diagnostic image or a description of the test that clinched the diagnosis, and then, most importantly, the lessons from that case, which are more widely applicable,” he said. “One of the things I love about Clinical Vignettes is it gives you a chance to highlight your best catches, but it also lets you, with humility, share your misses so that other people can learn from your experience.”
He said he hopes the sharing of research in formal oral presentations – and in the poster hall – continues to advance the hospital medicine literature.
“It’s come an incredible distance over the last 10 years, 15 years,” he said. “When I look at where we are heading next, I think it is into more multicenter research, multiple-institution quality improvement. I really see us graduating from proof-of-concept and pilot work into the kind of trials which answer questions – the big questions that face medicine.”
Best of Research and Innovations in 2018
8-9 a.m., Palms Ballroom
Clinical Vignettes #2
Poster Competition
12-1:30 p.m., Cypress Ballroom
The best research in hospital medicine will be front and center today in the “Best of Research and Innovations in 2018” part of this morning’s plenary session.
New research also will figure prominently in the second “Clinical Vignettes Poster Competition” at lunchtime today.
During the plenary session, investigators will present the top-rated research among hundreds of submissions, said Ethan Cumbler, MD, FHM, chair of the Research, Innovations, Vignettes (RIV) competition and professor of medicine and medical director of the University of Colorado Acute Care Center for the Elderly, Denver, unit. Three independent, blinded reviewers chose the abstracts for oral presentation after rating them based on originality, scientific rigor, and importance to hospital medicine, he said. These oral presentations are meant not only to provide information to other hospitalists but also to inspire hospitalists to engage in research themselves.
“I think about it as a collective celebration of how far the field of hospital medicine has advanced in the last year and its potential moving forward,” Dr. Cumbler said. “My fundamental view is every hospitalist can and should be examining what they’re doing, thinking about how to do it better, and, by God, sharing it with the rest of us when they figure out that something can work better.”
Even as a veteran hospitalist, Dr. Cumbler said he still gets inspired by research presented in the RIV.
“When I see the RIV posters or come to hear the oral presentations, I get inspired, because I get to see what other people are doing in their local microenvironments, in their laboratories, in their hospitals,” he said. “And often I think ... ‘We could be doing stuff just that exciting.’ Often, it’s a chance to collaborate with the people whose work is inspiring you, or to take a great idea and run with it.”
At the Clinical Vignettes competition, research will focus on lessons learned from specific cases, Dr. Cumbler said.
“A typical clinical vignette would be a case presentation, maybe a diagnostic image or a description of the test that clinched the diagnosis, and then, most importantly, the lessons from that case, which are more widely applicable,” he said. “One of the things I love about Clinical Vignettes is it gives you a chance to highlight your best catches, but it also lets you, with humility, share your misses so that other people can learn from your experience.”
He said he hopes the sharing of research in formal oral presentations – and in the poster hall – continues to advance the hospital medicine literature.
“It’s come an incredible distance over the last 10 years, 15 years,” he said. “When I look at where we are heading next, I think it is into more multicenter research, multiple-institution quality improvement. I really see us graduating from proof-of-concept and pilot work into the kind of trials which answer questions – the big questions that face medicine.”
Best of Research and Innovations in 2018
8-9 a.m., Palms Ballroom
Clinical Vignettes #2
Poster Competition
12-1:30 p.m., Cypress Ballroom
Genes, Cigarettes, and Blood Pressure: a Revealing Relationship
Using a technique called “gene-environment interaction analysis,” National Institutes of Health researchers zeroed in on areas of the genome associated with blood pressure. Cigarette smoking, known to raise blood pressure (BP), was an environmental “marker.”
The researchers tested different points of the genome in > 610,000 people from 5 ancestry groups to find where cigarette smoking and BP interacted.
They confirmed 56 known genetic regions and identified 83 new ones. Ten of the newly discovered genes seemed to have a much larger impact on smokers’ BP vs that of nonsmokers, the researchers say—levels were as much as 8 times higher.
Previous genetic studies have identified genes and genetic regions associated with BP but have not explored the interplay between genes and environmental factors, the researchers say. Moreover, the broad cohort of the new study makes the findings widely useful. Most of the known genetic regions linked to BP were identified through European descendants. In this study, several novel regions were identified through African ancestry analysis, “highlighting the importance of pursuing genetic studies in diverse populations.”
Using a technique called “gene-environment interaction analysis,” National Institutes of Health researchers zeroed in on areas of the genome associated with blood pressure. Cigarette smoking, known to raise blood pressure (BP), was an environmental “marker.”
The researchers tested different points of the genome in > 610,000 people from 5 ancestry groups to find where cigarette smoking and BP interacted.
They confirmed 56 known genetic regions and identified 83 new ones. Ten of the newly discovered genes seemed to have a much larger impact on smokers’ BP vs that of nonsmokers, the researchers say—levels were as much as 8 times higher.
Previous genetic studies have identified genes and genetic regions associated with BP but have not explored the interplay between genes and environmental factors, the researchers say. Moreover, the broad cohort of the new study makes the findings widely useful. Most of the known genetic regions linked to BP were identified through European descendants. In this study, several novel regions were identified through African ancestry analysis, “highlighting the importance of pursuing genetic studies in diverse populations.”
Using a technique called “gene-environment interaction analysis,” National Institutes of Health researchers zeroed in on areas of the genome associated with blood pressure. Cigarette smoking, known to raise blood pressure (BP), was an environmental “marker.”
The researchers tested different points of the genome in > 610,000 people from 5 ancestry groups to find where cigarette smoking and BP interacted.
They confirmed 56 known genetic regions and identified 83 new ones. Ten of the newly discovered genes seemed to have a much larger impact on smokers’ BP vs that of nonsmokers, the researchers say—levels were as much as 8 times higher.
Previous genetic studies have identified genes and genetic regions associated with BP but have not explored the interplay between genes and environmental factors, the researchers say. Moreover, the broad cohort of the new study makes the findings widely useful. Most of the known genetic regions linked to BP were identified through European descendants. In this study, several novel regions were identified through African ancestry analysis, “highlighting the importance of pursuing genetic studies in diverse populations.”
Health Canada approves product for adult ALL

Health Canada has approved inotuzumab ozogamicin (Besponsa™) as monotherapy for adults with relapsed or refractory, CD22-positive, B-cell precursor acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin is the first and only CD22-directed antibody-drug conjugate approved for this indication.
The product consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
Health Canada’s approval of inotuzumab ozogamicin is based on results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell precursor ALL.
Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens—high-dose cytarabine; cytarabine plus mitoxantrone; or fludarabine, cytarabine, and granulocyte colony-stimulating factor.
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were deemed related to chemotherapy.
Health Canada has approved inotuzumab ozogamicin (Besponsa™) as monotherapy for adults with relapsed or refractory, CD22-positive, B-cell precursor acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin is the first and only CD22-directed antibody-drug conjugate approved for this indication.
The product consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
Health Canada’s approval of inotuzumab ozogamicin is based on results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell precursor ALL.
Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens—high-dose cytarabine; cytarabine plus mitoxantrone; or fludarabine, cytarabine, and granulocyte colony-stimulating factor.
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were deemed related to chemotherapy.
Health Canada has approved inotuzumab ozogamicin (Besponsa™) as monotherapy for adults with relapsed or refractory, CD22-positive, B-cell precursor acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin is the first and only CD22-directed antibody-drug conjugate approved for this indication.
The product consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
Health Canada’s approval of inotuzumab ozogamicin is based on results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell precursor ALL.
Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens—high-dose cytarabine; cytarabine plus mitoxantrone; or fludarabine, cytarabine, and granulocyte colony-stimulating factor.
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were deemed related to chemotherapy.
Combo improves outcomes in MCL
A 2-drug combination can improve outcomes in patients with mantle cell lymphoma (MCL), according to researchers.
In a phase 2 trial of MCL patients, the BTK inhibitor ibrutinib and the BCL2 inhibitor venetoclax produced an overall response rate of 71% and a complete response (CR) rate of 62%.
“This was in patients who we expected to have a poor outcome on conventional therapy, and in which treatment with either ibrutinib or venetoclax alone was expected to see only 21% of patients show a complete response,” said Constantine Tam, MBBS, MD, of the Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
The most common adverse events (AEs) in patients receiving venetoclax and ibrutinib were diarrhea (83%), fatigue (75%), and nausea/vomiting (71%). Fourteen patients (58%) had serious AEs, including 2 with tumor lysis syndrome.
Dr Tam and his colleagues reported these results in NEJM.
The study included 24 patients—23 with relapsed/refractory MCL and 1 with previously untreated MCL. They had a median age of 68 (range, 47-81).
Most patients had high-risk (75%) or intermediate-risk (21%) disease, according to the MCL International Prognostic Index. Half of patients (including the previously untreated patient) had a TP53 aberration, and 25% had an NF-κB pathway mutation.
The relapsed/refractory patients had a median of 2 prior therapies (range, 1-6), and 48% were refractory to their most recent therapy.
Patients received ibrutinib monotherapy at 560 mg daily for 4 weeks. Then, patients began receiving venetoclax as well, in increasing doses, up to 400 mg per day. Patients received treatment until progression or unacceptable toxicity.
Efficacy
The study’s primary endpoint was CR at week 16, as assessed without PET. This was to allow the researchers to compare CR results in this trial to CR results in the PCYC-1104-CA study, a phase 2 trial of ibrutinib monotherapy in MCL.
According to CT, the CR rate was 42% in patients who received venetoclax and ibrutinib. This is significantly higher than the 9% CR rate observed in the patients treated with ibrutinib alone (P<0.001).
However, according to PET/CT, the 16-week CR rate was 62% in patients who received venetoclax and ibrutinib, and the overall response rate was 71%.
Overall, the rate of minimal residual disease (MRD) negativity was 67% (n=16) in the bone marrow according to flow cytometry and 38% (n=9) in the blood according to allele-specific oligonucleotide-polymerase chain reaction (ASO-PCR). However, not all patients were evaluable for MRD.
Among evaluable patients with a CR, 93% (14/15) were MRD negative according to flow cytometry, and 82% (9/11) were negative according to ASO-PCR.
The median progression-free survival was not reached at a median follow-up of 15.9 months. The estimated progression-free survival was 75% at 12 months and 57% at 18 months.
The rate of overall survival was 79% at 12 months and 74% at 18 months.
“These very promising results have triggered additional and larger studies to better understand the synergistic benefits of the venetoclax-ibrutinib treatment combination in MCL patients,” Dr Tam said.
Safety
The most common AEs were diarrhea (83%); fatigue (75%); nausea/vomiting (71%); bleeding, bruising, or postoperative hemorrhage (54%); musculoskeletal or connective-tissue pain (50%); cough or dyspnea (46%); soft-tissue infection (42%); upper respiratory tract infection (42%); neutropenia (33%); and lower respiratory tract infection (33%).
Grade 3/4 AEs included neutropenia (33%), thrombocytopenia (17%), anemia (12%), diarrhea (12%), tumor lysis syndrome (8%), atrial fibrillation (8%), lower respiratory tract infection (8%), soft-tissue infection (8%), cough or dyspnea (4%), musculoskeletal or connective-tissue pain (4%), and bleeding, bruising, or postoperative hemorrhage (4%).
Serious AEs included diarrhea (12%), tumor lysis syndrome (8%), atrial fibrillation (8%), pyrexia (8%), pleural effusion (8%), cardiac failure (4%), and soft-tissue infection (4%).
The patients who developed tumor lysis syndrome were among the first 15 patients who started venetoclax at a dose of 50 mg per day. Because of these cases, the study protocol was amended to lower the starting dose of venetoclax to 20 mg daily. After that, there were no additional cases of tumor lysis syndrome.
There were 6 deaths during the study. Four were attributed to disease progression, 1 to malignant otitis externa, and 1 to cardiac failure in a patient in CR.
A 2-drug combination can improve outcomes in patients with mantle cell lymphoma (MCL), according to researchers.
In a phase 2 trial of MCL patients, the BTK inhibitor ibrutinib and the BCL2 inhibitor venetoclax produced an overall response rate of 71% and a complete response (CR) rate of 62%.
“This was in patients who we expected to have a poor outcome on conventional therapy, and in which treatment with either ibrutinib or venetoclax alone was expected to see only 21% of patients show a complete response,” said Constantine Tam, MBBS, MD, of the Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
The most common adverse events (AEs) in patients receiving venetoclax and ibrutinib were diarrhea (83%), fatigue (75%), and nausea/vomiting (71%). Fourteen patients (58%) had serious AEs, including 2 with tumor lysis syndrome.
Dr Tam and his colleagues reported these results in NEJM.
The study included 24 patients—23 with relapsed/refractory MCL and 1 with previously untreated MCL. They had a median age of 68 (range, 47-81).
Most patients had high-risk (75%) or intermediate-risk (21%) disease, according to the MCL International Prognostic Index. Half of patients (including the previously untreated patient) had a TP53 aberration, and 25% had an NF-κB pathway mutation.
The relapsed/refractory patients had a median of 2 prior therapies (range, 1-6), and 48% were refractory to their most recent therapy.
Patients received ibrutinib monotherapy at 560 mg daily for 4 weeks. Then, patients began receiving venetoclax as well, in increasing doses, up to 400 mg per day. Patients received treatment until progression or unacceptable toxicity.
Efficacy
The study’s primary endpoint was CR at week 16, as assessed without PET. This was to allow the researchers to compare CR results in this trial to CR results in the PCYC-1104-CA study, a phase 2 trial of ibrutinib monotherapy in MCL.
According to CT, the CR rate was 42% in patients who received venetoclax and ibrutinib. This is significantly higher than the 9% CR rate observed in the patients treated with ibrutinib alone (P<0.001).
However, according to PET/CT, the 16-week CR rate was 62% in patients who received venetoclax and ibrutinib, and the overall response rate was 71%.
Overall, the rate of minimal residual disease (MRD) negativity was 67% (n=16) in the bone marrow according to flow cytometry and 38% (n=9) in the blood according to allele-specific oligonucleotide-polymerase chain reaction (ASO-PCR). However, not all patients were evaluable for MRD.
Among evaluable patients with a CR, 93% (14/15) were MRD negative according to flow cytometry, and 82% (9/11) were negative according to ASO-PCR.
The median progression-free survival was not reached at a median follow-up of 15.9 months. The estimated progression-free survival was 75% at 12 months and 57% at 18 months.
The rate of overall survival was 79% at 12 months and 74% at 18 months.
“These very promising results have triggered additional and larger studies to better understand the synergistic benefits of the venetoclax-ibrutinib treatment combination in MCL patients,” Dr Tam said.
Safety
The most common AEs were diarrhea (83%); fatigue (75%); nausea/vomiting (71%); bleeding, bruising, or postoperative hemorrhage (54%); musculoskeletal or connective-tissue pain (50%); cough or dyspnea (46%); soft-tissue infection (42%); upper respiratory tract infection (42%); neutropenia (33%); and lower respiratory tract infection (33%).
Grade 3/4 AEs included neutropenia (33%), thrombocytopenia (17%), anemia (12%), diarrhea (12%), tumor lysis syndrome (8%), atrial fibrillation (8%), lower respiratory tract infection (8%), soft-tissue infection (8%), cough or dyspnea (4%), musculoskeletal or connective-tissue pain (4%), and bleeding, bruising, or postoperative hemorrhage (4%).
Serious AEs included diarrhea (12%), tumor lysis syndrome (8%), atrial fibrillation (8%), pyrexia (8%), pleural effusion (8%), cardiac failure (4%), and soft-tissue infection (4%).
The patients who developed tumor lysis syndrome were among the first 15 patients who started venetoclax at a dose of 50 mg per day. Because of these cases, the study protocol was amended to lower the starting dose of venetoclax to 20 mg daily. After that, there were no additional cases of tumor lysis syndrome.
There were 6 deaths during the study. Four were attributed to disease progression, 1 to malignant otitis externa, and 1 to cardiac failure in a patient in CR.
A 2-drug combination can improve outcomes in patients with mantle cell lymphoma (MCL), according to researchers.
In a phase 2 trial of MCL patients, the BTK inhibitor ibrutinib and the BCL2 inhibitor venetoclax produced an overall response rate of 71% and a complete response (CR) rate of 62%.
“This was in patients who we expected to have a poor outcome on conventional therapy, and in which treatment with either ibrutinib or venetoclax alone was expected to see only 21% of patients show a complete response,” said Constantine Tam, MBBS, MD, of the Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
The most common adverse events (AEs) in patients receiving venetoclax and ibrutinib were diarrhea (83%), fatigue (75%), and nausea/vomiting (71%). Fourteen patients (58%) had serious AEs, including 2 with tumor lysis syndrome.
Dr Tam and his colleagues reported these results in NEJM.
The study included 24 patients—23 with relapsed/refractory MCL and 1 with previously untreated MCL. They had a median age of 68 (range, 47-81).
Most patients had high-risk (75%) or intermediate-risk (21%) disease, according to the MCL International Prognostic Index. Half of patients (including the previously untreated patient) had a TP53 aberration, and 25% had an NF-κB pathway mutation.
The relapsed/refractory patients had a median of 2 prior therapies (range, 1-6), and 48% were refractory to their most recent therapy.
Patients received ibrutinib monotherapy at 560 mg daily for 4 weeks. Then, patients began receiving venetoclax as well, in increasing doses, up to 400 mg per day. Patients received treatment until progression or unacceptable toxicity.
Efficacy
The study’s primary endpoint was CR at week 16, as assessed without PET. This was to allow the researchers to compare CR results in this trial to CR results in the PCYC-1104-CA study, a phase 2 trial of ibrutinib monotherapy in MCL.
According to CT, the CR rate was 42% in patients who received venetoclax and ibrutinib. This is significantly higher than the 9% CR rate observed in the patients treated with ibrutinib alone (P<0.001).
However, according to PET/CT, the 16-week CR rate was 62% in patients who received venetoclax and ibrutinib, and the overall response rate was 71%.
Overall, the rate of minimal residual disease (MRD) negativity was 67% (n=16) in the bone marrow according to flow cytometry and 38% (n=9) in the blood according to allele-specific oligonucleotide-polymerase chain reaction (ASO-PCR). However, not all patients were evaluable for MRD.
Among evaluable patients with a CR, 93% (14/15) were MRD negative according to flow cytometry, and 82% (9/11) were negative according to ASO-PCR.
The median progression-free survival was not reached at a median follow-up of 15.9 months. The estimated progression-free survival was 75% at 12 months and 57% at 18 months.
The rate of overall survival was 79% at 12 months and 74% at 18 months.
“These very promising results have triggered additional and larger studies to better understand the synergistic benefits of the venetoclax-ibrutinib treatment combination in MCL patients,” Dr Tam said.
Safety
The most common AEs were diarrhea (83%); fatigue (75%); nausea/vomiting (71%); bleeding, bruising, or postoperative hemorrhage (54%); musculoskeletal or connective-tissue pain (50%); cough or dyspnea (46%); soft-tissue infection (42%); upper respiratory tract infection (42%); neutropenia (33%); and lower respiratory tract infection (33%).
Grade 3/4 AEs included neutropenia (33%), thrombocytopenia (17%), anemia (12%), diarrhea (12%), tumor lysis syndrome (8%), atrial fibrillation (8%), lower respiratory tract infection (8%), soft-tissue infection (8%), cough or dyspnea (4%), musculoskeletal or connective-tissue pain (4%), and bleeding, bruising, or postoperative hemorrhage (4%).
Serious AEs included diarrhea (12%), tumor lysis syndrome (8%), atrial fibrillation (8%), pyrexia (8%), pleural effusion (8%), cardiac failure (4%), and soft-tissue infection (4%).
The patients who developed tumor lysis syndrome were among the first 15 patients who started venetoclax at a dose of 50 mg per day. Because of these cases, the study protocol was amended to lower the starting dose of venetoclax to 20 mg daily. After that, there were no additional cases of tumor lysis syndrome.
There were 6 deaths during the study. Four were attributed to disease progression, 1 to malignant otitis externa, and 1 to cardiac failure in a patient in CR.
Tenalisib receives orphan designation for CTCL

The US Food and Drug Administration (FDA) has granted orphan drug designation to tenalisib for treatment of cutaneous T-cell lymphoma (CTCL).
Tenalisib (formerly RP6530) is a dual PI3K delta/gamma inhibitor under development by Rhizen Pharmaceuticals SA.
The FDA previously granted tenalisib fast track and orphan drug designations for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib has been investigated in a phase 1 trial of patients with relapsed/refractory PTCL and CTCL. Results from this trial were presented at the 10th Annual T-cell Lymphoma Forum in February.
The data included 55 patients—28 with CTCL and 27 with PTCL—who received varying doses of tenalisib. The maximum tolerated dose was an 800 mg daily fasting dose.
Fourteen PTCL patients were evaluable for efficacy, and 7 responded (50%) to treatment. Three patients had a complete response, and 4 had a partial response.
Eighteen CTCL patients were evaluable for efficacy. Eight patients responded (44%), all with partial responses.
In the entire cohort, treatment-related adverse events (AEs) of grade 3 or higher included transaminitis (20%), rash (5%), neutropenia (2%), hypophosphatemia (2%), international normalized ratio increase (2%), sepsis (2%), pyrexia (2%), and diplopia secondary to neuropathy (2%).
Four CTCL patients stopped tenalisib due to a treatment-related AE—transaminitis, sepsis, diarrhea, and diplopia secondary to neuropathy. One PTCL patient stopped treatment due to a related AE, which was transaminitis.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available.
The US Food and Drug Administration (FDA) has granted orphan drug designation to tenalisib for treatment of cutaneous T-cell lymphoma (CTCL).
Tenalisib (formerly RP6530) is a dual PI3K delta/gamma inhibitor under development by Rhizen Pharmaceuticals SA.
The FDA previously granted tenalisib fast track and orphan drug designations for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib has been investigated in a phase 1 trial of patients with relapsed/refractory PTCL and CTCL. Results from this trial were presented at the 10th Annual T-cell Lymphoma Forum in February.
The data included 55 patients—28 with CTCL and 27 with PTCL—who received varying doses of tenalisib. The maximum tolerated dose was an 800 mg daily fasting dose.
Fourteen PTCL patients were evaluable for efficacy, and 7 responded (50%) to treatment. Three patients had a complete response, and 4 had a partial response.
Eighteen CTCL patients were evaluable for efficacy. Eight patients responded (44%), all with partial responses.
In the entire cohort, treatment-related adverse events (AEs) of grade 3 or higher included transaminitis (20%), rash (5%), neutropenia (2%), hypophosphatemia (2%), international normalized ratio increase (2%), sepsis (2%), pyrexia (2%), and diplopia secondary to neuropathy (2%).
Four CTCL patients stopped tenalisib due to a treatment-related AE—transaminitis, sepsis, diarrhea, and diplopia secondary to neuropathy. One PTCL patient stopped treatment due to a related AE, which was transaminitis.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available.
The US Food and Drug Administration (FDA) has granted orphan drug designation to tenalisib for treatment of cutaneous T-cell lymphoma (CTCL).
Tenalisib (formerly RP6530) is a dual PI3K delta/gamma inhibitor under development by Rhizen Pharmaceuticals SA.
The FDA previously granted tenalisib fast track and orphan drug designations for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib has been investigated in a phase 1 trial of patients with relapsed/refractory PTCL and CTCL. Results from this trial were presented at the 10th Annual T-cell Lymphoma Forum in February.
The data included 55 patients—28 with CTCL and 27 with PTCL—who received varying doses of tenalisib. The maximum tolerated dose was an 800 mg daily fasting dose.
Fourteen PTCL patients were evaluable for efficacy, and 7 responded (50%) to treatment. Three patients had a complete response, and 4 had a partial response.
Eighteen CTCL patients were evaluable for efficacy. Eight patients responded (44%), all with partial responses.
In the entire cohort, treatment-related adverse events (AEs) of grade 3 or higher included transaminitis (20%), rash (5%), neutropenia (2%), hypophosphatemia (2%), international normalized ratio increase (2%), sepsis (2%), pyrexia (2%), and diplopia secondary to neuropathy (2%).
Four CTCL patients stopped tenalisib due to a treatment-related AE—transaminitis, sepsis, diarrhea, and diplopia secondary to neuropathy. One PTCL patient stopped treatment due to a related AE, which was transaminitis.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available.
Duvelisib NDA granted priority review

The US Food and Drug Administration (FDA) has accepted for priority review the new drug application (NDA) for duvelisib, a dual PI3K delta/gamma inhibitor.
With this NDA, Verastem, Inc., is seeking full approval of duvelisib for the treatment of relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and accelerated approval of the drug for the treatment of relapsed or refractory follicular lymphoma (FL).
The FDA expects to make a decision on the NDA by October 5, 2018.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The application for duvelisib is supported by data from DUO™, a randomized, phase 3 study of patients with relapsed or refractory CLL/SLL, and DYNAMO™, a phase 2 study of patients with refractory indolent non-Hodgkin lymphoma.
Phase 3 DUO trial
Results from DUO were presented at the 2017 ASH Annual Meeting in December.
This study included 319 CLL/SLL patients who were randomized 1:1 to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2000 mg).
The study’s primary endpoint was met, as duvelisib conferred a significant improvement in median progression-free survival (PFS) over ofatumumab.
Per an independent review committee, the median PFS was 13.3 months with duvelisib and 9.9 months with ofatumumab (hazard ratio=0.52; P<0.0001). Duvelisib maintained a PFS advantage in all patient subgroups analyzed.
The overall response rate was 73.8% with duvelisib and 45.3% with ofatumumab (P<0.0001). The complete response rate was 0.6% in both arms.
Overall survival (OS) was similar in the duvelisib and ofatumumab arms (hazard ratio=0.99; P=0.4807). The median OS was not reached in either arm.
The most common grade 3 or higher adverse events (AEs)—in the duvelisib and ofatumumab arms, respectively—were neutropenia (30% vs 17%), anemia (13% vs 5%), diarrhea (15% vs 1%), pneumonia (14% vs 1%), and colitis (12% vs 1%).
Thirty-five percent of patients discontinued duvelisib due to an AE.
Severe opportunistic infections occurred in 6% of duvelisib recipients—bronchopulmonary aspergillosis (n=4), fungal infection (n=2), Pneumocystis jirovecii pneumonia (n=2), and cytomegalovirus colitis (n=1).
There were 4 deaths related to duvelisib—staphylococcal pneumonia (n=2), general physical health deterioration (n=1), and sepsis (n=1).
Phase 2 DYNAMO trial
Results from DYNAMO were presented at the 22nd EHA Congress (abstract S777) in June 2017.
This trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy.
There were 83 patients with FL. They had a median of 3 prior anticancer regimens (range, 1-10).
The patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate, per an independent review committee, was 43%. One patient achieved a complete response, and 35 had a partial response. The median duration of response was 7.9 months.
The median PFS was 8.3 months, and the median OS was 27.8 months.
The most common grade 3 or higher AEs were neutropenia (22%), anemia (13%), diarrhea (16%), lipase increase (10%), and thrombocytopenia (9%).
There were 2 serious opportunistic infections—Pneumocystis pneumonia and fungal pneumonia.
There were 3 deaths attributed to duvelisib—toxic epidermal necrolysis/sepsis syndrome (n=1), drug reaction/eosinophilia/systemic symptoms (n=1), and pneumonitis/pneumonia (n=1).
The US Food and Drug Administration (FDA) has accepted for priority review the new drug application (NDA) for duvelisib, a dual PI3K delta/gamma inhibitor.
With this NDA, Verastem, Inc., is seeking full approval of duvelisib for the treatment of relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and accelerated approval of the drug for the treatment of relapsed or refractory follicular lymphoma (FL).
The FDA expects to make a decision on the NDA by October 5, 2018.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The application for duvelisib is supported by data from DUO™, a randomized, phase 3 study of patients with relapsed or refractory CLL/SLL, and DYNAMO™, a phase 2 study of patients with refractory indolent non-Hodgkin lymphoma.
Phase 3 DUO trial
Results from DUO were presented at the 2017 ASH Annual Meeting in December.
This study included 319 CLL/SLL patients who were randomized 1:1 to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2000 mg).
The study’s primary endpoint was met, as duvelisib conferred a significant improvement in median progression-free survival (PFS) over ofatumumab.
Per an independent review committee, the median PFS was 13.3 months with duvelisib and 9.9 months with ofatumumab (hazard ratio=0.52; P<0.0001). Duvelisib maintained a PFS advantage in all patient subgroups analyzed.
The overall response rate was 73.8% with duvelisib and 45.3% with ofatumumab (P<0.0001). The complete response rate was 0.6% in both arms.
Overall survival (OS) was similar in the duvelisib and ofatumumab arms (hazard ratio=0.99; P=0.4807). The median OS was not reached in either arm.
The most common grade 3 or higher adverse events (AEs)—in the duvelisib and ofatumumab arms, respectively—were neutropenia (30% vs 17%), anemia (13% vs 5%), diarrhea (15% vs 1%), pneumonia (14% vs 1%), and colitis (12% vs 1%).
Thirty-five percent of patients discontinued duvelisib due to an AE.
Severe opportunistic infections occurred in 6% of duvelisib recipients—bronchopulmonary aspergillosis (n=4), fungal infection (n=2), Pneumocystis jirovecii pneumonia (n=2), and cytomegalovirus colitis (n=1).
There were 4 deaths related to duvelisib—staphylococcal pneumonia (n=2), general physical health deterioration (n=1), and sepsis (n=1).
Phase 2 DYNAMO trial
Results from DYNAMO were presented at the 22nd EHA Congress (abstract S777) in June 2017.
This trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy.
There were 83 patients with FL. They had a median of 3 prior anticancer regimens (range, 1-10).
The patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate, per an independent review committee, was 43%. One patient achieved a complete response, and 35 had a partial response. The median duration of response was 7.9 months.
The median PFS was 8.3 months, and the median OS was 27.8 months.
The most common grade 3 or higher AEs were neutropenia (22%), anemia (13%), diarrhea (16%), lipase increase (10%), and thrombocytopenia (9%).
There were 2 serious opportunistic infections—Pneumocystis pneumonia and fungal pneumonia.
There were 3 deaths attributed to duvelisib—toxic epidermal necrolysis/sepsis syndrome (n=1), drug reaction/eosinophilia/systemic symptoms (n=1), and pneumonitis/pneumonia (n=1).
The US Food and Drug Administration (FDA) has accepted for priority review the new drug application (NDA) for duvelisib, a dual PI3K delta/gamma inhibitor.
With this NDA, Verastem, Inc., is seeking full approval of duvelisib for the treatment of relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and accelerated approval of the drug for the treatment of relapsed or refractory follicular lymphoma (FL).
The FDA expects to make a decision on the NDA by October 5, 2018.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The application for duvelisib is supported by data from DUO™, a randomized, phase 3 study of patients with relapsed or refractory CLL/SLL, and DYNAMO™, a phase 2 study of patients with refractory indolent non-Hodgkin lymphoma.
Phase 3 DUO trial
Results from DUO were presented at the 2017 ASH Annual Meeting in December.
This study included 319 CLL/SLL patients who were randomized 1:1 to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2000 mg).
The study’s primary endpoint was met, as duvelisib conferred a significant improvement in median progression-free survival (PFS) over ofatumumab.
Per an independent review committee, the median PFS was 13.3 months with duvelisib and 9.9 months with ofatumumab (hazard ratio=0.52; P<0.0001). Duvelisib maintained a PFS advantage in all patient subgroups analyzed.
The overall response rate was 73.8% with duvelisib and 45.3% with ofatumumab (P<0.0001). The complete response rate was 0.6% in both arms.
Overall survival (OS) was similar in the duvelisib and ofatumumab arms (hazard ratio=0.99; P=0.4807). The median OS was not reached in either arm.
The most common grade 3 or higher adverse events (AEs)—in the duvelisib and ofatumumab arms, respectively—were neutropenia (30% vs 17%), anemia (13% vs 5%), diarrhea (15% vs 1%), pneumonia (14% vs 1%), and colitis (12% vs 1%).
Thirty-five percent of patients discontinued duvelisib due to an AE.
Severe opportunistic infections occurred in 6% of duvelisib recipients—bronchopulmonary aspergillosis (n=4), fungal infection (n=2), Pneumocystis jirovecii pneumonia (n=2), and cytomegalovirus colitis (n=1).
There were 4 deaths related to duvelisib—staphylococcal pneumonia (n=2), general physical health deterioration (n=1), and sepsis (n=1).
Phase 2 DYNAMO trial
Results from DYNAMO were presented at the 22nd EHA Congress (abstract S777) in June 2017.
This trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy.
There were 83 patients with FL. They had a median of 3 prior anticancer regimens (range, 1-10).
The patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate, per an independent review committee, was 43%. One patient achieved a complete response, and 35 had a partial response. The median duration of response was 7.9 months.
The median PFS was 8.3 months, and the median OS was 27.8 months.
The most common grade 3 or higher AEs were neutropenia (22%), anemia (13%), diarrhea (16%), lipase increase (10%), and thrombocytopenia (9%).
There were 2 serious opportunistic infections—Pneumocystis pneumonia and fungal pneumonia.
There were 3 deaths attributed to duvelisib—toxic epidermal necrolysis/sepsis syndrome (n=1), drug reaction/eosinophilia/systemic symptoms (n=1), and pneumonitis/pneumonia (n=1).
Alzheimer’s: Biomarkers, not cognition, will now define disorder
A new definition of Alzheimer’s disease based solely on biomarkers has the potential to strengthen clinical trials and change the way physicians talk to patients.
AB is the key to this classification paradigm – any patient with it (A+) is on the Alzheimer’s continuum. But only those with both amyloid and tau in the brain (A+T+) receive the “Alzheimer’s disease” classification. A third biomarker, neurodegeneration, may be either present or absent for an Alzheimer’s disease profile (N+ or N-). Cognitive staging adds important details, but remains secondary to the biomarker classification.

Jointly created by National Institute on Aging and the Alzheimer’s Association, the system – dubbed the NIA-AA Research Framework – represents a new, common language that researchers around the world may now use to generate and test Alzheimer’s hypotheses, and to optimize both epidemiologic studies and interventional trials. It will be especially important as Alzheimer’s prevention trials seek to target patients who are cognitively normal, yet harbor the neuropathological hallmarks of the disease.
This recasting adds Alzheimer’s to the list of biomarker-defined disorders, including hypertension, diabetes, and hyperlipidemia. It is a timely and necessary reframing, said Clifford Jack, MD, chair of the 20-member committee that created the paradigm. It appears in the April 10 issue of Alzheimer’s & Dementia.
“This is a fundamental change in the definition of Alzheimer’s disease,” Dr. Jack said in an interview. “We are advocating the disease be defined by its neuropathology [of plaques and tangles], which is specific to Alzheimer’s, and no longer by clinical symptoms which are not specific for any disease.”
One of the primary intents is to refine AD research cohorts, allowing pure stratification of patients who actually have the intended therapeutic targets of amyloid beta or tau. Without biomarker screening, up to 30% of subjects who enroll in AD drug trials don’t have the target pathologies – a situation researchers say contributes to the long string of failed Alzheimer’s drug studies.
For now, the system is intended only for research settings said Dr. Jack, an Alzheimer’s investigator at the Mayo Clinic, Rochester, Minn. But as biomarker testing comes of age and new less-expensive markers are discovered, the paradigm will likely be incorporated into clinical practice. The process can begin even now with a simple change in the way doctors talk to patients about Alzheimer’s, he said in an interview.
“We advocate people stop using the terms ‘probable or possible AD.’ A better term is ‘Alzheimer’s clinical syndrome.’ Without biomarkers, the clinical syndrome is the only thing you can know. What you can’t know is whether they do or don’t have Alzheimer’s disease. When I’m asked by physicians, ‘What do I tell my patients now?’ my very direct answer is ‘Tell them the truth.’ And the truth is that they have Alzheimer’s clinical syndrome and may or may not have Alzheimer’s disease.”
A reflection of evolving science
The research framework reflects advances in Alzheimer’s science that have occurred since the NIA last updated it AD diagnostic criteria in 2011. Those criteria divided the disease continuum into three phases largely based on cognitive symptoms, but were the first to recognize a presymptomatic AD phase.
- Preclinical: Brain changes, including amyloid buildup and other nerve cell changes already may be in progress but significant clinical symptoms are not yet evident.
- Mild cognitive impairment (MCI): A stage marked by symptoms of memory and/or other thinking problems that are greater than normal for a person’s age and education but that do not interfere with his or her independence. MCI may or may not progress to Alzheimer’s dementia.
- Alzheimer’s dementia: The final stage of the disease in which the symptoms of Alzheimer’s, such as memory loss, word-finding difficulties, and visual/spatial problems, are significant enough to impair a person’s ability to function independently.
The next 6 years brought striking advances in understanding the biology and pathology of AD, as well as technical advances in biomarker measurements. It became possible not only to measure AB and tau in cerebrospinal fluid but also to see these proteins in living brains with specialized PET ligands. It also became obvious that about a third of subjects in any given AD study didn’t have the disease-defining brain plaques and tangles – the therapeutic targets of all the largest drug studies to date. And while it’s clear that none of the interventions that have been through trials have exerted a significant benefit yet, “Treating people for a disease they don’t have can’t possibly help the results,” Dr. Jack said.
These research observations and revolutionary biomarker advances have reshaped the way researchers think about AD. To maximize research potential and to create a global classification standard that would unify studies as well, NIA and the Alzheimer’s Association convened several meetings to redefine Alzheimer’s disease biologically, by pathologic brain changes as measured by biomarkers. In this paradigm, cognitive dysfunction steps aside as the primary classification driver, becoming a symptom of AD rather than its definition.
“The way AD has historically been defined is by clinical symptoms: a progressive amnestic dementia was Alzheimer’s, and if there was no progressive amnestic dementia, it wasn’t,” Dr. Jack said. “Well, it turns out that both of those statements are wrong. About 30% of people with progressive amnestic dementia have other things causing it.”
It makes much more sense, he said, to define the disease based on its unique neuropathologic signature: amyloid beta plaques and tau neurofibrillary tangles in the brain.
The three-part key: A/T(N)
The NIA-AA research framework yields eight biomarker profiles with different combinations of amyloid (A), tau (T), and neuropathologic damage (N).
“Different measures have different roles,” Dr. Jack and his colleagues wrote in Alzheimer’s & Dementia. “Amyloid beta biomarkers determine whether or not an individual is in the Alzheimer’s continuum. Pathologic tau biomarkers determine if someone who is in the Alzheimer’s continuum has AD, because both amyloid beta and tau are required for a neuropathologic diagnosis of the disease. Neurodegenerative/neuronal injury biomarkers and cognitive symptoms, neither of which is specific for AD, are used only to stage severity not to define the presence of the Alzheimer’s continuum.”
The “N” category is not as cut and dried at the other biomarkers, the paper noted.
“Biomarkers in the (N) group are indicators of neurodegeneration or neuronal injury resulting from many causes; they are not specific for neurodegeneration due to AD. In any individual, the proportion of observed neurodegeneration/injury that can be attributed to AD versus other possible comorbid conditions (most of which have no extant biomarker) is unknown.”
The biomarker profiles are:
- A-T-(N): Normal AD biomarkers
- A+T-(N): Alzheimer’s pathologic change; Alzheimer’s continuum
- A+T+(N): Alzheimer’s disease; Alzheimer’s continuum
- A+T-(N)+: Alzheimer’s with suspected non Alzheimer’s pathologic change; Alzheimer’s continuum
- A-T+(N)-: Non-AD pathologic change
- A-T-(N)+: Non-AD pathologic change
- A-T+(N)+: Non-AD pathologic change
“This latter biomarker profile implies evidence of one or more neuropathologic processes other than AD and has been labeled ‘suspected non-Alzheimer’s pathophysiology, or SNAP,” according to the paper.
Cognitive staging further refines each person’s status. There are two clinical staging schemes in the framework. One is the familiar syndromal staging system of cognitively unimpaired, MCI, and dementia, which can be subdivided into mild, moderate, and severe. This can be applied to anyone with a biomarker profile.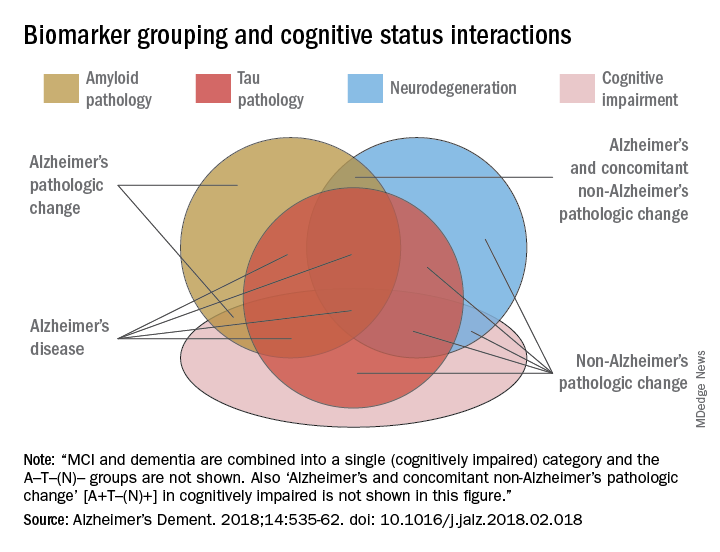
The second, a six-stage numerical clinical staging scheme, will apply only to those who are amyloid-positive and on the Alzheimer’s continuum. Stages run from 1 (unimpaired) to 6 (severe dementia). The numeric staging does not concentrate solely on cognition but also takes into account neurobehavioral and functional symptoms. It includes a transitional stage during which measures may be within population norms but have declined relative to the individual’s past performance.
The numeric staging scheme is intended to mesh with FDA guidance for clinical trials outcomes, the committee noted.
“A useful application envisioned for this numeric cognitive staging scheme is interventional trials. Indeed, the NIA-AA numeric staging scheme is intentionally very similar to the categorical system for staging AD outlined in recent FDA guidance for industry pertaining to developing drugs for treatment of early AD … it was our belief that harmonizing this aspect of the framework with FDA guidance would enhance cross fertilization between observational and interventional studies, which in turn would facilitate conduct of interventional clinical trials early in the disease process.”
The entire system yields a shorthand biomarker profile entirely unique to each subject. For example an A+T-(N)+ MCI profile suggests that both Alzheimer’s and non-Alzheimer’s pathologic change may be contributing to the cognitive impairment. A cognitive staging number could also be added.
This biomarker profile introduces the option of completely avoiding traditional AD nomenclature, the committee noted.
“Some investigators may prefer to not use the biomarker category terminology but instead simply report biomarker profile, i.e., A+T+(N)+ instead of ‘Alzheimer’s disease.’ An alternative is to combine the biomarker profile with a descriptive term – for example, ‘A+T+(N)+ with dementia’ instead of ‘Alzheimer’s disease with dementia’.”
Again, Dr. Jack cautioned, the paradigm is not intended for clinical use – at least not now. It relies entirely on biomarkers obtained by methods that are either invasive (lumbar puncture), unavailable outside research settings (tau scans), or very expensive when privately obtained (amyloid scans). Until this situation changes, the biomarker profile paradigm has little clinical impact.
IDEAS on the horizon
Change may be coming, however. The Alzheimer’s Association-sponsored Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study is assessing the clinical usefulness of amyloid PET scans and their impact on patient outcomes. The goal is to accumulate enough data to prove that amyloid scans are a cost-effective addition to the management of dementia patients. If federal payers agree and decide to cover amyloid scans, advocates hope that private insurers might follow suit.
An interim analysis of 4,000 scans, presented at the 2017 Alzheimer’s Association International Conference, was quite positive. Scan results changed patient management in 68% of cases, including refining dementia diagnoses, adding, stopping, or switching medications, and altering patient counseling.
IDEAS uses an FDA-approved amyloid imaging agent. But although several are under investigation, there are no approved tau PET ligands. However, other less-invasive and less-costly options may soon be developed, the committee noted. The search continues for a validated blood-based biomarker, including neurofilament light protein, plasma amyloid beta, and plasma tau.
“In the future, less-invasive/less-expensive blood-based biomarker tests - along with genetics, clinical, and demographic information - will likely play an important screening role in selecting individuals for more-expensive/more-invasive biomarker testing. This has been the history in other biologically defined diseases such as cardiovascular disease,” Dr. Jack and his colleagues noted in the paper.
In any case, however, without an effective treatment, much of the information conveyed by the biomarker profile paradigm remains, literally, academic, Dr. Jack said.
“If [the biomarker profile] were easy to determine and inexpensive, I imagine a lot of people would ask for it. Certainly many people would want to know, especially if they have a cognitive problem. People who have a family history, who may have Alzheimer’s pathology without the symptoms, might want to know. But the reality is that, until there’s a treatment that alters the course of this disease, finding out that you actually have Alzheimer’s is not going to enable you to change anything.”
The editors of Alzheimer’s & Dementia are seeking comment on the research framework. Letters and commentary can be submitted through June and will be considered for publication in an e-book, to be published sometime this summer, according to an accompanying editorial (https://doi.org/10/1016/j.jalz.2018.03.003).
Alzheimer’s & Dementia is the official journal of the Alzheimer’s Association. Dr. Jack has served on scientific advisory boards for Elan/Janssen AI, Bristol-Meyers Squibb, Eli Lilly, GE Healthcare, Siemens, and Eisai; received research support from Baxter International, Allon Therapeutics; and holds stock in Johnson & Johnson. Disclosures for other committee members can be found here.
SOURCE: Jack CR et al. Alzheimer’s Dement. 2018;14:535-62. doi: 10.1016/j.jalz.2018.02.018.
The biologically defined amyloid beta–tau–neuronal damage (ATN) framework is a logical and modern approach to Alzheimer’s disease (AD) diagnosis. It is hard to argue that more data are bad. Having such data on every patient would certainly be a luxury, but, with a few notable exceptions, the context in which this will most frequently occur is within the context of clinical trials.
While having this information does provide a biological basis for diagnosis, it does not account for non-AD contributions to the patient’s symptoms, which are found in more than half of all AD patients at autopsy; these non-AD pathologies also can influence clinical trial outcomes.

It also seems a bit ironic that the only meaningful manifestation of AD is now essentially left out of the diagnostic framework or relegated to nothing more than an adjective. Yet having a head full of amyloid means little if a person does not express symptoms (and vice versa), and we know that all people do not progress in the same way.
In the future, genomic and exposomic profiles may provide an even-more-nuanced picture, but further work is needed before that becomes a clinical reality. For now, the ATN biomarker framework represents the state of the art, though not an end.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale. He is also associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He has no relevant disclosures.
The biologically defined amyloid beta–tau–neuronal damage (ATN) framework is a logical and modern approach to Alzheimer’s disease (AD) diagnosis. It is hard to argue that more data are bad. Having such data on every patient would certainly be a luxury, but, with a few notable exceptions, the context in which this will most frequently occur is within the context of clinical trials.
While having this information does provide a biological basis for diagnosis, it does not account for non-AD contributions to the patient’s symptoms, which are found in more than half of all AD patients at autopsy; these non-AD pathologies also can influence clinical trial outcomes.
It also seems a bit ironic that the only meaningful manifestation of AD is now essentially left out of the diagnostic framework or relegated to nothing more than an adjective. Yet having a head full of amyloid means little if a person does not express symptoms (and vice versa), and we know that all people do not progress in the same way.
In the future, genomic and exposomic profiles may provide an even-more-nuanced picture, but further work is needed before that becomes a clinical reality. For now, the ATN biomarker framework represents the state of the art, though not an end.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale. He is also associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He has no relevant disclosures.
The biologically defined amyloid beta–tau–neuronal damage (ATN) framework is a logical and modern approach to Alzheimer’s disease (AD) diagnosis. It is hard to argue that more data are bad. Having such data on every patient would certainly be a luxury, but, with a few notable exceptions, the context in which this will most frequently occur is within the context of clinical trials.
While having this information does provide a biological basis for diagnosis, it does not account for non-AD contributions to the patient’s symptoms, which are found in more than half of all AD patients at autopsy; these non-AD pathologies also can influence clinical trial outcomes.
It also seems a bit ironic that the only meaningful manifestation of AD is now essentially left out of the diagnostic framework or relegated to nothing more than an adjective. Yet having a head full of amyloid means little if a person does not express symptoms (and vice versa), and we know that all people do not progress in the same way.
In the future, genomic and exposomic profiles may provide an even-more-nuanced picture, but further work is needed before that becomes a clinical reality. For now, the ATN biomarker framework represents the state of the art, though not an end.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale. He is also associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He has no relevant disclosures.
A new definition of Alzheimer’s disease based solely on biomarkers has the potential to strengthen clinical trials and change the way physicians talk to patients.
AB is the key to this classification paradigm – any patient with it (A+) is on the Alzheimer’s continuum. But only those with both amyloid and tau in the brain (A+T+) receive the “Alzheimer’s disease” classification. A third biomarker, neurodegeneration, may be either present or absent for an Alzheimer’s disease profile (N+ or N-). Cognitive staging adds important details, but remains secondary to the biomarker classification.
Jointly created by National Institute on Aging and the Alzheimer’s Association, the system – dubbed the NIA-AA Research Framework – represents a new, common language that researchers around the world may now use to generate and test Alzheimer’s hypotheses, and to optimize both epidemiologic studies and interventional trials. It will be especially important as Alzheimer’s prevention trials seek to target patients who are cognitively normal, yet harbor the neuropathological hallmarks of the disease.
This recasting adds Alzheimer’s to the list of biomarker-defined disorders, including hypertension, diabetes, and hyperlipidemia. It is a timely and necessary reframing, said Clifford Jack, MD, chair of the 20-member committee that created the paradigm. It appears in the April 10 issue of Alzheimer’s & Dementia.
“This is a fundamental change in the definition of Alzheimer’s disease,” Dr. Jack said in an interview. “We are advocating the disease be defined by its neuropathology [of plaques and tangles], which is specific to Alzheimer’s, and no longer by clinical symptoms which are not specific for any disease.”
One of the primary intents is to refine AD research cohorts, allowing pure stratification of patients who actually have the intended therapeutic targets of amyloid beta or tau. Without biomarker screening, up to 30% of subjects who enroll in AD drug trials don’t have the target pathologies – a situation researchers say contributes to the long string of failed Alzheimer’s drug studies.
For now, the system is intended only for research settings said Dr. Jack, an Alzheimer’s investigator at the Mayo Clinic, Rochester, Minn. But as biomarker testing comes of age and new less-expensive markers are discovered, the paradigm will likely be incorporated into clinical practice. The process can begin even now with a simple change in the way doctors talk to patients about Alzheimer’s, he said in an interview.
“We advocate people stop using the terms ‘probable or possible AD.’ A better term is ‘Alzheimer’s clinical syndrome.’ Without biomarkers, the clinical syndrome is the only thing you can know. What you can’t know is whether they do or don’t have Alzheimer’s disease. When I’m asked by physicians, ‘What do I tell my patients now?’ my very direct answer is ‘Tell them the truth.’ And the truth is that they have Alzheimer’s clinical syndrome and may or may not have Alzheimer’s disease.”
A reflection of evolving science
The research framework reflects advances in Alzheimer’s science that have occurred since the NIA last updated it AD diagnostic criteria in 2011. Those criteria divided the disease continuum into three phases largely based on cognitive symptoms, but were the first to recognize a presymptomatic AD phase.
- Preclinical: Brain changes, including amyloid buildup and other nerve cell changes already may be in progress but significant clinical symptoms are not yet evident.
- Mild cognitive impairment (MCI): A stage marked by symptoms of memory and/or other thinking problems that are greater than normal for a person’s age and education but that do not interfere with his or her independence. MCI may or may not progress to Alzheimer’s dementia.
- Alzheimer’s dementia: The final stage of the disease in which the symptoms of Alzheimer’s, such as memory loss, word-finding difficulties, and visual/spatial problems, are significant enough to impair a person’s ability to function independently.
The next 6 years brought striking advances in understanding the biology and pathology of AD, as well as technical advances in biomarker measurements. It became possible not only to measure AB and tau in cerebrospinal fluid but also to see these proteins in living brains with specialized PET ligands. It also became obvious that about a third of subjects in any given AD study didn’t have the disease-defining brain plaques and tangles – the therapeutic targets of all the largest drug studies to date. And while it’s clear that none of the interventions that have been through trials have exerted a significant benefit yet, “Treating people for a disease they don’t have can’t possibly help the results,” Dr. Jack said.
These research observations and revolutionary biomarker advances have reshaped the way researchers think about AD. To maximize research potential and to create a global classification standard that would unify studies as well, NIA and the Alzheimer’s Association convened several meetings to redefine Alzheimer’s disease biologically, by pathologic brain changes as measured by biomarkers. In this paradigm, cognitive dysfunction steps aside as the primary classification driver, becoming a symptom of AD rather than its definition.
“The way AD has historically been defined is by clinical symptoms: a progressive amnestic dementia was Alzheimer’s, and if there was no progressive amnestic dementia, it wasn’t,” Dr. Jack said. “Well, it turns out that both of those statements are wrong. About 30% of people with progressive amnestic dementia have other things causing it.”
It makes much more sense, he said, to define the disease based on its unique neuropathologic signature: amyloid beta plaques and tau neurofibrillary tangles in the brain.
The three-part key: A/T(N)
The NIA-AA research framework yields eight biomarker profiles with different combinations of amyloid (A), tau (T), and neuropathologic damage (N).
“Different measures have different roles,” Dr. Jack and his colleagues wrote in Alzheimer’s & Dementia. “Amyloid beta biomarkers determine whether or not an individual is in the Alzheimer’s continuum. Pathologic tau biomarkers determine if someone who is in the Alzheimer’s continuum has AD, because both amyloid beta and tau are required for a neuropathologic diagnosis of the disease. Neurodegenerative/neuronal injury biomarkers and cognitive symptoms, neither of which is specific for AD, are used only to stage severity not to define the presence of the Alzheimer’s continuum.”
The “N” category is not as cut and dried at the other biomarkers, the paper noted.
“Biomarkers in the (N) group are indicators of neurodegeneration or neuronal injury resulting from many causes; they are not specific for neurodegeneration due to AD. In any individual, the proportion of observed neurodegeneration/injury that can be attributed to AD versus other possible comorbid conditions (most of which have no extant biomarker) is unknown.”
The biomarker profiles are:
- A-T-(N): Normal AD biomarkers
- A+T-(N): Alzheimer’s pathologic change; Alzheimer’s continuum
- A+T+(N): Alzheimer’s disease; Alzheimer’s continuum
- A+T-(N)+: Alzheimer’s with suspected non Alzheimer’s pathologic change; Alzheimer’s continuum
- A-T+(N)-: Non-AD pathologic change
- A-T-(N)+: Non-AD pathologic change
- A-T+(N)+: Non-AD pathologic change
“This latter biomarker profile implies evidence of one or more neuropathologic processes other than AD and has been labeled ‘suspected non-Alzheimer’s pathophysiology, or SNAP,” according to the paper.
Cognitive staging further refines each person’s status. There are two clinical staging schemes in the framework. One is the familiar syndromal staging system of cognitively unimpaired, MCI, and dementia, which can be subdivided into mild, moderate, and severe. This can be applied to anyone with a biomarker profile.
The second, a six-stage numerical clinical staging scheme, will apply only to those who are amyloid-positive and on the Alzheimer’s continuum. Stages run from 1 (unimpaired) to 6 (severe dementia). The numeric staging does not concentrate solely on cognition but also takes into account neurobehavioral and functional symptoms. It includes a transitional stage during which measures may be within population norms but have declined relative to the individual’s past performance.
The numeric staging scheme is intended to mesh with FDA guidance for clinical trials outcomes, the committee noted.
“A useful application envisioned for this numeric cognitive staging scheme is interventional trials. Indeed, the NIA-AA numeric staging scheme is intentionally very similar to the categorical system for staging AD outlined in recent FDA guidance for industry pertaining to developing drugs for treatment of early AD … it was our belief that harmonizing this aspect of the framework with FDA guidance would enhance cross fertilization between observational and interventional studies, which in turn would facilitate conduct of interventional clinical trials early in the disease process.”
The entire system yields a shorthand biomarker profile entirely unique to each subject. For example an A+T-(N)+ MCI profile suggests that both Alzheimer’s and non-Alzheimer’s pathologic change may be contributing to the cognitive impairment. A cognitive staging number could also be added.
This biomarker profile introduces the option of completely avoiding traditional AD nomenclature, the committee noted.
“Some investigators may prefer to not use the biomarker category terminology but instead simply report biomarker profile, i.e., A+T+(N)+ instead of ‘Alzheimer’s disease.’ An alternative is to combine the biomarker profile with a descriptive term – for example, ‘A+T+(N)+ with dementia’ instead of ‘Alzheimer’s disease with dementia’.”
Again, Dr. Jack cautioned, the paradigm is not intended for clinical use – at least not now. It relies entirely on biomarkers obtained by methods that are either invasive (lumbar puncture), unavailable outside research settings (tau scans), or very expensive when privately obtained (amyloid scans). Until this situation changes, the biomarker profile paradigm has little clinical impact.
IDEAS on the horizon
Change may be coming, however. The Alzheimer’s Association-sponsored Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study is assessing the clinical usefulness of amyloid PET scans and their impact on patient outcomes. The goal is to accumulate enough data to prove that amyloid scans are a cost-effective addition to the management of dementia patients. If federal payers agree and decide to cover amyloid scans, advocates hope that private insurers might follow suit.
An interim analysis of 4,000 scans, presented at the 2017 Alzheimer’s Association International Conference, was quite positive. Scan results changed patient management in 68% of cases, including refining dementia diagnoses, adding, stopping, or switching medications, and altering patient counseling.
IDEAS uses an FDA-approved amyloid imaging agent. But although several are under investigation, there are no approved tau PET ligands. However, other less-invasive and less-costly options may soon be developed, the committee noted. The search continues for a validated blood-based biomarker, including neurofilament light protein, plasma amyloid beta, and plasma tau.
“In the future, less-invasive/less-expensive blood-based biomarker tests - along with genetics, clinical, and demographic information - will likely play an important screening role in selecting individuals for more-expensive/more-invasive biomarker testing. This has been the history in other biologically defined diseases such as cardiovascular disease,” Dr. Jack and his colleagues noted in the paper.
In any case, however, without an effective treatment, much of the information conveyed by the biomarker profile paradigm remains, literally, academic, Dr. Jack said.
“If [the biomarker profile] were easy to determine and inexpensive, I imagine a lot of people would ask for it. Certainly many people would want to know, especially if they have a cognitive problem. People who have a family history, who may have Alzheimer’s pathology without the symptoms, might want to know. But the reality is that, until there’s a treatment that alters the course of this disease, finding out that you actually have Alzheimer’s is not going to enable you to change anything.”
The editors of Alzheimer’s & Dementia are seeking comment on the research framework. Letters and commentary can be submitted through June and will be considered for publication in an e-book, to be published sometime this summer, according to an accompanying editorial (https://doi.org/10/1016/j.jalz.2018.03.003).
Alzheimer’s & Dementia is the official journal of the Alzheimer’s Association. Dr. Jack has served on scientific advisory boards for Elan/Janssen AI, Bristol-Meyers Squibb, Eli Lilly, GE Healthcare, Siemens, and Eisai; received research support from Baxter International, Allon Therapeutics; and holds stock in Johnson & Johnson. Disclosures for other committee members can be found here.
SOURCE: Jack CR et al. Alzheimer’s Dement. 2018;14:535-62. doi: 10.1016/j.jalz.2018.02.018.
A new definition of Alzheimer’s disease based solely on biomarkers has the potential to strengthen clinical trials and change the way physicians talk to patients.
AB is the key to this classification paradigm – any patient with it (A+) is on the Alzheimer’s continuum. But only those with both amyloid and tau in the brain (A+T+) receive the “Alzheimer’s disease” classification. A third biomarker, neurodegeneration, may be either present or absent for an Alzheimer’s disease profile (N+ or N-). Cognitive staging adds important details, but remains secondary to the biomarker classification.
Jointly created by National Institute on Aging and the Alzheimer’s Association, the system – dubbed the NIA-AA Research Framework – represents a new, common language that researchers around the world may now use to generate and test Alzheimer’s hypotheses, and to optimize both epidemiologic studies and interventional trials. It will be especially important as Alzheimer’s prevention trials seek to target patients who are cognitively normal, yet harbor the neuropathological hallmarks of the disease.
This recasting adds Alzheimer’s to the list of biomarker-defined disorders, including hypertension, diabetes, and hyperlipidemia. It is a timely and necessary reframing, said Clifford Jack, MD, chair of the 20-member committee that created the paradigm. It appears in the April 10 issue of Alzheimer’s & Dementia.
“This is a fundamental change in the definition of Alzheimer’s disease,” Dr. Jack said in an interview. “We are advocating the disease be defined by its neuropathology [of plaques and tangles], which is specific to Alzheimer’s, and no longer by clinical symptoms which are not specific for any disease.”
One of the primary intents is to refine AD research cohorts, allowing pure stratification of patients who actually have the intended therapeutic targets of amyloid beta or tau. Without biomarker screening, up to 30% of subjects who enroll in AD drug trials don’t have the target pathologies – a situation researchers say contributes to the long string of failed Alzheimer’s drug studies.
For now, the system is intended only for research settings said Dr. Jack, an Alzheimer’s investigator at the Mayo Clinic, Rochester, Minn. But as biomarker testing comes of age and new less-expensive markers are discovered, the paradigm will likely be incorporated into clinical practice. The process can begin even now with a simple change in the way doctors talk to patients about Alzheimer’s, he said in an interview.
“We advocate people stop using the terms ‘probable or possible AD.’ A better term is ‘Alzheimer’s clinical syndrome.’ Without biomarkers, the clinical syndrome is the only thing you can know. What you can’t know is whether they do or don’t have Alzheimer’s disease. When I’m asked by physicians, ‘What do I tell my patients now?’ my very direct answer is ‘Tell them the truth.’ And the truth is that they have Alzheimer’s clinical syndrome and may or may not have Alzheimer’s disease.”
A reflection of evolving science
The research framework reflects advances in Alzheimer’s science that have occurred since the NIA last updated it AD diagnostic criteria in 2011. Those criteria divided the disease continuum into three phases largely based on cognitive symptoms, but were the first to recognize a presymptomatic AD phase.
- Preclinical: Brain changes, including amyloid buildup and other nerve cell changes already may be in progress but significant clinical symptoms are not yet evident.
- Mild cognitive impairment (MCI): A stage marked by symptoms of memory and/or other thinking problems that are greater than normal for a person’s age and education but that do not interfere with his or her independence. MCI may or may not progress to Alzheimer’s dementia.
- Alzheimer’s dementia: The final stage of the disease in which the symptoms of Alzheimer’s, such as memory loss, word-finding difficulties, and visual/spatial problems, are significant enough to impair a person’s ability to function independently.
The next 6 years brought striking advances in understanding the biology and pathology of AD, as well as technical advances in biomarker measurements. It became possible not only to measure AB and tau in cerebrospinal fluid but also to see these proteins in living brains with specialized PET ligands. It also became obvious that about a third of subjects in any given AD study didn’t have the disease-defining brain plaques and tangles – the therapeutic targets of all the largest drug studies to date. And while it’s clear that none of the interventions that have been through trials have exerted a significant benefit yet, “Treating people for a disease they don’t have can’t possibly help the results,” Dr. Jack said.
These research observations and revolutionary biomarker advances have reshaped the way researchers think about AD. To maximize research potential and to create a global classification standard that would unify studies as well, NIA and the Alzheimer’s Association convened several meetings to redefine Alzheimer’s disease biologically, by pathologic brain changes as measured by biomarkers. In this paradigm, cognitive dysfunction steps aside as the primary classification driver, becoming a symptom of AD rather than its definition.
“The way AD has historically been defined is by clinical symptoms: a progressive amnestic dementia was Alzheimer’s, and if there was no progressive amnestic dementia, it wasn’t,” Dr. Jack said. “Well, it turns out that both of those statements are wrong. About 30% of people with progressive amnestic dementia have other things causing it.”
It makes much more sense, he said, to define the disease based on its unique neuropathologic signature: amyloid beta plaques and tau neurofibrillary tangles in the brain.
The three-part key: A/T(N)
The NIA-AA research framework yields eight biomarker profiles with different combinations of amyloid (A), tau (T), and neuropathologic damage (N).
“Different measures have different roles,” Dr. Jack and his colleagues wrote in Alzheimer’s & Dementia. “Amyloid beta biomarkers determine whether or not an individual is in the Alzheimer’s continuum. Pathologic tau biomarkers determine if someone who is in the Alzheimer’s continuum has AD, because both amyloid beta and tau are required for a neuropathologic diagnosis of the disease. Neurodegenerative/neuronal injury biomarkers and cognitive symptoms, neither of which is specific for AD, are used only to stage severity not to define the presence of the Alzheimer’s continuum.”
The “N” category is not as cut and dried at the other biomarkers, the paper noted.
“Biomarkers in the (N) group are indicators of neurodegeneration or neuronal injury resulting from many causes; they are not specific for neurodegeneration due to AD. In any individual, the proportion of observed neurodegeneration/injury that can be attributed to AD versus other possible comorbid conditions (most of which have no extant biomarker) is unknown.”
The biomarker profiles are:
- A-T-(N): Normal AD biomarkers
- A+T-(N): Alzheimer’s pathologic change; Alzheimer’s continuum
- A+T+(N): Alzheimer’s disease; Alzheimer’s continuum
- A+T-(N)+: Alzheimer’s with suspected non Alzheimer’s pathologic change; Alzheimer’s continuum
- A-T+(N)-: Non-AD pathologic change
- A-T-(N)+: Non-AD pathologic change
- A-T+(N)+: Non-AD pathologic change
“This latter biomarker profile implies evidence of one or more neuropathologic processes other than AD and has been labeled ‘suspected non-Alzheimer’s pathophysiology, or SNAP,” according to the paper.
Cognitive staging further refines each person’s status. There are two clinical staging schemes in the framework. One is the familiar syndromal staging system of cognitively unimpaired, MCI, and dementia, which can be subdivided into mild, moderate, and severe. This can be applied to anyone with a biomarker profile.
The second, a six-stage numerical clinical staging scheme, will apply only to those who are amyloid-positive and on the Alzheimer’s continuum. Stages run from 1 (unimpaired) to 6 (severe dementia). The numeric staging does not concentrate solely on cognition but also takes into account neurobehavioral and functional symptoms. It includes a transitional stage during which measures may be within population norms but have declined relative to the individual’s past performance.
The numeric staging scheme is intended to mesh with FDA guidance for clinical trials outcomes, the committee noted.
“A useful application envisioned for this numeric cognitive staging scheme is interventional trials. Indeed, the NIA-AA numeric staging scheme is intentionally very similar to the categorical system for staging AD outlined in recent FDA guidance for industry pertaining to developing drugs for treatment of early AD … it was our belief that harmonizing this aspect of the framework with FDA guidance would enhance cross fertilization between observational and interventional studies, which in turn would facilitate conduct of interventional clinical trials early in the disease process.”
The entire system yields a shorthand biomarker profile entirely unique to each subject. For example an A+T-(N)+ MCI profile suggests that both Alzheimer’s and non-Alzheimer’s pathologic change may be contributing to the cognitive impairment. A cognitive staging number could also be added.
This biomarker profile introduces the option of completely avoiding traditional AD nomenclature, the committee noted.
“Some investigators may prefer to not use the biomarker category terminology but instead simply report biomarker profile, i.e., A+T+(N)+ instead of ‘Alzheimer’s disease.’ An alternative is to combine the biomarker profile with a descriptive term – for example, ‘A+T+(N)+ with dementia’ instead of ‘Alzheimer’s disease with dementia’.”
Again, Dr. Jack cautioned, the paradigm is not intended for clinical use – at least not now. It relies entirely on biomarkers obtained by methods that are either invasive (lumbar puncture), unavailable outside research settings (tau scans), or very expensive when privately obtained (amyloid scans). Until this situation changes, the biomarker profile paradigm has little clinical impact.
IDEAS on the horizon
Change may be coming, however. The Alzheimer’s Association-sponsored Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study is assessing the clinical usefulness of amyloid PET scans and their impact on patient outcomes. The goal is to accumulate enough data to prove that amyloid scans are a cost-effective addition to the management of dementia patients. If federal payers agree and decide to cover amyloid scans, advocates hope that private insurers might follow suit.
An interim analysis of 4,000 scans, presented at the 2017 Alzheimer’s Association International Conference, was quite positive. Scan results changed patient management in 68% of cases, including refining dementia diagnoses, adding, stopping, or switching medications, and altering patient counseling.
IDEAS uses an FDA-approved amyloid imaging agent. But although several are under investigation, there are no approved tau PET ligands. However, other less-invasive and less-costly options may soon be developed, the committee noted. The search continues for a validated blood-based biomarker, including neurofilament light protein, plasma amyloid beta, and plasma tau.
“In the future, less-invasive/less-expensive blood-based biomarker tests - along with genetics, clinical, and demographic information - will likely play an important screening role in selecting individuals for more-expensive/more-invasive biomarker testing. This has been the history in other biologically defined diseases such as cardiovascular disease,” Dr. Jack and his colleagues noted in the paper.
In any case, however, without an effective treatment, much of the information conveyed by the biomarker profile paradigm remains, literally, academic, Dr. Jack said.
“If [the biomarker profile] were easy to determine and inexpensive, I imagine a lot of people would ask for it. Certainly many people would want to know, especially if they have a cognitive problem. People who have a family history, who may have Alzheimer’s pathology without the symptoms, might want to know. But the reality is that, until there’s a treatment that alters the course of this disease, finding out that you actually have Alzheimer’s is not going to enable you to change anything.”
The editors of Alzheimer’s & Dementia are seeking comment on the research framework. Letters and commentary can be submitted through June and will be considered for publication in an e-book, to be published sometime this summer, according to an accompanying editorial (https://doi.org/10/1016/j.jalz.2018.03.003).
Alzheimer’s & Dementia is the official journal of the Alzheimer’s Association. Dr. Jack has served on scientific advisory boards for Elan/Janssen AI, Bristol-Meyers Squibb, Eli Lilly, GE Healthcare, Siemens, and Eisai; received research support from Baxter International, Allon Therapeutics; and holds stock in Johnson & Johnson. Disclosures for other committee members can be found here.
SOURCE: Jack CR et al. Alzheimer’s Dement. 2018;14:535-62. doi: 10.1016/j.jalz.2018.02.018.
FROM ALZHEIMER’S & DEMENTIA
An Easy Approach to Obtaining Clean-catch Urine From Infants

A fussy 6-month-old infant is brought to the emergency department (ED) with a rectal temperature of 101.5°F. She is consolable,
A febrile infant in a family practice office or ED is a familiar clinical situation that may require an invasive diagnostic workup. Up to 7% of infants ages 2 to 24 months with fever of unknown origin may have a UTI.2 Collecting a urine sample from pre–toilet-trained children can be time consuming. In fact, in one RCT, obtaining a clean-catch urine sample in this age group took more than an hour, on average.3 But more convenient methods of urine collection, such as placing a cotton ball in the diaper or using a perineal collection bag, have contamination rates of up to 63%.4
In its guidelines for evaluating possible UTI in a febrile child younger than age 2, the American Academy of Pediatrics (AAP) recommends obtaining a sample for urinalysis “through the most convenient means.”5 If urinalysis is positive, only urine obtained by catheterization or suprapubic aspiration should be cultured. Guidelines from the National Institute for Health and Care Excellence in the United Kingdom are similar, but allow for culture of clean-catch urine samples.6
A recent prospective cohort study examined a noninvasive alternating lumbar-bladder tapping method to stimulate voiding in infants ages 6 months or younger.7 Within five minutes, 49% of the infants provided a clean-catch sample, with contamination rates similar to those of samples obtained using invasive methods.7 Younger infants were more likely to void within the time allotted. Another trial of bladder tapping conducted in hospitalized infants younger than 30 days old showed similar results.8 There are, however, no previously reported randomized trials demonstrating the efficacy of a noninvasive urine collection technique in the outpatient setting.
Use of invasive collection methods requires skilled personnel and may cause significant discomfort for patients (and parents). Noninvasive methods, such as bag urine collection, have unacceptable contamination rates. In addition, waiting to catch a potentially cleaner urine sample is time consuming, so better strategies to collect urine from infants are needed. This RCT is the first to examine the efficacy of a unique stimulation technique to obtain a clean-catch urine sample from infants ages 1 to 12 months.
STUDY SUMMARY
Noninvasive stimulation triggers faster samples
A nonblinded, single-center RCT conducted in Australia compared two methods for obtaining a clean-catch urine sample within five minutes: the Quick-Wee method (suprapubic stimulation with gauze soaked in cold fluid) or usual care (waiting for spontaneous voiding with no stimulation).1 A total of 354 infants (ages 1-12 mo) who required urine sample collection were randomized in a 1:1 ratio; allocation was concealed. Infants with anatomic or neurologic abnormalities and those needing immediate antibiotic therapy were excluded.
The most common reasons for obtaining the urine sample were fever of unknown origin and “unsettled baby,” followed by poor feeding and suspected UTI. The primary outcome was voiding within five minutes; secondary outcomes included time to void, whether urine was successfully caught, contamination rate, and parent/clinician satisfaction.
Study personnel removed the diaper, then cleaned the genitals of all patients with room temperature sterile water. A caregiver or clinician was ready and waiting to catch urine when the patient voided. In the Quick-Wee group, a clinician rubbed the patient’s suprapubic area in a circular fashion with gauze soaked in refrigerated saline (2.8°C). At five minutes, clinicians recorded the voiding status and decided how to proceed.
Using intention-to-treat analysis, 31% of the patients in the Quick-Wee group voided within five minutes, compared with 12% of the usual-care patients. Similarly, 30% of patients in the Quick-Wee group provided a successful clean-catch sample within five minutes, compared with 9% in the usual-care group (number needed to treat, 4.7).
Contamination rates were no different between the Quick-Wee and usual-care samples. Both parents and clinicians were more satisfied with the Quick-Wee method than with usual care (median score of 2 vs 3 on a 5-point Likert scale, in which 1 is “most satisfied”). There was no difference when results were adjusted for age or sex. No adverse events occurred.
Continue to: WHAT'S NEW
WHAT’S NEW
Method could reduce need for invasive sampling
A simple suprapubic stimulation technique increased the number of infants who provided a clean-catch voided urine sample within five minutes—a clinically relevant and satisfying outcome. In appropriate patients, use of the Quick-Wee method to obtain a clean-catch voided sample for initial urinalysis, rather than attempting methods with known high contamination rates, may potentially reduce the need for invasive sampling using catheterization or suprapubic aspiration.

CAVEATS
Complete age range & ideal storage temperature are unknown
Neonates and precontinent children older than 12 months were not included in this trial, so these conclusions do not apply to those groups. The intervention period lasted only five minutes, but other published studies suggest that this amount of time is adequate for voiding to occur.6,7 Although this study used soaking fluid stored at 2.8°C, the ideal storage temperature is unknown.
CHALLENGES TO IMPLEMENTATION
AAP doesn’t endorse clean-catch urine samples
The Quick-Wee method is simple and easy to implement, and requires no specialized training or equipment. AAP guidelines do not endorse the use of clean-catch voided urine for culture, which may be a barrier to changing urine collection practices in some settings.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018;67[3]: 166, 168-169).
1. Kaufman J, Fitzpatrick P, Tosif S, et al. Faster clean catch urine collection (Quick-Wee method) from infants: randomised controlled trial. BMJ. 2017;357:j1341.
2. Shaikh N, Morone NE, Bost JE, Farrell MH. Prevalence of urinary tract infection in childhood: a meta-analysis. Pediatr Infect Dis J. 2008;27(4):302-308.
3. Davies P, Greenwood R, Benger J. Randomised trial of a vibrating bladder stimulator—the time to pee study. Arch Dis Child. 2008;93(5):423-424.
4. Al-Orifi F, McGillivray D, Tange S, Kramer MS. Urine culture from bag specimens in young children: are the risks too high? J Pediatr. 2000;137(2):221-226.
5. Roberts KB, Downs SM, Finnell SM, et al; Subcommittee on Urinary Tract Infection. Reaffirmation of AAP clinical practice guideline: the diagnosis and management of the initial urinary tract infection in febrile infants and young children 2-24 months of age. Pediatrics. 2016;138(6): e20163026.
6. National Institute for Health and Care Excellence. Urinary tract infection in under 16s: diagnosis and management [clinical guideline CG54]. www.nice.org.uk/guidance/cg54/chapter/1-guidance. Accessed March 1, 2018.
7. Labrosse M, Levy A, Autmizguine J, Gravel J. Evaluation of a new strategy for clean-catch urine in infants. Pediatrics. 2016;138(3):e20160573.
8. Herreros Fernández ML, González Merino N, Tagarro García A, et al. A new technique for fast and safe collection of urine in newborns. Arch Dis Child. 2013;98(1):27-29.
A fussy 6-month-old infant is brought to the emergency department (ED) with a rectal temperature of 101.5°F. She is consolable,
A febrile infant in a family practice office or ED is a familiar clinical situation that may require an invasive diagnostic workup. Up to 7% of infants ages 2 to 24 months with fever of unknown origin may have a UTI.2 Collecting a urine sample from pre–toilet-trained children can be time consuming. In fact, in one RCT, obtaining a clean-catch urine sample in this age group took more than an hour, on average.3 But more convenient methods of urine collection, such as placing a cotton ball in the diaper or using a perineal collection bag, have contamination rates of up to 63%.4
In its guidelines for evaluating possible UTI in a febrile child younger than age 2, the American Academy of Pediatrics (AAP) recommends obtaining a sample for urinalysis “through the most convenient means.”5 If urinalysis is positive, only urine obtained by catheterization or suprapubic aspiration should be cultured. Guidelines from the National Institute for Health and Care Excellence in the United Kingdom are similar, but allow for culture of clean-catch urine samples.6
A recent prospective cohort study examined a noninvasive alternating lumbar-bladder tapping method to stimulate voiding in infants ages 6 months or younger.7 Within five minutes, 49% of the infants provided a clean-catch sample, with contamination rates similar to those of samples obtained using invasive methods.7 Younger infants were more likely to void within the time allotted. Another trial of bladder tapping conducted in hospitalized infants younger than 30 days old showed similar results.8 There are, however, no previously reported randomized trials demonstrating the efficacy of a noninvasive urine collection technique in the outpatient setting.
Use of invasive collection methods requires skilled personnel and may cause significant discomfort for patients (and parents). Noninvasive methods, such as bag urine collection, have unacceptable contamination rates. In addition, waiting to catch a potentially cleaner urine sample is time consuming, so better strategies to collect urine from infants are needed. This RCT is the first to examine the efficacy of a unique stimulation technique to obtain a clean-catch urine sample from infants ages 1 to 12 months.
STUDY SUMMARY
Noninvasive stimulation triggers faster samples
A nonblinded, single-center RCT conducted in Australia compared two methods for obtaining a clean-catch urine sample within five minutes: the Quick-Wee method (suprapubic stimulation with gauze soaked in cold fluid) or usual care (waiting for spontaneous voiding with no stimulation).1 A total of 354 infants (ages 1-12 mo) who required urine sample collection were randomized in a 1:1 ratio; allocation was concealed. Infants with anatomic or neurologic abnormalities and those needing immediate antibiotic therapy were excluded.
The most common reasons for obtaining the urine sample were fever of unknown origin and “unsettled baby,” followed by poor feeding and suspected UTI. The primary outcome was voiding within five minutes; secondary outcomes included time to void, whether urine was successfully caught, contamination rate, and parent/clinician satisfaction.
Study personnel removed the diaper, then cleaned the genitals of all patients with room temperature sterile water. A caregiver or clinician was ready and waiting to catch urine when the patient voided. In the Quick-Wee group, a clinician rubbed the patient’s suprapubic area in a circular fashion with gauze soaked in refrigerated saline (2.8°C). At five minutes, clinicians recorded the voiding status and decided how to proceed.
Using intention-to-treat analysis, 31% of the patients in the Quick-Wee group voided within five minutes, compared with 12% of the usual-care patients. Similarly, 30% of patients in the Quick-Wee group provided a successful clean-catch sample within five minutes, compared with 9% in the usual-care group (number needed to treat, 4.7).
Contamination rates were no different between the Quick-Wee and usual-care samples. Both parents and clinicians were more satisfied with the Quick-Wee method than with usual care (median score of 2 vs 3 on a 5-point Likert scale, in which 1 is “most satisfied”). There was no difference when results were adjusted for age or sex. No adverse events occurred.
Continue to: WHAT'S NEW
WHAT’S NEW
Method could reduce need for invasive sampling
A simple suprapubic stimulation technique increased the number of infants who provided a clean-catch voided urine sample within five minutes—a clinically relevant and satisfying outcome. In appropriate patients, use of the Quick-Wee method to obtain a clean-catch voided sample for initial urinalysis, rather than attempting methods with known high contamination rates, may potentially reduce the need for invasive sampling using catheterization or suprapubic aspiration.
CAVEATS
Complete age range & ideal storage temperature are unknown
Neonates and precontinent children older than 12 months were not included in this trial, so these conclusions do not apply to those groups. The intervention period lasted only five minutes, but other published studies suggest that this amount of time is adequate for voiding to occur.6,7 Although this study used soaking fluid stored at 2.8°C, the ideal storage temperature is unknown.
CHALLENGES TO IMPLEMENTATION
AAP doesn’t endorse clean-catch urine samples
The Quick-Wee method is simple and easy to implement, and requires no specialized training or equipment. AAP guidelines do not endorse the use of clean-catch voided urine for culture, which may be a barrier to changing urine collection practices in some settings.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018;67[3]: 166, 168-169).
A fussy 6-month-old infant is brought to the emergency department (ED) with a rectal temperature of 101.5°F. She is consolable,
A febrile infant in a family practice office or ED is a familiar clinical situation that may require an invasive diagnostic workup. Up to 7% of infants ages 2 to 24 months with fever of unknown origin may have a UTI.2 Collecting a urine sample from pre–toilet-trained children can be time consuming. In fact, in one RCT, obtaining a clean-catch urine sample in this age group took more than an hour, on average.3 But more convenient methods of urine collection, such as placing a cotton ball in the diaper or using a perineal collection bag, have contamination rates of up to 63%.4
In its guidelines for evaluating possible UTI in a febrile child younger than age 2, the American Academy of Pediatrics (AAP) recommends obtaining a sample for urinalysis “through the most convenient means.”5 If urinalysis is positive, only urine obtained by catheterization or suprapubic aspiration should be cultured. Guidelines from the National Institute for Health and Care Excellence in the United Kingdom are similar, but allow for culture of clean-catch urine samples.6
A recent prospective cohort study examined a noninvasive alternating lumbar-bladder tapping method to stimulate voiding in infants ages 6 months or younger.7 Within five minutes, 49% of the infants provided a clean-catch sample, with contamination rates similar to those of samples obtained using invasive methods.7 Younger infants were more likely to void within the time allotted. Another trial of bladder tapping conducted in hospitalized infants younger than 30 days old showed similar results.8 There are, however, no previously reported randomized trials demonstrating the efficacy of a noninvasive urine collection technique in the outpatient setting.
Use of invasive collection methods requires skilled personnel and may cause significant discomfort for patients (and parents). Noninvasive methods, such as bag urine collection, have unacceptable contamination rates. In addition, waiting to catch a potentially cleaner urine sample is time consuming, so better strategies to collect urine from infants are needed. This RCT is the first to examine the efficacy of a unique stimulation technique to obtain a clean-catch urine sample from infants ages 1 to 12 months.
STUDY SUMMARY
Noninvasive stimulation triggers faster samples
A nonblinded, single-center RCT conducted in Australia compared two methods for obtaining a clean-catch urine sample within five minutes: the Quick-Wee method (suprapubic stimulation with gauze soaked in cold fluid) or usual care (waiting for spontaneous voiding with no stimulation).1 A total of 354 infants (ages 1-12 mo) who required urine sample collection were randomized in a 1:1 ratio; allocation was concealed. Infants with anatomic or neurologic abnormalities and those needing immediate antibiotic therapy were excluded.
The most common reasons for obtaining the urine sample were fever of unknown origin and “unsettled baby,” followed by poor feeding and suspected UTI. The primary outcome was voiding within five minutes; secondary outcomes included time to void, whether urine was successfully caught, contamination rate, and parent/clinician satisfaction.
Study personnel removed the diaper, then cleaned the genitals of all patients with room temperature sterile water. A caregiver or clinician was ready and waiting to catch urine when the patient voided. In the Quick-Wee group, a clinician rubbed the patient’s suprapubic area in a circular fashion with gauze soaked in refrigerated saline (2.8°C). At five minutes, clinicians recorded the voiding status and decided how to proceed.
Using intention-to-treat analysis, 31% of the patients in the Quick-Wee group voided within five minutes, compared with 12% of the usual-care patients. Similarly, 30% of patients in the Quick-Wee group provided a successful clean-catch sample within five minutes, compared with 9% in the usual-care group (number needed to treat, 4.7).
Contamination rates were no different between the Quick-Wee and usual-care samples. Both parents and clinicians were more satisfied with the Quick-Wee method than with usual care (median score of 2 vs 3 on a 5-point Likert scale, in which 1 is “most satisfied”). There was no difference when results were adjusted for age or sex. No adverse events occurred.
Continue to: WHAT'S NEW
WHAT’S NEW
Method could reduce need for invasive sampling
A simple suprapubic stimulation technique increased the number of infants who provided a clean-catch voided urine sample within five minutes—a clinically relevant and satisfying outcome. In appropriate patients, use of the Quick-Wee method to obtain a clean-catch voided sample for initial urinalysis, rather than attempting methods with known high contamination rates, may potentially reduce the need for invasive sampling using catheterization or suprapubic aspiration.
CAVEATS
Complete age range & ideal storage temperature are unknown
Neonates and precontinent children older than 12 months were not included in this trial, so these conclusions do not apply to those groups. The intervention period lasted only five minutes, but other published studies suggest that this amount of time is adequate for voiding to occur.6,7 Although this study used soaking fluid stored at 2.8°C, the ideal storage temperature is unknown.
CHALLENGES TO IMPLEMENTATION
AAP doesn’t endorse clean-catch urine samples
The Quick-Wee method is simple and easy to implement, and requires no specialized training or equipment. AAP guidelines do not endorse the use of clean-catch voided urine for culture, which may be a barrier to changing urine collection practices in some settings.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018;67[3]: 166, 168-169).
1. Kaufman J, Fitzpatrick P, Tosif S, et al. Faster clean catch urine collection (Quick-Wee method) from infants: randomised controlled trial. BMJ. 2017;357:j1341.
2. Shaikh N, Morone NE, Bost JE, Farrell MH. Prevalence of urinary tract infection in childhood: a meta-analysis. Pediatr Infect Dis J. 2008;27(4):302-308.
3. Davies P, Greenwood R, Benger J. Randomised trial of a vibrating bladder stimulator—the time to pee study. Arch Dis Child. 2008;93(5):423-424.
4. Al-Orifi F, McGillivray D, Tange S, Kramer MS. Urine culture from bag specimens in young children: are the risks too high? J Pediatr. 2000;137(2):221-226.
5. Roberts KB, Downs SM, Finnell SM, et al; Subcommittee on Urinary Tract Infection. Reaffirmation of AAP clinical practice guideline: the diagnosis and management of the initial urinary tract infection in febrile infants and young children 2-24 months of age. Pediatrics. 2016;138(6): e20163026.
6. National Institute for Health and Care Excellence. Urinary tract infection in under 16s: diagnosis and management [clinical guideline CG54]. www.nice.org.uk/guidance/cg54/chapter/1-guidance. Accessed March 1, 2018.
7. Labrosse M, Levy A, Autmizguine J, Gravel J. Evaluation of a new strategy for clean-catch urine in infants. Pediatrics. 2016;138(3):e20160573.
8. Herreros Fernández ML, González Merino N, Tagarro García A, et al. A new technique for fast and safe collection of urine in newborns. Arch Dis Child. 2013;98(1):27-29.
1. Kaufman J, Fitzpatrick P, Tosif S, et al. Faster clean catch urine collection (Quick-Wee method) from infants: randomised controlled trial. BMJ. 2017;357:j1341.
2. Shaikh N, Morone NE, Bost JE, Farrell MH. Prevalence of urinary tract infection in childhood: a meta-analysis. Pediatr Infect Dis J. 2008;27(4):302-308.
3. Davies P, Greenwood R, Benger J. Randomised trial of a vibrating bladder stimulator—the time to pee study. Arch Dis Child. 2008;93(5):423-424.
4. Al-Orifi F, McGillivray D, Tange S, Kramer MS. Urine culture from bag specimens in young children: are the risks too high? J Pediatr. 2000;137(2):221-226.
5. Roberts KB, Downs SM, Finnell SM, et al; Subcommittee on Urinary Tract Infection. Reaffirmation of AAP clinical practice guideline: the diagnosis and management of the initial urinary tract infection in febrile infants and young children 2-24 months of age. Pediatrics. 2016;138(6): e20163026.
6. National Institute for Health and Care Excellence. Urinary tract infection in under 16s: diagnosis and management [clinical guideline CG54]. www.nice.org.uk/guidance/cg54/chapter/1-guidance. Accessed March 1, 2018.
7. Labrosse M, Levy A, Autmizguine J, Gravel J. Evaluation of a new strategy for clean-catch urine in infants. Pediatrics. 2016;138(3):e20160573.
8. Herreros Fernández ML, González Merino N, Tagarro García A, et al. A new technique for fast and safe collection of urine in newborns. Arch Dis Child. 2013;98(1):27-29.

Why do people act that way?
You can try all you want, but you never really figure people out.
Take Brendan. He spent a good few minutes chewing out my front desk staffer Linda. “You’re incompetent!” he shouted at her.
Linda tried to explain. “We can’t administer the Botox for your sweating today,” she said, “because it hasn’t come in yet.”
No, Brendan certainly didn’t want to hear that. Nor did he want to hear that we had called in advance to ask him to actually wait until we let him know that the Botox was in. But here he was, hyperinflating his lungs.
“You’re just damned useless!” he explained.
OK, we all have unreasonable patients from time to time. But what I’m describing is behavior that’s not just aggressive and unpleasant but incomprehensible. Would you like to know why I call it incomprehensible?
Because Brendan has been coming in for axillary Botox injections every 6 months for 5 years! He knows the drill. He knows the staff. He’s always pleasant as punch. Just not today. Why? Who knows?
What’s true for patients can of course also be true for employees. Model workers, superb team players, reliable folks who show up in snowstorms, who come right back to work after major surgery, who deliver heartfelt speeches at holiday parties about their good luck in overcoming adversity to be able to work. Contributors who share their pleasant disposition and can-do energy year after year ...
Until one day that they don’t show up, send a text to say they quit, no warning or explanation, then apply for Workmen’s Compensation, and at the magistrate’s hearing to which they’ve dragged my staff and our HR attorney, lie right to the face of the manager who was their best friend and confidante until the day before yesterday.
Get it? I don’t. Never will.
Two pieces of advice: 1) Don’t ever take things like that personally; and 2) Hire a good human resources attorney.
**********************
On the other hand ...
Jeralyn is 27 years old. She lists her chief complaint as “dark spots on my back.”
“How did you notice these?” I ask. “Did you see them, or did you doctor point them out?”
“It was when I tried on my wedding dress,” she says.
I understand, or think I do. Her dress exposed her back, her mother noticed the spots ...
“When is the wedding?” I ask.
“The wedding? Oh, the wedding already was. A year and a half ago.”
“You saw the spots when you tried on your wedding dress, and you’re coming in a year and half later?” I ask, trying not to sound incredulous. I’ve met enough nervous brides – and brides’ mothers – to find her account astonishing. “You must be a very patient person,” I say.
“Well,” says Jeralyn, “the spots didn’t spread or get any worse. And I am patient. I teach kindergarten. You have to be patient with little ones.”
“If you decide to have children, they’ll benefit from that,” I reply.
“That’s actually why I’m here,” says Jeralyn. “My husband and I want to start a family, and we want to be sure my skin condition wouldn’t affect that.”
A bride so unconcerned with herself that she wears a wedding dress that reveals a rash she doesn’t go running to a doctor to fix? Who comes only when the rash might affect someone else?
What is the matter with Jeralyn? Doesn’t she know she’s a millennial?
**********************
Kidding aside, wouldn’t it be nice if everyone acted like Jeralyn, and acted in a measured manner, appropriate to the concerns and circumstances? Wouldn’t it be nice if no one acted like Brendan?
That’s not how it is, though, is it? As professionals who deal with the public (as patients, coworkers, employees), we take all comers and roll with them: tolerate the annoyances and celebrate the (much larger, if less individually memorable) group who are pleasant, often delightful, sometimes inspiring.
And every once in a while, someone like Jeralyn comes along, upending all those negative expectations and reminding us that even if you can never really figure people out, it’s still worth the effort to keep trying.

Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
You can try all you want, but you never really figure people out.
Take Brendan. He spent a good few minutes chewing out my front desk staffer Linda. “You’re incompetent!” he shouted at her.
Linda tried to explain. “We can’t administer the Botox for your sweating today,” she said, “because it hasn’t come in yet.”
No, Brendan certainly didn’t want to hear that. Nor did he want to hear that we had called in advance to ask him to actually wait until we let him know that the Botox was in. But here he was, hyperinflating his lungs.
“You’re just damned useless!” he explained.
OK, we all have unreasonable patients from time to time. But what I’m describing is behavior that’s not just aggressive and unpleasant but incomprehensible. Would you like to know why I call it incomprehensible?
Because Brendan has been coming in for axillary Botox injections every 6 months for 5 years! He knows the drill. He knows the staff. He’s always pleasant as punch. Just not today. Why? Who knows?
What’s true for patients can of course also be true for employees. Model workers, superb team players, reliable folks who show up in snowstorms, who come right back to work after major surgery, who deliver heartfelt speeches at holiday parties about their good luck in overcoming adversity to be able to work. Contributors who share their pleasant disposition and can-do energy year after year ...
Until one day that they don’t show up, send a text to say they quit, no warning or explanation, then apply for Workmen’s Compensation, and at the magistrate’s hearing to which they’ve dragged my staff and our HR attorney, lie right to the face of the manager who was their best friend and confidante until the day before yesterday.
Get it? I don’t. Never will.
Two pieces of advice: 1) Don’t ever take things like that personally; and 2) Hire a good human resources attorney.
**********************
On the other hand ...
Jeralyn is 27 years old. She lists her chief complaint as “dark spots on my back.”
“How did you notice these?” I ask. “Did you see them, or did you doctor point them out?”
“It was when I tried on my wedding dress,” she says.
I understand, or think I do. Her dress exposed her back, her mother noticed the spots ...
“When is the wedding?” I ask.
“The wedding? Oh, the wedding already was. A year and a half ago.”
“You saw the spots when you tried on your wedding dress, and you’re coming in a year and half later?” I ask, trying not to sound incredulous. I’ve met enough nervous brides – and brides’ mothers – to find her account astonishing. “You must be a very patient person,” I say.
“Well,” says Jeralyn, “the spots didn’t spread or get any worse. And I am patient. I teach kindergarten. You have to be patient with little ones.”
“If you decide to have children, they’ll benefit from that,” I reply.
“That’s actually why I’m here,” says Jeralyn. “My husband and I want to start a family, and we want to be sure my skin condition wouldn’t affect that.”
A bride so unconcerned with herself that she wears a wedding dress that reveals a rash she doesn’t go running to a doctor to fix? Who comes only when the rash might affect someone else?
What is the matter with Jeralyn? Doesn’t she know she’s a millennial?
**********************
Kidding aside, wouldn’t it be nice if everyone acted like Jeralyn, and acted in a measured manner, appropriate to the concerns and circumstances? Wouldn’t it be nice if no one acted like Brendan?
That’s not how it is, though, is it? As professionals who deal with the public (as patients, coworkers, employees), we take all comers and roll with them: tolerate the annoyances and celebrate the (much larger, if less individually memorable) group who are pleasant, often delightful, sometimes inspiring.
And every once in a while, someone like Jeralyn comes along, upending all those negative expectations and reminding us that even if you can never really figure people out, it’s still worth the effort to keep trying.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
You can try all you want, but you never really figure people out.
Take Brendan. He spent a good few minutes chewing out my front desk staffer Linda. “You’re incompetent!” he shouted at her.
Linda tried to explain. “We can’t administer the Botox for your sweating today,” she said, “because it hasn’t come in yet.”
No, Brendan certainly didn’t want to hear that. Nor did he want to hear that we had called in advance to ask him to actually wait until we let him know that the Botox was in. But here he was, hyperinflating his lungs.
“You’re just damned useless!” he explained.
OK, we all have unreasonable patients from time to time. But what I’m describing is behavior that’s not just aggressive and unpleasant but incomprehensible. Would you like to know why I call it incomprehensible?
Because Brendan has been coming in for axillary Botox injections every 6 months for 5 years! He knows the drill. He knows the staff. He’s always pleasant as punch. Just not today. Why? Who knows?
What’s true for patients can of course also be true for employees. Model workers, superb team players, reliable folks who show up in snowstorms, who come right back to work after major surgery, who deliver heartfelt speeches at holiday parties about their good luck in overcoming adversity to be able to work. Contributors who share their pleasant disposition and can-do energy year after year ...
Until one day that they don’t show up, send a text to say they quit, no warning or explanation, then apply for Workmen’s Compensation, and at the magistrate’s hearing to which they’ve dragged my staff and our HR attorney, lie right to the face of the manager who was their best friend and confidante until the day before yesterday.
Get it? I don’t. Never will.
Two pieces of advice: 1) Don’t ever take things like that personally; and 2) Hire a good human resources attorney.
**********************
On the other hand ...
Jeralyn is 27 years old. She lists her chief complaint as “dark spots on my back.”
“How did you notice these?” I ask. “Did you see them, or did you doctor point them out?”
“It was when I tried on my wedding dress,” she says.
I understand, or think I do. Her dress exposed her back, her mother noticed the spots ...
“When is the wedding?” I ask.
“The wedding? Oh, the wedding already was. A year and a half ago.”
“You saw the spots when you tried on your wedding dress, and you’re coming in a year and half later?” I ask, trying not to sound incredulous. I’ve met enough nervous brides – and brides’ mothers – to find her account astonishing. “You must be a very patient person,” I say.
“Well,” says Jeralyn, “the spots didn’t spread or get any worse. And I am patient. I teach kindergarten. You have to be patient with little ones.”
“If you decide to have children, they’ll benefit from that,” I reply.
“That’s actually why I’m here,” says Jeralyn. “My husband and I want to start a family, and we want to be sure my skin condition wouldn’t affect that.”
A bride so unconcerned with herself that she wears a wedding dress that reveals a rash she doesn’t go running to a doctor to fix? Who comes only when the rash might affect someone else?
What is the matter with Jeralyn? Doesn’t she know she’s a millennial?
**********************
Kidding aside, wouldn’t it be nice if everyone acted like Jeralyn, and acted in a measured manner, appropriate to the concerns and circumstances? Wouldn’t it be nice if no one acted like Brendan?
That’s not how it is, though, is it? As professionals who deal with the public (as patients, coworkers, employees), we take all comers and roll with them: tolerate the annoyances and celebrate the (much larger, if less individually memorable) group who are pleasant, often delightful, sometimes inspiring.
And every once in a while, someone like Jeralyn comes along, upending all those negative expectations and reminding us that even if you can never really figure people out, it’s still worth the effort to keep trying.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].